
ヘテロオリゴマー蛋白質の生産方法
【課題】 ヘテロオリゴマー蛋白質を安定に発現し得る形質転換昆虫細胞を作製し、組換えヘテロオリゴマー蛋白質を安定に生産する方法を提供すること。
【解決手段】 本発明は、昆虫細胞を用いて異種サブユニットから構成されるオリゴマー蛋白質を生産する方法を提供し、該方法は、該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで該昆虫細胞を形質転換する工程;該形質転換された昆虫細胞を培養する工程;および培養物から該オリゴマー蛋白質を回収する工程を含む。
【解決手段】 本発明は、昆虫細胞を用いて異種サブユニットから構成されるオリゴマー蛋白質を生産する方法を提供し、該方法は、該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで該昆虫細胞を形質転換する工程;該形質転換された昆虫細胞を培養する工程;および培養物から該オリゴマー蛋白質を回収する工程を含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、昆虫細胞を用いてヘテロオリゴマー蛋白質を安定に生産する方法に関する。
【背景技術】
【0002】
遺伝子組換え技術を用いて、微生物、動物細胞などで蛋白質を高発現させる方法が種々検討されている。高等真核生物の蛋白質は、発現に際し、翻訳後修飾(例えば、糖鎖、脂肪酸などの付加、リン酸化)を受け、高次構造を保持するとともに、生物学的活性(機能)を有する。したがって、高等真核生物の蛋白質をDNA組換え技術で生産する場合、宿主として、高等真核生物である動物細胞を用いることが好ましい。しかし、動物細胞は取扱いが難しく、生育が遅いため、物質生産にはあまり適していない。そこで、翻訳後修飾活性を有する昆虫細胞を宿主として用いる蛋白質の発現系が検討されている。
【0003】
昆虫細胞を宿主とする蛋白質生産には、一般的に、昆虫細胞−バキュロウイルス系が用いられている。バキュロウイルス(核多角体病ウイルス)は、感染した昆虫細胞内で、ポリヘドリンとよばれるタンパク質を大量に生産するウイルスである。この昆虫細胞−バキュロウイルス系は、ポリヘドリンプロモーターの下流に外来遺伝子を挿入した組換えバキュロウイルスを培養昆虫細胞に感染させ、ウイルス感染した細胞に外来タンパク質を生産させる組換えタンパク質発現系である。しかし、ウイルスの感染により細胞が死滅してしまうため、目的タンパク質を連続的に生産することはできない。
【0004】
この昆虫細胞−バキュロウイルス系以外にも、種々の昆虫宿主−ベクター系が開発されている。例えば、カイコガ由来のアクチンプロモーターの上流にバキュロウイルスBmNPV由来のエンハンサーIE1とBmNPV由来のトランス作用因子HR3を有するベクターにヒト組織プラスミノーゲンアクチベーター(tPA)遺伝子を導入し、昆虫細胞を形質転換してtPAを発現させた例があるが(非特許文献1)、蛋白質は必ずしも安定には生産されていない。
【0005】
蛋白質の中でも、異種サブユニットから構成されるオリゴマータンパク質(以下、ヘテロオリゴマー蛋白質という)を発現する場合、複数のベクターを使用することが多く、さらに、選択マーカーを有するプラスミドを導入する必要があるため、目的とする遺伝子を有する形質転換体が得られにくい。また、得られた形質転換体におけるベクターの保持が困難であり、安定にヘテロオリゴマー蛋白質を生産することは困難である。
【非特許文献1】Farrellら、Biotechnol. Bioeng.、64巻、426−433頁 (1999)
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、ヘテロオリゴマー蛋白質を安定に発現し得る形質転換昆虫細胞を作製し、組換えヘテロオリゴマー蛋白質を安定に生産する方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明は、昆虫細胞を用いて異種サブユニットから構成されるオリゴマー蛋白質を生産する方法を提供し、該方法は、該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで該昆虫細胞を形質転換する工程;該形質転換された昆虫細胞を培養する工程;および培養物から該オリゴマー蛋白質を回収する工程を含む。
【0008】
好適な実施態様では、上記オリゴマー蛋白質は抗体である。
【0009】
より好適な実施態様では、上記異種サブユニットは、それぞれ、抗体を構成するH鎖の少なくとも一部のペプチドおよび該抗体を構成するL鎖の少なくとも一部のペプチドである。
【0010】
他の好適な実施形態では、上記複数のベクターは、それぞれ同じまたは異なるプロモーターを有する。
【0011】
さらに他の好適な実施態様では、上記複数のベクターの少なくとも1つは、上記昆虫細胞の選択マーカーを有する。
【0012】
本発明はまた、異種サブユニットから構成されるオリゴマー蛋白質を安定に発現する形質転換昆虫細胞を提供し、該昆虫細胞は該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターを有する。
【0013】
好適な実施態様では、上記複数のベクターの少なくとも1つは、上記昆虫細胞の選択マーカーを有する。
【発明の効果】
【0014】
異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで昆虫細胞を形質転換することにより、効率よく、ヘテロオリゴマー蛋白質が生産される。さらに、複数のベクターの少なくとも1つが、昆虫細胞の選択マーカーを有することにより、形質転換効率が向上するとともに、形質転換細胞は、安定にヘテロオリゴマー蛋白質を生産し得る。
【発明を実施するための最良の形態】
【0015】
まず、本発明に用いる種々の材料について、説明する。
【0016】
(宿主細胞)
宿主細胞としては、昆虫細胞であれば特に制限なく利用できる。市販されている昆虫細胞株も利用される。例えば、Trichoplusia ni由来のBTI-TN5B1-4 (High Five)株、Spodoptera frugiperda由来のSf9株などが好ましく用いられる。これらの株は、Invitrogenから入手可能である。
【0017】
(プラスミド、ベクター)
本発明においては、昆虫細胞内で発現し得るプラスミドであれば特に制限なく使用される。好ましくは、バキュロウイルス系のプラスミドが用いられる。発現効率が高い点で、カイコガ由来のアクチンプロモーターの上流に、バキュロウイルスBmNPV由来のエンハンサーIE1およびBmNPV由来のトランス作用因子HR3を有するプラスミドが、より好ましい。このようなプラスミドとしては、pXINSECT-DEST38、pXINSECT-DEST39などが挙げられる。これらのプラスミドは、Invitrogenから入手可能である。
【0018】
また、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターを有するプラスミドも好ましく用いられる。このようなプラスミドとしては、例えば、pIB/V5-Hisが挙げられる。このプラスミドは、Invitrogenから入手可能である。
【0019】
なお、形質転換細胞の選択効率、および目的遺伝子の発現の安定性を向上させ得る点で、選択マーカーを有するプラスミドが好ましく用いられる。前述のpIB/V5-Hisは、ブラスチシジン(Blasticidin)に対する耐性遺伝子を有しており、選択マーカーを有するプラスミドとして、好ましい。
【0020】
(ヘテロオリゴマー蛋白質)
本発明において、ヘテロオリゴマー蛋白質とは、異種サブユニットから構成されるオリゴマー蛋白質をいう。ヘテロオリゴマー蛋白質としては、抗体、酵素、レセプター、イオン・チャンネル、ヘモグロビン、血液凝固因子などが挙げられる。抗体を例に説明すると、抗体はH鎖とL鎖とから構成されるヘテロオリゴマー蛋白質である。抗体を構成するH鎖は、全長であってもよく、H鎖の少なくとも一部の配列、例えば、VHドメイン、Fdフラグメント、FcフラグメントなどのH鎖の一部分であってもよい。さらに、これらを任意に含むフラグメントであってもよい。抗体を構成するL鎖は、全長であってもよく、H鎖と相互作用し得るVLドメインなどであってもよい。H鎖とL鎖とが相互作用し得るように、H鎖およびL鎖が選択される。
【0021】
(ベクターの構築)
本発明においては、ヘテロオリゴマー蛋白質を生産するために、異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターを用いて、形質転換(コトランスフェクション)する。本発明において用いられる複数のベクターは、それぞれ、プロモーター、および発現されるべきヘテロオリゴマー蛋白質を構成する異なる1つのサブユニットをコードするDNA配列を含む。これらのベクターはさらに、分泌のためのシグナル配列を含み得る。例えば、2つのサブユニットから構成されるヘテロオリゴマー蛋白質の場合を例に挙げると、一方のサブユニットを発現し得るベクターおよび他方のサブユニットを発現し得るベクターの、2つのベクターが用いられる。複数のベクターは、それぞれ、分泌のためのシグナル配列と、1つのサブユニットをコードするDNA配列とを有するように構築される。これらの複数のベクターは、同じ構成のプラスミドに、それぞれ異なるサブユニットをコードするDNA配列が挿入されたものであってもよく、異なる構成のプラスミドに、それぞれ異なるサブユニットをコードするDNA配列が挿入されたものであってもよい。
【0022】
異なる構成のプラスミドを用いてベクターを構築する場合、それぞれ異なるプロモーターを有するプラスミドを用いてもよい。各サブユニットをそれぞれ異なるプロモーターの制御下で発現させることにより、発現効率が高められ得る。2つの異なるサブユニットから構成されるヘテロオリゴマー蛋白質を発現させる場合、好ましい実施態様では、2つのベクターのいずれか一方は、カイコガ由来のアクチンプロモーターを有し、そして他方は、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターを有する。
【0023】
また、形質転換株の選択効率および目的遺伝子の発現の安定性を高める目的で、少なくとも1つのベクターは選択マーカー遺伝子を含むように構築することが好ましい。プラスミドが本来有する選択マーカー遺伝子を利用しても、または外部から選択マーカー遺伝子を挿入してもよい。選択マーカー遺伝子としては、ネオマイシン耐性遺伝子、ブラスチシジン耐性遺伝子、ハイグロマイシン耐性遺伝子、ピューロマイシン耐性遺伝子、ブレオマイシン耐性遺伝子などが挙げられる。例えば、前述のpIB/V5-Hisは、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターおよびブラスチシジン(Blasticidin)に対する耐性遺伝子を有しているので、本発明において用いられるベクターを構築するために好ましく利用され得る。さらに、本発明において用いられるベクターが、アクチンプロモーター、IE1エンハンサー、およびHR3トランスアクチベーター、ならびに選択マーカー遺伝子(例えば、ネオマイシン耐性遺伝子、ブラスチシジン耐性遺伝子など)を有するように構築され得ることは、形質転換株の選択効率およびベクターの安定性を高め得る点で、好ましい。
【0024】
好ましくは、本発明において用いられる複数のベクターのそれぞれが、選択マーカー遺伝子を含む。これらの選択マーカー遺伝子は、ベクター間で同じであっても、または異なっていてもよい。複数のベクターが、それぞれ、異なる選択マーカー遺伝子を含むことは、形質転換株の選択効率を高める点で、より好ましい。
【0025】
(昆虫細胞の形質転換)
昆虫細胞の形質転換について、抗体を発現させる場合を例に挙げて、具体的に説明する。以下の説明において、例えば、カイコガ由来のアクチンプロモーターを有するpXINSECT-DEST38のプラスミドに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−1およびベクターL−1とする。あるいは、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターとブラスチシジン耐性遺伝子とを有するpIB/V5-Hisのプラスミドに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−2およびベクターL−2とする。
【0026】
形質転換には、同じプラスミドに由来するベクターH−1とベクターL−1との組合わせ、あるいはベクターH−2とベクターL−2との組合わせが用いられ得る。この場合、H鎖を発現するベクターH−1(またはH−2)とL鎖を発現するベクターL−1(またはL−2)とは、質量比で2:1以上の比率であることが好ましい。また、形質転換には、異なるプラスミドに由来するベクターの組合わせである、ベクターH−1とベクターL−2との組合わせ、あるいはベクターH−2とベクターL−1との組合わせを用いてもよい。この異なるプラスミドに由来するベクターの組合わせを用いる場合は、それぞれのベクターに含まれるプロモーターも異なり、抗体の発現効率を高め得る。
【0027】
さらに、pIB/V5-Hisに由来するベクターH−2とベクターL−2との組合わせでは、ベクターは両方とも選択マーカーであるブラスチシジン耐性遺伝子を有している点で有利である。例えば、市販の昆虫細胞形質転換キットを用いて抗体生産株を取得する場合、2つのpXINSECT-DEST38にそれぞれH鎖およびL鎖を導入した2つの異なるベクター(ベクターH−1およびL−1)と、ネオマイシン耐性遺伝子を有するプラスミドとをコトランスフェクションし、ネオマイシン存在下で生育する細胞を選択する。しかし、ネオマイシン耐性遺伝子のみが導入された細胞を含めて、ネオマイシン耐性株がすべて生育するため、3つの遺伝子が同時に導入された細胞の選択効率が悪くなる。しかし、選択マーカー(例えば、ブラスチシジン耐性遺伝子)を有するベクターH−2またはベクターL−2を使用すると、ブラスチシジン耐性株は、少なくともH鎖またはL鎖を発現するので、形質転換株の選択効率が向上する。
【0028】
ベクターH−1とベクターL−2との組合わせ、すなわち、カイコガ由来のアクチンプロモーターを有するプラスミドにH鎖(またはその一部)をコードするDNA配列を挿入したベクター(ベクターH−1)と、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターとブラスチシジン耐性遺伝子とを有するプラスミドにL鎖(またはその一部)をコードするDNA配列を挿入したベクター(ベクターL−2)との組合わせは、好ましく用いられる。この場合、ベクターH−1とベクターL−2との比率は、重量比で10:1以上であることが好ましく、20:1以上であることがより好ましい。50:1以上で行ってもよい。
【0029】
上述のように、形質転換株の選択効率および目的遺伝子の発現の安定性を高める目的で、選択マーカー遺伝子を含むベクターが構築され得る。このようなベクターの例を以下に示す。カイコガ由来のアクチンプロモーターと、選択マーカー遺伝子であるネオマイシン耐性遺伝子とを有するベクターに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−3およびベクターL−3とする。一方、カイコガ由来のアクチンプロモーターと、選択マーカー遺伝子であるブラスチシジン耐性遺伝子とを有するベクターに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−4およびベクターL−4とする。
【0030】
カイコガ由来のアクチンプロモーターと選択マーカー遺伝子とを含む複数のベクターを用いる場合、形質転換には、異なるサブユニットのそれぞれが(例えば、H鎖とL鎖とが)発現して相互作用する組合わせであれば、いずれの組合わせもが用いられ得る。上記の場合、ベクターH−3とベクターL−3、ベクターH−3とベクターL−4、ベクターH−4とベクターL−3、あるいはベクターH−4とベクターL−4とが組み合わされ得る。コトランスフェクションにおいて、H鎖を発現するベクターとL鎖を発現するベクターとの比率は、任意の重量比であり得る。好ましくは、重量比で、1:1以上、より好ましくは2:1以上、さらに好ましくは10:1以上、なおさらに好ましくは20:1以上である。50:1以上で行ってもよい。
【0031】
これらの複数のベクターで昆虫細胞を形質転換する手段は、特に限定されず、昆虫細胞について当業者が通常用いる手段により行われ得る。具体的には、リン酸カルシウム法、リポフェクション(リポソーム法)、エレクトロポレーション(電気穿孔法)などにより行われる。このようにして、ヘテロオリゴマー蛋白質を安定に生産し得る形質転換昆虫細胞株が得られる。
【0032】
形質転換された昆虫細胞は、選択培地を利用して選択され得る。この選択培地を用いる選択は、発現されるべきサブユニットをコードするDNAを含むベクター中に含まれた選択マーカー遺伝子を用いても、選択マーカー遺伝子を有する別のベクターとのコトランスフェクションによってもよい。形質転換された昆虫細胞を選択するために使用される選択培地には、導入された選択マーカー遺伝子に対応するネオマイシン、ブラスチシジンなどの抗生物質が添加され得る。
【0033】
安定形質転換細胞を効率良く取得するためには、抗生物質の耐性遺伝子が導入されなかった細胞が完全に死滅してしまうような抗生物質濃度の存在下で細胞を培養する必要がある。通常、安定形質転換細胞を選択するための抗生物質濃度は、一週間程度で完全に細胞が死滅してしまう濃度が最適である。このことを考慮して、形質転換細胞の選択のために異なる抗生物質を細胞に与える濃度は、当業者によって適宜決定され得る。
【0034】
例えば、選択マーカー遺伝子としてブラスチシジン耐性遺伝子を含む少なくとも1つのベクターをコトランスフェクションに用いる場合、ブラスチシジンの毒性が強いため、ブラスチシジン耐性遺伝子を有するベクターの比率を小さくすると、形成されるコロニーの数が減少するので、コロニーの選択がより容易になる。コトランスフェクションの際の他の抗生物質耐性遺伝子(例えば、ネオマイシン耐性遺伝子)を有するベクターとブラスチシジン耐性遺伝子を有するベクターとの比率は、任意の重量比であり得るが、好ましくは1:1以上、より好ましくは2:1以上、さらに好ましくは10:1以上、なおさらに好ましくは20:1以上である。50:1以上で行ってもよい。
【0035】
異なるサブユニットを発現するためのベクターが、それぞれ異なる選択マーカー遺伝子を含む場合、それぞれの選択マーカー遺伝子に対応する抗生物質を用いて、発現されるべき遺伝子が全て導入されている細胞を選択し得る。例えば、ベクターH−3は、ネオマイシン耐性遺伝子を選択マーカー遺伝子として含み、ベクターL−4は、ブラスチシジン耐性遺伝子を選択マーカー遺伝子として含むので、ネオマイシン処理に対して耐性である細胞は、H−3が導入され、ブラスチシジン処理に対して耐性である細胞は、L−4が導入されている。ネオマイシンおよびブラスチシジンの両方の処理に対して耐性である細胞は、H−3およびL−4の両方が、すなわち、H鎖およびL鎖の両方の遺伝子が導入されている。このように、異なるサブユニットの全てが発現している細胞の選択がより効率的になる。もちろん、ベクターH−4とベクターL−3との組合わせにおいても同様の効果が期待され得る。
【0036】
得られた形質転換細胞がヘテロオリゴマー蛋白質を生産することは、ウエスタンブロッティング、ELISAなどの当業者に周知の技術を用いて確認され得る。
【0037】
(ヘテロオリゴマー蛋白質の生産)
ヘテロオリゴマー蛋白質の生産が確認された形質転換昆虫細胞は、当業者が通常用いる培養条件下で培養される。培養期間後、生産されたヘテロオリゴマー蛋白質は、蛋白質に応じた適切な手段によって培養物(培養上清)から回収される。回収された蛋白質は、必要に応じて、適宜精製され得る。
【実施例】
【0038】
以下、実施例に基づいて本発明を説明するが、本発明はこの実施例に制限されない。
【0039】
以下の実施例において、ヘテロオリゴマー蛋白質として、マウス由来の触媒抗体6D9のFabフラグメントを用いた。触媒抗体6D9のFabフラグメントは、そのH鎖部分であるFdフラグメントおよびL鎖から構成されており、クロラムフェニコールモノエステルの加水分解反応を触媒する。このFdフラグメントおよびL鎖をコードするDNA配列を含むプラスミドpARA7Fabの構築に関しては、Miyashitaら,J. Mol. Biol.、267、1247-1257 (1997)に記載されている。このプラスミドpARA7Fabは、大阪府立大学先端科学研究所の藤井郁雄教授から分与された。
【0040】
(実施例1)
(H鎖を発現するプラスミドの構築)
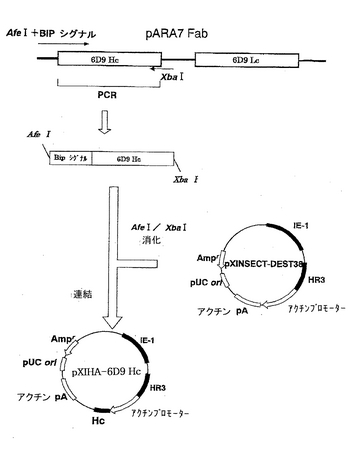
H鎖を発現するプラスミドpXIHA-6D9 Hcの構築を図1に示す。まず、上流部分に制限酵素AfeI認識部位およびDrosophila由来のBiPシグナル配列を有し、下流部分に制限酵素XbaI認識部位を有する6D9 Hc遺伝子を調製した。この遺伝子は、プラスミドpARA7Fabを鋳型とし、配列番号1および2のプライマーを用いてPCR法で増幅した。配列番号1のプライマーは、AfeI認識部位およびBiPシグナル配列を有し、そして配列番号2のプライマーは、XbaI認識部位を有する。PCRは、Thermal cycler(BIO-RAD)を用いて、以下のサイクルで行った。まず、94℃にて2分、72℃にて2分処理し、次いで、94℃にて30秒、52.9℃にて30秒、および72℃にて2分の処理を30サイクル行い、最後に72℃にて5分の処理を行った。アガロースゲル電気泳動で目的のフラグメントが増幅されたことを確認し、当業者が通常用いる方法により6D9 Hc遺伝子を回収した。
【0041】
次に、pXINSECT-DEST38(Invitrogen)を、制限酵素AfeIおよびXbaIで消化し、CONCERT Gel Extraction Systems(Invitrogen)を用いてDNAを精製した。このDNAと制限酵素AfeIおよびXbaIで切断した6D9 Hc遺伝子とを混合して連結(ライゲーション)を行い、E.coli K12株(NovaBlue,メルク (株))において形質転換を行って、当業者が通常用いる方法により、プラスミドpXIHA-6D9 Hcを得た。なお、pXINSECT-DEST38のカタログには認識部位AfeIが存在しないが、pXINSECT-DEST38のEco47III部位は、AfeIと同一認識配列であるので、このプラスミドはAfeIで切断される。
【0042】
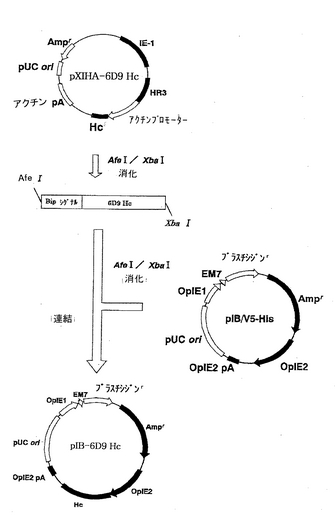
一方、H鎖を発現する別のプラスミドpIB-6D9 Hcの構築を図2に示す。pXINSECT-DEST38の代わりにpIB/V5-His(Invitrogen)を用いたこと以外は、同様に操作を行って、プラスミドpIB-6D9 Hcを得た。
【0043】
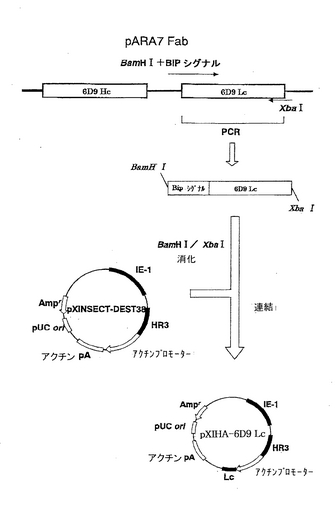
(L鎖を発現するプラスミドの調製)
L鎖を発現するプラスミドpXIHA-6D9 Lcの構築を図3に示す。まず、上流部分に制限酵素BamHI認識部位およびDrosophila由来のBiPシグナル配列を有し、下流部分に制限酵素XbaI認識部位を有する6D9 Lc遺伝子を調製した。この遺伝子は、プラスミドpARA7Fabを鋳型とし、配列番号3および4のプライマーを用いてPCR法で増幅した。配列番号3のプライマーは、BamHI認識部位およびBiPシグナル配列を有し、そして配列番号4のプライマーは、XbaI認識部位を有する。PCRは、Thermal cycler(BIO-RAD)を用いて、以下のサイクルで行った。まず、96℃にて2分、72℃にて2分処理し、次いで、96℃にて30秒、57.4℃にて30秒、および72℃にて2分の処理を35サイクル行い、最後に72℃にて5分の処理を行った。アガロースゲル電気泳動で目的のフラグメントが増幅されたことを確認し、当業者が通常用いる方法により6D9 Lc遺伝子を回収した。
【0044】
次に、pXINSECT-DEST38を、制限酵素BamHIおよびXbaIで消化し、CONCERT Gel Extraction Systems(Invitrogen)を用いてDNAを精製した。このDNAと制限酵素BamHIおよびXbaIで切断した6D9 Lc遺伝子とを混合してライゲーションを行い、E.coli K12株において形質転換を行って、当業者が通常用いる方法により、プラスミドpXIHA-6D9 Lcを得た。
【0045】
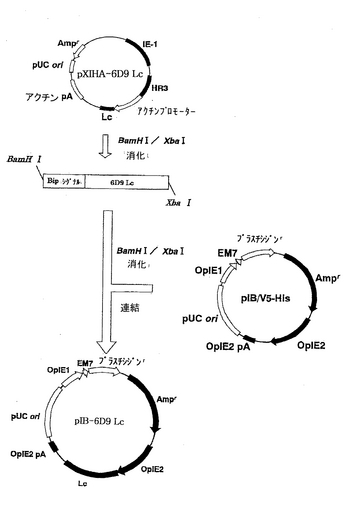
一方、L鎖を発現する別のプラスミドpIB-6D9 Lcの構築を図4に示す。pXINSECT-DEST38の代わりにpIB/V5-Hisを用いたこと以外は、同様に操作を行って、プラスミドpIB-6D9 Lcを得た。
【0046】
(昆虫細胞の形質転換)
宿主細胞としてTrichoplusia ni由来のBTI-TN5B1-4株(Invitrogen)(以下、High Five株という)およびSpodoptera frugiperda由来のSf9株(Invitrogen)を使用した。対数増殖期にあるSf9株またはHigh Five株を、それぞれ生細胞密度が4×105細胞/cm3となるように新鮮培地に懸濁し、直径35mmのディッシュ7枚にそれぞれ細胞懸濁培地2mLを播種した。なお、細胞密度の測定は血球計算盤を使用し、トリパンブルーを用いた色素排除法によって、生細胞のみを計数した。なお、Sf9株には無血清培地EX-CELL 420(JRH Biosciences)を用い、High Five株にはExpress Five SFM(Invitrogen)を用いた。
【0047】
構築したプラスミドpXIHA-6D9 HcおよびpXIHA-6D9 Lcを、質量比で2:1となるように混合した。この混合プラスミドDNA2μgに陽電荷脂質からなるFuGENE6(Roche)を6μl加え、室温で45分間インキュベートした。インキュベート後に、培地を加え、全量を100μlとして、High Five株およびSf9株を含む各ディッシュに加え、形質転換を行った。27℃で72時間インキュベートして上清を回収し、アルカリホスファターゼ標識抗マウスIgG(H+L)を用いるウエスタンブロッティングで抗体の生産を確認した。
【0048】
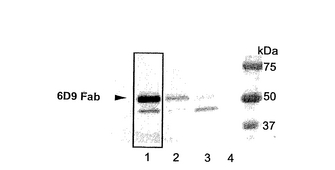
次に、pIB-6D9 HcおよびpIB-6D9 Lcの2:1混合プラスミドDNA2μgに陽電荷脂質からなるFuGENE6(Roche)を6μl加え、室温で45分間インキュベートした。インキュベート後に、培地を加え、全量を100μlとして、High Five株およびSf9株を含む各ディッシュに加え、形質転換を行った。上記と同様にウエスタンブロッティングで抗体の生産を確認した。結果を図5に示す。図5において、各レーンは以下の場合を示す。レーン1:pXIHA-6D9 HcおよびpXIHA-6D9 Lcを用いHigh Five株を宿主とした場合;レーン2:pXIHA-6D9 HcおよびpXIHA-6D9 Lcを用いSf9株を宿主とした場合;レーン3:pIB-6D9 HcおよびpIB-6D9 Lcを用いHigh Five株を宿主とした場合;レーン4:pIB-6D9 HcおよびpIB-6D9 Lcを用いSf9株を宿主とした場合。
【0049】
図5に示すように、ウエスタンブロッティングによる検出で、レーン1〜3の組合わせにおいて抗体の生産が認められた。特に、pXIHA-6D9 HcおよびpXIHA-6D9 Lcを用いHigh Five株を宿主とした場合(レーン1)に、顕著な抗体の生産が認められた。
【0050】
なお、各上清について、触媒抗体6D9の活性をELISAで測定した。ELISAは、以下の方法で行った。96ウェルプレート(Corning製)の各ウェルに、クロラムフェニコール・モノエステルの加水分解反応の遷移状態アナログであるホスホン酸エステルと牛血清アルブミンとの縮合物(ハプテン−BSA)を抗原として添加し(50μl)、4℃にて16時間静置し、常法によりブロッキングした。各ウェルを十分に洗浄した後、上記各上清を50μl加えて、室温で2時間インキュベートした。洗浄後、洗浄液で5μg/mLに希釈したヤギ抗マウスIgG(κ鎖特異的)HRP conjugate(Exalpha Biologicals)を100μl加えて、室温で1時間インキュベートした。ウェルを洗浄し、発色液を100μlずつ加えて遮光し、室温で20分インキュベートした後、各ウェルの405nmにおける吸光度をマイクロプレートリーダー(Bio-Rad)を用いて測定した。
【0051】
この方法により、ウエスタンブロッティングでバンドが検出できなかった上清のいずれにも、抗原結合活性があること、すなわち、触媒抗体6D9生産されたことが認められた(データは示さず)。また、High Five株を宿主とした場合の方が、Sf9株を宿主とした場合よりも、抗体の生産効率が高いことがわかった。しかし、これらの形質転換株は、一過的に抗体を発現したにすぎなかった。
【0052】
(実施例2)
上記で得られたベクターを用いて、(1)pIB-6D9 HcとpXIHA-6D9 Lcとの混合物、(2)pXIHA-6D9 HcとpIB-6D9 Lcとの混合物、(3)pIB-6D9 HcとpIB-6D9 Lcとの混合物、および(4)pXIHA-6D9 HcとpXIHA-6D9 Lcとの混合物(それぞれ、1:1の混合物)を用いて、実施例1と同様にして、High Five株を形質転換した。
【0053】
得られた形質転換株の抗体の発現についてのウエスタンブロッティングの結果を図6に示す。レーン1〜4は、上記(1)〜(4)のそれぞれの場合の結果を示す。図6に示すように、(1)、(2)、および(4)の組合わせで、抗体の生産が確認された。このうち、(1)および(2)のように、異種プロモーターの組合せでも、抗体を発現し得ることが確認された。しかし、これらの形質転換株も、一過的に抗体を発現したにすぎなかった。
【0054】
(実施例3)
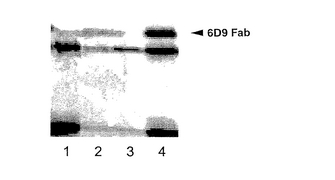
実施例2の (1) pIB-6D9 HcとpXIHA-6D9 Lcとの組合わせと (2) pXIHA-6D9 HcとpIB-6D9 Lcの組合わせにおいて、抗体の発現量を比較すると、図6に示すように、(2) の組合わせの場合、発現量が多いことがわかった。そこで、次に、pXIHA-6D9 HcとpIB-6D9 Lcの混合比を1:1、10:1、50:1および100:1として、実施例2と同様にHigh Five株を形質転換した後、27℃で48時間インキュベートした。次に、細胞を12ウェルプレートに移し、80μg/mlのブラスチシジンを含む培地でインキュベートした。3日毎または4日毎にブラスチシジンを含む培地で培地交換しながら,2週間インキュベートした。プラスミドを1:1または10:1の質量比で混合して形質転換した場合、ウェル内の至るところに多数の細胞が生育し、一個の細胞に由来するコロニーとして分離することはできなかった。また、プラスミドの質量比が100:1の場合、形質転換した細胞の生育は認められなかった。これに対し、プラスミド比が50:1の場合、各ウェル内にコロニーが数個ずつ得られた。これらのコロニーをマイクロピペットで吸い取り、1つのウェルにコロニーが1個ずつ入るように12ウェルプレートに移し、ブラスチシジン存在下、継代培養した。形質転換後50日経過した12ウェルプレートの各ウェルの培養上清中の触媒抗体6D9について、ウエスタンブロッティングを行った。結果を図7に示す。図7に示すように、形質転換して50日経過した後も形質転換株は抗体を生産しており、長時間安定に抗体を生産していることがわかる。また、プラスミドを50:1の質量比で混合した場合、効率よく形質転換株が得られたことがわかる。
【0055】
(実施例4)
(ブラスチシジン耐性遺伝子を有するプラスミドの構築)
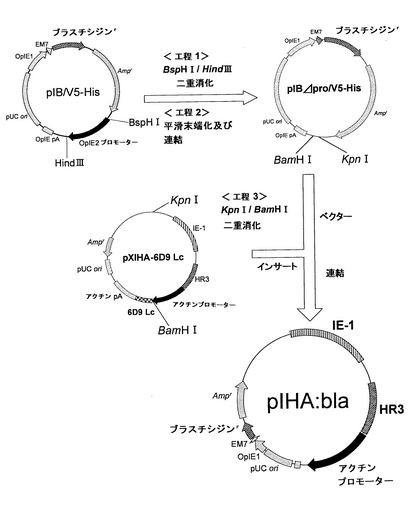
ブラスチシジン耐性遺伝子を有するプラスミドpIHA:blaの構築を図8に示す。
【0056】
まず、pIB/V5-HisのOpIE-2プロモーターを削除したプラスミドpIBΔpro/V5-Hisの構築を、以下のようにして行った。pIB/V5-Hisを、BspHIおよびHindIIIで消化して、アガロースゲル電気泳動で精製して、突出末端DNA断片を得た。次に、この突出末端DNA断片の末端平滑化を、DNA Blunting Kit(TaKaRa)を使用して行った。次いで、平滑末端DNAを含む溶液に、2×Ligation Mix(Ligation-Convenience Kit;和光純薬工業株式会社)10μlを加え、16℃にて20分間インキュベートした。これをE.coli K12株に形質転換し、ミニプレップおよびアルカリ1+2+3法を行い、pIBΔpro/V5-Hisを得た。
【0057】
次に、インサートとしてIE−1+HR3+アクチンプロモーターのDNA断片を得るために、上記実施例1で得たpXIHA-6D9 Lcを制限酵素KpnIおよびBamHIで消化した。また、ベクターとして、上記pIBΔpro/V5-Hisも制限酵素KpnIおよびBamHIで消化した。アガロース電気泳動により、ベクター側pIBΔpro/V5-Hisの約3000bpおよびインサート側IE−1+HR3+アクチンプロモーターの約6600bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、ベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップおよびアルカリ1+2+3法を行い、pIHA:blaを得た。
【0058】
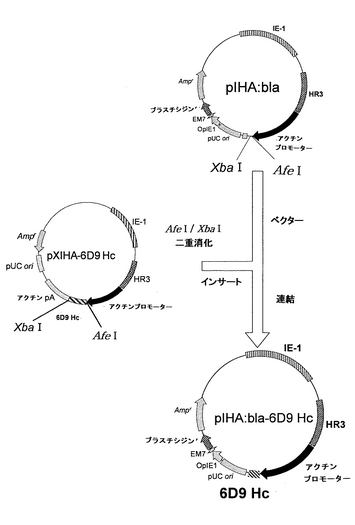
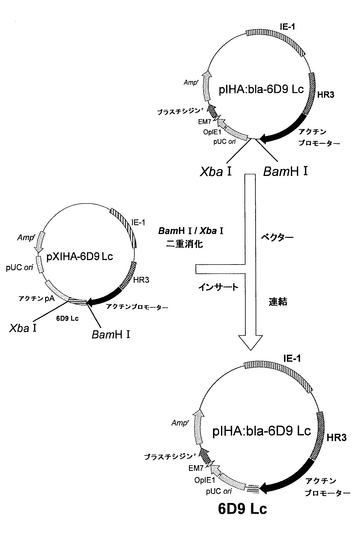
(ブラスチシジン耐性遺伝子およびH鎖遺伝子を有するプラスミドの構築)
上記ベクターpIHA:blaを用いて、H鎖を発現するためのベクターpIHA:bla-6D9 Hcの構築を、以下に記載するようにして行った。このブラスチシジン耐性遺伝子およびH鎖遺伝子を有するプラスミドpIHA:bla-6D9 Hcの構築を図9に示す。
【0059】
ベクターとして、上記pIHA:blaをAfeIおよびXbaIで消化した。また、インサート断片を得るために、上記実施例1で得たpXIHA-6D9 HcもAfeIおよびXbaIで消化した。アガロース電気泳動により、ベクター側pIHA:blaの約9600bpおよびインサート側pXIHA-6D9 Hcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、ベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップおよびアルカリ1+2+3法を行い、pIHA:bla-6D9 Hcを得た。
【0060】
(ブラスチシジン耐性遺伝子およびL鎖遺伝子を有するプラスミドの構築)
上記ベクターpIHA:blaを用いて、L鎖を発現するためのベクターpIHA:bla-6D9 Lcの構築を、以下に記載のように行った。このブラスチシジン耐性遺伝子およびL鎖遺伝子を有するプラスミドpIHA:bla-6D9 Lcの構築を図10に示す。
【0061】
ベクターとして、上記pIHA:blaをBamHIおよびXbaIで消化した。また、インサート断片を得るために、上記実施例1で得たpXIHA-6D9 LcもBamHIおよびXbaIで消化した。アガロース電気泳動により、ベクター側pIHA:blaの約9600bpおよびインサート側pXIHA-6D9 Lcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、ベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:bla-6D9 Lcを得た。
【0062】
(実施例5)
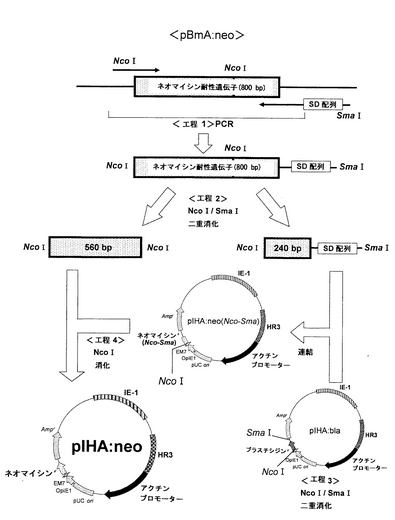
(ネオマイシン耐性遺伝子を有するプラスミドの構築)
上記実施例4で得たpIHA:blaのブラスチシジン耐性遺伝子をネオマイシン耐性遺伝子に置換し、pIHA:neoを構築した。ネオマイシン耐性遺伝子を有するプラスミドpIHA:neoの構築を図11に示す。
【0063】
ネオマイシン耐性遺伝子を、プラスミドpBmA:neoを鋳型とし、配列表の配列番号5および6に示すプライマーを用いて、PCR法で増幅した。これらのプライマーは、pBmA:neoから、ネオマイシン耐性遺伝子の一方の末端にNcoIおよび他方の末端にSmaIの認識塩基配列が位置するように設計した。配列番号5のプライマーは、NcoI認識部位を有し、配列番号6のプライマーは、SmaI認識部位を有する。
【0064】
PCRは、Thermal cycler(BIO-RAD)を用いて、以下の反応サイクルにより行った。まず、98℃にて10秒、98℃にて2分処理し、次いで、98℃にて2秒、60℃にて30秒、および72℃にて1分の処理を30サイクル行い、最後に4℃にて保持した。その後、アガロース電気泳動で目的の断片が増幅されたことを確認し、当業者が通常用いる方法によりネオマイシン耐性遺伝子を回収した。
【0065】
次に、この作製したネオマイシン耐性遺伝子を制限酵素NcoIおよびSmaIで消化した。アガロース電気泳動により、ネオマイシン耐性遺伝子の約560bpのDNA断片(ネオマイシンr Nco-Nco)および約260bpのDNA断片(ネオマイシンr Nco-Sma)を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次に、実施例4において構築したpIHA:blaを制限酵素NcoIおよびSmaIで消化した。アガロース電気泳動により、約9100bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。これにより、pIHA:blaからブラスチシジン耐性遺伝子を削除したDNA断片pIHA:Δblaを得た。
【0066】
次に、上記のベクター側DNA断片のpIHA:Δblaとインサート側DNA断片のネオマイシンr (Nco-Sma)とを、2×Ligation Mixを用いてライゲーションした。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:neo(Nco-Sma)を得た。
【0067】
次に、上記ベクターpIHA:neo(Nco-Sma)をNcoIで消化した。アガロース電気泳動によりベクター側pIHA:neo(Nco-Sma)の約9,400bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、得られたベクター側pIHA:neo(Nco-Sma)DNA断片とインサート側ネオマイシンr(Nco-Nco)DNA断片とのライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:neoを得た。
【0068】
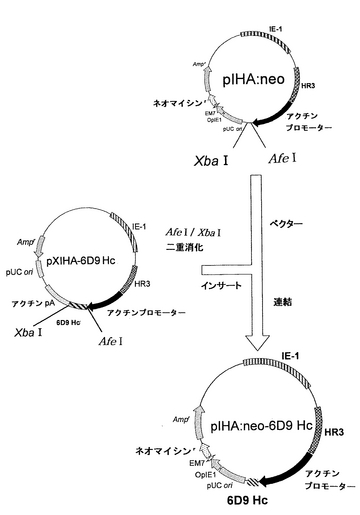
(ネオマイシン耐性遺伝子およびH鎖遺伝子を有するプラスミドの構築)
以下に記載のように、上記pIHA:neoを用いて、H鎖を発現するためのベクターpIHA:neo-6D9 Hcを構築した。このネオマイシン耐性遺伝子およびH鎖遺伝子を有するプラスミドpIHA:neo-6D9 Hcの構築を図12に示す。
【0069】
ベクターとして、上記pIHA:neoをAfeIおよびXbaIで消化した。また、インサート断片を得るために、上記実施例1で得たpXIHA-6D9 HcもAfeIおよびXbaIで消化した。アガロース電気泳動によりベクター側pIHA:neoの約10,000bpおよびインサート側pXIHA-6D9 Hcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いてベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:neo-6D9 Hcを得た。
【0070】
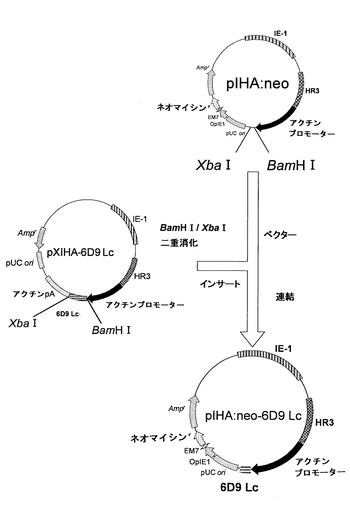
(ネオマイシン耐性遺伝子およびL鎖遺伝子を有するプラスミドの構築)
以下に記載のように、上記pIHA:neoを用いて、L鎖を発現するためのベクターpIHA:neo-6D9 Lcを構築した。このネオマイシン耐性遺伝子およびL鎖遺伝子を有するプラスミドpIHA:neo-6D9 Lcの構築を図13に示す。
【0071】
ベクターとして、上記pIHA:neoを、BamHI-XbaIで消化した。インサート断片を得るために、上記実施例1で得たpXIHA-6D9 LcもBamHI-XbaIで消化した。アガロース電気泳動により、ベクター側pIHA:neoの約10,000bpおよびインサート側pXIHA-6D9 Lcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いてベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、IHA:neo-6D9 Lcを得た。
【0072】
(実施例6)
(pIHA:blaおよびpIHA:neoを用いた一過性発現による6D9 Fabの発現確認)
上記実施例4および5で構築したベクターの組合わせ:(1)pIHA:bla-6D9 HcとpIHA:bla-6D9 Lcとの混合物、(2)pIHA:bla-6D9 HcとpIHA:neo-6D9 Lcとの混合物、(3)pIHA:neo-6D9 HcとpIHA:neo-6D9 Lcとの混合物、(4)pIHA:neo-6D9 HcとpIHA:bla-6D9 Lcとの混合物、および(5)pXIHA-6D9 HcとpXIHA-6D9 Lcとの混合物(それぞれ、1:1の混合物)を用いて、上記実施例1と同様にして、High Five株を形質転換した。上記実施例1と同様にして、ウエスタンブロッティングで抗体の生産を確認した。
【0073】
この結果を図14に示す。図中のレーン2〜5(上記の(1)〜(4)のそれぞれの結果を示す)において、選択マーカー耐性遺伝子を持たないpXIHAの場合(上記(5);レーン6)と同等以上のFabが分泌発現していることがわかった(図14中、レーン1はコントロールである)。これは、pIHA由来のベクター(約10500bp)はpXIHA(約11500bp)よりベクターサイズが小さいため、昆虫細胞へのトランスフェクション効率が若干上昇したことによると考えられる。
【0074】
(実施例7)
(安定形質転換細胞の選択のための最適な抗生物質濃度の検討)
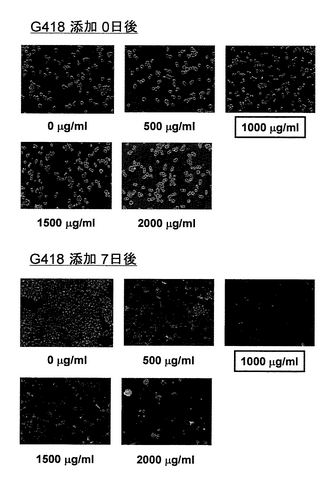
本実施例において、2種類の抗生物質(ブラスチシジンおよびG418)による選択の最適濃度の検討を行った。本実施例では、実施例3において80μg/mlのブラスチシジンを含む培地により樹立した安定形質転換細胞(pXIHA-6D9 Hc/pIB-6D9 Lc)を用いた。この細胞を12ウェルプレートに移し、Express Five SFMに80μg/mlのブラスチシジン、およびG418を0μg/ml、500μg/ml、1000μg/ml、1500μg/ml、または2000μg/mlを添加した培地で、27℃にて7日間インキュベートした。これら抗生物質を添加する前および添加して7日後の細胞の様子を、微分干渉顕微鏡を用いて観察した。この結果を図15に示す。図15の上方に、これらの両抗生物質を添加する前の細胞の様子を示し、図15の下方に、80μg/mlのブラスチシジンと、0μg/ml、500μg/ml、1000μg/ml、1500μg/ml、または2000μg/mlのG418とを添加して7日後の細胞の様子を示す(図中の濃度は、G418の濃度を表す)。図15より明らかなように、G418が1000μg/ml以上添加された培地では、細胞は完全に死滅していた。このことから、2種類の抗生物質を用いて安定形質転換細胞を効率的に得るためには、培地中のブラスチシジン濃度を80μg/mlおよびG418濃度を1000μg/mlで選択圧をかけることが必要であることがわかった。
【0075】
(実施例8)
(pIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcを用いた安定形質転換細胞の作製)
実施例5で作製したpIHA:neo-6D9 Hcと、実施例4で作製したpIHA:bla-6D9 Lcとを50:1、10:1、または1:1とで混合して、High Fiveにコトランスフェクションし、2種類の抗生物質(ブラスチシジンおよびG418)により選択圧をかけることにより、安定形質転換細胞の作製を試みた。
【0076】
35mmディッシュに、初期細胞密度が2.0×105細胞/cm3となるように培地2mlに懸濁した細胞を播種し、27℃にて24時間インキュベートした。細胞密度測定のために、血球計算盤を使用し、トリパンブルーを用いた色素排除法によって生細胞のみを計数した。3μl のFuGENE 6および1μgの全DNAを培地に添加して全量100μlとし、室温にて45分間インキュベートした。この反応液を上記の35mmディッシュに加え、27℃で細胞がコンフルエントになるまで静置培養した。その後、細胞をピペッティングにより剥離し、1.0×105細胞/cm3の細胞密度で、培地10mlに懸濁して100mmディッシュに播種した。播種24時間後に、ブラスチシジン(終濃度80μg/ml)およびG418(終濃度1000μg/ml)を添加した。その後、4日ごとに、ブラスチシジン80μg/mlおよびG418 1000μg/mlを添加したExpress Five SFMで培地交換を行った。細胞増殖が遅いときは、馴化培地と新鮮培地とを等量で混合した培地を用いた。抗生物質の添加から約2週間後、コロニー形成を目視で確認し、ピペットでコロニーをピックアップし、96ウェルプレートにコロニーを分注し(1ウェル当たり1コロニー)、27℃にて24時間静置培養した。ただし、培地は抗生物質を含まないものを1ウェル当たり100μlとした。培養24時間後にブラスチシジン(終濃度80μg/ml)およびG418(終濃度1000μg/ml)を添加し、細胞がコンフルエントになるまで静置培養を続けた。実施例1と同様にして、各ウェル中の培養上清をウエスタンブロッティングおよびELISAで分析して、Fab発現量が多いものを数十ウェル程度選択した。選択したウェル内の細胞を穏やかにピペッティングして細胞を懸濁させ、12ウェルプレートに移し変えた後、27℃にて24時間静置培養した。ただし、培地は、抗生物質を含まないものを1ウェル当たり1mlとした。培養24時間後にブラスチシジン(終濃度80μg/ml)およびG418(終濃度1000μg/ml)を添加し、細胞がコンフルエントになるまで静置培養を続けた。各ウェル中の培養上清を、上記のようにウエスタンブロッティングおよびELISAで分析して、Fab発現量が多いものを数ウェル程度選択した。選択したウェル内の細胞を穏やかにピペッティングして細胞を懸濁させ、100mmディッシュあるいは25cm2 Tフラスコに移し変えた後、27℃にて24時間静置培養した。培養24時間後にブラスチシジン(終濃度20μg/ml)およびG418(終濃度200μg/ml)を添加し、細胞がコンフルエントになるまで静置培養を続けた。以降、同様の操作で継代を行った。
【0077】
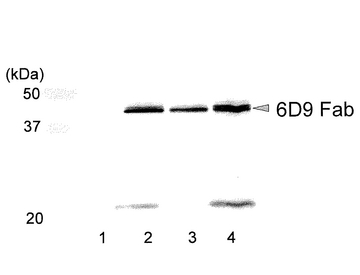
図16に、発現ベクターをトランスフェクションし、抗生物質存在下で16日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す。pIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcのコトランスフェクションの比率にかかわらず、比較的長期間安定にFabを分泌発現していることが確認された(レーン2〜4;それぞれ、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが50/1、10/1、1/1であることを示す;ここでレーン1はコントロールである)。また、実施例6における一過性発現での発現確認の結果に比べ、L鎖あるいはL鎖の二量体の発現が減少していた。このことから、HcおよびLcの遺伝子がともに導入された細胞のみが効率的に選択されていることが示唆された。
【0078】
(実施例9)
2種類の抗生物質の存在下で30日間培養を行ったこと以外は、実施例8と同様にして昆虫細胞へのコトランスフェクションおよび安定形質転換細胞の選択を行った。
【0079】
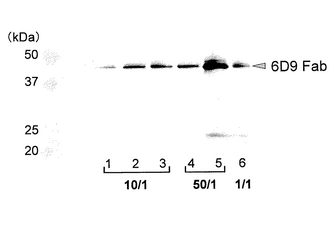
図17に、発現ベクターをトランスフェクションし、抗生物質存在下で30日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す(レーン1〜3、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが10/1;レーン4〜5、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが50/1;レーン6、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが1/1、これらにおいて、総DNA量は一定である)。図17から明らかなように、トランスフェクション30日後においても、Fabフラグメントが大量に分泌発現されていることがわかる。また、顕微鏡下における観察により、ネオマイシン耐性遺伝子を有するベクターの比率を高くすると(言い換えると、ブラスチシジン耐性遺伝子を有するベクターの比率を低くすると)、形成されるコロニーの数が減少し、コロニーを選択しやすくなることがわかった。レーン5では、Fabフラグメントの発現量も多く、高発現株が効率よく取得できることがわかる。
【産業上の利用可能性】
【0080】
本発明の方法は、昆虫細胞でヘテロオリゴマー蛋白質を効率的にかつ安定に分泌生産できるため、組換え蛋白質の生産の分野で広く利用され得る。特に、高等真核生物由来の抗体や酵素の製造に有用であり得る。
【図面の簡単な説明】
【0081】
【図1】ベクターpXIHA-6D9 Hcの構築を示す模式図である。
【図2】ベクターpIB-6D9 Hcの構築を示す模式図である。
【図3】ベクターpXIHA-6D9 Lcの構築を示す模式図である。
【図4】ベクターpIB-6D9 Lcの構築を示す模式図である。
【図5】形質転換昆虫細胞における抗体の産生についてのウエスタンブロッティングの結果を示す写真である。
【図6】種々のベクターの組合わせで形質転換した昆虫細胞における抗体の産生についてのウエスタンブロッティングの結果を示す写真である。
【図7】ベクターpXIHA-6D9 HcとpIB-6D9 Lcとで形質転換した昆虫細胞を50日間培養した場合の、培養上清中の抗体についてのウエスタンブロッティングの結果を示す写真である。
【図8】ベクターpIHA:blaの構築を示す模式図である。
【図9】ベクターpIHA:bla-6D9 Hcの構築を示す模式図である。
【図10】ベクターpIHA:bla-6D9 Lcの構築を示す模式図である。
【図11】ベクターpIHA:neoの構築を示す模式図である。
【図12】ベクターpIHA:neo-6D9 Hcの構築を示す模式図である。
【図13】ベクターpIHA:neo-6D9 Lcの構築を示す模式図である。
【図14】pIHA:blaおよびpIHA:neoを用いた種々のベクターの組合わせで形質転換した昆虫細胞における抗体の産生についてのウエスタンブロッティングの結果を示す写真である。
【図15】80μg/mlのブラスチシジンおよび種々の濃度のG418を含む選択培地で培養した場合のpXIHA-6D9 HcとpIB-6D9 Lcとで形質転換した昆虫細胞の様子を示す微分干渉顕微鏡写真である。
【図16】発現ベクターpIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcをトランスフェクションし、抗生物質存在下で16日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す写真である。
【図17】発現ベクターpIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcをトランスフェクションし、抗生物質存在下で30日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す写真である。
【技術分野】
【0001】
本発明は、昆虫細胞を用いてヘテロオリゴマー蛋白質を安定に生産する方法に関する。
【背景技術】
【0002】
遺伝子組換え技術を用いて、微生物、動物細胞などで蛋白質を高発現させる方法が種々検討されている。高等真核生物の蛋白質は、発現に際し、翻訳後修飾(例えば、糖鎖、脂肪酸などの付加、リン酸化)を受け、高次構造を保持するとともに、生物学的活性(機能)を有する。したがって、高等真核生物の蛋白質をDNA組換え技術で生産する場合、宿主として、高等真核生物である動物細胞を用いることが好ましい。しかし、動物細胞は取扱いが難しく、生育が遅いため、物質生産にはあまり適していない。そこで、翻訳後修飾活性を有する昆虫細胞を宿主として用いる蛋白質の発現系が検討されている。
【0003】
昆虫細胞を宿主とする蛋白質生産には、一般的に、昆虫細胞−バキュロウイルス系が用いられている。バキュロウイルス(核多角体病ウイルス)は、感染した昆虫細胞内で、ポリヘドリンとよばれるタンパク質を大量に生産するウイルスである。この昆虫細胞−バキュロウイルス系は、ポリヘドリンプロモーターの下流に外来遺伝子を挿入した組換えバキュロウイルスを培養昆虫細胞に感染させ、ウイルス感染した細胞に外来タンパク質を生産させる組換えタンパク質発現系である。しかし、ウイルスの感染により細胞が死滅してしまうため、目的タンパク質を連続的に生産することはできない。
【0004】
この昆虫細胞−バキュロウイルス系以外にも、種々の昆虫宿主−ベクター系が開発されている。例えば、カイコガ由来のアクチンプロモーターの上流にバキュロウイルスBmNPV由来のエンハンサーIE1とBmNPV由来のトランス作用因子HR3を有するベクターにヒト組織プラスミノーゲンアクチベーター(tPA)遺伝子を導入し、昆虫細胞を形質転換してtPAを発現させた例があるが(非特許文献1)、蛋白質は必ずしも安定には生産されていない。
【0005】
蛋白質の中でも、異種サブユニットから構成されるオリゴマータンパク質(以下、ヘテロオリゴマー蛋白質という)を発現する場合、複数のベクターを使用することが多く、さらに、選択マーカーを有するプラスミドを導入する必要があるため、目的とする遺伝子を有する形質転換体が得られにくい。また、得られた形質転換体におけるベクターの保持が困難であり、安定にヘテロオリゴマー蛋白質を生産することは困難である。
【非特許文献1】Farrellら、Biotechnol. Bioeng.、64巻、426−433頁 (1999)
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、ヘテロオリゴマー蛋白質を安定に発現し得る形質転換昆虫細胞を作製し、組換えヘテロオリゴマー蛋白質を安定に生産する方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明は、昆虫細胞を用いて異種サブユニットから構成されるオリゴマー蛋白質を生産する方法を提供し、該方法は、該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで該昆虫細胞を形質転換する工程;該形質転換された昆虫細胞を培養する工程;および培養物から該オリゴマー蛋白質を回収する工程を含む。
【0008】
好適な実施態様では、上記オリゴマー蛋白質は抗体である。
【0009】
より好適な実施態様では、上記異種サブユニットは、それぞれ、抗体を構成するH鎖の少なくとも一部のペプチドおよび該抗体を構成するL鎖の少なくとも一部のペプチドである。
【0010】
他の好適な実施形態では、上記複数のベクターは、それぞれ同じまたは異なるプロモーターを有する。
【0011】
さらに他の好適な実施態様では、上記複数のベクターの少なくとも1つは、上記昆虫細胞の選択マーカーを有する。
【0012】
本発明はまた、異種サブユニットから構成されるオリゴマー蛋白質を安定に発現する形質転換昆虫細胞を提供し、該昆虫細胞は該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターを有する。
【0013】
好適な実施態様では、上記複数のベクターの少なくとも1つは、上記昆虫細胞の選択マーカーを有する。
【発明の効果】
【0014】
異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで昆虫細胞を形質転換することにより、効率よく、ヘテロオリゴマー蛋白質が生産される。さらに、複数のベクターの少なくとも1つが、昆虫細胞の選択マーカーを有することにより、形質転換効率が向上するとともに、形質転換細胞は、安定にヘテロオリゴマー蛋白質を生産し得る。
【発明を実施するための最良の形態】
【0015】
まず、本発明に用いる種々の材料について、説明する。
【0016】
(宿主細胞)
宿主細胞としては、昆虫細胞であれば特に制限なく利用できる。市販されている昆虫細胞株も利用される。例えば、Trichoplusia ni由来のBTI-TN5B1-4 (High Five)株、Spodoptera frugiperda由来のSf9株などが好ましく用いられる。これらの株は、Invitrogenから入手可能である。
【0017】
(プラスミド、ベクター)
本発明においては、昆虫細胞内で発現し得るプラスミドであれば特に制限なく使用される。好ましくは、バキュロウイルス系のプラスミドが用いられる。発現効率が高い点で、カイコガ由来のアクチンプロモーターの上流に、バキュロウイルスBmNPV由来のエンハンサーIE1およびBmNPV由来のトランス作用因子HR3を有するプラスミドが、より好ましい。このようなプラスミドとしては、pXINSECT-DEST38、pXINSECT-DEST39などが挙げられる。これらのプラスミドは、Invitrogenから入手可能である。
【0018】
また、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターを有するプラスミドも好ましく用いられる。このようなプラスミドとしては、例えば、pIB/V5-Hisが挙げられる。このプラスミドは、Invitrogenから入手可能である。
【0019】
なお、形質転換細胞の選択効率、および目的遺伝子の発現の安定性を向上させ得る点で、選択マーカーを有するプラスミドが好ましく用いられる。前述のpIB/V5-Hisは、ブラスチシジン(Blasticidin)に対する耐性遺伝子を有しており、選択マーカーを有するプラスミドとして、好ましい。
【0020】
(ヘテロオリゴマー蛋白質)
本発明において、ヘテロオリゴマー蛋白質とは、異種サブユニットから構成されるオリゴマー蛋白質をいう。ヘテロオリゴマー蛋白質としては、抗体、酵素、レセプター、イオン・チャンネル、ヘモグロビン、血液凝固因子などが挙げられる。抗体を例に説明すると、抗体はH鎖とL鎖とから構成されるヘテロオリゴマー蛋白質である。抗体を構成するH鎖は、全長であってもよく、H鎖の少なくとも一部の配列、例えば、VHドメイン、Fdフラグメント、FcフラグメントなどのH鎖の一部分であってもよい。さらに、これらを任意に含むフラグメントであってもよい。抗体を構成するL鎖は、全長であってもよく、H鎖と相互作用し得るVLドメインなどであってもよい。H鎖とL鎖とが相互作用し得るように、H鎖およびL鎖が選択される。
【0021】
(ベクターの構築)
本発明においては、ヘテロオリゴマー蛋白質を生産するために、異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターを用いて、形質転換(コトランスフェクション)する。本発明において用いられる複数のベクターは、それぞれ、プロモーター、および発現されるべきヘテロオリゴマー蛋白質を構成する異なる1つのサブユニットをコードするDNA配列を含む。これらのベクターはさらに、分泌のためのシグナル配列を含み得る。例えば、2つのサブユニットから構成されるヘテロオリゴマー蛋白質の場合を例に挙げると、一方のサブユニットを発現し得るベクターおよび他方のサブユニットを発現し得るベクターの、2つのベクターが用いられる。複数のベクターは、それぞれ、分泌のためのシグナル配列と、1つのサブユニットをコードするDNA配列とを有するように構築される。これらの複数のベクターは、同じ構成のプラスミドに、それぞれ異なるサブユニットをコードするDNA配列が挿入されたものであってもよく、異なる構成のプラスミドに、それぞれ異なるサブユニットをコードするDNA配列が挿入されたものであってもよい。
【0022】
異なる構成のプラスミドを用いてベクターを構築する場合、それぞれ異なるプロモーターを有するプラスミドを用いてもよい。各サブユニットをそれぞれ異なるプロモーターの制御下で発現させることにより、発現効率が高められ得る。2つの異なるサブユニットから構成されるヘテロオリゴマー蛋白質を発現させる場合、好ましい実施態様では、2つのベクターのいずれか一方は、カイコガ由来のアクチンプロモーターを有し、そして他方は、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターを有する。
【0023】
また、形質転換株の選択効率および目的遺伝子の発現の安定性を高める目的で、少なくとも1つのベクターは選択マーカー遺伝子を含むように構築することが好ましい。プラスミドが本来有する選択マーカー遺伝子を利用しても、または外部から選択マーカー遺伝子を挿入してもよい。選択マーカー遺伝子としては、ネオマイシン耐性遺伝子、ブラスチシジン耐性遺伝子、ハイグロマイシン耐性遺伝子、ピューロマイシン耐性遺伝子、ブレオマイシン耐性遺伝子などが挙げられる。例えば、前述のpIB/V5-Hisは、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターおよびブラスチシジン(Blasticidin)に対する耐性遺伝子を有しているので、本発明において用いられるベクターを構築するために好ましく利用され得る。さらに、本発明において用いられるベクターが、アクチンプロモーター、IE1エンハンサー、およびHR3トランスアクチベーター、ならびに選択マーカー遺伝子(例えば、ネオマイシン耐性遺伝子、ブラスチシジン耐性遺伝子など)を有するように構築され得ることは、形質転換株の選択効率およびベクターの安定性を高め得る点で、好ましい。
【0024】
好ましくは、本発明において用いられる複数のベクターのそれぞれが、選択マーカー遺伝子を含む。これらの選択マーカー遺伝子は、ベクター間で同じであっても、または異なっていてもよい。複数のベクターが、それぞれ、異なる選択マーカー遺伝子を含むことは、形質転換株の選択効率を高める点で、より好ましい。
【0025】
(昆虫細胞の形質転換)
昆虫細胞の形質転換について、抗体を発現させる場合を例に挙げて、具体的に説明する。以下の説明において、例えば、カイコガ由来のアクチンプロモーターを有するpXINSECT-DEST38のプラスミドに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−1およびベクターL−1とする。あるいは、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターとブラスチシジン耐性遺伝子とを有するpIB/V5-Hisのプラスミドに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−2およびベクターL−2とする。
【0026】
形質転換には、同じプラスミドに由来するベクターH−1とベクターL−1との組合わせ、あるいはベクターH−2とベクターL−2との組合わせが用いられ得る。この場合、H鎖を発現するベクターH−1(またはH−2)とL鎖を発現するベクターL−1(またはL−2)とは、質量比で2:1以上の比率であることが好ましい。また、形質転換には、異なるプラスミドに由来するベクターの組合わせである、ベクターH−1とベクターL−2との組合わせ、あるいはベクターH−2とベクターL−1との組合わせを用いてもよい。この異なるプラスミドに由来するベクターの組合わせを用いる場合は、それぞれのベクターに含まれるプロモーターも異なり、抗体の発現効率を高め得る。
【0027】
さらに、pIB/V5-Hisに由来するベクターH−2とベクターL−2との組合わせでは、ベクターは両方とも選択マーカーであるブラスチシジン耐性遺伝子を有している点で有利である。例えば、市販の昆虫細胞形質転換キットを用いて抗体生産株を取得する場合、2つのpXINSECT-DEST38にそれぞれH鎖およびL鎖を導入した2つの異なるベクター(ベクターH−1およびL−1)と、ネオマイシン耐性遺伝子を有するプラスミドとをコトランスフェクションし、ネオマイシン存在下で生育する細胞を選択する。しかし、ネオマイシン耐性遺伝子のみが導入された細胞を含めて、ネオマイシン耐性株がすべて生育するため、3つの遺伝子が同時に導入された細胞の選択効率が悪くなる。しかし、選択マーカー(例えば、ブラスチシジン耐性遺伝子)を有するベクターH−2またはベクターL−2を使用すると、ブラスチシジン耐性株は、少なくともH鎖またはL鎖を発現するので、形質転換株の選択効率が向上する。
【0028】
ベクターH−1とベクターL−2との組合わせ、すなわち、カイコガ由来のアクチンプロモーターを有するプラスミドにH鎖(またはその一部)をコードするDNA配列を挿入したベクター(ベクターH−1)と、Orgya pseudotsugata核多角体病ウイルス由来のIE−2プロモーターとブラスチシジン耐性遺伝子とを有するプラスミドにL鎖(またはその一部)をコードするDNA配列を挿入したベクター(ベクターL−2)との組合わせは、好ましく用いられる。この場合、ベクターH−1とベクターL−2との比率は、重量比で10:1以上であることが好ましく、20:1以上であることがより好ましい。50:1以上で行ってもよい。
【0029】
上述のように、形質転換株の選択効率および目的遺伝子の発現の安定性を高める目的で、選択マーカー遺伝子を含むベクターが構築され得る。このようなベクターの例を以下に示す。カイコガ由来のアクチンプロモーターと、選択マーカー遺伝子であるネオマイシン耐性遺伝子とを有するベクターに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−3およびベクターL−3とする。一方、カイコガ由来のアクチンプロモーターと、選択マーカー遺伝子であるブラスチシジン耐性遺伝子とを有するベクターに、H鎖(またはその一部)をコードするDNA配列、またはL鎖(またはその一部)をコードするDNA配列を挿入して構築したベクターを、それぞれベクターH−4およびベクターL−4とする。
【0030】
カイコガ由来のアクチンプロモーターと選択マーカー遺伝子とを含む複数のベクターを用いる場合、形質転換には、異なるサブユニットのそれぞれが(例えば、H鎖とL鎖とが)発現して相互作用する組合わせであれば、いずれの組合わせもが用いられ得る。上記の場合、ベクターH−3とベクターL−3、ベクターH−3とベクターL−4、ベクターH−4とベクターL−3、あるいはベクターH−4とベクターL−4とが組み合わされ得る。コトランスフェクションにおいて、H鎖を発現するベクターとL鎖を発現するベクターとの比率は、任意の重量比であり得る。好ましくは、重量比で、1:1以上、より好ましくは2:1以上、さらに好ましくは10:1以上、なおさらに好ましくは20:1以上である。50:1以上で行ってもよい。
【0031】
これらの複数のベクターで昆虫細胞を形質転換する手段は、特に限定されず、昆虫細胞について当業者が通常用いる手段により行われ得る。具体的には、リン酸カルシウム法、リポフェクション(リポソーム法)、エレクトロポレーション(電気穿孔法)などにより行われる。このようにして、ヘテロオリゴマー蛋白質を安定に生産し得る形質転換昆虫細胞株が得られる。
【0032】
形質転換された昆虫細胞は、選択培地を利用して選択され得る。この選択培地を用いる選択は、発現されるべきサブユニットをコードするDNAを含むベクター中に含まれた選択マーカー遺伝子を用いても、選択マーカー遺伝子を有する別のベクターとのコトランスフェクションによってもよい。形質転換された昆虫細胞を選択するために使用される選択培地には、導入された選択マーカー遺伝子に対応するネオマイシン、ブラスチシジンなどの抗生物質が添加され得る。
【0033】
安定形質転換細胞を効率良く取得するためには、抗生物質の耐性遺伝子が導入されなかった細胞が完全に死滅してしまうような抗生物質濃度の存在下で細胞を培養する必要がある。通常、安定形質転換細胞を選択するための抗生物質濃度は、一週間程度で完全に細胞が死滅してしまう濃度が最適である。このことを考慮して、形質転換細胞の選択のために異なる抗生物質を細胞に与える濃度は、当業者によって適宜決定され得る。
【0034】
例えば、選択マーカー遺伝子としてブラスチシジン耐性遺伝子を含む少なくとも1つのベクターをコトランスフェクションに用いる場合、ブラスチシジンの毒性が強いため、ブラスチシジン耐性遺伝子を有するベクターの比率を小さくすると、形成されるコロニーの数が減少するので、コロニーの選択がより容易になる。コトランスフェクションの際の他の抗生物質耐性遺伝子(例えば、ネオマイシン耐性遺伝子)を有するベクターとブラスチシジン耐性遺伝子を有するベクターとの比率は、任意の重量比であり得るが、好ましくは1:1以上、より好ましくは2:1以上、さらに好ましくは10:1以上、なおさらに好ましくは20:1以上である。50:1以上で行ってもよい。
【0035】
異なるサブユニットを発現するためのベクターが、それぞれ異なる選択マーカー遺伝子を含む場合、それぞれの選択マーカー遺伝子に対応する抗生物質を用いて、発現されるべき遺伝子が全て導入されている細胞を選択し得る。例えば、ベクターH−3は、ネオマイシン耐性遺伝子を選択マーカー遺伝子として含み、ベクターL−4は、ブラスチシジン耐性遺伝子を選択マーカー遺伝子として含むので、ネオマイシン処理に対して耐性である細胞は、H−3が導入され、ブラスチシジン処理に対して耐性である細胞は、L−4が導入されている。ネオマイシンおよびブラスチシジンの両方の処理に対して耐性である細胞は、H−3およびL−4の両方が、すなわち、H鎖およびL鎖の両方の遺伝子が導入されている。このように、異なるサブユニットの全てが発現している細胞の選択がより効率的になる。もちろん、ベクターH−4とベクターL−3との組合わせにおいても同様の効果が期待され得る。
【0036】
得られた形質転換細胞がヘテロオリゴマー蛋白質を生産することは、ウエスタンブロッティング、ELISAなどの当業者に周知の技術を用いて確認され得る。
【0037】
(ヘテロオリゴマー蛋白質の生産)
ヘテロオリゴマー蛋白質の生産が確認された形質転換昆虫細胞は、当業者が通常用いる培養条件下で培養される。培養期間後、生産されたヘテロオリゴマー蛋白質は、蛋白質に応じた適切な手段によって培養物(培養上清)から回収される。回収された蛋白質は、必要に応じて、適宜精製され得る。
【実施例】
【0038】
以下、実施例に基づいて本発明を説明するが、本発明はこの実施例に制限されない。
【0039】
以下の実施例において、ヘテロオリゴマー蛋白質として、マウス由来の触媒抗体6D9のFabフラグメントを用いた。触媒抗体6D9のFabフラグメントは、そのH鎖部分であるFdフラグメントおよびL鎖から構成されており、クロラムフェニコールモノエステルの加水分解反応を触媒する。このFdフラグメントおよびL鎖をコードするDNA配列を含むプラスミドpARA7Fabの構築に関しては、Miyashitaら,J. Mol. Biol.、267、1247-1257 (1997)に記載されている。このプラスミドpARA7Fabは、大阪府立大学先端科学研究所の藤井郁雄教授から分与された。
【0040】
(実施例1)
(H鎖を発現するプラスミドの構築)
H鎖を発現するプラスミドpXIHA-6D9 Hcの構築を図1に示す。まず、上流部分に制限酵素AfeI認識部位およびDrosophila由来のBiPシグナル配列を有し、下流部分に制限酵素XbaI認識部位を有する6D9 Hc遺伝子を調製した。この遺伝子は、プラスミドpARA7Fabを鋳型とし、配列番号1および2のプライマーを用いてPCR法で増幅した。配列番号1のプライマーは、AfeI認識部位およびBiPシグナル配列を有し、そして配列番号2のプライマーは、XbaI認識部位を有する。PCRは、Thermal cycler(BIO-RAD)を用いて、以下のサイクルで行った。まず、94℃にて2分、72℃にて2分処理し、次いで、94℃にて30秒、52.9℃にて30秒、および72℃にて2分の処理を30サイクル行い、最後に72℃にて5分の処理を行った。アガロースゲル電気泳動で目的のフラグメントが増幅されたことを確認し、当業者が通常用いる方法により6D9 Hc遺伝子を回収した。
【0041】
次に、pXINSECT-DEST38(Invitrogen)を、制限酵素AfeIおよびXbaIで消化し、CONCERT Gel Extraction Systems(Invitrogen)を用いてDNAを精製した。このDNAと制限酵素AfeIおよびXbaIで切断した6D9 Hc遺伝子とを混合して連結(ライゲーション)を行い、E.coli K12株(NovaBlue,メルク (株))において形質転換を行って、当業者が通常用いる方法により、プラスミドpXIHA-6D9 Hcを得た。なお、pXINSECT-DEST38のカタログには認識部位AfeIが存在しないが、pXINSECT-DEST38のEco47III部位は、AfeIと同一認識配列であるので、このプラスミドはAfeIで切断される。
【0042】
一方、H鎖を発現する別のプラスミドpIB-6D9 Hcの構築を図2に示す。pXINSECT-DEST38の代わりにpIB/V5-His(Invitrogen)を用いたこと以外は、同様に操作を行って、プラスミドpIB-6D9 Hcを得た。
【0043】
(L鎖を発現するプラスミドの調製)
L鎖を発現するプラスミドpXIHA-6D9 Lcの構築を図3に示す。まず、上流部分に制限酵素BamHI認識部位およびDrosophila由来のBiPシグナル配列を有し、下流部分に制限酵素XbaI認識部位を有する6D9 Lc遺伝子を調製した。この遺伝子は、プラスミドpARA7Fabを鋳型とし、配列番号3および4のプライマーを用いてPCR法で増幅した。配列番号3のプライマーは、BamHI認識部位およびBiPシグナル配列を有し、そして配列番号4のプライマーは、XbaI認識部位を有する。PCRは、Thermal cycler(BIO-RAD)を用いて、以下のサイクルで行った。まず、96℃にて2分、72℃にて2分処理し、次いで、96℃にて30秒、57.4℃にて30秒、および72℃にて2分の処理を35サイクル行い、最後に72℃にて5分の処理を行った。アガロースゲル電気泳動で目的のフラグメントが増幅されたことを確認し、当業者が通常用いる方法により6D9 Lc遺伝子を回収した。
【0044】
次に、pXINSECT-DEST38を、制限酵素BamHIおよびXbaIで消化し、CONCERT Gel Extraction Systems(Invitrogen)を用いてDNAを精製した。このDNAと制限酵素BamHIおよびXbaIで切断した6D9 Lc遺伝子とを混合してライゲーションを行い、E.coli K12株において形質転換を行って、当業者が通常用いる方法により、プラスミドpXIHA-6D9 Lcを得た。
【0045】
一方、L鎖を発現する別のプラスミドpIB-6D9 Lcの構築を図4に示す。pXINSECT-DEST38の代わりにpIB/V5-Hisを用いたこと以外は、同様に操作を行って、プラスミドpIB-6D9 Lcを得た。
【0046】
(昆虫細胞の形質転換)
宿主細胞としてTrichoplusia ni由来のBTI-TN5B1-4株(Invitrogen)(以下、High Five株という)およびSpodoptera frugiperda由来のSf9株(Invitrogen)を使用した。対数増殖期にあるSf9株またはHigh Five株を、それぞれ生細胞密度が4×105細胞/cm3となるように新鮮培地に懸濁し、直径35mmのディッシュ7枚にそれぞれ細胞懸濁培地2mLを播種した。なお、細胞密度の測定は血球計算盤を使用し、トリパンブルーを用いた色素排除法によって、生細胞のみを計数した。なお、Sf9株には無血清培地EX-CELL 420(JRH Biosciences)を用い、High Five株にはExpress Five SFM(Invitrogen)を用いた。
【0047】
構築したプラスミドpXIHA-6D9 HcおよびpXIHA-6D9 Lcを、質量比で2:1となるように混合した。この混合プラスミドDNA2μgに陽電荷脂質からなるFuGENE6(Roche)を6μl加え、室温で45分間インキュベートした。インキュベート後に、培地を加え、全量を100μlとして、High Five株およびSf9株を含む各ディッシュに加え、形質転換を行った。27℃で72時間インキュベートして上清を回収し、アルカリホスファターゼ標識抗マウスIgG(H+L)を用いるウエスタンブロッティングで抗体の生産を確認した。
【0048】
次に、pIB-6D9 HcおよびpIB-6D9 Lcの2:1混合プラスミドDNA2μgに陽電荷脂質からなるFuGENE6(Roche)を6μl加え、室温で45分間インキュベートした。インキュベート後に、培地を加え、全量を100μlとして、High Five株およびSf9株を含む各ディッシュに加え、形質転換を行った。上記と同様にウエスタンブロッティングで抗体の生産を確認した。結果を図5に示す。図5において、各レーンは以下の場合を示す。レーン1:pXIHA-6D9 HcおよびpXIHA-6D9 Lcを用いHigh Five株を宿主とした場合;レーン2:pXIHA-6D9 HcおよびpXIHA-6D9 Lcを用いSf9株を宿主とした場合;レーン3:pIB-6D9 HcおよびpIB-6D9 Lcを用いHigh Five株を宿主とした場合;レーン4:pIB-6D9 HcおよびpIB-6D9 Lcを用いSf9株を宿主とした場合。
【0049】
図5に示すように、ウエスタンブロッティングによる検出で、レーン1〜3の組合わせにおいて抗体の生産が認められた。特に、pXIHA-6D9 HcおよびpXIHA-6D9 Lcを用いHigh Five株を宿主とした場合(レーン1)に、顕著な抗体の生産が認められた。
【0050】
なお、各上清について、触媒抗体6D9の活性をELISAで測定した。ELISAは、以下の方法で行った。96ウェルプレート(Corning製)の各ウェルに、クロラムフェニコール・モノエステルの加水分解反応の遷移状態アナログであるホスホン酸エステルと牛血清アルブミンとの縮合物(ハプテン−BSA)を抗原として添加し(50μl)、4℃にて16時間静置し、常法によりブロッキングした。各ウェルを十分に洗浄した後、上記各上清を50μl加えて、室温で2時間インキュベートした。洗浄後、洗浄液で5μg/mLに希釈したヤギ抗マウスIgG(κ鎖特異的)HRP conjugate(Exalpha Biologicals)を100μl加えて、室温で1時間インキュベートした。ウェルを洗浄し、発色液を100μlずつ加えて遮光し、室温で20分インキュベートした後、各ウェルの405nmにおける吸光度をマイクロプレートリーダー(Bio-Rad)を用いて測定した。
【0051】
この方法により、ウエスタンブロッティングでバンドが検出できなかった上清のいずれにも、抗原結合活性があること、すなわち、触媒抗体6D9生産されたことが認められた(データは示さず)。また、High Five株を宿主とした場合の方が、Sf9株を宿主とした場合よりも、抗体の生産効率が高いことがわかった。しかし、これらの形質転換株は、一過的に抗体を発現したにすぎなかった。
【0052】
(実施例2)
上記で得られたベクターを用いて、(1)pIB-6D9 HcとpXIHA-6D9 Lcとの混合物、(2)pXIHA-6D9 HcとpIB-6D9 Lcとの混合物、(3)pIB-6D9 HcとpIB-6D9 Lcとの混合物、および(4)pXIHA-6D9 HcとpXIHA-6D9 Lcとの混合物(それぞれ、1:1の混合物)を用いて、実施例1と同様にして、High Five株を形質転換した。
【0053】
得られた形質転換株の抗体の発現についてのウエスタンブロッティングの結果を図6に示す。レーン1〜4は、上記(1)〜(4)のそれぞれの場合の結果を示す。図6に示すように、(1)、(2)、および(4)の組合わせで、抗体の生産が確認された。このうち、(1)および(2)のように、異種プロモーターの組合せでも、抗体を発現し得ることが確認された。しかし、これらの形質転換株も、一過的に抗体を発現したにすぎなかった。
【0054】
(実施例3)
実施例2の (1) pIB-6D9 HcとpXIHA-6D9 Lcとの組合わせと (2) pXIHA-6D9 HcとpIB-6D9 Lcの組合わせにおいて、抗体の発現量を比較すると、図6に示すように、(2) の組合わせの場合、発現量が多いことがわかった。そこで、次に、pXIHA-6D9 HcとpIB-6D9 Lcの混合比を1:1、10:1、50:1および100:1として、実施例2と同様にHigh Five株を形質転換した後、27℃で48時間インキュベートした。次に、細胞を12ウェルプレートに移し、80μg/mlのブラスチシジンを含む培地でインキュベートした。3日毎または4日毎にブラスチシジンを含む培地で培地交換しながら,2週間インキュベートした。プラスミドを1:1または10:1の質量比で混合して形質転換した場合、ウェル内の至るところに多数の細胞が生育し、一個の細胞に由来するコロニーとして分離することはできなかった。また、プラスミドの質量比が100:1の場合、形質転換した細胞の生育は認められなかった。これに対し、プラスミド比が50:1の場合、各ウェル内にコロニーが数個ずつ得られた。これらのコロニーをマイクロピペットで吸い取り、1つのウェルにコロニーが1個ずつ入るように12ウェルプレートに移し、ブラスチシジン存在下、継代培養した。形質転換後50日経過した12ウェルプレートの各ウェルの培養上清中の触媒抗体6D9について、ウエスタンブロッティングを行った。結果を図7に示す。図7に示すように、形質転換して50日経過した後も形質転換株は抗体を生産しており、長時間安定に抗体を生産していることがわかる。また、プラスミドを50:1の質量比で混合した場合、効率よく形質転換株が得られたことがわかる。
【0055】
(実施例4)
(ブラスチシジン耐性遺伝子を有するプラスミドの構築)
ブラスチシジン耐性遺伝子を有するプラスミドpIHA:blaの構築を図8に示す。
【0056】
まず、pIB/V5-HisのOpIE-2プロモーターを削除したプラスミドpIBΔpro/V5-Hisの構築を、以下のようにして行った。pIB/V5-Hisを、BspHIおよびHindIIIで消化して、アガロースゲル電気泳動で精製して、突出末端DNA断片を得た。次に、この突出末端DNA断片の末端平滑化を、DNA Blunting Kit(TaKaRa)を使用して行った。次いで、平滑末端DNAを含む溶液に、2×Ligation Mix(Ligation-Convenience Kit;和光純薬工業株式会社)10μlを加え、16℃にて20分間インキュベートした。これをE.coli K12株に形質転換し、ミニプレップおよびアルカリ1+2+3法を行い、pIBΔpro/V5-Hisを得た。
【0057】
次に、インサートとしてIE−1+HR3+アクチンプロモーターのDNA断片を得るために、上記実施例1で得たpXIHA-6D9 Lcを制限酵素KpnIおよびBamHIで消化した。また、ベクターとして、上記pIBΔpro/V5-Hisも制限酵素KpnIおよびBamHIで消化した。アガロース電気泳動により、ベクター側pIBΔpro/V5-Hisの約3000bpおよびインサート側IE−1+HR3+アクチンプロモーターの約6600bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、ベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップおよびアルカリ1+2+3法を行い、pIHA:blaを得た。
【0058】
(ブラスチシジン耐性遺伝子およびH鎖遺伝子を有するプラスミドの構築)
上記ベクターpIHA:blaを用いて、H鎖を発現するためのベクターpIHA:bla-6D9 Hcの構築を、以下に記載するようにして行った。このブラスチシジン耐性遺伝子およびH鎖遺伝子を有するプラスミドpIHA:bla-6D9 Hcの構築を図9に示す。
【0059】
ベクターとして、上記pIHA:blaをAfeIおよびXbaIで消化した。また、インサート断片を得るために、上記実施例1で得たpXIHA-6D9 HcもAfeIおよびXbaIで消化した。アガロース電気泳動により、ベクター側pIHA:blaの約9600bpおよびインサート側pXIHA-6D9 Hcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、ベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップおよびアルカリ1+2+3法を行い、pIHA:bla-6D9 Hcを得た。
【0060】
(ブラスチシジン耐性遺伝子およびL鎖遺伝子を有するプラスミドの構築)
上記ベクターpIHA:blaを用いて、L鎖を発現するためのベクターpIHA:bla-6D9 Lcの構築を、以下に記載のように行った。このブラスチシジン耐性遺伝子およびL鎖遺伝子を有するプラスミドpIHA:bla-6D9 Lcの構築を図10に示す。
【0061】
ベクターとして、上記pIHA:blaをBamHIおよびXbaIで消化した。また、インサート断片を得るために、上記実施例1で得たpXIHA-6D9 LcもBamHIおよびXbaIで消化した。アガロース電気泳動により、ベクター側pIHA:blaの約9600bpおよびインサート側pXIHA-6D9 Lcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、ベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:bla-6D9 Lcを得た。
【0062】
(実施例5)
(ネオマイシン耐性遺伝子を有するプラスミドの構築)
上記実施例4で得たpIHA:blaのブラスチシジン耐性遺伝子をネオマイシン耐性遺伝子に置換し、pIHA:neoを構築した。ネオマイシン耐性遺伝子を有するプラスミドpIHA:neoの構築を図11に示す。
【0063】
ネオマイシン耐性遺伝子を、プラスミドpBmA:neoを鋳型とし、配列表の配列番号5および6に示すプライマーを用いて、PCR法で増幅した。これらのプライマーは、pBmA:neoから、ネオマイシン耐性遺伝子の一方の末端にNcoIおよび他方の末端にSmaIの認識塩基配列が位置するように設計した。配列番号5のプライマーは、NcoI認識部位を有し、配列番号6のプライマーは、SmaI認識部位を有する。
【0064】
PCRは、Thermal cycler(BIO-RAD)を用いて、以下の反応サイクルにより行った。まず、98℃にて10秒、98℃にて2分処理し、次いで、98℃にて2秒、60℃にて30秒、および72℃にて1分の処理を30サイクル行い、最後に4℃にて保持した。その後、アガロース電気泳動で目的の断片が増幅されたことを確認し、当業者が通常用いる方法によりネオマイシン耐性遺伝子を回収した。
【0065】
次に、この作製したネオマイシン耐性遺伝子を制限酵素NcoIおよびSmaIで消化した。アガロース電気泳動により、ネオマイシン耐性遺伝子の約560bpのDNA断片(ネオマイシンr Nco-Nco)および約260bpのDNA断片(ネオマイシンr Nco-Sma)を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次に、実施例4において構築したpIHA:blaを制限酵素NcoIおよびSmaIで消化した。アガロース電気泳動により、約9100bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。これにより、pIHA:blaからブラスチシジン耐性遺伝子を削除したDNA断片pIHA:Δblaを得た。
【0066】
次に、上記のベクター側DNA断片のpIHA:Δblaとインサート側DNA断片のネオマイシンr (Nco-Sma)とを、2×Ligation Mixを用いてライゲーションした。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:neo(Nco-Sma)を得た。
【0067】
次に、上記ベクターpIHA:neo(Nco-Sma)をNcoIで消化した。アガロース電気泳動によりベクター側pIHA:neo(Nco-Sma)の約9,400bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いて、得られたベクター側pIHA:neo(Nco-Sma)DNA断片とインサート側ネオマイシンr(Nco-Nco)DNA断片とのライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:neoを得た。
【0068】
(ネオマイシン耐性遺伝子およびH鎖遺伝子を有するプラスミドの構築)
以下に記載のように、上記pIHA:neoを用いて、H鎖を発現するためのベクターpIHA:neo-6D9 Hcを構築した。このネオマイシン耐性遺伝子およびH鎖遺伝子を有するプラスミドpIHA:neo-6D9 Hcの構築を図12に示す。
【0069】
ベクターとして、上記pIHA:neoをAfeIおよびXbaIで消化した。また、インサート断片を得るために、上記実施例1で得たpXIHA-6D9 HcもAfeIおよびXbaIで消化した。アガロース電気泳動によりベクター側pIHA:neoの約10,000bpおよびインサート側pXIHA-6D9 Hcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いてベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、pIHA:neo-6D9 Hcを得た。
【0070】
(ネオマイシン耐性遺伝子およびL鎖遺伝子を有するプラスミドの構築)
以下に記載のように、上記pIHA:neoを用いて、L鎖を発現するためのベクターpIHA:neo-6D9 Lcを構築した。このネオマイシン耐性遺伝子およびL鎖遺伝子を有するプラスミドpIHA:neo-6D9 Lcの構築を図13に示す。
【0071】
ベクターとして、上記pIHA:neoを、BamHI-XbaIで消化した。インサート断片を得るために、上記実施例1で得たpXIHA-6D9 LcもBamHI-XbaIで消化した。アガロース電気泳動により、ベクター側pIHA:neoの約10,000bpおよびインサート側pXIHA-6D9 Lcの約700bpのDNA断片を分離し、Wizard SV Gel and PCR Clean-UP System(Promega)を用いて精製した。次いで、2×Ligation Mixを用いてベクター側とインサート側とのDNA断片のライゲーションを行った。この連結物をE.coli K12株に形質転換し、ミニプレップ、アルカリ1+2+3法を行い、IHA:neo-6D9 Lcを得た。
【0072】
(実施例6)
(pIHA:blaおよびpIHA:neoを用いた一過性発現による6D9 Fabの発現確認)
上記実施例4および5で構築したベクターの組合わせ:(1)pIHA:bla-6D9 HcとpIHA:bla-6D9 Lcとの混合物、(2)pIHA:bla-6D9 HcとpIHA:neo-6D9 Lcとの混合物、(3)pIHA:neo-6D9 HcとpIHA:neo-6D9 Lcとの混合物、(4)pIHA:neo-6D9 HcとpIHA:bla-6D9 Lcとの混合物、および(5)pXIHA-6D9 HcとpXIHA-6D9 Lcとの混合物(それぞれ、1:1の混合物)を用いて、上記実施例1と同様にして、High Five株を形質転換した。上記実施例1と同様にして、ウエスタンブロッティングで抗体の生産を確認した。
【0073】
この結果を図14に示す。図中のレーン2〜5(上記の(1)〜(4)のそれぞれの結果を示す)において、選択マーカー耐性遺伝子を持たないpXIHAの場合(上記(5);レーン6)と同等以上のFabが分泌発現していることがわかった(図14中、レーン1はコントロールである)。これは、pIHA由来のベクター(約10500bp)はpXIHA(約11500bp)よりベクターサイズが小さいため、昆虫細胞へのトランスフェクション効率が若干上昇したことによると考えられる。
【0074】
(実施例7)
(安定形質転換細胞の選択のための最適な抗生物質濃度の検討)
本実施例において、2種類の抗生物質(ブラスチシジンおよびG418)による選択の最適濃度の検討を行った。本実施例では、実施例3において80μg/mlのブラスチシジンを含む培地により樹立した安定形質転換細胞(pXIHA-6D9 Hc/pIB-6D9 Lc)を用いた。この細胞を12ウェルプレートに移し、Express Five SFMに80μg/mlのブラスチシジン、およびG418を0μg/ml、500μg/ml、1000μg/ml、1500μg/ml、または2000μg/mlを添加した培地で、27℃にて7日間インキュベートした。これら抗生物質を添加する前および添加して7日後の細胞の様子を、微分干渉顕微鏡を用いて観察した。この結果を図15に示す。図15の上方に、これらの両抗生物質を添加する前の細胞の様子を示し、図15の下方に、80μg/mlのブラスチシジンと、0μg/ml、500μg/ml、1000μg/ml、1500μg/ml、または2000μg/mlのG418とを添加して7日後の細胞の様子を示す(図中の濃度は、G418の濃度を表す)。図15より明らかなように、G418が1000μg/ml以上添加された培地では、細胞は完全に死滅していた。このことから、2種類の抗生物質を用いて安定形質転換細胞を効率的に得るためには、培地中のブラスチシジン濃度を80μg/mlおよびG418濃度を1000μg/mlで選択圧をかけることが必要であることがわかった。
【0075】
(実施例8)
(pIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcを用いた安定形質転換細胞の作製)
実施例5で作製したpIHA:neo-6D9 Hcと、実施例4で作製したpIHA:bla-6D9 Lcとを50:1、10:1、または1:1とで混合して、High Fiveにコトランスフェクションし、2種類の抗生物質(ブラスチシジンおよびG418)により選択圧をかけることにより、安定形質転換細胞の作製を試みた。
【0076】
35mmディッシュに、初期細胞密度が2.0×105細胞/cm3となるように培地2mlに懸濁した細胞を播種し、27℃にて24時間インキュベートした。細胞密度測定のために、血球計算盤を使用し、トリパンブルーを用いた色素排除法によって生細胞のみを計数した。3μl のFuGENE 6および1μgの全DNAを培地に添加して全量100μlとし、室温にて45分間インキュベートした。この反応液を上記の35mmディッシュに加え、27℃で細胞がコンフルエントになるまで静置培養した。その後、細胞をピペッティングにより剥離し、1.0×105細胞/cm3の細胞密度で、培地10mlに懸濁して100mmディッシュに播種した。播種24時間後に、ブラスチシジン(終濃度80μg/ml)およびG418(終濃度1000μg/ml)を添加した。その後、4日ごとに、ブラスチシジン80μg/mlおよびG418 1000μg/mlを添加したExpress Five SFMで培地交換を行った。細胞増殖が遅いときは、馴化培地と新鮮培地とを等量で混合した培地を用いた。抗生物質の添加から約2週間後、コロニー形成を目視で確認し、ピペットでコロニーをピックアップし、96ウェルプレートにコロニーを分注し(1ウェル当たり1コロニー)、27℃にて24時間静置培養した。ただし、培地は抗生物質を含まないものを1ウェル当たり100μlとした。培養24時間後にブラスチシジン(終濃度80μg/ml)およびG418(終濃度1000μg/ml)を添加し、細胞がコンフルエントになるまで静置培養を続けた。実施例1と同様にして、各ウェル中の培養上清をウエスタンブロッティングおよびELISAで分析して、Fab発現量が多いものを数十ウェル程度選択した。選択したウェル内の細胞を穏やかにピペッティングして細胞を懸濁させ、12ウェルプレートに移し変えた後、27℃にて24時間静置培養した。ただし、培地は、抗生物質を含まないものを1ウェル当たり1mlとした。培養24時間後にブラスチシジン(終濃度80μg/ml)およびG418(終濃度1000μg/ml)を添加し、細胞がコンフルエントになるまで静置培養を続けた。各ウェル中の培養上清を、上記のようにウエスタンブロッティングおよびELISAで分析して、Fab発現量が多いものを数ウェル程度選択した。選択したウェル内の細胞を穏やかにピペッティングして細胞を懸濁させ、100mmディッシュあるいは25cm2 Tフラスコに移し変えた後、27℃にて24時間静置培養した。培養24時間後にブラスチシジン(終濃度20μg/ml)およびG418(終濃度200μg/ml)を添加し、細胞がコンフルエントになるまで静置培養を続けた。以降、同様の操作で継代を行った。
【0077】
図16に、発現ベクターをトランスフェクションし、抗生物質存在下で16日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す。pIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcのコトランスフェクションの比率にかかわらず、比較的長期間安定にFabを分泌発現していることが確認された(レーン2〜4;それぞれ、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが50/1、10/1、1/1であることを示す;ここでレーン1はコントロールである)。また、実施例6における一過性発現での発現確認の結果に比べ、L鎖あるいはL鎖の二量体の発現が減少していた。このことから、HcおよびLcの遺伝子がともに導入された細胞のみが効率的に選択されていることが示唆された。
【0078】
(実施例9)
2種類の抗生物質の存在下で30日間培養を行ったこと以外は、実施例8と同様にして昆虫細胞へのコトランスフェクションおよび安定形質転換細胞の選択を行った。
【0079】
図17に、発現ベクターをトランスフェクションし、抗生物質存在下で30日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す(レーン1〜3、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが10/1;レーン4〜5、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが50/1;レーン6、pIHA:neo-6D9 Hc/pIHA:bla-6D9 Lcが1/1、これらにおいて、総DNA量は一定である)。図17から明らかなように、トランスフェクション30日後においても、Fabフラグメントが大量に分泌発現されていることがわかる。また、顕微鏡下における観察により、ネオマイシン耐性遺伝子を有するベクターの比率を高くすると(言い換えると、ブラスチシジン耐性遺伝子を有するベクターの比率を低くすると)、形成されるコロニーの数が減少し、コロニーを選択しやすくなることがわかった。レーン5では、Fabフラグメントの発現量も多く、高発現株が効率よく取得できることがわかる。
【産業上の利用可能性】
【0080】
本発明の方法は、昆虫細胞でヘテロオリゴマー蛋白質を効率的にかつ安定に分泌生産できるため、組換え蛋白質の生産の分野で広く利用され得る。特に、高等真核生物由来の抗体や酵素の製造に有用であり得る。
【図面の簡単な説明】
【0081】
【図1】ベクターpXIHA-6D9 Hcの構築を示す模式図である。
【図2】ベクターpIB-6D9 Hcの構築を示す模式図である。
【図3】ベクターpXIHA-6D9 Lcの構築を示す模式図である。
【図4】ベクターpIB-6D9 Lcの構築を示す模式図である。
【図5】形質転換昆虫細胞における抗体の産生についてのウエスタンブロッティングの結果を示す写真である。
【図6】種々のベクターの組合わせで形質転換した昆虫細胞における抗体の産生についてのウエスタンブロッティングの結果を示す写真である。
【図7】ベクターpXIHA-6D9 HcとpIB-6D9 Lcとで形質転換した昆虫細胞を50日間培養した場合の、培養上清中の抗体についてのウエスタンブロッティングの結果を示す写真である。
【図8】ベクターpIHA:blaの構築を示す模式図である。
【図9】ベクターpIHA:bla-6D9 Hcの構築を示す模式図である。
【図10】ベクターpIHA:bla-6D9 Lcの構築を示す模式図である。
【図11】ベクターpIHA:neoの構築を示す模式図である。
【図12】ベクターpIHA:neo-6D9 Hcの構築を示す模式図である。
【図13】ベクターpIHA:neo-6D9 Lcの構築を示す模式図である。
【図14】pIHA:blaおよびpIHA:neoを用いた種々のベクターの組合わせで形質転換した昆虫細胞における抗体の産生についてのウエスタンブロッティングの結果を示す写真である。
【図15】80μg/mlのブラスチシジンおよび種々の濃度のG418を含む選択培地で培養した場合のpXIHA-6D9 HcとpIB-6D9 Lcとで形質転換した昆虫細胞の様子を示す微分干渉顕微鏡写真である。
【図16】発現ベクターpIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcをトランスフェクションし、抗生物質存在下で16日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す写真である。
【図17】発現ベクターpIHA:neo-6D9 HcおよびpIHA:bla-6D9 Lcをトランスフェクションし、抗生物質存在下で30日間培養を行った後の培養上清をウエスタンブロッティングにより分析した結果を示す写真である。
【特許請求の範囲】
【請求項1】
昆虫細胞を用いて異種サブユニットから構成されるオリゴマー蛋白質を生産する方法であって、
該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで該昆虫細胞を形質転換する工程;
該形質転換された昆虫細胞を培養する工程;および
培養物から該オリゴマー蛋白質を回収する工程
を含む、方法。
【請求項2】
前記オリゴマー蛋白質が抗体である、請求項1に記載の方法。
【請求項3】
前記異種サブユニットが、それぞれ、抗体を構成するH鎖の少なくとも一部のペプチドおよび該抗体を構成するL鎖の少なくとも一部のペプチドである、請求項2に記載の方法。
【請求項4】
前記複数のベクターが、それぞれ同じまたは異なるプロモーターを有する、請求項1から3のいずれかの項に記載の方法。
【請求項5】
前記複数のベクターの少なくとも1つが、前記昆虫細胞の選択マーカーを有する、請求項1から4のいずれかの項に記載の方法。
【請求項6】
異種サブユニットから構成されるオリゴマー蛋白質を安定に発現する形質転換昆虫細胞であって、該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターを有する、形質転換昆虫細胞。
【請求項7】
前記複数のベクターの少なくとも1つが、前記昆虫細胞の選択マーカーを有する、請求項6に記載の形質転換昆虫細胞。
【請求項1】
昆虫細胞を用いて異種サブユニットから構成されるオリゴマー蛋白質を生産する方法であって、
該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターで該昆虫細胞を形質転換する工程;
該形質転換された昆虫細胞を培養する工程;および
培養物から該オリゴマー蛋白質を回収する工程
を含む、方法。
【請求項2】
前記オリゴマー蛋白質が抗体である、請求項1に記載の方法。
【請求項3】
前記異種サブユニットが、それぞれ、抗体を構成するH鎖の少なくとも一部のペプチドおよび該抗体を構成するL鎖の少なくとも一部のペプチドである、請求項2に記載の方法。
【請求項4】
前記複数のベクターが、それぞれ同じまたは異なるプロモーターを有する、請求項1から3のいずれかの項に記載の方法。
【請求項5】
前記複数のベクターの少なくとも1つが、前記昆虫細胞の選択マーカーを有する、請求項1から4のいずれかの項に記載の方法。
【請求項6】
異種サブユニットから構成されるオリゴマー蛋白質を安定に発現する形質転換昆虫細胞であって、該異種サブユニットのうちのそれぞれ1つを発現し得る複数のベクターを有する、形質転換昆虫細胞。
【請求項7】
前記複数のベクターの少なくとも1つが、前記昆虫細胞の選択マーカーを有する、請求項6に記載の形質転換昆虫細胞。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】

【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】

【図15】
【図16】
【図17】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公開番号】特開2006−87427(P2006−87427A)
【公開日】平成18年4月6日(2006.4.6)
【国際特許分類】
【出願番号】特願2005−241472(P2005−241472)
【出願日】平成17年8月23日(2005.8.23)
【出願人】(502059825)バイオ・エナジー株式会社 (16)
【Fターム(参考)】
【公開日】平成18年4月6日(2006.4.6)
【国際特許分類】
【出願日】平成17年8月23日(2005.8.23)
【出願人】(502059825)バイオ・エナジー株式会社 (16)
【Fターム(参考)】
[ Back to top ]