
プロリン誘導体の塩、またはその溶媒和物
【課題】ジペプチジルペプチダーゼ−IV阻害薬として有用な3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン(化合物I)の安定性や吸湿性の面で優れた特性および再生可能な結晶構造を有
する塩を提供する。
【解決手段】3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンと有機または無機の一ないし三塩基酸との塩、またはその溶媒和物(但し、3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭
化水素酸塩、またはその溶媒和物は除く)。
する塩を提供する。
【解決手段】3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンと有機または無機の一ないし三塩基酸との塩、またはその溶媒和物(但し、3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭
化水素酸塩、またはその溶媒和物は除く)。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ジペプチジルペプチダーゼ−IV(以下、DPP-IVと記す)阻害薬として有用な3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンの新規な塩、またはその溶媒和物に関する。
【背景技術】
【0002】
DPP-IV阻害薬は、血漿中のグルカゴン様ペプチド-1(以下、GLP-1と記す)の不活性化を
阻害し、そのインクレチン作用を増強するため、糖尿病治療薬等として有用であり、糖尿病、特に2型糖尿病の治療において有効であり得る薬剤として研究開発段階にある(特許文献1ないし6、非特許文献1参照)。
【0003】
有用なチアゾリジン誘導体についても一連の化合物が報告されている。 (特許文献7参
照)。この文献に記載された実施例化合物の中でも、注目すべきものとして、3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンがある(以下、化合物Iと記す)。化合物Iは3塩酸塩の形態で記載されているが、この塩は安定性や吸湿性の面で薬学的に望ましくない特性を有し、同一形態で再現性をもって製造するのが困難であることが認められた。医薬品の開発における規制要件を満たすためには、特に、一定の品質の化合物を、再現性をもって製造しなければならないので、化合物Iの3塩酸塩に認められるこれらの特性は医薬品の開発には不利なものと考えられる。
【0004】
また、この文献(特許文献7)には「化合物I」ならびにその他のチアゾリジン誘導体における特定の塩が実施例化合物として開示されているが、いずれの実施例化合物においても結晶多形に関する議論はなされていない。
【0005】
ある物質が2種以上の結晶構造に結晶化する能力はポリモルフィズム(polymorphism)と
して知れらており、個々の結晶形は結晶多形と呼ばれている。同一化合物の異なる結晶多形は、保存安定性や溶解性等の物性が全く異なる場合がある。これらの物性の違いは作用効果における差に結びつく場合がある。これらの相違により、個々の結晶多形や結晶多形の混合物の検討は、医薬品開発において特に有用である。
【0006】
なお、結晶多形は命名方法により、A形、B形、I形、II形、α形、β形等複数の表記が
存在する。また、これらの表記において、「形」の代わりに「型」(A型等)を使用する場
合もあるが、いずれの表記であっても同様の意味で用いられる。
【0007】
しかしながら、ある化合物において種々の結晶多形を見つけることは、必ずしも容易に検討できるとは限らない。さらに、ひとたび特定の結晶多形の存在が認識され、その特徴が好ましいものであるとされた場合、 研究者はその結晶多形を常に一定に単一結晶とし
て多量に供給する方法を見出さなければならない。ある結晶多形の単一結晶あるいは実質的な単一結晶を供給する方法を確立することは容易ではなく、鋭意検討を必要とする。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第97/040832号パンフレット
【特許文献2】国際公開第98/019998号パンフレット
【特許文献3】米国特許第5939560号
【特許文献4】国際公開第01/055105号パンフレット
【特許文献5】国際公開第02/002560号パンフレット
【特許文献6】国際公開第02/062764号パンフレット
【特許文献7】国際公開第02/014271号パンフレット
【非特許文献】
【0009】
【非特許文献1】J. Med. Chem., 47(17), 4135-4141(2004)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明における課題は、化合物Iに関して、医薬品とするために所望される、安定性、
溶解性、吸湿性、または生物学的利用能等の面で優れた特性および再生可能な結晶構造を有する化合物を見出すこと、並びにそれらを製造する方法を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、化合物Iの一ないし三塩基酸塩を調製し、それぞれの塩またはそれらの
溶媒和物の結晶を特徴付け、安定性並びに吸湿性の点で好ましい特性を有する化合物Iの
新規塩を見出した。さらに鋭意研究を重ねた結果、本発明の新規塩の安定的な工業的調製方法を見出し、本発明を完成するに至った。
【0012】
すなわち、本発明の要旨は以下の(1)ないし(33)に記す塩またはその溶媒和物、および
それらの製造方法に存する。
(1) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンと有機または無機の一ないし三塩基酸との塩、またはその溶媒和物、
(2) 有機または無機の一塩基酸が塩酸、臭化水素酸、硝酸、メシル酸、トシル酸、ベシル酸、塩酸、ナフタレン-1-スルホン酸、ナフタレン-2-スルホン酸、没食子酸、またはカンファースルホン酸である前記(1)に記載の塩、またはその溶媒和物(ただし、一塩基酸が塩酸の場合、塩は2または2.5塩酸塩である)、
(3) 有機または無機の二塩基酸がフマル酸、マレイン酸、硫酸、コハク酸、L-酒石酸、エタンジスルホン酸、またはクエン酸である前記(1)に記載の塩、またはその溶媒和物、
(4) 有機または無機の三塩基酸がリン酸である前記(1)に記載の塩、またはその溶媒和物
、
(5) 2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2ベシル酸、2塩酸、2.5
塩酸、2ナフタレン-1-スルホン酸、2ナフタレン-2-スルホン酸、2カンファースルホン酸
、フマル酸、硫酸、コハク酸、L-酒石酸、またはクエン酸との塩である、前記(1)に記載
の塩、またはその溶媒和物、
(6) 2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2.5塩酸、2ナフタレン-1-スルホン酸、2メシル酸、3メシル酸、または2ナフタレン-2-スルホン酸との塩である前
記(5)記載の塩、またはその溶媒和物、
(7)周囲温度での水への溶解度が7mg/mLないし2g/mLである一塩基酸と3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカル
ボニル}チアゾリジンとの塩、またはその溶媒和物、
(8) 37℃での水への溶解度が20 mg/mL以上である前記(7)に記載の塩、またはその溶媒和
物、
(9) pH9から12での水への溶解度が7 mg/mLである前記(7)に記載の記載の塩、またはその
溶媒和物、
(10) 25℃で測定した吸湿性が6%以下である前記(1)に記載の塩、またはその溶媒和物、
(11) 25℃で測定した相対湿度0%から50%の範囲における吸湿性が5%である前記(10)に記載の塩、またはその溶媒和物、
(12) 25℃で測定した相対湿度5%から90%の範囲における吸湿性が2%である前記(10)に記
載の塩、またはその溶媒和物、
(13) 塩が前記(2)記載の一塩基性酸との塩である前記(10)ないし(12)いずれかに記載の塩、またはその溶媒和物、
(14) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物、
(15) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°及び14.4°(それぞれ±0.2°) にピークを有することを特徴とする前記(14)に記載の塩、またはその水和物、
(16) 1.0ないし2.0水和物である前記(15)に記載の水和物、
(17) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°, 14.4°, 22.6°及び26.5° (それぞれ±0.2°) にピークを有することを特徴とする前記(14)に記載の塩、またはその水和物、
(18) 1.0ないし2.0水和物である前記(17)に記載の水和物、
(19) 図1に例示される粉末X線回折パターンを示す、前記(14)に記載の塩、またはその水
和物、
(20) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0臭化水素酸塩、またはその溶媒和物、(21) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.7°, 7.7°, 11.3°, 16.2°及び17.0° (それぞれ±0.2°) にピークを有することを特徴とする前記(20)に記載の塩の水和物、
(22) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.2°, 10.4°, 19.1°, 19.8°及び20.7° (それぞれ±0.2°) にピークを有することを特徴とする前記(20)に記載の塩の水和物、
(23) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.5°, 13.4°, 14.3°, 21.4°及び26.7° (それぞれ±0.2°) にピークを有することを特徴とする前記(20)に記載の塩の水和物、
(24) 図2に例示される粉末X線回折パターンを示す、前記(20)に記載の塩の水和物、
(25) 図3に例示される粉末X線回折パターンを示す、前記(20)に記載の塩の水和物、
(26) 図4に例示される粉末X線回折パターンを示す、前記(20)に記載の塩の水和物、
(27) 3-{(2S,4S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンに対して臭化水素酸で脱1,1-ジメチルエチルオキシカルボニル反応を行うと共に同時に造塩を行うことを特徴とする3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イ
ル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法、
(28) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩を、許容される溶媒を用いて結晶化する工程を含む3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イ
ル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法、
(29) 許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”において1日摂取許容量(“PDE”)が10 mg/dayを越える溶媒から選ばれる溶媒である前記(28)に記
載の方法、
(30) 許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”におけるクラス3の溶媒から選ばれる溶媒である前記(28)に記載の方法、
(31) 許容される溶媒が、エタノール、1-プロパノール、2-プロパノール、酢酸エチル、
アセトンから選ばれる溶媒である前記(28)に記載の方法、
(32) 許容される溶媒がエタノール及び/または水である前記(28)に記載の方法、または
(33) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩の1.0ないし2.0水和物。
【発明の効果】
【0013】
化合物Iの塩若しくはその溶媒和物、あるいはそれぞれの新規の結晶多形は、安定性の
改善、吸湿性(潮解性)の改善、溶媒からの迅速な単離、及び製剤化の容易性から選択される1つ以上の特性を有しており、化合物Iの医薬品としての開発を促進する。
【図面の簡単な説明】
【0014】
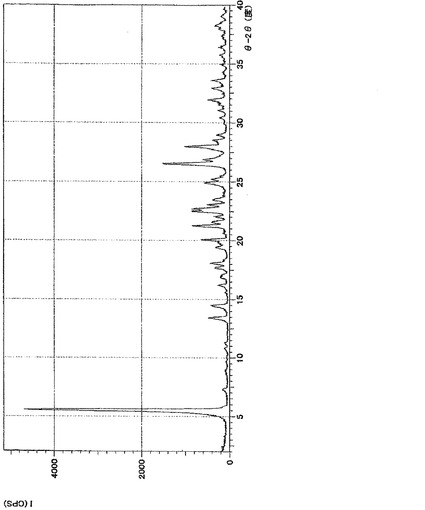
【図1】実施例4の表題化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
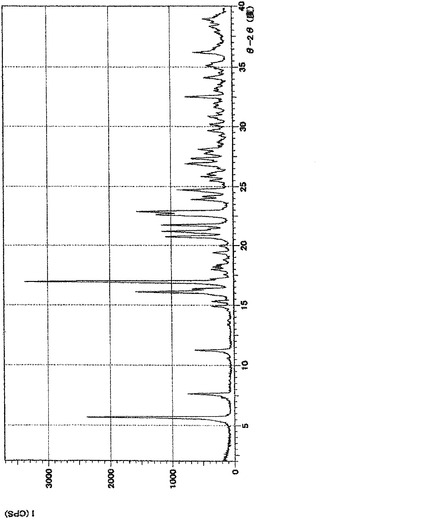
【図2】実施例5の(1)の化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
【図3】実施例5の(2)の化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
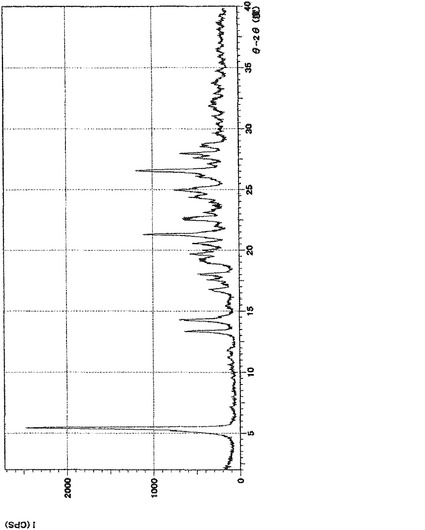
【図4】実施例5の(3)の化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
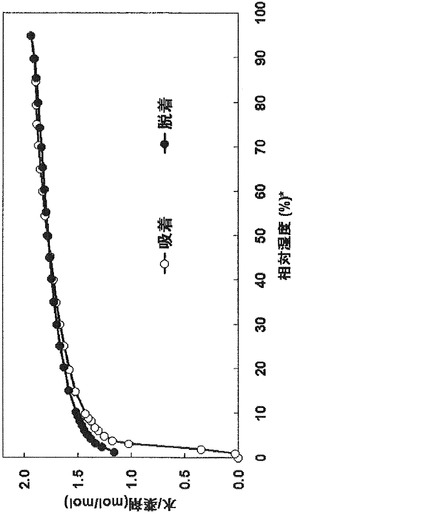
【図5】実施例3の表題化合物の吸湿性の測定結果である。―○―は横軸の湿度における化合物への水の吸着、―●―は横軸の湿度における化合物への水の脱着をプロットしたものである。
【発明を実施するための形態】
【0015】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン(化合物I)を以下に示す。
【0016】
【化1】

【0017】
化合物Iの3塩酸塩は、国際公開第02/14271号パンフレットの実施例222として記載され
ている合成方法に準じて製造することができる。このものは適当な塩基を用いて遊離塩基とすることができる。用いる塩基としては、アルカリ金属またはアルカリ土類金属炭酸塩(炭酸水素ナトリウム、炭酸ナトリウム、または炭酸カリウム等)あるいはアルカリ金属またはアルカリ土類金属の水酸化物(水酸化ナトリウム、または水酸化カリウム等)等が挙げられる。
【0018】
例えば、実施例222の化合物を上記いずれかの塩基の水溶液に加えた後に、炭化水素系
溶媒(ベンゼン、またはトルエン等)、ハロゲン系炭化水素溶媒(ジクロロメタン、ジクロ
ロエタン、クロロホルム、または四塩化炭素等)、酢酸エチル等で抽出することにより化
合物Iを得ることができる。
【0019】
さらに、化合物Iの2.5臭化水素酸塩は以下のスキームによっても製造することが出来る。
【0020】
【化2】
【0021】
1-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン(II)またはその塩と3-[(2S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-オキソピロリジン-2-イルカルボニル]
チアゾリジン(III)を還元的アミノ化反応させた後、臭化水素酸により脱1,1-ジメチルエチルオキシカルボニル反応させると化合物Iの2.5臭化水素酸塩(IV)を得ることができる。
この還元的アミノ化反応は、式(II)で表される化合物またはその塩の1モルに対して、式(III)で表される化合物を約0.5ないし10モル、好ましくは約1ないし2モル用い、金属水素錯化合物(水素化ほう素ナトリウム、シアノ水素化ほう素ナトリウム、トリアセトキシ
水素化ほう素ナトリウム等の複合水素化合物、またはジボラン等)を約0.5ないし10モル
、好ましくは約1ないし2モル用い、必要に応じて酸性触媒(酢酸、p-トルエンスルホン酸、または三フッ化ホウ素・ジエチルエーテル錯体等)の存在下、不活性な溶媒中で行われる。不活性な溶媒としては、アルコール類(メタノール、エタノール、1-プロパノール、2-プロパノール(以下、IPAと記す)、またはブタノール等)、ニトリル類(アセトニト
リル、またはプロピオニトリル等)、アミド類(ホルムアミド、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、または1,3-ジメチル-2-イミダゾリジノン等)、エーテ
ル類(ジエチルエーテル、ジイソプロピルエーテル、t-ブチルメチルエーテル、1,4-ジオキサン、またはテトラヒドロフラン(以下、THFと記す)等)、ハロゲン化炭化水素類(ジクロロメタン、クロロホルム、または1,2-ジクロロエタン等)、炭化水素類(n-ヘキサン、シクロヘキサン、ベンゼン、またはトルエン等)、またはこれらの任意の混合溶媒等が挙
げられる。
【0022】
このとき反応温度は-20ないし200℃、好ましくは0ないし80℃であり、反応時間は約0.5ないし96時間、好ましくは0.5ないし24時間である。
この反応の生成物を単離精製せず、水、アルコール類(メタノール、エタノール、またはIPA等)、エーテル類(THF、ジオキサン等)、ハロゲン化炭化水素類(ジクロロメタン、ジクロロエタン、クロロホルム等)、酢酸エチル、アセトニトリル等、またはこれらの任意の混合溶媒中、式(II)の化合物またはその塩の1モルに対して、臭化水素酸を1ないし20モル、好ましくは約2.5ないし5モルを反応させると化合物Iの2.5臭化水素酸塩を得ることができる。
【0023】
このとき反応温度は-20ないし200℃、好ましくは0ないし100℃であり、反応時間は約0.5ないし48時間、好ましくは0.5ないし24時間である。反応後、析出物をろ取することにより式(IV)で表される塩を得ることができる。
【0024】
本発明の化合物Iの塩酸塩、臭化水素酸塩、硝酸塩、メシル酸塩、マレイン酸塩、トシ
ル酸塩、ベシル酸塩、ナフタレン-1-スルホン酸塩、ナフタレン-2-スルホン酸塩、没食子酸塩、(+)-カンファースルホン酸塩、(-)-カンファースルホン酸塩、フマル酸塩、硫酸塩、コハク酸塩、L-酒石酸塩、エタンジスルホン酸塩、クエン酸塩、またはリン酸塩(以下、「本発明の塩」と略すこともある)は、光学的に純粋であり、例えば(2S,4S) -エナン
チオマーの光学純度は、90%以上のエナンチオマー過剰(以下、e.e.と記す)、好ましく
は95%e.e. 以上、より好ましくは99%e.e. 以上である。
【0025】
本発明の塩の形態は特に制限されず、油状物、非晶質(アモルファス)、結晶であってもよい。好ましい塩の形態は結晶である。
【0026】
前記結晶の形態の塩としては、2.0塩酸塩、2.5 塩酸塩、2臭化水素酸塩、2.5臭化水素
酸塩、2メシル酸塩、3メシル酸塩、2トシル酸塩、2ベシル酸塩、2ナフタレン-1-スルホン酸塩、2ナフタレン-2-スルホン酸塩、2(+)-カンファースルホン酸塩、2マレイン酸塩、2
フマル酸塩、または2L-酒石酸塩等が例示できる。これらの塩は粉末X線回折パターンにおける回折ピークによっても特徴付けることが出来る。
【0027】
本発明において、2.0臭化水素酸塩の結晶多形を、A形、B形およびC形と称する。前記結晶の形態の塩も本明細書において適当ならば2.0臭化水素酸塩と称する。
また、2トシル酸塩の結晶多形をA形、B形およびC形と、2トシル酸塩の結晶多形をA形、B形およびC形と、2ベシル酸塩の結晶多形をA形およびB形と、2マレイン酸塩の結晶多形をA形およびB形と、2フマル酸塩の結晶多形をA形およびB形とそれぞれ称する。
【0028】
本発明の塩の溶媒和物は、へミ−、モノ−、ジ−、トリ−、テトラ−、ペンタ−、ヘキサ−等の溶媒和物として存在し得る。結晶化に使用する溶媒(アルコール(メタノール、エタノール、またはIPA等)、アルデヒド、ケトン(アセトン等)、またはエステル(酢酸エチ
ル等)等)およびこれらの溶媒に含まれる水は、結晶格子中に取り込まれ得る。一般的に、溶媒が結晶化およびその後の製造工程において、溶媒和物となるか非溶媒和物となるかを予測することは不可能である。それらは化合物、製造条件ならびに選択された溶媒、とり
わけ水との間のさまざまな相互作用の組合せに依存する。さらに、ある化合物の塩、またはその溶媒和物の結晶または非晶質のそれぞれの安定性は、実測値によってのみ確認され得るものである。
【0029】
本発明の塩は、溶媒(水、または有機溶媒等)の溶媒和物であってもよく、非溶媒和物であってもよい。すなわち、本発明の塩は、水和物であってもよく、非水和物であってもよい。水和物である場合、水和する水の量は種々の条件によって変動することもあるが、好ましくは2.0以下の水和物であり、より好ましくは1.0ないし2.0水和物である。
【0030】
本発明の塩は、哺乳動物に対して安全な(薬学上、薬理学上、または生理学上に許容さ
れるもの等)溶媒を含んでいてもよく、または溶媒との溶媒和であってもよい。「溶媒」
としては、“残留溶媒Q3CのICHガイドライン”において1日摂取許容量(“PDE”)が10 mg/dayを越えるもの、及び/または、“残留溶媒Q3CのICHガイドライン”におけるクラス3の
ものから選ばれる。具体的には、エタノール、1-プロパノール、IPA、1-ブタノール、2-
ブタノール、1-ペンタノール、酢酸、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸n-ブチル、酢酸イソブチル、ギ酸、ギ酸エチル、アセトン、メチルエチルケトン、メチルイソブチルケトン、ヘプタン、ペンタン、ジエチルエーテル、t-ブチルメチルエーテル、THF、アニソール、クメン、またはジメチルスルホキシド等が挙げられる
。これらの溶媒のうち、エタノールが好ましい。「溶媒」の含有量は、50000 ppm以下、
好ましくは5000 ppm以下である。
【0031】
本発明の塩は自体公知の方法により製造することができ、例えば、化合物Iと、塩酸、
臭化水素酸、硝酸、メシル酸、マレイン酸、トシル酸、ベシル酸、ナフタレン-1-スルホ
ン酸、ナフタレン-2-スルホン酸、没食子酸、(+)-カンファースルホン酸、(-)-カンファ
ースルホン酸、フマル酸、硫酸、コハク酸、L-酒石酸、エタンジスルホン酸、クエン酸、またはリン酸から選択された有機酸または無機酸とを反応させることにより、本発明の塩を得ることができる。
【0032】
本反応は、一般に無溶媒または不活性な溶媒中で行われる。「不活性な溶媒」としては、水、アルコール類(メタノール、エタノール、1-プロパノール、IPA、またはブタノール等)、ケトン類(アセトン、またはメチルエチルケトン等)、ニトリル類(アセトニトリ
ル、またはプロピオニトリル等)、アミド類(ホルムアミド、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、または1,3-ジメチル-2-イミダゾリジノン等)、エーテル
類(ジエチルエーテル、ジイソプロピルエーテル、t-ブチルメチルエーテル、1,4-ジオキサン、またはTHF等)、エステル類(ギ酸エチル、酢酸エチル、または酢酸プロピルなど)
、ハロゲン化炭化水素類(ジクロロメタン、クロロホルム、または1,2-ジクロロエタン等)、炭化水素類(n-ヘキサン、シクロヘキサン、ベンゼン、またはトルエン等)、スルホキシド類(ジメチルスルホキシド等)、極性溶媒(スルホラン、またはヘキサメチルホスホリルアミド等)またはこれらの任意の混合溶媒等が挙げられる。これらの溶媒の中でも、水、水とアルコールとの混合溶媒(水とメタノールとの混合溶媒、水とエタノールとの混
合溶媒、水と1-プロパノールとの混合溶媒、または水とIPAとの混合溶媒等)が好ましい。
【0033】
「不活性な溶媒」は、化合物Iに対し、通常、1ないし100w/v%、好ましくは2ないし50 w/v%が用いられる。
【0034】
反応温度は、通常、-20℃ないし溶媒の還流温度、好ましくは0℃ないし溶媒の還流温度である。反応時間は、通常、約1分ないし24時間、好ましくは約10分ないし6時間、さらに好ましくは約30分ないし3時間である。
【0035】
このようにして得られた塩は、自体公知の分離手段(濃縮、減圧濃縮、溶媒抽出、結晶
化、再結晶、転溶、またはクロマトグラフィー等)により、反応混合物から単離、精製す
ることができる。
【0036】
このようにして得られた塩を結晶化に付すことにより、結晶の状態で化合物Iの塩を得
ることができる。この結晶化方法としては自体公知の方法が挙げられ、溶液からの結晶化、蒸気からの結晶化、溶融体からの結晶化が挙げられる(A. S. Myerson Ed., Handbook of Industrial Crystallization Second Edition, Butterworth-Heinemann 2002 参照)。
【0037】
「溶液からの結晶化」の方法としては、濃縮法、徐冷法、反応法(拡散法、または電解
法)、水熱育成法、または融剤法等が挙げられる。用いられる溶媒としては、前記「不活
性な溶媒」と同様の溶媒が挙げられる。
【0038】
「蒸気からの結晶化」の方法としては、気化法(封管法、または気流法)、気相反応法、または化学輸送法等が挙げられる。
【0039】
「溶融体からの結晶化」の方法としては、ノルマルフリージング法(引上げ法、温度傾
斜法、またはブリッジマン法)、帯融解法(ゾーンレベリング法、またはフロートゾーン法)、または特殊成長法(VLS法、または液相エピタキシー法)等が挙げられる。なお、化合物Iの塩の結晶化には、一般的に、40℃ないし使用する溶媒の還流温度程度に加熱して化合
物Iの塩を溶解した溶液の冷却による晶析、または化合物Iの塩を溶解した溶液(特に濃縮
液)への貧溶媒の添加による晶析等が利用される。得られた結晶の解析方法としては、X線解析の方法が一般的である。X線解析の測定結果は、Y軸を回折強度とし、横軸を回折角(2θ)として表されるが、2θ値は同一の結晶形を測定した場合であってもある程度の範囲でバラツキが観察される。具体的には±0.2°が一般的なバラツキの程度であるが、測定条
件などによって、より大きな誤差を生じる場合もある。2θ値により結晶形を比較する際
、当業者においてこれらのバラツキを考慮して結晶形の比較が行われる。さらに、本発明の塩またはその溶媒和物は水分含量によって回折角に多少のずれを生じることがあるが、これらも本発明の範疇に含まれる。
【0040】
本発明の塩またはその溶媒和物(以下、単に本発明の塩と記す)は、安定性に優れているため、長期にわたって室温で保存できるだけでなく、製造工程や保管において煩雑な操作を必要としないこと、また、製剤化が容易なことから医薬品の原薬として有用である。さらには本発明の塩は水への溶解度が高いため、注射用製剤としてより自由度の高い投与形態を開発可能である。
本発明の塩を医薬として用いる場合、本発明の塩を製剤上許容しうる担体(賦形剤、結
合剤、崩壊剤、矯味剤、矯臭剤、乳化剤、希釈剤、または溶解補助剤等)と混合して得ら
れる医薬組成物あるいは製剤(錠剤、ピル剤、カプセル剤、顆粒剤、散剤、シロップ剤、
エマルジョン剤、エリキシル剤、懸濁剤、溶液剤、注射剤、点滴剤、または坐剤等)の形態で経口的または非経口的に投与することができる。医薬組成物は通常の方法にしたがって製剤化することができる。
【0041】
本明細書において、非経口とは、皮下注射、静脈内注射、筋肉内注射、腹腔内注射あるいは点滴法等を含むものである。注射用調剤は当該分野で知られた方法で調製することができる。直腸投与用の坐剤は、その薬物と適当な補形剤等と混合して製造することができる。経口投与用の固形投与剤型としては、粉剤、顆粒剤、錠剤、ピル剤、またはカプセル剤等の上記したものが挙げられる。経口投与用の液剤は、医薬として許容されるエマルジョン剤、シロップ剤、エリキシル剤、懸濁剤、または溶液剤等が挙げられる。
【0042】
本発明の塩の投与量は年齢、体重、一般的健康状態、性別、食事、投与時間、投与方法、排泄速度、薬物の組合せ、または患者のその時に治療を行っている病状の程度に応じ、
それらあるいはその他の要因を考慮して決められる。本発明の塩は、低毒性で安全に使用することができ、その1日の投与量は、患者の状態や体重、塩の種類、投与経路等によっ
て異なるが、例えば非経口的には皮下、静脈内、筋肉内または直腸内に、0.01ないし100 mg/kg体重/日、好ましくは0.05ないし50 mg/kg体重/日を投与され、また経口的には0.01
ないし100 mg/kg体重/日、好ましくは0.05ないし50 mg/kg体重/日を、一日1ないし数回
に分けて投与するのが好ましい。
【実施例】
【0043】
以下に参考例及び実施例を挙げて本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。
【0044】
なお、抽出における有機溶液の乾燥には、特に明記しない限り、無水硫酸ナトリウムまたは無水硫酸マグネシウムを使用した。カラムクロマトグラフィーは富士シリシア化学社製のシリカゲルを用いて行った。
また、熱分析(DSC)は、熱量曲線における融解開始前の直線部分と、融解中の直線部分
との延長上の交点の温度(onset値)及び、熱量曲線における融点付近の変極点の温度(peak
top値)を示した。粉末X線回折パターン(XRD)は角度2θの特徴的なピークを示した(±0.2°)。1H-NMRは300 MHz核磁気共鳴分光装置で測定した。1H-NMRのケミカルシフトは、内部標準としてテトラメチルシラン(TMS)を用い、相対的なデルタ(δ)値をパーツパーミリオ
ン(ppm)で表した。カップリング定数は自明な多重度をヘルツ(Hz)で示し、s(シングレッ
ト)、d(ダブレット)、t(トリプレット)、m(マルチプレット)等と表した。赤外分光(IR)の吸光度の強度をst(強)、m(中)、w(弱)で表した。
【0045】
なお、以下の参考例、実施例の表題化合物は非溶媒和物として表記するが、それぞれ塩は調製時の条件等により溶媒和物(特に水和物)の形態をとることもある。
【0046】
参考例1
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン
3-{(2S,4S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン(国際公開第02/14271号パンフレットの実施例222に記載された化合物に準じて合成)25.45 g
をジクロロメタン200 mLに溶解し、室温下トリフルオロ酢酸50 mLを加え、19時間攪拌し
た。反応液を減圧下濃縮し、残渣に飽和炭酸水素ナトリウム水溶液を加え、クロロホルムで抽出した。抽出液を飽和食塩水で洗浄、乾燥後、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィーで精製して表題化合物を固体として得た(19.28g、収率93%)
。
【0047】
実施例1
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5塩酸塩
(1)参考例1の化合物 2.50 gをTHF 100 mLに溶解し、室温下4 mol/L塩酸の酢酸エチル溶
液3.0 mLを加えた後、1時間攪拌した。析出物を濾取し、減圧下50℃で乾燥して固体2.69 gを得た。
(2)上記生成物300 mgを水150μLとエタノール 1.0 mLの混合溶媒に加熱溶解し、氷冷下1時間攪拌した。析出物を濾取し、減圧下50℃にて乾燥して表題化合物を結晶として得た(144 mg、収率48%)。
XRD: 5.2°, 14.3°, 16.2°, 21.8°, 25.2°.
Anal. calcd for C22H30N6OS・2.3HCl・2H2O: C, 48.35; H, 6.69; N, 15.38; found: C,48.02; H, 6.60; N, 15.20.
【0048】
実施例2
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0塩酸塩
実施例1の表題化合物60 mgを酢酸エチル3.0 mLに懸濁し、13時間加熱還流した。室温
で冷却し、析出物を濾取し、40℃の温風下乾燥して表題化合物を結晶として得た(50 mg収率85%)。
XRD: 5.0°, 14.8°, 21.0°, 21.5°, 25.2°.
Anal. calcd for C22H30N6OS・2.0HCl・H2O: C, 51.05; H, 6.62; N, 16.24; found: C, 50.89; H, 6.58; N, 16.12.
【0049】
実施例3
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩
参考例1の化合物5.09 gをエタノール50.9 mLに溶解し、還流温度にて48%臭化水素酸5.03 gを加えた後、攪拌しながら約1時間かけて室温まで冷却し、室温にてさらに1時間攪拌した。析出物を濾取後、エタノール5 mLで洗浄し、45℃の温風下乾燥して表題化合物を結晶として得た(6.76 g)。
融点: 202.0℃(分解)
IR(KBr): 3600-3300 (st), 3116-2850 (st), 2800-2400 (st), 1647 (st), 1592 (m), 1572 (m), 1496 (m), 1450 (m), 1385 (m), 1361 (w), 768 (m), 692 (w) .
【0050】
実施例4
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩(実施例3の表題化合物の別途
合成方法)
(1)水素化トリアセトキシホウ素ナトリウム13.68 kgのトルエン300 L懸濁液に1-[4-(3-
メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン・酢酸塩15.00 kg、3-[(2S) -1-(1,1-ジメチルエチルオキシカルボニル)-4-オキソピロリジン-2-イルカルボニル] チアゾリジン14.90 kgを加えて、室温で2.5時間攪拌した。反応液に水90 Lを滴下し、0.5時間攪拌した後、トルエン層を分離した。このトルエン層を5%炭酸水素ナトリウム水溶液90 L、水90 Lの順で洗浄し、減圧下濃縮、乾固した。この残渣にIPA 224 Lを加え、約80℃で48%臭化水素酸25.08 kgを滴下後、2.5時間還流した。反応液を冷却し、約60℃で1.5時間、次いで約40℃で2時間、さらに室温で2時間攪拌した。析出物を濾取後、IPA 30 Lで洗浄し、温風下乾燥して表題化合物の固体を得た(29.76 kg、収率91%)。
(2)(1)で得た固体28.00 kgにエタノール168 Lを加え、加熱溶解後、熱時濾過し、反応
容器をエタノール28 Lで洗浄した。この濾液及び洗液を合わせ、67 ℃で水3 Lを加えた後に冷却し、49 ℃で1時間、次いで20〜15 ℃で1時間攪拌した。析出物を濾取後、エタノール28 Lで洗浄し、温風下乾燥して表題化合物を結晶として得た(25.84 kg、収率 92%)。
XRD: 5.4°, 13.4°, 14.4°, 22.6°, 26.5°.
【0051】
実施例5
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0臭化水素酸塩
(1)実施例4の表題化合物130 gを室温にて水260 mLに加え、攪拌により溶解させた後に析出物を濾取し、乾燥して表題化合物のA形結晶の3.5水和物を得た(53.58 g)。
XRD: 5.7°, 7.7°, 11.3°, 16.2°, 17.0°.
(2)(1)で得たA形結晶の水和物8.5 gを28〜30℃にて2%の水を含むエタノール100 mLに加え、攪拌により溶解した後に析出物を濾取し、乾燥して表題化合物のB形結晶の水和物を
得た(4.56 g)。
XRD: 5.2°, 10.4°, 19.1°, 19.8°, 20.7°.
(3)(1)で得たA形結晶の水和物8.5 gを15〜18℃にて2%の水を含むエタノール100 mLに加え、攪拌により溶解させた後に析出物を濾取し、乾燥して表題化合物のC形結晶の水和物
を得た(6.21 g)。
XRD: 5.5°, 13.4°, 14.3°, 21.4°, 26.7°.
【0052】
実施例6
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・n硝酸塩
参考例1の化合物200 mgをエタノール2 mLに溶解し、室温下硝酸0.07 mLを加え、4時間攪拌した。溶媒を留去後、酢酸エチル3 mLを加え、析出物を濾取し、減圧下にて乾燥して表題化合物を非晶質として得た(208 mg、収率80%)。(nは1ないし3を示す)
【0053】
実施例7
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・3メシル酸塩
(1)参考例1の化合物2.64 gをTHF25 mLに溶解し、室温下メシル酸1.32 mLを加え、1.5時間攪拌した。析出物を濾取し、減圧下にて乾燥して結晶を得た(3.52 g、収率80%)。
(2)上記結晶1.76 gをエタノール10 mLに加熱溶解し、室温にて17時間攪拌した。析出物
を濾取し、減圧下にて乾燥して表題化合物を結晶として得た(1.26 g、収率72%)。
DSC:193-197℃
Anal. calcd for C22H30N6OS・3CH4O3S・H2O: C, 40.97%; H, 6.05%; N, 11.47%; found: C, 41.05%; H, 5.72%; N, 11.48%.
【0054】
実施例8
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2メシル酸塩
(1)参考例1の化合物7.96 gをIPA60 mLに溶解し、室温下メシル酸3.59 gのIPA 20 mL溶
液を加え、2時間攪拌した。析出物を濾取し、減圧下にて乾燥して固体を得た(9.03 g、収率78%)。
(2)上記固体1000 mgをアセトニトリル20 mLに懸濁し、30分間加熱還流した。室温で冷却し、析出物を濾取して固体847 mgを得た。この固体813 mgをさらにアセトニトリル16mLに懸濁させ、1時間加熱還流した。室温で冷却し、析出物を濾取し、減圧下にて乾燥して表
題化合物を結晶として得た(690 mg、収率72%)。
DSC: 213-216℃
Anal. calcd for C22H30N6OS・2CH4O3S・0.5H2O: C, 45.92; H, 6.26; N, 13.39; found: C, 45.96; H, 6.17; N, 13.37.
【0055】
実施例9
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2トシル酸塩
(1)参考例1の化合物5.28 gをIPA 30 mLに溶解し、室温下トシル酸一水和物4.94 gを加
え、1.5時間攪拌した。析出物を濾取し、減圧下にて乾燥して表題化合物のA形結晶を得た(7.84 g、収率82%)。
XRD: 5.3°, 6.0°, 14.8°, 16.4°, 20.8°.
Anal. calcd for C22H30N6OS・2C7H8O3S・0.25H2O: C, 55.76%; H, 6.04%; N, 10.84%; found: C, 55.71%; H, 6.06%; N, 10.80%.
(2)上記生成物1.5 gを水20 mLに加熱溶解した。室温にて1時間攪拌し、析出物を濾取し
、減圧下にて乾燥して表題化合物のB形結晶を得た(1.2 g、収率80%)。
XRD: 5.7°, 11.4°, 14.0°, 18.2°, 19.7°.
Anal. calcd for C22H30N6OS・2C7H8O3S・0.5H2O: C, 55.43%; H, 6.07%; N, 10.77%; found: C, 55.14%; H, 6.09%; N, 10.73%.
(3)(1)の生成物1.4 gをIPA 100 mLに懸濁させ1時間加熱還流した。室温で冷却し、析出物を濾取し、減圧下にて乾燥して表題化合物のC形結晶を得た(1.1 g、収率81%)。
DSC: 227-230℃
XRD: 4.7°, 5.7°, 11.3°, 19.8°, 21.4°.
Anal. calcd for C22H30N6OS・2C7H8O3S: C, 56.08%; H, 6.01%; N, 10.90%; found: C, 55.83%; H, 6.11%; N, 10.87%.
【0056】
実施例10
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2べシル酸塩
(1)参考例1の化合物4.36 gをIPA 70 mLに溶解し、室温下ベシル酸3.78 gを加え、1時間攪拌した。析出物を濾取し、減圧下にて乾燥して表題化合物のA形結晶を得た(6.05 g、収率80%)。
XRD: 5.7°, 8.9°, 19.4°, 20.2°, 21.6°.
1H-NMR (DMSO-d6):δ 1.82-2.10 (1H, m), 2.17 (3H, s), 2.60-4.20 (16H, m), 4.11-4.72 (3H, m), 5.91 (1H, s), 7.31-7.35 (7H, m), 7.45-7.50 (2H, m), 7.59-7.62 (4H, m), 7.75 (2H, d, J = 7.8 Hz).
(2)上記生成物1.81 gをエタノール25 mLに加熱溶解した。室温にて1時間攪拌し、析出物を濾取し、減圧下にて乾燥して表題化合物のB形結晶を得た(1.25 g、収率69%)。
XRD: 5.6°, 6.7°, 19.3°, 22.9°, 23.2°.
Anal. calcd for C22H30N6OS・2C6H6O3S: C, 54.97%; H, 5.70%; N, 11.31%; found: C, 54.67%; H, 5.61%; N, 11.25%.
【0057】
実施例11
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2ナフタレン-1-スルホン酸塩
(1)参考例1の化合物4.01 gをTHF80 mLに溶解し、室温下ナフタレン-1-スルホン酸4.11 gのTHF 40 mL溶液を加え、3時間攪拌した。析出物を濾取し、減圧下乾燥して固体を得た(5.96 g、収率75%)。
(2)上記固体500 mgをエタノール25 mLに加熱溶解させ、30分間還流した。室温にて冷却
し、析出物を濾取し、減圧下乾燥して表題化合物を結晶として得た(445mg、収率89%)。
DSC: 184-189℃
Anal. calcd for C22H30N6OS・2C10H8O3S・0.25H2O: C, 59.52; H, 5.52; N, 9.92; found: C, 59.32; H, 5.46; N, 9.88.
【0058】
実施例12
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2ナフタレン-2-スルホン酸塩
(1)参考例1の化合物2.57 gを酢酸エチル50 mLに溶解し、室温下ナフタレン-2-スルホン酸1水和物2.86 gの酢酸エチル25 mL溶液を加え、12時間攪拌した。析出物を濾取し、減
圧下乾燥して固体を得た(4.60 g、収率91%)。
(2)上記固体500 mgをエタノール25 mLに加熱溶解した。室温にて攪拌し、析出物を濾取
し、減圧下乾燥して表題化合物を結晶として得た(372 mg、収率74%)。
DSC: 205-211℃
Anal. calcd for C22H30N6OS・2C10H8O3S・0.75H2O: C, 58.89; H, 5.59; N, 9.81; found: C, 58.96; H, 5.49; N, 9.76.
【0059】
実施例13
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・没食子酸塩
参考例1の化合物4.05 gをIPA 30 mLに溶解し、室温下没食子酸1水和物1.96 gのIPA溶
液30 mLを加え、1時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を固体として得た(4.84 g、収率85%)。
1H-NMR (DMSO-d6): δ1.43-1.62 (1H, m), 2.14 (3H, s), 2.19-3.08 (13H, m), 3.55-3.96(4H, m), 4.20-4.69 (2H, m), 5.91 (1H, s), 7.31-7.35 (7H, m), 7.45-7.50 (2H, m), 7.59-7.62 (4H, m), 7.75 (2H, d, J = 7.8 Hz).
【0060】
実施例14
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2(+)-カンファースルホン酸塩
(1)参考例1の化合物3.00 gをTHF 72.5 mLとt-ブチルメチルエーテル52.5 mLの混合溶媒に溶解し、室温下(+)-カンファースルホン酸3.25 gを加え、5時間攪拌した。析出物を
濾取し、減圧下乾燥して固体を得た(5.65 g、収率90%)。
(2)上記固体650 mgをエタノール7.0 mLとジエチルエーテル15.0mLの混合溶媒に加熱溶解した。室温にて一晩攪拌し、析出物を濾取し、温風下乾燥してエタノールを含有した表題化合物を結晶として得た(380mg、収率58%)。
TG/DTA: 142-156℃, 200-205℃
Anal. calcd for C22H30N6OS・2C10H16O4S・0.22C2H6O・2.5H2O: C, 53.66; H, 7.29; N, 8.86; found: C, 53.82; H, 7.27; N, 8.88.
【0061】
実施例15
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2(-)-カンファースルホン酸塩
参考例1の化合物3.00 gをTHF/t-ブチルメチルエーテルの1:3混合溶媒70 mLに溶解し
、室温下(-)-カンファースルホン酸3.25 gのTHF/t-ブチルメチルエーテルの1:3混合溶液を加え、1時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を固体として得た(5.66 g、収率91%)。
【0062】
実施例16
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2マレイン酸塩
(1)参考例1の化合物1.70 gをエタノール50 mLに溶解し、室温下マレイン酸0.98 gを加
え、1時間攪拌した。析出物を濾取し、減圧下にて乾燥して表題化合物のA形結晶を得た(1.87 g、収率71%)。
Anal. calcd for C22H30N6OS・2C4H4O4: C, 54.70%; H, 5.81%; N, 12.76%; found: C,
54.42%; H, 5.76%; N, 12.57%.
XRD: 8.6°, 15.8°, 17.8°, 18.6°, 23.4°.
(2)上記結晶3.0 gを水15 mLに加熱溶解した。室温にて1時間攪拌し、析出物を濾取し、
減圧下にて乾燥して表題化合物のB形結晶を得た(1.83 g、収率61%)。
XRD: 5.9°, 13.4°, 16.3°, 17.6°, 23.9°.
Anal. calcd for C22H30N6OS・2C4H4O4・2H2O: C, 51.86%; H, 6.09%; N, 12.10%; found: C, 51.80%; H, 5.84%; N, 12.10%.
【0063】
実施例17
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2フマル酸塩
(1)参考例1の化合物1.50 gをエタノール20 mLに溶解し、室温下フマル酸814 mgのエタ
ノール25mL溶液を加え、室温下1時間攪拌後氷冷下1時間攪拌した。反応溶媒を1/3留去し
、析出物を濾取し、減圧下にて乾燥して固体を得た(1.77 g、収率77%)。
(2)(1)の固体200 mgをアセトニトリル5 mLに懸濁させ4時間加熱還流した。室温にて冷
却後、析出物を濾取して表題化合物の結晶を得た(141 mg、収率71%)。この結晶はDSCで2
つの吸熱ピークを観測したことから2つの結晶形(A、B)の混合物、あるいは熱により結晶
形AがBに転移するものと推測した。
DSC: 128-(135及び142)℃
XRD: 3.1°, 15.2°, 17.4°, 23.4°, 25.5°.
Anal. calcd for C22H30N6OS・2C4H4O4: C, 54.70; H, 5.81; N, 12.76; found: C, 54.40; H, 5.88; N, 12.63
1H-NMR (DMSO-d6): δ1.50-1.78 (1H, m), 2.14 (3H, m), 2.37-3.90 (16H, m), 4.10-4.72 (3H, m), 5.79 (1H, s), 6.57 (4H, s), 7.27 (1H, t, J = 7.2 Hz), 7.46 (2H, t, J = 8.1 Hz), 7.74 (2H, d, J = 7.7 Hz).
(3)(1)の固体200 mgを水2 mLに溶解させた。室温にて攪拌し、析出物を濾取することにより表題化合物を結晶として得た(47.5mg、収率24%)。この結晶は(2)で得た結晶とは異
なる粉末X線のパターンを示した。
XRD: 9.4°, 17.8°, 19.6°, 21.0°, 23.5°, 24.3°.
【0064】
実施例18
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・1.6硫酸塩
参考例1の化合物2.00gをTHF 40 mLに溶解し、室温下0.5 mol/L硫酸水溶液14.5 mLを加え、0.5時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を固体として得た(2.57 g、収率94%)。
Anal. calcd for C22H30N6OS・1.6H2O4S・2H2O: C,42.65; H,6.05; N, 13.57; found: C, 42.56; H, 5.67; N, 13.44.
【0065】
実施例19
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2 L-酒石酸塩
(1)参考例1の化合物1.17 gをIPA 30 mLに溶解し、室温下L-酒石酸823 mgを加え、3時間攪拌した。析出物を濾取し、減圧下乾燥して結晶を得た(1.55 g、収率78%)。
(2)上記生成物254 mgを酢酸エチル10 mLに懸濁させ1.5時間加熱還流した。室温にて冷却し、析出物を濾取し、減圧下にて乾燥して表題化合物を結晶として得た(250 mg、収率98%)。
1H-NMR (DMSO-d6): δ1.50-1.69 (1H, m), 2.14 (3H, m), 2.40-3.90 (16H, m), 4.08 (2H, s), 4.30-4.70 (3H, m), 5.79 (1H, s), 7.27 (1H, t, J = 7.3 Hz), 7.46 (2H, m),
7.73 (2H, d, J = 7.8 Hz).
【0066】
実施例20
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・mリン酸塩
参考例1の化合物100 mgをTHF2 mLに溶解し、室温下リン酸0.032 mLを加え、1時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を非晶質として得た(144 mg、収率93%)。(mは1ないし3を示す)
【0067】
試験例1 粉末X線回折の測定
以下の測定条件で実施例4および5の表題化合物の粉末X線回折を測定した。
装置:島津製作所製 XRD-6000
対陰極:Cu
モノクロメーター:Graphite
管電圧:40 kV
管電流:40 mA
発散スリット:1°
受光スリット:0.15 mm
散乱スリット:1°
測定範囲:2〜40°(2θ)
検体回転速度:60 rpm
実施例4の表題化合物の、粉末X線回折の測定結果を図1に示す。
実施例5の表題化合物のA形結晶の粉末X線回折の測定の結果を図2に示す。
実施例5の表題化合物のB形結晶の粉末X線回折の測定の結果を図3に示す。
実施例5の表題化合物のC形結晶の粉末X線回折の測定の結果を図4に示す。
【0068】
試験例2 吸湿性の測定
水分吸着測定装置を用いて以下の測定条件で実施例3の表題化合物の吸湿性を測定した
。
装置:VTI社製 MB-300G
測定温度:25℃
測定範囲:0〜95% RH
吸湿性の測定結果を図5に示した。
実施例3の表題化合物を、減圧型の水分吸着測定装置を用いて水分吸着測定を行ったとこ
ろ、50%RHで1.8水和相当の水を保持し、0%RHではほぼ完全に乾燥していることが分かった。
【0069】
試験例3 溶解度の測定
(1)水への溶解度の測定
測定法として、少量の試料で簡便に大まかな溶解性を把握できる目視法を用いた。温度は37℃で測定した。実施例3の表題化合物約3 mgをスクリューキャップ付きサンプル瓶に
取り、試験液0.15 mLを加えて密栓した。1分間超音波をかけて試料を分散させた後、こ
れをあらかじめ37℃に安定させた恒温振とう水槽に入れ、1時間振とうした後、目視観察
で溶解を確認した。その結果、実施例3の表題化合物の水に対する溶解度は37℃において20 mg/mL以上であった。
(2)pH9〜13における溶解度の測定
試験液として0.2 mol/L NaOH/0.1 mol/L NaCl混合溶液を用いて、pHを9〜13に調整した時の室温での実施例3の表題化合物の溶解度について、液体クロマトグラフ法(HPLC)によ
り分析した(n=3)。 その結果、溶解度は6.4 mg/mL〜8.4 mg/mLであった。以上より実施
例3の表題化合物の溶解度は約7 mg/mLと結論付けた。
【0070】
【表1】
【産業上の利用可能性】
【0071】
化合物Iの塩若しくはその溶媒和物、あるいはそれぞれの新規の結晶多形は、安定性の
改善、吸湿性(潮解性)の改善、溶媒からの迅速な単離、及び製剤化の容易性から選択される1つ以上の特性を有しており、化合物Iの医薬品としての開発を促進する。
【0072】
なお、本願は特願2005-041851号を優先権主張して出願されたものである。
【技術分野】
【0001】
本発明は、ジペプチジルペプチダーゼ−IV(以下、DPP-IVと記す)阻害薬として有用な3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンの新規な塩、またはその溶媒和物に関する。
【背景技術】
【0002】
DPP-IV阻害薬は、血漿中のグルカゴン様ペプチド-1(以下、GLP-1と記す)の不活性化を
阻害し、そのインクレチン作用を増強するため、糖尿病治療薬等として有用であり、糖尿病、特に2型糖尿病の治療において有効であり得る薬剤として研究開発段階にある(特許文献1ないし6、非特許文献1参照)。
【0003】
有用なチアゾリジン誘導体についても一連の化合物が報告されている。 (特許文献7参
照)。この文献に記載された実施例化合物の中でも、注目すべきものとして、3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンがある(以下、化合物Iと記す)。化合物Iは3塩酸塩の形態で記載されているが、この塩は安定性や吸湿性の面で薬学的に望ましくない特性を有し、同一形態で再現性をもって製造するのが困難であることが認められた。医薬品の開発における規制要件を満たすためには、特に、一定の品質の化合物を、再現性をもって製造しなければならないので、化合物Iの3塩酸塩に認められるこれらの特性は医薬品の開発には不利なものと考えられる。
【0004】
また、この文献(特許文献7)には「化合物I」ならびにその他のチアゾリジン誘導体における特定の塩が実施例化合物として開示されているが、いずれの実施例化合物においても結晶多形に関する議論はなされていない。
【0005】
ある物質が2種以上の結晶構造に結晶化する能力はポリモルフィズム(polymorphism)と
して知れらており、個々の結晶形は結晶多形と呼ばれている。同一化合物の異なる結晶多形は、保存安定性や溶解性等の物性が全く異なる場合がある。これらの物性の違いは作用効果における差に結びつく場合がある。これらの相違により、個々の結晶多形や結晶多形の混合物の検討は、医薬品開発において特に有用である。
【0006】
なお、結晶多形は命名方法により、A形、B形、I形、II形、α形、β形等複数の表記が
存在する。また、これらの表記において、「形」の代わりに「型」(A型等)を使用する場
合もあるが、いずれの表記であっても同様の意味で用いられる。
【0007】
しかしながら、ある化合物において種々の結晶多形を見つけることは、必ずしも容易に検討できるとは限らない。さらに、ひとたび特定の結晶多形の存在が認識され、その特徴が好ましいものであるとされた場合、 研究者はその結晶多形を常に一定に単一結晶とし
て多量に供給する方法を見出さなければならない。ある結晶多形の単一結晶あるいは実質的な単一結晶を供給する方法を確立することは容易ではなく、鋭意検討を必要とする。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第97/040832号パンフレット
【特許文献2】国際公開第98/019998号パンフレット
【特許文献3】米国特許第5939560号
【特許文献4】国際公開第01/055105号パンフレット
【特許文献5】国際公開第02/002560号パンフレット
【特許文献6】国際公開第02/062764号パンフレット
【特許文献7】国際公開第02/014271号パンフレット
【非特許文献】
【0009】
【非特許文献1】J. Med. Chem., 47(17), 4135-4141(2004)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明における課題は、化合物Iに関して、医薬品とするために所望される、安定性、
溶解性、吸湿性、または生物学的利用能等の面で優れた特性および再生可能な結晶構造を有する化合物を見出すこと、並びにそれらを製造する方法を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、化合物Iの一ないし三塩基酸塩を調製し、それぞれの塩またはそれらの
溶媒和物の結晶を特徴付け、安定性並びに吸湿性の点で好ましい特性を有する化合物Iの
新規塩を見出した。さらに鋭意研究を重ねた結果、本発明の新規塩の安定的な工業的調製方法を見出し、本発明を完成するに至った。
【0012】
すなわち、本発明の要旨は以下の(1)ないし(33)に記す塩またはその溶媒和物、および
それらの製造方法に存する。
(1) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンと有機または無機の一ないし三塩基酸との塩、またはその溶媒和物、
(2) 有機または無機の一塩基酸が塩酸、臭化水素酸、硝酸、メシル酸、トシル酸、ベシル酸、塩酸、ナフタレン-1-スルホン酸、ナフタレン-2-スルホン酸、没食子酸、またはカンファースルホン酸である前記(1)に記載の塩、またはその溶媒和物(ただし、一塩基酸が塩酸の場合、塩は2または2.5塩酸塩である)、
(3) 有機または無機の二塩基酸がフマル酸、マレイン酸、硫酸、コハク酸、L-酒石酸、エタンジスルホン酸、またはクエン酸である前記(1)に記載の塩、またはその溶媒和物、
(4) 有機または無機の三塩基酸がリン酸である前記(1)に記載の塩、またはその溶媒和物
、
(5) 2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2ベシル酸、2塩酸、2.5
塩酸、2ナフタレン-1-スルホン酸、2ナフタレン-2-スルホン酸、2カンファースルホン酸
、フマル酸、硫酸、コハク酸、L-酒石酸、またはクエン酸との塩である、前記(1)に記載
の塩、またはその溶媒和物、
(6) 2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2.5塩酸、2ナフタレン-1-スルホン酸、2メシル酸、3メシル酸、または2ナフタレン-2-スルホン酸との塩である前
記(5)記載の塩、またはその溶媒和物、
(7)周囲温度での水への溶解度が7mg/mLないし2g/mLである一塩基酸と3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカル
ボニル}チアゾリジンとの塩、またはその溶媒和物、
(8) 37℃での水への溶解度が20 mg/mL以上である前記(7)に記載の塩、またはその溶媒和
物、
(9) pH9から12での水への溶解度が7 mg/mLである前記(7)に記載の記載の塩、またはその
溶媒和物、
(10) 25℃で測定した吸湿性が6%以下である前記(1)に記載の塩、またはその溶媒和物、
(11) 25℃で測定した相対湿度0%から50%の範囲における吸湿性が5%である前記(10)に記載の塩、またはその溶媒和物、
(12) 25℃で測定した相対湿度5%から90%の範囲における吸湿性が2%である前記(10)に記
載の塩、またはその溶媒和物、
(13) 塩が前記(2)記載の一塩基性酸との塩である前記(10)ないし(12)いずれかに記載の塩、またはその溶媒和物、
(14) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物、
(15) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°及び14.4°(それぞれ±0.2°) にピークを有することを特徴とする前記(14)に記載の塩、またはその水和物、
(16) 1.0ないし2.0水和物である前記(15)に記載の水和物、
(17) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°, 14.4°, 22.6°及び26.5° (それぞれ±0.2°) にピークを有することを特徴とする前記(14)に記載の塩、またはその水和物、
(18) 1.0ないし2.0水和物である前記(17)に記載の水和物、
(19) 図1に例示される粉末X線回折パターンを示す、前記(14)に記載の塩、またはその水
和物、
(20) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0臭化水素酸塩、またはその溶媒和物、(21) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.7°, 7.7°, 11.3°, 16.2°及び17.0° (それぞれ±0.2°) にピークを有することを特徴とする前記(20)に記載の塩の水和物、
(22) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.2°, 10.4°, 19.1°, 19.8°及び20.7° (それぞれ±0.2°) にピークを有することを特徴とする前記(20)に記載の塩の水和物、
(23) 粉末X線回折パターンにおいて、2θで表される回折角度として、5.5°, 13.4°, 14.3°, 21.4°及び26.7° (それぞれ±0.2°) にピークを有することを特徴とする前記(20)に記載の塩の水和物、
(24) 図2に例示される粉末X線回折パターンを示す、前記(20)に記載の塩の水和物、
(25) 図3に例示される粉末X線回折パターンを示す、前記(20)に記載の塩の水和物、
(26) 図4に例示される粉末X線回折パターンを示す、前記(20)に記載の塩の水和物、
(27) 3-{(2S,4S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンに対して臭化水素酸で脱1,1-ジメチルエチルオキシカルボニル反応を行うと共に同時に造塩を行うことを特徴とする3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イ
ル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法、
(28) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩を、許容される溶媒を用いて結晶化する工程を含む3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イ
ル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法、
(29) 許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”において1日摂取許容量(“PDE”)が10 mg/dayを越える溶媒から選ばれる溶媒である前記(28)に記
載の方法、
(30) 許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”におけるクラス3の溶媒から選ばれる溶媒である前記(28)に記載の方法、
(31) 許容される溶媒が、エタノール、1-プロパノール、2-プロパノール、酢酸エチル、
アセトンから選ばれる溶媒である前記(28)に記載の方法、
(32) 許容される溶媒がエタノール及び/または水である前記(28)に記載の方法、または
(33) 3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩の1.0ないし2.0水和物。
【発明の効果】
【0013】
化合物Iの塩若しくはその溶媒和物、あるいはそれぞれの新規の結晶多形は、安定性の
改善、吸湿性(潮解性)の改善、溶媒からの迅速な単離、及び製剤化の容易性から選択される1つ以上の特性を有しており、化合物Iの医薬品としての開発を促進する。
【図面の簡単な説明】
【0014】
【図1】実施例4の表題化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
【図2】実施例5の(1)の化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
【図3】実施例5の(2)の化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
【図4】実施例5の(3)の化合物の粉末X線回折の測定結果である。Y軸を回折強度とし、横軸を回折角(2θ)で表した。
【図5】実施例3の表題化合物の吸湿性の測定結果である。―○―は横軸の湿度における化合物への水の吸着、―●―は横軸の湿度における化合物への水の脱着をプロットしたものである。
【発明を実施するための形態】
【0015】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン(化合物I)を以下に示す。
【0016】
【化1】
【0017】
化合物Iの3塩酸塩は、国際公開第02/14271号パンフレットの実施例222として記載され
ている合成方法に準じて製造することができる。このものは適当な塩基を用いて遊離塩基とすることができる。用いる塩基としては、アルカリ金属またはアルカリ土類金属炭酸塩(炭酸水素ナトリウム、炭酸ナトリウム、または炭酸カリウム等)あるいはアルカリ金属またはアルカリ土類金属の水酸化物(水酸化ナトリウム、または水酸化カリウム等)等が挙げられる。
【0018】
例えば、実施例222の化合物を上記いずれかの塩基の水溶液に加えた後に、炭化水素系
溶媒(ベンゼン、またはトルエン等)、ハロゲン系炭化水素溶媒(ジクロロメタン、ジクロ
ロエタン、クロロホルム、または四塩化炭素等)、酢酸エチル等で抽出することにより化
合物Iを得ることができる。
【0019】
さらに、化合物Iの2.5臭化水素酸塩は以下のスキームによっても製造することが出来る。
【0020】
【化2】
【0021】
1-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン(II)またはその塩と3-[(2S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-オキソピロリジン-2-イルカルボニル]
チアゾリジン(III)を還元的アミノ化反応させた後、臭化水素酸により脱1,1-ジメチルエチルオキシカルボニル反応させると化合物Iの2.5臭化水素酸塩(IV)を得ることができる。
この還元的アミノ化反応は、式(II)で表される化合物またはその塩の1モルに対して、式(III)で表される化合物を約0.5ないし10モル、好ましくは約1ないし2モル用い、金属水素錯化合物(水素化ほう素ナトリウム、シアノ水素化ほう素ナトリウム、トリアセトキシ
水素化ほう素ナトリウム等の複合水素化合物、またはジボラン等)を約0.5ないし10モル
、好ましくは約1ないし2モル用い、必要に応じて酸性触媒(酢酸、p-トルエンスルホン酸、または三フッ化ホウ素・ジエチルエーテル錯体等)の存在下、不活性な溶媒中で行われる。不活性な溶媒としては、アルコール類(メタノール、エタノール、1-プロパノール、2-プロパノール(以下、IPAと記す)、またはブタノール等)、ニトリル類(アセトニト
リル、またはプロピオニトリル等)、アミド類(ホルムアミド、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、または1,3-ジメチル-2-イミダゾリジノン等)、エーテ
ル類(ジエチルエーテル、ジイソプロピルエーテル、t-ブチルメチルエーテル、1,4-ジオキサン、またはテトラヒドロフラン(以下、THFと記す)等)、ハロゲン化炭化水素類(ジクロロメタン、クロロホルム、または1,2-ジクロロエタン等)、炭化水素類(n-ヘキサン、シクロヘキサン、ベンゼン、またはトルエン等)、またはこれらの任意の混合溶媒等が挙
げられる。
【0022】
このとき反応温度は-20ないし200℃、好ましくは0ないし80℃であり、反応時間は約0.5ないし96時間、好ましくは0.5ないし24時間である。
この反応の生成物を単離精製せず、水、アルコール類(メタノール、エタノール、またはIPA等)、エーテル類(THF、ジオキサン等)、ハロゲン化炭化水素類(ジクロロメタン、ジクロロエタン、クロロホルム等)、酢酸エチル、アセトニトリル等、またはこれらの任意の混合溶媒中、式(II)の化合物またはその塩の1モルに対して、臭化水素酸を1ないし20モル、好ましくは約2.5ないし5モルを反応させると化合物Iの2.5臭化水素酸塩を得ることができる。
【0023】
このとき反応温度は-20ないし200℃、好ましくは0ないし100℃であり、反応時間は約0.5ないし48時間、好ましくは0.5ないし24時間である。反応後、析出物をろ取することにより式(IV)で表される塩を得ることができる。
【0024】
本発明の化合物Iの塩酸塩、臭化水素酸塩、硝酸塩、メシル酸塩、マレイン酸塩、トシ
ル酸塩、ベシル酸塩、ナフタレン-1-スルホン酸塩、ナフタレン-2-スルホン酸塩、没食子酸塩、(+)-カンファースルホン酸塩、(-)-カンファースルホン酸塩、フマル酸塩、硫酸塩、コハク酸塩、L-酒石酸塩、エタンジスルホン酸塩、クエン酸塩、またはリン酸塩(以下、「本発明の塩」と略すこともある)は、光学的に純粋であり、例えば(2S,4S) -エナン
チオマーの光学純度は、90%以上のエナンチオマー過剰(以下、e.e.と記す)、好ましく
は95%e.e. 以上、より好ましくは99%e.e. 以上である。
【0025】
本発明の塩の形態は特に制限されず、油状物、非晶質(アモルファス)、結晶であってもよい。好ましい塩の形態は結晶である。
【0026】
前記結晶の形態の塩としては、2.0塩酸塩、2.5 塩酸塩、2臭化水素酸塩、2.5臭化水素
酸塩、2メシル酸塩、3メシル酸塩、2トシル酸塩、2ベシル酸塩、2ナフタレン-1-スルホン酸塩、2ナフタレン-2-スルホン酸塩、2(+)-カンファースルホン酸塩、2マレイン酸塩、2
フマル酸塩、または2L-酒石酸塩等が例示できる。これらの塩は粉末X線回折パターンにおける回折ピークによっても特徴付けることが出来る。
【0027】
本発明において、2.0臭化水素酸塩の結晶多形を、A形、B形およびC形と称する。前記結晶の形態の塩も本明細書において適当ならば2.0臭化水素酸塩と称する。
また、2トシル酸塩の結晶多形をA形、B形およびC形と、2トシル酸塩の結晶多形をA形、B形およびC形と、2ベシル酸塩の結晶多形をA形およびB形と、2マレイン酸塩の結晶多形をA形およびB形と、2フマル酸塩の結晶多形をA形およびB形とそれぞれ称する。
【0028】
本発明の塩の溶媒和物は、へミ−、モノ−、ジ−、トリ−、テトラ−、ペンタ−、ヘキサ−等の溶媒和物として存在し得る。結晶化に使用する溶媒(アルコール(メタノール、エタノール、またはIPA等)、アルデヒド、ケトン(アセトン等)、またはエステル(酢酸エチ
ル等)等)およびこれらの溶媒に含まれる水は、結晶格子中に取り込まれ得る。一般的に、溶媒が結晶化およびその後の製造工程において、溶媒和物となるか非溶媒和物となるかを予測することは不可能である。それらは化合物、製造条件ならびに選択された溶媒、とり
わけ水との間のさまざまな相互作用の組合せに依存する。さらに、ある化合物の塩、またはその溶媒和物の結晶または非晶質のそれぞれの安定性は、実測値によってのみ確認され得るものである。
【0029】
本発明の塩は、溶媒(水、または有機溶媒等)の溶媒和物であってもよく、非溶媒和物であってもよい。すなわち、本発明の塩は、水和物であってもよく、非水和物であってもよい。水和物である場合、水和する水の量は種々の条件によって変動することもあるが、好ましくは2.0以下の水和物であり、より好ましくは1.0ないし2.0水和物である。
【0030】
本発明の塩は、哺乳動物に対して安全な(薬学上、薬理学上、または生理学上に許容さ
れるもの等)溶媒を含んでいてもよく、または溶媒との溶媒和であってもよい。「溶媒」
としては、“残留溶媒Q3CのICHガイドライン”において1日摂取許容量(“PDE”)が10 mg/dayを越えるもの、及び/または、“残留溶媒Q3CのICHガイドライン”におけるクラス3の
ものから選ばれる。具体的には、エタノール、1-プロパノール、IPA、1-ブタノール、2-
ブタノール、1-ペンタノール、酢酸、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸n-ブチル、酢酸イソブチル、ギ酸、ギ酸エチル、アセトン、メチルエチルケトン、メチルイソブチルケトン、ヘプタン、ペンタン、ジエチルエーテル、t-ブチルメチルエーテル、THF、アニソール、クメン、またはジメチルスルホキシド等が挙げられる
。これらの溶媒のうち、エタノールが好ましい。「溶媒」の含有量は、50000 ppm以下、
好ましくは5000 ppm以下である。
【0031】
本発明の塩は自体公知の方法により製造することができ、例えば、化合物Iと、塩酸、
臭化水素酸、硝酸、メシル酸、マレイン酸、トシル酸、ベシル酸、ナフタレン-1-スルホ
ン酸、ナフタレン-2-スルホン酸、没食子酸、(+)-カンファースルホン酸、(-)-カンファ
ースルホン酸、フマル酸、硫酸、コハク酸、L-酒石酸、エタンジスルホン酸、クエン酸、またはリン酸から選択された有機酸または無機酸とを反応させることにより、本発明の塩を得ることができる。
【0032】
本反応は、一般に無溶媒または不活性な溶媒中で行われる。「不活性な溶媒」としては、水、アルコール類(メタノール、エタノール、1-プロパノール、IPA、またはブタノール等)、ケトン類(アセトン、またはメチルエチルケトン等)、ニトリル類(アセトニトリ
ル、またはプロピオニトリル等)、アミド類(ホルムアミド、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、または1,3-ジメチル-2-イミダゾリジノン等)、エーテル
類(ジエチルエーテル、ジイソプロピルエーテル、t-ブチルメチルエーテル、1,4-ジオキサン、またはTHF等)、エステル類(ギ酸エチル、酢酸エチル、または酢酸プロピルなど)
、ハロゲン化炭化水素類(ジクロロメタン、クロロホルム、または1,2-ジクロロエタン等)、炭化水素類(n-ヘキサン、シクロヘキサン、ベンゼン、またはトルエン等)、スルホキシド類(ジメチルスルホキシド等)、極性溶媒(スルホラン、またはヘキサメチルホスホリルアミド等)またはこれらの任意の混合溶媒等が挙げられる。これらの溶媒の中でも、水、水とアルコールとの混合溶媒(水とメタノールとの混合溶媒、水とエタノールとの混
合溶媒、水と1-プロパノールとの混合溶媒、または水とIPAとの混合溶媒等)が好ましい。
【0033】
「不活性な溶媒」は、化合物Iに対し、通常、1ないし100w/v%、好ましくは2ないし50 w/v%が用いられる。
【0034】
反応温度は、通常、-20℃ないし溶媒の還流温度、好ましくは0℃ないし溶媒の還流温度である。反応時間は、通常、約1分ないし24時間、好ましくは約10分ないし6時間、さらに好ましくは約30分ないし3時間である。
【0035】
このようにして得られた塩は、自体公知の分離手段(濃縮、減圧濃縮、溶媒抽出、結晶
化、再結晶、転溶、またはクロマトグラフィー等)により、反応混合物から単離、精製す
ることができる。
【0036】
このようにして得られた塩を結晶化に付すことにより、結晶の状態で化合物Iの塩を得
ることができる。この結晶化方法としては自体公知の方法が挙げられ、溶液からの結晶化、蒸気からの結晶化、溶融体からの結晶化が挙げられる(A. S. Myerson Ed., Handbook of Industrial Crystallization Second Edition, Butterworth-Heinemann 2002 参照)。
【0037】
「溶液からの結晶化」の方法としては、濃縮法、徐冷法、反応法(拡散法、または電解
法)、水熱育成法、または融剤法等が挙げられる。用いられる溶媒としては、前記「不活
性な溶媒」と同様の溶媒が挙げられる。
【0038】
「蒸気からの結晶化」の方法としては、気化法(封管法、または気流法)、気相反応法、または化学輸送法等が挙げられる。
【0039】
「溶融体からの結晶化」の方法としては、ノルマルフリージング法(引上げ法、温度傾
斜法、またはブリッジマン法)、帯融解法(ゾーンレベリング法、またはフロートゾーン法)、または特殊成長法(VLS法、または液相エピタキシー法)等が挙げられる。なお、化合物Iの塩の結晶化には、一般的に、40℃ないし使用する溶媒の還流温度程度に加熱して化合
物Iの塩を溶解した溶液の冷却による晶析、または化合物Iの塩を溶解した溶液(特に濃縮
液)への貧溶媒の添加による晶析等が利用される。得られた結晶の解析方法としては、X線解析の方法が一般的である。X線解析の測定結果は、Y軸を回折強度とし、横軸を回折角(2θ)として表されるが、2θ値は同一の結晶形を測定した場合であってもある程度の範囲でバラツキが観察される。具体的には±0.2°が一般的なバラツキの程度であるが、測定条
件などによって、より大きな誤差を生じる場合もある。2θ値により結晶形を比較する際
、当業者においてこれらのバラツキを考慮して結晶形の比較が行われる。さらに、本発明の塩またはその溶媒和物は水分含量によって回折角に多少のずれを生じることがあるが、これらも本発明の範疇に含まれる。
【0040】
本発明の塩またはその溶媒和物(以下、単に本発明の塩と記す)は、安定性に優れているため、長期にわたって室温で保存できるだけでなく、製造工程や保管において煩雑な操作を必要としないこと、また、製剤化が容易なことから医薬品の原薬として有用である。さらには本発明の塩は水への溶解度が高いため、注射用製剤としてより自由度の高い投与形態を開発可能である。
本発明の塩を医薬として用いる場合、本発明の塩を製剤上許容しうる担体(賦形剤、結
合剤、崩壊剤、矯味剤、矯臭剤、乳化剤、希釈剤、または溶解補助剤等)と混合して得ら
れる医薬組成物あるいは製剤(錠剤、ピル剤、カプセル剤、顆粒剤、散剤、シロップ剤、
エマルジョン剤、エリキシル剤、懸濁剤、溶液剤、注射剤、点滴剤、または坐剤等)の形態で経口的または非経口的に投与することができる。医薬組成物は通常の方法にしたがって製剤化することができる。
【0041】
本明細書において、非経口とは、皮下注射、静脈内注射、筋肉内注射、腹腔内注射あるいは点滴法等を含むものである。注射用調剤は当該分野で知られた方法で調製することができる。直腸投与用の坐剤は、その薬物と適当な補形剤等と混合して製造することができる。経口投与用の固形投与剤型としては、粉剤、顆粒剤、錠剤、ピル剤、またはカプセル剤等の上記したものが挙げられる。経口投与用の液剤は、医薬として許容されるエマルジョン剤、シロップ剤、エリキシル剤、懸濁剤、または溶液剤等が挙げられる。
【0042】
本発明の塩の投与量は年齢、体重、一般的健康状態、性別、食事、投与時間、投与方法、排泄速度、薬物の組合せ、または患者のその時に治療を行っている病状の程度に応じ、
それらあるいはその他の要因を考慮して決められる。本発明の塩は、低毒性で安全に使用することができ、その1日の投与量は、患者の状態や体重、塩の種類、投与経路等によっ
て異なるが、例えば非経口的には皮下、静脈内、筋肉内または直腸内に、0.01ないし100 mg/kg体重/日、好ましくは0.05ないし50 mg/kg体重/日を投与され、また経口的には0.01
ないし100 mg/kg体重/日、好ましくは0.05ないし50 mg/kg体重/日を、一日1ないし数回
に分けて投与するのが好ましい。
【実施例】
【0043】
以下に参考例及び実施例を挙げて本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。
【0044】
なお、抽出における有機溶液の乾燥には、特に明記しない限り、無水硫酸ナトリウムまたは無水硫酸マグネシウムを使用した。カラムクロマトグラフィーは富士シリシア化学社製のシリカゲルを用いて行った。
また、熱分析(DSC)は、熱量曲線における融解開始前の直線部分と、融解中の直線部分
との延長上の交点の温度(onset値)及び、熱量曲線における融点付近の変極点の温度(peak
top値)を示した。粉末X線回折パターン(XRD)は角度2θの特徴的なピークを示した(±0.2°)。1H-NMRは300 MHz核磁気共鳴分光装置で測定した。1H-NMRのケミカルシフトは、内部標準としてテトラメチルシラン(TMS)を用い、相対的なデルタ(δ)値をパーツパーミリオ
ン(ppm)で表した。カップリング定数は自明な多重度をヘルツ(Hz)で示し、s(シングレッ
ト)、d(ダブレット)、t(トリプレット)、m(マルチプレット)等と表した。赤外分光(IR)の吸光度の強度をst(強)、m(中)、w(弱)で表した。
【0045】
なお、以下の参考例、実施例の表題化合物は非溶媒和物として表記するが、それぞれ塩は調製時の条件等により溶媒和物(特に水和物)の形態をとることもある。
【0046】
参考例1
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン
3-{(2S,4S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン(国際公開第02/14271号パンフレットの実施例222に記載された化合物に準じて合成)25.45 g
をジクロロメタン200 mLに溶解し、室温下トリフルオロ酢酸50 mLを加え、19時間攪拌し
た。反応液を減圧下濃縮し、残渣に飽和炭酸水素ナトリウム水溶液を加え、クロロホルムで抽出した。抽出液を飽和食塩水で洗浄、乾燥後、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィーで精製して表題化合物を固体として得た(19.28g、収率93%)
。
【0047】
実施例1
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5塩酸塩
(1)参考例1の化合物 2.50 gをTHF 100 mLに溶解し、室温下4 mol/L塩酸の酢酸エチル溶
液3.0 mLを加えた後、1時間攪拌した。析出物を濾取し、減圧下50℃で乾燥して固体2.69 gを得た。
(2)上記生成物300 mgを水150μLとエタノール 1.0 mLの混合溶媒に加熱溶解し、氷冷下1時間攪拌した。析出物を濾取し、減圧下50℃にて乾燥して表題化合物を結晶として得た(144 mg、収率48%)。
XRD: 5.2°, 14.3°, 16.2°, 21.8°, 25.2°.
Anal. calcd for C22H30N6OS・2.3HCl・2H2O: C, 48.35; H, 6.69; N, 15.38; found: C,48.02; H, 6.60; N, 15.20.
【0048】
実施例2
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0塩酸塩
実施例1の表題化合物60 mgを酢酸エチル3.0 mLに懸濁し、13時間加熱還流した。室温
で冷却し、析出物を濾取し、40℃の温風下乾燥して表題化合物を結晶として得た(50 mg収率85%)。
XRD: 5.0°, 14.8°, 21.0°, 21.5°, 25.2°.
Anal. calcd for C22H30N6OS・2.0HCl・H2O: C, 51.05; H, 6.62; N, 16.24; found: C, 50.89; H, 6.58; N, 16.12.
【0049】
実施例3
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩
参考例1の化合物5.09 gをエタノール50.9 mLに溶解し、還流温度にて48%臭化水素酸5.03 gを加えた後、攪拌しながら約1時間かけて室温まで冷却し、室温にてさらに1時間攪拌した。析出物を濾取後、エタノール5 mLで洗浄し、45℃の温風下乾燥して表題化合物を結晶として得た(6.76 g)。
融点: 202.0℃(分解)
IR(KBr): 3600-3300 (st), 3116-2850 (st), 2800-2400 (st), 1647 (st), 1592 (m), 1572 (m), 1496 (m), 1450 (m), 1385 (m), 1361 (w), 768 (m), 692 (w) .
【0050】
実施例4
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩(実施例3の表題化合物の別途
合成方法)
(1)水素化トリアセトキシホウ素ナトリウム13.68 kgのトルエン300 L懸濁液に1-[4-(3-
メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン・酢酸塩15.00 kg、3-[(2S) -1-(1,1-ジメチルエチルオキシカルボニル)-4-オキソピロリジン-2-イルカルボニル] チアゾリジン14.90 kgを加えて、室温で2.5時間攪拌した。反応液に水90 Lを滴下し、0.5時間攪拌した後、トルエン層を分離した。このトルエン層を5%炭酸水素ナトリウム水溶液90 L、水90 Lの順で洗浄し、減圧下濃縮、乾固した。この残渣にIPA 224 Lを加え、約80℃で48%臭化水素酸25.08 kgを滴下後、2.5時間還流した。反応液を冷却し、約60℃で1.5時間、次いで約40℃で2時間、さらに室温で2時間攪拌した。析出物を濾取後、IPA 30 Lで洗浄し、温風下乾燥して表題化合物の固体を得た(29.76 kg、収率91%)。
(2)(1)で得た固体28.00 kgにエタノール168 Lを加え、加熱溶解後、熱時濾過し、反応
容器をエタノール28 Lで洗浄した。この濾液及び洗液を合わせ、67 ℃で水3 Lを加えた後に冷却し、49 ℃で1時間、次いで20〜15 ℃で1時間攪拌した。析出物を濾取後、エタノール28 Lで洗浄し、温風下乾燥して表題化合物を結晶として得た(25.84 kg、収率 92%)。
XRD: 5.4°, 13.4°, 14.4°, 22.6°, 26.5°.
【0051】
実施例5
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0臭化水素酸塩
(1)実施例4の表題化合物130 gを室温にて水260 mLに加え、攪拌により溶解させた後に析出物を濾取し、乾燥して表題化合物のA形結晶の3.5水和物を得た(53.58 g)。
XRD: 5.7°, 7.7°, 11.3°, 16.2°, 17.0°.
(2)(1)で得たA形結晶の水和物8.5 gを28〜30℃にて2%の水を含むエタノール100 mLに加え、攪拌により溶解した後に析出物を濾取し、乾燥して表題化合物のB形結晶の水和物を
得た(4.56 g)。
XRD: 5.2°, 10.4°, 19.1°, 19.8°, 20.7°.
(3)(1)で得たA形結晶の水和物8.5 gを15〜18℃にて2%の水を含むエタノール100 mLに加え、攪拌により溶解させた後に析出物を濾取し、乾燥して表題化合物のC形結晶の水和物
を得た(6.21 g)。
XRD: 5.5°, 13.4°, 14.3°, 21.4°, 26.7°.
【0052】
実施例6
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・n硝酸塩
参考例1の化合物200 mgをエタノール2 mLに溶解し、室温下硝酸0.07 mLを加え、4時間攪拌した。溶媒を留去後、酢酸エチル3 mLを加え、析出物を濾取し、減圧下にて乾燥して表題化合物を非晶質として得た(208 mg、収率80%)。(nは1ないし3を示す)
【0053】
実施例7
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・3メシル酸塩
(1)参考例1の化合物2.64 gをTHF25 mLに溶解し、室温下メシル酸1.32 mLを加え、1.5時間攪拌した。析出物を濾取し、減圧下にて乾燥して結晶を得た(3.52 g、収率80%)。
(2)上記結晶1.76 gをエタノール10 mLに加熱溶解し、室温にて17時間攪拌した。析出物
を濾取し、減圧下にて乾燥して表題化合物を結晶として得た(1.26 g、収率72%)。
DSC:193-197℃
Anal. calcd for C22H30N6OS・3CH4O3S・H2O: C, 40.97%; H, 6.05%; N, 11.47%; found: C, 41.05%; H, 5.72%; N, 11.48%.
【0054】
実施例8
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2メシル酸塩
(1)参考例1の化合物7.96 gをIPA60 mLに溶解し、室温下メシル酸3.59 gのIPA 20 mL溶
液を加え、2時間攪拌した。析出物を濾取し、減圧下にて乾燥して固体を得た(9.03 g、収率78%)。
(2)上記固体1000 mgをアセトニトリル20 mLに懸濁し、30分間加熱還流した。室温で冷却し、析出物を濾取して固体847 mgを得た。この固体813 mgをさらにアセトニトリル16mLに懸濁させ、1時間加熱還流した。室温で冷却し、析出物を濾取し、減圧下にて乾燥して表
題化合物を結晶として得た(690 mg、収率72%)。
DSC: 213-216℃
Anal. calcd for C22H30N6OS・2CH4O3S・0.5H2O: C, 45.92; H, 6.26; N, 13.39; found: C, 45.96; H, 6.17; N, 13.37.
【0055】
実施例9
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2トシル酸塩
(1)参考例1の化合物5.28 gをIPA 30 mLに溶解し、室温下トシル酸一水和物4.94 gを加
え、1.5時間攪拌した。析出物を濾取し、減圧下にて乾燥して表題化合物のA形結晶を得た(7.84 g、収率82%)。
XRD: 5.3°, 6.0°, 14.8°, 16.4°, 20.8°.
Anal. calcd for C22H30N6OS・2C7H8O3S・0.25H2O: C, 55.76%; H, 6.04%; N, 10.84%; found: C, 55.71%; H, 6.06%; N, 10.80%.
(2)上記生成物1.5 gを水20 mLに加熱溶解した。室温にて1時間攪拌し、析出物を濾取し
、減圧下にて乾燥して表題化合物のB形結晶を得た(1.2 g、収率80%)。
XRD: 5.7°, 11.4°, 14.0°, 18.2°, 19.7°.
Anal. calcd for C22H30N6OS・2C7H8O3S・0.5H2O: C, 55.43%; H, 6.07%; N, 10.77%; found: C, 55.14%; H, 6.09%; N, 10.73%.
(3)(1)の生成物1.4 gをIPA 100 mLに懸濁させ1時間加熱還流した。室温で冷却し、析出物を濾取し、減圧下にて乾燥して表題化合物のC形結晶を得た(1.1 g、収率81%)。
DSC: 227-230℃
XRD: 4.7°, 5.7°, 11.3°, 19.8°, 21.4°.
Anal. calcd for C22H30N6OS・2C7H8O3S: C, 56.08%; H, 6.01%; N, 10.90%; found: C, 55.83%; H, 6.11%; N, 10.87%.
【0056】
実施例10
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2べシル酸塩
(1)参考例1の化合物4.36 gをIPA 70 mLに溶解し、室温下ベシル酸3.78 gを加え、1時間攪拌した。析出物を濾取し、減圧下にて乾燥して表題化合物のA形結晶を得た(6.05 g、収率80%)。
XRD: 5.7°, 8.9°, 19.4°, 20.2°, 21.6°.
1H-NMR (DMSO-d6):δ 1.82-2.10 (1H, m), 2.17 (3H, s), 2.60-4.20 (16H, m), 4.11-4.72 (3H, m), 5.91 (1H, s), 7.31-7.35 (7H, m), 7.45-7.50 (2H, m), 7.59-7.62 (4H, m), 7.75 (2H, d, J = 7.8 Hz).
(2)上記生成物1.81 gをエタノール25 mLに加熱溶解した。室温にて1時間攪拌し、析出物を濾取し、減圧下にて乾燥して表題化合物のB形結晶を得た(1.25 g、収率69%)。
XRD: 5.6°, 6.7°, 19.3°, 22.9°, 23.2°.
Anal. calcd for C22H30N6OS・2C6H6O3S: C, 54.97%; H, 5.70%; N, 11.31%; found: C, 54.67%; H, 5.61%; N, 11.25%.
【0057】
実施例11
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2ナフタレン-1-スルホン酸塩
(1)参考例1の化合物4.01 gをTHF80 mLに溶解し、室温下ナフタレン-1-スルホン酸4.11 gのTHF 40 mL溶液を加え、3時間攪拌した。析出物を濾取し、減圧下乾燥して固体を得た(5.96 g、収率75%)。
(2)上記固体500 mgをエタノール25 mLに加熱溶解させ、30分間還流した。室温にて冷却
し、析出物を濾取し、減圧下乾燥して表題化合物を結晶として得た(445mg、収率89%)。
DSC: 184-189℃
Anal. calcd for C22H30N6OS・2C10H8O3S・0.25H2O: C, 59.52; H, 5.52; N, 9.92; found: C, 59.32; H, 5.46; N, 9.88.
【0058】
実施例12
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2ナフタレン-2-スルホン酸塩
(1)参考例1の化合物2.57 gを酢酸エチル50 mLに溶解し、室温下ナフタレン-2-スルホン酸1水和物2.86 gの酢酸エチル25 mL溶液を加え、12時間攪拌した。析出物を濾取し、減
圧下乾燥して固体を得た(4.60 g、収率91%)。
(2)上記固体500 mgをエタノール25 mLに加熱溶解した。室温にて攪拌し、析出物を濾取
し、減圧下乾燥して表題化合物を結晶として得た(372 mg、収率74%)。
DSC: 205-211℃
Anal. calcd for C22H30N6OS・2C10H8O3S・0.75H2O: C, 58.89; H, 5.59; N, 9.81; found: C, 58.96; H, 5.49; N, 9.76.
【0059】
実施例13
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・没食子酸塩
参考例1の化合物4.05 gをIPA 30 mLに溶解し、室温下没食子酸1水和物1.96 gのIPA溶
液30 mLを加え、1時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を固体として得た(4.84 g、収率85%)。
1H-NMR (DMSO-d6): δ1.43-1.62 (1H, m), 2.14 (3H, s), 2.19-3.08 (13H, m), 3.55-3.96(4H, m), 4.20-4.69 (2H, m), 5.91 (1H, s), 7.31-7.35 (7H, m), 7.45-7.50 (2H, m), 7.59-7.62 (4H, m), 7.75 (2H, d, J = 7.8 Hz).
【0060】
実施例14
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2(+)-カンファースルホン酸塩
(1)参考例1の化合物3.00 gをTHF 72.5 mLとt-ブチルメチルエーテル52.5 mLの混合溶媒に溶解し、室温下(+)-カンファースルホン酸3.25 gを加え、5時間攪拌した。析出物を
濾取し、減圧下乾燥して固体を得た(5.65 g、収率90%)。
(2)上記固体650 mgをエタノール7.0 mLとジエチルエーテル15.0mLの混合溶媒に加熱溶解した。室温にて一晩攪拌し、析出物を濾取し、温風下乾燥してエタノールを含有した表題化合物を結晶として得た(380mg、収率58%)。
TG/DTA: 142-156℃, 200-205℃
Anal. calcd for C22H30N6OS・2C10H16O4S・0.22C2H6O・2.5H2O: C, 53.66; H, 7.29; N, 8.86; found: C, 53.82; H, 7.27; N, 8.88.
【0061】
実施例15
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2(-)-カンファースルホン酸塩
参考例1の化合物3.00 gをTHF/t-ブチルメチルエーテルの1:3混合溶媒70 mLに溶解し
、室温下(-)-カンファースルホン酸3.25 gのTHF/t-ブチルメチルエーテルの1:3混合溶液を加え、1時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を固体として得た(5.66 g、収率91%)。
【0062】
実施例16
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2マレイン酸塩
(1)参考例1の化合物1.70 gをエタノール50 mLに溶解し、室温下マレイン酸0.98 gを加
え、1時間攪拌した。析出物を濾取し、減圧下にて乾燥して表題化合物のA形結晶を得た(1.87 g、収率71%)。
Anal. calcd for C22H30N6OS・2C4H4O4: C, 54.70%; H, 5.81%; N, 12.76%; found: C,
54.42%; H, 5.76%; N, 12.57%.
XRD: 8.6°, 15.8°, 17.8°, 18.6°, 23.4°.
(2)上記結晶3.0 gを水15 mLに加熱溶解した。室温にて1時間攪拌し、析出物を濾取し、
減圧下にて乾燥して表題化合物のB形結晶を得た(1.83 g、収率61%)。
XRD: 5.9°, 13.4°, 16.3°, 17.6°, 23.9°.
Anal. calcd for C22H30N6OS・2C4H4O4・2H2O: C, 51.86%; H, 6.09%; N, 12.10%; found: C, 51.80%; H, 5.84%; N, 12.10%.
【0063】
実施例17
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2フマル酸塩
(1)参考例1の化合物1.50 gをエタノール20 mLに溶解し、室温下フマル酸814 mgのエタ
ノール25mL溶液を加え、室温下1時間攪拌後氷冷下1時間攪拌した。反応溶媒を1/3留去し
、析出物を濾取し、減圧下にて乾燥して固体を得た(1.77 g、収率77%)。
(2)(1)の固体200 mgをアセトニトリル5 mLに懸濁させ4時間加熱還流した。室温にて冷
却後、析出物を濾取して表題化合物の結晶を得た(141 mg、収率71%)。この結晶はDSCで2
つの吸熱ピークを観測したことから2つの結晶形(A、B)の混合物、あるいは熱により結晶
形AがBに転移するものと推測した。
DSC: 128-(135及び142)℃
XRD: 3.1°, 15.2°, 17.4°, 23.4°, 25.5°.
Anal. calcd for C22H30N6OS・2C4H4O4: C, 54.70; H, 5.81; N, 12.76; found: C, 54.40; H, 5.88; N, 12.63
1H-NMR (DMSO-d6): δ1.50-1.78 (1H, m), 2.14 (3H, m), 2.37-3.90 (16H, m), 4.10-4.72 (3H, m), 5.79 (1H, s), 6.57 (4H, s), 7.27 (1H, t, J = 7.2 Hz), 7.46 (2H, t, J = 8.1 Hz), 7.74 (2H, d, J = 7.7 Hz).
(3)(1)の固体200 mgを水2 mLに溶解させた。室温にて攪拌し、析出物を濾取することにより表題化合物を結晶として得た(47.5mg、収率24%)。この結晶は(2)で得た結晶とは異
なる粉末X線のパターンを示した。
XRD: 9.4°, 17.8°, 19.6°, 21.0°, 23.5°, 24.3°.
【0064】
実施例18
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・1.6硫酸塩
参考例1の化合物2.00gをTHF 40 mLに溶解し、室温下0.5 mol/L硫酸水溶液14.5 mLを加え、0.5時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を固体として得た(2.57 g、収率94%)。
Anal. calcd for C22H30N6OS・1.6H2O4S・2H2O: C,42.65; H,6.05; N, 13.57; found: C, 42.56; H, 5.67; N, 13.44.
【0065】
実施例19
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2 L-酒石酸塩
(1)参考例1の化合物1.17 gをIPA 30 mLに溶解し、室温下L-酒石酸823 mgを加え、3時間攪拌した。析出物を濾取し、減圧下乾燥して結晶を得た(1.55 g、収率78%)。
(2)上記生成物254 mgを酢酸エチル10 mLに懸濁させ1.5時間加熱還流した。室温にて冷却し、析出物を濾取し、減圧下にて乾燥して表題化合物を結晶として得た(250 mg、収率98%)。
1H-NMR (DMSO-d6): δ1.50-1.69 (1H, m), 2.14 (3H, m), 2.40-3.90 (16H, m), 4.08 (2H, s), 4.30-4.70 (3H, m), 5.79 (1H, s), 7.27 (1H, t, J = 7.3 Hz), 7.46 (2H, m),
7.73 (2H, d, J = 7.8 Hz).
【0066】
実施例20
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・mリン酸塩
参考例1の化合物100 mgをTHF2 mLに溶解し、室温下リン酸0.032 mLを加え、1時間攪拌した。析出物を濾取し、減圧下乾燥して表題化合物を非晶質として得た(144 mg、収率93%)。(mは1ないし3を示す)
【0067】
試験例1 粉末X線回折の測定
以下の測定条件で実施例4および5の表題化合物の粉末X線回折を測定した。
装置:島津製作所製 XRD-6000
対陰極:Cu
モノクロメーター:Graphite
管電圧:40 kV
管電流:40 mA
発散スリット:1°
受光スリット:0.15 mm
散乱スリット:1°
測定範囲:2〜40°(2θ)
検体回転速度:60 rpm
実施例4の表題化合物の、粉末X線回折の測定結果を図1に示す。
実施例5の表題化合物のA形結晶の粉末X線回折の測定の結果を図2に示す。
実施例5の表題化合物のB形結晶の粉末X線回折の測定の結果を図3に示す。
実施例5の表題化合物のC形結晶の粉末X線回折の測定の結果を図4に示す。
【0068】
試験例2 吸湿性の測定
水分吸着測定装置を用いて以下の測定条件で実施例3の表題化合物の吸湿性を測定した
。
装置:VTI社製 MB-300G
測定温度:25℃
測定範囲:0〜95% RH
吸湿性の測定結果を図5に示した。
実施例3の表題化合物を、減圧型の水分吸着測定装置を用いて水分吸着測定を行ったとこ
ろ、50%RHで1.8水和相当の水を保持し、0%RHではほぼ完全に乾燥していることが分かった。
【0069】
試験例3 溶解度の測定
(1)水への溶解度の測定
測定法として、少量の試料で簡便に大まかな溶解性を把握できる目視法を用いた。温度は37℃で測定した。実施例3の表題化合物約3 mgをスクリューキャップ付きサンプル瓶に
取り、試験液0.15 mLを加えて密栓した。1分間超音波をかけて試料を分散させた後、こ
れをあらかじめ37℃に安定させた恒温振とう水槽に入れ、1時間振とうした後、目視観察
で溶解を確認した。その結果、実施例3の表題化合物の水に対する溶解度は37℃において20 mg/mL以上であった。
(2)pH9〜13における溶解度の測定
試験液として0.2 mol/L NaOH/0.1 mol/L NaCl混合溶液を用いて、pHを9〜13に調整した時の室温での実施例3の表題化合物の溶解度について、液体クロマトグラフ法(HPLC)によ
り分析した(n=3)。 その結果、溶解度は6.4 mg/mL〜8.4 mg/mLであった。以上より実施
例3の表題化合物の溶解度は約7 mg/mLと結論付けた。
【0070】
【表1】
【産業上の利用可能性】
【0071】
化合物Iの塩若しくはその溶媒和物、あるいはそれぞれの新規の結晶多形は、安定性の
改善、吸湿性(潮解性)の改善、溶媒からの迅速な単離、及び製剤化の容易性から選択される1つ以上の特性を有しており、化合物Iの医薬品としての開発を促進する。
【0072】
なお、本願は特願2005-041851号を優先権主張して出願されたものである。
【特許請求の範囲】
【請求項1】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンと有機または無機の一ないし三塩基酸との塩、またはその溶媒和物。
【請求項2】
有機または無機の一塩基酸が塩酸、臭化水素酸、硝酸、メシル酸、トシル酸、ベシル酸、塩酸、ナフタレン-1-スルホン酸、ナフタレン-2-スルホン酸、没食子酸、またはカンファースルホン酸である請求項1に記載の塩、またはその溶媒和物(ただし、一塩基酸が塩酸の場合、塩は2または2.5塩酸塩である)。
【請求項3】
有機または無機の二塩基酸がフマル酸、マレイン酸、硫酸、コハク酸、L-酒石酸、エタンジスルホン酸、またはクエン酸である請求項1に記載の塩、またはその溶媒和物。
【請求項4】
有機または無機の三塩基酸がリン酸である請求項1に記載の塩、またはその溶媒和物。
【請求項5】
2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2ベシル酸、2塩酸、2.5塩酸、2ナフタレン-1-スルホン酸、2ナフタレン-2-スルホン酸、2カンファースルホン酸、フマル酸、硫酸、コハク酸、L-酒石酸、またはクエン酸との塩である、請求項1に記載の塩、またはその溶媒和物。
【請求項6】
2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2.5塩酸、2ナフタレン-1-スルホン酸、2メシル酸、3メシル酸、または2ナフタレン-2-スルホン酸との塩である請求項5記載の塩、またはその溶媒和物。
【請求項7】
周囲温度での水への溶解度が7mg/mLないし2g/mLである一塩基酸と3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンとの塩、またはその溶媒和物。
【請求項8】
37℃での水への溶解度が20 mg/mL以上である請求項7に記載の塩、またはその溶媒和物。
【請求項9】
pH9から12での水への溶解度が7 mg/mLである請求項7に記載の記載の塩、またはその溶媒和物。
【請求項10】
25℃で測定した吸湿性が6%以下である請求項1に記載の塩、またはその溶媒和物。
【請求項11】
25℃で測定した相対湿度0%から50%の範囲における吸湿性が5%である請求項10に記載の塩、またはその溶媒和物。
【請求項12】
25℃で測定した相対湿度5%から90%の範囲における吸湿性が2%である請求項10に記載の塩、またはその溶媒和物。
【請求項13】
塩が請求項2記載の一塩基性酸との塩である請求項10ない12いずれかに記載の塩、またはその溶媒和物。
【請求項14】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物。
【請求項15】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°及び14.4°(それぞれ±0.2°) にピークを有することを特徴とする請求項14に記載の塩、またはその水和物。
【請求項16】
1.0ないし2.0水和物である請求項15に記載の水和物。
【請求項17】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°, 14.4°, 22.6°及び26.5° (それぞれ±0.2°) にピークを有することを特徴とする請求項14に記載の塩、またはその水和物。
【請求項18】
1.0ないし2.0水和物である請求項17に記載の水和物。
【請求項19】
図1に例示される粉末X線回折パターンを示す、請求項14に記載の塩、またはその水和物。
【請求項20】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0臭化水素酸塩、またはその溶媒和物。
【請求項21】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.7°, 7.7°, 11.3°, 16.2°及び17.0° (それぞれ±0.2°) にピークを有することを特徴とする請求項20に記載の塩の水和物。
【請求項22】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.2°, 10.4°, 19.1°, 19.8°及び20.7° (それぞれ±0.2°) にピークを有することを特徴とする請求項20に記載の塩の水和物。
【請求項23】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.5°, 13.4°, 14.3°, 21.4°及び26.7° (それぞれ±0.2°) にピークを有することを特徴とする請求項20に記載の塩の水和物。
【請求項24】
図2に例示される粉末X線回折パターンを示す、請求項20に記載の塩の水和物。
【請求項25】
図3に例示される粉末X線回折パターンを示す、請求項20に記載の塩の水和物。
【請求項26】
図4に例示される粉末X線回折パターンを示す、請求項20に記載の塩の水和物。
【請求項27】
3-{(2S,4S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンに対して臭化水素酸で脱1,1-ジメチルエチルオキシカルボニル反応を行うと共に同時に造塩を行うことを特徴とする3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法。
【請求項28】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩を、許容される溶媒を用いて結晶化する工程を含む3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法。
【請求項29】
許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”において1日摂取許容量(“PDE”)が10 mg/dayを越える溶媒から選ばれる溶媒である請求項28に記載の方法。
【請求項30】
許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”におけるクラス3の溶媒から選ばれる溶媒である請求項28に記載の方法。
【請求項31】
許容される溶媒が、エタノール、1-プロパノール、2-プロパノール、酢酸エチル、アセトンから選ばれる溶媒である請求項28に記載の方法。
【請求項32】
許容される溶媒がエタノール及び/または水である請求項28に記載の方法。
【請求項33】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩の1.0ないし2.0水和物。
【請求項1】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンと有機または無機の一ないし三塩基酸との塩、またはその溶媒和物。
【請求項2】
有機または無機の一塩基酸が塩酸、臭化水素酸、硝酸、メシル酸、トシル酸、ベシル酸、塩酸、ナフタレン-1-スルホン酸、ナフタレン-2-スルホン酸、没食子酸、またはカンファースルホン酸である請求項1に記載の塩、またはその溶媒和物(ただし、一塩基酸が塩酸の場合、塩は2または2.5塩酸塩である)。
【請求項3】
有機または無機の二塩基酸がフマル酸、マレイン酸、硫酸、コハク酸、L-酒石酸、エタンジスルホン酸、またはクエン酸である請求項1に記載の塩、またはその溶媒和物。
【請求項4】
有機または無機の三塩基酸がリン酸である請求項1に記載の塩、またはその溶媒和物。
【請求項5】
2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2ベシル酸、2塩酸、2.5塩酸、2ナフタレン-1-スルホン酸、2ナフタレン-2-スルホン酸、2カンファースルホン酸、フマル酸、硫酸、コハク酸、L-酒石酸、またはクエン酸との塩である、請求項1に記載の塩、またはその溶媒和物。
【請求項6】
2.0臭化水素酸、2.5臭化水素酸、2マレイン酸、2トシル酸、2.5塩酸、2ナフタレン-1-スルホン酸、2メシル酸、3メシル酸、または2ナフタレン-2-スルホン酸との塩である請求項5記載の塩、またはその溶媒和物。
【請求項7】
周囲温度での水への溶解度が7mg/mLないし2g/mLである一塩基酸と3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンとの塩、またはその溶媒和物。
【請求項8】
37℃での水への溶解度が20 mg/mL以上である請求項7に記載の塩、またはその溶媒和物。
【請求項9】
pH9から12での水への溶解度が7 mg/mLである請求項7に記載の記載の塩、またはその溶媒和物。
【請求項10】
25℃で測定した吸湿性が6%以下である請求項1に記載の塩、またはその溶媒和物。
【請求項11】
25℃で測定した相対湿度0%から50%の範囲における吸湿性が5%である請求項10に記載の塩、またはその溶媒和物。
【請求項12】
25℃で測定した相対湿度5%から90%の範囲における吸湿性が2%である請求項10に記載の塩、またはその溶媒和物。
【請求項13】
塩が請求項2記載の一塩基性酸との塩である請求項10ない12いずれかに記載の塩、またはその溶媒和物。
【請求項14】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物。
【請求項15】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°及び14.4°(それぞれ±0.2°) にピークを有することを特徴とする請求項14に記載の塩、またはその水和物。
【請求項16】
1.0ないし2.0水和物である請求項15に記載の水和物。
【請求項17】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.4°, 13.4°, 14.4°, 22.6°及び26.5° (それぞれ±0.2°) にピークを有することを特徴とする請求項14に記載の塩、またはその水和物。
【請求項18】
1.0ないし2.0水和物である請求項17に記載の水和物。
【請求項19】
図1に例示される粉末X線回折パターンを示す、請求項14に記載の塩、またはその水和物。
【請求項20】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.0臭化水素酸塩、またはその溶媒和物。
【請求項21】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.7°, 7.7°, 11.3°, 16.2°及び17.0° (それぞれ±0.2°) にピークを有することを特徴とする請求項20に記載の塩の水和物。
【請求項22】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.2°, 10.4°, 19.1°, 19.8°及び20.7° (それぞれ±0.2°) にピークを有することを特徴とする請求項20に記載の塩の水和物。
【請求項23】
粉末X線回折パターンにおいて、2θで表される回折角度として、5.5°, 13.4°, 14.3°, 21.4°及び26.7° (それぞれ±0.2°) にピークを有することを特徴とする請求項20に記載の塩の水和物。
【請求項24】
図2に例示される粉末X線回折パターンを示す、請求項20に記載の塩の水和物。
【請求項25】
図3に例示される粉末X線回折パターンを示す、請求項20に記載の塩の水和物。
【請求項26】
図4に例示される粉末X線回折パターンを示す、請求項20に記載の塩の水和物。
【請求項27】
3-{(2S,4S)-1-(1,1-ジメチルエチルオキシカルボニル)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジンに対して臭化水素酸で脱1,1-ジメチルエチルオキシカルボニル反応を行うと共に同時に造塩を行うことを特徴とする3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法。
【請求項28】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩を、許容される溶媒を用いて結晶化する工程を含む3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩、またはその溶媒和物の製造方法。
【請求項29】
許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”において1日摂取許容量(“PDE”)が10 mg/dayを越える溶媒から選ばれる溶媒である請求項28に記載の方法。
【請求項30】
許容される溶媒が、水、及び/または、“残留溶媒Q3CのICHガイドライン”におけるクラス3の溶媒から選ばれる溶媒である請求項28に記載の方法。
【請求項31】
許容される溶媒が、エタノール、1-プロパノール、2-プロパノール、酢酸エチル、アセトンから選ばれる溶媒である請求項28に記載の方法。
【請求項32】
許容される溶媒がエタノール及び/または水である請求項28に記載の方法。
【請求項33】
3-{(2S,4S)-4-[4-(3-メチル-1-フェニル-1H-ピラゾール-5-イル)ピペラジン-1-イル]ピロリジン-2-イルカルボニル}チアゾリジン・2.5臭化水素酸塩の1.0ないし2.0水和物。
【図1】

【図2】
【図3】

【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2013−32376(P2013−32376A)
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願番号】特願2012−224923(P2012−224923)
【出願日】平成24年10月10日(2012.10.10)
【分割の表示】特願2008−119236(P2008−119236)の分割
【原出願日】平成18年2月17日(2006.2.17)
【出願人】(000002956)田辺三菱製薬株式会社 (225)
【Fターム(参考)】
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願日】平成24年10月10日(2012.10.10)
【分割の表示】特願2008−119236(P2008−119236)の分割
【原出願日】平成18年2月17日(2006.2.17)
【出願人】(000002956)田辺三菱製薬株式会社 (225)
【Fターム(参考)】
[ Back to top ]