
プロリル水酸化酵素阻害剤
【課題】 低酸素状態を改善し、低酸素症によるダメージから細胞や組織を保護ために有用なプロリル水酸化酵素阻害剤(PHD阻害剤)、並びに細胞や組織の低酸素状態の改善を目的とする医薬組成物の提供。PHD阻害剤として有効な新規化合物、並びにPHD阻害剤として有効な化合物を探索するためのスクリーニング方法の提供。
【解決手段】下式(A)または(B)で示される骨格を有する基の少なくとも1つを一分子中に含む化合物またはその塩をプロリル水酸化酵素阻害剤や細胞や組織の低酸素状態の改善を目的とする医薬組成物の有効成分とする:
【化1】

(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
【解決手段】下式(A)または(B)で示される骨格を有する基の少なくとも1つを一分子中に含む化合物またはその塩をプロリル水酸化酵素阻害剤や細胞や組織の低酸素状態の改善を目的とする医薬組成物の有効成分とする:
【化1】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、プロリル水酸化酵素阻害剤(以下、「PHD阻害剤」ともいう)に関する。より詳細には、本発明は低酸素誘導因子(以下、「HIF」ともいう)とプロリル水酸化酵素(PHD)との結合を阻害してPHDの働きを抑える作用を有し、その結果、PHDによるHIFの分解を抑制してHIFを安定化することのできるPHD阻害剤に関する。また本発明は、PHD阻害作用を有し、HIFを安定化させることで、低酸素状態が関連して生じる各種疾患や病態(障害)を予防または治療するのに有効な医薬組成物に関する。さらに、上記PHD阻害剤として有用な新規化合物、ならびにPHD阻害剤として有用な化合物を取得するのに有効なスクリーニング方法に関する。
【背景技術】
【0002】
虚血性心疾患、慢性腎不全および脳梗塞などの疾患では細胞や組織への酸素供給が減少する。かかる酸素供給の減少によって低酸素状態が続くと細胞や組織に機能的障害が生じるため、生体内では、血管新生、赤血球生成、解糖および酸化酵素阻害など、広範囲にわたって細胞防御機構が働くようになっている。
【0003】
低酸素誘導因子(HIF)は、これらの生体防御機構を媒介する重要なタンパク質であり、細胞や組織が低酸素状態に陥った際に発現が亢進し、細胞や組織がより多くの酸素を得られるように血管新生を促進することが知られている(例えば、非特許文献1〜3参照)。かかる低酸素誘導因子(HIF)は、α鎖とβ鎖のヘテロ二量体からなるタンパク質であり、低酸素状態では安定であるが、酸素存在下ではその安定性が著しく低下する。具体的には、HIFは、酸素存在下で酸素依存的なプロリル水酸化酵素(PHD)によってα鎖のプロリンが水酸化され(例えば、非特許文献4〜9参照)、その結果、von hipple lindau蛋白と結合してユビキチン化され(例えば、非特許文献10〜11参照)、その結果、プロテアソームの標的とされて分解される(例えば、非特許文献12〜13参照)。
【0004】
しかし、低酸素状態の下では、HIFはα鎖がPHDによる水酸化を受けないため分解されず、α鎖とβ鎖の二量体の状態で、核内でHRE(hypoxia response element)と結合し、その下流に位置する低酸素ストレスの順応に関わる多くの遺伝子〔例えばエリスロポエチン(赤血球産生ホルモン)、血管内皮増殖因子(VEGF)、解糖酵素、グルコーストランスポーター、ヘムオキシゲナーゼ〕の転写を促進して生体を低酸素改善へと導くように機能する(例えば、非特許文献14参照)。このためHIFを安定化することによって、広く下流反応を調整する「マスター遺伝子」のスイッチを活性化することができ、低酸素の影響から組織や細胞を保護することが可能となると考えられる。
【0005】
現在、虚血などの低酸素状態を招く疾患の治療法の多くは、例えば急性虚血時の血流を改善するために血栓溶解薬が使用されるなど、症状の改善や病因因子の改善に集中しているのが実情であり、他の角度から治療法を検討することが求められている。
【非特許文献1】Marx, J. Cell biology. How cells endure low oxygen. Science 303. 1454-1456 (2004):
【非特許文献2】Semenza, G.L. & Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 12. 5447-5454 (1992):
【非特許文献3】Wang, G.L. & Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. 90. 4304-4308 (1993)
【非特許文献4】Epstein, A.C.R. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107. 43-54 (2001)
【非特許文献5】Schofield, C.J. & Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5. 343-354 (2004)
【非特許文献6】Semenza, G.L. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda). 19. 176-182 (2004)
【非特許文献7】Masson, N. & Ratcliffe, P.J. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O(2) levels. J. Cell Sci. 116. 3041-3049 (2003)
【非特許文献8】Bruick, R.K. & McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294. 1337-1340 (2001)
【非特許文献9】Semenza, G.L. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 107. 1-3 (2001)
【非特許文献10】Maxwell, P.H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399. 271-275 (1999)
【非特許文献11】Ohh, M. et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2. 423-427 (2000)
【非特許文献12】Maxwell, P.H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399. 271-275 (1999)
【非特許文献13】Ohh, M. et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2. 423-427 (2000)
【非特許文献14】Pugh, C.W. & Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9. 677-684 (2003)
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、虚血性疾患などによって生じる低酸素状態を改善し、低酸素症によるダメージから細胞や組織を保護するために有効に用いられるプロリル水酸化酵素阻害剤(PHD阻害剤)を提供することを目的とする。本発明が提供するPHD阻害剤は、低酸素誘導因子(HIF)のα鎖プロリンとプロリル水酸化酵素(PHD)との結合を競合的に阻害することで、HIFα鎖のプロリンを水酸化するPHDの機能を阻害する作用を有するものである。また本発明は、かかるPHD阻害剤を用いた、細胞や組織の低酸素状態の改善を目的とする医薬組成物を提供することを目的とする。さらに本発明は、PHD阻害剤として有効な新規化合物、ならびにPHD阻害剤として有効な化合物を探索するためのスクリーニング方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
前述するように、低酸素誘導因子(HIF)は低酸素による細胞や組織のダメージに極めて重要な役割を果たしている。その活性は、α鎖のプロリン残基が酸素依存的にプロリル水酸化酵素(PHD)によって水酸化されることによって制御されている。このため、PHDを阻害することによってHIFが安定化され、これによって低酸素状態に対する生体防御機能が活性化され、低酸素によるダメージから細胞や組織を保護することができる。
【0008】
本発明者らは、この考えのもと、新規なプロリル水酸化酵素阻害剤を開発するために、プロリル水酸化酵素の三次元構造および構造をベースとしたドラッグデザインソフトウェアを用いて、まず公知のプロリル水酸化酵素阻害剤を用いてドッキング・シミュレーション研究を行った。その結果、プロリル水酸化酵素には、HIFαプロリンと結合する部位と2−オキシグルターゼと結合する部位の両方があり、そのうちHIFαプロリンと結合する部位を介してHIFとプロリン水酸化酵素との結合を競合的に阻害する作用を有する特定の基を有する化合物が、HIFを安定活性化ないしは安定化するプロリル水酸化酵素阻害剤として有効であることを見出した。さらにこの特定の基を有する化合物は、in vivo試験により、HIF活性を安定化し、血管新生形成を促進して虚血による神経死を防止できることを確認した。本発明はかかる知見に基づいて完成したものである。
【0009】
すなわち、本発明には下記の態様が含まれる:
(I)プロリル水酸化酵素阻害剤(PHD阻害剤)
項1.下式(A)または(B)で示される骨格を有する基の少なくとも1つを一分子中に含む化合物またはその塩を有効成分とするプロリル水酸化酵素阻害剤:
【0010】
【化1】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
項2.骨格Aを有する基が、下式:
【0011】
【化2】
(R1は水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、保護基を有していてもよいカルボキシアルキル基、またはアミノ基:R2は水素原子、水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、またはアミノ基:R3は水素原子、アミノ基、または低級アルキルアミノ基を意味する)
で示される基である、項1記載のプロリル水酸化酵素阻害剤。
項3.骨格Bを有する基が、下式:
【0012】
【化3】
(R4、R5、R6およびR7は、同一または異なって、水素原子または低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
で示される基である、項1または2に記載するプロリル水酸化酵素阻害剤。
項4.下式で示される化合物(I):
【0013】
【化4】
〔R1は炭素数1〜6の低級アルキル基または保護基を有していてもよい炭素数1〜4のカルボキルアルキル基:R2は水素原子、炭素数1〜6の低級アルキル基:R10は、R11−S−CH2−(式中、R11は置換基を有していてもよいヘテロアリール基、または下式:
【0014】
【化5】
(R4、R5、R6およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。)で示される基を意味する。〕
および下式で示される化合物(II):
【0015】
【化6】
(R5およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。)
からなる群から選択される少なくとも1つの化合物を有効成分とする項1乃至3のいずれかに記載するプロリル水酸化酵素阻害剤。
項5.下式で示される化合物1〜4:
(化合物1)
【0016】
【化7】
(化合物2)
【0017】
【化8】
(化合物3)
【0018】
【化9】
(化合物4)
【0019】
【化10】
からなる群から選択される少なくとも1つを有効成分とする項1乃至4のいずれかに記載するプロリル水酸化酵素阻害剤。
項6.プロリル水酸化酵素と低酸素誘導因子との結合阻害剤である項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
項7.プロリル水酸化酵素に結合して、プロリル水酸化酵素と低酸素誘導因子との結合を阻害する剤である項6に記載するプロリル水酸化酵素阻害剤。
項8.低酸素誘導因子分解阻害剤である項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
【0020】
(II)医薬組成物
項9.項1乃至8のいずれかに記載するプロリル水酸化酵素阻害剤および薬学的に許容される担体または添加剤を含む、医薬組成物。
項10.細胞または組織の低酸素状態を改善するための項9記載の医薬組成物。
項11.細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬である項9または10記載の医薬組成物。
項12.細胞または組織の低酸素状態に関連して生じる疾患また病態が、慢性または急性の虚血性疾患またはそれに起因して生じる病態である項10に記載する医薬組成物。
項13.経口投与形態を有する項9乃至12のいずれかに記載する医薬組成物。
【0021】
(III)プロリル水酸化酵素阻害剤のスクリーニング方法
項14.被験化合物の中から、プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物を選択する工程を有するプロリル水酸化酵素阻害剤のスクリーニング方法。
項15.プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物が,下式(A)または(B)で示され骨格を有する基の少なくとも1つを一分子中に含む化合物である項14記載のスクリーニング方法:
【0022】
【化11】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
項16.細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬の有効成分のスクリーニング方法である、項14または15に記載するスクリーニング方法。
【0023】
(III)新規化合物
項17.下式で示される化合物またはその塩:
【0024】
【化12】
(式中、R12は水素原子または低級アルキル基を意味する。)
【発明の効果】
【0025】
本発明により、HIFを安定化するために有効なPHD阻害剤を提供することができる。当該PHD阻害剤は、PHDによるHIFの水酸化による不活性化を阻止してHIFを安定化することにより、虚血などによる低酸素状態によって生じるダメージから生体を保護することができる。このため、本発明のPHD阻害剤は、低酸素状態に関連して生じる疾患や病態(障害)を予防または改善するための医薬組成物として有効に利用することができる。特に、本発明のPHD阻害剤は、腎臓、心臓、下肢または脳の各種器官の虚血性疾患やそれから進展する病態(障害)の予防または改善に有効に使用することができる。また本発明は、かかるPHD阻害剤として有効な新規な化合物を提供する。
【0026】
さらに本発明のスクリーニング方法によれば、上記作用を有し虚血性疾患やそれから進展する病態(障害)の予防または改善に有用な新規なPHD阻害剤を選別し取得することが可能となる。
【発明を実施するための最良の形態】
【0027】
(I)プロリル水酸化酵素阻害剤(PHD阻害剤)
本発明が提供するPHD阻害剤は、下式(A)または(B):
【0028】
【化13】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
で示される骨格を有する基の少なくとも1つを、一分子中に含む化合物またはその塩を有効成分とするものである。
【0029】
本発明が対象とする化合物(以下、「本発明化合物」という)は、上記骨格Aを有する基または骨格Bを有する基の少なくとも1つを一分子中に含み、HIFのα鎖のプロリンを水酸化するPHDの機能を妨げる作用を有するものである。本発明化合物は、結果としてPHDの機能を妨げる作用を有するものであればよく、PHDのFe(II)にキレートしてPHDの機能を妨げるもの、低酸素誘導因子(HIF)のα鎖のプロリン残基(以下、単に「HIFαプロリン」という)に対するPHDの結合を競合的に阻害するもの、PHDの水酸化活性を阻害するものなど、その作用機序は特に制限されない。特に本発明が対象とする化合物として好ましくは、HIF/PHD結合阻害作用を有するものである。
【0030】
本発明化合物のFe(II)に対するキレート作用は、制限されないが、例えばPriceらの方法(Price DL, et al.,” Chelating activity of advanced glycation end-product (AGE) inhibitors.” J Biol Chem 272: 5430-5437, 1997)に従って、または準じて測定し評価することができる。
【0031】
その一例を挙げると、後述する実験例2に記載するように、遷移金属である銅とのキレート作用を評価する方法を用いることができる。当該方法は、具体的には、被験化合物の存在下で塩化銅とアスコルビン酸をインキュベーションして、反応後のアスコルビン酸の残存量を測定する方法であり、被験化合物の存在によってアスコルビル酸の自動酸化が防止できれば、被験化合物について金属キレート能(Fe(II)に対するキレート作用)があると評価することができる。
【0032】
また本発明化合物のHIF/PHD結合阻害作用は、制限されないが、例えば実験例4に詳述するように、被験化合物の存在下で、放射性同位体等で標識したPHDとHIFのα鎖ドメイン(好ましくはHIF-1αのアミノ酸557-576領域)をインキュベーションし、HIFのα鎖に結合したPHD量を測定する方法であり、被験化合物の存在によってHIFのα鎖に対するPHDの結合が防止できれば、被験化合物についてHIF/PHD結合阻害作用があると評価することができる。
【0033】
さらに本発明化合物のPHD阻害活性は、制限されないが、例えばKauleらの方法(Cunhild Kaule and Volkmar Gunzler,” Assay for 2-Oxoglutarate Decarboxylating Enzymes Based on the Determination of [1-14C] Succinate: Application to Prolyl 4- Hydroxylase”, Analytical Biochemistry 184, 291-297 (1990))に従ってまたは準じて測定し評価することができる。
【0034】
その一例を挙げると、後述する実験例3に記載するように、2-oxyglutarate(2-OG)とHIFのプロリンからcis-3-hydroxyprolineを産生する触媒能力を評価する方法を用いることができる。当該方法は、具体的には、被験化合物の存在下で、PHD、[5-14C]2-oxyglutarate(2-OG)およびHIFのα鎖ドメイン(好ましくはHIF-1αのアミノ酸557-576領域)をインキュベーションして、反応後の[1-14C]コハク酸を測定する方法であり、被験化合物の存在によって [1-14C]コハク酸の生成が抑制できれば、被験化合物についてPHD阻害活性があると評価することができる。
【0035】
上記骨格Aを有する基として、より好ましくは下式(A’)で示される基である:
【0036】
【化14】
(式中、R1は水酸基、低級アルキル基、保護基を有していてもよいカルボキシアルキル基、アミノ基:R2は水素原子、水酸基、低級アルキル基、保護基を有していてもよいカルボキシアルキル基、アミノ基:R3は水素原子、アミノ基または低級アルキルアミノ基を意味する)
式(A’)において、R1およびR2の定義でみられる低級アルキル基としては、炭素数1〜6の直鎖または分枝鎖状のアルキル基を挙げることができる。具体的には、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、ter−ブチル基、ペンチル基、1,2−ジメチルプロピル基、1,1−ジメチルプロピル基、2,2−ジメチルプロピル基、2−エチルプロピル基、n−ヘキシル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1,3−ジメチルブチル基、1−エチル−2−メチルプロピル基、1−メチル−2−エチルプロピル基などを挙げることができる。好ましくは炭素数1〜4の直鎖または分枝鎖状のアルキル基であり、より好ましくはメチル基である。
【0037】
式(A’)において、R1の定義でみられるカルボキシアルキル基とは、上記低級アルキル基のいずれかに炭素原子にカルボキシル基が結合しているものを意味する。この場合のカルボキシルは保護基を有していてもよい。ここで保護基としては、例えばメチル基、エチル基、tert−ブチル基等の低級アルキル基;p−メトキシベンジル基、p−ニトロベンジル基、3,4−ジメトキシベンジル基、ジフェニルメチル基、トリチル基、フェネチル基等の置換基を有していてもよいフェニル基で置換された低級アルキル基;2,2,2−トリクロロエチル基、2−ヨードエチル基などのハロゲン化低級アルキル基;ピバロイルオキシメチル基、アセトキシメチル基、プオピオニルオキシメチル基、ブチリルオキシメチル基、バレリルオキシメチル基、1−アセトキシエチル基、2−アセトキシエチル基、1−ピバロイルオキシエチル基、2−ピバロイルオキシエチル基などの低級アルカノイルオキシ低級アルキル基;パルミトイルオキシエチル基、ヘプタデカノイルオキシメチル基、1−パルミトイルオキシエチル基などの高級アルカノイルオキシ低級アルキル基、メトキシカルボニルオキシメチル基、1−ブトキシカルボニルオキシエチル基、1−(イソプロポキシカルボニルオキシ)エチル基等の低級アルコキシカルボニルオキシ低級アルキル基;カルボキシメチル基、2−カルボキシエチル基などのカルボキシ低級アルキル基;3−フタリジル基等のヘテロアリール基、4−グリシルオキシベンゾイルオキシメチル基などの置換基を有していてもよいベンゾイルオキシ低級アルキル基、(5−メチル−2−オキソ−1,3−ジオキソレン−4−イル)メチル基などの(置換ジオキソレン)低級アルキル基;1−シクロヘキシルアセチルオキシエチル基などのシクロアルキル置換低級アルカノイルオキシ低級アルキル基、1−シクロヘキシルオキシカルボニルオキシエチル基などのシクロアルキルオキシカルボニルオキシ低級アルキル基などをあげることができる。更に、種々の酸アミドも本発明における保護基を有しているカルボキシル基に含まれる。要するに生体内で何らかの手段で分解されて、カルボン酸となりうるものであれば、いかなるものもカルボキシル基の保護基となり得る。
【0038】
R3の定義でみられる低級アルキルアミノ基のうち低級アルキル基としては、上記の低級アルキルを同様に挙げることができる。
【0039】
上記骨格A’を有する基を有する化合物として、下式で示される化合物(I)を挙げることができる。
【0040】
【化15】
ここで、R1として前述する基を挙げることができるが、好ましくは炭素数1〜6の低級アルキル基、炭素数1〜4のカルボキシアルキル基、または炭素数1〜6の低級アルキル基で保護されている炭素数1〜4のカルボキシアルキル基を挙げることができる。炭素数1〜6の低級アルキル基としてより好ましくはメチル基、カルボキシアルキル基としてより好ましくはカルボキシエチル基、アルキル基で保護されているカルボキシアルキル基としてより好ましくは、メチル基で保護されたカルボキシエチル基を挙げることができる。
【0041】
またR2としても前述の基を挙げることができるが、好ましくは炭素数1〜4の低級アルキル基、より好ましくはメチル基である。
【0042】
R10としては、R11−S−CH2−(式中、R11は置換基を有していてもよいヘテロアリール基を意味する)または下式で示される基を挙げることができる:
【0043】
【化16】
(式中、R4、R5、R6およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の、置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。)で示される基を意味する。〕
R11の定義でみられるヘテロアリール基とは、酸素原子、窒素原子または硫黄原子の少なくとも1つを1〜4個含む単環または縮合環から誘導される基を意味する。例えば、チェニル基、フリル基、ベンゾチェニル基、ベンゾフラニル基、イソベンゾフラニル基、ピロリル基、イミダゾリル基、ピラゾリル基、イソチアゾリル基、イソキサゾリル基、テトラゾリル基、ピリジル基、ピラジニル基、ピリミジニル基、ピリダジニル基、インドリル基、イソインドリル基、キノリル基、イソキノリル基、フタラジル基、キノキサリル基、ナフチリジル基、キナゾリル基、アクリジニル基、イミダゾピリジル基などを挙げることができる。かかる基の置換基としては、水酸基:メチル基、エチル基、n−プロピル基、イソプロピル基などの低級アルキル基;ハロゲン化アルキル基;メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基などの低級アルコキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子などのハロゲン原子;シアノ基、アセチル基、プロピオニル基、ベンゾイル基等のアシル基;アミノ基;ニトロ基;保護基を有していてもよいカルボキシル基;アシルアミノ基;スルホニルアミノ基;カルバモイル基;スルファモイル基;アミノスルホニル基;シアノアルキル基;ヘテロアリール基;カルボキシアルキル基;カルボキシアルコキシ基;ヘテロアリールアルキル基;ヘテロアリールアルコキシ基などを挙げることができる。R11として好ましくはピリジル基を挙げることができる。
【0044】
R4、R5、R6およびR7の定義でみられる低級アルキル基としては、R1およびR2に関して説明した炭素数1〜6の直鎖または分枝鎖状のアルキル基を同様に挙げることができる。
【0045】
R4、R5、R6およびR7の定義でみられる置換基を有していてもよいシクロ環としては、シクロペンタンやシクロヘキサンなどの5または6員の飽和単環:シクロペンテン、シクロヘキセン、1,3−シクロヘキサジエン、2,5−シクロヘキサジエン、ベンゼンなどの5または6員の不飽和単環を挙げることができる。R4、R5、R6およびR7の定義でみられる置換基を有していてもよいヘテロ環としては、酸素原子、窒素原子または硫黄原子の少なくとも1つを1乃至2含む、5または6員の飽和または不飽和の単環を意味する。かかる単環として、具体的にはフラン、チオフェン、ピロール、チアゾール、イソチアゾール、オキサゾール、イソオキサゾール、イミダゾール、ピラゾール、フラザン、ピラン、ピリジン、ピリダジン、ピリミジン、ピラジン、ピロリジン、イミダゾリジン、ピラゾリジン、ピペリジン、ピペラジン、およびモルホリンを挙げることができる。なお、これらのシクロ環およびヘテロ環において置換基としては、水酸基:メチル基、エチル基、n−プロピル基、イソプロピル基などの低級アルキル基;ハロゲン化アルキル基;メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基などの低級アルコキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子などのハロゲン原子;シアノ基、アセチル基、プロピオニル基、ベンゾイル基等のアシル基;アミノ基;ニトロ基;保護基を有していてもよいカルボキシル基;アシルアミノ基;スルホニルアミノ基;カルバモイル基;スルファモイル基;アミノスルホニル基;シアノアルキル基;ヘテロアリール基;カルボキシアルキル基;カルボキシアルコキシ基;ヘテロアリールアルキル基;ヘテロアリールアルコキシ基などを挙げることができる。
【0046】
化合物(I)として、具体的は下式で示される化合物1〜3を挙げることができる。本明細書において、便宜上、化合物1(6-amuno-1,3-dimethyl-5-(2-pyridin-2-yl-quinoline-4-carbonyl)-1H-pyrimidine-2、4-dione)をTM-6008、化合物2(6-amino-1,3-di-methyl-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-1H-pyrimidine-2,4-dione)をTM-6089、および化合物3(3-{4-Amino-3-methyl-2,6-dioxo-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-3,6-dihydro-2H-pyrimidin-1-yl}-propionic acid methyl ester)をTM-6101と称する場合がある。
【0047】
(1)化合物1:
6-amuno-1,3-dimethyl-5-(2-pyridin-2-yl-quinoline-4-carbonyl)-1H-pyrimidine-2,4-dione(TM-6008)
【0048】
【化17】
(2)化合物2:6-amino-1,3-di-methyl-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-1H-pyrimidine-2,4-dione(TM-6089);
【0049】
【化18】
(3)化合物3:
3-{4-Amino-3-methyl-2,6-dioxo-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-3,6-dihydro-2H-pyrimidin-1-yl}-propionic acid methyl ester(TM-6101)
【0050】
【化19】
また上記骨格Bを有する基として、より好ましくは下式(B’)で示される基である:
【0051】
【化20】
(R4、R5、R6およびR7は、同一または異なって、水素原子または低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の、置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
式(B’)中、R4、R5、R6、およびR7の定義にみられる低級アルキル基としては、R1およびR2に関して説明した炭素数1〜6の直鎖または分枝鎖状のアルキル基を同様に挙げることができる。好ましくは炭素数1〜4の直鎖または分枝鎖状の低級アルキル基であり、より好ましくはメチル基である。
【0052】
R4、R5、R6およびR7の定義でみられる置換基を有していてもよいシクロ環またはヘテロ環、ならびにその置換基は、前述の通りである。好ましくはベンゼン環である。
【0053】
R8およびR9の定義でみられるアルキレン鎖としては炭素数1〜6の直鎖または分岐状のアルキレン鎖を意味する。例えば、メチレン鎖、エチレン鎖、プロピレン鎖、ブチレン鎖、エチルメチレン鎖などを挙げることができる。好ましくはメチレン鎖である。R8およびR9は、互いに水素原子であるか、またはそれらが結合する酸素原子ととも環を形成していることが好ましい。
【0054】
上記骨格B’を有する基を有する化合物として、下式で示される化合物(II)を挙げることができる。
【0055】
【化21】
(R5およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。)
より好ましくは、下式で示される化合物4(3,7-Dimethy-furo[3,2-b;4,5-b]dipyridine)である。本明細書において、便宜上、化合物4をTM-6010と称する場合がある。
【0056】
(4)化合物4:3,7-Dimethy-furo[3,2-b;4,5-b]dipyridine(TM-6010)
【0057】
【化22】
また、前述する化合物1(TM-6008)も骨格B’を有する基を有する化合物Iに該当する。
【0058】
これらの化合物は、遊離または塩の形態を有するもののいずれであってもよい。ここで塩としては、通常、医薬上許容される塩、たとえば無機塩基または有機塩基との塩、あるいは無機酸、有機酸、または塩基性若しくは酸性アミノ酸などの酸付加塩等を挙げることができる。無機塩基としては、たとえば、ナトリウムやカリウム等のアルカリ金属;カルシウムやマグネシウム等のアルカリ土類金属;アルミニウムやアンモニウム等を挙げることができる。有機塩基としては、たとえば、エタノールアミン等の第一級アミン;ジエチルアミン、ジエタノールアミン、ジシクロヘキシルアミン、N,N'−ジベンジルエチレンジアミン等の第二級アミン;トリメチルアミン、トリエチルアミン、ピリジン、ピコリン、トリエタノールアミン等の第三級アミン等を挙げることができる。
【0059】
酸付加塩を形成する無機酸としては、たとえば、塩酸、臭化水素酸、硝酸、硫酸、リン酸等を挙げることができる。有機酸としては、たとえば、ギ酸、酢酸、乳酸、トリフルオロ酢酸、フマール酸、シュウ酸、酒石酸、マレイン酸、安息香酸、クエン酸、コハク酸、リンゴ酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸等を挙げることができる。塩基性アミノ酸としては、たとえば、アルギニン、リジン、オルニチン等を挙げることができる。酸性アミノ酸としては、たとえば、アスパラギン酸、グルタミン酸等を挙げることができる。
【0060】
上記化合物、特に化合物1(TM-6008)、2(TM-6089)および4(TM-6010)は商業的に入手可能であり〔例えば、Maybrige(英国)、ASINEX(ロシア)、LaboTest(ドイツ)、Specs(オランダ)、ENAMINE(ウクライナ)、Ambinter(フランス)、ASDI(米国)、Sigma(米国)、Pharmeks(ロシア)、Life Chemical(ウクライナ)、Key Organics(英国)など〕、また自体公知の方法により製造することができる。
【0061】
また、化合物3(TM-6101)は、後述する製造例1に記載する方法で製造することができる。その製造工程のうち、工程1において、Ethyl 3-Bromopropinateに代えてIodomethaneを用いることによって化合物2(TM-6089)を合成することができる。また、工程1において、Ethyl 3-Bromopropinateに代えてIodomethaneを用い、かつ工程4において2-mercaptopyridineに代えて2-Pyridine-2-yl-quinoline-4-carbonylchlorideを用いることによって化合物1(TM-6008)を合成することができる。
【0062】
本発明化合物は、前述するように、HIFのα鎖のプロリンを水酸化するPHDの機能を妨げる作用を有するものである。かかる作用は、金属キレート作用、HIF/PHD結合阻害作用、およびPHD活性阻害作用など、結果としてPHDの機能を妨げる作用であればよい。これらはいずれも前述するインビトロアッセイ系で評価することができる(詳細は実験例を参照のこと)。
【0063】
さらに本発明化合物は、PHDとHIFの結合を阻害する作用を有する結果、HIFの水酸化によるVHLとの結合および分解を阻害し、HIFを安定化する作用を有している。かかるHIF促進作用は、制限されないが、後述する実験例1の方法に従って、またはこの方法に準じて測定し、評価することができる。当該インビトロアッセイは、HRE(hypoxia responsive element (低酸素応答要素))を7回タンデムに含むプロモーターの下流にルシフェラーゼ遺伝子を有するベクターを導入したPHD発現細胞を被験化合物とともにインキュベーションし、細胞溶解物のルシフェラーゼ活性を測定する方法である。当該アッセイ系において、被験化合物の存在によってルシフェラーゼ活性が増加すれば、被験化合物の存在によってHIFが安定化しHREシステムが活性化していること、すなわち被験化合物にHIF安定化作用があると評価することができる。
【0064】
また本発明化合物は、PHDの機能を阻害しHIFを安定化させた結果、血管新生を促進する作用を有している。かかる血管新生促進作用は、制限されないが、後述する実験例5の方法(スポンジ血管新生分析)に従って、またはこの方法に準じて測定し、評価することができる。当該インビボアッセイは、被験動物の皮下に埋設したスポンジ・ディスクを介して被験化合物を皮下投与し、経時的にヘモグロビン量と血管数を測定する方法である。当該インビボアッセイ系において、被験化合物の投与によってヘモグロビン量と血管数が増加すれば、被験化合物に血管新生作用があると評価することができる。
【0065】
本発明化合物はPHDの機能を阻害する作用を有する。中でも化合物1〜4、好ましくは化合物1は、後述する実験例で示すように優れたPHD阻害作用を有している。当該PHD阻害作用により、本発明化合物はHIFの分解を阻止して安定化し、HIF−HREシステムを安定化することにより低酸素状態を改善し、その結果、虚血などの低酸素状態によって生じるダメージから細胞や組織を防御することができる。従って、本発明化合物によれば、下記の作用が期待される。
【0066】
(i) 腎慢性虚血改善作用
腎臓において、尿細管周囲毛細血管の血流量の停滞は慢性低酸素を誘発し、それに伴い尿細管間質障害・尿細管周囲毛細血管の喪失が促進される。公知のPHD阻害剤であるコバルトによってHIFの発現が亢進し、その結果、腎虚血性障害である尿細管間質性障害の改善が認められることが報告されている(Matsumoto M, Tanaka T, Yamamoto T, Noiri E, Miyata T, Inagi R, Fujita T, Nangaku M. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol 15: 1574-1581 (2004))。このことから、HIFを安定化することによりVEGFやエリスロポエチンの転写が促進されて、血管新生や血流の改善が惹起され、慢性虚血を改善して腎不全への進展が抑制できる可能性がある。
【0067】
(ii) 脳虚血改善作用
脳梗塞に陥った脳梗塞領域は虚血になっており、組織への虚血障害が深刻な問題となっている。よって、脳梗塞領域に起こる活発な脈管形成は、虚血の下で組織に有益であることが証明されている(Krupinski J, Kaluza J, Kumar P, Kumar S & Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 25: 1794?1798 (1994))。また、VEGFが、一時的な中大脳動脈閉塞の後、脳梗塞サイズ・浮腫形成・脳血液関門破壊を、虚血に起因する神経細胞障害を減少することによって減らすことが証明されている(Hayashi, T., Abe, K., Itoyama, Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J. Cereb. Blood Flow Metab. 18: 887-895 (1998))。これらのことより、HIFを安定化することによるVEGFの発現の亢進は、脳梗塞および虚血性脳卒中に対する効果的な治療になると考えられる。
【0068】
(iii) 虚血性心疾患改善作用
虚血性心疾患には狭心症・心筋梗塞がある。いずれも適切な治療が遅れると心不全や重度の不整脈を惹き起こし直接生死にかかわってくる。このため心筋虚血を改善方法の開発が喫緊の課題とされている。最近の研究では、ブタの進行性冠動脈閉塞モデルにVEGFを投与すると虚血領域の改善が見られ、また大量投与では心筋還流血液量の改善が見られることが証明されている(Pearlman JD, Hibberd MG, Chuang ML, Harada K, Lopez JJ, Glad-stone SR, Friedman M, Sellke FW, Simons M. Magnetic resonance mapping demonstrates benefits of VEGF-induced myocardial angiogenesis. Nat Med.1:1085-1089. (1995))。また、ブタの慢性心筋虚血モデルにVEGFを投与すると血管形成が促進されることが証明されている(Lopez JJ, Laham RJ, Stamler A, Pearlman JD, Bunting S, Kaplan A, Carrozza JP, Sellke FW, Simons.M. VEGF administration in chronic myocardial ischemia in pigs. Cardiovasc Res.;40: 272-281. (1998))。これらのことから、HIFの安定化によるVEGFの発現亢進は、心筋梗塞や狭心症など虚血性心疾患の効果的な治療になると考えられる。
【0069】
(iv) 下肢虚血改善作用
下肢虚血患者では症状が重度化すると下肢切断を余儀なくされ、患者のADLを極度に低下させることから医学的のみならず社会的にも、深刻な問題となっている。最近の研究では、下肢虚血患者において、VEGFが治療に対し有効と考えられている。VEGF、FGF-2遺伝子を虚血筋組織に導入すると、FGF-2遺伝子導入で内因性VEGF, HGF発現が亢進し、組織学的検査ならびに血流測定により新生血管の増加と側副血行路形成が認められ、明らかな下肢虚血の改善が認められることが報告されている(Onimaru M, Yonemitsu Y, Tanii M, Nakagawa K, Masaki I, Okano S, Ishibashi H, Shirasuna K, Hasegawa M, Sueishi K. Fibroblast growth factor-2 gene transfer can stimulate hepatocyte growth factor expression irrespective of hypoxia-mediated downregulation in ischemic limbs. Circ. Res, 91: 723-730 (2002))。これらのことから、HIFの安定化によるVEGFの発現亢進は、下肢虚血の効果的な治療になると考えられる。
【0070】
本発明のPHD阻害剤は、前述する本発明化合物またはその塩を有効成分とするものである。本発明のPHD阻害剤は、前述する化合物またはその塩100重量%からなるものであっても、またそうでなくてもPHD阻害作用を発揮する有効量の化合物を含有するものであればよい。制限されないが、PHD阻害剤には、通常、化合物またはその塩が10〜100重量%の割合で含まれる。
【0071】
(II)医薬組成物
本発明は、前述するPHD阻害剤を有効成分として含有する医薬組成物を提供する。言い換えれば、本発明の医薬組成物は、上記本発明化合物を有効成分として含有するものである。本発明の医薬組成物は、PHD阻害剤(本発明化合物)を、PHDを阻害する有効量含むことによって、HIFを安定化する作用を有している。その結果、本発明の医薬組成物は、生体のHIF−HREシステムを活性化することにより低酸素状態を改善し、その結果、虚血などの低酸素状態によって生じるダメージから細胞や組織を防御することができる。
【0072】
このため、本発明の医薬組成物は細胞や組織の低酸素状態に関連して生じる疾患や病態(障害)の予防または治療剤として有用である。かかる疾患または病態として、例えば、各種の虚血性疾患ならびに虚血から生じる障害を挙げることができる。虚血性疾患としては、腎虚血性障害(尿細管間質性障害)、虚血性脳血管障害(脳塞栓症、脳梗塞、一過性脳虚血発作等)、虚血性心疾患(狭心症、心筋梗塞)、下肢虚血性障害などを挙げることができる。また、これらの虚血性疾患の進展によって生じる病態として、慢性腎不全、慢性心不全、下肢壊疽、などを挙げることができる。
【0073】
本発明の医薬組成物は、通常、PHD阻害に有効量の、ひいては低酸素状態の改善に有効量の本発明化合物に加えて、薬学的に許容される担体または添加剤を配合して調製される。医薬組成物中の本発明化合物の配合量は、対象とする疾患や病態の種類や投与形態に応じて適宜選択されるが、通常、全身投与製剤の場合には、医薬組成物の全体重量(100重量%)の0.001〜50重量%、特に0.01〜10重量%とすることができる。
【0074】
本発明の医薬組成物の投与方法として、経口投与、ならびに静脈内投与、筋肉内投与、皮下投与、経粘膜投与、経皮投与、および直腸内投与等の非経口投与を挙げることができる。好ましくは経口投与および静脈内投与であり、より好ましくは経口投与である。本発明の医薬組成物は、かかる投与方法に応じて、種々の形態の製剤(剤型)に調製することができる。以下に、各製剤(剤型)について説明するが、本発明において用いられる剤型はこれらに限定されるものではなく、医薬製剤分野において通常用いられる各種剤型を用いることができる。
【0075】
経口投与を行う場合の剤型として、散剤、顆粒剤、カプセル剤、丸剤、錠剤、エリキシル剤、懸濁剤、乳剤およびシロップ剤を挙げることができ、これらの中から適宜選択することができる。また、それらの製剤について徐放化、安定化、易崩壊化、難崩壊化、腸溶性化、易吸収化等の修飾を施すことができる。
【0076】
また、静脈内投与、筋肉内投与、または皮下投与を行う場合の剤型として、注射剤または点滴剤(用時調製の乾燥品を含む)等があり、適宜選択することができる。
【0077】
また、経粘膜投与、経皮投与、または直腸内投与を行う場合の剤型として、咀嚼剤、舌下剤、パッカル剤、トローチ剤、軟膏剤、貼布剤、液剤等があり、適応場所に応じて適宜選択するここができる。また、それらの製剤について徐放化、安定化、易崩壊化、難崩壊化、易吸収化等の修飾を施すことができる。
【0078】
本発明の医薬組成物にはその剤形(経口投与または各種の非経口投与の剤形)に応じて、薬学的に許容される担体および添加剤を配合することができる。薬学的に許容される担体及び添加剤としては、溶剤、賦形剤、コーティング剤、基剤、結合剤、滑沢剤、崩壊剤、溶解補助剤、懸濁化剤、粘稠剤、乳化剤、安定剤、緩衝剤、等張化剤、無痛化剤、保存剤、矯味剤、芳香剤、着色剤が挙げられる。以下に、医薬上許容される担体および添加剤の具体例を列挙するが、本発明はこれらに制限されるものではない。
【0079】
溶剤としては、精製水、滅菌精製水、注射用水、生理食塩液、ラッカセイ油、エタノール、グリセリン等を挙げることができる。賦形剤としては、デンプン類(例えばバレイショデンプン、コムギデンプン、トウモロコシデンプン)、乳糖、ブドウ糖、白糖、結晶セルロース、硫酸カルシウム、炭酸カルシウム、炭酸水素ナトリウム、塩化ナトリウム、タルク、酸化チタン、トレハロース、キシリトール等を挙げることができる。
【0080】
結合剤としては、デンプンおよびその誘導体、セルロースおよびその誘導体(たとえばメチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロース)、ゼラチン、アルギン酸ナトリウム、トラガント、アラビアゴム等の天然高分子化合物、ポリビニルピロリドン、ポリビニルアルコール等の合成高分子化合物、デキストリン、ヒドロキシプロピルスターチ等を挙げることができる。
【0081】
滑沢剤としては、軽質無水ケイ酸、ステアリン酸およびその塩類(たとえばステアリン酸マグネシウム)、タルク、ワックス類、コムギデンブン、マクロゴール、水素添加植物油、ショ糖脂肪酸エステル、ポリエチレングリコール、シリコン油等を挙げることができる。
【0082】
崩壊剤としては、デンプンおよびその誘導体、寒天、ゼラチン末、炭酸水素ナトリウム、炭酸カルシウム、セルロースおよびその誘導体、ヒドロキシプロピルスターチ、カルボキシメチルセルロースおよびその塩類ならびにその架橋体、低置換型ヒドロキシプロピルセルロース等を挙げることができる。
【0083】
溶解補助剤としては、シクロデキストリン、エタノール、プロピレングリコール、ポリエチレングリコール等を挙げることができる。懸濁化剤としては、カルボキシメチルセルロースナトリウム、ポリピニルピロリドン、アラビアゴム、トラガント、アルギン酸ナトリウム、モノステアリン酸アルミニウム、クエン酸、各種界面活性剤等を挙げることができる。
【0084】
粘稠剤としては、カルボキシメチルセルロースナトリウム、ポリピニルピロリドン、メチルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、トラガント、アラビアゴム、アルギン酸ナトリウム等を挙げることができる。
【0085】
乳化剤は、アラビアゴム、コレステロール、トラガント、メチルセルロース、レシチン、各種界面活性剤(たとえば、ステアリン酸ポリオキシル40、セスキオレイン酸ソルビタン、ポリソルベート80、ラウリル硫酸ナトリウム)等を挙げることができる。
【0086】
安定剤としては、トコフェロール、キレート剤(たとえばEDTA、チオグリコール酸)、不活性ガス(たとえば窒素、二酸化炭素)、還元性物質(たとえば亜硫酸水素ナトリウム、チオ硫酸ナトリウム、アスコルビン酸、ロンガリット)等を挙げることができる。
【0087】
緩衝剤としては、リン酸水素ナトリウム、酢酸ナトリウム、クエン酸ナトリウム、ホウ酸等を挙げることができる。
【0088】
等張化剤としては、塩化ナトリウム、ブドウ糖等を挙げることができる。無痛化剤こしては、局所麻酔剤(塩酸プロカイン、リドカイン)、ペンジルアルコール、ブドウ糖、ソルビトール、アミノ酸等を挙げることができる。
【0089】
矯味剤としては、白糖、サッカリン、カンゾウエキス、ソルビトール、キシリトール、グリセリン等を挙げることができる。芳香剤としては、トウヒチンキ、ローズ油等を挙げることができる。着色剤としては、水溶性食用色素、レーキ色素等を挙げることができる。
【0090】
保存剤としては、安息香酸およびその塩類、パラオキシ安息香酸エステル類、クロロブタノール、逆性石けん、ベンジルアルコール、フェノール、チロメサール、デヒドロ酢酸、ホウ酸、等を挙げることができる。
【0091】
コーティング剤としては、白糖、ヒドロキシプロピルセルロース(HPC)、セラック、ゼラチン、グリセリン、ソルビトール、ヒドロキシプロピルメチルセルロース(HPMC)、エチルセルロース、ポリビニルピロリドン(PVP)、ヒドロキシプロピルメチルセルロースフタレート(HPMCP)、セルロースアセテートフタレート(CAP)、メチルメタアクリレート−メタアクリル酸共重合体および上記記載した高分子等を挙げることができる。
【0092】
基剤としては、ワセリン、流動パラフィン、カルナウバロウ、牛脂、硬化油、パラフィン、ミツロウ、植物油、マクロゴール、マクロゴール脂肪酸エステル、ステアリン酸、カルボキシメチルセルロースナトリウム、ベントナイト、カカオ脂、ウイテップゾール、ゼラチン、ステアリルアルコール、加水ラノリン、セタノール、軽質流動パラフィン、親水ワセリン、単軟膏、白色軟膏、親水軟膏、マクロゴール軟膏、ハードファット、水中油型乳剤性基剤、油中水型乳剤性碁剤等を挙げることができる。
【0093】
なお、上記の各剤型について、公知のドラッグデリバリーシステム(DDS)の技術を採用することができる。本明細書にいうDDS製剤とは、徐放化製剤、局所適用製剤(トローチ、バッカル錠、舌下錠等)、薬物放出制御製剤、腸溶性製剤および胃溶性製剤等、投与経路、バイオアベイラビリティー、副作用等を勘案した上で、最適の製剤形態にした製剤である。
【0094】
本発明の医薬組成物を、低酸素状態に関連する疾患や病態に対する予防薬または治療薬として用いる場合、その経口投与量として、本発明化合物の量に換算して0.03〜300mg/kgの範囲が好ましく、より好ましくは0.1〜50mg/kgである。静脈内投与をする場合、本発明化合物の有効血中濃度が0.2〜50μg/mL、より好ましくは0.5〜20μg/mLの範囲となるような投与量を挙げることができる。
【0095】
なお、これらの投与量は、年齢、性別、体型等により変動し得る。
【0096】
(III)スクリーニング方法
本発明は被験物質の中から、細胞や組織の低酸素状態の改善に有効に利用できるプロリル水酸化酵素阻害剤(PHD阻害剤)を選別する方法を提供する。本発明の方法で探索取得されるPHD阻害剤は、HIFにPHDが結合することを阻害して、HIFのαプロリンが水酸化されて分解することを防止する作用を有しており、その結果、HIF−HREシステムを活性化して細胞や組織の低酸素状態を改善することができる。このため当該物質によれば、細胞や組織の低酸素状態に関連して生じる疾患や病態(障害)を予防または治療することができると期待される。従って、本発明の方法は、細胞や組織の低酸素状態に関連して生じる疾患や病態(障害)の予防または治療するための医薬組成物の有効成分をスクリーニングする方法であるともいえる。
【0097】
本発明のスクリーニング方法は、基本的にはプロリル水酸化酵素(PHD)と低酸素誘導因子(HIF)のα鎖プロリンとの競合的結合を阻害する作用を有する物質を探索することからなるが、具体的には下記の(a)〜(c)の工程を有する方法を例示することができる:
(a) 被験物質の存在下で、PHDとHIFα鎖プロリンとを接触させる工程、
(b) PHDとHIFα鎖プロリンとの結合を測定する工程、および
(c) PHDとHIFα鎖プロリンとの結合を阻害する被験物質をPHD阻害剤として選択する工程。
【0098】
かかる工程により、PHDとHIFのα鎖プロリンとの競合的結合を阻害して、HIFのα鎖プロリンを水酸化するPHDの働きを抑制する物質を取得することができる。
【0099】
被験物質としては、制限はされないが、核酸、ペプチド、タンパク質、有機化合物、または無機化合物などであり、スクリーニングは、具体的には、これらの被験物質またはこれらを含む組成物(例えば、細胞抽出物、遺伝子ライブラリーの発現産物等を含む)を、対象のc-SrcおよびADAPと接触させることにより行うことができる。
【0100】
スクリーニングに際して採用される被験物質とPHDとHIFα鎖プロリンとの接触条件は、特に制限されないが、通常、生理的環境下におけるin vitro実験系、または細胞内で行うことが好ましい。
【0101】
本発明において用いるPHDは、その由来を特に制限されず、ヒト由来のもののほか、ヒト以外の哺乳動物を含む脊椎動物(例えば、マウス、ラット、ニワトリなど)または各種微生物(ストレプトマイシス)に由来するPHDを用いることができる。PHDは全長のタンパク質であってもよいが、HIFαプロリンとの結合に関わるドメインを含む部分断片であってもよい。
【0102】
本発明において用いるHIFα鎖も、その由来を特に制限されない。ヒト由来のもののほか、ヒト以外の哺乳動物を含む脊椎動物(例えば、マウス、ラットやニワトリなど)に由来するHIFを用いることができるが、一緒に使用するPHDと同一の動物に由来するものを使用することが好ましい。HIFα鎖は全長のタンパク質であってもよいが、PHDとの結合に関わるα鎖の564位に位置するプロリン残基を含む部分断片であってもよい。具体的には、HIFα鎖の557〜576または531〜652の領域を有する部分断片を用いることもできる。
【0103】
PHDとHIFα鎖との結合の測定は、PHDとHIFα鎖との結合の有無が評価できる方法であれば特に制限されない。例えば、被験物質の存在下でPHD(またはその部分断片)とHIFα鎖(又はプロリン残基を含む部分断片)とを接触させた後、その反応液(混合液)を電気泳動にかけて分子量から結合の有無を評価する方法、PHD(又はPHDの部分断片)あるいはHIFα鎖(又はプロリン残基を含む部分断片)の抗体を用いて他のタンパク質の共沈の有無を評価する方法などを例示することができる。
【0104】
かかるスクリーニング方法によるPHD阻害剤(候補物質)の選別は、cPHD(またはその部分断片)とHIFα鎖(又はプロリン残基を含む部分断片)との結合の有無を評価することによって行うことができる。この場合、被験物質の非存在下でPHD(またはその部分断片)とHIFα鎖(又はプロリン残基を含む部分断片)とを接触させて生じるPHDとHIFα鎖との結合(コントロール)と対比することによって評価することもできる。PHDとHIFα鎖とが全く結合しない場合、およびコントロールに比して結合が低減した場合に、使用された被験物質を、PHD阻害剤の候補物質として選別することができる。
【0105】
上記スクリーニングで選別される物質は、HIFのα鎖プロリンを水酸化するPHDの働きを阻害することによって、HIFを安定化する作用を有するものであり、HIF−HREシステムの活性化を介して低酸素状態を改善し、低酸素状態に関連して生じる疾患やそれから進展する病態(障害)を予防または治療する医薬組成物の有効成分として使用することが可能である。
【0106】
なお、低酸素状態に関連して生じる疾患としては、例えば各種の虚血性疾患ならびに虚血から生じる障害、具体的には、腎虚血性障害(尿細管間質性障害)、虚血性脳血管障害(脳塞栓症、脳梗塞、一過性脳虚血発作等)、虚血性心疾患(狭心症、心筋梗塞)、下肢虚血性障害などを挙げることができる。また、これらの虚血性疾患の進展によって生じる病態として、慢性腎不全、慢性心不全、下肢壊疽、などを挙げることができる。
【0107】
上記のスクリーニング方法によって選別された候補物質は、さらにその低酸素状態を改善する一つの作用として血管新生作用を評価するために、スポンジ血管新生分析や、低酸素状態にあるモデル非ヒト動物を用いてスクリーニングをかけることもできる。かくして選別される候補物質は、さらに虚血性疾患を有する病態非ヒト動物を用いた薬効試験、安全性試験、さらに虚血性疾患などの低酸素状態にある患者(ヒト)もしくはその前状態にある患者(ヒト)への臨床試験に供してもよく、これらの試験を実施することによって、より実用的な低酸素状態に関連する疾患の予防または治療用医薬組成物の有効成分を選別取得することができる。
【0108】
このようにして選別された物質は、必要に応じて構造解析を行った後、その物質の種類に応じて、化学的合成、生物学的合成(発酵を含む)または遺伝子工学的操作によって、工業的に製造することができ、低酸素状態に関連する疾患の予防・治療用医薬組成物の調製に使用することができる。
【0109】
上記のスクリーニング方法は、前述するように、その基本はPHDとHIFαプロリンとの結合を阻害する作用を有する物質を探索することからなる。従って、上記のスクリーニング方法は、別の角度から、PHDとHIFαプロリンとの結合を阻害する作用を有する物質(PHD/HIF結合阻害剤)をスクリーニングする方法と規定することができる。
【0110】
斯くして得られるプロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物には、下式(A)または(B)で示され骨格を有する基の少なくとも1つを一分子中に含む化合物が含まれる。
【0111】
【化23】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
好ましくは、本発明化合物、特に化合物1〜3を含む化合物(I)ならびに化合物4を含む化合物(II)が含まれる。
【実験例】
【0112】
以下、本発明を製造例、ならびに実験例によりさらに具体的に説明するが、本発明はこれらの例に制限されるものではない。
【0113】
製造例1 TM-6101の合成
下式に示すように、6-アミノ-1-メチルウラシル(化合物(a))を出発原料として、目的とするTM-6101(化合物4)(分子量:378.41)を製造した。
【0114】
【化24】
(1)工程1
アルゴン雰囲気下、6-アミノ-1-メチルウラシル(化合物(a))3.53g(25.0mmol)とN,N’-ジメチルホルムアミドジメチルアセチル11.9g(0.10mmol)の脱水DMF懸濁溶液(125mL)を、40℃にて3時間攪拌した。炭酸カリウム5.18g(37.5mmol)とエチル-3-ブロモプロピネート6.79g(37.5mmol)を加え、80℃にて3時間攪拌した。減圧下濃縮し無機化合物を濾過した。酢酸エチル洗浄濾液を、減圧下濃縮し、スラリー状でヘキサンを加えて析出結晶を濾過し、化合物(b)を3.00g取得した(収率40%)。
【0115】
(2)工程2
化合物(b)(2.50g、8.44mmol)の脱水メタノール溶液(127mL)に塩化亜鉛4.89g(35.9mmol)を加え、アルゴン雰囲気下で5日間還流した。冷却後水を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下で濃縮した。濃縮残渣を塩化メチレンで洗浄し、化合物(c)を0.93g取得した(収率48%)。
【0116】
(3)工程3
アルゴン雰囲気下、化合物(c)(0.69g、3.04mmol)の脱水ジオキサン溶液(15mL)にピリジン0.36g(4.55mmol)とクロロアセチルクロライド0.51g(4.52mmol)を加え、室温で30分間攪拌した。原料の消失を確認した後、これを水に注ぎ、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下で濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィーで精製し(溶離液:メタノール/塩化メチレン=1/9)、無色結晶の化合物(d)を0.88g取得した(収率95%)。
【0117】
(4)工程4
化合物(d)(0.80g、2.63mmol)のトルエン溶液(80mL)に、2-メルカプトピリジン0.44g(3.95mmol)とトリエチルアミン0.53g(5.27mmol)を加え、室温で24時間攪拌した。その後、40℃にて7時間、室温で19時間攪拌した後、トリエチルアミン0.53g(5.27mmol)を加え、さらに室温で16時間攪拌した。水を加え酢酸エチルで熱抽出した。有機層を減圧下濃縮し、濃縮残渣をシリカゲルカラムクロマトグラフィーで精製した後(溶離液:酢酸エチル)、再度作戦エチルで洗浄し、無色〜淡黄色結晶の化合物4(TM-6101)を0.60g取得した(収率60%)。
【0118】
TM-6101(化合物4)のNMRデータは下記の通りである:
1H NMR (CDCl3) δ=2.68 (2H, t, J=9 Hz), 3.46 (3H, s), 3.70 (3H, s), 4.29 (2H, t, J=9 Hz), 4.81 (2H, s), 6.93-6.97 (1H, m), 7.26-7.29 (1H, m) ,7.44-7.50 (1H, m), 8.37-8.40 (1H, m)。
【0119】
なお、化合物4をTHF:水の1対1溶液中で1規定のNaOHを用いて、NaOHとして化合物4の等モル〜3倍モルを加え、室温で撹拌処理することによりそのカルボン酸である化合物(e)を調製することができる。
【0120】
実験例1
(1)Prolyl-3-hydroxylase (PHD3)の三次元構造の分析
Prolyl-3-hydroxylase(PHD3)は、Fe(II)の存在下で2-oxyglutarate(2-OG)とHIFプロリンからcis-3-hydroxyprolineを産生する酵素である。そのX線結晶構造はストレプトマイシス(Streptomyces sp.)由来のPHD3について既に報告されている(Clifton, I.J. et al. Structure of proline 3-hydroxylase. Evolution of the family of 2-oxoglutarate dependent oxygenases. Eur. J. Biochem. 268. 6625-6636 (2001))。しかし、PHD3分子内の、2-OGおよびHIFプロリンの結合部位は、未だ不明である。そこで、これらの結合部位を決定するために下記の実験を行った。
【0121】
ストレプトマイシスに由来するPHD3のX線結晶構造はプロテインデータバンクから取得した(PDBコード: 1E5RS)(Bernstein, F.C. et al. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112. 535-542 (1977))。水分子および硫酸イオンを除去した後、水素原子を、タンパク質中の酸性および塩基性残基の標準プロトン化状態に合わせて添加し、それらの位置を最適化した。なお研究には、ソフトウェア・システムMOE(Molecular Operating Environment, version 2004.04)とMMFF94s力場(Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem. 17. 490-519 (1996))を採用した。
【0122】
結合部位は、MOEのalpha site finder function (Edelsbrunner, H., Facello, M., Fu, R., & Liang, J. Measuring Proteins and Voids in Proteins. Proceedings of the 28th Hawaii International Conference on Systems Science, 256-264 (1995))を用いて決定した。小分子と標的部位のドッキングはプログラムPh4Dock(Goto, J., Kataoka, R. & Hirayama, N. Ph4Dock-Pharmacophore-based protein-ligand docking. J. Med. Chem. 47. 6804-6811 (2004))により行った。ドッキングの最終段階で、PHD3分子内の水素原子と小分子内の全原子の位置を最適化した。得られたドッキング結果を、下記の相互作用エネルギーにより判断した。
Utotal = Uele + Uvdw+ Uligand
ここで、UeleとUvdwは、PHD3と小分子間の、静電気的な相互作用とワンデルワールスの相互作用をそれぞれ意味する。また、Uligandは小分子の構成エネルギーを意味する。
【0123】
上記ドッキングシミュレーションから、PHD3分子内に比較的大きなポケットがあり、その中にFe(II) が存在していることがわかった(図1a参照)。図1a中の球体(青)はFe(II)を示す。このFe(II)の周囲に2-OGとHIFのプロリン残基の結合部位があると思われる。図1aにおいて、2-OGとHIFのプロリン残基を枝状モデル(緑)で示すが、2-OGとHIFプロリンは、Fe(II)に対して互いに反対の部位に結合する〔図1a中、Fe(II)の上部(B部位)にHIFプロリンが結合、下部(A部位)に2-OGが結合〕。なお、2-OGはカルボキシル基を介してFe(II)と結合している。
【0124】
(2)既知のPHD抑制剤の分子ドッキング
PHD3分子内のFe(II)周囲のポケット内の結合部位(A部位:2-OG結合部位、B部位:HIFのプロリン残基結合部位)を標的にして、既知のPHD抑制剤を用いて、ドッキングシミュレーションを行った。
【0125】
既知のPHD抑制剤として、下式に示す3,4-dehydroxybenzoate(以下、「3,4-DHB」という。Warnecke, C. et al. Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB. J. 17. 1186-1188 (2003))、3-hydroxychinolin 2-carbonic acidの誘導体である6-chloro-3-hydroxychinoline-2-carbonic acid-N-(carboxymethyl)-amide (以下「S956711」という。Warnecke, C. et al. Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB. J. 17. 1186-1188 (2003))、および4-Oxo-1,4-dihydro-[1,10]phenanthroline-3-carboxylic acid (以下、「FG-0041」という。Ivan, M. et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. 99. 13459-13464 (2002))を用いた。
【0126】
【化25】
その結果、3,4-DHBは、そのカルボキシル基を介して、PHD3分子内のFe(II)周囲のA部位(2-OG結合部位)と結合することがわかった(図1b)。S956711は、比較的分子構造が大きくまた環構造を有することから、A部位とB部位の両方(2-OG結合部位とHIFのプロリン残基結合部位)に結合すると考えられた。さらにFG-0041は、B部位(HIFのプロリン残基結合部位)に結合することが明らかになった。
【0127】
なお、これらの3つのPHD抑制剤はいずれも、A部位または/およびB部位に結合する能力の他に、鉄イオンとキレートを形成するモチーフを有している。しかしながら、鉄は、酸化的リン酸化とアラキドン酸のシグナリングに関係して、多くの重要な細胞機能を発揮するのに必要なコファクターであるので、そのキレート化剤は長期的治療用のPHD抑制剤として適切ではないと考えられる。このため、鉄のキレート化とは無関係に作用するPHD抑制剤を探索した。PHD3分子内のA部位(2-OG結合部位)は、Arg168およびHis135といった嵩高い残基で囲まれているため、PHD抑制剤がアクセスしにくい可能性がある。このため、PHD抑制剤として、HIFのプロリン残基と競合してB部位に結合する分子を探索することにした。
【0128】
(3)新規なHIF安定化化合物の同定
上記コンセプトの下で、PHD3分子内のB部位に結合するFG-0041の誘導体を19つ購入し、HIFの安定化を調べた。19つの誘導体のうち12つは、[2、2]bipyridinyl骨格を、残りの7つは、他の部位(4-oxo-1、4-dihydro-quinoline-3-carboxylic acid)を有している。これら19つの誘導体のHIF安定化によるルシフェラーゼ産生促進活性を、後述するin vitroスクリーニングアッセイにより測定した。その際、HIF-1α(HIFのα鎖)を安定化することによって低酸素状態をもたらす公知の化合物であるコバルト(塩化コバルト)を、陽性コントロールとして使用した(Wang, G.L. & Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 20. 1230-1237 (1995))。
【0129】
<in vitroスクリーニングアッセイ>
不死化した腎近位尿細管 (IRPTC)細胞に、HREを7回タンデムに反復して有するプロモーターの下流にルシフェラーゼ遺伝子を有するリポーター・ベクターを導入し、これを上記19つの誘導体(被験化合物)のHIF安定化によるルシフェラーゼ産生促進活性の測定に使用した。具体的には、上記リポーター・ベクターを導入したIRPTC細胞を、被験化合物(最終濃度:6.7〜100μM)とともに、37℃で24時間インキュベーションした後溶解し、細胞溶解物中のルシフェラーゼ活性を、Lumat 9507照度計(EG and Berthold、ドイツ)を用いて測定した。なお、被験化合物の非存在下で、リポーター・ベクターを導入したIRPTC細胞を同様にインキュベーションした細胞溶解物中のルシフェラーゼ活性をコントロール(陰性コントロール)とした。
【0130】
陰性コントロールのルシフェラーゼ活性を1とし、これに対する各被験化合物のルシフェラーゼ活性の相対比を、各被験化合物のHIF安定化によるルシフェラーゼ産生促進活性として評価した。結果を図2に示す。図2に示すように、[2、2]bipyridinyl骨格を有する誘導体の中で、下式に示す2つの化合物:6-amino-1,3-dimethyl-5-(2-pyridin-2-yl-quinoline-4-carbonyl)-1H-pyridine-2,4-dione(TM-6008)(分子量:387.40)
【0131】
【化26】
および3,7-Dimethy-furo[3,2-b;4,5-b]dipyridine(TM-6010) (分子量:198.23)
【0132】
【化27】
が、HIF安定化によるルシフェラーゼ産生促進活性を示した。
【0133】
これらTM-6008およびTM-6010はいずれも、鉄のキレートモチーフとなる[2、2]bipyridinylを有している。このため、TM6008からこのモチーフを除去し、5-acetyl-6-amino-1,3-di-methyl-1H-pyrimidine-2,4-dione構造を有する18つの化合物をライブラリーから取得した。
【0134】
それら化合物のうちの1つ、6-amino-1,3-di-methyl-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-1H-pyrimidine-2,4-dione(TM-6089) (分子量:306.35)
【0135】
【化28】
は、鉄のキレート化モチーフ部([2、2]bipyridinyl)を欠如しているにもかかわらず、強いHIF安定化によるルシフェラーゼ産生促進活性を示した(図2)。
【0136】
これらのTM-6008、TM-6010およびTM-6089のドッッキング構造を図1c示す(図1c中、黄はTM-6008、緑はTM-6010および赤はTM-6089をそれぞれ示す)。これから、これらの化合物はいずれも、PHD3分子内のB部位に結合することがわかる。
【0137】
(4)TM6101のHIF安定化によるルシフェラーゼ産生促進活性
製造例1で合成した、TM-6089のカルボン酸誘導体TM6101(化合物5)について、上記(3)と同様にしてin vitroスクリーニングアッセイによりルシフェラーゼ活性を測定し、HIF安定化によるルシフェラーゼ産生促進活性を評価した。陽性コントロールおよび比較のため、それぞれ塩化コバルトおよびTM6008を被験化合物として用いて同様にルシフェラーゼ活性を測定し、HIF安定化によるルシフェラーゼ産生促進活性を評価した。結果を図3に示す。図3は、図2と同様に陰性コントロールのルシフェラーゼ活性を1とし、これに対する各被験化合物(塩化コバルト、TM6008、TM6101)のルシフェラーゼ活性の相対比を示す。その結果、TM6101は、公知のPHD抑制剤である塩化コバルトよりも低いものの、TM6008よりも高いHIF安定化によるルシフェラーゼ産生促進活性を示した。
【0138】
実験例2 金属キレート活性
前述するように、化学構造から、3,4-DHB、S965711、FG-0041、TM-6008およびTM-6010中に鉄イオンとキレートするモチーフが存在することが示唆された。そこで、アスコルビン酸の銅触媒酸化を用いたin vitroアッセイ(遷移金属キレート化の評価系アッセイ)で、これを確認した。
【0139】
具体的には、上記実験例1でHIF促進作用が認められた化合物(TM-6008、TM-6010およびTM-6089)および公知のPHD抑制剤である3,4-DHBおよびS965711について、遷移金属イオンに対するキレート活性を、Priceらの方法(Price DL, et al., “Chelating activity of advanced glycation end-product (AGE) inhibitors.” J Biol Chem 272: 5430-5437, 1997)に幾つか修正を加えて測定した。簡単に説明すると、50μM CuCl2 15μlと様々な濃度の被験化合物30μlを、1.38mlのリン酸緩衝液中で、30℃、5分間インキュベーションした。この反応は、10mMアスコルビン酸(CuCl2およびアスコルビン酸の最終濃度:それぞれ500 nMおよび500μM)を75μl添加することで開始した。30℃で0〜60分間インキュベーションを行った。反応液のアスコルビン酸量を、逆相高速液体クロマトグラフィー(HPLC)を用いて244nmでの吸光度検出により測定した。
【0140】
その結果、3,4-DHB、S965711、TM-6008およびTM-6010は遷移金属(銅)にキレートし、用量依存的にアスコルビン酸の自動酸化を阻害した(IC50はそれぞれ330μM、31.4μM、0.57μMおよび4.84μM)。一方、TM-6089は100μMの濃度でさえ遷移金属(銅)とキレートしなかった。 このことから、TM-6008およびTM-6010は確かに金属とキレートすることが判明した。一方、TM-6089には、金属キレート能がなく、同様にTM-6089と構造的に類似するTM6101も金属キレート能がないと思われる。
【0141】
実験例3 PHD阻害活性
上記実験例1でHIF促進作用が認められた化合物(TM-6008、TM-6010およびTM-6089、並びに陽性コントロールとして塩化コバルト)について、PHD阻害活性を調べた。
【0142】
PHD阻害活性は、IRPTC細胞(腎近位尿細管細胞)のPHDを豊富に含むミトコンドリア画分のホモジネートを用いて、Kauleらの方法(Cunhild Kaule and Volkmar Gunzler, “Assay for 2-Oxoglutarate Decarboxylating Enzymes Based on the Determination of [1-14C] Succinate: Application to Prolyl 4- Hydroxylase”,Analytical Biochemistry 184, 291 - 297, (1990))
に従って測定した。具体的には、まず0.15mM MgCl2と10mM KClを含む10mMトリス-HCl(pH 6.7)中で懸濁したIRPTC細胞(腎近位尿細管細胞)を、氷上で、デキストロース(最終濃度:0.25M)とともにホモジナイズし、4℃で15分間1500xgで遠心分離して、核画分を除去した。引き続き上清を4℃で10分間20000xgで遠心分離して、ミトコンドリア画分を取得した(30μg)。ミトコンドリア画分は様々な濃度の被験化合物(TM-6008、TM-6010、TM-6089および塩化コバルト)と37℃で30分間反応させ、さらに2.7mMトリス-HCl(pH 7.5)、3.3μM FeSO4、6.7μM [5-14C]-2-OG(アマシャム)、66μMアスコビル酸塩、27μg/mlカタラーゼ、および33μM ジチオスレイトールを含む緩衝液に溶解した13.3μMの酸素依存性分解ドメイン(ODD)ペプチド(HIF-1αのアミノ酸557-576領域)と、37℃で1時間反応させた。1時間インキュベーションした後、3.3mMコハク酸塩、3.3mM 2-OGおよび27mM 2,4-ジニトロフェニルヒドラジンを添加した。 混合液を室温で30分間放置して、更に、0.3Mの2-OGを添加した。7000xgで5分間遠心分離して上清を分離し、液体シンチレーションカウンタによって[1-14C]コハク酸塩の放射能を測定した。なお、コントロール(陰性コントロール)として、被験化合物なしで上記反応を行い、同様にして[1-14C]コハク酸塩の放射能を測定し、これをヒドロキシラーゼ活性100%とした。
【0143】
結果を図4に示す。図4で示す各被験化合物のヒドロキシラーゼ活性は、陰性コントロールから得られる[1-14C]コハク酸塩の放射能(ヒドロキシラーゼ活性100%)に対する相対比である。
【0144】
図4に示すように、被験化合物(TM-6008、TM-6010およびTM-6089、並びに陽性コントロールとして塩化コバルト)はすべて用量依存的にPHD活性を阻害した。TM-6008とTM-6089は、公知のPHD阻害剤である塩化コバルトよりもPHD阻害活性が高く、中でもTM-6008が最もPHD阻害活性が高かった。
【0145】
実験例4 HIF-1α(α鎖)ペプチドとPHDとのin vitro結合アッセイ
実験例1のドッキングシミュレーションにより、3つの化合物(TM-6008、TM-6010およびTM-6089)がHIFのプロリン残基と競合して、PHD3分子内のB部位に結合することがわかった。これを確認するために、上記化合物(TM-6008、TM-6010およびTM-6089)ならびに公知のPHD抑制剤である3,4-DHBおよびS965711を用いて、下記の方法に従ってHIF-1αペプチドとPHDのin vitro結合アッセイを行なった。
【0146】
<in vitro結合アッセイ>
PHD3を、発現プラスミドpBOS-FLAG-mEGLN3からin vitroで転写し、網状赤血球溶血液TnTクイックキット(Promega社製)を使用して、[35S]メチオニン(Amersham社製)の存在下で形質転換した。 pGEX4T-1ベクター中でクローン化されたGST-HIF-1(531-652)を大腸菌(DH5α)内で発現させ、GST結合ビーズ(Glutathion Sepharose 4B: Amersham社製)によりアフィニティー精製を行った。35Sで標識したPHD3を、100mM KCl、10%のグリセリン、1.4mMのβ-メルカプトエタノール、100mMのFeCl2、0.2%のNP-40および0.5mg/mlのウシ血清アルブミンを含む20mMのトリス-HCl(pH 8.0)の50μl中で、被験化合物(最終濃度:50および500μM)と15分間、あらかじめインキュベーションした。次いで、その混合液に、GST-HIF-1α(531-652)結合ビーズ(12.5μg)を添加し、4℃で2時間インキュベーションした。洗浄後、GST-HIF-1α(531-652)に結合した35S標識PHD3を、120mM SDS、12%のグリセリン、240mM β-メルカプトエタノールおよび230μMブロモフェノール・ブルーを含む120mMトリス-HCl(pH 6.8)で溶出した。その溶出液を、SDS-PAGEにかけ、オートラジオグラフィーに供した。
【0147】
GST-HIF-1α(531-652)に結合する35S標識PHD3に対する被験化合物の阻害作用を、プルダウンアッセイにより評価した。被験化合物非存在下で、上記結合アッセイを行った結果(結合活性)を100%とし(コントロール)、これに対する各被験化合物の結合活性を%として示した。
【0148】
結果を図5に示す。公知のPHD抑制剤であるS965711と同様に、3つの新規なPHD抑制剤(TM-6008、TM-6010およびTM-6089)はいずれも、HIFペプチドと競合し、GST-HIF-1α(531-652)との結合を阻害した(図5)。一方、公知のPHD抑制剤である3,4-DHBは、HIFペプチドと競合せず、GST-HIF-1α(531-652)との結合を阻害しなかった(図5)。また過剰量の2-OGを添加しても、この結果は変わらなかった。
【0149】
実験例5 血管新生促進作用のin vivo試験
(1)スポンジ血管新生分析
上記被験化合物(TM-6008、TM-6010およびTM-6089)の局所投与によって血管新生が促進されるかどうかをin vivo試験した。この目的のために、マウスの皮下に小さなスポンジ・ディスクを挿入し、このスポンジを介して被験化合物を皮下投与して、10日後にヘモグロビン量と血管数を測定し、HIF-HREシステムの促進を評価した(スポンジ血管新生分析)。
【0150】
スポンジ血管新生分析は、村松らの文献の記載に従って行った(Muramatsu, M., Katada, J., Hayashi, I. & Majima, M. Chymase as a proangiogenic factor. A possible involvement of chymase-angiotensin-dependent pathway in the hamster sponge angiogenesis model. J. Biol. Chem. 275. 5545-5552 (2000))。具体的には、円形のスポンジ・ディスク(厚さ5mmおよび直径12mm)を、エーテル麻酔した6週齢の雄ICRマウス(n=2)の背部の皮下空気嚢に移植した。移植5日後に、DMSOに溶解し生理食塩水で希釈した被験化合物(1マウスあたり、50μg のTM-6008、TM-6010またはTM-6089、あるいは30μgのコバルト)を、1週間に亘って一日おき4回、スポンジに皮下投与した。bFGF(10μg)(ジェンザイム製)を陽性コントロールとして用いた。また、DMSOを生理食塩水で希釈した液(図中「vehicle」と記載する)のみを投与したものを陰性コントロールとして使用した。
【0151】
マウスは、実験後に殺して、ただちにスポンジ移植片とともに肉芽腫組織を切除した。キット(ヘモグロビンBテスト: 和光純薬製)を用いて採取した組織中のヘモグロビン量を測定し、また抗マウスCD31抗体(Pharmingen社製)を用いて、組織中の血管数(CD31陽性血管数)を免疫組織学的手法により定量測定した。なお、血管数はマウス一匹あたり任意に選択された重複しない5箇所の400×field内で測定した。
【0152】
ヘモグロビン量を図6aに、血管数に対応する相対蛍光強度を図6bに示す。また図6cに内皮細胞の免疫染色画像を示す。図6aの結果からわかるように、TM-6008、TM-6010およびTM-6089の投与によって、bFGFと同様にヘモグロビン量が増加することが認められた。また図6cから、TM-6008、TM-6010およびTM-6089の投与によって血管数も増加し、血管新生が認められた。特に、TM-6008の投与によって顕著な血管新生が認められた(図5b、c)。
【0153】
(2)HIF-HREシステムの活性化
被験化合物(TM-6008、TM-6089)によって、様々な器官においてHIF-HREシステムが促進されるかどうかを、HREルシフェラーゼを発現する低酸素症の遺伝子組み換えマウス(Tanaka, T. et al. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am. J. Pathol. 165. 1979-1992 (2004))を用いて調べた。
【0154】
マウスには、屠殺前の2又は4時間前に、0.5%のカルボキシメチルセルロース・ナトリウム塩の水溶液に懸濁したTM-6008あるいはTM-6089を、100mg/kgの割合で経口投与した(各々n=3)。コントロール用のマウスには、0.5%のカルボキシメチルセルロース溶液だけを経口投与した(n=3)。マウスは殺した後、レポーター遺伝子の発現を調べるために組織を採取した。低酸素症応答ルシフェラーゼ遺伝子の発現は、以前報告した方法に従って半定量的なRT-PCRによって評価した(Tanaka, T. et al. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am. J. Pathol. 165. 1979-1992 (2004))。 プロトコールは、東京大学の動物実験ガイドラインに従って行った。
【0155】
その結果、腎臓におけるレポーター遺伝子の発現は、基本条件下では認められなかったが(40回増幅)、TM-6008またはTM-6089を100mg/kgの割合で単回経口投与することによって認められた(それぞれ31.0±0.85回および31.0±2.05回で検出)。肝臓におけるレポーター遺伝子の発現は、基本条件下では認められなかったが、TM-6008の投与により検知された(32.3±0.35回で検出)。しかしTM-6089の投与では効果がなかった。心臓におけるレポーター遺伝子の発現は、基本条件で検出され、TM-6008またはTM-6089の投与によりその増加が認められた (各々1.37±1.00倍と6.69±5.45倍)。
【0156】
以上のことから、TM-6008はあらゆる器官で、HIF-HREシステムを活性化し、血管ネットワークの形成に重要な役割を果たしていることが示唆された。
【0157】
実験例6 低酸素症による神経細胞死の防止
PHD抑制剤(TM-6008)について、低酸素症による細胞のダメージに対する保護作用を評価した。
【0158】
実験は、アレチネズミ科マウスの遅延神経死モデル(雄、70-80g)を用いて行った。マウスを3つの実験群にランダムに分け、グループ1と2のマウスには、人為的に一過性の虚血を生じさせた。
【0159】
具体的には、グループ1と2のマウスについて、麻酔下で、2本の総頸動脈を小動脈瘤クリップ(杉田クリップ#07-940-12: みずほ医科製)で5分間閉塞し、広範囲に亘って一過性の虚血を引き起こした(Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 239. 57-69 (1982).)。次いでクリップを解除し、表層の微還流をレーザー・ドップラー・フローメトリー(Omega Flow FLO-C1: Neurosci)によって確認した。グループ1のマウス(n=20)には、頚動脈閉塞の後、7日間に亘って0.5%のカルボキシメチルセルロース・ナトリウムに溶解したTM-6008(100mg/kg/日)を強制経口投与した。グループ2のマウス(n=18)には、頚動脈閉塞の後、7日間に亘って0.5%のカルボキシメチルセルロース溶液だけを経口投与した。 グループ3(n=8)には偽物操作し、コントロールとして使用した。
【0160】
全マウスはすべて閉塞後7日目に、ペントバルビタールを注入して殺し、脳を取り出して4%のパラホルムアルデヒドにより冠状脳セクションを固定し、ヘマトキシロンとエオシンで染色した。間接的な免疫組織化学的染色法(in situ アポトーシス検出キットのTdT-Bule label:Trevigen社製)を用いて、TUNEL染色を行った。CA1海馬の神経細胞を光学顕微鏡を用いて観察し、海馬CA1内の虚血によるダメージを評価した。当該評価は、高性能のフィールド(400 x)について予め決めた領域(0.62 mm2)に沿って、海馬中に存在するピラミッド形をした正常の神経数を数えることにより評価した。
【0161】
CA1海馬中の代表的な顕微鏡写真(ヘマトキシロン・エオシン染色)を図7に示す。 非虚血のマウスの神経細胞(図7a)に比べて、虚血状態に曝された後に、カルボキシメチルセルロース(図中「vehicle」と記載する)のみを投与されたマウスは、ピラミッド形の神経の多くに、萎んで暗く染まった細胞質やグリア細胞が濃縮して蓄積しているのが認められ、虚血によって細胞がダメージを受けていることがわかった (図7b)。一方、虚血後、TM-6008で処理したマウスでは、少数の神経だけが虚血状態を示した(図7c)。
【0162】
また核と核小体を示す細胞を生存神経として検出した。CA1海馬中に生存する神経数は、虚血後カルボキシメチルセルロースだけで処理したマウスよりも(61±55個)、TM6008で処理したマウスにおいて有意に多く(166±73個)、非虚血コントロール群中で観察された生存神経数(227±50)と統計的に有意差はなかった(図7d)。
【0163】
これら実験におけるTM-6008の最終血漿濃度は7.8±2.9μg/mlであった。以上の結果から TM-6008は、虚血によって生じる神経死に対して有意に保護作用を示すことが明らかになった。
【0164】
実験例7 毒性
(1)細胞毒性
HeLa細胞を、被験化合物(TM6008、TM6010およびTM6089、50-250μM)の存在下で、ダルベッコのMEM培地中で24時間培養した。細胞毒性は、キット(Promega社製)を使用して培地中の乳酸塩脱水素酵素(LDH)の放出量を測定し、全細胞が溶解した後に放出される全LDH活性に対する割合(パーセンテージ)で示した。TM-6008、TM-6010およびTM-6089は、それぞれ50μM、250μMおよび100μMより低い濃度では、細胞毒性を示さなかった。
【0165】
(2)In vivo急性または亜急性毒性
急性毒性は、30-35gのICRマウス(クレアジャパンから入手)を用いて評価した。TM6008、TM6010およびTM6089を、ICRマウスに単回投与量500、1000、1500および2000mg/kgの割合で強制経口投与した。この急性投与の毒性結果を2週間モニターした。TM-6008については2000mg/kg、TM-6010については1500mg/kgを2週間投与しても急性毒性は観察されなかった。一方、TM-6089の50%致死量は500mg/kgであった。また亜急性毒性の試験は、マウスに30日間に亘って被験化合物(TM6008、TM6010またはTM6089)100mg/kgを強制経口投与することにより実施した。30日間に亘って各被験化合物を100mg/kg/日の割合で投与しても、マウスに亜急性毒性は殆ど認められなかった。
【0166】
(3)生化学分析
血液と尿のサンプルを回収して、生化学分析(血漿中のグルコース、総コレステロール、トリグリセリド、GOT、GPT、クレアチニン、尿素窒素、総タンパク質、およびアルブミン;血液中のヘモグロビン、赤血球数およびヘマトクリット;尿中の蛋白質およびクレアチニン)を行った。 血漿エリスロポエチン量は、キット(F.Hoffmann-La,ロシュ製)を用いて測定した。脳、肺、心臓、腎臓および肝臓の組織学分析および血清試験の結果、1か月後も異常を示さなかった。 血漿のエリトロポイエチン・レベルも著しい変化がなかった。
【0167】
(4)薬物速度論
被験化合物(TM6008、TM6010およびTM6089、50mg/kg)を、180-200gの雄のWisterラットに強制経口投与した。投与前(0h)、投与後1、2、6および24hに、ヘパリン添加血液サンプルを回収した。血漿濃度を逆相HPLCにより分析し、最高薬物濃度時間(Tmax)、最高薬物濃度(Cmax)、および半減期(T1/2)を求めた。
【0168】
各化合物を50mg/kgの割合で経口投与したマウス中の薬物速度論〔最高薬物濃度時間(Tmax)、最高薬物濃度(Cmax)、および半減期(T1/2)〕の結果は下記の通りであった:
【0169】
【表1】
TM-6010は平面構造を有している。ドッキングシミュレーションはDNAとのインターカレーションの可能性を示した。 このため、in vivoでの実験はTM-6008とTM-6089でのみ行なった。
【図面の簡単な説明】
【0170】
【図1】図aは、ストレプトマイシスに由来するPHD3のX線結晶構造を示す図である。図中、真ん中のポケット内にある球体(青)はFe(II)を示す。図中、このFe(II)のまわりに位置するスティックモデル(枝状物)は、上がHIFのプロリン残基、下が2-OGを示す。図bは、公知のPDH抑制剤である3, 4-DHBの、PHD3中での結合様式を示す。2-OG の結合部位であるA部位に結合することがわかる。図cは、TM6008(図中、黄色で示す)、TM6010(図中、緑で示す)およびTM6089(図中、赤で示す)の、PHD3中での結合様式を示す。なお、図はソフトウェアPyMOL version0.97(DeLano Scientific LLC社製)によって描いた。
【図2】HIF依存のルシフェラーゼ・リポーター遺伝子を有する7xHRE/Lucプラスミドで形質転換したIRPTC細胞を、被験化合物(陽性コントロール:塩化コバルト、TM6008、TM6010およびTM6089、最終濃度:20μMあるいは60μM)とともに24時間インキュベーションした際の、ルシフェラーゼ活性を示す。結果は3つの独立した実験の平均値として、コントロールの結果(1とする)の何倍増加したかで評価する。データは、平均値±SDとして表現する(* P<0.05、**P<0.01対コントロール)。
【図3】被験化合物として、塩化コバルト、TM6008およびTM6101を用いて24時間インキュベーションした際の、ルシフェラーゼ活性を示す。結果は3つの独立した実験の平均値として、コントロールの結果(1とする)の何倍増加したかで評価する。データは、平均値±SDとして表現する(* P<0.05、**P<0.01対コントロール)。
【図4】IRPTC細胞ホモジェネート(30μg)のミトコンドリア画分を、被験化合物およびHIF-1αの酸素依存性分解ドメイン(ODD)ペプチド(アミノ酸556-575)と反応させた。ODD依存の水酸化酵素活性を、[5-14C]-2-OGから変換された[1-14C]コハク酸塩の放射活性をカウントすることにより評価した。結果は3つの独立した実験の平均値として、ODD依存性水酸化酵素活性の阻害%として示す。データは、平均値±SDとして表現した。 *P<0.01対コントロール。
【図5】GST-HIF-1α(531-652)に結合する、35S標識PHD3に対する被験化合物の阻害作用を、プルダウンアッセイにより評価した。3つの独立した実験結果から平均値を出し、結合活性の%として示した。データは平均値±SDとして示す。*P<0.01 v.s. コントロール
【図6】ポリウレタン・スポンジを、マウスの皮下に移植し、被験化合物(マウス1匹当たり、48μg TM6008あるいは30μgコバルト)を、一日おきに1週間スポンジに注入した。 血管新生の程度を評価するために、スポンジ(a)中のヘモグロビン量を測定し、内皮細胞マーカーCD31(b、c)を染色した。 bは、CD31の免疫染色の代表写真である (x 200)。 cは、各試薬によって引き起こされた結果の平均数を示す。
【図7】非虚血動物(a)、虚血状態後、ベヒクル(カルボキシメチルセルロース)だけで処理された動物(b)、および拒絶状態後、TM-6008で処理されや動物(c)の、CA1海馬の、ヘマトキシロン-エオシン染色の代表的な顕微鏡写真を示す。 図中、バーは0.1mmを示す。dは、非虚血群(No ischemia)、虚血後ベヒクル(カルボキシメチルセルロース)処理群(Ischemia treated with vehicle)、および虚血後TM6008処理群(Ischemia treated with TM6008)の、CA1海馬中の正常神経の数を示す。
【技術分野】
【0001】
本発明は、プロリル水酸化酵素阻害剤(以下、「PHD阻害剤」ともいう)に関する。より詳細には、本発明は低酸素誘導因子(以下、「HIF」ともいう)とプロリル水酸化酵素(PHD)との結合を阻害してPHDの働きを抑える作用を有し、その結果、PHDによるHIFの分解を抑制してHIFを安定化することのできるPHD阻害剤に関する。また本発明は、PHD阻害作用を有し、HIFを安定化させることで、低酸素状態が関連して生じる各種疾患や病態(障害)を予防または治療するのに有効な医薬組成物に関する。さらに、上記PHD阻害剤として有用な新規化合物、ならびにPHD阻害剤として有用な化合物を取得するのに有効なスクリーニング方法に関する。
【背景技術】
【0002】
虚血性心疾患、慢性腎不全および脳梗塞などの疾患では細胞や組織への酸素供給が減少する。かかる酸素供給の減少によって低酸素状態が続くと細胞や組織に機能的障害が生じるため、生体内では、血管新生、赤血球生成、解糖および酸化酵素阻害など、広範囲にわたって細胞防御機構が働くようになっている。
【0003】
低酸素誘導因子(HIF)は、これらの生体防御機構を媒介する重要なタンパク質であり、細胞や組織が低酸素状態に陥った際に発現が亢進し、細胞や組織がより多くの酸素を得られるように血管新生を促進することが知られている(例えば、非特許文献1〜3参照)。かかる低酸素誘導因子(HIF)は、α鎖とβ鎖のヘテロ二量体からなるタンパク質であり、低酸素状態では安定であるが、酸素存在下ではその安定性が著しく低下する。具体的には、HIFは、酸素存在下で酸素依存的なプロリル水酸化酵素(PHD)によってα鎖のプロリンが水酸化され(例えば、非特許文献4〜9参照)、その結果、von hipple lindau蛋白と結合してユビキチン化され(例えば、非特許文献10〜11参照)、その結果、プロテアソームの標的とされて分解される(例えば、非特許文献12〜13参照)。
【0004】
しかし、低酸素状態の下では、HIFはα鎖がPHDによる水酸化を受けないため分解されず、α鎖とβ鎖の二量体の状態で、核内でHRE(hypoxia response element)と結合し、その下流に位置する低酸素ストレスの順応に関わる多くの遺伝子〔例えばエリスロポエチン(赤血球産生ホルモン)、血管内皮増殖因子(VEGF)、解糖酵素、グルコーストランスポーター、ヘムオキシゲナーゼ〕の転写を促進して生体を低酸素改善へと導くように機能する(例えば、非特許文献14参照)。このためHIFを安定化することによって、広く下流反応を調整する「マスター遺伝子」のスイッチを活性化することができ、低酸素の影響から組織や細胞を保護することが可能となると考えられる。
【0005】
現在、虚血などの低酸素状態を招く疾患の治療法の多くは、例えば急性虚血時の血流を改善するために血栓溶解薬が使用されるなど、症状の改善や病因因子の改善に集中しているのが実情であり、他の角度から治療法を検討することが求められている。
【非特許文献1】Marx, J. Cell biology. How cells endure low oxygen. Science 303. 1454-1456 (2004):
【非特許文献2】Semenza, G.L. & Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 12. 5447-5454 (1992):
【非特許文献3】Wang, G.L. & Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. 90. 4304-4308 (1993)
【非特許文献4】Epstein, A.C.R. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107. 43-54 (2001)
【非特許文献5】Schofield, C.J. & Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5. 343-354 (2004)
【非特許文献6】Semenza, G.L. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda). 19. 176-182 (2004)
【非特許文献7】Masson, N. & Ratcliffe, P.J. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O(2) levels. J. Cell Sci. 116. 3041-3049 (2003)
【非特許文献8】Bruick, R.K. & McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294. 1337-1340 (2001)
【非特許文献9】Semenza, G.L. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 107. 1-3 (2001)
【非特許文献10】Maxwell, P.H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399. 271-275 (1999)
【非特許文献11】Ohh, M. et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2. 423-427 (2000)
【非特許文献12】Maxwell, P.H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399. 271-275 (1999)
【非特許文献13】Ohh, M. et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2. 423-427 (2000)
【非特許文献14】Pugh, C.W. & Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9. 677-684 (2003)
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、虚血性疾患などによって生じる低酸素状態を改善し、低酸素症によるダメージから細胞や組織を保護するために有効に用いられるプロリル水酸化酵素阻害剤(PHD阻害剤)を提供することを目的とする。本発明が提供するPHD阻害剤は、低酸素誘導因子(HIF)のα鎖プロリンとプロリル水酸化酵素(PHD)との結合を競合的に阻害することで、HIFα鎖のプロリンを水酸化するPHDの機能を阻害する作用を有するものである。また本発明は、かかるPHD阻害剤を用いた、細胞や組織の低酸素状態の改善を目的とする医薬組成物を提供することを目的とする。さらに本発明は、PHD阻害剤として有効な新規化合物、ならびにPHD阻害剤として有効な化合物を探索するためのスクリーニング方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
前述するように、低酸素誘導因子(HIF)は低酸素による細胞や組織のダメージに極めて重要な役割を果たしている。その活性は、α鎖のプロリン残基が酸素依存的にプロリル水酸化酵素(PHD)によって水酸化されることによって制御されている。このため、PHDを阻害することによってHIFが安定化され、これによって低酸素状態に対する生体防御機能が活性化され、低酸素によるダメージから細胞や組織を保護することができる。
【0008】
本発明者らは、この考えのもと、新規なプロリル水酸化酵素阻害剤を開発するために、プロリル水酸化酵素の三次元構造および構造をベースとしたドラッグデザインソフトウェアを用いて、まず公知のプロリル水酸化酵素阻害剤を用いてドッキング・シミュレーション研究を行った。その結果、プロリル水酸化酵素には、HIFαプロリンと結合する部位と2−オキシグルターゼと結合する部位の両方があり、そのうちHIFαプロリンと結合する部位を介してHIFとプロリン水酸化酵素との結合を競合的に阻害する作用を有する特定の基を有する化合物が、HIFを安定活性化ないしは安定化するプロリル水酸化酵素阻害剤として有効であることを見出した。さらにこの特定の基を有する化合物は、in vivo試験により、HIF活性を安定化し、血管新生形成を促進して虚血による神経死を防止できることを確認した。本発明はかかる知見に基づいて完成したものである。
【0009】
すなわち、本発明には下記の態様が含まれる:
(I)プロリル水酸化酵素阻害剤(PHD阻害剤)
項1.下式(A)または(B)で示される骨格を有する基の少なくとも1つを一分子中に含む化合物またはその塩を有効成分とするプロリル水酸化酵素阻害剤:
【0010】
【化1】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
項2.骨格Aを有する基が、下式:
【0011】
【化2】
(R1は水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、保護基を有していてもよいカルボキシアルキル基、またはアミノ基:R2は水素原子、水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、またはアミノ基:R3は水素原子、アミノ基、または低級アルキルアミノ基を意味する)
で示される基である、項1記載のプロリル水酸化酵素阻害剤。
項3.骨格Bを有する基が、下式:
【0012】
【化3】
(R4、R5、R6およびR7は、同一または異なって、水素原子または低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
で示される基である、項1または2に記載するプロリル水酸化酵素阻害剤。
項4.下式で示される化合物(I):
【0013】
【化4】
〔R1は炭素数1〜6の低級アルキル基または保護基を有していてもよい炭素数1〜4のカルボキルアルキル基:R2は水素原子、炭素数1〜6の低級アルキル基:R10は、R11−S−CH2−(式中、R11は置換基を有していてもよいヘテロアリール基、または下式:
【0014】
【化5】
(R4、R5、R6およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。)で示される基を意味する。〕
および下式で示される化合物(II):
【0015】
【化6】
(R5およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。)
からなる群から選択される少なくとも1つの化合物を有効成分とする項1乃至3のいずれかに記載するプロリル水酸化酵素阻害剤。
項5.下式で示される化合物1〜4:
(化合物1)
【0016】
【化7】
(化合物2)
【0017】
【化8】
(化合物3)
【0018】
【化9】
(化合物4)
【0019】
【化10】
からなる群から選択される少なくとも1つを有効成分とする項1乃至4のいずれかに記載するプロリル水酸化酵素阻害剤。
項6.プロリル水酸化酵素と低酸素誘導因子との結合阻害剤である項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
項7.プロリル水酸化酵素に結合して、プロリル水酸化酵素と低酸素誘導因子との結合を阻害する剤である項6に記載するプロリル水酸化酵素阻害剤。
項8.低酸素誘導因子分解阻害剤である項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
【0020】
(II)医薬組成物
項9.項1乃至8のいずれかに記載するプロリル水酸化酵素阻害剤および薬学的に許容される担体または添加剤を含む、医薬組成物。
項10.細胞または組織の低酸素状態を改善するための項9記載の医薬組成物。
項11.細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬である項9または10記載の医薬組成物。
項12.細胞または組織の低酸素状態に関連して生じる疾患また病態が、慢性または急性の虚血性疾患またはそれに起因して生じる病態である項10に記載する医薬組成物。
項13.経口投与形態を有する項9乃至12のいずれかに記載する医薬組成物。
【0021】
(III)プロリル水酸化酵素阻害剤のスクリーニング方法
項14.被験化合物の中から、プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物を選択する工程を有するプロリル水酸化酵素阻害剤のスクリーニング方法。
項15.プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物が,下式(A)または(B)で示され骨格を有する基の少なくとも1つを一分子中に含む化合物である項14記載のスクリーニング方法:
【0022】
【化11】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
項16.細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬の有効成分のスクリーニング方法である、項14または15に記載するスクリーニング方法。
【0023】
(III)新規化合物
項17.下式で示される化合物またはその塩:
【0024】
【化12】
(式中、R12は水素原子または低級アルキル基を意味する。)
【発明の効果】
【0025】
本発明により、HIFを安定化するために有効なPHD阻害剤を提供することができる。当該PHD阻害剤は、PHDによるHIFの水酸化による不活性化を阻止してHIFを安定化することにより、虚血などによる低酸素状態によって生じるダメージから生体を保護することができる。このため、本発明のPHD阻害剤は、低酸素状態に関連して生じる疾患や病態(障害)を予防または改善するための医薬組成物として有効に利用することができる。特に、本発明のPHD阻害剤は、腎臓、心臓、下肢または脳の各種器官の虚血性疾患やそれから進展する病態(障害)の予防または改善に有効に使用することができる。また本発明は、かかるPHD阻害剤として有効な新規な化合物を提供する。
【0026】
さらに本発明のスクリーニング方法によれば、上記作用を有し虚血性疾患やそれから進展する病態(障害)の予防または改善に有用な新規なPHD阻害剤を選別し取得することが可能となる。
【発明を実施するための最良の形態】
【0027】
(I)プロリル水酸化酵素阻害剤(PHD阻害剤)
本発明が提供するPHD阻害剤は、下式(A)または(B):
【0028】
【化13】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
で示される骨格を有する基の少なくとも1つを、一分子中に含む化合物またはその塩を有効成分とするものである。
【0029】
本発明が対象とする化合物(以下、「本発明化合物」という)は、上記骨格Aを有する基または骨格Bを有する基の少なくとも1つを一分子中に含み、HIFのα鎖のプロリンを水酸化するPHDの機能を妨げる作用を有するものである。本発明化合物は、結果としてPHDの機能を妨げる作用を有するものであればよく、PHDのFe(II)にキレートしてPHDの機能を妨げるもの、低酸素誘導因子(HIF)のα鎖のプロリン残基(以下、単に「HIFαプロリン」という)に対するPHDの結合を競合的に阻害するもの、PHDの水酸化活性を阻害するものなど、その作用機序は特に制限されない。特に本発明が対象とする化合物として好ましくは、HIF/PHD結合阻害作用を有するものである。
【0030】
本発明化合物のFe(II)に対するキレート作用は、制限されないが、例えばPriceらの方法(Price DL, et al.,” Chelating activity of advanced glycation end-product (AGE) inhibitors.” J Biol Chem 272: 5430-5437, 1997)に従って、または準じて測定し評価することができる。
【0031】
その一例を挙げると、後述する実験例2に記載するように、遷移金属である銅とのキレート作用を評価する方法を用いることができる。当該方法は、具体的には、被験化合物の存在下で塩化銅とアスコルビン酸をインキュベーションして、反応後のアスコルビン酸の残存量を測定する方法であり、被験化合物の存在によってアスコルビル酸の自動酸化が防止できれば、被験化合物について金属キレート能(Fe(II)に対するキレート作用)があると評価することができる。
【0032】
また本発明化合物のHIF/PHD結合阻害作用は、制限されないが、例えば実験例4に詳述するように、被験化合物の存在下で、放射性同位体等で標識したPHDとHIFのα鎖ドメイン(好ましくはHIF-1αのアミノ酸557-576領域)をインキュベーションし、HIFのα鎖に結合したPHD量を測定する方法であり、被験化合物の存在によってHIFのα鎖に対するPHDの結合が防止できれば、被験化合物についてHIF/PHD結合阻害作用があると評価することができる。
【0033】
さらに本発明化合物のPHD阻害活性は、制限されないが、例えばKauleらの方法(Cunhild Kaule and Volkmar Gunzler,” Assay for 2-Oxoglutarate Decarboxylating Enzymes Based on the Determination of [1-14C] Succinate: Application to Prolyl 4- Hydroxylase”, Analytical Biochemistry 184, 291-297 (1990))に従ってまたは準じて測定し評価することができる。
【0034】
その一例を挙げると、後述する実験例3に記載するように、2-oxyglutarate(2-OG)とHIFのプロリンからcis-3-hydroxyprolineを産生する触媒能力を評価する方法を用いることができる。当該方法は、具体的には、被験化合物の存在下で、PHD、[5-14C]2-oxyglutarate(2-OG)およびHIFのα鎖ドメイン(好ましくはHIF-1αのアミノ酸557-576領域)をインキュベーションして、反応後の[1-14C]コハク酸を測定する方法であり、被験化合物の存在によって [1-14C]コハク酸の生成が抑制できれば、被験化合物についてPHD阻害活性があると評価することができる。
【0035】
上記骨格Aを有する基として、より好ましくは下式(A’)で示される基である:
【0036】
【化14】
(式中、R1は水酸基、低級アルキル基、保護基を有していてもよいカルボキシアルキル基、アミノ基:R2は水素原子、水酸基、低級アルキル基、保護基を有していてもよいカルボキシアルキル基、アミノ基:R3は水素原子、アミノ基または低級アルキルアミノ基を意味する)
式(A’)において、R1およびR2の定義でみられる低級アルキル基としては、炭素数1〜6の直鎖または分枝鎖状のアルキル基を挙げることができる。具体的には、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、ter−ブチル基、ペンチル基、1,2−ジメチルプロピル基、1,1−ジメチルプロピル基、2,2−ジメチルプロピル基、2−エチルプロピル基、n−ヘキシル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1,3−ジメチルブチル基、1−エチル−2−メチルプロピル基、1−メチル−2−エチルプロピル基などを挙げることができる。好ましくは炭素数1〜4の直鎖または分枝鎖状のアルキル基であり、より好ましくはメチル基である。
【0037】
式(A’)において、R1の定義でみられるカルボキシアルキル基とは、上記低級アルキル基のいずれかに炭素原子にカルボキシル基が結合しているものを意味する。この場合のカルボキシルは保護基を有していてもよい。ここで保護基としては、例えばメチル基、エチル基、tert−ブチル基等の低級アルキル基;p−メトキシベンジル基、p−ニトロベンジル基、3,4−ジメトキシベンジル基、ジフェニルメチル基、トリチル基、フェネチル基等の置換基を有していてもよいフェニル基で置換された低級アルキル基;2,2,2−トリクロロエチル基、2−ヨードエチル基などのハロゲン化低級アルキル基;ピバロイルオキシメチル基、アセトキシメチル基、プオピオニルオキシメチル基、ブチリルオキシメチル基、バレリルオキシメチル基、1−アセトキシエチル基、2−アセトキシエチル基、1−ピバロイルオキシエチル基、2−ピバロイルオキシエチル基などの低級アルカノイルオキシ低級アルキル基;パルミトイルオキシエチル基、ヘプタデカノイルオキシメチル基、1−パルミトイルオキシエチル基などの高級アルカノイルオキシ低級アルキル基、メトキシカルボニルオキシメチル基、1−ブトキシカルボニルオキシエチル基、1−(イソプロポキシカルボニルオキシ)エチル基等の低級アルコキシカルボニルオキシ低級アルキル基;カルボキシメチル基、2−カルボキシエチル基などのカルボキシ低級アルキル基;3−フタリジル基等のヘテロアリール基、4−グリシルオキシベンゾイルオキシメチル基などの置換基を有していてもよいベンゾイルオキシ低級アルキル基、(5−メチル−2−オキソ−1,3−ジオキソレン−4−イル)メチル基などの(置換ジオキソレン)低級アルキル基;1−シクロヘキシルアセチルオキシエチル基などのシクロアルキル置換低級アルカノイルオキシ低級アルキル基、1−シクロヘキシルオキシカルボニルオキシエチル基などのシクロアルキルオキシカルボニルオキシ低級アルキル基などをあげることができる。更に、種々の酸アミドも本発明における保護基を有しているカルボキシル基に含まれる。要するに生体内で何らかの手段で分解されて、カルボン酸となりうるものであれば、いかなるものもカルボキシル基の保護基となり得る。
【0038】
R3の定義でみられる低級アルキルアミノ基のうち低級アルキル基としては、上記の低級アルキルを同様に挙げることができる。
【0039】
上記骨格A’を有する基を有する化合物として、下式で示される化合物(I)を挙げることができる。
【0040】
【化15】
ここで、R1として前述する基を挙げることができるが、好ましくは炭素数1〜6の低級アルキル基、炭素数1〜4のカルボキシアルキル基、または炭素数1〜6の低級アルキル基で保護されている炭素数1〜4のカルボキシアルキル基を挙げることができる。炭素数1〜6の低級アルキル基としてより好ましくはメチル基、カルボキシアルキル基としてより好ましくはカルボキシエチル基、アルキル基で保護されているカルボキシアルキル基としてより好ましくは、メチル基で保護されたカルボキシエチル基を挙げることができる。
【0041】
またR2としても前述の基を挙げることができるが、好ましくは炭素数1〜4の低級アルキル基、より好ましくはメチル基である。
【0042】
R10としては、R11−S−CH2−(式中、R11は置換基を有していてもよいヘテロアリール基を意味する)または下式で示される基を挙げることができる:
【0043】
【化16】
(式中、R4、R5、R6およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の、置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。)で示される基を意味する。〕
R11の定義でみられるヘテロアリール基とは、酸素原子、窒素原子または硫黄原子の少なくとも1つを1〜4個含む単環または縮合環から誘導される基を意味する。例えば、チェニル基、フリル基、ベンゾチェニル基、ベンゾフラニル基、イソベンゾフラニル基、ピロリル基、イミダゾリル基、ピラゾリル基、イソチアゾリル基、イソキサゾリル基、テトラゾリル基、ピリジル基、ピラジニル基、ピリミジニル基、ピリダジニル基、インドリル基、イソインドリル基、キノリル基、イソキノリル基、フタラジル基、キノキサリル基、ナフチリジル基、キナゾリル基、アクリジニル基、イミダゾピリジル基などを挙げることができる。かかる基の置換基としては、水酸基:メチル基、エチル基、n−プロピル基、イソプロピル基などの低級アルキル基;ハロゲン化アルキル基;メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基などの低級アルコキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子などのハロゲン原子;シアノ基、アセチル基、プロピオニル基、ベンゾイル基等のアシル基;アミノ基;ニトロ基;保護基を有していてもよいカルボキシル基;アシルアミノ基;スルホニルアミノ基;カルバモイル基;スルファモイル基;アミノスルホニル基;シアノアルキル基;ヘテロアリール基;カルボキシアルキル基;カルボキシアルコキシ基;ヘテロアリールアルキル基;ヘテロアリールアルコキシ基などを挙げることができる。R11として好ましくはピリジル基を挙げることができる。
【0044】
R4、R5、R6およびR7の定義でみられる低級アルキル基としては、R1およびR2に関して説明した炭素数1〜6の直鎖または分枝鎖状のアルキル基を同様に挙げることができる。
【0045】
R4、R5、R6およびR7の定義でみられる置換基を有していてもよいシクロ環としては、シクロペンタンやシクロヘキサンなどの5または6員の飽和単環:シクロペンテン、シクロヘキセン、1,3−シクロヘキサジエン、2,5−シクロヘキサジエン、ベンゼンなどの5または6員の不飽和単環を挙げることができる。R4、R5、R6およびR7の定義でみられる置換基を有していてもよいヘテロ環としては、酸素原子、窒素原子または硫黄原子の少なくとも1つを1乃至2含む、5または6員の飽和または不飽和の単環を意味する。かかる単環として、具体的にはフラン、チオフェン、ピロール、チアゾール、イソチアゾール、オキサゾール、イソオキサゾール、イミダゾール、ピラゾール、フラザン、ピラン、ピリジン、ピリダジン、ピリミジン、ピラジン、ピロリジン、イミダゾリジン、ピラゾリジン、ピペリジン、ピペラジン、およびモルホリンを挙げることができる。なお、これらのシクロ環およびヘテロ環において置換基としては、水酸基:メチル基、エチル基、n−プロピル基、イソプロピル基などの低級アルキル基;ハロゲン化アルキル基;メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基などの低級アルコキシ基;フッ素原子、塩素原子、臭素原子、ヨウ素原子などのハロゲン原子;シアノ基、アセチル基、プロピオニル基、ベンゾイル基等のアシル基;アミノ基;ニトロ基;保護基を有していてもよいカルボキシル基;アシルアミノ基;スルホニルアミノ基;カルバモイル基;スルファモイル基;アミノスルホニル基;シアノアルキル基;ヘテロアリール基;カルボキシアルキル基;カルボキシアルコキシ基;ヘテロアリールアルキル基;ヘテロアリールアルコキシ基などを挙げることができる。
【0046】
化合物(I)として、具体的は下式で示される化合物1〜3を挙げることができる。本明細書において、便宜上、化合物1(6-amuno-1,3-dimethyl-5-(2-pyridin-2-yl-quinoline-4-carbonyl)-1H-pyrimidine-2、4-dione)をTM-6008、化合物2(6-amino-1,3-di-methyl-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-1H-pyrimidine-2,4-dione)をTM-6089、および化合物3(3-{4-Amino-3-methyl-2,6-dioxo-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-3,6-dihydro-2H-pyrimidin-1-yl}-propionic acid methyl ester)をTM-6101と称する場合がある。
【0047】
(1)化合物1:
6-amuno-1,3-dimethyl-5-(2-pyridin-2-yl-quinoline-4-carbonyl)-1H-pyrimidine-2,4-dione(TM-6008)
【0048】
【化17】
(2)化合物2:6-amino-1,3-di-methyl-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-1H-pyrimidine-2,4-dione(TM-6089);
【0049】
【化18】
(3)化合物3:
3-{4-Amino-3-methyl-2,6-dioxo-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-3,6-dihydro-2H-pyrimidin-1-yl}-propionic acid methyl ester(TM-6101)
【0050】
【化19】
また上記骨格Bを有する基として、より好ましくは下式(B’)で示される基である:
【0051】
【化20】
(R4、R5、R6およびR7は、同一または異なって、水素原子または低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の、置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
式(B’)中、R4、R5、R6、およびR7の定義にみられる低級アルキル基としては、R1およびR2に関して説明した炭素数1〜6の直鎖または分枝鎖状のアルキル基を同様に挙げることができる。好ましくは炭素数1〜4の直鎖または分枝鎖状の低級アルキル基であり、より好ましくはメチル基である。
【0052】
R4、R5、R6およびR7の定義でみられる置換基を有していてもよいシクロ環またはヘテロ環、ならびにその置換基は、前述の通りである。好ましくはベンゼン環である。
【0053】
R8およびR9の定義でみられるアルキレン鎖としては炭素数1〜6の直鎖または分岐状のアルキレン鎖を意味する。例えば、メチレン鎖、エチレン鎖、プロピレン鎖、ブチレン鎖、エチルメチレン鎖などを挙げることができる。好ましくはメチレン鎖である。R8およびR9は、互いに水素原子であるか、またはそれらが結合する酸素原子ととも環を形成していることが好ましい。
【0054】
上記骨格B’を有する基を有する化合物として、下式で示される化合物(II)を挙げることができる。
【0055】
【化21】
(R5およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。)
より好ましくは、下式で示される化合物4(3,7-Dimethy-furo[3,2-b;4,5-b]dipyridine)である。本明細書において、便宜上、化合物4をTM-6010と称する場合がある。
【0056】
(4)化合物4:3,7-Dimethy-furo[3,2-b;4,5-b]dipyridine(TM-6010)
【0057】
【化22】
また、前述する化合物1(TM-6008)も骨格B’を有する基を有する化合物Iに該当する。
【0058】
これらの化合物は、遊離または塩の形態を有するもののいずれであってもよい。ここで塩としては、通常、医薬上許容される塩、たとえば無機塩基または有機塩基との塩、あるいは無機酸、有機酸、または塩基性若しくは酸性アミノ酸などの酸付加塩等を挙げることができる。無機塩基としては、たとえば、ナトリウムやカリウム等のアルカリ金属;カルシウムやマグネシウム等のアルカリ土類金属;アルミニウムやアンモニウム等を挙げることができる。有機塩基としては、たとえば、エタノールアミン等の第一級アミン;ジエチルアミン、ジエタノールアミン、ジシクロヘキシルアミン、N,N'−ジベンジルエチレンジアミン等の第二級アミン;トリメチルアミン、トリエチルアミン、ピリジン、ピコリン、トリエタノールアミン等の第三級アミン等を挙げることができる。
【0059】
酸付加塩を形成する無機酸としては、たとえば、塩酸、臭化水素酸、硝酸、硫酸、リン酸等を挙げることができる。有機酸としては、たとえば、ギ酸、酢酸、乳酸、トリフルオロ酢酸、フマール酸、シュウ酸、酒石酸、マレイン酸、安息香酸、クエン酸、コハク酸、リンゴ酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸等を挙げることができる。塩基性アミノ酸としては、たとえば、アルギニン、リジン、オルニチン等を挙げることができる。酸性アミノ酸としては、たとえば、アスパラギン酸、グルタミン酸等を挙げることができる。
【0060】
上記化合物、特に化合物1(TM-6008)、2(TM-6089)および4(TM-6010)は商業的に入手可能であり〔例えば、Maybrige(英国)、ASINEX(ロシア)、LaboTest(ドイツ)、Specs(オランダ)、ENAMINE(ウクライナ)、Ambinter(フランス)、ASDI(米国)、Sigma(米国)、Pharmeks(ロシア)、Life Chemical(ウクライナ)、Key Organics(英国)など〕、また自体公知の方法により製造することができる。
【0061】
また、化合物3(TM-6101)は、後述する製造例1に記載する方法で製造することができる。その製造工程のうち、工程1において、Ethyl 3-Bromopropinateに代えてIodomethaneを用いることによって化合物2(TM-6089)を合成することができる。また、工程1において、Ethyl 3-Bromopropinateに代えてIodomethaneを用い、かつ工程4において2-mercaptopyridineに代えて2-Pyridine-2-yl-quinoline-4-carbonylchlorideを用いることによって化合物1(TM-6008)を合成することができる。
【0062】
本発明化合物は、前述するように、HIFのα鎖のプロリンを水酸化するPHDの機能を妨げる作用を有するものである。かかる作用は、金属キレート作用、HIF/PHD結合阻害作用、およびPHD活性阻害作用など、結果としてPHDの機能を妨げる作用であればよい。これらはいずれも前述するインビトロアッセイ系で評価することができる(詳細は実験例を参照のこと)。
【0063】
さらに本発明化合物は、PHDとHIFの結合を阻害する作用を有する結果、HIFの水酸化によるVHLとの結合および分解を阻害し、HIFを安定化する作用を有している。かかるHIF促進作用は、制限されないが、後述する実験例1の方法に従って、またはこの方法に準じて測定し、評価することができる。当該インビトロアッセイは、HRE(hypoxia responsive element (低酸素応答要素))を7回タンデムに含むプロモーターの下流にルシフェラーゼ遺伝子を有するベクターを導入したPHD発現細胞を被験化合物とともにインキュベーションし、細胞溶解物のルシフェラーゼ活性を測定する方法である。当該アッセイ系において、被験化合物の存在によってルシフェラーゼ活性が増加すれば、被験化合物の存在によってHIFが安定化しHREシステムが活性化していること、すなわち被験化合物にHIF安定化作用があると評価することができる。
【0064】
また本発明化合物は、PHDの機能を阻害しHIFを安定化させた結果、血管新生を促進する作用を有している。かかる血管新生促進作用は、制限されないが、後述する実験例5の方法(スポンジ血管新生分析)に従って、またはこの方法に準じて測定し、評価することができる。当該インビボアッセイは、被験動物の皮下に埋設したスポンジ・ディスクを介して被験化合物を皮下投与し、経時的にヘモグロビン量と血管数を測定する方法である。当該インビボアッセイ系において、被験化合物の投与によってヘモグロビン量と血管数が増加すれば、被験化合物に血管新生作用があると評価することができる。
【0065】
本発明化合物はPHDの機能を阻害する作用を有する。中でも化合物1〜4、好ましくは化合物1は、後述する実験例で示すように優れたPHD阻害作用を有している。当該PHD阻害作用により、本発明化合物はHIFの分解を阻止して安定化し、HIF−HREシステムを安定化することにより低酸素状態を改善し、その結果、虚血などの低酸素状態によって生じるダメージから細胞や組織を防御することができる。従って、本発明化合物によれば、下記の作用が期待される。
【0066】
(i) 腎慢性虚血改善作用
腎臓において、尿細管周囲毛細血管の血流量の停滞は慢性低酸素を誘発し、それに伴い尿細管間質障害・尿細管周囲毛細血管の喪失が促進される。公知のPHD阻害剤であるコバルトによってHIFの発現が亢進し、その結果、腎虚血性障害である尿細管間質性障害の改善が認められることが報告されている(Matsumoto M, Tanaka T, Yamamoto T, Noiri E, Miyata T, Inagi R, Fujita T, Nangaku M. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol 15: 1574-1581 (2004))。このことから、HIFを安定化することによりVEGFやエリスロポエチンの転写が促進されて、血管新生や血流の改善が惹起され、慢性虚血を改善して腎不全への進展が抑制できる可能性がある。
【0067】
(ii) 脳虚血改善作用
脳梗塞に陥った脳梗塞領域は虚血になっており、組織への虚血障害が深刻な問題となっている。よって、脳梗塞領域に起こる活発な脈管形成は、虚血の下で組織に有益であることが証明されている(Krupinski J, Kaluza J, Kumar P, Kumar S & Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 25: 1794?1798 (1994))。また、VEGFが、一時的な中大脳動脈閉塞の後、脳梗塞サイズ・浮腫形成・脳血液関門破壊を、虚血に起因する神経細胞障害を減少することによって減らすことが証明されている(Hayashi, T., Abe, K., Itoyama, Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J. Cereb. Blood Flow Metab. 18: 887-895 (1998))。これらのことより、HIFを安定化することによるVEGFの発現の亢進は、脳梗塞および虚血性脳卒中に対する効果的な治療になると考えられる。
【0068】
(iii) 虚血性心疾患改善作用
虚血性心疾患には狭心症・心筋梗塞がある。いずれも適切な治療が遅れると心不全や重度の不整脈を惹き起こし直接生死にかかわってくる。このため心筋虚血を改善方法の開発が喫緊の課題とされている。最近の研究では、ブタの進行性冠動脈閉塞モデルにVEGFを投与すると虚血領域の改善が見られ、また大量投与では心筋還流血液量の改善が見られることが証明されている(Pearlman JD, Hibberd MG, Chuang ML, Harada K, Lopez JJ, Glad-stone SR, Friedman M, Sellke FW, Simons M. Magnetic resonance mapping demonstrates benefits of VEGF-induced myocardial angiogenesis. Nat Med.1:1085-1089. (1995))。また、ブタの慢性心筋虚血モデルにVEGFを投与すると血管形成が促進されることが証明されている(Lopez JJ, Laham RJ, Stamler A, Pearlman JD, Bunting S, Kaplan A, Carrozza JP, Sellke FW, Simons.M. VEGF administration in chronic myocardial ischemia in pigs. Cardiovasc Res.;40: 272-281. (1998))。これらのことから、HIFの安定化によるVEGFの発現亢進は、心筋梗塞や狭心症など虚血性心疾患の効果的な治療になると考えられる。
【0069】
(iv) 下肢虚血改善作用
下肢虚血患者では症状が重度化すると下肢切断を余儀なくされ、患者のADLを極度に低下させることから医学的のみならず社会的にも、深刻な問題となっている。最近の研究では、下肢虚血患者において、VEGFが治療に対し有効と考えられている。VEGF、FGF-2遺伝子を虚血筋組織に導入すると、FGF-2遺伝子導入で内因性VEGF, HGF発現が亢進し、組織学的検査ならびに血流測定により新生血管の増加と側副血行路形成が認められ、明らかな下肢虚血の改善が認められることが報告されている(Onimaru M, Yonemitsu Y, Tanii M, Nakagawa K, Masaki I, Okano S, Ishibashi H, Shirasuna K, Hasegawa M, Sueishi K. Fibroblast growth factor-2 gene transfer can stimulate hepatocyte growth factor expression irrespective of hypoxia-mediated downregulation in ischemic limbs. Circ. Res, 91: 723-730 (2002))。これらのことから、HIFの安定化によるVEGFの発現亢進は、下肢虚血の効果的な治療になると考えられる。
【0070】
本発明のPHD阻害剤は、前述する本発明化合物またはその塩を有効成分とするものである。本発明のPHD阻害剤は、前述する化合物またはその塩100重量%からなるものであっても、またそうでなくてもPHD阻害作用を発揮する有効量の化合物を含有するものであればよい。制限されないが、PHD阻害剤には、通常、化合物またはその塩が10〜100重量%の割合で含まれる。
【0071】
(II)医薬組成物
本発明は、前述するPHD阻害剤を有効成分として含有する医薬組成物を提供する。言い換えれば、本発明の医薬組成物は、上記本発明化合物を有効成分として含有するものである。本発明の医薬組成物は、PHD阻害剤(本発明化合物)を、PHDを阻害する有効量含むことによって、HIFを安定化する作用を有している。その結果、本発明の医薬組成物は、生体のHIF−HREシステムを活性化することにより低酸素状態を改善し、その結果、虚血などの低酸素状態によって生じるダメージから細胞や組織を防御することができる。
【0072】
このため、本発明の医薬組成物は細胞や組織の低酸素状態に関連して生じる疾患や病態(障害)の予防または治療剤として有用である。かかる疾患または病態として、例えば、各種の虚血性疾患ならびに虚血から生じる障害を挙げることができる。虚血性疾患としては、腎虚血性障害(尿細管間質性障害)、虚血性脳血管障害(脳塞栓症、脳梗塞、一過性脳虚血発作等)、虚血性心疾患(狭心症、心筋梗塞)、下肢虚血性障害などを挙げることができる。また、これらの虚血性疾患の進展によって生じる病態として、慢性腎不全、慢性心不全、下肢壊疽、などを挙げることができる。
【0073】
本発明の医薬組成物は、通常、PHD阻害に有効量の、ひいては低酸素状態の改善に有効量の本発明化合物に加えて、薬学的に許容される担体または添加剤を配合して調製される。医薬組成物中の本発明化合物の配合量は、対象とする疾患や病態の種類や投与形態に応じて適宜選択されるが、通常、全身投与製剤の場合には、医薬組成物の全体重量(100重量%)の0.001〜50重量%、特に0.01〜10重量%とすることができる。
【0074】
本発明の医薬組成物の投与方法として、経口投与、ならびに静脈内投与、筋肉内投与、皮下投与、経粘膜投与、経皮投与、および直腸内投与等の非経口投与を挙げることができる。好ましくは経口投与および静脈内投与であり、より好ましくは経口投与である。本発明の医薬組成物は、かかる投与方法に応じて、種々の形態の製剤(剤型)に調製することができる。以下に、各製剤(剤型)について説明するが、本発明において用いられる剤型はこれらに限定されるものではなく、医薬製剤分野において通常用いられる各種剤型を用いることができる。
【0075】
経口投与を行う場合の剤型として、散剤、顆粒剤、カプセル剤、丸剤、錠剤、エリキシル剤、懸濁剤、乳剤およびシロップ剤を挙げることができ、これらの中から適宜選択することができる。また、それらの製剤について徐放化、安定化、易崩壊化、難崩壊化、腸溶性化、易吸収化等の修飾を施すことができる。
【0076】
また、静脈内投与、筋肉内投与、または皮下投与を行う場合の剤型として、注射剤または点滴剤(用時調製の乾燥品を含む)等があり、適宜選択することができる。
【0077】
また、経粘膜投与、経皮投与、または直腸内投与を行う場合の剤型として、咀嚼剤、舌下剤、パッカル剤、トローチ剤、軟膏剤、貼布剤、液剤等があり、適応場所に応じて適宜選択するここができる。また、それらの製剤について徐放化、安定化、易崩壊化、難崩壊化、易吸収化等の修飾を施すことができる。
【0078】
本発明の医薬組成物にはその剤形(経口投与または各種の非経口投与の剤形)に応じて、薬学的に許容される担体および添加剤を配合することができる。薬学的に許容される担体及び添加剤としては、溶剤、賦形剤、コーティング剤、基剤、結合剤、滑沢剤、崩壊剤、溶解補助剤、懸濁化剤、粘稠剤、乳化剤、安定剤、緩衝剤、等張化剤、無痛化剤、保存剤、矯味剤、芳香剤、着色剤が挙げられる。以下に、医薬上許容される担体および添加剤の具体例を列挙するが、本発明はこれらに制限されるものではない。
【0079】
溶剤としては、精製水、滅菌精製水、注射用水、生理食塩液、ラッカセイ油、エタノール、グリセリン等を挙げることができる。賦形剤としては、デンプン類(例えばバレイショデンプン、コムギデンプン、トウモロコシデンプン)、乳糖、ブドウ糖、白糖、結晶セルロース、硫酸カルシウム、炭酸カルシウム、炭酸水素ナトリウム、塩化ナトリウム、タルク、酸化チタン、トレハロース、キシリトール等を挙げることができる。
【0080】
結合剤としては、デンプンおよびその誘導体、セルロースおよびその誘導体(たとえばメチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロース)、ゼラチン、アルギン酸ナトリウム、トラガント、アラビアゴム等の天然高分子化合物、ポリビニルピロリドン、ポリビニルアルコール等の合成高分子化合物、デキストリン、ヒドロキシプロピルスターチ等を挙げることができる。
【0081】
滑沢剤としては、軽質無水ケイ酸、ステアリン酸およびその塩類(たとえばステアリン酸マグネシウム)、タルク、ワックス類、コムギデンブン、マクロゴール、水素添加植物油、ショ糖脂肪酸エステル、ポリエチレングリコール、シリコン油等を挙げることができる。
【0082】
崩壊剤としては、デンプンおよびその誘導体、寒天、ゼラチン末、炭酸水素ナトリウム、炭酸カルシウム、セルロースおよびその誘導体、ヒドロキシプロピルスターチ、カルボキシメチルセルロースおよびその塩類ならびにその架橋体、低置換型ヒドロキシプロピルセルロース等を挙げることができる。
【0083】
溶解補助剤としては、シクロデキストリン、エタノール、プロピレングリコール、ポリエチレングリコール等を挙げることができる。懸濁化剤としては、カルボキシメチルセルロースナトリウム、ポリピニルピロリドン、アラビアゴム、トラガント、アルギン酸ナトリウム、モノステアリン酸アルミニウム、クエン酸、各種界面活性剤等を挙げることができる。
【0084】
粘稠剤としては、カルボキシメチルセルロースナトリウム、ポリピニルピロリドン、メチルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、トラガント、アラビアゴム、アルギン酸ナトリウム等を挙げることができる。
【0085】
乳化剤は、アラビアゴム、コレステロール、トラガント、メチルセルロース、レシチン、各種界面活性剤(たとえば、ステアリン酸ポリオキシル40、セスキオレイン酸ソルビタン、ポリソルベート80、ラウリル硫酸ナトリウム)等を挙げることができる。
【0086】
安定剤としては、トコフェロール、キレート剤(たとえばEDTA、チオグリコール酸)、不活性ガス(たとえば窒素、二酸化炭素)、還元性物質(たとえば亜硫酸水素ナトリウム、チオ硫酸ナトリウム、アスコルビン酸、ロンガリット)等を挙げることができる。
【0087】
緩衝剤としては、リン酸水素ナトリウム、酢酸ナトリウム、クエン酸ナトリウム、ホウ酸等を挙げることができる。
【0088】
等張化剤としては、塩化ナトリウム、ブドウ糖等を挙げることができる。無痛化剤こしては、局所麻酔剤(塩酸プロカイン、リドカイン)、ペンジルアルコール、ブドウ糖、ソルビトール、アミノ酸等を挙げることができる。
【0089】
矯味剤としては、白糖、サッカリン、カンゾウエキス、ソルビトール、キシリトール、グリセリン等を挙げることができる。芳香剤としては、トウヒチンキ、ローズ油等を挙げることができる。着色剤としては、水溶性食用色素、レーキ色素等を挙げることができる。
【0090】
保存剤としては、安息香酸およびその塩類、パラオキシ安息香酸エステル類、クロロブタノール、逆性石けん、ベンジルアルコール、フェノール、チロメサール、デヒドロ酢酸、ホウ酸、等を挙げることができる。
【0091】
コーティング剤としては、白糖、ヒドロキシプロピルセルロース(HPC)、セラック、ゼラチン、グリセリン、ソルビトール、ヒドロキシプロピルメチルセルロース(HPMC)、エチルセルロース、ポリビニルピロリドン(PVP)、ヒドロキシプロピルメチルセルロースフタレート(HPMCP)、セルロースアセテートフタレート(CAP)、メチルメタアクリレート−メタアクリル酸共重合体および上記記載した高分子等を挙げることができる。
【0092】
基剤としては、ワセリン、流動パラフィン、カルナウバロウ、牛脂、硬化油、パラフィン、ミツロウ、植物油、マクロゴール、マクロゴール脂肪酸エステル、ステアリン酸、カルボキシメチルセルロースナトリウム、ベントナイト、カカオ脂、ウイテップゾール、ゼラチン、ステアリルアルコール、加水ラノリン、セタノール、軽質流動パラフィン、親水ワセリン、単軟膏、白色軟膏、親水軟膏、マクロゴール軟膏、ハードファット、水中油型乳剤性基剤、油中水型乳剤性碁剤等を挙げることができる。
【0093】
なお、上記の各剤型について、公知のドラッグデリバリーシステム(DDS)の技術を採用することができる。本明細書にいうDDS製剤とは、徐放化製剤、局所適用製剤(トローチ、バッカル錠、舌下錠等)、薬物放出制御製剤、腸溶性製剤および胃溶性製剤等、投与経路、バイオアベイラビリティー、副作用等を勘案した上で、最適の製剤形態にした製剤である。
【0094】
本発明の医薬組成物を、低酸素状態に関連する疾患や病態に対する予防薬または治療薬として用いる場合、その経口投与量として、本発明化合物の量に換算して0.03〜300mg/kgの範囲が好ましく、より好ましくは0.1〜50mg/kgである。静脈内投与をする場合、本発明化合物の有効血中濃度が0.2〜50μg/mL、より好ましくは0.5〜20μg/mLの範囲となるような投与量を挙げることができる。
【0095】
なお、これらの投与量は、年齢、性別、体型等により変動し得る。
【0096】
(III)スクリーニング方法
本発明は被験物質の中から、細胞や組織の低酸素状態の改善に有効に利用できるプロリル水酸化酵素阻害剤(PHD阻害剤)を選別する方法を提供する。本発明の方法で探索取得されるPHD阻害剤は、HIFにPHDが結合することを阻害して、HIFのαプロリンが水酸化されて分解することを防止する作用を有しており、その結果、HIF−HREシステムを活性化して細胞や組織の低酸素状態を改善することができる。このため当該物質によれば、細胞や組織の低酸素状態に関連して生じる疾患や病態(障害)を予防または治療することができると期待される。従って、本発明の方法は、細胞や組織の低酸素状態に関連して生じる疾患や病態(障害)の予防または治療するための医薬組成物の有効成分をスクリーニングする方法であるともいえる。
【0097】
本発明のスクリーニング方法は、基本的にはプロリル水酸化酵素(PHD)と低酸素誘導因子(HIF)のα鎖プロリンとの競合的結合を阻害する作用を有する物質を探索することからなるが、具体的には下記の(a)〜(c)の工程を有する方法を例示することができる:
(a) 被験物質の存在下で、PHDとHIFα鎖プロリンとを接触させる工程、
(b) PHDとHIFα鎖プロリンとの結合を測定する工程、および
(c) PHDとHIFα鎖プロリンとの結合を阻害する被験物質をPHD阻害剤として選択する工程。
【0098】
かかる工程により、PHDとHIFのα鎖プロリンとの競合的結合を阻害して、HIFのα鎖プロリンを水酸化するPHDの働きを抑制する物質を取得することができる。
【0099】
被験物質としては、制限はされないが、核酸、ペプチド、タンパク質、有機化合物、または無機化合物などであり、スクリーニングは、具体的には、これらの被験物質またはこれらを含む組成物(例えば、細胞抽出物、遺伝子ライブラリーの発現産物等を含む)を、対象のc-SrcおよびADAPと接触させることにより行うことができる。
【0100】
スクリーニングに際して採用される被験物質とPHDとHIFα鎖プロリンとの接触条件は、特に制限されないが、通常、生理的環境下におけるin vitro実験系、または細胞内で行うことが好ましい。
【0101】
本発明において用いるPHDは、その由来を特に制限されず、ヒト由来のもののほか、ヒト以外の哺乳動物を含む脊椎動物(例えば、マウス、ラット、ニワトリなど)または各種微生物(ストレプトマイシス)に由来するPHDを用いることができる。PHDは全長のタンパク質であってもよいが、HIFαプロリンとの結合に関わるドメインを含む部分断片であってもよい。
【0102】
本発明において用いるHIFα鎖も、その由来を特に制限されない。ヒト由来のもののほか、ヒト以外の哺乳動物を含む脊椎動物(例えば、マウス、ラットやニワトリなど)に由来するHIFを用いることができるが、一緒に使用するPHDと同一の動物に由来するものを使用することが好ましい。HIFα鎖は全長のタンパク質であってもよいが、PHDとの結合に関わるα鎖の564位に位置するプロリン残基を含む部分断片であってもよい。具体的には、HIFα鎖の557〜576または531〜652の領域を有する部分断片を用いることもできる。
【0103】
PHDとHIFα鎖との結合の測定は、PHDとHIFα鎖との結合の有無が評価できる方法であれば特に制限されない。例えば、被験物質の存在下でPHD(またはその部分断片)とHIFα鎖(又はプロリン残基を含む部分断片)とを接触させた後、その反応液(混合液)を電気泳動にかけて分子量から結合の有無を評価する方法、PHD(又はPHDの部分断片)あるいはHIFα鎖(又はプロリン残基を含む部分断片)の抗体を用いて他のタンパク質の共沈の有無を評価する方法などを例示することができる。
【0104】
かかるスクリーニング方法によるPHD阻害剤(候補物質)の選別は、cPHD(またはその部分断片)とHIFα鎖(又はプロリン残基を含む部分断片)との結合の有無を評価することによって行うことができる。この場合、被験物質の非存在下でPHD(またはその部分断片)とHIFα鎖(又はプロリン残基を含む部分断片)とを接触させて生じるPHDとHIFα鎖との結合(コントロール)と対比することによって評価することもできる。PHDとHIFα鎖とが全く結合しない場合、およびコントロールに比して結合が低減した場合に、使用された被験物質を、PHD阻害剤の候補物質として選別することができる。
【0105】
上記スクリーニングで選別される物質は、HIFのα鎖プロリンを水酸化するPHDの働きを阻害することによって、HIFを安定化する作用を有するものであり、HIF−HREシステムの活性化を介して低酸素状態を改善し、低酸素状態に関連して生じる疾患やそれから進展する病態(障害)を予防または治療する医薬組成物の有効成分として使用することが可能である。
【0106】
なお、低酸素状態に関連して生じる疾患としては、例えば各種の虚血性疾患ならびに虚血から生じる障害、具体的には、腎虚血性障害(尿細管間質性障害)、虚血性脳血管障害(脳塞栓症、脳梗塞、一過性脳虚血発作等)、虚血性心疾患(狭心症、心筋梗塞)、下肢虚血性障害などを挙げることができる。また、これらの虚血性疾患の進展によって生じる病態として、慢性腎不全、慢性心不全、下肢壊疽、などを挙げることができる。
【0107】
上記のスクリーニング方法によって選別された候補物質は、さらにその低酸素状態を改善する一つの作用として血管新生作用を評価するために、スポンジ血管新生分析や、低酸素状態にあるモデル非ヒト動物を用いてスクリーニングをかけることもできる。かくして選別される候補物質は、さらに虚血性疾患を有する病態非ヒト動物を用いた薬効試験、安全性試験、さらに虚血性疾患などの低酸素状態にある患者(ヒト)もしくはその前状態にある患者(ヒト)への臨床試験に供してもよく、これらの試験を実施することによって、より実用的な低酸素状態に関連する疾患の予防または治療用医薬組成物の有効成分を選別取得することができる。
【0108】
このようにして選別された物質は、必要に応じて構造解析を行った後、その物質の種類に応じて、化学的合成、生物学的合成(発酵を含む)または遺伝子工学的操作によって、工業的に製造することができ、低酸素状態に関連する疾患の予防・治療用医薬組成物の調製に使用することができる。
【0109】
上記のスクリーニング方法は、前述するように、その基本はPHDとHIFαプロリンとの結合を阻害する作用を有する物質を探索することからなる。従って、上記のスクリーニング方法は、別の角度から、PHDとHIFαプロリンとの結合を阻害する作用を有する物質(PHD/HIF結合阻害剤)をスクリーニングする方法と規定することができる。
【0110】
斯くして得られるプロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物には、下式(A)または(B)で示され骨格を有する基の少なくとも1つを一分子中に含む化合物が含まれる。
【0111】
【化23】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
好ましくは、本発明化合物、特に化合物1〜3を含む化合物(I)ならびに化合物4を含む化合物(II)が含まれる。
【実験例】
【0112】
以下、本発明を製造例、ならびに実験例によりさらに具体的に説明するが、本発明はこれらの例に制限されるものではない。
【0113】
製造例1 TM-6101の合成
下式に示すように、6-アミノ-1-メチルウラシル(化合物(a))を出発原料として、目的とするTM-6101(化合物4)(分子量:378.41)を製造した。
【0114】
【化24】
(1)工程1
アルゴン雰囲気下、6-アミノ-1-メチルウラシル(化合物(a))3.53g(25.0mmol)とN,N’-ジメチルホルムアミドジメチルアセチル11.9g(0.10mmol)の脱水DMF懸濁溶液(125mL)を、40℃にて3時間攪拌した。炭酸カリウム5.18g(37.5mmol)とエチル-3-ブロモプロピネート6.79g(37.5mmol)を加え、80℃にて3時間攪拌した。減圧下濃縮し無機化合物を濾過した。酢酸エチル洗浄濾液を、減圧下濃縮し、スラリー状でヘキサンを加えて析出結晶を濾過し、化合物(b)を3.00g取得した(収率40%)。
【0115】
(2)工程2
化合物(b)(2.50g、8.44mmol)の脱水メタノール溶液(127mL)に塩化亜鉛4.89g(35.9mmol)を加え、アルゴン雰囲気下で5日間還流した。冷却後水を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下で濃縮した。濃縮残渣を塩化メチレンで洗浄し、化合物(c)を0.93g取得した(収率48%)。
【0116】
(3)工程3
アルゴン雰囲気下、化合物(c)(0.69g、3.04mmol)の脱水ジオキサン溶液(15mL)にピリジン0.36g(4.55mmol)とクロロアセチルクロライド0.51g(4.52mmol)を加え、室温で30分間攪拌した。原料の消失を確認した後、これを水に注ぎ、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下で濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィーで精製し(溶離液:メタノール/塩化メチレン=1/9)、無色結晶の化合物(d)を0.88g取得した(収率95%)。
【0117】
(4)工程4
化合物(d)(0.80g、2.63mmol)のトルエン溶液(80mL)に、2-メルカプトピリジン0.44g(3.95mmol)とトリエチルアミン0.53g(5.27mmol)を加え、室温で24時間攪拌した。その後、40℃にて7時間、室温で19時間攪拌した後、トリエチルアミン0.53g(5.27mmol)を加え、さらに室温で16時間攪拌した。水を加え酢酸エチルで熱抽出した。有機層を減圧下濃縮し、濃縮残渣をシリカゲルカラムクロマトグラフィーで精製した後(溶離液:酢酸エチル)、再度作戦エチルで洗浄し、無色〜淡黄色結晶の化合物4(TM-6101)を0.60g取得した(収率60%)。
【0118】
TM-6101(化合物4)のNMRデータは下記の通りである:
1H NMR (CDCl3) δ=2.68 (2H, t, J=9 Hz), 3.46 (3H, s), 3.70 (3H, s), 4.29 (2H, t, J=9 Hz), 4.81 (2H, s), 6.93-6.97 (1H, m), 7.26-7.29 (1H, m) ,7.44-7.50 (1H, m), 8.37-8.40 (1H, m)。
【0119】
なお、化合物4をTHF:水の1対1溶液中で1規定のNaOHを用いて、NaOHとして化合物4の等モル〜3倍モルを加え、室温で撹拌処理することによりそのカルボン酸である化合物(e)を調製することができる。
【0120】
実験例1
(1)Prolyl-3-hydroxylase (PHD3)の三次元構造の分析
Prolyl-3-hydroxylase(PHD3)は、Fe(II)の存在下で2-oxyglutarate(2-OG)とHIFプロリンからcis-3-hydroxyprolineを産生する酵素である。そのX線結晶構造はストレプトマイシス(Streptomyces sp.)由来のPHD3について既に報告されている(Clifton, I.J. et al. Structure of proline 3-hydroxylase. Evolution of the family of 2-oxoglutarate dependent oxygenases. Eur. J. Biochem. 268. 6625-6636 (2001))。しかし、PHD3分子内の、2-OGおよびHIFプロリンの結合部位は、未だ不明である。そこで、これらの結合部位を決定するために下記の実験を行った。
【0121】
ストレプトマイシスに由来するPHD3のX線結晶構造はプロテインデータバンクから取得した(PDBコード: 1E5RS)(Bernstein, F.C. et al. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112. 535-542 (1977))。水分子および硫酸イオンを除去した後、水素原子を、タンパク質中の酸性および塩基性残基の標準プロトン化状態に合わせて添加し、それらの位置を最適化した。なお研究には、ソフトウェア・システムMOE(Molecular Operating Environment, version 2004.04)とMMFF94s力場(Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem. 17. 490-519 (1996))を採用した。
【0122】
結合部位は、MOEのalpha site finder function (Edelsbrunner, H., Facello, M., Fu, R., & Liang, J. Measuring Proteins and Voids in Proteins. Proceedings of the 28th Hawaii International Conference on Systems Science, 256-264 (1995))を用いて決定した。小分子と標的部位のドッキングはプログラムPh4Dock(Goto, J., Kataoka, R. & Hirayama, N. Ph4Dock-Pharmacophore-based protein-ligand docking. J. Med. Chem. 47. 6804-6811 (2004))により行った。ドッキングの最終段階で、PHD3分子内の水素原子と小分子内の全原子の位置を最適化した。得られたドッキング結果を、下記の相互作用エネルギーにより判断した。
Utotal = Uele + Uvdw+ Uligand
ここで、UeleとUvdwは、PHD3と小分子間の、静電気的な相互作用とワンデルワールスの相互作用をそれぞれ意味する。また、Uligandは小分子の構成エネルギーを意味する。
【0123】
上記ドッキングシミュレーションから、PHD3分子内に比較的大きなポケットがあり、その中にFe(II) が存在していることがわかった(図1a参照)。図1a中の球体(青)はFe(II)を示す。このFe(II)の周囲に2-OGとHIFのプロリン残基の結合部位があると思われる。図1aにおいて、2-OGとHIFのプロリン残基を枝状モデル(緑)で示すが、2-OGとHIFプロリンは、Fe(II)に対して互いに反対の部位に結合する〔図1a中、Fe(II)の上部(B部位)にHIFプロリンが結合、下部(A部位)に2-OGが結合〕。なお、2-OGはカルボキシル基を介してFe(II)と結合している。
【0124】
(2)既知のPHD抑制剤の分子ドッキング
PHD3分子内のFe(II)周囲のポケット内の結合部位(A部位:2-OG結合部位、B部位:HIFのプロリン残基結合部位)を標的にして、既知のPHD抑制剤を用いて、ドッキングシミュレーションを行った。
【0125】
既知のPHD抑制剤として、下式に示す3,4-dehydroxybenzoate(以下、「3,4-DHB」という。Warnecke, C. et al. Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB. J. 17. 1186-1188 (2003))、3-hydroxychinolin 2-carbonic acidの誘導体である6-chloro-3-hydroxychinoline-2-carbonic acid-N-(carboxymethyl)-amide (以下「S956711」という。Warnecke, C. et al. Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB. J. 17. 1186-1188 (2003))、および4-Oxo-1,4-dihydro-[1,10]phenanthroline-3-carboxylic acid (以下、「FG-0041」という。Ivan, M. et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. 99. 13459-13464 (2002))を用いた。
【0126】
【化25】
その結果、3,4-DHBは、そのカルボキシル基を介して、PHD3分子内のFe(II)周囲のA部位(2-OG結合部位)と結合することがわかった(図1b)。S956711は、比較的分子構造が大きくまた環構造を有することから、A部位とB部位の両方(2-OG結合部位とHIFのプロリン残基結合部位)に結合すると考えられた。さらにFG-0041は、B部位(HIFのプロリン残基結合部位)に結合することが明らかになった。
【0127】
なお、これらの3つのPHD抑制剤はいずれも、A部位または/およびB部位に結合する能力の他に、鉄イオンとキレートを形成するモチーフを有している。しかしながら、鉄は、酸化的リン酸化とアラキドン酸のシグナリングに関係して、多くの重要な細胞機能を発揮するのに必要なコファクターであるので、そのキレート化剤は長期的治療用のPHD抑制剤として適切ではないと考えられる。このため、鉄のキレート化とは無関係に作用するPHD抑制剤を探索した。PHD3分子内のA部位(2-OG結合部位)は、Arg168およびHis135といった嵩高い残基で囲まれているため、PHD抑制剤がアクセスしにくい可能性がある。このため、PHD抑制剤として、HIFのプロリン残基と競合してB部位に結合する分子を探索することにした。
【0128】
(3)新規なHIF安定化化合物の同定
上記コンセプトの下で、PHD3分子内のB部位に結合するFG-0041の誘導体を19つ購入し、HIFの安定化を調べた。19つの誘導体のうち12つは、[2、2]bipyridinyl骨格を、残りの7つは、他の部位(4-oxo-1、4-dihydro-quinoline-3-carboxylic acid)を有している。これら19つの誘導体のHIF安定化によるルシフェラーゼ産生促進活性を、後述するin vitroスクリーニングアッセイにより測定した。その際、HIF-1α(HIFのα鎖)を安定化することによって低酸素状態をもたらす公知の化合物であるコバルト(塩化コバルト)を、陽性コントロールとして使用した(Wang, G.L. & Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 20. 1230-1237 (1995))。
【0129】
<in vitroスクリーニングアッセイ>
不死化した腎近位尿細管 (IRPTC)細胞に、HREを7回タンデムに反復して有するプロモーターの下流にルシフェラーゼ遺伝子を有するリポーター・ベクターを導入し、これを上記19つの誘導体(被験化合物)のHIF安定化によるルシフェラーゼ産生促進活性の測定に使用した。具体的には、上記リポーター・ベクターを導入したIRPTC細胞を、被験化合物(最終濃度:6.7〜100μM)とともに、37℃で24時間インキュベーションした後溶解し、細胞溶解物中のルシフェラーゼ活性を、Lumat 9507照度計(EG and Berthold、ドイツ)を用いて測定した。なお、被験化合物の非存在下で、リポーター・ベクターを導入したIRPTC細胞を同様にインキュベーションした細胞溶解物中のルシフェラーゼ活性をコントロール(陰性コントロール)とした。
【0130】
陰性コントロールのルシフェラーゼ活性を1とし、これに対する各被験化合物のルシフェラーゼ活性の相対比を、各被験化合物のHIF安定化によるルシフェラーゼ産生促進活性として評価した。結果を図2に示す。図2に示すように、[2、2]bipyridinyl骨格を有する誘導体の中で、下式に示す2つの化合物:6-amino-1,3-dimethyl-5-(2-pyridin-2-yl-quinoline-4-carbonyl)-1H-pyridine-2,4-dione(TM-6008)(分子量:387.40)
【0131】
【化26】
および3,7-Dimethy-furo[3,2-b;4,5-b]dipyridine(TM-6010) (分子量:198.23)
【0132】
【化27】
が、HIF安定化によるルシフェラーゼ産生促進活性を示した。
【0133】
これらTM-6008およびTM-6010はいずれも、鉄のキレートモチーフとなる[2、2]bipyridinylを有している。このため、TM6008からこのモチーフを除去し、5-acetyl-6-amino-1,3-di-methyl-1H-pyrimidine-2,4-dione構造を有する18つの化合物をライブラリーから取得した。
【0134】
それら化合物のうちの1つ、6-amino-1,3-di-methyl-5-[2-(pyridin-2-ylsulfanyl)-acetyl]-1H-pyrimidine-2,4-dione(TM-6089) (分子量:306.35)
【0135】
【化28】
は、鉄のキレート化モチーフ部([2、2]bipyridinyl)を欠如しているにもかかわらず、強いHIF安定化によるルシフェラーゼ産生促進活性を示した(図2)。
【0136】
これらのTM-6008、TM-6010およびTM-6089のドッッキング構造を図1c示す(図1c中、黄はTM-6008、緑はTM-6010および赤はTM-6089をそれぞれ示す)。これから、これらの化合物はいずれも、PHD3分子内のB部位に結合することがわかる。
【0137】
(4)TM6101のHIF安定化によるルシフェラーゼ産生促進活性
製造例1で合成した、TM-6089のカルボン酸誘導体TM6101(化合物5)について、上記(3)と同様にしてin vitroスクリーニングアッセイによりルシフェラーゼ活性を測定し、HIF安定化によるルシフェラーゼ産生促進活性を評価した。陽性コントロールおよび比較のため、それぞれ塩化コバルトおよびTM6008を被験化合物として用いて同様にルシフェラーゼ活性を測定し、HIF安定化によるルシフェラーゼ産生促進活性を評価した。結果を図3に示す。図3は、図2と同様に陰性コントロールのルシフェラーゼ活性を1とし、これに対する各被験化合物(塩化コバルト、TM6008、TM6101)のルシフェラーゼ活性の相対比を示す。その結果、TM6101は、公知のPHD抑制剤である塩化コバルトよりも低いものの、TM6008よりも高いHIF安定化によるルシフェラーゼ産生促進活性を示した。
【0138】
実験例2 金属キレート活性
前述するように、化学構造から、3,4-DHB、S965711、FG-0041、TM-6008およびTM-6010中に鉄イオンとキレートするモチーフが存在することが示唆された。そこで、アスコルビン酸の銅触媒酸化を用いたin vitroアッセイ(遷移金属キレート化の評価系アッセイ)で、これを確認した。
【0139】
具体的には、上記実験例1でHIF促進作用が認められた化合物(TM-6008、TM-6010およびTM-6089)および公知のPHD抑制剤である3,4-DHBおよびS965711について、遷移金属イオンに対するキレート活性を、Priceらの方法(Price DL, et al., “Chelating activity of advanced glycation end-product (AGE) inhibitors.” J Biol Chem 272: 5430-5437, 1997)に幾つか修正を加えて測定した。簡単に説明すると、50μM CuCl2 15μlと様々な濃度の被験化合物30μlを、1.38mlのリン酸緩衝液中で、30℃、5分間インキュベーションした。この反応は、10mMアスコルビン酸(CuCl2およびアスコルビン酸の最終濃度:それぞれ500 nMおよび500μM)を75μl添加することで開始した。30℃で0〜60分間インキュベーションを行った。反応液のアスコルビン酸量を、逆相高速液体クロマトグラフィー(HPLC)を用いて244nmでの吸光度検出により測定した。
【0140】
その結果、3,4-DHB、S965711、TM-6008およびTM-6010は遷移金属(銅)にキレートし、用量依存的にアスコルビン酸の自動酸化を阻害した(IC50はそれぞれ330μM、31.4μM、0.57μMおよび4.84μM)。一方、TM-6089は100μMの濃度でさえ遷移金属(銅)とキレートしなかった。 このことから、TM-6008およびTM-6010は確かに金属とキレートすることが判明した。一方、TM-6089には、金属キレート能がなく、同様にTM-6089と構造的に類似するTM6101も金属キレート能がないと思われる。
【0141】
実験例3 PHD阻害活性
上記実験例1でHIF促進作用が認められた化合物(TM-6008、TM-6010およびTM-6089、並びに陽性コントロールとして塩化コバルト)について、PHD阻害活性を調べた。
【0142】
PHD阻害活性は、IRPTC細胞(腎近位尿細管細胞)のPHDを豊富に含むミトコンドリア画分のホモジネートを用いて、Kauleらの方法(Cunhild Kaule and Volkmar Gunzler, “Assay for 2-Oxoglutarate Decarboxylating Enzymes Based on the Determination of [1-14C] Succinate: Application to Prolyl 4- Hydroxylase”,Analytical Biochemistry 184, 291 - 297, (1990))
に従って測定した。具体的には、まず0.15mM MgCl2と10mM KClを含む10mMトリス-HCl(pH 6.7)中で懸濁したIRPTC細胞(腎近位尿細管細胞)を、氷上で、デキストロース(最終濃度:0.25M)とともにホモジナイズし、4℃で15分間1500xgで遠心分離して、核画分を除去した。引き続き上清を4℃で10分間20000xgで遠心分離して、ミトコンドリア画分を取得した(30μg)。ミトコンドリア画分は様々な濃度の被験化合物(TM-6008、TM-6010、TM-6089および塩化コバルト)と37℃で30分間反応させ、さらに2.7mMトリス-HCl(pH 7.5)、3.3μM FeSO4、6.7μM [5-14C]-2-OG(アマシャム)、66μMアスコビル酸塩、27μg/mlカタラーゼ、および33μM ジチオスレイトールを含む緩衝液に溶解した13.3μMの酸素依存性分解ドメイン(ODD)ペプチド(HIF-1αのアミノ酸557-576領域)と、37℃で1時間反応させた。1時間インキュベーションした後、3.3mMコハク酸塩、3.3mM 2-OGおよび27mM 2,4-ジニトロフェニルヒドラジンを添加した。 混合液を室温で30分間放置して、更に、0.3Mの2-OGを添加した。7000xgで5分間遠心分離して上清を分離し、液体シンチレーションカウンタによって[1-14C]コハク酸塩の放射能を測定した。なお、コントロール(陰性コントロール)として、被験化合物なしで上記反応を行い、同様にして[1-14C]コハク酸塩の放射能を測定し、これをヒドロキシラーゼ活性100%とした。
【0143】
結果を図4に示す。図4で示す各被験化合物のヒドロキシラーゼ活性は、陰性コントロールから得られる[1-14C]コハク酸塩の放射能(ヒドロキシラーゼ活性100%)に対する相対比である。
【0144】
図4に示すように、被験化合物(TM-6008、TM-6010およびTM-6089、並びに陽性コントロールとして塩化コバルト)はすべて用量依存的にPHD活性を阻害した。TM-6008とTM-6089は、公知のPHD阻害剤である塩化コバルトよりもPHD阻害活性が高く、中でもTM-6008が最もPHD阻害活性が高かった。
【0145】
実験例4 HIF-1α(α鎖)ペプチドとPHDとのin vitro結合アッセイ
実験例1のドッキングシミュレーションにより、3つの化合物(TM-6008、TM-6010およびTM-6089)がHIFのプロリン残基と競合して、PHD3分子内のB部位に結合することがわかった。これを確認するために、上記化合物(TM-6008、TM-6010およびTM-6089)ならびに公知のPHD抑制剤である3,4-DHBおよびS965711を用いて、下記の方法に従ってHIF-1αペプチドとPHDのin vitro結合アッセイを行なった。
【0146】
<in vitro結合アッセイ>
PHD3を、発現プラスミドpBOS-FLAG-mEGLN3からin vitroで転写し、網状赤血球溶血液TnTクイックキット(Promega社製)を使用して、[35S]メチオニン(Amersham社製)の存在下で形質転換した。 pGEX4T-1ベクター中でクローン化されたGST-HIF-1(531-652)を大腸菌(DH5α)内で発現させ、GST結合ビーズ(Glutathion Sepharose 4B: Amersham社製)によりアフィニティー精製を行った。35Sで標識したPHD3を、100mM KCl、10%のグリセリン、1.4mMのβ-メルカプトエタノール、100mMのFeCl2、0.2%のNP-40および0.5mg/mlのウシ血清アルブミンを含む20mMのトリス-HCl(pH 8.0)の50μl中で、被験化合物(最終濃度:50および500μM)と15分間、あらかじめインキュベーションした。次いで、その混合液に、GST-HIF-1α(531-652)結合ビーズ(12.5μg)を添加し、4℃で2時間インキュベーションした。洗浄後、GST-HIF-1α(531-652)に結合した35S標識PHD3を、120mM SDS、12%のグリセリン、240mM β-メルカプトエタノールおよび230μMブロモフェノール・ブルーを含む120mMトリス-HCl(pH 6.8)で溶出した。その溶出液を、SDS-PAGEにかけ、オートラジオグラフィーに供した。
【0147】
GST-HIF-1α(531-652)に結合する35S標識PHD3に対する被験化合物の阻害作用を、プルダウンアッセイにより評価した。被験化合物非存在下で、上記結合アッセイを行った結果(結合活性)を100%とし(コントロール)、これに対する各被験化合物の結合活性を%として示した。
【0148】
結果を図5に示す。公知のPHD抑制剤であるS965711と同様に、3つの新規なPHD抑制剤(TM-6008、TM-6010およびTM-6089)はいずれも、HIFペプチドと競合し、GST-HIF-1α(531-652)との結合を阻害した(図5)。一方、公知のPHD抑制剤である3,4-DHBは、HIFペプチドと競合せず、GST-HIF-1α(531-652)との結合を阻害しなかった(図5)。また過剰量の2-OGを添加しても、この結果は変わらなかった。
【0149】
実験例5 血管新生促進作用のin vivo試験
(1)スポンジ血管新生分析
上記被験化合物(TM-6008、TM-6010およびTM-6089)の局所投与によって血管新生が促進されるかどうかをin vivo試験した。この目的のために、マウスの皮下に小さなスポンジ・ディスクを挿入し、このスポンジを介して被験化合物を皮下投与して、10日後にヘモグロビン量と血管数を測定し、HIF-HREシステムの促進を評価した(スポンジ血管新生分析)。
【0150】
スポンジ血管新生分析は、村松らの文献の記載に従って行った(Muramatsu, M., Katada, J., Hayashi, I. & Majima, M. Chymase as a proangiogenic factor. A possible involvement of chymase-angiotensin-dependent pathway in the hamster sponge angiogenesis model. J. Biol. Chem. 275. 5545-5552 (2000))。具体的には、円形のスポンジ・ディスク(厚さ5mmおよび直径12mm)を、エーテル麻酔した6週齢の雄ICRマウス(n=2)の背部の皮下空気嚢に移植した。移植5日後に、DMSOに溶解し生理食塩水で希釈した被験化合物(1マウスあたり、50μg のTM-6008、TM-6010またはTM-6089、あるいは30μgのコバルト)を、1週間に亘って一日おき4回、スポンジに皮下投与した。bFGF(10μg)(ジェンザイム製)を陽性コントロールとして用いた。また、DMSOを生理食塩水で希釈した液(図中「vehicle」と記載する)のみを投与したものを陰性コントロールとして使用した。
【0151】
マウスは、実験後に殺して、ただちにスポンジ移植片とともに肉芽腫組織を切除した。キット(ヘモグロビンBテスト: 和光純薬製)を用いて採取した組織中のヘモグロビン量を測定し、また抗マウスCD31抗体(Pharmingen社製)を用いて、組織中の血管数(CD31陽性血管数)を免疫組織学的手法により定量測定した。なお、血管数はマウス一匹あたり任意に選択された重複しない5箇所の400×field内で測定した。
【0152】
ヘモグロビン量を図6aに、血管数に対応する相対蛍光強度を図6bに示す。また図6cに内皮細胞の免疫染色画像を示す。図6aの結果からわかるように、TM-6008、TM-6010およびTM-6089の投与によって、bFGFと同様にヘモグロビン量が増加することが認められた。また図6cから、TM-6008、TM-6010およびTM-6089の投与によって血管数も増加し、血管新生が認められた。特に、TM-6008の投与によって顕著な血管新生が認められた(図5b、c)。
【0153】
(2)HIF-HREシステムの活性化
被験化合物(TM-6008、TM-6089)によって、様々な器官においてHIF-HREシステムが促進されるかどうかを、HREルシフェラーゼを発現する低酸素症の遺伝子組み換えマウス(Tanaka, T. et al. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am. J. Pathol. 165. 1979-1992 (2004))を用いて調べた。
【0154】
マウスには、屠殺前の2又は4時間前に、0.5%のカルボキシメチルセルロース・ナトリウム塩の水溶液に懸濁したTM-6008あるいはTM-6089を、100mg/kgの割合で経口投与した(各々n=3)。コントロール用のマウスには、0.5%のカルボキシメチルセルロース溶液だけを経口投与した(n=3)。マウスは殺した後、レポーター遺伝子の発現を調べるために組織を採取した。低酸素症応答ルシフェラーゼ遺伝子の発現は、以前報告した方法に従って半定量的なRT-PCRによって評価した(Tanaka, T. et al. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am. J. Pathol. 165. 1979-1992 (2004))。 プロトコールは、東京大学の動物実験ガイドラインに従って行った。
【0155】
その結果、腎臓におけるレポーター遺伝子の発現は、基本条件下では認められなかったが(40回増幅)、TM-6008またはTM-6089を100mg/kgの割合で単回経口投与することによって認められた(それぞれ31.0±0.85回および31.0±2.05回で検出)。肝臓におけるレポーター遺伝子の発現は、基本条件下では認められなかったが、TM-6008の投与により検知された(32.3±0.35回で検出)。しかしTM-6089の投与では効果がなかった。心臓におけるレポーター遺伝子の発現は、基本条件で検出され、TM-6008またはTM-6089の投与によりその増加が認められた (各々1.37±1.00倍と6.69±5.45倍)。
【0156】
以上のことから、TM-6008はあらゆる器官で、HIF-HREシステムを活性化し、血管ネットワークの形成に重要な役割を果たしていることが示唆された。
【0157】
実験例6 低酸素症による神経細胞死の防止
PHD抑制剤(TM-6008)について、低酸素症による細胞のダメージに対する保護作用を評価した。
【0158】
実験は、アレチネズミ科マウスの遅延神経死モデル(雄、70-80g)を用いて行った。マウスを3つの実験群にランダムに分け、グループ1と2のマウスには、人為的に一過性の虚血を生じさせた。
【0159】
具体的には、グループ1と2のマウスについて、麻酔下で、2本の総頸動脈を小動脈瘤クリップ(杉田クリップ#07-940-12: みずほ医科製)で5分間閉塞し、広範囲に亘って一過性の虚血を引き起こした(Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 239. 57-69 (1982).)。次いでクリップを解除し、表層の微還流をレーザー・ドップラー・フローメトリー(Omega Flow FLO-C1: Neurosci)によって確認した。グループ1のマウス(n=20)には、頚動脈閉塞の後、7日間に亘って0.5%のカルボキシメチルセルロース・ナトリウムに溶解したTM-6008(100mg/kg/日)を強制経口投与した。グループ2のマウス(n=18)には、頚動脈閉塞の後、7日間に亘って0.5%のカルボキシメチルセルロース溶液だけを経口投与した。 グループ3(n=8)には偽物操作し、コントロールとして使用した。
【0160】
全マウスはすべて閉塞後7日目に、ペントバルビタールを注入して殺し、脳を取り出して4%のパラホルムアルデヒドにより冠状脳セクションを固定し、ヘマトキシロンとエオシンで染色した。間接的な免疫組織化学的染色法(in situ アポトーシス検出キットのTdT-Bule label:Trevigen社製)を用いて、TUNEL染色を行った。CA1海馬の神経細胞を光学顕微鏡を用いて観察し、海馬CA1内の虚血によるダメージを評価した。当該評価は、高性能のフィールド(400 x)について予め決めた領域(0.62 mm2)に沿って、海馬中に存在するピラミッド形をした正常の神経数を数えることにより評価した。
【0161】
CA1海馬中の代表的な顕微鏡写真(ヘマトキシロン・エオシン染色)を図7に示す。 非虚血のマウスの神経細胞(図7a)に比べて、虚血状態に曝された後に、カルボキシメチルセルロース(図中「vehicle」と記載する)のみを投与されたマウスは、ピラミッド形の神経の多くに、萎んで暗く染まった細胞質やグリア細胞が濃縮して蓄積しているのが認められ、虚血によって細胞がダメージを受けていることがわかった (図7b)。一方、虚血後、TM-6008で処理したマウスでは、少数の神経だけが虚血状態を示した(図7c)。
【0162】
また核と核小体を示す細胞を生存神経として検出した。CA1海馬中に生存する神経数は、虚血後カルボキシメチルセルロースだけで処理したマウスよりも(61±55個)、TM6008で処理したマウスにおいて有意に多く(166±73個)、非虚血コントロール群中で観察された生存神経数(227±50)と統計的に有意差はなかった(図7d)。
【0163】
これら実験におけるTM-6008の最終血漿濃度は7.8±2.9μg/mlであった。以上の結果から TM-6008は、虚血によって生じる神経死に対して有意に保護作用を示すことが明らかになった。
【0164】
実験例7 毒性
(1)細胞毒性
HeLa細胞を、被験化合物(TM6008、TM6010およびTM6089、50-250μM)の存在下で、ダルベッコのMEM培地中で24時間培養した。細胞毒性は、キット(Promega社製)を使用して培地中の乳酸塩脱水素酵素(LDH)の放出量を測定し、全細胞が溶解した後に放出される全LDH活性に対する割合(パーセンテージ)で示した。TM-6008、TM-6010およびTM-6089は、それぞれ50μM、250μMおよび100μMより低い濃度では、細胞毒性を示さなかった。
【0165】
(2)In vivo急性または亜急性毒性
急性毒性は、30-35gのICRマウス(クレアジャパンから入手)を用いて評価した。TM6008、TM6010およびTM6089を、ICRマウスに単回投与量500、1000、1500および2000mg/kgの割合で強制経口投与した。この急性投与の毒性結果を2週間モニターした。TM-6008については2000mg/kg、TM-6010については1500mg/kgを2週間投与しても急性毒性は観察されなかった。一方、TM-6089の50%致死量は500mg/kgであった。また亜急性毒性の試験は、マウスに30日間に亘って被験化合物(TM6008、TM6010またはTM6089)100mg/kgを強制経口投与することにより実施した。30日間に亘って各被験化合物を100mg/kg/日の割合で投与しても、マウスに亜急性毒性は殆ど認められなかった。
【0166】
(3)生化学分析
血液と尿のサンプルを回収して、生化学分析(血漿中のグルコース、総コレステロール、トリグリセリド、GOT、GPT、クレアチニン、尿素窒素、総タンパク質、およびアルブミン;血液中のヘモグロビン、赤血球数およびヘマトクリット;尿中の蛋白質およびクレアチニン)を行った。 血漿エリスロポエチン量は、キット(F.Hoffmann-La,ロシュ製)を用いて測定した。脳、肺、心臓、腎臓および肝臓の組織学分析および血清試験の結果、1か月後も異常を示さなかった。 血漿のエリトロポイエチン・レベルも著しい変化がなかった。
【0167】
(4)薬物速度論
被験化合物(TM6008、TM6010およびTM6089、50mg/kg)を、180-200gの雄のWisterラットに強制経口投与した。投与前(0h)、投与後1、2、6および24hに、ヘパリン添加血液サンプルを回収した。血漿濃度を逆相HPLCにより分析し、最高薬物濃度時間(Tmax)、最高薬物濃度(Cmax)、および半減期(T1/2)を求めた。
【0168】
各化合物を50mg/kgの割合で経口投与したマウス中の薬物速度論〔最高薬物濃度時間(Tmax)、最高薬物濃度(Cmax)、および半減期(T1/2)〕の結果は下記の通りであった:
【0169】
【表1】
TM-6010は平面構造を有している。ドッキングシミュレーションはDNAとのインターカレーションの可能性を示した。 このため、in vivoでの実験はTM-6008とTM-6089でのみ行なった。
【図面の簡単な説明】
【0170】
【図1】図aは、ストレプトマイシスに由来するPHD3のX線結晶構造を示す図である。図中、真ん中のポケット内にある球体(青)はFe(II)を示す。図中、このFe(II)のまわりに位置するスティックモデル(枝状物)は、上がHIFのプロリン残基、下が2-OGを示す。図bは、公知のPDH抑制剤である3, 4-DHBの、PHD3中での結合様式を示す。2-OG の結合部位であるA部位に結合することがわかる。図cは、TM6008(図中、黄色で示す)、TM6010(図中、緑で示す)およびTM6089(図中、赤で示す)の、PHD3中での結合様式を示す。なお、図はソフトウェアPyMOL version0.97(DeLano Scientific LLC社製)によって描いた。
【図2】HIF依存のルシフェラーゼ・リポーター遺伝子を有する7xHRE/Lucプラスミドで形質転換したIRPTC細胞を、被験化合物(陽性コントロール:塩化コバルト、TM6008、TM6010およびTM6089、最終濃度:20μMあるいは60μM)とともに24時間インキュベーションした際の、ルシフェラーゼ活性を示す。結果は3つの独立した実験の平均値として、コントロールの結果(1とする)の何倍増加したかで評価する。データは、平均値±SDとして表現する(* P<0.05、**P<0.01対コントロール)。
【図3】被験化合物として、塩化コバルト、TM6008およびTM6101を用いて24時間インキュベーションした際の、ルシフェラーゼ活性を示す。結果は3つの独立した実験の平均値として、コントロールの結果(1とする)の何倍増加したかで評価する。データは、平均値±SDとして表現する(* P<0.05、**P<0.01対コントロール)。
【図4】IRPTC細胞ホモジェネート(30μg)のミトコンドリア画分を、被験化合物およびHIF-1αの酸素依存性分解ドメイン(ODD)ペプチド(アミノ酸556-575)と反応させた。ODD依存の水酸化酵素活性を、[5-14C]-2-OGから変換された[1-14C]コハク酸塩の放射活性をカウントすることにより評価した。結果は3つの独立した実験の平均値として、ODD依存性水酸化酵素活性の阻害%として示す。データは、平均値±SDとして表現した。 *P<0.01対コントロール。
【図5】GST-HIF-1α(531-652)に結合する、35S標識PHD3に対する被験化合物の阻害作用を、プルダウンアッセイにより評価した。3つの独立した実験結果から平均値を出し、結合活性の%として示した。データは平均値±SDとして示す。*P<0.01 v.s. コントロール
【図6】ポリウレタン・スポンジを、マウスの皮下に移植し、被験化合物(マウス1匹当たり、48μg TM6008あるいは30μgコバルト)を、一日おきに1週間スポンジに注入した。 血管新生の程度を評価するために、スポンジ(a)中のヘモグロビン量を測定し、内皮細胞マーカーCD31(b、c)を染色した。 bは、CD31の免疫染色の代表写真である (x 200)。 cは、各試薬によって引き起こされた結果の平均数を示す。
【図7】非虚血動物(a)、虚血状態後、ベヒクル(カルボキシメチルセルロース)だけで処理された動物(b)、および拒絶状態後、TM-6008で処理されや動物(c)の、CA1海馬の、ヘマトキシロン-エオシン染色の代表的な顕微鏡写真を示す。 図中、バーは0.1mmを示す。dは、非虚血群(No ischemia)、虚血後ベヒクル(カルボキシメチルセルロース)処理群(Ischemia treated with vehicle)、および虚血後TM6008処理群(Ischemia treated with TM6008)の、CA1海馬中の正常神経の数を示す。
【特許請求の範囲】
【請求項1】
下式(A)または(B)で示される骨格を有する基の少なくとも1つを一分子中に含む化合物またはその塩を有効成分とするプロリル水酸化酵素阻害剤:
【化1】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
【請求項2】
骨格Aを有する基が、下式:
【化2】
(R1は水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、保護基を有していてもよいカルボキシアルキル基、またはアミノ基:R2は水素原子、水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、またはアミノ基:R3は水素原子、アミノ基、または低級アルキルアミノ基を意味する)
で示される基である、請求項1記載のプロリル水酸化酵素阻害剤。
【請求項3】
骨格Bを有する基が、下式:
【化3】
(R4、R5、R6およびR7は、同一または異なって、水素原子または低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
で示される基である、請求項1または2に記載するプロリル水酸化酵素阻害剤。
【請求項4】
下式で示される化合物(I):
【化4】
〔R1は炭素数1〜6の低級アルキル基または保護基を有していてもよい炭素数1〜4のカルボキルアルキル基:R2は水素原子、炭素数1〜6の低級アルキル基:R10は、R11−S−CH2−(式中、R11は置換基を有していてもよいヘテロアリール基、または下式:
【化5】
(R4、R5、R6およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。)で示される基を意味する。〕
および下式で示される化合物(II):
【化6】
(R5およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。)
からなる群から選択される少なくとも1つの化合物を有効成分とする請求項1乃至3のいずれかに記載するプロリル水酸化酵素阻害剤。
【請求項5】
下式で示される化合物1〜4:
(化合物1)
【化7】
(化合物2)
【化8】
(化合物3)
【化9】
(化合物4)
【化10】
からなる群から選択される少なくとも1つを有効成分とする請求項1乃至4のいずれかに記載するプロリル水酸化酵素阻害剤。
【請求項6】
プロリル水酸化酵素と低酸素誘導因子との結合阻害剤である請求項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
【請求項7】
低酸素誘導因子分解阻害剤である請求項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
【請求項8】
請求項1乃至7のいずれかに記載するプロリル水酸化酵素阻害剤および薬学的に許容される担体または添加剤を含む、医薬組成物。
【請求項9】
細胞または組織の低酸素状態を改善するための請求項8記載の医薬組成物。
【請求項10】
細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬である請求項8または9記載の医薬組成物。
【請求項11】
細胞または組織の低酸素状態に関連して生じる疾患また病態が、慢性または急性の虚血性疾患またはそれに起因して生じる病態である請求項9に記載する医薬組成物。
【請求項12】
経口投与形態を有する請求項8乃至11のいずれかに記載する医薬組成物。
【請求項13】
被験化合物の中から、プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物を選択する工程を有するプロリル水酸化酵素阻害剤のスクリーニング方法。
【請求項14】
プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物が,下式(A)または(B)で示され骨格を有する基の少なくとも1つを一分子中に含む化合物である請求項13記載のスクリーニング方法:
【化11】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
【請求項15】
細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬の有効成分のスクリーニング方法である、請求項13または14に記載するスクリーニング方法。
【請求項16】
下式で示される化合物またはその塩:
【化12】
(式中、R12は水素原子または低級アルキル基を意味する。)
【請求項1】
下式(A)または(B)で示される骨格を有する基の少なくとも1つを一分子中に含む化合物またはその塩を有効成分とするプロリル水酸化酵素阻害剤:
【化1】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
【請求項2】
骨格Aを有する基が、下式:
【化2】
(R1は水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、保護基を有していてもよいカルボキシアルキル基、またはアミノ基:R2は水素原子、水酸基、低級アルキル基、カルボキシル基で置換されたアルキル基、またはアミノ基:R3は水素原子、アミノ基、または低級アルキルアミノ基を意味する)
で示される基である、請求項1記載のプロリル水酸化酵素阻害剤。
【請求項3】
骨格Bを有する基が、下式:
【化3】
(R4、R5、R6およびR7は、同一または異なって、水素原子または低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)
で示される基である、請求項1または2に記載するプロリル水酸化酵素阻害剤。
【請求項4】
下式で示される化合物(I):
【化4】
〔R1は炭素数1〜6の低級アルキル基または保護基を有していてもよい炭素数1〜4のカルボキルアルキル基:R2は水素原子、炭素数1〜6の低級アルキル基:R10は、R11−S−CH2−(式中、R11は置換基を有していてもよいヘテロアリール基、または下式:
【化5】
(R4、R5、R6およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。R4とR5またはR6とR7は互いに結合して、それらが結合する炭素原子と共に、5または6員の飽和もしくは不飽和の置換基を有していてもよいシクロ環またはヘテロ環を形成してもよい。)で示される基を意味する。〕
および下式で示される化合物(II):
【化6】
(R5およびR7は、同一または異なって、水素原子または炭素数1〜6の低級アルキル基を意味する。)
からなる群から選択される少なくとも1つの化合物を有効成分とする請求項1乃至3のいずれかに記載するプロリル水酸化酵素阻害剤。
【請求項5】
下式で示される化合物1〜4:
(化合物1)
【化7】
(化合物2)
【化8】
(化合物3)
【化9】
(化合物4)
【化10】
からなる群から選択される少なくとも1つを有効成分とする請求項1乃至4のいずれかに記載するプロリル水酸化酵素阻害剤。
【請求項6】
プロリル水酸化酵素と低酸素誘導因子との結合阻害剤である請求項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
【請求項7】
低酸素誘導因子分解阻害剤である請求項1乃至5のいずかに記載するプロリル水酸化酵素阻害剤。
【請求項8】
請求項1乃至7のいずれかに記載するプロリル水酸化酵素阻害剤および薬学的に許容される担体または添加剤を含む、医薬組成物。
【請求項9】
細胞または組織の低酸素状態を改善するための請求項8記載の医薬組成物。
【請求項10】
細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬である請求項8または9記載の医薬組成物。
【請求項11】
細胞または組織の低酸素状態に関連して生じる疾患また病態が、慢性または急性の虚血性疾患またはそれに起因して生じる病態である請求項9に記載する医薬組成物。
【請求項12】
経口投与形態を有する請求項8乃至11のいずれかに記載する医薬組成物。
【請求項13】
被験化合物の中から、プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物を選択する工程を有するプロリル水酸化酵素阻害剤のスクリーニング方法。
【請求項14】
プロリル水酸化酵素と低酸素誘導因子のα鎖プロリンとの競合的結合阻害作用を有する化合物が,下式(A)または(B)で示され骨格を有する基の少なくとも1つを一分子中に含む化合物である請求項13記載のスクリーニング方法:
【化11】
(式中、R8およびR9は、互いに水素原子であるか、またはこれらが結合するアルキレン鎖、酸素原子、窒素原子または硫黄原子とともに環を形成してもよい。)。
【請求項15】
細胞または組織の慢性または急性の低酸素状態に関連して生じる疾患または病態の予防または治療薬の有効成分のスクリーニング方法である、請求項13または14に記載するスクリーニング方法。
【請求項16】
下式で示される化合物またはその塩:
【化12】
(式中、R12は水素原子または低級アルキル基を意味する。)
【図1】


【図2】
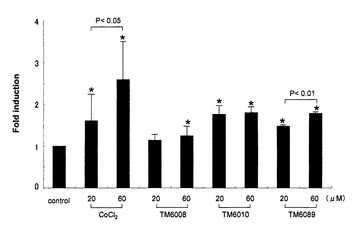
【図3】
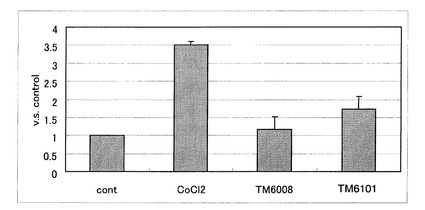
【図4】
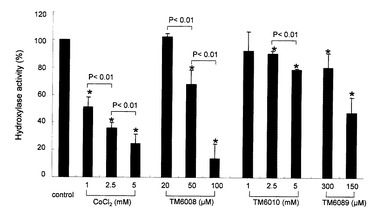
【図5】
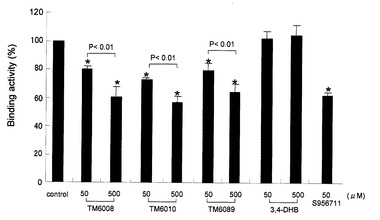
【図6】
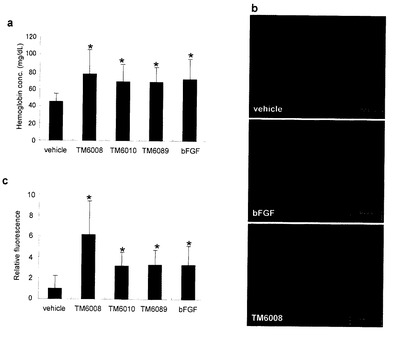
【図7】
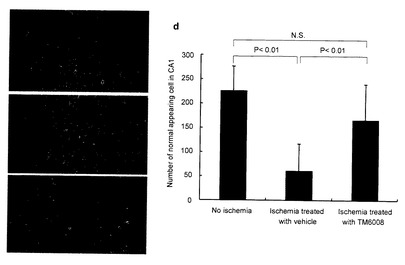
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2007−191451(P2007−191451A)
【公開日】平成19年8月2日(2007.8.2)
【国際特許分類】
【出願番号】特願2006−13252(P2006−13252)
【出願日】平成18年1月20日(2006.1.20)
【出願人】(000125369)学校法人東海大学 (352)
【Fターム(参考)】
【公開日】平成19年8月2日(2007.8.2)
【国際特許分類】
【出願日】平成18年1月20日(2006.1.20)
【出願人】(000125369)学校法人東海大学 (352)
【Fターム(参考)】
[ Back to top ]