
ヘテロアセン誘導体及びその用途
【課題】優れた耐酸化性を有し、塗布法による半導体活性相形成が可能な、ヘテロアセン誘導体、及びそれを用いた耐酸化性有機半導体材料並びに有機薄膜を提供する。
【解決手段】一般式(1)で示されるヘテロアセン誘導体。

(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、炭素数4〜30のアリール基、炭素数6〜20のアルキル基、を示し、T1及びT2は同一又は異なって、硫黄、セレン、テルル、リン、ホウ素を示し、l及びmは、各々0又は1の整数)
【解決手段】一般式(1)で示されるヘテロアセン誘導体。
(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、炭素数4〜30のアリール基、炭素数6〜20のアルキル基、を示し、T1及びT2は同一又は異なって、硫黄、セレン、テルル、リン、ホウ素を示し、l及びmは、各々0又は1の整数)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、有機半導体等の電子材料への展開が可能なヘテロアセン誘導体及びその用途に関する。
【背景技術】
【0002】
有機薄膜トランジスタに代表される有機半導体デバイスは、省エネルギー、低コスト及びフレキシブルといった無機半導体デバイスにはない特徴を有することから近年注目されるようになった。この有機半導体デバイスは有機半導体活性相、基板、絶縁相、電極等数種類の材料から構成されるが、中でも電荷のキャリアー移動を担う有機半導体活性相は該デバイスの中心的な役割を有している。この有機半導体活性相を構成する有機材料のキャリアー移動能により有機半導体デバイス性能が左右される。
【0003】
有機半導体活性相を作製する方法としては一般的に、高温真空下、有機材料を気化させて実施する真空蒸着法及び有機材料を適当な溶媒に溶解させその溶液を塗布する塗布法が知られている。塗布法においては、塗布は高温高真空条件を用いることなく印刷技術を用いても実施することができる。そのため、塗布法は印刷によりデバイス作製の大幅な製造コストの削減を図ることができることから、経済的に好ましいプロセスである。しかし、従来、有機半導体デバイスとして性能が高い材料ほど塗布法で有機半導体活性相を形成することが困難になるという問題があった。
【0004】
例えば、ペンタセン等の結晶性材料はアモルファスシリコン並みの高いキャリアー移動度を有し、優れた有機半導体デバイス特性を発現することが報告されている(例えば、非特許文献1参照)。又、ペンタセン等のポリアセンを溶解させ塗布法で有機半導体デバイスを製造する試みも報告されている(例えば、特許文献1参照)。しかしながら、ペンタセンはその強い凝集性のため溶解性が低く、塗布法を適用するためには高温加熱等の条件が必要とされ、さらにペンタセンの溶液は極めて容易に空気酸化されることから、塗布法の適用はプロセス的、経済的に困難を伴うものであった。また、ポリ−(3−ヘキシルチオフェン)等の自己組織化材料は溶媒に可溶であり、塗布法による有機半導体デバイス作製が報告されてはいるが、キャリアー移動度が結晶性化合物より1桁低いことから(例えば、非特許文献2参照)、得られた有機半導体デバイスの特性が低いという問題があった。
【0005】
またベンゼン環とチオフェン環が縮環したヘテロアセン誘導体は、ペンタセンに比べ耐酸化性が向上しているが、トルエン等の汎用溶媒に対して溶解度が低いという問題があった(例えば、特許文献2参照)。さらに、表面粗さについては記載されていない。
【0006】
【非特許文献1】「ジャーナル オブ アプライドフィジックス」、(米国)、2002年、92巻、5259−5263頁
【非特許文献2】「サイエンス」、(米国)、1998年、280巻、1741−1744頁
【特許文献1】WO2003/016599号
【特許文献2】特開2008−81494号
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明は上記の従来技術が有する問題点に鑑み、優れた耐酸化性を有し、塗布法による有機半導体活性相形成が可能な、ヘテロアセン誘導体及びそれを用いた耐酸化性有機半導体材料並びに表面粗さの小さな有機薄膜を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは上記課題を解決するため鋭意検討の結果、本発明の新規なヘテロアセン誘導体を見出した。加えて、該ヘテロアセン誘導体が耐酸化性に優れ、塗布法の適用が可能であるため結晶性の薄膜を容易に安定して作製することができることから、該ヘテロアセン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜を見出し、本発明を完成するに到った。
【0009】
以下に本発明を詳細に説明する。説明はヘテロアセン誘導体及びその製造方法、該ヘテロアセン誘導体の前駆体であるテトラハロターフェニル誘導体及びその製造方法、並びに該ヘテロアセン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜について述べる。
【0010】
(ヘテロアセン誘導体)
本発明のヘテロアセン誘導体は下記一般式(1)で示される。
【0011】
【化1】
【0012】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、炭素数4〜30のアリール基、炭素数6〜20のアルキル基、を示し、T1及びT2は同一又は異なって、硫黄、セレン、テルル、リン、ホウ素を示し、l及びmは、各々0又は1の整数であり、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有する。)
【0013】
【化2】
【0014】
【化3】
【0015】
(ここで、置換基R5〜R12は同一又は異なって、水素原子、フッ素原子、炭素数6〜20のアルキル基を示し、置換基T3は、硫黄、セレン、テルルを示し、nは0又は1の整数である。但し、置換基R5〜R12は同時に水素原子であることはなく、また環A及びBが(A−2)で示される構造である場合、何れか一方の(A−2)中のnは1である。))]
本発明の一般式(1)で示されるヘテロアセン誘導体の置換基について述べる。
【0016】
置換基R1〜R4における炭素数4〜30のアリール基は、特に限定はなく、例えばフェニル基、p−トリル基、p−(n−ヘキシル)フェニル基、p−(n−オクチル)フェニル基、p−(シクロヘキシル)フェニル基、m−(n−オクチル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(n−パーフルオロオクチル)フェニル基、2−チエニル基、5−(n−ヘキシル)−2−チエニル基、2,2’−ビチエニル−5−基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、アントラセニル基、2−フルオレニル基、9,9−ジメチル−2−フルオレニル基、1−ビフェニレノ基、2−ビフェニレノ基、ターフェニル基、2−ピリジル基、テトラフルオロピリジル基、ビピリジル基、(ジフェニルアミノ)フェニル基、(ジフェニルアミノ)ビフェニル基等を挙げることができ、好ましくはフェニル基、p−(n−オクチル)フェニル基、p−(n−パーフルオロオクチル)フェニル基、5−(n−ヘキシル)−2−チエニル基等である。
【0017】
置換基R1〜R4における炭素数6〜20のアルキル基は、特に限定はなく、例えばヘキシル基、へプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等のアルキル基;パーフルオロヘキシル基、パーフルオロヘプチル基、パーフルオロオクチル基、パーフルオロノニル基、パーフルオロデシル基、パーフルオロウンデシル基、パーフルオロドデシル基、パーフルオロトリデシル基、パーフルオロテトラデシル基、パーフルオロペンタデシル基、パーフルオロヘキサデシル基、パーフルオロヘプタデシル基、パーフルオロオクタデシル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくはアルキル基であり、特に好ましくはオクチル基、デシル基、ウンデシル基、トリデシル基、テトラデシル基、ペンタデシル基である。
【0018】
これらの置換基R1〜R4の中でも、特に水素原子、炭素数4〜30のアリール基が好ましく、さらに水素原子、フェニル基が好ましい。
【0019】
置換基T1及びT2は、硫黄、セレン、テルル、リン、ホウ素であり、その中でも好ましくは硫黄、セレン、リン、ホウ素であり、さらに好ましくは硫黄である。
【0020】
l及びmは、各々0又は1の整数である。ただし、置換基T1、T2が、硫黄、セレン、テルルの場合は、l、mは0であり、置換基T1、T2が、リン、ホウ素の場合は、l、mは1である。
【0021】
次に、一般式(A−1)及び(A−2)で示される、環A及びBについて述べる。
【0022】
本発明のヘテロアセン誘導体は、環A、Bを有する誘導体であり、環A,Bは一般式(A−1)又は(A−2)で示される構造を有するものである。
【0023】
置換基R5〜R12における炭素数6〜20のアルキル基は、特に限定はなく、例えばヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等のアルキル基;パーフルオロヘキシル基、パーフルオロヘプチル基、パーフルオロオクチル基、パーフルオロノニル基、パーフルオロデシル基、パーフルオロウンデシル基、パーフルオロドデシル基、パーフルオロトリデシル基、パーフルオロテトラデシル基、パーフルオロペンタデシル基、パーフルオロヘキサデシル基、パーフルオロヘプタデシル基、パーフルオロオクタデシル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくはアルキル基であり、特に好ましくはノニル基、デシル基、ウンデシル基、トリデシル基、テトラデシル基、ペンタデシル基、オクタデシル基である。
【0024】
置換基R5〜R12が炭素数5以下であるアルキル基を有するヘテロセン誘導体では、有機薄膜とした際に表面粗さが大きくなる。
【0025】
これらアルキル基においては、環(A−1)における置換基R5〜R8は、炭素数6〜20のアルキル基であり、特に炭素数7〜20のアルキル基が好ましく、環(A−2)における置換基R9〜R12は、炭素数6〜20のアルキル基であり、特に炭素数13〜20のアルキル基が好ましい。
【0026】
置換基T3は、硫黄、セレン、テルルであり、その中でも好ましくは硫黄である。
【0027】
nは0又は1の整数であり、好ましくは1である。また環A及びBが(A−2)で示される構造である場合、何れか一方の(A−2)中のnは1である。
【0028】
これらの中でも本発明の一般式(1)で示されるヘテロアセン誘導体は、該ヘテロアセン誘導体及び該ヘテロアセン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜が、高い耐酸化性及びキャリアー移動度を発現することから、以下の化合物が好ましく、
【0029】
【化4】
【0030】
【化5】
【0031】
【化6】
【0032】
【化7】
【0033】
【化8】
【0034】
【化9】
【0035】
【化10】
【0036】
【化11】
【0037】
【化12】
【0038】
【化13】
【0039】
【化14】
【0040】
【化15】
【0041】
【化16】
【0042】
【化17】
【0043】
【化18】
【0044】
【化19】
【0045】
特に好ましくは
【0046】
【化20】
【0047】
である。
【0048】
(テトラハロターフェニル誘導体)
次に、本発明の一般式(1)で示されるヘテロアセン誘導体の前駆化合物であるテトラハロターフェニル誘導体について述べる。
【0049】
本発明の一般式(1)で示されるヘテロアセン誘導体の前駆化合物であるテトラハロターフェニル誘導体は下記一般式(2)で示される。
【0050】
【化21】
【0051】
(ここで、置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、置換基R1及びR2並びに環A及びBは一般式(1)で示される置換基及び環と同意義を示す。)
置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子、ヨウ素原子であり、特に好ましくはいずれも臭素原子である。
【0052】
置換基R1及びR2は、一般式(1)で示される置換基と同意義を示し、その中でも特に水素原子が好ましい。
【0053】
環A及びBは一般式(1)で示される環と同意義を示す。すなわち、一般式(A−1)又は一般式(A−2)と同意義を示す。そして、その中でも一般式(A−1)においては、T3が硫黄であることが好ましい。
【0054】
本発明の一般式(2)で示されるテトラハロターフェニル誘導体としては、以下の化合物が好ましく、
【0055】
【化22】
【0056】
【化23】
【0057】
【化24】
【0058】
【化25】
【0059】
【化26】
【0060】
【化27】
【0061】
【化28】
【0062】
【化29】
【0063】
【化30】
【0064】
【化31】
【0065】
【化32】
【0066】
【化33】
【0067】
特に好ましくは
【0068】
【化34】
【0069】
である。
【0070】
(ヘテロアセン誘導体の製造方法)
本発明の一般式(1)で示されるヘテロアセン誘導体の製造方法について述べる。
【0071】
本発明の一般式(1)で示されるヘテロアセン誘導体は、一般式(2)で示されるテトラハロターフェニル誘導体をメタル化剤を用いてテトラメタル化し、下記一般式(3)及び/又は下記一般式(4)で示される反応剤と反応させることにより製造することができる。なお、一般式(3)、一般式(4)で示される反応剤が同じ化合物であっても良い。
(R3)lT1(L1)p (3)
(R4)mT2(L2)q (4)
(ここで、置換基T1、T2、R3、R4及び記号lとmは一般式(1)で示される置換基及び記号と同意義を示し、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、フェニルスルホニル基を示し、p及びqは0又は2の整数を示す。)
なお、ここでテトラメタル化とは、一般式(2)におけるX1〜X4をそれぞれメタルに置換することを意味する。
【0072】
一般式(2)で示されるテトラハロターフェニル誘導体をテトラメタル化する場合、用いるメタル化剤は、一般式(2)におけるX1〜X4をメタルに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属;メチルマグネシウムブロマイド、エチルマグネシウムブロマイド、イソプロピルマグネシウムクロライド、tert−ブチルマグネシウムクロライド、フェニルマグネシウムブロマイド等のグリニャール試薬;マグネシウム金属;亜鉛金属等を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウムである。
【0073】
該メタル化剤の使用量は一般式(2)で示されるテトラハロターフェニル誘導体1当量に対し、3〜20当量が好ましく、特に好ましくは4〜15当量、さらに好ましくは5〜10当量である。
【0074】
該テトラメタル化は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばテトラヒドロフラン(以下、THFと略す)、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジオキサン、トルエン、ヘキサン、シクロヘキサン等であり、特に好ましくはTHF、ジエチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。該テトラメタル化の温度は−100〜50℃で行うことが好ましく、特に好ましくは−90〜30℃である。反応時間は1〜120分が好ましく、特に好ましくは5〜60分である。なお、テトラメタル化の進行は、反応液の一部を取り出し、水で反応を停止させた後、ガスクロマトグラフィーで分析することで監視することができる。
【0075】
該テトラメタル化により生成したテトラメタル塩は、次いで一般式(3)及び一般式(4)で示される反応剤と反応させることにより、一般式(1)で示されるヘテロアセン誘導体が得られるものである。係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を直接用いて反応させる方法、生成したテトラメタル塩を一度単離した後、前記反応剤と反応させる方法のいずれを用いてもよい。
【0076】
ここで、一般式(3)、一般式(4)における置換基T1、T2、R3、R4及び記号lとmは一般式(1)で示される置換基及び記号と同意義を示す。
【0077】
また、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、好ましくは塩素原子、臭素原子、炭素数1〜20のオキシ基、アリールスルホニル基である。炭素数1〜20のオキシ基は特に限定はなく、例えばメトキシ基、エトキシ基、n−ブトキシ基、フェノキシ基、(2−メトキシ)フェノキシ基等を挙げることができ、アリールスルホニル基は特に限定はなく、例えばフェニルスルホニル基、p−トリルスルホニル基等を挙げることができる。これらの中でも特にフェニルスルホニル基が好ましい。
【0078】
そして、具体的な一般式(3)、一般式(4)で示される反応剤としては、例えば2塩化硫黄;2臭化硫黄;ビス(フェニルスルホニル)スルフィド、ビス(p−トリルスルホニル)スルフィド等のビス(アリールスルホニル)スルフィド類;硫黄;2塩化セレン;セレン;2塩化テルル;テルル;ジクロロフェニルホスフィン、ジメトキシフェニルホスフィン、ジフェノキシフェニルホスフィン、ジクロロ{4−(n−オクチル)フェニル}ホスフィン等のアリールホスフィン類;ジクロロ(n−ヘキシル)ホスフィン、ジクロロ(n−オクチル)ホスフィン、ジメトキシ(n−ヘキシル)ホスフィン等のアルキルホスフィン類;ジクロロフェニルボラン、ジメトキシフェニルボラン、ジメトキシ{4−(n−ヘキシル)フェニル}ボラン、ジフェノキシフェニルボラン、ジクロロ{4−(n−オクチル)フェニル}ボラン等のアリールボラン類;ジクロロ(n−ヘキシル)ボラン、ジクロロ(n−オクチル)ボラン、ジメトキシ(n−ヘキシル)ボラン等のアルキルボラン類等を挙げることができ、好ましくはビス(フェニルスルホニル)スルフィド、ジクロロフェニルホスフィン、ジクロロフェニルボラン等である。
【0079】
テトラメタル化により生成したテトラメタル塩と一般式(3)及び一般式(4)で示される反応剤と反応させる際には、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン、トルエン、ヘキサン、シクロヘキサン等であり、好ましくはTHF、ジエチルエーテルである。用いる反応剤の量は、一般式(2)で示されるテトラハロターフェニル誘導体1当量に対し、1.2〜10当量が好ましく、特に好ましくは2〜8当量である。該反応剤との反応温度は−100〜50℃が好ましく、特に好ましくは−90〜30℃であり、反応時間は0.5〜30時間が好ましく、特に好ましくは1〜18時間である。
【0080】
本発明の一般式(1)で示されるヘテロアセン誘導体の製造は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0081】
本発明の一般式(1)で示されるヘテロアセン誘導体の製造方法では、一般式(2)で示されるテトラハロターフェニル誘導体をテトラメタル化した後、塩化マグネシウムと反応させ、その後に一般式(3)及び一般式(4)で示される反応剤で処理することもできる。
【0082】
かくして得られた、本発明の一般式(1)で示されるヘテロアセン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0083】
(テトラハロターフェニル誘導体の製造方法)
次に、本発明の一般式(1)で示されるヘテロアセン誘導体の前駆体として用いられる一般式(2)で示されるテトラハロターフェニル誘導体の製造方法について述べる。
【0084】
本発明の一般式(2)で示されるテトラハロターフェニル誘導体は下記一般式(5)で示されるテトラハロベンゼンと下記一般式(6)及び/又は下記一般式(7)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させることにより製造することができる。なお、一般式(6)、一般式(7)で示される反応剤が同じ化合物であっても良い。
【0085】
【化35】
【0086】
(ここで、置換基X5及びX6は臭素原子、ヨウ素原子、塩素原子を示す。置換基R1、R2、X2及びX3は一般式(2)で示される置換基と同意義を示す。)
【0087】
【化36】
【0088】
(ここで、M1はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X1並びに環Aは、一般式(2)で示される置換基並びに環と同意義を示す。)
【0089】
【化37】
【0090】
(ここで、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X4並びにB環は、一般式(2)で示される置換基並びに環と同意義を示す。)
本発明の一般式(5)、(6)及び(7)について、さらに述べる。
【0091】
一般式(5)の置換基X5及びX6は、臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子及びヨウ素原子であり、さらに好ましくはヨウ素原子である。
【0092】
置換基R1、R2、X2及びX3は、一般式(2)で示される置換基と同意義を示す。
【0093】
そして、具体的な一般式(5)で示される化合物としては、例えば1,4−ジブロモ−2,5−ジヨードベンゼンが挙げられる。
【0094】
一般式(6)、(7)の置換基M1、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物であり、上記のパラジウム及び/又はニッケル触媒により脱離され、パラジウム及び/又はニッケルと置換できる基である限り特に限定はなく、例えばMgCl、MgBr、B(OH)2、B(OMe)2、テトラメチルジオキサボロラニル基、ZnCl、ZnBr、ZnI、Sn(Bu−n)3、Si(Me)3、Si(Bu−n)3、Si(OMe)3、Si(OEt)3等を挙げることができ、好ましくはZnCl、B(OH)2、Si(OMe)3である。
【0095】
置換基X1、X4並びに環A、Bは、一般式(2)で示される置換基並びに環と同意義を示す。
【0096】
そして、具体的な一般式(6)、一般式(7)で示される化合物としては、例えば3−ブロモ−6−ドデシルベンゾチエニル−2−ジンククロライド、3−ブロモ−6−ドデシルベンゾチエニル−2−トリメトキシシラン、3−ブロモ−6,7−ジペンタデシルアントラセニル−2−ボロン酸、3−ブロモ−6,7−ペンタデシル−9,10−ジフェニルアントラセニル−2−ボロン酸等が挙げられる。
【0097】
なお、一般式(6)、一般式(7)で示される2−ハロアリール金属試薬は、例えば、それらの原料となるアリールジハロゲン置換体をイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬によりハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン、トリ(イソプロポキシ)ボラン、2−イソプロポキシ−4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン、テトラメトキシシラン等と反応させることで好適に調製することができる。また、一般式(6)、(7)の置換基M1、M2がSi(OMe)3又はSi(OEt)3の場合、それらの原料となるアリールジハロゲン置換体とPd又はRh触媒を用いたトリアルコキシシランとの反応によっても調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁」に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている方法を用いることもできる。
【0098】
一般式(5)で示されるテトラハロベンゼンと一般式(6)及び/又は一般式(7)で示される2−ハロアリール金属試薬の反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ジクロロビス(トリフェニルホスフィン)パラジウム、ビス(トリ−tert−ブチルホスフィン)パラジウム、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/トリ−tert−ブチルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等のパラジウム触媒;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物、ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等のニッケル触媒;を挙げることができる。中でも、好ましい触媒は0価のパラジウム化合物であり、特に好ましい触媒はテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。又、これら触媒は1種若しくは2種以上の混合物を用いても良い。
【0099】
一般式(5)で示されるテトラハロベンゼンと一般式(6)及び/又は一般式(7)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させる際には、好ましくは溶媒中で実施する。用いる溶媒に特に限定はなく、例えばTHF、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水、N,N−ジメチルホルムアミド、N−メチルピロリドン、トリエチルアミン、ピペリジン、ピロリジン、ジイソプロピルアミン等を挙げることができ、又、これら溶剤は1種若しくは2種以上の混合物を用いても良く、例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0100】
パラジウム触媒、ニッケル触媒の使用量は一般式(5)で示されるテトラハロベンゼン1モルに対し、0.1〜20モル%が好ましく、特に好ましくは0.5〜10モル%の範囲である。
【0101】
一般式(6)、一般式(7)で示される2−ハロアリール金属試薬の使用量は一般式(5)で示されるテトラハロベンゼン1当量に対し、0.8〜3.2当量が好ましく、特に好ましくは1.0〜2.8当量、さらに好ましくは1.1〜2.5当量である。
【0102】
反応の際の温度は10〜120℃が好ましく、さらに好ましくは30〜100℃、特に好ましくは40〜90℃であり、反応時間は1〜100時間が好ましく、特に好ましくは2〜85時間である。
【0103】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、りん酸カリウム、りん酸ナトリウム、ナトリウムtert−ブトキサイド、フッ化カリウム、フッ化ナトリウム、フッ化セシウム等の無機塩基;トリエチルアミン、トリメチルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン、テトラブチルアンモニウムフルオライド等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(5)で示されるテトラハロベンゼン1当量に対し、0.5〜10.0当量が好ましく、特に好ましくは2.0〜8.0当量である。さらにこれらの塩基と併用し、相間移動触媒を用いることもできる。相間移動触媒の種類は特に限定はなく、例えばトリオクチルメチルアンモニウムクロライド、テトラブチルアンモニウムクロライド、セチルピリジニウムクロライド、ベンジルトリメチルアンモニウムクロライド等を好適なものとして挙げることができる。これらの相間移動触媒の使用量は一般式(5)で示されるテトラハロベンゼン1当量に対し、0.1〜1.5当量が好ましく、特に好ましくは0.1〜0.8当量である。
【0104】
さらに反応系中にトリフェニルホスフィン等のホスフィンを存在させることもできる。これらのホスフィンの使用量は、該パラジウム及び/又はニッケル触媒1当量に対し、0.9〜8.0当量が好ましく、特に好ましくは1.0〜3.0当量である。
【0105】
なお、反応系中に銅化合物を存在させることもできる。該銅化合物しては特に限定はなく、例えば塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、酢酸銅(I)等の1価銅;塩化銅(II)、臭化銅(II)、ヨウ化銅(II)、酢酸銅(II)、アセチルアセトナート銅(II)等の2価銅等を挙げることができる。その中でも好ましくは1価銅であり、特に好ましくはヨウ化銅(I)である。これらの銅化合物の使用量は該パラジウム及び/又はニッケル触媒1当量に対し、0.3〜10.0当量が好ましく、特に好ましくは0.6〜6.0当量である。
【0106】
また、一般式(5)で示されるテトラハロベンゼンと一般式(6)及び/又は(7)で示される2−ハロアリール金属試薬の反応により炭素−炭素結合が形成される位置はハロゲンの種類により制御することができる。
【0107】
即ち、ヨウ素原子の反応性が最も高く、臭素原子、塩素原子の順に反応性が低下することから、これらハロゲンの種類の反応性を利用することで反応する位置を任意に決めることができる。そのため、一般式(2)で示されるテトロハロターフェニル誘導体の製造は、例えば一般式(5)のX5及びX6をヨウ素原子とし、X2及びX3を臭素原子及び/又は塩素原子とすることにより、製造することができる。
【0108】
かくして得られた、本発明の一般式(2)で示されるテトラハロターフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0109】
(耐酸化性有機半導体材料)
次に、本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料について述べる。該耐酸化性有機半導体材料は溶剤への溶解性、耐酸化性に優れ、好適な塗布性を有する。該耐酸化性有機半導体材料は本発明の一般式(1)で示されるヘテロアセン誘導体を溶剤に溶解することにより製造することができる。
【0110】
耐酸化性有機半導体材料としては、本発明の一般式(1)で示されるヘテロアセン誘導体の中でも溶解性に優れたものとなることから、環(A−1)における置換基R5〜R8が炭素数7〜20のアルキル基であるもの又は環(A−2)における置換基R9〜R12が炭素数13〜20のアルキル基であるものが好ましい。
【0111】
本発明の一般式(1)で示されるヘテロアセン誘導体の溶解に用いる溶剤は、特に限定はなく、例えばo−ジクロロベンゼン、クロロベンゼン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、クロロホルム等のハロゲン系溶剤;THF、ジオキサン等のエーテル系溶剤;トルエン、キシレン、メシチレン、エチルベンゼン、n−プロピルベンゼン、n−ヘプチルベンゼン、n−オクチルベンゼン、n−ノニルベンゼン等の芳香族化合物の炭化水素系溶剤;酢酸エチル、γ−ブチロラクトン等のエステル系溶剤;N,N−ジメチルホルムアミド、N−メチルピロリドン等のアミド系溶剤;等が挙げられる。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。中でも、好ましくはトルエンである。
【0112】
上記に挙げた溶剤と一般式(1)で示されるヘテロアセン誘導体を混合攪拌することにより、一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料となるものである。混合攪拌する際の温度は10〜200℃が好ましく、特に好ましくは20〜190℃である。混合攪拌する際の一般式(1)で示されるヘテロアセン誘導体の濃度は、溶剤及び温度により変えることができ、0.01〜10.0重量%であることが好ましい。溶液の調製は空気中でも実施することができるが、好ましくは窒素、アルゴン等の不活性雰囲気下で調製する。
【0113】
一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料の耐酸化性の評価は、該溶液を所定時間、空気と接触させる方法で実施することができる。まず用いる溶剤は予め脱気しておき、溶存酸素を除去する。空気との接触時間は、温度により適宜選択することができ、0.5分〜3時間が好適である。酸化の進行は、溶液の色の変化並びにガスクロマトグラフィー及びガスクロマトグラフィー(GC)−マススペクトル(GCMS)分析による酸化物の検出により行うことができる。
【0114】
本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料は、用いられる一般式(1)で示されるヘテロアセン誘導体自体が適度の凝集性を有することから比較的に低温で溶剤へ溶解でき、且つ耐酸化性があることから、塗布法による有機薄膜の製造に好適に適用できる。即ち、雰囲気から厳密に空気を除く必要がないことから塗布工程を簡略化することができる。塗布は空気中でも実施できるが、好ましくは溶剤の乾燥を考慮して窒素気流下で行う。なお、好適な塗布性を得るために、本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料の粘度は、0.005〜20ポアズの範囲にあることが好ましい。
【0115】
(有機薄膜)
次に本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料を用いた有機薄膜について述べる。係る有機薄膜は上記の耐酸化性有機半導体材料(溶液)の再結晶化若しくは基板への塗布により製造することができ、特に基板への塗布により製造することが好ましい。そして、基板への塗布により製造することにより、基板上に形成される有機薄膜となるものである。
【0116】
有機薄膜としては、本発明の一般式(1)で示されるヘテロアセン誘導体の中でも表面粗さの小さい有機薄膜となることから、環(A−1)における置換基R5〜R8が炭素数7〜20のアルキル基であるもの又は環(A−2)における置換基R9〜R12が炭素数13〜20のアルキル基であるものが好ましい。
【0117】
再結晶化による薄膜は、前記耐酸化性有機半導体材料を冷却することで形成することができる。有機薄膜を製造する時の雰囲気は、窒素、アルゴン等の不活性ガス又は空気下で行うことが好ましく、特に窒素、アルゴン等の不活性ガス下で行うことが好ましい。該溶液中の一般式(1)で示されるヘテロアセン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%である。冷却は60〜200℃の温度から−20〜60℃が好ましく、特に好ましくは−10℃〜40℃の間に冷却することにより好適に実施することができる。またこのようにして製造した結晶状の有機薄膜を適当な基板の上に張り合わせる、即ちラミネーション等により基板上に製造することもできる。再結晶化により得られる有機薄膜の膜厚は特に限定はなく、好ましくは50nm〜2mm、特に好ましくは1〜500μmである。
【0118】
基板への塗布による有機薄膜の製造は、前記耐酸化性有機半導体材料を基板上に塗布した後、加熱、気流及び自然乾燥等の方法により溶剤を気化させることで実施することができる。該溶液中の一般式(1)で示されるヘテロアセン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%であることが好ましい。塗布温度は特に限定はなく、例えば20〜200℃の間で好適に実施することができる。塗布の具体的方法は特に限定はなく、公知の方法、例えばスピンコート、キャストコート及びディップコート等を用いることができる。さらにスクリーン印刷、インクジェット印刷、グラビア印刷等の印刷技術を用いても作製することが可能である。使用する基板の材料は特に限定はなく、結晶性、非結晶性の種々の材料を用いることができる。基板の具体例としては、例えばポリエチレンテレフタレート、ポリエチレンナフタレート、ポリメチルメタクリレート、ポリエチレン、ポリプロピレン、ポリスチレン、環状ポリオレフィン、ポリイミド、ポリカーボネート、ポリビニルフェノール、ポリビニルアルコール、ポリ(ジイソプロピルフマル酸)、ポリ(ジエチルフマル酸)、ポリ(ジイソプロピルマレイン酸)等のプラスチック基板;ガラス、石英、酸化アルミニウム、シリコン、酸化シリコン、二酸化タンタル、五酸化タンタル、インジウム錫酸化物等の無機材料基板;金、銅、クロム、チタン等の金属基板を好適に用いることができる。またこれらの基板の表面は、例えばオクタデシルトリクロロシラン、オクタデシルトリメトキシシラン、β−フェネチルトリクロロシラン等のシラン類;ヘキサメチルジシラザン等のシリルアミン類で修飾処理したものであっても使用することができる。さらに、基板は絶縁性あるいは誘電性を有する材料であっても良い。塗布した後の溶剤の乾燥は、常圧若しくは減圧で除去することができる、又、加熱、窒素気流により乾燥してもよい。さらに、溶剤の気化速度を調節することで本発明の一般式(1)で示されるヘテロアセン誘導体の結晶成長を制御することができる。基板への塗布により得られる有機薄膜の膜厚は特に限定はなく、好ましくは1nm〜100μm、特に好ましくは10nm〜20μmである。
【0119】
有機薄膜のキャリアー移動度は、有機薄膜の表面粗さと密接な関係があり、表面粗さが小さい程、高いキャリアー移動度を期待することができる。本発明の一般式(1)で示されるヘテロアセン誘導体からなる有機薄膜の表面粗さは、40nm以下が好ましく、特に好ましくは30nm以下であり、表面の平滑性が高いことから、高いキャリアー移動度を期待することができる。
【0120】
本発明の一般式(1)で示されるヘテロアセン誘導体は平面剛直性の高い分子構造を有することから、優れた半導体特性を与えることが期待できる。又、該ヘテロアセン誘導体はトルエンあるいはn−オクチルベンゼン等の溶媒に溶解し、溶液状態にあっても容易に空気酸化されることはない。従って、塗布法により半導体薄膜を容易に作成できる。したがって、本発明の一般式(1)で示されるヘテロアセン誘導体は、電子ペーパー、有機ELディスプレイ、液晶ディスプレイ、ICタグ用等のトランジスタの有機半導体活性相用途;有機ELディスプレイ材料;有機半導体レーザー材料;有機薄膜太陽電池材料;フォトニック結晶材料等の電子材料に利用することができる。
【発明の効果】
【0121】
優れた耐酸化性を有し、塗布法による有機半導体活性相形成が可能な、ヘテロアセン誘導体及びその用途を提供する。さらに、本発明のヘテロアセン誘導体からなる有機薄膜はその表面粗さが40nmと小さいことから、高いキャリアー移動度を有することが期待できる。
【実施例】
【0122】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例に限定されるものではない。
【0123】
生成物の同定には1H−NMRスペクトル及びマススペクトルを用いた。なお、1H−NMRスペクトルの測定は日本電子製JEOL GSX−270WB(270MHz)を用いた。マススペクトル(MS)は日本電子製JEOL JMS−700を用いて、試料を直接導入し、電子衝突(EI)法(70エレクトロンボルト)あるいはFAB法(6キロエレクトロンボルト、キセノンガス、マトリックス(ジチオスレイトール:ジチオエリスリトール=3:1))(FABMS)で測定した。
【0124】
反応の進行の確認等はガスクロマトグラフィー(GC)及びガスクロマトグラフィー−マススペクトル(GCMS)分析を用いた。
【0125】
ガスクロマトグラフィー分析
装置 島津GC14B
カラム J&Wサイエンティフィック社製、DB−1,30m
ガスクロマトグラフィー−マススペクトル分析
装置 パーキンエルマーオートシステムXL(MS部;ターボマスゴールド)
カラム J&Wサイエンティフィック社製、DB−1,30m
反応用の試薬及び溶媒は、断りのない限り市販品を用いた。なお、グリニャール試薬あるいはブチルリチウム等の有機金属試薬を用いた場合は、市販の脱水溶媒をそのまま用いた。
【0126】
有機薄膜の表面粗さは原子間力顕微鏡を用いて測定した。なお、原子間力顕微鏡はディジタルインスツルメンツ製のナノスコープIIIa D3100を用いた。
【0127】
合成例1 (1,4−ジブロモ−2,5−ジヨードベンゼンの合成)
1,4−ジブロモ−2,5−ジヨードベンゼンはジャーナル オブ アメリカン ケミカル ソサイエティー、1997年、119巻、4578−4593頁に記載されている方法を参考に合成を行った。
【0128】
メカニカルスターラー付き1lの三口フラスコに過ヨウ素酸16.7g(73.0mmol)及び硫酸525mlを加えた。過ヨウ素酸が溶解した後、ヨウ化カリウム36.4g(219mmol)を少しずつ添加した。その内容物の温度を−30℃に冷却し、1,4−ジブロモベンゼン34.5g(146mmol)を5分間かけて添加した。得られた混合物を−25℃で36時間撹拌した。反応混合物を氷(2kg)中へ注いだ後、濾過し固体を取り出した。その固体をクロロホルムに溶解させ、5%苛性ソーダ水溶液及び水で洗浄し、有機相を無水硫酸マグネシウムで乾燥した。減圧濃縮後、残渣をクロロホルムから再結晶化し、白色結晶を得た(36.0g、収率50%)。
1H−NMR(CDCl3,21℃):δ=8.02(s,2H)。
【0129】
1H−NMRスペクトルが文献値と一致したことより、1,4−ジブロモ−2,5−ジヨードベンゼンが得られたことを確認した。
【0130】
合成例2 [1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成(一般式(2)のテトラハロターフェニル誘導体の合成)]
1)2,3−ジブロモ−6−ドデカノイルベンゾチオフェンの合成
窒素雰囲気下、300ml二口ナスに2,3−ジブロモベンゾチオフェン(シグマ−アルドリッチ製)10.33g(35.37mmol)及びジクロロメタン(脱水品)150mlを加えた。−25℃に冷却した後、ドデカノイルクロライド(和光純薬工業製)9.00ml(8.28g、37.8mmol)、次いで塩化アルミニウム(和光純薬工業製)5.05g(37.8mmol)を添加した。−25℃で2日間反応させた後、水を加えて反応を停止させた。分相し、有機相に飽和炭酸ナトリウム水溶液を添加し、一昼夜撹拌した(過剰のドデカン酸除去のため)。分相、水洗し、有機相を無水硫酸ナトリウムで乾燥した。濾過減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィーで精製した。溶離液をヘキサンとすることで、未反応の2,3−ジブロモベンゾチオフェン及びトリブロモベンゾチオフェンが溶出し、溶離液をヘキサン:トルエン=10:1とすると異性体である2,3−ジブロモ−4−ドデカノイルベンゾチオフェンが溶出し、溶離液をヘキサン:トルエン=2:1とすると目的物を含む成分が溶出した。この最後の成分をヘプタン30mlで再結晶精製し、目的物である2,3−ジブロモ−6−ドデカノイルベンゾチオフェンの白色固体4.39g(9.25mmol)得た(収率26%)。
1H NMR(CDCl3,23℃):δ=8.35(s,1H),8.01(dd,J=8.5Hz,1.4Hz,1H),7.80(d,J=8.4Hz,1H),3.03(t,J=7.6Hz,2H),1.77(m,2H),1.26(m,16H),0.88(t、J=8.1Hz,3H)。
MS m/z: 474(M+,11%),394(M+−Br,2%),334(M+−C10H21+1,100%),319(M+−C11H23,58%)。
【0131】
2)2,3−ジブロモ−6−ドデシルベンゾチオフェンの合成
窒素雰囲気下、100mlシュレンク反応容器に合成例2の1)で合成した2,3−ジブロモ−6−ドデカノイルベンゾチオフェン3.85g(8.12mmol)及びトリフルオロ酢酸(和光純薬工業製)6.3mlを添加した。氷冷後、トリエチルシラン(信越化学製)2.9mlを滴下した。その後、40℃で6時間反応後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製した後(溶離液;ヘキサン)、目的物を含むフラクションを130パスカル、90℃で真空加熱し、低沸分を除去した。残渣に2,3−ジブロモ−6−ドデシルベンゾチオフェンの白色固体を3.35g(7.28mmol)得た(収率90%)。
1H NMR(CDCl3,23℃):δ=7.63(d,J=8.1Hz,1H),7.50(d,J=1.1Hz,1H),7.24(dd,J=8.1Hz,1.4Hz,1H),2.71(t,J=8.1Hz,2H),1.65(m,2H),1.25(m,18H),0.88(t、J=8.1Hz,3H)。
MS m/z: 460(M+,65%),380(M+−Br,4%),305(M+−C11H23,100%)。
【0132】
3)1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成
100mlシュレンク反応容器に合成例2の2)で合成した2,3−ジブロモ−6−ドデシルベンゾチオフェン984mg(2.13mmol)及びTHF24mlを添加した。−74℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.78M)のTHF溶液5.5ml(4.3mmol)を滴下した。−74℃、15分間、グリニャール化の熟成後、−85℃とし、塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液4.3ml(4.3mmol)を添加した。1時間熟成後、冷バスを外し、室温で30分間反応後、減圧濃縮した。得られた白色固体[(3−ブロモ−6−ドデシルベンゾチエニル−2−ジンククロライド)(一般式(6)及び(7)の化合物)]に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン428mg(0.877mmol)(一般式(5)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)38.8mg(0.0335mmol))及びTHF8mlを加え、60℃で、6時間反応させた。氷冷後、容器を水冷し3N塩酸4mlを添加することで反応を停止させた。トルエンを添加し、分相後、有機相を2回飽和食塩水で洗浄した。有機相に70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.2ml添加し、1時間撹拌した。水洗2回し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製した(溶離液;ヘキサン)。さらにヘプタンから再結晶精製し、目的物の淡黄固体380mgを得た(収率43%)。
1H NMR(CDCl3,23℃):δ=7.79(d,J=8.1Hz,2H),7.78(s,2H),7.66(s,2H),7.34(dd,J=8.1Hz,1.4Hz,2H),2.78(t,J=8.1Hz,4H),1.70(m,4H),1.27(m,36H),0.88(t,J=8.1Hz,6H)。
MS m/z: 994(M+,100%),914(M+−Br,4%),839(M+−C11H23,36%)。
【0133】
1H NMR及びMS測定より、1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0134】
【化38】
【0135】
実施例1 (ジドデシル)ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、THF21mlを添加し−74℃に冷却した。メタル化剤としてsec−ブチルリチウム(関東化学製1.0M)のシクロヘキサン/ヘキサン溶液3.0ml(3.0mmol)を添加した。さらに合成例2で合成した1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの粉体379mg(0.381mmol)を−74℃下で少しずつ添加した。この得られた溶液を−74℃〜−70℃で30分間熟成し、テトラメタル化を行った。次に−83℃とし、反応剤としてビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3)及び(4)の化合物)477mg(1.51mmol)を一気に投入した。一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、分相し、さらに有機相を飽和食塩水で洗浄した。有機相を減圧濃縮した後、残渣をヘキサンを用いて洗浄した。その残渣のトルエン再結晶精製を3回繰り返し、目的物の鮮黄固体39mgを得た(収率14%)。
1H NMR(重トルエン,105℃):δ=8.00(s,2H),7.60(d,J=8.3Hz,2H),7.54(d,J=1.5Hz,2H),7.10(dd,J=8.3Hz,1.5Hz,2H),2.64(t,J=8.1Hz,4H),1.66(m,4H),1.34(m,36H),0.88(t,J=8.1Hz,6H)。
MS m/z: 738(M+−1,100%),583(M+−C11H23−1,33%)。
【0136】
1H NMR及びMS測定より、(ジドデシル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0137】
【化39】
【0138】
合成例3 [1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成(一般式(2)のテトラハロターフェニル誘導体の合成)]
1)2,3−ジブロモ−6−デカノイルベンゾチオフェンの合成
ドデカノイルクロライドの代わりに、デカノイルクロライド(和光純薬工業製)を用いた以外は合成例2の1)と同じ操作を繰り返して2,3−ジブロモ−6−デカノイルベンゾチオフェンを収率28%で合成した。
【0139】
2)2,3−ジブロモ−6−デシルベンゾチオフェンの合成
合成例2の2)と同じ操作を繰り返して2,3−ジブロモ−6−デシルベンゾチオフェンを収率96%で合成した。
【0140】
3)1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成
合成例2の3)と同じ操作を繰り返して1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを収率46%で合成した。
1H NMR(CDCl3,23℃):δ=7.80(d,J=8.1Hz,2H),7.78(s,2H),7.67(s,2H),7.35(dd,J=8.1Hz,1.4Hz,2H),2.78(t,J=8.1Hz,4H),1.70(m,4H),1.27(m,28H),0.88(t,J=8.1Hz,6H)。
MS m/z: 938(M+,100%),858(M+−Br,3%),811(M+−C9H19,33%)。
1H NMR及びMS測定より、1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0141】
【化40】
【0142】
実施例2 (ジデシルジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、合成例3で合成した1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返して(ジデシル)ジベンゾテトラチエノアセンを収率17%で合成した。
1H NMR(重トルエン,105℃):δ=7.99(s,2H),7.60(d,J=8.3Hz,2H),7.53(d,J=1.5Hz,2H),7.11(dd,J=8.3Hz,1.5Hz,2H),2.64(t,J=8.1Hz,4H),1.66(m,4H),1.34(m,28H),0.88(t,J=8.1Hz,6H)。
MS m/z: 682(M+−1,100%),555(M+−C9H19−1,35%)。
【0143】
1H NMR及びMS測定より、(ジデシル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0144】
【化41】
【0145】
合成例4 [1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成(一般式(2)のテトラハロターフェニル誘導体の合成)]
1)2,3−ジブロモ−6−オクタデカノイルベンゾチオフェンの合成
ドデカノイルクロライドの代わりに、オクタデカノイルクロライド(和光純薬工業製)を用い、さらに反応を−25℃で3日間実施した以外は合成例2の1)と同じ操作を繰り返して2,3−ジブロモ−6−オクタデカノイルベンゾチオフェンを収率20%で合成した。
【0146】
2)2,3−ジブロモ−6−オクタデシルベンゾチオフェンの合成
40℃で6時間反応させる代わりに、60℃で4時間反応させた以外は合成例2の2)と同じ操作を繰り返して2,3−ジブロモ−6−オクタデシルベンゾチオフェンを収率99%で合成した。
【0147】
3)1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成
グリニャール化の熟成を『−74℃、15分間』から、『−74℃から−28℃まで4時間かけて行った』以外は合成例2の3)と同じ操作を繰り返して1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを収率33%で合成した。
1H NMR(CDCl3,23℃):δ=7.80(d,J=8.1Hz,2H),7.79(s,2H),7.67(s,2H),7.34(dd,J=8.1Hz,1.4Hz,2H),2.78(t,J=8.1Hz,4H),1.70(m,4H),1.27(m,60H),0.88(t,J=8.1Hz,6H)。
MS m/z: 1163(M+,100%),1083(M+−Br,4%),924(M+−C17H35,38%)。
1H NMR及びMS測定より、1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0148】
【化42】
【0149】
実施例3 (ジオクタデシル)ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、合成例4で合成した1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返して(ジオクタデシル)ジベンゾテトラチエノアセンを収率12%で合成した。
1H NMR(重トルエン,105℃):δ=8.00(s,2H),7.61(d,J=8.3Hz,2H),7.53(d,J=1.5Hz,2H),7.10(dd,J=8.3Hz,1.5Hz,2H),2.64(t,J=8.1Hz,4H),1.66(m,4H),1.34(m,60H),0.88(t,J=8.1Hz,6H)。
MS m/z: 907(M+,100%),667(M+−C17H35−1,29%)。
【0150】
1H NMR及びMS測定より、(ジオクタデシル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0151】
【化43】
【0152】
比較例1 (ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体においてR5〜R12が同時に水素である化合物)の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返してジベンゾテトラチエノアセンを収率21%で合成した。
MS m/z: 402(M+,100%),370(M+−S,16%),201(M+/2,74%)。
【0153】
MS測定より、ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0154】
【化44】
【0155】
窒素雰囲気下、得られた(ジベンゾ)テトラチエノアセン2.5mgをトルエン35gと混合し、100℃で1時間撹拌したが、全く溶解せず、溶液を調製することが困難であったことから、溶解性に劣るものであった。
【0156】
合成例5 (2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンの合成)
1)1,2−ジペンタデシルベンゼンの合成
1,2−ジペンタデシルベンゼンは、「シンセシス」、1993年、387−390頁の方法を参考に1,2−ジクロロベンゼンとn−ペンタデシルマグネシウムブロマイドから次のように合成した。
【0157】
窒素雰囲気下、300mlシュレンク反応容器に1,2−ジクロロベンゼン3.4ml(30.3mmol)、塩化ニッケル{ビス(ジフェニルホスフィノ)プロパン}164.2mg(0.303mmol)、及びジエチルエーテル30mlを添加した。0℃に冷却し、n−ペンタデシルマグネシウムブロマイド(シグマ−アルドリッチ製、0.5M)のジエチルエーテル溶液150ml(75mmol)を滴下した。35℃で16時間反応後、3N塩酸を加えて反応を停止させた。ジエチルエーテルで抽出し、有機相を水及び飽和炭酸水素ナトリウム水溶液で洗浄した。塩化カルシウムで乾燥し、溶媒を減圧濃縮した。さらに残渣を加熱減圧乾燥し(10パスカル、100℃)、残渣に1,2−ジペンタデシルベンゼンの粘性固体を得た(11.63g、収率77%)。
【0158】
2)4−ブロモ−5−ヨード無水フタル酸の合成
4−ブロモ−5−ヨード無水フタル酸は「ジャーナル オブ オルガニック ケミストリィー」(米国)、1951年、16巻、1577−1581頁に記載されている方法を参考に4−ブロモ無水フタル酸を原料に用いて次のように合成を行った。
【0159】
窒素雰囲気下、100mlの二口ナスフラスコに4−ブロモフタルイミド(東京化成工業製)13.3g(58.8mmol)を入れた。次いでヨウ素7.78g(30.6mmol)及び10%発煙硫酸(ヨツハタ化学工業製)50mlを加え、90℃で23時間反応を行った。反応混合物を室温に冷やして氷に注ぎ入れた後、ガラスフィルターでろ過し、黄色固体17.1gを得た。得られた固体を濃硫酸47mlに溶解させ、130℃で5時間反応を行った。反応混合物を氷冷後、氷水を加えて析出した固体をろ過し、フタル酸誘導体の固体18.5gを得た。次に水酸化ナトリウム4.8gを水24mlに溶かした水溶液に得られた固体を室温で溶かした。この塩基性水溶液に酢酸を加えpHを3〜4に中和し、析出するフタル酸誘導体のモノナトリウム塩の白色沈殿をろ過した。得られた白色沈殿を水に懸濁させ、濃塩酸でpHを1以下にし、再びフタル酸誘導体として白色固体7.75gを得た。この固体をトルエン70mlに溶かし、無水酢酸13ml(138mmol)を加え、105℃で4時間反応を行った。反応液を減圧濃縮して白色固体6.9gを得た。この固体を加熱トルエンで再結晶精製し、目的の4−ブロモ−5−ヨードフタル酸無水物を6.79g(19.2mmol)を得た(収率32.7%)。
【0160】
3)2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンの合成
2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンは「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁に記載されている方法を参考に次のように合成を行った。
【0161】
100mlの三口フラスコに合成例5の2)で合成した4−ブロモ−5−ヨード無水フタル酸2.40g(6.80mmol)、合成例5の1)で合成した1,2−ジペンタデシルベンゼン4.16g(8.34mmol)、及び1,2−ジクロロエタン7.0mlを加えた。そこへ塩化アルミニウム1.90g(14.3mmol)を添加し、加熱還流下で1.5時間撹拌した。得られた反応混合物に氷を少しずつ加えてクエンチした後、ジエチルエーテルで抽出した。無水硫酸マグネシウムで乾燥後、濾過、減圧濃縮した後、得られた残渣をシリカゲルカラムクロマトグラフィーで精製し(溶離液;ヘキサン:酢酸エチル=1:1)、5.55g(6.52mmol)の安息香酸誘導体を得た。この安息香酸誘導体に塩化チオニル5.0ml、N,N−ジメチルホルムアミド0.05ml、塩化アルミニウム1.74g(13.0mmol)、及びテトラクロロエタン25mlを添加し、50℃で5時間撹拌した。得られた反応混合物を室温まで冷却し、氷を加えた。トルエンで抽出し、有機相を無水硫酸マグネシウムで乾燥後、濾過、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーで精製し(溶離液;ヘキサン:クロロホルム=4:1)、3.02gの2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンを得た(収率53%)。
【0162】
4)2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンの合成
合成例5の3)で得た2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノン2.96g(3.55mmol)にTHF40mlを加えた後、水素化ジイソブチルアルミニウム(関東化学製、0.99M)トルエン溶液9.0ml(8.9mmol)を添加し、室温で2時間撹拌した。氷冷後、6N塩酸24mlを添加した後、65℃に加熱し、4時間反応を行った。トルエン及び食塩水を添加し、分相した。さらに有機相を食塩水で洗浄し、減圧濃縮及び真空乾燥した。得られた残渣に、水素化ジイソブチルアルミニウムを用いた還元、6N塩酸による脱水操作をさらに2回繰り返した。粗生成物をトルエンから再結晶精製し、2.27gの薄黄色固体である2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンを得た(収率80%)。
【0163】
合成例6 (3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニル(一般式(2)のテトラハロターフェニル誘導体)の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例5で合成した2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセン210mg(0.261mmol)及びTHF10mlを添加した。この溶液を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.78M)のTHF溶液0.73ml(0.57mmol)を滴下した。60分間かけて−40℃まで昇温することで熟成させた後、−83℃に冷却し、トリメトキシボラン(和光純薬工業製)67.7mg(0.651mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた3−ブロモ−6,7−ジペンタデシルアントラセニル−2−ボロン酸(一般式(6)及び(7)の化合物)に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン50.7mg(0.104mmol)(一般式(5)の化合物)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)7.6mg(0.0066mmol)、トルエン3.3ml、及びエタノール0.8mlを添加した。さらに炭酸ナトリウム66.4mg(0.626mmol)と水1.0mlからなる水溶液を加え、60℃で78時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーで精製した(溶離液:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の薄黄色固体43.7mgを得た(収率26%)。
1H NMR(CDCl3,23℃):δ=8.35(s,2H),8.33(s,2H),8.28(s,2H),7.96(s,1H),7.92(s,1H),7.79(s,2H),7.78(s,2H),7.72(s,1H),7.71(s,1H),2.81(m,8H),1.71(m,8H),1.28(m,96H),0.88(m,12H)。
FABMS m/z: 1587(M+,100%),1507(M+−Br,10)。
【0164】
1H NMR及びMS測定より、3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0165】
【化45】
【0166】
実施例4 (テトラペンタデシル)ジアントラジチエノアセン(一般式(1)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例6で合成した3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニル42.8mg(0.027mmol)及びジエチルエーテル6mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.57M)のヘキサン溶液0.10ml(0.15mmol)を滴下した。60分間かけて5℃まで昇温することで熟成させた後、−70℃に冷却し、ビス(フェニルスルホニル)スルフィド(アクロス製)23.6mg(0.075mmol)(一般式(3)及び(4)の化合物)を一気に投入した。徐々に昇温し、一晩かけて室温まで反応温度を上げた。飽和食塩水及びトルエンを添加した後、固体を濾過し、水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的物の黄色結晶12mgを得た(収率33%)。
1H NMR(重トルエン,100℃):δ=8.50(s,2H),8.43(s,2H),8.26(s,2H),8.20(s,2H),8.14(s,2H),7.85(s,2H),7.78(s,2H),2.87(t,J=7.8Hz,8H),1.77(m,8H),1.31(m,96H),0.88(m,J=7.8Hz,12H)。
【0167】
1H NMR測定より、(テトラペンタデシル)ジアントラジチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0168】
【化46】
【0169】
合成例7 (2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンの合成)
2−ジウンデシルベンゼンの合成
1)1,2−ジウンデシルベンゼンの合成
n−ペンタデシルマグネシウムブロマイド(シグマ−アルドリッチ製、0.5M)のジエチルエーテル溶液の代わりに、n−ウンデシルマグネシウムブロマイドのジエチルエーテル溶液(ジエチルエーテル中、1−ブロモウンデカン(和光純薬工業製)とマグネシウムから調製)を用いた以外は合成例5の1)と同じ操作を繰り返して1,2−ジウンデシルベンゼンを収率87%で合成した。
【0170】
2)2−ブロモ−3−ヨード−6,7−ジウンデシルアントラキノンの合成
1,2−ジペンタデシルベンゼンの代わりに、合成例7の1)で合成した1,2−ジウンデシルベンゼンを用いた以外は合成例5の3)と同じ操作を繰り返して2−ブロモ−3−ヨード−6,7−ジウンデシルアントラキノンを収率65%で合成した。
【0171】
3)2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンの合成
2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンの代わりに、合成例7の2)で合成した2−ブロモ−3−ヨード−6,7−ジウンデシルアントラキノンを用いた以外は合成例5の4)と同じ操作を繰り返して2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンを収率87%で合成した。
【0172】
合成例8 (3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニル(一般式(2)のテトラハロターフェニル誘導体)の合成)
2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンの代わりに、合成例7で合成した2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンを用いた以外は合成例6と同じ操作を繰り返して3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニルを収率58%で合成した。
1H NMR(CDCl3,23℃):δ=8.36(s,2H),8.34(s,2H),8.28(s,2H),7.96(s,1H),7.91(s,1H),7.79(s,2H),7.78(s,2H),7.71(s,1H),7.70(s,1H),2.81(m,8H),1.71(m,8H),1.28(m,64H),0.88(m,12H)。
FABMS m/z: 1363(M+,100%),1283(M+−Br,12)。
【0173】
1H NMR及びMS測定より、3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0174】
【化47】
【0175】
実施例5 (テトラウンデシル)ジアントラジチエノアセン(一般式(1)のヘテロアセン誘導体の合成)
3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニルの代わりに合成例8で合成した3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニルを用いた以外は実施例4と同じ操作を繰り返して、(テトラウンデシル)ジアントラジチエノアセンを収率39%で合成した。
1H NMR(重トルエン,100℃):δ=8.49(s,2H),8.44(s,2H),8.25(s,2H),8.20(s,2H),8.13(s,2H),7.85(s,2H),7.79(s,2H),2.87(t,J=7.8Hz,8H),1.77(m,8H),1.31(m,64H),0.88(m,J=7.8Hz,12H)。
【0176】
1H NMR測定より、(テトラウンデシル)ジアントラジチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0177】
【化48】
【0178】
実施例6 (耐酸化性有機半導体材料の合成及びその耐酸化性評価)
窒素雰囲気下、100mlシュレンク容器にトルエン5.4gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ実施例1で得られた(ジドデシル)ジベンゾテトラチエノアセンの固体5.1mgを添加し、100℃に加熱し溶解させ、(ジドデシル)ジベンゾテトラチエノアセンを含む耐酸化性有機半導体材料を合成した(山吹色溶液)。次に、このシュレンク容器の上部の栓を開け、1分間、外気に接触させることで空気を導入(耐酸化性評価)し、さらに90℃で撹拌したが、色の変化は見られなかった。したがって、色の変化が見られなかったことから、耐酸化性に優れるものであった。
【0179】
実施例7 (有機薄膜の作製)
窒素雰囲気下、実施例1で得られた(ジドデシル)ジベンゾテトラチエノアセン2.5mgをトルエン25gと混合し、100℃で1時間撹拌し、(ジドデシル)ジベンゾテトラチエノアセンの山吹色溶液を調製した(ジドデシル)ジベンゾテトラチエノアセンを含む耐酸化性有機半導体材料の合成)。
【0180】
窒素雰囲気下、凹面のあるガラス基板を90℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚260nmの有機薄膜を作製した。
【0181】
実施例8 (耐酸化性有機半導体材料の合成及びその耐酸化性評価)
窒素雰囲気下、100mlシュレンク容器にトルエン5.4gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ実施例4で得られた(テトラペンタデシル)ジアントラジチエノアセンの固体6.2mgを添加し、70℃に加熱し溶解させ、(テトラペンタデシル)ジアントラジチエノアセンを含む耐酸化性有機半導体材料を合成した(黄橙色溶液)。次に、このシュレンク容器の上部の栓を開け、1分間、外気に接触させることで空気を導入(耐酸化性評価)し、さらに70℃で撹拌したが、色の変化は見られなかった。したがって、色の変化が見られなかったことから、耐酸化性に優れるものであった。
【0182】
さらにこの溶液を70℃。1時間、撹拌下で空気と接触させても溶液の色の変化は見られず、耐酸化性に優れるものであった。
【0183】
実施例9 (有機薄膜の作製)
窒素雰囲気下、実施例4で得られた(テトラペンタデシル)ジアントラジチエノアセン4.7mgをトルエン15gと混合し、100℃で1時間撹拌し、(テトラペンタデシル)ジアントラジチエノアセンの黄橙色溶液を調製した(テトラペンタデシル)ジアントラジチエノアセンを含む耐酸化性有機半導体材料の合成)。
【0184】
窒素雰囲気下、凹面のあるガラス基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚320nmの有機薄膜を作製した。
【0185】
比較例2 (耐酸化性評価)
ペンタセンを用いて耐酸化性を評価した。
【0186】
窒素雰囲気下、20mlシュレンク容器にo−ジクロロベンゼン2.9gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへペンタセン(東京化成工業製)2.5mgを添加し、120℃に加熱し溶解させると赤紫色溶液となった。次にこのシュレンク容器の上部の栓を開け、1分間、外気に接触させることで空気を導入し、さらに120℃で撹拌した。ガスクロマトグラフィー及びガスクロマトグラフィー−マススペクトル(GCMS)分析から、6,13−ペンタセンキノンが生成していることがわかった。
【0187】
さらにこの溶液を120℃、1時間、撹拌下で空気と接触させると溶液の色が黄に変化していた。ガスクロマトグラフィー分析から、6,13−ペンタセンキノンの生成が増加していることがわかった。
【0188】
したがって、溶液の色の変化及び6,13−ペンタセンキノンが生成していることから、酸化が進行しており、耐酸化性に劣るものであった。
【0189】
実施例10 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、実施例1で得られた(ジドデシル)ジベンゾテトラチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(ジドデシル)ジベンゾテトラチエノアセンの黄色溶液を調製した。
【0190】
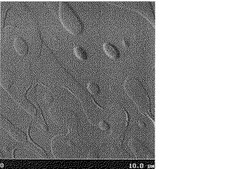
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは13nmであった。得られた原子間力顕微鏡写真を図1に示した。
【0191】
実施例11 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、実施例4で得られた(テトラペンタデシル)ジアントラジチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(テトラペンタデシル)ジアントラジチエノアセンの黄色溶液を調製した。
【0192】
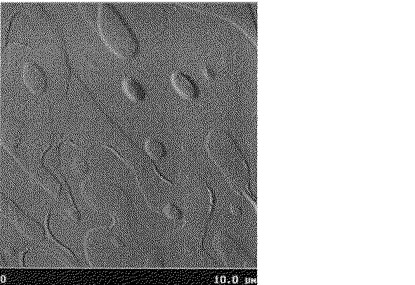
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板を165℃に加熱し、上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは26nmであった。得られた原子間力顕微鏡写真を図2に示した。
【0193】
実施例12 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、(テトラドデシル)ジアントラジチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(テトラドデシル)ジアントラジチエノアセンの黄色溶液を調製した。
【0194】
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板を165℃に加熱し、上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは31nmであった。得られた原子間力顕微鏡写真を図3に示した。
【0195】
比較例3 ((ジペンチル)ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体においてR5、R7、及びR8が同時に水素であり、R6が炭素数5のアルキル基である化合物)の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、1,4−ビス(6−ペンチル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返して(ジペンチル)ジベンゾテトラチエノアセンを収率18%で合成した。
MS m/z: 542(M+,100%),485(M+−C4H9−1,26%)。
【0196】
MS測定より、(ジペンチル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0197】
【化49】
【0198】
比較例4 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、比較例3で得られた(ジペンチル)ジベンゾテトラチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(ジペンチル)ジベンゾテトラチエノアセンの黄色溶液を調製した。
【0199】
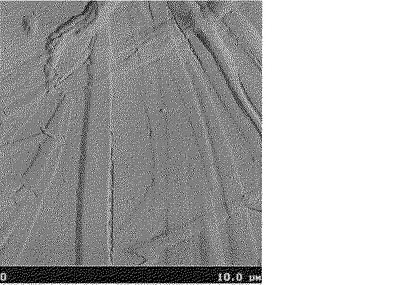
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板を165℃に加熱し、上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは142nmであった。得られた原子間力顕微鏡写真を図4に示した。
【図面の簡単な説明】
【0200】
【図1】実施例10で得られた(ジドデシル)ジベンゾテトラチエノアセンの原子間力顕微鏡写真
【図2】実施例11で得られた(ジペンタデシル)ジベンゾテトラチエノアセンの原子間力顕微鏡写真
【図3】実施例12参考例1で得られた(テトラドデシル)ジアントラジチエノアセンの原子間力顕微鏡写真
【図4】比較例4で得られた(テトラペンチル)ジアントラジチエノアセンの原子間力顕微鏡写真
【技術分野】
【0001】
本発明は、有機半導体等の電子材料への展開が可能なヘテロアセン誘導体及びその用途に関する。
【背景技術】
【0002】
有機薄膜トランジスタに代表される有機半導体デバイスは、省エネルギー、低コスト及びフレキシブルといった無機半導体デバイスにはない特徴を有することから近年注目されるようになった。この有機半導体デバイスは有機半導体活性相、基板、絶縁相、電極等数種類の材料から構成されるが、中でも電荷のキャリアー移動を担う有機半導体活性相は該デバイスの中心的な役割を有している。この有機半導体活性相を構成する有機材料のキャリアー移動能により有機半導体デバイス性能が左右される。
【0003】
有機半導体活性相を作製する方法としては一般的に、高温真空下、有機材料を気化させて実施する真空蒸着法及び有機材料を適当な溶媒に溶解させその溶液を塗布する塗布法が知られている。塗布法においては、塗布は高温高真空条件を用いることなく印刷技術を用いても実施することができる。そのため、塗布法は印刷によりデバイス作製の大幅な製造コストの削減を図ることができることから、経済的に好ましいプロセスである。しかし、従来、有機半導体デバイスとして性能が高い材料ほど塗布法で有機半導体活性相を形成することが困難になるという問題があった。
【0004】
例えば、ペンタセン等の結晶性材料はアモルファスシリコン並みの高いキャリアー移動度を有し、優れた有機半導体デバイス特性を発現することが報告されている(例えば、非特許文献1参照)。又、ペンタセン等のポリアセンを溶解させ塗布法で有機半導体デバイスを製造する試みも報告されている(例えば、特許文献1参照)。しかしながら、ペンタセンはその強い凝集性のため溶解性が低く、塗布法を適用するためには高温加熱等の条件が必要とされ、さらにペンタセンの溶液は極めて容易に空気酸化されることから、塗布法の適用はプロセス的、経済的に困難を伴うものであった。また、ポリ−(3−ヘキシルチオフェン)等の自己組織化材料は溶媒に可溶であり、塗布法による有機半導体デバイス作製が報告されてはいるが、キャリアー移動度が結晶性化合物より1桁低いことから(例えば、非特許文献2参照)、得られた有機半導体デバイスの特性が低いという問題があった。
【0005】
またベンゼン環とチオフェン環が縮環したヘテロアセン誘導体は、ペンタセンに比べ耐酸化性が向上しているが、トルエン等の汎用溶媒に対して溶解度が低いという問題があった(例えば、特許文献2参照)。さらに、表面粗さについては記載されていない。
【0006】
【非特許文献1】「ジャーナル オブ アプライドフィジックス」、(米国)、2002年、92巻、5259−5263頁
【非特許文献2】「サイエンス」、(米国)、1998年、280巻、1741−1744頁
【特許文献1】WO2003/016599号
【特許文献2】特開2008−81494号
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明は上記の従来技術が有する問題点に鑑み、優れた耐酸化性を有し、塗布法による有機半導体活性相形成が可能な、ヘテロアセン誘導体及びそれを用いた耐酸化性有機半導体材料並びに表面粗さの小さな有機薄膜を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは上記課題を解決するため鋭意検討の結果、本発明の新規なヘテロアセン誘導体を見出した。加えて、該ヘテロアセン誘導体が耐酸化性に優れ、塗布法の適用が可能であるため結晶性の薄膜を容易に安定して作製することができることから、該ヘテロアセン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜を見出し、本発明を完成するに到った。
【0009】
以下に本発明を詳細に説明する。説明はヘテロアセン誘導体及びその製造方法、該ヘテロアセン誘導体の前駆体であるテトラハロターフェニル誘導体及びその製造方法、並びに該ヘテロアセン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜について述べる。
【0010】
(ヘテロアセン誘導体)
本発明のヘテロアセン誘導体は下記一般式(1)で示される。
【0011】
【化1】
【0012】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、炭素数4〜30のアリール基、炭素数6〜20のアルキル基、を示し、T1及びT2は同一又は異なって、硫黄、セレン、テルル、リン、ホウ素を示し、l及びmは、各々0又は1の整数であり、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有する。)
【0013】
【化2】
【0014】
【化3】
【0015】
(ここで、置換基R5〜R12は同一又は異なって、水素原子、フッ素原子、炭素数6〜20のアルキル基を示し、置換基T3は、硫黄、セレン、テルルを示し、nは0又は1の整数である。但し、置換基R5〜R12は同時に水素原子であることはなく、また環A及びBが(A−2)で示される構造である場合、何れか一方の(A−2)中のnは1である。))]
本発明の一般式(1)で示されるヘテロアセン誘導体の置換基について述べる。
【0016】
置換基R1〜R4における炭素数4〜30のアリール基は、特に限定はなく、例えばフェニル基、p−トリル基、p−(n−ヘキシル)フェニル基、p−(n−オクチル)フェニル基、p−(シクロヘキシル)フェニル基、m−(n−オクチル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(n−パーフルオロオクチル)フェニル基、2−チエニル基、5−(n−ヘキシル)−2−チエニル基、2,2’−ビチエニル−5−基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、アントラセニル基、2−フルオレニル基、9,9−ジメチル−2−フルオレニル基、1−ビフェニレノ基、2−ビフェニレノ基、ターフェニル基、2−ピリジル基、テトラフルオロピリジル基、ビピリジル基、(ジフェニルアミノ)フェニル基、(ジフェニルアミノ)ビフェニル基等を挙げることができ、好ましくはフェニル基、p−(n−オクチル)フェニル基、p−(n−パーフルオロオクチル)フェニル基、5−(n−ヘキシル)−2−チエニル基等である。
【0017】
置換基R1〜R4における炭素数6〜20のアルキル基は、特に限定はなく、例えばヘキシル基、へプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等のアルキル基;パーフルオロヘキシル基、パーフルオロヘプチル基、パーフルオロオクチル基、パーフルオロノニル基、パーフルオロデシル基、パーフルオロウンデシル基、パーフルオロドデシル基、パーフルオロトリデシル基、パーフルオロテトラデシル基、パーフルオロペンタデシル基、パーフルオロヘキサデシル基、パーフルオロヘプタデシル基、パーフルオロオクタデシル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくはアルキル基であり、特に好ましくはオクチル基、デシル基、ウンデシル基、トリデシル基、テトラデシル基、ペンタデシル基である。
【0018】
これらの置換基R1〜R4の中でも、特に水素原子、炭素数4〜30のアリール基が好ましく、さらに水素原子、フェニル基が好ましい。
【0019】
置換基T1及びT2は、硫黄、セレン、テルル、リン、ホウ素であり、その中でも好ましくは硫黄、セレン、リン、ホウ素であり、さらに好ましくは硫黄である。
【0020】
l及びmは、各々0又は1の整数である。ただし、置換基T1、T2が、硫黄、セレン、テルルの場合は、l、mは0であり、置換基T1、T2が、リン、ホウ素の場合は、l、mは1である。
【0021】
次に、一般式(A−1)及び(A−2)で示される、環A及びBについて述べる。
【0022】
本発明のヘテロアセン誘導体は、環A、Bを有する誘導体であり、環A,Bは一般式(A−1)又は(A−2)で示される構造を有するものである。
【0023】
置換基R5〜R12における炭素数6〜20のアルキル基は、特に限定はなく、例えばヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等のアルキル基;パーフルオロヘキシル基、パーフルオロヘプチル基、パーフルオロオクチル基、パーフルオロノニル基、パーフルオロデシル基、パーフルオロウンデシル基、パーフルオロドデシル基、パーフルオロトリデシル基、パーフルオロテトラデシル基、パーフルオロペンタデシル基、パーフルオロヘキサデシル基、パーフルオロヘプタデシル基、パーフルオロオクタデシル基等のパーフルオロアルキル基;ペンタデカフルオロオクチル基、オクタデカフルオロデシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくはアルキル基であり、特に好ましくはノニル基、デシル基、ウンデシル基、トリデシル基、テトラデシル基、ペンタデシル基、オクタデシル基である。
【0024】
置換基R5〜R12が炭素数5以下であるアルキル基を有するヘテロセン誘導体では、有機薄膜とした際に表面粗さが大きくなる。
【0025】
これらアルキル基においては、環(A−1)における置換基R5〜R8は、炭素数6〜20のアルキル基であり、特に炭素数7〜20のアルキル基が好ましく、環(A−2)における置換基R9〜R12は、炭素数6〜20のアルキル基であり、特に炭素数13〜20のアルキル基が好ましい。
【0026】
置換基T3は、硫黄、セレン、テルルであり、その中でも好ましくは硫黄である。
【0027】
nは0又は1の整数であり、好ましくは1である。また環A及びBが(A−2)で示される構造である場合、何れか一方の(A−2)中のnは1である。
【0028】
これらの中でも本発明の一般式(1)で示されるヘテロアセン誘導体は、該ヘテロアセン誘導体及び該ヘテロアセン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜が、高い耐酸化性及びキャリアー移動度を発現することから、以下の化合物が好ましく、
【0029】
【化4】
【0030】
【化5】
【0031】
【化6】
【0032】
【化7】
【0033】
【化8】
【0034】
【化9】
【0035】
【化10】
【0036】
【化11】
【0037】
【化12】
【0038】
【化13】
【0039】
【化14】
【0040】
【化15】
【0041】
【化16】
【0042】
【化17】
【0043】
【化18】
【0044】
【化19】
【0045】
特に好ましくは
【0046】
【化20】
【0047】
である。
【0048】
(テトラハロターフェニル誘導体)
次に、本発明の一般式(1)で示されるヘテロアセン誘導体の前駆化合物であるテトラハロターフェニル誘導体について述べる。
【0049】
本発明の一般式(1)で示されるヘテロアセン誘導体の前駆化合物であるテトラハロターフェニル誘導体は下記一般式(2)で示される。
【0050】
【化21】
【0051】
(ここで、置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、置換基R1及びR2並びに環A及びBは一般式(1)で示される置換基及び環と同意義を示す。)
置換基X1〜X4は臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子、ヨウ素原子であり、特に好ましくはいずれも臭素原子である。
【0052】
置換基R1及びR2は、一般式(1)で示される置換基と同意義を示し、その中でも特に水素原子が好ましい。
【0053】
環A及びBは一般式(1)で示される環と同意義を示す。すなわち、一般式(A−1)又は一般式(A−2)と同意義を示す。そして、その中でも一般式(A−1)においては、T3が硫黄であることが好ましい。
【0054】
本発明の一般式(2)で示されるテトラハロターフェニル誘導体としては、以下の化合物が好ましく、
【0055】
【化22】
【0056】
【化23】
【0057】
【化24】
【0058】
【化25】
【0059】
【化26】
【0060】
【化27】
【0061】
【化28】
【0062】
【化29】
【0063】
【化30】
【0064】
【化31】
【0065】
【化32】
【0066】
【化33】
【0067】
特に好ましくは
【0068】
【化34】
【0069】
である。
【0070】
(ヘテロアセン誘導体の製造方法)
本発明の一般式(1)で示されるヘテロアセン誘導体の製造方法について述べる。
【0071】
本発明の一般式(1)で示されるヘテロアセン誘導体は、一般式(2)で示されるテトラハロターフェニル誘導体をメタル化剤を用いてテトラメタル化し、下記一般式(3)及び/又は下記一般式(4)で示される反応剤と反応させることにより製造することができる。なお、一般式(3)、一般式(4)で示される反応剤が同じ化合物であっても良い。
(R3)lT1(L1)p (3)
(R4)mT2(L2)q (4)
(ここで、置換基T1、T2、R3、R4及び記号lとmは一般式(1)で示される置換基及び記号と同意義を示し、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、フェニルスルホニル基を示し、p及びqは0又は2の整数を示す。)
なお、ここでテトラメタル化とは、一般式(2)におけるX1〜X4をそれぞれメタルに置換することを意味する。
【0072】
一般式(2)で示されるテトラハロターフェニル誘導体をテトラメタル化する場合、用いるメタル化剤は、一般式(2)におけるX1〜X4をメタルに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属;メチルマグネシウムブロマイド、エチルマグネシウムブロマイド、イソプロピルマグネシウムクロライド、tert−ブチルマグネシウムクロライド、フェニルマグネシウムブロマイド等のグリニャール試薬;マグネシウム金属;亜鉛金属等を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウムである。
【0073】
該メタル化剤の使用量は一般式(2)で示されるテトラハロターフェニル誘導体1当量に対し、3〜20当量が好ましく、特に好ましくは4〜15当量、さらに好ましくは5〜10当量である。
【0074】
該テトラメタル化は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばテトラヒドロフラン(以下、THFと略す)、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジオキサン、トルエン、ヘキサン、シクロヘキサン等であり、特に好ましくはTHF、ジエチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。該テトラメタル化の温度は−100〜50℃で行うことが好ましく、特に好ましくは−90〜30℃である。反応時間は1〜120分が好ましく、特に好ましくは5〜60分である。なお、テトラメタル化の進行は、反応液の一部を取り出し、水で反応を停止させた後、ガスクロマトグラフィーで分析することで監視することができる。
【0075】
該テトラメタル化により生成したテトラメタル塩は、次いで一般式(3)及び一般式(4)で示される反応剤と反応させることにより、一般式(1)で示されるヘテロアセン誘導体が得られるものである。係る反応剤との反応は、前記テトラメタル化により生成したテトラメタル塩を含む反応混合物に前記反応剤を直接用いて反応させる方法、生成したテトラメタル塩を一度単離した後、前記反応剤と反応させる方法のいずれを用いてもよい。
【0076】
ここで、一般式(3)、一般式(4)における置換基T1、T2、R3、R4及び記号lとmは一般式(1)で示される置換基及び記号と同意義を示す。
【0077】
また、置換基L1、L2は塩素原子、臭素原子、ヨウ素原子、炭素数1〜20のオキシ基、アセトキシ基、アリールスルホニル基を示し、好ましくは塩素原子、臭素原子、炭素数1〜20のオキシ基、アリールスルホニル基である。炭素数1〜20のオキシ基は特に限定はなく、例えばメトキシ基、エトキシ基、n−ブトキシ基、フェノキシ基、(2−メトキシ)フェノキシ基等を挙げることができ、アリールスルホニル基は特に限定はなく、例えばフェニルスルホニル基、p−トリルスルホニル基等を挙げることができる。これらの中でも特にフェニルスルホニル基が好ましい。
【0078】
そして、具体的な一般式(3)、一般式(4)で示される反応剤としては、例えば2塩化硫黄;2臭化硫黄;ビス(フェニルスルホニル)スルフィド、ビス(p−トリルスルホニル)スルフィド等のビス(アリールスルホニル)スルフィド類;硫黄;2塩化セレン;セレン;2塩化テルル;テルル;ジクロロフェニルホスフィン、ジメトキシフェニルホスフィン、ジフェノキシフェニルホスフィン、ジクロロ{4−(n−オクチル)フェニル}ホスフィン等のアリールホスフィン類;ジクロロ(n−ヘキシル)ホスフィン、ジクロロ(n−オクチル)ホスフィン、ジメトキシ(n−ヘキシル)ホスフィン等のアルキルホスフィン類;ジクロロフェニルボラン、ジメトキシフェニルボラン、ジメトキシ{4−(n−ヘキシル)フェニル}ボラン、ジフェノキシフェニルボラン、ジクロロ{4−(n−オクチル)フェニル}ボラン等のアリールボラン類;ジクロロ(n−ヘキシル)ボラン、ジクロロ(n−オクチル)ボラン、ジメトキシ(n−ヘキシル)ボラン等のアルキルボラン類等を挙げることができ、好ましくはビス(フェニルスルホニル)スルフィド、ジクロロフェニルホスフィン、ジクロロフェニルボラン等である。
【0079】
テトラメタル化により生成したテトラメタル塩と一般式(3)及び一般式(4)で示される反応剤と反応させる際には、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン、トルエン、ヘキサン、シクロヘキサン等であり、好ましくはTHF、ジエチルエーテルである。用いる反応剤の量は、一般式(2)で示されるテトラハロターフェニル誘導体1当量に対し、1.2〜10当量が好ましく、特に好ましくは2〜8当量である。該反応剤との反応温度は−100〜50℃が好ましく、特に好ましくは−90〜30℃であり、反応時間は0.5〜30時間が好ましく、特に好ましくは1〜18時間である。
【0080】
本発明の一般式(1)で示されるヘテロアセン誘導体の製造は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0081】
本発明の一般式(1)で示されるヘテロアセン誘導体の製造方法では、一般式(2)で示されるテトラハロターフェニル誘導体をテトラメタル化した後、塩化マグネシウムと反応させ、その後に一般式(3)及び一般式(4)で示される反応剤で処理することもできる。
【0082】
かくして得られた、本発明の一般式(1)で示されるヘテロアセン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0083】
(テトラハロターフェニル誘導体の製造方法)
次に、本発明の一般式(1)で示されるヘテロアセン誘導体の前駆体として用いられる一般式(2)で示されるテトラハロターフェニル誘導体の製造方法について述べる。
【0084】
本発明の一般式(2)で示されるテトラハロターフェニル誘導体は下記一般式(5)で示されるテトラハロベンゼンと下記一般式(6)及び/又は下記一般式(7)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させることにより製造することができる。なお、一般式(6)、一般式(7)で示される反応剤が同じ化合物であっても良い。
【0085】
【化35】
【0086】
(ここで、置換基X5及びX6は臭素原子、ヨウ素原子、塩素原子を示す。置換基R1、R2、X2及びX3は一般式(2)で示される置換基と同意義を示す。)
【0087】
【化36】
【0088】
(ここで、M1はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X1並びに環Aは、一般式(2)で示される置換基並びに環と同意義を示す。)
【0089】
【化37】
【0090】
(ここで、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物を示し、置換基X4並びにB環は、一般式(2)で示される置換基並びに環と同意義を示す。)
本発明の一般式(5)、(6)及び(7)について、さらに述べる。
【0091】
一般式(5)の置換基X5及びX6は、臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子及びヨウ素原子であり、さらに好ましくはヨウ素原子である。
【0092】
置換基R1、R2、X2及びX3は、一般式(2)で示される置換基と同意義を示す。
【0093】
そして、具体的な一般式(5)で示される化合物としては、例えば1,4−ジブロモ−2,5−ジヨードベンゼンが挙げられる。
【0094】
一般式(6)、(7)の置換基M1、M2はマグネシウム、ホウ素、亜鉛、錫、ケイ素のハロゲン化物;ハイドロオキサイド;アルコキサイド;アルキル化物であり、上記のパラジウム及び/又はニッケル触媒により脱離され、パラジウム及び/又はニッケルと置換できる基である限り特に限定はなく、例えばMgCl、MgBr、B(OH)2、B(OMe)2、テトラメチルジオキサボロラニル基、ZnCl、ZnBr、ZnI、Sn(Bu−n)3、Si(Me)3、Si(Bu−n)3、Si(OMe)3、Si(OEt)3等を挙げることができ、好ましくはZnCl、B(OH)2、Si(OMe)3である。
【0095】
置換基X1、X4並びに環A、Bは、一般式(2)で示される置換基並びに環と同意義を示す。
【0096】
そして、具体的な一般式(6)、一般式(7)で示される化合物としては、例えば3−ブロモ−6−ドデシルベンゾチエニル−2−ジンククロライド、3−ブロモ−6−ドデシルベンゾチエニル−2−トリメトキシシラン、3−ブロモ−6,7−ジペンタデシルアントラセニル−2−ボロン酸、3−ブロモ−6,7−ペンタデシル−9,10−ジフェニルアントラセニル−2−ボロン酸等が挙げられる。
【0097】
なお、一般式(6)、一般式(7)で示される2−ハロアリール金属試薬は、例えば、それらの原料となるアリールジハロゲン置換体をイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬によりハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン、トリ(イソプロポキシ)ボラン、2−イソプロポキシ−4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン、テトラメトキシシラン等と反応させることで好適に調製することができる。また、一般式(6)、(7)の置換基M1、M2がSi(OMe)3又はSi(OEt)3の場合、それらの原料となるアリールジハロゲン置換体とPd又はRh触媒を用いたトリアルコキシシランとの反応によっても調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁」に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている方法を用いることもできる。
【0098】
一般式(5)で示されるテトラハロベンゼンと一般式(6)及び/又は一般式(7)で示される2−ハロアリール金属試薬の反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ジクロロビス(トリフェニルホスフィン)パラジウム、ビス(トリ−tert−ブチルホスフィン)パラジウム、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/トリ−tert−ブチルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等のパラジウム触媒;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物、ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等のニッケル触媒;を挙げることができる。中でも、好ましい触媒は0価のパラジウム化合物であり、特に好ましい触媒はテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。又、これら触媒は1種若しくは2種以上の混合物を用いても良い。
【0099】
一般式(5)で示されるテトラハロベンゼンと一般式(6)及び/又は一般式(7)で示される2−ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させる際には、好ましくは溶媒中で実施する。用いる溶媒に特に限定はなく、例えばTHF、ジエチルエーテル、メチル−tert−ブチルエーテル、エチル−tert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水、N,N−ジメチルホルムアミド、N−メチルピロリドン、トリエチルアミン、ピペリジン、ピロリジン、ジイソプロピルアミン等を挙げることができ、又、これら溶剤は1種若しくは2種以上の混合物を用いても良く、例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0100】
パラジウム触媒、ニッケル触媒の使用量は一般式(5)で示されるテトラハロベンゼン1モルに対し、0.1〜20モル%が好ましく、特に好ましくは0.5〜10モル%の範囲である。
【0101】
一般式(6)、一般式(7)で示される2−ハロアリール金属試薬の使用量は一般式(5)で示されるテトラハロベンゼン1当量に対し、0.8〜3.2当量が好ましく、特に好ましくは1.0〜2.8当量、さらに好ましくは1.1〜2.5当量である。
【0102】
反応の際の温度は10〜120℃が好ましく、さらに好ましくは30〜100℃、特に好ましくは40〜90℃であり、反応時間は1〜100時間が好ましく、特に好ましくは2〜85時間である。
【0103】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、りん酸カリウム、りん酸ナトリウム、ナトリウムtert−ブトキサイド、フッ化カリウム、フッ化ナトリウム、フッ化セシウム等の無機塩基;トリエチルアミン、トリメチルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン、テトラブチルアンモニウムフルオライド等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(5)で示されるテトラハロベンゼン1当量に対し、0.5〜10.0当量が好ましく、特に好ましくは2.0〜8.0当量である。さらにこれらの塩基と併用し、相間移動触媒を用いることもできる。相間移動触媒の種類は特に限定はなく、例えばトリオクチルメチルアンモニウムクロライド、テトラブチルアンモニウムクロライド、セチルピリジニウムクロライド、ベンジルトリメチルアンモニウムクロライド等を好適なものとして挙げることができる。これらの相間移動触媒の使用量は一般式(5)で示されるテトラハロベンゼン1当量に対し、0.1〜1.5当量が好ましく、特に好ましくは0.1〜0.8当量である。
【0104】
さらに反応系中にトリフェニルホスフィン等のホスフィンを存在させることもできる。これらのホスフィンの使用量は、該パラジウム及び/又はニッケル触媒1当量に対し、0.9〜8.0当量が好ましく、特に好ましくは1.0〜3.0当量である。
【0105】
なお、反応系中に銅化合物を存在させることもできる。該銅化合物しては特に限定はなく、例えば塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、酢酸銅(I)等の1価銅;塩化銅(II)、臭化銅(II)、ヨウ化銅(II)、酢酸銅(II)、アセチルアセトナート銅(II)等の2価銅等を挙げることができる。その中でも好ましくは1価銅であり、特に好ましくはヨウ化銅(I)である。これらの銅化合物の使用量は該パラジウム及び/又はニッケル触媒1当量に対し、0.3〜10.0当量が好ましく、特に好ましくは0.6〜6.0当量である。
【0106】
また、一般式(5)で示されるテトラハロベンゼンと一般式(6)及び/又は(7)で示される2−ハロアリール金属試薬の反応により炭素−炭素結合が形成される位置はハロゲンの種類により制御することができる。
【0107】
即ち、ヨウ素原子の反応性が最も高く、臭素原子、塩素原子の順に反応性が低下することから、これらハロゲンの種類の反応性を利用することで反応する位置を任意に決めることができる。そのため、一般式(2)で示されるテトロハロターフェニル誘導体の製造は、例えば一般式(5)のX5及びX6をヨウ素原子とし、X2及びX3を臭素原子及び/又は塩素原子とすることにより、製造することができる。
【0108】
かくして得られた、本発明の一般式(2)で示されるテトラハロターフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0109】
(耐酸化性有機半導体材料)
次に、本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料について述べる。該耐酸化性有機半導体材料は溶剤への溶解性、耐酸化性に優れ、好適な塗布性を有する。該耐酸化性有機半導体材料は本発明の一般式(1)で示されるヘテロアセン誘導体を溶剤に溶解することにより製造することができる。
【0110】
耐酸化性有機半導体材料としては、本発明の一般式(1)で示されるヘテロアセン誘導体の中でも溶解性に優れたものとなることから、環(A−1)における置換基R5〜R8が炭素数7〜20のアルキル基であるもの又は環(A−2)における置換基R9〜R12が炭素数13〜20のアルキル基であるものが好ましい。
【0111】
本発明の一般式(1)で示されるヘテロアセン誘導体の溶解に用いる溶剤は、特に限定はなく、例えばo−ジクロロベンゼン、クロロベンゼン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、クロロホルム等のハロゲン系溶剤;THF、ジオキサン等のエーテル系溶剤;トルエン、キシレン、メシチレン、エチルベンゼン、n−プロピルベンゼン、n−ヘプチルベンゼン、n−オクチルベンゼン、n−ノニルベンゼン等の芳香族化合物の炭化水素系溶剤;酢酸エチル、γ−ブチロラクトン等のエステル系溶剤;N,N−ジメチルホルムアミド、N−メチルピロリドン等のアミド系溶剤;等が挙げられる。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。中でも、好ましくはトルエンである。
【0112】
上記に挙げた溶剤と一般式(1)で示されるヘテロアセン誘導体を混合攪拌することにより、一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料となるものである。混合攪拌する際の温度は10〜200℃が好ましく、特に好ましくは20〜190℃である。混合攪拌する際の一般式(1)で示されるヘテロアセン誘導体の濃度は、溶剤及び温度により変えることができ、0.01〜10.0重量%であることが好ましい。溶液の調製は空気中でも実施することができるが、好ましくは窒素、アルゴン等の不活性雰囲気下で調製する。
【0113】
一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料の耐酸化性の評価は、該溶液を所定時間、空気と接触させる方法で実施することができる。まず用いる溶剤は予め脱気しておき、溶存酸素を除去する。空気との接触時間は、温度により適宜選択することができ、0.5分〜3時間が好適である。酸化の進行は、溶液の色の変化並びにガスクロマトグラフィー及びガスクロマトグラフィー(GC)−マススペクトル(GCMS)分析による酸化物の検出により行うことができる。
【0114】
本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料は、用いられる一般式(1)で示されるヘテロアセン誘導体自体が適度の凝集性を有することから比較的に低温で溶剤へ溶解でき、且つ耐酸化性があることから、塗布法による有機薄膜の製造に好適に適用できる。即ち、雰囲気から厳密に空気を除く必要がないことから塗布工程を簡略化することができる。塗布は空気中でも実施できるが、好ましくは溶剤の乾燥を考慮して窒素気流下で行う。なお、好適な塗布性を得るために、本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料の粘度は、0.005〜20ポアズの範囲にあることが好ましい。
【0115】
(有機薄膜)
次に本発明の一般式(1)で示されるヘテロアセン誘導体を含む耐酸化性有機半導体材料を用いた有機薄膜について述べる。係る有機薄膜は上記の耐酸化性有機半導体材料(溶液)の再結晶化若しくは基板への塗布により製造することができ、特に基板への塗布により製造することが好ましい。そして、基板への塗布により製造することにより、基板上に形成される有機薄膜となるものである。
【0116】
有機薄膜としては、本発明の一般式(1)で示されるヘテロアセン誘導体の中でも表面粗さの小さい有機薄膜となることから、環(A−1)における置換基R5〜R8が炭素数7〜20のアルキル基であるもの又は環(A−2)における置換基R9〜R12が炭素数13〜20のアルキル基であるものが好ましい。
【0117】
再結晶化による薄膜は、前記耐酸化性有機半導体材料を冷却することで形成することができる。有機薄膜を製造する時の雰囲気は、窒素、アルゴン等の不活性ガス又は空気下で行うことが好ましく、特に窒素、アルゴン等の不活性ガス下で行うことが好ましい。該溶液中の一般式(1)で示されるヘテロアセン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%である。冷却は60〜200℃の温度から−20〜60℃が好ましく、特に好ましくは−10℃〜40℃の間に冷却することにより好適に実施することができる。またこのようにして製造した結晶状の有機薄膜を適当な基板の上に張り合わせる、即ちラミネーション等により基板上に製造することもできる。再結晶化により得られる有機薄膜の膜厚は特に限定はなく、好ましくは50nm〜2mm、特に好ましくは1〜500μmである。
【0118】
基板への塗布による有機薄膜の製造は、前記耐酸化性有機半導体材料を基板上に塗布した後、加熱、気流及び自然乾燥等の方法により溶剤を気化させることで実施することができる。該溶液中の一般式(1)で示されるヘテロアセン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%であることが好ましい。塗布温度は特に限定はなく、例えば20〜200℃の間で好適に実施することができる。塗布の具体的方法は特に限定はなく、公知の方法、例えばスピンコート、キャストコート及びディップコート等を用いることができる。さらにスクリーン印刷、インクジェット印刷、グラビア印刷等の印刷技術を用いても作製することが可能である。使用する基板の材料は特に限定はなく、結晶性、非結晶性の種々の材料を用いることができる。基板の具体例としては、例えばポリエチレンテレフタレート、ポリエチレンナフタレート、ポリメチルメタクリレート、ポリエチレン、ポリプロピレン、ポリスチレン、環状ポリオレフィン、ポリイミド、ポリカーボネート、ポリビニルフェノール、ポリビニルアルコール、ポリ(ジイソプロピルフマル酸)、ポリ(ジエチルフマル酸)、ポリ(ジイソプロピルマレイン酸)等のプラスチック基板;ガラス、石英、酸化アルミニウム、シリコン、酸化シリコン、二酸化タンタル、五酸化タンタル、インジウム錫酸化物等の無機材料基板;金、銅、クロム、チタン等の金属基板を好適に用いることができる。またこれらの基板の表面は、例えばオクタデシルトリクロロシラン、オクタデシルトリメトキシシラン、β−フェネチルトリクロロシラン等のシラン類;ヘキサメチルジシラザン等のシリルアミン類で修飾処理したものであっても使用することができる。さらに、基板は絶縁性あるいは誘電性を有する材料であっても良い。塗布した後の溶剤の乾燥は、常圧若しくは減圧で除去することができる、又、加熱、窒素気流により乾燥してもよい。さらに、溶剤の気化速度を調節することで本発明の一般式(1)で示されるヘテロアセン誘導体の結晶成長を制御することができる。基板への塗布により得られる有機薄膜の膜厚は特に限定はなく、好ましくは1nm〜100μm、特に好ましくは10nm〜20μmである。
【0119】
有機薄膜のキャリアー移動度は、有機薄膜の表面粗さと密接な関係があり、表面粗さが小さい程、高いキャリアー移動度を期待することができる。本発明の一般式(1)で示されるヘテロアセン誘導体からなる有機薄膜の表面粗さは、40nm以下が好ましく、特に好ましくは30nm以下であり、表面の平滑性が高いことから、高いキャリアー移動度を期待することができる。
【0120】
本発明の一般式(1)で示されるヘテロアセン誘導体は平面剛直性の高い分子構造を有することから、優れた半導体特性を与えることが期待できる。又、該ヘテロアセン誘導体はトルエンあるいはn−オクチルベンゼン等の溶媒に溶解し、溶液状態にあっても容易に空気酸化されることはない。従って、塗布法により半導体薄膜を容易に作成できる。したがって、本発明の一般式(1)で示されるヘテロアセン誘導体は、電子ペーパー、有機ELディスプレイ、液晶ディスプレイ、ICタグ用等のトランジスタの有機半導体活性相用途;有機ELディスプレイ材料;有機半導体レーザー材料;有機薄膜太陽電池材料;フォトニック結晶材料等の電子材料に利用することができる。
【発明の効果】
【0121】
優れた耐酸化性を有し、塗布法による有機半導体活性相形成が可能な、ヘテロアセン誘導体及びその用途を提供する。さらに、本発明のヘテロアセン誘導体からなる有機薄膜はその表面粗さが40nmと小さいことから、高いキャリアー移動度を有することが期待できる。
【実施例】
【0122】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例に限定されるものではない。
【0123】
生成物の同定には1H−NMRスペクトル及びマススペクトルを用いた。なお、1H−NMRスペクトルの測定は日本電子製JEOL GSX−270WB(270MHz)を用いた。マススペクトル(MS)は日本電子製JEOL JMS−700を用いて、試料を直接導入し、電子衝突(EI)法(70エレクトロンボルト)あるいはFAB法(6キロエレクトロンボルト、キセノンガス、マトリックス(ジチオスレイトール:ジチオエリスリトール=3:1))(FABMS)で測定した。
【0124】
反応の進行の確認等はガスクロマトグラフィー(GC)及びガスクロマトグラフィー−マススペクトル(GCMS)分析を用いた。
【0125】
ガスクロマトグラフィー分析
装置 島津GC14B
カラム J&Wサイエンティフィック社製、DB−1,30m
ガスクロマトグラフィー−マススペクトル分析
装置 パーキンエルマーオートシステムXL(MS部;ターボマスゴールド)
カラム J&Wサイエンティフィック社製、DB−1,30m
反応用の試薬及び溶媒は、断りのない限り市販品を用いた。なお、グリニャール試薬あるいはブチルリチウム等の有機金属試薬を用いた場合は、市販の脱水溶媒をそのまま用いた。
【0126】
有機薄膜の表面粗さは原子間力顕微鏡を用いて測定した。なお、原子間力顕微鏡はディジタルインスツルメンツ製のナノスコープIIIa D3100を用いた。
【0127】
合成例1 (1,4−ジブロモ−2,5−ジヨードベンゼンの合成)
1,4−ジブロモ−2,5−ジヨードベンゼンはジャーナル オブ アメリカン ケミカル ソサイエティー、1997年、119巻、4578−4593頁に記載されている方法を参考に合成を行った。
【0128】
メカニカルスターラー付き1lの三口フラスコに過ヨウ素酸16.7g(73.0mmol)及び硫酸525mlを加えた。過ヨウ素酸が溶解した後、ヨウ化カリウム36.4g(219mmol)を少しずつ添加した。その内容物の温度を−30℃に冷却し、1,4−ジブロモベンゼン34.5g(146mmol)を5分間かけて添加した。得られた混合物を−25℃で36時間撹拌した。反応混合物を氷(2kg)中へ注いだ後、濾過し固体を取り出した。その固体をクロロホルムに溶解させ、5%苛性ソーダ水溶液及び水で洗浄し、有機相を無水硫酸マグネシウムで乾燥した。減圧濃縮後、残渣をクロロホルムから再結晶化し、白色結晶を得た(36.0g、収率50%)。
1H−NMR(CDCl3,21℃):δ=8.02(s,2H)。
【0129】
1H−NMRスペクトルが文献値と一致したことより、1,4−ジブロモ−2,5−ジヨードベンゼンが得られたことを確認した。
【0130】
合成例2 [1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成(一般式(2)のテトラハロターフェニル誘導体の合成)]
1)2,3−ジブロモ−6−ドデカノイルベンゾチオフェンの合成
窒素雰囲気下、300ml二口ナスに2,3−ジブロモベンゾチオフェン(シグマ−アルドリッチ製)10.33g(35.37mmol)及びジクロロメタン(脱水品)150mlを加えた。−25℃に冷却した後、ドデカノイルクロライド(和光純薬工業製)9.00ml(8.28g、37.8mmol)、次いで塩化アルミニウム(和光純薬工業製)5.05g(37.8mmol)を添加した。−25℃で2日間反応させた後、水を加えて反応を停止させた。分相し、有機相に飽和炭酸ナトリウム水溶液を添加し、一昼夜撹拌した(過剰のドデカン酸除去のため)。分相、水洗し、有機相を無水硫酸ナトリウムで乾燥した。濾過減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィーで精製した。溶離液をヘキサンとすることで、未反応の2,3−ジブロモベンゾチオフェン及びトリブロモベンゾチオフェンが溶出し、溶離液をヘキサン:トルエン=10:1とすると異性体である2,3−ジブロモ−4−ドデカノイルベンゾチオフェンが溶出し、溶離液をヘキサン:トルエン=2:1とすると目的物を含む成分が溶出した。この最後の成分をヘプタン30mlで再結晶精製し、目的物である2,3−ジブロモ−6−ドデカノイルベンゾチオフェンの白色固体4.39g(9.25mmol)得た(収率26%)。
1H NMR(CDCl3,23℃):δ=8.35(s,1H),8.01(dd,J=8.5Hz,1.4Hz,1H),7.80(d,J=8.4Hz,1H),3.03(t,J=7.6Hz,2H),1.77(m,2H),1.26(m,16H),0.88(t、J=8.1Hz,3H)。
MS m/z: 474(M+,11%),394(M+−Br,2%),334(M+−C10H21+1,100%),319(M+−C11H23,58%)。
【0131】
2)2,3−ジブロモ−6−ドデシルベンゾチオフェンの合成
窒素雰囲気下、100mlシュレンク反応容器に合成例2の1)で合成した2,3−ジブロモ−6−ドデカノイルベンゾチオフェン3.85g(8.12mmol)及びトリフルオロ酢酸(和光純薬工業製)6.3mlを添加した。氷冷後、トリエチルシラン(信越化学製)2.9mlを滴下した。その後、40℃で6時間反応後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製した後(溶離液;ヘキサン)、目的物を含むフラクションを130パスカル、90℃で真空加熱し、低沸分を除去した。残渣に2,3−ジブロモ−6−ドデシルベンゾチオフェンの白色固体を3.35g(7.28mmol)得た(収率90%)。
1H NMR(CDCl3,23℃):δ=7.63(d,J=8.1Hz,1H),7.50(d,J=1.1Hz,1H),7.24(dd,J=8.1Hz,1.4Hz,1H),2.71(t,J=8.1Hz,2H),1.65(m,2H),1.25(m,18H),0.88(t、J=8.1Hz,3H)。
MS m/z: 460(M+,65%),380(M+−Br,4%),305(M+−C11H23,100%)。
【0132】
3)1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成
100mlシュレンク反応容器に合成例2の2)で合成した2,3−ジブロモ−6−ドデシルベンゾチオフェン984mg(2.13mmol)及びTHF24mlを添加した。−74℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.78M)のTHF溶液5.5ml(4.3mmol)を滴下した。−74℃、15分間、グリニャール化の熟成後、−85℃とし、塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液4.3ml(4.3mmol)を添加した。1時間熟成後、冷バスを外し、室温で30分間反応後、減圧濃縮した。得られた白色固体[(3−ブロモ−6−ドデシルベンゾチエニル−2−ジンククロライド)(一般式(6)及び(7)の化合物)]に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン428mg(0.877mmol)(一般式(5)の化合物)、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)38.8mg(0.0335mmol))及びTHF8mlを加え、60℃で、6時間反応させた。氷冷後、容器を水冷し3N塩酸4mlを添加することで反応を停止させた。トルエンを添加し、分相後、有機相を2回飽和食塩水で洗浄した。有機相に70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.2ml添加し、1時間撹拌した。水洗2回し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製した(溶離液;ヘキサン)。さらにヘプタンから再結晶精製し、目的物の淡黄固体380mgを得た(収率43%)。
1H NMR(CDCl3,23℃):δ=7.79(d,J=8.1Hz,2H),7.78(s,2H),7.66(s,2H),7.34(dd,J=8.1Hz,1.4Hz,2H),2.78(t,J=8.1Hz,4H),1.70(m,4H),1.27(m,36H),0.88(t,J=8.1Hz,6H)。
MS m/z: 994(M+,100%),914(M+−Br,4%),839(M+−C11H23,36%)。
【0133】
1H NMR及びMS測定より、1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0134】
【化38】
【0135】
実施例1 (ジドデシル)ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、THF21mlを添加し−74℃に冷却した。メタル化剤としてsec−ブチルリチウム(関東化学製1.0M)のシクロヘキサン/ヘキサン溶液3.0ml(3.0mmol)を添加した。さらに合成例2で合成した1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの粉体379mg(0.381mmol)を−74℃下で少しずつ添加した。この得られた溶液を−74℃〜−70℃で30分間熟成し、テトラメタル化を行った。次に−83℃とし、反応剤としてビス(フェニルスルホニル)スルフィド(アクロス製)(一般式(3)及び(4)の化合物)477mg(1.51mmol)を一気に投入した。一晩かけて室温まで温度を上げた。飽和食塩水を添加した後、分相し、さらに有機相を飽和食塩水で洗浄した。有機相を減圧濃縮した後、残渣をヘキサンを用いて洗浄した。その残渣のトルエン再結晶精製を3回繰り返し、目的物の鮮黄固体39mgを得た(収率14%)。
1H NMR(重トルエン,105℃):δ=8.00(s,2H),7.60(d,J=8.3Hz,2H),7.54(d,J=1.5Hz,2H),7.10(dd,J=8.3Hz,1.5Hz,2H),2.64(t,J=8.1Hz,4H),1.66(m,4H),1.34(m,36H),0.88(t,J=8.1Hz,6H)。
MS m/z: 738(M+−1,100%),583(M+−C11H23−1,33%)。
【0136】
1H NMR及びMS測定より、(ジドデシル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0137】
【化39】
【0138】
合成例3 [1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成(一般式(2)のテトラハロターフェニル誘導体の合成)]
1)2,3−ジブロモ−6−デカノイルベンゾチオフェンの合成
ドデカノイルクロライドの代わりに、デカノイルクロライド(和光純薬工業製)を用いた以外は合成例2の1)と同じ操作を繰り返して2,3−ジブロモ−6−デカノイルベンゾチオフェンを収率28%で合成した。
【0139】
2)2,3−ジブロモ−6−デシルベンゾチオフェンの合成
合成例2の2)と同じ操作を繰り返して2,3−ジブロモ−6−デシルベンゾチオフェンを収率96%で合成した。
【0140】
3)1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成
合成例2の3)と同じ操作を繰り返して1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを収率46%で合成した。
1H NMR(CDCl3,23℃):δ=7.80(d,J=8.1Hz,2H),7.78(s,2H),7.67(s,2H),7.35(dd,J=8.1Hz,1.4Hz,2H),2.78(t,J=8.1Hz,4H),1.70(m,4H),1.27(m,28H),0.88(t,J=8.1Hz,6H)。
MS m/z: 938(M+,100%),858(M+−Br,3%),811(M+−C9H19,33%)。
1H NMR及びMS測定より、1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0141】
【化40】
【0142】
実施例2 (ジデシルジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、合成例3で合成した1,4−ビス(6−デシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返して(ジデシル)ジベンゾテトラチエノアセンを収率17%で合成した。
1H NMR(重トルエン,105℃):δ=7.99(s,2H),7.60(d,J=8.3Hz,2H),7.53(d,J=1.5Hz,2H),7.11(dd,J=8.3Hz,1.5Hz,2H),2.64(t,J=8.1Hz,4H),1.66(m,4H),1.34(m,28H),0.88(t,J=8.1Hz,6H)。
MS m/z: 682(M+−1,100%),555(M+−C9H19−1,35%)。
【0143】
1H NMR及びMS測定より、(ジデシル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0144】
【化41】
【0145】
合成例4 [1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成(一般式(2)のテトラハロターフェニル誘導体の合成)]
1)2,3−ジブロモ−6−オクタデカノイルベンゾチオフェンの合成
ドデカノイルクロライドの代わりに、オクタデカノイルクロライド(和光純薬工業製)を用い、さらに反応を−25℃で3日間実施した以外は合成例2の1)と同じ操作を繰り返して2,3−ジブロモ−6−オクタデカノイルベンゾチオフェンを収率20%で合成した。
【0146】
2)2,3−ジブロモ−6−オクタデシルベンゾチオフェンの合成
40℃で6時間反応させる代わりに、60℃で4時間反応させた以外は合成例2の2)と同じ操作を繰り返して2,3−ジブロモ−6−オクタデシルベンゾチオフェンを収率99%で合成した。
【0147】
3)1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの合成
グリニャール化の熟成を『−74℃、15分間』から、『−74℃から−28℃まで4時間かけて行った』以外は合成例2の3)と同じ操作を繰り返して1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを収率33%で合成した。
1H NMR(CDCl3,23℃):δ=7.80(d,J=8.1Hz,2H),7.79(s,2H),7.67(s,2H),7.34(dd,J=8.1Hz,1.4Hz,2H),2.78(t,J=8.1Hz,4H),1.70(m,4H),1.27(m,60H),0.88(t,J=8.1Hz,6H)。
MS m/z: 1163(M+,100%),1083(M+−Br,4%),924(M+−C17H35,38%)。
1H NMR及びMS測定より、1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
【0148】
【化42】
【0149】
実施例3 (ジオクタデシル)ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、合成例4で合成した1,4−ビス(6−オクタデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返して(ジオクタデシル)ジベンゾテトラチエノアセンを収率12%で合成した。
1H NMR(重トルエン,105℃):δ=8.00(s,2H),7.61(d,J=8.3Hz,2H),7.53(d,J=1.5Hz,2H),7.10(dd,J=8.3Hz,1.5Hz,2H),2.64(t,J=8.1Hz,4H),1.66(m,4H),1.34(m,60H),0.88(t,J=8.1Hz,6H)。
MS m/z: 907(M+,100%),667(M+−C17H35−1,29%)。
【0150】
1H NMR及びMS測定より、(ジオクタデシル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0151】
【化43】
【0152】
比較例1 (ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体においてR5〜R12が同時に水素である化合物)の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、1,4−ビス(3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返してジベンゾテトラチエノアセンを収率21%で合成した。
MS m/z: 402(M+,100%),370(M+−S,16%),201(M+/2,74%)。
【0153】
MS測定より、ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0154】
【化44】
【0155】
窒素雰囲気下、得られた(ジベンゾ)テトラチエノアセン2.5mgをトルエン35gと混合し、100℃で1時間撹拌したが、全く溶解せず、溶液を調製することが困難であったことから、溶解性に劣るものであった。
【0156】
合成例5 (2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンの合成)
1)1,2−ジペンタデシルベンゼンの合成
1,2−ジペンタデシルベンゼンは、「シンセシス」、1993年、387−390頁の方法を参考に1,2−ジクロロベンゼンとn−ペンタデシルマグネシウムブロマイドから次のように合成した。
【0157】
窒素雰囲気下、300mlシュレンク反応容器に1,2−ジクロロベンゼン3.4ml(30.3mmol)、塩化ニッケル{ビス(ジフェニルホスフィノ)プロパン}164.2mg(0.303mmol)、及びジエチルエーテル30mlを添加した。0℃に冷却し、n−ペンタデシルマグネシウムブロマイド(シグマ−アルドリッチ製、0.5M)のジエチルエーテル溶液150ml(75mmol)を滴下した。35℃で16時間反応後、3N塩酸を加えて反応を停止させた。ジエチルエーテルで抽出し、有機相を水及び飽和炭酸水素ナトリウム水溶液で洗浄した。塩化カルシウムで乾燥し、溶媒を減圧濃縮した。さらに残渣を加熱減圧乾燥し(10パスカル、100℃)、残渣に1,2−ジペンタデシルベンゼンの粘性固体を得た(11.63g、収率77%)。
【0158】
2)4−ブロモ−5−ヨード無水フタル酸の合成
4−ブロモ−5−ヨード無水フタル酸は「ジャーナル オブ オルガニック ケミストリィー」(米国)、1951年、16巻、1577−1581頁に記載されている方法を参考に4−ブロモ無水フタル酸を原料に用いて次のように合成を行った。
【0159】
窒素雰囲気下、100mlの二口ナスフラスコに4−ブロモフタルイミド(東京化成工業製)13.3g(58.8mmol)を入れた。次いでヨウ素7.78g(30.6mmol)及び10%発煙硫酸(ヨツハタ化学工業製)50mlを加え、90℃で23時間反応を行った。反応混合物を室温に冷やして氷に注ぎ入れた後、ガラスフィルターでろ過し、黄色固体17.1gを得た。得られた固体を濃硫酸47mlに溶解させ、130℃で5時間反応を行った。反応混合物を氷冷後、氷水を加えて析出した固体をろ過し、フタル酸誘導体の固体18.5gを得た。次に水酸化ナトリウム4.8gを水24mlに溶かした水溶液に得られた固体を室温で溶かした。この塩基性水溶液に酢酸を加えpHを3〜4に中和し、析出するフタル酸誘導体のモノナトリウム塩の白色沈殿をろ過した。得られた白色沈殿を水に懸濁させ、濃塩酸でpHを1以下にし、再びフタル酸誘導体として白色固体7.75gを得た。この固体をトルエン70mlに溶かし、無水酢酸13ml(138mmol)を加え、105℃で4時間反応を行った。反応液を減圧濃縮して白色固体6.9gを得た。この固体を加熱トルエンで再結晶精製し、目的の4−ブロモ−5−ヨードフタル酸無水物を6.79g(19.2mmol)を得た(収率32.7%)。
【0160】
3)2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンの合成
2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンは「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁に記載されている方法を参考に次のように合成を行った。
【0161】
100mlの三口フラスコに合成例5の2)で合成した4−ブロモ−5−ヨード無水フタル酸2.40g(6.80mmol)、合成例5の1)で合成した1,2−ジペンタデシルベンゼン4.16g(8.34mmol)、及び1,2−ジクロロエタン7.0mlを加えた。そこへ塩化アルミニウム1.90g(14.3mmol)を添加し、加熱還流下で1.5時間撹拌した。得られた反応混合物に氷を少しずつ加えてクエンチした後、ジエチルエーテルで抽出した。無水硫酸マグネシウムで乾燥後、濾過、減圧濃縮した後、得られた残渣をシリカゲルカラムクロマトグラフィーで精製し(溶離液;ヘキサン:酢酸エチル=1:1)、5.55g(6.52mmol)の安息香酸誘導体を得た。この安息香酸誘導体に塩化チオニル5.0ml、N,N−ジメチルホルムアミド0.05ml、塩化アルミニウム1.74g(13.0mmol)、及びテトラクロロエタン25mlを添加し、50℃で5時間撹拌した。得られた反応混合物を室温まで冷却し、氷を加えた。トルエンで抽出し、有機相を無水硫酸マグネシウムで乾燥後、濾過、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーで精製し(溶離液;ヘキサン:クロロホルム=4:1)、3.02gの2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンを得た(収率53%)。
【0162】
4)2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンの合成
合成例5の3)で得た2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノン2.96g(3.55mmol)にTHF40mlを加えた後、水素化ジイソブチルアルミニウム(関東化学製、0.99M)トルエン溶液9.0ml(8.9mmol)を添加し、室温で2時間撹拌した。氷冷後、6N塩酸24mlを添加した後、65℃に加熱し、4時間反応を行った。トルエン及び食塩水を添加し、分相した。さらに有機相を食塩水で洗浄し、減圧濃縮及び真空乾燥した。得られた残渣に、水素化ジイソブチルアルミニウムを用いた還元、6N塩酸による脱水操作をさらに2回繰り返した。粗生成物をトルエンから再結晶精製し、2.27gの薄黄色固体である2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンを得た(収率80%)。
【0163】
合成例6 (3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニル(一般式(2)のテトラハロターフェニル誘導体)の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例5で合成した2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセン210mg(0.261mmol)及びTHF10mlを添加した。この溶液を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.78M)のTHF溶液0.73ml(0.57mmol)を滴下した。60分間かけて−40℃まで昇温することで熟成させた後、−83℃に冷却し、トリメトキシボラン(和光純薬工業製)67.7mg(0.651mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた3−ブロモ−6,7−ジペンタデシルアントラセニル−2−ボロン酸(一般式(6)及び(7)の化合物)に、合成例1で合成した1,4−ジブロモ−2,5−ジヨードベンゼン50.7mg(0.104mmol)(一般式(5)の化合物)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)7.6mg(0.0066mmol)、トルエン3.3ml、及びエタノール0.8mlを添加した。さらに炭酸ナトリウム66.4mg(0.626mmol)と水1.0mlからなる水溶液を加え、60℃で78時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーで精製した(溶離液:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の薄黄色固体43.7mgを得た(収率26%)。
1H NMR(CDCl3,23℃):δ=8.35(s,2H),8.33(s,2H),8.28(s,2H),7.96(s,1H),7.92(s,1H),7.79(s,2H),7.78(s,2H),7.72(s,1H),7.71(s,1H),2.81(m,8H),1.71(m,8H),1.28(m,96H),0.88(m,12H)。
FABMS m/z: 1587(M+,100%),1507(M+−Br,10)。
【0164】
1H NMR及びMS測定より、3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0165】
【化45】
【0166】
実施例4 (テトラペンタデシル)ジアントラジチエノアセン(一般式(1)のヘテロアセン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に、合成例6で合成した3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニル42.8mg(0.027mmol)及びジエチルエーテル6mlを添加した。この混合物を0℃に冷却し、メタル化剤としてn−ブチルリチウム(関東化学製1.57M)のヘキサン溶液0.10ml(0.15mmol)を滴下した。60分間かけて5℃まで昇温することで熟成させた後、−70℃に冷却し、ビス(フェニルスルホニル)スルフィド(アクロス製)23.6mg(0.075mmol)(一般式(3)及び(4)の化合物)を一気に投入した。徐々に昇温し、一晩かけて室温まで反応温度を上げた。飽和食塩水及びトルエンを添加した後、固体を濾過し、水、ヘキサンで洗浄した。固体を乾燥後、トルエンから再結晶精製し、目的物の黄色結晶12mgを得た(収率33%)。
1H NMR(重トルエン,100℃):δ=8.50(s,2H),8.43(s,2H),8.26(s,2H),8.20(s,2H),8.14(s,2H),7.85(s,2H),7.78(s,2H),2.87(t,J=7.8Hz,8H),1.77(m,8H),1.31(m,96H),0.88(m,J=7.8Hz,12H)。
【0167】
1H NMR測定より、(テトラペンタデシル)ジアントラジチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0168】
【化46】
【0169】
合成例7 (2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンの合成)
2−ジウンデシルベンゼンの合成
1)1,2−ジウンデシルベンゼンの合成
n−ペンタデシルマグネシウムブロマイド(シグマ−アルドリッチ製、0.5M)のジエチルエーテル溶液の代わりに、n−ウンデシルマグネシウムブロマイドのジエチルエーテル溶液(ジエチルエーテル中、1−ブロモウンデカン(和光純薬工業製)とマグネシウムから調製)を用いた以外は合成例5の1)と同じ操作を繰り返して1,2−ジウンデシルベンゼンを収率87%で合成した。
【0170】
2)2−ブロモ−3−ヨード−6,7−ジウンデシルアントラキノンの合成
1,2−ジペンタデシルベンゼンの代わりに、合成例7の1)で合成した1,2−ジウンデシルベンゼンを用いた以外は合成例5の3)と同じ操作を繰り返して2−ブロモ−3−ヨード−6,7−ジウンデシルアントラキノンを収率65%で合成した。
【0171】
3)2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンの合成
2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラキノンの代わりに、合成例7の2)で合成した2−ブロモ−3−ヨード−6,7−ジウンデシルアントラキノンを用いた以外は合成例5の4)と同じ操作を繰り返して2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンを収率87%で合成した。
【0172】
合成例8 (3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニル(一般式(2)のテトラハロターフェニル誘導体)の合成)
2−ブロモ−3−ヨード−6,7−ジペンタデシルアントラセンの代わりに、合成例7で合成した2−ブロモ−3−ヨード−6,7−ジウンデシルアントラセンを用いた以外は合成例6と同じ操作を繰り返して3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニルを収率58%で合成した。
1H NMR(CDCl3,23℃):δ=8.36(s,2H),8.34(s,2H),8.28(s,2H),7.96(s,1H),7.91(s,1H),7.79(s,2H),7.78(s,2H),7.71(s,1H),7.70(s,1H),2.81(m,8H),1.71(m,8H),1.28(m,64H),0.88(m,12H)。
FABMS m/z: 1363(M+,100%),1283(M+−Br,12)。
【0173】
1H NMR及びMS測定より、3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
【0174】
【化47】
【0175】
実施例5 (テトラウンデシル)ジアントラジチエノアセン(一般式(1)のヘテロアセン誘導体の合成)
3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラペンタデシル)−2,1’,4’,2”−ジナフトターフェニルの代わりに合成例8で合成した3,2’,5’,3”−テトラブロモ−6,7,6”,7”−(テトラウンデシル)−2,1’,4’,2”−ジナフトターフェニルを用いた以外は実施例4と同じ操作を繰り返して、(テトラウンデシル)ジアントラジチエノアセンを収率39%で合成した。
1H NMR(重トルエン,100℃):δ=8.49(s,2H),8.44(s,2H),8.25(s,2H),8.20(s,2H),8.13(s,2H),7.85(s,2H),7.79(s,2H),2.87(t,J=7.8Hz,8H),1.77(m,8H),1.31(m,64H),0.88(m,J=7.8Hz,12H)。
【0176】
1H NMR測定より、(テトラウンデシル)ジアントラジチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0177】
【化48】
【0178】
実施例6 (耐酸化性有機半導体材料の合成及びその耐酸化性評価)
窒素雰囲気下、100mlシュレンク容器にトルエン5.4gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ実施例1で得られた(ジドデシル)ジベンゾテトラチエノアセンの固体5.1mgを添加し、100℃に加熱し溶解させ、(ジドデシル)ジベンゾテトラチエノアセンを含む耐酸化性有機半導体材料を合成した(山吹色溶液)。次に、このシュレンク容器の上部の栓を開け、1分間、外気に接触させることで空気を導入(耐酸化性評価)し、さらに90℃で撹拌したが、色の変化は見られなかった。したがって、色の変化が見られなかったことから、耐酸化性に優れるものであった。
【0179】
実施例7 (有機薄膜の作製)
窒素雰囲気下、実施例1で得られた(ジドデシル)ジベンゾテトラチエノアセン2.5mgをトルエン25gと混合し、100℃で1時間撹拌し、(ジドデシル)ジベンゾテトラチエノアセンの山吹色溶液を調製した(ジドデシル)ジベンゾテトラチエノアセンを含む耐酸化性有機半導体材料の合成)。
【0180】
窒素雰囲気下、凹面のあるガラス基板を90℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚260nmの有機薄膜を作製した。
【0181】
実施例8 (耐酸化性有機半導体材料の合成及びその耐酸化性評価)
窒素雰囲気下、100mlシュレンク容器にトルエン5.4gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ実施例4で得られた(テトラペンタデシル)ジアントラジチエノアセンの固体6.2mgを添加し、70℃に加熱し溶解させ、(テトラペンタデシル)ジアントラジチエノアセンを含む耐酸化性有機半導体材料を合成した(黄橙色溶液)。次に、このシュレンク容器の上部の栓を開け、1分間、外気に接触させることで空気を導入(耐酸化性評価)し、さらに70℃で撹拌したが、色の変化は見られなかった。したがって、色の変化が見られなかったことから、耐酸化性に優れるものであった。
【0182】
さらにこの溶液を70℃。1時間、撹拌下で空気と接触させても溶液の色の変化は見られず、耐酸化性に優れるものであった。
【0183】
実施例9 (有機薄膜の作製)
窒素雰囲気下、実施例4で得られた(テトラペンタデシル)ジアントラジチエノアセン4.7mgをトルエン15gと混合し、100℃で1時間撹拌し、(テトラペンタデシル)ジアントラジチエノアセンの黄橙色溶液を調製した(テトラペンタデシル)ジアントラジチエノアセンを含む耐酸化性有機半導体材料の合成)。
【0184】
窒素雰囲気下、凹面のあるガラス基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚320nmの有機薄膜を作製した。
【0185】
比較例2 (耐酸化性評価)
ペンタセンを用いて耐酸化性を評価した。
【0186】
窒素雰囲気下、20mlシュレンク容器にo−ジクロロベンゼン2.9gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへペンタセン(東京化成工業製)2.5mgを添加し、120℃に加熱し溶解させると赤紫色溶液となった。次にこのシュレンク容器の上部の栓を開け、1分間、外気に接触させることで空気を導入し、さらに120℃で撹拌した。ガスクロマトグラフィー及びガスクロマトグラフィー−マススペクトル(GCMS)分析から、6,13−ペンタセンキノンが生成していることがわかった。
【0187】
さらにこの溶液を120℃、1時間、撹拌下で空気と接触させると溶液の色が黄に変化していた。ガスクロマトグラフィー分析から、6,13−ペンタセンキノンの生成が増加していることがわかった。
【0188】
したがって、溶液の色の変化及び6,13−ペンタセンキノンが生成していることから、酸化が進行しており、耐酸化性に劣るものであった。
【0189】
実施例10 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、実施例1で得られた(ジドデシル)ジベンゾテトラチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(ジドデシル)ジベンゾテトラチエノアセンの黄色溶液を調製した。
【0190】
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは13nmであった。得られた原子間力顕微鏡写真を図1に示した。
【0191】
実施例11 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、実施例4で得られた(テトラペンタデシル)ジアントラジチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(テトラペンタデシル)ジアントラジチエノアセンの黄色溶液を調製した。
【0192】
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板を165℃に加熱し、上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは26nmであった。得られた原子間力顕微鏡写真を図2に示した。
【0193】
実施例12 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、(テトラドデシル)ジアントラジチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(テトラドデシル)ジアントラジチエノアセンの黄色溶液を調製した。
【0194】
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板を165℃に加熱し、上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは31nmであった。得られた原子間力顕微鏡写真を図3に示した。
【0195】
比較例3 ((ジペンチル)ジベンゾテトラチエノアセンの合成(一般式(1)のヘテロアセン誘導体においてR5、R7、及びR8が同時に水素であり、R6が炭素数5のアルキル基である化合物)の合成)
1,4−ビス(6−ドデシル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンの代わりに、1,4−ビス(6−ペンチル−3−ブロモベンゾチエニル−2−)−2,5−ジブロモベンゼンを用いた以外は実施例1と同じ操作を繰り返して(ジペンチル)ジベンゾテトラチエノアセンを収率18%で合成した。
MS m/z: 542(M+,100%),485(M+−C4H9−1,26%)。
【0196】
MS測定より、(ジペンチル)ジベンゾテトラチエノアセンが得られたことを確認した。なお、その構造式を下記に示す。
【0197】
【化49】
【0198】
比較例4 (有機薄膜の作製及び表面粗さの測定)
窒素雰囲気下、比較例3で得られた(ジペンチル)ジベンゾテトラチエノアセン10.0mgをn−オクチルベンゼン5gと混合し、165℃で1時間撹拌し、(ジペンチル)ジベンゾテトラチエノアセンの黄色溶液を調製した。
【0199】
窒素雰囲気下、直径2インチのシリコン基板(セミテック製、抵抗値;0.001〜0.004Ω)をオクタデシルトリクロロシランの5mmol/l濃度のクロロホルム溶液に室温で、48時間、浸漬させることで表面処理を行った。この基板を165℃に加熱し、上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、有機薄膜を作製した。この有機薄膜の表面を原子間力顕微鏡で測定したところ、その表面粗さは142nmであった。得られた原子間力顕微鏡写真を図4に示した。
【図面の簡単な説明】
【0200】
【図1】実施例10で得られた(ジドデシル)ジベンゾテトラチエノアセンの原子間力顕微鏡写真
【図2】実施例11で得られた(ジペンタデシル)ジベンゾテトラチエノアセンの原子間力顕微鏡写真
【図3】実施例12参考例1で得られた(テトラドデシル)ジアントラジチエノアセンの原子間力顕微鏡写真
【図4】比較例4で得られた(テトラペンチル)ジアントラジチエノアセンの原子間力顕微鏡写真
【特許請求の範囲】
【請求項1】
下記一般式(1)で示されることを特徴とするヘテロアセン誘導体。
【化1】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、炭素数4〜30のアリール基、炭素数6〜20のアルキル基、を示し、T1及びT2は同一又は異なって、硫黄、セレン、テルル、リン、ホウ素を示し、l及びmは、各々0又は1の整数であり、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有する。)
【化2】
【化3】
(ここで、置換基R5〜R12は同一又は異なって、水素原子、フッ素原子、炭素数6〜20のアルキル基、を示し、置換基T3は、硫黄、セレン、テルルを示し、nは0又は1の整数である。但し、置換基R5〜R12は同時に水素原子であることはなく、また環A及びBが(A−2)で示される構造である場合、何れか一方の(A−2)中のnは1である。))]
【請求項2】
l及びmが各々0であり、且つT1及びT2は同一又は異なって、硫黄、セレン、テルルであることを特徴とする請求項1に記載のヘテロアセン誘導体。
【請求項3】
l及びmが各々1であり、且つT1及びT2は同一又は異なって、リン、ホウ素であることを特徴とする請求項1に記載のヘテロアセン誘導体。
【請求項4】
nが1であることを特徴とする請求項1〜3のいずれかに記載のヘテロアセン誘導体。
【請求項5】
請求項1〜4のいずれかに記載のヘテロアセン誘導体を含むことを特徴とする耐酸化性有機半導体材料。
【請求項6】
請求項5に記載の耐酸化性有機半導体材料を用いることを特徴とする有機薄膜。
【請求項7】
有機薄膜が基板上に形成されることを特徴とする請求項6に記載の有機薄膜。
【請求項8】
有機薄膜の表面粗さが40nm以下であることを特徴とする請求項7に記載の有機薄膜。
【請求項1】
下記一般式(1)で示されることを特徴とするヘテロアセン誘導体。
【化1】
[(ここで、置換基R1〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、炭素数4〜30のアリール基、炭素数6〜20のアルキル基、を示し、T1及びT2は同一又は異なって、硫黄、セレン、テルル、リン、ホウ素を示し、l及びmは、各々0又は1の整数であり、環A及びBは同一又は異なって、下記一般式(A−1)又は(A−2)で示される構造を有する。)
【化2】
【化3】
(ここで、置換基R5〜R12は同一又は異なって、水素原子、フッ素原子、炭素数6〜20のアルキル基、を示し、置換基T3は、硫黄、セレン、テルルを示し、nは0又は1の整数である。但し、置換基R5〜R12は同時に水素原子であることはなく、また環A及びBが(A−2)で示される構造である場合、何れか一方の(A−2)中のnは1である。))]
【請求項2】
l及びmが各々0であり、且つT1及びT2は同一又は異なって、硫黄、セレン、テルルであることを特徴とする請求項1に記載のヘテロアセン誘導体。
【請求項3】
l及びmが各々1であり、且つT1及びT2は同一又は異なって、リン、ホウ素であることを特徴とする請求項1に記載のヘテロアセン誘導体。
【請求項4】
nが1であることを特徴とする請求項1〜3のいずれかに記載のヘテロアセン誘導体。
【請求項5】
請求項1〜4のいずれかに記載のヘテロアセン誘導体を含むことを特徴とする耐酸化性有機半導体材料。
【請求項6】
請求項5に記載の耐酸化性有機半導体材料を用いることを特徴とする有機薄膜。
【請求項7】
有機薄膜が基板上に形成されることを特徴とする請求項6に記載の有機薄膜。
【請求項8】
有機薄膜の表面粗さが40nm以下であることを特徴とする請求項7に記載の有機薄膜。
【図1】

【図2】

【図3】
【図4】

【図2】
【図3】
【図4】
【公開番号】特開2010−138077(P2010−138077A)
【公開日】平成22年6月24日(2010.6.24)
【国際特許分類】
【出願番号】特願2008−313210(P2008−313210)
【出願日】平成20年12月9日(2008.12.9)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成22年6月24日(2010.6.24)
【国際特許分類】
【出願日】平成20年12月9日(2008.12.9)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]