
プロテインAアフィニティークロマトグラフィーを利用した抗IL−13抗体の単離精製
本願はアフィニティークロマトグラフィー工程を利用することにより医薬用として十分な純度の抗体組成物が得られる抗IL−13抗体の単離精製方法を開示する。本願に記載する方法はpHウイルス低減/不活性化、限外濾過/透析濾過、アフィニティークロマトグラフィー(例えばプロテインAアフィニティークロマトグラフィー)、イオン交換クロマトグラフィー及び疎水性クロマトグラフィーを含む。更に、本発明は本発明の1種以上の抗体を含有する医薬組成物に関する。

【発明の詳細な説明】
【背景技術】
【0001】
ヒトIL−13は、活性化T細胞からクローニングされた17kDa糖蛋白質であり、Th2系統の活性化T細胞、Th0及びTh1 CD4+T細胞、CD8+T細胞、並びに数種の非T細胞集団(例えば肥満細胞)により産生される(Zurawski and de Vries,1994 Immunol Today,15,19−26)。IL−13はヒトB細胞においてIgEへの免疫グロブリンアイソタイプスイッチを促進し(Punnonen,Aversa et al.1993 Proc Natl Acad Sci U S A 90 3730−4)、ヒト及びマウスの両者において炎症性サイトカイン産生を抑制する(de Waal Malefyt et al.,1993,J Immunol,151,6370−81;Doherty et al.,1993,J Immunol,151,7151−60)。IL−13はその細胞表面受容体であるIL−13Rα1及びIL−13Rα2と結合する。IL−13Rα1は低親和性(KD〜10nM)でIL−13と相互作用した後、IL−4Rを動員し、高親和性(KD〜0.4nM)シグナル伝達ヘテロ二量体受容体複合体を形成する(Aman et al.,1996,J Biol Chem,271,29265−70;Hilton et al.,1996,Proc Natl Acad Sci USA,93,497−501)。IL−4R/IL−13Rα1複合体はB細胞、単球/マクロファージ、樹状細胞、好酸球、好塩基球、線維芽細胞、内皮細胞、気道上皮細胞及び気道平滑筋細胞等の多数の細胞種で発現される(Graber et al.,1998,Eur J Immunol,28,4286−98;Murata et al.,1998,Int Immunol,10,1103−10;Akaiwa et al.,2001,Cytokine,13,75−84)。IL−13Rα1/IL−4R受容体複合体のライゲーションの結果、シグナル伝達性転写因子(STAT6)及びインスリン受容体基質−2(IRS−2)経路を含む各種シグナル伝達経路が活性化される(Wang et al.,1995,Blood,864218−27;Takeda et al.,1996,J Immunol,157,3220−2)。IL−13Rα2鎖自体はIL−13に対する親和性が高く(KD〜0.25〜0.4nM)、IL−13との結合を負に制御するデコイ受容体として機能する(Donaldson et al.,1998,J Immunol,161,2317−24)と同時に、マクロファージと恐らく他の細胞種でAP−1経路を介してTGF−β合成及び線維化を誘導するシグナル伝達受容体としても機能する(Fichtner−Feigl,Strober et al.2006 Nat Med 12 99−106)。
【0002】
喘息の前臨床動物モデルで実施された数件の試験によると、IL−13は、喘息において重要な役割を果たすことが分かっている。これらのデータによると、IL−13ノックアウトマウスは、喘息にかかりにくいことや、喘息表現型は、各種マウスモデルでIL−13アンタゴニスト(可溶性IL−13受容体、抗IL−13mAb等)により阻害されることが立証されている(Wills−Karp and Chiaramonte,2003,Curr Opin Pulm Med,9 21−7;Wills−Karp,2004,Immunol Rev,202 175−90)。組換えIL−13をマウスとモルモットの肺に薬理投与すると、気道粘液過剰分泌、好酸球増加及び気道過敏反応(「AHR」;Grunig et al.,1998,Science,282,2261−3;Wills−Karp et al.,1998,Science,282,2258−61;Kibe et al.,2003,Am J Respir Crit Care Med,167,50−6;Vargaftig and Singer,2003,Am J Physiol Lung Cell Mol Physiol,284,L260−9;Vargaftig and Singer,2003,Am J Respir Cell Mol Biol,28,410−9)を誘発することは複数の研究で実証されている。IL−13のこれらの作用はIL−13を構成的又は誘導的に発現させたトランスジェニックマウスシステムで再現される(Zhu et al.,1999,J Clin Invest,103,779−88;Zhu et al.,2001,Am J Respir Crit Care Med,164,S67−70;Lanone et al.,2002,J Clin Invest,110463−74)。IL−13の慢性トランスジェニック過剰発現は、上皮下線維化及び気腫も誘発する。IL−13(及びIL−4)シグナル伝達分子STAT6を欠損するマウスはアレルゲンにより誘発されるAHR及び粘液過剰産生を発現することができない(Kuperman et al.,2002,Nat Med,8,885−9)。可溶性IL−13受容体融合蛋白質(sIL−13Rα2Fc)を使用した試験によると、このサイトカインはアレルゲンであるオボアルブミン(OVA)により実験的に誘発した気道疾患において中枢的役割を果たすことが判明した(Grunig et al.,1998,Science,282,2261−3;Wilis−Karp et al.,1998,Science,282,2258−61;Taube et al.,2002,J Immunol,169,6482−9)。抗IL−13治療が有効であることもマウス喘息の慢性モデルで実証された。粘液過剰分泌及びAHRの特徴を示すことに加え、慢性喘息のこのモデルは急性度の高いモデルには認められないヒト疾患の数種の特徴を示す。これらの特徴としては、上皮細胞間スペースに位置する肺組織の好酸球増加と、コラーゲン沈着の増加により確認される平滑筋線維化が挙げられる。慢性喘息モデルはOVA感作マウスに週1回ずつ合計4週間OVAを反復エアゾール攻撃することにより誘導される。OVA攻撃の最後の2週間に抗IL−13抗体を投与する(36日目から投与して53日目の試験日に効力計測値を測定する)と、AHR、肺炎症、杯細胞異常増殖、粘液過剰分泌及び気道線維化を有意に阻害した(Yang et al.,2005,J Pharmacol Exp Ther,313,8−15)。喘息患者の肺で高濃度のIL−13 mRNA及び蛋白質が検出されており、疾患の重篤度に相関することから、IL−13はヒト喘息の病因に関与していると考えられている(Huang et al.,1995,J Immunol,155,2688−94)。更に、高濃度IL−13に繋がるヒトIL−3遺伝子多型が同定され、喘息とアトピーに関係があるとされ(Heinzmann et al.,2000,Hum Mol Genet,9,549−59;Hoerauf et al.,2002,Microbes Infect,4,37−42;Vercelli,2002,Curr Opin Allergy Clin Immunol,2,389−93;Heinzmann et al.,2003,J Allergy Clin Immunol,112,735−9;Chen et al.,2004,J Allergy Clin Immunol,114,553−60;Vladich et al.,2005,J Clin Invest,115,747−54)、喘息患者の肺で高濃度のIL−13が検出されている(Huang et al.,1995,J Immunol,155,2688−94;Arima et al.,2002,J Allergy Clin Immunol,109,980−7;Berry et al.,2004,J Allergy Clin Immunol,114,1106−9)。血漿IL−13濃度の上昇に繋がるIL−13遺伝子の多型を示す個体はアトピーと喘息の危険が高いことから、IL−13と喘息の遺伝的連鎖も立証されている(Wills−Karp,2000,Respir Res,1,19−23)。
【先行技術文献】
【非特許文献】
【0003】
【非特許文献1】Zurawski and de Vries,1994 Immunol Today,15,19−26
【非特許文献2】Punnonen,Aversa et al.1993 Proc Natl Acad Sci U S A 90 3730−4
【非特許文献3】de Waal Malefyt et al.,1993,J Immunol,151,6370−81
【非特許文献4】Doherty et al.,1993,J Immunol,151,7151−60
【非特許文献5】Aman et al.,1996,J Biol Chem,271,29265−70
【非特許文献6】Hilton et al.,1996,Proc Natl Acad Sci USA,93,497−501
【非特許文献7】Graber et al.,1998,Eur J Immunol,28,4286−98
【非特許文献8】Murata et al.,1998,Int Immunol,10,1103−10
【非特許文献9】Akaiwa et al.,2001,Cytokine,13,75−84
【非特許文献10】Wang et al.,1995,Blood,864218−27
【非特許文献11】Takeda et al.,1996,J Immunol,157,3220−2
【非特許文献12】Donaldson et al.,1998,J Immunol,161,2317−24
【非特許文献13】Fichtner−Feigl,Strober et al.2006 Nat Med 12 99−106
【非特許文献14】Wills−Karp and Chiaramonte,2003,Curr Opin Pulm Med,9 21−7
【非特許文献15】Wills−Karp,2004,Immunol Rev,202 175−90
【非特許文献16】Grunig et al.,1998,Science,282,2261−3
【非特許文献17】Wills−Karp et al.,1998,Science,282,2258−61
【非特許文献18】Kibe et al.,2003,Am J Respir Crit Care Med,167,50−6
【非特許文献19】Vargaftig and Singer,2003,Am J Physiol Lung Cell Mol Physiol,284,L260−9
【非特許文献20】Vargaftig and Singer,2003,Am J Respir Cell Mol Biol,28,410−9
【非特許文献21】Zhu et al.,1999,J Clin Invest,103,779−88
【非特許文献22】Zhu et al.,2001,Am J Respir Crit Care Med,164,S67−70
【非特許文献23】Lanone et al.,2002,J Clin Invest,110463−74
【非特許文献24】Kuperman et al.,2002,Nat Med,8,885−9
【非特許文献25】Taube et al.,2002,J Immunol,169,6482−9
【非特許文献26】Yang et al.,2005,J Pharmacol Exp Ther,313,8−15
【非特許文献27】Huang et al.,1995,J Immunol,155,2688−94
【非特許文献28】Heinzmann et al.,2000,Hum Mol Genet,9,549−59
【非特許文献29】Hoerauf et al.,2002,Microbes Infect,4,37−42
【非特許文献30】Vercelli,2002,Curr Opin Allergy Clin Immunol,2,389−93
【非特許文献31】Heinzmann et al.,2003,J Allergy Clin Immunol,112,735−9
【非特許文献32】Chen et al.,2004,J Allergy Clin Immunol,114,553−60
【非特許文献33】Vladich et al.,2005,J Clin Invest,115,747−54
【非特許文献34】Arima et al.,2002,J Allergy Clin Immunol,109,980−7
【非特許文献35】Berry et al.,2004,J Allergy Clin Immunol,114,1106−9
【非特許文献36】Wills−Karp,2000,Respir Res,1,19−23
【発明の概要】
【発明が解決しようとする課題】
【0004】
ヒトIL−13は、様々なヒトの障害に関与しているため、IL−13活性を阻害又は抑制するための治療方法が設計されてきた。特に、IL−13活性を阻害するための手段として、IL−13と結合してこれを中和する抗体が求められてきた。しかし、医薬用としてのこのような抗体を製造・精製する改良型の方法が当分野で必要とされている。本発明はこの必要に対処する。
【課題を解決するための手段】
【0005】
ある実施形態においては、本発明はIL−13と結合する精製・単離抗体及び抗体フラグメントと、このような抗体及びフラグメントを含有する医薬組成物に関する。ある実施形態においては、本発明はヒトIL−13と結合する単離抗体又はその抗原結合部分に関する。本発明の単離抗IL−13抗体は臨床環境及び研究・開発で使用することができる。ある実施形態においては、本発明は、図1に示す重鎖及び軽鎖配列を含む抗IL−13抗体に関する。
【0006】
本発明のある実施形態は、抗体が宿主細胞蛋白質(「HCP」)と浸出したプロテインAを実質的に含まないように抗IL−13抗体又はその抗原結合部分をサンプルマトリックスから精製する方法に関する。ある態様において、サンプルマトリックス(又は単に「サンプル」)は本発明の抗IL−13抗体を産生させるために利用される細胞株を含む。特定の態様において、サンプルはヒト抗IL−13抗体を産生させるために利用される細胞株を含む。
【0007】
ある実施形態において、本発明は、特に細胞及び細胞破片を除去するための一次回収工程を含むIL−13抗体の精製方法を提供する。前記方法のある実施形態において、一次回収工程は、1以上の遠心又は深層濾過工程を含む。限定するものではないが、例えば、このような遠心工程は、約7000×g〜約11,000×gで実施することができる。更に、上記方法のある実施形態は、デリピッド深層濾過工程等の深層濾過工程を含む。
【0008】
ある実施形態では、一次回収サンプルをアフィニティークロマトグラフィー工程に供する。アフィニティークロマトグラフィー工程は適切なアフィニティークロマトグラフィー担体を含むカラムに一次回収サンプルを供する段階を含む。このようなクロマトグラフィー担体の非限定的な例としては、限定されないが、プロテインA樹脂、プロテインG樹脂、目的抗体に特異的な抗原を含むアフィニティー担体、及びFc結合蛋白質を含むアフィニティー担体が挙げられる。プロテインA樹脂は抗体(IgG)のアフィニティー精製・単離に有用である。ある態様では、サンプルをロードする前にプロテインAカラムを適切な緩衝液で平衡化させる。適切な緩衝液の1例はpH約7.2のトリス/NaClバッファーである。平衡化後、サンプルをカラムにロードすることができる。カラムにロードした後、例えば平衡化バッファーを使用してカラムを1回以上洗浄することができる。カラムから溶出させる前に、別の緩衝液を利用する洗浄を含む他の洗浄を使用することができる。その後、適当な溶出バッファーを使用してプロテインAカラムから溶出させることができる。適切な溶出バッファーの1例はpH約3.5の酢酸/NaClバッファーである。当業者に周知の技術を使用して溶出液をモニターすることができる。例えば、OD280の吸光度を追跡することができる。次に、後続処理に備えて目的溶出フラクションを調製することができる。
【0009】
本発明のある実施形態では、プロテインAアフィニティークロマトグラフィー後に低pH調整工程を行う。このような態様では、推定抗IL−13抗体又はその抗原結合部分を含有するプロテインA溶出液を約3〜約4のpHへのpH調整に供する。ある態様では、pHを約3.5に調整する。特に、低いpHはサンプルを汚染している可能性のあるpH感受性ウイルスの低減及び/又は不活性化を促進する。適切な時間後、pHを約4.5〜約6.0(限定されないが、例えば約5.0)に調整し、サンプルを後続精製工程に供する。
【0010】
ある実施形態では、プロテインAアフィニティークロマトグラフィー又は低pH調整工程後にイオン交換工程を実施する。このイオン交換工程は、カチオン交換でもアニオン交換でもよいし、両者の順次併用でもよい。この工程は単一のイオン交換法でもよいし、カチオン交換工程後にアニオン交換工程又はアニオン交換工程後にカチオン交換工程を実施する等の複数のイオン交換工程でもよい。ある態様において、イオン交換工程は1段階法である。別の態様において、イオン交換工程は2段階イオン交換法である。適切なカチオン交換カラムは固定相にアニオン基を含むカラムである。このようなカラムの1例はFractogel(登録商標)SO3−である。このイオン交換捕集液クロマトグラフィー工程はサンプルからの抗体の単離を容易にする。適切なアニオン交換カラム固定相にカチオン基を含むカラムである。このようなカラムの1例はQ Sepharose(登録商標)カラムである。別の例はPall Mustang Qメンブレンカートリッジである。1以上のイオン交換工程は宿主細胞蛋白質及びDNAや、適用可能な場合には、アフィニティーマトリックス蛋白質等の不純物を低減することにより更に抗体を単離する。このアニオン交換法は目的抗体がアニオン交換樹脂(ないし固相)と相互作用又は結合しないフロースルーモードのクロマトグラフィーである。しかし、多くの不純物はアニオン交換樹脂と相互作用し、結合する。特定の態様において、イオン交換工程はアニオン交換クロマトグラフィーである。
【0011】
サンプル緩衝液のpH及びイオン強度を調整することにより、アフィニティークロマトグラフィー溶出液をイオン交換クロマトグラフィーに備えて調製する。例えば、アフィニティー溶出液を1Mトリスバッファーで約4.5〜約8.5のpHに調整することができる。サンプル(アフィニティー溶出液)をイオン交換カラムにロードした後、適切な緩衝液を使用してカラムを平衡化することができる。適切な緩衝液の1例はpH約4.5〜約8のトリス/NaClバッファーである。平衡化後、カラムにアフィニティー溶出液をロードすることができる。ロード後、カラムを適切な緩衝液で1回以上洗浄することができる。適切な緩衝液の1例は平衡化バッファー自体である。例えば吸光度(OD280)が約0.2AUよりも上昇すると、フロースルー捕集を開始することができる。
【0012】
ある実施形態では、一次回収後又はアフィニティークロマトグラフィー工程の不在下に第1及び第2イオン交換工程を実施する。このような態様の所定のものでは、第1イオン交換工程の前、2回のイオン交換工程の間、又はその両方にイオン交換サンプルを中間濾過工程に供する。ある態様において、この濾過工程は捕集液限外濾過/透析濾過(「UF/DF」)を含む。特に、このような濾過は抗IL−13抗体及びその抗原結合部分の濃縮と緩衝液交換を容易にする。
【0013】
本発明のある実施形態は、1以上の疎水性相互作用クロマトグラフィー(「HIC」)工程を含む方法を提供する。適切なHICカラムは固定相に疎水基を含むカラムである。このようなカラムの非限定的な1例はPhenyl HP Sepharose(登録商標)カラムである。ある状況においては、抗IL−13抗体は単離/精製工程中に凝集物を形成する。1以上のHIC工程を含むことにより、このような凝集物を低減又は排除し易くする。HICは更に不純物の除去にも役立つ。ある実施形態においては、HIC工程は抗IL−13抗体(又はその凝集物)と疎水性カラムの相互作用を促進するために高塩濃度緩衝液を利用する。その後、低濃度塩を使用して抗IL−13抗体を溶出させることができる。
【0014】
ある実施形態では、限定されないが、Ultipor DV50(登録商標)フィルター(Pall Corporation,East Hills,N.Y.)等のウイルス除去フィルターを使用してHIC溶出液を濾過する。このような態様ではViresolve(登録商標)フィルター(Millipore,Billerica,Mass.);Zeta Plus VR(登録商標)フィルター(CUNO;Meriden,Conn.);及びPlanova(登録商標)フィルター(Asahi Kasei Pharma,Planova Division,Buffalo Grove,Ill.)等の他のフィルターも使用することができる。
【0015】
ある実施形態においては、本発明は単離抗IL−13抗体又はその抗原結合部分と許容可能な担体を含有する1種以上の医薬組成物に関する。ある態様において、組成物は抗IL−13抗体以外に、更に1種以上の抗体又はその抗原結合部分を含有する。別の態様において、組成物は更に1種以上の薬剤を含有する。
【0016】
得られたサンプル生成物中の目的抗体の純度は、当業者に周知の方法(例えばサイズ排除クロマトグラフィー、Poros(登録商標)A HPLCアッセイ、HCP ELISA、プロテインA ELISA及びウェスタンブロット分析)を使用して分析することができる。
【図面の簡単な説明】
【0017】
【図1】抗IL−13抗体の非限定的な1例の重鎖及び軽鎖可変領域配列を示す。
【図2−1】設定点、インプロセス制御試験及び作用限界を含む典型的細胞培養工程流れ図を示す。
【図2−2】設定点、インプロセス制御試験及び作用限界を含む典型的細胞培養工程流れ図を示す。
【図3】代替細胞培養工程フローストラテジーの比較を示す。
【図4】設定点、インプロセス制御試験及び作用限界を含む一次回収・捕集クロマトグラフィー工程流れ図を示す。
【図5】代替一次回収・捕集フローストラテジーの比較を示す。
【図6−1】設定点、インプロセス制御試験及び作用限界を含む精密精製工程流れ図を示す。
【図6−2】設定点、インプロセス制御試験及び作用限界を含む精密精製工程流れ図を示す。
【図7】代替精密精製フローストラテジーの比較を示す。
【発明を実施するための形態】
【0018】
本発明は、IL−13と結合する抗体に関する。ある態様において、本発明は、ヒトIL−13と結合する単離抗体又はその抗原結合部分に関する。本発明の単離抗IL−13抗体は、臨床環境及び研究・開発で使用することができる。本発明は、抗IL−13抗体又はその抗原結合部分の精製方法にも関する。本発明の関連で精製することができる適切な抗IL−13抗体はその開示内容全体を本願に援用するPCT出願第PCT/US2007/019660号に開示されており、その後、ABT−308として同定された抗体を含む。典型的な抗IL−13抗体重鎖及び軽鎖配列を図1に示す。本発明は、本願に記載する抗IL−13抗体又はその抗原結合部分を含有する医薬組成物にも関する。
【0019】
限定するものではないが、分かり易くするために、以下の詳細な説明は次の項目に分けて説明する。
1.定義;
2.抗体作製;
3.抗体産生;
4.抗体精製;
5.サンプル純度の検定方法;
6.付加的修飾;
7.医薬組成物;及び
8.抗体使用。
【0020】
1.定義;
本発明を理解し易くするために、先ず所定の用語を定義する。
【0021】
「抗体」なる用語は、ジスルフィド結合により相互に結合された2本の重(H)鎖と2本の軽(L)鎖の4本のポリペプチド鎖から構成される免疫グロブリン分子を包含する。各重鎖は、重鎖可変領域(本願ではHCVR又はVHと略称する)と重鎖定常領域(CH)から構成される。重鎖定常領域は、CH1、CH2及びCH3の3個のドメインから構成される。各軽鎖は、軽鎖可変領域(本願ではLCVR又はVLと略称する)と軽鎖定常領域から構成される。軽鎖定常領域は、1個のドメインCLから構成される。VH及びVL領域は更に相補性決定領域(CDR)と呼ばれる超可変領域とその間に配置されたフレームワーク領域(FR)と呼ばれる保存度の高い領域に区分される。各VH及びVLはアミノ末端からカルボキシ末端に向かってFR1、CDR1、FR2、CDR2、FR3、CDR3、FR4の順に配置された3個のCDRと4個のFRから構成される。
【0022】
抗体の「抗原結合部分」(又は抗体部分」)なる用語は、抗原(例えばhIL−13)と特異的に結合する能力を保持する抗体の1個以上のフラグメントを包含する。抗体の抗原結合機能は、全長抗体のフラグメントにより実施可能であることが分かっている。抗体の「抗原結合部分」なる用語に含まれる結合フラグメントの例としては、(i)VL、VH、CL及びCH1ドメインから構成される1価フラグメントであるFabフラグメント;(ii)ヒンジ領域でジスルフィド架橋により結合された2個のFabフラグメントから構成される2価フラグメントであるF(ab’)2フラグメント;(iii)VHドメインとCH1ドメインから構成されるFdフラグメント;(iv)抗体の単一アームのVLドメインとVHドメインから構成されるFvフラグメント;(v)VHドメインから構成されるdAbフラグメント(その教示内容全体を本願に援用するWard et al.,(1989)Nature 341:544−546);並びに(vi)単離相補性決定領域(CDR)が挙げられる。更に、Fvフラグメントの2個のドメインVL及びVHは別々の遺伝子によりコードされるが、VL領域とVH領域が対合して1価分子を形成する1本の蛋白鎖(1本鎖Fv(scFv)と言う;例えばその教示内容全体を本願に援用するBird et al.(1988)Science 242:423−426;及びHuston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883参照)としての作製を可能にする合成リンカーにより組換え法を使用して結合することができる。このような1本鎖抗体も抗体の「抗原結合部分」なる用語に含むものとする。ダイアボディ等の他の形態の1本鎖抗体も含む。ダイアボディは、VH領域とVL領域が1本のポリペプチド鎖で発現される2価の二重特異性抗体であるが、非常に短いリンカーを使用するため、同一鎖の2領域間で対合することができず、これらの領域は別の鎖の相補性領域と対合し、2個の抗原結合部位を形成する(例えばその教示内容全体を本願に援用するHolliger,P.,et al.(1993)Proc.Natl.Acad.Sci.USA 90:6444−6448;Poljak,R.J.,et al.(1994)Structure 2:1121−1123参照)。更に、抗体又はその抗原結合部分は、抗体又は抗体部分と1種以上の他の蛋白質又はペプチドの共有的又は非共有的結合により形成される大きな免疫接着分子の一部でもよい。このような免疫接着分子の例としては、ストレプトアビジンコア領域を使用した四量体scFv分子の作製(その教示内容全体を本願に援用するKipriyanov,S.M.,et al.(1995)Human Antibodies and Hybridomas 6:93−101)や、システイン残基、マーカーペプチド及びC末端ポリヒスチジンタグを使用した2価ビオチン化scFv分子の作製(その教示内容全体を本願に援用するKipriyanov,S.M.,et al.(1994)Mol.Immunol.31:1047−1058)が挙げられる。Fab及びF(ab’)2フラグメント等の抗体部分は、夫々全長抗体のパパイン又はペプシン消化等の慣用技術を使用して全長抗体から作製することができる。更に、抗体、抗体部分及び免疫接着分子は本願に記載するように、標準組換えDNA技術を使用して取得することができる。ある態様において、抗原結合部分は競合領域又は競合領域対である。
【0023】
本願で使用する「ヒトインターロイキン13」(本願ではhIL−13又はIL−13と略称する)なる用語は、活性化T細胞(Zurawski and de Vries,1994 Immunol Today 15 19−26)からクローニングされ、Th2系統の活性化T細胞により産生される17kDa糖蛋白質を意味する。Th0及びTh1 CD4+T細胞、CD8+T細胞、並びに数種の非T細胞集団(例えば肥満細胞)もIL−13を産生する(Zurawski and de Vries,1994 Immunol Today 15 19−26)。IL−13の機能としてとしては、ヒトB細胞におけるIgEへの免疫グロブリンアイソタイプスイッチの促進(Punnonen,Aversa et al.1993 Proc Natl Acad Sci U S A 90 3730−4)と、ヒト及びマウスの両者における炎症性サイトカイン産生の抑制(de Waal et al.,1993 J Immunol 151 6370−81;Doherty et al.,1993 J Immunol 151 7151−60)が挙げられる。IL−13はIL−13Rα1及びIL−13Rα2と呼ばれる細胞表面受容体と結合する。IL−13Rα1受容体は低親和性(KD〜10nM)でIL−13と相互作用した後、IL−4Rを動員し、高親和性(KD〜0.4nM)シグナル伝達ヘテロ二量体受容体複合体を形成する(Aman et al.,1996 J Biol Chem 271 29265−70;Hilton et al.,1996 Proc Natl Acad Sci U S A 93 497−501)。IL−4R/IL−13Rα1複合体はB細胞、単球/マクロファージ、樹状細胞、好酸球、好塩基球、線維芽細胞、内皮細胞、気道上皮細胞及び気道平滑筋細胞等の多数の細胞種で発現される(Graber et al,1998 Eur J Immunol 28 4286−98;Murata et al.,1998 Int Immunol 10 1103−10;Akaiwa et al.,2001 Cytokine 13 75−84)。IL−13Rα1/IL−4R受容体複合体のライゲーションの結果、シグナル伝達性転写因子(STAT6)及びインスリン受容体基質−2(IRS−2)経路を含む各種シグナル伝達経路が活性化される(Wang et al.,1995 Blood 864218−27;Takeda et al.,1996 J Immunol 157 3220−2)。IL−13Rα2鎖自体はIL−13に対する親和性が高く(KD〜0.25〜0.4nM)、IL−13との結合を負に制御するデコイ受容体として機能する(Donaldson,Whitters et al.1 98 J Immunol 161 2317−24)と同時に、マクロファージと恐らく他の細胞種でAP−1経路を介してTGF−b合成及び線維化を誘導するシグナル伝達受容体としても機能する(Fichtner−Feigl et al.,2006 Nat Med 12 99−106)。IL−13をコードする核酸はGenBank Accession No.NM_002188として入手可能であり、ポリペプチド配列はGenBank Accession No.NP_002179として入手可能である。ヒトIL−13なる用語は、標準組換え発現法により作製することができる組換えヒトIL−13(rh IL−13)を包含するものとする。
【0024】
「Kabatナンバリング」、「Kabat定義」及び「Kabatラベリング」なる用語は、本願では同義に使用する。これらの用語は、当分野で認識されている通り、抗体又はその抗原結合部分の重鎖及び軽鎖可変領域の他のアミノ酸残基よりも可変性(即ち超可変性)のアミノ酸残基のナンバリングシステムを意味する(その教示内容全体を本願に援用するKabat et al.(1971)Ann.NY Acad,Sci.190:382−391及びKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242)。重鎖可変領域では、超可変領域はCDR1がアミノ酸31〜35位、CDR2がアミノ酸50〜65位、CDR3がアミノ酸95〜102位に位置する。軽鎖可変領域では、超可変領域はCDR1がアミノ酸24〜34位、CDR2がアミノ酸50〜56位、CDR3がアミノ酸89〜97位に位置する。
【0025】
「ヒト抗体」なる用語は、Kabatらに記載されているように、ヒト生殖細胞系列免疫グロブリン配列に対応する可変領域と定常領域をもつ抗体を包含する(Kabat,et al.(1991)Sequences of proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)。本発明のヒト抗体は、ヒト生殖細胞系列免疫グロブリン配列によりコードされないアミノ酸残基(例えばランダムもしくは部位特異的突然変異誘発によりインビトロ又は体細胞突然変異によりインビボで導入される突然変異)を例えばCDR、特にCDR3に含むことができる。突然変異は、「選択的突然変異誘発アプローチ」を使用して導入することができる。ヒト抗体は少なくとも1カ所をアミノ酸残基(例えばヒト生殖細胞系列免疫グロブリン配列によりコードされない活性強化アミノ酸残基)で置換することができる。ヒト抗体は20カ所までをヒト生殖細胞系列免疫グロブリン配列に含まれないアミノ酸残基で置換することができる。他の態様では、10カ所まで、5カ所まで、3カ所まで又は2カ所までが置換されている。1態様において、これらの置換はCDR領域内に存在する。他方、本願で使用する「ヒト抗体」なる用語は、別の哺乳動物種(例えばマウス)の生殖細胞系列に由来するCDR配列をヒトフレームワーク配列に移植した抗体を包含するものではない。
【0026】
「選択的突然変異誘発アプローチ」なる用語は、少なくとも1カ所の適切な選択的突然変異誘発位置、超変異位置及び/又は接触位置のCDRアミノ酸を選択し、個々に突然変異させることにより、抗体の活性を改善する方法を包含する。「選択的に突然変異された」ヒト抗体とは、選択的突然変異誘発アプローチを使用して選択された位置に突然変異を含む抗体である。別の態様において、選択的突然変異誘発アプローチは抗体の重鎖可変領域のCDR1、CDR2もしくはCDR3(以下、夫々H1、H2及びH3と言う)、又は軽鎖可変領域のCDR1、CDR2もしくはCDR3(以下、夫々L1、L2及びL3と言う)における選択された個々のアミノ酸残基を優先的に突然変異させる方法を提供するものとする。アミノ酸残基は、選択的突然変異誘発位置、接触位置又は超変異位置から選択することができる。個々のアミノ酸は、軽鎖又は重鎖可変領域におけるそれらの位置に基づいて選択される。当然のことながら、超変異位置は接触位置でもよい。ある態様において、選択的突然変異誘発アプローチは「標的アプローチ」である。「標的アプローチ」なる用語は、抗体の重鎖可変領域のCDR1、CDR2もしくはCDR3又は軽鎖可変領域のCDR1、CDR2もしくはCDR3における選択された個々のアミノ酸残基を標的として突然変異させる方法(例えば「グループ標的アプローチ」又は「CDR標的アプローチ」)を包含するものとする。「グループ標的アプローチ」では、標的の優先順にグループI(L3及びH3を含む)、II(H2及びL1を含む)及びIII(L2及びH1を含む)を含む選択的突然変異の標的として特定グループにおける個々のアミノ酸残基を選択する。「CDR標的アプローチ」では、H3、L3、H2、L1、H1及びL2の標的の優先順で特定CDRにおける個々のアミノ酸残基を選択的突然変異の標的として選択する。選択されたアミノ酸残基を例えば少なくとも2個の他のアミノ酸残基に突然変異させ、抗体の活性に及ぼす突然変異の影響を判定する。活性は、抗体の結合特異性/親和性、及び/又は抗体の中和力価の変化として測定される。当然のことながら、選択的突然変異誘発アプローチは、ファージディスプレイ、ヒトIgG生殖細胞系列遺伝子をもつトランスジェニック動物、ヒトB細胞から単離されたヒト抗体を含む任意起源に由来する任意抗体の最適化に使用することができる。選択的突然変異誘発アプローチは、ファージディスプレイ技術を使用してそれ以上最適化することができない抗体で使用することができる。当然のことながら、ファージディスプレイ、ヒトIgG生殖細胞系列遺伝子をもつトランスジェニック動物、ヒトB細胞から単離されたヒト抗体を含む任意起源に由来する抗体を選択的突然変異誘発アプローチの前又は後に復帰突然変異に供することができる。
【0027】
「組換えヒト抗体」なる用語は、組換え手段により作製、発現、創製又は単離したヒト抗体を包含し、例えば、宿主細胞にトランスフェクトした組換え発現ベクターを使用して発現させた抗体、組換えコンビナトリアルヒト抗体ライブラリーから単離した抗体、ヒト免疫グロブリン遺伝子を導入したトランスジェニック動物(例えばマウス)から単離した抗体(例えばその教示内容全体を本願に援用するTaylor,L,D.,et al.(1992)Nucl.Acids Res.20:6287−6295参照)、あるいはヒト免疫グロブリン遺伝子配列を他のDNA配列にスプライスする操作を含む他の任意手段により作製、発現、創製又は単離した抗体が挙げられる。このような組換えヒト抗体は、可変領域と定常領域がヒト生殖細胞系列免疫グロブリン配列に由来する(Kabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)。しかし、ある実施形態では、このような組換えヒト抗体をインビトロ突然変異誘発(又は、ヒトIg配列を導入したトランスジェニック動物を使用する場合には、インビボ体細胞突然変異誘発)に供するため、組換え抗体のVH領域とVL領域のアミノ酸配列は、ヒト生殖細胞系列VH及びVL配列に由来し、これらの配列に近縁であるが、天然にヒト抗体生殖細胞系列レパートリー内にインビボで存在していなくてもよい配列である。他方、ある実施形態において、このような組換え抗体は選択的突然変異誘発アプローチ又は復帰突然変異又は両者の産物である。
【0028】
「単離抗体」とは、異なる抗原特異性をもつ他の抗体を実質的に含まない抗体を包含する(例えばhIL−13と特異的に結合する単離抗体はhIL−13以外の抗原と特異的に結合する抗体を実質的に含まない)。hIL−13と特異的に結合する単離抗体は他の種に由来するIL−13分子と結合する場合がある。更に、単離抗体は他の細胞材料及び/又は化学物質を実質的に含まない場合がある。
【0029】
「中和抗体」(又は「hIL−13活性を中和した抗体」)とは、hIL−13と結合する結果としてhIL−13の生物活性を阻害する抗体を包含する。hIL−13の生物活性のこの阻害はIL−13生物活性の1種以上の指標を測定することにより判定することができる。hIL−13生物活性のこれらの指標は、当分野で公知の数種の標準インビトロ又はインビボアッセイの1種以上により判定することができる。
【0030】
「活性」なる用語は、抗原に対する抗体(例えばIL−13抗原と結合する抗hIL−13抗体)の結合特異性/親和性及び/又は抗体(例えばhIL−13と結合してhIL−13の生物活性を阻害する抗hIL−13抗体)の中和力価等の活性を包含する。
【0031】
「表面プラズモン共鳴」なる用語は、例えばBIAcoreシステム(Pharmacia Biosensor AB,Uppsala,Sweden及びPiscataway,N.J.)を使用してバイオセンサーマトリックス内の蛋白質濃度の変化の検出によりリアルタイム生体特異的相互作用の分析を可能にする光学現象を意味する。更に詳細については、その教示内容全体を本願に援用するJonsson,U.,et al.(1993)Ann.Biol.Clin.51:19−26;Jonsson,U.,et al.(1991)Biotechniques 11:620−627;Johnsson,B.,el al.(1995)J.Mol.Recognit.8:125−131;及びJohnnson,B.,et al.(1991)Anal.Biochem.198:268−277参照。
【0032】
本願で使用する「Koff」なる用語は、抗体/抗原複合体から抗体が解離する解離速度定数を意味する。
【0033】
本願で使用する「Kd」なる用語は、特定抗体−抗原相互作用の解離定数を意味する。
【0034】
「核酸分子」なる用語は、DNA分子とRNA分子を包含する。核酸分子は1本鎖でも2本鎖でもよいが、ある態様では2本鎖DNAである。
【0035】
抗体又は抗体部分(例えばVH、VL、CDR3)(例えばhIL−13と結合するもの)をコードする核酸に関して本願で使用する「単離核酸分子」なる用語は、抗体又は抗体部分をコードするヌクレオチド配列がhIL−13以外の抗原と結合する抗体又は抗体部分をコードする他のヌクレオチド配列を含まない核酸分子を包含し、前記他の配列は、天然でヒトゲノムDNAにおいて前記核酸のフランキング配列であり得る。従って、例えば、抗hIL−13抗体のVH領域をコードする単離核酸は例えばhIL−13以外の抗原と結合する他のVH領域をコードする他の配列を含まない。「単離核酸分子」なる用語は、VH領域とVL領域がダイアボディの配列以外の他の配列を含まないダイアボディ等の2価の二重特異性抗体をコードする配列も包含するものとする。
【0036】
「組換え宿主細胞」(又は単に「宿主細胞」)なる用語は、組換え発現ベクターが導入された細胞を包含する。当然のことながら、このような用語は特定対象細胞のみならず、このような細胞の子孫も意味するものとする。後続世代には、突然変異又は環境影響により所定の変異が生じる場合があるので、このような子孫は実際に親細胞と一致しない場合もあるが、本願で使用する「宿主細胞」の用語の範囲に含まれる。
【0037】
本願で使用する「改変」なる用語は、抗体又はその抗原結合部分の1個以上のアミノ酸を変異させることを意味する。変異は1以上の位置でアミノ酸を付加、置換又は欠失させることにより生じさせることができる。変異はPCR突然変異誘発法等の公知技術を使用して生じさせることができる。
【0038】
本願で使用する「約」なる用語は、指定値の前後約10〜20%の範囲を意味する。所定状況では、当業者に自明の通り、指定値の種類により、「約」なる用語は指定値から10〜20%を上回る偏差又は下回る偏差を意味する場合もある。
【0039】
本願で使用する「ウイルス低減/不活性化」なる用語は、特定サンプルにおけるウイルス粒子数の低下(「低減」)と、特定サンプルにおけるウイルス粒子の活性(限定されないが、例えば感染性又は複製能)の低下(「不活性化」)を意味する。このようなウイルス粒子数及び/又は活性の低下は、約1%〜約99%のオーダーとすることができ、例えば約20%〜約99%、例えば約30%〜約99%、例えば約40%〜約99%、例えば約50%〜約99%、例えば約60%〜約99%、例えば約70%〜約99%、例えば約80%〜99%、例えば約90%〜約99%が挙げられる。所定の非限定的な態様において、精製抗体産物中にウイルスが存在する場合、その量はこのウイルスのID50(対象集団の50%に感染するウイルスの量)未満であり、このウイルスのID50の最大で10分の1、又はこのウイルスのID50の最大で100分の1、又はこのウイルスのID50の最大で1000分の1である。
【0040】
「接触位置」なる用語は、26種類の公知抗体−抗原構造の1種において抗原と接触するアミノ酸が抗体の重鎖可変領域又は軽鎖可変領域のCDR1、CDR2又はCDR3において占めるアミノ酸位置を包含する。抗体−抗原複合体の26種類の公知解明構造のいずれかにおけるCDRアミノ酸が抗原と接触するならば、このアミノ酸は接触位置を占めるとみなすことができる。接触位置は非接触位置よりも抗原と接触するアミノ酸により占められる確率が高い。ある態様において、接触位置は26種類の構造のうちの4種類以上(>1.5%)で抗原と接触するアミノ酸を含むCDR位置である。別の態様において、接触位置は25種類の構造のうちの9種類以上(>32%)で抗原と接触するアミノ酸を含むCDR位置である。
【0041】
2.抗体作製
本セクションで使用する「抗体」なる用語は、無傷抗体又はその抗原結合フラグメントを意味する。
【0042】
本発明の抗体は、各種技術により作製することができ、例えば、動物に目的抗原を免疫した後、従来のモノクローナル抗体法(例えばKohler and Milstein(1975)Nature 256:495の標準体細胞ハイブリダイゼーション法)を実施すればよい。原則として、体細胞ハイブリダイゼーション法が好ましいが、他のモノクローナル抗体作製技術(例えばBリンパ球のウイルス性又は発癌性形質転換)も利用できる。
【0043】
ハイブリドーマを作製するための動物システムの1例はマウスシステムである。ハイブリドーマ作製は非常によく知られた方法である。免疫プロトコルと融合用免疫脾細胞の単離技術は当分野で公知である。融合パートナー(例えばマウス骨髄腫細胞)と融合手順も公知である。
【0044】
抗体はヒト抗体、キメラ抗体又はヒト化抗体とすることができる。本発明のキメラ抗体又はヒト化抗体は上記のように作製した非ヒトモノクローナル抗体の配列に基づいて作製することができる。目的の非ヒトハイブリドーマから重鎖及び軽鎖免疫グロブリンをコードするDNAを取得し、標準分子生物学技術を使用して非マウス(例えばヒト)免疫グロブリン配列を含むように組換えることができる。例えば、キメラ抗体を創製するためには、当分野で公知の方法を使用してマウス可変領域をヒト定常領域と連結すればよい(例えばCabillyらの米国特許第4,816,567号参照)。ヒト化抗体を創製するためには、当分野で公知の方法を使用してマウスCDR領域をヒトフレームワークに挿入すればよい(例えばWinterの米国特許第5,225,539号、並びにQueenらの米国特許第5,530,101号、5,585,089号、5,693,762号及び6,180,370号参照)。
【0045】
非限定的な1態様において、本発明の抗体はヒトモノクローナル抗体である。IL−13に対するこのようなヒトモノクローナル抗体はマウス免疫系ではなくヒト免疫系の部分を導入したトランスジェニックマウス又はトランスクロモソミックマウスを使用して作製することができる。これらのトランスジェニックマウス及びトランスクロモソミックマウスとしては、本願でHuMAb Mouse(登録商標)(Medarex,Inc.)、KM Mouse(登録商標)(Medarex,Inc.)及びXenoMouse(登録商標)(Amgen)と呼ぶマウスが挙げられる。
【0046】
更に、ヒト免疫グロブリン遺伝子を発現する他のトランスクロモソミック動物システムも当分野で入手可能であり、本発明の抗体(例えば抗IL−13抗体)を産生させるために使用することができる。例えば、ヒト重鎖トランスクロモソームとヒト軽鎖トランスクロモソームを併有するマウス(「TC マウス」と呼ぶ)を使用することができ、このようなマウスはTomizuka et al.(2000)Proc.Natl.Acad.Sci.USA 97:722−727に記載されている。更に、ヒト重鎖及び軽鎖トランスクロモソームを導入したウシも当分野で記載されており(例えばKuroiwa et al.(2002)Nature Biotechnology 20:889−894及びPCT出願第WO2002/092812号)、本発明の抗IL−13抗体を産生させるために使用することができる。
【0047】
本発明の組換えヒト抗体としては、限定されないが、本願に開示する抗IL−13抗体、その抗原結合部分又は抗IL−13関連抗体が挙げられ、例えばヒトリンパ球に由来するmRNAから作製されたヒトVL及びVH cDNAを使用して作製された組換えコンビナトリアル抗体ライブラリー(例えばscFvファージディスプレイライブラリー)のスクリーニングにより単離することができる。このようなライブラリーの作製及びスクリーニング方法は当分野で公知である。このようなライブラリーの作製及びスクリーニング方法は当分野で公知である。ファージディスプレイライブラリーの作製用市販キット(例えばその教示内容全体を本願に援用するPharmacia組換えファージ抗体システム,カタログ番号27−9400−01;及びStratagene SurfZAPTMファージディスプレイキット,カタログ番号240612参照)に加え、抗体ディスプレイライブラリーを作製及びスクリーニングするために特に使用可能な方法及び試薬の例は例えばLadnerらの米国特許第5,223,409号;KangらのPCT公開第WO92/18619号;DowerらのPCT公開第WO91/17271号;WinterらのPCT公開第WO92/20791号;MarklandらのPCT公開第WO92/15679号;BreitlingらのPCT公開第WO93/01288号;McCaffertyらのPCT公開第WO92/01047号;GarrardらのPCT公開第WO92/09690号;Fuchs et al.(1991)Bio/Technology 9:1370−1372;Hay et al.(1992)Hum Antibod Hybridomas 3:81−85;Huse et al.(1989)Science 246:1275−1281;McCafferty et al.,Nature(1990)348:552−554;Griffiths et al.(1993)EMBO J 12:725−734;Hawkins et al.(1992)J Mol Biol 226:889−896;Clackson et al.(1991)Nature 352:624−628;Gram et al.(1992)PNAS 89:3576−3580;Garrard et al.(1991)Bio/Technology 9:1373−1377;Hoogenboom et al.(1991)Nuc Acid Res 19:4133−4137;及びBarbas et al.(1991)PNAS 88:7978−7982に記載されており、その教示内容全体を本願に援用する。
【0048】
本発明のヒトモノクローナル抗体は、免疫後にヒト抗体応答を生じることができるようにヒト免疫細胞を再構成したSCIDマウスを使用して作製することもできる。このようなマウスは例えばWilsonらの米国特許第5,476,996号及び5,698,767号に記載されている。
【0049】
ある実施形態においては、本発明の方法は、抗IL−13抗体及び抗体部分、抗IL−13関連抗体及び抗体部分、並びに抗IL−13抗体と同等の性質(例えば低い解離速度と高い中和能でhIL−13と高親和性結合する性質)をもつヒト抗体及び抗体部分を包含する。ある態様において、本発明はいずれも表面プラズモン共鳴法により測定した場合に約1×10−8M以下のKdと1×10−3s−1以下のKoff速度定数でhIL−13から解離する単離ヒト抗体又はその抗原結合部分による治療法を提供する。非限定的な特定態様において、本発明に従って精製した抗IL−13抗体は生理的条件下でABT−308とIL−13の結合を競合的に阻害する。
【0050】
本発明の更に別の態様では、少なくとも1種の定常領域介在性生物エフェクター機能を非改変抗体に比較して低下させるように抗体の定常領域を改変させるように、抗体又はそのフラグメント(限定されないが、例えば抗IL−13抗体又はそのフラグメント)を改変させることができる。Fc受容体との結合の低下を示すように本発明の抗体を改変させるためには、Fc受容体(FcR)相互作用に必要な特定領域で抗体の免疫グロブリン定常領域セグメントを突然変異させればよい(例えばその教示内容全体を本願に援用するCanfield and Morrison(1991)J.Exp.Med.173:1483−1491;及びLund et al.(1991)J.of Immunol.147:2657−2662参照)。抗体のFcR結合能の低下はFcR相互作用に依存する他のエフェクター機能(例えばオプソニン化及び貪食作用及び抗原依存性細胞傷害作用)も低下させる可能性がある。
【0051】
3.抗体産生
3.1 一般産生ストラテジー
本発明の抗体を発現させるためには、遺伝子を転写及び翻訳制御配列と機能的に連結するように、部分又は全長軽鎖及び重鎖をコードするDNAを1以上の発現ベクターに挿入する(例えばその教示内容全体を本願に援用する米国特許第6,914,128号参照)。この関連で「機能的に連結」なる用語はベクター内の転写及び翻訳制御配列が抗体遺伝子の転写と翻訳を制御するというその所期機能を発揮するように抗体遺伝子をベクターにライゲーションすることを意味する。発現ベクターと発現制御配列は、使用される発現宿主細胞と適合可能となるように選択される。抗体軽鎖遺伝子と抗体重鎖遺伝子は、別々のベクターに挿入してもよいが、より典型的には、両方の遺伝子を同一発現ベクターに挿入する。抗体遺伝子は、標準方法(例えば抗体遺伝子フラグメントとベクター上の相補的制限部位のライゲーション又は制限部位が存在しない場合には平滑末端ライゲーション)により発現ベクターに挿入される。抗体又は抗体関連軽鎖及び重鎖配列を挿入する前に、発現ベクターに予め定常領域配列を保持させてもよい。例えば、抗IL−13抗体又は抗IL−13抗体関連VH及びVL配列から全長抗体遺伝子を作製する1つのアプローチはVHセグメントをベクター内のCHセグメントと機能的に連結し、VLセグメントをベクター内のCLセグメントと機能的に連結するように、夫々重鎖定常領域と軽鎖定常領域を予めコードする発現ベクターにこれらの配列を挿入する方法である。上記に加え、又は上記の代わりに、組換え発現ベクターは宿主細胞からの抗体鎖の分泌を助長するシグナルペプチドをコードすることができる。シグナルペプチドを抗体鎖遺伝子のアミノ末端にインフレームで連結するように抗体鎖遺伝子をベクターにクローニングすることができる。シグナルペプチドは免疫グロブリンシグナルペプチド又は異種シグナルペプチド(即ち、非免疫グロブリン蛋白質に由来するシグナルペプチド)とすることができる。
【0052】
抗体鎖遺伝子に加え、本発明の組換え発現ベクターは、宿主細胞における抗体鎖遺伝子の発現を制御する1個以上の調節配列を保持することができる。「調節配列」なる用語は、プロモーター、エンハンサー及び抗体鎖遺伝子の転写又は翻訳を制御する他の発現制御エレメント(例えばポリアデニル化シグナル)を包含するものとする。このような調節配列は、例えばその教示内容全体を本願に援用するGoeddel;Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,CA(1990)に記載されている。当業者に自明の通り、調節配列の選択を含む発現ベクターの設計は、形質転換しようとする宿主細胞の選択、所望される蛋白質発現レベル等の因子により異なる。哺乳動物宿主細胞発現に適した調節配列としては、哺乳動物細胞で高レベルの蛋白質発現を誘導するウイルスエレメントが挙げられ、例えばサイトメガロウイルス(CMV)(例えばCMVプロモーター/エンハンサー)、サルウイルス40(SV40)(例えばSV40プロモーター/エンハンサー)、アデノウイルス(例えばアデノウイルス主要後期プロモーター(AdMLP))及びポリオーマに由来するプロモーター及び/又はエンハンサーが挙げられる。ウイルス調節エレメントとその配列に関する更に詳細な説明については、例えばその教示内容全体を本願に援用するStinskiの米国特許第5,168,062号、Bellらの米国特許第4,510,245号、及びSchaffnerらの米国特許第4,968,615号参照。
【0053】
抗体鎖遺伝子と調節配列に加え、本発明の組換え発現ベクターは、1個以上の他の配列(例えば宿主細胞におけるベクターの複製を調節する配列(例えば複製起点)及び/又は選択マーカー遺伝子)を保持することができる。選択マーカー遺伝子は、ベクターを導入した宿主細胞の選択を容易にする(例えばその教示内容全体を本願に援用するいずれもAxelらの米国特許第4,399,216号、4,634,665号及び5,179,017号参照)。例えば、典型的には選択マーカー遺伝子はベクターを導入した宿主細胞にG418、ハイグロマイシン又はメトトレキサート等の薬剤に対する耐性を付与する。適切な選択マーカー遺伝子としては、ジヒドロ葉酸還元酵素(DHFR)遺伝子(メトトレキサート選択/増幅下にdhfr−宿主細胞で使用)とneo遺伝子(G418選択用)が挙げられる。
【0054】
本発明の抗体又は抗体部分は、宿主細胞で免疫グロブリン軽鎖及び重鎖遺伝子を組換え発現させることにより作製することができる。抗体を組換え発現させるためには、抗体の免疫グロブリン軽鎖及び重鎖を宿主細胞で発現させ、宿主細胞を培養する培地中に分泌させるように、前記軽鎖及び重鎖をコードするDNAフラグメントを保持する1以上の組換え発現ベクターを宿主細胞にトランスフェクトし、培地から抗体を回収することができる。標準組換えDNA法を使用して抗体重鎖及び軽鎖遺伝子を得、これらの遺伝子を組換え発現ベクターに導入し、その教示内容全体を本願に援用するSambrook,Fritsch and Maniatis(eds),Molecular Cloning;A Laboratory Manual,Second Edition,Cold Spring Harbor,N.Y.,(1989)、Ausubel et al.(eds.)Current Protocols in Molecular Biology,Greene Publishing Associates(1989)、並びに米国特許第4,816,397号及び6,914,128号に記載されているもの等の宿主細胞にベクターを導入する。
【0055】
軽鎖と重鎖を発現させるためには、重鎖と軽鎖をコードする発現ベクターを標準技術により宿主細胞にトランスフェクトする。各種語形の「トランスフェクション」なる用語は外来DNAを原核又は真核宿主細胞に導入するために広く使用されている多様な技術を包含するものとし、例えばエレクトロポレーション、リン酸カルシウム沈殿法、DEAE−デキストラントランスフェクション等が挙げられる。理論的には、原核宿主細胞でも真核宿主細胞でも本発明の抗体を発現させることが可能であるが、真核細胞、特に哺乳動物細胞は適正に折り畳まれた免疫活性抗体を組立てて分泌する可能性が原核細胞よりも高いため、哺乳動物宿主細胞等の真核細胞で抗体を発現させると適切である。抗体遺伝子の原核細胞発現は、活性な抗体の高収率産生には有効でないと報告されている(その教示内容全体を本願に援用するBoss and Wood(1985)Immunology Today 6:12−13)。
【0056】
本願のベクターでDNAをクローニング又は発現させるのに適した宿主細胞は上記原核細胞、酵母細胞又は高等真核細胞である。この目的に適した原核生物としては、グラム陰性菌やグラム陽性菌等の真正細菌が挙げられ、例えばEscherichia(例えば大腸菌)、Enterobacter、Erwinia、Klebsiella、Proteus、Salmonella(例えばSalmonella typhimurium)、Serratia(例えばSerratia marcescans)及びShigella等の腸内細菌や、桿菌(例えばB.subtilis及びB.licheniformis(例えば1989年4月12日発行のDD 266,710に開示されているB.licheniformis 41P))、Pseudomonas(例えばP.aeruginosa)及びStreptomycesが挙げられる。適切な大腸菌クローニング宿主の1例は大腸菌294株(ATCC31,446)であるが、大腸菌B株、大腸菌X1776株(ATCC31,537)、及び大腸菌W3110株(ATCC27,325)等の他の株も適切である。これらの例は例示であり、非限定的である。
【0057】
原核生物に加え、糸状菌や酵母等の真核微生物もポリペプチドをコードするベクターの適切なクローニング又は発現宿主である。下等真核宿主微生物としてはSaccharomyces cerevisiaeないしパン酵母が最も広く使用されている。この他、Schizosaccharomyces pombe;Kluyveromyces宿主(例えばK.lactis、K.fragilis(ATCC12,424)、K.bulgaricus(ATCC16,045)、K.wickeramii(ATCC24,178)、K.waltii(ATCC56,500)、K.drosophilarum(ATCC36,906)、K.thermotolerans及びK.marxianus);Yarrowia(EP402,226);Pichia pastoris(EP183,070);Candida;Trichoderma reesia(EP244,234);Neurospora crassa;Schwanniomyces(例えばSchwanniomyces occidentalis);及び糸状菌(例えばNeurospora、Penicillium、Tolypocladium及びAspergillus宿主(例えばA.nidulans及びA.niger))等の多数の他の属、種及び株も広く入手可能であり、本願で有用である。
【0058】
グリコシル化抗体の発現に適した宿主細胞は、多細胞生物に由来する。無脊椎動物細胞の例としては植物細胞と昆虫細胞が挙げられる。多数のバキュロウイルス株及び変異株と、Spodoptera frugiperda(ヨトウムシ)、Aedes aegypti(蚊)、Aedes albopictus(蚊)、Drosophila melanogaster(果実蝿)、及びBombyx mori等の宿主に由来する対応する許容昆虫宿主細胞が同定されている。トランスフェクション用の各種ウイルス株(例えばAutographa californica NPVのL−l変異株及びBombyx mori NPVのBm−5株)が公共入手可能であり、このようなウイルスを特にSpodoptera frugiperda細胞のトランスフェクション用として、本発明に従って本願のウイルスとして使用することができる。ワタ、トウモロコシ、ジャガイモ、ダイズ、ペチュニア、トマト及びタバコの植物細胞培養物も宿主として利用できる。
【0059】
本発明の組換え抗体の発現に適した哺乳動物宿主細胞としては、チャイニーズハムスター卵巣(CHO細胞)(例えばその教示内容全体を本願に援用するUrlaub and Chasin,(1980)PNAS USA 77:4216−4220に記載のdhfr−CHO細胞を例えばKaufman and Sharp(1982)Mol.Biol.159:601−621に記載されているようにDHFR選択マーカーと併用する)、NS0骨髄腫細胞、COS細胞及びSP2細胞が挙げられる。抗体遺伝子をコードする組換え発現ベクターを哺乳動物宿主細胞に導入する場合には、宿主細胞で抗体を発現させるため又は宿主細胞を増殖させる培養培地に抗体を分泌させるために十分な時間にわたって宿主細胞を培養することにより抗体を産生させる。有用な哺乳動物宿主細胞株の他の例はSV40で形質転換させたサル腎細胞CV1株(COS−7,ATCC CRL 1651);ヒト胎児腎細胞株(293又は懸濁培養増殖用にサブクローニングした293細胞,Graham et al.,J.Gen Virol:36:59(1977));ベビーハムスター腎細胞(BHK,ATCC CCL 10);チャイニーズハムスター卵巣細胞/−DHFR(CHO,Urlaub et al.,Proc.Natl.Acad.Sci.USA 77:4216(1980));マウスセルトリ細胞(TM4,Mather,Biol.Reprod.23:243−251(1980));サル腎細胞(CV1 ATCC CCL 70);アフリカミドリザル腎細胞(VERO−76,ATCC CRL−1587);ヒト子宮頸癌細胞(HELA,ATCC CCL 2);イヌ腎細胞(MDCK,ATCC CCL 34);バッファローラット肝細胞(BRL 3A,ATCC CRL 1442);ヒト肺細胞(W138,ATCC CCL 75);ヒト肝細胞(Hep G2,HB 8065);マウス乳癌(MMT 060562,ATCC CCL51);TRI細胞(Mather et al.,Annals N.Y.Acad.Sci.383:44−68(1982));MRC 5細胞;FS4細胞;及びヒト肝細胞癌株(Hep G2)であり、上記文献はその教示内容全体を本願に援用する。
【0060】
宿主細胞を上記抗体産生用発現又はクローニングベクターで形質転換させ、プロモーターの誘導、形質転換細胞の選択又は所望配列をコードする遺伝子の増幅の必要に応じて改変した慣用栄養培地で培養する。
【0061】
抗体を産生させるために使用する宿主細胞は、各種培地で培養することができる。ハムF10(登録商標)(Sigma)、最少必須培地Minimal Essential Medium(登録商標)((MEM)(Sigma))、RPMI−1640(Sigma)及びダルベッコ改変イーグル培地Dulbecco’s Modified Eagle’s Medium(登録商標)((DMEM),Sigma)等の市販培地が宿主細胞を培養するのに適している。更に、その教示内容全体を本願に援用するHam et al.,Meth.Enz.58:44(1979),Barnes et al.,Anal.Biochem.102:255(1980)、米国特許第4,767,704号、4,657,866号、4,927,762号、4,560,655号もしくは5,122,469号、WO90/03430、WO87/00195、又は米国再発行特許第30,985号に記載されている培地の任意のものを宿主細胞の培養培地として使用することができる。必要に応じてホルモン及び/又は他の成長因子(例えばインスリン、トランスフェリン又は上皮成長因子)、塩(例えば塩化ナトリウム、カルシウム、マグネシウム及びリン酸塩)、緩衝液(例えばHEPES)、ヌクレオチド(例えばアデノシン及びチミジン)、抗生物質(例えばゲンタマイシン薬物)、(通常ではマイクロモル範囲の最終濃度で存在する無機化合物として定義される)微量元素、及びグルコース又は同等のエネルギー源をこれらの培地の任意のものに添加することができる。当業者に公知の任意の他の必要な助剤も適当な濃度で添加することができる。温度、pH等の培養条件は発現に選択される宿主細胞で従来使用されている条件であり、当業者に容易に理解されよう。
【0062】
無傷の抗体の部分(例えばFabフラグメントやscFv分子)を作製するために宿主細胞を使用することもできる。当然のことながら、上記手順の変形も本発明の範囲内である。例えば、ある実施形態では、本発明の抗体の軽鎖又は重鎖の(両方ではなく)いずれかをコードするDNAを宿主細胞にトランスフェクトすることが望ましい場合がある。IL−13、特にhIL−13との結合に不要な軽鎖及び重鎖のいずれか又は両方をコードするDNAの一部又は全部を除去するために組換えDNA技術を使用してもよい。このような短縮型DNA分子から発現される分子も本発明の抗体に含まれる。更に、本発明の抗体を標準化学架橋法により第2の抗体と架橋することにより、一方の重鎖と一方の軽鎖が本発明の抗体であり、他方の重鎖及び軽鎖がIL−13以外の抗原に特異的である二機能性抗体を作製することができる。
【0063】
本発明の抗体又はその抗原結合部分の組換え発現に適したシステムでは、抗体重鎖及び抗体軽鎖の両方をコードする組換え発現ベクターをリン酸カルシウムトランスフェクションによりdhfr−CHO細胞に導入する。組換え発現ベクターの内側で、遺伝子の高レベルの転写を誘導するように、抗体重鎖及び軽鎖遺伝子を各々CMVエンハンサー/AdMLPプロモーター調節エレメントと機能的に連結する。組換え発現ベクターは更にメトトレキサート選択/増幅を使用してベクターをトランスフェクトしたCHO細胞の選択を可能にするDHFR遺伝子を保持する。選択した形質転換宿主細胞を培養し、抗体重鎖及び軽鎖を発現させ、無傷抗体を培養培地から回収する。標準分子生物学技術を使用して組換え発現ベクターを作製し、宿主細胞にトランスフェクトし、形質転換細胞を選択し、宿主細胞を培養し、抗体を培養培地から回収する。
【0064】
組換え技術を使用する場合には、抗体をペリプラズム空間で細胞内産生させることもできるし、培地に直接分泌させることもできる。ある態様において、抗体を細胞内産生させる場合には、第1段階として、宿主細胞又は(例えば均質化による)溶解細胞のいずれかであるかに関係なく、粒状破片を例えば遠心又は限外濾過により除去することができる。抗体を培地に分泌させる場合には、市販蛋白質濃縮フィルター(例えばAmicon(登録商標)又はMillipore Pellicon(登録商標)限外濾過ユニット)を使用して、先ずこのような発現システムからの上清を濃縮することができる。
【0065】
本発明の方法の前に、細胞破片から抗体を精製する手順は、まず第1に抗体の発現部位によって異なる。抗体によっては細胞から周囲の増殖培地に直接分泌させることもできるし、細胞内に産生するものもある。後者抗体では、精製工程の第1段階は典型的には細胞の溶解を含み、これは機械的剪断、浸透圧ショック法又は酵素処理を含む各種方法により実施できる。このような破砕により細胞の全内容物はホモジネート中に放出され、更に、寸法が小さいために除去しにくい細胞以下のフラグメントが生成される。これらは一般に分画遠心法又は濾過により除去される。抗体が分泌される場合には、一般に先ず市販蛋白質濃縮フィルター(例えばAmicon(登録商標)又はMillipore Pellicon(登録商標)限外濾過ユニット)を使用してこのような発現システムからの上清を濃縮する。抗体が培地中に分泌される場合には、組換え宿主細胞も、例えば、タンジェンシャルフロー濾過により細胞培養培地から分離することができる。本発明の抗体精製法を使用して抗体を更に培養培地から回収することができる。
【0066】
3.2.典型的産生ストラテジー
ある実施形態において、抗IL−13抗体産生の初期工程は、スピナーフラスコとバイオウェーブバッグの操作を使用し、抗IL−13抗体を発現するCHO細胞を凍結バイアル1本から110Lシードバイオリアクターの接種に望ましいバイオマスまで増殖させる。マスターセルバンクCHO細胞の凍結バイアルを解凍し、増殖培地(SR−512)に加え、遠心する。細胞を増殖培地に再懸濁し、容積を増加させながら使い捨てスピナーフラスコ、振盪フラスコ及び/又はバイオウェーブバッグで37℃及び5% CO2にて増殖させる。シードバイオリアクターに接種する前に20Lウェーブバッグ2個を使用して最終細胞塊増殖を最大にする。両方の20Lウェーブバッグからの細胞密度が約15〜17日目に生存細胞≧2.0×106個/mLに達したら、更に増殖させるために、増殖培地SR−520を充填した110シードバイオリアクターに培養液を移す。接種後、目標温度を37℃とし、pHを7.1の目標値に設定し、NaOHの添加とCO2スパージングにより制御する。空気と酸素のスパージングによりバイオリアクター内の溶存酸素(DO)を40%の目標値に制御する。約2〜4日後に細胞密度が生存細胞≧2.6×106個/mLに達したら、培養液を3000L産生用バイオリアクターに移す。
【0067】
ある実施形態では、3000L産生用バイオリアクターの部分フィルを使用して細胞培養物を更に増殖させる。先ず、リアクターに増殖培地(SR−520)を仕込み、110Lシードバイオリアクターからのバッチを接種する。このショートフィル段階中は、温度、溶存酸素及びpHを夫々37℃、40%及び7.1に制御する。培養pHはCO2スパージングとNaOH添加により制御する。典型的には、細胞は生存細胞≧1.6×106個/mLの産生段階密度に達する前に2〜4日間増殖する。
【0068】
産生培地SR−521(1950L)を3000Lバイオリアクター内の細胞培養液に加え、産生段階を開始する。消泡剤Cを加え、発泡を抑える。CO2スパージングとNaOH添加のオンオフにより培養pHを6.9の目標値に制御する。温度と溶存酸素を夫々35℃及び40%の目標値に制御する。バイオリアクター内のDOは先ず空気スパージングにより所望値に制御し、必要に応じて純酸素を補充する。ある実施形態では、生存細胞密度が≧3.0×106個/mLに達したら、温度を33℃の目標値まで下げ、pHとDOを夫々6.9と40%の目標値に維持するが、他の態様では、35℃の目標値に維持する。必要に応じてグルコース(SR−334)を添加する。細胞生存率が≦50%まで低下したら、培養液を回収し、以下に概説するように精製する。
【0069】
4.抗体精製
4.1 抗体精製概論
本発明は、抗体と少なくとも1種のHCPを含有する混合物から精製(ないし「HCP低減」)抗体調製物を生成する方法を提供する。本発明は、最終精製調製物の浸出プロテインAを低減する方法も提供する。本発明の精製工程は、上記方法及び当分野の慣用方法を使用して抗体が産生された時点で分離工程から開始する。表1は精製スキームの1態様を要約する。このスキームの変形も考えられ、限定されないが、プロテインAアフィニティークロマトグラフィー工程を省略したり、イオン交換工程の順序を逆にした変形が考えられ、本発明の範囲に含まれる。
【0070】
【表1】
【0071】
抗体を含有する清澄化溶液又は混合物が得られたら、イオン交換分離工程と疎水性相互作用分離工程を含む異なる精製技術の組み合わせを使用し、細胞により産生された他の蛋白質(例えばHCP)からの抗体の分離を実施する。分離工程は蛋白質の電荷、疎水度又は寸法に基づいて蛋白質混合物を分離する。本発明のある態様では、カチオン、アニオン及び疎水性相互作用を含むクロマトグラフィーを使用して分離を実施する。これらの技術の各々に数種の異なるクロマトグラフィー樹脂が利用可能であり、該当する特定蛋白質に合わせて精製スキームを正確に適応させることができる。分離方法の各々の要点は、蛋白質を異なる速度でカラムに流し、更にカラムを通過するにつれて増加する物理的分離を達成すること、又は蛋白質を選択的に分離媒体に接着させた後、異なる溶媒により分画溶出させることである。場合により、不純物がカラムと特異的に接着し、抗体が接着しない場合、即ち抗体がフロースルー中に存在する場合には、抗体を不純物から分離する。
【0072】
上記のように、精製スキームの正確な適応は、精製しようとする蛋白質の要件に依存する。ある実施形態では、1種以上のHCPから抗体を分離するために本発明の分離工程を利用する。本願に記載する方法を使用して有効に精製することができる抗体としては、限定されないが、ヒトIgA1、IgA2、IgD、IgE、IgG1、IgG2、IgG3、IgG4及びIgM抗体が挙げられる。ある実施形態において、本発明の精製ストラテジーはプロテインAアフィニティークロマトグラフィーの使用を除外し、例えばIgG3抗体はプロテインAと効率的に結合しないので、IgG3抗体の精製がこれに該当する。精製スキームを個別に適応させる他の因子としては、限定されないが、プロテインAはFc領域と結合するので(例えばそのFabフラグメントに対する全長抗体のコンテキストにおける)Fc領域の有無、目的抗体の作製に利用される特定生殖細胞系列配列、及び抗体のアミノ酸組成(例えば抗体の一次配列と分子の総電荷/疎水度)が挙げられる。1種以上の特徴を共有する抗体はこの特徴を利用するように適応させた精製ストラテジーを使用して精製することができる。
【0073】
4.2 一次回収
本発明の精製方法の初期工程は、サンプルマトリックスからの抗体の第1段階の清澄化及び一次回収を含む。更に、一次回収工程はサンプルマトリックスに存在する可能性のあるウイルスを低減又は不活性化する点でもあり得る。例えば、熱不活性化(低温殺菌)、pH不活性化、溶媒/デタージェント処理、UV及びγ線照射、並びに所定の化学不活性化剤(例えばβ−プロピオラクトン又は例えばその教示内容全体を本願に援用する米国特許第4,534,972号におけるように銅フェナントロリン)の添加を含む各種ウイルス低減/不活性化方法の任意の1種以上を精製の一次回収段階中に使用することができる。本発明のある実施形態では、一次回収段階中にサンプルマトリックスをpHウイルス低減/不活性化に供する。
【0074】
pHウイルス低減/不活性化方法としては、限定されないが、混合物を低pHで所定時間インキュベートした後、pHを中和させ、粒状物を濾過により除去する。ある実施形態では、混合物を約2〜5のpH、約3〜4のpH、限定されないが、例えば約3.5のpHでインキュベートする。サンプル混合物のpHは任意の適切な酸により低下させることができ、限定されないが、クエン酸、酢酸、カプリル酸又は他の適切な酸が挙げられる。pHレベルの選択は抗体産物と緩衝液成分の安定性プロファイルに大きく依存する。低pHウイルス低減/不活性化中の目的抗体の品質がpHと低pHインキュベーション時間に影響されることは知られている。ある実施形態において、低pHインキュベーション時間は0.5時間〜2時間となり、限定されないが、例えば0.5時間〜1.5時間、限定されないが、例えば約1時間が挙げられる。ウイルス低減/不活性化は蛋白質濃度に依存し、高濃度の低減/不活性化が制限される可能性があるが、その他にこれらの同一パラメーターにも依存する。従って、所望レベルのウイルス低減/不活性化を達成するように蛋白質濃度、pH及び低減/不活性化時間の適正なパラメーターを選択することができる。
【0075】
ある実施形態では、適切なフィルターの使用によりウイルス低減/不活性化を達成することができる。適切なフィルターの非限定的な1例はPall Corporation製品であるUltipor DV50(登録商標)フィルターである。本発明のある実施形態は、一次回収段階中にこのような濾過を利用するが、他の態様では、精製工程の他の段階(例えば最後から2番目又は最終精製工程)に利用する。ある実施形態では、別のフィルターをウイルス低減/不活性化に利用し、限定されないが、Viresolve(登録商標)フィルター(Millipore,Billerica,Mass.);Zeta Plus VR(登録商標)フィルター(CUNO;Meriden,Conn.);及びPlanova(登録商標)フィルター(Asahi Kasei Pharma,Planova Division,Buffalo Grove,Ill.)が挙げられる。
【0076】
ウイルス低減/不活性化を利用する態様では、必要に応じて後続精製工程に備えてサンプル混合物を調整することができる。例えば、低pHウイルス低減/不活性化後に、精製工程を続ける前にサンプル混合物のpHを典型的にはより中性のpH(例えば約4.5〜約8.5、限定されないが、例えば約4.9)に調整する。更に、所望の導電率を得るように混合物を注射用水(WFI)でフラッシュしてもよい。
【0077】
ある実施形態においては、一次回収はサンプルマトリックスを更に清澄化することにより、抗IL−13抗体を精製し易くするための1以上の遠心工程を含む。サンプルの遠心は限定されないが、例えば7,000×g〜約12,750×gで実施することができる。大規模精製の場合には、このような遠心は限定されないが、例えば得られる上清中で150NTUの濁度レベルを達成するように設定された流速を使用してオンラインで実施することができる。次に後続精製に備えてこのような上清を採取することができる。
【0078】
ある実施形態においては、一次回収は、サンプルマトリックスを更に清澄化することにより、本発明の抗体を精製し易くするための1以上の深層濾過工程の使用を含む。デプスフィルターは段階的密度の濾過媒体を含む。このような段階的密度により、フィルターの表面付近に大きい粒子をトラップすると共に、フィルターの表面の広い開口領域を通過する小さい粒子のみをフィルターの中心に近い狭い開口にトラップすることができる。ある実施形態において、深層濾過工程はデリピッド深層濾過工程とすることができる。ある実施形態では、一次回収段階中のみに深層濾過工程を利用するが、他の態様は、1以上の他の精製段階中にもデリピッドデプスフィルター等のデプスフィルターを利用する。本発明に関連して使用することができるデプスフィルターの非限定的な例としては、Cuno(登録商標)モデル30/60ZAデプスフィルター(3M Corp.)と、0.45/0.2μm Sartopore(登録商標)二重層フィルターカートリッジが挙げられる。
【0079】
4.3 アフィニティークロマトグラフィー
ある実施形態では、一次回収サンプルをアフィニティークロマトグラフィーに供し、更に目的抗体をHCPから精製する。ある実施形態において、クロマトグラフィー材料は目的抗体と選択的又は特異的に結合することが可能である。このようなクロマトグラフィー材料の非限定的な例としては、プロテインA、プロテインG、目的抗体と結合する抗原を含むクロマトグラフィー材料、及びFc結合蛋白質を含むクロマトグラフィー材料が挙げられる。特定態様において、アフィニティークロマトグラフィー工程は適切なプロテインA樹脂を充填したカラムに一次回収サンプルを供する段階を含む。プロテインA樹脂は各種抗体、特にIgG1、IgG2及びIgG4のアフィニティー精製・単離に有用である。プロテインAは主にそのFc領域を介して哺乳動物IgGと結合する細菌細胞壁蛋白質である。その天然状態において、プロテインAは5個のIgG結合ドメインと機能の不明な他のドメインをもつ。
【0080】
プロテインA樹脂には、数種類の市販品が存在する。適切な樹脂としては、限定されないが、GE Healthcare製品であるMabSelect(登録商標)とMillipore製品であるProSep Ultra Plus(登録商標)が挙げられる。MabSelect(登録商標)を充填した適切なカラムの非限定的な1例は直径約1.0cm×長さ約21.6cm(ベッドボリューム〜17mL)のカラムである。このサイズのカラムは小規模精製に使用することができ、大規模化に使用される他のカラムと比較することができる。例えば、より大規模の精製にはベッドボリューム約6.6Lの20cm×21cmカラムを使用することができる。カラムに関係なく、MabSelect(登録商標)やProSep Ultra Plus(登録商標)等の適切な樹脂を使用してカラムに充填することができる。
【0081】
ある実施形態では、精製を特定の目的抗体に適応させるために、プロテインA樹脂の動的結合容量(DBC)を測定すると有利であろう。例えば、限定されないが、MabSelect(登録商標)又はProSept Ultra Plus(登録商標)カラムのDBCは一段階流速ロード又は二段階流速ロードストラテジーにより測定することができる。一段階流速ロードは全ロード時間を通して約300cm/時の速度で評価することができる。二段階流速ロードストラテジーは樹脂1mL当たり蛋白質約35mgまでを約300cm/時の線速度でカラムにロードした後、ロードの最後の部分の滞留時間を長くするように線速度を半減させることにより測定することができる。
【0082】
ある実施形態では、サンプルロードの前に適切な緩衝液でプロテインAカラムを平衡化することができる。適切な緩衝液の非限定的な1例はpH約7.2のトリス/NaClバッファーである。適切な平衡化条件の非限定的な1例は25mMトリス,100mM NaCl,pH約7.2である。この平衡化後、サンプルをカラムにロードすることができる。カラムにロードした後、例えば平衡化バッファーを使用してカラムを1回以上洗浄することができる。カラムから溶出させる前に、別の緩衝液を利用する洗浄を含む他の洗浄を使用することができる。例えば、1カラム体積以上の20mMクエン酸/クエン酸ナトリウム,0.5M NaCl,pH約6.0を使用してカラムを洗浄することができる。この洗浄後、場合により平衡化バッファーを使用して1回以上洗浄してもよい。その後、適当な溶出バッファーを使用してプロテインAカラムから溶出させることができる。適切な溶出バッファーの非限定的な1例はpH約3.5の酢酸/NaClバッファーである。適切な条件は例えば0.1M酢酸,pH約3.5である。当業者に周知の技術を使用して溶出液をモニターすることができる。例えば、OD280の吸光度を追跡することができる。約0.5AUの初期偏光から出発して溶出ピークの立下りの約0.5AUの読取値までカラム溶出液を採取することができる。次に、後続処理に備えて目的の溶出画分を調製することができる。例えば、採取したサンプルをpH約10のトリス(例えば1.0M)で約5.0のpHまで滴定することができる。場合により、この滴定後のサンプルを濾過し、更に処理してもよい。
【0083】
4.4 イオン交換クロマトグラフィー
ある実施形態においては、本発明は抗体を含有する溶出液が得られるように、抗体と少なくとも1種のHCPを含有する混合物を少なくとも1種のイオン交換分離工程に供することにより、前記混合物からHCP低減抗体調製物を生成する方法を提供する。イオン交換分離としては、2種類の物質を夫々のイオン電荷の差に基づいて分離する任意方法が挙げられ、カチオン交換材料又はアニオン交換材料を利用することができる。
【0084】
カチオン交換材料とアニオン交換材料のどちらを使用するかは蛋白質の総電荷による。従って、カチオン交換工程を使用する前にアニオン交換工程を使用することも、アニオン交換工程を使用する前にカチオン交換工程を使用することも本発明の範囲に含まれる。更に、カチオン交換工程のみを使用することも、アニオン交換工程のみを使用することも、両者の任意の順次併用も本発明の範囲に含まれる。
【0085】
分離を実施するには、各種技術のいずれかを使用(例えばバッチ精製技術やクロマトグラフィー技術を使用)することにより、初期抗体混合物をイオン交換材料と接触させればよい。
【0086】
例えば、バッチ精製の場合には、所望の出発バッファー中でイオン交換材料を調製又は平衡化する。調製又は平衡化後、イオン交換材料のスラリーが得られる。抗体溶液をスラリーと接触させ、分離しようとする抗体をイオン交換材料に吸着させる。例えばスラリーを沈殿させ、上清を除去することにより、イオン交換材料と結合しないHCPを含有する溶液をスラリーから分離する。スラリーを1回以上の洗浄工程に供することができる。必要に応じて、スラリーを高導電率の溶液と接触させ、イオン交換材料と結合したHCPを脱着させてもよい。結合したポリペプチドを溶出させるためには、緩衝液の塩濃度を上げればよい。
【0087】
イオン交換分離法としてイオン交換クロマトグラフィーを使用してもよい。イオン交換クロマトグラフィーは分子の総電荷の差に基づいて分子を分離する。抗体の精製では、抗体はイオン交換材料(例えば樹脂)に付加した官能基と結合するためにこの官能基に対して逆電荷でなければならない。例えば、そのpI未満のpHの緩衝液中で一般に総電荷がプラスの抗体は負電荷の官能基を含むカチオン交換材料と結合する。
【0088】
イオン交換クロマトグラフィーでは、周囲の緩衝液のイオン強度が低いならば、溶質の表面の帯電パッチはクロマトグラフィーマトリックスに付加した逆電荷により吸引される。溶出は一般にイオン交換マトリックスの帯電部位について溶質と競合するように緩衝液のイオン強度(即ち導電率)を上げることにより行われる。溶質の溶出を行う別の方法はpHを変化させることにより、溶質の電荷を変化させる方法である。導電率又はpHの変化は漸次(グラジエント溶出)でも段階的(ステップワイズ溶出)でもよい。
【0089】
クロマトグラフィーのアニオン性又はカチオン性担体を形成するためにはアニオン性又はカチオン性置換基をマトリックスに付加すればよい。アニオン交換置換基の非限定的な例としては、ジエチルアミノエチル(DEAE)、第四級アミノエチル(QAE)及び第四級アミン(Q)基が挙げられる。カチオン置換基としては、カルボキシメチル(CM)、スルホエチル(SE)、スルホプロピル(SP)、リン酸(P)及びスルホン酸(S)が挙げられる。DE23(登録商標)、DE32(登録商標)、DE52(登録商標)、CM−23(登録商標)、CM−32(登録商標)及びCM−52(登録商標)等のセルロースイオン交換樹脂がWhatman Ltd.Maidstone,Kent,U.K.から市販されている。SEPHADEX(登録商標)系架橋結合イオン交換体も知られている。例えば、DEAE−、QAE−、CM−及びSP−SEPHADEX(登録商標)、DEAE−、Q−、CM−及びS−SEPHAROSE(登録商標)、並びにSEPHAROSE(登録商標)ファストフローがいずれもPharmacia ABから市販されている。更に、TOYOPEARL(登録商標)DEAE−650S又はM及びTOYOPEARL(登録商標)CM−650S又はM等のDEAE及びCMの両者で誘導体化したエチレングリコール−メタクリレートコポリマーもToso Haas Co.,Philadelphia,Paから市販されている。ある実施形態では、Pall Mustang Qメンブレンカートリッジを使用してアニオン交換工程を実施する。
【0090】
抗体と不純物(例えばHCP)を含有する混合物をイオン交換カラム(例えばカチオン交換カラム)にロードする。例えば、限定されないが、使用するカラムに応じて蛋白質約80g/L樹脂のロード率で混合物をロードすることができる。適切なカチオン交換カラムの1例は直径80cm×長さ23cm、ベッドボリューム約116Lのカラムである。このカチオンカラムにロードした混合物を次に洗浄バッファー(平衡化バッファー)で洗浄することができる。その後、抗体をカラムから溶出させ、第1の溶出液を得る。
【0091】
このイオン交換工程は、HCP等の不純物を低減しながら目的抗体の捕集を容易にする。ある態様において、イオン交換カラムはカチオン交換カラムである。例えば、限定されないが、このようなカチオン交換カラムに適した樹脂はCM HyperDF(登録商標)樹脂である。これらの樹脂はPall Corporation等の業者から入手可能である。このカチオン交換法は室温又は室温付近で実施することができる。
【0092】
4.5 限外濾過/透析濾過
本発明のある実施形態では抗体サンプルを更に精製・濃縮するために限外濾過及び/又は透析濾過工程を利用する。限外濾過はMicrofiltration and Ultrafiltration:Principles and Applications,L.Zeman and A.Zydney(Marcel Dekker,Inc.,New York,N.Y.,1996)及びUltrafiltration Handbook,Munir Cheryan(Technomic Publishing,1986;ISBN No.87762−456−9)に詳細に記載されている。濾過工程の1例は表題“Pharmaceutical Process Filtration Catalogue”
のMilliporeカタログ177〜202頁(Bedford,Mass.,1995/96)に記載されているようなタンジェンシャルフロー濾過である。限外濾過は一般に0.1μm未満の細孔サイズのフィルターを使用する濾過を意味するとみなされる。このような細孔サイズの小さいフィルターを利用することにより、抗体をフィルターに保持しながら、サンプルバッファーをフィルターに透過させることによりサンプルの体積を減少させることができる。
【0093】
透析濾過は、塩、糖及び非水性溶媒を除去及び交換するため、遊離種を結合種から分離するため、低分子量材料を除去するため、並びに/又はイオン及び/もしくはpH環境の迅速な変化を誘導するために限外濾過フィルターを使用する方法である。限外濾過中の溶液に溶媒を限外濾過速度とほぼ等しい速度で加えることにより、微小溶質は最も効率的に除去される。こうして微小種が一定流量で溶液から洗い流され、保持された抗体が有効に精製される。本発明のある実施形態では、場合により後続クロマトグラフィー又は他の精製工程の前に本発明に関連して使用される各種緩衝液を交換するため、あるいは抗体調製物から不純物を除去するために透析濾過工程を利用する。
【0094】
4.6 疎水性相互作用クロマトグラフィー
本発明は、抗体と少なくとも1種のHCPを含有する混合物からHCP低減抗体調製物を生成する方法であって、更に疎水性相互作用分離工程を含む方法にも関する。例えば、HCP濃度の低下した第2の溶出液が得られるように、イオン交換カラムから得られた第1の溶出液を疎水性相互作用材料に供することができる。本願に記載する工程等の疎水性相互作用クロマトグラフィー工程を一般に実施し、抗体凝集物等の蛋白質凝集物と工程関連不純物を除去する。
【0095】
分離を実施するには、例えばバッチ精製技術又はカラムを使用してサンプル混合物をHIC材料と接触させる。HIC精製の前に、例えば混合物をプレカラムに通すことにより、カオトロピック剤又は超疎水性物質を除去することが望ましい場合もある。
【0096】
例えば、バッチ精製の場合には、所望平衡化バッファー中でHIC材料を調製又は平衡化する。HIC材料のスラリーが得られる。抗体溶液をスラリーと接触させ、分離しようとする抗体をHIC材料に吸着させる。例えばスラリーを沈殿させ、上清を除去することにより、HIC材料と結合しないHCPを含有する溶液をスラリーから分離する。スラリーを1回以上の洗浄工程に供することができる。必要に応じて、スラリーを導電率の低い溶液と接触させ、HIC材料と結合した抗体を脱着させてもよい。結合した抗体を溶出させるためには、塩濃度を下げればよい。
【0097】
イオン交換クロマトグラフィーは、抗体を単離するために抗体の電荷に依存し、疎水性相互作用クロマトグラフィーは抗体の疎水性を利用する。抗体上の疎水基がカラム上の疎水基と相互作用する。疎水度が高いほど、蛋白質はカラムと強く相互作用する。従って、HIC工程は宿主細胞に由来する不純物(例えばDNAと他の高分子量及び低分子量産物関連種)を除去する。
【0098】
疎水性相互作用は、イオン強度が高いときに最強であるため、この型の分離は塩沈殿法又はイオン交換法に従って適切に実施される。HICカラムへの抗体の吸着は高い塩濃度により助長されるが、実際の濃度は抗体の種類と選択される特定HICリガンドに応じて広い範囲を取ることができる。疎水性相互作用を促進する(塩析効果)か又は水の構造を破壊する(カオトロピック効果)かに応じて所謂疎溶媒性系列に各種イオンを配置し、疎水性相互作用を弱化させることができる。カチオンを塩析効果の昇順に並べると、Ba++、Ca++、Mg++、Li+、Cs+、Na+、K+、Rb+、NH4+であり、アニオンをカオトロピック効果の昇順に並べると、PO−−−、SO4−−、CH3CO3−、Cl−、Br−、NO3−、ClO4−、I−、SCN−となる。
【0099】
一般に、硫酸Na、硫酸K又は、硫酸NH4はHICにおいてリガンド−蛋白質相互作用を有効に促進する。この相互作用の強度に影響を与える塩を体系化すると、以下の関係:(NH4)2SO4>Na2SO4>NaCl>NH4Cl>NaBr>NaSCNにより示すことができる。一般に、約0.75〜約2M硫酸アンモニウム又は約1〜4M NaClの塩濃度が有用である。
【0100】
HICカラムは、通例では疎水性リガンド(例えばアルキル又はアリール基)をカップリングさせる塩基マトリックス(例えば架橋アガロース又は合成コポリマー材料)を含む。適切なHICカラムは、フェニル基で置換されたアガロース樹脂を含む(例えばPhenyl Sepharose(登録商標)カラム)。多数のHICカラムが市販されている。例としては、限定されないが、低置換度又は高置換度のPhenyl Sepharose(登録商標)6ファストフローカラム(Pharmacia LKB Biotechnology,AB,Sweden)、Phenyl Sepharose(登録商標)高性能カラム(Pharmacia LKB Biotechnology,AB,Sweden)、Octyl Sepharose(登録商標)高性能カラム(Pharmacia LKB Biotechnology,AB,Sweden)、Fractogel(登録商標)EMDプロピル又はFractogel(登録商標)EMDフェニルカラム(E.Merck,Germany)、Macro−Prep(登録商標)メチル又はMacro−Prep(登録商標)t−ブチル担体(Bio−Rad,California)、WP HI−Propyl(C3)(登録商標)カラム(J.T.Baker,New Jersey)、及びToyopearl(登録商標)エーテル、フェニル又はブチルカラム(TosoHaas,PA)が挙げられる。
【0101】
4.7 典型的精製ストラテジー
ある実施形態では、一次回収の前に先ず遠心及び濾過工程を利用して産生用バイオリアクター回収液から細胞及び細胞破片(HCPを含む)を除去する。例えば、非限定的な例として、抗体と培地と細胞を含む培養液を約7000×g〜約11,000×gで遠心することができる。ある実施形態では、得られたサンプル上清を次に複数のデプスフィルターを含むフィルター列に通す。ある実施形態においては、フィルター列は、12個前後の16インチCuno(登録商標)モデル30/60ZAデプスフィルター(3M Corp.)と、3個の30インチ0.45/0.2μm Sartopore(登録商標)2フィルターカートリッジ(Sartorius)を装着した3個前後のフィルターハウジングから構成される。清澄化した上清を予め滅菌した回収液容器等の容器に採取し、約8℃に維持する。この温度を次に、以下に概説する1以上の捕集クロマトグラフィー工程の前に約20℃に調整する。当然のことながら、当業者は上記条件を変更することができ、このような変更も本発明の範囲内に含まれる。
【0102】
ある実施形態では、一次回収後に、プロテインA樹脂を使用してアフィニティークロマトグラフィーを行う。プロテインA樹脂は数件の業者から市販されている。適切な樹脂の1例は、GE Healthcare製品であるMabSelect(登録商標)である。MabSelect(登録商標)を充填した適切なカラムの1例は直径約1.0cm×長さ約21.6cm(ベッドボリューム〜17mL)のカラムである。このサイズのカラムはベンチスケール用に使用することができる。これを大規模化に使用される他のカラムと比較することができる。例えば、商業的生産にはベッドボリューム約6.6Lの20cm×21cmカラムを使用することができる。カラムに関係なく、MabSelect(登録商標)等の適切な樹脂を使用してカラムに充填することができる。
【0103】
ある態様では、サンプルをロードする前に、プロテインAカラムを適切な緩衝液で平衡化することができる。適切な緩衝液の1例は、pH約6〜8のトリス/NaClバッファーであり、限定されないが、約7.2が挙げられる。適切な条件の特定例は、25mMトリス,100mM NaCl,pH7.2である。この平衡化後、サンプルをカラムにロードすることができる。カラムにロードした後、例えば平衡化バッファーを使用してカラムを1回以上洗浄することができる。カラムから溶出させる前に、別の緩衝液を利用する洗浄を含む他の洗浄を使用することができる。例えば、1カラム体積以上の20mMクエン酸/クエン酸ナトリウム,0.5M NaCl,pH約6.0を使用してカラムを洗浄することができる。この洗浄後、場合により平衡化バッファーを使用して1回以上洗浄してもよい。その後、適切な溶出バッファーを使用してプロテインAカラムから溶出させることができる。適切な溶出バッファーの1例はpH約3.5の酢酸/NaClバッファーである。適切な条件は例えば0.1M酢酸,pH3.5である。当業者に周知の技術を使用して溶出液をモニターすることができる。例えば、OD280の吸光度を追跡することができる。約0.5AUの初期偏光から出発して溶出ピークの立下りの約0.5AUの読取値までカラム溶出液を採取することができる。次に、後続処理に備えて目的の溶出画分を調製することができる。例えば、採取したサンプルをpH約10のトリス(例えば1.0M)で約5.0のpHまで滴定することができる。場合により、この滴定後のサンプルを濾過し、更に処理してもよい。
【0104】
MabSelect(登録商標)カラムの動的結合容量(DBC)は、一段階流速ロード又は二段階流速ロードストラテジーにより測定することができる。一段階流速ロードは、全ロード時間を通して約300cm/時の速度で評価することができる。二段階流速ロードストラテジーは樹脂1mL当たり蛋白質約35mgまでを約300cm/時の線速度でカラムにロードした後、ロードの最後の部分の滞留時間を長くするように線速度を半減させることにより測定することができる。
【0105】
次に、pH介在ウイルス低減/不活性化工程を利用することによりプロテインA溶出液を更に精製することができる。ある実施形態において、この工程は溶出液のpHを約3〜約5(限定されないが、例えば約3.5)に約1時間調整する。クエン酸(例えば3Mクエン酸)等の公知酸調製物を使用してpH低下を助長することができる。酸性pHに暴露すると、pH感受性ウイルス汚染物質は完全には除去されないとしても、低減し、多少の培地/細胞汚染物質が沈殿する。このウイルス低減/不活性化工程後、水酸化ナトリウム(例えば3M水酸化ナトリウム)等の塩基を使用して約20〜約40分間pHを約4.9又は5.0に調整する。この調整は約20℃で実施することができる。
【0106】
ある実施形態では、pHを調整した培地をアニオン交換カラムで更に精製する。この工程に適したカラムの非限定的な1例は、直径60cm×長さ30cm、ベッドボリューム約85Lのカラムである。GE Healthcare製品であるQ Sepharose(登録商標)ファストフロー等のアニオン交換樹脂をカラムに充填する。約7カラム体積の適切な緩衝液(例えばトリス/塩化ナトリウム)を使用してカラムを平衡化することができる。適切な条件の1例は、25mMトリス,50mM塩化ナトリウム,pH8.0である。当業者は上記条件を変更することができ、このような変更も本発明の範囲内に含まれる。上記に概説したプロテインA精製工程から採取したサンプルをカラムにロードする。別の側面では、カチオン交換中に採取した溶出液からカラムにロードする。カラムにロードした後、カラムを平衡化バッファー(例えばトリス/塩化ナトリウムバッファー)で洗浄する。OD280nmでUV分光光度計を使用して抗体を含有するフロースルーをモニターすることができる。このアニオン交換工程は工程関連不純物(例えばDNA等の核酸や宿主細胞蛋白質)を低減させる。目的抗体は、カラムの固相(例えばQ Sepharose(登録商標))と実質的に相互作用も結合もしないが、多くの不純物は、カラムの固相と相互作用し、結合するので、分離が行われる。アニオン交換は約12℃で実施することができる。
【0107】
ある実施形態では、pHを調整した培地をカチオン交換カラムで更に精製する。ある実施形態においては、カチオン交換カラムで使用される平衡化バッファーは、pH約5.0の緩衝液である。適切な緩衝液の1例は約210mM酢酸ナトリウム,pH5.0である。平衡化後、上記一次回収工程から調製したサンプルをカラムにロードする。GE Healthcare製品であるCM Sepharose(登録商標)ファストフロー等のカチオン交換樹脂をカラムに充填する。次に、平衡化バッファーを使用してカラムを洗浄する。次に、平衡化又は洗浄バッファーに比較してイオン強度の高い緩衝液を使用する溶出工程にカラムを供する。例えば、適切な溶出バッファーとしては、約790mM酢酸ナトリウム,pH5.0が挙げられる。抗体を溶出させ、OD280nmに設定したUV分光光度計を使用してモニターすることができる。特定例において、溶出液採取は、上側3OD280nmから下側8OD280nmまでとすることができる。当然のことながら、当業者は上記条件を変更することができ、このような変更も本発明の範囲内に含まれる。
【0108】
ある実施形態では、pHを調整した培地、カチオン交換溶出液又はアニオン交換溶出液を例えば16インチCuno(登録商標)デリピッドフィルターで濾過する。デリピッドフィルターを使用するこの濾過後、例えば30インチ0.45/0.2μm Sartopore(登録商標)二重層フィルターカートリッジで濾過することができる。イオン交換溶出バッファーを使用してフィルターに残留している残量分をフラッシュし、限外濾過/透析濾過に備えることができる。
【0109】
限外濾過/透析濾過工程を実施するためには、適切な緩衝液(例えば20mMリン酸ナトリウム,pH7.0)で濾過媒体を調製する。イオン強度を増すために塩化ナトリウム(例えば100mM塩化ナトリウム)等の塩を加えることができる。この限外濾過/透析濾過工程は抗IL−13抗体を濃縮し、酢酸ナトリウムを除去し、pHを調整するように機能する。この工程を実施するには市販フィルターが利用可能である。例えば、Milliporeは30kD分子量カットオフ(MWCO)セルロース限外濾過膜カセットを製造している。この濾過法は室温又は室温付近で実施することができる。
【0110】
ある実施形態では、上記捕集濾過工程からのサンプルを第2のイオン交換分離工程に供する。この第2のイオン交換分離はある実施形態においては、第1のイオン交換分離の逆電荷に基づく分離を含む。例えば、一次回収後にアニオン交換工程を利用する場合には、第2のイオン交換クロマトグラフィー工程をカチオン交換工程とすることができる。逆に、一次回収工程後にカチオン交換工程を行う場合には、この工程後にアニオン交換工程を実施する。ある実施形態では、第1のイオン交換溶出液を第2のイオン交換クロマトグラフィー工程に直接供することができ、第1のイオン交換溶出液を適切な緩衝液条件に調整する。適切なアニオン及びカチオン分離材料及び条件は上記の通りである。
【0111】
本発明のある実施形態では、抗体を含有するサンプルを疎水性相互作用分離工程で更に処理する。このような工程に適したカラムの非限定的な1例は、直径80cm×長さ15cm、ベッドボリューム約75Lのカラムであり、HICに使用するのに適した樹脂(限定されないが、例えばAmersham Biosciences,Upsala,Sweden製品であるPhenyl HP Sepharose(登録商標))を充填する。前のアニオン交換クロマトグラフィー工程から得られた目的抗体を含有するフロースルーサンプルを等体積の約1.7M硫酸アンモニウム,50mMリン酸ナトリウム,pH7.0で希釈することができる。その後、0.45/0.2μm Sartopore(登録商標)2二重層フィルター又は同等手段を使用してこれを濾過することができる。ある実施形態においては、疎水性クロマトグラフィー法は2サイクル以上を含む。
【0112】
ある実施形態では、先ず適切な緩衝液を使用してHICカラムを平衡化する。適切な緩衝液の非限定的な1例は0.85M硫酸アンモニウム,50mMリン酸ナトリウム,pH7.0である。当業者は、緩衝剤の濃度を変更及び/又は同等の緩衝液に置換することにより平衡化バッファーを変更することができ、このような変更も本発明の範囲内である。ある実施形態では、次にカラムにアニオン交換フロースルーサンプルをロードし、硫酸アンモニウム/リン酸ナトリウム等の適切な緩衝液系で複数回(例えば3回)洗浄する。適切な緩衝液の1例としては、pH約7.0の1.1M硫酸アンモニウム,50mMリン酸ナトリウムバッファーが挙げられる。場合により、カラムを更に洗浄サイクルに供することができる。例えば、第2の洗浄サイクルでは、適切な緩衝液系を使用してカラムを複数回(例えば1〜7回)洗浄することができる。適切な緩衝液系の非限定的な1例としては、0.85M硫酸アンモニウム,50mMリン酸ナトリウム,pH7.0が挙げられる。ある態様では、ロードしたカラムに対して適切な緩衝液系を使用して3回目の洗浄を行う。pH約7.0の1.1M硫酸アンモニウム,50mMリン酸ナトリウム等の適切な緩衝液系を使用してカラムを複数回(例えば1〜3回)洗浄することができる。この場合も、当業者は緩衝条件を変更することができ、このような変更も本発明の範囲内に含まれる。
【0113】
適当な溶出バッファーを使用してカラムから溶出させる。このような溶出バッファーの適切な1例は、pH約7.0の0.5M硫酸アンモニウム,15mMリン酸ナトリウムである。従来の分光光度計を使用して3OD280nmの上側から3OD280nmのピークの下側まで目的抗体を検出・採取することができる。
【0114】
本発明のある態様では、無傷ウイルスを含むウイルス粒子が残存する場合にはこれを除去するために疎水性クロマトグラフィー工程からの溶出液を濾過する。適切なフィルターの非限定的な1例は、Pall Corporation製品であるUltipor DV50(登録商標)フィルターである。他のウイルスフィルターもこの濾過工程で使用することができ、当業者に周知である。予め湿潤させた約0.1μmのフィルターと2×30インチUltipor DV50(登録商標)フィルター列に約34psigでHIC溶出液を通す。ある実施形態では、濾過工程後に、フィルターハウジングに抗体が保持されている場合にはこれを除去するために例えばHIC溶出バッファーを使用してフィルターを洗浄する。予め滅菌した容器に濾液を約12℃で保存することができる。
【0115】
ある実施形態では、上記からの濾液を再び限外濾過/透析濾過に供する。実施者の最終目的が例えば医薬製剤中で抗体を使用することである場合にはこの工程が重要である。この工程を利用する場合には、抗体の濃縮、先に使用した緩衝塩の除去及び特定の製剤化バッファーとの交換を容易にすることができる。ある実施形態では、複数倍容量(例えば2倍容量)の製剤化バッファーを使用して連続透析濾過を実施する。適切な製剤化バッファーの非限定的な1例は5mMメチオニン,2%マンニトール,0.5%スクロース,pH5.9バッファー(Tween不含)である。このダイアボリューム交換が完了したら、抗体を濃縮する。所定の抗体濃度に達したら、実施者は約0.005%(v/v)の最終Tween濃度に達するために加えるべき10% Tweenの量を計算することができる。
【0116】
本発明のある実施形態は、付加的な精製工程を含む。イオン交換クロマトグラフィー法の前、その間、又はその後に実施することができる付加的な精製法の例としては、エタノール沈殿、等電点電気泳動、逆相HPLC、シリカクロマトグラフィー、ヘパリンSepharose(登録商標)クロマトグラフィー、付加的なアニオン交換クロマトグラフィー及び/又は付加的なカチオン交換クロマトグラフィー、クロマトフォーカシング、SDS−PAGE、硫酸アンモニウム沈殿、ヒドロキシアパタイトクロマトグラフィー、ゲル電気泳動、透析濾過、並びに(例えば捕集試薬としてプロテインG、抗体、特異的基質、リガンド又は抗原を使用する)アフィニティークロマトグラフィーが挙げられる。
【0117】
本発明のある実施形態においては、抗IL−13抗体は図1にまとめた重鎖及び軽鎖可変領域配列を含むIgA1、IgA2、IgD、IgE、IgG1、IgG2、IgG3、IgG4又はIgMアイソタイプ抗体である。ある実施形態においては、抗IL−13抗体は図1にまとめた重鎖及び軽鎖可変領域配列を含むIgG1、IgG2、IgG3又はIgG4アイソタイプ抗体である。
【0118】
5.サンプル純度検定方法
5.1 宿主細胞蛋白質の検定
本発明は、単離/精製抗体組成物中の残留レベルの宿主細胞蛋白質(HCP)濃度の測定方法も提供する。上記のように、最終目的物質(例えば抗IL−13抗体)からHCPを排除することが望ましい。典型的なHCPとしては、抗体産生源に由来する蛋白質が挙げられる。目的抗体からHCPを同定し、十分に除去することができないと、効力低下及び/又は有害な対象反応に繋がる恐れがある。
【0119】
本願で使用する「HCP ELISA」なる用語は、アッセイで使用される第2の抗体が抗体(例えば抗IL−13抗体)を作製するために使用される細胞(例えばCHO細胞)から産生されるHCPに特異的であるELISAを意味する。第2の抗体は当業者に公知の従来方法に従って産生させることができる。例えば、第2の抗体はsham産生及び精製試験で得られるHCPを使用して産生させることができ、即ち目的抗体を産生させるために使用されると同一の細胞株を使用するが、細胞株には抗体DNAをトランスフェクトしない。典型的な態様では、選択細胞発現系、即ち目的抗体を産生させるために使用される細胞発現系で発現されると同様のHPCを使用して第2の抗体を産生させる。
【0120】
一般に、HCP ELISAはHCPを含有する液体サンプルを2層の抗体、即ち第1の抗体と第2の抗体の間にサンドイッチする。サンプルをインキュベートすると、その間にサンプル内のHCPは第1の抗体(限定されないが、例えばアフィニティー精製したヤギ抗CHO(Cygnus))により捕集される。抗体を作製するために使用される細胞から産生されるHCPに特異的な第2の標識抗体又は抗体ブレンド(例えばビオチン化抗CHO HCP)を加えると、サンプル内のHCPと結合する。ある実施形態においては、第1の抗体と第2の抗体はポリクローナル抗体である。ある態様においては、第1の抗体と第2の抗体はHCPに対するポリクローナル抗体のブレンドであり、限定されないが、例えばビオチン化ヤギ抗宿主細胞蛋白質混合物599/626/748である。第2の抗体のラベルに基づく適切な試験を使用してサンプルに含まれるHCPの量を測定する。
【0121】
HCP ELISAは、上記方法を使用して得られる溶出液又はフロースルー等の抗体組成物中のHCP濃度を測定するために使用することができる。本発明は、抗体を含有する組成物も提供し、前記組成物はHCP酵素免疫測定法(「ELISA」)により測定した場合に検出可能な濃度のHCPを含有しない。
【0122】
5.2 アフィニティークロマトグラフィー材料の検定
ある実施形態においては、本発明は、単離/精製抗体組成物中のアフィニティークロマトグラフィー材料の残留濃度の測定方法も提供する。所定状況において、このような材料は精製工程中に抗体組成物中に浸出する。ある実施形態では、単離/精製抗体組成物中のプロテインA濃度を測定するアッセイを利用する。本願で使用する「プロテインA ELISA」なる用語はアッセイで使用される第2の抗体が目的抗体(例えば抗IL−13抗体)を精製するために利用されるプロテインAに特異的であるELISAを意味する。第2の抗体は、当業者に公知の従来方法に従って産生させることができる。例えば、第2の抗体は従来の抗体作製及び産生方法のコンテキストで天然又は組換えプロテインAを使用して産生させることができる。
【0123】
一般に、プロテインA ELISAはプロテインAを含有する(又はプロテインAを含有すると予想される)液体サンプルを2層の抗プロテインA抗体、即ち第1の抗プロテインA抗体と第2の抗プロテインA抗体の間にサンドイッチする。サンプルを第1層の抗プロテインA抗体(限定されないが、例えば、ポリクローナル抗体又はポリクローナル抗体のブレンド)に暴露し、サンプル内のプロテインAが第1の抗体により捕集されるために十分な時間インキュベートする。次にプロテインAに特異的な第2の標識抗体(限定されないが、例えば、ポリクローナル抗体又はポリクローナル抗体のブレンド)を加えると、サンプル内の捕集されたプロテインAと結合する。本発明に関連して有用な抗プロテインA抗体の他の非限定的な例としては、ニワトリ抗プロテインA抗体とビオチン化抗プロテインA抗体が挙げられる。第2の抗体のラベルに基づく適切な試験を使用してサンプルに含まれるプロテインAの量を測定する。別のアフィニティークロマトグラフィー材料の濃度を測定するために同様のアッセイを利用することができる。
【0124】
プロテインA ELISAは、上記方法を使用して得られる溶出液又はフロースルー等の抗体組成物中のプロテインA濃度を測定するために使用することができる。本発明は抗体を含有する組成物も提供し、前記組成物はプロテインA酵素免疫測定法(「ELISA」)により測定した場合に検出可能な濃度のプロテインAを含有しない。
【0125】
6.付加的修飾
本発明の抗体は、修飾することができる。ある実施形態では、所望効果を提供するために抗体又はその抗原結合フラグメントを化学的に修飾する。例えば、本発明の抗体又は抗体フラグメントのペグ化は例えば各々その開示内容全体を本願に援用するFocus on Growth Factors 3:4−10(1992);EP0 154 316;及びEP0 401 384に記載されているような当分野で公知のペグ化反応のいずれかにより実施することができる。ある態様において、ペグ化は反応性ポリエチレングリコール分子(又は同様の反応性水溶性ポリマー)とのアシル化反応又はアルキル化反応により実施される。本発明の抗体及び抗体フラグメントのペグ化に適した水溶性ポリマーはポリエチレングリコール(PEG)である。本願で使用する「ポリエチレングリコール」とは、モノ(C1−C10)アルコキシ−又はアリールオキシ−ポリエチレングリコール等の他の蛋白質を誘導体化するために従来から使用されている型のPEGの任意のものを包含する。
【0126】
本発明のペグ化抗体及び抗体フラグメントの作製方法は、一般に、(a)抗体又は抗体フラグメントが1個以上のPEG基と結合する適切な条件下で抗体又は抗体フラグメントをポリエチレングリコール(例えばPEGの反応性エステル又はアルデヒド誘導体)と反応させる段階と、(b)反応生成物を得る段階を含む。公知パラメーターと所望結果に基づいて最適反応条件又はアシル化反応を選択することは当業者に容易に理解されよう。
【0127】
本願に記載する抗IL−13抗体及び抗体フラグメントの投与により、本発明のIL−13関連障害を治療するためには、一般に、IL−13に特異的なペグ化抗体及び抗体フラグメントを使用することができる。一般に、ペグ化抗体及び抗体フラグメントは非ペグ化抗体及び抗体フラグメントに比較して半減期が長い。ペグ化抗体及び抗体フラグメントは単独で使用してもよいし、一緒に使用してもよいし、他の医薬組成物と併用してもよい。
【0128】
本発明の抗体又は抗体部分は、誘導体化又は別の機能分子(例えば別のペプチド又は蛋白質)と連結することができる。従って、本発明の抗体及び抗体部分は免疫接着分子を含めて本願に記載するヒト抗hIL−13抗体の誘導体化及び他の修飾形態を包含するものとする。例えば、別の抗体(例えば二重特異性抗体又はダイアボディ)、検出可能な物質、細胞傷害性物質、薬剤、及び/又は抗体もしくは抗体部分と別の分子(例えばストレプトアビジンコア領域又はポリヒスチジンタグ)との結合に介在することができる蛋白質もしくはペプチド等の1個以上の他の分子体と本発明の抗体又は抗体部分を(化学的カップリング、遺伝子融合、非共有的結合又は他の方法により)機能的に連結することができる。
【0129】
ある種の誘導体化抗体は、(例えば二重特異性抗体を創製するために同一型又は異なる型の)2個以上の抗体を架橋することにより作製される。適切な架橋剤としては、適当なスペーサーにより分離された2個の非常に反応性の基をもつヘテロ二価性架橋剤(例えばm−マレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル)又はホモ二価性架橋剤(例えばスベリン酸ジスクシンイミジル)が挙げられる。このようなリンカーはPierce Chemical Company,Rockford,ILから市販されている。
【0130】
本発明の抗体又は抗体部分を誘導体化することができる有用な検出可能な物質としては、蛍光化合物が挙げられる。検出可能な蛍光物質の例としてはフルオレセイン、イソチオシアン酸フルオレセイン、ローダミン、5−ジメチルアミン−1−ナフタレンスルホニルクロリド、フィコエリスリン等が挙げられる。アルカリホスファターゼ、西洋ワサビペルオキシダーゼ、グルコースオキシダーゼ等の検出可能な酵素で抗体を誘導体化することもできる。検出可能な酵素で抗体を誘導体化する場合には、検出可能な反応生成物を生成するために酵素により使用される他の試薬を加えることにより抗体を検出する。例えば、検出可能な物質である西洋ワサビペルオキシダーゼが存在する場合には、過酸化水素とジアミノベンジジンを加えると、検出可能な着色反応生成物が得られる。抗体をビオチンで誘導体化し、アビジン又はストレプトアビジン結合の間接測定により検出することもできる。
【0131】
7.医薬組成物
本発明の抗体及び抗体部分は、対象に投与するのに適した医薬組成物に配合することができる。典型的には、医薬組成物は本発明の抗体又は抗体部分と、医薬的に許容可能な担体を含有する。本願で使用する「医薬的に許容可能な担体」とは、生理的に適合可能な全溶媒、分散媒、コーティング、抗細菌剤及び抗真菌剤、等張剤及び吸収遅延剤等を包含する。医薬的に許容可能な担体の例としては水、生理食塩水、リン酸緩衝生理食塩水、デキストロース、グリセロール、エタノール等の1種以上とその組み合わせが挙げられる。多くの場合には、例えば糖、多価アルコール(例えばマンニトール、ソルビトール)、又は塩化ナトリウム等の等張剤を組成物に加えることが望ましい。医薬的に許容可能な担体としては更に抗体又は抗体部分の保存期間又は効力を増す微量の助剤(例えば湿潤剤、乳化剤、防腐剤又は緩衝剤)も挙げられる。
【0132】
本発明の抗体及び抗体部分は、非経口投与に適した医薬組成物に配合することができる。例えば抗体0.1〜250mg/mlを含有する注射溶液として抗体又は抗体部分を製造することができる。注射溶液はフリントもしくはアンバーバイアル、アンプル又はプレフィルドシリンジに収容した液体又は凍結乾燥剤形から構成することができる。緩衝液はL−ヒスチジン約1〜50mM(最適値5〜10mM),pH5.0〜7.0(最適値pH6.0)とすることができる。他の適切な緩衝液としては限定されないが、琥珀酸ナトリウム、クエン酸ナトリウム、リン酸ナトリウム又はリン酸カリウムが挙げられる。溶液の毒性を緩和するためには濃度0〜300mM(液体剤形には150mMが最適)の塩化ナトリウムを使用することができる。凍結乾燥剤形には凍結保護剤、主に0〜10%スクロース(最適値0.5〜1.0%)を添加することができる。他の適切な凍結保護剤としてはトレハロースとラクトースが挙げられる。凍結乾燥剤形にはバルク剤、主に1〜10%マンニトール(最適値2〜4%)を添加することができる。液体及び凍結乾燥剤形の両者で安定剤、主に1〜50mM L−メチオニン(最適値5〜10mM)を使用することができる。他の適切なバルク剤としてはグリシン、アルギニンが挙げられ、0〜0.05%ポリソルベート80(最適値0.005〜0.01%)として界面活性剤も添加することができる。その他の界面活性剤としては限定されないが、ポリソルベート20及びBRIJ界面活性剤が挙げられる。
【0133】
ある態様において、医薬組成物は、約0.01mg/kg〜10mg/kgの用量の抗体を含有する。別の態様において、抗体の用量としては、約1mg/kgの隔週投与や、約0.3mg/kgの毎週投与が挙げられる。当業者は適正な用量と対象への投与レジメンを決定することができる。
【0134】
本発明の組成物は、各種形態を取ることができる。これらの形態としては、例えば液体、半固体及び固体剤形が挙げられ、例えば溶液(例えば注射溶液及び輸液溶液)、分散液又は懸濁液、錠剤、ピル、散剤、リポソーム及び座剤が挙げられる。剤形は例えば所期投与方法及び治療用途により異なる。典型的な組成物は他の抗体をヒトに受動免疫するために使用されるものと同様の組成物等の注射溶液又は輸液溶液の形態である。投与方法の1例は非経口(例えば静脈内、皮下、腹膜内、筋肉内)である。ある態様では、静脈内輸液又は注射により抗体を投与する。別の側面では、筋肉内又は皮下注射により抗体を投与する。
【0135】
治療用組成物は、典型的には製造及び保存条件下で無菌・安定でなければならない。組成物は溶液、マイクロエマルション、分散液、リポソーム、又は高薬剤濃度に適した他の注文構造として製剤化することができる。滅菌注射溶液は必要量の活性化合物(即ち抗体又は抗体部分)を必要に応じて上記成分の1種又はその組み合わせと共に適切な溶媒に加えた後に濾過滅菌することにより製造することができる。一般に、分散液は塩基性分散媒と上記から選択される必要な他の成分を含有する滅菌担体に活性化合物を加えることにより製造される。滅菌注射溶液の調製用滅菌凍結乾燥粉末の場合には、製造方法は滅菌濾過溶液から活性成分と任意の他の所望成分の粉末を生成する真空乾燥と噴霧乾燥である。例えばレシチン等のコーティングの使用、分散液の場合には必要な粒度の維持及び界面活性剤の使用により、溶液の適正な流動性を維持することができる。吸収を遅らせる物質(例えばモノステアリン酸塩やゼラチン)を組成物に加えることにより、注射用組成物の長期吸収を実現することができる。
【0136】
本発明の抗体及び抗体部分は、当分野で公知の各種方法により投与することができるが、投与経路/方法の1例は皮下注射、静脈内注射又は輸液である。当業者に自明の通り、投与経路及び/又は方法は所望結果により異なる。ある実施形態では、制御放出製剤のように、化合物の迅速な放出を防止する担体を活性化合物に配合することができ、例えばインプラント、経皮パッチ及びマイクロカプセル送達システムが挙げられる。エチレン酢酸ビニル、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル及びポリ乳酸等の生分解性生体適合性ポリマーを使用することができる。このような製剤の多数の製造方法が特許登録されており、あるいは一般に当業者に公知である。例えば、その教示内容全体を本願に援用するSustained and Controlled Release Drug Delivery Systems,J.R.Robinson,ed.,Marcel Dekker,Inc.,New York,1978参照。
【0137】
ある態様では、本発明の抗体又は抗体部分を例えば不活性希釈剤又は同化可食性担体と共に経口投与することができる。化合物(及び必要に応じて他の成分)をハード又はソフトシェルゼラチンカプセルに封入してもよいし、錠剤に圧縮してもよいし、対象の食事に直接配合してもよい。経口治療投与には、化合物に賦形剤を添加し、内服錠、口腔錠、トローチ剤、カプセル剤、エリキシル剤、懸濁液剤、シロップ剤、ウェハース剤等の形態で使用することができる。本発明の化合物を非経口投与以外の方法で投与するためには、その不活性化を防止するための材料を化合物にコーティングしたり、化合物と併用投与することが必要な場合がある。
【0138】
補助活性化合物も組成物に配合することができる。ある態様では、IL−13活性が有害である障害の治療に有用な1種以上の他の治療剤と本発明の抗体又は抗体部分を合剤化及び/又は併用投与する。例えば、他のターゲットと結合する1種以上の他の抗体(例えば他のサイトカインと結合する抗体又は細胞表面分子と結合する抗体)と本発明の抗hIL−13抗体又は抗体部分を合剤化及び/又は併用投与することができる。更に、1種以上の本発明の抗体を上記治療剤の2種以上と併用することもできる。このような併用療法は治療剤の投与用量を減らし、各種単独療法に伴う可能性のある毒性や合併症を避けることができるという利点がある。熟練した実施者には自明のことながら、本発明の抗体を併用療法の一部として使用する場合には、抗体を単独で対象に投与する場合よりも抗体の用量を減らすことが望ましいと思われる(例えば併用療法の使用により相乗治療効果を達成することができ、その結果、低用量の抗体を使用して所望の治療効果を達成することが可能になる)。
【0139】
当然のことながら、本発明の抗体又はその抗原結合部分は単独で使用してもよいし、別の物質(例えば治療剤)と併用してもよく、前記別の物質はその所期目的に合わせて当業者により選択される。例えば、別の物質は本発明の抗体により治療される疾患又は病態を治療するために有用であると当分野で認められている治療剤とすることができる。別の物質は治療用組成物に有益な属性を付与する物質(例えば組成物の粘性に作用する物質)でもよい。
【0140】
更に当然のことながら、本発明に含まれる併用剤は、その所期目的に有用な併用剤である。以下に挙げる物質は例示であり、限定的ではない。本発明に含まれる併用剤は本発明の抗体と下記から選択される少なくとも1種の他の物質から構成することができる。形成される組成物がその所期機能を実施できるような併用剤であるならば、併用剤は2種以上の他の物質、例えば2種又は3種の他の物質を含有することもできる。
【0141】
所定の併用剤は、非ステロイド性抗炎症薬(別称NSAIDS)であり、イブプロフェン等の薬剤を含む。他の併用剤はプレドニゾロン等のコルチコステロイドであり、本発明の抗体と併用して患者を治療する際に必要なステロイド用量を徐々に減らすことによりステロイド使用の周知副作用を低減又は解消することさえできる。本発明の抗体又は抗体部分と併用することができる関節リウマチ治療剤の非限定的な例としては、サイトカイン抑制性抗炎症薬(CSAID);他のヒトサイトカイン又は成長因子(例えばTNF、LT、IL−1、IL−2、IL−6、IL−7、IL−8、IL−15、IL−16、IL−18、EMAP−II、GM−CSF、FGF及びPDGF)の抗体又はアンタゴニストが挙げられる。本発明の抗体又はその抗原結合部分はCD2、CD3、CD4、CD8、CD25、CD28、CD30、CD40、CD45、CD69、CD80(B7.1)、CD86(B7.2)、CD90等の細胞表面分子又はそのリガンド(例えばCD154(gp39又はCD40L))に対する抗体と併用することができる。
【0142】
治療剤の組合せによっては、自己免疫とそれに続く炎症カスケードにおける種々の点に介入する場合があり、例としては、キメラ、ヒト化又はヒトTNF抗体、D2E7(その教示内容全体を本願に援用する1996年2月9日出願米国出願第08/599,226号)、cA2(Remicade(登録商標))、CDP571、抗TNF抗体フラグメント(例えばCDP870)、及び可溶性p55又はp75 TNF受容体、その誘導体(p75TNFRIgG(Enbrel(登録商標))又はp55TNFRIgG(レネルセプト)、可溶性IL−13受容体(sIL−13)、並びにTNFα変換酵素(TACE)阻害剤等のTNFアンタゴニストが挙げられ、同様にIL−1阻害剤(例えばVx740又はIL−1RA等のインターロイキン−1変換酵素阻害剤)も同じ理由から有効であると思われる。他の併用剤としては、インターロイキン11、抗P7及びp−セレクチン糖蛋白質リガンド(PSGL)が挙げられる。更に他の併用剤としては、IL−13機能と平行、依存又は協調して作用し得る自己免疫反応の他の主要素が挙げられる。更に別の併用剤としては、非枯渇性抗CD4阻害剤が挙げられる。更に他の併用剤としては、抗体、可溶性受容体又はアンタゴニストリガンドを含む共刺激経路CD80(B7.1)又はCD86(B7.2)のアンタゴニストが挙げられる。
【0143】
本発明の抗体又はその抗原結合部分は、メトトレキサート、6−MP、アザチオプリンスルファサラジン、メサラジン、オルサラジンクロロキン/ヒドロキシクロロキン、ペニシラミン、金チオリンゴ酸塩(筋肉内及び経口)、アザチオプリン、コルヒチン、コルチコステロイド(経口、吸入及び局所注射)、β2アドレナリン受容体アゴニスト(サルブタモール、テルブタリン、サルメテロール)、キサンチン(テオフィリン、アミノフィリン)、クロモグリケート、ネドクロミル、ケトチフェン、イプラトロピウム及びオキシトロピウム、シクロスポリン、FK506、ラパマイシン、ミコフェノール酸モフェチル、レフルノミド、NSAID(例えばイブプロフェン)、コルチコステロイド(例えばプレドニゾロン)、ホスホジエステラーゼ阻害剤、アデノシンアゴニスト、抗血栓剤、補体阻害剤、アドレナリン作動薬、炎症誘発性サイトカイン(例えばTNFα又はIL−1)によるシグナル伝達を妨害する物質(例えばIRAK、NIK、IKK、p38又はMAPキナーゼ阻害剤)、IL−1β変換酵素阻害剤(例えばVx740)、抗P7、p−セレクチン糖蛋白質リガンド(PSGL)、TNFα変換酵素(TACE)阻害剤、T細胞シグナル伝達阻害剤(例えばキナーゼ阻害剤)、メタロプロテイナーゼ阻害剤、スルファサラジン、アザチオプリン、6−メルカプトプリン、アンギオテンシン変換酵素阻害剤、可溶性サイトカイン受容体とその誘導体(例えば可溶性p55又はp75 TNF受容体と誘導体p75TNFRIgG(ENBREL(登録商標))及びp55TNFRIgG(レネルセプト)、sIL−1RI、sIL−1RII、sIL−6R、可溶性IL−13受容体(sIL−13))、並びに抗炎症性サイトカイン(例えばIL−4、IL−10、IL−11、IL−13及びTGF)等の物質と併用することもできる。所定の併用剤はメトトレキサート又はレフルノミドを含み、中等度又は重度関節リウマチの場合にはシクロスポリンを含む。
【0144】
本発明の医薬組成物は「治療有効量」又は「予防有効量」の本発明の抗体又は抗体部分を含有することができる。「治療有効量」とは必要な用量と期間で所望治療結果を達成するために有効な量を意味する。抗体又は抗体部分の治療有効量は個体の疾患状態、年齢、性別及び体重や、抗体又は抗体合部分が個体に所望応答を誘発する能力等の因子により異なる。治療有効量は抗体又は抗体部分の有益な治療作用が毒性又は有害作用を上回る量でもある。「予防有効量」とは必要な用量と期間で所望予防結果を達成するために有効な量を意味する。典型的には、予防用量は初期疾患ステージ又はそれ以前の対象で使用するので、予防有効量は治療有効量よりも少ない。
【0145】
最適所望応答(例えば治療又は予防応答)が得られるように投与レジメンを調節することができる。例えば、単回ボーラスを投与してもよいし、時間をかけて用量を数回に分けて投与してもよいし、治療状況の緊急性に応じて用量を比例的に増減してもよい。ある実施形態では、投与し易く、用量を均一にするために非経口組成物を用量単位形態に製剤化すると特に有利である。本願で使用する用量単位形態とは治療する哺乳動物対象に単位用量として適した物理的に別個の単位を意味し、各単位は所望治療効果を生じるように計算された所定量の活性化合物を必要な医薬担体と共に含有する。本発明の用量単位形態の仕様は(a)活性化合物の固有特性及び達成すべき特定治療又は予防効果と、(b)個体の過敏症の治療用としてこのような活性化合物を配合する技術に内在する問題により決定され、これらの因子に直接依存する。
【0146】
本発明の抗体又は抗体部分の治療有効量又は予防有効量の非限定的な範囲の例は0.01〜20mg/kg、又は1〜10mg/kg、又は0.3〜1mg/kgである。なお、用量値は緩和しようとする病態の種類と重篤度により異なる。更に、当然のことながら、任意特定対象の特定投与レジメンは個体の必要と組成物の投与者又は投与監理者の専門的判断に従って時間と共に調節すべきであり、本願に記載する用量範囲は例示に過ぎず、特許請求の範囲に記載する組成物の範囲又は実施を制限するものではない。
【0147】
8.本発明の抗体の使用
8.1 抗IL−13抗体の使用概論
本発明の抗IL−13抗体又はその抗原結合部分は、IL−13と結合することができるため、酵素免疫測定法(ELISA)、放射免疫測定法(RIA)又は組織免疫組織化学等の従来のイムノアッセイを使用して(例えばサンプルマトリックス中、ある態様では血清や血漿等の生体サンプル中で)IL−13、ある態様ではhIL−13を検出するために使用することができる。本発明は生体サンプル中のIL−13の検出方法として、サンプルを本発明の抗体又は抗体部分と接触させる段階と、IL−13と結合した抗体又は未結合抗体を検出することにより、サンプル中のIL−13を検出する段階を含む方法を提供する。結合した抗体又は未結合抗体の検出を容易にするために検出可能な物質で抗体を直接又は間接的に標識する。適切な検出可能な物質としては各種酵素、補欠分子族、蛍光材料、発光材料及び放射性材料が挙げられる。適切な酵素の例としては西洋ワサビペルオキシダーゼ、アルカリホスファターゼ、β−ガラクトシダーゼ又はアセチルコリンエステラーゼが挙げられ、適切な補欠分子族複合体の例としてはストレプトアビジン/ビオチンとアビジン/ビオチンが挙げられ、適切な蛍光材料の例としてはウンベリフェロン、フルオレセイン、イソチオシアン酸フルオレセイン、ローダミン、ジクロロトリアジニルアミンフルオレセイン、ダンシルクロリド又はフィコエリスリンが挙げられ、発光材料の1例としてはルミノールが挙げられ、適切な放射性材料の例としては、125I、131I、35S又は3Hが挙げられる。診断関連、例えばIL−13の上昇を伴う病態の診断にはサンプル中のIL−13の検出が有用であると思われ、及び/又は抗IL−13抗体による治療が有効であり得る対象を同定するのに有用であると思われる。
【0148】
標識抗IL−13抗体を利用する検出アッセイの代法として、例えば検出可能な物質で標識したrhIL−13標準と未標識抗IL−13抗体(例えば抗hIL−13抗体)を利用する競合イムノアッセイによりサンプル中のIL−13を検出することができる。このアッセイでは、サンプルと、標識rhIL−13標準と、抗hIL−13抗体を混合し、未標識抗体に結合した標識rhIL−13標準の量を測定する。サンプル中のhIL−13の量は抗hIL−13抗体と結合した標識rhIL−13標準の量に反比例する。
【0149】
本発明の抗体及び抗体部分は、IL−13活性、ある態様ではhIL−13活性をインビトロ及びインビボで中和することができる。従って、本発明の抗体及び抗体部分は例えばIL−13を含有する細胞培養液や、本発明の抗体と交差反応するIL−13をもつヒト対象又は他の哺乳動物対象(例えばヒヒ、カニクイザル及びアカゲザル等の霊長類)においてIL−13活性を阻害するために使用することができる。ある態様において、本発明はヒトIL−13と、ヒヒIL−13、マーモセットIL−13、チンパンジーIL−13、カニクイザルIL−13及びアカゲザルIL−13から構成される群から選択される少なくとも1種の他の霊長類IL−13の活性を中和し且つマウスIL−13の活性を阻害しない単離ヒト抗体又はその抗原結合部分を提供する。ある態様において、IL−13はヒトIL−13である。例えば、hIL−13を含有する細胞培養液又は含有する疑いのある細胞培養液において、本発明の抗体又は抗体部分を培養培地に加え、培養液中のhIL−13活性を阻害することができる。
【0150】
別の態様において、本発明はIL−13活性が有害である障害に罹患した対象におけるIL−13活性の阻害方法を提供する。本願で使用する「IL−13活性が有害である障害」なる用語はこの障害に罹患した対象におけるIL−13の存在が障害の病態生理の原因であるか又は障害の増悪に寄与する因子であることが分かっているか又はその疑いのある疾患及び他の障害を包含するものとする。従って、IL−13活性が有害である障害は、IL−13活性の阻害が障害の症状及び/又は進行を緩和すると予想される障害である。このような障害は例えば障害に罹患した対象の体液中のIL−13濃度の上昇(例えば対象の血清、血漿、滑液等におけるIL−13濃度の上昇)により判断することができ、例えば上記のような抗IL−13抗体を使用して検出することができる。ある態様において、抗体又はその抗原結合部分は本願に記載する疾患又は障害を治療するための療法で使用することができる。別の態様において、抗体又はその抗原結合部分は本願に記載する疾患又は障害の治療用医薬を製造するために使用することができる。IL−13活性が有害である障害としては多数の例が挙げられる。例えば、IL−13は免疫及び炎症要素を伴う各種疾患(限定されないが、例えば喘息や慢性閉塞性肺疾患等の呼吸器障害)に関連する病理に重要な役割を果たす。その他のIL−13関連障害としては、限定されないが、アトピー性障害(例えばアトピー性皮膚炎及びアレルギー性鼻炎);皮膚、胃腸臓器(例えば潰瘍性大腸炎及び/又はクローン病等の炎症性腸疾患(IBD))、及び肝臓(例えば肝硬変、線維症)の炎症性及び/又は自己免疫性病態;強皮症;腫瘍又は癌(例えばホジキンリンパ腫)が挙げられる。
【0151】
従って、抗IL−13抗体もしくはその抗原結合部分又はこれらをインビボ発現するベクターはIL−13の異常発現の結果としてIL−13の過剰を生じる疾患(例えば喘息又は他の炎症性及び/又は自己免疫性病態)の治療又は体外から投与したIL−13に起因する合併症の場合に処方される。
【0152】
8.2 呼吸器障害における抗IL−13抗体の使用
本発明のある実施形態において、抗IL−13抗体又はその抗原結合部分は1種以上のIL−13関連障害の治療に利用され、このような障害としては、限定されないが、呼吸器障害(例えば喘息(例えばアレルギー性及び非アレルギー性喘息(例えば、例えば幼児における例えば呼吸器合胞体ウイルス(RSV)感染に起因する喘息))、慢性閉塞性肺疾患(COPD)、並びに気道炎症、好酸球増加、線維化及び粘液過剰産生を伴う他の病態(例えば嚢胞性線維症及び肺線維症))が挙げられる。
【0153】
ある実施形態において、本願は呼吸器障害、例えば喘息(例えばアレルギー性及び非アレルギー性喘息);アレルギー;慢性閉塞性肺疾患(COPD);気道炎症、好酸球増加、線維化及び粘液過剰産生を伴う病態(例えば嚢胞性線維症及び肺線維症)に関連する1種以上の症状の治療(例えば抑制、改善)又は予防方法を提供する。例えば、喘息の症状としては、限定されないが、喘鳴、息切れ、気道狭窄、気道過敏、肺活量低下、線維化、気道炎症及び粘液産生が挙げられる。前記方法は1種以上の症状を治療(例えば抑制、改善)又は予防するために十分な量のIL−13抗体又はそのフラグメントを対象に投与する段階を含む。IL−13抗体は治療用又は予防用又はその両方に投与することができる。IL−13アンタゴニスト(例えば抗IL−13抗体)又はそのフラグメントは対象に単独で投与してもよいし、本願に記載する他の治療手段と併用投与してもよい。ある実施形態において、対象は哺乳動物(例えば本願に記載するようなIL−13関連障害に罹患したヒト)である。
【0154】
上記のように、IL−13は喘息に関連する病理応答の誘導に中枢的役割を果たすと考えられている。しかし、別の免疫経路の他のメディエーターも喘息病因に関与しているので、IL−13に加えてこれらのメディエーターを遮断すると、他の治療効果が得られると思われる。従って、本発明の結合蛋白質は、限定されないが、例えばIL−13と炎症誘発性サイトカイン(例えば腫瘍壊死因子α(TNFα))等の標的対と結合することが可能な二重特異性抗体に配合することができる。TNFαは喘息において炎症応答を増幅し、疾患の重篤度に関係があるらしい(McDonnell et al.,Progress in Respiratory Research(2001),31(New Drugs for Asthma,Allergy and COPD),247−250.)。従って、IL−13とTNFαを同時に遮断すると、特に重度気道疾患において有益な効果があると思われる。非限定的な1態様において、本発明の二重特異性抗体はIL−13とTNFαの標的に結合し、喘息を治療するために使用される。
【0155】
別の態様において、 本発明の結合蛋白質はIL−13とIL−1β、IL−13とIL−9、IL−13とIL−4、IL−13とIL−5、IL−13とDL−25、IL−13とTARC、EL−13とMDC、IL−13とMIF、IL−13とTGF−β、EL−13とLHRアゴニスト、DL−13とCL25、IL−13とSPRR2a、EL−13とSPRR2b、及びDL−13とADAM8に結合する二重特異性抗体分子を作製するために使用することができる。本発明は更に、CSF1(MCSF)、CSF2(GM−CSF)、CSF3(GCSF)、FGF2、IFNA1、IFNB1、IFNG、ヒスタミン及びヒスタミン受容体、EL1A、DL1B、BL2、IL3、EL4、IL5、IL6、IL7、IL8、IL9、IL10、EL11、IL12A、IL12B、IL14、IL15、IL16、IL17、IL18、EL19、IL−20、IL−21、IL−22、EL−23、EL−24、EL−25、IL−26、IL−27、EL−28、IL−30、EL−31、EL−32、IL−33、KtTLG、PDGFB、IL2RA、EL4R、IL5RA、IL8RA、DL8RB、IL12RB1、IL12RB2、EL13RA1、IL13RA2、IL18R1、TSLP、CCL1、CCL2、CCL3、CCL4、CCL5、CCL7、CCL8、CCL13、CCL17、CCL18、CCL19、CCL20、CCL22、CCL24.CX3CL1、CXCL1、CXCL2、CXCL3、XCL1、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、CX3CR1、GPR2、XCR1、FOS、GATA3、JAK1、JAK3、STAT6、TBX21、TGFB1、TNFSF6、YY1、CYSLTR1、FCER1A、FCER2、LTB4R、TB4R2、LTBR並びにキチナーゼから構成される群から選択される喘息に関与する1種以上の標的とIL−13に結合することが可能な二重特異性抗体も提供する。
【実施例】
【0156】
1.抗IL−13抗体の産生
原薬の産生バッチは産生用リアクターの1サイクルに由来する原薬のシード継代培養、産生、一次回収・捕集、及び精密精製から得られたABT−308モノクローナル抗体の溶液である。
【0157】
1.1 培地調製
USP/EP/JP標準に準拠する精製水を使用し、GMP Solution Recordsに従って溶液を調製する。調合培地溶液を0.1μmフィルターで濾過し、予め滅菌した適当な寸法の容器、バッグ又はバイオリアクターに捕集する。使用後に0.1μnmフィルターの完全性を試験する。増殖培地と産生培地の組成を表2に示す。
【0158】
【表2】
【0159】
1.2 接種材料増殖
スピナーフラスコとバイオウェーブバッグの操作を利用してCHO細胞をMCBの凍結バイアル1本から110Lシードバイオリアクターの接種に望ましいバイオマスまで増殖させる。マスターセルバンクの凍結バイアルを解凍し、増殖培地(SR−512)に加え、遠心する。細胞を増殖培地に再懸濁し、容積を増加させながら使い捨てスピナーフラスコ又はバイオウェーブバッグで37℃、5% CO2にて増殖させる。20Lウェーブバッグ2個を使用し、シードバイオリアクターに接種する前に20Lウェーブバッグ2個を使用して最終細胞塊増殖を最大にする。両方の20Lウェーブバッグからの細胞密度が約15〜17日目に生存細胞≧2.0×106個/mLに達したら、更に増殖させるために、増殖培地SR−520を充填した110シードバイオリアクターに培養液を移す。接種後、目標温度を37℃とし、pHを7.1の目標値に設定し、NaOH添加とCO2スパージングにより制御する。空気と酸素のスパージングによりバイオリアクター内の溶存酸素(DO)を40%の目標値に制御する。約2〜4日後に細胞密度が生存細胞≧2.6×106個/mLに達したら、培養液を3000L産生用バイオリアクターに移す。
【0160】
1.3 ショートフィルバイオリアクター
3000L産生用バイオリアクターの部分フィルを使用して細胞培養液を更に増殖させる。先ず、リアクターに増殖培地(SR−520)を仕込み、110Lシードバイオリアクターからのバッチを接種する。
【0161】
このショートフィル段階中は、温度、溶存酸素及びpHを夫々37℃、40%及び7.1に制御する。培養pHはCO2スパージングとNaOH添加により制御する。典型的には、細胞は必要な密度である生存細胞≧1.6×106個/mLに達する前に2〜4日間増殖する。
【0162】
1.4 産生用バイオリアクター
産生培地SR−521(1950L)を3000Lバイオリアクター内の細胞培養液に加え、産生段階を開始する。消泡剤Cを加えて発泡を抑える。CO2スパージングとNaOH添加のオンオフにより培養pHを6.9の目標値に制御する。温度と溶存酸素を夫々35℃と40%の目標値に制御する。バイオリアクター内のDOは先ず空気スパージングにより所望値に制御した後、必要に応じて純水素を補充する。生存細胞密度が≧3.0×106個/mLに達したら、温度を33℃の目標値まで下げ、pHとDOを夫々6.9と40%の目標値に維持する。必要に応じてグルコース(SR−334)を加える。細胞生存率が≦50%まで低下したら、培養液を回収する。
【0163】
1.5 工程性能
工程性能及びインプロセス試験結果を表3及び表4に示す。
【0164】
【表3】
【0165】
【表4】
【0166】
2.抗IL−13抗体の単離・精製
一次回収・捕集操作は、濾過による回収液の清澄化、プロテインAアフィニティークロマトグラフィーによる抗体の捕集、及び低pHウイルス不活性化と、その後の深層濾過を含む。精密精製操作はアニオン交換クロマトグラフィー、疎水性相互作用クロマトグラフィー、ウイルス濾過、限外濾過/透析濾過、並びに最終濾過、ボトリング及び凍結を含む。
【0167】
2.1 溶液の調製
USP精製水(USP−PW)又は注射用水(WFI)を使用し、GMP Solution Recordsに従って溶液を調製する。大半の溶液を0.2μmフィルターで濾過し、放射線照射済みバッグ、オートクレーブ滅菌済み又は現場蒸気処理済み容器に捕集する。
【0168】
2.2 一次回収及び清澄化
濾過による一次回収の目的は、産生用バイオリアクター回収液から細胞及び細胞破片を除去することである。未処理回収液をデプスフィルター、デリピッドデプスフィルター及びメンブレンフィルターから構成されるフィルター列に通す。清澄化上清を回収液槽に捕集し、2〜8℃に維持する。清澄化した回収液のインプロセス制御はPoros AクロマトグラフィーによるABT−308濃縮、バイオバーデン及び内毒素試験を含む。
【0169】
2.3 プロテインAアフィニティークロマトグラフィー
プロテインAアフィニティークロマトグラフィーの目的は、清澄化した回収液からABT−308を捕集し、工程関連不純物を低減することである。回収液全体を処理するために典型的には3クロマトグラフィーサイクルを実施する。後続処理のために3サイクルからの生成物プールを集める。
【0170】
直径45cm×長さ22cmのカラム(35L)にMabSelect(登録商標)プロテインA樹脂(GE Healthcare)又はProSep Ultra Plus(登録商標)(Millipore)を充填し、使用できるようにする。USP精製水(USP−PW)後に0.2M酢酸を使用し、最後にUSP PWでリンスすることにより保存バッファーをカラムから除去する。カラムを25mMトリス,100mM NaCl,pH7.2で平衡化後、MabSelect(登録商標)プロテインA樹脂(GE Healthcare)の場合には蛋白質32g/L樹脂、ProSep Ultra Plus(登録商標)(Millipore)樹脂の場合には蛋白質45g/L樹脂の最大値まで清澄化した回収液をロードする。カラムを25mMトリス,100mM NaCl,pH7.2、次いで20mMクエン酸ナトリウム,0.5M NaCl,pH6.0で洗浄後、最後に25mMトリス,100mM NaCl,pH7.2で再び洗浄する。0.1M酢酸,pH3.5でカラムから抗体を溶出させる。各サイクル後、必要に応じて溶出液プールのpHを4.1の目標値に調整する。インプロセス制御はA280による蛋白質濃度の測定、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0171】
第1サイクル後、カラムを0.2M酢酸で再生し、USP−PWでリンスする。第2サイクル後、カラムを0.2M酢酸で再生し、USP−PWでリンスした後、0.1M酢酸、20%エタノールで消毒後、50mM酢酸ナトリウム、20%エタノール,pH5で洗浄・短期間保存する。第3サイクル後、カラムを0M酢酸で再生し、USP−PWでリンスする。その後、0.4M酢酸,0.5M NaCl,0.1% Tween 80で洗浄後、USP−PWで洗浄し、次いで50mM NaOH 1.0M NaClで洗浄後、USP−PWで洗浄する。最後に、0.1M酢酸,20%エタノールで消毒後、50mM酢酸,20%エタノール,pH5.0で洗浄・保存する。
【0172】
2.4 低pHインキュベーション及び濾過
低pHインキュベーションは、プロテインA溶出液中に存在する可能性のある外来エンベロープウイルスの不活性化によりウイルス安全性を更に確実にする専用ウイルス低減工程である。低pHインキュベーション後の濾過の目的は低pH処理中に形成される可能性のある沈殿を除去することである。
【0173】
集めたプロテインAクロマトグラフィー溶出液のpHを0.5Mリン酸で3.5の目標値に調整し、18〜25℃に60〜70分間維持する。次に、混合物を1Mトリス,pH10でpH5に調整し、デプスフィルターとメンブレンフィルターの併用により清澄化した後、10〜14℃まで冷却する。低pH処理・濾過工程のインプロセス制御はA280による蛋白質濃度の測定、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0174】
2.5 一次回収・捕集工程性能
一次回収・捕集操作の工程性能を表5に示し、インプロセス制御の結果を表6に示す。
【0175】
【表5】
【0176】
【表6】
【0177】
2.6 強アニオン交換クロマトグラフィー
強アニオン交換クロマトグラフィー工程の目的は、宿主細胞蛋白質、DNA及び内毒素等の工程関連不純物を低減することである。この工程は、ウイルス除去工程も兼用できる。ある実施形態では、抗体がカラムを通過し、不純物が樹脂に保持されるフロースルーモードでQ Sepharose(登録商標)FF樹脂カラムを操作し、代替態様では、Q Sepharose FF樹脂カラムの代わりにMustang Q(登録商標)メンブレン(Pall Corp.)を利用する。操作は10〜14℃で実施する。
【0178】
直径45cm×長さ22cmのカラム(35L)にQ Sepharose(登録商標)FF樹脂(GE Healthcare)を充填し、使用できるようにする。カラムを25mMトリス,50mM NaCl,pH8.0で平衡化する。中和・濾過した不活性化溶液のpHを1Mトリス,pH10で8.0に調整し、導電率を5.0〜6.5に調整し、溶液をデリピッド及びメンブレンフィルターで濾過する。Q Sepharose(登録商標)ロードを蛋白質80g/L樹脂の最大ロードでカラムにポンプ導入する。ロード後、カラムを25mMトリス,50mM NaCl,pH8.0で洗浄し、フロースルーと洗浄液を合わせる。これをQ Sepharose(登録商標)フロースルー・洗浄液(QFTW)プールとする。Q Sepharose(登録商標)工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0179】
カラムを25mMリン酸ナトリウム,1.0M NaCl,pH7.0で再生した後、WFIでリンスし、1.0M NaOHで消毒後、WFIでリンスする。次にカラムを25mMリン酸ナトリウム,1.0M NaCl,pH7.0で中和し、25mMリン酸ナトリウム,20%イソプロパノール,pH7.0中で保存する。
【0180】
2.7 疎水性相互作用クロマトグラフィー
Phenyl Sepharose(登録商標)工程の目的は、ABT−308凝集物、フラグメント及び工程関連不純物を除去することである。操作は10〜14℃で実施する。
【0181】
直径60cm×長さ15cmのカラム(42L)にPhenyl Sepharose(登録商標)HP樹脂(GE Healthcare)を充填し、使用できるようにする。カラムをWFI、次いで20mMリン酸ナトリウム,1.1M硫酸アンモニウム,pH7.0で平衡化する。
【0182】
Q Sepharose(登録商標)フロースルー及び洗浄液を40mMリン酸ナトリウム,2.2M硫酸アンモニウム,pH7.0で1:1(v/v)に希釈する。この溶液即ちPhenyl Sepharose(登録商標)ロードを0.2μmフィルターで濾過し、蛋白質64g/L樹脂の最大ロードでカラムにロードする。カラムを25mMリン酸ナトリウム,1.4M硫酸アンモニウム,pH7.0で洗浄し、ABT−308を11mMリン酸ナトリウム,0.625M硫酸アンモニウム,pH7.0でカラムから溶出させる。Phenyl Sepharose(登録商標)工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0183】
カラムをWFIで再生した後、1M NaOHで消毒し、WFIでリンスし、25mMリン酸ナトリウム,20%イソプロパノール,pH7中で保存する。
【0184】
2.8 ナノ濾過
ナノ濾過は、Phenyl Sepharose(登録商標)HPカラム溶出液中に存在する可能性のある直径≧20nmの外来ウイルスの物理的除去によりウイルス安全性を更に確実にする専用ウイルス除去工程である。操作は10〜14℃で実施する。
【0185】
15mMヒスチジン,pH5.6で予め湿潤させた0.1μmフィルターとUltipor DV20 フィルター列にPhenyl Sepharose(登録商標)HPカラム溶出液を通す。濾過後、フィルター列を15mMヒスチジン,pH5.6でフラッシュし、保持されたABT−308を回収する。使用後、DV20フィルターで完全性試験を実施し、フィルターを捨てる。フィルターが完全性試験に合格しない場合には、溶液を上記のように再濾過すればよい。ナノ濾過工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0186】
2.9 限外濾過/透析濾過によるABT−308原薬の製剤化
UF/DF工程の目的は、原薬を最終製剤化バッファーである15mMヒスチジン,pH5.6で透析濾過し、ABT−308を濃縮することである。これらの操作は10〜14℃で実施する。
【0187】
30kDa MWCOポリエーテルスルホンメンブレンを使用してナノ濾液を約50g/Lまで濃縮し、製剤化バッファーで透析濾過した後、約180g/Lまで濃縮する。UFシステムから生成物を取出し、透析濾過バッファーでリンスし、システム内に生成物が残っている場合には回収する。濃縮液と洗浄液を合わせ、約120〜160g/Lの濃度の透析濾過ABT−308を生成する。濃縮したABT−308をメンブレンフィルターで濾過する。限外濾過/透析濾過工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0188】
各回の試験後、限外濾過システムをWFIでフラッシュし、250ppm次亜塩素酸ナトリウム溶液で洗浄後、0.1M水酸化ナトリウムで消毒・保存する。
【0189】
2.10 最終濾過、ボトリング及び凍結
濾過・ボトリング操作は、クラス100層流フード下にクラス100領域で2〜8℃にて実施する。製剤化したABT−308を0.2μmフィルターで濾過し、予め滅菌したパイロジェンフリーPETGボトルに捕集する。ラベルを付けたボトルを凍結するまで空の−80℃(公称値)冷凍庫に入れた後、−80℃(公称値)に維持した保存用冷凍庫に移す。最終濾過・ボトリング工程のインプロセス制御はA280、バイオバーデン及び内毒素試験(原薬試験結果)を含む。
【0190】
2.11 精密精製工程性能
一次回収・捕集操作の工程性能を表7に示し、インプロセス制御結果を表8に示す。
【0191】
【表7】
【0192】
【表8】
【0193】
3.抗体組成物中の宿主細胞蛋白質濃度の測定
この工程は、抗体サンプル中の残留宿主細胞蛋白質濃度の測定試験方法に関する。酵素免疫測定法(ELISA)を使用して宿主細胞蛋白質(抗原)を2層の特異的抗体の間にサンドイッチする。その後、非特異的部位をカゼインでブロックする。次に宿主細胞蛋白質をインキュベートすると、この間に抗原分子は第1の抗体(コーティング抗体)により捕集される。次に抗原(宿主細胞蛋白質)と結合する第2の抗体(ビオチン化抗宿主細胞蛋白質)を加える。ビオチン化抗宿主細胞蛋白質と結合するニュートラアビジンHRP−コンジュゲートを加える。次にK−Blue基質を加える。発色基質は結合した酵素共役抗体により加水分解され、青色を発色する。2M H3PO4で反応を停止すると、黄色に変色する。色強度はウェル内で結合した抗原の量に正比例する。
【0194】
50mM重炭酸ナトリウム(コーティングバッファー),pH9.4の調製。1LビーカーにミリQ水900mL、重炭酸ナトリウム4.20g±0.01gを加える。完全に溶解するまで撹拌する。pHを1N NaOHで9.4に調整する。1Lメスフラスコに移し、ミリQ水を補充する。均質になるまで反転により混合する。0.22μm滅菌フィルターユニットで濾過する。調製日から7日間まで公称値4℃で保存する。
【0195】
0.104M Na2HPO4・7H2O,1.37M NaCl,0.027M KCl,0.0176M KH2PO4,pH=6.8〜6.9(10×PBS)の調製。ミリQ水約400mLをガラスビーカーに加える。Na2HPO4・7H2O 13.94g±0.01gを加える。NaCl 40.0g±0.1gを加える。KCl 1.00g±0.01を加える。KH2PO4 1.20g±0.01gを加える。均質になるまで撹拌する。500mLメスフラスコに移す。ミリQ水を補充して500mLにする。反転により混合する。0.2μm滅菌フィルターユニットで濾過する。室温で7日間まで保存する。
【0196】
1×PBS+0.1%トリトンX−100,pH7.40(プレート洗浄バッファー)の調製。4Lメスシリンダー内で10×PBS(ステップ5.2)400mLをミリQ水3500mLと混合する。pHをチェックし、必要に応じて1N HCl又は1N NaOHで7.40±0.05に調整する。ミリQ水を補充する。シリンダーをパラフィルムで密閉し、均質になるまで反転により混合する。4Lボトルに移す。1×PBS 4mLを取り出し、捨てる。4mLのトリトンX−100を3996mの1×PBSに加える。撹拌プレートにセットし、完全に溶解するまで撹拌する。希釈バッファー調製に必要な量のプレート洗浄バッファーを0.22μm滅菌フィルターユニットで濾過する。室温で7日間まで保存する。
【0197】
コーティング抗体混合物:アフィニティー精製ヤギ抗CHO 599/626/748(ロット#G11201@1.534mg/mL)の調製(注:バイアルに入れて−80℃で保存したストック)。アリコートを調製する。使用時にプレート1枚当たりアリコート1個を取出す。使用直前に、抗体混合物を最終濃度4μg/mLとなるように冷温50mM重炭酸ナトリウムで次のように希釈する。例えば、コーティング抗体混合物31μLを冷温コーティングバッファー11969μLに加える。反転により穏やかに混合する。
【0198】
ビオチン化ヤギ抗宿主細胞蛋白質混合物599/626/748(ロット#G11202@0.822mg/mL)の調製(注:バイアルに入れて−80℃で保存したストック)。アリコートを調製する。使用時にプレート1枚当たりアリコート1個を取出す。使用直前に、ビオチン化抗体混合物を最終濃度1μg/mLとなるように37℃±2℃カゼインで次のように希釈する。例えば、ビオチン化抗体混合物14.6μLを37℃±2℃カゼイン11985μLに加える。反転により穏やかに混合する。
【0199】
ニュートラアビジン−HRPの調製。新規ロット(2mg/バイアル)を次のように1mg/mLまで再構成する。ミリQ水400μLをバイアルに加えた後、1×PBS 1600μLを加え、合計2mLとする。穏やかにボルテックスして混合する。公称値−20℃で保存する。プレート1枚当たりアリコート1個を使用するように所望体積のアリコートを調製する。ポリプロピレンチューブ内で調製する。ワーキング濃度を決定するように新規ロットを適格化する。調製日から6カ月間の期日を指定する。例えば、ワーキング濃度が0.2μg/mLと決定されたならば、以下のように調製する。使用直前にニュートラアビジン−HRPのアリコートを室温で解凍する。1mg/mLニュートラアビジン溶液を37℃±2℃カゼインで0.1mg/mL(100μg/mL)まで希釈する。例えば、10倍に希釈し、ニュートラアビジン50μLをカゼイン450μLに加える。穏やかにボルテックスして混合する。更に100μg/mL溶液を37℃±2℃カゼインで0.2μg/mLまで希釈する。例えば、500倍に希釈し、ニュートラアビジン(100μg/mL)24μLをカゼイン11976μLに加える。穏やかにボルテックスして混合する。
【0200】
5.7 2Mリン酸(停止溶液)の調製。濃リン酸から2Mリン酸溶液を次のように調製する。ラベルに記載されたリン酸%、密度(1.685g/mL)及び製剤重量(98g/モル)から、2Mリン酸500mLを調製するために必要な濃リン酸の体積を計算する。上記のように計算した体積の濃リン酸をフラスコに加える。ミリQ水を補充し、均質になるまで反転により混合する。調製日から6カ月間まで周囲温度で保存する。
【0201】
希釈バッファー(1×PBS+0.1%トリトンX100,pH7.4で100倍に希釈したカゼイン)の調製。(上記からの)0.22μmで滅菌濾過した1×PBS+0.1%トリトンX100,pH7.4で37℃±2℃カゼインを100倍に希釈する。例えば、0.22μmで滅菌濾過した1×PBS+0.1%トリトンX100(pH7.4)99mLに37℃±2℃カゼイン1mLを加える。よく混合する。使用毎に新たに調製する。
【0202】
標準の調製。宿主細胞蛋白質標準(抗原標準)(ロット#G11203@1.218mg/mL)(注:70μLアリコートとして公称値−80℃で保存したストック)。アリコートを室温で解凍する。希釈バッファーを使用してポリプロピレンチューブ内で系列希釈を実施する。
【0203】
サンプルの調製。ポリプロピレンチューブ内で最終バルクサンプルを希釈バッファーで24mg/mLまで希釈する。濃度を記録する。注:下記溶液を使用してスパイクサンプルを調製し、下記12mg/mL溶液を調製する。ポリプロピレンマイクロチューブ内で更に24mg/mL溶液を希釈バッファーで12mg/mLまで希釈する。12mg/mL溶液を各々プレート上の3個のウェルにロードし、合計6個のウェルにロードする。
【0204】
スパイクの調製。ポリプロピレンマイクロチューブ内で希釈バッファーで2倍に希釈することにより上記に調製した20ng/mL標準から10ng/mL宿主細胞蛋白質スパイクを調製する。10ng/mLスパイク溶液を各々プレート上の3個のウェルにロードする。サンプルをスパイクするためにステップ6.1からの20ng/mL標準溶液を使用する。
【0205】
スパイクサンプルの調製。ポリプロピレンマイクロチューブ内で24mg/mL最終バルク溶液各300μLを20ng/mLスパイク溶液(6.1)300μLでスパイクする。スパイクサンプル溶液を各々3個のウェルにロードし、合計6個のウェルにロードする。
【0206】
対照の調製。日常的試験で使用する前に全新規対照ストック溶液に対照範囲を設定する必要がある。対照ストック:ABT−308原薬濃縮液バッチの150μLアリコートを調製し、3年間まで公称値−80℃で凍結保存する。
【0207】
ワーキング対照の調製。対照のアリコートを室温で解凍する。ポリプロピレンチューブ内で対照を希釈バッファーで24mg/mLまで希釈する。ポリプロピレンマイクロチューブ内で更に24mg/mL対照溶液を希釈バッファーで12mg/mLまで希釈する。単一希釈液を調製し、対照をプレートの3個のウェルにロードする。
【0208】
ELISA手順。プレート洗浄ボトルにプレート洗浄バッファーを充填する(ステップ5.3参照,1×PBS+0.1%トリトンX−100)。プレートウォッシャーを起動する。以下のパラメーターをチェックする。パラメーターは以下のように設定すべきである:プレートタイプ:各サイクルで1(合計5サイクル);体積:400μL;浸漬時間:10秒;吸引時間:4秒。
【0209】
アッセイ手順。冷温50mM重炭酸ナトリウム中4μg/mLヤギコーティング抗体混合物100μL/ウェルでプレートをコーティングする。コーティング溶液がウェルの底を均等に覆うまでプレートの側面をタッピングし、シーリングテープを貼り付け、プレートシェーカー(又は同等手段)速度3にて18時間±1時間振盪しながら公称温度4℃でインキュベートする。一晩インキュベーション後、プレートを冷蔵庫から取り出し、室温で平衡化させる。コーティングを振り払う。プレートをペーパータオルで拭う。37℃±2℃カゼイン300μL/ウェルでブロックし、シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。ブロッキングインキュベーション中に標準、サンプル、対照、スパイク及びスパイクサンプルを調製する。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。8チャネルピペットを使用し、標準、サンプル、スパイク、スパイクサンプル及び対照100μL/ウェルをプレートの3個のウェルに分注する。ブランクとしてプレートの全ての空のウェルに希釈バッファー100μL/ウェルをピペットで分注する。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。テンプレートを作成し、プレートにロードする際のガイドとして使用する。
【0210】
プレートリーダー設定。テンプレートを設定し、標準用濃度を入力する。サンプル、対照、スパイク又はスパイクサンプルの希釈係数は入力しない。希釈剤を加えたウェルを全ウェルから差し引くブランクとして割り当てる。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。ビオチン化ヤギ抗体100μL/ウェルを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。ニュートラアビジン−HRPコンジュゲート溶液100μL/ウェルを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。冷温K−Blue基質100μL/ウェルを加え、シーリングテープを貼り付け、Lab−lineタイタープレートシェーカー(又は同等手段)で速度3にて振盪しながら室温で10分間インキュベートする(基質を最初の行に添加後、すぐにタイマーをスタートする)。2Mリン酸100μL/ウェルを加えることにより反応を停止する(ステップ5.7)。プレートをプレートシェーカーにセットし、速度3で3〜5分間振盪する。プレートを450nmで読取る。
【0211】
データ解析及び計算。注:光学密度が標準曲線の実際の定量限界(2.5ng/mL標準)内にあり且つ下記%CV又は%差条件を満たすサンプル、スパイク、スパイクサンプル及び対照のみが許容される。サンプルODが2.5ng/mL標準に達しない場合には、結果を2.5ng/mL未満と報告する。その後、この数値を希釈後のサンプル濃度(12mg/mL)で割り、数値(ng/mg)を報告する。サンプルの宿主細胞濃度が高く、非スパイク及び/又はスパイクサンプルが標準曲線を上回る場合には、数値を>100ng/mLと報告する。その後、この数値を希釈後のサンプル濃度(12mg/mL)で割り、数値(ng/mg)を報告する。サンプルが2.5ng/mL標準を下回る場合には、スパイク回収率計算のサンプル値をゼロとみなす。
【0212】
標準曲線。標準濃度をプロトコルテンプレートに入力する。二次曲線フィットを使用する。測定係数は0.99でなければならず、3個のウェル間の%CVは20%でなければならない。この条件を満たさない場合には、標準を1本(1レベル,ウェル3個)下げることができる。1.25ng/mL下げる場合には、光学密度が2.5ng/mL及び100ng/mL(残りの標準曲線の点)光学密度以内のサンプル及びスパイクサンプルのみが許容可能である。更に、各標準レベルの3個について、1個のウェルが明白に汚染されているか又は結合度が低い場合には、このウェルを下げることができる。1個のウェルを標準レベルから下げる場合には、残りの2個は%差=20%でなければならない。プレートのバックグラウンド(ブランク)に近いOD値を示す最低標準の%CVは30%でなければならない。ウェル1個を下げる場合には、残りの2個の%差は35%でなければならない。最低標準を下げる場合には、光学密度が残りの標準曲線レベル光学密度以内にあるサンプル及びスパイクサンプルのみが許容可能である。
【0213】
サンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを報告する。各サンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。注:非スパイクサンプルODが2.5ng/mL標準ODを下回る場合には、%差条件は非スパイク結果に適用されない。上記計算参照。
【0214】
平均(ng/mL)値から実際の宿主細胞濃度(ng/mg)を次のように計算する:CHO宿主細胞蛋白質(ng/mg)=平均「非スパイクサンプル結果(ng/mL)」/希釈後のサンプル濃度(12mg/mL)。
【0215】
スパイク。%CVは3個のウェル間で20%でなければならない。%CVを記録する。スパイクから1個のウェルを下げることができる。残りの点は%差=20%でなければならない。上記計算参照。宿主細胞濃度(ng/mL)を報告する。この結果をスパイク回収率計算で使用する。各スパイクに得られる濃度(ng/mL)は理論スパイク濃度の±20%でなければならない。結果を記録し、合否を判定する。スパイク結果が理論値の20%以内にない場合には、アッセイを繰返す必要がある。平均スパイク濃度(ng/mL)×100は100%±20% 10ng/mLでなければならない。
【0216】
スパイクサンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを記録する。各スパイクサンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。上記計算参照。各希釈液の「スパイクサンプル結果」(ng/mL)を報告する。2種類の希釈液間の%差を記録する。希釈液間の%差は25%でなければならない。これらの結果をスパイク回収率計算で使用する。
【0217】
下式を使用して各希釈液セットの%スパイク回収率を計算する:%スパイク回収率=スパイクサンプル値−非スパイクサンプル値×100。スパイク値。注:(1)非スパイクサンプル値ODが2.5ng/mL標準を下回る場合には、%スパイク回収率計算において数値をゼロとみなす。各サンプルの各希釈液の%スパイク回収率は100%±50%(50%〜150%)でなければならない。結果と合否を記録する。
【0218】
対照。%CVは3個のウェル間で20%でなければならない。%CV結果を記録する。対照からの1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。上記計算参照。対照の宿主細胞濃度を報告する(ng/mL)。宿主細胞濃度(ng/mg)を次のように計算する:宿主細胞蛋白質(ng/mg)=対照宿主細胞蛋白質結果(ng/mL)。
【0219】
4.抗体組成物中のプロテインA濃度の測定
このELISAでは、プレートをニワトリ抗プロテインAでコーティングし、インキュベートする。非特異的部位をPBS中カゼインでブロックする。プレートを1×PBS+0.1%トリトンX−100で洗浄し、未結合材料を除去する。サンプルとCys−プロテインA標準を1×PBS+4.1%トリトンX+10%カゼインで希釈する。95℃±2℃に加熱することにより溶液を変性させ、プロテインAを抗体から分離する。ある実施形態において、例えば(GE Healthcare)を使用する場合には、溶液をプレートに加え、インキュベートする。代替態様において、例えば、プロテインAアフィニティー工程がProSep Ultra Plus(登録商標)(Milipore)を使用する場合には、溶液を冷却し、0.85% NaCl+12.5% 1N酢酸+0.1% Tween 20を各チューブに加え(1:1)、サンプル蛋白質からプロテインAを更に分離し易くする。チューブを激しくボルテックスし、インキュベートし、遠心する。上清を取り出し、更に処理する。未結合材料を1×PBS+0.1%トリトンX−I00で洗い流す。ビオチン化ニワトリ抗プロテインAをマイクロタイタープレートに加え、インキュベートする。プレートを洗浄して未結合材料を除去し、ニュートラアビジン−ペルオキシダーゼコンジュゲートを加える。
【0220】
ニュートラアビジンはウェルと結合したビオチン化ニワトリ抗プロテインAと結合する。プレートを再び洗浄し、未結合ニュートラアビジンを除去し、K−Blue(テトラメチルベンジジン(TMB))基質をプレートに加える。基質は結合したニュートラアビジンにより加水分解され、青色を発色する。リン酸で反応を停止すると、黄色に変色する。ウェル内の黄色の強度はウェル内に存在するプロテインAの濃度に正比例する。
【0221】
試薬及び溶液の調製。カゼインボトルを37℃±2℃に加温し、2分間超音波処理し、分取する。アリコートを公称値4℃で保存する。アッセイを実施する場合には、必要数のカゼインアリコートを37℃±2℃にする必要がある。コーティングバッファーと基質は冷温で使用する(使用直前に公称値4℃から取出す)。
【0222】
50mM重炭酸ナトリウム(コーティングバッファー),pH9.4。1LビーカーにミリQ水900mL、重炭酸ナトリウム4.20g±0.01gを加える。完全に溶解するまで撹拌する。pHを1N NaOHで9.4に調整する。1Lメスフラスコに移し、ミリQ水を補充する。均質になるまで反転により混合する。0.22CAμm滅菌フィルターユニットで濾過する。調製日から7日間まで公称値4℃で保存する。
【0223】
104M Na2HPO4・7H2O,1.37M NaCl,0.027M KCl,0.0176M KH2PO4,pH=6.8〜6.9(10×PBS)の調製。ミリQ水約400mLをガラスビーカーに加える。Na2HPO4・7H2O 13.94g±0.01gを加える。NaCl 40.0g±0.1gを加える。KCl 1.00g±0.01gを加える。KH2PO4 1.20g±0.01gを加える。均質になるまで撹拌する。500mLメスフラスコに移す。ミリQ水を補充して500mLにする。反転により混合する。0.2CAμm滅菌フィルターユニットで濾過する。調製日から7日間まで室温で保存する。
【0224】
1×PBS+0.1%トリトンX−100,pH7.40(プレート洗浄バッファー)。4Lメスシリンダー内で10×PBS(上記参照)400mLをミリQ水3500mLと混合する。pHをチェックし、必要に応じて1N HCl又は1N NaOHで7.40±0.05に調整する。ミリQ水を補充する。シリンダーをパラフィルムで密閉し、均質になるまで反転により混合する。4Lボトルに移す。1×PBS 4mLを取り出し、捨てる。4mLのトリトンX−100を3996mLの1×PBSに加える。撹拌プレートにセットして撹拌し、完全に溶解させる。室温で7日間まで保存する。
【0225】
ニワトリ抗プロテインAコーティング抗体。使用時にプレート1枚当たり抗体アリコート1個を取出す。新規ロットのニワトリ抗プロテインAを適格化するためには、(同一ロットのコーティングから調製した)ニワトリ抗プロテインA−ビオチンコンジュゲートを同時に使用・適格化する必要があると思われる。使用直前に抗体混合物をコーティング適格化中に決定された濃度まで冷温50mM重炭酸ナトリウムで希釈する。例えば、適格化中にプレートにロードするコーティング濃度が6μg/mLと決定され、ストック濃度が3000μg/mLであるならば、コーティング抗体24μLを冷温コーティングバッファー11976μLに加える。反転により穏やかに混合する。
【0226】
ビオチン化ニワトリ抗プロテインA。使用時にプレート1枚当たり抗体アリコート1個を取出す。新規ロットのニワトリ抗プロテインA−ビオチンコンジュゲートを適格化するためには、コンジュゲートの作製に使用した同一ロットのニワトリ抗プロテインAと同時に使用・適格化する必要があると思われる。使用直前にビオチン化抗体をビオチン化抗体適格化中に決定された濃度まで37℃±2℃カゼインで希釈する。例えば、適格化中にプレートにロードするビオチン化抗体濃度が4μg/mLと決定され、ストック濃度が1000μg/mLであるならば、ビオチン化抗体48μLを37℃±2℃カゼイン11952μLに加える。反転により穏やかに混合する。
【0227】
ニュートラアビジン−HRP。新規ロット(2mg/バイアル)を次のように1mg/mLまで再構成する。ミリQ水400μLをバイアルに加えた後、1×PBS 1600μLを加え、合計2mLとする。穏やかにボルテックスして混合する。公称値−80℃で保存する。プレート1枚当たりアリコート1個を使用するように所望体積のアリコートを調製する。ポリプロピレンチューブ内で調製する。調製日から6カ月間の期日を指定する。例えば、ワーキング濃度が0.1μg/mLと決定されたならば、以下のように調製する。使用直前にニュートラアビジン−HRPのアリコートを室温で解凍する。1mg/mLニュートラアビジン溶液を37℃±2℃カゼインで0.01mg/mL(10μg/mL)まで希釈する。例えば、10倍に希釈し、ニュートラアビジン50μLをカゼイン450μLに加える。穏やかにボルテックスして混合し、再び10倍に希釈し、10倍ニュートラアビジン希釈液100μLをカゼイン900μLに加える。穏やかにボルテックスして混合する。更に10μg/mL溶液を37℃±2℃カゼインで0.1μg/mLまで希釈する。例えば、100倍に希釈し、ニュートラアビジン(10μg/mL)120μLをカゼイン11880μLに加える。数回穏やかに反転して混合する。
【0228】
停止溶液(購入した1Nリン酸を使用する)。入手日から1年間まで周囲温度で保存する。希釈バッファー(1×PBS+4.1%トリトンX100+10%カゼイン,pH7.4)。(ステップ5.3からの)1×PBS+0.1%トリトンX100(pH7.4)86mLをビーカー又はフラスコに加え、4mLのトリトンX−100を加え、ブロッカーカゼインPBS溶液10mLを加え、撹拌し、溶解/混合する。トリトンを溶解させるには20〜30分間かかると思われる。これは1×PBS+4.1%トリトンX100+10%カゼイン,pH7.4溶液に等しい。0.22CAμm滅菌フィルターユニットで濾過する。使用毎に新たに調製する。プレート1枚にはこれで十分である。
【0229】
プロテインA標準(抗原標準)。注:70μLアリコートとして公称値−20℃で保存したストック。アリコートを氷上で解凍する。製造業者のCOAに記載されている濃度で希釈バッファー(上記参照)を使用してポリプロピレンチューブ内で下表の例に従って系列希釈を実施する。例えば、ストック濃度が2.1mg/mL(2100000ng/mL)であるとCOAに記載されている場合には、サンプルを氷上で解凍する。ポリプロピレンマイクロ遠心管内で最終バルクサンプルを希釈バッファー(上記)で20mg/mLまで希釈する。別々に2回希釈を行う。濃度を記録する。下記溶液を使用してスパイクサンプルを調製し、10mg/mL溶液を調製する。例えば、濃度(mg/mL)、体積μLのXmg/mL溶液、体積(μL)の希釈剤で120個のストックサンプルからの系列希釈。ポリプロピレンマイクロ遠心管内で、20mg/mL溶液を更に希釈バッファーで10mg/mLまで希釈する。
【0230】
スパイクの調製。ポリプロピレンマイクロ遠心管内で、希釈バッファーで2倍に希釈することにより、上記ステップ6.1で調製した0.593ng/mL標準から0.296ng/mLプロテインAスパイクを調製する。希釈を1回行う。0.296ng/mLスパイク溶液をプレートの3個のウェルにロードする。サンプルをスパイクするにはステップ6.1からの0.593ng/mL標準溶液を使用する。
【0231】
スパイクサンプルの調製。ポリプロピレンマイクロ遠心管内で、20mg/mL最終バルク溶液各500μLを0.593ng/mLスパイク溶液500μLでスパイクする。変性用に保持する。各スパイクサンプル溶液をプレートの3個のウェルにロードし、合計6個のウェルにロードする。
【0232】
対照の調製。ABT−308原薬のロットを入手する。150μLアリコートを分取し、分取日から3年間まで公称値−80℃で凍結保存する。
【0233】
ワーキング対照:対照のアリコートを氷上で解凍する。ポリプロピレンマイクロ遠心管内で対照を最終体積1000μLとなるように希釈バッファーで10mg/mLまで希釈する。単一希釈液を調製する。変性用に保持する。プレートの3個のウェルに対照をロードする。
【0234】
変性。プレートブランクでは、プレートで試験するブランク数と同数のマイクロ遠心管に希釈バッファー1000μLを加える。加熱中にチューブのキャップが開かないようにパラフィルムで密閉するか、又はキャップを閉じた状態に維持するように第2のラックをキャップの上に配置することができる。標準、非スパイクサンプル、スパイクサンプル、スパイク、ブランク及び対照を95℃±2℃に15分間加熱する。パラフィルムを使用した場合には冷却中にチューブから剥がす。15分間冷却し、約10000rpmで5分間遠心する。上清700μLをマイクロチューブに移し、プレートにロードする。トリトン/蛋白質ペレットに支障のないように注意する。
【0235】
プレートウォッシャー指示及び水浴設定。プレート洗浄ボトルにプレート洗浄バッファー(ステップ5.3参照,1×PBS+0.1%トリトンX−100)を充填する。プレートウォッシャーを起動する。以下のパラメーターをチェックする。パラメーターは以下のように設定すべきである:プレートタイプ:各サイクルで1(合計4サイクル);吸引速度:10mm/s;体積:400μl;浸漬時間:5秒;吸引時間6秒。水浴をオンにし、95℃に設定する。水浴温度を95℃±2℃に少なくとも30分間平衡化させる。
【0236】
アッセイ手順:ステップを完了するにつれて外していくことによりチェックリストをガイドとして使用することができる。更に、アッセイ中に使用される全機器を記録する。アッセイを実施する日毎に使用する量のカゼインアリコートを37℃±2℃にする必要がある。コーティングバッファーと基質は冷温で使用する。ブロッキングインキュベーションの前とその間に標準、サンプル、対照、スパイク及びスパイクサンプルを調製する。希釈液を調製し、エッペンドルフチューブに移し、15分間変性させ、15分間冷却し、5分間遠心し、マイクロチューブに移すためには、1時間を越えるブロックインキュベーションが必要になる場合がある。プレートをブロックする前に少なくとも40分間放置する。12チャネルピペットを使用してサンプル、スパイクサンプル、標準、対照、アッセイスパイク及びブランクを水平方向にプレートのB〜G行にロードする。標準は高濃度から低濃度の順にロードする。プレートコーティング、ビオチン添加、ニュートラアビジン添加、基質添加、及び停止溶液添加はカラム2〜11に垂直に実施する。
【0237】
冷温50mM重炭酸ナトリウム中コーティング抗体100μL/ウェルでプレートをコーティングする。コーティング溶液がウェルの底を均等に覆うまでプレートの側面をタッピングし、シーリングテープを貼り付け、プレートシェーカー(又は同等手段)速度3にて振盪しながら公称温度4℃でインキュベートする。
【0238】
一晩インキュベーション後、プレートを冷蔵庫から取り出し、室温で平衡化させる。コーティングを振り払う。プレートをペーパータオルで拭う。37℃±2℃カゼイン300μL/ウェルでブロックし、シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間±10分間振盪しながら37℃±2℃でインキュベートする。
【0239】
ブロッキングインキュベーション前とその間に標準、サンプル、対照、スパイク及びスパイクサンプルを調製する。プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。8チャネルピペットを使用し、変性した標準、サンプル、スパイク、スパイクサンプル、ブランク及び対照100μL/ウェルをプレートの3個のウェルに分注する。プレートの外側のウェルは使用せず、これらのウェルには非処理希釈バッファーを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて2時間振盪しながら37℃±2℃でインキュベートする。テンプレートを作成し、プレートにロードする際のガイドとして使用する。
【0240】
プレートリーダー設定。プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。ビオチン化抗体100μL/ウェルを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。
【0241】
プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。ニュートラアビジン−HRPコンジュゲート溶液100μL/ウェルを加える。ニュートラアビジンを最後の行に添加後、すぐにタイマーをスタートする。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて30分間振盪しながら37℃±2℃でインキュベートする。プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。冷温K−Blue基質100μL/ウェルを加え、シーリングテープを貼り付け、Lab−lineタイタープレートシェーカー(又は同等手段)で速度3にて振盪しながら室温で10分間インキュベートする(基質を最初の行に添加後、すぐにタイマーをスタートする)。1Nリン酸100μL/ウェルを加えることにより反応を停止する。プレートをプレートシェーカーにセットし、速度3で3分間振盪する。プレートを450nmで読取る。
【0242】
データ解析及び計算。注:光学密度が標準曲線の実際の定量限界内にあり且つ下記%CV又は%差条件を満たすサンプル、スパイク、スパイクサンプル及び対照のみが許容される。サンプルODが標準曲線に達しない場合には、結果を0.18ng/mL未満と報告する(アッセイLOQ)。その後、この数値を希釈後のサンプル濃度(10mg/mL)で割り、数値(ng/mg)を報告する。サンプルのプロテインA濃度が高く、非スパイク及び/又はスパイクサンプルが標準曲線(2ng/mL)を上回る場合には、標準曲線内となるように更に希釈する。その後、この数値を希釈後のサンプル濃度で割り、数値(ng/mg)を報告する。スパイク回収率計算では、非スパイクサンプル値(ng/mL)が曲線に達しない場合でも、スパイクサンプル値(ng/mL)から非スパイクサンプル値(ng/mL)を差し引く。数値がマイナスの場合又は「範囲」が得られる場合には、スパイク回収率計算の非スパイクサンプルをゼロとみなす。
【0243】
標準曲線。標準濃度をプロトコルテンプレートに入力する。二次曲線フィットを使用する。測定係数は0.99でなければならず、3個のウェル間の%CVは20%でなければならない。この条件を満たさない場合には、標準を1本(1レベル,ウェル3個)下げることができる。0.18ng/mL下げる場合には、光学密度が0.26ng/mL及び2ng/mL(残りの標準曲線の点)光学密度以内のサンプル及びスパイクサンプルのみが許容可能である。更に、各標準レベルの3個について、1個のウェルが明白に汚染されているか又は結合度が低い場合には、このウェルを下げることができる。1個のウェルを標準レベルから下げる場合には、残りの2個は%差=20%でなければならない。プレートのバックグラウンド(ブランク)に近いOD値を示す最低標準の%CVは30%でなければならない。ウェル1個を下げる場合には、残りの2個の%差は35%でなければならない。最低標準を下げる場合には、光学密度が残りの標準曲線レベル光学密度以内にあるサンプル及びスパイクサンプルのみが許容可能である。
【0244】
%差を次のように計算する:%差=(絶対値(希釈液1の結果−希釈液2の結果)/平均値)×100%。標準が上記条件を満たさない場合には、アッセイを繰返す必要がある。%CV及び/又は%差の数値と標準曲線測定係数結果を報告する。
【0245】
サンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを報告する。各サンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。注:非スパイクサンプルODが最低標準ODを下回る場合には、%差条件は非スパイク結果に適用されない。上記計算参照。
【0246】
各希釈液の「非スパイクサンプル結果」(ng/mL)を報告する。これらの数値をスパイク回収率計算で使用する。平均「非スパイクサンプル結果(ng/mL)」と希釈液間の%差を計算する。結果を報告する。希釈液間の%差は25%でなければならない。平均(ng/mL)値から実際のプロテインA濃度ng/mgを次のように計算する:プロテインA(ng/mg)=平均「非スパイクサンプル結果(ng/mL)」/希釈後のサンプル濃度(10mg/mL)。結果を記録する。
【0247】
スパイク。%CVは3個のウェル間で20%でなければならない。%CVを記録する。スパイクから1個のウェルを下げることができる。残りの点は%差=20%でなければならない。上記計算参照。プロテインA濃度(ng/mL)を報告する。この結果をスパイク回収率計算で使用する。各スパイクに得られる濃度(ng/mL)は理論スパイク濃度の±20%でなければならない。結果を記録し、合否を判定する。スパイク結果が理論値の20%以内にない場合には、アッセイを繰返す必要がある。平均スパイク濃度(ng/mL)×100は100%±20% 0.296ng/mLでなければならない。
【0248】
スパイクサンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを記録する。各スパイクサンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。上記計算参照。各希釈液の「スパイクサンプル結果」(ng/mL)を報告する。2種類の希釈液間の%差を記録する。希釈液間の%差は25%でなければならない。これらの結果をスパイク回収率計算で使用する。下式を使用して各希釈液セットの%スパイク回収率を計算する:%スパイク回収率=スパイクサンプル値−非スパイクサンプル値×100。スパイク値。注:スパイク回収率計算では、非スパイクサンプル値(ng/mL)が曲線を下回る場合でも、スパイクサンプル値(ng/mL)から非スパイクサンプル値(ng/mL)を差し引く。数値がマイナスの場合又は「範囲」が得られる場合には、スパイク回収率計算では非スパイクサンプルをゼロとみなす。各サンプルの各希釈液の%スパイク回収率は100%±50%(50%〜150%)でなければならない。結果と合否を記録する。
【0249】
対照。%CVは3個のウェル間で20%でなければならない。%CV結果を記録する。対照から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。
【0250】
【表9】
【0251】
【表10】
【0252】
本願では種々の刊行物を引用するが、その開示内容全体を本願に援用する。
【背景技術】
【0001】
ヒトIL−13は、活性化T細胞からクローニングされた17kDa糖蛋白質であり、Th2系統の活性化T細胞、Th0及びTh1 CD4+T細胞、CD8+T細胞、並びに数種の非T細胞集団(例えば肥満細胞)により産生される(Zurawski and de Vries,1994 Immunol Today,15,19−26)。IL−13はヒトB細胞においてIgEへの免疫グロブリンアイソタイプスイッチを促進し(Punnonen,Aversa et al.1993 Proc Natl Acad Sci U S A 90 3730−4)、ヒト及びマウスの両者において炎症性サイトカイン産生を抑制する(de Waal Malefyt et al.,1993,J Immunol,151,6370−81;Doherty et al.,1993,J Immunol,151,7151−60)。IL−13はその細胞表面受容体であるIL−13Rα1及びIL−13Rα2と結合する。IL−13Rα1は低親和性(KD〜10nM)でIL−13と相互作用した後、IL−4Rを動員し、高親和性(KD〜0.4nM)シグナル伝達ヘテロ二量体受容体複合体を形成する(Aman et al.,1996,J Biol Chem,271,29265−70;Hilton et al.,1996,Proc Natl Acad Sci USA,93,497−501)。IL−4R/IL−13Rα1複合体はB細胞、単球/マクロファージ、樹状細胞、好酸球、好塩基球、線維芽細胞、内皮細胞、気道上皮細胞及び気道平滑筋細胞等の多数の細胞種で発現される(Graber et al.,1998,Eur J Immunol,28,4286−98;Murata et al.,1998,Int Immunol,10,1103−10;Akaiwa et al.,2001,Cytokine,13,75−84)。IL−13Rα1/IL−4R受容体複合体のライゲーションの結果、シグナル伝達性転写因子(STAT6)及びインスリン受容体基質−2(IRS−2)経路を含む各種シグナル伝達経路が活性化される(Wang et al.,1995,Blood,864218−27;Takeda et al.,1996,J Immunol,157,3220−2)。IL−13Rα2鎖自体はIL−13に対する親和性が高く(KD〜0.25〜0.4nM)、IL−13との結合を負に制御するデコイ受容体として機能する(Donaldson et al.,1998,J Immunol,161,2317−24)と同時に、マクロファージと恐らく他の細胞種でAP−1経路を介してTGF−β合成及び線維化を誘導するシグナル伝達受容体としても機能する(Fichtner−Feigl,Strober et al.2006 Nat Med 12 99−106)。
【0002】
喘息の前臨床動物モデルで実施された数件の試験によると、IL−13は、喘息において重要な役割を果たすことが分かっている。これらのデータによると、IL−13ノックアウトマウスは、喘息にかかりにくいことや、喘息表現型は、各種マウスモデルでIL−13アンタゴニスト(可溶性IL−13受容体、抗IL−13mAb等)により阻害されることが立証されている(Wills−Karp and Chiaramonte,2003,Curr Opin Pulm Med,9 21−7;Wills−Karp,2004,Immunol Rev,202 175−90)。組換えIL−13をマウスとモルモットの肺に薬理投与すると、気道粘液過剰分泌、好酸球増加及び気道過敏反応(「AHR」;Grunig et al.,1998,Science,282,2261−3;Wills−Karp et al.,1998,Science,282,2258−61;Kibe et al.,2003,Am J Respir Crit Care Med,167,50−6;Vargaftig and Singer,2003,Am J Physiol Lung Cell Mol Physiol,284,L260−9;Vargaftig and Singer,2003,Am J Respir Cell Mol Biol,28,410−9)を誘発することは複数の研究で実証されている。IL−13のこれらの作用はIL−13を構成的又は誘導的に発現させたトランスジェニックマウスシステムで再現される(Zhu et al.,1999,J Clin Invest,103,779−88;Zhu et al.,2001,Am J Respir Crit Care Med,164,S67−70;Lanone et al.,2002,J Clin Invest,110463−74)。IL−13の慢性トランスジェニック過剰発現は、上皮下線維化及び気腫も誘発する。IL−13(及びIL−4)シグナル伝達分子STAT6を欠損するマウスはアレルゲンにより誘発されるAHR及び粘液過剰産生を発現することができない(Kuperman et al.,2002,Nat Med,8,885−9)。可溶性IL−13受容体融合蛋白質(sIL−13Rα2Fc)を使用した試験によると、このサイトカインはアレルゲンであるオボアルブミン(OVA)により実験的に誘発した気道疾患において中枢的役割を果たすことが判明した(Grunig et al.,1998,Science,282,2261−3;Wilis−Karp et al.,1998,Science,282,2258−61;Taube et al.,2002,J Immunol,169,6482−9)。抗IL−13治療が有効であることもマウス喘息の慢性モデルで実証された。粘液過剰分泌及びAHRの特徴を示すことに加え、慢性喘息のこのモデルは急性度の高いモデルには認められないヒト疾患の数種の特徴を示す。これらの特徴としては、上皮細胞間スペースに位置する肺組織の好酸球増加と、コラーゲン沈着の増加により確認される平滑筋線維化が挙げられる。慢性喘息モデルはOVA感作マウスに週1回ずつ合計4週間OVAを反復エアゾール攻撃することにより誘導される。OVA攻撃の最後の2週間に抗IL−13抗体を投与する(36日目から投与して53日目の試験日に効力計測値を測定する)と、AHR、肺炎症、杯細胞異常増殖、粘液過剰分泌及び気道線維化を有意に阻害した(Yang et al.,2005,J Pharmacol Exp Ther,313,8−15)。喘息患者の肺で高濃度のIL−13 mRNA及び蛋白質が検出されており、疾患の重篤度に相関することから、IL−13はヒト喘息の病因に関与していると考えられている(Huang et al.,1995,J Immunol,155,2688−94)。更に、高濃度IL−13に繋がるヒトIL−3遺伝子多型が同定され、喘息とアトピーに関係があるとされ(Heinzmann et al.,2000,Hum Mol Genet,9,549−59;Hoerauf et al.,2002,Microbes Infect,4,37−42;Vercelli,2002,Curr Opin Allergy Clin Immunol,2,389−93;Heinzmann et al.,2003,J Allergy Clin Immunol,112,735−9;Chen et al.,2004,J Allergy Clin Immunol,114,553−60;Vladich et al.,2005,J Clin Invest,115,747−54)、喘息患者の肺で高濃度のIL−13が検出されている(Huang et al.,1995,J Immunol,155,2688−94;Arima et al.,2002,J Allergy Clin Immunol,109,980−7;Berry et al.,2004,J Allergy Clin Immunol,114,1106−9)。血漿IL−13濃度の上昇に繋がるIL−13遺伝子の多型を示す個体はアトピーと喘息の危険が高いことから、IL−13と喘息の遺伝的連鎖も立証されている(Wills−Karp,2000,Respir Res,1,19−23)。
【先行技術文献】
【非特許文献】
【0003】
【非特許文献1】Zurawski and de Vries,1994 Immunol Today,15,19−26
【非特許文献2】Punnonen,Aversa et al.1993 Proc Natl Acad Sci U S A 90 3730−4
【非特許文献3】de Waal Malefyt et al.,1993,J Immunol,151,6370−81
【非特許文献4】Doherty et al.,1993,J Immunol,151,7151−60
【非特許文献5】Aman et al.,1996,J Biol Chem,271,29265−70
【非特許文献6】Hilton et al.,1996,Proc Natl Acad Sci USA,93,497−501
【非特許文献7】Graber et al.,1998,Eur J Immunol,28,4286−98
【非特許文献8】Murata et al.,1998,Int Immunol,10,1103−10
【非特許文献9】Akaiwa et al.,2001,Cytokine,13,75−84
【非特許文献10】Wang et al.,1995,Blood,864218−27
【非特許文献11】Takeda et al.,1996,J Immunol,157,3220−2
【非特許文献12】Donaldson et al.,1998,J Immunol,161,2317−24
【非特許文献13】Fichtner−Feigl,Strober et al.2006 Nat Med 12 99−106
【非特許文献14】Wills−Karp and Chiaramonte,2003,Curr Opin Pulm Med,9 21−7
【非特許文献15】Wills−Karp,2004,Immunol Rev,202 175−90
【非特許文献16】Grunig et al.,1998,Science,282,2261−3
【非特許文献17】Wills−Karp et al.,1998,Science,282,2258−61
【非特許文献18】Kibe et al.,2003,Am J Respir Crit Care Med,167,50−6
【非特許文献19】Vargaftig and Singer,2003,Am J Physiol Lung Cell Mol Physiol,284,L260−9
【非特許文献20】Vargaftig and Singer,2003,Am J Respir Cell Mol Biol,28,410−9
【非特許文献21】Zhu et al.,1999,J Clin Invest,103,779−88
【非特許文献22】Zhu et al.,2001,Am J Respir Crit Care Med,164,S67−70
【非特許文献23】Lanone et al.,2002,J Clin Invest,110463−74
【非特許文献24】Kuperman et al.,2002,Nat Med,8,885−9
【非特許文献25】Taube et al.,2002,J Immunol,169,6482−9
【非特許文献26】Yang et al.,2005,J Pharmacol Exp Ther,313,8−15
【非特許文献27】Huang et al.,1995,J Immunol,155,2688−94
【非特許文献28】Heinzmann et al.,2000,Hum Mol Genet,9,549−59
【非特許文献29】Hoerauf et al.,2002,Microbes Infect,4,37−42
【非特許文献30】Vercelli,2002,Curr Opin Allergy Clin Immunol,2,389−93
【非特許文献31】Heinzmann et al.,2003,J Allergy Clin Immunol,112,735−9
【非特許文献32】Chen et al.,2004,J Allergy Clin Immunol,114,553−60
【非特許文献33】Vladich et al.,2005,J Clin Invest,115,747−54
【非特許文献34】Arima et al.,2002,J Allergy Clin Immunol,109,980−7
【非特許文献35】Berry et al.,2004,J Allergy Clin Immunol,114,1106−9
【非特許文献36】Wills−Karp,2000,Respir Res,1,19−23
【発明の概要】
【発明が解決しようとする課題】
【0004】
ヒトIL−13は、様々なヒトの障害に関与しているため、IL−13活性を阻害又は抑制するための治療方法が設計されてきた。特に、IL−13活性を阻害するための手段として、IL−13と結合してこれを中和する抗体が求められてきた。しかし、医薬用としてのこのような抗体を製造・精製する改良型の方法が当分野で必要とされている。本発明はこの必要に対処する。
【課題を解決するための手段】
【0005】
ある実施形態においては、本発明はIL−13と結合する精製・単離抗体及び抗体フラグメントと、このような抗体及びフラグメントを含有する医薬組成物に関する。ある実施形態においては、本発明はヒトIL−13と結合する単離抗体又はその抗原結合部分に関する。本発明の単離抗IL−13抗体は臨床環境及び研究・開発で使用することができる。ある実施形態においては、本発明は、図1に示す重鎖及び軽鎖配列を含む抗IL−13抗体に関する。
【0006】
本発明のある実施形態は、抗体が宿主細胞蛋白質(「HCP」)と浸出したプロテインAを実質的に含まないように抗IL−13抗体又はその抗原結合部分をサンプルマトリックスから精製する方法に関する。ある態様において、サンプルマトリックス(又は単に「サンプル」)は本発明の抗IL−13抗体を産生させるために利用される細胞株を含む。特定の態様において、サンプルはヒト抗IL−13抗体を産生させるために利用される細胞株を含む。
【0007】
ある実施形態において、本発明は、特に細胞及び細胞破片を除去するための一次回収工程を含むIL−13抗体の精製方法を提供する。前記方法のある実施形態において、一次回収工程は、1以上の遠心又は深層濾過工程を含む。限定するものではないが、例えば、このような遠心工程は、約7000×g〜約11,000×gで実施することができる。更に、上記方法のある実施形態は、デリピッド深層濾過工程等の深層濾過工程を含む。
【0008】
ある実施形態では、一次回収サンプルをアフィニティークロマトグラフィー工程に供する。アフィニティークロマトグラフィー工程は適切なアフィニティークロマトグラフィー担体を含むカラムに一次回収サンプルを供する段階を含む。このようなクロマトグラフィー担体の非限定的な例としては、限定されないが、プロテインA樹脂、プロテインG樹脂、目的抗体に特異的な抗原を含むアフィニティー担体、及びFc結合蛋白質を含むアフィニティー担体が挙げられる。プロテインA樹脂は抗体(IgG)のアフィニティー精製・単離に有用である。ある態様では、サンプルをロードする前にプロテインAカラムを適切な緩衝液で平衡化させる。適切な緩衝液の1例はpH約7.2のトリス/NaClバッファーである。平衡化後、サンプルをカラムにロードすることができる。カラムにロードした後、例えば平衡化バッファーを使用してカラムを1回以上洗浄することができる。カラムから溶出させる前に、別の緩衝液を利用する洗浄を含む他の洗浄を使用することができる。その後、適当な溶出バッファーを使用してプロテインAカラムから溶出させることができる。適切な溶出バッファーの1例はpH約3.5の酢酸/NaClバッファーである。当業者に周知の技術を使用して溶出液をモニターすることができる。例えば、OD280の吸光度を追跡することができる。次に、後続処理に備えて目的溶出フラクションを調製することができる。
【0009】
本発明のある実施形態では、プロテインAアフィニティークロマトグラフィー後に低pH調整工程を行う。このような態様では、推定抗IL−13抗体又はその抗原結合部分を含有するプロテインA溶出液を約3〜約4のpHへのpH調整に供する。ある態様では、pHを約3.5に調整する。特に、低いpHはサンプルを汚染している可能性のあるpH感受性ウイルスの低減及び/又は不活性化を促進する。適切な時間後、pHを約4.5〜約6.0(限定されないが、例えば約5.0)に調整し、サンプルを後続精製工程に供する。
【0010】
ある実施形態では、プロテインAアフィニティークロマトグラフィー又は低pH調整工程後にイオン交換工程を実施する。このイオン交換工程は、カチオン交換でもアニオン交換でもよいし、両者の順次併用でもよい。この工程は単一のイオン交換法でもよいし、カチオン交換工程後にアニオン交換工程又はアニオン交換工程後にカチオン交換工程を実施する等の複数のイオン交換工程でもよい。ある態様において、イオン交換工程は1段階法である。別の態様において、イオン交換工程は2段階イオン交換法である。適切なカチオン交換カラムは固定相にアニオン基を含むカラムである。このようなカラムの1例はFractogel(登録商標)SO3−である。このイオン交換捕集液クロマトグラフィー工程はサンプルからの抗体の単離を容易にする。適切なアニオン交換カラム固定相にカチオン基を含むカラムである。このようなカラムの1例はQ Sepharose(登録商標)カラムである。別の例はPall Mustang Qメンブレンカートリッジである。1以上のイオン交換工程は宿主細胞蛋白質及びDNAや、適用可能な場合には、アフィニティーマトリックス蛋白質等の不純物を低減することにより更に抗体を単離する。このアニオン交換法は目的抗体がアニオン交換樹脂(ないし固相)と相互作用又は結合しないフロースルーモードのクロマトグラフィーである。しかし、多くの不純物はアニオン交換樹脂と相互作用し、結合する。特定の態様において、イオン交換工程はアニオン交換クロマトグラフィーである。
【0011】
サンプル緩衝液のpH及びイオン強度を調整することにより、アフィニティークロマトグラフィー溶出液をイオン交換クロマトグラフィーに備えて調製する。例えば、アフィニティー溶出液を1Mトリスバッファーで約4.5〜約8.5のpHに調整することができる。サンプル(アフィニティー溶出液)をイオン交換カラムにロードした後、適切な緩衝液を使用してカラムを平衡化することができる。適切な緩衝液の1例はpH約4.5〜約8のトリス/NaClバッファーである。平衡化後、カラムにアフィニティー溶出液をロードすることができる。ロード後、カラムを適切な緩衝液で1回以上洗浄することができる。適切な緩衝液の1例は平衡化バッファー自体である。例えば吸光度(OD280)が約0.2AUよりも上昇すると、フロースルー捕集を開始することができる。
【0012】
ある実施形態では、一次回収後又はアフィニティークロマトグラフィー工程の不在下に第1及び第2イオン交換工程を実施する。このような態様の所定のものでは、第1イオン交換工程の前、2回のイオン交換工程の間、又はその両方にイオン交換サンプルを中間濾過工程に供する。ある態様において、この濾過工程は捕集液限外濾過/透析濾過(「UF/DF」)を含む。特に、このような濾過は抗IL−13抗体及びその抗原結合部分の濃縮と緩衝液交換を容易にする。
【0013】
本発明のある実施形態は、1以上の疎水性相互作用クロマトグラフィー(「HIC」)工程を含む方法を提供する。適切なHICカラムは固定相に疎水基を含むカラムである。このようなカラムの非限定的な1例はPhenyl HP Sepharose(登録商標)カラムである。ある状況においては、抗IL−13抗体は単離/精製工程中に凝集物を形成する。1以上のHIC工程を含むことにより、このような凝集物を低減又は排除し易くする。HICは更に不純物の除去にも役立つ。ある実施形態においては、HIC工程は抗IL−13抗体(又はその凝集物)と疎水性カラムの相互作用を促進するために高塩濃度緩衝液を利用する。その後、低濃度塩を使用して抗IL−13抗体を溶出させることができる。
【0014】
ある実施形態では、限定されないが、Ultipor DV50(登録商標)フィルター(Pall Corporation,East Hills,N.Y.)等のウイルス除去フィルターを使用してHIC溶出液を濾過する。このような態様ではViresolve(登録商標)フィルター(Millipore,Billerica,Mass.);Zeta Plus VR(登録商標)フィルター(CUNO;Meriden,Conn.);及びPlanova(登録商標)フィルター(Asahi Kasei Pharma,Planova Division,Buffalo Grove,Ill.)等の他のフィルターも使用することができる。
【0015】
ある実施形態においては、本発明は単離抗IL−13抗体又はその抗原結合部分と許容可能な担体を含有する1種以上の医薬組成物に関する。ある態様において、組成物は抗IL−13抗体以外に、更に1種以上の抗体又はその抗原結合部分を含有する。別の態様において、組成物は更に1種以上の薬剤を含有する。
【0016】
得られたサンプル生成物中の目的抗体の純度は、当業者に周知の方法(例えばサイズ排除クロマトグラフィー、Poros(登録商標)A HPLCアッセイ、HCP ELISA、プロテインA ELISA及びウェスタンブロット分析)を使用して分析することができる。
【図面の簡単な説明】
【0017】
【図1】抗IL−13抗体の非限定的な1例の重鎖及び軽鎖可変領域配列を示す。
【図2−1】設定点、インプロセス制御試験及び作用限界を含む典型的細胞培養工程流れ図を示す。
【図2−2】設定点、インプロセス制御試験及び作用限界を含む典型的細胞培養工程流れ図を示す。
【図3】代替細胞培養工程フローストラテジーの比較を示す。
【図4】設定点、インプロセス制御試験及び作用限界を含む一次回収・捕集クロマトグラフィー工程流れ図を示す。
【図5】代替一次回収・捕集フローストラテジーの比較を示す。
【図6−1】設定点、インプロセス制御試験及び作用限界を含む精密精製工程流れ図を示す。
【図6−2】設定点、インプロセス制御試験及び作用限界を含む精密精製工程流れ図を示す。
【図7】代替精密精製フローストラテジーの比較を示す。
【発明を実施するための形態】
【0018】
本発明は、IL−13と結合する抗体に関する。ある態様において、本発明は、ヒトIL−13と結合する単離抗体又はその抗原結合部分に関する。本発明の単離抗IL−13抗体は、臨床環境及び研究・開発で使用することができる。本発明は、抗IL−13抗体又はその抗原結合部分の精製方法にも関する。本発明の関連で精製することができる適切な抗IL−13抗体はその開示内容全体を本願に援用するPCT出願第PCT/US2007/019660号に開示されており、その後、ABT−308として同定された抗体を含む。典型的な抗IL−13抗体重鎖及び軽鎖配列を図1に示す。本発明は、本願に記載する抗IL−13抗体又はその抗原結合部分を含有する医薬組成物にも関する。
【0019】
限定するものではないが、分かり易くするために、以下の詳細な説明は次の項目に分けて説明する。
1.定義;
2.抗体作製;
3.抗体産生;
4.抗体精製;
5.サンプル純度の検定方法;
6.付加的修飾;
7.医薬組成物;及び
8.抗体使用。
【0020】
1.定義;
本発明を理解し易くするために、先ず所定の用語を定義する。
【0021】
「抗体」なる用語は、ジスルフィド結合により相互に結合された2本の重(H)鎖と2本の軽(L)鎖の4本のポリペプチド鎖から構成される免疫グロブリン分子を包含する。各重鎖は、重鎖可変領域(本願ではHCVR又はVHと略称する)と重鎖定常領域(CH)から構成される。重鎖定常領域は、CH1、CH2及びCH3の3個のドメインから構成される。各軽鎖は、軽鎖可変領域(本願ではLCVR又はVLと略称する)と軽鎖定常領域から構成される。軽鎖定常領域は、1個のドメインCLから構成される。VH及びVL領域は更に相補性決定領域(CDR)と呼ばれる超可変領域とその間に配置されたフレームワーク領域(FR)と呼ばれる保存度の高い領域に区分される。各VH及びVLはアミノ末端からカルボキシ末端に向かってFR1、CDR1、FR2、CDR2、FR3、CDR3、FR4の順に配置された3個のCDRと4個のFRから構成される。
【0022】
抗体の「抗原結合部分」(又は抗体部分」)なる用語は、抗原(例えばhIL−13)と特異的に結合する能力を保持する抗体の1個以上のフラグメントを包含する。抗体の抗原結合機能は、全長抗体のフラグメントにより実施可能であることが分かっている。抗体の「抗原結合部分」なる用語に含まれる結合フラグメントの例としては、(i)VL、VH、CL及びCH1ドメインから構成される1価フラグメントであるFabフラグメント;(ii)ヒンジ領域でジスルフィド架橋により結合された2個のFabフラグメントから構成される2価フラグメントであるF(ab’)2フラグメント;(iii)VHドメインとCH1ドメインから構成されるFdフラグメント;(iv)抗体の単一アームのVLドメインとVHドメインから構成されるFvフラグメント;(v)VHドメインから構成されるdAbフラグメント(その教示内容全体を本願に援用するWard et al.,(1989)Nature 341:544−546);並びに(vi)単離相補性決定領域(CDR)が挙げられる。更に、Fvフラグメントの2個のドメインVL及びVHは別々の遺伝子によりコードされるが、VL領域とVH領域が対合して1価分子を形成する1本の蛋白鎖(1本鎖Fv(scFv)と言う;例えばその教示内容全体を本願に援用するBird et al.(1988)Science 242:423−426;及びHuston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883参照)としての作製を可能にする合成リンカーにより組換え法を使用して結合することができる。このような1本鎖抗体も抗体の「抗原結合部分」なる用語に含むものとする。ダイアボディ等の他の形態の1本鎖抗体も含む。ダイアボディは、VH領域とVL領域が1本のポリペプチド鎖で発現される2価の二重特異性抗体であるが、非常に短いリンカーを使用するため、同一鎖の2領域間で対合することができず、これらの領域は別の鎖の相補性領域と対合し、2個の抗原結合部位を形成する(例えばその教示内容全体を本願に援用するHolliger,P.,et al.(1993)Proc.Natl.Acad.Sci.USA 90:6444−6448;Poljak,R.J.,et al.(1994)Structure 2:1121−1123参照)。更に、抗体又はその抗原結合部分は、抗体又は抗体部分と1種以上の他の蛋白質又はペプチドの共有的又は非共有的結合により形成される大きな免疫接着分子の一部でもよい。このような免疫接着分子の例としては、ストレプトアビジンコア領域を使用した四量体scFv分子の作製(その教示内容全体を本願に援用するKipriyanov,S.M.,et al.(1995)Human Antibodies and Hybridomas 6:93−101)や、システイン残基、マーカーペプチド及びC末端ポリヒスチジンタグを使用した2価ビオチン化scFv分子の作製(その教示内容全体を本願に援用するKipriyanov,S.M.,et al.(1994)Mol.Immunol.31:1047−1058)が挙げられる。Fab及びF(ab’)2フラグメント等の抗体部分は、夫々全長抗体のパパイン又はペプシン消化等の慣用技術を使用して全長抗体から作製することができる。更に、抗体、抗体部分及び免疫接着分子は本願に記載するように、標準組換えDNA技術を使用して取得することができる。ある態様において、抗原結合部分は競合領域又は競合領域対である。
【0023】
本願で使用する「ヒトインターロイキン13」(本願ではhIL−13又はIL−13と略称する)なる用語は、活性化T細胞(Zurawski and de Vries,1994 Immunol Today 15 19−26)からクローニングされ、Th2系統の活性化T細胞により産生される17kDa糖蛋白質を意味する。Th0及びTh1 CD4+T細胞、CD8+T細胞、並びに数種の非T細胞集団(例えば肥満細胞)もIL−13を産生する(Zurawski and de Vries,1994 Immunol Today 15 19−26)。IL−13の機能としてとしては、ヒトB細胞におけるIgEへの免疫グロブリンアイソタイプスイッチの促進(Punnonen,Aversa et al.1993 Proc Natl Acad Sci U S A 90 3730−4)と、ヒト及びマウスの両者における炎症性サイトカイン産生の抑制(de Waal et al.,1993 J Immunol 151 6370−81;Doherty et al.,1993 J Immunol 151 7151−60)が挙げられる。IL−13はIL−13Rα1及びIL−13Rα2と呼ばれる細胞表面受容体と結合する。IL−13Rα1受容体は低親和性(KD〜10nM)でIL−13と相互作用した後、IL−4Rを動員し、高親和性(KD〜0.4nM)シグナル伝達ヘテロ二量体受容体複合体を形成する(Aman et al.,1996 J Biol Chem 271 29265−70;Hilton et al.,1996 Proc Natl Acad Sci U S A 93 497−501)。IL−4R/IL−13Rα1複合体はB細胞、単球/マクロファージ、樹状細胞、好酸球、好塩基球、線維芽細胞、内皮細胞、気道上皮細胞及び気道平滑筋細胞等の多数の細胞種で発現される(Graber et al,1998 Eur J Immunol 28 4286−98;Murata et al.,1998 Int Immunol 10 1103−10;Akaiwa et al.,2001 Cytokine 13 75−84)。IL−13Rα1/IL−4R受容体複合体のライゲーションの結果、シグナル伝達性転写因子(STAT6)及びインスリン受容体基質−2(IRS−2)経路を含む各種シグナル伝達経路が活性化される(Wang et al.,1995 Blood 864218−27;Takeda et al.,1996 J Immunol 157 3220−2)。IL−13Rα2鎖自体はIL−13に対する親和性が高く(KD〜0.25〜0.4nM)、IL−13との結合を負に制御するデコイ受容体として機能する(Donaldson,Whitters et al.1 98 J Immunol 161 2317−24)と同時に、マクロファージと恐らく他の細胞種でAP−1経路を介してTGF−b合成及び線維化を誘導するシグナル伝達受容体としても機能する(Fichtner−Feigl et al.,2006 Nat Med 12 99−106)。IL−13をコードする核酸はGenBank Accession No.NM_002188として入手可能であり、ポリペプチド配列はGenBank Accession No.NP_002179として入手可能である。ヒトIL−13なる用語は、標準組換え発現法により作製することができる組換えヒトIL−13(rh IL−13)を包含するものとする。
【0024】
「Kabatナンバリング」、「Kabat定義」及び「Kabatラベリング」なる用語は、本願では同義に使用する。これらの用語は、当分野で認識されている通り、抗体又はその抗原結合部分の重鎖及び軽鎖可変領域の他のアミノ酸残基よりも可変性(即ち超可変性)のアミノ酸残基のナンバリングシステムを意味する(その教示内容全体を本願に援用するKabat et al.(1971)Ann.NY Acad,Sci.190:382−391及びKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242)。重鎖可変領域では、超可変領域はCDR1がアミノ酸31〜35位、CDR2がアミノ酸50〜65位、CDR3がアミノ酸95〜102位に位置する。軽鎖可変領域では、超可変領域はCDR1がアミノ酸24〜34位、CDR2がアミノ酸50〜56位、CDR3がアミノ酸89〜97位に位置する。
【0025】
「ヒト抗体」なる用語は、Kabatらに記載されているように、ヒト生殖細胞系列免疫グロブリン配列に対応する可変領域と定常領域をもつ抗体を包含する(Kabat,et al.(1991)Sequences of proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)。本発明のヒト抗体は、ヒト生殖細胞系列免疫グロブリン配列によりコードされないアミノ酸残基(例えばランダムもしくは部位特異的突然変異誘発によりインビトロ又は体細胞突然変異によりインビボで導入される突然変異)を例えばCDR、特にCDR3に含むことができる。突然変異は、「選択的突然変異誘発アプローチ」を使用して導入することができる。ヒト抗体は少なくとも1カ所をアミノ酸残基(例えばヒト生殖細胞系列免疫グロブリン配列によりコードされない活性強化アミノ酸残基)で置換することができる。ヒト抗体は20カ所までをヒト生殖細胞系列免疫グロブリン配列に含まれないアミノ酸残基で置換することができる。他の態様では、10カ所まで、5カ所まで、3カ所まで又は2カ所までが置換されている。1態様において、これらの置換はCDR領域内に存在する。他方、本願で使用する「ヒト抗体」なる用語は、別の哺乳動物種(例えばマウス)の生殖細胞系列に由来するCDR配列をヒトフレームワーク配列に移植した抗体を包含するものではない。
【0026】
「選択的突然変異誘発アプローチ」なる用語は、少なくとも1カ所の適切な選択的突然変異誘発位置、超変異位置及び/又は接触位置のCDRアミノ酸を選択し、個々に突然変異させることにより、抗体の活性を改善する方法を包含する。「選択的に突然変異された」ヒト抗体とは、選択的突然変異誘発アプローチを使用して選択された位置に突然変異を含む抗体である。別の態様において、選択的突然変異誘発アプローチは抗体の重鎖可変領域のCDR1、CDR2もしくはCDR3(以下、夫々H1、H2及びH3と言う)、又は軽鎖可変領域のCDR1、CDR2もしくはCDR3(以下、夫々L1、L2及びL3と言う)における選択された個々のアミノ酸残基を優先的に突然変異させる方法を提供するものとする。アミノ酸残基は、選択的突然変異誘発位置、接触位置又は超変異位置から選択することができる。個々のアミノ酸は、軽鎖又は重鎖可変領域におけるそれらの位置に基づいて選択される。当然のことながら、超変異位置は接触位置でもよい。ある態様において、選択的突然変異誘発アプローチは「標的アプローチ」である。「標的アプローチ」なる用語は、抗体の重鎖可変領域のCDR1、CDR2もしくはCDR3又は軽鎖可変領域のCDR1、CDR2もしくはCDR3における選択された個々のアミノ酸残基を標的として突然変異させる方法(例えば「グループ標的アプローチ」又は「CDR標的アプローチ」)を包含するものとする。「グループ標的アプローチ」では、標的の優先順にグループI(L3及びH3を含む)、II(H2及びL1を含む)及びIII(L2及びH1を含む)を含む選択的突然変異の標的として特定グループにおける個々のアミノ酸残基を選択する。「CDR標的アプローチ」では、H3、L3、H2、L1、H1及びL2の標的の優先順で特定CDRにおける個々のアミノ酸残基を選択的突然変異の標的として選択する。選択されたアミノ酸残基を例えば少なくとも2個の他のアミノ酸残基に突然変異させ、抗体の活性に及ぼす突然変異の影響を判定する。活性は、抗体の結合特異性/親和性、及び/又は抗体の中和力価の変化として測定される。当然のことながら、選択的突然変異誘発アプローチは、ファージディスプレイ、ヒトIgG生殖細胞系列遺伝子をもつトランスジェニック動物、ヒトB細胞から単離されたヒト抗体を含む任意起源に由来する任意抗体の最適化に使用することができる。選択的突然変異誘発アプローチは、ファージディスプレイ技術を使用してそれ以上最適化することができない抗体で使用することができる。当然のことながら、ファージディスプレイ、ヒトIgG生殖細胞系列遺伝子をもつトランスジェニック動物、ヒトB細胞から単離されたヒト抗体を含む任意起源に由来する抗体を選択的突然変異誘発アプローチの前又は後に復帰突然変異に供することができる。
【0027】
「組換えヒト抗体」なる用語は、組換え手段により作製、発現、創製又は単離したヒト抗体を包含し、例えば、宿主細胞にトランスフェクトした組換え発現ベクターを使用して発現させた抗体、組換えコンビナトリアルヒト抗体ライブラリーから単離した抗体、ヒト免疫グロブリン遺伝子を導入したトランスジェニック動物(例えばマウス)から単離した抗体(例えばその教示内容全体を本願に援用するTaylor,L,D.,et al.(1992)Nucl.Acids Res.20:6287−6295参照)、あるいはヒト免疫グロブリン遺伝子配列を他のDNA配列にスプライスする操作を含む他の任意手段により作製、発現、創製又は単離した抗体が挙げられる。このような組換えヒト抗体は、可変領域と定常領域がヒト生殖細胞系列免疫グロブリン配列に由来する(Kabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)。しかし、ある実施形態では、このような組換えヒト抗体をインビトロ突然変異誘発(又は、ヒトIg配列を導入したトランスジェニック動物を使用する場合には、インビボ体細胞突然変異誘発)に供するため、組換え抗体のVH領域とVL領域のアミノ酸配列は、ヒト生殖細胞系列VH及びVL配列に由来し、これらの配列に近縁であるが、天然にヒト抗体生殖細胞系列レパートリー内にインビボで存在していなくてもよい配列である。他方、ある実施形態において、このような組換え抗体は選択的突然変異誘発アプローチ又は復帰突然変異又は両者の産物である。
【0028】
「単離抗体」とは、異なる抗原特異性をもつ他の抗体を実質的に含まない抗体を包含する(例えばhIL−13と特異的に結合する単離抗体はhIL−13以外の抗原と特異的に結合する抗体を実質的に含まない)。hIL−13と特異的に結合する単離抗体は他の種に由来するIL−13分子と結合する場合がある。更に、単離抗体は他の細胞材料及び/又は化学物質を実質的に含まない場合がある。
【0029】
「中和抗体」(又は「hIL−13活性を中和した抗体」)とは、hIL−13と結合する結果としてhIL−13の生物活性を阻害する抗体を包含する。hIL−13の生物活性のこの阻害はIL−13生物活性の1種以上の指標を測定することにより判定することができる。hIL−13生物活性のこれらの指標は、当分野で公知の数種の標準インビトロ又はインビボアッセイの1種以上により判定することができる。
【0030】
「活性」なる用語は、抗原に対する抗体(例えばIL−13抗原と結合する抗hIL−13抗体)の結合特異性/親和性及び/又は抗体(例えばhIL−13と結合してhIL−13の生物活性を阻害する抗hIL−13抗体)の中和力価等の活性を包含する。
【0031】
「表面プラズモン共鳴」なる用語は、例えばBIAcoreシステム(Pharmacia Biosensor AB,Uppsala,Sweden及びPiscataway,N.J.)を使用してバイオセンサーマトリックス内の蛋白質濃度の変化の検出によりリアルタイム生体特異的相互作用の分析を可能にする光学現象を意味する。更に詳細については、その教示内容全体を本願に援用するJonsson,U.,et al.(1993)Ann.Biol.Clin.51:19−26;Jonsson,U.,et al.(1991)Biotechniques 11:620−627;Johnsson,B.,el al.(1995)J.Mol.Recognit.8:125−131;及びJohnnson,B.,et al.(1991)Anal.Biochem.198:268−277参照。
【0032】
本願で使用する「Koff」なる用語は、抗体/抗原複合体から抗体が解離する解離速度定数を意味する。
【0033】
本願で使用する「Kd」なる用語は、特定抗体−抗原相互作用の解離定数を意味する。
【0034】
「核酸分子」なる用語は、DNA分子とRNA分子を包含する。核酸分子は1本鎖でも2本鎖でもよいが、ある態様では2本鎖DNAである。
【0035】
抗体又は抗体部分(例えばVH、VL、CDR3)(例えばhIL−13と結合するもの)をコードする核酸に関して本願で使用する「単離核酸分子」なる用語は、抗体又は抗体部分をコードするヌクレオチド配列がhIL−13以外の抗原と結合する抗体又は抗体部分をコードする他のヌクレオチド配列を含まない核酸分子を包含し、前記他の配列は、天然でヒトゲノムDNAにおいて前記核酸のフランキング配列であり得る。従って、例えば、抗hIL−13抗体のVH領域をコードする単離核酸は例えばhIL−13以外の抗原と結合する他のVH領域をコードする他の配列を含まない。「単離核酸分子」なる用語は、VH領域とVL領域がダイアボディの配列以外の他の配列を含まないダイアボディ等の2価の二重特異性抗体をコードする配列も包含するものとする。
【0036】
「組換え宿主細胞」(又は単に「宿主細胞」)なる用語は、組換え発現ベクターが導入された細胞を包含する。当然のことながら、このような用語は特定対象細胞のみならず、このような細胞の子孫も意味するものとする。後続世代には、突然変異又は環境影響により所定の変異が生じる場合があるので、このような子孫は実際に親細胞と一致しない場合もあるが、本願で使用する「宿主細胞」の用語の範囲に含まれる。
【0037】
本願で使用する「改変」なる用語は、抗体又はその抗原結合部分の1個以上のアミノ酸を変異させることを意味する。変異は1以上の位置でアミノ酸を付加、置換又は欠失させることにより生じさせることができる。変異はPCR突然変異誘発法等の公知技術を使用して生じさせることができる。
【0038】
本願で使用する「約」なる用語は、指定値の前後約10〜20%の範囲を意味する。所定状況では、当業者に自明の通り、指定値の種類により、「約」なる用語は指定値から10〜20%を上回る偏差又は下回る偏差を意味する場合もある。
【0039】
本願で使用する「ウイルス低減/不活性化」なる用語は、特定サンプルにおけるウイルス粒子数の低下(「低減」)と、特定サンプルにおけるウイルス粒子の活性(限定されないが、例えば感染性又は複製能)の低下(「不活性化」)を意味する。このようなウイルス粒子数及び/又は活性の低下は、約1%〜約99%のオーダーとすることができ、例えば約20%〜約99%、例えば約30%〜約99%、例えば約40%〜約99%、例えば約50%〜約99%、例えば約60%〜約99%、例えば約70%〜約99%、例えば約80%〜99%、例えば約90%〜約99%が挙げられる。所定の非限定的な態様において、精製抗体産物中にウイルスが存在する場合、その量はこのウイルスのID50(対象集団の50%に感染するウイルスの量)未満であり、このウイルスのID50の最大で10分の1、又はこのウイルスのID50の最大で100分の1、又はこのウイルスのID50の最大で1000分の1である。
【0040】
「接触位置」なる用語は、26種類の公知抗体−抗原構造の1種において抗原と接触するアミノ酸が抗体の重鎖可変領域又は軽鎖可変領域のCDR1、CDR2又はCDR3において占めるアミノ酸位置を包含する。抗体−抗原複合体の26種類の公知解明構造のいずれかにおけるCDRアミノ酸が抗原と接触するならば、このアミノ酸は接触位置を占めるとみなすことができる。接触位置は非接触位置よりも抗原と接触するアミノ酸により占められる確率が高い。ある態様において、接触位置は26種類の構造のうちの4種類以上(>1.5%)で抗原と接触するアミノ酸を含むCDR位置である。別の態様において、接触位置は25種類の構造のうちの9種類以上(>32%)で抗原と接触するアミノ酸を含むCDR位置である。
【0041】
2.抗体作製
本セクションで使用する「抗体」なる用語は、無傷抗体又はその抗原結合フラグメントを意味する。
【0042】
本発明の抗体は、各種技術により作製することができ、例えば、動物に目的抗原を免疫した後、従来のモノクローナル抗体法(例えばKohler and Milstein(1975)Nature 256:495の標準体細胞ハイブリダイゼーション法)を実施すればよい。原則として、体細胞ハイブリダイゼーション法が好ましいが、他のモノクローナル抗体作製技術(例えばBリンパ球のウイルス性又は発癌性形質転換)も利用できる。
【0043】
ハイブリドーマを作製するための動物システムの1例はマウスシステムである。ハイブリドーマ作製は非常によく知られた方法である。免疫プロトコルと融合用免疫脾細胞の単離技術は当分野で公知である。融合パートナー(例えばマウス骨髄腫細胞)と融合手順も公知である。
【0044】
抗体はヒト抗体、キメラ抗体又はヒト化抗体とすることができる。本発明のキメラ抗体又はヒト化抗体は上記のように作製した非ヒトモノクローナル抗体の配列に基づいて作製することができる。目的の非ヒトハイブリドーマから重鎖及び軽鎖免疫グロブリンをコードするDNAを取得し、標準分子生物学技術を使用して非マウス(例えばヒト)免疫グロブリン配列を含むように組換えることができる。例えば、キメラ抗体を創製するためには、当分野で公知の方法を使用してマウス可変領域をヒト定常領域と連結すればよい(例えばCabillyらの米国特許第4,816,567号参照)。ヒト化抗体を創製するためには、当分野で公知の方法を使用してマウスCDR領域をヒトフレームワークに挿入すればよい(例えばWinterの米国特許第5,225,539号、並びにQueenらの米国特許第5,530,101号、5,585,089号、5,693,762号及び6,180,370号参照)。
【0045】
非限定的な1態様において、本発明の抗体はヒトモノクローナル抗体である。IL−13に対するこのようなヒトモノクローナル抗体はマウス免疫系ではなくヒト免疫系の部分を導入したトランスジェニックマウス又はトランスクロモソミックマウスを使用して作製することができる。これらのトランスジェニックマウス及びトランスクロモソミックマウスとしては、本願でHuMAb Mouse(登録商標)(Medarex,Inc.)、KM Mouse(登録商標)(Medarex,Inc.)及びXenoMouse(登録商標)(Amgen)と呼ぶマウスが挙げられる。
【0046】
更に、ヒト免疫グロブリン遺伝子を発現する他のトランスクロモソミック動物システムも当分野で入手可能であり、本発明の抗体(例えば抗IL−13抗体)を産生させるために使用することができる。例えば、ヒト重鎖トランスクロモソームとヒト軽鎖トランスクロモソームを併有するマウス(「TC マウス」と呼ぶ)を使用することができ、このようなマウスはTomizuka et al.(2000)Proc.Natl.Acad.Sci.USA 97:722−727に記載されている。更に、ヒト重鎖及び軽鎖トランスクロモソームを導入したウシも当分野で記載されており(例えばKuroiwa et al.(2002)Nature Biotechnology 20:889−894及びPCT出願第WO2002/092812号)、本発明の抗IL−13抗体を産生させるために使用することができる。
【0047】
本発明の組換えヒト抗体としては、限定されないが、本願に開示する抗IL−13抗体、その抗原結合部分又は抗IL−13関連抗体が挙げられ、例えばヒトリンパ球に由来するmRNAから作製されたヒトVL及びVH cDNAを使用して作製された組換えコンビナトリアル抗体ライブラリー(例えばscFvファージディスプレイライブラリー)のスクリーニングにより単離することができる。このようなライブラリーの作製及びスクリーニング方法は当分野で公知である。このようなライブラリーの作製及びスクリーニング方法は当分野で公知である。ファージディスプレイライブラリーの作製用市販キット(例えばその教示内容全体を本願に援用するPharmacia組換えファージ抗体システム,カタログ番号27−9400−01;及びStratagene SurfZAPTMファージディスプレイキット,カタログ番号240612参照)に加え、抗体ディスプレイライブラリーを作製及びスクリーニングするために特に使用可能な方法及び試薬の例は例えばLadnerらの米国特許第5,223,409号;KangらのPCT公開第WO92/18619号;DowerらのPCT公開第WO91/17271号;WinterらのPCT公開第WO92/20791号;MarklandらのPCT公開第WO92/15679号;BreitlingらのPCT公開第WO93/01288号;McCaffertyらのPCT公開第WO92/01047号;GarrardらのPCT公開第WO92/09690号;Fuchs et al.(1991)Bio/Technology 9:1370−1372;Hay et al.(1992)Hum Antibod Hybridomas 3:81−85;Huse et al.(1989)Science 246:1275−1281;McCafferty et al.,Nature(1990)348:552−554;Griffiths et al.(1993)EMBO J 12:725−734;Hawkins et al.(1992)J Mol Biol 226:889−896;Clackson et al.(1991)Nature 352:624−628;Gram et al.(1992)PNAS 89:3576−3580;Garrard et al.(1991)Bio/Technology 9:1373−1377;Hoogenboom et al.(1991)Nuc Acid Res 19:4133−4137;及びBarbas et al.(1991)PNAS 88:7978−7982に記載されており、その教示内容全体を本願に援用する。
【0048】
本発明のヒトモノクローナル抗体は、免疫後にヒト抗体応答を生じることができるようにヒト免疫細胞を再構成したSCIDマウスを使用して作製することもできる。このようなマウスは例えばWilsonらの米国特許第5,476,996号及び5,698,767号に記載されている。
【0049】
ある実施形態においては、本発明の方法は、抗IL−13抗体及び抗体部分、抗IL−13関連抗体及び抗体部分、並びに抗IL−13抗体と同等の性質(例えば低い解離速度と高い中和能でhIL−13と高親和性結合する性質)をもつヒト抗体及び抗体部分を包含する。ある態様において、本発明はいずれも表面プラズモン共鳴法により測定した場合に約1×10−8M以下のKdと1×10−3s−1以下のKoff速度定数でhIL−13から解離する単離ヒト抗体又はその抗原結合部分による治療法を提供する。非限定的な特定態様において、本発明に従って精製した抗IL−13抗体は生理的条件下でABT−308とIL−13の結合を競合的に阻害する。
【0050】
本発明の更に別の態様では、少なくとも1種の定常領域介在性生物エフェクター機能を非改変抗体に比較して低下させるように抗体の定常領域を改変させるように、抗体又はそのフラグメント(限定されないが、例えば抗IL−13抗体又はそのフラグメント)を改変させることができる。Fc受容体との結合の低下を示すように本発明の抗体を改変させるためには、Fc受容体(FcR)相互作用に必要な特定領域で抗体の免疫グロブリン定常領域セグメントを突然変異させればよい(例えばその教示内容全体を本願に援用するCanfield and Morrison(1991)J.Exp.Med.173:1483−1491;及びLund et al.(1991)J.of Immunol.147:2657−2662参照)。抗体のFcR結合能の低下はFcR相互作用に依存する他のエフェクター機能(例えばオプソニン化及び貪食作用及び抗原依存性細胞傷害作用)も低下させる可能性がある。
【0051】
3.抗体産生
3.1 一般産生ストラテジー
本発明の抗体を発現させるためには、遺伝子を転写及び翻訳制御配列と機能的に連結するように、部分又は全長軽鎖及び重鎖をコードするDNAを1以上の発現ベクターに挿入する(例えばその教示内容全体を本願に援用する米国特許第6,914,128号参照)。この関連で「機能的に連結」なる用語はベクター内の転写及び翻訳制御配列が抗体遺伝子の転写と翻訳を制御するというその所期機能を発揮するように抗体遺伝子をベクターにライゲーションすることを意味する。発現ベクターと発現制御配列は、使用される発現宿主細胞と適合可能となるように選択される。抗体軽鎖遺伝子と抗体重鎖遺伝子は、別々のベクターに挿入してもよいが、より典型的には、両方の遺伝子を同一発現ベクターに挿入する。抗体遺伝子は、標準方法(例えば抗体遺伝子フラグメントとベクター上の相補的制限部位のライゲーション又は制限部位が存在しない場合には平滑末端ライゲーション)により発現ベクターに挿入される。抗体又は抗体関連軽鎖及び重鎖配列を挿入する前に、発現ベクターに予め定常領域配列を保持させてもよい。例えば、抗IL−13抗体又は抗IL−13抗体関連VH及びVL配列から全長抗体遺伝子を作製する1つのアプローチはVHセグメントをベクター内のCHセグメントと機能的に連結し、VLセグメントをベクター内のCLセグメントと機能的に連結するように、夫々重鎖定常領域と軽鎖定常領域を予めコードする発現ベクターにこれらの配列を挿入する方法である。上記に加え、又は上記の代わりに、組換え発現ベクターは宿主細胞からの抗体鎖の分泌を助長するシグナルペプチドをコードすることができる。シグナルペプチドを抗体鎖遺伝子のアミノ末端にインフレームで連結するように抗体鎖遺伝子をベクターにクローニングすることができる。シグナルペプチドは免疫グロブリンシグナルペプチド又は異種シグナルペプチド(即ち、非免疫グロブリン蛋白質に由来するシグナルペプチド)とすることができる。
【0052】
抗体鎖遺伝子に加え、本発明の組換え発現ベクターは、宿主細胞における抗体鎖遺伝子の発現を制御する1個以上の調節配列を保持することができる。「調節配列」なる用語は、プロモーター、エンハンサー及び抗体鎖遺伝子の転写又は翻訳を制御する他の発現制御エレメント(例えばポリアデニル化シグナル)を包含するものとする。このような調節配列は、例えばその教示内容全体を本願に援用するGoeddel;Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,CA(1990)に記載されている。当業者に自明の通り、調節配列の選択を含む発現ベクターの設計は、形質転換しようとする宿主細胞の選択、所望される蛋白質発現レベル等の因子により異なる。哺乳動物宿主細胞発現に適した調節配列としては、哺乳動物細胞で高レベルの蛋白質発現を誘導するウイルスエレメントが挙げられ、例えばサイトメガロウイルス(CMV)(例えばCMVプロモーター/エンハンサー)、サルウイルス40(SV40)(例えばSV40プロモーター/エンハンサー)、アデノウイルス(例えばアデノウイルス主要後期プロモーター(AdMLP))及びポリオーマに由来するプロモーター及び/又はエンハンサーが挙げられる。ウイルス調節エレメントとその配列に関する更に詳細な説明については、例えばその教示内容全体を本願に援用するStinskiの米国特許第5,168,062号、Bellらの米国特許第4,510,245号、及びSchaffnerらの米国特許第4,968,615号参照。
【0053】
抗体鎖遺伝子と調節配列に加え、本発明の組換え発現ベクターは、1個以上の他の配列(例えば宿主細胞におけるベクターの複製を調節する配列(例えば複製起点)及び/又は選択マーカー遺伝子)を保持することができる。選択マーカー遺伝子は、ベクターを導入した宿主細胞の選択を容易にする(例えばその教示内容全体を本願に援用するいずれもAxelらの米国特許第4,399,216号、4,634,665号及び5,179,017号参照)。例えば、典型的には選択マーカー遺伝子はベクターを導入した宿主細胞にG418、ハイグロマイシン又はメトトレキサート等の薬剤に対する耐性を付与する。適切な選択マーカー遺伝子としては、ジヒドロ葉酸還元酵素(DHFR)遺伝子(メトトレキサート選択/増幅下にdhfr−宿主細胞で使用)とneo遺伝子(G418選択用)が挙げられる。
【0054】
本発明の抗体又は抗体部分は、宿主細胞で免疫グロブリン軽鎖及び重鎖遺伝子を組換え発現させることにより作製することができる。抗体を組換え発現させるためには、抗体の免疫グロブリン軽鎖及び重鎖を宿主細胞で発現させ、宿主細胞を培養する培地中に分泌させるように、前記軽鎖及び重鎖をコードするDNAフラグメントを保持する1以上の組換え発現ベクターを宿主細胞にトランスフェクトし、培地から抗体を回収することができる。標準組換えDNA法を使用して抗体重鎖及び軽鎖遺伝子を得、これらの遺伝子を組換え発現ベクターに導入し、その教示内容全体を本願に援用するSambrook,Fritsch and Maniatis(eds),Molecular Cloning;A Laboratory Manual,Second Edition,Cold Spring Harbor,N.Y.,(1989)、Ausubel et al.(eds.)Current Protocols in Molecular Biology,Greene Publishing Associates(1989)、並びに米国特許第4,816,397号及び6,914,128号に記載されているもの等の宿主細胞にベクターを導入する。
【0055】
軽鎖と重鎖を発現させるためには、重鎖と軽鎖をコードする発現ベクターを標準技術により宿主細胞にトランスフェクトする。各種語形の「トランスフェクション」なる用語は外来DNAを原核又は真核宿主細胞に導入するために広く使用されている多様な技術を包含するものとし、例えばエレクトロポレーション、リン酸カルシウム沈殿法、DEAE−デキストラントランスフェクション等が挙げられる。理論的には、原核宿主細胞でも真核宿主細胞でも本発明の抗体を発現させることが可能であるが、真核細胞、特に哺乳動物細胞は適正に折り畳まれた免疫活性抗体を組立てて分泌する可能性が原核細胞よりも高いため、哺乳動物宿主細胞等の真核細胞で抗体を発現させると適切である。抗体遺伝子の原核細胞発現は、活性な抗体の高収率産生には有効でないと報告されている(その教示内容全体を本願に援用するBoss and Wood(1985)Immunology Today 6:12−13)。
【0056】
本願のベクターでDNAをクローニング又は発現させるのに適した宿主細胞は上記原核細胞、酵母細胞又は高等真核細胞である。この目的に適した原核生物としては、グラム陰性菌やグラム陽性菌等の真正細菌が挙げられ、例えばEscherichia(例えば大腸菌)、Enterobacter、Erwinia、Klebsiella、Proteus、Salmonella(例えばSalmonella typhimurium)、Serratia(例えばSerratia marcescans)及びShigella等の腸内細菌や、桿菌(例えばB.subtilis及びB.licheniformis(例えば1989年4月12日発行のDD 266,710に開示されているB.licheniformis 41P))、Pseudomonas(例えばP.aeruginosa)及びStreptomycesが挙げられる。適切な大腸菌クローニング宿主の1例は大腸菌294株(ATCC31,446)であるが、大腸菌B株、大腸菌X1776株(ATCC31,537)、及び大腸菌W3110株(ATCC27,325)等の他の株も適切である。これらの例は例示であり、非限定的である。
【0057】
原核生物に加え、糸状菌や酵母等の真核微生物もポリペプチドをコードするベクターの適切なクローニング又は発現宿主である。下等真核宿主微生物としてはSaccharomyces cerevisiaeないしパン酵母が最も広く使用されている。この他、Schizosaccharomyces pombe;Kluyveromyces宿主(例えばK.lactis、K.fragilis(ATCC12,424)、K.bulgaricus(ATCC16,045)、K.wickeramii(ATCC24,178)、K.waltii(ATCC56,500)、K.drosophilarum(ATCC36,906)、K.thermotolerans及びK.marxianus);Yarrowia(EP402,226);Pichia pastoris(EP183,070);Candida;Trichoderma reesia(EP244,234);Neurospora crassa;Schwanniomyces(例えばSchwanniomyces occidentalis);及び糸状菌(例えばNeurospora、Penicillium、Tolypocladium及びAspergillus宿主(例えばA.nidulans及びA.niger))等の多数の他の属、種及び株も広く入手可能であり、本願で有用である。
【0058】
グリコシル化抗体の発現に適した宿主細胞は、多細胞生物に由来する。無脊椎動物細胞の例としては植物細胞と昆虫細胞が挙げられる。多数のバキュロウイルス株及び変異株と、Spodoptera frugiperda(ヨトウムシ)、Aedes aegypti(蚊)、Aedes albopictus(蚊)、Drosophila melanogaster(果実蝿)、及びBombyx mori等の宿主に由来する対応する許容昆虫宿主細胞が同定されている。トランスフェクション用の各種ウイルス株(例えばAutographa californica NPVのL−l変異株及びBombyx mori NPVのBm−5株)が公共入手可能であり、このようなウイルスを特にSpodoptera frugiperda細胞のトランスフェクション用として、本発明に従って本願のウイルスとして使用することができる。ワタ、トウモロコシ、ジャガイモ、ダイズ、ペチュニア、トマト及びタバコの植物細胞培養物も宿主として利用できる。
【0059】
本発明の組換え抗体の発現に適した哺乳動物宿主細胞としては、チャイニーズハムスター卵巣(CHO細胞)(例えばその教示内容全体を本願に援用するUrlaub and Chasin,(1980)PNAS USA 77:4216−4220に記載のdhfr−CHO細胞を例えばKaufman and Sharp(1982)Mol.Biol.159:601−621に記載されているようにDHFR選択マーカーと併用する)、NS0骨髄腫細胞、COS細胞及びSP2細胞が挙げられる。抗体遺伝子をコードする組換え発現ベクターを哺乳動物宿主細胞に導入する場合には、宿主細胞で抗体を発現させるため又は宿主細胞を増殖させる培養培地に抗体を分泌させるために十分な時間にわたって宿主細胞を培養することにより抗体を産生させる。有用な哺乳動物宿主細胞株の他の例はSV40で形質転換させたサル腎細胞CV1株(COS−7,ATCC CRL 1651);ヒト胎児腎細胞株(293又は懸濁培養増殖用にサブクローニングした293細胞,Graham et al.,J.Gen Virol:36:59(1977));ベビーハムスター腎細胞(BHK,ATCC CCL 10);チャイニーズハムスター卵巣細胞/−DHFR(CHO,Urlaub et al.,Proc.Natl.Acad.Sci.USA 77:4216(1980));マウスセルトリ細胞(TM4,Mather,Biol.Reprod.23:243−251(1980));サル腎細胞(CV1 ATCC CCL 70);アフリカミドリザル腎細胞(VERO−76,ATCC CRL−1587);ヒト子宮頸癌細胞(HELA,ATCC CCL 2);イヌ腎細胞(MDCK,ATCC CCL 34);バッファローラット肝細胞(BRL 3A,ATCC CRL 1442);ヒト肺細胞(W138,ATCC CCL 75);ヒト肝細胞(Hep G2,HB 8065);マウス乳癌(MMT 060562,ATCC CCL51);TRI細胞(Mather et al.,Annals N.Y.Acad.Sci.383:44−68(1982));MRC 5細胞;FS4細胞;及びヒト肝細胞癌株(Hep G2)であり、上記文献はその教示内容全体を本願に援用する。
【0060】
宿主細胞を上記抗体産生用発現又はクローニングベクターで形質転換させ、プロモーターの誘導、形質転換細胞の選択又は所望配列をコードする遺伝子の増幅の必要に応じて改変した慣用栄養培地で培養する。
【0061】
抗体を産生させるために使用する宿主細胞は、各種培地で培養することができる。ハムF10(登録商標)(Sigma)、最少必須培地Minimal Essential Medium(登録商標)((MEM)(Sigma))、RPMI−1640(Sigma)及びダルベッコ改変イーグル培地Dulbecco’s Modified Eagle’s Medium(登録商標)((DMEM),Sigma)等の市販培地が宿主細胞を培養するのに適している。更に、その教示内容全体を本願に援用するHam et al.,Meth.Enz.58:44(1979),Barnes et al.,Anal.Biochem.102:255(1980)、米国特許第4,767,704号、4,657,866号、4,927,762号、4,560,655号もしくは5,122,469号、WO90/03430、WO87/00195、又は米国再発行特許第30,985号に記載されている培地の任意のものを宿主細胞の培養培地として使用することができる。必要に応じてホルモン及び/又は他の成長因子(例えばインスリン、トランスフェリン又は上皮成長因子)、塩(例えば塩化ナトリウム、カルシウム、マグネシウム及びリン酸塩)、緩衝液(例えばHEPES)、ヌクレオチド(例えばアデノシン及びチミジン)、抗生物質(例えばゲンタマイシン薬物)、(通常ではマイクロモル範囲の最終濃度で存在する無機化合物として定義される)微量元素、及びグルコース又は同等のエネルギー源をこれらの培地の任意のものに添加することができる。当業者に公知の任意の他の必要な助剤も適当な濃度で添加することができる。温度、pH等の培養条件は発現に選択される宿主細胞で従来使用されている条件であり、当業者に容易に理解されよう。
【0062】
無傷の抗体の部分(例えばFabフラグメントやscFv分子)を作製するために宿主細胞を使用することもできる。当然のことながら、上記手順の変形も本発明の範囲内である。例えば、ある実施形態では、本発明の抗体の軽鎖又は重鎖の(両方ではなく)いずれかをコードするDNAを宿主細胞にトランスフェクトすることが望ましい場合がある。IL−13、特にhIL−13との結合に不要な軽鎖及び重鎖のいずれか又は両方をコードするDNAの一部又は全部を除去するために組換えDNA技術を使用してもよい。このような短縮型DNA分子から発現される分子も本発明の抗体に含まれる。更に、本発明の抗体を標準化学架橋法により第2の抗体と架橋することにより、一方の重鎖と一方の軽鎖が本発明の抗体であり、他方の重鎖及び軽鎖がIL−13以外の抗原に特異的である二機能性抗体を作製することができる。
【0063】
本発明の抗体又はその抗原結合部分の組換え発現に適したシステムでは、抗体重鎖及び抗体軽鎖の両方をコードする組換え発現ベクターをリン酸カルシウムトランスフェクションによりdhfr−CHO細胞に導入する。組換え発現ベクターの内側で、遺伝子の高レベルの転写を誘導するように、抗体重鎖及び軽鎖遺伝子を各々CMVエンハンサー/AdMLPプロモーター調節エレメントと機能的に連結する。組換え発現ベクターは更にメトトレキサート選択/増幅を使用してベクターをトランスフェクトしたCHO細胞の選択を可能にするDHFR遺伝子を保持する。選択した形質転換宿主細胞を培養し、抗体重鎖及び軽鎖を発現させ、無傷抗体を培養培地から回収する。標準分子生物学技術を使用して組換え発現ベクターを作製し、宿主細胞にトランスフェクトし、形質転換細胞を選択し、宿主細胞を培養し、抗体を培養培地から回収する。
【0064】
組換え技術を使用する場合には、抗体をペリプラズム空間で細胞内産生させることもできるし、培地に直接分泌させることもできる。ある態様において、抗体を細胞内産生させる場合には、第1段階として、宿主細胞又は(例えば均質化による)溶解細胞のいずれかであるかに関係なく、粒状破片を例えば遠心又は限外濾過により除去することができる。抗体を培地に分泌させる場合には、市販蛋白質濃縮フィルター(例えばAmicon(登録商標)又はMillipore Pellicon(登録商標)限外濾過ユニット)を使用して、先ずこのような発現システムからの上清を濃縮することができる。
【0065】
本発明の方法の前に、細胞破片から抗体を精製する手順は、まず第1に抗体の発現部位によって異なる。抗体によっては細胞から周囲の増殖培地に直接分泌させることもできるし、細胞内に産生するものもある。後者抗体では、精製工程の第1段階は典型的には細胞の溶解を含み、これは機械的剪断、浸透圧ショック法又は酵素処理を含む各種方法により実施できる。このような破砕により細胞の全内容物はホモジネート中に放出され、更に、寸法が小さいために除去しにくい細胞以下のフラグメントが生成される。これらは一般に分画遠心法又は濾過により除去される。抗体が分泌される場合には、一般に先ず市販蛋白質濃縮フィルター(例えばAmicon(登録商標)又はMillipore Pellicon(登録商標)限外濾過ユニット)を使用してこのような発現システムからの上清を濃縮する。抗体が培地中に分泌される場合には、組換え宿主細胞も、例えば、タンジェンシャルフロー濾過により細胞培養培地から分離することができる。本発明の抗体精製法を使用して抗体を更に培養培地から回収することができる。
【0066】
3.2.典型的産生ストラテジー
ある実施形態において、抗IL−13抗体産生の初期工程は、スピナーフラスコとバイオウェーブバッグの操作を使用し、抗IL−13抗体を発現するCHO細胞を凍結バイアル1本から110Lシードバイオリアクターの接種に望ましいバイオマスまで増殖させる。マスターセルバンクCHO細胞の凍結バイアルを解凍し、増殖培地(SR−512)に加え、遠心する。細胞を増殖培地に再懸濁し、容積を増加させながら使い捨てスピナーフラスコ、振盪フラスコ及び/又はバイオウェーブバッグで37℃及び5% CO2にて増殖させる。シードバイオリアクターに接種する前に20Lウェーブバッグ2個を使用して最終細胞塊増殖を最大にする。両方の20Lウェーブバッグからの細胞密度が約15〜17日目に生存細胞≧2.0×106個/mLに達したら、更に増殖させるために、増殖培地SR−520を充填した110シードバイオリアクターに培養液を移す。接種後、目標温度を37℃とし、pHを7.1の目標値に設定し、NaOHの添加とCO2スパージングにより制御する。空気と酸素のスパージングによりバイオリアクター内の溶存酸素(DO)を40%の目標値に制御する。約2〜4日後に細胞密度が生存細胞≧2.6×106個/mLに達したら、培養液を3000L産生用バイオリアクターに移す。
【0067】
ある実施形態では、3000L産生用バイオリアクターの部分フィルを使用して細胞培養物を更に増殖させる。先ず、リアクターに増殖培地(SR−520)を仕込み、110Lシードバイオリアクターからのバッチを接種する。このショートフィル段階中は、温度、溶存酸素及びpHを夫々37℃、40%及び7.1に制御する。培養pHはCO2スパージングとNaOH添加により制御する。典型的には、細胞は生存細胞≧1.6×106個/mLの産生段階密度に達する前に2〜4日間増殖する。
【0068】
産生培地SR−521(1950L)を3000Lバイオリアクター内の細胞培養液に加え、産生段階を開始する。消泡剤Cを加え、発泡を抑える。CO2スパージングとNaOH添加のオンオフにより培養pHを6.9の目標値に制御する。温度と溶存酸素を夫々35℃及び40%の目標値に制御する。バイオリアクター内のDOは先ず空気スパージングにより所望値に制御し、必要に応じて純酸素を補充する。ある実施形態では、生存細胞密度が≧3.0×106個/mLに達したら、温度を33℃の目標値まで下げ、pHとDOを夫々6.9と40%の目標値に維持するが、他の態様では、35℃の目標値に維持する。必要に応じてグルコース(SR−334)を添加する。細胞生存率が≦50%まで低下したら、培養液を回収し、以下に概説するように精製する。
【0069】
4.抗体精製
4.1 抗体精製概論
本発明は、抗体と少なくとも1種のHCPを含有する混合物から精製(ないし「HCP低減」)抗体調製物を生成する方法を提供する。本発明は、最終精製調製物の浸出プロテインAを低減する方法も提供する。本発明の精製工程は、上記方法及び当分野の慣用方法を使用して抗体が産生された時点で分離工程から開始する。表1は精製スキームの1態様を要約する。このスキームの変形も考えられ、限定されないが、プロテインAアフィニティークロマトグラフィー工程を省略したり、イオン交換工程の順序を逆にした変形が考えられ、本発明の範囲に含まれる。
【0070】
【表1】
【0071】
抗体を含有する清澄化溶液又は混合物が得られたら、イオン交換分離工程と疎水性相互作用分離工程を含む異なる精製技術の組み合わせを使用し、細胞により産生された他の蛋白質(例えばHCP)からの抗体の分離を実施する。分離工程は蛋白質の電荷、疎水度又は寸法に基づいて蛋白質混合物を分離する。本発明のある態様では、カチオン、アニオン及び疎水性相互作用を含むクロマトグラフィーを使用して分離を実施する。これらの技術の各々に数種の異なるクロマトグラフィー樹脂が利用可能であり、該当する特定蛋白質に合わせて精製スキームを正確に適応させることができる。分離方法の各々の要点は、蛋白質を異なる速度でカラムに流し、更にカラムを通過するにつれて増加する物理的分離を達成すること、又は蛋白質を選択的に分離媒体に接着させた後、異なる溶媒により分画溶出させることである。場合により、不純物がカラムと特異的に接着し、抗体が接着しない場合、即ち抗体がフロースルー中に存在する場合には、抗体を不純物から分離する。
【0072】
上記のように、精製スキームの正確な適応は、精製しようとする蛋白質の要件に依存する。ある実施形態では、1種以上のHCPから抗体を分離するために本発明の分離工程を利用する。本願に記載する方法を使用して有効に精製することができる抗体としては、限定されないが、ヒトIgA1、IgA2、IgD、IgE、IgG1、IgG2、IgG3、IgG4及びIgM抗体が挙げられる。ある実施形態において、本発明の精製ストラテジーはプロテインAアフィニティークロマトグラフィーの使用を除外し、例えばIgG3抗体はプロテインAと効率的に結合しないので、IgG3抗体の精製がこれに該当する。精製スキームを個別に適応させる他の因子としては、限定されないが、プロテインAはFc領域と結合するので(例えばそのFabフラグメントに対する全長抗体のコンテキストにおける)Fc領域の有無、目的抗体の作製に利用される特定生殖細胞系列配列、及び抗体のアミノ酸組成(例えば抗体の一次配列と分子の総電荷/疎水度)が挙げられる。1種以上の特徴を共有する抗体はこの特徴を利用するように適応させた精製ストラテジーを使用して精製することができる。
【0073】
4.2 一次回収
本発明の精製方法の初期工程は、サンプルマトリックスからの抗体の第1段階の清澄化及び一次回収を含む。更に、一次回収工程はサンプルマトリックスに存在する可能性のあるウイルスを低減又は不活性化する点でもあり得る。例えば、熱不活性化(低温殺菌)、pH不活性化、溶媒/デタージェント処理、UV及びγ線照射、並びに所定の化学不活性化剤(例えばβ−プロピオラクトン又は例えばその教示内容全体を本願に援用する米国特許第4,534,972号におけるように銅フェナントロリン)の添加を含む各種ウイルス低減/不活性化方法の任意の1種以上を精製の一次回収段階中に使用することができる。本発明のある実施形態では、一次回収段階中にサンプルマトリックスをpHウイルス低減/不活性化に供する。
【0074】
pHウイルス低減/不活性化方法としては、限定されないが、混合物を低pHで所定時間インキュベートした後、pHを中和させ、粒状物を濾過により除去する。ある実施形態では、混合物を約2〜5のpH、約3〜4のpH、限定されないが、例えば約3.5のpHでインキュベートする。サンプル混合物のpHは任意の適切な酸により低下させることができ、限定されないが、クエン酸、酢酸、カプリル酸又は他の適切な酸が挙げられる。pHレベルの選択は抗体産物と緩衝液成分の安定性プロファイルに大きく依存する。低pHウイルス低減/不活性化中の目的抗体の品質がpHと低pHインキュベーション時間に影響されることは知られている。ある実施形態において、低pHインキュベーション時間は0.5時間〜2時間となり、限定されないが、例えば0.5時間〜1.5時間、限定されないが、例えば約1時間が挙げられる。ウイルス低減/不活性化は蛋白質濃度に依存し、高濃度の低減/不活性化が制限される可能性があるが、その他にこれらの同一パラメーターにも依存する。従って、所望レベルのウイルス低減/不活性化を達成するように蛋白質濃度、pH及び低減/不活性化時間の適正なパラメーターを選択することができる。
【0075】
ある実施形態では、適切なフィルターの使用によりウイルス低減/不活性化を達成することができる。適切なフィルターの非限定的な1例はPall Corporation製品であるUltipor DV50(登録商標)フィルターである。本発明のある実施形態は、一次回収段階中にこのような濾過を利用するが、他の態様では、精製工程の他の段階(例えば最後から2番目又は最終精製工程)に利用する。ある実施形態では、別のフィルターをウイルス低減/不活性化に利用し、限定されないが、Viresolve(登録商標)フィルター(Millipore,Billerica,Mass.);Zeta Plus VR(登録商標)フィルター(CUNO;Meriden,Conn.);及びPlanova(登録商標)フィルター(Asahi Kasei Pharma,Planova Division,Buffalo Grove,Ill.)が挙げられる。
【0076】
ウイルス低減/不活性化を利用する態様では、必要に応じて後続精製工程に備えてサンプル混合物を調整することができる。例えば、低pHウイルス低減/不活性化後に、精製工程を続ける前にサンプル混合物のpHを典型的にはより中性のpH(例えば約4.5〜約8.5、限定されないが、例えば約4.9)に調整する。更に、所望の導電率を得るように混合物を注射用水(WFI)でフラッシュしてもよい。
【0077】
ある実施形態においては、一次回収はサンプルマトリックスを更に清澄化することにより、抗IL−13抗体を精製し易くするための1以上の遠心工程を含む。サンプルの遠心は限定されないが、例えば7,000×g〜約12,750×gで実施することができる。大規模精製の場合には、このような遠心は限定されないが、例えば得られる上清中で150NTUの濁度レベルを達成するように設定された流速を使用してオンラインで実施することができる。次に後続精製に備えてこのような上清を採取することができる。
【0078】
ある実施形態においては、一次回収は、サンプルマトリックスを更に清澄化することにより、本発明の抗体を精製し易くするための1以上の深層濾過工程の使用を含む。デプスフィルターは段階的密度の濾過媒体を含む。このような段階的密度により、フィルターの表面付近に大きい粒子をトラップすると共に、フィルターの表面の広い開口領域を通過する小さい粒子のみをフィルターの中心に近い狭い開口にトラップすることができる。ある実施形態において、深層濾過工程はデリピッド深層濾過工程とすることができる。ある実施形態では、一次回収段階中のみに深層濾過工程を利用するが、他の態様は、1以上の他の精製段階中にもデリピッドデプスフィルター等のデプスフィルターを利用する。本発明に関連して使用することができるデプスフィルターの非限定的な例としては、Cuno(登録商標)モデル30/60ZAデプスフィルター(3M Corp.)と、0.45/0.2μm Sartopore(登録商標)二重層フィルターカートリッジが挙げられる。
【0079】
4.3 アフィニティークロマトグラフィー
ある実施形態では、一次回収サンプルをアフィニティークロマトグラフィーに供し、更に目的抗体をHCPから精製する。ある実施形態において、クロマトグラフィー材料は目的抗体と選択的又は特異的に結合することが可能である。このようなクロマトグラフィー材料の非限定的な例としては、プロテインA、プロテインG、目的抗体と結合する抗原を含むクロマトグラフィー材料、及びFc結合蛋白質を含むクロマトグラフィー材料が挙げられる。特定態様において、アフィニティークロマトグラフィー工程は適切なプロテインA樹脂を充填したカラムに一次回収サンプルを供する段階を含む。プロテインA樹脂は各種抗体、特にIgG1、IgG2及びIgG4のアフィニティー精製・単離に有用である。プロテインAは主にそのFc領域を介して哺乳動物IgGと結合する細菌細胞壁蛋白質である。その天然状態において、プロテインAは5個のIgG結合ドメインと機能の不明な他のドメインをもつ。
【0080】
プロテインA樹脂には、数種類の市販品が存在する。適切な樹脂としては、限定されないが、GE Healthcare製品であるMabSelect(登録商標)とMillipore製品であるProSep Ultra Plus(登録商標)が挙げられる。MabSelect(登録商標)を充填した適切なカラムの非限定的な1例は直径約1.0cm×長さ約21.6cm(ベッドボリューム〜17mL)のカラムである。このサイズのカラムは小規模精製に使用することができ、大規模化に使用される他のカラムと比較することができる。例えば、より大規模の精製にはベッドボリューム約6.6Lの20cm×21cmカラムを使用することができる。カラムに関係なく、MabSelect(登録商標)やProSep Ultra Plus(登録商標)等の適切な樹脂を使用してカラムに充填することができる。
【0081】
ある実施形態では、精製を特定の目的抗体に適応させるために、プロテインA樹脂の動的結合容量(DBC)を測定すると有利であろう。例えば、限定されないが、MabSelect(登録商標)又はProSept Ultra Plus(登録商標)カラムのDBCは一段階流速ロード又は二段階流速ロードストラテジーにより測定することができる。一段階流速ロードは全ロード時間を通して約300cm/時の速度で評価することができる。二段階流速ロードストラテジーは樹脂1mL当たり蛋白質約35mgまでを約300cm/時の線速度でカラムにロードした後、ロードの最後の部分の滞留時間を長くするように線速度を半減させることにより測定することができる。
【0082】
ある実施形態では、サンプルロードの前に適切な緩衝液でプロテインAカラムを平衡化することができる。適切な緩衝液の非限定的な1例はpH約7.2のトリス/NaClバッファーである。適切な平衡化条件の非限定的な1例は25mMトリス,100mM NaCl,pH約7.2である。この平衡化後、サンプルをカラムにロードすることができる。カラムにロードした後、例えば平衡化バッファーを使用してカラムを1回以上洗浄することができる。カラムから溶出させる前に、別の緩衝液を利用する洗浄を含む他の洗浄を使用することができる。例えば、1カラム体積以上の20mMクエン酸/クエン酸ナトリウム,0.5M NaCl,pH約6.0を使用してカラムを洗浄することができる。この洗浄後、場合により平衡化バッファーを使用して1回以上洗浄してもよい。その後、適当な溶出バッファーを使用してプロテインAカラムから溶出させることができる。適切な溶出バッファーの非限定的な1例はpH約3.5の酢酸/NaClバッファーである。適切な条件は例えば0.1M酢酸,pH約3.5である。当業者に周知の技術を使用して溶出液をモニターすることができる。例えば、OD280の吸光度を追跡することができる。約0.5AUの初期偏光から出発して溶出ピークの立下りの約0.5AUの読取値までカラム溶出液を採取することができる。次に、後続処理に備えて目的の溶出画分を調製することができる。例えば、採取したサンプルをpH約10のトリス(例えば1.0M)で約5.0のpHまで滴定することができる。場合により、この滴定後のサンプルを濾過し、更に処理してもよい。
【0083】
4.4 イオン交換クロマトグラフィー
ある実施形態においては、本発明は抗体を含有する溶出液が得られるように、抗体と少なくとも1種のHCPを含有する混合物を少なくとも1種のイオン交換分離工程に供することにより、前記混合物からHCP低減抗体調製物を生成する方法を提供する。イオン交換分離としては、2種類の物質を夫々のイオン電荷の差に基づいて分離する任意方法が挙げられ、カチオン交換材料又はアニオン交換材料を利用することができる。
【0084】
カチオン交換材料とアニオン交換材料のどちらを使用するかは蛋白質の総電荷による。従って、カチオン交換工程を使用する前にアニオン交換工程を使用することも、アニオン交換工程を使用する前にカチオン交換工程を使用することも本発明の範囲に含まれる。更に、カチオン交換工程のみを使用することも、アニオン交換工程のみを使用することも、両者の任意の順次併用も本発明の範囲に含まれる。
【0085】
分離を実施するには、各種技術のいずれかを使用(例えばバッチ精製技術やクロマトグラフィー技術を使用)することにより、初期抗体混合物をイオン交換材料と接触させればよい。
【0086】
例えば、バッチ精製の場合には、所望の出発バッファー中でイオン交換材料を調製又は平衡化する。調製又は平衡化後、イオン交換材料のスラリーが得られる。抗体溶液をスラリーと接触させ、分離しようとする抗体をイオン交換材料に吸着させる。例えばスラリーを沈殿させ、上清を除去することにより、イオン交換材料と結合しないHCPを含有する溶液をスラリーから分離する。スラリーを1回以上の洗浄工程に供することができる。必要に応じて、スラリーを高導電率の溶液と接触させ、イオン交換材料と結合したHCPを脱着させてもよい。結合したポリペプチドを溶出させるためには、緩衝液の塩濃度を上げればよい。
【0087】
イオン交換分離法としてイオン交換クロマトグラフィーを使用してもよい。イオン交換クロマトグラフィーは分子の総電荷の差に基づいて分子を分離する。抗体の精製では、抗体はイオン交換材料(例えば樹脂)に付加した官能基と結合するためにこの官能基に対して逆電荷でなければならない。例えば、そのpI未満のpHの緩衝液中で一般に総電荷がプラスの抗体は負電荷の官能基を含むカチオン交換材料と結合する。
【0088】
イオン交換クロマトグラフィーでは、周囲の緩衝液のイオン強度が低いならば、溶質の表面の帯電パッチはクロマトグラフィーマトリックスに付加した逆電荷により吸引される。溶出は一般にイオン交換マトリックスの帯電部位について溶質と競合するように緩衝液のイオン強度(即ち導電率)を上げることにより行われる。溶質の溶出を行う別の方法はpHを変化させることにより、溶質の電荷を変化させる方法である。導電率又はpHの変化は漸次(グラジエント溶出)でも段階的(ステップワイズ溶出)でもよい。
【0089】
クロマトグラフィーのアニオン性又はカチオン性担体を形成するためにはアニオン性又はカチオン性置換基をマトリックスに付加すればよい。アニオン交換置換基の非限定的な例としては、ジエチルアミノエチル(DEAE)、第四級アミノエチル(QAE)及び第四級アミン(Q)基が挙げられる。カチオン置換基としては、カルボキシメチル(CM)、スルホエチル(SE)、スルホプロピル(SP)、リン酸(P)及びスルホン酸(S)が挙げられる。DE23(登録商標)、DE32(登録商標)、DE52(登録商標)、CM−23(登録商標)、CM−32(登録商標)及びCM−52(登録商標)等のセルロースイオン交換樹脂がWhatman Ltd.Maidstone,Kent,U.K.から市販されている。SEPHADEX(登録商標)系架橋結合イオン交換体も知られている。例えば、DEAE−、QAE−、CM−及びSP−SEPHADEX(登録商標)、DEAE−、Q−、CM−及びS−SEPHAROSE(登録商標)、並びにSEPHAROSE(登録商標)ファストフローがいずれもPharmacia ABから市販されている。更に、TOYOPEARL(登録商標)DEAE−650S又はM及びTOYOPEARL(登録商標)CM−650S又はM等のDEAE及びCMの両者で誘導体化したエチレングリコール−メタクリレートコポリマーもToso Haas Co.,Philadelphia,Paから市販されている。ある実施形態では、Pall Mustang Qメンブレンカートリッジを使用してアニオン交換工程を実施する。
【0090】
抗体と不純物(例えばHCP)を含有する混合物をイオン交換カラム(例えばカチオン交換カラム)にロードする。例えば、限定されないが、使用するカラムに応じて蛋白質約80g/L樹脂のロード率で混合物をロードすることができる。適切なカチオン交換カラムの1例は直径80cm×長さ23cm、ベッドボリューム約116Lのカラムである。このカチオンカラムにロードした混合物を次に洗浄バッファー(平衡化バッファー)で洗浄することができる。その後、抗体をカラムから溶出させ、第1の溶出液を得る。
【0091】
このイオン交換工程は、HCP等の不純物を低減しながら目的抗体の捕集を容易にする。ある態様において、イオン交換カラムはカチオン交換カラムである。例えば、限定されないが、このようなカチオン交換カラムに適した樹脂はCM HyperDF(登録商標)樹脂である。これらの樹脂はPall Corporation等の業者から入手可能である。このカチオン交換法は室温又は室温付近で実施することができる。
【0092】
4.5 限外濾過/透析濾過
本発明のある実施形態では抗体サンプルを更に精製・濃縮するために限外濾過及び/又は透析濾過工程を利用する。限外濾過はMicrofiltration and Ultrafiltration:Principles and Applications,L.Zeman and A.Zydney(Marcel Dekker,Inc.,New York,N.Y.,1996)及びUltrafiltration Handbook,Munir Cheryan(Technomic Publishing,1986;ISBN No.87762−456−9)に詳細に記載されている。濾過工程の1例は表題“Pharmaceutical Process Filtration Catalogue”
のMilliporeカタログ177〜202頁(Bedford,Mass.,1995/96)に記載されているようなタンジェンシャルフロー濾過である。限外濾過は一般に0.1μm未満の細孔サイズのフィルターを使用する濾過を意味するとみなされる。このような細孔サイズの小さいフィルターを利用することにより、抗体をフィルターに保持しながら、サンプルバッファーをフィルターに透過させることによりサンプルの体積を減少させることができる。
【0093】
透析濾過は、塩、糖及び非水性溶媒を除去及び交換するため、遊離種を結合種から分離するため、低分子量材料を除去するため、並びに/又はイオン及び/もしくはpH環境の迅速な変化を誘導するために限外濾過フィルターを使用する方法である。限外濾過中の溶液に溶媒を限外濾過速度とほぼ等しい速度で加えることにより、微小溶質は最も効率的に除去される。こうして微小種が一定流量で溶液から洗い流され、保持された抗体が有効に精製される。本発明のある実施形態では、場合により後続クロマトグラフィー又は他の精製工程の前に本発明に関連して使用される各種緩衝液を交換するため、あるいは抗体調製物から不純物を除去するために透析濾過工程を利用する。
【0094】
4.6 疎水性相互作用クロマトグラフィー
本発明は、抗体と少なくとも1種のHCPを含有する混合物からHCP低減抗体調製物を生成する方法であって、更に疎水性相互作用分離工程を含む方法にも関する。例えば、HCP濃度の低下した第2の溶出液が得られるように、イオン交換カラムから得られた第1の溶出液を疎水性相互作用材料に供することができる。本願に記載する工程等の疎水性相互作用クロマトグラフィー工程を一般に実施し、抗体凝集物等の蛋白質凝集物と工程関連不純物を除去する。
【0095】
分離を実施するには、例えばバッチ精製技術又はカラムを使用してサンプル混合物をHIC材料と接触させる。HIC精製の前に、例えば混合物をプレカラムに通すことにより、カオトロピック剤又は超疎水性物質を除去することが望ましい場合もある。
【0096】
例えば、バッチ精製の場合には、所望平衡化バッファー中でHIC材料を調製又は平衡化する。HIC材料のスラリーが得られる。抗体溶液をスラリーと接触させ、分離しようとする抗体をHIC材料に吸着させる。例えばスラリーを沈殿させ、上清を除去することにより、HIC材料と結合しないHCPを含有する溶液をスラリーから分離する。スラリーを1回以上の洗浄工程に供することができる。必要に応じて、スラリーを導電率の低い溶液と接触させ、HIC材料と結合した抗体を脱着させてもよい。結合した抗体を溶出させるためには、塩濃度を下げればよい。
【0097】
イオン交換クロマトグラフィーは、抗体を単離するために抗体の電荷に依存し、疎水性相互作用クロマトグラフィーは抗体の疎水性を利用する。抗体上の疎水基がカラム上の疎水基と相互作用する。疎水度が高いほど、蛋白質はカラムと強く相互作用する。従って、HIC工程は宿主細胞に由来する不純物(例えばDNAと他の高分子量及び低分子量産物関連種)を除去する。
【0098】
疎水性相互作用は、イオン強度が高いときに最強であるため、この型の分離は塩沈殿法又はイオン交換法に従って適切に実施される。HICカラムへの抗体の吸着は高い塩濃度により助長されるが、実際の濃度は抗体の種類と選択される特定HICリガンドに応じて広い範囲を取ることができる。疎水性相互作用を促進する(塩析効果)か又は水の構造を破壊する(カオトロピック効果)かに応じて所謂疎溶媒性系列に各種イオンを配置し、疎水性相互作用を弱化させることができる。カチオンを塩析効果の昇順に並べると、Ba++、Ca++、Mg++、Li+、Cs+、Na+、K+、Rb+、NH4+であり、アニオンをカオトロピック効果の昇順に並べると、PO−−−、SO4−−、CH3CO3−、Cl−、Br−、NO3−、ClO4−、I−、SCN−となる。
【0099】
一般に、硫酸Na、硫酸K又は、硫酸NH4はHICにおいてリガンド−蛋白質相互作用を有効に促進する。この相互作用の強度に影響を与える塩を体系化すると、以下の関係:(NH4)2SO4>Na2SO4>NaCl>NH4Cl>NaBr>NaSCNにより示すことができる。一般に、約0.75〜約2M硫酸アンモニウム又は約1〜4M NaClの塩濃度が有用である。
【0100】
HICカラムは、通例では疎水性リガンド(例えばアルキル又はアリール基)をカップリングさせる塩基マトリックス(例えば架橋アガロース又は合成コポリマー材料)を含む。適切なHICカラムは、フェニル基で置換されたアガロース樹脂を含む(例えばPhenyl Sepharose(登録商標)カラム)。多数のHICカラムが市販されている。例としては、限定されないが、低置換度又は高置換度のPhenyl Sepharose(登録商標)6ファストフローカラム(Pharmacia LKB Biotechnology,AB,Sweden)、Phenyl Sepharose(登録商標)高性能カラム(Pharmacia LKB Biotechnology,AB,Sweden)、Octyl Sepharose(登録商標)高性能カラム(Pharmacia LKB Biotechnology,AB,Sweden)、Fractogel(登録商標)EMDプロピル又はFractogel(登録商標)EMDフェニルカラム(E.Merck,Germany)、Macro−Prep(登録商標)メチル又はMacro−Prep(登録商標)t−ブチル担体(Bio−Rad,California)、WP HI−Propyl(C3)(登録商標)カラム(J.T.Baker,New Jersey)、及びToyopearl(登録商標)エーテル、フェニル又はブチルカラム(TosoHaas,PA)が挙げられる。
【0101】
4.7 典型的精製ストラテジー
ある実施形態では、一次回収の前に先ず遠心及び濾過工程を利用して産生用バイオリアクター回収液から細胞及び細胞破片(HCPを含む)を除去する。例えば、非限定的な例として、抗体と培地と細胞を含む培養液を約7000×g〜約11,000×gで遠心することができる。ある実施形態では、得られたサンプル上清を次に複数のデプスフィルターを含むフィルター列に通す。ある実施形態においては、フィルター列は、12個前後の16インチCuno(登録商標)モデル30/60ZAデプスフィルター(3M Corp.)と、3個の30インチ0.45/0.2μm Sartopore(登録商標)2フィルターカートリッジ(Sartorius)を装着した3個前後のフィルターハウジングから構成される。清澄化した上清を予め滅菌した回収液容器等の容器に採取し、約8℃に維持する。この温度を次に、以下に概説する1以上の捕集クロマトグラフィー工程の前に約20℃に調整する。当然のことながら、当業者は上記条件を変更することができ、このような変更も本発明の範囲内に含まれる。
【0102】
ある実施形態では、一次回収後に、プロテインA樹脂を使用してアフィニティークロマトグラフィーを行う。プロテインA樹脂は数件の業者から市販されている。適切な樹脂の1例は、GE Healthcare製品であるMabSelect(登録商標)である。MabSelect(登録商標)を充填した適切なカラムの1例は直径約1.0cm×長さ約21.6cm(ベッドボリューム〜17mL)のカラムである。このサイズのカラムはベンチスケール用に使用することができる。これを大規模化に使用される他のカラムと比較することができる。例えば、商業的生産にはベッドボリューム約6.6Lの20cm×21cmカラムを使用することができる。カラムに関係なく、MabSelect(登録商標)等の適切な樹脂を使用してカラムに充填することができる。
【0103】
ある態様では、サンプルをロードする前に、プロテインAカラムを適切な緩衝液で平衡化することができる。適切な緩衝液の1例は、pH約6〜8のトリス/NaClバッファーであり、限定されないが、約7.2が挙げられる。適切な条件の特定例は、25mMトリス,100mM NaCl,pH7.2である。この平衡化後、サンプルをカラムにロードすることができる。カラムにロードした後、例えば平衡化バッファーを使用してカラムを1回以上洗浄することができる。カラムから溶出させる前に、別の緩衝液を利用する洗浄を含む他の洗浄を使用することができる。例えば、1カラム体積以上の20mMクエン酸/クエン酸ナトリウム,0.5M NaCl,pH約6.0を使用してカラムを洗浄することができる。この洗浄後、場合により平衡化バッファーを使用して1回以上洗浄してもよい。その後、適切な溶出バッファーを使用してプロテインAカラムから溶出させることができる。適切な溶出バッファーの1例はpH約3.5の酢酸/NaClバッファーである。適切な条件は例えば0.1M酢酸,pH3.5である。当業者に周知の技術を使用して溶出液をモニターすることができる。例えば、OD280の吸光度を追跡することができる。約0.5AUの初期偏光から出発して溶出ピークの立下りの約0.5AUの読取値までカラム溶出液を採取することができる。次に、後続処理に備えて目的の溶出画分を調製することができる。例えば、採取したサンプルをpH約10のトリス(例えば1.0M)で約5.0のpHまで滴定することができる。場合により、この滴定後のサンプルを濾過し、更に処理してもよい。
【0104】
MabSelect(登録商標)カラムの動的結合容量(DBC)は、一段階流速ロード又は二段階流速ロードストラテジーにより測定することができる。一段階流速ロードは、全ロード時間を通して約300cm/時の速度で評価することができる。二段階流速ロードストラテジーは樹脂1mL当たり蛋白質約35mgまでを約300cm/時の線速度でカラムにロードした後、ロードの最後の部分の滞留時間を長くするように線速度を半減させることにより測定することができる。
【0105】
次に、pH介在ウイルス低減/不活性化工程を利用することによりプロテインA溶出液を更に精製することができる。ある実施形態において、この工程は溶出液のpHを約3〜約5(限定されないが、例えば約3.5)に約1時間調整する。クエン酸(例えば3Mクエン酸)等の公知酸調製物を使用してpH低下を助長することができる。酸性pHに暴露すると、pH感受性ウイルス汚染物質は完全には除去されないとしても、低減し、多少の培地/細胞汚染物質が沈殿する。このウイルス低減/不活性化工程後、水酸化ナトリウム(例えば3M水酸化ナトリウム)等の塩基を使用して約20〜約40分間pHを約4.9又は5.0に調整する。この調整は約20℃で実施することができる。
【0106】
ある実施形態では、pHを調整した培地をアニオン交換カラムで更に精製する。この工程に適したカラムの非限定的な1例は、直径60cm×長さ30cm、ベッドボリューム約85Lのカラムである。GE Healthcare製品であるQ Sepharose(登録商標)ファストフロー等のアニオン交換樹脂をカラムに充填する。約7カラム体積の適切な緩衝液(例えばトリス/塩化ナトリウム)を使用してカラムを平衡化することができる。適切な条件の1例は、25mMトリス,50mM塩化ナトリウム,pH8.0である。当業者は上記条件を変更することができ、このような変更も本発明の範囲内に含まれる。上記に概説したプロテインA精製工程から採取したサンプルをカラムにロードする。別の側面では、カチオン交換中に採取した溶出液からカラムにロードする。カラムにロードした後、カラムを平衡化バッファー(例えばトリス/塩化ナトリウムバッファー)で洗浄する。OD280nmでUV分光光度計を使用して抗体を含有するフロースルーをモニターすることができる。このアニオン交換工程は工程関連不純物(例えばDNA等の核酸や宿主細胞蛋白質)を低減させる。目的抗体は、カラムの固相(例えばQ Sepharose(登録商標))と実質的に相互作用も結合もしないが、多くの不純物は、カラムの固相と相互作用し、結合するので、分離が行われる。アニオン交換は約12℃で実施することができる。
【0107】
ある実施形態では、pHを調整した培地をカチオン交換カラムで更に精製する。ある実施形態においては、カチオン交換カラムで使用される平衡化バッファーは、pH約5.0の緩衝液である。適切な緩衝液の1例は約210mM酢酸ナトリウム,pH5.0である。平衡化後、上記一次回収工程から調製したサンプルをカラムにロードする。GE Healthcare製品であるCM Sepharose(登録商標)ファストフロー等のカチオン交換樹脂をカラムに充填する。次に、平衡化バッファーを使用してカラムを洗浄する。次に、平衡化又は洗浄バッファーに比較してイオン強度の高い緩衝液を使用する溶出工程にカラムを供する。例えば、適切な溶出バッファーとしては、約790mM酢酸ナトリウム,pH5.0が挙げられる。抗体を溶出させ、OD280nmに設定したUV分光光度計を使用してモニターすることができる。特定例において、溶出液採取は、上側3OD280nmから下側8OD280nmまでとすることができる。当然のことながら、当業者は上記条件を変更することができ、このような変更も本発明の範囲内に含まれる。
【0108】
ある実施形態では、pHを調整した培地、カチオン交換溶出液又はアニオン交換溶出液を例えば16インチCuno(登録商標)デリピッドフィルターで濾過する。デリピッドフィルターを使用するこの濾過後、例えば30インチ0.45/0.2μm Sartopore(登録商標)二重層フィルターカートリッジで濾過することができる。イオン交換溶出バッファーを使用してフィルターに残留している残量分をフラッシュし、限外濾過/透析濾過に備えることができる。
【0109】
限外濾過/透析濾過工程を実施するためには、適切な緩衝液(例えば20mMリン酸ナトリウム,pH7.0)で濾過媒体を調製する。イオン強度を増すために塩化ナトリウム(例えば100mM塩化ナトリウム)等の塩を加えることができる。この限外濾過/透析濾過工程は抗IL−13抗体を濃縮し、酢酸ナトリウムを除去し、pHを調整するように機能する。この工程を実施するには市販フィルターが利用可能である。例えば、Milliporeは30kD分子量カットオフ(MWCO)セルロース限外濾過膜カセットを製造している。この濾過法は室温又は室温付近で実施することができる。
【0110】
ある実施形態では、上記捕集濾過工程からのサンプルを第2のイオン交換分離工程に供する。この第2のイオン交換分離はある実施形態においては、第1のイオン交換分離の逆電荷に基づく分離を含む。例えば、一次回収後にアニオン交換工程を利用する場合には、第2のイオン交換クロマトグラフィー工程をカチオン交換工程とすることができる。逆に、一次回収工程後にカチオン交換工程を行う場合には、この工程後にアニオン交換工程を実施する。ある実施形態では、第1のイオン交換溶出液を第2のイオン交換クロマトグラフィー工程に直接供することができ、第1のイオン交換溶出液を適切な緩衝液条件に調整する。適切なアニオン及びカチオン分離材料及び条件は上記の通りである。
【0111】
本発明のある実施形態では、抗体を含有するサンプルを疎水性相互作用分離工程で更に処理する。このような工程に適したカラムの非限定的な1例は、直径80cm×長さ15cm、ベッドボリューム約75Lのカラムであり、HICに使用するのに適した樹脂(限定されないが、例えばAmersham Biosciences,Upsala,Sweden製品であるPhenyl HP Sepharose(登録商標))を充填する。前のアニオン交換クロマトグラフィー工程から得られた目的抗体を含有するフロースルーサンプルを等体積の約1.7M硫酸アンモニウム,50mMリン酸ナトリウム,pH7.0で希釈することができる。その後、0.45/0.2μm Sartopore(登録商標)2二重層フィルター又は同等手段を使用してこれを濾過することができる。ある実施形態においては、疎水性クロマトグラフィー法は2サイクル以上を含む。
【0112】
ある実施形態では、先ず適切な緩衝液を使用してHICカラムを平衡化する。適切な緩衝液の非限定的な1例は0.85M硫酸アンモニウム,50mMリン酸ナトリウム,pH7.0である。当業者は、緩衝剤の濃度を変更及び/又は同等の緩衝液に置換することにより平衡化バッファーを変更することができ、このような変更も本発明の範囲内である。ある実施形態では、次にカラムにアニオン交換フロースルーサンプルをロードし、硫酸アンモニウム/リン酸ナトリウム等の適切な緩衝液系で複数回(例えば3回)洗浄する。適切な緩衝液の1例としては、pH約7.0の1.1M硫酸アンモニウム,50mMリン酸ナトリウムバッファーが挙げられる。場合により、カラムを更に洗浄サイクルに供することができる。例えば、第2の洗浄サイクルでは、適切な緩衝液系を使用してカラムを複数回(例えば1〜7回)洗浄することができる。適切な緩衝液系の非限定的な1例としては、0.85M硫酸アンモニウム,50mMリン酸ナトリウム,pH7.0が挙げられる。ある態様では、ロードしたカラムに対して適切な緩衝液系を使用して3回目の洗浄を行う。pH約7.0の1.1M硫酸アンモニウム,50mMリン酸ナトリウム等の適切な緩衝液系を使用してカラムを複数回(例えば1〜3回)洗浄することができる。この場合も、当業者は緩衝条件を変更することができ、このような変更も本発明の範囲内に含まれる。
【0113】
適当な溶出バッファーを使用してカラムから溶出させる。このような溶出バッファーの適切な1例は、pH約7.0の0.5M硫酸アンモニウム,15mMリン酸ナトリウムである。従来の分光光度計を使用して3OD280nmの上側から3OD280nmのピークの下側まで目的抗体を検出・採取することができる。
【0114】
本発明のある態様では、無傷ウイルスを含むウイルス粒子が残存する場合にはこれを除去するために疎水性クロマトグラフィー工程からの溶出液を濾過する。適切なフィルターの非限定的な1例は、Pall Corporation製品であるUltipor DV50(登録商標)フィルターである。他のウイルスフィルターもこの濾過工程で使用することができ、当業者に周知である。予め湿潤させた約0.1μmのフィルターと2×30インチUltipor DV50(登録商標)フィルター列に約34psigでHIC溶出液を通す。ある実施形態では、濾過工程後に、フィルターハウジングに抗体が保持されている場合にはこれを除去するために例えばHIC溶出バッファーを使用してフィルターを洗浄する。予め滅菌した容器に濾液を約12℃で保存することができる。
【0115】
ある実施形態では、上記からの濾液を再び限外濾過/透析濾過に供する。実施者の最終目的が例えば医薬製剤中で抗体を使用することである場合にはこの工程が重要である。この工程を利用する場合には、抗体の濃縮、先に使用した緩衝塩の除去及び特定の製剤化バッファーとの交換を容易にすることができる。ある実施形態では、複数倍容量(例えば2倍容量)の製剤化バッファーを使用して連続透析濾過を実施する。適切な製剤化バッファーの非限定的な1例は5mMメチオニン,2%マンニトール,0.5%スクロース,pH5.9バッファー(Tween不含)である。このダイアボリューム交換が完了したら、抗体を濃縮する。所定の抗体濃度に達したら、実施者は約0.005%(v/v)の最終Tween濃度に達するために加えるべき10% Tweenの量を計算することができる。
【0116】
本発明のある実施形態は、付加的な精製工程を含む。イオン交換クロマトグラフィー法の前、その間、又はその後に実施することができる付加的な精製法の例としては、エタノール沈殿、等電点電気泳動、逆相HPLC、シリカクロマトグラフィー、ヘパリンSepharose(登録商標)クロマトグラフィー、付加的なアニオン交換クロマトグラフィー及び/又は付加的なカチオン交換クロマトグラフィー、クロマトフォーカシング、SDS−PAGE、硫酸アンモニウム沈殿、ヒドロキシアパタイトクロマトグラフィー、ゲル電気泳動、透析濾過、並びに(例えば捕集試薬としてプロテインG、抗体、特異的基質、リガンド又は抗原を使用する)アフィニティークロマトグラフィーが挙げられる。
【0117】
本発明のある実施形態においては、抗IL−13抗体は図1にまとめた重鎖及び軽鎖可変領域配列を含むIgA1、IgA2、IgD、IgE、IgG1、IgG2、IgG3、IgG4又はIgMアイソタイプ抗体である。ある実施形態においては、抗IL−13抗体は図1にまとめた重鎖及び軽鎖可変領域配列を含むIgG1、IgG2、IgG3又はIgG4アイソタイプ抗体である。
【0118】
5.サンプル純度検定方法
5.1 宿主細胞蛋白質の検定
本発明は、単離/精製抗体組成物中の残留レベルの宿主細胞蛋白質(HCP)濃度の測定方法も提供する。上記のように、最終目的物質(例えば抗IL−13抗体)からHCPを排除することが望ましい。典型的なHCPとしては、抗体産生源に由来する蛋白質が挙げられる。目的抗体からHCPを同定し、十分に除去することができないと、効力低下及び/又は有害な対象反応に繋がる恐れがある。
【0119】
本願で使用する「HCP ELISA」なる用語は、アッセイで使用される第2の抗体が抗体(例えば抗IL−13抗体)を作製するために使用される細胞(例えばCHO細胞)から産生されるHCPに特異的であるELISAを意味する。第2の抗体は当業者に公知の従来方法に従って産生させることができる。例えば、第2の抗体はsham産生及び精製試験で得られるHCPを使用して産生させることができ、即ち目的抗体を産生させるために使用されると同一の細胞株を使用するが、細胞株には抗体DNAをトランスフェクトしない。典型的な態様では、選択細胞発現系、即ち目的抗体を産生させるために使用される細胞発現系で発現されると同様のHPCを使用して第2の抗体を産生させる。
【0120】
一般に、HCP ELISAはHCPを含有する液体サンプルを2層の抗体、即ち第1の抗体と第2の抗体の間にサンドイッチする。サンプルをインキュベートすると、その間にサンプル内のHCPは第1の抗体(限定されないが、例えばアフィニティー精製したヤギ抗CHO(Cygnus))により捕集される。抗体を作製するために使用される細胞から産生されるHCPに特異的な第2の標識抗体又は抗体ブレンド(例えばビオチン化抗CHO HCP)を加えると、サンプル内のHCPと結合する。ある実施形態においては、第1の抗体と第2の抗体はポリクローナル抗体である。ある態様においては、第1の抗体と第2の抗体はHCPに対するポリクローナル抗体のブレンドであり、限定されないが、例えばビオチン化ヤギ抗宿主細胞蛋白質混合物599/626/748である。第2の抗体のラベルに基づく適切な試験を使用してサンプルに含まれるHCPの量を測定する。
【0121】
HCP ELISAは、上記方法を使用して得られる溶出液又はフロースルー等の抗体組成物中のHCP濃度を測定するために使用することができる。本発明は、抗体を含有する組成物も提供し、前記組成物はHCP酵素免疫測定法(「ELISA」)により測定した場合に検出可能な濃度のHCPを含有しない。
【0122】
5.2 アフィニティークロマトグラフィー材料の検定
ある実施形態においては、本発明は、単離/精製抗体組成物中のアフィニティークロマトグラフィー材料の残留濃度の測定方法も提供する。所定状況において、このような材料は精製工程中に抗体組成物中に浸出する。ある実施形態では、単離/精製抗体組成物中のプロテインA濃度を測定するアッセイを利用する。本願で使用する「プロテインA ELISA」なる用語はアッセイで使用される第2の抗体が目的抗体(例えば抗IL−13抗体)を精製するために利用されるプロテインAに特異的であるELISAを意味する。第2の抗体は、当業者に公知の従来方法に従って産生させることができる。例えば、第2の抗体は従来の抗体作製及び産生方法のコンテキストで天然又は組換えプロテインAを使用して産生させることができる。
【0123】
一般に、プロテインA ELISAはプロテインAを含有する(又はプロテインAを含有すると予想される)液体サンプルを2層の抗プロテインA抗体、即ち第1の抗プロテインA抗体と第2の抗プロテインA抗体の間にサンドイッチする。サンプルを第1層の抗プロテインA抗体(限定されないが、例えば、ポリクローナル抗体又はポリクローナル抗体のブレンド)に暴露し、サンプル内のプロテインAが第1の抗体により捕集されるために十分な時間インキュベートする。次にプロテインAに特異的な第2の標識抗体(限定されないが、例えば、ポリクローナル抗体又はポリクローナル抗体のブレンド)を加えると、サンプル内の捕集されたプロテインAと結合する。本発明に関連して有用な抗プロテインA抗体の他の非限定的な例としては、ニワトリ抗プロテインA抗体とビオチン化抗プロテインA抗体が挙げられる。第2の抗体のラベルに基づく適切な試験を使用してサンプルに含まれるプロテインAの量を測定する。別のアフィニティークロマトグラフィー材料の濃度を測定するために同様のアッセイを利用することができる。
【0124】
プロテインA ELISAは、上記方法を使用して得られる溶出液又はフロースルー等の抗体組成物中のプロテインA濃度を測定するために使用することができる。本発明は抗体を含有する組成物も提供し、前記組成物はプロテインA酵素免疫測定法(「ELISA」)により測定した場合に検出可能な濃度のプロテインAを含有しない。
【0125】
6.付加的修飾
本発明の抗体は、修飾することができる。ある実施形態では、所望効果を提供するために抗体又はその抗原結合フラグメントを化学的に修飾する。例えば、本発明の抗体又は抗体フラグメントのペグ化は例えば各々その開示内容全体を本願に援用するFocus on Growth Factors 3:4−10(1992);EP0 154 316;及びEP0 401 384に記載されているような当分野で公知のペグ化反応のいずれかにより実施することができる。ある態様において、ペグ化は反応性ポリエチレングリコール分子(又は同様の反応性水溶性ポリマー)とのアシル化反応又はアルキル化反応により実施される。本発明の抗体及び抗体フラグメントのペグ化に適した水溶性ポリマーはポリエチレングリコール(PEG)である。本願で使用する「ポリエチレングリコール」とは、モノ(C1−C10)アルコキシ−又はアリールオキシ−ポリエチレングリコール等の他の蛋白質を誘導体化するために従来から使用されている型のPEGの任意のものを包含する。
【0126】
本発明のペグ化抗体及び抗体フラグメントの作製方法は、一般に、(a)抗体又は抗体フラグメントが1個以上のPEG基と結合する適切な条件下で抗体又は抗体フラグメントをポリエチレングリコール(例えばPEGの反応性エステル又はアルデヒド誘導体)と反応させる段階と、(b)反応生成物を得る段階を含む。公知パラメーターと所望結果に基づいて最適反応条件又はアシル化反応を選択することは当業者に容易に理解されよう。
【0127】
本願に記載する抗IL−13抗体及び抗体フラグメントの投与により、本発明のIL−13関連障害を治療するためには、一般に、IL−13に特異的なペグ化抗体及び抗体フラグメントを使用することができる。一般に、ペグ化抗体及び抗体フラグメントは非ペグ化抗体及び抗体フラグメントに比較して半減期が長い。ペグ化抗体及び抗体フラグメントは単独で使用してもよいし、一緒に使用してもよいし、他の医薬組成物と併用してもよい。
【0128】
本発明の抗体又は抗体部分は、誘導体化又は別の機能分子(例えば別のペプチド又は蛋白質)と連結することができる。従って、本発明の抗体及び抗体部分は免疫接着分子を含めて本願に記載するヒト抗hIL−13抗体の誘導体化及び他の修飾形態を包含するものとする。例えば、別の抗体(例えば二重特異性抗体又はダイアボディ)、検出可能な物質、細胞傷害性物質、薬剤、及び/又は抗体もしくは抗体部分と別の分子(例えばストレプトアビジンコア領域又はポリヒスチジンタグ)との結合に介在することができる蛋白質もしくはペプチド等の1個以上の他の分子体と本発明の抗体又は抗体部分を(化学的カップリング、遺伝子融合、非共有的結合又は他の方法により)機能的に連結することができる。
【0129】
ある種の誘導体化抗体は、(例えば二重特異性抗体を創製するために同一型又は異なる型の)2個以上の抗体を架橋することにより作製される。適切な架橋剤としては、適当なスペーサーにより分離された2個の非常に反応性の基をもつヘテロ二価性架橋剤(例えばm−マレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル)又はホモ二価性架橋剤(例えばスベリン酸ジスクシンイミジル)が挙げられる。このようなリンカーはPierce Chemical Company,Rockford,ILから市販されている。
【0130】
本発明の抗体又は抗体部分を誘導体化することができる有用な検出可能な物質としては、蛍光化合物が挙げられる。検出可能な蛍光物質の例としてはフルオレセイン、イソチオシアン酸フルオレセイン、ローダミン、5−ジメチルアミン−1−ナフタレンスルホニルクロリド、フィコエリスリン等が挙げられる。アルカリホスファターゼ、西洋ワサビペルオキシダーゼ、グルコースオキシダーゼ等の検出可能な酵素で抗体を誘導体化することもできる。検出可能な酵素で抗体を誘導体化する場合には、検出可能な反応生成物を生成するために酵素により使用される他の試薬を加えることにより抗体を検出する。例えば、検出可能な物質である西洋ワサビペルオキシダーゼが存在する場合には、過酸化水素とジアミノベンジジンを加えると、検出可能な着色反応生成物が得られる。抗体をビオチンで誘導体化し、アビジン又はストレプトアビジン結合の間接測定により検出することもできる。
【0131】
7.医薬組成物
本発明の抗体及び抗体部分は、対象に投与するのに適した医薬組成物に配合することができる。典型的には、医薬組成物は本発明の抗体又は抗体部分と、医薬的に許容可能な担体を含有する。本願で使用する「医薬的に許容可能な担体」とは、生理的に適合可能な全溶媒、分散媒、コーティング、抗細菌剤及び抗真菌剤、等張剤及び吸収遅延剤等を包含する。医薬的に許容可能な担体の例としては水、生理食塩水、リン酸緩衝生理食塩水、デキストロース、グリセロール、エタノール等の1種以上とその組み合わせが挙げられる。多くの場合には、例えば糖、多価アルコール(例えばマンニトール、ソルビトール)、又は塩化ナトリウム等の等張剤を組成物に加えることが望ましい。医薬的に許容可能な担体としては更に抗体又は抗体部分の保存期間又は効力を増す微量の助剤(例えば湿潤剤、乳化剤、防腐剤又は緩衝剤)も挙げられる。
【0132】
本発明の抗体及び抗体部分は、非経口投与に適した医薬組成物に配合することができる。例えば抗体0.1〜250mg/mlを含有する注射溶液として抗体又は抗体部分を製造することができる。注射溶液はフリントもしくはアンバーバイアル、アンプル又はプレフィルドシリンジに収容した液体又は凍結乾燥剤形から構成することができる。緩衝液はL−ヒスチジン約1〜50mM(最適値5〜10mM),pH5.0〜7.0(最適値pH6.0)とすることができる。他の適切な緩衝液としては限定されないが、琥珀酸ナトリウム、クエン酸ナトリウム、リン酸ナトリウム又はリン酸カリウムが挙げられる。溶液の毒性を緩和するためには濃度0〜300mM(液体剤形には150mMが最適)の塩化ナトリウムを使用することができる。凍結乾燥剤形には凍結保護剤、主に0〜10%スクロース(最適値0.5〜1.0%)を添加することができる。他の適切な凍結保護剤としてはトレハロースとラクトースが挙げられる。凍結乾燥剤形にはバルク剤、主に1〜10%マンニトール(最適値2〜4%)を添加することができる。液体及び凍結乾燥剤形の両者で安定剤、主に1〜50mM L−メチオニン(最適値5〜10mM)を使用することができる。他の適切なバルク剤としてはグリシン、アルギニンが挙げられ、0〜0.05%ポリソルベート80(最適値0.005〜0.01%)として界面活性剤も添加することができる。その他の界面活性剤としては限定されないが、ポリソルベート20及びBRIJ界面活性剤が挙げられる。
【0133】
ある態様において、医薬組成物は、約0.01mg/kg〜10mg/kgの用量の抗体を含有する。別の態様において、抗体の用量としては、約1mg/kgの隔週投与や、約0.3mg/kgの毎週投与が挙げられる。当業者は適正な用量と対象への投与レジメンを決定することができる。
【0134】
本発明の組成物は、各種形態を取ることができる。これらの形態としては、例えば液体、半固体及び固体剤形が挙げられ、例えば溶液(例えば注射溶液及び輸液溶液)、分散液又は懸濁液、錠剤、ピル、散剤、リポソーム及び座剤が挙げられる。剤形は例えば所期投与方法及び治療用途により異なる。典型的な組成物は他の抗体をヒトに受動免疫するために使用されるものと同様の組成物等の注射溶液又は輸液溶液の形態である。投与方法の1例は非経口(例えば静脈内、皮下、腹膜内、筋肉内)である。ある態様では、静脈内輸液又は注射により抗体を投与する。別の側面では、筋肉内又は皮下注射により抗体を投与する。
【0135】
治療用組成物は、典型的には製造及び保存条件下で無菌・安定でなければならない。組成物は溶液、マイクロエマルション、分散液、リポソーム、又は高薬剤濃度に適した他の注文構造として製剤化することができる。滅菌注射溶液は必要量の活性化合物(即ち抗体又は抗体部分)を必要に応じて上記成分の1種又はその組み合わせと共に適切な溶媒に加えた後に濾過滅菌することにより製造することができる。一般に、分散液は塩基性分散媒と上記から選択される必要な他の成分を含有する滅菌担体に活性化合物を加えることにより製造される。滅菌注射溶液の調製用滅菌凍結乾燥粉末の場合には、製造方法は滅菌濾過溶液から活性成分と任意の他の所望成分の粉末を生成する真空乾燥と噴霧乾燥である。例えばレシチン等のコーティングの使用、分散液の場合には必要な粒度の維持及び界面活性剤の使用により、溶液の適正な流動性を維持することができる。吸収を遅らせる物質(例えばモノステアリン酸塩やゼラチン)を組成物に加えることにより、注射用組成物の長期吸収を実現することができる。
【0136】
本発明の抗体及び抗体部分は、当分野で公知の各種方法により投与することができるが、投与経路/方法の1例は皮下注射、静脈内注射又は輸液である。当業者に自明の通り、投与経路及び/又は方法は所望結果により異なる。ある実施形態では、制御放出製剤のように、化合物の迅速な放出を防止する担体を活性化合物に配合することができ、例えばインプラント、経皮パッチ及びマイクロカプセル送達システムが挙げられる。エチレン酢酸ビニル、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル及びポリ乳酸等の生分解性生体適合性ポリマーを使用することができる。このような製剤の多数の製造方法が特許登録されており、あるいは一般に当業者に公知である。例えば、その教示内容全体を本願に援用するSustained and Controlled Release Drug Delivery Systems,J.R.Robinson,ed.,Marcel Dekker,Inc.,New York,1978参照。
【0137】
ある態様では、本発明の抗体又は抗体部分を例えば不活性希釈剤又は同化可食性担体と共に経口投与することができる。化合物(及び必要に応じて他の成分)をハード又はソフトシェルゼラチンカプセルに封入してもよいし、錠剤に圧縮してもよいし、対象の食事に直接配合してもよい。経口治療投与には、化合物に賦形剤を添加し、内服錠、口腔錠、トローチ剤、カプセル剤、エリキシル剤、懸濁液剤、シロップ剤、ウェハース剤等の形態で使用することができる。本発明の化合物を非経口投与以外の方法で投与するためには、その不活性化を防止するための材料を化合物にコーティングしたり、化合物と併用投与することが必要な場合がある。
【0138】
補助活性化合物も組成物に配合することができる。ある態様では、IL−13活性が有害である障害の治療に有用な1種以上の他の治療剤と本発明の抗体又は抗体部分を合剤化及び/又は併用投与する。例えば、他のターゲットと結合する1種以上の他の抗体(例えば他のサイトカインと結合する抗体又は細胞表面分子と結合する抗体)と本発明の抗hIL−13抗体又は抗体部分を合剤化及び/又は併用投与することができる。更に、1種以上の本発明の抗体を上記治療剤の2種以上と併用することもできる。このような併用療法は治療剤の投与用量を減らし、各種単独療法に伴う可能性のある毒性や合併症を避けることができるという利点がある。熟練した実施者には自明のことながら、本発明の抗体を併用療法の一部として使用する場合には、抗体を単独で対象に投与する場合よりも抗体の用量を減らすことが望ましいと思われる(例えば併用療法の使用により相乗治療効果を達成することができ、その結果、低用量の抗体を使用して所望の治療効果を達成することが可能になる)。
【0139】
当然のことながら、本発明の抗体又はその抗原結合部分は単独で使用してもよいし、別の物質(例えば治療剤)と併用してもよく、前記別の物質はその所期目的に合わせて当業者により選択される。例えば、別の物質は本発明の抗体により治療される疾患又は病態を治療するために有用であると当分野で認められている治療剤とすることができる。別の物質は治療用組成物に有益な属性を付与する物質(例えば組成物の粘性に作用する物質)でもよい。
【0140】
更に当然のことながら、本発明に含まれる併用剤は、その所期目的に有用な併用剤である。以下に挙げる物質は例示であり、限定的ではない。本発明に含まれる併用剤は本発明の抗体と下記から選択される少なくとも1種の他の物質から構成することができる。形成される組成物がその所期機能を実施できるような併用剤であるならば、併用剤は2種以上の他の物質、例えば2種又は3種の他の物質を含有することもできる。
【0141】
所定の併用剤は、非ステロイド性抗炎症薬(別称NSAIDS)であり、イブプロフェン等の薬剤を含む。他の併用剤はプレドニゾロン等のコルチコステロイドであり、本発明の抗体と併用して患者を治療する際に必要なステロイド用量を徐々に減らすことによりステロイド使用の周知副作用を低減又は解消することさえできる。本発明の抗体又は抗体部分と併用することができる関節リウマチ治療剤の非限定的な例としては、サイトカイン抑制性抗炎症薬(CSAID);他のヒトサイトカイン又は成長因子(例えばTNF、LT、IL−1、IL−2、IL−6、IL−7、IL−8、IL−15、IL−16、IL−18、EMAP−II、GM−CSF、FGF及びPDGF)の抗体又はアンタゴニストが挙げられる。本発明の抗体又はその抗原結合部分はCD2、CD3、CD4、CD8、CD25、CD28、CD30、CD40、CD45、CD69、CD80(B7.1)、CD86(B7.2)、CD90等の細胞表面分子又はそのリガンド(例えばCD154(gp39又はCD40L))に対する抗体と併用することができる。
【0142】
治療剤の組合せによっては、自己免疫とそれに続く炎症カスケードにおける種々の点に介入する場合があり、例としては、キメラ、ヒト化又はヒトTNF抗体、D2E7(その教示内容全体を本願に援用する1996年2月9日出願米国出願第08/599,226号)、cA2(Remicade(登録商標))、CDP571、抗TNF抗体フラグメント(例えばCDP870)、及び可溶性p55又はp75 TNF受容体、その誘導体(p75TNFRIgG(Enbrel(登録商標))又はp55TNFRIgG(レネルセプト)、可溶性IL−13受容体(sIL−13)、並びにTNFα変換酵素(TACE)阻害剤等のTNFアンタゴニストが挙げられ、同様にIL−1阻害剤(例えばVx740又はIL−1RA等のインターロイキン−1変換酵素阻害剤)も同じ理由から有効であると思われる。他の併用剤としては、インターロイキン11、抗P7及びp−セレクチン糖蛋白質リガンド(PSGL)が挙げられる。更に他の併用剤としては、IL−13機能と平行、依存又は協調して作用し得る自己免疫反応の他の主要素が挙げられる。更に別の併用剤としては、非枯渇性抗CD4阻害剤が挙げられる。更に他の併用剤としては、抗体、可溶性受容体又はアンタゴニストリガンドを含む共刺激経路CD80(B7.1)又はCD86(B7.2)のアンタゴニストが挙げられる。
【0143】
本発明の抗体又はその抗原結合部分は、メトトレキサート、6−MP、アザチオプリンスルファサラジン、メサラジン、オルサラジンクロロキン/ヒドロキシクロロキン、ペニシラミン、金チオリンゴ酸塩(筋肉内及び経口)、アザチオプリン、コルヒチン、コルチコステロイド(経口、吸入及び局所注射)、β2アドレナリン受容体アゴニスト(サルブタモール、テルブタリン、サルメテロール)、キサンチン(テオフィリン、アミノフィリン)、クロモグリケート、ネドクロミル、ケトチフェン、イプラトロピウム及びオキシトロピウム、シクロスポリン、FK506、ラパマイシン、ミコフェノール酸モフェチル、レフルノミド、NSAID(例えばイブプロフェン)、コルチコステロイド(例えばプレドニゾロン)、ホスホジエステラーゼ阻害剤、アデノシンアゴニスト、抗血栓剤、補体阻害剤、アドレナリン作動薬、炎症誘発性サイトカイン(例えばTNFα又はIL−1)によるシグナル伝達を妨害する物質(例えばIRAK、NIK、IKK、p38又はMAPキナーゼ阻害剤)、IL−1β変換酵素阻害剤(例えばVx740)、抗P7、p−セレクチン糖蛋白質リガンド(PSGL)、TNFα変換酵素(TACE)阻害剤、T細胞シグナル伝達阻害剤(例えばキナーゼ阻害剤)、メタロプロテイナーゼ阻害剤、スルファサラジン、アザチオプリン、6−メルカプトプリン、アンギオテンシン変換酵素阻害剤、可溶性サイトカイン受容体とその誘導体(例えば可溶性p55又はp75 TNF受容体と誘導体p75TNFRIgG(ENBREL(登録商標))及びp55TNFRIgG(レネルセプト)、sIL−1RI、sIL−1RII、sIL−6R、可溶性IL−13受容体(sIL−13))、並びに抗炎症性サイトカイン(例えばIL−4、IL−10、IL−11、IL−13及びTGF)等の物質と併用することもできる。所定の併用剤はメトトレキサート又はレフルノミドを含み、中等度又は重度関節リウマチの場合にはシクロスポリンを含む。
【0144】
本発明の医薬組成物は「治療有効量」又は「予防有効量」の本発明の抗体又は抗体部分を含有することができる。「治療有効量」とは必要な用量と期間で所望治療結果を達成するために有効な量を意味する。抗体又は抗体部分の治療有効量は個体の疾患状態、年齢、性別及び体重や、抗体又は抗体合部分が個体に所望応答を誘発する能力等の因子により異なる。治療有効量は抗体又は抗体部分の有益な治療作用が毒性又は有害作用を上回る量でもある。「予防有効量」とは必要な用量と期間で所望予防結果を達成するために有効な量を意味する。典型的には、予防用量は初期疾患ステージ又はそれ以前の対象で使用するので、予防有効量は治療有効量よりも少ない。
【0145】
最適所望応答(例えば治療又は予防応答)が得られるように投与レジメンを調節することができる。例えば、単回ボーラスを投与してもよいし、時間をかけて用量を数回に分けて投与してもよいし、治療状況の緊急性に応じて用量を比例的に増減してもよい。ある実施形態では、投与し易く、用量を均一にするために非経口組成物を用量単位形態に製剤化すると特に有利である。本願で使用する用量単位形態とは治療する哺乳動物対象に単位用量として適した物理的に別個の単位を意味し、各単位は所望治療効果を生じるように計算された所定量の活性化合物を必要な医薬担体と共に含有する。本発明の用量単位形態の仕様は(a)活性化合物の固有特性及び達成すべき特定治療又は予防効果と、(b)個体の過敏症の治療用としてこのような活性化合物を配合する技術に内在する問題により決定され、これらの因子に直接依存する。
【0146】
本発明の抗体又は抗体部分の治療有効量又は予防有効量の非限定的な範囲の例は0.01〜20mg/kg、又は1〜10mg/kg、又は0.3〜1mg/kgである。なお、用量値は緩和しようとする病態の種類と重篤度により異なる。更に、当然のことながら、任意特定対象の特定投与レジメンは個体の必要と組成物の投与者又は投与監理者の専門的判断に従って時間と共に調節すべきであり、本願に記載する用量範囲は例示に過ぎず、特許請求の範囲に記載する組成物の範囲又は実施を制限するものではない。
【0147】
8.本発明の抗体の使用
8.1 抗IL−13抗体の使用概論
本発明の抗IL−13抗体又はその抗原結合部分は、IL−13と結合することができるため、酵素免疫測定法(ELISA)、放射免疫測定法(RIA)又は組織免疫組織化学等の従来のイムノアッセイを使用して(例えばサンプルマトリックス中、ある態様では血清や血漿等の生体サンプル中で)IL−13、ある態様ではhIL−13を検出するために使用することができる。本発明は生体サンプル中のIL−13の検出方法として、サンプルを本発明の抗体又は抗体部分と接触させる段階と、IL−13と結合した抗体又は未結合抗体を検出することにより、サンプル中のIL−13を検出する段階を含む方法を提供する。結合した抗体又は未結合抗体の検出を容易にするために検出可能な物質で抗体を直接又は間接的に標識する。適切な検出可能な物質としては各種酵素、補欠分子族、蛍光材料、発光材料及び放射性材料が挙げられる。適切な酵素の例としては西洋ワサビペルオキシダーゼ、アルカリホスファターゼ、β−ガラクトシダーゼ又はアセチルコリンエステラーゼが挙げられ、適切な補欠分子族複合体の例としてはストレプトアビジン/ビオチンとアビジン/ビオチンが挙げられ、適切な蛍光材料の例としてはウンベリフェロン、フルオレセイン、イソチオシアン酸フルオレセイン、ローダミン、ジクロロトリアジニルアミンフルオレセイン、ダンシルクロリド又はフィコエリスリンが挙げられ、発光材料の1例としてはルミノールが挙げられ、適切な放射性材料の例としては、125I、131I、35S又は3Hが挙げられる。診断関連、例えばIL−13の上昇を伴う病態の診断にはサンプル中のIL−13の検出が有用であると思われ、及び/又は抗IL−13抗体による治療が有効であり得る対象を同定するのに有用であると思われる。
【0148】
標識抗IL−13抗体を利用する検出アッセイの代法として、例えば検出可能な物質で標識したrhIL−13標準と未標識抗IL−13抗体(例えば抗hIL−13抗体)を利用する競合イムノアッセイによりサンプル中のIL−13を検出することができる。このアッセイでは、サンプルと、標識rhIL−13標準と、抗hIL−13抗体を混合し、未標識抗体に結合した標識rhIL−13標準の量を測定する。サンプル中のhIL−13の量は抗hIL−13抗体と結合した標識rhIL−13標準の量に反比例する。
【0149】
本発明の抗体及び抗体部分は、IL−13活性、ある態様ではhIL−13活性をインビトロ及びインビボで中和することができる。従って、本発明の抗体及び抗体部分は例えばIL−13を含有する細胞培養液や、本発明の抗体と交差反応するIL−13をもつヒト対象又は他の哺乳動物対象(例えばヒヒ、カニクイザル及びアカゲザル等の霊長類)においてIL−13活性を阻害するために使用することができる。ある態様において、本発明はヒトIL−13と、ヒヒIL−13、マーモセットIL−13、チンパンジーIL−13、カニクイザルIL−13及びアカゲザルIL−13から構成される群から選択される少なくとも1種の他の霊長類IL−13の活性を中和し且つマウスIL−13の活性を阻害しない単離ヒト抗体又はその抗原結合部分を提供する。ある態様において、IL−13はヒトIL−13である。例えば、hIL−13を含有する細胞培養液又は含有する疑いのある細胞培養液において、本発明の抗体又は抗体部分を培養培地に加え、培養液中のhIL−13活性を阻害することができる。
【0150】
別の態様において、本発明はIL−13活性が有害である障害に罹患した対象におけるIL−13活性の阻害方法を提供する。本願で使用する「IL−13活性が有害である障害」なる用語はこの障害に罹患した対象におけるIL−13の存在が障害の病態生理の原因であるか又は障害の増悪に寄与する因子であることが分かっているか又はその疑いのある疾患及び他の障害を包含するものとする。従って、IL−13活性が有害である障害は、IL−13活性の阻害が障害の症状及び/又は進行を緩和すると予想される障害である。このような障害は例えば障害に罹患した対象の体液中のIL−13濃度の上昇(例えば対象の血清、血漿、滑液等におけるIL−13濃度の上昇)により判断することができ、例えば上記のような抗IL−13抗体を使用して検出することができる。ある態様において、抗体又はその抗原結合部分は本願に記載する疾患又は障害を治療するための療法で使用することができる。別の態様において、抗体又はその抗原結合部分は本願に記載する疾患又は障害の治療用医薬を製造するために使用することができる。IL−13活性が有害である障害としては多数の例が挙げられる。例えば、IL−13は免疫及び炎症要素を伴う各種疾患(限定されないが、例えば喘息や慢性閉塞性肺疾患等の呼吸器障害)に関連する病理に重要な役割を果たす。その他のIL−13関連障害としては、限定されないが、アトピー性障害(例えばアトピー性皮膚炎及びアレルギー性鼻炎);皮膚、胃腸臓器(例えば潰瘍性大腸炎及び/又はクローン病等の炎症性腸疾患(IBD))、及び肝臓(例えば肝硬変、線維症)の炎症性及び/又は自己免疫性病態;強皮症;腫瘍又は癌(例えばホジキンリンパ腫)が挙げられる。
【0151】
従って、抗IL−13抗体もしくはその抗原結合部分又はこれらをインビボ発現するベクターはIL−13の異常発現の結果としてIL−13の過剰を生じる疾患(例えば喘息又は他の炎症性及び/又は自己免疫性病態)の治療又は体外から投与したIL−13に起因する合併症の場合に処方される。
【0152】
8.2 呼吸器障害における抗IL−13抗体の使用
本発明のある実施形態において、抗IL−13抗体又はその抗原結合部分は1種以上のIL−13関連障害の治療に利用され、このような障害としては、限定されないが、呼吸器障害(例えば喘息(例えばアレルギー性及び非アレルギー性喘息(例えば、例えば幼児における例えば呼吸器合胞体ウイルス(RSV)感染に起因する喘息))、慢性閉塞性肺疾患(COPD)、並びに気道炎症、好酸球増加、線維化及び粘液過剰産生を伴う他の病態(例えば嚢胞性線維症及び肺線維症))が挙げられる。
【0153】
ある実施形態において、本願は呼吸器障害、例えば喘息(例えばアレルギー性及び非アレルギー性喘息);アレルギー;慢性閉塞性肺疾患(COPD);気道炎症、好酸球増加、線維化及び粘液過剰産生を伴う病態(例えば嚢胞性線維症及び肺線維症)に関連する1種以上の症状の治療(例えば抑制、改善)又は予防方法を提供する。例えば、喘息の症状としては、限定されないが、喘鳴、息切れ、気道狭窄、気道過敏、肺活量低下、線維化、気道炎症及び粘液産生が挙げられる。前記方法は1種以上の症状を治療(例えば抑制、改善)又は予防するために十分な量のIL−13抗体又はそのフラグメントを対象に投与する段階を含む。IL−13抗体は治療用又は予防用又はその両方に投与することができる。IL−13アンタゴニスト(例えば抗IL−13抗体)又はそのフラグメントは対象に単独で投与してもよいし、本願に記載する他の治療手段と併用投与してもよい。ある実施形態において、対象は哺乳動物(例えば本願に記載するようなIL−13関連障害に罹患したヒト)である。
【0154】
上記のように、IL−13は喘息に関連する病理応答の誘導に中枢的役割を果たすと考えられている。しかし、別の免疫経路の他のメディエーターも喘息病因に関与しているので、IL−13に加えてこれらのメディエーターを遮断すると、他の治療効果が得られると思われる。従って、本発明の結合蛋白質は、限定されないが、例えばIL−13と炎症誘発性サイトカイン(例えば腫瘍壊死因子α(TNFα))等の標的対と結合することが可能な二重特異性抗体に配合することができる。TNFαは喘息において炎症応答を増幅し、疾患の重篤度に関係があるらしい(McDonnell et al.,Progress in Respiratory Research(2001),31(New Drugs for Asthma,Allergy and COPD),247−250.)。従って、IL−13とTNFαを同時に遮断すると、特に重度気道疾患において有益な効果があると思われる。非限定的な1態様において、本発明の二重特異性抗体はIL−13とTNFαの標的に結合し、喘息を治療するために使用される。
【0155】
別の態様において、 本発明の結合蛋白質はIL−13とIL−1β、IL−13とIL−9、IL−13とIL−4、IL−13とIL−5、IL−13とDL−25、IL−13とTARC、EL−13とMDC、IL−13とMIF、IL−13とTGF−β、EL−13とLHRアゴニスト、DL−13とCL25、IL−13とSPRR2a、EL−13とSPRR2b、及びDL−13とADAM8に結合する二重特異性抗体分子を作製するために使用することができる。本発明は更に、CSF1(MCSF)、CSF2(GM−CSF)、CSF3(GCSF)、FGF2、IFNA1、IFNB1、IFNG、ヒスタミン及びヒスタミン受容体、EL1A、DL1B、BL2、IL3、EL4、IL5、IL6、IL7、IL8、IL9、IL10、EL11、IL12A、IL12B、IL14、IL15、IL16、IL17、IL18、EL19、IL−20、IL−21、IL−22、EL−23、EL−24、EL−25、IL−26、IL−27、EL−28、IL−30、EL−31、EL−32、IL−33、KtTLG、PDGFB、IL2RA、EL4R、IL5RA、IL8RA、DL8RB、IL12RB1、IL12RB2、EL13RA1、IL13RA2、IL18R1、TSLP、CCL1、CCL2、CCL3、CCL4、CCL5、CCL7、CCL8、CCL13、CCL17、CCL18、CCL19、CCL20、CCL22、CCL24.CX3CL1、CXCL1、CXCL2、CXCL3、XCL1、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、CX3CR1、GPR2、XCR1、FOS、GATA3、JAK1、JAK3、STAT6、TBX21、TGFB1、TNFSF6、YY1、CYSLTR1、FCER1A、FCER2、LTB4R、TB4R2、LTBR並びにキチナーゼから構成される群から選択される喘息に関与する1種以上の標的とIL−13に結合することが可能な二重特異性抗体も提供する。
【実施例】
【0156】
1.抗IL−13抗体の産生
原薬の産生バッチは産生用リアクターの1サイクルに由来する原薬のシード継代培養、産生、一次回収・捕集、及び精密精製から得られたABT−308モノクローナル抗体の溶液である。
【0157】
1.1 培地調製
USP/EP/JP標準に準拠する精製水を使用し、GMP Solution Recordsに従って溶液を調製する。調合培地溶液を0.1μmフィルターで濾過し、予め滅菌した適当な寸法の容器、バッグ又はバイオリアクターに捕集する。使用後に0.1μnmフィルターの完全性を試験する。増殖培地と産生培地の組成を表2に示す。
【0158】
【表2】
【0159】
1.2 接種材料増殖
スピナーフラスコとバイオウェーブバッグの操作を利用してCHO細胞をMCBの凍結バイアル1本から110Lシードバイオリアクターの接種に望ましいバイオマスまで増殖させる。マスターセルバンクの凍結バイアルを解凍し、増殖培地(SR−512)に加え、遠心する。細胞を増殖培地に再懸濁し、容積を増加させながら使い捨てスピナーフラスコ又はバイオウェーブバッグで37℃、5% CO2にて増殖させる。20Lウェーブバッグ2個を使用し、シードバイオリアクターに接種する前に20Lウェーブバッグ2個を使用して最終細胞塊増殖を最大にする。両方の20Lウェーブバッグからの細胞密度が約15〜17日目に生存細胞≧2.0×106個/mLに達したら、更に増殖させるために、増殖培地SR−520を充填した110シードバイオリアクターに培養液を移す。接種後、目標温度を37℃とし、pHを7.1の目標値に設定し、NaOH添加とCO2スパージングにより制御する。空気と酸素のスパージングによりバイオリアクター内の溶存酸素(DO)を40%の目標値に制御する。約2〜4日後に細胞密度が生存細胞≧2.6×106個/mLに達したら、培養液を3000L産生用バイオリアクターに移す。
【0160】
1.3 ショートフィルバイオリアクター
3000L産生用バイオリアクターの部分フィルを使用して細胞培養液を更に増殖させる。先ず、リアクターに増殖培地(SR−520)を仕込み、110Lシードバイオリアクターからのバッチを接種する。
【0161】
このショートフィル段階中は、温度、溶存酸素及びpHを夫々37℃、40%及び7.1に制御する。培養pHはCO2スパージングとNaOH添加により制御する。典型的には、細胞は必要な密度である生存細胞≧1.6×106個/mLに達する前に2〜4日間増殖する。
【0162】
1.4 産生用バイオリアクター
産生培地SR−521(1950L)を3000Lバイオリアクター内の細胞培養液に加え、産生段階を開始する。消泡剤Cを加えて発泡を抑える。CO2スパージングとNaOH添加のオンオフにより培養pHを6.9の目標値に制御する。温度と溶存酸素を夫々35℃と40%の目標値に制御する。バイオリアクター内のDOは先ず空気スパージングにより所望値に制御した後、必要に応じて純水素を補充する。生存細胞密度が≧3.0×106個/mLに達したら、温度を33℃の目標値まで下げ、pHとDOを夫々6.9と40%の目標値に維持する。必要に応じてグルコース(SR−334)を加える。細胞生存率が≦50%まで低下したら、培養液を回収する。
【0163】
1.5 工程性能
工程性能及びインプロセス試験結果を表3及び表4に示す。
【0164】
【表3】
【0165】
【表4】
【0166】
2.抗IL−13抗体の単離・精製
一次回収・捕集操作は、濾過による回収液の清澄化、プロテインAアフィニティークロマトグラフィーによる抗体の捕集、及び低pHウイルス不活性化と、その後の深層濾過を含む。精密精製操作はアニオン交換クロマトグラフィー、疎水性相互作用クロマトグラフィー、ウイルス濾過、限外濾過/透析濾過、並びに最終濾過、ボトリング及び凍結を含む。
【0167】
2.1 溶液の調製
USP精製水(USP−PW)又は注射用水(WFI)を使用し、GMP Solution Recordsに従って溶液を調製する。大半の溶液を0.2μmフィルターで濾過し、放射線照射済みバッグ、オートクレーブ滅菌済み又は現場蒸気処理済み容器に捕集する。
【0168】
2.2 一次回収及び清澄化
濾過による一次回収の目的は、産生用バイオリアクター回収液から細胞及び細胞破片を除去することである。未処理回収液をデプスフィルター、デリピッドデプスフィルター及びメンブレンフィルターから構成されるフィルター列に通す。清澄化上清を回収液槽に捕集し、2〜8℃に維持する。清澄化した回収液のインプロセス制御はPoros AクロマトグラフィーによるABT−308濃縮、バイオバーデン及び内毒素試験を含む。
【0169】
2.3 プロテインAアフィニティークロマトグラフィー
プロテインAアフィニティークロマトグラフィーの目的は、清澄化した回収液からABT−308を捕集し、工程関連不純物を低減することである。回収液全体を処理するために典型的には3クロマトグラフィーサイクルを実施する。後続処理のために3サイクルからの生成物プールを集める。
【0170】
直径45cm×長さ22cmのカラム(35L)にMabSelect(登録商標)プロテインA樹脂(GE Healthcare)又はProSep Ultra Plus(登録商標)(Millipore)を充填し、使用できるようにする。USP精製水(USP−PW)後に0.2M酢酸を使用し、最後にUSP PWでリンスすることにより保存バッファーをカラムから除去する。カラムを25mMトリス,100mM NaCl,pH7.2で平衡化後、MabSelect(登録商標)プロテインA樹脂(GE Healthcare)の場合には蛋白質32g/L樹脂、ProSep Ultra Plus(登録商標)(Millipore)樹脂の場合には蛋白質45g/L樹脂の最大値まで清澄化した回収液をロードする。カラムを25mMトリス,100mM NaCl,pH7.2、次いで20mMクエン酸ナトリウム,0.5M NaCl,pH6.0で洗浄後、最後に25mMトリス,100mM NaCl,pH7.2で再び洗浄する。0.1M酢酸,pH3.5でカラムから抗体を溶出させる。各サイクル後、必要に応じて溶出液プールのpHを4.1の目標値に調整する。インプロセス制御はA280による蛋白質濃度の測定、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0171】
第1サイクル後、カラムを0.2M酢酸で再生し、USP−PWでリンスする。第2サイクル後、カラムを0.2M酢酸で再生し、USP−PWでリンスした後、0.1M酢酸、20%エタノールで消毒後、50mM酢酸ナトリウム、20%エタノール,pH5で洗浄・短期間保存する。第3サイクル後、カラムを0M酢酸で再生し、USP−PWでリンスする。その後、0.4M酢酸,0.5M NaCl,0.1% Tween 80で洗浄後、USP−PWで洗浄し、次いで50mM NaOH 1.0M NaClで洗浄後、USP−PWで洗浄する。最後に、0.1M酢酸,20%エタノールで消毒後、50mM酢酸,20%エタノール,pH5.0で洗浄・保存する。
【0172】
2.4 低pHインキュベーション及び濾過
低pHインキュベーションは、プロテインA溶出液中に存在する可能性のある外来エンベロープウイルスの不活性化によりウイルス安全性を更に確実にする専用ウイルス低減工程である。低pHインキュベーション後の濾過の目的は低pH処理中に形成される可能性のある沈殿を除去することである。
【0173】
集めたプロテインAクロマトグラフィー溶出液のpHを0.5Mリン酸で3.5の目標値に調整し、18〜25℃に60〜70分間維持する。次に、混合物を1Mトリス,pH10でpH5に調整し、デプスフィルターとメンブレンフィルターの併用により清澄化した後、10〜14℃まで冷却する。低pH処理・濾過工程のインプロセス制御はA280による蛋白質濃度の測定、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0174】
2.5 一次回収・捕集工程性能
一次回収・捕集操作の工程性能を表5に示し、インプロセス制御の結果を表6に示す。
【0175】
【表5】
【0176】
【表6】
【0177】
2.6 強アニオン交換クロマトグラフィー
強アニオン交換クロマトグラフィー工程の目的は、宿主細胞蛋白質、DNA及び内毒素等の工程関連不純物を低減することである。この工程は、ウイルス除去工程も兼用できる。ある実施形態では、抗体がカラムを通過し、不純物が樹脂に保持されるフロースルーモードでQ Sepharose(登録商標)FF樹脂カラムを操作し、代替態様では、Q Sepharose FF樹脂カラムの代わりにMustang Q(登録商標)メンブレン(Pall Corp.)を利用する。操作は10〜14℃で実施する。
【0178】
直径45cm×長さ22cmのカラム(35L)にQ Sepharose(登録商標)FF樹脂(GE Healthcare)を充填し、使用できるようにする。カラムを25mMトリス,50mM NaCl,pH8.0で平衡化する。中和・濾過した不活性化溶液のpHを1Mトリス,pH10で8.0に調整し、導電率を5.0〜6.5に調整し、溶液をデリピッド及びメンブレンフィルターで濾過する。Q Sepharose(登録商標)ロードを蛋白質80g/L樹脂の最大ロードでカラムにポンプ導入する。ロード後、カラムを25mMトリス,50mM NaCl,pH8.0で洗浄し、フロースルーと洗浄液を合わせる。これをQ Sepharose(登録商標)フロースルー・洗浄液(QFTW)プールとする。Q Sepharose(登録商標)工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0179】
カラムを25mMリン酸ナトリウム,1.0M NaCl,pH7.0で再生した後、WFIでリンスし、1.0M NaOHで消毒後、WFIでリンスする。次にカラムを25mMリン酸ナトリウム,1.0M NaCl,pH7.0で中和し、25mMリン酸ナトリウム,20%イソプロパノール,pH7.0中で保存する。
【0180】
2.7 疎水性相互作用クロマトグラフィー
Phenyl Sepharose(登録商標)工程の目的は、ABT−308凝集物、フラグメント及び工程関連不純物を除去することである。操作は10〜14℃で実施する。
【0181】
直径60cm×長さ15cmのカラム(42L)にPhenyl Sepharose(登録商標)HP樹脂(GE Healthcare)を充填し、使用できるようにする。カラムをWFI、次いで20mMリン酸ナトリウム,1.1M硫酸アンモニウム,pH7.0で平衡化する。
【0182】
Q Sepharose(登録商標)フロースルー及び洗浄液を40mMリン酸ナトリウム,2.2M硫酸アンモニウム,pH7.0で1:1(v/v)に希釈する。この溶液即ちPhenyl Sepharose(登録商標)ロードを0.2μmフィルターで濾過し、蛋白質64g/L樹脂の最大ロードでカラムにロードする。カラムを25mMリン酸ナトリウム,1.4M硫酸アンモニウム,pH7.0で洗浄し、ABT−308を11mMリン酸ナトリウム,0.625M硫酸アンモニウム,pH7.0でカラムから溶出させる。Phenyl Sepharose(登録商標)工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0183】
カラムをWFIで再生した後、1M NaOHで消毒し、WFIでリンスし、25mMリン酸ナトリウム,20%イソプロパノール,pH7中で保存する。
【0184】
2.8 ナノ濾過
ナノ濾過は、Phenyl Sepharose(登録商標)HPカラム溶出液中に存在する可能性のある直径≧20nmの外来ウイルスの物理的除去によりウイルス安全性を更に確実にする専用ウイルス除去工程である。操作は10〜14℃で実施する。
【0185】
15mMヒスチジン,pH5.6で予め湿潤させた0.1μmフィルターとUltipor DV20 フィルター列にPhenyl Sepharose(登録商標)HPカラム溶出液を通す。濾過後、フィルター列を15mMヒスチジン,pH5.6でフラッシュし、保持されたABT−308を回収する。使用後、DV20フィルターで完全性試験を実施し、フィルターを捨てる。フィルターが完全性試験に合格しない場合には、溶液を上記のように再濾過すればよい。ナノ濾過工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0186】
2.9 限外濾過/透析濾過によるABT−308原薬の製剤化
UF/DF工程の目的は、原薬を最終製剤化バッファーである15mMヒスチジン,pH5.6で透析濾過し、ABT−308を濃縮することである。これらの操作は10〜14℃で実施する。
【0187】
30kDa MWCOポリエーテルスルホンメンブレンを使用してナノ濾液を約50g/Lまで濃縮し、製剤化バッファーで透析濾過した後、約180g/Lまで濃縮する。UFシステムから生成物を取出し、透析濾過バッファーでリンスし、システム内に生成物が残っている場合には回収する。濃縮液と洗浄液を合わせ、約120〜160g/Lの濃度の透析濾過ABT−308を生成する。濃縮したABT−308をメンブレンフィルターで濾過する。限外濾過/透析濾過工程のインプロセス制御はA280による濃度、SE−HPLC、バイオバーデン及び内毒素試験を含む。
【0188】
各回の試験後、限外濾過システムをWFIでフラッシュし、250ppm次亜塩素酸ナトリウム溶液で洗浄後、0.1M水酸化ナトリウムで消毒・保存する。
【0189】
2.10 最終濾過、ボトリング及び凍結
濾過・ボトリング操作は、クラス100層流フード下にクラス100領域で2〜8℃にて実施する。製剤化したABT−308を0.2μmフィルターで濾過し、予め滅菌したパイロジェンフリーPETGボトルに捕集する。ラベルを付けたボトルを凍結するまで空の−80℃(公称値)冷凍庫に入れた後、−80℃(公称値)に維持した保存用冷凍庫に移す。最終濾過・ボトリング工程のインプロセス制御はA280、バイオバーデン及び内毒素試験(原薬試験結果)を含む。
【0190】
2.11 精密精製工程性能
一次回収・捕集操作の工程性能を表7に示し、インプロセス制御結果を表8に示す。
【0191】
【表7】
【0192】
【表8】
【0193】
3.抗体組成物中の宿主細胞蛋白質濃度の測定
この工程は、抗体サンプル中の残留宿主細胞蛋白質濃度の測定試験方法に関する。酵素免疫測定法(ELISA)を使用して宿主細胞蛋白質(抗原)を2層の特異的抗体の間にサンドイッチする。その後、非特異的部位をカゼインでブロックする。次に宿主細胞蛋白質をインキュベートすると、この間に抗原分子は第1の抗体(コーティング抗体)により捕集される。次に抗原(宿主細胞蛋白質)と結合する第2の抗体(ビオチン化抗宿主細胞蛋白質)を加える。ビオチン化抗宿主細胞蛋白質と結合するニュートラアビジンHRP−コンジュゲートを加える。次にK−Blue基質を加える。発色基質は結合した酵素共役抗体により加水分解され、青色を発色する。2M H3PO4で反応を停止すると、黄色に変色する。色強度はウェル内で結合した抗原の量に正比例する。
【0194】
50mM重炭酸ナトリウム(コーティングバッファー),pH9.4の調製。1LビーカーにミリQ水900mL、重炭酸ナトリウム4.20g±0.01gを加える。完全に溶解するまで撹拌する。pHを1N NaOHで9.4に調整する。1Lメスフラスコに移し、ミリQ水を補充する。均質になるまで反転により混合する。0.22μm滅菌フィルターユニットで濾過する。調製日から7日間まで公称値4℃で保存する。
【0195】
0.104M Na2HPO4・7H2O,1.37M NaCl,0.027M KCl,0.0176M KH2PO4,pH=6.8〜6.9(10×PBS)の調製。ミリQ水約400mLをガラスビーカーに加える。Na2HPO4・7H2O 13.94g±0.01gを加える。NaCl 40.0g±0.1gを加える。KCl 1.00g±0.01を加える。KH2PO4 1.20g±0.01gを加える。均質になるまで撹拌する。500mLメスフラスコに移す。ミリQ水を補充して500mLにする。反転により混合する。0.2μm滅菌フィルターユニットで濾過する。室温で7日間まで保存する。
【0196】
1×PBS+0.1%トリトンX−100,pH7.40(プレート洗浄バッファー)の調製。4Lメスシリンダー内で10×PBS(ステップ5.2)400mLをミリQ水3500mLと混合する。pHをチェックし、必要に応じて1N HCl又は1N NaOHで7.40±0.05に調整する。ミリQ水を補充する。シリンダーをパラフィルムで密閉し、均質になるまで反転により混合する。4Lボトルに移す。1×PBS 4mLを取り出し、捨てる。4mLのトリトンX−100を3996mの1×PBSに加える。撹拌プレートにセットし、完全に溶解するまで撹拌する。希釈バッファー調製に必要な量のプレート洗浄バッファーを0.22μm滅菌フィルターユニットで濾過する。室温で7日間まで保存する。
【0197】
コーティング抗体混合物:アフィニティー精製ヤギ抗CHO 599/626/748(ロット#G11201@1.534mg/mL)の調製(注:バイアルに入れて−80℃で保存したストック)。アリコートを調製する。使用時にプレート1枚当たりアリコート1個を取出す。使用直前に、抗体混合物を最終濃度4μg/mLとなるように冷温50mM重炭酸ナトリウムで次のように希釈する。例えば、コーティング抗体混合物31μLを冷温コーティングバッファー11969μLに加える。反転により穏やかに混合する。
【0198】
ビオチン化ヤギ抗宿主細胞蛋白質混合物599/626/748(ロット#G11202@0.822mg/mL)の調製(注:バイアルに入れて−80℃で保存したストック)。アリコートを調製する。使用時にプレート1枚当たりアリコート1個を取出す。使用直前に、ビオチン化抗体混合物を最終濃度1μg/mLとなるように37℃±2℃カゼインで次のように希釈する。例えば、ビオチン化抗体混合物14.6μLを37℃±2℃カゼイン11985μLに加える。反転により穏やかに混合する。
【0199】
ニュートラアビジン−HRPの調製。新規ロット(2mg/バイアル)を次のように1mg/mLまで再構成する。ミリQ水400μLをバイアルに加えた後、1×PBS 1600μLを加え、合計2mLとする。穏やかにボルテックスして混合する。公称値−20℃で保存する。プレート1枚当たりアリコート1個を使用するように所望体積のアリコートを調製する。ポリプロピレンチューブ内で調製する。ワーキング濃度を決定するように新規ロットを適格化する。調製日から6カ月間の期日を指定する。例えば、ワーキング濃度が0.2μg/mLと決定されたならば、以下のように調製する。使用直前にニュートラアビジン−HRPのアリコートを室温で解凍する。1mg/mLニュートラアビジン溶液を37℃±2℃カゼインで0.1mg/mL(100μg/mL)まで希釈する。例えば、10倍に希釈し、ニュートラアビジン50μLをカゼイン450μLに加える。穏やかにボルテックスして混合する。更に100μg/mL溶液を37℃±2℃カゼインで0.2μg/mLまで希釈する。例えば、500倍に希釈し、ニュートラアビジン(100μg/mL)24μLをカゼイン11976μLに加える。穏やかにボルテックスして混合する。
【0200】
5.7 2Mリン酸(停止溶液)の調製。濃リン酸から2Mリン酸溶液を次のように調製する。ラベルに記載されたリン酸%、密度(1.685g/mL)及び製剤重量(98g/モル)から、2Mリン酸500mLを調製するために必要な濃リン酸の体積を計算する。上記のように計算した体積の濃リン酸をフラスコに加える。ミリQ水を補充し、均質になるまで反転により混合する。調製日から6カ月間まで周囲温度で保存する。
【0201】
希釈バッファー(1×PBS+0.1%トリトンX100,pH7.4で100倍に希釈したカゼイン)の調製。(上記からの)0.22μmで滅菌濾過した1×PBS+0.1%トリトンX100,pH7.4で37℃±2℃カゼインを100倍に希釈する。例えば、0.22μmで滅菌濾過した1×PBS+0.1%トリトンX100(pH7.4)99mLに37℃±2℃カゼイン1mLを加える。よく混合する。使用毎に新たに調製する。
【0202】
標準の調製。宿主細胞蛋白質標準(抗原標準)(ロット#G11203@1.218mg/mL)(注:70μLアリコートとして公称値−80℃で保存したストック)。アリコートを室温で解凍する。希釈バッファーを使用してポリプロピレンチューブ内で系列希釈を実施する。
【0203】
サンプルの調製。ポリプロピレンチューブ内で最終バルクサンプルを希釈バッファーで24mg/mLまで希釈する。濃度を記録する。注:下記溶液を使用してスパイクサンプルを調製し、下記12mg/mL溶液を調製する。ポリプロピレンマイクロチューブ内で更に24mg/mL溶液を希釈バッファーで12mg/mLまで希釈する。12mg/mL溶液を各々プレート上の3個のウェルにロードし、合計6個のウェルにロードする。
【0204】
スパイクの調製。ポリプロピレンマイクロチューブ内で希釈バッファーで2倍に希釈することにより上記に調製した20ng/mL標準から10ng/mL宿主細胞蛋白質スパイクを調製する。10ng/mLスパイク溶液を各々プレート上の3個のウェルにロードする。サンプルをスパイクするためにステップ6.1からの20ng/mL標準溶液を使用する。
【0205】
スパイクサンプルの調製。ポリプロピレンマイクロチューブ内で24mg/mL最終バルク溶液各300μLを20ng/mLスパイク溶液(6.1)300μLでスパイクする。スパイクサンプル溶液を各々3個のウェルにロードし、合計6個のウェルにロードする。
【0206】
対照の調製。日常的試験で使用する前に全新規対照ストック溶液に対照範囲を設定する必要がある。対照ストック:ABT−308原薬濃縮液バッチの150μLアリコートを調製し、3年間まで公称値−80℃で凍結保存する。
【0207】
ワーキング対照の調製。対照のアリコートを室温で解凍する。ポリプロピレンチューブ内で対照を希釈バッファーで24mg/mLまで希釈する。ポリプロピレンマイクロチューブ内で更に24mg/mL対照溶液を希釈バッファーで12mg/mLまで希釈する。単一希釈液を調製し、対照をプレートの3個のウェルにロードする。
【0208】
ELISA手順。プレート洗浄ボトルにプレート洗浄バッファーを充填する(ステップ5.3参照,1×PBS+0.1%トリトンX−100)。プレートウォッシャーを起動する。以下のパラメーターをチェックする。パラメーターは以下のように設定すべきである:プレートタイプ:各サイクルで1(合計5サイクル);体積:400μL;浸漬時間:10秒;吸引時間:4秒。
【0209】
アッセイ手順。冷温50mM重炭酸ナトリウム中4μg/mLヤギコーティング抗体混合物100μL/ウェルでプレートをコーティングする。コーティング溶液がウェルの底を均等に覆うまでプレートの側面をタッピングし、シーリングテープを貼り付け、プレートシェーカー(又は同等手段)速度3にて18時間±1時間振盪しながら公称温度4℃でインキュベートする。一晩インキュベーション後、プレートを冷蔵庫から取り出し、室温で平衡化させる。コーティングを振り払う。プレートをペーパータオルで拭う。37℃±2℃カゼイン300μL/ウェルでブロックし、シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。ブロッキングインキュベーション中に標準、サンプル、対照、スパイク及びスパイクサンプルを調製する。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。8チャネルピペットを使用し、標準、サンプル、スパイク、スパイクサンプル及び対照100μL/ウェルをプレートの3個のウェルに分注する。ブランクとしてプレートの全ての空のウェルに希釈バッファー100μL/ウェルをピペットで分注する。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。テンプレートを作成し、プレートにロードする際のガイドとして使用する。
【0210】
プレートリーダー設定。テンプレートを設定し、標準用濃度を入力する。サンプル、対照、スパイク又はスパイクサンプルの希釈係数は入力しない。希釈剤を加えたウェルを全ウェルから差し引くブランクとして割り当てる。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。ビオチン化ヤギ抗体100μL/ウェルを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。ニュートラアビジン−HRPコンジュゲート溶液100μL/ウェルを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。プレートを洗浄バッファーで5回洗浄する。プレートをペーパータオルで拭う。冷温K−Blue基質100μL/ウェルを加え、シーリングテープを貼り付け、Lab−lineタイタープレートシェーカー(又は同等手段)で速度3にて振盪しながら室温で10分間インキュベートする(基質を最初の行に添加後、すぐにタイマーをスタートする)。2Mリン酸100μL/ウェルを加えることにより反応を停止する(ステップ5.7)。プレートをプレートシェーカーにセットし、速度3で3〜5分間振盪する。プレートを450nmで読取る。
【0211】
データ解析及び計算。注:光学密度が標準曲線の実際の定量限界(2.5ng/mL標準)内にあり且つ下記%CV又は%差条件を満たすサンプル、スパイク、スパイクサンプル及び対照のみが許容される。サンプルODが2.5ng/mL標準に達しない場合には、結果を2.5ng/mL未満と報告する。その後、この数値を希釈後のサンプル濃度(12mg/mL)で割り、数値(ng/mg)を報告する。サンプルの宿主細胞濃度が高く、非スパイク及び/又はスパイクサンプルが標準曲線を上回る場合には、数値を>100ng/mLと報告する。その後、この数値を希釈後のサンプル濃度(12mg/mL)で割り、数値(ng/mg)を報告する。サンプルが2.5ng/mL標準を下回る場合には、スパイク回収率計算のサンプル値をゼロとみなす。
【0212】
標準曲線。標準濃度をプロトコルテンプレートに入力する。二次曲線フィットを使用する。測定係数は0.99でなければならず、3個のウェル間の%CVは20%でなければならない。この条件を満たさない場合には、標準を1本(1レベル,ウェル3個)下げることができる。1.25ng/mL下げる場合には、光学密度が2.5ng/mL及び100ng/mL(残りの標準曲線の点)光学密度以内のサンプル及びスパイクサンプルのみが許容可能である。更に、各標準レベルの3個について、1個のウェルが明白に汚染されているか又は結合度が低い場合には、このウェルを下げることができる。1個のウェルを標準レベルから下げる場合には、残りの2個は%差=20%でなければならない。プレートのバックグラウンド(ブランク)に近いOD値を示す最低標準の%CVは30%でなければならない。ウェル1個を下げる場合には、残りの2個の%差は35%でなければならない。最低標準を下げる場合には、光学密度が残りの標準曲線レベル光学密度以内にあるサンプル及びスパイクサンプルのみが許容可能である。
【0213】
サンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを報告する。各サンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。注:非スパイクサンプルODが2.5ng/mL標準ODを下回る場合には、%差条件は非スパイク結果に適用されない。上記計算参照。
【0214】
平均(ng/mL)値から実際の宿主細胞濃度(ng/mg)を次のように計算する:CHO宿主細胞蛋白質(ng/mg)=平均「非スパイクサンプル結果(ng/mL)」/希釈後のサンプル濃度(12mg/mL)。
【0215】
スパイク。%CVは3個のウェル間で20%でなければならない。%CVを記録する。スパイクから1個のウェルを下げることができる。残りの点は%差=20%でなければならない。上記計算参照。宿主細胞濃度(ng/mL)を報告する。この結果をスパイク回収率計算で使用する。各スパイクに得られる濃度(ng/mL)は理論スパイク濃度の±20%でなければならない。結果を記録し、合否を判定する。スパイク結果が理論値の20%以内にない場合には、アッセイを繰返す必要がある。平均スパイク濃度(ng/mL)×100は100%±20% 10ng/mLでなければならない。
【0216】
スパイクサンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを記録する。各スパイクサンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。上記計算参照。各希釈液の「スパイクサンプル結果」(ng/mL)を報告する。2種類の希釈液間の%差を記録する。希釈液間の%差は25%でなければならない。これらの結果をスパイク回収率計算で使用する。
【0217】
下式を使用して各希釈液セットの%スパイク回収率を計算する:%スパイク回収率=スパイクサンプル値−非スパイクサンプル値×100。スパイク値。注:(1)非スパイクサンプル値ODが2.5ng/mL標準を下回る場合には、%スパイク回収率計算において数値をゼロとみなす。各サンプルの各希釈液の%スパイク回収率は100%±50%(50%〜150%)でなければならない。結果と合否を記録する。
【0218】
対照。%CVは3個のウェル間で20%でなければならない。%CV結果を記録する。対照からの1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。上記計算参照。対照の宿主細胞濃度を報告する(ng/mL)。宿主細胞濃度(ng/mg)を次のように計算する:宿主細胞蛋白質(ng/mg)=対照宿主細胞蛋白質結果(ng/mL)。
【0219】
4.抗体組成物中のプロテインA濃度の測定
このELISAでは、プレートをニワトリ抗プロテインAでコーティングし、インキュベートする。非特異的部位をPBS中カゼインでブロックする。プレートを1×PBS+0.1%トリトンX−100で洗浄し、未結合材料を除去する。サンプルとCys−プロテインA標準を1×PBS+4.1%トリトンX+10%カゼインで希釈する。95℃±2℃に加熱することにより溶液を変性させ、プロテインAを抗体から分離する。ある実施形態において、例えば(GE Healthcare)を使用する場合には、溶液をプレートに加え、インキュベートする。代替態様において、例えば、プロテインAアフィニティー工程がProSep Ultra Plus(登録商標)(Milipore)を使用する場合には、溶液を冷却し、0.85% NaCl+12.5% 1N酢酸+0.1% Tween 20を各チューブに加え(1:1)、サンプル蛋白質からプロテインAを更に分離し易くする。チューブを激しくボルテックスし、インキュベートし、遠心する。上清を取り出し、更に処理する。未結合材料を1×PBS+0.1%トリトンX−I00で洗い流す。ビオチン化ニワトリ抗プロテインAをマイクロタイタープレートに加え、インキュベートする。プレートを洗浄して未結合材料を除去し、ニュートラアビジン−ペルオキシダーゼコンジュゲートを加える。
【0220】
ニュートラアビジンはウェルと結合したビオチン化ニワトリ抗プロテインAと結合する。プレートを再び洗浄し、未結合ニュートラアビジンを除去し、K−Blue(テトラメチルベンジジン(TMB))基質をプレートに加える。基質は結合したニュートラアビジンにより加水分解され、青色を発色する。リン酸で反応を停止すると、黄色に変色する。ウェル内の黄色の強度はウェル内に存在するプロテインAの濃度に正比例する。
【0221】
試薬及び溶液の調製。カゼインボトルを37℃±2℃に加温し、2分間超音波処理し、分取する。アリコートを公称値4℃で保存する。アッセイを実施する場合には、必要数のカゼインアリコートを37℃±2℃にする必要がある。コーティングバッファーと基質は冷温で使用する(使用直前に公称値4℃から取出す)。
【0222】
50mM重炭酸ナトリウム(コーティングバッファー),pH9.4。1LビーカーにミリQ水900mL、重炭酸ナトリウム4.20g±0.01gを加える。完全に溶解するまで撹拌する。pHを1N NaOHで9.4に調整する。1Lメスフラスコに移し、ミリQ水を補充する。均質になるまで反転により混合する。0.22CAμm滅菌フィルターユニットで濾過する。調製日から7日間まで公称値4℃で保存する。
【0223】
104M Na2HPO4・7H2O,1.37M NaCl,0.027M KCl,0.0176M KH2PO4,pH=6.8〜6.9(10×PBS)の調製。ミリQ水約400mLをガラスビーカーに加える。Na2HPO4・7H2O 13.94g±0.01gを加える。NaCl 40.0g±0.1gを加える。KCl 1.00g±0.01gを加える。KH2PO4 1.20g±0.01gを加える。均質になるまで撹拌する。500mLメスフラスコに移す。ミリQ水を補充して500mLにする。反転により混合する。0.2CAμm滅菌フィルターユニットで濾過する。調製日から7日間まで室温で保存する。
【0224】
1×PBS+0.1%トリトンX−100,pH7.40(プレート洗浄バッファー)。4Lメスシリンダー内で10×PBS(上記参照)400mLをミリQ水3500mLと混合する。pHをチェックし、必要に応じて1N HCl又は1N NaOHで7.40±0.05に調整する。ミリQ水を補充する。シリンダーをパラフィルムで密閉し、均質になるまで反転により混合する。4Lボトルに移す。1×PBS 4mLを取り出し、捨てる。4mLのトリトンX−100を3996mLの1×PBSに加える。撹拌プレートにセットして撹拌し、完全に溶解させる。室温で7日間まで保存する。
【0225】
ニワトリ抗プロテインAコーティング抗体。使用時にプレート1枚当たり抗体アリコート1個を取出す。新規ロットのニワトリ抗プロテインAを適格化するためには、(同一ロットのコーティングから調製した)ニワトリ抗プロテインA−ビオチンコンジュゲートを同時に使用・適格化する必要があると思われる。使用直前に抗体混合物をコーティング適格化中に決定された濃度まで冷温50mM重炭酸ナトリウムで希釈する。例えば、適格化中にプレートにロードするコーティング濃度が6μg/mLと決定され、ストック濃度が3000μg/mLであるならば、コーティング抗体24μLを冷温コーティングバッファー11976μLに加える。反転により穏やかに混合する。
【0226】
ビオチン化ニワトリ抗プロテインA。使用時にプレート1枚当たり抗体アリコート1個を取出す。新規ロットのニワトリ抗プロテインA−ビオチンコンジュゲートを適格化するためには、コンジュゲートの作製に使用した同一ロットのニワトリ抗プロテインAと同時に使用・適格化する必要があると思われる。使用直前にビオチン化抗体をビオチン化抗体適格化中に決定された濃度まで37℃±2℃カゼインで希釈する。例えば、適格化中にプレートにロードするビオチン化抗体濃度が4μg/mLと決定され、ストック濃度が1000μg/mLであるならば、ビオチン化抗体48μLを37℃±2℃カゼイン11952μLに加える。反転により穏やかに混合する。
【0227】
ニュートラアビジン−HRP。新規ロット(2mg/バイアル)を次のように1mg/mLまで再構成する。ミリQ水400μLをバイアルに加えた後、1×PBS 1600μLを加え、合計2mLとする。穏やかにボルテックスして混合する。公称値−80℃で保存する。プレート1枚当たりアリコート1個を使用するように所望体積のアリコートを調製する。ポリプロピレンチューブ内で調製する。調製日から6カ月間の期日を指定する。例えば、ワーキング濃度が0.1μg/mLと決定されたならば、以下のように調製する。使用直前にニュートラアビジン−HRPのアリコートを室温で解凍する。1mg/mLニュートラアビジン溶液を37℃±2℃カゼインで0.01mg/mL(10μg/mL)まで希釈する。例えば、10倍に希釈し、ニュートラアビジン50μLをカゼイン450μLに加える。穏やかにボルテックスして混合し、再び10倍に希釈し、10倍ニュートラアビジン希釈液100μLをカゼイン900μLに加える。穏やかにボルテックスして混合する。更に10μg/mL溶液を37℃±2℃カゼインで0.1μg/mLまで希釈する。例えば、100倍に希釈し、ニュートラアビジン(10μg/mL)120μLをカゼイン11880μLに加える。数回穏やかに反転して混合する。
【0228】
停止溶液(購入した1Nリン酸を使用する)。入手日から1年間まで周囲温度で保存する。希釈バッファー(1×PBS+4.1%トリトンX100+10%カゼイン,pH7.4)。(ステップ5.3からの)1×PBS+0.1%トリトンX100(pH7.4)86mLをビーカー又はフラスコに加え、4mLのトリトンX−100を加え、ブロッカーカゼインPBS溶液10mLを加え、撹拌し、溶解/混合する。トリトンを溶解させるには20〜30分間かかると思われる。これは1×PBS+4.1%トリトンX100+10%カゼイン,pH7.4溶液に等しい。0.22CAμm滅菌フィルターユニットで濾過する。使用毎に新たに調製する。プレート1枚にはこれで十分である。
【0229】
プロテインA標準(抗原標準)。注:70μLアリコートとして公称値−20℃で保存したストック。アリコートを氷上で解凍する。製造業者のCOAに記載されている濃度で希釈バッファー(上記参照)を使用してポリプロピレンチューブ内で下表の例に従って系列希釈を実施する。例えば、ストック濃度が2.1mg/mL(2100000ng/mL)であるとCOAに記載されている場合には、サンプルを氷上で解凍する。ポリプロピレンマイクロ遠心管内で最終バルクサンプルを希釈バッファー(上記)で20mg/mLまで希釈する。別々に2回希釈を行う。濃度を記録する。下記溶液を使用してスパイクサンプルを調製し、10mg/mL溶液を調製する。例えば、濃度(mg/mL)、体積μLのXmg/mL溶液、体積(μL)の希釈剤で120個のストックサンプルからの系列希釈。ポリプロピレンマイクロ遠心管内で、20mg/mL溶液を更に希釈バッファーで10mg/mLまで希釈する。
【0230】
スパイクの調製。ポリプロピレンマイクロ遠心管内で、希釈バッファーで2倍に希釈することにより、上記ステップ6.1で調製した0.593ng/mL標準から0.296ng/mLプロテインAスパイクを調製する。希釈を1回行う。0.296ng/mLスパイク溶液をプレートの3個のウェルにロードする。サンプルをスパイクするにはステップ6.1からの0.593ng/mL標準溶液を使用する。
【0231】
スパイクサンプルの調製。ポリプロピレンマイクロ遠心管内で、20mg/mL最終バルク溶液各500μLを0.593ng/mLスパイク溶液500μLでスパイクする。変性用に保持する。各スパイクサンプル溶液をプレートの3個のウェルにロードし、合計6個のウェルにロードする。
【0232】
対照の調製。ABT−308原薬のロットを入手する。150μLアリコートを分取し、分取日から3年間まで公称値−80℃で凍結保存する。
【0233】
ワーキング対照:対照のアリコートを氷上で解凍する。ポリプロピレンマイクロ遠心管内で対照を最終体積1000μLとなるように希釈バッファーで10mg/mLまで希釈する。単一希釈液を調製する。変性用に保持する。プレートの3個のウェルに対照をロードする。
【0234】
変性。プレートブランクでは、プレートで試験するブランク数と同数のマイクロ遠心管に希釈バッファー1000μLを加える。加熱中にチューブのキャップが開かないようにパラフィルムで密閉するか、又はキャップを閉じた状態に維持するように第2のラックをキャップの上に配置することができる。標準、非スパイクサンプル、スパイクサンプル、スパイク、ブランク及び対照を95℃±2℃に15分間加熱する。パラフィルムを使用した場合には冷却中にチューブから剥がす。15分間冷却し、約10000rpmで5分間遠心する。上清700μLをマイクロチューブに移し、プレートにロードする。トリトン/蛋白質ペレットに支障のないように注意する。
【0235】
プレートウォッシャー指示及び水浴設定。プレート洗浄ボトルにプレート洗浄バッファー(ステップ5.3参照,1×PBS+0.1%トリトンX−100)を充填する。プレートウォッシャーを起動する。以下のパラメーターをチェックする。パラメーターは以下のように設定すべきである:プレートタイプ:各サイクルで1(合計4サイクル);吸引速度:10mm/s;体積:400μl;浸漬時間:5秒;吸引時間6秒。水浴をオンにし、95℃に設定する。水浴温度を95℃±2℃に少なくとも30分間平衡化させる。
【0236】
アッセイ手順:ステップを完了するにつれて外していくことによりチェックリストをガイドとして使用することができる。更に、アッセイ中に使用される全機器を記録する。アッセイを実施する日毎に使用する量のカゼインアリコートを37℃±2℃にする必要がある。コーティングバッファーと基質は冷温で使用する。ブロッキングインキュベーションの前とその間に標準、サンプル、対照、スパイク及びスパイクサンプルを調製する。希釈液を調製し、エッペンドルフチューブに移し、15分間変性させ、15分間冷却し、5分間遠心し、マイクロチューブに移すためには、1時間を越えるブロックインキュベーションが必要になる場合がある。プレートをブロックする前に少なくとも40分間放置する。12チャネルピペットを使用してサンプル、スパイクサンプル、標準、対照、アッセイスパイク及びブランクを水平方向にプレートのB〜G行にロードする。標準は高濃度から低濃度の順にロードする。プレートコーティング、ビオチン添加、ニュートラアビジン添加、基質添加、及び停止溶液添加はカラム2〜11に垂直に実施する。
【0237】
冷温50mM重炭酸ナトリウム中コーティング抗体100μL/ウェルでプレートをコーティングする。コーティング溶液がウェルの底を均等に覆うまでプレートの側面をタッピングし、シーリングテープを貼り付け、プレートシェーカー(又は同等手段)速度3にて振盪しながら公称温度4℃でインキュベートする。
【0238】
一晩インキュベーション後、プレートを冷蔵庫から取り出し、室温で平衡化させる。コーティングを振り払う。プレートをペーパータオルで拭う。37℃±2℃カゼイン300μL/ウェルでブロックし、シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間±10分間振盪しながら37℃±2℃でインキュベートする。
【0239】
ブロッキングインキュベーション前とその間に標準、サンプル、対照、スパイク及びスパイクサンプルを調製する。プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。8チャネルピペットを使用し、変性した標準、サンプル、スパイク、スパイクサンプル、ブランク及び対照100μL/ウェルをプレートの3個のウェルに分注する。プレートの外側のウェルは使用せず、これらのウェルには非処理希釈バッファーを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて2時間振盪しながら37℃±2℃でインキュベートする。テンプレートを作成し、プレートにロードする際のガイドとして使用する。
【0240】
プレートリーダー設定。プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。ビオチン化抗体100μL/ウェルを加える。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて1時間振盪しながら37℃±2℃でインキュベートする。
【0241】
プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。ニュートラアビジン−HRPコンジュゲート溶液100μL/ウェルを加える。ニュートラアビジンを最後の行に添加後、すぐにタイマーをスタートする。シーリングテープを貼り付け、Lab−line Environプレートシェーカー(又は同等手段)で80rpm±5rpmにて30分間振盪しながら37℃±2℃でインキュベートする。プレートを洗浄バッファーで4回洗浄する。プレートをペーパータオルで拭う。冷温K−Blue基質100μL/ウェルを加え、シーリングテープを貼り付け、Lab−lineタイタープレートシェーカー(又は同等手段)で速度3にて振盪しながら室温で10分間インキュベートする(基質を最初の行に添加後、すぐにタイマーをスタートする)。1Nリン酸100μL/ウェルを加えることにより反応を停止する。プレートをプレートシェーカーにセットし、速度3で3分間振盪する。プレートを450nmで読取る。
【0242】
データ解析及び計算。注:光学密度が標準曲線の実際の定量限界内にあり且つ下記%CV又は%差条件を満たすサンプル、スパイク、スパイクサンプル及び対照のみが許容される。サンプルODが標準曲線に達しない場合には、結果を0.18ng/mL未満と報告する(アッセイLOQ)。その後、この数値を希釈後のサンプル濃度(10mg/mL)で割り、数値(ng/mg)を報告する。サンプルのプロテインA濃度が高く、非スパイク及び/又はスパイクサンプルが標準曲線(2ng/mL)を上回る場合には、標準曲線内となるように更に希釈する。その後、この数値を希釈後のサンプル濃度で割り、数値(ng/mg)を報告する。スパイク回収率計算では、非スパイクサンプル値(ng/mL)が曲線に達しない場合でも、スパイクサンプル値(ng/mL)から非スパイクサンプル値(ng/mL)を差し引く。数値がマイナスの場合又は「範囲」が得られる場合には、スパイク回収率計算の非スパイクサンプルをゼロとみなす。
【0243】
標準曲線。標準濃度をプロトコルテンプレートに入力する。二次曲線フィットを使用する。測定係数は0.99でなければならず、3個のウェル間の%CVは20%でなければならない。この条件を満たさない場合には、標準を1本(1レベル,ウェル3個)下げることができる。0.18ng/mL下げる場合には、光学密度が0.26ng/mL及び2ng/mL(残りの標準曲線の点)光学密度以内のサンプル及びスパイクサンプルのみが許容可能である。更に、各標準レベルの3個について、1個のウェルが明白に汚染されているか又は結合度が低い場合には、このウェルを下げることができる。1個のウェルを標準レベルから下げる場合には、残りの2個は%差=20%でなければならない。プレートのバックグラウンド(ブランク)に近いOD値を示す最低標準の%CVは30%でなければならない。ウェル1個を下げる場合には、残りの2個の%差は35%でなければならない。最低標準を下げる場合には、光学密度が残りの標準曲線レベル光学密度以内にあるサンプル及びスパイクサンプルのみが許容可能である。
【0244】
%差を次のように計算する:%差=(絶対値(希釈液1の結果−希釈液2の結果)/平均値)×100%。標準が上記条件を満たさない場合には、アッセイを繰返す必要がある。%CV及び/又は%差の数値と標準曲線測定係数結果を報告する。
【0245】
サンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを報告する。各サンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。注:非スパイクサンプルODが最低標準ODを下回る場合には、%差条件は非スパイク結果に適用されない。上記計算参照。
【0246】
各希釈液の「非スパイクサンプル結果」(ng/mL)を報告する。これらの数値をスパイク回収率計算で使用する。平均「非スパイクサンプル結果(ng/mL)」と希釈液間の%差を計算する。結果を報告する。希釈液間の%差は25%でなければならない。平均(ng/mL)値から実際のプロテインA濃度ng/mgを次のように計算する:プロテインA(ng/mg)=平均「非スパイクサンプル結果(ng/mL)」/希釈後のサンプル濃度(10mg/mL)。結果を記録する。
【0247】
スパイク。%CVは3個のウェル間で20%でなければならない。%CVを記録する。スパイクから1個のウェルを下げることができる。残りの点は%差=20%でなければならない。上記計算参照。プロテインA濃度(ng/mL)を報告する。この結果をスパイク回収率計算で使用する。各スパイクに得られる濃度(ng/mL)は理論スパイク濃度の±20%でなければならない。結果を記録し、合否を判定する。スパイク結果が理論値の20%以内にない場合には、アッセイを繰返す必要がある。平均スパイク濃度(ng/mL)×100は100%±20% 0.296ng/mLでなければならない。
【0248】
スパイクサンプル。%CVは3個のウェル間で20%でなければならない。3個のウェル間の%CVを記録する。各スパイクサンプル希釈液から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。上記計算参照。各希釈液の「スパイクサンプル結果」(ng/mL)を報告する。2種類の希釈液間の%差を記録する。希釈液間の%差は25%でなければならない。これらの結果をスパイク回収率計算で使用する。下式を使用して各希釈液セットの%スパイク回収率を計算する:%スパイク回収率=スパイクサンプル値−非スパイクサンプル値×100。スパイク値。注:スパイク回収率計算では、非スパイクサンプル値(ng/mL)が曲線を下回る場合でも、スパイクサンプル値(ng/mL)から非スパイクサンプル値(ng/mL)を差し引く。数値がマイナスの場合又は「範囲」が得られる場合には、スパイク回収率計算では非スパイクサンプルをゼロとみなす。各サンプルの各希釈液の%スパイク回収率は100%±50%(50%〜150%)でなければならない。結果と合否を記録する。
【0249】
対照。%CVは3個のウェル間で20%でなければならない。%CV結果を記録する。対照から1個のウェルを下げることができる。残りの2個は%差=20%でなければならない。
【0250】
【表9】
【0251】
【表10】
【0252】
本願では種々の刊行物を引用するが、その開示内容全体を本願に援用する。
【特許請求の範囲】
【請求項1】
抗IL−13抗体又はその抗原結合フラグメントと少なくとも1種の宿主細胞蛋白質(HCP)を含有するサンプル混合物からHCP低減抗IL−13抗体又はその抗原結合フラグメント調製物を生成する方法であって、前記方法が、
(a)前記サンプル混合物をアフィニティークロマトグラフィー樹脂と接触させ、アフィニティークロマトグラフィーサンプルを捕集する工程と;
(c)前記アフィニティークロマトグラフィーサンプルをpH低下に供し、pHを約3〜約4低下させ、pH低下サンプルを形成する工程と;
(d)前記pH低下サンプルを約4.5〜約6.5のpHに調整し、前記pH調整サンプルをイオン交換樹脂と接触させ、イオン交換サンプルを捕集する工程と;
(e)前記イオン交換サンプルを疎水性相互作用クロマトグラフィー(HIC)樹脂と接触させ、前記HCP低減抗体調製物を含有するHICサンプルを捕集する工程
を含む、前記方法。
【請求項2】
前記pH低下が適切な酸を前記サンプル混合物と混合することにより実施され、前記適切な酸がクエン酸、酢酸、カプリル酸等から構成される群から選択される、請求項1に記載の方法。
【請求項3】
前記アフィニティークロマトグラフィー樹脂が、プロテインA樹脂である、請求項1に記載の方法。
【請求項4】
前記プロテインA樹脂が、MabSelect(登録商標)樹脂である、請求項3に記載の方法。
【請求項5】
前記イオン交換樹脂が、アニオン交換樹脂又はカチオン交換樹脂である、請求項1に記載の方法。
【請求項6】
前記イオン交換樹脂が、カチオン交換樹脂である、請求項5に記載の方法。
【請求項7】
前記カチオン交換樹脂が、Fractogel、カルボキシメチル(CM)、スルホエチル(SE)、スルホプロピル(SP)、リン酸(P)及びスルホン酸(S)から構成される群から選択される、請求項6に記載の方法。
【請求項8】
前記カチオン交換樹脂が、Fractogel(登録商標)SO3−である、請求項7に記載の方法。
【請求項9】
前記イオン交換樹脂が、アニオン交換樹脂である、請求項5に記載の方法。
【請求項10】
前記アニオン交換樹脂が、Qセファロース、ジエチルアミノエチル(DEAE)、第四級アミノエチル(QAE)及び第四級アミン(Q)基から構成される群から選択される、請求項9に記載の方法。
【請求項11】
前記アニオン交換樹脂が、Qセファロースである、請求項11に記載の方法。
【請求項12】
前記イオン交換工程が、第1イオン交換工程と第2イオン交換工程を含む、請求項1に記載の方法。
【請求項13】
前記HICが、1種以上の疎水基を含むカラムを使用して実施される、請求項1に記載の方法。
【請求項14】
前記1種以上の疎水基が、アルキル基、アリール基及びその組合せから構成される群から選択される、請求項13に記載の方法。
【請求項15】
前記カラムが、フェニルセファロース(例えばPhenyl Sepharose(登録商標)6ファストフローカラム、Phenyl Sepharose(登録商標)高性能カラム)、Octyl Sepharose(登録商標)高性能カラム、Fractogel(登録商標)EMDプロピル、Fractogel(登録商標)EMDフェニルカラム、Macro−Prep(登録商標)メチル、Macro−Prep(登録商標)t−ブチル担体、WP HI−Propyl(C3)(登録商標)カラム、及びToyopearl(登録商標)エーテル、フェニル又はブチルカラムから構成される群から選択される、請求項14に記載の方法。
【請求項16】
前記カラムが、フェニルセファロースを含む、請求項15に記載の方法。
【請求項17】
更に濾過工程を含み、前記HICサンプルを濾過に供してウイルス粒子を除去し、バッファー交換を容易にする、請求項1に記載の方法。
【請求項18】
前記抗IL−13抗体又はその抗原結合部分が、ヒト化抗体、キメラ抗体又は多価抗体である、請求項11に記載の方法。
【請求項19】
前記抗IL−13抗体又はその抗原結合部分が、ヒト化抗体である、請求項18に記載の方法。
【請求項20】
前記調製物が、HCPを実質的に含まない、請求項1に記載の方法。
【請求項21】
更に深層濾過工程を含む、請求項1に記載の方法。
【請求項22】
請求項1に記載の方法により生成されたHCP低減抗体調製物と医薬的に許容可能な担体を含有する、医薬組成物。
【請求項23】
前記組成物が、HCPを実質的に含まない、請求項22に記載の医薬組成物。
【請求項1】
抗IL−13抗体又はその抗原結合フラグメントと少なくとも1種の宿主細胞蛋白質(HCP)を含有するサンプル混合物からHCP低減抗IL−13抗体又はその抗原結合フラグメント調製物を生成する方法であって、前記方法が、
(a)前記サンプル混合物をアフィニティークロマトグラフィー樹脂と接触させ、アフィニティークロマトグラフィーサンプルを捕集する工程と;
(c)前記アフィニティークロマトグラフィーサンプルをpH低下に供し、pHを約3〜約4低下させ、pH低下サンプルを形成する工程と;
(d)前記pH低下サンプルを約4.5〜約6.5のpHに調整し、前記pH調整サンプルをイオン交換樹脂と接触させ、イオン交換サンプルを捕集する工程と;
(e)前記イオン交換サンプルを疎水性相互作用クロマトグラフィー(HIC)樹脂と接触させ、前記HCP低減抗体調製物を含有するHICサンプルを捕集する工程
を含む、前記方法。
【請求項2】
前記pH低下が適切な酸を前記サンプル混合物と混合することにより実施され、前記適切な酸がクエン酸、酢酸、カプリル酸等から構成される群から選択される、請求項1に記載の方法。
【請求項3】
前記アフィニティークロマトグラフィー樹脂が、プロテインA樹脂である、請求項1に記載の方法。
【請求項4】
前記プロテインA樹脂が、MabSelect(登録商標)樹脂である、請求項3に記載の方法。
【請求項5】
前記イオン交換樹脂が、アニオン交換樹脂又はカチオン交換樹脂である、請求項1に記載の方法。
【請求項6】
前記イオン交換樹脂が、カチオン交換樹脂である、請求項5に記載の方法。
【請求項7】
前記カチオン交換樹脂が、Fractogel、カルボキシメチル(CM)、スルホエチル(SE)、スルホプロピル(SP)、リン酸(P)及びスルホン酸(S)から構成される群から選択される、請求項6に記載の方法。
【請求項8】
前記カチオン交換樹脂が、Fractogel(登録商標)SO3−である、請求項7に記載の方法。
【請求項9】
前記イオン交換樹脂が、アニオン交換樹脂である、請求項5に記載の方法。
【請求項10】
前記アニオン交換樹脂が、Qセファロース、ジエチルアミノエチル(DEAE)、第四級アミノエチル(QAE)及び第四級アミン(Q)基から構成される群から選択される、請求項9に記載の方法。
【請求項11】
前記アニオン交換樹脂が、Qセファロースである、請求項11に記載の方法。
【請求項12】
前記イオン交換工程が、第1イオン交換工程と第2イオン交換工程を含む、請求項1に記載の方法。
【請求項13】
前記HICが、1種以上の疎水基を含むカラムを使用して実施される、請求項1に記載の方法。
【請求項14】
前記1種以上の疎水基が、アルキル基、アリール基及びその組合せから構成される群から選択される、請求項13に記載の方法。
【請求項15】
前記カラムが、フェニルセファロース(例えばPhenyl Sepharose(登録商標)6ファストフローカラム、Phenyl Sepharose(登録商標)高性能カラム)、Octyl Sepharose(登録商標)高性能カラム、Fractogel(登録商標)EMDプロピル、Fractogel(登録商標)EMDフェニルカラム、Macro−Prep(登録商標)メチル、Macro−Prep(登録商標)t−ブチル担体、WP HI−Propyl(C3)(登録商標)カラム、及びToyopearl(登録商標)エーテル、フェニル又はブチルカラムから構成される群から選択される、請求項14に記載の方法。
【請求項16】
前記カラムが、フェニルセファロースを含む、請求項15に記載の方法。
【請求項17】
更に濾過工程を含み、前記HICサンプルを濾過に供してウイルス粒子を除去し、バッファー交換を容易にする、請求項1に記載の方法。
【請求項18】
前記抗IL−13抗体又はその抗原結合部分が、ヒト化抗体、キメラ抗体又は多価抗体である、請求項11に記載の方法。
【請求項19】
前記抗IL−13抗体又はその抗原結合部分が、ヒト化抗体である、請求項18に記載の方法。
【請求項20】
前記調製物が、HCPを実質的に含まない、請求項1に記載の方法。
【請求項21】
更に深層濾過工程を含む、請求項1に記載の方法。
【請求項22】
請求項1に記載の方法により生成されたHCP低減抗体調製物と医薬的に許容可能な担体を含有する、医薬組成物。
【請求項23】
前記組成物が、HCPを実質的に含まない、請求項22に記載の医薬組成物。
【図1】


【図2−1】

【図2−2】

【図3】
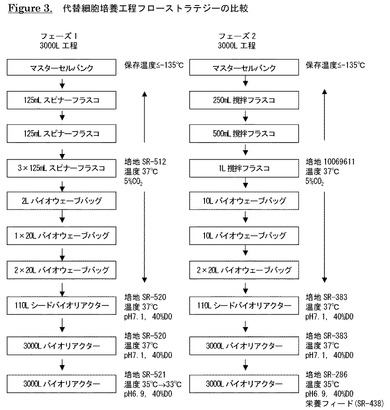
【図4】

【図5】
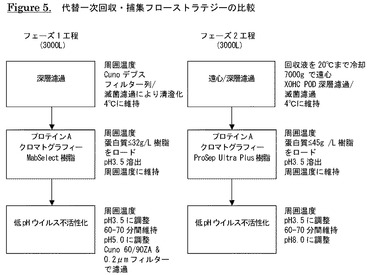
【図6−1】

【図6−2】

【図7】
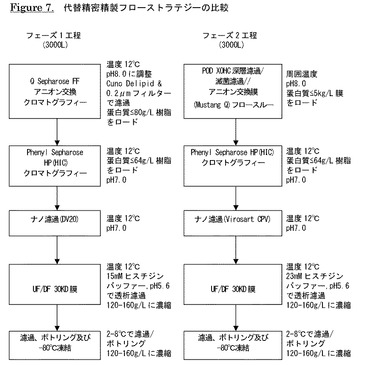
【図2−1】
【図2−2】
【図3】
【図4】
【図5】
【図6−1】
【図6−2】
【図7】
【公表番号】特表2013−508387(P2013−508387A)
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願番号】特願2012−535337(P2012−535337)
【出願日】平成22年10月20日(2010.10.20)
【国際出願番号】PCT/US2010/053388
【国際公開番号】WO2011/050071
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(391008788)アボット・ラボラトリーズ (650)
【氏名又は名称原語表記】ABBOTT LABORATORIES
【Fターム(参考)】
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願日】平成22年10月20日(2010.10.20)
【国際出願番号】PCT/US2010/053388
【国際公開番号】WO2011/050071
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(391008788)アボット・ラボラトリーズ (650)
【氏名又は名称原語表記】ABBOTT LABORATORIES
【Fターム(参考)】
[ Back to top ]