
プロスタグランジン作動薬の多形体およびそれらを製造するための方法
【課題】緑内障、高眼圧症または低骨量を示す状態を有する哺乳類を治療するための薬物である(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸に関して、塩、水和物、多形体、結晶性および非結晶性形態など複数の物理的形態を提供する。
【解決手段】(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩もしくはその水和物の多形性の結晶性形態もしくは非結晶性形態もしくは非晶質性、それを調製するための方法、それを使用するための方法、およびそれを含有する医薬組成物。
【解決手段】(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩もしくはその水和物の多形性の結晶性形態もしくは非結晶性形態もしくは非晶質性、それを調製するための方法、それを使用するための方法、およびそれを含有する医薬組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、プロスタグランジンの治療上活性かつ選択的なモジュレーター、具体的には、EP2の作動薬の多形体、これらの化合物を含む医薬組成物、これらの化合物を調製するための方法ならびに骨障害、緑内障および高眼圧症などのプロスタグランジンの調節に関連している状態を治療するためのそのような化合物の使用に関する。より具体的には、本発明は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形体、この化合物の多形体を含む医薬組成物、これらの多形体を調製するための方法ならびにプロスタグランジンの調節に関連している状態を治療するためのこの化合物の多形体の使用に関する。
【背景技術】
【0002】
薬物開発の過程において、薬物の最も安定な結晶性形態を発見することが重要であると一般的に考えられている。この最も安定な結晶性形態は、最良の化学的安定性、すなわち、製剤における最長の貯蔵寿命を有する可能性が高い形態である。しかしながら、薬物の複数の形態、例えば、塩、水和物、多形体、結晶性および非結晶性形態を有することが有利でもある。異なる物理的形態は異なる利点を提供するため、薬物の理想的な物理的形態は1つではない。最も安定な形態およびそのような他の形態の探索は困難であり、その成果は予測できない。
【0003】
薬物開発を成功させるには、患者にとって治療上有効な治療であるという特定の要件を満たすことが必要である。これらの要件は、2つの範疇、すなわち、(1)剤形の製造を成功させるための要件および(2)薬物製剤が患者に投与された後の薬物の送達および体内動態を成功させるための要件に分類される。
【0004】
様々な経路による投与のための多種類の薬物製剤が存在し、異なる製剤にとって最適な薬物形態は、異なっている可能性が高い。上述のように、薬物製剤は、治療を必要としている患者への流通を成功させるのに十分な貯蔵寿命を有していなければならない。さらに、経口用薬物製剤は、経口的に投与された場合に患者の胃腸管で溶解するであろう形態で薬物を提供しなければならない。即時放出性の錠剤、カプセル、懸濁液、またはサシェなどの即時放出性剤形で経口的に投与する場合、投与量の完全な溶解および最適な生物学的利用能を保証するために、高い溶解性を有する薬物塩または薬物形態を有することが一般的に望ましい。一部の薬物、特に、低い溶解性の薬物または湿潤性に乏しい薬物の場合、胃腸管内に投与された場合に結晶性形態よりも高い初期溶解性を一般的に有するであろう非結晶性薬物形態を利用することが有利であることがある。薬物の非結晶性形態は、結晶性形態よりも化学的に安定性が低いことが多い。したがって、剤形製造、包装、貯蔵、および世界中の患者への流通を可能にするのに十分な時間にわたってその効力を維持するのに十分に安定である実用的な製品を提供するのに十分に化学的に安定である非結晶性薬物形態を識別することが有利である。
【0005】
一方、薬物形態があまり可溶性でない場合によりよく動作する剤形が存在する。例えば、チュアブル錠剤または懸濁液またはサシェの剤形は、薬物に直接舌をさらす。そのような剤形の場合、薬物の部分を固体の状態に保ち、悪い味を最小限に抑えるために、口における薬物の溶解性を最小限に抑えることが望ましい。そのような剤形の場合、低い溶解性の塩または結晶性形態を使用することが望ましいことが多い。
【0006】
制御放出性の経口用または注射用の、例えば、皮下または筋肉内の剤形の場合、望ましい薬物溶解性は、送達経路、投与量、剤形設計および望ましい放出継続時間の複雑な関数である。高い溶解性を有する薬物の場合、遅い溶解による遅い放出の実現を助けるために、制御放出性剤形のためのより低い溶解性の結晶性の塩または多形体を利用することが望ましいことがある。低い溶解性を有する薬物の場合、制御放出性剤形からの望ましい薬物放出速度を支えるのに十分な溶解速度を実現するために、より高い溶解性の結晶性の塩もしくは多形体、または非結晶性形態を利用することが必要であることがある。
【0007】
ソフトゼラチンカプセル剤形(「ソフトゲル」)において、薬物は、トリグリセリド油またはポリエチレングリコールなどの少量の溶媒またはビヒクルに溶かされ、ゼラチンカプセルに封入される。この剤形にとって最適の薬物形態は、適切なソフトゲルビヒクルにおいて高い溶解性を有する薬物形態である。一般に、トリグリセリド油により可溶性である薬物形態は、水にあまり可溶性でないであろう。ソフトゲル剤形に適している薬物形態の識別には、様々な塩、多形体、結晶性および非結晶性の形態の研究が必要である。
【0008】
したがって、薬物形態の望ましい溶解性は、使用目的によって異なり、薬物形態がすべて等価であるとは限らないことが分かる。
【0009】
ヒトまたは動物の治療法にとって実際に有用である薬物形態の場合、薬物形態は、最小限の吸湿性を示すことが望ましい。極めて吸湿性の薬物を含有する剤形は、保護包装を必要とし、湿潤環境で貯蔵される場合に、変化した溶解を示すことがある。したがって、薬物の非吸湿性の結晶性の塩および多形体を識別することが望ましい。薬物が非結晶性である場合、または溶解性および溶解速度を改善するために非結晶性形態が望まれる場合、他の非結晶性の塩または形態に比べて低い吸湿性を有する非結晶性の塩または形態を識別することが望ましい。
【0010】
結晶性または非結晶性の薬物は、無水形態で、または水和物もしくは溶媒和物もしくは水和物/溶媒和物として存在することがある。薬物の水和状態および溶媒和状態は、その溶解性および溶解挙動に影響を及ぼす。
【0011】
薬物の融点は、異なる塩、多形体、結晶性および非結晶性の形態について変わることがある。商業用錠剤プレスでの錠剤の製造を可能にするため、薬物融点は、錠剤製造中の薬物溶融を防ぐために、約60℃を超え、好ましくは100℃を超えていることが望ましい。この場合の好ましい薬物形態は、最も高い融点を有する薬物形態である。さらに、直射日光および赤道近くなどの地理的領域で起きる高い環境貯蔵温度において固体剤形中の固体薬物の化学的安定性を保証するために高い融点を有することが望ましい。ソフトゲル剤形が望まれる場合、剤形における薬物の結晶化を最小限に抑えるために、低い融点を有する薬物形態を有することが好ましい。したがって、薬物形態の望ましい融点は、使用目的によって異なり、薬物形態がすべて等価であるとは限らないことが分かる。
【0012】
薬物の投与量が高い場合、または小さな剤形が望まれる場合、塩、水和物または溶媒和物の選択は、単位重量当たりの効力に影響を及ぼす。例えば、より高い分子量の対イオンを有する薬物塩は、より低い分子量の対イオンを有する薬物塩よりも低いグラム当たりの薬物効力を有するであろう。単位重量当たり最も高い効力を有する薬物形態を選択することが望ましい。
【0013】
異なる結晶性多形体および非結晶性形態を調製する方法は、薬物によって大きく異なる。これらの方法において、特に、最終合成ステップについて、特に、薬物が、合成の最終ステップで利用される溶媒との溶媒和物として存在する傾向がある場合、毒性が最も少ない溶媒が使用されることが望ましい。好ましい薬物形態は、それらの合成において毒性が少ない溶媒を利用する薬物形態である。
【0014】
商業規模で良好な錠剤を形成する薬物の能力は、Hiestand H、Smith D.Indices of tableting performance.Powder Technology、1984;38:145〜159に記載されている錠剤化指数(Tableting Indices)などの様々な薬物物理的特性によって異なる。これらの指数を用い、優れた錠剤化性能を有する薬物、例えば、アトルバスタチンカルシウムの形態を識別することができる。そのような指数の1つは、脆性を反映する脆性破壊指数(Brittle Fracture Index)(BFI)であり、0(良好−低脆性)から1(不良−高脆性)まで様々である。機械的特性、流動特性および錠剤化性能の他の有用な指数または尺度は、圧縮応力、絶対密度、固相率(solid fraction)、動的圧入硬度、延性、弾性率、低減(reduced)弾性率、準静的圧入硬度、剛性率、引張強度、妥協(compromised)引張強度、最高結合指数、最低結合指数、脆性/粘弾性結合指数、歪指数、粘弾性数、内部摩擦の有効角(シェアセル試験から)、凝集性(粉末雪崩試験から)および流動変動性を包含する。多くのこれらの尺度は、3軸油圧プレスを用いて調製されることが好ましい薬物成形体で得られる。これらの尺度の多くは、Hancock B、Carlson G、Ladipo D、Langdon B、およびMullarney M.Comparison of the Mechanical Properties of the Crystalline and Amorphous Forms of a Drug Substance.International Journal of Pharmaceutics、2002;241:73〜85にさらに記載されている。
【0015】
流動に影響を及ぼす薬物形態特性は、錠剤剤形製造にとってだけではなく、カプセル剤、懸濁剤、およびサシェ剤の製造にとっても重要である。
【0016】
薬物粉末の粒径分布も、特に、粉末流動に対する影響により、製造プロセスに大きな影響を有することがある。異なる薬物形態は、異なる特徴的な粒径分布を有する。
【0017】
上記の論議から、すべての治療的応用に理想的である薬物形態は存在しないことは明らかである。したがって、様々な製剤において使用することができる様々な独特の薬物形態、例えば、塩、多形体、非結晶性形態を探し求めることが重要である。具体的な製剤または治療的応用のための薬物形態の選択には、上記に記載されているような様々な特性を考慮することが必要であり、特定の応用のための最良の形態は、1つの具体的な重要で良好な性質を有する形態であってよく、一方、他の特性は、許容できるか、または辛うじて許容できるかであってよい。
【0018】
米国特許第6,498,172B1号は、例えば、骨障害の治療に有用であるような化合物(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸を開示している。この出願は、一般論として、薬学的に許容できる塩に言及しており、ナトリウム塩の調製が開示されている。しかしながら、‘172特許は、結晶化手順について何ら記載せず、特許は、この化合物の多形体形態についても何ら論議していない。
【0019】
公開された国際特許出願WO99/19300およびWO98/28264は、プロスタグランジン作動薬ならびに局所適用(例えば、骨折または骨切り術の部位への)による骨折および骨切り術の治癒を治療および促進するためのそれらの使用を開示している。
【0020】
S.C.MillerおよびS.C.Marks,Jr.、Bone 14、143〜151(1993)は、プロスタグランジンE1(PGE1)によるイヌ下顎骨の骨膜表面上の新生骨形成の局所刺激を研究し、外側下顎皮質に隣接して骨膜下に埋め込まれた浸透圧ミニポンプと制御放出性ペレット剤による送達を比較した。
【0021】
S.C.Marks,Jr.およびS.C.Miler、J.Oral Pathol.17:500〜505(1988)は、1週当たり500〜2000μgの投与量で3週間にわたるPGE1の局所注入が、イヌの下顎骨における歯槽骨の劇的で局所的な形成を生み出すことを報告した。
【0022】
M−S.ShihおよびR.W.Norrdin、Am.J.Vet.Res.48:828〜830(1986)では、横骨折を、成体ビーグル犬の肋骨で外科的に作製し、10%エタノールTris−緩衝液ビヒクル0.5mlまたはPGE1(10%エタノールTris−緩衝液中にPGE10.2mgを含有する)0.5mlを10日間にわたって1日2回、骨折部位に直接注射した。PGE1の投与は、骨折部位およびその対側整合部位に隣接する骨膜外層上の骨基質形成を誘導するという結論であった。
【0023】
M−S.ShihおよびR.W.Norrdin、Calcif.Tissue Int.(1986)39:191〜197は、手術後10日間にわたって1日2回、ビーグル犬の頸骨における欠損部位に注射されたPGE1(10%エタノール中0.2mg/kg)の影響を研究した。局所的にPGE1を投与されたイヌは、存在する類骨量の増加と共に、より多くの骨膜および皮質の骨内膜性骨形成を有することが判明した。
【0024】
R.Yang、T.LiuおよびS.Lin−Shiau、Calcif.Tissue Int.、52:57〜61(1993)は、14日間にわたる左頸骨の骨幹端中への骨内経路を介するプロスタグランジンE2の連日注射の影響を検討した。この参考文献によれば、この投与計画は、骨幹端における骨梁の有意な増加をもたらした。
【0025】
K.Notoya他、The Journal of Pharmacology and Experimental Therapeutics、290:1054〜1064(1999)は、in vivoにおける骨格再生および骨修復に対して局所的に適用された持続放出性マイクロカプセル剤で、新規な骨芽細胞分化促進化合物であるTAK−778の影響を調べた。
【発明の開示】
【発明が解決しようとする課題】
【0026】
本発明の他の実施形態によれば、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態が提供される。
【課題を解決するための手段】
【0027】
本発明のさらに他の実施形態によれば、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態、および薬学的に許容できる希釈剤または担体を含む医薬組成物が提供される。
【0028】
本発明の別の実施形態によれば、緑内障、高眼圧症または低骨量を示す状態を有する哺乳類を治療するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態を哺乳類に投与することを含む方法が提供される。
【0029】
本発明のさらに他の実施形態によれば、哺乳類において骨量を増加および維持するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態を哺乳類に投与することを含む方法が提供される。
【0030】
本発明のさらに別の実施形態によれば、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物の結晶性形態Aを調製するための方法であって、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸を、遊離の酸として、有機溶媒中で水酸化ナトリウムと接触させて反応混合物を形成すること、反応混合物を約50C〜約90Cの第1の温度まで温め、少なくとも1時間にわたって第1の温度を保つこと、反応混合物を約15C〜約25Cの第2の温度まで冷却すること、および反応混合物から固体を集めることを含む方法が提供される。この方法において使用される有機溶媒は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約8〜約12mlで存在する酢酸イソプロピルであってよい。この方法において使用される水酸化ナトリウムは、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約1〜約1.3mmolで存在する水酸化ナトリウムの50%水溶液であってよい。
【0031】
本発明の他の実施形態によれば、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態が提供される。
【0032】
本発明のさらに他の実施形態によれば、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態、および薬学的に許容できる希釈剤または担体を含む医薬組成物が提供される。
【0033】
本発明の別の実施形態によれば、緑内障、高眼圧症または低骨量を示す状態を有する哺乳類を治療するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態を哺乳類に投与することを含む方法が提供される。
【0034】
本発明のさらに他の実施形態によれば、哺乳類において骨量を増加および維持するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態を哺乳類に投与することを含む方法が提供される。
【0035】
化合物「(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸」は、化合物の遊離の酸を指す。
【0036】
「哺乳類」という用語は、例えば、イヌ、ネコ、雌ウシ、ヒツジ、ウマ、およびヒトを包含する動物を意味する。好ましい哺乳類は、ヒトを包含する。
【0037】
「実質的に純粋な」という語句は、化合物の総量(すなわち、望ましい多形体および/または塩+その他の多形体および非晶質性形態、塩および遊離の酸の量)に関して望ましい多形体および/または塩の相対的純度を指す。「実質的に純粋な」多形性形態は、化合物の総量に対して望ましい多形体を少なくとも約90%含有するべきである。「実質的に純粋な」多形性形態は、望ましい多形体を少なくとも約95%含有するべきであることが好ましい。本発明の一部の実施形態において、「実質的に純粋な」多形性形態は、望ましい多形体を少なくとも約99%含有することがある。
【0038】
「低骨量を示す1種または複数の状態」という語句は、骨量のレベルが、World Health Organization「Assessment of Fracture Risk and its Application to Screening for Postmenopausal Osteoporosis(1994)、Report of a World Health Organization Study Group、World Health Organization Technical Series 843」による基準で定義されているような年齢特異的な正常以下である状態を指す。原発性および続発性骨粗鬆症は、「低骨量を示す1種または複数の状態」に包含される。続発性骨粗鬆症は、グルココルチコイド誘発性骨粗鬆症、甲状腺機能亢進症誘発性骨粗鬆症、不動化(immobilization)誘発性骨粗鬆症、ヘパリン誘発性骨粗鬆症および免疫抑制誘発性骨粗鬆症を包含する。歯周疾患、歯槽骨喪失、骨切り術後および小児期特発性骨喪失も包含される。「低骨量を示す1種または複数の状態」という語句は、背骨の湾曲、身長の減少および補綴手術などの骨粗鬆症の長期合併症も包含する。
【0039】
「低骨量を示す1種または複数の状態」という語句は、骨粗鬆症を包含する上記に記載されているような疾患を発症する平均よりも有意に高い確率を有することが知られている哺乳類、例えば、哺乳類(例えば、閉経後の女性、および60歳を超える男性)も指す。
【0040】
他の骨量を増加または増強する用途は、骨修復、骨折治癒速度の増加、骨移植手術の完全な置き換え、骨移植成功率の向上、顔面再建、上顎再建、下顎再建、頭蓋顔面再建後の骨治癒、補綴内方成長、椎骨癒合、長骨伸長および脊椎固定術を包含する。
【0041】
本発明の医薬組成物は、脊椎固定ケージ、脊椎固定金属製品、内部および外部の骨固定装置、ネジならびにピンなどの当業者に知られている整形外科用装置と併せて使用することもできる。
【0042】
当業者は、骨量という用語が、実際には、時に(厳密には正しくないが)骨塩密度(BMD)と呼ばれる単位面積当たりの骨量を指すことを理解するであろう。
【0043】
本明細書で使用する「治療すること」、「治療する」または「治療」という用語は、予防的(preventative)(例えば、予防的(prophylactic))、姑息的および治癒的治療を包含する。
【0044】
「有効量」という用語は、特定の疾患もしくは状態または特定の疾患もしくは状態の症状を改善、軽減もしくは除去するか、または特定の疾患もしくは状態または特定の疾患もしくは状態の症状の発現を予防もしくは遅延する化合物または化合物の組合せの量を意味する。
【0045】
「患者」という用語は、ヒト、イヌ、ネコおよびウマなどのコンパニオンアニマル、ならびにウシ、ブタおよびヒツジなどの家畜などの動物を意味する。特に好ましい患者は、雄と雌の両方を包含する哺乳類であり、ヒトがより好ましい。
【0046】
本明細書で使用する「薬学的に許容できる」という用語は、担体、ビヒクル、希釈剤、賦形剤および/または塩が、製剤の他の成分と適合していなければならず、そのレシピエントに対して有害でないことを意味する。
【0047】
「プロドラッグ」という表現は、投与後、いくつかの化学的または生理学的プロセスを介してin vivoで薬物を放出する薬物前駆体である化合物を指す(例えば、生理学的pHにされるか、または酵素作用によりプロドラッグは、望ましい薬物形態へ変換される)。例示的プロドラッグは、切断により、対応する薬物化合物を放出する。
【0048】
「薬学的に許容できる塩」という表現は、塩化物、臭化物、ヨウ化物、硫酸塩、重硫酸塩、リン酸塩、酢酸塩、マレイン酸塩、フマル酸塩、シュウ酸塩、酪酸塩、酒石酸塩、クエン酸塩、グルコン酸塩、メタンスルホン酸塩および4−トルエン−スルホン酸塩など(であるが、これらに限定されない)アニオン性の塩を指す。この表現は、ナトリウム、カリウム、カルシウム、マグネシウム、アンモニウムまたはプロトン化したベンザチン(N,N’−ジベンジルエチレンジアミン)、コリン、エタノールアミン、ジエタノールアミン、エチレンジアミン、メグラミン(meglamine)(N−メチル−グルカミン)、ベネタミン(N−ベンジルフェネチルアミン)、ピペラジンおよびトロメタミン(2−アミノ−2−ヒドロキシメチル−1,3−プロパンジオール)など(であるが、これらに限定されない)カチオン性の塩も指す。
【0049】
本発明のこれらおよび他の特徴、態様および利点は、以下の図面、説明および特許請求の範囲を参照してよりよく理解されていくであろう。
【発明を実施するための最良の形態】
【0050】
一般に、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、米国特許第6,498,172B1号に開示されている方法により調製することができ、その主題は、参照により全体として本明細書に組み込まれるものとする。代替方法として、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、以下のスキームIに記載されている新規な方法により調製することができる。これらの化合物の具体的な多形体を製造するための特定のプロセスは、実験の項に示される。すべての出発化合物は、文献手順によるか、またはSigma−Aldrich Corporation、St.Louis、MOなどの一般的商業ソースから得ることができる。
【0051】
考察、実施例および調製において、以下の略語が使用される。2B EtOH−変性エタノール、br−幅広いピーク、℃−セ氏温度、d−二重線、DMSO−d6−重水素化ジメチルスルホキシド、EtOAc−酢酸エチル、equiv−当量、g−グラム、1H NMR−プロトン核磁気共鳴、H2O−水、HCl−塩化水素、Hz−ヘルツ、iPr2Net−ジイソプロピルエチルアミン(ヒューニッヒ塩基)、iPrOAc−酢酸イソプロピル、J−結合定数(多重線の間隔)、kg−キログラム、L−リットル、m−多重線、M−モル、MeCl2−ジクロロメタン、mg(s)−ミリグラム、min−分、mL−ミリリットル、mmol−ミリモル、mp−融点、MPa−メガパスカル、MS−マススペクトル、N−規定濃度、NaHCO3−炭酸水素ナトリウム、NaOH−水酸化ナトリウム、psi−毎平方インチ当たりポンド、Pt/C−炭素上白金、RT−室温、s−一重線ピークおよびt−三重線ピーク。さらに、以下の考察、実施例および調製において、Brukerへの言及は、Bruker AXS,Inc.、Madison、WIの製品を指し、Kevexへの言及は、Thermo Electron Corporation、Waltham、MAの製品を指す。
【0052】
【化1】

【0053】
ステップ1:{3−[(4−tert−ブチル−ベンジルアミノ)−メチル]−フェノキシ}−酢酸エチルエステルコハク酸塩(II)の調製
500mLのParr瓶に、2Bエタノール(180mL)および5%炭素上白金、50%水湿(2.00g)を充填した。2Bエタノール(20mL)中の3−ホルミルフェノキシ酢酸エチル(20.0g、96.06mmol、1.0当量)の溶液と、続いて、4−tert−ブチルベンジルアミン(16.86mL、96.06mmol、1.0当量)を加えた。混合物を5時間にわたって窒素雰囲気下で室温において攪拌し、その時点で、式Iの中間体の生成が完了した。反応容器を50psi(0.3447MPa)の水素雰囲気下に置き、18時間にわたって室温において振盪させた。混合物を珪藻土に通して濾過し、濾過ケーキを2Bエタノール(40mL)で1回洗浄した。濾液を、添加ロートおよびメカニカルスターラーを取り付けた1リットルのフラスコに移した。2Bエタノール(120mL)中のコハク酸(11.34g、96.06mmol、1.0当量)の温かい溶液を15分かけて加えた。得られたスラリーを19時間にわたって室温において攪拌し、次いで、濾過した。固体を2Bエタノール(50mL)で1回洗浄し、乾燥すると、II(33.10g、収率73%)が得られた。融点=138〜139℃。元素分析 C22H29NO3・C4H6O4の計算値:C 65.94;H 7.45;N 2.96。実測値:C 65.74;H 7.62;N 2.99。1H NMR(400MHz,CD3OD):δ 1.28(t,3H,J=7.0Hz)、1.32(s,9H)、2.50(s,4H)、4.08(s,2H)、4.10(s,2H)、4.23(q,2H,J=7.0Hz)、4.73(s,2H)、6.96〜6.98(m,1H)、7.04〜7.06(m,2H)、7.33〜7.38(m,3H)、7.46〜7.48(m,2H)。
【0054】
ステップ2および3:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(V)の調製
化合物II(48.00g、101mmol、1.0当量)を、ジクロロメタン(360mL)と1M水性重炭酸ナトリウム(240mL)の間で分配することにより遊離塩基にした。層を分離し、有機層を水(240mL)で1回洗浄した。層を分離し、N,N−ジイソプロピルエチルアミン(53.4mL、304mmol、3.0当量)を有機層に加えた。塩化ピリジン−3−スルホニル塩酸塩(III)(26.4g、123mmol、1.2当量)およびジクロロメタン(240mL)を別の反応容器に充填し、窒素雰囲気下で0℃まで冷却した。遊離塩基/N,N−ジイソプロピルエチルアミン溶液を1.8時間かけて加えた。反応混合物を室温まで温め、19時間にわたって保ち、その時点で、式IV((3−{[(4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ]−メチル}−フェノキシ)−酢酸エチルエステル)の中間体の生成が完了した。反応混合物を1N塩酸(336mL)、次いで、1M水性重炭酸ナトリウム(336mL)で洗浄した。2Bエタノール(384mL)を有機層に加え、混合物を、ポット容積が約400mLになるまで大気圧蒸留した。6N水酸化ナトリウム(19.9mL、119mmol、1.2当量)を加え、混合物を19時間にわたって室温に保った。濃塩酸(11.0mL、134mmol、1.3当量)と、続いて、水(192mL)を加えた。スラリーを室温において5時間にわたって顆粒化し、濾過した。固体を50:50 2Bエタノール:水(144mL)、水(144mL)、2Bエタノール(144mL)で洗浄し、次いで、乾燥すると、化合物V(34.78g、収率73%)が得られた。融点=159〜160℃。元素分析 C25H28N2O5Sの計算値:C 64.08;H 6.02;N 5.98。実測値 C 64.13;H 6.11;N 5.99。1H NMR(400MHz,DMSO−d6):δ 1.21(s,9H)、4.31(s,2H)、4.33(s,2H)、4.54(s,2H)、6.65〜6.69(m,1H)、6.71〜6.74(m,1H)、6.75〜6.76(m,1H)、7.01(d,2H,J=8.1Hz)、7.13(t,1H,J=7.9Hz)、7.21(d,2H,J=8.3Hz)、7.56〜7.59(m,1H)、8.16〜8.19(m,1H)、8.80〜8.82(m,1H)、8.97〜8.98(m,1H)、13.04(br s,1H)。
【0055】
ステップ3R:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(V)の再結晶
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(V)(20.0g、42.7mmol、1.0当量)およびメチルエチルケトン(300mL)を混ぜ合わせ、すべての固体が溶けるまで60℃まで加熱した。溶液を35℃まで冷却し、0.45ミクロンのナイロンフィルターに通して濾過した。フィルターをメチルエチルケトン(20mL)ですすいだ。濾液を混ぜ合わせ、約160mLのポット容積まで大気圧蒸留により濃縮した。混合物を室温まで冷却し、その時点で、結晶化が始まった。混合物を室温において19時間にわたって攪拌し、次いで、濾過した。固体をメチルエチルケトン(20mLで2回、次いで、40mLで1回)で洗浄し、乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(15.30g、収率77%)が得られた。融点=159.0〜159.5℃。元素分析 C25H28N2O5Sの計算値:C 64.08;H 6.02;N 5.98。実測値 C 63.89;H 5.82;N 5.91。1H NMR(400MHz,DMSO−d6):上記のステップ2および3と同じ。1H NMRスペクトルは、メチルエチルケトン約1.4%の存在も示した。
【0056】
ステップ4:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII)の調製
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(20.0g、42.7mmol、1.0当量)および酢酸イソプロピル(400mL)を反応容器に充填した。50%水酸化ナトリウム(3.520g、44.0mmol、1.03当量)と、続いて、脱イオン水(4.165g)を加えた。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII、200mg)をシード材料として加えた。混合物を30分かけて70℃まで加熱し、5時間にわたってこの温度に保った。混合物を4時間かけて40℃まで冷却し、次いで、1時間かけて20℃まで冷却し、19時間にわたって顆粒化し、次いで、濾過した。固体を1%水性酢酸イソプロピル(各洗浄液20mL)で2回洗浄し、次いで、乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII、18.22g、収率85%)が得られた。半水和物は、約120℃に脱水温度を有し、融点は有していない。元素分析 C25H27N2O5S・Na・1/2H2Oの計算値:C 61.21;H 5.55;N 5.71。実測値 C 60.19;H 5.55;N 5.56。1H NMR(400MHz,DMSO−d6):δ 1.21(s,9H)、4.07(s,2H)、4.31(s,4H)、6.59〜6.61(m,1H)、6.64(m,1H)、6.71〜6.74(m,1H)、7.01〜7.09(m,3H)、7.22(d,2H,J=8.4Hz)、7.56〜7.59(m,1H)、8.14〜8.17(m,1H)、8.79〜8.80(m,1H)、8.94(m,1H)。
【0057】
代替方法として、ステップ4におけるナトリウム塩生成および再結晶は、以下のスキームIIに示す手順を用いて行うことができる。
【0058】
【化2】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(47.7g)およびメタノール(500mL)を混ぜ合わせた。1N水酸化ナトリウム(1.01当量、103mL)を、室温において攪拌しながらゆっくりと加えた。2時間にわたって攪拌した後、混合物を濃縮し、アセトンで5回(毎回300mL)およびジクロロメタンで2回(毎回200mL)と一緒に共沸させると、非晶質性の泡が得られた。これに、アセトン(500mL)および水(15mL)を加え、混合物を50℃まで加熱した。20分後、材料は極めて粘稠になり、追加のアセトン(200mL)を加えた。混合物を室温まで冷却し、濾過した。濾過ケーキを2−プロパノールで洗浄し、高真空下で乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII、収率77%)39gが得られた。
【0059】
粉末X線回折
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態A、B、C、E、F、およびGを、それらのX線粉末回折パターンにより特徴付けた。すなわち、形態A、B、C、E、F、およびGのX線回折パターンを、銅放射線(波長:1.54056Å)を用いるBruker D5000回折計で行った。管の電圧およびアンペア数は、それぞれ40kVおよび50mAに設定した。発散スリットおよび散乱スリットは、1mmに設定し、受光スリットは、0.6mmに設定した。回折された放射線は、Kevex PSI検出器により検出した。3.0から40°2θまでの2.4°/分(1秒/0.04°ステップ)におけるθ−2θ連続スキャンを用いた。アルミナ標準品を分析し、機器アラインメントをチェックした。データを集め、Bruker AXSソフトウェアバージョン7.0を用いて分析した。試料は、石英ホルダーに試料を入れることにより調製した。
【0060】
本明細書で報告される測定のために使用されるBrukerシステムのようなBragg−Brentano機器でX線回折測定を行うため、試料を、通常、空洞を有するホルダーの中に入れる。試料粉末をガラススライドまたは同等物により押し付け、ランダムな表面および適切な試料高を確保する。次いで、試料ホルダーを機器の中に入れる。入射X線ビームを、初めはホルダー面に対して小さな角度で試料に向け、次いで、入射ビームとホルダー面の間の角度を連続的に増加させる円弧に沿って移動させる。そのようなX線粉末分析に関連した測定差は、(a)試料調製における誤差(例えば、試料高)、(b)機器誤差(例えば、平面試料誤差)、(c)較正誤差、(d)操作者誤差(ピーク位置を決定する場合に存在する誤差を包含する)、および(e)材料の性質(例えば、好ましい配向性および透明度誤差)を包含する様々な要因に起因する。較正誤差および試料高誤差は、同じ方向ですべてのピークのシフトをもたらすことが多い。平面ホルダーを用いる場合の試料高における小さな差は、XRPDピーク位置における大きな移動につながるであろう。系統的な研究は、典型的なBragg−Brentano配置でShimadzu XRD−6000を用いると、1mmの試料高差が、1°2θと同じ高さのピークシフトにつながることを明らかにした(Chen他;J Pharmaceutical and Biomedical Analysis、2001;26、63)。これらのシフトは、X線ディフラクトグラムから識別することができ、シフトについて補正すること(系統的な補正係数をすべてのピーク位置値に適用すること)、または機器を再較正することにより除去することができる。上述のように、ピーク位置を一致させるために系統的な補正係数を適用することにより、様々な機械からの測定値を修正することが可能である。一般に、この補正係数は、Brukerからの測定ピーク位置を、期待されるピーク位置と一致させるであろうし、0〜0.2°2θの範囲であってよい。
【0061】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Hを、そのX線粉末回折パターンにより特徴付けた。形態HについてのX線粉末回折パターンを、銅放射線を用いるSiemens D5000回折計で作成した。機器に、直線集束X線管を取り付けた。管の電圧およびアンペア数は、それぞれ38kVおよび38mAに設定した。発散スリットおよび散乱スリットは、1mmに設定し、受光スリットは、0.6mmに設定した。回折されたCu Kα1放射線(λ=1.54056Å)は、Kevex PSI検出器を用いて検出した。3.0から40°2θまでの2.4°2θ/分(1秒/0.04°2θステップ)におけるθ2θ連続スキャンを用いた。アルミナ標準品(NIST標準基準材料1976)を分析し、機器アラインメントをチェックした。データを集め、BRUKER AXS DIFFRAC PLUSソフトウェアバージョン2.0を用いて分析した。試料は、石英ホルダーに試料を入れることにより調製した。PXRDピークは、最大ピーク高を用いて手動で選択した。
【0062】
図1を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aは、CuKα放射線によりBruker D5000回折計で測定された7%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0063】
【表1】
【0064】
図2を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Bのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Bは、CuKα放射線によりBruker D5000回折計で測定された3%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0065】
【表2】
【0066】
図3を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Cのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Cは、CuKα放射線によりBruker D5000回折計で測定された5%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0067】
【表3】
【0068】
図4を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Eのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Eは、CuKα放射線によりBruker D5000回折計で測定された4%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0069】
【表4】
【0070】
図5を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Fのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Fは、CuKα放射線によりBruker D5000回折計で測定された5%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0071】
【表5】
【0072】
次に、図6を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Gのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Gは、CuKα放射線によりBruker D5000回折計で測定された6%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0073】
【表6】
【0074】
要約すれば、表7は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態A、B、C、E、F、およびGについて試料における回折線の度2θおよび相対強度を列挙している:
【0075】
【表7】
【0076】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の6つの結晶性形態のみが知られているため、各形態は、単一のX線粉末回折線、線の組合せまたは他の形態のX線粉末回折と異なるパターンにより、他の結晶性形態から識別および区別することができる。
【0077】
例えば、表8は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態A、B、C、E、F、およびGについての独特なピーク、ならびに2θピークの組合せ、すなわち、各形態に独特である一連のX線回折線を列挙している。
【0078】
【表8】
【0079】
次に、図6Bを参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Hのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Hは、CuKα放射線によりBruker D5000回折計で測定された6%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0080】
【表9】
【0081】
次に、図6Cを参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非晶質性形態Iのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Iは、図に示されるようなX線粉末回折パターンにより特徴付けられる。
【0082】
固体核磁気共鳴
次に、図7を参照すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aを、その固体核磁気共鳴スペクトル(SSNMR)により特徴付けることもできる。すなわち、形態Aの固体核磁気共鳴スペクトルを、Bruker−Biospin(Buker−Biospin、Billerica、MA)Avance DSX 500MHz NMR分光計で行った。
【0083】
13C SSNMR
化合物約70mgを、分析される試料のための4mmのZrOスピナーに固く詰め込んだ。一次元13Cスペクトルは、ワイドボアBruker−Biospin Avance DSX 500MHz NMR分光計内に配置されたBruker 4mm BL CPMASプローブで293Kにおいて1H−13C交差分極マジック角回転(CPMAS)を用いて周囲圧力にて集めた。試料は、4mmのスピナーにとっての最大規定回転速度に相当する15.0kHzにて回転させた。速い回転速度は、スピニングサイドバンドの強度を最小限に抑えた。シグナル感度を最適化するため、交差分極接触時間を2.0msに調整し、デカップリングパワーを85kHzに設定した。炭素スペクトルは、3秒のリサイクルディレイで2,820のスキャンを取得した。炭素スペクトルは、その高磁場共鳴が29.5ppmに設定されているアダマンタンの外部試料を用いて参照した。
【0084】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aは、化学シフトが百万分率(ppm)で表される以下の固体13C核磁気共鳴(SSNMR)スペクトルにより特徴付けられた:
【0085】
【表10】
【0086】
次に、図8を参照すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Hを、その固体核磁気共鳴スペクトル(SSNMR)により特徴付けることもできる。すなわち、形態Aの固体核磁気共鳴スペクトルを、Bruker−Biospin(Buker−Biospin、Billerica、MA)Avance DSX 500MHz NMR分光計で行った。
【0087】
形態Hのための13C SSNMR法
化合物約70mgを、4mmのZrO2ローターに固く詰め込んだ。一次元13Cスペクトルは、ワイドボアBruker−Biospin Avance DSX 500MHz NMR分光計内に配置されたBruker 4mm BL CPMASプローブで294Kにおいて1H−13C交差分極マジック角回転(CPMAS)を用いて周囲圧力にて集めた。試料は、4mmのローターにとっての最大規定回転速度に相当する15.0kHzにて回転させた。速い回転速度は、スピニングサイドバンドの強度を最小限に抑えた。シグナル感度を最適化するため、交差分極接触時間を2.0msに調整し、デカップリングパワーを85kHzに設定した。炭素スペクトルは、5秒のリサイクルディレイで5400のスキャンを取得した。炭素スペクトルは、その高磁場共鳴が29.5ppmに設定されているアダマンタンの外部試料を用いて参照した。
【0088】
【表11】
【0089】
本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態または非結晶性形態または非晶質性形態は、無水形態ならびに水和および溶媒和形態で存在することができる。一般に、水和形態は、非水和形態と等価であり、本発明の範囲内に包含されることが意図されている。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態A、B、およびCは、水和物として生じることが好ましいが、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態E、F、およびGは、無水形態として生じることが好ましい。
【0090】
本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態または非結晶性形態または非晶質性形態は、等価なX線粉末ディフラクトグラム、または固体NMRスペクトルを有する水和および/または溶媒和の程度にかかわらず、本発明の範囲内にある。
【0091】
以下の非限定的な実施例は、本発明の化合物を調製するための好ましい方法を例示している。
【実施例】
【0092】
形態A:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0093】
【化3】
手順:500mLのジャケット付き反応器に、酢酸イソプロピル(400mL)、50%水性水酸化ナトリウム(3.520g)および水(4.165g)を充填した。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(20.0g)と、続いて、シードとしての(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸の形態A(200mg)(例えば、上記で、スキームIIのプロセスにより調製される)を混合物に充填した。混合物を30分かけて70℃まで加熱し、5時間にわたってこの温度において攪拌した。混合物を4時間かけて40℃まで冷却し、次いで、1時間かけて20℃まで冷却した。次いで、スラリーを12時間にわたって20℃において顆粒化した。スラリーを、ブフナーロート(濾紙)に通して濾過した。濾過ケーキを1%水性酢酸イソプロピル20mLで2回洗浄し、次いで、窒素を抽気しながら40〜45℃において一夜にわたって真空オーブン中で乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物形態A(VIII、収率79%)16.84gが得られた。
【0094】
形態B:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0095】
【化4】
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(4.98g)、酢酸エチル(59.8mL)および水(1.3mL)を混ぜ合わせ、55℃まで加熱した。酢酸エチル(20mL)および水(0.44mL)中の2−エチルヘキサン酸ナトリウム(97%、2.025g)の溶液を加えると、澄明な溶液が得られた。溶液を、55°〜60℃において15分間にわたって活性炭で処理し、珪藻土に通して濾過した。濾過ケーキを酢酸エチル/2.2%水5mLで1回洗浄した。濾液は、40℃であった。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(VIII)50mgをシードとして加え、混合物を室温まで冷却し、18時間にわたって攪拌した。スラリーを濾過すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩3.1gが得られた。
【0096】
濾液を20mLまで濃縮し、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(VIII)をシードし、室温において3時間にわたって攪拌し、濾過すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩0.1gが得られた。
【0097】
第2の濾液を濃縮乾固し、室温において水40mL中で顆粒化し、濾過し、空気乾燥すると、固体2.4gが得られた。これらを酢酸エチル30mLおよび酢酸エチル10mL中の2−エチルヘキサン酸ナトリウム(97%、1g)の溶液と混ぜ合わせた。混合物を18時間にわたって室温において攪拌し、濾過し、乾燥すると、非結晶性の固体が得られた。この材料を酢酸エチル40mLおよび水1mLに溶かし、18時間まで室温において攪拌した。ヘキサンを、かすみ点に達するまで加え、次いで、混合物を24時間にわたって攪拌した。粘稠な沈殿が形成し、濾過し、乾燥酢酸エチルで洗浄し、乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩一水和物形態B(IX)1.27gが得られた。
【0098】
形態C:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0099】
【化5】
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(501mg)および含水THF(2.2%水)5.0mLを混ぜ合わせ、窒素雰囲気下で1時間かけて50°〜55℃まで加熱した。懸濁液に、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩4mgをシードし、次いで、室温まで冷却し、66時間にわたって攪拌した。溶媒を完全に蒸発させると、白色の固体が残った。追加の含水THF(2.2%水)5mLを加え、混合物を5時間にわたって攪拌した。固体を濾過し、45°〜50℃において真空下で乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩一水和物形態C(X、収率65.3%)333mgが得られた。
【0100】
形態EおよびF:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0101】
【化6】
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩をエチレングリコールに溶かし、溶液を、室温においてゆっくりと蒸発させた。1週間後、微細針状晶が生成し、濾過し、空気乾燥し、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩無水形態E(XI)として特徴付けた。
【0102】
濾液を低容積まで蒸発させ、濾過した。単離された生成物は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩無水形態F(XII)として特徴付けた。
【0103】
形態G:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(50mg)を、1時間かけて155℃まで加熱し、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩形態Gを製造した。
【0104】
本明細書において識別および記載された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の6つの結晶性形態(形態A〜G)の他に、形態Hと命名された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態も識別された。
【0105】
非結晶性(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(形態H)の調製
以下の2つの方法を用い、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Hを調製することができる。
【0106】
形態H、方法1。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の20〜100mg/mLの溶液を、注射用の水に薬物物質を溶かすことにより作製した。溶液を0.22μの滅菌フィルターで濾過した。溶液をバイアル/シリンジに充填した。バイアル/シリンジを−45℃において冷凍し、2時間にわたってその温度に保った。凍結乾燥は、150mτにて(−20〜25℃)における一次乾燥と、続く、(20〜30℃)における二次乾燥により完了した。得られる固体を貯蔵する。
【0107】
形態H、方法2。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩1グラムを注射用の水10mLに溶かし、100mg/mL溶液を作製する。溶液を0.22μの滅菌フィルターで濾過する。溶液0.5mLのアリコートをガラスバイアルに充填する。バイアルを凍結乾燥機に装入する。保管温度を、0.5℃/分の速度で−45℃まで下げる。保管温度を2時間にわたって−45℃に保つ。150mτの真空をかける。保管温度を、0.5℃/分の速度で−20℃まで上げる。保管温度を20時間にわたって−10℃に保つ。保管温度を、0.5℃/分の速度で30℃まで上げる。10時間にわたって30℃に保つ。凍結乾燥プロセスを通して、150mτの真空を維持する。
【0108】
本明細書において識別および記載された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の6つの結晶性形態(形態A〜G)および非結晶性形態Hの他に、形態Iと命名された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非晶質性形態も識別された。
【0109】
以下の2つの方法を用い、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Iを調製することができる。
【0110】
形態I、方法1。非晶質性の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、まず、過剰の結晶性形態Aを有機溶媒(アセトニトリルまたはジクロロメタン)に加え、72時間にわたって室温において攪拌することにより調製した。次いで、試料を濾過し、溶液を周囲条件下で室温において蒸発させると、白色の固体が得られ、PXRD分析により非晶質性固体であると決定された。
【0111】
形態I、方法2。非晶質性の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、まず、過剰の結晶性形態Aを以下の溶媒系(イソプロピルエーテル、メチルt−ブチルエーテル、または97%酢酸エチル/3%水)のうちの1つに加え、72時間にわたって40℃において攪拌することにより調製した。次いで、試料を濾過し、溶液を40℃において蒸発させると、白色の固体が得られ、PXRD分析により非晶質性固体であると決定された。
【0112】
上記に記載されている(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形性形態は、プロスタグランジン作動薬として有用であり、したがって、そのようなプロスタグランジン作動薬を用いる方法およびそのようなプロスタグランジン作動薬を含有する医薬組成物に有用である。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形性形態は、プロスタグランジンの調節により仲介される状態、特に、EP2受容体の作動薬により仲介される状態の治療および/または予防に有用である。
【0113】
本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形性形態が、放射標識形態で存在することができること、すなわち、前記化合物が、天然において通常見出される原子量または質量数と異なる原子量または質量数を含有する1個または複数の原子を含有することがあることは理解されるであろう。水素、炭素、リン、フッ素および塩素の放射性同位元素は、それぞれ3H、14C、32P、35S、18Fおよび36Clを包含する。これらの放射性同位元素および/または他の原子の他の放射性同位元素を含有する本発明の化合物は、本発明の範囲内にある。トリチウム化、すなわち3H、および炭素−14、すなわち14C、放射性同位元素は、それらの調製の容易さおよび検出能のため特に好ましい。本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の放射標識多形性形態は、一般的に、当業者によく知られている方法で調製することができる。好都合には、そのような放射標識化合物は、上記のスキームおよび/または実施例に開示されている手順を行い、非放射標識試薬の代わりに、容易に入手可能な放射標識試薬を用いることにより調製することができる。
【0114】
当業者は、抗再吸収薬(例えば、プロゲスチン、ポリホスホネート、ビスホスホネート、エストロゲン作動薬/拮抗薬、エストロゲン、エストロゲン/プロゲスチン合剤、Premarin(登録商標)、エストロン、エストリオールまたは17α−もしくは17β−エチニルエストラジオール)を、本発明の化合物と併せて使用することができることを理解するであろう。
【0115】
例示的プロゲスチンは、商業ソースから入手可能であり、アルゲストンアセトフェニド、アルトレノゲスト、酢酸アマジノン、酢酸アナゲストン、酢酸クロルマジノン、シンゲストール、酢酸クロゲストン、酢酸クロメゲストン、酢酸デルマジノン、デソゲストレル、ジメチステロン、ジドロゲステロン、エチネロン、二酢酸エチノジオール、エトノゲストレル、酢酸フルロゲストン、ゲスタクロン、ゲストデン、カプロン酸ゲストノロン、ゲストリノン、ハロプロゲステロン、カプロン酸ヒドロキシプロゲステロン、レボノルゲストレル、リネストレノール、メドロゲストン、酢酸メドロキシプロゲステロン、酢酸メレンゲストロール、二酢酸メチノジオール、ノルエチンドロン、酢酸ノルエチンドロン、ノルエチノドレル、ノルゲスチメート、ノルゲストメト、ノルゲストレル、フェンプロピオン酸オキソゲストン、プロゲステロン、酢酸キンゲスタノール、キンゲストロン、およびチゲストールを包含する。
【0116】
好ましいプロゲスチンは、メドロキシプロゲステロン、ノルエチンドロンおよびノルエチノドレルである。
【0117】
例示的な骨再吸収阻害性ポリホスホネートは、米国特許第3,683,080号に開示されているタイプのポリホスホネートを包含し、その開示は、参照により本明細書に組み込まれるものとする。好ましいポリホスホネートは、ジェミナルジホスホネート(ビス−ホスホネートとも呼ばれる)である。チルドロネート二ナトリウムは、特に好ましいポリホスホネートである。イバンドロン酸は、特に好ましいポリホスホネートである。アレンドロネートは、特に好ましいポリホスホネートである。ゾレドロン酸は、特に好ましいポリホスホネートである。他の好ましいポリホスホネートは、6−アミノ−1−ヒドロキシ−ヘキシリデン−ビスホスホン酸および1−ヒドロキシ−3(メチルペンチルアミノ)−プロピリデン−ビスホスホン酸である。ポリホスホネートは、酸、または可溶性のアルカリ金属塩もしくはアルカリ土類金属塩の形態で投与することができる。ポリホスホネートの加水分解性エステルは、同様に包含される。具体例は、エタン−1−ヒドロキシ1,1−ジホスホン酸、メタンジホスホン酸、ペンタン−1−ヒドロキシ−1,1−ジホスホン酸、メタンジクロロジホスホン酸、メタンヒドロキシジホスホン酸、エタン−1−アミノ−1,1−ジホスホン酸、エタン−2−アミノ−1,1−ジホスホン酸、プロパン−3−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、プロパン−N,N−ジメチル−3−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、プロパン−3,3−ジメチル−3−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、フェニルアミノメタンジホスホン酸、N,N−ジメチルアミノメタンジホスホン酸、N(2−ヒドロキシエチル)アミノメタンジホスホン酸、ブタン−4−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、ペンタン−5−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、ヘキサン−6−アミノ−1−ヒドロキシ−1,1−ジホスホン酸ならびに薬学的に許容できるそれらのエステルおよび塩を包含する。
【0118】
特に、本発明の化合物は、哺乳類のエストロゲン作動薬/拮抗薬と組み合わせることができる。本発明の第2の化合物として、任意のエストロゲン作動薬/拮抗薬を使用することができる。エストロゲン作動薬/拮抗薬という用語は、エストロゲン受容体と結合し、骨代謝回転を阻害しかつ/または骨喪失を予防する化合物を指す。特に、エストロゲン作動薬は、本明細書において、哺乳類組織におけるエストロゲン受容体部位と結合し、1つまたは複数の組織においてエストロゲンの作用を模倣することができる化合物と定義される。エストロゲン拮抗薬は、本明細書において、哺乳類組織におけるエストロゲン受容体部位と結合し、1つまたは複数の組織においてエストロゲンの作用をブロックすることができる化合物と定義される。そのような活性は、エストロゲン受容体結合アッセイ、標準的な骨の組織形態計測法および密度測定法、ならびにEriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London 1994、1〜296ページを包含する標準的アッセイで当業者により容易に決定される。様々なこれらの化合物について、以下に記載および言及する。
【0119】
好ましいエストロゲン作動薬/拮抗薬は、米国特許第5,047,431号に開示されているドロロキシフェン:(フェノール、3(1−(4−(2−(ジメチルアミノ)エトキシ)フェニル)−2−フェニル−1−ブテニル)−、(E)−)および関連化合物であり、その開示は、参照により本明細書に組み込まれるものとする。
【0120】
別の好ましいエストロゲン作動薬/拮抗薬は、Willson他、Endocrinology、1997、138、3901〜3911に開示されている3−(4−(1,2−ジフェニル−ブタ−1−エニル)−フェニル)−アクリル酸である。
【0121】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,536,516号に開示されているタモキシフェン:(エタナミン、2−(−4−(1,2−ジフェニル−1−ブテニル)フェノキシ)−N,N−ジメチル、(Z)−2−、2−ヒドロキシ−1,2,3−プロパントリカルボキシレート(1:1))および関連化合物であり、その開示は、参照により本明細書に組み込まれるものとする。
【0122】
別の関連化合物は、米国特許第4,623,660号に開示されている4−ヒドロキシタモキシフェンであり、その開示は、参照により本明細書に組み込まれるものとする。
【0123】
好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,418,068号に開示されているラロキシフェン:(メタノン、(6−ヒドロキシ−2−(4−ヒドロキシフェニル)ベンゾ[b]チエン−3−イル)(4−(2−(1−ピペリジニル)エトキシ)フェニル)−塩酸塩)であり、その開示は、参照により本明細書に組み込まれるものとする。
【0124】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,996,225号に開示されているトレミフェン:(エタナミン、2−(4−(4−クロロ−1,2−ジフェニル−1−ブテニル)フェノキシ)−N,N−ジメチル−、(Z)−、2−ヒドロキシ−1,2,3−プロパントリカルボキシレート(1:1)であり、その開示は、参照により本明細書に組み込まれるものとする。
【0125】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第3,822,287号に開示されているセントクロマン(centchroman):(1−(2−((4−(−メトキシ−2,2,ジメチル−3−フェニル−クロマン−4−イル)−フェノキシ)−エチル)−ピロリジンであり、その開示は、参照により本明細書に組み込まれるものとする。レボルメロキシフェンも好ましい。
【0126】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,839,155号に開示されているイドキシフェン:(E)−1−(2−(4−(1−(4−ヨード−フェニル)−2−フェニル−ブタ−1−エニル)−フェノキシ)−エチル)−ピロリジノンであり、その開示は、参照により本明細書に組み込まれるものとする。
【0127】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第5,488,058号に開示されている2−(4−メトキシ−フェニル)−3−[4−(2−ピペリジン−1−イル−エトキシ)−フェノキシ]−ベンゾ[b]チオフェン−6−オールであり、その開示は、参照により本明細書に組み込まれるものとする。
【0128】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第5,484,795号に開示されている6−(4−ヒドロキシ−フェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−ベンジル)−ナフタレン−2−オールであり、その開示は、参照により本明細書に組み込まれるものとする。
【0129】
別の好ましいエストロゲン作動薬/拮抗薬は、Pfizer Inc.に譲渡されたPCT公開WO95/10513に、調製の方法と一緒に開示されている4−(2−(2−アザ−ビシクロ[2.2.1]ヘプタ−2−イル)−エトキシ)−フェニル)―(6−ヒドロキシ−2−(4−ヒドロキシ−フェニル)−ベンゾ[b]チオフェン−3−イル)−メタノンである。
【0130】
他の好ましいエストロゲン作動薬/拮抗薬は、本発明の譲受人に譲渡された米国特許第5,552,412号に記載されているような化合物を包含し、その開示は、参照により本明細書に組み込まれるものとする。その中に記載されている特に好ましい化合物は、
cis−6−(4−フルオロ−フェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
(−)−cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−1−(6’−ピロロジノエトキシ−3’−ピリジル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロナフタレン;
1−(4’−ピロリジノエトキシフェニル)−2−(4”−フルオロフェニル)−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン;
cis−6−(4−ヒドロキシフェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;および
1−(4’−ピロリジノレトキシフェニル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン
である。
【0131】
他のエストロゲン作動薬/拮抗薬は、米国特許第4,133,814号に記載されている(その開示は、参照により本明細書に組み込まれるものとする)。米国特許第4,133,814号は、2−フェニル−3−アロイル−ベンゾチオフェンおよび2−フェニル−3−アロイルベンゾチオフェン−1−オキシドの誘導体を開示している。
【0132】
当業者は、骨量増加薬とも呼ばれる他の骨同化薬を、本発明の化合物と併せて使用することができることを理解するであろう。骨量増加薬は、World Health Organization Study World Health Organization、「Assessment of Fracture Risk and its Application to Screening for Postmenopausal Osteoporosis(1994).Report of a WHO Study Group.World Health Organization Technical Series 843」に詳述されているような骨折閾値以上であるレベルまで骨量を増加させる化合物である。
【0133】
任意のプロスタグランジン、またはプロスタグランジン作動薬/拮抗薬を、本発明の特定の態様における第2の化合物として使用することができる。当業者は、IGF−1、フッ化ナトリウム、副甲状腺ホルモン(PTH)、副甲状腺ホルモンの活性フラグメント、成長ホルモンまたは成長ホルモン分泌促進薬も使用できることを理解するであろう。以下の段落は、例示的な本発明の第2の化合物についてより詳細に記載している。
【0134】
任意のプロスタグランジンを、本発明の特定の態様における第2の化合物として使用することができる。プロスタグランジンという用語は、骨粗鬆症の治療に有用である天然プロスタグランジンPGD1、PGD2、PGE2、PGE1およびPGF2の類似体である化合物を指す。これらの化合物は、プロスタグランジン受容体に結合する。そのような結合は、標準的アッセイ(例えば、An S.他、Cloning and Expression of EP2 Subtype of Human Receptors for Prostaglandin E2、Biochemical and Biophysical Research Communications、1993、197(1):263〜270)で当業者により容易に決定される。
【0135】
プロスタグランジンは、基本化合物プロスタン酸に関連する脂環式化合物である。基本プロスタグランジンの炭素原子は、カルボン酸炭素原子からシクロペンチル環を経て隣接する側鎖上の末端炭素原子まで連続して付番される。普通、隣接する側鎖は、トランス配向である。シクロペンチル部分のC−9におけるオキソ基の存在は、Eクラス内のプロスタグランジンを示すが、PGE2は、C13−C14におけるトランス不飽和二重結合およびC5−C6におけるシス二重結合を含有する。
【0136】
様々なプロスタグランジンについて以下に記載および言及する。しかしながら、他のプロスタグランジンは当業者に知られているであろう。例示的なプロスタグランジンは、米国特許第4,171,331号および第3,927,197号に開示されており、その各々の開示は、参照により本明細書に組み込まれるものとする。
【0137】
Norrdin他、The Role of Prostaglandins in Bone In Vivo、Prostaglandins Leukotriene Essential Fatty Acids 41、139〜150、1990は、骨同化性プロスタグランジンの総説である。
【0138】
任意のプロスタグランジン作動薬/拮抗薬を、本発明の特定の態様における第2の化合物として使用することができる。プロスタグランジン作動薬/拮抗薬という用語は、プロスタグランジン受容体に結合し(例えば、An S.他、Cloning and Expression of EP2 Subtype of Human Receptors for Prostaglandin E2、Biochemical and Biophysical Research Communications、1993、197(1):263〜270)、in vivoでプロスタグランジンの作用を模倣する(例えば、骨形成を刺激し、骨量を増加させる)化合物を指す。そのような作用は、標準的アッセイで当業者により容易に決定される。Eriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London 1994、1〜296ページ)。様々なこれらの化合物について以下に記載および言及する。しかしながら、他のプロスタグランジン作動薬/拮抗薬は当業者に知られているであろう。例示的なプロスタグランジン作動薬/拮抗薬を以下の通り開示する。
【0139】
本発明の譲受人に譲渡された米国特許第3,932,389号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な2−デスカルボキシ−2−(テトラゾール−5−イル)−11−デスオキシ−15−置換−ω−ペンタノルプロスタグランジンを開示している。
【0140】
本発明の譲受人に譲渡された米国特許第4,018,892号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な16−アリール−13,14−ジヒドロ−PGE2 p−ビフェニルエステルを開示している。
【0141】
本発明の譲受人に譲渡された米国特許第4,219,483号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な2,3,6−置換−4−ピロンを開示している。
【0142】
本発明の譲受人に譲渡された米国特許第4,132,847号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な2,3,6−置換−4−ピロンを開示している。
【0143】
米国特許第4,000,309号(その開示は、参照により本明細書に組み込まれるものとする)に、骨形成活性に有用な16−アリール−13,14−ジヒドロ−PGE2 p−ビフェニルエステルを開示している。
【0144】
米国特許第3,982,016号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な16−アリール−13,14−ジヒドロ−PGE2 p−ビフェニルエステルを開示している。
【0145】
米国特許第4,621,100号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な置換シクロペンタンを開示している。
【0146】
米国特許第5,216,183号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用なシクロペンタノンを開示している。
【0147】
フッ化ナトリウムを、本発明の特定の態様における第2の化合物として使用することができる。フッ化ナトリウムという用語は、そのすべての形態のフッ化ナトリウム(例えば、徐放性フッ化ナトリウム、持続放出性フッ化ナトリウム)を指す。持続放出性フッ化ナトリウムは、米国特許第4,904,478号に開示されており、その開示は、参照により本明細書に組み込まれるものとする。フッ化ナトリウムの活性は、生物学的プロトコル(例えば、Eriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London1994、1〜296を参照)で当業者により容易に決定される。
【0148】
骨形態形成タンパク質を、本発明の第2の化合物として使用することができる(例えば、Ono、他、Promotion of the Osteogenetic Activity of Recombinant Human Bone Morphogenetic Protein by Prostaglandin E1、Bone、1996、19(6)、581〜588を参照)。
【0149】
任意の副甲状腺ホルモン(PTH)を、本発明の特定の態様における第2の化合物として使用することができる。副甲状腺ホルモンという用語は、骨形成を刺激し、骨量を増加させる副甲状腺ホルモン、そのフラグメントまたは代謝産物およびその構造類似体を指す。副甲状腺ホルモン関連ペプチド、ならびに副甲状腺関連ペプチドの活性フラグメントおよび類似体も包含される(PCT公開WO94/01460を参照)。そのような骨同化機能活性は、標準的アッセイ(例えば、Eriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London 1994、1〜296を参照)で当業者により容易に決定される。様々なこれらの化合物について以下に記載および言及する。しかしながら、他の副甲状腺ホルモンは当業者に知られているであろう。例示的な副甲状腺ホルモンは、以下の参考文献に開示されている。
【0150】
「Human Parathyroid Peptide Treatment of Vertebral Osteoporosis」、Osteoporosis Int.、3、(Supp 1):199〜203。
【0151】
「PTH 1−34 Treatment of Osteoporosis with Added Hormone Replacement Therapy:Biochemical,Kinetic and Histological Responses」Osteoporosis Int.1:162〜170。
【0152】
任意の成長ホルモンまたは成長ホルモン分泌促進薬を、本発明の特定の態様における第2の化合物として使用することができる。成長ホルモン分泌促進薬という用語は、成長ホルモンの放出を刺激するか、または成長ホルモンの作用を模倣する(例えば、骨量の増加につながる骨形成を高める)化合物を指す。そのような作用は、当業者によく知られている標準的アッセイで当業者により容易に決定される。様々なこれらの化合物は、以下の公開されたPCT特許出願:WO95/14666;WO95/13069;WO94/19367;WO94/13696;およびWO95/34311に開示されている。しかしながら、他の成長ホルモンまたは成長ホルモン分泌促進薬は当業者に知られているであろう。
【0153】
特に、好ましい成長ホルモン分泌促進薬は、N−[1(R)−[1,2−ジヒドロ−1−メタンスルホニルスピロ[3H−インドール−3,4’−ピペリジン]−1’−イル]カルボニル]−2−(フェニルメチルオキシ)エチル]−2−アミノ−2−メチルプロパンアミド:MK−677である。
【0154】
他の好ましい成長ホルモン分泌促進薬は、
2−アミノ−N−(2−(3a−(R)−ベンジル−2−メチル−3−オキソ−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ−[4,3−c]ピリジン−5−イル)−1−(R)−ベンジルオキシメチル−2−オキソ−エチル)−イソブチルアミドまたはそのL−酒石酸塩;
2−アミノ−N−(1−(R)−ベンジルオキシメチル−2−(3a−(R)−(4−フルオロ−ベンジル)−2−メチル−3−オキソ−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ[4,3−c]ピリジン−5−イル)−2−オキソ−エチル)イソブチルアミド;
2−アミノ−N−(2−(3a−(R)−ベンジル−3−オキソ−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ[4,3−c]ピリジン−5−イル)−1−(R)ベンジルオキシメチル−2−オキソ−エチル)イソブチルアミド;および
2−アミノ−N−(1−(2,4−ジフルオロ−ベンジルオキシメチル)−2−オキソ−2−(3−オキソ−3a−ピリジン−2−イルメチル−2−(2,2,2−トリフルオロ−エチル)−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ[4,3−c]ピリジン−5−イル)−エチル)−2−メチル−プロピオンアミド
を包含する。
【0155】
本明細書に記載されている化合物を調製するのに有用な調製方法の一部は、遠く離れた官能基(例えば、式Iの前駆体における第一級アミン、第二級アミン、カルボキシル)の保護を必要とすることがある。そのような保護の必要性は、遠く離れた官能基の性質および調製方法の条件に応じて異なるであろう。そのような保護の必要性は、当業者により容易に決定される。そのような保護/脱保護方法の使用も、当業者の範囲内にある。保護基およびそれらの使用の一般的な説明については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照されたい。
【0156】
本発明の化合物、それらのプロドラッグならびに薬学的に許容できる前記化合物およびプロドラッグの塩はすべて、脊椎動物、例えば、哺乳動物、特にヒトにおいて骨形成を刺激し、骨量を増加させる薬剤として治療的使用に適応している。骨形成は、骨粗鬆症および骨関連障害の発症と密接に関連していることから、これらの化合物、それらのプロドラッグならびに薬学的に許容できる前記化合物および前記プロドラッグの塩は、骨に対するそれらの作用により、骨粗鬆症を予防、阻止および/または回復させる。
【0157】
脊椎動物、例えば、哺乳類(例えば、ヒト、特に女性)において低骨量を示す状態(例えば、骨粗鬆症)を治療する際の医薬としての本発明化合物の有用性は、in vivoアッセイ、受容体結合アッセイ、サイクリックAMPアッセイおよび骨折治癒アッセイ(それらはすべて、以下に記載される)を包含する従来のアッセイにおいて本発明の化合物の活性により証明される。in viovアッセイ(当業者の範囲内にある適切な改変を含む)を用い、他の同化薬ならびに本発明のプロスタグランジン作動薬の活性を決定することができる。エストロゲン作動薬/拮抗薬プロトコルを用い、特にエストロゲン作動薬/拮抗薬および他の抗吸収薬の活性を決定することができる(当業者の範囲内にある適切な改変を含む)。以下に記載される組合せおよび逐次処理プロトコルは、本明細書に記載されている同化薬(例えば、本発明の化合物)と抗吸収薬(例えば、エストロゲン作動薬/拮抗薬)の組合せの有用性を証明するのに有用である。そのようなアッセイは、それにより、本発明の化合物(または、本明細書に記載されている他の同化薬および抗吸収薬)の活性を、お互いに対して、および他の知られている化合物の活性と比較することができる手段も提供する。これらの比較の結果は、そのような疾患を治療するために、脊椎動物、例えば、ヒトを包含する哺乳類における用量レベルを決定するのに有用である。
【0158】
同化薬in vivoアッセイ
骨形成を刺激し、骨量を増加させる同化骨薬の活性は、無処置の雄性または雌性ラット、性ホルモン欠乏の雄性(精巣摘出)または雌性(卵巣切除)ラットでテストすることができる。
【0159】
様々な齢(3カ月齢など)の雄性または雌性ラットを試験で使用することができる。ラットは、無処置であるか、または去勢(卵巣切除または精巣摘出)されており、30日間にわたって様々な投与量(1、3または10mg/kg/日)でプロスタグランジン作動薬を皮下注射または胃管投与される。去勢ラットにおいて、治療は、手術の翌日に(骨喪失を予防する目的で)または骨喪失が既に起きた時点で(骨量を回復する目的で)開始する。試験中、すべてのラットに、水ならびにカルシウム1.46%、リン0.99%およびビタミンD34.96IU/gを含有するペレット市販飼料(Teklad Rodent Diet #8064、Harlan Teklad、Madison、WI)を自由に摂取させる。すべてのラットに屠殺前12日目および2日目にカルセイン10mg/kgを皮下注射する。ラットを屠殺する。以下のエンドポイントを決定する:
【0160】
大腿骨塩測定:
各ラットからの右大腿骨を、剖検時に摘出し、「Regional High Resolution Scan」ソフトウェア(Hologic Inc.、Waltham、MA)を備えた二重エネルギーX線吸収法(DXA、QDR1000/W、Hologic Inc.、Waltham、MA)を用いてスキャンする。スキャン領域サイズは、5.08×1.902cmであり、解像度は、0.0254×0.0127cmであり、スキャン速度は、7.25mm/秒である。大腿骨スキャン画像を分析し、全大腿骨(WF)、遠位大腿骨幹端(DFM)、大腿骨幹軸(FS)、および近位大腿骨(PF)の、骨面積、骨塩量(BMC)、および骨塩密度(BMD)を決定する。
【0161】
脛骨組織形態計測分析:
右脛骨を、剖検時に摘出し、筋肉を切り離し、3つの部分に切り分ける。近位脛骨および脛骨幹軸を、70%エタノール中で固定し、段階的濃度のエタノール中で脱水し、アセトン中で脱脂し、次いで、メタクリル酸メチル(Eastman Organic Chemicals、Rochester、NY)に包埋する。
【0162】
厚さ4および10μmで、近位脛骨幹端の前部切片を、Reichert−Jung Polycut Sミクロトームを用いて切り取る。4μmの切片を、変法マッソントリクローム染色で染色し、一方、10μmの切片は、未染色のままとする。各ラットからの1つの4μmの切片および1つの10μmの切片を、海綿骨組織形態計測のために使用する。
【0163】
厚さ10μmで、脛骨幹の断面切片を、Reichert−Jung Polycut Sミクロトームを用いて切り取る。これらの切片を、皮質骨組織形態計測分析のために使用する。
【0164】
海綿骨組織形態計測:Bioquant OS/2組織形態計測システム(R&M Biometrics,Inc.、Nashville、TN)を、成長板−骨端接合部より1.2mm〜3.6mm遠位の近位脛骨幹端の二次海綿骨の静的および動的組織形態計測測定のために使用する。測定を二次海綿骨に限定するため、脛骨幹端領域の最初の1.2mmを除外することが必要である。4μmの切片を用い、骨体積、骨構造、および骨吸収に関連する指数を測定し、一方、10μmの切片を用い、骨形成および骨代謝回転に関連する指数を測定する。
【0165】
I)骨梁の体積および構造に関連する測定および計算:(1)総骨幹端面積(TV、mm2):成長板−骨端接合部より1.2mm〜3.6mm遠位の骨幹端面積。(2)骨梁面積(BV、mm2):TV内の柵状織の総面積。(3)骨梁周囲長(BS、mm):柵状織の総周囲長の長さ。(4)骨梁体積(BV/TV、%):BV/TV×100。(5)骨梁数(TBN、#/mm):1.199/2×BS/TV。(6)骨梁厚(TBT、μm):(2000/1.199)×(BV/BS)。(7)骨梁間隔(TBS、μm):(2000/1.199)×(TV−BV)。
【0166】
II)骨吸収に関連する測定および計算:(1)破骨細胞数(OCN、#):総骨幹端面積内の破骨細胞の総数。(2)破骨細胞周囲長(OCP、mm):破骨細胞により覆われた骨梁周囲長の長さ。(3)破骨細胞数/mm(OCN/mm、#/mm):OCN/BS。(4)破骨細胞周囲長率(%OCP、%):OCP/BS×100。
【0167】
III)骨の形成および代謝回転に関連する測定および計算:(1)単一カルセイン標識周囲長(SLS、mm):1回のカルセイン標識で標識された骨梁周囲長の全長。(2)二重カルセイン標識周囲長(DLS、mm):2回のカルセイン標識で標識された骨梁周囲長の全長。(3)標識間幅(ILW、μm):2回のカルセイン標識間の平均距離。(4)石灰化周囲長率(PMS、%):(SLS/2+DLS)/BS×100。(5)石灰化速度(mineral apposition rate)(MAR、μm/日):ILW/標識間隔。(6)骨形成速度/表面基準(ref.)(BFR/BS、μm2/d/μm):(SLS/2+DLS)×MAR/BS。(7)骨代謝回転速度(BTR、%/y):(SLS/2+DLS)×MAR/BV×100。
【0168】
皮質骨組織形態計測:BioquantOS/2組織形態計測システム(R&M Biometrics,Inc.、Nashville、TN)を、脛骨幹軸皮質骨の静的および動的組織形態計測のために用いる。総組織面積、骨髄腔面積、骨膜周囲長、内皮質周囲長、単一標識周囲長、二重標識周囲長、ならびに骨膜表面と内皮質表面の両方の標識間幅を測定し、皮質骨面積(総組織面積−骨髄腔面積)、皮質骨面積率(皮質面積/総組織面積×100)、骨髄面積率(骨髄腔面積/総組織面積×100)、骨膜および内皮質の標識周囲長率[(単一標識周囲長/2+二重標識周囲長)/総周囲長×100]、石灰化速度(標識間幅/間隔)、ならびに骨形成速度[石灰化速度×[(単一標識周囲長/2+二重標識周囲長)/総周囲長]を計算する。
【0169】
統計値
統計値は、StatView4.0パッケージ(Abacus Concepts,Inc.、Berkeley、CA)を用いて計算することができる。分散分析(ANOVA)検定と、続く、フィッシャーのPLSD(Stat View、Abacus Concepts Inc.、1918 Bonita Ave、Berkeley、CA94704−1014)を用い、群間の差を比較する。
【0170】
組換えヒトEP2およびEP4受容体を安定に過剰発現する293−S細胞系におけるcAMP上昇の測定
ヒトEP2およびEP4受容体の完全オープンリーディングフレームに相当するcDNAを、公表された配列(Regan,J.W.Bailey,T.J.Pepperl,D.J.Pierce,K.L.Bogardus,A.M.Donello,J.E.Fairbairn,C.E.Kedzie,K.M.Woodward,D.F.およびGil,D.W.1994 Cloning of a Novel Human Prostaglandin Receptor with Characteristics of the Pharmacologically Defined EP2 Subtype。Mol.Pharmacology 46:213〜220;およびBastien,L.、Sawyer,N.、Grygorczyk,R.、Metters,K.、およびAdam,M.1994 Cloning,Functional Expression,and Characterization of the Human Prostagrandin E2 Receptor EP2 Subtype。J.Biol.Chem.Vol269、16:11873〜11877)および一次ヒト腎細胞(EP2)または一次ヒト肺細胞(EP4)由来のRNAに基づくオリゴヌクレオチドプライマーを鋳型として用いる逆転写酵素ポリメラーゼ連鎖反応により作製する。cDNAを、pcDNA3(Invitrogen Corporation、3985B Sorrento Valley Blvd.、San Diego、CA92121)の多重クローニング部位中にクローニングし、それを用い、リン酸カルシウム共沈を介して293−Sヒト胚性腎細胞にトランスフェクトする。G418耐性コロニーを拡張させ、特異的[3H]PGE2結合について調べる。高レベルの特異的[3H]PGE2結合を示すトランスフェクタントを、スキャッチャード解析によりさらに特徴付け、PGE2についてBmaxおよびKdを決定する。化合物スクリーニングのために選択された系は、PGE2(EP2)の場合、細胞当たり約338,400個の受容体およびKd=12nMを、およびPGE2(EP4)の場合、細胞当たり約256,400個の受容体およびKd=2.9nMを有する。親の293−S細胞における両受容体の構成的発現は無視できる。細胞は、ウシ胎児血清(最終10%)およびG418(最終700ug/ml)を添加したRPMI中に維持する。
【0171】
293−S/EP2系および293−S/EP4系におけるcAMP応答は、Ca++およびMg++欠乏のPBS1ml中で激しく叩くことにより細胞を培養フラスコから引き離し、1×106細胞/mlの最終濃度まで無血清RPMIを加え、3−イソブチル−1−メチルキサンチン(IBMX)を1mMの最終濃度まで加えることにより測定する。直ちに、細胞懸濁液1ミリリットルを、個々の2mlのスクリューキャップ微小遠心管に等分し、37℃、5%CO2、相対湿度95%にて、蓋を取って10分間にわたってインキュベートする。次いで、DMSOまたはエタノールの最終濃度が1%になるように、テストされる化合物を1:100の希釈度で細胞に加える。化合物を加えた直後に、管に蓋をし、2回逆さにすることにより混ぜ、12分間にわたって37℃においてインキュベートする。次いで、試料を、10分間にわたって100℃においてインキュベートすることにより溶解し、直ちに、5分間にわたって氷上で冷却する。細胞残屑を、5分間にわたって1000×gにて遠心分離することによりペレット化し、澄明にしたライセートを新たな試験管に移す。cAMP濃度は、澄明にしたライセートをcAMP RIAアッセイ緩衝液(キットに包含されている)中で1:10に希釈した後、市販のcAMPラジオイムノアッセイキットRIA(NEK−033、DuPont/NEN Research Products、549 Albany St.、Boston、MA02118)を用いて測定する。通常、1 logの増分で、6〜8種類の濃度のテストすべき化合物で細胞を処理する。EC50の計算は、用量反応曲線の直線部分に対する線形回帰分析を用い、計算機で行う。
【0172】
プロスタグランジンE2受容体との結合についてのアッセイ
膜調製:すべての操作は4℃において行う。プロスタグランジンE21型受容体(EP1)、2型(EP2)、3型(EP3)または4型(EP4)受容体を発現するトランスフェクトされた細胞を収集し、緩衝液A[50mM Tris−HCl(pH7.4)、10mM MgCl2、1mM EDTA、1mM ペファブロックペプチド、(Boehringer Mannheim Corp.、Indianapolis、IN)、10uM ホスポラミドン(Phosporamidon)ペプチド、(Sigma、St.Louis、MO)、1uMペプスタチンAペプチド、(Sigma、St.Louis、MO)、10uM エラスタチナールペプチド、(Sigma、St.Louis、MO)、100uMアンチパインペプチド、(Sigma、St.Louis、MO)]中に1ml当たり200万個の細胞になるように懸濁する。細胞を、2回の15秒バーストでBranson Sonifier(Model #250、Branson Ultrasonics Corporation、Danbury、CT)による超音波処理により溶解する。溶解しない細胞および残屑を、10分間にわたって100×gにて遠心分離することにより除去する。次いで、膜を、30分間にわたって45,000×gにて遠心分離することにより収集する。ペレット化した膜を、1ml当たりタンパク質3〜10mgになるように再懸濁し、ブラッドフォードの方法[Bradford,M.、Anal.Biochem.、72、248(1976)]でタンパク質濃度を決定する。次いで、再懸濁した膜を、使用するまで−80℃において凍結保存する。
【0173】
結合アッセイ:上記のように調製される凍結膜を解凍し、上記の緩衝液A中で1ml当たりタンパク質1mgになるように希釈する。1容積の膜調製物を、緩衝液A中で、0.05容積のテスト化合物または緩衝液および1容積の3nM 3H−プロスタグランジンE2(#TRK431、Amersham、Arlington Heights、IL)と混ぜ合わせる。この混合物(全容積205μL)を、25℃において1時間にわたってインキュベートする。次いで、膜を、Tomtecハーベスター(Model Mach II/96、Tomtec、Orange、CT)を用い、タイプGF/Cガラス繊維フィルター(#1205−401、Wallac、Gaithersburg、MD)に通して濾過することにより回収する。結合した3H−プロスタグランジンE2を有する膜は、フィルターにより捕捉されるが、緩衝液および結合していない3H−プロスタグランジンE2は、フィルターを通過して廃液中に入る。次いで、各試料を[50mM Tris−HCl(pH7.4)、10mM MgCl2、1mM EDTA]3mlで3回洗浄する。次いで、フィルターを、電子レンジ内で加熱することにより乾燥させる。膜に結合している3H−プロスタグランジンの量を測定するため、乾燥したフィルターを、シンチレーション液と一緒にプラスチックバッグに入れ、LKB 1205 Betaplateリーダー(Wallac、Gaithersburg、MD)でカウントする。IC50は、特異的に結合した3H−プロスタグランジンE2の50%を置き換えるのに必要とされるテスト化合物の濃度から決定される。
【0174】
完全長EP1受容体は、Funk他、Journal of Biological Chemistry、1993、268、26767〜26772に開示されているように作製する。完全長EP2受容体は、Regan他、Molecular Pharmacology、1994、46、213〜220に開示されているように作製する。完全長EP3受容体は、Regan他、British Journal of Pharmacology、1994、112、377〜385に開示されているように作製する。完全長EP4受容体は、Bastien、Journal of Biological Chemistry、1994、269、11873〜11877に開示されているように作製する。これらの完全長受容体を用い、EP1、EP2、EP3およびEP4受容体を発現する293S細胞を調製する。
【0175】
ヒトEP1、EP2、EP3またはEP4プロスタグランジンE2受容体を発現する293S細胞は、当業者に知られている方法に従って作製する。通常、公表されている完全長受容体の5’および3’末端に対応するPCR(ポリメラーゼ連鎖反応)プライマーを、上記に開示されているよく知られている方法に従って作製し、ソースとしてのヒト腎(EP1用)、ヒト肺(EP2用)、ヒト肺(EP3用)またはヒト白血球(EP4用)に由来する全RNAを用いるRT−PCRで使用する。PCR産物を、TAオーバーハング法によりpCR2.1(Invitrogen、Carlsbad、CA)中にクローニングし、クローン化された受容体の同一性をDNA配列決定により確認する。
【0176】
293S細胞(Mayo、Dept.of Biochemistry、Northwestern Univ.)に、エレクトロポレーションにより、pcDNA3中のクローン化された受容体をトランスフェクトする。受容体を発現する安定な細胞系は、G418をトランスフェクトした細胞の選択に続いて確立される。
【0177】
最大数の受容体を発現するクローン細胞系は、コンペティターとして非標識PGE2を用いるホールセルの3H−PGE2結合アッセイに続いて選択される。
【0178】
骨折治癒アッセイ
全身投与後の骨折治癒に対する影響についてのアッセイ
骨折技法:3カ月齢のSprage−Dawleyラットを、ケタミンで麻酔する。右の脛骨または大腿骨の近位部分の前内側面に1cmの切開を作製する。以下は、脛骨手術技法について記載している。切開を骨まで到達させ、1mmの孔を、前縁から2mm内側の脛骨粗面の遠位面から4mm近位に開ける。骨髄内釘固定は、0.8mmのステンレス鋼管で行う(最大点荷重36.3N、最大点剛性61.8N/mm、骨と同じ条件下でテストする)。髄管のリーミングは行わない。標準化された閉鎖骨折を、顎が尖っていない特別に設計された調整可能な鉗子を用いる三点曲げにより、脛腓接合部の2mm上方に作製する。軟組織の損傷を最小限に抑えるため、骨折の位置をずらさないように注意する。皮膚を、モノフィラメントナイロン縫合糸で閉じる。手術は、無菌条件下で行う。釘固定直後にすべての骨折のラジオグラフをとり、指定した骨幹領域外に骨折のある、または釘の位置がずれているラットは除外する。残りの動物を、骨折治癒をテストするために、1時点につき各サブグループ当たり10〜12匹の以下のグループに無作為に分ける。第1グループには、1ml/ラットでビヒクル(水:100%エタノール=95:5)を連日胃管投与し、一方、他のラットには、10、20、40および80日間にわたって、テストすべき化合物0.01〜100mg/kg/日(1ml/ラット)を連日胃管投与する。
【0179】
10、20、40および80日目に、各グループからのラット10〜12匹を、ケタミンで麻酔し、放血により屠殺する。両方の脛腓骨を切開により摘出し、すべての軟組織をはぎ取る。各グループにつき5〜6匹のラットからの骨を、組織学的分析のために70%エタノール中に保存し、各グループにつき別の5〜6匹のラットからの骨を、行われるラジオグラフおよび生体力学的テストのために緩衝リンゲル液(+4℃、pH7.4)中に保存する。
【0180】
組織学的分析:骨折した骨の組織学的分析のための方法は、MosekildeおよびBak(The Effects of Growth Hormone on Fracture Healing in Rats:A Histological Description。Bone、14:19〜27、1993)により以前に公表されている。手短に言うと、骨折側を、骨折線の各側までのこぎりで8mm切り、脱灰せずにメタクリル酸メチルに包埋し、8μmの厚さでReichert−Jung Polycutミクロトームで前部切片を切り取る。マッソントリクローム染色した中前部切片(脛骨と腓骨の両方を包含する)を、治療の有無による骨折治癒に対する細胞および組織の応答を可視化するために使用する。シリウスレッド染色した切片を用い、仮骨構造の特徴を立証し、骨折部位における線維性骨と層板骨を区別する。以下の測定を行う:(1)骨折間隙−骨折における皮質骨末端間の最短距離として測定される、(2)仮骨長および仮骨直径、(3)仮骨の総骨体積面積、(4)仮骨面積内組織面積当たりの骨組織、(5)仮骨における線維組織、および(6)仮骨における軟骨面積。
【0181】
生体力学的分析:生体力学的分析のための方法は、BakおよびAndreassen(The Effects of Aging on Fracture Healing in Rats。Calcif Tissue Int 45:292〜297、1989)により以前に公表されている。手短に言うと、生体力学的テストに先立って、すべての骨折のラジオグラフをとる。治癒しつつある骨折の力学的特性を、破壊的三点または四点曲げ手順により分析する。最大点荷重、剛性、最大点荷重時のエネルギー、最大点荷重時の歪み、および最大点応力を測定する。
【0182】
局所投与後の骨折治癒に対する影響についてのアッセイ
骨折技法:約2歳の雌性または雄性ビーグル犬を、麻酔下で試験に用いる。横方向の橈骨骨折を、Lenehan他(Lenehan,T.M.;Balligand,M.;Nunamaker,D.M.;Wood,F.E.:Effects of EHDP on Fracture Healing in Dogs。J Orthop Res 3:499〜507;1985)により記載されているような三点曲げの緩徐な連続荷重により作製する。ワイヤを骨折部位に通して引っ張り、骨の完全な解剖学的破壊を保証する。その後、骨折部位へのプロスタグランジン作動薬の局所送達を、10、15または20週間にわたって、徐放性ペレットにより送達される化合物の徐放により、またはペーストゲルの溶液または懸濁液などの適当な製剤中の化合物の投与により行う。
【0183】
組織学的分析:骨折した骨の組織学的分析のための方法は、Peter他(Peter,C.P.;Cook,W.O.;Nunamaker,D.M.;Provost,M.T.;Seedor,J.G.;Rodan,G.A.Effects of alendronate on fracture healing and bone remodeling in dogs。J.Orthop.Res.14:74〜70、1996)ならびにMosekildeおよびBak(The Effects of Growth Hormone on Fracture Healing in Rats:A Histological Description。Bone、14:19〜27、1993)により以前に公表されている。手短に言うと、屠殺後、骨折した側を、骨折線の各側までのこぎりで3cm切り、脱灰せずにメタクリル酸メチルに包埋し、8μmの厚さでReichert−Jung Polycutミクロトームで前部切片を切り取る。マッソントリクローム染色した中前部切片(脛骨と腓骨の両方を包含する)を、治療の有無による骨折治癒に対する細胞および組織の応答を可視化するために使用する。シリウスレッド染色した切片を用い、仮骨構造の特徴を立証し、骨折部位における線維性骨と層板骨を区別する。以下の測定を行う:(1)骨折間隙−骨折における皮質骨末端間の最短距離として測定される、(2)仮骨長および仮骨直径、(3)仮骨の総骨体積面積、(4)仮骨面積内組織面積当たりの骨組織、(5)仮骨における線維組織、および(6)仮骨における軟骨面積。
【0184】
生体力学的分析:生体力学的分析のための方法は、BakおよびAndreassen(The Effects of Aging on Fracture Healing in Rats。Calcif Tissue Int 45:292〜297、1989)およびPeter他(Peter,C.P.;Cook,W.O.;Nunamaker,D.M.;Provost,M.T.;Seedor,J.G.;Rodan,G.A.Effects of Alendronate On Fracture Healing And Bone Remodeling In Dogs。J.Orthop.Res.14:74〜70、1996)により以前に公表されている。手短に言うと、生体力学的テストに先立って、すべての骨折のラジオグラフをとる。治癒しつつある骨折の力学的特性を、破壊的三点または四点曲げ手順により分析する。最大点荷重、剛性、最大点荷重時のエネルギー、最大点荷重時の歪み、および最大点応力を測定する。
【0185】
エストロゲン作動薬/拮抗薬プロトコル
エストロゲン作動薬/拮抗薬は、骨代謝回転を阻害し、エストロゲン欠乏誘発性骨喪失を予防する化合物の一種である。卵巣切除ラット骨喪失モデルは、閉経後骨喪失のモデルとして広く使用されてきた。このモデルを使用して、骨喪失を予防し骨吸収を阻害する際のエストロゲン作動薬/拮抗薬化合物の有効性をテスト試験することができる。
【0186】
様々な齢(5カ月齢など)のSprague−Dawley雌性ラット(Charles River、Wilmington、MA)を、これらの試験で使用する。実験期間中、ラットを、1匹ずつ20cm×32cm×20cmのケージに収容する。すべてのラットに、水ならびにカルシウム0.97%、リン0.85%、およびビタミンD31.05IU/gを含有するペレット市販飼料(Agway ProLab 3000、Agway Country Food,Inc.、Syracuse、NY)を自由に摂取させる。
【0187】
一群のラット(8〜10匹)に偽手術をし、ビヒクル(10%エタノールおよび90%食塩水、1ml/日)で経口的に治療し、一方、残りのラットは、両側卵巣切除し(OVX)、一定期間(4週間など)にわたってビヒクル(経口)、17β−エストラジオール(Sigma、E−8876、E2、30μg/kg、連日皮下注射)、またはエストロゲン作動薬/拮抗薬(連日経口的に5、10または20mg/kgドロロキシフェンなど)で治療する。骨組織における動的変化を調べるため、すべてのラットに屠殺する12日および2日前にカルセイン(蛍光色素骨マーカー)10mg/kgを皮下注射する。4週間の治療後、ラットを屠殺し、剖検する。以下のエンドポイントを決定する。
【0188】
体重増加:剖検時の体重−手術時の体重。
【0189】
子宮の重量および組織学:剖検中に各ラットから子宮を摘出し、直ちに秤量する。その後、子宮断面組織面積、間質厚、および内腔上皮厚などの組織学的測定のために子宮を処理する。
【0190】
総血清コレステロール:血液を、心臓穿刺により採取し、4℃において凝固させ、次いで、10分間にわたって2,000gにて遠心分離する。血清試料を、高性能コレステロール熱量測定アッセイ(Boehringer Mannheim Biochemicals、Indianapolis、IN)を用いて総血清コレステロールについて分析する。
【0191】
大腿骨塩測定:各ラットからの右大腿骨を、剖検時に摘出し、「Regional High Resolution Scan」ソフトウェア(Hologic Inc.、Waltham、MA)を備えた二重エネルギーX線吸収法(DEXA、QDR 1000/W、Hologic Inc.、Waltham、MA)を用いてスキャンする。スキャン範囲サイズは、5.08×1.902cmであり、解像度は、0.0254×0.0127cmであり、スキャン速度は、7.25mm/秒である。大腿骨スキャン画像を分析し、全大腿骨(WF)、遠位大腿骨幹端(DFM)、大腿骨幹軸(FS)、および近位大腿骨(PF)の、骨面積、骨塩量(BMC)、および骨塩密度(BMD)を決定する。
【0192】
近位脛骨幹端海綿骨組織形態計測分析:右脛骨を、剖検時に摘出し、筋肉を切り離し、3つの部分に切り分ける。近位脛骨を、70%エタノール中で固定し、段階的濃度のエタノール中で脱水し、アセトン中で脱脂し、次いで、メタクリル酸メチル(Eastman Organic Chemicals、Rochester,NY)に包埋する。厚さ4および10μmで、近位脛骨幹端の前部切片を、Reichert−Jung Polycut Sミクロトームを用いて切り取る。各ラットからの1つの4μmの切片および1つの10μmの切片を、海綿骨組織形態計測のために用いる。4μmの切片は、変法マッソントリクローム染料で染色し、一方、10μmの切片は、未染色のままとする。
【0193】
Bioquant OS/2組織形態計測システム(R&M Biometrics,Inc.、Nashville、TN)を、成長板−骨端接合部より1.2mm〜3.6mm遠位の近位脛骨幹端の二次海綿骨の静的および動的組織形態計測測定のために使用する。測定を二次海綿骨に限定するため、脛骨幹端領域の最初の1.2mmを除外することが必要である。4μmの切片を用い、骨体積、骨構造、および骨吸収に関連する指数を測定し、一方、10μmの切片を用い、骨形成および骨代謝回転に関連する指数を測定する。
【0194】
I.骨梁の体積および構造に関連する測定および計算:
1.総骨幹端面積(TV、mm2):成長板−骨端接合部より1.2mm〜3.6mm遠位の骨幹端面積。
2.骨梁面積(BV、mm2):TV内の柵状織の総面積。
3.骨梁周囲長(BS、mm):柵状織の総周囲長の長さ。
4.骨梁体積(BV/TV、%):BV/TV×100。
5.骨梁数(TBN、#/mm):1.199/2×BS/TV。
6.骨梁厚(TBT、μm):(2000/1.199)×(BV/BS)。
7.骨梁間隔(TBS、μm):(2000/1.199)×(TV−BV)。
【0195】
II.骨吸収に関連する測定および計算:
1.破骨細胞数(OCN、#):総骨幹端面積内の破骨細胞の総数。
2.破骨細胞周囲長(OCP、mm):破骨細胞により覆われた骨梁周囲長の長さ。
3.破骨細胞数/mm(OCN/mm、#/mm):OCN/BS。
4.破骨細胞周囲長率(%OCP、%):OCP/BS×100。
【0196】
III.骨の形成および代謝回転に関連する測定および計算:
1.単一カルセイン標識周囲長(SLS、mm):1回のカルセイン標識で標識された骨梁周囲長の全長。
2.二重カルセイン標識周囲長(DLS、mm):2回のカルセイン標識で標識された骨梁周囲長の全長。
3.標識間幅(ILW、μm):2回のカルセイン標識間の平均距離。
4.石灰化周囲長率(PMS、%):(SLS/2+DLS)/BS×100。
5.石灰化速度(MAR、μm/日):ILW/標識間隔。
6.骨形成速度/表面基準(ref.)(BFR/BS、μm2/d/μm):(SLS/2+DLS)×MAR/BS。
7.骨代謝回転速度(BTR、%/y):(SLS/2+DLS)×MAR/BV×100。
【0197】
統計値
統計値は、StatView4.0パッケージ(Abacus Concepts,Inc.、Berkeley、CA)を用いて計算することができる。分散分析(ANOVA)検定と、続く、フィッシャーのPLSD(Stat View、Abacus Concepts,Inc.、1918 Bonita Ave、Berkeley、CA94704−1014)を用い、群間の差を比較する。
【0198】
組合せ治療および順次治療プロトコル
以下のプロトコルは、当業者により変えることができることは言うまでもない。例えば、無処置の雄性または雌性ラット、性ホルモン欠乏の雄性(精巣摘出)または雌性(卵巣切除)ラットを使用することができる。さらに、様々な齢(12カ月齢など)の雄性または雌性ラットを試験で使用することができる。ラットは、無処置であるか、または去勢(卵巣切除または精巣摘出)されており、一定期間(2週〜2カ月など)にわたって様々な投与量(1、3または6mg/kg/日など)で本発明の化合物などの同化薬を投与し、続いて、一定期間(2週〜2カ月など)にわたって様々な投与量(1、5、10mg/kg/日など)でドロロキシフェンなどの抗吸収薬を投与するか、または一定期間(2週〜2カ月など)にわたって様々な投与量で同化薬と抗吸収薬の組合せ治療を行うことができる。去勢ラットにおいて、治療は、手術の翌日(骨喪失を予防する目的で)または骨喪失が既に起きた時点(骨量を回復する目的で)で開始することができる。
【0199】
ラットを、ケタミン麻酔下で屠殺する。以下のエンドポイントを決定する。
【0200】
大腿骨塩測定は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。
【0201】
腰椎骨塩測定:「Regional High Resolution Scan」ソフトウェア(Hologic Inc.、Waltham、MA)を備えた二重エネルギーX線吸収法(QDR 1000/W、Hologic Inc.、Waltham、MA)を用い、麻酔ラットにおける全腰椎および6個の腰椎(LV1〜6)の各々の骨面積、骨塩量(BMC)、および骨塩密度(BMD)を測定。ラットを、ケタミン/ロンパン(4対3の比)の混合物1ml/kgの腹腔内注射により麻酔し、ラットプラットホーム上に置く。スキャン領域サイズは、6×1.9cmであり、解像度は、0.0254×0.0127cmであり、スキャン速度は、7.25mm/秒である。全腰椎スキャン画像を取得し分析する。骨面積(BA)、および骨塩量(BMC)を測定し、骨塩密度は、全腰椎および6個の腰椎(LV1〜6)の各々について計算する(MBCをBAで除す)。
【0202】
近位脛骨幹端海綿骨組織形態計測分析は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。
【0203】
骨梁の体積および構造に関連する測定および計算は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。さらに、骨吸収に関連する測定および計算も、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。さらに、骨の形成および代謝回転に関連する測定および計算は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。さらに、得られるデータは、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されている統計操作を用いて分析する。
【0204】
腎臓再生アッセイ
腎臓再生におけるプロスタグランジン作動薬の役割は、野生型293S細胞およびEP2をトランスフェクトした293S細胞における骨形態形成タンパク質7(BMP−7)の発現を誘導するプロスタグランジンE2(PGE2)またはプロスタグランジン作動薬の能力により検討する。
【0205】
方法:293S細胞およびEP2 293S細胞を、ダルベッコ変法イーグル培地(DMEM、Gibco、BRL;Gaithersburg、MD)中で培養する。PGE2またはプロスタグランジン作動薬で処理する1日前に、細胞を、10cmの皿当たり1.5×106個の細胞の密度でプレートする。一般的に、約16〜24時間後、細胞単層を、OptiMEM(Gibco、BRL;Gaithersburg、MD)で1回洗浄し、続いて、ビヒクル(DMSO)、PGE2(10−6M)またはプロスタグランジン作動薬(10−6M)の存在下および非存在下で皿当たりOptiMEM10mLを加える。8、16および24時間目に細胞を収集し、RNAを抽出する。全RNA(20mg/レーン)のノーザンブロット分析を、32P−標識BMP−7プローブでブロットを探索することにより行う。ブロットを、32P−標識18sリボソームRNAプローブとのハイブリダイゼーションによりRNA負荷について正規化する。PGE2およびプロスタグランジン作動薬は、時間依存的にEP2 293S細胞におけるBMP−7の発現を誘導する。そのような発現誘導は、親細胞系では一般的に観察されない。腎臓再生におけるBMP−7の知られている役割を考えると、時間および受容体特異的に293S腎臓細胞におけるBMP−7発現を誘導するプロスタグランジン作動薬の能力は、腎臓再生におけるプロスタグランジン作動薬の役割を示している。
【0206】
本発明の化合物の投与は、本発明の化合物を全身的および/または局所的に(例えば、骨折、骨切り術または整形手術の部位において)送達する任意の方法を介することができる。これらの方法は、経口経路、非経口経路、十二指腸内経路などを包含する。一般的に、本発明の化合物は、経口的に投与されるが、例えば、経口投与が標的にとって不適切である場合、または患者が薬物を経口摂取することができない場合、非経口投与(例えば、静脈内、筋肉内、経皮、皮下、経直腸または髄内)を利用することができる。
【0207】
化合物は、本発明の化合物またはそれらの組成物の局所適用(例えば、骨折または骨切り術の部位への)により、骨折および骨切り術を治療することおよびそれらの治癒を促進することのために使用される。本発明の化合物は、例えば、適当な溶媒(例えば、ラッカセイ油などの油性溶媒)中の本発明化合物の軟骨成長板への注射によるか、または、切開手術の場合には、骨蝋、脱灰骨粉、ポリマー骨セメント、骨シーラントなどの適当な担体または希釈剤中のそのような化合物のそこへの局所適用により、骨折または骨切り術の部位に適用される。代替方法として、局所適用は、適当な担体または希釈剤中の化合物の溶液もしくは分散液を、ダクロン−メッシュ、ゲル−フォームおよびキールボーン(kiel bone)、もしくは補綴などの整形外科手術において従来から使用されている固体もしくは半固体のインプラントの表面上または中に、塗布または組み入れることにより行うことができる。
【0208】
本発明の化合物は、上記に記載されている同化薬または骨抗吸収薬のうちの1種または複数と組み合わせて、適当な担体または希釈剤中で骨折または骨切り術の部位に局所的に適用することもできる。
【0209】
本発明の範囲内にあるそのような組合せは、任意の順序で同時に、または順次に同時投与することができ、または薬学的に許容できる担体または希釈剤中に上記に記載されているような(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸およびそのナトリウム塩の多形性形態、そのプロドラッグまたは前記化合物もしくは前記プロドラッグの薬学的塩を含む単一医薬組成物を投与することができる。
【0210】
例えば、本発明において、骨同化薬を、3カ月〜3年にわたって単独で、または抗吸収薬と組み合わせて、続いて、3カ月〜3年にわたって抗吸収薬を単独で使用することができ、全治療サイクルを繰り返してもよい。代替方法として、骨同化薬を、3カ月〜3年にわたって単独でまたは抗吸収薬と組み合わせて、続いて、患者の人生の残りの期間にわたって抗吸収薬を単独で使用することができる。例えば、1つの好ましい投与様式において、上記に記載されているような(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸およびそのナトリウム塩の多形性形態、またはそのプロドラッグもしくは薬学的に許容できるプロドラッグの塩を1日1回投与することができ、上記に記載されているような第2の化合物(例えば、エストロゲン作動薬/拮抗薬)を、単回投与または複数回投与で連日投与することができる。代替方法として、例えば、別の好ましい投与様式において、2つの化合物を順次に投与することができ、上記に記載されているような(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸およびそのナトリウム塩化合物の多形性形態、そのプロドラッグまたは薬学的に許容できるプロドラッグの塩を、骨折閾値(World Health Organization Study「Assessment of Fracture Risk and its Application to Screening for Postmenopausal Osteoporosis(1994)。Report of a World Health Organization Study Group。World Health Organization Technical Series 843」)以上であるレベルまで骨量を増加させるのに十分な期間にわたって1日1回投与することができ、続いて、上記に記載されているような第2の化合物(例えば、エストロゲン作動薬/拮抗薬)を、単回投与または複数回投与で連日投与する。上記に記載されているような第1の化合物は、経口送達などの迅速な送達形態で1日1回投与されることが好ましい。
【0211】
いずれにしても、投与される化合物の量およびタイミングは、治療されている対象、苦痛の重症度、投与の様式および処方医師の判断により左右されるであろうことは言うまでもない。したがって、患者間の変動性のため、以下に示す用量は、指針であり、医師は、医師がその患者に適切であると考える治療(例えば、骨量増加)を達成するために薬物の投与量を設定することができる。望まれる治療の程度を考慮する際、医師は、骨量出発レベル、患者の年齢、既存疾患の存在、ならびに他の疾患(例えば、心血管疾患)の存在などの様々な要因のバランスをとらなければならない。
【0212】
一般に、骨量を骨折閾値(本明細書で以前に引用されているWorld Health Organization Studyに詳述されているような)以上であるレベルまで増加させるのに十分な量の本発明の化合物が使用される。
【0213】
一般に、上記に記載されている本発明において使用される同化薬の有効用量は、0.001〜100mg/kg/日、好ましくは0.01〜50mg/kg/日の範囲である。
【0214】
以下の段落は、様々な抗吸収薬についての好ましい用量範囲を提供する。
【0215】
使用すべき抗吸収薬の量は、骨喪失阻害薬としてのその活性により決定される。この活性は、個々の化合物の薬物動態および上記に記載されているようなプロトコル(例えば、エストロゲン作動薬/拮抗薬プロトコル)を用いる骨喪失の阻害におけるその最小有効投与量対最大有効投与量により決定される。
【0216】
一般に、抗吸収薬についての有効用量は、約0.001mg/kg/日〜約20mg/kg/日である。
【0217】
一般に、プロゲスチンについての有効用量は、1日当たり約0.1〜10mgであり、好ましい投与量は、1日当たり約0.25〜5mgである。
【0218】
一般に、ポリホスホネートについての有効用量は、標準的アッセイの骨吸収阻害薬としてのその効力により決定される。
【0219】
一部のポリホスホネートの連日投与のための範囲は、約0.001mg/kg/日〜約20mg/kg/日である。
【0220】
一般に、本発明の治療、例えば、本発明の骨吸収治療についての有効用量は、本発明のエストロゲン作動薬/拮抗薬について、0.01〜200mg/kg/日、好ましくは0.5〜100mg/kg/日の範囲である。
【0221】
特に、ドロロキシフェンについての有効用量は、0.1〜40mg/kg/日、好ましくは0.1〜5mg/kg/日である。
【0222】
特に、ラロキシフェンについての有効用量は、0.1〜100mg/kg/日、好ましくは0.1〜10mg/kg/日である。
【0223】
特に、タモキシフェンについての有効用量は、0.1〜100mg/kg/日、好ましくは0.1〜5mg/kg/日である。
【0224】
特に、2−(4−メトキシ−フェニル)−3−[4−(2−ピペリジン−1−イル−エトキシ)−フェノキシ]−ベンゾ[b]チオフェン−6−オールについての有効用量は、0.001〜1mg/kg/日である。
【0225】
特に、
cis−6−(4−フルオロ−フェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
(−)−cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−1−(6’−ピロロジノエトキシ−3’−ピリジル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロナフタレン;
1−(4’−ピロリジノエトキシフェニル)−2−(4”−フルオロフェニル)−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン;
cis−6−(4−ヒドロキシフェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;または
1−(4’−ピロリジノレトキシフェニル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン
についての有効用量は、0.0001〜100mg/kg/日、好ましくは0.001〜10mg/kg/日の範囲である。
【0226】
特に、4−ヒドロキシタモキシフェンについての有効用量は、0.0001〜100mg/kg/日、好ましくは0.001〜10mg/kg/日の範囲である。
【0227】
本発明の化合物は、一般的に、薬学的に許容できるビヒクルまたは希釈剤と一緒に本発明の化合物のうちの少なくとも1つを含む医薬組成物の形態で投与される。したがって、本発明の化合物は、任意の従来型の経口、非経口、直腸または経皮剤形で個々に、または一緒に投与することができる。
【0228】
経口投与の場合、医薬組成物は、液剤、懸濁剤、錠剤、丸剤、カプセル剤、散剤などの形態をとることができる。クエン酸ナトリウム、炭酸カルシウムおよびリン酸カルシウムなどの様々な賦形剤を含有する錠剤は、ポリビニルピロリドン、スクロース、ゼラチンおよびアカシアなどの結合剤に加えて、デンプン、好ましくはジャガイモまたはタピオカデンプンおよび特定の複雑なケイ酸塩などの様々な崩壊剤と一緒に用いられる。さらに、ステアリン酸マグネシウム、ラウリル硫酸ナトリウムおよびタルクなどの滑沢剤は、錠剤化目的に極めて有用であることが多い。類似のタイプの固体組成物も、軟質および硬質充填ゼラチンカプセルにおける充填剤として用いられ、これに関する好ましい材料は、ラクトースすなわち乳糖ならびに高分子量ポリエチレングリコールも包含する。水性懸濁剤および/またはエリキシル剤が経口投与にとって望ましい場合には、本発明の化合物を、様々な甘味剤、矯味剤、着色剤、乳化剤および/または懸濁化剤、ならびに水、エタノール、プロピレングリコール、グリセリンおよびそれらの様々な同類の組合せなどの希釈剤と組み合わせることができる。
【0229】
非経口投与の目的には、ゴマもしくはピーナッツ油または水性プロピレングリコールの溶液、ならびに対応する水溶性の塩の滅菌水溶液を用いることができる。そのような水溶液は、必要ならば、適切に緩衝されていてもよく、液体希釈剤は、まず初めに十分な食塩水またはグルコースで等張にされる。これらの水溶液は、静脈内、筋肉内、皮下および腹腔内注射目的に特に適している。これに関して、用いられる滅菌水性媒体はすべて、当業者によく知られている標準的技法により容易に得ることができる。
【0230】
経皮(例えば、局所)投与の目的には、その他の点では上記の非経口溶液に類似した希釈滅菌、水性または部分的水性水溶液(通常、約0.1%〜5%濃度で)が調製される。
【0231】
特定の量の活性成分を含む様々な医薬組成物を調製する方法は、当業者に知られているか、または本開示を踏まえて当業者に明らかであろう。医薬組成物を調製する方法の例については、Remington’s Pharmaceutical Sciences、Mack Publishing Company、Easter、Pa.、第15版(1975)を参照されたい。
【0232】
本発明の医薬組成物は、本発明の1つまたは複数の化合物を0.1%〜95%、好ましくは1%〜70%含有することができる。いずれにしても、投与すべき組成物または製剤は、治療されている対象の疾患/状態、例えば骨障害を治療するのに有効な量で、ある量の本発明の1つまたは複数の化合物を含有するであろう。
【0233】
本発明は、別々に投与することができる活性成分の組合せで治療することによる骨量の増加および維持に関する態様を有することから、本発明は、キット形態で別々の医薬組成物を組み合わせることにも関する。キットは、2つの別々の医薬組成物、すなわち、上記に記載されているような式Iの化合物、そのプロドラッグまたは薬学的に許容できる前記化合物もしくは前記プロドラッグの塩および第2の化合物を含む。キットは、分割されたボトルまたは分割されたホイルパケットなどの別々の組成物を含有するための容器手段を含むが、別々の組成物は、単一の分割されていない容器内に含有されていてもよい。通常、キットは、別々の成分を投与するための説明書を含む。キット形態は、別々の成分が、異なる剤形(例えば、経口および非経口)で投与されることが好ましい場合、異なる投与間隔で投与される場合、または処方医師が、組合せの個々の成分の用量設定を望む場合は特に有利である。
【0234】
そのようなキットの一例は、いわゆるブリスターパックである。ブリスターパックは、包装産業においてよく知られており、医薬品単位剤形(錠剤、カプセル剤など)を包装するために広く使用されている。一般的に、ブリスターパックは、好ましくは透明なプラスチック材料のホイルで覆われている比較的硬い材料のシートからなる。包装プロセス中に、プラスチックホイルに凹部が形成される。凹部は、包装すべき錠剤またはカプセル剤のサイズおよび形状を有する。次に、錠剤またはカプセル剤は、凹部に入れられ、比較的硬い材料のシートは、凹部が形成された方向から反対のホイルの表面でプラスチックホイルに対して密閉される。結果として、錠剤またはカプセル剤は、プラスチックホイルとシートの間の凹部に密閉される。シートの強度は、凹部上に手で圧力をかけ、それにより凹部の場所で開口部がシートに形成されることにより、錠剤またはカプセル剤をブリスターパックから取り出すことができるような強度であることが好ましい。次いで、錠剤またはカプセル剤は、前記開口部を介して取り出すことができる。
【0235】
キット上の記憶補助を、例えば、数字が、そのように定められた剤形を摂取するべき投与計画の日数と一致する錠剤またはカプセル剤に隣接する数字の形態で提供することが望ましいことがある。そのような記憶補助の別の例は、例えば、以下の通り「First Week,Monday,Tuesday,..etc....Second Week,Monday、Tuesday,...」など、カード上に印刷されたカレンダーである。記憶補助の他の変形例は、容易に明らかであろう。「1日投与量」は、所与の日に摂取すべき単一の錠剤もしくはカプセル剤またはいくつかの錠剤もしくはカプセル剤であってよい。また、式I化合物、そのプロドラッグまたは薬学的に許容できる前記化合物もしくは前記プロドラッグの塩の1日投与量は、1個の錠剤またはカプセルからなっていてもよく、一方、第2の化合物の1日投与量は、いくつかの錠剤またはカプセルからなっていてもよく、逆もまた同様である。記憶補助は、これを反映するべきである。
【0236】
本発明の別の具体的実施形態において、それらの意図した使用の順序で一度に1日投与量を投薬するように設計されたディスペンサーが提供される。ディスペンサーは、投与計画の順守をさらに容易にするように、記憶補助を備えていることが好ましい。そのような記憶補助の一例は、投薬された1日投与量の数を示すメカニカルカウンターである。そのような記憶補助の別の例は、例えば、最後の1日投与量が摂取された日付を読み出し、かつ/または次の投与量を摂取するべき日付を指摘する液晶読み出し、または可聴式注意喚起シグナルと一体となった電池式マイクロチップメモリーである。
【0237】
一般的に、本発明の化合物は、単独で、またはお互いと、もしくは他の化合物と組み合わせて好都合な製剤で投与されるであろう。以下の製剤例は、例示的であるに過ぎず、本発明の範囲を制限することは意図されていない。
【0238】
続く製剤において、「活性成分」は、本発明の1種または複数の化合物を意味する。
【0239】
製剤1:ゼラチンカプセル剤
硬質ゼラチンカプセル剤は、以下を用いて調製する:
【0240】
【表12】
【0241】
錠剤製剤は、以下の成分を用いて調製する:
製剤2:錠剤
【0242】
【表13】
【0243】
成分を混ぜ合わせて圧縮し、錠剤を形成する。
【0244】
代替方法として、各々が活性成分を0.25〜100mg含有する錠剤は、以下のように作製する:
製剤3:錠剤
【0245】
【表14】
【0246】
活性成分、デンプン、およびセルロースを、No.45メッシュU.S.シーブに通し、十分に混合する。ポリビニルピロリドンの溶液を、得られる粉末と混合し、次いで、No.14メッシュU.S.シーブに通す。そのようにして製造した顆粒を、50℃〜60℃において乾燥し、No.18メッシュU.S.シーブに通す。次いで、前もってNo.60U.S.シーブに通したカルボキシメチルセルロースナトリウム、ステアリン酸マグネシウム、およびタルクを顆粒に加え、混合した後、錠剤機で圧縮すると、錠剤が得られる。
【0247】
各々が5ml投与量当たり活性成分を0.25〜100mg含有する懸濁剤は、以下のように製造する:
製剤4:懸濁剤
【0248】
【表15】
【0249】
活性成分を、No.45メッシュU.S.シーブに通し、カルボキシメチルセルロースナトリウムおよびシロップと混合し、滑らかなペーストを形成する。安息香酸溶液、香料、および着色料を、水の一部で希釈し、攪拌しながら加える。次いで、十分な水を加え、必要な容積にする。
【0250】
以下の成分を含有するエアロゾル溶液を調製する:
製剤5:エアロゾル
【0251】
【表16】
【0252】
活性成分をエタノールと混合し、混合物をプロペラントの一部に加え、30℃まで冷却し、充填装置に移す。次いで、必要量をステンレス鋼の容器に送り込み、残りのプロペラントで希釈する。次いで、バルブユニットを容器に取り付ける。
【0253】
坐剤は、以下のように調製する:
製剤6:坐剤
【0254】
【表17】
【0255】
活性成分を、No.60メッシュU.S.シーブに通し、最低限必要な熱を用いて前もって溶融させた飽和脂肪酸グリセリドに懸濁させる。次いで、混合物を、公称2g容量の坐剤鋳型中に注ぎ込み、冷却させる。
【0256】
静脈内製剤は、以下のように調製する:
製剤7:静脈内溶液
【0257】
【表18】
【0258】
上記成分の溶液は、1分当たり約1mLの速度で患者に静脈内投与される。
【0259】
上記の活性成分は、薬剤の組合せであってもよい。
【0260】
上記は、本発明の例示的実施形態に関しており、以下の特許請求の範囲に示されるような本発明の精神および範囲を逸脱することなく改変を行うことができることが理解されるべきであることは言うまでもない。
【図面の簡単な説明】
【0261】
【図1】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aのディフラクトグラムを示す図である。
【図2】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Bのディフラクトグラムを示す図である。
【図3】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Cのディフラクトグラムを示す図である。
【図4】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Eのディフラクトグラムを示す図である。
【図5】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Fのディフラクトグラムを示す図である。
【図6】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Gのディフラクトグラムを示す図である。
【図6B】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Hのディフラクトグラムを示す図である。
【図6C】本発明による非晶質性の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩のディフラクトグラムを示す図である。
【図7】(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩、形態Aの固体13C核磁気共鳴スペクトルを示す図である。
【図8】(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩、形態Hの固体13C核磁気共鳴スペクトルを示す図である。
【技術分野】
【0001】
本発明は、プロスタグランジンの治療上活性かつ選択的なモジュレーター、具体的には、EP2の作動薬の多形体、これらの化合物を含む医薬組成物、これらの化合物を調製するための方法ならびに骨障害、緑内障および高眼圧症などのプロスタグランジンの調節に関連している状態を治療するためのそのような化合物の使用に関する。より具体的には、本発明は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形体、この化合物の多形体を含む医薬組成物、これらの多形体を調製するための方法ならびにプロスタグランジンの調節に関連している状態を治療するためのこの化合物の多形体の使用に関する。
【背景技術】
【0002】
薬物開発の過程において、薬物の最も安定な結晶性形態を発見することが重要であると一般的に考えられている。この最も安定な結晶性形態は、最良の化学的安定性、すなわち、製剤における最長の貯蔵寿命を有する可能性が高い形態である。しかしながら、薬物の複数の形態、例えば、塩、水和物、多形体、結晶性および非結晶性形態を有することが有利でもある。異なる物理的形態は異なる利点を提供するため、薬物の理想的な物理的形態は1つではない。最も安定な形態およびそのような他の形態の探索は困難であり、その成果は予測できない。
【0003】
薬物開発を成功させるには、患者にとって治療上有効な治療であるという特定の要件を満たすことが必要である。これらの要件は、2つの範疇、すなわち、(1)剤形の製造を成功させるための要件および(2)薬物製剤が患者に投与された後の薬物の送達および体内動態を成功させるための要件に分類される。
【0004】
様々な経路による投与のための多種類の薬物製剤が存在し、異なる製剤にとって最適な薬物形態は、異なっている可能性が高い。上述のように、薬物製剤は、治療を必要としている患者への流通を成功させるのに十分な貯蔵寿命を有していなければならない。さらに、経口用薬物製剤は、経口的に投与された場合に患者の胃腸管で溶解するであろう形態で薬物を提供しなければならない。即時放出性の錠剤、カプセル、懸濁液、またはサシェなどの即時放出性剤形で経口的に投与する場合、投与量の完全な溶解および最適な生物学的利用能を保証するために、高い溶解性を有する薬物塩または薬物形態を有することが一般的に望ましい。一部の薬物、特に、低い溶解性の薬物または湿潤性に乏しい薬物の場合、胃腸管内に投与された場合に結晶性形態よりも高い初期溶解性を一般的に有するであろう非結晶性薬物形態を利用することが有利であることがある。薬物の非結晶性形態は、結晶性形態よりも化学的に安定性が低いことが多い。したがって、剤形製造、包装、貯蔵、および世界中の患者への流通を可能にするのに十分な時間にわたってその効力を維持するのに十分に安定である実用的な製品を提供するのに十分に化学的に安定である非結晶性薬物形態を識別することが有利である。
【0005】
一方、薬物形態があまり可溶性でない場合によりよく動作する剤形が存在する。例えば、チュアブル錠剤または懸濁液またはサシェの剤形は、薬物に直接舌をさらす。そのような剤形の場合、薬物の部分を固体の状態に保ち、悪い味を最小限に抑えるために、口における薬物の溶解性を最小限に抑えることが望ましい。そのような剤形の場合、低い溶解性の塩または結晶性形態を使用することが望ましいことが多い。
【0006】
制御放出性の経口用または注射用の、例えば、皮下または筋肉内の剤形の場合、望ましい薬物溶解性は、送達経路、投与量、剤形設計および望ましい放出継続時間の複雑な関数である。高い溶解性を有する薬物の場合、遅い溶解による遅い放出の実現を助けるために、制御放出性剤形のためのより低い溶解性の結晶性の塩または多形体を利用することが望ましいことがある。低い溶解性を有する薬物の場合、制御放出性剤形からの望ましい薬物放出速度を支えるのに十分な溶解速度を実現するために、より高い溶解性の結晶性の塩もしくは多形体、または非結晶性形態を利用することが必要であることがある。
【0007】
ソフトゼラチンカプセル剤形(「ソフトゲル」)において、薬物は、トリグリセリド油またはポリエチレングリコールなどの少量の溶媒またはビヒクルに溶かされ、ゼラチンカプセルに封入される。この剤形にとって最適の薬物形態は、適切なソフトゲルビヒクルにおいて高い溶解性を有する薬物形態である。一般に、トリグリセリド油により可溶性である薬物形態は、水にあまり可溶性でないであろう。ソフトゲル剤形に適している薬物形態の識別には、様々な塩、多形体、結晶性および非結晶性の形態の研究が必要である。
【0008】
したがって、薬物形態の望ましい溶解性は、使用目的によって異なり、薬物形態がすべて等価であるとは限らないことが分かる。
【0009】
ヒトまたは動物の治療法にとって実際に有用である薬物形態の場合、薬物形態は、最小限の吸湿性を示すことが望ましい。極めて吸湿性の薬物を含有する剤形は、保護包装を必要とし、湿潤環境で貯蔵される場合に、変化した溶解を示すことがある。したがって、薬物の非吸湿性の結晶性の塩および多形体を識別することが望ましい。薬物が非結晶性である場合、または溶解性および溶解速度を改善するために非結晶性形態が望まれる場合、他の非結晶性の塩または形態に比べて低い吸湿性を有する非結晶性の塩または形態を識別することが望ましい。
【0010】
結晶性または非結晶性の薬物は、無水形態で、または水和物もしくは溶媒和物もしくは水和物/溶媒和物として存在することがある。薬物の水和状態および溶媒和状態は、その溶解性および溶解挙動に影響を及ぼす。
【0011】
薬物の融点は、異なる塩、多形体、結晶性および非結晶性の形態について変わることがある。商業用錠剤プレスでの錠剤の製造を可能にするため、薬物融点は、錠剤製造中の薬物溶融を防ぐために、約60℃を超え、好ましくは100℃を超えていることが望ましい。この場合の好ましい薬物形態は、最も高い融点を有する薬物形態である。さらに、直射日光および赤道近くなどの地理的領域で起きる高い環境貯蔵温度において固体剤形中の固体薬物の化学的安定性を保証するために高い融点を有することが望ましい。ソフトゲル剤形が望まれる場合、剤形における薬物の結晶化を最小限に抑えるために、低い融点を有する薬物形態を有することが好ましい。したがって、薬物形態の望ましい融点は、使用目的によって異なり、薬物形態がすべて等価であるとは限らないことが分かる。
【0012】
薬物の投与量が高い場合、または小さな剤形が望まれる場合、塩、水和物または溶媒和物の選択は、単位重量当たりの効力に影響を及ぼす。例えば、より高い分子量の対イオンを有する薬物塩は、より低い分子量の対イオンを有する薬物塩よりも低いグラム当たりの薬物効力を有するであろう。単位重量当たり最も高い効力を有する薬物形態を選択することが望ましい。
【0013】
異なる結晶性多形体および非結晶性形態を調製する方法は、薬物によって大きく異なる。これらの方法において、特に、最終合成ステップについて、特に、薬物が、合成の最終ステップで利用される溶媒との溶媒和物として存在する傾向がある場合、毒性が最も少ない溶媒が使用されることが望ましい。好ましい薬物形態は、それらの合成において毒性が少ない溶媒を利用する薬物形態である。
【0014】
商業規模で良好な錠剤を形成する薬物の能力は、Hiestand H、Smith D.Indices of tableting performance.Powder Technology、1984;38:145〜159に記載されている錠剤化指数(Tableting Indices)などの様々な薬物物理的特性によって異なる。これらの指数を用い、優れた錠剤化性能を有する薬物、例えば、アトルバスタチンカルシウムの形態を識別することができる。そのような指数の1つは、脆性を反映する脆性破壊指数(Brittle Fracture Index)(BFI)であり、0(良好−低脆性)から1(不良−高脆性)まで様々である。機械的特性、流動特性および錠剤化性能の他の有用な指数または尺度は、圧縮応力、絶対密度、固相率(solid fraction)、動的圧入硬度、延性、弾性率、低減(reduced)弾性率、準静的圧入硬度、剛性率、引張強度、妥協(compromised)引張強度、最高結合指数、最低結合指数、脆性/粘弾性結合指数、歪指数、粘弾性数、内部摩擦の有効角(シェアセル試験から)、凝集性(粉末雪崩試験から)および流動変動性を包含する。多くのこれらの尺度は、3軸油圧プレスを用いて調製されることが好ましい薬物成形体で得られる。これらの尺度の多くは、Hancock B、Carlson G、Ladipo D、Langdon B、およびMullarney M.Comparison of the Mechanical Properties of the Crystalline and Amorphous Forms of a Drug Substance.International Journal of Pharmaceutics、2002;241:73〜85にさらに記載されている。
【0015】
流動に影響を及ぼす薬物形態特性は、錠剤剤形製造にとってだけではなく、カプセル剤、懸濁剤、およびサシェ剤の製造にとっても重要である。
【0016】
薬物粉末の粒径分布も、特に、粉末流動に対する影響により、製造プロセスに大きな影響を有することがある。異なる薬物形態は、異なる特徴的な粒径分布を有する。
【0017】
上記の論議から、すべての治療的応用に理想的である薬物形態は存在しないことは明らかである。したがって、様々な製剤において使用することができる様々な独特の薬物形態、例えば、塩、多形体、非結晶性形態を探し求めることが重要である。具体的な製剤または治療的応用のための薬物形態の選択には、上記に記載されているような様々な特性を考慮することが必要であり、特定の応用のための最良の形態は、1つの具体的な重要で良好な性質を有する形態であってよく、一方、他の特性は、許容できるか、または辛うじて許容できるかであってよい。
【0018】
米国特許第6,498,172B1号は、例えば、骨障害の治療に有用であるような化合物(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸を開示している。この出願は、一般論として、薬学的に許容できる塩に言及しており、ナトリウム塩の調製が開示されている。しかしながら、‘172特許は、結晶化手順について何ら記載せず、特許は、この化合物の多形体形態についても何ら論議していない。
【0019】
公開された国際特許出願WO99/19300およびWO98/28264は、プロスタグランジン作動薬ならびに局所適用(例えば、骨折または骨切り術の部位への)による骨折および骨切り術の治癒を治療および促進するためのそれらの使用を開示している。
【0020】
S.C.MillerおよびS.C.Marks,Jr.、Bone 14、143〜151(1993)は、プロスタグランジンE1(PGE1)によるイヌ下顎骨の骨膜表面上の新生骨形成の局所刺激を研究し、外側下顎皮質に隣接して骨膜下に埋め込まれた浸透圧ミニポンプと制御放出性ペレット剤による送達を比較した。
【0021】
S.C.Marks,Jr.およびS.C.Miler、J.Oral Pathol.17:500〜505(1988)は、1週当たり500〜2000μgの投与量で3週間にわたるPGE1の局所注入が、イヌの下顎骨における歯槽骨の劇的で局所的な形成を生み出すことを報告した。
【0022】
M−S.ShihおよびR.W.Norrdin、Am.J.Vet.Res.48:828〜830(1986)では、横骨折を、成体ビーグル犬の肋骨で外科的に作製し、10%エタノールTris−緩衝液ビヒクル0.5mlまたはPGE1(10%エタノールTris−緩衝液中にPGE10.2mgを含有する)0.5mlを10日間にわたって1日2回、骨折部位に直接注射した。PGE1の投与は、骨折部位およびその対側整合部位に隣接する骨膜外層上の骨基質形成を誘導するという結論であった。
【0023】
M−S.ShihおよびR.W.Norrdin、Calcif.Tissue Int.(1986)39:191〜197は、手術後10日間にわたって1日2回、ビーグル犬の頸骨における欠損部位に注射されたPGE1(10%エタノール中0.2mg/kg)の影響を研究した。局所的にPGE1を投与されたイヌは、存在する類骨量の増加と共に、より多くの骨膜および皮質の骨内膜性骨形成を有することが判明した。
【0024】
R.Yang、T.LiuおよびS.Lin−Shiau、Calcif.Tissue Int.、52:57〜61(1993)は、14日間にわたる左頸骨の骨幹端中への骨内経路を介するプロスタグランジンE2の連日注射の影響を検討した。この参考文献によれば、この投与計画は、骨幹端における骨梁の有意な増加をもたらした。
【0025】
K.Notoya他、The Journal of Pharmacology and Experimental Therapeutics、290:1054〜1064(1999)は、in vivoにおける骨格再生および骨修復に対して局所的に適用された持続放出性マイクロカプセル剤で、新規な骨芽細胞分化促進化合物であるTAK−778の影響を調べた。
【発明の開示】
【発明が解決しようとする課題】
【0026】
本発明の他の実施形態によれば、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態が提供される。
【課題を解決するための手段】
【0027】
本発明のさらに他の実施形態によれば、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態、および薬学的に許容できる希釈剤または担体を含む医薬組成物が提供される。
【0028】
本発明の別の実施形態によれば、緑内障、高眼圧症または低骨量を示す状態を有する哺乳類を治療するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態を哺乳類に投与することを含む方法が提供される。
【0029】
本発明のさらに他の実施形態によれば、哺乳類において骨量を増加および維持するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態を哺乳類に投与することを含む方法が提供される。
【0030】
本発明のさらに別の実施形態によれば、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物の結晶性形態Aを調製するための方法であって、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸を、遊離の酸として、有機溶媒中で水酸化ナトリウムと接触させて反応混合物を形成すること、反応混合物を約50C〜約90Cの第1の温度まで温め、少なくとも1時間にわたって第1の温度を保つこと、反応混合物を約15C〜約25Cの第2の温度まで冷却すること、および反応混合物から固体を集めることを含む方法が提供される。この方法において使用される有機溶媒は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約8〜約12mlで存在する酢酸イソプロピルであってよい。この方法において使用される水酸化ナトリウムは、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約1〜約1.3mmolで存在する水酸化ナトリウムの50%水溶液であってよい。
【0031】
本発明の他の実施形態によれば、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態が提供される。
【0032】
本発明のさらに他の実施形態によれば、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態、および薬学的に許容できる希釈剤または担体を含む医薬組成物が提供される。
【0033】
本発明の別の実施形態によれば、緑内障、高眼圧症または低骨量を示す状態を有する哺乳類を治療するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態を哺乳類に投与することを含む方法が提供される。
【0034】
本発明のさらに他の実施形態によれば、哺乳類において骨量を増加および維持するための方法であって、治療有効量の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態または非晶質性形態を哺乳類に投与することを含む方法が提供される。
【0035】
化合物「(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸」は、化合物の遊離の酸を指す。
【0036】
「哺乳類」という用語は、例えば、イヌ、ネコ、雌ウシ、ヒツジ、ウマ、およびヒトを包含する動物を意味する。好ましい哺乳類は、ヒトを包含する。
【0037】
「実質的に純粋な」という語句は、化合物の総量(すなわち、望ましい多形体および/または塩+その他の多形体および非晶質性形態、塩および遊離の酸の量)に関して望ましい多形体および/または塩の相対的純度を指す。「実質的に純粋な」多形性形態は、化合物の総量に対して望ましい多形体を少なくとも約90%含有するべきである。「実質的に純粋な」多形性形態は、望ましい多形体を少なくとも約95%含有するべきであることが好ましい。本発明の一部の実施形態において、「実質的に純粋な」多形性形態は、望ましい多形体を少なくとも約99%含有することがある。
【0038】
「低骨量を示す1種または複数の状態」という語句は、骨量のレベルが、World Health Organization「Assessment of Fracture Risk and its Application to Screening for Postmenopausal Osteoporosis(1994)、Report of a World Health Organization Study Group、World Health Organization Technical Series 843」による基準で定義されているような年齢特異的な正常以下である状態を指す。原発性および続発性骨粗鬆症は、「低骨量を示す1種または複数の状態」に包含される。続発性骨粗鬆症は、グルココルチコイド誘発性骨粗鬆症、甲状腺機能亢進症誘発性骨粗鬆症、不動化(immobilization)誘発性骨粗鬆症、ヘパリン誘発性骨粗鬆症および免疫抑制誘発性骨粗鬆症を包含する。歯周疾患、歯槽骨喪失、骨切り術後および小児期特発性骨喪失も包含される。「低骨量を示す1種または複数の状態」という語句は、背骨の湾曲、身長の減少および補綴手術などの骨粗鬆症の長期合併症も包含する。
【0039】
「低骨量を示す1種または複数の状態」という語句は、骨粗鬆症を包含する上記に記載されているような疾患を発症する平均よりも有意に高い確率を有することが知られている哺乳類、例えば、哺乳類(例えば、閉経後の女性、および60歳を超える男性)も指す。
【0040】
他の骨量を増加または増強する用途は、骨修復、骨折治癒速度の増加、骨移植手術の完全な置き換え、骨移植成功率の向上、顔面再建、上顎再建、下顎再建、頭蓋顔面再建後の骨治癒、補綴内方成長、椎骨癒合、長骨伸長および脊椎固定術を包含する。
【0041】
本発明の医薬組成物は、脊椎固定ケージ、脊椎固定金属製品、内部および外部の骨固定装置、ネジならびにピンなどの当業者に知られている整形外科用装置と併せて使用することもできる。
【0042】
当業者は、骨量という用語が、実際には、時に(厳密には正しくないが)骨塩密度(BMD)と呼ばれる単位面積当たりの骨量を指すことを理解するであろう。
【0043】
本明細書で使用する「治療すること」、「治療する」または「治療」という用語は、予防的(preventative)(例えば、予防的(prophylactic))、姑息的および治癒的治療を包含する。
【0044】
「有効量」という用語は、特定の疾患もしくは状態または特定の疾患もしくは状態の症状を改善、軽減もしくは除去するか、または特定の疾患もしくは状態または特定の疾患もしくは状態の症状の発現を予防もしくは遅延する化合物または化合物の組合せの量を意味する。
【0045】
「患者」という用語は、ヒト、イヌ、ネコおよびウマなどのコンパニオンアニマル、ならびにウシ、ブタおよびヒツジなどの家畜などの動物を意味する。特に好ましい患者は、雄と雌の両方を包含する哺乳類であり、ヒトがより好ましい。
【0046】
本明細書で使用する「薬学的に許容できる」という用語は、担体、ビヒクル、希釈剤、賦形剤および/または塩が、製剤の他の成分と適合していなければならず、そのレシピエントに対して有害でないことを意味する。
【0047】
「プロドラッグ」という表現は、投与後、いくつかの化学的または生理学的プロセスを介してin vivoで薬物を放出する薬物前駆体である化合物を指す(例えば、生理学的pHにされるか、または酵素作用によりプロドラッグは、望ましい薬物形態へ変換される)。例示的プロドラッグは、切断により、対応する薬物化合物を放出する。
【0048】
「薬学的に許容できる塩」という表現は、塩化物、臭化物、ヨウ化物、硫酸塩、重硫酸塩、リン酸塩、酢酸塩、マレイン酸塩、フマル酸塩、シュウ酸塩、酪酸塩、酒石酸塩、クエン酸塩、グルコン酸塩、メタンスルホン酸塩および4−トルエン−スルホン酸塩など(であるが、これらに限定されない)アニオン性の塩を指す。この表現は、ナトリウム、カリウム、カルシウム、マグネシウム、アンモニウムまたはプロトン化したベンザチン(N,N’−ジベンジルエチレンジアミン)、コリン、エタノールアミン、ジエタノールアミン、エチレンジアミン、メグラミン(meglamine)(N−メチル−グルカミン)、ベネタミン(N−ベンジルフェネチルアミン)、ピペラジンおよびトロメタミン(2−アミノ−2−ヒドロキシメチル−1,3−プロパンジオール)など(であるが、これらに限定されない)カチオン性の塩も指す。
【0049】
本発明のこれらおよび他の特徴、態様および利点は、以下の図面、説明および特許請求の範囲を参照してよりよく理解されていくであろう。
【発明を実施するための最良の形態】
【0050】
一般に、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、米国特許第6,498,172B1号に開示されている方法により調製することができ、その主題は、参照により全体として本明細書に組み込まれるものとする。代替方法として、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、以下のスキームIに記載されている新規な方法により調製することができる。これらの化合物の具体的な多形体を製造するための特定のプロセスは、実験の項に示される。すべての出発化合物は、文献手順によるか、またはSigma−Aldrich Corporation、St.Louis、MOなどの一般的商業ソースから得ることができる。
【0051】
考察、実施例および調製において、以下の略語が使用される。2B EtOH−変性エタノール、br−幅広いピーク、℃−セ氏温度、d−二重線、DMSO−d6−重水素化ジメチルスルホキシド、EtOAc−酢酸エチル、equiv−当量、g−グラム、1H NMR−プロトン核磁気共鳴、H2O−水、HCl−塩化水素、Hz−ヘルツ、iPr2Net−ジイソプロピルエチルアミン(ヒューニッヒ塩基)、iPrOAc−酢酸イソプロピル、J−結合定数(多重線の間隔)、kg−キログラム、L−リットル、m−多重線、M−モル、MeCl2−ジクロロメタン、mg(s)−ミリグラム、min−分、mL−ミリリットル、mmol−ミリモル、mp−融点、MPa−メガパスカル、MS−マススペクトル、N−規定濃度、NaHCO3−炭酸水素ナトリウム、NaOH−水酸化ナトリウム、psi−毎平方インチ当たりポンド、Pt/C−炭素上白金、RT−室温、s−一重線ピークおよびt−三重線ピーク。さらに、以下の考察、実施例および調製において、Brukerへの言及は、Bruker AXS,Inc.、Madison、WIの製品を指し、Kevexへの言及は、Thermo Electron Corporation、Waltham、MAの製品を指す。
【0052】
【化1】
【0053】
ステップ1:{3−[(4−tert−ブチル−ベンジルアミノ)−メチル]−フェノキシ}−酢酸エチルエステルコハク酸塩(II)の調製
500mLのParr瓶に、2Bエタノール(180mL)および5%炭素上白金、50%水湿(2.00g)を充填した。2Bエタノール(20mL)中の3−ホルミルフェノキシ酢酸エチル(20.0g、96.06mmol、1.0当量)の溶液と、続いて、4−tert−ブチルベンジルアミン(16.86mL、96.06mmol、1.0当量)を加えた。混合物を5時間にわたって窒素雰囲気下で室温において攪拌し、その時点で、式Iの中間体の生成が完了した。反応容器を50psi(0.3447MPa)の水素雰囲気下に置き、18時間にわたって室温において振盪させた。混合物を珪藻土に通して濾過し、濾過ケーキを2Bエタノール(40mL)で1回洗浄した。濾液を、添加ロートおよびメカニカルスターラーを取り付けた1リットルのフラスコに移した。2Bエタノール(120mL)中のコハク酸(11.34g、96.06mmol、1.0当量)の温かい溶液を15分かけて加えた。得られたスラリーを19時間にわたって室温において攪拌し、次いで、濾過した。固体を2Bエタノール(50mL)で1回洗浄し、乾燥すると、II(33.10g、収率73%)が得られた。融点=138〜139℃。元素分析 C22H29NO3・C4H6O4の計算値:C 65.94;H 7.45;N 2.96。実測値:C 65.74;H 7.62;N 2.99。1H NMR(400MHz,CD3OD):δ 1.28(t,3H,J=7.0Hz)、1.32(s,9H)、2.50(s,4H)、4.08(s,2H)、4.10(s,2H)、4.23(q,2H,J=7.0Hz)、4.73(s,2H)、6.96〜6.98(m,1H)、7.04〜7.06(m,2H)、7.33〜7.38(m,3H)、7.46〜7.48(m,2H)。
【0054】
ステップ2および3:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(V)の調製
化合物II(48.00g、101mmol、1.0当量)を、ジクロロメタン(360mL)と1M水性重炭酸ナトリウム(240mL)の間で分配することにより遊離塩基にした。層を分離し、有機層を水(240mL)で1回洗浄した。層を分離し、N,N−ジイソプロピルエチルアミン(53.4mL、304mmol、3.0当量)を有機層に加えた。塩化ピリジン−3−スルホニル塩酸塩(III)(26.4g、123mmol、1.2当量)およびジクロロメタン(240mL)を別の反応容器に充填し、窒素雰囲気下で0℃まで冷却した。遊離塩基/N,N−ジイソプロピルエチルアミン溶液を1.8時間かけて加えた。反応混合物を室温まで温め、19時間にわたって保ち、その時点で、式IV((3−{[(4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ]−メチル}−フェノキシ)−酢酸エチルエステル)の中間体の生成が完了した。反応混合物を1N塩酸(336mL)、次いで、1M水性重炭酸ナトリウム(336mL)で洗浄した。2Bエタノール(384mL)を有機層に加え、混合物を、ポット容積が約400mLになるまで大気圧蒸留した。6N水酸化ナトリウム(19.9mL、119mmol、1.2当量)を加え、混合物を19時間にわたって室温に保った。濃塩酸(11.0mL、134mmol、1.3当量)と、続いて、水(192mL)を加えた。スラリーを室温において5時間にわたって顆粒化し、濾過した。固体を50:50 2Bエタノール:水(144mL)、水(144mL)、2Bエタノール(144mL)で洗浄し、次いで、乾燥すると、化合物V(34.78g、収率73%)が得られた。融点=159〜160℃。元素分析 C25H28N2O5Sの計算値:C 64.08;H 6.02;N 5.98。実測値 C 64.13;H 6.11;N 5.99。1H NMR(400MHz,DMSO−d6):δ 1.21(s,9H)、4.31(s,2H)、4.33(s,2H)、4.54(s,2H)、6.65〜6.69(m,1H)、6.71〜6.74(m,1H)、6.75〜6.76(m,1H)、7.01(d,2H,J=8.1Hz)、7.13(t,1H,J=7.9Hz)、7.21(d,2H,J=8.3Hz)、7.56〜7.59(m,1H)、8.16〜8.19(m,1H)、8.80〜8.82(m,1H)、8.97〜8.98(m,1H)、13.04(br s,1H)。
【0055】
ステップ3R:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(V)の再結晶
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(V)(20.0g、42.7mmol、1.0当量)およびメチルエチルケトン(300mL)を混ぜ合わせ、すべての固体が溶けるまで60℃まで加熱した。溶液を35℃まで冷却し、0.45ミクロンのナイロンフィルターに通して濾過した。フィルターをメチルエチルケトン(20mL)ですすいだ。濾液を混ぜ合わせ、約160mLのポット容積まで大気圧蒸留により濃縮した。混合物を室温まで冷却し、その時点で、結晶化が始まった。混合物を室温において19時間にわたって攪拌し、次いで、濾過した。固体をメチルエチルケトン(20mLで2回、次いで、40mLで1回)で洗浄し、乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(15.30g、収率77%)が得られた。融点=159.0〜159.5℃。元素分析 C25H28N2O5Sの計算値:C 64.08;H 6.02;N 5.98。実測値 C 63.89;H 5.82;N 5.91。1H NMR(400MHz,DMSO−d6):上記のステップ2および3と同じ。1H NMRスペクトルは、メチルエチルケトン約1.4%の存在も示した。
【0056】
ステップ4:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII)の調製
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(20.0g、42.7mmol、1.0当量)および酢酸イソプロピル(400mL)を反応容器に充填した。50%水酸化ナトリウム(3.520g、44.0mmol、1.03当量)と、続いて、脱イオン水(4.165g)を加えた。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII、200mg)をシード材料として加えた。混合物を30分かけて70℃まで加熱し、5時間にわたってこの温度に保った。混合物を4時間かけて40℃まで冷却し、次いで、1時間かけて20℃まで冷却し、19時間にわたって顆粒化し、次いで、濾過した。固体を1%水性酢酸イソプロピル(各洗浄液20mL)で2回洗浄し、次いで、乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII、18.22g、収率85%)が得られた。半水和物は、約120℃に脱水温度を有し、融点は有していない。元素分析 C25H27N2O5S・Na・1/2H2Oの計算値:C 61.21;H 5.55;N 5.71。実測値 C 60.19;H 5.55;N 5.56。1H NMR(400MHz,DMSO−d6):δ 1.21(s,9H)、4.07(s,2H)、4.31(s,4H)、6.59〜6.61(m,1H)、6.64(m,1H)、6.71〜6.74(m,1H)、7.01〜7.09(m,3H)、7.22(d,2H,J=8.4Hz)、7.56〜7.59(m,1H)、8.14〜8.17(m,1H)、8.79〜8.80(m,1H)、8.94(m,1H)。
【0057】
代替方法として、ステップ4におけるナトリウム塩生成および再結晶は、以下のスキームIIに示す手順を用いて行うことができる。
【0058】
【化2】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(47.7g)およびメタノール(500mL)を混ぜ合わせた。1N水酸化ナトリウム(1.01当量、103mL)を、室温において攪拌しながらゆっくりと加えた。2時間にわたって攪拌した後、混合物を濃縮し、アセトンで5回(毎回300mL)およびジクロロメタンで2回(毎回200mL)と一緒に共沸させると、非晶質性の泡が得られた。これに、アセトン(500mL)および水(15mL)を加え、混合物を50℃まで加熱した。20分後、材料は極めて粘稠になり、追加のアセトン(200mL)を加えた。混合物を室温まで冷却し、濾過した。濾過ケーキを2−プロパノールで洗浄し、高真空下で乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸半水和物形態A(VII、収率77%)39gが得られた。
【0059】
粉末X線回折
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態A、B、C、E、F、およびGを、それらのX線粉末回折パターンにより特徴付けた。すなわち、形態A、B、C、E、F、およびGのX線回折パターンを、銅放射線(波長:1.54056Å)を用いるBruker D5000回折計で行った。管の電圧およびアンペア数は、それぞれ40kVおよび50mAに設定した。発散スリットおよび散乱スリットは、1mmに設定し、受光スリットは、0.6mmに設定した。回折された放射線は、Kevex PSI検出器により検出した。3.0から40°2θまでの2.4°/分(1秒/0.04°ステップ)におけるθ−2θ連続スキャンを用いた。アルミナ標準品を分析し、機器アラインメントをチェックした。データを集め、Bruker AXSソフトウェアバージョン7.0を用いて分析した。試料は、石英ホルダーに試料を入れることにより調製した。
【0060】
本明細書で報告される測定のために使用されるBrukerシステムのようなBragg−Brentano機器でX線回折測定を行うため、試料を、通常、空洞を有するホルダーの中に入れる。試料粉末をガラススライドまたは同等物により押し付け、ランダムな表面および適切な試料高を確保する。次いで、試料ホルダーを機器の中に入れる。入射X線ビームを、初めはホルダー面に対して小さな角度で試料に向け、次いで、入射ビームとホルダー面の間の角度を連続的に増加させる円弧に沿って移動させる。そのようなX線粉末分析に関連した測定差は、(a)試料調製における誤差(例えば、試料高)、(b)機器誤差(例えば、平面試料誤差)、(c)較正誤差、(d)操作者誤差(ピーク位置を決定する場合に存在する誤差を包含する)、および(e)材料の性質(例えば、好ましい配向性および透明度誤差)を包含する様々な要因に起因する。較正誤差および試料高誤差は、同じ方向ですべてのピークのシフトをもたらすことが多い。平面ホルダーを用いる場合の試料高における小さな差は、XRPDピーク位置における大きな移動につながるであろう。系統的な研究は、典型的なBragg−Brentano配置でShimadzu XRD−6000を用いると、1mmの試料高差が、1°2θと同じ高さのピークシフトにつながることを明らかにした(Chen他;J Pharmaceutical and Biomedical Analysis、2001;26、63)。これらのシフトは、X線ディフラクトグラムから識別することができ、シフトについて補正すること(系統的な補正係数をすべてのピーク位置値に適用すること)、または機器を再較正することにより除去することができる。上述のように、ピーク位置を一致させるために系統的な補正係数を適用することにより、様々な機械からの測定値を修正することが可能である。一般に、この補正係数は、Brukerからの測定ピーク位置を、期待されるピーク位置と一致させるであろうし、0〜0.2°2θの範囲であってよい。
【0061】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Hを、そのX線粉末回折パターンにより特徴付けた。形態HについてのX線粉末回折パターンを、銅放射線を用いるSiemens D5000回折計で作成した。機器に、直線集束X線管を取り付けた。管の電圧およびアンペア数は、それぞれ38kVおよび38mAに設定した。発散スリットおよび散乱スリットは、1mmに設定し、受光スリットは、0.6mmに設定した。回折されたCu Kα1放射線(λ=1.54056Å)は、Kevex PSI検出器を用いて検出した。3.0から40°2θまでの2.4°2θ/分(1秒/0.04°2θステップ)におけるθ2θ連続スキャンを用いた。アルミナ標準品(NIST標準基準材料1976)を分析し、機器アラインメントをチェックした。データを集め、BRUKER AXS DIFFRAC PLUSソフトウェアバージョン2.0を用いて分析した。試料は、石英ホルダーに試料を入れることにより調製した。PXRDピークは、最大ピーク高を用いて手動で選択した。
【0062】
図1を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aは、CuKα放射線によりBruker D5000回折計で測定された7%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0063】
【表1】
【0064】
図2を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Bのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Bは、CuKα放射線によりBruker D5000回折計で測定された3%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0065】
【表2】
【0066】
図3を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Cのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Cは、CuKα放射線によりBruker D5000回折計で測定された5%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0067】
【表3】
【0068】
図4を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Eのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Eは、CuKα放射線によりBruker D5000回折計で測定された4%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0069】
【表4】
【0070】
図5を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Fのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Fは、CuKα放射線によりBruker D5000回折計で測定された5%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0071】
【表5】
【0072】
次に、図6を参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Gのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Gは、CuKα放射線によりBruker D5000回折計で測定された6%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0073】
【表6】
【0074】
要約すれば、表7は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態A、B、C、E、F、およびGについて試料における回折線の度2θおよび相対強度を列挙している:
【0075】
【表7】
【0076】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の6つの結晶性形態のみが知られているため、各形態は、単一のX線粉末回折線、線の組合せまたは他の形態のX線粉末回折と異なるパターンにより、他の結晶性形態から識別および区別することができる。
【0077】
例えば、表8は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態A、B、C、E、F、およびGについての独特なピーク、ならびに2θピークの組合せ、すなわち、各形態に独特である一連のX線回折線を列挙している。
【0078】
【表8】
【0079】
次に、図6Bを参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Hのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Hは、CuKα放射線によりBruker D5000回折計で測定された6%以上の相対強度を有する度2θ、d間隔、および相対強度に関して表される以下のX線粉末回折パターンにより特徴付けられる:
【0080】
【表9】
【0081】
次に、図6Cを参照すると、本発明のある実施形態による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非晶質性形態Iのディフラクトグラムが示される。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Iは、図に示されるようなX線粉末回折パターンにより特徴付けられる。
【0082】
固体核磁気共鳴
次に、図7を参照すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aを、その固体核磁気共鳴スペクトル(SSNMR)により特徴付けることもできる。すなわち、形態Aの固体核磁気共鳴スペクトルを、Bruker−Biospin(Buker−Biospin、Billerica、MA)Avance DSX 500MHz NMR分光計で行った。
【0083】
13C SSNMR
化合物約70mgを、分析される試料のための4mmのZrOスピナーに固く詰め込んだ。一次元13Cスペクトルは、ワイドボアBruker−Biospin Avance DSX 500MHz NMR分光計内に配置されたBruker 4mm BL CPMASプローブで293Kにおいて1H−13C交差分極マジック角回転(CPMAS)を用いて周囲圧力にて集めた。試料は、4mmのスピナーにとっての最大規定回転速度に相当する15.0kHzにて回転させた。速い回転速度は、スピニングサイドバンドの強度を最小限に抑えた。シグナル感度を最適化するため、交差分極接触時間を2.0msに調整し、デカップリングパワーを85kHzに設定した。炭素スペクトルは、3秒のリサイクルディレイで2,820のスキャンを取得した。炭素スペクトルは、その高磁場共鳴が29.5ppmに設定されているアダマンタンの外部試料を用いて参照した。
【0084】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aは、化学シフトが百万分率(ppm)で表される以下の固体13C核磁気共鳴(SSNMR)スペクトルにより特徴付けられた:
【0085】
【表10】
【0086】
次に、図8を参照すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Hを、その固体核磁気共鳴スペクトル(SSNMR)により特徴付けることもできる。すなわち、形態Aの固体核磁気共鳴スペクトルを、Bruker−Biospin(Buker−Biospin、Billerica、MA)Avance DSX 500MHz NMR分光計で行った。
【0087】
形態Hのための13C SSNMR法
化合物約70mgを、4mmのZrO2ローターに固く詰め込んだ。一次元13Cスペクトルは、ワイドボアBruker−Biospin Avance DSX 500MHz NMR分光計内に配置されたBruker 4mm BL CPMASプローブで294Kにおいて1H−13C交差分極マジック角回転(CPMAS)を用いて周囲圧力にて集めた。試料は、4mmのローターにとっての最大規定回転速度に相当する15.0kHzにて回転させた。速い回転速度は、スピニングサイドバンドの強度を最小限に抑えた。シグナル感度を最適化するため、交差分極接触時間を2.0msに調整し、デカップリングパワーを85kHzに設定した。炭素スペクトルは、5秒のリサイクルディレイで5400のスキャンを取得した。炭素スペクトルは、その高磁場共鳴が29.5ppmに設定されているアダマンタンの外部試料を用いて参照した。
【0088】
【表11】
【0089】
本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態または非結晶性形態または非晶質性形態は、無水形態ならびに水和および溶媒和形態で存在することができる。一般に、水和形態は、非水和形態と等価であり、本発明の範囲内に包含されることが意図されている。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態A、B、およびCは、水和物として生じることが好ましいが、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態E、F、およびGは、無水形態として生じることが好ましい。
【0090】
本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の結晶性形態または非結晶性形態または非晶質性形態は、等価なX線粉末ディフラクトグラム、または固体NMRスペクトルを有する水和および/または溶媒和の程度にかかわらず、本発明の範囲内にある。
【0091】
以下の非限定的な実施例は、本発明の化合物を調製するための好ましい方法を例示している。
【実施例】
【0092】
形態A:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0093】
【化3】
手順:500mLのジャケット付き反応器に、酢酸イソプロピル(400mL)、50%水性水酸化ナトリウム(3.520g)および水(4.165g)を充填した。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(20.0g)と、続いて、シードとしての(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸の形態A(200mg)(例えば、上記で、スキームIIのプロセスにより調製される)を混合物に充填した。混合物を30分かけて70℃まで加熱し、5時間にわたってこの温度において攪拌した。混合物を4時間かけて40℃まで冷却し、次いで、1時間かけて20℃まで冷却した。次いで、スラリーを12時間にわたって20℃において顆粒化した。スラリーを、ブフナーロート(濾紙)に通して濾過した。濾過ケーキを1%水性酢酸イソプロピル20mLで2回洗浄し、次いで、窒素を抽気しながら40〜45℃において一夜にわたって真空オーブン中で乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物形態A(VIII、収率79%)16.84gが得られた。
【0094】
形態B:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0095】
【化4】
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸(VI)(4.98g)、酢酸エチル(59.8mL)および水(1.3mL)を混ぜ合わせ、55℃まで加熱した。酢酸エチル(20mL)および水(0.44mL)中の2−エチルヘキサン酸ナトリウム(97%、2.025g)の溶液を加えると、澄明な溶液が得られた。溶液を、55°〜60℃において15分間にわたって活性炭で処理し、珪藻土に通して濾過した。濾過ケーキを酢酸エチル/2.2%水5mLで1回洗浄した。濾液は、40℃であった。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(VIII)50mgをシードとして加え、混合物を室温まで冷却し、18時間にわたって攪拌した。スラリーを濾過すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩3.1gが得られた。
【0096】
濾液を20mLまで濃縮し、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(VIII)をシードし、室温において3時間にわたって攪拌し、濾過すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩0.1gが得られた。
【0097】
第2の濾液を濃縮乾固し、室温において水40mL中で顆粒化し、濾過し、空気乾燥すると、固体2.4gが得られた。これらを酢酸エチル30mLおよび酢酸エチル10mL中の2−エチルヘキサン酸ナトリウム(97%、1g)の溶液と混ぜ合わせた。混合物を18時間にわたって室温において攪拌し、濾過し、乾燥すると、非結晶性の固体が得られた。この材料を酢酸エチル40mLおよび水1mLに溶かし、18時間まで室温において攪拌した。ヘキサンを、かすみ点に達するまで加え、次いで、混合物を24時間にわたって攪拌した。粘稠な沈殿が形成し、濾過し、乾燥酢酸エチルで洗浄し、乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩一水和物形態B(IX)1.27gが得られた。
【0098】
形態C:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0099】
【化5】
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(501mg)および含水THF(2.2%水)5.0mLを混ぜ合わせ、窒素雰囲気下で1時間かけて50°〜55℃まで加熱した。懸濁液に、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩4mgをシードし、次いで、室温まで冷却し、66時間にわたって攪拌した。溶媒を完全に蒸発させると、白色の固体が残った。追加の含水THF(2.2%水)5mLを加え、混合物を5時間にわたって攪拌した。固体を濾過し、45°〜50℃において真空下で乾燥すると、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩一水和物形態C(X、収率65.3%)333mgが得られた。
【0100】
形態EおよびF:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
【0101】
【化6】
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩をエチレングリコールに溶かし、溶液を、室温においてゆっくりと蒸発させた。1週間後、微細針状晶が生成し、濾過し、空気乾燥し、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩無水形態E(XI)として特徴付けた。
【0102】
濾液を低容積まで蒸発させ、濾過した。単離された生成物は、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩無水形態F(XII)として特徴付けた。
【0103】
形態G:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩
手順:(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(50mg)を、1時間かけて155℃まで加熱し、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩形態Gを製造した。
【0104】
本明細書において識別および記載された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の6つの結晶性形態(形態A〜G)の他に、形態Hと命名された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態も識別された。
【0105】
非結晶性(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩(形態H)の調製
以下の2つの方法を用い、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Hを調製することができる。
【0106】
形態H、方法1。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の20〜100mg/mLの溶液を、注射用の水に薬物物質を溶かすことにより作製した。溶液を0.22μの滅菌フィルターで濾過した。溶液をバイアル/シリンジに充填した。バイアル/シリンジを−45℃において冷凍し、2時間にわたってその温度に保った。凍結乾燥は、150mτにて(−20〜25℃)における一次乾燥と、続く、(20〜30℃)における二次乾燥により完了した。得られる固体を貯蔵する。
【0107】
形態H、方法2。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩1グラムを注射用の水10mLに溶かし、100mg/mL溶液を作製する。溶液を0.22μの滅菌フィルターで濾過する。溶液0.5mLのアリコートをガラスバイアルに充填する。バイアルを凍結乾燥機に装入する。保管温度を、0.5℃/分の速度で−45℃まで下げる。保管温度を2時間にわたって−45℃に保つ。150mτの真空をかける。保管温度を、0.5℃/分の速度で−20℃まで上げる。保管温度を20時間にわたって−10℃に保つ。保管温度を、0.5℃/分の速度で30℃まで上げる。10時間にわたって30℃に保つ。凍結乾燥プロセスを通して、150mτの真空を維持する。
【0108】
本明細書において識別および記載された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の6つの結晶性形態(形態A〜G)および非結晶性形態Hの他に、形態Iと命名された(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非晶質性形態も識別された。
【0109】
以下の2つの方法を用い、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の非結晶性形態Iを調製することができる。
【0110】
形態I、方法1。非晶質性の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、まず、過剰の結晶性形態Aを有機溶媒(アセトニトリルまたはジクロロメタン)に加え、72時間にわたって室温において攪拌することにより調製した。次いで、試料を濾過し、溶液を周囲条件下で室温において蒸発させると、白色の固体が得られ、PXRD分析により非晶質性固体であると決定された。
【0111】
形態I、方法2。非晶質性の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩は、まず、過剰の結晶性形態Aを以下の溶媒系(イソプロピルエーテル、メチルt−ブチルエーテル、または97%酢酸エチル/3%水)のうちの1つに加え、72時間にわたって40℃において攪拌することにより調製した。次いで、試料を濾過し、溶液を40℃において蒸発させると、白色の固体が得られ、PXRD分析により非晶質性固体であると決定された。
【0112】
上記に記載されている(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形性形態は、プロスタグランジン作動薬として有用であり、したがって、そのようなプロスタグランジン作動薬を用いる方法およびそのようなプロスタグランジン作動薬を含有する医薬組成物に有用である。(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形性形態は、プロスタグランジンの調節により仲介される状態、特に、EP2受容体の作動薬により仲介される状態の治療および/または予防に有用である。
【0113】
本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の多形性形態が、放射標識形態で存在することができること、すなわち、前記化合物が、天然において通常見出される原子量または質量数と異なる原子量または質量数を含有する1個または複数の原子を含有することがあることは理解されるであろう。水素、炭素、リン、フッ素および塩素の放射性同位元素は、それぞれ3H、14C、32P、35S、18Fおよび36Clを包含する。これらの放射性同位元素および/または他の原子の他の放射性同位元素を含有する本発明の化合物は、本発明の範囲内にある。トリチウム化、すなわち3H、および炭素−14、すなわち14C、放射性同位元素は、それらの調製の容易さおよび検出能のため特に好ましい。本発明の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の放射標識多形性形態は、一般的に、当業者によく知られている方法で調製することができる。好都合には、そのような放射標識化合物は、上記のスキームおよび/または実施例に開示されている手順を行い、非放射標識試薬の代わりに、容易に入手可能な放射標識試薬を用いることにより調製することができる。
【0114】
当業者は、抗再吸収薬(例えば、プロゲスチン、ポリホスホネート、ビスホスホネート、エストロゲン作動薬/拮抗薬、エストロゲン、エストロゲン/プロゲスチン合剤、Premarin(登録商標)、エストロン、エストリオールまたは17α−もしくは17β−エチニルエストラジオール)を、本発明の化合物と併せて使用することができることを理解するであろう。
【0115】
例示的プロゲスチンは、商業ソースから入手可能であり、アルゲストンアセトフェニド、アルトレノゲスト、酢酸アマジノン、酢酸アナゲストン、酢酸クロルマジノン、シンゲストール、酢酸クロゲストン、酢酸クロメゲストン、酢酸デルマジノン、デソゲストレル、ジメチステロン、ジドロゲステロン、エチネロン、二酢酸エチノジオール、エトノゲストレル、酢酸フルロゲストン、ゲスタクロン、ゲストデン、カプロン酸ゲストノロン、ゲストリノン、ハロプロゲステロン、カプロン酸ヒドロキシプロゲステロン、レボノルゲストレル、リネストレノール、メドロゲストン、酢酸メドロキシプロゲステロン、酢酸メレンゲストロール、二酢酸メチノジオール、ノルエチンドロン、酢酸ノルエチンドロン、ノルエチノドレル、ノルゲスチメート、ノルゲストメト、ノルゲストレル、フェンプロピオン酸オキソゲストン、プロゲステロン、酢酸キンゲスタノール、キンゲストロン、およびチゲストールを包含する。
【0116】
好ましいプロゲスチンは、メドロキシプロゲステロン、ノルエチンドロンおよびノルエチノドレルである。
【0117】
例示的な骨再吸収阻害性ポリホスホネートは、米国特許第3,683,080号に開示されているタイプのポリホスホネートを包含し、その開示は、参照により本明細書に組み込まれるものとする。好ましいポリホスホネートは、ジェミナルジホスホネート(ビス−ホスホネートとも呼ばれる)である。チルドロネート二ナトリウムは、特に好ましいポリホスホネートである。イバンドロン酸は、特に好ましいポリホスホネートである。アレンドロネートは、特に好ましいポリホスホネートである。ゾレドロン酸は、特に好ましいポリホスホネートである。他の好ましいポリホスホネートは、6−アミノ−1−ヒドロキシ−ヘキシリデン−ビスホスホン酸および1−ヒドロキシ−3(メチルペンチルアミノ)−プロピリデン−ビスホスホン酸である。ポリホスホネートは、酸、または可溶性のアルカリ金属塩もしくはアルカリ土類金属塩の形態で投与することができる。ポリホスホネートの加水分解性エステルは、同様に包含される。具体例は、エタン−1−ヒドロキシ1,1−ジホスホン酸、メタンジホスホン酸、ペンタン−1−ヒドロキシ−1,1−ジホスホン酸、メタンジクロロジホスホン酸、メタンヒドロキシジホスホン酸、エタン−1−アミノ−1,1−ジホスホン酸、エタン−2−アミノ−1,1−ジホスホン酸、プロパン−3−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、プロパン−N,N−ジメチル−3−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、プロパン−3,3−ジメチル−3−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、フェニルアミノメタンジホスホン酸、N,N−ジメチルアミノメタンジホスホン酸、N(2−ヒドロキシエチル)アミノメタンジホスホン酸、ブタン−4−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、ペンタン−5−アミノ−1−ヒドロキシ−1,1−ジホスホン酸、ヘキサン−6−アミノ−1−ヒドロキシ−1,1−ジホスホン酸ならびに薬学的に許容できるそれらのエステルおよび塩を包含する。
【0118】
特に、本発明の化合物は、哺乳類のエストロゲン作動薬/拮抗薬と組み合わせることができる。本発明の第2の化合物として、任意のエストロゲン作動薬/拮抗薬を使用することができる。エストロゲン作動薬/拮抗薬という用語は、エストロゲン受容体と結合し、骨代謝回転を阻害しかつ/または骨喪失を予防する化合物を指す。特に、エストロゲン作動薬は、本明細書において、哺乳類組織におけるエストロゲン受容体部位と結合し、1つまたは複数の組織においてエストロゲンの作用を模倣することができる化合物と定義される。エストロゲン拮抗薬は、本明細書において、哺乳類組織におけるエストロゲン受容体部位と結合し、1つまたは複数の組織においてエストロゲンの作用をブロックすることができる化合物と定義される。そのような活性は、エストロゲン受容体結合アッセイ、標準的な骨の組織形態計測法および密度測定法、ならびにEriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London 1994、1〜296ページを包含する標準的アッセイで当業者により容易に決定される。様々なこれらの化合物について、以下に記載および言及する。
【0119】
好ましいエストロゲン作動薬/拮抗薬は、米国特許第5,047,431号に開示されているドロロキシフェン:(フェノール、3(1−(4−(2−(ジメチルアミノ)エトキシ)フェニル)−2−フェニル−1−ブテニル)−、(E)−)および関連化合物であり、その開示は、参照により本明細書に組み込まれるものとする。
【0120】
別の好ましいエストロゲン作動薬/拮抗薬は、Willson他、Endocrinology、1997、138、3901〜3911に開示されている3−(4−(1,2−ジフェニル−ブタ−1−エニル)−フェニル)−アクリル酸である。
【0121】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,536,516号に開示されているタモキシフェン:(エタナミン、2−(−4−(1,2−ジフェニル−1−ブテニル)フェノキシ)−N,N−ジメチル、(Z)−2−、2−ヒドロキシ−1,2,3−プロパントリカルボキシレート(1:1))および関連化合物であり、その開示は、参照により本明細書に組み込まれるものとする。
【0122】
別の関連化合物は、米国特許第4,623,660号に開示されている4−ヒドロキシタモキシフェンであり、その開示は、参照により本明細書に組み込まれるものとする。
【0123】
好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,418,068号に開示されているラロキシフェン:(メタノン、(6−ヒドロキシ−2−(4−ヒドロキシフェニル)ベンゾ[b]チエン−3−イル)(4−(2−(1−ピペリジニル)エトキシ)フェニル)−塩酸塩)であり、その開示は、参照により本明細書に組み込まれるものとする。
【0124】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,996,225号に開示されているトレミフェン:(エタナミン、2−(4−(4−クロロ−1,2−ジフェニル−1−ブテニル)フェノキシ)−N,N−ジメチル−、(Z)−、2−ヒドロキシ−1,2,3−プロパントリカルボキシレート(1:1)であり、その開示は、参照により本明細書に組み込まれるものとする。
【0125】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第3,822,287号に開示されているセントクロマン(centchroman):(1−(2−((4−(−メトキシ−2,2,ジメチル−3−フェニル−クロマン−4−イル)−フェノキシ)−エチル)−ピロリジンであり、その開示は、参照により本明細書に組み込まれるものとする。レボルメロキシフェンも好ましい。
【0126】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第4,839,155号に開示されているイドキシフェン:(E)−1−(2−(4−(1−(4−ヨード−フェニル)−2−フェニル−ブタ−1−エニル)−フェノキシ)−エチル)−ピロリジノンであり、その開示は、参照により本明細書に組み込まれるものとする。
【0127】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第5,488,058号に開示されている2−(4−メトキシ−フェニル)−3−[4−(2−ピペリジン−1−イル−エトキシ)−フェノキシ]−ベンゾ[b]チオフェン−6−オールであり、その開示は、参照により本明細書に組み込まれるものとする。
【0128】
別の好ましいエストロゲン作動薬/拮抗薬は、米国特許第5,484,795号に開示されている6−(4−ヒドロキシ−フェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−ベンジル)−ナフタレン−2−オールであり、その開示は、参照により本明細書に組み込まれるものとする。
【0129】
別の好ましいエストロゲン作動薬/拮抗薬は、Pfizer Inc.に譲渡されたPCT公開WO95/10513に、調製の方法と一緒に開示されている4−(2−(2−アザ−ビシクロ[2.2.1]ヘプタ−2−イル)−エトキシ)−フェニル)―(6−ヒドロキシ−2−(4−ヒドロキシ−フェニル)−ベンゾ[b]チオフェン−3−イル)−メタノンである。
【0130】
他の好ましいエストロゲン作動薬/拮抗薬は、本発明の譲受人に譲渡された米国特許第5,552,412号に記載されているような化合物を包含し、その開示は、参照により本明細書に組み込まれるものとする。その中に記載されている特に好ましい化合物は、
cis−6−(4−フルオロ−フェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
(−)−cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−1−(6’−ピロロジノエトキシ−3’−ピリジル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロナフタレン;
1−(4’−ピロリジノエトキシフェニル)−2−(4”−フルオロフェニル)−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン;
cis−6−(4−ヒドロキシフェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;および
1−(4’−ピロリジノレトキシフェニル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン
である。
【0131】
他のエストロゲン作動薬/拮抗薬は、米国特許第4,133,814号に記載されている(その開示は、参照により本明細書に組み込まれるものとする)。米国特許第4,133,814号は、2−フェニル−3−アロイル−ベンゾチオフェンおよび2−フェニル−3−アロイルベンゾチオフェン−1−オキシドの誘導体を開示している。
【0132】
当業者は、骨量増加薬とも呼ばれる他の骨同化薬を、本発明の化合物と併せて使用することができることを理解するであろう。骨量増加薬は、World Health Organization Study World Health Organization、「Assessment of Fracture Risk and its Application to Screening for Postmenopausal Osteoporosis(1994).Report of a WHO Study Group.World Health Organization Technical Series 843」に詳述されているような骨折閾値以上であるレベルまで骨量を増加させる化合物である。
【0133】
任意のプロスタグランジン、またはプロスタグランジン作動薬/拮抗薬を、本発明の特定の態様における第2の化合物として使用することができる。当業者は、IGF−1、フッ化ナトリウム、副甲状腺ホルモン(PTH)、副甲状腺ホルモンの活性フラグメント、成長ホルモンまたは成長ホルモン分泌促進薬も使用できることを理解するであろう。以下の段落は、例示的な本発明の第2の化合物についてより詳細に記載している。
【0134】
任意のプロスタグランジンを、本発明の特定の態様における第2の化合物として使用することができる。プロスタグランジンという用語は、骨粗鬆症の治療に有用である天然プロスタグランジンPGD1、PGD2、PGE2、PGE1およびPGF2の類似体である化合物を指す。これらの化合物は、プロスタグランジン受容体に結合する。そのような結合は、標準的アッセイ(例えば、An S.他、Cloning and Expression of EP2 Subtype of Human Receptors for Prostaglandin E2、Biochemical and Biophysical Research Communications、1993、197(1):263〜270)で当業者により容易に決定される。
【0135】
プロスタグランジンは、基本化合物プロスタン酸に関連する脂環式化合物である。基本プロスタグランジンの炭素原子は、カルボン酸炭素原子からシクロペンチル環を経て隣接する側鎖上の末端炭素原子まで連続して付番される。普通、隣接する側鎖は、トランス配向である。シクロペンチル部分のC−9におけるオキソ基の存在は、Eクラス内のプロスタグランジンを示すが、PGE2は、C13−C14におけるトランス不飽和二重結合およびC5−C6におけるシス二重結合を含有する。
【0136】
様々なプロスタグランジンについて以下に記載および言及する。しかしながら、他のプロスタグランジンは当業者に知られているであろう。例示的なプロスタグランジンは、米国特許第4,171,331号および第3,927,197号に開示されており、その各々の開示は、参照により本明細書に組み込まれるものとする。
【0137】
Norrdin他、The Role of Prostaglandins in Bone In Vivo、Prostaglandins Leukotriene Essential Fatty Acids 41、139〜150、1990は、骨同化性プロスタグランジンの総説である。
【0138】
任意のプロスタグランジン作動薬/拮抗薬を、本発明の特定の態様における第2の化合物として使用することができる。プロスタグランジン作動薬/拮抗薬という用語は、プロスタグランジン受容体に結合し(例えば、An S.他、Cloning and Expression of EP2 Subtype of Human Receptors for Prostaglandin E2、Biochemical and Biophysical Research Communications、1993、197(1):263〜270)、in vivoでプロスタグランジンの作用を模倣する(例えば、骨形成を刺激し、骨量を増加させる)化合物を指す。そのような作用は、標準的アッセイで当業者により容易に決定される。Eriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London 1994、1〜296ページ)。様々なこれらの化合物について以下に記載および言及する。しかしながら、他のプロスタグランジン作動薬/拮抗薬は当業者に知られているであろう。例示的なプロスタグランジン作動薬/拮抗薬を以下の通り開示する。
【0139】
本発明の譲受人に譲渡された米国特許第3,932,389号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な2−デスカルボキシ−2−(テトラゾール−5−イル)−11−デスオキシ−15−置換−ω−ペンタノルプロスタグランジンを開示している。
【0140】
本発明の譲受人に譲渡された米国特許第4,018,892号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な16−アリール−13,14−ジヒドロ−PGE2 p−ビフェニルエステルを開示している。
【0141】
本発明の譲受人に譲渡された米国特許第4,219,483号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な2,3,6−置換−4−ピロンを開示している。
【0142】
本発明の譲受人に譲渡された米国特許第4,132,847号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な2,3,6−置換−4−ピロンを開示している。
【0143】
米国特許第4,000,309号(その開示は、参照により本明細書に組み込まれるものとする)に、骨形成活性に有用な16−アリール−13,14−ジヒドロ−PGE2 p−ビフェニルエステルを開示している。
【0144】
米国特許第3,982,016号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な16−アリール−13,14−ジヒドロ−PGE2 p−ビフェニルエステルを開示している。
【0145】
米国特許第4,621,100号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用な置換シクロペンタンを開示している。
【0146】
米国特許第5,216,183号(その開示は、参照により本明細書に組み込まれるものとする)は、骨形成活性に有用なシクロペンタノンを開示している。
【0147】
フッ化ナトリウムを、本発明の特定の態様における第2の化合物として使用することができる。フッ化ナトリウムという用語は、そのすべての形態のフッ化ナトリウム(例えば、徐放性フッ化ナトリウム、持続放出性フッ化ナトリウム)を指す。持続放出性フッ化ナトリウムは、米国特許第4,904,478号に開示されており、その開示は、参照により本明細書に組み込まれるものとする。フッ化ナトリウムの活性は、生物学的プロトコル(例えば、Eriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London1994、1〜296を参照)で当業者により容易に決定される。
【0148】
骨形態形成タンパク質を、本発明の第2の化合物として使用することができる(例えば、Ono、他、Promotion of the Osteogenetic Activity of Recombinant Human Bone Morphogenetic Protein by Prostaglandin E1、Bone、1996、19(6)、581〜588を参照)。
【0149】
任意の副甲状腺ホルモン(PTH)を、本発明の特定の態様における第2の化合物として使用することができる。副甲状腺ホルモンという用語は、骨形成を刺激し、骨量を増加させる副甲状腺ホルモン、そのフラグメントまたは代謝産物およびその構造類似体を指す。副甲状腺ホルモン関連ペプチド、ならびに副甲状腺関連ペプチドの活性フラグメントおよび類似体も包含される(PCT公開WO94/01460を参照)。そのような骨同化機能活性は、標準的アッセイ(例えば、Eriksen E.F.他、Bone Histomorphometry、Raven Press、New York、1994、1〜74ページ;Grier S.J.他、The Use of Dual−Energy X−Ray Absorptiometry In Animals、Inv.Radiol.、1996、31(1):50〜62;Wahner H.W.およびFogelman I.、The Evaluation of Osteoporosis:Dual Energy X−Ray Absorptiometry in Clinical Practice.、Martin Dunitz Ltd.、London 1994、1〜296を参照)で当業者により容易に決定される。様々なこれらの化合物について以下に記載および言及する。しかしながら、他の副甲状腺ホルモンは当業者に知られているであろう。例示的な副甲状腺ホルモンは、以下の参考文献に開示されている。
【0150】
「Human Parathyroid Peptide Treatment of Vertebral Osteoporosis」、Osteoporosis Int.、3、(Supp 1):199〜203。
【0151】
「PTH 1−34 Treatment of Osteoporosis with Added Hormone Replacement Therapy:Biochemical,Kinetic and Histological Responses」Osteoporosis Int.1:162〜170。
【0152】
任意の成長ホルモンまたは成長ホルモン分泌促進薬を、本発明の特定の態様における第2の化合物として使用することができる。成長ホルモン分泌促進薬という用語は、成長ホルモンの放出を刺激するか、または成長ホルモンの作用を模倣する(例えば、骨量の増加につながる骨形成を高める)化合物を指す。そのような作用は、当業者によく知られている標準的アッセイで当業者により容易に決定される。様々なこれらの化合物は、以下の公開されたPCT特許出願:WO95/14666;WO95/13069;WO94/19367;WO94/13696;およびWO95/34311に開示されている。しかしながら、他の成長ホルモンまたは成長ホルモン分泌促進薬は当業者に知られているであろう。
【0153】
特に、好ましい成長ホルモン分泌促進薬は、N−[1(R)−[1,2−ジヒドロ−1−メタンスルホニルスピロ[3H−インドール−3,4’−ピペリジン]−1’−イル]カルボニル]−2−(フェニルメチルオキシ)エチル]−2−アミノ−2−メチルプロパンアミド:MK−677である。
【0154】
他の好ましい成長ホルモン分泌促進薬は、
2−アミノ−N−(2−(3a−(R)−ベンジル−2−メチル−3−オキソ−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ−[4,3−c]ピリジン−5−イル)−1−(R)−ベンジルオキシメチル−2−オキソ−エチル)−イソブチルアミドまたはそのL−酒石酸塩;
2−アミノ−N−(1−(R)−ベンジルオキシメチル−2−(3a−(R)−(4−フルオロ−ベンジル)−2−メチル−3−オキソ−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ[4,3−c]ピリジン−5−イル)−2−オキソ−エチル)イソブチルアミド;
2−アミノ−N−(2−(3a−(R)−ベンジル−3−オキソ−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ[4,3−c]ピリジン−5−イル)−1−(R)ベンジルオキシメチル−2−オキソ−エチル)イソブチルアミド;および
2−アミノ−N−(1−(2,4−ジフルオロ−ベンジルオキシメチル)−2−オキソ−2−(3−オキソ−3a−ピリジン−2−イルメチル−2−(2,2,2−トリフルオロ−エチル)−2,3,3a,4,6,7−ヘキサヒドロ−ピラゾロ[4,3−c]ピリジン−5−イル)−エチル)−2−メチル−プロピオンアミド
を包含する。
【0155】
本明細書に記載されている化合物を調製するのに有用な調製方法の一部は、遠く離れた官能基(例えば、式Iの前駆体における第一級アミン、第二級アミン、カルボキシル)の保護を必要とすることがある。そのような保護の必要性は、遠く離れた官能基の性質および調製方法の条件に応じて異なるであろう。そのような保護の必要性は、当業者により容易に決定される。そのような保護/脱保護方法の使用も、当業者の範囲内にある。保護基およびそれらの使用の一般的な説明については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照されたい。
【0156】
本発明の化合物、それらのプロドラッグならびに薬学的に許容できる前記化合物およびプロドラッグの塩はすべて、脊椎動物、例えば、哺乳動物、特にヒトにおいて骨形成を刺激し、骨量を増加させる薬剤として治療的使用に適応している。骨形成は、骨粗鬆症および骨関連障害の発症と密接に関連していることから、これらの化合物、それらのプロドラッグならびに薬学的に許容できる前記化合物および前記プロドラッグの塩は、骨に対するそれらの作用により、骨粗鬆症を予防、阻止および/または回復させる。
【0157】
脊椎動物、例えば、哺乳類(例えば、ヒト、特に女性)において低骨量を示す状態(例えば、骨粗鬆症)を治療する際の医薬としての本発明化合物の有用性は、in vivoアッセイ、受容体結合アッセイ、サイクリックAMPアッセイおよび骨折治癒アッセイ(それらはすべて、以下に記載される)を包含する従来のアッセイにおいて本発明の化合物の活性により証明される。in viovアッセイ(当業者の範囲内にある適切な改変を含む)を用い、他の同化薬ならびに本発明のプロスタグランジン作動薬の活性を決定することができる。エストロゲン作動薬/拮抗薬プロトコルを用い、特にエストロゲン作動薬/拮抗薬および他の抗吸収薬の活性を決定することができる(当業者の範囲内にある適切な改変を含む)。以下に記載される組合せおよび逐次処理プロトコルは、本明細書に記載されている同化薬(例えば、本発明の化合物)と抗吸収薬(例えば、エストロゲン作動薬/拮抗薬)の組合せの有用性を証明するのに有用である。そのようなアッセイは、それにより、本発明の化合物(または、本明細書に記載されている他の同化薬および抗吸収薬)の活性を、お互いに対して、および他の知られている化合物の活性と比較することができる手段も提供する。これらの比較の結果は、そのような疾患を治療するために、脊椎動物、例えば、ヒトを包含する哺乳類における用量レベルを決定するのに有用である。
【0158】
同化薬in vivoアッセイ
骨形成を刺激し、骨量を増加させる同化骨薬の活性は、無処置の雄性または雌性ラット、性ホルモン欠乏の雄性(精巣摘出)または雌性(卵巣切除)ラットでテストすることができる。
【0159】
様々な齢(3カ月齢など)の雄性または雌性ラットを試験で使用することができる。ラットは、無処置であるか、または去勢(卵巣切除または精巣摘出)されており、30日間にわたって様々な投与量(1、3または10mg/kg/日)でプロスタグランジン作動薬を皮下注射または胃管投与される。去勢ラットにおいて、治療は、手術の翌日に(骨喪失を予防する目的で)または骨喪失が既に起きた時点で(骨量を回復する目的で)開始する。試験中、すべてのラットに、水ならびにカルシウム1.46%、リン0.99%およびビタミンD34.96IU/gを含有するペレット市販飼料(Teklad Rodent Diet #8064、Harlan Teklad、Madison、WI)を自由に摂取させる。すべてのラットに屠殺前12日目および2日目にカルセイン10mg/kgを皮下注射する。ラットを屠殺する。以下のエンドポイントを決定する:
【0160】
大腿骨塩測定:
各ラットからの右大腿骨を、剖検時に摘出し、「Regional High Resolution Scan」ソフトウェア(Hologic Inc.、Waltham、MA)を備えた二重エネルギーX線吸収法(DXA、QDR1000/W、Hologic Inc.、Waltham、MA)を用いてスキャンする。スキャン領域サイズは、5.08×1.902cmであり、解像度は、0.0254×0.0127cmであり、スキャン速度は、7.25mm/秒である。大腿骨スキャン画像を分析し、全大腿骨(WF)、遠位大腿骨幹端(DFM)、大腿骨幹軸(FS)、および近位大腿骨(PF)の、骨面積、骨塩量(BMC)、および骨塩密度(BMD)を決定する。
【0161】
脛骨組織形態計測分析:
右脛骨を、剖検時に摘出し、筋肉を切り離し、3つの部分に切り分ける。近位脛骨および脛骨幹軸を、70%エタノール中で固定し、段階的濃度のエタノール中で脱水し、アセトン中で脱脂し、次いで、メタクリル酸メチル(Eastman Organic Chemicals、Rochester、NY)に包埋する。
【0162】
厚さ4および10μmで、近位脛骨幹端の前部切片を、Reichert−Jung Polycut Sミクロトームを用いて切り取る。4μmの切片を、変法マッソントリクローム染色で染色し、一方、10μmの切片は、未染色のままとする。各ラットからの1つの4μmの切片および1つの10μmの切片を、海綿骨組織形態計測のために使用する。
【0163】
厚さ10μmで、脛骨幹の断面切片を、Reichert−Jung Polycut Sミクロトームを用いて切り取る。これらの切片を、皮質骨組織形態計測分析のために使用する。
【0164】
海綿骨組織形態計測:Bioquant OS/2組織形態計測システム(R&M Biometrics,Inc.、Nashville、TN)を、成長板−骨端接合部より1.2mm〜3.6mm遠位の近位脛骨幹端の二次海綿骨の静的および動的組織形態計測測定のために使用する。測定を二次海綿骨に限定するため、脛骨幹端領域の最初の1.2mmを除外することが必要である。4μmの切片を用い、骨体積、骨構造、および骨吸収に関連する指数を測定し、一方、10μmの切片を用い、骨形成および骨代謝回転に関連する指数を測定する。
【0165】
I)骨梁の体積および構造に関連する測定および計算:(1)総骨幹端面積(TV、mm2):成長板−骨端接合部より1.2mm〜3.6mm遠位の骨幹端面積。(2)骨梁面積(BV、mm2):TV内の柵状織の総面積。(3)骨梁周囲長(BS、mm):柵状織の総周囲長の長さ。(4)骨梁体積(BV/TV、%):BV/TV×100。(5)骨梁数(TBN、#/mm):1.199/2×BS/TV。(6)骨梁厚(TBT、μm):(2000/1.199)×(BV/BS)。(7)骨梁間隔(TBS、μm):(2000/1.199)×(TV−BV)。
【0166】
II)骨吸収に関連する測定および計算:(1)破骨細胞数(OCN、#):総骨幹端面積内の破骨細胞の総数。(2)破骨細胞周囲長(OCP、mm):破骨細胞により覆われた骨梁周囲長の長さ。(3)破骨細胞数/mm(OCN/mm、#/mm):OCN/BS。(4)破骨細胞周囲長率(%OCP、%):OCP/BS×100。
【0167】
III)骨の形成および代謝回転に関連する測定および計算:(1)単一カルセイン標識周囲長(SLS、mm):1回のカルセイン標識で標識された骨梁周囲長の全長。(2)二重カルセイン標識周囲長(DLS、mm):2回のカルセイン標識で標識された骨梁周囲長の全長。(3)標識間幅(ILW、μm):2回のカルセイン標識間の平均距離。(4)石灰化周囲長率(PMS、%):(SLS/2+DLS)/BS×100。(5)石灰化速度(mineral apposition rate)(MAR、μm/日):ILW/標識間隔。(6)骨形成速度/表面基準(ref.)(BFR/BS、μm2/d/μm):(SLS/2+DLS)×MAR/BS。(7)骨代謝回転速度(BTR、%/y):(SLS/2+DLS)×MAR/BV×100。
【0168】
皮質骨組織形態計測:BioquantOS/2組織形態計測システム(R&M Biometrics,Inc.、Nashville、TN)を、脛骨幹軸皮質骨の静的および動的組織形態計測のために用いる。総組織面積、骨髄腔面積、骨膜周囲長、内皮質周囲長、単一標識周囲長、二重標識周囲長、ならびに骨膜表面と内皮質表面の両方の標識間幅を測定し、皮質骨面積(総組織面積−骨髄腔面積)、皮質骨面積率(皮質面積/総組織面積×100)、骨髄面積率(骨髄腔面積/総組織面積×100)、骨膜および内皮質の標識周囲長率[(単一標識周囲長/2+二重標識周囲長)/総周囲長×100]、石灰化速度(標識間幅/間隔)、ならびに骨形成速度[石灰化速度×[(単一標識周囲長/2+二重標識周囲長)/総周囲長]を計算する。
【0169】
統計値
統計値は、StatView4.0パッケージ(Abacus Concepts,Inc.、Berkeley、CA)を用いて計算することができる。分散分析(ANOVA)検定と、続く、フィッシャーのPLSD(Stat View、Abacus Concepts Inc.、1918 Bonita Ave、Berkeley、CA94704−1014)を用い、群間の差を比較する。
【0170】
組換えヒトEP2およびEP4受容体を安定に過剰発現する293−S細胞系におけるcAMP上昇の測定
ヒトEP2およびEP4受容体の完全オープンリーディングフレームに相当するcDNAを、公表された配列(Regan,J.W.Bailey,T.J.Pepperl,D.J.Pierce,K.L.Bogardus,A.M.Donello,J.E.Fairbairn,C.E.Kedzie,K.M.Woodward,D.F.およびGil,D.W.1994 Cloning of a Novel Human Prostaglandin Receptor with Characteristics of the Pharmacologically Defined EP2 Subtype。Mol.Pharmacology 46:213〜220;およびBastien,L.、Sawyer,N.、Grygorczyk,R.、Metters,K.、およびAdam,M.1994 Cloning,Functional Expression,and Characterization of the Human Prostagrandin E2 Receptor EP2 Subtype。J.Biol.Chem.Vol269、16:11873〜11877)および一次ヒト腎細胞(EP2)または一次ヒト肺細胞(EP4)由来のRNAに基づくオリゴヌクレオチドプライマーを鋳型として用いる逆転写酵素ポリメラーゼ連鎖反応により作製する。cDNAを、pcDNA3(Invitrogen Corporation、3985B Sorrento Valley Blvd.、San Diego、CA92121)の多重クローニング部位中にクローニングし、それを用い、リン酸カルシウム共沈を介して293−Sヒト胚性腎細胞にトランスフェクトする。G418耐性コロニーを拡張させ、特異的[3H]PGE2結合について調べる。高レベルの特異的[3H]PGE2結合を示すトランスフェクタントを、スキャッチャード解析によりさらに特徴付け、PGE2についてBmaxおよびKdを決定する。化合物スクリーニングのために選択された系は、PGE2(EP2)の場合、細胞当たり約338,400個の受容体およびKd=12nMを、およびPGE2(EP4)の場合、細胞当たり約256,400個の受容体およびKd=2.9nMを有する。親の293−S細胞における両受容体の構成的発現は無視できる。細胞は、ウシ胎児血清(最終10%)およびG418(最終700ug/ml)を添加したRPMI中に維持する。
【0171】
293−S/EP2系および293−S/EP4系におけるcAMP応答は、Ca++およびMg++欠乏のPBS1ml中で激しく叩くことにより細胞を培養フラスコから引き離し、1×106細胞/mlの最終濃度まで無血清RPMIを加え、3−イソブチル−1−メチルキサンチン(IBMX)を1mMの最終濃度まで加えることにより測定する。直ちに、細胞懸濁液1ミリリットルを、個々の2mlのスクリューキャップ微小遠心管に等分し、37℃、5%CO2、相対湿度95%にて、蓋を取って10分間にわたってインキュベートする。次いで、DMSOまたはエタノールの最終濃度が1%になるように、テストされる化合物を1:100の希釈度で細胞に加える。化合物を加えた直後に、管に蓋をし、2回逆さにすることにより混ぜ、12分間にわたって37℃においてインキュベートする。次いで、試料を、10分間にわたって100℃においてインキュベートすることにより溶解し、直ちに、5分間にわたって氷上で冷却する。細胞残屑を、5分間にわたって1000×gにて遠心分離することによりペレット化し、澄明にしたライセートを新たな試験管に移す。cAMP濃度は、澄明にしたライセートをcAMP RIAアッセイ緩衝液(キットに包含されている)中で1:10に希釈した後、市販のcAMPラジオイムノアッセイキットRIA(NEK−033、DuPont/NEN Research Products、549 Albany St.、Boston、MA02118)を用いて測定する。通常、1 logの増分で、6〜8種類の濃度のテストすべき化合物で細胞を処理する。EC50の計算は、用量反応曲線の直線部分に対する線形回帰分析を用い、計算機で行う。
【0172】
プロスタグランジンE2受容体との結合についてのアッセイ
膜調製:すべての操作は4℃において行う。プロスタグランジンE21型受容体(EP1)、2型(EP2)、3型(EP3)または4型(EP4)受容体を発現するトランスフェクトされた細胞を収集し、緩衝液A[50mM Tris−HCl(pH7.4)、10mM MgCl2、1mM EDTA、1mM ペファブロックペプチド、(Boehringer Mannheim Corp.、Indianapolis、IN)、10uM ホスポラミドン(Phosporamidon)ペプチド、(Sigma、St.Louis、MO)、1uMペプスタチンAペプチド、(Sigma、St.Louis、MO)、10uM エラスタチナールペプチド、(Sigma、St.Louis、MO)、100uMアンチパインペプチド、(Sigma、St.Louis、MO)]中に1ml当たり200万個の細胞になるように懸濁する。細胞を、2回の15秒バーストでBranson Sonifier(Model #250、Branson Ultrasonics Corporation、Danbury、CT)による超音波処理により溶解する。溶解しない細胞および残屑を、10分間にわたって100×gにて遠心分離することにより除去する。次いで、膜を、30分間にわたって45,000×gにて遠心分離することにより収集する。ペレット化した膜を、1ml当たりタンパク質3〜10mgになるように再懸濁し、ブラッドフォードの方法[Bradford,M.、Anal.Biochem.、72、248(1976)]でタンパク質濃度を決定する。次いで、再懸濁した膜を、使用するまで−80℃において凍結保存する。
【0173】
結合アッセイ:上記のように調製される凍結膜を解凍し、上記の緩衝液A中で1ml当たりタンパク質1mgになるように希釈する。1容積の膜調製物を、緩衝液A中で、0.05容積のテスト化合物または緩衝液および1容積の3nM 3H−プロスタグランジンE2(#TRK431、Amersham、Arlington Heights、IL)と混ぜ合わせる。この混合物(全容積205μL)を、25℃において1時間にわたってインキュベートする。次いで、膜を、Tomtecハーベスター(Model Mach II/96、Tomtec、Orange、CT)を用い、タイプGF/Cガラス繊維フィルター(#1205−401、Wallac、Gaithersburg、MD)に通して濾過することにより回収する。結合した3H−プロスタグランジンE2を有する膜は、フィルターにより捕捉されるが、緩衝液および結合していない3H−プロスタグランジンE2は、フィルターを通過して廃液中に入る。次いで、各試料を[50mM Tris−HCl(pH7.4)、10mM MgCl2、1mM EDTA]3mlで3回洗浄する。次いで、フィルターを、電子レンジ内で加熱することにより乾燥させる。膜に結合している3H−プロスタグランジンの量を測定するため、乾燥したフィルターを、シンチレーション液と一緒にプラスチックバッグに入れ、LKB 1205 Betaplateリーダー(Wallac、Gaithersburg、MD)でカウントする。IC50は、特異的に結合した3H−プロスタグランジンE2の50%を置き換えるのに必要とされるテスト化合物の濃度から決定される。
【0174】
完全長EP1受容体は、Funk他、Journal of Biological Chemistry、1993、268、26767〜26772に開示されているように作製する。完全長EP2受容体は、Regan他、Molecular Pharmacology、1994、46、213〜220に開示されているように作製する。完全長EP3受容体は、Regan他、British Journal of Pharmacology、1994、112、377〜385に開示されているように作製する。完全長EP4受容体は、Bastien、Journal of Biological Chemistry、1994、269、11873〜11877に開示されているように作製する。これらの完全長受容体を用い、EP1、EP2、EP3およびEP4受容体を発現する293S細胞を調製する。
【0175】
ヒトEP1、EP2、EP3またはEP4プロスタグランジンE2受容体を発現する293S細胞は、当業者に知られている方法に従って作製する。通常、公表されている完全長受容体の5’および3’末端に対応するPCR(ポリメラーゼ連鎖反応)プライマーを、上記に開示されているよく知られている方法に従って作製し、ソースとしてのヒト腎(EP1用)、ヒト肺(EP2用)、ヒト肺(EP3用)またはヒト白血球(EP4用)に由来する全RNAを用いるRT−PCRで使用する。PCR産物を、TAオーバーハング法によりpCR2.1(Invitrogen、Carlsbad、CA)中にクローニングし、クローン化された受容体の同一性をDNA配列決定により確認する。
【0176】
293S細胞(Mayo、Dept.of Biochemistry、Northwestern Univ.)に、エレクトロポレーションにより、pcDNA3中のクローン化された受容体をトランスフェクトする。受容体を発現する安定な細胞系は、G418をトランスフェクトした細胞の選択に続いて確立される。
【0177】
最大数の受容体を発現するクローン細胞系は、コンペティターとして非標識PGE2を用いるホールセルの3H−PGE2結合アッセイに続いて選択される。
【0178】
骨折治癒アッセイ
全身投与後の骨折治癒に対する影響についてのアッセイ
骨折技法:3カ月齢のSprage−Dawleyラットを、ケタミンで麻酔する。右の脛骨または大腿骨の近位部分の前内側面に1cmの切開を作製する。以下は、脛骨手術技法について記載している。切開を骨まで到達させ、1mmの孔を、前縁から2mm内側の脛骨粗面の遠位面から4mm近位に開ける。骨髄内釘固定は、0.8mmのステンレス鋼管で行う(最大点荷重36.3N、最大点剛性61.8N/mm、骨と同じ条件下でテストする)。髄管のリーミングは行わない。標準化された閉鎖骨折を、顎が尖っていない特別に設計された調整可能な鉗子を用いる三点曲げにより、脛腓接合部の2mm上方に作製する。軟組織の損傷を最小限に抑えるため、骨折の位置をずらさないように注意する。皮膚を、モノフィラメントナイロン縫合糸で閉じる。手術は、無菌条件下で行う。釘固定直後にすべての骨折のラジオグラフをとり、指定した骨幹領域外に骨折のある、または釘の位置がずれているラットは除外する。残りの動物を、骨折治癒をテストするために、1時点につき各サブグループ当たり10〜12匹の以下のグループに無作為に分ける。第1グループには、1ml/ラットでビヒクル(水:100%エタノール=95:5)を連日胃管投与し、一方、他のラットには、10、20、40および80日間にわたって、テストすべき化合物0.01〜100mg/kg/日(1ml/ラット)を連日胃管投与する。
【0179】
10、20、40および80日目に、各グループからのラット10〜12匹を、ケタミンで麻酔し、放血により屠殺する。両方の脛腓骨を切開により摘出し、すべての軟組織をはぎ取る。各グループにつき5〜6匹のラットからの骨を、組織学的分析のために70%エタノール中に保存し、各グループにつき別の5〜6匹のラットからの骨を、行われるラジオグラフおよび生体力学的テストのために緩衝リンゲル液(+4℃、pH7.4)中に保存する。
【0180】
組織学的分析:骨折した骨の組織学的分析のための方法は、MosekildeおよびBak(The Effects of Growth Hormone on Fracture Healing in Rats:A Histological Description。Bone、14:19〜27、1993)により以前に公表されている。手短に言うと、骨折側を、骨折線の各側までのこぎりで8mm切り、脱灰せずにメタクリル酸メチルに包埋し、8μmの厚さでReichert−Jung Polycutミクロトームで前部切片を切り取る。マッソントリクローム染色した中前部切片(脛骨と腓骨の両方を包含する)を、治療の有無による骨折治癒に対する細胞および組織の応答を可視化するために使用する。シリウスレッド染色した切片を用い、仮骨構造の特徴を立証し、骨折部位における線維性骨と層板骨を区別する。以下の測定を行う:(1)骨折間隙−骨折における皮質骨末端間の最短距離として測定される、(2)仮骨長および仮骨直径、(3)仮骨の総骨体積面積、(4)仮骨面積内組織面積当たりの骨組織、(5)仮骨における線維組織、および(6)仮骨における軟骨面積。
【0181】
生体力学的分析:生体力学的分析のための方法は、BakおよびAndreassen(The Effects of Aging on Fracture Healing in Rats。Calcif Tissue Int 45:292〜297、1989)により以前に公表されている。手短に言うと、生体力学的テストに先立って、すべての骨折のラジオグラフをとる。治癒しつつある骨折の力学的特性を、破壊的三点または四点曲げ手順により分析する。最大点荷重、剛性、最大点荷重時のエネルギー、最大点荷重時の歪み、および最大点応力を測定する。
【0182】
局所投与後の骨折治癒に対する影響についてのアッセイ
骨折技法:約2歳の雌性または雄性ビーグル犬を、麻酔下で試験に用いる。横方向の橈骨骨折を、Lenehan他(Lenehan,T.M.;Balligand,M.;Nunamaker,D.M.;Wood,F.E.:Effects of EHDP on Fracture Healing in Dogs。J Orthop Res 3:499〜507;1985)により記載されているような三点曲げの緩徐な連続荷重により作製する。ワイヤを骨折部位に通して引っ張り、骨の完全な解剖学的破壊を保証する。その後、骨折部位へのプロスタグランジン作動薬の局所送達を、10、15または20週間にわたって、徐放性ペレットにより送達される化合物の徐放により、またはペーストゲルの溶液または懸濁液などの適当な製剤中の化合物の投与により行う。
【0183】
組織学的分析:骨折した骨の組織学的分析のための方法は、Peter他(Peter,C.P.;Cook,W.O.;Nunamaker,D.M.;Provost,M.T.;Seedor,J.G.;Rodan,G.A.Effects of alendronate on fracture healing and bone remodeling in dogs。J.Orthop.Res.14:74〜70、1996)ならびにMosekildeおよびBak(The Effects of Growth Hormone on Fracture Healing in Rats:A Histological Description。Bone、14:19〜27、1993)により以前に公表されている。手短に言うと、屠殺後、骨折した側を、骨折線の各側までのこぎりで3cm切り、脱灰せずにメタクリル酸メチルに包埋し、8μmの厚さでReichert−Jung Polycutミクロトームで前部切片を切り取る。マッソントリクローム染色した中前部切片(脛骨と腓骨の両方を包含する)を、治療の有無による骨折治癒に対する細胞および組織の応答を可視化するために使用する。シリウスレッド染色した切片を用い、仮骨構造の特徴を立証し、骨折部位における線維性骨と層板骨を区別する。以下の測定を行う:(1)骨折間隙−骨折における皮質骨末端間の最短距離として測定される、(2)仮骨長および仮骨直径、(3)仮骨の総骨体積面積、(4)仮骨面積内組織面積当たりの骨組織、(5)仮骨における線維組織、および(6)仮骨における軟骨面積。
【0184】
生体力学的分析:生体力学的分析のための方法は、BakおよびAndreassen(The Effects of Aging on Fracture Healing in Rats。Calcif Tissue Int 45:292〜297、1989)およびPeter他(Peter,C.P.;Cook,W.O.;Nunamaker,D.M.;Provost,M.T.;Seedor,J.G.;Rodan,G.A.Effects of Alendronate On Fracture Healing And Bone Remodeling In Dogs。J.Orthop.Res.14:74〜70、1996)により以前に公表されている。手短に言うと、生体力学的テストに先立って、すべての骨折のラジオグラフをとる。治癒しつつある骨折の力学的特性を、破壊的三点または四点曲げ手順により分析する。最大点荷重、剛性、最大点荷重時のエネルギー、最大点荷重時の歪み、および最大点応力を測定する。
【0185】
エストロゲン作動薬/拮抗薬プロトコル
エストロゲン作動薬/拮抗薬は、骨代謝回転を阻害し、エストロゲン欠乏誘発性骨喪失を予防する化合物の一種である。卵巣切除ラット骨喪失モデルは、閉経後骨喪失のモデルとして広く使用されてきた。このモデルを使用して、骨喪失を予防し骨吸収を阻害する際のエストロゲン作動薬/拮抗薬化合物の有効性をテスト試験することができる。
【0186】
様々な齢(5カ月齢など)のSprague−Dawley雌性ラット(Charles River、Wilmington、MA)を、これらの試験で使用する。実験期間中、ラットを、1匹ずつ20cm×32cm×20cmのケージに収容する。すべてのラットに、水ならびにカルシウム0.97%、リン0.85%、およびビタミンD31.05IU/gを含有するペレット市販飼料(Agway ProLab 3000、Agway Country Food,Inc.、Syracuse、NY)を自由に摂取させる。
【0187】
一群のラット(8〜10匹)に偽手術をし、ビヒクル(10%エタノールおよび90%食塩水、1ml/日)で経口的に治療し、一方、残りのラットは、両側卵巣切除し(OVX)、一定期間(4週間など)にわたってビヒクル(経口)、17β−エストラジオール(Sigma、E−8876、E2、30μg/kg、連日皮下注射)、またはエストロゲン作動薬/拮抗薬(連日経口的に5、10または20mg/kgドロロキシフェンなど)で治療する。骨組織における動的変化を調べるため、すべてのラットに屠殺する12日および2日前にカルセイン(蛍光色素骨マーカー)10mg/kgを皮下注射する。4週間の治療後、ラットを屠殺し、剖検する。以下のエンドポイントを決定する。
【0188】
体重増加:剖検時の体重−手術時の体重。
【0189】
子宮の重量および組織学:剖検中に各ラットから子宮を摘出し、直ちに秤量する。その後、子宮断面組織面積、間質厚、および内腔上皮厚などの組織学的測定のために子宮を処理する。
【0190】
総血清コレステロール:血液を、心臓穿刺により採取し、4℃において凝固させ、次いで、10分間にわたって2,000gにて遠心分離する。血清試料を、高性能コレステロール熱量測定アッセイ(Boehringer Mannheim Biochemicals、Indianapolis、IN)を用いて総血清コレステロールについて分析する。
【0191】
大腿骨塩測定:各ラットからの右大腿骨を、剖検時に摘出し、「Regional High Resolution Scan」ソフトウェア(Hologic Inc.、Waltham、MA)を備えた二重エネルギーX線吸収法(DEXA、QDR 1000/W、Hologic Inc.、Waltham、MA)を用いてスキャンする。スキャン範囲サイズは、5.08×1.902cmであり、解像度は、0.0254×0.0127cmであり、スキャン速度は、7.25mm/秒である。大腿骨スキャン画像を分析し、全大腿骨(WF)、遠位大腿骨幹端(DFM)、大腿骨幹軸(FS)、および近位大腿骨(PF)の、骨面積、骨塩量(BMC)、および骨塩密度(BMD)を決定する。
【0192】
近位脛骨幹端海綿骨組織形態計測分析:右脛骨を、剖検時に摘出し、筋肉を切り離し、3つの部分に切り分ける。近位脛骨を、70%エタノール中で固定し、段階的濃度のエタノール中で脱水し、アセトン中で脱脂し、次いで、メタクリル酸メチル(Eastman Organic Chemicals、Rochester,NY)に包埋する。厚さ4および10μmで、近位脛骨幹端の前部切片を、Reichert−Jung Polycut Sミクロトームを用いて切り取る。各ラットからの1つの4μmの切片および1つの10μmの切片を、海綿骨組織形態計測のために用いる。4μmの切片は、変法マッソントリクローム染料で染色し、一方、10μmの切片は、未染色のままとする。
【0193】
Bioquant OS/2組織形態計測システム(R&M Biometrics,Inc.、Nashville、TN)を、成長板−骨端接合部より1.2mm〜3.6mm遠位の近位脛骨幹端の二次海綿骨の静的および動的組織形態計測測定のために使用する。測定を二次海綿骨に限定するため、脛骨幹端領域の最初の1.2mmを除外することが必要である。4μmの切片を用い、骨体積、骨構造、および骨吸収に関連する指数を測定し、一方、10μmの切片を用い、骨形成および骨代謝回転に関連する指数を測定する。
【0194】
I.骨梁の体積および構造に関連する測定および計算:
1.総骨幹端面積(TV、mm2):成長板−骨端接合部より1.2mm〜3.6mm遠位の骨幹端面積。
2.骨梁面積(BV、mm2):TV内の柵状織の総面積。
3.骨梁周囲長(BS、mm):柵状織の総周囲長の長さ。
4.骨梁体積(BV/TV、%):BV/TV×100。
5.骨梁数(TBN、#/mm):1.199/2×BS/TV。
6.骨梁厚(TBT、μm):(2000/1.199)×(BV/BS)。
7.骨梁間隔(TBS、μm):(2000/1.199)×(TV−BV)。
【0195】
II.骨吸収に関連する測定および計算:
1.破骨細胞数(OCN、#):総骨幹端面積内の破骨細胞の総数。
2.破骨細胞周囲長(OCP、mm):破骨細胞により覆われた骨梁周囲長の長さ。
3.破骨細胞数/mm(OCN/mm、#/mm):OCN/BS。
4.破骨細胞周囲長率(%OCP、%):OCP/BS×100。
【0196】
III.骨の形成および代謝回転に関連する測定および計算:
1.単一カルセイン標識周囲長(SLS、mm):1回のカルセイン標識で標識された骨梁周囲長の全長。
2.二重カルセイン標識周囲長(DLS、mm):2回のカルセイン標識で標識された骨梁周囲長の全長。
3.標識間幅(ILW、μm):2回のカルセイン標識間の平均距離。
4.石灰化周囲長率(PMS、%):(SLS/2+DLS)/BS×100。
5.石灰化速度(MAR、μm/日):ILW/標識間隔。
6.骨形成速度/表面基準(ref.)(BFR/BS、μm2/d/μm):(SLS/2+DLS)×MAR/BS。
7.骨代謝回転速度(BTR、%/y):(SLS/2+DLS)×MAR/BV×100。
【0197】
統計値
統計値は、StatView4.0パッケージ(Abacus Concepts,Inc.、Berkeley、CA)を用いて計算することができる。分散分析(ANOVA)検定と、続く、フィッシャーのPLSD(Stat View、Abacus Concepts,Inc.、1918 Bonita Ave、Berkeley、CA94704−1014)を用い、群間の差を比較する。
【0198】
組合せ治療および順次治療プロトコル
以下のプロトコルは、当業者により変えることができることは言うまでもない。例えば、無処置の雄性または雌性ラット、性ホルモン欠乏の雄性(精巣摘出)または雌性(卵巣切除)ラットを使用することができる。さらに、様々な齢(12カ月齢など)の雄性または雌性ラットを試験で使用することができる。ラットは、無処置であるか、または去勢(卵巣切除または精巣摘出)されており、一定期間(2週〜2カ月など)にわたって様々な投与量(1、3または6mg/kg/日など)で本発明の化合物などの同化薬を投与し、続いて、一定期間(2週〜2カ月など)にわたって様々な投与量(1、5、10mg/kg/日など)でドロロキシフェンなどの抗吸収薬を投与するか、または一定期間(2週〜2カ月など)にわたって様々な投与量で同化薬と抗吸収薬の組合せ治療を行うことができる。去勢ラットにおいて、治療は、手術の翌日(骨喪失を予防する目的で)または骨喪失が既に起きた時点(骨量を回復する目的で)で開始することができる。
【0199】
ラットを、ケタミン麻酔下で屠殺する。以下のエンドポイントを決定する。
【0200】
大腿骨塩測定は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。
【0201】
腰椎骨塩測定:「Regional High Resolution Scan」ソフトウェア(Hologic Inc.、Waltham、MA)を備えた二重エネルギーX線吸収法(QDR 1000/W、Hologic Inc.、Waltham、MA)を用い、麻酔ラットにおける全腰椎および6個の腰椎(LV1〜6)の各々の骨面積、骨塩量(BMC)、および骨塩密度(BMD)を測定。ラットを、ケタミン/ロンパン(4対3の比)の混合物1ml/kgの腹腔内注射により麻酔し、ラットプラットホーム上に置く。スキャン領域サイズは、6×1.9cmであり、解像度は、0.0254×0.0127cmであり、スキャン速度は、7.25mm/秒である。全腰椎スキャン画像を取得し分析する。骨面積(BA)、および骨塩量(BMC)を測定し、骨塩密度は、全腰椎および6個の腰椎(LV1〜6)の各々について計算する(MBCをBAで除す)。
【0202】
近位脛骨幹端海綿骨組織形態計測分析は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。
【0203】
骨梁の体積および構造に関連する測定および計算は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。さらに、骨吸収に関連する測定および計算も、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。さらに、骨の形成および代謝回転に関連する測定および計算は、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されているように行う。さらに、得られるデータは、エストロゲン作動薬/拮抗薬プロトコルで上記に記載されている統計操作を用いて分析する。
【0204】
腎臓再生アッセイ
腎臓再生におけるプロスタグランジン作動薬の役割は、野生型293S細胞およびEP2をトランスフェクトした293S細胞における骨形態形成タンパク質7(BMP−7)の発現を誘導するプロスタグランジンE2(PGE2)またはプロスタグランジン作動薬の能力により検討する。
【0205】
方法:293S細胞およびEP2 293S細胞を、ダルベッコ変法イーグル培地(DMEM、Gibco、BRL;Gaithersburg、MD)中で培養する。PGE2またはプロスタグランジン作動薬で処理する1日前に、細胞を、10cmの皿当たり1.5×106個の細胞の密度でプレートする。一般的に、約16〜24時間後、細胞単層を、OptiMEM(Gibco、BRL;Gaithersburg、MD)で1回洗浄し、続いて、ビヒクル(DMSO)、PGE2(10−6M)またはプロスタグランジン作動薬(10−6M)の存在下および非存在下で皿当たりOptiMEM10mLを加える。8、16および24時間目に細胞を収集し、RNAを抽出する。全RNA(20mg/レーン)のノーザンブロット分析を、32P−標識BMP−7プローブでブロットを探索することにより行う。ブロットを、32P−標識18sリボソームRNAプローブとのハイブリダイゼーションによりRNA負荷について正規化する。PGE2およびプロスタグランジン作動薬は、時間依存的にEP2 293S細胞におけるBMP−7の発現を誘導する。そのような発現誘導は、親細胞系では一般的に観察されない。腎臓再生におけるBMP−7の知られている役割を考えると、時間および受容体特異的に293S腎臓細胞におけるBMP−7発現を誘導するプロスタグランジン作動薬の能力は、腎臓再生におけるプロスタグランジン作動薬の役割を示している。
【0206】
本発明の化合物の投与は、本発明の化合物を全身的および/または局所的に(例えば、骨折、骨切り術または整形手術の部位において)送達する任意の方法を介することができる。これらの方法は、経口経路、非経口経路、十二指腸内経路などを包含する。一般的に、本発明の化合物は、経口的に投与されるが、例えば、経口投与が標的にとって不適切である場合、または患者が薬物を経口摂取することができない場合、非経口投与(例えば、静脈内、筋肉内、経皮、皮下、経直腸または髄内)を利用することができる。
【0207】
化合物は、本発明の化合物またはそれらの組成物の局所適用(例えば、骨折または骨切り術の部位への)により、骨折および骨切り術を治療することおよびそれらの治癒を促進することのために使用される。本発明の化合物は、例えば、適当な溶媒(例えば、ラッカセイ油などの油性溶媒)中の本発明化合物の軟骨成長板への注射によるか、または、切開手術の場合には、骨蝋、脱灰骨粉、ポリマー骨セメント、骨シーラントなどの適当な担体または希釈剤中のそのような化合物のそこへの局所適用により、骨折または骨切り術の部位に適用される。代替方法として、局所適用は、適当な担体または希釈剤中の化合物の溶液もしくは分散液を、ダクロン−メッシュ、ゲル−フォームおよびキールボーン(kiel bone)、もしくは補綴などの整形外科手術において従来から使用されている固体もしくは半固体のインプラントの表面上または中に、塗布または組み入れることにより行うことができる。
【0208】
本発明の化合物は、上記に記載されている同化薬または骨抗吸収薬のうちの1種または複数と組み合わせて、適当な担体または希釈剤中で骨折または骨切り術の部位に局所的に適用することもできる。
【0209】
本発明の範囲内にあるそのような組合せは、任意の順序で同時に、または順次に同時投与することができ、または薬学的に許容できる担体または希釈剤中に上記に記載されているような(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸およびそのナトリウム塩の多形性形態、そのプロドラッグまたは前記化合物もしくは前記プロドラッグの薬学的塩を含む単一医薬組成物を投与することができる。
【0210】
例えば、本発明において、骨同化薬を、3カ月〜3年にわたって単独で、または抗吸収薬と組み合わせて、続いて、3カ月〜3年にわたって抗吸収薬を単独で使用することができ、全治療サイクルを繰り返してもよい。代替方法として、骨同化薬を、3カ月〜3年にわたって単独でまたは抗吸収薬と組み合わせて、続いて、患者の人生の残りの期間にわたって抗吸収薬を単独で使用することができる。例えば、1つの好ましい投与様式において、上記に記載されているような(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸およびそのナトリウム塩の多形性形態、またはそのプロドラッグもしくは薬学的に許容できるプロドラッグの塩を1日1回投与することができ、上記に記載されているような第2の化合物(例えば、エストロゲン作動薬/拮抗薬)を、単回投与または複数回投与で連日投与することができる。代替方法として、例えば、別の好ましい投与様式において、2つの化合物を順次に投与することができ、上記に記載されているような(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸およびそのナトリウム塩化合物の多形性形態、そのプロドラッグまたは薬学的に許容できるプロドラッグの塩を、骨折閾値(World Health Organization Study「Assessment of Fracture Risk and its Application to Screening for Postmenopausal Osteoporosis(1994)。Report of a World Health Organization Study Group。World Health Organization Technical Series 843」)以上であるレベルまで骨量を増加させるのに十分な期間にわたって1日1回投与することができ、続いて、上記に記載されているような第2の化合物(例えば、エストロゲン作動薬/拮抗薬)を、単回投与または複数回投与で連日投与する。上記に記載されているような第1の化合物は、経口送達などの迅速な送達形態で1日1回投与されることが好ましい。
【0211】
いずれにしても、投与される化合物の量およびタイミングは、治療されている対象、苦痛の重症度、投与の様式および処方医師の判断により左右されるであろうことは言うまでもない。したがって、患者間の変動性のため、以下に示す用量は、指針であり、医師は、医師がその患者に適切であると考える治療(例えば、骨量増加)を達成するために薬物の投与量を設定することができる。望まれる治療の程度を考慮する際、医師は、骨量出発レベル、患者の年齢、既存疾患の存在、ならびに他の疾患(例えば、心血管疾患)の存在などの様々な要因のバランスをとらなければならない。
【0212】
一般に、骨量を骨折閾値(本明細書で以前に引用されているWorld Health Organization Studyに詳述されているような)以上であるレベルまで増加させるのに十分な量の本発明の化合物が使用される。
【0213】
一般に、上記に記載されている本発明において使用される同化薬の有効用量は、0.001〜100mg/kg/日、好ましくは0.01〜50mg/kg/日の範囲である。
【0214】
以下の段落は、様々な抗吸収薬についての好ましい用量範囲を提供する。
【0215】
使用すべき抗吸収薬の量は、骨喪失阻害薬としてのその活性により決定される。この活性は、個々の化合物の薬物動態および上記に記載されているようなプロトコル(例えば、エストロゲン作動薬/拮抗薬プロトコル)を用いる骨喪失の阻害におけるその最小有効投与量対最大有効投与量により決定される。
【0216】
一般に、抗吸収薬についての有効用量は、約0.001mg/kg/日〜約20mg/kg/日である。
【0217】
一般に、プロゲスチンについての有効用量は、1日当たり約0.1〜10mgであり、好ましい投与量は、1日当たり約0.25〜5mgである。
【0218】
一般に、ポリホスホネートについての有効用量は、標準的アッセイの骨吸収阻害薬としてのその効力により決定される。
【0219】
一部のポリホスホネートの連日投与のための範囲は、約0.001mg/kg/日〜約20mg/kg/日である。
【0220】
一般に、本発明の治療、例えば、本発明の骨吸収治療についての有効用量は、本発明のエストロゲン作動薬/拮抗薬について、0.01〜200mg/kg/日、好ましくは0.5〜100mg/kg/日の範囲である。
【0221】
特に、ドロロキシフェンについての有効用量は、0.1〜40mg/kg/日、好ましくは0.1〜5mg/kg/日である。
【0222】
特に、ラロキシフェンについての有効用量は、0.1〜100mg/kg/日、好ましくは0.1〜10mg/kg/日である。
【0223】
特に、タモキシフェンについての有効用量は、0.1〜100mg/kg/日、好ましくは0.1〜5mg/kg/日である。
【0224】
特に、2−(4−メトキシ−フェニル)−3−[4−(2−ピペリジン−1−イル−エトキシ)−フェノキシ]−ベンゾ[b]チオフェン−6−オールについての有効用量は、0.001〜1mg/kg/日である。
【0225】
特に、
cis−6−(4−フルオロ−フェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
(−)−cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−6−フェニル−5−(4−(2−ピロリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;
cis−1−(6’−ピロロジノエトキシ−3’−ピリジル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロナフタレン;
1−(4’−ピロリジノエトキシフェニル)−2−(4”−フルオロフェニル)−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン;
cis−6−(4−ヒドロキシフェニル)−5−(4−(2−ピペリジン−1−イル−エトキシ)−フェニル)−5,6,7,8−テトラヒドロ−ナフタレン−2−オール;または
1−(4’−ピロリジノレトキシフェニル)−2−フェニル−6−ヒドロキシ−1,2,3,4−テトラヒドロイソキノリン
についての有効用量は、0.0001〜100mg/kg/日、好ましくは0.001〜10mg/kg/日の範囲である。
【0226】
特に、4−ヒドロキシタモキシフェンについての有効用量は、0.0001〜100mg/kg/日、好ましくは0.001〜10mg/kg/日の範囲である。
【0227】
本発明の化合物は、一般的に、薬学的に許容できるビヒクルまたは希釈剤と一緒に本発明の化合物のうちの少なくとも1つを含む医薬組成物の形態で投与される。したがって、本発明の化合物は、任意の従来型の経口、非経口、直腸または経皮剤形で個々に、または一緒に投与することができる。
【0228】
経口投与の場合、医薬組成物は、液剤、懸濁剤、錠剤、丸剤、カプセル剤、散剤などの形態をとることができる。クエン酸ナトリウム、炭酸カルシウムおよびリン酸カルシウムなどの様々な賦形剤を含有する錠剤は、ポリビニルピロリドン、スクロース、ゼラチンおよびアカシアなどの結合剤に加えて、デンプン、好ましくはジャガイモまたはタピオカデンプンおよび特定の複雑なケイ酸塩などの様々な崩壊剤と一緒に用いられる。さらに、ステアリン酸マグネシウム、ラウリル硫酸ナトリウムおよびタルクなどの滑沢剤は、錠剤化目的に極めて有用であることが多い。類似のタイプの固体組成物も、軟質および硬質充填ゼラチンカプセルにおける充填剤として用いられ、これに関する好ましい材料は、ラクトースすなわち乳糖ならびに高分子量ポリエチレングリコールも包含する。水性懸濁剤および/またはエリキシル剤が経口投与にとって望ましい場合には、本発明の化合物を、様々な甘味剤、矯味剤、着色剤、乳化剤および/または懸濁化剤、ならびに水、エタノール、プロピレングリコール、グリセリンおよびそれらの様々な同類の組合せなどの希釈剤と組み合わせることができる。
【0229】
非経口投与の目的には、ゴマもしくはピーナッツ油または水性プロピレングリコールの溶液、ならびに対応する水溶性の塩の滅菌水溶液を用いることができる。そのような水溶液は、必要ならば、適切に緩衝されていてもよく、液体希釈剤は、まず初めに十分な食塩水またはグルコースで等張にされる。これらの水溶液は、静脈内、筋肉内、皮下および腹腔内注射目的に特に適している。これに関して、用いられる滅菌水性媒体はすべて、当業者によく知られている標準的技法により容易に得ることができる。
【0230】
経皮(例えば、局所)投与の目的には、その他の点では上記の非経口溶液に類似した希釈滅菌、水性または部分的水性水溶液(通常、約0.1%〜5%濃度で)が調製される。
【0231】
特定の量の活性成分を含む様々な医薬組成物を調製する方法は、当業者に知られているか、または本開示を踏まえて当業者に明らかであろう。医薬組成物を調製する方法の例については、Remington’s Pharmaceutical Sciences、Mack Publishing Company、Easter、Pa.、第15版(1975)を参照されたい。
【0232】
本発明の医薬組成物は、本発明の1つまたは複数の化合物を0.1%〜95%、好ましくは1%〜70%含有することができる。いずれにしても、投与すべき組成物または製剤は、治療されている対象の疾患/状態、例えば骨障害を治療するのに有効な量で、ある量の本発明の1つまたは複数の化合物を含有するであろう。
【0233】
本発明は、別々に投与することができる活性成分の組合せで治療することによる骨量の増加および維持に関する態様を有することから、本発明は、キット形態で別々の医薬組成物を組み合わせることにも関する。キットは、2つの別々の医薬組成物、すなわち、上記に記載されているような式Iの化合物、そのプロドラッグまたは薬学的に許容できる前記化合物もしくは前記プロドラッグの塩および第2の化合物を含む。キットは、分割されたボトルまたは分割されたホイルパケットなどの別々の組成物を含有するための容器手段を含むが、別々の組成物は、単一の分割されていない容器内に含有されていてもよい。通常、キットは、別々の成分を投与するための説明書を含む。キット形態は、別々の成分が、異なる剤形(例えば、経口および非経口)で投与されることが好ましい場合、異なる投与間隔で投与される場合、または処方医師が、組合せの個々の成分の用量設定を望む場合は特に有利である。
【0234】
そのようなキットの一例は、いわゆるブリスターパックである。ブリスターパックは、包装産業においてよく知られており、医薬品単位剤形(錠剤、カプセル剤など)を包装するために広く使用されている。一般的に、ブリスターパックは、好ましくは透明なプラスチック材料のホイルで覆われている比較的硬い材料のシートからなる。包装プロセス中に、プラスチックホイルに凹部が形成される。凹部は、包装すべき錠剤またはカプセル剤のサイズおよび形状を有する。次に、錠剤またはカプセル剤は、凹部に入れられ、比較的硬い材料のシートは、凹部が形成された方向から反対のホイルの表面でプラスチックホイルに対して密閉される。結果として、錠剤またはカプセル剤は、プラスチックホイルとシートの間の凹部に密閉される。シートの強度は、凹部上に手で圧力をかけ、それにより凹部の場所で開口部がシートに形成されることにより、錠剤またはカプセル剤をブリスターパックから取り出すことができるような強度であることが好ましい。次いで、錠剤またはカプセル剤は、前記開口部を介して取り出すことができる。
【0235】
キット上の記憶補助を、例えば、数字が、そのように定められた剤形を摂取するべき投与計画の日数と一致する錠剤またはカプセル剤に隣接する数字の形態で提供することが望ましいことがある。そのような記憶補助の別の例は、例えば、以下の通り「First Week,Monday,Tuesday,..etc....Second Week,Monday、Tuesday,...」など、カード上に印刷されたカレンダーである。記憶補助の他の変形例は、容易に明らかであろう。「1日投与量」は、所与の日に摂取すべき単一の錠剤もしくはカプセル剤またはいくつかの錠剤もしくはカプセル剤であってよい。また、式I化合物、そのプロドラッグまたは薬学的に許容できる前記化合物もしくは前記プロドラッグの塩の1日投与量は、1個の錠剤またはカプセルからなっていてもよく、一方、第2の化合物の1日投与量は、いくつかの錠剤またはカプセルからなっていてもよく、逆もまた同様である。記憶補助は、これを反映するべきである。
【0236】
本発明の別の具体的実施形態において、それらの意図した使用の順序で一度に1日投与量を投薬するように設計されたディスペンサーが提供される。ディスペンサーは、投与計画の順守をさらに容易にするように、記憶補助を備えていることが好ましい。そのような記憶補助の一例は、投薬された1日投与量の数を示すメカニカルカウンターである。そのような記憶補助の別の例は、例えば、最後の1日投与量が摂取された日付を読み出し、かつ/または次の投与量を摂取するべき日付を指摘する液晶読み出し、または可聴式注意喚起シグナルと一体となった電池式マイクロチップメモリーである。
【0237】
一般的に、本発明の化合物は、単独で、またはお互いと、もしくは他の化合物と組み合わせて好都合な製剤で投与されるであろう。以下の製剤例は、例示的であるに過ぎず、本発明の範囲を制限することは意図されていない。
【0238】
続く製剤において、「活性成分」は、本発明の1種または複数の化合物を意味する。
【0239】
製剤1:ゼラチンカプセル剤
硬質ゼラチンカプセル剤は、以下を用いて調製する:
【0240】
【表12】
【0241】
錠剤製剤は、以下の成分を用いて調製する:
製剤2:錠剤
【0242】
【表13】
【0243】
成分を混ぜ合わせて圧縮し、錠剤を形成する。
【0244】
代替方法として、各々が活性成分を0.25〜100mg含有する錠剤は、以下のように作製する:
製剤3:錠剤
【0245】
【表14】
【0246】
活性成分、デンプン、およびセルロースを、No.45メッシュU.S.シーブに通し、十分に混合する。ポリビニルピロリドンの溶液を、得られる粉末と混合し、次いで、No.14メッシュU.S.シーブに通す。そのようにして製造した顆粒を、50℃〜60℃において乾燥し、No.18メッシュU.S.シーブに通す。次いで、前もってNo.60U.S.シーブに通したカルボキシメチルセルロースナトリウム、ステアリン酸マグネシウム、およびタルクを顆粒に加え、混合した後、錠剤機で圧縮すると、錠剤が得られる。
【0247】
各々が5ml投与量当たり活性成分を0.25〜100mg含有する懸濁剤は、以下のように製造する:
製剤4:懸濁剤
【0248】
【表15】
【0249】
活性成分を、No.45メッシュU.S.シーブに通し、カルボキシメチルセルロースナトリウムおよびシロップと混合し、滑らかなペーストを形成する。安息香酸溶液、香料、および着色料を、水の一部で希釈し、攪拌しながら加える。次いで、十分な水を加え、必要な容積にする。
【0250】
以下の成分を含有するエアロゾル溶液を調製する:
製剤5:エアロゾル
【0251】
【表16】
【0252】
活性成分をエタノールと混合し、混合物をプロペラントの一部に加え、30℃まで冷却し、充填装置に移す。次いで、必要量をステンレス鋼の容器に送り込み、残りのプロペラントで希釈する。次いで、バルブユニットを容器に取り付ける。
【0253】
坐剤は、以下のように調製する:
製剤6:坐剤
【0254】
【表17】
【0255】
活性成分を、No.60メッシュU.S.シーブに通し、最低限必要な熱を用いて前もって溶融させた飽和脂肪酸グリセリドに懸濁させる。次いで、混合物を、公称2g容量の坐剤鋳型中に注ぎ込み、冷却させる。
【0256】
静脈内製剤は、以下のように調製する:
製剤7:静脈内溶液
【0257】
【表18】
【0258】
上記成分の溶液は、1分当たり約1mLの速度で患者に静脈内投与される。
【0259】
上記の活性成分は、薬剤の組合せであってもよい。
【0260】
上記は、本発明の例示的実施形態に関しており、以下の特許請求の範囲に示されるような本発明の精神および範囲を逸脱することなく改変を行うことができることが理解されるべきであることは言うまでもない。
【図面の簡単な説明】
【0261】
【図1】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Aのディフラクトグラムを示す図である。
【図2】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Bのディフラクトグラムを示す図である。
【図3】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Cのディフラクトグラムを示す図である。
【図4】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Eのディフラクトグラムを示す図である。
【図5】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Fのディフラクトグラムを示す図である。
【図6】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Gのディフラクトグラムを示す図である。
【図6B】本発明による(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩の形態Hのディフラクトグラムを示す図である。
【図6C】本発明による非晶質性の(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩のディフラクトグラムを示す図である。
【図7】(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩、形態Aの固体13C核磁気共鳴スペクトルを示す図である。
【図8】(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩、形態Hの固体13C核磁気共鳴スペクトルを示す図である。
【特許請求の範囲】
【請求項1】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態。
【請求項2】
結晶性形態が、結晶性形態Aであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.1、20.2および20.8を含有する請求項1に記載の結晶性形態。
【請求項3】
結晶性形態が、結晶性形態Aであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.1、16.1、20.2および20.8を含有する請求項1に記載の結晶性形態。
【請求項4】
結晶性形態が、結晶性形態Aであり、
結晶性形態が、結晶性形態1モル当たり水0.5モルを含有する請求項1に記載の結晶性形態。
【請求項5】
結晶性形態が、結晶性形態Bであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.3および3.6を含有する請求項1に記載の結晶性形態。
【請求項6】
結晶性形態が、結晶性形態Bであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.3、3.6、7.1、8.4および13.8を含有する請求項1に記載の結晶性形態。
【請求項7】
結晶性形態が、結晶性形態Bであり、
結晶性形態が、結晶性形態1モル当たり水1.0モルを含有する請求項1に記載の結晶性形態。
【請求項8】
結晶性形態が、結晶性形態Cであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.6、4.0および13.3を含有する請求項1に記載の結晶性形態。
【請求項9】
結晶性形態が、結晶性形態Cであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.5、4.0、5.9、13.3および16.9を含有する請求項1に記載の結晶性形態。
【請求項10】
結晶性形態が、結晶性形態Cであり、
結晶性形態が、結晶性形態1モル当たり水1.0モルを含有する請求項1に記載の結晶性形態。
【請求項11】
結晶性形態が、結晶性形態Eであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、7.0および21.2を含有する請求項1に記載の結晶性形態。
【請求項12】
結晶性形態が、結晶性形態Eであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、7.0、14.1、21.2、22.3および28.5を含有する請求項1に記載の結晶性形態。
【請求項13】
結晶性形態が、結晶性形態Fであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、5.5、8.2および11.0を含有する請求項1に記載の結晶性形態。
【請求項14】
結晶性形態が、結晶性形態Fであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、5.5、8.2、11.0、19.2および27.5を含有する請求項1に記載の結晶性形態。
【請求項15】
結晶性形態が、結晶性形態Gであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.0および7.9を含有する請求項1に記載の結晶性形態。
【請求項16】
結晶性形態が、結晶性形態Gであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.0、7.9、10.0および13.1を含有する請求項1に記載の結晶性形態。
【請求項17】
結晶性形態が、結晶性形態Aであり、
結晶性形態が、百万分率で表される以下の化学シフト、31.2、32.1、33.7および34.5を有する固体13C核磁気共鳴を有する請求項1に記載の結晶性形態。
【請求項18】
結晶性形態が、百万分率で表される以下の化学シフト、31.2、32.1、33.7、34.5、173.2、176.3、および178.2を有する固体13C核磁気共鳴を有する請求項17に記載の結晶性形態。
【請求項19】
結晶性形態が、結晶性形態Aであり、
結晶性形態が、百万分率で表される以下の化学シフト、173.2、176.3、および178.2を有する固体13C核磁気共鳴を有する請求項1に記載の結晶性形態。
【請求項20】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の実質的に純粋な結晶性形態。
【請求項21】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の単離された結晶性形態。
【請求項22】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物の結晶性形態A。
【請求項23】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物の結晶性形態Aを調製するための方法であって、
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸を、遊離の酸として、有機溶媒中の水酸化ナトリウムと接触させて、反応混合物を生成すること、
反応混合物を約50C〜約90Cの第1の温度まで温め、少なくとも1時間にわたって第1の温度を保つこと、
反応混合物を約15C〜約25Cの第2の温度まで冷却すること、および
反応混合物から固体を集めることを含む方法。
【請求項24】
有機溶媒が、酢酸イソプロピルである請求項23に記載の方法。
【請求項25】
酢酸イソプロピルが、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約8〜約12mlで存在し、
水酸化ナトリウムが、水酸化ナトリウムの50%水溶液であり、
水酸化ナトリウムが、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約1〜約1.3mmolで存在する請求項24に記載の方法。
【請求項26】
非結晶性形態が、非結晶性形態Hであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.1を含有する(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態。
【請求項27】
非結晶性形態が、百万分率で表される以下の化学シフト、125.7、151.2、および175.8を有する固体13C核磁気共鳴を有する請求項26に記載の非結晶性形態。
【請求項28】
非晶質性形態が、非晶質性形態Iであり、
X線粉末回折パターンが、実質的に、図6Cに示す通りである(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非晶質性形態。
【請求項1】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の結晶性形態。
【請求項2】
結晶性形態が、結晶性形態Aであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.1、20.2および20.8を含有する請求項1に記載の結晶性形態。
【請求項3】
結晶性形態が、結晶性形態Aであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.1、16.1、20.2および20.8を含有する請求項1に記載の結晶性形態。
【請求項4】
結晶性形態が、結晶性形態Aであり、
結晶性形態が、結晶性形態1モル当たり水0.5モルを含有する請求項1に記載の結晶性形態。
【請求項5】
結晶性形態が、結晶性形態Bであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.3および3.6を含有する請求項1に記載の結晶性形態。
【請求項6】
結晶性形態が、結晶性形態Bであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.3、3.6、7.1、8.4および13.8を含有する請求項1に記載の結晶性形態。
【請求項7】
結晶性形態が、結晶性形態Bであり、
結晶性形態が、結晶性形態1モル当たり水1.0モルを含有する請求項1に記載の結晶性形態。
【請求項8】
結晶性形態が、結晶性形態Cであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.6、4.0および13.3を含有する請求項1に記載の結晶性形態。
【請求項9】
結晶性形態が、結晶性形態Cであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.5、4.0、5.9、13.3および16.9を含有する請求項1に記載の結晶性形態。
【請求項10】
結晶性形態が、結晶性形態Cであり、
結晶性形態が、結晶性形態1モル当たり水1.0モルを含有する請求項1に記載の結晶性形態。
【請求項11】
結晶性形態が、結晶性形態Eであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、7.0および21.2を含有する請求項1に記載の結晶性形態。
【請求項12】
結晶性形態が、結晶性形態Eであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、7.0、14.1、21.2、22.3および28.5を含有する請求項1に記載の結晶性形態。
【請求項13】
結晶性形態が、結晶性形態Fであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、5.5、8.2および11.0を含有する請求項1に記載の結晶性形態。
【請求項14】
結晶性形態が、結晶性形態Fであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、5.5、8.2、11.0、19.2および27.5を含有する請求項1に記載の結晶性形態。
【請求項15】
結晶性形態が、結晶性形態Gであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.0および7.9を含有する請求項1に記載の結晶性形態。
【請求項16】
結晶性形態が、結晶性形態Gであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.2、4.0、7.9、10.0および13.1を含有する請求項1に記載の結晶性形態。
【請求項17】
結晶性形態が、結晶性形態Aであり、
結晶性形態が、百万分率で表される以下の化学シフト、31.2、32.1、33.7および34.5を有する固体13C核磁気共鳴を有する請求項1に記載の結晶性形態。
【請求項18】
結晶性形態が、百万分率で表される以下の化学シフト、31.2、32.1、33.7、34.5、173.2、176.3、および178.2を有する固体13C核磁気共鳴を有する請求項17に記載の結晶性形態。
【請求項19】
結晶性形態が、結晶性形態Aであり、
結晶性形態が、百万分率で表される以下の化学シフト、173.2、176.3、および178.2を有する固体13C核磁気共鳴を有する請求項1に記載の結晶性形態。
【請求項20】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の実質的に純粋な結晶性形態。
【請求項21】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の単離された結晶性形態。
【請求項22】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物の結晶性形態A。
【請求項23】
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩半水和物の結晶性形態Aを調製するための方法であって、
(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸を、遊離の酸として、有機溶媒中の水酸化ナトリウムと接触させて、反応混合物を生成すること、
反応混合物を約50C〜約90Cの第1の温度まで温め、少なくとも1時間にわたって第1の温度を保つこと、
反応混合物を約15C〜約25Cの第2の温度まで冷却すること、および
反応混合物から固体を集めることを含む方法。
【請求項24】
有機溶媒が、酢酸イソプロピルである請求項23に記載の方法。
【請求項25】
酢酸イソプロピルが、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約8〜約12mlで存在し、
水酸化ナトリウムが、水酸化ナトリウムの50%水溶液であり、
水酸化ナトリウムが、(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸1mmol当たり約1〜約1.3mmolで存在する請求項24に記載の方法。
【請求項26】
非結晶性形態が、非結晶性形態Hであり、
X線粉末回折パターンが、CuKα放射線を用いて測定される以下の2θ値、3.1を含有する(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非結晶性形態。
【請求項27】
非結晶性形態が、百万分率で表される以下の化学シフト、125.7、151.2、および175.8を有する固体13C核磁気共鳴を有する請求項26に記載の非結晶性形態。
【請求項28】
非晶質性形態が、非晶質性形態Iであり、
X線粉末回折パターンが、実質的に、図6Cに示す通りである(3−(((4−tert−ブチル−ベンジル)−(ピリジン−3−スルホニル)−アミノ)−メチル)−フェノキシ)−酢酸ナトリウム塩またはその水和物の非晶質性形態。
【図1】

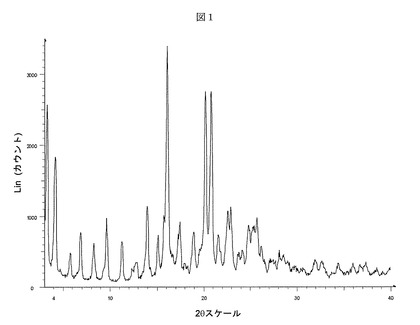
【図2】
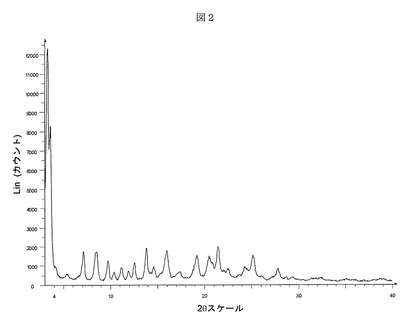
【図3】
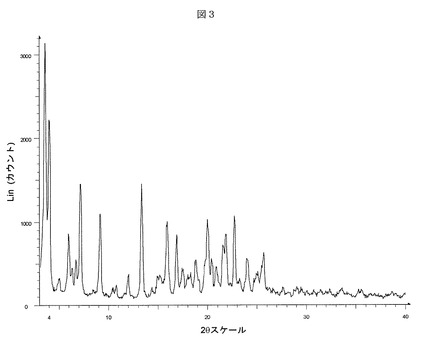
【図4】
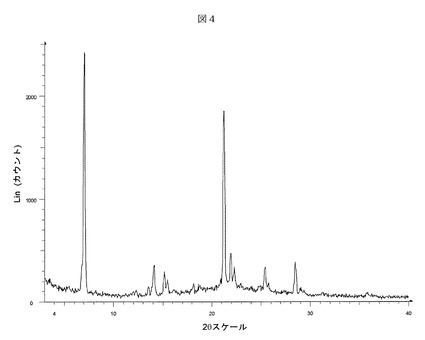
【図5】
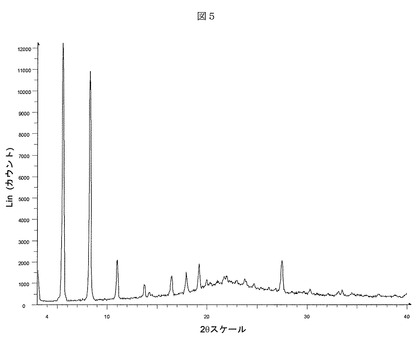
【図6】
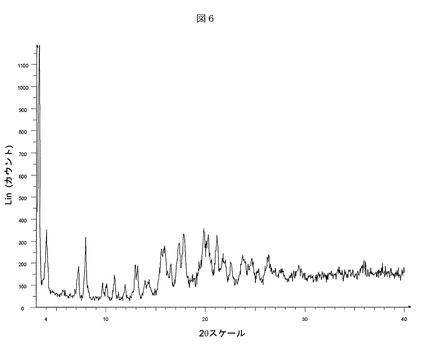
【図6B】
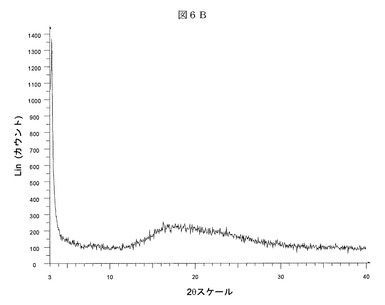
【図6C】
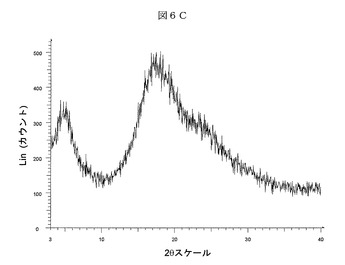
【図7】
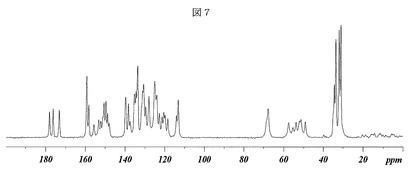
【図8】
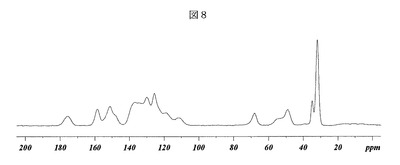
【図2】
【図3】
【図4】
【図5】
【図6】
【図6B】
【図6C】
【図7】
【図8】
【公開番号】特開2009−137934(P2009−137934A)
【公開日】平成21年6月25日(2009.6.25)
【国際特許分類】
【外国語出願】
【出願番号】特願2008−218939(P2008−218939)
【出願日】平成20年8月28日(2008.8.28)
【出願人】(397067152)ファイザー・プロダクツ・インク (504)
【Fターム(参考)】
【公開日】平成21年6月25日(2009.6.25)
【国際特許分類】
【出願番号】特願2008−218939(P2008−218939)
【出願日】平成20年8月28日(2008.8.28)
【出願人】(397067152)ファイザー・プロダクツ・インク (504)
【Fターム(参考)】
[ Back to top ]