
プロトン導電性酸化物膜−水素透過膜複合膜型電解質およびこれを用いた電気化学デバイス
【課題】 AECeO3のプロトン導電性は維持しつつも、当該AECeO3のCO2に対する脆弱性を克服してCO2を含む混合ガス雰囲気環境下でも使用できるようにする。
【解決手段】 バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造とし、プロトン(H+)が通過可能でありCO2を含むガス3中から水素のみを分離可能なプロトン導電性酸化物膜1と、プロトン(H+)が通過可能でありCO2を含むガス3中から水素のみを分離可能な水素透過膜2との複合膜からなる。これら両膜の複合界面が連続接合した状態で、プロトン導電性酸化物膜1の少なくとも前記ガス側の表面に水素透過膜2が成膜され、尚かつ当該複合膜1,2が多孔質支持体Sによって支持されている。
【解決手段】 バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造とし、プロトン(H+)が通過可能でありCO2を含むガス3中から水素のみを分離可能なプロトン導電性酸化物膜1と、プロトン(H+)が通過可能でありCO2を含むガス3中から水素のみを分離可能な水素透過膜2との複合膜からなる。これら両膜の複合界面が連続接合した状態で、プロトン導電性酸化物膜1の少なくとも前記ガス側の表面に水素透過膜2が成膜され、尚かつ当該複合膜1,2が多孔質支持体Sによって支持されている。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、プロトン導電性酸化物膜−水素透過膜複合膜型電解質およびこれを用いた電気化学デバイスに関する。さらに詳述すると、本発明は、CO2を含むガス中におけるプロトン導電性酸化物電解質の脆弱性の回避法に関する。
【背景技術】
【0002】
本件出願人たる電力中央研究所においては、これまで、既存の4つの燃料電池(PAFC(リン酸型燃料電池)、MCFC(溶融炭酸塩型燃料電池)、SOFC(固体電解質型燃料電池)、PEFC(固体高分子型燃料電池))の性能を同一の評価手法で分析することにより燃料電池の出力電圧と運転温度との一般化された関係を明らかにする、という研究が続けられている。また、かかる燃料電池の研究により、プロトン(H+)移動型の電解質(つまり当該電解質中をプロトンが移動可能なもの)を用いて250〜500℃程度の中温域で作動する燃料電池を開発できれば、既存の燃料電池よりも性能面、材料面で優位な燃料電池になる可能性があることが示されている(特許文献1参照)。ここでいうプロトン移動型の電解質としては、溶融塩や水溶液系の電解質に可能性がある。しかし、これら電解質は、250℃〜500℃程度の中温域では化学的に不安定であったり、あるいは内在する他のイオンも電極反応に関わったりすることがあるため、燃料電池に適用できなかった。
【0003】
一方、プロトン移動型電解質のその他の具体例として、例えば「高温型プロトン導電体を用いた炭化水素改質ガス類から水素を分離する方法」が記載された特許文献2においてはプロトン導電性酸化物(プロトン導電性酸化物固体電解質)が開示されている(特許文献2参照)。このプロトン導電性酸化物は、当該酸化物がプロトン導電性機能を有していることから、ガス(例えば天然ガス改質ガス)中の水素を電気化学的に分離する技術、さらには種々の電気化学デバイスに関する技術に応用することが考えられあるいは期待されているものである(特許文献2参照)。
【0004】
上述のプロトン導電性酸化物固体電解質の一例として、SrCeO3に代表されるペロブスカイト構造を持つ酸化物AECeO3(ただしAEはBa または Sr )が存在する。AECeO3は、図35に示すように水素気流中、中高温域において良好なプロトン導電性を示し、尚かつ高い導電率を示すことが知られている。このため、これら酸化物を、水素のみを用いた電気化学デバイス(例えば燃料電池、水素センサー、水素ポンプ)の電解質に適用した場合には、高い性能が発揮されるものと考えられる。
【0005】
例えば図36に示すプロトン導電性のSrCeO3膜101は、図中の改質ガス102のような水素や二酸化炭素などが混成したガスの流路に設けられて水素透過膜として機能する。この場合、1次側(この場合であれば改質ガス102側)の電極104と2次側(この場合であれば分離後の水素103側)の電極105との間の外部電圧106によって印加される電圧がプロトンH+の推進力(ドライビングフォース)となる。このようなプロトン導電性のSrCeO3膜(水素透過膜)101を用いた場合には、1次側圧力を固定しながら様々な2次側水素圧力を設定して供給することが可能になるという長所がある。また、常圧の改質ガス102を使用しながら加圧することが可能であるため、貯蔵水素用の昇圧装置やそのための設置スペースなどが不要だという長所もある。
【0006】
【特許文献1】特開2004−119077号公報
【特許文献2】特開2003−2610号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、通常、水素分離の対象となるガス(上述の例でいえば改質ガス102)は、CO2など他のガスも含まれている混合ガスである場合がほとんどであり、このことから、上述したSrCeO3膜101のようなペロブスカイト構造を持つ酸化物(AECeO3)を電気化学デバイスの電解質に適用すると特有の問題が生じてしまう。
【0008】
すなわち、酸化物AECeO3には、CO2を含む雰囲気に曝されると、
AECeO3+CO2→AECO3+CeO2
というようにCO2と反応を起こし、容易に分解してしまうという性質がある。このため、例えば特許文献1に見られるように、CO2を含んだガスが流れる電気化学デバイス(例えば水素ポンプなど)の電解質にこの酸化物AECeO3を適用した場合、当初の段階では特許文献1にも記されているような初期の性能が発揮されるものの、長時間は耐用できない、つまりこのような混合ガス中のCO2に対して脆弱である、という点で問題がある(例えば、第27回電池討論会要旨集(3B10, p226-227)参照)。
【0009】
そこで、本発明は、かかる従来技術が有していた課題に鑑み、AECeO3のプロトン導電性は維持しつつも、当該AECeO3のCO2に対する脆弱性を克服してCO2を含むガス雰囲気下でも使用可能としたプロトン導電性酸化物膜−水素透過膜複合膜型電解質およびこれを用いた電気化学デバイスを提供することを目的とする。
【課題を解決するための手段】
【0010】
かかる目的を達成するため、本発明者は種々の検討と実験を行った。かかる検討と実験の過程および概要について以下に説明する。
【0011】
まず、本発明者は従来における別の水素分離法について検討した(図37参照)。水素分離に用いられる水素透過膜(Pd金属膜など、図中で符号201で示す)は、例えば改質ガスといったようなH2(水素)やCO2(二酸化炭素)などの混成ガス(図中、符号202で示す)の流路に設けられ、水素分子が分離した状態(つまりプロトン(H+)と電子(e-)になった状態)で透過させることが可能なもので、これにより、改質ガスから水素を分離することを可能としている(図37参照、分離後の水素を符号203で示す)。このような水素分離法の長所としては水素分離のための電力量が不要だという点が挙げられる。したがって推進力(ドライビングフォース)は水素分圧差のみである。ただし、効率のよい分離を行うためには非常に高い水素分圧差が必要になる(つまり、図37中に示す圧力High PH2と圧力Low PH2と間に高い分圧差が必要になる)。したがって、目的とする水素圧に応じて1次側(改質ガス側)と2次側(分離された水素側)の圧力が固定されることになるから貯蔵用昇圧装置が必要となってくる。また、常圧の改質ガスが使用できないという短所もある。
【0012】
ここで検討した水素透過膜(Pd金属膜など)を利用した水素分離法(図37参照)には上述のように長所と短所とがあるが、かかる長所・短所を、ペロブスカイト構造を持つ酸化物(AECeO3)を利用した水素分離法の長所・短所と比較すると、両者が互いに入れ替わった関係(つまり一方が長所とする点が他方においては短所になっている関係)にあることがわかる。そこで、本発明者はかかる関係に着目しつつ、上記課題を解決すべく鋭意検討を重ねた結果、プロトン導電性酸化物(AECeO3)と水素透過膜(例えばPd金属)とを複合膜化した電解質を用いると、水素のみが(具体的にはプロトンの形で)Pd膜を透過しAECeO3膜に達するため、AECeO3のCO2に対する脆弱性を克服でき、CO2を含むガス環境下でも使用できる電気化学デバイスの電解質として使用可能となることを知見し、この電解質を用いればCO2を含むガス環境下でも上記課題を解決できることを見出した。
【0013】
本発明はかかる知見に基づくものであり、請求項1に記載の発明であるプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造とし、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能なプロトン導電性酸化物膜と、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な水素透過膜との複合膜からなり、これら両膜の複合界面が連続接合した状態で、プロトン導電性酸化物膜の少なくともガス側の表面に水素透過膜が成膜され、尚かつ当該複合膜が多孔質支持体によって支持されているというものである。
【0014】
本発明においては、上述した知見の下、水素分離に好適である新規な複合膜型電解質構造を構築している点が特徴的である。すなわち、プロトン導電性酸化物膜と水素透過膜とが互いの短所を補完し合う複合膜型構造とすることにより、AECeO3のプロトン導電性は維持しつつも、AECeO3のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することを可能としている。この複合膜型電解質はいわばセラミックス(AECeO3)と改質ガスとの間に別の膜が介在している状態、別の表現をするならばセラミックス(AECeO3)の表面に水素透過性の保護膜的要素が形成された状態にあり、このような複合膜型構造が、CO2によるセラミックス分解を防ぐ。
【0015】
また、この複合膜型電解質構造においては、水素が固相から固相へと相間を移動する際に、水素元素ないしは水素分子として移動するのではなく、プロトンとして連続して移動する現象を呈することを利用している(図1参照)。この場合、水素透過膜(例えばPd膜)は、水素(H2)のみ選択的に透過させ、他の分子(N2, CO, CO2など)は透過させないという性質を持つことから、この水素透過膜をプロトン−電子混合導電体とみなすことができる(図1参照)。したがって、この水素透過膜が電極と電解質の役目を果たすことになる。上述の保護膜として水素透過膜をプロトン導電性酸化物膜の一部と見なすと、プロトン導電性酸化物膜に接触(あるいは通過)するガスは水素(H2)のみということになる。
【0016】
しかも、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質においては、プロトン導電性酸化物膜と水素透過膜とからなる複合膜が多孔質支持体によって支持されている。この場合における多孔質支持体は、ガス中から水素を分離するという複合膜の機能に影響を及ぼすものではない。要は、多孔質支持体は多孔質体であるので、水素が水素透過膜からプロトンとして膜中に入る際にこの反応を阻害しない程度にガスの拡散性に優れていればよい。しかも、この多孔質支持体が上記複合膜を強度的に支持する基材として働くことから、複合膜(プロトン導電性酸化物膜および水素透過膜)はそれ自体が基材に要する強度を有していなくても足りる。つまり、複合膜自体で所定の強度を確保する必要がなくなることから、この複合膜を構成するプロトン導電性酸化物膜と水素透過膜のそれぞれを必要十分な厚さにまで薄膜化することが可能となり設計の自由度が広がるという利点がある。また、プロトン導電性酸化物膜および水素透過膜の薄膜化によりコスト面でも有利となる。
【0017】
また、請求項2に記載の発明は、請求項1に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質におけるプロトン導電性酸化物膜が、希土類元素セリウム(Ce)の一部が当該セリウム(Ce4+)とは異なる他の低原子価希土類元素で置換されている構造となっているというものである。低原子価元素で置換することにより、例えば、Ce4+→Yb3+とすると、結晶格子中の電気的中性が崩れ、これを補うため格子中の酸素1/2個が抜ける。そうすると格子中の電気的中性が保たれる。このように意図的に酸化物イオン空孔を生成させること(ドーピング)ができ、よりプロトンの生成が促進されることになる。
【0018】
請求項3に記載の発明は、請求項1または2に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における他の希土類元素が、イッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素であるであるというものである。
【0019】
請求項4に記載の発明は、請求項1から3のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における水素透過膜が、水素透過性を示す金属または合金であるというものである。金属または合金のうち、水素透過性を示すものがこの発明における水素透過膜に適用される。
【0020】
請求項5に記載の発明は、請求項4に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とするものである。
【0021】
請求項6に記載の発明は、請求項4もしくは5に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とするものである。
【0022】
請求項7に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、多孔質支持体が複合膜のガス側の表面に形成されているというものである。
【0023】
請求項8に記載の発明は、請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面に多孔質カソードが形成されているというものである。
【0024】
請求項9に記載の発明は、請求項8に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、多孔質カソードが少なくともペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物および銀(Ag)のうちいずれか一つを含んでいることを特徴とするものである。
【0025】
請求項10に記載の発明は、請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面に、プロトン(H+)が通過可能な別の水素透過膜が形成されているというものである。
【0026】
請求項11に記載の発明は、請求項10に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における別の水素透過膜が、水素透過性を示す金属または合金であるというものである。
【0027】
請求項12に記載の発明は、請求項11に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とするものである。
【0028】
請求項13に記載の発明は、請求項11もしくは12に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とするものである。
【0029】
請求項14に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、多孔質支持体が、複合膜のガス側とは反対側の表面に形成された多孔質カソードであるというものである。
【0030】
請求項15に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面に多孔質カソードが形成され、さらに該多孔質カソードの表面に多孔質支持体が形成されているというものである。
【0031】
請求項16に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜が形成され、さらに該別の水素透過膜の表面に多孔質支持体が形成されているというものである。
【0032】
請求項17に記載の発明は、請求項1から16のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質を用いた電気化学デバイスであって、燃料電池、水素センサー、水素ポンプ、水蒸気センサーおよび排ガス浄化デバイスのいずれかに応用されるというものである。
【0033】
この電気化学デバイスは、請求項18に記載のように少なくともH2とCO2とを含有する混合ガス中の水素濃度を定量できる水素センサーに適用することができる。
【0034】
また、電気化学デバイスは、請求項19に記載のように少なくともH2とCO2とを含有する混合ガス中から水素のみを分離できる水素ポンプに適用することもできる。
【0035】
さらに、電気化学デバイスは、請求項20に記載のように少なくとも水蒸気とCO2とを含有する混合ガス中から水蒸気濃度を定量できる水蒸気センサーに適用することもできる。
【0036】
さらに、電気化学デバイスを、請求項21に記載のように、少なくとも水蒸気、CO2およびNOx を含有する排ガスからNOxを除去するための排ガス浄化用デバイスに適用することもできる。
【0037】
ここで、以下、金属/酸化物複合膜型電解質の概念について説明し、続いてAECeO3のCO2に対する脆弱性を克服することについての検討内容を説明することにより、本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質の内容について更に詳細に説明することとする。以下では、「1.ペロブスカイト型プロトン導電体酸化物について」、「2.水素透過膜について」、「3.水素透過膜とプロトン導電性酸化物を用いた燃料電池について」、そして「4.金属/酸化物複合膜型電解質を用いた燃料電池について」という項目順で説明していく。
【0038】
1.ペロブスカイト型プロトン導電体酸化物について
従来技術の課題を解決するにあたり、本発明者はまずペロブスカイト構造を持つ酸化物AECeO3について検討した。例えば、中温域で作動する燃料電池には、当該中温域で高いプロトン導電率を有する電解質が必要であり、燃料電池として十分に作動するには、以下に示す化学式1,2の各電極反応が円滑に進行する必要がある。
【0039】
【化1】

【化2】
【0040】
しかし、一般的に低温域で高いプロトン導電率を有する電解質は、中温域においては分解反応が進行するため、化学的安定性に欠けるという問題を有している。また、仮に中温域で安定に存在しても、実際に上述した化学式1,2のような燃料電池反応を発現する電解質は皆無である。
【0041】
一方、600℃以上の中高温域では、図35に示すようなSrCeO3やBaCeO3などの高いプロトン導電率を有するペロブスカイト型構造のセレイト系酸化物(AECeO3)が開示されている。また、前述のようなペロブスカイト型構造(ABO3)を持つ酸化物は、水素源が全くない高温雰囲気において、酸素分圧が高くなるほど正孔(ホール)による伝導(p型電子導電性)が増大することが知られている。この場合、導電率が酸素分圧の1/4乗に比例することから、酸化物イオン空格子点の関与した格子欠陥平衡にあるものと考えられている。
【0042】
このような酸化物を水素ガスや水蒸気のような水素源を含む雰囲気に曝すとプロトン導電性が発現する。このことは、これら酸化物が雰囲気中の水素源から水素を取り込み、水素がプロトンの形で存在することを意味する。このような酸化物の格子欠陥とプロトン生成の関係を模式的に図2に示す。図2において、(a)は化学量論組成を、(b)は湿潤酸素中におけるプロトン又はホール伝導機構を、(c)は乾燥酸素中におけるホール電子伝導機構を、(d)は水素中におけるプロトン伝導機構を示している。尚、図2中における正方形(□)は酸化物イオン空孔を表している。ここでは、格子欠陥が直接プロトンを生成するわけではない。まず雰囲気中の酸素と空孔が欠陥反応式(図中の式参照)に従いホール(h)を生成し、これらが平衡になっているところへ水素が導入されるとホールと水素とが反応してプロトンが生成する。つまり、酸化物イオン空孔が多数存在しなければプロトン導電性は発現しない。ただ、導電率はプロトンの濃度と易動度に比例するので、酸化物イオン空孔濃度が高い(=プロトン濃度が高い)からといって導電率が高くなるわけではない。高い濃度と易動度の両方が揃ってはじめて高い導電率を得ることができる。
【0043】
これまで多くの研究から、母体酸化物のみではプロトン導電性を示さず、プロトン導電性の発現には三価カチオンによるBサイトへのドーピングが必須であることが確認されている。例えばSrCeO3のBサイトカチオンのCe4+の一部をYb3+で置換固溶させると、結晶内では電気的中性条件を満たすため酸化物イオン空格子点を生じる。
【0044】
【化3】
【0045】
この空格子点は高温において正孔(化学式4中においては、右肩に傍点が付された記号hで示す)および雰囲気酸素と化学式4に示す平衡関係にある。したがって、水素源のない雰囲気ではp型電子導電性のみを示すことになる。これに対して、水素源、例えば水蒸気を雰囲気に導入すると、水蒸気分子と正孔または酸化物イオン空格子点との間に化学式5,6に示す平衡関係が化学式4とともに成立し、水素が酸化物中に取り込まれてプロトン導電性が発現する。
【0046】
【化4】
【化5】
【化6】
【化7】
【0047】
ここで、K1,K2,K3は化学式4〜6中のそれぞれにおける平衡定数である。化学式4、化学式5および化学式6はそれぞれ独立ではなく、化学式7によって関係づけられているのでこれら化学式のうちの二つの式を用いて平衡関係を表すことができる。
【0048】
しかし、アルカリ土類金属をAサイト、CeをBサイトに配するペロブスカイト型酸化物(AECeO3)は、プロトン導電性を有する一方で、雰囲気中にCO2が存在すると、熱力学的平衡により化学式8のようにCeO2とACO3とに容易に分解してしまうというように化学的安定性に欠ける。このため、SrCeO3やBaCeO3を電気化学デバイスに用いるには雰囲気中のCO2に注意を要する。
【0049】
【化8】
【0050】
また、仮にCO2を含む雰囲気でもSrCeO3やBaCeO3を電解質として利用するためには、電解質膜表面を雰囲気中のCO2から遮断する必要がある。
【0051】
2.水素透過膜について
水素がプロトンとして金属膜を透過する現象はパラジウム(Pd)膜において見いだされている。Pdおよびその合金膜の水素透過機構は、定性的には図3に示すような原理によって水素が透過する機構になっていると考えられている。水素透過能を有するPd膜の両側において水素分圧差が生じると、高水素分圧側の水素分子が金属表面で解離し水素原子となる(図3参照)。水素原子は、Pdによって電子を奪われプロトンとなり、濃度勾配によりプロトンとして金属中を拡散する。拡散したプロトンは、低水素分圧側で自由電子と再結合し水素原子となり、さらにPdの触媒能によってこれら水素原子が会合して分子となり、最終的に水素分子として低水素分圧側に放出される。このように金属Pd膜は、プロトンと電子を導電する一種の混合導電体と見なすことができる。本発明者は、特にPd膜において顕著なこのような機能を利用することとした。尚、Pd膜単独では、水素脆化が生じる虞があるため、PdとAgを合金化したものがより好適である。
【0052】
3.水素透過膜とプロトン導電性酸化物を用いた燃料電池について
上述の「2.水素透過膜について」で述べた水素透過能を持つ金属PdをAECeO3とガス雰囲気の間に配置できれば、CO2を含むガス環境下においてもAECeO3を使用することができると考えられる。これは、図4に示すように、CO2を含むガス(例えば改質ガス)側からH2がPd膜を透過することにより、酸化物(AECeO3)の膜側ではH2のみを燃料ガスとして利用できるようになるためである。
【0053】
ただし、このような二重膜構造(Pd膜およびAECeO3膜からなる二重構造)が構成された場合(図4参照)、以下の点があることが判った。すなわち、第一に、Pd膜を透過してきた水素によって水素分圧が極度に高くなり、酸化物中のCe4+がCe3+へと容易に還元されてしまうことがある。これに対しては例えば室温飽和程度の水蒸気を加えれば水蒸気との平衡によって水素分圧が若干低下し、Ce4+が還元されるのを防止することが可能となると考えられた。ただし、この場合、水蒸気はPd膜に阻害され酸化物膜まで届かないということが生じる。第二に、例えばPd膜を厚くするとコストに大きく影響することから、ある程度薄膜化する必要があるが、Pd金属等の水素透過膜単独の薄膜化は強度的に問題があると考えられた。
【0054】
4.金属/酸化物複合膜型電解質を用いた燃料電池について
種々の検討と実験の結果、上述のようなプロトン導電性酸化物膜および水素透過膜の両者を例えば燃料電池に適用するにあたっては、図5に示すような金属/酸化物複合膜型電解質、すなわち金属Pd膜と酸化物AECeO3膜とが重ね合わされた状態で複合した形態の電解質とすることが望ましいという知見が得られた(図5参照)。このように金属(Pd)膜と酸化物(AECeO3)膜とを接合して複合化することにより、CO2を含むガス環境から酸化物(AECeO3)を保護することが可能となる。ここで、図4に示した「水素透過膜とプロトン導電性セラミックスを用いた燃料電池」と比較すると、図5に示す電解質においては、バルク(この場合、Pd膜→AECeO3膜へと続く一連の部分のこと)内をプロトンがPd膜→AECeO3膜というように直接的かつ連続的に通過することができるという点が特徴的である。この場合、Pd膜と酸化物(AECeO3)膜の界面で電子の授受(電極反応)が生じないため、酸化物(AECeO3)は発生したHまたはH2によって還元されるということがなくなる。つまり、プロトン導電性を維持しつつ、CO2に対する脆弱性を克服することが可能となる。
【0055】
以上のように金属(Pd)膜と酸化物(AECeO3)膜とを接合して理想的界面を作成するにあたっては、以下のような手法によるコーティングが考えられる。
【0056】
まず、酸化物(AECeO3)膜に金属(Pd)膜を成膜する方法としては、以下の方法が挙げられる。
(1) 化学的…無電解めっき法、電解めっき法、MOD法、CVD法、スラリーコート法、スピンコーティング法、ディップ法
(2) 物理的…PVD法{スパッタ、蒸着、MBE(Molecular Beam Epitaxy)法、プラズマ溶射法、プラズマスプレー法}
上記の中で、無電解めっき法は、緻密薄膜を均一に作製可能で、酸化物(AECeO3)膜と金属(Pd)膜の複合界面の連続接合性を向上させる上で好適な方法である。また、プラズマ溶射法およびプラズマスプレー法も、緻密薄膜を作製可能である。本発明においては、緻密膜を複合する必要があるため、工業的に量産化をターゲットにおけば、自動成膜化が容易なプラズマ溶射法およびプラズマスプレー法が複合膜を成膜することに好適である。
【0057】
また、多孔質支持体上に酸化物(AECeO3)膜を薄く成膜する方法としては、以下の方法が挙げられる。
(1) 化学的…スラリーコート法、スピンコーティング法、ディップ法
(2) 物理的…PVD法{スパッタ、蒸着、MBE(Molecular Beam Epitaxy)法、プラズマ溶射法、プラズマスプレー法}
上記の方法を採用することで、酸化物(AECeO3)膜の薄膜化が可能となり、さらに、多孔質支持体上に酸化物(AECeO3)薄膜を成膜し、さらにその上に金属(Pd)薄膜を成膜することで低コスト化につながる。
【0058】
なお、ここではPdやPd-Ag合金を例に検討したが、上述したような水素の透過形態からすると、複合する膜として、水素を透過するPd以外の金属(例えばニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)など)の膜、これらの元素のうち2種以上からなる合金膜、あるいは酸化物(例えばSrZrO3、BaZrO3)の膜等を適用した場合にも同様の効果が得られると考えられた。中でも、金属膜あるいは合金膜を用いる場合には、それ自体がプロトン−電子混合導電体と見なすことができるため、電極が不必要となる利点がある。
【0059】
また、Pd膜を使用することから、コスト的にある程度薄膜化する必要があるが、Pd膜等の水素透過膜単独の薄膜化は強度的に問題があるという点に関しては、以下のように考えた。すなわち、まずPd膜自体を厚くして当該Pd膜自体を基材としても機能させることを検討してみたが、コスト面を考慮すると到底実用的ではない(Pdの使用量が実用レベルを超える)と考えられた。そうすると、このように複合膜自体で強度を確保するよりも、水素透過に影響を及ぼさない材質によって基材(支持体)を構成することが目的に適うと考えられた。具体的には、プロトン導電性酸化物膜と水素透過膜とからなる複合膜を多孔質支持体によって支持するという構造が適すると考えられた。この場合における多孔質支持体は、ガス中から水素を分離するという複合膜の機能に影響を及ぼすものではない。しかも、この多孔質支持体が上記複合膜を強度的に支持する基材として働くことから、複合膜自体で所定の強度を確保する必要がなくなることから、この複合膜を構成するプロトン導電性酸化物膜と水素透過膜のそれぞれを必要十分な厚さにまで薄膜化することが可能となる。
【0060】
ここまで説明したように、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質は燃料電池における電解質として特有の効果を奏しうるものであるが、適用可能な電気化学デバイスはこれに限らない。以下に発明者による実験結果を実施例1として示すように、水素センサー、水素ポンプ、水蒸気センサーといった電気化学デバイスにも好適なものである。さらには、以下に説明するように、排ガス浄化用電気化学デバイスにおける電解質としてもきわめて好適なものである。
【0061】
すなわち、本発明者は、水素センサー、水素ポンプ、水蒸気センサー以外にも、本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質の適用の可能性について検討した。一例として、具体的には、このプロトン導電性酸化物膜−水素透過膜複合膜型電解質を排ガス浄化用電気化学デバイスとして適用することについて詳細な検討をした。その背景は以下のとおりである。
【0062】
すなわち、従来、自動車など内燃機関の排気ガス中に含まれる窒素酸化物(NOx)の除去方法としては、活性成分として貴金属を担持した触媒による接触還元法が用いられてきている。つまり、上記した自動車排気ガス浄化用の触媒として、白金やロジウムなどの貴金属を比表面積の大きいアルミナなどの担体に担持した触媒が用いられている。また、ボイラなどの各種プラントの排ガス中に含まれる窒素酸化物(NOx)は、NH3 添加による無触媒脱硝法、触媒を用いたNH3 による選択接触還元法によってN2 およびO2 に変換され、無害化されている。しかし、このような従来の窒素酸化物の除去方法には下記(1)、(2) の問題点がある。
【0063】
(1) 自動車排気ガス浄化用触媒の問題点
自動車排気ガス浄化用の貴金属を担持した触媒は、貴金属の埋蔵量が少なく、価格が高く、さらに貴金属は、価格変動が大きく、安定して確保することが難しい問題がある。近年の自動車はリーンバーン域での燃焼にシフトする方向にあり、この結果、不完全燃焼を伴い、NOx が増加する傾向にあり、貴金属担持量の増加を伴うことなくNOx 除去率を向上することが可能な技術開発が必要である。なお、窒素酸化物の他の分解法として、アンモニアによる還元法が挙げられ、この方法は貴金属を必要としない点で優れているが、アンモニアを供給するための設備を必要とし、自動車のようにスペースに余裕がない場合、アンモニアを供給するためのボンベなどの装置を自動車に搭載することは困難である。
【0064】
(2) ボイラなど各種プラントにおけるアンモニアを用いた還元法の問題点
ボイラなどの各種プラントにNOx のアンモニアによる還元法を適用する場合、未反応のアンモニアが、排ガス中に僅かに含まれるSOx と反応し、硫安類を生成し、ボイラの熱回収装置である熱交換器の内部の低温部に付着し、熱交換器の圧力損失が経時的に増加する問題があり、アンモニア添加量の低減が望まれる。
【0065】
以上の問題に鑑み本発明者は本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質が排ガス浄化用電気化学デバイスに適用された場合に特有の効果を奏することを知見するに至った。すなわち、本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、基本的には水素ポンプの動作原理により、雰囲気中の水蒸気から化学式4〜7で示される欠陥平衡によってプロトンを取り込み、カソード側に水素を生成する。この生成された水素によってNOxが以下の化学式9,10に基づいて還元される。さらに、このNOxの還元反応は、デバイスに通電する電流量によって制御することができるので、NOxセンサーによるフィードバック制御により、よりきめ細かいNOx還元が実現できる。
【0066】
【化9】
【化10】
【0067】
例えば自動車の排気ガス中には水蒸気が存在し、走行中における排ガス浄化用触媒の温度は数百度にも達する。このような状況下におかれた自動車の排ガス浄化用電気化学デバイスは、排ガス中の水蒸気を分解し、排ガス中にNOx の還元剤である水素を供給し、化学式9,10に基づいてNOx を分解する。
【0068】
また、上述したボイラなど各種プラントにおける排ガスも多くが水蒸気を含有しているため、これら各種プラントにおけるアンモニアによる無触媒脱硝もしくは触媒を用いたNOx のアンモニア選択接触還元においても、ボイラの高温部に本発明に係る排ガス浄化用電気化学デバイスを配設することによって、排ガス中に水素を供給することが可能となり、還元剤としてのアンモニアを低減できる。
【発明の効果】
【0069】
以上のように、請求項1のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によると、AECeO3のプロトン導電性は維持しつつも、AECeO3のCO2に対する脆弱性を克服してCO2を含む混合ガス雰囲気環境下でも使用することが可能となる。
【0070】
また、請求項2のプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、希土類元素セリウム(Ce)の一部を当該セリウム(Ce4+)とは異なる他の低原子価希土類元素で置換したというもので、AECeO3のプロトン導電性は維持しつつもAECeO3のCO2に対する脆弱性を克服することができる。低原子価元素で置換することにより、例えば、Ce4+→Yb3+とすると、結晶格子中の電気的中性が崩れ、これを補うため格子中の酸素1/2個が抜ける。そうすると格子中の電気的中性が保たれる。このように意図的に酸化物イオン空孔を生成させること(ドーピング)ができ、よりプロトンの生成が促進されることになる。また、他の希土類元素としては、請求項3に記載のようにイッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素を用いることができる。
【0071】
水素透過膜が金属または合金である請求項4のプロトン導電性酸化物膜−水素透過膜複合膜型電解質の場合、当該金属膜自体をプロトン−電子混合導電体と見なすことができるため燃料極が不必要になるという利点がある。この場合、水素透過性を示す金属または合金としては、請求項5に記載のごとくパラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とするものを用いることができる。尚、請求項7に記載したように、これらの金属または合金を無電解めっき法によりプロトン導電性酸化物膜表面に成膜することで、より緻密かつ均一な膜が成膜可能となり、プロトン導電性酸化物膜−水素透過膜複合膜型電解質における複合界面の連続接合性が向上し、AECeO3のCO2に対する脆弱性を克服しつつ、アノード過電圧を低減することが可能となる。
【0072】
また、多孔質支持体が複合膜のガス側の表面に形成されている請求項7のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によると、水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となることから、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。請求項8のように複合膜のガス側とは反対側の表面に多孔質カソードが形成されている場合においても同様である。さらに、請求項9に記載したように多孔質カソードに銀(Ag)を含むことにより、カソード過電圧を低減することが可能となる。
【0073】
また、複合膜のガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜が形成されている請求項10のプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、プロトン導電性酸化物膜の両面に保護膜が形成された状態となっている。したがって、カソード側を流れるガス(酸化剤ガス)中にCO2が含まれていても、プロトン導電性酸化物膜(AECeO3)のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することが可能となる。別の水素透過膜が金属または合金である請求項11のプロトン導電性酸化物膜−水素透過膜複合膜型電解質の場合、当該金属膜自体をプロトン−電子混合導電体と見なすことができるため電極が不必要になるという利点がある。この場合、水素透過性を示す金属または合金としては、請求項11に記載のごとくパラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金をを成分とするものを用いることができる。尚、請求項13に記載したように、これらの金属または合金を無電解めっき法によりプロトン導電性酸化物膜表面に成膜することで、より緻密かつ均一な膜が成膜可能となり、プロトン導電性酸化物膜−水素透過膜複合膜型電解質における複合界面の連続接合性が向上し、AECeO3のCO2に対する脆弱性を克服することができる
【0074】
多孔質支持体が、複合膜のガス側とは反対側の表面に形成された多孔質カソードである請求項14のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によると、多孔質カソードが基材としても機能し、複合膜を強度的に支持する。したがってこの場合においても水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となることから、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。
【0075】
複合膜のガス側とは反対側の表面に多孔質カソードが形成され、さらに該多孔質カソードの表面に多孔質支持体が形成されている請求項15のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によっても、水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となることから、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。
【0076】
また、複合膜のガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜が形成され、さらに該別の水素透過膜の表面に多孔質支持体が形成されている請求項16のプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、プロトン導電性酸化物膜の両面に保護膜が形成された状態となっている。したがって、カソード側を流れるガス(酸化剤ガス)中にCO2が含まれていても、プロトン導電性酸化物膜(AECeO3)のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することが可能となる。
【0077】
また請求項17の発明によれば、燃料装置等の各種装置において、AECeO3のプロトン導電性は維持しつつもAECeO3のCO2に対する脆弱性を克服してCO2を含む混合ガス雰囲気環境下で使用することが可能となる。例えば、請求項18のように水素センサーに適用したり、請求項19のように水素ポンプに適用したり、請求項20のように水蒸気センサーに適用したりすることができる。あるいは、請求項21のように排ガス浄化用デバイスに適用することもできる。
【発明を実施するための最良の形態】
【0078】
以下、本発明を図面に示す実施の形態に基づいて詳細に説明する。
【0079】
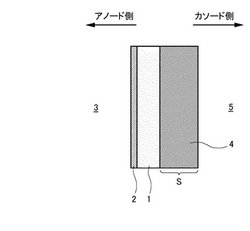
本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、図1に示すようにCO2を含むガス3中から水素を分離可能な水素透過性あるいはプロトン(H+)透過性の電解質であって、プロトン(H+)が通過可能なプロトン導電性酸化物膜1と、プロトン(H+)が通過可能な水素透過膜2との複合膜からなり、これら両膜の界面が連続接合した状態で、プロトン導電性酸化物膜1の少なくともガス3側の表面に水素透過膜2が形成されているものである。
【0080】
プロトン導電性酸化物膜1としては、バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造としたものを採用できる。
【0081】
Aサイト元素は、電解質抵抗低減の観点からバリウム(Ba)を採用することがより好ましい。
【0082】
また、酸化物イオン空格子点を発生させてプロトン導電性を発現させるために、Bサイトのセリウム(Ce)はイッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素で一部置換することが好ましいが、これらの元素に限定されるものではなく、セリウム(Ce)とは異なる他の低原子価希土類元素を採用することも可能である。
【0083】
尚、本発明のプロトン導電性酸化物膜のような固体電解質は、焼結性が低減するほど電解質抵抗は高くなる。かかる観点から、焼結性を高めることが好ましい。焼結性を高めるための手法としては、焼結温度を上昇させる、最終成形時に成形加重を増大する等が挙げられるがこれらに限定されるものではない。好ましい焼結度としては90%以上、さらに好ましくは95%以上である。
【0084】
水素透過膜2は、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な膜であり、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とすることが好ましいが、これらに限定されるものではなく、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な材料であれば、金属もしくは合金等に限らず、酸化物等の材料も採用することが可能である。尚、水素脆化を抑える効果のあるAgと合金化したものがより好適である。
【0085】
ここで、金属もしくは合金膜を水素透過膜として採用する場合には、それ自体がプロトン−電子混合導電体とみなせるため電極として作用する。したがって、燃料極を必要としなくなるという観点から金属もしくは合金膜を採用することが好ましい。尚、酸化物等の材料を用いる場合には、燃料極を併用すれば良く、また、金属もしくは合金膜を用いた水素透過膜が薄いために電極としてうまく作用しない場合にも燃料極を併用すればよい。
【0086】
燃料極としては、燃料ガスを透過させ、かつ電極として作用する材料が好ましく、例えば多孔質金属、格子状に形成した電極もしくはガスを透過させるための穴を複数あけた電極等を採用することができる。水素透過膜が酸化膜の場合には酸化膜をガス側に配置し、燃料極をプロトン導電性酸化物に接触させて用いるようにすればよい。また、水素透過膜が金属もしくは合金の場合には、燃料極をプロトン導電性酸化物に接触させて用いるようにしてもよいし、水素透過膜をプロトン導電性酸化物に接触させ、ガス側で燃料極を水素透過膜に接触させるようにしても良い。
【0087】
次に、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質においては、アノード過電圧を低減させるために、複合界面が連続接合させることを特徴とするものである。
【0088】
複合界面の連続接合は、プロトン導電性酸化物膜を基材として緻密膜な水素透過膜を形成するか、もしくは水素透過膜を基材として緻密膜なプロトン導電性酸化物膜を形成することにより達成される。本発明では、プロトン導電性酸化物膜を基材として緻密膜な水素透過膜を形成している。
【0089】
緻密膜を形成するための方法としてペースト法が挙げられる。この場合には、ペースト材料の粒子径が大きいため、ペーストを基板に塗布した後に高温処理することで、基材との密着性が向上し、アノード過電圧を低減することが可能となる。
【0090】
ペースト剤には、例えば、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)のうち1つもしくは2つ以上の元素を粒子状にしたものを含ませて、良好な水素透過性を示す膜を形成することが好ましい。
尚、水素脆化を抑える効果のある銀(Ag)を含ませることがより好ましい。
【0091】
また、ペースト法を用いる場合、金属レジネートをペースト剤に添加して基材との密着性を向上させることが好ましい。これにより基板とペースト法により形成した膜が剥離するのを防ぐことが可能となる。
【0092】
金属レジネートは、金属樹脂酸塩であり、金属レジネートを含まない金属を直接熱処理する場合と比べて、樹脂が熱処理中に燃えることにより細かい金属が発生するようになる。この細かい金属がセラミックスへミクロ的に食い込む、すなわち、セラミックスへのアンカー効果を期待して配合されるものであり、例えば、硫化テレピネオールPdを挙げることができるが、これに限定されるものではない。
【0093】
基板に金属レジネートを含むペースト剤を塗布した後、熱処理をおこなうとポア(穴)が発生する。このポアは、CO2を通過させてしまい、CO2に対して脆弱な本発明のプロトン導電性酸化物膜をCO2に曝すことになるため好ましくない。そこで、当該ポアを埋めるために、金属レジネートを含まないペーストをさらに重ね塗りして前記熱処理温度よりも高温で熱処理して水素透過膜をより緻密にすることが好ましい。
【0094】
Pd-Ag合金膜を形成する場合を例に挙げて説明すると以下のようになる。まず金属レジネートを含むペーストを基板に塗布し、200℃程度でペースト剤に含まれるバインダーを焼失させる。次いで、熱処理を行う。熱処理温度は900℃程度ではポアが無数に発生し、熱処理温度を上昇させるにつれてポア数は減少する。ただし、1300℃以上ではPd-Agが焼結し始めてポア径が増加する。よって、熱処理温度をxとすると、900℃<x<1300℃であればよいが、1100℃<x<1300℃であることが好ましく、最も好ましくはx≒1200℃である。
【0095】
上記の最も好ましい熱処理温度である1200℃で熱処理を施してもポアは発生する。そこで、当該ポアを埋めるため、ペーストを重ね塗りする。ここで、重ね塗りをおこなうためのペーストとしてはポアが発生しにくい金属レジネートを含まないペーストが好ましい。これを一層目の膜を熱処理した温度よりも高温の1300℃程度で熱処理することにより、ポアが焼失し、かつ緻密な膜を形成することが可能となる。
【0096】
次に、さらに緻密な膜を形成して複合界面の連続接合状態をより良好なものとするための好ましい手段としては、物理堆積法(PVD法)、化学堆積法(CVD法)、放電重合法、ゾル−ゲル法、電解めっき法、無電解めっき法、スクリーン法等が挙げられるが、コスト面、成膜簡易性および成膜均一性の観点から、無電解めっき法を採用することが最も好ましい。
【0097】
ここで、プロトン導電性酸化物膜はプロトン導電性は有するものの、電子導電性は有しないため、めっきされにくい。そこで無電解めっき処理を施す前に基板にめっき反応に対する活性を付与する必要がある。非導電性基板に無電解めっきを施す方法としては、湿式法による代表的な活性触媒化前処理である二液法(センシタイゼーション−アクチベーション法)を採用して、基板にめっき反応に対する活性を付与する触媒活性化前処理を行うようにすればよいが、めっき反応に対する活性を付与できるならば、この方法に限られるものではない。
【0098】
ここで、無電解めっき法の採用にあたって、プロトン導電性酸化物膜上に膜形成するためにめっき浴を準備する必要がある。当該めっき浴中には、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)、銀(Ag)等を含ませることで、目的とする水素透過膜の構成成分がめっきされて膜形成が可能となる。
【0099】
例として、Pd-Ag合金を無電解めっき法により形成する方法について説明する。プロトン導電性酸化物膜を基板としてめっき反応に対する活性を付与する。具体的にはSnCl2塩酸溶液とPdCl2塩酸溶液それぞれの溶液に基板を交互に浸漬し、Pd核を発生させる。次に、Pdめっき浴として、[Pd(NH3)4]Cl2・H2Oを含むアルカリ性溶液中にめっき反応に対する活性を付与した基板を浸漬し、Pd核を触媒としてその周囲にPdを析出させる。次に、Agめっき浴として[Pd(NH3)4]Cl2、AgNO3を含む溶液中にPdを析出させた基板を浸漬し、Agを析出させる。最後に、N2雰囲気にて熱処理することでプロトン導電性酸化物膜1と非常に良好に連続接合したPd-Ag緻密合金膜を得ることができる。
【0100】
次に、プロトン導電性酸化物膜を薄膜化することがコスト低減の観点から好ましい。プロトン導電性酸化物薄膜を成膜する方法としてはスラリーコート法、スピンコーティング法、ディップ法、物理堆積法(PVD法)が挙げられるが、薄膜を形成することが可能であればこれらに限定されるものではない。尚、プロトン導電性酸化物膜はコストな問題を考えた場合、その厚さを50μm以下にするのがよく、35μm以下にするのがより好ましく、20μm以下にするのがさらに好ましい。ただし、プロトン導電性酸化物膜をここまで薄膜化すると電解質自立型構造をとることが難しくなるので、多孔質支持型構造とすることが好ましい。具体的には、多孔質支持体にプロトン導電性酸化物薄膜を成膜し、さらに水素透過薄膜を形成する。これにより低コストな電気化学デバイスの作製が可能となる。
【0101】
尚、コスト面を考えた場合の水素透過膜の好適な膜厚は、1μm以下、より好ましくは0.5μm以下、さらに好ましくは0.1μm以下である。
【0102】
以上、プロトン導電性酸化物膜1と水素透過膜2とを複合膜化した電解質の最も好適な組み合わせは以下のようになる。すなわち、プロトン導電性酸化物膜1としてAサイトをバリウム(Ba)とし、Bサイトのセリウム(Ce)をイッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素で一部置換したものを用い、水素透過膜2として無電解めっき法により形成したパラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とする膜を用いることが最も好適である。
【0103】
このように、プロトン導電性酸化物膜1と水素透過膜2とを複合膜化した電解質においては、水素のみが(具体的にはプロトンの形で)Pd膜を透過しAECeO3膜に達する。このため、AECeO3のCO2に対する脆弱性を克服でき、CO2を含むガス3の環境下でも使用できる電気化学デバイスの電解質として使用可能となる。このようなプロトン導電性酸化物膜−水素透過膜複合膜型電解質は電気化学デバイスに適用して好適なものであり、一例を挙げれば、自動車排ガスのような水蒸気およびNOx を含有する排ガスを浄化するための排ガス浄化用デバイスとして適用することができる。こうした場合には、排ガス中の水蒸気を分解し、排ガス中にNOx の還元剤である水素を供給して当該NOx を分解することができる。
【0104】
さらに、プロトン導電性酸化物膜−水素透過膜複合膜型電解質におけるカソード電極としては、銀(Ag)を用いることが好ましい。従来、カソード電極としては高コスト材料である白金(Pt)が用いられてきたが、白金(Pt)よりも銀(Ag)を用いた方が、コストを低減でき、カソード過電圧も低減できる点で好ましい。銀(Ag)の成膜法としてはペースト法等が挙げられるが、これらに限定されるものではない。
【0105】
また、多孔質カソードをペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物単体もしくはペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物と銀(Ag)のサーメットとするのが好ましい。この場合にも白金(Pt)に比べコストを低減できる。ペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物としては、例えばLaMnO3やSrMnO3が挙げられるが、これらに限定されるものではない。プロトン導電性酸化物膜にこれらを良好に接触させるためにはプロトン導電性酸化物膜に対して電子−酸化物イオン混合導電性酸化物を押しつけながら熱処理を施すか、もしくは単にこれらの膜同士を接触させて熱処理すればよいがこれらの方法に限定されるものではない。
【0106】
上述した好適例にさらにカソード電極として銀(Ag)、ペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物単体もしくはペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物と銀(Ag)のサーメットを用いることで、電気化学デバイスとして非常に優れた構成となる。
【0107】
また、本実施形態における複合膜は多孔質支持体Sによって強度的に支持され、これによって薄膜化することが可能となっている。多孔質支持体Sは、例えば多孔質Niによって構成される。このように多孔質支持体Sによって複合膜を支持する場合の具体的な形態を以下に説明する(図30〜図34参照)。
【0108】
まず、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体Sが形成されているというものである(図30参照)。この複合膜のアノードガス側と反対側の表面には多孔質カソード(図30〜図34中において符号4で示す)が形成されている(図30参照)。ちなみに、本実施形態における多孔質カソード4の厚さは40μmであるがこれは一例に過ぎずこの厚さに限定されるわけではない。また、本実施形態においては多孔質支持体Sと多孔質カソード4とが別個に設けられているが(図30参照)、多孔質カソード4自体に多孔質支持体としての機能を備えさせることも可能である(図32参照)。多孔質支持体Sはガスの拡散を妨げることなく、0.002〜3μm、好ましくは0.004〜0.5μmの最高を有するものを適宜選択して使用することが可能である。具体的には、例えば、0.1〜0.5μmの細孔を有するNi-YSZサーメット多孔体またはNi-Al2O3サーメット多孔体などを例示できる。これにより、水素透過膜(例えばPd金属)2の薄膜化を可能とするため、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。また、600℃以下の低温になれば、アノード側に設けられる多孔質支持体Sには金属NiまたはNi合金、カソード側に設けられる多孔質支持体SにはSUS316L、SUS310SなどのSUS材が使用可能となる。
【0109】
また、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体Sを形成し、尚かつアノードガスとは反対側の面にプロトン(H+)が通過可能な別の水素透過膜2’を形成することもできる(図31参照)。多孔質支持体Sは水素透過を妨げることなく水素透過膜(例えばPd金属)2,2’の薄膜化を可能とする。また、カソード側を流れるガス(酸化剤ガス)5中にCO2が含まれている場合にも、プロトン導電性酸化物膜1を覆う水素透過膜2’がこのプロトン導電性酸化物膜1のCO2に対する脆弱性を克服する。
【0110】
図32に示すように、複合膜のアノードガス側とは反対側の表面に形成された多孔質カソード4を多孔質支持体Sとして機能させることもできる(図32参照)。この場合、多孔質カソード4が基材としても機能し、複合膜を強度的に支持するため、水素透過を妨げることなくPd等の水素透過膜2を薄膜化することが可能となる。
【0111】
図33に示すように、複合膜のうちアノードガス側とは反対側の表面に多孔質カソード4を形成し、さらにこの多孔質カソード4のカソード側表面に多孔質支持体Sを形成することもできる(図33参照)。この場合にも、水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となる。
【0112】
図34に示すように、複合膜のアノードガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜2’を形成し、さらにこの水素透過膜2’のカソード側表面に多孔質支持体Sを形成することもできる(図34参照)。この場合、プロトン導電性酸化物膜1の両面に保護膜が形成された状態となっている。したがって、カソード側を流れるガス(酸化剤ガス)5中にCO2が含まれていても、プロトン導電性酸化物膜(AECeO3)1のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することが可能となる。
【0113】
以上が本発明の好適な実施形態であるが、ここで説明したのは好適な実施の一例に過ぎず、これに限定されることなく本発明の要旨を逸脱しない範囲において種々変形実施が可能である。例えば、上述したような排ガス浄化用デバイスに限らず、燃料電池、水素センサー、水素ポンプ、水蒸気センサーなどの電気化学デバイスに適用して好適なものである。
【0114】
なお、本明細書では「プロトン導電性酸化物膜−水素透過膜複合膜型電解質」を中心に説明しているが、ここでいう「複合膜型」としては、物理的に接合されている場合と化学的に接合されている場合の両方が考えられる。仮に化学的な接合であると考えると、両膜の間に中間生成物が存在しこれがプロトン移動の高抵抗層になる可能性がある一方で、中間生成物が逆に高プロトン導電層になる可能性も存在する。以上を考慮すると、化学的接合の場合にはプロトン移動の高抵抗層が存在しないことが条件になる。
【0115】
また、本発明を例えば排ガス浄化用電気化学デバイスに適用する場合にあっては、以下に述べるPd/SCO複合膜(SCOはSrCe0.95Yb0.05O3-aのことであり、以下単にSCOと呼ぶ)が、Pd薄膜によって両側が覆われる構造を有するPd/SCO/Pd電解質膜であることが望ましい。電気化学デバイスを排ガス中で使用する際にはアノードおよびカソードともに排ガス中に曝されているので、カソード側もPd電解質膜で覆うことによってこちら側がCO2と反応して分解してしまうことが防止できる。
【実施例】
【0116】
続いて、以下では実際に行った実験および検討の具体的内容を実施例として示しつつ、まず金属/酸化物複合膜型電解質の概念について説明し、その後、本発明にかかるAECeO3のCO2に対する脆弱性克服の確認をするための実験内容を説明していく。
【0117】
[実施例1]
本発明者は、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質の有効性を検証するため、Pd/AECeO3複合膜を成膜し、CO2を含む燃料を用いる燃料電池での発電の可能性を確認した。これは、主として、「CO2に対する脆弱性の克服の確認」や「電気化学デバイスのセル構造」について検証するとともに、水素センサー、水素ポンプ、燃料電池といった各種電気化学デバイスへの適用を検討したものである。
【0118】
なお、本実施例においては、固相である金属(Pd)と固相である酸化物(AECeO3)との界面(固/固界面)を連続とし、この固/固界面を、固相と液相との界面(固/液界面)と同様の接合状態とすることを念頭においた。これは、マクロレベルでは両者に差異はないもののミクロレベルにおいて比較すると大きな隔たりが生じること、より具体的には、電解質に液相が使用されている場合には固/液界面においてプロトンが連続移動できる一方で、固/固界面においては図29に示すように不連続面が生じ、
・界面に生じるミクロな空間が電気化学的抵抗となって性能が低下する
・界面では極度な還元雰囲気となり、酸化物が還元されて材料が劣化する
といったことが生じるおそれがあることによる。つまり、固/固界面においては特にプロトンの連続した導電が重要であり、固/液界面と同様の接合状態を実現する必要がある。ちなみに、固/液界面では、液体を保持する容器として金属Pd膜が必要とされているのに対し、固/固界面ではセラミックのCO2耐性を金属Pd膜で覆い確保するという点で差異がある。
【0119】
以下では、
(1)金属/酸化物複合膜型電解質の作製
(2)CO2雰囲気連続暴露試験による複合膜のCO2耐性の確認
(3)Pd/SCO複合膜のプロトン導電性
(4)Pd/SCO複合膜を用いた燃料電池発電
という項目に分け、この項目に沿って説明する。
【0120】
(1)金属/酸化物複合膜型電解質の作製
(1.1)水素透過膜2の検討
水素透過膜2として必要な水素透過能力をもつ金属材料のほとんどは、水素雰囲気下では自己崩壊を起こすことが知られている。これらの金属は、水素の圧力と温度に応じて多量の水素を吸収し、相変態する性質がある。相変態は、金属格子を拡張するため金属内に歪みが生じ、塑性変形(水素脆化)する。水素透過膜2として、注目されるPd膜も例外ではなく、水素脆化による膜の寿命が問題となる。しかし、Pdは、Agが置換型固溶し合金化した状態では、Agが水素原子と相互作用し、相変態を緩和するとされている。また、Agが適量固溶すると水素の拡散係数は減少するが、水素の溶解度は増加し、合金化により水素透過係数が増加する。このため、近年、燃料電池や半導体産業など、高純度水素を必要とする分野において、Pd−Ag合金膜を利用した水素分離膜の研究が盛んに行われている。そこで本発明者は、酸化物の複合材として上記のPd−Ag合金に着目した。
【0121】
(1.2)ペースト組成
本実施例では、Pd−Ag合金膜をペースト法にて作製した。Pd−Agペーストは、表1に示す性状のPdおよびAg金属粉末、溶剤(αテレピネオール)、バインダー(エチルセルロース、および酸化物とのアンカー効果を期待して金属レジネート:硫化テレピネオールPd)を適量混練し作製した。なお、金属レジネートは、金属樹脂酸塩であり、金属レジネートを含まないPdを直接熱処理する場合と比べて、樹脂が熱処理中に燃えて細かいPd金属が発生するようになる。この細かいPd金属がセラミックスへミクロ的に食い込む、すなわち、セラミックスへのアンカー効果を期待して配合したものである。Pd/Ag混合比(wt%)は、水素透過能が最も優れると報告されているPd/Ag=77/23を仕込み組成とし、金属レジネートを含むペースト(AR)および金属レジネートを含まないペースト(AN)の2種類を作製した。
【0122】
【表1】
【0123】
(1.3)アルミナ基板上への成膜
熱処理温度を決定するために、アルミナ基板上にPd−Agの成膜を試みた。アルミナ基板にペースト(AR)を塗布し、雰囲気炉にて、200℃、空気中でバインダーを焼失させた後、900, 1000, 1100, 1200, 1300℃でアニール処理を1時間施し、熱処理温度を比較した。その結果を図6に示す。900℃では、膜表面に無数の穴が空いているが、アニール温度を上昇すると徐々にその数が減少した。しかし、1300℃以上になると逆に穴の径が大きくなった。これは、Pd−Agが焼結し始めたことを意味する。また、金属レジネートを添加しないペースト(AN)を使用したところ、1200℃以下では、膜の剥離が観察された。金属レジネートを添加したペーストを使用した場合には膜の剥離は起こらなかったことから、金属レジネートを用いることでアンカー効果を有するようになることが確認された。さらに、金属レジネートを添加しないペースト(AN)を使用して1300℃で熱処理した場合には剥離はなく、また、ARほどポア数は多くならなかった。
【0124】
ARでは、熱処理温度1200℃において、最もポア数が少ない結果となったが、依然として、ポアが観察され、完全な緻密膜ではなかった。そこで、残ったポアを埋めつつ、より高温で熱処理して膜の緻密化を図るため、成膜した前述の膜の上に1300℃でも熱処理しても金属レジネートを添加したペースト(AR)に比べてポアの発生量が少ない金属レジネートを添加しないペースト(AN)を重ね塗りし、1300℃でアニール処理を施した。図7にその結果を示す。重ね塗り処理により、ほとんどのポアが消失した。以上より、金属レジネートを添加したペースト(AR)をアルミナ基板上に塗布して1200℃で熱処理しPd−Ag膜を形成した後に、金属レジネートを添加しないペースト(AN)を当該Pd−Ag膜上に重ね塗りして1300℃で熱処理を施す成膜法が成膜条件として好ましいと判断した。
【0125】
(1.4)SrCeO3基板上への成膜
次にAECeO3基板上へのPd膜の成膜を試みた。AECeO3基板は、まず入手が容易なTYK社製のSrCe0.95Yb0.05O3-a(SCO)のうち、φ16.8mm、厚さ1.0μm、相対密度95%以上のものの緻密体を選定した。成膜条件には、前節(1.3)のアルミナ上への成膜条件を用いた。結果を図8に示す。SCO基板においても、アルミナ基板と同様の成膜条件、すなわち、金属レジネートを添加したペースト(AR)をSCO基板上に塗布して1200℃で熱処理しPd−Ag膜を形成した後に、金属レジネートを添加しないペースト(AN)を当該Pd−Ag膜上に重ね塗りして1300℃で熱処理することで、緻密かつほとんどのポアが消失した膜の形成が可能であることが確認された。
【0126】
(1.5)Pd/SCO複合膜の密着性
Pd-Ag膜とSCO基板との密着性を確認するため、試料断面を鏡面仕上げして光学顕微鏡によりその断面を観察(観察倍率×500)した。結果を図9に示す。Pd-Ag膜は、SCO基板に非常に良く密着しており、引っ掻き試験においてもPd膜が剥がれないことを確認した。更に、SCO基板が相対密度約95%以上の緻密膜でありながらクローズドポアを含有していることも確認された。一方、Pd膜にはほとんどポアはなく、非常に緻密な膜として成膜していることが確認できた。膜厚は、約65μmであった。
【0127】
(1.6)EPMAによる複合膜の断面観察
膜の元素分析をEPMA(Electron Probe MicroAnalyzer: JEOL, JXA-8900R)によって行った。EPMAによるPd、Ag、Ce、Srの特性X線像を図10に示す。各膜成分元素の拡散は確認されず、成膜した膜は、化学拡散を伴わずに物理的に良く密着して複合化していることがわかった。また、1300℃という高温での熱処理において元素相互の拡散がなかったことは、Pd-Ag相とSCO相との間に化学的反応が全くないことを示した結果とも言える。したがって、Pd/SCO複合膜は、Pd-Ag膜およびSCO基板各々の物性を維持して複合していると考えられる。更に、Pd-Ag膜は、ペースト作製仕込み初期には、それぞれPd粒子、Ag粒子であったが、Pd, Ag元素の偏在化なしに、均一に合金化していることが確認できた。
【0128】
次に、EDX解析より、Pd-Ag膜組成の分析を行った。図11に、標準試料を用いたEDX測定(Energy Dispersive X-ray Analysis:エネルギー分散型X線解析の略)の実測値による組成補正を行った結果を示す。仕込み組成はPd/Ag=77/23であったのに対して、成膜後の組成はPd/Ag=84.9/15.1となり、Ag成分がかなり減少した結果となった。これはアニール処理温度が大きく影響していると考えられる。Pd-Ag二元系相図を図12に示す。PdとAgは互いに同じ立方晶の結晶構造を持ち、全率固溶体を形成することが知られている。Agの融点(962℃)は、Pd(約1555℃)と比較してかなり低く、アニール処理昇温中に、まずAg粒子が最初に溶解すると考えられる。EPMAの結果から、SCO膜内へのAgの拡散は確認できなかったが、Pd-Ag膜ではPd元素に関しては均一に分布しているが、Ag元素に関しては、表面からSCO膜界面へ僅かに濃度分布が確認された。このことは溶解したAg粒子が膜表面より昇温とともに蒸発するため、表面側に向かうにつれて僅かにAg濃度が低くなり、最終的にPdと合金化するAgの濃度分布が生じたものと考えられ、トータルのAg量も減少したものと考えられる。
【0129】
(2)CO2雰囲気連続暴露試験による複合膜のCO2耐性の確認
作製した複合膜のCO2に対する耐性を確認するために、CO2雰囲気における連続暴露試験を行った。
【0130】
(2.1)実験方法
試験に供した試料は、2種類(SCO基板、Pd/SCO複合膜基板)である。Pd/SCO基板は、上述した(1.1)〜(1.6)における成膜条件、すなわち、SCO基板全体に金属レジネートを添加したペースト(AR)を塗布して1200℃で熱処理しPd−Ag膜を形成した後に、金属レジネートを添加しないペースト(AN)を当該Pd−Ag膜上に重ね塗りして1300℃で熱処理することで、SCO相の露出面をなくした。双方の試料をアルミナフェルトの上に置き、管状炉内にセットした。CO2耐性試験は、管状炉内を真空にした後、CO2雰囲気で行った。CO2暴露時間は10時間とし、温度は650℃で一定とした。
【0131】
(2.2)XRDによる複合膜の表面組成解析
CO2曝露試験後のXRD解析結果を図13に示す。Pd/SCO板は、SCO層の分析を行うために表面のPd膜を剥がした。熱処理前のSCO基板のピークと比較して、CO2暴露後のSCO基板のピークでは、ほぼCeO2およびSrCO3に帰属する回折ピークを示し、SCO相が完全に分解したことを確認した。一方、Pd/SCO基板では、ほぼSCO相のみに帰属する回折ピークを示しており、Pd膜との複合化により、SCO相のCO2に対する脆弱性が克服されることを明らかとした。
【0132】
(3)Pd/SCO複合膜のプロトン導電性
(3.1)実験方法
Pd/SCO複合膜のプロトン導電性を確認するために、(1)水素濃淡電池法により得られる起電力から、導電種がイオンであるか否かを判断し、(2)水素の電気化学的透過法により、直接、プロトンを導電種として同定した。以下に、上記2種類の測定法を説明する。
【0133】
(3.1.1)水素濃淡電池法
プロトン導電体を挟む両側に水素の活量差(濃度差)が生じると、その活量差に応じた起電力を示す。水素の濃淡差を用いた水素濃淡電池の作動原理を図14に示す。プロトン導電性固体を電解質隔壁とし、その両面に多孔質電極を取り付けて二つの電極室とし、一方に高水素分圧のガスを、もう一方に低水素分圧のガスを導入すると、高水素分圧側を負極として起電力が発生する。このときの電池構成は、式(化学式)11のように表され、負極(アノード)と正極(カソード)で化学式12,13のような電極反応が生じようとして、ネルンストの式(化学式14)から導出される起電力E(V)が発生する。
【0134】
【化11】
【化12】
【化13】
【化14】
ここで、Rは気体定数(8.314JK-1mol-1)、Tは絶対温度(K)、Fはファラデー定数(96485Cmol-1)、PH2(1)とPH2(2)は、アノードおよびカソードの水素分圧である。化学式14において一方の水素分圧(例えばPH2(1))を固定すれば、図15に示すように、起電力はもう一方の水素分圧(この場合、PH2(2))の対数に対して直線関係となる。また、測定した起電力Eを化学式14より求めた理論起電力E0で除した値は、その条件におけるイオン輸率とみなすことができる。尚、プロトン導電体を挟む両側に水素の活量差(濃度差)が生じると、その活量差に応じた起電力を示すことを利用して、これを水素センサーに適用することが可能である。
【0135】
(3.1.2)水素の電気化学的透過法
水素の電気化学的透過法(水素ポンプ)の作動原理を図16に示す。プロトン導電性固体を隔壁として一方に水素源を導入し、他方に不活性ガス、例えばアルゴンなどを導入する。アルゴン側をカソードとして直流を通電する時、もしプロトン導電体であれば、それぞれの極で化学式12,13のような電極反応が起きる。この場合、プロトン以外のイオンは、この隔壁内を通過できないので、水素のみを透過(ポンプ)することができる。このように水素ポンプは、電気化学反応を利用して、強制的に水素を透過させるために、供した電解質のプロトン導電性を直接確認することができる。更に、カソード出口ガス分析から得られる水素濃度から、水素透過速度を計算した後、ファラデーの法則から計算される理論水素透過速度と比較すれば、プロトン輸率(全導電種に占めるプロトンの占める割合)が計算できる。
【0136】
(3.2)Pd/SCO複合膜を用いた水素濃淡電池
作製した電解質膜は、図17、図18に示すようにSrCe0.95Yb0.05O3-a(厚さ1mm、φ16.8mm)基板5に、片側にはPd-Ag膜6(φ13mm、厚さ65μm)が成膜されている。他方には中央部に円状(φ12mm)に白金ペーストを塗布し、900℃において30分空気中で焼き付け多孔質電極Pt7(φ12mm)として試験に供した。実験に使用した装置概略を図19に示す。両面の電極部分に集電体として白金ネットを取り付けた試料を、パイレックス(登録商標)ガラスリングを介してφ17×13mmのアルミナ管で上下から押さえつけた。それぞれのガス室は、1000℃まで昇温することによりパイレックス(登録商標)ガラスを軟化させてガスタイト(ガスシールが完全に効いている状態)にした。アノード6およびカソード7とも、膜流量計を用いて入出口の流量に差がないことで、ガスタイトであることを確認した。その後、約30分間空気を導入して電極の状態を安定させた後、所定のガスに切り替えて実験を開始した。図19において、ガス流量はMFC8により設定し、加湿器9を経由してアノード6およびカソード7それぞれに目的のガスが供給され、アノード排ガス10、カソード排ガス11が排出されるようにしている。ここで、符号5は電解質を表している。カソード排ガス11はマイクロガスクロマトグラフィー装置13により、排ガス組成を測定可能となるよう配置している。尚、反応温度は電気炉12により調整するようにしている。ガスは100ml/minの流量で導入した。アノード6とカソード7間に発生する起電力は、エレクトロメータ(北斗電工製 HE-106)を用いて測定した。ガスは、室温加湿(3%)した100%H2を、アノード6となるPd膜側に導入し、カソード7にはそれぞれ1,5,10,20,50,100%H2をArベースの混合ガスとして導入した。測定前にカソード入口ガスをマイクロガスクロマトグラフィー(HP社製 M-200、モルキュラシーブカラム:MS-20)を用いて、目的ガス組成になっていることを確認した。
【0137】
650,750,850,950℃において、それぞれのガス組成における起電力を測定した結果を図20に示す。測定条件範囲内では、ネルンストの式(化学式14)から計算される理論値にほぼ一致した。この結果から、Pd/SCO複合膜の導電種はイオンであることが確認された。
【0138】
(3.3)Pd/SCO複合膜を用いた水素の電気化学的透過実験
水素濃淡電池法においては、導電種がイオンであるか否かは判断できるものの、プロトンが導電するか否かを直接判断することはできない。そこで、アノード、カソード間に直流を通電し、カソード側に水素が発生するか否かを直接確認した(水素ポンプ)。上述の場合、Pd/SCO複合膜(図17、図18参照)を挟んでPd-Ag膜側に水素を、他方にアルゴンを、双方とも室温でバブリングし加湿して、100ml/minで流通した。装置は、上述の実験装置(図19参照)を使用した。直流通電にはソーラートロン社製ポテンショガルバノスタットSI1286 を使用し、アノード−カソード間に定電流を流した。その際、カソード出口ではマイクロガスクロマトグラフィー(HP社製 M-200、モルキュラシーブカラム:MS-20)を用いて発生水素量を定量した。測定は、650, 750, 850, 950℃において実施した。
【0139】
通電電流に対する水素の発生速度を図21に示す。破線はファラデーの法則に従って計算した理論水素発生速度(ml/min)である。650℃では、ほぼ理論値通りの水素発生速度を示し、Pd/SCO複合膜はプロトン輸率tH+が1に極めて近いプロトン導電体であることがわかった。しかし、温度が上昇するに従い、高電流において破線からはずれ、水素発生速度が低下した。低電流ではほぼプロトン輸率が1であるが、高電流ではプロトン以外の電荷担体が存在することになる。この点については、電流量を増やすとカソード中の水蒸気が分解して化学式15のように酸化物イオン導電性が発現する旨の報告もなされている(S.Hmajima, H.Matsumoto, H.Iwahara, 第26回固体イオニクス討論会予稿集P154, 2000)。この報告によれば、高電流ではプロトン輸率が低下し、電流効率が低下した可能性がある。
【0140】
【化15】
【0141】
したがって、Pd/SCO複合膜は、ほぼSCO膜と同様のプロトン導電機能を有すると考えられる。以上より、SCO膜は、Pdと複合化することによって、プロトン導電機能を損なうことなく、SCOと同様のプロトン導電機能を有することが明らかとなった。
【0142】
(4)Pd/SCO複合膜を用いた燃料電池発電
(4.1)燃料電池の発電原理
プロトン導電体を用いる水素酸素燃料電池の原理を説明する。作動原理を図22に示す。今、化学式16のような水素と酸素の燃焼反応を考えてみる。
【0143】
【化16】
水素の燃焼反応は、水素の酸素による酸化反応であり、通常、熱エネルギーを放出する。この燃焼反応は酸素の立場から見ると、燃焼による酸素の還元反応である。従って、この両反応が同一の場で起こる時に熱エネルギーが放出される。そこで、例えば図22に示すように、プロトン導電性電解質を介して水素をアノード活物質として酸化反応を行わせ、酸素を正極(カソード)活物質として還元反応を行わせることができれば、水素の燃焼反応の化学エネルギーを、乾電池と同様の原理で直接電気エネルギーに変換することができる(化学式17,18)。
【0144】
【化17】
【化18】
【0145】
この場合、電極間の起電力Eは、水素の酸化反応より電子が放出されるアノードと酸素の還元反応により電子が不足するカソードとの間の電位差として発生する。水素酸素燃料電池の場合、起電力Eは以下の式によって表すことができる。
【0146】
【化19】
ここで、Kは化学式16の平衡定数を、piは各ガス分圧を、添字a、cはそれぞれアノードおよびカソードを、R、FおよびTはそれぞれ気体定数、ファラデー定数および絶対温度を表す。
【0147】
(4.2)実験方法
実験で用いた装置構成を図23に示す。図23の基本構成は図19と同様であるが、アノード側に供給できるガスがH2ではなくO2である点で異なっている。電解質には、(3.2)と同様の試料(図17、図18参照)を使用した。試料電解質を電解質隔壁として、Pd-Ag膜側をアノード、多孔質白金側をカソードとしてアルミナ管で挟み込んだ。アノードには燃料ガスとして室温飽和した湿潤水素または水素と二酸化炭素の湿潤混合ガスを、カソードには酸化剤ガス5として室温飽和した湿潤酸素をそれぞれ100ml/min流通した。このように燃料電池を構成することによって、ネルンストの式(化学式19)から計算される起電力を発生するようになる。発生した端子電圧はエレクトロメータ(北斗電工製 HE-106)を用いて測定した。外部負荷装置としてソーラートロン社製ポテンショガルバノスタットSI1286 をアノードとカソードとの間に接続し発電性能を確認した。
【0148】
(4.3)Pd/SCO複合膜を用いた燃料電池の特性
測定温度が650,750,850,950℃における水素−酸素燃料電池の起電力を図24に示す。同時にネルンストの式(化学式19)より計算される理論起電力を同図に示した。起電力は、650℃でほぼ理論起電力に近い値を示した。試験前には、必ずガスタイトとクロスリークがないことを確認しているので、起電力の実測値と理論値の比は、イオン輸率(実測値/理論値):tion(0<tion<1)と表され、逆に電子輸率(この場合、ホール伝導)はth(=1−tion)として表される。起電力から計算されるtionおよびthを図25に示す。tionは、950℃において0.88であったが、650℃において0.99となった。この結果、今回作製したPd/SCO複合膜は、650℃よりも低温の燃料電池雰囲気条件において、プロトン輸率が1の純粋なプロトン導電体として機能することがわかった。
【0149】
次に、実際に電流を取り出し発電を試みた。結果を図26に示す。発電時に安定な電圧および電流を示し、本発明に係るPd-Ag膜とSCO膜との複合膜型電解質を用いて、燃料電池発電が可能であることがわかった。
【0150】
(4.4)CO2を含んだ燃料ガスを用いた長時間発電特性
前節(4.3)までは、Pd/SCO複合膜を用いて発電の可能性を確認したが、本来の目的は、CO2を含むアノードガス条件における発電の可能性を確認することである。これを受け、以下に説明するように、CO2を含むアノードガスを導入した場合、Pd膜によってSCO膜のCO2による変質がなく、安定に燃料電池発電ができることを確認することにした。
【0151】
図27にCO2とH2の混合ガスをアノードに導入した場合の発電時の電圧経時変化を示す。電圧は、初期にはしばらく減少した後、一定となり安定した。また、連続負荷試験を行ったところ、少なくともCO2曝露試験と同じ10日間は安定して発電できることが確認できた。以上の結果より、本発明に係るプロトン導電性を維持し、かつCO2に対する脆弱性を克服できる金属/酸化物複合膜型電解質(Pd/SCO)は、CO2を含む燃料を用いる燃料電池に適用可能であることが明らかとなった。
【0152】
[実施例2]
実施例1では、Pd/SCO膜を燃料電池の電解質に用いた場合の発電の可能性に検討ついて検討し、理論通り実証することができた。しかし、実際に燃料電池の電解質として適用するには、動作する温度において種々の検討を加えなければならない。そこで、以下に実施例2として、金属/酸化物複合膜型電解質を用いた中温形燃料電池に対して、性能面および電解質の材料コスト面から金属/酸化物複合膜型電解質についてさらに検討した。
【0153】
燃料電池発電試験は、[実施例1]と同様の手順で行った。尚、実施例2では、SCOよりも導電率が高く、コスト低減につながるBaCe0.8Y0.2O3-a(以下BCOと呼ぶ)についても検討した。作製した複合膜型電解質は、SCOまたはBCO基板片側中央部に円形状にPd-Ag合金膜(φ13mm、厚さ65mm)が成膜されている。他方には中央部に円形状(φ13mm)に白金ペーストを塗布し、900℃において30分空気中で焼き付け多孔質電極として試験に供した。
【0154】
電解質にSCOおよびBCOを用いた場合の中温域(500〜600℃)における電流―電圧特性を図39に示す。発電試験温度は、Pd/SCO複合膜では600℃とし、Pd/BCO複合膜では500および600℃とした。600℃の電流―電圧特性から、短絡電流密度は、プロトン導電率の高いBCOを用いることによって、約2倍となった。また、Pd/BCOの500℃での電池性能は、Pd/SCOの600℃での電池性能レベルの3分の2程度まで向上した。しかしながら、Pd/BCOセルにおいても、短絡電流密度が約7mAcm-2であるため、更なる性能向上が期待される。そこで、次にBCOセルの性能を支配する要因について分析を行い、性能向上への課題を抽出するための実験を以下の手法により行った。
【0155】
まず、電流―電圧特性および過電圧特性を評価した。これらを評価することで、BCOセルの発電時の電圧降下要因を電解質抵抗、アノード反応抵抗、カソード反応抵抗に分離することができる。したがって、この結果から電池性能がどの要因(構成部材)でどのくらい低下しているか割合を把握することが可能となる。過電圧特性は、参照電極16を設け、カレントインターラプション(電流遮断)法により測定した。参照電極15は、図38に示すように電解質14の側面に、Ptペーストを塗布、乾燥後、900℃において焼き付け、作製した。その上からφ0.3mmの白金線を巻き付け白金リード線16に取り付けた。符号19はアノード電極であり、電解質14を介して反対側の面にはカソード電極が形成されている。カソード電極側、アノード電極側には集電ネット17が配置され、ガラスパッキン18によりこれらを挟むようにしている(アノード電極側の集電ネット17およびガラスパッキン18は図示を省略してある)。
【0156】
ここで、カレントインターラプション(電流遮断)法について説明する。電極と電解質からなる電気化学セルは最も簡略化された電気的な等価回路で表すと図40のようになる。高温においては、電解質のキャパシター成分のコンダクタンスは無視できるほど小さい(Cb=0)と考えられ、電解質に関しては通常オーム抵抗(Rb)のみで表される。電極―電解質界面に関しては電極反応抵抗Reと二重層容量Cdlから成る並列回路で表すものとする。カレントインターラプション法は、実際の電池に数msec程度のサイクルによるパルス電流で電気二重層を充電し、その後観察される過渡曲線から過電圧を求める方法である。図41(b)に装置構成を示す。具体的には、ファンクションジェネレーター20(NF回路ブロック WF1256)およびポテンショガルバノスタット23(SolarTron社 SI1286)を用いて、測定セル24のカソード、アノード間に2msec/サイクル、0.1msecのオフパルス幅でのパルス電流を印加する。この際、各電極―参照電極間の電位の変化は、デジタルオシロスコープ21を用いて同期させ、エレクトロメーター22で測定した。この場合、図41(a)に示すように電流が遮断されたときにオーム抵抗に相当する電位分だけ急激に変化する(図41、iRbドロップ)。次にCdlに充電された電荷は、キャパシタンスと並列にある抵抗(電極反応抵抗Re)を通って放電し、その分の電位が過渡的に変化する。このように電位差を遮断する前後の電位を測定することにより、過電圧(h=iRe)を求めることが可能である。
【0157】
尚、試験中には経時的に電解質が劣化していないか確認するために、交流法にてセル端子間に1kHz, 5mVの交流信号を入力し、そのインピーダンスを電解質抵抗としてモニターした。
【0158】
500℃における燃料電池の電流―電圧特性を図42に示す。また、図42には、電流を取り出すことによる電圧降下の内訳をBCO膜による過電圧(電解質過電圧)、Pd膜による過電圧(アノード過電圧)および多孔質Ptによる過電圧(カソード過電圧)から求めた。アノード過電圧が一番小さく、次いでカソード過電圧、電解質過電圧の順に過電圧が大きくなっており、500℃において、短絡電流密度は、7mA/cm2以下であった。そこで、各電池部材を材質および作製法から見直すことにより、過電圧を低減し、性能向上を図ることを試みた。
【0159】
(1)電解質緻密化による過電圧低減
電解質過電圧は、電解質抵抗に比例する。そこで、図42における電解質過電圧から初期BCOの電解質抵抗を求め、図43に示した。図43に示されるBCOの500℃における導電率から換算される電解質抵抗と比較して、初期BCOの電解質抵抗がかなり大きいことがわかった。この原因としては、1電解質の反応劣化および2電解質の焼結性が悪いことが挙げられる。1の原因については、これまで試験後のBCO表面をX線回折解析を行っているが、BCO単相のピークしか得られていないことから、BCOの反応劣化は起こっていないと考えられる。このため、過電圧のほとんどは2の電解質の焼結性に起因していると考えられる。BCOのような固体電解質は、その焼結度が95%以上ならば、導電率から換算される電解質抵抗を示し、逆に焼結度が低くなるにつれて、電解質抵抗は増大する。BCOでは、導電率から換算される理論電解質抵抗は、500℃で約6Ωとして得られるはずであるが、今回、試験中に測定した電解質抵抗は、500℃で約80Ω程度であった。電解質抵抗には、電解質の正味の抵抗以外に、集電ネットとの接触抵抗、リード線抵抗が含まれている。しかしながら、接触抵抗およびリード線抵抗が数10Ω以上あることは考えられないことから、80Ωのほとんどが電解質抵抗に起因することがわかる。つまり、初期BCOは、その焼結性が悪く、BCOの緻密化が必要であることがわかった。そこで、BCOの焼結工程を見直した。具体的には、焼結熱処理前の最終成形において、成形加重を増大したところ、同重量の焼結体において、約8%体積が減少し、緻密化することがわかった。この緻密化したBCO試料を試験に供し、初期のBCOによる電解質抵抗、改良後のBCOの電解質抵抗およびBCOの導電率から換算されるBCOの電解質抵抗を物性値として比較した結果を図43に示す。初期のBCOの電解質抵抗は、BCO導電率から計算される電解質抵抗より約20倍程度大きく、性能低下要因になっていた。しかし、今回、BCO電解質の緻密化を図ったことにより、初期BCO電解質抵抗と比較して、約7分の1程度まで電解質抵抗を低減できた。電解質の緻密性は電気化学デバイス性能を左右するものであるため、焼結度を95%以上とすることでより好ましい電気化学デバイス性能が得られるようになることが期待された。
【0160】
(2)Pd膜成膜法見直しによる過電圧低
(2.1)Pd成膜法の検討
前述したようにアノード過電圧の低減も一つの課題として挙げられた。以下、アノード過電圧の低減方法について膜の緻密性および電解質への密着性の観点から検討した。
【0161】
基板に対する膜の密着性をさらに向上させ、緻密膜を成膜するためには、成膜温度を十分高くする必要がある。しかしながら、Pdペースト膜は、実施例1で上述したように、Agとの合金膜であるために、処理温度を高くすることはできない。そこで、膜を構成する出発粒子を細かくすることによって、処理温度を高くすることと同等の効果が得られると考えた。かかる観点から、基板との密着性が良く、均一な緻密膜が成膜できる可能性の高い無電解めっき法について検討した。
【0162】
(2.2)無電解めっき法によるPd合金緻密膜の成膜
セラミックス、プラスチック、ガラスなどの非導電性基板に無電解めっきを施すためには、基板にめっき反応に対する活性を付与する触媒活性化前処理を施す必要があることが知られている。また、この時の処理によって、基板表面に形成される触媒核の分布や活性が変化し、表面形態や物性が異なるめっき膜が析出することが知られている。ここでは、湿式法による代表的な触媒活性化前処理である二液法(センシタイゼイション―アクチベーション(sensitization - activation)法)を用いて無電解めっきを施した。二液法活性化前処理は、一般に次の反応式で表される。
Pd2++Sn2+=Pd+Sn4
以下、ここで行った無電解めっき法を図44で示す無電解めっきフロー図に基づき説明する。
準備段階として、基板をアセトン洗浄しアルコール置換後に乾燥した。次に触媒活性化前処理(二液法)として、SnCl2塩酸溶液を一液、PdCl2塩酸溶液を二液として、一液と二液へ交互に10回浸漬を繰り返し、Pd核を析出させた。次に、[Pd(NH3)4]Cl2・H2O、EDTA・2Na、NH3(28%水溶液)、H2NNH2・H2Oを含み、アンモニア・アルカリ性とした無電解めっき浴に1時間浸漬して、Pd核を触媒としてその周囲にPdを析出させ、Pdの薄膜を形成した。これを、蒸留水にて洗浄し、[Pd(NH3)4]Cl2 、AgNO3、EDTA・2Na、NH3(28%水溶液)、H2NNH2・H2Oを含む溶液に1時間浸漬して、パラジウム薄膜上に銀を析出させて銀の薄膜を形成した。さらにこれを蒸留水にて洗浄し、N2気流中にて、850℃、10時間熱処理を施し、Pd合金緻密膜を得た。
【0163】
(2.3)EPMAによるPd合金膜の断面観察
無電解めっき基板には、Al2O3を選定し、めっき膜性状を確認した。基板上にめっきされたPd合金緻密膜の断面分析をEPMA(Electron Probe MicroAnaly- zer: JEOL, JXA-8900R)によって行った。EPMAによるPd, Ag, Alの特性X線像を図45に示す。図中において、(a)はPd、(b)はAg、(c)はAlの特性X線像である。PdおよびAgは相互によく拡散し、合金化している様子が観察された。一方、基板からのAl元素のPd膜側への拡散は確認されなかった。
次に、EPMA付属のEDX(Energy Dispersive X-ray)解析より、Pd合金膜組成の分析を行った。表2は、PdおよびAg標準試料を用いて、膜の各部位におけるEDXによる組成比(wt%)を定量した結果である。組成比は、平均でPd/Ag=76.6/23.4となり、良好な結果を得た。以上より、無電解めっき法を選定することにより、Pd, Ag元素の偏在化なしに、均一に目的組成に合金化が可能であることを確認できた。
【0164】
【表2】
【0165】
(2.4)無電解めっきを適用した場合のアノード過電圧
BCO上に無電解めっきによりPd合金緻密膜を施した複合膜型電解質とペースト法を用いた複合膜型電解質を用いて、アノード過電圧を比較した。測定法は、カレントインターラプション法を用いた。500℃におけるアノード過電圧を測定した結果を図46に示す。無電解めっき法を用いたPd合金緻密膜では、ペースト法により作製されたPd合金緻密膜と比較してアノード過電圧をさらに低減できることが確認された。したがって、無電解めっき法のPd合金緻密膜は、ペースト法により作製されたPd合金緻密膜と比較してBCOとより強固に密着し、かつ緻密膜となっていることが示唆された。
【0166】
ここで、複合膜型電解質のアノード反応について、以下のように考察した。1水素の緻密膜上への拡散、吸着、2水素分子の水素原子へ解離、3水素原子のPd緻密膜への溶解、4水素の拡散、5Pd/BCO界面における水素のプロトンへのイオン化、6プロトンのBCOへの溶解である。アノード過電圧が大きくなる要因を以下に考察した。1〜4の過程については、Pd合金膜の物性に依存すると考えられ、成膜法による差異は生じないと考えられる。しかしながら、5、6の過程には、膜の密着性の優劣に依存する。これらの観点からも、無電解めっき法のPd合金緻密膜は、ペースト法により作製されたPd合金緻密膜と比較してBCOとより強固に密着していることが示唆された。
【0167】
(3)カソード材見直しによるカソード過電圧低減
これまでカソード材として使用してきた多孔質Ptは、図47に示す過電圧の温度依存性に現れているように温度の低下に伴う電極触媒能の低下により、カソード過電圧がかなり増大している。また、Pt自体高価な材料であり、今後の中温形燃料電池のカソードとして適切ではないと考えられる。そこで、カソード材について検討をおこなった。
【0168】
中温域においては、電極活性を示し、かつ安価なカソード材料を選択しなければならない。最近、安価なAgを用いたカソードにおいて、良好な中温域の電極活性が示されている。Ptが約3,000円/gであるのに対し、Agは約26円/g(26,000円/kg)とコスト的に優位である。そこで、本研究では、Agをカソード目標材として、Pt, Pdを比較カソード材として、500℃における過電圧の比較を行った。Pdは、アノード材として、中温域で使用できているために、カソードにおいて適用可能か確認するために選択した。過電圧の比較結果を図48に示す。それぞれペーストを塗布後、乾燥し、Pt, Pdは900℃において焼き付け、Agは融点が962℃と低いために、800℃で焼き付けを行った。図48より、カソード過電圧は、多孔質Agカソードを適用することで大幅に低減できることが明らかとなった。一方、Pdは、Ptと同程度のカソード過電圧を示した。
【0169】
試験後のカソード表面のSEM写真を図49に示す。図中において(a)はPd、(b)はPt、(c)はAgカソード表面のSEM観察結果である。Agの表面形態は、Pt, Pdと比較して、Agが凝集しているように観察された。通常、多孔質電極では、電極反応が起こる三相(ガス-電極-電解質)界面の面積が狭いほど過電圧が大きくなるが、図48では、上記とは逆に過電圧が低くなる結果を示した。この結果については、唯一Pt, PdにはないAgの特性として、酸素の透過能を示すことが電極反応に関与している可能性が示唆される。以上より、Agの中温域でのカソード材としての可能性が示された。
【0170】
(4)性能向上対策を施したセルによる電池性能の向上
電解質の緻密化、Pd成膜法の見直し、カソード材の変更により過電圧が低減され、セル性能の向上が見込めるため、それぞれの改善策を適用して単セルを再構成し、500℃で発電試験を行った結果を図50に示す。無電解Pd合金めっき緻密膜+BCO膜+多孔質Agカソードを選定した結果、最大出力密度は、500℃でも0.037Wcm-2を示し、実施例1のPdペースト法を用いたPd/SrCeO3セルの電池性能よりも約15倍性能を向上させることに成功した。
【0171】
次に、中温形燃料電池において到達可能な電池性能を試算し、その電池性能レベルを検討した。中温形燃料電池の電池性能試算は以下のように行った。BCOの導電率、Pd緻密膜とAg多孔質カソードの過電圧特性から、電解質抵抗、アノードおよびカソード反応抵抗が得られることから、電池出力密度つまり電池性能を試算することができる。試算条件として、水素酸素燃料電池において、動作温度500℃、電解質の薄膜化が成功したとして、BCO膜厚20mmと設定し、アノードには無電解Pd合金メッキ緻密薄膜を、カソードには多孔質Agを選定し、全抵抗を算出した後、電流-電圧特性から出力密度特性を試算した。試算結果を図51に示す。比較のためにこれまでの結果も同図に示す。500℃における試算性能は、無電解Pd合金メッキ緻密薄膜+BCO薄膜+多孔質Agカソードを選定した場合、現行の高温SOFC 性能(0.22 Wcm-2, 300mAcm-2, 0.74Vにおける出力密度)に匹敵する0.24 Wcm-2(BaCeO3膜厚20mm, 300mAcm-2における出力密度)と試算され、高温SOFCに匹敵する電池性能レベルであることが示された。
[実施例3]
ここまでは、Pd/SCO膜およびPd/BCO膜を燃料電池の電解質に用いた場合の発電の可能性を検討し、理論通り実証することができた。そこで、以下に材料コスト面からさらに考察を加え、燃料電池としての可能性を試算した。
【0172】
比較対象はSOFCとした。また、コスト試算では、SCOよりも導電率が高く、コスト低減につながるBCOを比較対象とした。本発明に係る複合膜型電解質は、金属と酸化物電解質を複合化することにより、金属部がアノード機能も持ち合わせるため、アノード材が不要となるメリットがある。一方、適用したパラジウム(Pd)合金膜は、高い水素選択透過性を示し、半導体製造に必要な高純度水素の製造に使用されるポテンシャルをもつが、金属Pdは金や白金と同様に極めて高価な金属であり、実用化を妨げる大きな障害となる。水素透過膜2の材質としては、現在、これに代わる安価な金属膜材料の開発が活発に行われており、その中で、Zr-Ni系、V-Ni系、Nb-Ni系などの安価な合金材料に注目が集まっている。これら透過膜の水素透過能は、Pd膜(10-7mol/(m2・s・Pa))より1桁劣るものの、例えば、Zr-Ni系では、Pd膜に比べて原材料費は数百分の一と大変安価となる。したがって、実用化を目指すためには、Pd使用量を極力減らすか、安価な水素透過膜2の開発が不可欠であると考えられる。
【0173】
実際に、材料コスト面から金属/酸化物複合膜型電解質の膜厚を試算した。試算式には、次の数式1を用いた。また、材料単価および密度は、表3の値を用いた。
【0174】
【数1】
【0175】
【表3】
【0176】
まず、SOFCにおける電解質(YSZ)+アノード(Ni-YSZ)分のコストを試算した。試算性能は、電解質厚さ40μm、アノード厚さ100μmとし、300mA/cm2において0.74Vで行った(結果については表3参照)。YSZを用いたSOFCの(電解質+アノード)の1kWあたりの原材料コストは約1,785円となった。実際に試算されるコストには、製造に要した様々なコストをトータルに考慮する必要があり、月産ベースで試算されている平板型SOFCを例にとってみると、約68,000円/kWであり、電解質+アノード個別のコストでみると約11,000円/kWと試算された。次に、YSZの原材料コストをベースにPd-BCOのコストを試算した。Pd-BCOでは、Pdの原価が極端に高いために、Pd膜厚をパラメータとし、YSZの原材料コストと比較することで、最適なPd膜厚およびBCO膜厚を求めた。なお、BCO膜厚は、現行のSOFCの電解質膜厚が数10μmであり、製法上からも10μm以下の電解質膜厚を作製することは難しいと考えられるため、YSZの膜厚オーダーに揃えた。結果を図28に示す。図28よりPd膜厚が0.1μmよりも厚い場合、コスト的に実用化は難しいと思われる。また、Pd膜厚が0.1μm以下でも、BCO膜厚が20μmより厚くなると、YSZよりも割高になってしまうと考えられる。また、Pd膜厚は、現在報告されているPd水素透過膜2の成膜技術で0.1μmまでは支持膜方式を用いて薄膜化が可能であることから、0.1μmまでのPd膜厚を試算対象とすると、コスト的にBCO膜厚は20μm以下が望ましいと試算される。
【0177】
以上より、Pd/BCO電解質膜は、最も厚くて約20μmであるために、構造は電解質自立膜型では、到底機械的強度を保つことができない。従って、カソードあるいはアノードによる多孔質体支持膜型構造が望ましい。多孔質体支持膜型構造はセンサーで使用できる構造である。電流を流さない起電力式のセンサーの場合はオーミックロスによる電圧降下がないので、電解質膜自立型構造をとることもできる。他のデバイスについては、通電するため電解質はどうしても極力薄くする必要がある。このため、電解質自身では強度が足りなくなるため多孔質体支持膜型構造が望ましい。
【0178】
以上、原材料コスト面から、金属/酸化物複合膜型電解質の今後の開発目標仕様を試算した。電解質は、SCOから導電率の高いBCOに変え膜厚を20μm以下に、Pd膜厚は0.1μm以下とすることが望ましい。
【図面の簡単な説明】
【0179】
【図1】本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質の構造を簡単に示した概略図である。
【図2】AECe1-xMxO3-aの欠陥構造とプロトンの生成を、(a)化学量論組成と比較し(Stoichiometry)、(b)湿潤酸素中、(c)乾燥酸素中、(d)水素中のそれぞれについて概略的に示した図である。
【図3】Pd膜における水素透過の原理を示した図である。
【図4】水素透過膜とプロトン導電性セラミックスを用いた燃料電池の構造を示す図である。
【図5】金属/酸化物複合膜型電解質を用いた燃料電池の構造を示す図である。
【図6】金属レジネートを含むペースト(AR)および金属レジネートを含まないペースト(AN)につき、各温度(900℃、1000℃、1100℃、1200℃、1300℃)においてアルミナ板上に成膜されたPd膜の様子を示す画像である。(a)AR 900℃ in air、(b)AR 1000℃ in air、(c)AR 1100℃ in air、(d)AR 1200℃ in air、(e)AR 1300℃ in air、(f)AN 1300℃ in air、である。
【図7】二度の熱処理によってアルミナ上に緻密に成膜されたPd膜の様子を示す画像である。
【図8】二度の熱処理によってSCO基板上に緻密に成膜されたPd膜の様子を示す画像である。
【図9】試料断面の光学顕微鏡写真(×500)である。
【図10】試料断面のEPMAによる各元素の特性X線像を示す図である。
【図11】標準物質を用いたEDX実測値の補正内容を示すグラフである。
【図12】Pd-Agの二元系相図である。
【図13】XRD解析結果から見た曝露試験前後のSrCeO3相の比較結果を示すグラフである。
【図14】プロトン導電体を利用した水素濃淡電池の原理を示す図である。
【図15】水素濃淡電池における水素分圧と起電力との概略関係を示すグラフである。
【図16】プロトン導電体を利用した水素ポンプの原理を示す図である。
【図17】本実施例において作製した複合膜型電解質膜の平面図である。
【図18】本実施例において作製した複合膜型電解質膜の側面図である。
【図19】水素濃淡電池法の装置概略図である。
【図20】水素濃淡電池の起電力と水素分圧の関係(実戦:理論値)を示すグラフである。
【図21】電流に対する水素発生速度を示すグラフである。
【図22】プロトン導電体を利用した水素酸素燃料電池の原理を示す図である。
【図23】燃料電池実験に用いた装置の概略図である。
【図24】各温度における燃料電池の起電力を示すグラフである。
【図25】起電力から求めたPd/SCOの燃料電池雰囲気におけるプロトン輸率の温度依存性を示すグラフである。
【図26】Pd/SCO膜を用いた燃料電池の電流−電圧特性を示すグラフである。
【図27】CO2を含む燃料を用いた燃料電池における放電時の電圧の経時変化を示すグラフである。
【図28】Pd-BCO膜材料コストのPd膜厚依存性を示すグラフである。
【図29】固/液界面における接合の様子と固/固界面における接合の様子を示す図である。
【図30】本発明の一実施形態を示す図で、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体が形成されている複合膜型電解質を示したものである。
【図31】本発明の一実施形態を示す図で、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体を形成し、尚かつアノードガスとは反対側の面にプロトン(H+)が通過可能な別の水素透過膜を形成したものである。
【図32】本発明の一実施形態を示す図で、複合膜のアノードガス側とは反対側の表面に形成された多孔質カソードを多孔質支持体として機能させたものである。
【図33】本発明の一実施形態を示す図で、複合膜のうちアノードガス側とは反対側の表面に多孔質カソードを形成し、さらにこの多孔質カソードのカソード側表面に多孔質支持体を形成したものである。
【図34】本発明の一実施形態を示す図で、複合膜のアノードガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜を形成し、さらにこの水素透過膜のカソード側表面に多孔質支持体を形成したものである。
【図35】各種ペロブスカイト型酸化物の水素中のプロトン導電率を示すグラフである。
【図36】SrCeO3膜を使った従来の水素分離法の概念を示す図である。
【図37】Pd膜を使った従来の水素分離法の概念を示す図である。
【図38】単セルのセッティング状態を示す図である。
【図39】電解質にSCOおよびBCOを用いた場合の中温域(500〜600℃)における電流―電圧特性を示す図である。
【図40】電極と電解質からなる電気化学セルの電気的な等価回路を示す図である。尚、Rb:電解質抵抗、Re:反応抵抗、Cb:電解質キャパシター容量、Cdl:二重層容量である。
【図41】カレントインターラプション(電流遮断)法について示す図である。(a)はオシロスコープで観測される波形、(b)はカレントインターラプション(電流遮断)法の装置概略図である。
【図42】500℃における燃料電池の電流―電圧特性を示す図である。図中でOCVは開回路電圧を表している。
【図43】初期のBCOによる電解質抵抗、改良後のBCOの電解質抵抗およびBCOの導電率から換算されるBCOの電解質抵抗を物性値として比較した結果を示す図である。
【図44】無電解めっき工程のフロー図である。
【図45】Al2O3基板上にめっきされたPd合金緻密膜の断面をEPMAにより分析した結果である、Pd, Ag, Alの特性X線像を示す図である。(a)はPd、(b)はAg、(c)はAlの特性X線像である。
【図46】BCO上に無電解めっきによりPd合金緻密膜を施した複合膜型電解質とペースト法を用いた複合膜型電解質を用いて、カレントインターラプション法により、500℃におけるアノード過電圧を測定した結果を示す図である。
【図47】多孔質Ptをカソード材として用いたときの過電圧の温度依存性を示す図である。
【図48】Ag、PtおよびPdをカソード材として用いたときの過電圧の比較結果を示す図である。
【図49】Ag、PtおよびPdカソード表面のSEM観察結果を示す図である。(a)はPd、(b)はPt、(c)はAgカソード表面のSEM観察結果である。
【図50】電解質の緻密化、Pd成膜法の見直しおよびカソード材の変更により過電圧を低減させるための改善策を適用して単セルを再構成し、500℃で発電試験を行った結果を示す図である。
【図51】アノードに無電解Pd合金メッキ緻密薄膜を、カソードに多孔質Agを選定し、全抵抗を算出した後、電流-電圧特性から出力密度特性を試算した結果を示す図である。
【符号の説明】
【0180】
1 SrCeO3膜(プロトン導電性酸化物膜)
2 Pd膜(水素透過膜)
3 酸化剤ガス(CO2を含むガス)
S 多孔質支持体
【技術分野】
【0001】
本発明は、プロトン導電性酸化物膜−水素透過膜複合膜型電解質およびこれを用いた電気化学デバイスに関する。さらに詳述すると、本発明は、CO2を含むガス中におけるプロトン導電性酸化物電解質の脆弱性の回避法に関する。
【背景技術】
【0002】
本件出願人たる電力中央研究所においては、これまで、既存の4つの燃料電池(PAFC(リン酸型燃料電池)、MCFC(溶融炭酸塩型燃料電池)、SOFC(固体電解質型燃料電池)、PEFC(固体高分子型燃料電池))の性能を同一の評価手法で分析することにより燃料電池の出力電圧と運転温度との一般化された関係を明らかにする、という研究が続けられている。また、かかる燃料電池の研究により、プロトン(H+)移動型の電解質(つまり当該電解質中をプロトンが移動可能なもの)を用いて250〜500℃程度の中温域で作動する燃料電池を開発できれば、既存の燃料電池よりも性能面、材料面で優位な燃料電池になる可能性があることが示されている(特許文献1参照)。ここでいうプロトン移動型の電解質としては、溶融塩や水溶液系の電解質に可能性がある。しかし、これら電解質は、250℃〜500℃程度の中温域では化学的に不安定であったり、あるいは内在する他のイオンも電極反応に関わったりすることがあるため、燃料電池に適用できなかった。
【0003】
一方、プロトン移動型電解質のその他の具体例として、例えば「高温型プロトン導電体を用いた炭化水素改質ガス類から水素を分離する方法」が記載された特許文献2においてはプロトン導電性酸化物(プロトン導電性酸化物固体電解質)が開示されている(特許文献2参照)。このプロトン導電性酸化物は、当該酸化物がプロトン導電性機能を有していることから、ガス(例えば天然ガス改質ガス)中の水素を電気化学的に分離する技術、さらには種々の電気化学デバイスに関する技術に応用することが考えられあるいは期待されているものである(特許文献2参照)。
【0004】
上述のプロトン導電性酸化物固体電解質の一例として、SrCeO3に代表されるペロブスカイト構造を持つ酸化物AECeO3(ただしAEはBa または Sr )が存在する。AECeO3は、図35に示すように水素気流中、中高温域において良好なプロトン導電性を示し、尚かつ高い導電率を示すことが知られている。このため、これら酸化物を、水素のみを用いた電気化学デバイス(例えば燃料電池、水素センサー、水素ポンプ)の電解質に適用した場合には、高い性能が発揮されるものと考えられる。
【0005】
例えば図36に示すプロトン導電性のSrCeO3膜101は、図中の改質ガス102のような水素や二酸化炭素などが混成したガスの流路に設けられて水素透過膜として機能する。この場合、1次側(この場合であれば改質ガス102側)の電極104と2次側(この場合であれば分離後の水素103側)の電極105との間の外部電圧106によって印加される電圧がプロトンH+の推進力(ドライビングフォース)となる。このようなプロトン導電性のSrCeO3膜(水素透過膜)101を用いた場合には、1次側圧力を固定しながら様々な2次側水素圧力を設定して供給することが可能になるという長所がある。また、常圧の改質ガス102を使用しながら加圧することが可能であるため、貯蔵水素用の昇圧装置やそのための設置スペースなどが不要だという長所もある。
【0006】
【特許文献1】特開2004−119077号公報
【特許文献2】特開2003−2610号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、通常、水素分離の対象となるガス(上述の例でいえば改質ガス102)は、CO2など他のガスも含まれている混合ガスである場合がほとんどであり、このことから、上述したSrCeO3膜101のようなペロブスカイト構造を持つ酸化物(AECeO3)を電気化学デバイスの電解質に適用すると特有の問題が生じてしまう。
【0008】
すなわち、酸化物AECeO3には、CO2を含む雰囲気に曝されると、
AECeO3+CO2→AECO3+CeO2
というようにCO2と反応を起こし、容易に分解してしまうという性質がある。このため、例えば特許文献1に見られるように、CO2を含んだガスが流れる電気化学デバイス(例えば水素ポンプなど)の電解質にこの酸化物AECeO3を適用した場合、当初の段階では特許文献1にも記されているような初期の性能が発揮されるものの、長時間は耐用できない、つまりこのような混合ガス中のCO2に対して脆弱である、という点で問題がある(例えば、第27回電池討論会要旨集(3B10, p226-227)参照)。
【0009】
そこで、本発明は、かかる従来技術が有していた課題に鑑み、AECeO3のプロトン導電性は維持しつつも、当該AECeO3のCO2に対する脆弱性を克服してCO2を含むガス雰囲気下でも使用可能としたプロトン導電性酸化物膜−水素透過膜複合膜型電解質およびこれを用いた電気化学デバイスを提供することを目的とする。
【課題を解決するための手段】
【0010】
かかる目的を達成するため、本発明者は種々の検討と実験を行った。かかる検討と実験の過程および概要について以下に説明する。
【0011】
まず、本発明者は従来における別の水素分離法について検討した(図37参照)。水素分離に用いられる水素透過膜(Pd金属膜など、図中で符号201で示す)は、例えば改質ガスといったようなH2(水素)やCO2(二酸化炭素)などの混成ガス(図中、符号202で示す)の流路に設けられ、水素分子が分離した状態(つまりプロトン(H+)と電子(e-)になった状態)で透過させることが可能なもので、これにより、改質ガスから水素を分離することを可能としている(図37参照、分離後の水素を符号203で示す)。このような水素分離法の長所としては水素分離のための電力量が不要だという点が挙げられる。したがって推進力(ドライビングフォース)は水素分圧差のみである。ただし、効率のよい分離を行うためには非常に高い水素分圧差が必要になる(つまり、図37中に示す圧力High PH2と圧力Low PH2と間に高い分圧差が必要になる)。したがって、目的とする水素圧に応じて1次側(改質ガス側)と2次側(分離された水素側)の圧力が固定されることになるから貯蔵用昇圧装置が必要となってくる。また、常圧の改質ガスが使用できないという短所もある。
【0012】
ここで検討した水素透過膜(Pd金属膜など)を利用した水素分離法(図37参照)には上述のように長所と短所とがあるが、かかる長所・短所を、ペロブスカイト構造を持つ酸化物(AECeO3)を利用した水素分離法の長所・短所と比較すると、両者が互いに入れ替わった関係(つまり一方が長所とする点が他方においては短所になっている関係)にあることがわかる。そこで、本発明者はかかる関係に着目しつつ、上記課題を解決すべく鋭意検討を重ねた結果、プロトン導電性酸化物(AECeO3)と水素透過膜(例えばPd金属)とを複合膜化した電解質を用いると、水素のみが(具体的にはプロトンの形で)Pd膜を透過しAECeO3膜に達するため、AECeO3のCO2に対する脆弱性を克服でき、CO2を含むガス環境下でも使用できる電気化学デバイスの電解質として使用可能となることを知見し、この電解質を用いればCO2を含むガス環境下でも上記課題を解決できることを見出した。
【0013】
本発明はかかる知見に基づくものであり、請求項1に記載の発明であるプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造とし、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能なプロトン導電性酸化物膜と、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な水素透過膜との複合膜からなり、これら両膜の複合界面が連続接合した状態で、プロトン導電性酸化物膜の少なくともガス側の表面に水素透過膜が成膜され、尚かつ当該複合膜が多孔質支持体によって支持されているというものである。
【0014】
本発明においては、上述した知見の下、水素分離に好適である新規な複合膜型電解質構造を構築している点が特徴的である。すなわち、プロトン導電性酸化物膜と水素透過膜とが互いの短所を補完し合う複合膜型構造とすることにより、AECeO3のプロトン導電性は維持しつつも、AECeO3のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することを可能としている。この複合膜型電解質はいわばセラミックス(AECeO3)と改質ガスとの間に別の膜が介在している状態、別の表現をするならばセラミックス(AECeO3)の表面に水素透過性の保護膜的要素が形成された状態にあり、このような複合膜型構造が、CO2によるセラミックス分解を防ぐ。
【0015】
また、この複合膜型電解質構造においては、水素が固相から固相へと相間を移動する際に、水素元素ないしは水素分子として移動するのではなく、プロトンとして連続して移動する現象を呈することを利用している(図1参照)。この場合、水素透過膜(例えばPd膜)は、水素(H2)のみ選択的に透過させ、他の分子(N2, CO, CO2など)は透過させないという性質を持つことから、この水素透過膜をプロトン−電子混合導電体とみなすことができる(図1参照)。したがって、この水素透過膜が電極と電解質の役目を果たすことになる。上述の保護膜として水素透過膜をプロトン導電性酸化物膜の一部と見なすと、プロトン導電性酸化物膜に接触(あるいは通過)するガスは水素(H2)のみということになる。
【0016】
しかも、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質においては、プロトン導電性酸化物膜と水素透過膜とからなる複合膜が多孔質支持体によって支持されている。この場合における多孔質支持体は、ガス中から水素を分離するという複合膜の機能に影響を及ぼすものではない。要は、多孔質支持体は多孔質体であるので、水素が水素透過膜からプロトンとして膜中に入る際にこの反応を阻害しない程度にガスの拡散性に優れていればよい。しかも、この多孔質支持体が上記複合膜を強度的に支持する基材として働くことから、複合膜(プロトン導電性酸化物膜および水素透過膜)はそれ自体が基材に要する強度を有していなくても足りる。つまり、複合膜自体で所定の強度を確保する必要がなくなることから、この複合膜を構成するプロトン導電性酸化物膜と水素透過膜のそれぞれを必要十分な厚さにまで薄膜化することが可能となり設計の自由度が広がるという利点がある。また、プロトン導電性酸化物膜および水素透過膜の薄膜化によりコスト面でも有利となる。
【0017】
また、請求項2に記載の発明は、請求項1に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質におけるプロトン導電性酸化物膜が、希土類元素セリウム(Ce)の一部が当該セリウム(Ce4+)とは異なる他の低原子価希土類元素で置換されている構造となっているというものである。低原子価元素で置換することにより、例えば、Ce4+→Yb3+とすると、結晶格子中の電気的中性が崩れ、これを補うため格子中の酸素1/2個が抜ける。そうすると格子中の電気的中性が保たれる。このように意図的に酸化物イオン空孔を生成させること(ドーピング)ができ、よりプロトンの生成が促進されることになる。
【0018】
請求項3に記載の発明は、請求項1または2に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における他の希土類元素が、イッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素であるであるというものである。
【0019】
請求項4に記載の発明は、請求項1から3のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における水素透過膜が、水素透過性を示す金属または合金であるというものである。金属または合金のうち、水素透過性を示すものがこの発明における水素透過膜に適用される。
【0020】
請求項5に記載の発明は、請求項4に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とするものである。
【0021】
請求項6に記載の発明は、請求項4もしくは5に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とするものである。
【0022】
請求項7に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、多孔質支持体が複合膜のガス側の表面に形成されているというものである。
【0023】
請求項8に記載の発明は、請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面に多孔質カソードが形成されているというものである。
【0024】
請求項9に記載の発明は、請求項8に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、多孔質カソードが少なくともペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物および銀(Ag)のうちいずれか一つを含んでいることを特徴とするものである。
【0025】
請求項10に記載の発明は、請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面に、プロトン(H+)が通過可能な別の水素透過膜が形成されているというものである。
【0026】
請求項11に記載の発明は、請求項10に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における別の水素透過膜が、水素透過性を示す金属または合金であるというものである。
【0027】
請求項12に記載の発明は、請求項11に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とするものである。
【0028】
請求項13に記載の発明は、請求項11もしくは12に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質における別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とするものである。
【0029】
請求項14に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、多孔質支持体が、複合膜のガス側とは反対側の表面に形成された多孔質カソードであるというものである。
【0030】
請求項15に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面に多孔質カソードが形成され、さらに該多孔質カソードの表面に多孔質支持体が形成されているというものである。
【0031】
請求項16に記載の発明は、請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質において、複合膜のガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜が形成され、さらに該別の水素透過膜の表面に多孔質支持体が形成されているというものである。
【0032】
請求項17に記載の発明は、請求項1から16のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質を用いた電気化学デバイスであって、燃料電池、水素センサー、水素ポンプ、水蒸気センサーおよび排ガス浄化デバイスのいずれかに応用されるというものである。
【0033】
この電気化学デバイスは、請求項18に記載のように少なくともH2とCO2とを含有する混合ガス中の水素濃度を定量できる水素センサーに適用することができる。
【0034】
また、電気化学デバイスは、請求項19に記載のように少なくともH2とCO2とを含有する混合ガス中から水素のみを分離できる水素ポンプに適用することもできる。
【0035】
さらに、電気化学デバイスは、請求項20に記載のように少なくとも水蒸気とCO2とを含有する混合ガス中から水蒸気濃度を定量できる水蒸気センサーに適用することもできる。
【0036】
さらに、電気化学デバイスを、請求項21に記載のように、少なくとも水蒸気、CO2およびNOx を含有する排ガスからNOxを除去するための排ガス浄化用デバイスに適用することもできる。
【0037】
ここで、以下、金属/酸化物複合膜型電解質の概念について説明し、続いてAECeO3のCO2に対する脆弱性を克服することについての検討内容を説明することにより、本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質の内容について更に詳細に説明することとする。以下では、「1.ペロブスカイト型プロトン導電体酸化物について」、「2.水素透過膜について」、「3.水素透過膜とプロトン導電性酸化物を用いた燃料電池について」、そして「4.金属/酸化物複合膜型電解質を用いた燃料電池について」という項目順で説明していく。
【0038】
1.ペロブスカイト型プロトン導電体酸化物について
従来技術の課題を解決するにあたり、本発明者はまずペロブスカイト構造を持つ酸化物AECeO3について検討した。例えば、中温域で作動する燃料電池には、当該中温域で高いプロトン導電率を有する電解質が必要であり、燃料電池として十分に作動するには、以下に示す化学式1,2の各電極反応が円滑に進行する必要がある。
【0039】
【化1】
【化2】
【0040】
しかし、一般的に低温域で高いプロトン導電率を有する電解質は、中温域においては分解反応が進行するため、化学的安定性に欠けるという問題を有している。また、仮に中温域で安定に存在しても、実際に上述した化学式1,2のような燃料電池反応を発現する電解質は皆無である。
【0041】
一方、600℃以上の中高温域では、図35に示すようなSrCeO3やBaCeO3などの高いプロトン導電率を有するペロブスカイト型構造のセレイト系酸化物(AECeO3)が開示されている。また、前述のようなペロブスカイト型構造(ABO3)を持つ酸化物は、水素源が全くない高温雰囲気において、酸素分圧が高くなるほど正孔(ホール)による伝導(p型電子導電性)が増大することが知られている。この場合、導電率が酸素分圧の1/4乗に比例することから、酸化物イオン空格子点の関与した格子欠陥平衡にあるものと考えられている。
【0042】
このような酸化物を水素ガスや水蒸気のような水素源を含む雰囲気に曝すとプロトン導電性が発現する。このことは、これら酸化物が雰囲気中の水素源から水素を取り込み、水素がプロトンの形で存在することを意味する。このような酸化物の格子欠陥とプロトン生成の関係を模式的に図2に示す。図2において、(a)は化学量論組成を、(b)は湿潤酸素中におけるプロトン又はホール伝導機構を、(c)は乾燥酸素中におけるホール電子伝導機構を、(d)は水素中におけるプロトン伝導機構を示している。尚、図2中における正方形(□)は酸化物イオン空孔を表している。ここでは、格子欠陥が直接プロトンを生成するわけではない。まず雰囲気中の酸素と空孔が欠陥反応式(図中の式参照)に従いホール(h)を生成し、これらが平衡になっているところへ水素が導入されるとホールと水素とが反応してプロトンが生成する。つまり、酸化物イオン空孔が多数存在しなければプロトン導電性は発現しない。ただ、導電率はプロトンの濃度と易動度に比例するので、酸化物イオン空孔濃度が高い(=プロトン濃度が高い)からといって導電率が高くなるわけではない。高い濃度と易動度の両方が揃ってはじめて高い導電率を得ることができる。
【0043】
これまで多くの研究から、母体酸化物のみではプロトン導電性を示さず、プロトン導電性の発現には三価カチオンによるBサイトへのドーピングが必須であることが確認されている。例えばSrCeO3のBサイトカチオンのCe4+の一部をYb3+で置換固溶させると、結晶内では電気的中性条件を満たすため酸化物イオン空格子点を生じる。
【0044】
【化3】
【0045】
この空格子点は高温において正孔(化学式4中においては、右肩に傍点が付された記号hで示す)および雰囲気酸素と化学式4に示す平衡関係にある。したがって、水素源のない雰囲気ではp型電子導電性のみを示すことになる。これに対して、水素源、例えば水蒸気を雰囲気に導入すると、水蒸気分子と正孔または酸化物イオン空格子点との間に化学式5,6に示す平衡関係が化学式4とともに成立し、水素が酸化物中に取り込まれてプロトン導電性が発現する。
【0046】
【化4】
【化5】
【化6】
【化7】
【0047】
ここで、K1,K2,K3は化学式4〜6中のそれぞれにおける平衡定数である。化学式4、化学式5および化学式6はそれぞれ独立ではなく、化学式7によって関係づけられているのでこれら化学式のうちの二つの式を用いて平衡関係を表すことができる。
【0048】
しかし、アルカリ土類金属をAサイト、CeをBサイトに配するペロブスカイト型酸化物(AECeO3)は、プロトン導電性を有する一方で、雰囲気中にCO2が存在すると、熱力学的平衡により化学式8のようにCeO2とACO3とに容易に分解してしまうというように化学的安定性に欠ける。このため、SrCeO3やBaCeO3を電気化学デバイスに用いるには雰囲気中のCO2に注意を要する。
【0049】
【化8】
【0050】
また、仮にCO2を含む雰囲気でもSrCeO3やBaCeO3を電解質として利用するためには、電解質膜表面を雰囲気中のCO2から遮断する必要がある。
【0051】
2.水素透過膜について
水素がプロトンとして金属膜を透過する現象はパラジウム(Pd)膜において見いだされている。Pdおよびその合金膜の水素透過機構は、定性的には図3に示すような原理によって水素が透過する機構になっていると考えられている。水素透過能を有するPd膜の両側において水素分圧差が生じると、高水素分圧側の水素分子が金属表面で解離し水素原子となる(図3参照)。水素原子は、Pdによって電子を奪われプロトンとなり、濃度勾配によりプロトンとして金属中を拡散する。拡散したプロトンは、低水素分圧側で自由電子と再結合し水素原子となり、さらにPdの触媒能によってこれら水素原子が会合して分子となり、最終的に水素分子として低水素分圧側に放出される。このように金属Pd膜は、プロトンと電子を導電する一種の混合導電体と見なすことができる。本発明者は、特にPd膜において顕著なこのような機能を利用することとした。尚、Pd膜単独では、水素脆化が生じる虞があるため、PdとAgを合金化したものがより好適である。
【0052】
3.水素透過膜とプロトン導電性酸化物を用いた燃料電池について
上述の「2.水素透過膜について」で述べた水素透過能を持つ金属PdをAECeO3とガス雰囲気の間に配置できれば、CO2を含むガス環境下においてもAECeO3を使用することができると考えられる。これは、図4に示すように、CO2を含むガス(例えば改質ガス)側からH2がPd膜を透過することにより、酸化物(AECeO3)の膜側ではH2のみを燃料ガスとして利用できるようになるためである。
【0053】
ただし、このような二重膜構造(Pd膜およびAECeO3膜からなる二重構造)が構成された場合(図4参照)、以下の点があることが判った。すなわち、第一に、Pd膜を透過してきた水素によって水素分圧が極度に高くなり、酸化物中のCe4+がCe3+へと容易に還元されてしまうことがある。これに対しては例えば室温飽和程度の水蒸気を加えれば水蒸気との平衡によって水素分圧が若干低下し、Ce4+が還元されるのを防止することが可能となると考えられた。ただし、この場合、水蒸気はPd膜に阻害され酸化物膜まで届かないということが生じる。第二に、例えばPd膜を厚くするとコストに大きく影響することから、ある程度薄膜化する必要があるが、Pd金属等の水素透過膜単独の薄膜化は強度的に問題があると考えられた。
【0054】
4.金属/酸化物複合膜型電解質を用いた燃料電池について
種々の検討と実験の結果、上述のようなプロトン導電性酸化物膜および水素透過膜の両者を例えば燃料電池に適用するにあたっては、図5に示すような金属/酸化物複合膜型電解質、すなわち金属Pd膜と酸化物AECeO3膜とが重ね合わされた状態で複合した形態の電解質とすることが望ましいという知見が得られた(図5参照)。このように金属(Pd)膜と酸化物(AECeO3)膜とを接合して複合化することにより、CO2を含むガス環境から酸化物(AECeO3)を保護することが可能となる。ここで、図4に示した「水素透過膜とプロトン導電性セラミックスを用いた燃料電池」と比較すると、図5に示す電解質においては、バルク(この場合、Pd膜→AECeO3膜へと続く一連の部分のこと)内をプロトンがPd膜→AECeO3膜というように直接的かつ連続的に通過することができるという点が特徴的である。この場合、Pd膜と酸化物(AECeO3)膜の界面で電子の授受(電極反応)が生じないため、酸化物(AECeO3)は発生したHまたはH2によって還元されるということがなくなる。つまり、プロトン導電性を維持しつつ、CO2に対する脆弱性を克服することが可能となる。
【0055】
以上のように金属(Pd)膜と酸化物(AECeO3)膜とを接合して理想的界面を作成するにあたっては、以下のような手法によるコーティングが考えられる。
【0056】
まず、酸化物(AECeO3)膜に金属(Pd)膜を成膜する方法としては、以下の方法が挙げられる。
(1) 化学的…無電解めっき法、電解めっき法、MOD法、CVD法、スラリーコート法、スピンコーティング法、ディップ法
(2) 物理的…PVD法{スパッタ、蒸着、MBE(Molecular Beam Epitaxy)法、プラズマ溶射法、プラズマスプレー法}
上記の中で、無電解めっき法は、緻密薄膜を均一に作製可能で、酸化物(AECeO3)膜と金属(Pd)膜の複合界面の連続接合性を向上させる上で好適な方法である。また、プラズマ溶射法およびプラズマスプレー法も、緻密薄膜を作製可能である。本発明においては、緻密膜を複合する必要があるため、工業的に量産化をターゲットにおけば、自動成膜化が容易なプラズマ溶射法およびプラズマスプレー法が複合膜を成膜することに好適である。
【0057】
また、多孔質支持体上に酸化物(AECeO3)膜を薄く成膜する方法としては、以下の方法が挙げられる。
(1) 化学的…スラリーコート法、スピンコーティング法、ディップ法
(2) 物理的…PVD法{スパッタ、蒸着、MBE(Molecular Beam Epitaxy)法、プラズマ溶射法、プラズマスプレー法}
上記の方法を採用することで、酸化物(AECeO3)膜の薄膜化が可能となり、さらに、多孔質支持体上に酸化物(AECeO3)薄膜を成膜し、さらにその上に金属(Pd)薄膜を成膜することで低コスト化につながる。
【0058】
なお、ここではPdやPd-Ag合金を例に検討したが、上述したような水素の透過形態からすると、複合する膜として、水素を透過するPd以外の金属(例えばニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)など)の膜、これらの元素のうち2種以上からなる合金膜、あるいは酸化物(例えばSrZrO3、BaZrO3)の膜等を適用した場合にも同様の効果が得られると考えられた。中でも、金属膜あるいは合金膜を用いる場合には、それ自体がプロトン−電子混合導電体と見なすことができるため、電極が不必要となる利点がある。
【0059】
また、Pd膜を使用することから、コスト的にある程度薄膜化する必要があるが、Pd膜等の水素透過膜単独の薄膜化は強度的に問題があるという点に関しては、以下のように考えた。すなわち、まずPd膜自体を厚くして当該Pd膜自体を基材としても機能させることを検討してみたが、コスト面を考慮すると到底実用的ではない(Pdの使用量が実用レベルを超える)と考えられた。そうすると、このように複合膜自体で強度を確保するよりも、水素透過に影響を及ぼさない材質によって基材(支持体)を構成することが目的に適うと考えられた。具体的には、プロトン導電性酸化物膜と水素透過膜とからなる複合膜を多孔質支持体によって支持するという構造が適すると考えられた。この場合における多孔質支持体は、ガス中から水素を分離するという複合膜の機能に影響を及ぼすものではない。しかも、この多孔質支持体が上記複合膜を強度的に支持する基材として働くことから、複合膜自体で所定の強度を確保する必要がなくなることから、この複合膜を構成するプロトン導電性酸化物膜と水素透過膜のそれぞれを必要十分な厚さにまで薄膜化することが可能となる。
【0060】
ここまで説明したように、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質は燃料電池における電解質として特有の効果を奏しうるものであるが、適用可能な電気化学デバイスはこれに限らない。以下に発明者による実験結果を実施例1として示すように、水素センサー、水素ポンプ、水蒸気センサーといった電気化学デバイスにも好適なものである。さらには、以下に説明するように、排ガス浄化用電気化学デバイスにおける電解質としてもきわめて好適なものである。
【0061】
すなわち、本発明者は、水素センサー、水素ポンプ、水蒸気センサー以外にも、本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質の適用の可能性について検討した。一例として、具体的には、このプロトン導電性酸化物膜−水素透過膜複合膜型電解質を排ガス浄化用電気化学デバイスとして適用することについて詳細な検討をした。その背景は以下のとおりである。
【0062】
すなわち、従来、自動車など内燃機関の排気ガス中に含まれる窒素酸化物(NOx)の除去方法としては、活性成分として貴金属を担持した触媒による接触還元法が用いられてきている。つまり、上記した自動車排気ガス浄化用の触媒として、白金やロジウムなどの貴金属を比表面積の大きいアルミナなどの担体に担持した触媒が用いられている。また、ボイラなどの各種プラントの排ガス中に含まれる窒素酸化物(NOx)は、NH3 添加による無触媒脱硝法、触媒を用いたNH3 による選択接触還元法によってN2 およびO2 に変換され、無害化されている。しかし、このような従来の窒素酸化物の除去方法には下記(1)、(2) の問題点がある。
【0063】
(1) 自動車排気ガス浄化用触媒の問題点
自動車排気ガス浄化用の貴金属を担持した触媒は、貴金属の埋蔵量が少なく、価格が高く、さらに貴金属は、価格変動が大きく、安定して確保することが難しい問題がある。近年の自動車はリーンバーン域での燃焼にシフトする方向にあり、この結果、不完全燃焼を伴い、NOx が増加する傾向にあり、貴金属担持量の増加を伴うことなくNOx 除去率を向上することが可能な技術開発が必要である。なお、窒素酸化物の他の分解法として、アンモニアによる還元法が挙げられ、この方法は貴金属を必要としない点で優れているが、アンモニアを供給するための設備を必要とし、自動車のようにスペースに余裕がない場合、アンモニアを供給するためのボンベなどの装置を自動車に搭載することは困難である。
【0064】
(2) ボイラなど各種プラントにおけるアンモニアを用いた還元法の問題点
ボイラなどの各種プラントにNOx のアンモニアによる還元法を適用する場合、未反応のアンモニアが、排ガス中に僅かに含まれるSOx と反応し、硫安類を生成し、ボイラの熱回収装置である熱交換器の内部の低温部に付着し、熱交換器の圧力損失が経時的に増加する問題があり、アンモニア添加量の低減が望まれる。
【0065】
以上の問題に鑑み本発明者は本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質が排ガス浄化用電気化学デバイスに適用された場合に特有の効果を奏することを知見するに至った。すなわち、本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、基本的には水素ポンプの動作原理により、雰囲気中の水蒸気から化学式4〜7で示される欠陥平衡によってプロトンを取り込み、カソード側に水素を生成する。この生成された水素によってNOxが以下の化学式9,10に基づいて還元される。さらに、このNOxの還元反応は、デバイスに通電する電流量によって制御することができるので、NOxセンサーによるフィードバック制御により、よりきめ細かいNOx還元が実現できる。
【0066】
【化9】
【化10】
【0067】
例えば自動車の排気ガス中には水蒸気が存在し、走行中における排ガス浄化用触媒の温度は数百度にも達する。このような状況下におかれた自動車の排ガス浄化用電気化学デバイスは、排ガス中の水蒸気を分解し、排ガス中にNOx の還元剤である水素を供給し、化学式9,10に基づいてNOx を分解する。
【0068】
また、上述したボイラなど各種プラントにおける排ガスも多くが水蒸気を含有しているため、これら各種プラントにおけるアンモニアによる無触媒脱硝もしくは触媒を用いたNOx のアンモニア選択接触還元においても、ボイラの高温部に本発明に係る排ガス浄化用電気化学デバイスを配設することによって、排ガス中に水素を供給することが可能となり、還元剤としてのアンモニアを低減できる。
【発明の効果】
【0069】
以上のように、請求項1のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によると、AECeO3のプロトン導電性は維持しつつも、AECeO3のCO2に対する脆弱性を克服してCO2を含む混合ガス雰囲気環境下でも使用することが可能となる。
【0070】
また、請求項2のプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、希土類元素セリウム(Ce)の一部を当該セリウム(Ce4+)とは異なる他の低原子価希土類元素で置換したというもので、AECeO3のプロトン導電性は維持しつつもAECeO3のCO2に対する脆弱性を克服することができる。低原子価元素で置換することにより、例えば、Ce4+→Yb3+とすると、結晶格子中の電気的中性が崩れ、これを補うため格子中の酸素1/2個が抜ける。そうすると格子中の電気的中性が保たれる。このように意図的に酸化物イオン空孔を生成させること(ドーピング)ができ、よりプロトンの生成が促進されることになる。また、他の希土類元素としては、請求項3に記載のようにイッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素を用いることができる。
【0071】
水素透過膜が金属または合金である請求項4のプロトン導電性酸化物膜−水素透過膜複合膜型電解質の場合、当該金属膜自体をプロトン−電子混合導電体と見なすことができるため燃料極が不必要になるという利点がある。この場合、水素透過性を示す金属または合金としては、請求項5に記載のごとくパラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とするものを用いることができる。尚、請求項7に記載したように、これらの金属または合金を無電解めっき法によりプロトン導電性酸化物膜表面に成膜することで、より緻密かつ均一な膜が成膜可能となり、プロトン導電性酸化物膜−水素透過膜複合膜型電解質における複合界面の連続接合性が向上し、AECeO3のCO2に対する脆弱性を克服しつつ、アノード過電圧を低減することが可能となる。
【0072】
また、多孔質支持体が複合膜のガス側の表面に形成されている請求項7のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によると、水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となることから、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。請求項8のように複合膜のガス側とは反対側の表面に多孔質カソードが形成されている場合においても同様である。さらに、請求項9に記載したように多孔質カソードに銀(Ag)を含むことにより、カソード過電圧を低減することが可能となる。
【0073】
また、複合膜のガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜が形成されている請求項10のプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、プロトン導電性酸化物膜の両面に保護膜が形成された状態となっている。したがって、カソード側を流れるガス(酸化剤ガス)中にCO2が含まれていても、プロトン導電性酸化物膜(AECeO3)のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することが可能となる。別の水素透過膜が金属または合金である請求項11のプロトン導電性酸化物膜−水素透過膜複合膜型電解質の場合、当該金属膜自体をプロトン−電子混合導電体と見なすことができるため電極が不必要になるという利点がある。この場合、水素透過性を示す金属または合金としては、請求項11に記載のごとくパラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金をを成分とするものを用いることができる。尚、請求項13に記載したように、これらの金属または合金を無電解めっき法によりプロトン導電性酸化物膜表面に成膜することで、より緻密かつ均一な膜が成膜可能となり、プロトン導電性酸化物膜−水素透過膜複合膜型電解質における複合界面の連続接合性が向上し、AECeO3のCO2に対する脆弱性を克服することができる
【0074】
多孔質支持体が、複合膜のガス側とは反対側の表面に形成された多孔質カソードである請求項14のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によると、多孔質カソードが基材としても機能し、複合膜を強度的に支持する。したがってこの場合においても水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となることから、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。
【0075】
複合膜のガス側とは反対側の表面に多孔質カソードが形成され、さらに該多孔質カソードの表面に多孔質支持体が形成されている請求項15のプロトン導電性酸化物膜−水素透過膜複合膜型電解質によっても、水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となることから、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。
【0076】
また、複合膜のガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜が形成され、さらに該別の水素透過膜の表面に多孔質支持体が形成されている請求項16のプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、プロトン導電性酸化物膜の両面に保護膜が形成された状態となっている。したがって、カソード側を流れるガス(酸化剤ガス)中にCO2が含まれていても、プロトン導電性酸化物膜(AECeO3)のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することが可能となる。
【0077】
また請求項17の発明によれば、燃料装置等の各種装置において、AECeO3のプロトン導電性は維持しつつもAECeO3のCO2に対する脆弱性を克服してCO2を含む混合ガス雰囲気環境下で使用することが可能となる。例えば、請求項18のように水素センサーに適用したり、請求項19のように水素ポンプに適用したり、請求項20のように水蒸気センサーに適用したりすることができる。あるいは、請求項21のように排ガス浄化用デバイスに適用することもできる。
【発明を実施するための最良の形態】
【0078】
以下、本発明を図面に示す実施の形態に基づいて詳細に説明する。
【0079】
本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質は、図1に示すようにCO2を含むガス3中から水素を分離可能な水素透過性あるいはプロトン(H+)透過性の電解質であって、プロトン(H+)が通過可能なプロトン導電性酸化物膜1と、プロトン(H+)が通過可能な水素透過膜2との複合膜からなり、これら両膜の界面が連続接合した状態で、プロトン導電性酸化物膜1の少なくともガス3側の表面に水素透過膜2が形成されているものである。
【0080】
プロトン導電性酸化物膜1としては、バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造としたものを採用できる。
【0081】
Aサイト元素は、電解質抵抗低減の観点からバリウム(Ba)を採用することがより好ましい。
【0082】
また、酸化物イオン空格子点を発生させてプロトン導電性を発現させるために、Bサイトのセリウム(Ce)はイッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素で一部置換することが好ましいが、これらの元素に限定されるものではなく、セリウム(Ce)とは異なる他の低原子価希土類元素を採用することも可能である。
【0083】
尚、本発明のプロトン導電性酸化物膜のような固体電解質は、焼結性が低減するほど電解質抵抗は高くなる。かかる観点から、焼結性を高めることが好ましい。焼結性を高めるための手法としては、焼結温度を上昇させる、最終成形時に成形加重を増大する等が挙げられるがこれらに限定されるものではない。好ましい焼結度としては90%以上、さらに好ましくは95%以上である。
【0084】
水素透過膜2は、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な膜であり、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とすることが好ましいが、これらに限定されるものではなく、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な材料であれば、金属もしくは合金等に限らず、酸化物等の材料も採用することが可能である。尚、水素脆化を抑える効果のあるAgと合金化したものがより好適である。
【0085】
ここで、金属もしくは合金膜を水素透過膜として採用する場合には、それ自体がプロトン−電子混合導電体とみなせるため電極として作用する。したがって、燃料極を必要としなくなるという観点から金属もしくは合金膜を採用することが好ましい。尚、酸化物等の材料を用いる場合には、燃料極を併用すれば良く、また、金属もしくは合金膜を用いた水素透過膜が薄いために電極としてうまく作用しない場合にも燃料極を併用すればよい。
【0086】
燃料極としては、燃料ガスを透過させ、かつ電極として作用する材料が好ましく、例えば多孔質金属、格子状に形成した電極もしくはガスを透過させるための穴を複数あけた電極等を採用することができる。水素透過膜が酸化膜の場合には酸化膜をガス側に配置し、燃料極をプロトン導電性酸化物に接触させて用いるようにすればよい。また、水素透過膜が金属もしくは合金の場合には、燃料極をプロトン導電性酸化物に接触させて用いるようにしてもよいし、水素透過膜をプロトン導電性酸化物に接触させ、ガス側で燃料極を水素透過膜に接触させるようにしても良い。
【0087】
次に、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質においては、アノード過電圧を低減させるために、複合界面が連続接合させることを特徴とするものである。
【0088】
複合界面の連続接合は、プロトン導電性酸化物膜を基材として緻密膜な水素透過膜を形成するか、もしくは水素透過膜を基材として緻密膜なプロトン導電性酸化物膜を形成することにより達成される。本発明では、プロトン導電性酸化物膜を基材として緻密膜な水素透過膜を形成している。
【0089】
緻密膜を形成するための方法としてペースト法が挙げられる。この場合には、ペースト材料の粒子径が大きいため、ペーストを基板に塗布した後に高温処理することで、基材との密着性が向上し、アノード過電圧を低減することが可能となる。
【0090】
ペースト剤には、例えば、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)のうち1つもしくは2つ以上の元素を粒子状にしたものを含ませて、良好な水素透過性を示す膜を形成することが好ましい。
尚、水素脆化を抑える効果のある銀(Ag)を含ませることがより好ましい。
【0091】
また、ペースト法を用いる場合、金属レジネートをペースト剤に添加して基材との密着性を向上させることが好ましい。これにより基板とペースト法により形成した膜が剥離するのを防ぐことが可能となる。
【0092】
金属レジネートは、金属樹脂酸塩であり、金属レジネートを含まない金属を直接熱処理する場合と比べて、樹脂が熱処理中に燃えることにより細かい金属が発生するようになる。この細かい金属がセラミックスへミクロ的に食い込む、すなわち、セラミックスへのアンカー効果を期待して配合されるものであり、例えば、硫化テレピネオールPdを挙げることができるが、これに限定されるものではない。
【0093】
基板に金属レジネートを含むペースト剤を塗布した後、熱処理をおこなうとポア(穴)が発生する。このポアは、CO2を通過させてしまい、CO2に対して脆弱な本発明のプロトン導電性酸化物膜をCO2に曝すことになるため好ましくない。そこで、当該ポアを埋めるために、金属レジネートを含まないペーストをさらに重ね塗りして前記熱処理温度よりも高温で熱処理して水素透過膜をより緻密にすることが好ましい。
【0094】
Pd-Ag合金膜を形成する場合を例に挙げて説明すると以下のようになる。まず金属レジネートを含むペーストを基板に塗布し、200℃程度でペースト剤に含まれるバインダーを焼失させる。次いで、熱処理を行う。熱処理温度は900℃程度ではポアが無数に発生し、熱処理温度を上昇させるにつれてポア数は減少する。ただし、1300℃以上ではPd-Agが焼結し始めてポア径が増加する。よって、熱処理温度をxとすると、900℃<x<1300℃であればよいが、1100℃<x<1300℃であることが好ましく、最も好ましくはx≒1200℃である。
【0095】
上記の最も好ましい熱処理温度である1200℃で熱処理を施してもポアは発生する。そこで、当該ポアを埋めるため、ペーストを重ね塗りする。ここで、重ね塗りをおこなうためのペーストとしてはポアが発生しにくい金属レジネートを含まないペーストが好ましい。これを一層目の膜を熱処理した温度よりも高温の1300℃程度で熱処理することにより、ポアが焼失し、かつ緻密な膜を形成することが可能となる。
【0096】
次に、さらに緻密な膜を形成して複合界面の連続接合状態をより良好なものとするための好ましい手段としては、物理堆積法(PVD法)、化学堆積法(CVD法)、放電重合法、ゾル−ゲル法、電解めっき法、無電解めっき法、スクリーン法等が挙げられるが、コスト面、成膜簡易性および成膜均一性の観点から、無電解めっき法を採用することが最も好ましい。
【0097】
ここで、プロトン導電性酸化物膜はプロトン導電性は有するものの、電子導電性は有しないため、めっきされにくい。そこで無電解めっき処理を施す前に基板にめっき反応に対する活性を付与する必要がある。非導電性基板に無電解めっきを施す方法としては、湿式法による代表的な活性触媒化前処理である二液法(センシタイゼーション−アクチベーション法)を採用して、基板にめっき反応に対する活性を付与する触媒活性化前処理を行うようにすればよいが、めっき反応に対する活性を付与できるならば、この方法に限られるものではない。
【0098】
ここで、無電解めっき法の採用にあたって、プロトン導電性酸化物膜上に膜形成するためにめっき浴を準備する必要がある。当該めっき浴中には、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)、銀(Ag)等を含ませることで、目的とする水素透過膜の構成成分がめっきされて膜形成が可能となる。
【0099】
例として、Pd-Ag合金を無電解めっき法により形成する方法について説明する。プロトン導電性酸化物膜を基板としてめっき反応に対する活性を付与する。具体的にはSnCl2塩酸溶液とPdCl2塩酸溶液それぞれの溶液に基板を交互に浸漬し、Pd核を発生させる。次に、Pdめっき浴として、[Pd(NH3)4]Cl2・H2Oを含むアルカリ性溶液中にめっき反応に対する活性を付与した基板を浸漬し、Pd核を触媒としてその周囲にPdを析出させる。次に、Agめっき浴として[Pd(NH3)4]Cl2、AgNO3を含む溶液中にPdを析出させた基板を浸漬し、Agを析出させる。最後に、N2雰囲気にて熱処理することでプロトン導電性酸化物膜1と非常に良好に連続接合したPd-Ag緻密合金膜を得ることができる。
【0100】
次に、プロトン導電性酸化物膜を薄膜化することがコスト低減の観点から好ましい。プロトン導電性酸化物薄膜を成膜する方法としてはスラリーコート法、スピンコーティング法、ディップ法、物理堆積法(PVD法)が挙げられるが、薄膜を形成することが可能であればこれらに限定されるものではない。尚、プロトン導電性酸化物膜はコストな問題を考えた場合、その厚さを50μm以下にするのがよく、35μm以下にするのがより好ましく、20μm以下にするのがさらに好ましい。ただし、プロトン導電性酸化物膜をここまで薄膜化すると電解質自立型構造をとることが難しくなるので、多孔質支持型構造とすることが好ましい。具体的には、多孔質支持体にプロトン導電性酸化物薄膜を成膜し、さらに水素透過薄膜を形成する。これにより低コストな電気化学デバイスの作製が可能となる。
【0101】
尚、コスト面を考えた場合の水素透過膜の好適な膜厚は、1μm以下、より好ましくは0.5μm以下、さらに好ましくは0.1μm以下である。
【0102】
以上、プロトン導電性酸化物膜1と水素透過膜2とを複合膜化した電解質の最も好適な組み合わせは以下のようになる。すなわち、プロトン導電性酸化物膜1としてAサイトをバリウム(Ba)とし、Bサイトのセリウム(Ce)をイッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素で一部置換したものを用い、水素透過膜2として無電解めっき法により形成したパラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とする膜を用いることが最も好適である。
【0103】
このように、プロトン導電性酸化物膜1と水素透過膜2とを複合膜化した電解質においては、水素のみが(具体的にはプロトンの形で)Pd膜を透過しAECeO3膜に達する。このため、AECeO3のCO2に対する脆弱性を克服でき、CO2を含むガス3の環境下でも使用できる電気化学デバイスの電解質として使用可能となる。このようなプロトン導電性酸化物膜−水素透過膜複合膜型電解質は電気化学デバイスに適用して好適なものであり、一例を挙げれば、自動車排ガスのような水蒸気およびNOx を含有する排ガスを浄化するための排ガス浄化用デバイスとして適用することができる。こうした場合には、排ガス中の水蒸気を分解し、排ガス中にNOx の還元剤である水素を供給して当該NOx を分解することができる。
【0104】
さらに、プロトン導電性酸化物膜−水素透過膜複合膜型電解質におけるカソード電極としては、銀(Ag)を用いることが好ましい。従来、カソード電極としては高コスト材料である白金(Pt)が用いられてきたが、白金(Pt)よりも銀(Ag)を用いた方が、コストを低減でき、カソード過電圧も低減できる点で好ましい。銀(Ag)の成膜法としてはペースト法等が挙げられるが、これらに限定されるものではない。
【0105】
また、多孔質カソードをペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物単体もしくはペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物と銀(Ag)のサーメットとするのが好ましい。この場合にも白金(Pt)に比べコストを低減できる。ペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物としては、例えばLaMnO3やSrMnO3が挙げられるが、これらに限定されるものではない。プロトン導電性酸化物膜にこれらを良好に接触させるためにはプロトン導電性酸化物膜に対して電子−酸化物イオン混合導電性酸化物を押しつけながら熱処理を施すか、もしくは単にこれらの膜同士を接触させて熱処理すればよいがこれらの方法に限定されるものではない。
【0106】
上述した好適例にさらにカソード電極として銀(Ag)、ペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物単体もしくはペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物と銀(Ag)のサーメットを用いることで、電気化学デバイスとして非常に優れた構成となる。
【0107】
また、本実施形態における複合膜は多孔質支持体Sによって強度的に支持され、これによって薄膜化することが可能となっている。多孔質支持体Sは、例えば多孔質Niによって構成される。このように多孔質支持体Sによって複合膜を支持する場合の具体的な形態を以下に説明する(図30〜図34参照)。
【0108】
まず、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体Sが形成されているというものである(図30参照)。この複合膜のアノードガス側と反対側の表面には多孔質カソード(図30〜図34中において符号4で示す)が形成されている(図30参照)。ちなみに、本実施形態における多孔質カソード4の厚さは40μmであるがこれは一例に過ぎずこの厚さに限定されるわけではない。また、本実施形態においては多孔質支持体Sと多孔質カソード4とが別個に設けられているが(図30参照)、多孔質カソード4自体に多孔質支持体としての機能を備えさせることも可能である(図32参照)。多孔質支持体Sはガスの拡散を妨げることなく、0.002〜3μm、好ましくは0.004〜0.5μmの最高を有するものを適宜選択して使用することが可能である。具体的には、例えば、0.1〜0.5μmの細孔を有するNi-YSZサーメット多孔体またはNi-Al2O3サーメット多孔体などを例示できる。これにより、水素透過膜(例えばPd金属)2の薄膜化を可能とするため、各種電気化学デバイスとして好適な膜厚を設定することが可能となる。また、600℃以下の低温になれば、アノード側に設けられる多孔質支持体Sには金属NiまたはNi合金、カソード側に設けられる多孔質支持体SにはSUS316L、SUS310SなどのSUS材が使用可能となる。
【0109】
また、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体Sを形成し、尚かつアノードガスとは反対側の面にプロトン(H+)が通過可能な別の水素透過膜2’を形成することもできる(図31参照)。多孔質支持体Sは水素透過を妨げることなく水素透過膜(例えばPd金属)2,2’の薄膜化を可能とする。また、カソード側を流れるガス(酸化剤ガス)5中にCO2が含まれている場合にも、プロトン導電性酸化物膜1を覆う水素透過膜2’がこのプロトン導電性酸化物膜1のCO2に対する脆弱性を克服する。
【0110】
図32に示すように、複合膜のアノードガス側とは反対側の表面に形成された多孔質カソード4を多孔質支持体Sとして機能させることもできる(図32参照)。この場合、多孔質カソード4が基材としても機能し、複合膜を強度的に支持するため、水素透過を妨げることなくPd等の水素透過膜2を薄膜化することが可能となる。
【0111】
図33に示すように、複合膜のうちアノードガス側とは反対側の表面に多孔質カソード4を形成し、さらにこの多孔質カソード4のカソード側表面に多孔質支持体Sを形成することもできる(図33参照)。この場合にも、水素透過を妨げることなくPd等の水素透過膜単独の薄膜化が可能となる。
【0112】
図34に示すように、複合膜のアノードガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜2’を形成し、さらにこの水素透過膜2’のカソード側表面に多孔質支持体Sを形成することもできる(図34参照)。この場合、プロトン導電性酸化物膜1の両面に保護膜が形成された状態となっている。したがって、カソード側を流れるガス(酸化剤ガス)5中にCO2が含まれていても、プロトン導電性酸化物膜(AECeO3)1のCO2に対する脆弱性を克服してCO2を含むガス雰囲気環境下でも使用することが可能となる。
【0113】
以上が本発明の好適な実施形態であるが、ここで説明したのは好適な実施の一例に過ぎず、これに限定されることなく本発明の要旨を逸脱しない範囲において種々変形実施が可能である。例えば、上述したような排ガス浄化用デバイスに限らず、燃料電池、水素センサー、水素ポンプ、水蒸気センサーなどの電気化学デバイスに適用して好適なものである。
【0114】
なお、本明細書では「プロトン導電性酸化物膜−水素透過膜複合膜型電解質」を中心に説明しているが、ここでいう「複合膜型」としては、物理的に接合されている場合と化学的に接合されている場合の両方が考えられる。仮に化学的な接合であると考えると、両膜の間に中間生成物が存在しこれがプロトン移動の高抵抗層になる可能性がある一方で、中間生成物が逆に高プロトン導電層になる可能性も存在する。以上を考慮すると、化学的接合の場合にはプロトン移動の高抵抗層が存在しないことが条件になる。
【0115】
また、本発明を例えば排ガス浄化用電気化学デバイスに適用する場合にあっては、以下に述べるPd/SCO複合膜(SCOはSrCe0.95Yb0.05O3-aのことであり、以下単にSCOと呼ぶ)が、Pd薄膜によって両側が覆われる構造を有するPd/SCO/Pd電解質膜であることが望ましい。電気化学デバイスを排ガス中で使用する際にはアノードおよびカソードともに排ガス中に曝されているので、カソード側もPd電解質膜で覆うことによってこちら側がCO2と反応して分解してしまうことが防止できる。
【実施例】
【0116】
続いて、以下では実際に行った実験および検討の具体的内容を実施例として示しつつ、まず金属/酸化物複合膜型電解質の概念について説明し、その後、本発明にかかるAECeO3のCO2に対する脆弱性克服の確認をするための実験内容を説明していく。
【0117】
[実施例1]
本発明者は、本発明にかかるプロトン導電性酸化物膜−水素透過膜複合膜型電解質の有効性を検証するため、Pd/AECeO3複合膜を成膜し、CO2を含む燃料を用いる燃料電池での発電の可能性を確認した。これは、主として、「CO2に対する脆弱性の克服の確認」や「電気化学デバイスのセル構造」について検証するとともに、水素センサー、水素ポンプ、燃料電池といった各種電気化学デバイスへの適用を検討したものである。
【0118】
なお、本実施例においては、固相である金属(Pd)と固相である酸化物(AECeO3)との界面(固/固界面)を連続とし、この固/固界面を、固相と液相との界面(固/液界面)と同様の接合状態とすることを念頭においた。これは、マクロレベルでは両者に差異はないもののミクロレベルにおいて比較すると大きな隔たりが生じること、より具体的には、電解質に液相が使用されている場合には固/液界面においてプロトンが連続移動できる一方で、固/固界面においては図29に示すように不連続面が生じ、
・界面に生じるミクロな空間が電気化学的抵抗となって性能が低下する
・界面では極度な還元雰囲気となり、酸化物が還元されて材料が劣化する
といったことが生じるおそれがあることによる。つまり、固/固界面においては特にプロトンの連続した導電が重要であり、固/液界面と同様の接合状態を実現する必要がある。ちなみに、固/液界面では、液体を保持する容器として金属Pd膜が必要とされているのに対し、固/固界面ではセラミックのCO2耐性を金属Pd膜で覆い確保するという点で差異がある。
【0119】
以下では、
(1)金属/酸化物複合膜型電解質の作製
(2)CO2雰囲気連続暴露試験による複合膜のCO2耐性の確認
(3)Pd/SCO複合膜のプロトン導電性
(4)Pd/SCO複合膜を用いた燃料電池発電
という項目に分け、この項目に沿って説明する。
【0120】
(1)金属/酸化物複合膜型電解質の作製
(1.1)水素透過膜2の検討
水素透過膜2として必要な水素透過能力をもつ金属材料のほとんどは、水素雰囲気下では自己崩壊を起こすことが知られている。これらの金属は、水素の圧力と温度に応じて多量の水素を吸収し、相変態する性質がある。相変態は、金属格子を拡張するため金属内に歪みが生じ、塑性変形(水素脆化)する。水素透過膜2として、注目されるPd膜も例外ではなく、水素脆化による膜の寿命が問題となる。しかし、Pdは、Agが置換型固溶し合金化した状態では、Agが水素原子と相互作用し、相変態を緩和するとされている。また、Agが適量固溶すると水素の拡散係数は減少するが、水素の溶解度は増加し、合金化により水素透過係数が増加する。このため、近年、燃料電池や半導体産業など、高純度水素を必要とする分野において、Pd−Ag合金膜を利用した水素分離膜の研究が盛んに行われている。そこで本発明者は、酸化物の複合材として上記のPd−Ag合金に着目した。
【0121】
(1.2)ペースト組成
本実施例では、Pd−Ag合金膜をペースト法にて作製した。Pd−Agペーストは、表1に示す性状のPdおよびAg金属粉末、溶剤(αテレピネオール)、バインダー(エチルセルロース、および酸化物とのアンカー効果を期待して金属レジネート:硫化テレピネオールPd)を適量混練し作製した。なお、金属レジネートは、金属樹脂酸塩であり、金属レジネートを含まないPdを直接熱処理する場合と比べて、樹脂が熱処理中に燃えて細かいPd金属が発生するようになる。この細かいPd金属がセラミックスへミクロ的に食い込む、すなわち、セラミックスへのアンカー効果を期待して配合したものである。Pd/Ag混合比(wt%)は、水素透過能が最も優れると報告されているPd/Ag=77/23を仕込み組成とし、金属レジネートを含むペースト(AR)および金属レジネートを含まないペースト(AN)の2種類を作製した。
【0122】
【表1】
【0123】
(1.3)アルミナ基板上への成膜
熱処理温度を決定するために、アルミナ基板上にPd−Agの成膜を試みた。アルミナ基板にペースト(AR)を塗布し、雰囲気炉にて、200℃、空気中でバインダーを焼失させた後、900, 1000, 1100, 1200, 1300℃でアニール処理を1時間施し、熱処理温度を比較した。その結果を図6に示す。900℃では、膜表面に無数の穴が空いているが、アニール温度を上昇すると徐々にその数が減少した。しかし、1300℃以上になると逆に穴の径が大きくなった。これは、Pd−Agが焼結し始めたことを意味する。また、金属レジネートを添加しないペースト(AN)を使用したところ、1200℃以下では、膜の剥離が観察された。金属レジネートを添加したペーストを使用した場合には膜の剥離は起こらなかったことから、金属レジネートを用いることでアンカー効果を有するようになることが確認された。さらに、金属レジネートを添加しないペースト(AN)を使用して1300℃で熱処理した場合には剥離はなく、また、ARほどポア数は多くならなかった。
【0124】
ARでは、熱処理温度1200℃において、最もポア数が少ない結果となったが、依然として、ポアが観察され、完全な緻密膜ではなかった。そこで、残ったポアを埋めつつ、より高温で熱処理して膜の緻密化を図るため、成膜した前述の膜の上に1300℃でも熱処理しても金属レジネートを添加したペースト(AR)に比べてポアの発生量が少ない金属レジネートを添加しないペースト(AN)を重ね塗りし、1300℃でアニール処理を施した。図7にその結果を示す。重ね塗り処理により、ほとんどのポアが消失した。以上より、金属レジネートを添加したペースト(AR)をアルミナ基板上に塗布して1200℃で熱処理しPd−Ag膜を形成した後に、金属レジネートを添加しないペースト(AN)を当該Pd−Ag膜上に重ね塗りして1300℃で熱処理を施す成膜法が成膜条件として好ましいと判断した。
【0125】
(1.4)SrCeO3基板上への成膜
次にAECeO3基板上へのPd膜の成膜を試みた。AECeO3基板は、まず入手が容易なTYK社製のSrCe0.95Yb0.05O3-a(SCO)のうち、φ16.8mm、厚さ1.0μm、相対密度95%以上のものの緻密体を選定した。成膜条件には、前節(1.3)のアルミナ上への成膜条件を用いた。結果を図8に示す。SCO基板においても、アルミナ基板と同様の成膜条件、すなわち、金属レジネートを添加したペースト(AR)をSCO基板上に塗布して1200℃で熱処理しPd−Ag膜を形成した後に、金属レジネートを添加しないペースト(AN)を当該Pd−Ag膜上に重ね塗りして1300℃で熱処理することで、緻密かつほとんどのポアが消失した膜の形成が可能であることが確認された。
【0126】
(1.5)Pd/SCO複合膜の密着性
Pd-Ag膜とSCO基板との密着性を確認するため、試料断面を鏡面仕上げして光学顕微鏡によりその断面を観察(観察倍率×500)した。結果を図9に示す。Pd-Ag膜は、SCO基板に非常に良く密着しており、引っ掻き試験においてもPd膜が剥がれないことを確認した。更に、SCO基板が相対密度約95%以上の緻密膜でありながらクローズドポアを含有していることも確認された。一方、Pd膜にはほとんどポアはなく、非常に緻密な膜として成膜していることが確認できた。膜厚は、約65μmであった。
【0127】
(1.6)EPMAによる複合膜の断面観察
膜の元素分析をEPMA(Electron Probe MicroAnalyzer: JEOL, JXA-8900R)によって行った。EPMAによるPd、Ag、Ce、Srの特性X線像を図10に示す。各膜成分元素の拡散は確認されず、成膜した膜は、化学拡散を伴わずに物理的に良く密着して複合化していることがわかった。また、1300℃という高温での熱処理において元素相互の拡散がなかったことは、Pd-Ag相とSCO相との間に化学的反応が全くないことを示した結果とも言える。したがって、Pd/SCO複合膜は、Pd-Ag膜およびSCO基板各々の物性を維持して複合していると考えられる。更に、Pd-Ag膜は、ペースト作製仕込み初期には、それぞれPd粒子、Ag粒子であったが、Pd, Ag元素の偏在化なしに、均一に合金化していることが確認できた。
【0128】
次に、EDX解析より、Pd-Ag膜組成の分析を行った。図11に、標準試料を用いたEDX測定(Energy Dispersive X-ray Analysis:エネルギー分散型X線解析の略)の実測値による組成補正を行った結果を示す。仕込み組成はPd/Ag=77/23であったのに対して、成膜後の組成はPd/Ag=84.9/15.1となり、Ag成分がかなり減少した結果となった。これはアニール処理温度が大きく影響していると考えられる。Pd-Ag二元系相図を図12に示す。PdとAgは互いに同じ立方晶の結晶構造を持ち、全率固溶体を形成することが知られている。Agの融点(962℃)は、Pd(約1555℃)と比較してかなり低く、アニール処理昇温中に、まずAg粒子が最初に溶解すると考えられる。EPMAの結果から、SCO膜内へのAgの拡散は確認できなかったが、Pd-Ag膜ではPd元素に関しては均一に分布しているが、Ag元素に関しては、表面からSCO膜界面へ僅かに濃度分布が確認された。このことは溶解したAg粒子が膜表面より昇温とともに蒸発するため、表面側に向かうにつれて僅かにAg濃度が低くなり、最終的にPdと合金化するAgの濃度分布が生じたものと考えられ、トータルのAg量も減少したものと考えられる。
【0129】
(2)CO2雰囲気連続暴露試験による複合膜のCO2耐性の確認
作製した複合膜のCO2に対する耐性を確認するために、CO2雰囲気における連続暴露試験を行った。
【0130】
(2.1)実験方法
試験に供した試料は、2種類(SCO基板、Pd/SCO複合膜基板)である。Pd/SCO基板は、上述した(1.1)〜(1.6)における成膜条件、すなわち、SCO基板全体に金属レジネートを添加したペースト(AR)を塗布して1200℃で熱処理しPd−Ag膜を形成した後に、金属レジネートを添加しないペースト(AN)を当該Pd−Ag膜上に重ね塗りして1300℃で熱処理することで、SCO相の露出面をなくした。双方の試料をアルミナフェルトの上に置き、管状炉内にセットした。CO2耐性試験は、管状炉内を真空にした後、CO2雰囲気で行った。CO2暴露時間は10時間とし、温度は650℃で一定とした。
【0131】
(2.2)XRDによる複合膜の表面組成解析
CO2曝露試験後のXRD解析結果を図13に示す。Pd/SCO板は、SCO層の分析を行うために表面のPd膜を剥がした。熱処理前のSCO基板のピークと比較して、CO2暴露後のSCO基板のピークでは、ほぼCeO2およびSrCO3に帰属する回折ピークを示し、SCO相が完全に分解したことを確認した。一方、Pd/SCO基板では、ほぼSCO相のみに帰属する回折ピークを示しており、Pd膜との複合化により、SCO相のCO2に対する脆弱性が克服されることを明らかとした。
【0132】
(3)Pd/SCO複合膜のプロトン導電性
(3.1)実験方法
Pd/SCO複合膜のプロトン導電性を確認するために、(1)水素濃淡電池法により得られる起電力から、導電種がイオンであるか否かを判断し、(2)水素の電気化学的透過法により、直接、プロトンを導電種として同定した。以下に、上記2種類の測定法を説明する。
【0133】
(3.1.1)水素濃淡電池法
プロトン導電体を挟む両側に水素の活量差(濃度差)が生じると、その活量差に応じた起電力を示す。水素の濃淡差を用いた水素濃淡電池の作動原理を図14に示す。プロトン導電性固体を電解質隔壁とし、その両面に多孔質電極を取り付けて二つの電極室とし、一方に高水素分圧のガスを、もう一方に低水素分圧のガスを導入すると、高水素分圧側を負極として起電力が発生する。このときの電池構成は、式(化学式)11のように表され、負極(アノード)と正極(カソード)で化学式12,13のような電極反応が生じようとして、ネルンストの式(化学式14)から導出される起電力E(V)が発生する。
【0134】
【化11】
【化12】
【化13】
【化14】
ここで、Rは気体定数(8.314JK-1mol-1)、Tは絶対温度(K)、Fはファラデー定数(96485Cmol-1)、PH2(1)とPH2(2)は、アノードおよびカソードの水素分圧である。化学式14において一方の水素分圧(例えばPH2(1))を固定すれば、図15に示すように、起電力はもう一方の水素分圧(この場合、PH2(2))の対数に対して直線関係となる。また、測定した起電力Eを化学式14より求めた理論起電力E0で除した値は、その条件におけるイオン輸率とみなすことができる。尚、プロトン導電体を挟む両側に水素の活量差(濃度差)が生じると、その活量差に応じた起電力を示すことを利用して、これを水素センサーに適用することが可能である。
【0135】
(3.1.2)水素の電気化学的透過法
水素の電気化学的透過法(水素ポンプ)の作動原理を図16に示す。プロトン導電性固体を隔壁として一方に水素源を導入し、他方に不活性ガス、例えばアルゴンなどを導入する。アルゴン側をカソードとして直流を通電する時、もしプロトン導電体であれば、それぞれの極で化学式12,13のような電極反応が起きる。この場合、プロトン以外のイオンは、この隔壁内を通過できないので、水素のみを透過(ポンプ)することができる。このように水素ポンプは、電気化学反応を利用して、強制的に水素を透過させるために、供した電解質のプロトン導電性を直接確認することができる。更に、カソード出口ガス分析から得られる水素濃度から、水素透過速度を計算した後、ファラデーの法則から計算される理論水素透過速度と比較すれば、プロトン輸率(全導電種に占めるプロトンの占める割合)が計算できる。
【0136】
(3.2)Pd/SCO複合膜を用いた水素濃淡電池
作製した電解質膜は、図17、図18に示すようにSrCe0.95Yb0.05O3-a(厚さ1mm、φ16.8mm)基板5に、片側にはPd-Ag膜6(φ13mm、厚さ65μm)が成膜されている。他方には中央部に円状(φ12mm)に白金ペーストを塗布し、900℃において30分空気中で焼き付け多孔質電極Pt7(φ12mm)として試験に供した。実験に使用した装置概略を図19に示す。両面の電極部分に集電体として白金ネットを取り付けた試料を、パイレックス(登録商標)ガラスリングを介してφ17×13mmのアルミナ管で上下から押さえつけた。それぞれのガス室は、1000℃まで昇温することによりパイレックス(登録商標)ガラスを軟化させてガスタイト(ガスシールが完全に効いている状態)にした。アノード6およびカソード7とも、膜流量計を用いて入出口の流量に差がないことで、ガスタイトであることを確認した。その後、約30分間空気を導入して電極の状態を安定させた後、所定のガスに切り替えて実験を開始した。図19において、ガス流量はMFC8により設定し、加湿器9を経由してアノード6およびカソード7それぞれに目的のガスが供給され、アノード排ガス10、カソード排ガス11が排出されるようにしている。ここで、符号5は電解質を表している。カソード排ガス11はマイクロガスクロマトグラフィー装置13により、排ガス組成を測定可能となるよう配置している。尚、反応温度は電気炉12により調整するようにしている。ガスは100ml/minの流量で導入した。アノード6とカソード7間に発生する起電力は、エレクトロメータ(北斗電工製 HE-106)を用いて測定した。ガスは、室温加湿(3%)した100%H2を、アノード6となるPd膜側に導入し、カソード7にはそれぞれ1,5,10,20,50,100%H2をArベースの混合ガスとして導入した。測定前にカソード入口ガスをマイクロガスクロマトグラフィー(HP社製 M-200、モルキュラシーブカラム:MS-20)を用いて、目的ガス組成になっていることを確認した。
【0137】
650,750,850,950℃において、それぞれのガス組成における起電力を測定した結果を図20に示す。測定条件範囲内では、ネルンストの式(化学式14)から計算される理論値にほぼ一致した。この結果から、Pd/SCO複合膜の導電種はイオンであることが確認された。
【0138】
(3.3)Pd/SCO複合膜を用いた水素の電気化学的透過実験
水素濃淡電池法においては、導電種がイオンであるか否かは判断できるものの、プロトンが導電するか否かを直接判断することはできない。そこで、アノード、カソード間に直流を通電し、カソード側に水素が発生するか否かを直接確認した(水素ポンプ)。上述の場合、Pd/SCO複合膜(図17、図18参照)を挟んでPd-Ag膜側に水素を、他方にアルゴンを、双方とも室温でバブリングし加湿して、100ml/minで流通した。装置は、上述の実験装置(図19参照)を使用した。直流通電にはソーラートロン社製ポテンショガルバノスタットSI1286 を使用し、アノード−カソード間に定電流を流した。その際、カソード出口ではマイクロガスクロマトグラフィー(HP社製 M-200、モルキュラシーブカラム:MS-20)を用いて発生水素量を定量した。測定は、650, 750, 850, 950℃において実施した。
【0139】
通電電流に対する水素の発生速度を図21に示す。破線はファラデーの法則に従って計算した理論水素発生速度(ml/min)である。650℃では、ほぼ理論値通りの水素発生速度を示し、Pd/SCO複合膜はプロトン輸率tH+が1に極めて近いプロトン導電体であることがわかった。しかし、温度が上昇するに従い、高電流において破線からはずれ、水素発生速度が低下した。低電流ではほぼプロトン輸率が1であるが、高電流ではプロトン以外の電荷担体が存在することになる。この点については、電流量を増やすとカソード中の水蒸気が分解して化学式15のように酸化物イオン導電性が発現する旨の報告もなされている(S.Hmajima, H.Matsumoto, H.Iwahara, 第26回固体イオニクス討論会予稿集P154, 2000)。この報告によれば、高電流ではプロトン輸率が低下し、電流効率が低下した可能性がある。
【0140】
【化15】
【0141】
したがって、Pd/SCO複合膜は、ほぼSCO膜と同様のプロトン導電機能を有すると考えられる。以上より、SCO膜は、Pdと複合化することによって、プロトン導電機能を損なうことなく、SCOと同様のプロトン導電機能を有することが明らかとなった。
【0142】
(4)Pd/SCO複合膜を用いた燃料電池発電
(4.1)燃料電池の発電原理
プロトン導電体を用いる水素酸素燃料電池の原理を説明する。作動原理を図22に示す。今、化学式16のような水素と酸素の燃焼反応を考えてみる。
【0143】
【化16】
水素の燃焼反応は、水素の酸素による酸化反応であり、通常、熱エネルギーを放出する。この燃焼反応は酸素の立場から見ると、燃焼による酸素の還元反応である。従って、この両反応が同一の場で起こる時に熱エネルギーが放出される。そこで、例えば図22に示すように、プロトン導電性電解質を介して水素をアノード活物質として酸化反応を行わせ、酸素を正極(カソード)活物質として還元反応を行わせることができれば、水素の燃焼反応の化学エネルギーを、乾電池と同様の原理で直接電気エネルギーに変換することができる(化学式17,18)。
【0144】
【化17】
【化18】
【0145】
この場合、電極間の起電力Eは、水素の酸化反応より電子が放出されるアノードと酸素の還元反応により電子が不足するカソードとの間の電位差として発生する。水素酸素燃料電池の場合、起電力Eは以下の式によって表すことができる。
【0146】
【化19】
ここで、Kは化学式16の平衡定数を、piは各ガス分圧を、添字a、cはそれぞれアノードおよびカソードを、R、FおよびTはそれぞれ気体定数、ファラデー定数および絶対温度を表す。
【0147】
(4.2)実験方法
実験で用いた装置構成を図23に示す。図23の基本構成は図19と同様であるが、アノード側に供給できるガスがH2ではなくO2である点で異なっている。電解質には、(3.2)と同様の試料(図17、図18参照)を使用した。試料電解質を電解質隔壁として、Pd-Ag膜側をアノード、多孔質白金側をカソードとしてアルミナ管で挟み込んだ。アノードには燃料ガスとして室温飽和した湿潤水素または水素と二酸化炭素の湿潤混合ガスを、カソードには酸化剤ガス5として室温飽和した湿潤酸素をそれぞれ100ml/min流通した。このように燃料電池を構成することによって、ネルンストの式(化学式19)から計算される起電力を発生するようになる。発生した端子電圧はエレクトロメータ(北斗電工製 HE-106)を用いて測定した。外部負荷装置としてソーラートロン社製ポテンショガルバノスタットSI1286 をアノードとカソードとの間に接続し発電性能を確認した。
【0148】
(4.3)Pd/SCO複合膜を用いた燃料電池の特性
測定温度が650,750,850,950℃における水素−酸素燃料電池の起電力を図24に示す。同時にネルンストの式(化学式19)より計算される理論起電力を同図に示した。起電力は、650℃でほぼ理論起電力に近い値を示した。試験前には、必ずガスタイトとクロスリークがないことを確認しているので、起電力の実測値と理論値の比は、イオン輸率(実測値/理論値):tion(0<tion<1)と表され、逆に電子輸率(この場合、ホール伝導)はth(=1−tion)として表される。起電力から計算されるtionおよびthを図25に示す。tionは、950℃において0.88であったが、650℃において0.99となった。この結果、今回作製したPd/SCO複合膜は、650℃よりも低温の燃料電池雰囲気条件において、プロトン輸率が1の純粋なプロトン導電体として機能することがわかった。
【0149】
次に、実際に電流を取り出し発電を試みた。結果を図26に示す。発電時に安定な電圧および電流を示し、本発明に係るPd-Ag膜とSCO膜との複合膜型電解質を用いて、燃料電池発電が可能であることがわかった。
【0150】
(4.4)CO2を含んだ燃料ガスを用いた長時間発電特性
前節(4.3)までは、Pd/SCO複合膜を用いて発電の可能性を確認したが、本来の目的は、CO2を含むアノードガス条件における発電の可能性を確認することである。これを受け、以下に説明するように、CO2を含むアノードガスを導入した場合、Pd膜によってSCO膜のCO2による変質がなく、安定に燃料電池発電ができることを確認することにした。
【0151】
図27にCO2とH2の混合ガスをアノードに導入した場合の発電時の電圧経時変化を示す。電圧は、初期にはしばらく減少した後、一定となり安定した。また、連続負荷試験を行ったところ、少なくともCO2曝露試験と同じ10日間は安定して発電できることが確認できた。以上の結果より、本発明に係るプロトン導電性を維持し、かつCO2に対する脆弱性を克服できる金属/酸化物複合膜型電解質(Pd/SCO)は、CO2を含む燃料を用いる燃料電池に適用可能であることが明らかとなった。
【0152】
[実施例2]
実施例1では、Pd/SCO膜を燃料電池の電解質に用いた場合の発電の可能性に検討ついて検討し、理論通り実証することができた。しかし、実際に燃料電池の電解質として適用するには、動作する温度において種々の検討を加えなければならない。そこで、以下に実施例2として、金属/酸化物複合膜型電解質を用いた中温形燃料電池に対して、性能面および電解質の材料コスト面から金属/酸化物複合膜型電解質についてさらに検討した。
【0153】
燃料電池発電試験は、[実施例1]と同様の手順で行った。尚、実施例2では、SCOよりも導電率が高く、コスト低減につながるBaCe0.8Y0.2O3-a(以下BCOと呼ぶ)についても検討した。作製した複合膜型電解質は、SCOまたはBCO基板片側中央部に円形状にPd-Ag合金膜(φ13mm、厚さ65mm)が成膜されている。他方には中央部に円形状(φ13mm)に白金ペーストを塗布し、900℃において30分空気中で焼き付け多孔質電極として試験に供した。
【0154】
電解質にSCOおよびBCOを用いた場合の中温域(500〜600℃)における電流―電圧特性を図39に示す。発電試験温度は、Pd/SCO複合膜では600℃とし、Pd/BCO複合膜では500および600℃とした。600℃の電流―電圧特性から、短絡電流密度は、プロトン導電率の高いBCOを用いることによって、約2倍となった。また、Pd/BCOの500℃での電池性能は、Pd/SCOの600℃での電池性能レベルの3分の2程度まで向上した。しかしながら、Pd/BCOセルにおいても、短絡電流密度が約7mAcm-2であるため、更なる性能向上が期待される。そこで、次にBCOセルの性能を支配する要因について分析を行い、性能向上への課題を抽出するための実験を以下の手法により行った。
【0155】
まず、電流―電圧特性および過電圧特性を評価した。これらを評価することで、BCOセルの発電時の電圧降下要因を電解質抵抗、アノード反応抵抗、カソード反応抵抗に分離することができる。したがって、この結果から電池性能がどの要因(構成部材)でどのくらい低下しているか割合を把握することが可能となる。過電圧特性は、参照電極16を設け、カレントインターラプション(電流遮断)法により測定した。参照電極15は、図38に示すように電解質14の側面に、Ptペーストを塗布、乾燥後、900℃において焼き付け、作製した。その上からφ0.3mmの白金線を巻き付け白金リード線16に取り付けた。符号19はアノード電極であり、電解質14を介して反対側の面にはカソード電極が形成されている。カソード電極側、アノード電極側には集電ネット17が配置され、ガラスパッキン18によりこれらを挟むようにしている(アノード電極側の集電ネット17およびガラスパッキン18は図示を省略してある)。
【0156】
ここで、カレントインターラプション(電流遮断)法について説明する。電極と電解質からなる電気化学セルは最も簡略化された電気的な等価回路で表すと図40のようになる。高温においては、電解質のキャパシター成分のコンダクタンスは無視できるほど小さい(Cb=0)と考えられ、電解質に関しては通常オーム抵抗(Rb)のみで表される。電極―電解質界面に関しては電極反応抵抗Reと二重層容量Cdlから成る並列回路で表すものとする。カレントインターラプション法は、実際の電池に数msec程度のサイクルによるパルス電流で電気二重層を充電し、その後観察される過渡曲線から過電圧を求める方法である。図41(b)に装置構成を示す。具体的には、ファンクションジェネレーター20(NF回路ブロック WF1256)およびポテンショガルバノスタット23(SolarTron社 SI1286)を用いて、測定セル24のカソード、アノード間に2msec/サイクル、0.1msecのオフパルス幅でのパルス電流を印加する。この際、各電極―参照電極間の電位の変化は、デジタルオシロスコープ21を用いて同期させ、エレクトロメーター22で測定した。この場合、図41(a)に示すように電流が遮断されたときにオーム抵抗に相当する電位分だけ急激に変化する(図41、iRbドロップ)。次にCdlに充電された電荷は、キャパシタンスと並列にある抵抗(電極反応抵抗Re)を通って放電し、その分の電位が過渡的に変化する。このように電位差を遮断する前後の電位を測定することにより、過電圧(h=iRe)を求めることが可能である。
【0157】
尚、試験中には経時的に電解質が劣化していないか確認するために、交流法にてセル端子間に1kHz, 5mVの交流信号を入力し、そのインピーダンスを電解質抵抗としてモニターした。
【0158】
500℃における燃料電池の電流―電圧特性を図42に示す。また、図42には、電流を取り出すことによる電圧降下の内訳をBCO膜による過電圧(電解質過電圧)、Pd膜による過電圧(アノード過電圧)および多孔質Ptによる過電圧(カソード過電圧)から求めた。アノード過電圧が一番小さく、次いでカソード過電圧、電解質過電圧の順に過電圧が大きくなっており、500℃において、短絡電流密度は、7mA/cm2以下であった。そこで、各電池部材を材質および作製法から見直すことにより、過電圧を低減し、性能向上を図ることを試みた。
【0159】
(1)電解質緻密化による過電圧低減
電解質過電圧は、電解質抵抗に比例する。そこで、図42における電解質過電圧から初期BCOの電解質抵抗を求め、図43に示した。図43に示されるBCOの500℃における導電率から換算される電解質抵抗と比較して、初期BCOの電解質抵抗がかなり大きいことがわかった。この原因としては、1電解質の反応劣化および2電解質の焼結性が悪いことが挙げられる。1の原因については、これまで試験後のBCO表面をX線回折解析を行っているが、BCO単相のピークしか得られていないことから、BCOの反応劣化は起こっていないと考えられる。このため、過電圧のほとんどは2の電解質の焼結性に起因していると考えられる。BCOのような固体電解質は、その焼結度が95%以上ならば、導電率から換算される電解質抵抗を示し、逆に焼結度が低くなるにつれて、電解質抵抗は増大する。BCOでは、導電率から換算される理論電解質抵抗は、500℃で約6Ωとして得られるはずであるが、今回、試験中に測定した電解質抵抗は、500℃で約80Ω程度であった。電解質抵抗には、電解質の正味の抵抗以外に、集電ネットとの接触抵抗、リード線抵抗が含まれている。しかしながら、接触抵抗およびリード線抵抗が数10Ω以上あることは考えられないことから、80Ωのほとんどが電解質抵抗に起因することがわかる。つまり、初期BCOは、その焼結性が悪く、BCOの緻密化が必要であることがわかった。そこで、BCOの焼結工程を見直した。具体的には、焼結熱処理前の最終成形において、成形加重を増大したところ、同重量の焼結体において、約8%体積が減少し、緻密化することがわかった。この緻密化したBCO試料を試験に供し、初期のBCOによる電解質抵抗、改良後のBCOの電解質抵抗およびBCOの導電率から換算されるBCOの電解質抵抗を物性値として比較した結果を図43に示す。初期のBCOの電解質抵抗は、BCO導電率から計算される電解質抵抗より約20倍程度大きく、性能低下要因になっていた。しかし、今回、BCO電解質の緻密化を図ったことにより、初期BCO電解質抵抗と比較して、約7分の1程度まで電解質抵抗を低減できた。電解質の緻密性は電気化学デバイス性能を左右するものであるため、焼結度を95%以上とすることでより好ましい電気化学デバイス性能が得られるようになることが期待された。
【0160】
(2)Pd膜成膜法見直しによる過電圧低
(2.1)Pd成膜法の検討
前述したようにアノード過電圧の低減も一つの課題として挙げられた。以下、アノード過電圧の低減方法について膜の緻密性および電解質への密着性の観点から検討した。
【0161】
基板に対する膜の密着性をさらに向上させ、緻密膜を成膜するためには、成膜温度を十分高くする必要がある。しかしながら、Pdペースト膜は、実施例1で上述したように、Agとの合金膜であるために、処理温度を高くすることはできない。そこで、膜を構成する出発粒子を細かくすることによって、処理温度を高くすることと同等の効果が得られると考えた。かかる観点から、基板との密着性が良く、均一な緻密膜が成膜できる可能性の高い無電解めっき法について検討した。
【0162】
(2.2)無電解めっき法によるPd合金緻密膜の成膜
セラミックス、プラスチック、ガラスなどの非導電性基板に無電解めっきを施すためには、基板にめっき反応に対する活性を付与する触媒活性化前処理を施す必要があることが知られている。また、この時の処理によって、基板表面に形成される触媒核の分布や活性が変化し、表面形態や物性が異なるめっき膜が析出することが知られている。ここでは、湿式法による代表的な触媒活性化前処理である二液法(センシタイゼイション―アクチベーション(sensitization - activation)法)を用いて無電解めっきを施した。二液法活性化前処理は、一般に次の反応式で表される。
Pd2++Sn2+=Pd+Sn4
以下、ここで行った無電解めっき法を図44で示す無電解めっきフロー図に基づき説明する。
準備段階として、基板をアセトン洗浄しアルコール置換後に乾燥した。次に触媒活性化前処理(二液法)として、SnCl2塩酸溶液を一液、PdCl2塩酸溶液を二液として、一液と二液へ交互に10回浸漬を繰り返し、Pd核を析出させた。次に、[Pd(NH3)4]Cl2・H2O、EDTA・2Na、NH3(28%水溶液)、H2NNH2・H2Oを含み、アンモニア・アルカリ性とした無電解めっき浴に1時間浸漬して、Pd核を触媒としてその周囲にPdを析出させ、Pdの薄膜を形成した。これを、蒸留水にて洗浄し、[Pd(NH3)4]Cl2 、AgNO3、EDTA・2Na、NH3(28%水溶液)、H2NNH2・H2Oを含む溶液に1時間浸漬して、パラジウム薄膜上に銀を析出させて銀の薄膜を形成した。さらにこれを蒸留水にて洗浄し、N2気流中にて、850℃、10時間熱処理を施し、Pd合金緻密膜を得た。
【0163】
(2.3)EPMAによるPd合金膜の断面観察
無電解めっき基板には、Al2O3を選定し、めっき膜性状を確認した。基板上にめっきされたPd合金緻密膜の断面分析をEPMA(Electron Probe MicroAnaly- zer: JEOL, JXA-8900R)によって行った。EPMAによるPd, Ag, Alの特性X線像を図45に示す。図中において、(a)はPd、(b)はAg、(c)はAlの特性X線像である。PdおよびAgは相互によく拡散し、合金化している様子が観察された。一方、基板からのAl元素のPd膜側への拡散は確認されなかった。
次に、EPMA付属のEDX(Energy Dispersive X-ray)解析より、Pd合金膜組成の分析を行った。表2は、PdおよびAg標準試料を用いて、膜の各部位におけるEDXによる組成比(wt%)を定量した結果である。組成比は、平均でPd/Ag=76.6/23.4となり、良好な結果を得た。以上より、無電解めっき法を選定することにより、Pd, Ag元素の偏在化なしに、均一に目的組成に合金化が可能であることを確認できた。
【0164】
【表2】
【0165】
(2.4)無電解めっきを適用した場合のアノード過電圧
BCO上に無電解めっきによりPd合金緻密膜を施した複合膜型電解質とペースト法を用いた複合膜型電解質を用いて、アノード過電圧を比較した。測定法は、カレントインターラプション法を用いた。500℃におけるアノード過電圧を測定した結果を図46に示す。無電解めっき法を用いたPd合金緻密膜では、ペースト法により作製されたPd合金緻密膜と比較してアノード過電圧をさらに低減できることが確認された。したがって、無電解めっき法のPd合金緻密膜は、ペースト法により作製されたPd合金緻密膜と比較してBCOとより強固に密着し、かつ緻密膜となっていることが示唆された。
【0166】
ここで、複合膜型電解質のアノード反応について、以下のように考察した。1水素の緻密膜上への拡散、吸着、2水素分子の水素原子へ解離、3水素原子のPd緻密膜への溶解、4水素の拡散、5Pd/BCO界面における水素のプロトンへのイオン化、6プロトンのBCOへの溶解である。アノード過電圧が大きくなる要因を以下に考察した。1〜4の過程については、Pd合金膜の物性に依存すると考えられ、成膜法による差異は生じないと考えられる。しかしながら、5、6の過程には、膜の密着性の優劣に依存する。これらの観点からも、無電解めっき法のPd合金緻密膜は、ペースト法により作製されたPd合金緻密膜と比較してBCOとより強固に密着していることが示唆された。
【0167】
(3)カソード材見直しによるカソード過電圧低減
これまでカソード材として使用してきた多孔質Ptは、図47に示す過電圧の温度依存性に現れているように温度の低下に伴う電極触媒能の低下により、カソード過電圧がかなり増大している。また、Pt自体高価な材料であり、今後の中温形燃料電池のカソードとして適切ではないと考えられる。そこで、カソード材について検討をおこなった。
【0168】
中温域においては、電極活性を示し、かつ安価なカソード材料を選択しなければならない。最近、安価なAgを用いたカソードにおいて、良好な中温域の電極活性が示されている。Ptが約3,000円/gであるのに対し、Agは約26円/g(26,000円/kg)とコスト的に優位である。そこで、本研究では、Agをカソード目標材として、Pt, Pdを比較カソード材として、500℃における過電圧の比較を行った。Pdは、アノード材として、中温域で使用できているために、カソードにおいて適用可能か確認するために選択した。過電圧の比較結果を図48に示す。それぞれペーストを塗布後、乾燥し、Pt, Pdは900℃において焼き付け、Agは融点が962℃と低いために、800℃で焼き付けを行った。図48より、カソード過電圧は、多孔質Agカソードを適用することで大幅に低減できることが明らかとなった。一方、Pdは、Ptと同程度のカソード過電圧を示した。
【0169】
試験後のカソード表面のSEM写真を図49に示す。図中において(a)はPd、(b)はPt、(c)はAgカソード表面のSEM観察結果である。Agの表面形態は、Pt, Pdと比較して、Agが凝集しているように観察された。通常、多孔質電極では、電極反応が起こる三相(ガス-電極-電解質)界面の面積が狭いほど過電圧が大きくなるが、図48では、上記とは逆に過電圧が低くなる結果を示した。この結果については、唯一Pt, PdにはないAgの特性として、酸素の透過能を示すことが電極反応に関与している可能性が示唆される。以上より、Agの中温域でのカソード材としての可能性が示された。
【0170】
(4)性能向上対策を施したセルによる電池性能の向上
電解質の緻密化、Pd成膜法の見直し、カソード材の変更により過電圧が低減され、セル性能の向上が見込めるため、それぞれの改善策を適用して単セルを再構成し、500℃で発電試験を行った結果を図50に示す。無電解Pd合金めっき緻密膜+BCO膜+多孔質Agカソードを選定した結果、最大出力密度は、500℃でも0.037Wcm-2を示し、実施例1のPdペースト法を用いたPd/SrCeO3セルの電池性能よりも約15倍性能を向上させることに成功した。
【0171】
次に、中温形燃料電池において到達可能な電池性能を試算し、その電池性能レベルを検討した。中温形燃料電池の電池性能試算は以下のように行った。BCOの導電率、Pd緻密膜とAg多孔質カソードの過電圧特性から、電解質抵抗、アノードおよびカソード反応抵抗が得られることから、電池出力密度つまり電池性能を試算することができる。試算条件として、水素酸素燃料電池において、動作温度500℃、電解質の薄膜化が成功したとして、BCO膜厚20mmと設定し、アノードには無電解Pd合金メッキ緻密薄膜を、カソードには多孔質Agを選定し、全抵抗を算出した後、電流-電圧特性から出力密度特性を試算した。試算結果を図51に示す。比較のためにこれまでの結果も同図に示す。500℃における試算性能は、無電解Pd合金メッキ緻密薄膜+BCO薄膜+多孔質Agカソードを選定した場合、現行の高温SOFC 性能(0.22 Wcm-2, 300mAcm-2, 0.74Vにおける出力密度)に匹敵する0.24 Wcm-2(BaCeO3膜厚20mm, 300mAcm-2における出力密度)と試算され、高温SOFCに匹敵する電池性能レベルであることが示された。
[実施例3]
ここまでは、Pd/SCO膜およびPd/BCO膜を燃料電池の電解質に用いた場合の発電の可能性を検討し、理論通り実証することができた。そこで、以下に材料コスト面からさらに考察を加え、燃料電池としての可能性を試算した。
【0172】
比較対象はSOFCとした。また、コスト試算では、SCOよりも導電率が高く、コスト低減につながるBCOを比較対象とした。本発明に係る複合膜型電解質は、金属と酸化物電解質を複合化することにより、金属部がアノード機能も持ち合わせるため、アノード材が不要となるメリットがある。一方、適用したパラジウム(Pd)合金膜は、高い水素選択透過性を示し、半導体製造に必要な高純度水素の製造に使用されるポテンシャルをもつが、金属Pdは金や白金と同様に極めて高価な金属であり、実用化を妨げる大きな障害となる。水素透過膜2の材質としては、現在、これに代わる安価な金属膜材料の開発が活発に行われており、その中で、Zr-Ni系、V-Ni系、Nb-Ni系などの安価な合金材料に注目が集まっている。これら透過膜の水素透過能は、Pd膜(10-7mol/(m2・s・Pa))より1桁劣るものの、例えば、Zr-Ni系では、Pd膜に比べて原材料費は数百分の一と大変安価となる。したがって、実用化を目指すためには、Pd使用量を極力減らすか、安価な水素透過膜2の開発が不可欠であると考えられる。
【0173】
実際に、材料コスト面から金属/酸化物複合膜型電解質の膜厚を試算した。試算式には、次の数式1を用いた。また、材料単価および密度は、表3の値を用いた。
【0174】
【数1】
【0175】
【表3】
【0176】
まず、SOFCにおける電解質(YSZ)+アノード(Ni-YSZ)分のコストを試算した。試算性能は、電解質厚さ40μm、アノード厚さ100μmとし、300mA/cm2において0.74Vで行った(結果については表3参照)。YSZを用いたSOFCの(電解質+アノード)の1kWあたりの原材料コストは約1,785円となった。実際に試算されるコストには、製造に要した様々なコストをトータルに考慮する必要があり、月産ベースで試算されている平板型SOFCを例にとってみると、約68,000円/kWであり、電解質+アノード個別のコストでみると約11,000円/kWと試算された。次に、YSZの原材料コストをベースにPd-BCOのコストを試算した。Pd-BCOでは、Pdの原価が極端に高いために、Pd膜厚をパラメータとし、YSZの原材料コストと比較することで、最適なPd膜厚およびBCO膜厚を求めた。なお、BCO膜厚は、現行のSOFCの電解質膜厚が数10μmであり、製法上からも10μm以下の電解質膜厚を作製することは難しいと考えられるため、YSZの膜厚オーダーに揃えた。結果を図28に示す。図28よりPd膜厚が0.1μmよりも厚い場合、コスト的に実用化は難しいと思われる。また、Pd膜厚が0.1μm以下でも、BCO膜厚が20μmより厚くなると、YSZよりも割高になってしまうと考えられる。また、Pd膜厚は、現在報告されているPd水素透過膜2の成膜技術で0.1μmまでは支持膜方式を用いて薄膜化が可能であることから、0.1μmまでのPd膜厚を試算対象とすると、コスト的にBCO膜厚は20μm以下が望ましいと試算される。
【0177】
以上より、Pd/BCO電解質膜は、最も厚くて約20μmであるために、構造は電解質自立膜型では、到底機械的強度を保つことができない。従って、カソードあるいはアノードによる多孔質体支持膜型構造が望ましい。多孔質体支持膜型構造はセンサーで使用できる構造である。電流を流さない起電力式のセンサーの場合はオーミックロスによる電圧降下がないので、電解質膜自立型構造をとることもできる。他のデバイスについては、通電するため電解質はどうしても極力薄くする必要がある。このため、電解質自身では強度が足りなくなるため多孔質体支持膜型構造が望ましい。
【0178】
以上、原材料コスト面から、金属/酸化物複合膜型電解質の今後の開発目標仕様を試算した。電解質は、SCOから導電率の高いBCOに変え膜厚を20μm以下に、Pd膜厚は0.1μm以下とすることが望ましい。
【図面の簡単な説明】
【0179】
【図1】本発明に係るプロトン導電性酸化物膜−水素透過膜複合膜型電解質の構造を簡単に示した概略図である。
【図2】AECe1-xMxO3-aの欠陥構造とプロトンの生成を、(a)化学量論組成と比較し(Stoichiometry)、(b)湿潤酸素中、(c)乾燥酸素中、(d)水素中のそれぞれについて概略的に示した図である。
【図3】Pd膜における水素透過の原理を示した図である。
【図4】水素透過膜とプロトン導電性セラミックスを用いた燃料電池の構造を示す図である。
【図5】金属/酸化物複合膜型電解質を用いた燃料電池の構造を示す図である。
【図6】金属レジネートを含むペースト(AR)および金属レジネートを含まないペースト(AN)につき、各温度(900℃、1000℃、1100℃、1200℃、1300℃)においてアルミナ板上に成膜されたPd膜の様子を示す画像である。(a)AR 900℃ in air、(b)AR 1000℃ in air、(c)AR 1100℃ in air、(d)AR 1200℃ in air、(e)AR 1300℃ in air、(f)AN 1300℃ in air、である。
【図7】二度の熱処理によってアルミナ上に緻密に成膜されたPd膜の様子を示す画像である。
【図8】二度の熱処理によってSCO基板上に緻密に成膜されたPd膜の様子を示す画像である。
【図9】試料断面の光学顕微鏡写真(×500)である。
【図10】試料断面のEPMAによる各元素の特性X線像を示す図である。
【図11】標準物質を用いたEDX実測値の補正内容を示すグラフである。
【図12】Pd-Agの二元系相図である。
【図13】XRD解析結果から見た曝露試験前後のSrCeO3相の比較結果を示すグラフである。
【図14】プロトン導電体を利用した水素濃淡電池の原理を示す図である。
【図15】水素濃淡電池における水素分圧と起電力との概略関係を示すグラフである。
【図16】プロトン導電体を利用した水素ポンプの原理を示す図である。
【図17】本実施例において作製した複合膜型電解質膜の平面図である。
【図18】本実施例において作製した複合膜型電解質膜の側面図である。
【図19】水素濃淡電池法の装置概略図である。
【図20】水素濃淡電池の起電力と水素分圧の関係(実戦:理論値)を示すグラフである。
【図21】電流に対する水素発生速度を示すグラフである。
【図22】プロトン導電体を利用した水素酸素燃料電池の原理を示す図である。
【図23】燃料電池実験に用いた装置の概略図である。
【図24】各温度における燃料電池の起電力を示すグラフである。
【図25】起電力から求めたPd/SCOの燃料電池雰囲気におけるプロトン輸率の温度依存性を示すグラフである。
【図26】Pd/SCO膜を用いた燃料電池の電流−電圧特性を示すグラフである。
【図27】CO2を含む燃料を用いた燃料電池における放電時の電圧の経時変化を示すグラフである。
【図28】Pd-BCO膜材料コストのPd膜厚依存性を示すグラフである。
【図29】固/液界面における接合の様子と固/固界面における接合の様子を示す図である。
【図30】本発明の一実施形態を示す図で、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体が形成されている複合膜型電解質を示したものである。
【図31】本発明の一実施形態を示す図で、複合膜の面のうちアノードガス(CO2を含むガス)側の表面に多孔質支持体を形成し、尚かつアノードガスとは反対側の面にプロトン(H+)が通過可能な別の水素透過膜を形成したものである。
【図32】本発明の一実施形態を示す図で、複合膜のアノードガス側とは反対側の表面に形成された多孔質カソードを多孔質支持体として機能させたものである。
【図33】本発明の一実施形態を示す図で、複合膜のうちアノードガス側とは反対側の表面に多孔質カソードを形成し、さらにこの多孔質カソードのカソード側表面に多孔質支持体を形成したものである。
【図34】本発明の一実施形態を示す図で、複合膜のアノードガス側とは反対側の表面にプロトン(H+)が通過可能な別の水素透過膜を形成し、さらにこの水素透過膜のカソード側表面に多孔質支持体を形成したものである。
【図35】各種ペロブスカイト型酸化物の水素中のプロトン導電率を示すグラフである。
【図36】SrCeO3膜を使った従来の水素分離法の概念を示す図である。
【図37】Pd膜を使った従来の水素分離法の概念を示す図である。
【図38】単セルのセッティング状態を示す図である。
【図39】電解質にSCOおよびBCOを用いた場合の中温域(500〜600℃)における電流―電圧特性を示す図である。
【図40】電極と電解質からなる電気化学セルの電気的な等価回路を示す図である。尚、Rb:電解質抵抗、Re:反応抵抗、Cb:電解質キャパシター容量、Cdl:二重層容量である。
【図41】カレントインターラプション(電流遮断)法について示す図である。(a)はオシロスコープで観測される波形、(b)はカレントインターラプション(電流遮断)法の装置概略図である。
【図42】500℃における燃料電池の電流―電圧特性を示す図である。図中でOCVは開回路電圧を表している。
【図43】初期のBCOによる電解質抵抗、改良後のBCOの電解質抵抗およびBCOの導電率から換算されるBCOの電解質抵抗を物性値として比較した結果を示す図である。
【図44】無電解めっき工程のフロー図である。
【図45】Al2O3基板上にめっきされたPd合金緻密膜の断面をEPMAにより分析した結果である、Pd, Ag, Alの特性X線像を示す図である。(a)はPd、(b)はAg、(c)はAlの特性X線像である。
【図46】BCO上に無電解めっきによりPd合金緻密膜を施した複合膜型電解質とペースト法を用いた複合膜型電解質を用いて、カレントインターラプション法により、500℃におけるアノード過電圧を測定した結果を示す図である。
【図47】多孔質Ptをカソード材として用いたときの過電圧の温度依存性を示す図である。
【図48】Ag、PtおよびPdをカソード材として用いたときの過電圧の比較結果を示す図である。
【図49】Ag、PtおよびPdカソード表面のSEM観察結果を示す図である。(a)はPd、(b)はPt、(c)はAgカソード表面のSEM観察結果である。
【図50】電解質の緻密化、Pd成膜法の見直しおよびカソード材の変更により過電圧を低減させるための改善策を適用して単セルを再構成し、500℃で発電試験を行った結果を示す図である。
【図51】アノードに無電解Pd合金メッキ緻密薄膜を、カソードに多孔質Agを選定し、全抵抗を算出した後、電流-電圧特性から出力密度特性を試算した結果を示す図である。
【符号の説明】
【0180】
1 SrCeO3膜(プロトン導電性酸化物膜)
2 Pd膜(水素透過膜)
3 酸化剤ガス(CO2を含むガス)
S 多孔質支持体
【特許請求の範囲】
【請求項1】
バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造とし、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能なプロトン導電性酸化物膜と、前記プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な水素透過膜との複合膜からなり、これら両膜の複合界面が連続接合した状態で、前記プロトン導電性酸化物膜の少なくとも前記ガス側の表面に前記水素透過膜が成膜され、尚かつ当該複合膜が多孔質支持体によって支持されていることを特徴とするプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項2】
前記プロトン導電性酸化物膜は、希土類元素セリウム(Ce)の一部が当該セリウム(Ce4+)とは異なる他の低原子価希土類元素で置換されている構造となっていることを特徴とする請求項1に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項3】
前記他の希土類元素は、イッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素である請求項2に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項4】
前記水素透過膜は水素透過性を示す金属または合金である請求項1から3のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項5】
前記水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とする請求項4に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項6】
前記水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とする請求項4もしくは5に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項7】
多孔質支持体が前記複合膜の前記ガス側の表面に形成されていることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項8】
前記複合膜の前記ガス側とは反対側の表面に多孔質カソードが形成されていることを特徴とする請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項9】
前記多孔質カソードが少なくともペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物および銀(Ag)のうちいずれか一つを含んでいることを特徴とする請求項8に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項10】
前記複合膜の前記ガス側とは反対側の表面に、前記プロトン(H+)が通過可能な別の水素透過膜が形成されていることを特徴とする請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項11】
前記別の水素透過膜は水素透過性を示す金属または合金である請求項10に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項12】
前記別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とする請求項11に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項13】
前記別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とする請求項11もしくは12に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項14】
前記多孔質支持体は、前記複合膜の前記ガス側とは反対側の表面に形成された多孔質カソードであることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項15】
前記複合膜の前記ガス側とは反対側の表面に多孔質カソードが形成され、さらに該多孔質カソードの表面に前記多孔質支持体が形成されていることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項16】
前記複合膜の前記ガス側とは反対側の表面に前記プロトン(H+)が通過可能な別の水素透過膜が形成され、さらに該別の水素透過膜の表面に前記多孔質支持体が形成されていることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項17】
請求項1から16のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質を用いた電気化学デバイスであって、燃料電池、水素センサー、水素ポンプ、水蒸気センサーおよび排ガス浄化デバイスのいずれかに適用されるものであることを特徴とする電気化学デバイス。
【請求項18】
少なくともH2とCO2とを含有する混合ガス中の水素濃度を定量できる水素センサーとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【請求項19】
少なくともH2とCO2とを含有する混合ガス中から水素のみを分離できる水素ポンプとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【請求項20】
少なくとも水蒸気とCO2とを含有する混合ガス中から水蒸気濃度を定量できる水蒸気センサーとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【請求項21】
少なくとも水蒸気、CO2およびNOx を含有する排ガスからNOxを除去するための排ガス浄化用デバイスとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【請求項1】
バリウム(Ba)またはストロンチウム(Sr)をAサイトに配し、希土類元素セリウム(Ce)をBサイトに配するペロブスカイト型酸化物(ABO3)を基本構造とし、プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能なプロトン導電性酸化物膜と、前記プロトン(H+)が通過可能でありCO2を含むガス中から水素のみを分離可能な水素透過膜との複合膜からなり、これら両膜の複合界面が連続接合した状態で、前記プロトン導電性酸化物膜の少なくとも前記ガス側の表面に前記水素透過膜が成膜され、尚かつ当該複合膜が多孔質支持体によって支持されていることを特徴とするプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項2】
前記プロトン導電性酸化物膜は、希土類元素セリウム(Ce)の一部が当該セリウム(Ce4+)とは異なる他の低原子価希土類元素で置換されている構造となっていることを特徴とする請求項1に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項3】
前記他の希土類元素は、イッテリビウム(Yb)、イットリウム(Y)、ディスプロシウム(Dy)、ガドリニウム(Gd)、サマリウム(Sm)、ネオジム(Nd)からなる群より選ばれた1種の希土類元素である請求項2に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項4】
前記水素透過膜は水素透過性を示す金属または合金である請求項1から3のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項5】
前記水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とする請求項4に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項6】
前記水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とする請求項4もしくは5に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項7】
多孔質支持体が前記複合膜の前記ガス側の表面に形成されていることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項8】
前記複合膜の前記ガス側とは反対側の表面に多孔質カソードが形成されていることを特徴とする請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項9】
前記多孔質カソードが少なくともペロブスカイト構造を有する電子−酸化物イオン混合導電性酸化物および銀(Ag)のうちいずれか一つを含んでいることを特徴とする請求項8に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項10】
前記複合膜の前記ガス側とは反対側の表面に、前記プロトン(H+)が通過可能な別の水素透過膜が形成されていることを特徴とする請求項7に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項11】
前記別の水素透過膜は水素透過性を示す金属または合金である請求項10に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項12】
前記別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分とする請求項11に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項13】
前記別の水素透過膜を構成する水素透過性を示す金属または合金が、パラジウム(Pd)、ニオブ(Nb)、タンタル(Ta)、ランタン(La)、チタン(Ti)、ジルコニウム(Zr)、銅(Cu)、ニッケル(Ni)からなる群より選ばれた1種または2種以上、あるいはそれらと銀(Ag)の合金を成分として無電解めっき法により形成されることを特徴とする請求項11もしくは12に記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項14】
前記多孔質支持体は、前記複合膜の前記ガス側とは反対側の表面に形成された多孔質カソードであることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項15】
前記複合膜の前記ガス側とは反対側の表面に多孔質カソードが形成され、さらに該多孔質カソードの表面に前記多孔質支持体が形成されていることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項16】
前記複合膜の前記ガス側とは反対側の表面に前記プロトン(H+)が通過可能な別の水素透過膜が形成され、さらに該別の水素透過膜の表面に前記多孔質支持体が形成されていることを特徴とする請求項1から6のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質。
【請求項17】
請求項1から16のいずれかひとつに記載のプロトン導電性酸化物膜−水素透過膜複合膜型電解質を用いた電気化学デバイスであって、燃料電池、水素センサー、水素ポンプ、水蒸気センサーおよび排ガス浄化デバイスのいずれかに適用されるものであることを特徴とする電気化学デバイス。
【請求項18】
少なくともH2とCO2とを含有する混合ガス中の水素濃度を定量できる水素センサーとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【請求項19】
少なくともH2とCO2とを含有する混合ガス中から水素のみを分離できる水素ポンプとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【請求項20】
少なくとも水蒸気とCO2とを含有する混合ガス中から水蒸気濃度を定量できる水蒸気センサーとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【請求項21】
少なくとも水蒸気、CO2およびNOx を含有する排ガスからNOxを除去するための排ガス浄化用デバイスとして適用されることを特徴とする請求項17に記載の電気化学デバイス。
【図1】

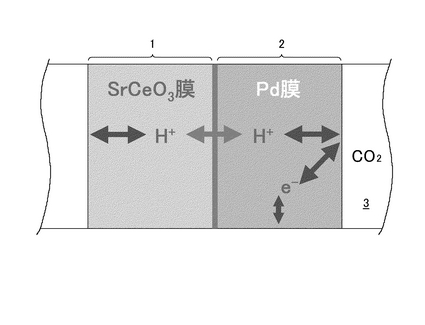
【図2】
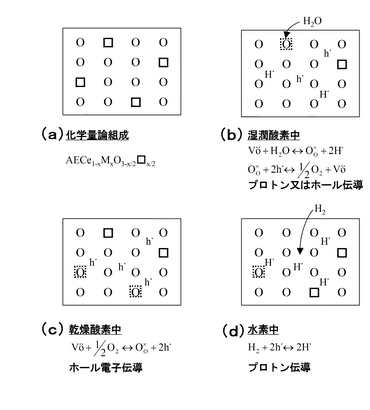
【図3】
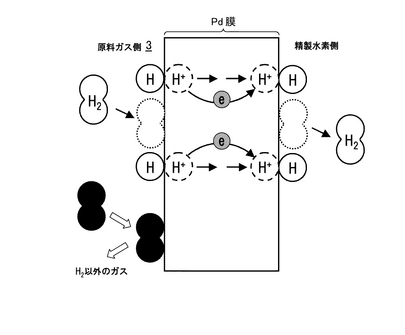
【図4】
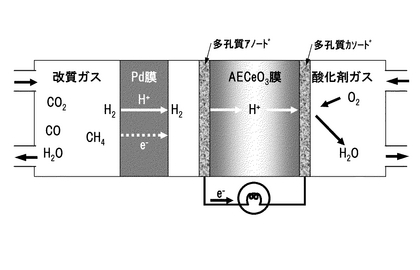
【図5】
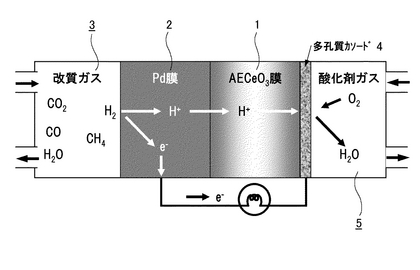
【図11】
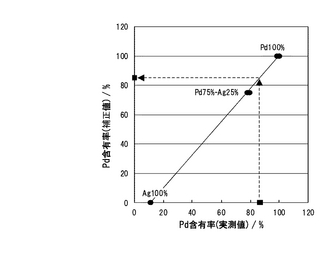
【図12】
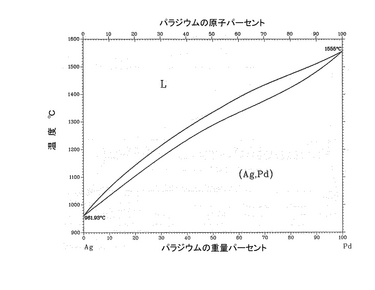
【図13】
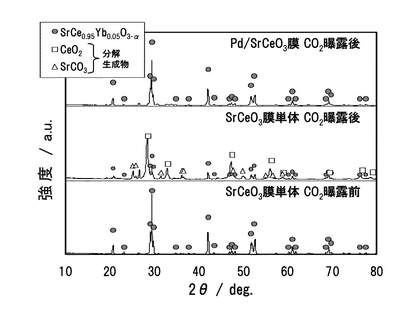
【図14】
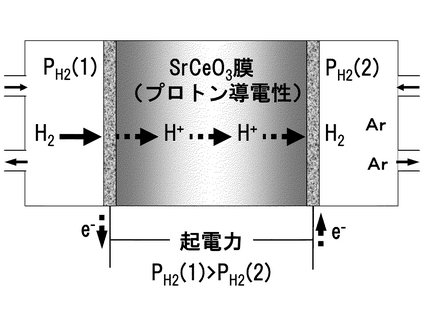
【図15】
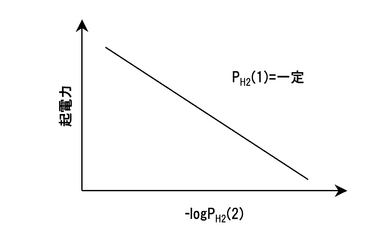
【図16】
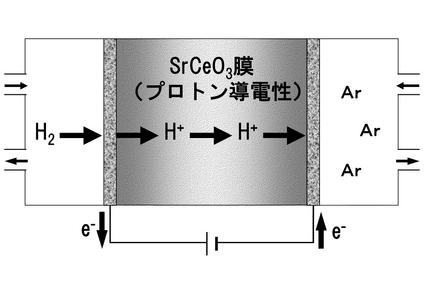
【図17】
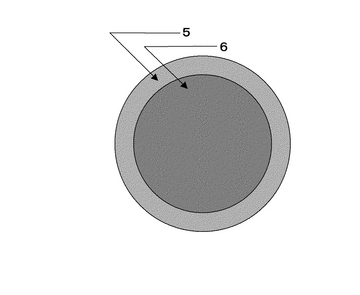
【図18】
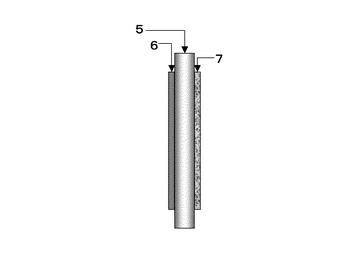
【図19】
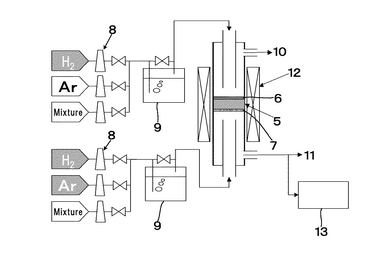
【図20】
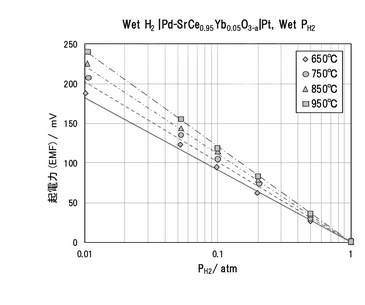
【図21】
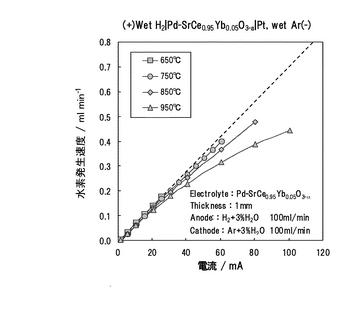
【図22】
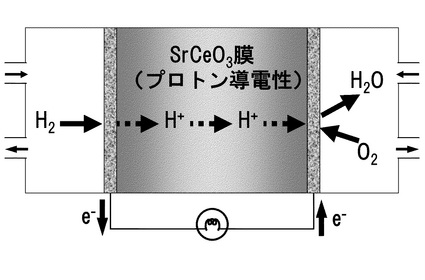
【図23】
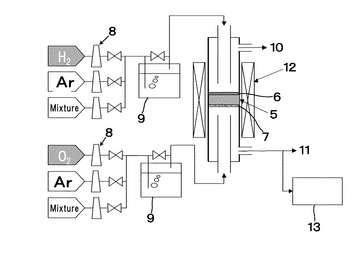
【図24】
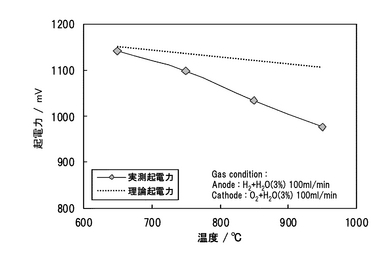
【図25】
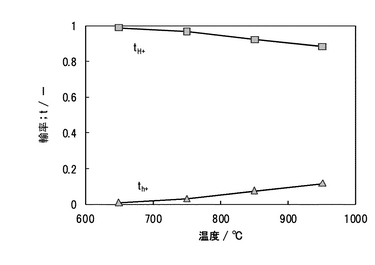
【図26】
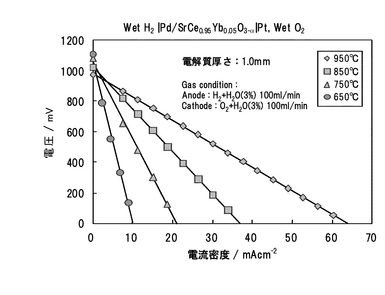
【図27】
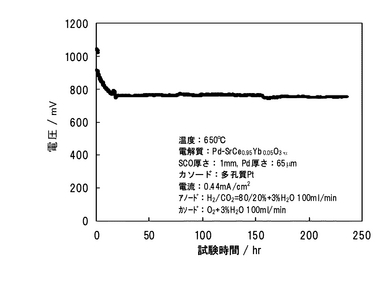
【図28】
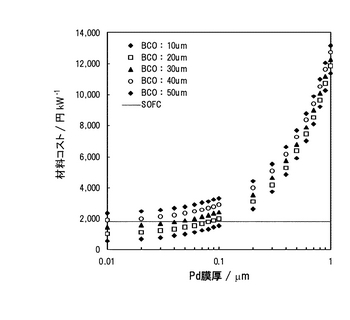
【図29】
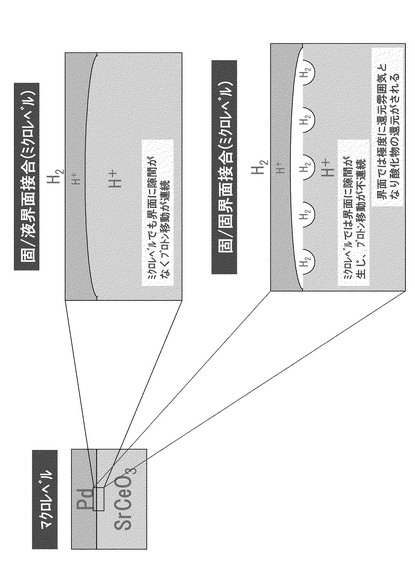
【図30】
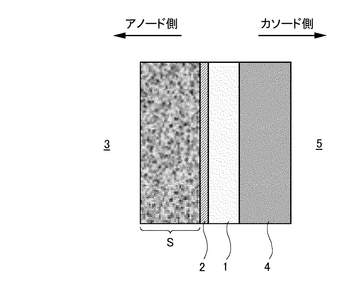
【図31】
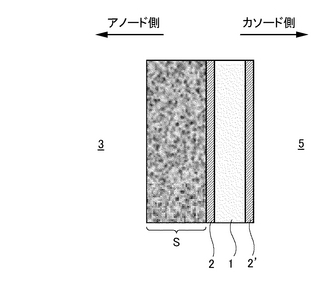
【図32】
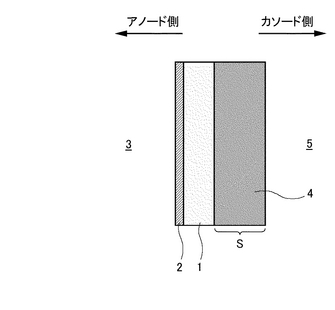
【図33】
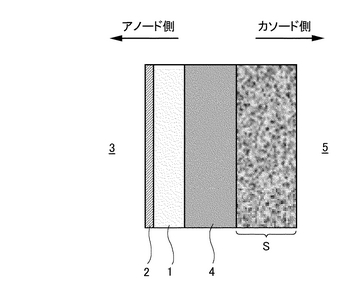
【図34】
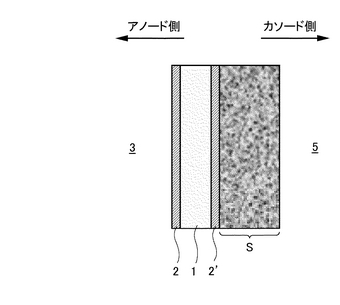
【図35】
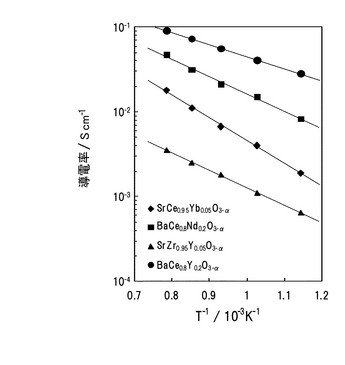
【図36】
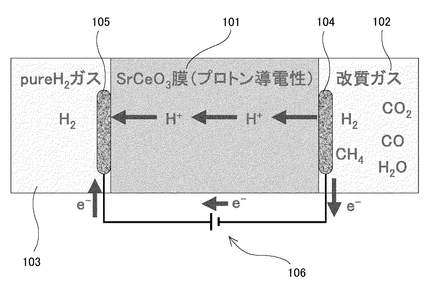
【図37】
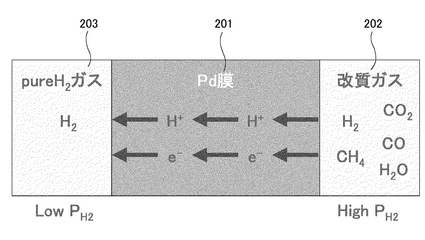
【図38】
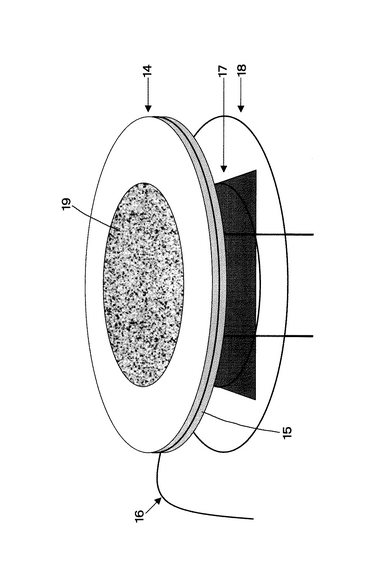
【図39】
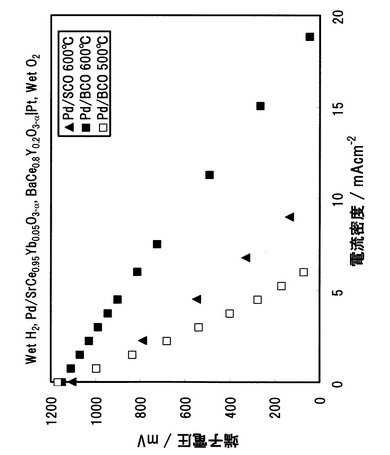
【図40】
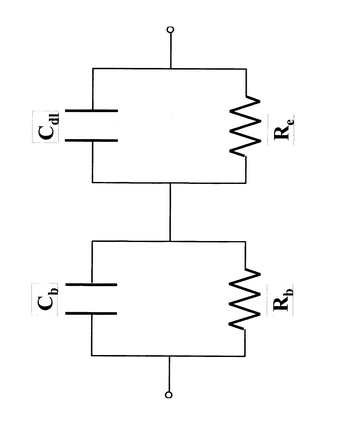
【図42】
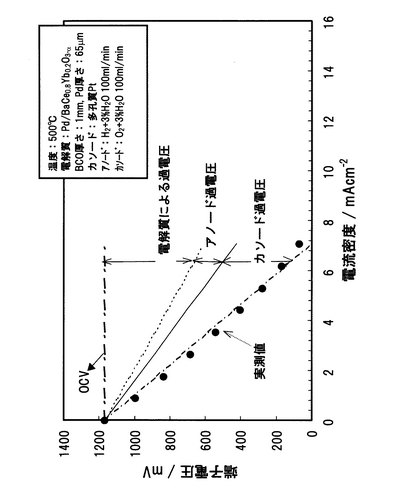
【図43】
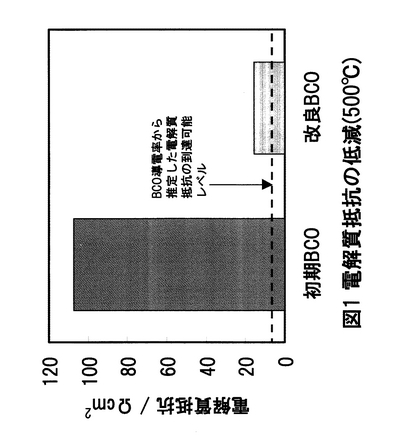
【図44】
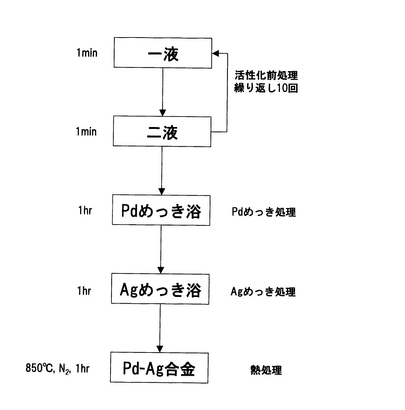
【図46】
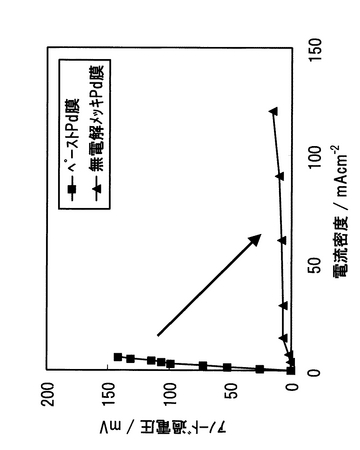
【図47】
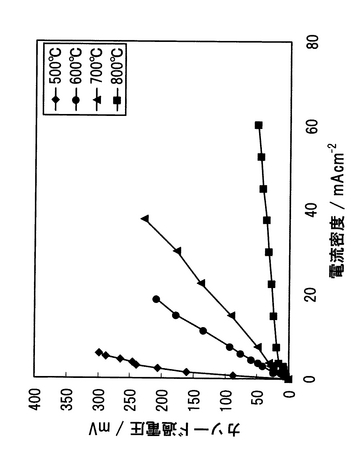
【図48】
【図50】
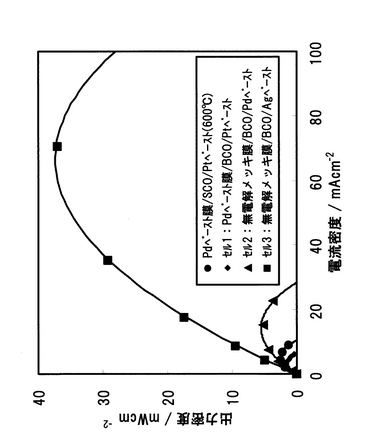
【図51】
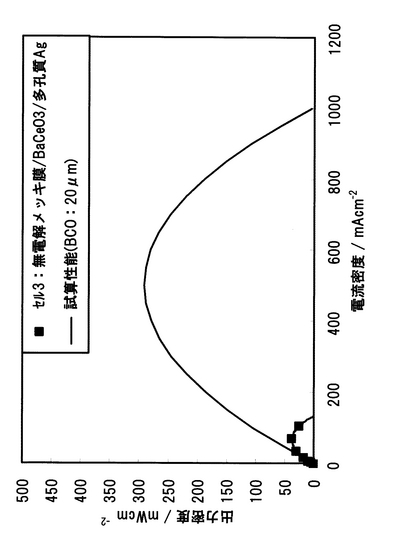
【図6】

【図7】

【図8】

【図9】

【図10】

【図41】
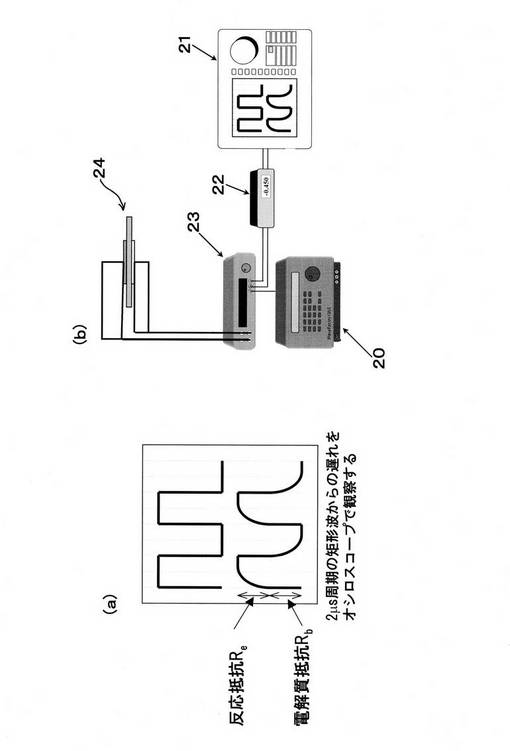
【図45】

【図49】
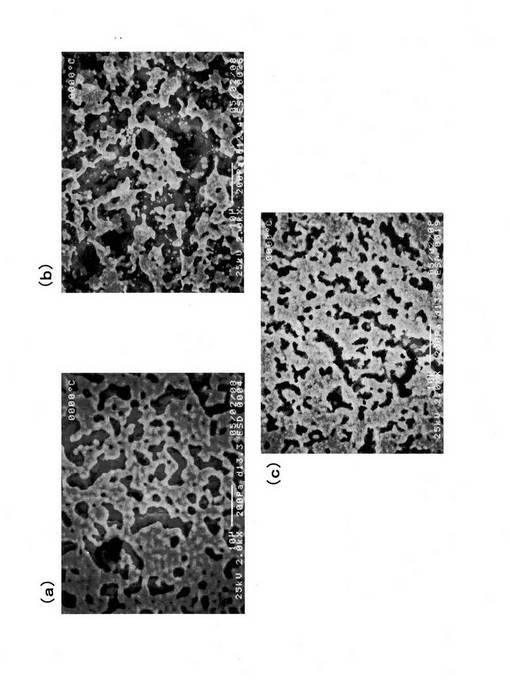
【図2】
【図3】
【図4】
【図5】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図42】
【図43】
【図44】
【図46】
【図47】
【図48】
【図50】
【図51】
【図6】
【図7】
【図8】
【図9】
【図10】
【図41】
【図45】
【図49】
【公開番号】特開2006−54170(P2006−54170A)
【公開日】平成18年2月23日(2006.2.23)
【国際特許分類】
【出願番号】特願2005−199456(P2005−199456)
【出願日】平成17年7月7日(2005.7.7)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成17年4月1日 社団法人電気化学会発行の「電気化学会第72回大会 講演要旨集」に発表
【出願人】(000173809)財団法人電力中央研究所 (1,040)
【Fターム(参考)】
【公開日】平成18年2月23日(2006.2.23)
【国際特許分類】
【出願日】平成17年7月7日(2005.7.7)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成17年4月1日 社団法人電気化学会発行の「電気化学会第72回大会 講演要旨集」に発表
【出願人】(000173809)財団法人電力中央研究所 (1,040)
【Fターム(参考)】
[ Back to top ]