
プリプレグ及びこれを硬化した繊維強化複合材料
【課題】いわゆる部分含浸プリプレグの欠点である、マトリクス樹脂本来の耐熱性が成形品において得られないという不具合を解決する。
【解決手段】エポキシ樹脂(A)、硬化剤として塩化ホウ素アミン錯体(B)、繊維基材(C)からなるプリプレグであり、エポキシ樹脂(A)中に含まれるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B)中のホウ素のモル数比が4〜7モル%であること、エポキシ樹脂(A)が(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラック型エポキシ樹脂(3)であることが好ましい。
【解決手段】エポキシ樹脂(A)、硬化剤として塩化ホウ素アミン錯体(B)、繊維基材(C)からなるプリプレグであり、エポキシ樹脂(A)中に含まれるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B)中のホウ素のモル数比が4〜7モル%であること、エポキシ樹脂(A)が(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラック型エポキシ樹脂(3)であることが好ましい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特にオーブン成形において優れた耐熱性を有する繊維強化複合材料が得られるプリプレグ及びこれを硬化してなる繊維強化複合材に関する。
【背景技術】
【0002】
繊維強化複合材料は、軽量かつ高強度で高剛性の特徴を生かし、スポーツ・レジャー用途から自動車や航空機等の産業用途まで、幅広く用いられている。特に近年では、より軽量でかつより高強度・高剛性の炭素繊維強化複合材料が産業用途に用いられることが多くなってきた。
【0003】
産業用途の中でも列車や航空機の機体などの構造部材に用いられる炭素繊維強化複合材料は、プリプレグを中間材料として用い、オートクレーブ成形で製造されることが一般的である。これはオートクレーブを用いて高圧下で成形することにより、成形体中のボイドを低減し、成形体の特性を期待された通りに発現させることを目的としている。
【0004】
しかしながら、オートクレーブの設備は非常に高価なため、新規に導入することは困難であるばかりでなく、一旦導入するとそのオートクレーブの大きさにより成形体の大きさが制限され、それより大きな成形体の製造が事実上不可能となる。このような問題に対し、脱オートクレーブ、低コスト成形の開発が盛んに行われており、その代表的なものとしては、真空、大気圧のみの低圧下で成形する、オーブン成形(または真空バッグ成形などとも呼ばれる)やRFI成形がある。オーブン成形やRFI成形は大気圧以外に圧力を加えないので、オートクレーブのようなしっかりした耐圧力容器でなくても良く、温度さえ上げることができる炉(オーブン)があれば成形でき、断熱ボードと熱風ヒーターといった簡便な設備でも成形可能である。ただし圧力を加えないので、成形体中にボイドが残りやすく、成形体はオートクレーブでの成形体に比べて強度が低い、あるいは表面にピンホールが発生するという問題があった。
【0005】
そこで、特許文献1には、部分含浸させたプリプレグを使用しボイドの少ない成形品が
例示されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特表2003−513110号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
しかしながら、この様な部分含浸プリプレグを用いると、マトリクス樹脂本来の耐熱性が、成形品において得られないという問題点がある。マトリクス樹脂単体の硬化物のガラス転移点温度(Tg)よりも、成形品のTgが低くなってしまう。低コスト化への要求から選択される強化繊維の目付が高いプリプレグにおいては、この問題点が顕著に現れる。
【0008】
本発明の課題は、成形法に左右されること無く本来の樹脂の耐熱性を発現するプリプレグを提供することである。
【課題を解決するための手段】
【0009】
本発明者は鋭意検討した結果、プリプレグにおいて硬化剤として塩化ホウ素アミン錯体を用いることで課題を解決できることを見出し本発明に至った。
【0010】
すなわち、本発明の要旨は、エポキシ樹脂(A)、硬化剤として塩化ホウ素アミン錯体(B)、繊維基材(C)からなるプリプレグである。
【0011】
エポキシ樹脂(A)中に含まれるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B)中のホウ素のモル数比が4〜7モル%であることが好ましい。エポキシ樹脂(A)が(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラック型エポキシ樹脂(3)であることが好ましい。
【0012】
【化3】

【0013】
【化4】
【発明の効果】
【0014】
本発明は、成形方法によらず所望の耐熱性を発現するプリプレグを提供し、繊維強化複合材料の成形コストあるいは成形設備投資を削減でき、有用である。
【図面の簡単な説明】
【0015】
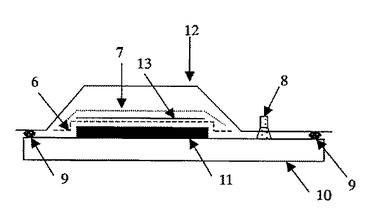
【図1】本発明の繊維強化複合材料のパネル製造方法を示す図でバギングの構成を示した断面図である。
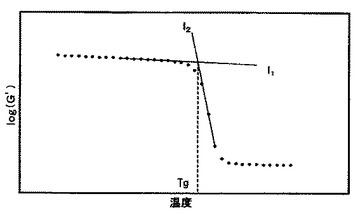
【図2】硬化物のlogG´の転移する前の平坦領域の近似直線とlogG´が転移する領域の近似直線との交点からG´−Tgを求めるときの一例を示す。
【発明を実施するための形態】
【0016】
「エポキシ樹脂(A)」
本発明のエポキシ樹脂(A)として用いるエポキシ樹脂は、公知の各種のものが使用でき、その分子中にエポキシ基を少なくとも2個有するものであれば分子構造、分子量等に特に制限はない。例えばビスフェノール型、フェノールノボラック型、クレゾールノボラック型、ジシクロペンタジエン型、ナフタレン型、ビフェニル型、オキサゾリドン型などの各種エポキシ樹脂を単独または2種以上併用して用いることができる。また、必要に応じて、単官能エポキシ樹脂、ビニル重合性樹脂、フェノール樹脂、ビスマレイミド樹脂、BT樹脂、シアネートエステル樹脂、ビニルエステル樹脂、ベンゾオキサジン樹脂、不飽和ポリエステル樹脂等配合することができる。好ましいエポキシ樹脂としてはオキサゾリドン型、ビスフェノール型、フェノールノボラック型である。
【0017】
エポキシ樹脂の組み合わせとして(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)とビスフェノール型エポキシ樹脂(2)と(II)で表されるフェノールノボラック型エポキシ樹脂(3)の組み合わせが好ましい。
【0018】
(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)は工業的に入手でき、例えば、旭化成ケミカルズ株式会社からAER4152やXAC4151として入手できる。
【0019】
ビスフェノール型エポキシ樹脂(2)はビスフェノールA型、ビスフェノールF型、ビスフェノールS型等が挙げられる。好ましくはビスフェノールA型、ビスフェノールS型である。これらは工業的にたやすく入手可能である。
【0020】
(II)で表されるフェノールノボラック型エポキシ樹脂(3)は工業的に入手でき、例えば、日本化薬株式会社からEPPN−501H、EPPN−502HY、EPPN−502H等が、ジャパンエポキシレジン株式会社からjER1032H60や、ハンツマンからTactix742等が挙げられる。好ましいエポキシ当量は150〜180g/eqである。
【0021】
(I)のオキサゾリドン環構造を有する二官能エポキシ樹脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラック型エポキシ樹脂(3)の合計100質量%に対し、(I)のオキサゾリドン環構造を有する二官能エポキシ樹脂(1)が20〜40質量%、ビスフェノール型エポキシ樹脂(2)が45〜65質量%、(II)で表されるフェノールノボラック型エポキシ樹脂(3)が5〜25質量%の配合比が好ましい。
【0022】
(I)のオキサゾリドン環構造を有する二官能エポキシ樹脂(1)が20質量%未満の場合、繊維強化複合材料での機械特性が低下し、40質量%を超えると繊維強化複合材料での耐熱性が低下する。更に好ましくは25〜35質量%である。また、ビスフェノール型エポキシ樹脂(2)が45質量%以上とすれば、プリプレグのドレープ性が保たれ、65質量%以下であれば、硬化樹脂の架橋密度が低くなりすぎず耐熱性を損なわない。更に好ましくは50〜60質量%である。ビスフェノール型エポキシ樹脂(2)をビスフェノールA型とビスフェノールS型の組み合わせにすることでプリプレグのドレープ性と繊維強化複合材料の耐熱性が高度にバランスがとれさらに好ましい。(II)で表されるフェノールノボラック型エポキシ樹脂(3)が5質量%以上であれば、硬化樹脂の架橋密度が低くなりすぎず、耐熱性が損なわれない、25質量%以下であれば繊維強化複合材料での機械特性が損なわれない。更に好ましくは10〜20質量%である。
【0023】
本発明のプリプレグは硬化剤として塩化ホウ素アミン錯体(B)を用いる。塩化ホウ素アミン錯体(B)を用いることでプリプレグの製造方法や形態、繊維強化複合材料の成形方法に影響されずマトリクス樹脂本来の耐熱性を発揮するプリプレグを提供することができる。塩化ホウ素アミン錯体(B)は工業的に入手できる。好ましい配合量はエポキシ樹脂(A)のエポキシ当量から計算されるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B)中のホウ素原子のモル数比が4〜7モル%となる配合量である。この範囲でであれば耐熱性の発現性に優れ好ましい。
【0024】
本発明のエポキシ樹脂(A)と硬化剤として塩化ホウ素アミン錯体(B)からなるマトリクス樹脂には熱可塑性樹脂が配合されても良い。ポリアミド、ポリエステル、ポリカーボネート、ポリエーテルスルフォン、ポリフェニレンエーテル、ポリフェニレンスルフィド、ポリエーテルエーテルケトン、ポリイミド、ポリテトラフルオロエチレン、ポリエーテル、ポリオレフィン、液晶ポリマー、ポリアリレート、ポリスルフォン、ポリアクリロニトリルスチレン、ポリスチレン、ポリアクリロニトリル、ポリメタクリレート、ABS、AES、ASA、ポリ塩化ビニル、ポリビニルホルマール、フェノキシ樹脂等が挙げられる。好ましい熱可塑性樹脂はポリビニルホルマール、フェノキシ樹脂、ポリエーテルスルフォン等が挙げられる。更に好ましくはフェノキシ樹脂である。本発明のエポキシ樹脂(A)と硬化剤として塩化ホウ素アミン錯体(B)からなるマトリクス樹脂には必要に応じて公知の様々な添加剤を併用することができる。例えば、種々の硬化促進剤、シリコーンオイル、天然ワックス類、合成ワックス類、直鎖脂肪酸の金属塩、酸アミド、エステル類、パラフィン類等の離型剤、結晶質シリカ、溶融シリカ、ケイ酸カルシウム、アルミナ、炭酸カルシウム、タルク、硫酸バリウム等の粉体やガラス繊維、炭素繊維等の無機充填剤、塩素化パラフィン、ブロムトルエン、ヘキサブロムベンゼン、三酸化アンチモン等の難燃剤、カーボンブラック、ベンガラ等の着色剤、シランカップリング剤等を使用することができる。
【0025】
「繊維基材(C)」
本発明の繊維基材(C)は一般に繊維強化複合材料として用いられる強化繊維を用いることができる。炭素繊維、黒鉛繊維、アラミド繊維、炭化珪素繊維、アルミナ繊維、ボロン繊維、高強度ポリエチレン繊維、タングステンカーバイド繊維、PBO繊維、ガラス繊維等などが挙げられ、これらを単独で、または2種以上を組合して用いてもかまわない。好ましくは炭素繊維である。繊維基材(C)は、そのままのトウの形態で、強化繊維トウを一方向に引き揃えた一方向材の形態で、製織した織物の形態で、短く裁断した強化繊維からなる不織布の形態などで使用される。織物の場合は、平織、綾織、朱子織、若しくはノンクリンプファブリックに代表される繊維束を一方向に引き揃えたシートや角度を変えて積層したようなシートをほぐれないようにステッチしたステッチングシート等が例示できる。得られる繊維強化複合材料の機械特性が優れるため一方向材が好ましい。取り扱い性からは織物が好ましい。本発明のプリプレグは繊維目付けに制限はないが、繊維目付けが大きいほどその性能優位性を発揮できる。
【0026】
本発明のプリプレグの製造方法に特段の制限はない。一般的な方法で製造できる。マトリクス樹脂はガラスフラスコ、ニーダー、プラネタリーミキサー、一般的な撹拌加熱釜、攪拌加圧加熱釜等で調製ができる。マトリクス樹脂の繊維基材への付与方法としてホットメルトフィルム法、ラッカー法等が挙げられる。
【0027】
本発明のプリプレグの成形方法、つまり、本発明の繊維強化複合材料の製造方法への特段の制限はない。オートクレーブ成形法、オーブン成形法、プレス成形法、連続プレス成形法、引き抜き成形法、内圧成形法等一般的な成形方法が適用できる。好ましい硬化温度は130℃〜200℃である。更に好ましくは145℃〜185である。
【0028】
以下、実施例により本発明を説明するが、これにより本発明が何らかの制限を受けるものではない。
【0029】
樹脂組成物の原材料および繊維材料を表1に示した。
【0030】
【表1】
【0031】
樹脂組成物Aの調製
jER828とYP−70を160℃で溶解させマスターバッチを調製した。このマスターバッチを用い表2の組成でDY9577以外の成分を120℃で混合した。これを60℃にしDY9577を所定量添加混合し樹脂組成物Aを調製した。
【0032】
樹脂組成物Bの調製
jER828とYP−70を160℃で溶解させマスターバッチを調製した。また、jER828とDicy15及びDCMUを3本ロールミルで均一に分散させてマスターバッチを調製した。YP−70マスターバッチを用い、表2の組成でDicy15/DCMUマスターバッチ以外の成分を100℃で混合した。これを60℃にしDicy15/DCMUマスターバッチを所定量添加混合し樹脂組成物Bを調製した。
【0033】
樹脂組成物Cの調製
jER828とYP−70を160℃で溶解させマスターバッチを調製した。また、jER828と2MA−OK及びL−07Nを3本ロールミルで均一に分散させてマスターバッチを調製した。YP−70マスターバッチを用い、表2の組成で2MA−OK/L−07Nマスターバッチ以外の成分を100℃で混合した。これを60℃にし2MA−OK/L−07Nマスターバッチを所定量添加混合し樹脂組成物Cを調製した。
【0034】
DMAによるTgの測定
樹脂板および繊維強化複合材料のパネルを試験片(長さ50mm×幅12mm、樹脂板は厚み2mm、繊維強化複合材料は厚み2.8mm)に加工した。繊維強化複合材料のパネルの場合は長さ方向と繊維経糸方向が一致するように試験片を加工した。測定装置はレオメトリクス社製レオメーターRDA700またはTAインスツルメント社製ARES−RDAを使用した。測定周波数は1Hz、RDA700の昇温速度は5℃ステップ昇温で、ARES−RDAは5℃/min昇温で測定した。logG´を温度に対してプロットし、logG´の転移する前の平坦領域の近似直線とG´が転移する領域の近似直線との交点から求まる温度をG´−Tgとして記録した。また、tanδを温度に対してプロットし、tanδの極大を示す温度をtanδmaxとして記録した。測定装置差として、RDA700によるG´−Tg及びtanδmaxの測定値は、ARES−RDAによる測定値よりも1.05倍高い値となる。
【0035】
DSCによるTgの測定
TAインスツルメント社製Q100を用いサンプルをアルミパンに入れ、昇温速度10℃/分にて測定した。
【0036】
コンポジット層間せん断強度の測定
繊維強化複合材料の層間せん断強度は、繊維強化複合材料から、試験片の長手方向に対して補強繊維の経糸方向が0°に配向するように試験片(長さ25mm×幅6.3mm×厚さ2.8mm)を切り出し、該試験片について、3点曲げ治具(圧子3.2mmR、サポート1.6mmR、サポート間距離 試験片厚さの4倍、クロスヘッドスピード サポート間距離の2乗×0.01/6×試験片の厚み)を設置したインストロン社製の万能試験機を用い、層間せん断特性を測定した。
【0037】
コンポジット曲げ強度
繊維強化複合材料から、試験片の長手方向に対して補強繊維の経糸方向が0゜に配向するように試験片(長さ130mm×幅12mm×厚さ2.8mm)を切り出し、該試験片について、3点曲げ治具(圧子、サポートとも3.2mmR、サポート間距離 試験片厚さの40倍、クロスヘッドスピード サポート間距離の2乗×0.01/6×試験片の厚み)を設置したインストロン社製の万能試験機を用い、曲げ特性を測定した。
【実施例】
【0038】
<実施例1>
【0039】
繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Aをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に貼り合わせ、温度40℃、圧力0.05MPa、送り速度1.6m/分の条件でフュージングプレス(アサヒ繊維機械工業(株)、JR−600S、処理長1340mm、圧力はシリンダー圧)を通しプリプレグ1を得た。プリプレグの樹脂含有率は45質量%であった。得られたプリプレグをカットし断面を目視観察すると内部に樹脂が含浸していない部分が観察された。得られたプレプレグを、経糸方向を揃えて、積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて4時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら150℃で2時間加熱硬化させてパネルを得た。
【0040】
得られたパネルより試験片を切り出しDMAによる測定をした。結果を表2に示す。測定装置はRDA700を用いた。
【0041】
得られた樹脂組成物Aを60℃に加熱して脱泡した後、離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Aを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。
【0042】
得られた硬化樹脂板より試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例1>
【0043】
繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Bをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に手で貼り合わせプリプレグ2を得た。プリプレグの樹脂含有率は45質量%であった。得られたプリプレグをカットし断面を目視観察すると内部に樹脂が含浸していない部分が観察された。得られたプレプレグを積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて6時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら150℃で2時間加熱硬化させてパネルを得た。
【0044】
得られたパネルより試験片を切り出しDMAによる測定をした。結果を表2に示す。測定装置はRDA700を用いた。
【0045】
得られた樹脂組成物Bを60℃に加熱して脱泡した後、離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Bを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。
【0046】
得られた硬化樹脂板より試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例2>
【0047】
繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Bをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に貼り合わせ、温度100℃、圧力0.4MPa、送り速度1m/分の条件でフュージングプレス(アサヒ繊維機械工業(株)、JR−600S、処理長1340mm、圧力はシリンダー圧)を通しプリプレグ3を得た。得られたプリプレグをカットし断面を目視観察すると繊維基材の大部分に樹脂が含浸しているのが観察された。得られたプレプレグを積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて6時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら150℃で2時間加熱硬化させてパネルを得た。
【0048】
得られたパネルより試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例3>
【0049】
オーブンを使わずにオートクレーブを用い圧力0.3MPa、150℃で2時間加熱成形した以外は比較例1と同様に行った。
【0050】
得られたパネルより試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例4>
【0051】
樹脂組成物Bの代わりに樹脂組成物Cを用いた以外は比較例1と同様に行いプレプレグ4及びそのパネルを得た。
【0052】
得られたパネルより試験片を切り出しDMAによる測定をした。結果を表2に示す。得られた樹脂組成物Cを60℃に加熱して脱泡した後、離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Aを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。
【0053】
得られた硬化樹脂板より試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<実施例2>
【0054】
実施例1と同様にして得た樹脂組成物Aの硬化樹脂板からサンプルを取り出しDSCによるTgを測定した。結果を表3に示す。
<実施例3>
【0055】
jER828とDY9577を表3の組成で室温にて混合し樹脂組成物Dを得た。得られた樹脂組成物をアルミ皿内に置きオーブンにて150℃、2時間加熱硬化させ硬化樹脂を得た。得られた硬化樹脂からサンプルを取り出しDSCによるTgを測定した。結果を表3に示す。
<実施例4〜8>
【0056】
組成を変更する以外は実施例3と同様に行いDSCによるTgを測定した。結果を表3に示す。
<実施例9>
【0057】
jER828とjER1001及びDY9577を表3の組成で70℃にて混合及び脱泡を行い、樹脂組成物Jを得た。離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Jを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。得られた硬化樹脂からサンプルを取り出しDSCによるTgを測定した。結果を表3に示す。
<実施例10>
【0058】
組成を変更する以外は実施例9と同様に行いDSCによるTgを測定した。結果を表3に示す。
【0059】
表2、3に示した様に本発明のプリプレグはマトリクス樹脂本来のTgを示している。従来の技術では比較例1〜4に示した様にマトリクス樹脂本来のTgを発現できていない。
【0060】
【表2】
【表3】
<実施例11>
【0061】
組成を変更する以外は樹脂組成物Aの調製と同様に行い、樹脂組成物Lを得た。繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Lをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に手で貼り合わせプリプレグ5を得た。プリプレグの樹脂含有率は45質量%であった。得られたプリプレグをカットし断面を目視観察すると内部に樹脂が含浸していない部分が観察された。得られたプレプレグを積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて12時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら95℃で1時間加熱硬化後、150℃で2時間加熱硬化させてパネルを得た。
【0062】
得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
<実施例12>
【0063】
組成を変更する以外は実施例11と同様に行い、樹脂組成物Mからプリプレグ6およびパネルを得た。得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
<実施例13>
【0064】
組成を変更する以外は実施例11と同様に行い、樹脂組成物Nからプリプレグ7およびパネルを得た。得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
<実施例14>
【0065】
組成を変更する以外は実施例11と同様に行い、樹脂組成物Oからプリプレグ8およびパネルを得た。得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
【0066】
表4に示した様に、本発明のプリプレグはマトリクス樹脂本来のG´−Tgを示している。
【0067】
【表4】
【符号の説明】
【0068】
1 繊維基材
2 樹脂が含浸されていない繊維基材
3 樹脂が含浸された繊維基材
4,5 樹脂フィルム
6 離型フィルム
7 不織布
8 真空引き口
9 シーラント
10 ツール
11 積層体
12 ナイロンバッグフィルム
13 プレッシャープレート
【技術分野】
【0001】
本発明は、特にオーブン成形において優れた耐熱性を有する繊維強化複合材料が得られるプリプレグ及びこれを硬化してなる繊維強化複合材に関する。
【背景技術】
【0002】
繊維強化複合材料は、軽量かつ高強度で高剛性の特徴を生かし、スポーツ・レジャー用途から自動車や航空機等の産業用途まで、幅広く用いられている。特に近年では、より軽量でかつより高強度・高剛性の炭素繊維強化複合材料が産業用途に用いられることが多くなってきた。
【0003】
産業用途の中でも列車や航空機の機体などの構造部材に用いられる炭素繊維強化複合材料は、プリプレグを中間材料として用い、オートクレーブ成形で製造されることが一般的である。これはオートクレーブを用いて高圧下で成形することにより、成形体中のボイドを低減し、成形体の特性を期待された通りに発現させることを目的としている。
【0004】
しかしながら、オートクレーブの設備は非常に高価なため、新規に導入することは困難であるばかりでなく、一旦導入するとそのオートクレーブの大きさにより成形体の大きさが制限され、それより大きな成形体の製造が事実上不可能となる。このような問題に対し、脱オートクレーブ、低コスト成形の開発が盛んに行われており、その代表的なものとしては、真空、大気圧のみの低圧下で成形する、オーブン成形(または真空バッグ成形などとも呼ばれる)やRFI成形がある。オーブン成形やRFI成形は大気圧以外に圧力を加えないので、オートクレーブのようなしっかりした耐圧力容器でなくても良く、温度さえ上げることができる炉(オーブン)があれば成形でき、断熱ボードと熱風ヒーターといった簡便な設備でも成形可能である。ただし圧力を加えないので、成形体中にボイドが残りやすく、成形体はオートクレーブでの成形体に比べて強度が低い、あるいは表面にピンホールが発生するという問題があった。
【0005】
そこで、特許文献1には、部分含浸させたプリプレグを使用しボイドの少ない成形品が
例示されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特表2003−513110号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
しかしながら、この様な部分含浸プリプレグを用いると、マトリクス樹脂本来の耐熱性が、成形品において得られないという問題点がある。マトリクス樹脂単体の硬化物のガラス転移点温度(Tg)よりも、成形品のTgが低くなってしまう。低コスト化への要求から選択される強化繊維の目付が高いプリプレグにおいては、この問題点が顕著に現れる。
【0008】
本発明の課題は、成形法に左右されること無く本来の樹脂の耐熱性を発現するプリプレグを提供することである。
【課題を解決するための手段】
【0009】
本発明者は鋭意検討した結果、プリプレグにおいて硬化剤として塩化ホウ素アミン錯体を用いることで課題を解決できることを見出し本発明に至った。
【0010】
すなわち、本発明の要旨は、エポキシ樹脂(A)、硬化剤として塩化ホウ素アミン錯体(B)、繊維基材(C)からなるプリプレグである。
【0011】
エポキシ樹脂(A)中に含まれるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B)中のホウ素のモル数比が4〜7モル%であることが好ましい。エポキシ樹脂(A)が(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラック型エポキシ樹脂(3)であることが好ましい。
【0012】
【化3】
【0013】
【化4】
【発明の効果】
【0014】
本発明は、成形方法によらず所望の耐熱性を発現するプリプレグを提供し、繊維強化複合材料の成形コストあるいは成形設備投資を削減でき、有用である。
【図面の簡単な説明】
【0015】
【図1】本発明の繊維強化複合材料のパネル製造方法を示す図でバギングの構成を示した断面図である。
【図2】硬化物のlogG´の転移する前の平坦領域の近似直線とlogG´が転移する領域の近似直線との交点からG´−Tgを求めるときの一例を示す。
【発明を実施するための形態】
【0016】
「エポキシ樹脂(A)」
本発明のエポキシ樹脂(A)として用いるエポキシ樹脂は、公知の各種のものが使用でき、その分子中にエポキシ基を少なくとも2個有するものであれば分子構造、分子量等に特に制限はない。例えばビスフェノール型、フェノールノボラック型、クレゾールノボラック型、ジシクロペンタジエン型、ナフタレン型、ビフェニル型、オキサゾリドン型などの各種エポキシ樹脂を単独または2種以上併用して用いることができる。また、必要に応じて、単官能エポキシ樹脂、ビニル重合性樹脂、フェノール樹脂、ビスマレイミド樹脂、BT樹脂、シアネートエステル樹脂、ビニルエステル樹脂、ベンゾオキサジン樹脂、不飽和ポリエステル樹脂等配合することができる。好ましいエポキシ樹脂としてはオキサゾリドン型、ビスフェノール型、フェノールノボラック型である。
【0017】
エポキシ樹脂の組み合わせとして(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)とビスフェノール型エポキシ樹脂(2)と(II)で表されるフェノールノボラック型エポキシ樹脂(3)の組み合わせが好ましい。
【0018】
(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹脂(1)は工業的に入手でき、例えば、旭化成ケミカルズ株式会社からAER4152やXAC4151として入手できる。
【0019】
ビスフェノール型エポキシ樹脂(2)はビスフェノールA型、ビスフェノールF型、ビスフェノールS型等が挙げられる。好ましくはビスフェノールA型、ビスフェノールS型である。これらは工業的にたやすく入手可能である。
【0020】
(II)で表されるフェノールノボラック型エポキシ樹脂(3)は工業的に入手でき、例えば、日本化薬株式会社からEPPN−501H、EPPN−502HY、EPPN−502H等が、ジャパンエポキシレジン株式会社からjER1032H60や、ハンツマンからTactix742等が挙げられる。好ましいエポキシ当量は150〜180g/eqである。
【0021】
(I)のオキサゾリドン環構造を有する二官能エポキシ樹脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラック型エポキシ樹脂(3)の合計100質量%に対し、(I)のオキサゾリドン環構造を有する二官能エポキシ樹脂(1)が20〜40質量%、ビスフェノール型エポキシ樹脂(2)が45〜65質量%、(II)で表されるフェノールノボラック型エポキシ樹脂(3)が5〜25質量%の配合比が好ましい。
【0022】
(I)のオキサゾリドン環構造を有する二官能エポキシ樹脂(1)が20質量%未満の場合、繊維強化複合材料での機械特性が低下し、40質量%を超えると繊維強化複合材料での耐熱性が低下する。更に好ましくは25〜35質量%である。また、ビスフェノール型エポキシ樹脂(2)が45質量%以上とすれば、プリプレグのドレープ性が保たれ、65質量%以下であれば、硬化樹脂の架橋密度が低くなりすぎず耐熱性を損なわない。更に好ましくは50〜60質量%である。ビスフェノール型エポキシ樹脂(2)をビスフェノールA型とビスフェノールS型の組み合わせにすることでプリプレグのドレープ性と繊維強化複合材料の耐熱性が高度にバランスがとれさらに好ましい。(II)で表されるフェノールノボラック型エポキシ樹脂(3)が5質量%以上であれば、硬化樹脂の架橋密度が低くなりすぎず、耐熱性が損なわれない、25質量%以下であれば繊維強化複合材料での機械特性が損なわれない。更に好ましくは10〜20質量%である。
【0023】
本発明のプリプレグは硬化剤として塩化ホウ素アミン錯体(B)を用いる。塩化ホウ素アミン錯体(B)を用いることでプリプレグの製造方法や形態、繊維強化複合材料の成形方法に影響されずマトリクス樹脂本来の耐熱性を発揮するプリプレグを提供することができる。塩化ホウ素アミン錯体(B)は工業的に入手できる。好ましい配合量はエポキシ樹脂(A)のエポキシ当量から計算されるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B)中のホウ素原子のモル数比が4〜7モル%となる配合量である。この範囲でであれば耐熱性の発現性に優れ好ましい。
【0024】
本発明のエポキシ樹脂(A)と硬化剤として塩化ホウ素アミン錯体(B)からなるマトリクス樹脂には熱可塑性樹脂が配合されても良い。ポリアミド、ポリエステル、ポリカーボネート、ポリエーテルスルフォン、ポリフェニレンエーテル、ポリフェニレンスルフィド、ポリエーテルエーテルケトン、ポリイミド、ポリテトラフルオロエチレン、ポリエーテル、ポリオレフィン、液晶ポリマー、ポリアリレート、ポリスルフォン、ポリアクリロニトリルスチレン、ポリスチレン、ポリアクリロニトリル、ポリメタクリレート、ABS、AES、ASA、ポリ塩化ビニル、ポリビニルホルマール、フェノキシ樹脂等が挙げられる。好ましい熱可塑性樹脂はポリビニルホルマール、フェノキシ樹脂、ポリエーテルスルフォン等が挙げられる。更に好ましくはフェノキシ樹脂である。本発明のエポキシ樹脂(A)と硬化剤として塩化ホウ素アミン錯体(B)からなるマトリクス樹脂には必要に応じて公知の様々な添加剤を併用することができる。例えば、種々の硬化促進剤、シリコーンオイル、天然ワックス類、合成ワックス類、直鎖脂肪酸の金属塩、酸アミド、エステル類、パラフィン類等の離型剤、結晶質シリカ、溶融シリカ、ケイ酸カルシウム、アルミナ、炭酸カルシウム、タルク、硫酸バリウム等の粉体やガラス繊維、炭素繊維等の無機充填剤、塩素化パラフィン、ブロムトルエン、ヘキサブロムベンゼン、三酸化アンチモン等の難燃剤、カーボンブラック、ベンガラ等の着色剤、シランカップリング剤等を使用することができる。
【0025】
「繊維基材(C)」
本発明の繊維基材(C)は一般に繊維強化複合材料として用いられる強化繊維を用いることができる。炭素繊維、黒鉛繊維、アラミド繊維、炭化珪素繊維、アルミナ繊維、ボロン繊維、高強度ポリエチレン繊維、タングステンカーバイド繊維、PBO繊維、ガラス繊維等などが挙げられ、これらを単独で、または2種以上を組合して用いてもかまわない。好ましくは炭素繊維である。繊維基材(C)は、そのままのトウの形態で、強化繊維トウを一方向に引き揃えた一方向材の形態で、製織した織物の形態で、短く裁断した強化繊維からなる不織布の形態などで使用される。織物の場合は、平織、綾織、朱子織、若しくはノンクリンプファブリックに代表される繊維束を一方向に引き揃えたシートや角度を変えて積層したようなシートをほぐれないようにステッチしたステッチングシート等が例示できる。得られる繊維強化複合材料の機械特性が優れるため一方向材が好ましい。取り扱い性からは織物が好ましい。本発明のプリプレグは繊維目付けに制限はないが、繊維目付けが大きいほどその性能優位性を発揮できる。
【0026】
本発明のプリプレグの製造方法に特段の制限はない。一般的な方法で製造できる。マトリクス樹脂はガラスフラスコ、ニーダー、プラネタリーミキサー、一般的な撹拌加熱釜、攪拌加圧加熱釜等で調製ができる。マトリクス樹脂の繊維基材への付与方法としてホットメルトフィルム法、ラッカー法等が挙げられる。
【0027】
本発明のプリプレグの成形方法、つまり、本発明の繊維強化複合材料の製造方法への特段の制限はない。オートクレーブ成形法、オーブン成形法、プレス成形法、連続プレス成形法、引き抜き成形法、内圧成形法等一般的な成形方法が適用できる。好ましい硬化温度は130℃〜200℃である。更に好ましくは145℃〜185である。
【0028】
以下、実施例により本発明を説明するが、これにより本発明が何らかの制限を受けるものではない。
【0029】
樹脂組成物の原材料および繊維材料を表1に示した。
【0030】
【表1】
【0031】
樹脂組成物Aの調製
jER828とYP−70を160℃で溶解させマスターバッチを調製した。このマスターバッチを用い表2の組成でDY9577以外の成分を120℃で混合した。これを60℃にしDY9577を所定量添加混合し樹脂組成物Aを調製した。
【0032】
樹脂組成物Bの調製
jER828とYP−70を160℃で溶解させマスターバッチを調製した。また、jER828とDicy15及びDCMUを3本ロールミルで均一に分散させてマスターバッチを調製した。YP−70マスターバッチを用い、表2の組成でDicy15/DCMUマスターバッチ以外の成分を100℃で混合した。これを60℃にしDicy15/DCMUマスターバッチを所定量添加混合し樹脂組成物Bを調製した。
【0033】
樹脂組成物Cの調製
jER828とYP−70を160℃で溶解させマスターバッチを調製した。また、jER828と2MA−OK及びL−07Nを3本ロールミルで均一に分散させてマスターバッチを調製した。YP−70マスターバッチを用い、表2の組成で2MA−OK/L−07Nマスターバッチ以外の成分を100℃で混合した。これを60℃にし2MA−OK/L−07Nマスターバッチを所定量添加混合し樹脂組成物Cを調製した。
【0034】
DMAによるTgの測定
樹脂板および繊維強化複合材料のパネルを試験片(長さ50mm×幅12mm、樹脂板は厚み2mm、繊維強化複合材料は厚み2.8mm)に加工した。繊維強化複合材料のパネルの場合は長さ方向と繊維経糸方向が一致するように試験片を加工した。測定装置はレオメトリクス社製レオメーターRDA700またはTAインスツルメント社製ARES−RDAを使用した。測定周波数は1Hz、RDA700の昇温速度は5℃ステップ昇温で、ARES−RDAは5℃/min昇温で測定した。logG´を温度に対してプロットし、logG´の転移する前の平坦領域の近似直線とG´が転移する領域の近似直線との交点から求まる温度をG´−Tgとして記録した。また、tanδを温度に対してプロットし、tanδの極大を示す温度をtanδmaxとして記録した。測定装置差として、RDA700によるG´−Tg及びtanδmaxの測定値は、ARES−RDAによる測定値よりも1.05倍高い値となる。
【0035】
DSCによるTgの測定
TAインスツルメント社製Q100を用いサンプルをアルミパンに入れ、昇温速度10℃/分にて測定した。
【0036】
コンポジット層間せん断強度の測定
繊維強化複合材料の層間せん断強度は、繊維強化複合材料から、試験片の長手方向に対して補強繊維の経糸方向が0°に配向するように試験片(長さ25mm×幅6.3mm×厚さ2.8mm)を切り出し、該試験片について、3点曲げ治具(圧子3.2mmR、サポート1.6mmR、サポート間距離 試験片厚さの4倍、クロスヘッドスピード サポート間距離の2乗×0.01/6×試験片の厚み)を設置したインストロン社製の万能試験機を用い、層間せん断特性を測定した。
【0037】
コンポジット曲げ強度
繊維強化複合材料から、試験片の長手方向に対して補強繊維の経糸方向が0゜に配向するように試験片(長さ130mm×幅12mm×厚さ2.8mm)を切り出し、該試験片について、3点曲げ治具(圧子、サポートとも3.2mmR、サポート間距離 試験片厚さの40倍、クロスヘッドスピード サポート間距離の2乗×0.01/6×試験片の厚み)を設置したインストロン社製の万能試験機を用い、曲げ特性を測定した。
【実施例】
【0038】
<実施例1>
【0039】
繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Aをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に貼り合わせ、温度40℃、圧力0.05MPa、送り速度1.6m/分の条件でフュージングプレス(アサヒ繊維機械工業(株)、JR−600S、処理長1340mm、圧力はシリンダー圧)を通しプリプレグ1を得た。プリプレグの樹脂含有率は45質量%であった。得られたプリプレグをカットし断面を目視観察すると内部に樹脂が含浸していない部分が観察された。得られたプレプレグを、経糸方向を揃えて、積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて4時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら150℃で2時間加熱硬化させてパネルを得た。
【0040】
得られたパネルより試験片を切り出しDMAによる測定をした。結果を表2に示す。測定装置はRDA700を用いた。
【0041】
得られた樹脂組成物Aを60℃に加熱して脱泡した後、離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Aを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。
【0042】
得られた硬化樹脂板より試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例1>
【0043】
繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Bをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に手で貼り合わせプリプレグ2を得た。プリプレグの樹脂含有率は45質量%であった。得られたプリプレグをカットし断面を目視観察すると内部に樹脂が含浸していない部分が観察された。得られたプレプレグを積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて6時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら150℃で2時間加熱硬化させてパネルを得た。
【0044】
得られたパネルより試験片を切り出しDMAによる測定をした。結果を表2に示す。測定装置はRDA700を用いた。
【0045】
得られた樹脂組成物Bを60℃に加熱して脱泡した後、離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Bを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。
【0046】
得られた硬化樹脂板より試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例2>
【0047】
繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Bをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に貼り合わせ、温度100℃、圧力0.4MPa、送り速度1m/分の条件でフュージングプレス(アサヒ繊維機械工業(株)、JR−600S、処理長1340mm、圧力はシリンダー圧)を通しプリプレグ3を得た。得られたプリプレグをカットし断面を目視観察すると繊維基材の大部分に樹脂が含浸しているのが観察された。得られたプレプレグを積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて6時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら150℃で2時間加熱硬化させてパネルを得た。
【0048】
得られたパネルより試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例3>
【0049】
オーブンを使わずにオートクレーブを用い圧力0.3MPa、150℃で2時間加熱成形した以外は比較例1と同様に行った。
【0050】
得られたパネルより試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<比較例4>
【0051】
樹脂組成物Bの代わりに樹脂組成物Cを用いた以外は比較例1と同様に行いプレプレグ4及びそのパネルを得た。
【0052】
得られたパネルより試験片を切り出しDMAによる測定をした。結果を表2に示す。得られた樹脂組成物Cを60℃に加熱して脱泡した後、離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Aを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。
【0053】
得られた硬化樹脂板より試験片を切り出しDMAによるTgの測定をした。結果を表2に示す。測定装置はRDA700を用いた。
<実施例2>
【0054】
実施例1と同様にして得た樹脂組成物Aの硬化樹脂板からサンプルを取り出しDSCによるTgを測定した。結果を表3に示す。
<実施例3>
【0055】
jER828とDY9577を表3の組成で室温にて混合し樹脂組成物Dを得た。得られた樹脂組成物をアルミ皿内に置きオーブンにて150℃、2時間加熱硬化させ硬化樹脂を得た。得られた硬化樹脂からサンプルを取り出しDSCによるTgを測定した。結果を表3に示す。
<実施例4〜8>
【0056】
組成を変更する以外は実施例3と同様に行いDSCによるTgを測定した。結果を表3に示す。
<実施例9>
【0057】
jER828とjER1001及びDY9577を表3の組成で70℃にて混合及び脱泡を行い、樹脂組成物Jを得た。離型処理を施してある2枚のガラス板で2mm厚のスペーサーを介して樹脂組成物Jを挟みオーブンにて150℃、2時間加熱硬化させ2mm厚の硬化樹脂板を得た。得られた硬化樹脂からサンプルを取り出しDSCによるTgを測定した。結果を表3に示す。
<実施例10>
【0058】
組成を変更する以外は実施例9と同様に行いDSCによるTgを測定した。結果を表3に示す。
【0059】
表2、3に示した様に本発明のプリプレグはマトリクス樹脂本来のTgを示している。従来の技術では比較例1〜4に示した様にマトリクス樹脂本来のTgを発現できていない。
【0060】
【表2】
【表3】
<実施例11>
【0061】
組成を変更する以外は樹脂組成物Aの調製と同様に行い、樹脂組成物Lを得た。繊維基材として、三菱レイヨン株式会社製のTRK510を用意した。プリプレグの樹脂含有率が45質量%となるように樹脂フィルム目付けを設定し60℃の条件で樹脂組成物Lをフィルムコーターにて離型紙に塗布し樹脂フィルムを得た。得られた樹脂フィルムを繊維基材の両面に手で貼り合わせプリプレグ5を得た。プリプレグの樹脂含有率は45質量%であった。得られたプリプレグをカットし断面を目視観察すると内部に樹脂が含浸していない部分が観察された。得られたプレプレグを積層し積層体を図1に示した構成でバギングを行った。更に引き口に真空ポンプを接続させて室温にて12時間予備脱気させた。オーブン内にバギングした積層体を入れて引き口に真空ポンプを接続させて脱気しながら95℃で1時間加熱硬化後、150℃で2時間加熱硬化させてパネルを得た。
【0062】
得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
<実施例12>
【0063】
組成を変更する以外は実施例11と同様に行い、樹脂組成物Mからプリプレグ6およびパネルを得た。得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
<実施例13>
【0064】
組成を変更する以外は実施例11と同様に行い、樹脂組成物Nからプリプレグ7およびパネルを得た。得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
<実施例14>
【0065】
組成を変更する以外は実施例11と同様に行い、樹脂組成物Oからプリプレグ8およびパネルを得た。得られたパネルより試験片を切り出しDMAによるG´−Tgおよびtanδmaxの測定、コンポジット層間せん断強度およびコンポジット曲げによる強度、弾性率の測定を実施した。結果を表4に示す。DMAの測定はARES−RDAを用いた。
【0066】
表4に示した様に、本発明のプリプレグはマトリクス樹脂本来のG´−Tgを示している。
【0067】
【表4】
【符号の説明】
【0068】
1 繊維基材
2 樹脂が含浸されていない繊維基材
3 樹脂が含浸された繊維基材
4,5 樹脂フィルム
6 離型フィルム
7 不織布
8 真空引き口
9 シーラント
10 ツール
11 積層体
12 ナイロンバッグフィルム
13 プレッシャープレート
【特許請求の範囲】
【請求項1】
エポキシ樹脂(A)、硬化剤として塩化ホウ素アミン錯体(B)、繊維基材(C)から
なるプリプレグ。
【請求項2】
エポキシ樹脂(A)中に含まれるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B
)中のホウ素のモル数比が4〜7モル%である請求項1記載のプリプレグ。
【請求項3】
エポキシ樹脂(A)が(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹
脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラッ
ク型エポキシ樹脂(3)からなる請求項1及び2記載のプリプレグ。
【化1】
【化2】
【請求項4】
請求項1〜3記載のプリプレグを硬化してなる繊維強化複合材料。
【請求項1】
エポキシ樹脂(A)、硬化剤として塩化ホウ素アミン錯体(B)、繊維基材(C)から
なるプリプレグ。
【請求項2】
エポキシ樹脂(A)中に含まれるエポキシ基のモル数に対し塩化ホウ素アミン錯体(B
)中のホウ素のモル数比が4〜7モル%である請求項1記載のプリプレグ。
【請求項3】
エポキシ樹脂(A)が(I)で表されるオキサゾリドン構造を有する二官能エポキシ樹
脂(1)、ビスフェノール型エポキシ樹脂(2)、(II)で表されるフェノールノボラッ
ク型エポキシ樹脂(3)からなる請求項1及び2記載のプリプレグ。
【化1】
【化2】
【請求項4】
請求項1〜3記載のプリプレグを硬化してなる繊維強化複合材料。
【図1】

【図2】
【図2】
【公開番号】特開2010−241845(P2010−241845A)
【公開日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願番号】特願2009−84953(P2009−84953)
【出願日】平成21年3月31日(2009.3.31)
【出願人】(000006035)三菱レイヨン株式会社 (2,875)
【Fターム(参考)】
【公開日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願日】平成21年3月31日(2009.3.31)
【出願人】(000006035)三菱レイヨン株式会社 (2,875)
【Fターム(参考)】
[ Back to top ]