
プロパン及び/又はイソブタンの不均一系触媒作用による部分直接酸化方法
反応段階において得られた生成物ガス混合物から後処理段階において目的生成物を分離する、プロパン及び/又はイソブタンの不均一系触媒作用による部分直接酸化の方法であって、その際に残っている残留生成物ガス混合物を同じ組成の2つの部分量へ分割し、一方の部分量を反応段階へ返送し、かつ他方の部分量を外へ移し、かつ反応段階並びに後処理段階を高められた圧力で操作する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、プロパン及び/又はイソブタンを、少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法に関するものであり、前記方法の場合に、反応ガス出発混合物用の入口、場合により補助ガス用の別の入口及び生成物ガス混合物用の出口を除いてガス側で封止されている反応段階に、プロパン及び/又はイソブタン、分子酸素及び少なくとも1つの不活性な希釈ガスを含有し、入口圧力P1を有する反応出発混合物を供給し、反応段階において高められた温度で反応ガス出発混合物を固体の集合状態である触媒に通過させることにより反応ガス出発混合物中に含まれるプロパン及び/又はイソブタンを少なくとも1つの目的生成物に直接部分酸化及び/又はアンモ酸化し、かつ反応ガス混合物を少なくとも1つの目的生成物として含有し、出口圧力P2を有する生成物ガス混合物を、反応段階から外へ及びこの圧力P2で、ガス側で生成物ガス混合物用の入口、場合により補助ガス用の別の入口及び残留生成物ガス混合物用の出口を除いて封止されている後処理段階へ導き、後処理段階において反応段階の生成物ガス混合物からその中に含まれる目的生成物を液相中へ基本分離し(grundabgetrennt)、かつその際に残っている、プロパン及び/又はイソブタンだけでなく並びに場合によりプロペン及び/又はイソブテンを含有し、出口圧力P3[ここでP3<P1である]を有する残留生成物ガス混合物を、後処理段階から外へ導き(herausfuehrt)、かつ残留生成物ガス混合物中に含まれるプロパン及び/又はイソブタンを反応段階へ返送する。
【0002】
アクリル酸及びメタクリル酸は、例えばポリマーを製造するための、重要な中間生成物である。
【0003】
反応段階においてプロパン及び/又はイソブタンの不均一系触媒作用による部分直接酸化によるアクリル酸及びメタクリル酸の製造は公知である(例えばEP-A 529853、EP-A 603836、EP-A 608838、EP-A 895809、DE-A 19835247、DE-A 10051419、DE-A 10122027、EP-A 1254707、EP-A 1254709、EP-A 1192987、EPA 1090684、DE-A 10254279及びこれらの明細書に引用された文献参照)。
【0004】
酸化剤として通常、分子酸素が使用され、これは例えば純粋な形でか又は部分酸化に関して本質的に不活性に挙動するガス(例えば空気中のN2)との混合物で反応ガス出発混合物に添加されることができる。不活性な希釈ガス、例えばN2、H2O、CO、CO2、He及び/又はAr等は、反応熱を吸収し、かつ反応ガス混合物を爆発範囲の外側に保持する。一般的に、本明細書において不活性な希釈ガスは、部分酸化を通過する反応ガス混合物のワンパス(einmaligen Durchgang)の際に5mol%未満、好ましくは3mol%未満及び特に好ましくは2mol%未満が反応するようなガスであると理解される。触媒として通例、固体の集合状態である多元素酸化物(Multielementoxide)が使用される。反応段階の実施は、固体の集合状態である多元素酸化物については触媒固定床、−渦動床(-wirbelbett)又は−移動床(−流動床)において可能である。
【0005】
反応段階における使用圧力は、技術水準の教示によれば原則的に常圧(=1bar)を下回るだけではなく1barを上回っていてよい(例えばDE-A 19835247、EP-A 895809及びDE-A 10261186参照)。通例、この圧力は反応段階における流れ抵抗を克服する目的のために常圧をやや上回っている。
【0006】
プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化することにとって不利であるのは、プロパン及びイソブタンの比較的際立つ反応不活性である。これは、相応する反応段階を通過する反応ガス混合物のワンパスの際に高められた温度でさえ通例、プロパン及び/又はイソブタンの部分転化のみが達成される原因となる。
【0007】
プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸不均一系触媒作用により部分直接酸化するという目標設定は、故に、反応段階を通過する反応ガス混合物のワンパスの際にできるだけ低い温度で、プロパン及び/又はイソブタンのできるだけ高い転化率を同時に目的生成物形成のできるだけ高い選択率で、すなわち、目的生成物のできるだけ高い空時収量をできるだけ僅かなエネルギー消費で、達成することにある。
【0008】
さらに、プロパン及び/又はイソブタンを所望の目的生成物の少なくとも1つに不均一系触媒作用により部分直接酸化する経済的な実施のための別の必要条件は、生成物ガス混合物中に含まれる未反応のプロパン及び/又はイソブタンを広範囲に亘って反応段階へ返送することにある。技術水準においてこのためには次の提案がなされている。
【0009】
DE-A 10119933は、生成物ガス混合物からのプロパンの不均一系触媒作用による部分直接酸化によるアクリル酸の製造の際に、その中に含まれるアクリル酸を液状吸収剤中への吸収により基本分離し、かつその際に生じる吸収剤及びアクリル酸からなる液状混合物を引き続いてそれ自体として公知の方法で精留により、抽出により及び/又は結晶化により純アクリル酸まで後処理するか又は分別凝縮による液相中への生成物ガス混合物からのアクリル酸の基本分離を、例えばDE-A 19924532に記載されているように、実施し、かつその際に生じる水性のアクリル酸凝縮物を例えば分別晶出によりさらに精製することを推奨する。
【0010】
液相中へのアクリル酸のそのような基本分離の際に残っている未反応のプロパンを含有する残留生成物ガス混合物に関して、DE-A 10119933は、プロパンを残留生成物ガス混合物から分離し、かつ分離されたプロパンをアクリル酸への部分直接酸化へ返送することを推奨する。分離法として、そのためには例えば分別加圧精留又は疎水性有機溶剤(プロパンを好ましくは吸収することができる)での抽出及びプロパンの遊離の目的のためのその後の脱着及び/又は空気でのストリッピングが推奨される。
【0011】
完全な対応関係において、EP-A 1193240も、プロパンのようなアルカンの部分直接酸化の場合に、残留生成物ガス混合物中に含まれる未反応のアルカンを(前記の技術水準の明細書と同じように、好ましくは副生物として形成されたアルケンと一緒に)それから分離し(例えば吸収又は吸着により)、かつ部分酸化へ返送することを推奨する。
【0012】
しかしながら、技術水準の推奨による反応段階への未反応のアルカン及び場合により副生物として形成されたアルケンの返送様式にとって不利であるのは、未反応のアルカンが通常、比較的高い希釈で含まれている残留生成物ガス混合物からのアルカン及び場合によりアルケンの分離が、比較的費用がかかり、かつ特に高い圧力損失と結びついていることである。後者は反応段階における高められた圧力の適用を退ける、それというのも、分離されたプロパンを返送する際に常にこの圧力に再圧縮されなければならないからである。
【0013】
さらにそのために不利であるのは、残留生成物ガス混合物中にそれ以外にさらに含まれる、不均一系触媒作用による部分直接酸化に有利に作用する成分(例えば、通例、触媒活性材料の活性及び選択率を向上させる水蒸気、又は高圧縮されてはいけない残留酸素)が反応段階へ返送されず(むしろ流出され)、かつそれに必要に応じて再三再四新たに供給されなければならないことである。
【0014】
残留生成物ガス混合物中に含まれる未反応の炭化水素を別個に、すなわち、分離されて反応段階へ返送するという提案の原因となることに加えて、とりわけ、循環すべきガス量(及びこれと共に反応ガス出発混合物量も)をできるだけ僅かに保持して、このようにしてこれに関して適用すべき移送出力及び圧縮機出力(循環されるガスは反応段階への入口の前に反応ガス混合物の入口圧力に再圧縮されなければならない、それというのも、反応段階、後処理段階及び残留生成物ガス混合物からの分離を通過する経路で、移送抵抗を克服するために消費され、かつ再供給されなければならない圧力損失を受けるからである)並びに必要な体積を最小限にすることを考慮すべきである。別の目標設定はその際に、供給原料損失を最小限に保持すべきである。
【0015】
本発明の課題は故に、プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法を提供することにあり、前記方法の場合に、消費する圧縮機出力及び供給原料損失は他のより有利な方法で最小限にされ、かつ同時に空時収量が最小限にされたエネルギー消費で最適化される。
【0016】
それに応じて、プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法であって、
反応ガス出発混合物用の入口、場合により補助ガス用の別の入口及び生成物ガス混合物用の出口は別としてガス側で封止されている反応段階に、プロパン及び/又はイソブタン、分子酸素及び少なくとも1つの不活性な希釈ガスを含有し、入口圧力P1を有する反応ガス出発混合物を供給し、
反応段階において反応ガス出発混合物を高められた温度で固体の集合状態である触媒に通過させることにより反応ガス出発混合物中に含まれるプロパン及び/又はイソブタンを少なくとも1つの目的生成物に直接部分酸化させ、かつ
反応ガス混合物を少なくとも1つの目的生成物として含有し、出口圧力P2を有する生成物ガス混合物を、反応段階の中から外へ及びこの圧力P2で、ガス側で生成物ガス混合物用の入口、場合により補助ガス用の別の入口及び残留生成物ガス混合物用の出口を除いて封止されている後処理段階中へ導き、後処理段階において反応段階の生成物ガス混合物からその中に含まれる目的生成物を液相中へ基本分離し、かつ
その際に残っている、プロパン及び/又はイソブタンだけでなく並びに場合によりプロペン及び/又はイソブテンを含有し、出口圧力P3[ここでP3<P1である]を有する残留生成物ガス混合物を、後処理段階から外へ導き、かつ残留生成物ガス混合物中に含まれるプロパン及び/又はイソブタンを反応段階へ返送する
方法が見出され、前記方法は、
P1が、P3が≧1.5barであり、かつ残留生成物ガス混合物を同じ組成の2つの部分量へ分割し、その一方の部分量を流出物として外へ移し、他方の部分量を循環ガスとして返送し、かつ反応出発混合物の成分として入口圧力P1に圧縮して再び反応段階に供給するように選択される
ことにより特徴付けられている。それゆえ、本発明による方法はとりわけEP-A 495504に開示された手法とは、後処理段階中及び/又は後処理段階後に流出の前に接触一酸化炭素酸化を有しないことが異なる。本発明による方法の場合に残留生成物ガス混合物の二酸化炭素洗浄も必要としない。
【0017】
反応段階において副生物としてプロペン及び/又はイソブテンが生じる場合には、これらの化合物は後処理段階を経て通常、プロパン及び/又はイソブタンと共存したままであり、かつ一緒に循環ガス中に存在して反応段階へ返送される。
【0018】
本明細書における全ての圧力の記載は、他に明示的に記載されない限り、絶対圧力であると理解される。
【0019】
通常、残留生成物ガス混合物は、プロパン及び/又はイソブタンとは異なる並びにプロペン及び/又はイソブテンとは異なる並びに場合により酸素とは異なる(例えばCO、CO2、H2O及び/又はN2等)成分(通例これらは生成物ガス混合物中に含まれる、目的生成物よりも容易に(常圧で)沸騰する成分である)を少なくとも2体積%、又は少なくとも5体積%、たいてい少なくとも10体積%含有する。
【0020】
本発明による方法の場合に部分酸化すべき有機の前駆物質化合物(すなわち、プロパン及び/又はイソブタン)は実地においてしばしば液状で貯蔵される一方で、しかし常圧及び常温でガス状であり、通例、有機の前駆物質化合物を反応段階入口圧力にするためには単純な蒸発で十分である。不活性な希釈ガスとして場合により併用される水蒸気は、多種多様な源からたいてい同様に十分な過圧で入手できる。
【0021】
しかしながらこのことは、通例少なくとも酸素源(例えば空気又は窒素の減損された空気)、場合によりその他の不活性な希釈ガスに当てはまらず、かつ、とりわけプロパンを含有する循環ガスには当てはまらない(この循環ガスは通常、反応段階及び後処理段階を通過する経路での圧力損失を差し引いて反応段階入口圧力P1並びに双方の部分量への除数を有し;反応段階へのその返送は本発明による方法の場合に通常、より大きな付加的な圧力損失を有しない内部構造物が含まれない管を経て行われる)。
【0022】
故に実地において、反応ガス出発混合物の成分の少なくとも1つの部分量(少なくとも循環ガス)を、圧縮機を用いて低い出発圧力からより高い最終圧力(反応段階への入口圧力P1)にすることが通常必要である。
【0023】
その際に、これらの成分の圧縮(例えば酸素源である空気及び循環ガス)は空間的に別個の圧縮機中で又は単一の圧縮機中で行われることができる。
【0024】
原則的に、挙げられたガスのこの圧縮に多種多様な種類の圧縮機は使用されることができる。押しのけ式圧縮機(例えば往復ピストン圧縮機、スクリュー圧縮機及び回転ピストン圧縮機)、流動式圧縮機(例えばターボ圧縮機、円心圧縮機、軸流圧縮機及び半径流圧縮機)及びジェット圧縮機を例示的に挙げることができる。本発明によれば好ましくは、例えばDE-A 10259023に記載されているような半径流圧縮機が使用される。
【0025】
さらに本発明によれば好ましくは、多様な源に由来し、本質的には反応段階入口圧力である(反応段階入口圧力にされた)反応ガス出発混合物の部分量を、別個の管路から来て、まず最初にたいてい、例えばスタティックミキサー(空の管に対して高められた混合作用を有する内部構造物を有する空間)中で混合し、次に、場合により入口温度に加熱し、反応段階に供給するようにして行われる。
【0026】
スタティックミキサーに供給された管路への個々のガスの進入はその際に好都合にはしばしば、爆発性混合物の発生が回避されるように選択される(本発明による部分酸化の場合にこの進入順序は好都合には例えば次のような内容であってよい:まず最初に循環ガス及び/又は蒸気、ついで空気及び最終的に有機の前駆物質化合物)。もちろん、水蒸気含量は反応ガス出発混合物に、生成物ガス流との間接熱交換により本質的に反応温度に加熱された反応ガス出発前駆物質混合物中へ微細に分配された液状の水滴を計量供給し、前記水滴は吸熱下に自発的に蒸発し、その際に反応ガス出発混合物が生じるように添加されることもできる。選択的に、予熱された反応ガス出発前駆物質混合物はガス飽和器を経て(ガス混合物及び水は並流又は向流で大表面積に導かれる)導かれることができる。
【0027】
それゆえ、本発明により選択された出口圧力P3は本質的には、循環ガス及び酸素源の圧縮のための圧縮機出力に影響を及ぼす。
【0028】
応用技術的に好都合には、残留生成物ガス混合物が本発明による方法の場合に後処理段階を去る圧力P3は、通例30又は25bar以下、しばしば20bar以下である。本発明によれば好都合には出口圧力P3は1.5bar以上かつ10bar以下、好ましくは2bar以上かつ8bar以下、しばしば3bar以上及び6bar以下もしくは5bar以下(例えば4bar)である。
【0029】
すなわち、本発明による方法を特徴付ける特徴は、反応段階並びに後処理段階を高められた圧力で運転することである。
【0030】
そのような手法は次の理由から有利である:
・ 意外にも、プロパン及び/又はイソブタンの不均一系触媒作用による部分直接酸化が高められた圧力で、それ以外は同じ反応条件下で及びワンパスに基づいて高められた転化率を、そのために目的生成物形成の選択率の本質的な減少が付随することなく、もたらすことが見出された;
・ 高められた圧力での後処理段階運転もまた、比較的僅かなそのために必要な体積で及び比較的僅かな受けた圧力損失で高められた循環ガス量の移送も可能にする、それというのも、所定のガス量の移送体積並びにその移送に付随している圧力損失は、増大する圧力と共に通例低下するからであり;後者は、反応段階の入口圧力P1への循環ガス圧縮に必要な圧縮機出力を減少させ;同時に、流出物量と比較して高められた循環ガス量は、流出物中に含まれる未反応のプロパン及び/又はイソブタンの損失を最小限にする;
・ 残留生成物ガス混合物からのプロパンの予備分離なしでのプロパンの返送は、そのような分離に必然的に付随している圧力損失を回避し、かつ残留生成物ガス混合物中に場合により含まれる他の成分、例えば水蒸気及びO2の同時で及びエネルギー的に好都合な返送を保証する。
【0031】
すなわち、全体として、本発明によれば、圧力上昇という比較的単純な措置を通じて、技術水準による方法の本質的に全ての有利な本質的特徴が、そのために残留生成物ガス混合物からの未反応のアルカン及び場合によりアルケンの費用のかかる分離を必要とすることなく(このことは付加的に望ましくない欠点、例えば水蒸気の完全な不返送又はエネルギー的に費用のかかる返送を回避する)、同様に達成されることができ、かつ同時に圧力上昇の措置は、目的生成物選択率の本質的な減少なしに反応段階を通過する単一処理量に基づく反応物転化率の増大を引き起こす。
【0032】
“反応段階”もしくは“後処理段階”という概念は、本明細書において特に、ガス側で、入口及び出口並びに場合により補助ガス用の別の入口を除いて封止されている1つ又は複数の直列に接続された装置ユニット(apparative Vorrichtungen)を表すので、ガス混合物がそのような装置ユニットもしくはそのような装置ユニットの直列接続を通過する際に受ける圧力損失は、流れ抵抗の克服を制限する。
【0033】
例えば、装置ユニット(又はそのようなユニットの直列接続)は、管束反応器、渦動床反応器、流動床反応器、そのような反応器の直列接続、吸収塔、精留塔、凝縮塔又はそのような塔もしくは個々の急冷段階の直列接続であってよい。しかしもちろん、前記のような反応器は、本発明による方法を実施する間に、例えばWO 02/081421に記載されているように、例えば反応器中へ触媒活性剤を添加する可能性を含むことができる。補助ガスの概念は、本発明による方法の場合に例えば、反応器の直列接続の場合に反応器の間で不活性ガス及び/又は酸素(例えば空気)を補充するか、又は後処理段階において例えば重合防止の理由から分子酸素を含有するガス(例えば空気)を生成物ガス混合物と一緒に後処理段階へ導く可能性を含むべきである。典型的には圧力損失は本発明による方法の場合に反応段階に亘って0.1〜3bar、しばしば0.3〜1又は0.5bar、及び後処理段階に亘って0.5〜3bar、しばしば1〜2barである。
【0034】
特に高い圧力の範囲内で本発明による方法を実施する場合に、圧力損失は反応段階において並びに後処理段階においてさらに明らかにより低くなりうるものであり、かつ例えば0.05bar及びそれ未満までの値を達成することができる。
【0035】
反応段階への入口での圧力P1は、本発明による方法の場合に、適用される後処理段階の方法に応じて、後処理段階の出口での圧力P3を0.5又は1〜4bar、たいてい1.5〜3.5bar、多くの場合に2〜3bar上回っている。
【0036】
特に高い圧力の範囲内で本発明による方法を実施する際に、反応段階への入口での圧力P1は通例、後処理段階の出口での圧力P3を0.5bar未満(例えば0.1又は0.01bar)上回っている。
【0037】
反応段階への入口での典型的な圧力P1は故に2.5〜25barである。通例、反応段階への入口での圧力P1は3〜10bar及び本発明によれば好都合には4〜8barである。
【0038】
後処理段階への入口での典型的な圧力P2は3〜25bar、しばしば3〜20bar、又は3〜15bar又は3〜8barである。
【0039】
圧力比の制御は、本発明による方法の場合に単純な方法で、例えば、残留生成物ガス混合物の外へ移すべき部分量についての流出物の絞り装置を用いて可能である。絞り装置の代わりに後接続されたエクスパンダー(外へ移すことが行われる逆圧縮機)を用いて、本発明による方法の場合に、その他の点では既に記載された利点に加えて、残留生成物ガス混合物の一部の流出の際に常圧へのその制御された放圧により入口圧力P1への残留生成物ガス混合物及び/又は酸素源(例えば空気)の循環される他方の部分を圧縮するために必要な圧縮機出力の一部が回収されることができる。
【0040】
流出物として外へ移される残留生成物ガス混合物の部分量に対する本発明による方法の場合に循環ガスとして返送される残留生成物ガス混合物の部分量の量比は、反応ガス出発混合物の組成に個別に依存する。しかしながら、通例、Vは0.5以上又は1以上、たいてい1.5以上、好ましくは2以上、特に好ましくは3以上である。もちろんVは本発明による方法の場合に8以上又は10以上であってもよい。通常、Vは本発明による方法の場合に30以下、たいてい25以下、しばしば20以下である。しばしばVは15以下又は10以下、好ましくは2〜8である。
【0041】
その他の点では、本発明による方法は、プロパン及び/又はイソブタンを少なくとも1つの目的生成物に不均一系触媒作用により部分酸化する技術水準から公知の方法のように実施されることができる。
【0042】
すなわち、本発明による方法の範囲内で必要な分子酸素のための源として、空気、並びに分子窒素の減損された空気(例えば≧90体積%−O2、≦10体積%−N2)、並びに純粋な分子酸素又は分子酸素と他の不活性ガスとからなる混合物が考慮される。
【0043】
触媒として、本発明による方法のためには、技術水準においてプロパン及び/又はイソブタンを少なくとも1つの目的生成物に不均一系触媒作用により部分直接酸化するのに推奨される原則的に全ての触媒が考慮される。
【0044】
これらは例えば明細書JP-A 3-170445、EP-A 609122及びEP-A 747349の触媒である。
【0045】
その際に、本質的に全ての触媒が本発明により可能な不均一系触媒作用による部分直接酸化のそれぞれに使用されることができることが本発明によれば本質的である。
【0046】
これらの触媒の活性材料は、通例、多元素酸化物、たいてい多金属酸化物である。
【0047】
多金属酸化物の中では、本発明による方法のためには特に、明細書EP-A 608838、EP-A 529853、DE-A 10254279、DE-A 19835247、EP-A 895809、JP-A 7-232071、JP-A 11-169716、DE-A 10261186、EP-A 1192987、JP-A 10-57813、JP-A 2000-37623、JP-A 10-36311、WO 00/29105、EP-A 767164、DE-A 10029338、JP-A 8-57319、JP-A 10-28862、JP-A 11-43314、JP-A 11-574719、WO 00/29106、JP-A 10-330343、JP-A 11-285637、JP-A 310539、JP-A 11-42434、JP-A 11-343261、JP-A 3423262、WO 99/03825、JP-A 7-53448、JP-A 2000-51693、JP-A 11-263745、DE-A 10046672、DE-A 10118814、DE-A 10119933、JP-A 2000/143244、EP-A 318295、EP-A 603836、DE-A 19832033、DE-A 19836359、EP-A 962253、DE-A 10119933、DE-A 10051419、DE-A 10046672、DE-A 10033121、DE-A 101 459 58、DE-A 10122027、EP-A 1193240及びこれらの明細書に引用された文献の多金属酸化物材料が適している。
【0048】
使用すべき触媒装入物の活性材料は、前記の場合に本質的には、元素Mo、V、双方の元素Te及びSbの少なくとも1つ、及びNb、Ta、W、Ti、Al、Zr、Cr、Mn、Ga、Fe、Ru、Co、Cs、Ca、Sr、Ba、Rh、Ni、Pd、Pt、La、Pb、Cu、Re、Ir、Y、Pr、Nd、Tb、Bi、B、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの元素の少なくとも1つを組み合わせて含有する多金属酸化物材料である。
【0049】
好ましくはその際に最後の元素の群からの組合せは元素Nb、Ta、W及び/又はTi及び特に好ましくは元素Nbを含有する。
【0050】
好ましくは、該当する多金属酸化物活性材料は、化学量論I
Mo1VbM1cM2d (I)
[ここで、
M1=Te及び/又はSb、
M2=Nb、Ta、W、Ti、Al、Zr、Cs、Ca、Sr、Ba、Cr、Mn、Ga、Fe、Ru、Co、Rh、Ni、Pd、Pt、La、Bi、Pb、Cu、Re、Ir、V、Pr、Nd、Tb、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの元素の少なくとも1つ、
b=0.01〜1、
c=>0〜1、及び
d=>0〜1]
における前記の元素の組合せを含有する。
【0051】
本発明によれば好ましくはM1=Te及びM2=Nb、Ta、W及び/又はTiである。特にM2=Nbである。
【0052】
化学量論係数bは、有利には0.1〜0.6である。相応して、化学量論係数cの好ましい範囲は0.01〜1もしくは0.05〜0.4に達し、かつdの好都合な値は0.001〜1もしくは0.01〜0.6である。
【0053】
本発明によれば、化学量論係数b、c及びdが同時に前記の好ましい範囲内である場合に特に好都合である。
【0054】
前記のことは、触媒装入物の活性材料が酸素とは異なるその元素に関して前記の元素の組合せからなる場合に特に当てはまる。
【0055】
これらは特に、一般的な化学量論II
Mo1VbM1cM2dOn (II)
[ここで、変数は化学量論Iに関して記載された意味を有し、かつnは(II)中の酸素とは異なる元素の原子価及び頻度により決定される数である]
で示される多金属酸化物活性材料である。
【0056】
好ましくは 該当する多金属酸化物材料は、化学量論III
Mo1Va′M4b′M5c′M6d′ (III)
[ここで、
M4=Te及びSbを含む群からの元素の少なくとも1つ;
M5=Nb、Ti、W、Ta及びCeを含む群からの元素の少なくとも1つ;
M6=Pb、Ni、Co、Bi、Pd、Cs、Ca、Sr、Ba、Ag、Pt、Cu、Au、Ga、Zn、Sn、In、Re、Ir、Sm、Sc、Y、Pr、Nd及びTbを含む群からの元素の少なくとも1つ;
a′=0.01〜1;
b′=>0〜1;
c′=>0〜1;及び
d′=0〜0.5]
における冒頭に挙げられた元素の組合せを含有する。
【0057】
好ましくはa′は0.05〜0.6、特に好ましくは0.1〜0.6又は0.5である。好ましくはb′は0.01〜1、特に好ましくは0.01もしくは0.1〜0.5又は0.4である。好ましくはc′は0.01〜1、特に好ましくは0.01もしくは0.1〜0.5又は0.4である。好ましくはd′は0.00005もしくは0.0005〜0.5、特に好ましくは0.001〜0.5、しばしば0.002〜0.3及びより頻繁に0.005もしくは0.01〜0.1である。
【0058】
M4は好ましくはTeである。
M5は好ましくは、その全量の少なくとも50mol%、より好ましくは少なくとも75mol%及び極めて特に好ましくは少なくとも100mol%がNbである。
M6は好ましくはNi、Co、Bi、Pd、Ag、Au、Pb及びGaを含む群からの少なくとも1つの元素、特に好ましくはNi、Co、Pd及びBiを含む群からの少なくとも1つの元素である。
【0059】
極めて特に好ましくは、M5はその全量の少なくとも50mol%、又は少なくとも75mol%、又は100mol%がNbであり、かつM6はNi、Co、Pd及びBiを含む群からの少なくとも1つの元素である。
【0060】
本発明によれば傑出しているのは、M4=Te、M5=Nb及びM6がNi、Co及びPdを含む群からの少なくとも1つの元素である。
【0061】
前記のことは特に、触媒装入物の活性材料が酸素とは異なるその元素に関して化学量論(III)の元素の組合せからなる場合に当てはまる。これらは特に、一般的な化学量論(IV)
Mo1Va′M4b′M5c′M6d′On′ (IV)
[ここで、変数は化学量論IIIに関して記載された意味を有し、かつn′は、(IV)中の酸素とは異なる元素の原子価及び頻度により決定される数である]
で示される多金属酸化物活性材料である。
【0062】
さらに本発明による方法のためには好ましくは、一方では前記の元素の組合せ(I)、(III)の1つを含有するか又は、酸素とは異なる元素に関して、それらからなり、かつその都度同時に、回折反射h及びiを示し、それらのピークが回折角(2Θ)22.2±0.5゜(h)及び27.3±0.5゜(i)であるX線回折図を有するような多金属酸化物活性材料が使用される(本明細書においてX線回折図に関する全ての記載は、X線としてのCu−Kα−線の使用下に発生されたX線回折図に関する(Siemens-回折計Theta-Theta D-5000、管電圧:40kV、管電流:40mA、絞りV20(可変)、散乱線絞りV20(可変)、二次単色光器絞り(0.1mm)、検出器絞り(0.6mm)、測定間隔(2Θ):0.02゜、1工程あたりの測定時間:2.4s、検出器:シンチレーション計数管)。
【0063】
これらの回折反射の半値幅はその際に極めて小さくても、又は極めて際立っていてもよい。
【0064】
本発明による方法のためには、前記の多金属酸化物活性材料の、X線回折図が回折反射h及びiに加えて回折反射kを有し、そのピークが28.2±0.5゜(k)であるものが好都合である。
【0065】
後者の中では、そしてまた本発明によれば、回折反射hがX線回折図の中で強度が最も強く、並びに多くとも0.5゜の半値幅を有するものが好ましく、かつ極めて特に好ましくは、回折反射i及び回折反射kの半値幅が同時にその都度1゜以下であり、かつ回折反射kの強度Pk及び回折反射iの強度Piが関係0.2≦R≦0.85、より良好には0.3≦R≦0.85、好ましくは0.4≦R≦0.85、特に好ましくは0.65≦R≦0.85、さらにより好ましくは0.67≦R≦0.75及び極めて特に好ましくはR=0.70〜0.75もしくはR=0.72を満たし、ここでRは式
R=Pi/(Pi+Pk)
により定義された強度比であるものである。好ましくは前記のX線回折図は回折反射を有さず、その最大は2Θ=50±0.3゜である。
【0066】
X線回折図における回折反射の強度の定義は、本明細書においてDE-A 19835247、DE-A 10122027、並びにDE-A 10051419及びDE-A 10046672において記載された定義に関連している。同じことは半値幅の定義に当てはまる。
【0067】
回折反射h、i及びkに加えて、本発明によれば有利には使用すべきそのような多金属酸化物活性材料の前記のX線回折図は、さらに別の回折反射を有し、それらのピークは次の回折角(2Θ)である:
9.0±0.4゜(l)
6.7±0.4゜(o)及び
7.9±0.4゜(p)。
【0068】
さらに、X線回折図が付加的に、ピークが回折角(2Θ)=45.2±0.4゜(q)である回折反射を有する場合に好都合である。
【0069】
しばしば、X線回折図はさらに反射29.2±0.4゜(m)及び35.4±0.4゜(n)も有する。
【0070】
さらに、式(I)、(II)、(III)及び(IV)において定義された元素の組合せが純粋なi−相として存在する場合に好都合である。触媒活性な酸化物材料がさらにk−相も含有する場合には、それらのX線回折図は前記のものに加えてさらに別の回折反射を有し、それらのピークは次の回折角(2Θ)である:36.2±0.4゜及び50±0.4゜(i−及びk−相という概念は本明細書においてDE-A 10122027及びDE-A 10119933に規定されたように使用される)。
【0071】
回折反射hに強度100が割り当てられる場合に、本発明によれば、回折反射i、l、m、n、o、p、qが同じ強度スケールにおいて次の強度を有する場合に好都合である:
i:5〜95、しばしば5〜80、部分的には10〜60;
l:1〜30;
m:1〜40;
n:1〜40;
o:1〜30;
p:1〜30及び
q:5〜60。
【0072】
X線回折図が前記の付加的な回折反射を有する場合には、その半値幅は通例1゜以下である。
【0073】
本発明により使用すべき一般式(II)又は(IV)の多金属酸化物活性材料又は一般式(I)又は(III)の元素の組合せを有する多金属酸化物活性材料の比表面積は、とりわけ、それらのX線回折図が記載されたように構成されている場合には、多くの場合に1〜40m2/gもしくは10〜30m2/g(BET表面積、窒素)である。
【0074】
記載された多金属酸化物活性材料の製造は、これらの引用された技術水準に関連して見出される。これらには、特にDE-A 10303526、DE-A 10261186、DE-A 10254279、DE-A 10254278、DE-A 10122027、DE-A 10119933、DE-A 10033121、EP-A 1192987、DE-A 10029338、JP-A 2000-143244、EP-A 962253、EP-A 895809、DE-A 19835247、WO 00/29105、WO 00/29106、EP-A 529853及びEP-A 608838が含まれる(最後に挙げた2つの明細書の全ての実施例において乾燥法として噴霧乾燥が適用されうる;例えば300〜350℃の入口温度及び100〜150℃の出口温度で;向流又は並流)。
【0075】
記載された多金属酸化物活性材料は、それ自体として(すなわち粉末形で)又は適しているジオメトリーに成形されて(例えばDE-A 10051419のシェル触媒(Schalenkatalysatoren)又はDE-A 10122027の幾何学的な変型)、本発明による方法に使用されることができる。これらは、プロパンからのアクリル酸の製造に、しかしまたイソブタンからのメタクリル酸の製造にも卓越して適している。
【0076】
本発明による方法を実施するためには、全ての挙げられた触媒は、希釈されずに、並びに不活性な粒子及び/又は成形体(これらは活性材料を有しない)で希釈されて使用されることができる。適している希釈材料は例えばステアタイトである。
【0077】
希釈成形体のジオメトリーはその際に触媒成形体のそれと好ましくは同一である。
【0078】
本発明による方法に適している多金属酸化物活性材料触媒に関して明細書に記載されたように、本発明による方法は固定床触媒装入物並びに渦動床−又は流動床触媒装入物上で実施されることができる。その際に本発明により適用可能な反応段階入口圧力は既に記載されている。
【0079】
反応温度は、特に本明細書に推奨された触媒を使用する場合に、200〜700℃、好ましくは200〜550℃又は230〜480℃又は300〜440℃であってよい。
【0080】
プロパン及び/又はイソブタンでの触媒装入物の負荷は50〜3000Nl/l(触媒装入物)/h、又は80〜1500Nl/l/h、又は100〜1000Nl/l/h、又は120〜600Nl/l/h、又は140〜300Nl/l/hであってよい。
【0081】
反応ガス出発混合物での触媒装入物の負荷はその際に100〜10000Nl/l/h、又は300〜6000Nl/l/h、又は300〜2000Nl/l/hであってよい。触媒装入物中での平均滞留時間は0.01〜10s、又は0.1〜10s、又は2〜6sであってよい。
【0082】
プロパンからのアクリル酸もしくはイソブタンからのメタクリル酸の製造の場合に、反応ガス出発混合物は例えば:
プロパン、もしくはイソブタン 0.5〜15体積%、しばしば1〜7体積%、
空気 10〜90体積%、しばしば20〜50体積%、
水蒸気 0〜50体積%及び
残りの量として循環ガス
を含有していてよい。
【0083】
反応ガス出発混合物はしかしまた、本発明による方法のためのプロパンからのアクリル酸もしくはイソブタンからのメタクリル酸の製造の場合に:
プロパン、もしくはイソブタン 0.6〜1.2体積%、
空気 65〜95体積%、
窒素 2〜30体積%、
COx 0.05〜0.8体積%及び
水蒸気 2〜3体積%
を含有していてよい。
【0084】
水蒸気(新鮮)10〜50体積%を含有する反応ガス出発混合物が好ましい。
【0085】
反応ガス出発混合物の他の可能な組成は、例えば
プロパン又はイソブタン 70〜90体積%、
分子酸素 5〜25体積%、
水蒸気 0〜25体積%及び
残りの量として循環ガス
を含有していてよい。
【0086】
ここでも、全体として水蒸気を10〜50体積%含有する反応ガス出発混合物が好ましい。すなわち、典型的には反応ガス出発混合物の組成は、本発明による方法のためにはプロパン又はイソブタン部分酸化の場合に次の範囲内で変わる(モル比):
イソブタン又はプロパン: 酸素: H2O:その他の希釈ガス=
1:(0.1〜10): (0〜50): (0〜50)、
好ましくは 1:(0.1〜10):(0.1〜50): (1〜50)、
特に好ましくは 1: (0.5〜5): (1〜30): (1〜30)。
【0087】
前記の範囲は特に、その他の希釈ガスとして主に分子窒素が使用される場合に当てはまる。他の可能な希釈ガスは、例えばHe、Ar、CO及び/又はCO2等である。
【0088】
かなり一般的に反応ガス出発混合物はその組成において、好ましくは爆発ガス範囲の外側にあるように選択される。
【0089】
本発明による方法が部分酸化として実施される場合には、これは例えば、EP-A 700714及びEP-A 700893に記載されているような、一帯域−管束反応器中で行われることができる。しかしまた、DE-A 19927624、DE-A 19948242、DE-A 19948241、DE-A 19910508及びDE-A 19910506に記載されているような、多帯域−管束反応器中で実施されることもできる。渦動床における本発明による方法の実施は、例えばWO 02/0811421に記載されたように実施されることができる。
【0090】
反応ガス出発混合物中に含まれるプロパン及び/又はイソブタンに対して、プロパン及び/又はイソブタン転化率は、本発明による方法の場合に、反応段階を通過する反応ガス混合物のワンパスに基づいて、通例10又は20〜70mol%、しばしば30〜60mol%及び多くの場合に40〜60mol%もしくは45〜55mol%である。目的生成物形成の選択率は、通常、40〜98又は95又は〜90mol%、多くの場合に50〜80mol%、しばしば60〜80mol%である。
【0091】
本発明による方法の反応段階において生じる生成物ガス混合物からの少なくとも1つの目的生成物の基本分離は、本発明による後処理段階において原則的に、技術水準による方法から公知であるように実施されることができる。特に、本発明によれば、プロペン及び/又はイソブテンの不均一系触媒作用による部分酸化による目的生成物の製造の際に公知であるような、技術水準において生成物ガス混合物からの同じ目的生成物の基本分離に公知である後処理法も使用されることができる。
【0092】
通例、本発明による方法の場合に反応段階において生じる生成物ガス混合物は本発明による後処理段階への進入の際にまず最初に間接及び/又は直接の冷却にかけられる。
【0093】
液相中への生成物ガス混合物中に含まれる目的生成物の基本分離の目的のためには、こうして冷却された(アクリル酸の場合に例えば典型的には150〜250℃に)又はまた冷却されない生成物ガス混合物は例えば吸収塔中で、少なくとも1つの目的生成物を本質的に選択的に生成物ガス混合物から吸収する下降する液状吸収剤に対して向流で導かれることができ、例えばJP-A 2001/0026269、EP-A 990636、JP-A 2000/327651、EP-A 925272、EP-A 695736、EP-A 778255、DE-A 2136396、DE-A 2449780、DE-A 4308087、EP-A 982287、EP-A 982289、EP-A 982288及びDE-A 19631645には目的生成物及び多様な吸収剤について記載されている。
【0094】
本質的には双方の目的生成物のためには、吸収剤として、水(又は水性混合物、例えば水性カセイソーダ液又は水性のアクリル酸もしくはメタクリル酸)、アクリル酸及びメタクリル酸のエステル化に使用されるアルコール、例えば2−エチルヘキサノール、並びにより高い沸点を有する有機溶剤が考慮される。本発明によれば好ましくは、有機溶剤の沸点は、生成物ガス混合物から分離すべき目的生成物(アクリル酸及び/又はメタクリル酸)の沸点を少なくとも20℃、特に少なくとも50℃及び特に好ましくは少なくとも70℃上回っている。本発明によれば好ましい有機吸収剤は180〜400℃、特に220〜360℃の沸点(常圧で)を有する。本発明によれば特に適している吸収剤は、目的生成物であるアクリル酸及びメタクリル酸の場合に、外に向けて作用する極性基を有しない、高沸点で過度に疎水性の溶剤、例えば脂肪族又は芳香族の炭化水素、例えばパラフィン蒸留からの中油留分、又は酸素原子上にかさばる基を有するエーテル、又はそれらの混合物であり、その際にこれらに有利には極性溶剤、例えばDE-A 4308087に開示された1,2−ジメチルフタレートが添加される。さらに、炭素原子1〜8個を有する直鎖状のアルカノールとの安息香酸及びフタル酸のエステル、例えば安息香酸−n−ブチルエステル、安息香酸メチルエステル、安息香酸エチルエステル、フタル酸ジメチルエステル、フタル酸ジエチルエステル、並びにいわゆる熱媒体油、例えばジフェニル、ジフェニルエーテル及びジフェニルとジフェニルエーテルとからなる混合物又はそれらの塩素誘導体及びトリアリールアルカン、例えば4−メチル−4′−ベンジル−ジフェニルメタン及びその異性体2−メチル−2′−ベンジル−ジフェニルメタン、2−メチル−4′−ベンジル−ジフェニルメタン及び4−メチル−2′−ベンジル−ジフェニルメタン及びそのような異性体の混合物が適している。
【0095】
アクリル酸に特に好ましい吸収剤は(メタクリル酸は好ましくは水中に吸収される)、ジフェニル及びジフェニルエーテルからなり、好ましくは共沸組成物で、特にジフェニル及びジフェニルエーテル100質量%に対してジフェニル(ビフェニル)約25質量%及びジフェニルエーテル約75質量%からなる溶剤混合物、例えば商業的に入手可能なDiphyl(登録商標)である。好ましくはこの溶剤混合物はさらに、極性溶剤、例えばジメチルフタレートを全ての溶剤混合物に対して0.1〜25質量%の量で含有する。有利には生成物ガス混合物は、吸収剤として高沸点の有機溶剤の使用の際に、吸収前に直接凝縮器又は急冷装置中での吸収剤の部分蒸発により冷却される。このためには特にベンチュリスクラバー、気泡塔又はスプレー凝縮器が適している。
【0096】
高沸成分、重沸成分、中沸成分及び低沸成分という概念は、本明細書において、目的化合物よりも高い沸点(高沸成分)、目的化合物とほぼ同じ沸点(中沸成分)もしくは目的化合物よりも低い沸点(低沸成分)を有する化合物を意味する。特に、目的化合物がアクリル酸である場合である。
【0097】
かなり一般的に、向流吸収は好ましくは、規則充填物又は不規則充填積重ね物を備えた塔中で又は好ましくはデュアルフロートレイ及び/又はバルブトレイが設けられており、かつ上から溶剤が装入される、段塔中で行われる。生成物ガス混合物(及び場合により急冷装置からの蒸発された吸収剤)は下から塔中へ導通され、引き続いて吸収温度に冷却される。冷却は有利には冷却回路により行われる。すなわち、加熱され、塔中で下降する吸収剤は塔から取り出され、熱交換器中で冷却され、取り出し位置を上回る位置で吸収塔へ再び返送される。吸収後に、本質的には全ての重沸成分、大部分の目的化合物(例えばアクリル酸)及び一部の低沸成分は吸収剤中に存在する。目的化合物(例えばアクリル酸)を基本分離されて含有する被吸収剤から、ついで目的生成物は技術水準において(例えば吸収の際に引用された)記載されたように任意の純度でさらに分離されることができ(例えばDE-A 19606877又はDE-A 19838845のように)、かつ目的生成物を含まない吸収剤は吸収において再び使用されることができる(例えば有機の被吸収剤からアクリル酸を頂部を経て有機吸収剤から分離し、かつ精留により及び/又は結晶化により(例えば融成物洗浄塔中での結晶分離を伴う懸濁晶出)更に精製するか又は水性被吸収剤から水を有機共沸剤を用いて精留により頂部を経て及びアクリル酸をアクリル酸を含有する缶出液から精留により及び/又は結晶化により任意の純度で更に分離し;最後の場合に留出液は通例2つの相へ分けられ(冷却により);有機相は精留塔へ及び水相は吸収塔へ返送される(その都度塔頂部で))。
【0098】
残っている、吸収されない残留生成物ガス混合物はさらに、単純に凝縮可能な部分の低沸点副成分(例えば水、ホルムアルデヒド及び酢酸)を分離するために、冷却されることができる(通例、酸性水が挙げられる)。残っている残留生成物ガス混合物は、本発明によれば2つの部分へ分割され、かつ双方の部分の一方は循環ガスとして返送されることができ(反応段階へ)、他方の部分は流出されることができる。好ましくは本発明によれば酸性水分離は行われない。特に本発明による方法の反応段階において水蒸気が希釈ガスとして併用される(多金属酸化物材料(I)、(II)、(III)又は(IV)の使用の際に、このことは通例、目的生成物形成の選択率のために有利である)場合に、液相中への反応段階の生成物ガス混合物中に含まれる目的生成物の本発明による基本分離は(それとは独立して、特に本明細書に記載された分離方法が使用される)、好ましくは、残っている、本発明によれば少なくとも部分量として(循環ガスとして)反応段階へ返送すべき残留生成物ガス混合物中に、その中に含まれるプロパンに対するその中に含まれる水蒸気のモル比Wが、反応段階の生成物ガス混合物中の相応するモル比W′よりも多くとも50%、より良好には多くとも40又は30%、さらにより良好には多くとも20又は10%、好ましくは多くとも5%少ないように行われる。極端な場合には本発明による方法の場合に前記の比W及びW′は同一であってもよい。
【0099】
残留生成物ガス混合物中にできるだけ多くの水蒸気をそのままにしておく試みは、ここでは、反応ガス出発混合物中への水蒸気の新鮮なフィード(もしくは凝縮及び新たな蒸発)をできるだけ広範囲に亘って放棄することができるという目的を追求する。
【0100】
しかしながら、本発明による方法の場合に、流出物として外へ移される残留生成物ガス混合物の部分量は、反応ガス混合物中での水の水準上昇(Aufpegeln)を防止するために反応段階中に副生物として形成されるのと少なくとも同じ水を含有していなければならない(反応段階における反応がより選択的に導かれるほど、流出すべき水量はより僅かになる)。このことは相応して、反応段階において形成された他の副成分にも当てはまる。本発明による方法の場合に酸素源として空気が使用される場合には、残留生成物ガス混合物の流出量は同時に、その中に含まれる窒素量が、空気フィード中に含まれている窒素量に少なくとも相当するように、見積もられなければならない。
【0101】
吸収剤として高沸点有機溶剤の1つが使用される場合には、吸収は前記の場合に好ましくは(特にアクリル酸の吸収の場合に)、吸収塔からの排出が単相であるように実施される。その他の点では、吸収の際に残っている残留生成物ガス混合物中の水蒸気含量は、選択された吸収剤から独立して、吸収温度の適している選択により調節されることができる。
【0102】
溶剤中への吸収による液相中への目的生成物の基本分離に選択的に、この基本分離は(特にアクリル酸の場合に)、凝縮、特に分別凝縮によっても行われることができ、例えばDE-A 19924532、DE-A 10200583、DE-A 10053086、DE-A 19627847、DE-A 19740253、DE-A 19740252、DE-A 19740253、DE-A 19814387及びDE-A 10247240に記載されている。
【0103】
その際に、反応段階の生成物ガス混合物は、場合により直接及び/又は間接の予備冷却後に、分離効果のある内部構造物が設けられた分離塔中で自体が上昇して分別凝縮にかけられ、かつ目的生成物は分離塔の側部取出しを経て取り出され、かつ技術水準において記載されたように別の結晶化による及び/又は精留による分離工程にかけられる。
【0104】
分離効果のある内部構造物を有する凝縮塔としてその際に、下から上へまず最初にデュアルフロートレイ及びそれに引き続いて水力学的に密封された十字流トレイを分離効果のある内部構造物として有する段塔が好ましく、これらは例えば前記の技術水準に記載されている。
【0105】
本発明によれば好ましくは、前記の分別凝縮も、本質的には生成物ガス混合物中に含まれる水の分離が行われないように実施される。すなわち、好ましくはここでも酸性水分離は行われない。
【0106】
しかしながら、流出物として外へ移される残留生成物ガス混合物の部分量は、それらの流出の前に、メタクリル酸もしくはアクリル酸損失を回避するために、さらに水で洗浄されることができる。その際に生じるメタクリル酸もしくはアクリル酸を含有する酸性水から、アクリル酸もしくはメタクリル酸及び場合により他の有機の価値のある生成物は、例えば吸収に使用される有機吸収剤で抽出され、かつ被吸収剤と合一されることができる。
【0107】
吸収塔並びに分別凝縮用の塔は、高められた圧力損失の回避の目的のために、原則的に吸収剤又は凝縮物を用いて運転される急冷段階の直列接続によっても置換されることができる。
【0108】
かなり一般的に、重合防止は液相中への生成物ガス混合物からの目的生成物の基本分離の範囲内で、技術水準に記載されたように相応する重合防止剤の添加により行われる。
【0109】
本発明によれば、目的生成物の基本分離の際に残っている残留生成物ガス混合物が、プロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンに加えて、常圧で目的生成物よりも容易に沸騰する成分を通常少なくとも5体積%、たいてい少なくとも10体積%含有することが本質的である。これらは特に、生成物ガス混合物の常圧で水よりも容易に沸騰する成分(例えばN2、CO、CO2)並びに好ましくは水自体である。
【0110】
本発明による方法の一利点は、循環ガスが通例、送風機を用いて(ほぼ出口圧力P3から)入口圧力P1に再圧縮されることができることにある。送風機はその際に、低い圧力比(1.1:1〜3:1のような最終圧力対出口圧力)を有する圧縮機(通常、軸流圧縮機)であると理解される。それとは異なり、酸素源として使用される空気は通常、半径流圧縮機を用いて(ほぼ周囲圧力から)入口圧力P1に圧縮される。原則的に本明細書において言及された圧縮は等温的に又はポリトロープに実施されることができる。後者は本発明によれば好ましい。
【0111】
本発明による手法の有利な点の一部は、残留生成物ガス混合物の外へ移すべき部分から、場合によりその中に含まれる水含分(反応段階へ返送されることができる)の予めの凝縮による分離後に、その中に含まれるプロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンが分離され、かつ入口圧力P1に再圧縮されて反応段階へ返送される場合にも、依然として得られたままである。
【0112】
この分離(技術水準において残留生成物ガス混合物の全量のために推奨され、かつ原則的に(特に次に記載されたように)使用されることもできる)は、例えば、残留生成物ガス混合物の外へ移すべき部分に含まれるプロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンが吸収的及び/又は吸着的に分離され、かつ脱離(Desorbtion)及び/又は脱着により次に再び遊離されるようにして行われる(WO 0196271参照)。例えば被吸収剤からのこの遊離は、空気でのストリッピングにより実施されることができ、これは例えばWO 0196271に記載されている。空気は、次に共に圧縮され、かつ反応段階へ共に返送されることができる。
【0113】
選択的に残留生成物ガス混合物の外へ移すべき部分は、別の酸化反応器にも、場合により酸素の補充後に、炭化水素転化率を高めるために供給されることができる。
【0114】
通例、既に述べたように、本発明による方法の場合に循環ガスの再圧縮の目的のためには送風機で十分である。
【0115】
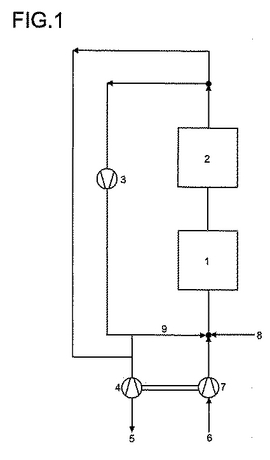
本明細書に添付している図1は本発明による方法の例示的な実施変法を示す。
【0116】
その際に、多様な数字は以下の意味を有する:
1=反応段階
2=後処理段階
3=循環ガス圧縮機(送風機)
4=流出物エクスパンダー
5=流出物
6=酸素源としての空気
7=空気圧縮機
8=新鮮なプロパン及び/又はイソブタン並びに場合により水蒸気
9=再圧縮された循環ガス。
【0117】
実施例
1.多金属酸化物活性材料Mo1V0.29Te0.13Nb0.13Ox(純粋なi−相)を有する触媒の製造
メタバナジン酸アンモニウム87.61g(V2O5 78.55質量%、G.f.E.社、ニュルンベルク)を80℃で水3040ml中に溶解させた(撹拌機、温度計及び還流冷却器を備えた三つ口フラスコ)。澄明な帯黄色溶液が生じた。この溶液を60℃に冷却し、ついで60℃に維持しながら連続してテルル酸117.03g(H6TeO6 99質量%、Aldrich社)及び七モリブデン酸アンモニウム400.00g(MoO3 82.52質量%、Starck社、Goslar)を撹拌混入した。生じる深紅色の溶液を30℃に冷却した(溶液A)。
【0118】
ビーカー中で別個にシュウ酸ニオブアンモニウム112.67g(Nb 20.8質量%、Starck社、Goslar)を60℃で水500ml中に溶解させた(溶液B)。
【0119】
溶液Bを30℃に冷却し、この温度で溶液Aと合一し、その際に溶液Bを溶液Aに添加した。添加を連続的に約5分の期間に亘って行った。浮遊沈殿を有する橙色の水性懸濁液が生じた。引き続いてこの懸濁液を噴霧乾燥した(Tリザーバ=30℃、T入=320℃、T出=110℃、t=1.5h、Niro社のアトマイザー型のスプレー塔)。生じた噴霧物は同様に橙色であり、かつ秤量化学量論Mo1V0.33Te0.22Nb0.11を有していた。
【0120】
スプレー粉末100g×2を、DE-A 10119933の図1による回転球面炉(内容積1 l)中で、まず最初に27.5minかけて線形加熱傾斜で50Nl/hの空気流下に25℃から275℃に加熱し、引き続いてこの温度を1h保持することにより熱処理した。その後、線形加熱傾斜で32.5minかけて275℃から600℃に加熱し、その際に空気流を窒素流50Nl/hにより置換した。窒素流を保持しながら600℃を2h保持し、次に炉全体を室温に冷却した。得られた酸化物材料100gを10質量%のHNO3−水溶液1000ml中で70℃で7h還流下に撹拌し、残っている固体を、生じたスラリーからろ別し、水(25℃)を用いて硝酸塩不含に洗浄した。得られたフィルターケーキをマッフル炉中で一晩に亘り空気下に110℃で乾燥させた。
【0121】
得られた固体の化学分析は、組成Mo1V0.29Te0.13Nb0.13Oxとなった。属するX線回折図は純粋なi−相を有していた。
【0122】
引き続いて、乾燥させた材料をDE-A 10119933に記載されたようにRetschミル中で粉砕し(粒度≦0.12mm)、DE-A 10119933の例A)a)のようにシェル触媒に加工した:
粉砕した活性材料38g;2.2〜3.2mmの直径を有する球状担体150g(担持材料=CeramTec社、DEのステアタイトC-220、45μmの表面粗面度Rzを有する)、粘着剤=グリセリン及び水からなる混合物30ml(グリセリン:水の質量比=1:3)、乾燥期間=150℃で16h;生じるシェル触媒の活性材料含分は20質量%であった(シェル触媒の質量に対して)。
【0123】
2.異なる圧力でのアクリル酸へのプロパンの不均一系触媒作用による部分直接酸化
1.からのシェル触媒を、V2A鋼(外径=60mm、内径=8.5mm)からなる反応管(長さ140cm)に装入した。装入物長さは53cmを選択した(=シェル触媒約35.0g)。担持材料として使用されるステアタイト球からなる長さ30cmの予備積重ね物を触媒帯域の位置調節に利用した。同じステアタイト球を、反応管に触媒帯域の後に最終的に充填した(反応ガス出発混合物を温め始めるための予備加熱帯域)。反応管を、外側からその長さ全体で電気加熱マットを用いて加熱した。反応ガス出発混合物のモル組成はプロパン:空気:水=1:15:14であった。次の表は、ワンパスでの生じたプロパン転化率(UPAN、mol%)並びにこの転化率に付随しているアクリル酸形成の選択率(SACS、mol%)を、選択された入口圧力並びに加熱マットの属する温度に応じて示す。滞留時間(触媒積重ね物体積に対して)は全ての場合に2.4sであった。付加的に表はSPENでプロペン副生物形成の選択率を示す。
【0124】
【表1】

【0125】
より高い使用圧力の場合に、より低い温度でより高いプロパン転化率が、目的生成物化合物の本質的には不変の選択率で生じる。
その他の点では、生成物ガス混合物は全ての場合に僅かな量の別の酸、例えば酢酸並びにCOxを含有していた。
【0126】
3.所定の循環ガス比での出口圧力P3に依存した本発明による方法の場合の等温圧縮機出力
2.による方法を図2による装置配置中で実施する。40mol%の転化率及び70mol%のアクリル酸形成の選択率に基づいている。プロパン:酸素:水のモル組成は2.における組成に相応する。循環ガス操作は窒素含量に影響を及ぼす。その際に多様な数字は次の意味を有する:
1=反応段階
2=後処理段階
3=残留生成物ガス混合物の流出された部分
4=循環ガス圧縮機(送風機)
5=空気圧縮機
6=酸素源としての空気
7=新鮮なプロパン及び新鮮な水蒸気
8=再圧縮された循環ガス
9=出口圧力P3の制御のための絞り装置
10=入口圧力P1を有する反応ガス出発混合物。
【0127】
新鮮なプロパン及び新鮮な水蒸気は、その都度必要な入口圧力で入手できる。
【0128】
すなわち、P3の変化は循環ガス圧縮機の出力及び空気圧縮機の出力にのみ影響を及ぼす。
【0129】
単純さのために、双方の圧縮機が等温圧縮であると仮定する。
【0130】
VDI-Waermeatlas、Verlag des Vereins Deutscher Ingenieure、Duesseldorf、第5版、1988、シートLa 1によれば、空気圧縮機の等温圧縮機出力(VL)は次の通りである:
【0131】
【数1】
[式中、
【0132】
【数2】
nL=空気圧縮機の効率;
ZL=空気についての実在気体ファクター;
R=比気体定数=モル質量により除した理想の気体定数;
TL=新鮮な空気が周囲から吸い込まれる温度;
l=常圧(周囲圧力)=空気が吸い込まれる1bar;
P1=空気が圧縮される反応段階への入口圧力;
すなわち、
【0133】
【数3】
ここで、Aは定数である]。
【0134】
相応して、循環ガス圧縮機VKの等温圧縮機出力は次の通りである:
【0135】
【数4】
【0136】
すなわち、
【0137】
【数5】
ここで、A′は定数であり、ここで:A′≒Aである。
【0138】
それゆえ、適用すべき全圧縮機出力Vges=VL+VKは次の通りである:
【0139】
【数6】
【0140】
ここで、
【0141】
【数7】
であり、その際に、Cは使用される反応装置及び後処理装置に特徴的な定数であり、かつ
【0142】
【数8】
は次の通りとなる:
【0143】
【数9】
【0144】
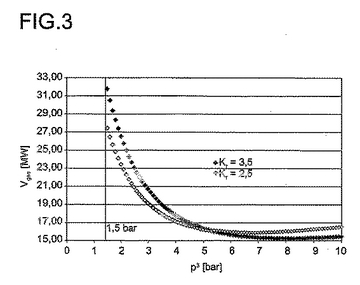
循環ガス比を一度2.5に及び一度3.5と選択する場合に、かつ定数CをC=(1.5bar+ΔP1.5)・ΔP1.5と決定し、
その際に、ΔP1.5は1.5barの出口圧力(後処理段階の出口)で反応段階及び後処理段階を経て全体として受けた圧力損失であり、本発明による方法に代表的な2barの圧力損失の場合にC=(1.5bar+2bar)・2bar=7bar2となり、
【0145】
【数10】
Kr=2.5で
【0146】
【数11】
及びKr=3.5で
【0147】
【数12】
双方の選択された循環ガス比で、P3に依存した図3に示されたVgesの経過となる(増大する圧力と共に同じままである転化率及び選択率の2.の結果による保存的な上昇のもとで)。
【0148】
P3=1.5barから出発して、増大するP3と共に双方の場合に全体として適用すべき圧縮機出力は減少し、このことは本発明による手法の有利な点を有する。
【図面の簡単な説明】
【0149】
【図1】本発明による方法の例示的な実施変法を示す略示図。
【図2】本発明による方法の例示的な実施変法を示す略示図。
【図3】P3に依存した図3に示されたVgesの経過を示すグラフ。
【符号の説明】
【0150】
1 反応段階、 2 後処理段階、 3 循環ガス圧縮機(送風機)、 4 流出物エクスパンダー、 5 流出物、 6 酸素源としての空気、 7 空気圧縮機、 8 新鮮なプロパン及び/又はイソブタン並びに場合により水蒸気、 9 再圧縮された循環ガス
【技術分野】
【0001】
本発明は、プロパン及び/又はイソブタンを、少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法に関するものであり、前記方法の場合に、反応ガス出発混合物用の入口、場合により補助ガス用の別の入口及び生成物ガス混合物用の出口を除いてガス側で封止されている反応段階に、プロパン及び/又はイソブタン、分子酸素及び少なくとも1つの不活性な希釈ガスを含有し、入口圧力P1を有する反応出発混合物を供給し、反応段階において高められた温度で反応ガス出発混合物を固体の集合状態である触媒に通過させることにより反応ガス出発混合物中に含まれるプロパン及び/又はイソブタンを少なくとも1つの目的生成物に直接部分酸化及び/又はアンモ酸化し、かつ反応ガス混合物を少なくとも1つの目的生成物として含有し、出口圧力P2を有する生成物ガス混合物を、反応段階から外へ及びこの圧力P2で、ガス側で生成物ガス混合物用の入口、場合により補助ガス用の別の入口及び残留生成物ガス混合物用の出口を除いて封止されている後処理段階へ導き、後処理段階において反応段階の生成物ガス混合物からその中に含まれる目的生成物を液相中へ基本分離し(grundabgetrennt)、かつその際に残っている、プロパン及び/又はイソブタンだけでなく並びに場合によりプロペン及び/又はイソブテンを含有し、出口圧力P3[ここでP3<P1である]を有する残留生成物ガス混合物を、後処理段階から外へ導き(herausfuehrt)、かつ残留生成物ガス混合物中に含まれるプロパン及び/又はイソブタンを反応段階へ返送する。
【0002】
アクリル酸及びメタクリル酸は、例えばポリマーを製造するための、重要な中間生成物である。
【0003】
反応段階においてプロパン及び/又はイソブタンの不均一系触媒作用による部分直接酸化によるアクリル酸及びメタクリル酸の製造は公知である(例えばEP-A 529853、EP-A 603836、EP-A 608838、EP-A 895809、DE-A 19835247、DE-A 10051419、DE-A 10122027、EP-A 1254707、EP-A 1254709、EP-A 1192987、EPA 1090684、DE-A 10254279及びこれらの明細書に引用された文献参照)。
【0004】
酸化剤として通常、分子酸素が使用され、これは例えば純粋な形でか又は部分酸化に関して本質的に不活性に挙動するガス(例えば空気中のN2)との混合物で反応ガス出発混合物に添加されることができる。不活性な希釈ガス、例えばN2、H2O、CO、CO2、He及び/又はAr等は、反応熱を吸収し、かつ反応ガス混合物を爆発範囲の外側に保持する。一般的に、本明細書において不活性な希釈ガスは、部分酸化を通過する反応ガス混合物のワンパス(einmaligen Durchgang)の際に5mol%未満、好ましくは3mol%未満及び特に好ましくは2mol%未満が反応するようなガスであると理解される。触媒として通例、固体の集合状態である多元素酸化物(Multielementoxide)が使用される。反応段階の実施は、固体の集合状態である多元素酸化物については触媒固定床、−渦動床(-wirbelbett)又は−移動床(−流動床)において可能である。
【0005】
反応段階における使用圧力は、技術水準の教示によれば原則的に常圧(=1bar)を下回るだけではなく1barを上回っていてよい(例えばDE-A 19835247、EP-A 895809及びDE-A 10261186参照)。通例、この圧力は反応段階における流れ抵抗を克服する目的のために常圧をやや上回っている。
【0006】
プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化することにとって不利であるのは、プロパン及びイソブタンの比較的際立つ反応不活性である。これは、相応する反応段階を通過する反応ガス混合物のワンパスの際に高められた温度でさえ通例、プロパン及び/又はイソブタンの部分転化のみが達成される原因となる。
【0007】
プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸不均一系触媒作用により部分直接酸化するという目標設定は、故に、反応段階を通過する反応ガス混合物のワンパスの際にできるだけ低い温度で、プロパン及び/又はイソブタンのできるだけ高い転化率を同時に目的生成物形成のできるだけ高い選択率で、すなわち、目的生成物のできるだけ高い空時収量をできるだけ僅かなエネルギー消費で、達成することにある。
【0008】
さらに、プロパン及び/又はイソブタンを所望の目的生成物の少なくとも1つに不均一系触媒作用により部分直接酸化する経済的な実施のための別の必要条件は、生成物ガス混合物中に含まれる未反応のプロパン及び/又はイソブタンを広範囲に亘って反応段階へ返送することにある。技術水準においてこのためには次の提案がなされている。
【0009】
DE-A 10119933は、生成物ガス混合物からのプロパンの不均一系触媒作用による部分直接酸化によるアクリル酸の製造の際に、その中に含まれるアクリル酸を液状吸収剤中への吸収により基本分離し、かつその際に生じる吸収剤及びアクリル酸からなる液状混合物を引き続いてそれ自体として公知の方法で精留により、抽出により及び/又は結晶化により純アクリル酸まで後処理するか又は分別凝縮による液相中への生成物ガス混合物からのアクリル酸の基本分離を、例えばDE-A 19924532に記載されているように、実施し、かつその際に生じる水性のアクリル酸凝縮物を例えば分別晶出によりさらに精製することを推奨する。
【0010】
液相中へのアクリル酸のそのような基本分離の際に残っている未反応のプロパンを含有する残留生成物ガス混合物に関して、DE-A 10119933は、プロパンを残留生成物ガス混合物から分離し、かつ分離されたプロパンをアクリル酸への部分直接酸化へ返送することを推奨する。分離法として、そのためには例えば分別加圧精留又は疎水性有機溶剤(プロパンを好ましくは吸収することができる)での抽出及びプロパンの遊離の目的のためのその後の脱着及び/又は空気でのストリッピングが推奨される。
【0011】
完全な対応関係において、EP-A 1193240も、プロパンのようなアルカンの部分直接酸化の場合に、残留生成物ガス混合物中に含まれる未反応のアルカンを(前記の技術水準の明細書と同じように、好ましくは副生物として形成されたアルケンと一緒に)それから分離し(例えば吸収又は吸着により)、かつ部分酸化へ返送することを推奨する。
【0012】
しかしながら、技術水準の推奨による反応段階への未反応のアルカン及び場合により副生物として形成されたアルケンの返送様式にとって不利であるのは、未反応のアルカンが通常、比較的高い希釈で含まれている残留生成物ガス混合物からのアルカン及び場合によりアルケンの分離が、比較的費用がかかり、かつ特に高い圧力損失と結びついていることである。後者は反応段階における高められた圧力の適用を退ける、それというのも、分離されたプロパンを返送する際に常にこの圧力に再圧縮されなければならないからである。
【0013】
さらにそのために不利であるのは、残留生成物ガス混合物中にそれ以外にさらに含まれる、不均一系触媒作用による部分直接酸化に有利に作用する成分(例えば、通例、触媒活性材料の活性及び選択率を向上させる水蒸気、又は高圧縮されてはいけない残留酸素)が反応段階へ返送されず(むしろ流出され)、かつそれに必要に応じて再三再四新たに供給されなければならないことである。
【0014】
残留生成物ガス混合物中に含まれる未反応の炭化水素を別個に、すなわち、分離されて反応段階へ返送するという提案の原因となることに加えて、とりわけ、循環すべきガス量(及びこれと共に反応ガス出発混合物量も)をできるだけ僅かに保持して、このようにしてこれに関して適用すべき移送出力及び圧縮機出力(循環されるガスは反応段階への入口の前に反応ガス混合物の入口圧力に再圧縮されなければならない、それというのも、反応段階、後処理段階及び残留生成物ガス混合物からの分離を通過する経路で、移送抵抗を克服するために消費され、かつ再供給されなければならない圧力損失を受けるからである)並びに必要な体積を最小限にすることを考慮すべきである。別の目標設定はその際に、供給原料損失を最小限に保持すべきである。
【0015】
本発明の課題は故に、プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法を提供することにあり、前記方法の場合に、消費する圧縮機出力及び供給原料損失は他のより有利な方法で最小限にされ、かつ同時に空時収量が最小限にされたエネルギー消費で最適化される。
【0016】
それに応じて、プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法であって、
反応ガス出発混合物用の入口、場合により補助ガス用の別の入口及び生成物ガス混合物用の出口は別としてガス側で封止されている反応段階に、プロパン及び/又はイソブタン、分子酸素及び少なくとも1つの不活性な希釈ガスを含有し、入口圧力P1を有する反応ガス出発混合物を供給し、
反応段階において反応ガス出発混合物を高められた温度で固体の集合状態である触媒に通過させることにより反応ガス出発混合物中に含まれるプロパン及び/又はイソブタンを少なくとも1つの目的生成物に直接部分酸化させ、かつ
反応ガス混合物を少なくとも1つの目的生成物として含有し、出口圧力P2を有する生成物ガス混合物を、反応段階の中から外へ及びこの圧力P2で、ガス側で生成物ガス混合物用の入口、場合により補助ガス用の別の入口及び残留生成物ガス混合物用の出口を除いて封止されている後処理段階中へ導き、後処理段階において反応段階の生成物ガス混合物からその中に含まれる目的生成物を液相中へ基本分離し、かつ
その際に残っている、プロパン及び/又はイソブタンだけでなく並びに場合によりプロペン及び/又はイソブテンを含有し、出口圧力P3[ここでP3<P1である]を有する残留生成物ガス混合物を、後処理段階から外へ導き、かつ残留生成物ガス混合物中に含まれるプロパン及び/又はイソブタンを反応段階へ返送する
方法が見出され、前記方法は、
P1が、P3が≧1.5barであり、かつ残留生成物ガス混合物を同じ組成の2つの部分量へ分割し、その一方の部分量を流出物として外へ移し、他方の部分量を循環ガスとして返送し、かつ反応出発混合物の成分として入口圧力P1に圧縮して再び反応段階に供給するように選択される
ことにより特徴付けられている。それゆえ、本発明による方法はとりわけEP-A 495504に開示された手法とは、後処理段階中及び/又は後処理段階後に流出の前に接触一酸化炭素酸化を有しないことが異なる。本発明による方法の場合に残留生成物ガス混合物の二酸化炭素洗浄も必要としない。
【0017】
反応段階において副生物としてプロペン及び/又はイソブテンが生じる場合には、これらの化合物は後処理段階を経て通常、プロパン及び/又はイソブタンと共存したままであり、かつ一緒に循環ガス中に存在して反応段階へ返送される。
【0018】
本明細書における全ての圧力の記載は、他に明示的に記載されない限り、絶対圧力であると理解される。
【0019】
通常、残留生成物ガス混合物は、プロパン及び/又はイソブタンとは異なる並びにプロペン及び/又はイソブテンとは異なる並びに場合により酸素とは異なる(例えばCO、CO2、H2O及び/又はN2等)成分(通例これらは生成物ガス混合物中に含まれる、目的生成物よりも容易に(常圧で)沸騰する成分である)を少なくとも2体積%、又は少なくとも5体積%、たいてい少なくとも10体積%含有する。
【0020】
本発明による方法の場合に部分酸化すべき有機の前駆物質化合物(すなわち、プロパン及び/又はイソブタン)は実地においてしばしば液状で貯蔵される一方で、しかし常圧及び常温でガス状であり、通例、有機の前駆物質化合物を反応段階入口圧力にするためには単純な蒸発で十分である。不活性な希釈ガスとして場合により併用される水蒸気は、多種多様な源からたいてい同様に十分な過圧で入手できる。
【0021】
しかしながらこのことは、通例少なくとも酸素源(例えば空気又は窒素の減損された空気)、場合によりその他の不活性な希釈ガスに当てはまらず、かつ、とりわけプロパンを含有する循環ガスには当てはまらない(この循環ガスは通常、反応段階及び後処理段階を通過する経路での圧力損失を差し引いて反応段階入口圧力P1並びに双方の部分量への除数を有し;反応段階へのその返送は本発明による方法の場合に通常、より大きな付加的な圧力損失を有しない内部構造物が含まれない管を経て行われる)。
【0022】
故に実地において、反応ガス出発混合物の成分の少なくとも1つの部分量(少なくとも循環ガス)を、圧縮機を用いて低い出発圧力からより高い最終圧力(反応段階への入口圧力P1)にすることが通常必要である。
【0023】
その際に、これらの成分の圧縮(例えば酸素源である空気及び循環ガス)は空間的に別個の圧縮機中で又は単一の圧縮機中で行われることができる。
【0024】
原則的に、挙げられたガスのこの圧縮に多種多様な種類の圧縮機は使用されることができる。押しのけ式圧縮機(例えば往復ピストン圧縮機、スクリュー圧縮機及び回転ピストン圧縮機)、流動式圧縮機(例えばターボ圧縮機、円心圧縮機、軸流圧縮機及び半径流圧縮機)及びジェット圧縮機を例示的に挙げることができる。本発明によれば好ましくは、例えばDE-A 10259023に記載されているような半径流圧縮機が使用される。
【0025】
さらに本発明によれば好ましくは、多様な源に由来し、本質的には反応段階入口圧力である(反応段階入口圧力にされた)反応ガス出発混合物の部分量を、別個の管路から来て、まず最初にたいてい、例えばスタティックミキサー(空の管に対して高められた混合作用を有する内部構造物を有する空間)中で混合し、次に、場合により入口温度に加熱し、反応段階に供給するようにして行われる。
【0026】
スタティックミキサーに供給された管路への個々のガスの進入はその際に好都合にはしばしば、爆発性混合物の発生が回避されるように選択される(本発明による部分酸化の場合にこの進入順序は好都合には例えば次のような内容であってよい:まず最初に循環ガス及び/又は蒸気、ついで空気及び最終的に有機の前駆物質化合物)。もちろん、水蒸気含量は反応ガス出発混合物に、生成物ガス流との間接熱交換により本質的に反応温度に加熱された反応ガス出発前駆物質混合物中へ微細に分配された液状の水滴を計量供給し、前記水滴は吸熱下に自発的に蒸発し、その際に反応ガス出発混合物が生じるように添加されることもできる。選択的に、予熱された反応ガス出発前駆物質混合物はガス飽和器を経て(ガス混合物及び水は並流又は向流で大表面積に導かれる)導かれることができる。
【0027】
それゆえ、本発明により選択された出口圧力P3は本質的には、循環ガス及び酸素源の圧縮のための圧縮機出力に影響を及ぼす。
【0028】
応用技術的に好都合には、残留生成物ガス混合物が本発明による方法の場合に後処理段階を去る圧力P3は、通例30又は25bar以下、しばしば20bar以下である。本発明によれば好都合には出口圧力P3は1.5bar以上かつ10bar以下、好ましくは2bar以上かつ8bar以下、しばしば3bar以上及び6bar以下もしくは5bar以下(例えば4bar)である。
【0029】
すなわち、本発明による方法を特徴付ける特徴は、反応段階並びに後処理段階を高められた圧力で運転することである。
【0030】
そのような手法は次の理由から有利である:
・ 意外にも、プロパン及び/又はイソブタンの不均一系触媒作用による部分直接酸化が高められた圧力で、それ以外は同じ反応条件下で及びワンパスに基づいて高められた転化率を、そのために目的生成物形成の選択率の本質的な減少が付随することなく、もたらすことが見出された;
・ 高められた圧力での後処理段階運転もまた、比較的僅かなそのために必要な体積で及び比較的僅かな受けた圧力損失で高められた循環ガス量の移送も可能にする、それというのも、所定のガス量の移送体積並びにその移送に付随している圧力損失は、増大する圧力と共に通例低下するからであり;後者は、反応段階の入口圧力P1への循環ガス圧縮に必要な圧縮機出力を減少させ;同時に、流出物量と比較して高められた循環ガス量は、流出物中に含まれる未反応のプロパン及び/又はイソブタンの損失を最小限にする;
・ 残留生成物ガス混合物からのプロパンの予備分離なしでのプロパンの返送は、そのような分離に必然的に付随している圧力損失を回避し、かつ残留生成物ガス混合物中に場合により含まれる他の成分、例えば水蒸気及びO2の同時で及びエネルギー的に好都合な返送を保証する。
【0031】
すなわち、全体として、本発明によれば、圧力上昇という比較的単純な措置を通じて、技術水準による方法の本質的に全ての有利な本質的特徴が、そのために残留生成物ガス混合物からの未反応のアルカン及び場合によりアルケンの費用のかかる分離を必要とすることなく(このことは付加的に望ましくない欠点、例えば水蒸気の完全な不返送又はエネルギー的に費用のかかる返送を回避する)、同様に達成されることができ、かつ同時に圧力上昇の措置は、目的生成物選択率の本質的な減少なしに反応段階を通過する単一処理量に基づく反応物転化率の増大を引き起こす。
【0032】
“反応段階”もしくは“後処理段階”という概念は、本明細書において特に、ガス側で、入口及び出口並びに場合により補助ガス用の別の入口を除いて封止されている1つ又は複数の直列に接続された装置ユニット(apparative Vorrichtungen)を表すので、ガス混合物がそのような装置ユニットもしくはそのような装置ユニットの直列接続を通過する際に受ける圧力損失は、流れ抵抗の克服を制限する。
【0033】
例えば、装置ユニット(又はそのようなユニットの直列接続)は、管束反応器、渦動床反応器、流動床反応器、そのような反応器の直列接続、吸収塔、精留塔、凝縮塔又はそのような塔もしくは個々の急冷段階の直列接続であってよい。しかしもちろん、前記のような反応器は、本発明による方法を実施する間に、例えばWO 02/081421に記載されているように、例えば反応器中へ触媒活性剤を添加する可能性を含むことができる。補助ガスの概念は、本発明による方法の場合に例えば、反応器の直列接続の場合に反応器の間で不活性ガス及び/又は酸素(例えば空気)を補充するか、又は後処理段階において例えば重合防止の理由から分子酸素を含有するガス(例えば空気)を生成物ガス混合物と一緒に後処理段階へ導く可能性を含むべきである。典型的には圧力損失は本発明による方法の場合に反応段階に亘って0.1〜3bar、しばしば0.3〜1又は0.5bar、及び後処理段階に亘って0.5〜3bar、しばしば1〜2barである。
【0034】
特に高い圧力の範囲内で本発明による方法を実施する場合に、圧力損失は反応段階において並びに後処理段階においてさらに明らかにより低くなりうるものであり、かつ例えば0.05bar及びそれ未満までの値を達成することができる。
【0035】
反応段階への入口での圧力P1は、本発明による方法の場合に、適用される後処理段階の方法に応じて、後処理段階の出口での圧力P3を0.5又は1〜4bar、たいてい1.5〜3.5bar、多くの場合に2〜3bar上回っている。
【0036】
特に高い圧力の範囲内で本発明による方法を実施する際に、反応段階への入口での圧力P1は通例、後処理段階の出口での圧力P3を0.5bar未満(例えば0.1又は0.01bar)上回っている。
【0037】
反応段階への入口での典型的な圧力P1は故に2.5〜25barである。通例、反応段階への入口での圧力P1は3〜10bar及び本発明によれば好都合には4〜8barである。
【0038】
後処理段階への入口での典型的な圧力P2は3〜25bar、しばしば3〜20bar、又は3〜15bar又は3〜8barである。
【0039】
圧力比の制御は、本発明による方法の場合に単純な方法で、例えば、残留生成物ガス混合物の外へ移すべき部分量についての流出物の絞り装置を用いて可能である。絞り装置の代わりに後接続されたエクスパンダー(外へ移すことが行われる逆圧縮機)を用いて、本発明による方法の場合に、その他の点では既に記載された利点に加えて、残留生成物ガス混合物の一部の流出の際に常圧へのその制御された放圧により入口圧力P1への残留生成物ガス混合物及び/又は酸素源(例えば空気)の循環される他方の部分を圧縮するために必要な圧縮機出力の一部が回収されることができる。
【0040】
流出物として外へ移される残留生成物ガス混合物の部分量に対する本発明による方法の場合に循環ガスとして返送される残留生成物ガス混合物の部分量の量比は、反応ガス出発混合物の組成に個別に依存する。しかしながら、通例、Vは0.5以上又は1以上、たいてい1.5以上、好ましくは2以上、特に好ましくは3以上である。もちろんVは本発明による方法の場合に8以上又は10以上であってもよい。通常、Vは本発明による方法の場合に30以下、たいてい25以下、しばしば20以下である。しばしばVは15以下又は10以下、好ましくは2〜8である。
【0041】
その他の点では、本発明による方法は、プロパン及び/又はイソブタンを少なくとも1つの目的生成物に不均一系触媒作用により部分酸化する技術水準から公知の方法のように実施されることができる。
【0042】
すなわち、本発明による方法の範囲内で必要な分子酸素のための源として、空気、並びに分子窒素の減損された空気(例えば≧90体積%−O2、≦10体積%−N2)、並びに純粋な分子酸素又は分子酸素と他の不活性ガスとからなる混合物が考慮される。
【0043】
触媒として、本発明による方法のためには、技術水準においてプロパン及び/又はイソブタンを少なくとも1つの目的生成物に不均一系触媒作用により部分直接酸化するのに推奨される原則的に全ての触媒が考慮される。
【0044】
これらは例えば明細書JP-A 3-170445、EP-A 609122及びEP-A 747349の触媒である。
【0045】
その際に、本質的に全ての触媒が本発明により可能な不均一系触媒作用による部分直接酸化のそれぞれに使用されることができることが本発明によれば本質的である。
【0046】
これらの触媒の活性材料は、通例、多元素酸化物、たいてい多金属酸化物である。
【0047】
多金属酸化物の中では、本発明による方法のためには特に、明細書EP-A 608838、EP-A 529853、DE-A 10254279、DE-A 19835247、EP-A 895809、JP-A 7-232071、JP-A 11-169716、DE-A 10261186、EP-A 1192987、JP-A 10-57813、JP-A 2000-37623、JP-A 10-36311、WO 00/29105、EP-A 767164、DE-A 10029338、JP-A 8-57319、JP-A 10-28862、JP-A 11-43314、JP-A 11-574719、WO 00/29106、JP-A 10-330343、JP-A 11-285637、JP-A 310539、JP-A 11-42434、JP-A 11-343261、JP-A 3423262、WO 99/03825、JP-A 7-53448、JP-A 2000-51693、JP-A 11-263745、DE-A 10046672、DE-A 10118814、DE-A 10119933、JP-A 2000/143244、EP-A 318295、EP-A 603836、DE-A 19832033、DE-A 19836359、EP-A 962253、DE-A 10119933、DE-A 10051419、DE-A 10046672、DE-A 10033121、DE-A 101 459 58、DE-A 10122027、EP-A 1193240及びこれらの明細書に引用された文献の多金属酸化物材料が適している。
【0048】
使用すべき触媒装入物の活性材料は、前記の場合に本質的には、元素Mo、V、双方の元素Te及びSbの少なくとも1つ、及びNb、Ta、W、Ti、Al、Zr、Cr、Mn、Ga、Fe、Ru、Co、Cs、Ca、Sr、Ba、Rh、Ni、Pd、Pt、La、Pb、Cu、Re、Ir、Y、Pr、Nd、Tb、Bi、B、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの元素の少なくとも1つを組み合わせて含有する多金属酸化物材料である。
【0049】
好ましくはその際に最後の元素の群からの組合せは元素Nb、Ta、W及び/又はTi及び特に好ましくは元素Nbを含有する。
【0050】
好ましくは、該当する多金属酸化物活性材料は、化学量論I
Mo1VbM1cM2d (I)
[ここで、
M1=Te及び/又はSb、
M2=Nb、Ta、W、Ti、Al、Zr、Cs、Ca、Sr、Ba、Cr、Mn、Ga、Fe、Ru、Co、Rh、Ni、Pd、Pt、La、Bi、Pb、Cu、Re、Ir、V、Pr、Nd、Tb、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの元素の少なくとも1つ、
b=0.01〜1、
c=>0〜1、及び
d=>0〜1]
における前記の元素の組合せを含有する。
【0051】
本発明によれば好ましくはM1=Te及びM2=Nb、Ta、W及び/又はTiである。特にM2=Nbである。
【0052】
化学量論係数bは、有利には0.1〜0.6である。相応して、化学量論係数cの好ましい範囲は0.01〜1もしくは0.05〜0.4に達し、かつdの好都合な値は0.001〜1もしくは0.01〜0.6である。
【0053】
本発明によれば、化学量論係数b、c及びdが同時に前記の好ましい範囲内である場合に特に好都合である。
【0054】
前記のことは、触媒装入物の活性材料が酸素とは異なるその元素に関して前記の元素の組合せからなる場合に特に当てはまる。
【0055】
これらは特に、一般的な化学量論II
Mo1VbM1cM2dOn (II)
[ここで、変数は化学量論Iに関して記載された意味を有し、かつnは(II)中の酸素とは異なる元素の原子価及び頻度により決定される数である]
で示される多金属酸化物活性材料である。
【0056】
好ましくは 該当する多金属酸化物材料は、化学量論III
Mo1Va′M4b′M5c′M6d′ (III)
[ここで、
M4=Te及びSbを含む群からの元素の少なくとも1つ;
M5=Nb、Ti、W、Ta及びCeを含む群からの元素の少なくとも1つ;
M6=Pb、Ni、Co、Bi、Pd、Cs、Ca、Sr、Ba、Ag、Pt、Cu、Au、Ga、Zn、Sn、In、Re、Ir、Sm、Sc、Y、Pr、Nd及びTbを含む群からの元素の少なくとも1つ;
a′=0.01〜1;
b′=>0〜1;
c′=>0〜1;及び
d′=0〜0.5]
における冒頭に挙げられた元素の組合せを含有する。
【0057】
好ましくはa′は0.05〜0.6、特に好ましくは0.1〜0.6又は0.5である。好ましくはb′は0.01〜1、特に好ましくは0.01もしくは0.1〜0.5又は0.4である。好ましくはc′は0.01〜1、特に好ましくは0.01もしくは0.1〜0.5又は0.4である。好ましくはd′は0.00005もしくは0.0005〜0.5、特に好ましくは0.001〜0.5、しばしば0.002〜0.3及びより頻繁に0.005もしくは0.01〜0.1である。
【0058】
M4は好ましくはTeである。
M5は好ましくは、その全量の少なくとも50mol%、より好ましくは少なくとも75mol%及び極めて特に好ましくは少なくとも100mol%がNbである。
M6は好ましくはNi、Co、Bi、Pd、Ag、Au、Pb及びGaを含む群からの少なくとも1つの元素、特に好ましくはNi、Co、Pd及びBiを含む群からの少なくとも1つの元素である。
【0059】
極めて特に好ましくは、M5はその全量の少なくとも50mol%、又は少なくとも75mol%、又は100mol%がNbであり、かつM6はNi、Co、Pd及びBiを含む群からの少なくとも1つの元素である。
【0060】
本発明によれば傑出しているのは、M4=Te、M5=Nb及びM6がNi、Co及びPdを含む群からの少なくとも1つの元素である。
【0061】
前記のことは特に、触媒装入物の活性材料が酸素とは異なるその元素に関して化学量論(III)の元素の組合せからなる場合に当てはまる。これらは特に、一般的な化学量論(IV)
Mo1Va′M4b′M5c′M6d′On′ (IV)
[ここで、変数は化学量論IIIに関して記載された意味を有し、かつn′は、(IV)中の酸素とは異なる元素の原子価及び頻度により決定される数である]
で示される多金属酸化物活性材料である。
【0062】
さらに本発明による方法のためには好ましくは、一方では前記の元素の組合せ(I)、(III)の1つを含有するか又は、酸素とは異なる元素に関して、それらからなり、かつその都度同時に、回折反射h及びiを示し、それらのピークが回折角(2Θ)22.2±0.5゜(h)及び27.3±0.5゜(i)であるX線回折図を有するような多金属酸化物活性材料が使用される(本明細書においてX線回折図に関する全ての記載は、X線としてのCu−Kα−線の使用下に発生されたX線回折図に関する(Siemens-回折計Theta-Theta D-5000、管電圧:40kV、管電流:40mA、絞りV20(可変)、散乱線絞りV20(可変)、二次単色光器絞り(0.1mm)、検出器絞り(0.6mm)、測定間隔(2Θ):0.02゜、1工程あたりの測定時間:2.4s、検出器:シンチレーション計数管)。
【0063】
これらの回折反射の半値幅はその際に極めて小さくても、又は極めて際立っていてもよい。
【0064】
本発明による方法のためには、前記の多金属酸化物活性材料の、X線回折図が回折反射h及びiに加えて回折反射kを有し、そのピークが28.2±0.5゜(k)であるものが好都合である。
【0065】
後者の中では、そしてまた本発明によれば、回折反射hがX線回折図の中で強度が最も強く、並びに多くとも0.5゜の半値幅を有するものが好ましく、かつ極めて特に好ましくは、回折反射i及び回折反射kの半値幅が同時にその都度1゜以下であり、かつ回折反射kの強度Pk及び回折反射iの強度Piが関係0.2≦R≦0.85、より良好には0.3≦R≦0.85、好ましくは0.4≦R≦0.85、特に好ましくは0.65≦R≦0.85、さらにより好ましくは0.67≦R≦0.75及び極めて特に好ましくはR=0.70〜0.75もしくはR=0.72を満たし、ここでRは式
R=Pi/(Pi+Pk)
により定義された強度比であるものである。好ましくは前記のX線回折図は回折反射を有さず、その最大は2Θ=50±0.3゜である。
【0066】
X線回折図における回折反射の強度の定義は、本明細書においてDE-A 19835247、DE-A 10122027、並びにDE-A 10051419及びDE-A 10046672において記載された定義に関連している。同じことは半値幅の定義に当てはまる。
【0067】
回折反射h、i及びkに加えて、本発明によれば有利には使用すべきそのような多金属酸化物活性材料の前記のX線回折図は、さらに別の回折反射を有し、それらのピークは次の回折角(2Θ)である:
9.0±0.4゜(l)
6.7±0.4゜(o)及び
7.9±0.4゜(p)。
【0068】
さらに、X線回折図が付加的に、ピークが回折角(2Θ)=45.2±0.4゜(q)である回折反射を有する場合に好都合である。
【0069】
しばしば、X線回折図はさらに反射29.2±0.4゜(m)及び35.4±0.4゜(n)も有する。
【0070】
さらに、式(I)、(II)、(III)及び(IV)において定義された元素の組合せが純粋なi−相として存在する場合に好都合である。触媒活性な酸化物材料がさらにk−相も含有する場合には、それらのX線回折図は前記のものに加えてさらに別の回折反射を有し、それらのピークは次の回折角(2Θ)である:36.2±0.4゜及び50±0.4゜(i−及びk−相という概念は本明細書においてDE-A 10122027及びDE-A 10119933に規定されたように使用される)。
【0071】
回折反射hに強度100が割り当てられる場合に、本発明によれば、回折反射i、l、m、n、o、p、qが同じ強度スケールにおいて次の強度を有する場合に好都合である:
i:5〜95、しばしば5〜80、部分的には10〜60;
l:1〜30;
m:1〜40;
n:1〜40;
o:1〜30;
p:1〜30及び
q:5〜60。
【0072】
X線回折図が前記の付加的な回折反射を有する場合には、その半値幅は通例1゜以下である。
【0073】
本発明により使用すべき一般式(II)又は(IV)の多金属酸化物活性材料又は一般式(I)又は(III)の元素の組合せを有する多金属酸化物活性材料の比表面積は、とりわけ、それらのX線回折図が記載されたように構成されている場合には、多くの場合に1〜40m2/gもしくは10〜30m2/g(BET表面積、窒素)である。
【0074】
記載された多金属酸化物活性材料の製造は、これらの引用された技術水準に関連して見出される。これらには、特にDE-A 10303526、DE-A 10261186、DE-A 10254279、DE-A 10254278、DE-A 10122027、DE-A 10119933、DE-A 10033121、EP-A 1192987、DE-A 10029338、JP-A 2000-143244、EP-A 962253、EP-A 895809、DE-A 19835247、WO 00/29105、WO 00/29106、EP-A 529853及びEP-A 608838が含まれる(最後に挙げた2つの明細書の全ての実施例において乾燥法として噴霧乾燥が適用されうる;例えば300〜350℃の入口温度及び100〜150℃の出口温度で;向流又は並流)。
【0075】
記載された多金属酸化物活性材料は、それ自体として(すなわち粉末形で)又は適しているジオメトリーに成形されて(例えばDE-A 10051419のシェル触媒(Schalenkatalysatoren)又はDE-A 10122027の幾何学的な変型)、本発明による方法に使用されることができる。これらは、プロパンからのアクリル酸の製造に、しかしまたイソブタンからのメタクリル酸の製造にも卓越して適している。
【0076】
本発明による方法を実施するためには、全ての挙げられた触媒は、希釈されずに、並びに不活性な粒子及び/又は成形体(これらは活性材料を有しない)で希釈されて使用されることができる。適している希釈材料は例えばステアタイトである。
【0077】
希釈成形体のジオメトリーはその際に触媒成形体のそれと好ましくは同一である。
【0078】
本発明による方法に適している多金属酸化物活性材料触媒に関して明細書に記載されたように、本発明による方法は固定床触媒装入物並びに渦動床−又は流動床触媒装入物上で実施されることができる。その際に本発明により適用可能な反応段階入口圧力は既に記載されている。
【0079】
反応温度は、特に本明細書に推奨された触媒を使用する場合に、200〜700℃、好ましくは200〜550℃又は230〜480℃又は300〜440℃であってよい。
【0080】
プロパン及び/又はイソブタンでの触媒装入物の負荷は50〜3000Nl/l(触媒装入物)/h、又は80〜1500Nl/l/h、又は100〜1000Nl/l/h、又は120〜600Nl/l/h、又は140〜300Nl/l/hであってよい。
【0081】
反応ガス出発混合物での触媒装入物の負荷はその際に100〜10000Nl/l/h、又は300〜6000Nl/l/h、又は300〜2000Nl/l/hであってよい。触媒装入物中での平均滞留時間は0.01〜10s、又は0.1〜10s、又は2〜6sであってよい。
【0082】
プロパンからのアクリル酸もしくはイソブタンからのメタクリル酸の製造の場合に、反応ガス出発混合物は例えば:
プロパン、もしくはイソブタン 0.5〜15体積%、しばしば1〜7体積%、
空気 10〜90体積%、しばしば20〜50体積%、
水蒸気 0〜50体積%及び
残りの量として循環ガス
を含有していてよい。
【0083】
反応ガス出発混合物はしかしまた、本発明による方法のためのプロパンからのアクリル酸もしくはイソブタンからのメタクリル酸の製造の場合に:
プロパン、もしくはイソブタン 0.6〜1.2体積%、
空気 65〜95体積%、
窒素 2〜30体積%、
COx 0.05〜0.8体積%及び
水蒸気 2〜3体積%
を含有していてよい。
【0084】
水蒸気(新鮮)10〜50体積%を含有する反応ガス出発混合物が好ましい。
【0085】
反応ガス出発混合物の他の可能な組成は、例えば
プロパン又はイソブタン 70〜90体積%、
分子酸素 5〜25体積%、
水蒸気 0〜25体積%及び
残りの量として循環ガス
を含有していてよい。
【0086】
ここでも、全体として水蒸気を10〜50体積%含有する反応ガス出発混合物が好ましい。すなわち、典型的には反応ガス出発混合物の組成は、本発明による方法のためにはプロパン又はイソブタン部分酸化の場合に次の範囲内で変わる(モル比):
イソブタン又はプロパン: 酸素: H2O:その他の希釈ガス=
1:(0.1〜10): (0〜50): (0〜50)、
好ましくは 1:(0.1〜10):(0.1〜50): (1〜50)、
特に好ましくは 1: (0.5〜5): (1〜30): (1〜30)。
【0087】
前記の範囲は特に、その他の希釈ガスとして主に分子窒素が使用される場合に当てはまる。他の可能な希釈ガスは、例えばHe、Ar、CO及び/又はCO2等である。
【0088】
かなり一般的に反応ガス出発混合物はその組成において、好ましくは爆発ガス範囲の外側にあるように選択される。
【0089】
本発明による方法が部分酸化として実施される場合には、これは例えば、EP-A 700714及びEP-A 700893に記載されているような、一帯域−管束反応器中で行われることができる。しかしまた、DE-A 19927624、DE-A 19948242、DE-A 19948241、DE-A 19910508及びDE-A 19910506に記載されているような、多帯域−管束反応器中で実施されることもできる。渦動床における本発明による方法の実施は、例えばWO 02/0811421に記載されたように実施されることができる。
【0090】
反応ガス出発混合物中に含まれるプロパン及び/又はイソブタンに対して、プロパン及び/又はイソブタン転化率は、本発明による方法の場合に、反応段階を通過する反応ガス混合物のワンパスに基づいて、通例10又は20〜70mol%、しばしば30〜60mol%及び多くの場合に40〜60mol%もしくは45〜55mol%である。目的生成物形成の選択率は、通常、40〜98又は95又は〜90mol%、多くの場合に50〜80mol%、しばしば60〜80mol%である。
【0091】
本発明による方法の反応段階において生じる生成物ガス混合物からの少なくとも1つの目的生成物の基本分離は、本発明による後処理段階において原則的に、技術水準による方法から公知であるように実施されることができる。特に、本発明によれば、プロペン及び/又はイソブテンの不均一系触媒作用による部分酸化による目的生成物の製造の際に公知であるような、技術水準において生成物ガス混合物からの同じ目的生成物の基本分離に公知である後処理法も使用されることができる。
【0092】
通例、本発明による方法の場合に反応段階において生じる生成物ガス混合物は本発明による後処理段階への進入の際にまず最初に間接及び/又は直接の冷却にかけられる。
【0093】
液相中への生成物ガス混合物中に含まれる目的生成物の基本分離の目的のためには、こうして冷却された(アクリル酸の場合に例えば典型的には150〜250℃に)又はまた冷却されない生成物ガス混合物は例えば吸収塔中で、少なくとも1つの目的生成物を本質的に選択的に生成物ガス混合物から吸収する下降する液状吸収剤に対して向流で導かれることができ、例えばJP-A 2001/0026269、EP-A 990636、JP-A 2000/327651、EP-A 925272、EP-A 695736、EP-A 778255、DE-A 2136396、DE-A 2449780、DE-A 4308087、EP-A 982287、EP-A 982289、EP-A 982288及びDE-A 19631645には目的生成物及び多様な吸収剤について記載されている。
【0094】
本質的には双方の目的生成物のためには、吸収剤として、水(又は水性混合物、例えば水性カセイソーダ液又は水性のアクリル酸もしくはメタクリル酸)、アクリル酸及びメタクリル酸のエステル化に使用されるアルコール、例えば2−エチルヘキサノール、並びにより高い沸点を有する有機溶剤が考慮される。本発明によれば好ましくは、有機溶剤の沸点は、生成物ガス混合物から分離すべき目的生成物(アクリル酸及び/又はメタクリル酸)の沸点を少なくとも20℃、特に少なくとも50℃及び特に好ましくは少なくとも70℃上回っている。本発明によれば好ましい有機吸収剤は180〜400℃、特に220〜360℃の沸点(常圧で)を有する。本発明によれば特に適している吸収剤は、目的生成物であるアクリル酸及びメタクリル酸の場合に、外に向けて作用する極性基を有しない、高沸点で過度に疎水性の溶剤、例えば脂肪族又は芳香族の炭化水素、例えばパラフィン蒸留からの中油留分、又は酸素原子上にかさばる基を有するエーテル、又はそれらの混合物であり、その際にこれらに有利には極性溶剤、例えばDE-A 4308087に開示された1,2−ジメチルフタレートが添加される。さらに、炭素原子1〜8個を有する直鎖状のアルカノールとの安息香酸及びフタル酸のエステル、例えば安息香酸−n−ブチルエステル、安息香酸メチルエステル、安息香酸エチルエステル、フタル酸ジメチルエステル、フタル酸ジエチルエステル、並びにいわゆる熱媒体油、例えばジフェニル、ジフェニルエーテル及びジフェニルとジフェニルエーテルとからなる混合物又はそれらの塩素誘導体及びトリアリールアルカン、例えば4−メチル−4′−ベンジル−ジフェニルメタン及びその異性体2−メチル−2′−ベンジル−ジフェニルメタン、2−メチル−4′−ベンジル−ジフェニルメタン及び4−メチル−2′−ベンジル−ジフェニルメタン及びそのような異性体の混合物が適している。
【0095】
アクリル酸に特に好ましい吸収剤は(メタクリル酸は好ましくは水中に吸収される)、ジフェニル及びジフェニルエーテルからなり、好ましくは共沸組成物で、特にジフェニル及びジフェニルエーテル100質量%に対してジフェニル(ビフェニル)約25質量%及びジフェニルエーテル約75質量%からなる溶剤混合物、例えば商業的に入手可能なDiphyl(登録商標)である。好ましくはこの溶剤混合物はさらに、極性溶剤、例えばジメチルフタレートを全ての溶剤混合物に対して0.1〜25質量%の量で含有する。有利には生成物ガス混合物は、吸収剤として高沸点の有機溶剤の使用の際に、吸収前に直接凝縮器又は急冷装置中での吸収剤の部分蒸発により冷却される。このためには特にベンチュリスクラバー、気泡塔又はスプレー凝縮器が適している。
【0096】
高沸成分、重沸成分、中沸成分及び低沸成分という概念は、本明細書において、目的化合物よりも高い沸点(高沸成分)、目的化合物とほぼ同じ沸点(中沸成分)もしくは目的化合物よりも低い沸点(低沸成分)を有する化合物を意味する。特に、目的化合物がアクリル酸である場合である。
【0097】
かなり一般的に、向流吸収は好ましくは、規則充填物又は不規則充填積重ね物を備えた塔中で又は好ましくはデュアルフロートレイ及び/又はバルブトレイが設けられており、かつ上から溶剤が装入される、段塔中で行われる。生成物ガス混合物(及び場合により急冷装置からの蒸発された吸収剤)は下から塔中へ導通され、引き続いて吸収温度に冷却される。冷却は有利には冷却回路により行われる。すなわち、加熱され、塔中で下降する吸収剤は塔から取り出され、熱交換器中で冷却され、取り出し位置を上回る位置で吸収塔へ再び返送される。吸収後に、本質的には全ての重沸成分、大部分の目的化合物(例えばアクリル酸)及び一部の低沸成分は吸収剤中に存在する。目的化合物(例えばアクリル酸)を基本分離されて含有する被吸収剤から、ついで目的生成物は技術水準において(例えば吸収の際に引用された)記載されたように任意の純度でさらに分離されることができ(例えばDE-A 19606877又はDE-A 19838845のように)、かつ目的生成物を含まない吸収剤は吸収において再び使用されることができる(例えば有機の被吸収剤からアクリル酸を頂部を経て有機吸収剤から分離し、かつ精留により及び/又は結晶化により(例えば融成物洗浄塔中での結晶分離を伴う懸濁晶出)更に精製するか又は水性被吸収剤から水を有機共沸剤を用いて精留により頂部を経て及びアクリル酸をアクリル酸を含有する缶出液から精留により及び/又は結晶化により任意の純度で更に分離し;最後の場合に留出液は通例2つの相へ分けられ(冷却により);有機相は精留塔へ及び水相は吸収塔へ返送される(その都度塔頂部で))。
【0098】
残っている、吸収されない残留生成物ガス混合物はさらに、単純に凝縮可能な部分の低沸点副成分(例えば水、ホルムアルデヒド及び酢酸)を分離するために、冷却されることができる(通例、酸性水が挙げられる)。残っている残留生成物ガス混合物は、本発明によれば2つの部分へ分割され、かつ双方の部分の一方は循環ガスとして返送されることができ(反応段階へ)、他方の部分は流出されることができる。好ましくは本発明によれば酸性水分離は行われない。特に本発明による方法の反応段階において水蒸気が希釈ガスとして併用される(多金属酸化物材料(I)、(II)、(III)又は(IV)の使用の際に、このことは通例、目的生成物形成の選択率のために有利である)場合に、液相中への反応段階の生成物ガス混合物中に含まれる目的生成物の本発明による基本分離は(それとは独立して、特に本明細書に記載された分離方法が使用される)、好ましくは、残っている、本発明によれば少なくとも部分量として(循環ガスとして)反応段階へ返送すべき残留生成物ガス混合物中に、その中に含まれるプロパンに対するその中に含まれる水蒸気のモル比Wが、反応段階の生成物ガス混合物中の相応するモル比W′よりも多くとも50%、より良好には多くとも40又は30%、さらにより良好には多くとも20又は10%、好ましくは多くとも5%少ないように行われる。極端な場合には本発明による方法の場合に前記の比W及びW′は同一であってもよい。
【0099】
残留生成物ガス混合物中にできるだけ多くの水蒸気をそのままにしておく試みは、ここでは、反応ガス出発混合物中への水蒸気の新鮮なフィード(もしくは凝縮及び新たな蒸発)をできるだけ広範囲に亘って放棄することができるという目的を追求する。
【0100】
しかしながら、本発明による方法の場合に、流出物として外へ移される残留生成物ガス混合物の部分量は、反応ガス混合物中での水の水準上昇(Aufpegeln)を防止するために反応段階中に副生物として形成されるのと少なくとも同じ水を含有していなければならない(反応段階における反応がより選択的に導かれるほど、流出すべき水量はより僅かになる)。このことは相応して、反応段階において形成された他の副成分にも当てはまる。本発明による方法の場合に酸素源として空気が使用される場合には、残留生成物ガス混合物の流出量は同時に、その中に含まれる窒素量が、空気フィード中に含まれている窒素量に少なくとも相当するように、見積もられなければならない。
【0101】
吸収剤として高沸点有機溶剤の1つが使用される場合には、吸収は前記の場合に好ましくは(特にアクリル酸の吸収の場合に)、吸収塔からの排出が単相であるように実施される。その他の点では、吸収の際に残っている残留生成物ガス混合物中の水蒸気含量は、選択された吸収剤から独立して、吸収温度の適している選択により調節されることができる。
【0102】
溶剤中への吸収による液相中への目的生成物の基本分離に選択的に、この基本分離は(特にアクリル酸の場合に)、凝縮、特に分別凝縮によっても行われることができ、例えばDE-A 19924532、DE-A 10200583、DE-A 10053086、DE-A 19627847、DE-A 19740253、DE-A 19740252、DE-A 19740253、DE-A 19814387及びDE-A 10247240に記載されている。
【0103】
その際に、反応段階の生成物ガス混合物は、場合により直接及び/又は間接の予備冷却後に、分離効果のある内部構造物が設けられた分離塔中で自体が上昇して分別凝縮にかけられ、かつ目的生成物は分離塔の側部取出しを経て取り出され、かつ技術水準において記載されたように別の結晶化による及び/又は精留による分離工程にかけられる。
【0104】
分離効果のある内部構造物を有する凝縮塔としてその際に、下から上へまず最初にデュアルフロートレイ及びそれに引き続いて水力学的に密封された十字流トレイを分離効果のある内部構造物として有する段塔が好ましく、これらは例えば前記の技術水準に記載されている。
【0105】
本発明によれば好ましくは、前記の分別凝縮も、本質的には生成物ガス混合物中に含まれる水の分離が行われないように実施される。すなわち、好ましくはここでも酸性水分離は行われない。
【0106】
しかしながら、流出物として外へ移される残留生成物ガス混合物の部分量は、それらの流出の前に、メタクリル酸もしくはアクリル酸損失を回避するために、さらに水で洗浄されることができる。その際に生じるメタクリル酸もしくはアクリル酸を含有する酸性水から、アクリル酸もしくはメタクリル酸及び場合により他の有機の価値のある生成物は、例えば吸収に使用される有機吸収剤で抽出され、かつ被吸収剤と合一されることができる。
【0107】
吸収塔並びに分別凝縮用の塔は、高められた圧力損失の回避の目的のために、原則的に吸収剤又は凝縮物を用いて運転される急冷段階の直列接続によっても置換されることができる。
【0108】
かなり一般的に、重合防止は液相中への生成物ガス混合物からの目的生成物の基本分離の範囲内で、技術水準に記載されたように相応する重合防止剤の添加により行われる。
【0109】
本発明によれば、目的生成物の基本分離の際に残っている残留生成物ガス混合物が、プロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンに加えて、常圧で目的生成物よりも容易に沸騰する成分を通常少なくとも5体積%、たいてい少なくとも10体積%含有することが本質的である。これらは特に、生成物ガス混合物の常圧で水よりも容易に沸騰する成分(例えばN2、CO、CO2)並びに好ましくは水自体である。
【0110】
本発明による方法の一利点は、循環ガスが通例、送風機を用いて(ほぼ出口圧力P3から)入口圧力P1に再圧縮されることができることにある。送風機はその際に、低い圧力比(1.1:1〜3:1のような最終圧力対出口圧力)を有する圧縮機(通常、軸流圧縮機)であると理解される。それとは異なり、酸素源として使用される空気は通常、半径流圧縮機を用いて(ほぼ周囲圧力から)入口圧力P1に圧縮される。原則的に本明細書において言及された圧縮は等温的に又はポリトロープに実施されることができる。後者は本発明によれば好ましい。
【0111】
本発明による手法の有利な点の一部は、残留生成物ガス混合物の外へ移すべき部分から、場合によりその中に含まれる水含分(反応段階へ返送されることができる)の予めの凝縮による分離後に、その中に含まれるプロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンが分離され、かつ入口圧力P1に再圧縮されて反応段階へ返送される場合にも、依然として得られたままである。
【0112】
この分離(技術水準において残留生成物ガス混合物の全量のために推奨され、かつ原則的に(特に次に記載されたように)使用されることもできる)は、例えば、残留生成物ガス混合物の外へ移すべき部分に含まれるプロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンが吸収的及び/又は吸着的に分離され、かつ脱離(Desorbtion)及び/又は脱着により次に再び遊離されるようにして行われる(WO 0196271参照)。例えば被吸収剤からのこの遊離は、空気でのストリッピングにより実施されることができ、これは例えばWO 0196271に記載されている。空気は、次に共に圧縮され、かつ反応段階へ共に返送されることができる。
【0113】
選択的に残留生成物ガス混合物の外へ移すべき部分は、別の酸化反応器にも、場合により酸素の補充後に、炭化水素転化率を高めるために供給されることができる。
【0114】
通例、既に述べたように、本発明による方法の場合に循環ガスの再圧縮の目的のためには送風機で十分である。
【0115】
本明細書に添付している図1は本発明による方法の例示的な実施変法を示す。
【0116】
その際に、多様な数字は以下の意味を有する:
1=反応段階
2=後処理段階
3=循環ガス圧縮機(送風機)
4=流出物エクスパンダー
5=流出物
6=酸素源としての空気
7=空気圧縮機
8=新鮮なプロパン及び/又はイソブタン並びに場合により水蒸気
9=再圧縮された循環ガス。
【0117】
実施例
1.多金属酸化物活性材料Mo1V0.29Te0.13Nb0.13Ox(純粋なi−相)を有する触媒の製造
メタバナジン酸アンモニウム87.61g(V2O5 78.55質量%、G.f.E.社、ニュルンベルク)を80℃で水3040ml中に溶解させた(撹拌機、温度計及び還流冷却器を備えた三つ口フラスコ)。澄明な帯黄色溶液が生じた。この溶液を60℃に冷却し、ついで60℃に維持しながら連続してテルル酸117.03g(H6TeO6 99質量%、Aldrich社)及び七モリブデン酸アンモニウム400.00g(MoO3 82.52質量%、Starck社、Goslar)を撹拌混入した。生じる深紅色の溶液を30℃に冷却した(溶液A)。
【0118】
ビーカー中で別個にシュウ酸ニオブアンモニウム112.67g(Nb 20.8質量%、Starck社、Goslar)を60℃で水500ml中に溶解させた(溶液B)。
【0119】
溶液Bを30℃に冷却し、この温度で溶液Aと合一し、その際に溶液Bを溶液Aに添加した。添加を連続的に約5分の期間に亘って行った。浮遊沈殿を有する橙色の水性懸濁液が生じた。引き続いてこの懸濁液を噴霧乾燥した(Tリザーバ=30℃、T入=320℃、T出=110℃、t=1.5h、Niro社のアトマイザー型のスプレー塔)。生じた噴霧物は同様に橙色であり、かつ秤量化学量論Mo1V0.33Te0.22Nb0.11を有していた。
【0120】
スプレー粉末100g×2を、DE-A 10119933の図1による回転球面炉(内容積1 l)中で、まず最初に27.5minかけて線形加熱傾斜で50Nl/hの空気流下に25℃から275℃に加熱し、引き続いてこの温度を1h保持することにより熱処理した。その後、線形加熱傾斜で32.5minかけて275℃から600℃に加熱し、その際に空気流を窒素流50Nl/hにより置換した。窒素流を保持しながら600℃を2h保持し、次に炉全体を室温に冷却した。得られた酸化物材料100gを10質量%のHNO3−水溶液1000ml中で70℃で7h還流下に撹拌し、残っている固体を、生じたスラリーからろ別し、水(25℃)を用いて硝酸塩不含に洗浄した。得られたフィルターケーキをマッフル炉中で一晩に亘り空気下に110℃で乾燥させた。
【0121】
得られた固体の化学分析は、組成Mo1V0.29Te0.13Nb0.13Oxとなった。属するX線回折図は純粋なi−相を有していた。
【0122】
引き続いて、乾燥させた材料をDE-A 10119933に記載されたようにRetschミル中で粉砕し(粒度≦0.12mm)、DE-A 10119933の例A)a)のようにシェル触媒に加工した:
粉砕した活性材料38g;2.2〜3.2mmの直径を有する球状担体150g(担持材料=CeramTec社、DEのステアタイトC-220、45μmの表面粗面度Rzを有する)、粘着剤=グリセリン及び水からなる混合物30ml(グリセリン:水の質量比=1:3)、乾燥期間=150℃で16h;生じるシェル触媒の活性材料含分は20質量%であった(シェル触媒の質量に対して)。
【0123】
2.異なる圧力でのアクリル酸へのプロパンの不均一系触媒作用による部分直接酸化
1.からのシェル触媒を、V2A鋼(外径=60mm、内径=8.5mm)からなる反応管(長さ140cm)に装入した。装入物長さは53cmを選択した(=シェル触媒約35.0g)。担持材料として使用されるステアタイト球からなる長さ30cmの予備積重ね物を触媒帯域の位置調節に利用した。同じステアタイト球を、反応管に触媒帯域の後に最終的に充填した(反応ガス出発混合物を温め始めるための予備加熱帯域)。反応管を、外側からその長さ全体で電気加熱マットを用いて加熱した。反応ガス出発混合物のモル組成はプロパン:空気:水=1:15:14であった。次の表は、ワンパスでの生じたプロパン転化率(UPAN、mol%)並びにこの転化率に付随しているアクリル酸形成の選択率(SACS、mol%)を、選択された入口圧力並びに加熱マットの属する温度に応じて示す。滞留時間(触媒積重ね物体積に対して)は全ての場合に2.4sであった。付加的に表はSPENでプロペン副生物形成の選択率を示す。
【0124】
【表1】
【0125】
より高い使用圧力の場合に、より低い温度でより高いプロパン転化率が、目的生成物化合物の本質的には不変の選択率で生じる。
その他の点では、生成物ガス混合物は全ての場合に僅かな量の別の酸、例えば酢酸並びにCOxを含有していた。
【0126】
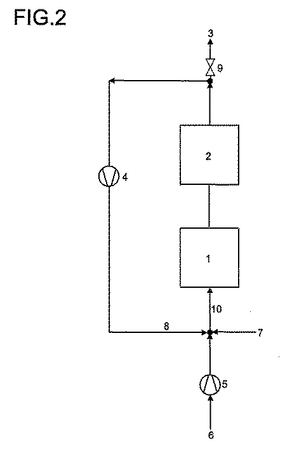
3.所定の循環ガス比での出口圧力P3に依存した本発明による方法の場合の等温圧縮機出力
2.による方法を図2による装置配置中で実施する。40mol%の転化率及び70mol%のアクリル酸形成の選択率に基づいている。プロパン:酸素:水のモル組成は2.における組成に相応する。循環ガス操作は窒素含量に影響を及ぼす。その際に多様な数字は次の意味を有する:
1=反応段階
2=後処理段階
3=残留生成物ガス混合物の流出された部分
4=循環ガス圧縮機(送風機)
5=空気圧縮機
6=酸素源としての空気
7=新鮮なプロパン及び新鮮な水蒸気
8=再圧縮された循環ガス
9=出口圧力P3の制御のための絞り装置
10=入口圧力P1を有する反応ガス出発混合物。
【0127】
新鮮なプロパン及び新鮮な水蒸気は、その都度必要な入口圧力で入手できる。
【0128】
すなわち、P3の変化は循環ガス圧縮機の出力及び空気圧縮機の出力にのみ影響を及ぼす。
【0129】
単純さのために、双方の圧縮機が等温圧縮であると仮定する。
【0130】
VDI-Waermeatlas、Verlag des Vereins Deutscher Ingenieure、Duesseldorf、第5版、1988、シートLa 1によれば、空気圧縮機の等温圧縮機出力(VL)は次の通りである:
【0131】
【数1】
[式中、
【0132】
【数2】
nL=空気圧縮機の効率;
ZL=空気についての実在気体ファクター;
R=比気体定数=モル質量により除した理想の気体定数;
TL=新鮮な空気が周囲から吸い込まれる温度;
l=常圧(周囲圧力)=空気が吸い込まれる1bar;
P1=空気が圧縮される反応段階への入口圧力;
すなわち、
【0133】
【数3】
ここで、Aは定数である]。
【0134】
相応して、循環ガス圧縮機VKの等温圧縮機出力は次の通りである:
【0135】
【数4】
【0136】
すなわち、
【0137】
【数5】
ここで、A′は定数であり、ここで:A′≒Aである。
【0138】
それゆえ、適用すべき全圧縮機出力Vges=VL+VKは次の通りである:
【0139】
【数6】
【0140】
ここで、
【0141】
【数7】
であり、その際に、Cは使用される反応装置及び後処理装置に特徴的な定数であり、かつ
【0142】
【数8】
は次の通りとなる:
【0143】
【数9】
【0144】
循環ガス比を一度2.5に及び一度3.5と選択する場合に、かつ定数CをC=(1.5bar+ΔP1.5)・ΔP1.5と決定し、
その際に、ΔP1.5は1.5barの出口圧力(後処理段階の出口)で反応段階及び後処理段階を経て全体として受けた圧力損失であり、本発明による方法に代表的な2barの圧力損失の場合にC=(1.5bar+2bar)・2bar=7bar2となり、
【0145】
【数10】
Kr=2.5で
【0146】
【数11】
及びKr=3.5で
【0147】
【数12】
双方の選択された循環ガス比で、P3に依存した図3に示されたVgesの経過となる(増大する圧力と共に同じままである転化率及び選択率の2.の結果による保存的な上昇のもとで)。
【0148】
P3=1.5barから出発して、増大するP3と共に双方の場合に全体として適用すべき圧縮機出力は減少し、このことは本発明による手法の有利な点を有する。
【図面の簡単な説明】
【0149】
【図1】本発明による方法の例示的な実施変法を示す略示図。
【図2】本発明による方法の例示的な実施変法を示す略示図。
【図3】P3に依存した図3に示されたVgesの経過を示すグラフ。
【符号の説明】
【0150】
1 反応段階、 2 後処理段階、 3 循環ガス圧縮機(送風機)、 4 流出物エクスパンダー、 5 流出物、 6 酸素源としての空気、 7 空気圧縮機、 8 新鮮なプロパン及び/又はイソブタン並びに場合により水蒸気、 9 再圧縮された循環ガス
【特許請求の範囲】
【請求項1】
プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法であって、
反応ガス出発混合物用の入口、場合により補助ガス用の別の入口及び生成物ガス混合物用の出口を除いてガス側で封止されている反応段階に、プロパン及び/又はイソブタン、分子酸素及び少なくとも1つの不活性な希釈ガスを含有し、入口圧力P1を有する反応ガス出発混合物を供給し、反応段階において反応ガス出発混合物を高められた温度で固体の集合状態である触媒に通過させることにより反応出発混合物中に含まれるプロパン及び/又はイソブタンを少なくとも1つの目的生成物に直接部分酸化させ、かつ反応ガス混合物を少なくとも1つの目的生成物として含有し、出口圧力P2を有する生成物ガス混合物を、反応段階から外へ及びこの圧力P2で、ガス側で生成物ガス混合物用の入口、場合により補助ガス用の別の入口及び残留生成物ガス混合物用の出口を除いて封止されている後処理段階へ導き、後処理段階において反応段階の生成物ガス混合物からその中に含まれる目的生成物を液相中へ基本分離し、かつその際に残っている、プロパン及び/又はイソブタンだけでなく並びに場合によりプロペン及び/又はイソブテンを含有し、出口圧力P3[ここでP3<P1である]を有する残留生成物ガス混合物を、後処理段階から外へ導き、かつ残留生成物ガス混合物中に含まれるプロパン及び/又はイソブタンを反応段階へ返送する方法において、
P1を、P3が1.5bar以上であり、かつ残留生成物ガス混合物を同じ組成の2つの部分量に分割し、かつその中から一方の部分量を流出物として外へ移し、かつ他方の部分量を循環ガスとして返送し、かつ反応ガス出発混合物の成分として入口圧力P1に圧縮して再び反応段階に供給する
ように選択する
ことを特徴とする、プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法。
【請求項2】
残留生成物ガス混合物が、プロパン及び/又はイソブタンとは異なる並びにプロペン及び/又はイソブテンとは異なる成分を少なくとも5体積%含有する、請求項1記載の方法。
【請求項3】
残留生成物ガス混合物が、プロパン及び/又はイソブタンとは異なる並びにプロペン及び/又はイソブテンとは異なる成分を少なくとも10体積%含有する、請求項1記載の方法。
【請求項4】
圧力P3が1.5bar以上かつ25bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項5】
圧力P3が1.5bar以上かつ20bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項6】
圧力P3が1.5bar以上かつ10bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項7】
圧力P3が2bar以上かつ8bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項8】
圧力P1が圧力P3を1〜4bar上回っている、請求項1から7までのいずれか1項記載の方法。
【請求項9】
圧力P1が圧力P3を1.5〜3.5bar上回っている、請求項1から7までのいずれか1項記載の方法。
【請求項10】
P1が3〜10barである、請求項1から9までのいずれか1項記載の方法。
【請求項11】
P1が4〜8barである、請求項1から9までのいずれか1項記載の方法。
【請求項12】
流出物として外へ移される残留生成物ガス混合物の部分量を、エクスパンダーを経て外へ移す、請求項1から11までのいずれか1項記載の方法。
【請求項13】
反応段階が、触媒が装入された管束反応器又は渦動床反応器である、請求項1から12までのいずれか1項記載の方法。
【請求項14】
後処理段階が、吸収塔又は分別凝縮用の塔又は急冷段階の直列接続である、請求項1から13までのいずれか1項記載の方法。
【請求項15】
触媒が活性材料として、元素Mo、V、双方の元素Te及びSbの少なくとも1つ及びNb、Ta、W、Ti、Al、Zr、Cr、Mn、Ga、Fe、Ru、Co、Cs、Ca、Sr、Ba、Rh、Ni、Pd、Pt、La、Pb、Cu、Re、Ir、Y、Pr、Nd、Tb、Bi、B、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの少なくとも1つの元素を組み合わせて含有する多金属酸化物材料を有する、請求項1から14までのいずれか1項記載の方法。
【請求項16】
触媒が活性材料として、化学量論I
Mo1VbM1cM2d (I)
[ここで、
M1=Te及び/又はSb、
M2=Nb、Ta、W、Ti、Al、Zr、Cs、Ca、Sr、Ba、Cr、Mn、Ga、Fe、Ru、Co、Rh、Ni、Pd、Pt、La、Bi、Pb、Cu、Re、Ir、Y、Pr、Nd、Tb、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの少なくとも1つの元素、
b=0.01〜1
c=>0〜1及び
d=>0〜1]
を有する元素の組合せを含有する多金属酸化物材料を有する、請求項1から14までのいずれか1項記載の方法。
【請求項17】
酸素源として空気を使用する、請求項1から16までのいずれか1項記載の方法。
【請求項18】
反応温度が200〜700℃である、請求項1から17までのいずれか1項記載の方法。
【請求項19】
反応ガス出発混合物が、
プロパン又はイソブタン 0.5〜15体積%、
空気 10〜90体積%、
水蒸気 0〜50体積%及び
残りの量として循環ガス
を含有する、請求項1から18までのいずれか1項記載の方法。
【請求項20】
反応ガス出発混合物が、
プロパン又はイソブタン 0.5〜15体積%、
空気 10〜90体積%、
水蒸気 10〜50体積%及び
残りの量として循環ガス
を含有する、請求項1から18までのいずれか1項記載の方法。
【請求項21】
反応ガス出発混合物が、
プロパン又はイソブタン 70〜90体積%、
分子酸素 5〜25体積%、
水蒸気 0〜25体積%及び
残りの量として循環ガス
を含有する、請求項1から18までのいずれか1項記載の方法。
【請求項22】
反応段階を通過する反応ガス混合物のワンパスに基づくプロパン及び/又はイソブタンからなる転化率が10〜70mol%である、請求項1から21までのいずれか1項記載の方法。
【請求項23】
目的生成物形成の選択率が40〜98mol%である、請求項22記載の方法。
【請求項24】
液相中への反応段階の生成物ガス混合物中に含まれる目的生成物の基本分離を、残っている残留生成物ガス混合物中で、その中に含まれるプロパンに対するその中に含まれる水蒸気のモル比Wが、反応段階の生成物ガス混合物中の相応するモル比W′よりも多くとも50%少ないように行う、請求項1から23までのいずれか1項記載の方法。
【請求項25】
吸収塔中で有機溶剤中への吸収による液相中への反応段階の生成物ガス混合物中に含まれる目的生成物の基本分離を、吸収塔からの排出が単相であるように行う、請求項1から24までのいずれか1項記載の方法。
【請求項26】
流出物として外へ移される残留生成物ガス混合物の部分量から、その中に含まれるプロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンを分離し、かつ入口圧力P1に再圧縮して反応段階へ返送する、請求項1から25までのいずれか1項記載の方法。
【請求項27】
残留生成物ガス混合物の流出物として外に移される部分量に対する残留生成物ガス混合物の循環ガスとして返送される部分量の量比Vが、0.5以上かつ30以下である、請求項1から26までのいずれか1項記載の方法。
【請求項28】
残留生成物ガス混合物の流出物として外へ移される部分量に対する残留生成物ガス混合物の循環ガスとして返送される部分量の量比Vが、2以上かつ25以下である、請求項1から26までのいずれか1項記載の方法。
【請求項29】
残留生成物ガス混合物の流出物として外へ移される部分量に対する残留生成物ガス混合物の循環ガスとして返送される部分量の量比Vが、3以上かつ20以下である、請求項1から26までのいずれか1項記載の方法。
【請求項30】
循環ガスを、送風機を用いて入口圧力P1に再圧縮する、請求項1から29までのいずれか1項記載の方法。
【請求項31】
酸素源として空気を使用し、前記空気を、半径流圧縮機を用いて入口圧力P1に圧縮する、請求項1から30までのいずれか1項記載の方法。
【請求項32】
プロパンをアクリル酸に部分直接酸化する方法である、請求項1から31までのいずれか1項記載の方法。
【請求項1】
プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法であって、
反応ガス出発混合物用の入口、場合により補助ガス用の別の入口及び生成物ガス混合物用の出口を除いてガス側で封止されている反応段階に、プロパン及び/又はイソブタン、分子酸素及び少なくとも1つの不活性な希釈ガスを含有し、入口圧力P1を有する反応ガス出発混合物を供給し、反応段階において反応ガス出発混合物を高められた温度で固体の集合状態である触媒に通過させることにより反応出発混合物中に含まれるプロパン及び/又はイソブタンを少なくとも1つの目的生成物に直接部分酸化させ、かつ反応ガス混合物を少なくとも1つの目的生成物として含有し、出口圧力P2を有する生成物ガス混合物を、反応段階から外へ及びこの圧力P2で、ガス側で生成物ガス混合物用の入口、場合により補助ガス用の別の入口及び残留生成物ガス混合物用の出口を除いて封止されている後処理段階へ導き、後処理段階において反応段階の生成物ガス混合物からその中に含まれる目的生成物を液相中へ基本分離し、かつその際に残っている、プロパン及び/又はイソブタンだけでなく並びに場合によりプロペン及び/又はイソブテンを含有し、出口圧力P3[ここでP3<P1である]を有する残留生成物ガス混合物を、後処理段階から外へ導き、かつ残留生成物ガス混合物中に含まれるプロパン及び/又はイソブタンを反応段階へ返送する方法において、
P1を、P3が1.5bar以上であり、かつ残留生成物ガス混合物を同じ組成の2つの部分量に分割し、かつその中から一方の部分量を流出物として外へ移し、かつ他方の部分量を循環ガスとして返送し、かつ反応ガス出発混合物の成分として入口圧力P1に圧縮して再び反応段階に供給する
ように選択する
ことを特徴とする、プロパン及び/又はイソブタンを少なくとも1つの目的生成物であるアクリル酸、メタクリル酸に不均一系触媒作用により部分直接酸化する方法。
【請求項2】
残留生成物ガス混合物が、プロパン及び/又はイソブタンとは異なる並びにプロペン及び/又はイソブテンとは異なる成分を少なくとも5体積%含有する、請求項1記載の方法。
【請求項3】
残留生成物ガス混合物が、プロパン及び/又はイソブタンとは異なる並びにプロペン及び/又はイソブテンとは異なる成分を少なくとも10体積%含有する、請求項1記載の方法。
【請求項4】
圧力P3が1.5bar以上かつ25bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項5】
圧力P3が1.5bar以上かつ20bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項6】
圧力P3が1.5bar以上かつ10bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項7】
圧力P3が2bar以上かつ8bar以下である、請求項1から3までのいずれか1項記載の方法。
【請求項8】
圧力P1が圧力P3を1〜4bar上回っている、請求項1から7までのいずれか1項記載の方法。
【請求項9】
圧力P1が圧力P3を1.5〜3.5bar上回っている、請求項1から7までのいずれか1項記載の方法。
【請求項10】
P1が3〜10barである、請求項1から9までのいずれか1項記載の方法。
【請求項11】
P1が4〜8barである、請求項1から9までのいずれか1項記載の方法。
【請求項12】
流出物として外へ移される残留生成物ガス混合物の部分量を、エクスパンダーを経て外へ移す、請求項1から11までのいずれか1項記載の方法。
【請求項13】
反応段階が、触媒が装入された管束反応器又は渦動床反応器である、請求項1から12までのいずれか1項記載の方法。
【請求項14】
後処理段階が、吸収塔又は分別凝縮用の塔又は急冷段階の直列接続である、請求項1から13までのいずれか1項記載の方法。
【請求項15】
触媒が活性材料として、元素Mo、V、双方の元素Te及びSbの少なくとも1つ及びNb、Ta、W、Ti、Al、Zr、Cr、Mn、Ga、Fe、Ru、Co、Cs、Ca、Sr、Ba、Rh、Ni、Pd、Pt、La、Pb、Cu、Re、Ir、Y、Pr、Nd、Tb、Bi、B、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの少なくとも1つの元素を組み合わせて含有する多金属酸化物材料を有する、請求項1から14までのいずれか1項記載の方法。
【請求項16】
触媒が活性材料として、化学量論I
Mo1VbM1cM2d (I)
[ここで、
M1=Te及び/又はSb、
M2=Nb、Ta、W、Ti、Al、Zr、Cs、Ca、Sr、Ba、Cr、Mn、Ga、Fe、Ru、Co、Rh、Ni、Pd、Pt、La、Bi、Pb、Cu、Re、Ir、Y、Pr、Nd、Tb、Ce、Sn、Zn、Si、Na、Li、K、Mg、Ag、Au及びInを含む群からの少なくとも1つの元素、
b=0.01〜1
c=>0〜1及び
d=>0〜1]
を有する元素の組合せを含有する多金属酸化物材料を有する、請求項1から14までのいずれか1項記載の方法。
【請求項17】
酸素源として空気を使用する、請求項1から16までのいずれか1項記載の方法。
【請求項18】
反応温度が200〜700℃である、請求項1から17までのいずれか1項記載の方法。
【請求項19】
反応ガス出発混合物が、
プロパン又はイソブタン 0.5〜15体積%、
空気 10〜90体積%、
水蒸気 0〜50体積%及び
残りの量として循環ガス
を含有する、請求項1から18までのいずれか1項記載の方法。
【請求項20】
反応ガス出発混合物が、
プロパン又はイソブタン 0.5〜15体積%、
空気 10〜90体積%、
水蒸気 10〜50体積%及び
残りの量として循環ガス
を含有する、請求項1から18までのいずれか1項記載の方法。
【請求項21】
反応ガス出発混合物が、
プロパン又はイソブタン 70〜90体積%、
分子酸素 5〜25体積%、
水蒸気 0〜25体積%及び
残りの量として循環ガス
を含有する、請求項1から18までのいずれか1項記載の方法。
【請求項22】
反応段階を通過する反応ガス混合物のワンパスに基づくプロパン及び/又はイソブタンからなる転化率が10〜70mol%である、請求項1から21までのいずれか1項記載の方法。
【請求項23】
目的生成物形成の選択率が40〜98mol%である、請求項22記載の方法。
【請求項24】
液相中への反応段階の生成物ガス混合物中に含まれる目的生成物の基本分離を、残っている残留生成物ガス混合物中で、その中に含まれるプロパンに対するその中に含まれる水蒸気のモル比Wが、反応段階の生成物ガス混合物中の相応するモル比W′よりも多くとも50%少ないように行う、請求項1から23までのいずれか1項記載の方法。
【請求項25】
吸収塔中で有機溶剤中への吸収による液相中への反応段階の生成物ガス混合物中に含まれる目的生成物の基本分離を、吸収塔からの排出が単相であるように行う、請求項1から24までのいずれか1項記載の方法。
【請求項26】
流出物として外へ移される残留生成物ガス混合物の部分量から、その中に含まれるプロパン及び/又はイソブタン並びに場合によりプロペン及び/又はイソブテンを分離し、かつ入口圧力P1に再圧縮して反応段階へ返送する、請求項1から25までのいずれか1項記載の方法。
【請求項27】
残留生成物ガス混合物の流出物として外に移される部分量に対する残留生成物ガス混合物の循環ガスとして返送される部分量の量比Vが、0.5以上かつ30以下である、請求項1から26までのいずれか1項記載の方法。
【請求項28】
残留生成物ガス混合物の流出物として外へ移される部分量に対する残留生成物ガス混合物の循環ガスとして返送される部分量の量比Vが、2以上かつ25以下である、請求項1から26までのいずれか1項記載の方法。
【請求項29】
残留生成物ガス混合物の流出物として外へ移される部分量に対する残留生成物ガス混合物の循環ガスとして返送される部分量の量比Vが、3以上かつ20以下である、請求項1から26までのいずれか1項記載の方法。
【請求項30】
循環ガスを、送風機を用いて入口圧力P1に再圧縮する、請求項1から29までのいずれか1項記載の方法。
【請求項31】
酸素源として空気を使用し、前記空気を、半径流圧縮機を用いて入口圧力P1に圧縮する、請求項1から30までのいずれか1項記載の方法。
【請求項32】
プロパンをアクリル酸に部分直接酸化する方法である、請求項1から31までのいずれか1項記載の方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公表番号】特表2006−522764(P2006−522764A)
【公表日】平成18年10月5日(2006.10.5)
【国際特許分類】
【出願番号】特願2006−505033(P2006−505033)
【出願日】平成16年4月7日(2004.4.7)
【国際出願番号】PCT/EP2004/003690
【国際公開番号】WO2004/089856
【国際公開日】平成16年10月21日(2004.10.21)
【出願人】(595123069)ビーエーエスエフ アクチェンゲゼルシャフト (847)
【氏名又は名称原語表記】BASF Aktiengesellschaft
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
【公表日】平成18年10月5日(2006.10.5)
【国際特許分類】
【出願日】平成16年4月7日(2004.4.7)
【国際出願番号】PCT/EP2004/003690
【国際公開番号】WO2004/089856
【国際公開日】平成16年10月21日(2004.10.21)
【出願人】(595123069)ビーエーエスエフ アクチェンゲゼルシャフト (847)
【氏名又は名称原語表記】BASF Aktiengesellschaft
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
[ Back to top ]