
プロカスパーゼ−3の活性化を含む、癌細胞における選択的アポトーシス誘導
【課題】癌細胞におけるアポトーシスを選択的に誘導する方法の提供。
【解決手段】癌細胞のプロカスパーゼ−3分子を修飾することができる化合物を当該癌細胞に投与する。プロカスパーゼ−3分子を修飾できる化合物として、次式

[式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。]で示される化合物が用いられる。
【解決手段】癌細胞のプロカスパーゼ−3分子を修飾することができる化合物を当該癌細胞に投与する。プロカスパーゼ−3分子を修飾できる化合物として、次式
[式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。]で示される化合物が用いられる。
【発明の詳細な説明】
【関連出願の相互参照】
【0001】
[0001] 本出願は、2005年5月26日に提出された米国仮出願第60/684807号及び2006年3月28日に提出された米国仮出願第60743878号の米国特許法第119条(e)の下の利益を主張し、それぞれの全体が参照により援用される。
【連邦政府による資金提供を受けた研究開発の記載】
【0002】
[0002] 本発明は、米国国立科学財団により与えられたNSF助成金/契約CHE−0134779の下、政府援助でなされた。政府は、本発明における一定の権利を有している。
【発明の背景】
【0003】
[0003] アポトーシス又はプログラム細胞死はすべての多細胞生物の発生及びホメオスタシスで中心的な役割を果たしている(Shi Y,2002,Molecular Cell 9:459−470)。よくある癌の特徴は、本来のアポトーシスシグナルに対する抵抗性である。癌型によっては、この抵抗性は、通常、アポトーシスカスケードにおける重要なタンパク質のアップレギュレーション若しくはダウンレギュレーション又はこれらのタンパク質をコードする遺伝子内の変異による。上記変化は、ミトコンドリア及びカスパーゼ−9を経る内因性アポトーシス経路並びにデスレセプター及びカスパーゼ−8の作用が関与する外因性アポトーシス経路の両方で起こる。例えば、p53、Bim、Bax、Apaf−1、FLIP、その他多くのタンパク質の適切なレベルの変化が癌で観察された。この変化は、欠陥のあるアポトーシスカスケードにつながる可能性があり、上流のアポトーシス促進シグナルが十分に伝達されず、実行因子カスパーゼであるカスパーゼ−3及びカスパーゼ−7が活性化されない。
【0004】
[0004] ほとんどのアポトーシス経路がプロカスパーゼ−3の活性化を最終的に伴うので、上流の遺伝的異常は、有効に、アポトーシス回路における「断線箇所(break)」となり、その結果、上記細胞は異常に増殖する。癌におけるアポトーシスの中心的な役割から判断して、アポトーシスカスケードにおける特定のタンパク質を標的にする治療を開発するための試みがなされた。例えば、p53及びBclファミリーのタンパク質のカスケードメンバーに対する、又はアポトーシス抑制因子(IAP)ファミリーのタンパク質に対するペプチド結合剤又は低分子結合剤は、Apaf−1のオリゴマー化を促進する化合物のようにアポトーシス促進活性を有している。しかしながら、上記化合物が、アポトーシスカスケード上の早期に(又は中間から高所に)位置するものを標的にするので、それらのメンバーのうちの下流のタンパク質における変異を有する癌は、それらの化合物の可能な有益である効果になお抵抗性がある可能性がある。
【0005】
[0005] 治療目的として、アポトーシスカスケードにおけるはるか下流のアポトーシス促進タンパク質を直接活性化する低分子を同定することは有利であろう。我々の発明へのアプローチは、カスケードにおける上記の比較的低位置を含み、したがって、上流のアポトーシス機構における変異を有するそれらの細胞でさえ殺すことを可能にする。さらに、本明細書中に開示された治療戦略は、アポトーシス促進タンパク質が癌細胞内でアップレギュレートした場合、成功の可能性がより高い可能性がある。本発明において、低分子を同定するための我々の試みは、アポトーシスの重要な下流のエフェクタータンパク質であるプロカスパーゼ−3を標的にすることから始まった。
【0006】
[0006] プロカスパーゼ−3のカスパーゼ−3への変換又は活性化により、多数のタンパク質基質の加水分解を続いて触媒する活性「実行因子」カスパーゼ形態の生成がもたらされる。活性カスパーゼ−3は、ヘテロダイマーのホモダイマーであり、プロカスパーゼ−3のタンパク質分解により産生される。生体内では、このタンパク質分解性の活性化は、通常、カスパーゼ−8又はカスパーゼ−9の作用を介して生じる。プロ酵素又は酵素前駆体が早まって活性化されないことを確実にするために、プロカスパーゼ−3は、タンパク質分解のIETD部位(アミノ酸配列、ile−glu−thr−asp)への到達をブロックする12個のアミノ酸「安全装置」を有している。Roy,S.ら;Maintenance of caspase−3 proenzyme dormancy by an intrinsic“safety catch”regulatory tripeptide,Proc.Natl.Acad.Sci.98,6132−6137(2001)参照。
【0007】
[0007] この安全装置は、プロカスパーゼ−3が、自己触媒活性化及びカスパーゼ−9によるタンパク質分解に抵抗することを可能にする。突然変異誘発研究は、3つの連続したアスパラギン酸残基が安全装置の重要な成分であると思われることを示す。安全装置の部位はpHに感受性である。したがって、細胞の酸性化(アポトーシス中に起こる)の際、安全装置は、タンパク質分解の部位への到達を許可すると思われ、活性カスパーゼ−3は、カスパーゼ−9の作用により又は自己活性化機構を介して産生することができる。
【0008】
[0008] 特定の癌では、プロカスパーゼ−3の発現はアップレギュレートされる。20人の大腸癌患者からの一次単離物に関する研究は、平均して、プロカスパーゼ−3が、隣接した非癌性組織に比べて上記試料中で6倍アップレギュレートされたことを明らかにした(Royら,2001)。さらに、プロカスパーゼ−3は、ある種の神経芽細胞腫、リンパ腫、及び肝臓癌においてアップレギュレートされる(Nakagawara,A.ら,1997,Cancer Res.57:4578−4584;lzban,K.F.ら,Am.J.Pathol.154:1439−1447;Persad,R.ら,Modem Patholo.17:861−867)。さらに、国立癌研究所(NCI)の開発治療プログラムによる癌スクリーニングに使用された60枚の細胞株パネル中のプロカスパーゼ−3レベルについて系統的評価を行った。評価は、ある種の肺癌、黒色腫、腎臓癌、及び乳癌が著しく増強されたプロカスパーゼ−3発現レベルを示すことを明らかにした(Svingen,P.A.ら,Clin.Cancer Res.10:6807−6820)。
【0009】
[0009] アポトーシスの達成における活性カスパーゼ−3の役割、ある種の癌性細胞型におけるプロカスパーゼ−3の比較的高度な発現レベル、及び興味深い、安全装置媒介性のその自己活性化の抑制により、プロカスパーゼ−3を直接修飾する低分子が同定される可能性があるということ及び上記分子は、標的癌治療法において顕著な適用可能性を有する可能性があるということを我々は推論した。
【0010】
[0010] 本明細書で、我々は、プロカスパーゼ−3をそのエフェクター形態に変換することができる活性化剤を特に含む、プロカスパーゼの低分子修飾因子を開示する。さらに、我々は、ある種の薬物低分子がプロカスパーゼ−3を直接、即時活性化し、生体内において癌細胞におけるアポトーシス促進効果を達成することができることを実証する。我々は、これらがプロカスパーゼ−3を直接活性化することで知られている最初の低分子であると考える。実行因子カスパーゼの直接的な活性化は新規で価値のある抗癌戦略である。
【発明の概要】
【0011】
[0011] 全体として、本明細書で使用される用語及び語句は、それらの技術的に認識された意味を有しており、当業者に知られている標準教科書、雑誌論文、及び文脈の参照により見つけることができる。
【0012】
[0012] 以下の略語が適用可能である。IAP、アポトーシス抑制因子;PAC−1、プロカスパーゼ活性化化合物1;PARP、ポリ(ADPリボース)ポリメラーゼ。
【0013】
[0013] 本発明は、化合物、治療処置のための方法、化合物をスクリーニングするための方法、並びにプロカスパーゼの修飾因子に関連した処置に対する細胞及び患者適性をスクリーニングするための方法を広く提供する。一実施形態では、修飾因子は、阻害剤又は活性化剤である。一実施形態では、本発明は、プロカスパーゼ−3及びプロカスパーゼ−7の活性化剤に関連した上記化合物及び方法を提供する。一実施形態では、本発明は、当技術分野で知られている、乳房、リンパ腫、副腎、腎臓、黒色腫、白血病、神経芽細胞腫、肺、脳等、様々な癌疾患及び癌細胞型の状況に適用可能である。
【0014】
[0014] さらなる概要として、我々は、癌細胞内で不活性形態で過剰発現することが多い酵素を活性化することができる化合物を発見した。化合物は、アップレギュレートされたプロカスパーゼ−3を有する癌細胞を含む癌細胞におけるプログラム細胞死(アポトーシス)を誘導する。癌は増大する大きな課題であり、現在、米国における第1位の死因である。多くの癌は、標準的な化学療法に抵抗する。本発明の化合物は、癌細胞内でアップレギュレートされることがある生物学的標的を利用することができ、したがって、アポトーシス機構に欠陥を有する細胞内でさえ有効であると証明することができる。これらの化合物は、さらに、標的癌治療法においても成功する可能性があり、ここでは、より低レベルのプロカスパーゼ−3を有する非癌性細胞に対して比較的毒性が低下した状態で癌細胞を死滅させる点での選択性の利点がある可能性がある。
【0015】
[0015] 特定の理論に束縛されるものではないが、本発明の化合物は、アポトーシス又はプログラム細胞死の調節の機構を介して作用して、癌細胞の処置において有効となることが可能であると考えられる。好ましい実施形態では、アポトーシスの調節はアポトーシスの誘導による。他の実施形態では、アポトーシスの調節はアポトーシスの阻害による。
【0016】
[0016] 一実施形態では、本発明は、癌細胞におけるアポトーシスを選択的に誘導する方法であって、(a)上述の癌細胞のプロカスパーゼ−3分子を修飾することができる化合物を上述の癌細胞に投与することと、(b)アポトーシスを誘導するように上述のプロカスパーゼ−3分子を修飾すること、を含む方法を提供する。一実施形態では、上述の癌細胞は、処置を必要とする患者に存在する。
【0017】
[0017] 一実施形態では、上述の化合物は下記式ZZである。
【化1】
式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。一実施形態では、上述の化合物は、式ZZ、PAC−1及び構造5からなる群より選択される。一実施形態では、上述の化合物はPAC−1である。
【0018】
[0018] 一実施形態では、化合物は、式ZZを有する化合物から選択され、式中、R4及びR5は共にNであり、Aは酸素であり、他の可変基は上記に定義した通りである。一実施形態では、化合物は、式ZZを有する化合物から選択され、式中、R4及びR5は共にNであり、Aは酸素であり、R2は水素であり、他の可変基は上記に定義した通りである。一実施形態では、化合物は、式ZZを有する化合物から選択され、式中、R4及びR5は共にNであり、Aは酸素であり、R2は水素であり、R3はアリルであり、他の可変基は上記に定義した通りである。
【0019】
[0019] 一実施形態では、方法は、癌細胞内のプロカスパーゼ−3又はカスパーゼ−3のパラメーターを評価するステップをさらに含み、上述のパラメーターは、半定量的若しくは定量的な量、機能的な量、又は上述のプロカスパーゼ−3又はカスパーゼ−3の活性レベルの1つ又は複数である。
【0020】
[0020] 一実施形態では、本発明は、プロカスパーゼ−3分子を修飾することができる化合物を直接、in vitroでスクリーニングするための方法であって、(a)試験化合物を提供することと、(b)精製したプロカスパーゼ−3を提供することと、(c)精製したプロカスパーゼ−3に試験化合物を暴露することと、(d)試験化合物への暴露の後でプロカスパーゼ−3活性を測定することと、(e)試験化合物への暴露の際の試験活性を試験化合物への暴露なしでの無修飾活性と比較することにより修飾化合物を同定すること、を含み、それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法を提供する。一実施形態では、方法は、上述の修飾活性又は上述の無修飾活性を基準活性と比較することをさらに含み、上述の基準活性は、構造式ZZ又は上記式の化合物のサブセット、PAC−1及び構造5からなる群より選択される化合物へのプロカスパーゼ−3の暴露による。
【0021】
[0021] 一実施形態では、本発明は、プロカスパーゼ−3を活性化することができる化合物のスクリーニングのための方法であって、a)プロカスパーゼ−3を提供し、試験化合物、好ましくは低分子を提供することと、b)プロカスパーゼ−3を試験化合物と反応させること(これにより、カスパーゼ−3が生成すると推定される)と、c)カスパーゼ−3活性を測定することと、を含む方法を提供する。特定の実施形態では、カスパーゼ−3活性を測定することに基質Ac−DEVD−pNAを使用する。特定の実施形態では、測定することに、約410nmの波長読み出しパラメーターを使用する。特定の実施形態では、スクリーニングは、多数の試験化合物を使用して並行して実行される。
【0022】
[0022] 一実施形態では、本発明は、プロカスパーゼ−3のサブユニットの検出を完全長(不活性)プロカスパーゼ−3がカスパーゼ−3に処理されたという指標として使用するスクリーニングの方法を提供する。特定の実施形態では、サブユニットは、タンパク質ゲル移動技術、例えばウエスタンブロットにより測定される約19kDの分子量を有している。
【0023】
[0023] 一実施形態では、本発明は、プロカスパーゼ−3分子を修飾することができる化合物の細胞スクリーニングのための方法であって、(a)試験化合物を提供することと、(b)プロカスパーゼ−3を推定上発現する細胞を提供することと、(c)試験化合物に細胞を暴露することと、(d)試験化合物への暴露の後で、1つ又は複数の細胞生存率、アポトーシス指標、及び他のパラメーターを含む細胞パラメーターを測定することと、(e)試験化合物への暴露の際の試験細胞パラメーターを試験化合物への暴露なしでの無修飾細胞パラメーターと比較することにより修飾化合物を同定することと、を含み、それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法を提供する。一実施形態では、方法は、上述の修飾活性又は上述の無修飾活性を基準活性と比較することをさらに含み、上述の基準活性は、式ZZ又は上記式の化合物のサブセット、PAC−1及び構造5からなる群より選択される化合物への暴露による。
【0024】
[0024] 一実施形態では、本発明は、プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定又は診断するための方法であって、(a)上述の癌細胞内のプロカスパーゼパラメーターを評価することと、(b)上述のパラメーターによりプロカスパーゼの活性化に対する感受性が上昇したかどうかを判定すること、を含む方法を提供する。一実施形態では、上述のプロカスパーゼパラメーターはプロカスパーゼ−3レベルであり、上述のプロカスパーゼはプロカスパーゼ−3である。一実施形態では、上述のプロカスパーゼパラメーターはプロカスパーゼ−7レベルであり、上述のプロカスパーゼはプロカスパーゼ−7である。レベルは、半定量的若しくは定量的な量、又は機能的な量(例えば活性ベースの量、例えば規格単位又は国際単位)とすることができる。
【0025】
[0025] 一実施形態では、本発明は、癌細胞を処置するための方法であって、(a)プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定することと、(b)有効な量のプロカスパーゼ活性化化合物に上述の癌細胞を暴露すること、を含む方法を提供する。一実施形態では、プロカスパーゼ活性化化合物は、式ZZ又は上記式の化合物のサブセット、PAC−1及び構造5からなる群より選択される。一実施形態では、上述のプロカスパーゼ活性化化合物は、プロカスパーゼ−3、プロカスパーゼ−7、又はプロカスパーゼ−3及びプロカスパーゼ−7の両方を活性化することができる請求項16に記載の方法。
【0026】
[0026] 一実施形態では、本発明は、スキーム1のステップを含む、PAC−1を合成するための方法を提供する。一実施形態では、本発明は、適切な変更を有するスキーム1のステップを含む、化合物5を合成するための方法を提供する。一実施形態では、本発明は、本明細書で開示されており、当技術分野で理解されるであろう式ZZの化合物を合成するための方法を提供する。
【0027】
[0027] 一実施形態では、本発明は、下記式ZZの化合物を提供する。
【化2】
式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。
【0028】
[0028] 一実施形態では、本発明は、構造が
【化3】
であるPAC−1を除く、式ZZの化合物を提供する。
【0029】
[0029] 一実施形態では、本発明は、構造5の化合物を提供する。構造は次の通りである。
【化4】
【0030】
[0030] 一実施形態では、本発明の組成物は化学療法剤である。
【0031】
[0031] 一実施形態では、本発明は、開示された構造式の好ましくは約10nM〜約100μMの有効濃度を伴う化合物及び方法を提供する。他の好ましい実施形態では、有効な濃度は約200nM〜約5μMである。一実施形態では、有効な濃度は、直接的なプロカスパーゼ活性化アッセイ、細胞アポトーシス誘導アッセイ、又は動物の臨床的治療評価での50%活性濃度等の値であると考えられる。好ましい実施形態では、上記値は、約200μM未満である。好ましい実施形態では、上記値は約10μM未満である。
【0032】
[0032] 本発明の化合物及び本発明の方法において有用な化合物は、開示された式の化合物、並びに好ましくは薬学的に許容し得る塩及びエステルを含む、それらの化合物の塩及びエステルを含む。
【0033】
[0033] 一実施形態では、本発明は、組成物のプロドラッグ形態を提供する。本発明の化合物のプロドラッグは本発明の方法において有用である。生体内で変換され、本発明の化合物の生物学的、薬学的、又は治療的な活性形態をもたらすであろういかなる化合物もプロドラッグである。プロドラッグの様々な例及び形態が当技術分野でよく知られている。前駆体タンパク質、前駆体核酸等の生体分子をプロドラッグとすることができる。プロドラッグの例は、Design of Prodrugs,H.Bundgaard編(Elsevier,1985)、Methods in Enzymology,Vol.42,pp.309−396,K.Widderら編(Academic Press,1985)、A Textbook of Drug Design and Development,Krosgaard−Larsen及びH.Bundgaard編,Chapter 5,「Design and Application of Prodrugs」H.Bundgaard編,pp.113−191,1991)、H.Bundgaard,Advanced Drug Delivery Reviews,Vol.8,p.1−38(1992),H.Bundgaardら,Journal of Pharmaceutical Sciences,Vol.77,p.285(1988)、並びにNogrady(1985)Medicinal Chemistry A Biochemical Approach,Oxford University Press,New York,pages388−392)にとりわけ見つけられる。
【0034】
[0034] 置換基のグループが本明細書で開示される場合、グループのメンバーのいかなる異性体及び鏡像異性体をも含む、そのグループ及びすべてのサブグループの個々のメンバーがすべて、個別に開示されることが理解される。マーカッシュグループ又は他のグループ分けが本明細書で使用される場合、グループ並びにグループのすべての組み合わせ及び下位組み合わせの可能性がある個々のメンバーがすべて、本開示に個々に含まれることが意図される。明細書で提供された任意のマーカッシュグループ又は一覧の任意の1つ又は複数のメンバーは、所望により、本発明から除外することができることが意図される。化合物が、化合物の特定の異性体又は鏡像異性体が、例えば式又は化学名で明記されないように本明細書で記載される場合、その記載は、個々に又は任意の組み合わせで記載された化合物のそれぞれの異性体及び鏡像異性体を含むことが意図される。さらに、特に明記されていない限り、本明細書で開示された化合物の同位体の変形形態はすべて、本開示により包含されることが意図される。例えば、開示された分子内の任意の1つ又は複数の水素が重水素又は三重水素で置換することができることが理解されるであろう。分子の同位体の変形形態は、分子のアッセイ並びに分子又はその使用に関連する化学的研究及び生物学的研究で標準物質として一般に有用である。化合物の特定の名称は、当業者が、同一の化合物をさまざまに称することができることが知られているように、例示的であることが意図される。
【0035】
[0035] 本明細書で開示された分子は1つ又は複数のイオン化できるグループ[プロトンを除去する(例えばOH、−COOH、等)若しくは加える(例えばアミン)ことができる又は四級化することができる(例えばアミン)グループ]を含んでいてもよい。可能な上記分子のイオン形態及びそれらの塩はすべて、本明細書中の本開示に個々に含まれることが意図される。本明細書中の化合物の塩に関して、当業者は、種々様々の利用可能な対イオンの中から、所定の用途に対する本発明の塩の調製に適切な対イオンを選択することができる。例えば、一般に、任意のアニオンが、本明細書中の化合物の塩、例えば、ハロゲン化物、硫酸塩、カルボン酸塩、酢酸塩、リン酸塩、硝酸塩、トリフルオロ酢酸塩、グリコール酸塩、ピルビン酸塩、シュウ酸塩、リンゴ酸塩、コハク酸塩、フマル酸塩、酒石酸塩、クエン酸塩、安息香酸塩、メタンスルホン酸塩、エタンスルホン酸塩、p−トルエンスルホン酸塩、サリチル酸塩、等の形成に使用することができる。
【0036】
[0036] 本発明の化合物及びそれらの塩又はエステルは、それらの互変異性形態で存在してもよく、水素原子は、分子の他の部分に転位され、分子の原子間の化学結合は結果的に再配列される。互変異性形態はすべて、存在することがある限り、本発明の範囲内に含まれることが理解されるべきである。さらに、化合物は、トランス異性体及びシス異性体を有していてもよく、1つ又は複数のキラル中心を含み、したがって、鏡像異性体形態及びジアステレオマー形態で存在してもよい。本発明は、特に本明細書で述べられていない限り、上記すべての異性体及び個々の鏡像異性体と同様にシス異性体及びトランス異性体の混合物、ジアステレオマーの混合物、鏡像異性体(光学異性体)の非ラセミ混合物及びラセミ混合物、並びに1つ又は複数の形態について豊富な前述の混合物を包含することができる。化合物の(又は不斉炭素の)配置(シス若しくはトランス又はR若しくはS)について特定の言及がなされていない場合、そのとき、任意の1つの異性体又は1つを超える異性体の混合物が意図される。調製のプロセスは、ラセミ化合物、鏡像異性体、又はジアステレオマーを出発物質として使用することができる。鏡像異性体又はジアステレオマーの生成物が調製される場合、それらは、従来の方法により、例えばクロマトグラフィー又は分別結晶により分離することができる。発明の化合物は、遊離形態又は水和物形態であってもよい。
【0037】
[0037] 本明細書中に記載された又は例示された成分のすべての処方又は組み合わせは、特に述べられていない限り、本発明を実行するために使用することができる。
【0038】
[0038] 範囲、例えば温度範囲、時間範囲、組成範囲、又は濃度範囲が本出願中に記載されるときには必ず、すべて中間範囲及び部分範囲並びに所定の範囲に含まれるすべての個々の値が本開示に含まれることが意図される。
【0039】
[0039] 本明細書中に開示された任意の参考文献中の情報は、場合により、例えば、特許文献についてのそれらの有効出願日時点での技術水準を示すことができ、上記情報は、必要であれば、実際に先行技術にあると認められる特定の実施形態を除外するために本明細書中で使用することができることが意図される。例えば、化合物が、開示及び/又は特許請求される場合、実施可能な程度の開示が参考文献中に提供されている化合物を含む、本発明に関して先行技術としてみなされている化合物は、本出願中の物質の請求項の組成物に含まれることが意図されないことが理解されるべきである。
【0040】
[0040] 本明細書中で提供されるいくつかの参考文献は、出発物質、補足的な出発物質、補足的な試薬、補足的な合成の方法、補足的な分析の方法、及び補足的な本発明の使用についての情報源に関する細部を提供するように参照により援用される。具体的に例示されたもの以外の出発物質、試薬、固体基質、合成方法、精製方法、及び分析方法は、当技術分野の知識に基づき、過度の実験に頼ることなく、本発明の実行に使用することができることを当業者は十分に理解するであろう。
【0041】
[0041] 一実施形態では、本発明は、1つ又は複数の化合物及び各化合物について薬学的に許容し得るそれらの塩又はエステルを含み、化合物は、所望される治療上の有益性を得るために有効な量又は総量で組成物中に存在する治療用組成物を提供する。本発明の治療用組成物は、1つ又は複数の薬学的に許容し得る成分、例えば、当技術分野で知られている担体及び賦形剤を任意選択でさらに含む。
【0042】
[0042] 一実施形態では、本発明は、下記式ZZ2を有する化合物を提供する。
【化5】
式中、R1及びR2は、それぞれ独立して、水素、ハロゲン、アルキル、アリル、ハロアルキル、アルケニル、アルケノール、アルカノール又はハロアルケニルである。一実施形態では、R1及びR2は、それぞれ独立して、水素、ハロゲン、アリル又は短鎖アルキルである。
【0043】
[0043] 一実施形態では、本発明は、アルデヒド化合物と結合したヒドラジド化合物を含むPAC−1誘導体コンビナトリアルライブラリーからなる群より選択される化合物を提供する。一実施形態では、ヒドラジド化合物は、本明細書中に記載されたAX化合物から生成されたヒドラジドからなる群より選択される。
【0044】
[0044] 一実施形態では、アルデヒド化合物は、本明細書中に記載されたBX化合物からなる群より選択される。一実施形態では、ヒドラジド化合物は、本明細書中に記載されたAX化合物からなる群より選択され、アルデヒド化合物は、本明細書中に記載されたBX化合物からなる群より選択される。
【0045】
[0045] 一実施形態では、本発明は、PAC−1誘導体化合物を合成するための方法であって、ヒドラジド化合物を提供することと、アルデヒド化合物を提供することと、ヒドラジド化合物をアルデヒド化合物と反応させることと、を含み、それにより、PAC−1誘導体化合物を合成する方法を提供する。
【0046】
[0046] 一実施形態では、ヒドラジド化合物は下記式ZZ3を有する。
【化6】
【0047】
[0047] 一実施形態では、アルデヒド化合物は下記式ZZ4を有する。
【化7】
【0048】
[0048] 一実施形態では、ヒドラジド化合物は式ZZ3を有し、アルデヒド化合物は式ZZ4を有する。
【0049】
[0049] 一実施形態では、本発明は、L01R06、L02R03、L02R06、L08R06、L09R03、L09R06及びL09R08からなる群より選択される化合物を提供する。
【0050】
[0050] 一実施形態では、本発明は、癌患者の候補者を、候補者においてプロカスパーゼレベルの上昇を同定することによりプロカスパーゼ活性化剤による可能な処置についてスクリーニングするための方法であって、候補者からの細胞又は組織の試験サンプルを得ることと、試験サンプル中のプロカスパーゼレベルを評価することと、プロカスパーゼレベルは基準レベルに比べて試験サンプル中で上昇したかどうかを判定することと、を含み、それにより、癌患者の候補者をプロカスパーゼ活性化剤による可能な処置についてスクリーニングする方法を提供する。一実施形態では、プロカスパーゼは、プロカスパーゼ−2、プロカスパーゼ−3、プロカスパーゼ−6、プロカスパーゼ−7、プロカスパーゼ−8及びプロカスパーゼ−9からなる群より選択される。特定の実施形態では、プロカスパーゼはプロカスパーゼ−3である。
【0051】
[0051] 一実施形態では、試験サンプルの上昇レベルは、基準レベルを超えた少なくとも約2倍である。一実施形態では、試験サンプルの上昇レベルは、基準レベルを超えた少なくとも約4倍である。一実施形態では、基準レベルは、同一患者からの第2の試験サンプルからのものである。一実施形態では、基準レベルは、正常細胞又は正常組織のサンプルからのものである。基準レベルは、癌細胞株、正常細胞株等の細胞株からのものとすることができる。一実施形態では、基準レベルは絶対閾値の量である。細胞当たりの分子数を含むプロカスパーゼのレベルについての様々な量を記載しているSvingen,P.A.ら,Clin.Cancer Res.10:6807−6820を参照のこと。
【0052】
[0052] 一実施形態では、本発明は、上述の癌細胞中のプロカスパーゼ分子を活性化することができる化合物を上述の癌細胞に投与することを含む、癌細胞における死を誘導するための方法を提供する。一実施形態では、プロカスパーゼは、1つ又は複数のプロカスパーゼ−3及びプロカスパーゼ−7である。好ましい実施形態では、プロカスパーゼはプロカスパーゼ3である。一実施形態では、化合物は構造式ZZを有する。一実施形態では、化合物は構造式ZZ2を有する。
【0053】
[0053] 本明細書で考えられた又は開示されたいかなる機構的な説明又は仮説についての最終的な正確さに関係なく、本発明の一実施形態は、それでもなお、効果があり、有用である可能性があると認識される。
【図面の簡単な説明】
【0054】
【図1A】in vitroでのPAC−1によるプロカスパーゼ−3の活性化及び活性カスパーゼ−3。PAC−1は、EC50=0.22μMでプロカスパーゼ−3を活性化する。エラーバーは、平均値からの標準偏差を表す。
【図1B】PAC−1により誘導される活性カスパーゼ−3へのプロカスパーゼ−3の切断。プロカスパーゼ−3をN末端His−6タグを用いて大腸菌中で組換え発現させ、精製した。免疫ブロットは抗His−6抗体を用いて行った。PAC−1非存在下でプロカスパーゼ−3の成熟は観察されない。100μM PAC−1存在下で、p19フラグメントを生成する切断が1時間以内に観察され、>50%の切断が4時間後に観察される。
【図2A】プロカスパーゼ−3の「安全装置」領域における変異体のPAC−1による活性化。PAC−1は、野性型プロカスパーゼ−3(DDD)に対して0.22μMの活性化のためのEC50並びにある種の変異体については2.77μM(DAD)、113μM(DDA)、及び131μM(ADD)の対応するEC50値を有している。
【図2B】PAC−1は、4.5μMのEC50でプロカスパーゼ−7を活性化する。
【図2C】プロカスパーゼ−3のPAC−1の活性化についてのpHへの依存性。低pHでは、安全装置は「オフ」の状態にあり、プロカスパーゼ−3は本質的に最大限に活性化される。エラーバーは、平均値からの標準偏差を表す。
【図3A】PAC−1は、HL−60細胞におけるアポトーシスを誘導する。A)100μM PAC−1での20時間の処置の後のホスファチジルセリン露出(アネキシン−V染色により測定される)。
【図3B】100μM PAC−1での20時間の処置の後のヘキスト染色により可視化されるクロマチン凝縮。
【図4A】10μMエトポシドで処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。
【図4B】100μM PAC−1で処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。
【図4C】PAC−1処置(100μM)は、細胞内カスパーゼ−3/−7の即時活性化と一致する、HL−60細胞における細胞内PARP活性の急速な減少を誘導する。対照的に、エトポシド(10μM)処置細胞は、ずっと後の時点でのPARP活性の減少を示す。
【図4D】PAC−1は、プロカスパーゼ−3依存的に細胞死を誘導する。多くの多様な癌細胞株について、プロカスパーゼ−3レベルを求め(フローサイトメトリーにより)、PAC−1のIC50を測定した(R2=0.9822)。PAC−1は、高度なレベルのプロカスパーゼ−3を有するとして知られているNCI−H226肺癌細胞株において相当効力がある(IC50=0.35μM)が、健常なヒトドナーの骨髄に由来する正常な白血球において効力が著しく少ない。
【図4E】PAC−1は、プロカスパーゼ−3依存的に細胞死を誘導する。多くの多様な癌細胞株について、プロカスパーゼ−3レベルを求め(フローサイトメトリーにより)、PAC−1のIC50を測定した(R2=0.9822)。PAC−1は、高度なレベルのプロカスパーゼ−3を有するとして知られているNCI−H226肺癌細胞株において相当効力がある(IC50=0.35μM)が、健常なヒトドナーの骨髄に由来する正常な白血球において効力が著しく少ない。
【図5A】数名の患者からの正常細胞及び癌性細胞中の相対的なプロカスパーゼ−3レベルを示す。
【図5B】様々な相対的なプロカスパーゼ−3レベルを有する様々な細胞型におけるPAC−1のIC50レベルを示す。
【図5C】腫瘍成長の結果に対するPAC−1での動物の処置の効果を示す。
【図5D】腫瘍成長の結果に対するPAC−1での動物の経口処置の効果を示す。
【図5E】対照処置群、PAC−1処置群、及びゲフィチニブ(イレッサ(商標);アストラゼネカ社)処置群についての肺癌モデルにおける癌の進行の結果を示す図である。腫瘍細胞をi.v.投与によりマウスに注射し、イレッサ及びPAC−1を100mg/kgで経口的に与えた。
【図6A】3名の患者の正常細胞及び癌性細胞中の相対的なプロカスパーゼ−3レベルを示す。
【図6B】患者3からの正常細胞及び癌性細胞のPAC−1での処置に対する感受性を示す。
【図7】肺癌のマウスモデルという状況におけるPAC−1の腹腔内投与の結果を示す。
【図8A】PAC−1誘導体及びコンビナトリアルライブラリーの化合物の構造を示す。
【図8B】PAC−1誘導体及びコンビナトリアルライブラリーの化合物の構造を示す。
【図9】ホモサピエンス カスパーゼ3、アポトーシス関連システインペプチダーゼ(CASP3)、転写バリアントアルファ、mRNAのヌクレオチド配列を示す。(受託番号NM_004346;2689bp mRNA直鎖状;http://www.ncbi.nlm.nih.gov/entrezから入手)。
【図10】ホモサピエンス カスパーゼ7、アポトーシス関連システインペプチダーゼ(CASP7)、転写バリアントアルファ、mRNAのヌクレオチド配列を示す。(受託番号NM_001227;2605bp;mRNA直鎖状;http://www.ncbi.nlm.nih.gov/entrezから入手)。
【発明の詳細な説明】
【0055】
[0069] 以下の定義は、本発明の文脈中でのそれらの特定の用法を明確にするために提供される。
【0056】
[0070] 用語「化学療法剤」は、本明細書中で使用される場合、癌細胞、癌細胞の集団、腫瘍、又は他の悪性組織の成長、増殖、又は拡散を低下又は阻止することができる任意の物質を指す。用語はまた、任意の抗腫瘍剤又は抗癌剤を包含することも意図される。
【0057】
[0071] 用語「有効な量」は、本明細書中で使用される場合、薬学的に有効な量、治療的に有効な量、等の文脈を包含することが意図される。例えば、実施形態ではこの量は、有利な状態、有利な結果、スクリーニングアッセイにおける機能的な活性、又は臨床症状の改善を達成することができる。
【0058】
[0072] 用語「癌細胞」は、本明細書中で使用される場合、当技術分野で広く理解されている定義を包含することが意図される。一実施形態では、この用語は、ヒト又は動物における癌の臨床症状の一因となる可能性がある異常に制御された細胞を指す。一実施形態では、この用語は、培養された細胞株或いはヒト若しくは動物の体内の又はヒト若しくは動物に由来する細胞を指すことができる。癌細胞は、当技術分野で理解されているように、種々様々な分化した細胞型、組織型、又は器官型になる可能性がある。
【0059】
[0073] 用語「アルキル」は、好ましくは1〜22個の炭素原子を有するモノラジカルの分枝又は非分枝の飽和炭化水素鎖、及び3〜22個の炭素原子を有する1つ又は複数の環を有するシクロアルキル基を指す。短鎖アルキル基は、メチル基、エチル基、プロピル基、ブチル基、ペンチル基及びヘキシル基を含む、1〜6個の炭素原子を有する基であり、それらのすべての異性体を含む。長鎖アルキル基は、8〜22個の炭素原子を有する基、好ましくは、12〜22個の炭素原子を有する基、12〜20個の炭素原子を有する基、及び16〜18個の炭素原子を有する基である。
【0060】
[0074] 用語「シクロアルキル」は、単環式環又は多縮合環を有する、3〜22個の炭素原子の環式のアルキル基を指す。シクロアルキル基は、例として、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロオクチル等の単環状構造、又はアダマンタニル等の多環状構造を含む。
【0061】
[0075] 用語「アルケニル」は、好ましくは2〜22個の炭素原子を有する分枝又は非分枝の不飽和炭化水素基のモノラジカル、及び3〜22個の炭素原子を有する1つ又は複数の環を有するシクロアルケニル基であって、少なくとも1つの環は二重結合を含むシクロアルケニル基を指す。アルケニル基は、共役していてもよい1つ又は複数の二重結合(C=C)を含んでいてもよい。好ましいアルケニル基は、1つ又は2つの二重結合を有する基である。短鎖アルケニル基は、エチレン(ビニル)プロピレン基、ブチレン基、ペンチレン基及びヘキシレン基を含み、それらのすべての異性体を含む2〜6個の炭素原子を有する基である。長鎖アルケニル基は、8〜22個の炭素原子を有する基、好ましくは、12〜22個の炭素原子を有する基、12〜20個の炭素原子を有する基、及び16〜18個の炭素原子を有する基である。用語「シクロアルケニル」は、少なくとも1つの環が二重結合(C=C)を含む単環式環又は多縮合環を有する3〜22個の炭素原子の環式のアルケニル基を指す。シクロアルケニル基は、例として、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロオクテニル、シクロオクタジエニル、シクロオクタトリエニル等の単環状構造を含む。
【0062】
[0076] 用語「アルキニル」は、好ましくは2〜22個の炭素原子を有し、1つ又は複数の三重結合(C□C)を有する不飽和炭化水素のモノラジカルを指す。アルキニル基は、エチニル、プロパルギル等を含む。短鎖アルキニル基は、2〜6個の炭素原子を有する基であり、それらの異性体をすべて含む。長鎖アルキニル基は、8〜22個の炭素原子を有する基、好ましくは、12〜22個の炭素原子を有する基、12〜20個の炭素原子を有する基、及び16〜18個の炭素原子を有する基である。
【0063】
[0077] 用語「アリール」は、単環(例えばフェニル)、1つ又は複数の環(例えばビフェニル)、又は多縮合(融合)環を有する6〜22個の炭素原子の不飽和芳香族炭素環式基を含む基であって、少なくとも1つの環は芳香族である(例えば、ナフチル、ジヒドロフェナントレニル(dihydrophenanthrenyl)、フルオレニル、又はアントリル)基を指す。アリールは、フェニル、ナフチル等を含む。アリール基は、不飽和の芳香環(複数可)に加えて、アルキル、アルケニル又はアキニルである部分を含んでいてもよい。用語「アルカリル」は、アルキル部分、つまり、−アルキレン−アリール、及び−置換されたアルキレン−アリール、を含むアリール基を指す。上記アルカリル基は、ベンジル、フェネチル等により例示される。
【0064】
[0078] 本明細書中に記載されるように、アルキル基、アルケニル基、アルキニル基及びアリール基は任意選択で置換され(用語(複数可)は置換された変形形態を含むことができる)、基中の炭素原子数及び基の不飽和度に依存した1〜8個の非水素置換基を含んでいてもよい。
【0065】
[0079] 用語「アルキレン」は、好ましくは1〜10個、より好ましくは1〜6個、より好ましくは2〜4個の炭素原子を有する分枝又は非分枝の飽和炭化水素鎖のジラジカルのことを言い、この用語は、置換された変形形態を含むことができる。この用語は、メチレン(−CH2−)、エチレン(−CH2CH2−)、より概略的には−(CH2)n−[式中、nは1〜10、より好ましくは1〜6、又は2、3若しくは4である]、等の基により例示される。アルキレン基は分枝していてもよい。アルキレン基は任意選択で置換されてもよい。アルキレン基は、炭素原子当たり2つまでの非水素置換基を有していてもよい。好ましい置換されたアルキレン基は1つ、2つ、3つ又は4つの非水素置換基を有している。
【0066】
[0080] 用語「アリーレン」は、上記に定義されるアリール(置換されたアリールを含む)に由来するジラジカルのことを言い、1,2−フェニレン、1,3−フェニレン、1,4−フェニレン、1,2−ナフチレン等により例示される。
【0067】
[0081] 用語「アミノ」は、基−NH2又は基−NRRを指し、式中、両方のRが水素ではないとすれば、各Rは、水素、アルキル、置換されたアルキル、シクロアルキル、置換されたシクロアルキル、アルケニル、置換されたアルケニル、シクロアルケニル、置換されたシクロアルケニル、アルキニル、置換されたアルキニル、アリール、ヘテロアリール、及び複素環からなる基から独立して選択される。
【0068】
[0082] 本明細書中に論じられるように、アルキル基は任意選択で置換され、アルキル基の大きさに依存して、好ましくは1〜10個の置換基を有していてもよい。置換されたアルキル基は、1〜8個の置換基、1〜5個の置換基、1〜3個の置換基及び1個若しくは2個の置換基を伴う基を含む。
【0069】
[0083] 「ハロアルキル」は、同じ又は異なっていてもよい、本明細書中に定義される1つ又は複数のハロ基により置換された、本明細書中に定義されるアルキルを指す。代表的なハロアルキル基は、例として、トリフルオロメチル、3−フルオロドデシル、12,12,12−トリフルオロドデシル、2−ブロモオクチル、3−ブロモ−6−クロロヘプチル等を含む。
【0070】
[0084] 用語「ヘテロアリール」は、少なくとも1つの環内に酸素、窒素、及び硫黄から選択される1〜4個のヘテロ原子を有する(環が1個を超える場合)、2〜22個の炭素原子の芳香族基を指す。ヘテロアリール基は任意選択で置換されてもよい。
【0071】
[0085] 用語「ヘテロ環」又は「複素環」は、単環又は多縮合環、2〜22個の炭素原子、並びに少なくとも1つの環内における窒素、硫黄、リン及び/又は酸素から選択される1〜6個、好ましくは1〜4個のヘテロ原子、を有するモノラジカルの飽和基又は不飽和基を指す。複素環基は置換されてもよい。
【0072】
[0086] 用語「エステル」は、当技術分野で理解されている化学物質のことを言い、特に形態(RCO−)の基を含むことができる。
【0073】
[0087] 1つ又は複数の置換基を含む上記基のいずれに関しても、上記基は、立体的に非実用的である及び/又は合成的に実現不可能であるいかなる置換又は置換パターンも含まないことが理解される。本発明の化合物は、開示された化合物の置換から生じる新規な立体化学的異性体をすべて含む。
【実施例】
【0074】
[0088] 本発明は、以下の非限定的な実施例によりさらに理解されるであろう。
【0075】
〔実施例1:プロカスパーゼ活性化化合物〕
[0089] アポトーシスカスケードにおけるタンパク質の変異又は異常発現はよくある癌の特徴である。これらの変化は、アポトーシス促進シグナルが実行因子カスパーゼに伝達されるのを阻止し、したがってアポトーシス細胞死を阻止し細胞増殖が許可される可能性がある。カスパーゼ−3及びカスパーゼ−7は重要な実行因子カスパーゼであり、上流のシグナルにより活性化される不活性な酵素前駆体として存在する。重要なことには、プロカスパーゼ−3の発現レベルは、ある種の癌性細胞内では、非癌性対照に比べて著しく高度である。本明細書で、我々は、プロカスパーゼ−3を活性カスパーゼ−3に直接活性化する低分子の同定について報告する。特定の化合物であるPAC−1は、約220ナノモル濃度の桁でEC50のin vitroでの活性化を引き起こし、多数の癌性細胞株におけるアポトーシスを誘導する。
【0076】
[0090] 多くの知られている抗癌薬とは対照的に、PAC−1で処置した細胞はプロカスパーゼ−3の即時活性化を示し、PAC−1の毒性は、細胞中に含まれるプロカスパーゼ−3の量に正比例することが示される。したがって、PAC−1は、生体内で、プロカスパーゼ−3をカスパーゼ−3に直接活性化し、欠陥のあるアポトーシス機構を有する細胞においてでさえこの化合物がアポトーシスを誘導することを可能にする。PAC−1は、プロカスパーゼ−3を直接活性化することで知られている最初の低分子であり、実行因子カスパーゼの直接的な活性化は、プロカスパーゼ−3がアップレギュレートされる多くの癌を含む様々な癌において有利であると証明することが可能である新規な抗癌戦略である。
【0077】
[0091] 約20,000種の構造的に多様な低分子の集合体を、in vitroのプロカスパーゼ−3を活性化する能力に対してスクリーニングした。プロカスパーゼ−3を大腸菌中で発現させ、精製した(Royら,2001)。プロカスパーゼ−3(50ng/mLの濃度)を384ウェルプレートのウェルに加え、化合物を約40μMの最終濃度まで加えた。次いで、各プレートを37℃で2時間インキュベートし、その後、カスパーゼ−3ペプチド基質Ac−Asp−Glu−Val−Asp−p−ニトロアニリド(Ac−DEVD−pNa)を200μMの濃度まで加えた。2時間にわたりp−ニトロアニリン発色団の形成を405nmで追跡した。
【0078】
[0092] 評価した化合物のうち、4種が、バックグラウンドに対してカスパーゼ−3ペプチド基質の加水分解の著しい増加を誘導した。それらの4種のうち、1種は、in vitroのプロカスパーゼ−3活性化に対する強い用量依存効果を示した。図1Aに示すように、この最初のプロカスパーゼ活性化化合物(PAC−1)は、0.22μMの濃度で、プロカスパーゼ−3の最大活性化の2分の1をもたらす。この化合物は、完全に活性化されたカスパーゼ−3酵素の触媒活性に対する効果を有していないので、カスパーゼ−3自体の活性を単に上昇させているのではない(図1A)。
【0079】
[0093] プロカスパーゼ−3はN末端プロドメイン(1〜28残基)、それに続く、サブユニット間のリンカーにより分けられた大サブユニット(17kDa)及び小サブユニット(12kDa)を有している(Popら,2003)。生体内では、2つのプロカスパーゼ−3モノマーが集合して、サブユニット間のリンカー中のD175での切断により活性化することができる触媒的に不活性なホモダイマーを形成する。プロドメインの明確な役割は不明であり、最高の触媒活性には、サブユニット間の領域の切断のみで十分であることが示された(Stennicke,H.R.ら,1998)。プロカスパーゼ−3は触媒的に有能であるが、12個のアミノ酸の安全装置の存在により自己活性化に対して高度に抵抗性がある。しかしながら、安全装置が変異すると、プロカスパーゼ−3の著しい自己活性化が観察される(Royら,2001)。この重要な調節領域と、又は他の部位と相互に作用する化合物は、プロカスパーゼ−3の自己活性化を可能にすることができる。
【0080】
[0094] プロカスパーゼ−3の自己活性化を触媒するPAC−1の能力を直接評価するために、プロカスパーゼ−3タンパク質を100μM PAC−1と共に1〜5時間インキュベートした。図1Bでウエスタンブロットにより示すように、PAC−1は、時間依存的にプロカスパーゼ−3の切断を誘導し、4時間後に>50%の処理が観察された。対照的に、緩衝液中でインキュベートしたプロカスパーゼ−3は、同一の時間にわたり事実上自己活性化を示さない。PAC−1が相互に作用しているプロカスパーゼ−3の領域を正確に示すための試みでは、安全装置領域中の重要なアスパラギン酸の三つ組である残基Asp179、Asp180、及びAsp181においてアラニン置換を行った。これらの部位での変異はすべて、プロカスパーゼ−3を活性化するPAC−1の能力を劇的に低下させ、ある種の変異はPAC−1によるプロカスパーゼ−3の活性化に対してより有害であった(図2A)。
【0081】
[0095] カスパーゼ−3と同様に、カスパーゼ−7もまた、タンパク質分解により活性化される不活性な酵素前駆体として存在する。カスパーゼ−3及びカスパーゼ−7は共に実行因子カスパーゼであり、重要な配列及び構造上の相同性を有している(Denault,J.−Bら,2003)。プロカスパーゼ−7はまた、重要な三つ組中に3つのアスパラギン酸の代わりに2つのアスパラギン酸(Asp−Thr−Asp)しか有していないが、類似する安全装置領域を有するかもしれない。図2B中のデータにより示すように、PAC−1はまた、プロカスパーゼ−3のその活性化よりも効果が低いが、プロカスパーゼ−7を活性化することもできる(EC50はプロカスパーゼ−3活性化の0.22μMに対して4.5μM)。PAC−1によるプロカスパーゼ−7活性化の効力は、プロカスパーゼ−3のAsp−Ala−Asp変異体に対するその効果に類似している(EC50=2.77μM)。PAC−1の効果は、プロカスパーゼ−3が急速な自己活性化を起こす低pH値で無効にされる(図2C)。
【0082】
[0096] プロカスパーゼ−3を活性化してヒト細胞株におけるアポトーシスを誘導する低分子の能力について試験し、PAC−1は、様々な癌細胞株におけるアポトーシスを誘導することが分かった。HL−60細胞ではPAC−1の添加は、著しいクロマチン凝縮を伴う細胞膜上への多量のホスファチジルセリンの露出につながる(図3A及びB)。さらに、化合物は、PARP−1の切断を誘導し(生体内でのPARP活性アッセイによる評価;Putt KSら,2005)、ミトコンドリア膜脱分極を引き起こす(以下参照)。著しい細胞の小疱形成も顕微鏡検査により観察された。さらに、PAC−1の毒性は、カスパーゼ阻害剤z−VAD−fmk存在下で無効にすることができるかもしれない(データ掲載せず;Sleeら,1996参照)。
【0083】
[0097] PAC−1がプロカスパーゼ−3の直接的な活性化を介してアポトーシスを実際に誘導している場合、アポトーシス現象の時間的経過が、標準的なアポトーシス促進剤を用いて観察されるものに比べて変化するはずである。エトポシドは、内因性の経路を介してアポトーシスを誘導することでよく知られており、したがって、エトポシド処置細胞において、ミトコンドリア膜脱分極の後、プロカスパーゼ−3が活性化される。実際に、10μMエトポシドで処置したHL−60細胞では、ミトコンドリア膜脱分極が観察された後、カスパーゼ−3様活性が検出される(図4A)。対照的に、PAC−1での細胞の処置は著しく異なる結果をもたらす。PAC−1では、最初に観察されたアポトーシスの生化学的特徴はカスパーゼ−3様酵素活性である。この活性は、化合物添加の数分以内に認められ、50%の活性化が、2時間余りで、いかなる著しいミトコンドリア膜脱分極の十分前に起こる(図4B)。さらに、PARP活性は、PAC−1で処置した細胞では急速に低下するが、この低下は、エトポシド処置細胞ではより遅い時点で観察される(図4C)。対照実験は、PAC−1がPARP−1の酵素活性を直接抑制しないことを示す。アポトーシス現象の典型的な順序では、ミトコンドリア膜が脱分極し、カスパーゼが活性化され、カスパーゼ基質(PARP−1等)が切断される。PAC−1で処置した細胞が、カスパーゼ−3/−7の急速な活性化(ミトコンドリア膜脱分極前)及びカスパーゼ基質の急速な切断を示すといった観察は、この化合物が、プロカスパーゼ−3の直接的な活性化を介したその細胞毒性を発揮することを示す。
【0084】
[0098] さらにPAC−1の効力を明示するために、様々なレベルのプロカスパーゼ−3を有する癌細胞株における細胞死を誘導するこの化合物の能力を評価した。決定は、多数の癌細胞株(白血病、リンパ腫、黒色腫、神経芽細胞腫、乳癌、肺癌、及び腎臓癌)及び健常なドナーの骨髄から単離した白血球中に存在するプロカスパーゼ−3の量に関して行った。細胞死誘導に対するIC50値はこれらの細胞株におけるPAC−1に関して得られた。組み合わせたデータは、プロカスパーゼ−3の細胞内濃度及びPAC−1に対する感受性の間に強い相関を示す(図4D)。特に、健常なヒトドナーの骨髄に由来する白血球は、最低量のプロカスパーゼ−3を有するものに含まれ、PAC−1は、これらの細胞に対して比較的低毒性である。PAC−1は、肺癌細胞株NCI−H226に対して最も効力があり、0.35μMのIC50である。文献(Svingenら,2004)のデータに一致して、我々は、この細胞株が、非癌性対照のプロカスパーゼ−3の濃度の5倍を超える濃度を有することが分かった。
【0085】
[0099] PAC−1を用いたこれらの実験とは対照的に、エトポシドは、細胞培養中の効力及びプロカスパーゼ−3の細胞内レベルの間の上記相関を示さなかった。例えば、エトポシドは黒色腫細胞株のうちの3種(UACC−62、CRL−1872、及びB16−F10)、乳癌細胞株(Hs 578t)、及び肺癌細胞株(NCI−H226)での死の誘導において効果がなく(IC50>50μM)、これらの細胞株は、1.0、2.4、1.9、3.7、及び5.3のプロカスパーゼ−3レベルをそれぞれ有している。エトポシドは、4.3、4.0、4.7、及び4.4のプロカスパーゼ−3レベルをそれぞれ有するHL−60、U−937、SK−N−SH、及びPC−12に対して有効であった(IC50<1μM)。したがって、全体として、プロカスパーゼ−3レベルとエトポシドについてのIC50の間に相関はない。
【0086】
[00100] 癌性細胞は、通常、アポトーシスカスケードにおける種々様々なタンパク質の変異又は異常発現によりアポトーシス促進シグナルに対する感受性が低下している。そのため、癌の多くの型は、周知のように、アポトーシス細胞死に対する内在性シグナルだけでなく類似した機構を介して作用する化学療法剤にも抵抗性がある。ある種の癌におけるプロカスパーゼ−3発現レベルの奇異なアップレギュレーションは、この存在するタンパク質の細胞内プールを使用する機会を提供して、アポトーシスを直接誘導し、それにより、機能しない又は損なわれることが多いカスケードの上流の部分を回避する。プロカスパーゼ−3は、自己活性化を起こすことが相対的にできないことが周知であるが、それは、不活性状態に自身を維持するための12個のアミノ酸の安全装置に依存している。PAC−1は、in vitroで、プロカスパーゼ−3の自己活性化を誘導し、この活性化は、安全装置の重要なトリ−アスパラギン酸領域の変異により著しく減少する。このデータは、PAC−1が、プロカスパーゼ−3の休止状態を維持するための安全装置の能力を直接妨害するという見解と一致している。
【0087】
[00101] 細胞培養中、PAC−1処置は、急速なカスパーゼ−3様活性を誘導する。次いで、抗アポトーシスタンパク質(Bcl−2、Bcl−XL等)のカスパーゼ−3媒介性の切断は、ミトコンドリア膜の脱分極を誘導し、アポトーシスを増強すると思われる。さらに、様々な癌細胞株に対するPAC−1の効力は細胞中のプロカスパーゼ−3の濃度に正比例する。PAC−1が有効ないくつかの細胞株は、アポトーシスに対して抵抗性にする欠陥のあるアポトーシス経路を有していることに注目する価値がある。例えば、Apaf−1発現はSK−MEL−5細胞中で劇的に減少し、Bcl−2は、NCI−H226肺癌細胞株中で過剰発現する。
【0088】
[00102] 本明細書中に提示したデータは、プロカスパーゼ−3活性化化合物が一般的な癌に対して非常に有効となる可能性があるという見解を十分に支持する。有効性は、プロカスパーゼ−3レベルが異常に高度な状態に対して高めることができる。
【0089】
[00103] 癌生検材料のプロカスパーゼ−3レベルの評価は単純で迅速にすることができる。そのため、PAC−1等の化合物の考えられる有効性は、高度な精度で推測的に評価することができる。したがって、本明細書中のプロカスパーゼ−3活性化剤及び方法は、抗癌処置の分野で、一般的な細胞毒素に頼る治療法に優先される可能性があるオーダーメイド医療戦略を提供する。
【0090】
材料及び方法:
[00104] 材料:Ni−NTA樹脂及び抗Penta His Alexa Fluor 647抗体はQiagen社(バレンシア、CA)から購入した。ブラッドフォード染料はBio−Rad社(ハーキュリーズ、CA)から購入した。ピン移動装置はV&P Scientific社(サンディエゴ、CA)から購入した。試薬z−vad−fmkはCalbiochem社(サンディエゴ、CA)から購入した。大腸菌RosettaはNovagen社(マディソン、WI)から購入した。抗カスパーゼ−3抗体はSigma社(セントルイス、MO)から購入した。アネキシンV Alexa Fluor 488複合体、JC−9、及びヨウ化プロピジウムはMolecular Probes社(ユージーン、OR)から購入した。IPTG及びMTS/PMS CellTiter 96細胞増殖アッセイ試薬はPromega社(マディソン、WI)から購入した。ウシ胎児血清はBiomeda社(フォスターシティ、CA)から購入した。96及び384ウェルマイクロタイタープレート、顕微鏡用スライドガラス、顕微鏡用カバーガラス、ウマ血清、並びに他のすべての試薬はFisher社(シカゴ、IL)から購入した。
【0091】
[00105] 方法:細胞培養条件。U−937細胞、HL−60細胞、CRL−1872細胞、ACHN細胞、NCI−H226細胞、SK−MEL−5細胞、及びUACC−62細胞を10%FBSを添加したRPMI1640培地中で成長させた。SK−N−SH細胞、B16−F10細胞、及びHs 578t細胞を、アールのBSS、1.5g/L炭酸水素ナトリウムを有し、10%FBSを添加したイーグルの最小必須培地中で成長させた。PC−12細胞は、5%FBS及び10%ウマ血清を添加したRPMI1640培地中で成長させた。ヒト骨髄を、40%FBSを添加したIDMEM中で成長させた。すべての細胞株を、5%CO2、95%空気雰囲気中37℃でインキュベートした。U−937細胞及びHL−60細胞は必要に応じて2〜3日ごとに継代した。ヒト骨髄は、冷凍ストックから解凍し、直ちに希釈して実験に使用した。他のすべての細胞は、約80%の培養密度に達した時に継代した。
【0092】
[00106] タンパク質の発現及び精製。プロカスパーゼ−3又はプロカスパーゼ−7の発現プラスミドを含む大腸菌Rosettaの終夜培養物1mLを適切な抗生物質を含むLB培地1Lに接種した。細胞を1mM IPTGで30分間誘導した。次いで、細胞を遠心沈殿し、NTA結合緩衝液中に再懸濁した(150mM NaCl、50mM Tris、10mMイミダゾール、pH7.9)。フレンチプレスに2回通過させることにより細胞を溶解した。次いで、細胞溶解物を14,000×gで30分間回転させた。上清をデカントし、1mLのニッケル−NTA樹脂を加えた。細胞溶解物を4℃で1時間インキュベートした。樹脂をカラムに装填し、10mL NTA結合緩衝液、次に10mL NTA洗浄緩衝液で洗浄した(150mM NaCl、50mM Tris、20mMイミダゾール、pH7.9)。タンパク質を10mL NTA溶出緩衝液で1mL画分中に溶出した(150mM NaCl、50mM Tris、250mMイミダゾール、pH7.9)。タンパク質を含む画分をプールし、タンパク質の量をブラッドフォードアッセイを使用して測定した。
【0093】
[00107] ライブラリースクリーニング。単離したプロカスパーゼ−3をカスパーゼアッセイ緩衝液中に50ng/mLまで希釈した(50mM HEPES、100mM NaCl、10mM DTT、0.1mM EDTA、0.1%CHAPS、及び10%グリセロール、pH7.4)。45μLのプロカスパーゼ−3溶液をNunc社製384ウェル平底マイクロタイタープレートの各ウェルに加えた。約20,000種の化合物をスクリーニングした。約6,000種の化合物をイリノイ大学の化学科内の様々な供給源から収集した。それらの構造は、http://www.scs.uiuc.edu/〜phgroup/comcollections.html.から入手可能である。他の約14,000種の化合物をChembridge社(サンディエゴ、カリフォルニア)から購入した。PAC−1は、Chembridge社から購入した化合物のメンバーの1種であった。
【0094】
[00108] DMSO中10mMストック溶液として作製した化合物を、0.2μLの化合物を移動させる384ピン移動器具を使用してウェル中に移動させた。これにより、約40μMの最終化合物濃度を得た。DMSOのみ(化合物を含まず)をピン移動させて対照実験を行った。次いで、プレートを37℃で2時間インキュベートした。カスパーゼアッセイ緩衝液中Ac−DEVD−pNA(N−アセチル−ASP−Glu−Val−Asp−p−ニトロアニリド)の2mM溶液の5μLを各ウェルに加えた。次いで、プレートを、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きをカスパーゼ−3の活性を測定するために使用した。
【0095】
[00109] 活性化曲線。プロカスパーゼ−3活性化剤の用量依存性は、96ウェルプレート中のカスパーゼアッセイ緩衝液中50ng/mLのプロカスパーゼ−3、活性カスパーゼ−3、プロカスパーゼ−7、又は活性カスパーゼ−7の90μLに様々な濃度の化合物を加えることにより測定した。次いで、プレートを37℃で12時間インキュベートした。次いで、カスパーゼアッセイ緩衝液中Ac−DEVD−pNAの2mM溶液の10μLを各ウェルに加えた。プレートを、Spectra Max Plus 384ウェルプレートリーダー中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、非処置対照ウェルからの活性化における変化倍率を算出した。
【0096】
[00110] PAC−1活性化ゲル。プロカスパーゼ−3を正確に上記の通りに発現させ単離した。プロカスパーゼ−3をカスパーゼアッセイ緩衝液中約50μg/mLに希釈した。次いで、プロカスパーゼ−3を100μM PAC−1存在下又は非存在下で37℃で様々な時間インキュベートした。この培養の後、等量のローディング緩衝液(150mM NaCl、50mM Tris、2%SDS、20%グリセロール、pH8.0)を各プロカスパーゼ−3サンプルに加えた。次いで、時間的経過が終了するまで、すべてのサンプルを−80℃で保存した。次いで、すべてのサンプルを95℃で5分間インキュベートし、12%SDS−PAGEゲル上で泳動した。次いで、タンパク質を、ニトロセルロース紙に一晩移した。ブロットをTTBS中で洗浄し(150mM NaCl、50mM Tris、0.1%Tween−20、pH7.4)、10%ミルク溶液で2時間ブロックした。次いで、ブロットを、抗Penta His Alexa Fluor 647抗体の1:5000希釈液中で2時間インキュベートした。次いで、ブロットをTTBSで洗浄し、Typhoon蛍光スキャナーでスキャンした(Amersham Biosciences社、サニーベール CA)。
【0097】
[00111] 安全装置の変異。DDDプロカスパーゼ−3安全装置(配列番号1;配列番号2;配列番号9)を、ADD(配列番号3、配列番号4、配列番号10)、DAD(配列番号5、配列番号6、配列番号11)、及びDDA(配列番号7、配列番号8、配列番号12)に以下のプライマー、それぞれ、gacagacagtggtgttgCGgatgacatggcgtgtcataaaatacc(配列番号13)、gacagacagtggtgttgatgCtgacatggcgtgtcataaaatacc(配列番号14)、及びgacagacagtggtgttgatgatgCcatggcgtgtcataaaatacc(配列番号15)を用いたQuickchange法を使用して変異させた。図9及び図10も参照すること。変異した塩基は下線を引き、大文字で示す。遺伝子の全体にわたる適切な配列を保証するためにすべての変異体プラスミドについて配列を決定した。すべての変異体プラスミドは、上記の野生型プロカスパーゼ−3の通りに正確に発現した。96ウェルプレート中のカスパーゼアッセイ緩衝液中50ng/mLの野性型プロカスパーゼ−3及び変異体プロカスパーゼ−3の90μLに様々な濃度のPAC−1を加えることにより、各プロカスパーゼ−3変異体を活性化するPAC−1の能力を測定した。次いで、プレートを37℃で12時間インキュベートした。次いで、カスパーゼアッセイ緩衝液中Ac−DEVD−pNAの2mM溶液の10μLを各ウェルに加えた。プレートを、Spectra Max Plus 384ウェルプレートリーダー中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、各変異体についての活性の変化倍率を算出した。
【0098】
[00112] PAC−1のプロカスパーゼ−3の活性化に対するpHの影響。PAC−1によるプロカスパーゼ−3活性化に対するpHの影響は、pHカスパーゼアッセイ緩衝液(25mM MES、25mM Tris、25mM HEPES、25mM PIPES、100mM NaCl、10mM DTT、0.1mM EDTA、0.1%CHAPS、及び10%グリセロール)中にプロカスパーゼ−3を50ng/mLの濃度に希釈することにより測定した。次いで、緩衝液を様々なpH値に変化させ、90μLを96ウェルプレートの各ウェルに加えた。各pH値に対して、PAC−1を100μMの濃度まで加え、又は対照としてDMSOを加えた。次いで、プレートを37℃で12時間インキュベートした。次いで、カスパーゼアッセイ緩衝液中Ac−DEVD−pNAの2mM溶液の10μLを各ウェルに加えた。プレートを、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、各pH値に対する活性化における変化倍率を算出した。
【0099】
[00113] アネキシンV染色。200μM PAC−1又は対照としてDMSOのみを含む培地500μLを24ウェルプレートのウェルに加えた。次いで、2×106細胞/mLの濃度のHL−60細胞500μLを24ウェルプレートに加えた。プレートを37℃で20時間インキュベートした。細胞を遠心分離により回収し、PBS中で2回洗浄した。次いで、細胞をアネキシンV結合緩衝液(10mM HEPES、140mM NaCl、2.5mM CaCl2、pH7.4)中で洗浄し、100μLのアネキシンV結合緩衝液中に再懸濁した。5μLのアネキシンV Alexa Fluor 488複合体を加え、チューブを、光から保護して、室温で15分間インキュベートした。次いで、400μLのアネキシンV結合緩衝液を加え、次に、ヨウ化プロピジウムの1mg/mL溶液を1μL加えた。各細胞の蛍光強度を、525nm(緑色チャンネル)及び675nm(赤色チャンネル)でフローサイトメトリーにより測定した。少なくとも50,000個の細胞を各実験で分析した。
【0100】
[00114] 凝縮したクロマチンの染色。200μM PAC−1又は対照としてDMSOのみを含む培地500μLを24ウェルプレートのウェルに加えた。次いで、2×106細胞/mLの濃度のHL−60細胞500μLを24ウェルプレートに加えた。細胞を20時間インキュベートし、遠心分離により回収した。次いで、細胞をPBS緩衝液中で洗浄し、次に氷冷100%エタノールを加えた。細胞を一晩4℃で固定した。固定細胞を2μg/mLヘキスト−33258と共に室温で30分間インキュベートした。次いで、1滴の細胞を顕微鏡用スライドガラスに加え、厚さNo.1のカバーガラスでカバーした。凝縮したクロマチンをZeiss社製Axiovert 100顕微鏡上で400×倍率で観察した。
【0101】
[00115] z−vad−fmkによる細胞死阻害。5×105細胞/mLの濃度のHL−60細胞100μLを96ウェルプレートのウェルに加えた。次いで、細胞を、細胞透過性全カスパーゼ阻害剤であるz−vad−fmk 100μM存在下又は非存在下で1時間インキュベートした。次いで、PAC−1を様々な濃度で加え、細胞をさらに24時間インキュベートした。MTS/PMS CellTiter 96細胞増殖アッセイ試薬20μLの各ウェルへの添加により細胞死を定量した。プレートを、37℃で、約45分間、有色生成物が形成されるまでインキュベートした。次いで、吸光度を、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)で490nmで測定した。
【0102】
[00116] 生体内でのミトコンドリア膜電位の測定。1×106細胞/mLの濃度のHL−60細胞1mLを24ウェルプレートのウェルに加えた。次いで、PAC−1を100μMの濃度まで加え又は対照としてDMSOのみを加えた。細胞を様々な時間インキュベートし、次いで、細胞を遠心分離により回収した。細胞をPBS中で洗浄し、1mL PBS中で再懸濁した。JC−9染料10μgを加え、細胞を光から保護して、室温で10分間インキュベートした。次いで、細胞をPBSで2回洗浄し、500μL PBS中に調製した。各細胞の蛍光強度を、525nm(緑色チャンネル)及び675nm(赤色チャンネル)でフローサイトメトリーにより測定した。各実験で50,000個の細胞を分析した。次いで、赤色チャンネルにおけるシフトを使用してミトコンドリア膜脱分極の量を測定した。
【0103】
[00117] 生体内でのカスパーゼ−3様活性の測定。カスパーゼ−3様プロテアーゼ活性の量を、細胞溶解物により毎分切断されるAc−DEVD−pNA(N−アセチル−ASP−Glu−Val−Asp−p−ニトロアニリド)の量により測定した。これを達成するために、様々な濃度のPAC−1を含む培地50μLを96ウェルプレートのウェルに加えた。5×106細胞/mLの濃度のHL−60細胞50μLをプレートに加え、様々な時間インキュベートした。インキュベーション期間の後、プレートを1000×gで5分間回転させ、細胞をペレットにした。次いで、100μL PBS中で細胞を洗浄し、150μL氷冷カスパーゼアッセイ緩衝液中に再懸濁した。次いで、各ウェルを超音波処理して細胞を溶解させた。細胞溶解物90μLを各ウェルから新しいプレート中に移動した。200μMの最終濃度となるよう各ウェル中にAc−DEVD−pNAを加えた。次いで、プレートを、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、毎分切断されたAc−DEVD−pNAの量を算出した。
【0104】
[00118] 生体内でのPARP切断の測定。生体内でのPARP活性アッセイを使用することによりPARP切断の量を測定した。これを達成するために、200μM NAD+を含む培地50μLを96ウェルプレートの対照ウェルに加えた。次いで、200μM PAC−1及び200μM NAD+を含む培地50μLを試験ウェルに加えた。次いで、5×106細胞/mLの濃度のHL−60細胞25μLを各ウェルに加えた。細胞を様々な時間インキュベートし、次いで、1000×gで5分間回転させた。細胞培地を除去し、25mM H2O2を含む50μL PARP溶解緩衝液(50mM Tris、10mM MgCl2、pH8.0、1%Triton X−100)と置換した。次いで、プレートを37℃で60分間インキュベートした。まだ存在しているNAD+の量を測定するために、20μLの2M KOH及び20μLの20%(v/v)アセトフェノン(エタノール中)溶液を96ウェルプレートの各ウェルに加えた。次いで、プレートを4℃で10分間インキュベートした。90μLの88%(v/v)ギ酸溶液を96ウェルプレートの各ウェルに加えた。次いで、プレートを、110℃に設定したオーブン中で5分間インキュベートした。プレートを冷却し、次いで、Criterion Analyst AD(Molecular Devices社、サニーベール、CA)上で励起360nm、発光445nm、及び400nmカットオフ ダイクロイックミラーで読み取った。ウェル当たり5回の読み取りを実行しながら1.6×105μsの1000W連続ランプを使用して蛍光体を励起させた。次いで、毎分切断されたNAD+のモル数を算出し、対照ウェルと比較した残存するPARP活性を測定した。
【0105】
[00119] 様々な細胞株中のプロカスパーゼ−3の相対濃度。U−937細胞、HL−60細胞、及びヒト骨髄細胞は遠心分離により回収し、一方、他のすべての細胞株は、最初にトリプシン処理して細胞を遊離し、次いで、遠心分離により回収した。PBS中ですべての細胞を洗浄し、1mL氷冷100%エタノール中に再懸濁した。細胞を一晩4℃で固定した。細胞を1000×gで5分間回転させ、PBSで洗浄し、次いで、PBS中の抗カスパーゼ−3抗体の1:100希釈液100μLを加えた。細胞を室温で2時間インキュベートし、次に、5回PBS洗浄した。次いで、細胞を、光から保護して、抗マウスAb Cy3標識抗体の1:10,000希釈液1mL中に室温で2時間再懸濁した。PBSで細胞を5回洗浄し、500μL PBS中に再懸濁した。フローサイトメトリーにより675nm(赤色チャンネル)で各細胞の蛍光強度を測定した。少なくとも20,000個の細胞を各実験で分析した。集団の中央値を使用して各細胞株中のプロカスパーゼ−3の相対濃度を測定した。
【0106】
[00120] 様々な細胞株におけるIC50値の測定。様々な濃度のPAC−1又はエトポシドを含む培地50μLを、DMSOのみを含む対照ウェル以外の96ウェルプレートの各ウェルに加えた。U−937細胞、HL−60細胞、及びヒト骨髄細胞は遠心分離により回収し、一方、他のすべての細胞株は遠心分離前に最初にトリプシン処理した。次いで、細胞を培地中に再懸濁し、U−937細胞、HL−60細胞、及びヒト骨髄細胞については1×106細胞/mL又は他のすべての細胞株については50,000細胞/mLに希釈した。次いで、50μL細胞溶液を各ウェルに加え、エトポシド及びPAC−1に対してそれぞれ24時間又は72時間プレートをインキュベートした。MTS/PMS CellTiter 96細胞増殖アッセイ試薬20μLの各ウェルへの添加により細胞死を定量した。次いで、プレートを、37℃で、約1時間、有色生成物が形成されるまでインキュベートした。Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)で490nmで吸光度を測定した。
【0107】
[00121] データ分析:Summitソフトウェア(Cytomation社、フォートコリンズ CO)を使用して、すべてのフローサイトメトリー実験のデータを分析した。すべてのグラフを、Table Curve 2Dを使用して、分析した。
【0108】
[00122] Ronald Hoffman教授(イリノイ大学−シカゴ癌センター(Chicago Cancer Center))はヒト骨髄を提供した。Guy Salvesen教授(バーナム研究所)はプロカスパーゼ−3及びプロカスパーゼ−7の発現ベクターを提供した。
【0109】
[00123] 配列表の参照−添付A:添付Aと指定した個別に添付する配列表情報は、本明細書と共に明細書の一部とみなされ、明細書の一部として組み込まれるものとする。
【表1】
【0110】
〔実施例2:プロカスパーゼ活性化化合物の合成〕
[00124] PAC−1及び他の化合物は、以下のスキーム、例えば下記スキーム1及び/又はスキーム2に従って調製した。さらなる変形形態は、当技術分野で知られている方法に従って調製する。
【化8】
【0111】
[00125] 特定の実施例では、PAC−1は、下記スキーム2に従って調製する。
【化9】
【0112】
〔実施例3:PAC−1の類似体〕
[00126] PAC−1の類似(アナログ)化合物を調製し、精製したプロカスパーゼ−3をin vitroで直接活性化する能力について評価した。
【表2】
【0113】
〔実施例4:医薬品実施形態〕
[00127] 以下は、医薬品実施形態及び薬理学的実施形態に関連する情報を記載し、当業者にとって入手可能な当技術分野における情報によりさらに補足される。厳密な処方、投与経路、及び用量は、個々の医師が、患者の症状を考慮して選択することができる(例えばFinglら,in The Pharmacological Basis of Therapeutics,1975,Ch.1 p.1参照)。
【0114】
[00128] 毒性又は器官の機能不全等により、どのようにそしていつ投与を終了すべきか、中断すべきか、又は調整すべきかについて主治医が知っていたであろうということに注目するべきである。反対に、臨床反応が十分でなかった場合(毒性の側面を考慮して又は毒性の側面を防止する中で)、主治医はまた高度なレベルに処置を調整することも知っていたであろう。関心のある疾患の管理における投薬量の大きさは、処置される症状の重症度及び投与経路によって変動する可能性がある。症状の重症度は、例えば、部分的に、標準的な予後評価法により評価してもよい。さらに、投与量及びおそらく投与回数はまた、状況、例えば個々の患者の年齢、体重、及び反応に従って変動する可能性がある。上記に論じたものに相当するプログラムはまた獣医学に使用してもよい。
【0115】
[00129] 処置されている特定の症状及び選択された標的法によっては、上記薬剤を全身に又は局所的に処方及び投与してもよい。処方及び投与の技術は、Alfonso及びGennaro(1995)並びに当技術分野の他のものに認められてもよい。適切な経路は、例えば、経口投与、直腸投与、経皮投与、膣投与、経粘膜投与、若しくは腸管投与又は筋肉注射、皮下注射、若しくは髄内注射を含む非経口送達と同様に眼内投与、鞘内投与、静脈内投与、又は腹腔内投与を含んでいてもよい。
【0116】
[00130] 注射又は他の経路については、本発明の薬剤を水溶液中に、好ましくは、ハンクス溶液、リンガー溶液、注射用水、生理的食塩緩衝液、等の生理学的に適合性のある緩衝液中に処方してもよい。経粘膜投与については、浸透するバリアに適切な浸透剤を製剤に使用することができる。上記浸透剤は、当技術分野で一般に知られている。
【0117】
[00131] 本発明の実行に関して本明細書中に開示された化合物を全身性の又は他の投与に適した用量に処方するための薬学的に許容し得る担体の使用は本発明の範囲内である。担体が適切に選択され、かつ適正な製造が実施されれば、本発明の組成物、特に、溶液として処方される組成物は、静脈注射等の経路によって非経口的に投与されてもよい。適切な化合物は、当技術分野でよく知られている薬学的に許容し得る担体を使用して、経口投与に適した用量に容易に処方することができる。上記担体は、本発明の化合物を、錠剤、丸剤、カプセル剤、液剤、ゲル、シロップ剤、スラリー、エリキシル剤、溶液剤、懸濁剤等として、例えば処置される患者による経口摂取のために処方することを可能にする。他の経路については、クリーム剤、軟膏剤、ローション剤等の製剤を調製することができる。
【0118】
[00132] 細胞内に投与することを意図した薬剤は、当業者によく知られている技術を使用して投与されてもよい。例えば、上記薬剤を、リポソーム、他の膜移動促進部分、又は他の標的部分中にカプセル化し、次いで、上記のように投与してもよい。リポソームは、内部が水性の球状の脂質二重層を含むことができる。リポソーム形成時に水溶液中に存在するすべての分子を水性内部に取り込むことができる。リポソーム内容物は外部の微小環境から保護されるのみならず、リポソームが細胞膜と融合するので、細胞の細胞質に効率的に送達される。さらに、疎水性の特性により、有機低分子を直接細胞内に投与してもよい。
【0119】
[00133] 本発明で使用するのに適した医薬組成物は、意図された目的を達成するための有効な量の活性成分を含んでいる組成物を含む。有効な量の決定は、特に本明細書中に提供された本開示及び当技術分野における他の情報を考慮すれば十分に当業者の能力の範囲内である。
【0120】
[00134] 活性成分に加えて、これらの医薬組成物は、薬学的に使用することができる調製物への活性化合物の加工を促進する賦形剤及び助剤を含む、薬学的に許容し得る適当な担体を含んでいてもよい。経口投与のために処方された調製物は、錠剤、糖衣錠、カプセル剤、又は溶液剤の形態をしていてもよく、遅延して放出されるために処方された調製物又は医薬が小腸又は大腸に到達する場合に単に放出されるために処方された調製物を含む。
【0121】
[00135] 本発明の医薬組成物は、それ自体知られている方法、例えば従来の混合プロセス、溶解プロセス、懸濁プロセス、造粒プロセス、糖衣錠製造プロセス、浮上プロセス、乳化プロセス、カプセル化プロセス、封入プロセス、凍結乾燥プロセス及び他のプロセスによって製造してもよい。
【0122】
[00136] 非経口投与のための医薬製剤は水溶性形態の活性化合物の水溶液を含む。さらに、活性化合物の懸濁剤を適切な油性注射懸濁剤として調製してもよい。適切な親油性の溶媒又は賦形剤は、ゴマ油等の脂肪油;オレイン酸エチル、トリグリセリド等の合成脂肪酸エステル;又はリポソームを含む。水性注射懸濁剤は、カルボキシルメチルセルロースナトリウム、ソルビトール、デキストラン等の、懸濁剤の粘度を上昇させる物質を含んでいてもよい。任意選択で、高度に濃縮された溶液の調製を考慮に入れるために、懸濁剤は、化合物の溶解度を上昇させる適切な安定剤又は薬剤をさらに含んでいてもよい。
【0123】
[00137] 経口使用のための医薬調製物は、所望により、錠剤又は糖衣錠コアを得るために、活性化合物を固体賦形剤と混合し、任意選択で、結果として生じる混合物を粉砕し、適当な助剤を加えた後、顆粒の混合物を加工することにより得ることができる。適切な賦形剤は、特に、ラクトース、スクロース、マンニトール又はソルビトールを含む糖のような充填剤、並びに例えば、トウモロコシデンプン、コムギデンプン、コメデンプン、バレイショデンプン、ゼラチン、トラガカントゴム、メチルセルロース、ヒドロキシプロピルメチル−セルロース、カルボキシルメチルセルロースナトリウム及び/又はポリビニルピロリドン(PVP)のようなセルロース調製物である。所望により、架橋ポリビニルピロリドン、寒天、アルギン酸、又はアルギン酸ナトリウム等の塩、のような崩壊剤を加えてもよい。
【0124】
[00138] 適切なコーティングを糖衣錠コアに任意選択で施す。この目的のために、濃縮糖溶液を使用してもよく、濃縮糖溶液は、任意選択で、アラビアゴム、タルク、ポリビニルピロリドン、カーボポールゲル、ポリエチレングリコール、及び/又は二酸化チタン、ラッカー溶液、並びに適当な有機溶媒又は混合溶媒を含んでいてもよい。染料又は色素を、錠剤又は糖衣錠のコーティングに、識別のために又は活性化合物投与量の異なる組み合わせを特徴づけるために加えてもよい。
【0125】
[00139] 経口的に使用することができる医薬調製物は、ゼラチンで作製されたプッシュフィットカプセル剤並びにゼラチンで作製された軟質の密閉カプセル剤、及びグリセロール、ソルビトール等の可塑剤を含む。プッシュフィットカプセル剤は、ラクトース等の充填剤、デンプン等の結合剤、及び/又はタルク、ステアリン酸マグネシウム等の潤滑剤、並びに任意選択で安定剤と混合した活性成分を含むことができる。軟質のカプセル剤では、脂肪油、流動パラフィン、液体ポリエチレングリコール等の適当な液剤中に活性化合物を溶解又は懸濁してもよい。さらに、安定剤を加えてもよい。
【0126】
〔実施例5:プロカスパーゼ−3の低分子活性化剤での癌細胞におけるアポトーシスの直接的な誘導〕
[00140] 要約:アポトーシスカスケードにおけるタンパク質の変異又は異常発現は癌の特徴である。これらの変化は、アポトーシス促進シグナルが実行因子カスパーゼに伝達されるのを阻止し、したがってアポトーシス細胞死を阻止し、細胞増殖が許可される。カスパーゼ−3及びカスパーゼ−7は重要な実行因子カスパーゼであり、上流のシグナルにより活性化される不活性な酵素前駆体として存在する。重要なことには、プロカスパーゼ−3のレベルは非癌性対照に比べてある種の癌性細胞内で著しく高度である。本明細書で、我々は、プロカスパーゼ−3を活性カスパーゼ−3に220ナノモル濃度のEC50でin vitroで直接活性化し、様々な癌細胞株におけるアポトーシスを誘導する低分子(PAC−1)の同定について報告する。多くの知られている抗癌薬とは対照的に、PAC−1で処置した細胞はプロカスパーゼ−3の即時活性化を示し、PAC−1の有効性は、細胞に含まれるプロカスパーゼ−3の量に比例することが示される。in vitroでプロカスパーゼ−3を活性化しないPAC−1の誘導体はアポトーシス促進活性も有していない。一次大腸腫瘍から単離した癌性細胞は、同一患者からの隣接した非癌性組織の細胞よりも、PAC−1によるアポトーシス誘導にかなりの感受性がある。これらの癌性細胞は、隣接した非癌性一次組織の細胞よりも平均して約7倍のプロカスパーゼ−3を含んでいる。さらに、大腸癌腫瘍からの一次細胞のPAC−1に対する感受性は、プロカスパーゼ−3標的のレベルと強く関連している。最終的に、単一の実体としてのPAC−1は、PAC−1を経口的に投与した2つのモデルを含む3つの異なるマウスモデルにおいて腫瘍の成長を遅延させるのに活性のあるものとして示された。したがって、PAC−1は、生体内で、プロカスパーゼ−3をカスパーゼ−3に直接活性化し、それによって、欠陥のあるアポトーシス機構を有する細胞においてでさえこの化合物がアポトーシスを誘導することを可能にする。PAC−1は、プロカスパーゼ−3を直接活性化することで知られている最初の低分子であり、実行因子カスパーゼの直接的な活性化は、プロカスパーゼ−3レベルが上昇した多くの癌において有利であると証明することが可能である抗癌戦略である。
【0127】
[00141] 導入。癌の特徴は、本来のアポトーシスシグナルに対するその抵抗性である。癌型によっては、この抵抗性は、通常、アポトーシスカスケードにおける重要なタンパク質のアップレギュレーション若しくはダウンレギュレーション又はこれらのタンパク質をコードする遺伝子内の変異のいずれかによる。上記変化は、ミトコンドリア及びカスパーゼ−9を通る内因性アポトーシス経路並びにデスレセプター及びカスパーゼ−8の作用を伴う外因性アポトーシス経路の両方で起こる。例えば、p53、Bim、Bax、Apaf−1、FLIP、及びその他多くのものの適切なレベルの変化が癌で観察され、欠陥のあるアポトーシスカスケードにつながり、上流のアポトーシス促進シグナルが適切に伝達されず、実行因子カスパーゼであるカスパーゼ−3及びカスパーゼ−7が活性化されない。ほとんどのアポトーシス経路がプロカスパーゼ−3の活性化を最終的に伴うので、これらの遺伝的異常は、有効に、アポトーシス回路における「断線箇所」となり、その結果、上記細胞は制御されずに増殖する。
【0128】
[00142] 癌におけるアポトーシスの中心的な役割から判断して、アポトーシスカスケードにおける特定のタンパク質を標的にする治療を開発するための試みがなされた。例えば、p53、Bclファミリーのタンパク質、又はIAPに対するペプチド結合剤又は低分子結合剤は、Apaf−1のオリゴマー化を促進する化合物のように、アポトーシス促進活性を有している。しかしながら、これらの化合物の多くは、アポトーシスカスケード上の早期に又は中間に位置するものを標的にするので、下流のタンパク質における変異を有する癌は、それらの効果におそらく抵抗性があるであろう。治療目的のために、アポトーシスカスケードにおけるはるか下流のアポトーシス促進タンパク質を直接活性化する低分子を同定すること理想的であろう。さらに、上記治療戦略は、アポトーシス促進タンパク質のレベルが癌細胞内で上昇した場合、成功の可能性がより高いであろう。
【0129】
[00143] プロカスパーゼ−3のカスパーゼ−3への変換により、多数のタンパク質基質の加水分解を続いて触媒する活性「実行因子」カスパーゼの生成がもたらされる。活性カスパーゼ−3は、ヘテロダイマーのホモダイマーで、プロカスパーゼ−3のタンパク質分解により産生される。生体内では、このタンパク質分解性の活性化は、通常、カスパーゼ−8又はカスパーゼ−9の作用を介して生じる。この酵素前駆体が早まって活性化されないことを確実にするために、プロカスパーゼ−3は、タンパク質分解のIETD部位への到達をブロックするトリ−アスパラギン酸「安全装置」を有している。この安全装置は、プロカスパーゼ−3が、自己触媒活性化及びカスパーゼ−9によるタンパク質分解に抵抗することを可能にする。安全装置の部位はpHに感受性である。したがって、細胞の酸性化(アポトーシス中に起こる)の際、安全装置は、タンパク質分解の部位への到達を許可すると思われ、活性カスパーゼ−3は、カスパーゼ−9の作用により又は自己活性化機構を介して産生することができる。
【0130】
[00144] 癌性組織のある種の型からの細胞はプロカスパーゼ−3のレベルが上昇している。20人の大腸癌患者からの一次試料に関する研究は、平均して、プロカスパーゼ−3が、隣接した非癌性組織に比べて上記試料中で6倍上昇したことを明らかにした。さらに、プロカスパーゼ−3レベルは、ある種の神経芽細胞腫、リンパ腫、及び肝臓癌において上昇する。実際、NCIにより使用された60枚の細胞株パネルにおけるプロカスパーゼ−3レベルの系統的評価は、特定の肺癌、黒色腫、腎臓癌、及び乳癌がプロカスパーゼ−3の著しく増強したレベルを示すことを明らかにした。アポトーシスを成功させる活性カスパーゼ−3の中心的な重要性、ある種の癌性細胞型におけるプロカスパーゼ−3の高度なレベル、及び興味深い、安全装置媒介性のその自己活性化の抑制から判断して、プロカスパーゼ−3を直接活性化する低分子が同定される可能性があるということ及び上記分子は、標的癌治療法において多大な可能性を有する可能性があるということを我々は推論した。この原稿で、我々は、プロカスパーゼ−3の低分子活性化剤であるPAC−1のin vitroの同定について報告する。PAC−1は、癌細胞株において、プロカスパーゼ−3レベルに比例してアポトーシス促進性に効果的であり、そのアポトーシス促進効果はプロカスパーゼ−3のその直接的で即時の活性化によるものであり、一次大腸癌試料に対して及び3つの異なる癌のマウスモデルにおいて有効である。
【0131】
[00145] 約20,500種の構造的に多様な低分子を、in vitroのプロカスパーゼ−3を活性化する能力に対してスクリーニングした。プロカスパーゼ−3を、標準的な手順に従って、大腸菌中で発現させ、精製した。プロカスパーゼ−3を、384ウェルプレートのウェルに加え、化合物を、約40μMの最終濃度まで加えた(プロカスパーゼ−3の最終濃度は50ng/mLであった)。次いで、各プレートを37℃で2時間インキュベートし、その後、カスパーゼ−3ペプチド基質Ac−Asp−Glu−Val−Asp−p−ニトロアニリド(Ac−DEVD−pNa)を200μMの濃度まで加えた。p−ニトロアニリン発色団の形成を405nmで2時間にわたり追跡した。評価した〜20,500種の化合物のうち、4種が、バックグラウンドに対してカスパーゼ−3ペプチド基質の加水分解の著しい増加を誘導した。それらの4種のうち、1種は、in vitroのプロカスパーゼ−3活性化に対する強い用量依存効果を示した。図1Aに示すように、この最初のプロカスパーゼ活性化化合物(PAC−1)は、0.22μMの濃度で、プロカスパーゼ−3の最大活性化の2分の1をもたらす。この化合物は、完全に活性化されたカスパーゼ−3酵素の触媒活性に対する効果を有していないので、カスパーゼ−3自体の活性を単に上昇させているのではない(図1A)。
【0132】
[00146] プロカスパーゼ−3はN末端プロドメイン(残基1〜28)、それに続く、サブユニット間のリンカーにより分けられた大サブユニット(17kDa)及び小サブユニット(12kDa)からなる22。生体内では、2つのプロカスパーゼ−3モノマーが集合して、サブユニット間のリンカー中のD175での切断により活性化することができるホモダイマーを形成する。プロドメインの明確な役割は不明であり、最高の触媒活性には、サブユニット間の領域の切断のみで十分であることが示された。プロカスパーゼ−3は、それ自身のタンパク質分解性の成熟を推進するための十分な触媒活性を有しているが、3つのアミノ酸の安全装置の存在によりこの自己活性化に対して高度に抵抗性がある。しかしながら、安全装置が変異した場合、プロカスパーゼ−3の著しい自己活性化が観察される。プロカスパーゼ−3の活性カスパーゼ−3への成熟を触媒するPAC−1の能力を直接評価するために、プロカスパーゼ−3タンパク質を100μM PAC−1と共に1〜5時間インキュベートした。図1Bでウエスタンブロットにより示すように、PAC−1は時間依存的にプロカスパーゼ−3の切断を誘導し、4時間後に>50%の処理が観察された。対照的に、緩衝液中でインキュベートしたプロカスパーゼ−3は、同一の時間にわたり実質的に自己活性化を示さない。PAC−1はまた、このアッセイにおいて、5μMの濃度でも有効であった。
【0133】
[00147] 次いで、プロカスパーゼ−3の安全装置領域中の重要なアスパラギン酸の三つ組である残基Asp179、Asp180、及びAsp181においてアラニン置換を行った。これらの部位での変異はすべて、プロカスパーゼ−3を活性化するPAC−1の能力を劇的に低下させ、ある種の変異はPAC−1によるプロカスパーゼ−3の活性化に対してより有害であった(図2A)。カスパーゼ−3と同様に、カスパーゼ−7もまた、タンパク質分解により活性化される不活性な酵素前駆体として存在する。カスパーゼ−3及びカスパーゼ−7は共に実行因子カスパーゼであり、かなりの構造上の相同性を有している。プロカスパーゼ−7はまた、重要な三つ組中に3つのアスパラギン酸の代わりに2つのアスパラギン酸(Asp−Thr−Asp)しか有していないが、類似する安全装置領域を有することが予測される。図2B中のデータにより示すように、PAC−1は、さらに、プロカスパーゼ−3のその活性化よりも効果が低いが、プロカスパーゼ−7を活性化することもできる(EC50はプロカスパーゼ−3活性化の0.22μMに対して4.5μM)。PAC−1によるプロカスパーゼ−7活性化の効力は、プロカスパーゼ−3のAsp−Ala−Asp変異体に対するその効果に類似している(EC50=2.77μM)。予想どおり、PAC−1の効果は、プロカスパーゼ−3が急速な自己活性化を起こす低pH値で無効にされる(図2C)。
【0134】
[00148] PAC−1は、様々な癌細胞株におけるアポトーシスを誘導することが分かった。HL−60細胞では、PAC−1の添加は、著しいクロマチン凝縮を伴う細胞膜上への多量のホスファチジルセリンの露出につながる(図3A、3B)。さらに、化合物は、カスパーゼ基質PARP−1の切断を誘導し(生体内でのPARP活性アッセイによる評価)、ミトコンドリア膜脱分極を引き起こす(以下参照)。PAC−1処置細胞の著しい細胞の小疱形成も顕微鏡検査により観察された。さらに、PAC−1の毒性は、カスパーゼ阻害剤z−VAD−fmk存在下で無効にすることができるかもしれない。
【0135】
[00149] PAC−1がプロカスパーゼ−3の直接的な活性化を介してアポトーシスを実際に誘導している場合、アポトーシス現象の時間的経過が、標準的なアポトーシス促進剤を用いて観察される現象に比べて変化するはずである。エトポシドは、内因性の経路を介してアポトーシスを誘導することでよく知られており、したがって、エトポシド処置細胞において、ミトコンドリア膜脱分極の後、プロカスパーゼ−3が活性化される。実際に、10μMエトポシドで処置したHL−60細胞では、ミトコンドリア膜脱分極が観察された後、カスパーゼ−3様活性が検出される(図4A)。対照的に、PAC−1での細胞の処置は著しく異なる結果をもたらす。この化合物では、最初に観察されたアポトーシスの生化学的特徴はカスパーゼ−3様酵素活性であり、活性はPAC−1添加の数分以内に認められ、50%の活性化が、2時間余りで、いかなる著しいミトコンドリア膜脱分極の十分前に起こる(図4B)。さらに、PARP−1活性は、PAC−1で処置した細胞では急速に低下するが、この低下は、エトポシド処置細胞ではより遅い時点で観察され(図4C)、対照実験は、PAC−1がPARP−1の酵素活性を直接抑制しないことを示す。アポトーシス現象の典型的な順序では、ミトコンドリア膜が脱分極し、カスパーゼが活性化され、カスパーゼ基質(PARP−1等)が切断される。PAC−1で処置した細胞がプロカスパーゼ−3/−7の急速な活性化(ミトコンドリア膜脱分極前)及びカスパーゼ基質(PARP−1)の急速な切断を示すといった観察は、PAC−1がプロカスパーゼ−3の直接的な活性化を介してその細胞毒性を発揮していることを示す。
【0136】
[00150] さらにPAC−1の効力を明示するために、様々なレベルのプロカスパーゼ−3を有する癌細胞株における細胞死を誘導するこの化合物の能力を評価した。決定は、多数の癌細胞株(白血病、リンパ腫、黒色腫、神経芽細胞腫、乳癌、肺癌、副腎癌、及び腎臓癌)に存在するプロカスパーゼ−3のレベルに関して最初に行った。細胞死誘導に対するIC50値はこれらの細胞株に対するPAC−1に関して得られた。組み合わせたデータは、プロカスパーゼ−3の細胞内濃度及びPAC−1に対する感受性の間に強い相関を示す(図4D、図4E)。PAC−1は、肺癌細胞株NCI−H226に対して最も効力があり、0.35μMのIC50である。我々は、この細胞株が、ベースラインレベルのプロカスパーゼ−3の濃度の5倍を超える濃度を有することが分かった。重要なことには、プロカスパーゼ−3の発現を有していないことで知られているある癌細胞株(MCF−7、乳癌細胞)がある。PAC−1はMCF−7細胞に事実上効果がなく、IC50>75μMで死を誘導する。
【0137】
[00151] 対照的に、エトポシドは、細胞培養中の効力及びプロカスパーゼ−3の細胞内レベルの間の上記相関を示さなかった。例えば、エトポシドは黒色腫細胞株のうちの3種(UACC−62、CRL−1872、及びB16−F10)、乳癌細胞株(Hs 578t)、及び肺癌細胞株(NCI−H226)での死の誘導において効果がなく(IC50>50μM)、これらの細胞株は、1.0、2.4、1.9、3.7、及び5.3のプロカスパーゼ−3レベルをそれぞれ有している。エトポシドは、4.3、4.0、4.7、及び4.4のプロカスパーゼ−3レベルをそれぞれ有するHL−60、U−937、SK−N−SH、及びPC−12に対して有効であった(IC50<1μM)。したがって、全体として、プロカスパーゼ−3レベル及びエトポシドについてのIC50の間に相関はない。
【0138】
[00152] PAC−1のいくつかの誘導体を合成し、それらのプロカスパーゼ−3活性化特性及び細胞培養中の癌細胞に対するそれらの効果の両方を評価した(表3)。アリル基を欠くPAC−1誘導体(非アリルPAC−1)はPAC−1と同様のレベルでプロカスパーゼ−3活性化及び細胞死を誘導することができる。しかしながら、他のすべての誘導体はいずれのアッセイにおいても活性を示さなかった。したがってアリル基は生物活性には重要でないと思われる一方で、フェノール性水酸基の環及び芳香環はすべて、PAC−1活性に重要である。このデータはまた、提案されたPAC−1の作用機構とも一致しており、in vitroで、プロカスパーゼ−3を活性化しない化合物は培養中の癌細胞に対するアポトーシス促進効果を有していない。
【0139】
[00153] 癌の臨床試料におけるこの直接的な低分子媒介性のプロカスパーゼ−3活性化戦略を試験するために、我々は、新たに切除した大腸腫瘍(隣接した非癌性組織と共に)を、カール財団病院(Carle Foundation Hospital)(アーバナ、IL)の18人の患者から得た。癌性組織及び非癌性組織を分け、これらに由来する細胞を、それらのプロカスパーゼ−3のレベル及びPAC−1に対するそれらの感受性について評価した。図5Aに示すように、すべての患者で、癌性細胞は、同一患者の隣接した非癌性組織からの細胞に比べて、プロカスパーゼ−3のレベルが上昇していた(1.7倍〜17.2倍、平均で7.6倍の上昇)。さらに、これらの癌性細胞は、PAC−1による死の誘導にかなり影響を受けやすかった。PAC−1は、0.007〜1.41μMのIC50値で一次癌性細胞における細胞死を誘導し、一方、PAC−1は、5.02〜9.98μMのIC50値で隣接した非癌性組織における細胞死を誘導した(図5B及び表4)。プロカスパーゼ−3のレベルが上昇した癌性組織はPAC−1に非常に感受性があった。例えば、PAC−1は、7nMのIC50で、患者17からの癌細胞における死を誘導した。これらの細胞は、隣接した正常組織からの細胞よりもPAC−1に対して700倍を超える感受性があった。患者1、2、及び3からの正常サンプル及び癌性サンプル中の約54日の期間にわたる相対的なプロカスパーゼ−3濃度を示す図6Aもまた参照のこと。図6Bは、癌性組織における細胞が、正常組織と比較してPAC−1に対して約80倍を超える感受性がある可能性があることを示す。
【0140】
[00154] 18人の患者の非癌性組織からの細胞に加えて、4種の他の非癌性細胞型に対してもPAC−1を評価した:健常なドナーの骨髄から単離した白血球、Hs888Lu(肺繊維芽細胞)、MCF−10A(乳房の繊維芽細胞)、及びHs578Bst(乳房上皮細胞)。特に、非癌性細胞型は、最低量のプロカスパーゼ−3を有する細胞型に含まれ、PAC−1は、これらの細胞における死を比較的誘導することができず、IC50値は3.2〜8.5μMであった(図5B、緑色の菱形)。図5Bから明らかなように、PAC−1は、プロカスパーゼ−3のレベルと直接関連して、種々様々な細胞型(非癌性細胞株、非癌性一次細胞、癌性細胞株、一次癌性細胞)における死を誘導する。癌性細胞におけるプロカスパーゼ−3の上昇は、PAC−1が選択的にこれらの細胞型における死を誘導することを可能にする。
【0141】
[00155] 薬物送達の徐放法を使用して、マウス異種移植モデルにおいてPAC−1を評価した。このモデルにおいて、ACHN(腎臓癌)細胞株を使用して、卵巣切除した雌の無胸腺BALB/c(ヌード)マウスに皮下腫瘍を形成させた。腫瘍を測定して約30mm2よりも大きければ、PAC−1及びコレステロールのペレットの移植を介して薬物を投与し、遅く安定したレベルで化合物を放出させた。3グループのマウスを使用し、ペレットは0mg、1mg、及び5mgのPAC−1を含み、グループ当たり6匹のマウス、マウス当たり4つの腫瘍とした。腫瘍の大きさを約8週間監視した。図5Cに示すように、腫瘍成長は、5mgのPAC−1を含むペレットを移植したマウスで著しく遅延した。実験の最後の週の摂食評価は、マウスの3グループ間での摂食量における差異を示さなかった。マウスを屠殺した後、各マウスから血漿サンプルを得、それぞれのPAC−1含有量を分析した。PAC−1の5mgのペレットを与えたマウスについては、この分析は、PAC−1が、血漿中に5nMの濃度で54日の実験の後に存在することを明らかにした。
【0142】
[00156] 第2のマウス異種移植モデルにおいてPAC−1を評価し、このモデルには薬物送達法として経口投与を使用した。このモデルにおいて、NCI−H226(肺癌)細胞株を使用して、雄の無胸腺BALB/c−nu/nuマウス(5週令、SLC、Hamamaysu、日本)に皮下異種移植腫瘍を形成させ、グループ当たり8匹のマウス、マウス当たり3つの腫瘍とした。マウスに腫瘍を形成させた後、経口栄養で、1日1回、21日間、0、50、又は100mg/kgの濃度でマウスをPAC−1で処置し、1週目後に屠殺した。図5D中のグラフにより明確に示されるように、PAC−1の経口投与は用量依存的に腫瘍成長を著しく遅延させる。
【0143】
[00157] 最終的に、雄の無胸腺BALB/c−nu/nuマウスに尾静脈注射を介してNCI−H226細胞を注射したマウスモデルでPAC−1を評価した。全実験は28日間続いた。1日1回、PAC−1(100mg/kg)で、経口栄養により、1〜4日目及び7〜11日目にマウスを処置した。他の日は、マウスにPAC−1を与えなかった。マウスの第2のグループに賦形剤のみを与えた。28日後にマウスを屠殺し、それらの肺を検査した。図5Eに示すように、対照マウスの肺(明白な灰色の腫瘤を有する)及びPAC−1処置マウスの肺の間に明らかな相違がある。ゲフィチニブ(イレッサ(商標);アストラゼネカ社)で処置した動物からのパネルにおいても結果を示す。
【0144】
[00158] 癌性細胞は、アポトーシスカスケードにおける種々様々のタンパク質の変異又は異常発現により、通常、アポトーシス促進シグナルに対する感受性が低下している。そのため、癌の多くの型は、周知のように、アポトーシス細胞死に対する内在性シグナルだけでなく類似した機構を介して作用する化学療法剤にも抵抗性がある。ある種の癌におけるプロカスパーゼ−3レベルの奇異な上昇は、この存在するタンパク質の細胞内プールを使用する機会を提供して、アポトーシスを直接誘導し、それにより、機能しないことが多いカスケードの上流の部分を回避する。PAC−1は、in vitroで、プロカスパーゼ−3の自己活性化を誘導する。細胞培養中、PAC−1処置は、急速なカスパーゼ−3様活性を誘導する。次いで、抗アポトーシスタンパク質(Bcl−2、Bcl−XL等)のカスパーゼ−3媒介性の切断は、ミトコンドリア膜の脱分極を誘導し、アポトーシスを増強すると思われる。さらに、様々な癌性細胞型及び非癌性細胞型に対するPAC−1の効力は細胞中のプロカスパーゼ−3の濃度に比例する。切除した大腸腫瘍から単離した一次癌性細胞は、プロカスパーゼ−3のレベルが上昇しているので、これらの細胞は、隣接した非癌性組織からの細胞よりもPAC−1に対してかなり感受性がある。PAC−1が有効ないくつかの細胞株は、アポトーシスに対して抵抗性にする欠陥のあるアポトーシス経路を有していることに注目する価値がある。例えば、Apaf−1発現はSK−MEL−5細胞中で劇的に減少し、Bcl−2は、NCI−H226肺癌細胞株中で過剰発現する。最終的に、PAC−1は、PAC−1を経口的に投与した2つのモデルを含む癌の3つの異なるマウスモデルで有効である。
【0145】
[00159] 本明細書中に提示したデータは、プロカスパーゼ−3活性化化合物が、プロカスパーゼ−3レベルが異常に高度な様々な一般的な癌に対して非常に有効となる可能性があるという見解を支持する。癌生検材料のプロカスパーゼ−3レベルの評価は単純で迅速であり、そのため、PAC−1等の化合物の考えられる有効性は、高度な精度で推測的に評価することができる。上記オーダーメイド医療戦略は一般的な細胞毒素に頼る治療法に優先される可能性があり、抗癌治療法に価値がある可能性がある。
【0146】
[00160] Guy Salvesen教授(バーナム研究所)はプロカスパーゼ−3及びプロカスパーゼ−7の発現ベクターを提供した。
【0147】
〔図の説明〕
[00161] 図1及び図2:PAC−1の構造は明細書の他のところで示す。図1A)in vitroでのPAC−1によるプロカスパーゼ−3の活性化及び活性カスパーゼ−3。PAC−1は、EC50=0.22μMでプロカスパーゼ−3を活性化する。図1B)PAC−1により誘導される活性カスパーゼ−3へのプロカスパーゼ−3の切断。プロカスパーゼ−3をN末端His−6タグを用いて大腸菌中で組換え発現させ、精製した。免疫ブロットは抗His−6抗体を用いて行った。PAC−1非存在下でプロカスパーゼ−3の成熟は観察されない。100μM PAC−1存在下で、p19フラグメントを生成する切断が1時間以内に観察され、>50%の切断が4時間後に観察される。PAC−1はまた、このアッセイにおいて5μMでも有効である。図2A)プロカスパーゼ−3の「安全装置」領域における変異体のPAC−1による活性化。PAC−1は、野性型プロカスパーゼ−3(DDD)に対して0.22μMの活性化のためのEC50並びに変異体については2.77μM(DAD)、113μM(DDA)、及び131μM(ADD)の対応するEC50値を有している。図2B)PAC−1は、4.5μMのEC50でプロカスパーゼ−7を活性化する。図2C)プロカスパーゼ−3のPAC−1による活性化についてのpHへの依存性。低pHでは、安全装置はオフの状態にあり、プロカスパーゼ−3は本質的に最大限に活性化される。エラーバーは、平均値からの標準偏差を表す。
【0148】
[00162] 図3及び図4:PAC−1は、HL−60細胞におけるアポトーシスを誘導する。図3A)100μM PAC−1での20時間の処置の後のホスファチジルセリン露出(アネキシン−V染色により測定される)。PAC−1はまた、このアッセイにおいて5μMでも有効である(裏付けとなる図2を参照)。図3B)100μM PAC−1での20時間の処置の後のヘキスト染色により可視化されるクロマチン凝縮。図4A)10μMエトポシドで処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。図4B)100μM PAC−1で処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。図4C)PAC−1処置(100μM)は、細胞内カスパーゼ−3/−7の即時活性化と一致する、HL−60細胞における細胞内PARP活性の急速な減少を誘導する。対照的に、エトポシド(10μM)処置細胞は、ずっと後の時点でのPARP活性の減少を示す。図4D及び図4E)PAC−1は、プロカスパーゼ−3依存的に細胞死を誘導する。多くの多様な癌性細胞株について、プロカスパーゼ−3レベルを求め(プロカスパーゼ−3に対する抗体を用いたフローサイトメトリーにより)そしてPAC−1のIC50を測定した(様々なPAC−1濃度での72時間の処置及びMTSアッセイを使用した定量化により)。PAC−1は、高度なレベルのプロカスパーゼ−3を有するとして知られているNCI−H226肺癌細胞株において相当効力がある(IC50=0.35μM)。エラーバーは、平均値からの標準偏差を表す。
【0149】
[00163] 表3:PAC−1及び非アリルPAC−1はプロカスパーゼ−3をin vitroで活性化し、細胞培養中の癌細胞における死を誘導するが、他の構造類似体は、in vitroでのプロカスパーゼ−3活性化効果を有しておらず、細胞培養中に死の誘導をもたらさない。
【0150】
[00164] 図5:図5A)プロカスパーゼ−3レベルは、新たに切除した大腸癌組織に由来する細胞内で上昇する。新たに切除した一次大腸腫瘍(隣接した非癌性組織と共に)を18人の異なる患者から得、癌性組織及び非癌性組織を分け、プロカスパーゼ−3に対する抗体及びフローサイトメトリーを使用して、プロカスパーゼ−3レベルをそれぞれについて測定した。平均して、癌性組織からの細胞は、同一患者からの隣接した非癌性組織に由来する細胞と比較してプロカスパーゼ−3が7.6倍上昇している。図5B)PAC−1は、プロカスパーゼ−3の細胞内レベルに比例して細胞死を誘導する。赤色の円は、18個の大腸腫瘍からの一次癌性細胞を表す。黒色の三角は、図4D中に示した同一の癌細胞株を表す。緑色の菱形は4種の非癌性細胞型である:Hs888Lu(肺繊維芽細胞)、MCF−10A(乳房の繊維芽細胞)、Hs578Bst(乳房上皮細胞)、及び健常なドナーの骨髄から単離した白血球。青色の四角は、18人の患者の腫瘍縁から単離した一次非癌性細胞である。表4)一次大腸癌組織に由来する細胞は、同一患者からの隣接した非癌性組織に由来する細胞よりも、PAC−1による死の誘導にかなりの感受性がある。図5C)PAC−1は、癌の異種移植モデルにおける腫瘍の成長を低下させる。ACHN(腎臓癌)細胞株を用いて皮下注射により腫瘍を形成させ、各グループに6匹のマウス、マウス当たり4つの腫瘍とした。腫瘍が約30mm2まで成長したら、PAC−1を、コレステロールペレットとして移植した。エラーバーは、平均値からの標準誤差を表す。図5D)PAC−1の経口投与はマウス異種移植モデルにおける腫瘍成長を著しく遅延させる。NCI−H226(肺癌)細胞株を使用して皮下注射により腫瘍を形成させ、各グループに8匹のマウス、マウス当たり3つの腫瘍とした。1日1回、経口栄養により、1〜21日目に、PAC−1又は賦形剤を投与した。エラーバーは、平均値からの標準誤差を表す。図5E)PAC−1の経口投与は、i.v.注射モデルにおける腫瘍成長を著しく遅延させる。マウスに、NCI−H226(肺癌)細胞株をi.v.注射した。本文中に記載されるプロトコールに従って経口栄養によりPAC−1(100mg/kg)でマウスを処置した。画像は、PAC−1を与えられず、肺に大量の灰色の腫瘤を有するマウスの肺を示す。対照的に、PAC−1を与えたマウスは目に見える灰色の物体をほとんど有していない。
【0151】
【表3】
【0152】
【表4】
【0153】
〔実施例6:肺癌のマウスモデルにおけるPAC−1の試験〕
[00165] NCI−H226(肺癌)細胞を使用して、異種移植モデルを用いた。PAC−1を腹腔内に(i.p.)10mg/kg与えた。40mg/kgのゲフィチニブ(イレッサ(商標);アストラゼネカ社、ウィルミントン、デラウェア)を用い、グループ当たり5匹のマウスを使用して有効性の比較を行った。図7に結果を示すが、結果は、PAC−1が、腫瘍容積という点で成長を低下させることと関連したことを示す。
【0154】
〔実施例7:コンビナトリアル誘導体、コンビナトリアル合成、及び治療上の使用〕
[00166] 多くの化合物をPAC−1構造の誘導体として調製する。ヒドラジド基をアルデヒド基と反応させ、誘導体化合物のコンビナトリアルライブラリーを得る。
【0155】
[00167] L1〜L20と示したヒドラジド前駆物質グループ(AX)の任意の1つを使用して、1〜28と示したアルデヒドグループ(BX)の任意の1つと反応するヒドラジドを生成し、それによって、560種のPAC−1誘導体化合物を得る。本明細書中に記載される方法を使用し、当技術分野で入手可能な知識に従って、誘導体化合物を合成する。図8A及び8Bに加えて以下のスキーム及び成分の構造を参照のこと。
【0156】
【化10】
【0157】
【化11】
【0158】
【化12】
【0159】
[00168] 一実施形態では、上記誘導体化合物を、例えば、活性、溶解度、毒性、安定性、及び/又は医薬品の用途に関連したその他の特性を変化させるためにさらに修飾する。
【0160】
[00169] 誘導体化合物を抗癌薬剤として使用する。化合物は、抗腫瘍活性、アポトーシス制御、及び/又はプロカスパーゼ−3活性化を有することが可能であるとして確認されている。例えば、新たに除去した大腸癌の一次試料を、プロカスパーゼ−3レベル及び試験化合物レベルに対する細胞の感受性を評価するために使用する。ここで試験化合物はPAC−1又は誘導体化合物である。正常細胞に対する、癌性細胞における細胞死を誘導する傾向に関して、化合物を分類する。
【0161】
[00170] 誘導体化合物のさらなる評価において、in vitro及び生体内での試験を行う。肝臓ミクロソームへの暴露に関する安定性を評価する。
【0162】
〔実施例8:PAC−1と比べたある種の誘導体の活性〕
[00171] PAC−1及びある種の誘導体をHL−60細胞株で試験し、IC50値を測定した。表5に結果を示す(L−及びR−の指定は上記のAX及びBXのシリーズに示した構造をそれぞれ指す)。PAC−1誘導体のいくつかは、概してPAC−1化合物の活性レベルよりも約1桁大きい活性レベルを示した。
【0163】
【表5】
【参照による援用及び変形形態に関する記載】
【0164】
[00172] 本出願にわたるすべての参考文献、例えば発行された若しくは許可された特許又は均等物を含む特許文献、特許出願公開、非公開特許出願、及び非特許文献又は他の資料は、それぞれの参考文献が本出願における本開示と少なくとも部分的に矛盾していない程度に個々に引用されるように(例えば、部分的に矛盾した参考文献の部分を除いて、部分的に矛盾した参考文献が参照により援用される)、これによってそれらの全体が参照により本明細書中に援用される。
【0165】
[00173] 本明細書に対するいかなる添付又は付録をも明細書及び/又は図面の一部として参照により援用される。
【0166】
[00174] 用語「〜を含む(comprise)」、「〜を含む(comprises)」、「〜を含んだ(comprised)」、又は「〜を含む(comprising)」が本明細書中で使用される場合、それらは、言及された、定まった特徴、完全体、ステップ、又は成分の存在を明示するとして解釈され、1つ又は複数の他の特徴、完全体、ステップ、成分、又はそれらのグループの存在又は追加を排除するものではないと解釈される。用語「〜を含む(comprising)」、「〜を含む(comprise(s))」、又は「〜を含んだ(comprised)」が文法上類似する用語、例えば、「〜からなる(consisting/consist(s))」又は「本質的に〜からなる(consisting essentially of/consist(s) essentially of)」と任意選択で置換される本発明の個々の実施形態もまた包含されることが意図され、それにより、必ずしも同延でないさらなる実施形態を記載する。説明として、本明細書中に使用されるように、「〜を含む(comprising)」は、「〜を有する(having)」、「〜を含む(including)」、「〜を含む(containing)」、又は「〜を特徴とする(characterized by)」と同義であり、包括的又は無制限であり、追加の、記載されなかった要素又は方法ステップを除外しない。本明細書中に使用されるように、「〜からなる(consisting of)」は、請求項の要素に明示されていないいかなる要素、ステップ、成分、又は原料をも除外する。本明細書中に使用されるように、「本質的に〜なる(consisting essentially of)」は、請求項の基本的で新規な特徴に実質的に影響しない(例えば、活性成分に影響しない)材料又はステップを除外しない。本明細書中の各例において、用語「〜を含む(comprising)」、「本質的に〜からなる(consisting essentially of)」、及び「〜からなる(consisting of)」のいずれも他の2つの用語のうちのいずれかと置換してもよい。例証的に本明細書中に記載された本発明は、具体的に本明細書中に開示されていない任意の要素又は要素(複数)、限定又は限定(複数)の非存在下で適切に実行してもよい。
【0167】
[00175] 本発明は、様々な特定の好ましい実施形態及び技術に関して記載された。しかしながら、本発明の精神及び範囲内にとどまる一方で、多くの変形形態及び修飾形態を作製してもよいことが理解されるべきである。具体的に本明細書中に記載されたもの以外の組成物、方法、装置、装置要素、材料、任意の特徴、手順、及び技術は、広く本明細書中に開示されるように、過度の実験に頼ることなく本発明の実行に用いることができることを当業者は十分に理解するであろう。本明細書中に記載された組成物、方法、装置、装置要素、材料、手順、及び技術並びにそれらの部分の、技術的に知られている機能的な均等物はすべて、本発明により包含されることが意図される。範囲を開示する場合は常に、部分範囲及び個々の値すべてが包含されることが意図される。本発明は、図面に示された又は明細書に例示されたいかなるものをも含む開示された実施形態により限定されず、実施形態は、限定としてではなく実施例又は例証として与えられる。本発明の範囲は請求項のみにより限定されるものとする。
【参考文献】
【0168】
[00176] 次の出願は、特に、その全体が参照により援用される。2003年10月30日に提出されたHergenrotherらによる米国仮特許出願第60/516556号;2004年8月20日に提出されたHergenrotherらによる米国仮特許出願第60/603246号;2004年10月27日に提出されたHergenrotherらによる米国特許出願第10/976186号。
【0169】
[00177] 米国特許第6762045号 Membrane derived caspase−3,compositions comprising the same and methods of use therefore;米国特許第6534267号 Polynucleotides encoding activators of caspases;米国特許第6403765号 Truncated Apaf−1 and methods of use thereof;2005年4月12日に発行されたChoongらによる米国特許第6303329号及び第6878743号;2004年4月22日に公開されたWang,Xiaodongらによる米国特許公開第20040077542号;2004年9月16日に公開されたShi,Yigongによる米国特許公開第20040180828号。
【0170】
Slee EAら,Benzyloxycarbonyl−Val−Ala−Asp(OMe)fluoromethylketone(Z−VAD.FMK)inhibits apoptosis by blocking the processing of CPP32,Biochem J.1996 APR 1;315(Pt1):21−4.
【0171】
1. Hanahan,D. & Weinberg,R.A. The hallmarks of cancer. Cell 100,57−70(2000).
2. Okada,H. & Mak,T.W. Pathways of apoptotic and non−apoptotic death in tumour cells. Nature Rev. Cancer 4,592−603(2004).
3. Roy,S. et al. Maintenance of caspase−3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc.Natl.Acad.Sci.98,6132−6137(2001).
4. Svingen,P.A. et al. Components of the cell death machine and drug sensitivity of the National Cancer Institute Cell Line Panel. Clin.Cancer Res.10,6807−6820(2004).
5. Lowe,S.W., Cepero,E. & Evan,G. Intrinsic tumor suppression. Nature 432,307−315 (2004).
6. Vogelstein,B. & Kinzler,K.W. Achilles’ heel of cancer. Nature 412,865−866(2001).
7. Traven,A., Huang,D.C. & Lithgow,T. Protein hijacking: key proteins held captive against their will. Cancer Cell 5,107−108(2004).
8. Soengas,M.S. et al. Inactivation of the apoptosis effector Apaf−1 in malignant melanoma. Nature 409,207−211(2001).
9. Wajant,H. Targeting the FLICE inhibitory protein(FLIP)in cancer therapy. Mol. Interv. 3,124−127(2003).
10. Denicourt,C. & Dowdy,S.F. Targeting apoptotic pathways in cancer cells. Science 305,1411−1413(2004).
11. Vassilev,L.T. et al. In vivo activation of the p53 pathway by small−molecule antagonists of MDM2. Science 303,844−848(2004).
12. Degterev,A. et al. Identification of small−molecule inhibitors of interaction between the BH3 domain and Bcl−XL. Nature Cell Biol. 3,173−182(2001).
13. Becattini,B. et al. Rational design and real time, in−cell detection of the proapoptotic activity of a novel compound targeting Bcl−XL. Chem.Biol. 11,389−395(2004).
14. Wang,J.−L. et al. Structure−based discovery of an organic compound that binds Bcl−2 protein and induces apoptosis of tumor cells. Proc.Natl.Acad.Sci. 97,7124−7129(2000).
15. Li,L. et al. A small molecule Smac mimic potentiates TRAIL−and TNFa−mediated cell death. Science 305,1471−1474(2004).
16. Nguyen,J.T. & Wells,J.A. Direct activation of the apoptosis machinery as a mechanism to target cancer cells. Proc.Natl.Acad.Sci.U.S.A. 100,7533−7538(2003).
17. Jiang,X. et al. Dstincitive roles of PHAP proteins and prothymosin−α in a death regulatory pathway. Science 299,223−226(2003).
18. Boatright,K.M. & Salvesen,G.S. Mechanisms of caspase activation. Curr.Opin.Cell.Biol. 15,725−731(2003).
19. Nakagawara,A. et al. High levels of expression and nuclear localization of interleukin−1β converting enzyme (ICE) and CPP32 in favorable human neuroblastomas. Cancer Res. 57,4578−4584(1997).
20. Izban,K.F. et al. Characterization of the interleukin−1β−converting enzyme/Ced−3−family protease, caspase−3/CPP32, in Hodgkin’s disease. Am.J.Pathol. 154,1439−1447(1999).
21. Persad,R. et al. Overexpression of caspase−3 in hepatocellular carcinomas. Modern Patholo. 17,861−867(2004).
22. Pop,C., Feeney,B., Tripathy,A. & Clark,A.C. Mutations in the procaspase−3 dimer interface affect the activity of the zymogen. Biochemistry 42,12311−12320(2003).
23. Stennicke,H.R. et al. J.Biol.Chem. 273,27084−27090(1998).
24. Denault,J.−B & Salvesen, G.S. Human caspase−7 activity and regulation by its N−terminal peptide. J.Biol.Chem. 278,34042−24050(2003).
25. Putt,K.S., Beilman,G.J. & Hergenrother,P.J. Direct quantitation of Poly(ADP−ribose) polymerase (PARP) activity as a means to distinguish necrotic and apoptotic death in cell and tissue samples. ChemBioChem 6,53−55(2005).
26. Liang,Y., Nylander, K.D., Yan,C. & Schor,N.F. Role of caspase 3−dependent Bcl−2 cleavage in potentiation of apoptosis by Bcl−2. Mol.Pharmacol. 61,142−149(2002).
27. Fujita,N., Nagahshi,A., Nagashima,K., Rokudai,S. & Tsuruo,T. Acceleration of apoptotic cell death after the cleavage of Bcl−XL protein by caspase−3−like proteases. Oncogene 17,1295−1304(1998).
28. Earnshaw,W.C., Martins,L.M. & Kaufmann,S.H. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu.Rev.Biochem. 68,383−424(1999).
29. Koty,P.P., Zhang,H. & Levitt,M.L. Antisense bcl−2 treatment increases programmed cell death in non−small cell lung cancer cell lines. Lung Cancer 23,115−127(1999).
【0172】
国立医学図書館/国立衛生研究所(NIH)の国立バイオテクノロジー情報センター(NCBI)のデータベース、ウェブサイト:http://www.ncbi.nlm.nih.gov/ CASP3(カスパーゼ3、アポトーシス関連システインプロテアーゼ[ホモサピエンス] 遺伝子ID:836 遺伝子座タグ:HGNC:1504;MIM:600636 2005年5月15日に更新。他の別名:HGNC:1504、アポパイン、CPP32、CPP32B、SCA−1;他の指定:ヒトプロカスパーゼ3コード配列;PARP切断プロテアーゼ;SREBP切断活性1;Yama;カスパーゼ3;システインプロテアーゼCPP32)を検索するために遺伝子データベースを使用。
【0173】
Hergenrother PJ.Obtaining and screening compound collections:a user’s guide and a call to chemists.Curr Opin Chem Biol.2006.
Silverman SK,Hergenrother PJ.Combinatorial chemistry and molecular diversity Tools for molecular diversification and their applications in chemical biology.Curr Opin Chem Biol.2006.
Goode DR,Sharma AK,Hergenrother PJ.Using peptidic inhibitors to systematically probe the S1’site of caspase−3 and caspase−7.Org Lett.2005 Aug 4;7(16):3529−32.PMID:16048334.
Dothager RS,Putt KS,Allen BJ,Leslie BJ,Nesterenko V,Hergenrother PJ.Synthesis and identification of small molecules that potently induce apoptosis in melanoma cells through G1 cell cycle arrest.J Am Chem Soc.2005 Jun 22;127(24):8686−96.PMID:15954774.
Putt KS,Hergenrother PJ.A nonradiometric,high−throughput assay for poly(ADP−ribose) glycohydrolase(PARG):application to inhibitor identification and evaluation.Anal Biochem.2004 Oct 15;333(2):256−64.PMID:15450800.
Putt KS,Hergenrother PJ.An enzymatic assay for poly(ADP−ribose) polymerase−1(PARP−1) via the chemical quantitation of NAD(+):application to the high−throughput screening of small molecules as potential inhibitors.Anal Biochem.2004 Mar 1;326(1):78−86.PMID:14769338.
Nesterenko V,Putt KS,Hergenrother PJ.Identification from a combinatorial library of a small molecule that selectively induces apoptosis in cancer cells.J Am Chem Soc.2003 Dec 3;125(48):14672−3.PMID:14640619.
【関連出願の相互参照】
【0001】
[0001] 本出願は、2005年5月26日に提出された米国仮出願第60/684807号及び2006年3月28日に提出された米国仮出願第60743878号の米国特許法第119条(e)の下の利益を主張し、それぞれの全体が参照により援用される。
【連邦政府による資金提供を受けた研究開発の記載】
【0002】
[0002] 本発明は、米国国立科学財団により与えられたNSF助成金/契約CHE−0134779の下、政府援助でなされた。政府は、本発明における一定の権利を有している。
【発明の背景】
【0003】
[0003] アポトーシス又はプログラム細胞死はすべての多細胞生物の発生及びホメオスタシスで中心的な役割を果たしている(Shi Y,2002,Molecular Cell 9:459−470)。よくある癌の特徴は、本来のアポトーシスシグナルに対する抵抗性である。癌型によっては、この抵抗性は、通常、アポトーシスカスケードにおける重要なタンパク質のアップレギュレーション若しくはダウンレギュレーション又はこれらのタンパク質をコードする遺伝子内の変異による。上記変化は、ミトコンドリア及びカスパーゼ−9を経る内因性アポトーシス経路並びにデスレセプター及びカスパーゼ−8の作用が関与する外因性アポトーシス経路の両方で起こる。例えば、p53、Bim、Bax、Apaf−1、FLIP、その他多くのタンパク質の適切なレベルの変化が癌で観察された。この変化は、欠陥のあるアポトーシスカスケードにつながる可能性があり、上流のアポトーシス促進シグナルが十分に伝達されず、実行因子カスパーゼであるカスパーゼ−3及びカスパーゼ−7が活性化されない。
【0004】
[0004] ほとんどのアポトーシス経路がプロカスパーゼ−3の活性化を最終的に伴うので、上流の遺伝的異常は、有効に、アポトーシス回路における「断線箇所(break)」となり、その結果、上記細胞は異常に増殖する。癌におけるアポトーシスの中心的な役割から判断して、アポトーシスカスケードにおける特定のタンパク質を標的にする治療を開発するための試みがなされた。例えば、p53及びBclファミリーのタンパク質のカスケードメンバーに対する、又はアポトーシス抑制因子(IAP)ファミリーのタンパク質に対するペプチド結合剤又は低分子結合剤は、Apaf−1のオリゴマー化を促進する化合物のようにアポトーシス促進活性を有している。しかしながら、上記化合物が、アポトーシスカスケード上の早期に(又は中間から高所に)位置するものを標的にするので、それらのメンバーのうちの下流のタンパク質における変異を有する癌は、それらの化合物の可能な有益である効果になお抵抗性がある可能性がある。
【0005】
[0005] 治療目的として、アポトーシスカスケードにおけるはるか下流のアポトーシス促進タンパク質を直接活性化する低分子を同定することは有利であろう。我々の発明へのアプローチは、カスケードにおける上記の比較的低位置を含み、したがって、上流のアポトーシス機構における変異を有するそれらの細胞でさえ殺すことを可能にする。さらに、本明細書中に開示された治療戦略は、アポトーシス促進タンパク質が癌細胞内でアップレギュレートした場合、成功の可能性がより高い可能性がある。本発明において、低分子を同定するための我々の試みは、アポトーシスの重要な下流のエフェクタータンパク質であるプロカスパーゼ−3を標的にすることから始まった。
【0006】
[0006] プロカスパーゼ−3のカスパーゼ−3への変換又は活性化により、多数のタンパク質基質の加水分解を続いて触媒する活性「実行因子」カスパーゼ形態の生成がもたらされる。活性カスパーゼ−3は、ヘテロダイマーのホモダイマーであり、プロカスパーゼ−3のタンパク質分解により産生される。生体内では、このタンパク質分解性の活性化は、通常、カスパーゼ−8又はカスパーゼ−9の作用を介して生じる。プロ酵素又は酵素前駆体が早まって活性化されないことを確実にするために、プロカスパーゼ−3は、タンパク質分解のIETD部位(アミノ酸配列、ile−glu−thr−asp)への到達をブロックする12個のアミノ酸「安全装置」を有している。Roy,S.ら;Maintenance of caspase−3 proenzyme dormancy by an intrinsic“safety catch”regulatory tripeptide,Proc.Natl.Acad.Sci.98,6132−6137(2001)参照。
【0007】
[0007] この安全装置は、プロカスパーゼ−3が、自己触媒活性化及びカスパーゼ−9によるタンパク質分解に抵抗することを可能にする。突然変異誘発研究は、3つの連続したアスパラギン酸残基が安全装置の重要な成分であると思われることを示す。安全装置の部位はpHに感受性である。したがって、細胞の酸性化(アポトーシス中に起こる)の際、安全装置は、タンパク質分解の部位への到達を許可すると思われ、活性カスパーゼ−3は、カスパーゼ−9の作用により又は自己活性化機構を介して産生することができる。
【0008】
[0008] 特定の癌では、プロカスパーゼ−3の発現はアップレギュレートされる。20人の大腸癌患者からの一次単離物に関する研究は、平均して、プロカスパーゼ−3が、隣接した非癌性組織に比べて上記試料中で6倍アップレギュレートされたことを明らかにした(Royら,2001)。さらに、プロカスパーゼ−3は、ある種の神経芽細胞腫、リンパ腫、及び肝臓癌においてアップレギュレートされる(Nakagawara,A.ら,1997,Cancer Res.57:4578−4584;lzban,K.F.ら,Am.J.Pathol.154:1439−1447;Persad,R.ら,Modem Patholo.17:861−867)。さらに、国立癌研究所(NCI)の開発治療プログラムによる癌スクリーニングに使用された60枚の細胞株パネル中のプロカスパーゼ−3レベルについて系統的評価を行った。評価は、ある種の肺癌、黒色腫、腎臓癌、及び乳癌が著しく増強されたプロカスパーゼ−3発現レベルを示すことを明らかにした(Svingen,P.A.ら,Clin.Cancer Res.10:6807−6820)。
【0009】
[0009] アポトーシスの達成における活性カスパーゼ−3の役割、ある種の癌性細胞型におけるプロカスパーゼ−3の比較的高度な発現レベル、及び興味深い、安全装置媒介性のその自己活性化の抑制により、プロカスパーゼ−3を直接修飾する低分子が同定される可能性があるということ及び上記分子は、標的癌治療法において顕著な適用可能性を有する可能性があるということを我々は推論した。
【0010】
[0010] 本明細書で、我々は、プロカスパーゼ−3をそのエフェクター形態に変換することができる活性化剤を特に含む、プロカスパーゼの低分子修飾因子を開示する。さらに、我々は、ある種の薬物低分子がプロカスパーゼ−3を直接、即時活性化し、生体内において癌細胞におけるアポトーシス促進効果を達成することができることを実証する。我々は、これらがプロカスパーゼ−3を直接活性化することで知られている最初の低分子であると考える。実行因子カスパーゼの直接的な活性化は新規で価値のある抗癌戦略である。
【発明の概要】
【0011】
[0011] 全体として、本明細書で使用される用語及び語句は、それらの技術的に認識された意味を有しており、当業者に知られている標準教科書、雑誌論文、及び文脈の参照により見つけることができる。
【0012】
[0012] 以下の略語が適用可能である。IAP、アポトーシス抑制因子;PAC−1、プロカスパーゼ活性化化合物1;PARP、ポリ(ADPリボース)ポリメラーゼ。
【0013】
[0013] 本発明は、化合物、治療処置のための方法、化合物をスクリーニングするための方法、並びにプロカスパーゼの修飾因子に関連した処置に対する細胞及び患者適性をスクリーニングするための方法を広く提供する。一実施形態では、修飾因子は、阻害剤又は活性化剤である。一実施形態では、本発明は、プロカスパーゼ−3及びプロカスパーゼ−7の活性化剤に関連した上記化合物及び方法を提供する。一実施形態では、本発明は、当技術分野で知られている、乳房、リンパ腫、副腎、腎臓、黒色腫、白血病、神経芽細胞腫、肺、脳等、様々な癌疾患及び癌細胞型の状況に適用可能である。
【0014】
[0014] さらなる概要として、我々は、癌細胞内で不活性形態で過剰発現することが多い酵素を活性化することができる化合物を発見した。化合物は、アップレギュレートされたプロカスパーゼ−3を有する癌細胞を含む癌細胞におけるプログラム細胞死(アポトーシス)を誘導する。癌は増大する大きな課題であり、現在、米国における第1位の死因である。多くの癌は、標準的な化学療法に抵抗する。本発明の化合物は、癌細胞内でアップレギュレートされることがある生物学的標的を利用することができ、したがって、アポトーシス機構に欠陥を有する細胞内でさえ有効であると証明することができる。これらの化合物は、さらに、標的癌治療法においても成功する可能性があり、ここでは、より低レベルのプロカスパーゼ−3を有する非癌性細胞に対して比較的毒性が低下した状態で癌細胞を死滅させる点での選択性の利点がある可能性がある。
【0015】
[0015] 特定の理論に束縛されるものではないが、本発明の化合物は、アポトーシス又はプログラム細胞死の調節の機構を介して作用して、癌細胞の処置において有効となることが可能であると考えられる。好ましい実施形態では、アポトーシスの調節はアポトーシスの誘導による。他の実施形態では、アポトーシスの調節はアポトーシスの阻害による。
【0016】
[0016] 一実施形態では、本発明は、癌細胞におけるアポトーシスを選択的に誘導する方法であって、(a)上述の癌細胞のプロカスパーゼ−3分子を修飾することができる化合物を上述の癌細胞に投与することと、(b)アポトーシスを誘導するように上述のプロカスパーゼ−3分子を修飾すること、を含む方法を提供する。一実施形態では、上述の癌細胞は、処置を必要とする患者に存在する。
【0017】
[0017] 一実施形態では、上述の化合物は下記式ZZである。
【化1】
式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。一実施形態では、上述の化合物は、式ZZ、PAC−1及び構造5からなる群より選択される。一実施形態では、上述の化合物はPAC−1である。
【0018】
[0018] 一実施形態では、化合物は、式ZZを有する化合物から選択され、式中、R4及びR5は共にNであり、Aは酸素であり、他の可変基は上記に定義した通りである。一実施形態では、化合物は、式ZZを有する化合物から選択され、式中、R4及びR5は共にNであり、Aは酸素であり、R2は水素であり、他の可変基は上記に定義した通りである。一実施形態では、化合物は、式ZZを有する化合物から選択され、式中、R4及びR5は共にNであり、Aは酸素であり、R2は水素であり、R3はアリルであり、他の可変基は上記に定義した通りである。
【0019】
[0019] 一実施形態では、方法は、癌細胞内のプロカスパーゼ−3又はカスパーゼ−3のパラメーターを評価するステップをさらに含み、上述のパラメーターは、半定量的若しくは定量的な量、機能的な量、又は上述のプロカスパーゼ−3又はカスパーゼ−3の活性レベルの1つ又は複数である。
【0020】
[0020] 一実施形態では、本発明は、プロカスパーゼ−3分子を修飾することができる化合物を直接、in vitroでスクリーニングするための方法であって、(a)試験化合物を提供することと、(b)精製したプロカスパーゼ−3を提供することと、(c)精製したプロカスパーゼ−3に試験化合物を暴露することと、(d)試験化合物への暴露の後でプロカスパーゼ−3活性を測定することと、(e)試験化合物への暴露の際の試験活性を試験化合物への暴露なしでの無修飾活性と比較することにより修飾化合物を同定すること、を含み、それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法を提供する。一実施形態では、方法は、上述の修飾活性又は上述の無修飾活性を基準活性と比較することをさらに含み、上述の基準活性は、構造式ZZ又は上記式の化合物のサブセット、PAC−1及び構造5からなる群より選択される化合物へのプロカスパーゼ−3の暴露による。
【0021】
[0021] 一実施形態では、本発明は、プロカスパーゼ−3を活性化することができる化合物のスクリーニングのための方法であって、a)プロカスパーゼ−3を提供し、試験化合物、好ましくは低分子を提供することと、b)プロカスパーゼ−3を試験化合物と反応させること(これにより、カスパーゼ−3が生成すると推定される)と、c)カスパーゼ−3活性を測定することと、を含む方法を提供する。特定の実施形態では、カスパーゼ−3活性を測定することに基質Ac−DEVD−pNAを使用する。特定の実施形態では、測定することに、約410nmの波長読み出しパラメーターを使用する。特定の実施形態では、スクリーニングは、多数の試験化合物を使用して並行して実行される。
【0022】
[0022] 一実施形態では、本発明は、プロカスパーゼ−3のサブユニットの検出を完全長(不活性)プロカスパーゼ−3がカスパーゼ−3に処理されたという指標として使用するスクリーニングの方法を提供する。特定の実施形態では、サブユニットは、タンパク質ゲル移動技術、例えばウエスタンブロットにより測定される約19kDの分子量を有している。
【0023】
[0023] 一実施形態では、本発明は、プロカスパーゼ−3分子を修飾することができる化合物の細胞スクリーニングのための方法であって、(a)試験化合物を提供することと、(b)プロカスパーゼ−3を推定上発現する細胞を提供することと、(c)試験化合物に細胞を暴露することと、(d)試験化合物への暴露の後で、1つ又は複数の細胞生存率、アポトーシス指標、及び他のパラメーターを含む細胞パラメーターを測定することと、(e)試験化合物への暴露の際の試験細胞パラメーターを試験化合物への暴露なしでの無修飾細胞パラメーターと比較することにより修飾化合物を同定することと、を含み、それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法を提供する。一実施形態では、方法は、上述の修飾活性又は上述の無修飾活性を基準活性と比較することをさらに含み、上述の基準活性は、式ZZ又は上記式の化合物のサブセット、PAC−1及び構造5からなる群より選択される化合物への暴露による。
【0024】
[0024] 一実施形態では、本発明は、プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定又は診断するための方法であって、(a)上述の癌細胞内のプロカスパーゼパラメーターを評価することと、(b)上述のパラメーターによりプロカスパーゼの活性化に対する感受性が上昇したかどうかを判定すること、を含む方法を提供する。一実施形態では、上述のプロカスパーゼパラメーターはプロカスパーゼ−3レベルであり、上述のプロカスパーゼはプロカスパーゼ−3である。一実施形態では、上述のプロカスパーゼパラメーターはプロカスパーゼ−7レベルであり、上述のプロカスパーゼはプロカスパーゼ−7である。レベルは、半定量的若しくは定量的な量、又は機能的な量(例えば活性ベースの量、例えば規格単位又は国際単位)とすることができる。
【0025】
[0025] 一実施形態では、本発明は、癌細胞を処置するための方法であって、(a)プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定することと、(b)有効な量のプロカスパーゼ活性化化合物に上述の癌細胞を暴露すること、を含む方法を提供する。一実施形態では、プロカスパーゼ活性化化合物は、式ZZ又は上記式の化合物のサブセット、PAC−1及び構造5からなる群より選択される。一実施形態では、上述のプロカスパーゼ活性化化合物は、プロカスパーゼ−3、プロカスパーゼ−7、又はプロカスパーゼ−3及びプロカスパーゼ−7の両方を活性化することができる請求項16に記載の方法。
【0026】
[0026] 一実施形態では、本発明は、スキーム1のステップを含む、PAC−1を合成するための方法を提供する。一実施形態では、本発明は、適切な変更を有するスキーム1のステップを含む、化合物5を合成するための方法を提供する。一実施形態では、本発明は、本明細書で開示されており、当技術分野で理解されるであろう式ZZの化合物を合成するための方法を提供する。
【0027】
[0027] 一実施形態では、本発明は、下記式ZZの化合物を提供する。
【化2】
式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。
【0028】
[0028] 一実施形態では、本発明は、構造が
【化3】
であるPAC−1を除く、式ZZの化合物を提供する。
【0029】
[0029] 一実施形態では、本発明は、構造5の化合物を提供する。構造は次の通りである。
【化4】
【0030】
[0030] 一実施形態では、本発明の組成物は化学療法剤である。
【0031】
[0031] 一実施形態では、本発明は、開示された構造式の好ましくは約10nM〜約100μMの有効濃度を伴う化合物及び方法を提供する。他の好ましい実施形態では、有効な濃度は約200nM〜約5μMである。一実施形態では、有効な濃度は、直接的なプロカスパーゼ活性化アッセイ、細胞アポトーシス誘導アッセイ、又は動物の臨床的治療評価での50%活性濃度等の値であると考えられる。好ましい実施形態では、上記値は、約200μM未満である。好ましい実施形態では、上記値は約10μM未満である。
【0032】
[0032] 本発明の化合物及び本発明の方法において有用な化合物は、開示された式の化合物、並びに好ましくは薬学的に許容し得る塩及びエステルを含む、それらの化合物の塩及びエステルを含む。
【0033】
[0033] 一実施形態では、本発明は、組成物のプロドラッグ形態を提供する。本発明の化合物のプロドラッグは本発明の方法において有用である。生体内で変換され、本発明の化合物の生物学的、薬学的、又は治療的な活性形態をもたらすであろういかなる化合物もプロドラッグである。プロドラッグの様々な例及び形態が当技術分野でよく知られている。前駆体タンパク質、前駆体核酸等の生体分子をプロドラッグとすることができる。プロドラッグの例は、Design of Prodrugs,H.Bundgaard編(Elsevier,1985)、Methods in Enzymology,Vol.42,pp.309−396,K.Widderら編(Academic Press,1985)、A Textbook of Drug Design and Development,Krosgaard−Larsen及びH.Bundgaard編,Chapter 5,「Design and Application of Prodrugs」H.Bundgaard編,pp.113−191,1991)、H.Bundgaard,Advanced Drug Delivery Reviews,Vol.8,p.1−38(1992),H.Bundgaardら,Journal of Pharmaceutical Sciences,Vol.77,p.285(1988)、並びにNogrady(1985)Medicinal Chemistry A Biochemical Approach,Oxford University Press,New York,pages388−392)にとりわけ見つけられる。
【0034】
[0034] 置換基のグループが本明細書で開示される場合、グループのメンバーのいかなる異性体及び鏡像異性体をも含む、そのグループ及びすべてのサブグループの個々のメンバーがすべて、個別に開示されることが理解される。マーカッシュグループ又は他のグループ分けが本明細書で使用される場合、グループ並びにグループのすべての組み合わせ及び下位組み合わせの可能性がある個々のメンバーがすべて、本開示に個々に含まれることが意図される。明細書で提供された任意のマーカッシュグループ又は一覧の任意の1つ又は複数のメンバーは、所望により、本発明から除外することができることが意図される。化合物が、化合物の特定の異性体又は鏡像異性体が、例えば式又は化学名で明記されないように本明細書で記載される場合、その記載は、個々に又は任意の組み合わせで記載された化合物のそれぞれの異性体及び鏡像異性体を含むことが意図される。さらに、特に明記されていない限り、本明細書で開示された化合物の同位体の変形形態はすべて、本開示により包含されることが意図される。例えば、開示された分子内の任意の1つ又は複数の水素が重水素又は三重水素で置換することができることが理解されるであろう。分子の同位体の変形形態は、分子のアッセイ並びに分子又はその使用に関連する化学的研究及び生物学的研究で標準物質として一般に有用である。化合物の特定の名称は、当業者が、同一の化合物をさまざまに称することができることが知られているように、例示的であることが意図される。
【0035】
[0035] 本明細書で開示された分子は1つ又は複数のイオン化できるグループ[プロトンを除去する(例えばOH、−COOH、等)若しくは加える(例えばアミン)ことができる又は四級化することができる(例えばアミン)グループ]を含んでいてもよい。可能な上記分子のイオン形態及びそれらの塩はすべて、本明細書中の本開示に個々に含まれることが意図される。本明細書中の化合物の塩に関して、当業者は、種々様々の利用可能な対イオンの中から、所定の用途に対する本発明の塩の調製に適切な対イオンを選択することができる。例えば、一般に、任意のアニオンが、本明細書中の化合物の塩、例えば、ハロゲン化物、硫酸塩、カルボン酸塩、酢酸塩、リン酸塩、硝酸塩、トリフルオロ酢酸塩、グリコール酸塩、ピルビン酸塩、シュウ酸塩、リンゴ酸塩、コハク酸塩、フマル酸塩、酒石酸塩、クエン酸塩、安息香酸塩、メタンスルホン酸塩、エタンスルホン酸塩、p−トルエンスルホン酸塩、サリチル酸塩、等の形成に使用することができる。
【0036】
[0036] 本発明の化合物及びそれらの塩又はエステルは、それらの互変異性形態で存在してもよく、水素原子は、分子の他の部分に転位され、分子の原子間の化学結合は結果的に再配列される。互変異性形態はすべて、存在することがある限り、本発明の範囲内に含まれることが理解されるべきである。さらに、化合物は、トランス異性体及びシス異性体を有していてもよく、1つ又は複数のキラル中心を含み、したがって、鏡像異性体形態及びジアステレオマー形態で存在してもよい。本発明は、特に本明細書で述べられていない限り、上記すべての異性体及び個々の鏡像異性体と同様にシス異性体及びトランス異性体の混合物、ジアステレオマーの混合物、鏡像異性体(光学異性体)の非ラセミ混合物及びラセミ混合物、並びに1つ又は複数の形態について豊富な前述の混合物を包含することができる。化合物の(又は不斉炭素の)配置(シス若しくはトランス又はR若しくはS)について特定の言及がなされていない場合、そのとき、任意の1つの異性体又は1つを超える異性体の混合物が意図される。調製のプロセスは、ラセミ化合物、鏡像異性体、又はジアステレオマーを出発物質として使用することができる。鏡像異性体又はジアステレオマーの生成物が調製される場合、それらは、従来の方法により、例えばクロマトグラフィー又は分別結晶により分離することができる。発明の化合物は、遊離形態又は水和物形態であってもよい。
【0037】
[0037] 本明細書中に記載された又は例示された成分のすべての処方又は組み合わせは、特に述べられていない限り、本発明を実行するために使用することができる。
【0038】
[0038] 範囲、例えば温度範囲、時間範囲、組成範囲、又は濃度範囲が本出願中に記載されるときには必ず、すべて中間範囲及び部分範囲並びに所定の範囲に含まれるすべての個々の値が本開示に含まれることが意図される。
【0039】
[0039] 本明細書中に開示された任意の参考文献中の情報は、場合により、例えば、特許文献についてのそれらの有効出願日時点での技術水準を示すことができ、上記情報は、必要であれば、実際に先行技術にあると認められる特定の実施形態を除外するために本明細書中で使用することができることが意図される。例えば、化合物が、開示及び/又は特許請求される場合、実施可能な程度の開示が参考文献中に提供されている化合物を含む、本発明に関して先行技術としてみなされている化合物は、本出願中の物質の請求項の組成物に含まれることが意図されないことが理解されるべきである。
【0040】
[0040] 本明細書中で提供されるいくつかの参考文献は、出発物質、補足的な出発物質、補足的な試薬、補足的な合成の方法、補足的な分析の方法、及び補足的な本発明の使用についての情報源に関する細部を提供するように参照により援用される。具体的に例示されたもの以外の出発物質、試薬、固体基質、合成方法、精製方法、及び分析方法は、当技術分野の知識に基づき、過度の実験に頼ることなく、本発明の実行に使用することができることを当業者は十分に理解するであろう。
【0041】
[0041] 一実施形態では、本発明は、1つ又は複数の化合物及び各化合物について薬学的に許容し得るそれらの塩又はエステルを含み、化合物は、所望される治療上の有益性を得るために有効な量又は総量で組成物中に存在する治療用組成物を提供する。本発明の治療用組成物は、1つ又は複数の薬学的に許容し得る成分、例えば、当技術分野で知られている担体及び賦形剤を任意選択でさらに含む。
【0042】
[0042] 一実施形態では、本発明は、下記式ZZ2を有する化合物を提供する。
【化5】
式中、R1及びR2は、それぞれ独立して、水素、ハロゲン、アルキル、アリル、ハロアルキル、アルケニル、アルケノール、アルカノール又はハロアルケニルである。一実施形態では、R1及びR2は、それぞれ独立して、水素、ハロゲン、アリル又は短鎖アルキルである。
【0043】
[0043] 一実施形態では、本発明は、アルデヒド化合物と結合したヒドラジド化合物を含むPAC−1誘導体コンビナトリアルライブラリーからなる群より選択される化合物を提供する。一実施形態では、ヒドラジド化合物は、本明細書中に記載されたAX化合物から生成されたヒドラジドからなる群より選択される。
【0044】
[0044] 一実施形態では、アルデヒド化合物は、本明細書中に記載されたBX化合物からなる群より選択される。一実施形態では、ヒドラジド化合物は、本明細書中に記載されたAX化合物からなる群より選択され、アルデヒド化合物は、本明細書中に記載されたBX化合物からなる群より選択される。
【0045】
[0045] 一実施形態では、本発明は、PAC−1誘導体化合物を合成するための方法であって、ヒドラジド化合物を提供することと、アルデヒド化合物を提供することと、ヒドラジド化合物をアルデヒド化合物と反応させることと、を含み、それにより、PAC−1誘導体化合物を合成する方法を提供する。
【0046】
[0046] 一実施形態では、ヒドラジド化合物は下記式ZZ3を有する。
【化6】
【0047】
[0047] 一実施形態では、アルデヒド化合物は下記式ZZ4を有する。
【化7】
【0048】
[0048] 一実施形態では、ヒドラジド化合物は式ZZ3を有し、アルデヒド化合物は式ZZ4を有する。
【0049】
[0049] 一実施形態では、本発明は、L01R06、L02R03、L02R06、L08R06、L09R03、L09R06及びL09R08からなる群より選択される化合物を提供する。
【0050】
[0050] 一実施形態では、本発明は、癌患者の候補者を、候補者においてプロカスパーゼレベルの上昇を同定することによりプロカスパーゼ活性化剤による可能な処置についてスクリーニングするための方法であって、候補者からの細胞又は組織の試験サンプルを得ることと、試験サンプル中のプロカスパーゼレベルを評価することと、プロカスパーゼレベルは基準レベルに比べて試験サンプル中で上昇したかどうかを判定することと、を含み、それにより、癌患者の候補者をプロカスパーゼ活性化剤による可能な処置についてスクリーニングする方法を提供する。一実施形態では、プロカスパーゼは、プロカスパーゼ−2、プロカスパーゼ−3、プロカスパーゼ−6、プロカスパーゼ−7、プロカスパーゼ−8及びプロカスパーゼ−9からなる群より選択される。特定の実施形態では、プロカスパーゼはプロカスパーゼ−3である。
【0051】
[0051] 一実施形態では、試験サンプルの上昇レベルは、基準レベルを超えた少なくとも約2倍である。一実施形態では、試験サンプルの上昇レベルは、基準レベルを超えた少なくとも約4倍である。一実施形態では、基準レベルは、同一患者からの第2の試験サンプルからのものである。一実施形態では、基準レベルは、正常細胞又は正常組織のサンプルからのものである。基準レベルは、癌細胞株、正常細胞株等の細胞株からのものとすることができる。一実施形態では、基準レベルは絶対閾値の量である。細胞当たりの分子数を含むプロカスパーゼのレベルについての様々な量を記載しているSvingen,P.A.ら,Clin.Cancer Res.10:6807−6820を参照のこと。
【0052】
[0052] 一実施形態では、本発明は、上述の癌細胞中のプロカスパーゼ分子を活性化することができる化合物を上述の癌細胞に投与することを含む、癌細胞における死を誘導するための方法を提供する。一実施形態では、プロカスパーゼは、1つ又は複数のプロカスパーゼ−3及びプロカスパーゼ−7である。好ましい実施形態では、プロカスパーゼはプロカスパーゼ3である。一実施形態では、化合物は構造式ZZを有する。一実施形態では、化合物は構造式ZZ2を有する。
【0053】
[0053] 本明細書で考えられた又は開示されたいかなる機構的な説明又は仮説についての最終的な正確さに関係なく、本発明の一実施形態は、それでもなお、効果があり、有用である可能性があると認識される。
【図面の簡単な説明】
【0054】
【図1A】in vitroでのPAC−1によるプロカスパーゼ−3の活性化及び活性カスパーゼ−3。PAC−1は、EC50=0.22μMでプロカスパーゼ−3を活性化する。エラーバーは、平均値からの標準偏差を表す。
【図1B】PAC−1により誘導される活性カスパーゼ−3へのプロカスパーゼ−3の切断。プロカスパーゼ−3をN末端His−6タグを用いて大腸菌中で組換え発現させ、精製した。免疫ブロットは抗His−6抗体を用いて行った。PAC−1非存在下でプロカスパーゼ−3の成熟は観察されない。100μM PAC−1存在下で、p19フラグメントを生成する切断が1時間以内に観察され、>50%の切断が4時間後に観察される。
【図2A】プロカスパーゼ−3の「安全装置」領域における変異体のPAC−1による活性化。PAC−1は、野性型プロカスパーゼ−3(DDD)に対して0.22μMの活性化のためのEC50並びにある種の変異体については2.77μM(DAD)、113μM(DDA)、及び131μM(ADD)の対応するEC50値を有している。
【図2B】PAC−1は、4.5μMのEC50でプロカスパーゼ−7を活性化する。
【図2C】プロカスパーゼ−3のPAC−1の活性化についてのpHへの依存性。低pHでは、安全装置は「オフ」の状態にあり、プロカスパーゼ−3は本質的に最大限に活性化される。エラーバーは、平均値からの標準偏差を表す。
【図3A】PAC−1は、HL−60細胞におけるアポトーシスを誘導する。A)100μM PAC−1での20時間の処置の後のホスファチジルセリン露出(アネキシン−V染色により測定される)。
【図3B】100μM PAC−1での20時間の処置の後のヘキスト染色により可視化されるクロマチン凝縮。
【図4A】10μMエトポシドで処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。
【図4B】100μM PAC−1で処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。
【図4C】PAC−1処置(100μM)は、細胞内カスパーゼ−3/−7の即時活性化と一致する、HL−60細胞における細胞内PARP活性の急速な減少を誘導する。対照的に、エトポシド(10μM)処置細胞は、ずっと後の時点でのPARP活性の減少を示す。
【図4D】PAC−1は、プロカスパーゼ−3依存的に細胞死を誘導する。多くの多様な癌細胞株について、プロカスパーゼ−3レベルを求め(フローサイトメトリーにより)、PAC−1のIC50を測定した(R2=0.9822)。PAC−1は、高度なレベルのプロカスパーゼ−3を有するとして知られているNCI−H226肺癌細胞株において相当効力がある(IC50=0.35μM)が、健常なヒトドナーの骨髄に由来する正常な白血球において効力が著しく少ない。
【図4E】PAC−1は、プロカスパーゼ−3依存的に細胞死を誘導する。多くの多様な癌細胞株について、プロカスパーゼ−3レベルを求め(フローサイトメトリーにより)、PAC−1のIC50を測定した(R2=0.9822)。PAC−1は、高度なレベルのプロカスパーゼ−3を有するとして知られているNCI−H226肺癌細胞株において相当効力がある(IC50=0.35μM)が、健常なヒトドナーの骨髄に由来する正常な白血球において効力が著しく少ない。
【図5A】数名の患者からの正常細胞及び癌性細胞中の相対的なプロカスパーゼ−3レベルを示す。
【図5B】様々な相対的なプロカスパーゼ−3レベルを有する様々な細胞型におけるPAC−1のIC50レベルを示す。
【図5C】腫瘍成長の結果に対するPAC−1での動物の処置の効果を示す。
【図5D】腫瘍成長の結果に対するPAC−1での動物の経口処置の効果を示す。
【図5E】対照処置群、PAC−1処置群、及びゲフィチニブ(イレッサ(商標);アストラゼネカ社)処置群についての肺癌モデルにおける癌の進行の結果を示す図である。腫瘍細胞をi.v.投与によりマウスに注射し、イレッサ及びPAC−1を100mg/kgで経口的に与えた。
【図6A】3名の患者の正常細胞及び癌性細胞中の相対的なプロカスパーゼ−3レベルを示す。
【図6B】患者3からの正常細胞及び癌性細胞のPAC−1での処置に対する感受性を示す。
【図7】肺癌のマウスモデルという状況におけるPAC−1の腹腔内投与の結果を示す。
【図8A】PAC−1誘導体及びコンビナトリアルライブラリーの化合物の構造を示す。
【図8B】PAC−1誘導体及びコンビナトリアルライブラリーの化合物の構造を示す。
【図9】ホモサピエンス カスパーゼ3、アポトーシス関連システインペプチダーゼ(CASP3)、転写バリアントアルファ、mRNAのヌクレオチド配列を示す。(受託番号NM_004346;2689bp mRNA直鎖状;http://www.ncbi.nlm.nih.gov/entrezから入手)。
【図10】ホモサピエンス カスパーゼ7、アポトーシス関連システインペプチダーゼ(CASP7)、転写バリアントアルファ、mRNAのヌクレオチド配列を示す。(受託番号NM_001227;2605bp;mRNA直鎖状;http://www.ncbi.nlm.nih.gov/entrezから入手)。
【発明の詳細な説明】
【0055】
[0069] 以下の定義は、本発明の文脈中でのそれらの特定の用法を明確にするために提供される。
【0056】
[0070] 用語「化学療法剤」は、本明細書中で使用される場合、癌細胞、癌細胞の集団、腫瘍、又は他の悪性組織の成長、増殖、又は拡散を低下又は阻止することができる任意の物質を指す。用語はまた、任意の抗腫瘍剤又は抗癌剤を包含することも意図される。
【0057】
[0071] 用語「有効な量」は、本明細書中で使用される場合、薬学的に有効な量、治療的に有効な量、等の文脈を包含することが意図される。例えば、実施形態ではこの量は、有利な状態、有利な結果、スクリーニングアッセイにおける機能的な活性、又は臨床症状の改善を達成することができる。
【0058】
[0072] 用語「癌細胞」は、本明細書中で使用される場合、当技術分野で広く理解されている定義を包含することが意図される。一実施形態では、この用語は、ヒト又は動物における癌の臨床症状の一因となる可能性がある異常に制御された細胞を指す。一実施形態では、この用語は、培養された細胞株或いはヒト若しくは動物の体内の又はヒト若しくは動物に由来する細胞を指すことができる。癌細胞は、当技術分野で理解されているように、種々様々な分化した細胞型、組織型、又は器官型になる可能性がある。
【0059】
[0073] 用語「アルキル」は、好ましくは1〜22個の炭素原子を有するモノラジカルの分枝又は非分枝の飽和炭化水素鎖、及び3〜22個の炭素原子を有する1つ又は複数の環を有するシクロアルキル基を指す。短鎖アルキル基は、メチル基、エチル基、プロピル基、ブチル基、ペンチル基及びヘキシル基を含む、1〜6個の炭素原子を有する基であり、それらのすべての異性体を含む。長鎖アルキル基は、8〜22個の炭素原子を有する基、好ましくは、12〜22個の炭素原子を有する基、12〜20個の炭素原子を有する基、及び16〜18個の炭素原子を有する基である。
【0060】
[0074] 用語「シクロアルキル」は、単環式環又は多縮合環を有する、3〜22個の炭素原子の環式のアルキル基を指す。シクロアルキル基は、例として、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロオクチル等の単環状構造、又はアダマンタニル等の多環状構造を含む。
【0061】
[0075] 用語「アルケニル」は、好ましくは2〜22個の炭素原子を有する分枝又は非分枝の不飽和炭化水素基のモノラジカル、及び3〜22個の炭素原子を有する1つ又は複数の環を有するシクロアルケニル基であって、少なくとも1つの環は二重結合を含むシクロアルケニル基を指す。アルケニル基は、共役していてもよい1つ又は複数の二重結合(C=C)を含んでいてもよい。好ましいアルケニル基は、1つ又は2つの二重結合を有する基である。短鎖アルケニル基は、エチレン(ビニル)プロピレン基、ブチレン基、ペンチレン基及びヘキシレン基を含み、それらのすべての異性体を含む2〜6個の炭素原子を有する基である。長鎖アルケニル基は、8〜22個の炭素原子を有する基、好ましくは、12〜22個の炭素原子を有する基、12〜20個の炭素原子を有する基、及び16〜18個の炭素原子を有する基である。用語「シクロアルケニル」は、少なくとも1つの環が二重結合(C=C)を含む単環式環又は多縮合環を有する3〜22個の炭素原子の環式のアルケニル基を指す。シクロアルケニル基は、例として、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロオクテニル、シクロオクタジエニル、シクロオクタトリエニル等の単環状構造を含む。
【0062】
[0076] 用語「アルキニル」は、好ましくは2〜22個の炭素原子を有し、1つ又は複数の三重結合(C□C)を有する不飽和炭化水素のモノラジカルを指す。アルキニル基は、エチニル、プロパルギル等を含む。短鎖アルキニル基は、2〜6個の炭素原子を有する基であり、それらの異性体をすべて含む。長鎖アルキニル基は、8〜22個の炭素原子を有する基、好ましくは、12〜22個の炭素原子を有する基、12〜20個の炭素原子を有する基、及び16〜18個の炭素原子を有する基である。
【0063】
[0077] 用語「アリール」は、単環(例えばフェニル)、1つ又は複数の環(例えばビフェニル)、又は多縮合(融合)環を有する6〜22個の炭素原子の不飽和芳香族炭素環式基を含む基であって、少なくとも1つの環は芳香族である(例えば、ナフチル、ジヒドロフェナントレニル(dihydrophenanthrenyl)、フルオレニル、又はアントリル)基を指す。アリールは、フェニル、ナフチル等を含む。アリール基は、不飽和の芳香環(複数可)に加えて、アルキル、アルケニル又はアキニルである部分を含んでいてもよい。用語「アルカリル」は、アルキル部分、つまり、−アルキレン−アリール、及び−置換されたアルキレン−アリール、を含むアリール基を指す。上記アルカリル基は、ベンジル、フェネチル等により例示される。
【0064】
[0078] 本明細書中に記載されるように、アルキル基、アルケニル基、アルキニル基及びアリール基は任意選択で置換され(用語(複数可)は置換された変形形態を含むことができる)、基中の炭素原子数及び基の不飽和度に依存した1〜8個の非水素置換基を含んでいてもよい。
【0065】
[0079] 用語「アルキレン」は、好ましくは1〜10個、より好ましくは1〜6個、より好ましくは2〜4個の炭素原子を有する分枝又は非分枝の飽和炭化水素鎖のジラジカルのことを言い、この用語は、置換された変形形態を含むことができる。この用語は、メチレン(−CH2−)、エチレン(−CH2CH2−)、より概略的には−(CH2)n−[式中、nは1〜10、より好ましくは1〜6、又は2、3若しくは4である]、等の基により例示される。アルキレン基は分枝していてもよい。アルキレン基は任意選択で置換されてもよい。アルキレン基は、炭素原子当たり2つまでの非水素置換基を有していてもよい。好ましい置換されたアルキレン基は1つ、2つ、3つ又は4つの非水素置換基を有している。
【0066】
[0080] 用語「アリーレン」は、上記に定義されるアリール(置換されたアリールを含む)に由来するジラジカルのことを言い、1,2−フェニレン、1,3−フェニレン、1,4−フェニレン、1,2−ナフチレン等により例示される。
【0067】
[0081] 用語「アミノ」は、基−NH2又は基−NRRを指し、式中、両方のRが水素ではないとすれば、各Rは、水素、アルキル、置換されたアルキル、シクロアルキル、置換されたシクロアルキル、アルケニル、置換されたアルケニル、シクロアルケニル、置換されたシクロアルケニル、アルキニル、置換されたアルキニル、アリール、ヘテロアリール、及び複素環からなる基から独立して選択される。
【0068】
[0082] 本明細書中に論じられるように、アルキル基は任意選択で置換され、アルキル基の大きさに依存して、好ましくは1〜10個の置換基を有していてもよい。置換されたアルキル基は、1〜8個の置換基、1〜5個の置換基、1〜3個の置換基及び1個若しくは2個の置換基を伴う基を含む。
【0069】
[0083] 「ハロアルキル」は、同じ又は異なっていてもよい、本明細書中に定義される1つ又は複数のハロ基により置換された、本明細書中に定義されるアルキルを指す。代表的なハロアルキル基は、例として、トリフルオロメチル、3−フルオロドデシル、12,12,12−トリフルオロドデシル、2−ブロモオクチル、3−ブロモ−6−クロロヘプチル等を含む。
【0070】
[0084] 用語「ヘテロアリール」は、少なくとも1つの環内に酸素、窒素、及び硫黄から選択される1〜4個のヘテロ原子を有する(環が1個を超える場合)、2〜22個の炭素原子の芳香族基を指す。ヘテロアリール基は任意選択で置換されてもよい。
【0071】
[0085] 用語「ヘテロ環」又は「複素環」は、単環又は多縮合環、2〜22個の炭素原子、並びに少なくとも1つの環内における窒素、硫黄、リン及び/又は酸素から選択される1〜6個、好ましくは1〜4個のヘテロ原子、を有するモノラジカルの飽和基又は不飽和基を指す。複素環基は置換されてもよい。
【0072】
[0086] 用語「エステル」は、当技術分野で理解されている化学物質のことを言い、特に形態(RCO−)の基を含むことができる。
【0073】
[0087] 1つ又は複数の置換基を含む上記基のいずれに関しても、上記基は、立体的に非実用的である及び/又は合成的に実現不可能であるいかなる置換又は置換パターンも含まないことが理解される。本発明の化合物は、開示された化合物の置換から生じる新規な立体化学的異性体をすべて含む。
【実施例】
【0074】
[0088] 本発明は、以下の非限定的な実施例によりさらに理解されるであろう。
【0075】
〔実施例1:プロカスパーゼ活性化化合物〕
[0089] アポトーシスカスケードにおけるタンパク質の変異又は異常発現はよくある癌の特徴である。これらの変化は、アポトーシス促進シグナルが実行因子カスパーゼに伝達されるのを阻止し、したがってアポトーシス細胞死を阻止し細胞増殖が許可される可能性がある。カスパーゼ−3及びカスパーゼ−7は重要な実行因子カスパーゼであり、上流のシグナルにより活性化される不活性な酵素前駆体として存在する。重要なことには、プロカスパーゼ−3の発現レベルは、ある種の癌性細胞内では、非癌性対照に比べて著しく高度である。本明細書で、我々は、プロカスパーゼ−3を活性カスパーゼ−3に直接活性化する低分子の同定について報告する。特定の化合物であるPAC−1は、約220ナノモル濃度の桁でEC50のin vitroでの活性化を引き起こし、多数の癌性細胞株におけるアポトーシスを誘導する。
【0076】
[0090] 多くの知られている抗癌薬とは対照的に、PAC−1で処置した細胞はプロカスパーゼ−3の即時活性化を示し、PAC−1の毒性は、細胞中に含まれるプロカスパーゼ−3の量に正比例することが示される。したがって、PAC−1は、生体内で、プロカスパーゼ−3をカスパーゼ−3に直接活性化し、欠陥のあるアポトーシス機構を有する細胞においてでさえこの化合物がアポトーシスを誘導することを可能にする。PAC−1は、プロカスパーゼ−3を直接活性化することで知られている最初の低分子であり、実行因子カスパーゼの直接的な活性化は、プロカスパーゼ−3がアップレギュレートされる多くの癌を含む様々な癌において有利であると証明することが可能である新規な抗癌戦略である。
【0077】
[0091] 約20,000種の構造的に多様な低分子の集合体を、in vitroのプロカスパーゼ−3を活性化する能力に対してスクリーニングした。プロカスパーゼ−3を大腸菌中で発現させ、精製した(Royら,2001)。プロカスパーゼ−3(50ng/mLの濃度)を384ウェルプレートのウェルに加え、化合物を約40μMの最終濃度まで加えた。次いで、各プレートを37℃で2時間インキュベートし、その後、カスパーゼ−3ペプチド基質Ac−Asp−Glu−Val−Asp−p−ニトロアニリド(Ac−DEVD−pNa)を200μMの濃度まで加えた。2時間にわたりp−ニトロアニリン発色団の形成を405nmで追跡した。
【0078】
[0092] 評価した化合物のうち、4種が、バックグラウンドに対してカスパーゼ−3ペプチド基質の加水分解の著しい増加を誘導した。それらの4種のうち、1種は、in vitroのプロカスパーゼ−3活性化に対する強い用量依存効果を示した。図1Aに示すように、この最初のプロカスパーゼ活性化化合物(PAC−1)は、0.22μMの濃度で、プロカスパーゼ−3の最大活性化の2分の1をもたらす。この化合物は、完全に活性化されたカスパーゼ−3酵素の触媒活性に対する効果を有していないので、カスパーゼ−3自体の活性を単に上昇させているのではない(図1A)。
【0079】
[0093] プロカスパーゼ−3はN末端プロドメイン(1〜28残基)、それに続く、サブユニット間のリンカーにより分けられた大サブユニット(17kDa)及び小サブユニット(12kDa)を有している(Popら,2003)。生体内では、2つのプロカスパーゼ−3モノマーが集合して、サブユニット間のリンカー中のD175での切断により活性化することができる触媒的に不活性なホモダイマーを形成する。プロドメインの明確な役割は不明であり、最高の触媒活性には、サブユニット間の領域の切断のみで十分であることが示された(Stennicke,H.R.ら,1998)。プロカスパーゼ−3は触媒的に有能であるが、12個のアミノ酸の安全装置の存在により自己活性化に対して高度に抵抗性がある。しかしながら、安全装置が変異すると、プロカスパーゼ−3の著しい自己活性化が観察される(Royら,2001)。この重要な調節領域と、又は他の部位と相互に作用する化合物は、プロカスパーゼ−3の自己活性化を可能にすることができる。
【0080】
[0094] プロカスパーゼ−3の自己活性化を触媒するPAC−1の能力を直接評価するために、プロカスパーゼ−3タンパク質を100μM PAC−1と共に1〜5時間インキュベートした。図1Bでウエスタンブロットにより示すように、PAC−1は、時間依存的にプロカスパーゼ−3の切断を誘導し、4時間後に>50%の処理が観察された。対照的に、緩衝液中でインキュベートしたプロカスパーゼ−3は、同一の時間にわたり事実上自己活性化を示さない。PAC−1が相互に作用しているプロカスパーゼ−3の領域を正確に示すための試みでは、安全装置領域中の重要なアスパラギン酸の三つ組である残基Asp179、Asp180、及びAsp181においてアラニン置換を行った。これらの部位での変異はすべて、プロカスパーゼ−3を活性化するPAC−1の能力を劇的に低下させ、ある種の変異はPAC−1によるプロカスパーゼ−3の活性化に対してより有害であった(図2A)。
【0081】
[0095] カスパーゼ−3と同様に、カスパーゼ−7もまた、タンパク質分解により活性化される不活性な酵素前駆体として存在する。カスパーゼ−3及びカスパーゼ−7は共に実行因子カスパーゼであり、重要な配列及び構造上の相同性を有している(Denault,J.−Bら,2003)。プロカスパーゼ−7はまた、重要な三つ組中に3つのアスパラギン酸の代わりに2つのアスパラギン酸(Asp−Thr−Asp)しか有していないが、類似する安全装置領域を有するかもしれない。図2B中のデータにより示すように、PAC−1はまた、プロカスパーゼ−3のその活性化よりも効果が低いが、プロカスパーゼ−7を活性化することもできる(EC50はプロカスパーゼ−3活性化の0.22μMに対して4.5μM)。PAC−1によるプロカスパーゼ−7活性化の効力は、プロカスパーゼ−3のAsp−Ala−Asp変異体に対するその効果に類似している(EC50=2.77μM)。PAC−1の効果は、プロカスパーゼ−3が急速な自己活性化を起こす低pH値で無効にされる(図2C)。
【0082】
[0096] プロカスパーゼ−3を活性化してヒト細胞株におけるアポトーシスを誘導する低分子の能力について試験し、PAC−1は、様々な癌細胞株におけるアポトーシスを誘導することが分かった。HL−60細胞ではPAC−1の添加は、著しいクロマチン凝縮を伴う細胞膜上への多量のホスファチジルセリンの露出につながる(図3A及びB)。さらに、化合物は、PARP−1の切断を誘導し(生体内でのPARP活性アッセイによる評価;Putt KSら,2005)、ミトコンドリア膜脱分極を引き起こす(以下参照)。著しい細胞の小疱形成も顕微鏡検査により観察された。さらに、PAC−1の毒性は、カスパーゼ阻害剤z−VAD−fmk存在下で無効にすることができるかもしれない(データ掲載せず;Sleeら,1996参照)。
【0083】
[0097] PAC−1がプロカスパーゼ−3の直接的な活性化を介してアポトーシスを実際に誘導している場合、アポトーシス現象の時間的経過が、標準的なアポトーシス促進剤を用いて観察されるものに比べて変化するはずである。エトポシドは、内因性の経路を介してアポトーシスを誘導することでよく知られており、したがって、エトポシド処置細胞において、ミトコンドリア膜脱分極の後、プロカスパーゼ−3が活性化される。実際に、10μMエトポシドで処置したHL−60細胞では、ミトコンドリア膜脱分極が観察された後、カスパーゼ−3様活性が検出される(図4A)。対照的に、PAC−1での細胞の処置は著しく異なる結果をもたらす。PAC−1では、最初に観察されたアポトーシスの生化学的特徴はカスパーゼ−3様酵素活性である。この活性は、化合物添加の数分以内に認められ、50%の活性化が、2時間余りで、いかなる著しいミトコンドリア膜脱分極の十分前に起こる(図4B)。さらに、PARP活性は、PAC−1で処置した細胞では急速に低下するが、この低下は、エトポシド処置細胞ではより遅い時点で観察される(図4C)。対照実験は、PAC−1がPARP−1の酵素活性を直接抑制しないことを示す。アポトーシス現象の典型的な順序では、ミトコンドリア膜が脱分極し、カスパーゼが活性化され、カスパーゼ基質(PARP−1等)が切断される。PAC−1で処置した細胞が、カスパーゼ−3/−7の急速な活性化(ミトコンドリア膜脱分極前)及びカスパーゼ基質の急速な切断を示すといった観察は、この化合物が、プロカスパーゼ−3の直接的な活性化を介したその細胞毒性を発揮することを示す。
【0084】
[0098] さらにPAC−1の効力を明示するために、様々なレベルのプロカスパーゼ−3を有する癌細胞株における細胞死を誘導するこの化合物の能力を評価した。決定は、多数の癌細胞株(白血病、リンパ腫、黒色腫、神経芽細胞腫、乳癌、肺癌、及び腎臓癌)及び健常なドナーの骨髄から単離した白血球中に存在するプロカスパーゼ−3の量に関して行った。細胞死誘導に対するIC50値はこれらの細胞株におけるPAC−1に関して得られた。組み合わせたデータは、プロカスパーゼ−3の細胞内濃度及びPAC−1に対する感受性の間に強い相関を示す(図4D)。特に、健常なヒトドナーの骨髄に由来する白血球は、最低量のプロカスパーゼ−3を有するものに含まれ、PAC−1は、これらの細胞に対して比較的低毒性である。PAC−1は、肺癌細胞株NCI−H226に対して最も効力があり、0.35μMのIC50である。文献(Svingenら,2004)のデータに一致して、我々は、この細胞株が、非癌性対照のプロカスパーゼ−3の濃度の5倍を超える濃度を有することが分かった。
【0085】
[0099] PAC−1を用いたこれらの実験とは対照的に、エトポシドは、細胞培養中の効力及びプロカスパーゼ−3の細胞内レベルの間の上記相関を示さなかった。例えば、エトポシドは黒色腫細胞株のうちの3種(UACC−62、CRL−1872、及びB16−F10)、乳癌細胞株(Hs 578t)、及び肺癌細胞株(NCI−H226)での死の誘導において効果がなく(IC50>50μM)、これらの細胞株は、1.0、2.4、1.9、3.7、及び5.3のプロカスパーゼ−3レベルをそれぞれ有している。エトポシドは、4.3、4.0、4.7、及び4.4のプロカスパーゼ−3レベルをそれぞれ有するHL−60、U−937、SK−N−SH、及びPC−12に対して有効であった(IC50<1μM)。したがって、全体として、プロカスパーゼ−3レベルとエトポシドについてのIC50の間に相関はない。
【0086】
[00100] 癌性細胞は、通常、アポトーシスカスケードにおける種々様々なタンパク質の変異又は異常発現によりアポトーシス促進シグナルに対する感受性が低下している。そのため、癌の多くの型は、周知のように、アポトーシス細胞死に対する内在性シグナルだけでなく類似した機構を介して作用する化学療法剤にも抵抗性がある。ある種の癌におけるプロカスパーゼ−3発現レベルの奇異なアップレギュレーションは、この存在するタンパク質の細胞内プールを使用する機会を提供して、アポトーシスを直接誘導し、それにより、機能しない又は損なわれることが多いカスケードの上流の部分を回避する。プロカスパーゼ−3は、自己活性化を起こすことが相対的にできないことが周知であるが、それは、不活性状態に自身を維持するための12個のアミノ酸の安全装置に依存している。PAC−1は、in vitroで、プロカスパーゼ−3の自己活性化を誘導し、この活性化は、安全装置の重要なトリ−アスパラギン酸領域の変異により著しく減少する。このデータは、PAC−1が、プロカスパーゼ−3の休止状態を維持するための安全装置の能力を直接妨害するという見解と一致している。
【0087】
[00101] 細胞培養中、PAC−1処置は、急速なカスパーゼ−3様活性を誘導する。次いで、抗アポトーシスタンパク質(Bcl−2、Bcl−XL等)のカスパーゼ−3媒介性の切断は、ミトコンドリア膜の脱分極を誘導し、アポトーシスを増強すると思われる。さらに、様々な癌細胞株に対するPAC−1の効力は細胞中のプロカスパーゼ−3の濃度に正比例する。PAC−1が有効ないくつかの細胞株は、アポトーシスに対して抵抗性にする欠陥のあるアポトーシス経路を有していることに注目する価値がある。例えば、Apaf−1発現はSK−MEL−5細胞中で劇的に減少し、Bcl−2は、NCI−H226肺癌細胞株中で過剰発現する。
【0088】
[00102] 本明細書中に提示したデータは、プロカスパーゼ−3活性化化合物が一般的な癌に対して非常に有効となる可能性があるという見解を十分に支持する。有効性は、プロカスパーゼ−3レベルが異常に高度な状態に対して高めることができる。
【0089】
[00103] 癌生検材料のプロカスパーゼ−3レベルの評価は単純で迅速にすることができる。そのため、PAC−1等の化合物の考えられる有効性は、高度な精度で推測的に評価することができる。したがって、本明細書中のプロカスパーゼ−3活性化剤及び方法は、抗癌処置の分野で、一般的な細胞毒素に頼る治療法に優先される可能性があるオーダーメイド医療戦略を提供する。
【0090】
材料及び方法:
[00104] 材料:Ni−NTA樹脂及び抗Penta His Alexa Fluor 647抗体はQiagen社(バレンシア、CA)から購入した。ブラッドフォード染料はBio−Rad社(ハーキュリーズ、CA)から購入した。ピン移動装置はV&P Scientific社(サンディエゴ、CA)から購入した。試薬z−vad−fmkはCalbiochem社(サンディエゴ、CA)から購入した。大腸菌RosettaはNovagen社(マディソン、WI)から購入した。抗カスパーゼ−3抗体はSigma社(セントルイス、MO)から購入した。アネキシンV Alexa Fluor 488複合体、JC−9、及びヨウ化プロピジウムはMolecular Probes社(ユージーン、OR)から購入した。IPTG及びMTS/PMS CellTiter 96細胞増殖アッセイ試薬はPromega社(マディソン、WI)から購入した。ウシ胎児血清はBiomeda社(フォスターシティ、CA)から購入した。96及び384ウェルマイクロタイタープレート、顕微鏡用スライドガラス、顕微鏡用カバーガラス、ウマ血清、並びに他のすべての試薬はFisher社(シカゴ、IL)から購入した。
【0091】
[00105] 方法:細胞培養条件。U−937細胞、HL−60細胞、CRL−1872細胞、ACHN細胞、NCI−H226細胞、SK−MEL−5細胞、及びUACC−62細胞を10%FBSを添加したRPMI1640培地中で成長させた。SK−N−SH細胞、B16−F10細胞、及びHs 578t細胞を、アールのBSS、1.5g/L炭酸水素ナトリウムを有し、10%FBSを添加したイーグルの最小必須培地中で成長させた。PC−12細胞は、5%FBS及び10%ウマ血清を添加したRPMI1640培地中で成長させた。ヒト骨髄を、40%FBSを添加したIDMEM中で成長させた。すべての細胞株を、5%CO2、95%空気雰囲気中37℃でインキュベートした。U−937細胞及びHL−60細胞は必要に応じて2〜3日ごとに継代した。ヒト骨髄は、冷凍ストックから解凍し、直ちに希釈して実験に使用した。他のすべての細胞は、約80%の培養密度に達した時に継代した。
【0092】
[00106] タンパク質の発現及び精製。プロカスパーゼ−3又はプロカスパーゼ−7の発現プラスミドを含む大腸菌Rosettaの終夜培養物1mLを適切な抗生物質を含むLB培地1Lに接種した。細胞を1mM IPTGで30分間誘導した。次いで、細胞を遠心沈殿し、NTA結合緩衝液中に再懸濁した(150mM NaCl、50mM Tris、10mMイミダゾール、pH7.9)。フレンチプレスに2回通過させることにより細胞を溶解した。次いで、細胞溶解物を14,000×gで30分間回転させた。上清をデカントし、1mLのニッケル−NTA樹脂を加えた。細胞溶解物を4℃で1時間インキュベートした。樹脂をカラムに装填し、10mL NTA結合緩衝液、次に10mL NTA洗浄緩衝液で洗浄した(150mM NaCl、50mM Tris、20mMイミダゾール、pH7.9)。タンパク質を10mL NTA溶出緩衝液で1mL画分中に溶出した(150mM NaCl、50mM Tris、250mMイミダゾール、pH7.9)。タンパク質を含む画分をプールし、タンパク質の量をブラッドフォードアッセイを使用して測定した。
【0093】
[00107] ライブラリースクリーニング。単離したプロカスパーゼ−3をカスパーゼアッセイ緩衝液中に50ng/mLまで希釈した(50mM HEPES、100mM NaCl、10mM DTT、0.1mM EDTA、0.1%CHAPS、及び10%グリセロール、pH7.4)。45μLのプロカスパーゼ−3溶液をNunc社製384ウェル平底マイクロタイタープレートの各ウェルに加えた。約20,000種の化合物をスクリーニングした。約6,000種の化合物をイリノイ大学の化学科内の様々な供給源から収集した。それらの構造は、http://www.scs.uiuc.edu/〜phgroup/comcollections.html.から入手可能である。他の約14,000種の化合物をChembridge社(サンディエゴ、カリフォルニア)から購入した。PAC−1は、Chembridge社から購入した化合物のメンバーの1種であった。
【0094】
[00108] DMSO中10mMストック溶液として作製した化合物を、0.2μLの化合物を移動させる384ピン移動器具を使用してウェル中に移動させた。これにより、約40μMの最終化合物濃度を得た。DMSOのみ(化合物を含まず)をピン移動させて対照実験を行った。次いで、プレートを37℃で2時間インキュベートした。カスパーゼアッセイ緩衝液中Ac−DEVD−pNA(N−アセチル−ASP−Glu−Val−Asp−p−ニトロアニリド)の2mM溶液の5μLを各ウェルに加えた。次いで、プレートを、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きをカスパーゼ−3の活性を測定するために使用した。
【0095】
[00109] 活性化曲線。プロカスパーゼ−3活性化剤の用量依存性は、96ウェルプレート中のカスパーゼアッセイ緩衝液中50ng/mLのプロカスパーゼ−3、活性カスパーゼ−3、プロカスパーゼ−7、又は活性カスパーゼ−7の90μLに様々な濃度の化合物を加えることにより測定した。次いで、プレートを37℃で12時間インキュベートした。次いで、カスパーゼアッセイ緩衝液中Ac−DEVD−pNAの2mM溶液の10μLを各ウェルに加えた。プレートを、Spectra Max Plus 384ウェルプレートリーダー中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、非処置対照ウェルからの活性化における変化倍率を算出した。
【0096】
[00110] PAC−1活性化ゲル。プロカスパーゼ−3を正確に上記の通りに発現させ単離した。プロカスパーゼ−3をカスパーゼアッセイ緩衝液中約50μg/mLに希釈した。次いで、プロカスパーゼ−3を100μM PAC−1存在下又は非存在下で37℃で様々な時間インキュベートした。この培養の後、等量のローディング緩衝液(150mM NaCl、50mM Tris、2%SDS、20%グリセロール、pH8.0)を各プロカスパーゼ−3サンプルに加えた。次いで、時間的経過が終了するまで、すべてのサンプルを−80℃で保存した。次いで、すべてのサンプルを95℃で5分間インキュベートし、12%SDS−PAGEゲル上で泳動した。次いで、タンパク質を、ニトロセルロース紙に一晩移した。ブロットをTTBS中で洗浄し(150mM NaCl、50mM Tris、0.1%Tween−20、pH7.4)、10%ミルク溶液で2時間ブロックした。次いで、ブロットを、抗Penta His Alexa Fluor 647抗体の1:5000希釈液中で2時間インキュベートした。次いで、ブロットをTTBSで洗浄し、Typhoon蛍光スキャナーでスキャンした(Amersham Biosciences社、サニーベール CA)。
【0097】
[00111] 安全装置の変異。DDDプロカスパーゼ−3安全装置(配列番号1;配列番号2;配列番号9)を、ADD(配列番号3、配列番号4、配列番号10)、DAD(配列番号5、配列番号6、配列番号11)、及びDDA(配列番号7、配列番号8、配列番号12)に以下のプライマー、それぞれ、gacagacagtggtgttgCGgatgacatggcgtgtcataaaatacc(配列番号13)、gacagacagtggtgttgatgCtgacatggcgtgtcataaaatacc(配列番号14)、及びgacagacagtggtgttgatgatgCcatggcgtgtcataaaatacc(配列番号15)を用いたQuickchange法を使用して変異させた。図9及び図10も参照すること。変異した塩基は下線を引き、大文字で示す。遺伝子の全体にわたる適切な配列を保証するためにすべての変異体プラスミドについて配列を決定した。すべての変異体プラスミドは、上記の野生型プロカスパーゼ−3の通りに正確に発現した。96ウェルプレート中のカスパーゼアッセイ緩衝液中50ng/mLの野性型プロカスパーゼ−3及び変異体プロカスパーゼ−3の90μLに様々な濃度のPAC−1を加えることにより、各プロカスパーゼ−3変異体を活性化するPAC−1の能力を測定した。次いで、プレートを37℃で12時間インキュベートした。次いで、カスパーゼアッセイ緩衝液中Ac−DEVD−pNAの2mM溶液の10μLを各ウェルに加えた。プレートを、Spectra Max Plus 384ウェルプレートリーダー中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、各変異体についての活性の変化倍率を算出した。
【0098】
[00112] PAC−1のプロカスパーゼ−3の活性化に対するpHの影響。PAC−1によるプロカスパーゼ−3活性化に対するpHの影響は、pHカスパーゼアッセイ緩衝液(25mM MES、25mM Tris、25mM HEPES、25mM PIPES、100mM NaCl、10mM DTT、0.1mM EDTA、0.1%CHAPS、及び10%グリセロール)中にプロカスパーゼ−3を50ng/mLの濃度に希釈することにより測定した。次いで、緩衝液を様々なpH値に変化させ、90μLを96ウェルプレートの各ウェルに加えた。各pH値に対して、PAC−1を100μMの濃度まで加え、又は対照としてDMSOを加えた。次いで、プレートを37℃で12時間インキュベートした。次いで、カスパーゼアッセイ緩衝液中Ac−DEVD−pNAの2mM溶液の10μLを各ウェルに加えた。プレートを、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、各pH値に対する活性化における変化倍率を算出した。
【0099】
[00113] アネキシンV染色。200μM PAC−1又は対照としてDMSOのみを含む培地500μLを24ウェルプレートのウェルに加えた。次いで、2×106細胞/mLの濃度のHL−60細胞500μLを24ウェルプレートに加えた。プレートを37℃で20時間インキュベートした。細胞を遠心分離により回収し、PBS中で2回洗浄した。次いで、細胞をアネキシンV結合緩衝液(10mM HEPES、140mM NaCl、2.5mM CaCl2、pH7.4)中で洗浄し、100μLのアネキシンV結合緩衝液中に再懸濁した。5μLのアネキシンV Alexa Fluor 488複合体を加え、チューブを、光から保護して、室温で15分間インキュベートした。次いで、400μLのアネキシンV結合緩衝液を加え、次に、ヨウ化プロピジウムの1mg/mL溶液を1μL加えた。各細胞の蛍光強度を、525nm(緑色チャンネル)及び675nm(赤色チャンネル)でフローサイトメトリーにより測定した。少なくとも50,000個の細胞を各実験で分析した。
【0100】
[00114] 凝縮したクロマチンの染色。200μM PAC−1又は対照としてDMSOのみを含む培地500μLを24ウェルプレートのウェルに加えた。次いで、2×106細胞/mLの濃度のHL−60細胞500μLを24ウェルプレートに加えた。細胞を20時間インキュベートし、遠心分離により回収した。次いで、細胞をPBS緩衝液中で洗浄し、次に氷冷100%エタノールを加えた。細胞を一晩4℃で固定した。固定細胞を2μg/mLヘキスト−33258と共に室温で30分間インキュベートした。次いで、1滴の細胞を顕微鏡用スライドガラスに加え、厚さNo.1のカバーガラスでカバーした。凝縮したクロマチンをZeiss社製Axiovert 100顕微鏡上で400×倍率で観察した。
【0101】
[00115] z−vad−fmkによる細胞死阻害。5×105細胞/mLの濃度のHL−60細胞100μLを96ウェルプレートのウェルに加えた。次いで、細胞を、細胞透過性全カスパーゼ阻害剤であるz−vad−fmk 100μM存在下又は非存在下で1時間インキュベートした。次いで、PAC−1を様々な濃度で加え、細胞をさらに24時間インキュベートした。MTS/PMS CellTiter 96細胞増殖アッセイ試薬20μLの各ウェルへの添加により細胞死を定量した。プレートを、37℃で、約45分間、有色生成物が形成されるまでインキュベートした。次いで、吸光度を、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)で490nmで測定した。
【0102】
[00116] 生体内でのミトコンドリア膜電位の測定。1×106細胞/mLの濃度のHL−60細胞1mLを24ウェルプレートのウェルに加えた。次いで、PAC−1を100μMの濃度まで加え又は対照としてDMSOのみを加えた。細胞を様々な時間インキュベートし、次いで、細胞を遠心分離により回収した。細胞をPBS中で洗浄し、1mL PBS中で再懸濁した。JC−9染料10μgを加え、細胞を光から保護して、室温で10分間インキュベートした。次いで、細胞をPBSで2回洗浄し、500μL PBS中に調製した。各細胞の蛍光強度を、525nm(緑色チャンネル)及び675nm(赤色チャンネル)でフローサイトメトリーにより測定した。各実験で50,000個の細胞を分析した。次いで、赤色チャンネルにおけるシフトを使用してミトコンドリア膜脱分極の量を測定した。
【0103】
[00117] 生体内でのカスパーゼ−3様活性の測定。カスパーゼ−3様プロテアーゼ活性の量を、細胞溶解物により毎分切断されるAc−DEVD−pNA(N−アセチル−ASP−Glu−Val−Asp−p−ニトロアニリド)の量により測定した。これを達成するために、様々な濃度のPAC−1を含む培地50μLを96ウェルプレートのウェルに加えた。5×106細胞/mLの濃度のHL−60細胞50μLをプレートに加え、様々な時間インキュベートした。インキュベーション期間の後、プレートを1000×gで5分間回転させ、細胞をペレットにした。次いで、100μL PBS中で細胞を洗浄し、150μL氷冷カスパーゼアッセイ緩衝液中に再懸濁した。次いで、各ウェルを超音波処理して細胞を溶解させた。細胞溶解物90μLを各ウェルから新しいプレート中に移動した。200μMの最終濃度となるよう各ウェル中にAc−DEVD−pNAを加えた。次いで、プレートを、Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)中で2時間の間、405nmで2分ごとに読み取った。各ウェルに対する直線部分の傾きを決定し、毎分切断されたAc−DEVD−pNAの量を算出した。
【0104】
[00118] 生体内でのPARP切断の測定。生体内でのPARP活性アッセイを使用することによりPARP切断の量を測定した。これを達成するために、200μM NAD+を含む培地50μLを96ウェルプレートの対照ウェルに加えた。次いで、200μM PAC−1及び200μM NAD+を含む培地50μLを試験ウェルに加えた。次いで、5×106細胞/mLの濃度のHL−60細胞25μLを各ウェルに加えた。細胞を様々な時間インキュベートし、次いで、1000×gで5分間回転させた。細胞培地を除去し、25mM H2O2を含む50μL PARP溶解緩衝液(50mM Tris、10mM MgCl2、pH8.0、1%Triton X−100)と置換した。次いで、プレートを37℃で60分間インキュベートした。まだ存在しているNAD+の量を測定するために、20μLの2M KOH及び20μLの20%(v/v)アセトフェノン(エタノール中)溶液を96ウェルプレートの各ウェルに加えた。次いで、プレートを4℃で10分間インキュベートした。90μLの88%(v/v)ギ酸溶液を96ウェルプレートの各ウェルに加えた。次いで、プレートを、110℃に設定したオーブン中で5分間インキュベートした。プレートを冷却し、次いで、Criterion Analyst AD(Molecular Devices社、サニーベール、CA)上で励起360nm、発光445nm、及び400nmカットオフ ダイクロイックミラーで読み取った。ウェル当たり5回の読み取りを実行しながら1.6×105μsの1000W連続ランプを使用して蛍光体を励起させた。次いで、毎分切断されたNAD+のモル数を算出し、対照ウェルと比較した残存するPARP活性を測定した。
【0105】
[00119] 様々な細胞株中のプロカスパーゼ−3の相対濃度。U−937細胞、HL−60細胞、及びヒト骨髄細胞は遠心分離により回収し、一方、他のすべての細胞株は、最初にトリプシン処理して細胞を遊離し、次いで、遠心分離により回収した。PBS中ですべての細胞を洗浄し、1mL氷冷100%エタノール中に再懸濁した。細胞を一晩4℃で固定した。細胞を1000×gで5分間回転させ、PBSで洗浄し、次いで、PBS中の抗カスパーゼ−3抗体の1:100希釈液100μLを加えた。細胞を室温で2時間インキュベートし、次に、5回PBS洗浄した。次いで、細胞を、光から保護して、抗マウスAb Cy3標識抗体の1:10,000希釈液1mL中に室温で2時間再懸濁した。PBSで細胞を5回洗浄し、500μL PBS中に再懸濁した。フローサイトメトリーにより675nm(赤色チャンネル)で各細胞の蛍光強度を測定した。少なくとも20,000個の細胞を各実験で分析した。集団の中央値を使用して各細胞株中のプロカスパーゼ−3の相対濃度を測定した。
【0106】
[00120] 様々な細胞株におけるIC50値の測定。様々な濃度のPAC−1又はエトポシドを含む培地50μLを、DMSOのみを含む対照ウェル以外の96ウェルプレートの各ウェルに加えた。U−937細胞、HL−60細胞、及びヒト骨髄細胞は遠心分離により回収し、一方、他のすべての細胞株は遠心分離前に最初にトリプシン処理した。次いで、細胞を培地中に再懸濁し、U−937細胞、HL−60細胞、及びヒト骨髄細胞については1×106細胞/mL又は他のすべての細胞株については50,000細胞/mLに希釈した。次いで、50μL細胞溶液を各ウェルに加え、エトポシド及びPAC−1に対してそれぞれ24時間又は72時間プレートをインキュベートした。MTS/PMS CellTiter 96細胞増殖アッセイ試薬20μLの各ウェルへの添加により細胞死を定量した。次いで、プレートを、37℃で、約1時間、有色生成物が形成されるまでインキュベートした。Spectra Max Plus 384プレートリーダー(Molecular Devices社、サニーベール CA)で490nmで吸光度を測定した。
【0107】
[00121] データ分析:Summitソフトウェア(Cytomation社、フォートコリンズ CO)を使用して、すべてのフローサイトメトリー実験のデータを分析した。すべてのグラフを、Table Curve 2Dを使用して、分析した。
【0108】
[00122] Ronald Hoffman教授(イリノイ大学−シカゴ癌センター(Chicago Cancer Center))はヒト骨髄を提供した。Guy Salvesen教授(バーナム研究所)はプロカスパーゼ−3及びプロカスパーゼ−7の発現ベクターを提供した。
【0109】
[00123] 配列表の参照−添付A:添付Aと指定した個別に添付する配列表情報は、本明細書と共に明細書の一部とみなされ、明細書の一部として組み込まれるものとする。
【表1】
【0110】
〔実施例2:プロカスパーゼ活性化化合物の合成〕
[00124] PAC−1及び他の化合物は、以下のスキーム、例えば下記スキーム1及び/又はスキーム2に従って調製した。さらなる変形形態は、当技術分野で知られている方法に従って調製する。
【化8】
【0111】
[00125] 特定の実施例では、PAC−1は、下記スキーム2に従って調製する。
【化9】
【0112】
〔実施例3:PAC−1の類似体〕
[00126] PAC−1の類似(アナログ)化合物を調製し、精製したプロカスパーゼ−3をin vitroで直接活性化する能力について評価した。
【表2】
【0113】
〔実施例4:医薬品実施形態〕
[00127] 以下は、医薬品実施形態及び薬理学的実施形態に関連する情報を記載し、当業者にとって入手可能な当技術分野における情報によりさらに補足される。厳密な処方、投与経路、及び用量は、個々の医師が、患者の症状を考慮して選択することができる(例えばFinglら,in The Pharmacological Basis of Therapeutics,1975,Ch.1 p.1参照)。
【0114】
[00128] 毒性又は器官の機能不全等により、どのようにそしていつ投与を終了すべきか、中断すべきか、又は調整すべきかについて主治医が知っていたであろうということに注目するべきである。反対に、臨床反応が十分でなかった場合(毒性の側面を考慮して又は毒性の側面を防止する中で)、主治医はまた高度なレベルに処置を調整することも知っていたであろう。関心のある疾患の管理における投薬量の大きさは、処置される症状の重症度及び投与経路によって変動する可能性がある。症状の重症度は、例えば、部分的に、標準的な予後評価法により評価してもよい。さらに、投与量及びおそらく投与回数はまた、状況、例えば個々の患者の年齢、体重、及び反応に従って変動する可能性がある。上記に論じたものに相当するプログラムはまた獣医学に使用してもよい。
【0115】
[00129] 処置されている特定の症状及び選択された標的法によっては、上記薬剤を全身に又は局所的に処方及び投与してもよい。処方及び投与の技術は、Alfonso及びGennaro(1995)並びに当技術分野の他のものに認められてもよい。適切な経路は、例えば、経口投与、直腸投与、経皮投与、膣投与、経粘膜投与、若しくは腸管投与又は筋肉注射、皮下注射、若しくは髄内注射を含む非経口送達と同様に眼内投与、鞘内投与、静脈内投与、又は腹腔内投与を含んでいてもよい。
【0116】
[00130] 注射又は他の経路については、本発明の薬剤を水溶液中に、好ましくは、ハンクス溶液、リンガー溶液、注射用水、生理的食塩緩衝液、等の生理学的に適合性のある緩衝液中に処方してもよい。経粘膜投与については、浸透するバリアに適切な浸透剤を製剤に使用することができる。上記浸透剤は、当技術分野で一般に知られている。
【0117】
[00131] 本発明の実行に関して本明細書中に開示された化合物を全身性の又は他の投与に適した用量に処方するための薬学的に許容し得る担体の使用は本発明の範囲内である。担体が適切に選択され、かつ適正な製造が実施されれば、本発明の組成物、特に、溶液として処方される組成物は、静脈注射等の経路によって非経口的に投与されてもよい。適切な化合物は、当技術分野でよく知られている薬学的に許容し得る担体を使用して、経口投与に適した用量に容易に処方することができる。上記担体は、本発明の化合物を、錠剤、丸剤、カプセル剤、液剤、ゲル、シロップ剤、スラリー、エリキシル剤、溶液剤、懸濁剤等として、例えば処置される患者による経口摂取のために処方することを可能にする。他の経路については、クリーム剤、軟膏剤、ローション剤等の製剤を調製することができる。
【0118】
[00132] 細胞内に投与することを意図した薬剤は、当業者によく知られている技術を使用して投与されてもよい。例えば、上記薬剤を、リポソーム、他の膜移動促進部分、又は他の標的部分中にカプセル化し、次いで、上記のように投与してもよい。リポソームは、内部が水性の球状の脂質二重層を含むことができる。リポソーム形成時に水溶液中に存在するすべての分子を水性内部に取り込むことができる。リポソーム内容物は外部の微小環境から保護されるのみならず、リポソームが細胞膜と融合するので、細胞の細胞質に効率的に送達される。さらに、疎水性の特性により、有機低分子を直接細胞内に投与してもよい。
【0119】
[00133] 本発明で使用するのに適した医薬組成物は、意図された目的を達成するための有効な量の活性成分を含んでいる組成物を含む。有効な量の決定は、特に本明細書中に提供された本開示及び当技術分野における他の情報を考慮すれば十分に当業者の能力の範囲内である。
【0120】
[00134] 活性成分に加えて、これらの医薬組成物は、薬学的に使用することができる調製物への活性化合物の加工を促進する賦形剤及び助剤を含む、薬学的に許容し得る適当な担体を含んでいてもよい。経口投与のために処方された調製物は、錠剤、糖衣錠、カプセル剤、又は溶液剤の形態をしていてもよく、遅延して放出されるために処方された調製物又は医薬が小腸又は大腸に到達する場合に単に放出されるために処方された調製物を含む。
【0121】
[00135] 本発明の医薬組成物は、それ自体知られている方法、例えば従来の混合プロセス、溶解プロセス、懸濁プロセス、造粒プロセス、糖衣錠製造プロセス、浮上プロセス、乳化プロセス、カプセル化プロセス、封入プロセス、凍結乾燥プロセス及び他のプロセスによって製造してもよい。
【0122】
[00136] 非経口投与のための医薬製剤は水溶性形態の活性化合物の水溶液を含む。さらに、活性化合物の懸濁剤を適切な油性注射懸濁剤として調製してもよい。適切な親油性の溶媒又は賦形剤は、ゴマ油等の脂肪油;オレイン酸エチル、トリグリセリド等の合成脂肪酸エステル;又はリポソームを含む。水性注射懸濁剤は、カルボキシルメチルセルロースナトリウム、ソルビトール、デキストラン等の、懸濁剤の粘度を上昇させる物質を含んでいてもよい。任意選択で、高度に濃縮された溶液の調製を考慮に入れるために、懸濁剤は、化合物の溶解度を上昇させる適切な安定剤又は薬剤をさらに含んでいてもよい。
【0123】
[00137] 経口使用のための医薬調製物は、所望により、錠剤又は糖衣錠コアを得るために、活性化合物を固体賦形剤と混合し、任意選択で、結果として生じる混合物を粉砕し、適当な助剤を加えた後、顆粒の混合物を加工することにより得ることができる。適切な賦形剤は、特に、ラクトース、スクロース、マンニトール又はソルビトールを含む糖のような充填剤、並びに例えば、トウモロコシデンプン、コムギデンプン、コメデンプン、バレイショデンプン、ゼラチン、トラガカントゴム、メチルセルロース、ヒドロキシプロピルメチル−セルロース、カルボキシルメチルセルロースナトリウム及び/又はポリビニルピロリドン(PVP)のようなセルロース調製物である。所望により、架橋ポリビニルピロリドン、寒天、アルギン酸、又はアルギン酸ナトリウム等の塩、のような崩壊剤を加えてもよい。
【0124】
[00138] 適切なコーティングを糖衣錠コアに任意選択で施す。この目的のために、濃縮糖溶液を使用してもよく、濃縮糖溶液は、任意選択で、アラビアゴム、タルク、ポリビニルピロリドン、カーボポールゲル、ポリエチレングリコール、及び/又は二酸化チタン、ラッカー溶液、並びに適当な有機溶媒又は混合溶媒を含んでいてもよい。染料又は色素を、錠剤又は糖衣錠のコーティングに、識別のために又は活性化合物投与量の異なる組み合わせを特徴づけるために加えてもよい。
【0125】
[00139] 経口的に使用することができる医薬調製物は、ゼラチンで作製されたプッシュフィットカプセル剤並びにゼラチンで作製された軟質の密閉カプセル剤、及びグリセロール、ソルビトール等の可塑剤を含む。プッシュフィットカプセル剤は、ラクトース等の充填剤、デンプン等の結合剤、及び/又はタルク、ステアリン酸マグネシウム等の潤滑剤、並びに任意選択で安定剤と混合した活性成分を含むことができる。軟質のカプセル剤では、脂肪油、流動パラフィン、液体ポリエチレングリコール等の適当な液剤中に活性化合物を溶解又は懸濁してもよい。さらに、安定剤を加えてもよい。
【0126】
〔実施例5:プロカスパーゼ−3の低分子活性化剤での癌細胞におけるアポトーシスの直接的な誘導〕
[00140] 要約:アポトーシスカスケードにおけるタンパク質の変異又は異常発現は癌の特徴である。これらの変化は、アポトーシス促進シグナルが実行因子カスパーゼに伝達されるのを阻止し、したがってアポトーシス細胞死を阻止し、細胞増殖が許可される。カスパーゼ−3及びカスパーゼ−7は重要な実行因子カスパーゼであり、上流のシグナルにより活性化される不活性な酵素前駆体として存在する。重要なことには、プロカスパーゼ−3のレベルは非癌性対照に比べてある種の癌性細胞内で著しく高度である。本明細書で、我々は、プロカスパーゼ−3を活性カスパーゼ−3に220ナノモル濃度のEC50でin vitroで直接活性化し、様々な癌細胞株におけるアポトーシスを誘導する低分子(PAC−1)の同定について報告する。多くの知られている抗癌薬とは対照的に、PAC−1で処置した細胞はプロカスパーゼ−3の即時活性化を示し、PAC−1の有効性は、細胞に含まれるプロカスパーゼ−3の量に比例することが示される。in vitroでプロカスパーゼ−3を活性化しないPAC−1の誘導体はアポトーシス促進活性も有していない。一次大腸腫瘍から単離した癌性細胞は、同一患者からの隣接した非癌性組織の細胞よりも、PAC−1によるアポトーシス誘導にかなりの感受性がある。これらの癌性細胞は、隣接した非癌性一次組織の細胞よりも平均して約7倍のプロカスパーゼ−3を含んでいる。さらに、大腸癌腫瘍からの一次細胞のPAC−1に対する感受性は、プロカスパーゼ−3標的のレベルと強く関連している。最終的に、単一の実体としてのPAC−1は、PAC−1を経口的に投与した2つのモデルを含む3つの異なるマウスモデルにおいて腫瘍の成長を遅延させるのに活性のあるものとして示された。したがって、PAC−1は、生体内で、プロカスパーゼ−3をカスパーゼ−3に直接活性化し、それによって、欠陥のあるアポトーシス機構を有する細胞においてでさえこの化合物がアポトーシスを誘導することを可能にする。PAC−1は、プロカスパーゼ−3を直接活性化することで知られている最初の低分子であり、実行因子カスパーゼの直接的な活性化は、プロカスパーゼ−3レベルが上昇した多くの癌において有利であると証明することが可能である抗癌戦略である。
【0127】
[00141] 導入。癌の特徴は、本来のアポトーシスシグナルに対するその抵抗性である。癌型によっては、この抵抗性は、通常、アポトーシスカスケードにおける重要なタンパク質のアップレギュレーション若しくはダウンレギュレーション又はこれらのタンパク質をコードする遺伝子内の変異のいずれかによる。上記変化は、ミトコンドリア及びカスパーゼ−9を通る内因性アポトーシス経路並びにデスレセプター及びカスパーゼ−8の作用を伴う外因性アポトーシス経路の両方で起こる。例えば、p53、Bim、Bax、Apaf−1、FLIP、及びその他多くのものの適切なレベルの変化が癌で観察され、欠陥のあるアポトーシスカスケードにつながり、上流のアポトーシス促進シグナルが適切に伝達されず、実行因子カスパーゼであるカスパーゼ−3及びカスパーゼ−7が活性化されない。ほとんどのアポトーシス経路がプロカスパーゼ−3の活性化を最終的に伴うので、これらの遺伝的異常は、有効に、アポトーシス回路における「断線箇所」となり、その結果、上記細胞は制御されずに増殖する。
【0128】
[00142] 癌におけるアポトーシスの中心的な役割から判断して、アポトーシスカスケードにおける特定のタンパク質を標的にする治療を開発するための試みがなされた。例えば、p53、Bclファミリーのタンパク質、又はIAPに対するペプチド結合剤又は低分子結合剤は、Apaf−1のオリゴマー化を促進する化合物のように、アポトーシス促進活性を有している。しかしながら、これらの化合物の多くは、アポトーシスカスケード上の早期に又は中間に位置するものを標的にするので、下流のタンパク質における変異を有する癌は、それらの効果におそらく抵抗性があるであろう。治療目的のために、アポトーシスカスケードにおけるはるか下流のアポトーシス促進タンパク質を直接活性化する低分子を同定すること理想的であろう。さらに、上記治療戦略は、アポトーシス促進タンパク質のレベルが癌細胞内で上昇した場合、成功の可能性がより高いであろう。
【0129】
[00143] プロカスパーゼ−3のカスパーゼ−3への変換により、多数のタンパク質基質の加水分解を続いて触媒する活性「実行因子」カスパーゼの生成がもたらされる。活性カスパーゼ−3は、ヘテロダイマーのホモダイマーで、プロカスパーゼ−3のタンパク質分解により産生される。生体内では、このタンパク質分解性の活性化は、通常、カスパーゼ−8又はカスパーゼ−9の作用を介して生じる。この酵素前駆体が早まって活性化されないことを確実にするために、プロカスパーゼ−3は、タンパク質分解のIETD部位への到達をブロックするトリ−アスパラギン酸「安全装置」を有している。この安全装置は、プロカスパーゼ−3が、自己触媒活性化及びカスパーゼ−9によるタンパク質分解に抵抗することを可能にする。安全装置の部位はpHに感受性である。したがって、細胞の酸性化(アポトーシス中に起こる)の際、安全装置は、タンパク質分解の部位への到達を許可すると思われ、活性カスパーゼ−3は、カスパーゼ−9の作用により又は自己活性化機構を介して産生することができる。
【0130】
[00144] 癌性組織のある種の型からの細胞はプロカスパーゼ−3のレベルが上昇している。20人の大腸癌患者からの一次試料に関する研究は、平均して、プロカスパーゼ−3が、隣接した非癌性組織に比べて上記試料中で6倍上昇したことを明らかにした。さらに、プロカスパーゼ−3レベルは、ある種の神経芽細胞腫、リンパ腫、及び肝臓癌において上昇する。実際、NCIにより使用された60枚の細胞株パネルにおけるプロカスパーゼ−3レベルの系統的評価は、特定の肺癌、黒色腫、腎臓癌、及び乳癌がプロカスパーゼ−3の著しく増強したレベルを示すことを明らかにした。アポトーシスを成功させる活性カスパーゼ−3の中心的な重要性、ある種の癌性細胞型におけるプロカスパーゼ−3の高度なレベル、及び興味深い、安全装置媒介性のその自己活性化の抑制から判断して、プロカスパーゼ−3を直接活性化する低分子が同定される可能性があるということ及び上記分子は、標的癌治療法において多大な可能性を有する可能性があるということを我々は推論した。この原稿で、我々は、プロカスパーゼ−3の低分子活性化剤であるPAC−1のin vitroの同定について報告する。PAC−1は、癌細胞株において、プロカスパーゼ−3レベルに比例してアポトーシス促進性に効果的であり、そのアポトーシス促進効果はプロカスパーゼ−3のその直接的で即時の活性化によるものであり、一次大腸癌試料に対して及び3つの異なる癌のマウスモデルにおいて有効である。
【0131】
[00145] 約20,500種の構造的に多様な低分子を、in vitroのプロカスパーゼ−3を活性化する能力に対してスクリーニングした。プロカスパーゼ−3を、標準的な手順に従って、大腸菌中で発現させ、精製した。プロカスパーゼ−3を、384ウェルプレートのウェルに加え、化合物を、約40μMの最終濃度まで加えた(プロカスパーゼ−3の最終濃度は50ng/mLであった)。次いで、各プレートを37℃で2時間インキュベートし、その後、カスパーゼ−3ペプチド基質Ac−Asp−Glu−Val−Asp−p−ニトロアニリド(Ac−DEVD−pNa)を200μMの濃度まで加えた。p−ニトロアニリン発色団の形成を405nmで2時間にわたり追跡した。評価した〜20,500種の化合物のうち、4種が、バックグラウンドに対してカスパーゼ−3ペプチド基質の加水分解の著しい増加を誘導した。それらの4種のうち、1種は、in vitroのプロカスパーゼ−3活性化に対する強い用量依存効果を示した。図1Aに示すように、この最初のプロカスパーゼ活性化化合物(PAC−1)は、0.22μMの濃度で、プロカスパーゼ−3の最大活性化の2分の1をもたらす。この化合物は、完全に活性化されたカスパーゼ−3酵素の触媒活性に対する効果を有していないので、カスパーゼ−3自体の活性を単に上昇させているのではない(図1A)。
【0132】
[00146] プロカスパーゼ−3はN末端プロドメイン(残基1〜28)、それに続く、サブユニット間のリンカーにより分けられた大サブユニット(17kDa)及び小サブユニット(12kDa)からなる22。生体内では、2つのプロカスパーゼ−3モノマーが集合して、サブユニット間のリンカー中のD175での切断により活性化することができるホモダイマーを形成する。プロドメインの明確な役割は不明であり、最高の触媒活性には、サブユニット間の領域の切断のみで十分であることが示された。プロカスパーゼ−3は、それ自身のタンパク質分解性の成熟を推進するための十分な触媒活性を有しているが、3つのアミノ酸の安全装置の存在によりこの自己活性化に対して高度に抵抗性がある。しかしながら、安全装置が変異した場合、プロカスパーゼ−3の著しい自己活性化が観察される。プロカスパーゼ−3の活性カスパーゼ−3への成熟を触媒するPAC−1の能力を直接評価するために、プロカスパーゼ−3タンパク質を100μM PAC−1と共に1〜5時間インキュベートした。図1Bでウエスタンブロットにより示すように、PAC−1は時間依存的にプロカスパーゼ−3の切断を誘導し、4時間後に>50%の処理が観察された。対照的に、緩衝液中でインキュベートしたプロカスパーゼ−3は、同一の時間にわたり実質的に自己活性化を示さない。PAC−1はまた、このアッセイにおいて、5μMの濃度でも有効であった。
【0133】
[00147] 次いで、プロカスパーゼ−3の安全装置領域中の重要なアスパラギン酸の三つ組である残基Asp179、Asp180、及びAsp181においてアラニン置換を行った。これらの部位での変異はすべて、プロカスパーゼ−3を活性化するPAC−1の能力を劇的に低下させ、ある種の変異はPAC−1によるプロカスパーゼ−3の活性化に対してより有害であった(図2A)。カスパーゼ−3と同様に、カスパーゼ−7もまた、タンパク質分解により活性化される不活性な酵素前駆体として存在する。カスパーゼ−3及びカスパーゼ−7は共に実行因子カスパーゼであり、かなりの構造上の相同性を有している。プロカスパーゼ−7はまた、重要な三つ組中に3つのアスパラギン酸の代わりに2つのアスパラギン酸(Asp−Thr−Asp)しか有していないが、類似する安全装置領域を有することが予測される。図2B中のデータにより示すように、PAC−1は、さらに、プロカスパーゼ−3のその活性化よりも効果が低いが、プロカスパーゼ−7を活性化することもできる(EC50はプロカスパーゼ−3活性化の0.22μMに対して4.5μM)。PAC−1によるプロカスパーゼ−7活性化の効力は、プロカスパーゼ−3のAsp−Ala−Asp変異体に対するその効果に類似している(EC50=2.77μM)。予想どおり、PAC−1の効果は、プロカスパーゼ−3が急速な自己活性化を起こす低pH値で無効にされる(図2C)。
【0134】
[00148] PAC−1は、様々な癌細胞株におけるアポトーシスを誘導することが分かった。HL−60細胞では、PAC−1の添加は、著しいクロマチン凝縮を伴う細胞膜上への多量のホスファチジルセリンの露出につながる(図3A、3B)。さらに、化合物は、カスパーゼ基質PARP−1の切断を誘導し(生体内でのPARP活性アッセイによる評価)、ミトコンドリア膜脱分極を引き起こす(以下参照)。PAC−1処置細胞の著しい細胞の小疱形成も顕微鏡検査により観察された。さらに、PAC−1の毒性は、カスパーゼ阻害剤z−VAD−fmk存在下で無効にすることができるかもしれない。
【0135】
[00149] PAC−1がプロカスパーゼ−3の直接的な活性化を介してアポトーシスを実際に誘導している場合、アポトーシス現象の時間的経過が、標準的なアポトーシス促進剤を用いて観察される現象に比べて変化するはずである。エトポシドは、内因性の経路を介してアポトーシスを誘導することでよく知られており、したがって、エトポシド処置細胞において、ミトコンドリア膜脱分極の後、プロカスパーゼ−3が活性化される。実際に、10μMエトポシドで処置したHL−60細胞では、ミトコンドリア膜脱分極が観察された後、カスパーゼ−3様活性が検出される(図4A)。対照的に、PAC−1での細胞の処置は著しく異なる結果をもたらす。この化合物では、最初に観察されたアポトーシスの生化学的特徴はカスパーゼ−3様酵素活性であり、活性はPAC−1添加の数分以内に認められ、50%の活性化が、2時間余りで、いかなる著しいミトコンドリア膜脱分極の十分前に起こる(図4B)。さらに、PARP−1活性は、PAC−1で処置した細胞では急速に低下するが、この低下は、エトポシド処置細胞ではより遅い時点で観察され(図4C)、対照実験は、PAC−1がPARP−1の酵素活性を直接抑制しないことを示す。アポトーシス現象の典型的な順序では、ミトコンドリア膜が脱分極し、カスパーゼが活性化され、カスパーゼ基質(PARP−1等)が切断される。PAC−1で処置した細胞がプロカスパーゼ−3/−7の急速な活性化(ミトコンドリア膜脱分極前)及びカスパーゼ基質(PARP−1)の急速な切断を示すといった観察は、PAC−1がプロカスパーゼ−3の直接的な活性化を介してその細胞毒性を発揮していることを示す。
【0136】
[00150] さらにPAC−1の効力を明示するために、様々なレベルのプロカスパーゼ−3を有する癌細胞株における細胞死を誘導するこの化合物の能力を評価した。決定は、多数の癌細胞株(白血病、リンパ腫、黒色腫、神経芽細胞腫、乳癌、肺癌、副腎癌、及び腎臓癌)に存在するプロカスパーゼ−3のレベルに関して最初に行った。細胞死誘導に対するIC50値はこれらの細胞株に対するPAC−1に関して得られた。組み合わせたデータは、プロカスパーゼ−3の細胞内濃度及びPAC−1に対する感受性の間に強い相関を示す(図4D、図4E)。PAC−1は、肺癌細胞株NCI−H226に対して最も効力があり、0.35μMのIC50である。我々は、この細胞株が、ベースラインレベルのプロカスパーゼ−3の濃度の5倍を超える濃度を有することが分かった。重要なことには、プロカスパーゼ−3の発現を有していないことで知られているある癌細胞株(MCF−7、乳癌細胞)がある。PAC−1はMCF−7細胞に事実上効果がなく、IC50>75μMで死を誘導する。
【0137】
[00151] 対照的に、エトポシドは、細胞培養中の効力及びプロカスパーゼ−3の細胞内レベルの間の上記相関を示さなかった。例えば、エトポシドは黒色腫細胞株のうちの3種(UACC−62、CRL−1872、及びB16−F10)、乳癌細胞株(Hs 578t)、及び肺癌細胞株(NCI−H226)での死の誘導において効果がなく(IC50>50μM)、これらの細胞株は、1.0、2.4、1.9、3.7、及び5.3のプロカスパーゼ−3レベルをそれぞれ有している。エトポシドは、4.3、4.0、4.7、及び4.4のプロカスパーゼ−3レベルをそれぞれ有するHL−60、U−937、SK−N−SH、及びPC−12に対して有効であった(IC50<1μM)。したがって、全体として、プロカスパーゼ−3レベル及びエトポシドについてのIC50の間に相関はない。
【0138】
[00152] PAC−1のいくつかの誘導体を合成し、それらのプロカスパーゼ−3活性化特性及び細胞培養中の癌細胞に対するそれらの効果の両方を評価した(表3)。アリル基を欠くPAC−1誘導体(非アリルPAC−1)はPAC−1と同様のレベルでプロカスパーゼ−3活性化及び細胞死を誘導することができる。しかしながら、他のすべての誘導体はいずれのアッセイにおいても活性を示さなかった。したがってアリル基は生物活性には重要でないと思われる一方で、フェノール性水酸基の環及び芳香環はすべて、PAC−1活性に重要である。このデータはまた、提案されたPAC−1の作用機構とも一致しており、in vitroで、プロカスパーゼ−3を活性化しない化合物は培養中の癌細胞に対するアポトーシス促進効果を有していない。
【0139】
[00153] 癌の臨床試料におけるこの直接的な低分子媒介性のプロカスパーゼ−3活性化戦略を試験するために、我々は、新たに切除した大腸腫瘍(隣接した非癌性組織と共に)を、カール財団病院(Carle Foundation Hospital)(アーバナ、IL)の18人の患者から得た。癌性組織及び非癌性組織を分け、これらに由来する細胞を、それらのプロカスパーゼ−3のレベル及びPAC−1に対するそれらの感受性について評価した。図5Aに示すように、すべての患者で、癌性細胞は、同一患者の隣接した非癌性組織からの細胞に比べて、プロカスパーゼ−3のレベルが上昇していた(1.7倍〜17.2倍、平均で7.6倍の上昇)。さらに、これらの癌性細胞は、PAC−1による死の誘導にかなり影響を受けやすかった。PAC−1は、0.007〜1.41μMのIC50値で一次癌性細胞における細胞死を誘導し、一方、PAC−1は、5.02〜9.98μMのIC50値で隣接した非癌性組織における細胞死を誘導した(図5B及び表4)。プロカスパーゼ−3のレベルが上昇した癌性組織はPAC−1に非常に感受性があった。例えば、PAC−1は、7nMのIC50で、患者17からの癌細胞における死を誘導した。これらの細胞は、隣接した正常組織からの細胞よりもPAC−1に対して700倍を超える感受性があった。患者1、2、及び3からの正常サンプル及び癌性サンプル中の約54日の期間にわたる相対的なプロカスパーゼ−3濃度を示す図6Aもまた参照のこと。図6Bは、癌性組織における細胞が、正常組織と比較してPAC−1に対して約80倍を超える感受性がある可能性があることを示す。
【0140】
[00154] 18人の患者の非癌性組織からの細胞に加えて、4種の他の非癌性細胞型に対してもPAC−1を評価した:健常なドナーの骨髄から単離した白血球、Hs888Lu(肺繊維芽細胞)、MCF−10A(乳房の繊維芽細胞)、及びHs578Bst(乳房上皮細胞)。特に、非癌性細胞型は、最低量のプロカスパーゼ−3を有する細胞型に含まれ、PAC−1は、これらの細胞における死を比較的誘導することができず、IC50値は3.2〜8.5μMであった(図5B、緑色の菱形)。図5Bから明らかなように、PAC−1は、プロカスパーゼ−3のレベルと直接関連して、種々様々な細胞型(非癌性細胞株、非癌性一次細胞、癌性細胞株、一次癌性細胞)における死を誘導する。癌性細胞におけるプロカスパーゼ−3の上昇は、PAC−1が選択的にこれらの細胞型における死を誘導することを可能にする。
【0141】
[00155] 薬物送達の徐放法を使用して、マウス異種移植モデルにおいてPAC−1を評価した。このモデルにおいて、ACHN(腎臓癌)細胞株を使用して、卵巣切除した雌の無胸腺BALB/c(ヌード)マウスに皮下腫瘍を形成させた。腫瘍を測定して約30mm2よりも大きければ、PAC−1及びコレステロールのペレットの移植を介して薬物を投与し、遅く安定したレベルで化合物を放出させた。3グループのマウスを使用し、ペレットは0mg、1mg、及び5mgのPAC−1を含み、グループ当たり6匹のマウス、マウス当たり4つの腫瘍とした。腫瘍の大きさを約8週間監視した。図5Cに示すように、腫瘍成長は、5mgのPAC−1を含むペレットを移植したマウスで著しく遅延した。実験の最後の週の摂食評価は、マウスの3グループ間での摂食量における差異を示さなかった。マウスを屠殺した後、各マウスから血漿サンプルを得、それぞれのPAC−1含有量を分析した。PAC−1の5mgのペレットを与えたマウスについては、この分析は、PAC−1が、血漿中に5nMの濃度で54日の実験の後に存在することを明らかにした。
【0142】
[00156] 第2のマウス異種移植モデルにおいてPAC−1を評価し、このモデルには薬物送達法として経口投与を使用した。このモデルにおいて、NCI−H226(肺癌)細胞株を使用して、雄の無胸腺BALB/c−nu/nuマウス(5週令、SLC、Hamamaysu、日本)に皮下異種移植腫瘍を形成させ、グループ当たり8匹のマウス、マウス当たり3つの腫瘍とした。マウスに腫瘍を形成させた後、経口栄養で、1日1回、21日間、0、50、又は100mg/kgの濃度でマウスをPAC−1で処置し、1週目後に屠殺した。図5D中のグラフにより明確に示されるように、PAC−1の経口投与は用量依存的に腫瘍成長を著しく遅延させる。
【0143】
[00157] 最終的に、雄の無胸腺BALB/c−nu/nuマウスに尾静脈注射を介してNCI−H226細胞を注射したマウスモデルでPAC−1を評価した。全実験は28日間続いた。1日1回、PAC−1(100mg/kg)で、経口栄養により、1〜4日目及び7〜11日目にマウスを処置した。他の日は、マウスにPAC−1を与えなかった。マウスの第2のグループに賦形剤のみを与えた。28日後にマウスを屠殺し、それらの肺を検査した。図5Eに示すように、対照マウスの肺(明白な灰色の腫瘤を有する)及びPAC−1処置マウスの肺の間に明らかな相違がある。ゲフィチニブ(イレッサ(商標);アストラゼネカ社)で処置した動物からのパネルにおいても結果を示す。
【0144】
[00158] 癌性細胞は、アポトーシスカスケードにおける種々様々のタンパク質の変異又は異常発現により、通常、アポトーシス促進シグナルに対する感受性が低下している。そのため、癌の多くの型は、周知のように、アポトーシス細胞死に対する内在性シグナルだけでなく類似した機構を介して作用する化学療法剤にも抵抗性がある。ある種の癌におけるプロカスパーゼ−3レベルの奇異な上昇は、この存在するタンパク質の細胞内プールを使用する機会を提供して、アポトーシスを直接誘導し、それにより、機能しないことが多いカスケードの上流の部分を回避する。PAC−1は、in vitroで、プロカスパーゼ−3の自己活性化を誘導する。細胞培養中、PAC−1処置は、急速なカスパーゼ−3様活性を誘導する。次いで、抗アポトーシスタンパク質(Bcl−2、Bcl−XL等)のカスパーゼ−3媒介性の切断は、ミトコンドリア膜の脱分極を誘導し、アポトーシスを増強すると思われる。さらに、様々な癌性細胞型及び非癌性細胞型に対するPAC−1の効力は細胞中のプロカスパーゼ−3の濃度に比例する。切除した大腸腫瘍から単離した一次癌性細胞は、プロカスパーゼ−3のレベルが上昇しているので、これらの細胞は、隣接した非癌性組織からの細胞よりもPAC−1に対してかなり感受性がある。PAC−1が有効ないくつかの細胞株は、アポトーシスに対して抵抗性にする欠陥のあるアポトーシス経路を有していることに注目する価値がある。例えば、Apaf−1発現はSK−MEL−5細胞中で劇的に減少し、Bcl−2は、NCI−H226肺癌細胞株中で過剰発現する。最終的に、PAC−1は、PAC−1を経口的に投与した2つのモデルを含む癌の3つの異なるマウスモデルで有効である。
【0145】
[00159] 本明細書中に提示したデータは、プロカスパーゼ−3活性化化合物が、プロカスパーゼ−3レベルが異常に高度な様々な一般的な癌に対して非常に有効となる可能性があるという見解を支持する。癌生検材料のプロカスパーゼ−3レベルの評価は単純で迅速であり、そのため、PAC−1等の化合物の考えられる有効性は、高度な精度で推測的に評価することができる。上記オーダーメイド医療戦略は一般的な細胞毒素に頼る治療法に優先される可能性があり、抗癌治療法に価値がある可能性がある。
【0146】
[00160] Guy Salvesen教授(バーナム研究所)はプロカスパーゼ−3及びプロカスパーゼ−7の発現ベクターを提供した。
【0147】
〔図の説明〕
[00161] 図1及び図2:PAC−1の構造は明細書の他のところで示す。図1A)in vitroでのPAC−1によるプロカスパーゼ−3の活性化及び活性カスパーゼ−3。PAC−1は、EC50=0.22μMでプロカスパーゼ−3を活性化する。図1B)PAC−1により誘導される活性カスパーゼ−3へのプロカスパーゼ−3の切断。プロカスパーゼ−3をN末端His−6タグを用いて大腸菌中で組換え発現させ、精製した。免疫ブロットは抗His−6抗体を用いて行った。PAC−1非存在下でプロカスパーゼ−3の成熟は観察されない。100μM PAC−1存在下で、p19フラグメントを生成する切断が1時間以内に観察され、>50%の切断が4時間後に観察される。PAC−1はまた、このアッセイにおいて5μMでも有効である。図2A)プロカスパーゼ−3の「安全装置」領域における変異体のPAC−1による活性化。PAC−1は、野性型プロカスパーゼ−3(DDD)に対して0.22μMの活性化のためのEC50並びに変異体については2.77μM(DAD)、113μM(DDA)、及び131μM(ADD)の対応するEC50値を有している。図2B)PAC−1は、4.5μMのEC50でプロカスパーゼ−7を活性化する。図2C)プロカスパーゼ−3のPAC−1による活性化についてのpHへの依存性。低pHでは、安全装置はオフの状態にあり、プロカスパーゼ−3は本質的に最大限に活性化される。エラーバーは、平均値からの標準偏差を表す。
【0148】
[00162] 図3及び図4:PAC−1は、HL−60細胞におけるアポトーシスを誘導する。図3A)100μM PAC−1での20時間の処置の後のホスファチジルセリン露出(アネキシン−V染色により測定される)。PAC−1はまた、このアッセイにおいて5μMでも有効である(裏付けとなる図2を参照)。図3B)100μM PAC−1での20時間の処置の後のヘキスト染色により可視化されるクロマチン凝縮。図4A)10μMエトポシドで処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。図4B)100μM PAC−1で処置したHL−60細胞におけるミトコンドリア膜脱分極(MMP)及びカスパーゼ−3様活性。図4C)PAC−1処置(100μM)は、細胞内カスパーゼ−3/−7の即時活性化と一致する、HL−60細胞における細胞内PARP活性の急速な減少を誘導する。対照的に、エトポシド(10μM)処置細胞は、ずっと後の時点でのPARP活性の減少を示す。図4D及び図4E)PAC−1は、プロカスパーゼ−3依存的に細胞死を誘導する。多くの多様な癌性細胞株について、プロカスパーゼ−3レベルを求め(プロカスパーゼ−3に対する抗体を用いたフローサイトメトリーにより)そしてPAC−1のIC50を測定した(様々なPAC−1濃度での72時間の処置及びMTSアッセイを使用した定量化により)。PAC−1は、高度なレベルのプロカスパーゼ−3を有するとして知られているNCI−H226肺癌細胞株において相当効力がある(IC50=0.35μM)。エラーバーは、平均値からの標準偏差を表す。
【0149】
[00163] 表3:PAC−1及び非アリルPAC−1はプロカスパーゼ−3をin vitroで活性化し、細胞培養中の癌細胞における死を誘導するが、他の構造類似体は、in vitroでのプロカスパーゼ−3活性化効果を有しておらず、細胞培養中に死の誘導をもたらさない。
【0150】
[00164] 図5:図5A)プロカスパーゼ−3レベルは、新たに切除した大腸癌組織に由来する細胞内で上昇する。新たに切除した一次大腸腫瘍(隣接した非癌性組織と共に)を18人の異なる患者から得、癌性組織及び非癌性組織を分け、プロカスパーゼ−3に対する抗体及びフローサイトメトリーを使用して、プロカスパーゼ−3レベルをそれぞれについて測定した。平均して、癌性組織からの細胞は、同一患者からの隣接した非癌性組織に由来する細胞と比較してプロカスパーゼ−3が7.6倍上昇している。図5B)PAC−1は、プロカスパーゼ−3の細胞内レベルに比例して細胞死を誘導する。赤色の円は、18個の大腸腫瘍からの一次癌性細胞を表す。黒色の三角は、図4D中に示した同一の癌細胞株を表す。緑色の菱形は4種の非癌性細胞型である:Hs888Lu(肺繊維芽細胞)、MCF−10A(乳房の繊維芽細胞)、Hs578Bst(乳房上皮細胞)、及び健常なドナーの骨髄から単離した白血球。青色の四角は、18人の患者の腫瘍縁から単離した一次非癌性細胞である。表4)一次大腸癌組織に由来する細胞は、同一患者からの隣接した非癌性組織に由来する細胞よりも、PAC−1による死の誘導にかなりの感受性がある。図5C)PAC−1は、癌の異種移植モデルにおける腫瘍の成長を低下させる。ACHN(腎臓癌)細胞株を用いて皮下注射により腫瘍を形成させ、各グループに6匹のマウス、マウス当たり4つの腫瘍とした。腫瘍が約30mm2まで成長したら、PAC−1を、コレステロールペレットとして移植した。エラーバーは、平均値からの標準誤差を表す。図5D)PAC−1の経口投与はマウス異種移植モデルにおける腫瘍成長を著しく遅延させる。NCI−H226(肺癌)細胞株を使用して皮下注射により腫瘍を形成させ、各グループに8匹のマウス、マウス当たり3つの腫瘍とした。1日1回、経口栄養により、1〜21日目に、PAC−1又は賦形剤を投与した。エラーバーは、平均値からの標準誤差を表す。図5E)PAC−1の経口投与は、i.v.注射モデルにおける腫瘍成長を著しく遅延させる。マウスに、NCI−H226(肺癌)細胞株をi.v.注射した。本文中に記載されるプロトコールに従って経口栄養によりPAC−1(100mg/kg)でマウスを処置した。画像は、PAC−1を与えられず、肺に大量の灰色の腫瘤を有するマウスの肺を示す。対照的に、PAC−1を与えたマウスは目に見える灰色の物体をほとんど有していない。
【0151】
【表3】
【0152】
【表4】
【0153】
〔実施例6:肺癌のマウスモデルにおけるPAC−1の試験〕
[00165] NCI−H226(肺癌)細胞を使用して、異種移植モデルを用いた。PAC−1を腹腔内に(i.p.)10mg/kg与えた。40mg/kgのゲフィチニブ(イレッサ(商標);アストラゼネカ社、ウィルミントン、デラウェア)を用い、グループ当たり5匹のマウスを使用して有効性の比較を行った。図7に結果を示すが、結果は、PAC−1が、腫瘍容積という点で成長を低下させることと関連したことを示す。
【0154】
〔実施例7:コンビナトリアル誘導体、コンビナトリアル合成、及び治療上の使用〕
[00166] 多くの化合物をPAC−1構造の誘導体として調製する。ヒドラジド基をアルデヒド基と反応させ、誘導体化合物のコンビナトリアルライブラリーを得る。
【0155】
[00167] L1〜L20と示したヒドラジド前駆物質グループ(AX)の任意の1つを使用して、1〜28と示したアルデヒドグループ(BX)の任意の1つと反応するヒドラジドを生成し、それによって、560種のPAC−1誘導体化合物を得る。本明細書中に記載される方法を使用し、当技術分野で入手可能な知識に従って、誘導体化合物を合成する。図8A及び8Bに加えて以下のスキーム及び成分の構造を参照のこと。
【0156】
【化10】
【0157】
【化11】
【0158】
【化12】
【0159】
[00168] 一実施形態では、上記誘導体化合物を、例えば、活性、溶解度、毒性、安定性、及び/又は医薬品の用途に関連したその他の特性を変化させるためにさらに修飾する。
【0160】
[00169] 誘導体化合物を抗癌薬剤として使用する。化合物は、抗腫瘍活性、アポトーシス制御、及び/又はプロカスパーゼ−3活性化を有することが可能であるとして確認されている。例えば、新たに除去した大腸癌の一次試料を、プロカスパーゼ−3レベル及び試験化合物レベルに対する細胞の感受性を評価するために使用する。ここで試験化合物はPAC−1又は誘導体化合物である。正常細胞に対する、癌性細胞における細胞死を誘導する傾向に関して、化合物を分類する。
【0161】
[00170] 誘導体化合物のさらなる評価において、in vitro及び生体内での試験を行う。肝臓ミクロソームへの暴露に関する安定性を評価する。
【0162】
〔実施例8:PAC−1と比べたある種の誘導体の活性〕
[00171] PAC−1及びある種の誘導体をHL−60細胞株で試験し、IC50値を測定した。表5に結果を示す(L−及びR−の指定は上記のAX及びBXのシリーズに示した構造をそれぞれ指す)。PAC−1誘導体のいくつかは、概してPAC−1化合物の活性レベルよりも約1桁大きい活性レベルを示した。
【0163】
【表5】
【参照による援用及び変形形態に関する記載】
【0164】
[00172] 本出願にわたるすべての参考文献、例えば発行された若しくは許可された特許又は均等物を含む特許文献、特許出願公開、非公開特許出願、及び非特許文献又は他の資料は、それぞれの参考文献が本出願における本開示と少なくとも部分的に矛盾していない程度に個々に引用されるように(例えば、部分的に矛盾した参考文献の部分を除いて、部分的に矛盾した参考文献が参照により援用される)、これによってそれらの全体が参照により本明細書中に援用される。
【0165】
[00173] 本明細書に対するいかなる添付又は付録をも明細書及び/又は図面の一部として参照により援用される。
【0166】
[00174] 用語「〜を含む(comprise)」、「〜を含む(comprises)」、「〜を含んだ(comprised)」、又は「〜を含む(comprising)」が本明細書中で使用される場合、それらは、言及された、定まった特徴、完全体、ステップ、又は成分の存在を明示するとして解釈され、1つ又は複数の他の特徴、完全体、ステップ、成分、又はそれらのグループの存在又は追加を排除するものではないと解釈される。用語「〜を含む(comprising)」、「〜を含む(comprise(s))」、又は「〜を含んだ(comprised)」が文法上類似する用語、例えば、「〜からなる(consisting/consist(s))」又は「本質的に〜からなる(consisting essentially of/consist(s) essentially of)」と任意選択で置換される本発明の個々の実施形態もまた包含されることが意図され、それにより、必ずしも同延でないさらなる実施形態を記載する。説明として、本明細書中に使用されるように、「〜を含む(comprising)」は、「〜を有する(having)」、「〜を含む(including)」、「〜を含む(containing)」、又は「〜を特徴とする(characterized by)」と同義であり、包括的又は無制限であり、追加の、記載されなかった要素又は方法ステップを除外しない。本明細書中に使用されるように、「〜からなる(consisting of)」は、請求項の要素に明示されていないいかなる要素、ステップ、成分、又は原料をも除外する。本明細書中に使用されるように、「本質的に〜なる(consisting essentially of)」は、請求項の基本的で新規な特徴に実質的に影響しない(例えば、活性成分に影響しない)材料又はステップを除外しない。本明細書中の各例において、用語「〜を含む(comprising)」、「本質的に〜からなる(consisting essentially of)」、及び「〜からなる(consisting of)」のいずれも他の2つの用語のうちのいずれかと置換してもよい。例証的に本明細書中に記載された本発明は、具体的に本明細書中に開示されていない任意の要素又は要素(複数)、限定又は限定(複数)の非存在下で適切に実行してもよい。
【0167】
[00175] 本発明は、様々な特定の好ましい実施形態及び技術に関して記載された。しかしながら、本発明の精神及び範囲内にとどまる一方で、多くの変形形態及び修飾形態を作製してもよいことが理解されるべきである。具体的に本明細書中に記載されたもの以外の組成物、方法、装置、装置要素、材料、任意の特徴、手順、及び技術は、広く本明細書中に開示されるように、過度の実験に頼ることなく本発明の実行に用いることができることを当業者は十分に理解するであろう。本明細書中に記載された組成物、方法、装置、装置要素、材料、手順、及び技術並びにそれらの部分の、技術的に知られている機能的な均等物はすべて、本発明により包含されることが意図される。範囲を開示する場合は常に、部分範囲及び個々の値すべてが包含されることが意図される。本発明は、図面に示された又は明細書に例示されたいかなるものをも含む開示された実施形態により限定されず、実施形態は、限定としてではなく実施例又は例証として与えられる。本発明の範囲は請求項のみにより限定されるものとする。
【参考文献】
【0168】
[00176] 次の出願は、特に、その全体が参照により援用される。2003年10月30日に提出されたHergenrotherらによる米国仮特許出願第60/516556号;2004年8月20日に提出されたHergenrotherらによる米国仮特許出願第60/603246号;2004年10月27日に提出されたHergenrotherらによる米国特許出願第10/976186号。
【0169】
[00177] 米国特許第6762045号 Membrane derived caspase−3,compositions comprising the same and methods of use therefore;米国特許第6534267号 Polynucleotides encoding activators of caspases;米国特許第6403765号 Truncated Apaf−1 and methods of use thereof;2005年4月12日に発行されたChoongらによる米国特許第6303329号及び第6878743号;2004年4月22日に公開されたWang,Xiaodongらによる米国特許公開第20040077542号;2004年9月16日に公開されたShi,Yigongによる米国特許公開第20040180828号。
【0170】
Slee EAら,Benzyloxycarbonyl−Val−Ala−Asp(OMe)fluoromethylketone(Z−VAD.FMK)inhibits apoptosis by blocking the processing of CPP32,Biochem J.1996 APR 1;315(Pt1):21−4.
【0171】
1. Hanahan,D. & Weinberg,R.A. The hallmarks of cancer. Cell 100,57−70(2000).
2. Okada,H. & Mak,T.W. Pathways of apoptotic and non−apoptotic death in tumour cells. Nature Rev. Cancer 4,592−603(2004).
3. Roy,S. et al. Maintenance of caspase−3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc.Natl.Acad.Sci.98,6132−6137(2001).
4. Svingen,P.A. et al. Components of the cell death machine and drug sensitivity of the National Cancer Institute Cell Line Panel. Clin.Cancer Res.10,6807−6820(2004).
5. Lowe,S.W., Cepero,E. & Evan,G. Intrinsic tumor suppression. Nature 432,307−315 (2004).
6. Vogelstein,B. & Kinzler,K.W. Achilles’ heel of cancer. Nature 412,865−866(2001).
7. Traven,A., Huang,D.C. & Lithgow,T. Protein hijacking: key proteins held captive against their will. Cancer Cell 5,107−108(2004).
8. Soengas,M.S. et al. Inactivation of the apoptosis effector Apaf−1 in malignant melanoma. Nature 409,207−211(2001).
9. Wajant,H. Targeting the FLICE inhibitory protein(FLIP)in cancer therapy. Mol. Interv. 3,124−127(2003).
10. Denicourt,C. & Dowdy,S.F. Targeting apoptotic pathways in cancer cells. Science 305,1411−1413(2004).
11. Vassilev,L.T. et al. In vivo activation of the p53 pathway by small−molecule antagonists of MDM2. Science 303,844−848(2004).
12. Degterev,A. et al. Identification of small−molecule inhibitors of interaction between the BH3 domain and Bcl−XL. Nature Cell Biol. 3,173−182(2001).
13. Becattini,B. et al. Rational design and real time, in−cell detection of the proapoptotic activity of a novel compound targeting Bcl−XL. Chem.Biol. 11,389−395(2004).
14. Wang,J.−L. et al. Structure−based discovery of an organic compound that binds Bcl−2 protein and induces apoptosis of tumor cells. Proc.Natl.Acad.Sci. 97,7124−7129(2000).
15. Li,L. et al. A small molecule Smac mimic potentiates TRAIL−and TNFa−mediated cell death. Science 305,1471−1474(2004).
16. Nguyen,J.T. & Wells,J.A. Direct activation of the apoptosis machinery as a mechanism to target cancer cells. Proc.Natl.Acad.Sci.U.S.A. 100,7533−7538(2003).
17. Jiang,X. et al. Dstincitive roles of PHAP proteins and prothymosin−α in a death regulatory pathway. Science 299,223−226(2003).
18. Boatright,K.M. & Salvesen,G.S. Mechanisms of caspase activation. Curr.Opin.Cell.Biol. 15,725−731(2003).
19. Nakagawara,A. et al. High levels of expression and nuclear localization of interleukin−1β converting enzyme (ICE) and CPP32 in favorable human neuroblastomas. Cancer Res. 57,4578−4584(1997).
20. Izban,K.F. et al. Characterization of the interleukin−1β−converting enzyme/Ced−3−family protease, caspase−3/CPP32, in Hodgkin’s disease. Am.J.Pathol. 154,1439−1447(1999).
21. Persad,R. et al. Overexpression of caspase−3 in hepatocellular carcinomas. Modern Patholo. 17,861−867(2004).
22. Pop,C., Feeney,B., Tripathy,A. & Clark,A.C. Mutations in the procaspase−3 dimer interface affect the activity of the zymogen. Biochemistry 42,12311−12320(2003).
23. Stennicke,H.R. et al. J.Biol.Chem. 273,27084−27090(1998).
24. Denault,J.−B & Salvesen, G.S. Human caspase−7 activity and regulation by its N−terminal peptide. J.Biol.Chem. 278,34042−24050(2003).
25. Putt,K.S., Beilman,G.J. & Hergenrother,P.J. Direct quantitation of Poly(ADP−ribose) polymerase (PARP) activity as a means to distinguish necrotic and apoptotic death in cell and tissue samples. ChemBioChem 6,53−55(2005).
26. Liang,Y., Nylander, K.D., Yan,C. & Schor,N.F. Role of caspase 3−dependent Bcl−2 cleavage in potentiation of apoptosis by Bcl−2. Mol.Pharmacol. 61,142−149(2002).
27. Fujita,N., Nagahshi,A., Nagashima,K., Rokudai,S. & Tsuruo,T. Acceleration of apoptotic cell death after the cleavage of Bcl−XL protein by caspase−3−like proteases. Oncogene 17,1295−1304(1998).
28. Earnshaw,W.C., Martins,L.M. & Kaufmann,S.H. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu.Rev.Biochem. 68,383−424(1999).
29. Koty,P.P., Zhang,H. & Levitt,M.L. Antisense bcl−2 treatment increases programmed cell death in non−small cell lung cancer cell lines. Lung Cancer 23,115−127(1999).
【0172】
国立医学図書館/国立衛生研究所(NIH)の国立バイオテクノロジー情報センター(NCBI)のデータベース、ウェブサイト:http://www.ncbi.nlm.nih.gov/ CASP3(カスパーゼ3、アポトーシス関連システインプロテアーゼ[ホモサピエンス] 遺伝子ID:836 遺伝子座タグ:HGNC:1504;MIM:600636 2005年5月15日に更新。他の別名:HGNC:1504、アポパイン、CPP32、CPP32B、SCA−1;他の指定:ヒトプロカスパーゼ3コード配列;PARP切断プロテアーゼ;SREBP切断活性1;Yama;カスパーゼ3;システインプロテアーゼCPP32)を検索するために遺伝子データベースを使用。
【0173】
Hergenrother PJ.Obtaining and screening compound collections:a user’s guide and a call to chemists.Curr Opin Chem Biol.2006.
Silverman SK,Hergenrother PJ.Combinatorial chemistry and molecular diversity Tools for molecular diversification and their applications in chemical biology.Curr Opin Chem Biol.2006.
Goode DR,Sharma AK,Hergenrother PJ.Using peptidic inhibitors to systematically probe the S1’site of caspase−3 and caspase−7.Org Lett.2005 Aug 4;7(16):3529−32.PMID:16048334.
Dothager RS,Putt KS,Allen BJ,Leslie BJ,Nesterenko V,Hergenrother PJ.Synthesis and identification of small molecules that potently induce apoptosis in melanoma cells through G1 cell cycle arrest.J Am Chem Soc.2005 Jun 22;127(24):8686−96.PMID:15954774.
Putt KS,Hergenrother PJ.A nonradiometric,high−throughput assay for poly(ADP−ribose) glycohydrolase(PARG):application to inhibitor identification and evaluation.Anal Biochem.2004 Oct 15;333(2):256−64.PMID:15450800.
Putt KS,Hergenrother PJ.An enzymatic assay for poly(ADP−ribose) polymerase−1(PARP−1) via the chemical quantitation of NAD(+):application to the high−throughput screening of small molecules as potential inhibitors.Anal Biochem.2004 Mar 1;326(1):78−86.PMID:14769338.
Nesterenko V,Putt KS,Hergenrother PJ.Identification from a combinatorial library of a small molecule that selectively induces apoptosis in cancer cells.J Am Chem Soc.2003 Dec 3;125(48):14672−3.PMID:14640619.
【特許請求の範囲】
【請求項1】
癌細胞におけるアポトーシスを選択的に誘導する方法であって、
(a)前記癌細胞のプロカスパーゼ−3分子を修飾することができる化合物を当該癌細胞に投与するステップと、
(b)アポトーシスを誘導するように前記プロカスパーゼ−3分子を修飾するステップと、
を含む方法。
【請求項2】
前記癌細胞が、処置を必要とする患者に存在する、請求項1に記載の方法。
【請求項3】
前記化合物が下記式ZZを有する、請求項1又は2に記載の方法。
【化1】
[式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。]
【請求項4】
前記化合物が、式ZZ、PAC−1及び構造5からなる群より選択される、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記化合物がPAC−1である、請求項1〜3のいずれか一項に記載の方法。
【請求項6】
癌細胞におけるプロカスパーゼ−3又はカスパーゼ−3のパラメーターを評価するステップをさらに含み、
前記パラメーターが、半定量的若しくは定量的な量、機能的な量、又は前記プロカスパーゼ−3若しくはカスパーゼ−3の活性レベル、の1つ又は複数である、
請求項1〜3のいずれか一項に記載の方法。
【請求項7】
下記構造式ZZの化合物。
【化2】
[式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。]
【請求項8】
下記構造のPAC−1の化合物を除く、請求項7に記載の化合物。
【化3】
【請求項9】
プロカスパーゼ−3分子を修飾することができる化合物を直接、in vitroでスクリーニングするための方法であって、
(a)試験化合物を提供するステップと、
(b)精製したプロカスパーゼ−3を提供するステップと、
(c)前記精製したプロカスパーゼ−3に前記試験化合物を暴露するステップと、
(d)前記試験化合物への暴露の後でプロカスパーゼ−3活性を測定するステップと、
(e)前記試験化合物への前記暴露の際の試験活性を、前記試験化合物への暴露なしでの無修飾活性と比較することにより、修飾化合物を同定するステップと、
を含み、
それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法。
【請求項10】
前記修飾活性又は前記無修飾活性を基準活性と比較するステップをさらに含み、
前記基準活性が、構造式ZZ、PAC−1及び構造5からなる群より選択される化合物への暴露によるものである、
請求項9に記載の方法。
【請求項11】
プロカスパーゼ−3分子を修飾することができる化合物の細胞スクリーニングのための方法であって、
(a)試験化合物を提供するステップと、
(b)プロカスパーゼ−3を推定上発現する細胞を提供するステップと、
(c)前記試験化合物に前記細胞を暴露するステップと、
(d)前記試験化合物への暴露の後で、細胞生存率、アポトーシス指標、及び他のパラメーターを含む細胞パラメーターの、1つ又は複数を測定するステップと、
(e)前記試験化合物への前記暴露の際の試験細胞パラメーターを、前記試験化合物への暴露なしでの無修飾細胞パラメーターと比較することにより、修飾化合物を同定するステップと、
を含み、
それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法。
【請求項12】
前記修飾活性又は前記無修飾活性を基準活性と比較するステップをさらに含み、
前記基準活性が、ZZ、PAC−1及び構造5からなる群より選択される化合物への暴露によるものである、
請求項11に記載の方法。
【請求項13】
プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定又は診断するための方法であって、
(a)前記癌細胞内のプロカスパーゼパラメーターを評価するステップと、
(b)前記パラメーターによりプロカスパーゼの活性化に対する感受性が上昇したかどうかを判定するステップと、
を含む方法。
【請求項14】
前記プロカスパーゼパラメーターがプロカスパーゼ−3レベルであり、前記プロカスパーゼがプロカスパーゼ−3である、請求項13に記載の方法。
【請求項15】
前記プロカスパーゼパラメーターがプロカスパーゼ−7レベルであり、前記プロカスパーゼがプロカスパーゼ−7である、請求項13に記載の方法。
【請求項16】
癌細胞を処置するための方法であって、
(a)プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定するステップと、
(b)有効な量の前記プロカスパーゼ活性化化合物に前記癌細胞を暴露するステップと、
を含む方法。
【請求項17】
前記プロカスパーゼ活性化化合物が、式ZZ、PAC−1及び構造5からなる群より選択される、請求項16に記載の方法。
【請求項18】
前記プロカスパーゼ活性化化合物が、プロカスパーゼ−3、プロカスパーゼ−7、又はプロカスパーゼ−3及びプロカスパーゼ−7の両方を活性化することができる、請求項16又は17に記載の方法。
【請求項19】
PAC−1を合成するための方法であって、
スキーム1のステップを含む方法。
【請求項20】
化合物5又は式ZZの化合物を合成するための方法であって、
適切な変更を有するスキーム1のステップを含む方法。
【請求項21】
構造式が
【化4】
である構造5の化合物。
【請求項22】
下記式ZZ2を有する化合物。
【化5】
[式中、R1及びR2は、それぞれ独立して、水素、ハロゲン、アルキル、アリル、ハロアルキル、アルケニル、アルケノール、アルカノール又はハロアルケニルである。]
【請求項23】
R1及びR2が、それぞれ独立して、水素、ハロゲン、アリル又は短鎖アルキルである、請求項22に記載の化合物。
【請求項24】
アルデヒド化合物と結合したヒドラジド化合物を含むPAC−1誘導体コンビナトリアルライブラリーからなる群より選択される化合物。
【請求項25】
前記ヒドラジド化合物が、本明細書中に記載されたAX化合物から生成されたヒドラジドからなる群より選択され、
前記アルデヒド化合物が、本明細書中に記載されたBX化合物からなる群より選択される、
請求項24に記載の化合物。
【請求項26】
PAC−1誘導体化合物を合成するための方法であって、
ヒドラジド化合物を提供するステップと、
アルデヒド化合物を提供するステップと、
前記ヒドラジド化合物を前記アルデヒド化合物と反応させるステップと、
を含み、
それによりPAC−1誘導体化合物を合成する方法。
【請求項27】
前記ヒドラジド化合物が下記式ZZ3を有する、請求項26に記載の方法。
【化6】
【請求項28】
前記アルデヒド化合物が下記式ZZ4を有する、請求項26又は27に記載の方法。
【化7】
【請求項29】
前記ヒドラジド化合物が前記式ZZ3を有し、
前記アルデヒド化合物が前記式ZZ4を有する、
請求項26〜28のいずれか一項に記載の方法。
【請求項30】
L01R06、L02R03、L02R06、L08R06、L09R03、L09R06及びL09R08からなる群より選択される化合物。
【請求項31】
癌患者の候補者を、前記候補者においてプロカスパーゼレベルの上昇を同定することにより、プロカスパーゼ活性化剤による可能な処置についてスクリーニングするための方法であって、
前記候補者から細胞又は組織の試験サンプルを得るステップと、
前記試験サンプル中のプロカスパーゼレベルを評価するステップと、
前記プロカスパーゼレベルが、基準レベルに比べて前記試験サンプル中で上昇したかどうかを判定するステップと、
を含み、
それにより、癌患者の候補者を、プロカスパーゼ活性化剤による可能な処置についてスクリーニングする方法。
【請求項32】
前記プロカスパーゼが、プロカスパーゼ−2、プロカスパーゼ−3、プロカスパーゼ−6、プロカスパーゼ−7、プロカスパーゼ−8及びプロカスパーゼ−9からなる群より選択される、請求項31に記載の方法。
【請求項33】
前記プロカスパーゼがプロカスパーゼ−3である、請求項31又は32に記載の方法。
【請求項34】
前記試験サンプルの前記上昇レベルが前記基準レベルの少なくとも約2倍である、請求項31〜33のいずれか一項に記載の方法。
【請求項35】
前記試験サンプルの前記上昇レベルが前記基準レベルの少なくとも約4倍である、請求項31〜34のいずれか一項に記載の方法。
【請求項36】
前記基準レベルが、同一患者からの第2の試験サンプルからのものである、請求項31〜35のいずれか一項に記載の方法。
【請求項37】
前記基準レベルが正常細胞又は正常組織のサンプルからのものである、請求項31〜36のいずれか一項に記載の方法。
【請求項38】
前記基準レベルが細胞株からのものである、請求項31〜37のいずれか一項に記載の方法。
【請求項39】
前記基準レベルが癌細胞株からのものである、請求項31〜38のいずれか一項に記載の方法。
【請求項40】
前記基準レベルが正常細胞株からのものである、請求項31〜39のいずれか一項に記載の方法。
【請求項41】
前記基準レベルが絶対閾値の量である、請求項31〜40のいずれか一項に記載の方法。
【請求項42】
癌細胞における死を誘導するための方法であって、
当該癌細胞のプロカスパーゼ−3分子を活性化することができる化合物を当該癌細胞に投与するステップを含む方法。
【請求項43】
前記化合物が構造式ZZを有する、請求項42に記載の方法。
【請求項44】
前記化合物が構造式ZZ2を有する、請求項42に記載の方法。
【請求項1】
癌細胞におけるアポトーシスを選択的に誘導する方法であって、
(a)前記癌細胞のプロカスパーゼ−3分子を修飾することができる化合物を当該癌細胞に投与するステップと、
(b)アポトーシスを誘導するように前記プロカスパーゼ−3分子を修飾するステップと、
を含む方法。
【請求項2】
前記癌細胞が、処置を必要とする患者に存在する、請求項1に記載の方法。
【請求項3】
前記化合物が下記式ZZを有する、請求項1又は2に記載の方法。
【化1】
[式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。]
【請求項4】
前記化合物が、式ZZ、PAC−1及び構造5からなる群より選択される、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記化合物がPAC−1である、請求項1〜3のいずれか一項に記載の方法。
【請求項6】
癌細胞におけるプロカスパーゼ−3又はカスパーゼ−3のパラメーターを評価するステップをさらに含み、
前記パラメーターが、半定量的若しくは定量的な量、機能的な量、又は前記プロカスパーゼ−3若しくはカスパーゼ−3の活性レベル、の1つ又は複数である、
請求項1〜3のいずれか一項に記載の方法。
【請求項7】
下記構造式ZZの化合物。
【化2】
[式中、n=1又は2であり、Rは、他のRとは独立して、水素、ハロゲン、アリル又は短鎖アルキルであり、R2=水素、短鎖アルキル、エステル、又は生理学的条件下で除去可能な他の部分であり、R3=水素、ハロゲン、アルキル、ハロアルキル、アリル、アルケニル、アルケノール、アルカノール又はハロアルケニルであり、R4及びR5は、いずれもNであるか、R4=N及びR5=Cであるか、R4及びR5=Cであり、A=酸素又は硫黄である。]
【請求項8】
下記構造のPAC−1の化合物を除く、請求項7に記載の化合物。
【化3】
【請求項9】
プロカスパーゼ−3分子を修飾することができる化合物を直接、in vitroでスクリーニングするための方法であって、
(a)試験化合物を提供するステップと、
(b)精製したプロカスパーゼ−3を提供するステップと、
(c)前記精製したプロカスパーゼ−3に前記試験化合物を暴露するステップと、
(d)前記試験化合物への暴露の後でプロカスパーゼ−3活性を測定するステップと、
(e)前記試験化合物への前記暴露の際の試験活性を、前記試験化合物への暴露なしでの無修飾活性と比較することにより、修飾化合物を同定するステップと、
を含み、
それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法。
【請求項10】
前記修飾活性又は前記無修飾活性を基準活性と比較するステップをさらに含み、
前記基準活性が、構造式ZZ、PAC−1及び構造5からなる群より選択される化合物への暴露によるものである、
請求項9に記載の方法。
【請求項11】
プロカスパーゼ−3分子を修飾することができる化合物の細胞スクリーニングのための方法であって、
(a)試験化合物を提供するステップと、
(b)プロカスパーゼ−3を推定上発現する細胞を提供するステップと、
(c)前記試験化合物に前記細胞を暴露するステップと、
(d)前記試験化合物への暴露の後で、細胞生存率、アポトーシス指標、及び他のパラメーターを含む細胞パラメーターの、1つ又は複数を測定するステップと、
(e)前記試験化合物への前記暴露の際の試験細胞パラメーターを、前記試験化合物への暴露なしでの無修飾細胞パラメーターと比較することにより、修飾化合物を同定するステップと、
を含み、
それにより、プロカスパーゼ−3分子を修飾することができる化合物をスクリーニングする方法。
【請求項12】
前記修飾活性又は前記無修飾活性を基準活性と比較するステップをさらに含み、
前記基準活性が、ZZ、PAC−1及び構造5からなる群より選択される化合物への暴露によるものである、
請求項11に記載の方法。
【請求項13】
プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定又は診断するための方法であって、
(a)前記癌細胞内のプロカスパーゼパラメーターを評価するステップと、
(b)前記パラメーターによりプロカスパーゼの活性化に対する感受性が上昇したかどうかを判定するステップと、
を含む方法。
【請求項14】
前記プロカスパーゼパラメーターがプロカスパーゼ−3レベルであり、前記プロカスパーゼがプロカスパーゼ−3である、請求項13に記載の方法。
【請求項15】
前記プロカスパーゼパラメーターがプロカスパーゼ−7レベルであり、前記プロカスパーゼがプロカスパーゼ−7である、請求項13に記載の方法。
【請求項16】
癌細胞を処置するための方法であって、
(a)プロカスパーゼ活性化化合物による癌細胞の処置に対する潜在的感受性を同定するステップと、
(b)有効な量の前記プロカスパーゼ活性化化合物に前記癌細胞を暴露するステップと、
を含む方法。
【請求項17】
前記プロカスパーゼ活性化化合物が、式ZZ、PAC−1及び構造5からなる群より選択される、請求項16に記載の方法。
【請求項18】
前記プロカスパーゼ活性化化合物が、プロカスパーゼ−3、プロカスパーゼ−7、又はプロカスパーゼ−3及びプロカスパーゼ−7の両方を活性化することができる、請求項16又は17に記載の方法。
【請求項19】
PAC−1を合成するための方法であって、
スキーム1のステップを含む方法。
【請求項20】
化合物5又は式ZZの化合物を合成するための方法であって、
適切な変更を有するスキーム1のステップを含む方法。
【請求項21】
構造式が
【化4】
である構造5の化合物。
【請求項22】
下記式ZZ2を有する化合物。
【化5】
[式中、R1及びR2は、それぞれ独立して、水素、ハロゲン、アルキル、アリル、ハロアルキル、アルケニル、アルケノール、アルカノール又はハロアルケニルである。]
【請求項23】
R1及びR2が、それぞれ独立して、水素、ハロゲン、アリル又は短鎖アルキルである、請求項22に記載の化合物。
【請求項24】
アルデヒド化合物と結合したヒドラジド化合物を含むPAC−1誘導体コンビナトリアルライブラリーからなる群より選択される化合物。
【請求項25】
前記ヒドラジド化合物が、本明細書中に記載されたAX化合物から生成されたヒドラジドからなる群より選択され、
前記アルデヒド化合物が、本明細書中に記載されたBX化合物からなる群より選択される、
請求項24に記載の化合物。
【請求項26】
PAC−1誘導体化合物を合成するための方法であって、
ヒドラジド化合物を提供するステップと、
アルデヒド化合物を提供するステップと、
前記ヒドラジド化合物を前記アルデヒド化合物と反応させるステップと、
を含み、
それによりPAC−1誘導体化合物を合成する方法。
【請求項27】
前記ヒドラジド化合物が下記式ZZ3を有する、請求項26に記載の方法。
【化6】
【請求項28】
前記アルデヒド化合物が下記式ZZ4を有する、請求項26又は27に記載の方法。
【化7】
【請求項29】
前記ヒドラジド化合物が前記式ZZ3を有し、
前記アルデヒド化合物が前記式ZZ4を有する、
請求項26〜28のいずれか一項に記載の方法。
【請求項30】
L01R06、L02R03、L02R06、L08R06、L09R03、L09R06及びL09R08からなる群より選択される化合物。
【請求項31】
癌患者の候補者を、前記候補者においてプロカスパーゼレベルの上昇を同定することにより、プロカスパーゼ活性化剤による可能な処置についてスクリーニングするための方法であって、
前記候補者から細胞又は組織の試験サンプルを得るステップと、
前記試験サンプル中のプロカスパーゼレベルを評価するステップと、
前記プロカスパーゼレベルが、基準レベルに比べて前記試験サンプル中で上昇したかどうかを判定するステップと、
を含み、
それにより、癌患者の候補者を、プロカスパーゼ活性化剤による可能な処置についてスクリーニングする方法。
【請求項32】
前記プロカスパーゼが、プロカスパーゼ−2、プロカスパーゼ−3、プロカスパーゼ−6、プロカスパーゼ−7、プロカスパーゼ−8及びプロカスパーゼ−9からなる群より選択される、請求項31に記載の方法。
【請求項33】
前記プロカスパーゼがプロカスパーゼ−3である、請求項31又は32に記載の方法。
【請求項34】
前記試験サンプルの前記上昇レベルが前記基準レベルの少なくとも約2倍である、請求項31〜33のいずれか一項に記載の方法。
【請求項35】
前記試験サンプルの前記上昇レベルが前記基準レベルの少なくとも約4倍である、請求項31〜34のいずれか一項に記載の方法。
【請求項36】
前記基準レベルが、同一患者からの第2の試験サンプルからのものである、請求項31〜35のいずれか一項に記載の方法。
【請求項37】
前記基準レベルが正常細胞又は正常組織のサンプルからのものである、請求項31〜36のいずれか一項に記載の方法。
【請求項38】
前記基準レベルが細胞株からのものである、請求項31〜37のいずれか一項に記載の方法。
【請求項39】
前記基準レベルが癌細胞株からのものである、請求項31〜38のいずれか一項に記載の方法。
【請求項40】
前記基準レベルが正常細胞株からのものである、請求項31〜39のいずれか一項に記載の方法。
【請求項41】
前記基準レベルが絶対閾値の量である、請求項31〜40のいずれか一項に記載の方法。
【請求項42】
癌細胞における死を誘導するための方法であって、
当該癌細胞のプロカスパーゼ−3分子を活性化することができる化合物を当該癌細胞に投与するステップを含む方法。
【請求項43】
前記化合物が構造式ZZを有する、請求項42に記載の方法。
【請求項44】
前記化合物が構造式ZZ2を有する、請求項42に記載の方法。
【図1A】


【図1B】
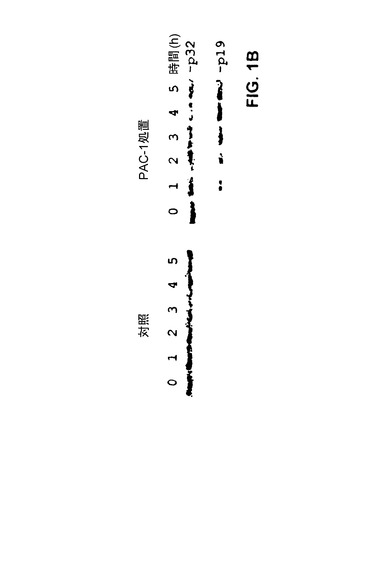
【図2A】
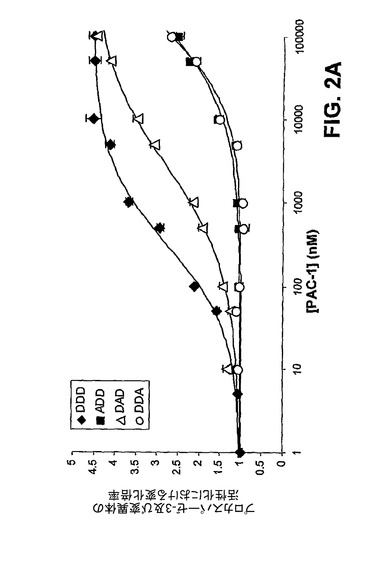
【図2B】
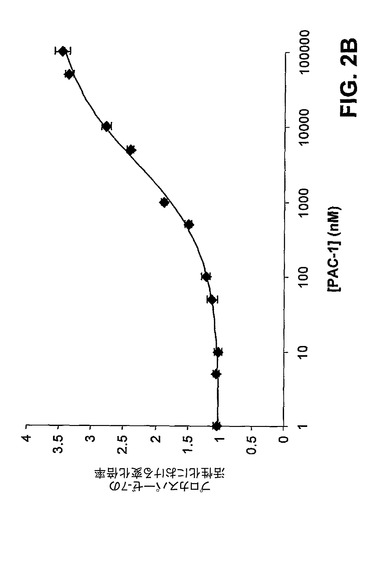
【図2C】
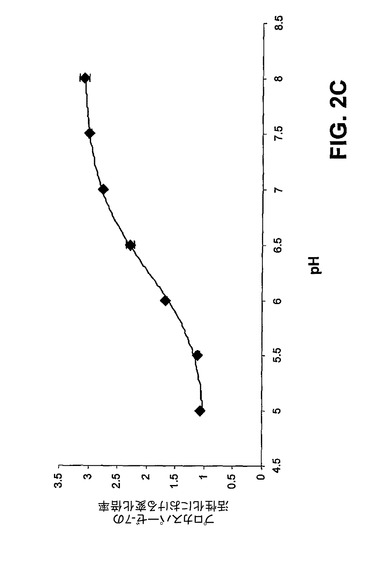
【図3A】
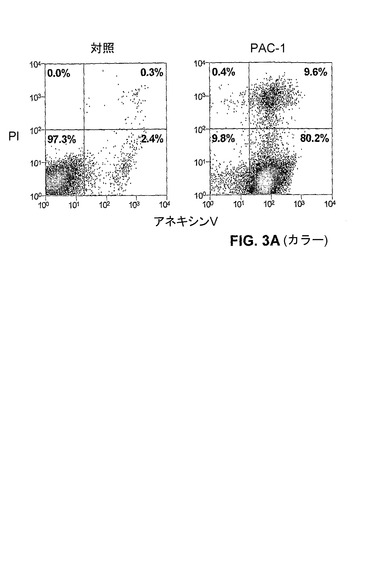
【図3B】

【図4A】

【図4B】
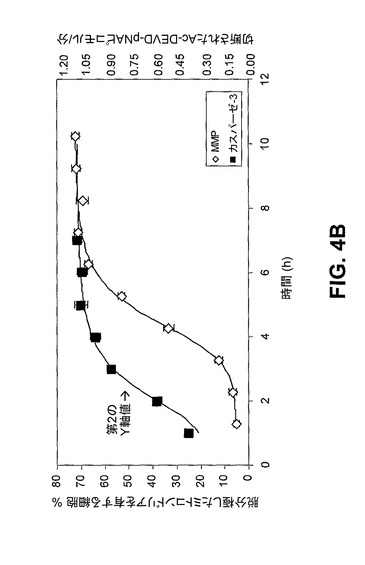
【図4C】
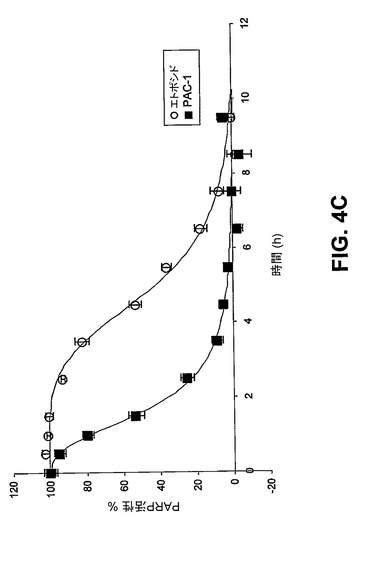
【図4D】

【図4E】
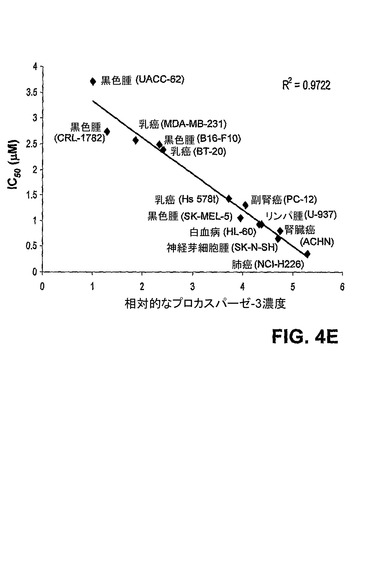
【図5A】

【図5B】

【図5C】
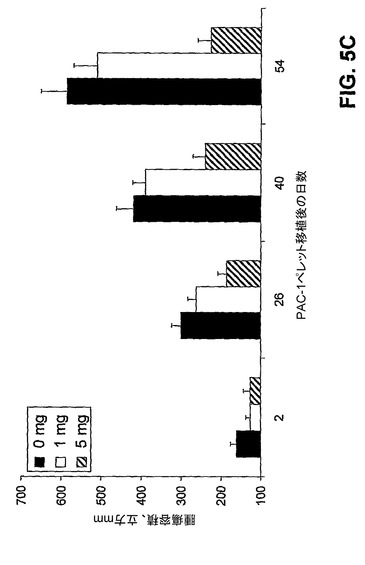
【図5D】
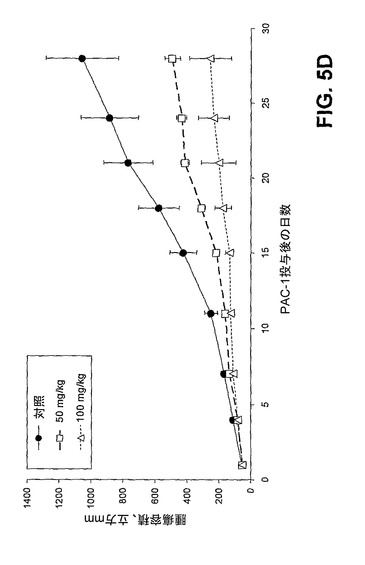
【図5E】

【図6A】

【図6B】

【図7】
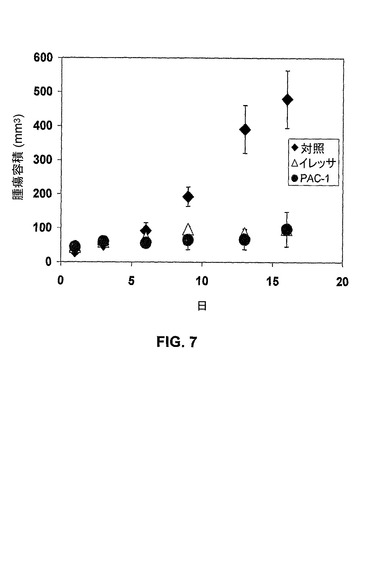
【図8A】
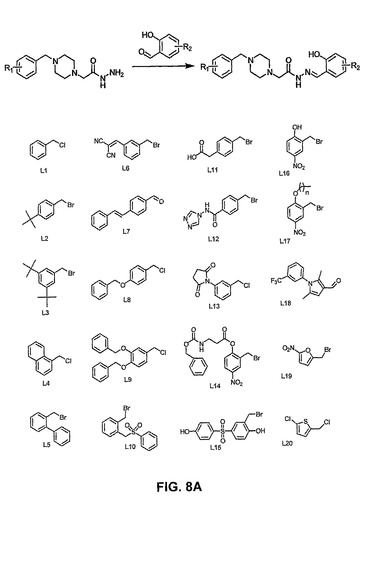
【図8B】
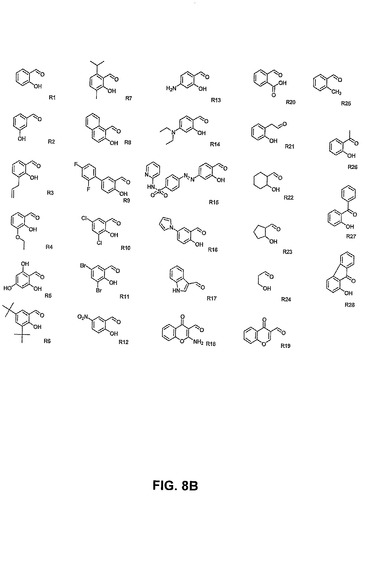
【図9】

【図10】

【図1B】
【図2A】
【図2B】
【図2C】
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図6A】
【図6B】
【図7】
【図8A】
【図8B】
【図9】
【図10】
【公開番号】特開2013−100285(P2013−100285A)
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−251584(P2012−251584)
【出願日】平成24年11月15日(2012.11.15)
【分割の表示】特願2008−513829(P2008−513829)の分割
【原出願日】平成18年5月26日(2006.5.26)
【出願人】(506175840)ザ ボード オブ トラスティーズ オブ ザ ユニヴァーシティー オブ イリノイ (30)
【Fターム(参考)】
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願番号】特願2012−251584(P2012−251584)
【出願日】平成24年11月15日(2012.11.15)
【分割の表示】特願2008−513829(P2008−513829)の分割
【原出願日】平成18年5月26日(2006.5.26)
【出願人】(506175840)ザ ボード オブ トラスティーズ オブ ザ ユニヴァーシティー オブ イリノイ (30)
【Fターム(参考)】
[ Back to top ]