
プロテアーゼインヒビター及びそれらの変異体のバクテリア発現

【課題】バクテリア種におけるプロテアーゼインヒビター及びそれらの変異体の発現に関係する組成物及び方法を提供する。
【解決手段】融合タンパク質を含む組成物であって、前記融合タンパク質がプロテアーゼインヒビター及び所望のペプチドを含むことを特徴とする組成物。
【解決手段】融合タンパク質を含む組成物であって、前記融合タンパク質がプロテアーゼインヒビター及び所望のペプチドを含むことを特徴とする組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、バクテリア種内でプロテアーゼインヒビター及びそれらの変異体の発現に関係する組成物及び方法を提供する。本発明は更に、融合核酸、ベクター、融合ポリペプチド、及びプロテアーゼインヒビターを得るための工程を提供する。
【背景技術】
【0002】
プロテアーゼは幅広い各種生体内作用に含まれている酵素である。プロテアーゼ及びプロテアーゼインヒビターのバランスを阻害することは病原性組織崩壊に関係する。すなわち、多くの研究は、組織障害におけるプロテアーゼの役割に焦点をあてており、プロテアーゼ及びプロテアーゼインヒビターの間のバランスが組織の完全性の維持における主要な決定要因であると言われてきた。好中球を含む炎症細胞由来のセリンプロテアーゼは、肺気腫、関節炎、アトピー性皮膚炎及び乾癬等の各種炎症性の病気に関係している。
【0003】
プロテアーゼは特定の癌を転移させるために機能しているとも考えられている。正常な細胞は、細胞外マトリクス(ECM)と呼ばれている、タンパク質ネットワークと接触して存在している。ECMは細胞の移動に対するバリアであり、癌細胞は、それらのアタッチメントを壊し、分解し、転移するためにECMに沿って移動する。プロテアーゼはタンパク質を分解する酵素であり、ECMを壊すことにより元の位置から腫瘍細胞を遊離させることを補助すると考えられてきた。近年の研究において、プロテアーゼは細胞形態変化およびプロテアーゼ活性化レセプター(PAR2)といわれる腫瘍細胞膜においてタンパク質を活性化することによる細胞の運動を促進することが示唆されている。このことは、細胞の運動装置を活性化する細胞内反応のカスケードとなる。従って、腫瘍の転移における第一のステップは移動方向に面した1の縁に明瞭な突起を形成するように細胞形状が再構成されることであると考えられている。形状を再構築した細胞はその後、血管壁を通じて移動し、離れた位置に移動し、最終的には、再付着し、転移腫瘍を形成する。例えば、ヒトの前立腺上皮細胞は、精漿の一成分のカリクレン様セリンプロテアーゼである前立腺特異抗原(PSA)を構造的に分泌する。このプロテアーゼは細胞外マトリクスを分解し、癌細胞の侵入を促進させる作用をする。
【0004】
合成及び天然のプロテアーゼインヒビターはインビボ(in vivo)及びインビトロ(in vitro)において癌の促進を抑制することが示唆されている。研究の結果、セリンプロテアーゼインヒビター又はセルピンズ(SERPINS)として分類される構造的に類似したタンパク質のファミリーに属している特定のプロテアーゼインヒビターは、好中球エラスターゼだけでなくトリプシン、カテプシンG、トロンビン、及び組織性カリクレインを含む幾つかのプロテアーゼを抑制することが示唆されている。セルピンズ(SERPINS)はインビトロ(invitro)において発癌物質により誘発された形質転換、及び実験動物モデルにおける発癌の予防/抑制に効果的である。精製されたプロテアーゼインヒビターの全身への投与は関節炎及び軟骨及び骨の崩壊を予防することが明らかにされている。
【0005】
プロテアーゼインヒビターの局所投与は、身体に位置するわずかな斑及び身体の大部分を含むアトピー性皮膚炎及び通常の皮膚炎症等を治療するために使用されている。プロテアーゼインヒビターの脱色活性及び紫外線誘導着色を防止する能力は、インビトロ(invitro)及びインビボ(in vivo)にて報告されている(Paine et al., J. Invest. Dermatol., 116: 587-595[2001]参照のこと)。プロテアーゼインヒビターは外傷の治癒も促進することが報告されている。例えば、分泌白血球プロテアーゼインヒビターは局所的に適用したときに組織の傷を修復し、外傷治癒を早めることが報告されている。更に、セリンプロテアーゼインヒビターは紅斑性狼瘡患者において痛みを減らすことが報告されている(米国特許6537968参照)。
【0006】
上に記載したように、プロテアーゼインヒビターはプロテアーゼの作用を阻害する。天然のプロテアーゼインヒビターは、穀物顆粒(オーツ麦、大麦、及びメイズ)、ブリュッセル芽キャベツ、たまねぎ、大根、小麦、シコクビエ(fingermillet)、及び落花生等の各種食物の中に存在する。特に興味のある供給源はダイズである。ダイズの中にある2の最も重要なプロテアーゼインヒビターの平均レベルはクニッツ(Kunitz)インヒビターにおいて約1.4%及びボウマン-バーク(Bowman-Birk)において約0.6 %である。これらの含有量が低い原材料は医薬品組成物のための天然プロテアーゼインヒビターを単離するのに実用的でない。
【0007】
従って、プロテアーゼインヒビター及びその変異体の大規模生産スケールに適した方法及びそれに用いる組成物に対する需要が存在する。特に、これらのプロテアーゼが哺乳類の組織培養細胞において製造されるときに生ずる、骨-血液感染因子に関する危険性を減らす又は除く方法及び組成物に対する需要も存在する。
【発明の概要】
【0008】
本発明はバクテリア種の中でプロテアーゼインヒビター及びそれらの変異体の発現に関係する方法及び組成物を提供する。本発明は更に、融合核酸、ベクター、融合ポリペプチド及びプロテアーゼインヒビターを得る工程を提供する。
【0009】
本明細書において提供されるものは、核酸、細胞、及びプロテアーゼインヒビター、これらの変異体、並びにその生産方法である。
【0010】
本発明は少なくとも1の機能的なプロテアーゼインヒビターをコードする核酸を提供する。一の側面において、第一、第二、第三及び第四の核酸配列に作動可能に結合している調節配列を含む核酸が提供される。幾つかの態様において、転写終了配列は、第四の核酸配列の後に付加されている。
【0011】
代替的な態様において、第一の核酸配列は、第一宿主微生物の中の分泌配列として機能するシグナルペプチドをコードする。第二の核酸は、通常第一又は第二宿主微生物から分泌される分泌ポリペプチド又はそれらの機能部分をコードする。第三の核酸は切断可能リンカーをコードし、第四の核酸は、プロテアーゼインヒビター又はそれらのフラグメントをコードする。更なる態様において、本発明は少なくとも1のプロテアーゼインヒビターをコードしている核酸配列を含む少なくとも1の発現カセットを提供する。
【0012】
追加的な態様において、本発明は少なくとも1のプロテアーゼインヒビター変異体をコードするポリヌクレオチドを提供する。幾つかの好ましい態様において、このポリヌクレオチドは、野生型ボウマン-バーク(Bowman-Birk)インヒビター(BBI)の少なくとも1のループが修飾されているBBI変異体をコードしている。
【0013】
本発明は機能的なプロテアーゼインヒビター又はそれらの変異体を発現する方法も提供する。幾つかの好ましい態様において、機能的なプロテアーゼインヒビター及び/又はそれらの変異体に適した宿主細胞が提供される。幾つかの態様において、(i)少なくとも1のプロテアーゼインヒビター又はそれらの変異体をコードする核酸配列を含む少なくとも1の発現カセットで宿主細胞を形質転換し、及び(ii)少なくとも1のプロテアーゼインヒビター又はそれらの変異体の発現に適した条件下で宿主細胞を培養する。幾つかの態様において、この方法は更にプロテアーゼインヒビター又はこれらの変異体の回収工程を含む。
【0014】
代替的な態様において、 (i)プロテアーゼインヒビター又はそれらの変異体をコードする少なくとも1の核酸配列を含む第一の発現カセットで宿主細胞を形質転換し、更に(ii)少なくとも1のチオールジスルフィド酸化還元酵素又はシャペロンをコードする核酸配列を含む第二の発現カセットで宿主細胞を形質転換し、そして(iii)プロテアーゼインヒビター又はそれらの変異体の発現に適した環境下において宿主細胞を培養する。幾つかの態様において、プロテアーゼインヒビター又はそれらの変異体は培地から回収される。幾つかの好ましい態様において、プロテアーゼインヒビター又はそれらの変異体は融合タンパク質として発現される。更なる態様において、該方法は更にプロテアーゼインヒビター又はそれらの変異体の回収工程を含む。
【0015】
更なる態様において、機能的なプロテアーゼインヒビター又はそれらの変異体はシグナル配列、セルラーゼ触媒ドメイン、切断可能リンカー領域を含む融合タンパク質として発現され、その後、発現されたタンパク質は、プロテアーゼインヒビター又はそれらの変異体の成熟型になる。
【0016】
追加的な態様において、発現されたタンパク質はプロテアーゼ及び/又は酸/熱で、プロテアーゼインヒビター又はそれらの変異体を融合タンパク質から遊離するために処理される。
【0017】
更なる態様において、本発明は、
a) Bowman-Birk インヒビター変異体、
b) Bowman-Birk インヒビター、及び
c) 少なくとも1の変異体配列を含むスキャッフォールド
から成る群より選択されるプロテアーゼインヒビター活性を有するポリペプチドを提供する。
【0018】
本発明は、プロテアーゼインヒビター及び所望のペプチドを含む融合タンパク質を含む組成物を提供する。幾つかの態様において、融合タンパク質は配列番号2、4、6、及び8から成る群より選択されるアミノ酸配列を含む。代替的な態様において、融合タンパク質は、配列番号1、3、5及び7から成る群より選択される核酸配列によりコードされている。幾つかの好ましい態様において、プロテアーゼインヒビターはボウマン-バーク(Bowman-Birk)インヒビター、ダイズトリプシンインヒビター(STI)、及びエグリン(eglin)Cから成る群より選択される。幾つかの特定の好ましい態様において、このプロテアーゼインヒビターはトリプシンループ及びキモトリプシンループから成る群より選択される少なくとも1のループを含むBBIである。更なる態様において、プロテアーゼインヒビターは所望のペプチドに対するスキャッフォールドである。幾つかの好ましい態様において、このループは少なくとも1の所望のペプチドを含む。幾つかの好ましい態様において、所望のペプチドは、配列番号9で定義されるアミノ酸配列を含む。幾つかの好ましい態様において、融合タンパク質は配列番号4で定義されるアミノ酸配列を含む。
【0019】
本発明は、a)ボウマン-バーク(Bowman-Birk)インヒビター(BBI)又はそれらの変異体由来のプロテアーゼインヒビターをコードする異種DNA配列を含むDNA構築体を宿主細胞内に導入する工程、b)異種DNA配列を発現させるために適切な培養条件下でバクテリア細胞を培養する工程、及びc)プロテアーゼインヒビターを生産する工程を含むことを特徴とするバクテリア細胞内で少なくとも1のプロテアーゼインヒビターを生産する方法を提供する。幾つかの好ましい態様において、構築体は更に少なくとも1の触媒ドメインを有する。幾つかの態様において、触媒ドメインはセルラーゼ、クチナーゼ、及びジスルフィドイソメラーゼから成る群より選択される。幾つかの好ましい態様において、この方法は更にプロテアーゼインヒビターを回収する工程を含む。幾つかの好ましい態様において、この方法は更にプロテアーゼインヒビターを活性化する工程を含む。最も好ましい態様において、この活性化は、ジスルフィド結合を還元する組成物、ジスルフィド結合を酸化する組成物、及び酸化還元電位を変更する組成物から成る群から選択される少なくとも1の試薬にプロテアーゼインヒビターを曝すことにより実施される。幾つかの態様において、バクテリア細胞はバチルス(Bacillus)属のメンバーである。追加的な態様において、プロテアーゼインヒビターはi)配列番号11と少なくとも90%の配列同一性を有するプロテアーゼインヒビター、及びii)配列番号13と少なくとも90 %の配列同一性を有するプロテアーゼインヒビターから成る群より選択される。更なる態様において、プロテアーゼインヒビターは変異体配列を含む。追加的な態様において、プロテアーゼインヒビターは、少なくとも1のループが配列番号9で置換されており、このループがトリプシンループ及びキモトリプシンループから成る群より選択されることを特徴とするボウマン-バーク(Bowman-Birk)インヒビターである。幾つかの態様において、この方法は更にチオールジスルフィド酸化還元酵素又はシャペロンをコードする第二の核酸配列をバクテリア細胞内に導入する工程を含む。幾つかの好ましい態様において、プロテアーゼインヒビターは融合タンパク質として発現される。幾つかの好ましい態様において、前記融合タンパク質は更にセルラーゼ触媒ドメイン、切断可能部位、及びプロテアーゼインヒビターを含む。幾つかの特定の好ましい態様において、融合タンパク質は、プロテアーゼインヒビターを遊離するためにプロテアーゼ及び/又は酸/熱で処理される。更なる好ましい態様において、融合タンパク質は少なくとも1のリンカー配列を更に含む。幾つかの態様において、このリンカー配列は配列番号141乃至143から成る群より選択される。
【0020】
本発明は、本明細書で説明する方法のいずれか又は全てに従って製造されたプロテアーゼインヒビターを含む組成物も提供する。幾つかの態様において、本明細書で開示する各種プライマー及びオリゴヌクレオチドは本発明の融合タンパク質の製造に用いる。更に、本発明は本発明の使用に適した多数のペプチドを提供する。
【0021】
本発明は本明細書で開示するプロテアーゼインヒビター組成物のいずれかを標的タンパク質に接触させる工程、及び標的タンパク質のタンパク質分解活性を抑制するために標的タンパク質と前記プロテアーゼインヒビター組成物を結合させる工程を含む標的タンパク質のタンパク質分解活性を抑制する方法を提供する。
【0022】
本発明は更に配列番号10及び12で定義されるポリペプチド配列から成る群より選択されるプロテアーゼインヒビターをコードする単離ポリヌクレオチドを提供する。
【0023】
本発明は、ポリヌクレオチド配列を含む発現ベクターも提供する。前記ポリヌクレオチド配列は、a) 少なくとも1のループ残渣が変異体配列で置換されていることを特徴とし、配列番号11と少なくとも90%の配列同一性を有するプロテアーゼインヒビターをコードしているポリヌクレオチド配列、及びb) 少なくとも1のループ残渣が変異体配列で置換されていることを特徴とし、配列番号13と少なくとも90%の配列同一性を有するプロテアーゼインヒビターをコードしているポリヌクレオチド配列から成る群より選択される。幾つかの態様において、この発現ベクターは更に5’末端から3’末端に向けて、バクテリア細胞内において分泌配列として機能するシグナルペプチドをコードする第一の核酸配列、分泌ポリペプチド又はそれらの機能部分をコードする第二の核酸配列、切断可能リンカーをコードする第三の核酸配列及びプロモーターインヒビターをコードするDNA配列を更に含む。本発明は、本発明の発現ベクターで形質転換させた宿主細胞も提供する。幾つかの好ましい態様において、宿主細胞はバチルス(Bacillus)属の細胞である。
【0024】
本発明は少なくとも1のスキャッフォールドタンパク質及び少なくとも1のペプチドを含む組成物を提供する。前記スキャッフォールドは、ボウマン-バーク(Bowman-Birk)インヒビターを含む。幾つかの好ましい態様において、このペプチドはVegFに結合するペプチドを含む。
【0025】
本発明の他の目的、技術的特徴、及び利点は以下の詳細な説明に明確に記載する。しかしながら、本発明の好ましい態様を示す詳細な説明及び特定の実施例は説明の用途にのみ用い、詳細な説明から当業者にとって自明である各種変更及び修正は本発明の範囲及び本発明の精神の範囲内であることが理解される。
【発明を実施するための最良の形態】
【0026】
発明の開示
本発明はバクテリア種の中でプロテアーゼインヒビター及びそれらの変異体を発現することに関係する組成物及び方法を提供する。本発明は更に融合核酸、ベクター、融合ポリペプチド、及びプロテアーゼインヒビターを得る工程を提供する。
【0027】
他の方法で示さない限り、本発明の実施は、分子生物学、微生物学、組換えDNA技術に関する従来技術を含む。そのような技術は、当業者に知られており、多くのテキスト及び参考文献に記載されている(Sambrooket al,. “Molecular Cloning: A Laboratory Manual”, Second Edition (Cold SpringHarbor), [1989]及びAusbel et al., “Current Protocols in Molecular Biology” [1987]参照のこと)。本明細書中で言及するすべての特許、特許出願、文献及び刊行物は参照により本明細書に援用する。
【0028】
他の違った方法で定義しない限り、本明細書で用いられる全ての技術的及び化学的言語は、本出願に関係する技術分野において当業者に通常理解されているものと同じ意味を有する。例えば、Singletonand Sainsbury, Dictionary of Microbiology and Molecular Biology, 2d Ed.,JohnWiley and Sons, NY (1994)及び Hale and Marham, The Harper Collins Dictionaryof Biology, Harper Perennial, NY (1991)は、本発明の技術分野の当業者が利用する、そして本発明において多く用いられている用語の一般的な辞書である。本明細書で述べられているのと同じかあるいは類似の多くの方法及び物質は本発明の実施に用いることができるけれども、本明細書中では特定の好ましい方法及び物質にのみ言及する。従って、以下で定義される文言は、本明細書全体を参照することで十分に説明される。本明細書で用いる記号、「一つの」及び「この」という語は、他に違った形で定義されていない限り、複数を示す場合にも用いる。数値範囲は、範囲を定義している上限及び下限の数値も含ものとする。違った方法で示さない限り、核酸は左から右に向けて5’から3’に向けて表記されるものとし、アミノ酸配列は、左から右に向けて、アミノ末端からカルボキシ末端として表記する。本発明は記載した特定の方法、手順及び試薬に限定されないことは当然でありこれらは変更可能である。
【0029】
更に、本明細書で付す標題は本発明の各種側面を限定するものではない。発明は明細書全体を参照して理解されるべきである。従って、以下で示す定義は、明細書全体を参照して完全に定義される。しかしながら、本発明の迅速な理解のために多数の文言を以下で定義する。
【0030】
定義
本明細書で用いる、「発現カセット」及び「発現ベクター」の語は、標的細胞の中で特定の核酸を転写させる一連の特定の核酸エレメントを持つ、組換え又は合成により生成された核酸構築体を意味する。組換え発現カセットは、プラスミド、染色体、ミトコンドリアDNA、色素体DNA、ウイスル又は核酸フラグメントに取り込ませることができる。通常、発現ベクターの組換え発現カセット部分には、転写される核酸配列及びプロモーターが含まれる。「発現カセット」の語は、本明細書において、「DNA構築体」及びこの等価物を指す場合に互換的に使用される。
【0031】
本明細書において、「ベクター」及び「クローニングベクター」の語は細胞内へ核酸配列を輸送することを意図する核酸構築体を意味する。
【0032】
本明細書において、「発現ベクター」の語は、異種細胞の中で異種DNAフラグメントを取り込み発現することができるベクターを意味する。多くの真核及び原核生物の発現ベクターが市販されている。適切な発現ベクターを選択することは当業者の知識の範囲内である。
【0033】
本明細書において、「プラスミド」の語は、クローニングベクターとして用いる環状二本鎖(ds)DNA構築体であり、種々の原核及び真核生物内で染色体外自己複製遺伝子要素を形成し、又は他の宿主細胞に組み込むことができるものを意味する。
【0034】
本明細書において、「核酸分子」及び「核酸配列」の語は、RNA、DNA、及びcDNA分子を含むがこれらに限定されない、任意の核酸の形態が含まれる。遺伝子コードの縮退の結果として、所定のタンパク質をコード化する多数のヌクレオチド配列が生産され得ることは当該技術分野において理解されている。
【0035】
本明細書において、「融合DNA配列」の語は、5’末端から3’末端に向けて、第一、第二、第三、及び第四DNA配列を含む。
【0036】
本明細書において、「第一核酸配列」又は「第一DNA配列」は、バクテリア種の中の分泌配列として機能するシグナルペプチドをコードしている。特に好ましいシグナル配列は融合ポリペプチドを発現及び分泌するために発現宿主により分泌されるポリペプチド由来の配列である。本明細書で用いるように、第一アミノ酸配列は、バクテリア種内で機能する分泌配列に対応している。そのようなアミノ酸配列は上に述べた第一DNA配列によりコードされる。
【0037】
本明細書において、「第二DNA配列」は選択されたバクテリア種により発現される「分泌ポリペプチド」(すなわち、「所望の分泌ポリペプチド」)をコードする。第一DNA配列と同様に、分泌ポリペプチドはバクテリア発現宿主が自然に分泌するものであることが好ましい。しかしながら、幾つかの態様において、この語は、異種タンパク質(すなわち、特定のバクテリア宿主が通常分泌しないタンパク質)も意味する。例えば、幾つかの好ましい態様において、バチルススブチルス(B.subtilis)以外のバシルス(Bacillus)属により発現されるセルラーゼは所望の分泌ポリペプチドとして用いられ、バチルススブチルス(B.subtilis)は、発現宿主として用いられる。
【0038】
本明細書で用いる、「分泌ポリペプチドの機能部分」及びこの同義語は、切り離された形状をしていても、正常な構造に折りたたまれる能力を有する切断された分泌ポリペプチドを意味する。幾つかの態様において、所望のポリペプチドに付着しているものとは関係なく、ポリペプチドが正規構造にホールドされるためには分泌ポリペプチドドメイン残渣の十分量が必要となる。しかしながら、大部分のケースにおいて、分泌ポリペプチドの一部のみでも正しく折りたたまれ、分泌ポリペプチドドメイン残渣欠失体と比較して、分泌が高まることが知られている。
【0039】
同様に、大部分の場合において、分泌ポリペプチドの切断体はその機能部分が生物学的機能を保持することを意味する。他の機能的なドメイン(例えば、基質結合ドメイン)も用いることができるけれども、本発明には分泌ポリペプチドの触媒ドメインを用いることが好ましい。加えて、好ましい態様において、触媒ドメイン及び全リンカー領域又は一部のリンカー領域が用いられる。
【0040】
本明細書で用いる、「第三のDNA配列」は、切断可能なリンカーポリペプチドをコードするDNA配列を含む。第三のDNA配列は、融合ポリペプチドを切断するための特定の酵素又は試薬により認識されるのに必要なアミノ酸配列のみをコードすることが必要であると解される。従って、適切な酵素による認識及び切断に必要なリンカー部分のみが必要とされる。
【0041】
本明細書で用いる、「第四のDNA配列」は「所望のポリペプチド」をコードする。そのような所望のポリペプチドは成熟型、又は未成熟型のいずれかのプロテアーゼインヒビター及びそれらの変異体を含む。
【0042】
対応する4のアミノ酸配列をコードする上で定義された4つのDNA配列は、「融合DNA配列」を形成するために結合される。そのような融合DNA配列は、固有のリーディングフレームの5’末端から3’末端に向かって、第一、第二、第三、第四DNA配列の順に結合される。そのように結合されているので、融合DNA配列は、そのアミノ末端から、バクテリア種の中で分泌配列として機能するシグナルペプチド、バクテリア種により通常分泌される分泌ポリペプチド又はその一部分、切断可能リンカーポリペプチド及び所望のポリペプチドをコードする「融合ポリペプチド」、「融合タンパク質」及び「融合アナログ」をコードする。
【0043】
本明細書で用いる、「所望のタンパク質」および、「所望のポリペプチド」の語は、分泌増強構築体に融合されない、成熟型又は未成熟型ポリペプチド又はタンパク質を意味する。従って、「所望のタンパク質」及び「所望のポリペプチド」の語は、非融合形態で宿主細胞により発現され、分泌されるものを意味する。
【0044】
本明細書で用いる、「融合ポリペプチド」、「融合タンパク質」及び「融合アナログ」は、アミノ末端から順に、宿主細胞の中で分泌配列として機能するシグナルペプチド、宿主細胞から通常分泌される分泌ポリペプチド又はその一部分、切断可能リンカーポリペプチド及び所望のポリペプチドをコードしている。幾つかの態様において、この融合タンパク質は宿主細胞酵素(例えば、プロテアーゼ)により、融合タンパク質の中の他のタンパク質配列から遊離した所望のタンパク質を生産するために処理される。本明細書において、「融合アナログ」、「融合ポリペプチド」及び「融合タンパク質」は互換的に使用する。
【0045】
本明細書で用いる、「プロモーター配列」の語は、発現の目的で、バクテリア宿主細胞により認識されるDNA配列を意味する。好ましい態様において、それは、融合ポリペプチドをコードしているDNA配列に作動可能に結合している。そのような結合は、融合DNA配列をコードしているDNA配列の翻訳開始コドンに関係するプロモーター位置を含む。特に好ましい態様において、このプロモーター配列は、融合DNA配列の発現を仲介する翻訳及び転写制御配列を含む。
【0046】
本明細書で用いる「ターミネーター配列」の語は、転写を終了させるために発現宿主により認識されるDNA配列を意味する。この配列は、発現される融合タンパク質をコードしている融合DNAの3’末端に作動可能に結合している。
【0047】
本明細書で用いる、「選択マーカーコード核酸配列」の語は、バクテリア細胞において発現できるヌクレオチド配列をいう。選択マーカーの発現は、対応する選択条件下において、発現している遺伝子を含む細胞に成長能力を付与する。核酸は、他の核酸配列と機能的な位置にある場合に「作動可能に連結する」という。例えば、分泌リーダー(すなわち、シグナルペプチド)をコード化するDNAは、ポリペプチドの分泌に関与するプレタンパク質として発現する場合に、当該ポリペプチドについてのDNAに作動可能に連結し、プロモーター又はエンハンサーは、配列の転写に影響を与える場合に、コード配列に作動可能に連結し、又は、リボソーム結合部位は、翻訳を促進する位置に存在する場合にコード配列に作動可能に連結するという。一般に、「作動可能に連結」とは、結合するDNA配列が隣接することを意味し、分泌リーダーの場合には、隣接しかつリーディング・フェーズ(reading
phase)中にあることを意味する。しかし、エンハンサーは隣接している必要はない。結合は、適宜の制限部位における連結反応(ligation)により達成される。そのような部位が存在しない場合には、従来法にしたがって合成オリゴヌクレオチド・アダプタ、又はリンカーを用いる。
【0048】
ここで用いる「組換え体」は、非相同核酸配列の導入により修飾された、またはそのように修飾された細胞に由来する細胞またはベクターに関するものを含む。従って、例えば組換え細胞は、天然型細胞(非組換え体)内に見られない遺伝子を発現し、あるいは、意図的な人為的介入により、そうでなければ異常に発現し、発現不足であり又は全く発現しない天然遺伝子を発現させる。
【0049】
本明細書において、「発現」という語は、ポリペプチドが遺伝子の核酸配列に基づいて生成される工程をいう。この工程には、転写及び翻訳の両方が含まれる。従って、「プロテアーゼインヒビター発現」は、発現される特定のプロテアーゼインヒビター及びそれらの変異体の転写及び翻訳を意味する。その製品は、前駆体RNA、mRNA、ポリペプチド、翻訳後処理を受けたポリペプチド及びそれらの誘導体を含む。同様に、「プロテアーゼインヒビター発現」の語は、図1で示す発現カセットにより示されるプロテアーゼインヒビター及びそれらの変異体の転写、翻訳、及び構築を意味する。プロテアーゼインヒビター発現のアッセイの例は、特定の条件に曝したときのバクテリアコロニーを用いる試験及びプロテアーゼインヒビターのウエスタンブロットだけでなくノザンブロット解析及びプロテアーゼインヒビターmRNAの逆転写ポリメラーゼ連鎖反応アッセイも含む。
【0050】
本明細書で用いる「単離」及び「精製」の語は、少なくとも1の天然由来成分が除去されている核酸又はポリペプチドを意味する。
【0051】
本明細書において用いる、「実質的に含まない(free)」の語は、乾燥重量で20%未満、10%未満、5%未満、又は1%未満の他のタンパク質(すなわち、汚染タンパク質)を含む所望のポリペプチド調製物を意味する。
【0052】
本明細書で用いる「実質的に精製された」の語は、本発明のタンパク質又はそれらのフラグメントについて使用するときは、意図する使用のための実用的及び適切な他の物質が実質的に存在しないことを言う。例えば、タンパク質はタンパク質配列決定及び/又は医薬品組成物の製造に対して使用するときは、宿主細胞の他の生物成分から十分に精製されており、このような成分を実質的に含まない。
【0053】
本明細書で用いる、「標的タンパク質」の語は、本明細書で提供される変異体インヒビターを結合することにより活性が阻害されるタンパク質(例えば、酵素、ホルモン等)を意味する。
【0054】
本明細書で用いる「変異体配列」の語は、野生型プロテアーゼインヒビター又は他のスキャッフォールドの結合ループと置き換わる短いペプチドループを意味する。この変異体配列はスキャッフォールドにおいて置き換わる場合には結合ループと同じ長さである必要はない。
【0055】
本明細書で用いる「スキャッフォールド」の語は、変異体配列が導入される野生型タンパク質配列を意味する。幾つかの態様において、スキャッフォールドは置き換わる部分(例えば、ループ)を有する。例えば、本明細書で提供されるBBI配列は変異体配列に対するスキャッフォールドとして使用することができる。
【0056】
プロテアーゼインヒビター
2のタンパク質プロテアーゼインヒビター、クニッツ(Kunitz)タイプトリプシンインヒビター(ダイズトリプシンインヒビター、STI)及び「ボウマン-バーク(Bowman-Birk)インヒビター(BBI)が大豆から単離されている(Birk,Int. J. Pept. Protein Res., 25:113-131 [1985]及びKennedy, Am. J. Clin. Neutr.,68:1406S-1412S [1998] 参照のこと)。これらのインヒビターは、変異体配列に対するスキャッフォールドとして提供される。加えて、変異体配列を含むスキャッフォールドの修飾に加えて、本明細書で用いられる他の所望のタンパク質はC末端に6のヒスチジン残基を含む(図1及び2を参照のこと)。
【0057】
ダイズトリプシンインヒビター(STI)
STIは、適切な化学量論的複合体を形成することによりトリプシンのタンパク質分解活性を抑制する(Liu, Chemistry and NutritionalValue of Soybean Components, In : Soybeans, Chemistry. Technology and Utilization, pp. 32-35, Aspen Publishers, Inc., Gaithersburg, Md., [1999] 参照のこと)。STIは2のジスルフィド結合を有する181アミノ酸残基からなり、荒い球形をしている(Song et al., J. Mol. Biol., 275: 347-63
[1998] 参照のこと)。トリプシン抑制ループは第一ジスルフィド結合の中に存在する。クニッツ(Kunitz)タイプのダイズトリプシンインヒビター(STI)は、プロテイナーゼインヒビターの活性の標準的メカニズムの定義を導く生化学及び動態研究における主要な物質として用いられ、プロテイナーゼの初期の研究に重要な役割を果たしてきた。
【0058】
ボウマン-バーク(Bowman-Birk)インヒビター(BBI)
ボウマン-バーク(Bowman-Birk)インヒビター(BBI)タンパク質は、豆科の種及び各種草を含む他の植物から単離された、動態学的及び構造学的によく知られているスモールタンパク質(60-90残基)のファミリーである。それらは、通常、それぞれ独立に結合ループを含んでいる2の三環式ドメインを含む左右対称構造をしている。しかしながら、1のドメインを有するもの、及び2より多いドメインを有するものもある。ダイズ(BBI)から単離された主に8kDa以下のボウマン-バーク(Bowman-Birk)インヒビター(BBI)は、2の分離した活性サイトループを有する。ループIはトリプシン様特異性を有するプロテアーゼを抑制し、ループIIは、キモトリプシン様特異性を有するプロテアーゼを抑制する(Chen etal., J. Biol. Chem., 267:1990 1994 [1992]、Wernerand Wemmer, Biochem., 31:999-1010 [1992]、Lin et al., Eur. J. Biochem., 212: 549-555 [1993]、Voss et al.,Eur. J. Biochem. , 242: 122-131 [1996]及びBillings et al., Pro. Natl. Acad. Sci.,
89: 3120-3124 [1992] 参照のこと)。これらの結合領域はそれぞれ、各種セリンプロテイナーゼインヒビターに見られる構造である「カノニカルループ(canonicalloop)」構造を含む(Bode and Huber, Eur. J. Biochem., 204: 433-451 [1992])。STI及びBBIはダイズの種の中のみに見られ、その植物の他の部分には見られない(Birk,Int. J. Pept.Protein Res., 25: 113-131 [1985] 参照のこと)。
【0059】
多くのBBIのイソ型が特徴付けられているけれども、配列番号13は、約71アミノ鎖残基を含むBBI主鎖のアミノ酸配列を示す(実施例1を参照のこと)。
【0060】
ダイズにおいて、BBIは19アミノ酸長のN末端プロペプチドを有するプロタンパク質として生産される。従って、幾つかの態様において、BBIは、完全なプロペプチド又は少なくともプロペプチドの1部を用いて生産される。幾つかの態様において、BBIはN-又はC-末端のいずれかから10ものアミノ残渣が除去されて、切断されている。例えば、種子を乾燥するときに、幾つかのBBI分子は、9又は10アミノ酸残基が取り除かれたC末端を有する。従って、タンパク質分解は、通常、最初のジスルフィド結合の前及び最後のジスルフィド結合の直後が高くなる。この影響は標的タンパク質への結合にとり通常マイナスとはならない。しかしながら、異性体又は切断形態のいずれか1が本発明の各種態様で使用されることは評価されるであろう。
【0061】
プロテアーゼインヒビター変異体
上で説明するように、STI及びBBIプロテアーゼインヒビターはプロテアーゼを抑制する結合ループを有する。本発明は、1以上の活性部位が修飾されている(例えば、BBIのループI及び/又はループII)プロテアーゼインヒビター変異体を提供する。幾つかの好ましい態様において、標的タンパク質と相互作用する配列でこのループを置換する。
【0062】
例えば、幾つかの態様において、このループは、VEGF結合タンパク質、C2、C3、C4及びC5インヒビター等の補体経路のインヒビター、コンプスタチン(Compstatin)、サイトカイン、他の所望のタンパク質等に由来する配列で置換される。すなわち、任意の適切な配列を本発明に用いることから、本発明はこれらのループのいずれかの中で置換された特定の配列に限定することは意図していない。
【0063】
幾つかの態様において、変異体配列は、ファージディスプレイ法及び他の適切な方法を含むがこれらに限定されない本技術分野で知られた各種方法により選択される。例えば、ランダムペプチド遺伝子ライブラリーは、ファージPIII遺伝子で融合され、このペプチドライブラリーは、ファージの表面に提示される。引き続き、このファージディスプレイライブラリーは、標的タンパク質に曝され、非特異結合を除去するために緩衝液で洗浄される(この工程はパニングと称される)。最終的に、結合しているファージ及びこのペプチドをコードするDNA配列のPCR産物が単離される。
【0064】
幾つかの態様において、このループの1は、変異体配列で置換される(すなわち、ペプチド、3から14のアミノ酸長、5から10のアミノ酸長が好ましい)。それらが所望の結合及び/又は抑制を示す限り、より長い配列を本発明に用いることができる。加えて、結合ループの置換に適したペプチドが固定ループ内(すなわち、2のシステイン残基の間のジスルフィド結合の存在により形成されいているループ)に含まれる場合には、機能的な構造を採用することが好ましい。幾つかの態様において、このペプチドは7から9アミノ酸長である。これらの置き換え配列もまたプロテアーゼインヒビター又は標的配列への結合も提供する。幾つかの態様において、1のアミノ酸が置換されていることが好ましい。
【0065】
融合タンパク質
好ましい態様において、各プロテインプロテアーゼインヒビター又はそれらの変異体は宿主バクテリア細胞により融合タンパク質として発現される。所望のタンパク質を遊離するために融合タンパク質を切断することは有用であるが、必要ではない。融合タンパク質として発現及び分泌されたプロテアーゼインヒビター及びそれらの変異体は、驚くべきことにそれらの機能を保持している。
【0066】
対応するアミノ配列をコードする、上で定義するDNA配列は「融合DNA配列」を形成するように結合される。そのようなDNA配列は、第一、第二、第三及び第四DNA配列の順で、5’末端から3’末端に向けて固有のリーディングフレーム中に構築される。そのように構築されるので、このDNA配列は、アミノ末端からバクテリア種の中で分泌配列として機能するシグナルペプチド、バクテリア種から通常分泌される分泌ポリペプチド又はそれらの一部、切断リンカーペプチド及び所望のタンパク質(例えば、プロテアーゼインヒビター及びそれらの変異体)をコードしている「融合ポリペプチド」をコードする。融合タンパク質を生産するための各種方法は本技術分野で知られている(米国特許5,411,873、5,429,950及び5,679,543参照のこと。これらの文献を参照により本明細書に援用する)。従って、任意の適切な方法を本発明に用いることができる。
【0067】
組換えプロテアーゼインヒビターの発現
本発明は融合タンパク質の生産に依存しているので、本発明を実施するには組み換え遺伝子の分野の既存技術を用いる。本発明に用いる一般的方法が開示されている基本テキストには、Sambrooket al., Molecular Cloning, A Laboratory Manual (2nd ed. 1989);Kriegler, GeneTransfer and Expression : A Laboratory Manual (1990); 及びAusubel et al., eds.,Current Protocols in Molecular Biology(1994)がある。
【0068】
本発明は、プロテアーゼインヒビターをコードする核酸配列を含む発現ベクターで形質導入、形質転換又は形質感染させたバクテリア宿主細胞を提供する。温度、pH等の培養条件は形質導入、形質転換、又は形質感染以前に親宿主細胞に用いられるものと同様であり、当業者にとって自明な事項である。
【0069】
基本的に、融合タンパク質をコードしているヌクレオチド配列は宿主細胞の中で機能するプロモーター配列に作動可能に結合している。このプロモーター遺伝子単位は、複製及び/又は発現のために宿主細胞の中で形質転換する前に仲介ベクター内でクローンされる。これらの仲介ベクターは、通常原核細胞ベクター(例えば、プラスミド、又はシャトルベクター)である。しかしながら、この工程は幾つかの好ましい態様では省略することができるので、本発明は仲介ベクターを使用する方法に限定されない。
【0070】
一のアプローチにおいて、プロテアーゼをこの細胞の中で発現させるために、プロテアーゼインヒビターをコードする核酸配列に作動可能に結合する、プロモーター、生物学的に活性なプロモーターフラグメント、又は宿主細胞の中で機能する1以上の(例えば、連続する)エンハンサーを有する発現ベクターによりバクテリア培地を形質転換する。幾つかの好ましい態様において、このDNA配列はプロテアーゼインヒビター又はそれらの変異体をコードする。他の好ましい態様において、プロモーターは調節可能なものである。
【0071】
核酸構築体/発現ベクター
プロテアーゼインヒビター(すなわち、「PI-コード核酸配列」)をコードしている天然又は合成のポリヌクレオチドフラグメントは、異種の核酸構築体又はベクターに取り込まれ、バクテリア細胞の中に導入され、複製される。本明細書で開示するベクター及び方法は、プロテアーゼインヒビター及びそれらの変異体を発現させる各種宿主細胞に用いるのに適している。導入された細胞内で複製され、利用可能である限り、どのようなベクターでも用いることができる。本技術分野において数多くの適したベクター及びプロモーターが知られており、市販されている。適したクローニング及び発現ベクターは、本技術分野においてよく知られている文献に記載されている(上記のSambrooket al.,及びAusbel et al.,を参照のこと。これらの文献を参照により本明細書に援用する)。適切なDNA配列は適切な方法を用いてプラスミド又はベクター(これらを集合的に「ベクター等」と言う)内に挿入される。一般的にDNA配列は当該技術分野分野において既知の標準的方法を用いて適した制限エンドヌクレアーゼ内に挿入される。
【0072】
適切なベクターは、通常、選択マーカーコード核酸配列、挿入部位、及び転写終了配列等の適切な制御エレメントを備えている。幾つかの態様において、ベクターは例えば、コード配列に作動可能に結合している宿主細胞の中の(及び/又は修飾された可溶性タンパク質コード配列が通常発現しないベクター又は宿主細胞の環境内で)コード配列の発現に効果的な制御エレメント(すなわち、プロモーター及びターミネーターエレメント又は5’及び/又は3’非翻訳領域)を含む調節配列を含む。数多くの適切なベクター及びプロモーターは本発明の技術分野において知られており、それらの多くは市販されている。
【0073】
典型的なプロモーターは、構造プロモーター及び誘導プロモーターの両方を含む。そのようなプロモーターは、当該技術分野で知られている。当業者は、天然のプロモーターが有する機能を変えることなく1以上のヌクレオチドの交換、置換、付加又は排除により修飾することができることを知っている。本発明はプロモーターに対するそのような変更を包含するけれども、このことに限定されるものではない。遺伝子構築体に用いるプロモーターの選択は当業者の知識の範囲内である。
【0074】
適正な選択マーカーを選択することは宿主細胞に依存している。異なるバクテリア宿主に対して適切な選択マーカーを選択することは当業者の知識の範囲内である。典型的な選択マーカー遺伝子は(a)抗生物質又は他の毒性(例えば、アンピリシン、メトキサレート、テトラサイクリン、ネオマイシン マイコフェノール酸、ピューロマイシン、ゼノマイシン又はヒドロマイシン)に対する耐性を付与するタンパク質、又は(b)宿主細胞における栄養素要求性変異又は自然発生栄養欠失を補うタンパク質をコードする。
【0075】
幾つかの態様において、選択されたPIコード配列は良く知られた組み換え技術に従って適切なベクター内に挿入され、PI発現が可能な細胞系統を用いた形質転換に用いられる。遺伝子コード固有の退縮により、実質的に同じ又は機能的に等しいアミノ酸配列をコードする他の核酸配列はクローニングに用いられ、及び上で詳述したように特定のプロテアーゼインヒビターを発現する。それゆえコード領域において、そのような置換をすることは本発明でカバーされる配列変異体に含まれる。これらの配列変異体のいずれか及びすべては、親PIコード核酸配列に対するのと同じ方法で本発明に用いることができる。当業者は異なるPIは異なった核酸配列によりコードされることを認識している。
【0076】
幾つかの態様において、プロテアーゼインヒビター核酸配列の所望の形態、相同体、変異体又はそれらのフラグメントが一旦形成されると、多くの方法により修飾される。非コードフランキング領域を含む配列の場合、このフランキング領域は、切除、変異等を受ける。従って、塩基転位、塩基転移、欠失及び挿入が天然配列上で起こる。
【0077】
幾つかの好ましい態様において、異種核酸構築体は少なくとも1のプロテアーゼインヒビター、又はそれらの変異体、フラグメント、又はスプライス変異体のコード配列を、(i)単独で、(ii)PIコード配列が主なコード配列である場には、融合タンパク質又はシグナルペプチドコード配列のような追加的なコード配列と組み合わせて、(iii)適切な宿主細胞におけるコード配列の発現に有効な、プロモーター及び終了エレメント又は5’及び/又は3’非翻訳領域等の調節エレメントのような非コード配列と組み合わせて、及び/又は(iv)PIコード配列が異種の遺伝子となるようなベクター又は宿主細胞環境下に含んでいる。
【0078】
幾つかの態様において、適切なプロモーター及び制御配列と一緒に適切な核酸コード配列を含む異種の核酸は、少なくとも1のプロテアーゼインヒビター又はそれらの変異体を発現させるためにバクテリア宿主細胞内へ導入される。
【0079】
本発明の幾つかの態様において、異種の核酸構築体はインビトロ(in vitro)において細胞内へPIコード核酸配列を輸送するために用いられる。幾つかの好ましい態様において、この宿主細胞は本発明の核酸配列を安定に組み込む。従って、安定な形質転換体を効果的に生成するための任意の適切な方法を本発明に用いる。
【0080】
本発明の追加的な態様において、第一及び第二発現カセットは単一ベクター上に存在する。一方、他の態様においては、これらのカセットは分離したベクター上に存在する。
【0081】
幾つかの好ましい態様において、プロモーター配列に加えて、効果的な転写終了を提供するために、発現カセットは構造遺伝子組成物の下流に転写終了領域を含む。幾つかの態様において、転写終了領域は、プロモーター配列と同じ遺伝子から得られるが、他の態様においては、他の遺伝子から得られる。適切な転写終了シグナルの選択は当該技術分野においてよく知られている。
【0082】
更に、任意の適切な発現ベクターを本発明に用いることができる。すなわち、原核又は真核生物の細胞中の発現に用いる各種簡便なベクターが適しており、本発明に用いることができる。標準的なバクテリア発現ベクターは、pBR322ベースプラスミド、pSKF、pET23D等のプラスミド及びMBP、GST及びLacZ等の融合発現システムだけでなくバクテリオファージλ及びM13も含む。更なる態様において、単離を簡便にする目的でエピトープタグが組換えタンパク質に添加される(例えばc-myc)。
【0083】
発現ベクターに通常含まれる追加的なエレメントは、レプリコン、組換えプラスミドを有するバクテリアを選択するのための抗生物質耐性をコードする遺伝子、及び異種配列を挿入するためのプラスミドの非必須領域における特徴的な制限部位である。特定の抗生物質耐性遺伝子を選択することは決定的な要素ではない。任意の多くの知られた抗生物質遺伝子が適している。
【0084】
宿主細胞へのプロテアーゼインヒビターコード核酸配列の導入
幾つかの好ましい態様において、本発明の方法は、安定に組み込まれた所望の配列(すなわち、PIコード核酸)を含む宿主細胞を提供する。しかしながら、代替的な態様において、本発明の方法は、自己複製染色体外形質転換ベクターの維持も提供する。
【0085】
本発明は、外部から提供されたPIコード核酸配列を包含させるために遺伝的に修飾された細胞及び細胞組成物を提供する。幾つかの態様において、親宿主細胞は、発現ベクターで遺伝的に修飾されている。幾つかの態様において、このベクターはプラスミドであり、他の態様においてはこのベクターはウイス粒子、ファージ、ネイキッドDNA等である。従って、各種ベクターを本発明に用いることができるので、ベクターの形態を特定のベクターに限定することは意図しない。
【0086】
インビトロ(in vitro)において発現ベクターの輸送のために各種方法が用いられる。異種核酸配列の発現のために細胞内へ核酸を導入する方法は、当業者に知られており、エレクトロポレーション、無傷細胞によるプロトプラスト融合、形質感染、DNAコート-マイクロプロジェクタイル法を用いた高速照射、修飾ウイスル(例えばファージ)核酸を用いた感染、化学媒介形質転換、コンピテンス等を含むがこれらに限定されない。更に、幾つかの態様において、PIコード核酸を含む異種核酸構築体は、インビトロ(in
vivo)で転写され、得られたRNAは宿主細胞内へ本技術分野でよく知られた方法を用いて導入される。
【0087】
プロテアーゼインヒビターのコード配列を含む異種核酸構築体の導入に続いて、遺伝的に修飾された細胞は、プロモーターの活性化、形質転換体の選択及び/又はPIコード核酸配列の発現の増幅のために適切に調整された栄養培地中で培養される。温度、pH等の培養条件は発現のために選択された宿主細胞に対して用いている条件と同じであり、当業者に明らかである。
【0088】
このようにして異種核酸構築体が導入された細胞の子孫は、異種核酸構築体の中に存在したPIコード核酸配列を含むと考えられる。
【0089】
バクテリア宿主細胞及び発現
適切な宿主細胞は任意の好適なバクテリア種を含む。幾つかの態様において、バクテリア宿主細胞は発現宿主細胞及び第一及び第二核酸の供給源として機能する。本発明の方法及び宿主細胞を用いて、驚くべきレベルの発現を得ることができる。本明細書で用いるシステムを用いて達成されるプロテアーゼインヒビターの発現及び分泌レベルは0.5
g/l以上である。
【0090】
発現ベクターが宿主細胞に導入された後で、形質転換された宿主細胞は、所望のタンパク質をコードする遺伝子の発現に好ましい環境下で培養される。形質転換細胞の大容量バッチを上で述べたように培養することができる。最終的に、生産物は当該技術分野において既知の方法により回収される。
【0091】
チオールジスルフィド酸化還元酵素又はシャペロンのような補足タンパク質は分泌されたタンパク質を活性構造へと折りたたむことを助けることから、本発明の幾つかの態様にこれらの補足的なタンパク質を用いることができる。チオールジスルフィド酸化還元酵素及びタンパク質ジスルフィドイソメラーゼは、タンパク質の中の正確なジスルフィド結合の形成を促進する。バチルススブチルス(B.subtilis)の中のbdbDCオペロンの過剰発現は、ジスルフィド結合を有するタンパク質の生成に有用であることが証明されている(Meima etal.,J. Biol. Chem., 277: 6994-7001, [2002] 参照のこと)。シャペロンは、折りたたまれていない状態中の、曝されている疎水性の領域に結合することによって、分泌タンパク質が折りたたまれるのを助け、好ましくない相互作用を防ぎ、プロピル-ペプチドイル シス-トランスイソメメラーゼはプロリン残基に隣接するペプチド鎖の適正な立体構造の形成を補助する。
【0092】
本発明の幾つかの態様において、宿主細胞は少なくとも1のチオールジスルフィド酸化還元酵素又はシャペロンをコードする発現ベクターで形質転換される。本発明は特定のチオールジスルフィド酸化還元酵素又はシャペロンに限定することを意図していないので、本発明の技術分野で知られている任意の適切なチオールジスルフィド酸化還元酵素又はシャペロンが本発明に用いられる。
【0093】
本発明の幾つかの態様において、適切に折りたたまれた分泌タンパク質の分画は、成長培地にジスルフィド結合を酸化及び/又は還元する試薬、及び/又は通常の酸化還元電位を変更する試薬、及び/又は溶媒の性質を変更し、タンパク質の構造及び凝集に影響を与える薬品を添加することにより増やすことができる。特に好ましい態様において、2-メルカプトエタノールのようなジスルフィド結合を還元する試薬は、適切に折りたたまれたタンパク質の分画を高めるために好ましい。しかしながら、他の態様及び使用する培地によっては、他のジスルフィド還元及び酸化試薬(例えば、DTT、TCEP、酸化及び還元型グルタチオン,システイン、シスチン、 システアミン、 チオグリコレート、S2O32-、S2O42-、S2O52-、SO32-、S2O72-、Cu+等)を単独で、又は組み合わせて本発明に用いることがきでる。単独で、又は、βME等のジスルフィド酸化還元試薬と組み合わせて成長培地に添加する溶媒の性質を変更する他のアジュバンド(例えば、尿素、DMSO、TWEEN(登録商標)-80等)も、適切に折りたたまれた分泌ペプチドの分画を増やし、本発明の各種態様に用いることができる。幾つかの好ましい態様において、βMEは0.5mM乃至4 mMの濃度の幅で、他の態様においては、0.1 mMから10 mMの濃度で用いられる。すなわち、当業者は、用いるべきチオール酸化還元試薬及び他のアジュバンドの濃度だけでなく、添加するチオール酸化還元試薬及び/又は他のアジュバンドの影響を最適化するための最良の成長培地及び最良の成長条件をどのように選択するかを知っている。本発明は特定のジスルフィド還元/酸化試薬又はアジュバンドに限定することを意図していないので、本発明の技術分野において知られている任意の適切な試薬を本発明に用いることができる。
【0094】
発酵指標
本発明は培養されたバクテリア種に対する発酵方法に関係する。バクテリア種による異種タンパク質の生産に対する発酵方法は当該技術分野でよく知られている。培養は、水溶性ミネラル塩、培地、有機成長因子、炭素源及び栄養源となる物質、酸素分子(好気性及び嫌気性バクテリアに対する)を含み、用いられる1以上の特定の微生物種の開始接種をした培地中で行われる。
【0095】
適切な微生物の成長を確実にし、この微生物の転化工程における細胞による炭素及びエネルギー源の同化を最大限にし、発酵培地の中で最大細胞濃度で、最大細胞生産を達成するために、炭素及び栄養源、同化窒素並びに微生物の接種に加えて、適切な割合のミネラルを適切な量供給することが必要である。
【0096】
当該技術分野で知られているように、各種培養培地を本発明に用いることができる。しかしながら、標準的なバクテリア培養培地も本発明に用いられる。幾つかの好ましい態様において、培地は、窒素に加えて、適切な量のリン酸、マグネシウム、カルシウム、カリウム、硫黄、及びナトリウムを適切な可溶性同化イオン及び結合状態で含み、銅、マンガン、モリブデン、亜鉛、鉄、ホウ素、及びヨウ素等の微量元素も、適切な可溶性同化形態で培地中に含まれる。
【0097】
幾つかの態様において、発酵反応は、微生物を勢い良く成長させるために有効な適切な酸素分圧力において、発酵管の内容物を維持するために空気、酸素高含量空気、又は実質的に純粋な酸素分子等の酸素分子含有ガスにより必要な酸素分子が供給される好気プロセスを含む。実際には、酸素化炭化水素基質を用いることにより、微生物の成長に必要な酸素を減らすことができる。しかしながら、好気性微生物及びより程度は低いが嫌気性微生物の生長為に酸素分子の供給は必要である。
【0098】
通気割合は非常に広範囲にわたり変動させることができが、通常の通気速度は、一分間に発酵槽中の液体当たりに供給される酸素含有気体の容量は、(採用圧力及び25℃において)約0.5乃至10容量、好ましくは約0.5乃至7容量である。この量は反応器に供給される通常の酸素含量の空気に基づいている、そして、精製酸素に換算すると、前記割合は、(採用された圧力及び25℃の温度において)1分間に発酵槽の中の液体当たりに供給される酸素は約0.1乃至1.7容量又は好ましくは0.1乃至1.3容量である。
【0099】
微生物転化プロセスに対して用いられる圧力は幅広く変動する。通常の圧力は、装置及び操作コストに対して達成される溶存酸素の理由から、0乃至50 psig、好ましくは0乃至30psig、より好ましくは少なくとも大気圧よりわずかに高い圧力である。大気圧より高い圧力は水性発酵槽内の溶存酸素を増やす傾向があり、それによって、細胞の成長速度を高めることから大気圧より高い圧力は有利である。同時に、高い大気圧は装置及び操作のコストを高めるという事実とバランスする必要がある。
【0100】
発酵温度は若干変動させることができるが、大部分のバクテリア種にとって、本発明の技術分野において知られているように、バクテリア種にもよるけれども、通常20℃乃至40 ℃、一般的に好まれている温度は、28 ℃乃至37 ℃である。
【0101】
微生物は、同化窒素源も必要とする。同化窒素の源は、微生物による代謝の利用に適した形態で窒素を放出することができる任意の窒素含有化合物である。タンパク質の加水分解物等の各種有機窒素化合物を用いることができるけれども、通常、アンモニア、水酸化アンモニウム、尿素、リン酸アンモニウム、硫酸アンモニウム、ピロリン酸アンモニウム、塩化アンモニウム等の各種アンモニウム塩、又は、安価な他の各種アンモニウム化合物を用いることができる。アンモニアガスは大規模操作に対して便利であり、、水性発酵槽(発酵培地)中への通気により適切な量を用いることができる。同時に、そのようなアンモニアはpH調節にも用いることができる。
【0102】
水溶性微生物発酵物(発酵混合物)の中のこのpHの幅は、約2.0乃至8.0の典型的な幅である。しかしながら、特定の微生物に対して適したpH幅は、微生物だけでなく、用いる培地にも依存する。従って、当該技術分野で知られているように、培地組成によりpHを幾分変更することも可能である。
【0103】
発酵槽内での発酵混合物の平均滞留時間は、本発明の技術分野で知られているように、発酵温度及び用いられる培地に部分的に依存して変動する。
【0104】
幾つかの態様において、発酵は、炭素含有基質が制限因子として制御でき、それゆえ、良い炭素含有基質の転化が細胞に提供され、非常に大量の転化されない基質による細胞の汚染を避けるような方法が好ましい。残留微量元素は容易に取り除くことができるので、汚染は水溶性基質では問題とならない。しかしながら、非水溶性基質の場合には問題となるので、適切な洗浄工程等の追加的な生産工程が必要とされる。この制限された基質レベルを達成するために必要な時間は、重要ではなく、実施される特定の微生物及び発酵工程により変動する。しかしながら、発酵培地の中の炭素源濃度及び望ましい炭素源レベルが達成されたか否かの決定方法は本発明の技術分野において知られている。
【0105】
幾つかの態様において、発酵はバッチ又は連続操作で実施されるけれども、フェドバッチ操作は制御が容易で、生成物の質が均一であり、全装置の使用が経済的であることから好ましい。
【0106】
必要ならば、炭素及び栄養源物質の一部又は全部及び/又はアンモニア等の同化窒素源は発酵槽内に水溶性ミネラル培地を供給する前に添加することができる。すなわち、反応器に導入される原材料流れの各々は、予め決められた速度で、又は発酵槽から出てくるオフガス中の炭素源及びエネルギー基質、pH、溶存酸素、酸素又は二酸化炭素、光透過度により測定される細胞密度等の濃度モニタリングで決定される必要性に応じて制御される。各種物質の供給速度は、チャージされた基質に対してできる限り高い微生物細胞の産生を達成し、効果的な炭素及びエネルギー源の利用と矛盾しないようにできるだけ早い細胞成長を得るために変動させることができる、しかし、より重要なのは、容量単位当たり最も多い生成物を得ることである。
【0107】
各バッチにおいて、好ましくはフェドバッチ操作において、全部の装置、反応器、又は発酵手段、発酵管又はコンテナ、配管、付属の循環又は冷却装置等は、初めに通常約121℃で約15分間蒸気に曝すことにより滅菌される。滅菌された反応器では、酸素を含む必要な全ての栄養素及び炭素含有基質の存在下で選択された微生物を培地に接種する。用いられる発酵槽のタイプは、重要ではないが、幾つかの態様において、15Lのバイオラフィッテ(Biolafitte)(Saint-Germain-en-Laye、France)が好ましい。
【0108】
タンパク質の分離
好ましい態様において、一旦所望のタンパク質が発現すると、分泌タンパク質が分離される。本発明はその融合類似体から所望のタンパク質を分離する方法を提供する。本明細書で述べる方法はプロテアーゼインヒビター及びその変異体を融合類似体から分離することに用いることができる。
【0109】
発酵ブロスから所望のタンパク質を収集及び精製することは、本発明の技術分野で知られている方法を用いて行うことができる。発酵ブロスは通常、所望のタンパク様物質のほかに、細胞を含む細胞の破片、各種懸濁された固形物、及び他の生物汚染物質を含む。これらは、本発明の分野で既知の方法により発酵ブロスから除去することが望ましい。そのような除去に適した工程は、細胞を含まないろ液を生産するための従来の固体-液体分離技術である(例えば、遠心分離、ろ過、透析、精密ろ過、ロータリー吸引ろ過、又は他の既知の方法)。幾つかの態様において、発酵ブロス又は他の無細胞ろ液を精製及び/又は結晶化工程に用いる前に、精密ろ過、エバポレーション、及び/又は透析を用いて、更に濃縮することが好ましい。
【0110】
上清中のタンパク様成分の沈殿又はろ過は、塩(例えば、硫酸アンモニウム)又は低いpH(通常3未満)を用いて行い、次いで、各種クロマトグラフ手段(例えば、イオン交換クロマトグラフィー、親和クロマトグラフィー、疎水相互作用クロマトグラフィー、疎水電化誘導クロマトグラフィー(hydrophobiccharge induction chromatography)等)又はこれと同様の方法による精製が続く。本発明は特定の分離方法に限定されないので、任意の分離方法を本発明に用いることができる。
【0111】
特に好ましい態様において、発現された所望のポリペプチドがバクテリア細胞から分泌されると、このポリペプチドは成長培地から精製される。好ましい態様において、発現宿主細胞はこのポリペプチドの精製(例えば、遠心分離)の前に、培地から除去される。
【0112】
発現された所望の組換えペプチドが宿主細胞から分泌されない場合には、宿主細胞を崩壊させ、及び水相に放出されたポリペプチドを「抽出」することが精製の第一工程となることが好ましい。この場合、発現宿主細胞は、細胞を崩壊させる前に培地から集められることが好ましい(例えば、遠心分離)。細胞破壊は、リゾチーム、又はベータグルカナーゼ消化により、又は、細胞を高圧で処理すること等の本発明の技術分野で知られている方法を用いて行われる(Scobes,Protein Purification. Second edition, Springer-Verlag参照のこと)。
【0113】
幾つかの態様において、6のヒスチジン残基(すなわちヒスタグ)をC末端に付加することは所望のタンパク質及びその融合アナログの精製における補助として用いられる。精製の補助としてのヒスタグの使用は、当該技術分野においてよく知られている(Hengen,TIBS
20: 285-286 [1995] 参照のこと)。6のヒスタグを付加されたタンパク質は、当該技術分野で知られている固定金属イオン親和クロマトグラフィー(IMAC)を用いて容易に精製することができる。
【0114】
精製
特定の使用目的のためには、本発明を用いて製造されたプロテアーゼインヒビターは高度に精製されていることが重要となる(例えば、99 %以上の精製度)。精製タンパク質が治療に用いられるときには高度に精製されていることは特に重要であるが、他の使用目的においても必要である。本発明の明細書で述べる方法は、実質的に精製された所望のタンパク質を製造する方法である。本明細書で述べる所望のタンパク質は、医薬品及びパーソナルケア製品に有用である。しかしながら、各種精製レベルのタンパク質が本発明の方法を用いて製造され、本発明の方法を用いて精製したタンパク質が特定の精製レベルであることは意図しない。
【0115】
精製の間のBBIの活性化
本発明の幾つかの態様において、精製工程における育成の後、ジスルフィド結合を酸化及び/又は還元する組成物、及び/又は通常の酸化還元電位を変更する組成物、及び/又は溶媒の性質を変更しタンパク質の構造及び凝集に影響を与える組成物を添加することによりタンパク質の活性が増強される。幾つかの特定の好ましい態様において、2-メルカプトエタノールのようなジスルフィド結合を還元する試薬の添加は、タンパク質の活性を高めるのに使用される。しかしながら、当該技術分野において知られているように、精製度及び緩衝液の組成に依存して、他のジスルフィド還元又は酸化試薬(例えば、DTT、
TCEP、酸化及び還元グルタチオン、システイン、 シスチン、 システアミン、 チオグリコレート、S2O32-、S2O42-、S2O52-、SO32-、S2O72-、Cu+、タンパク質ジスルフィド イソメラーゼ、タンパク質チオールジスルフィド オキシドレダクターゼ等)を単独で、又は組み合わせて本発明に用いることができる。単独で、又はβME等のジスルフィド酸化還元試薬と組み合わせて溶媒の性質を変更する他のアジュバンド(例えば、エタノールアミン、DMSO、TWEEN(登録商標)-80、アルギニン、尿素、等)も、タンパク質の活性を高めるために本発明に用いることができる。特定の好ましい態様において、部分的に精製されたタンパク質が緩衝液の中に溶解され(特に好ましい態様において、塩基性pHにおけるTWEEN(登録商標)-80を用いた両性イオン緩衝液)bME及びジスルフィド酸化試薬(代替的な好ましい態様は、酸化グルタチオン又は硫酸ナトリウム)を用いて活性化される。
【0116】
更に、必要ならば、タンパク質の最適な活性を決定するための条件が選択される。例えば、各種βME濃度(0.1乃至10 mM)、酸化試薬濃度(βMEの濃度の0乃至1/20乃至20倍)、pH(7.5乃至9.5)、温度(15乃至40℃)、希釈倍率(1乃至20倍)、インキュベーション時間(12乃至72時間)、通気(酸素含有ガスの下で激しく攪拌するための挿入ガス下におけるインキュベーション)、緩衝液タイプ(トリス、CHES、CAPS、トリシン(Tricine)、TAPS、他の両性イオン緩衝液等)、緩衝液濃度(0.1乃至1M)、及び溶媒の性質を変え、それゆえタンパク質の立体構造に影響を与え、凝集をさせることが知られている各種アジュバンド(例えば、エタノールアミン、DMSO、TWEEN(登録商標)-80、アルギニン、尿素、等)の添加、が用いられる発現システムの最適な条件を決定するために試験される。本発明は特定の還元/酸化試薬、希釈倍率、温度、pH、緩衝液タイプ、又は組成物、並びにアジュバンドに限定することを意図していないので、当該技術分野で既知の好適な試薬を本発明に用いることができる。
【0117】
実施例
本発明は、以下の実施例においてより詳細に説明されるが、これら実施例に本発明を限定することは意図していない。添付の図は、明細書及び本発明の不可欠な部分である。引用された全ての文献は本発明の開示のために参照により本明細書に援用される。以下の実施例は、本発明を説明するためのものであり、本発明を限定するためのものではない。
【0118】
以下の実施例において、以下の略字を用いる。
【0119】
PI(プロテアイナーゼインヒビター)、BBI(Bowman-Brikインヒビター)、STI (ダイズトリプシンインヒビター)、 VEGF 及びVegF (血管内皮成長因子)、ppm(100万分の1)、 M(モルの)、mM(ミリモルの)、μM(マイクロモルの)、nM(ナノモルの)、mol(モル)、 mmol(ミリモル)、μmol(マイクロモル)、nmol(ナノモル)、gm(グラム)、mg(ミリグラム)、μg(マイクログラム)、pg(ピコグラム)、L(リットル)、ml 及び mL(ミリリットル)、 μl及びμL (マイクロリットル)、cm(センチメートル)、mm(ミリメートル)、μm(マイクロメートル)、nm(ナノメートル)、 U(単位)、V(ボルト)、MW(分子量)、sec(秒)、min(s)(分)、h(s)及びhr(s)(時間)、℃(摂氏)、QS(十分量)、ND(検出不能)、NA(適用不能)、rpm(1分間あたりの回転数)、H2O(水)、dH2O(脱イオン水)、HCl(塩酸)、aa(アミノ酸)、bp(ベースペア)、
kb(キロベースペア)、kD (キロダルトン)、cDNA(転写又は相補DNA)、DNA(デオキシリボ核酸)、ssDNA(一本鎖 DNA)、dsDNA(二本鎖DNA)、dNTP(デオキシリボヌクレオチド三リン酸)、RNA(リボ核酸)、MgCl2(塩化マグネシウム)、NaCl(塩化ナトリウム)、w/v(容量対重量)、v/v(重量対重量)、g(比重)、OD(光学密度)、ダルベッコリン酸緩衝溶液(DPBS)、SOC(2% バクト-トリプトン、0.5 % バクトイースト抽出物、10 mM NaCl、2.5 mM KCl),テリフィックブロス(TerrificBroth)(TB;12 g/l バクト-トリプトン, 24 g/lグリセロール、2.31 g/l KH2PO4、及び12.54g/l K2HPO4) 、OD280(280 nmにおける光学密度) 、OD600(600nmにおける光学密度) 、A405 (405 nmにおける吸光度)、Vmax(酵素触媒反応における最大速度)、PAGE(ポリアクリルアミドゲル電気泳動)、PBS(リン酸緩衝生理食塩水 [150 mM NaCl、10 mM リン酸緩衝液、pH 7.2]) 、PBST(PBS+0.25% TWEEN(登録商標)20)、PEG(ポリエチレングリコール)、PCR(ポリメラーゼ連鎖反応)、RT-PCR(逆転写PCR)、SDS(ドデシル硫酸ナトリウム)、Tris(トリス(ヒドロキシメチル)アミノメタン)、HEPES(N-[2-ヒドロキシエチル] ピペラジン-N-[2-エタンスルホン酸] )、HBS(HEPES 緩衝生理食塩水)、SDS(ドデシル硫酸ナトリウム)、bME,BME 及びβME (ベータメルカプトエタノール又は2-メルカプトエタノール)、Tris-HCl(トリス [ヒドロキシメチル]アミノメタン-クロライド)、Tricine(トリシン)(N-[トリス-(ヒドロキシメチル)-メチル]グリシン)、CHES(2-(N-シクロ-ヘキシルアミノ)エタン-スルホン酸)、TAPS(3-{[トリス-(ヒドロキシメチル)-メチル]-アミノ}-プロパンスルホン酸)、CAPS(3-
(シクロ-ヘキシルアミノ)-プロパンスルホン酸)、DMSO(ジメチルスルホキシド)、DTT(1,4-ジチオ-DL-トレイトール)、Glut 及び GSH (還元グルタチオン)、GSSG(酸化グルタチオン)、TCEP(トリス[2-カルボキシメチル]ホスフィン)、Ci(キューリ)、mCi(ミリキューリ)、μCi(マイクロキューリ)、TLC(薄層クロマトグラフィー)、Ts(トシル)、Bn(ベンジル)、Ph(フェニル)、Ms(メシル)、Et(エチル)、Me(メチル)、Taq(サーマスアクアチクス(Thermusaquaticus)DNA ポリメラーゼ)、Klenow(DNA ポリメラーゼ I ラージフラグメント(Klenow(クレノウ))フラグメント )、rpm(一分当たりの回転数)、EGTA(エチレングリコール-ビス(β-アミノメチル エーテル)N, N, N', N'-四酢酸)、EDTA(エチレンジアミン四酢酸)、bla(β-ラクタマーゼ 又は アンピシリン耐性遺伝子)、GEHealthcare (GEヘルスケア、チャルフォントストリート、ジャイルズ、UK)、DNA2.0(DNA2.0、メロンパーク、カリフォルニア)、OXOID(オキシドベージングストーク、ハンプシャー、UK)、Megazyme(メガザイム インターナショナル アイルランド社、ブレービジネスパーク、ブレー、ウィックロー、アイルランド)、Coming(コーニングライフサイエンス、コーニング、ニューヨーク)、NEN(NENライフサイエンスプロダクツ、ボストン、マサチューセッツ)、Pharma AS(ファルマAS, オスロ、ノルウェイ)、Dynal(ダイナル、オスロ、ノルウェイ)、Bio-Synthesis(バイオ-シンセシス、ルイスビル、テキサス)、ATCC(アメリカンタイプカルチャーコレクション、ロックイビル、メリーランド)、Gibco/BRL(ギブコ/BRL、グランドアイランド、ニューヨーク)、Sigma(シグマケミカル社、セントルイス、ミズリー)、Pharmacia(ファルマシア、ピスカタウェイ、ニュージャージー)、NCBI(全米バイオテクノロジー情報センター)、Applied Biosystems(アプライドバイオシステム、フォスターシティー、カリフォルニア)、Clontech(クローンテックラボラトリーズ、パロアルト、カリフォルニア)、OperonTechnologies(オペロンテクノロジイーズ社、アラメダ、カリフォルニア)、MWG Biotech(エムダブリュジーバイオテック、ハイポイント、ノースカロライナ)、Oligos
Etc(オリゴイーティーシ社、ウィルスンビル、オーランド)、Bachem (ベイケムバイオサイエンス社、キングオブプルシア、ペンシルバニア)、Difco(ディフコラボラトリーズ、デトロイト、ミシガン)、Mediatech(メディアテック、ハードン、バージニア)、Santa Cruz(サンタクルーズバイオテクノロジー、カリフォルニア)、BioVeris(バイオベリス社、ゲーサーズバーグ、メリーランド)、Oxoid(オキシド社、オグデンスブルグ、ニューヨーク)、Worthington(オージントンバイオケミカル社、フリーホールド、ニュージャージー) 、GIBCO BRL 又は Gibco BRL (ライフテクノロジー社、ゲーサーズバーグ、メリーランド)、Millipore(ミリポア、ビルリカ、マサチューセッツ)、Bio-Rad(バイオラド、ハーリーキューズ、カリフォルニア)、Invitrogen(インビトロジェン社、サンディエゴ、カリフォルニア)、NEB(ニューイングランドバイオラドズ、ベバリー、マサチューセッツ)、Sigma(シグマケミカル社、セントルイス、ミズリー)、Pierce(ピアスバイオテクノロジー、ロックフォード、イリノイ)、Takara(タカラ・バイオ社、大津、日本)、Roche(ホフマン-ラ・ロッシュ、バーゼル、スイス)、EM Science(エムイーサイエン、ギブスタウン、ニュージャージー)、Qiagen(キアゲン社、バレンシア、カリフォルニア)、Biodesign(バイオデザイン社、ソーコ、メイン)、Aptagen(アプタゲン社、ハーンダン、バージニア)、MolecularDevices(モレキュラーデバイス社、サニーベール、カリフォルニア)、R & D Systems(R & D システムズ、ミネアポリ、ミネソタ)、Stratagene(ストラタジェンクローニングシステムズ、ラホーヤ、カリフォルニア)、Marsh(マーシュバイオサイエンス、ロチェスター、ニューヨーク)、Bio-Tek(バイオテック社、ウィヌースキー、 バーモント)、Biacore(ビアコア社、ピスカタウェイ、ニュージャージー)、PeproTech(ペプロテック、ロッキーヒル、ニュージャージー)、SynPep(シンペップ、ダブリン、カリフォルニア)、及びMicrosoft(マイクロソフト社、レドモンド、ワシントン)。
【実施例1】
【0120】
バチルススブチルス(B. subtilis)におけるBCE103-BBI融合タンパク質の生産
この実施例では、バチルススブチルス(B. subtilis)中にBCE103-BBI融合タンパク質を生産するために行った試験を説明する。この試験に用いられるC末端にヘキサヒスチジンタグを有するプロ-BBIタンパク質をコードする合成遺伝子(Operon
Technologies)のDNA配列は以下である。
【表1】

上記合成遺伝子によりコードされるC末端にヘキサヒスチジンタグを有するプロ-BBIタンパク質配列は以下である。
【表2】
成熟型BBIをコードする合成遺伝子のDNA配列の一部は以下である。
【表3】
上の合成遺伝子によりコードされる成熟型BBIのタンパク質配列は以下である。
【表4】
pJ10ベクター中のBCE103セルラーゼ発現カセットへ融合させるBBI遺伝子の増幅に用いるPCRプライマーは以下である。
【表5】
pJM103組込みベクター内へクローンされるaprE-BCE103-BBI-ヒスタグ発現カセット(EocRI-HindIII)の配列を図1に示す。PJM103BBIヒス発現ベクターのプラスミドマップの概略を図2に示す。
【0121】
バチルススブチルス(B. subutilis)aprEプロモーター及びシグナル配列に融合されたアルカリセルラーゼ(BCE103)遺伝子(vanSoligen、米国特許6,063,611を参照のこと。この文献を参照により本明細書に援用する)は、pUCAPR103(Shaw et al., J.Mol.Biol., 320: 303-309 [2002])をEcoRI-BamHIフラグメントとして(すなわち、BCE103触媒ドメイン及び第一セルロース結合ドメインリンカーのみのコード配列を運搬するフラグメント)pJM103内でクローニングした(Perego,"Integrational vectors for genetic manipulation in Bacillussubtilis"In, Bacillus subtilis and Other Gram-positive Bacteria:
Biochemistry, Physiology, and Molecular Genetics, Sonenshein, Hoch, and Losick
(eds), American Society for Microbiology, Washington D.C., pp. 615-624 [1993])。C末端にヒスチジンタグ(His-tag)を有するダイズボウマン-バーク(Bowman-Birk)プロテアーゼインヒビター(BBI)(Swiss-ProtAccession #P01055;Odani and Ikenaka, J. Biochem., 71: 839-848 [1972]参照のこと)は、オペロンテクノロジー(OperonTechnologis)(上記DNA配列を参照のこと)により合成された。3’HindIII部位を用いてBBI遺伝子をpJM103ベクター内でクローンさせた後に、BCE103遺伝子を用いる正確なりーディングフレーム中に5’BamHI部位及び3’末端に導入された強力な転写終了因子を生成するプライマーを用いてBBI遺伝子をPCRにより増幅した(全プライマーはMWG Biotech、OligosEtc.又はOperon Technologiesにより合成された)。
【0122】
合成BBI遺伝子をテンプレートとして用いて、BBIfusion_FW (配列番号14) 及びBBIHISHindIII_RV (配列番号16)、又はBBIfusion_FW(配列番号14) 及びBBI-HindIII_RV (配列番号15)のプライマーをそれぞれ用いてC末端にHis-Tagを有する、又は有しないPCRフラグメントを生成した。他の方法で示さない限り、製造会社の説明書に従いハイフェデリティープラチナTaqポリメラーゼ(HighFidelity PlatinumTaq polymerase) (Invitrogen)を用いてPCRを30サイクル行った(55℃のアニーリング温度)。aprE-BCE発現カセットを運ぶBamHI-HindIIIフラグメントとして、PCRフラグメントをpJM103内でクローニングした。正確な遺伝子配列をDNA配列決定により確認した。
【0123】
従って、図1に示すように、BCE103セルラーゼ遺伝子によりコードされる第一CBD(セルロース結合ドメイン)リンカー配列のC末端コード領域に対するフレーム中にBBI遺伝子(His-Tagを有するもの又は有しないもの)の成熟コード領域のN末端を融合した。それゆえ、2のBCE103のCBD(Shaw et al.,上記)は、最終的な発現カセットの中のBBIにより置き換えられ、pJM103BBI又はpJM103BBIhisとなった(図2参照)。このaprEプロモーターはBCE103-BBI遺伝子融合体の発現を制御する(Ferrariet al., J. Bact., 170: 289-295 [1988]及びHenner et al., J. Bact., 170: 296-300[1988]参照のこと)。
【0124】
有用なバチルススブチルス(Bacillus subtilis)細胞、BG3934comK(degUHY32、oppA、△spoIIE3501、△aprE、△npfE、△epr、△ispA、△bpr、amyE : : xylRPxylAcomK-phleo)を発現プラスミド、pJM103BBI又はpJM103BBIhisで形質転換した。バクテリアは、キシロース誘発プロモーター(Halmet al., Mol. Microbiol., 21: 763-775 [1996])の存在下でcomK遺伝子により誘発させることにより有用にすることができる。形質転換体を、5μg/mlクロラムフェニコールを含むウリアブロスアガー(Luria Broth agar)(LA)プレート上で選択した。遺伝子増幅での発現を高めるために、コロニーが縞状になり、抗生物質を含む培地における成長率がクロラムフェニコールが欠失している培地における成長率と同じになるまで、25μg/mlのクロラムフェニコールを用いて、LAプレート上で育成した。BCE-BBI融合タンパク質は、TSB培地(OXOIDより市販されているトリプトンダイズブロス(TryptoneSoya Broth)、30 g/L)又はMOPSベース合成培地中であるMBD培地において、37 ℃の振とうフラスコの中で育成することにより生成された。NH4Cl2、FeSO4、及びCaCl2を培地から除き、3mMのK2HPO4を用い、60 mMの尿素、75 g/Lのグルコース及び1 %のソイトン(soytone)を供給した以外は、(Neidhardtet al., J. Bacteriol., 119: 736-747 [1974] )に記載されたようにMBD培地を調製した。微量栄養素は、100倍濃度の貯蔵溶液として調製された。この溶液は1リットル中400mgのFeSO4-7H2O、100 mgのMnSO4-H2O、100mgのZnSO4-7H2O、50 mgのCuCl2-2H2O、100mgのCoCl2-6H2O, 100 mgのNaMoO4-2H2O、100mgのNa2B4O7-1OH2O、10 mlの1M CaCl2、及び10mlの0.5 M クエン酸ナトリウムを含む。
【0125】
BCE103-BBI融合タンパク質は、標準的なタンパク質染色方法(例えば、ゲルコードブルー染色試薬(GelCode Blue StainReagent)Pierce)を用いて44 kDa付近のSDS-PAGEゲル上の主要なタンパク質バンドとして視認することができる(EMS緩衝液中10 %のSDS-PAGE、製造会社の説明書に従い操作。Invitrogen)。BCE103-BBI融合タンパク質は製造会社の説明書に記載の手順を用いたSDS-PAGEゲルの免役ブロットにより確認された(BM染色体ウエスタンブロットキット(BMChromogenic Western Blotting Kit)、Roche Applied Science抗ヒスタグ抗体又は抗BCE103セルラーゼポリクローナル抗体を検出に用いた)。
【0126】
BCE103活性は、LAセルラーゼ指標プレート(0.2 %のコンゴレッド又は0.5 %のアゾ-CMセルロースを用いて染色された1 %のカルボキシメチルセルロース、Megazyme)上でセルラーゼ消化を定性的に評価し、当該技術分野で知られている方法を用いて、合成基質、4-ニトロフェニルβ-D-セロビオース(Sigma)を用いた培養ブロス中のアッセイ緩衝液(AssayBuffre)(100 mMとリスpH 8.6、0.005 % Tween-80)を直接アッセイすることにより定量的に評価された(例えば vanTilbeurgh etal., Meth. Enzymol., 160: 45-59 [1988]参照のこと)。
【0127】
BBI活性を決定するのアッセイ緩衝液の中でトリプシン抑制アッセイを行った。特に、標準曲線は、2 μg/mlの標準BBI溶液を1:1の連続希釈(100 μLBBI+100 mlアッセイ緩衝液)することにより作製した。疎水性相互作用カラム(POROSHP2、フェニルカラム、Applied Biosystems)を用いて、20mM MES pH 6.0中1 mg/mlトリプシン-キモトリプシンインヒビター(シグマカタログ#T-9777)からこの標準BBIを精製した。このカラムを20mM MES pH 6.0で平衡化し、5 mgのインヒビターを負荷し、平衡化に用いた緩衝液で洗浄した、その後、BBIを水で溶出した。抑制アッセイで試験する未知のBBIサンプルを、2以上のデータポイントが標準曲線の範囲内に入るように必要に応じて希釈した(通常、1:10、1:100、1:200、1:1000、1:2000の希釈のサンプルの希釈率が試験され、必要に応じて希釈率を微調整した)。希釈した各標準BBI及びサンプル20
μLに、80 μLの5 ng/mlのウシ脾臓トリプシン(Worthington)(アッセイ緩衝液の中へ、1 mg/mlトリプシンストック溶液を希釈することにより作製された)。便宜上、この標準物質とサンプルを96ウェルマイクロタイタープレート中に整列させた。混合及び25℃15分間のインキュベーションにより反応を行った。インキュベーションの後、0.5
mg/mlのトリプシン基質100 μL(DMSO中100 mg/mlの溶液からアッセイ緩衝液の希釈により調整)、Suc-AAPR-pNA(スクシニル-アラニン-アラインヒビター-プロリン-アルギニン-パラニトロアニリド、Bachem)を添加し、混合し、OD(A405)を15分間モニターした。ペクトラマックス250(SpectraMax250)(Moleculaer Devices)を用いて少なくとも12秒毎に一回記録をとった。記録されたデータのポイントは、各反応に対するVmaxの決定に用いた。標準曲線は、BBI濃度に対するVmaxのプロットにより作製し、4の指標曲線に当てはめた。全てのデータのフィッティングは製造会社(MolecularDevices)が提供するソフトウェアを用いて行われた。未知サンプルのBBI濃度を標準曲線を用いて計算した。代替的に、BBI活性は同じ手順を用いてウシ脾臓キモトリプシン(Worthington)抑制を検出することにより測定された(キモトリプシンは、トリプシンと同じ濃度で用い、キモトリプシン活性は、100μLの0.4 mg/mlキモトリプシン基質、スクシニル-アラニン-アラニン-プロリン-パラニトロアニリド、(Bachem)の添加により測定された)。
【0128】
振とうフラスコサンプル(2.8 Lのフェルバッハ6 バッフルフラスコ中 500 mM MBD培地、37 ℃、225 rpm、60時間後に収穫する)の力価が、通常、0.4乃至0.9mg/ml BCE活性及び40乃至150 μg/ml BBIトリプシン抑制活性になるように培養が行われる。しかしながら、力価はバクテリア系統、培養培地及び育成条件(通気、温度、収穫時間等)の最適化により、更に改善することができる。
【0129】
野生型BBIに対するBCE103融合に加えて、BBIに対する融合タンパク質変異体及びBCE103及びBBIの間の各種リンカーを有する融合タンパク質は上で概説した方法を用いて、以下の実施例のように製造することができる。更に、融合タンパク質はBBIを精製工程において第一CBDを用いることの助けとなる第二CBDリンカー(BCE-cbdD-BBI;実施例4を参照のこと)に結合させたときにも生成される。
【実施例2】
【0130】
BCE103-BBI融合タンパク質としてのBBI反応部位ループのペプチド置換体の生成
この実施例では、BCE103-BBIのようにBBI反応部位へ置換するペプチドを生成するための試験を説明する。これらの試験の各工程に用いるプライマー及び他の配列を以下に記載する。BCE103融合部位(BamIIIの位置)から遺伝子の終わりまでの12BBIck81配列を図3に示す。CK37281ペプチド配列(ACYNLYGWTC(配列番号9))をトリプシン及びキモトリプシン抑制ループの両方に挿入した。
【0131】
QuikChange(登録商標)部位直接変異誘発(Stratagene)を用いたBBI遺伝子中にEcoRI部位を導入するために用いるプライマーは以下である。
【表6】
VegF結合ペプチドCK37281でトリプシン抑制部位を置換するためにアニールされ、BBI遺伝子(SacI-EcoRI)の中でクローニングされるDNAオリゴヌクレオチド配列は以下である。
【表7】
VegF結合ペプチドCK37281でキモトリプシン抑制部位を置換するためにアニールされ、BBI遺伝子(EcoRI−SaclI)の中でクローニングされるDNAオリゴヌクレオチド配列は以下である。
【表8】
合成BBI遺伝子のトリプシン(SacI及びEcoRI制限部位)又はキモトリプシン(EcoRI及びSaII制限部位)反応部位ループに中にペプチドを導入するためのカセットを作るために用いるオリゴヌクレオチドペアのDNA配列を以下に記載する。これらのペプチドコード配列はp2JM103発現ベクター内にSacI-SalIフラグメントとして導入された。
【表9】
【表10】
QuickChange(登録商標)II-XL部位直接変異誘発キット(Stratagene)を用いてトリプシン又はキモトリプシン反応部位へペプチド配列を導入するために用いるオリゴヌクレオチドプライマーペアのDNA配列を以下に記載する。製造会社の説明書及びこの実施例に記載の通りに反応を行った。68℃で6分間の伸長を行い、95 ℃で50秒間の変性を行い、55 ℃で50秒間のアニーリングを20サイクル行った。20サイクル終了後、68 ℃で20分間の最終伸長を行った。
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【0132】
p2JM103-lnk2-BBI発現ベクターのキモトリプシン反応部位ループへMM021ペプチドを導入するためのカセットを作るために用いるオリゴヌクレオチドペアのDNA配列を以下に記載する。このカセットはベクター内のSphI及びSalI制限部位へ結合された。
【表18】
【0133】
システインコンストレインドペプチド(cysteine constrained peptides)からできているライブラリーは所望の基質に結合するペプチドの選択のための良く知られた試薬(例えば、市販のPhD-C7Cファージディスプレイライブラリーキット、NEB)である。BBIはペプチド結合を選択するために用いられる各種方法で提示されるペプチドループに似た構造を有する2のシステインコンストレインド(cysteineconstrained )反応部位ループを有する。従って、一旦システインコンストレインド(cysteine constrained )結合ペプチドが選択されると、BBIは結合反応におけるペプチドを提供するためのスキャッフォールドとして使用するのに適している。
【0134】
DNAオリゴヌクレオチドカセットを用いて、CK37281 ペプチド配列(ACYNLYGWTC)(配列番号9)で、トリプシン、キモトリプシン、又は両方の活性部位ループを置換することによりVegF結合ペプチド CK37281 (例えば、2003年9月13日に提出された米国継続出願 60/520,403, 参照のこと。該出願を参照により本明細書に援用する。)
をBBIの中へ移植した。構築を促進させるために、上で示すBowBeco_F 及びBowBeco_Rプライマーを用いたQuikChange(登録商標)部位直接変異誘発を用いて製造会社の説明書に記載従い、トリプシン及びキモトリプシン反応部位ループの間のBBI遺伝子(OperonTechnologiesの合成品 ; 実施例1を参照のこと)のコード領域にEcoRIサイトを導入した (0.5 pmol の各プライマーを QuikChange(登録商標)
反応に用いた。97 ℃で3分間の第一変性工程の後、68 ℃で12分間、95 ℃で30秒間及び55 ℃で1分間のPCRサイクルを18回行い、その後に68 ℃で15分間の最終伸長を行った)。
【0135】
トリプシン抑制ペプチドループの置換のために、2のDNAオリゴヌクレオチド(1BBCK81+及び1BBCk81-)をアニールし、SacI及びEcoRI反応部位に結合した。同様に、キモトリプシン抑制ループの置換のために、EcoRI及びSalIサイトをオリゴヌクレオチド(2BBck81+及び2BBck81-)のアニーリングにより作られたDNAカセットの挿入に用いた。CK37281ペプチドは、トリプシンループを初めにCK37281ペプチドで置換した後に、オリゴヌクレオチド(2BBck81+及び2BBck81-)を用いてキモトリプシンループの中にCK37281を挿入することにより両ループの中に移植した。トリプシンループ(1BBIck81)の中CK37281を有するBBIをSacI-SphIフラグメントとしてpJM103BB発現カセット内に移動した。キモトリプシンループ(2BBIck81)又は両者のループ(12BBIck81)の中のCK37281を有するBBIを、SacI-SalIフラグメントとしてpJM103BBIの中に挿入した。正確な配列はDNA配列決定を用いて確認した(12BBIck81遺伝子の配列は図3に示す)。得られたベクター、pJK103-1BBIck81、pJK103-2BBIck81又はpJK103-12BBIck81をバチルススブチルス(B.subtilis)BG3934comKの形質転換に用い、上の実施例1に示すように、BCE融合タンパク質の生産を確認した。
【0136】
無細胞ブロスに存在する主要タンパク質である44kDa付近の融合タンパク質がSDS-PAGEで検出された。幾つかのケースにおいて、BCE触媒コアに対応する34
kDa付近の主要タンパク質バンドの存在により視認することができる有意なBBI部分の退縮(degradation)(50 %まで)(特にMBD倍地中で48時間以上育成した場合)が見られた。このケースにおいて、BCE103セルラーゼの力価は、野生型BBI(実施例1)に融合させて測定したの同様であるが、BBIの活性は通常の半分以下であった。
【0137】
育成の間のBBI変異体のタンパク質分解を減らすために(すなわち、融合タンパク質との比較においてSDS-PAGE上に存在するBCE103セルラーぜ触媒コアの量を減らすために)、9のプロテアーゼ遺伝子(degUHY32、oppA、△spoIIE3501、△aprE、△npf
E、△epr、△ispA、△bpr、△vpr、△wprA、△mpr-ybiF、△nprB、amyE : : xylRPxylAcomK-phleo)を欠失させたバチルススブチルス(Bacillussubtilis)系統を発現宿主細胞として用いた。育成温度(35 ℃)及び通気量(200 rpm)を減らした。これらの変更により、主要融合タンパク質バンド(〜44kDa)はBCE触媒コア(〜34 kDa)と思われる分子量で存在するバンドと大差なく、SDS-PAGE上で観察された。
【0138】
BCE103セルラーゼのC末端に融合させたBBIのトリプシン及び/又はキモトリプシン活性部位ループへの置換を行ったときに、CK37281ペプチドに加えて、多くのシステインコンストレインドペプチド(cysteineconstrained peptides)が生産された。特定の例を以下に示す。
【0139】
(1) 相補的な抑制因子として設計又は選択されたペプチド、第一又は第二反応部位ループへ導入されたコンプスタチン(compstatin)(Sahu etal.,J. Immunol., 157: 884-891, [1996] 参照のこと)、C2c(第一ループ)、C3c(第一ループ)、C4c(第一ループ)、及びC5c(第一ループ)、又は1B、2B、4A、5A、6-1A、7A、8B、9A、10B、11-1A、12B、13A、及び15-1A(全て第二ループ)へ結合する反応においてFactor Bにより選択されたペプチド
(2) プロテアーゼファクターXa、又は角質溶解キモトリプシンXal(第二ループ)又はhSCC1(第一ループ)に結合するためのペプチド
(3) FGF5結合反応により選択されたペプチド; 1A6 (第一又は第二ループ)、1C2 (第一又は第二ループ)、2E2 (第一又は第二ループ)、2E5(第一、第二、又は両ループ),FGFns(第一又は第二ループ)、FGFkr(第一又は第二ループ)、FGFhl(第一又は第二ループ)、FGFgy (第一又は第二ループ)、MM005(第一又は第二ループ)、MM007(第一、第二、又は両ループ)、MM009 (第二ループ)、MM010(第一、第二、又は両ループ)、MM017 (第二ループ)、FGFps1(第二ループ)、FGFps2(第一、第二、又は両ループ)、及びFGFpsB(第二ループ) 、及び
(4) TGF-1結合反応により選択されたペプチド;1A8(第二ループ)、lA12 (第二ループ), lE11 (第二ループ)、TGFpsl(第二ループ)及びMM021(第二ループ)。
【0140】
これらのペプチドをトリプシン(第一ループ)又はキモトリプシン(第二ループ)活性部位ループのいずれかへ導入するために用いるオリゴヌクレオチド及びこれらのペプチドをBBIに移植するために用いる方法は上に記載されている。全てのケースにおいて、生産された融合タンパク質をDSD-PAGEゲルにより評価した。CK37281置換ペプチドに対して説明したのと同様に、幾つかの置換ペプチドをもちいて、タンパク質分解を減少させることによりにより無傷の融合タンパク質の生成量が高められた。
【実施例3】
【0141】
チオール還元/酸化試薬によるBBIの活性化
育成の後の(トリプシン又はキモトリプシンの抑制による)BBIの活性は、(2の分子が融合タンパク質の中で1:1のモル比で存在する)無細胞上清中で測定されるBCE103セルラーゼの活性から予想されるものよりも通常5乃至20倍低い。BCE103-BBI融合タンパク質中のBBI活性の増加(トリプシン又はキモトリプシン抑制により測定される)は、接種の後、1-4mMの濃度のbMEをMBD成長培地に添加した後14時間育成することにより確実に得ることができる。融合タンパク質中のBBIのトリプシン又はキモトリプシン抑制活性は、結合ペプチド(例えば、VergF結合ペプチドCK37281)をキモトリプシン又はトリプシン反応部位ループにそれぞれ置換したときよりも低くなる。野生型BBIを用いて、bMEで処理することによって、抑制活性は増加させることができる。予想しないことに、これらの試験においてBBI育成の間の活性における他のチオール還元試薬(例えば、システイン、還元グルタチオン、DL-ジチオスレイトール(dithiothreitol)及びトリス[2-カルボキシメチル]フォスフィン)の影響は小さいか、無視できる程度であった。抗酸化剤(例えば、アスコルビン酸又はDL-α酢酸トコフェロール)、他のアジュバンド(例えば、イソロイシン、ダイズオイル、Tween(登録商標)-80)を培地に添加すること、又は30℃における育成も、BCE103:BBI活性の比を改善しなかった。
【0142】
特に育成の間のBBI活性を決定するために、p2JM103-E3-2BBIcK81(以下実施例4を参照のこと)で形質転換させたバチルススブチルス(B.subtilis)BG6006を含む培地を、250mlの振とうフラスコ内で37 ℃、13時間、40 mlのMBD中で育成した。その後、図4で示すチオール還元試薬を添加し、62時間の育成の後に細胞の上清を収穫した。2-メルカプトエタノール(BME)、システイン(Cys)、還元グルタチオン(Glut)及びDL-ジチオスレイトール(DTT)を図4に提供されているグラフで示す最終濃度になるように成長培地に添加した。5mM βMEの濃度はより良いBCE103:BBI活性比となったが、通常は、試薬の添加がバクテリア成長を減少させることから、BCE103及びBBI力価は全体的に減少した(図4参照のこと)。BCE103及び2BBIck81の力価は実施例1で説明したアッセイを用いて測定した。
【0143】
Qセファロースイオン交換クロマトグラフィーによる融合タンパク質(例えば、BCE-lnk2-2BBIck81、実施例4を参照にこと)の部分的な精製の後にもBBIの活性化を行うことがきでる。
【0144】
融合タンパク質を作動している振とうタンク又は発酵槽から得られた無細胞ブロスから精製した。このブロスを、ろ過し、5乃至10倍の水で希釈し、pHを7.5乃至8.0に調製した。希釈サンプルはQセファロースレジン(GHhealthcare)を充填したカラムに負荷した。このカラムを50 mMトリスpH 7.5で洗浄し、300 mM NaClを含む同じ緩衝液で再度洗浄した。融合タンパク質は、700mMのNaClで溶出した。
【0145】
BBIを活性化させるために収集された融合タンパク質分画を10倍のアッセイ緩衝液の中で溶出し、その後、2 mMのβME及び0.2 mMの酸化グルタチオン(GSSG)を添加し約24時間室温で、スターラープレート又はロックプラットフォーム上で攪拌を続けることにより処理した。BBIは、BCE103セルラーゼで測定される濃度にもとづき予測されるトリプシン抑制活性の約70乃至100%を達成した。上で概説した活性化方法は、通常最良の結果を出すけれども、幾つかのケースにおいて、所与のサンプルの活性を最大化するために96ウェルプレートにおいて、スクリーニングを行った。最初にスクリーニングする典型的な条件は、アッセイ緩衝液による希釈倍率(例えば、2乃至50倍の連続希釈)、βME濃度(例えば、連続する0.5乃至5mM)及び酸化グルタチオン濃度(例えば、0 mM、1/20乃至1/2 βME濃度)である。
【0146】
BCE-lnk2-2BBIck81融合タンパク質の活性化を図5に示す。この特定の態様において、Qセファロース精製後の融合タンパク質を0.005 %のTween(登録商標)-80含むダルベッコPBS(Mediatech)を用いて1:10に希釈した。βメルカプトエタノールを3
mMの最終濃度で添加し、振とう機上で室温、一晩インキュベートした。このサンプルは、更に磁気攪拌プレート上で激しく攪拌しながら60時間室温でインキュベートした。(βMEで処置する前後の)BCE103及び2BBIck81の力価を、それぞれ、セルラーゼアッセイ及びトリプシン抑制アッセイにより決定した。
【0147】
幾つかの態様において、大容量発酵槽のからの無細胞ブロス中のBBI又はその変異体の活性化のような、活性化反応において、希釈及びβME濃度を減少させることが好ましい。このことは、高濃度緩衝液(500mMトリスpH 6.8)、又はCHES等(CAPS、トリシン、TAPS及び他の適切な両性イオン緩衝液)の両性イオン緩衝液に変更することにより達成できる。例えば、無細胞ブロス(又はイオン交換クロマトグラファイーにより精製された融合タンパク質分画)は375mMのCHES pH 6.8中、0.005 %のTWEEN(登録商標)-80で希釈し、その後に、1 mMのβME及び10 mMのNa2SO3で活性化し、攪拌しながら室温で約24時間インキュベーションする。BCE103セルラーゼ融合変異体等のBBIあるいはこの変異体の力価はこの方法により、(BCE103セルラーゼ活性に基づく)予測値に対して70乃至100%まで活性化することができる。
【実施例4】
【0148】
BCE103-BBI融合タンパク質の切断による遊離BBI/変異体の放出
この実施例は、BCE103-BBI融合タンパク質を切断による遊離BBI又はその変異体の放出について説明する。
【0149】
培地育成の間にBCE103触媒ドメイン及びBBIの間に切断可能部位を作り出すために、アニールされ、pJM103のBamHI及びSacIサイトへ結合するDNAオリゴヌクレオチドペアの配列を以下に示す。
【表19】
【0150】
BCE103セルラーゼの第二CBDリンカーへBBIを融合するために、へアニールされ、p2JM103-BBIのBamHI及びSacI部位へ結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表20】
【0151】
アスパラギン酸及びプロリン残渣の間の酸切断をするのに適したペプチド配列を以下に示す。
【表21】
【0152】
QuikChange(登録商標)部位直接変異誘発によりBssHII部位をpJM103BBIへ導入するために用いるオリゴヌクレオチドプライマーを以下に示す。
【表22】
【0153】
BBIの停止コドンの後にHIndIII及びXhoI部位を導入し、LATの後にPacI部位を導入し、本来のHindIII部位を除去するためのカセット(SalI-HindIII)としてアニールされるDNAオリゴヌクレオチドの配列を以下に示す。
【表23】
【0154】
BCE103触媒ドメイン及びBBIの間に挿入される上で提供される酸ラベルリンカー(すなわち、リンカー1、リンカー2及びリンカー3)を生成するために用いるPCRプライマーを以下に示す。
【表24】
【0155】
上で説明する第一CBDリンカーに挿入する酸ラベルリンカーを生成するために用いるPCRプライマーを以下に示す。
【表25】
【0156】
BBI及びBCE103触媒ドメインの間に挿入された酸ラベルリンカーのタンパク質配列を以下に示す。酸ラベルリンカーを太字で示し、第一CBDドメイン配列を下線で示す。
【表26】
【0157】
精製工程の間にBBI及びBCE103触媒ドメインの間に切断可能部位を生成するために、アニールされ、pJM103-BBIのBamHI及びSacIへ結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表27】
【0158】
精製工程の間にBBI及びBCE103触媒ドメインの間に挿入される切断可能部位を生成するために、へアニールされ、pJM103-lnk2-1BBIck81のBamHI及びSacI結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表28】
【0159】
QuikChange部位直接変異誘発(Stratagen)を用いてリンカー2へE及びE3リンカーを導入するために用いるオリゴヌクレオチドプライマーペアの配列を以下に示す。
【表29】
【0160】
BCE103触媒ドメイン及びBBIの間のE3リンカーのタンパク質配列はDNNDPIPEP[DDESFNMPIPEP] (配列番号 174)である。この配列において、Eリンカーを下線で示し、大腸菌における間違った組換えにより生成された配列を太字([]ので囲む配列)で示す。Mpr(又はV8プロテアーゼ)による切断はE3リンカーの中にある3のグルタミン酸のいずれかの後に引き起こすことができる。従って、この構築体は、BCE-(配列番号174)-BBIとなる。
【0161】
BBI及びBCE103触媒ドメインの間に挿入されるGenenase I切断可能部位を生成するために、へアニールされ、p2JM103-lnk2-2BBIck81のBamHI及びSacIへ結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表30】
【0162】
BDE103触媒ドメイン及びBBIの間に挿入されるGenenase I感受性切断部位(酸感受性及びMpr感受性でもある)のタンパク質配列は、DNNDPIPDP[GAAHYVEFQ]
(配列番号 179)である。GeneaseI部位(Gen4 リンカーは太字([]ので囲む配列)で(チロシン及びバリンの間に発生する切断)(NEB)、リンカー2は下線で示。Mprによる切断もGen4リンカーの中のバリンの後のグルタミンの後に発生させることができる。本発明で用いられる配列はBCE-(配列番号179)-BBI である。
【0163】
BCE103-lnk2-2BBIck81融合タンパク質における切断部位を以下に示す。BCE103触媒ドメイン(下線)のC末端のアミノ酸、リンカー2配列(太字)及び2BBIck81配列を示す。酸/熱に不安定なAsp-Pro結合は塗りつぶし矢印、グルタミン酸の後のMpr感受性結合は線矢印で示した。
【表31】
【0164】
遊離BBI又はその変異体を単離するために、BCE103-BBI融合タンパク質からBBI部分を切り離すことが必要になる。幾つかの態様において、バチルススブチルス(B.
subtilis)により製造される固有のプロテアーゼを用いてこれは育成の間に行われる。幾つかの代替的な態様において、この切断は育成の後の精製工程(例えば、酸/熱又はタンパク質分解切断)dで行われる。リンカーは育成の間の切断に感受性を有する可能性のあるものが設計され(上記、sub、cbdl、pro、shortpro、及びcbdD)、BamHI-SacIカセットとして、pJM103BBI又はp2JM103BBI発現ベクター内でクローニングされる。融合タンパク質に対するBCE103触媒ドメインの生成は実施例1で説明されるSDS-PAGEゲル上で解析された。
【0165】
proリンカーを除く、全てのリンカーにおいて、BCE103触媒コアに対応する大きなバンドのほかに、ほぼ完全に切断されていると考えられる融合タンパク質の小さな切断が観察された。不幸なことに、育成の間に切断を行うと無細胞上清中で測定されるBBI活性は無視できるほどとなり、SDS-PAGEゲル上で同定するされるBBIバンドを生じなかった。本発明を特定の方法又は理論に制限することは意図しないけれども、BBIはその不活性形態において、タンパク質分解に影響を受けやすいことが明らかでる。従って、活性化後の精製工程の間に切断することが通常好ましい。
【0166】
幾つかの態様において、アスパラギン酸及びプロリン残渣の間の結合は、当該技術分野で知られている様に、酸性pHにおける熱処理により切断される(LandonMeth. Enzymol., 47: 145-149 [1977] )。BCE103セルラーゼの中の第一CBDリンカーは酸/熱処理により切断される可能性のある3のAsp-Proペプチド配列を有する(図1を参照のこと)。しかしながら、これらの部位において、酸/熱処理による切断は効率が悪いことがわかった。特に酸/熱に不安定なタンパク質配列は文献に記載されている。そのような3の配列は、WGDPHY(配列番号 141)、DNNDPI (配列番号 142)、及びWADPN (配列番号 143) (すなわち、リンカー1、2、及び3)である。
【0167】
これらの酸に不安定なリンカーが、BCE103-BBI発現ベクター、pJM103BBI、へ導入される前に、QuikChange(登録商標)XL(Stratagene)変異誘発部位を用いて(8分間の伸長時間、及び1分間の変性工程を用いる以外は、製造会社の説明書に従い、実施例2で述べた方法を用いて)、BCEbss_F及びBCEbss_R(上記)のオリゴヌクレオチドを用いて、aprEシグナル配列コード領域にaBssHII部位を挿入した。その後、HindIII及びXhoI部位をLATターミネーター(BBI停止コドンの後の)の前に挿入し、SalI及び元のHindII部位へのオリゴヌクレオチドカセット(BCEterm+及びBCEterm-;上記)の挿入により、このターミネーター(LATターミネーターの後のオリジナルHindIII部位)の後にPacI部位を付加した。この新しいベクターを「p2JM103BBI」と称した。
【0168】
前方向プライマー、BCE103coreBssHII_Fw、リンカーWGDPHY_R、リンカーDNNDPI_RV又はリンカーVVADPN_RV(配列は上記)の各リバースプライマーを用いて、及びpJM103BBIをテンプレートとして用いたPCRにより、酸に不安定なリンカーフラグメントを製造した(PCR手順については実施例1を参照のこと)。970bpのPCRフラグメントをBanHI及びPstIで消化し、酸リンカーフラグメントをコードしている154 bpのフラグメントを電気泳動の後、アガロースから単離し、BanHI及びPstIで消化ししたpJM103ベクター内へ結合した。最終発現ベクターにおけるリンカー配列、p2LM103lnk-BBI、p2JM103lnk2-BBI及びp2JM103lnk-BBIをDNA配列決定により確認した。
【0169】
有能なバチルススブチルス(B.subtilis)系統、BG3934comK又はBG6006を、プラスミドを用いて形質転換し、コロニーを5 μg/mlクロラムフェニコールを含むLAプレート上で選択し、実施例1で説明する様に、25μg/mlクロラムフェニコールを用いて増幅した。
【0170】
同様に、第一CBDリンカーに挿入した。特に、PCRフラグメントは、前方向プライマー、BCE103corePstI_FW、LplusDNNDPI_RV又はLplusVVADPN_RVのリバースプライマー(配列を上に記載)を用いて、テンプレートをp2JM103BBIとして酸に不安定なリンカーを生成した。約150
bpのフラグメントを、BamHI及びPstIを用いて消化し、精製し、BanHI及びPstIを用いて消化したp2JM103BBIに結合した。正確な配列をDNA配列決定法により確認し、上で述べたように、p2JM103plnk-BBI、p2JM103plnk2-BBI及びp2JM103pllnk3-BBIは、バチルススブチルス(B.subtilis)の形質転換に用いた。
【0171】
MBD培地での育成の後、上で説明した(実施例2を参照のこと)イオン交換クロマトグラフィーにより融合タンパク質を精製した。融合タンパク質は10 %のギ酸中、55℃、16時間での処理することにより切断された。BCE103触媒ドメインは、酸処理の間に沈殿し、遠心分離により分離された。上清中の遊離BBIをスピードバク(SpeedVac)で一晩乾燥した。SDS-PAGEゲルに負荷する前に50mMトリスpH 8の中にサンプルを懸濁した。SDS-PAGEのタンパク質染色解析により、酸切断は、リンカー2がBCE103触媒ドメイン及びBBI(BCE-DNNDPI-PDP-BBI)の間に挿入されている場合には、融合タンパク質において有効であることが観察された。このリンカーは、pH2、75 ℃、20 mMのグリシン中で数時間処理することで切断されることがわかった。
【0172】
代替的な態様において、精製工程の間にプロテアーゼで処理することにより融合タンパク質を切断した。グルタミン酸特異プロテアーゼ(例えば、Mpr又はV8プロテアーゼ)、フリン/ブリスターゼ、GenenaseI及びエンテロペプチダーゼ(エンテロキナーゼ)に対する切断部位を用いてリンカーを消化した。リンカーは、BenHI及びSacI部位を用いて、発現ベクター中のBCE103触媒コア及びBBIの間にオリゴヌクレオチドカセットとして(配列は上に記載)導入された。オリジナルの発現ベクター(pJM103BBI)のコード領域において、第一CBDドメインの中及びBBIの中の第三残渣にバチルススブチルス(B.
subutilis)Mpr又はV8プロテアーゼ等のグルタミン酸特異プロテアーゼにより切断されやすいと予測されるグルタミン酸残渣がある(図1)。しかしながら、BsMpr又はV8プロテアーゼのいずれもがこれらの部位でBCE-BBI融合タンパク質を切断することが困難であることがわかった。従って、これらのプロテアーゼにより切断されやすい他のリンカーを設計することが必要であった。
【0173】
上で説明した6の不安定なリンカーの切断をBsMprにより試験した。これらの融合タンパク質は室温で、16 μgのBsMprを用いて16時間処理することにより切断された。切断の後、BCE103触媒ドメインを10%のギ酸を添加することにで乾燥させた。DSD-PAGEに負荷する前に50 mMトリスpH 8にサンプルを懸濁した。酸切断と同様、BCE-DNNDPI-PDP-BBI(リンカー2)融合タンパク質は、他のリンカーのどれよりもBsMprで効果的に切断された。それゆえ、BBI及びその変異体は、酸/熱処理又はBsMpr等のグルタミン酸特異プロテアーゼを用いたタンパク質分解のいずれかによりBCE-DNNDPD-PDP-BBI融合タンパク質から、効果的に放出されていることがわかった。Mprにより切断されるように設計された幾つかの他のリンカーも試験されたが、リンカー2よりも効果的なこのはなかった(E3リンカーは、Eリンカーを構築するために意図されたQuikChange(登録商標)部位切断変異誘発を用いた形質転換の後に大腸菌内の誤った組換えにより生成された)。上に示す様に、リンカー2の中に、酸/熱に不安定な切断部位と、Mrpにより切断されやすい3の部位がある。
【0174】
フリン又はブリスターゼ(NBE)(BCEfurinBBI)又は、エンテロペプチダーゼ(エンテロキナーゼ、NEB)(BCEentBBI)による切断のために構築されたリンカーが試験されたが、これらのプロテアーゼより効果的に切断された配列はなかった。4のリンカー(BCEgenen1BBI、BCEgenen2BBI、BCEgenen3BBI、及びBCEgenen4BBI)も消化され、GenenaseI(NEB)による切断について試験された。融合タンパク質の効果的な切断は、Gene4リンカー(BCEgenen4BBI)を用いたときに観察された。BsMprもまた、Gene4リンカーを効果的に切断した。
【0175】
精製されたBCE-lnk2-2BBIck81融合タンパク質の活性化の後の、BsMprによる切断はSDS-PAGE上では観察されなかった。しかしながら、リンカー2(又はGen4リンカー)を有するBCE-BBI融合タンパク質の活性化の後の切断は、バチルスリケニフォルミス(Bacillus lichenifornais)から単離されたMprプロテアーゼ(BIMpr)を用いて成し遂げることができる。本発明を特定のメカニズムに限定することは意図しないけれども、成熟BBIの中の3番目のアミノ酸の切断はBIMprに対してより感受性があることが明らかである一方で、BBIのC末端から6番目のアミノ酸はBsMpr切断に対してより敏感である。
【0176】
幾つかの態様において、切断の後、BBIは、上で述べるBCE103触媒ドメインの選択的酸沈殿(pH3以下)により、イオン交換クロマトグラフィー(実施例5を参照のこと)、又は、50mMトリスpH8.0を用いた無水トリプシンアガロースに負荷し、150mMNaClで50mMトリスpH8.0を洗浄し、その後、300mMNaClde,pH2.2 50mMグリシンで結合BBIを溶出することにより、BCE103触媒ドメインから単離精製された。
【実施例5】
【0177】
BBIck81のVegFへの結合
本実施例は、BBIcK81のVegFへの結合を評価するために行う試験について述べる。BCE103-lnk2-2BBIck81融合タンパク質は実施例2で述べたように、バチルススブチルス(B. subtilis)を用いて生成された。融合タンパク質を生成し、実施例3に記載されているように、βME及び酸化グルタチオンを用いた処理により、BBIトリプシン抑制活性が増加された。融合タンパク質はBsMprプロテアーゼ(実施例4参照のこと)により切断され、Qセファロースカラムを用いたイオン交換クロマトグラフィーによりBCE103触媒ドメインから2BBIcK81を精製した。
【0178】
すなわち、切断の後、切断されたサンプルのpHを5.5に調製し、このサンプルをその後、(pH5.5 25mMMESを用いて平衡化した)カラムに負荷した。遊離2BBIck81は、25mM酢酸ナトリウムをカラムに通すことにより洗浄されるが、BCE103触媒コアは樹脂に結合したままであった。2BBIcK81分画は精密ろ過により濃縮され、結合アッセイ(Bio
Veris)に基づく、電気化学発光(ECL)分析を用いて解析された。抗-VegF抗体(Santa Cruz)及びVegF(Pepro Tech)は、製造会社(BioVeris)の説明書に従って電気化学発光ダイ及びビオチンでそれぞれ標識した。全ての材料は0.1 %Tween(登録商標)-80を用いてダルベッコPBS(Mediatech)中に溶解した。抗-VegF抗体の一連の希釈物(125、250、及び500ng/ml)及びVegFの一連の希釈物(100、150、200及び250 ng/ml)は、強固なECLシグナルを与える濃度を決定するためにアッセイされた。
【0179】
2BBIck81結合テストのために、50 μLの500 ng/ml ECL標識抗-VerF抗体、50 μLの250ng/mlビオチン標識VegF及び100 μL2BBIcL81(12.5、15、31.25、62.5、125、250又は500 ng/ml)を振とうしながら、室温で2時間インキュベーションした。その後、50μLの0.2 mg/mlストレプトアビジンコートビーズを添加し、室温で30分インキュベートした。ECLシグナルは、製造会社(Bio Veris)の説明書に従い、BioVeris M8/384解析を用いて測定した。図6に示すように、ECLシグナルは2BBIck81濃度が増加する毎に減少した。標識された抗-VegF抗体のより多くが磁気ビースが付着しているVegFに結合したことを示す。
【0180】
従って、CK37281ペプチドは、BBIキモトリプシン抑制ループ(2BBIcK81)上に移植されたときに、μモルの濃度で抗-VergF抗体とVegFの結合について競合することがわかった。実際には、2BBIck81はVegF結合に対して、合成CK3781ペプチド自身よりも強固に競合することがわかった(図6参照のこと)。CK37281ペプチドは、トリプシン抑制ループ、1BBIc81内に挿入され、BioVerisアッセイにおいて、抗-VegF抗体と競合した。従って、BBIは各種スクリーニング法により選択される本発明の活性結合ペプチドのスキャッフォールドとして有用である。
【実施例6】
【0181】
2BBIck81の生産に対する融合代替パートナーの使用
本実施例は、融合代替パートナーを評価するための試験を説明する。pET-40(+)由来のdsbc遺伝子(大腸菌)を増殖させるのに用いるオリゴヌクレオチドプライマーのDNA配列を以下に示す。このプライマーはp2JM103-Gen4-2BBIck81内へのクローニングのために5’末端にBssHII部位を、3’末端にBanHI部位を生成する。
【表32】
【0182】
リンカー2を有するBBIへ、シュードモナスメンドセラ(P.mendocina)クチナーゼ遺伝子の融合のためのカセット(Alw44I-BanHI)を作るために共にアニールされるオリゴヌクレオチドのDNA配列を以下に示す。
【表33】
【0183】
BBI部分は7個のジスルフィド結合を有することから、BCE103セルロース触媒ドメイン以外の融合タンパク質を用いて、活性BBIの高い力価が得られることが予測される。例えば、幾つかの態様において、チオールジスルフィド酸化還元酵素及び/又はタンパク質ジスルフィドイソメラーゼは正しく折りたたまれたBBIを生産するための融合タンパク質として用いられる。この態様において、追加的な活性化ステップが大部分の環境下において必要とされない。追加的な態様において、バチルススブチルス(B.
sbutilis)の中で高い力価で製造される他のタンパク質も融合パートナーとして用いることができる。例えば、フミコーラインソレンス(Humicolainsolens)由来の熱安定タンパク質ジスルフィドイソメラーゼ(hiPDI)は、バチスルスブレビス(Bacillus brevis)の中の免役グロブリンの軽鎖(2ジスルフィド)の融合パターンとして用いられている(Kajinoet al., Appl. Env. Microbiol. , 66:638-642 [2000])参照のこと。
【0184】
BBI生産に関して、hiPDIがBCE103よりも良い融合パートナーであるかどうかを決定するために、hiPDI遺伝子を合成し(DNA2.0)、発現ベクター、p2JM103-lnk2-2BBIck81(実施例4を参照のこと)、の中で、BssHII-SacIフラグメントとしてクローニングした。制限部位が導入された又は欠失されている場合を除き、高頻度で、高発現するバチルススブチルス(B.subtilis)遺伝子の発生コドンが合成遺伝子の設計のために選択された。最終濃度において、成熟hiPDI遺伝子のN末端をAprEシグナル配列に融合し、C末端はエンテロペプチダーゼ切断部位を有するリンカーに融合した(Kajinoet al., Appl. Env. Microbiol. , 66: 638- 642 [2000])。それらを順番に、2BBIck81(図7参照のこと)に融合する。この発現ベクター、p2JM-PDI-EK-2BBIck81はバチルススブチルス(B.subtilis)BG6006を形質転換するために用いた。融合タンパク質の生成はMBD培地中で(実施例1で説明したように)2mMβMEを添加、あるいは無添加した状態で、接種後14時間で評価に用いた。
【0185】
SDS-PAGEゲルにより評価されるように、PDI-2BBIck81融合タンパク質の生産は、通常同じ条件で育成されたBCE-2BBIck81によりわずかに少ない。PDI-2BBIck81細胞を含まない上清を用いて測定された(トリプシン抑制)BBI力価も又、通常BCE-2BBIck81融合細胞よりもわずかに少なかった。BCE103への融合と同様に、PDIへ融合させたときに測定されるBBI力価は、2mMβME中で育成したときに高くなり、BBI活性はβMEフリー培地中で育成したときの育成の後、細胞フリー上清にβMEを添加することにより高められた(実施例3に記載されているように)。したがって、PDIのチオールジスルフィド酸化還元活性は、融合タンパク質の中の活性か2BBIck81の力価を有意に改善しなようであり、BBI分子を活性化する必要を排除した。
【0186】
育成の間のBBI力価を改善すると思われる融合タンパク質の還元電位を増加させるために、大腸菌(Escherichia coli)由来のDsbCが2BBIcK81に対する融合パートナーとして用いられた。dsbC遺伝子は、DsbCBBI-F及びDsbCBBI-Rをプライマーとして用いて(配列は上に記載)、pET-40(+)(Novagen)をテンプレートとして用いて、製造会社(Stratagen)の説明書に従い、HerculaseEnhanced DNA ポリメラーゼを用いたPCRにより増幅させた。単離されたPCRフラグメントはベクターp2JM103-Gen4-2BBIck81(実施例4を参照のこと)の中で、BssHII-BsmHIフラグメントとして用いてクローニングした。融合遺伝子の正確な配列はDNA配列決定により確認された。このケースにおいて、DSbC-2BBIck81融合タンパク質の力価はSDS-PAGEゲル上で判断した時に、BCE-2BBIck81融合タンパク質よりも有意に低かった。トリプシン抑制により測定される活性2BBIck81力価も又十分に低かった。
【0187】
バチルススブチルス(B. subutilis)の中で高い力価を伴い生産される他のタンパク質は、BBIの生産に対する融合パートナーとして用いることができる。そのようなタンパク質の一つは、バチルススブチルス(B.subutilis)由来のaprEプロモーターを用いて高力価が発現されるシュードモナスメンドシナ(Pseudomonas mendocina)由来のクチナーゼである(参照により本発明の明細書に援用される米国特許5,429,950参照のこと)。apr-Eクチナーゼ遺伝子融合(体)は、EcoR1-Aln44Iフラグメント(pAK-15由来)として、Alw44I-BamHIリンカーオリゴヌクレオチドカセット(配列は上記)を用いて、EcoRI及びBamHIを用いて切断されたp2JM103-lnk2-2BBIck81(実施例4参照のこと)へ結合した。このクチナーゼ-リンカー2-2BBIcK81発現ベクターは、バチルススブチルスBG6006細胞の形質転換に用いて、融合タンパク質はMBD培地の中で他の融合タンパク質に対して記載したのと同様に生産された(実施例1参照のこと)。このケースにおいて、クチナーゼリンカ−2-2BBIck融合タンパク質はSDS-PAGE上で見られる主要バンドではなく、測定されたリパーゼ力価(米国特許5,429,950に記載の方法を用いて測定された)及びBBIの力価はBCE-2BBIck81融合タンパク質で見られたものよりも大部低くなっていた(約20倍)。クチナーゼ融合タンパク質中のBBI力価は育成培地に、3mMのβMEを添加したときにも改善されなかった。したがって、活性2BBIck81の最高力価はBCE-2BBIck融合タンパク質の活性化により、確実に得られるのである。しかしながら、各種融合タンパク質も本発明に用いることができるだろう。
【0188】
本明細書中で言及する全ての特許及び文献は本発明の技術分野における出願時の技術常識を示している。全ての特許及び文献が明確かつ別個に示している内容を参照により本発明の明細書に援用する。
【0189】
本明細書に開示されいてる好ましい態様を用いて、当業者が開示されている態様の変法をなすことができることは明らかであり、そのような変更は発明の範囲内である。
【0190】
本発明は課題を実行するために最適化されており、本明細書に内在するものだけでなく、目標及び有利な結果を得るために適していることを当業者は容易に理解する。本明細書で開示されている組成物及び方法は好ましい態様の代表例であり、例示であり、本発明を限定するものではない。当業者が本明細書に開示されている発明の各種置換、及び変更を本発明の範囲及び精神から逸脱することなくなしうることは明らかである。
【0191】
本明細書で例示する発明は、本明細書で特に開示されていない、構成要素、限定を伴わずに実行することができる。採用された文言及び表現は説明のために用い、限定のために用いるものではない。しかしながら、それらが示す、あるいは、それらの一部の特徴を有するあらゆる等価物を含む文言及び表現の使用を意図するものではない。したがって、本発明は好ましい態様を追加的な特徴により構成されるものであり、そのような変更及び変法は明示の請求項により定義される発明の範囲内である。
【0192】
この発明は、本明細書に幅広く一般的に記載されている。開示されている属のより狭義の又は亜種グループも本発明の一部を構成する。これは、削除された物質がとくに本発明に引用されたかどうかは別として、この属から除去する旨の規定又は負の限定を伴う本発明の包括的な記載を含む。
【図面の簡単な説明】
【0193】
【図1A】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図1B】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図1C】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図1D】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図2】図2はpJM103BBIhis発現ベクターの概略図を示す。
【図3】図3はBCE103融合部位(BamHIの位置)から端部までの12BBIcK81のDNA配列及びアミノ酸配列を示す(配列番号3及び4)。CK37281ペプチド配列(ACYNLYGWTC(配列番号9))は、トリプシン及びキモトリプシン抑制ループ内へ挿入される。
【図4】図4は、トリプシンによる活性2BBIck81及び不活性2BBIck81の力価及び培地の育成中に各種チオール還元試薬を添加した2BBck81に対するBCE103セルラーゼの活性比を示す。この図において、BME=2-メルカプトエタノール、Cyt=システイン、Glut=還元グルタチオン、DTT=ジチオスレイトールをそれぞれ示す。
【図5】図5はイオン交換クロマトグラフィーによる部分的な精製の後で、2-メルカプトエタノール(bME)で処理したときのBCE-lnk2-2BBIck81の活性を示すグラフである。
【図6】図6は、VegFに結合する2BBIck81と抗-VegF抗体の比較解析の結果を示す。
【図7A】図7はバチルススブチルス(B. subtilis)BBI発現ベクター内に挿入されたフミコーラインソレンス(H. insolens)PDI(hiPDI)を運ぶ合成DNAフラグメントの配列及びこのアミノ酸配列を示す(配列番号5及び6)。
【図7B】図7はバチルススブチルス(B. subtilis)BBI発現ベクター内に挿入されたフミコーラインソレンス(H. insolens)PDI(hiPDI)を運ぶ合成DNAフラグメントの配列及びこのアミノ酸配列を示す(配列番号5及び6)。
【図7C】図7はバチルススブチルス(B. subtilis)BBI発現ベクター内に挿入されたフミコーラインソレンス(H. insolens)PDI(hiPDI)を運ぶ合成DNAフラグメントの配列及びこのアミノ酸配列を示す(配列番号5及び6)。
【図8A】図8はp2JM103-lnk2-2BBIck81のEcoRI-BamHI部位へ結合されるaprE-クチナーゼ発現カセットのDNA配列及びアミノ酸配列である(配列番号7及び8)。
【図8B】図8はp2JM103-lnk2-2BBIck81のEcoRI-BamHI部位へ結合されるaprE-クチナーゼ発現カセットのDNA配列及びアミノ酸配列である(配列番号7及び8)。
【図8C】図8はp2JM103-lnk2-2BBIck81のEcoRI-BamHI部位へ結合されるaprE-クチナーゼ発現カセットのDNA配列及びアミノ酸配列である(配列番号7及び8)。
【技術分野】
【0001】
本発明は、バクテリア種内でプロテアーゼインヒビター及びそれらの変異体の発現に関係する組成物及び方法を提供する。本発明は更に、融合核酸、ベクター、融合ポリペプチド、及びプロテアーゼインヒビターを得るための工程を提供する。
【背景技術】
【0002】
プロテアーゼは幅広い各種生体内作用に含まれている酵素である。プロテアーゼ及びプロテアーゼインヒビターのバランスを阻害することは病原性組織崩壊に関係する。すなわち、多くの研究は、組織障害におけるプロテアーゼの役割に焦点をあてており、プロテアーゼ及びプロテアーゼインヒビターの間のバランスが組織の完全性の維持における主要な決定要因であると言われてきた。好中球を含む炎症細胞由来のセリンプロテアーゼは、肺気腫、関節炎、アトピー性皮膚炎及び乾癬等の各種炎症性の病気に関係している。
【0003】
プロテアーゼは特定の癌を転移させるために機能しているとも考えられている。正常な細胞は、細胞外マトリクス(ECM)と呼ばれている、タンパク質ネットワークと接触して存在している。ECMは細胞の移動に対するバリアであり、癌細胞は、それらのアタッチメントを壊し、分解し、転移するためにECMに沿って移動する。プロテアーゼはタンパク質を分解する酵素であり、ECMを壊すことにより元の位置から腫瘍細胞を遊離させることを補助すると考えられてきた。近年の研究において、プロテアーゼは細胞形態変化およびプロテアーゼ活性化レセプター(PAR2)といわれる腫瘍細胞膜においてタンパク質を活性化することによる細胞の運動を促進することが示唆されている。このことは、細胞の運動装置を活性化する細胞内反応のカスケードとなる。従って、腫瘍の転移における第一のステップは移動方向に面した1の縁に明瞭な突起を形成するように細胞形状が再構成されることであると考えられている。形状を再構築した細胞はその後、血管壁を通じて移動し、離れた位置に移動し、最終的には、再付着し、転移腫瘍を形成する。例えば、ヒトの前立腺上皮細胞は、精漿の一成分のカリクレン様セリンプロテアーゼである前立腺特異抗原(PSA)を構造的に分泌する。このプロテアーゼは細胞外マトリクスを分解し、癌細胞の侵入を促進させる作用をする。
【0004】
合成及び天然のプロテアーゼインヒビターはインビボ(in vivo)及びインビトロ(in vitro)において癌の促進を抑制することが示唆されている。研究の結果、セリンプロテアーゼインヒビター又はセルピンズ(SERPINS)として分類される構造的に類似したタンパク質のファミリーに属している特定のプロテアーゼインヒビターは、好中球エラスターゼだけでなくトリプシン、カテプシンG、トロンビン、及び組織性カリクレインを含む幾つかのプロテアーゼを抑制することが示唆されている。セルピンズ(SERPINS)はインビトロ(invitro)において発癌物質により誘発された形質転換、及び実験動物モデルにおける発癌の予防/抑制に効果的である。精製されたプロテアーゼインヒビターの全身への投与は関節炎及び軟骨及び骨の崩壊を予防することが明らかにされている。
【0005】
プロテアーゼインヒビターの局所投与は、身体に位置するわずかな斑及び身体の大部分を含むアトピー性皮膚炎及び通常の皮膚炎症等を治療するために使用されている。プロテアーゼインヒビターの脱色活性及び紫外線誘導着色を防止する能力は、インビトロ(invitro)及びインビボ(in vivo)にて報告されている(Paine et al., J. Invest. Dermatol., 116: 587-595[2001]参照のこと)。プロテアーゼインヒビターは外傷の治癒も促進することが報告されている。例えば、分泌白血球プロテアーゼインヒビターは局所的に適用したときに組織の傷を修復し、外傷治癒を早めることが報告されている。更に、セリンプロテアーゼインヒビターは紅斑性狼瘡患者において痛みを減らすことが報告されている(米国特許6537968参照)。
【0006】
上に記載したように、プロテアーゼインヒビターはプロテアーゼの作用を阻害する。天然のプロテアーゼインヒビターは、穀物顆粒(オーツ麦、大麦、及びメイズ)、ブリュッセル芽キャベツ、たまねぎ、大根、小麦、シコクビエ(fingermillet)、及び落花生等の各種食物の中に存在する。特に興味のある供給源はダイズである。ダイズの中にある2の最も重要なプロテアーゼインヒビターの平均レベルはクニッツ(Kunitz)インヒビターにおいて約1.4%及びボウマン-バーク(Bowman-Birk)において約0.6 %である。これらの含有量が低い原材料は医薬品組成物のための天然プロテアーゼインヒビターを単離するのに実用的でない。
【0007】
従って、プロテアーゼインヒビター及びその変異体の大規模生産スケールに適した方法及びそれに用いる組成物に対する需要が存在する。特に、これらのプロテアーゼが哺乳類の組織培養細胞において製造されるときに生ずる、骨-血液感染因子に関する危険性を減らす又は除く方法及び組成物に対する需要も存在する。
【発明の概要】
【0008】
本発明はバクテリア種の中でプロテアーゼインヒビター及びそれらの変異体の発現に関係する方法及び組成物を提供する。本発明は更に、融合核酸、ベクター、融合ポリペプチド及びプロテアーゼインヒビターを得る工程を提供する。
【0009】
本明細書において提供されるものは、核酸、細胞、及びプロテアーゼインヒビター、これらの変異体、並びにその生産方法である。
【0010】
本発明は少なくとも1の機能的なプロテアーゼインヒビターをコードする核酸を提供する。一の側面において、第一、第二、第三及び第四の核酸配列に作動可能に結合している調節配列を含む核酸が提供される。幾つかの態様において、転写終了配列は、第四の核酸配列の後に付加されている。
【0011】
代替的な態様において、第一の核酸配列は、第一宿主微生物の中の分泌配列として機能するシグナルペプチドをコードする。第二の核酸は、通常第一又は第二宿主微生物から分泌される分泌ポリペプチド又はそれらの機能部分をコードする。第三の核酸は切断可能リンカーをコードし、第四の核酸は、プロテアーゼインヒビター又はそれらのフラグメントをコードする。更なる態様において、本発明は少なくとも1のプロテアーゼインヒビターをコードしている核酸配列を含む少なくとも1の発現カセットを提供する。
【0012】
追加的な態様において、本発明は少なくとも1のプロテアーゼインヒビター変異体をコードするポリヌクレオチドを提供する。幾つかの好ましい態様において、このポリヌクレオチドは、野生型ボウマン-バーク(Bowman-Birk)インヒビター(BBI)の少なくとも1のループが修飾されているBBI変異体をコードしている。
【0013】
本発明は機能的なプロテアーゼインヒビター又はそれらの変異体を発現する方法も提供する。幾つかの好ましい態様において、機能的なプロテアーゼインヒビター及び/又はそれらの変異体に適した宿主細胞が提供される。幾つかの態様において、(i)少なくとも1のプロテアーゼインヒビター又はそれらの変異体をコードする核酸配列を含む少なくとも1の発現カセットで宿主細胞を形質転換し、及び(ii)少なくとも1のプロテアーゼインヒビター又はそれらの変異体の発現に適した条件下で宿主細胞を培養する。幾つかの態様において、この方法は更にプロテアーゼインヒビター又はこれらの変異体の回収工程を含む。
【0014】
代替的な態様において、 (i)プロテアーゼインヒビター又はそれらの変異体をコードする少なくとも1の核酸配列を含む第一の発現カセットで宿主細胞を形質転換し、更に(ii)少なくとも1のチオールジスルフィド酸化還元酵素又はシャペロンをコードする核酸配列を含む第二の発現カセットで宿主細胞を形質転換し、そして(iii)プロテアーゼインヒビター又はそれらの変異体の発現に適した環境下において宿主細胞を培養する。幾つかの態様において、プロテアーゼインヒビター又はそれらの変異体は培地から回収される。幾つかの好ましい態様において、プロテアーゼインヒビター又はそれらの変異体は融合タンパク質として発現される。更なる態様において、該方法は更にプロテアーゼインヒビター又はそれらの変異体の回収工程を含む。
【0015】
更なる態様において、機能的なプロテアーゼインヒビター又はそれらの変異体はシグナル配列、セルラーゼ触媒ドメイン、切断可能リンカー領域を含む融合タンパク質として発現され、その後、発現されたタンパク質は、プロテアーゼインヒビター又はそれらの変異体の成熟型になる。
【0016】
追加的な態様において、発現されたタンパク質はプロテアーゼ及び/又は酸/熱で、プロテアーゼインヒビター又はそれらの変異体を融合タンパク質から遊離するために処理される。
【0017】
更なる態様において、本発明は、
a) Bowman-Birk インヒビター変異体、
b) Bowman-Birk インヒビター、及び
c) 少なくとも1の変異体配列を含むスキャッフォールド
から成る群より選択されるプロテアーゼインヒビター活性を有するポリペプチドを提供する。
【0018】
本発明は、プロテアーゼインヒビター及び所望のペプチドを含む融合タンパク質を含む組成物を提供する。幾つかの態様において、融合タンパク質は配列番号2、4、6、及び8から成る群より選択されるアミノ酸配列を含む。代替的な態様において、融合タンパク質は、配列番号1、3、5及び7から成る群より選択される核酸配列によりコードされている。幾つかの好ましい態様において、プロテアーゼインヒビターはボウマン-バーク(Bowman-Birk)インヒビター、ダイズトリプシンインヒビター(STI)、及びエグリン(eglin)Cから成る群より選択される。幾つかの特定の好ましい態様において、このプロテアーゼインヒビターはトリプシンループ及びキモトリプシンループから成る群より選択される少なくとも1のループを含むBBIである。更なる態様において、プロテアーゼインヒビターは所望のペプチドに対するスキャッフォールドである。幾つかの好ましい態様において、このループは少なくとも1の所望のペプチドを含む。幾つかの好ましい態様において、所望のペプチドは、配列番号9で定義されるアミノ酸配列を含む。幾つかの好ましい態様において、融合タンパク質は配列番号4で定義されるアミノ酸配列を含む。
【0019】
本発明は、a)ボウマン-バーク(Bowman-Birk)インヒビター(BBI)又はそれらの変異体由来のプロテアーゼインヒビターをコードする異種DNA配列を含むDNA構築体を宿主細胞内に導入する工程、b)異種DNA配列を発現させるために適切な培養条件下でバクテリア細胞を培養する工程、及びc)プロテアーゼインヒビターを生産する工程を含むことを特徴とするバクテリア細胞内で少なくとも1のプロテアーゼインヒビターを生産する方法を提供する。幾つかの好ましい態様において、構築体は更に少なくとも1の触媒ドメインを有する。幾つかの態様において、触媒ドメインはセルラーゼ、クチナーゼ、及びジスルフィドイソメラーゼから成る群より選択される。幾つかの好ましい態様において、この方法は更にプロテアーゼインヒビターを回収する工程を含む。幾つかの好ましい態様において、この方法は更にプロテアーゼインヒビターを活性化する工程を含む。最も好ましい態様において、この活性化は、ジスルフィド結合を還元する組成物、ジスルフィド結合を酸化する組成物、及び酸化還元電位を変更する組成物から成る群から選択される少なくとも1の試薬にプロテアーゼインヒビターを曝すことにより実施される。幾つかの態様において、バクテリア細胞はバチルス(Bacillus)属のメンバーである。追加的な態様において、プロテアーゼインヒビターはi)配列番号11と少なくとも90%の配列同一性を有するプロテアーゼインヒビター、及びii)配列番号13と少なくとも90 %の配列同一性を有するプロテアーゼインヒビターから成る群より選択される。更なる態様において、プロテアーゼインヒビターは変異体配列を含む。追加的な態様において、プロテアーゼインヒビターは、少なくとも1のループが配列番号9で置換されており、このループがトリプシンループ及びキモトリプシンループから成る群より選択されることを特徴とするボウマン-バーク(Bowman-Birk)インヒビターである。幾つかの態様において、この方法は更にチオールジスルフィド酸化還元酵素又はシャペロンをコードする第二の核酸配列をバクテリア細胞内に導入する工程を含む。幾つかの好ましい態様において、プロテアーゼインヒビターは融合タンパク質として発現される。幾つかの好ましい態様において、前記融合タンパク質は更にセルラーゼ触媒ドメイン、切断可能部位、及びプロテアーゼインヒビターを含む。幾つかの特定の好ましい態様において、融合タンパク質は、プロテアーゼインヒビターを遊離するためにプロテアーゼ及び/又は酸/熱で処理される。更なる好ましい態様において、融合タンパク質は少なくとも1のリンカー配列を更に含む。幾つかの態様において、このリンカー配列は配列番号141乃至143から成る群より選択される。
【0020】
本発明は、本明細書で説明する方法のいずれか又は全てに従って製造されたプロテアーゼインヒビターを含む組成物も提供する。幾つかの態様において、本明細書で開示する各種プライマー及びオリゴヌクレオチドは本発明の融合タンパク質の製造に用いる。更に、本発明は本発明の使用に適した多数のペプチドを提供する。
【0021】
本発明は本明細書で開示するプロテアーゼインヒビター組成物のいずれかを標的タンパク質に接触させる工程、及び標的タンパク質のタンパク質分解活性を抑制するために標的タンパク質と前記プロテアーゼインヒビター組成物を結合させる工程を含む標的タンパク質のタンパク質分解活性を抑制する方法を提供する。
【0022】
本発明は更に配列番号10及び12で定義されるポリペプチド配列から成る群より選択されるプロテアーゼインヒビターをコードする単離ポリヌクレオチドを提供する。
【0023】
本発明は、ポリヌクレオチド配列を含む発現ベクターも提供する。前記ポリヌクレオチド配列は、a) 少なくとも1のループ残渣が変異体配列で置換されていることを特徴とし、配列番号11と少なくとも90%の配列同一性を有するプロテアーゼインヒビターをコードしているポリヌクレオチド配列、及びb) 少なくとも1のループ残渣が変異体配列で置換されていることを特徴とし、配列番号13と少なくとも90%の配列同一性を有するプロテアーゼインヒビターをコードしているポリヌクレオチド配列から成る群より選択される。幾つかの態様において、この発現ベクターは更に5’末端から3’末端に向けて、バクテリア細胞内において分泌配列として機能するシグナルペプチドをコードする第一の核酸配列、分泌ポリペプチド又はそれらの機能部分をコードする第二の核酸配列、切断可能リンカーをコードする第三の核酸配列及びプロモーターインヒビターをコードするDNA配列を更に含む。本発明は、本発明の発現ベクターで形質転換させた宿主細胞も提供する。幾つかの好ましい態様において、宿主細胞はバチルス(Bacillus)属の細胞である。
【0024】
本発明は少なくとも1のスキャッフォールドタンパク質及び少なくとも1のペプチドを含む組成物を提供する。前記スキャッフォールドは、ボウマン-バーク(Bowman-Birk)インヒビターを含む。幾つかの好ましい態様において、このペプチドはVegFに結合するペプチドを含む。
【0025】
本発明の他の目的、技術的特徴、及び利点は以下の詳細な説明に明確に記載する。しかしながら、本発明の好ましい態様を示す詳細な説明及び特定の実施例は説明の用途にのみ用い、詳細な説明から当業者にとって自明である各種変更及び修正は本発明の範囲及び本発明の精神の範囲内であることが理解される。
【発明を実施するための最良の形態】
【0026】
発明の開示
本発明はバクテリア種の中でプロテアーゼインヒビター及びそれらの変異体を発現することに関係する組成物及び方法を提供する。本発明は更に融合核酸、ベクター、融合ポリペプチド、及びプロテアーゼインヒビターを得る工程を提供する。
【0027】
他の方法で示さない限り、本発明の実施は、分子生物学、微生物学、組換えDNA技術に関する従来技術を含む。そのような技術は、当業者に知られており、多くのテキスト及び参考文献に記載されている(Sambrooket al,. “Molecular Cloning: A Laboratory Manual”, Second Edition (Cold SpringHarbor), [1989]及びAusbel et al., “Current Protocols in Molecular Biology” [1987]参照のこと)。本明細書中で言及するすべての特許、特許出願、文献及び刊行物は参照により本明細書に援用する。
【0028】
他の違った方法で定義しない限り、本明細書で用いられる全ての技術的及び化学的言語は、本出願に関係する技術分野において当業者に通常理解されているものと同じ意味を有する。例えば、Singletonand Sainsbury, Dictionary of Microbiology and Molecular Biology, 2d Ed.,JohnWiley and Sons, NY (1994)及び Hale and Marham, The Harper Collins Dictionaryof Biology, Harper Perennial, NY (1991)は、本発明の技術分野の当業者が利用する、そして本発明において多く用いられている用語の一般的な辞書である。本明細書で述べられているのと同じかあるいは類似の多くの方法及び物質は本発明の実施に用いることができるけれども、本明細書中では特定の好ましい方法及び物質にのみ言及する。従って、以下で定義される文言は、本明細書全体を参照することで十分に説明される。本明細書で用いる記号、「一つの」及び「この」という語は、他に違った形で定義されていない限り、複数を示す場合にも用いる。数値範囲は、範囲を定義している上限及び下限の数値も含ものとする。違った方法で示さない限り、核酸は左から右に向けて5’から3’に向けて表記されるものとし、アミノ酸配列は、左から右に向けて、アミノ末端からカルボキシ末端として表記する。本発明は記載した特定の方法、手順及び試薬に限定されないことは当然でありこれらは変更可能である。
【0029】
更に、本明細書で付す標題は本発明の各種側面を限定するものではない。発明は明細書全体を参照して理解されるべきである。従って、以下で示す定義は、明細書全体を参照して完全に定義される。しかしながら、本発明の迅速な理解のために多数の文言を以下で定義する。
【0030】
定義
本明細書で用いる、「発現カセット」及び「発現ベクター」の語は、標的細胞の中で特定の核酸を転写させる一連の特定の核酸エレメントを持つ、組換え又は合成により生成された核酸構築体を意味する。組換え発現カセットは、プラスミド、染色体、ミトコンドリアDNA、色素体DNA、ウイスル又は核酸フラグメントに取り込ませることができる。通常、発現ベクターの組換え発現カセット部分には、転写される核酸配列及びプロモーターが含まれる。「発現カセット」の語は、本明細書において、「DNA構築体」及びこの等価物を指す場合に互換的に使用される。
【0031】
本明細書において、「ベクター」及び「クローニングベクター」の語は細胞内へ核酸配列を輸送することを意図する核酸構築体を意味する。
【0032】
本明細書において、「発現ベクター」の語は、異種細胞の中で異種DNAフラグメントを取り込み発現することができるベクターを意味する。多くの真核及び原核生物の発現ベクターが市販されている。適切な発現ベクターを選択することは当業者の知識の範囲内である。
【0033】
本明細書において、「プラスミド」の語は、クローニングベクターとして用いる環状二本鎖(ds)DNA構築体であり、種々の原核及び真核生物内で染色体外自己複製遺伝子要素を形成し、又は他の宿主細胞に組み込むことができるものを意味する。
【0034】
本明細書において、「核酸分子」及び「核酸配列」の語は、RNA、DNA、及びcDNA分子を含むがこれらに限定されない、任意の核酸の形態が含まれる。遺伝子コードの縮退の結果として、所定のタンパク質をコード化する多数のヌクレオチド配列が生産され得ることは当該技術分野において理解されている。
【0035】
本明細書において、「融合DNA配列」の語は、5’末端から3’末端に向けて、第一、第二、第三、及び第四DNA配列を含む。
【0036】
本明細書において、「第一核酸配列」又は「第一DNA配列」は、バクテリア種の中の分泌配列として機能するシグナルペプチドをコードしている。特に好ましいシグナル配列は融合ポリペプチドを発現及び分泌するために発現宿主により分泌されるポリペプチド由来の配列である。本明細書で用いるように、第一アミノ酸配列は、バクテリア種内で機能する分泌配列に対応している。そのようなアミノ酸配列は上に述べた第一DNA配列によりコードされる。
【0037】
本明細書において、「第二DNA配列」は選択されたバクテリア種により発現される「分泌ポリペプチド」(すなわち、「所望の分泌ポリペプチド」)をコードする。第一DNA配列と同様に、分泌ポリペプチドはバクテリア発現宿主が自然に分泌するものであることが好ましい。しかしながら、幾つかの態様において、この語は、異種タンパク質(すなわち、特定のバクテリア宿主が通常分泌しないタンパク質)も意味する。例えば、幾つかの好ましい態様において、バチルススブチルス(B.subtilis)以外のバシルス(Bacillus)属により発現されるセルラーゼは所望の分泌ポリペプチドとして用いられ、バチルススブチルス(B.subtilis)は、発現宿主として用いられる。
【0038】
本明細書で用いる、「分泌ポリペプチドの機能部分」及びこの同義語は、切り離された形状をしていても、正常な構造に折りたたまれる能力を有する切断された分泌ポリペプチドを意味する。幾つかの態様において、所望のポリペプチドに付着しているものとは関係なく、ポリペプチドが正規構造にホールドされるためには分泌ポリペプチドドメイン残渣の十分量が必要となる。しかしながら、大部分のケースにおいて、分泌ポリペプチドの一部のみでも正しく折りたたまれ、分泌ポリペプチドドメイン残渣欠失体と比較して、分泌が高まることが知られている。
【0039】
同様に、大部分の場合において、分泌ポリペプチドの切断体はその機能部分が生物学的機能を保持することを意味する。他の機能的なドメイン(例えば、基質結合ドメイン)も用いることができるけれども、本発明には分泌ポリペプチドの触媒ドメインを用いることが好ましい。加えて、好ましい態様において、触媒ドメイン及び全リンカー領域又は一部のリンカー領域が用いられる。
【0040】
本明細書で用いる、「第三のDNA配列」は、切断可能なリンカーポリペプチドをコードするDNA配列を含む。第三のDNA配列は、融合ポリペプチドを切断するための特定の酵素又は試薬により認識されるのに必要なアミノ酸配列のみをコードすることが必要であると解される。従って、適切な酵素による認識及び切断に必要なリンカー部分のみが必要とされる。
【0041】
本明細書で用いる、「第四のDNA配列」は「所望のポリペプチド」をコードする。そのような所望のポリペプチドは成熟型、又は未成熟型のいずれかのプロテアーゼインヒビター及びそれらの変異体を含む。
【0042】
対応する4のアミノ酸配列をコードする上で定義された4つのDNA配列は、「融合DNA配列」を形成するために結合される。そのような融合DNA配列は、固有のリーディングフレームの5’末端から3’末端に向かって、第一、第二、第三、第四DNA配列の順に結合される。そのように結合されているので、融合DNA配列は、そのアミノ末端から、バクテリア種の中で分泌配列として機能するシグナルペプチド、バクテリア種により通常分泌される分泌ポリペプチド又はその一部分、切断可能リンカーポリペプチド及び所望のポリペプチドをコードする「融合ポリペプチド」、「融合タンパク質」及び「融合アナログ」をコードする。
【0043】
本明細書で用いる、「所望のタンパク質」および、「所望のポリペプチド」の語は、分泌増強構築体に融合されない、成熟型又は未成熟型ポリペプチド又はタンパク質を意味する。従って、「所望のタンパク質」及び「所望のポリペプチド」の語は、非融合形態で宿主細胞により発現され、分泌されるものを意味する。
【0044】
本明細書で用いる、「融合ポリペプチド」、「融合タンパク質」及び「融合アナログ」は、アミノ末端から順に、宿主細胞の中で分泌配列として機能するシグナルペプチド、宿主細胞から通常分泌される分泌ポリペプチド又はその一部分、切断可能リンカーポリペプチド及び所望のポリペプチドをコードしている。幾つかの態様において、この融合タンパク質は宿主細胞酵素(例えば、プロテアーゼ)により、融合タンパク質の中の他のタンパク質配列から遊離した所望のタンパク質を生産するために処理される。本明細書において、「融合アナログ」、「融合ポリペプチド」及び「融合タンパク質」は互換的に使用する。
【0045】
本明細書で用いる、「プロモーター配列」の語は、発現の目的で、バクテリア宿主細胞により認識されるDNA配列を意味する。好ましい態様において、それは、融合ポリペプチドをコードしているDNA配列に作動可能に結合している。そのような結合は、融合DNA配列をコードしているDNA配列の翻訳開始コドンに関係するプロモーター位置を含む。特に好ましい態様において、このプロモーター配列は、融合DNA配列の発現を仲介する翻訳及び転写制御配列を含む。
【0046】
本明細書で用いる「ターミネーター配列」の語は、転写を終了させるために発現宿主により認識されるDNA配列を意味する。この配列は、発現される融合タンパク質をコードしている融合DNAの3’末端に作動可能に結合している。
【0047】
本明細書で用いる、「選択マーカーコード核酸配列」の語は、バクテリア細胞において発現できるヌクレオチド配列をいう。選択マーカーの発現は、対応する選択条件下において、発現している遺伝子を含む細胞に成長能力を付与する。核酸は、他の核酸配列と機能的な位置にある場合に「作動可能に連結する」という。例えば、分泌リーダー(すなわち、シグナルペプチド)をコード化するDNAは、ポリペプチドの分泌に関与するプレタンパク質として発現する場合に、当該ポリペプチドについてのDNAに作動可能に連結し、プロモーター又はエンハンサーは、配列の転写に影響を与える場合に、コード配列に作動可能に連結し、又は、リボソーム結合部位は、翻訳を促進する位置に存在する場合にコード配列に作動可能に連結するという。一般に、「作動可能に連結」とは、結合するDNA配列が隣接することを意味し、分泌リーダーの場合には、隣接しかつリーディング・フェーズ(reading
phase)中にあることを意味する。しかし、エンハンサーは隣接している必要はない。結合は、適宜の制限部位における連結反応(ligation)により達成される。そのような部位が存在しない場合には、従来法にしたがって合成オリゴヌクレオチド・アダプタ、又はリンカーを用いる。
【0048】
ここで用いる「組換え体」は、非相同核酸配列の導入により修飾された、またはそのように修飾された細胞に由来する細胞またはベクターに関するものを含む。従って、例えば組換え細胞は、天然型細胞(非組換え体)内に見られない遺伝子を発現し、あるいは、意図的な人為的介入により、そうでなければ異常に発現し、発現不足であり又は全く発現しない天然遺伝子を発現させる。
【0049】
本明細書において、「発現」という語は、ポリペプチドが遺伝子の核酸配列に基づいて生成される工程をいう。この工程には、転写及び翻訳の両方が含まれる。従って、「プロテアーゼインヒビター発現」は、発現される特定のプロテアーゼインヒビター及びそれらの変異体の転写及び翻訳を意味する。その製品は、前駆体RNA、mRNA、ポリペプチド、翻訳後処理を受けたポリペプチド及びそれらの誘導体を含む。同様に、「プロテアーゼインヒビター発現」の語は、図1で示す発現カセットにより示されるプロテアーゼインヒビター及びそれらの変異体の転写、翻訳、及び構築を意味する。プロテアーゼインヒビター発現のアッセイの例は、特定の条件に曝したときのバクテリアコロニーを用いる試験及びプロテアーゼインヒビターのウエスタンブロットだけでなくノザンブロット解析及びプロテアーゼインヒビターmRNAの逆転写ポリメラーゼ連鎖反応アッセイも含む。
【0050】
本明細書で用いる「単離」及び「精製」の語は、少なくとも1の天然由来成分が除去されている核酸又はポリペプチドを意味する。
【0051】
本明細書において用いる、「実質的に含まない(free)」の語は、乾燥重量で20%未満、10%未満、5%未満、又は1%未満の他のタンパク質(すなわち、汚染タンパク質)を含む所望のポリペプチド調製物を意味する。
【0052】
本明細書で用いる「実質的に精製された」の語は、本発明のタンパク質又はそれらのフラグメントについて使用するときは、意図する使用のための実用的及び適切な他の物質が実質的に存在しないことを言う。例えば、タンパク質はタンパク質配列決定及び/又は医薬品組成物の製造に対して使用するときは、宿主細胞の他の生物成分から十分に精製されており、このような成分を実質的に含まない。
【0053】
本明細書で用いる、「標的タンパク質」の語は、本明細書で提供される変異体インヒビターを結合することにより活性が阻害されるタンパク質(例えば、酵素、ホルモン等)を意味する。
【0054】
本明細書で用いる「変異体配列」の語は、野生型プロテアーゼインヒビター又は他のスキャッフォールドの結合ループと置き換わる短いペプチドループを意味する。この変異体配列はスキャッフォールドにおいて置き換わる場合には結合ループと同じ長さである必要はない。
【0055】
本明細書で用いる「スキャッフォールド」の語は、変異体配列が導入される野生型タンパク質配列を意味する。幾つかの態様において、スキャッフォールドは置き換わる部分(例えば、ループ)を有する。例えば、本明細書で提供されるBBI配列は変異体配列に対するスキャッフォールドとして使用することができる。
【0056】
プロテアーゼインヒビター
2のタンパク質プロテアーゼインヒビター、クニッツ(Kunitz)タイプトリプシンインヒビター(ダイズトリプシンインヒビター、STI)及び「ボウマン-バーク(Bowman-Birk)インヒビター(BBI)が大豆から単離されている(Birk,Int. J. Pept. Protein Res., 25:113-131 [1985]及びKennedy, Am. J. Clin. Neutr.,68:1406S-1412S [1998] 参照のこと)。これらのインヒビターは、変異体配列に対するスキャッフォールドとして提供される。加えて、変異体配列を含むスキャッフォールドの修飾に加えて、本明細書で用いられる他の所望のタンパク質はC末端に6のヒスチジン残基を含む(図1及び2を参照のこと)。
【0057】
ダイズトリプシンインヒビター(STI)
STIは、適切な化学量論的複合体を形成することによりトリプシンのタンパク質分解活性を抑制する(Liu, Chemistry and NutritionalValue of Soybean Components, In : Soybeans, Chemistry. Technology and Utilization, pp. 32-35, Aspen Publishers, Inc., Gaithersburg, Md., [1999] 参照のこと)。STIは2のジスルフィド結合を有する181アミノ酸残基からなり、荒い球形をしている(Song et al., J. Mol. Biol., 275: 347-63
[1998] 参照のこと)。トリプシン抑制ループは第一ジスルフィド結合の中に存在する。クニッツ(Kunitz)タイプのダイズトリプシンインヒビター(STI)は、プロテイナーゼインヒビターの活性の標準的メカニズムの定義を導く生化学及び動態研究における主要な物質として用いられ、プロテイナーゼの初期の研究に重要な役割を果たしてきた。
【0058】
ボウマン-バーク(Bowman-Birk)インヒビター(BBI)
ボウマン-バーク(Bowman-Birk)インヒビター(BBI)タンパク質は、豆科の種及び各種草を含む他の植物から単離された、動態学的及び構造学的によく知られているスモールタンパク質(60-90残基)のファミリーである。それらは、通常、それぞれ独立に結合ループを含んでいる2の三環式ドメインを含む左右対称構造をしている。しかしながら、1のドメインを有するもの、及び2より多いドメインを有するものもある。ダイズ(BBI)から単離された主に8kDa以下のボウマン-バーク(Bowman-Birk)インヒビター(BBI)は、2の分離した活性サイトループを有する。ループIはトリプシン様特異性を有するプロテアーゼを抑制し、ループIIは、キモトリプシン様特異性を有するプロテアーゼを抑制する(Chen etal., J. Biol. Chem., 267:1990 1994 [1992]、Wernerand Wemmer, Biochem., 31:999-1010 [1992]、Lin et al., Eur. J. Biochem., 212: 549-555 [1993]、Voss et al.,Eur. J. Biochem. , 242: 122-131 [1996]及びBillings et al., Pro. Natl. Acad. Sci.,
89: 3120-3124 [1992] 参照のこと)。これらの結合領域はそれぞれ、各種セリンプロテイナーゼインヒビターに見られる構造である「カノニカルループ(canonicalloop)」構造を含む(Bode and Huber, Eur. J. Biochem., 204: 433-451 [1992])。STI及びBBIはダイズの種の中のみに見られ、その植物の他の部分には見られない(Birk,Int. J. Pept.Protein Res., 25: 113-131 [1985] 参照のこと)。
【0059】
多くのBBIのイソ型が特徴付けられているけれども、配列番号13は、約71アミノ鎖残基を含むBBI主鎖のアミノ酸配列を示す(実施例1を参照のこと)。
【0060】
ダイズにおいて、BBIは19アミノ酸長のN末端プロペプチドを有するプロタンパク質として生産される。従って、幾つかの態様において、BBIは、完全なプロペプチド又は少なくともプロペプチドの1部を用いて生産される。幾つかの態様において、BBIはN-又はC-末端のいずれかから10ものアミノ残渣が除去されて、切断されている。例えば、種子を乾燥するときに、幾つかのBBI分子は、9又は10アミノ酸残基が取り除かれたC末端を有する。従って、タンパク質分解は、通常、最初のジスルフィド結合の前及び最後のジスルフィド結合の直後が高くなる。この影響は標的タンパク質への結合にとり通常マイナスとはならない。しかしながら、異性体又は切断形態のいずれか1が本発明の各種態様で使用されることは評価されるであろう。
【0061】
プロテアーゼインヒビター変異体
上で説明するように、STI及びBBIプロテアーゼインヒビターはプロテアーゼを抑制する結合ループを有する。本発明は、1以上の活性部位が修飾されている(例えば、BBIのループI及び/又はループII)プロテアーゼインヒビター変異体を提供する。幾つかの好ましい態様において、標的タンパク質と相互作用する配列でこのループを置換する。
【0062】
例えば、幾つかの態様において、このループは、VEGF結合タンパク質、C2、C3、C4及びC5インヒビター等の補体経路のインヒビター、コンプスタチン(Compstatin)、サイトカイン、他の所望のタンパク質等に由来する配列で置換される。すなわち、任意の適切な配列を本発明に用いることから、本発明はこれらのループのいずれかの中で置換された特定の配列に限定することは意図していない。
【0063】
幾つかの態様において、変異体配列は、ファージディスプレイ法及び他の適切な方法を含むがこれらに限定されない本技術分野で知られた各種方法により選択される。例えば、ランダムペプチド遺伝子ライブラリーは、ファージPIII遺伝子で融合され、このペプチドライブラリーは、ファージの表面に提示される。引き続き、このファージディスプレイライブラリーは、標的タンパク質に曝され、非特異結合を除去するために緩衝液で洗浄される(この工程はパニングと称される)。最終的に、結合しているファージ及びこのペプチドをコードするDNA配列のPCR産物が単離される。
【0064】
幾つかの態様において、このループの1は、変異体配列で置換される(すなわち、ペプチド、3から14のアミノ酸長、5から10のアミノ酸長が好ましい)。それらが所望の結合及び/又は抑制を示す限り、より長い配列を本発明に用いることができる。加えて、結合ループの置換に適したペプチドが固定ループ内(すなわち、2のシステイン残基の間のジスルフィド結合の存在により形成されいているループ)に含まれる場合には、機能的な構造を採用することが好ましい。幾つかの態様において、このペプチドは7から9アミノ酸長である。これらの置き換え配列もまたプロテアーゼインヒビター又は標的配列への結合も提供する。幾つかの態様において、1のアミノ酸が置換されていることが好ましい。
【0065】
融合タンパク質
好ましい態様において、各プロテインプロテアーゼインヒビター又はそれらの変異体は宿主バクテリア細胞により融合タンパク質として発現される。所望のタンパク質を遊離するために融合タンパク質を切断することは有用であるが、必要ではない。融合タンパク質として発現及び分泌されたプロテアーゼインヒビター及びそれらの変異体は、驚くべきことにそれらの機能を保持している。
【0066】
対応するアミノ配列をコードする、上で定義するDNA配列は「融合DNA配列」を形成するように結合される。そのようなDNA配列は、第一、第二、第三及び第四DNA配列の順で、5’末端から3’末端に向けて固有のリーディングフレーム中に構築される。そのように構築されるので、このDNA配列は、アミノ末端からバクテリア種の中で分泌配列として機能するシグナルペプチド、バクテリア種から通常分泌される分泌ポリペプチド又はそれらの一部、切断リンカーペプチド及び所望のタンパク質(例えば、プロテアーゼインヒビター及びそれらの変異体)をコードしている「融合ポリペプチド」をコードする。融合タンパク質を生産するための各種方法は本技術分野で知られている(米国特許5,411,873、5,429,950及び5,679,543参照のこと。これらの文献を参照により本明細書に援用する)。従って、任意の適切な方法を本発明に用いることができる。
【0067】
組換えプロテアーゼインヒビターの発現
本発明は融合タンパク質の生産に依存しているので、本発明を実施するには組み換え遺伝子の分野の既存技術を用いる。本発明に用いる一般的方法が開示されている基本テキストには、Sambrooket al., Molecular Cloning, A Laboratory Manual (2nd ed. 1989);Kriegler, GeneTransfer and Expression : A Laboratory Manual (1990); 及びAusubel et al., eds.,Current Protocols in Molecular Biology(1994)がある。
【0068】
本発明は、プロテアーゼインヒビターをコードする核酸配列を含む発現ベクターで形質導入、形質転換又は形質感染させたバクテリア宿主細胞を提供する。温度、pH等の培養条件は形質導入、形質転換、又は形質感染以前に親宿主細胞に用いられるものと同様であり、当業者にとって自明な事項である。
【0069】
基本的に、融合タンパク質をコードしているヌクレオチド配列は宿主細胞の中で機能するプロモーター配列に作動可能に結合している。このプロモーター遺伝子単位は、複製及び/又は発現のために宿主細胞の中で形質転換する前に仲介ベクター内でクローンされる。これらの仲介ベクターは、通常原核細胞ベクター(例えば、プラスミド、又はシャトルベクター)である。しかしながら、この工程は幾つかの好ましい態様では省略することができるので、本発明は仲介ベクターを使用する方法に限定されない。
【0070】
一のアプローチにおいて、プロテアーゼをこの細胞の中で発現させるために、プロテアーゼインヒビターをコードする核酸配列に作動可能に結合する、プロモーター、生物学的に活性なプロモーターフラグメント、又は宿主細胞の中で機能する1以上の(例えば、連続する)エンハンサーを有する発現ベクターによりバクテリア培地を形質転換する。幾つかの好ましい態様において、このDNA配列はプロテアーゼインヒビター又はそれらの変異体をコードする。他の好ましい態様において、プロモーターは調節可能なものである。
【0071】
核酸構築体/発現ベクター
プロテアーゼインヒビター(すなわち、「PI-コード核酸配列」)をコードしている天然又は合成のポリヌクレオチドフラグメントは、異種の核酸構築体又はベクターに取り込まれ、バクテリア細胞の中に導入され、複製される。本明細書で開示するベクター及び方法は、プロテアーゼインヒビター及びそれらの変異体を発現させる各種宿主細胞に用いるのに適している。導入された細胞内で複製され、利用可能である限り、どのようなベクターでも用いることができる。本技術分野において数多くの適したベクター及びプロモーターが知られており、市販されている。適したクローニング及び発現ベクターは、本技術分野においてよく知られている文献に記載されている(上記のSambrooket al.,及びAusbel et al.,を参照のこと。これらの文献を参照により本明細書に援用する)。適切なDNA配列は適切な方法を用いてプラスミド又はベクター(これらを集合的に「ベクター等」と言う)内に挿入される。一般的にDNA配列は当該技術分野分野において既知の標準的方法を用いて適した制限エンドヌクレアーゼ内に挿入される。
【0072】
適切なベクターは、通常、選択マーカーコード核酸配列、挿入部位、及び転写終了配列等の適切な制御エレメントを備えている。幾つかの態様において、ベクターは例えば、コード配列に作動可能に結合している宿主細胞の中の(及び/又は修飾された可溶性タンパク質コード配列が通常発現しないベクター又は宿主細胞の環境内で)コード配列の発現に効果的な制御エレメント(すなわち、プロモーター及びターミネーターエレメント又は5’及び/又は3’非翻訳領域)を含む調節配列を含む。数多くの適切なベクター及びプロモーターは本発明の技術分野において知られており、それらの多くは市販されている。
【0073】
典型的なプロモーターは、構造プロモーター及び誘導プロモーターの両方を含む。そのようなプロモーターは、当該技術分野で知られている。当業者は、天然のプロモーターが有する機能を変えることなく1以上のヌクレオチドの交換、置換、付加又は排除により修飾することができることを知っている。本発明はプロモーターに対するそのような変更を包含するけれども、このことに限定されるものではない。遺伝子構築体に用いるプロモーターの選択は当業者の知識の範囲内である。
【0074】
適正な選択マーカーを選択することは宿主細胞に依存している。異なるバクテリア宿主に対して適切な選択マーカーを選択することは当業者の知識の範囲内である。典型的な選択マーカー遺伝子は(a)抗生物質又は他の毒性(例えば、アンピリシン、メトキサレート、テトラサイクリン、ネオマイシン マイコフェノール酸、ピューロマイシン、ゼノマイシン又はヒドロマイシン)に対する耐性を付与するタンパク質、又は(b)宿主細胞における栄養素要求性変異又は自然発生栄養欠失を補うタンパク質をコードする。
【0075】
幾つかの態様において、選択されたPIコード配列は良く知られた組み換え技術に従って適切なベクター内に挿入され、PI発現が可能な細胞系統を用いた形質転換に用いられる。遺伝子コード固有の退縮により、実質的に同じ又は機能的に等しいアミノ酸配列をコードする他の核酸配列はクローニングに用いられ、及び上で詳述したように特定のプロテアーゼインヒビターを発現する。それゆえコード領域において、そのような置換をすることは本発明でカバーされる配列変異体に含まれる。これらの配列変異体のいずれか及びすべては、親PIコード核酸配列に対するのと同じ方法で本発明に用いることができる。当業者は異なるPIは異なった核酸配列によりコードされることを認識している。
【0076】
幾つかの態様において、プロテアーゼインヒビター核酸配列の所望の形態、相同体、変異体又はそれらのフラグメントが一旦形成されると、多くの方法により修飾される。非コードフランキング領域を含む配列の場合、このフランキング領域は、切除、変異等を受ける。従って、塩基転位、塩基転移、欠失及び挿入が天然配列上で起こる。
【0077】
幾つかの好ましい態様において、異種核酸構築体は少なくとも1のプロテアーゼインヒビター、又はそれらの変異体、フラグメント、又はスプライス変異体のコード配列を、(i)単独で、(ii)PIコード配列が主なコード配列である場には、融合タンパク質又はシグナルペプチドコード配列のような追加的なコード配列と組み合わせて、(iii)適切な宿主細胞におけるコード配列の発現に有効な、プロモーター及び終了エレメント又は5’及び/又は3’非翻訳領域等の調節エレメントのような非コード配列と組み合わせて、及び/又は(iv)PIコード配列が異種の遺伝子となるようなベクター又は宿主細胞環境下に含んでいる。
【0078】
幾つかの態様において、適切なプロモーター及び制御配列と一緒に適切な核酸コード配列を含む異種の核酸は、少なくとも1のプロテアーゼインヒビター又はそれらの変異体を発現させるためにバクテリア宿主細胞内へ導入される。
【0079】
本発明の幾つかの態様において、異種の核酸構築体はインビトロ(in vitro)において細胞内へPIコード核酸配列を輸送するために用いられる。幾つかの好ましい態様において、この宿主細胞は本発明の核酸配列を安定に組み込む。従って、安定な形質転換体を効果的に生成するための任意の適切な方法を本発明に用いる。
【0080】
本発明の追加的な態様において、第一及び第二発現カセットは単一ベクター上に存在する。一方、他の態様においては、これらのカセットは分離したベクター上に存在する。
【0081】
幾つかの好ましい態様において、プロモーター配列に加えて、効果的な転写終了を提供するために、発現カセットは構造遺伝子組成物の下流に転写終了領域を含む。幾つかの態様において、転写終了領域は、プロモーター配列と同じ遺伝子から得られるが、他の態様においては、他の遺伝子から得られる。適切な転写終了シグナルの選択は当該技術分野においてよく知られている。
【0082】
更に、任意の適切な発現ベクターを本発明に用いることができる。すなわち、原核又は真核生物の細胞中の発現に用いる各種簡便なベクターが適しており、本発明に用いることができる。標準的なバクテリア発現ベクターは、pBR322ベースプラスミド、pSKF、pET23D等のプラスミド及びMBP、GST及びLacZ等の融合発現システムだけでなくバクテリオファージλ及びM13も含む。更なる態様において、単離を簡便にする目的でエピトープタグが組換えタンパク質に添加される(例えばc-myc)。
【0083】
発現ベクターに通常含まれる追加的なエレメントは、レプリコン、組換えプラスミドを有するバクテリアを選択するのための抗生物質耐性をコードする遺伝子、及び異種配列を挿入するためのプラスミドの非必須領域における特徴的な制限部位である。特定の抗生物質耐性遺伝子を選択することは決定的な要素ではない。任意の多くの知られた抗生物質遺伝子が適している。
【0084】
宿主細胞へのプロテアーゼインヒビターコード核酸配列の導入
幾つかの好ましい態様において、本発明の方法は、安定に組み込まれた所望の配列(すなわち、PIコード核酸)を含む宿主細胞を提供する。しかしながら、代替的な態様において、本発明の方法は、自己複製染色体外形質転換ベクターの維持も提供する。
【0085】
本発明は、外部から提供されたPIコード核酸配列を包含させるために遺伝的に修飾された細胞及び細胞組成物を提供する。幾つかの態様において、親宿主細胞は、発現ベクターで遺伝的に修飾されている。幾つかの態様において、このベクターはプラスミドであり、他の態様においてはこのベクターはウイス粒子、ファージ、ネイキッドDNA等である。従って、各種ベクターを本発明に用いることができるので、ベクターの形態を特定のベクターに限定することは意図しない。
【0086】
インビトロ(in vitro)において発現ベクターの輸送のために各種方法が用いられる。異種核酸配列の発現のために細胞内へ核酸を導入する方法は、当業者に知られており、エレクトロポレーション、無傷細胞によるプロトプラスト融合、形質感染、DNAコート-マイクロプロジェクタイル法を用いた高速照射、修飾ウイスル(例えばファージ)核酸を用いた感染、化学媒介形質転換、コンピテンス等を含むがこれらに限定されない。更に、幾つかの態様において、PIコード核酸を含む異種核酸構築体は、インビトロ(in
vivo)で転写され、得られたRNAは宿主細胞内へ本技術分野でよく知られた方法を用いて導入される。
【0087】
プロテアーゼインヒビターのコード配列を含む異種核酸構築体の導入に続いて、遺伝的に修飾された細胞は、プロモーターの活性化、形質転換体の選択及び/又はPIコード核酸配列の発現の増幅のために適切に調整された栄養培地中で培養される。温度、pH等の培養条件は発現のために選択された宿主細胞に対して用いている条件と同じであり、当業者に明らかである。
【0088】
このようにして異種核酸構築体が導入された細胞の子孫は、異種核酸構築体の中に存在したPIコード核酸配列を含むと考えられる。
【0089】
バクテリア宿主細胞及び発現
適切な宿主細胞は任意の好適なバクテリア種を含む。幾つかの態様において、バクテリア宿主細胞は発現宿主細胞及び第一及び第二核酸の供給源として機能する。本発明の方法及び宿主細胞を用いて、驚くべきレベルの発現を得ることができる。本明細書で用いるシステムを用いて達成されるプロテアーゼインヒビターの発現及び分泌レベルは0.5
g/l以上である。
【0090】
発現ベクターが宿主細胞に導入された後で、形質転換された宿主細胞は、所望のタンパク質をコードする遺伝子の発現に好ましい環境下で培養される。形質転換細胞の大容量バッチを上で述べたように培養することができる。最終的に、生産物は当該技術分野において既知の方法により回収される。
【0091】
チオールジスルフィド酸化還元酵素又はシャペロンのような補足タンパク質は分泌されたタンパク質を活性構造へと折りたたむことを助けることから、本発明の幾つかの態様にこれらの補足的なタンパク質を用いることができる。チオールジスルフィド酸化還元酵素及びタンパク質ジスルフィドイソメラーゼは、タンパク質の中の正確なジスルフィド結合の形成を促進する。バチルススブチルス(B.subtilis)の中のbdbDCオペロンの過剰発現は、ジスルフィド結合を有するタンパク質の生成に有用であることが証明されている(Meima etal.,J. Biol. Chem., 277: 6994-7001, [2002] 参照のこと)。シャペロンは、折りたたまれていない状態中の、曝されている疎水性の領域に結合することによって、分泌タンパク質が折りたたまれるのを助け、好ましくない相互作用を防ぎ、プロピル-ペプチドイル シス-トランスイソメメラーゼはプロリン残基に隣接するペプチド鎖の適正な立体構造の形成を補助する。
【0092】
本発明の幾つかの態様において、宿主細胞は少なくとも1のチオールジスルフィド酸化還元酵素又はシャペロンをコードする発現ベクターで形質転換される。本発明は特定のチオールジスルフィド酸化還元酵素又はシャペロンに限定することを意図していないので、本発明の技術分野で知られている任意の適切なチオールジスルフィド酸化還元酵素又はシャペロンが本発明に用いられる。
【0093】
本発明の幾つかの態様において、適切に折りたたまれた分泌タンパク質の分画は、成長培地にジスルフィド結合を酸化及び/又は還元する試薬、及び/又は通常の酸化還元電位を変更する試薬、及び/又は溶媒の性質を変更し、タンパク質の構造及び凝集に影響を与える薬品を添加することにより増やすことができる。特に好ましい態様において、2-メルカプトエタノールのようなジスルフィド結合を還元する試薬は、適切に折りたたまれたタンパク質の分画を高めるために好ましい。しかしながら、他の態様及び使用する培地によっては、他のジスルフィド還元及び酸化試薬(例えば、DTT、TCEP、酸化及び還元型グルタチオン,システイン、シスチン、 システアミン、 チオグリコレート、S2O32-、S2O42-、S2O52-、SO32-、S2O72-、Cu+等)を単独で、又は組み合わせて本発明に用いることがきでる。単独で、又は、βME等のジスルフィド酸化還元試薬と組み合わせて成長培地に添加する溶媒の性質を変更する他のアジュバンド(例えば、尿素、DMSO、TWEEN(登録商標)-80等)も、適切に折りたたまれた分泌ペプチドの分画を増やし、本発明の各種態様に用いることができる。幾つかの好ましい態様において、βMEは0.5mM乃至4 mMの濃度の幅で、他の態様においては、0.1 mMから10 mMの濃度で用いられる。すなわち、当業者は、用いるべきチオール酸化還元試薬及び他のアジュバンドの濃度だけでなく、添加するチオール酸化還元試薬及び/又は他のアジュバンドの影響を最適化するための最良の成長培地及び最良の成長条件をどのように選択するかを知っている。本発明は特定のジスルフィド還元/酸化試薬又はアジュバンドに限定することを意図していないので、本発明の技術分野において知られている任意の適切な試薬を本発明に用いることができる。
【0094】
発酵指標
本発明は培養されたバクテリア種に対する発酵方法に関係する。バクテリア種による異種タンパク質の生産に対する発酵方法は当該技術分野でよく知られている。培養は、水溶性ミネラル塩、培地、有機成長因子、炭素源及び栄養源となる物質、酸素分子(好気性及び嫌気性バクテリアに対する)を含み、用いられる1以上の特定の微生物種の開始接種をした培地中で行われる。
【0095】
適切な微生物の成長を確実にし、この微生物の転化工程における細胞による炭素及びエネルギー源の同化を最大限にし、発酵培地の中で最大細胞濃度で、最大細胞生産を達成するために、炭素及び栄養源、同化窒素並びに微生物の接種に加えて、適切な割合のミネラルを適切な量供給することが必要である。
【0096】
当該技術分野で知られているように、各種培養培地を本発明に用いることができる。しかしながら、標準的なバクテリア培養培地も本発明に用いられる。幾つかの好ましい態様において、培地は、窒素に加えて、適切な量のリン酸、マグネシウム、カルシウム、カリウム、硫黄、及びナトリウムを適切な可溶性同化イオン及び結合状態で含み、銅、マンガン、モリブデン、亜鉛、鉄、ホウ素、及びヨウ素等の微量元素も、適切な可溶性同化形態で培地中に含まれる。
【0097】
幾つかの態様において、発酵反応は、微生物を勢い良く成長させるために有効な適切な酸素分圧力において、発酵管の内容物を維持するために空気、酸素高含量空気、又は実質的に純粋な酸素分子等の酸素分子含有ガスにより必要な酸素分子が供給される好気プロセスを含む。実際には、酸素化炭化水素基質を用いることにより、微生物の成長に必要な酸素を減らすことができる。しかしながら、好気性微生物及びより程度は低いが嫌気性微生物の生長為に酸素分子の供給は必要である。
【0098】
通気割合は非常に広範囲にわたり変動させることができが、通常の通気速度は、一分間に発酵槽中の液体当たりに供給される酸素含有気体の容量は、(採用圧力及び25℃において)約0.5乃至10容量、好ましくは約0.5乃至7容量である。この量は反応器に供給される通常の酸素含量の空気に基づいている、そして、精製酸素に換算すると、前記割合は、(採用された圧力及び25℃の温度において)1分間に発酵槽の中の液体当たりに供給される酸素は約0.1乃至1.7容量又は好ましくは0.1乃至1.3容量である。
【0099】
微生物転化プロセスに対して用いられる圧力は幅広く変動する。通常の圧力は、装置及び操作コストに対して達成される溶存酸素の理由から、0乃至50 psig、好ましくは0乃至30psig、より好ましくは少なくとも大気圧よりわずかに高い圧力である。大気圧より高い圧力は水性発酵槽内の溶存酸素を増やす傾向があり、それによって、細胞の成長速度を高めることから大気圧より高い圧力は有利である。同時に、高い大気圧は装置及び操作のコストを高めるという事実とバランスする必要がある。
【0100】
発酵温度は若干変動させることができるが、大部分のバクテリア種にとって、本発明の技術分野において知られているように、バクテリア種にもよるけれども、通常20℃乃至40 ℃、一般的に好まれている温度は、28 ℃乃至37 ℃である。
【0101】
微生物は、同化窒素源も必要とする。同化窒素の源は、微生物による代謝の利用に適した形態で窒素を放出することができる任意の窒素含有化合物である。タンパク質の加水分解物等の各種有機窒素化合物を用いることができるけれども、通常、アンモニア、水酸化アンモニウム、尿素、リン酸アンモニウム、硫酸アンモニウム、ピロリン酸アンモニウム、塩化アンモニウム等の各種アンモニウム塩、又は、安価な他の各種アンモニウム化合物を用いることができる。アンモニアガスは大規模操作に対して便利であり、、水性発酵槽(発酵培地)中への通気により適切な量を用いることができる。同時に、そのようなアンモニアはpH調節にも用いることができる。
【0102】
水溶性微生物発酵物(発酵混合物)の中のこのpHの幅は、約2.0乃至8.0の典型的な幅である。しかしながら、特定の微生物に対して適したpH幅は、微生物だけでなく、用いる培地にも依存する。従って、当該技術分野で知られているように、培地組成によりpHを幾分変更することも可能である。
【0103】
発酵槽内での発酵混合物の平均滞留時間は、本発明の技術分野で知られているように、発酵温度及び用いられる培地に部分的に依存して変動する。
【0104】
幾つかの態様において、発酵は、炭素含有基質が制限因子として制御でき、それゆえ、良い炭素含有基質の転化が細胞に提供され、非常に大量の転化されない基質による細胞の汚染を避けるような方法が好ましい。残留微量元素は容易に取り除くことができるので、汚染は水溶性基質では問題とならない。しかしながら、非水溶性基質の場合には問題となるので、適切な洗浄工程等の追加的な生産工程が必要とされる。この制限された基質レベルを達成するために必要な時間は、重要ではなく、実施される特定の微生物及び発酵工程により変動する。しかしながら、発酵培地の中の炭素源濃度及び望ましい炭素源レベルが達成されたか否かの決定方法は本発明の技術分野において知られている。
【0105】
幾つかの態様において、発酵はバッチ又は連続操作で実施されるけれども、フェドバッチ操作は制御が容易で、生成物の質が均一であり、全装置の使用が経済的であることから好ましい。
【0106】
必要ならば、炭素及び栄養源物質の一部又は全部及び/又はアンモニア等の同化窒素源は発酵槽内に水溶性ミネラル培地を供給する前に添加することができる。すなわち、反応器に導入される原材料流れの各々は、予め決められた速度で、又は発酵槽から出てくるオフガス中の炭素源及びエネルギー基質、pH、溶存酸素、酸素又は二酸化炭素、光透過度により測定される細胞密度等の濃度モニタリングで決定される必要性に応じて制御される。各種物質の供給速度は、チャージされた基質に対してできる限り高い微生物細胞の産生を達成し、効果的な炭素及びエネルギー源の利用と矛盾しないようにできるだけ早い細胞成長を得るために変動させることができる、しかし、より重要なのは、容量単位当たり最も多い生成物を得ることである。
【0107】
各バッチにおいて、好ましくはフェドバッチ操作において、全部の装置、反応器、又は発酵手段、発酵管又はコンテナ、配管、付属の循環又は冷却装置等は、初めに通常約121℃で約15分間蒸気に曝すことにより滅菌される。滅菌された反応器では、酸素を含む必要な全ての栄養素及び炭素含有基質の存在下で選択された微生物を培地に接種する。用いられる発酵槽のタイプは、重要ではないが、幾つかの態様において、15Lのバイオラフィッテ(Biolafitte)(Saint-Germain-en-Laye、France)が好ましい。
【0108】
タンパク質の分離
好ましい態様において、一旦所望のタンパク質が発現すると、分泌タンパク質が分離される。本発明はその融合類似体から所望のタンパク質を分離する方法を提供する。本明細書で述べる方法はプロテアーゼインヒビター及びその変異体を融合類似体から分離することに用いることができる。
【0109】
発酵ブロスから所望のタンパク質を収集及び精製することは、本発明の技術分野で知られている方法を用いて行うことができる。発酵ブロスは通常、所望のタンパク様物質のほかに、細胞を含む細胞の破片、各種懸濁された固形物、及び他の生物汚染物質を含む。これらは、本発明の分野で既知の方法により発酵ブロスから除去することが望ましい。そのような除去に適した工程は、細胞を含まないろ液を生産するための従来の固体-液体分離技術である(例えば、遠心分離、ろ過、透析、精密ろ過、ロータリー吸引ろ過、又は他の既知の方法)。幾つかの態様において、発酵ブロス又は他の無細胞ろ液を精製及び/又は結晶化工程に用いる前に、精密ろ過、エバポレーション、及び/又は透析を用いて、更に濃縮することが好ましい。
【0110】
上清中のタンパク様成分の沈殿又はろ過は、塩(例えば、硫酸アンモニウム)又は低いpH(通常3未満)を用いて行い、次いで、各種クロマトグラフ手段(例えば、イオン交換クロマトグラフィー、親和クロマトグラフィー、疎水相互作用クロマトグラフィー、疎水電化誘導クロマトグラフィー(hydrophobiccharge induction chromatography)等)又はこれと同様の方法による精製が続く。本発明は特定の分離方法に限定されないので、任意の分離方法を本発明に用いることができる。
【0111】
特に好ましい態様において、発現された所望のポリペプチドがバクテリア細胞から分泌されると、このポリペプチドは成長培地から精製される。好ましい態様において、発現宿主細胞はこのポリペプチドの精製(例えば、遠心分離)の前に、培地から除去される。
【0112】
発現された所望の組換えペプチドが宿主細胞から分泌されない場合には、宿主細胞を崩壊させ、及び水相に放出されたポリペプチドを「抽出」することが精製の第一工程となることが好ましい。この場合、発現宿主細胞は、細胞を崩壊させる前に培地から集められることが好ましい(例えば、遠心分離)。細胞破壊は、リゾチーム、又はベータグルカナーゼ消化により、又は、細胞を高圧で処理すること等の本発明の技術分野で知られている方法を用いて行われる(Scobes,Protein Purification. Second edition, Springer-Verlag参照のこと)。
【0113】
幾つかの態様において、6のヒスチジン残基(すなわちヒスタグ)をC末端に付加することは所望のタンパク質及びその融合アナログの精製における補助として用いられる。精製の補助としてのヒスタグの使用は、当該技術分野においてよく知られている(Hengen,TIBS
20: 285-286 [1995] 参照のこと)。6のヒスタグを付加されたタンパク質は、当該技術分野で知られている固定金属イオン親和クロマトグラフィー(IMAC)を用いて容易に精製することができる。
【0114】
精製
特定の使用目的のためには、本発明を用いて製造されたプロテアーゼインヒビターは高度に精製されていることが重要となる(例えば、99 %以上の精製度)。精製タンパク質が治療に用いられるときには高度に精製されていることは特に重要であるが、他の使用目的においても必要である。本発明の明細書で述べる方法は、実質的に精製された所望のタンパク質を製造する方法である。本明細書で述べる所望のタンパク質は、医薬品及びパーソナルケア製品に有用である。しかしながら、各種精製レベルのタンパク質が本発明の方法を用いて製造され、本発明の方法を用いて精製したタンパク質が特定の精製レベルであることは意図しない。
【0115】
精製の間のBBIの活性化
本発明の幾つかの態様において、精製工程における育成の後、ジスルフィド結合を酸化及び/又は還元する組成物、及び/又は通常の酸化還元電位を変更する組成物、及び/又は溶媒の性質を変更しタンパク質の構造及び凝集に影響を与える組成物を添加することによりタンパク質の活性が増強される。幾つかの特定の好ましい態様において、2-メルカプトエタノールのようなジスルフィド結合を還元する試薬の添加は、タンパク質の活性を高めるのに使用される。しかしながら、当該技術分野において知られているように、精製度及び緩衝液の組成に依存して、他のジスルフィド還元又は酸化試薬(例えば、DTT、
TCEP、酸化及び還元グルタチオン、システイン、 シスチン、 システアミン、 チオグリコレート、S2O32-、S2O42-、S2O52-、SO32-、S2O72-、Cu+、タンパク質ジスルフィド イソメラーゼ、タンパク質チオールジスルフィド オキシドレダクターゼ等)を単独で、又は組み合わせて本発明に用いることができる。単独で、又はβME等のジスルフィド酸化還元試薬と組み合わせて溶媒の性質を変更する他のアジュバンド(例えば、エタノールアミン、DMSO、TWEEN(登録商標)-80、アルギニン、尿素、等)も、タンパク質の活性を高めるために本発明に用いることができる。特定の好ましい態様において、部分的に精製されたタンパク質が緩衝液の中に溶解され(特に好ましい態様において、塩基性pHにおけるTWEEN(登録商標)-80を用いた両性イオン緩衝液)bME及びジスルフィド酸化試薬(代替的な好ましい態様は、酸化グルタチオン又は硫酸ナトリウム)を用いて活性化される。
【0116】
更に、必要ならば、タンパク質の最適な活性を決定するための条件が選択される。例えば、各種βME濃度(0.1乃至10 mM)、酸化試薬濃度(βMEの濃度の0乃至1/20乃至20倍)、pH(7.5乃至9.5)、温度(15乃至40℃)、希釈倍率(1乃至20倍)、インキュベーション時間(12乃至72時間)、通気(酸素含有ガスの下で激しく攪拌するための挿入ガス下におけるインキュベーション)、緩衝液タイプ(トリス、CHES、CAPS、トリシン(Tricine)、TAPS、他の両性イオン緩衝液等)、緩衝液濃度(0.1乃至1M)、及び溶媒の性質を変え、それゆえタンパク質の立体構造に影響を与え、凝集をさせることが知られている各種アジュバンド(例えば、エタノールアミン、DMSO、TWEEN(登録商標)-80、アルギニン、尿素、等)の添加、が用いられる発現システムの最適な条件を決定するために試験される。本発明は特定の還元/酸化試薬、希釈倍率、温度、pH、緩衝液タイプ、又は組成物、並びにアジュバンドに限定することを意図していないので、当該技術分野で既知の好適な試薬を本発明に用いることができる。
【0117】
実施例
本発明は、以下の実施例においてより詳細に説明されるが、これら実施例に本発明を限定することは意図していない。添付の図は、明細書及び本発明の不可欠な部分である。引用された全ての文献は本発明の開示のために参照により本明細書に援用される。以下の実施例は、本発明を説明するためのものであり、本発明を限定するためのものではない。
【0118】
以下の実施例において、以下の略字を用いる。
【0119】
PI(プロテアイナーゼインヒビター)、BBI(Bowman-Brikインヒビター)、STI (ダイズトリプシンインヒビター)、 VEGF 及びVegF (血管内皮成長因子)、ppm(100万分の1)、 M(モルの)、mM(ミリモルの)、μM(マイクロモルの)、nM(ナノモルの)、mol(モル)、 mmol(ミリモル)、μmol(マイクロモル)、nmol(ナノモル)、gm(グラム)、mg(ミリグラム)、μg(マイクログラム)、pg(ピコグラム)、L(リットル)、ml 及び mL(ミリリットル)、 μl及びμL (マイクロリットル)、cm(センチメートル)、mm(ミリメートル)、μm(マイクロメートル)、nm(ナノメートル)、 U(単位)、V(ボルト)、MW(分子量)、sec(秒)、min(s)(分)、h(s)及びhr(s)(時間)、℃(摂氏)、QS(十分量)、ND(検出不能)、NA(適用不能)、rpm(1分間あたりの回転数)、H2O(水)、dH2O(脱イオン水)、HCl(塩酸)、aa(アミノ酸)、bp(ベースペア)、
kb(キロベースペア)、kD (キロダルトン)、cDNA(転写又は相補DNA)、DNA(デオキシリボ核酸)、ssDNA(一本鎖 DNA)、dsDNA(二本鎖DNA)、dNTP(デオキシリボヌクレオチド三リン酸)、RNA(リボ核酸)、MgCl2(塩化マグネシウム)、NaCl(塩化ナトリウム)、w/v(容量対重量)、v/v(重量対重量)、g(比重)、OD(光学密度)、ダルベッコリン酸緩衝溶液(DPBS)、SOC(2% バクト-トリプトン、0.5 % バクトイースト抽出物、10 mM NaCl、2.5 mM KCl),テリフィックブロス(TerrificBroth)(TB;12 g/l バクト-トリプトン, 24 g/lグリセロール、2.31 g/l KH2PO4、及び12.54g/l K2HPO4) 、OD280(280 nmにおける光学密度) 、OD600(600nmにおける光学密度) 、A405 (405 nmにおける吸光度)、Vmax(酵素触媒反応における最大速度)、PAGE(ポリアクリルアミドゲル電気泳動)、PBS(リン酸緩衝生理食塩水 [150 mM NaCl、10 mM リン酸緩衝液、pH 7.2]) 、PBST(PBS+0.25% TWEEN(登録商標)20)、PEG(ポリエチレングリコール)、PCR(ポリメラーゼ連鎖反応)、RT-PCR(逆転写PCR)、SDS(ドデシル硫酸ナトリウム)、Tris(トリス(ヒドロキシメチル)アミノメタン)、HEPES(N-[2-ヒドロキシエチル] ピペラジン-N-[2-エタンスルホン酸] )、HBS(HEPES 緩衝生理食塩水)、SDS(ドデシル硫酸ナトリウム)、bME,BME 及びβME (ベータメルカプトエタノール又は2-メルカプトエタノール)、Tris-HCl(トリス [ヒドロキシメチル]アミノメタン-クロライド)、Tricine(トリシン)(N-[トリス-(ヒドロキシメチル)-メチル]グリシン)、CHES(2-(N-シクロ-ヘキシルアミノ)エタン-スルホン酸)、TAPS(3-{[トリス-(ヒドロキシメチル)-メチル]-アミノ}-プロパンスルホン酸)、CAPS(3-
(シクロ-ヘキシルアミノ)-プロパンスルホン酸)、DMSO(ジメチルスルホキシド)、DTT(1,4-ジチオ-DL-トレイトール)、Glut 及び GSH (還元グルタチオン)、GSSG(酸化グルタチオン)、TCEP(トリス[2-カルボキシメチル]ホスフィン)、Ci(キューリ)、mCi(ミリキューリ)、μCi(マイクロキューリ)、TLC(薄層クロマトグラフィー)、Ts(トシル)、Bn(ベンジル)、Ph(フェニル)、Ms(メシル)、Et(エチル)、Me(メチル)、Taq(サーマスアクアチクス(Thermusaquaticus)DNA ポリメラーゼ)、Klenow(DNA ポリメラーゼ I ラージフラグメント(Klenow(クレノウ))フラグメント )、rpm(一分当たりの回転数)、EGTA(エチレングリコール-ビス(β-アミノメチル エーテル)N, N, N', N'-四酢酸)、EDTA(エチレンジアミン四酢酸)、bla(β-ラクタマーゼ 又は アンピシリン耐性遺伝子)、GEHealthcare (GEヘルスケア、チャルフォントストリート、ジャイルズ、UK)、DNA2.0(DNA2.0、メロンパーク、カリフォルニア)、OXOID(オキシドベージングストーク、ハンプシャー、UK)、Megazyme(メガザイム インターナショナル アイルランド社、ブレービジネスパーク、ブレー、ウィックロー、アイルランド)、Coming(コーニングライフサイエンス、コーニング、ニューヨーク)、NEN(NENライフサイエンスプロダクツ、ボストン、マサチューセッツ)、Pharma AS(ファルマAS, オスロ、ノルウェイ)、Dynal(ダイナル、オスロ、ノルウェイ)、Bio-Synthesis(バイオ-シンセシス、ルイスビル、テキサス)、ATCC(アメリカンタイプカルチャーコレクション、ロックイビル、メリーランド)、Gibco/BRL(ギブコ/BRL、グランドアイランド、ニューヨーク)、Sigma(シグマケミカル社、セントルイス、ミズリー)、Pharmacia(ファルマシア、ピスカタウェイ、ニュージャージー)、NCBI(全米バイオテクノロジー情報センター)、Applied Biosystems(アプライドバイオシステム、フォスターシティー、カリフォルニア)、Clontech(クローンテックラボラトリーズ、パロアルト、カリフォルニア)、OperonTechnologies(オペロンテクノロジイーズ社、アラメダ、カリフォルニア)、MWG Biotech(エムダブリュジーバイオテック、ハイポイント、ノースカロライナ)、Oligos
Etc(オリゴイーティーシ社、ウィルスンビル、オーランド)、Bachem (ベイケムバイオサイエンス社、キングオブプルシア、ペンシルバニア)、Difco(ディフコラボラトリーズ、デトロイト、ミシガン)、Mediatech(メディアテック、ハードン、バージニア)、Santa Cruz(サンタクルーズバイオテクノロジー、カリフォルニア)、BioVeris(バイオベリス社、ゲーサーズバーグ、メリーランド)、Oxoid(オキシド社、オグデンスブルグ、ニューヨーク)、Worthington(オージントンバイオケミカル社、フリーホールド、ニュージャージー) 、GIBCO BRL 又は Gibco BRL (ライフテクノロジー社、ゲーサーズバーグ、メリーランド)、Millipore(ミリポア、ビルリカ、マサチューセッツ)、Bio-Rad(バイオラド、ハーリーキューズ、カリフォルニア)、Invitrogen(インビトロジェン社、サンディエゴ、カリフォルニア)、NEB(ニューイングランドバイオラドズ、ベバリー、マサチューセッツ)、Sigma(シグマケミカル社、セントルイス、ミズリー)、Pierce(ピアスバイオテクノロジー、ロックフォード、イリノイ)、Takara(タカラ・バイオ社、大津、日本)、Roche(ホフマン-ラ・ロッシュ、バーゼル、スイス)、EM Science(エムイーサイエン、ギブスタウン、ニュージャージー)、Qiagen(キアゲン社、バレンシア、カリフォルニア)、Biodesign(バイオデザイン社、ソーコ、メイン)、Aptagen(アプタゲン社、ハーンダン、バージニア)、MolecularDevices(モレキュラーデバイス社、サニーベール、カリフォルニア)、R & D Systems(R & D システムズ、ミネアポリ、ミネソタ)、Stratagene(ストラタジェンクローニングシステムズ、ラホーヤ、カリフォルニア)、Marsh(マーシュバイオサイエンス、ロチェスター、ニューヨーク)、Bio-Tek(バイオテック社、ウィヌースキー、 バーモント)、Biacore(ビアコア社、ピスカタウェイ、ニュージャージー)、PeproTech(ペプロテック、ロッキーヒル、ニュージャージー)、SynPep(シンペップ、ダブリン、カリフォルニア)、及びMicrosoft(マイクロソフト社、レドモンド、ワシントン)。
【実施例1】
【0120】
バチルススブチルス(B. subtilis)におけるBCE103-BBI融合タンパク質の生産
この実施例では、バチルススブチルス(B. subtilis)中にBCE103-BBI融合タンパク質を生産するために行った試験を説明する。この試験に用いられるC末端にヘキサヒスチジンタグを有するプロ-BBIタンパク質をコードする合成遺伝子(Operon
Technologies)のDNA配列は以下である。
【表1】
上記合成遺伝子によりコードされるC末端にヘキサヒスチジンタグを有するプロ-BBIタンパク質配列は以下である。
【表2】
成熟型BBIをコードする合成遺伝子のDNA配列の一部は以下である。
【表3】
上の合成遺伝子によりコードされる成熟型BBIのタンパク質配列は以下である。
【表4】
pJ10ベクター中のBCE103セルラーゼ発現カセットへ融合させるBBI遺伝子の増幅に用いるPCRプライマーは以下である。
【表5】
pJM103組込みベクター内へクローンされるaprE-BCE103-BBI-ヒスタグ発現カセット(EocRI-HindIII)の配列を図1に示す。PJM103BBIヒス発現ベクターのプラスミドマップの概略を図2に示す。
【0121】
バチルススブチルス(B. subutilis)aprEプロモーター及びシグナル配列に融合されたアルカリセルラーゼ(BCE103)遺伝子(vanSoligen、米国特許6,063,611を参照のこと。この文献を参照により本明細書に援用する)は、pUCAPR103(Shaw et al., J.Mol.Biol., 320: 303-309 [2002])をEcoRI-BamHIフラグメントとして(すなわち、BCE103触媒ドメイン及び第一セルロース結合ドメインリンカーのみのコード配列を運搬するフラグメント)pJM103内でクローニングした(Perego,"Integrational vectors for genetic manipulation in Bacillussubtilis"In, Bacillus subtilis and Other Gram-positive Bacteria:
Biochemistry, Physiology, and Molecular Genetics, Sonenshein, Hoch, and Losick
(eds), American Society for Microbiology, Washington D.C., pp. 615-624 [1993])。C末端にヒスチジンタグ(His-tag)を有するダイズボウマン-バーク(Bowman-Birk)プロテアーゼインヒビター(BBI)(Swiss-ProtAccession #P01055;Odani and Ikenaka, J. Biochem., 71: 839-848 [1972]参照のこと)は、オペロンテクノロジー(OperonTechnologis)(上記DNA配列を参照のこと)により合成された。3’HindIII部位を用いてBBI遺伝子をpJM103ベクター内でクローンさせた後に、BCE103遺伝子を用いる正確なりーディングフレーム中に5’BamHI部位及び3’末端に導入された強力な転写終了因子を生成するプライマーを用いてBBI遺伝子をPCRにより増幅した(全プライマーはMWG Biotech、OligosEtc.又はOperon Technologiesにより合成された)。
【0122】
合成BBI遺伝子をテンプレートとして用いて、BBIfusion_FW (配列番号14) 及びBBIHISHindIII_RV (配列番号16)、又はBBIfusion_FW(配列番号14) 及びBBI-HindIII_RV (配列番号15)のプライマーをそれぞれ用いてC末端にHis-Tagを有する、又は有しないPCRフラグメントを生成した。他の方法で示さない限り、製造会社の説明書に従いハイフェデリティープラチナTaqポリメラーゼ(HighFidelity PlatinumTaq polymerase) (Invitrogen)を用いてPCRを30サイクル行った(55℃のアニーリング温度)。aprE-BCE発現カセットを運ぶBamHI-HindIIIフラグメントとして、PCRフラグメントをpJM103内でクローニングした。正確な遺伝子配列をDNA配列決定により確認した。
【0123】
従って、図1に示すように、BCE103セルラーゼ遺伝子によりコードされる第一CBD(セルロース結合ドメイン)リンカー配列のC末端コード領域に対するフレーム中にBBI遺伝子(His-Tagを有するもの又は有しないもの)の成熟コード領域のN末端を融合した。それゆえ、2のBCE103のCBD(Shaw et al.,上記)は、最終的な発現カセットの中のBBIにより置き換えられ、pJM103BBI又はpJM103BBIhisとなった(図2参照)。このaprEプロモーターはBCE103-BBI遺伝子融合体の発現を制御する(Ferrariet al., J. Bact., 170: 289-295 [1988]及びHenner et al., J. Bact., 170: 296-300[1988]参照のこと)。
【0124】
有用なバチルススブチルス(Bacillus subtilis)細胞、BG3934comK(degUHY32、oppA、△spoIIE3501、△aprE、△npfE、△epr、△ispA、△bpr、amyE : : xylRPxylAcomK-phleo)を発現プラスミド、pJM103BBI又はpJM103BBIhisで形質転換した。バクテリアは、キシロース誘発プロモーター(Halmet al., Mol. Microbiol., 21: 763-775 [1996])の存在下でcomK遺伝子により誘発させることにより有用にすることができる。形質転換体を、5μg/mlクロラムフェニコールを含むウリアブロスアガー(Luria Broth agar)(LA)プレート上で選択した。遺伝子増幅での発現を高めるために、コロニーが縞状になり、抗生物質を含む培地における成長率がクロラムフェニコールが欠失している培地における成長率と同じになるまで、25μg/mlのクロラムフェニコールを用いて、LAプレート上で育成した。BCE-BBI融合タンパク質は、TSB培地(OXOIDより市販されているトリプトンダイズブロス(TryptoneSoya Broth)、30 g/L)又はMOPSベース合成培地中であるMBD培地において、37 ℃の振とうフラスコの中で育成することにより生成された。NH4Cl2、FeSO4、及びCaCl2を培地から除き、3mMのK2HPO4を用い、60 mMの尿素、75 g/Lのグルコース及び1 %のソイトン(soytone)を供給した以外は、(Neidhardtet al., J. Bacteriol., 119: 736-747 [1974] )に記載されたようにMBD培地を調製した。微量栄養素は、100倍濃度の貯蔵溶液として調製された。この溶液は1リットル中400mgのFeSO4-7H2O、100 mgのMnSO4-H2O、100mgのZnSO4-7H2O、50 mgのCuCl2-2H2O、100mgのCoCl2-6H2O, 100 mgのNaMoO4-2H2O、100mgのNa2B4O7-1OH2O、10 mlの1M CaCl2、及び10mlの0.5 M クエン酸ナトリウムを含む。
【0125】
BCE103-BBI融合タンパク質は、標準的なタンパク質染色方法(例えば、ゲルコードブルー染色試薬(GelCode Blue StainReagent)Pierce)を用いて44 kDa付近のSDS-PAGEゲル上の主要なタンパク質バンドとして視認することができる(EMS緩衝液中10 %のSDS-PAGE、製造会社の説明書に従い操作。Invitrogen)。BCE103-BBI融合タンパク質は製造会社の説明書に記載の手順を用いたSDS-PAGEゲルの免役ブロットにより確認された(BM染色体ウエスタンブロットキット(BMChromogenic Western Blotting Kit)、Roche Applied Science抗ヒスタグ抗体又は抗BCE103セルラーゼポリクローナル抗体を検出に用いた)。
【0126】
BCE103活性は、LAセルラーゼ指標プレート(0.2 %のコンゴレッド又は0.5 %のアゾ-CMセルロースを用いて染色された1 %のカルボキシメチルセルロース、Megazyme)上でセルラーゼ消化を定性的に評価し、当該技術分野で知られている方法を用いて、合成基質、4-ニトロフェニルβ-D-セロビオース(Sigma)を用いた培養ブロス中のアッセイ緩衝液(AssayBuffre)(100 mMとリスpH 8.6、0.005 % Tween-80)を直接アッセイすることにより定量的に評価された(例えば vanTilbeurgh etal., Meth. Enzymol., 160: 45-59 [1988]参照のこと)。
【0127】
BBI活性を決定するのアッセイ緩衝液の中でトリプシン抑制アッセイを行った。特に、標準曲線は、2 μg/mlの標準BBI溶液を1:1の連続希釈(100 μLBBI+100 mlアッセイ緩衝液)することにより作製した。疎水性相互作用カラム(POROSHP2、フェニルカラム、Applied Biosystems)を用いて、20mM MES pH 6.0中1 mg/mlトリプシン-キモトリプシンインヒビター(シグマカタログ#T-9777)からこの標準BBIを精製した。このカラムを20mM MES pH 6.0で平衡化し、5 mgのインヒビターを負荷し、平衡化に用いた緩衝液で洗浄した、その後、BBIを水で溶出した。抑制アッセイで試験する未知のBBIサンプルを、2以上のデータポイントが標準曲線の範囲内に入るように必要に応じて希釈した(通常、1:10、1:100、1:200、1:1000、1:2000の希釈のサンプルの希釈率が試験され、必要に応じて希釈率を微調整した)。希釈した各標準BBI及びサンプル20
μLに、80 μLの5 ng/mlのウシ脾臓トリプシン(Worthington)(アッセイ緩衝液の中へ、1 mg/mlトリプシンストック溶液を希釈することにより作製された)。便宜上、この標準物質とサンプルを96ウェルマイクロタイタープレート中に整列させた。混合及び25℃15分間のインキュベーションにより反応を行った。インキュベーションの後、0.5
mg/mlのトリプシン基質100 μL(DMSO中100 mg/mlの溶液からアッセイ緩衝液の希釈により調整)、Suc-AAPR-pNA(スクシニル-アラニン-アラインヒビター-プロリン-アルギニン-パラニトロアニリド、Bachem)を添加し、混合し、OD(A405)を15分間モニターした。ペクトラマックス250(SpectraMax250)(Moleculaer Devices)を用いて少なくとも12秒毎に一回記録をとった。記録されたデータのポイントは、各反応に対するVmaxの決定に用いた。標準曲線は、BBI濃度に対するVmaxのプロットにより作製し、4の指標曲線に当てはめた。全てのデータのフィッティングは製造会社(MolecularDevices)が提供するソフトウェアを用いて行われた。未知サンプルのBBI濃度を標準曲線を用いて計算した。代替的に、BBI活性は同じ手順を用いてウシ脾臓キモトリプシン(Worthington)抑制を検出することにより測定された(キモトリプシンは、トリプシンと同じ濃度で用い、キモトリプシン活性は、100μLの0.4 mg/mlキモトリプシン基質、スクシニル-アラニン-アラニン-プロリン-パラニトロアニリド、(Bachem)の添加により測定された)。
【0128】
振とうフラスコサンプル(2.8 Lのフェルバッハ6 バッフルフラスコ中 500 mM MBD培地、37 ℃、225 rpm、60時間後に収穫する)の力価が、通常、0.4乃至0.9mg/ml BCE活性及び40乃至150 μg/ml BBIトリプシン抑制活性になるように培養が行われる。しかしながら、力価はバクテリア系統、培養培地及び育成条件(通気、温度、収穫時間等)の最適化により、更に改善することができる。
【0129】
野生型BBIに対するBCE103融合に加えて、BBIに対する融合タンパク質変異体及びBCE103及びBBIの間の各種リンカーを有する融合タンパク質は上で概説した方法を用いて、以下の実施例のように製造することができる。更に、融合タンパク質はBBIを精製工程において第一CBDを用いることの助けとなる第二CBDリンカー(BCE-cbdD-BBI;実施例4を参照のこと)に結合させたときにも生成される。
【実施例2】
【0130】
BCE103-BBI融合タンパク質としてのBBI反応部位ループのペプチド置換体の生成
この実施例では、BCE103-BBIのようにBBI反応部位へ置換するペプチドを生成するための試験を説明する。これらの試験の各工程に用いるプライマー及び他の配列を以下に記載する。BCE103融合部位(BamIIIの位置)から遺伝子の終わりまでの12BBIck81配列を図3に示す。CK37281ペプチド配列(ACYNLYGWTC(配列番号9))をトリプシン及びキモトリプシン抑制ループの両方に挿入した。
【0131】
QuikChange(登録商標)部位直接変異誘発(Stratagene)を用いたBBI遺伝子中にEcoRI部位を導入するために用いるプライマーは以下である。
【表6】
VegF結合ペプチドCK37281でトリプシン抑制部位を置換するためにアニールされ、BBI遺伝子(SacI-EcoRI)の中でクローニングされるDNAオリゴヌクレオチド配列は以下である。
【表7】
VegF結合ペプチドCK37281でキモトリプシン抑制部位を置換するためにアニールされ、BBI遺伝子(EcoRI−SaclI)の中でクローニングされるDNAオリゴヌクレオチド配列は以下である。
【表8】
合成BBI遺伝子のトリプシン(SacI及びEcoRI制限部位)又はキモトリプシン(EcoRI及びSaII制限部位)反応部位ループに中にペプチドを導入するためのカセットを作るために用いるオリゴヌクレオチドペアのDNA配列を以下に記載する。これらのペプチドコード配列はp2JM103発現ベクター内にSacI-SalIフラグメントとして導入された。
【表9】
【表10】
QuickChange(登録商標)II-XL部位直接変異誘発キット(Stratagene)を用いてトリプシン又はキモトリプシン反応部位へペプチド配列を導入するために用いるオリゴヌクレオチドプライマーペアのDNA配列を以下に記載する。製造会社の説明書及びこの実施例に記載の通りに反応を行った。68℃で6分間の伸長を行い、95 ℃で50秒間の変性を行い、55 ℃で50秒間のアニーリングを20サイクル行った。20サイクル終了後、68 ℃で20分間の最終伸長を行った。
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【0132】
p2JM103-lnk2-BBI発現ベクターのキモトリプシン反応部位ループへMM021ペプチドを導入するためのカセットを作るために用いるオリゴヌクレオチドペアのDNA配列を以下に記載する。このカセットはベクター内のSphI及びSalI制限部位へ結合された。
【表18】
【0133】
システインコンストレインドペプチド(cysteine constrained peptides)からできているライブラリーは所望の基質に結合するペプチドの選択のための良く知られた試薬(例えば、市販のPhD-C7Cファージディスプレイライブラリーキット、NEB)である。BBIはペプチド結合を選択するために用いられる各種方法で提示されるペプチドループに似た構造を有する2のシステインコンストレインド(cysteineconstrained )反応部位ループを有する。従って、一旦システインコンストレインド(cysteine constrained )結合ペプチドが選択されると、BBIは結合反応におけるペプチドを提供するためのスキャッフォールドとして使用するのに適している。
【0134】
DNAオリゴヌクレオチドカセットを用いて、CK37281 ペプチド配列(ACYNLYGWTC)(配列番号9)で、トリプシン、キモトリプシン、又は両方の活性部位ループを置換することによりVegF結合ペプチド CK37281 (例えば、2003年9月13日に提出された米国継続出願 60/520,403, 参照のこと。該出願を参照により本明細書に援用する。)
をBBIの中へ移植した。構築を促進させるために、上で示すBowBeco_F 及びBowBeco_Rプライマーを用いたQuikChange(登録商標)部位直接変異誘発を用いて製造会社の説明書に記載従い、トリプシン及びキモトリプシン反応部位ループの間のBBI遺伝子(OperonTechnologiesの合成品 ; 実施例1を参照のこと)のコード領域にEcoRIサイトを導入した (0.5 pmol の各プライマーを QuikChange(登録商標)
反応に用いた。97 ℃で3分間の第一変性工程の後、68 ℃で12分間、95 ℃で30秒間及び55 ℃で1分間のPCRサイクルを18回行い、その後に68 ℃で15分間の最終伸長を行った)。
【0135】
トリプシン抑制ペプチドループの置換のために、2のDNAオリゴヌクレオチド(1BBCK81+及び1BBCk81-)をアニールし、SacI及びEcoRI反応部位に結合した。同様に、キモトリプシン抑制ループの置換のために、EcoRI及びSalIサイトをオリゴヌクレオチド(2BBck81+及び2BBck81-)のアニーリングにより作られたDNAカセットの挿入に用いた。CK37281ペプチドは、トリプシンループを初めにCK37281ペプチドで置換した後に、オリゴヌクレオチド(2BBck81+及び2BBck81-)を用いてキモトリプシンループの中にCK37281を挿入することにより両ループの中に移植した。トリプシンループ(1BBIck81)の中CK37281を有するBBIをSacI-SphIフラグメントとしてpJM103BB発現カセット内に移動した。キモトリプシンループ(2BBIck81)又は両者のループ(12BBIck81)の中のCK37281を有するBBIを、SacI-SalIフラグメントとしてpJM103BBIの中に挿入した。正確な配列はDNA配列決定を用いて確認した(12BBIck81遺伝子の配列は図3に示す)。得られたベクター、pJK103-1BBIck81、pJK103-2BBIck81又はpJK103-12BBIck81をバチルススブチルス(B.subtilis)BG3934comKの形質転換に用い、上の実施例1に示すように、BCE融合タンパク質の生産を確認した。
【0136】
無細胞ブロスに存在する主要タンパク質である44kDa付近の融合タンパク質がSDS-PAGEで検出された。幾つかのケースにおいて、BCE触媒コアに対応する34
kDa付近の主要タンパク質バンドの存在により視認することができる有意なBBI部分の退縮(degradation)(50 %まで)(特にMBD倍地中で48時間以上育成した場合)が見られた。このケースにおいて、BCE103セルラーゼの力価は、野生型BBI(実施例1)に融合させて測定したの同様であるが、BBIの活性は通常の半分以下であった。
【0137】
育成の間のBBI変異体のタンパク質分解を減らすために(すなわち、融合タンパク質との比較においてSDS-PAGE上に存在するBCE103セルラーぜ触媒コアの量を減らすために)、9のプロテアーゼ遺伝子(degUHY32、oppA、△spoIIE3501、△aprE、△npf
E、△epr、△ispA、△bpr、△vpr、△wprA、△mpr-ybiF、△nprB、amyE : : xylRPxylAcomK-phleo)を欠失させたバチルススブチルス(Bacillussubtilis)系統を発現宿主細胞として用いた。育成温度(35 ℃)及び通気量(200 rpm)を減らした。これらの変更により、主要融合タンパク質バンド(〜44kDa)はBCE触媒コア(〜34 kDa)と思われる分子量で存在するバンドと大差なく、SDS-PAGE上で観察された。
【0138】
BCE103セルラーゼのC末端に融合させたBBIのトリプシン及び/又はキモトリプシン活性部位ループへの置換を行ったときに、CK37281ペプチドに加えて、多くのシステインコンストレインドペプチド(cysteineconstrained peptides)が生産された。特定の例を以下に示す。
【0139】
(1) 相補的な抑制因子として設計又は選択されたペプチド、第一又は第二反応部位ループへ導入されたコンプスタチン(compstatin)(Sahu etal.,J. Immunol., 157: 884-891, [1996] 参照のこと)、C2c(第一ループ)、C3c(第一ループ)、C4c(第一ループ)、及びC5c(第一ループ)、又は1B、2B、4A、5A、6-1A、7A、8B、9A、10B、11-1A、12B、13A、及び15-1A(全て第二ループ)へ結合する反応においてFactor Bにより選択されたペプチド
(2) プロテアーゼファクターXa、又は角質溶解キモトリプシンXal(第二ループ)又はhSCC1(第一ループ)に結合するためのペプチド
(3) FGF5結合反応により選択されたペプチド; 1A6 (第一又は第二ループ)、1C2 (第一又は第二ループ)、2E2 (第一又は第二ループ)、2E5(第一、第二、又は両ループ),FGFns(第一又は第二ループ)、FGFkr(第一又は第二ループ)、FGFhl(第一又は第二ループ)、FGFgy (第一又は第二ループ)、MM005(第一又は第二ループ)、MM007(第一、第二、又は両ループ)、MM009 (第二ループ)、MM010(第一、第二、又は両ループ)、MM017 (第二ループ)、FGFps1(第二ループ)、FGFps2(第一、第二、又は両ループ)、及びFGFpsB(第二ループ) 、及び
(4) TGF-1結合反応により選択されたペプチド;1A8(第二ループ)、lA12 (第二ループ), lE11 (第二ループ)、TGFpsl(第二ループ)及びMM021(第二ループ)。
【0140】
これらのペプチドをトリプシン(第一ループ)又はキモトリプシン(第二ループ)活性部位ループのいずれかへ導入するために用いるオリゴヌクレオチド及びこれらのペプチドをBBIに移植するために用いる方法は上に記載されている。全てのケースにおいて、生産された融合タンパク質をDSD-PAGEゲルにより評価した。CK37281置換ペプチドに対して説明したのと同様に、幾つかの置換ペプチドをもちいて、タンパク質分解を減少させることによりにより無傷の融合タンパク質の生成量が高められた。
【実施例3】
【0141】
チオール還元/酸化試薬によるBBIの活性化
育成の後の(トリプシン又はキモトリプシンの抑制による)BBIの活性は、(2の分子が融合タンパク質の中で1:1のモル比で存在する)無細胞上清中で測定されるBCE103セルラーゼの活性から予想されるものよりも通常5乃至20倍低い。BCE103-BBI融合タンパク質中のBBI活性の増加(トリプシン又はキモトリプシン抑制により測定される)は、接種の後、1-4mMの濃度のbMEをMBD成長培地に添加した後14時間育成することにより確実に得ることができる。融合タンパク質中のBBIのトリプシン又はキモトリプシン抑制活性は、結合ペプチド(例えば、VergF結合ペプチドCK37281)をキモトリプシン又はトリプシン反応部位ループにそれぞれ置換したときよりも低くなる。野生型BBIを用いて、bMEで処理することによって、抑制活性は増加させることができる。予想しないことに、これらの試験においてBBI育成の間の活性における他のチオール還元試薬(例えば、システイン、還元グルタチオン、DL-ジチオスレイトール(dithiothreitol)及びトリス[2-カルボキシメチル]フォスフィン)の影響は小さいか、無視できる程度であった。抗酸化剤(例えば、アスコルビン酸又はDL-α酢酸トコフェロール)、他のアジュバンド(例えば、イソロイシン、ダイズオイル、Tween(登録商標)-80)を培地に添加すること、又は30℃における育成も、BCE103:BBI活性の比を改善しなかった。
【0142】
特に育成の間のBBI活性を決定するために、p2JM103-E3-2BBIcK81(以下実施例4を参照のこと)で形質転換させたバチルススブチルス(B.subtilis)BG6006を含む培地を、250mlの振とうフラスコ内で37 ℃、13時間、40 mlのMBD中で育成した。その後、図4で示すチオール還元試薬を添加し、62時間の育成の後に細胞の上清を収穫した。2-メルカプトエタノール(BME)、システイン(Cys)、還元グルタチオン(Glut)及びDL-ジチオスレイトール(DTT)を図4に提供されているグラフで示す最終濃度になるように成長培地に添加した。5mM βMEの濃度はより良いBCE103:BBI活性比となったが、通常は、試薬の添加がバクテリア成長を減少させることから、BCE103及びBBI力価は全体的に減少した(図4参照のこと)。BCE103及び2BBIck81の力価は実施例1で説明したアッセイを用いて測定した。
【0143】
Qセファロースイオン交換クロマトグラフィーによる融合タンパク質(例えば、BCE-lnk2-2BBIck81、実施例4を参照にこと)の部分的な精製の後にもBBIの活性化を行うことがきでる。
【0144】
融合タンパク質を作動している振とうタンク又は発酵槽から得られた無細胞ブロスから精製した。このブロスを、ろ過し、5乃至10倍の水で希釈し、pHを7.5乃至8.0に調製した。希釈サンプルはQセファロースレジン(GHhealthcare)を充填したカラムに負荷した。このカラムを50 mMトリスpH 7.5で洗浄し、300 mM NaClを含む同じ緩衝液で再度洗浄した。融合タンパク質は、700mMのNaClで溶出した。
【0145】
BBIを活性化させるために収集された融合タンパク質分画を10倍のアッセイ緩衝液の中で溶出し、その後、2 mMのβME及び0.2 mMの酸化グルタチオン(GSSG)を添加し約24時間室温で、スターラープレート又はロックプラットフォーム上で攪拌を続けることにより処理した。BBIは、BCE103セルラーゼで測定される濃度にもとづき予測されるトリプシン抑制活性の約70乃至100%を達成した。上で概説した活性化方法は、通常最良の結果を出すけれども、幾つかのケースにおいて、所与のサンプルの活性を最大化するために96ウェルプレートにおいて、スクリーニングを行った。最初にスクリーニングする典型的な条件は、アッセイ緩衝液による希釈倍率(例えば、2乃至50倍の連続希釈)、βME濃度(例えば、連続する0.5乃至5mM)及び酸化グルタチオン濃度(例えば、0 mM、1/20乃至1/2 βME濃度)である。
【0146】
BCE-lnk2-2BBIck81融合タンパク質の活性化を図5に示す。この特定の態様において、Qセファロース精製後の融合タンパク質を0.005 %のTween(登録商標)-80含むダルベッコPBS(Mediatech)を用いて1:10に希釈した。βメルカプトエタノールを3
mMの最終濃度で添加し、振とう機上で室温、一晩インキュベートした。このサンプルは、更に磁気攪拌プレート上で激しく攪拌しながら60時間室温でインキュベートした。(βMEで処置する前後の)BCE103及び2BBIck81の力価を、それぞれ、セルラーゼアッセイ及びトリプシン抑制アッセイにより決定した。
【0147】
幾つかの態様において、大容量発酵槽のからの無細胞ブロス中のBBI又はその変異体の活性化のような、活性化反応において、希釈及びβME濃度を減少させることが好ましい。このことは、高濃度緩衝液(500mMトリスpH 6.8)、又はCHES等(CAPS、トリシン、TAPS及び他の適切な両性イオン緩衝液)の両性イオン緩衝液に変更することにより達成できる。例えば、無細胞ブロス(又はイオン交換クロマトグラファイーにより精製された融合タンパク質分画)は375mMのCHES pH 6.8中、0.005 %のTWEEN(登録商標)-80で希釈し、その後に、1 mMのβME及び10 mMのNa2SO3で活性化し、攪拌しながら室温で約24時間インキュベーションする。BCE103セルラーゼ融合変異体等のBBIあるいはこの変異体の力価はこの方法により、(BCE103セルラーゼ活性に基づく)予測値に対して70乃至100%まで活性化することができる。
【実施例4】
【0148】
BCE103-BBI融合タンパク質の切断による遊離BBI/変異体の放出
この実施例は、BCE103-BBI融合タンパク質を切断による遊離BBI又はその変異体の放出について説明する。
【0149】
培地育成の間にBCE103触媒ドメイン及びBBIの間に切断可能部位を作り出すために、アニールされ、pJM103のBamHI及びSacIサイトへ結合するDNAオリゴヌクレオチドペアの配列を以下に示す。
【表19】
【0150】
BCE103セルラーゼの第二CBDリンカーへBBIを融合するために、へアニールされ、p2JM103-BBIのBamHI及びSacI部位へ結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表20】
【0151】
アスパラギン酸及びプロリン残渣の間の酸切断をするのに適したペプチド配列を以下に示す。
【表21】
【0152】
QuikChange(登録商標)部位直接変異誘発によりBssHII部位をpJM103BBIへ導入するために用いるオリゴヌクレオチドプライマーを以下に示す。
【表22】
【0153】
BBIの停止コドンの後にHIndIII及びXhoI部位を導入し、LATの後にPacI部位を導入し、本来のHindIII部位を除去するためのカセット(SalI-HindIII)としてアニールされるDNAオリゴヌクレオチドの配列を以下に示す。
【表23】
【0154】
BCE103触媒ドメイン及びBBIの間に挿入される上で提供される酸ラベルリンカー(すなわち、リンカー1、リンカー2及びリンカー3)を生成するために用いるPCRプライマーを以下に示す。
【表24】
【0155】
上で説明する第一CBDリンカーに挿入する酸ラベルリンカーを生成するために用いるPCRプライマーを以下に示す。
【表25】
【0156】
BBI及びBCE103触媒ドメインの間に挿入された酸ラベルリンカーのタンパク質配列を以下に示す。酸ラベルリンカーを太字で示し、第一CBDドメイン配列を下線で示す。
【表26】
【0157】
精製工程の間にBBI及びBCE103触媒ドメインの間に切断可能部位を生成するために、アニールされ、pJM103-BBIのBamHI及びSacIへ結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表27】
【0158】
精製工程の間にBBI及びBCE103触媒ドメインの間に挿入される切断可能部位を生成するために、へアニールされ、pJM103-lnk2-1BBIck81のBamHI及びSacI結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表28】
【0159】
QuikChange部位直接変異誘発(Stratagen)を用いてリンカー2へE及びE3リンカーを導入するために用いるオリゴヌクレオチドプライマーペアの配列を以下に示す。
【表29】
【0160】
BCE103触媒ドメイン及びBBIの間のE3リンカーのタンパク質配列はDNNDPIPEP[DDESFNMPIPEP] (配列番号 174)である。この配列において、Eリンカーを下線で示し、大腸菌における間違った組換えにより生成された配列を太字([]ので囲む配列)で示す。Mpr(又はV8プロテアーゼ)による切断はE3リンカーの中にある3のグルタミン酸のいずれかの後に引き起こすことができる。従って、この構築体は、BCE-(配列番号174)-BBIとなる。
【0161】
BBI及びBCE103触媒ドメインの間に挿入されるGenenase I切断可能部位を生成するために、へアニールされ、p2JM103-lnk2-2BBIck81のBamHI及びSacIへ結合されるDNAオリゴヌクレオチドペアの配列を以下に示す。
【表30】
【0162】
BDE103触媒ドメイン及びBBIの間に挿入されるGenenase I感受性切断部位(酸感受性及びMpr感受性でもある)のタンパク質配列は、DNNDPIPDP[GAAHYVEFQ]
(配列番号 179)である。GeneaseI部位(Gen4 リンカーは太字([]ので囲む配列)で(チロシン及びバリンの間に発生する切断)(NEB)、リンカー2は下線で示。Mprによる切断もGen4リンカーの中のバリンの後のグルタミンの後に発生させることができる。本発明で用いられる配列はBCE-(配列番号179)-BBI である。
【0163】
BCE103-lnk2-2BBIck81融合タンパク質における切断部位を以下に示す。BCE103触媒ドメイン(下線)のC末端のアミノ酸、リンカー2配列(太字)及び2BBIck81配列を示す。酸/熱に不安定なAsp-Pro結合は塗りつぶし矢印、グルタミン酸の後のMpr感受性結合は線矢印で示した。
【表31】
【0164】
遊離BBI又はその変異体を単離するために、BCE103-BBI融合タンパク質からBBI部分を切り離すことが必要になる。幾つかの態様において、バチルススブチルス(B.
subtilis)により製造される固有のプロテアーゼを用いてこれは育成の間に行われる。幾つかの代替的な態様において、この切断は育成の後の精製工程(例えば、酸/熱又はタンパク質分解切断)dで行われる。リンカーは育成の間の切断に感受性を有する可能性のあるものが設計され(上記、sub、cbdl、pro、shortpro、及びcbdD)、BamHI-SacIカセットとして、pJM103BBI又はp2JM103BBI発現ベクター内でクローニングされる。融合タンパク質に対するBCE103触媒ドメインの生成は実施例1で説明されるSDS-PAGEゲル上で解析された。
【0165】
proリンカーを除く、全てのリンカーにおいて、BCE103触媒コアに対応する大きなバンドのほかに、ほぼ完全に切断されていると考えられる融合タンパク質の小さな切断が観察された。不幸なことに、育成の間に切断を行うと無細胞上清中で測定されるBBI活性は無視できるほどとなり、SDS-PAGEゲル上で同定するされるBBIバンドを生じなかった。本発明を特定の方法又は理論に制限することは意図しないけれども、BBIはその不活性形態において、タンパク質分解に影響を受けやすいことが明らかでる。従って、活性化後の精製工程の間に切断することが通常好ましい。
【0166】
幾つかの態様において、アスパラギン酸及びプロリン残渣の間の結合は、当該技術分野で知られている様に、酸性pHにおける熱処理により切断される(LandonMeth. Enzymol., 47: 145-149 [1977] )。BCE103セルラーゼの中の第一CBDリンカーは酸/熱処理により切断される可能性のある3のAsp-Proペプチド配列を有する(図1を参照のこと)。しかしながら、これらの部位において、酸/熱処理による切断は効率が悪いことがわかった。特に酸/熱に不安定なタンパク質配列は文献に記載されている。そのような3の配列は、WGDPHY(配列番号 141)、DNNDPI (配列番号 142)、及びWADPN (配列番号 143) (すなわち、リンカー1、2、及び3)である。
【0167】
これらの酸に不安定なリンカーが、BCE103-BBI発現ベクター、pJM103BBI、へ導入される前に、QuikChange(登録商標)XL(Stratagene)変異誘発部位を用いて(8分間の伸長時間、及び1分間の変性工程を用いる以外は、製造会社の説明書に従い、実施例2で述べた方法を用いて)、BCEbss_F及びBCEbss_R(上記)のオリゴヌクレオチドを用いて、aprEシグナル配列コード領域にaBssHII部位を挿入した。その後、HindIII及びXhoI部位をLATターミネーター(BBI停止コドンの後の)の前に挿入し、SalI及び元のHindII部位へのオリゴヌクレオチドカセット(BCEterm+及びBCEterm-;上記)の挿入により、このターミネーター(LATターミネーターの後のオリジナルHindIII部位)の後にPacI部位を付加した。この新しいベクターを「p2JM103BBI」と称した。
【0168】
前方向プライマー、BCE103coreBssHII_Fw、リンカーWGDPHY_R、リンカーDNNDPI_RV又はリンカーVVADPN_RV(配列は上記)の各リバースプライマーを用いて、及びpJM103BBIをテンプレートとして用いたPCRにより、酸に不安定なリンカーフラグメントを製造した(PCR手順については実施例1を参照のこと)。970bpのPCRフラグメントをBanHI及びPstIで消化し、酸リンカーフラグメントをコードしている154 bpのフラグメントを電気泳動の後、アガロースから単離し、BanHI及びPstIで消化ししたpJM103ベクター内へ結合した。最終発現ベクターにおけるリンカー配列、p2LM103lnk-BBI、p2JM103lnk2-BBI及びp2JM103lnk-BBIをDNA配列決定により確認した。
【0169】
有能なバチルススブチルス(B.subtilis)系統、BG3934comK又はBG6006を、プラスミドを用いて形質転換し、コロニーを5 μg/mlクロラムフェニコールを含むLAプレート上で選択し、実施例1で説明する様に、25μg/mlクロラムフェニコールを用いて増幅した。
【0170】
同様に、第一CBDリンカーに挿入した。特に、PCRフラグメントは、前方向プライマー、BCE103corePstI_FW、LplusDNNDPI_RV又はLplusVVADPN_RVのリバースプライマー(配列を上に記載)を用いて、テンプレートをp2JM103BBIとして酸に不安定なリンカーを生成した。約150
bpのフラグメントを、BamHI及びPstIを用いて消化し、精製し、BanHI及びPstIを用いて消化したp2JM103BBIに結合した。正確な配列をDNA配列決定法により確認し、上で述べたように、p2JM103plnk-BBI、p2JM103plnk2-BBI及びp2JM103pllnk3-BBIは、バチルススブチルス(B.subtilis)の形質転換に用いた。
【0171】
MBD培地での育成の後、上で説明した(実施例2を参照のこと)イオン交換クロマトグラフィーにより融合タンパク質を精製した。融合タンパク質は10 %のギ酸中、55℃、16時間での処理することにより切断された。BCE103触媒ドメインは、酸処理の間に沈殿し、遠心分離により分離された。上清中の遊離BBIをスピードバク(SpeedVac)で一晩乾燥した。SDS-PAGEゲルに負荷する前に50mMトリスpH 8の中にサンプルを懸濁した。SDS-PAGEのタンパク質染色解析により、酸切断は、リンカー2がBCE103触媒ドメイン及びBBI(BCE-DNNDPI-PDP-BBI)の間に挿入されている場合には、融合タンパク質において有効であることが観察された。このリンカーは、pH2、75 ℃、20 mMのグリシン中で数時間処理することで切断されることがわかった。
【0172】
代替的な態様において、精製工程の間にプロテアーゼで処理することにより融合タンパク質を切断した。グルタミン酸特異プロテアーゼ(例えば、Mpr又はV8プロテアーゼ)、フリン/ブリスターゼ、GenenaseI及びエンテロペプチダーゼ(エンテロキナーゼ)に対する切断部位を用いてリンカーを消化した。リンカーは、BenHI及びSacI部位を用いて、発現ベクター中のBCE103触媒コア及びBBIの間にオリゴヌクレオチドカセットとして(配列は上に記載)導入された。オリジナルの発現ベクター(pJM103BBI)のコード領域において、第一CBDドメインの中及びBBIの中の第三残渣にバチルススブチルス(B.
subutilis)Mpr又はV8プロテアーゼ等のグルタミン酸特異プロテアーゼにより切断されやすいと予測されるグルタミン酸残渣がある(図1)。しかしながら、BsMpr又はV8プロテアーゼのいずれもがこれらの部位でBCE-BBI融合タンパク質を切断することが困難であることがわかった。従って、これらのプロテアーゼにより切断されやすい他のリンカーを設計することが必要であった。
【0173】
上で説明した6の不安定なリンカーの切断をBsMprにより試験した。これらの融合タンパク質は室温で、16 μgのBsMprを用いて16時間処理することにより切断された。切断の後、BCE103触媒ドメインを10%のギ酸を添加することにで乾燥させた。DSD-PAGEに負荷する前に50 mMトリスpH 8にサンプルを懸濁した。酸切断と同様、BCE-DNNDPI-PDP-BBI(リンカー2)融合タンパク質は、他のリンカーのどれよりもBsMprで効果的に切断された。それゆえ、BBI及びその変異体は、酸/熱処理又はBsMpr等のグルタミン酸特異プロテアーゼを用いたタンパク質分解のいずれかによりBCE-DNNDPD-PDP-BBI融合タンパク質から、効果的に放出されていることがわかった。Mprにより切断されるように設計された幾つかの他のリンカーも試験されたが、リンカー2よりも効果的なこのはなかった(E3リンカーは、Eリンカーを構築するために意図されたQuikChange(登録商標)部位切断変異誘発を用いた形質転換の後に大腸菌内の誤った組換えにより生成された)。上に示す様に、リンカー2の中に、酸/熱に不安定な切断部位と、Mrpにより切断されやすい3の部位がある。
【0174】
フリン又はブリスターゼ(NBE)(BCEfurinBBI)又は、エンテロペプチダーゼ(エンテロキナーゼ、NEB)(BCEentBBI)による切断のために構築されたリンカーが試験されたが、これらのプロテアーゼより効果的に切断された配列はなかった。4のリンカー(BCEgenen1BBI、BCEgenen2BBI、BCEgenen3BBI、及びBCEgenen4BBI)も消化され、GenenaseI(NEB)による切断について試験された。融合タンパク質の効果的な切断は、Gene4リンカー(BCEgenen4BBI)を用いたときに観察された。BsMprもまた、Gene4リンカーを効果的に切断した。
【0175】
精製されたBCE-lnk2-2BBIck81融合タンパク質の活性化の後の、BsMprによる切断はSDS-PAGE上では観察されなかった。しかしながら、リンカー2(又はGen4リンカー)を有するBCE-BBI融合タンパク質の活性化の後の切断は、バチルスリケニフォルミス(Bacillus lichenifornais)から単離されたMprプロテアーゼ(BIMpr)を用いて成し遂げることができる。本発明を特定のメカニズムに限定することは意図しないけれども、成熟BBIの中の3番目のアミノ酸の切断はBIMprに対してより感受性があることが明らかである一方で、BBIのC末端から6番目のアミノ酸はBsMpr切断に対してより敏感である。
【0176】
幾つかの態様において、切断の後、BBIは、上で述べるBCE103触媒ドメインの選択的酸沈殿(pH3以下)により、イオン交換クロマトグラフィー(実施例5を参照のこと)、又は、50mMトリスpH8.0を用いた無水トリプシンアガロースに負荷し、150mMNaClで50mMトリスpH8.0を洗浄し、その後、300mMNaClde,pH2.2 50mMグリシンで結合BBIを溶出することにより、BCE103触媒ドメインから単離精製された。
【実施例5】
【0177】
BBIck81のVegFへの結合
本実施例は、BBIcK81のVegFへの結合を評価するために行う試験について述べる。BCE103-lnk2-2BBIck81融合タンパク質は実施例2で述べたように、バチルススブチルス(B. subtilis)を用いて生成された。融合タンパク質を生成し、実施例3に記載されているように、βME及び酸化グルタチオンを用いた処理により、BBIトリプシン抑制活性が増加された。融合タンパク質はBsMprプロテアーゼ(実施例4参照のこと)により切断され、Qセファロースカラムを用いたイオン交換クロマトグラフィーによりBCE103触媒ドメインから2BBIcK81を精製した。
【0178】
すなわち、切断の後、切断されたサンプルのpHを5.5に調製し、このサンプルをその後、(pH5.5 25mMMESを用いて平衡化した)カラムに負荷した。遊離2BBIck81は、25mM酢酸ナトリウムをカラムに通すことにより洗浄されるが、BCE103触媒コアは樹脂に結合したままであった。2BBIcK81分画は精密ろ過により濃縮され、結合アッセイ(Bio
Veris)に基づく、電気化学発光(ECL)分析を用いて解析された。抗-VegF抗体(Santa Cruz)及びVegF(Pepro Tech)は、製造会社(BioVeris)の説明書に従って電気化学発光ダイ及びビオチンでそれぞれ標識した。全ての材料は0.1 %Tween(登録商標)-80を用いてダルベッコPBS(Mediatech)中に溶解した。抗-VegF抗体の一連の希釈物(125、250、及び500ng/ml)及びVegFの一連の希釈物(100、150、200及び250 ng/ml)は、強固なECLシグナルを与える濃度を決定するためにアッセイされた。
【0179】
2BBIck81結合テストのために、50 μLの500 ng/ml ECL標識抗-VerF抗体、50 μLの250ng/mlビオチン標識VegF及び100 μL2BBIcL81(12.5、15、31.25、62.5、125、250又は500 ng/ml)を振とうしながら、室温で2時間インキュベーションした。その後、50μLの0.2 mg/mlストレプトアビジンコートビーズを添加し、室温で30分インキュベートした。ECLシグナルは、製造会社(Bio Veris)の説明書に従い、BioVeris M8/384解析を用いて測定した。図6に示すように、ECLシグナルは2BBIck81濃度が増加する毎に減少した。標識された抗-VegF抗体のより多くが磁気ビースが付着しているVegFに結合したことを示す。
【0180】
従って、CK37281ペプチドは、BBIキモトリプシン抑制ループ(2BBIcK81)上に移植されたときに、μモルの濃度で抗-VergF抗体とVegFの結合について競合することがわかった。実際には、2BBIck81はVegF結合に対して、合成CK3781ペプチド自身よりも強固に競合することがわかった(図6参照のこと)。CK37281ペプチドは、トリプシン抑制ループ、1BBIc81内に挿入され、BioVerisアッセイにおいて、抗-VegF抗体と競合した。従って、BBIは各種スクリーニング法により選択される本発明の活性結合ペプチドのスキャッフォールドとして有用である。
【実施例6】
【0181】
2BBIck81の生産に対する融合代替パートナーの使用
本実施例は、融合代替パートナーを評価するための試験を説明する。pET-40(+)由来のdsbc遺伝子(大腸菌)を増殖させるのに用いるオリゴヌクレオチドプライマーのDNA配列を以下に示す。このプライマーはp2JM103-Gen4-2BBIck81内へのクローニングのために5’末端にBssHII部位を、3’末端にBanHI部位を生成する。
【表32】
【0182】
リンカー2を有するBBIへ、シュードモナスメンドセラ(P.mendocina)クチナーゼ遺伝子の融合のためのカセット(Alw44I-BanHI)を作るために共にアニールされるオリゴヌクレオチドのDNA配列を以下に示す。
【表33】
【0183】
BBI部分は7個のジスルフィド結合を有することから、BCE103セルロース触媒ドメイン以外の融合タンパク質を用いて、活性BBIの高い力価が得られることが予測される。例えば、幾つかの態様において、チオールジスルフィド酸化還元酵素及び/又はタンパク質ジスルフィドイソメラーゼは正しく折りたたまれたBBIを生産するための融合タンパク質として用いられる。この態様において、追加的な活性化ステップが大部分の環境下において必要とされない。追加的な態様において、バチルススブチルス(B.
sbutilis)の中で高い力価で製造される他のタンパク質も融合パートナーとして用いることができる。例えば、フミコーラインソレンス(Humicolainsolens)由来の熱安定タンパク質ジスルフィドイソメラーゼ(hiPDI)は、バチスルスブレビス(Bacillus brevis)の中の免役グロブリンの軽鎖(2ジスルフィド)の融合パターンとして用いられている(Kajinoet al., Appl. Env. Microbiol. , 66:638-642 [2000])参照のこと。
【0184】
BBI生産に関して、hiPDIがBCE103よりも良い融合パートナーであるかどうかを決定するために、hiPDI遺伝子を合成し(DNA2.0)、発現ベクター、p2JM103-lnk2-2BBIck81(実施例4を参照のこと)、の中で、BssHII-SacIフラグメントとしてクローニングした。制限部位が導入された又は欠失されている場合を除き、高頻度で、高発現するバチルススブチルス(B.subtilis)遺伝子の発生コドンが合成遺伝子の設計のために選択された。最終濃度において、成熟hiPDI遺伝子のN末端をAprEシグナル配列に融合し、C末端はエンテロペプチダーゼ切断部位を有するリンカーに融合した(Kajinoet al., Appl. Env. Microbiol. , 66: 638- 642 [2000])。それらを順番に、2BBIck81(図7参照のこと)に融合する。この発現ベクター、p2JM-PDI-EK-2BBIck81はバチルススブチルス(B.subtilis)BG6006を形質転換するために用いた。融合タンパク質の生成はMBD培地中で(実施例1で説明したように)2mMβMEを添加、あるいは無添加した状態で、接種後14時間で評価に用いた。
【0185】
SDS-PAGEゲルにより評価されるように、PDI-2BBIck81融合タンパク質の生産は、通常同じ条件で育成されたBCE-2BBIck81によりわずかに少ない。PDI-2BBIck81細胞を含まない上清を用いて測定された(トリプシン抑制)BBI力価も又、通常BCE-2BBIck81融合細胞よりもわずかに少なかった。BCE103への融合と同様に、PDIへ融合させたときに測定されるBBI力価は、2mMβME中で育成したときに高くなり、BBI活性はβMEフリー培地中で育成したときの育成の後、細胞フリー上清にβMEを添加することにより高められた(実施例3に記載されているように)。したがって、PDIのチオールジスルフィド酸化還元活性は、融合タンパク質の中の活性か2BBIck81の力価を有意に改善しなようであり、BBI分子を活性化する必要を排除した。
【0186】
育成の間のBBI力価を改善すると思われる融合タンパク質の還元電位を増加させるために、大腸菌(Escherichia coli)由来のDsbCが2BBIcK81に対する融合パートナーとして用いられた。dsbC遺伝子は、DsbCBBI-F及びDsbCBBI-Rをプライマーとして用いて(配列は上に記載)、pET-40(+)(Novagen)をテンプレートとして用いて、製造会社(Stratagen)の説明書に従い、HerculaseEnhanced DNA ポリメラーゼを用いたPCRにより増幅させた。単離されたPCRフラグメントはベクターp2JM103-Gen4-2BBIck81(実施例4を参照のこと)の中で、BssHII-BsmHIフラグメントとして用いてクローニングした。融合遺伝子の正確な配列はDNA配列決定により確認された。このケースにおいて、DSbC-2BBIck81融合タンパク質の力価はSDS-PAGEゲル上で判断した時に、BCE-2BBIck81融合タンパク質よりも有意に低かった。トリプシン抑制により測定される活性2BBIck81力価も又十分に低かった。
【0187】
バチルススブチルス(B. subutilis)の中で高い力価を伴い生産される他のタンパク質は、BBIの生産に対する融合パートナーとして用いることができる。そのようなタンパク質の一つは、バチルススブチルス(B.subutilis)由来のaprEプロモーターを用いて高力価が発現されるシュードモナスメンドシナ(Pseudomonas mendocina)由来のクチナーゼである(参照により本発明の明細書に援用される米国特許5,429,950参照のこと)。apr-Eクチナーゼ遺伝子融合(体)は、EcoR1-Aln44Iフラグメント(pAK-15由来)として、Alw44I-BamHIリンカーオリゴヌクレオチドカセット(配列は上記)を用いて、EcoRI及びBamHIを用いて切断されたp2JM103-lnk2-2BBIck81(実施例4参照のこと)へ結合した。このクチナーゼ-リンカー2-2BBIcK81発現ベクターは、バチルススブチルスBG6006細胞の形質転換に用いて、融合タンパク質はMBD培地の中で他の融合タンパク質に対して記載したのと同様に生産された(実施例1参照のこと)。このケースにおいて、クチナーゼリンカ−2-2BBIck融合タンパク質はSDS-PAGE上で見られる主要バンドではなく、測定されたリパーゼ力価(米国特許5,429,950に記載の方法を用いて測定された)及びBBIの力価はBCE-2BBIck81融合タンパク質で見られたものよりも大部低くなっていた(約20倍)。クチナーゼ融合タンパク質中のBBI力価は育成培地に、3mMのβMEを添加したときにも改善されなかった。したがって、活性2BBIck81の最高力価はBCE-2BBIck融合タンパク質の活性化により、確実に得られるのである。しかしながら、各種融合タンパク質も本発明に用いることができるだろう。
【0188】
本明細書中で言及する全ての特許及び文献は本発明の技術分野における出願時の技術常識を示している。全ての特許及び文献が明確かつ別個に示している内容を参照により本発明の明細書に援用する。
【0189】
本明細書に開示されいてる好ましい態様を用いて、当業者が開示されている態様の変法をなすことができることは明らかであり、そのような変更は発明の範囲内である。
【0190】
本発明は課題を実行するために最適化されており、本明細書に内在するものだけでなく、目標及び有利な結果を得るために適していることを当業者は容易に理解する。本明細書で開示されている組成物及び方法は好ましい態様の代表例であり、例示であり、本発明を限定するものではない。当業者が本明細書に開示されている発明の各種置換、及び変更を本発明の範囲及び精神から逸脱することなくなしうることは明らかである。
【0191】
本明細書で例示する発明は、本明細書で特に開示されていない、構成要素、限定を伴わずに実行することができる。採用された文言及び表現は説明のために用い、限定のために用いるものではない。しかしながら、それらが示す、あるいは、それらの一部の特徴を有するあらゆる等価物を含む文言及び表現の使用を意図するものではない。したがって、本発明は好ましい態様を追加的な特徴により構成されるものであり、そのような変更及び変法は明示の請求項により定義される発明の範囲内である。
【0192】
この発明は、本明細書に幅広く一般的に記載されている。開示されている属のより狭義の又は亜種グループも本発明の一部を構成する。これは、削除された物質がとくに本発明に引用されたかどうかは別として、この属から除去する旨の規定又は負の限定を伴う本発明の包括的な記載を含む。
【図面の簡単な説明】
【0193】
【図1A】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図1B】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図1C】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図1D】図1は、pJM103インテグレーションベクター内でクローンされたaprE-BCE103-BBI-ヒスタグ発現カセット(EcoRI-HindIII)のDNA配列及びアミノ酸配列を示す(配列番号1及び配列番号2)。
【図2】図2はpJM103BBIhis発現ベクターの概略図を示す。
【図3】図3はBCE103融合部位(BamHIの位置)から端部までの12BBIcK81のDNA配列及びアミノ酸配列を示す(配列番号3及び4)。CK37281ペプチド配列(ACYNLYGWTC(配列番号9))は、トリプシン及びキモトリプシン抑制ループ内へ挿入される。
【図4】図4は、トリプシンによる活性2BBIck81及び不活性2BBIck81の力価及び培地の育成中に各種チオール還元試薬を添加した2BBck81に対するBCE103セルラーゼの活性比を示す。この図において、BME=2-メルカプトエタノール、Cyt=システイン、Glut=還元グルタチオン、DTT=ジチオスレイトールをそれぞれ示す。
【図5】図5はイオン交換クロマトグラフィーによる部分的な精製の後で、2-メルカプトエタノール(bME)で処理したときのBCE-lnk2-2BBIck81の活性を示すグラフである。
【図6】図6は、VegFに結合する2BBIck81と抗-VegF抗体の比較解析の結果を示す。
【図7A】図7はバチルススブチルス(B. subtilis)BBI発現ベクター内に挿入されたフミコーラインソレンス(H. insolens)PDI(hiPDI)を運ぶ合成DNAフラグメントの配列及びこのアミノ酸配列を示す(配列番号5及び6)。
【図7B】図7はバチルススブチルス(B. subtilis)BBI発現ベクター内に挿入されたフミコーラインソレンス(H. insolens)PDI(hiPDI)を運ぶ合成DNAフラグメントの配列及びこのアミノ酸配列を示す(配列番号5及び6)。
【図7C】図7はバチルススブチルス(B. subtilis)BBI発現ベクター内に挿入されたフミコーラインソレンス(H. insolens)PDI(hiPDI)を運ぶ合成DNAフラグメントの配列及びこのアミノ酸配列を示す(配列番号5及び6)。
【図8A】図8はp2JM103-lnk2-2BBIck81のEcoRI-BamHI部位へ結合されるaprE-クチナーゼ発現カセットのDNA配列及びアミノ酸配列である(配列番号7及び8)。
【図8B】図8はp2JM103-lnk2-2BBIck81のEcoRI-BamHI部位へ結合されるaprE-クチナーゼ発現カセットのDNA配列及びアミノ酸配列である(配列番号7及び8)。
【図8C】図8はp2JM103-lnk2-2BBIck81のEcoRI-BamHI部位へ結合されるaprE-クチナーゼ発現カセットのDNA配列及びアミノ酸配列である(配列番号7及び8)。
【特許請求の範囲】
【請求項1】
融合タンパク質を含む組成物であって、前記融合タンパク質がプロテアーゼインヒビター及び所望のペプチドを含むことを特徴とする組成物。
【請求項2】
請求項1の組成物であって、前記融合タンパク質が配列番号2,4,6及び8から成る群より選択されたアミノ酸を含むことを特徴とする組成物。
【請求項3】
請求項2に記載の組成物であって、前記融合タンパク質が配列番号1,3,5及び7から成る群より選択される核酸配列によりコードされることを特徴とする組成物。
【請求項4】
請求項1に記載の組成物であって、前記プロテアーゼインヒビターがボウマン−バーク(Bowman-Brik)インヒビター(BBI)、ダイズトリプシンインヒビター(STI)、及びエグリン(eglin)Cから成る群より選択されるプロテアーゼインヒビターであることを特徴とする組成物。
【請求項5】
請求項4に記載の組成物であって、前記プロテゼーゼインヒビターがBBIであり、前記BBIがトリプシンループ及びキモトリプシンループから成る群より選択される少なくとも1のループを含むことを特徴とする組成物。
【請求項6】
請求項4に記載の組成物であって、前記プロテアーゼインヒビターが前記所望のペプチドのスキャッフォールドであることを特徴とする組成物。
【請求項7】
請求項5に記載の組成物であって、前記ループが前記所望のペプチドを含むことを特徴とする組成物。
【請求項8】
請求項5に記載の組成物であって、前記所望のペプチドが配列番号9で定義されるアミノ酸配列を含むことを特徴とする組成物。
【請求項9】
請求項5に記載の組成物であって、前記融合タンパク質が配列番号4で定義されるアミノ酸配列を有することを特徴とする組成物。
【請求項10】
配列番号10及び12で定義されるポリペプチド配列から成る群から選択されるプロテアーゼインヒビターをコードする単離ポリヌクレオチド。
【請求項11】
スキャッフォールドタンパク質及び少なくとも1のペプチドを含む組成物であって、前記スキャッフォールドタンパク質がボウマン-バーク(Bowman-Brik)インヒビターを含むことを特徴とする組成物。
【請求項12】
請求項1乃至9及び11のいずれか1項に記載の組成物であって、前記少なくとも1のペプチドが血管内皮成長因子に結合することを特徴とする組成物。
【請求項1】
融合タンパク質を含む組成物であって、前記融合タンパク質がプロテアーゼインヒビター及び所望のペプチドを含むことを特徴とする組成物。
【請求項2】
請求項1の組成物であって、前記融合タンパク質が配列番号2,4,6及び8から成る群より選択されたアミノ酸を含むことを特徴とする組成物。
【請求項3】
請求項2に記載の組成物であって、前記融合タンパク質が配列番号1,3,5及び7から成る群より選択される核酸配列によりコードされることを特徴とする組成物。
【請求項4】
請求項1に記載の組成物であって、前記プロテアーゼインヒビターがボウマン−バーク(Bowman-Brik)インヒビター(BBI)、ダイズトリプシンインヒビター(STI)、及びエグリン(eglin)Cから成る群より選択されるプロテアーゼインヒビターであることを特徴とする組成物。
【請求項5】
請求項4に記載の組成物であって、前記プロテゼーゼインヒビターがBBIであり、前記BBIがトリプシンループ及びキモトリプシンループから成る群より選択される少なくとも1のループを含むことを特徴とする組成物。
【請求項6】
請求項4に記載の組成物であって、前記プロテアーゼインヒビターが前記所望のペプチドのスキャッフォールドであることを特徴とする組成物。
【請求項7】
請求項5に記載の組成物であって、前記ループが前記所望のペプチドを含むことを特徴とする組成物。
【請求項8】
請求項5に記載の組成物であって、前記所望のペプチドが配列番号9で定義されるアミノ酸配列を含むことを特徴とする組成物。
【請求項9】
請求項5に記載の組成物であって、前記融合タンパク質が配列番号4で定義されるアミノ酸配列を有することを特徴とする組成物。
【請求項10】
配列番号10及び12で定義されるポリペプチド配列から成る群から選択されるプロテアーゼインヒビターをコードする単離ポリヌクレオチド。
【請求項11】
スキャッフォールドタンパク質及び少なくとも1のペプチドを含む組成物であって、前記スキャッフォールドタンパク質がボウマン-バーク(Bowman-Brik)インヒビターを含むことを特徴とする組成物。
【請求項12】
請求項1乃至9及び11のいずれか1項に記載の組成物であって、前記少なくとも1のペプチドが血管内皮成長因子に結合することを特徴とする組成物。
【図1A】


【図1B】

【図1C】

【図1D】

【図2】

【図3】

【図4】
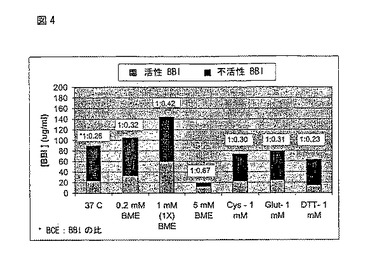
【図5】
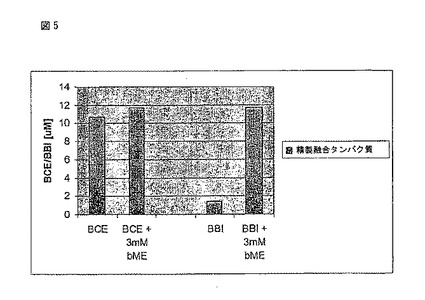
【図6】

【図7A】

【図7B】

【図7C】

【図8A】

【図8B】

【図8C】
【図1B】
【図1C】
【図1D】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7A】
【図7B】
【図7C】
【図8A】
【図8B】
【図8C】
【公開番号】特開2012−24091(P2012−24091A)
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【出願番号】特願2011−179629(P2011−179629)
【出願日】平成23年8月19日(2011.8.19)
【分割の表示】特願2006−539676(P2006−539676)の分割
【原出願日】平成16年11月6日(2004.11.6)
【出願人】(500284580)ジェネンコー・インターナショナル・インク (67)
【Fターム(参考)】
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【出願日】平成23年8月19日(2011.8.19)
【分割の表示】特願2006−539676(P2006−539676)の分割
【原出願日】平成16年11月6日(2004.11.6)
【出願人】(500284580)ジェネンコー・インターナショナル・インク (67)
【Fターム(参考)】
[ Back to top ]