
プロテインキナーゼの阻害剤
本発明は、サイクリン依存性キナーゼの阻害剤及びその治療的適用に関する。さらに、本発明は、サイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤の投与を含む、いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を予防し及び/又は治療する方法に関する。
【化1】

【化1】
【発明の詳細な説明】
【背景技術】
【0001】
(発明の背景)
サイクリン依存性プロテインキナーゼ(「CDK」)は、細胞周期の制御及び転写調節等の、細胞内での複数の役割を担う十分に保存された酵素のファミリーを構成する(Science 1996, Vol. 274:1643-1677;Ann. Rev. Cell Dev. Biol., 1997, 13:261-291)。
【0002】
CDK1、2、3、4、及び6等の、ファミリーのいくつかのメンバーは、G1の静止期(細胞分裂の新たな一区切りについての有糸分裂とDNA複製の開始との間のギャップ)からS期(活発なDNA合成の期間)への進行、又はG2期から、活発な有糸分裂及び細胞分裂が生じるM期への進行など、細胞周期の異なる相間の移行を制御する。CDK7、8、及び9を含むこのファミリーのタンパク質の他のメンバーは、転写周期における重要な点を制御するのに対し、CDK5は、神経細胞及び分泌細胞の機能における役割を担う。
【0003】
CDK複合体は、制御サイクリンサブユニット(例えば、サイクリンA、B1、B2、D1、D2、D3、及びE)と触媒キナーゼサブユニット(例えば、cdc2(CDK1)、CDK2、CDK4、CDK5、及びCDK6)との会合を通じて形成される。名称が含意するように、CDKは、CDKの標的基質をリン酸化するために、サイクリンサブユニットへの絶対的な依存性を示し、異なるキナーゼ/サイクリン対は、細胞周期の特定の部分を通じて進行を制御するよう機能する。
【0004】
CDK9のサイクリンパートナー(サイクリンT1、T2a、T2b、又はK)との会合にあるCDK9は、RNAポリメラーゼIIの最大のサブユニットのカルボキシル末端ドメイン(CTD)をリン酸化することによって、転写の伸長相の間に機能する正のP-TEFbプロテインキナーゼ複合体の触媒性構成要素を構成する。P-TEFbは、正の転写因子NfκB及び負の転写因子に関して作用し、このように転写伸長の遮断を克服する(Liu及びHerrmann 2005)。
【0005】
腫瘍細胞の中心的な特徴の1つである細胞周期の制御障害が、CDK及びその制御因子の遺伝的な変化及び制御障害と密接に関連していることは公知であり、CDKの阻害剤が、癌等の増殖性疾患のための治療薬として有用であり得ることを示唆する。このように、CDKを標的とする小分子阻害剤はこれまで、癌治療法における甚大な関心対象の焦点であった(Current Opinion in Pharmacology, 2003(3): 362-370)。細胞周期の進行を阻害する能力は、癌等の増殖性疾患のための治療薬としての、CDKの小分子阻害剤についての一般的な役割を示唆する。細胞周期関連型CDKの阻害は、腫瘍学の適用において明確に関連性があるが、このことは、RNAポリメラーゼを制御するCDKの阻害に関する場合ではないかもしれない。近年、CDK9/サイクリンTの機能の阻害は、HIV複製の予防と連関し、このように、新たなCDK生物学に関する発見は、例えば、ウイルス感染(WO 02/100401)等のCDK阻害剤についての新たな治療上の効能を利用できるようにし続ける(Sausville, E.A.の文献(Trends Molec. Med. 2002, 8, S32-S37))。また、CDK阻害剤は、とりわけ免疫学的疾患及び神経変性疾患等の他の容態を治療するためにおそらく使用され得る。
【0006】
50を超える薬理学的CDK阻害剤が記載されており、そのうちのいくつかは、強力な抗腫瘍活性を有する(Current Opinion in Pharmacology, 2003(3): 362-370)。公知のCDK阻害剤についての包括的な総説は、Angew. Chem. Int. Ed. Engl. 2003, 42(19):2122-2138において見出され得る。
【0007】
例えば、癌の治療のためのサイクリン依存性キナーゼ阻害剤としての2-アニリノ-4-フェニルピリミジン誘導体の使用は、WO 2005/012262において報告されている。さらに、癌等の治療のための2-ピリジニルアミノ-4-チアゾリル-ピリミジン誘導体は、WO2005/012298に記載されている。プロテインキナーゼ阻害剤としての4,5-ジヒドロ-チアゾロ、オキサゾロ及びイミダゾロ[4,5-h]キナゾリン-8-イルアミンの使用は、WO2005/005438から公知である。さらに、サイクリン依存性キナーゼ阻害剤として有用なインドリノン誘導体及びインヅリビン(induribin)誘導体は、WO02/081445及びWO02/074742に開示されている。さらに、治療上の多様な適用のためのCDK阻害剤は、WO2005/026129に記載されている。
【0008】
公知のCDK阻害剤は、一般にCDKを阻害するCDK阻害剤の能力に従って、又は特定のCDKに対するCDK阻害剤の選択性に従って分類され得る。例えば、フラボピリドールは、「受け皿の」CDKアンタゴニストとして作用し、特定のCDKに対して特に選択的ではない(Current Opinion in Pharmacology, 2003(3): 362-370)。オロモウシン、ロスコビチン、プルバノロール(purvanolol)及びCGP74514A等のプリンベースのCDK阻害剤は、CDK1、2及び5に対するより大きな選択性を呈することが公知であるが、CDK4及び6に対する阻害活性を何ら示さない(Current Opinion in Pharmacology, 2003(3): 362-370)。さらに、ロスコビチン等のプリンベースのCDK阻害剤が、神経系において抗アポトーシス性効果を発揮でき(Pharmacol Ther 2002, 93:135-143)、又はアルツハイマー病等の神経変性疾患における神経細胞の死滅を予防できることが示されている(Biochem Biophys Res Commun 2002 (297):1154-1158;Trends Pharmacol Sci 2002 (23):417-425)。
【0009】
増殖性疾患、免疫学的疾患、感染性疾患、心血管疾患及び神経変性疾患等の容態の治療法のための標的指向化CDKのすばらしい潜在性を考慮すると、具体的なCDKの選択的阻害剤としての小分子の開発は、所望の目的の構成要素となる。
【0010】
本発明は、サイクリン依存性キナーゼの新規の小分子阻害剤を提供する。好ましくは、該小分子阻害剤は、具体的なCDK、特にCDK9を阻害する高い効能を示す。該小分子阻害剤は、増殖性疾患、免疫学的疾患、神経変性疾患、感染性疾患及び心血管疾患等の容態の治療のための治療的有用性を有し得る。さらに、本発明の小分子阻害剤は、炎症性疾患の及びいずれかの種類の疼痛の治療において有益な効果を発揮することが驚くべきことに示されている。
【0011】
疼痛についての現行の治療は、部分的に効果的であるに過ぎず、また多くは、衰弱させる又は危険な副作用を生じる。例えば、重度の疼痛を治療するのに使用される伝統的な鎮痛薬の多くは、吐き気、めまい、便秘、呼吸抑制、及び認知機能障害等の衰弱させる副作用を誘発する(Brower, 2000)。
【0012】
利用可能な非麻酔性鎮痛薬、オピオイド鎮痛薬、カルシウムチャネル遮断薬、筋弛緩薬、及び全身性コルチコステロイドのような認可された疼痛投薬の広範な名簿はすでに存在するが、該治療は、単に経験的なものであるままに過ぎず、及び該治療は、疼痛の症状を軽減し得るが、該治療は、ほとんどのケースにおいて軽減を完了させるには至らない。また、このことは、異なる種類の疼痛の発達に潜む機構が、なおもほとんど理解されていないに過ぎないという事実による。研究者は、各種類の疼痛についての神経インパルスを遅延させるのに使用されるシグナル伝達系の複雑性及び多様性をまさに正しく認識し始めつつあるに過ぎない。
【0013】
一般的に、疼痛は、国際疼痛学会(International Association for the Study of Pain(IASP))に従って、実際の又は潜在的な組織の障害と関連した不愉快な感覚的及び情動的経験として定義され、又はこのような障害の点で記載される。具体的には、疼痛は、急性又は慢性の疼痛として生じ得る。
【0014】
急性疼痛は、短時間、典型的には1ヶ月未満にわたって生じ、一時的な障害と関連する。急性疼痛は、短時間内でさらなる障害を結果的に生じ得る生理学的又は生化学的変化を、宿主に知らせるための自然な身体反応である。有害な刺激が、末梢神経終末における高い閾値の機械的侵害受容器及び/又は熱侵害受容器を活性化し、薄い有髄(Aδ)の及び/又は無髄(C)の求心性線維における誘発活動電位が知覚のある脳に到達する場合、急性疼痛が感じられる。該有害な刺激は、損傷、手術、病気、外傷又は有痛の医学的手法によって提供され得る。急性疼痛は通常、潜在的な原因が治療され又は治癒された場合に消失する。しかしながら、軽減されない急性疼痛は、長期の入院、再入院、外来診療所及び緊急部門への訪問、並びに高額な保健医療費用を結果として生じ得る慢性疼痛の問題に至り得る。
【0015】
急性疼痛とは対照的に、慢性疼痛は、初期損傷が治癒した後に長く持続し、しばしば身体の他の部分へ拡大し、それに伴って多様な病理学的及び精神医学的結果が生じる。慢性的な体性痛は、末梢組織における外傷に対する炎症反応から結果的に生じ(例えば、神経の絞扼、外科的手法、癌、又は関節炎)、それが侵害受容器の感作過剰、及び通常は有害ではない刺激に対する激しい灼熱性疼痛反応に至る(痛覚過敏)。慢性疼痛は、持続的かつ再発性があり、その強度は、軽度から、生活の質を有意に低下させ得る重度の身体障害疼痛まで変動するであろう。
【0016】
慢性疼痛は目下のところ、NSAID(イブプロフェン、ナプロキセン)、Cox-2阻害剤(セレコキシブ、バルデコキシブ、ロフェコキシブ)及びアヘン製剤(コデイン、モルヒネ、テバイン、パパベリン、ノスカピン)等の従来の鎮痛薬を使用して治療される。しかしながら、有意な数の患者に対して、これらの薬物は不十分な疼痛軽減しか提供しない。
【0017】
別のサブタイプの疼痛である炎症性疼痛は、急性及び慢性の疼痛として生じ得る。結果として生じる組織及び神経細胞の損傷は、これらの炎症性事象を継承して、長期間持続する慢性の神経因性疼痛効果へ発展してはならないが、発展し得る。
【0018】
炎症性疼痛は、例えば、組織損傷、疾患、又は炎症後に放出される炎症性仲介物質(例えば、TNFα等のサイトカイン、プロスタグランジン、P基質、ブラジキニン、プリン、ヒスタミン、及びセロトニン)のような有害刺激及び他の有害刺激(例えば、熱刺激、機械的刺激、又は化学的刺激)によって仲介される。さらに、サイトカイン及び増殖因子は、神経細胞の表現型及び機能に影響し得る(Besson 1999)。これらの仲介物質は、組織の周囲のいたるところに分布する侵害受容器(感覚受容器)によって検知される。該侵害受容器は、長期化した場合、組織に障害を与えるであろう(例えば、機械的、熱の、又は化学的)有害刺激に対して感受性がある(Koltzenburg 2000)。特別なクラスのいわゆるC型侵害受容器は、いずれのレベルの機械的刺激又は熱刺激にも反応しないが炎症のみの存在下で活性化されるあるクラスの「サイレントな」侵害受容器に相当する。
【0019】
特に炎症性疼痛の治療のための現行のアプローチは、サイトカイン阻害(例えば、IL1β)及び炎症誘発性TNFαの抑制を目的とする。認可された現行の抗サイトカイン/抗TNFα治療は、インフリキシマブ及びエタネルセプト等の、血流におけるTNFα循環を低下させるキメラ抗体に基づいている。TNFαは、COX-2、MMP、iNOS、cPLa2及びその他等の重要な酵素の合成を誘導する最も重要な炎症性仲介物質の1つである。しかしながら、これらの「生物製剤」の主要な欠点は、効能の損失の付随する生物製剤の免疫原性能力及び、循環しているTNFαの多かれ少なかれ全か無のデジタルな減少に至る生物製剤の動態に存する。後者は、重度の免疫抑制性の副作用を結果として生じ得る。
【0020】
別個の形態の慢性疼痛である神経因性(又は神経原性)疼痛は、末梢神経又は中枢神経の機能障害の結果として生じ、病因及び部位において異なる多様な容態を含む。一般的に、神経因性疼痛の原因は多様だが、末梢神経に対する又は中枢経路の構成要素に対する障害に関する普遍的な症状を共有する。原因となる因子は、代謝性の、ウイルス性の又は機械的な神経病変であり得る。神経因性疼痛は、末梢神経系、中枢神経系、又はその両者における異所性の体性感覚過程によって持続されると信じられている。神経因性疼痛は、侵害受容器の刺激と直接連関していないが、代わりに、例えば脊髄の灰白質(後角)にあるシナプス後神経線維におけるグルタミン酸受容体の感作過剰から生じると考えられる。
【0021】
神経因性疼痛は、糖尿病及び帯状疱疹後神経痛(帯状疱疹)における神経変性等の容態と関連している。神経因性疼痛の容態は、糖尿病、エイズ、多発性硬化症、切断後の断端痛及び幻肢痛、癌関連ニューロパチー、帯状疱疹後神経痛、外傷性神経損傷、虚血性ニューロパチー、神経圧迫、脳卒中、脊髄損傷を含む多くの疾患及び容態の結果である。
【0022】
神経因性疼痛の管理は、神経因性疼痛の発達及び維持に関与する機構に関する不適切な理解に一部起因して、臨床上の主要な挑戦の域を出ない。既存の多くの鎮痛薬は、神経因性疼痛を治療する上で効果がなく、現行の麻酔性薬物及び非麻酔性薬物のほとんどは疼痛を調節しない。現行の臨床的実践には、神経因性疼痛の管理のための多くの薬物クラス、例えば抗痙攣薬、三環系抗うつ薬、及び全身性局所麻酔薬の使用が含まれる。しかしながら、これらの治療の通常の結果は、部分的な又は不満足な疼痛軽減であり、いくつかの場合、これらの薬物の有害作用は、該薬物の有用性よりも重い。古典的な鎮痛薬は、神経因性疼痛の治療においてほとんど効果がなく又はまったく効果がないと広く信じられている。神経因性疼痛の治療における非ステロイド性消炎薬(NSAID)又はアヘン製剤の使用に関するいくつかの臨床研究が実施されたが、実施された該臨床研究において、結果は、NSAIDがほとんど効果的ではなく又はまったく効果的ではなく、及びアヘン製剤が高い用量でしか作用しないことを示すように思われる。末梢神経因性疼痛(PNP)についての調節された臨床データを分析する総説(Pain, November, 1997 73(2), 123-39)は、NSAIDがPNPのための鎮痛薬としておそらく効果的ではなく、薬物の鎮痛効果を支持する長期的なデータが全くないことを報告した。
【0023】
利用可能な鎮痛薬はしばしば、不十分な疼痛軽減を生じる。三環系抗うつ薬及びいくつかの抗てんかん薬、例えばガバペンチン、ラモトリギン及びカルバマゼピンは、患者によっては有効であるが、これらの容態の治療に有効な薬物について、まだ対処されていない需要が大きいままである。
結論として、疼痛治療、特に慢性の炎症性疼痛及び神経因性疼痛に関する安全でかつ効果的な方法について、まだ対処されていない高い需要がある。
【発明の概要】
【0024】
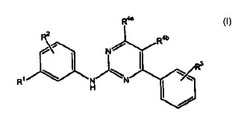
本発明は、サイクリン依存性キナーゼの阻害剤、並びにいずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療し及び/又は予防するための方法及び組成物であって、該方法又は組成物を必要とする対象へ、サイクリン依存性キナーゼ(cdk、CDK)の有効量の少なくとも1つの阻害剤を投与することを含む、前記方法及び組成物に関する。阻害剤は、一般式I記載の化合物:
【化1】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;及びこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)のうちで選択される。
(式中、
R1が、-XSO2NR5R6又は-XSO2R8であり;式中、
Xが、(分岐鎖アルキレンを含む)C1-4アルキレンであり、式中、該C1-4アルキレンが、R5又はR6へ結合して、5〜6員の複素環を形成でき;
R5及びR6が互いに独立して、水素、C1-4アルキル、ヒドロキシ-C1-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル若しくはC4-7-ヘテロシクロアルキル-C0-4アルキル、C4-7-アリール-C0-4アルキル、C4-7-ヘテロアリール-C0-4アルキル又は
式中、R5及びR6はそれらが結合するN原子とともにまた、5〜8員のヘテロシクロアルキルを形成し得、
式中、該シクロアルキル、ヘテロシクロアルキル、アリール、ヘテロアリール又はアルキルがさらに、ハロ、ヒドロキシ、アミノカルボニル、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O-及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R8が、C1-4アルキル、ヒドロキシ-C2-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル又はC4-7-ヘテロシクロアルキル-C0-4アルキルであり;
式中、該シクロアルキル、ヘテロシクロアルキル又はアルキルがさらに、ハロ、ヒドロキシ、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R2が、ハロゲン及び水素から独立して選択される1個又は2個の置換基であり;
R3が、水素、ハロ、ヒドロキシ、C1-4アルキル、C3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、アリール-C1-4アルコキシ、ヘテロアリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルキル、ヘテロアリール-C1-4-アルキル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から各々独立して選択される1〜3個の置換基であり得、この中で、上述の置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、アミノカルボニル、ハロ及びNR5R6の群から選択されるラジカルによってさらに置換でき;
R4a及びR4bが、同一又は異別であり、かつ各々が独立して、水素、C1-4アルキル又は-NR'R"であり、式中R'及びR"が各々独立して、水素又はC1-4アルキルである。)。
【0025】
本明細書に開示されるように、「C1-4アルキル」という用語は、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、イソブチル、tert-ブチル等の、1〜4個の炭素原子を有する直鎖又は分岐鎖の飽和脂肪族炭化水素を含むよう意味される。
「低級アルケニル」という用語は、例えば、ビニル、アリル、ブト-2-エニル、-ブト-3-エニル、又はイソプロペニル等の2〜6個の炭素原子を有する直鎖又は分岐鎖アルケン基を指し;「低級アルケニル」という用語は好ましくは、アリル、-ブト-2-エニル、又は-ブト-3-エニルを表す。
【0026】
本明細書に開示されるように、「ハロ」という用語は、フルオロ、クロロ、ブロモ、及びヨードを含むことを意味する。
C3-C8シクロアルキルという用語は、下記のシクロアルキル:
【化2】
を示す。
アリールという用語は、フェニル、ナフチル、3-クロロフェニル、2,6-ジブロモフェニル、2,4,6トリブロモフェニル、4,7-ジクロロナフチル、好ましくはフェニル又はナフチル等の芳香族単環式又は二環式の6〜10員の環系を示す。
【0027】
ヘテロシクロアルキルという用語は、酸素、硫黄又は窒素から独立して選択される1〜3個のヘテロ原子を含有する5〜10員の単環式又は二環式環系を含むことを意味し、好ましくはアジリジニル、アゼチジニル、ピロリジニル、テトラヒドロフラニル、テトラヒドロチオフェニル、ピペリジニル、ピペラジジニル、ピペラジニル、テトラヒドロピラニル、テトラヒドロチオピラニル又はモルフォリニルを含む群から選択される。
【0028】
ヘテロシクリルという用語はさらに、以下に定義されるすべてのヘテロアリールを含み、この中で、対応するヘテロアリールのすべての二重結合は、単結合によって置換される。
ヘテロアリールという用語は、酸素、硫黄又は窒素から独立して選択される1〜3個のヘテロ原子を含有する、部分的に又は完全に不飽和の5〜10員の単環式又は二環式の環系を示し、好ましくはピロリル、フラニル、チオフェニル、チエニル、イミダゾリル、ピラゾリル、チアゾリル、オキサゾリル、イソチアゾリル、イソキサゾリル、ピリジニル、ピリジル、ピリミジニル、ピリミジル、ピラジニル、ピラジル、ピラジジニル、ピラジジル、3-メチルピリジル、ベンゾチエニル、4-エチルベンゾチエニル、3,4-ジエチルフラニル、ピロリル、テトラヒドロキノリル、キノリル、テトラヒドロイソキノリニル、イソキノリニル、ベンゾイミダゾリル、ベンゾチアゾリル、ベンゾオキシゾリル、ベンゾ[1,3]ジオキソリル、インドリル、ベンゾフラニル、ベンゾチオフェニル、インダゾリル又はクロム-2-オニルからなる群から選択される。
【0029】
また、ヘテロアリールという用語が、部分的に不飽和の5〜10員の単環式又は二環式の環系を含み、この中で、該環系の1〜4個の二重結合が単結合によって置換され、及びこの中で、該環系が少なくとも1個の二重結合を含有することは理解されるべきである。
【0030】
変数が、式I又はいずれかの置換基において2回以上生じる場合、各発生に関する該変数の定義は、その他すべての発生における該変数の定義とは独立している。例えば、化合物が、2個以上のR5及び/又はR6置換基を含む場合、これらの置換基は同一又は異別であり得る。
【0031】
式Iの化合物において、
Xは、好ましくはメチレンである。
R2は、好ましくは水素である。
好ましくは、R6は、水素又はメチルであり、及びR5は、エチル、2-ヒドロキシエチル、イソプロピル、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシル、n-プロピル、t-ブチル、3-メトキシ-プロピル、2-ジメチルアミノエチル、3-ジメチルアミノプロピル、ピペリジニル及び特に4-ピペリジニル、ピリジニル及び特にピリジン-3-イル又はピリジン-4-イル、ピロリジニル及び特にピロリジン-3-イル、テトラヒドロフラニル及び特にテトラヒドロ-フラン-3-イル、テトラヒドロ-フラン-2-イルメチル、4-クロロ-ベンジル、チオフェン-2-イル-メチル、又はR5及びR6は両者とも、水素、メチル、エチル、又は
R5及びR6はそれらが結合するN原子とともに、モルフォリン、4-アミノカルボニル-ピペリジン又はアゼパンを形成し、又は-XSO2NR5R6は、
【化3】
である。
R5及びR6は、より好ましくは各々独立して、水素、メチル、シクロプロピル、2-ジメチルアミノエチル、3-ジメチルアミノプロピル及び2-ヒドロキシエチルから選択される。R5及びR6は、最も好ましくは水素である。
【0032】
R8は好ましくは、C1-4アルキル又はヒドロキシ-C-2-4-アルキルであり;R8はより好ましくは、メチル又は2-ヒドロキシエチルである。
R3は好ましくは、水素、ハロ、ヒドロキシ、C1-4アルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基であり;又はC3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、アリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4-アルキル、ヘテロアリール-C1-4アルコキシ、ヘテロアリール-C1-4-アルキルから選択される1個の置換基であり;この中で、該置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、ハロ、アミノカルボニル、及びNR5R6の群から選択される1個以上のラジカルによってさらに置換できる。R3はより好ましくは、メチル、エチル、ヒドロキシメチル、ヒドロキシ、メトキシ、エトキシ、イソプロポキシ、ベンジルオキシ、水素、フルオロ、クロロ、トリフルオロメチル、2-メトキシ-エトキシ、メトキシメチル、2-メトキシ-エチル、テトラヒドロ-フラン-3-イルオキシ、テトラヒドロ-フラン-2-イル-メトキシ、-N(CH3)SO2CH3、ピペリジン-1-イル-メチル、2-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-(2-ヒドロキシ-エチル)-ピペリジン-1-イル-メチル、3-アミノカルボニル-ピペリジン-1-イル-メチル、ジメチルアミノメチル、ジエチルアミノメチル、(エチル-イソプロピル-アミノ)-メチル、モルフォリン-4-イルメチル、4-メチル-ピペラジン-1-イル-メチル、[1,2,4]トリアゾール-1-イル-メチル、ピリジン-3-イル-メトキシ及びピリジン-4-イル-メトキシからなる群から独立して選択される1〜3個の置換基である。R3は最も好ましくは、メトキシ、イソプロポキシ及び/又はフルオロである。
【0033】
式Iの化合物の別の好ましい群は、式Iaの化合物
【化4】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;及びこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば水和物)を含む
(式中、
R2、R4a及びR4bは、上述に定義されるとおりであり;
R3a、R3b及びR3cは、R3に関して上述に定義されるとおりであり;及び
R9は、NR5R6又はR8であり、式中、R5、R6及びR8は、上述に定義されるとおりである。)。
【0034】
式Iaのこれらの化合物内で:
R2は好ましくは、水素又はハロゲンであり、より好ましくは水素である。
R3aは、好ましくは水素、ハロ又はC1-4アルコキシであり;より好ましくはC1-4アルコキシであり;最も好ましくはメトキシ又はイソプロポキシである。
R3bは、好ましくは水素である。
R3cは、好ましくは水素又はハロゲンであり、特にフルオロである。
R4aは、好ましくはC1-4アルキル又は水素であり;より好ましくは水素又はメチルであり;最も好ましくは水素である。
R4bは、好ましくは水素又はC1-4アルキルであり;より好ましくは水素又はメチルであり;最も好ましくは水素である。
R5及びR6は、好ましくは各々独立して、水素、ヒドロキシル又はジアルキルアミノによって任意に置換されるシクロアルキル又はC1-4アルキルであり;最も好ましくはR5及びR6は、各々独立して、水素、メチル、シクロプロピル、2-ヒドロキシエチル、2-ジメチルアミノエチル又は3-ジメチルアミノプロピルである。
R8は、好ましくはヒドロキシ-C-2-4-アルキルであり、より好ましくは2-ヒドロキシエチルである。
【0035】
下記の化合物が好ましい:
{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物1);
{3-[4-(2-イソプロポキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物2);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N,N-ジメチル-メタンスルホンアミド(化合物3);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物4);
{3-[4-(4-フルオロ-2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物5);
N-シクロプロピル-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物6);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物7);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物8);
2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物9);
{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物10);
-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物11);
2-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物12);
{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物13);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物14);及び
2-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物15)。
【0036】
さらなる実施態様において、本発明は、医療的使用のために、式I記載の上述の化合物を提供する。
さらなる実施態様において、本発明は、医薬として許容し得る担体とともに先に概略した化合物を含有する医薬組成物を提供する。
【0037】
さらなる実施態様において、本発明は、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための医薬組成物を調製するための先に概略した化合物の使用を提供する。
【0038】
さらなる実施態様において、本発明は、疼痛、慢性疼痛、及び/又は神経因性疼痛を治療するための医薬組成物を調製するための先に概略した化合物の使用を提供する。
さらなる実施態様において、本発明は、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための方法であって、必要とする対象に、有効量の上述の化合物の少なくとも1つの投与を含む、前記方法を提供する。
さらなる実施態様において、本発明は、疼痛、慢性疼痛及び/又は神経因性疼痛を治療するための方法であって、必要とする対象に、有効量の上述の化合物の少なくとも1つの投与を含む、前記方法を提供する。
【図面の簡単な説明】
【0039】
【図1】図1は、坐骨神経の2つの分岐(すなわち脛骨神経及び総腓骨神経)の結紮及び切断を特徴とする、腓腹神経を無処置のままにしている坐骨神経部分損傷モデル(SNIモデル、Decosterd及びWoolf(2000)によって開発)を模式的に図示する。
【図2】図2は、疼痛の発達における標的としてのCDK9の可能性のある役割を模式的に図示する。
【図3】図3は、LPS誘発性THP-1マクロファージを使用して実施されるサイトカイン測定の結果を図示する。TNF発現は、DMSO対照と比較した%阻害として示した。化合物1、5、6、8、又は9で処置した細胞は、媒体(DMSO)と比較して、上清におけるTNF発現の有意な阻害を示す。基準化合物SB203580(p38阻害剤)及びBMS345541(IKK阻害剤)と比較して、これらの化合物は、本アッセイにおけるTNF発現の同様の又はより良好な阻害を呈する。
【発明を実施するための形態】
【0040】
(発明の詳細な記述)
本発明によって提供されるCDKの阻害剤は、一般式I記載の化合物:
【化5】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;及びこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)である
(式中、
R1は、-XSO2NR5R6又は-XSO2R8であり;式中、
Xは、(分岐鎖アルキレンを含む)C1-4アルキレンであり、式中、該C1-4アルキレンが、R5又はR6へ結合して、5又は6員の複素環を形成でき;
R5及びR6が互いに独立して、水素、C1-4アルキル、ヒドロキシ-C1-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル若しくはC4-7-ヘテロシクロアルキル-C0-4アルキル、C4-7-アリール-C0-4アルキル、C4-7-ヘテロアリール-C0-4アルキル又は
式中、R5及びR6はそれらが結合するN原子とともにまた、5〜8員のヘテロシクロアルキルを形成し得、
式中、該シクロアルキル、ヘテロシクロアルキル、アリール、ヘテロアリール又はアルキルがさらに、ハロ、ヒドロキシ、アミノカルボニル、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O-及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R8が、C1-4アルキル、ヒドロキシ-C2-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル又はC4-7-ヘテロシクロアルキル-C0-4アルキルであり;
この中で、該シクロアルキル、ヘテロシクロアルキル又はアルキルがさらに、ハロ、ヒドロキシ、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R2が、ハロゲン及び水素から独立して選択される1個又は2個の置換基であり;
R3が、水素、ハロ、ヒドロキシ、C1-4アルキル、C3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、アリール-C1-4アルコキシ、ヘテロアリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルキル、ヘテロアリール-C1-4-アルキル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から各々独立して選択される1〜3個の置換基であり得、この中で、上述の置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、アミノカルボニル、ハロ及びNR5R6の群から選択されるラジカルによってさらに置換でき;
R4a及びR4bが、同一又は異別であり、かつ各々が独立して、水素、C1-4アルキル又は-NR'R"であり、式中R'及びR"が各々独立して、水素又はC1-4アルキルである。)。
【0041】
本発明の好ましい実施態様において、式I記載のサイクリン依存性キナーゼ阻害剤は、CDK1、CDK2、CDK3、CDK4、CDK5、CDK6、CDK7、CDK8、CDK9、CDK10、CDK11、CrkRS(CDC2関連プロテインキナーゼ7(Crk7))、CDKL1(サイクリン依存性キナーゼ様1型);KKIALRE、CDKL2(サイクリン依存性キナーゼ様2型)、KKIAMRE、CDKL3(サイクリン依存性キナーゼ様3型)、NKIAMRE、サイクリン依存性キナーゼ様1型と同様のCDKL4、CDC2L1(細胞分裂周期様2型)、PITSLRE A、CDC2L5(細胞分裂周期2型様5型)、PCTK1(PCTAIREプロテインキナーゼ1型)、PCTK2(PCTAIREプロテインキナーゼ2型)、PCTK3(PCTAIREプロテインキナーゼ3型)又はPFTK1(PFTAIREプロテインキナーゼ1型)からなる群から選択されるCDKを阻害する。
【0042】
また、阻害剤は、上述に列挙される群から選択される2つ以上のサイクリン依存性キナーゼを阻害し得る。
本発明の具体的な好ましい実施態様において、式I記載の化合物は、CDK9を阻害する。
【0043】
本発明のさらなる実施態様において、式I記載の化合物は、他の酵素又はタンパク質に及ぼす実質的な阻害効果を有さずに、1つ以上のCDKを選択的に阻害する。
好ましい実施態様において、これらの阻害化合物は、具体的なCDKに対する高い選択性を示す。本明細書で使用される「高い選択性」は、阻害化合物が、本明細書に列挙される上述のCDKの群から選択される具体的なCDKに対して少なくとも10〜100倍以上選択的である。本発明の好ましい実施態様において、阻害化合物は、具体的なCDKに対して20〜90倍以上選択的である。具体的な好ましい実施態様において、阻害化合物は、具体的なCDKに対して30〜80倍以上選択的である。
具体的な好ましい実施態様において、式I記載の化合物は、他のCDKに対するよりもCDK9に対して高い選択性を示す。
【0044】
本明細書で使用されるように、「阻害する」又は「阻害」という用語は、サイクリン依存性キナーゼの細胞機能、すなわち、サイクリン依存性キナーゼの活性又は発現を少なくとも部分的に下方制御し、低下させ、減少させ、抑制し、不活性化し、又は阻害する化合物の能力を指す。
【0045】
さらに、「サイクリン依存性キナーゼ阻害剤」という用語は、サイクリン依存性キナーゼの量及び/又は活性を下方制御でき、低下でき、抑制でき又はそれに代わるものとして制御できる化合物又は化合物の群を指す。該キナーゼの阻害は、キナーゼポリペプチドに直接結合すること、キナーゼを変性させること又はそれに代わるものとして不活性化させること、又はキナーゼをコードする遺伝子の発現(例えば、mRNAへの転写、新生ポリペプチドへの翻訳、及び/又は成熟タンパク質への最終的なポリペプチド修飾)を阻害することを含むがそれらに限定されるわけではない、当技術分野で公知の多様な機構によって達成できる。さらにまた、サイクリン依存性キナーゼ阻害剤は、CDK依存性経路におけるCDKの下流に作用する分子の発現、修飾、制御又は活性化に干渉し得る。一般的に、キナーゼ阻害剤は、タンパク質、ポリペプチド、核酸、小分子、又は他の化学的部分であり得る。特に、キナーゼ阻害剤にはまた、サイクリン依存性キナーゼに対して配向されるモノクローナル抗体又はポリクローナル抗体が含まれる。
好ましい実施態様において、サイクリン依存性キナーゼ阻害剤は、本明細書に開示される式Iによって表される化合物から選択される。
【0046】
(治療的使用)
式Iの化合物は、サイクリン依存性キナーゼの阻害剤である。このように、該化合物は、異常に分裂する細胞における細胞周期の調節を抑止し又は回復させる能力を有すると期待される。結果として、式I記載の化合物は、癌等の増殖性障害を治療し及び/又は予防する上で有用であると立証するであろう。
【0047】
CDKは、アポトーシス、増殖、分化及び転写における役割を担うことが公知であり、それゆえまた、式I記載の化合物は、感染性疾患、免疫学的疾患、神経変性疾患及び心血管疾患など、増殖性疾患以外の疾患の治療において有用であり得る。
【0048】
さらにまた、式I記載の化合物は、予期せぬ抗侵害受容効果及び抗炎症効果を示す。
このように、好ましい実施態様において、式Iの化合物は、慢性疼痛、神経因性疼痛及び/又は炎症性疼痛を含むいずれかの種類の疼痛の治療のための方法及び/又は医薬組成物において使用され得る。
【0049】
さらなる好ましい実施態様において、式Iの化合物は、炎症性障害の治療のための方法及び/又は医薬組成物において使用され得る。
具体的な好ましい実施態様において、いずれかの種類の疼痛の治療における又は炎症性障害の治療における使用のための式Iの化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0050】
(疼痛)
神経病変を罹患しているマウスへの式I記載のCDK阻害剤の投与が、特に炎症性及び神経因性疼痛のマウスモデルにおいて痛覚鈍麻効果を発揮することがわかっている。
サイクリン依存性キナーゼの阻害が、痛覚鈍麻効果を仲介することに関与するという発見は、予期されていなかった。
【0051】
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の阻害剤を投与することを含む、いずれかの種類の疼痛を治療する方法に関する。特に、式Iの化合物は、慢性、神経因性及び/又は炎症性疼痛の治療に使用され得る。具体的な好ましい実施態様において、いずれかの種類の疼痛の治療における使用のための式Iの化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0052】
疼痛の発達におけるCDK9の役割は、下記の作用機構に基づき得る:サイクリンT1及びCDK9の両者は、TNFαのプロモーターの基礎活性を刺激する。TNFαは、炎症誘発性サイトカインおよび炎症性の遺伝子ネットワークの発現を調節する疼痛仲介因子である。細胞性TNF受容体反応の仲介のために、核因子κB(NFκB)経路が非常に重要である。TNFαは、サイトカイン遺伝子へのNFκBの動員を惹起するのに対し、NfκBは、遺伝子転写の刺激のためにp-TEFb複合体と相互作用する(Barboric Mら, 2001)。
【0053】
さらに、CDK9が、TNFα受容体複合体の一員であるTRAF2の結合パートナーであることが示されている(MacLachlanら, 1998)のに対し、炎症誘発性IL6受容体複合体のサブユニットであるGP130が、CDK9の別のあり得る結合パートナーとして近年同定されている(Falcoら, 2002)。TNFα及びインターロイキンのシグナル伝達における、及びいくつかの遺伝子(例えば、疼痛仲介因子としてのサイトカイン)のNfκB仲介性発現における鍵となる担い手として、このように、CDK9は、炎症性疼痛など、いずれかの種類の疼痛の治療のための中心的な標的として考慮できる(図2参照)。
【0054】
神経因性疼痛の治療のために、薬理学的作用は、中枢神経系(CNS)における血液脳関門(BBB)を越えて生じなければならない。例えば、CNSにおける主要な免疫細胞としての小膠細胞は、活性化の際に、サイトカイン(TNFα、IL1β、IL6)及び他の炎症誘発性分子等の多様な有害な因子を放出する(Huwe 2003)。ミクログリアは、TNFα受容体又はトール様受容体の刺激によって活性化され、シグナルは、Iκキナーゼ(IKK)及び、上述のサイトカインの転写活性化に至るNfκBを介して仲介される。ミクログリアの関与は、慢性CNS疾患において助けになるものとして論議されてきており、疼痛知覚に関与し得る(Watkinsら, 2003)。
【0055】
近年、転写因子NfκBが、脊髄においてインターロイキン1β(IL1β)を介してシクロオキシゲナーゼ2(COX-2)の発現を制御することが示された(Leeら 2004)。脊髄プロスタグランジンE2の上昇に対する主要な関与因子として、疼痛仲介因子COX-2はすでに、多様な抗侵害受容薬/抗炎症薬のための標的として公知である。NfκB阻害剤は、動物モデルにおけるCOX-2レベル及び機械的アロディニア及び熱痛覚過敏を有意に減少させるNfκB阻害剤の能力を立証している。
【0056】
COX-2とは対照的に、CDK9作用の阻害は、単一の疼痛仲介因子のみの抑制の代わりに、多様な疼痛仲介因子の抑制に至るであろう。このように、CDK9阻害剤の抗侵害受容作用は、例えばCOX-2阻害剤と比較して優れ得る。
それゆえ、NfκB仲介性遺伝子転写とのCDK9阻害剤の関連性により、CDK9との阻害相互作用は、急性炎症性疼痛の治療のためのみならず、慢性疼痛の治療のための妥当なアプローチであり得る。
【0057】
本明細書で使用される「疼痛」という用語は一般的に、いずれかの種類の疼痛に関し、急性疼痛、慢性疼痛、炎症性及び神経障害性疼痛等の様々な種類の疼痛を広範に包含する。本発明の好ましい実施態様において、「疼痛」は、神経因性疼痛及び関連容態を含む。疼痛は、慢性、アロディニア(通常無害の刺激に由来する疼痛の知覚)、痛覚過敏(付与されたいずれかの疼痛刺激に対する過度の反応)及び受容野(すなわち、刺激が適用される場合に「痛い」領域)の拡大、幻肢痛又は炎症性疼痛であり得る。
【0058】
急性疼痛の種類は、組織障害と関連した疼痛、術後痛、外傷後の疼痛、熱傷によって生じる疼痛、局所感染又は全身感染によって生じる疼痛、下記を含む疾患と関連した内臓痛を含むが、これらに限定されるわけではない:膵炎、腸管膀胱炎、月経困難、過敏性腸症候群、クローン病、尿管仙痛及び心筋梗塞。
【0059】
さらに、「疼痛」という用語は、下記を含むCNS障害と関連した疼痛を含む:多発性硬化症、脊髄損傷、外傷性脳損傷、パーキンソン病及び脳卒中。
好ましい実施態様において、「疼痛」は、頭痛(例えば、片頭痛障害、偶発性及び慢性緊張型頭痛、緊張型様頭痛、群発頭痛、及び慢性発作性片側頭痛)、腰痛、癌性疼痛、骨関節炎疼痛及び神経因性疼痛を含む慢性疼痛の種類に関するが、それらに限定されるわけではない。
【0060】
本明細書で定義される炎症性疼痛(組織損傷及び結果として生じる炎症過程に応答した疼痛)は、結合組織疾患、関節リウマチ、全身性エリテマトーデス、多発性硬化症及び関節炎を含む疾患と関連した炎症性疼痛に関するが、それらに限定されるわけではない。
【0061】
神経因性疼痛(末梢神経又は中枢神経系自体に対する障害から結果として生じる疼痛)には、代謝性ニューロパチー(例えば、糖尿病性ニューロパチー)、帯状疱疹後神経痛、三叉神経痛、頭部神経痛、脳卒中後神経因性疼痛、多発性硬化症関連神経因性疼痛、HIV/エイズ関連神経因性疼痛、癌関連神経因性疼痛、手根管関連神経因性疼痛、脊髄損傷関連神経因性疼痛、複合性局所疼痛症候群、線維筋痛症関連神経因性疼痛、反射性交感神経性ジストロフィー、幻肢症候群又は末梢神経若しくは脊髄の外傷、外科的治療、体肢切断及び断端痛を含む神経離断、抗癌治療法及び抗エイズ治療法の副作用によって生じる疼痛、術後神経因性疼痛、特発性又は外傷後ニューロパチー及び単神経炎などにおけるニューロパチー関連疼痛、及び関節リウマチ、ワーレンベルグ症候群、全身性エリテマトーデス、多発性硬化症、又は結節性多発動脈炎等の結合組織疾患によって生じる神経因性疼痛を含む容態が含まれるが、それらに限定されるわけではない。ニューロパチーは、神経根障害、単ニューロパチー、多発性単神経炎、多発ニューロパチー又は神経叢障害として分類できる。
【0062】
「アロディニア」という用語は、通常は痛くない刺激から生じる疼痛を表す。アロディニア性疼痛は、刺激された領域以外で生じ得る。
「痛覚過敏」という用語は、痛い刺激に対する高い感度を表す。
「痛覚鈍麻」という用語は、痛い刺激に対する低い感度を表す。
【0063】
(炎症性疾患)
驚くべきことに、本明細書に開示される式I記載のCDK阻害化合物が、インビトロ及びインビボでの炎症アッセイにおいて抗炎症効果を発揮することを示すことができた。
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の阻害剤を投与することを含む炎症性疾患を治療する方法に関する。具体的な好ましい実施態様において、炎症性疾患の治療における使用のための式Iの化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0064】
炎症性疾患の発達におけるCDK9の役割は、下記の作用機構に基づき得る:関節リウマチ(RA)等の炎症性疾患;粥状硬化;喘息;炎症性腸疾患、全身性エリテマトーデス及びいくつかの他の自己免疫疾患は、該疾患における炎症性経路及び組織閉塞性経路の鍵となる制御因子である腫瘍壊死因子α(TNFα)によって仲介される。IκBタンパク質のリン酸化の際にNfκBから解離するIκBタンパク質をリン酸化するIκBキナーゼ(IKK)等のいくつかの伝達物質を介してTNFαシグナルが仲介されることは公知である。サイトカイン転写の正の制御因子である解離されたNfκBは、細胞核内に転位置し、そこで該NfκBはその認識部位に結合する。
【0065】
活性化されたNfκBは、RA患者の滑膜において発見されている[Hanらの文献(2003, Autoimmunity, 28, 197-208)]。該NfκBは、TNFα、IL-6、IL-8、NOS及びCOX2等の炎症誘発性遺伝子を制御する。標的指向化NfκB及びその上流のシグナル伝達パートナーであるIKKは、関節炎に関する多くの動物モデルにおける有効な治療戦略であることがすでに立証されている[Firesteinの文献(2003, Nature 423, 356-361)]。
【0066】
結合したNfκBは、RNA Pol IIのCTDのリン酸化を触媒するCDK9を動員し及び活性化するヒストンアセチルトランスフェラーゼ(CBP、p300、p/CAF、SRC-1、及びSRC-1関連タンパク質)を含有する活性化補助因子複合体と会合する[Westらの文献(2001, Journal of Virology 75(18), 85248537)]。結果として生じるRNA Pol II CTDの過剰リン酸化は、TNFαによって制御されるものとしても公知であるIL-1β、IL-6及びIL-8等の炎症誘発性サイトカインの転写の活性化に至る。
【0067】
いくつかの研究は、TNFαが、炎症誘発性サイトカイン発現を制御する自己シグナル伝達カスケードの「主要制御因子」であることを示した。この炎症誘発性カスケードを中断するために、特異的な抗体(Ab)をうまく使用して、TNFαシグナルを遮断できる。AbによるRAの抗TNFα治療は、いくつかの臨床研究におけるその治療的効能をすでに立証しており、インフリキシマブ及びエタネルセプト等の米国食品医薬品局認可薬は、市場に参入している[Feldmann及びMainiの文献(NatMed, 2003, 9 (10); 356-61)]。しかしながら、Abベースの治療法の不利には、該治療法の免疫原性能力、連続的な治療中の付随する効能の損失及び高い治療費が含まれる。さらに、Ab動態によって、循環しているTNFαの多かれ少なかれ全か無の減少が可能となる。結果としてまた、免疫応答の生理学的機能は抑制される[Lauferらの文献(免疫及びリウマチ疾患(Inflammation and Rheumatic Diseases), 2003; Thieme, pp. 104-5)]。
【0068】
P38 MAPK又はIKK等の標的を照準としたキナーゼ阻害剤を使用するTNFα仲介性シグナル伝達カスケードへの治療的介入は、重度の有害な効果を示しており、たいていの場合、個々の標的に対する特異性の欠失に起因する。
それとは対照的に、本明細書に提示される式I記載のCDK特異的阻害剤は、生理学的機能との相互作用を減少させるTNFαシグナル伝達経路の最下端で介入し得る。さらに、該化合物は、優れた特異性を介する有害な効果の回避によって、自己TNFα仲介性炎症ネットワークを中断できるであろう。それゆえ、式IのCDK特異的阻害剤を使用する治療は、炎症性疾患及び自己免疫疾患の治療のための有望な戦略を構成する。
このように、本明細書で提示される式I記載の化合物は、炎症性疾患の治療及び/又は予防のために使用され得る。
【0069】
本明細書で使用される「炎症性疾患」という用語は、身体組織の炎症を生じ、その後急性又は慢性の炎症容態を生じる免疫系又は組織の細胞性又は非細胞性仲介因子によって惹起される疾患に関する。
【0070】
これらの炎症性疾患の例は、I〜IV型の過敏性反応であり、例えば、喘息を含む肺の過敏性疾患、アトピー性疾患、アレルギー性鼻炎又は結膜炎、眼瞼の血管性浮腫、遺伝性血管性浮腫、抗受容体過敏性反応及び自己免疫疾患、橋本甲状腺炎、全身性エリテマトーデス、グッドパスチャー症候群、天疱瘡、重症筋無力症、グレーヴス及びレイノー病、 B型インスリン抵抗性糖尿病、関節リウマチ、乾癬、クローン病、皮膚硬化症、混合型結合組織疾患、多発性筋炎、サルコイドーシス、ウエゲナー肉芽腫、糸球体腎炎、急性又は慢性の宿主体移植片反応であるが、それらに限定されるわけではない。
【0071】
さらに、「炎症性疾患」という用語には、腹腔炎症、皮膚炎、消化管炎症(炎症性腸疾患、潰瘍性大腸炎を含む)、線維症、眼及び眼窩の炎症、眼乾燥症疾患及びシェーグレン症候群から結果的に生じる重度の眼乾燥症疾患、乳腺炎、耳炎、口腔炎症、筋骨格系の炎症(痛風、骨関節炎を含む)、中枢神経系の炎症性疾患(多発性硬化症、細菌性髄膜炎、髄膜炎を含む)、腎尿路生殖器の炎症(前立腺炎、糸球体腎炎を含む)、心血管の炎症(粥状硬化、心不全を含む)、気道の炎症(慢性気管支炎、慢性閉塞性肺疾患を含む)、甲状腺炎、糖尿病、骨炎、筋炎、多臓器不全(敗血症を含む)、多発性筋炎及び乾癬性関節炎が含まれるが、それらに限定されるわけではない。
【0072】
(免疫学的疾患)
また、式I記載の化合物は、例えば自己免疫疾患等の免疫学的疾患の治療及び/又は予防において有用であると考えられる。
従って、本発明は、免疫学的疾患の治療及び/又は予防のための方法であって、必要とする対象に、式I記載の有効量の少なくとも1つのCDK阻害剤の投与を含む、前記方法を提供する。
【0073】
本明細書で使用される「免疫学的疾患」という用語は、アレルギー、喘息、移植片対宿主病、免疫不全症及び自己免疫疾患を含むがそれらに限定されない疾患に関する。
特に、免疫学的疾患には、糖尿病、リウマチ性疾患、エイズ、慢性肉芽腫性疾患、移植された臓器及び組織の拒絶、鼻炎、慢性閉塞性肺疾患、骨粗鬆症、潰瘍性大腸炎、クローン病、副鼻腔炎、エリテマトーデス、乾癬、多発性硬化症、重症筋無力症、脱毛症、反復性感染症、アトピー性皮膚炎、湿疹及び重度のアナフィラキシー反応が含まれるが、それらに限定されるわけではない。さらに、「免疫学的疾患」にはまた、接触アレルギー、食物アレルギー又は薬剤アレルギー等のアレルギーが含まれる。
【0074】
(増殖性疾患)
式Iの化合物は、細胞周期の制御に関与する鍵分子を表すサイクリン依存性キナーゼの阻害剤である。細胞周期の制御障害は、腫瘍細胞の主要な特徴の1つである。このように、該化合物は、異常に分裂する細胞における細胞周期の調節を抑止し又は回復させる上で有用であることを立証すると期待される。このように、式I記載の化合物が、癌等の増殖性疾患の治療及び/又は予防において有用であることが期待される。
【0075】
従って、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む増殖性疾患の治療及び/又は予防のための方法を提供する。
本明細書で使用されるように、「増殖性疾患」という用語は、良性腫瘍、形成異常、過形成及び転移性増殖又は他の形質転換を示す腫瘍を含むがそれらに限定されるわけではない癌障害に関する。
【0076】
「癌」という用語には、癌腫、肉腫、癌肉腫、造血性組織の癌、脳を含む神経組織の腫瘍及び皮膚細胞の癌のような良性及び悪性腫瘍が含まれるが、それらに限定されるわけではない。
治療され得る癌の例には、癌腫、例えば、膀胱癌、乳癌、結腸癌(例えば、結腸腺癌及び結腸腺腫等の大腸癌)、腎癌、表皮癌、肝癌、肺癌、例えば腺癌、小細胞肺癌及び非小細胞肺癌、食道癌、胆嚢癌、卵巣癌、膵癌、例えば、膵外分泌腺癌、胃癌、子宮頚癌、甲状腺癌、前立腺癌、又は皮膚癌、例えば、扁平上皮細胞癌;リンパ系系統の造血器腫瘍、例えば、白血病、急性リンパ球性白血病、B細胞性リンパ腫、T細胞性リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、毛髪様細胞リンパ腫、又はバーケットリンパ腫;骨髄系系統の造血器腫瘍、例えば、急性及び慢性骨髄性白血病、骨髄異形成症候群、又は前骨髄球性白血病;甲状腺濾胞癌;間葉系起源の腫瘍、例えば、線維肉腫又は横紋筋肉腫;中枢神経系又は末梢神経系の腫瘍、例えば、星状細胞腫、神経芽腫、神経膠腫又は神経鞘腫;黒色腫;精上皮腫;悪性奇形腫;骨肉腫;色素性乾皮症;ケラトクタントーマ(keratoctanthoma);甲状腺濾胞癌;カポジ肉腫、星状細胞腫、基底細胞癌、小腸癌、小腸腫瘍、消化管腫瘍、膠芽腫、脂肪肉腫、胚細胞腫瘍、頭部及び頚部腫瘍(耳、鼻及び咽頭領域の腫瘍)、口腔、咽頭、喉頭、及び食道の癌、悪性又は良性骨腫瘍のような骨並びにその支持組織及び結合組織の癌、例えば、悪性骨肉腫、良性骨腫、軟骨腫瘍;悪性軟骨肉腫又は良性の軟骨腫、骨肉腫のようなもの;膀胱並びに男性及び女性の腎尿路生殖器系の内部及び外部の臓器及び構造の腫瘍、軟部組織腫瘍、軟部組織肉腫、ウィルムス腫瘍又は、例えば甲状腺、副甲状腺、下垂体、副腎、唾液腺のような内分泌腺及び外分泌腺の癌が含まれるが、それらに限定されるわけではない。
【0077】
(感染性疾患)
さらに、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む感染性疾患を治療し及び/又は予防する方法に関する。
ある宿主細胞CDKが、ウイルス複製に関与する、すなわちCDK2、CDK7、CDK8及びCDK9であることは公知である(J. Virol. 2001; 75: 7266-7279)。特に、HIV-1転写伸長及びヒストンのメチル化の制御におけるCDK9キナーゼ活性の役割が記載されている(J. Virol 2004, 78(24):13522-13533)。
【0078】
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む感染性疾患を治療し及び/又は予防する方法に関し、この中で、該化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0079】
本明細書で使用される「感染性疾患」という用語は、ウイルス、細菌、真菌及び/又は寄生虫等の病原体によって生じる感染を含む。
ウイルスによって誘発される感染性疾患には、レトロウイルス、ヒト内在性レトロウイルス、ヘパドナウイルス、ヘルペスウイルス、フラビウイルス、アデノウイルス、トガウイルス及びポックスウイルスによる感染によって生じる疾患が含まれる。特に、感染性疾患は、HIV-1、HIV-2、HTLV-1及びHTLV-II等のウイルス、HBV等のヘパドナウイルス、単純ヘルペスウイルスI型(HSV I)、単純ヘルペスウイルス11型(HSV II)、エプスタイン・バーウイルス(EBV)、水痘帯状疱疹ウイルス(VZV)、ヒトサイトメガロウイルス(HCMV)又はヒトヘルペスウイルス8型(HHV-8)等のヘルペスウイルス、HCV、ウェストナイル熱ウイルス又は黄熱ウイルス等のフラビウイルス、ヒトパピローマウイルウス、ポックスウイルス、シンドビスウイルス又はアデノウイルスを含むがそれらに限定されないウイルスによって生じる。
【0080】
感染性疾患の例には、エイズ、ボレリア症、ボツリヌス中毒、下痢、BSE(ウシ海綿状脳症)、チクングニヤ、コレラ、CJD(クロイツフェルト・ヤコブ病)、結膜炎、サイトメガロウイルス感染症、デング熱(dengue)/デング熱(dengue Fever)、脳炎、東部ウマ脳炎、西部ウマ脳炎、エプスタイン・バーウイルス感染症、大腸菌感染症、食物感染、口蹄疫、真菌性皮膚炎、胃腸炎、ヘリコバクター・ピロリ感染症、肝炎(HCV、HBV)、帯状疱疹(Herpes Zoster)(帯状疱疹(Shingles))、HIV感染症、インフルエンザ、マラリア、麻疹、髄膜炎、髄膜脳炎、伝染性軟属腫、蚊媒介疾患、パルボウイルス感染症、ペスト、PCP(ニューモシスチスカリニ肺炎)、ポリオ、原発性胃腸炎、Q熱、恐水病、呼吸シンチウムウイルス(RSV)感染症、リウマチ熱、鼻炎、リフトバレー熱、ロタウイルス感染症、サルモネラ症、サルモネラエンテリティデス、疥癬、細菌性赤痢、天然痘、連鎖球菌感染症、破傷風、毒素性ショック症候群、結核、潰瘍(消化性潰瘍疾患)、出血熱、痘瘡、疣贅、ウェストナイル熱ウイルス感染症(ウェストナイル脳炎)、百日咳、黄熱が含まれるが、それらに限定されるわけではない。
【0081】
(心血管疾患)
さらに、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む心血管疾患の治療及び/又は予防に関する。
心血管疾患の分野が、CDK阻害剤について起こり得る臨床的適用を構成することが報告されている(Pharmacol Ther 1999, 82(2-3):279-284)。さらに、サイクリンT/CDK9複合体の阻害、より具体的には、CDK9の阻害が、心不全等の心血管疾患の治療において有益な役割を担い得ることは公知である(WO2005/027902)。
【0082】
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む心血管疾患を治療し及び/又は予防する方法に関し、この中で、該化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0083】
「心血管疾患」という用語には、うっ血性心不全、心筋梗塞、安定狭心症、不安定狭心症及び無症候性虚血等の心臓の虚血性疾患、全ての種類の心房性及び心室性の不整脈、高血圧性血管疾患、末梢血管疾患、冠動脈性心疾患及び粥状硬化などの心臓及び血管系の障害が含まれるが、それらに限定されるわけではない。さらに、本明細書で使用されるように、該用語には、成人性先天性心疾患、動脈瘤、狭心症、血管神経性浮腫、大動脈弁狭窄、大動脈瘤、大動脈弁閉鎖不全症、不整脈原性右室異形成、動静脈奇形、心房細動、ベーチェット症候群、徐脈、心肥大、うっ血性、肥大型及び収縮性心筋症等の心筋症、頚動脈狭窄症、脳出血、チャーグ・ストラウス症候群、コレステロール塞栓症、細菌性心内膜炎、線維筋異形成症、うっ血性心不全、機能不全弁又は狭窄弁等の心臓弁疾患、心臓発作、硬膜外又は硬膜下血腫、フォンヒッペル・リンダウ病、充血、高血圧症、肺高血圧症、肥大性増殖、左室肥大、右室肥大、左心形成不全症候群、低血圧症、間欠跛行、虚血性心疾患、クリッペル・トレノネー・ウェーバー症候群、延髄外側症候群、僧帽弁逸脱症、QT延長症候群、僧帽弁逸脱症、心筋虚血、心筋炎、心膜の障害、心膜炎、末梢血管疾患、静脈炎、結節性多発動脈炎、肺動脈閉鎖症、レイノー病、再狭窄、リウマチ性心疾患、スネッドン症候群、狭窄、上大静脈症候群、症候群X、頻脈、遺伝性出血性末梢血管拡張症、毛細血管拡張症、側頭動脈炎、閉塞性血栓血管炎、血栓症、血栓塞栓症、静脈瘤、脈管疾患、脈管炎、血管攣縮、心室細動、ウィリアムス症候群、末梢血管疾患、静脈瘤及び下腿潰瘍、深部静脈血栓症及びウォルフ・パーキンソン・ホワイト症候群が含まれるが、それらに限定されるわけではない。
さらに、心血管疾患という用語には、先天性異常、遺伝的欠陥、環境の影響(すなわち、食事の影響、生活様式、ストレスなど)、及び他の欠陥又は影響から結果的に生じる疾患が含まれる。
【0084】
(神経変性疾患)
CDK阻害剤は、神経保護効果を発揮することが記載されている。特に、CDK阻害剤が、アルツハイマー病等の神経変性疾患における神経細胞の死滅を予防することが報告されている(Biochem Biophys Res Commun 2002 (297):1154-1158;Trends Pharmacol Sci 2002 (23):417-425; Pharmacol Ther 1999, 82(2-3):279-284)。
【0085】
このように、CDK阻害剤である式I記載の化合物は、神経変性疾患の治療管理において有益な効果を提供すると期待される。
従って、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む神経変性疾患を治療し及び/又は予防する方法に関する。
【0086】
本明細書で使用される「神経変性疾患」という用語には、脳損傷、脳血管障害及びその結果、パーキンソン病、皮質基底核変性症、運動ニューロン疾患、筋萎縮性側索硬化症、多発性硬化症、外傷性脳損傷、脳卒中、脳卒中後脳損傷、外傷後脳損傷、及び小血管性脳血管障害を含む認知症、アルツハイマー病等の認知症、脳血管性認知症、レヴィ小体型認知症、前頭側頭葉型認知症及び17番染色体と関連したパーキンソン症候、ピック病を含む前頭側頭葉型認知症、進行性核性麻痺、皮質基底核変性症、ハンチントン病、視床変性、クロイツフェルト・ヤコブ認知症、HIV認知症、認知症を伴う統合失調症、コルサコフ精神病及びエイズ関連認知症を含むがそれらに限定されるわけではない中枢神経系の障害並びに末梢神経系の障害が含まれる。
【0087】
同様にまた、軽度認知欠陥、加齢随伴性記憶欠陥、加齢関連性認知低下、血管性認知欠陥、注意欠陥障害、注意欠陥多動障害、及び学習障害を有する子供における記憶障害等の認知関連障害は、神経変性障害であると考慮される。
【0088】
特に、本発明は、上述の種類の疼痛及び随伴する容態並びに炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための方法に関し、この中で、「治療すること」という用語は、疼痛及び随伴する容態並びに炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患の予防、寛解又は治療することを含む。
【0089】
(医薬組成物)
本発明の好ましい実施態様には、医薬として許容し得る(すなわち非毒性の)少なくとも1つの担体、賦形剤及び/又は希釈剤とともに、活性成分としての式I記載の少なくとも1つのサイクリン依存性キナーゼ阻害剤を含む組成物の投与が含まれる。
好ましくは、組成物は、活性成分として式I記載の少なくとも1つのサイクリン依存性キナーゼ阻害剤を含み、この中で、該少なくとも1つのサイクリン依存性キナーゼ阻害剤は、他のCDKに対するよりもCDK9に対する高い選択性を有する。
【0090】
さらにまた、本発明は、CDKの少なくとも2つの阻害剤及び/又は医薬として許容し得るそれらの塩を組み合わせる組成物を含む。該少なくとも2つの阻害剤は、同一のサイクリン依存性キナーゼを阻害し得、又は異なる種類のサイクリン依存性キナーゼも阻害し得、例えば、組成物におけるある阻害剤は、CDK9を阻害し得るのに対し、他の阻害剤は、例えばCDK2を阻害できる。
【0091】
疼痛治療に関して、個々の疼痛投薬は、多くの中からたった1つの疼痛伝達経路に干渉するので、部分的に有効な疼痛軽減のみをしばしば提供する。このようにまた、疼痛知覚過程における異なる地点で作用する疼痛減少(鎮痛)薬との組み合わせで、式I記載のCDK阻害剤を投与することが企図される。
【0092】
「鎮痛薬」は、疼痛知覚における減少を生じる分子又は分子の組み合わせを含む。鎮痛薬は、CDKの阻害以外の作用の機構を採用する。
非ステロイド性抗炎症薬(NSAID)等のあるクラスの鎮痛薬は、侵害受容器によって検知される刺激の化学的メッセンジャーを下方制御し、オピオイド等の別のクラスの薬物は、CNSにおける侵害受容情報の処理を変化させる。他の鎮痛薬は、局所麻酔薬、抗痙攣薬及び三環系抗うつ薬等の抗うつ薬である。CDK阻害剤に加えて1つ以上のクラスの薬物を投与することは、より有効な疼痛の寛解を提供できる。
【0093】
本発明の方法及び組成物における使用のための好ましいNSAIDは、アスピリン、アセトアミノフェン、イブプロフェン、及びインドメタシンである。さらにまた、特異的シクロオキシゲナーゼ2(COX-2)阻害剤等のCOX-2阻害剤(例えば、セレコキシブ、COX189、及びロフェコキシブ)は、本発明の方法又は組成物における鎮痛薬として使用され得る。
【0094】
好ましい三環系抗うつ薬は、クロミプラミン、アモキサピン、ノルトリプチリン、アミトリプチリン、イミプラミン、デシプラミン、ドキセピン、トリミプラミン、プロトリプチリン、及びパモ酸イミプラミンからなる群から選択される。
さらにまた、鎮痛薬としての抗痙攣薬(例えば、ガバペンチン)、GABABアゴニスト(例えば、L-バクロフェン)、オピオイド、バニロイド受容体アンタゴニスト及びカンナビノイド(CB)受容体アゴニスト、例えばCB1受容体アゴニストの使用は、本発明における方法及び組成物において好ましい。
【0095】
本発明のサイクリン依存性キナーゼ阻害剤組成物を調製する上で、Remingtonの文献(薬剤学の科学及び実践第19版(The Science and Practice of Pharmacy, 19th ed.)(Mack Publishing, 1995))等の周知の製薬出典の標準的な推奨を手本とすることができる。
本発明の医薬組成物は、従来の固体又は液体の担体又は希釈剤及び医薬として調製される従来のアジュバントにおいて、公知の方法で適切な薬用量レベルで調製できる。好ましい調製物は、経口適用に適している。これらの投与形態には、例えば、丸剤、錠剤、フィルム錠、コート錠、カプセル、散剤及び沈着物が含まれる。
【0096】
さらにまた、本発明には、真皮、真皮内、胃内、皮内、静脈内、脈管内、静脈内、筋肉内、腹腔内、鼻内、膣内、頬内、経皮、直腸内、皮下、舌下、局所、又は経真皮適用を含む非経口適用のための医薬調製物が含まれ、この中で、該調製物は、典型的な媒体及び/又は希釈剤に加えて、本発明記載の少なくとも1つの阻害剤及び/又は医薬として許容し得るその塩を活性成分として含有する。
【0097】
本発明記載の少なくとも1つの阻害剤及び/又は医薬として許容し得るその塩を活性成分として含有する本発明記載の医薬組成物は典型的に、企図された投与の形態に関して、すなわち、錠剤、カプセル(固体充填、半固体充填又は液体充填のいずれか)、構成用散剤、ゲル、エリキシル剤、分散可能な顆粒、シロップ、懸濁液、及びそれらの類似物の形態での経口投与のために、従来の薬事的実践と合致して選択された適切な担体材料とともに投与されるであろう。例えば、錠剤又はカプセルの形態での経口投与のために、活性薬物構成要素は、医薬として許容し得るいずれかの経口用非毒性担体と、好ましくは乳糖、デンプン、ショ糖、セルロース、ステアリン酸マグネシウム、リン酸二カルシウム、硫酸カルシウム、滑石、マンニトール、エチルアルコール(液体充填カプセル)及びそれらの類似物のような不活性担体と組み合わせることができる。
【0098】
さらにまた、適切な結合剤、潤滑剤、崩壊剤及び着色料は、錠剤又はカプセルへ組み込まれ得る。散剤及び錠剤は、約5〜約95重量%の本明細書に列挙される式I記載のサイクリン依存性キナーゼ阻害剤又はそのアナログ又は医薬活性のある個々の塩を活性成分として含有し得る。
【0099】
適切な結合剤には、デンプン、ゼラチン、天然糖、トウモロコシ甘味料、アカシア等の天然及び合成ゴム、アルギン酸ナトリウム、カルボキシメチルセルロース、ポリエチレングリコール及び蝋が含まれる。適切な潤滑剤のうち、ホウ酸、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウム、及びそれらの類似物が挙げられ得る。
【0100】
適切な崩壊剤には、デンプン、メチルセルロース、グアーガム、及びそれらの類似物が含まれる。
また、甘味料及び調味料及び保存料は、適宜含まれ得る。崩壊剤、希釈剤、潤滑剤、結合剤等は、以下により詳細に論議される。
【0101】
さらに、本発明の医薬組成物は、いずれかの1つ以上の構成要素又は活性成分の速度調節放出を提供して治療効果、例えば抗ヒスタミン活性及びその類似項目を最適化するための徐放形態で製剤され得る。持続放出に適した剤形には、崩壊速度の変動する層を有する錠剤、又は活性成分の含浸され、かつ錠剤形態に成形された徐放性ポリマーマトリックス、又はこのような含浸され又は被包された多孔性ポリマーマトリックスを含有するカプセルが含まれる。
【0102】
液状製剤には、溶液、懸濁液、及び乳濁液が含まれる。一例として、非経口注射のための水又は水/プロピレングリコール溶液、又は経口用の溶液、懸濁液、及び乳濁液のための甘味料及び乳白剤の添加が挙げられ得る。また、液状調製物には、鼻内投与のための溶液が含まれ得る。
吸入に適したエアロゾル調製物には、不活性圧縮ガス、例えば窒素等の医薬として許容し得る担体との組み合わせで存在し得る溶液及び散剤形態の固体が含まれ得る。
【0103】
坐剤を調製するために、ココアバターのような脂肪酸グリセリドの混合物等の低融点蝋がまず融解され、次に、活性成分が、例えば撹拌によってその中に均質に分散する。次に、融解された均質な混合物が、便利な大きさの鋳型へ注入され、冷却され、それにより凝固する。
また、使用直前に経口又は非経口のいずれかの投与のための液状調製物へ変換されるよう企図された固体状調製物が含まれる。このような液状には、溶液、懸濁液、及び乳濁液が含まれる。
【0104】
また、本発明記載の化合物は、経真皮的に送達され得る。経真皮組成物は、クリーム、ローション、エアロゾル及び/又は乳濁液の形態を有し得、この目的のために当技術分野で公知のようなマトリックス又は貯蔵器の種類の経真皮パッチに含まれ得る。
【0105】
本明細書で列挙されるカプセルという用語は、活性成分を含む組成物を保持し又は含有するために、例えばメチルセルロース、ポリビニルアルコール、又は変性したゼラチン若しくはデンプンでできた特殊な容器又は被包体を指す。硬質シェルを有するカプセルは典型的には、骨又は豚肉の皮膚由来の比較的高いゲル強度のゼラチンでできており、又は該ゼラチンを配合する。カプセル自体は、少量の色素、不透明剤、可塑剤及び/又は保存料を含有し得る。錠剤のうち、活性成分を適切な希釈剤とともに含む圧縮され又は鋳造された固体剤形が理解される。錠剤は、湿式造粒、乾式造粒によって、又は当業者に周知の圧縮によって得られる混合物又は顆粒の圧縮によって調製され得る。
【0106】
経口用ゲルは、親水性の半固体マトリックスに分散し又は可溶化した活性成分を指す。
構成用散剤は、例えば水において又はジュースにおいて懸濁できる、活性成分と適切な希釈剤とを含有する散剤配合物を指す。
【0107】
適切な希釈剤は、組成物又は剤形の大部分を通常調合する物質である。適切な希釈剤には、乳糖、ショ糖、マンニトール、及びソルビトール等の糖、コムギ、トウモロコシ、コメ、及びジャガイモ由来のデンプン、及び微結晶性セルロース等のセルロースが含まれる。組成物における希釈剤の量は、組成物全体の約5〜約95重量%、好ましくは約25〜約75重量%、より好ましくは約30〜約60重量%の範囲とし得る。
【0108】
崩壊剤という用語は、薬剤の医薬活性成分の崩壊及び放出を支持するために組成物へ添加される材料を指す。適切な崩壊剤には、デンプン、カルボキシメチルナトリウムデンプン等の修飾された「冷水可溶性」デンプン、イナゴマメ、カラヤ、グアー、トラガカント及びアガー等の天然及び合成ゴム、メチルセルロース及びカルボキシメチルセルロースナトリウム等のセルロース誘導体、微結晶性セルロース、及びクロスカルメロースナトリウム等の架橋された微結晶性セルロース、アルギン酸及びアルギン酸ナトリウム等のアルギン酸塩、ベントナイト等の粘土、及び起沸性混合物が含まれる。組成物における崩壊剤の量は、組成物の約2〜約20重量%、より好ましくは約5〜約10重量%の範囲とし得る。
【0109】
結合剤は、散剤粒子と結合し又は「接着する」物質であり、顆粒を形成することによって散剤粒子を接着性にし、このように、製剤における「接着剤」として機能する。結合剤は、希釈剤又は増量剤においてすでに利用可能な接着力を付加する。適切な結合剤には、ショ糖等の糖、コムギ、トウモロコシ、コメ及びジャガイモ由来のデンプン、アカシア、ゼラチン及びトラガカント等の天然ゴム、アルギン酸、アルギン酸ナトリウム及びアルギン酸アンモニウムカルシウム等の海藻の誘導体、メチルセルロース、カルボキシメチルセルロースナトリウム及びヒドロキシプロピルメチルセルロース等のセルロース材料、ポリビニルピロリドン、及びケイ酸マグネシウムアルミニウム等の無機化合物が含まれる。組成物における結合剤の量は、組成物の約2〜約20重量%、好ましくは約3〜約10重量%、より好ましくは約3〜約6重量%の範囲とし得る。
【0110】
潤滑剤は、摩擦又は摩損を減少させることによって、鋳型又はダイから放出するよう圧縮された後、錠剤造粒等を可能にするよう剤形へ添加されるあるクラスの物質を指す。適切な潤滑剤には、ステアリン酸マグネシウム、ステアリン酸カルシウム、又はステアリン酸カリウム等の金属性ステアリン酸塩、ステアリン酸、高融点蝋、並びに塩化ナトリウム、安息香酸ナトリウム、酢酸ナトリウム、オレイン酸ナトリウム、ポリエチレングリコール及びD,L-ロイシン等の他の水溶性潤滑剤が含まれる。潤滑剤は、顆粒の表面に存在しなければならないので、通常、圧縮前のまさに最終工程で添加される。組成物における潤滑剤の量は、組成物の約0.2〜約5重量%、好ましくは約0.5〜約2重量%、より好ましくは約0.3〜約1.5重量%の範囲とし得る。
【0111】
滑走剤は、医薬組成物の構成要素の焼成を互いに防止し、流動が潤滑かつ均一であるよう顆粒の流動特徴を改善する材料である。適切な滑走剤には、二酸化ケイ素及び滑石が含まれる。
該組成物における滑走剤の量は、最終組成物の約0.1〜約5重量%、好ましくは約0.5〜約2重量%の範囲とし得る。
【0112】
着色料は、組成物又は剤形へ着色を提供する賦形剤である。このような賦形剤には、粘土又は酸化アルミニウム等の適切な吸着剤へ吸着された食品等級の色素が含まれ得る。着色料の量は、組成物の約0.1〜約5重量%、好ましくは約0.1〜約1重量%で変動し得る。
【0113】
本発明は、いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、心血管疾患又は神経変性疾患の治療のための、活性成分としてサイクリン依存性キナーゼ阻害剤を含有する組成物の投与を必要とする対象への該投与に関する。
【0114】
「該投与を必要とする対象」は、近い将来いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、心血管疾患又は神経変性疾患を経験すると期待され、又は該容態の経験が進行中である動物、好ましくは哺乳動物、最も好ましくはヒトを含む。例えば、このような動物又はヒトは、現に疼痛を生じている進行中の容態を有し得、疼痛を生じ続けるようであり、又は動物若しくはヒトは、痛い結果を通常有する手法若しくは事象に忍耐中であり又は忍耐中であろう。糖尿病性神経障害性痛覚過敏及びコラーゲン脈管疾患等の慢性の痛い容態は、第一の種類の例であり;特に炎症又は神経障害の一領域における歯科業務、及び毒素曝露(化学治療薬への曝露を含む)は、後者の種類の例である。
【0115】
所望の治療効果を達成するために、個々のサイクリン依存性キナーゼ阻害剤は、治療的有効量で投与されなければならない。
「治療的有効量」という用語は、示される生物学的又は医学的応答を誘発する活性化合物又は医薬の量を示すために使用される。この応答は、研究者、獣医、医師又は他の臨床家によって探求されている最中の組織、系、動物又はヒトにおいて生じ得、治療されている最中の疾患の症状の寛解を含む。本発明の文脈において、治療的有効量は、例えば、疼痛、特に炎症性又は神経因性疼痛を減少させる量を含む。特に、治療的有効量は、治療されるべき対象において痛覚鈍麻効果を発揮する量を示す。
【0116】
このような有効量は、例えば疼痛に対する対象の正常感度、対象の身長、体重、齢、及び健康状態、疼痛の起源、CDKの阻害剤を投与する様式、投与される具体的な阻害剤、及び他の因子に応じて、対象によって変動するであろう。結果として、具体的なセットの環境の下で具体的な対象のための有効量を経験的に決定することが賢明である。
本発明は、下記の制限的でない実施例によってさらに説明される。
【実施例】
【0117】
(実施例)
すべての試薬をACROS Organics、Aldrich、Lancaster、Maybridge及びBoron Molecularから購入した。
化合物についての液体クロマトグラフィー/質量分析を、APCIイオン化を備えたSurveyor MSQ(Thermo Finnigan, USA)で実施した。
【0118】
本発明の化合物は、当技術分野で公知のいずれかの方法によって調製できる。簡便な1つの合成経路を以下のスキーム1:
【化6】
に示す。
フェニルボロン酸(2、Y=B(OH)2)又はその誘導体と2,4-ジハロゲン化ピリミジン(1、例えば、X1=X2=Cl)とのパラジウム触媒性交差共役は、4-アリール化2-ハロゲノピリミジン(3)を生じ、該4-アリール化2-ハロゲノピリミジンは、アニリン(4)によってアミノ化される。
【0119】
(実施例1:{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物1)の合成)
【化7】
a.塩化(ニトロフェニル)メタンスルホニル(4.17g、20mM)(Ziegler, Carl及びSprague, James M.の文献(J. of Org. Chem. (1951), 16, 621-5))をアセトニトリル(20mL)に溶解し、炭酸アンモニウムで飽和した濃アンモニア水溶液(20mL)を添加し、混合物を室温で1時間激しく撹拌した。次に、アセトニトリルを蒸発させ、残渣を冷水(20mL)で希釈して沈殿の形成に至らせ、該沈殿を濾過し、水(2×5mL)及びエーテルで洗浄し、減圧下で乾燥させた。(3-ニトロ-フェニル)メタンスルホンアミドの収量:3.5g(80%)。
【0120】
b.(3-ニトロフェニル)メタンスルホニルアミド(3.5g、16mM)をメタノールにおけるラネーニッケル(0.5g)上で50℃及び70psiで4時間水素化した。次に、触媒を濾過し、温メタノールで洗浄した。組み合わせた濾液を蒸発させて、2.83g(95%)の(3-アミノ-フェニル)-メタンスルホンアミドを得た。
【0121】
c.4,6-ジクロロ-ピリミジン(6g、0.04mol)(Ranganathan, Subramaniaらの文献(予稿集−インド科学学会(Proceedings - Indian Academy of Sciences), Chemical Sciences (1994), 106(5), 1051-70);Maggiali, C.らの文献(Farmaco, Edizione Scientifica (1988), 43(3), 277-91);Gershon, Hermanらの文献(J. of Med. Chem. (1963), 6, 87-9))及び(2-メトキシ-フェニル)-ボロン酸(4.37g、0.029mol)を、ジメトキシエタン(120mL)及び水(18mL)に溶解した。この溶液に、NaHCO3(6.72g、0.08mol)、PdCl2(PPh3)2(0.84g)を添加し、7時間還流させた(薄層クロマトグラフィー対照)。次に、混合物を室温に冷却し、溶媒を減圧下で除去した。得られた粗固体をジクロロメタン(100mL)に溶解し、水(1×100mL)で洗浄し、有機層を分離し、K2CO3上で乾燥させ、濾過し、溶媒を減圧下で除去した。得られた固体をフラッシュクロマトグラフィー(溶離剤ジクロロメタン)によってさらに精製すると粗生成物を生じ、それをヘキサンから結晶化させて、2-クロロ-4-(2-メトキシ-フェニル)-ピリミジン(4.7g、73%)を得た。
【0122】
D.DMF(12mL)における2-クロロ-4-(2-メトキシ-フェニル)-ピリミジン(0.882g、0.004mol)及び(3-アミノ-フェニル)-メタンスルホンアミド(0.865g、0.005mol)の溶液を80℃で2時間撹拌した(薄層クロマトグラフィー対照)。溶媒を減圧下で除去すると、{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミドを得た。
【0123】
(実施例2:2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物9)の合成)
【化8】
a.水素化フラスコに、2-(3-ニトロ-フェニルメタンスルホニル)-エタノール(0.8g、3.3mmol)(Harms, Wolfgangらの文献(Ger. Offen. (1995))、独国特許第4402189 A1号、第19950727号)を配置し、メタノール(100mL)に溶解した。ラネーニッケル(200mg)の添加後、混合物を70psiの水素圧で50℃で4時間振盪させた。次に、濾過によって触媒を除去し、メタノールで洗浄した。メタノール溶液を組み合わせ、溶媒を減圧下で蒸発させた。粗生成物をフラッシュクロマトグラフィー(溶離系:ジクロロメタン/メタノール−10/1)によって精製し、所望の画分を組み合わせ、乾燥させ、蒸発させた。残渣をジエチルエーテルで洗浄して、2-(3-アミノ-フェニルメタンスルホニル)-エタノール(0.51g、収率73%)を得た。
【0124】
b.DMF(3mL)における2-(3-アミノ-フェニルメタンスルホニル)-エタノール(0.215g、1mmol)及び2-クロロ-4-(2-メトキシ-フェニル)-ピリミジン(0.22g、1mmol)の溶液を80℃で2時間撹拌した(薄層クロマトグラフィー対照)。溶媒を減圧下で除去すると、油状残渣を生じ、それをイソプロパノールから結晶化して、2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(0.3g、75%)を得た。
同様に、相応じて置換される出発材料を使用して、下記の化合物を合成した。
【表1】
【0125】
(実施例3)
I.炎症性及び神経因性疼痛の分析のための行動動物モデル
炎症性及び神経因性疼痛の分析のためのいくつかの動物モデルは公知である。該モデルは、例えば神経病変の誘発(例えば、坐骨神経部分損傷、SNI)後又は侵害刺激に対する実験動物の曝露(例えば、ホルマリン又はカラゲナンの注射)後、該介入によって誘発されるような疼痛の徴候は、例えばフォン・フレイ毛髪を使用する機械的刺激に対する(又は、レーザー源若しくは舐める行動を使用する熱刺激に対する)足引っ込め閾値等の行動の定量可能な構成要素によって測定される。これらの反応は、ヒトにおける機械的及び熱アロディニア(機械的刺激に対する過敏症)又は痛覚過敏と等価であるものとして解釈される。
【0126】
坐骨神経部分損傷モデル(SNIモデル、Decosterd及びWoolf(2000)によって開発、図1参照)は、臨床的に関連する神経病変の誘発及び外科的介入後のその後の行動実験(例えば、フォン・フレイアッセイ)によって特徴付けられる。該モデルは、腓腹神経を無傷のままにしておき坐骨神経の2つの分岐(すなわち、脛骨神経及び総腓骨神経)の結紮及び切断からなる共通神経損傷モデルを構成する。SNIモデルは、臨床的な神経因性疼痛の特徴を密接に模倣する機械的及び冷却感度における早期の(24時間未満)、長期の及び実質的な変化を結果的に生じる。これらの種類の神経損傷を有する動物は、神経因性疼痛患者によって報告されるものと同様の異常な疼痛感覚及び機械的刺激に対する過敏症(アロディニア)を発生することが示されている。
【0127】
或いは、マウスにおけるホルマリンアッセイは、炎症性及び神経因性疼痛における侵害受容の妥当なかつ信頼性のある行動モデルである。該アッセイは、多様なクラスの鎮痛薬に対して感受性がある(Hunskaar S及びHole Kの文献(Pain. 1987 Jul;30(1):103-14))。侵害刺激は、左後足の背側面の皮膚の下(左後足内への皮下又は足底間)での10μLの希釈したホルマリン(塩類溶液における2%)の注射液からなる。反応は、注射された足を舐めること及び畏縮させることである。
【0128】
カラゲナンアッセイについて、マウスの単一の後足(同側足)への(塩類溶液における)1%カラゲナン25μLの皮下注射が適用される。その後の炎症は結果的に、足の長期に持続する発汗及び(機械的及び熱刺激に対する)過敏症を生じる。カラゲナンアッセイは、検査化合物の抗炎症性活性を予測するために使用される標準的な実験室アッセイである。足の浮腫測定及びハーグリーブスアッセイ(Hargreaves Assay)(光源を介する熱刺激による足の引っ込め)は、読み出しに使用される。
【0129】
本発明に関して、炎症性及び神経因性疼痛の発生に及ぼす式I記載のサイクリン依存性キナーゼ(CDK)阻害化合物の投与の効果は、SNIモデルにおいて、カラゲナンにおいて及びホルマリンアッセイにおいてアッセイされる。実験手法及び結果を、以下に詳細に記載する。
【0130】
(実施例4)
A.坐骨神経部分損傷(SNI)−慢性神経因性疼痛のモデル
先に概略されるように、坐骨神経部分損傷(SNI)モデル(図1参照)は、腓腹神経を無傷のままにしておく実験動物の坐骨神経の3つの末端分岐のうちの2つ(脛骨神経及び総腓骨神経)の病変を包含する。SNIは、損傷を受けていない腓腹神経の皮膚での支配領域における機械的及び熱アロディニアを結果的に生じる(Decosterd及びWoolfの文献(Pain 2000; 87:149-158)、(2) Tsujinoらの文献(Mol. Cel. Neurosci. 2000; 15:170-182))。
【0131】
1.野生型マウスにおける坐骨神経部分損傷(神経病変)の誘発
野生型マウス(C3HeB/FeJ系)(齢、性別及び体重は対形成)を、外科的準備の前に4μL/gで1:1:2の比でHypnorm(0.315mg/mLクエン酸フェンタニル+10mg/mLフルアニソン;Janssen)/Hypnovel(5mg/mLミダゾラム;Roche Applied Sciences)/水で麻酔した。
【0132】
その後、坐骨神経の3つの末端分岐:総腓骨神経、脛骨神経及び腓腹神経を露出して、膝のレベルのちょうど上ですべてのマウスの同側右後脚において、無菌予防措置の下で病変を作製した。総腓骨神経及び脛骨神経を7/0絹糸でしっかり結紮し、結紮に対して遠位で切断し、遠位の神経断端約2mmを除去した。腓腹分岐は、手法の間触らないままでだった(本明細書で「SNI同側」と記載)。重なっている筋肉及び皮膚を縫合し、動物を回復させ、創傷治癒させた。同一のマウスにおいて、反対側の左後脚の坐骨神経分岐を露出させたが、病変形成しなかった(本明細書で「SNI反対側」と記載)。坐骨神経部分損傷を経験したマウスを、以後「SNIマウス」と記載する。
【0133】
2.SNIマウスに対するCDK阻害化合物の投与
手術からの回復及び創傷治癒後、SNIマウスはCDK阻害化合物の経口注入を受容した。
2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μLに溶解したCDK阻害剤30mg/kgを、フォン・フレイ測定(機械的アロディニア)の30分前に経口適用を介して投与した。ネガティブコントロールとして、同量(400μL)の2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)媒体を、フォン・フレイ測定の30分前に単回経口適用によって投与した。
【0134】
阻害剤又は媒体の注入及びその後のフォン・フレイアッセイにおける機械的刺激に対する足引っ込め閾値の測定を、SNI後107日目に実施した。機械的刺激に対する反射侵害受容反応を各注入の30分後にフォン・フレイアッセイにおいて測定した。
機械的アロディニアの発生に及ぼすSNIマウスへのCDK阻害剤の投与の効果を、以下に記載のとおり、フォン・フレイアッセイにおいて分析した。
【0135】
3.CDK阻害化合物の投与後のSNIマウスの行動検査(フォン・フレイアッセイ)
SNI及びその後の本発明の化合物の投与を経験するマウスを、フォン・フレイアッセイにおける神経損傷後及び投与後の機械的アロディニアの徴候について検査した(Decosterd及びWoolfの文献(Pain 2000; 87:149-158))。本アッセイは、通常では痛くない刺激が不快な又は痛いものとして動物によって認識される機械的閾値を決定する。SNI同側及びSNI反対側の基線を個々に確立した。
【0136】
Chaplanら(1994)並びにMalmberg及びBasbaum(1998)に基づいたアップダウン法を使用して、SNIマウスの機械的閾値を定量した。
直径約9.5cm、高さ14cmで上部に向いた4つの通気孔及びプレキシガラス蓋のついたプレキシガラスシリンダーにマウスを配置した。シリンダーを高架のメッシュ表面(7×7平方mm)の上に配置した。検査前日、マウスを検査シリンダーに1〜2時間順応させた。検査当日、マウスをシリンダーに約1時間順化させ、この中で、順化時間は、マウスの系及びマウスが以前検査された回数等の因子に依存する。一般的に、検査は、マウスが一旦落ち着いて新たな環境を探検するのを停止したら開始し得る。
【0137】
マウスを検査するために、フィラメント2.44、2.83、3.22、3.61、3.84、4.08、及び4.31(力の範囲=0.04〜2.0g)を使用した。3.61mNフィラメントを最初に適用した。該フィラメントを片足の足底面に穏やかに適用し、曲げさせ、しかるべき位置で2〜4秒間保持した。刺激に対する正の反応(屈曲反応)が生じた場合はいつでも、次のより弱いフォン・フレイ毛髪を適用し;負の反応(無反応)が生じた場合はいつでも、次のより強い力を適用した。反応における最初の変化後にさらに4回の刺激に対する反応が得られるまで、検査を続行した。検査した最大力は、4.31であった。カットオフ閾値は、2gであった。
【0138】
一連のスコア(すなわち、「屈曲反応」及び「無反応」)及び適用された最後のフィラメントの力を使用して、Chaplanらの文献(Journal of Neuroscience Methods. 53(1):55-63, 1994 Jul.)に記載される機械的閾値を決定した。決定された閾値は、動物が回数の50%に反応すると期待されるであろうものである。フォン・フレイ測定を達成した後に、マウスを屠殺した。
【0139】
4.神経因性疼痛の発生に及ぼすCDK阻害化合物の投与の効果
上述のとおり、CDK阻害化合物をSNIマウスへ投与した。上述のとおり、術後107日目に動物の同側及び反対側の足でフォン・フレイ測定を実施した。薬理学的処置をせずに、SNIマウスは、SNI手術後に安定したアロディニアを示す。化合物で処置された動物は、機械的刺激に対する低い感度(低いアロディニア)を示す閾値の有意な増大を示す。低いアロディニアの観察は、CDK阻害化合物が、慢性神経因性疼痛のモデルにおける痛覚鈍麻薬として効果的であることを意味する。
【0140】
(実施例5)
ホルマリンアッセイ−炎症過程/炎症性及び慢性神経因性疼痛のモデル
マウスにおけるホルマリンアッセイは、侵害受容の妥当かつ信頼性のある行動モデルであり、多様なクラスの鎮痛薬に対して感受性がある(Hunskaar S及びHole Kの文献(Pain. 1987 Jul;30(1):103-14))。有害な刺激は、10μLの希釈したホルマリン(塩類溶液における2%)の左後足への皮下又は足底内注射である。反応は、注射された足を舐めること及び畏縮させることである。反応は、炎症過程の異なる部分を反映する2つの相(Abbottら1995)、すなわち早期/急性相である注射0〜5分後、及び後期/慢性相である注射5〜30分後を示す。下記のプロトコールは、実験を実施するための可能な1つの方法を記載する:
【0141】
1.ホルマリンの注射及びCDK阻害化合物の投与
本アッセイにおいて、齢、性別及び体重の対形成した野生型マウス(C3HeB/FeJ)を使用する。ホルマリン注射の前に、動物を各群10匹の実験群へ無作為にさらに分ける。ホルマリン注射の30分前に、2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μLに溶解した適量のCDK阻害剤は、腹腔内注射により投与できる。同様に、2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μLにおけるIkキナーゼ(IKK)阻害剤(30mg/kg)(ポジティブコントロール)、又は媒体単独(2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μL)(ネガティブコントロール)は、ホルマリン注射の30分前に腹腔内注射によって投与できる。
【0142】
ホルマリン注射に向けて、移動による注射の妨害を回避するために、マウスをペーパータオルで保持する。注射した後足を親指と人差し指との間に保持し、ハミルトンシリンジを使用してホルマリン(2%)10μLを2つの隆起間で後足足底へと皮下注射する。ホルマリン及び阻害剤で処置したマウスの行動を以下に記載のとおり分析する。
【0143】
2.ホルマリンの注射及びCDK阻害化合物の投与後のマウスの行動分析
ホルマリン処置したマウスの行動、すなわち舐めること及び畏縮を、規定された時間にわたって自動追跡システム(Ethovision 3.0 Color Pro, Noldus, Wageningen, Netherlands)によってモニターする:ホルマリン注射5分後に測定を開始し、ホルマリン注射30分後に終了する。この時間枠は、痛覚過敏であるホルマリン誘発性侵害受容(疼痛)の位相IIにわたる。
【0144】
注射した後足(黄色色素)(Lumogenyellow; BASF Pigment, Cologne, Germany)及び反対側の足(青色色素)(Lumogenviolet; Kremer Pigmente, Aichstetten, Germany)をそれぞれ局所的に標識するために、2つの異なる染色色素を使用する。舐める行動を決定するために、マウスをCCDカメラでモニターする。モニタリング及び録画後、EthoVisionソフトウェア(Ethovision 3.0 Color Pro, Noldus, Wageningen, Netherlands)を使用して、又は手動分析によってビデオを分析する。蛍光点の大きさ及び蛍光強度を測定し、舐めること及び噛みつくことを通じての蛍光点の大きさの減少を算出した。処置された足と処置されていない足の点の大きさの減少の比較により、全体的な舐める時間の強度を自動的に算出した。
【0145】
アッセイの読み出しの別の変形として、個々の動物の舐める行動をビデオファイルに基づいて手動で追跡した。ホルマリン注射後30分間にわたって舐める時間を記録し、3つの異なる舐める区域(背面、足底、つま先)についてさらに分けた。全体的な舐める時間は各動物及び各実験群について算出でき、化合物の効能の決定のためのパラメータとして使用できる。
【0146】
結果として、ホルマリン注射前に媒体処置を受容するマウス(ネガティブコントロール)は、ホルマリン処置した足での長い舐める時間及び蛍光点の大きさの有意な減少を示すことが発見された。
対照的に、ホルマリン処置した足の舐める時間の短縮、及びその結果として蛍光点の大きさの有意な減少のないことは、検査化合物/ホルマリン処置したマウスにおいて観察できた。同一の効果、すなわち舐める時間における短縮及び蛍光点の大きさにおけるわずかな変化は、Iκキナーゼ阻害剤(IKK;IKKの機能については、図2参照、ポジティブコントロール)で処置したコントロールマウスにおいて観察された。
この観察は、CDK9阻害剤で処置したマウスにおける低い炎症性/慢性炎症性疼痛知覚を、及び検査された化合物の痛覚鈍麻効果を示す。
【0147】
(実施例6)
マウスにおけるカラゲナンアッセイ−炎症及び炎症性疼痛のモデル
カラゲナン誘発性の足の浮腫のモデルは、個々の化合物の抗炎症活性及び炎症誘発性疼痛知覚の減少を予測するために使用される標準的な実験室アッセイである。下記のプロトコールは、実験を実施するための可能な1つの方法を記載する。
【0148】
基本的な測定は、浮腫並びに、カラゲナン等の刺激物に応答した機械的及び熱による過敏症の測定において構成する。
炎症及び結果として生じる炎症性疼痛は、(塩類溶液における)1%カラゲナン25μLのマウス後足(同側足)への皮下注射によって誘発される。各群10匹のマウスは、カラゲナン注射の30分前の腹腔内注射による式I記載の化合物(30mg/体重kg)、媒体(2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μL)及び塩類溶液(生理的NaCl)の投与を受容する。反対側の足は、カラゲナン注射を受容しない。
【0149】
1.1 カラゲナン処置したマウスに及ぼすCDK阻害化合物の投与の効果
カラゲナン注射によって誘発される足の浮腫を、注射した(同側の)足の中足領域で背面から足底まで測定される大きくなった足のサイズによって検出する。同側及び反対側の足の大きさは、炎症についての代理マーカーとして機能し、カラゲナン注射後のいくつかの時点:注射前(-1)、注射2時間後(2)、3時間後(3)、4時間後(4)、5時間後(5)、6時間後(6)、24時間後(24)に測定される。
【0150】
すべてのマウスの足のサイズは、カラゲナンの30分前に注射される処置物質の種類とは関係なく、カラゲナン注射後の最初の1時間以内に、例えば2〜3mm(+10%)増大し得る。時間経過の間、カラゲナン注射前にCDK阻害化合物による処置を受容したマウスは、カラゲナン注射24時間後まで浮腫の減少を示し得:足の大きさの増大は、例えば10%から8%へと低下できた。対照的に、対照マウスの足の大きさは、この時点で平均して30%増大できた。カラゲナン注射の24時間後、カラゲナンで処置したすべての足の大きさは、注射96時間後で最大に到達するよう増大し得る。
【0151】
カラゲナンアッセイの読み出しとして、ハーグリーブスアッセイが実施され得、この中で、該アッセイによって輻射熱に対する熱感受性の測定が可能となる。ハーグリーブスアッセイ(Hargreavesら, 1988)は、動物がプレキシガラスチャンバーの中で立つ場合に、動物の後足の足底面に輻射熱源を焦点合わせすることによって、自由に移動する動物における侵害受容感受性を測定する。特に、例えば55℃の温度を生じる光源へ、足の下位側を曝露する。曝露の開始と曝露された足の挙上/引き抜きとの間の潜時として熱感受性を測定する。
【0152】
本明細書に開示されるCDK9阻害剤及びカラゲナンを使用して、又はナプロキセン及びカラゲナンを使用して、又は溶媒及びカラゲナンを使用してそれぞれ処置されたマウスを、ハーグリーブスアッセイに供する。CDK阻害剤及びカラゲナンを使用して処置されたマウスは、ネガティブコントロールマウスと比較してより長い潜時を示すことができた。この観察は、本明細書に開示されるCDK阻害剤の痛覚鈍麻効果を示すであろう。
【0153】
(実施例7)
ラットにおけるカラゲナンアッセイ−炎症及び炎症性疼痛のモデル
下記は、ラットにおいてカラゲナンアッセイを実施する可能な1つの方法を示す。
該アッセイは、Winterらの文献(Proc. Soc. Exp. Biol. Med., 111, 544-547, 1962)によって記載されるプロトコールに従って、炎症性疼痛を有するラットにおける鎮痛/抗炎症活性を検出する。
【0154】
ラット(200〜250g)の右後足の下位表面にカラゲナンの懸濁液を注射する(1足につき0.05mL生理塩類溶液における0.75mg)。2時間後、両足の触覚刺激及び熱刺激へ連続的にラットを供する。
触覚刺激のために、格子床上の反転したアクリル製プラスチック箱(18×11.5×13cm)の下に動物を配置する。次に、力を増大させながら、電子フォン・フレイプローブ(Bioseb, Model 1610)の先端をまず、炎症を生じていない後足へ、次に炎症を生じた後足へ適用し、足の引っ込めを誘発させるのに必要な力を自動的に記録する。この手法を3回実施し、1足あたりの平均の力を算出する。
【0155】
熱刺激のために、装置(Ugo Basile, 参考文献: 7371)は、高架のガラス床上に配置された個々のアクリル製プラスチック箱(17×11×13cm)からなる。ラットを箱の中に配置し、10分間自由に慣れさせる。次に、携帯赤外線源(96±10mW/cm2)をまず炎症を生じていない後足の下に、次に炎症を生じた後足の下に焦点合わせし、足引っ込め潜時を自動的に記録する。組織障害を防止するために、熱源を45秒後に自動的にオフにする。
【0156】
行動測定後、足の浸漬によって誘発される水置換(mL)を示すデジタル体積記録計(Letica, Model 7500)を使用して、各後足の体積を測定することによって、足の浮腫を評価する。
各群につき10匹のラットを研究する。検査を盲検で実施する。
【0157】
本明細書に呈される式I記載のCDK阻害剤等の検査物質は、検査60分前に経口投与される2用量(10及び30mg/kg)で評価され、媒体対照群と比較されるであろう。
同一実験条件下で投与されるモルヒネ(128mg/kg経口投与)及びアセチルサリチル酸(512mg/kg経口投与)は、基準物質として使用されるであろう。
【0158】
それゆえ、実験には6群が含まれるであろう。対応のないスチューデントのt検定を使用して、処置された群を媒体対照と比較することによって、データが分析されるであろう。
本明細書に開示されるCDK9阻害剤及びカラゲナンを使用して、又はナプロキセン及びカラゲナンを使用して、又は溶媒及びカラゲナンを使用してそれぞれ処置されたラットを、ハーグリーブスアッセイに供する。CDK阻害剤及びカラゲナンを使用して処置されたラットは、ネガティブコントロールラットと比較してより長い潜時を示すべきである。この観察は、本明細書に開示されるようにCDK阻害剤の痛覚鈍麻効果を示すであろう。
【0159】
(実施例8)
A.LPSインビボアッセイ(LPS)−インビボでのサイトカイン抑制のモデル
敗血症性ショックのLPS誘発性モデルのために、マウスは、塩類溶液における30μgの細菌リポ多糖(LPS;L2630 SIGMA)の腹腔内注射を受容する。炎症性シグナル伝達カスケードの該LPS仲介性の開始は、例えば、TNFα、IL-6及びIL1β等のサイトカインの高い血清濃度を結果的に生じる。血液は、規定された時点でこれらの動物から採取できる。その後、血清は分離されるであろうし、試料は、サイトカイン濃度が市販のELISAアッセイを使用して測定されるまで、-80℃で保存できる(AL Moreiraらの文献(Braz J Med Biol Res 1997; 30:1199-1207))。
【0160】
サイトカイン、TNFα、IL6及びIL1β等の炎症性仲介因子が、持続性の疼痛状態及び炎症性障害に関与する可能性があることは認識されている。末梢組織におけるマクロファージ及びCNS組織におけるミクログリアのような免疫細胞から放出された後、これらの仲介因子は、炎症性及び神経因性疼痛においてだけでなく、関節リウマチ等の炎症性障害においても中枢の役割を担うように見える(F Marchandらの文献(Nat Rev Neurosci 2005; 6 (7); 521-532))。このように、腫瘍壊死因子α(TNFα)の阻害は同様に、炎症性疾患の治療のための関連性のある標的を表す[Lavagnoらの文献(Eur J Pharmacol 2004; 501, 199-208)]。
【0161】
LPSインビボアッセイは、薬理学的治療によるサイトカインの発現の抑制を扱うための強力なモデルとして使用できる。
1.野生型マウスにおけるサイトカインの発現の誘導
野生型マウス(C3HeB/FeJ系)(齢、性別及び体重を対形成)に30μgLPS(SIGMA)を腹腔内注射した。LPS投与の90分後、これらの動物を0.1mL/体重10gのケタミン-ロムプン(Rompun)(50/20mg/mL)で麻酔し、心穿刺を介して、血清調製のための血液を採取した。
【0162】
2.LPSマウスに対するCDK阻害化合物の投与
LPSマウスの薬理学的処置群(n=4)は、CDK阻害化合物又は媒体(ネガティブコントロール)の経口注入をそれぞれ受容した。
1%カルボキシメチルセルロース(SIGMA)に溶解した10又は30mg/kg(体重あたりの化合物)のCDK阻害剤を単回経口薬用量として、LPS刺激の30分前に投与した。媒体コントロールを同一の様式で投与した。
【0163】
LPS刺激の90分後に、血液試料をマウスから採取した。あらかじめ、経時実験によって、この動物モデルにおけるTNFα発現のピークとして、90分の時点を同定した。
LPSマウスにおけるサイトカインレベルに及ぼすCDK阻害剤による薬理学的処置の効果を、以下に記載のとおり市販のELISAアッセイにおいて分析した。
【0164】
3.CDK阻害化合物の投与後のLPSマウスにおけるサイトカイン血清濃度の決定
心穿刺後、LPS動物由来の血液試料(〜500μL/個体)を濡れた氷の上で30分間インキュベートした。その後、試料を13,000rpmで15分間遠心分離した。血餅から血清を分離し、-80℃で凍結保存した。
製造元の説明書に従って市販のELISAキット(Natutec)を使用することによって、試料内のTNFα及びIL6の血清濃度を測定した。
【0165】
4.サイトカインのタンパク質発現に及ぼすCDK阻害化合物の投与の効果
上述のように、化合物をLPSマウスに投与した。サイトカイン血清濃度に関するELISAベースの決定を上述のとおり実施した。CDK阻害化合物で処置した動物と媒体で処置した対照動物との比較は、血清におけるTNFα及びIL6タンパク質濃度に及ぼす有意な抑制効果を示し、サイトカイン発現のモデルにおけるサイトカインTNFα及びIL6の抑制剤として、化合物が効果的であることを示した。
【0166】
(実施例9)
A.インビトロTHP-1アッセイ−サイトカイン阻害のインビトロモデル
ヒトTHP-1細胞系は、リポ多糖(LPS)又は腫瘍壊死因子α[TNFα]によって仲介される場合のサイトカイン発現のインビトロモデルとして利用できる。
単球性THP-1細胞(ATCC;TIB-202)は、LPSによる誘導の際に、又はTNFα(自己分泌誘導)自体によってTNFα、IL6及び1L1βのような炎症誘発性サイトカインを発現するマクロファージ様細胞へ分化できる。
【0167】
サイトカインであるTNFα、IL6及びIL1β等の炎症性仲介因子が、持続性の疼痛状態及び炎症性障害に関与する可能性があることは認識されている。末梢組織におけるマクロファージ及びCNS組織におけるミクログリアのような免疫細胞から放出された後、これらの仲介因子は、炎症性及び神経因性疼痛においてだけでなく、関節リウマチ等の炎症性障害においても中枢の役割を担うように見える(F Marchandらの文献(Nat Rev Neurosci 2005; 6 (7); 521-532))。それにより、腫瘍壊死因子α(TNFα)の阻害は同様に、炎症性障害の治療における関連性のある標的を表す[Lavagnoらの文献(Eur J Pharmacol 2004; 501, 199-208)]。
それゆえ、THP-1インビトロアッセイは、サイトカイン発現の薬理学的阻害を扱うための強力なスクリーニングモデルとして使用できる(U Singhらの文献(Clin Chem 2005; 51(12); 2252-6);K Rutaultらの文献(J Biol Chem 2001; 276(9); 6666-74))。
【0168】
1.THP-1細胞の増殖及び分化
10%ウシ胎児血清及び1%ペニシリン/ストレプトマイシンを補充した修正RPMI-1640培地(ATCC、カタログ番号30-2001)において、THP-1細胞を増殖させる。サイトカイン阻害アッセイのために、100ng/mL PMA(Sigma, P1585)を補充した標準的な増殖培地において、6ウェルプレートへ5×105個/mLの密度で細胞を播種し、マクロファージ様細胞への分化を誘導する。24時間後、培地を標準的な増殖培地(PMAなし)と交換し、細胞をさらに48時間インキュベートして、分化を完了させる。
【0169】
2.CDK阻害化合物及びLPSの刺激による分化したTHP-1細胞の処置
分化の72時間後、培地を無血清増殖培地と交換し、各々DMSOに溶解したCDK阻害化合物並びにポジティブ及びネガティブコントロール等の基準化合物を、0.5〜5μMの範囲の濃度で添加する(ウェルにおけるDMSOの終濃度は0.1%である。)。100ng/mLのLPS(Sigma, L2630)を使用してさらに4〜48時間刺激する前に、細胞を化合物とともに60分間インキュベートする。上清を回収し、サイトカイン発現について、例えば、TNFα、IL-6及びIL-1bについて、市販のサンドイッチELISAアッセイ(eBioscience, カタログ番号88-7346、88-7066、88-7010)を使用して即時アッセイし、又は評価するまで20℃で凍結保存した。
【0170】
3.CDK阻害化合物の投与後のTHP-1上清におけるサイトカイン濃度の決定
製造元の説明書に従って、市販のELISAキット(eBioscience)を使用することによって、細胞培養上清内のTNFα、IL6及びIL1βの濃度を測定する。
【0171】
4.THP-1細胞上清におけるサイトカインのタンパク質発現に及ぼすCDK阻害化合物による処置の効果
CDK阻害化合物1、5、6、8、及び9番を上述のとおり三つ組で、分化したTHP-1細胞へ投与した(第2節参照)。検査化合物又は基準化合物(p38阻害剤であるSB203580及びIKK阻害剤であるBMS345541)単独によるプレインキュベーションの60分後、細胞をLPSで刺激した。4〜48時間のインキュベーションの後、上清を回収し、ELISAベースのサイトカイン上清濃度の決定を、上述の第3節に記載されるとおり実施した。
【0172】
化合物1、5、6、8、及び9及び基準化合物で処置した細胞と、媒体(DMSO)で処置した細胞との比較は、細胞上清におけるTNFα及びIL6のタンパク質濃度に及ぼす化合物1、5、6、8、及び9番の有意な阻害効果を示した。基準化合物SB203580又はBMS3455541と比較して、これらの化合物は、TNFα/IL6発現の同様の又はより良好な阻害を呈した。
【0173】
LPSにより誘導されるTHP-1マクロファージにおけるTNFαの発現に及ぼす化合物1、5、6、8、及び9番の投与の効果を図3に示す。
これらの知見は、CDK阻害化合物1、5、6、8、及び9がサイトカインTNFαの発現の効果的なサプレッサーであることを示す。
【0174】
(実施例10)
A.インビトロでのキナーゼ阻害アッセイ
インビトロでの酵素キナーゼ阻害アッセイにおけるサイクリン依存性キナーゼCDK2/CycA、CDK4/CycD1、CDK5/p35NCK、CDK6/CycD1及びCDK9/CycTについて、化合物1〜15番のIC50特性を決定した。CDK9阻害に関する化合物の特異的選択性及び効力を評価するために、これらのアッセイにおいて得られるIC50値を使用した。
【0175】
これらのアッセイにおいて得られた結果を使用して、CDK9に対する特異性を示す化合物を選択した。特に、該アッセイは、CDK9特異的化合物を、他のCDKに関しても、すなわちCDK2、4、5、及び6のいくつか又はすべてに関して有意な阻害効力を有する他の化合物と識別するよう企図された。この分離は、細胞周期関連CDK2、4、5、及び6の阻害の際に生じ得る有害な(細胞分裂抑制/細胞毒性)効果を回避するために必須である。
さらに、これらのデータを使用して、効力及び選択性に関して新たなかつさらに改良された構造/化合物の設計を支持する構造活性関係性(SAR)を確立した。
【0176】
1.検査化合物
100%DMSOにおける1×10-02Mストック溶液として化合物を使用し、3つの96ウェルV字型マイクロタイタープレート(以下、該プレートを「マスタープレート」と呼ぶ。)の2列目に各100μLを使用した。
その後、100%DMSOを溶媒として使用する連続的な半対数希釈へマスタープレートの2列目における1×10-02Mストック溶液を供し、結果的に10個の異なる濃度を生じ、希釈終点は、12列目における3×10-07M/100%DMSOであった。対照として、1列目及び7列目に100%DMSOを充填した。その後、96チャネルピペッターを使用して、連続希釈したコピープレートの各ウェルの2×5μLを「化合物希釈プレート」の2つの同一セットに分注した。
【0177】
キナーゼ阻害アッセイ当日、45μLのH2Oを1セットの化合物希釈プレートの各ウェルに添加した。沈殿を最小化するために、アッセイプレートへ化合物溶液を転移させるほんの数分前に、H2Oをプレートに添加した。プレートを入念に振盪し、結果的に半対数工程で1×10-03M/10%DMSO〜3×10-08M/10%DMSOの濃度の「化合物希釈プレート/10%DMSO」を生じた。「アッセイプレート」への5μLの化合物溶液の転移のために、これらのプレートを使用した。作業日の終了時に、化合物希釈プレートを廃棄した。アッセイ(以下参照)のために、化合物希釈プレートの各ウェルから5μL溶液をアッセイプレートに転移させた。アッセイの最終容積は50μLであった。1×10-04M〜3×10-09Mの範囲の10個の最終アッセイ濃度で、すべての化合物を検査した。反応混合物における最終DMSO濃度は、すべての場合において1%であった。
【0178】
2.組換えプロテインキナーゼ
阻害特性の決定のために、下記の5個のプロテインキナーゼを使用した:CDK2/CycA、CDK4/CycD1、CDK5/p35NCK、CDK6/CycD1及びCDK9/CycT。バキュロウイルス発現系によって、ヒト組換えGST融合タンパク質又はHisタグつきタンパク質として、該プロテインキナーゼをSf9昆虫細胞において発現させた。GSH-アガロース(Sigma)又はNi-NTH-アガロース(Qiagen)のいずれかを使用するアフィニティクロマトグラフィーによって、キナーゼを精製した。各キナーゼの純度をSDS-PAGE/銀染色によって決定し、キナーゼ特異的抗体を使用するウェスタンブロット分析によって又は質量分析によって、各キナーゼの同一性を確認した。
【0179】
3.プロテインキナーゼアッセイ
50μL反応容積におけるPerkin Elmer/NEN(Boston, MA, USA)製の96ウェルFlashPlates(商標)において、すべてのキナーゼアッセイを実施した。反応混合物を下記の順序で4工程でピペッティングした:
・アッセイ緩衝液20μL(標準緩衝液)
・(H2Oにおける)ATP溶液5μL
・(10%DMSOにおける)検査化合物5μL
・基質10μL/酵素溶液10μL(あらかじめ混合)
【0180】
すべての酵素についてのアッセイは、60mM HEPES-NaOH(pH7.5)、3mM MgCl2、3mM MnCl2、3μMオルトバナジン酸ナトリウム、1.2mM DTT、50μg/mL PEG20000、1μM[-33P]-ATP(ウェルあたり約5×1005cpm)を含有した。
下記の量の酵素及び基質をウェルあたり使用した:
【表2】
【0181】
反応混合物を30℃で80分間インキュベートした。50μLの2%(v/v)H3PO4を使用して反応を停止させ、プレートを吸引し、200μLのH2O又は200μLの0.9%(w/v)NaClを使用して2回洗浄した。マイクロプレートシンチレーションカウンタ(Microbeta, Wallac)を使用して、33Pの組み込みを決定した。
BeckmanCoulter/Sagianロボットシステムを使用して、すべてのアッセイを実施した。
【0182】
4.生データの評価
各アッセイプレートの1列目(n=8)における計数の中央値を「低コントロール」と定義した。この値は、プロテインキナーゼの不在下だが基質の存在下にあるプレートに対する放射能の非特異的結合を反映する。各アッセイプレートの7列目(n=8)における計数の中央値を「高コントロール」、すなわち阻害剤の不在下での完全な活性とみなした。高コントロールと低コントロールとの間の差異を100%活性と呼んだ。データ評価の一部として、高コントロール値から、及び対応するプレートの80個の「化合物値」すべてから、具体的なプレート由来の低コントロール値を減算した。下記の式を使用することによって、具体的なプレートの各ウェルについての残効性(%)を算出した:
残効性(%)=100×[(化合物のcpm−低コントロール)/(高コントロール−低コントロール)]
【0183】
Quattro Workflow V2.0.1.3(Quattro Research GmbH, Munich, Germany; www.quattro-research.com)を使用して、各濃度についての残効性及び化合物のIC50値を算出した。使用したモデルは、「シグモイド反応(可変傾斜)」であり、パラメータの「上部」は100%に、「下部」は0%に固定した。
化合物1〜15のIC50値がすべて、1nM〜10μMの間に含まれることがわかる。
【先行技術文献】
【非特許文献】
【0184】
(参考文献)
【非特許文献1】Barboric M.らの文献(NfkBはP-TEFbと結合してRNAポリメラーゼIIによる転写伸長を刺激する(NfκB Binds P-TEFb to Stimulate Transcriptional Elongation by RNA Polymerase II.)Molecular Cell, 2001, Vol. 8, 327-337)
【非特許文献2】Besson J.M.の文献(疼痛の神経生物学(The neurobiology of pain). Lancet, 1999, 353(9164), 1610-1615)
【非特許文献3】Browerの文献(疼痛緩和のための新たな経路(New paths to pain relief). Nat Biotechnol, 2000, 18(4), 387-391)
【非特許文献4】Chao S.H.及びPrice D.H.の文献(フラボピリドールはインビボでP-TEFb を不活性化し、ほとんどのRNAポリメラーゼII転写を遮断する(Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo). J Biol Chem, 2001, 276(34),31793-9)
【非特許文献5】Chaplan SR, Bach FW, Pogrel JW, Chung JM, 及びYaksh, TLの文献((1994) ラットの足における触覚性アロディニアの定量的評価(Quantitative assessment of tactile allodynia in the rat paw). J Neurosci Methods 53: 55-63)
【非特許文献6】Dai Y.及びGrant S.の文献(サイクリン依存性キナーゼ阻害剤(Cyclin-dependent kinase inhibitors). Curr Opin Pharmacol, 2003, 3(4), 362-370)
【非特許文献7】Falco G.D.らの文献(cdc2様ファミリーのキナーゼの一員であるCDK9は、サイトカインのIL-6ファミリーの受容体であるgp130に結合する(CDK9, a member of the cdc2-like family of kinases, binds to gp130, the receptor of the IL-6 family of cytokines). Oncogene, 2002, 21(49), 7464-7470)
【非特許文献8】Feldmann及びMainiの文献(NatMed, 2003, 9 (10); 356-61)
【非特許文献9】Firesteinの文献(2003, Nature 423, 356-361)
【非特許文献10】Hanらの文献(2003, Autoimmunity, 28, 197-208)
【非特許文献11】Hargreaves, Kの文献(Pain 32(1) (1988 Jan) 77-88)
【非特許文献12】Huweらの文献(サイクリン依存性キナーゼの阻害剤としての小分子(Small molecules as inhibitors of cyclin-dependent kinases). Angew Chem Int Ed Engl, 2003, 42(19), 2122-2138)
【非特許文献13】Kimらの文献(CDK9によるRNAポリメラーゼIIカルボキシル末端ドメインのリン酸化は、ヒト免疫不全ウイルス1型Tatにより活性化される転写伸長の直接の原因である(Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation). Mol Cell Biol, 2002, 22(13), 4622-4637)
【非特許文献14】Koltzenburg Mの文献(皮膚侵害受容疼痛の神経性機構(Neural mechanisms of cutaneous nociceptive pain). Clin J Pain, 2000, 16(3 Suppl), 131-138)
【非特許文献15】Laufer S., Gay S.及びBrune K.の文献(炎症及びリウマチ疾患−新規の治療法の分子レベルの基礎(Inflammation and Rheumatic Diseases The molecular basis of novel therapies), Thieme, 2003)
【非特許文献16】Lee K.M.らの文献(脊髄性NfkBの活性化は、COX-2上方制御を誘導し、炎症性疼痛過敏症に関与する(Spinal NfκB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity). European Journal of Neuroscience, 2004, Vol. 19, 3375-3381)
【非特許文献17】Liu H.及びHerrmann C.の文献(CDK9の42k及び55k分子種の異なる局在性及び発現(Differential Localization and Expression of the CDK9 42k and 55k Isoforms). J Cell Physiol, 2004, 203, 251-260)
【非特許文献18】MacLachlan T.K.らの文献(TRAF2に対するCDK9の結合(Binding of CDK9 to TRAF2). J Cell Biochem, 1998, 71(4), 467-478)
【非特許文献19】Malmberg AB及びBasbaum AI.の文献((1998) 神経因性疼痛のモデルとしてのマウスにおける部分的坐骨神経損傷:行動相関及び神経解剖学的相関(Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates). Pain 76: 215-2)
【非特許文献20】Meijer L, Leclerc S.及びLeost M.の文献(サイクリン依存性キナーゼの化学的阻害剤の特性および可能性のある応用(Properties and potential applications of chemical inhibitors of cyclin-dependent kinases), Pharmacol Ther 1999, 82(2-3):279-284)
【非特許文献21】Sausville, E.A.の文献(Trends Molec. Med. 2002, 8, S32-S37)
【非特許文献22】Tian B.らの文献(TNFシグナル伝達を仲介するNfkB転写因子の下流の直接的なゲノム標的の同定(Identification of direct genomic targets downstream of the NfSYMBOL 107 \f "Symbol" \s 10.5B transcription factor mediating TNF signaling). JBC, 2005, 原稿M500437200として)
【非特許文献23】Wang Dらの文献(化学的サイクリン依存性キナーゼ阻害剤によるヒト免疫不全ウイルス1型の転写の阻害(Inhibition of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent kinase inhibitors). J. Virol. 2001; 75: 7266-7279)
【非特許文献24】Watkins L.R.らの文献(グリア炎症誘発性サイトカインは過度の疼痛状態を仲介する:臨床的疼痛についての含意(Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain). Adv Exp Med Biol., 2003, 521, 1-21)
【非特許文献25】Westらの文献(2001, Journal of Virology 75(18), 85248537)
【非特許文献26】Zhou M.らの文献(ヒト免疫不全ウイルス1型の転写の間のP-TEFbキナーゼによる転写因子のリン酸化及びヒストンのメチル化の協調(Coordination of transcription factor phosphorylation and histone methylation by the P-TEFb Kinase during human immunodeficiency virus type I transcription), J. Virol 2004, 78(24):13522-13533)
【背景技術】
【0001】
(発明の背景)
サイクリン依存性プロテインキナーゼ(「CDK」)は、細胞周期の制御及び転写調節等の、細胞内での複数の役割を担う十分に保存された酵素のファミリーを構成する(Science 1996, Vol. 274:1643-1677;Ann. Rev. Cell Dev. Biol., 1997, 13:261-291)。
【0002】
CDK1、2、3、4、及び6等の、ファミリーのいくつかのメンバーは、G1の静止期(細胞分裂の新たな一区切りについての有糸分裂とDNA複製の開始との間のギャップ)からS期(活発なDNA合成の期間)への進行、又はG2期から、活発な有糸分裂及び細胞分裂が生じるM期への進行など、細胞周期の異なる相間の移行を制御する。CDK7、8、及び9を含むこのファミリーのタンパク質の他のメンバーは、転写周期における重要な点を制御するのに対し、CDK5は、神経細胞及び分泌細胞の機能における役割を担う。
【0003】
CDK複合体は、制御サイクリンサブユニット(例えば、サイクリンA、B1、B2、D1、D2、D3、及びE)と触媒キナーゼサブユニット(例えば、cdc2(CDK1)、CDK2、CDK4、CDK5、及びCDK6)との会合を通じて形成される。名称が含意するように、CDKは、CDKの標的基質をリン酸化するために、サイクリンサブユニットへの絶対的な依存性を示し、異なるキナーゼ/サイクリン対は、細胞周期の特定の部分を通じて進行を制御するよう機能する。
【0004】
CDK9のサイクリンパートナー(サイクリンT1、T2a、T2b、又はK)との会合にあるCDK9は、RNAポリメラーゼIIの最大のサブユニットのカルボキシル末端ドメイン(CTD)をリン酸化することによって、転写の伸長相の間に機能する正のP-TEFbプロテインキナーゼ複合体の触媒性構成要素を構成する。P-TEFbは、正の転写因子NfκB及び負の転写因子に関して作用し、このように転写伸長の遮断を克服する(Liu及びHerrmann 2005)。
【0005】
腫瘍細胞の中心的な特徴の1つである細胞周期の制御障害が、CDK及びその制御因子の遺伝的な変化及び制御障害と密接に関連していることは公知であり、CDKの阻害剤が、癌等の増殖性疾患のための治療薬として有用であり得ることを示唆する。このように、CDKを標的とする小分子阻害剤はこれまで、癌治療法における甚大な関心対象の焦点であった(Current Opinion in Pharmacology, 2003(3): 362-370)。細胞周期の進行を阻害する能力は、癌等の増殖性疾患のための治療薬としての、CDKの小分子阻害剤についての一般的な役割を示唆する。細胞周期関連型CDKの阻害は、腫瘍学の適用において明確に関連性があるが、このことは、RNAポリメラーゼを制御するCDKの阻害に関する場合ではないかもしれない。近年、CDK9/サイクリンTの機能の阻害は、HIV複製の予防と連関し、このように、新たなCDK生物学に関する発見は、例えば、ウイルス感染(WO 02/100401)等のCDK阻害剤についての新たな治療上の効能を利用できるようにし続ける(Sausville, E.A.の文献(Trends Molec. Med. 2002, 8, S32-S37))。また、CDK阻害剤は、とりわけ免疫学的疾患及び神経変性疾患等の他の容態を治療するためにおそらく使用され得る。
【0006】
50を超える薬理学的CDK阻害剤が記載されており、そのうちのいくつかは、強力な抗腫瘍活性を有する(Current Opinion in Pharmacology, 2003(3): 362-370)。公知のCDK阻害剤についての包括的な総説は、Angew. Chem. Int. Ed. Engl. 2003, 42(19):2122-2138において見出され得る。
【0007】
例えば、癌の治療のためのサイクリン依存性キナーゼ阻害剤としての2-アニリノ-4-フェニルピリミジン誘導体の使用は、WO 2005/012262において報告されている。さらに、癌等の治療のための2-ピリジニルアミノ-4-チアゾリル-ピリミジン誘導体は、WO2005/012298に記載されている。プロテインキナーゼ阻害剤としての4,5-ジヒドロ-チアゾロ、オキサゾロ及びイミダゾロ[4,5-h]キナゾリン-8-イルアミンの使用は、WO2005/005438から公知である。さらに、サイクリン依存性キナーゼ阻害剤として有用なインドリノン誘導体及びインヅリビン(induribin)誘導体は、WO02/081445及びWO02/074742に開示されている。さらに、治療上の多様な適用のためのCDK阻害剤は、WO2005/026129に記載されている。
【0008】
公知のCDK阻害剤は、一般にCDKを阻害するCDK阻害剤の能力に従って、又は特定のCDKに対するCDK阻害剤の選択性に従って分類され得る。例えば、フラボピリドールは、「受け皿の」CDKアンタゴニストとして作用し、特定のCDKに対して特に選択的ではない(Current Opinion in Pharmacology, 2003(3): 362-370)。オロモウシン、ロスコビチン、プルバノロール(purvanolol)及びCGP74514A等のプリンベースのCDK阻害剤は、CDK1、2及び5に対するより大きな選択性を呈することが公知であるが、CDK4及び6に対する阻害活性を何ら示さない(Current Opinion in Pharmacology, 2003(3): 362-370)。さらに、ロスコビチン等のプリンベースのCDK阻害剤が、神経系において抗アポトーシス性効果を発揮でき(Pharmacol Ther 2002, 93:135-143)、又はアルツハイマー病等の神経変性疾患における神経細胞の死滅を予防できることが示されている(Biochem Biophys Res Commun 2002 (297):1154-1158;Trends Pharmacol Sci 2002 (23):417-425)。
【0009】
増殖性疾患、免疫学的疾患、感染性疾患、心血管疾患及び神経変性疾患等の容態の治療法のための標的指向化CDKのすばらしい潜在性を考慮すると、具体的なCDKの選択的阻害剤としての小分子の開発は、所望の目的の構成要素となる。
【0010】
本発明は、サイクリン依存性キナーゼの新規の小分子阻害剤を提供する。好ましくは、該小分子阻害剤は、具体的なCDK、特にCDK9を阻害する高い効能を示す。該小分子阻害剤は、増殖性疾患、免疫学的疾患、神経変性疾患、感染性疾患及び心血管疾患等の容態の治療のための治療的有用性を有し得る。さらに、本発明の小分子阻害剤は、炎症性疾患の及びいずれかの種類の疼痛の治療において有益な効果を発揮することが驚くべきことに示されている。
【0011】
疼痛についての現行の治療は、部分的に効果的であるに過ぎず、また多くは、衰弱させる又は危険な副作用を生じる。例えば、重度の疼痛を治療するのに使用される伝統的な鎮痛薬の多くは、吐き気、めまい、便秘、呼吸抑制、及び認知機能障害等の衰弱させる副作用を誘発する(Brower, 2000)。
【0012】
利用可能な非麻酔性鎮痛薬、オピオイド鎮痛薬、カルシウムチャネル遮断薬、筋弛緩薬、及び全身性コルチコステロイドのような認可された疼痛投薬の広範な名簿はすでに存在するが、該治療は、単に経験的なものであるままに過ぎず、及び該治療は、疼痛の症状を軽減し得るが、該治療は、ほとんどのケースにおいて軽減を完了させるには至らない。また、このことは、異なる種類の疼痛の発達に潜む機構が、なおもほとんど理解されていないに過ぎないという事実による。研究者は、各種類の疼痛についての神経インパルスを遅延させるのに使用されるシグナル伝達系の複雑性及び多様性をまさに正しく認識し始めつつあるに過ぎない。
【0013】
一般的に、疼痛は、国際疼痛学会(International Association for the Study of Pain(IASP))に従って、実際の又は潜在的な組織の障害と関連した不愉快な感覚的及び情動的経験として定義され、又はこのような障害の点で記載される。具体的には、疼痛は、急性又は慢性の疼痛として生じ得る。
【0014】
急性疼痛は、短時間、典型的には1ヶ月未満にわたって生じ、一時的な障害と関連する。急性疼痛は、短時間内でさらなる障害を結果的に生じ得る生理学的又は生化学的変化を、宿主に知らせるための自然な身体反応である。有害な刺激が、末梢神経終末における高い閾値の機械的侵害受容器及び/又は熱侵害受容器を活性化し、薄い有髄(Aδ)の及び/又は無髄(C)の求心性線維における誘発活動電位が知覚のある脳に到達する場合、急性疼痛が感じられる。該有害な刺激は、損傷、手術、病気、外傷又は有痛の医学的手法によって提供され得る。急性疼痛は通常、潜在的な原因が治療され又は治癒された場合に消失する。しかしながら、軽減されない急性疼痛は、長期の入院、再入院、外来診療所及び緊急部門への訪問、並びに高額な保健医療費用を結果として生じ得る慢性疼痛の問題に至り得る。
【0015】
急性疼痛とは対照的に、慢性疼痛は、初期損傷が治癒した後に長く持続し、しばしば身体の他の部分へ拡大し、それに伴って多様な病理学的及び精神医学的結果が生じる。慢性的な体性痛は、末梢組織における外傷に対する炎症反応から結果的に生じ(例えば、神経の絞扼、外科的手法、癌、又は関節炎)、それが侵害受容器の感作過剰、及び通常は有害ではない刺激に対する激しい灼熱性疼痛反応に至る(痛覚過敏)。慢性疼痛は、持続的かつ再発性があり、その強度は、軽度から、生活の質を有意に低下させ得る重度の身体障害疼痛まで変動するであろう。
【0016】
慢性疼痛は目下のところ、NSAID(イブプロフェン、ナプロキセン)、Cox-2阻害剤(セレコキシブ、バルデコキシブ、ロフェコキシブ)及びアヘン製剤(コデイン、モルヒネ、テバイン、パパベリン、ノスカピン)等の従来の鎮痛薬を使用して治療される。しかしながら、有意な数の患者に対して、これらの薬物は不十分な疼痛軽減しか提供しない。
【0017】
別のサブタイプの疼痛である炎症性疼痛は、急性及び慢性の疼痛として生じ得る。結果として生じる組織及び神経細胞の損傷は、これらの炎症性事象を継承して、長期間持続する慢性の神経因性疼痛効果へ発展してはならないが、発展し得る。
【0018】
炎症性疼痛は、例えば、組織損傷、疾患、又は炎症後に放出される炎症性仲介物質(例えば、TNFα等のサイトカイン、プロスタグランジン、P基質、ブラジキニン、プリン、ヒスタミン、及びセロトニン)のような有害刺激及び他の有害刺激(例えば、熱刺激、機械的刺激、又は化学的刺激)によって仲介される。さらに、サイトカイン及び増殖因子は、神経細胞の表現型及び機能に影響し得る(Besson 1999)。これらの仲介物質は、組織の周囲のいたるところに分布する侵害受容器(感覚受容器)によって検知される。該侵害受容器は、長期化した場合、組織に障害を与えるであろう(例えば、機械的、熱の、又は化学的)有害刺激に対して感受性がある(Koltzenburg 2000)。特別なクラスのいわゆるC型侵害受容器は、いずれのレベルの機械的刺激又は熱刺激にも反応しないが炎症のみの存在下で活性化されるあるクラスの「サイレントな」侵害受容器に相当する。
【0019】
特に炎症性疼痛の治療のための現行のアプローチは、サイトカイン阻害(例えば、IL1β)及び炎症誘発性TNFαの抑制を目的とする。認可された現行の抗サイトカイン/抗TNFα治療は、インフリキシマブ及びエタネルセプト等の、血流におけるTNFα循環を低下させるキメラ抗体に基づいている。TNFαは、COX-2、MMP、iNOS、cPLa2及びその他等の重要な酵素の合成を誘導する最も重要な炎症性仲介物質の1つである。しかしながら、これらの「生物製剤」の主要な欠点は、効能の損失の付随する生物製剤の免疫原性能力及び、循環しているTNFαの多かれ少なかれ全か無のデジタルな減少に至る生物製剤の動態に存する。後者は、重度の免疫抑制性の副作用を結果として生じ得る。
【0020】
別個の形態の慢性疼痛である神経因性(又は神経原性)疼痛は、末梢神経又は中枢神経の機能障害の結果として生じ、病因及び部位において異なる多様な容態を含む。一般的に、神経因性疼痛の原因は多様だが、末梢神経に対する又は中枢経路の構成要素に対する障害に関する普遍的な症状を共有する。原因となる因子は、代謝性の、ウイルス性の又は機械的な神経病変であり得る。神経因性疼痛は、末梢神経系、中枢神経系、又はその両者における異所性の体性感覚過程によって持続されると信じられている。神経因性疼痛は、侵害受容器の刺激と直接連関していないが、代わりに、例えば脊髄の灰白質(後角)にあるシナプス後神経線維におけるグルタミン酸受容体の感作過剰から生じると考えられる。
【0021】
神経因性疼痛は、糖尿病及び帯状疱疹後神経痛(帯状疱疹)における神経変性等の容態と関連している。神経因性疼痛の容態は、糖尿病、エイズ、多発性硬化症、切断後の断端痛及び幻肢痛、癌関連ニューロパチー、帯状疱疹後神経痛、外傷性神経損傷、虚血性ニューロパチー、神経圧迫、脳卒中、脊髄損傷を含む多くの疾患及び容態の結果である。
【0022】
神経因性疼痛の管理は、神経因性疼痛の発達及び維持に関与する機構に関する不適切な理解に一部起因して、臨床上の主要な挑戦の域を出ない。既存の多くの鎮痛薬は、神経因性疼痛を治療する上で効果がなく、現行の麻酔性薬物及び非麻酔性薬物のほとんどは疼痛を調節しない。現行の臨床的実践には、神経因性疼痛の管理のための多くの薬物クラス、例えば抗痙攣薬、三環系抗うつ薬、及び全身性局所麻酔薬の使用が含まれる。しかしながら、これらの治療の通常の結果は、部分的な又は不満足な疼痛軽減であり、いくつかの場合、これらの薬物の有害作用は、該薬物の有用性よりも重い。古典的な鎮痛薬は、神経因性疼痛の治療においてほとんど効果がなく又はまったく効果がないと広く信じられている。神経因性疼痛の治療における非ステロイド性消炎薬(NSAID)又はアヘン製剤の使用に関するいくつかの臨床研究が実施されたが、実施された該臨床研究において、結果は、NSAIDがほとんど効果的ではなく又はまったく効果的ではなく、及びアヘン製剤が高い用量でしか作用しないことを示すように思われる。末梢神経因性疼痛(PNP)についての調節された臨床データを分析する総説(Pain, November, 1997 73(2), 123-39)は、NSAIDがPNPのための鎮痛薬としておそらく効果的ではなく、薬物の鎮痛効果を支持する長期的なデータが全くないことを報告した。
【0023】
利用可能な鎮痛薬はしばしば、不十分な疼痛軽減を生じる。三環系抗うつ薬及びいくつかの抗てんかん薬、例えばガバペンチン、ラモトリギン及びカルバマゼピンは、患者によっては有効であるが、これらの容態の治療に有効な薬物について、まだ対処されていない需要が大きいままである。
結論として、疼痛治療、特に慢性の炎症性疼痛及び神経因性疼痛に関する安全でかつ効果的な方法について、まだ対処されていない高い需要がある。
【発明の概要】
【0024】
本発明は、サイクリン依存性キナーゼの阻害剤、並びにいずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療し及び/又は予防するための方法及び組成物であって、該方法又は組成物を必要とする対象へ、サイクリン依存性キナーゼ(cdk、CDK)の有効量の少なくとも1つの阻害剤を投与することを含む、前記方法及び組成物に関する。阻害剤は、一般式I記載の化合物:
【化1】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;及びこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)のうちで選択される。
(式中、
R1が、-XSO2NR5R6又は-XSO2R8であり;式中、
Xが、(分岐鎖アルキレンを含む)C1-4アルキレンであり、式中、該C1-4アルキレンが、R5又はR6へ結合して、5〜6員の複素環を形成でき;
R5及びR6が互いに独立して、水素、C1-4アルキル、ヒドロキシ-C1-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル若しくはC4-7-ヘテロシクロアルキル-C0-4アルキル、C4-7-アリール-C0-4アルキル、C4-7-ヘテロアリール-C0-4アルキル又は
式中、R5及びR6はそれらが結合するN原子とともにまた、5〜8員のヘテロシクロアルキルを形成し得、
式中、該シクロアルキル、ヘテロシクロアルキル、アリール、ヘテロアリール又はアルキルがさらに、ハロ、ヒドロキシ、アミノカルボニル、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O-及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R8が、C1-4アルキル、ヒドロキシ-C2-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル又はC4-7-ヘテロシクロアルキル-C0-4アルキルであり;
式中、該シクロアルキル、ヘテロシクロアルキル又はアルキルがさらに、ハロ、ヒドロキシ、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R2が、ハロゲン及び水素から独立して選択される1個又は2個の置換基であり;
R3が、水素、ハロ、ヒドロキシ、C1-4アルキル、C3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、アリール-C1-4アルコキシ、ヘテロアリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルキル、ヘテロアリール-C1-4-アルキル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から各々独立して選択される1〜3個の置換基であり得、この中で、上述の置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、アミノカルボニル、ハロ及びNR5R6の群から選択されるラジカルによってさらに置換でき;
R4a及びR4bが、同一又は異別であり、かつ各々が独立して、水素、C1-4アルキル又は-NR'R"であり、式中R'及びR"が各々独立して、水素又はC1-4アルキルである。)。
【0025】
本明細書に開示されるように、「C1-4アルキル」という用語は、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、イソブチル、tert-ブチル等の、1〜4個の炭素原子を有する直鎖又は分岐鎖の飽和脂肪族炭化水素を含むよう意味される。
「低級アルケニル」という用語は、例えば、ビニル、アリル、ブト-2-エニル、-ブト-3-エニル、又はイソプロペニル等の2〜6個の炭素原子を有する直鎖又は分岐鎖アルケン基を指し;「低級アルケニル」という用語は好ましくは、アリル、-ブト-2-エニル、又は-ブト-3-エニルを表す。
【0026】
本明細書に開示されるように、「ハロ」という用語は、フルオロ、クロロ、ブロモ、及びヨードを含むことを意味する。
C3-C8シクロアルキルという用語は、下記のシクロアルキル:
【化2】
を示す。
アリールという用語は、フェニル、ナフチル、3-クロロフェニル、2,6-ジブロモフェニル、2,4,6トリブロモフェニル、4,7-ジクロロナフチル、好ましくはフェニル又はナフチル等の芳香族単環式又は二環式の6〜10員の環系を示す。
【0027】
ヘテロシクロアルキルという用語は、酸素、硫黄又は窒素から独立して選択される1〜3個のヘテロ原子を含有する5〜10員の単環式又は二環式環系を含むことを意味し、好ましくはアジリジニル、アゼチジニル、ピロリジニル、テトラヒドロフラニル、テトラヒドロチオフェニル、ピペリジニル、ピペラジジニル、ピペラジニル、テトラヒドロピラニル、テトラヒドロチオピラニル又はモルフォリニルを含む群から選択される。
【0028】
ヘテロシクリルという用語はさらに、以下に定義されるすべてのヘテロアリールを含み、この中で、対応するヘテロアリールのすべての二重結合は、単結合によって置換される。
ヘテロアリールという用語は、酸素、硫黄又は窒素から独立して選択される1〜3個のヘテロ原子を含有する、部分的に又は完全に不飽和の5〜10員の単環式又は二環式の環系を示し、好ましくはピロリル、フラニル、チオフェニル、チエニル、イミダゾリル、ピラゾリル、チアゾリル、オキサゾリル、イソチアゾリル、イソキサゾリル、ピリジニル、ピリジル、ピリミジニル、ピリミジル、ピラジニル、ピラジル、ピラジジニル、ピラジジル、3-メチルピリジル、ベンゾチエニル、4-エチルベンゾチエニル、3,4-ジエチルフラニル、ピロリル、テトラヒドロキノリル、キノリル、テトラヒドロイソキノリニル、イソキノリニル、ベンゾイミダゾリル、ベンゾチアゾリル、ベンゾオキシゾリル、ベンゾ[1,3]ジオキソリル、インドリル、ベンゾフラニル、ベンゾチオフェニル、インダゾリル又はクロム-2-オニルからなる群から選択される。
【0029】
また、ヘテロアリールという用語が、部分的に不飽和の5〜10員の単環式又は二環式の環系を含み、この中で、該環系の1〜4個の二重結合が単結合によって置換され、及びこの中で、該環系が少なくとも1個の二重結合を含有することは理解されるべきである。
【0030】
変数が、式I又はいずれかの置換基において2回以上生じる場合、各発生に関する該変数の定義は、その他すべての発生における該変数の定義とは独立している。例えば、化合物が、2個以上のR5及び/又はR6置換基を含む場合、これらの置換基は同一又は異別であり得る。
【0031】
式Iの化合物において、
Xは、好ましくはメチレンである。
R2は、好ましくは水素である。
好ましくは、R6は、水素又はメチルであり、及びR5は、エチル、2-ヒドロキシエチル、イソプロピル、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシル、n-プロピル、t-ブチル、3-メトキシ-プロピル、2-ジメチルアミノエチル、3-ジメチルアミノプロピル、ピペリジニル及び特に4-ピペリジニル、ピリジニル及び特にピリジン-3-イル又はピリジン-4-イル、ピロリジニル及び特にピロリジン-3-イル、テトラヒドロフラニル及び特にテトラヒドロ-フラン-3-イル、テトラヒドロ-フラン-2-イルメチル、4-クロロ-ベンジル、チオフェン-2-イル-メチル、又はR5及びR6は両者とも、水素、メチル、エチル、又は
R5及びR6はそれらが結合するN原子とともに、モルフォリン、4-アミノカルボニル-ピペリジン又はアゼパンを形成し、又は-XSO2NR5R6は、
【化3】
である。
R5及びR6は、より好ましくは各々独立して、水素、メチル、シクロプロピル、2-ジメチルアミノエチル、3-ジメチルアミノプロピル及び2-ヒドロキシエチルから選択される。R5及びR6は、最も好ましくは水素である。
【0032】
R8は好ましくは、C1-4アルキル又はヒドロキシ-C-2-4-アルキルであり;R8はより好ましくは、メチル又は2-ヒドロキシエチルである。
R3は好ましくは、水素、ハロ、ヒドロキシ、C1-4アルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基であり;又はC3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、アリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4-アルキル、ヘテロアリール-C1-4アルコキシ、ヘテロアリール-C1-4-アルキルから選択される1個の置換基であり;この中で、該置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、ハロ、アミノカルボニル、及びNR5R6の群から選択される1個以上のラジカルによってさらに置換できる。R3はより好ましくは、メチル、エチル、ヒドロキシメチル、ヒドロキシ、メトキシ、エトキシ、イソプロポキシ、ベンジルオキシ、水素、フルオロ、クロロ、トリフルオロメチル、2-メトキシ-エトキシ、メトキシメチル、2-メトキシ-エチル、テトラヒドロ-フラン-3-イルオキシ、テトラヒドロ-フラン-2-イル-メトキシ、-N(CH3)SO2CH3、ピペリジン-1-イル-メチル、2-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-(2-ヒドロキシ-エチル)-ピペリジン-1-イル-メチル、3-アミノカルボニル-ピペリジン-1-イル-メチル、ジメチルアミノメチル、ジエチルアミノメチル、(エチル-イソプロピル-アミノ)-メチル、モルフォリン-4-イルメチル、4-メチル-ピペラジン-1-イル-メチル、[1,2,4]トリアゾール-1-イル-メチル、ピリジン-3-イル-メトキシ及びピリジン-4-イル-メトキシからなる群から独立して選択される1〜3個の置換基である。R3は最も好ましくは、メトキシ、イソプロポキシ及び/又はフルオロである。
【0033】
式Iの化合物の別の好ましい群は、式Iaの化合物
【化4】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;及びこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば水和物)を含む
(式中、
R2、R4a及びR4bは、上述に定義されるとおりであり;
R3a、R3b及びR3cは、R3に関して上述に定義されるとおりであり;及び
R9は、NR5R6又はR8であり、式中、R5、R6及びR8は、上述に定義されるとおりである。)。
【0034】
式Iaのこれらの化合物内で:
R2は好ましくは、水素又はハロゲンであり、より好ましくは水素である。
R3aは、好ましくは水素、ハロ又はC1-4アルコキシであり;より好ましくはC1-4アルコキシであり;最も好ましくはメトキシ又はイソプロポキシである。
R3bは、好ましくは水素である。
R3cは、好ましくは水素又はハロゲンであり、特にフルオロである。
R4aは、好ましくはC1-4アルキル又は水素であり;より好ましくは水素又はメチルであり;最も好ましくは水素である。
R4bは、好ましくは水素又はC1-4アルキルであり;より好ましくは水素又はメチルであり;最も好ましくは水素である。
R5及びR6は、好ましくは各々独立して、水素、ヒドロキシル又はジアルキルアミノによって任意に置換されるシクロアルキル又はC1-4アルキルであり;最も好ましくはR5及びR6は、各々独立して、水素、メチル、シクロプロピル、2-ヒドロキシエチル、2-ジメチルアミノエチル又は3-ジメチルアミノプロピルである。
R8は、好ましくはヒドロキシ-C-2-4-アルキルであり、より好ましくは2-ヒドロキシエチルである。
【0035】
下記の化合物が好ましい:
{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物1);
{3-[4-(2-イソプロポキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物2);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N,N-ジメチル-メタンスルホンアミド(化合物3);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物4);
{3-[4-(4-フルオロ-2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物5);
N-シクロプロピル-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物6);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物7);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物8);
2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物9);
{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物10);
-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物11);
2-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物12);
{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物13);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物14);及び
2-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物15)。
【0036】
さらなる実施態様において、本発明は、医療的使用のために、式I記載の上述の化合物を提供する。
さらなる実施態様において、本発明は、医薬として許容し得る担体とともに先に概略した化合物を含有する医薬組成物を提供する。
【0037】
さらなる実施態様において、本発明は、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための医薬組成物を調製するための先に概略した化合物の使用を提供する。
【0038】
さらなる実施態様において、本発明は、疼痛、慢性疼痛、及び/又は神経因性疼痛を治療するための医薬組成物を調製するための先に概略した化合物の使用を提供する。
さらなる実施態様において、本発明は、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための方法であって、必要とする対象に、有効量の上述の化合物の少なくとも1つの投与を含む、前記方法を提供する。
さらなる実施態様において、本発明は、疼痛、慢性疼痛及び/又は神経因性疼痛を治療するための方法であって、必要とする対象に、有効量の上述の化合物の少なくとも1つの投与を含む、前記方法を提供する。
【図面の簡単な説明】
【0039】
【図1】図1は、坐骨神経の2つの分岐(すなわち脛骨神経及び総腓骨神経)の結紮及び切断を特徴とする、腓腹神経を無処置のままにしている坐骨神経部分損傷モデル(SNIモデル、Decosterd及びWoolf(2000)によって開発)を模式的に図示する。
【図2】図2は、疼痛の発達における標的としてのCDK9の可能性のある役割を模式的に図示する。
【図3】図3は、LPS誘発性THP-1マクロファージを使用して実施されるサイトカイン測定の結果を図示する。TNF発現は、DMSO対照と比較した%阻害として示した。化合物1、5、6、8、又は9で処置した細胞は、媒体(DMSO)と比較して、上清におけるTNF発現の有意な阻害を示す。基準化合物SB203580(p38阻害剤)及びBMS345541(IKK阻害剤)と比較して、これらの化合物は、本アッセイにおけるTNF発現の同様の又はより良好な阻害を呈する。
【発明を実施するための形態】
【0040】
(発明の詳細な記述)
本発明によって提供されるCDKの阻害剤は、一般式I記載の化合物:
【化5】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;及びこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)である
(式中、
R1は、-XSO2NR5R6又は-XSO2R8であり;式中、
Xは、(分岐鎖アルキレンを含む)C1-4アルキレンであり、式中、該C1-4アルキレンが、R5又はR6へ結合して、5又は6員の複素環を形成でき;
R5及びR6が互いに独立して、水素、C1-4アルキル、ヒドロキシ-C1-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル若しくはC4-7-ヘテロシクロアルキル-C0-4アルキル、C4-7-アリール-C0-4アルキル、C4-7-ヘテロアリール-C0-4アルキル又は
式中、R5及びR6はそれらが結合するN原子とともにまた、5〜8員のヘテロシクロアルキルを形成し得、
式中、該シクロアルキル、ヘテロシクロアルキル、アリール、ヘテロアリール又はアルキルがさらに、ハロ、ヒドロキシ、アミノカルボニル、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O-及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R8が、C1-4アルキル、ヒドロキシ-C2-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル又はC4-7-ヘテロシクロアルキル-C0-4アルキルであり;
この中で、該シクロアルキル、ヘテロシクロアルキル又はアルキルがさらに、ハロ、ヒドロキシ、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R2が、ハロゲン及び水素から独立して選択される1個又は2個の置換基であり;
R3が、水素、ハロ、ヒドロキシ、C1-4アルキル、C3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、アリール-C1-4アルコキシ、ヘテロアリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルキル、ヘテロアリール-C1-4-アルキル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から各々独立して選択される1〜3個の置換基であり得、この中で、上述の置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、アミノカルボニル、ハロ及びNR5R6の群から選択されるラジカルによってさらに置換でき;
R4a及びR4bが、同一又は異別であり、かつ各々が独立して、水素、C1-4アルキル又は-NR'R"であり、式中R'及びR"が各々独立して、水素又はC1-4アルキルである。)。
【0041】
本発明の好ましい実施態様において、式I記載のサイクリン依存性キナーゼ阻害剤は、CDK1、CDK2、CDK3、CDK4、CDK5、CDK6、CDK7、CDK8、CDK9、CDK10、CDK11、CrkRS(CDC2関連プロテインキナーゼ7(Crk7))、CDKL1(サイクリン依存性キナーゼ様1型);KKIALRE、CDKL2(サイクリン依存性キナーゼ様2型)、KKIAMRE、CDKL3(サイクリン依存性キナーゼ様3型)、NKIAMRE、サイクリン依存性キナーゼ様1型と同様のCDKL4、CDC2L1(細胞分裂周期様2型)、PITSLRE A、CDC2L5(細胞分裂周期2型様5型)、PCTK1(PCTAIREプロテインキナーゼ1型)、PCTK2(PCTAIREプロテインキナーゼ2型)、PCTK3(PCTAIREプロテインキナーゼ3型)又はPFTK1(PFTAIREプロテインキナーゼ1型)からなる群から選択されるCDKを阻害する。
【0042】
また、阻害剤は、上述に列挙される群から選択される2つ以上のサイクリン依存性キナーゼを阻害し得る。
本発明の具体的な好ましい実施態様において、式I記載の化合物は、CDK9を阻害する。
【0043】
本発明のさらなる実施態様において、式I記載の化合物は、他の酵素又はタンパク質に及ぼす実質的な阻害効果を有さずに、1つ以上のCDKを選択的に阻害する。
好ましい実施態様において、これらの阻害化合物は、具体的なCDKに対する高い選択性を示す。本明細書で使用される「高い選択性」は、阻害化合物が、本明細書に列挙される上述のCDKの群から選択される具体的なCDKに対して少なくとも10〜100倍以上選択的である。本発明の好ましい実施態様において、阻害化合物は、具体的なCDKに対して20〜90倍以上選択的である。具体的な好ましい実施態様において、阻害化合物は、具体的なCDKに対して30〜80倍以上選択的である。
具体的な好ましい実施態様において、式I記載の化合物は、他のCDKに対するよりもCDK9に対して高い選択性を示す。
【0044】
本明細書で使用されるように、「阻害する」又は「阻害」という用語は、サイクリン依存性キナーゼの細胞機能、すなわち、サイクリン依存性キナーゼの活性又は発現を少なくとも部分的に下方制御し、低下させ、減少させ、抑制し、不活性化し、又は阻害する化合物の能力を指す。
【0045】
さらに、「サイクリン依存性キナーゼ阻害剤」という用語は、サイクリン依存性キナーゼの量及び/又は活性を下方制御でき、低下でき、抑制でき又はそれに代わるものとして制御できる化合物又は化合物の群を指す。該キナーゼの阻害は、キナーゼポリペプチドに直接結合すること、キナーゼを変性させること又はそれに代わるものとして不活性化させること、又はキナーゼをコードする遺伝子の発現(例えば、mRNAへの転写、新生ポリペプチドへの翻訳、及び/又は成熟タンパク質への最終的なポリペプチド修飾)を阻害することを含むがそれらに限定されるわけではない、当技術分野で公知の多様な機構によって達成できる。さらにまた、サイクリン依存性キナーゼ阻害剤は、CDK依存性経路におけるCDKの下流に作用する分子の発現、修飾、制御又は活性化に干渉し得る。一般的に、キナーゼ阻害剤は、タンパク質、ポリペプチド、核酸、小分子、又は他の化学的部分であり得る。特に、キナーゼ阻害剤にはまた、サイクリン依存性キナーゼに対して配向されるモノクローナル抗体又はポリクローナル抗体が含まれる。
好ましい実施態様において、サイクリン依存性キナーゼ阻害剤は、本明細書に開示される式Iによって表される化合物から選択される。
【0046】
(治療的使用)
式Iの化合物は、サイクリン依存性キナーゼの阻害剤である。このように、該化合物は、異常に分裂する細胞における細胞周期の調節を抑止し又は回復させる能力を有すると期待される。結果として、式I記載の化合物は、癌等の増殖性障害を治療し及び/又は予防する上で有用であると立証するであろう。
【0047】
CDKは、アポトーシス、増殖、分化及び転写における役割を担うことが公知であり、それゆえまた、式I記載の化合物は、感染性疾患、免疫学的疾患、神経変性疾患及び心血管疾患など、増殖性疾患以外の疾患の治療において有用であり得る。
【0048】
さらにまた、式I記載の化合物は、予期せぬ抗侵害受容効果及び抗炎症効果を示す。
このように、好ましい実施態様において、式Iの化合物は、慢性疼痛、神経因性疼痛及び/又は炎症性疼痛を含むいずれかの種類の疼痛の治療のための方法及び/又は医薬組成物において使用され得る。
【0049】
さらなる好ましい実施態様において、式Iの化合物は、炎症性障害の治療のための方法及び/又は医薬組成物において使用され得る。
具体的な好ましい実施態様において、いずれかの種類の疼痛の治療における又は炎症性障害の治療における使用のための式Iの化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0050】
(疼痛)
神経病変を罹患しているマウスへの式I記載のCDK阻害剤の投与が、特に炎症性及び神経因性疼痛のマウスモデルにおいて痛覚鈍麻効果を発揮することがわかっている。
サイクリン依存性キナーゼの阻害が、痛覚鈍麻効果を仲介することに関与するという発見は、予期されていなかった。
【0051】
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の阻害剤を投与することを含む、いずれかの種類の疼痛を治療する方法に関する。特に、式Iの化合物は、慢性、神経因性及び/又は炎症性疼痛の治療に使用され得る。具体的な好ましい実施態様において、いずれかの種類の疼痛の治療における使用のための式Iの化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0052】
疼痛の発達におけるCDK9の役割は、下記の作用機構に基づき得る:サイクリンT1及びCDK9の両者は、TNFαのプロモーターの基礎活性を刺激する。TNFαは、炎症誘発性サイトカインおよび炎症性の遺伝子ネットワークの発現を調節する疼痛仲介因子である。細胞性TNF受容体反応の仲介のために、核因子κB(NFκB)経路が非常に重要である。TNFαは、サイトカイン遺伝子へのNFκBの動員を惹起するのに対し、NfκBは、遺伝子転写の刺激のためにp-TEFb複合体と相互作用する(Barboric Mら, 2001)。
【0053】
さらに、CDK9が、TNFα受容体複合体の一員であるTRAF2の結合パートナーであることが示されている(MacLachlanら, 1998)のに対し、炎症誘発性IL6受容体複合体のサブユニットであるGP130が、CDK9の別のあり得る結合パートナーとして近年同定されている(Falcoら, 2002)。TNFα及びインターロイキンのシグナル伝達における、及びいくつかの遺伝子(例えば、疼痛仲介因子としてのサイトカイン)のNfκB仲介性発現における鍵となる担い手として、このように、CDK9は、炎症性疼痛など、いずれかの種類の疼痛の治療のための中心的な標的として考慮できる(図2参照)。
【0054】
神経因性疼痛の治療のために、薬理学的作用は、中枢神経系(CNS)における血液脳関門(BBB)を越えて生じなければならない。例えば、CNSにおける主要な免疫細胞としての小膠細胞は、活性化の際に、サイトカイン(TNFα、IL1β、IL6)及び他の炎症誘発性分子等の多様な有害な因子を放出する(Huwe 2003)。ミクログリアは、TNFα受容体又はトール様受容体の刺激によって活性化され、シグナルは、Iκキナーゼ(IKK)及び、上述のサイトカインの転写活性化に至るNfκBを介して仲介される。ミクログリアの関与は、慢性CNS疾患において助けになるものとして論議されてきており、疼痛知覚に関与し得る(Watkinsら, 2003)。
【0055】
近年、転写因子NfκBが、脊髄においてインターロイキン1β(IL1β)を介してシクロオキシゲナーゼ2(COX-2)の発現を制御することが示された(Leeら 2004)。脊髄プロスタグランジンE2の上昇に対する主要な関与因子として、疼痛仲介因子COX-2はすでに、多様な抗侵害受容薬/抗炎症薬のための標的として公知である。NfκB阻害剤は、動物モデルにおけるCOX-2レベル及び機械的アロディニア及び熱痛覚過敏を有意に減少させるNfκB阻害剤の能力を立証している。
【0056】
COX-2とは対照的に、CDK9作用の阻害は、単一の疼痛仲介因子のみの抑制の代わりに、多様な疼痛仲介因子の抑制に至るであろう。このように、CDK9阻害剤の抗侵害受容作用は、例えばCOX-2阻害剤と比較して優れ得る。
それゆえ、NfκB仲介性遺伝子転写とのCDK9阻害剤の関連性により、CDK9との阻害相互作用は、急性炎症性疼痛の治療のためのみならず、慢性疼痛の治療のための妥当なアプローチであり得る。
【0057】
本明細書で使用される「疼痛」という用語は一般的に、いずれかの種類の疼痛に関し、急性疼痛、慢性疼痛、炎症性及び神経障害性疼痛等の様々な種類の疼痛を広範に包含する。本発明の好ましい実施態様において、「疼痛」は、神経因性疼痛及び関連容態を含む。疼痛は、慢性、アロディニア(通常無害の刺激に由来する疼痛の知覚)、痛覚過敏(付与されたいずれかの疼痛刺激に対する過度の反応)及び受容野(すなわち、刺激が適用される場合に「痛い」領域)の拡大、幻肢痛又は炎症性疼痛であり得る。
【0058】
急性疼痛の種類は、組織障害と関連した疼痛、術後痛、外傷後の疼痛、熱傷によって生じる疼痛、局所感染又は全身感染によって生じる疼痛、下記を含む疾患と関連した内臓痛を含むが、これらに限定されるわけではない:膵炎、腸管膀胱炎、月経困難、過敏性腸症候群、クローン病、尿管仙痛及び心筋梗塞。
【0059】
さらに、「疼痛」という用語は、下記を含むCNS障害と関連した疼痛を含む:多発性硬化症、脊髄損傷、外傷性脳損傷、パーキンソン病及び脳卒中。
好ましい実施態様において、「疼痛」は、頭痛(例えば、片頭痛障害、偶発性及び慢性緊張型頭痛、緊張型様頭痛、群発頭痛、及び慢性発作性片側頭痛)、腰痛、癌性疼痛、骨関節炎疼痛及び神経因性疼痛を含む慢性疼痛の種類に関するが、それらに限定されるわけではない。
【0060】
本明細書で定義される炎症性疼痛(組織損傷及び結果として生じる炎症過程に応答した疼痛)は、結合組織疾患、関節リウマチ、全身性エリテマトーデス、多発性硬化症及び関節炎を含む疾患と関連した炎症性疼痛に関するが、それらに限定されるわけではない。
【0061】
神経因性疼痛(末梢神経又は中枢神経系自体に対する障害から結果として生じる疼痛)には、代謝性ニューロパチー(例えば、糖尿病性ニューロパチー)、帯状疱疹後神経痛、三叉神経痛、頭部神経痛、脳卒中後神経因性疼痛、多発性硬化症関連神経因性疼痛、HIV/エイズ関連神経因性疼痛、癌関連神経因性疼痛、手根管関連神経因性疼痛、脊髄損傷関連神経因性疼痛、複合性局所疼痛症候群、線維筋痛症関連神経因性疼痛、反射性交感神経性ジストロフィー、幻肢症候群又は末梢神経若しくは脊髄の外傷、外科的治療、体肢切断及び断端痛を含む神経離断、抗癌治療法及び抗エイズ治療法の副作用によって生じる疼痛、術後神経因性疼痛、特発性又は外傷後ニューロパチー及び単神経炎などにおけるニューロパチー関連疼痛、及び関節リウマチ、ワーレンベルグ症候群、全身性エリテマトーデス、多発性硬化症、又は結節性多発動脈炎等の結合組織疾患によって生じる神経因性疼痛を含む容態が含まれるが、それらに限定されるわけではない。ニューロパチーは、神経根障害、単ニューロパチー、多発性単神経炎、多発ニューロパチー又は神経叢障害として分類できる。
【0062】
「アロディニア」という用語は、通常は痛くない刺激から生じる疼痛を表す。アロディニア性疼痛は、刺激された領域以外で生じ得る。
「痛覚過敏」という用語は、痛い刺激に対する高い感度を表す。
「痛覚鈍麻」という用語は、痛い刺激に対する低い感度を表す。
【0063】
(炎症性疾患)
驚くべきことに、本明細書に開示される式I記載のCDK阻害化合物が、インビトロ及びインビボでの炎症アッセイにおいて抗炎症効果を発揮することを示すことができた。
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の阻害剤を投与することを含む炎症性疾患を治療する方法に関する。具体的な好ましい実施態様において、炎症性疾患の治療における使用のための式Iの化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0064】
炎症性疾患の発達におけるCDK9の役割は、下記の作用機構に基づき得る:関節リウマチ(RA)等の炎症性疾患;粥状硬化;喘息;炎症性腸疾患、全身性エリテマトーデス及びいくつかの他の自己免疫疾患は、該疾患における炎症性経路及び組織閉塞性経路の鍵となる制御因子である腫瘍壊死因子α(TNFα)によって仲介される。IκBタンパク質のリン酸化の際にNfκBから解離するIκBタンパク質をリン酸化するIκBキナーゼ(IKK)等のいくつかの伝達物質を介してTNFαシグナルが仲介されることは公知である。サイトカイン転写の正の制御因子である解離されたNfκBは、細胞核内に転位置し、そこで該NfκBはその認識部位に結合する。
【0065】
活性化されたNfκBは、RA患者の滑膜において発見されている[Hanらの文献(2003, Autoimmunity, 28, 197-208)]。該NfκBは、TNFα、IL-6、IL-8、NOS及びCOX2等の炎症誘発性遺伝子を制御する。標的指向化NfκB及びその上流のシグナル伝達パートナーであるIKKは、関節炎に関する多くの動物モデルにおける有効な治療戦略であることがすでに立証されている[Firesteinの文献(2003, Nature 423, 356-361)]。
【0066】
結合したNfκBは、RNA Pol IIのCTDのリン酸化を触媒するCDK9を動員し及び活性化するヒストンアセチルトランスフェラーゼ(CBP、p300、p/CAF、SRC-1、及びSRC-1関連タンパク質)を含有する活性化補助因子複合体と会合する[Westらの文献(2001, Journal of Virology 75(18), 85248537)]。結果として生じるRNA Pol II CTDの過剰リン酸化は、TNFαによって制御されるものとしても公知であるIL-1β、IL-6及びIL-8等の炎症誘発性サイトカインの転写の活性化に至る。
【0067】
いくつかの研究は、TNFαが、炎症誘発性サイトカイン発現を制御する自己シグナル伝達カスケードの「主要制御因子」であることを示した。この炎症誘発性カスケードを中断するために、特異的な抗体(Ab)をうまく使用して、TNFαシグナルを遮断できる。AbによるRAの抗TNFα治療は、いくつかの臨床研究におけるその治療的効能をすでに立証しており、インフリキシマブ及びエタネルセプト等の米国食品医薬品局認可薬は、市場に参入している[Feldmann及びMainiの文献(NatMed, 2003, 9 (10); 356-61)]。しかしながら、Abベースの治療法の不利には、該治療法の免疫原性能力、連続的な治療中の付随する効能の損失及び高い治療費が含まれる。さらに、Ab動態によって、循環しているTNFαの多かれ少なかれ全か無の減少が可能となる。結果としてまた、免疫応答の生理学的機能は抑制される[Lauferらの文献(免疫及びリウマチ疾患(Inflammation and Rheumatic Diseases), 2003; Thieme, pp. 104-5)]。
【0068】
P38 MAPK又はIKK等の標的を照準としたキナーゼ阻害剤を使用するTNFα仲介性シグナル伝達カスケードへの治療的介入は、重度の有害な効果を示しており、たいていの場合、個々の標的に対する特異性の欠失に起因する。
それとは対照的に、本明細書に提示される式I記載のCDK特異的阻害剤は、生理学的機能との相互作用を減少させるTNFαシグナル伝達経路の最下端で介入し得る。さらに、該化合物は、優れた特異性を介する有害な効果の回避によって、自己TNFα仲介性炎症ネットワークを中断できるであろう。それゆえ、式IのCDK特異的阻害剤を使用する治療は、炎症性疾患及び自己免疫疾患の治療のための有望な戦略を構成する。
このように、本明細書で提示される式I記載の化合物は、炎症性疾患の治療及び/又は予防のために使用され得る。
【0069】
本明細書で使用される「炎症性疾患」という用語は、身体組織の炎症を生じ、その後急性又は慢性の炎症容態を生じる免疫系又は組織の細胞性又は非細胞性仲介因子によって惹起される疾患に関する。
【0070】
これらの炎症性疾患の例は、I〜IV型の過敏性反応であり、例えば、喘息を含む肺の過敏性疾患、アトピー性疾患、アレルギー性鼻炎又は結膜炎、眼瞼の血管性浮腫、遺伝性血管性浮腫、抗受容体過敏性反応及び自己免疫疾患、橋本甲状腺炎、全身性エリテマトーデス、グッドパスチャー症候群、天疱瘡、重症筋無力症、グレーヴス及びレイノー病、 B型インスリン抵抗性糖尿病、関節リウマチ、乾癬、クローン病、皮膚硬化症、混合型結合組織疾患、多発性筋炎、サルコイドーシス、ウエゲナー肉芽腫、糸球体腎炎、急性又は慢性の宿主体移植片反応であるが、それらに限定されるわけではない。
【0071】
さらに、「炎症性疾患」という用語には、腹腔炎症、皮膚炎、消化管炎症(炎症性腸疾患、潰瘍性大腸炎を含む)、線維症、眼及び眼窩の炎症、眼乾燥症疾患及びシェーグレン症候群から結果的に生じる重度の眼乾燥症疾患、乳腺炎、耳炎、口腔炎症、筋骨格系の炎症(痛風、骨関節炎を含む)、中枢神経系の炎症性疾患(多発性硬化症、細菌性髄膜炎、髄膜炎を含む)、腎尿路生殖器の炎症(前立腺炎、糸球体腎炎を含む)、心血管の炎症(粥状硬化、心不全を含む)、気道の炎症(慢性気管支炎、慢性閉塞性肺疾患を含む)、甲状腺炎、糖尿病、骨炎、筋炎、多臓器不全(敗血症を含む)、多発性筋炎及び乾癬性関節炎が含まれるが、それらに限定されるわけではない。
【0072】
(免疫学的疾患)
また、式I記載の化合物は、例えば自己免疫疾患等の免疫学的疾患の治療及び/又は予防において有用であると考えられる。
従って、本発明は、免疫学的疾患の治療及び/又は予防のための方法であって、必要とする対象に、式I記載の有効量の少なくとも1つのCDK阻害剤の投与を含む、前記方法を提供する。
【0073】
本明細書で使用される「免疫学的疾患」という用語は、アレルギー、喘息、移植片対宿主病、免疫不全症及び自己免疫疾患を含むがそれらに限定されない疾患に関する。
特に、免疫学的疾患には、糖尿病、リウマチ性疾患、エイズ、慢性肉芽腫性疾患、移植された臓器及び組織の拒絶、鼻炎、慢性閉塞性肺疾患、骨粗鬆症、潰瘍性大腸炎、クローン病、副鼻腔炎、エリテマトーデス、乾癬、多発性硬化症、重症筋無力症、脱毛症、反復性感染症、アトピー性皮膚炎、湿疹及び重度のアナフィラキシー反応が含まれるが、それらに限定されるわけではない。さらに、「免疫学的疾患」にはまた、接触アレルギー、食物アレルギー又は薬剤アレルギー等のアレルギーが含まれる。
【0074】
(増殖性疾患)
式Iの化合物は、細胞周期の制御に関与する鍵分子を表すサイクリン依存性キナーゼの阻害剤である。細胞周期の制御障害は、腫瘍細胞の主要な特徴の1つである。このように、該化合物は、異常に分裂する細胞における細胞周期の調節を抑止し又は回復させる上で有用であることを立証すると期待される。このように、式I記載の化合物が、癌等の増殖性疾患の治療及び/又は予防において有用であることが期待される。
【0075】
従って、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む増殖性疾患の治療及び/又は予防のための方法を提供する。
本明細書で使用されるように、「増殖性疾患」という用語は、良性腫瘍、形成異常、過形成及び転移性増殖又は他の形質転換を示す腫瘍を含むがそれらに限定されるわけではない癌障害に関する。
【0076】
「癌」という用語には、癌腫、肉腫、癌肉腫、造血性組織の癌、脳を含む神経組織の腫瘍及び皮膚細胞の癌のような良性及び悪性腫瘍が含まれるが、それらに限定されるわけではない。
治療され得る癌の例には、癌腫、例えば、膀胱癌、乳癌、結腸癌(例えば、結腸腺癌及び結腸腺腫等の大腸癌)、腎癌、表皮癌、肝癌、肺癌、例えば腺癌、小細胞肺癌及び非小細胞肺癌、食道癌、胆嚢癌、卵巣癌、膵癌、例えば、膵外分泌腺癌、胃癌、子宮頚癌、甲状腺癌、前立腺癌、又は皮膚癌、例えば、扁平上皮細胞癌;リンパ系系統の造血器腫瘍、例えば、白血病、急性リンパ球性白血病、B細胞性リンパ腫、T細胞性リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、毛髪様細胞リンパ腫、又はバーケットリンパ腫;骨髄系系統の造血器腫瘍、例えば、急性及び慢性骨髄性白血病、骨髄異形成症候群、又は前骨髄球性白血病;甲状腺濾胞癌;間葉系起源の腫瘍、例えば、線維肉腫又は横紋筋肉腫;中枢神経系又は末梢神経系の腫瘍、例えば、星状細胞腫、神経芽腫、神経膠腫又は神経鞘腫;黒色腫;精上皮腫;悪性奇形腫;骨肉腫;色素性乾皮症;ケラトクタントーマ(keratoctanthoma);甲状腺濾胞癌;カポジ肉腫、星状細胞腫、基底細胞癌、小腸癌、小腸腫瘍、消化管腫瘍、膠芽腫、脂肪肉腫、胚細胞腫瘍、頭部及び頚部腫瘍(耳、鼻及び咽頭領域の腫瘍)、口腔、咽頭、喉頭、及び食道の癌、悪性又は良性骨腫瘍のような骨並びにその支持組織及び結合組織の癌、例えば、悪性骨肉腫、良性骨腫、軟骨腫瘍;悪性軟骨肉腫又は良性の軟骨腫、骨肉腫のようなもの;膀胱並びに男性及び女性の腎尿路生殖器系の内部及び外部の臓器及び構造の腫瘍、軟部組織腫瘍、軟部組織肉腫、ウィルムス腫瘍又は、例えば甲状腺、副甲状腺、下垂体、副腎、唾液腺のような内分泌腺及び外分泌腺の癌が含まれるが、それらに限定されるわけではない。
【0077】
(感染性疾患)
さらに、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む感染性疾患を治療し及び/又は予防する方法に関する。
ある宿主細胞CDKが、ウイルス複製に関与する、すなわちCDK2、CDK7、CDK8及びCDK9であることは公知である(J. Virol. 2001; 75: 7266-7279)。特に、HIV-1転写伸長及びヒストンのメチル化の制御におけるCDK9キナーゼ活性の役割が記載されている(J. Virol 2004, 78(24):13522-13533)。
【0078】
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む感染性疾患を治療し及び/又は予防する方法に関し、この中で、該化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0079】
本明細書で使用される「感染性疾患」という用語は、ウイルス、細菌、真菌及び/又は寄生虫等の病原体によって生じる感染を含む。
ウイルスによって誘発される感染性疾患には、レトロウイルス、ヒト内在性レトロウイルス、ヘパドナウイルス、ヘルペスウイルス、フラビウイルス、アデノウイルス、トガウイルス及びポックスウイルスによる感染によって生じる疾患が含まれる。特に、感染性疾患は、HIV-1、HIV-2、HTLV-1及びHTLV-II等のウイルス、HBV等のヘパドナウイルス、単純ヘルペスウイルスI型(HSV I)、単純ヘルペスウイルス11型(HSV II)、エプスタイン・バーウイルス(EBV)、水痘帯状疱疹ウイルス(VZV)、ヒトサイトメガロウイルス(HCMV)又はヒトヘルペスウイルス8型(HHV-8)等のヘルペスウイルス、HCV、ウェストナイル熱ウイルス又は黄熱ウイルス等のフラビウイルス、ヒトパピローマウイルウス、ポックスウイルス、シンドビスウイルス又はアデノウイルスを含むがそれらに限定されないウイルスによって生じる。
【0080】
感染性疾患の例には、エイズ、ボレリア症、ボツリヌス中毒、下痢、BSE(ウシ海綿状脳症)、チクングニヤ、コレラ、CJD(クロイツフェルト・ヤコブ病)、結膜炎、サイトメガロウイルス感染症、デング熱(dengue)/デング熱(dengue Fever)、脳炎、東部ウマ脳炎、西部ウマ脳炎、エプスタイン・バーウイルス感染症、大腸菌感染症、食物感染、口蹄疫、真菌性皮膚炎、胃腸炎、ヘリコバクター・ピロリ感染症、肝炎(HCV、HBV)、帯状疱疹(Herpes Zoster)(帯状疱疹(Shingles))、HIV感染症、インフルエンザ、マラリア、麻疹、髄膜炎、髄膜脳炎、伝染性軟属腫、蚊媒介疾患、パルボウイルス感染症、ペスト、PCP(ニューモシスチスカリニ肺炎)、ポリオ、原発性胃腸炎、Q熱、恐水病、呼吸シンチウムウイルス(RSV)感染症、リウマチ熱、鼻炎、リフトバレー熱、ロタウイルス感染症、サルモネラ症、サルモネラエンテリティデス、疥癬、細菌性赤痢、天然痘、連鎖球菌感染症、破傷風、毒素性ショック症候群、結核、潰瘍(消化性潰瘍疾患)、出血熱、痘瘡、疣贅、ウェストナイル熱ウイルス感染症(ウェストナイル脳炎)、百日咳、黄熱が含まれるが、それらに限定されるわけではない。
【0081】
(心血管疾患)
さらに、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む心血管疾患の治療及び/又は予防に関する。
心血管疾患の分野が、CDK阻害剤について起こり得る臨床的適用を構成することが報告されている(Pharmacol Ther 1999, 82(2-3):279-284)。さらに、サイクリンT/CDK9複合体の阻害、より具体的には、CDK9の阻害が、心不全等の心血管疾患の治療において有益な役割を担い得ることは公知である(WO2005/027902)。
【0082】
このように、好ましい実施態様において、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む心血管疾患を治療し及び/又は予防する方法に関し、この中で、該化合物は、他のCDKに対するよりもCDK9に対する高い選択性を示す。
【0083】
「心血管疾患」という用語には、うっ血性心不全、心筋梗塞、安定狭心症、不安定狭心症及び無症候性虚血等の心臓の虚血性疾患、全ての種類の心房性及び心室性の不整脈、高血圧性血管疾患、末梢血管疾患、冠動脈性心疾患及び粥状硬化などの心臓及び血管系の障害が含まれるが、それらに限定されるわけではない。さらに、本明細書で使用されるように、該用語には、成人性先天性心疾患、動脈瘤、狭心症、血管神経性浮腫、大動脈弁狭窄、大動脈瘤、大動脈弁閉鎖不全症、不整脈原性右室異形成、動静脈奇形、心房細動、ベーチェット症候群、徐脈、心肥大、うっ血性、肥大型及び収縮性心筋症等の心筋症、頚動脈狭窄症、脳出血、チャーグ・ストラウス症候群、コレステロール塞栓症、細菌性心内膜炎、線維筋異形成症、うっ血性心不全、機能不全弁又は狭窄弁等の心臓弁疾患、心臓発作、硬膜外又は硬膜下血腫、フォンヒッペル・リンダウ病、充血、高血圧症、肺高血圧症、肥大性増殖、左室肥大、右室肥大、左心形成不全症候群、低血圧症、間欠跛行、虚血性心疾患、クリッペル・トレノネー・ウェーバー症候群、延髄外側症候群、僧帽弁逸脱症、QT延長症候群、僧帽弁逸脱症、心筋虚血、心筋炎、心膜の障害、心膜炎、末梢血管疾患、静脈炎、結節性多発動脈炎、肺動脈閉鎖症、レイノー病、再狭窄、リウマチ性心疾患、スネッドン症候群、狭窄、上大静脈症候群、症候群X、頻脈、遺伝性出血性末梢血管拡張症、毛細血管拡張症、側頭動脈炎、閉塞性血栓血管炎、血栓症、血栓塞栓症、静脈瘤、脈管疾患、脈管炎、血管攣縮、心室細動、ウィリアムス症候群、末梢血管疾患、静脈瘤及び下腿潰瘍、深部静脈血栓症及びウォルフ・パーキンソン・ホワイト症候群が含まれるが、それらに限定されるわけではない。
さらに、心血管疾患という用語には、先天性異常、遺伝的欠陥、環境の影響(すなわち、食事の影響、生活様式、ストレスなど)、及び他の欠陥又は影響から結果的に生じる疾患が含まれる。
【0084】
(神経変性疾患)
CDK阻害剤は、神経保護効果を発揮することが記載されている。特に、CDK阻害剤が、アルツハイマー病等の神経変性疾患における神経細胞の死滅を予防することが報告されている(Biochem Biophys Res Commun 2002 (297):1154-1158;Trends Pharmacol Sci 2002 (23):417-425; Pharmacol Ther 1999, 82(2-3):279-284)。
【0085】
このように、CDK阻害剤である式I記載の化合物は、神経変性疾患の治療管理において有益な効果を提供すると期待される。
従って、本発明は、式I記載のサイクリン依存性キナーゼの有効量の少なくとも1つの阻害剤を投与することを含む神経変性疾患を治療し及び/又は予防する方法に関する。
【0086】
本明細書で使用される「神経変性疾患」という用語には、脳損傷、脳血管障害及びその結果、パーキンソン病、皮質基底核変性症、運動ニューロン疾患、筋萎縮性側索硬化症、多発性硬化症、外傷性脳損傷、脳卒中、脳卒中後脳損傷、外傷後脳損傷、及び小血管性脳血管障害を含む認知症、アルツハイマー病等の認知症、脳血管性認知症、レヴィ小体型認知症、前頭側頭葉型認知症及び17番染色体と関連したパーキンソン症候、ピック病を含む前頭側頭葉型認知症、進行性核性麻痺、皮質基底核変性症、ハンチントン病、視床変性、クロイツフェルト・ヤコブ認知症、HIV認知症、認知症を伴う統合失調症、コルサコフ精神病及びエイズ関連認知症を含むがそれらに限定されるわけではない中枢神経系の障害並びに末梢神経系の障害が含まれる。
【0087】
同様にまた、軽度認知欠陥、加齢随伴性記憶欠陥、加齢関連性認知低下、血管性認知欠陥、注意欠陥障害、注意欠陥多動障害、及び学習障害を有する子供における記憶障害等の認知関連障害は、神経変性障害であると考慮される。
【0088】
特に、本発明は、上述の種類の疼痛及び随伴する容態並びに炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための方法に関し、この中で、「治療すること」という用語は、疼痛及び随伴する容態並びに炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患の予防、寛解又は治療することを含む。
【0089】
(医薬組成物)
本発明の好ましい実施態様には、医薬として許容し得る(すなわち非毒性の)少なくとも1つの担体、賦形剤及び/又は希釈剤とともに、活性成分としての式I記載の少なくとも1つのサイクリン依存性キナーゼ阻害剤を含む組成物の投与が含まれる。
好ましくは、組成物は、活性成分として式I記載の少なくとも1つのサイクリン依存性キナーゼ阻害剤を含み、この中で、該少なくとも1つのサイクリン依存性キナーゼ阻害剤は、他のCDKに対するよりもCDK9に対する高い選択性を有する。
【0090】
さらにまた、本発明は、CDKの少なくとも2つの阻害剤及び/又は医薬として許容し得るそれらの塩を組み合わせる組成物を含む。該少なくとも2つの阻害剤は、同一のサイクリン依存性キナーゼを阻害し得、又は異なる種類のサイクリン依存性キナーゼも阻害し得、例えば、組成物におけるある阻害剤は、CDK9を阻害し得るのに対し、他の阻害剤は、例えばCDK2を阻害できる。
【0091】
疼痛治療に関して、個々の疼痛投薬は、多くの中からたった1つの疼痛伝達経路に干渉するので、部分的に有効な疼痛軽減のみをしばしば提供する。このようにまた、疼痛知覚過程における異なる地点で作用する疼痛減少(鎮痛)薬との組み合わせで、式I記載のCDK阻害剤を投与することが企図される。
【0092】
「鎮痛薬」は、疼痛知覚における減少を生じる分子又は分子の組み合わせを含む。鎮痛薬は、CDKの阻害以外の作用の機構を採用する。
非ステロイド性抗炎症薬(NSAID)等のあるクラスの鎮痛薬は、侵害受容器によって検知される刺激の化学的メッセンジャーを下方制御し、オピオイド等の別のクラスの薬物は、CNSにおける侵害受容情報の処理を変化させる。他の鎮痛薬は、局所麻酔薬、抗痙攣薬及び三環系抗うつ薬等の抗うつ薬である。CDK阻害剤に加えて1つ以上のクラスの薬物を投与することは、より有効な疼痛の寛解を提供できる。
【0093】
本発明の方法及び組成物における使用のための好ましいNSAIDは、アスピリン、アセトアミノフェン、イブプロフェン、及びインドメタシンである。さらにまた、特異的シクロオキシゲナーゼ2(COX-2)阻害剤等のCOX-2阻害剤(例えば、セレコキシブ、COX189、及びロフェコキシブ)は、本発明の方法又は組成物における鎮痛薬として使用され得る。
【0094】
好ましい三環系抗うつ薬は、クロミプラミン、アモキサピン、ノルトリプチリン、アミトリプチリン、イミプラミン、デシプラミン、ドキセピン、トリミプラミン、プロトリプチリン、及びパモ酸イミプラミンからなる群から選択される。
さらにまた、鎮痛薬としての抗痙攣薬(例えば、ガバペンチン)、GABABアゴニスト(例えば、L-バクロフェン)、オピオイド、バニロイド受容体アンタゴニスト及びカンナビノイド(CB)受容体アゴニスト、例えばCB1受容体アゴニストの使用は、本発明における方法及び組成物において好ましい。
【0095】
本発明のサイクリン依存性キナーゼ阻害剤組成物を調製する上で、Remingtonの文献(薬剤学の科学及び実践第19版(The Science and Practice of Pharmacy, 19th ed.)(Mack Publishing, 1995))等の周知の製薬出典の標準的な推奨を手本とすることができる。
本発明の医薬組成物は、従来の固体又は液体の担体又は希釈剤及び医薬として調製される従来のアジュバントにおいて、公知の方法で適切な薬用量レベルで調製できる。好ましい調製物は、経口適用に適している。これらの投与形態には、例えば、丸剤、錠剤、フィルム錠、コート錠、カプセル、散剤及び沈着物が含まれる。
【0096】
さらにまた、本発明には、真皮、真皮内、胃内、皮内、静脈内、脈管内、静脈内、筋肉内、腹腔内、鼻内、膣内、頬内、経皮、直腸内、皮下、舌下、局所、又は経真皮適用を含む非経口適用のための医薬調製物が含まれ、この中で、該調製物は、典型的な媒体及び/又は希釈剤に加えて、本発明記載の少なくとも1つの阻害剤及び/又は医薬として許容し得るその塩を活性成分として含有する。
【0097】
本発明記載の少なくとも1つの阻害剤及び/又は医薬として許容し得るその塩を活性成分として含有する本発明記載の医薬組成物は典型的に、企図された投与の形態に関して、すなわち、錠剤、カプセル(固体充填、半固体充填又は液体充填のいずれか)、構成用散剤、ゲル、エリキシル剤、分散可能な顆粒、シロップ、懸濁液、及びそれらの類似物の形態での経口投与のために、従来の薬事的実践と合致して選択された適切な担体材料とともに投与されるであろう。例えば、錠剤又はカプセルの形態での経口投与のために、活性薬物構成要素は、医薬として許容し得るいずれかの経口用非毒性担体と、好ましくは乳糖、デンプン、ショ糖、セルロース、ステアリン酸マグネシウム、リン酸二カルシウム、硫酸カルシウム、滑石、マンニトール、エチルアルコール(液体充填カプセル)及びそれらの類似物のような不活性担体と組み合わせることができる。
【0098】
さらにまた、適切な結合剤、潤滑剤、崩壊剤及び着色料は、錠剤又はカプセルへ組み込まれ得る。散剤及び錠剤は、約5〜約95重量%の本明細書に列挙される式I記載のサイクリン依存性キナーゼ阻害剤又はそのアナログ又は医薬活性のある個々の塩を活性成分として含有し得る。
【0099】
適切な結合剤には、デンプン、ゼラチン、天然糖、トウモロコシ甘味料、アカシア等の天然及び合成ゴム、アルギン酸ナトリウム、カルボキシメチルセルロース、ポリエチレングリコール及び蝋が含まれる。適切な潤滑剤のうち、ホウ酸、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウム、及びそれらの類似物が挙げられ得る。
【0100】
適切な崩壊剤には、デンプン、メチルセルロース、グアーガム、及びそれらの類似物が含まれる。
また、甘味料及び調味料及び保存料は、適宜含まれ得る。崩壊剤、希釈剤、潤滑剤、結合剤等は、以下により詳細に論議される。
【0101】
さらに、本発明の医薬組成物は、いずれかの1つ以上の構成要素又は活性成分の速度調節放出を提供して治療効果、例えば抗ヒスタミン活性及びその類似項目を最適化するための徐放形態で製剤され得る。持続放出に適した剤形には、崩壊速度の変動する層を有する錠剤、又は活性成分の含浸され、かつ錠剤形態に成形された徐放性ポリマーマトリックス、又はこのような含浸され又は被包された多孔性ポリマーマトリックスを含有するカプセルが含まれる。
【0102】
液状製剤には、溶液、懸濁液、及び乳濁液が含まれる。一例として、非経口注射のための水又は水/プロピレングリコール溶液、又は経口用の溶液、懸濁液、及び乳濁液のための甘味料及び乳白剤の添加が挙げられ得る。また、液状調製物には、鼻内投与のための溶液が含まれ得る。
吸入に適したエアロゾル調製物には、不活性圧縮ガス、例えば窒素等の医薬として許容し得る担体との組み合わせで存在し得る溶液及び散剤形態の固体が含まれ得る。
【0103】
坐剤を調製するために、ココアバターのような脂肪酸グリセリドの混合物等の低融点蝋がまず融解され、次に、活性成分が、例えば撹拌によってその中に均質に分散する。次に、融解された均質な混合物が、便利な大きさの鋳型へ注入され、冷却され、それにより凝固する。
また、使用直前に経口又は非経口のいずれかの投与のための液状調製物へ変換されるよう企図された固体状調製物が含まれる。このような液状には、溶液、懸濁液、及び乳濁液が含まれる。
【0104】
また、本発明記載の化合物は、経真皮的に送達され得る。経真皮組成物は、クリーム、ローション、エアロゾル及び/又は乳濁液の形態を有し得、この目的のために当技術分野で公知のようなマトリックス又は貯蔵器の種類の経真皮パッチに含まれ得る。
【0105】
本明細書で列挙されるカプセルという用語は、活性成分を含む組成物を保持し又は含有するために、例えばメチルセルロース、ポリビニルアルコール、又は変性したゼラチン若しくはデンプンでできた特殊な容器又は被包体を指す。硬質シェルを有するカプセルは典型的には、骨又は豚肉の皮膚由来の比較的高いゲル強度のゼラチンでできており、又は該ゼラチンを配合する。カプセル自体は、少量の色素、不透明剤、可塑剤及び/又は保存料を含有し得る。錠剤のうち、活性成分を適切な希釈剤とともに含む圧縮され又は鋳造された固体剤形が理解される。錠剤は、湿式造粒、乾式造粒によって、又は当業者に周知の圧縮によって得られる混合物又は顆粒の圧縮によって調製され得る。
【0106】
経口用ゲルは、親水性の半固体マトリックスに分散し又は可溶化した活性成分を指す。
構成用散剤は、例えば水において又はジュースにおいて懸濁できる、活性成分と適切な希釈剤とを含有する散剤配合物を指す。
【0107】
適切な希釈剤は、組成物又は剤形の大部分を通常調合する物質である。適切な希釈剤には、乳糖、ショ糖、マンニトール、及びソルビトール等の糖、コムギ、トウモロコシ、コメ、及びジャガイモ由来のデンプン、及び微結晶性セルロース等のセルロースが含まれる。組成物における希釈剤の量は、組成物全体の約5〜約95重量%、好ましくは約25〜約75重量%、より好ましくは約30〜約60重量%の範囲とし得る。
【0108】
崩壊剤という用語は、薬剤の医薬活性成分の崩壊及び放出を支持するために組成物へ添加される材料を指す。適切な崩壊剤には、デンプン、カルボキシメチルナトリウムデンプン等の修飾された「冷水可溶性」デンプン、イナゴマメ、カラヤ、グアー、トラガカント及びアガー等の天然及び合成ゴム、メチルセルロース及びカルボキシメチルセルロースナトリウム等のセルロース誘導体、微結晶性セルロース、及びクロスカルメロースナトリウム等の架橋された微結晶性セルロース、アルギン酸及びアルギン酸ナトリウム等のアルギン酸塩、ベントナイト等の粘土、及び起沸性混合物が含まれる。組成物における崩壊剤の量は、組成物の約2〜約20重量%、より好ましくは約5〜約10重量%の範囲とし得る。
【0109】
結合剤は、散剤粒子と結合し又は「接着する」物質であり、顆粒を形成することによって散剤粒子を接着性にし、このように、製剤における「接着剤」として機能する。結合剤は、希釈剤又は増量剤においてすでに利用可能な接着力を付加する。適切な結合剤には、ショ糖等の糖、コムギ、トウモロコシ、コメ及びジャガイモ由来のデンプン、アカシア、ゼラチン及びトラガカント等の天然ゴム、アルギン酸、アルギン酸ナトリウム及びアルギン酸アンモニウムカルシウム等の海藻の誘導体、メチルセルロース、カルボキシメチルセルロースナトリウム及びヒドロキシプロピルメチルセルロース等のセルロース材料、ポリビニルピロリドン、及びケイ酸マグネシウムアルミニウム等の無機化合物が含まれる。組成物における結合剤の量は、組成物の約2〜約20重量%、好ましくは約3〜約10重量%、より好ましくは約3〜約6重量%の範囲とし得る。
【0110】
潤滑剤は、摩擦又は摩損を減少させることによって、鋳型又はダイから放出するよう圧縮された後、錠剤造粒等を可能にするよう剤形へ添加されるあるクラスの物質を指す。適切な潤滑剤には、ステアリン酸マグネシウム、ステアリン酸カルシウム、又はステアリン酸カリウム等の金属性ステアリン酸塩、ステアリン酸、高融点蝋、並びに塩化ナトリウム、安息香酸ナトリウム、酢酸ナトリウム、オレイン酸ナトリウム、ポリエチレングリコール及びD,L-ロイシン等の他の水溶性潤滑剤が含まれる。潤滑剤は、顆粒の表面に存在しなければならないので、通常、圧縮前のまさに最終工程で添加される。組成物における潤滑剤の量は、組成物の約0.2〜約5重量%、好ましくは約0.5〜約2重量%、より好ましくは約0.3〜約1.5重量%の範囲とし得る。
【0111】
滑走剤は、医薬組成物の構成要素の焼成を互いに防止し、流動が潤滑かつ均一であるよう顆粒の流動特徴を改善する材料である。適切な滑走剤には、二酸化ケイ素及び滑石が含まれる。
該組成物における滑走剤の量は、最終組成物の約0.1〜約5重量%、好ましくは約0.5〜約2重量%の範囲とし得る。
【0112】
着色料は、組成物又は剤形へ着色を提供する賦形剤である。このような賦形剤には、粘土又は酸化アルミニウム等の適切な吸着剤へ吸着された食品等級の色素が含まれ得る。着色料の量は、組成物の約0.1〜約5重量%、好ましくは約0.1〜約1重量%で変動し得る。
【0113】
本発明は、いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、心血管疾患又は神経変性疾患の治療のための、活性成分としてサイクリン依存性キナーゼ阻害剤を含有する組成物の投与を必要とする対象への該投与に関する。
【0114】
「該投与を必要とする対象」は、近い将来いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、心血管疾患又は神経変性疾患を経験すると期待され、又は該容態の経験が進行中である動物、好ましくは哺乳動物、最も好ましくはヒトを含む。例えば、このような動物又はヒトは、現に疼痛を生じている進行中の容態を有し得、疼痛を生じ続けるようであり、又は動物若しくはヒトは、痛い結果を通常有する手法若しくは事象に忍耐中であり又は忍耐中であろう。糖尿病性神経障害性痛覚過敏及びコラーゲン脈管疾患等の慢性の痛い容態は、第一の種類の例であり;特に炎症又は神経障害の一領域における歯科業務、及び毒素曝露(化学治療薬への曝露を含む)は、後者の種類の例である。
【0115】
所望の治療効果を達成するために、個々のサイクリン依存性キナーゼ阻害剤は、治療的有効量で投与されなければならない。
「治療的有効量」という用語は、示される生物学的又は医学的応答を誘発する活性化合物又は医薬の量を示すために使用される。この応答は、研究者、獣医、医師又は他の臨床家によって探求されている最中の組織、系、動物又はヒトにおいて生じ得、治療されている最中の疾患の症状の寛解を含む。本発明の文脈において、治療的有効量は、例えば、疼痛、特に炎症性又は神経因性疼痛を減少させる量を含む。特に、治療的有効量は、治療されるべき対象において痛覚鈍麻効果を発揮する量を示す。
【0116】
このような有効量は、例えば疼痛に対する対象の正常感度、対象の身長、体重、齢、及び健康状態、疼痛の起源、CDKの阻害剤を投与する様式、投与される具体的な阻害剤、及び他の因子に応じて、対象によって変動するであろう。結果として、具体的なセットの環境の下で具体的な対象のための有効量を経験的に決定することが賢明である。
本発明は、下記の制限的でない実施例によってさらに説明される。
【実施例】
【0117】
(実施例)
すべての試薬をACROS Organics、Aldrich、Lancaster、Maybridge及びBoron Molecularから購入した。
化合物についての液体クロマトグラフィー/質量分析を、APCIイオン化を備えたSurveyor MSQ(Thermo Finnigan, USA)で実施した。
【0118】
本発明の化合物は、当技術分野で公知のいずれかの方法によって調製できる。簡便な1つの合成経路を以下のスキーム1:
【化6】
に示す。
フェニルボロン酸(2、Y=B(OH)2)又はその誘導体と2,4-ジハロゲン化ピリミジン(1、例えば、X1=X2=Cl)とのパラジウム触媒性交差共役は、4-アリール化2-ハロゲノピリミジン(3)を生じ、該4-アリール化2-ハロゲノピリミジンは、アニリン(4)によってアミノ化される。
【0119】
(実施例1:{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物1)の合成)
【化7】
a.塩化(ニトロフェニル)メタンスルホニル(4.17g、20mM)(Ziegler, Carl及びSprague, James M.の文献(J. of Org. Chem. (1951), 16, 621-5))をアセトニトリル(20mL)に溶解し、炭酸アンモニウムで飽和した濃アンモニア水溶液(20mL)を添加し、混合物を室温で1時間激しく撹拌した。次に、アセトニトリルを蒸発させ、残渣を冷水(20mL)で希釈して沈殿の形成に至らせ、該沈殿を濾過し、水(2×5mL)及びエーテルで洗浄し、減圧下で乾燥させた。(3-ニトロ-フェニル)メタンスルホンアミドの収量:3.5g(80%)。
【0120】
b.(3-ニトロフェニル)メタンスルホニルアミド(3.5g、16mM)をメタノールにおけるラネーニッケル(0.5g)上で50℃及び70psiで4時間水素化した。次に、触媒を濾過し、温メタノールで洗浄した。組み合わせた濾液を蒸発させて、2.83g(95%)の(3-アミノ-フェニル)-メタンスルホンアミドを得た。
【0121】
c.4,6-ジクロロ-ピリミジン(6g、0.04mol)(Ranganathan, Subramaniaらの文献(予稿集−インド科学学会(Proceedings - Indian Academy of Sciences), Chemical Sciences (1994), 106(5), 1051-70);Maggiali, C.らの文献(Farmaco, Edizione Scientifica (1988), 43(3), 277-91);Gershon, Hermanらの文献(J. of Med. Chem. (1963), 6, 87-9))及び(2-メトキシ-フェニル)-ボロン酸(4.37g、0.029mol)を、ジメトキシエタン(120mL)及び水(18mL)に溶解した。この溶液に、NaHCO3(6.72g、0.08mol)、PdCl2(PPh3)2(0.84g)を添加し、7時間還流させた(薄層クロマトグラフィー対照)。次に、混合物を室温に冷却し、溶媒を減圧下で除去した。得られた粗固体をジクロロメタン(100mL)に溶解し、水(1×100mL)で洗浄し、有機層を分離し、K2CO3上で乾燥させ、濾過し、溶媒を減圧下で除去した。得られた固体をフラッシュクロマトグラフィー(溶離剤ジクロロメタン)によってさらに精製すると粗生成物を生じ、それをヘキサンから結晶化させて、2-クロロ-4-(2-メトキシ-フェニル)-ピリミジン(4.7g、73%)を得た。
【0122】
D.DMF(12mL)における2-クロロ-4-(2-メトキシ-フェニル)-ピリミジン(0.882g、0.004mol)及び(3-アミノ-フェニル)-メタンスルホンアミド(0.865g、0.005mol)の溶液を80℃で2時間撹拌した(薄層クロマトグラフィー対照)。溶媒を減圧下で除去すると、{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミドを得た。
【0123】
(実施例2:2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物9)の合成)
【化8】
a.水素化フラスコに、2-(3-ニトロ-フェニルメタンスルホニル)-エタノール(0.8g、3.3mmol)(Harms, Wolfgangらの文献(Ger. Offen. (1995))、独国特許第4402189 A1号、第19950727号)を配置し、メタノール(100mL)に溶解した。ラネーニッケル(200mg)の添加後、混合物を70psiの水素圧で50℃で4時間振盪させた。次に、濾過によって触媒を除去し、メタノールで洗浄した。メタノール溶液を組み合わせ、溶媒を減圧下で蒸発させた。粗生成物をフラッシュクロマトグラフィー(溶離系:ジクロロメタン/メタノール−10/1)によって精製し、所望の画分を組み合わせ、乾燥させ、蒸発させた。残渣をジエチルエーテルで洗浄して、2-(3-アミノ-フェニルメタンスルホニル)-エタノール(0.51g、収率73%)を得た。
【0124】
b.DMF(3mL)における2-(3-アミノ-フェニルメタンスルホニル)-エタノール(0.215g、1mmol)及び2-クロロ-4-(2-メトキシ-フェニル)-ピリミジン(0.22g、1mmol)の溶液を80℃で2時間撹拌した(薄層クロマトグラフィー対照)。溶媒を減圧下で除去すると、油状残渣を生じ、それをイソプロパノールから結晶化して、2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(0.3g、75%)を得た。
同様に、相応じて置換される出発材料を使用して、下記の化合物を合成した。
【表1】
【0125】
(実施例3)
I.炎症性及び神経因性疼痛の分析のための行動動物モデル
炎症性及び神経因性疼痛の分析のためのいくつかの動物モデルは公知である。該モデルは、例えば神経病変の誘発(例えば、坐骨神経部分損傷、SNI)後又は侵害刺激に対する実験動物の曝露(例えば、ホルマリン又はカラゲナンの注射)後、該介入によって誘発されるような疼痛の徴候は、例えばフォン・フレイ毛髪を使用する機械的刺激に対する(又は、レーザー源若しくは舐める行動を使用する熱刺激に対する)足引っ込め閾値等の行動の定量可能な構成要素によって測定される。これらの反応は、ヒトにおける機械的及び熱アロディニア(機械的刺激に対する過敏症)又は痛覚過敏と等価であるものとして解釈される。
【0126】
坐骨神経部分損傷モデル(SNIモデル、Decosterd及びWoolf(2000)によって開発、図1参照)は、臨床的に関連する神経病変の誘発及び外科的介入後のその後の行動実験(例えば、フォン・フレイアッセイ)によって特徴付けられる。該モデルは、腓腹神経を無傷のままにしておき坐骨神経の2つの分岐(すなわち、脛骨神経及び総腓骨神経)の結紮及び切断からなる共通神経損傷モデルを構成する。SNIモデルは、臨床的な神経因性疼痛の特徴を密接に模倣する機械的及び冷却感度における早期の(24時間未満)、長期の及び実質的な変化を結果的に生じる。これらの種類の神経損傷を有する動物は、神経因性疼痛患者によって報告されるものと同様の異常な疼痛感覚及び機械的刺激に対する過敏症(アロディニア)を発生することが示されている。
【0127】
或いは、マウスにおけるホルマリンアッセイは、炎症性及び神経因性疼痛における侵害受容の妥当なかつ信頼性のある行動モデルである。該アッセイは、多様なクラスの鎮痛薬に対して感受性がある(Hunskaar S及びHole Kの文献(Pain. 1987 Jul;30(1):103-14))。侵害刺激は、左後足の背側面の皮膚の下(左後足内への皮下又は足底間)での10μLの希釈したホルマリン(塩類溶液における2%)の注射液からなる。反応は、注射された足を舐めること及び畏縮させることである。
【0128】
カラゲナンアッセイについて、マウスの単一の後足(同側足)への(塩類溶液における)1%カラゲナン25μLの皮下注射が適用される。その後の炎症は結果的に、足の長期に持続する発汗及び(機械的及び熱刺激に対する)過敏症を生じる。カラゲナンアッセイは、検査化合物の抗炎症性活性を予測するために使用される標準的な実験室アッセイである。足の浮腫測定及びハーグリーブスアッセイ(Hargreaves Assay)(光源を介する熱刺激による足の引っ込め)は、読み出しに使用される。
【0129】
本発明に関して、炎症性及び神経因性疼痛の発生に及ぼす式I記載のサイクリン依存性キナーゼ(CDK)阻害化合物の投与の効果は、SNIモデルにおいて、カラゲナンにおいて及びホルマリンアッセイにおいてアッセイされる。実験手法及び結果を、以下に詳細に記載する。
【0130】
(実施例4)
A.坐骨神経部分損傷(SNI)−慢性神経因性疼痛のモデル
先に概略されるように、坐骨神経部分損傷(SNI)モデル(図1参照)は、腓腹神経を無傷のままにしておく実験動物の坐骨神経の3つの末端分岐のうちの2つ(脛骨神経及び総腓骨神経)の病変を包含する。SNIは、損傷を受けていない腓腹神経の皮膚での支配領域における機械的及び熱アロディニアを結果的に生じる(Decosterd及びWoolfの文献(Pain 2000; 87:149-158)、(2) Tsujinoらの文献(Mol. Cel. Neurosci. 2000; 15:170-182))。
【0131】
1.野生型マウスにおける坐骨神経部分損傷(神経病変)の誘発
野生型マウス(C3HeB/FeJ系)(齢、性別及び体重は対形成)を、外科的準備の前に4μL/gで1:1:2の比でHypnorm(0.315mg/mLクエン酸フェンタニル+10mg/mLフルアニソン;Janssen)/Hypnovel(5mg/mLミダゾラム;Roche Applied Sciences)/水で麻酔した。
【0132】
その後、坐骨神経の3つの末端分岐:総腓骨神経、脛骨神経及び腓腹神経を露出して、膝のレベルのちょうど上ですべてのマウスの同側右後脚において、無菌予防措置の下で病変を作製した。総腓骨神経及び脛骨神経を7/0絹糸でしっかり結紮し、結紮に対して遠位で切断し、遠位の神経断端約2mmを除去した。腓腹分岐は、手法の間触らないままでだった(本明細書で「SNI同側」と記載)。重なっている筋肉及び皮膚を縫合し、動物を回復させ、創傷治癒させた。同一のマウスにおいて、反対側の左後脚の坐骨神経分岐を露出させたが、病変形成しなかった(本明細書で「SNI反対側」と記載)。坐骨神経部分損傷を経験したマウスを、以後「SNIマウス」と記載する。
【0133】
2.SNIマウスに対するCDK阻害化合物の投与
手術からの回復及び創傷治癒後、SNIマウスはCDK阻害化合物の経口注入を受容した。
2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μLに溶解したCDK阻害剤30mg/kgを、フォン・フレイ測定(機械的アロディニア)の30分前に経口適用を介して投与した。ネガティブコントロールとして、同量(400μL)の2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)媒体を、フォン・フレイ測定の30分前に単回経口適用によって投与した。
【0134】
阻害剤又は媒体の注入及びその後のフォン・フレイアッセイにおける機械的刺激に対する足引っ込め閾値の測定を、SNI後107日目に実施した。機械的刺激に対する反射侵害受容反応を各注入の30分後にフォン・フレイアッセイにおいて測定した。
機械的アロディニアの発生に及ぼすSNIマウスへのCDK阻害剤の投与の効果を、以下に記載のとおり、フォン・フレイアッセイにおいて分析した。
【0135】
3.CDK阻害化合物の投与後のSNIマウスの行動検査(フォン・フレイアッセイ)
SNI及びその後の本発明の化合物の投与を経験するマウスを、フォン・フレイアッセイにおける神経損傷後及び投与後の機械的アロディニアの徴候について検査した(Decosterd及びWoolfの文献(Pain 2000; 87:149-158))。本アッセイは、通常では痛くない刺激が不快な又は痛いものとして動物によって認識される機械的閾値を決定する。SNI同側及びSNI反対側の基線を個々に確立した。
【0136】
Chaplanら(1994)並びにMalmberg及びBasbaum(1998)に基づいたアップダウン法を使用して、SNIマウスの機械的閾値を定量した。
直径約9.5cm、高さ14cmで上部に向いた4つの通気孔及びプレキシガラス蓋のついたプレキシガラスシリンダーにマウスを配置した。シリンダーを高架のメッシュ表面(7×7平方mm)の上に配置した。検査前日、マウスを検査シリンダーに1〜2時間順応させた。検査当日、マウスをシリンダーに約1時間順化させ、この中で、順化時間は、マウスの系及びマウスが以前検査された回数等の因子に依存する。一般的に、検査は、マウスが一旦落ち着いて新たな環境を探検するのを停止したら開始し得る。
【0137】
マウスを検査するために、フィラメント2.44、2.83、3.22、3.61、3.84、4.08、及び4.31(力の範囲=0.04〜2.0g)を使用した。3.61mNフィラメントを最初に適用した。該フィラメントを片足の足底面に穏やかに適用し、曲げさせ、しかるべき位置で2〜4秒間保持した。刺激に対する正の反応(屈曲反応)が生じた場合はいつでも、次のより弱いフォン・フレイ毛髪を適用し;負の反応(無反応)が生じた場合はいつでも、次のより強い力を適用した。反応における最初の変化後にさらに4回の刺激に対する反応が得られるまで、検査を続行した。検査した最大力は、4.31であった。カットオフ閾値は、2gであった。
【0138】
一連のスコア(すなわち、「屈曲反応」及び「無反応」)及び適用された最後のフィラメントの力を使用して、Chaplanらの文献(Journal of Neuroscience Methods. 53(1):55-63, 1994 Jul.)に記載される機械的閾値を決定した。決定された閾値は、動物が回数の50%に反応すると期待されるであろうものである。フォン・フレイ測定を達成した後に、マウスを屠殺した。
【0139】
4.神経因性疼痛の発生に及ぼすCDK阻害化合物の投与の効果
上述のとおり、CDK阻害化合物をSNIマウスへ投与した。上述のとおり、術後107日目に動物の同側及び反対側の足でフォン・フレイ測定を実施した。薬理学的処置をせずに、SNIマウスは、SNI手術後に安定したアロディニアを示す。化合物で処置された動物は、機械的刺激に対する低い感度(低いアロディニア)を示す閾値の有意な増大を示す。低いアロディニアの観察は、CDK阻害化合物が、慢性神経因性疼痛のモデルにおける痛覚鈍麻薬として効果的であることを意味する。
【0140】
(実施例5)
ホルマリンアッセイ−炎症過程/炎症性及び慢性神経因性疼痛のモデル
マウスにおけるホルマリンアッセイは、侵害受容の妥当かつ信頼性のある行動モデルであり、多様なクラスの鎮痛薬に対して感受性がある(Hunskaar S及びHole Kの文献(Pain. 1987 Jul;30(1):103-14))。有害な刺激は、10μLの希釈したホルマリン(塩類溶液における2%)の左後足への皮下又は足底内注射である。反応は、注射された足を舐めること及び畏縮させることである。反応は、炎症過程の異なる部分を反映する2つの相(Abbottら1995)、すなわち早期/急性相である注射0〜5分後、及び後期/慢性相である注射5〜30分後を示す。下記のプロトコールは、実験を実施するための可能な1つの方法を記載する:
【0141】
1.ホルマリンの注射及びCDK阻害化合物の投与
本アッセイにおいて、齢、性別及び体重の対形成した野生型マウス(C3HeB/FeJ)を使用する。ホルマリン注射の前に、動物を各群10匹の実験群へ無作為にさらに分ける。ホルマリン注射の30分前に、2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μLに溶解した適量のCDK阻害剤は、腹腔内注射により投与できる。同様に、2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μLにおけるIkキナーゼ(IKK)阻害剤(30mg/kg)(ポジティブコントロール)、又は媒体単独(2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μL)(ネガティブコントロール)は、ホルマリン注射の30分前に腹腔内注射によって投与できる。
【0142】
ホルマリン注射に向けて、移動による注射の妨害を回避するために、マウスをペーパータオルで保持する。注射した後足を親指と人差し指との間に保持し、ハミルトンシリンジを使用してホルマリン(2%)10μLを2つの隆起間で後足足底へと皮下注射する。ホルマリン及び阻害剤で処置したマウスの行動を以下に記載のとおり分析する。
【0143】
2.ホルマリンの注射及びCDK阻害化合物の投与後のマウスの行動分析
ホルマリン処置したマウスの行動、すなわち舐めること及び畏縮を、規定された時間にわたって自動追跡システム(Ethovision 3.0 Color Pro, Noldus, Wageningen, Netherlands)によってモニターする:ホルマリン注射5分後に測定を開始し、ホルマリン注射30分後に終了する。この時間枠は、痛覚過敏であるホルマリン誘発性侵害受容(疼痛)の位相IIにわたる。
【0144】
注射した後足(黄色色素)(Lumogenyellow; BASF Pigment, Cologne, Germany)及び反対側の足(青色色素)(Lumogenviolet; Kremer Pigmente, Aichstetten, Germany)をそれぞれ局所的に標識するために、2つの異なる染色色素を使用する。舐める行動を決定するために、マウスをCCDカメラでモニターする。モニタリング及び録画後、EthoVisionソフトウェア(Ethovision 3.0 Color Pro, Noldus, Wageningen, Netherlands)を使用して、又は手動分析によってビデオを分析する。蛍光点の大きさ及び蛍光強度を測定し、舐めること及び噛みつくことを通じての蛍光点の大きさの減少を算出した。処置された足と処置されていない足の点の大きさの減少の比較により、全体的な舐める時間の強度を自動的に算出した。
【0145】
アッセイの読み出しの別の変形として、個々の動物の舐める行動をビデオファイルに基づいて手動で追跡した。ホルマリン注射後30分間にわたって舐める時間を記録し、3つの異なる舐める区域(背面、足底、つま先)についてさらに分けた。全体的な舐める時間は各動物及び各実験群について算出でき、化合物の効能の決定のためのパラメータとして使用できる。
【0146】
結果として、ホルマリン注射前に媒体処置を受容するマウス(ネガティブコントロール)は、ホルマリン処置した足での長い舐める時間及び蛍光点の大きさの有意な減少を示すことが発見された。
対照的に、ホルマリン処置した足の舐める時間の短縮、及びその結果として蛍光点の大きさの有意な減少のないことは、検査化合物/ホルマリン処置したマウスにおいて観察できた。同一の効果、すなわち舐める時間における短縮及び蛍光点の大きさにおけるわずかな変化は、Iκキナーゼ阻害剤(IKK;IKKの機能については、図2参照、ポジティブコントロール)で処置したコントロールマウスにおいて観察された。
この観察は、CDK9阻害剤で処置したマウスにおける低い炎症性/慢性炎症性疼痛知覚を、及び検査された化合物の痛覚鈍麻効果を示す。
【0147】
(実施例6)
マウスにおけるカラゲナンアッセイ−炎症及び炎症性疼痛のモデル
カラゲナン誘発性の足の浮腫のモデルは、個々の化合物の抗炎症活性及び炎症誘発性疼痛知覚の減少を予測するために使用される標準的な実験室アッセイである。下記のプロトコールは、実験を実施するための可能な1つの方法を記載する。
【0148】
基本的な測定は、浮腫並びに、カラゲナン等の刺激物に応答した機械的及び熱による過敏症の測定において構成する。
炎症及び結果として生じる炎症性疼痛は、(塩類溶液における)1%カラゲナン25μLのマウス後足(同側足)への皮下注射によって誘発される。各群10匹のマウスは、カラゲナン注射の30分前の腹腔内注射による式I記載の化合物(30mg/体重kg)、媒体(2%ヒドロクスプロリルセルロース;0.25%乳酸(85%溶液)400μL)及び塩類溶液(生理的NaCl)の投与を受容する。反対側の足は、カラゲナン注射を受容しない。
【0149】
1.1 カラゲナン処置したマウスに及ぼすCDK阻害化合物の投与の効果
カラゲナン注射によって誘発される足の浮腫を、注射した(同側の)足の中足領域で背面から足底まで測定される大きくなった足のサイズによって検出する。同側及び反対側の足の大きさは、炎症についての代理マーカーとして機能し、カラゲナン注射後のいくつかの時点:注射前(-1)、注射2時間後(2)、3時間後(3)、4時間後(4)、5時間後(5)、6時間後(6)、24時間後(24)に測定される。
【0150】
すべてのマウスの足のサイズは、カラゲナンの30分前に注射される処置物質の種類とは関係なく、カラゲナン注射後の最初の1時間以内に、例えば2〜3mm(+10%)増大し得る。時間経過の間、カラゲナン注射前にCDK阻害化合物による処置を受容したマウスは、カラゲナン注射24時間後まで浮腫の減少を示し得:足の大きさの増大は、例えば10%から8%へと低下できた。対照的に、対照マウスの足の大きさは、この時点で平均して30%増大できた。カラゲナン注射の24時間後、カラゲナンで処置したすべての足の大きさは、注射96時間後で最大に到達するよう増大し得る。
【0151】
カラゲナンアッセイの読み出しとして、ハーグリーブスアッセイが実施され得、この中で、該アッセイによって輻射熱に対する熱感受性の測定が可能となる。ハーグリーブスアッセイ(Hargreavesら, 1988)は、動物がプレキシガラスチャンバーの中で立つ場合に、動物の後足の足底面に輻射熱源を焦点合わせすることによって、自由に移動する動物における侵害受容感受性を測定する。特に、例えば55℃の温度を生じる光源へ、足の下位側を曝露する。曝露の開始と曝露された足の挙上/引き抜きとの間の潜時として熱感受性を測定する。
【0152】
本明細書に開示されるCDK9阻害剤及びカラゲナンを使用して、又はナプロキセン及びカラゲナンを使用して、又は溶媒及びカラゲナンを使用してそれぞれ処置されたマウスを、ハーグリーブスアッセイに供する。CDK阻害剤及びカラゲナンを使用して処置されたマウスは、ネガティブコントロールマウスと比較してより長い潜時を示すことができた。この観察は、本明細書に開示されるCDK阻害剤の痛覚鈍麻効果を示すであろう。
【0153】
(実施例7)
ラットにおけるカラゲナンアッセイ−炎症及び炎症性疼痛のモデル
下記は、ラットにおいてカラゲナンアッセイを実施する可能な1つの方法を示す。
該アッセイは、Winterらの文献(Proc. Soc. Exp. Biol. Med., 111, 544-547, 1962)によって記載されるプロトコールに従って、炎症性疼痛を有するラットにおける鎮痛/抗炎症活性を検出する。
【0154】
ラット(200〜250g)の右後足の下位表面にカラゲナンの懸濁液を注射する(1足につき0.05mL生理塩類溶液における0.75mg)。2時間後、両足の触覚刺激及び熱刺激へ連続的にラットを供する。
触覚刺激のために、格子床上の反転したアクリル製プラスチック箱(18×11.5×13cm)の下に動物を配置する。次に、力を増大させながら、電子フォン・フレイプローブ(Bioseb, Model 1610)の先端をまず、炎症を生じていない後足へ、次に炎症を生じた後足へ適用し、足の引っ込めを誘発させるのに必要な力を自動的に記録する。この手法を3回実施し、1足あたりの平均の力を算出する。
【0155】
熱刺激のために、装置(Ugo Basile, 参考文献: 7371)は、高架のガラス床上に配置された個々のアクリル製プラスチック箱(17×11×13cm)からなる。ラットを箱の中に配置し、10分間自由に慣れさせる。次に、携帯赤外線源(96±10mW/cm2)をまず炎症を生じていない後足の下に、次に炎症を生じた後足の下に焦点合わせし、足引っ込め潜時を自動的に記録する。組織障害を防止するために、熱源を45秒後に自動的にオフにする。
【0156】
行動測定後、足の浸漬によって誘発される水置換(mL)を示すデジタル体積記録計(Letica, Model 7500)を使用して、各後足の体積を測定することによって、足の浮腫を評価する。
各群につき10匹のラットを研究する。検査を盲検で実施する。
【0157】
本明細書に呈される式I記載のCDK阻害剤等の検査物質は、検査60分前に経口投与される2用量(10及び30mg/kg)で評価され、媒体対照群と比較されるであろう。
同一実験条件下で投与されるモルヒネ(128mg/kg経口投与)及びアセチルサリチル酸(512mg/kg経口投与)は、基準物質として使用されるであろう。
【0158】
それゆえ、実験には6群が含まれるであろう。対応のないスチューデントのt検定を使用して、処置された群を媒体対照と比較することによって、データが分析されるであろう。
本明細書に開示されるCDK9阻害剤及びカラゲナンを使用して、又はナプロキセン及びカラゲナンを使用して、又は溶媒及びカラゲナンを使用してそれぞれ処置されたラットを、ハーグリーブスアッセイに供する。CDK阻害剤及びカラゲナンを使用して処置されたラットは、ネガティブコントロールラットと比較してより長い潜時を示すべきである。この観察は、本明細書に開示されるようにCDK阻害剤の痛覚鈍麻効果を示すであろう。
【0159】
(実施例8)
A.LPSインビボアッセイ(LPS)−インビボでのサイトカイン抑制のモデル
敗血症性ショックのLPS誘発性モデルのために、マウスは、塩類溶液における30μgの細菌リポ多糖(LPS;L2630 SIGMA)の腹腔内注射を受容する。炎症性シグナル伝達カスケードの該LPS仲介性の開始は、例えば、TNFα、IL-6及びIL1β等のサイトカインの高い血清濃度を結果的に生じる。血液は、規定された時点でこれらの動物から採取できる。その後、血清は分離されるであろうし、試料は、サイトカイン濃度が市販のELISAアッセイを使用して測定されるまで、-80℃で保存できる(AL Moreiraらの文献(Braz J Med Biol Res 1997; 30:1199-1207))。
【0160】
サイトカイン、TNFα、IL6及びIL1β等の炎症性仲介因子が、持続性の疼痛状態及び炎症性障害に関与する可能性があることは認識されている。末梢組織におけるマクロファージ及びCNS組織におけるミクログリアのような免疫細胞から放出された後、これらの仲介因子は、炎症性及び神経因性疼痛においてだけでなく、関節リウマチ等の炎症性障害においても中枢の役割を担うように見える(F Marchandらの文献(Nat Rev Neurosci 2005; 6 (7); 521-532))。このように、腫瘍壊死因子α(TNFα)の阻害は同様に、炎症性疾患の治療のための関連性のある標的を表す[Lavagnoらの文献(Eur J Pharmacol 2004; 501, 199-208)]。
【0161】
LPSインビボアッセイは、薬理学的治療によるサイトカインの発現の抑制を扱うための強力なモデルとして使用できる。
1.野生型マウスにおけるサイトカインの発現の誘導
野生型マウス(C3HeB/FeJ系)(齢、性別及び体重を対形成)に30μgLPS(SIGMA)を腹腔内注射した。LPS投与の90分後、これらの動物を0.1mL/体重10gのケタミン-ロムプン(Rompun)(50/20mg/mL)で麻酔し、心穿刺を介して、血清調製のための血液を採取した。
【0162】
2.LPSマウスに対するCDK阻害化合物の投与
LPSマウスの薬理学的処置群(n=4)は、CDK阻害化合物又は媒体(ネガティブコントロール)の経口注入をそれぞれ受容した。
1%カルボキシメチルセルロース(SIGMA)に溶解した10又は30mg/kg(体重あたりの化合物)のCDK阻害剤を単回経口薬用量として、LPS刺激の30分前に投与した。媒体コントロールを同一の様式で投与した。
【0163】
LPS刺激の90分後に、血液試料をマウスから採取した。あらかじめ、経時実験によって、この動物モデルにおけるTNFα発現のピークとして、90分の時点を同定した。
LPSマウスにおけるサイトカインレベルに及ぼすCDK阻害剤による薬理学的処置の効果を、以下に記載のとおり市販のELISAアッセイにおいて分析した。
【0164】
3.CDK阻害化合物の投与後のLPSマウスにおけるサイトカイン血清濃度の決定
心穿刺後、LPS動物由来の血液試料(〜500μL/個体)を濡れた氷の上で30分間インキュベートした。その後、試料を13,000rpmで15分間遠心分離した。血餅から血清を分離し、-80℃で凍結保存した。
製造元の説明書に従って市販のELISAキット(Natutec)を使用することによって、試料内のTNFα及びIL6の血清濃度を測定した。
【0165】
4.サイトカインのタンパク質発現に及ぼすCDK阻害化合物の投与の効果
上述のように、化合物をLPSマウスに投与した。サイトカイン血清濃度に関するELISAベースの決定を上述のとおり実施した。CDK阻害化合物で処置した動物と媒体で処置した対照動物との比較は、血清におけるTNFα及びIL6タンパク質濃度に及ぼす有意な抑制効果を示し、サイトカイン発現のモデルにおけるサイトカインTNFα及びIL6の抑制剤として、化合物が効果的であることを示した。
【0166】
(実施例9)
A.インビトロTHP-1アッセイ−サイトカイン阻害のインビトロモデル
ヒトTHP-1細胞系は、リポ多糖(LPS)又は腫瘍壊死因子α[TNFα]によって仲介される場合のサイトカイン発現のインビトロモデルとして利用できる。
単球性THP-1細胞(ATCC;TIB-202)は、LPSによる誘導の際に、又はTNFα(自己分泌誘導)自体によってTNFα、IL6及び1L1βのような炎症誘発性サイトカインを発現するマクロファージ様細胞へ分化できる。
【0167】
サイトカインであるTNFα、IL6及びIL1β等の炎症性仲介因子が、持続性の疼痛状態及び炎症性障害に関与する可能性があることは認識されている。末梢組織におけるマクロファージ及びCNS組織におけるミクログリアのような免疫細胞から放出された後、これらの仲介因子は、炎症性及び神経因性疼痛においてだけでなく、関節リウマチ等の炎症性障害においても中枢の役割を担うように見える(F Marchandらの文献(Nat Rev Neurosci 2005; 6 (7); 521-532))。それにより、腫瘍壊死因子α(TNFα)の阻害は同様に、炎症性障害の治療における関連性のある標的を表す[Lavagnoらの文献(Eur J Pharmacol 2004; 501, 199-208)]。
それゆえ、THP-1インビトロアッセイは、サイトカイン発現の薬理学的阻害を扱うための強力なスクリーニングモデルとして使用できる(U Singhらの文献(Clin Chem 2005; 51(12); 2252-6);K Rutaultらの文献(J Biol Chem 2001; 276(9); 6666-74))。
【0168】
1.THP-1細胞の増殖及び分化
10%ウシ胎児血清及び1%ペニシリン/ストレプトマイシンを補充した修正RPMI-1640培地(ATCC、カタログ番号30-2001)において、THP-1細胞を増殖させる。サイトカイン阻害アッセイのために、100ng/mL PMA(Sigma, P1585)を補充した標準的な増殖培地において、6ウェルプレートへ5×105個/mLの密度で細胞を播種し、マクロファージ様細胞への分化を誘導する。24時間後、培地を標準的な増殖培地(PMAなし)と交換し、細胞をさらに48時間インキュベートして、分化を完了させる。
【0169】
2.CDK阻害化合物及びLPSの刺激による分化したTHP-1細胞の処置
分化の72時間後、培地を無血清増殖培地と交換し、各々DMSOに溶解したCDK阻害化合物並びにポジティブ及びネガティブコントロール等の基準化合物を、0.5〜5μMの範囲の濃度で添加する(ウェルにおけるDMSOの終濃度は0.1%である。)。100ng/mLのLPS(Sigma, L2630)を使用してさらに4〜48時間刺激する前に、細胞を化合物とともに60分間インキュベートする。上清を回収し、サイトカイン発現について、例えば、TNFα、IL-6及びIL-1bについて、市販のサンドイッチELISAアッセイ(eBioscience, カタログ番号88-7346、88-7066、88-7010)を使用して即時アッセイし、又は評価するまで20℃で凍結保存した。
【0170】
3.CDK阻害化合物の投与後のTHP-1上清におけるサイトカイン濃度の決定
製造元の説明書に従って、市販のELISAキット(eBioscience)を使用することによって、細胞培養上清内のTNFα、IL6及びIL1βの濃度を測定する。
【0171】
4.THP-1細胞上清におけるサイトカインのタンパク質発現に及ぼすCDK阻害化合物による処置の効果
CDK阻害化合物1、5、6、8、及び9番を上述のとおり三つ組で、分化したTHP-1細胞へ投与した(第2節参照)。検査化合物又は基準化合物(p38阻害剤であるSB203580及びIKK阻害剤であるBMS345541)単独によるプレインキュベーションの60分後、細胞をLPSで刺激した。4〜48時間のインキュベーションの後、上清を回収し、ELISAベースのサイトカイン上清濃度の決定を、上述の第3節に記載されるとおり実施した。
【0172】
化合物1、5、6、8、及び9及び基準化合物で処置した細胞と、媒体(DMSO)で処置した細胞との比較は、細胞上清におけるTNFα及びIL6のタンパク質濃度に及ぼす化合物1、5、6、8、及び9番の有意な阻害効果を示した。基準化合物SB203580又はBMS3455541と比較して、これらの化合物は、TNFα/IL6発現の同様の又はより良好な阻害を呈した。
【0173】
LPSにより誘導されるTHP-1マクロファージにおけるTNFαの発現に及ぼす化合物1、5、6、8、及び9番の投与の効果を図3に示す。
これらの知見は、CDK阻害化合物1、5、6、8、及び9がサイトカインTNFαの発現の効果的なサプレッサーであることを示す。
【0174】
(実施例10)
A.インビトロでのキナーゼ阻害アッセイ
インビトロでの酵素キナーゼ阻害アッセイにおけるサイクリン依存性キナーゼCDK2/CycA、CDK4/CycD1、CDK5/p35NCK、CDK6/CycD1及びCDK9/CycTについて、化合物1〜15番のIC50特性を決定した。CDK9阻害に関する化合物の特異的選択性及び効力を評価するために、これらのアッセイにおいて得られるIC50値を使用した。
【0175】
これらのアッセイにおいて得られた結果を使用して、CDK9に対する特異性を示す化合物を選択した。特に、該アッセイは、CDK9特異的化合物を、他のCDKに関しても、すなわちCDK2、4、5、及び6のいくつか又はすべてに関して有意な阻害効力を有する他の化合物と識別するよう企図された。この分離は、細胞周期関連CDK2、4、5、及び6の阻害の際に生じ得る有害な(細胞分裂抑制/細胞毒性)効果を回避するために必須である。
さらに、これらのデータを使用して、効力及び選択性に関して新たなかつさらに改良された構造/化合物の設計を支持する構造活性関係性(SAR)を確立した。
【0176】
1.検査化合物
100%DMSOにおける1×10-02Mストック溶液として化合物を使用し、3つの96ウェルV字型マイクロタイタープレート(以下、該プレートを「マスタープレート」と呼ぶ。)の2列目に各100μLを使用した。
その後、100%DMSOを溶媒として使用する連続的な半対数希釈へマスタープレートの2列目における1×10-02Mストック溶液を供し、結果的に10個の異なる濃度を生じ、希釈終点は、12列目における3×10-07M/100%DMSOであった。対照として、1列目及び7列目に100%DMSOを充填した。その後、96チャネルピペッターを使用して、連続希釈したコピープレートの各ウェルの2×5μLを「化合物希釈プレート」の2つの同一セットに分注した。
【0177】
キナーゼ阻害アッセイ当日、45μLのH2Oを1セットの化合物希釈プレートの各ウェルに添加した。沈殿を最小化するために、アッセイプレートへ化合物溶液を転移させるほんの数分前に、H2Oをプレートに添加した。プレートを入念に振盪し、結果的に半対数工程で1×10-03M/10%DMSO〜3×10-08M/10%DMSOの濃度の「化合物希釈プレート/10%DMSO」を生じた。「アッセイプレート」への5μLの化合物溶液の転移のために、これらのプレートを使用した。作業日の終了時に、化合物希釈プレートを廃棄した。アッセイ(以下参照)のために、化合物希釈プレートの各ウェルから5μL溶液をアッセイプレートに転移させた。アッセイの最終容積は50μLであった。1×10-04M〜3×10-09Mの範囲の10個の最終アッセイ濃度で、すべての化合物を検査した。反応混合物における最終DMSO濃度は、すべての場合において1%であった。
【0178】
2.組換えプロテインキナーゼ
阻害特性の決定のために、下記の5個のプロテインキナーゼを使用した:CDK2/CycA、CDK4/CycD1、CDK5/p35NCK、CDK6/CycD1及びCDK9/CycT。バキュロウイルス発現系によって、ヒト組換えGST融合タンパク質又はHisタグつきタンパク質として、該プロテインキナーゼをSf9昆虫細胞において発現させた。GSH-アガロース(Sigma)又はNi-NTH-アガロース(Qiagen)のいずれかを使用するアフィニティクロマトグラフィーによって、キナーゼを精製した。各キナーゼの純度をSDS-PAGE/銀染色によって決定し、キナーゼ特異的抗体を使用するウェスタンブロット分析によって又は質量分析によって、各キナーゼの同一性を確認した。
【0179】
3.プロテインキナーゼアッセイ
50μL反応容積におけるPerkin Elmer/NEN(Boston, MA, USA)製の96ウェルFlashPlates(商標)において、すべてのキナーゼアッセイを実施した。反応混合物を下記の順序で4工程でピペッティングした:
・アッセイ緩衝液20μL(標準緩衝液)
・(H2Oにおける)ATP溶液5μL
・(10%DMSOにおける)検査化合物5μL
・基質10μL/酵素溶液10μL(あらかじめ混合)
【0180】
すべての酵素についてのアッセイは、60mM HEPES-NaOH(pH7.5)、3mM MgCl2、3mM MnCl2、3μMオルトバナジン酸ナトリウム、1.2mM DTT、50μg/mL PEG20000、1μM[-33P]-ATP(ウェルあたり約5×1005cpm)を含有した。
下記の量の酵素及び基質をウェルあたり使用した:
【表2】
【0181】
反応混合物を30℃で80分間インキュベートした。50μLの2%(v/v)H3PO4を使用して反応を停止させ、プレートを吸引し、200μLのH2O又は200μLの0.9%(w/v)NaClを使用して2回洗浄した。マイクロプレートシンチレーションカウンタ(Microbeta, Wallac)を使用して、33Pの組み込みを決定した。
BeckmanCoulter/Sagianロボットシステムを使用して、すべてのアッセイを実施した。
【0182】
4.生データの評価
各アッセイプレートの1列目(n=8)における計数の中央値を「低コントロール」と定義した。この値は、プロテインキナーゼの不在下だが基質の存在下にあるプレートに対する放射能の非特異的結合を反映する。各アッセイプレートの7列目(n=8)における計数の中央値を「高コントロール」、すなわち阻害剤の不在下での完全な活性とみなした。高コントロールと低コントロールとの間の差異を100%活性と呼んだ。データ評価の一部として、高コントロール値から、及び対応するプレートの80個の「化合物値」すべてから、具体的なプレート由来の低コントロール値を減算した。下記の式を使用することによって、具体的なプレートの各ウェルについての残効性(%)を算出した:
残効性(%)=100×[(化合物のcpm−低コントロール)/(高コントロール−低コントロール)]
【0183】
Quattro Workflow V2.0.1.3(Quattro Research GmbH, Munich, Germany; www.quattro-research.com)を使用して、各濃度についての残効性及び化合物のIC50値を算出した。使用したモデルは、「シグモイド反応(可変傾斜)」であり、パラメータの「上部」は100%に、「下部」は0%に固定した。
化合物1〜15のIC50値がすべて、1nM〜10μMの間に含まれることがわかる。
【先行技術文献】
【非特許文献】
【0184】
(参考文献)
【非特許文献1】Barboric M.らの文献(NfkBはP-TEFbと結合してRNAポリメラーゼIIによる転写伸長を刺激する(NfκB Binds P-TEFb to Stimulate Transcriptional Elongation by RNA Polymerase II.)Molecular Cell, 2001, Vol. 8, 327-337)
【非特許文献2】Besson J.M.の文献(疼痛の神経生物学(The neurobiology of pain). Lancet, 1999, 353(9164), 1610-1615)
【非特許文献3】Browerの文献(疼痛緩和のための新たな経路(New paths to pain relief). Nat Biotechnol, 2000, 18(4), 387-391)
【非特許文献4】Chao S.H.及びPrice D.H.の文献(フラボピリドールはインビボでP-TEFb を不活性化し、ほとんどのRNAポリメラーゼII転写を遮断する(Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo). J Biol Chem, 2001, 276(34),31793-9)
【非特許文献5】Chaplan SR, Bach FW, Pogrel JW, Chung JM, 及びYaksh, TLの文献((1994) ラットの足における触覚性アロディニアの定量的評価(Quantitative assessment of tactile allodynia in the rat paw). J Neurosci Methods 53: 55-63)
【非特許文献6】Dai Y.及びGrant S.の文献(サイクリン依存性キナーゼ阻害剤(Cyclin-dependent kinase inhibitors). Curr Opin Pharmacol, 2003, 3(4), 362-370)
【非特許文献7】Falco G.D.らの文献(cdc2様ファミリーのキナーゼの一員であるCDK9は、サイトカインのIL-6ファミリーの受容体であるgp130に結合する(CDK9, a member of the cdc2-like family of kinases, binds to gp130, the receptor of the IL-6 family of cytokines). Oncogene, 2002, 21(49), 7464-7470)
【非特許文献8】Feldmann及びMainiの文献(NatMed, 2003, 9 (10); 356-61)
【非特許文献9】Firesteinの文献(2003, Nature 423, 356-361)
【非特許文献10】Hanらの文献(2003, Autoimmunity, 28, 197-208)
【非特許文献11】Hargreaves, Kの文献(Pain 32(1) (1988 Jan) 77-88)
【非特許文献12】Huweらの文献(サイクリン依存性キナーゼの阻害剤としての小分子(Small molecules as inhibitors of cyclin-dependent kinases). Angew Chem Int Ed Engl, 2003, 42(19), 2122-2138)
【非特許文献13】Kimらの文献(CDK9によるRNAポリメラーゼIIカルボキシル末端ドメインのリン酸化は、ヒト免疫不全ウイルス1型Tatにより活性化される転写伸長の直接の原因である(Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation). Mol Cell Biol, 2002, 22(13), 4622-4637)
【非特許文献14】Koltzenburg Mの文献(皮膚侵害受容疼痛の神経性機構(Neural mechanisms of cutaneous nociceptive pain). Clin J Pain, 2000, 16(3 Suppl), 131-138)
【非特許文献15】Laufer S., Gay S.及びBrune K.の文献(炎症及びリウマチ疾患−新規の治療法の分子レベルの基礎(Inflammation and Rheumatic Diseases The molecular basis of novel therapies), Thieme, 2003)
【非特許文献16】Lee K.M.らの文献(脊髄性NfkBの活性化は、COX-2上方制御を誘導し、炎症性疼痛過敏症に関与する(Spinal NfκB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity). European Journal of Neuroscience, 2004, Vol. 19, 3375-3381)
【非特許文献17】Liu H.及びHerrmann C.の文献(CDK9の42k及び55k分子種の異なる局在性及び発現(Differential Localization and Expression of the CDK9 42k and 55k Isoforms). J Cell Physiol, 2004, 203, 251-260)
【非特許文献18】MacLachlan T.K.らの文献(TRAF2に対するCDK9の結合(Binding of CDK9 to TRAF2). J Cell Biochem, 1998, 71(4), 467-478)
【非特許文献19】Malmberg AB及びBasbaum AI.の文献((1998) 神経因性疼痛のモデルとしてのマウスにおける部分的坐骨神経損傷:行動相関及び神経解剖学的相関(Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates). Pain 76: 215-2)
【非特許文献20】Meijer L, Leclerc S.及びLeost M.の文献(サイクリン依存性キナーゼの化学的阻害剤の特性および可能性のある応用(Properties and potential applications of chemical inhibitors of cyclin-dependent kinases), Pharmacol Ther 1999, 82(2-3):279-284)
【非特許文献21】Sausville, E.A.の文献(Trends Molec. Med. 2002, 8, S32-S37)
【非特許文献22】Tian B.らの文献(TNFシグナル伝達を仲介するNfkB転写因子の下流の直接的なゲノム標的の同定(Identification of direct genomic targets downstream of the NfSYMBOL 107 \f "Symbol" \s 10.5B transcription factor mediating TNF signaling). JBC, 2005, 原稿M500437200として)
【非特許文献23】Wang Dらの文献(化学的サイクリン依存性キナーゼ阻害剤によるヒト免疫不全ウイルス1型の転写の阻害(Inhibition of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent kinase inhibitors). J. Virol. 2001; 75: 7266-7279)
【非特許文献24】Watkins L.R.らの文献(グリア炎症誘発性サイトカインは過度の疼痛状態を仲介する:臨床的疼痛についての含意(Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain). Adv Exp Med Biol., 2003, 521, 1-21)
【非特許文献25】Westらの文献(2001, Journal of Virology 75(18), 85248537)
【非特許文献26】Zhou M.らの文献(ヒト免疫不全ウイルス1型の転写の間のP-TEFbキナーゼによる転写因子のリン酸化及びヒストンのメチル化の協調(Coordination of transcription factor phosphorylation and histone methylation by the P-TEFb Kinase during human immunodeficiency virus type I transcription), J. Virol 2004, 78(24):13522-13533)
【特許請求の範囲】
【請求項1】
一般式I記載の化合物
【化1】
及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;並びにこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)
(式中、
R1が、-XSO2NR5R6又は-XSO2R8であり;
Xが、(分岐鎖アルキレンを含む)C1-4アルキレンであり、式中、該C1-4アルキレンが、R5又はR6へ結合して、5〜6員の複素環を形成でき;
R5及びR6が互いに独立して、水素、C1-4アルキル、ヒドロキシ-C1-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル若しくはC4-7-ヘテロシクロアルキル-C0-4アルキル、C4-7-アリール-C0-4アルキル、C4-7-ヘテロアリール-C0-4アルキルであるか又は
式中、R5及びR6はそれらが結合するN原子とともにまた、5〜8員のヘテロシクロアルキルを形成し得、
この中で、該シクロアルキル、ヘテロシクロアルキル、アリール、ヘテロアリール又はアルキルがさらに、ハロ、ヒドロキシ、アミノカルボニル、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O-及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R8が、C1-4アルキル、ヒドロキシ-C2-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル又はC4-7-ヘテロシクロアルキル-C0-4アルキルであり;
この中で、該シクロアルキル、ヘテロシクロアルキル又はアルキルがさらに、ハロ、ヒドロキシ、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R2が、ハロゲン及び水素から独立して選択される1個又は2個の置換基であり;
R3が、水素、ハロ、ヒドロキシ、C1-4アルキル、C3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、アリール-C1-4アルコキシ、ヘテロアリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルキル、ヘテロアリール-C1-4-アルキル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から各々独立して選択される1〜3個の置換基であり得、この中で、上述の置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、アミノカルボニル、ハロ及びNR5R6の群から選択されるラジカルによってさらに置換でき;
R4a及びR4bが、同一又は異別であり、かつ各々が独立して、水素、C1-4アルキル又は-NR'R"であり、式中R'及びR"が各々独立して、水素又はC1-4アルキルである。)。
【請求項2】
R3が、メチル、エチル、ヒドロキシメチル、ヒドロキシ、メトキシ、エトキシ、イソプロポキシ、ベンジルオキシ、水素、フルオロ、クロロ、トリフルオロメチル、2-メトキシ-エトキシ、メトキシメチル、2-メトキシ-エチル、テトラヒドロ-フラン-3-イルオキシ、テトラヒドロ-フラン-2-イル-メトキシ、-N(CH3)SO2CH3、ピペリジン-1-イル-メチル、2-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-(2-ヒドロキシ-エチル)-ピペリジン-1-イル-メチル、3-アミノカルボニル-ピペリジン-1-イル-メチル、ジメチルアミノメチル、ジエチルアミノメチル、(エチル-イソプロピル-アミノ)-メチル、モルフォリン-4-イルメチル、4-メチル-ピペラジン-1-イル-メチル、[1,2,4]トリアゾール-1-イル-メチル、ピリジン-3-イル-メトキシ及びピリジン-4-イル-メトキシからなる群から独立して選択される1〜3個の置換基であることを特徴とする、請求項1記載の化合物。
【請求項3】
R1が、-XSO2NR5R6であり、かつR5及びR6が、水素、メチル、シクロプロピル、2-ジメチルアミノエチル、3-ジメチルアミノプロピル及び2-ヒドロキシエチルからなる群から各々独立して選択されることを特徴とする、請求項1又は請求項2記載の化合物。
【請求項4】
R1が、-XSO2R8であり、かつR8が、C1-4アルキル又はヒドロキシ-C2-4-アルキルであることを特徴とする、請求項1又は請求項2記載の化合物。
【請求項5】
Xがメチレンであることを特徴とする、請求項1〜4のいずれか一項記載の化合物。
【請求項6】
前記化合物が、式Ia記載の構造
【化2】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;並びにこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)を有することを特徴とする、請求項1記載の化合物
(式中、
R9が、-NR5R6又はR8であり、
R2が、水素又はハロゲンであり、
R3aが、水素、ハロゲン又はC1-4-アルコキシであり、
R3bが、水素であり、
R3cが、水素又はハロゲンであり、
R4a及びR4bが各々独立して、水素又はC1-4アルキルであり、
R5及びR6が各々独立して、水素、ヒドロキシル若しくはジアルキルアミノによって任意に置換されるシクロアルキル又はC1-4アルキルであり、
及びR8が、ヒドロキシ-C2-4-アルキルである。)。
【請求項7】
請求項1記載の化合物であって、下記からなる群から選択される該化合物:
{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物1);
{3-[4-(2-イソプロポキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物2);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N,N-ジメチル-メタンスルホンアミド(化合物3);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物4);
{3-[4-(4-フルオロ-2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物5);
N-シクロプロピル-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物6);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物7);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物8);
2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物9);
{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物10);
-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物11);
2-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物12);
{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物13);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物14);及び
2-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物15)。
【請求項8】
医学的使用のための、請求項1〜7のいずれか一項記載の化合物。
【請求項9】
医薬として許容し得る担体とともに請求項1〜7のいずれか一項記載の化合物を含有する医薬組成物。
【請求項10】
いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患の群から選択される疾患の治療のための方法であって、治療的有効量の請求項1〜7記載の化合物の少なくとも1つを投与することを含む、前記方法。
【請求項11】
いずれかの種類の疼痛が、慢性疼痛、炎症性及び/又は神経因性疼痛を含む、請求項10記載の方法。
【請求項12】
いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための医薬組成物を製造するための、請求項1〜7のいずれか一項記載の化合物の使用。
【請求項13】
いずれかの種類の疼痛が、慢性疼痛、炎症性及び/又は神経因性疼痛を含む、請求項12記載の使用。
【請求項1】
一般式I記載の化合物
【化1】
及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;並びにこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)
(式中、
R1が、-XSO2NR5R6又は-XSO2R8であり;
Xが、(分岐鎖アルキレンを含む)C1-4アルキレンであり、式中、該C1-4アルキレンが、R5又はR6へ結合して、5〜6員の複素環を形成でき;
R5及びR6が互いに独立して、水素、C1-4アルキル、ヒドロキシ-C1-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル若しくはC4-7-ヘテロシクロアルキル-C0-4アルキル、C4-7-アリール-C0-4アルキル、C4-7-ヘテロアリール-C0-4アルキルであるか又は
式中、R5及びR6はそれらが結合するN原子とともにまた、5〜8員のヘテロシクロアルキルを形成し得、
この中で、該シクロアルキル、ヘテロシクロアルキル、アリール、ヘテロアリール又はアルキルがさらに、ハロ、ヒドロキシ、アミノカルボニル、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O-及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R8が、C1-4アルキル、ヒドロキシ-C2-4アルキル若しくはC3-4アルケニル、C3-8-シクロアルキル、C3-8-シクロアルキル-C1-4アルキル又はC4-7-ヘテロシクロアルキル-C0-4アルキルであり;
この中で、該シクロアルキル、ヘテロシクロアルキル又はアルキルがさらに、ハロ、ヒドロキシ、C1-4アルキル、ヒドロキシ-C1-4アルキル、C1-4アルキル-O-C1-4アルキル、C1-4アルキル-O及び-NR5R6からなる群から選択される最大2個のラジカルによって任意に置換され;
R2が、ハロゲン及び水素から独立して選択される1個又は2個の置換基であり;
R3が、水素、ハロ、ヒドロキシ、C1-4アルキル、C3-7シクロアルキル、C1-4アルキル-シクロアルキル、C1-4アルキル-ヘテロシクロアルキル、-O-ヘテロシクロアルキル、C1-4アルコキシ、C2-4アルケニルオキシ、-OCF3、C2-4アルカノイル、C1-4アルキルスルホニル、モノ-及びジ-(C1-C4アルキル)スルホンアミド、アミノカルボニル、モノ-及びジ-(C1-C4アルキル)アミノカルボニル、アリール-C1-4アルコキシ、ヘテロアリール-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルコキシ、ヘテロシクロアルキル-C1-4アルキル、ヘテロアリール-C1-4-アルキル、C1-4アルキルオキシメチル、ヒドロキシ-C1-4アルキルオキシメチル、シアノ、-COOH及びC1-C4アルコキシカルボニルからなる群から各々独立して選択される1〜3個の置換基であり得、この中で、上述の置換基は、C1-4-アルキル、ヒドロキシル-C0-4-アルキル、C1-4-アルコキシ、アミノカルボニル、ハロ及びNR5R6の群から選択されるラジカルによってさらに置換でき;
R4a及びR4bが、同一又は異別であり、かつ各々が独立して、水素、C1-4アルキル又は-NR'R"であり、式中R'及びR"が各々独立して、水素又はC1-4アルキルである。)。
【請求項2】
R3が、メチル、エチル、ヒドロキシメチル、ヒドロキシ、メトキシ、エトキシ、イソプロポキシ、ベンジルオキシ、水素、フルオロ、クロロ、トリフルオロメチル、2-メトキシ-エトキシ、メトキシメチル、2-メトキシ-エチル、テトラヒドロ-フラン-3-イルオキシ、テトラヒドロ-フラン-2-イル-メトキシ、-N(CH3)SO2CH3、ピペリジン-1-イル-メチル、2-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-ヒドロキシメチル-ピペリジン-1-イル-メチル、3-(2-ヒドロキシ-エチル)-ピペリジン-1-イル-メチル、3-アミノカルボニル-ピペリジン-1-イル-メチル、ジメチルアミノメチル、ジエチルアミノメチル、(エチル-イソプロピル-アミノ)-メチル、モルフォリン-4-イルメチル、4-メチル-ピペラジン-1-イル-メチル、[1,2,4]トリアゾール-1-イル-メチル、ピリジン-3-イル-メトキシ及びピリジン-4-イル-メトキシからなる群から独立して選択される1〜3個の置換基であることを特徴とする、請求項1記載の化合物。
【請求項3】
R1が、-XSO2NR5R6であり、かつR5及びR6が、水素、メチル、シクロプロピル、2-ジメチルアミノエチル、3-ジメチルアミノプロピル及び2-ヒドロキシエチルからなる群から各々独立して選択されることを特徴とする、請求項1又は請求項2記載の化合物。
【請求項4】
R1が、-XSO2R8であり、かつR8が、C1-4アルキル又はヒドロキシ-C2-4-アルキルであることを特徴とする、請求項1又は請求項2記載の化合物。
【請求項5】
Xがメチレンであることを特徴とする、請求項1〜4のいずれか一項記載の化合物。
【請求項6】
前記化合物が、式Ia記載の構造
【化2】
、及びそのN酸化物誘導体、プロドラッグ誘導体、保護された誘導体、個々の異性体及びその異性体の混合物;並びにこれらの化合物の医薬として許容し得る塩及び溶媒和化合物(例えば、水和物)を有することを特徴とする、請求項1記載の化合物
(式中、
R9が、-NR5R6又はR8であり、
R2が、水素又はハロゲンであり、
R3aが、水素、ハロゲン又はC1-4-アルコキシであり、
R3bが、水素であり、
R3cが、水素又はハロゲンであり、
R4a及びR4bが各々独立して、水素又はC1-4アルキルであり、
R5及びR6が各々独立して、水素、ヒドロキシル若しくはジアルキルアミノによって任意に置換されるシクロアルキル又はC1-4アルキルであり、
及びR8が、ヒドロキシ-C2-4-アルキルである。)。
【請求項7】
請求項1記載の化合物であって、下記からなる群から選択される該化合物:
{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物1);
{3-[4-(2-イソプロポキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物2);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N,N-ジメチル-メタンスルホンアミド(化合物3);
C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物4);
{3-[4-(4-フルオロ-2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物5);
N-シクロプロピル-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物6);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物7);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物8);
2-{3-[4-(2-メトキシ-フェニル)-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物9);
{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物10);
-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物11);
2-{3-[4-(2-メトキシ-フェニル)-6-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物12);
{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-メタンスルホンアミド(化合物13);
N-(2-ヒドロキシ-エチル)-C-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニル}-N-メチル-メタンスルホンアミド(化合物14);及び
2-{3-[4-(2-メトキシ-フェニル)-5-メチル-ピリミジン-2-イルアミノ]-フェニルメタンスルホニル}-エタノール(化合物15)。
【請求項8】
医学的使用のための、請求項1〜7のいずれか一項記載の化合物。
【請求項9】
医薬として許容し得る担体とともに請求項1〜7のいずれか一項記載の化合物を含有する医薬組成物。
【請求項10】
いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患の群から選択される疾患の治療のための方法であって、治療的有効量の請求項1〜7記載の化合物の少なくとも1つを投与することを含む、前記方法。
【請求項11】
いずれかの種類の疼痛が、慢性疼痛、炎症性及び/又は神経因性疼痛を含む、請求項10記載の方法。
【請求項12】
いずれかの種類の疼痛、炎症性障害、免疫学的疾患、増殖性疾患、感染性疾患、心血管疾患及び神経変性疾患を治療するための医薬組成物を製造するための、請求項1〜7のいずれか一項記載の化合物の使用。
【請求項13】
いずれかの種類の疼痛が、慢性疼痛、炎症性及び/又は神経因性疼痛を含む、請求項12記載の使用。
【図1】

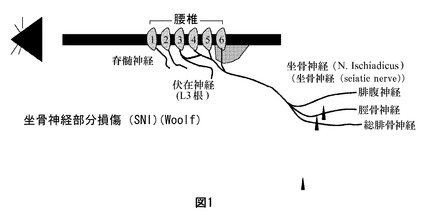
【図2】
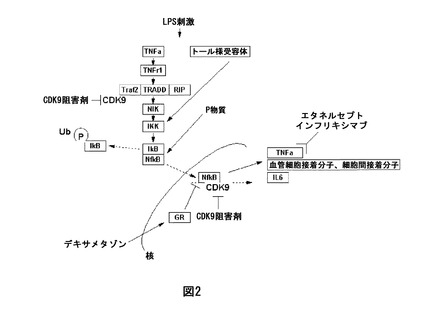
【図3】
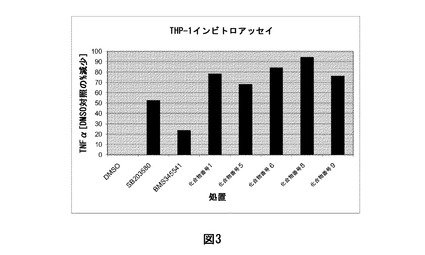
【図2】
【図3】
【公表番号】特表2011−502956(P2011−502956A)
【公表日】平成23年1月27日(2011.1.27)
【国際特許分類】
【出願番号】特願2010−504681(P2010−504681)
【出願日】平成20年4月24日(2008.4.24)
【国際出願番号】PCT/EP2008/054975
【国際公開番号】WO2008/129070
【国際公開日】平成20年10月30日(2008.10.30)
【出願人】(507387815)インゲニウム ファーマシューティカルズ ジーエムビーエイチ (7)
【Fターム(参考)】
【公表日】平成23年1月27日(2011.1.27)
【国際特許分類】
【出願日】平成20年4月24日(2008.4.24)
【国際出願番号】PCT/EP2008/054975
【国際公開番号】WO2008/129070
【国際公開日】平成20年10月30日(2008.10.30)
【出願人】(507387815)インゲニウム ファーマシューティカルズ ジーエムビーエイチ (7)
【Fターム(参考)】
[ Back to top ]