
ヘテロアセン誘導体の製造方法
【課題】優れた耐酸化性を有し、塗布法による半導体活性相形成が可能な、ヘテロアセン誘導体の製造方法を提供する。
【解決手段】一般式(1a)で示されるヘテロアセン誘導体の製造方法であり、一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3a)及び一般式(4a)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法;一般式(1b)で示されるヘテロアセン誘導体の製造方法であり、一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3b)及び一般式(4b)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法。
【解決手段】一般式(1a)で示されるヘテロアセン誘導体の製造方法であり、一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3a)及び一般式(4a)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法;一般式(1b)で示されるヘテロアセン誘導体の製造方法であり、一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3b)及び一般式(4b)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、有機半導体材料等の電子材料への展開が可能なヘテロアセン誘導体の製造方法に関する。
【背景技術】
【0002】
有機薄膜トランジスタに代表される有機半導体デバイスは、省エネルギー、低コスト及びフレキシブルといった無機半導体デバイスにはない特徴を有することから近年注目されるようになった。この有機半導体デバイスは有機半導体活性相、基板、絶縁相、電極等数種類の材料から構成されるが、中でも電荷のキャリアー移動を担う有機半導体活性相は該デバイスの中心的な役割を有している。この有機半導体活性相を構成する有機材料のキャリアー移動能により有機半導体デバイス性能が左右される。
【0003】
有機半導体活性相を作製する方法としては一般的に、高温真空下、有機材料を気化させて実施する真空蒸着法及び有機材料を適当な溶媒に溶解させその溶液を塗布する塗布法が知られている。塗布法においては、塗布は高温高真空条件を用いることなく印刷技術を用いても実施することができる。そのため、塗布法は印刷によりデバイス作製の大幅な製造コストの削減を図ることができることから、経済的に好ましいプロセスである。しかし、従来、有機半導体デバイスとして性能が高い材料ほど塗布法で有機半導体活性相を形成することが困難になるという問題があった。
【0004】
例えば、ペンタセン等の結晶性材料はアモルファスシリコン並みの高いキャリアー移動度を有し、優れた有機半導体デバイス特性を発現することが報告されている(例えば、非特許文献1参照)。又、ペンタセン等のポリアセンを溶解させ塗布法で有機半導体デバイスを製造する試みも報告されている(例えば、特許文献1参照)。しかしながら、ペンタセンはその強い凝集性のため溶解性が低く、塗布法を適用するためには高温加熱等の条件が必要とされ、さらにペンタセンの溶液は極めて容易に空気酸化されることから、塗布法の適用はプロセス的、経済的に困難を伴うものであった。また、ポリ−(3−ヘキシルチオフェン)等の自己組織化材料は溶媒に可溶であり、塗布法による有機半導体デバイス作製が報告されてはいるが、キャリアー移動度が結晶性低分子化合物より1桁低いことから(例えば、非特許文献2参照)、得られた有機半導体デバイスの特性が低いという問題があった。
【0005】
またチオフェン環が縮環したペンタチエノアセンはペンタセンに比べ耐酸化性が向上しているが、その合成に多工程を必要とすることから(例えば、非特許文献3参照)実用上好ましい材料ではなかった。また、ブチルリチウムによるジリチオ化/硫黄との反応でチオフェン環を形成する反応が知られているが、低収率で副生物が多く生成する問題があった(例えば、非特許文献4参照)。
【0006】
【非特許文献1】「ジャーナル オブ アプライドフィジックス」(米国)、2002年、92巻、5259−5263頁
【非特許文献2】「サイエンス」(米国)、1998年、280巻、1741−1744頁
【非特許文献3】「オルガニック レターズ」(米国)、2005年、7巻、5301−5304頁
【非特許文献4】「ジャーナル オブ ヘテロサイクリック ケミストリィー」、1991年、28巻、433−438頁
【特許文献1】WO2003/016599号
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明は上記の従来技術が有する問題点に鑑み、優れた半導体性能及び耐酸化性を有し、塗布法による有機半導体活性相形成が可能な、ヘテロアセン誘導体の製造方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは上記課題を解決するため鋭意検討の結果、テトラハロターフェニル誘導体を原料に用いる本発明のヘテロアセン誘導体の新規な製造方法を見出した。加えて、該ヘテロアセン誘導体が耐酸化性に優れ、塗布法の適用が可能であるため結晶性の薄膜を容易に安定して作製することができることから、本発明を完成するに到った。
【0009】
以下に本発明を詳細に説明する。説明はヘテロアセン誘導体の製造方法並びに該ヘテロアセン誘導体の前駆体であるテトラハロターフェニル誘導体の製造方法について述べる。
【0010】
(ヘテロアセン誘導体の製造方法)
最初に、下記一般式(1a)で示されるヘテロアセン誘導体の製造方法について説明する。本発明の下記一般式(1a)で示されるヘテロアセン誘導体は、下記一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、下記一般式(3a)及び下記一般式(4a)で示される反応剤と反応させることで製造する。
【0011】
【化1】

【0012】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T1及びT2は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有し、l及びmは、各々0又は1の整数を示す。)
【0013】
【化2】
【0014】
【化3】
【0015】
(ここで、置換基R5〜R9は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、炭素数2〜30のアルキニル基、炭素数2〜30のアルケニル基を示す。nは0〜2の整数であり、oは0又は1の整数である。)]
【0016】
【化4】
【0017】
(ここで、置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、置換基R1及びR2並びに環A及びBは一般式(1a)で示される置換基並びに環と同意義を示す。)
(R3)lT1(L1)p (3a)
(R4)mT2(L2)q (4a)
(ここで、置換基T1、T2、R3、R4及び記号lとmは一般式(1a)で示される置換基及び記号と同意義を示し、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、p及びqは0又は2の整数を示す。)
なお、ここでテトラメタル化とは、一般式(2a)におけるX1〜X4をそれぞれメタルに置換することを意味する。
【0018】
一般式(1a)で示されるヘテロアセン誘導体の置換基について述べる。
【0019】
置換基R1〜R4及びR5〜R9における炭素数1〜30のアルキル基は、特に限定はなく、例えばメチル基、プロピル基、ブチル基、イソブチル基、tert−ブチル基、ネオペンチル基、ヘキシル基、オクチル基、デシル基、ドデシル基、オクタデシル基、2−エチルヘキシル基、シクロヘキシル基、シクロオクチル基等のアルキル基;トリフルオロメチル基、ペンタフルオロエチル基、パーフルオロオクチル基、パーフルオロデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基、パーフルオロシクロヘキシル基、パーフルオロシクロオクチル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基、2−エチルパーフルオロヘキシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくは炭素数6〜30のアルキル基であり、より好ましくはドデシル基、オクタデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基であり、特に好ましくはドデシル基、パーフルオロドデシル基である。
【0020】
置換基R1〜R4及びR5〜R9における炭素数4〜30のアリール基は、特に限定はなく、例えばフェニル基、p−トリル基、p−(オクチル)フェニル基、p−(ドデシル)フェニル基、p−(シクロヘキシル)フェニル基、m−(オクチル)フェニル基、m−(ドデシル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基、p−(パーフルオロドデシル)フェニル基、m−(パーフルオロドデシル)フェニル基、2−チエニル基、5−(ドデシル)−2−チエニル基、ベンゾチエニル−2−基、6−ドデシルベンゾチエニル−2−基、2,2’−ビチエニル−5−基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、アントラセニル基等を挙げることができ、好ましくはフェニル基、p−(オクチル)フェニル基、p−(パーフルオロオクチル)フェニル基、5−(ドデシル)−2−チエニル基等であり、特に好ましくはフェニル基である。
【0021】
置換基R5〜R9における、炭素数2〜30のアルキニル基は、特に限定はなく、例えばエチニル基、メチルエチニル基、イソプロピルエチニル基、tert−ブチルエチニル基、(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(トリフルオロメチル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基、{p−(オクチル)フェニル}エチニル基、{p−(ドデシル)フェニル}エチニル基、{m−(ドデシル)フェニル}エチニル基、ナフチルエチニル基、アントラセニルエチニル基、ベンジルエチニル基、パーフルオロフェニルエチニル基、{p−(トリフルオロメチル)フェニル}エチニル基、{p−(パーフルオロオクチル)フェニル}エチニル基、{p−(パーフルオロドデシル)フェニル}エチニル基、{m−(パーフルオロドデシル)フェニル}エチニル基、5−(ヘキシル)チエニル−2−}エチニル基、{5−(パーフルオロヘキシル)チエニル−2−}エチニル基等を挙げることができ、好ましくは(オクチル)エチニル基、(デシル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基等である。
【0022】
置換基R5〜R9における、炭素数2〜30のアルケニル基は、特に限定はなく、例えばエテニル基、メチルエテニル基、イソプロピルエテニル基、tert−ブチルエテニル基、(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(トリフルオロメチル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、フェニルエテニル基、{p−(ヘキシル)フェニル}エテニル基、{p−(オクチル)フェニル}エテニル基、{p−(ドデシル)フェニル}エテニル基、{m−(ドデシル)フェニル}エテニル基、2−フェニル−1,2−ジフルオロエテニル基、2−フェニル−1,2−ジメチルエテニル基、ジフェニルエテニル基、トリフェニルエテニル基、ナフチルエテニル基、アントラセニルエテニル基、ベンジルエテニル基、フェニル(メチル)エテニル基、(パーフルオロフェニル)エテニル基、{p−(トリフルオロメチル)フェニル}エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基等を挙げることができ、好ましくは(オクチル)エテニル基、(デシル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基等を挙げることができる。なお、該炭素数2〜30のアルケニル基はトランス体及びシス体の何れであってもよく、またそれらの任意の割合の混合物であってもよい。
【0023】
これらの置換基の中でも、R1、R2としては、水素原子、フッ素原子、炭素数4〜30のアリール基が好ましく、特に好ましくは水素原子である。R3、R4としては、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、さらに好ましくは炭素数4〜30のアリール基であり、特に好ましくはフェニル基である。一方、R5〜R7としては、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数2〜30のアルキニル基が好ましく、さらに好ましくはフッ素原子、炭素数1〜30のアルキル基であり、特に好ましくはフッ素原子、ドデシル基であり、R8、R9としては、水素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、さらに好ましくは水素原子、炭素数4〜30のアリール基であり、特に好ましくは水素原子、フェニル基である。
【0024】
置換基T1及びT2は、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子であり、その中でも好ましくは硫黄原子、セレン原子、リン原子、ホウ素原子であり、さらに好ましくは硫黄原子、リン原子、ホウ素原子である。
【0025】
l及びmは各々0又は1の整数であり、T1及びT2が硫黄原子、セレン原子、テルル原子、酸素原子の場合0であり、T1及びT2がリン原子、ホウ素原子、アルミニウム原子の場合1である。
【0026】
次に、一般式(A−1)及び(A−2)で示される、環A及びBについて述べる。
【0027】
一般式(1a)で示されるヘテロアセン誘導体は、環A及びBを有する誘導体であり、環A及びBは一般式(A−1)又は(A−2)で示される構造を有するものである。
【0028】
一般式(A−1)中の記号nは、0〜2の整数であり、好ましくは1であり、一般式(A−2)中の記号oは、0又は1の整数であり、好ましくは1である。また、環A及びBが一般式(A−1)の構造の場合、一般式(1a)で示されるヘテロアセン誘導体の構造としては、下記一般式(1c)で示されるものであることが好ましい。
【0029】
【化5】
【0030】
(ここで、置換基R1〜R4、T1及びT2並びにl及びmは一般式(1a)で示される置換基並びに記号と同意義を示し、置換基R5は一般式(A−1)で示される置換基と同意義を示し、rは0〜2の整数である。)
本発明の一般式(1a)で示されるヘテロアセン誘導体の製造方法は、一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3a)及び一般式(4a)で示される反応剤と反応させることで達成することができる。
【0031】
一般式(2a)で示されるテトラハロターフェニル誘導体をテトラメタル化する場合、用いるメタル化剤は、一般式(2a)におけるX1〜X4をメタルに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属;メチルマグネシウムブロマイド、エチルマグネシウムブロマイド、イソプロピルマグネシウムクロライド、tert−ブチルマグネシウムクロライド、フェニルマグネシウムブロマイド等のグリニャール試薬;マグネシウム金属;亜鉛金属等を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムである。
【0032】
該メタル化剤の使用量は一般式(2a)で示されるテトラハロターフェニル誘導体1当量に対し、3.0〜10当量が好ましく、さらに好ましくは4.0〜8.0当量、特に好ましくは4.2〜6.0当量である。
【0033】
該テトラメタル化は、ジアルキルエーテル中で実施する。ジアルキルエーテル中で実施することにより、テトラヒドロフラン、ジオキサン、トルエン、ヘキサン、シクロヘキサン等の溶媒中で実施するよりも効率よく目的物が得られる。該ジアルキルエーテルとしては、例えばジメチルエーテル、メチルエチルエーテル、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル等が挙げられ、特に好ましくはジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。該テトラメタル化の温度は−80〜50℃が好ましく、特に好ましくは−20〜30℃である。反応時間は1〜120分が好ましく、特に好ましくは5〜90分である。本発明では、低溶解性のテトラハロターフェニル誘導体でもジアルキルエーテル中−20〜30℃でリチウム化合物と反応させることにより、収率良くテトラリチオ化できることを見出した。なお、テトラメタル化の進行は、反応液の一部を取り出し、水で反応を停止させた後、薄層クロマトグラフィー、ガスクロマトグラフィーで分析することで監視することができる。
【0034】
該テトラメタル化により生成したテトラメタル塩は、次いで一般式(3a)及び一般式(4a)で示される反応剤と反応させる。係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を直接用いて反応させる方法、生成したテトラメタル塩を一度単離した後、前記反応剤と反応させる方法のいずれを用いてもよい。
【0035】
また係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を添加する方法、生成したテトラメタル塩を含む反応混合物を前記反応剤に添加する方法のいずれを用いてもよい。
【0036】
ここで、一般式(3a)、(4a)における置換基T1、T2、R3、R4及び記号lとmは一般式(1a)で示される置換基及び記号と同意義を示す。
【0037】
また、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、好ましくは塩素原子、臭素原子、炭素数1〜20のオキシ基、アリールスルホニル基である。炭素数1〜20のオキシ基としては、特に限定はなく、例えばメトキシ基、エトキシ基、n−ブトキシ基、フェノキシ基、(2−メトキシ)フェノキシ基等を挙げることができ、アリールスルホニル基としては、特に限定はなく、例えばフェニルスルホニル基、p−トリルスルホニル基等を挙げることができる。これらの中でも特にフェニルスルホニル基が好ましい。
【0038】
そして、具体的な一般式(3a)、(4a)で示される反応剤としては、例えば2塩化硫黄;2臭化硫黄;ビス(フェニルスルホニル)スルフィド、ビス(p−トリルスルホニル)スルフィド等のビス(アリールスルホニル)スルフィド類;硫黄;2塩化セレン;セレン;2塩化テルル;テルル;ジクロロフェニルホスフィン、ジメトキシフェニルホスフィン、ジフェノキシフェニルホスフィン、ジクロロ{4−(オクチル)フェニル}ホスフィン、ジクロロ{4−(パーフルオロオクチル)フェニル}ホスフィン等のアリールホスフィン類;ジクロロ(ヘキシル)ホスフィン、ジクロロ(オクチル)ホスフィン、ジメトキシ(ヘキシル)ホスフィン等のアルキルホスフィン類;ジクロロフェニルボラン、ジメトキシフェニルボラン、ジメトキシ{4−(ヘキシル)フェニル}ボラン、ジフェノキシフェニルボラン、ジクロロ{4−(オクチル)フェニル}ボラン等のアリールボラン類;ジクロロ(ヘキシル)ボラン、ジクロロ(オクチル)ボラン、ジメトキシ(ヘキシル)ボラン等のアルキルボラン類;ジクロロフェニルアルミニウム、ジメトキシフェニルアルミニウム、ジメトキシ{4−(ヘキシル)フェニル}アルミニウム、ジフェノキシフェニルアルミニウム、ジクロロ{4−(オクチル)フェニル}アルミニウム等のアリールアルミニウム類;ジクロロ(ヘキシル)アルミニウム、ジクロロ(オクチル)アルミニウム、ジメトキシ(ヘキシル)アルミニウム等のアルキルアルミニウム類等を挙げることができ、好ましくはビス(フェニルスルホニル)スルフィド、ジクロロフェニルホスフィン、ジクロロフェニルボランである。なお、一般式(3a)及び一般式(4a)で示される反応剤は、同じ化合物であっても良い。
【0039】
テトラメタル化により生成したテトラメタル塩を一般式(3a)及び一般式(4a)で示される反応剤へ添加して反応させる際には、一般式(3a)及び一般式(4a)で示される反応剤は溶媒を存在させておくことが好ましい。用いる溶媒は特に限定はなく、例えばテトラヒドロフラン(以後THFと略す)、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン、トルエン、ヘキサン、シクロヘキサン等が挙げられ、好ましくはTHF、ジエチルエーテル、メチル−tert−ブチルエーテルである。用いる反応剤の量は、一般式(2a)で示されるテトラハロターフェニル誘導体1当量に対し、1.2〜5当量が好ましく、特に好ましくは2〜4当量である。テトラメタル塩とジアルキルエーテルの混合物はテフロン(登録商標)キャヌラーあるいはステンレス製のキャヌラーを用いて、一般式(3a)及び一般式(4a)の反応剤へ添加することが好ましい。該反応剤との反応温度は−90〜50℃が好ましく、特に好ましくは−80〜30℃であり、反応時間は0.5〜30時間が好ましく、特に好ましくは1〜18時間である。
【0040】
本発明の一般式(1a)で示されるヘテロアセン誘導体の製造方法では、一般式(2a)で示されるテトラハロターフェニル誘導体をテトラメタル化した後、塩化マグネシウムあるいは塩化亜鉛と反応させ、その後に一般式(3a)、(4a)に示される反応剤へ添加させ、処理することもできる。
【0041】
本発明の一般式(1a)で示されるヘテロアセン誘導体の製造方法は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0042】
かくして得られた、一般式(1a)で示されるヘテロアセン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0043】
本発明の製造方法で製造される一般式(1a)で示されるヘテロアセン誘導体は特に限定はなく、例えば以下の化合物を挙げることができ、
【0044】
【化6】
【0045】
【化7】
【0046】
【化8】
【0047】
【化9】
【0048】
【化10】
【0049】
特に好ましくは
【0050】
【化11】
【0051】
である。
【0052】
次に、下記一般式(1b)で示されるヘテロアセン誘導体の製造方法について説明する。
【0053】
本発明の下記一般式(1b)で示されるヘテロアセン誘導体は、下記一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3b)及び一般式(4b)で示される反応剤と反応させることで製造する。
【0054】
【化12】
【0055】
(ここで、置換基R10及びR11は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T3及びT4は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、上記一般式(A−1)又は(A−2)で示される構造を有し、v及びwは、各々0又は1の整数を示す。)
【0056】
【化13】
【0057】
(ここで、置換基X5〜X8は臭素原子、ヨウ素原子、塩素原子を示し、環A及びBは一般式(1b)で示される環と同意義を示す。)
(R10)vT3(L3)x (3b)
(R11)wT4(L4)y (4b)
(ここで、置換基T3、T4、R10、R11及び記号vとwは一般式(1b)で示される置換基及び記号と同意義を示し、置換基L3、L4は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、x及びyは0又は2の整数を示す。)
一般式(1b)で示されるヘテロアセン誘導体の置換基について述べる。
【0058】
置換基R10及びR11並びにR5〜R9における炭素数1〜30のアルキル基は、特に限定はなく、例えばメチル基、プロピル基、ブチル基、イソブチル基、tert−ブチル基、ネオペンチル基、ヘキシル基、オクチル基、デシル基、ドデシル基、オクタデシル基、2−エチルヘキシル基、シクロヘキシル基、シクロオクチル基等のアルキル基;トリフルオロメチル基、ペンタフルオロエチル基、パーフルオロオクチル基、パーフルオロデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基、パーフルオロシクロヘキシル基、パーフルオロシクロオクチル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基、2−エチルパーフルオロヘキシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくは炭素数6〜30のアルキル基であり、より好ましくはドデシル基、オクタデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基であり、特に好ましくはドデシル基、パーフルオロドデシル基である。
【0059】
置換基R10及びR11並びにR5〜R9における炭素数4〜30のアリール基は、特に限定はなく、例えばフェニル基、p−トリル基、p−(オクチル)フェニル基、p−(ドデシル)フェニル基、p−(シクロヘキシル)フェニル基、m−(オクチル)フェニル基、m−(ドデシル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基、p−(パーフルオロドデシル)フェニル基、m−(パーフルオロドデシル)フェニル基、2−チエニル基、5−(ドデシル)−2−チエニル基、ベンゾチエニル−2−基、6−ドデシルベンゾチエニル−2−基、2,2’−ビチエニル−5−基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、アントラセニル基等を挙げることができ、好ましくはフェニル基、p−(オクチル)フェニル基、p−(パーフルオロオクチル)フェニル基、5−(ドデシル)−2−チエニル基等であり、特に好ましくはフェニル基である。
【0060】
置換基R5〜R9における、炭素数2〜30のアルキニル基は、特に限定はなく、例えばエチニル基、メチルエチニル基、イソプロピルエチニル基、tert−ブチルエチニル基、(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(トリフルオロメチル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基、{p−(オクチル)フェニル}エチニル基、{p−(ドデシル)フェニル}エチニル基、{m−(ドデシル)フェニル}エチニル基、ナフチルエチニル基、アントラセニルエチニル基、ベンジルエチニル基、パーフルオロフェニルエチニル基、{p−(トリフルオロメチル)フェニル}エチニル基、{p−(パーフルオロオクチル)フェニル}エチニル基、{p−(パーフルオロドデシル)フェニル}エチニル基、{m−(パーフルオロドデシル)フェニル}エチニル基、5−(ヘキシル)チエニル−2−}エチニル基、{5−(パーフルオロヘキシル)チエニル−2−}エチニル基等を挙げることができ、好ましくは(オクチル)エチニル基、(デシル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基等である。
【0061】
置換基R5〜R9における、炭素数2〜30のアルケニル基は、特に限定はなく、例えばエテニル基、メチルエテニル基、イソプロピルエテニル基、tert−ブチルエテニル基、(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(トリフルオロメチル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、フェニルエテニル基、{p−(ヘキシル)フェニル}エテニル基、{p−(オクチル)フェニル}エテニル基、{p−(ドデシル)フェニル}エテニル基、{m−(ドデシル)フェニル}エテニル基、2−フェニル−1,2−ジフルオロエテニル基、2−フェニル−1,2−ジメチルエテニル基、ジフェニルエテニル基、トリフェニルエテニル基、ナフチルエテニル基、アントラセニルエテニル基、ベンジルエテニル基、フェニル(メチル)エテニル基、(パーフルオロフェニル)エテニル基、{p−(トリフルオロメチル)フェニル}エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基等を挙げることができ、好ましくは(オクチル)エテニル基、(デシル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基等を挙げることができる。なお、該炭素数2〜30のアルケニル基はトランス体及びシス体の何れであってもよく、またそれらの任意の割合の混合物であってもよい。
【0062】
これらの置換基の中でも、R10、R11としては、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、特に好ましくは炭素数4〜30のアリール基である。一方、R5〜R7としては、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数2〜30のアルキニル基が好ましく、さらに好ましくはフッ素原子、炭素数1〜30のアルキル基であり、特に好ましくはフッ素原子、ドデシル基であり、R8、R9としては、水素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、特に好ましくは水素原子、炭素数4〜30のアリール基である。
【0063】
置換基T3及びT4は、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子であり、その中でも好ましくは硫黄原子、セレン原子、リン原子、ホウ素原子であり、さらに好ましくは硫黄原子、リン原子、ホウ素原子である。
【0064】
v及びwは各々0又は1の整数であり、T3及びT4が硫黄原子、セレン原子、テルル原子、酸素原子の場合0であり、T3及びT4がリン原子、ホウ素原子、アルミニウム原子の場合1である。
【0065】
次に、一般式(A−1)及び(A−2)で示される、環A及びBについて述べる。
【0066】
一般式(1b)で示されるヘテロアセン誘導体は、環A及びBを有する誘導体であり、環A及びBは一般式(A−1)又は(A−2)で示される構造を有するものである。
【0067】
一般式(A−1)中の記号nは、0〜2の整数であり、好ましくは1であり、一般式(A−2)中の記号oは、0又は1の整数であり、好ましくは1である。また、環A及びBが一般式(A−1)の構造の場合、一般式(1b)で示されるヘテロアセン誘導体の構造としては、下記一般式(1d)で示されるものであることが好ましい。
【0068】
【化14】
【0069】
(ここで、置換基R10及びR11、T3及びT4並びにv及びwは一般式(1b)で示される置換基並びに記号と同意義を示し、置換基R5は一般式(A−1)で示され置換基と同意義を示し、tは0〜2の整数である。)
本発明の一般式(1b)で示されるヘテロアセン誘導体の製造方法は、一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3b)及び一般式(4b)で示される反応剤と反応させることで達成することができる。
【0070】
一般式(2b)で示されるテトラハロターフェニル誘導体をテトラメタル化する場合、用いるメタル化剤は、一般式(2b)におけるX5〜X8をメタルに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属;メチルマグネシウムブロマイド、エチルマグネシウムブロマイド、イソプロピルマグネシウムクロライド、tert−ブチルマグネシウムクロライド、フェニルマグネシウムブロマイド等のグリニャール試薬;マグネシウム金属;亜鉛金属等を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムである。
【0071】
該メタル化剤の使用量は一般式(2b)で示されるテトラハロターフェニル誘導体1当量に対し、3.0〜10当量が好ましく、さらに好ましくは4.0〜8.0当量、特に好ましくは4.2〜6.0当量である。
【0072】
該テトラメタル化は、ジアルキルエーテル中で実施する。ジアルキルエーテル中で実施することにより、テトラヒドロフラン、ジオキサン、トルエン、ヘキサン、シクロヘキサン等の溶媒中で実施するよりも効率よく目的物が得られる。該ジアルキルエーテルとしては、例えばジメチルエーテル、メチルエチルエーテル、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル等が挙げられ、特に好ましくはジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。該テトラメタル化の温度は−80〜50℃が好ましく、特に好ましくは−20〜30℃である。反応時間は1〜120分が好ましく、特に好ましくは5〜90分である。本発明では、低溶解性のテトラハロターフェニル誘導体でもジアルキルエーテル溶媒中−20〜30℃でリチウム化合物と反応させることにより、収率良くテトラリチオ化できることを見出した。なお、テトラメタル化の進行は、反応液の一部を取り出し、水で反応を停止させた後、薄層クロマトグラフィー、ガスクロマトグラフィーで分析することで監視することができる。
【0073】
該テトラメタル化により生成したテトラメタル塩は、次いで一般式(3b)及び一般式(4b)で示される反応剤と反応させる。係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を直接用いて反応させる方法、生成したテトラメタル塩を一度単離した後、前記反応剤と反応させる方法のいずれを用いてもよい。
【0074】
また係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を添加する方法、生成したテトラメタル塩を含む反応混合物を前記反応剤に添加する方法のいずれを用いてもよい。
【0075】
ここで、一般式(3b)、(4b)における置換基T3、T4、R10、R11及び記号vとwは一般式(1b)で示される置換基及び記号と同意義を示す。
【0076】
また、置換基L3、L4は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、好ましくは塩素原子、臭素原子、炭素数1〜20のオキシ基、アリールスルホニル基である。炭素数1〜20のオキシ基としては、特に限定はなく、例えばメトキシ基、エトキシ基、n−ブトキシ基、フェノキシ基、(2−メトキシ)フェノキシ基等を挙げることができ、アリールスルホニル基としては、特に限定はなく、例えばフェニルスルホニル基、p−トリルスルホニル基等を挙げることができる。これらの中でも特にフェニルスルホニル基が好ましい。
【0077】
そして、具体的な一般式(3b)、(4b)で示される反応剤としては、例えば2塩化硫黄;2臭化硫黄;ビス(フェニルスルホニル)スルフィド、ビス(p−トリルスルホニル)スルフィド等のビス(アリールスルホニル)スルフィド類;硫黄;2塩化セレン;セレン;2塩化テルル;テルル;ジクロロフェニルホスフィン、ジメトキシフェニルホスフィン、ジフェノキシフェニルホスフィン、ジクロロ{4−(オクチル)フェニル}ホスフィン、ジクロロ{4−(パーフルオロオクチル)フェニル}ホスフィン等のアリールホスフィン類;ジクロロ(ヘキシル)ホスフィン、ジクロロ(オクチル)ホスフィン、ジメトキシ(ヘキシル)ホスフィン等のアルキルホスフィン類;ジクロロフェニルボラン、ジメトキシフェニルボラン、ジメトキシ{4−(ヘキシル)フェニル}ボラン、ジフェノキシフェニルボラン、ジクロロ{4−(オクチル)フェニル}ボラン等のアリールボラン類;ジクロロ(ヘキシル)ボラン、ジクロロ(オクチル)ボラン、ジメトキシ(ヘキシル)ボラン等のアルキルボラン類;ジクロロフェニルアルミニウム、ジメトキシフェニルアルミニウム、ジメトキシ{4−(ヘキシル)フェニル}アルミニウム、ジフェノキシフェニルアルミニウム、ジクロロ{4−(オクチル)フェニル}アルミニウム等のアリールアルミニウム類;ジクロロ(ヘキシル)アルミニウム、ジクロロ(オクチル)アルミニウム、ジメトキシ(ヘキシル)アルミニウム等のアルキルアルミニウム類等を挙げることができ、好ましくはビス(フェニルスルホニル)スルフィド、ジクロロフェニルホスフィン、ジクロロフェニルボランである。
【0078】
テトラメタル化により生成したテトラメタル塩を一般式(3b)及び一般式(4b)で示される反応剤へ添加して反応させる際には、一般式(3b)及び一般式(4b)で示される反応剤は溶媒を存在させておくことが好ましい。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン、トルエン、ヘキサン、シクロヘキサン等が挙げられ、好ましくはTHF、ジエチルエーテル、メチル−tert−ブチルエーテルである。用いる反応剤の量は、一般式(2b)で示されるテトラハロターフェニル誘導体1当量に対し、1.2〜5当量が好ましく、特に好ましくは2〜4当量である。テトラメタル塩とジアルキルエーテルの混合物はテフロン(登録商標)キャヌラーあるいはステンレス製のキャヌラーを用いて、一般式(3b)及び一般式(4b)の反応剤へ添加することが好ましい。該反応剤との反応温度は−90〜50℃が好ましく、特に好ましくは−80〜30℃であり、反応時間は0.5〜30時間が好ましく、特に好ましくは1〜18時間である。
【0079】
本発明の一般式(1b)で示されるヘテロアセン誘導体の製造方法では、一般式(2b)で示されるテトラハロターフェニル誘導体をテトラメタル化した後、塩化マグネシウムあるいは塩化亜鉛と反応させ、その後に一般式(3b)又は一般式(4b)に示される反応剤へ添加させ、処理することもできる。
【0080】
本発明の一般式(1b)で示されるヘテロアセン誘導体の製造方法は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0081】
かくして得られた、一般式(1b)で示されるヘテロアセン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0082】
本発明の製造方法で製造される一般式(1b)で示されるヘテロアセン誘導体は特に限定はなく、例えば以下の化合物を挙げることができ、
【0083】
【化15】
【0084】
【化16】
【0085】
特に好ましくは
【0086】
【化17】
【0087】
である。
【0088】
(テトラハロターフェニル誘導体の製造方法)
最初に、本発明の本発明の一般式(1a)で示されるヘテロアセン誘導体の前駆化合物である一般式(2a)で示されるテトラハロターフェニル誘導体の製造方法について述べる。
【0089】
置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子、塩素原子である。
【0090】
一般式(2a)で示されるテトラハロターフェニル誘導体は下記一般式(5a)で示されるテトラハロベンゼンと下記一般式(6a)及び下記一般式(7a)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させることにより製造することができる。なお、一般式(6a)、(7a)で示される反応剤が同じ化合物であっても良い。
【0091】
【化18】
【0092】
(ここで、置換基X9及びX10は臭素原子、ヨウ素原子、塩素原子を示す。置換基R1、R2、X2及びX3は一般式(2a)で示される置換基と同意義を示す。)
【0093】
【化19】
【0094】
(ここで、M1はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X1は臭素原子、ヨウ素原子、塩素原子を示し、環Aは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
【0095】
【化20】
【0096】
(ここで、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X4は臭素原子、ヨウ素原子、塩素原子を示し、環Bは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
一般式(5a)の置換基X9及びX10は、臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子及び塩素原子である。
【0097】
そして、具体的な一般式(5a)で示されるテトラハロベンゼンとしては、1,4−ジブロモ−2,5−ジヨードベンゼンが挙げられる。
【0098】
一般式(6a)、(7a)の置換基M1、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物であり、上記のパラジウム及び/又はニッケル触媒により脱離され、パラジウム及び/又はニッケルと置換できる基である限り特に限定はなく、例えばMgCl、MgBr、B(OH)2、B(OMe)2、テトラメチルジオキサボロラニル基、ZnCl、ZnBr、ZnI、Sn(Bu−n)3、Si(Bu−n)3等を挙げることができ、好ましくはZnCl、B(OH)2である。
【0099】
そして、具体的な一般式(6a)、(7a)で示される化合物としては、例えば3−ブロモベンゾチエニル−2−ジンククロライド、6−ドデシル−3−ブロモベンゾチエニル−2−ジンククロライド、6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸、2−ブロモフェニルボロン酸、6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸等が挙げられる。
【0100】
なお、一般式(6a)、(7a)で示される2−ハロアリール金属試薬は、例えば、それらの原料となるアリールジハロゲン置換体をイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬によりハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン等と反応させることで好適に調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁」に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている方法を用いることもできる。
【0101】
一般式(5a)で示されるテトラハロベンゼンと一般式(6a)、(7a)で示される2−ハロアリール金属試薬の反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ジクロロビス(トリフェニルホスフィン)パラジウム、ビス(トリ−tert−ブチルホスフィン)パラジウム、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/トリ−tert−ブチルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等のパラジウム触媒;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物、ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等のニッケル触媒;を挙げることができる。中でも、好ましい触媒はテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。又、これら触媒は1種若しくは2種以上の混合物を用いても良い。
【0102】
一般式(5a)で示されるテトラハロベンゼンと一般式(6a)、(7a)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させる際には、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、エーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水、N,N−ジメチルホルムアミド、N−メチルピロリドン、トリエチルアミン、ピペリジン、ピロリジン、ジイソプロピルアミン等を挙げることができ、又、これら溶剤は1種若しくは2種以上の混合物を用いても良く、例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0103】
パラジウム触媒、ニッケル触媒の使用量は一般式(5a)で示されるテトラハロベンゼン1モルに対し、0.1〜20モル%が好ましく、特に好ましくは1〜10モル%である。
【0104】
一般式(6a)、(7a)で示される2−ハロアリール金属試薬の使用量は一般式(5a)で示されるテトラハロベンゼン1当量に対し、1.8〜3.2当量が好ましく、特に好ましくは1.9〜2.8である。
【0105】
反応の際の温度は10〜120℃が好ましく、さらに好ましくは30〜100℃、特に好ましくは40〜90℃であり、反応時間は1〜80時間が好ましく、特に好ましくは5〜70時間である。
【0106】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、水酸化セシウム、りん酸カリウム、りん酸ナトリウム、ナトリウムtert−ブトキサイド、フッ化カリウム等の無機塩基;トリエチルアミン、トリメチルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(5a)で示されるテトラハロベンゼン1当量に対し、2.0〜10.0当量が好ましく、特に好ましくは4.0〜8.0当量である。
【0107】
また、一般式(5a)で示されるテトラハロベンゼンと一般式(6a)、(7a)で示される2−ハロアリール金属試薬の反応により炭素−炭素結合が形成される位置はハロゲンの種類により制御することができる。
【0108】
即ち、ヨウ素原子の反応性が最も高く、臭素原子、塩素原子の順に反応性が低下することから、これらハロゲンの種類の反応性を利用することで反応する位置を任意に決めることができる。そのため、一般式(2a)で示されるテトロハロターフェニル誘導体の製造は、例えば一般式(5a)のX9及びX10をヨウ素原子とし、X2及びX3を臭素原子及び/又は塩素原子とすることにより、製造することができる。
【0109】
かくして得られた、本発明の一般式(2a)で示されるテトラハロターフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0110】
次に、本発明の一般式(1b)で示されるヘテロアセン誘導体の前駆化合物である一般式(2b)で示されるテトラハロターフェニル誘導体の製造方法について述べる。
【0111】
置換基X5〜X8は臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子、塩素原子である。
【0112】
一般式(2b)で示されるテトラハロターフェニル誘導体は下記一般式(5b)で示されるテトラハロチエノチオフェンと下記一般式(6b)及び下記一般式(7b)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させることにより製造することができる。なお、一般式(6b)、(7b)で示される反応剤が同じ化合物であっても良い。
【0113】
【化21】
【0114】
(ここで、置換基X11及びX12は臭素原子、ヨウ素原子、塩素原子を示す。置換基X6及びX7は一般式(2b)で示される置換基と同意義を示す。)
【0115】
【化22】
【0116】
(ここで、M3はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X5は臭素原子、ヨウ素原子、塩素原子を示し、環Aは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
【0117】
【化23】
【0118】
(ここで、M4はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X8は臭素原子、ヨウ素原子、塩素原子を示し、環Bは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
一般式(5b)の置換基X11及びX12は、臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子及び塩素原子である。
【0119】
そして、具体的な一般式(5b)で示されるテトラハロチエノチオフェンとしては、2,3,5,6−テトラブロモチエノチオフェンが挙げられる。
【0120】
一般式(6b)、(7b)の置換基M3、M4はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物であり、上記のパラジウム及び/又はニッケル触媒により脱離され、パラジウム及び/又はニッケルと置換できる基である限り特に限定はなく、例えばMgCl、MgBr、B(OH)2、B(OMe)2、テトラメチルジオキサボロラニル基、ZnCl、ZnBr、ZnI、Sn(Bu−n)3、Si(Bu−n)3等を挙げることができ、好ましくはZnCl、B(OH)2である。
【0121】
そして、具体的な一般式(6b)、(7b)で示される化合物としては、例えば3−ブロモベンゾチエニル−2−ジンククロライド、6−ドデシル−3−ブロモベンゾチエニル−2−ジンククロライド、6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸、2−ブロモフェニルボロン酸、6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸等が挙げられる。
【0122】
なお、一般式(6b)、(7b)で示される2−ハロアリール金属試薬は、例えば、それらの原料となるアリールジハロゲン置換体をイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬によりハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン等と反応させることで好適に調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁」に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている方法を用いることもできる。
【0123】
一般式(5b)で示されるテトラハロチエノチオフェンと一般式(6b)、(7b)で示される2−ハロアリール金属試薬の反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ジクロロビス(トリフェニルホスフィン)パラジウム、ビス(トリ−tert−ブチルホスフィン)パラジウム、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/トリ−tert−ブチルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等のパラジウム触媒;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物、ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等のニッケル触媒;を挙げることができる。中でも、好ましい触媒はテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。又、これら触媒は1種若しくは2種以上の混合物を用いても良い。
【0124】
一般式(5b)で示されるテトラハロチエノチオフェンと一般式(6b)、(7b)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させる際には、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、エーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水、N,N−ジメチルホルムアミド、N−メチルピロリドン、トリエチルアミン、ピペリジン、ピロリジン、ジイソプロピルアミン等を挙げることができ、又、これら溶剤は1種若しくは2種以上の混合物を用いても良く、例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0125】
パラジウム触媒、ニッケル触媒の使用量は一般式(5b)で示されるテトラハロチエノチオフェン1モルに対し、0.1〜20モル%が好ましく、特に好ましくは1〜10モル%である。
【0126】
一般式(6b)、(7b)で示される2−ハロアリール金属試薬の使用量は一般式(5b)で示されるテトラハロチエノチオフェン1当量に対し、1.8〜3.2当量が好ましく、特に好ましくは1.9〜2.8当量である。
【0127】
反応の際の温度は10〜120℃が好ましく、さらに好ましくは30〜100℃、特に好ましくは40〜90℃であり、反応時間は1〜80時間が好ましく、特に好ましくは5〜70時間である。
【0128】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、水酸化セシウム、りん酸カリウム、りん酸ナトリウム、ナトリウムtert−ブトキサイド、フッ化カリウム等の無機塩基;トリエチルアミン、トリメチルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(5b)で示されるテトラハロチエノチオフェン1当量に対し、2.0〜10.0当量が好ましく、特に好ましくは4.0〜8.0当量である。
【0129】
また、一般式(5b)で示されるテトラハロチエノチオフェンと一般式(6b)、(7b)で示される2−ハロアリール金属試薬の反応により炭素−炭素結合が形成される位置は硫黄原子の隣の位置が反応性に富むことから、2及び5位になる。
【0130】
かくして得られた、本発明の一般式(2b)で示されるテトラハロターフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【発明の効果】
【0131】
本発明は一般式(1a)、(1b)で示されるヘテロアセン誘導体を製造する新規な方法を提供する。さらに、本発明により低溶解性のテトラハロターフェニル誘導体を原料に用いても、収率良く目的のヘテロアセン誘導体を提供することができる。
【0132】
本発明の製造方法で得られるヘテロアセン誘導体は、塗布法の適用が可能であり且つ剛直棒状分子構造を有するため優れた半導体デバイス特性及び耐酸化性を有する有機半導体材料として期待される。
【実施例】
【0133】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例に限定されるものではない。
【0134】
生成物の同定には1H NMRスペクトル及びマススペクトルを用いた。なお、1H NMRスペクトルは日本電子製JEOL GSX−270WB(270MHz)を用いた。マススペクトル(MS)は日本電子製JEOL JMS−700を用いて、試料を直接導入し、電子衝突(EI)法(70エレクトロンボルト)あるいはFAB法(6キロエレクトロンボルト、キセノンガス、マトリックス:2−ニトロフェニルオクチルエーテル)(FABMS)で測定した。
【0135】
反応の進行の確認等は薄層クロマトグラフィー、ガスクロマトグラフィー(GC)及びガスクロマトグラフィー−マススペクトル(GCMS)分析を用いた。
【0136】
ガスクロマトグラフィー分析
装置 島津GC14B
カラム J&Wサイエンティフィック社製、DB−1,30m
ガスクロマトグラフィー−マススペクトル分析
装置 パーキンエルマーオートシステムXL(MS部;ターボマスゴールド)
カラム J&Wサイエンティフィック社製、DB−1,30m
反応用の試薬及び溶媒は、断りのない限り市販品を用いた。なお、グリニャール試薬あるいはブチルリチウム等の有機金属試薬を用いた場合は、市販の脱水溶媒をそのまま用いた。
【0137】
合成例1 (1,4−ジブロモ−2,5−ジヨードベンゼンの合成)(一般式(5a)の化合物の合成)
1,4−ジブロモ−2,5−ジヨードベンゼンはジャーナル オブ アメリカン ケミカル ソサイエティー、1997年、119巻、4578−4593頁に記載されている方法を参考に以下のように合成を行った。
【0138】
メカニカルスターラー付き1lの三口フラスコに過ヨウ素酸16.7g(73.0mmol)及び硫酸525mlを加えた。過ヨウ素酸が溶解した後、ヨウ化カリウム36.4g(219mmol)を少しずつ添加した。その内容物の温度を−30℃に冷却し、1,4−ジブロモベンゼン34.5g(146mmol)を5分間かけて添加した。得られた混合物を−25℃で36時間撹拌した。反応混合物を氷(2Kg)中へ注いだ後、濾過し固体を取り出した。その固体をクロロホルムに溶解させ、5%苛性ソーダ水溶液及び水で洗浄し、有機相を無水硫酸マグネシウムで乾燥した。減圧濃縮後、残渣をクロロホルムから再結晶化し、白色結晶を得た(36.0g、収率50%)。
1H NMR(CDCl3,21℃):δ=8.02(s,2H)。
【0139】
1H NMRスペクトルが文献値と一致したことより、1,4−ジブロモ−2,5−ジヨードベンゼンが得られたことを確認した。
【0140】
合成例2 ({1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモ}ベンゼンの合成(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に2,3−ジブロモベンゾチオフェン(シグマ−アルドリッチ製)886mg(3.03mmol)及びTHF8mlを添加した。この溶液を−30℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.80M)のTHF溶液3.8ml(3.0mmol)を滴下した。30分間熟成後、−50℃に冷却し、その温度で塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液3.0ml(3.0mmol)を滴下した。徐々に室温まで昇温した後、生成した白色スラリー液を減圧濃縮した。得られた白色固体[3−ブロモベンゾチエニル−2−ジンククロライド(一般式(6a)及び(7a)の化合物)]に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン492mg(1.01mmol)(一般式(5a)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)91.7mg(0.079mmol)及びTHF8mlを添加した。63℃で10時間反応を実施した後、容器を水冷し1M塩酸水溶液4mlを添加することで反応を停止させた。トルエンを添加し、得られた懸濁液を濾過し、濾板上の固体をトルエン及び水で洗浄した。固体を減圧乾燥し、白色固体292mgを得た。一方、濾液を分相し有機相を水洗した。有機相を減圧濃縮し、溶媒を留去した。得られた固体をヘキサン洗浄し(10ml)、残渣をトルエンから再結晶化した。析出した結晶を減圧乾燥後、206mgの白色固体を得た。先の濾過後の白色固体と合わせ、収率75%で目的物を得た。
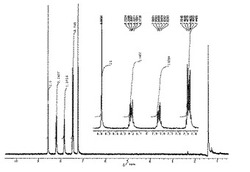
1H NMR(CDCl3,21℃):δ=7.95−7.84(m,4H),7.81(s,2H),7.58−7.44(m,4H)。
【0141】
1H NMRスペクトルを図1に示した。
MS m/z: 658(M+,44%),498(M+−2Br,34),338(M+−4Br,100),306(M+−4Br−S),9),169(M+−4Br)/2,66)。
【0142】
1H NMR及びMS測定より、{1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモ}ベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0143】
【化24】
【0144】
実施例1 (テトラチエノアセンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例2で合成した{1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモ}ベンゼン422mg(0.641mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.0ml(3.2mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)530mg(1.69mmol)とジエチルエーテル30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加えた。一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、分相し、さらに有機相を飽和食塩水で洗浄した。有機相は黄色懸濁液であったことから濾過し、黄色固体を取り出し、アセトン、メタノール、トルエンの順に洗浄後、真空乾燥し、目的物の黄色固体106mgを得た(収率41%)。
【0145】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
【0146】
MS m/z: 402(M+,100%),201(M+/2,14)。
【0147】
MS測定より、テトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0148】
【化25】
【0149】
合成例3 (2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニルの合成(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン4.39g(9.00mmol)(一般式(5a)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)974mg(0.84mmol)及び2−ブロモフェニルボロン酸(シグマ−アルドリッチ製)4.16g(一般式(6a)及び(7a)の化合物)(20.7mmol)を添加した。さらにトルエン72ml、エタノール18ml及び炭酸ナトリウム5.72g(54.0mmol)と水22mlからなる水溶液を添加した。85℃のオイルバスに浸し、15時間撹拌した。室温まで冷却後、ジクロロメタン及び飽和食塩水を添加し分相した。有機相を減圧濃縮し、残渣をトルエンから再結晶化し、白色針状晶を得た(3.68g、収率75%)。
融点:230−231℃。
1H NMR(CDCl3,21℃):δ=7.70(d,J=8.0Hz,2H),7.55(d,J=1.5Hz,2H),7.45−7.23(m,6H)。
MS m/z: 546(M+,92%),466(M+−Br,45),386(M+−2Br,53),226(M+−4Br,100)。
【0150】
1H NMR及びMS測定より、2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0151】
【化26】
【0152】
実施例2 (ベンゾビスベンゾチオフェンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル410mg(0.751mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.4ml(3.82mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)623mg(1.98mmol)とTHF30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、分相し、さらに有機相を飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的物の白色結晶を得た(79mg、収率36%)。
【0153】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
1H NMR(CDCl3,55℃):δ=8.58(s,2H),8.20(m,2H),7.85(m,2H),7.47(m,4H)。
【0154】
1H NMRスペクトルを図2に示した。
MS m/z: 290(M+,100%),145(M+/2,10)。
【0155】
1H NMR及びMS測定より、ベンゾビスベンゾチオフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0156】
【化27】
【0157】
実施例3 (P,P−ジフェニルベンゾホスホロジベンゾホスホールの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル410mg(0.751mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.4ml(3.82mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したジクロロフェニルホスフィン(東京化成工業製)355mg(1.98mmol)(一般式(3a)及び(4a)の化合物)とTHF30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄した。減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的の淡黄色結晶を得た(143mg、収率43%)。
【0158】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
1H NMRスペクトル(CDCl3,21℃):δ=8.26(s,2H),7.94(d,J=7.8Hz,2H),7.69(d,J=7.1Hz,2H),7.44(t,J=7.8Hz,2H),7.41−7.10(m,12H)。
【0159】
1H NMRスペクトルを図3に示した。
MS m/z: 442(M+,100%),364(M+−Ph−1,38),288(M+−2Ph,19),221(M+/2,10)。
【0160】
1H NMR及びMS測定より、P,P−ジフェニルベンゾホスホロジベンゾホスホールが得られたことを確認した。なお、その構造式を下記に示す。
【0161】
【化28】
【0162】
実施例4 (B,B−ジフェニルベンゾボロリルジベンゾボロール)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル425mg(0.778mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.5ml(3.98mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したジクロロフェニルボラン(シグマ−アルドリッチ製)326mg(2.05mmol)(一般式(3a)及び(4a)の化合物)とTHF30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄した。減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的の淡黄色結晶を得た(107mg、収率34%)。
【0163】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
MS m/z: 402(M+,100%),201(M+/2,14)。
【0164】
MS測定より、B,B−ジフェニルベンゾボロリルジベンゾボロールが得られたことを確認した。なお、その構造式を下記に示す。
【0165】
【化29】
【0166】
実施例5 (ベンゾビスベンゾテルロフェンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル382mg(0.70mmol)及びジエチルエーテル13.5mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.3ml(3.66mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したテルル粉末(シグマ−アルドリッチ製)242mg(1.9mmol)(一般式(3a)及び(4a)の化合物)とTHF27mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄した。減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的の淡黄色の結晶を得た(74mg、収率22%)。
【0167】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
MS m/z: 481(M+,100%),240(M+/2,13)。
【0168】
MS測定より、ベンゾビスベンゾテルロフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0169】
【化30】
【0170】
合成例4 (4−ブロモ−5−ヨード無水フタル酸の合成)
4−ブロモ−5−ヨード無水フタル酸は「ジャーナル オブ オーガニック ケミストリー」(米国)、1951年、16巻、1577−1581頁を参考に、以下の様に合成した。
【0171】
4−ブロモフタルイミド(東京化成工業製)13.3g(58.9mmol)を窒素ガスで置換した100mlの二口ナスフラスコに入れた。次いでヨウ素7.78g(30.6mmol)及び10%発煙硫酸(ヨツハタ化学工業製)50mlを加え、90℃で23時間反応を行った。
【0172】
反応混合物を室温に冷やして氷に注ぎ入れた後、ガラスフィルターでろ過し、黄色固体17.1gを得た。得られた固体を濃硫酸47mlに溶解させ、130℃で5時間反応を行った。反応混合物を氷冷後、氷水を加えて析出した固体をろ過し、フタル酸誘導体の固体18.5gを得た。次に水酸化ナトリウム4.8gを水24mlに溶かした水溶液に得られた固体を室温で溶かした。この塩基性水溶液に酢酸を加えpHを3〜4に調整し、析出するフタル酸誘導体のモノナトリウム塩の白色沈殿をろ過した。得られた白色沈殿を水に懸濁させ、濃塩酸でpHを1以下にし、再びフタル酸誘導体として白色固体7.75gを得た。この固体をトルエン70mlに溶かし、無水酢酸13ml(138mmol)を加え、105℃で4時間反応を行った。反応液を減圧濃縮して白色固体6.9gを得た。この固体を加熱トルエンで再結晶し、目的の4−ブロモ−5−ヨードフタル酸無水物6.79g(19.2mmol)を得た。(収率33%)。
1H NMR(CDCl3,22℃):δ=8.51(s,1H),8.23(s,1H)。
MS m/z: 353(M+,100%),309(M+−CO2,18%),282(M+−C2O3,10%),155(M+−C2O3−I,16%),74(M+−C2O3−I−Br,32%)。
【0173】
合成例5 (1,2―ジドデシルベンゼンの合成)
1,2−ジドデシルベンゼンは「日本化学会誌」1989年、983−987頁に従い以下の様に合成した。
【0174】
1,2−ジクロロベンゼン2.22g(15.1mmol)、ジクロロ〔1,3−ビス(ジフェニルホスフィノ)プロパン〕ニッケル(東京化成工業製)131mg(0.24mmol)、エーテル12mlの混合液にドデシルマグネシウムブロミド(シグマ−アルドリッチ製、1.0mol/lエーテル溶液)45ml(45.0mmol)を窒素雰囲気中0℃で滴下した。35℃で20時間反応を行い、反応混合物を0℃に冷やして希塩酸を加え、エーテルで抽出した。エーテル溶液を水、飽和炭酸水素ナトリウム水溶液、水の順に洗浄し、塩化カルシウムで乾燥させた。得られた液体をシリカゲルカラムクロマトグラフィー(溶離液:ヘキサン)及び減圧蒸留で精製し、目的の1,2―ジドデシルベンゼン5.56g(13.4mmol)を得た(収率88%)。
1H NMR(CDCl3,22℃):δ=7.11(m,4H),2.59(t,J=7.8Hz,4H),1.55(m,4H),1.26(m,36H),0.88(t,J=6.8Hz,6H)。
MS m/z: 414(M+,100%),260(M+−C11H23,71%),106(M+−C22H46,98%)。
【0175】
合成例6 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの合成)
2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンは「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁を参考に以下の様に合成した。
【0176】
合成例4で得られた4−ブロモ−5−ヨード無水フタル酸6.79g(19.2mmol)、合成例5で得られた1,2−ジドデシルベンゼン7.97g(19.2mmol)、テトラクロロエタン12.0mlの混合液に塩化アルミニウム5.78g(43.4mmol)を加え、室温で3時間反応を行った。水を加えてクエンチし、さらに水洗浄を行い、加熱真空乾燥後、白色固体15.0gを得た。得られた固体に濃硫酸39mlを添加し、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの固体2.95g(3.93mmol)を得た(収率60%)。
1H NMR(CDCl3,22℃):δ=8.73(s,1H),8.45(s,1H),8.05(s,2H),2.75(m,4H),1.62(m,4H),1.26(m,36H),0.88(m,6H)。
MS m/z: 750(M+,100%),440(M+−C22H46,8%),313(M+−C22H46I,2%),233(M+−C22H46IBr,1%)。
【0177】
合成例7 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの合成)
窒素雰囲気下、1lシュレンク反応容器に合成例6で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン8.92g(11.9mmol)を入れた。次いでTHF140mlを加え、水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)31ml(30.7mmol)を加え、室温で1.5時間反応を行った。次いで反応混合物に6M塩酸水溶液82mlを加え、65℃で3時間反応を行った。反応混合物を室温まで冷やし、エーテルで抽出した。エーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥、減圧濃縮し、得られた残渣に再びTHF140mlを加え、水素化ジイソブチルアルミニウム31ml(30.7mmol)を加え、室温で1.5時間反応を行った。次いで反応混合物に6M塩酸水溶液82mlを加え、3時間反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。エーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧乾燥した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの黄色固体7,11g(9.88mmol)を得た(収率83%)。
1H NMR(CDCl3,22℃):δ=8.55(s,1H),8.27(s,1H),8.16(s,1H),8.15(s,1H),7.72(s,2H),2.78(m,4H),1.71(m,4H),1.27(m,36H),0.88(m,6H)。
MS m/z: 720(M+,100%),410(M+−C22H46,16%),283(M+−C22H46−I,4%),203(M+−C22H46−I−Br,5%)。
【0178】
合成例8 (テトラブロモ(テトラドデシル)ジナフトターフェニルの合成)(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、1lシュレンク反応容器に合成例7で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン3.66g(5.08mmol)及びTHF167mlを添加した。この溶液を−52℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液13.0ml(10.2mmol)を滴下し、攪拌した。5分間熟成後、トリメトキシボラン(和光純薬工業製)1.2ml(10.8mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を添加し、40分間撹拌した。トルエンを加え、分相し、有機相を減圧濃縮し6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸を含む固体3.48gを得た(一般式(6a)及び(7a)の化合物の合成)。この内2.48g(3.62mmol)取り出し、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン(一般式(5a)の化合物)706mg(1.45mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)101mg(0.087mmol)、トルエン41ml、及びエタノール10mlを添加した。さらに炭酸ナトリウム922mg(8.70mmol)を含む水溶液13mlを加え、60℃で60時間反応を実施した。容器を水冷し3M塩酸水溶液30mlを添加することで反応を停止させた。トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(1.7ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン及びヘキサン:クロロホルム=10:1)、テトラブロモ(テトラドデシル)ジナフトターフェニルの薄黄色固体1.30gを得た(収率63%)。
1H NMR(CDCl3,22℃):δ=8.34(s,2H),8.32(s,2H),8.28(s,2H),7.97(s,1H),7.92(s,1H),7.79(s,2H),7.78(s,2H),7.73(s,1H),7.72(s,1H),2.81(m,8H),1.71(m,8H),1.28(m,72H),0.88(m,12H)。
【0179】
1H NMRスペクトルを図4に示した。
FABMS m/z: 1419(M+,100%),1340(M+−Br,10)。
【0180】
1H NMR及びMS測定より、テトラブロモ(テトラドデシル)ジナフトターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0181】
【化31】
【0182】
実施例6 (テトラドデシルジアントラジチエノベンゼンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、50mlシュレンク反応容器に、合成例8で合成したテトラブロモ(テトラドデシル)ジナフトターフェニル98mg(0.069mmol)及びジエチルエーテル3.4mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.22ml(0.35mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)57mg(0.18mmol)とTHF6.8mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的の黄色結晶33mgを得た(収率41%)。
【0183】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
1H NMR(重トルエン,100℃):δ=8.49(s,2H),8.44(s,2H),8.26(s,2H),8.21(s,2H),8.14(s,2H),7.85(s,2H),7.78(s,2H),2.87(t,J=7.8Hz,8H),1.77(m,8H),1.31(m,72H),0.88(m,J=7.8Hz,12H)。
【0184】
1H NMRスペクトルを図5に示した。
MS m/z: 1164(M+)。
【0185】
1H NMR及びMS測定より、テトラドデシルジアントラジチエノベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0186】
【化32】
【0187】
合成例9 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例6で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン541mg(0.722mmol)及びTHF20mlを添加した。−78℃に冷却後、フェニルリチウム(関東化学製、1.0mol/l、シクロヘキサン/エーテル溶液)1.5ml(1.5mmol)を加えた後、一晩かけて室温まで昇温した。次いで反応混合物に3M塩酸水溶液及びエーテルを加えた後、分相し、さらにエーテルで抽出した。エーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの黄色固体453mgを得た(収率72%)。
【0188】
合成例10 (テトラブロモ(テトラドデシル)(テトラフェニル)ジナフトターフェニルの合成)(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例9で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセン448mg(0.513mmol)及びTHF8mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液1.27ml(1.03mmol)を滴下した。10分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)107.0mg(1.03mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮し、6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸を得た(一般式(6a)及び(7a)の化合物の合成)。得られた6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン(一般式(5a)の化合物)117mg(0.240mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)16.6mg(0.014mmol)、トルエン4ml、及びエタノール1.0mlを添加した。さらに炭酸ナトリウム154mg(1.46mmol)を含む水溶液1.2mlを加え、60℃で60時間反応を実施した。容器を水冷し3M塩酸水溶液3mlを添加することで反応を停止させた。室温まで冷却後、トルエンを添加分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.05mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒;トルエン:ヘキサン=1:1)。得られた溶液を減圧濃縮し、得られた残渣をヘプタンから再結晶化し、目的物の薄黄色固体211mgを得た(収率51%)。
FABMS m/z: 1723(M+)。
【0189】
MS測定より、テトラブロモ(テトラドデシル)(テトラフェニル)ジナフトターフェニルが得られたことを確認した。なお。その構造式を下記に示す。
【0190】
【化33】
【0191】
実施例7 (テトラドデシルテトラフェニルジアントラジチエノベンゼンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例10で合成したテトラブロモ(テトラドデシル)(テトラフェニル)ジナフトターフェニル207mg(0.12mmol)及びジエチルエーテル6mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.38ml(0.60mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)101mg(0.32mmol)とTHF12mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルテトラフェニルジアントラジチエノベンゼンのオレンジ色固体56mgを得た(収率32%)。
【0192】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
FABMS m/z: 1468(M+)。
【0193】
MS測定より、テトラドデシルテトラフェニルジアントラジチエノベンゼンが得られたことを確認した。なお。その構造式を下記に示す。
【0194】
【化34】
【0195】
合成例11 (2,3,5,6−テトラブロモチエノチオフェンの合成)(一般式(5b)の化合物の合成)
2,3,5,6−テトラブロモチエノチオフェンはジャーナル オブ ケミカル ソサイエティー、パーキン トランザクション 1: 1997年、3465−3470頁に記載されている方法を参考に以下の様に合成を行った。
【0196】
1)エチル チエノ[3,2−b]チオフェン−2−カルボキシレートの合成
メカニカルスターラー付き1lの三口フラスコに、3−ブロモチオフェン−2−カルボキシアルデヒド(アルファアエサール製)26.8g(140mmol)、チオグリコール酸エチル(和光純薬工業製)16.9g(141mmol)、炭酸カリウム26g、及びジメチルホルムアルデヒド250mlを加え、室温で72時間撹拌した。反応終了後、水500mlを添加し、ジクロロメタンで抽出し、エチル チエノ[3,2−b]チオフェン−2−カルボキシレートの粗生成物23.8gを得た。
【0197】
2)チエノ[3,2−b]チオフェン−2−カルボン酸の合成
1l三口フラスコに1)で得られたエチル チエノ[3,2−b]チオフェン−2−カルボキシレート23.8g、THF300ml、及び水酸化リチウム水溶液(1.0M)300mlを加え、3時間加熱還流した。反応終了後、溶媒を減圧下濃縮し、塩酸150mlを添加すると結晶が析出した。得られた結晶を濾別し、水で3回洗浄し、乾燥後、チエノ[3,2−b]チオフェン−2−カルボン酸16.0gを得た。
【0198】
3)2,3,5−トリブロモ−チエノ[3,2−b]チオフェンの合成
窒素雰囲気下、1l三口フラスコに2)で得られたチエノ[3,2−b]チオフェン−2−カルボン酸16.0g及び酢酸水溶液500mlを加え、臭素15.2gを滴下した。5時間撹拌した後、水を加え、析出した固体を濾別し、固体を水洗し、2,3,5−トリブロモチエノ[3,2−b]チオフェン27.4gを得た。
【0199】
4)2,3,5,6−テトラブロモチエノチオフェンの合成
窒素雰囲気下、1l三口フラスコにジイソプロピルアミン2.85g(28.0mmol)及びTHF150mlを加え、0℃とした。n−ブチルリチウム(関東化学製、1.60M)ヘキサン溶液17.5ml(28.0mmol)を滴下した。得られた溶液を−66℃に冷却後、3)で得られた2,3,5−トリブロモ−チエノ[3,2−b]チオフェン10.3g(27.3mmol)を添加した。さらに臭素21gを滴下し、ゆっくりと室温まで上昇させた。飽和塩化アンモニウム水溶液を加えた後、水洗し、結晶物を濾別した。真空乾燥後、2,3,5,6−テトラブロモチエノチオフェンの固体2.1gを得た。
【0200】
合成例12 (テトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェンの合成)(一般式(2b)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例7で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン625mg(0.868mmol)及びTHF11mlを添加した。この溶液を−55℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液2.8ml(1.82mmol)を滴下し、攪拌した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)180.8mg(1.740mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を添加し、30分間撹拌した。トルエンを加え、分相し、有機相を減圧濃縮し、6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸を得た。(一般式(6b)及び(7b)の化合物の合成)。そこへ合成例11で合成した2,3,5,6−テトラブロモチエノチオフェン182mg(0.400mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)27.7mg(0.0240mmol)、トルエン6ml、及びエタノール1.6mlを添加した。さらに炭酸ナトリウム254mg(2.40mmol)を含む水溶液2mlを加え、60℃で56時間反応を実施した。容器を水冷し3M塩酸水溶液3mlを添加することで反応を停止させた。トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.06ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン及びヘキサン:クロロホルム=10:1)、テトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェンの薄黄色固体427mgを得た(収率72%)。
FABMS m/z: 1481(M+,100%),1401(M+−Br,10)。
【0201】
MS測定より、テトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0202】
【化35】
【0203】
実施例8 (テトラドデシルジアントラテトラチエノアセンの合成)(一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例12で合成したテトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェン204mg(0.137mmol)及びジエチルエーテル4.0mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.43ml(0.68mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3b)及び(4b)の化合物)113mg(0.36mmol)とTHF6.0mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルジアントラテトラチエノアセン70mgの黄色固体を得た(収率44%)。
【0204】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
FABMS m/z: 1162(M+)。
【0205】
MS測定より、テトラドデシルジアントラテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0206】
【化36】
【0207】
実施例9 (テトラドデシルジアントラジテルロジチエノアセンの合成)(一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例12で合成したテトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェン163mg(0.110mmol)及びジエチルエーテル3.2mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.34ml(0.54mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したテルル粉末(シグマ−アルドリッチ製)37mg(0.29mmol)(一般式(3b)及び(4b)の化合物)とTHF4.8mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルジアントラジテルロジチエノアセン43mgの黄色固体を得た(収率29%)。
【0208】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
【0209】
FABMS m/z: 1353(M+).
MS測定より、テトラドデシルジアントラテトラジテルロジチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0210】
【化37】
【0211】
合成例13 ({2,5−ビス(3−ブロモ−6−フルオロベンゾチエニル−2−)−3,6−ジブロモ}チエノチオフェンの合成)(一般式(2b)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に2,3−ジブロモ−6−フルオロベンゾチオフェン401mg(1.29mmol)及びTHF8mlを添加した。この溶液を−40℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.80M)のTHF溶液1.6ml(1.28mmol)を滴下した。30分間熟成後、−80℃に冷却し、その温度で塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液1.3ml(1.3mmol)を滴下した。徐々に室温まで昇温した後、生成した白色スラリー液を減圧濃縮した。得られた白色固体[3−ブロモ−6−フルオロベンゾチエニル−2−ジンククロライド、一般式(6b)及び(7b)の化合物]に、合成例11で合成した2,3,5,6−テトラブロモチエノチオフェン251mg(0.551mmol)(一般式(5b)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)38.2mg(0.033mmol)及びTHF7mlを添加した。63℃で10時間反応を実施した後、容器を水冷し1M塩酸水溶液4mlを添加することで反応を停止させた。トルエンを添加し、得られた懸濁液を濾過し、濾板上の固体をトルエン及び水で洗浄した。固体を減圧乾燥し、白色固体151mgを得た。一方、濾液を分相し有機相を水洗した。有機相を減圧濃縮し、溶媒を留去した。得られた固体をヘキサン洗浄し(10ml)、残渣をトルエンから再結晶化した。析出した結晶を減圧乾燥後、102mgの白色固体を得た。先の濾過後の白色固体と合わせ、収率61%で目的物を得た。
FABMS m/z: 756(M+,100%),676(M+−2Br,14)。
【0212】
MS測定より、{2,5−ビス(3−ブロモ−6−フルオロベンゾチエニル−2−)−3,6−ジブロモ}チエノチオフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0213】
【化38】
【0214】
実施例10 (ジフルオロジベンゾヘキサチエノアセンの合成)(一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例13で合成した{2,5−ビス(3−ブロモ−6−フルオロベンゾチエニル−2−)−3,6−ジブロモ}チエノチオフェン246mg(0.325mmol)及びジエチルエーテル4.4mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液1.0ml(1.59mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3b)及び(4b)の化合物)268mg(0.85mmol)とTHF8.8mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサン、メタノール、アセトン、トルエンの順にて洗浄した。固体を乾燥後、目的のジフルオロジベンゾヘキサチエノアセン65mgの黄色固体を得た(収率40%)。
【0215】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
MS m/z: 501(M+)。
【0216】
MS測定より、ジフルオロジベンゾヘキサチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0217】
【化39】
【0218】
参考例1 (テトラドデシルジアントラジチエノベンゼンの合成)(メタル化をTHF中で行った一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、50mlシュレンク反応容器に、合成例8で合成したテトラブロモ(テトラドデシル)ジナフトターフェニル243mg(0.171mmol)及びTHF15mlを添加した。この混合物を−72℃に冷却し、メタル化剤としてsec−ブチルリチウム(関東化学製1.0M)のシクロヘキサン/ヘキサン溶液1.4ml(1.40mmol)を滴下し、テトラメタル化を行った。30分間撹拌後、ビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)226mg(0.72mmol)を加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、黄色結晶40mgを得た(収率20%)。また、未反応の原料テトラブロモ(テトラドデシル)ジナフトターフェニルが120mg回収された。
【0219】
参考例2 (テトラドデシルジアントラテトラチエノアセンの合成)(メタル化をTHF中で行った一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例12で合成したテトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェン102mg(0.069mmol)及びTHF5.0mlを添加した。この混合物を−72℃に冷却し、メタル化剤としてsec−ブチルリチウム(関東化学製1.0M)のシクロヘキサン/ヘキサン溶液0.56ml(0.56mmol)を滴下し、テトラメタル化を行った。30分間撹拌後、ビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3b)及び(4b)の化合物)91mg(0.29mmol)を加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルジアントラテトラチエノアセン12mgの黄色固体を得た(収率15%)。また、未反応の原料テトラブロモ(テトラドデシル)ジナフトターフェニルが52mg回収された。
【図面の簡単な説明】
【0220】
【図1】合成例2で合成した{1,4−ビス(3−ブロモベンゾチエニル)−2,5−ジブロモ}ベンゼンの1H NMRスペクトル
【図2】実施例2で合成したベンゾビスベンゾチオフェンの1H NMRスペクトル
【図3】実施例3で合成したP,P−ジフェニルベンゾホスホロジベンゾホスホールの1H NMRスペクトル
【図4】合成例8で合成したテトラブロモ(テトラドデシル)ジナフトターフェニルの1H NMRスペクトル
【図5】実施例5で合成したテトラドデシルジアントラジチエノベンゼンの1H NMRスペクトル
【技術分野】
【0001】
本発明は、有機半導体材料等の電子材料への展開が可能なヘテロアセン誘導体の製造方法に関する。
【背景技術】
【0002】
有機薄膜トランジスタに代表される有機半導体デバイスは、省エネルギー、低コスト及びフレキシブルといった無機半導体デバイスにはない特徴を有することから近年注目されるようになった。この有機半導体デバイスは有機半導体活性相、基板、絶縁相、電極等数種類の材料から構成されるが、中でも電荷のキャリアー移動を担う有機半導体活性相は該デバイスの中心的な役割を有している。この有機半導体活性相を構成する有機材料のキャリアー移動能により有機半導体デバイス性能が左右される。
【0003】
有機半導体活性相を作製する方法としては一般的に、高温真空下、有機材料を気化させて実施する真空蒸着法及び有機材料を適当な溶媒に溶解させその溶液を塗布する塗布法が知られている。塗布法においては、塗布は高温高真空条件を用いることなく印刷技術を用いても実施することができる。そのため、塗布法は印刷によりデバイス作製の大幅な製造コストの削減を図ることができることから、経済的に好ましいプロセスである。しかし、従来、有機半導体デバイスとして性能が高い材料ほど塗布法で有機半導体活性相を形成することが困難になるという問題があった。
【0004】
例えば、ペンタセン等の結晶性材料はアモルファスシリコン並みの高いキャリアー移動度を有し、優れた有機半導体デバイス特性を発現することが報告されている(例えば、非特許文献1参照)。又、ペンタセン等のポリアセンを溶解させ塗布法で有機半導体デバイスを製造する試みも報告されている(例えば、特許文献1参照)。しかしながら、ペンタセンはその強い凝集性のため溶解性が低く、塗布法を適用するためには高温加熱等の条件が必要とされ、さらにペンタセンの溶液は極めて容易に空気酸化されることから、塗布法の適用はプロセス的、経済的に困難を伴うものであった。また、ポリ−(3−ヘキシルチオフェン)等の自己組織化材料は溶媒に可溶であり、塗布法による有機半導体デバイス作製が報告されてはいるが、キャリアー移動度が結晶性低分子化合物より1桁低いことから(例えば、非特許文献2参照)、得られた有機半導体デバイスの特性が低いという問題があった。
【0005】
またチオフェン環が縮環したペンタチエノアセンはペンタセンに比べ耐酸化性が向上しているが、その合成に多工程を必要とすることから(例えば、非特許文献3参照)実用上好ましい材料ではなかった。また、ブチルリチウムによるジリチオ化/硫黄との反応でチオフェン環を形成する反応が知られているが、低収率で副生物が多く生成する問題があった(例えば、非特許文献4参照)。
【0006】
【非特許文献1】「ジャーナル オブ アプライドフィジックス」(米国)、2002年、92巻、5259−5263頁
【非特許文献2】「サイエンス」(米国)、1998年、280巻、1741−1744頁
【非特許文献3】「オルガニック レターズ」(米国)、2005年、7巻、5301−5304頁
【非特許文献4】「ジャーナル オブ ヘテロサイクリック ケミストリィー」、1991年、28巻、433−438頁
【特許文献1】WO2003/016599号
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明は上記の従来技術が有する問題点に鑑み、優れた半導体性能及び耐酸化性を有し、塗布法による有機半導体活性相形成が可能な、ヘテロアセン誘導体の製造方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは上記課題を解決するため鋭意検討の結果、テトラハロターフェニル誘導体を原料に用いる本発明のヘテロアセン誘導体の新規な製造方法を見出した。加えて、該ヘテロアセン誘導体が耐酸化性に優れ、塗布法の適用が可能であるため結晶性の薄膜を容易に安定して作製することができることから、本発明を完成するに到った。
【0009】
以下に本発明を詳細に説明する。説明はヘテロアセン誘導体の製造方法並びに該ヘテロアセン誘導体の前駆体であるテトラハロターフェニル誘導体の製造方法について述べる。
【0010】
(ヘテロアセン誘導体の製造方法)
最初に、下記一般式(1a)で示されるヘテロアセン誘導体の製造方法について説明する。本発明の下記一般式(1a)で示されるヘテロアセン誘導体は、下記一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、下記一般式(3a)及び下記一般式(4a)で示される反応剤と反応させることで製造する。
【0011】
【化1】
【0012】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T1及びT2は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有し、l及びmは、各々0又は1の整数を示す。)
【0013】
【化2】
【0014】
【化3】
【0015】
(ここで、置換基R5〜R9は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、炭素数2〜30のアルキニル基、炭素数2〜30のアルケニル基を示す。nは0〜2の整数であり、oは0又は1の整数である。)]
【0016】
【化4】
【0017】
(ここで、置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、置換基R1及びR2並びに環A及びBは一般式(1a)で示される置換基並びに環と同意義を示す。)
(R3)lT1(L1)p (3a)
(R4)mT2(L2)q (4a)
(ここで、置換基T1、T2、R3、R4及び記号lとmは一般式(1a)で示される置換基及び記号と同意義を示し、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、p及びqは0又は2の整数を示す。)
なお、ここでテトラメタル化とは、一般式(2a)におけるX1〜X4をそれぞれメタルに置換することを意味する。
【0018】
一般式(1a)で示されるヘテロアセン誘導体の置換基について述べる。
【0019】
置換基R1〜R4及びR5〜R9における炭素数1〜30のアルキル基は、特に限定はなく、例えばメチル基、プロピル基、ブチル基、イソブチル基、tert−ブチル基、ネオペンチル基、ヘキシル基、オクチル基、デシル基、ドデシル基、オクタデシル基、2−エチルヘキシル基、シクロヘキシル基、シクロオクチル基等のアルキル基;トリフルオロメチル基、ペンタフルオロエチル基、パーフルオロオクチル基、パーフルオロデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基、パーフルオロシクロヘキシル基、パーフルオロシクロオクチル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基、2−エチルパーフルオロヘキシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくは炭素数6〜30のアルキル基であり、より好ましくはドデシル基、オクタデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基であり、特に好ましくはドデシル基、パーフルオロドデシル基である。
【0020】
置換基R1〜R4及びR5〜R9における炭素数4〜30のアリール基は、特に限定はなく、例えばフェニル基、p−トリル基、p−(オクチル)フェニル基、p−(ドデシル)フェニル基、p−(シクロヘキシル)フェニル基、m−(オクチル)フェニル基、m−(ドデシル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基、p−(パーフルオロドデシル)フェニル基、m−(パーフルオロドデシル)フェニル基、2−チエニル基、5−(ドデシル)−2−チエニル基、ベンゾチエニル−2−基、6−ドデシルベンゾチエニル−2−基、2,2’−ビチエニル−5−基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、アントラセニル基等を挙げることができ、好ましくはフェニル基、p−(オクチル)フェニル基、p−(パーフルオロオクチル)フェニル基、5−(ドデシル)−2−チエニル基等であり、特に好ましくはフェニル基である。
【0021】
置換基R5〜R9における、炭素数2〜30のアルキニル基は、特に限定はなく、例えばエチニル基、メチルエチニル基、イソプロピルエチニル基、tert−ブチルエチニル基、(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(トリフルオロメチル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基、{p−(オクチル)フェニル}エチニル基、{p−(ドデシル)フェニル}エチニル基、{m−(ドデシル)フェニル}エチニル基、ナフチルエチニル基、アントラセニルエチニル基、ベンジルエチニル基、パーフルオロフェニルエチニル基、{p−(トリフルオロメチル)フェニル}エチニル基、{p−(パーフルオロオクチル)フェニル}エチニル基、{p−(パーフルオロドデシル)フェニル}エチニル基、{m−(パーフルオロドデシル)フェニル}エチニル基、5−(ヘキシル)チエニル−2−}エチニル基、{5−(パーフルオロヘキシル)チエニル−2−}エチニル基等を挙げることができ、好ましくは(オクチル)エチニル基、(デシル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基等である。
【0022】
置換基R5〜R9における、炭素数2〜30のアルケニル基は、特に限定はなく、例えばエテニル基、メチルエテニル基、イソプロピルエテニル基、tert−ブチルエテニル基、(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(トリフルオロメチル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、フェニルエテニル基、{p−(ヘキシル)フェニル}エテニル基、{p−(オクチル)フェニル}エテニル基、{p−(ドデシル)フェニル}エテニル基、{m−(ドデシル)フェニル}エテニル基、2−フェニル−1,2−ジフルオロエテニル基、2−フェニル−1,2−ジメチルエテニル基、ジフェニルエテニル基、トリフェニルエテニル基、ナフチルエテニル基、アントラセニルエテニル基、ベンジルエテニル基、フェニル(メチル)エテニル基、(パーフルオロフェニル)エテニル基、{p−(トリフルオロメチル)フェニル}エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基等を挙げることができ、好ましくは(オクチル)エテニル基、(デシル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基等を挙げることができる。なお、該炭素数2〜30のアルケニル基はトランス体及びシス体の何れであってもよく、またそれらの任意の割合の混合物であってもよい。
【0023】
これらの置換基の中でも、R1、R2としては、水素原子、フッ素原子、炭素数4〜30のアリール基が好ましく、特に好ましくは水素原子である。R3、R4としては、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、さらに好ましくは炭素数4〜30のアリール基であり、特に好ましくはフェニル基である。一方、R5〜R7としては、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数2〜30のアルキニル基が好ましく、さらに好ましくはフッ素原子、炭素数1〜30のアルキル基であり、特に好ましくはフッ素原子、ドデシル基であり、R8、R9としては、水素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、さらに好ましくは水素原子、炭素数4〜30のアリール基であり、特に好ましくは水素原子、フェニル基である。
【0024】
置換基T1及びT2は、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子であり、その中でも好ましくは硫黄原子、セレン原子、リン原子、ホウ素原子であり、さらに好ましくは硫黄原子、リン原子、ホウ素原子である。
【0025】
l及びmは各々0又は1の整数であり、T1及びT2が硫黄原子、セレン原子、テルル原子、酸素原子の場合0であり、T1及びT2がリン原子、ホウ素原子、アルミニウム原子の場合1である。
【0026】
次に、一般式(A−1)及び(A−2)で示される、環A及びBについて述べる。
【0027】
一般式(1a)で示されるヘテロアセン誘導体は、環A及びBを有する誘導体であり、環A及びBは一般式(A−1)又は(A−2)で示される構造を有するものである。
【0028】
一般式(A−1)中の記号nは、0〜2の整数であり、好ましくは1であり、一般式(A−2)中の記号oは、0又は1の整数であり、好ましくは1である。また、環A及びBが一般式(A−1)の構造の場合、一般式(1a)で示されるヘテロアセン誘導体の構造としては、下記一般式(1c)で示されるものであることが好ましい。
【0029】
【化5】
【0030】
(ここで、置換基R1〜R4、T1及びT2並びにl及びmは一般式(1a)で示される置換基並びに記号と同意義を示し、置換基R5は一般式(A−1)で示される置換基と同意義を示し、rは0〜2の整数である。)
本発明の一般式(1a)で示されるヘテロアセン誘導体の製造方法は、一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3a)及び一般式(4a)で示される反応剤と反応させることで達成することができる。
【0031】
一般式(2a)で示されるテトラハロターフェニル誘導体をテトラメタル化する場合、用いるメタル化剤は、一般式(2a)におけるX1〜X4をメタルに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属;メチルマグネシウムブロマイド、エチルマグネシウムブロマイド、イソプロピルマグネシウムクロライド、tert−ブチルマグネシウムクロライド、フェニルマグネシウムブロマイド等のグリニャール試薬;マグネシウム金属;亜鉛金属等を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムである。
【0032】
該メタル化剤の使用量は一般式(2a)で示されるテトラハロターフェニル誘導体1当量に対し、3.0〜10当量が好ましく、さらに好ましくは4.0〜8.0当量、特に好ましくは4.2〜6.0当量である。
【0033】
該テトラメタル化は、ジアルキルエーテル中で実施する。ジアルキルエーテル中で実施することにより、テトラヒドロフラン、ジオキサン、トルエン、ヘキサン、シクロヘキサン等の溶媒中で実施するよりも効率よく目的物が得られる。該ジアルキルエーテルとしては、例えばジメチルエーテル、メチルエチルエーテル、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル等が挙げられ、特に好ましくはジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。該テトラメタル化の温度は−80〜50℃が好ましく、特に好ましくは−20〜30℃である。反応時間は1〜120分が好ましく、特に好ましくは5〜90分である。本発明では、低溶解性のテトラハロターフェニル誘導体でもジアルキルエーテル中−20〜30℃でリチウム化合物と反応させることにより、収率良くテトラリチオ化できることを見出した。なお、テトラメタル化の進行は、反応液の一部を取り出し、水で反応を停止させた後、薄層クロマトグラフィー、ガスクロマトグラフィーで分析することで監視することができる。
【0034】
該テトラメタル化により生成したテトラメタル塩は、次いで一般式(3a)及び一般式(4a)で示される反応剤と反応させる。係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を直接用いて反応させる方法、生成したテトラメタル塩を一度単離した後、前記反応剤と反応させる方法のいずれを用いてもよい。
【0035】
また係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を添加する方法、生成したテトラメタル塩を含む反応混合物を前記反応剤に添加する方法のいずれを用いてもよい。
【0036】
ここで、一般式(3a)、(4a)における置換基T1、T2、R3、R4及び記号lとmは一般式(1a)で示される置換基及び記号と同意義を示す。
【0037】
また、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、好ましくは塩素原子、臭素原子、炭素数1〜20のオキシ基、アリールスルホニル基である。炭素数1〜20のオキシ基としては、特に限定はなく、例えばメトキシ基、エトキシ基、n−ブトキシ基、フェノキシ基、(2−メトキシ)フェノキシ基等を挙げることができ、アリールスルホニル基としては、特に限定はなく、例えばフェニルスルホニル基、p−トリルスルホニル基等を挙げることができる。これらの中でも特にフェニルスルホニル基が好ましい。
【0038】
そして、具体的な一般式(3a)、(4a)で示される反応剤としては、例えば2塩化硫黄;2臭化硫黄;ビス(フェニルスルホニル)スルフィド、ビス(p−トリルスルホニル)スルフィド等のビス(アリールスルホニル)スルフィド類;硫黄;2塩化セレン;セレン;2塩化テルル;テルル;ジクロロフェニルホスフィン、ジメトキシフェニルホスフィン、ジフェノキシフェニルホスフィン、ジクロロ{4−(オクチル)フェニル}ホスフィン、ジクロロ{4−(パーフルオロオクチル)フェニル}ホスフィン等のアリールホスフィン類;ジクロロ(ヘキシル)ホスフィン、ジクロロ(オクチル)ホスフィン、ジメトキシ(ヘキシル)ホスフィン等のアルキルホスフィン類;ジクロロフェニルボラン、ジメトキシフェニルボラン、ジメトキシ{4−(ヘキシル)フェニル}ボラン、ジフェノキシフェニルボラン、ジクロロ{4−(オクチル)フェニル}ボラン等のアリールボラン類;ジクロロ(ヘキシル)ボラン、ジクロロ(オクチル)ボラン、ジメトキシ(ヘキシル)ボラン等のアルキルボラン類;ジクロロフェニルアルミニウム、ジメトキシフェニルアルミニウム、ジメトキシ{4−(ヘキシル)フェニル}アルミニウム、ジフェノキシフェニルアルミニウム、ジクロロ{4−(オクチル)フェニル}アルミニウム等のアリールアルミニウム類;ジクロロ(ヘキシル)アルミニウム、ジクロロ(オクチル)アルミニウム、ジメトキシ(ヘキシル)アルミニウム等のアルキルアルミニウム類等を挙げることができ、好ましくはビス(フェニルスルホニル)スルフィド、ジクロロフェニルホスフィン、ジクロロフェニルボランである。なお、一般式(3a)及び一般式(4a)で示される反応剤は、同じ化合物であっても良い。
【0039】
テトラメタル化により生成したテトラメタル塩を一般式(3a)及び一般式(4a)で示される反応剤へ添加して反応させる際には、一般式(3a)及び一般式(4a)で示される反応剤は溶媒を存在させておくことが好ましい。用いる溶媒は特に限定はなく、例えばテトラヒドロフラン(以後THFと略す)、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン、トルエン、ヘキサン、シクロヘキサン等が挙げられ、好ましくはTHF、ジエチルエーテル、メチル−tert−ブチルエーテルである。用いる反応剤の量は、一般式(2a)で示されるテトラハロターフェニル誘導体1当量に対し、1.2〜5当量が好ましく、特に好ましくは2〜4当量である。テトラメタル塩とジアルキルエーテルの混合物はテフロン(登録商標)キャヌラーあるいはステンレス製のキャヌラーを用いて、一般式(3a)及び一般式(4a)の反応剤へ添加することが好ましい。該反応剤との反応温度は−90〜50℃が好ましく、特に好ましくは−80〜30℃であり、反応時間は0.5〜30時間が好ましく、特に好ましくは1〜18時間である。
【0040】
本発明の一般式(1a)で示されるヘテロアセン誘導体の製造方法では、一般式(2a)で示されるテトラハロターフェニル誘導体をテトラメタル化した後、塩化マグネシウムあるいは塩化亜鉛と反応させ、その後に一般式(3a)、(4a)に示される反応剤へ添加させ、処理することもできる。
【0041】
本発明の一般式(1a)で示されるヘテロアセン誘導体の製造方法は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0042】
かくして得られた、一般式(1a)で示されるヘテロアセン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0043】
本発明の製造方法で製造される一般式(1a)で示されるヘテロアセン誘導体は特に限定はなく、例えば以下の化合物を挙げることができ、
【0044】
【化6】
【0045】
【化7】
【0046】
【化8】
【0047】
【化9】
【0048】
【化10】
【0049】
特に好ましくは
【0050】
【化11】
【0051】
である。
【0052】
次に、下記一般式(1b)で示されるヘテロアセン誘導体の製造方法について説明する。
【0053】
本発明の下記一般式(1b)で示されるヘテロアセン誘導体は、下記一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3b)及び一般式(4b)で示される反応剤と反応させることで製造する。
【0054】
【化12】
【0055】
(ここで、置換基R10及びR11は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T3及びT4は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、上記一般式(A−1)又は(A−2)で示される構造を有し、v及びwは、各々0又は1の整数を示す。)
【0056】
【化13】
【0057】
(ここで、置換基X5〜X8は臭素原子、ヨウ素原子、塩素原子を示し、環A及びBは一般式(1b)で示される環と同意義を示す。)
(R10)vT3(L3)x (3b)
(R11)wT4(L4)y (4b)
(ここで、置換基T3、T4、R10、R11及び記号vとwは一般式(1b)で示される置換基及び記号と同意義を示し、置換基L3、L4は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、x及びyは0又は2の整数を示す。)
一般式(1b)で示されるヘテロアセン誘導体の置換基について述べる。
【0058】
置換基R10及びR11並びにR5〜R9における炭素数1〜30のアルキル基は、特に限定はなく、例えばメチル基、プロピル基、ブチル基、イソブチル基、tert−ブチル基、ネオペンチル基、ヘキシル基、オクチル基、デシル基、ドデシル基、オクタデシル基、2−エチルヘキシル基、シクロヘキシル基、シクロオクチル基等のアルキル基;トリフルオロメチル基、ペンタフルオロエチル基、パーフルオロオクチル基、パーフルオロデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基、パーフルオロシクロヘキシル基、パーフルオロシクロオクチル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基、2−エチルパーフルオロヘキシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくは炭素数6〜30のアルキル基であり、より好ましくはドデシル基、オクタデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基であり、特に好ましくはドデシル基、パーフルオロドデシル基である。
【0059】
置換基R10及びR11並びにR5〜R9における炭素数4〜30のアリール基は、特に限定はなく、例えばフェニル基、p−トリル基、p−(オクチル)フェニル基、p−(ドデシル)フェニル基、p−(シクロヘキシル)フェニル基、m−(オクチル)フェニル基、m−(ドデシル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基、p−(パーフルオロドデシル)フェニル基、m−(パーフルオロドデシル)フェニル基、2−チエニル基、5−(ドデシル)−2−チエニル基、ベンゾチエニル−2−基、6−ドデシルベンゾチエニル−2−基、2,2’−ビチエニル−5−基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、アントラセニル基等を挙げることができ、好ましくはフェニル基、p−(オクチル)フェニル基、p−(パーフルオロオクチル)フェニル基、5−(ドデシル)−2−チエニル基等であり、特に好ましくはフェニル基である。
【0060】
置換基R5〜R9における、炭素数2〜30のアルキニル基は、特に限定はなく、例えばエチニル基、メチルエチニル基、イソプロピルエチニル基、tert−ブチルエチニル基、(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(トリフルオロメチル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基、{p−(オクチル)フェニル}エチニル基、{p−(ドデシル)フェニル}エチニル基、{m−(ドデシル)フェニル}エチニル基、ナフチルエチニル基、アントラセニルエチニル基、ベンジルエチニル基、パーフルオロフェニルエチニル基、{p−(トリフルオロメチル)フェニル}エチニル基、{p−(パーフルオロオクチル)フェニル}エチニル基、{p−(パーフルオロドデシル)フェニル}エチニル基、{m−(パーフルオロドデシル)フェニル}エチニル基、5−(ヘキシル)チエニル−2−}エチニル基、{5−(パーフルオロヘキシル)チエニル−2−}エチニル基等を挙げることができ、好ましくは(オクチル)エチニル基、(デシル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基等である。
【0061】
置換基R5〜R9における、炭素数2〜30のアルケニル基は、特に限定はなく、例えばエテニル基、メチルエテニル基、イソプロピルエテニル基、tert−ブチルエテニル基、(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(トリフルオロメチル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、フェニルエテニル基、{p−(ヘキシル)フェニル}エテニル基、{p−(オクチル)フェニル}エテニル基、{p−(ドデシル)フェニル}エテニル基、{m−(ドデシル)フェニル}エテニル基、2−フェニル−1,2−ジフルオロエテニル基、2−フェニル−1,2−ジメチルエテニル基、ジフェニルエテニル基、トリフェニルエテニル基、ナフチルエテニル基、アントラセニルエテニル基、ベンジルエテニル基、フェニル(メチル)エテニル基、(パーフルオロフェニル)エテニル基、{p−(トリフルオロメチル)フェニル}エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基等を挙げることができ、好ましくは(オクチル)エテニル基、(デシル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基等を挙げることができる。なお、該炭素数2〜30のアルケニル基はトランス体及びシス体の何れであってもよく、またそれらの任意の割合の混合物であってもよい。
【0062】
これらの置換基の中でも、R10、R11としては、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、特に好ましくは炭素数4〜30のアリール基である。一方、R5〜R7としては、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数2〜30のアルキニル基が好ましく、さらに好ましくはフッ素原子、炭素数1〜30のアルキル基であり、特に好ましくはフッ素原子、ドデシル基であり、R8、R9としては、水素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基が好ましく、特に好ましくは水素原子、炭素数4〜30のアリール基である。
【0063】
置換基T3及びT4は、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子であり、その中でも好ましくは硫黄原子、セレン原子、リン原子、ホウ素原子であり、さらに好ましくは硫黄原子、リン原子、ホウ素原子である。
【0064】
v及びwは各々0又は1の整数であり、T3及びT4が硫黄原子、セレン原子、テルル原子、酸素原子の場合0であり、T3及びT4がリン原子、ホウ素原子、アルミニウム原子の場合1である。
【0065】
次に、一般式(A−1)及び(A−2)で示される、環A及びBについて述べる。
【0066】
一般式(1b)で示されるヘテロアセン誘導体は、環A及びBを有する誘導体であり、環A及びBは一般式(A−1)又は(A−2)で示される構造を有するものである。
【0067】
一般式(A−1)中の記号nは、0〜2の整数であり、好ましくは1であり、一般式(A−2)中の記号oは、0又は1の整数であり、好ましくは1である。また、環A及びBが一般式(A−1)の構造の場合、一般式(1b)で示されるヘテロアセン誘導体の構造としては、下記一般式(1d)で示されるものであることが好ましい。
【0068】
【化14】
【0069】
(ここで、置換基R10及びR11、T3及びT4並びにv及びwは一般式(1b)で示される置換基並びに記号と同意義を示し、置換基R5は一般式(A−1)で示され置換基と同意義を示し、tは0〜2の整数である。)
本発明の一般式(1b)で示されるヘテロアセン誘導体の製造方法は、一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、一般式(3b)及び一般式(4b)で示される反応剤と反応させることで達成することができる。
【0070】
一般式(2b)で示されるテトラハロターフェニル誘導体をテトラメタル化する場合、用いるメタル化剤は、一般式(2b)におけるX5〜X8をメタルに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属;メチルマグネシウムブロマイド、エチルマグネシウムブロマイド、イソプロピルマグネシウムクロライド、tert−ブチルマグネシウムクロライド、フェニルマグネシウムブロマイド等のグリニャール試薬;マグネシウム金属;亜鉛金属等を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムである。
【0071】
該メタル化剤の使用量は一般式(2b)で示されるテトラハロターフェニル誘導体1当量に対し、3.0〜10当量が好ましく、さらに好ましくは4.0〜8.0当量、特に好ましくは4.2〜6.0当量である。
【0072】
該テトラメタル化は、ジアルキルエーテル中で実施する。ジアルキルエーテル中で実施することにより、テトラヒドロフラン、ジオキサン、トルエン、ヘキサン、シクロヘキサン等の溶媒中で実施するよりも効率よく目的物が得られる。該ジアルキルエーテルとしては、例えばジメチルエーテル、メチルエチルエーテル、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル等が挙げられ、特に好ましくはジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。該テトラメタル化の温度は−80〜50℃が好ましく、特に好ましくは−20〜30℃である。反応時間は1〜120分が好ましく、特に好ましくは5〜90分である。本発明では、低溶解性のテトラハロターフェニル誘導体でもジアルキルエーテル溶媒中−20〜30℃でリチウム化合物と反応させることにより、収率良くテトラリチオ化できることを見出した。なお、テトラメタル化の進行は、反応液の一部を取り出し、水で反応を停止させた後、薄層クロマトグラフィー、ガスクロマトグラフィーで分析することで監視することができる。
【0073】
該テトラメタル化により生成したテトラメタル塩は、次いで一般式(3b)及び一般式(4b)で示される反応剤と反応させる。係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を直接用いて反応させる方法、生成したテトラメタル塩を一度単離した後、前記反応剤と反応させる方法のいずれを用いてもよい。
【0074】
また係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を添加する方法、生成したテトラメタル塩を含む反応混合物を前記反応剤に添加する方法のいずれを用いてもよい。
【0075】
ここで、一般式(3b)、(4b)における置換基T3、T4、R10、R11及び記号vとwは一般式(1b)で示される置換基及び記号と同意義を示す。
【0076】
また、置換基L3、L4は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、好ましくは塩素原子、臭素原子、炭素数1〜20のオキシ基、アリールスルホニル基である。炭素数1〜20のオキシ基としては、特に限定はなく、例えばメトキシ基、エトキシ基、n−ブトキシ基、フェノキシ基、(2−メトキシ)フェノキシ基等を挙げることができ、アリールスルホニル基としては、特に限定はなく、例えばフェニルスルホニル基、p−トリルスルホニル基等を挙げることができる。これらの中でも特にフェニルスルホニル基が好ましい。
【0077】
そして、具体的な一般式(3b)、(4b)で示される反応剤としては、例えば2塩化硫黄;2臭化硫黄;ビス(フェニルスルホニル)スルフィド、ビス(p−トリルスルホニル)スルフィド等のビス(アリールスルホニル)スルフィド類;硫黄;2塩化セレン;セレン;2塩化テルル;テルル;ジクロロフェニルホスフィン、ジメトキシフェニルホスフィン、ジフェノキシフェニルホスフィン、ジクロロ{4−(オクチル)フェニル}ホスフィン、ジクロロ{4−(パーフルオロオクチル)フェニル}ホスフィン等のアリールホスフィン類;ジクロロ(ヘキシル)ホスフィン、ジクロロ(オクチル)ホスフィン、ジメトキシ(ヘキシル)ホスフィン等のアルキルホスフィン類;ジクロロフェニルボラン、ジメトキシフェニルボラン、ジメトキシ{4−(ヘキシル)フェニル}ボラン、ジフェノキシフェニルボラン、ジクロロ{4−(オクチル)フェニル}ボラン等のアリールボラン類;ジクロロ(ヘキシル)ボラン、ジクロロ(オクチル)ボラン、ジメトキシ(ヘキシル)ボラン等のアルキルボラン類;ジクロロフェニルアルミニウム、ジメトキシフェニルアルミニウム、ジメトキシ{4−(ヘキシル)フェニル}アルミニウム、ジフェノキシフェニルアルミニウム、ジクロロ{4−(オクチル)フェニル}アルミニウム等のアリールアルミニウム類;ジクロロ(ヘキシル)アルミニウム、ジクロロ(オクチル)アルミニウム、ジメトキシ(ヘキシル)アルミニウム等のアルキルアルミニウム類等を挙げることができ、好ましくはビス(フェニルスルホニル)スルフィド、ジクロロフェニルホスフィン、ジクロロフェニルボランである。
【0078】
テトラメタル化により生成したテトラメタル塩を一般式(3b)及び一般式(4b)で示される反応剤へ添加して反応させる際には、一般式(3b)及び一般式(4b)で示される反応剤は溶媒を存在させておくことが好ましい。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン、トルエン、ヘキサン、シクロヘキサン等が挙げられ、好ましくはTHF、ジエチルエーテル、メチル−tert−ブチルエーテルである。用いる反応剤の量は、一般式(2b)で示されるテトラハロターフェニル誘導体1当量に対し、1.2〜5当量が好ましく、特に好ましくは2〜4当量である。テトラメタル塩とジアルキルエーテルの混合物はテフロン(登録商標)キャヌラーあるいはステンレス製のキャヌラーを用いて、一般式(3b)及び一般式(4b)の反応剤へ添加することが好ましい。該反応剤との反応温度は−90〜50℃が好ましく、特に好ましくは−80〜30℃であり、反応時間は0.5〜30時間が好ましく、特に好ましくは1〜18時間である。
【0079】
本発明の一般式(1b)で示されるヘテロアセン誘導体の製造方法では、一般式(2b)で示されるテトラハロターフェニル誘導体をテトラメタル化した後、塩化マグネシウムあるいは塩化亜鉛と反応させ、その後に一般式(3b)又は一般式(4b)に示される反応剤へ添加させ、処理することもできる。
【0080】
本発明の一般式(1b)で示されるヘテロアセン誘導体の製造方法は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0081】
かくして得られた、一般式(1b)で示されるヘテロアセン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0082】
本発明の製造方法で製造される一般式(1b)で示されるヘテロアセン誘導体は特に限定はなく、例えば以下の化合物を挙げることができ、
【0083】
【化15】
【0084】
【化16】
【0085】
特に好ましくは
【0086】
【化17】
【0087】
である。
【0088】
(テトラハロターフェニル誘導体の製造方法)
最初に、本発明の本発明の一般式(1a)で示されるヘテロアセン誘導体の前駆化合物である一般式(2a)で示されるテトラハロターフェニル誘導体の製造方法について述べる。
【0089】
置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子、塩素原子である。
【0090】
一般式(2a)で示されるテトラハロターフェニル誘導体は下記一般式(5a)で示されるテトラハロベンゼンと下記一般式(6a)及び下記一般式(7a)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させることにより製造することができる。なお、一般式(6a)、(7a)で示される反応剤が同じ化合物であっても良い。
【0091】
【化18】
【0092】
(ここで、置換基X9及びX10は臭素原子、ヨウ素原子、塩素原子を示す。置換基R1、R2、X2及びX3は一般式(2a)で示される置換基と同意義を示す。)
【0093】
【化19】
【0094】
(ここで、M1はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X1は臭素原子、ヨウ素原子、塩素原子を示し、環Aは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
【0095】
【化20】
【0096】
(ここで、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X4は臭素原子、ヨウ素原子、塩素原子を示し、環Bは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
一般式(5a)の置換基X9及びX10は、臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子及び塩素原子である。
【0097】
そして、具体的な一般式(5a)で示されるテトラハロベンゼンとしては、1,4−ジブロモ−2,5−ジヨードベンゼンが挙げられる。
【0098】
一般式(6a)、(7a)の置換基M1、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物であり、上記のパラジウム及び/又はニッケル触媒により脱離され、パラジウム及び/又はニッケルと置換できる基である限り特に限定はなく、例えばMgCl、MgBr、B(OH)2、B(OMe)2、テトラメチルジオキサボロラニル基、ZnCl、ZnBr、ZnI、Sn(Bu−n)3、Si(Bu−n)3等を挙げることができ、好ましくはZnCl、B(OH)2である。
【0099】
そして、具体的な一般式(6a)、(7a)で示される化合物としては、例えば3−ブロモベンゾチエニル−2−ジンククロライド、6−ドデシル−3−ブロモベンゾチエニル−2−ジンククロライド、6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸、2−ブロモフェニルボロン酸、6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸等が挙げられる。
【0100】
なお、一般式(6a)、(7a)で示される2−ハロアリール金属試薬は、例えば、それらの原料となるアリールジハロゲン置換体をイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬によりハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン等と反応させることで好適に調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁」に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている方法を用いることもできる。
【0101】
一般式(5a)で示されるテトラハロベンゼンと一般式(6a)、(7a)で示される2−ハロアリール金属試薬の反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ジクロロビス(トリフェニルホスフィン)パラジウム、ビス(トリ−tert−ブチルホスフィン)パラジウム、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/トリ−tert−ブチルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等のパラジウム触媒;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物、ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等のニッケル触媒;を挙げることができる。中でも、好ましい触媒はテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。又、これら触媒は1種若しくは2種以上の混合物を用いても良い。
【0102】
一般式(5a)で示されるテトラハロベンゼンと一般式(6a)、(7a)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させる際には、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、エーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水、N,N−ジメチルホルムアミド、N−メチルピロリドン、トリエチルアミン、ピペリジン、ピロリジン、ジイソプロピルアミン等を挙げることができ、又、これら溶剤は1種若しくは2種以上の混合物を用いても良く、例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0103】
パラジウム触媒、ニッケル触媒の使用量は一般式(5a)で示されるテトラハロベンゼン1モルに対し、0.1〜20モル%が好ましく、特に好ましくは1〜10モル%である。
【0104】
一般式(6a)、(7a)で示される2−ハロアリール金属試薬の使用量は一般式(5a)で示されるテトラハロベンゼン1当量に対し、1.8〜3.2当量が好ましく、特に好ましくは1.9〜2.8である。
【0105】
反応の際の温度は10〜120℃が好ましく、さらに好ましくは30〜100℃、特に好ましくは40〜90℃であり、反応時間は1〜80時間が好ましく、特に好ましくは5〜70時間である。
【0106】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、水酸化セシウム、りん酸カリウム、りん酸ナトリウム、ナトリウムtert−ブトキサイド、フッ化カリウム等の無機塩基;トリエチルアミン、トリメチルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(5a)で示されるテトラハロベンゼン1当量に対し、2.0〜10.0当量が好ましく、特に好ましくは4.0〜8.0当量である。
【0107】
また、一般式(5a)で示されるテトラハロベンゼンと一般式(6a)、(7a)で示される2−ハロアリール金属試薬の反応により炭素−炭素結合が形成される位置はハロゲンの種類により制御することができる。
【0108】
即ち、ヨウ素原子の反応性が最も高く、臭素原子、塩素原子の順に反応性が低下することから、これらハロゲンの種類の反応性を利用することで反応する位置を任意に決めることができる。そのため、一般式(2a)で示されるテトロハロターフェニル誘導体の製造は、例えば一般式(5a)のX9及びX10をヨウ素原子とし、X2及びX3を臭素原子及び/又は塩素原子とすることにより、製造することができる。
【0109】
かくして得られた、本発明の一般式(2a)で示されるテトラハロターフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0110】
次に、本発明の一般式(1b)で示されるヘテロアセン誘導体の前駆化合物である一般式(2b)で示されるテトラハロターフェニル誘導体の製造方法について述べる。
【0111】
置換基X5〜X8は臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子、塩素原子である。
【0112】
一般式(2b)で示されるテトラハロターフェニル誘導体は下記一般式(5b)で示されるテトラハロチエノチオフェンと下記一般式(6b)及び下記一般式(7b)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させることにより製造することができる。なお、一般式(6b)、(7b)で示される反応剤が同じ化合物であっても良い。
【0113】
【化21】
【0114】
(ここで、置換基X11及びX12は臭素原子、ヨウ素原子、塩素原子を示す。置換基X6及びX7は一般式(2b)で示される置換基と同意義を示す。)
【0115】
【化22】
【0116】
(ここで、M3はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X5は臭素原子、ヨウ素原子、塩素原子を示し、環Aは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
【0117】
【化23】
【0118】
(ここで、M4はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X8は臭素原子、ヨウ素原子、塩素原子を示し、環Bは、上記一般式(A−1)又は(A−2)で示される構造を示す。)
一般式(5b)の置換基X11及びX12は、臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子及び塩素原子である。
【0119】
そして、具体的な一般式(5b)で示されるテトラハロチエノチオフェンとしては、2,3,5,6−テトラブロモチエノチオフェンが挙げられる。
【0120】
一般式(6b)、(7b)の置換基M3、M4はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物であり、上記のパラジウム及び/又はニッケル触媒により脱離され、パラジウム及び/又はニッケルと置換できる基である限り特に限定はなく、例えばMgCl、MgBr、B(OH)2、B(OMe)2、テトラメチルジオキサボロラニル基、ZnCl、ZnBr、ZnI、Sn(Bu−n)3、Si(Bu−n)3等を挙げることができ、好ましくはZnCl、B(OH)2である。
【0121】
そして、具体的な一般式(6b)、(7b)で示される化合物としては、例えば3−ブロモベンゾチエニル−2−ジンククロライド、6−ドデシル−3−ブロモベンゾチエニル−2−ジンククロライド、6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸、2−ブロモフェニルボロン酸、6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸等が挙げられる。
【0122】
なお、一般式(6b)、(7b)で示される2−ハロアリール金属試薬は、例えば、それらの原料となるアリールジハロゲン置換体をイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬によりハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン等と反応させることで好適に調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁」に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている方法を用いることもできる。
【0123】
一般式(5b)で示されるテトラハロチエノチオフェンと一般式(6b)、(7b)で示される2−ハロアリール金属試薬の反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ジクロロビス(トリフェニルホスフィン)パラジウム、ビス(トリ−tert−ブチルホスフィン)パラジウム、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/トリ−tert−ブチルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等のパラジウム触媒;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物、ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等のニッケル触媒;を挙げることができる。中でも、好ましい触媒はテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。又、これら触媒は1種若しくは2種以上の混合物を用いても良い。
【0124】
一般式(5b)で示されるテトラハロチエノチオフェンと一般式(6b)、(7b)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させる際には、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、エーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水、N,N−ジメチルホルムアミド、N−メチルピロリドン、トリエチルアミン、ピペリジン、ピロリジン、ジイソプロピルアミン等を挙げることができ、又、これら溶剤は1種若しくは2種以上の混合物を用いても良く、例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0125】
パラジウム触媒、ニッケル触媒の使用量は一般式(5b)で示されるテトラハロチエノチオフェン1モルに対し、0.1〜20モル%が好ましく、特に好ましくは1〜10モル%である。
【0126】
一般式(6b)、(7b)で示される2−ハロアリール金属試薬の使用量は一般式(5b)で示されるテトラハロチエノチオフェン1当量に対し、1.8〜3.2当量が好ましく、特に好ましくは1.9〜2.8当量である。
【0127】
反応の際の温度は10〜120℃が好ましく、さらに好ましくは30〜100℃、特に好ましくは40〜90℃であり、反応時間は1〜80時間が好ましく、特に好ましくは5〜70時間である。
【0128】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、水酸化セシウム、りん酸カリウム、りん酸ナトリウム、ナトリウムtert−ブトキサイド、フッ化カリウム等の無機塩基;トリエチルアミン、トリメチルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(5b)で示されるテトラハロチエノチオフェン1当量に対し、2.0〜10.0当量が好ましく、特に好ましくは4.0〜8.0当量である。
【0129】
また、一般式(5b)で示されるテトラハロチエノチオフェンと一般式(6b)、(7b)で示される2−ハロアリール金属試薬の反応により炭素−炭素結合が形成される位置は硫黄原子の隣の位置が反応性に富むことから、2及び5位になる。
【0130】
かくして得られた、本発明の一般式(2b)で示されるテトラハロターフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【発明の効果】
【0131】
本発明は一般式(1a)、(1b)で示されるヘテロアセン誘導体を製造する新規な方法を提供する。さらに、本発明により低溶解性のテトラハロターフェニル誘導体を原料に用いても、収率良く目的のヘテロアセン誘導体を提供することができる。
【0132】
本発明の製造方法で得られるヘテロアセン誘導体は、塗布法の適用が可能であり且つ剛直棒状分子構造を有するため優れた半導体デバイス特性及び耐酸化性を有する有機半導体材料として期待される。
【実施例】
【0133】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例に限定されるものではない。
【0134】
生成物の同定には1H NMRスペクトル及びマススペクトルを用いた。なお、1H NMRスペクトルは日本電子製JEOL GSX−270WB(270MHz)を用いた。マススペクトル(MS)は日本電子製JEOL JMS−700を用いて、試料を直接導入し、電子衝突(EI)法(70エレクトロンボルト)あるいはFAB法(6キロエレクトロンボルト、キセノンガス、マトリックス:2−ニトロフェニルオクチルエーテル)(FABMS)で測定した。
【0135】
反応の進行の確認等は薄層クロマトグラフィー、ガスクロマトグラフィー(GC)及びガスクロマトグラフィー−マススペクトル(GCMS)分析を用いた。
【0136】
ガスクロマトグラフィー分析
装置 島津GC14B
カラム J&Wサイエンティフィック社製、DB−1,30m
ガスクロマトグラフィー−マススペクトル分析
装置 パーキンエルマーオートシステムXL(MS部;ターボマスゴールド)
カラム J&Wサイエンティフィック社製、DB−1,30m
反応用の試薬及び溶媒は、断りのない限り市販品を用いた。なお、グリニャール試薬あるいはブチルリチウム等の有機金属試薬を用いた場合は、市販の脱水溶媒をそのまま用いた。
【0137】
合成例1 (1,4−ジブロモ−2,5−ジヨードベンゼンの合成)(一般式(5a)の化合物の合成)
1,4−ジブロモ−2,5−ジヨードベンゼンはジャーナル オブ アメリカン ケミカル ソサイエティー、1997年、119巻、4578−4593頁に記載されている方法を参考に以下のように合成を行った。
【0138】
メカニカルスターラー付き1lの三口フラスコに過ヨウ素酸16.7g(73.0mmol)及び硫酸525mlを加えた。過ヨウ素酸が溶解した後、ヨウ化カリウム36.4g(219mmol)を少しずつ添加した。その内容物の温度を−30℃に冷却し、1,4−ジブロモベンゼン34.5g(146mmol)を5分間かけて添加した。得られた混合物を−25℃で36時間撹拌した。反応混合物を氷(2Kg)中へ注いだ後、濾過し固体を取り出した。その固体をクロロホルムに溶解させ、5%苛性ソーダ水溶液及び水で洗浄し、有機相を無水硫酸マグネシウムで乾燥した。減圧濃縮後、残渣をクロロホルムから再結晶化し、白色結晶を得た(36.0g、収率50%)。
1H NMR(CDCl3,21℃):δ=8.02(s,2H)。
【0139】
1H NMRスペクトルが文献値と一致したことより、1,4−ジブロモ−2,5−ジヨードベンゼンが得られたことを確認した。
【0140】
合成例2 ({1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモ}ベンゼンの合成(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に2,3−ジブロモベンゾチオフェン(シグマ−アルドリッチ製)886mg(3.03mmol)及びTHF8mlを添加した。この溶液を−30℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.80M)のTHF溶液3.8ml(3.0mmol)を滴下した。30分間熟成後、−50℃に冷却し、その温度で塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液3.0ml(3.0mmol)を滴下した。徐々に室温まで昇温した後、生成した白色スラリー液を減圧濃縮した。得られた白色固体[3−ブロモベンゾチエニル−2−ジンククロライド(一般式(6a)及び(7a)の化合物)]に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン492mg(1.01mmol)(一般式(5a)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)91.7mg(0.079mmol)及びTHF8mlを添加した。63℃で10時間反応を実施した後、容器を水冷し1M塩酸水溶液4mlを添加することで反応を停止させた。トルエンを添加し、得られた懸濁液を濾過し、濾板上の固体をトルエン及び水で洗浄した。固体を減圧乾燥し、白色固体292mgを得た。一方、濾液を分相し有機相を水洗した。有機相を減圧濃縮し、溶媒を留去した。得られた固体をヘキサン洗浄し(10ml)、残渣をトルエンから再結晶化した。析出した結晶を減圧乾燥後、206mgの白色固体を得た。先の濾過後の白色固体と合わせ、収率75%で目的物を得た。
1H NMR(CDCl3,21℃):δ=7.95−7.84(m,4H),7.81(s,2H),7.58−7.44(m,4H)。
【0141】
1H NMRスペクトルを図1に示した。
MS m/z: 658(M+,44%),498(M+−2Br,34),338(M+−4Br,100),306(M+−4Br−S),9),169(M+−4Br)/2,66)。
【0142】
1H NMR及びMS測定より、{1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモ}ベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0143】
【化24】
【0144】
実施例1 (テトラチエノアセンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例2で合成した{1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモ}ベンゼン422mg(0.641mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.0ml(3.2mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)530mg(1.69mmol)とジエチルエーテル30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加えた。一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、分相し、さらに有機相を飽和食塩水で洗浄した。有機相は黄色懸濁液であったことから濾過し、黄色固体を取り出し、アセトン、メタノール、トルエンの順に洗浄後、真空乾燥し、目的物の黄色固体106mgを得た(収率41%)。
【0145】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
【0146】
MS m/z: 402(M+,100%),201(M+/2,14)。
【0147】
MS測定より、テトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0148】
【化25】
【0149】
合成例3 (2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニルの合成(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン4.39g(9.00mmol)(一般式(5a)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)974mg(0.84mmol)及び2−ブロモフェニルボロン酸(シグマ−アルドリッチ製)4.16g(一般式(6a)及び(7a)の化合物)(20.7mmol)を添加した。さらにトルエン72ml、エタノール18ml及び炭酸ナトリウム5.72g(54.0mmol)と水22mlからなる水溶液を添加した。85℃のオイルバスに浸し、15時間撹拌した。室温まで冷却後、ジクロロメタン及び飽和食塩水を添加し分相した。有機相を減圧濃縮し、残渣をトルエンから再結晶化し、白色針状晶を得た(3.68g、収率75%)。
融点:230−231℃。
1H NMR(CDCl3,21℃):δ=7.70(d,J=8.0Hz,2H),7.55(d,J=1.5Hz,2H),7.45−7.23(m,6H)。
MS m/z: 546(M+,92%),466(M+−Br,45),386(M+−2Br,53),226(M+−4Br,100)。
【0150】
1H NMR及びMS測定より、2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0151】
【化26】
【0152】
実施例2 (ベンゾビスベンゾチオフェンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル410mg(0.751mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.4ml(3.82mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)623mg(1.98mmol)とTHF30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、分相し、さらに有機相を飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的物の白色結晶を得た(79mg、収率36%)。
【0153】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
1H NMR(CDCl3,55℃):δ=8.58(s,2H),8.20(m,2H),7.85(m,2H),7.47(m,4H)。
【0154】
1H NMRスペクトルを図2に示した。
MS m/z: 290(M+,100%),145(M+/2,10)。
【0155】
1H NMR及びMS測定より、ベンゾビスベンゾチオフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0156】
【化27】
【0157】
実施例3 (P,P−ジフェニルベンゾホスホロジベンゾホスホールの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル410mg(0.751mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.4ml(3.82mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したジクロロフェニルホスフィン(東京化成工業製)355mg(1.98mmol)(一般式(3a)及び(4a)の化合物)とTHF30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄した。減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的の淡黄色結晶を得た(143mg、収率43%)。
【0158】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
1H NMRスペクトル(CDCl3,21℃):δ=8.26(s,2H),7.94(d,J=7.8Hz,2H),7.69(d,J=7.1Hz,2H),7.44(t,J=7.8Hz,2H),7.41−7.10(m,12H)。
【0159】
1H NMRスペクトルを図3に示した。
MS m/z: 442(M+,100%),364(M+−Ph−1,38),288(M+−2Ph,19),221(M+/2,10)。
【0160】
1H NMR及びMS測定より、P,P−ジフェニルベンゾホスホロジベンゾホスホールが得られたことを確認した。なお、その構造式を下記に示す。
【0161】
【化28】
【0162】
実施例4 (B,B−ジフェニルベンゾボロリルジベンゾボロール)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル425mg(0.778mmol)及びジエチルエーテル15mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.5ml(3.98mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したジクロロフェニルボラン(シグマ−アルドリッチ製)326mg(2.05mmol)(一般式(3a)及び(4a)の化合物)とTHF30mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄した。減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的の淡黄色結晶を得た(107mg、収率34%)。
【0163】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
MS m/z: 402(M+,100%),201(M+/2,14)。
【0164】
MS測定より、B,B−ジフェニルベンゾボロリルジベンゾボロールが得られたことを確認した。なお、その構造式を下記に示す。
【0165】
【化29】
【0166】
実施例5 (ベンゾビスベンゾテルロフェンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2,2’,5’,2”−テトラブロモ−1,1’,4’,1”−ターフェニル382mg(0.70mmol)及びジエチルエーテル13.5mlを添加した。この溶液を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液2.3ml(3.66mmol)を滴下し、テトラメタル化を行った。90分間撹拌後、−78℃に冷却したテルル粉末(シグマ−アルドリッチ製)242mg(1.9mmol)(一般式(3a)及び(4a)の化合物)とTHF27mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄した。減圧濃縮し、得られた残渣にヘキサンを添加し撹拌後静置し、上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、目的の淡黄色の結晶を得た(74mg、収率22%)。
【0167】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
MS m/z: 481(M+,100%),240(M+/2,13)。
【0168】
MS測定より、ベンゾビスベンゾテルロフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0169】
【化30】
【0170】
合成例4 (4−ブロモ−5−ヨード無水フタル酸の合成)
4−ブロモ−5−ヨード無水フタル酸は「ジャーナル オブ オーガニック ケミストリー」(米国)、1951年、16巻、1577−1581頁を参考に、以下の様に合成した。
【0171】
4−ブロモフタルイミド(東京化成工業製)13.3g(58.9mmol)を窒素ガスで置換した100mlの二口ナスフラスコに入れた。次いでヨウ素7.78g(30.6mmol)及び10%発煙硫酸(ヨツハタ化学工業製)50mlを加え、90℃で23時間反応を行った。
【0172】
反応混合物を室温に冷やして氷に注ぎ入れた後、ガラスフィルターでろ過し、黄色固体17.1gを得た。得られた固体を濃硫酸47mlに溶解させ、130℃で5時間反応を行った。反応混合物を氷冷後、氷水を加えて析出した固体をろ過し、フタル酸誘導体の固体18.5gを得た。次に水酸化ナトリウム4.8gを水24mlに溶かした水溶液に得られた固体を室温で溶かした。この塩基性水溶液に酢酸を加えpHを3〜4に調整し、析出するフタル酸誘導体のモノナトリウム塩の白色沈殿をろ過した。得られた白色沈殿を水に懸濁させ、濃塩酸でpHを1以下にし、再びフタル酸誘導体として白色固体7.75gを得た。この固体をトルエン70mlに溶かし、無水酢酸13ml(138mmol)を加え、105℃で4時間反応を行った。反応液を減圧濃縮して白色固体6.9gを得た。この固体を加熱トルエンで再結晶し、目的の4−ブロモ−5−ヨードフタル酸無水物6.79g(19.2mmol)を得た。(収率33%)。
1H NMR(CDCl3,22℃):δ=8.51(s,1H),8.23(s,1H)。
MS m/z: 353(M+,100%),309(M+−CO2,18%),282(M+−C2O3,10%),155(M+−C2O3−I,16%),74(M+−C2O3−I−Br,32%)。
【0173】
合成例5 (1,2―ジドデシルベンゼンの合成)
1,2−ジドデシルベンゼンは「日本化学会誌」1989年、983−987頁に従い以下の様に合成した。
【0174】
1,2−ジクロロベンゼン2.22g(15.1mmol)、ジクロロ〔1,3−ビス(ジフェニルホスフィノ)プロパン〕ニッケル(東京化成工業製)131mg(0.24mmol)、エーテル12mlの混合液にドデシルマグネシウムブロミド(シグマ−アルドリッチ製、1.0mol/lエーテル溶液)45ml(45.0mmol)を窒素雰囲気中0℃で滴下した。35℃で20時間反応を行い、反応混合物を0℃に冷やして希塩酸を加え、エーテルで抽出した。エーテル溶液を水、飽和炭酸水素ナトリウム水溶液、水の順に洗浄し、塩化カルシウムで乾燥させた。得られた液体をシリカゲルカラムクロマトグラフィー(溶離液:ヘキサン)及び減圧蒸留で精製し、目的の1,2―ジドデシルベンゼン5.56g(13.4mmol)を得た(収率88%)。
1H NMR(CDCl3,22℃):δ=7.11(m,4H),2.59(t,J=7.8Hz,4H),1.55(m,4H),1.26(m,36H),0.88(t,J=6.8Hz,6H)。
MS m/z: 414(M+,100%),260(M+−C11H23,71%),106(M+−C22H46,98%)。
【0175】
合成例6 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの合成)
2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンは「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁を参考に以下の様に合成した。
【0176】
合成例4で得られた4−ブロモ−5−ヨード無水フタル酸6.79g(19.2mmol)、合成例5で得られた1,2−ジドデシルベンゼン7.97g(19.2mmol)、テトラクロロエタン12.0mlの混合液に塩化アルミニウム5.78g(43.4mmol)を加え、室温で3時間反応を行った。水を加えてクエンチし、さらに水洗浄を行い、加熱真空乾燥後、白色固体15.0gを得た。得られた固体に濃硫酸39mlを添加し、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの固体2.95g(3.93mmol)を得た(収率60%)。
1H NMR(CDCl3,22℃):δ=8.73(s,1H),8.45(s,1H),8.05(s,2H),2.75(m,4H),1.62(m,4H),1.26(m,36H),0.88(m,6H)。
MS m/z: 750(M+,100%),440(M+−C22H46,8%),313(M+−C22H46I,2%),233(M+−C22H46IBr,1%)。
【0177】
合成例7 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの合成)
窒素雰囲気下、1lシュレンク反応容器に合成例6で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン8.92g(11.9mmol)を入れた。次いでTHF140mlを加え、水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)31ml(30.7mmol)を加え、室温で1.5時間反応を行った。次いで反応混合物に6M塩酸水溶液82mlを加え、65℃で3時間反応を行った。反応混合物を室温まで冷やし、エーテルで抽出した。エーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥、減圧濃縮し、得られた残渣に再びTHF140mlを加え、水素化ジイソブチルアルミニウム31ml(30.7mmol)を加え、室温で1.5時間反応を行った。次いで反応混合物に6M塩酸水溶液82mlを加え、3時間反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。エーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧乾燥した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの黄色固体7,11g(9.88mmol)を得た(収率83%)。
1H NMR(CDCl3,22℃):δ=8.55(s,1H),8.27(s,1H),8.16(s,1H),8.15(s,1H),7.72(s,2H),2.78(m,4H),1.71(m,4H),1.27(m,36H),0.88(m,6H)。
MS m/z: 720(M+,100%),410(M+−C22H46,16%),283(M+−C22H46−I,4%),203(M+−C22H46−I−Br,5%)。
【0178】
合成例8 (テトラブロモ(テトラドデシル)ジナフトターフェニルの合成)(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、1lシュレンク反応容器に合成例7で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン3.66g(5.08mmol)及びTHF167mlを添加した。この溶液を−52℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液13.0ml(10.2mmol)を滴下し、攪拌した。5分間熟成後、トリメトキシボラン(和光純薬工業製)1.2ml(10.8mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を添加し、40分間撹拌した。トルエンを加え、分相し、有機相を減圧濃縮し6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸を含む固体3.48gを得た(一般式(6a)及び(7a)の化合物の合成)。この内2.48g(3.62mmol)取り出し、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン(一般式(5a)の化合物)706mg(1.45mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)101mg(0.087mmol)、トルエン41ml、及びエタノール10mlを添加した。さらに炭酸ナトリウム922mg(8.70mmol)を含む水溶液13mlを加え、60℃で60時間反応を実施した。容器を水冷し3M塩酸水溶液30mlを添加することで反応を停止させた。トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(1.7ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン及びヘキサン:クロロホルム=10:1)、テトラブロモ(テトラドデシル)ジナフトターフェニルの薄黄色固体1.30gを得た(収率63%)。
1H NMR(CDCl3,22℃):δ=8.34(s,2H),8.32(s,2H),8.28(s,2H),7.97(s,1H),7.92(s,1H),7.79(s,2H),7.78(s,2H),7.73(s,1H),7.72(s,1H),2.81(m,8H),1.71(m,8H),1.28(m,72H),0.88(m,12H)。
【0179】
1H NMRスペクトルを図4に示した。
FABMS m/z: 1419(M+,100%),1340(M+−Br,10)。
【0180】
1H NMR及びMS測定より、テトラブロモ(テトラドデシル)ジナフトターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0181】
【化31】
【0182】
実施例6 (テトラドデシルジアントラジチエノベンゼンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、50mlシュレンク反応容器に、合成例8で合成したテトラブロモ(テトラドデシル)ジナフトターフェニル98mg(0.069mmol)及びジエチルエーテル3.4mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.22ml(0.35mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)57mg(0.18mmol)とTHF6.8mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的の黄色結晶33mgを得た(収率41%)。
【0183】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
1H NMR(重トルエン,100℃):δ=8.49(s,2H),8.44(s,2H),8.26(s,2H),8.21(s,2H),8.14(s,2H),7.85(s,2H),7.78(s,2H),2.87(t,J=7.8Hz,8H),1.77(m,8H),1.31(m,72H),0.88(m,J=7.8Hz,12H)。
【0184】
1H NMRスペクトルを図5に示した。
MS m/z: 1164(M+)。
【0185】
1H NMR及びMS測定より、テトラドデシルジアントラジチエノベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0186】
【化32】
【0187】
合成例9 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例6で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン541mg(0.722mmol)及びTHF20mlを添加した。−78℃に冷却後、フェニルリチウム(関東化学製、1.0mol/l、シクロヘキサン/エーテル溶液)1.5ml(1.5mmol)を加えた後、一晩かけて室温まで昇温した。次いで反応混合物に3M塩酸水溶液及びエーテルを加えた後、分相し、さらにエーテルで抽出した。エーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの黄色固体453mgを得た(収率72%)。
【0188】
合成例10 (テトラブロモ(テトラドデシル)(テトラフェニル)ジナフトターフェニルの合成)(一般式(2a)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例9で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセン448mg(0.513mmol)及びTHF8mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液1.27ml(1.03mmol)を滴下した。10分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)107.0mg(1.03mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮し、6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸を得た(一般式(6a)及び(7a)の化合物の合成)。得られた6,7−ジドデシル−9,10−ジフェニル−3−ブロモ−2−アントラセニルボロン酸に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン(一般式(5a)の化合物)117mg(0.240mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)16.6mg(0.014mmol)、トルエン4ml、及びエタノール1.0mlを添加した。さらに炭酸ナトリウム154mg(1.46mmol)を含む水溶液1.2mlを加え、60℃で60時間反応を実施した。容器を水冷し3M塩酸水溶液3mlを添加することで反応を停止させた。室温まで冷却後、トルエンを添加分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.05mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒;トルエン:ヘキサン=1:1)。得られた溶液を減圧濃縮し、得られた残渣をヘプタンから再結晶化し、目的物の薄黄色固体211mgを得た(収率51%)。
FABMS m/z: 1723(M+)。
【0189】
MS測定より、テトラブロモ(テトラドデシル)(テトラフェニル)ジナフトターフェニルが得られたことを確認した。なお。その構造式を下記に示す。
【0190】
【化33】
【0191】
実施例7 (テトラドデシルテトラフェニルジアントラジチエノベンゼンの合成)(一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例10で合成したテトラブロモ(テトラドデシル)(テトラフェニル)ジナフトターフェニル207mg(0.12mmol)及びジエチルエーテル6mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.38ml(0.60mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)101mg(0.32mmol)とTHF12mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルテトラフェニルジアントラジチエノベンゼンのオレンジ色固体56mgを得た(収率32%)。
【0192】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
FABMS m/z: 1468(M+)。
【0193】
MS測定より、テトラドデシルテトラフェニルジアントラジチエノベンゼンが得られたことを確認した。なお。その構造式を下記に示す。
【0194】
【化34】
【0195】
合成例11 (2,3,5,6−テトラブロモチエノチオフェンの合成)(一般式(5b)の化合物の合成)
2,3,5,6−テトラブロモチエノチオフェンはジャーナル オブ ケミカル ソサイエティー、パーキン トランザクション 1: 1997年、3465−3470頁に記載されている方法を参考に以下の様に合成を行った。
【0196】
1)エチル チエノ[3,2−b]チオフェン−2−カルボキシレートの合成
メカニカルスターラー付き1lの三口フラスコに、3−ブロモチオフェン−2−カルボキシアルデヒド(アルファアエサール製)26.8g(140mmol)、チオグリコール酸エチル(和光純薬工業製)16.9g(141mmol)、炭酸カリウム26g、及びジメチルホルムアルデヒド250mlを加え、室温で72時間撹拌した。反応終了後、水500mlを添加し、ジクロロメタンで抽出し、エチル チエノ[3,2−b]チオフェン−2−カルボキシレートの粗生成物23.8gを得た。
【0197】
2)チエノ[3,2−b]チオフェン−2−カルボン酸の合成
1l三口フラスコに1)で得られたエチル チエノ[3,2−b]チオフェン−2−カルボキシレート23.8g、THF300ml、及び水酸化リチウム水溶液(1.0M)300mlを加え、3時間加熱還流した。反応終了後、溶媒を減圧下濃縮し、塩酸150mlを添加すると結晶が析出した。得られた結晶を濾別し、水で3回洗浄し、乾燥後、チエノ[3,2−b]チオフェン−2−カルボン酸16.0gを得た。
【0198】
3)2,3,5−トリブロモ−チエノ[3,2−b]チオフェンの合成
窒素雰囲気下、1l三口フラスコに2)で得られたチエノ[3,2−b]チオフェン−2−カルボン酸16.0g及び酢酸水溶液500mlを加え、臭素15.2gを滴下した。5時間撹拌した後、水を加え、析出した固体を濾別し、固体を水洗し、2,3,5−トリブロモチエノ[3,2−b]チオフェン27.4gを得た。
【0199】
4)2,3,5,6−テトラブロモチエノチオフェンの合成
窒素雰囲気下、1l三口フラスコにジイソプロピルアミン2.85g(28.0mmol)及びTHF150mlを加え、0℃とした。n−ブチルリチウム(関東化学製、1.60M)ヘキサン溶液17.5ml(28.0mmol)を滴下した。得られた溶液を−66℃に冷却後、3)で得られた2,3,5−トリブロモ−チエノ[3,2−b]チオフェン10.3g(27.3mmol)を添加した。さらに臭素21gを滴下し、ゆっくりと室温まで上昇させた。飽和塩化アンモニウム水溶液を加えた後、水洗し、結晶物を濾別した。真空乾燥後、2,3,5,6−テトラブロモチエノチオフェンの固体2.1gを得た。
【0200】
合成例12 (テトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェンの合成)(一般式(2b)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例7で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン625mg(0.868mmol)及びTHF11mlを添加した。この溶液を−55℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液2.8ml(1.82mmol)を滴下し、攪拌した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)180.8mg(1.740mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を添加し、30分間撹拌した。トルエンを加え、分相し、有機相を減圧濃縮し、6,7−ジドデシル−3−ブロモ−2−アントラセニルボロン酸を得た。(一般式(6b)及び(7b)の化合物の合成)。そこへ合成例11で合成した2,3,5,6−テトラブロモチエノチオフェン182mg(0.400mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)27.7mg(0.0240mmol)、トルエン6ml、及びエタノール1.6mlを添加した。さらに炭酸ナトリウム254mg(2.40mmol)を含む水溶液2mlを加え、60℃で56時間反応を実施した。容器を水冷し3M塩酸水溶液3mlを添加することで反応を停止させた。トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.06ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン及びヘキサン:クロロホルム=10:1)、テトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェンの薄黄色固体427mgを得た(収率72%)。
FABMS m/z: 1481(M+,100%),1401(M+−Br,10)。
【0201】
MS測定より、テトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0202】
【化35】
【0203】
実施例8 (テトラドデシルジアントラテトラチエノアセンの合成)(一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例12で合成したテトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェン204mg(0.137mmol)及びジエチルエーテル4.0mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.43ml(0.68mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3b)及び(4b)の化合物)113mg(0.36mmol)とTHF6.0mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルジアントラテトラチエノアセン70mgの黄色固体を得た(収率44%)。
【0204】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
FABMS m/z: 1162(M+)。
【0205】
MS測定より、テトラドデシルジアントラテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0206】
【化36】
【0207】
実施例9 (テトラドデシルジアントラジテルロジチエノアセンの合成)(一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例12で合成したテトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェン163mg(0.110mmol)及びジエチルエーテル3.2mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液0.34ml(0.54mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したテルル粉末(シグマ−アルドリッチ製)37mg(0.29mmol)(一般式(3b)及び(4b)の化合物)とTHF4.8mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルジアントラジテルロジチエノアセン43mgの黄色固体を得た(収率29%)。
【0208】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
【0209】
FABMS m/z: 1353(M+).
MS測定より、テトラドデシルジアントラテトラジテルロジチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0210】
【化37】
【0211】
合成例13 ({2,5−ビス(3−ブロモ−6−フルオロベンゾチエニル−2−)−3,6−ジブロモ}チエノチオフェンの合成)(一般式(2b)のテトラハロターフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に2,3−ジブロモ−6−フルオロベンゾチオフェン401mg(1.29mmol)及びTHF8mlを添加した。この溶液を−40℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.80M)のTHF溶液1.6ml(1.28mmol)を滴下した。30分間熟成後、−80℃に冷却し、その温度で塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液1.3ml(1.3mmol)を滴下した。徐々に室温まで昇温した後、生成した白色スラリー液を減圧濃縮した。得られた白色固体[3−ブロモ−6−フルオロベンゾチエニル−2−ジンククロライド、一般式(6b)及び(7b)の化合物]に、合成例11で合成した2,3,5,6−テトラブロモチエノチオフェン251mg(0.551mmol)(一般式(5b)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)38.2mg(0.033mmol)及びTHF7mlを添加した。63℃で10時間反応を実施した後、容器を水冷し1M塩酸水溶液4mlを添加することで反応を停止させた。トルエンを添加し、得られた懸濁液を濾過し、濾板上の固体をトルエン及び水で洗浄した。固体を減圧乾燥し、白色固体151mgを得た。一方、濾液を分相し有機相を水洗した。有機相を減圧濃縮し、溶媒を留去した。得られた固体をヘキサン洗浄し(10ml)、残渣をトルエンから再結晶化した。析出した結晶を減圧乾燥後、102mgの白色固体を得た。先の濾過後の白色固体と合わせ、収率61%で目的物を得た。
FABMS m/z: 756(M+,100%),676(M+−2Br,14)。
【0212】
MS測定より、{2,5−ビス(3−ブロモ−6−フルオロベンゾチエニル−2−)−3,6−ジブロモ}チエノチオフェンが得られたことを確認した。なお、その構造式を下記に示す。
【0213】
【化38】
【0214】
実施例10 (ジフルオロジベンゾヘキサチエノアセンの合成)(一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例13で合成した{2,5−ビス(3−ブロモ−6−フルオロベンゾチエニル−2−)−3,6−ジブロモ}チエノチオフェン246mg(0.325mmol)及びジエチルエーテル4.4mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.59M)のヘキサン溶液1.0ml(1.59mmol)を滴下し、テトラメタル化を行った。60分間撹拌後、−78℃に冷却したビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3b)及び(4b)の化合物)268mg(0.85mmol)とTHF8.8mlの混合物へ先のテトラメタル化混合物をテフロン(登録商標)キャヌラーを用いて窒素下で加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサン、メタノール、アセトン、トルエンの順にて洗浄した。固体を乾燥後、目的のジフルオロジベンゾヘキサチエノアセン65mgの黄色固体を得た(収率40%)。
【0215】
ジアルキルエーテル中でテトラメタル化を行ったことから、高収率で目的物が得られた。
MS m/z: 501(M+)。
【0216】
MS測定より、ジフルオロジベンゾヘキサチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0217】
【化39】
【0218】
参考例1 (テトラドデシルジアントラジチエノベンゼンの合成)(メタル化をTHF中で行った一般式(1a)のヘテロアセン誘導体の合成)
窒素雰囲気下、50mlシュレンク反応容器に、合成例8で合成したテトラブロモ(テトラドデシル)ジナフトターフェニル243mg(0.171mmol)及びTHF15mlを添加した。この混合物を−72℃に冷却し、メタル化剤としてsec−ブチルリチウム(関東化学製1.0M)のシクロヘキサン/ヘキサン溶液1.4ml(1.40mmol)を滴下し、テトラメタル化を行った。30分間撹拌後、ビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3a)及び(4a)の化合物)226mg(0.72mmol)を加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、黄色結晶40mgを得た(収率20%)。また、未反応の原料テトラブロモ(テトラドデシル)ジナフトターフェニルが120mg回収された。
【0219】
参考例2 (テトラドデシルジアントラテトラチエノアセンの合成)(メタル化をTHF中で行った一般式(1b)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例12で合成したテトラブロモ(テトラドデシル)ジアントラセニルチエノチオフェン102mg(0.069mmol)及びTHF5.0mlを添加した。この混合物を−72℃に冷却し、メタル化剤としてsec−ブチルリチウム(関東化学製1.0M)のシクロヘキサン/ヘキサン溶液0.56ml(0.56mmol)を滴下し、テトラメタル化を行った。30分間撹拌後、ビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3b)及び(4b)の化合物)91mg(0.29mmol)を加え、一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、濾過した固体を水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的のテトラドデシルジアントラテトラチエノアセン12mgの黄色固体を得た(収率15%)。また、未反応の原料テトラブロモ(テトラドデシル)ジナフトターフェニルが52mg回収された。
【図面の簡単な説明】
【0220】
【図1】合成例2で合成した{1,4−ビス(3−ブロモベンゾチエニル)−2,5−ジブロモ}ベンゼンの1H NMRスペクトル
【図2】実施例2で合成したベンゾビスベンゾチオフェンの1H NMRスペクトル
【図3】実施例3で合成したP,P−ジフェニルベンゾホスホロジベンゾホスホールの1H NMRスペクトル
【図4】合成例8で合成したテトラブロモ(テトラドデシル)ジナフトターフェニルの1H NMRスペクトル
【図5】実施例5で合成したテトラドデシルジアントラジチエノベンゼンの1H NMRスペクトル
【特許請求の範囲】
【請求項1】
下記一般式(1a)で示されるヘテロアセン誘導体の製造方法であり、下記一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、下記一般式(3a)及び下記一般式(4a)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法。
【化1】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T1及びT2は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有し、l及びmは、各々0又は1の整数を示す。)
【化2】
【化3】
(ここで、置換基R5〜R9は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、炭素数2〜30のアルキニル基、炭素数2〜30のアルケニル基を示す。nは0〜2の整数であり、oは0又は1の整数である。)]
【化4】
(ここで、置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、置換基R1及びR2並びに環A及びBは一般式(1a)で示される置換基並びに環と同意義を示す。)
(R3)lT1(L1)p (3a)
(R4)mT2(L2)q (4a)
(ここで、置換基T1、T2、R3、R4及び記号lとmは一般式(1a)で示される置換基及び記号と同意義を示し、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、p及びqは0又は2の整数を示す。)
【請求項2】
下記一般式(1b)で示されるヘテロアセン誘導体の製造方法であり、下記一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、下記一般式(3b)及び下記一般式(4b)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法。
【化5】
(ここで、置換基R10及びR11は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T3及びT4は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、上記一般式(A−1)又は(A−2)で示される構造を有し、v及びwは、各々0又は1の整数を示す。)
【化6】
(ここで、置換基X5〜X8は臭素原子、ヨウ素原子、塩素原子を示し、環A及びBは一般式(1b)で示される環と同意義を示す。)
(R10)vT3(L3)x (3b)
(R11)wT4(L4)y (4b)
(ここで、置換基T3、T4、R10、R11及び記号vとwは一般式(1b)で示される置換基及び記号と同意義を示し、置換基L3、L4は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、x及びyは0又は2の整数を示す。)
【請求項3】
T1及びT2が同一原子であることを特徴とする請求項1に記載のヘテロアセン誘導体の製造方法。
【請求項4】
T1及びT2が硫黄原子であることを特徴とする請求項1又は3に記載のヘテロアセン誘導体の製造方法。
【請求項5】
T3及びT4が同一原子であることを特徴とする請求項2に記載のヘテロアセン誘導体の製造方法。
【請求項6】
T3及びT4が硫黄原子であることを特徴とする請求2又は5に記載のヘテロアセン誘導体の製造方法。
【請求項7】
メタル化剤としてアルキルリチウムを用いることを特徴とする請求項1〜6のいずれかに記載のヘテロアセン誘導体の製造方法。
【請求項8】
ジアルキルエーテルがジエチルエーテルであることを特徴とする請求項1〜7のいずれかに記載のヘテロアセン誘導体の製造方法。
【請求項9】
テトラメタル化の温度が−20〜30℃であることを特徴とする請求項1〜8のいずれかに記載のヘテロアセン誘導体の製造方法。
【請求項1】
下記一般式(1a)で示されるヘテロアセン誘導体の製造方法であり、下記一般式(2a)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、下記一般式(3a)及び下記一般式(4a)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法。
【化1】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T1及びT2は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有し、l及びmは、各々0又は1の整数を示す。)
【化2】
【化3】
(ここで、置換基R5〜R9は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、炭素数2〜30のアルキニル基、炭素数2〜30のアルケニル基を示す。nは0〜2の整数であり、oは0又は1の整数である。)]
【化4】
(ここで、置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、置換基R1及びR2並びに環A及びBは一般式(1a)で示される置換基並びに環と同意義を示す。)
(R3)lT1(L1)p (3a)
(R4)mT2(L2)q (4a)
(ここで、置換基T1、T2、R3、R4及び記号lとmは一般式(1a)で示される置換基及び記号と同意義を示し、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、p及びqは0又は2の整数を示す。)
【請求項2】
下記一般式(1b)で示されるヘテロアセン誘導体の製造方法であり、下記一般式(2b)で示されるテトラハロターフェニル誘導体をジアルキルエーテル中でメタル化剤を用いてテトラメタル化した後、下記一般式(3b)及び下記一般式(4b)で示される反応剤と反応させることを特徴とするヘテロアセン誘導体の製造方法。
【化5】
(ここで、置換基R10及びR11は同一又は異なって、水素原子、フッ素原子、炭素数1〜30のアルキル基、炭素数4〜30のアリール基、T3及びT4は同一又は異なって、硫黄原子、セレン原子、テルル原子、酸素原子、リン原子、ホウ素原子、アルミニウム原子を示し、環A及びBは同一又は異なって、上記一般式(A−1)又は(A−2)で示される構造を有し、v及びwは、各々0又は1の整数を示す。)
【化6】
(ここで、置換基X5〜X8は臭素原子、ヨウ素原子、塩素原子を示し、環A及びBは一般式(1b)で示される環と同意義を示す。)
(R10)vT3(L3)x (3b)
(R11)wT4(L4)y (4b)
(ここで、置換基T3、T4、R10、R11及び記号vとwは一般式(1b)で示される置換基及び記号と同意義を示し、置換基L3、L4は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、x及びyは0又は2の整数を示す。)
【請求項3】
T1及びT2が同一原子であることを特徴とする請求項1に記載のヘテロアセン誘導体の製造方法。
【請求項4】
T1及びT2が硫黄原子であることを特徴とする請求項1又は3に記載のヘテロアセン誘導体の製造方法。
【請求項5】
T3及びT4が同一原子であることを特徴とする請求項2に記載のヘテロアセン誘導体の製造方法。
【請求項6】
T3及びT4が硫黄原子であることを特徴とする請求2又は5に記載のヘテロアセン誘導体の製造方法。
【請求項7】
メタル化剤としてアルキルリチウムを用いることを特徴とする請求項1〜6のいずれかに記載のヘテロアセン誘導体の製造方法。
【請求項8】
ジアルキルエーテルがジエチルエーテルであることを特徴とする請求項1〜7のいずれかに記載のヘテロアセン誘導体の製造方法。
【請求項9】
テトラメタル化の温度が−20〜30℃であることを特徴とする請求項1〜8のいずれかに記載のヘテロアセン誘導体の製造方法。
【図1】

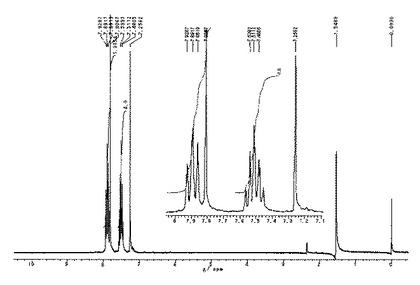
【図2】
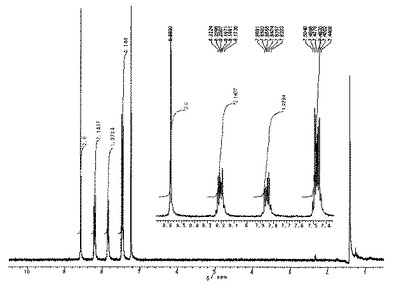
【図3】
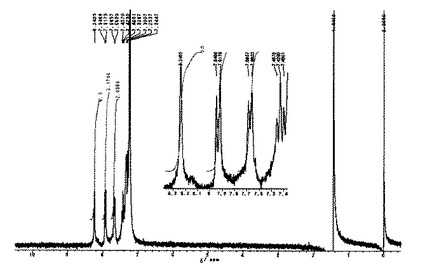
【図4】
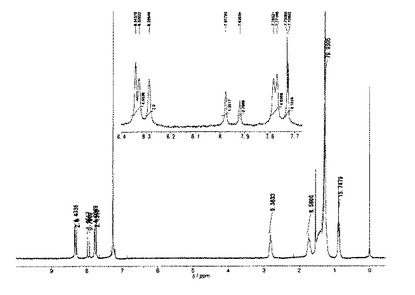
【図5】
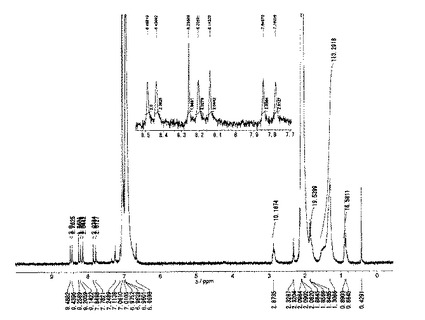
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2009−203183(P2009−203183A)
【公開日】平成21年9月10日(2009.9.10)
【国際特許分類】
【出願番号】特願2008−46436(P2008−46436)
【出願日】平成20年2月27日(2008.2.27)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成21年9月10日(2009.9.10)
【国際特許分類】
【出願日】平成20年2月27日(2008.2.27)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]