
プロスタグランジンD2受容体拮抗剤の二水素リン酸塩
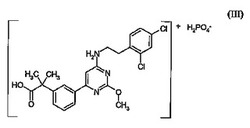
式(III):
【化1】

の2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩、式(III)の化合物の薬学的有効量及び薬学的に許容される担体を含む薬学的組成物、並びにアレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介障害を患う患者に、式(III)の化合物の薬学的有効量を投与することにより、該患者を治療するための方法に関する。
【化1】
の2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩、式(III)の化合物の薬学的有効量及び薬学的に許容される担体を含む薬学的組成物、並びにアレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介障害を患う患者に、式(III)の化合物の薬学的有効量を投与することにより、該患者を治療するための方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
薬学的組成物の大規模製造は化学者及び化学技術者に多くの挑戦を提起し得る。これらの挑戦の多くは大量の試薬の取り扱い及び大規模の反応の制御に関するが、最終生成物の取り扱いは、最終活性生成物自体の性質に関連する特別の挑戦をもたらす。生成物は高収率で製造され、安定でそして容易に単離できなければならないだけでなく、生成物は、最終的に使用される可能性のある製剤の種類に適した性質を有さなければならない。製剤の活性成分の安定性は、合成、単離、バルクの貯蔵、医薬製剤化及び長期用製剤化を含む製造工程の各ステップにおいて考慮されなければならない。これらのステップの各々は、温度及び湿気の種々の環境条件に影響され得る。
【0002】
薬学的組成物を製造するのに使用される薬学的に活性な物質はできるだけ純粋でなければならず、そして長期保蔵でのその化学的安定性は種々の環境条件下で保証されなければならない。これは、望ましくない分解生成物が薬学的組成物中に出現するのを防止するために絶対的に不可欠である。これらの分解生成物は潜在的に毒性であり得るか、或いは組成物の効能を単に減少させる結果となる。
【0003】
薬学的化合物の大規模製造についての主な懸案事項は、活性物質がその取り扱い中、一貫したプロセスパラメータと薬学的品質を確保するために多様な安定性を維持しなければならないことである。薬学的化合物の安定特性に依存して、それは製造及び/又は保存の間に変化して、品質管理上の問題及び製剤化での問題をもたらす可能性がある。かかる変化は製造工程の再現性に影響し、その結果薬学的組成物の製剤に対して監督官庁が課す高品質及び厳しい要件を満たさない最終製剤となる可能性がある。以上を念頭において、一般に、少なくとも物理的安定特性が改良された薬学的化合物の選択は、同じ化合物の安定性がより低い形態よりも著しい利点を与えられることを心に留めるべきである。
【0004】
本発明は、非常に好ましい物性を有する薬理学的に活性なプロスタグランジンD2受容体拮抗剤の二水素リン酸塩に関する。該化合物は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介病的(疾患)病状を患う又は該症状を被りやすい患者を治療するためのDP拮抗剤として有用である。
【背景技術】
【0005】
アレルギー性鼻炎、気管支喘息、アレルギー性結膜炎及びアトピー性皮膚炎をもつ患者で局所的にアレルゲンでチャレンジすると、鼻及び気管支洗浄液、涙及びスキンチャンバー液中のプロスタグランジンD2 “(PGD2)”濃度が急速に上昇する結果をもたらすことが示されている。PGD2は、結膜及び皮膚における血管透過性の増大、鼻腔抵抗の増加、気道縮小、並びに結膜及び気管への好酸性浸潤のような多くの炎症作用を有する。
【0006】
PGD2は、免疫的チャレンジにより肥満細胞から産生するアラキドン酸の主なシクロオキシゲナーゼ生成物である[非特許文献1]。活性化された肥満細胞、即ちPGD2の主な源は、喘息、アレルギー性鼻炎、アレルギー性結膜炎、アレルギー性皮膚炎、及びその他の疾患のようなアレルギー反応症状を駆動するキープレーヤーの1つである[非特許文献2]。
【0007】
PGD2の作用の多くはD−型プロスタグランジン(“DP”)受容体、即ち、上皮及び平滑筋に発現するGタンパク質共役受容体、に対するその作用を通して仲介される。喘息において、気道上皮は、病気の進行を促進する炎症サイトカイン及びケモカインの主な源と長い間認識されていた[非特許文献3]。喘息の実験マウスモデルで、DP受容体は抗原のチャレンジにより気道上皮において劇的に上方調節される[非特許文献4]。DP受容体を欠いたノックアウトマウスで、ヒト喘息の基本的な特徴の二つである、気道過敏性及び慢性炎症の著しい減少が存在する[非特許文献5]。
【0008】
DP受容体はまた、くしゃみ、かゆみ、鼻汁及び鼻詰りの症状により特徴付けられる、よくあるアレルギー性疾患である、ヒトアレルギー性鼻炎に関係するとも考えられている。鼻へのPGD2の局部投与は鼻詰りの用量依存的な増加を引き起こす[非特許文献6]。
【0009】
DP受容体拮抗剤は、モルモットの実験喘息モデルにおいて気道炎症を減少させることが示された[非特許文献7]。従って、PGD2はDP受容体に作用しそしてアレルギー性喘息のある重要な特徴を引き出すのに重要な役割を果たすようである。
【0010】
DP拮抗剤は、多くの種においてアレルギー性鼻炎の症状を軽減するのに有効であることが示され、更に詳しくは、アレルギー性鼻炎で最もよく現れる徴候である、抗原誘発性の鼻詰りを阻止することが示された[非特許文献8及び非特許文献9]。
【0011】
DP拮抗剤はまた、アレルギー性結膜炎及びアレルギー性皮膚炎の実験モデルにおいても有効である[非特許文献10及び非特許文献11]。
【0012】
参照することにより組み入れられる、特許文献1(以後、'732公報と云う)は、特にDP受容体と関連しそしてそれを調節する能力を含む、有効な薬学的性質を有するピリミジンを開示する。'732公報は式(I)のピリミジン:
【化1】
それらの調製、それらの化合物を含む薬学的組成物、及びプロスタグランジンD2受容体の阻害により調節することができる疾患状態の治療へのそれらの薬学的使用を開示する。更に、'732公報は具体的には2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸(以後、“遊離形”と云う)を開示しそしてクレームする。'732公報はまた、該発明の化合物の多様な酸付加塩及び塩基付加塩の一般的記述、並びに塩酸塩及びナトリウム塩の製造に限定された様々な実施例を提供する。更に詳しくは、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の塩酸塩及びナトリウム塩が開示される。しかしながら'732公報は、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩を具体的に開示しない。
【非特許文献1】Lewis, RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ II, Prostaglandin D2 generation after activation of rat and human mast cells with anti−IgE, J. Immunol. 129, 1627−1631, 1982
【非特許文献2】Brightling CE, Bradding P, Pavord ID, Wardlaw AJ, New Insights into the role of the mast cell in asthma, Clin. Exp. Allergy 33, 550−556, 2003
【非特許文献3】Holgate S, Lackie P, Wilson S, Roche W, Davies D, Bronchial Epithelium as a Key Regulator of Airway Allergen Sensisitzation and Remodelling in Asthma, Am J Respir Crit Care Med. 162, 113−117, 2000
【非特許文献4】Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S, prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013−2017, 2000
【非特許文献5】Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S, prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013−2017, 2000
【非特許文献6】Doyle WJ, Boehm S, Skoner DP, Physiologic responses to intranasal dose−response challenges with histamine, methacholine, bradykinin, and prostaglandin in adult volunteers with and without nasal allergy, J Allergy Clin Immunol. 86(6 Pt 1), 924−35, 1990
【非特許文献7】Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H, Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S−5751, J Pharmacol Exp Ther. 298(2), 411−9, 2001
【非特許文献8】Jones, T. R., Savoie, C., Robichaud, A., Sturino, C., Scheigetz, J., Lachance, N., Roy, B., Boyd, M., Abraham, W., Studies with a DP receptor antagonist in sheep and guinea pig models of allergic rhinitis, Am. J. Resp. Crit. Care Med. 167, A218, 2003
【非特許文献9】Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H, Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S−5751. J. Pharmacol. Exp. Ther. 298(2), 411−9, 2001
【非特許文献10】Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H, Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S−5751. J. Pharmacol. Exp. Ther. 298(2), 411−9, 2001
【非特許文献11】Torisu K, Kobayashi K, Iwahashi M, Nakai Y, Onoda T, Nagase T, Sugimoto I, Okada Y, Matsumoto R, Nanbu F, Ohuchida S, Nakai H, Toda M, Discovery of a new class of potent, selective, and orally active prostaglandin D2 receptor antagonists, Bioorg.. & Med. Chem. 12, 5361−5378, 2004
【特許文献1】特許出願WO 2006044732
【発明の開示】
【課題を解決するための手段】
【0013】
本発明は、式(III)の2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩(以後、“二水素リン酸塩”と云う)に関する:
【化2】
【0014】
本発明の別の側面は、薬学的有効量の二水素リン酸塩を含む薬学的組成物である。
【0015】
本発明の別の側面は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介障害を患う患者に、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩の薬学的有効量を投与することにより、該患者を治療するための方法である。
【0016】
本発明を下記の図及び下記の詳細な記述の助けにより更に充分に論じる。
【発明を実施するための最良の形態】
【0017】
略語の定義
上で使用し、そして本発明の記述の全体を通して、下記の略語は、外に指示がない限り、下記の意味を有すると理解されたい:
DMSO:ジメチルスルホキシド
cAMP:環式アデノシンモノホスフェート
IBMX: 3−イソブチル−1−メチルキサンチン
SPA:シンチレーション近接アッセイ
ATTC:アメリカ培養株保存機関
MEM:最小必須培地
FBS:ウシ胎仔血清
CPM;1分当りのカウント数
【0018】
上で使用し、そして本発明の記述の全体を通して、下記の用語は、外に指示がない限り、下記の意味を有すると理解されたい:
“治療する”又は“治療”とは、疾病の予防、部分的緩和、又は治癒を意味する。本発明の化合物及び組成物は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果、2次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介障害を、該障害を患う患者に、式(III)の化合物の薬学的有効量を投与することにより治療するのに有効である。
【0019】
“患者”はヒト及び他の哺乳動物を含む。
【0020】
“薬学的有効量”とは、所望の治療効果を生じるのに有効な化合物、組成物、医薬又は他の活性成分の量を云う。
【0021】
本発明の1つの特定の態様は、結晶形の式(III)の化合物である。
【0022】
本発明の化合物はプロスタグランジンD2受容体拮抗剤活性を示し、そして有効な薬理学的作用薬剤である。従って、該化合物は薬学的組成物に混入され、そしてある種の医学的障害を患う患者の治療に使用される。
【0023】
本発明の化合物は、文献に記載された試験、及び以下の薬学的試験の記載部分によれば、プロスタグランジンD2受容体の拮抗剤であり、その試験結果はヒト及び他の哺乳動物における薬理学的活性と関連していると信じられている。従って、更なる態様において、本発明は、本発明の化合物及び本発明の化合物を含む組成物を、PGD2拮抗剤の投与により改善することができる症状を患う又は該症状を被りやすい患者の治療へ使用することを提供する。例えば、本発明の化合物は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されない多様なPGD2媒介障害の治療に有用であり得る。
【0024】
本発明の治療方法の一つの特定の態様は、アレルギー性鼻炎の治療である。
本発明の治療方法の別の特定の態様は、気管支喘息の治療である。
本発明の治療方法の別の特定の態様は、COPDの治療である。
本発明の治療方法の別の特定の態様は、アレルギー性結膜炎の治療である。
本発明の治療方法の別の特定の態様は、アレルギー性皮膚炎の治療である。
本願で治療とは、確立した症状の治療と同時に予防的治療をも含むと理解されたい。
【0025】
本発明の化合物は更に、下記の少なくとも1種との併用療法を含む治療において有用である:
(i) アレルギー性鼻炎の治療に、抗ヒスタミン剤、例えばフェキソフェナジン(fexofenadine)、デスロラタジン(desloratadine)、ロラタジン(loratadine)及びシチリジン(citirizine);
(ii) アレルギー性鼻炎、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、ロイコトリエン拮抗剤、例えばモンテルカスト(montelukast)及びザフィルラスト(zafirulast)(詳しくはWO 01/78697 A2のクレーム参照);
(iii) 喘息、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、ベータ作用薬、例えばアルブテロール(albuterol)、サルブテロール(salbuterol)及びテルブタリン(terbutaline);
(iv) 喘息、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、抗ヒスタミン剤、例えばフェキソフェナジン、ロラタジン及びシチリジン;
(v) 喘息、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、PDE4 (ホスホジエステラーゼ4)阻害剤、例えばロフルミラスト(roflumilast)及びシロミラスト(cilomilast);又は
(vi) COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、TP(トロンボキサンA2受容体)又はCrTh2(Th2細胞に発現する化学誘引薬受容体に相同な分子)拮抗剤、例えばラマトロブラン(Ramatrobran)(BAY−u3405)。
【0026】
本発明はまたその範囲に、本発明の化合物の薬学的有効量を薬学的に許容される担体と混合して含む薬学的組成物を包含する。
【0027】
実際には、本発明の化合物は薬学的に許容される投与形体でヒト及び他の哺乳動物に、経口、吸入、直腸、鼻、頬、舌下、膣内、直腸内、非経口(皮下、筋肉内、静脈内、皮内、髄腔内、及び硬膜外を含む)、嚢内(intracisternal)及び腹膜内投与を含む局所的又は全身的投与により投与し得る。特定の経路は例えば受容者の生理的状態により変えることができることが理解されるであろう。
【0028】
“薬学的に許容される投与形態”とは、本発明の化合物の投与形態を云い、例えば錠剤、糖衣錠、粉末、エリキシル、シロップ、懸濁液を含む液体製剤、スプレー、吸入用錠剤、トローチ、乳濁液、溶液、顆粒、カプセル及び坐薬、及びリポソーム製剤を含む注射用液体製剤を含む。技術及び製剤は一般にRemington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, latest editionで見つけることができる。
【0029】
本発明の特定の側面は薬学的組成物の形態で投与する本発明の化合物を提供する。
【0030】
薬学的に許容される担体は、投与様式及び投与形体の性質に依存して、薬学的に許容される担体、希釈剤、被覆剤、補助剤、賦形剤、又はベヒクル、例えば保存剤、充填剤、崩壊剤、湿潤剤、乳化剤、エマルション安定剤、懸濁剤、等張剤、甘味剤、矯味矯臭剤、芳香剤、着色剤、抗菌剤、抗カビ剤、他の治療剤、潤滑剤、吸着遅延又は促進剤、及び調合剤、を含む群から選ばれる少なくとも1つの成分を含む。
【0031】
懸濁剤の例には、エトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトール及びソルビタンエステル、微結晶セルロース、メタ水酸化アルミニウム、ベントナイト、寒天及びトラガカント、又はこれらの物質の混合物が含まれる。
【0032】
微生物の作用を防止するための抗菌剤及び抗カビ剤の例には、パラベン、クロロブタノール、フェノール、ソルビン酸等が含まれる。
【0033】
等張剤の例には、糖類、塩化ナトリウム等が含まれる。
【0034】
吸収を遅延させる吸着遅延剤の例には、モノステアリン酸アルミニウム及びゼラチンが含まれる。
【0035】
吸収を促進するための吸着促進剤の例には、ジメチルスルホキシド及び関連する類似体が含まれる。
【0036】
希釈剤、溶剤、ベヒクル、可溶化剤、乳化剤及びエマルション安定剤の例には、水、クロロホルム、スクロース、エタノール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、テトラヒドロフルフリルアルコール、安息香酸ベンジル、ポリオール類、プロピレングリコール、1,3−ブチレングリコール、グリセロール、ポリエチレングリコール、ジメチルホルムアミド、トウィーン(Tween)(登録商標)60、スパン(Span)(登録商標)60、セトステアリルアルコール、ミリスチルアルコール、モノステアリン酸グリセリル、及びラウリル硫酸ナトリウム、ソルビタンの脂肪酸エステル、植物油(例えば綿実油、落花生油、オリーブ油、ひまし油、及びゴマ油)及び注射可能な有機エステル、例えばオレイン酸エチル等、又はこれらの物質の適当な混合物が含まれる。
【0037】
賦形剤の例にはラクト−ス、乳糖、クエン酸ナトリウム、炭酸カルシウム及びリン酸二カルシウムが含まれる。
【0038】
崩壊剤の例には、澱粉、アルギン酸、及びある種の複合ケイ酸塩が含まれる。
【0039】
潤滑剤の例には、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム、タルク、並びに高分子量ポリエチレングリコールが含まれる。
【0040】
薬学的に許容される担体の選択は一般に、溶解性のような活性化合物の化学的性質、投与の特定の様式、及び薬学的実務で順守される規定に従って決定される。
【0041】
経口投与に適した本発明の薬学的組成物は、固体調剤形体のような分離した単位として、例えばそれぞれ活性成分の所定量を含むカプセル、カシェット(cachets)又は錠剤として、又は粉末若しくは顆粒として;又は水性液体若しくは非水性液体の溶液若しくは懸濁液のような液体投与体として、又は水中油型乳濁液若しくは油中水型乳濁液として提供し得る。また活性成分は、ボーラス、舐剤又はペーストとして提供し得る。
【0042】
“固体調剤形体”とは、本発明の化合物の投与が固体形体、例えば、カプセル、錠剤、ピル、粉末、糖衣剤又は顆粒であることを意味する。かかる固体調剤形体において、本発明の化合物は少なくとも1種の不活性な慣用の賦形剤(又は担体)、例えばクエン酸ナトリウム又はリン酸二カルシウム又は:(a)充填剤又は増量剤、例えば澱粉、ラクトース、スクロース、グルコース、マンニトール及びケイ酸、(b)結合剤、例えばカルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロース及びアカシア、(c)保湿剤、例えばグリセロール、(d)崩壊剤、例えば寒天、炭酸カルシウム、馬鈴薯又はタピオカ澱粉、アルギン酸、ある種の複合ケイ酸塩及び炭酸ナトリウム、(e)溶解遅延剤、例えばパラフィン、(f)吸収促進剤、例えば第四級アンモニウム化合物、 (g)湿潤剤、例えばセチルアルコール及びモノステアリン酸グリセロール、(h)吸着剤、例えばカオリン及びベントナイト、(i)潤滑剤、例えばタルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、(j)不透明化剤、(k)緩衝剤、及び本発明の化合物を胃腸管のある部分に遅延して放出する薬剤と混合される。
【0043】
錠剤は、任意に1種又はそれ以上の付属成分と共に圧縮又は成形して製造し得る。圧縮錠剤は、適当な機械中で粉末又は顆粒のような自由流動形体の活性成分を、任意に結合剤、潤滑剤、不活性希釈剤、保存料、界面活性剤又は分散剤と混合して圧縮することにより製造し得る。賦形剤、例えばラクトース、クエン酸ナトリウム、炭酸カルシウム、リン酸二カルシウム、及び崩壊剤、例えば澱粉、アルギン酸、及び、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム及びタルクのような潤滑剤と組合わせたある種の複合ケイ酸塩、のような賦形剤を使用し得る。不活性液体希釈剤で湿らせた粉末化した化合物の混合物を適当な機械で成形して、成形した錠剤を製造し得る。該錠剤は場合によっては被覆又は切り込みを入れ、そしてその中の活性成分を遅延又は制御放出するように調合し得る。
【0044】
固体組成物はまた、ラクトース又は乳糖のような賦形剤、並びに高分子量ポリエチレングリコール等を使用して軟充填及び硬充填ゼラチンカプセル中の充填剤としても使用し得る。
【0045】
所望により、そして更に有効な分配のために、化合物を遅延放出又は標的伝達システム、 例えば生体適合性、生体分解性ポリマーマトリクス(例えばポリ(d,l−ラクチド・コ−グリコリド))、リポソーム及びミクロスフェア、の中にマイクロカプセル化して又はそれに付けて、皮下又は筋肉内デポ(deposit)と呼ばれる技術により皮下又は筋肉内注射して、該化合物を2週間又はそれ以上の期間、継続的にゆっくり放出させることができる。該化合物を、例えばバクテリア保持フィルターを通す濾過により、又は殺菌剤を、使用直前に殺菌水又は他の殺菌した注射可能な媒体に溶解することができる殺菌固体組成物の形で加えることにより、殺菌し得る。
【0046】
“液体投与形体”とは、患者に投与する活性化合物の投与体が液体の形体、例えば薬学的に許容される乳濁液、溶液、懸濁液、シロップ及びエリキシル、であることを意味する。活性化合物に加えて、液体投与形体は当業界で通常使用される不活性希釈剤、溶媒、可溶化剤及び乳化剤を含んでもよい。
【0047】
水性懸濁液が使用される場合、それらは乳化剤又は懸濁を容易にする薬剤を含むことができる。
【0048】
局部投与に適した薬学的組成物とは、患者に局部的に投与するのに適した形体にある配合物を意味する。該配合物は、当業界で一般に知られる局部軟膏、軟膏、粉末及び吸入剤、ゲル(水又はアルコールを基材とする)、クリームとして提供するか、又は経皮バリアーを通して化合物の制御放出を可能にするパッチに適用するためにマトリクスベースに混入し得る。軟膏に調合した場合、活性成分はパラフィン又は水混和性軟膏基材と共に使用しえる。或いは、活性成分は水中油型クリーム基材と共にクリームに調合し得る。目に局部投与するのに適した配合物には、活性成分が適当な担体、特に活性成分の水性溶媒、に溶解又は懸濁した点眼液が含まれる。口に局部投与するのに適した配合物には、活性成分を矯味矯臭基材、通常スクロース及びアカシア又はトラガカント、中に含む薬用キャンデー;活性成分を不活性基材、例えばゼラチン及びグリセリン、又はスクロース及びアカシア、中に含むトローチ;及び活性成分を適当な液体担体中に含むうがい薬が含まれる。
【0049】
乳化薬学的組成物の油相は、公知の成分から公知の方法で構成し得る。該相は乳化剤(或いはエマルゲントとして知られる)のみを含んでもよいが、少なくとも1種の乳化剤と脂肪又は油、又は脂肪及び油の両方との混合物を含むのが望ましい。特別の態様では、親水性乳化剤が、安定剤として作用する脂肪親和性乳化剤と共に含まれる。乳化剤は安定剤と共に又は安定剤なしで、乳化ワックスを形成し、そして油及び脂肪と共に、クリーム配合物の油分散相を形成する乳化軟膏基材を形成する。
【0050】
所望により、クリ−ム基材の水性相は、例えば少なくとも30% w/wの多価アルコール、即ち、2個又はそれ以上の水酸基を有するアルコール、例えばプロピレングリコール、ブタン1,3−ジオール、マンニトール、ソルビトール、グリセロール及びポリエチレングリコール(PEG 400を含む)及びそれらの混合物、を含んでもよい。局部配合物は、皮膚若しくは他の発症領域を通しての活性成分の吸収又は浸透を向上させる化合物を含むのが望ましいであろう。
【0051】
組成物に適した油又は脂肪の選択は所望の性質の達成に基づく。従って、クリームは特にベタベタせず、汚染性でなく、そして洗える製品で、チューブ又は他の容器から漏れるのを防ぐために適当な稠度を有していなければならない。直鎖若しくは分岐鎖一若しくは二塩基性アルキルエステル、例えばミリスチン酸ジイソプロピル、オレイン酸デシル、パルミチン酸イソプロピル、ステアリン酸ブチル、パルミチン酸2−エチルヘキシル、Crodamol CAPとして知られた分岐鎖エステルのブレンド物が使用し得る。これらは、要求される性質に依存して、単独で又は組み合わせて使用し得る。或いは、高融点脂質、例えば白色軟質パラフィン及び/又は液体パラフィン又は他の鉱油を使用することができる。
【0052】
直腸又は膣内投与に適した薬学的組成物とは、患者に直腸内又は膣内投与するのに適した形態でありそして少なくとも1種の本発明の化合物を含む製剤を意味する。坐薬はかかる製剤の特定の形態であり、本発明の化合物を適当な非刺激性賦形剤又は担体、例えば、通常の温度では固体であるが体温では液体であり、従って直腸又は膣腔内で融解して活性成分を放出するココアバター、ポリエチレングリコール又は坐薬ワックスと混合して調製することができる。
【0053】
注射により投与される薬学的組成物は、筋肉内、静脈内、腹膜内及び/又は皮下注射により注射し得る。本発明の組成物は溶液、特に生理学的適合性緩衝液、例えばハンク溶液(Hank's solution)又はリンガー溶液中に配合される。更に、該組成物は固体形体に製剤化されそして使用直前に再溶解又は懸濁され得る。凍結乾燥した形態もまた含まれる。製剤は無菌であり、そして乳濁液、懸濁液、水性及び非水性注射液を含み、それらは懸濁剤及び増粘剤及び酸化防止剤、緩衝剤、静菌剤、及び配合物を等張性にする溶質を含んでもよく、そして意図する受容者の血液に適するように調整したpHを有する。
【0054】
鼻又は吸入投与に適した本発明の薬学的組成物とは、患者に鼻に又は吸入により投与するのに適した形態の組成物を意味する。該組成物は、粒径が例えば1〜500ミクロンの範囲(30ミクロン、35ミクロン等、5ミクロン単位で増加する20〜500ミクロンの範囲の粒径を含む)の粉末形態の担体を含んでもよい。担体が例えば鼻腔スプレー又は点鼻薬として投与するために液体である、適当な組成物には、活性成分の水溶液又は油溶液が含まれる。エアロゾル投与に適した組成物は慣用の方法で調製することができ、そして他の治療剤と共に供給し得る。吸入療法は、特許出願WO2004/026380及び米国特許第5,176,132号に記載されたように、計量投与量吸入器により又は適当な乾燥粉末吸入器、例えばEclipse,
Spinhaler(登録商標)、又はUltrahaler(登録商標)により、容易に投与される。
【0055】
本発明の組成物中の活性成分の実際の投与量は、特定の組成物及び患者への投与方法について所望の治療反応を得るのに有効な活性成分の量を得るように、変えることができる。従って、あらゆる特定の患者への選択した投与量は、所望の治療効果、投与経路、所望する治療持続期間、疾病の病因及び重症度、患者の状態、体重、性別、食生活及び年齢、各活性成分の種類及び効能、吸収速度、代謝作用及び/又は排泄及び他の要因を含む多様の要因に依存する。
【0056】
患者に1回又は分割用量で投与される本発明の化合物の1日合計投与量は、例えば、1日約0.001〜約100mg/kg(体重)、特に0.01〜10mg/kg/日であろう。例えば成人では、投与量は一般に吸入による場合は1日当り約0.01〜約100mg/kg(体重)、特に約0.01〜約10mg/kg(体重)、経口投与では1日当り約0.01〜約100mg/kg(体重)、特に0.1〜70mg/kg(体重)、更に特に0.5〜10mg/kg(体重)、そして静脈投与では1日当り約0.01〜約50mg/kg(体重)、特に0.01〜10mg/kg(体重)である。組成物中の活性成分のパーセントは変化し得るが、適当な投与量が得られるような割合でなければならない。投与単位組成物は、1日の投与量を作り上げるために使用されるようなその約数(submultiples)の量を含み得る。明らかに、いくつかの単位投与形態をほぼ同時に投与してもよい。調剤は、所望の治療効果を得るために、必要な回数投与してもよい。幾人かの患者は高い又は低い投与量に迅速に反応し、ずっと低い維持投与量が適当であることが判明するかもしれない。他の患者には、各特定の患者の生理的要求に従って、1日当り1〜4回投与の割合で長期間治療するのが必要かもしれない。云うまでもなく、他の患者には、1日当り1又は2回以下の投与量を処方することが必要であろう。
【0057】
製剤は薬局業界でよく知られたあらゆる方法で単位投与形態で製造することができる。
【0058】
かかる方法は薬学的活性成分を1種又はそれ以上の補助成分を構成する担体と合わせるステップを含む。一般に製剤は、活性成分を液体担体又は微粉砕固体担体又は両方と均一に且つ密接に合わせ、次に必要な場合は製品に成形することにより製造される。
【0059】
製剤は単位用量又は多回用量容器、例えば密閉アンプル及び弾性栓を有する小瓶、で提供し得、そして使用直前に殺菌液体担体、例えば注射用の水、のみを必要とする凍結乾燥条件で保存し得る。即時注射溶液及び懸濁液は前述した種類の殺菌粉末、顆粒及び錠剤から調製し得る。
【0060】
本発明の化合物を下記の分析方法で分析する。
高圧液体クロマトグラフィー− 質量分光分析(LCMS)
保持時間(RT)及び関連する質量イオンを決定するためのLCMS実験を下記の方法を用いて行う。質量スペクトル(MS)をマイクロマス(Micromass)LCT質量分光計を用いて記録する。該方法は、質量m/zを100から1000まで走査するエレクトロスプレーイオン化法である。液体クロマトグラフィーは、ヒューレットパッカード(Hewlett Packard)1100シリーズのバイナリポンプ&デガッサー(Binary Pump & Degasser)で、静止相:フェノメネックス(phenomenex)のシナージ(Synergi)2μハイドロ(Hydro)−RP 20 X 4.0mmカラム、可動相:A=水中の0.1%蟻酸(FA)、B=アセトニトリル中の0.1% FAで行う。5μLの注入容量はCTC分析パルシステム(PAL System)による。流量は1mL/分である。勾配は3分間で10% Bから90% B、次いで2分間で90% Bから100% Bである。付属する検出器は:ヒューレットパッカード(Hewlett Packard)1100シリーズのUV検出器、波長=220nm、及びセデレセデックス(Sedere SEDEX)75蒸発性光散乱(ELS)検出器、温度=46℃、窒素圧=4バールである。
【0061】
1H核磁気共鳴スペクトル(NMR)
300MHz 1H NMRを、周囲温度で、ASW 5mmプローブを有するバリアンマーキュリー(Varian Mercury)(300MHz)分光計を用いて記録する。NMRにおいて、化学シフト(δ)はテトラメチルシランに対するppmで表す。化学シフト値は、内部標準としてテトタメチルシランを参照して、百万分の一単位(ppm)で示す。
【0062】
X−線粉末回折(XRPD)
XRPDは、シーメンズ−ブルッカー(Siemens−Bruker)D5000回折計で、パラ集束(parafocusing)ブラッグ−ブレンタノ(Bragg−Brentano)(シータ−2−シータ)型の構成を用いて行う。二水素リン酸塩を、 (510)結晶方向に従って切断した単結晶シリコンウェハー上に置く。銅対陰極管(45kV/40mA)から放出される銅K−アルファ放射線(1.54056オングストローム)をX線源として用い、Cu K−ベータ放射線を反射ビーム単色光分光器を用いてろ過除去する。シンチレーションカウンターを検出に用いる。発散スリット0.6mm、抗散乱スリット0.6mm、単色光分光器スリット0.1mm、及び検出器スリット0.6mmを使用する。回折パターンを下記の条件で得る:角度2シータで2〜40度の走査、1ステップ当り1秒のカウント時間、0.02度のステップサイズ、周囲条件の圧力、温度及び相対湿度。
【0063】
熱重量分析による溶媒和/水和状態
熱分析を、TA機器モデルQ−600同時示差走査熱量計/熱重量分析計(DSC/TGA)を用いて乾燥窒素雰囲気下で行う。TGA温度を、インジウム基準を用いて較正する。二水素リン酸塩をアルミニウムパン(TA機器部品番号900793.901)に移す。サーモグラムを、毎分10℃の線形加熱速度で得る。
【0064】
示差走査熱量測定(DSC)
DSCを、冷蔵型冷却システムを備えたTA機器モデルQ−1000 DSCを用いて乾燥窒素雰囲気下で行う。DSCを、インジウム基準を用いて較正する。二水素リン酸塩をアルミニウムパン(TA機器部品番号900793.901)に移し、レーザーで穴あけしたピンホールを有するふた(TA機器部品番号900860.901)を該パンに冷溶接する。DSCサーモグラムを、毎分10℃の線形加熱速度で得る。
【0065】
顕微鏡写真
顕微鏡写真を、交差極(cross polars)を備えたオリンパスBX−41顕微鏡を用いて得る。サンプルは、それを鉱油に分散することにより調製する。
【0066】
粒子サイズ分布
粒子サイズ分布は、R3測定レンズ、RODOS乾式分散器、及び632.8nmに合わせたレーザーを有する、シンパテック(Sympatec)HELOS−BFレーザー回折粒子サイズ分析器を用いて測定する。該システムを、炭化珪素基準を用いて較正する。粉末を、初期圧3.0バールを有するRODOS乾式分散付属部品を用いて分散させ、そして下降を最大にする。容積基準粒子サイズ分布を、シンパテックウィンドクス(Sympatec Windox)(バージョン4.0)ソフトウェアにより、フラウンホッファー(Fraunhofer)法を用いて計算する。
【0067】
動的水蒸気収着(DVS)
水収着プロフィールを、SMS機器動的蒸気収着分析器モデルDVS−1又はVTI機器モデルSGA−100 動的蒸気収着分析器を用いて測定する。RH及び質量を、基準を用いて較正する。二水素リン酸塩を載せ、そして実験開始前に1% RH以下で2.5時間乾燥する。RHを約0から95%RHに進める。試料質量は、質量変化パーセントが、最小絶対平衡時間が15分で、5分の間にわたって0.005%未満の場合、各ステップで一定と考える。
【0068】
フーリエ変換−赤外(FT−IR)分光法
FT−IRスペクトルを、ニコレット(Nicolet)Nic−Plan FT−IR顕微鏡に取り付けたニコレットマグナ(Magna)−IR 分光計55を用いて得る。固体サンプルをKBrディスク上で分析する。スペクトルが、4000−400cm-1から32走査後に4cm-1の解像度で得られる。
【0069】
本発明の化合物の調製及び性質を下記の実験部分に記載する。
【実施例】
【0070】
2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩
ステップ1. 4,6−ジクロロ−2−メトキシピリミジン(0.7g)、2,4−ジクロロフェネチルアミン(0.82g)及び重炭酸ナトリウム(0.88g)のエタノール(25mL)溶液を80℃に3時間加熱し、そして水(400mL)に注ぐ。得られた固体を濾過しそして空気乾燥して(6−クロロ−2−メトキシ−ピリミジン−4−イル)−[2−(2,4−ジクロロ−フェニル)−エチル]−アミンを得る。
ステップ2. リチウムジイソプロピルアミドのテトラヒドロフラン/n−ヘプタン/エチルベンゼン(1.8M, 17mL)溶液に2−(3−ブロモ−フェニル)−プロピオン酸(3g, 13.9ミリモル)のテトラヒドロフラン(5mL)溶液を0℃で15分間で滴下する。混合物を1時間攪拌し、次いでヨウ化メチル(4.93g, 34.8ミリモル)のテトラヒドロフラン(5mL)溶液を10分間で滴下する。反応混合物を15時間攪拌し、2N HClでクエンチし、真空中で濃縮し、そしてエーテル(150mL)で希釈する。エーテル層を2N HClで洗い、2N NaOH (50mL)で3回抽出する。合わせたNaOH層を6N HClでpH=1に酸性化し、そしてエーテル(75mL)で3回抽出する。合わせた有機層をブラインで洗い、硫酸ナトリウムで乾燥し、そして濃縮して2−(3−ブロモ−フェニル)−2−メチル−プロピオン酸を固体(3.08g, 91%)として得、それを更に精製することなく使用する。LC/MS:243 (M+H)。
ステップ3. 2−(3−ブロモ−フェニル)−2−メチル−プロピオン酸(2.18ミリモル)の無水エーテル(20mL)溶液にtert−ブチルリチウム(ペンタン中1.7M, 5.4mL, 9.16ミリモル)を−78℃で滴下し、そしてこの混合物をホウ酸トリブチル(2.34mL, 8.72ミリモル)で処理して30分間攪拌する。反応混合物を室温まで温め、15時間攪拌し、エーテルで希釈し、そして1M H3PO4でクエンチする。30分間攪拌した後、エーテル層を分離し、そして2N NaOH(20mL)で3回抽出する。合わせたNaOH抽出物を6N HClでpH=1に酸性化し、そしてエーテル(50mL)で3回抽出する。合わせた有機抽出物をブラインで洗い、硫酸ナトリウムで乾燥し、そして濃縮して3−(1−カルボキシ−1−メチル−エチル)−フェニルホウ酸を得、それを更に精製することなく使用する。MS:209 (M+H)。
ステップ4. (6−クロロ−2−メトキシ−ピリミジン−4−イル)−[2−(2,4−ジクロロ−フェニル)−エチル]−アミン(0.51ミリモル)及び3−(1−カルボキシ−1−メチル−エチル)−フェニルホウ酸(0.61ミリモル)をアセトニトリル(2.5mL)及び炭酸ナトリウム水溶液(0.4M, 2.5mL)中に含む溶液を、テトラキストリフェニルホスフィンパラジウム(0)(29.5mg, 5mol%)の添加前に窒素で5分間脱ガスする。反応容器を密閉しそしてマイクロ波下で130℃に30分間加熱する。反応混合物に水2mLを加え、2N HCl水溶液を使用してpHを約7に調節し、そしてこの混合物を酢酸エチル(30mL)で3回抽出する。合わせた抽出物をブラインで洗い、硫酸ナトリウムで乾燥し、そして濃縮する。残留物をシリカゲルクロマトグラフィーに付して2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸を固体(205mg, 75%)として得る。LC/MS: RT=2.39分, MS: 460 (M+H); 1H NMR [300 MHz, (CD3)2SO]: δ 12.38 (1H, s), 7.36−8.00 (7H, m), 6.58 (1H, s), 3.84 (3H, s), 3.58 (2H, m), 2.98 (2H, m), 1.54 (6H, s)。
ステップ5. リン酸(3.21mL, 1.49N水溶液)を、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸(2.1g, 4.56ミリモル)のテトラヒドロフラン(45mL)溶液に加え、そして混合物を10分間攪拌する。水を間隔をおいて該混合物が透明溶液になるまで滴下し、そして攪拌を1.5時間室温で続ける。該混合物を真空中で濃縮し、そして残留物をアセトンから再結晶させて2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩を粉末(2.4g, 94%)として得る。LCMS: RT=2.41分;MS: 462 (M+H);1H NMR [300 MHz, (CD3SO)2SO]: δ 7.95 (1H, b), 7.8 (1H, b), 7.6 (2H, b), 7.45 (2H, d, J=2Hz), 7.35 (2H, s), 6.55 (1H, s), 3.85 (3H, s), 3.55 (2H, b), 2.95 (2H, t, J=2Hz), 1.5
(6H, s)。
【0071】
薬理学的試験
本発明の化合物の阻害効果をヒトDP機能検定で評価する。cAMP検定を、内生DP受容体を発現するヒト細胞系LS174Tを使用して用いる。プロトコルは前に記載したもの(Wright DH, Ford−Hutchinson AW, Chadee K, Metters KM, The human prostanoid DP receptor stimulates mucin secretion in LS174T cells, Br J Pharmacol. 131(8):1537−45 (2000))と同様である。
【0072】
ヒトLS174 T細胞におけるSPA cAMP検定のプロトコル
材料
・PGD2 (カイマンケミカル(Cayman Chemical)Cat No.12010)
・IBMX (シグマ(Sigma)Cat No.5879)
・cAMP SPA直接スクリーニング検定システム(アマーシャム(Amersham)コードRPA 559)
・96−穴細胞プレート(ワラック(Wallac)Cat No.1450−516)
・ワラック1450マイクロプレートトリラックス(Trilux)シンチレーションカウンター(パーキンエルマー(PerkinElmer))
・プレート密封器
・エッペンドルフチューブ
・ダルベッコのリン酸塩緩衝食塩水(PBS) (インビトロゲン(Invitrogen)Cat No.14040−133)
・蒸留水
・ボルテックス(Vortex)
・磁気各攪拌器及び攪拌棒
【0073】
試薬の調製:
全試薬は、再構成前に室温に平衡させなければならない。
1Xアッセイ用緩衝液
瓶の内容物を500mL目盛り付きシリンダーに、繰り返し蒸留水で洗うことにより移す。最終容量を蒸留水で500mLに調整し、そして充分混合する。
溶解用試薬1及び2
溶解用試薬1及び2の各々を、夫々200mLのアッセイ用緩衝液に溶解させる。室温に20分間放置して溶解させる。
SPA抗ウサギビーズ
溶解用緩衝液2の30mLを瓶に加える。該瓶を5分間静かに振る。
抗血清
溶解用緩衝液2の15mLを各小瓶に加え、そして内容物が完全に溶解するまで静かに混ぜる。
トレーサー(I125−cAMP)
溶解用緩衝液2の14mLを各小瓶に加え、そして内容物が完全に溶解するまで静かに混ぜる。
免疫試薬の調製
1)トレーサー、抗血清及びSPA抗ウサギ試薬を同じ量瓶に加え、所望の数の穴(150μL/穴)に充分な量のこの混合物が確実に調製されるようにする。
2)充分に混合する。
3)この免疫試薬溶液は各アッセイ前に新たに調製し、再使用してはならない。
標準品
1)溶解用緩衝液1を1mL加え、そして内容物が完全に溶解するまで静かに混ぜる。
2)最終溶液はcAMPを濃度512pmol/mLで含む。
3)7個のポリプロピレン又はポリスチレンチューブに0.2pmol、0.4pmol、0.8pmol、1.6pmol、3.2pmol、6.4pmol及び12.8pmolとラベルを貼る。
4) 溶解用緩衝液1の500μLを全てのチューブにピペットで加える。
5)12.8pmolチューブに標準品原液(512pmol/mL)を500μLピペットで取り、そして充分混合する。12.8pmolチューブから500μLを6.4pmolチューブに移し、そして充分混合する。この2倍希釈を残りのチューブを用いて次々繰り返す。
6)各一連の希釈物及び標準品原液からの2本づつ50μLのアリコートは、0.2〜25.6pmol標準の範囲でcAMPの8個の標準濃度を与える。
化合物希釈用緩衝液
1mM IBMXの50μLをPBS 100mLに加えて最終濃度を100μMにし、そして30℃で20分間超音波処理する。
PGD2の調製
PGD2(FW, 352.5)1mgをDMSO 284μLに溶解させて10mMの原液を作り、20℃で保存する。各アッセイの前に新たに調製する。10mM原液の3μLをDMSO 20mLに加え、それを充分混合し、そして10mLをPBS 40mLに移す。
【0074】
化合物の希釈
本発明の化合物のいくつかのバッチを96穴プレートで試験する。化合物の各バッチは96穴プレートの1列を占める。
化合物の希釈はビオメックス(Biomex)2000(ベックマン(Beckman))中で方法 1_cAMP DP 11ポイントを使用して実施する。
10mM原液化合物プレートからの各化合物5μLを96−穴プレートの穴にそれぞれ以下のように移す。
【表1】
【0075】
カラム7に28μLのDMSOを満たす以外はプレートを45μLのDMSOで満たす。カラム1を充分ピペットで取り、そして12μLをカラム7に平行に移す。1:10連続希釈をカラム1からカラム6まで及びカラム7からカラム11まで、5μLを45μLのDMSOに移すことにより行って下記の濃度を作る。
【表2】
【0076】
新しい96−穴プレートを247.5μLの化合物希釈用緩衝液で満たす。上記プレートからの連続希釈化合物の2.5μLを新しいプレート(1:100希釈)に次のように移す。
【表3】
【0077】
細胞の成長
1.LS174 Tを常にMEM (ATCC Cat No.30−2003), 10% FBS (ATCC Cat No.30−2020)及び追加の2mM L−グルタミン中で、37℃及び5% CO2で増殖させる。
2.0.05%トリプシン及びベルシン(Versine)(インビトロゲン(Invitrogen) Cat No. 25300−054)を37℃水浴で温める。
3.細胞から増殖用培地を除く。T165フラスコ中の細胞をトリプシン4mLで2回洗い、次いで37℃及び5% CO2で3分間インキュベートする。
4.培地10mLを加え、ピペット操作で完全に細胞を分離し、そして細胞を数える。
5.細胞密度を2.25×105細胞/mlにし、そしてアッセイ1日前に96−穴プレート中に200μL細胞/穴(45,000細胞/穴)を蒔く。
【0078】
検定手順
1日目
96−穴プレート中の培地200μLに45,000細胞/穴を蒔く。細胞プレートを37℃、5% CO2及び95%湿度で1晩インキュベートする。
2日目
1.化合物の希釈を行う。
2.アッセイ用緩衝液、溶解用緩衝液1及び2、PGD2及び標準品を用意する。
3.細胞から培地を吸引し、そしてツィマーク(Zymark)のサイクローン(Sciclone)−ALH/FDプロトコルcAMP DPを用いて、化合物溶液100μLを加える。
4.該細胞を37℃、5%CO2及び95%湿度で15分間インキュベートする。
5.300nM PGD2(最終濃度20X 15 nM) 5μLを、各穴にツィマークプロトコルcAMP DP PGD2を用いて加え、そして該細胞を37℃、5%CO2及び95%湿度で更に15分間インキュベートする。
6.細胞から培地を吸引し、そしてツィマークプロトコルcAMP DP溶解を用いて、溶解用緩衝液50μLを加え、そして室温で振とうしながら30分間インキュベートする。
6.免疫試薬150μLを全穴に加える(合計容量200μL/穴)。
7.プレートを密閉し、そして2分間振り、ワラック(Wallac)マイクロタイタープレートμシンチレーションカウンターのチャンバーに16時間入れる。
3日目
[125I] cAMPの量を2分間1450トリラックス(Trilux) シンチレーションカウンター中で計測する。
【0079】
データ処理
cAMP対CPMの標準曲線をセットアップする。
【表4】
【0080】
未知のサンプルのcAMP濃度(pmol/mL)を、cAMP対CPMの標準曲線から計算する。%阻害を下記の式を用いて計算する:
%阻害=[(対照のpmol−サンプルのpmol)×100]/[対象のpmol(細胞+PGD2のみ)]
【0081】
結果
二水素リン酸塩は、0.3ナノモルの濃度で、ヒトLS174 T細胞におけるSPA cAMPアッセイで50%阻害を生じる。
二水素リン酸塩の水溶性は、遊離形及び塩酸塩よりも増加した。下記のチャートに、遊離形、塩酸塩及び二水素リン酸塩の25℃でのリン酸緩衝液(PBS pH 7.4、0.1M一塩基性リン酸ナトリウム溶液19%と0.1M二塩基性リン酸ナトリウム溶液81%との混合物)中の溶解度、並びに遊離形及び二水素リン酸塩の37℃での絶食状態の擬似胃腸液(“FaSSIF”、Dressman J.B., Amidon G.L., Reppas C. 及びShah V.P., Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms, Pharmaceutical Research: 1998, Vol. 15, No. 1, 11−22に従って調製)中の溶解度を記載する。
【表5】
【0082】
二水素リン酸塩は大規模製造及び薬学的製剤に有用な予期しない性質も有する。第1に、二水素リン酸塩は非常に良好な結晶化度を有する。図1に示す二水素リン酸塩のXRPDは中程度の強度と解像度を有するピークを含む。中央の2シータ領域にハロがないことは、無晶相がほとんどないことを示唆する。更に、図8に示された二水素リン酸塩の粉砕前及び粉砕後の顕微鏡写真は、物理化学的性質が変化することなく、粒子径が良好に減少したことを証明する。
【0083】
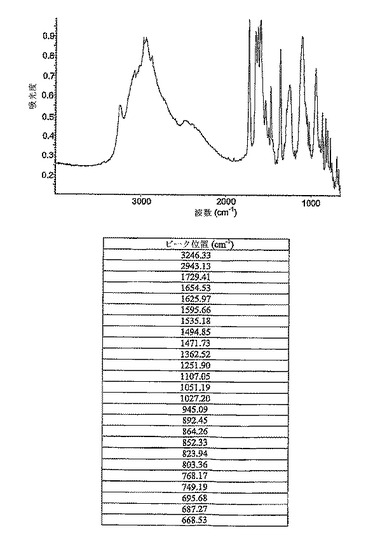
しかしながら、ナトリウム塩は低い結晶化度を有する。図11に示されたように、ナトリウム塩のバッチ2のXRPDは弱い強度の幅広いピークを含み、低い結晶化度を示す。ナトリウム塩のバッチ1のXRPDは、物質が殆ど無晶形であることを示す。更に、両バッチのXRPD中の中央の2シータ領域にハロがあることは、物質の非常に低い結晶化度を示唆する。
【0084】
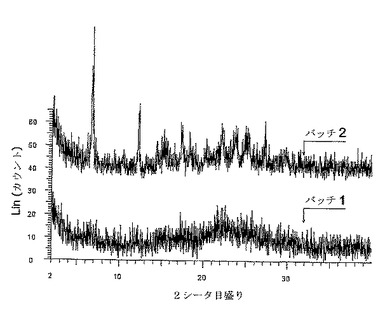
更に、二水素リン酸塩は今回、1つの多形型のみで存在することが示された。しかしながら塩酸塩は今回、3つの多形型で存在することが示され、3つの多形型はある条件下で相互変換に付される可能性がある。塩酸塩の多形型の1つである、図12のバッチ2は、XRPDが弱い強度の幅広いピークを有するので、低い結晶化度を有する。塩酸塩の多形型の別の1つである、図12のバッチ1は、良好な結晶化度を有する。
【図面の簡単な説明】
【0085】
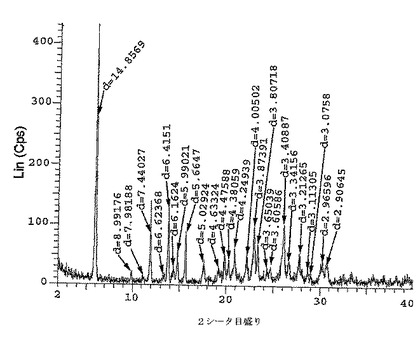
【図1】二水素リン酸塩のX線粉末回折パターン(XRPD)である。
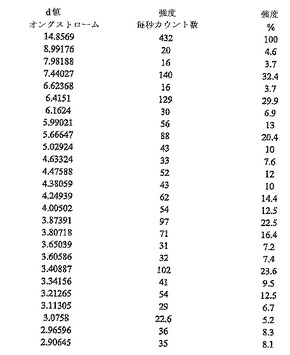
【図2】二水素リン酸塩の図1のX線粉末回折パターンについて、XRPD d−距離及び相対強度の対応する表である。
【図3】二水素リン酸塩の示差走査熱量計−熱重量分析器(DSC−TGA)サーモグラムである。TGAデータは、室温から175℃でほぼ0.4%の合計質量損失を示す。DSCは結晶相の融液のみを含む。分子の分解は融点で始まる。
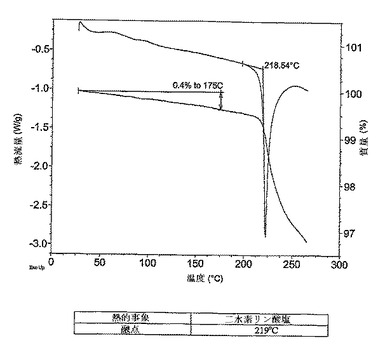
【図4】二水素リン酸塩のDSCサーモグラムである。溶融はサーモグラム中で219℃で開始することが観察された唯一の熱的事象である。
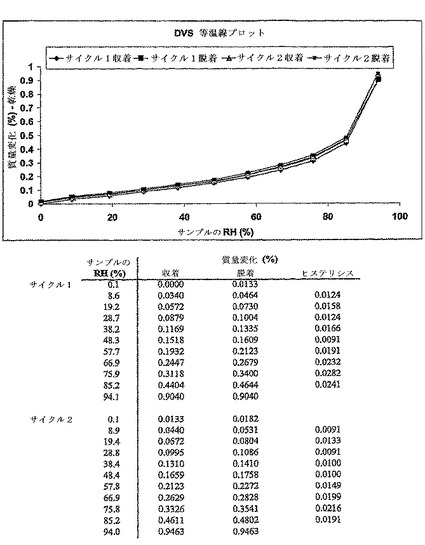
【図5】二水素リン酸塩の動的蒸気収着分析器(DVS)吸湿等温プロット、及び対応する水収着プロフィールの表を示す。データは、94%の相対湿度(RH)でヒステリシスがほとんどない、収着中の〜0.9%の重量増加を示し、これはゆるく結合した表面水収着を示唆する。
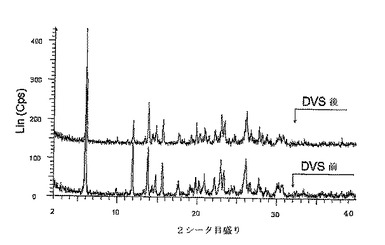
【図6】DVS前及び後の二水素リン酸塩のXRPDパターンを示す。試料を調製しそして分析を、0% RHで12時間置いたDVSから取り出してから2分以内に始める。データは、DVS実験の結果、強度及びd距離にほとんど変化がないことを示し、これは結晶構造に検出可能な変化が起きなかったことを示す。
【図7】二水素リン酸塩の顕微鏡写真を示す。このロットは主として長さが約30ミクロンまでの棒及び針形状の粒子から成る。
【図8】二水素リン酸塩の粉砕前及び粉砕後の顕微鏡写真、及び定量的粒子サイズ分布を示し、これは物理化学的性質に変化なく粒子サイズが良好に減少したことを示す。二水素リン酸塩の粉砕後の顕微鏡写真は、100倍の倍率で約30個の視野の検索の間、長さが10ミクロンよりも大きい粒子がない良く微粉化された結晶性物質を示す。微粉化二水素リン酸塩の粒子サイズ分布は単一モード曲線であると決定される。中央値[x(50)]は2.0ミクロンであり、粒子の90%は4.7ミクロン以下である。微粉化工程は中央値 (以前は5.8ミクロン)及びx(90)サイズ(以前は〜16ミクロン)の両方を減少させる。微粉化の前及び後の二水素リン酸塩のXRPD、即ちピーク強度、位置(d−距離)及び解像度、は同じである。
【図9】微粉化の後の二水素リン酸塩のDVS吸湿性等温線であり、無晶化が起きないことを示す。
【図10】二水素リン酸塩のフーリエ変換−赤外(FT−IR)スペクトル及び対応するFT−IRピークの表を示す。
【図11】ナトリウム塩の2つの別々の調製物、バッチ1及びバッチ2、についてのXRPDパターンの重ね合わせ図を示す。ナトリウム塩のバッチ1はエタノール:酢酸エチルから再結晶させた。ナトリウム塩のバッチ2はメタノール:酢酸エチルから再結晶させた。
【図12】アセトンから再結晶させた塩酸塩の2つの別々の調製物、バッチ1及びバッチ2、についてのXRPDパターンの重ね合わせ図を示す。
【技術分野】
【0001】
薬学的組成物の大規模製造は化学者及び化学技術者に多くの挑戦を提起し得る。これらの挑戦の多くは大量の試薬の取り扱い及び大規模の反応の制御に関するが、最終生成物の取り扱いは、最終活性生成物自体の性質に関連する特別の挑戦をもたらす。生成物は高収率で製造され、安定でそして容易に単離できなければならないだけでなく、生成物は、最終的に使用される可能性のある製剤の種類に適した性質を有さなければならない。製剤の活性成分の安定性は、合成、単離、バルクの貯蔵、医薬製剤化及び長期用製剤化を含む製造工程の各ステップにおいて考慮されなければならない。これらのステップの各々は、温度及び湿気の種々の環境条件に影響され得る。
【0002】
薬学的組成物を製造するのに使用される薬学的に活性な物質はできるだけ純粋でなければならず、そして長期保蔵でのその化学的安定性は種々の環境条件下で保証されなければならない。これは、望ましくない分解生成物が薬学的組成物中に出現するのを防止するために絶対的に不可欠である。これらの分解生成物は潜在的に毒性であり得るか、或いは組成物の効能を単に減少させる結果となる。
【0003】
薬学的化合物の大規模製造についての主な懸案事項は、活性物質がその取り扱い中、一貫したプロセスパラメータと薬学的品質を確保するために多様な安定性を維持しなければならないことである。薬学的化合物の安定特性に依存して、それは製造及び/又は保存の間に変化して、品質管理上の問題及び製剤化での問題をもたらす可能性がある。かかる変化は製造工程の再現性に影響し、その結果薬学的組成物の製剤に対して監督官庁が課す高品質及び厳しい要件を満たさない最終製剤となる可能性がある。以上を念頭において、一般に、少なくとも物理的安定特性が改良された薬学的化合物の選択は、同じ化合物の安定性がより低い形態よりも著しい利点を与えられることを心に留めるべきである。
【0004】
本発明は、非常に好ましい物性を有する薬理学的に活性なプロスタグランジンD2受容体拮抗剤の二水素リン酸塩に関する。該化合物は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介病的(疾患)病状を患う又は該症状を被りやすい患者を治療するためのDP拮抗剤として有用である。
【背景技術】
【0005】
アレルギー性鼻炎、気管支喘息、アレルギー性結膜炎及びアトピー性皮膚炎をもつ患者で局所的にアレルゲンでチャレンジすると、鼻及び気管支洗浄液、涙及びスキンチャンバー液中のプロスタグランジンD2 “(PGD2)”濃度が急速に上昇する結果をもたらすことが示されている。PGD2は、結膜及び皮膚における血管透過性の増大、鼻腔抵抗の増加、気道縮小、並びに結膜及び気管への好酸性浸潤のような多くの炎症作用を有する。
【0006】
PGD2は、免疫的チャレンジにより肥満細胞から産生するアラキドン酸の主なシクロオキシゲナーゼ生成物である[非特許文献1]。活性化された肥満細胞、即ちPGD2の主な源は、喘息、アレルギー性鼻炎、アレルギー性結膜炎、アレルギー性皮膚炎、及びその他の疾患のようなアレルギー反応症状を駆動するキープレーヤーの1つである[非特許文献2]。
【0007】
PGD2の作用の多くはD−型プロスタグランジン(“DP”)受容体、即ち、上皮及び平滑筋に発現するGタンパク質共役受容体、に対するその作用を通して仲介される。喘息において、気道上皮は、病気の進行を促進する炎症サイトカイン及びケモカインの主な源と長い間認識されていた[非特許文献3]。喘息の実験マウスモデルで、DP受容体は抗原のチャレンジにより気道上皮において劇的に上方調節される[非特許文献4]。DP受容体を欠いたノックアウトマウスで、ヒト喘息の基本的な特徴の二つである、気道過敏性及び慢性炎症の著しい減少が存在する[非特許文献5]。
【0008】
DP受容体はまた、くしゃみ、かゆみ、鼻汁及び鼻詰りの症状により特徴付けられる、よくあるアレルギー性疾患である、ヒトアレルギー性鼻炎に関係するとも考えられている。鼻へのPGD2の局部投与は鼻詰りの用量依存的な増加を引き起こす[非特許文献6]。
【0009】
DP受容体拮抗剤は、モルモットの実験喘息モデルにおいて気道炎症を減少させることが示された[非特許文献7]。従って、PGD2はDP受容体に作用しそしてアレルギー性喘息のある重要な特徴を引き出すのに重要な役割を果たすようである。
【0010】
DP拮抗剤は、多くの種においてアレルギー性鼻炎の症状を軽減するのに有効であることが示され、更に詳しくは、アレルギー性鼻炎で最もよく現れる徴候である、抗原誘発性の鼻詰りを阻止することが示された[非特許文献8及び非特許文献9]。
【0011】
DP拮抗剤はまた、アレルギー性結膜炎及びアレルギー性皮膚炎の実験モデルにおいても有効である[非特許文献10及び非特許文献11]。
【0012】
参照することにより組み入れられる、特許文献1(以後、'732公報と云う)は、特にDP受容体と関連しそしてそれを調節する能力を含む、有効な薬学的性質を有するピリミジンを開示する。'732公報は式(I)のピリミジン:
【化1】
それらの調製、それらの化合物を含む薬学的組成物、及びプロスタグランジンD2受容体の阻害により調節することができる疾患状態の治療へのそれらの薬学的使用を開示する。更に、'732公報は具体的には2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸(以後、“遊離形”と云う)を開示しそしてクレームする。'732公報はまた、該発明の化合物の多様な酸付加塩及び塩基付加塩の一般的記述、並びに塩酸塩及びナトリウム塩の製造に限定された様々な実施例を提供する。更に詳しくは、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の塩酸塩及びナトリウム塩が開示される。しかしながら'732公報は、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩を具体的に開示しない。
【非特許文献1】Lewis, RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ II, Prostaglandin D2 generation after activation of rat and human mast cells with anti−IgE, J. Immunol. 129, 1627−1631, 1982
【非特許文献2】Brightling CE, Bradding P, Pavord ID, Wardlaw AJ, New Insights into the role of the mast cell in asthma, Clin. Exp. Allergy 33, 550−556, 2003
【非特許文献3】Holgate S, Lackie P, Wilson S, Roche W, Davies D, Bronchial Epithelium as a Key Regulator of Airway Allergen Sensisitzation and Remodelling in Asthma, Am J Respir Crit Care Med. 162, 113−117, 2000
【非特許文献4】Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S, prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013−2017, 2000
【非特許文献5】Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S, prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013−2017, 2000
【非特許文献6】Doyle WJ, Boehm S, Skoner DP, Physiologic responses to intranasal dose−response challenges with histamine, methacholine, bradykinin, and prostaglandin in adult volunteers with and without nasal allergy, J Allergy Clin Immunol. 86(6 Pt 1), 924−35, 1990
【非特許文献7】Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H, Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S−5751, J Pharmacol Exp Ther. 298(2), 411−9, 2001
【非特許文献8】Jones, T. R., Savoie, C., Robichaud, A., Sturino, C., Scheigetz, J., Lachance, N., Roy, B., Boyd, M., Abraham, W., Studies with a DP receptor antagonist in sheep and guinea pig models of allergic rhinitis, Am. J. Resp. Crit. Care Med. 167, A218, 2003
【非特許文献9】Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H, Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S−5751. J. Pharmacol. Exp. Ther. 298(2), 411−9, 2001
【非特許文献10】Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H, Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S−5751. J. Pharmacol. Exp. Ther. 298(2), 411−9, 2001
【非特許文献11】Torisu K, Kobayashi K, Iwahashi M, Nakai Y, Onoda T, Nagase T, Sugimoto I, Okada Y, Matsumoto R, Nanbu F, Ohuchida S, Nakai H, Toda M, Discovery of a new class of potent, selective, and orally active prostaglandin D2 receptor antagonists, Bioorg.. & Med. Chem. 12, 5361−5378, 2004
【特許文献1】特許出願WO 2006044732
【発明の開示】
【課題を解決するための手段】
【0013】
本発明は、式(III)の2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩(以後、“二水素リン酸塩”と云う)に関する:
【化2】
【0014】
本発明の別の側面は、薬学的有効量の二水素リン酸塩を含む薬学的組成物である。
【0015】
本発明の別の側面は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介障害を患う患者に、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩の薬学的有効量を投与することにより、該患者を治療するための方法である。
【0016】
本発明を下記の図及び下記の詳細な記述の助けにより更に充分に論じる。
【発明を実施するための最良の形態】
【0017】
略語の定義
上で使用し、そして本発明の記述の全体を通して、下記の略語は、外に指示がない限り、下記の意味を有すると理解されたい:
DMSO:ジメチルスルホキシド
cAMP:環式アデノシンモノホスフェート
IBMX: 3−イソブチル−1−メチルキサンチン
SPA:シンチレーション近接アッセイ
ATTC:アメリカ培養株保存機関
MEM:最小必須培地
FBS:ウシ胎仔血清
CPM;1分当りのカウント数
【0018】
上で使用し、そして本発明の記述の全体を通して、下記の用語は、外に指示がない限り、下記の意味を有すると理解されたい:
“治療する”又は“治療”とは、疾病の予防、部分的緩和、又は治癒を意味する。本発明の化合物及び組成物は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果、2次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されないPGD2媒介障害を、該障害を患う患者に、式(III)の化合物の薬学的有効量を投与することにより治療するのに有効である。
【0019】
“患者”はヒト及び他の哺乳動物を含む。
【0020】
“薬学的有効量”とは、所望の治療効果を生じるのに有効な化合物、組成物、医薬又は他の活性成分の量を云う。
【0021】
本発明の1つの特定の態様は、結晶形の式(III)の化合物である。
【0022】
本発明の化合物はプロスタグランジンD2受容体拮抗剤活性を示し、そして有効な薬理学的作用薬剤である。従って、該化合物は薬学的組成物に混入され、そしてある種の医学的障害を患う患者の治療に使用される。
【0023】
本発明の化合物は、文献に記載された試験、及び以下の薬学的試験の記載部分によれば、プロスタグランジンD2受容体の拮抗剤であり、その試験結果はヒト及び他の哺乳動物における薬理学的活性と関連していると信じられている。従って、更なる態様において、本発明は、本発明の化合物及び本発明の化合物を含む組成物を、PGD2拮抗剤の投与により改善することができる症状を患う又は該症状を被りやすい患者の治療へ使用することを提供する。例えば、本発明の化合物は、アレルギー疾患(例えばアレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、及び食物アレルギー)、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患(例えばアトピー性皮膚炎及びじんましん)、かゆみに伴う行動(例えば掻爬及び殴打)の結果二次的に発生する疾患(例えば白内障、網膜剥離、炎症、感染症及び睡眠障害)、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、潰瘍性大腸炎等を含むがこれらに限定されない多様なPGD2媒介障害の治療に有用であり得る。
【0024】
本発明の治療方法の一つの特定の態様は、アレルギー性鼻炎の治療である。
本発明の治療方法の別の特定の態様は、気管支喘息の治療である。
本発明の治療方法の別の特定の態様は、COPDの治療である。
本発明の治療方法の別の特定の態様は、アレルギー性結膜炎の治療である。
本発明の治療方法の別の特定の態様は、アレルギー性皮膚炎の治療である。
本願で治療とは、確立した症状の治療と同時に予防的治療をも含むと理解されたい。
【0025】
本発明の化合物は更に、下記の少なくとも1種との併用療法を含む治療において有用である:
(i) アレルギー性鼻炎の治療に、抗ヒスタミン剤、例えばフェキソフェナジン(fexofenadine)、デスロラタジン(desloratadine)、ロラタジン(loratadine)及びシチリジン(citirizine);
(ii) アレルギー性鼻炎、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、ロイコトリエン拮抗剤、例えばモンテルカスト(montelukast)及びザフィルラスト(zafirulast)(詳しくはWO 01/78697 A2のクレーム参照);
(iii) 喘息、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、ベータ作用薬、例えばアルブテロール(albuterol)、サルブテロール(salbuterol)及びテルブタリン(terbutaline);
(iv) 喘息、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、抗ヒスタミン剤、例えばフェキソフェナジン、ロラタジン及びシチリジン;
(v) 喘息、COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、PDE4 (ホスホジエステラーゼ4)阻害剤、例えばロフルミラスト(roflumilast)及びシロミラスト(cilomilast);又は
(vi) COPD、アレルギー性皮膚炎、アレルギー性結膜炎等の治療に、TP(トロンボキサンA2受容体)又はCrTh2(Th2細胞に発現する化学誘引薬受容体に相同な分子)拮抗剤、例えばラマトロブラン(Ramatrobran)(BAY−u3405)。
【0026】
本発明はまたその範囲に、本発明の化合物の薬学的有効量を薬学的に許容される担体と混合して含む薬学的組成物を包含する。
【0027】
実際には、本発明の化合物は薬学的に許容される投与形体でヒト及び他の哺乳動物に、経口、吸入、直腸、鼻、頬、舌下、膣内、直腸内、非経口(皮下、筋肉内、静脈内、皮内、髄腔内、及び硬膜外を含む)、嚢内(intracisternal)及び腹膜内投与を含む局所的又は全身的投与により投与し得る。特定の経路は例えば受容者の生理的状態により変えることができることが理解されるであろう。
【0028】
“薬学的に許容される投与形態”とは、本発明の化合物の投与形態を云い、例えば錠剤、糖衣錠、粉末、エリキシル、シロップ、懸濁液を含む液体製剤、スプレー、吸入用錠剤、トローチ、乳濁液、溶液、顆粒、カプセル及び坐薬、及びリポソーム製剤を含む注射用液体製剤を含む。技術及び製剤は一般にRemington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, latest editionで見つけることができる。
【0029】
本発明の特定の側面は薬学的組成物の形態で投与する本発明の化合物を提供する。
【0030】
薬学的に許容される担体は、投与様式及び投与形体の性質に依存して、薬学的に許容される担体、希釈剤、被覆剤、補助剤、賦形剤、又はベヒクル、例えば保存剤、充填剤、崩壊剤、湿潤剤、乳化剤、エマルション安定剤、懸濁剤、等張剤、甘味剤、矯味矯臭剤、芳香剤、着色剤、抗菌剤、抗カビ剤、他の治療剤、潤滑剤、吸着遅延又は促進剤、及び調合剤、を含む群から選ばれる少なくとも1つの成分を含む。
【0031】
懸濁剤の例には、エトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトール及びソルビタンエステル、微結晶セルロース、メタ水酸化アルミニウム、ベントナイト、寒天及びトラガカント、又はこれらの物質の混合物が含まれる。
【0032】
微生物の作用を防止するための抗菌剤及び抗カビ剤の例には、パラベン、クロロブタノール、フェノール、ソルビン酸等が含まれる。
【0033】
等張剤の例には、糖類、塩化ナトリウム等が含まれる。
【0034】
吸収を遅延させる吸着遅延剤の例には、モノステアリン酸アルミニウム及びゼラチンが含まれる。
【0035】
吸収を促進するための吸着促進剤の例には、ジメチルスルホキシド及び関連する類似体が含まれる。
【0036】
希釈剤、溶剤、ベヒクル、可溶化剤、乳化剤及びエマルション安定剤の例には、水、クロロホルム、スクロース、エタノール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、テトラヒドロフルフリルアルコール、安息香酸ベンジル、ポリオール類、プロピレングリコール、1,3−ブチレングリコール、グリセロール、ポリエチレングリコール、ジメチルホルムアミド、トウィーン(Tween)(登録商標)60、スパン(Span)(登録商標)60、セトステアリルアルコール、ミリスチルアルコール、モノステアリン酸グリセリル、及びラウリル硫酸ナトリウム、ソルビタンの脂肪酸エステル、植物油(例えば綿実油、落花生油、オリーブ油、ひまし油、及びゴマ油)及び注射可能な有機エステル、例えばオレイン酸エチル等、又はこれらの物質の適当な混合物が含まれる。
【0037】
賦形剤の例にはラクト−ス、乳糖、クエン酸ナトリウム、炭酸カルシウム及びリン酸二カルシウムが含まれる。
【0038】
崩壊剤の例には、澱粉、アルギン酸、及びある種の複合ケイ酸塩が含まれる。
【0039】
潤滑剤の例には、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム、タルク、並びに高分子量ポリエチレングリコールが含まれる。
【0040】
薬学的に許容される担体の選択は一般に、溶解性のような活性化合物の化学的性質、投与の特定の様式、及び薬学的実務で順守される規定に従って決定される。
【0041】
経口投与に適した本発明の薬学的組成物は、固体調剤形体のような分離した単位として、例えばそれぞれ活性成分の所定量を含むカプセル、カシェット(cachets)又は錠剤として、又は粉末若しくは顆粒として;又は水性液体若しくは非水性液体の溶液若しくは懸濁液のような液体投与体として、又は水中油型乳濁液若しくは油中水型乳濁液として提供し得る。また活性成分は、ボーラス、舐剤又はペーストとして提供し得る。
【0042】
“固体調剤形体”とは、本発明の化合物の投与が固体形体、例えば、カプセル、錠剤、ピル、粉末、糖衣剤又は顆粒であることを意味する。かかる固体調剤形体において、本発明の化合物は少なくとも1種の不活性な慣用の賦形剤(又は担体)、例えばクエン酸ナトリウム又はリン酸二カルシウム又は:(a)充填剤又は増量剤、例えば澱粉、ラクトース、スクロース、グルコース、マンニトール及びケイ酸、(b)結合剤、例えばカルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロース及びアカシア、(c)保湿剤、例えばグリセロール、(d)崩壊剤、例えば寒天、炭酸カルシウム、馬鈴薯又はタピオカ澱粉、アルギン酸、ある種の複合ケイ酸塩及び炭酸ナトリウム、(e)溶解遅延剤、例えばパラフィン、(f)吸収促進剤、例えば第四級アンモニウム化合物、 (g)湿潤剤、例えばセチルアルコール及びモノステアリン酸グリセロール、(h)吸着剤、例えばカオリン及びベントナイト、(i)潤滑剤、例えばタルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、(j)不透明化剤、(k)緩衝剤、及び本発明の化合物を胃腸管のある部分に遅延して放出する薬剤と混合される。
【0043】
錠剤は、任意に1種又はそれ以上の付属成分と共に圧縮又は成形して製造し得る。圧縮錠剤は、適当な機械中で粉末又は顆粒のような自由流動形体の活性成分を、任意に結合剤、潤滑剤、不活性希釈剤、保存料、界面活性剤又は分散剤と混合して圧縮することにより製造し得る。賦形剤、例えばラクトース、クエン酸ナトリウム、炭酸カルシウム、リン酸二カルシウム、及び崩壊剤、例えば澱粉、アルギン酸、及び、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム及びタルクのような潤滑剤と組合わせたある種の複合ケイ酸塩、のような賦形剤を使用し得る。不活性液体希釈剤で湿らせた粉末化した化合物の混合物を適当な機械で成形して、成形した錠剤を製造し得る。該錠剤は場合によっては被覆又は切り込みを入れ、そしてその中の活性成分を遅延又は制御放出するように調合し得る。
【0044】
固体組成物はまた、ラクトース又は乳糖のような賦形剤、並びに高分子量ポリエチレングリコール等を使用して軟充填及び硬充填ゼラチンカプセル中の充填剤としても使用し得る。
【0045】
所望により、そして更に有効な分配のために、化合物を遅延放出又は標的伝達システム、 例えば生体適合性、生体分解性ポリマーマトリクス(例えばポリ(d,l−ラクチド・コ−グリコリド))、リポソーム及びミクロスフェア、の中にマイクロカプセル化して又はそれに付けて、皮下又は筋肉内デポ(deposit)と呼ばれる技術により皮下又は筋肉内注射して、該化合物を2週間又はそれ以上の期間、継続的にゆっくり放出させることができる。該化合物を、例えばバクテリア保持フィルターを通す濾過により、又は殺菌剤を、使用直前に殺菌水又は他の殺菌した注射可能な媒体に溶解することができる殺菌固体組成物の形で加えることにより、殺菌し得る。
【0046】
“液体投与形体”とは、患者に投与する活性化合物の投与体が液体の形体、例えば薬学的に許容される乳濁液、溶液、懸濁液、シロップ及びエリキシル、であることを意味する。活性化合物に加えて、液体投与形体は当業界で通常使用される不活性希釈剤、溶媒、可溶化剤及び乳化剤を含んでもよい。
【0047】
水性懸濁液が使用される場合、それらは乳化剤又は懸濁を容易にする薬剤を含むことができる。
【0048】
局部投与に適した薬学的組成物とは、患者に局部的に投与するのに適した形体にある配合物を意味する。該配合物は、当業界で一般に知られる局部軟膏、軟膏、粉末及び吸入剤、ゲル(水又はアルコールを基材とする)、クリームとして提供するか、又は経皮バリアーを通して化合物の制御放出を可能にするパッチに適用するためにマトリクスベースに混入し得る。軟膏に調合した場合、活性成分はパラフィン又は水混和性軟膏基材と共に使用しえる。或いは、活性成分は水中油型クリーム基材と共にクリームに調合し得る。目に局部投与するのに適した配合物には、活性成分が適当な担体、特に活性成分の水性溶媒、に溶解又は懸濁した点眼液が含まれる。口に局部投与するのに適した配合物には、活性成分を矯味矯臭基材、通常スクロース及びアカシア又はトラガカント、中に含む薬用キャンデー;活性成分を不活性基材、例えばゼラチン及びグリセリン、又はスクロース及びアカシア、中に含むトローチ;及び活性成分を適当な液体担体中に含むうがい薬が含まれる。
【0049】
乳化薬学的組成物の油相は、公知の成分から公知の方法で構成し得る。該相は乳化剤(或いはエマルゲントとして知られる)のみを含んでもよいが、少なくとも1種の乳化剤と脂肪又は油、又は脂肪及び油の両方との混合物を含むのが望ましい。特別の態様では、親水性乳化剤が、安定剤として作用する脂肪親和性乳化剤と共に含まれる。乳化剤は安定剤と共に又は安定剤なしで、乳化ワックスを形成し、そして油及び脂肪と共に、クリーム配合物の油分散相を形成する乳化軟膏基材を形成する。
【0050】
所望により、クリ−ム基材の水性相は、例えば少なくとも30% w/wの多価アルコール、即ち、2個又はそれ以上の水酸基を有するアルコール、例えばプロピレングリコール、ブタン1,3−ジオール、マンニトール、ソルビトール、グリセロール及びポリエチレングリコール(PEG 400を含む)及びそれらの混合物、を含んでもよい。局部配合物は、皮膚若しくは他の発症領域を通しての活性成分の吸収又は浸透を向上させる化合物を含むのが望ましいであろう。
【0051】
組成物に適した油又は脂肪の選択は所望の性質の達成に基づく。従って、クリームは特にベタベタせず、汚染性でなく、そして洗える製品で、チューブ又は他の容器から漏れるのを防ぐために適当な稠度を有していなければならない。直鎖若しくは分岐鎖一若しくは二塩基性アルキルエステル、例えばミリスチン酸ジイソプロピル、オレイン酸デシル、パルミチン酸イソプロピル、ステアリン酸ブチル、パルミチン酸2−エチルヘキシル、Crodamol CAPとして知られた分岐鎖エステルのブレンド物が使用し得る。これらは、要求される性質に依存して、単独で又は組み合わせて使用し得る。或いは、高融点脂質、例えば白色軟質パラフィン及び/又は液体パラフィン又は他の鉱油を使用することができる。
【0052】
直腸又は膣内投与に適した薬学的組成物とは、患者に直腸内又は膣内投与するのに適した形態でありそして少なくとも1種の本発明の化合物を含む製剤を意味する。坐薬はかかる製剤の特定の形態であり、本発明の化合物を適当な非刺激性賦形剤又は担体、例えば、通常の温度では固体であるが体温では液体であり、従って直腸又は膣腔内で融解して活性成分を放出するココアバター、ポリエチレングリコール又は坐薬ワックスと混合して調製することができる。
【0053】
注射により投与される薬学的組成物は、筋肉内、静脈内、腹膜内及び/又は皮下注射により注射し得る。本発明の組成物は溶液、特に生理学的適合性緩衝液、例えばハンク溶液(Hank's solution)又はリンガー溶液中に配合される。更に、該組成物は固体形体に製剤化されそして使用直前に再溶解又は懸濁され得る。凍結乾燥した形態もまた含まれる。製剤は無菌であり、そして乳濁液、懸濁液、水性及び非水性注射液を含み、それらは懸濁剤及び増粘剤及び酸化防止剤、緩衝剤、静菌剤、及び配合物を等張性にする溶質を含んでもよく、そして意図する受容者の血液に適するように調整したpHを有する。
【0054】
鼻又は吸入投与に適した本発明の薬学的組成物とは、患者に鼻に又は吸入により投与するのに適した形態の組成物を意味する。該組成物は、粒径が例えば1〜500ミクロンの範囲(30ミクロン、35ミクロン等、5ミクロン単位で増加する20〜500ミクロンの範囲の粒径を含む)の粉末形態の担体を含んでもよい。担体が例えば鼻腔スプレー又は点鼻薬として投与するために液体である、適当な組成物には、活性成分の水溶液又は油溶液が含まれる。エアロゾル投与に適した組成物は慣用の方法で調製することができ、そして他の治療剤と共に供給し得る。吸入療法は、特許出願WO2004/026380及び米国特許第5,176,132号に記載されたように、計量投与量吸入器により又は適当な乾燥粉末吸入器、例えばEclipse,
Spinhaler(登録商標)、又はUltrahaler(登録商標)により、容易に投与される。
【0055】
本発明の組成物中の活性成分の実際の投与量は、特定の組成物及び患者への投与方法について所望の治療反応を得るのに有効な活性成分の量を得るように、変えることができる。従って、あらゆる特定の患者への選択した投与量は、所望の治療効果、投与経路、所望する治療持続期間、疾病の病因及び重症度、患者の状態、体重、性別、食生活及び年齢、各活性成分の種類及び効能、吸収速度、代謝作用及び/又は排泄及び他の要因を含む多様の要因に依存する。
【0056】
患者に1回又は分割用量で投与される本発明の化合物の1日合計投与量は、例えば、1日約0.001〜約100mg/kg(体重)、特に0.01〜10mg/kg/日であろう。例えば成人では、投与量は一般に吸入による場合は1日当り約0.01〜約100mg/kg(体重)、特に約0.01〜約10mg/kg(体重)、経口投与では1日当り約0.01〜約100mg/kg(体重)、特に0.1〜70mg/kg(体重)、更に特に0.5〜10mg/kg(体重)、そして静脈投与では1日当り約0.01〜約50mg/kg(体重)、特に0.01〜10mg/kg(体重)である。組成物中の活性成分のパーセントは変化し得るが、適当な投与量が得られるような割合でなければならない。投与単位組成物は、1日の投与量を作り上げるために使用されるようなその約数(submultiples)の量を含み得る。明らかに、いくつかの単位投与形態をほぼ同時に投与してもよい。調剤は、所望の治療効果を得るために、必要な回数投与してもよい。幾人かの患者は高い又は低い投与量に迅速に反応し、ずっと低い維持投与量が適当であることが判明するかもしれない。他の患者には、各特定の患者の生理的要求に従って、1日当り1〜4回投与の割合で長期間治療するのが必要かもしれない。云うまでもなく、他の患者には、1日当り1又は2回以下の投与量を処方することが必要であろう。
【0057】
製剤は薬局業界でよく知られたあらゆる方法で単位投与形態で製造することができる。
【0058】
かかる方法は薬学的活性成分を1種又はそれ以上の補助成分を構成する担体と合わせるステップを含む。一般に製剤は、活性成分を液体担体又は微粉砕固体担体又は両方と均一に且つ密接に合わせ、次に必要な場合は製品に成形することにより製造される。
【0059】
製剤は単位用量又は多回用量容器、例えば密閉アンプル及び弾性栓を有する小瓶、で提供し得、そして使用直前に殺菌液体担体、例えば注射用の水、のみを必要とする凍結乾燥条件で保存し得る。即時注射溶液及び懸濁液は前述した種類の殺菌粉末、顆粒及び錠剤から調製し得る。
【0060】
本発明の化合物を下記の分析方法で分析する。
高圧液体クロマトグラフィー− 質量分光分析(LCMS)
保持時間(RT)及び関連する質量イオンを決定するためのLCMS実験を下記の方法を用いて行う。質量スペクトル(MS)をマイクロマス(Micromass)LCT質量分光計を用いて記録する。該方法は、質量m/zを100から1000まで走査するエレクトロスプレーイオン化法である。液体クロマトグラフィーは、ヒューレットパッカード(Hewlett Packard)1100シリーズのバイナリポンプ&デガッサー(Binary Pump & Degasser)で、静止相:フェノメネックス(phenomenex)のシナージ(Synergi)2μハイドロ(Hydro)−RP 20 X 4.0mmカラム、可動相:A=水中の0.1%蟻酸(FA)、B=アセトニトリル中の0.1% FAで行う。5μLの注入容量はCTC分析パルシステム(PAL System)による。流量は1mL/分である。勾配は3分間で10% Bから90% B、次いで2分間で90% Bから100% Bである。付属する検出器は:ヒューレットパッカード(Hewlett Packard)1100シリーズのUV検出器、波長=220nm、及びセデレセデックス(Sedere SEDEX)75蒸発性光散乱(ELS)検出器、温度=46℃、窒素圧=4バールである。
【0061】
1H核磁気共鳴スペクトル(NMR)
300MHz 1H NMRを、周囲温度で、ASW 5mmプローブを有するバリアンマーキュリー(Varian Mercury)(300MHz)分光計を用いて記録する。NMRにおいて、化学シフト(δ)はテトラメチルシランに対するppmで表す。化学シフト値は、内部標準としてテトタメチルシランを参照して、百万分の一単位(ppm)で示す。
【0062】
X−線粉末回折(XRPD)
XRPDは、シーメンズ−ブルッカー(Siemens−Bruker)D5000回折計で、パラ集束(parafocusing)ブラッグ−ブレンタノ(Bragg−Brentano)(シータ−2−シータ)型の構成を用いて行う。二水素リン酸塩を、 (510)結晶方向に従って切断した単結晶シリコンウェハー上に置く。銅対陰極管(45kV/40mA)から放出される銅K−アルファ放射線(1.54056オングストローム)をX線源として用い、Cu K−ベータ放射線を反射ビーム単色光分光器を用いてろ過除去する。シンチレーションカウンターを検出に用いる。発散スリット0.6mm、抗散乱スリット0.6mm、単色光分光器スリット0.1mm、及び検出器スリット0.6mmを使用する。回折パターンを下記の条件で得る:角度2シータで2〜40度の走査、1ステップ当り1秒のカウント時間、0.02度のステップサイズ、周囲条件の圧力、温度及び相対湿度。
【0063】
熱重量分析による溶媒和/水和状態
熱分析を、TA機器モデルQ−600同時示差走査熱量計/熱重量分析計(DSC/TGA)を用いて乾燥窒素雰囲気下で行う。TGA温度を、インジウム基準を用いて較正する。二水素リン酸塩をアルミニウムパン(TA機器部品番号900793.901)に移す。サーモグラムを、毎分10℃の線形加熱速度で得る。
【0064】
示差走査熱量測定(DSC)
DSCを、冷蔵型冷却システムを備えたTA機器モデルQ−1000 DSCを用いて乾燥窒素雰囲気下で行う。DSCを、インジウム基準を用いて較正する。二水素リン酸塩をアルミニウムパン(TA機器部品番号900793.901)に移し、レーザーで穴あけしたピンホールを有するふた(TA機器部品番号900860.901)を該パンに冷溶接する。DSCサーモグラムを、毎分10℃の線形加熱速度で得る。
【0065】
顕微鏡写真
顕微鏡写真を、交差極(cross polars)を備えたオリンパスBX−41顕微鏡を用いて得る。サンプルは、それを鉱油に分散することにより調製する。
【0066】
粒子サイズ分布
粒子サイズ分布は、R3測定レンズ、RODOS乾式分散器、及び632.8nmに合わせたレーザーを有する、シンパテック(Sympatec)HELOS−BFレーザー回折粒子サイズ分析器を用いて測定する。該システムを、炭化珪素基準を用いて較正する。粉末を、初期圧3.0バールを有するRODOS乾式分散付属部品を用いて分散させ、そして下降を最大にする。容積基準粒子サイズ分布を、シンパテックウィンドクス(Sympatec Windox)(バージョン4.0)ソフトウェアにより、フラウンホッファー(Fraunhofer)法を用いて計算する。
【0067】
動的水蒸気収着(DVS)
水収着プロフィールを、SMS機器動的蒸気収着分析器モデルDVS−1又はVTI機器モデルSGA−100 動的蒸気収着分析器を用いて測定する。RH及び質量を、基準を用いて較正する。二水素リン酸塩を載せ、そして実験開始前に1% RH以下で2.5時間乾燥する。RHを約0から95%RHに進める。試料質量は、質量変化パーセントが、最小絶対平衡時間が15分で、5分の間にわたって0.005%未満の場合、各ステップで一定と考える。
【0068】
フーリエ変換−赤外(FT−IR)分光法
FT−IRスペクトルを、ニコレット(Nicolet)Nic−Plan FT−IR顕微鏡に取り付けたニコレットマグナ(Magna)−IR 分光計55を用いて得る。固体サンプルをKBrディスク上で分析する。スペクトルが、4000−400cm-1から32走査後に4cm-1の解像度で得られる。
【0069】
本発明の化合物の調製及び性質を下記の実験部分に記載する。
【実施例】
【0070】
2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩
ステップ1. 4,6−ジクロロ−2−メトキシピリミジン(0.7g)、2,4−ジクロロフェネチルアミン(0.82g)及び重炭酸ナトリウム(0.88g)のエタノール(25mL)溶液を80℃に3時間加熱し、そして水(400mL)に注ぐ。得られた固体を濾過しそして空気乾燥して(6−クロロ−2−メトキシ−ピリミジン−4−イル)−[2−(2,4−ジクロロ−フェニル)−エチル]−アミンを得る。
ステップ2. リチウムジイソプロピルアミドのテトラヒドロフラン/n−ヘプタン/エチルベンゼン(1.8M, 17mL)溶液に2−(3−ブロモ−フェニル)−プロピオン酸(3g, 13.9ミリモル)のテトラヒドロフラン(5mL)溶液を0℃で15分間で滴下する。混合物を1時間攪拌し、次いでヨウ化メチル(4.93g, 34.8ミリモル)のテトラヒドロフラン(5mL)溶液を10分間で滴下する。反応混合物を15時間攪拌し、2N HClでクエンチし、真空中で濃縮し、そしてエーテル(150mL)で希釈する。エーテル層を2N HClで洗い、2N NaOH (50mL)で3回抽出する。合わせたNaOH層を6N HClでpH=1に酸性化し、そしてエーテル(75mL)で3回抽出する。合わせた有機層をブラインで洗い、硫酸ナトリウムで乾燥し、そして濃縮して2−(3−ブロモ−フェニル)−2−メチル−プロピオン酸を固体(3.08g, 91%)として得、それを更に精製することなく使用する。LC/MS:243 (M+H)。
ステップ3. 2−(3−ブロモ−フェニル)−2−メチル−プロピオン酸(2.18ミリモル)の無水エーテル(20mL)溶液にtert−ブチルリチウム(ペンタン中1.7M, 5.4mL, 9.16ミリモル)を−78℃で滴下し、そしてこの混合物をホウ酸トリブチル(2.34mL, 8.72ミリモル)で処理して30分間攪拌する。反応混合物を室温まで温め、15時間攪拌し、エーテルで希釈し、そして1M H3PO4でクエンチする。30分間攪拌した後、エーテル層を分離し、そして2N NaOH(20mL)で3回抽出する。合わせたNaOH抽出物を6N HClでpH=1に酸性化し、そしてエーテル(50mL)で3回抽出する。合わせた有機抽出物をブラインで洗い、硫酸ナトリウムで乾燥し、そして濃縮して3−(1−カルボキシ−1−メチル−エチル)−フェニルホウ酸を得、それを更に精製することなく使用する。MS:209 (M+H)。
ステップ4. (6−クロロ−2−メトキシ−ピリミジン−4−イル)−[2−(2,4−ジクロロ−フェニル)−エチル]−アミン(0.51ミリモル)及び3−(1−カルボキシ−1−メチル−エチル)−フェニルホウ酸(0.61ミリモル)をアセトニトリル(2.5mL)及び炭酸ナトリウム水溶液(0.4M, 2.5mL)中に含む溶液を、テトラキストリフェニルホスフィンパラジウム(0)(29.5mg, 5mol%)の添加前に窒素で5分間脱ガスする。反応容器を密閉しそしてマイクロ波下で130℃に30分間加熱する。反応混合物に水2mLを加え、2N HCl水溶液を使用してpHを約7に調節し、そしてこの混合物を酢酸エチル(30mL)で3回抽出する。合わせた抽出物をブラインで洗い、硫酸ナトリウムで乾燥し、そして濃縮する。残留物をシリカゲルクロマトグラフィーに付して2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸を固体(205mg, 75%)として得る。LC/MS: RT=2.39分, MS: 460 (M+H); 1H NMR [300 MHz, (CD3)2SO]: δ 12.38 (1H, s), 7.36−8.00 (7H, m), 6.58 (1H, s), 3.84 (3H, s), 3.58 (2H, m), 2.98 (2H, m), 1.54 (6H, s)。
ステップ5. リン酸(3.21mL, 1.49N水溶液)を、2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸(2.1g, 4.56ミリモル)のテトラヒドロフラン(45mL)溶液に加え、そして混合物を10分間攪拌する。水を間隔をおいて該混合物が透明溶液になるまで滴下し、そして攪拌を1.5時間室温で続ける。該混合物を真空中で濃縮し、そして残留物をアセトンから再結晶させて2−(3−{6−[2−(2,4−ジクロロ−フェニル)−エチルアミノ]−2−メトキシ−ピリミジン−4−イル}−フェニル)−2−メチル−プロピオン酸の二水素リン酸塩を粉末(2.4g, 94%)として得る。LCMS: RT=2.41分;MS: 462 (M+H);1H NMR [300 MHz, (CD3SO)2SO]: δ 7.95 (1H, b), 7.8 (1H, b), 7.6 (2H, b), 7.45 (2H, d, J=2Hz), 7.35 (2H, s), 6.55 (1H, s), 3.85 (3H, s), 3.55 (2H, b), 2.95 (2H, t, J=2Hz), 1.5
(6H, s)。
【0071】
薬理学的試験
本発明の化合物の阻害効果をヒトDP機能検定で評価する。cAMP検定を、内生DP受容体を発現するヒト細胞系LS174Tを使用して用いる。プロトコルは前に記載したもの(Wright DH, Ford−Hutchinson AW, Chadee K, Metters KM, The human prostanoid DP receptor stimulates mucin secretion in LS174T cells, Br J Pharmacol. 131(8):1537−45 (2000))と同様である。
【0072】
ヒトLS174 T細胞におけるSPA cAMP検定のプロトコル
材料
・PGD2 (カイマンケミカル(Cayman Chemical)Cat No.12010)
・IBMX (シグマ(Sigma)Cat No.5879)
・cAMP SPA直接スクリーニング検定システム(アマーシャム(Amersham)コードRPA 559)
・96−穴細胞プレート(ワラック(Wallac)Cat No.1450−516)
・ワラック1450マイクロプレートトリラックス(Trilux)シンチレーションカウンター(パーキンエルマー(PerkinElmer))
・プレート密封器
・エッペンドルフチューブ
・ダルベッコのリン酸塩緩衝食塩水(PBS) (インビトロゲン(Invitrogen)Cat No.14040−133)
・蒸留水
・ボルテックス(Vortex)
・磁気各攪拌器及び攪拌棒
【0073】
試薬の調製:
全試薬は、再構成前に室温に平衡させなければならない。
1Xアッセイ用緩衝液
瓶の内容物を500mL目盛り付きシリンダーに、繰り返し蒸留水で洗うことにより移す。最終容量を蒸留水で500mLに調整し、そして充分混合する。
溶解用試薬1及び2
溶解用試薬1及び2の各々を、夫々200mLのアッセイ用緩衝液に溶解させる。室温に20分間放置して溶解させる。
SPA抗ウサギビーズ
溶解用緩衝液2の30mLを瓶に加える。該瓶を5分間静かに振る。
抗血清
溶解用緩衝液2の15mLを各小瓶に加え、そして内容物が完全に溶解するまで静かに混ぜる。
トレーサー(I125−cAMP)
溶解用緩衝液2の14mLを各小瓶に加え、そして内容物が完全に溶解するまで静かに混ぜる。
免疫試薬の調製
1)トレーサー、抗血清及びSPA抗ウサギ試薬を同じ量瓶に加え、所望の数の穴(150μL/穴)に充分な量のこの混合物が確実に調製されるようにする。
2)充分に混合する。
3)この免疫試薬溶液は各アッセイ前に新たに調製し、再使用してはならない。
標準品
1)溶解用緩衝液1を1mL加え、そして内容物が完全に溶解するまで静かに混ぜる。
2)最終溶液はcAMPを濃度512pmol/mLで含む。
3)7個のポリプロピレン又はポリスチレンチューブに0.2pmol、0.4pmol、0.8pmol、1.6pmol、3.2pmol、6.4pmol及び12.8pmolとラベルを貼る。
4) 溶解用緩衝液1の500μLを全てのチューブにピペットで加える。
5)12.8pmolチューブに標準品原液(512pmol/mL)を500μLピペットで取り、そして充分混合する。12.8pmolチューブから500μLを6.4pmolチューブに移し、そして充分混合する。この2倍希釈を残りのチューブを用いて次々繰り返す。
6)各一連の希釈物及び標準品原液からの2本づつ50μLのアリコートは、0.2〜25.6pmol標準の範囲でcAMPの8個の標準濃度を与える。
化合物希釈用緩衝液
1mM IBMXの50μLをPBS 100mLに加えて最終濃度を100μMにし、そして30℃で20分間超音波処理する。
PGD2の調製
PGD2(FW, 352.5)1mgをDMSO 284μLに溶解させて10mMの原液を作り、20℃で保存する。各アッセイの前に新たに調製する。10mM原液の3μLをDMSO 20mLに加え、それを充分混合し、そして10mLをPBS 40mLに移す。
【0074】
化合物の希釈
本発明の化合物のいくつかのバッチを96穴プレートで試験する。化合物の各バッチは96穴プレートの1列を占める。
化合物の希釈はビオメックス(Biomex)2000(ベックマン(Beckman))中で方法 1_cAMP DP 11ポイントを使用して実施する。
10mM原液化合物プレートからの各化合物5μLを96−穴プレートの穴にそれぞれ以下のように移す。
【表1】
【0075】
カラム7に28μLのDMSOを満たす以外はプレートを45μLのDMSOで満たす。カラム1を充分ピペットで取り、そして12μLをカラム7に平行に移す。1:10連続希釈をカラム1からカラム6まで及びカラム7からカラム11まで、5μLを45μLのDMSOに移すことにより行って下記の濃度を作る。
【表2】
【0076】
新しい96−穴プレートを247.5μLの化合物希釈用緩衝液で満たす。上記プレートからの連続希釈化合物の2.5μLを新しいプレート(1:100希釈)に次のように移す。
【表3】
【0077】
細胞の成長
1.LS174 Tを常にMEM (ATCC Cat No.30−2003), 10% FBS (ATCC Cat No.30−2020)及び追加の2mM L−グルタミン中で、37℃及び5% CO2で増殖させる。
2.0.05%トリプシン及びベルシン(Versine)(インビトロゲン(Invitrogen) Cat No. 25300−054)を37℃水浴で温める。
3.細胞から増殖用培地を除く。T165フラスコ中の細胞をトリプシン4mLで2回洗い、次いで37℃及び5% CO2で3分間インキュベートする。
4.培地10mLを加え、ピペット操作で完全に細胞を分離し、そして細胞を数える。
5.細胞密度を2.25×105細胞/mlにし、そしてアッセイ1日前に96−穴プレート中に200μL細胞/穴(45,000細胞/穴)を蒔く。
【0078】
検定手順
1日目
96−穴プレート中の培地200μLに45,000細胞/穴を蒔く。細胞プレートを37℃、5% CO2及び95%湿度で1晩インキュベートする。
2日目
1.化合物の希釈を行う。
2.アッセイ用緩衝液、溶解用緩衝液1及び2、PGD2及び標準品を用意する。
3.細胞から培地を吸引し、そしてツィマーク(Zymark)のサイクローン(Sciclone)−ALH/FDプロトコルcAMP DPを用いて、化合物溶液100μLを加える。
4.該細胞を37℃、5%CO2及び95%湿度で15分間インキュベートする。
5.300nM PGD2(最終濃度20X 15 nM) 5μLを、各穴にツィマークプロトコルcAMP DP PGD2を用いて加え、そして該細胞を37℃、5%CO2及び95%湿度で更に15分間インキュベートする。
6.細胞から培地を吸引し、そしてツィマークプロトコルcAMP DP溶解を用いて、溶解用緩衝液50μLを加え、そして室温で振とうしながら30分間インキュベートする。
6.免疫試薬150μLを全穴に加える(合計容量200μL/穴)。
7.プレートを密閉し、そして2分間振り、ワラック(Wallac)マイクロタイタープレートμシンチレーションカウンターのチャンバーに16時間入れる。
3日目
[125I] cAMPの量を2分間1450トリラックス(Trilux) シンチレーションカウンター中で計測する。
【0079】
データ処理
cAMP対CPMの標準曲線をセットアップする。
【表4】
【0080】
未知のサンプルのcAMP濃度(pmol/mL)を、cAMP対CPMの標準曲線から計算する。%阻害を下記の式を用いて計算する:
%阻害=[(対照のpmol−サンプルのpmol)×100]/[対象のpmol(細胞+PGD2のみ)]
【0081】
結果
二水素リン酸塩は、0.3ナノモルの濃度で、ヒトLS174 T細胞におけるSPA cAMPアッセイで50%阻害を生じる。
二水素リン酸塩の水溶性は、遊離形及び塩酸塩よりも増加した。下記のチャートに、遊離形、塩酸塩及び二水素リン酸塩の25℃でのリン酸緩衝液(PBS pH 7.4、0.1M一塩基性リン酸ナトリウム溶液19%と0.1M二塩基性リン酸ナトリウム溶液81%との混合物)中の溶解度、並びに遊離形及び二水素リン酸塩の37℃での絶食状態の擬似胃腸液(“FaSSIF”、Dressman J.B., Amidon G.L., Reppas C. 及びShah V.P., Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms, Pharmaceutical Research: 1998, Vol. 15, No. 1, 11−22に従って調製)中の溶解度を記載する。
【表5】
【0082】
二水素リン酸塩は大規模製造及び薬学的製剤に有用な予期しない性質も有する。第1に、二水素リン酸塩は非常に良好な結晶化度を有する。図1に示す二水素リン酸塩のXRPDは中程度の強度と解像度を有するピークを含む。中央の2シータ領域にハロがないことは、無晶相がほとんどないことを示唆する。更に、図8に示された二水素リン酸塩の粉砕前及び粉砕後の顕微鏡写真は、物理化学的性質が変化することなく、粒子径が良好に減少したことを証明する。
【0083】
しかしながら、ナトリウム塩は低い結晶化度を有する。図11に示されたように、ナトリウム塩のバッチ2のXRPDは弱い強度の幅広いピークを含み、低い結晶化度を示す。ナトリウム塩のバッチ1のXRPDは、物質が殆ど無晶形であることを示す。更に、両バッチのXRPD中の中央の2シータ領域にハロがあることは、物質の非常に低い結晶化度を示唆する。
【0084】
更に、二水素リン酸塩は今回、1つの多形型のみで存在することが示された。しかしながら塩酸塩は今回、3つの多形型で存在することが示され、3つの多形型はある条件下で相互変換に付される可能性がある。塩酸塩の多形型の1つである、図12のバッチ2は、XRPDが弱い強度の幅広いピークを有するので、低い結晶化度を有する。塩酸塩の多形型の別の1つである、図12のバッチ1は、良好な結晶化度を有する。
【図面の簡単な説明】
【0085】
【図1】二水素リン酸塩のX線粉末回折パターン(XRPD)である。
【図2】二水素リン酸塩の図1のX線粉末回折パターンについて、XRPD d−距離及び相対強度の対応する表である。
【図3】二水素リン酸塩の示差走査熱量計−熱重量分析器(DSC−TGA)サーモグラムである。TGAデータは、室温から175℃でほぼ0.4%の合計質量損失を示す。DSCは結晶相の融液のみを含む。分子の分解は融点で始まる。
【図4】二水素リン酸塩のDSCサーモグラムである。溶融はサーモグラム中で219℃で開始することが観察された唯一の熱的事象である。
【図5】二水素リン酸塩の動的蒸気収着分析器(DVS)吸湿等温プロット、及び対応する水収着プロフィールの表を示す。データは、94%の相対湿度(RH)でヒステリシスがほとんどない、収着中の〜0.9%の重量増加を示し、これはゆるく結合した表面水収着を示唆する。
【図6】DVS前及び後の二水素リン酸塩のXRPDパターンを示す。試料を調製しそして分析を、0% RHで12時間置いたDVSから取り出してから2分以内に始める。データは、DVS実験の結果、強度及びd距離にほとんど変化がないことを示し、これは結晶構造に検出可能な変化が起きなかったことを示す。
【図7】二水素リン酸塩の顕微鏡写真を示す。このロットは主として長さが約30ミクロンまでの棒及び針形状の粒子から成る。
【図8】二水素リン酸塩の粉砕前及び粉砕後の顕微鏡写真、及び定量的粒子サイズ分布を示し、これは物理化学的性質に変化なく粒子サイズが良好に減少したことを示す。二水素リン酸塩の粉砕後の顕微鏡写真は、100倍の倍率で約30個の視野の検索の間、長さが10ミクロンよりも大きい粒子がない良く微粉化された結晶性物質を示す。微粉化二水素リン酸塩の粒子サイズ分布は単一モード曲線であると決定される。中央値[x(50)]は2.0ミクロンであり、粒子の90%は4.7ミクロン以下である。微粉化工程は中央値 (以前は5.8ミクロン)及びx(90)サイズ(以前は〜16ミクロン)の両方を減少させる。微粉化の前及び後の二水素リン酸塩のXRPD、即ちピーク強度、位置(d−距離)及び解像度、は同じである。
【図9】微粉化の後の二水素リン酸塩のDVS吸湿性等温線であり、無晶化が起きないことを示す。
【図10】二水素リン酸塩のフーリエ変換−赤外(FT−IR)スペクトル及び対応するFT−IRピークの表を示す。
【図11】ナトリウム塩の2つの別々の調製物、バッチ1及びバッチ2、についてのXRPDパターンの重ね合わせ図を示す。ナトリウム塩のバッチ1はエタノール:酢酸エチルから再結晶させた。ナトリウム塩のバッチ2はメタノール:酢酸エチルから再結晶させた。
【図12】アセトンから再結晶させた塩酸塩の2つの別々の調製物、バッチ1及びバッチ2、についてのXRPDパターンの重ね合わせ図を示す。
【特許請求の範囲】
【請求項1】
式(III):
【化1】
の化合物。
【請求項2】
結晶形である、請求項1記載の化合物。
【請求項3】
請求項1記載の化合物の薬学的有効量を、薬学的に許容される担体と混合して含む、薬学的組成物。
【請求項4】
アレルギー疾患、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患、かゆみに伴う行動の結果二次的に発生する疾患、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、又は潰瘍性大腸炎の治療方法であって、請求項1記載の化合物の薬学的有効量を、これらの治療が必要な患者に投与することを含む、上記治療方法。
【請求項5】
かゆみに伴う行動の結果二次的に発生する疾患が、白内障、網膜剥離、炎症、感染症又は睡眠障害である、請求項4記載の方法。
【請求項6】
アレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、食物アレルギー、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、アトピー性皮膚炎、じんましん、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、又は潰瘍性大腸炎の治療方法であって、請求項1記載の化合物の薬学的有効量を、これらの治療が必要な患者に投与することを含む上記治療方法。
【請求項7】
アレルギー疾患を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー患者の治療方法。
【請求項8】
気管支喘息を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、気管支喘息の患者の治療方法。
【請求項9】
アレルギー性鼻炎を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー性鼻炎の患者の治療方法。
【請求項10】
アレルギー性皮膚炎を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー性皮膚炎の患者の治療方法。
【請求項11】
アレルギー性結膜炎を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー性結膜炎の患者の治療方法。
【請求項12】
慢性閉塞性肺疾患を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、慢性閉塞性肺疾患の患者の治療方法。
【請求項13】
請求項1記載の化合物の薬学的有効量、並びに抗ヒスタミン剤、ロイコトリエン拮抗剤、ベータ作用薬、PDE4阻害剤、TP拮抗剤及びCrTh2拮抗剤から成る群から選ばれる化合物を、薬学的に許容される担体と混合して含む、薬学的組成物。
【請求項14】
抗ヒスタミン剤がフェキソフェナジン、デスロラタジン、ロラタジン又はシチリジンであり、ロイコトリエン拮抗剤がモンテルカスト又はザフィルラストであり、ベータ作用薬がアルブテロール、サルブテロール又はテルブタリンであり、PDE4阻害剤がロフルミラスト又はシロミラストであり、TP拮抗薬がラマトロブランであり、そしてCrTh2拮抗剤がラマトロブランである、請求項13記載の薬学的組成物。
【請求項1】
式(III):
【化1】
の化合物。
【請求項2】
結晶形である、請求項1記載の化合物。
【請求項3】
請求項1記載の化合物の薬学的有効量を、薬学的に許容される担体と混合して含む、薬学的組成物。
【請求項4】
アレルギー疾患、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、かゆみを伴う疾患、かゆみに伴う行動の結果二次的に発生する疾患、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、又は潰瘍性大腸炎の治療方法であって、請求項1記載の化合物の薬学的有効量を、これらの治療が必要な患者に投与することを含む、上記治療方法。
【請求項5】
かゆみに伴う行動の結果二次的に発生する疾患が、白内障、網膜剥離、炎症、感染症又は睡眠障害である、請求項4記載の方法。
【請求項6】
アレルギー性鼻炎、アレルギー性結膜炎、アトピー性皮膚炎、気管支喘息、食物アレルギー、全身性肥満細胞症、全身性肥満細胞活性化に伴う障害、過敏症ショック、気管支収縮、気管支炎、じんましん、湿疹、アトピー性皮膚炎、じんましん、炎症、慢性閉塞性肺疾患、虚血性再潅流傷害、脳血管障害、慢性関節リウマチ、肋膜炎、又は潰瘍性大腸炎の治療方法であって、請求項1記載の化合物の薬学的有効量を、これらの治療が必要な患者に投与することを含む上記治療方法。
【請求項7】
アレルギー疾患を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー患者の治療方法。
【請求項8】
気管支喘息を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、気管支喘息の患者の治療方法。
【請求項9】
アレルギー性鼻炎を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー性鼻炎の患者の治療方法。
【請求項10】
アレルギー性皮膚炎を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー性皮膚炎の患者の治療方法。
【請求項11】
アレルギー性結膜炎を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、アレルギー性結膜炎の患者の治療方法。
【請求項12】
慢性閉塞性肺疾患を患う患者に請求項1記載の化合物の薬学的有効量を投与することを含む、慢性閉塞性肺疾患の患者の治療方法。
【請求項13】
請求項1記載の化合物の薬学的有効量、並びに抗ヒスタミン剤、ロイコトリエン拮抗剤、ベータ作用薬、PDE4阻害剤、TP拮抗剤及びCrTh2拮抗剤から成る群から選ばれる化合物を、薬学的に許容される担体と混合して含む、薬学的組成物。
【請求項14】
抗ヒスタミン剤がフェキソフェナジン、デスロラタジン、ロラタジン又はシチリジンであり、ロイコトリエン拮抗剤がモンテルカスト又はザフィルラストであり、ベータ作用薬がアルブテロール、サルブテロール又はテルブタリンであり、PDE4阻害剤がロフルミラスト又はシロミラストであり、TP拮抗薬がラマトロブランであり、そしてCrTh2拮抗剤がラマトロブランである、請求項13記載の薬学的組成物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】

【図8】

【図9】
【図10】
【図11】
【図12】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2009−511591(P2009−511591A)
【公表日】平成21年3月19日(2009.3.19)
【国際特許分類】
【出願番号】特願2008−535672(P2008−535672)
【出願日】平成18年10月12日(2006.10.12)
【国際出願番号】PCT/US2006/039901
【国際公開番号】WO2007/047378
【国際公開日】平成19年4月26日(2007.4.26)
【出願人】(500137976)アベンティス・ファーマスーティカルズ・インコーポレイテツド (76)
【Fターム(参考)】
【公表日】平成21年3月19日(2009.3.19)
【国際特許分類】
【出願日】平成18年10月12日(2006.10.12)
【国際出願番号】PCT/US2006/039901
【国際公開番号】WO2007/047378
【国際公開日】平成19年4月26日(2007.4.26)
【出願人】(500137976)アベンティス・ファーマスーティカルズ・インコーポレイテツド (76)
【Fターム(参考)】
[ Back to top ]