
ヘテロ二官能性全セレクチン阻害剤
【課題】セレクチン媒介性機能(例えば、セレクチン依存性細胞接着)の阻害剤を同定し、並びに過剰なセレクチン活性に関連する病態を阻害するためにこのような化合物を使用する方法の提供。
【解決手段】セレクチン結合によって媒介されるin vitro及びin vivoプロセスを調節するための化合物及び方法を示す。より具体的には、セレクチンモジュレーター及びそれらの使用について記載し、セレクチン媒介性機能を調節(例えば、阻害又は増強)するこれらのセレクチンモジュレーターは、特定の糖模倣物を単独で、或いはBASA(ベンジルアミノスルホン酸)と呼ばれる化合物のクラスのメンバー又はBACA(ベンジルアミノカルボン酸)と呼ばれる化合物のクラスのメンバーと結合して含む。
【解決手段】セレクチン結合によって媒介されるin vitro及びin vivoプロセスを調節するための化合物及び方法を示す。より具体的には、セレクチンモジュレーター及びそれらの使用について記載し、セレクチン媒介性機能を調節(例えば、阻害又は増強)するこれらのセレクチンモジュレーターは、特定の糖模倣物を単独で、或いはBASA(ベンジルアミノスルホン酸)と呼ばれる化合物のクラスのメンバー又はBACA(ベンジルアミノカルボン酸)と呼ばれる化合物のクラスのメンバーと結合して含む。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は一般的に、セレクチン結合によって媒介されるプロセスを調節するための化合物、組成物、及び方法に関する。より具体的には、セレクチンモジュレーター及びそれらの使用に関し、セレクチン媒介性機能を調節するこれらのセレクチンモジュレーターは、特定の糖模倣物を単独で、或いはBASA(ベンジルアミノスルホン酸)と呼ばれる化合物のクラスのメンバー又はBACA(ベンジルアミノカルボン酸)と呼ばれる化合物のクラスのメンバーと結合して含む。
【背景技術】
【0002】
(関連技術の記載)
組織が感染又は損傷すると、炎症性プロセスによって、白血球及びその他の免疫系構成要素が感染又は損傷の部位に誘導される。このプロセスにおいては、白血球が、微生物の飲み込み及び消化において重要な役割を果たす。従って、感染又は損傷した組織への白血球の動員は、有効な免疫防御を備える上で極めて重要である。
【0003】
セレクチンは、内皮細胞への白血球の結合を媒介するために重要な、構造的に類似する細胞表面受容体の群である。これらのタンパク質は、I型膜タンパク質であり、アミノ末端レクチンドメイン、上皮成長因子(EGF)様ドメイン、種々の補体受容体関連反復、疎水性ドメインが広がる領域、及び細胞質ドメインから構成される。結合相互作用は、セレクチンのレクチンドメインと、種々の炭水化物リガンドとの接触によって媒介されると考えられている。
【0004】
既知のセレクチンには、E−セレクチン、P−セレクチン及びL−セレクチンの3種類が存在する。E−セレクチンは、毛細管の内部壁に沿って並ぶ活性化内皮細胞の表面に存在する。E−セレクチンは、炭水化物シアリル−ルイスX(SLeX)に結合する。これは、特定の白血球(単球及び好中球)の表面に糖タンパク質又は糖脂質として存在し、周囲組織が感染又は損傷している領域の毛細血管壁にこれらの細胞が接着するのを助ける。又、E−セレクチンは、多くの腫瘍細胞上に発現するシアリル−ルイスa(SLea)にも結合する。P−セレクチンは、炎症性内皮及び血小板上に発現し、これもSLeX及びSLeaを認識するが、硫酸化チロシンと相互作用する第2の部位も含む。一般的にE−セレクチン及びP−セレクチンの発現は、毛細血管に隣接する組織が感染又は損傷した場合に増加する。L−セレクチンは、白血球上に発現する。セレクチンにより媒介される細胞間接着は、セレクチン媒介性機能の一つの例である。
【0005】
セレクチン媒介性機能のモジュレーターには、PSGL−1タンパク質(及びより小さなペプチドフラグメント)、フコイダン(fucoidan)、グリチルリチン(及び誘導体)、抗セレクチン抗体、硫酸化ラクトース誘導体、並びにヘパリンが含まれる。これらは全て、不十分な活性、毒性、特異性の欠如、乏しいADME特徴、及び/又は材料の可用性によって、薬剤開発に不適切であることが示されている。
【0006】
セレクチン媒介性細胞接着は、感染と戦い、異物を破壊するために必要とされるが、このような細胞接着が望ましくないか又は過剰であり、組織の修復ではなく損傷が生じる状況もある。例えば、多くの病状(例えば、自己免疫疾患及び炎症性疾患、ショック症状、並びに再灌流障害)には、白血球の異常な接着が関与する。このような異常な細胞接着は、移植及び移植片の拒絶においても役割を果たす場合がある。更に、循環癌細胞の中には、活性化された内皮に結合するために炎症機序を利用するものもあると考えられている。このような状況において、セレクチン媒介性細胞間接着の調節が望まれる場合がある。
【発明の概要】
【発明が解決しようとする課題】
【0007】
従って、当該技術分野では、セレクチン媒介性機能(例えば、セレクチン依存性細胞接着)の阻害剤を同定し、並びに過剰なセレクチン活性に関連する病態を阻害するためにこのような化合物を使用する方法を開発することが必要とされている。本発明は、これらの必要性を満たす他、更にはその他の関連する利点も提供する。
【課題を解決するための手段】
【0008】
(発明の簡潔な要旨)
簡潔に述べると、本発明は、セレクチン媒介性プロセスを調節するための化合物、組成物、及び方法を提供する。本発明において、セレクチン媒介性機能を調節(例えば、阻害又は増強)する化合物は、特定の糖模倣物を単独で、或いはBASA又はBACAに結合して含む。このような化合物は、薬学的に許容される担体又は希釈剤と組み合わせて、薬学的組成物を形成する場合がある。本化合物又は組成物は、セレクチン媒介性機能(例えば、セレクチン媒介性細胞間接着の阻害)を調節(例えば、阻害又は増強)する方法で使用される場合がある。
【0009】
本発明の一態様においては、以下の化学式を有する化合物であって:
【0010】
【化17】

式中:
R1が、
【0011】
【化18】
(式中、nは0〜2であり、nが2の場合に、R8は独立して選択される)であり;
R2が、H、−C(=O)OX(式中、XはC1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)、−C(=O)NH(CH2)nNH2、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、nは0〜30であり、mは0〜1であり、Lはリンカーであり、Zはベンジルアミノスルホン酸、ベンジルアミノカルボン酸、ポリエチレングリコールである)、或いは二量体を形成するために上記の化学式を有する第二の化合物又はその塩であって、この場合、該第二の化合物又はその塩のR2がm=0であって、Zを有さず、且つ結合点であり;
R3が、−OH、
【0012】
【化19】
、−O−C(=O)−X、−NH2、−NH−C(=O)−NHX、又は−NH−C(=O)−X(式中、nは0〜2であり、Xは、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0013】
【化20】
【0014】
【化21】
(式中、nは0〜10である)からなる群から独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R4が、
【0015】
【化22】
、6’硫酸化GlcNAc、6’カルボキシル化GlcNAc、6’硫酸化GalNAc、6’硫酸化ガラクトース、6’カルボキシル化ガラクトース、又は
【0016】
【化23】
(式中、R9は、アリール、ヘテロアリール、シクロヘキサン、t−ブタン、アダマンタン、又はトリアゾールであり、且つR9の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C8アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R5がHであるか、或いはR4及びR5が一緒になって、
【0017】
【化24】
(式中、R10はアリール、ヘテロアリール、
【0018】
【化25】
(式中、nは0〜10である)であり、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)から独立して選択される1個〜3個で置換される場合がある)であり;
R6が、H、フコース、マンノース、アラビノース、ガラクトース、又はポリオールであり;
R7が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又は
【0019】
【化26】
であり;並びに
R8が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0020】
【化27】
(式中、nは0〜3であり、Xは、H、OH、Cl、F、N3、NH2、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、OC1−C8アルカニル、OC1−C8アルケニル、OC1−C8アルキニル、及びOC1−C14アリールから独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)である、
化合物が提供される。
【0021】
本発明の化合物は、その生理学的に許容される塩を含む。本発明の化合物は、薬学的に許容される担体又は希釈剤と組み合わせて、本発明の組成物を提供する。本明細書中の化学式において、記載された原子、又はその他2本の線の交差によって含意される炭素から伸長する線は、結合点を指す(メチル基を表さない)。
【0022】
本発明の一実施形態において、R6はフコース:
【0023】
【化28】
である。
【0024】
一実施形態において、R7はHである。
【0025】
一実施形態において、R4は
【0026】
【化29】
である。
【0027】
一実施形態において、R4は、
【0028】
【化30】
(式中、R9は上述の一般化学式のように定義される)である。
【0029】
一実施形態において、R9はシクロヘキサンである。
【0030】
一実施形態において、R6はガラクトースである。
【0031】
一実施形態において、R8は
【0032】
【化31】
である。
【0033】
一実施形態において、R2は、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、n、m、L及びZは、上述の一般化学式のように定義される)である。
【0034】
一実施形態において、Zは、ベンジルアミノスルホン酸、ベンジルアミノカルボン酸、又はポリエチレングリコールである。
【0035】
一実施形態において、R3は−O−C(=O)−X(式中、Xは上記の一般化学式のように定義される)である。
【0036】
一実施形態において、Xは、
【0037】
【化32】
である。
【0038】
一実施形態において、R5はHである。
【0039】
一実施形態において、Lは、ポリエチレングリコール又はチアジアゾールである。
【0040】
一実施形態において、化合物は、本発明による化合物を含み、更には診断薬又は治療薬も含む。このような化合物は、薬学的に許容される担体又は希釈剤と組み合わせて、本発明の組成物の一実施形態を形成する場合がある。
【0041】
本発明の別の態様においては、セレクチン媒介性機能を調節するために本発明の化合物又は組成物を使用する方法が提供される。このような化合物又は組成物は、例えば、セレクチン媒介性機能(例えば、セレクチン媒介性細胞間相互作用)を阻害又は増強するために使用される場合がある。化合物又は組成物は、セレクチンを発現する細胞を、セレクチンの機能を調節するのに有効な量で接触させる方法において使用される場合がある。化合物又は組成物は、過剰なセレクチン媒介性機能(例えば、過剰なセレクチン媒介細胞間接着)に関連する病態の発症を阻害する必要がある患者に、このような病態の発症を阻害するのに有効な量で投与する方法において使用される場合がある。このような病態の例には、炎症疾患、自己免疫疾患、感染症、癌、ショック、血栓症、創傷、火傷、再灌流傷害、血小板媒介性疾患、白血球媒介性肺傷害、脊髄損傷、消化管粘膜障害、骨粗鬆症、関節炎、喘息及びアレルギー反応が含まれる。化合物又は組成物は、移植した組織の受容者である患者に、移植した組織の拒絶を阻害するのに有効な量で投与する方法において使用される場合がある。化合物又は組成物は、セレクチンを発現する細胞を、この化合物又は組成物に結合した薬剤と接触させることにより、このような細胞にその薬剤(例えば、診断薬又は治療薬)を標的化するのに有効な量で使用する方法において使用される場合がある。化合物又は組成物は、例えば、上述の使用の何れかのための医薬品の製造において使用される場合がある。
【0042】
本発明のこれら及びその他の態様は、以下の詳細な説明及び添付の図面を参照することにより明らかになるであろう。本明細書に開示される全ての参考文献は、それぞれが個別に援用されるのと同じく、全体が参考として本明細書で援用される。
例えば、本願発明は以下の項目を提供する。
(項目1)
以下の化学式を有する化合物であって:
【化1】
式中:
R1が、
【化2】
(式中、nは0〜2であり、nが2の場合に、R8は独立して選択される)であり;
R2が、H、−C(=O)OX(式中、XはC1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)、−C(=O)NH(CH2)nNH2、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、nは0〜30であり、mは0〜1であり、Lはリンカーであり、Zはベンジルアミノスルホン酸、ベンジルアミノカルボン酸、ポリエチレングリコールである)、或いは二量体を形成するために上記の化学式を有する第二の化合物又はその塩であって、この場合、該第二の化合物又はその塩のR2がm=0であって、Zを有さず、且つ結合点であり;
R3が、−OH、
【化3】
、−O−C(=O)−X、−NH2、−NH−C(=O)−NHX、又は−NH−C(=O)−X(式中、nは0〜2であり、Xは、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【化4】
(式中、nは0〜10である)から独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R4が、
【化5】
、6’硫酸化GlcNAc、6’カルボキシル化GlcNAc、6’硫酸化GalNAc、6’硫酸化ガラクトース、6’カルボキシル化ガラクトース、又は
【化6】
(式中、R9は、アリール、ヘテロアリール、シクロヘキサン、t−ブタン、アダマンタン、又はトリアゾールであり、且つR9の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R5がHであるか、或いはR4及びR5が一緒になって、
【化7】
(式中、R10はアリール、ヘテロアリール、
【化8】
【化9】
(式中、nは0〜10である)を形成し、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)から独立して選択される1個〜3個で置換される場合がある)であり;
R6が、H、フコース、マンノース、アラビノース、ガラクトース、又はポリオールであり;
R7が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又は
【化10】
であり;並びに
R8が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【化11】
(式中、nは0〜3であり、Xは、H、OH、Cl、F、N3、NH2、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、OC1−C8アルカニル、OC1−C8アルケニル、OC1−C8アルキニル、及びOC1−C14アリールから独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)である、
化合物;或いはその生理学的に許容される塩。
(項目2)
R6が
【化12】
である、項目1に記載の化合物又はその塩。
(項目3)
R7がHである、項目1に記載の化合物又はその塩。
(項目4)
R4が
【化13】
である、項目1に記載の化合物又はその塩。
(項目5)
R4が
【化14】
(式中、R9は項目1に従って定義される)である、項目1に記載の化合物又はその塩。
(項目6)
R9がシクロヘキサンである、項目5に記載の化合物又はその塩。
(項目7)
R6がガラクトースである、項目1に記載の化合物又はその塩。
(項目8)
R8が
【化15】
である、項目1に記載の化合物又はその塩。
(項目9)
R2が、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、n、m、L及びZが項目1に従って定義される)である、項目1に記載の化合物又はその塩。
(項目10)
Zが、ベンジルアミノスルホン酸、ベンジルアミノカルボン酸、又はポリエチレングリコールである、項目9に記載の化合物又はその塩。
(項目11)
R3が、−O−C(=O)−X、又は−NH−C(=O)−X(式中、Xが項目1に従って定義される)である、項目1に記載の化合物又はその塩。
(項目12)
Xが
【化16】
である、項目11に記載の化合物又はその塩。
(項目13)
R5がHである、項目1に記載の化合物又はその塩。
(項目14)
Lがポリエチレングリコール又はチアジアゾールである、項目1〜13の何れか1項に記載の化合物又はその塩。
(項目15)
薬学的に許容される担体又は希釈剤と組み合わせて、項目1〜14の何れか1項に記載の化合物又はその塩を含む、組成物。
(項目16)
項目1〜14の何れか1項に記載の化合物又はその塩を含み、更に、診断薬又は治療薬も含む、化合物又はその生理学的に許容される塩。
(項目17)
薬学的に許容される担体又は希釈剤と組み合わせて、項目16に記載の化合物又はその塩を含む、組成物。
(項目18)
セレクチン媒介性機能を調節する方法であって、セレクチンを発現する細胞を、該セレクチンの機能を調節するのに有効な量で、項目1〜14の何れか1項に記載の化合物又はその塩と接触させることを含む、方法。
(項目19)
セレクチン媒介性機能を調節する方法であって、セレクチンを発現する細胞を、該セレクチンの機能を調節するのに有効な量で、項目15に記載の組成物と接触させることを含む、方法。
(項目20)
患者を処置する方法であって、過剰なセレクチン媒介性機能に関連する病態の発症を阻害する必要がある該患者に、当該病態の発症を阻害するのに有効な量で、項目1〜14の何れか1項に記載の化合物又はその塩を投与することを含む、方法。
(項目21)
患者を処置する方法であって、過剰なセレクチン媒介性機能に関連する病態の発症を阻害する必要がある該患者に、当該病態の発症を阻害するのに有効な量で、項目15に記載の組成物を投与することを含む、方法。
(項目22)
移植した組織の拒絶を阻害する方法であって、移植した組織の受容者である患者に、該移植した組織の拒絶を阻害するのに有効な量で、項目1〜14の何れか1項に記載の化合物又はその塩を投与することを含む、方法。
(項目23)
移植した組織の拒絶を阻害する方法であって、移植した組織の受容者である患者に、該移植した組織の拒絶を阻害するのに有効な量で、項目15に記載の組成物を投与することを含む、方法。
(項目24)
薬剤をセレクチン発現細胞に標的化する方法であって、セレクチンを発現する細胞を、該細胞に診断薬又は治療薬を標的化するのに有効な量で、項目16に記載の化合物又はその塩と接触させることを含む、方法。
(項目25)
薬剤をセレクチン発現細胞に標的化する方法であって、セレクチンを発現する細胞を、該細胞に診断薬又は治療薬を標的化するのに有効な量で、項目17に記載の組成物と接触させることを含む、方法。
(項目26)
セレクチン媒介性機能を調節する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目27)
過剰なセレクチン媒介性機能に関連する病態の発症を阻害する必要がある患者を処置する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目28)
移植した組織の拒絶を阻害する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目29)
診断薬又は治療薬を細胞に標的化する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目30)
セレクチン媒介性機能を調節するための医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
(項目31)
過剰なセレクチン媒介性機能に関連する病態の発症を阻害する医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
(項目32)
移植した組織の拒絶を阻害する医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
(項目33)
細胞に診断薬又は治療薬を標的化する医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
【図面の簡単な説明】
【0043】
【図1】図1A及び1Bは、BASAの合成を例示する図である。
【図2】図2は、BASAの合成を例示する図である。
【図3】図3は、糖模倣物(XIX)の合成を例示する図である。
【図4】図4は、糖模倣物(XXVIII)の合成を例示する図である。
【図5】図5は、PEG化糖模倣物(XXX)の合成を例示する図である。
【図6】図6は、PEG化糖模倣物(XXXI)の合成を例示する図である。
【図7A】図7A、7B及び7Cは、PEG化BASA(XXXII及びXXXIII)及びPEG化BACA(XXXVI及びXXXVIa)の合成を例示する図である。
【図7B】図7A、7B及び7Cは、PEG化BASA(XXXII及びXXXIII)及びPEG化BACA(XXXVI及びXXXVIa)の合成を例示する図である。
【図7C】図7A、7B及び7Cは、PEG化BASA(XXXII及びXXXIII)及びPEG化BACA(XXXVI及びXXXVIa)の合成を例示する図である。
【図8A】図8A、8B及び8Cは、糖模倣BASA(図8A及び8C)及び糖模倣BACA(図8B)の合成を例示する図である。
【図8B】図8A、8B及び8Cは、糖模倣BASA(図8A及び8C)及び糖模倣BACA(図8B)の合成を例示する図である。
【図8C】図8A、8B及び8Cは、糖模倣BASA(図8A及び8C)及び糖模倣BACA(図8B)の合成を例示する図である。
【図9A】図9A、9B及び9Cは、糖模倣BASA(図9A及び9C)及び糖模倣BACA(図9B)の合成を例示する図である。
【図9B】図9A、9B及び9Cは、糖模倣BASA(図9A及び9C)及び糖模倣BACA(図9B)の合成を例示する図である。
【図9C】図9A、9B及び9Cは、糖模倣BASA(図9A及び9C)及び糖模倣BACA(図9B)の合成を例示する図である。
【図10】図10は、糖模倣物の合成を例示する図である。
【図11】図11は、糖模倣物の合成を例示する図である。
【図12】図12は、糖模倣物の合成を例示する図である。
【図13】図13は、糖模倣BASAの合成を例示する図である。
【図14】図14は、糖模倣BASAの合成を例示する図である。
【図15】図15は、糖模倣BASAの合成を例示する図である。
【図16】図16は、糖模倣物の合成を例示する図である。
【図17】図17A及び17Bは、E−セレクチン(図17A)及びP−セレクチン(図17B)のin vitro結合検定における糖模倣阻害剤の活性の比較を示す。A及びBは、本発明以外の糖模倣BASAである。Cは、図8Cの糖模倣BASAである。
【図18】図18は、好中球遊走に対する図8Cの糖模倣BASAの効果を示す。Aはビヒクルのみであり、Eは陽性対照(混合抗体)である。B、C及びDは、5mg/kg、10mg/kg、20mg/kgの用量の図8Cの糖模倣BASAである。
【図19】図19は、マウス空気嚢モデルにおける好中球遊走に対する糖模倣阻害剤の効果の比較を示す。Aは、IL−1β+ビヒクルである。Bは、図13のIL−1β+糖模倣BASAである。Cは、図8CのIL−1β+糖模倣BASAである。Dは、IL−1β+混合抗体(陽性対照)である。*A(ビヒクル群)に対してP<0.05。
【発明を実施するための形態】
【0044】
(発明の詳細な説明)
上述の通り、本発明は、セレクチンモジュレーター、それらの組成物、及びセレクチン媒介性機能を調節する方法を提供する。このような調節因子は、以下で更に詳細に述べる種々の状況において、セレクチン媒介性機能を調節(例えば、阻害又は増強)するためにin vitro又はin vivoで使用される場合がある。セレクチン媒介性機能の例には、細胞間接着及び血管新生の間の新しい毛細管の形成が含まれる。
セレクチンモジュレーター
本明細書で使用される「セレクチンモジュレーター」という用語は、セレクチン媒介性機能(例えば、セレクチン媒介細胞間相互作用)を調節(例えば、阻害又は増強)する分子を指す。セレクチンモジュレーターは、本発明の糖模倣化合物から全体が構成される場合もあれば、BASA(ベンジルアミノスルホン酸)又はBACA(ベンジルアミノカルボン酸)に結合したこのような糖模倣物から構成される場合もあり、或いは上述の何れかの1つ以上の更なる分子構成要素を含む場合もある。
【0045】
BASA又はBACAを有さない本発明のセレクチンモジュレーターは、好ましくはE−セレクチン媒介性機能を阻害するために使用される。本発明の糖模倣物にBASA又はBACAを追加することにより、セレクチンモジュレーターは、P−セレクチン及びE−セレクチン媒介性機能を調節する能力も増大した。
【0046】
本発明のセレクチンモジュレーターは、以下の化学式を有する化合物であって:
【0047】
【化33】
式中:
R1が、
【0048】
【化34】
(式中、nは0〜2であり、nが2の場合に、R8は独立して選択される)であり;
R2が、H、−C(=O)OX(式中、XはC1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)、−C(=O)NH(CH2)nNH2、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、nは0〜30であり、mは0〜1であり、Lはリンカーであり、Zはベンジルアミノスルホン酸、ベンジルアミノカルボン酸、ポリエチレングリコールである)、或いは二量体を形成するために上記の化学式を有する第二の化合物又はその塩であって、この場合、該第二の化合物又はその塩のR2がm=0であって、Zを有さず、且つ結合点であり;
R3が、−OH、
【0049】
【化35】
、−O−C(=O)−X、−NH2、−NH−C(=O)−NHX、又は−NH−C(=O)−X(式中、nは0〜2であり、Xは、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0050】
【化36】
(式中、nは0〜10である)からなる群から独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R4が、
【0051】
【化37】
、6’硫酸化GlcNAc、6’カルボキシル化GlcNAc、6’硫酸化GalNAc、6’硫酸化ガラクトース、6’カルボキシル化ガラクトース、又は
【0052】
【化38】
(式中、R9は、アリール、ヘテロアリール、シクロヘキサン、t−ブタン、アダマンタン、又はトリアゾールであり、且つR9の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R5がHであるか、或いはR4及びR5が一緒になって、
【0053】
【化39】
(式中、R10はアリール、ヘテロアリール、
【0054】
【化40】
(式中、nは0〜10である)であり、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)から独立して選択される1個〜3個で置換される場合がある)であり;
R6が、H、フコース、マンノース、アラビノース、ガラクトース、又はポリオールであり;
R7が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又は
【0055】
【化41】
であり;並びに
R8が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0056】
【化42】
【0057】
【化43】
(式中、nは0〜3であり、Xは、H、OH、Cl、F、N3、NH2、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、OC1−C8アルカニル、OC1−C8アルケニル、OC1−C8アルキニル、及びOC1−C14アリールから独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)である、
化合物;或いはその生理学的に許容される塩である。
【0058】
本明細書で使用される「C1−C8アルカニル」という用語は、1〜8個の炭素原子を有するアルカン置換基を指し、直鎖又は分岐鎖である場合がある。例としては、メチル、エチル、プロピル、イソプロピル、ブチル、及びt−ブチルが含まれる。「C1−C8アルケニル」という用語は、1〜8個の炭素原子及び少なくとも1つの炭素−炭素二重結合を有するアルケン置換基を指し、直鎖又は分岐鎖である場合がある。例としては、少なくとも1つの炭素−炭素二重結合を有していることを除いて、「C1−C8アルカニル」と同様の例が含まれる。「C1−C8アルキニル」という用語は、1〜8個の炭素原子及び少なくとも1つの炭素−炭素三重結合を有するアルキン置換基を指し、直鎖又は分岐鎖である場合がある。例としては、少なくとも1つの炭素−炭素三重結合を有していることを除いて、「C1−C8アルカニル」と同様の例が含まれる。「アリール」という用語は、結合により分離される場合もあれば、縮合する場合もある1つ以上の環に、1〜14個の炭素原子を有する芳香族置換基を指す。「ヘテロアリール」は、環炭素に代わって少なくとも1つのヘテロ原子を有する芳香族置換基であることを除いて、「アリール」と同様である。アリール及びヘテロアリールの例には、フェニル、ナフチル、ピリジニル、ピリミジニル、トリアゾロ、フラニル、オキサゾリル、チオフェニル、キノリニル、及びジフェニルが含まれる。本明細書で使用される「独立して選択される」という用語は、同じ又は異なる置換基の選択を指す。
【0059】
本明細書で使用されるポリエチレングリコール(「PEG」)とは、エチレングリコールの複数のユニット、並びに1つ以上の置換基を有するもの(例えば、ジカルボン酸化PEG)を指す。置換基の有無にかかわらずPEGは、当業者にとって周知である。本発明において、PEGは、セレクチンモジュレーターの置換基として、或いはその他の基又は化合物をセレクチンモジュレーターに結合させるリンカーとしての役割を果たす場合がある。即ち、セレクチンモジュレーターは複数のPEGを有する場合がある。
【0060】
第二のセレクチンモジュレーターが第一のセレクチンモジュレーターに結合すると、セレクチンモジュレーターの二量体(即ち、二価の分子)が形成される。2つのセレクチンモジュレーターを接続するために、種々のリンカーが使用される場合がある。例えば、二量体を調製するリンカーとしてPEGが使用される場合がある。本明細書で使用される「二量体」という用語は、ホモ二量体又はヘテロ二量体である場合がある。ホモ二量体とは、(互いに結合するための置換基から独立した)共に結合する2つのセレクチンモジュレーターが同一である場合の二量体を指す。ヘテロ二量体は、(結合置換基から独立した)2つのセレクチンモジュレーターが同一でない場合の二量体を指す。
【0061】
本発明のセレクチンモジュレーターは、上述の通り、上記の化学式のR4において、シアル酸又はシアル酸模倣物を有する場合がある。例えば、シアル酸のヘキソース環をシクロヘキサンと置換する場合がある。セレクチンモジュレーターにシアル酸が存在すると、P−セレクチン結合が増強された。(E−及びP−セレクチン結合の両方ではなく)E−セレクチン結合のみが所望される場合、シアル酸模倣物がセレクチンモジュレーターのシアル酸と置き換わる。
【0062】
シアル酸模倣物とシアル酸とを置き換える代わりに(又はこの置換と組み合わせて)、BASA又はBACAを追加することによってP−セレクチン結合が増強される場合がある。上に開示した通り、本発明のセレクチンモジュレーター化合物は、R2で「Z」を有する場合があり、ZはBASA又はBACAである場合がある。シアル酸を有さない本発明のセレクチンモジュレーター化合物にBASA又はBACAを追加することで、セレクチンモジュレーターを、E−セレクチンに対して選択的に結合する化合物から、E−及びP−セレクチンの両方へ結合する化合物へと変換することができる。BASA又はBACAは、BASA又はBACAの一部或いは類似体を含む(但し、その化合物がセレクチン媒介性機能を調節する能力を維持する場合に限る)。PEGは、BASA(又はBACA)の有無にかかわらず、セレクチンモジュレーターに追加される場合がある。又、BASA又はBACAをセレクチンモジュレーターに結合するために、PEGが使用される場合がある。
【0063】
本発明において、BASAは、セレクチンと相互作用する能力を有する低分子量硫酸化合物である。この相互作用は、セレクチン媒介性機能(例えば、細胞間相互作用)を調節するか、又はその機能の調節(例えば、阻害又は増強)を助ける。BASAは、プロトン化された酸形態として、又はナトリウム塩としての何れかで存在するが、ナトリウムは、カリウム又はその他何れかの薬学的に許容される対イオンで置換される場合がある。代表的なBASAは、以下の構造式を有する:
【0064】
【化44】
。
【0065】
セレクチンと相互作用する(本明細書に記載のセレクチン媒介性機能を調節するか、又はその調節を助ける)能力を維持するBASAの部分も又、本発明のセレクチンモジュレーターのBASA成分である。このような部分は一般的に、BASA構造内に存在する少なくとも一つの芳香族環を含む。特定の実施形態において、一部分は、単一の芳香族環、又は複数のこのような環、或いは対称的なBASA分子の半分を含む場合がある。
【0066】
上述の通り、BASAの類似体及びそれらの部分(いずれも上述の生物学的特性を有する)も又、例えば、このセレクチンモジュレーターのBASA成分により、本発明に包含される。本明細書で使用される「類似体」とは、セレクチン媒介相互作用を阻害する類似体の能力が減少しないような化学的部分の1つ以上の追加、削除及び/又は置換によって、BASA又はその部分とは異なる化合物である。例えば、類似体は、SからPへの置換(例えば、硫酸基をリン酸基で置換)を含む場合がある。その他の可能な変更には:(a)環サイズの変更(例えば、何れの環も、4〜7個の炭素原子を含む場合がある);(b)縮合環の数の変化(例えば、単一の環が3つまでの縮合環を含む多環式部分で置換される場合もあれば、多環式部分が単一の非縮合環で置換される場合もあり、多環式部分内の縮合環の数が変更される場合もある);(c)環置換(芳香族環内の炭素原子に共有結合した水素原子又はその他の部分が、種々の部分(例えば、F、Cl、Br、I、OH、O−アルキル(C1〜8)、SH、NO2,CN、NH2,NH−アルキル(C1〜8)、N−(アルキル)2、SO3M(式中、MはH+、Na+、K+、又はその他の薬学的に許容される対イオンである)、CO2M、PO4M2、SO2NH2,アルキル(C1〜8)、アリール(C6〜10)、CO2−アルキル(C1〜8)、−CF2X(式中、Xは、H、F、アルキル基、アリール基又はアシル基である場合がある)、及び炭水化物)の何れかで置換される場合がある);並びに(d)結合部分(即ち、BASA分子中の環の間に存在する部分)の変更(例えば、アルキル、エステル、アミド、無水物、及びカルバミン酸基等の基が互いに置換される場合がある)、
が含まれる。
【0067】
特定のBASA部分及び類似体は、以下の一般構造式の1つを含む:
【0068】
【化45】
(構造式中、nは0又は1である場合があり、X1は−PO2M、−SO2M、又は−CF2(式中、Mは、例えば水素、ナトリウム、又はカリウム等の薬学的に許容される対イオンである)である場合があり、R1は−OH、−F、又は−CO2R4(式中、R4は−H又は−(CH2)m−CH3である場合があり、mは0〜3の範囲の数である)である場合があり、R2は−H、−PO3M2、−SO3M2、−CH2−PO3M2、−CH2−SO3M2、−CF3、−(CH2)m−C(R6)H−R5、又はR9−N(R10)−である場合があり、R3は−H、−(CH2)m−C(R6)H−R5、又はR9−N(R10)−(式中、R5及びR6は、−H、−CO2−R7、及び−NH−R8から独立して選択される場合があり、R7及びR8は、水素、並びに1つ以上のアルキル基、芳香族部分、アミノ基、又はカルボキシ基を含む部分から独立して選択される場合があり、R9及びR10は、−H、−(CH2)m−CH3、−CH2−Ar、−CO−Ar(式中、mは0〜3の範囲の数であり、Arは芳香族部分(即ち、少なくとも1つの置換又は非置換の芳香族環を含む何れかの部分であって、該環は上述の−CH2−又は−CO−基に直接結合する)である)から独立して選択される場合がある)である場合がある)。
【0069】
BASAのその他の部分及び類似体は、以下の一般構造式を含む:
【0070】
【化46】
(構造式中、R1及びR2は、(i)水素、(ii)1つ以上のアルキル基、芳香族部分、アミノ基、又はカルボキシ基を含む部分、及び(iii)−CO−R3(式中、R3は、上述のようなアルキル又は芳香族部分を含む)、から独立して選択される場合があり、Mは薬学的に許容される対イオンである)。
【0071】
本明細書に記載の構造式及び置換基の種々の組み合わせに由来する個々の化合物又は化合物群は、各化合物又は化合物群を個別に記載するのと同じように本明細書で開示される。従って、特定の構造式及び/又は特定の置換基の選択は、本発明の範囲内に含まれる。
【0072】
代表的なBASA部分及び類似体は、図1A〜1Bに示す化合物に含まれる。セレクチンモジュレーターとして機能する能力に悪影響を及ぼすことなく、これらの図に示す化合物に変更を加える場合があることは、当業者に明らかになるであろう。このような変更には、上述のような削除、追加、及び置換が含まれる。
【0073】
スルホン酸基の代わりに化合物がカルボン酸基を有することを除いて、BACAは、BASAと同様である。例えば、上記のBASA化合物のスルホン酸基をカルボン酸基に置換する場合がある。従って、上記のBASAの開示内容は、参考としてBACAの説明に援用される。
【0074】
BACAの例としては:
【0075】
【化47】
(式中、XはF又はClであり;YはH、−C(=O)(O−CH2CH2)n、又は−C(=0)(CH2)n(式中、nは0〜8である)であり;Zは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)が挙げられる。
【0076】
上述の通り、BASA又はBACAは、R2においてリンカー(「L」)を介して本発明の化合物に結合する場合がある。一般的に、リンカーは、最初に糖模倣物又はBASA/BACAの一方に結合し、次に他方と反応する。特定の糖模倣物へのBASA又はBACAの結合は、セレクチンモジュレーターを形成する種々の方法で達成することができる。BASA又はBACA、或いは糖模倣物によって保有される(又はこれらに付加する)リンカーは、例えば、−(CH2)n−又は−O(CH2)n−(式中、nは一般的に約1〜20である(その範囲内の任意の整数範囲を含む))等のスペーサー基を含む場合がある。リンカーの例には、糖模倣物上の−NH2(それが短いスペーサー基を含む場合は、例えば−CH2−NH2)がある。一実施形態において、−CH2−NH2は、BASA又はBACAを結合するために使用される場合があるR1において糖模倣物に結合する。最も簡単な結合方法には、還元末端(アノマー性ヒドロキシル/アルデヒド)を含む糖模倣物への、BASA又はBACAの還元アミノ化がある。これは、還元末端へのBASA又はBACAの簡単な反応、及び形成されたイミンのそれに続く還元(例えば、pH4.0においてNaCNBH3を使用)により達成される。最も一般的な手法は、活性化されたリンカーを、アノマー位置においてO、S又はNヘテロ原子(又はC原子)を介して糖模倣物に単に結合させる方法である。このような結合の方法論は、炭水化物において広く研究されており、アノマー選択性は、方法論及び/又は保護基の適切な選択によって容易に達成される。可能性のあるグリコシド合成方法の例には、ハロゲン又は過アセチル化糖でのルイス酸触媒結合形成(Koenigs Knorr)、トリクロロアセトアミデート結合形成、チオグリコシド活性化及びカップリング、グルカール(glucal)活性化及びカップリング、n−ペンテニルカップリング、ホスホン酸エステル同族体化(Horner−Wadsworth−Emmons reaction)、及びその他の多くのものが含まれる。或いは、リンカーは、このアノマー性以外の部分の位置に結合される場合がある。結合に最も利用しやすい部位は、糖模倣物(第一級アルコール)の6ヒドロキシル(6−OH)位置である。6−OHでリンカーを結合させるのは、種々の手段によって容易に達成される場合がある。例としては、オキシ−アニオン(塩基を使用した脱プロトン化によって形成されたアルコールアニオン)と適切な求電子剤(例えば、臭化アルキル/アシルエステル、塩化アルキル/アシルエステル又はスルホン酸アルキル/アシルエステル)との反応、スルホン酸エステル塩化物又はPOCl3との反応によるアルコールの活性化及びその後の求核剤による置換、カップリングのためのアルデヒド又はカルボン酸へのアルコールの酸化、又は更には、異なる官能性を導入するMitsunobu反応の使用が含まれる。リンカーが結合すると、次に、BASA又はBACAの好適な求核基との反応のために官能化される(又はその逆も同様)。これは、チオ尿素結合二官能性リガンドを作製するためのチオホスゲン及びアミンの使用、スクアリン酸ジエチルの結合(再度、アミンにより)及び/又は単純なアルキル化/アシル化反応により達成されることが多い。利用可能な更なる方法には、従来から炭水化物及びペプチドのカップリングに使用されているFMOC固相又は溶液相合成法、及びグリコシル/フコシルトランスフェラーゼ及び/又はオリゴサッカリルトランスフェラーゼ(OST)を利用する可能性がある化学−酵素合成法が含まれる。
【0077】
リンカーの実施形態には、以下が含まれる:
【0078】
【化48】
。
その他のリンカー(例えば、PEG)は、当業者又は本開示内容を保有する者にとって周知であろう。
【0079】
本発明の化合物又はその生理学的に許容される塩は、以下の化学式を有する:
【0080】
【化49】
(式中、R1〜R9は、上述の通り定義される)。
【0081】
好ましい実施形態において、R6はフコース:
【0082】
【化50】
である。好ましい実施形態において、R7はHである。好ましい実施形態において、R4は
【0083】
【化51】
である。好ましい実施形態において、R4は、
【0084】
【化52】
(式中、R9は上述の通り定義される)である。好ましい実施形態において、R9はシクロヘキサンである。好ましい実施形態において、R6はガラクトースである。好ましい実施形態において、R8は、
【0085】
【化53】
である。好ましい実施形態において、R2は、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、n、m、L及びZは、上述の通り定義される)である。好ましい実施形態において、Zは、ベンジルアミノスルホン酸、ベンジルアミノカルボン酸、又はポリエチレングリコールである。好ましい実施形態において、R3は−O−C(=O)−X(式中、Xは上述の通り定義される)である。好ましい実施形態において、Xは
【0086】
【化54】
である。好ましい実施形態において、R5はHである。好ましい実施形態において、Lはポリエチレングリコール又はチアジアゾールである。
【0087】
本明細書に記載のセレクチンモジュレーターは、in vivoで所望の部位を十分に標的化する場合があるが、1つ以上の特定の組織に対する標的化を容易にするための更なる標的化部分を含むことが、特定の用途において有利な場合がある。本明細書で使用される「標的化部分」とは、調節剤に結合する場合に、そのモジュレーターの標的組織への輸送を高め、それによって、モジュレーターの局所的濃度を増加させるような何れかの物質(例えば、化合物又は細胞)である場合がある。標的化部分には、抗体又はそのフラグメント、受容体、リガンド、及び標的組織の細胞に結合する又は標的組織付近にあるその他の分子が含まれる。結合は、一般的に共有結合であり、例えば、直接的縮合又はその他の反応によって、或いは二官能性リンカー又は多官能性リンカーを使用する方法によって達成される場合がある。
【0088】
特定の実施形態では、セレクチンモジュレーターに対して薬剤も結合させるか、又は代わりに結合させることが有益な場合がある。本明細書で使用される「薬剤」という用語は、疾患又はその他の望ましくない病態を予防又は処置するために哺乳動物に投与することを目的とした、何れかの生体活性剤を指す。薬剤には、ホルモン、成長因子、タンパク質、ペプチド及びその他の化合物が含まれる。可能性のある薬剤の例には、抗腫瘍性薬剤(例えば、5−フルオロウラシル及びジスタマイシン)、インテグリン作動薬/拮抗薬(例えば、環状RGDペプチド)、サイトカイン作動薬/拮抗薬、ヒスタミン作動薬/拮抗薬(例えば、ジフェンヒドラミン及びクロルフェニラミン)、抗生物質(例えば、アミノグリコシド及びセファロスポリン)並びに酸化還元活性生物学的薬剤(例えば、グルタチオン及びチオレドキシン)が含まれる。他の実施形態において、診断用放射性核種又は治療用放射性核種は、セレクチンモジュレーターに結合する場合がある。多くの実施形態において、本薬剤は、セレクチンモジュレーターへ直接的又は間接的に結合する場合がある。
【0089】
本明細書に記載のモジュレーターは、薬学的組成物内に存在する場合がある。薬学的組成物は、1つ以上の薬学的又は生理学的に許容される担体、希釈剤、又は賦形剤と組み合わせて、1つ以上のモジュレーターを含む。このような組成物は、緩衝剤(例えば、中性緩衝生理食塩水又はリン酸緩衝生理食塩水)、糖質(例えば、グルコース、マンノース、スクロース、又はデキストラン)、マンニトール、タンパク質、ポリペプチド、又はグリシンのようなアミノ酸、抗酸化剤、キレート剤(例えば、EDTA又はグルタチオン)、アジュバント(例えば、水酸化アルミニウム)、及び/又は保存剤を含む場合がある。更に他の実施形態では、本発明の組成物は、凍結乾燥物として処方される場合がある。本発明の化合物は、例えば、局所的、経口、経鼻、静脈内、頭蓋内、腹腔内、皮下又は筋肉内の投与を含めた何れかの適切な投与様式のために処方される場合がある。
【0090】
薬学的組成物は又、薬剤(例えば、上で示される薬剤))等のような1つ以上の活性薬剤も同時に又は代わりに含む場合もあり、それらの活性薬剤は、モジュレーターに結合する場合もあれば、組成物内で何れとも結合しない場合もある。
【0091】
本明細書に記載の組成物は、徐放性製剤(即ち、投与の後に、調節剤の持続放出をもたらすカプセル状又はスポンジ状のような製剤)の一部として、投与される場合がある。このような製剤は一般的に、周知の技法を使用して調製される場合があり、例えば、経口、直腸又は皮下移植によって、又は所望の標的部位における移植によって、投与される場合がある。このような製剤内で使用する担体は、生体適合性を有し、且つ生物分解性も有する場合がある。好ましくは、本製剤は、比較的一定のレベルでの調節剤の放出を提供する。徐放性製剤内に含まれる調節剤の量は、移植の部位、放出の速度、及び期待される放出持続時間、並びに処置又は予防する病態の性質により異なる。
【0092】
セレクチンモジュレーターは一般的に、治療有効量において薬学的組成物中に存在する。治療有効量は、識別可能な患者のベネフィット、例えば、下記のような過剰なセレクチン媒介性機能(例えば、細胞間接着)と関連する病態の治癒の向上等をもたらす量である。
【0093】
一般的に、本明細書に記載の調節剤及び組成物は、セレクチン媒介性機能を増強又は阻害するために使用される場合がある。このような増強又は阻害は、温血動物(好ましくはヒト等の哺乳動物)においてin vitro及び/又はin vivoで達成される場合がある。但し、セレクチンを発現する細胞は最終的に、セレクチン媒介性機能を増強又は阻害するのに十分な量で及び十分な時間にわたって、モジュレーターと接触する。
【0094】
特定の態様において、本発明は、セレクチン媒介性機能(例えば、細胞間接着)に関連する病態の進行を抑制する方法を提供する。一般的に、このような方法は、このような病態を予防、遅延又は処置するために使用される場合がある。換言すれば、本明細書に示される治療方法は、疾患を処置するために使用される場合もあれば、或いは疾患に罹患していない患者、又はセレクチン媒介性機能に関連しない疾患に罹患した患者において、このような疾患の発症を予防又は遅延するために使用される場合もある。例えば、本治療方法には、細胞成長の阻害、細胞の殺傷、細胞成長の防止、細胞成長の発生の遅延、或いは生物体の生存の延長が含まれる場合がある用途がある。
【0095】
セレクチン媒介性機能に関連する病態には、種々のものがある。このような病態には、例えば、組織移植片拒絶、血小板媒介性疾患(例えば、アテローム性動脈硬化症及び凝固)、過活動性冠状動脈循環、急性白血病媒介性肺損傷(例えば、成人呼吸窮迫症候群(ARDS))、クローン病、炎症性疾患(例えば、炎症性腸疾患)、自己免疫疾患(MS、重症筋無力症)、感染症、癌(及び転移)、血栓症、創傷(及び創傷関連の敗血症)、熱傷、脊髄損傷、消化管粘膜疾患(胃炎、潰瘍)、骨粗しょう症、慢性関節リウマチ、変形性関節症、喘息、アレルギー、乾癬、敗血症性ショック、外傷性ショック、脳卒中、腎炎、アトピー性皮膚炎、凍傷性損傷、成人型呼吸困難症候群、潰瘍性大腸炎、全身性エリテマトーデス、糖尿病、及び虚血性エピソード後の再灌流障害が含まれる。又、セレクチンモジュレーターは、回復を高めるために心臓手術の前に患者に投与される場合もある。その他の用途には、疼痛処理の用途、血管ステントに関連する再狭窄の予防、及び例えば癌に関連する望ましくない新脈管形成に対する用途が含まれる。
【0096】
本発明のセレクチンモジュレーターは、処置(又は予防)する疾患に適切な様式で投与される場合がある。適切な投薬量、並びに好適な投与持続期間及び投与頻度は、患者の病態、患者の疾患の型及び重篤度、並びに投与方法等の要因により決定される場合がある。一般的に、適切な投薬量及び処置レジメンは、治療効果及び/又は予防効果を提供するのに十分な量にて調節剤を提供する。本発明の特に好ましい実施形態では、セレクチンモジュレーターは、0.001〜1000mg/体重kg(より一般的には0.01〜1000mg/kg)の範囲の投与量で、毎日1回又は複数回の投与レジメンで投与される場合がある。適切な投与量は一般的に、実験モデル及び/又は臨床試験を使用して決定される場合がある。一般的に、有効な治療を提供するために十分な、最小の投薬量を使用することが好ましい。患者は一般的に、処置又は予防する病態に好適な、当業者に周知の検定を使用して、治療効果について監視される場合がある。
【0097】
セレクチンモジュレーターは又、セレクチンを発現する細胞に物質を標的化するために使用される場合もある。このような物質には、治療薬及び診断薬が含まれる。治療薬は、疾患の処置又は予防或いは患者の生理学的機能の調節に対して有用性を提供するように標的細胞の特性を変更することが実証できる分子、ウイルス、ウイルス成分、細胞、細胞成分、又はその他何れかの物質である場合がある。治療剤は又、in vivoにて生物学的活性を有する薬剤を生成するプロドラッグである場合もある。治療薬である場合がある分子は、例えば、ポリペプチド、アミノ酸、核酸、ポリヌクレオチド、ステロイド、多糖、又は無機化合物である場合がある。このような分子は、種々の方法(例えば、酵素、酵素阻害剤、ホルモン、受容体、アンチセンスオリゴヌクレオチド、触媒性ポリヌクレオチド、抗ウイルス剤、抗腫瘍剤、抗細菌剤、免疫調節剤及び細胞傷害性剤(例えば、ヨウ素、臭素、鉛、パラジウム又は銅のような放射性核種)を含む)の何れかにおいて機能する場合がある。診断薬には、金属及び放射性薬剤のような画像化剤(例えば、ガリウム、テクネチウム、インジウム、ストロンチウム、ヨウ素、バリウム、臭素及びリン含有化合物)、造影剤、色素(例えば、蛍光色素又は発蛍光団)、並びに比色定量的反応又は蛍光定量的反応を触媒する酵素が含まれる。一般的に、治療薬及び診断薬は、上記の技法のような種々の技法を使用してセレクチンモジュレーターへと結合される場合がある。標的化において、セレクチンモジュレーターは、本明細書に記載の通り、患者に投与される場合がある。セレクチンは、血管新生の間の新しい毛細管の形成に関与する内皮細胞上に発現することから、セレクチンモジュレーターは、腫瘍の脈管構造を殺傷するために、治療薬を標的とするために使用される場合がある。セレクチンモジュレーターは遺伝子標的化にも使用される場合がある。
【0098】
セレクチンモジュレーターは又、in vitroで(例えば、種々の周知の細胞培養物内及び細胞分離法において)使用される場合もある。例えば、モジュレーターは、培養におけるスクリーニング、検定及び成長に対し、セレクチンを発現する細胞を固定化する際に使用するために、組織培養プレート又はその他の細胞培養支持体の内部表面に結合される場合がある。このような結合は、上述に記載の方法のような任意の適切な技法、及びその他の標準的技法によって実施される場合がある。モジュレーターは又、例えば、in
vitroでの細胞の同定及び選別を容易にし、セレクチン(又は異なるセレクチンレベル)を発現する細胞の選択を可能にするために使用される場合もある。好ましくは、このような方法で使用するモジュレーターは、検出可能なマーカーに結合する。好適なマーカーは当該技術分野で周知であり、これらには、放射性核種、発光基、蛍光基、酵素、色素、免疫グロブリン定常ドメイン及びビオチンが含まれる。好ましい一実施形態では、蛍光マーカー(例えば、フルオレセイン)に結合したモジュレーターを、細胞と接触させ、次いでこれを、蛍光活性化細胞選別(FACS)によって分析する。
【0099】
上記のような調節剤は、例えば、セレクチン媒介性細胞接着を阻害することができる。この能力は一般的に、セレクチンを発現する細胞間の接着(例えば、白血球又は腫瘍細胞と血小板又は内皮細胞との間の接着)に対する効果を測定するために設計された種々のin vitro検定の何れかを使用して評価される場合がある。例えば、このような細胞は、モジュレーターの非存在下で、細胞接着を可能にする標準的な条件下で平板培養される場合がある。一般的に、試験細胞とモジュレーターとの接触により、細胞接着の識別可能な破壊が生じる場合、このモジュレーターは、セレクチン媒介性細胞接着の阻害剤である。例えば、モジュレーターの存在下(例えば、マイクロモル濃度レベル)では、白血球又は腫瘍細胞と血小板又は内皮細胞との間の接着の破壊は、互いに相互作用する細胞の減少を観察することによって、およそ数分内で視覚的に判定される場合がある。
【0100】
本発明の化合物又は本発明に有用な化合物は全て、それらの生理学的に許容される塩を含む。
【0101】
以下の実施例は、本発明を例示するために提供されるのであって、本発明を限定するために提供されるのではない。
【実施例】
【0102】
(実施例1)
BASA(図1A)の合成
化合物4の合成:市販の2(1g)のニトロ化は、記載の手順に従って行う(文献の条件については、米国特許第4,534,905号;Allison, F., et al., Helv. Chim. Acta 4:2139 (1952)を参照)。
【0103】
粗生成物3を水(40mL)に溶解させ、10%のPd/C(0.3g)を加える。混合物を48時間室温にて水素化(約45psi)する。触媒をセライトを通して濾過し、濾床を水で洗浄する。濾液を減圧濃縮し、桃色の固体を得る。触媒の除去後に、濾液を15mLまで濃縮し、等容積のエタノールを加える。沈殿物を濾過により収集し、不純物の非常に少ない化合物4を得る。
【0104】
化合物7aの合成:5(5g)及び8(4.45g、24.7mmol)、並びにK2CO3(H2O中に2M、24.7mL、49.4mmol)の10:1のトルエン/エタノール(70mL)中の溶液をPd(PPh3)4(1.43g、1.24mmol)で処理し、その混合物を20時間還流する。後処理の後、EtOH溶液中における粗生成物の再結晶化、及び再結晶化の濾液のクロマトグラフィー精製により、化合物9(2.9g、46%、>90%HPLC)を得て、2.2gの5を回収する。生成物を、1H−NMRによって同定する。
【0105】
THF/H2O(1:1)中の9(2.9g、11.3mmol)及びLiOH・H2O(1.43g、34.1mmol)の混合物を室温にて21時間攪拌する。後処理の後、この反応により7(2.58g、94%、>90%HPLC)を得る。生成物を、1H−NMRによって同定する。
【0106】
7(500mg、1.94mmol)、SOCl2(0.23mL、3.10mmol)、及びトルエン(3mL)の懸濁液にDMF(20μL)を加えた後、80℃に加熱する。20時間後、反応が終了し、酸塩化物(640mg)を得る。生成物7aを、IR及び1H−NMRによって同定する。
【0107】
化合物10の合成:H2O(2mL)及びジオキサン(18mL)中のアミンの4(268mg、0.641mmol)の溶液に、ジオキサン(16mL)中の7a(273mg、0.99mmol)の溶液を、30分かけて滴下する。滴下が進むにつれ、反応混合物のpHを0.25MのNaOHにより8.5に調整する。添加後、反応物を室温にて2.5時間攪拌する。カラムクロマトグラフィー(メタノール/トルエン1:1)により精製し、続いて分取TLC(メタノール/トルエン1:1)により精製して、50mgの化合物10を得る。得られた生成物を、1H−NMR及びMSによって同定する。
【0108】
化合物10の水素化:H2O(20mL)中の10(30mg、0.049mmol)及び炭素上の10%Pd(50mg)の懸濁液を、室温にて4時間水素化し(55psi)、図1AのBASAを得る。
【0109】
(実施例2)
BASA(図1B)の合成
化合物4の合成:3−ニトロベンジルヨウ化物(1)(48.3g)を、市販の8−アミノナフタレン−1,3,5−トリスルホン酸(2)(29.5g)の水溶液(pH11)に、室温にて攪拌しながら加える。その溶液のpHを1に調整し、その溶媒を蒸発除去した後、生成物3(6.4g)をエタノールから沈殿させる。
【0110】
化合物3を白金触媒により水素化して、化合物4(図1BのBASA)を収率96%で得る。
【0111】
(実施例3)
BACA(図2)の合成
1(8.9g)、パラホルムアルデヒド(8.9g)、及び硫酸(125mL)の懸濁液を90℃にて14時間加熱し、後処理の後に2の粗生成物(7.8g)を得る。その粗生成物をHPLCにより精製し(純度77%)、1H−NMRによって同定する。
【0112】
アセトン(30mL)中の2(1.0g)の溶液に、K2CO3(3.1g)及び硫酸ジメチル(1.4mL)を加え、反応物を24時間加熱還流する。後処理及び精製のため、反応物を次のバッチと混合する。
【0113】
アセトン(225mL)中の2(7.5g)の溶液に、K2CO3(23.2g)及び硫酸ジメチル(10.8mL)を加え、反応物を16時間加熱還流する。反応物を前のバッチと合わせ、後処理及びカラムクロマトグラフィー精製(酢酸エチル/ヘプタン=1:9)した後、3(7.3g、74%)を得る。生成物をHPLCにより精製し(純度80%)、1H−NMRによって同定する。
【0114】
3℃にて無水クロム酸(6.94g)を、無水酢酸(175mL)中の3(7.16g)の懸濁液に加えた後、室温にて15時間攪拌する。反応物を後処理及びカラム精製(100%のジクロロメタン)した後、4(5.89g)を得る。生成物をHPLCにより精製し(純度90%)、1H−NMRによって同定する。
【0115】
THF/H2O(300mL、1:1)中の4(5.89g)の懸濁液に、室温にてLiOH・H2O(1.74g)を加え、得られた混合物を14時間攪拌する。酸/塩基による後処理の後に、白色固体の生成物を得る。生成物を高真空下で乾燥させて、NMR及び質量分析により同定する。
【0116】
(実施例4)
糖模倣物(図3)の合成
中間体IIの合成:メタノール(200mL)及び硫酸(2mL、98%)中の(−)シキミ酸(20g)を、室温にて50時間攪拌する。反応混合物を2Nの冷NaOH水溶液によって中和する。蒸発乾固後、その残留物をシリカゲルクロマトグラフィーにより精製し、II(19.2g)を得る。
【0117】
中間体(III)の合成:シキミ酸メチル(II、10g)、2,2−ジメトキシプロパン(10mL)、及びp−TsOH(0.8g)をアセトニトリル(125mL)に溶解させ、室温にて1時間攪拌する。次に、反応混合物をトリエチルアミン(2mL)により中和し、蒸発乾固する。その残留物をシリカゲルクロマトグラフィーで処理して、III(11g)を得る。
【0118】
中間体IVの合成:MeOH(40mL)中のシキミ酸誘導体III(10g)及びPtO2/C(10%、250mg)を、室温にて激しく攪拌しながら水素化する。16時間後、反応混合物をセライトによって濾過し、蒸発乾固する。その残留物をシリカゲルクロマトグラフィーで処理して、IVを得る。
【0119】
中間体Vの合成:DCM(100mL)中のIV(8g)の溶液に、0℃にてピリジン(12mL)、無水酢酸(7mL)、及びDMAP(25mg)を加える。反応混合物を室温にて1時間攪拌し、EtOAc(250mL)で希釈する。0.5Mの塩酸(50mLで3回)、KHCO3の飽和溶液(50mLで3回)、及び塩水(50mLで3回)で洗浄後、収集した有機層を乾燥させ(Na2SO4)、蒸発乾固する。残留物をシリカゲルクロマトグラフィーにより精製し、V(6.8g)を得る。
【0120】
中間体VIの合成:酢酸(30mL、80%)中のV(6.0g)の溶液を80℃にて1時間攪拌する。溶媒を蒸発除去し、残留物をシリカゲルクロマトグラフィー(DCM/メタノール14:1)により精製し、VI(3.6g)を得る。
【0121】
中間体VIIの合成:ピリジン(30mL)中のVI(3g)及びp−TsCl(3.5g)の溶液を、室温にて6時間攪拌する。MeOH(5mL)を加え、溶媒を減圧蒸発させて、その残留物をEtOAcに溶解させ(150mLで3回)、その有機層を0.5Mの塩酸水溶液(0℃)、冷水、及び冷塩水で洗浄する。収集した有機層を乾燥させ(Na2SO4により)、セライトで濾過して、蒸発乾固する。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=4:1)により精製し、VII(3.7g)を得る。
【0122】
化合物VIIIの合成:DMF(20mL)中のVII(3g)及びNaN3(2.5g)の溶液を80℃にて攪拌する。反応混合物を室温に冷却し、EtOAc(200mL)及び水(50mL)で希釈する。有機層を更に水で2回(50mLで2回)、塩水で1回(50mL)洗浄する。全ての水層をEtOAcで2回抽出する(50mLで2回)。収集した有機層をNa2SO4で乾燥させ、濾過して、その溶媒を蒸発除去する。残留物をシリカゲルクロマトグラフィー(石油エーテル/EtOAc=5:2)により精製し、VIII(2.2g)を得る。
【0123】
化合物Xの合成:DCM(3mL)中のエチル−2,3,4−トリ−o−ベンジル−α−L−フコチオピラノシドIX(1.5g)の溶液に、0℃にてアルゴン雰囲気下で臭素(150μL)を加える。5分後、冷却槽を取り外して、反応混合物を更に室温にて25分間攪拌する。シクロヘキセン(200μL)を加え、その反応混合物をDCM(10mL)及びDMF(5mL)中のVIII(400mg)、(Et)4NBr(750mg)、及び粉末状の4Åモレキュラーシーブの溶液に加える。16時間後、トリエチルアミン(1.5mL)を加え、更に10分間攪拌し、EtOAc(50mL)で希釈して、飽和NaHCO3水溶液、水、塩水で洗浄する。水層をEtOAcで2回抽出する(50mLで2回)。収集した有機層を、乾燥させ(Na2SO4により)、濾過して蒸発乾固する。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、X(700mg)を得る。
【0124】
化合物XIの合成:メタノール(20mL)中のX(1.5g)の溶液に、新たに調製したNaOMe(80mg)を加え、その反応混合物を圧力管内にて80℃にて20時間攪拌する。反応混合物を室温に冷却し、酢酸によって中和する。溶媒を蒸発乾固して、残留物をエーテルに溶解させる。新たに調製したジアゾメタンを加え、過剰なジアゾメタンを酢酸によって中和する。溶媒を蒸発除去し、XI(1.25g)を得る。
【0125】
構築ブロックXVの合成:この合成は、既述のものと同じ方法で実施する(「Helvetica Chemica Acta 83:2893−2907 (2000))。
【0126】
化合物XVIの合成:DCM(17mL)中のXI(1.6g)、XV(3g)、及び粉末状の活性化モレキュラーシーブ4Å(1g)の混合物を、アルゴン雰囲気下にて室温にて2時間攪拌する。次いで、DMTST(2g)を等量ずつ4回に分けて1.5時間かけて加える。24時間後、反応混合物をセライトによって濾過し、その濾液をDCM(100mL)で希釈する。有機層を飽和NaHCO3水溶液及び塩水で洗浄し、その水層をDCMで2回抽出する。収集した有機層を乾燥させて(Na2SO4により)、濾過して蒸発乾固する。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=8:1)により精製し、XVI(1.5g)を得る。
【0127】
化合物XVIIの合成:ジクロロメタン(10mL)中のXVI(500mg)及びオロチン酸塩化物(500mL)の溶液に、トリフェニルホスフィンの溶液(5mLのジクロロメタン中に500mg)を10分間かけて滴下する。反応混合物を室温にて25時間攪拌し、溶媒を蒸発除去する。残留物を精製し(シリカゲルクロマトグラフィー、DCM/メタノール=19:1)、XVII(250mg)を得る。
【0128】
化合物XVIIIの合成:ジオキサン−水(5:1、12mL)中のXVII(200mg)の溶液に、10%Pd−C(100mg)を加え、その反応混合物を水素下(55psi)で24時間激しく攪拌する。触媒をセライトのベッドを通して濾過し、溶媒を蒸発除去する。残留物をシリカゲルクロマトグラフィーにより精製し、化合物XVIII(150mg)を得る。
【0129】
XIXの合成:メタノール(5mL)中の化合物XVIII(145mg)の溶液に、NaOMeのメタノール溶液(25%、0.025mL)を加え、反応混合物を室温にて4時間攪拌し、酢酸で中和して、溶媒を蒸発除去する。残留物を水に溶解させ、ダウエックス50wX−8(Na形態)樹脂のベッドを通して濾過する。水洗浄物を蒸発除去し、化合物XIX(100mg)を得る。
【0130】
EDA−XIXの合成:XIX(80mg)をエチレンジアミン(EDA)(1mL)と共に攪拌しながら70℃にて5時間加熱する。溶媒を蒸発除去し、セファデックスG−25カラムにより精製して、EDA−XIX(82mg)を得る。
【0131】
(実施例5)
糖模倣物(図4)の合成
化合物XXIの合成:ピリジン(60mL)中の化合物XX(1.5g、以前に公開された手順(Carbohydrate Chemistry and Biochemistry 2000, vol.1, page 345−365)に従って合成)の溶液に、無水安息香酸(0.73g)及びジメチルアミノピリジン(0.02g)を加える。反応混合物を室温にて20時間攪拌する。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させる。溶液を1Mの冷塩酸及び水で連続して洗浄する。溶液を乾燥させ(酢酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXI(1g)を得る。
【0132】
化合物XXIIの合成:ジクロロメタン(20mL)中の化合物XXIの溶液に、トリフルオロ酢酸(20mL)を0℃にて加え、反応混合物を同じ温度で1時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカゲル)により精製して、化合物XXII(0.6g)を得る。
【0133】
化合物XXIIIの合成:ジクロロメタン(40mL)中の化合物XXII(1g)の溶液に、DBU(0.05mL)及びトリクロロアセトニトリル(0.4g)を0℃にて加える。溶液を同じ温度で1.5時間攪拌する。溶媒を蒸発除去し、カラムクロマトグラフィー(シリカゲル)により精製して、化合物XXIII(0.6g)を得る。
【0134】
化合物XXIVの合成:化合物XI(0.7g)、ジクロロメタン(40mL)中の化合物XXIII(0.5g)、及びモレキュラーシーブ(4Å、5g)の混合物に、TMSOTf溶液(ジクロロメタン(5mL)中に0.15mL)を0℃にて攪拌しながら滴下する。同じ温度で2時間攪拌を継続する。トリエチルアミン(0.2mL)を加え、反応混合物をセライトベッドを通して濾過する。反応混合物を、冷飽和重炭酸ナトリウム液によって洗浄する。乾燥させて(酢酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXIV(0.7g)を得る。
【0135】
化合物XXVの合成:DMF(30mL)中の化合物XXIV(0.6g)の溶液に、DBU(30滴)及びdl−ジチオトイトール(DTT、0.28g)を加える。反応混合物を室温にて1時間攪拌する。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させて、水で洗浄する。有機層を乾燥させ(酢酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXV(0.45g)を得る。
【0136】
化合物XXVIの合成:DMF(10mL)中の化合物XXV(0.4g)の溶液に、オロチン酸(0.14g)、EEDQ(0.19g)、及び4−メチル−モルフォリン(0.09g)を加え、反応混合物を70℃にて20時間攪拌する。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させる。溶液を冷飽和重炭酸ナトリウム液及び水で洗浄する。有機層を乾燥させ(硫酸ソーダにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXVI(0.2g)を得る。
【0137】
化合物XXVIIの合成:化合物XXVI(0.2g)を、記載と同じ条件下で水素化し、記載の通りMeOH中のNaOMeを使用して中間体を部分的に脱ベンゾイル化し、クロマトグラフィーにより精製して、化合物XXVII(0.05Og)を得る。
【0138】
化合物XXVIIIの合成:化合物XXVIIを、記載の通りにエチレンジアミンで処理し、カラムクロマトグラフィー(シリカゲル及びゲル濾過セファデックスG−25)により精製して、化合物XXVIII(25mg)を得る。
【0139】
(実施例6)
PEG化BASA(図7B)の合成
DMF(1mL)中の3,6−ジオキサオクタン二酸(PEG、200mg、市販)の溶液にヒューニッヒ塩基(0.4mL)を加え、5分後にHATU(0.35g)を加える。溶液を室温にて10分間攪拌した後、DMF(0.1mL)中に溶解させた実施例2(50mg)のBASAの溶液を加える。反応混合物を室温にて4時間攪拌し、溶媒を蒸発除去する。残留物をHPLC(逆相C18カラム)により精製し、XXXIII(40mg)を得る。
【0140】
(実施例7)
ペグ化BASA(図7A)の合成
この合成は、実施例1のBASAを使用することを除き実施例6で説明したのと同じ方法で実施し、XXXII(50mg)を得る。
【0141】
(実施例8)
ペグ化BACA(図7C)の合成
中間体XXXIV(方法1)の合成:実施例3のBACA(0.5g)をメタノール−水(1mL、9:1)に懸濁させ、Cs2CO3の水溶液を加えてpHを8.2に調整する。溶媒をトルエンで共蒸発させて除去する。残留物をDMF(1mL)に溶解させる。臭化ベンジル(0.5mL)を加え、室温にて20時間攪拌する。ジクロロメタン(15mL)を加え、冷水で洗浄する。有機層を乾燥させ(無水硫酸ナトリウムにより)、溶媒を蒸発除去する。残留物をカラムクロマトグラフィー(シリカ)により精製し、XXXIV(0.48g)を得る。
【0142】
中間体XXXIV(方法2)の合成:DMF中に溶解させた実施例3のBACA(1g)の溶液にN,N−ジイソプロピルエチルアミン(1.5g)及び臭化ベンジル(1.5g)を加える。反応混合物を50℃にて20時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカ)により精製して、XXXIVを得る。
【0143】
中間体XXXVの合成:メタノール(10mL)中のXXXIV(0.2g)の溶液に、0℃にて水素化ホウ素ナトリウム(0.070g)を加え、反応混合物を0℃にて1時間攪拌する。酢酸を加えて反応物を急冷し、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカ)により精製し、XXXV(0.16g)を得る。
【0144】
XXXVbの中間体の合成:ジクロロメタン中のXXXV(0.36g)の溶液にトリエチルアミン(0.6mL)及びMeSO2Cl(0.29mL)を加える。反応混合物を室温にて21時間攪拌する。反応混合物をジクロロメタンで希釈し、水、1Mの塩酸、及び塩水で洗浄する。有機層を乾燥させ(Na2SO4により)、乾燥状態まで濃縮して、粗生成物XXXVaを得る。DMF(5mL)中の粗XXXVaの溶液に、NaN3(0.18g)を加える。反応混合物を100℃にて4時間攪拌し、溶媒を蒸発除去する。残留物をCH2Cl2に溶解させ、冷塩水、1Mの冷塩酸、冷炭酸水素ナトリウム溶液、及び冷水で洗浄する。有機層を乾燥させ(Na2SO4により)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカ)により精製して、XXXVb(0.14g)を得る。
【0145】
中間体XXXVcの合成:DMF(3mL)中のXXXVb(0.08g)の溶液にDTT(0.04g)及びDBU(0.02mL)を加え、室温にて1時間攪拌する。溶媒を蒸発除去し、残留物をEtOAcに溶解させ、H2Oで洗浄して、有機層を乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカ)により精製し、中間体XXXVc(0.061g)を得る。
【0146】
中間体XXXVdの合成:DMF(3mL)中のモノ保護PEGジカルボン酸(0.6g)の溶液に、ジイソプロピルエチルアミン(0.24mL)及びHATU(0.513g)を攪拌しながら加える。上記の溶液にDMF(3mL)中の中間体XXXVc(0.185g)の溶液を加える。反応混合物を室温にて1時間攪拌する。溶媒を蒸発除去し、残留物をEtOAcに溶解させる。EtOAc層を水で洗浄し、カラムクロマトグラフィーにより精製して、中間体XXXVdを得る。
【0147】
XXXVIの合成:氷酢酸(3mL)中のXXXVd(0.185g)の溶液にZn粉末(0.1g)を加え、反応物を40℃にて30分攪拌する。反応混合物をセライトベッドを通して濾過し、Zn固形物をメタノールによって洗浄する。濾液を乾固状態まで濃縮し、セップパックC18カラムにより精製し、XXXVI(0.050g)を得る。
【0148】
中間体XXXIVaの合成:THF(25mL)中のTiCl4−テトラヒドロフラン錯体(0.705g)及びZn粉末(0.28g)の懸濁液を、不活性雰囲気下で攪拌しながら75℃にて2時間還流する。この混合物に実施例3のBACA(0.3g)及びN−フルオレニルメトキシカルボニル−3−アミノプロパノール(0.312g、参考文献Casimiro−Garcia, et al., Bioorg. Med. Chem. 1979 (2001) 2827に記載の通りに調製)の溶液を加える。反応混合物を不活性雰囲気下で75℃にて2.5時間攪拌する(McMurrayカップリング)。反応物を室温に冷却し、H2O(30mL)を加え、セライトベッドを通して濾過し、濾液をEtOAcで3回洗浄する(各30mL)。有機層を収集し、乾燥させ(Na2SO4によって)、濾過して、乾固状態まで濃縮する。残留物をカラムクロマトグラフィーにより精製し、XXXIVaを得る。
【0149】
中間体XXXIVbの合成:無水THF(21mL)中のXXXIVa(0.444g)の溶液に、ピペリジン(6.25mL)を加え、反応混合物を室温にて3時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカ)により精製し、中間体XXXIVb(0.26g)を得る。
【0150】
中間体XXXIVcの固相合成:PS−カルボジイミド樹脂(0.20Og)、HOBt(0.03Og)、及びモノ保護PEG−COOH(0.08Og)のCH2Cl2(3mL)中での混合物を、シリンジ反応器中にて室温にて5分間攪拌する。その混合物に、CH2Cl2(3mL)中の中間体XXXIVb(0.08Og)の溶液を加え、反応混合物を室温にて3時間攪拌する。MP炭酸樹脂(0.216g)を加え、室温にて2時間攪拌する。樹脂を濾別した後、その樹脂をCH2Cl2で5回洗浄する。濾液を収集し、乾固状態まで濃縮して、中間体XXXIVcを得る。
【0151】
中間体XXXVIaの合成:中間体XXXVIの合成について説明したのと全く同じ方法で、XXXVIc(0.12g)をZn/酢酸で処理して、中間体XXXVIa(0.104g)を得る。
【0152】
(実施例9)
ペグ化BACA(図7C)の合成
中間体XXXIVaの合成:THF(25mL)中のTiCl4−テトラヒドロフラン錯体(0.705g)及びZn−粉末(0.28g)の懸濁液を、不活性雰囲気下で攪拌しながら75℃にて2時間還流する。この混合物に実施例3のBACA(0.3g)及びN−フルオレニルメトキシカルボニル3アミノプロパノール(0.312g、参考文献Casimiro−Garcia, et al., Bioorg. Med. hem. 1979 (2001) 2827に記載の通り調製)の溶液を加える。反応混合物を不活性雰囲気下で75℃にて2.5時間攪拌する(McMurrayカップリング)。室温まで反応物を冷却し、H2O(30mL)を加え、セライトベッドを通して濾過し、濾液をEtOAcで3回洗浄する(各30mL)。有機層を収集し、乾燥させて(Na2SO4により)、濾過して乾固状態まで濃縮する。残留物をカラムクロマトグラフィーにより精製し、XXXIVaを得る。
【0153】
中間体XXXIVbの合成:無水THF(21mL)中のXXXIVa(0.444g)の溶液に、ピペリジン(6.25mL)を加え、反応混合物を室温にて3時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカ)により精製し、中間体XXXIVb(0.26g)を得る。
【0154】
中間体XXXIVcの固相合成:PSカルボジイミド樹脂(0.200g)、HOBt(0.030g)及びモノ保護PEG−COOH(0.080g)のCH2Cl2(3mL)中での混合物を、シリンジ反応器中にて室温にて5分攪拌する。上記の混合物に、CH2Cl2(3mL)中の中間体XXXIVb(0.080g)の溶液を加え、反応混合物を室温にて3時間攪拌する。MP−炭酸樹脂(0.216g)を加え、室温にて2時間攪拌する。樹脂を濾別した後、その樹脂をCH2Cl2で5回洗浄する。濾液を収集し、乾固状態まで濃縮して、中間体XXXIVcを得る。
【0155】
XXXVIaの合成:中間体XXXVIの合成について説明したのと同じ方法で、XXXIVc(0.12g)をZn/酢酸で処理して、中間体XXXVIa(0.104g)を得る。
【0156】
(実施例10)
糖模倣BASA(図8A)の合成
実施例7(0.015g)のXXXIIのDMF(0.1mL)中の溶液に、ヒューニッヒ塩基(0.015mL)を加えた後、HATU(0.007g)を加える。反応混合物を室温にて10分攪拌する。実施例4のEDA−XIX(DMFmL中に0.010g)の溶液を加え、反応混合物を室温にて8時間攪拌する。溶媒を蒸発除去し、残留物をセファデックスG−25クロマトグラフィーにより精製し、図8Aの糖模倣BASA(0.008g)を得る。
【0157】
(実施例11)
糖模倣BACA(図8B)の合成
EDA−XIX及びXXXVI間のカップリング:実施例8のXXXVIのDMF中の溶液にジイソプロピルエチルアミンを加えた後、HATUを加える。溶液を室温にて3分間攪拌する。その後、円錐形のバイアル瓶中で攪拌しながら上記の溶液をEDA−XIXに加える。反応混合物を室温にて2時間攪拌する。溶媒を蒸発除去し、粗中間体XXXVIbを得る。これは精製せずに次の段階で使用する。
【0158】
粗XXXVIbをNaOMe−メタノール−H2Oで2時間処理した後、ゲル濾過により精製して、糖模倣BACAを得る。
【0159】
EDA−XIX及び実施例9のXXXVIa間のカップリング:このカップリング反応は、XXXVIbの合成について説明したのと全く同じ方法で実施し、粗生成物XXXVIcを得る。
【0160】
XXXVIcを、記載と同じ方法によりNaOMe−メタノール−H2Oで処理し、糖模倣BACAを得る。
【0161】
(実施例12)
糖模倣BASA(図8C)の合成
この合成は、実施例6のXXXIII及び実施例4のEDA−XIXを使用して、実施例10で説明したのと同じ方法で実施し、図8Cの糖模倣BASAを得る。或いは、EDA−XIXとの反応に、XXXIIIを実施例18のXLVに置き換えることも可能である。
【0162】
(実施例13)
糖模倣BASA(図9A)の合成
この合成は、実施例7のXXXII及び実施例5のEDA−XXVIIIを使用して、実施例10で説明したのと同じ方法で実施し、図9Aの糖模倣BASAを得る。
【0163】
(実施例14)
糖模倣BACA(図9B)の合成
EDA−XXVIII及びXXXVI間のカップリング:このカップリング反応は、XXXVIbの合成について説明したのと全く同じ方法で実施し、粗生成物XXXVIdを得る。
【0164】
記載と全く同じ方法でXXXVIdをNaOMe−メタノール−H2Oで処理し、糖模倣BACAを得る。
【0165】
EDA−XXVIII及びXXXVIa間のカップリング:このカップリング反応は、XXXVIbの合成について説明したのと全く同じ方法で実施し、粗生成物XXXVIeを得る。
【0166】
記載と全く同じ方法によりXXXVIeをNaOMe−メタノール−H2Oで処理し、糖模倣BACAを得る。
【0167】
(実施例15)
糖模倣BASA(図9C)の合成
この合成は、実施例6のXXXIII及び実施例5のEDA−XXVIIIを使用して、実施例10で説明したのと同じ方法で実施し、図9Cの糖模倣BASAを得る。或いは、EDA−XXVIIIとの反応に、XXXIIIを実施例18のXLVに置き換えることも可能である。
【0168】
(実施例16)
糖模倣物(図12)の合成
中間体XXXVIIの合成:中間体XXVの合成について実施例5で説明したのと同じ方法により、実施例5の中間体XXIV(0.1g)のピリジン溶液を、ピリジン中の塩化ニコチニル(0.08mL)及びジメチルアミノピリジン(0.04g)で処理して、中間体XXXVII(0.075g)を得る。
【0169】
中間体XXXVIIIの合成:化合物XVIIの合成について実施例4で説明したのと同じ方法により、中間体XXXVII(0.07g)をトリフェニルホスフィン及びオロチン酸塩化物で処理して、XXXVIII(0.048g)を得る。
【0170】
中間体XXXIXの合成:記載の通り、Pd/Cによる中間体XXXVIII(0.04g)の水素化により、化合物XXXIX(0.02g)を得る。
【0171】
化合物XLの合成:中間体XXXIX(0.015g)を60℃にて10分間0.5NのNaOHで処理し、セファデックスG−25ゲル濾過により精製して、化合物XL(0.010g)を得る。
【0172】
(実施例17)
糖模倣物(図13)の合成
XLIの合成:t−BuOH−水(4mL、1:1)中の化合物XVI(0.1g)の懸濁液に、1−エチニル−3−フルオロベンゼン(0.9g)、1%のCuSO4(0.1mL)、及びL−アスコルビン酸Na(4mg)を加える。混合物を20時間攪拌しながら加熱する(70℃)。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させる。有機層を水で洗浄し、乾燥させ(無水硫酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XLI(0.08g)を得る。
【0173】
XLIIの合成:化合物XLI(0.25g)をジオキサン−水(4:1、7.5mL)に溶解させる。10%のPd/C(0.25g)を加え、続いて酢酸(7滴)を加える。混合物を40psiにて15時間水素化する。反応混合物をセライトベッドを通して濾過し、乾固状態まで濃縮して、化合物XLII(0.2g)を得る。
【0174】
XLIIIの合成:メタノール(5mL)中の化合物XLII(0.2g)の溶液に、メタノール(0.05mL)中のNaOMeの溶液を加え、反応混合物を室温にて2時間攪拌する。反応混合物を、酢酸を数滴加えて中和し、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XLIII(0.15g)を得る。
【0175】
XLIVの合成:化合物XLIII(0.15g)をエチレンジアミン(7mL)に溶解させ、反応混合物を70℃にて9時間攪拌する。溶媒を蒸発除去し、残留物を、最初にカラムクロマトグラフィー(シリカゲル)により精製した後、逆相C18により精製し、化合物XLIV(0.11g)を得る。
【0176】
糖模倣BASA(化合物XLV)の合成:この合成は、実施例6のXXXIIIとXLIVを使用して、実施例10で説明したのと同じ方法で実施し、化合物XLVを得る。
【0177】
(実施例18)
糖模倣BASA(図14)の合成
化合物XLVの合成:DMF(1mL)中の3,6−ジオキサオクタン二酸(PEG、200mg、市販)の溶液にヒューニッヒ塩基(0.4mL)を加え、5分後にHATU(0.35g)を加える。溶液を室温にて10分間攪拌した後、8−アミノナフタレン−1,3,6トリスルホン酸(50mg、市販)のDMF溶液を加える。反応混合物を室温にて4時間攪拌して、溶媒を蒸発除去する。残留物をHPLC(逆相C18カラム)により精製し、XLV(25mg)を得る。
【0178】
化合物XLVIの合成:この合成は、実施例4のXLV及びEDA−XIXを使用して、実施例10で説明したのと同じ方法で実施し、化合物XLVI(4mg)を得る。
【0179】
(実施例19)
糖模倣BASA(図15)の合成
XLVIIの合成:この合成は、化合物XXIVを出発物質として、XLIの合成について説明したのと同じ方法で実施し、化合物XLVIを得る。
【0180】
XLVIIIの合成:この合成は、化合物XLVIIを出発物質として、化合物XLIII(XLIから)で説明したのと同じ方法で実施し、化合物XLVIIIを得る。
【0181】
XLIXの合成:この合成は、化合物XLVIIIを出発物質として、XLIVの合成について説明したのと同じ方法で実施し、化合物XLIXを得る。
【0182】
糖模倣BASA(化合物L)の合成:この合成は、実施例6のXXXIII及びXLIXを使用して、実施例10で説明したのと同じ方法で実施し、化合物Lを得る。或いは、XLIXとの反応のために、XXXIIIを実施例18のXLVに置き換えることも可能である。
【0183】
(実施例20)
糖模倣物(図16)の合成
化合物Iの合成:参考文献[J. Org. Chem. 54, 3738−3740 (1989);Liebigs Annalen der Chemie 575,
1 (1952)]に記載の通り合成する。
【0184】
中間体IIの合成:I(2.8g、15.04mmol)、ピリジン(4.8mL、60.15mmol)、塩化ベンゾイル(3.5mL、30.07mmol)、及び触媒量のジメチルアミノピリジンを、ジクロロメタン(6mL)中にて室温にて攪拌する。2時間後、TLC対照が反応の完了を示す。次に、反応混合物を酢酸エチル(200mL)で希釈し、水、1NのHCI水溶液(氷冷)、飽和炭酸水素ナトリウム水溶液、及び塩水で洗浄する(各50mL)。水層を、酢酸エチルで2回洗浄し(150mLで2回)、収集してNa2SO4で乾燥させる。溶媒を濾過し蒸発させた後、残留物をシリカゲルクロマトグラフィー(PE/EtOAc=4:1)により精製し、化合物II(3.78g、86%)を得る。
【0185】
中間体IIIの合成:II(3.78g、13.02mmol)及びNaBH4を、メタノール(35mL)中にて0℃にて攪拌する。30分後、反応混合物を水(15mL)で急冷し、1NのAcOH水溶液で中和する。再び水(10mL)を加え、混合物をジクロロメタンで3回抽出する(150mLで3回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾燥させる。残留物をシリカゲルクロマトグラフィー(PE/EtOAc=3:2)により処理し、化合物III(3.7g、97%)を得る。[α]D+79.78°(c=0.940、CH2Cl2)。
【0186】
中間体IVの合成:CHCl3(50mL、標準的アロックスで濾過)中のIII(3.4mg、11.64mmol)の溶液に、1−クロローN,N,2−トリメチルプロペニルアミン(4.94mL、34.93mmol)をアルゴン雰囲気下で室温にてシリンジによって加える。TLC対照が反応の完了を示すまで、反応混合物を還流しながら攪拌する(30分)。反応混合物を室温に冷却した後、トリエチルアミン(6mL)によって急冷して、蒸発乾固する(浴温30℃)。残留物をシリカゲルクロマトグラフィー(PE/EtOAc=9:1)により精製し、化合物IV(3.3g、92%)を得る。
【0187】
中間体Vの合成:乾燥トルエン中(40mL)におけるIV(3.25g、10.47mmol)の溶液に、新たに蒸留されたBu3SnH(30.58mL、115.14mmol)及びAIBN(1.7g、10.47mmol)をアルゴン雰囲気下で加える。反応物を還流する。75分後、TLC対照が反応の完了を示したら、反応混合物を室温に冷却した後、アセトニトリル(50mL)で希釈する。溶液をヘキサン(50mL)によって洗浄し、そのヘキサン層をアセトニトリル(50mL)によって再び抽出する。収集したアセトニトリル層を蒸発させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=14:1)で処理し、化合物V(2.66g、92%)を得る。[α]D−117.79°(c=1.810、CH2Cl2)。
【0188】
中間体VIの合成:酢酸80%水溶液中のV(2.63g、9.54mmol)の溶液を80℃にて攪拌する。TLC対照が反応の完了を示したら(30分)、反応混合物を室温に冷却する。NaOH水溶液で中和した後、混合物をジクロロメタンで3回抽出する(200mLで3回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾固する。残留物をシリカゲルクロマトグラフィー(PE/EtOAc=3:2)により精製して、化合物Vl(2.01g、89%)を得る。[α]D−54.86°(c=1.420、CH2Cl2)。
【0189】
中間体VIIの合成:乾燥ジクロロメタン(40mL)中の、Vl(1.98g、8.39mmol)、新たに再結晶したトルエン−4−スルホニルクロリド(1.9g、10.07mmol)、Bu2SnO(2.09g、8.39mmol)、及びトリエチルアミン(1.8mL、16.78mmol)を、アルゴン雰囲気下で室温にて攪拌する。20時間後、TLC対照が反応の完了を示す。その後、反応混合物をメタノール(15mL)で急冷し、蒸発させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=8:1)で処理し、化合物VII(2.65g、85%)を得る。[α]D−68.08°(c=0.448、CH2Cl2)
中間体VIIIの合成:DMF(30mL)中のVII(640mg、1.71mmol)及びNaN3(555mg、8.55mmol)の混合物を、アルゴン雰囲気下で80℃にて攪拌する。TLCにより反応の完了を示したら(1時間後)、反応混合物を室温に冷却し、ジクロロメタン(50mL)で希釈して、水(50mL)で洗浄する。次に、水層をジクロロメタンで2回抽出し(50mLで2回)、収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=6:1)により精製して、化合物VIII(391mg、87%)を得る。[α]D−69.39°(c=2.330、CH2Cl2)。
【0190】
中間体IXの合成:乾燥ジクロロメタン(1mL)中のエチル2,3,4−トリ−O−ベンジル−α−L−フコチオピラノシド(223mg、0.47mmol)の攪拌溶液に、アルゴン雰囲気下で0℃にて臭素(48μ、0.54mmol)を加える。5分後、冷却浴を取り外し、反応混合物を更に室温にて40分攪拌する。過剰の臭素を除去するために、反応混合物にシクロヘキセン(50μL)を加えると、反応混合物の色が消える。次に、この反応混合物を、VIII(61mg、0.23mmol)、(Et)4NBr(98mg、0.47mmol)、及び粉末状の4Åモレキュラーシーブ(100mg)をジクロロメタン(1.6mL)及びDMF(1mL)中にてあらかじめ攪拌しておいた溶液(室温にて1時間)に加える。18時間後、反応をピリジン(2mL)によって急冷し、更に15分間攪拌し、EtOAc(50mL)で希釈して、飽和KHCO3水溶液、水、及び塩水で洗浄する(各50mL)。水層をEtOAcで2回抽出する(50mLで2回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾固させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、IX(116mg、73%)を得る。[α]D−95.96°(c=1.040、CH2Cl2)。
【0191】
中間体Xの合成:メタノール(10mL)中のIX(530mg、0.78mmol)、及び新たに調製した触媒量のNaOMeを、アルゴン雰囲気下で室温にて攪拌する。反応混合物を、粉末状のアンバーリスト−15で中和し、セライトで濾過する。濾液を蒸発乾固させ、シリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、化合物X(380mg、85%)を得る。[α]D−76.425°(c=4.00、CH2Cl2)。
【0192】
中間体XIの合成:ジクロロメタン(3mL)中の、X(184mg、0.32mmol)、ガラクトース構築ブロック[375mg、0.4811mmol、HeIv. Chim. Acta 83:2893−2907 (2000)に既述の通り合成]、及び活性化粉末状のモレキュラーシーブ4Å(200mg)の混合物を、アルゴン雰囲気下で室温にて4時間攪拌する。その後、DMTST[B](165mg、0.64mmol)を4等分して1.5時間かけて、ジクロロメタン(3mL)中にて活性化粉末状のモレキュラーシーブ4Å(100mg)を予め攪拌しておいた混合物(室温にて4時間)に加える。92時間後、TLC対照が反応の完了を示したら、反応混合物をセライトで濾過し、濾液をジクロロメタン(50mL)で希釈する。有機層を飽和炭酸水素ナトリウム水溶液及び塩水(各20mL)で洗浄し、水層をジクロロメタンで2回抽出する(50mLで2回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾固させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、化合物XI(310mg、75%)を得る。[α]D−36.98°(c=2.32、CH2Cl2)。
基本手順A
中間体XIIの合成:乾燥ジクロロメタン(3mL)中のXl(100mg、0.077mol)の攪拌溶液に、塩化酢酸(27μL、0.387mmol)を加える。5分後、PPh3(101mg、0.387mmol)を加え、溶液を室温にて攪拌する。22時間後、TLC対照が反応の完了を示したら、溶媒を除去し、残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=2:1)で処理して、化合物XII(43mg、43%)を得る。[α]D−55.33°(c=2.185、CH2Cl2)。
【0193】
中間体XIIIの合成:手順Aに従って:Xl(80mg、0.062mol)及びオロチン酸塩化物(59mg、0.310mmol)、及びPPh3(81mg、0.310mmol)を使用して、4時間後、(ジクロロメタン/メタノール25:1)で処理して、化合物XIII(35mg、40%)を得る。[α]D−26.08°(c=1.48、CH2Cl2)。
【0194】
中間体XIVの合成:手順Aに従って:Xl(70mg、0.054mmol)及びビフェニル−4−カルボン酸塩化物(58mg、0.271mmol)、及びPPh3(71mg、0.271mmol)を使用して、4時間後、(トルエン/EtOAc=6:1)で処理して、XIV(22mg、28%)を得る。[α]D−32.11°(c=1.065、CH2Cl2)
中間体XVの合成:手順Aに従って:Xl(45mg、0.035mmol)及びビフェニル−2−カルボン酸塩化物(125mg0.577mmol)、及びPPh3(151mg、0.577mmol)を使用して、5時間後、(トルエン/EtOAc6:1)で処理して、化合物XV(22mg、44%)を得る。[α]D−19.54°(c=1.10、CH2Cl2)。
基本手順B
中間体XVIの合成:CH2Cl2(2mL)及び水(100μL)中のXl(120mg、0.093mmol)及びPPh3(30mg、0.116mmol)の混合物を、室温にて44時間攪拌する。次に、溶媒を除去し、残留物をCH2Cl2(2mL)に溶解させ、DIC(11.8mg、0.094mmol)及びバニリン酸(24mg、0.139mmol)を加える。混合物を室温にて更に62時間攪拌し、溶媒を除去して、粗生成物をシリカゲルクロマトグラフィー(トルエン/EtOAc=2.5:1)により精製し、化合物XVI(62mg、47%)を得る。[α]D−29.58°(c=2.86、CH2Cl2)。
基本手順C
中間体XVIIの合成:XII(40mg、0.0306mmol)、Pd(OH)2(30mg)、ジオキサン(2mL)、及び水(0.4mL)の混合物を、室温にて5barにてParrシェーカーで水素化する。20時間後、TLC対照が反応の完了を示す。反応混合物をセライトで濾過し、蒸発乾固する。クロマトグラフィー(CH2Cl2/メタノール=9:1)によって粗生成物を精製し、XVII(20mg、69%)を得る。[α]D−43.50°(c=1.00、MeOH)。
【0195】
中間体XVIIIの合成:手順Cに従って:XIV(26mg、0.018mmol)、Pd(OH)2(11mg)、ジオキサン(1.2mL)、及び水(0.25mL)を使用して、50時間後、(CH2Cl2/メタノール=2:1)で処理して、XVIII(10mg、50%)を得る。
【0196】
中間体XIXの合成:手順Cに従って:XV(22mg、0.015mmol)、Pd(OH)2(20mg)、ジオキサン(1mL)、及び水(0.25mL)を使用して、22時間後、(CH2Cl2/メタノール=15:1)で処理して、化合物XIX(13mg、82%)得る。[α]D−9.50°(c=1.21、MeOH)。
【0197】
中間体XXIの合成:手順Cに従って:XIII(21mg、0.015mmol)、Pd(OH)2(20mg)、ジオキサン(1mL)、及び水(0.25mL)を使用して、22時間後、(CH2Cl2/メタノール=15:1)で処理して、化合物XXI(13mg、86%)を得る。
【0198】
中間体XXの合成:XVI(20mg、0.0141mmol)、10%Pd/C(20mg)、メタノール(2mL)、及び酢酸(50μL)の混合物を、水素雰囲気下で攪拌する。TLC対照が反応の完了を示したら(22時間後)、混合物をセライトで濾過し、蒸発乾固する。残留物をシリカゲルクロマトグラフィー(CH2Cl2/メタノール=2:1)で処理し、XX(15mg、定量)を得る。[α]D−29.32°(c=1.105、MeOH)。
基本手順D
生成物XXIIの合成:乾燥メタノール(1mL)中のXVII(27mg、0.0278mmol)の溶液、及び新たに調製した触媒量のNaOMeを、アルゴン雰囲気下で室温にて攪拌する。1時間後、TLCにより反応の完了を確認する。反応混合物を粉末状のAmberlyst−15で中和し、セライトで濾過して、濾液を蒸発乾固する。残留物をメタノール(20mL)に溶解させて、イオン交換ダウエックスNa(登録商標)で濾過する。濾液を蒸発乾固して、粗生成物をクロマトグラフィー(CH2Cl2/メタノール/水=10:4:0.8)で処理し、XXII(12mg、56%)を得る。[α]D−97.49°(c=0.546、MeOH)。
【0199】
生成物XXIIIの合成:手順Dに従って:XVIII(9.0mg、0.0083mmol)を使用して、2時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理して、化合物XXIII(7.5mg、定量)を得る。[α]D−79.50°(c=0.400、MeOH)。
【0200】
生成物XXIVの合成:手順Dに従って:XIX(23mg、0.0212mmol)及びMeOH(2mL)を使用して、2.5時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理して、化合物XXIV(7.0mg、39%)を得る。[α]D−54.54°(c=0.586、MeOH)。
【0201】
生成物XXVの合成:手順Dに従って:XX(19mg、0.0180mmol)及びMeOH(2mL)を使用して、4時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理し、化合物XXV(10.5mg、70%)を得る。[α]D−69.96°(c=0.793、MeOH)。
【0202】
生成物XXVIの合成:手順Dに従って:XXI(19mg、0.0180mmol)及びメタノール(2mL)を使用して、4時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理して、化合物XXVI(10mg、65%)を得る。
【0203】
(実施例21)
E−セレクチン拮抗薬活性の検定(図17A)
マイクロタイタープレート(プレート1)のウェルをE−セレクチン/hIgキメラ(GlycoTech Corp.[米国メリーランド州ロックビル])により、37℃にて2時間インキュベートすることによってコーティングする。50mMのTris HCl、150mMのNaCl、2mMのCaCl2、pH7.4(Tris−Ca)でプレートを5回洗浄した後、Tris−Ca/Stabilcoat(SurModics[米国ミネソタ州エデン・プレーリー])(1:1,v/v)中の1%BSA(100μL)を、各ウェルに添加して、非特異的結合をブロッキングする。試験化合物を、第2の低結合丸底プレート(プレート2)において、Tris−Ca(60μL/ウェル)中、順次希釈する。ストレプトアビジン−HRP(Sigma[米国ミズーリ州セントルイス])をSLea−PAA−ビオチン(GlycoTech Corp.[米国メリーランド州ロックビル])と混合させて予め形成しておいた結合体を、プレート2の各ウェルに添加する(60μL/ウェルの1μg/mL)。プレート1を、Tris−Caで数回洗浄し、100μL/ウェルをプレート2からプレート1に移す。正確に2時間室温にてインキュベートした後、プレートを洗浄し、100μL/ウェルのTMB試薬(KPL labs[米国メリーランド州ゲイザースバーグ])を各ウェルに加える。3分間室温にてインキュベートした後、100μL/ウェルの1MのH3PO4を加えることによってこの反応を停止させ、450nmでの吸光度をマイクロタイタープレートリーダーによって測定する。
【0204】
(実施例22)
P−セレクチン拮抗薬活性の検定(図17B)
ネオ糖タンパク質であるシアリルLea−HSA(Isosep AB[スウェーデン])を、マイクロタイタープレート(プレート1)のウェル上にコーティングした後、そのウェルをダルベッコのリン酸緩衝化生理食塩水(DPBS)中で希釈された2%のウシ血清アルブミン(BSA)を添加することによりブロックする。第2のマイクロタイタープレート(プレート2)において、試験拮抗薬をDPBS中の1%BSAにおいて順次希釈する。ブロック後、プレート1を洗浄し、プレート2の内容物をプレート1に移す。P−セレクチン/hIg組換えキメラタンパク質(GlycoTech Corp.[米国メリーランド州ロックビル])をプレート1における各ウェルに更に加え、この結合プロセスを室温にて2時間インキュベートする。次いで、プレート1をDPBSで洗浄し、ペルオキシダーゼ標識ヤギ抗ヒトIg(γ)(KPL Labs[米国メリーランド州ゲイザースバーグ])の1μg/mLを各ウェルに加える。室温にて1時間インキュベーションした後、そのプレートをDBPSで洗浄した後、TMB基質(KPL Labs)を各ウェルに加える。5分後、1MのH3PO4を添加して反応を停止させる。その後、450nmでの吸光度をマイクロタイタープレートリーダーを使用して測定する。
【0205】
(実施例23)
抗炎症性マウスモデル。in vivoでの空気嚢に対するIL−1β−誘発性好中球遊走についての試験化合物の効果(図19)
動物
雄の非近交系スイスアルビノマウス(15〜18g体重)を、Bantin and Kingmanから購入し(T.O. stain;ハル、ハンバーサイド)、標準の固形飼料を与え、水道水を自由に摂取できるようにして、12時間の明/暗サイクルで飼育する。全ての動物は、実験前の7日間飼育し、実験日(6日目;以下を参照)には体重が約25gに達する。
実験設計
0日目及び3日目に空気注入(2.5mL、皮下注射)により、マウスの背部に空気嚢を形成する(Perretti & Flower, 1993)。カルボキシメチルセルロース(CMC)の均質な懸濁液を、PBSにおいて0.5%w/vで調製し、その懸濁液にマウス組換え型IL−1βを20ng/mLの濃度で加える。
【0206】
IL−1β投与の直前の0時間の時点で試験化合物を投与する。又、基底の陰性対照を提供するために、CMCだけ(IL−1βなし)を投与した比較用のマウスの群にも投与する。
【0207】
全ての場合において、IL−1β後4時間で3mMのEDTAを含むPBS(2mL)によって空気嚢を洗浄し、肺胞洗浄液の定比容量(100μL)を採取し、チュルク溶液(3%酢酸中の0.01%クリスタルバイオレット)で1:10に希釈して、遊走した白血球(>=90% 多形核白血球、PMN)の数を測定する。次に、サンプルをボルテックスで攪拌し、10μLの染色した細胞溶液をノイバウエル血液波長計にセットして光学顕微鏡(Olympus B061)を使用して好中球数を計数する。
化合物投与
実験当日、試験化合物の新鮮な溶液を、1mMのCaCl2及びMgCl2を補ったPBSのダルベッコ緩衝剤において調製する。マウスのP−セレクチン又はE−セレクチンに対するモノクローナル抗体(mAb)を、BD Pharmingenから購入し、一方、抗L−セレクチンmAbはSerotecから購入する。
【0208】
ラット抗マウスL−セレクチン(クローンMEL−14):1mg
ラット抗マウスP−セレクチン(クローンRB40.34):0.5mg/mL
ラット抗マウスE−セレクチン(クローン10E9.6):0.5mg/mL
図19
6日後のマウス空気嚢は、0時間の時点でIL−1β(10ng)によって炎症を起こしている。実施例17の糖模倣BASA(図13)及び実施例12の糖模倣BASA(図8C)を、0時間及び4時間の時点で静脈内に投与する。抗セレクチンmAbの混合物を0時間の時点で静脈内に投与する。
【0209】
8時間の時点で空気嚢を洗浄し、染色して光学顕微鏡検査により遊走したPMNの数を測定する。
【0210】
図19において、n数は、群A、B、C及びD当たり、それぞれマウス9、8、7及び8匹である。
【0211】
本明細書で引用し及び/又は出願データシートに列挙した上記の米国特許、米国特許出願公開、米国特許出願、外国特許、外国特許出願及び特許以外の刊行物は全て、全体が参考として本明細書で援用される。
【0212】
以上において本発明の特定の実施形態を例示のために本明細書で説明してきたが、本発明の趣旨及び適用範囲から逸脱することなく、種々の改変が行われる場合があることが、以上のことから理解されるであろう。
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は一般的に、セレクチン結合によって媒介されるプロセスを調節するための化合物、組成物、及び方法に関する。より具体的には、セレクチンモジュレーター及びそれらの使用に関し、セレクチン媒介性機能を調節するこれらのセレクチンモジュレーターは、特定の糖模倣物を単独で、或いはBASA(ベンジルアミノスルホン酸)と呼ばれる化合物のクラスのメンバー又はBACA(ベンジルアミノカルボン酸)と呼ばれる化合物のクラスのメンバーと結合して含む。
【背景技術】
【0002】
(関連技術の記載)
組織が感染又は損傷すると、炎症性プロセスによって、白血球及びその他の免疫系構成要素が感染又は損傷の部位に誘導される。このプロセスにおいては、白血球が、微生物の飲み込み及び消化において重要な役割を果たす。従って、感染又は損傷した組織への白血球の動員は、有効な免疫防御を備える上で極めて重要である。
【0003】
セレクチンは、内皮細胞への白血球の結合を媒介するために重要な、構造的に類似する細胞表面受容体の群である。これらのタンパク質は、I型膜タンパク質であり、アミノ末端レクチンドメイン、上皮成長因子(EGF)様ドメイン、種々の補体受容体関連反復、疎水性ドメインが広がる領域、及び細胞質ドメインから構成される。結合相互作用は、セレクチンのレクチンドメインと、種々の炭水化物リガンドとの接触によって媒介されると考えられている。
【0004】
既知のセレクチンには、E−セレクチン、P−セレクチン及びL−セレクチンの3種類が存在する。E−セレクチンは、毛細管の内部壁に沿って並ぶ活性化内皮細胞の表面に存在する。E−セレクチンは、炭水化物シアリル−ルイスX(SLeX)に結合する。これは、特定の白血球(単球及び好中球)の表面に糖タンパク質又は糖脂質として存在し、周囲組織が感染又は損傷している領域の毛細血管壁にこれらの細胞が接着するのを助ける。又、E−セレクチンは、多くの腫瘍細胞上に発現するシアリル−ルイスa(SLea)にも結合する。P−セレクチンは、炎症性内皮及び血小板上に発現し、これもSLeX及びSLeaを認識するが、硫酸化チロシンと相互作用する第2の部位も含む。一般的にE−セレクチン及びP−セレクチンの発現は、毛細血管に隣接する組織が感染又は損傷した場合に増加する。L−セレクチンは、白血球上に発現する。セレクチンにより媒介される細胞間接着は、セレクチン媒介性機能の一つの例である。
【0005】
セレクチン媒介性機能のモジュレーターには、PSGL−1タンパク質(及びより小さなペプチドフラグメント)、フコイダン(fucoidan)、グリチルリチン(及び誘導体)、抗セレクチン抗体、硫酸化ラクトース誘導体、並びにヘパリンが含まれる。これらは全て、不十分な活性、毒性、特異性の欠如、乏しいADME特徴、及び/又は材料の可用性によって、薬剤開発に不適切であることが示されている。
【0006】
セレクチン媒介性細胞接着は、感染と戦い、異物を破壊するために必要とされるが、このような細胞接着が望ましくないか又は過剰であり、組織の修復ではなく損傷が生じる状況もある。例えば、多くの病状(例えば、自己免疫疾患及び炎症性疾患、ショック症状、並びに再灌流障害)には、白血球の異常な接着が関与する。このような異常な細胞接着は、移植及び移植片の拒絶においても役割を果たす場合がある。更に、循環癌細胞の中には、活性化された内皮に結合するために炎症機序を利用するものもあると考えられている。このような状況において、セレクチン媒介性細胞間接着の調節が望まれる場合がある。
【発明の概要】
【発明が解決しようとする課題】
【0007】
従って、当該技術分野では、セレクチン媒介性機能(例えば、セレクチン依存性細胞接着)の阻害剤を同定し、並びに過剰なセレクチン活性に関連する病態を阻害するためにこのような化合物を使用する方法を開発することが必要とされている。本発明は、これらの必要性を満たす他、更にはその他の関連する利点も提供する。
【課題を解決するための手段】
【0008】
(発明の簡潔な要旨)
簡潔に述べると、本発明は、セレクチン媒介性プロセスを調節するための化合物、組成物、及び方法を提供する。本発明において、セレクチン媒介性機能を調節(例えば、阻害又は増強)する化合物は、特定の糖模倣物を単独で、或いはBASA又はBACAに結合して含む。このような化合物は、薬学的に許容される担体又は希釈剤と組み合わせて、薬学的組成物を形成する場合がある。本化合物又は組成物は、セレクチン媒介性機能(例えば、セレクチン媒介性細胞間接着の阻害)を調節(例えば、阻害又は増強)する方法で使用される場合がある。
【0009】
本発明の一態様においては、以下の化学式を有する化合物であって:
【0010】
【化17】
式中:
R1が、
【0011】
【化18】
(式中、nは0〜2であり、nが2の場合に、R8は独立して選択される)であり;
R2が、H、−C(=O)OX(式中、XはC1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)、−C(=O)NH(CH2)nNH2、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、nは0〜30であり、mは0〜1であり、Lはリンカーであり、Zはベンジルアミノスルホン酸、ベンジルアミノカルボン酸、ポリエチレングリコールである)、或いは二量体を形成するために上記の化学式を有する第二の化合物又はその塩であって、この場合、該第二の化合物又はその塩のR2がm=0であって、Zを有さず、且つ結合点であり;
R3が、−OH、
【0012】
【化19】
、−O−C(=O)−X、−NH2、−NH−C(=O)−NHX、又は−NH−C(=O)−X(式中、nは0〜2であり、Xは、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0013】
【化20】
【0014】
【化21】
(式中、nは0〜10である)からなる群から独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R4が、
【0015】
【化22】
、6’硫酸化GlcNAc、6’カルボキシル化GlcNAc、6’硫酸化GalNAc、6’硫酸化ガラクトース、6’カルボキシル化ガラクトース、又は
【0016】
【化23】
(式中、R9は、アリール、ヘテロアリール、シクロヘキサン、t−ブタン、アダマンタン、又はトリアゾールであり、且つR9の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C8アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R5がHであるか、或いはR4及びR5が一緒になって、
【0017】
【化24】
(式中、R10はアリール、ヘテロアリール、
【0018】
【化25】
(式中、nは0〜10である)であり、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)から独立して選択される1個〜3個で置換される場合がある)であり;
R6が、H、フコース、マンノース、アラビノース、ガラクトース、又はポリオールであり;
R7が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又は
【0019】
【化26】
であり;並びに
R8が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0020】
【化27】
(式中、nは0〜3であり、Xは、H、OH、Cl、F、N3、NH2、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、OC1−C8アルカニル、OC1−C8アルケニル、OC1−C8アルキニル、及びOC1−C14アリールから独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)である、
化合物が提供される。
【0021】
本発明の化合物は、その生理学的に許容される塩を含む。本発明の化合物は、薬学的に許容される担体又は希釈剤と組み合わせて、本発明の組成物を提供する。本明細書中の化学式において、記載された原子、又はその他2本の線の交差によって含意される炭素から伸長する線は、結合点を指す(メチル基を表さない)。
【0022】
本発明の一実施形態において、R6はフコース:
【0023】
【化28】
である。
【0024】
一実施形態において、R7はHである。
【0025】
一実施形態において、R4は
【0026】
【化29】
である。
【0027】
一実施形態において、R4は、
【0028】
【化30】
(式中、R9は上述の一般化学式のように定義される)である。
【0029】
一実施形態において、R9はシクロヘキサンである。
【0030】
一実施形態において、R6はガラクトースである。
【0031】
一実施形態において、R8は
【0032】
【化31】
である。
【0033】
一実施形態において、R2は、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、n、m、L及びZは、上述の一般化学式のように定義される)である。
【0034】
一実施形態において、Zは、ベンジルアミノスルホン酸、ベンジルアミノカルボン酸、又はポリエチレングリコールである。
【0035】
一実施形態において、R3は−O−C(=O)−X(式中、Xは上記の一般化学式のように定義される)である。
【0036】
一実施形態において、Xは、
【0037】
【化32】
である。
【0038】
一実施形態において、R5はHである。
【0039】
一実施形態において、Lは、ポリエチレングリコール又はチアジアゾールである。
【0040】
一実施形態において、化合物は、本発明による化合物を含み、更には診断薬又は治療薬も含む。このような化合物は、薬学的に許容される担体又は希釈剤と組み合わせて、本発明の組成物の一実施形態を形成する場合がある。
【0041】
本発明の別の態様においては、セレクチン媒介性機能を調節するために本発明の化合物又は組成物を使用する方法が提供される。このような化合物又は組成物は、例えば、セレクチン媒介性機能(例えば、セレクチン媒介性細胞間相互作用)を阻害又は増強するために使用される場合がある。化合物又は組成物は、セレクチンを発現する細胞を、セレクチンの機能を調節するのに有効な量で接触させる方法において使用される場合がある。化合物又は組成物は、過剰なセレクチン媒介性機能(例えば、過剰なセレクチン媒介細胞間接着)に関連する病態の発症を阻害する必要がある患者に、このような病態の発症を阻害するのに有効な量で投与する方法において使用される場合がある。このような病態の例には、炎症疾患、自己免疫疾患、感染症、癌、ショック、血栓症、創傷、火傷、再灌流傷害、血小板媒介性疾患、白血球媒介性肺傷害、脊髄損傷、消化管粘膜障害、骨粗鬆症、関節炎、喘息及びアレルギー反応が含まれる。化合物又は組成物は、移植した組織の受容者である患者に、移植した組織の拒絶を阻害するのに有効な量で投与する方法において使用される場合がある。化合物又は組成物は、セレクチンを発現する細胞を、この化合物又は組成物に結合した薬剤と接触させることにより、このような細胞にその薬剤(例えば、診断薬又は治療薬)を標的化するのに有効な量で使用する方法において使用される場合がある。化合物又は組成物は、例えば、上述の使用の何れかのための医薬品の製造において使用される場合がある。
【0042】
本発明のこれら及びその他の態様は、以下の詳細な説明及び添付の図面を参照することにより明らかになるであろう。本明細書に開示される全ての参考文献は、それぞれが個別に援用されるのと同じく、全体が参考として本明細書で援用される。
例えば、本願発明は以下の項目を提供する。
(項目1)
以下の化学式を有する化合物であって:
【化1】
式中:
R1が、
【化2】
(式中、nは0〜2であり、nが2の場合に、R8は独立して選択される)であり;
R2が、H、−C(=O)OX(式中、XはC1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)、−C(=O)NH(CH2)nNH2、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、nは0〜30であり、mは0〜1であり、Lはリンカーであり、Zはベンジルアミノスルホン酸、ベンジルアミノカルボン酸、ポリエチレングリコールである)、或いは二量体を形成するために上記の化学式を有する第二の化合物又はその塩であって、この場合、該第二の化合物又はその塩のR2がm=0であって、Zを有さず、且つ結合点であり;
R3が、−OH、
【化3】
、−O−C(=O)−X、−NH2、−NH−C(=O)−NHX、又は−NH−C(=O)−X(式中、nは0〜2であり、Xは、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【化4】
(式中、nは0〜10である)から独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R4が、
【化5】
、6’硫酸化GlcNAc、6’カルボキシル化GlcNAc、6’硫酸化GalNAc、6’硫酸化ガラクトース、6’カルボキシル化ガラクトース、又は
【化6】
(式中、R9は、アリール、ヘテロアリール、シクロヘキサン、t−ブタン、アダマンタン、又はトリアゾールであり、且つR9の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R5がHであるか、或いはR4及びR5が一緒になって、
【化7】
(式中、R10はアリール、ヘテロアリール、
【化8】
【化9】
(式中、nは0〜10である)を形成し、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)から独立して選択される1個〜3個で置換される場合がある)であり;
R6が、H、フコース、マンノース、アラビノース、ガラクトース、又はポリオールであり;
R7が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又は
【化10】
であり;並びに
R8が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【化11】
(式中、nは0〜3であり、Xは、H、OH、Cl、F、N3、NH2、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、OC1−C8アルカニル、OC1−C8アルケニル、OC1−C8アルキニル、及びOC1−C14アリールから独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)である、
化合物;或いはその生理学的に許容される塩。
(項目2)
R6が
【化12】
である、項目1に記載の化合物又はその塩。
(項目3)
R7がHである、項目1に記載の化合物又はその塩。
(項目4)
R4が
【化13】
である、項目1に記載の化合物又はその塩。
(項目5)
R4が
【化14】
(式中、R9は項目1に従って定義される)である、項目1に記載の化合物又はその塩。
(項目6)
R9がシクロヘキサンである、項目5に記載の化合物又はその塩。
(項目7)
R6がガラクトースである、項目1に記載の化合物又はその塩。
(項目8)
R8が
【化15】
である、項目1に記載の化合物又はその塩。
(項目9)
R2が、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、n、m、L及びZが項目1に従って定義される)である、項目1に記載の化合物又はその塩。
(項目10)
Zが、ベンジルアミノスルホン酸、ベンジルアミノカルボン酸、又はポリエチレングリコールである、項目9に記載の化合物又はその塩。
(項目11)
R3が、−O−C(=O)−X、又は−NH−C(=O)−X(式中、Xが項目1に従って定義される)である、項目1に記載の化合物又はその塩。
(項目12)
Xが
【化16】
である、項目11に記載の化合物又はその塩。
(項目13)
R5がHである、項目1に記載の化合物又はその塩。
(項目14)
Lがポリエチレングリコール又はチアジアゾールである、項目1〜13の何れか1項に記載の化合物又はその塩。
(項目15)
薬学的に許容される担体又は希釈剤と組み合わせて、項目1〜14の何れか1項に記載の化合物又はその塩を含む、組成物。
(項目16)
項目1〜14の何れか1項に記載の化合物又はその塩を含み、更に、診断薬又は治療薬も含む、化合物又はその生理学的に許容される塩。
(項目17)
薬学的に許容される担体又は希釈剤と組み合わせて、項目16に記載の化合物又はその塩を含む、組成物。
(項目18)
セレクチン媒介性機能を調節する方法であって、セレクチンを発現する細胞を、該セレクチンの機能を調節するのに有効な量で、項目1〜14の何れか1項に記載の化合物又はその塩と接触させることを含む、方法。
(項目19)
セレクチン媒介性機能を調節する方法であって、セレクチンを発現する細胞を、該セレクチンの機能を調節するのに有効な量で、項目15に記載の組成物と接触させることを含む、方法。
(項目20)
患者を処置する方法であって、過剰なセレクチン媒介性機能に関連する病態の発症を阻害する必要がある該患者に、当該病態の発症を阻害するのに有効な量で、項目1〜14の何れか1項に記載の化合物又はその塩を投与することを含む、方法。
(項目21)
患者を処置する方法であって、過剰なセレクチン媒介性機能に関連する病態の発症を阻害する必要がある該患者に、当該病態の発症を阻害するのに有効な量で、項目15に記載の組成物を投与することを含む、方法。
(項目22)
移植した組織の拒絶を阻害する方法であって、移植した組織の受容者である患者に、該移植した組織の拒絶を阻害するのに有効な量で、項目1〜14の何れか1項に記載の化合物又はその塩を投与することを含む、方法。
(項目23)
移植した組織の拒絶を阻害する方法であって、移植した組織の受容者である患者に、該移植した組織の拒絶を阻害するのに有効な量で、項目15に記載の組成物を投与することを含む、方法。
(項目24)
薬剤をセレクチン発現細胞に標的化する方法であって、セレクチンを発現する細胞を、該細胞に診断薬又は治療薬を標的化するのに有効な量で、項目16に記載の化合物又はその塩と接触させることを含む、方法。
(項目25)
薬剤をセレクチン発現細胞に標的化する方法であって、セレクチンを発現する細胞を、該細胞に診断薬又は治療薬を標的化するのに有効な量で、項目17に記載の組成物と接触させることを含む、方法。
(項目26)
セレクチン媒介性機能を調節する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目27)
過剰なセレクチン媒介性機能に関連する病態の発症を阻害する必要がある患者を処置する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目28)
移植した組織の拒絶を阻害する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目29)
診断薬又は治療薬を細胞に標的化する方法において使用する、項目1〜14の何れか1項に記載の化合物又はその塩。
(項目30)
セレクチン媒介性機能を調節するための医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
(項目31)
過剰なセレクチン媒介性機能に関連する病態の発症を阻害する医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
(項目32)
移植した組織の拒絶を阻害する医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
(項目33)
細胞に診断薬又は治療薬を標的化する医薬品を調製するための、項目1〜14の何れか1項に記載の化合物又はその塩の使用。
【図面の簡単な説明】
【0043】
【図1】図1A及び1Bは、BASAの合成を例示する図である。
【図2】図2は、BASAの合成を例示する図である。
【図3】図3は、糖模倣物(XIX)の合成を例示する図である。
【図4】図4は、糖模倣物(XXVIII)の合成を例示する図である。
【図5】図5は、PEG化糖模倣物(XXX)の合成を例示する図である。
【図6】図6は、PEG化糖模倣物(XXXI)の合成を例示する図である。
【図7A】図7A、7B及び7Cは、PEG化BASA(XXXII及びXXXIII)及びPEG化BACA(XXXVI及びXXXVIa)の合成を例示する図である。
【図7B】図7A、7B及び7Cは、PEG化BASA(XXXII及びXXXIII)及びPEG化BACA(XXXVI及びXXXVIa)の合成を例示する図である。
【図7C】図7A、7B及び7Cは、PEG化BASA(XXXII及びXXXIII)及びPEG化BACA(XXXVI及びXXXVIa)の合成を例示する図である。
【図8A】図8A、8B及び8Cは、糖模倣BASA(図8A及び8C)及び糖模倣BACA(図8B)の合成を例示する図である。
【図8B】図8A、8B及び8Cは、糖模倣BASA(図8A及び8C)及び糖模倣BACA(図8B)の合成を例示する図である。
【図8C】図8A、8B及び8Cは、糖模倣BASA(図8A及び8C)及び糖模倣BACA(図8B)の合成を例示する図である。
【図9A】図9A、9B及び9Cは、糖模倣BASA(図9A及び9C)及び糖模倣BACA(図9B)の合成を例示する図である。
【図9B】図9A、9B及び9Cは、糖模倣BASA(図9A及び9C)及び糖模倣BACA(図9B)の合成を例示する図である。
【図9C】図9A、9B及び9Cは、糖模倣BASA(図9A及び9C)及び糖模倣BACA(図9B)の合成を例示する図である。
【図10】図10は、糖模倣物の合成を例示する図である。
【図11】図11は、糖模倣物の合成を例示する図である。
【図12】図12は、糖模倣物の合成を例示する図である。
【図13】図13は、糖模倣BASAの合成を例示する図である。
【図14】図14は、糖模倣BASAの合成を例示する図である。
【図15】図15は、糖模倣BASAの合成を例示する図である。
【図16】図16は、糖模倣物の合成を例示する図である。
【図17】図17A及び17Bは、E−セレクチン(図17A)及びP−セレクチン(図17B)のin vitro結合検定における糖模倣阻害剤の活性の比較を示す。A及びBは、本発明以外の糖模倣BASAである。Cは、図8Cの糖模倣BASAである。
【図18】図18は、好中球遊走に対する図8Cの糖模倣BASAの効果を示す。Aはビヒクルのみであり、Eは陽性対照(混合抗体)である。B、C及びDは、5mg/kg、10mg/kg、20mg/kgの用量の図8Cの糖模倣BASAである。
【図19】図19は、マウス空気嚢モデルにおける好中球遊走に対する糖模倣阻害剤の効果の比較を示す。Aは、IL−1β+ビヒクルである。Bは、図13のIL−1β+糖模倣BASAである。Cは、図8CのIL−1β+糖模倣BASAである。Dは、IL−1β+混合抗体(陽性対照)である。*A(ビヒクル群)に対してP<0.05。
【発明を実施するための形態】
【0044】
(発明の詳細な説明)
上述の通り、本発明は、セレクチンモジュレーター、それらの組成物、及びセレクチン媒介性機能を調節する方法を提供する。このような調節因子は、以下で更に詳細に述べる種々の状況において、セレクチン媒介性機能を調節(例えば、阻害又は増強)するためにin vitro又はin vivoで使用される場合がある。セレクチン媒介性機能の例には、細胞間接着及び血管新生の間の新しい毛細管の形成が含まれる。
セレクチンモジュレーター
本明細書で使用される「セレクチンモジュレーター」という用語は、セレクチン媒介性機能(例えば、セレクチン媒介細胞間相互作用)を調節(例えば、阻害又は増強)する分子を指す。セレクチンモジュレーターは、本発明の糖模倣化合物から全体が構成される場合もあれば、BASA(ベンジルアミノスルホン酸)又はBACA(ベンジルアミノカルボン酸)に結合したこのような糖模倣物から構成される場合もあり、或いは上述の何れかの1つ以上の更なる分子構成要素を含む場合もある。
【0045】
BASA又はBACAを有さない本発明のセレクチンモジュレーターは、好ましくはE−セレクチン媒介性機能を阻害するために使用される。本発明の糖模倣物にBASA又はBACAを追加することにより、セレクチンモジュレーターは、P−セレクチン及びE−セレクチン媒介性機能を調節する能力も増大した。
【0046】
本発明のセレクチンモジュレーターは、以下の化学式を有する化合物であって:
【0047】
【化33】
式中:
R1が、
【0048】
【化34】
(式中、nは0〜2であり、nが2の場合に、R8は独立して選択される)であり;
R2が、H、−C(=O)OX(式中、XはC1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)、−C(=O)NH(CH2)nNH2、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、nは0〜30であり、mは0〜1であり、Lはリンカーであり、Zはベンジルアミノスルホン酸、ベンジルアミノカルボン酸、ポリエチレングリコールである)、或いは二量体を形成するために上記の化学式を有する第二の化合物又はその塩であって、この場合、該第二の化合物又はその塩のR2がm=0であって、Zを有さず、且つ結合点であり;
R3が、−OH、
【0049】
【化35】
、−O−C(=O)−X、−NH2、−NH−C(=O)−NHX、又は−NH−C(=O)−X(式中、nは0〜2であり、Xは、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0050】
【化36】
(式中、nは0〜10である)からなる群から独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R4が、
【0051】
【化37】
、6’硫酸化GlcNAc、6’カルボキシル化GlcNAc、6’硫酸化GalNAc、6’硫酸化ガラクトース、6’カルボキシル化ガラクトース、又は
【0052】
【化38】
(式中、R9は、アリール、ヘテロアリール、シクロヘキサン、t−ブタン、アダマンタン、又はトリアゾールであり、且つR9の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)であり;
R5がHであるか、或いはR4及びR5が一緒になって、
【0053】
【化39】
(式中、R10はアリール、ヘテロアリール、
【0054】
【化40】
(式中、nは0〜10である)であり、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)から独立して選択される1個〜3個で置換される場合がある)であり;
R6が、H、フコース、マンノース、アラビノース、ガラクトース、又はポリオールであり;
R7が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又は
【0055】
【化41】
であり;並びに
R8が、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、
【0056】
【化42】
【0057】
【化43】
(式中、nは0〜3であり、Xは、H、OH、Cl、F、N3、NH2、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、OC1−C8アルカニル、OC1−C8アルケニル、OC1−C8アルキニル、及びOC1−C14アリールから独立して選択され、上記の環式化合物の何れもが、Cl、F、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、C1−C14アリール、又はOY(式中、Yは、H、C1−C8アルカニル、C1−C8アルケニル、C1−C8アルキニル、又はC1−C14アリールである)から独立して選択される1個〜3個で置換される場合がある)である、
化合物;或いはその生理学的に許容される塩である。
【0058】
本明細書で使用される「C1−C8アルカニル」という用語は、1〜8個の炭素原子を有するアルカン置換基を指し、直鎖又は分岐鎖である場合がある。例としては、メチル、エチル、プロピル、イソプロピル、ブチル、及びt−ブチルが含まれる。「C1−C8アルケニル」という用語は、1〜8個の炭素原子及び少なくとも1つの炭素−炭素二重結合を有するアルケン置換基を指し、直鎖又は分岐鎖である場合がある。例としては、少なくとも1つの炭素−炭素二重結合を有していることを除いて、「C1−C8アルカニル」と同様の例が含まれる。「C1−C8アルキニル」という用語は、1〜8個の炭素原子及び少なくとも1つの炭素−炭素三重結合を有するアルキン置換基を指し、直鎖又は分岐鎖である場合がある。例としては、少なくとも1つの炭素−炭素三重結合を有していることを除いて、「C1−C8アルカニル」と同様の例が含まれる。「アリール」という用語は、結合により分離される場合もあれば、縮合する場合もある1つ以上の環に、1〜14個の炭素原子を有する芳香族置換基を指す。「ヘテロアリール」は、環炭素に代わって少なくとも1つのヘテロ原子を有する芳香族置換基であることを除いて、「アリール」と同様である。アリール及びヘテロアリールの例には、フェニル、ナフチル、ピリジニル、ピリミジニル、トリアゾロ、フラニル、オキサゾリル、チオフェニル、キノリニル、及びジフェニルが含まれる。本明細書で使用される「独立して選択される」という用語は、同じ又は異なる置換基の選択を指す。
【0059】
本明細書で使用されるポリエチレングリコール(「PEG」)とは、エチレングリコールの複数のユニット、並びに1つ以上の置換基を有するもの(例えば、ジカルボン酸化PEG)を指す。置換基の有無にかかわらずPEGは、当業者にとって周知である。本発明において、PEGは、セレクチンモジュレーターの置換基として、或いはその他の基又は化合物をセレクチンモジュレーターに結合させるリンカーとしての役割を果たす場合がある。即ち、セレクチンモジュレーターは複数のPEGを有する場合がある。
【0060】
第二のセレクチンモジュレーターが第一のセレクチンモジュレーターに結合すると、セレクチンモジュレーターの二量体(即ち、二価の分子)が形成される。2つのセレクチンモジュレーターを接続するために、種々のリンカーが使用される場合がある。例えば、二量体を調製するリンカーとしてPEGが使用される場合がある。本明細書で使用される「二量体」という用語は、ホモ二量体又はヘテロ二量体である場合がある。ホモ二量体とは、(互いに結合するための置換基から独立した)共に結合する2つのセレクチンモジュレーターが同一である場合の二量体を指す。ヘテロ二量体は、(結合置換基から独立した)2つのセレクチンモジュレーターが同一でない場合の二量体を指す。
【0061】
本発明のセレクチンモジュレーターは、上述の通り、上記の化学式のR4において、シアル酸又はシアル酸模倣物を有する場合がある。例えば、シアル酸のヘキソース環をシクロヘキサンと置換する場合がある。セレクチンモジュレーターにシアル酸が存在すると、P−セレクチン結合が増強された。(E−及びP−セレクチン結合の両方ではなく)E−セレクチン結合のみが所望される場合、シアル酸模倣物がセレクチンモジュレーターのシアル酸と置き換わる。
【0062】
シアル酸模倣物とシアル酸とを置き換える代わりに(又はこの置換と組み合わせて)、BASA又はBACAを追加することによってP−セレクチン結合が増強される場合がある。上に開示した通り、本発明のセレクチンモジュレーター化合物は、R2で「Z」を有する場合があり、ZはBASA又はBACAである場合がある。シアル酸を有さない本発明のセレクチンモジュレーター化合物にBASA又はBACAを追加することで、セレクチンモジュレーターを、E−セレクチンに対して選択的に結合する化合物から、E−及びP−セレクチンの両方へ結合する化合物へと変換することができる。BASA又はBACAは、BASA又はBACAの一部或いは類似体を含む(但し、その化合物がセレクチン媒介性機能を調節する能力を維持する場合に限る)。PEGは、BASA(又はBACA)の有無にかかわらず、セレクチンモジュレーターに追加される場合がある。又、BASA又はBACAをセレクチンモジュレーターに結合するために、PEGが使用される場合がある。
【0063】
本発明において、BASAは、セレクチンと相互作用する能力を有する低分子量硫酸化合物である。この相互作用は、セレクチン媒介性機能(例えば、細胞間相互作用)を調節するか、又はその機能の調節(例えば、阻害又は増強)を助ける。BASAは、プロトン化された酸形態として、又はナトリウム塩としての何れかで存在するが、ナトリウムは、カリウム又はその他何れかの薬学的に許容される対イオンで置換される場合がある。代表的なBASAは、以下の構造式を有する:
【0064】
【化44】
。
【0065】
セレクチンと相互作用する(本明細書に記載のセレクチン媒介性機能を調節するか、又はその調節を助ける)能力を維持するBASAの部分も又、本発明のセレクチンモジュレーターのBASA成分である。このような部分は一般的に、BASA構造内に存在する少なくとも一つの芳香族環を含む。特定の実施形態において、一部分は、単一の芳香族環、又は複数のこのような環、或いは対称的なBASA分子の半分を含む場合がある。
【0066】
上述の通り、BASAの類似体及びそれらの部分(いずれも上述の生物学的特性を有する)も又、例えば、このセレクチンモジュレーターのBASA成分により、本発明に包含される。本明細書で使用される「類似体」とは、セレクチン媒介相互作用を阻害する類似体の能力が減少しないような化学的部分の1つ以上の追加、削除及び/又は置換によって、BASA又はその部分とは異なる化合物である。例えば、類似体は、SからPへの置換(例えば、硫酸基をリン酸基で置換)を含む場合がある。その他の可能な変更には:(a)環サイズの変更(例えば、何れの環も、4〜7個の炭素原子を含む場合がある);(b)縮合環の数の変化(例えば、単一の環が3つまでの縮合環を含む多環式部分で置換される場合もあれば、多環式部分が単一の非縮合環で置換される場合もあり、多環式部分内の縮合環の数が変更される場合もある);(c)環置換(芳香族環内の炭素原子に共有結合した水素原子又はその他の部分が、種々の部分(例えば、F、Cl、Br、I、OH、O−アルキル(C1〜8)、SH、NO2,CN、NH2,NH−アルキル(C1〜8)、N−(アルキル)2、SO3M(式中、MはH+、Na+、K+、又はその他の薬学的に許容される対イオンである)、CO2M、PO4M2、SO2NH2,アルキル(C1〜8)、アリール(C6〜10)、CO2−アルキル(C1〜8)、−CF2X(式中、Xは、H、F、アルキル基、アリール基又はアシル基である場合がある)、及び炭水化物)の何れかで置換される場合がある);並びに(d)結合部分(即ち、BASA分子中の環の間に存在する部分)の変更(例えば、アルキル、エステル、アミド、無水物、及びカルバミン酸基等の基が互いに置換される場合がある)、
が含まれる。
【0067】
特定のBASA部分及び類似体は、以下の一般構造式の1つを含む:
【0068】
【化45】
(構造式中、nは0又は1である場合があり、X1は−PO2M、−SO2M、又は−CF2(式中、Mは、例えば水素、ナトリウム、又はカリウム等の薬学的に許容される対イオンである)である場合があり、R1は−OH、−F、又は−CO2R4(式中、R4は−H又は−(CH2)m−CH3である場合があり、mは0〜3の範囲の数である)である場合があり、R2は−H、−PO3M2、−SO3M2、−CH2−PO3M2、−CH2−SO3M2、−CF3、−(CH2)m−C(R6)H−R5、又はR9−N(R10)−である場合があり、R3は−H、−(CH2)m−C(R6)H−R5、又はR9−N(R10)−(式中、R5及びR6は、−H、−CO2−R7、及び−NH−R8から独立して選択される場合があり、R7及びR8は、水素、並びに1つ以上のアルキル基、芳香族部分、アミノ基、又はカルボキシ基を含む部分から独立して選択される場合があり、R9及びR10は、−H、−(CH2)m−CH3、−CH2−Ar、−CO−Ar(式中、mは0〜3の範囲の数であり、Arは芳香族部分(即ち、少なくとも1つの置換又は非置換の芳香族環を含む何れかの部分であって、該環は上述の−CH2−又は−CO−基に直接結合する)である)から独立して選択される場合がある)である場合がある)。
【0069】
BASAのその他の部分及び類似体は、以下の一般構造式を含む:
【0070】
【化46】
(構造式中、R1及びR2は、(i)水素、(ii)1つ以上のアルキル基、芳香族部分、アミノ基、又はカルボキシ基を含む部分、及び(iii)−CO−R3(式中、R3は、上述のようなアルキル又は芳香族部分を含む)、から独立して選択される場合があり、Mは薬学的に許容される対イオンである)。
【0071】
本明細書に記載の構造式及び置換基の種々の組み合わせに由来する個々の化合物又は化合物群は、各化合物又は化合物群を個別に記載するのと同じように本明細書で開示される。従って、特定の構造式及び/又は特定の置換基の選択は、本発明の範囲内に含まれる。
【0072】
代表的なBASA部分及び類似体は、図1A〜1Bに示す化合物に含まれる。セレクチンモジュレーターとして機能する能力に悪影響を及ぼすことなく、これらの図に示す化合物に変更を加える場合があることは、当業者に明らかになるであろう。このような変更には、上述のような削除、追加、及び置換が含まれる。
【0073】
スルホン酸基の代わりに化合物がカルボン酸基を有することを除いて、BACAは、BASAと同様である。例えば、上記のBASA化合物のスルホン酸基をカルボン酸基に置換する場合がある。従って、上記のBASAの開示内容は、参考としてBACAの説明に援用される。
【0074】
BACAの例としては:
【0075】
【化47】
(式中、XはF又はClであり;YはH、−C(=O)(O−CH2CH2)n、又は−C(=0)(CH2)n(式中、nは0〜8である)であり;Zは、H、C1−C8アルカニル、C1−C8アルケニル、又はC1−C8アルキニルである)が挙げられる。
【0076】
上述の通り、BASA又はBACAは、R2においてリンカー(「L」)を介して本発明の化合物に結合する場合がある。一般的に、リンカーは、最初に糖模倣物又はBASA/BACAの一方に結合し、次に他方と反応する。特定の糖模倣物へのBASA又はBACAの結合は、セレクチンモジュレーターを形成する種々の方法で達成することができる。BASA又はBACA、或いは糖模倣物によって保有される(又はこれらに付加する)リンカーは、例えば、−(CH2)n−又は−O(CH2)n−(式中、nは一般的に約1〜20である(その範囲内の任意の整数範囲を含む))等のスペーサー基を含む場合がある。リンカーの例には、糖模倣物上の−NH2(それが短いスペーサー基を含む場合は、例えば−CH2−NH2)がある。一実施形態において、−CH2−NH2は、BASA又はBACAを結合するために使用される場合があるR1において糖模倣物に結合する。最も簡単な結合方法には、還元末端(アノマー性ヒドロキシル/アルデヒド)を含む糖模倣物への、BASA又はBACAの還元アミノ化がある。これは、還元末端へのBASA又はBACAの簡単な反応、及び形成されたイミンのそれに続く還元(例えば、pH4.0においてNaCNBH3を使用)により達成される。最も一般的な手法は、活性化されたリンカーを、アノマー位置においてO、S又はNヘテロ原子(又はC原子)を介して糖模倣物に単に結合させる方法である。このような結合の方法論は、炭水化物において広く研究されており、アノマー選択性は、方法論及び/又は保護基の適切な選択によって容易に達成される。可能性のあるグリコシド合成方法の例には、ハロゲン又は過アセチル化糖でのルイス酸触媒結合形成(Koenigs Knorr)、トリクロロアセトアミデート結合形成、チオグリコシド活性化及びカップリング、グルカール(glucal)活性化及びカップリング、n−ペンテニルカップリング、ホスホン酸エステル同族体化(Horner−Wadsworth−Emmons reaction)、及びその他の多くのものが含まれる。或いは、リンカーは、このアノマー性以外の部分の位置に結合される場合がある。結合に最も利用しやすい部位は、糖模倣物(第一級アルコール)の6ヒドロキシル(6−OH)位置である。6−OHでリンカーを結合させるのは、種々の手段によって容易に達成される場合がある。例としては、オキシ−アニオン(塩基を使用した脱プロトン化によって形成されたアルコールアニオン)と適切な求電子剤(例えば、臭化アルキル/アシルエステル、塩化アルキル/アシルエステル又はスルホン酸アルキル/アシルエステル)との反応、スルホン酸エステル塩化物又はPOCl3との反応によるアルコールの活性化及びその後の求核剤による置換、カップリングのためのアルデヒド又はカルボン酸へのアルコールの酸化、又は更には、異なる官能性を導入するMitsunobu反応の使用が含まれる。リンカーが結合すると、次に、BASA又はBACAの好適な求核基との反応のために官能化される(又はその逆も同様)。これは、チオ尿素結合二官能性リガンドを作製するためのチオホスゲン及びアミンの使用、スクアリン酸ジエチルの結合(再度、アミンにより)及び/又は単純なアルキル化/アシル化反応により達成されることが多い。利用可能な更なる方法には、従来から炭水化物及びペプチドのカップリングに使用されているFMOC固相又は溶液相合成法、及びグリコシル/フコシルトランスフェラーゼ及び/又はオリゴサッカリルトランスフェラーゼ(OST)を利用する可能性がある化学−酵素合成法が含まれる。
【0077】
リンカーの実施形態には、以下が含まれる:
【0078】
【化48】
。
その他のリンカー(例えば、PEG)は、当業者又は本開示内容を保有する者にとって周知であろう。
【0079】
本発明の化合物又はその生理学的に許容される塩は、以下の化学式を有する:
【0080】
【化49】
(式中、R1〜R9は、上述の通り定義される)。
【0081】
好ましい実施形態において、R6はフコース:
【0082】
【化50】
である。好ましい実施形態において、R7はHである。好ましい実施形態において、R4は
【0083】
【化51】
である。好ましい実施形態において、R4は、
【0084】
【化52】
(式中、R9は上述の通り定義される)である。好ましい実施形態において、R9はシクロヘキサンである。好ましい実施形態において、R6はガラクトースである。好ましい実施形態において、R8は、
【0085】
【化53】
である。好ましい実施形態において、R2は、−[C(=O)NH(CH2)nNHC(=O)]m(L)mZ(式中、n、m、L及びZは、上述の通り定義される)である。好ましい実施形態において、Zは、ベンジルアミノスルホン酸、ベンジルアミノカルボン酸、又はポリエチレングリコールである。好ましい実施形態において、R3は−O−C(=O)−X(式中、Xは上述の通り定義される)である。好ましい実施形態において、Xは
【0086】
【化54】
である。好ましい実施形態において、R5はHである。好ましい実施形態において、Lはポリエチレングリコール又はチアジアゾールである。
【0087】
本明細書に記載のセレクチンモジュレーターは、in vivoで所望の部位を十分に標的化する場合があるが、1つ以上の特定の組織に対する標的化を容易にするための更なる標的化部分を含むことが、特定の用途において有利な場合がある。本明細書で使用される「標的化部分」とは、調節剤に結合する場合に、そのモジュレーターの標的組織への輸送を高め、それによって、モジュレーターの局所的濃度を増加させるような何れかの物質(例えば、化合物又は細胞)である場合がある。標的化部分には、抗体又はそのフラグメント、受容体、リガンド、及び標的組織の細胞に結合する又は標的組織付近にあるその他の分子が含まれる。結合は、一般的に共有結合であり、例えば、直接的縮合又はその他の反応によって、或いは二官能性リンカー又は多官能性リンカーを使用する方法によって達成される場合がある。
【0088】
特定の実施形態では、セレクチンモジュレーターに対して薬剤も結合させるか、又は代わりに結合させることが有益な場合がある。本明細書で使用される「薬剤」という用語は、疾患又はその他の望ましくない病態を予防又は処置するために哺乳動物に投与することを目的とした、何れかの生体活性剤を指す。薬剤には、ホルモン、成長因子、タンパク質、ペプチド及びその他の化合物が含まれる。可能性のある薬剤の例には、抗腫瘍性薬剤(例えば、5−フルオロウラシル及びジスタマイシン)、インテグリン作動薬/拮抗薬(例えば、環状RGDペプチド)、サイトカイン作動薬/拮抗薬、ヒスタミン作動薬/拮抗薬(例えば、ジフェンヒドラミン及びクロルフェニラミン)、抗生物質(例えば、アミノグリコシド及びセファロスポリン)並びに酸化還元活性生物学的薬剤(例えば、グルタチオン及びチオレドキシン)が含まれる。他の実施形態において、診断用放射性核種又は治療用放射性核種は、セレクチンモジュレーターに結合する場合がある。多くの実施形態において、本薬剤は、セレクチンモジュレーターへ直接的又は間接的に結合する場合がある。
【0089】
本明細書に記載のモジュレーターは、薬学的組成物内に存在する場合がある。薬学的組成物は、1つ以上の薬学的又は生理学的に許容される担体、希釈剤、又は賦形剤と組み合わせて、1つ以上のモジュレーターを含む。このような組成物は、緩衝剤(例えば、中性緩衝生理食塩水又はリン酸緩衝生理食塩水)、糖質(例えば、グルコース、マンノース、スクロース、又はデキストラン)、マンニトール、タンパク質、ポリペプチド、又はグリシンのようなアミノ酸、抗酸化剤、キレート剤(例えば、EDTA又はグルタチオン)、アジュバント(例えば、水酸化アルミニウム)、及び/又は保存剤を含む場合がある。更に他の実施形態では、本発明の組成物は、凍結乾燥物として処方される場合がある。本発明の化合物は、例えば、局所的、経口、経鼻、静脈内、頭蓋内、腹腔内、皮下又は筋肉内の投与を含めた何れかの適切な投与様式のために処方される場合がある。
【0090】
薬学的組成物は又、薬剤(例えば、上で示される薬剤))等のような1つ以上の活性薬剤も同時に又は代わりに含む場合もあり、それらの活性薬剤は、モジュレーターに結合する場合もあれば、組成物内で何れとも結合しない場合もある。
【0091】
本明細書に記載の組成物は、徐放性製剤(即ち、投与の後に、調節剤の持続放出をもたらすカプセル状又はスポンジ状のような製剤)の一部として、投与される場合がある。このような製剤は一般的に、周知の技法を使用して調製される場合があり、例えば、経口、直腸又は皮下移植によって、又は所望の標的部位における移植によって、投与される場合がある。このような製剤内で使用する担体は、生体適合性を有し、且つ生物分解性も有する場合がある。好ましくは、本製剤は、比較的一定のレベルでの調節剤の放出を提供する。徐放性製剤内に含まれる調節剤の量は、移植の部位、放出の速度、及び期待される放出持続時間、並びに処置又は予防する病態の性質により異なる。
【0092】
セレクチンモジュレーターは一般的に、治療有効量において薬学的組成物中に存在する。治療有効量は、識別可能な患者のベネフィット、例えば、下記のような過剰なセレクチン媒介性機能(例えば、細胞間接着)と関連する病態の治癒の向上等をもたらす量である。
【0093】
一般的に、本明細書に記載の調節剤及び組成物は、セレクチン媒介性機能を増強又は阻害するために使用される場合がある。このような増強又は阻害は、温血動物(好ましくはヒト等の哺乳動物)においてin vitro及び/又はin vivoで達成される場合がある。但し、セレクチンを発現する細胞は最終的に、セレクチン媒介性機能を増強又は阻害するのに十分な量で及び十分な時間にわたって、モジュレーターと接触する。
【0094】
特定の態様において、本発明は、セレクチン媒介性機能(例えば、細胞間接着)に関連する病態の進行を抑制する方法を提供する。一般的に、このような方法は、このような病態を予防、遅延又は処置するために使用される場合がある。換言すれば、本明細書に示される治療方法は、疾患を処置するために使用される場合もあれば、或いは疾患に罹患していない患者、又はセレクチン媒介性機能に関連しない疾患に罹患した患者において、このような疾患の発症を予防又は遅延するために使用される場合もある。例えば、本治療方法には、細胞成長の阻害、細胞の殺傷、細胞成長の防止、細胞成長の発生の遅延、或いは生物体の生存の延長が含まれる場合がある用途がある。
【0095】
セレクチン媒介性機能に関連する病態には、種々のものがある。このような病態には、例えば、組織移植片拒絶、血小板媒介性疾患(例えば、アテローム性動脈硬化症及び凝固)、過活動性冠状動脈循環、急性白血病媒介性肺損傷(例えば、成人呼吸窮迫症候群(ARDS))、クローン病、炎症性疾患(例えば、炎症性腸疾患)、自己免疫疾患(MS、重症筋無力症)、感染症、癌(及び転移)、血栓症、創傷(及び創傷関連の敗血症)、熱傷、脊髄損傷、消化管粘膜疾患(胃炎、潰瘍)、骨粗しょう症、慢性関節リウマチ、変形性関節症、喘息、アレルギー、乾癬、敗血症性ショック、外傷性ショック、脳卒中、腎炎、アトピー性皮膚炎、凍傷性損傷、成人型呼吸困難症候群、潰瘍性大腸炎、全身性エリテマトーデス、糖尿病、及び虚血性エピソード後の再灌流障害が含まれる。又、セレクチンモジュレーターは、回復を高めるために心臓手術の前に患者に投与される場合もある。その他の用途には、疼痛処理の用途、血管ステントに関連する再狭窄の予防、及び例えば癌に関連する望ましくない新脈管形成に対する用途が含まれる。
【0096】
本発明のセレクチンモジュレーターは、処置(又は予防)する疾患に適切な様式で投与される場合がある。適切な投薬量、並びに好適な投与持続期間及び投与頻度は、患者の病態、患者の疾患の型及び重篤度、並びに投与方法等の要因により決定される場合がある。一般的に、適切な投薬量及び処置レジメンは、治療効果及び/又は予防効果を提供するのに十分な量にて調節剤を提供する。本発明の特に好ましい実施形態では、セレクチンモジュレーターは、0.001〜1000mg/体重kg(より一般的には0.01〜1000mg/kg)の範囲の投与量で、毎日1回又は複数回の投与レジメンで投与される場合がある。適切な投与量は一般的に、実験モデル及び/又は臨床試験を使用して決定される場合がある。一般的に、有効な治療を提供するために十分な、最小の投薬量を使用することが好ましい。患者は一般的に、処置又は予防する病態に好適な、当業者に周知の検定を使用して、治療効果について監視される場合がある。
【0097】
セレクチンモジュレーターは又、セレクチンを発現する細胞に物質を標的化するために使用される場合もある。このような物質には、治療薬及び診断薬が含まれる。治療薬は、疾患の処置又は予防或いは患者の生理学的機能の調節に対して有用性を提供するように標的細胞の特性を変更することが実証できる分子、ウイルス、ウイルス成分、細胞、細胞成分、又はその他何れかの物質である場合がある。治療剤は又、in vivoにて生物学的活性を有する薬剤を生成するプロドラッグである場合もある。治療薬である場合がある分子は、例えば、ポリペプチド、アミノ酸、核酸、ポリヌクレオチド、ステロイド、多糖、又は無機化合物である場合がある。このような分子は、種々の方法(例えば、酵素、酵素阻害剤、ホルモン、受容体、アンチセンスオリゴヌクレオチド、触媒性ポリヌクレオチド、抗ウイルス剤、抗腫瘍剤、抗細菌剤、免疫調節剤及び細胞傷害性剤(例えば、ヨウ素、臭素、鉛、パラジウム又は銅のような放射性核種)を含む)の何れかにおいて機能する場合がある。診断薬には、金属及び放射性薬剤のような画像化剤(例えば、ガリウム、テクネチウム、インジウム、ストロンチウム、ヨウ素、バリウム、臭素及びリン含有化合物)、造影剤、色素(例えば、蛍光色素又は発蛍光団)、並びに比色定量的反応又は蛍光定量的反応を触媒する酵素が含まれる。一般的に、治療薬及び診断薬は、上記の技法のような種々の技法を使用してセレクチンモジュレーターへと結合される場合がある。標的化において、セレクチンモジュレーターは、本明細書に記載の通り、患者に投与される場合がある。セレクチンは、血管新生の間の新しい毛細管の形成に関与する内皮細胞上に発現することから、セレクチンモジュレーターは、腫瘍の脈管構造を殺傷するために、治療薬を標的とするために使用される場合がある。セレクチンモジュレーターは遺伝子標的化にも使用される場合がある。
【0098】
セレクチンモジュレーターは又、in vitroで(例えば、種々の周知の細胞培養物内及び細胞分離法において)使用される場合もある。例えば、モジュレーターは、培養におけるスクリーニング、検定及び成長に対し、セレクチンを発現する細胞を固定化する際に使用するために、組織培養プレート又はその他の細胞培養支持体の内部表面に結合される場合がある。このような結合は、上述に記載の方法のような任意の適切な技法、及びその他の標準的技法によって実施される場合がある。モジュレーターは又、例えば、in
vitroでの細胞の同定及び選別を容易にし、セレクチン(又は異なるセレクチンレベル)を発現する細胞の選択を可能にするために使用される場合もある。好ましくは、このような方法で使用するモジュレーターは、検出可能なマーカーに結合する。好適なマーカーは当該技術分野で周知であり、これらには、放射性核種、発光基、蛍光基、酵素、色素、免疫グロブリン定常ドメイン及びビオチンが含まれる。好ましい一実施形態では、蛍光マーカー(例えば、フルオレセイン)に結合したモジュレーターを、細胞と接触させ、次いでこれを、蛍光活性化細胞選別(FACS)によって分析する。
【0099】
上記のような調節剤は、例えば、セレクチン媒介性細胞接着を阻害することができる。この能力は一般的に、セレクチンを発現する細胞間の接着(例えば、白血球又は腫瘍細胞と血小板又は内皮細胞との間の接着)に対する効果を測定するために設計された種々のin vitro検定の何れかを使用して評価される場合がある。例えば、このような細胞は、モジュレーターの非存在下で、細胞接着を可能にする標準的な条件下で平板培養される場合がある。一般的に、試験細胞とモジュレーターとの接触により、細胞接着の識別可能な破壊が生じる場合、このモジュレーターは、セレクチン媒介性細胞接着の阻害剤である。例えば、モジュレーターの存在下(例えば、マイクロモル濃度レベル)では、白血球又は腫瘍細胞と血小板又は内皮細胞との間の接着の破壊は、互いに相互作用する細胞の減少を観察することによって、およそ数分内で視覚的に判定される場合がある。
【0100】
本発明の化合物又は本発明に有用な化合物は全て、それらの生理学的に許容される塩を含む。
【0101】
以下の実施例は、本発明を例示するために提供されるのであって、本発明を限定するために提供されるのではない。
【実施例】
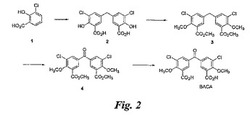
【0102】
(実施例1)
BASA(図1A)の合成
化合物4の合成:市販の2(1g)のニトロ化は、記載の手順に従って行う(文献の条件については、米国特許第4,534,905号;Allison, F., et al., Helv. Chim. Acta 4:2139 (1952)を参照)。
【0103】
粗生成物3を水(40mL)に溶解させ、10%のPd/C(0.3g)を加える。混合物を48時間室温にて水素化(約45psi)する。触媒をセライトを通して濾過し、濾床を水で洗浄する。濾液を減圧濃縮し、桃色の固体を得る。触媒の除去後に、濾液を15mLまで濃縮し、等容積のエタノールを加える。沈殿物を濾過により収集し、不純物の非常に少ない化合物4を得る。
【0104】
化合物7aの合成:5(5g)及び8(4.45g、24.7mmol)、並びにK2CO3(H2O中に2M、24.7mL、49.4mmol)の10:1のトルエン/エタノール(70mL)中の溶液をPd(PPh3)4(1.43g、1.24mmol)で処理し、その混合物を20時間還流する。後処理の後、EtOH溶液中における粗生成物の再結晶化、及び再結晶化の濾液のクロマトグラフィー精製により、化合物9(2.9g、46%、>90%HPLC)を得て、2.2gの5を回収する。生成物を、1H−NMRによって同定する。
【0105】
THF/H2O(1:1)中の9(2.9g、11.3mmol)及びLiOH・H2O(1.43g、34.1mmol)の混合物を室温にて21時間攪拌する。後処理の後、この反応により7(2.58g、94%、>90%HPLC)を得る。生成物を、1H−NMRによって同定する。
【0106】
7(500mg、1.94mmol)、SOCl2(0.23mL、3.10mmol)、及びトルエン(3mL)の懸濁液にDMF(20μL)を加えた後、80℃に加熱する。20時間後、反応が終了し、酸塩化物(640mg)を得る。生成物7aを、IR及び1H−NMRによって同定する。
【0107】
化合物10の合成:H2O(2mL)及びジオキサン(18mL)中のアミンの4(268mg、0.641mmol)の溶液に、ジオキサン(16mL)中の7a(273mg、0.99mmol)の溶液を、30分かけて滴下する。滴下が進むにつれ、反応混合物のpHを0.25MのNaOHにより8.5に調整する。添加後、反応物を室温にて2.5時間攪拌する。カラムクロマトグラフィー(メタノール/トルエン1:1)により精製し、続いて分取TLC(メタノール/トルエン1:1)により精製して、50mgの化合物10を得る。得られた生成物を、1H−NMR及びMSによって同定する。
【0108】
化合物10の水素化:H2O(20mL)中の10(30mg、0.049mmol)及び炭素上の10%Pd(50mg)の懸濁液を、室温にて4時間水素化し(55psi)、図1AのBASAを得る。
【0109】
(実施例2)
BASA(図1B)の合成
化合物4の合成:3−ニトロベンジルヨウ化物(1)(48.3g)を、市販の8−アミノナフタレン−1,3,5−トリスルホン酸(2)(29.5g)の水溶液(pH11)に、室温にて攪拌しながら加える。その溶液のpHを1に調整し、その溶媒を蒸発除去した後、生成物3(6.4g)をエタノールから沈殿させる。
【0110】
化合物3を白金触媒により水素化して、化合物4(図1BのBASA)を収率96%で得る。
【0111】
(実施例3)
BACA(図2)の合成
1(8.9g)、パラホルムアルデヒド(8.9g)、及び硫酸(125mL)の懸濁液を90℃にて14時間加熱し、後処理の後に2の粗生成物(7.8g)を得る。その粗生成物をHPLCにより精製し(純度77%)、1H−NMRによって同定する。
【0112】
アセトン(30mL)中の2(1.0g)の溶液に、K2CO3(3.1g)及び硫酸ジメチル(1.4mL)を加え、反応物を24時間加熱還流する。後処理及び精製のため、反応物を次のバッチと混合する。
【0113】
アセトン(225mL)中の2(7.5g)の溶液に、K2CO3(23.2g)及び硫酸ジメチル(10.8mL)を加え、反応物を16時間加熱還流する。反応物を前のバッチと合わせ、後処理及びカラムクロマトグラフィー精製(酢酸エチル/ヘプタン=1:9)した後、3(7.3g、74%)を得る。生成物をHPLCにより精製し(純度80%)、1H−NMRによって同定する。
【0114】
3℃にて無水クロム酸(6.94g)を、無水酢酸(175mL)中の3(7.16g)の懸濁液に加えた後、室温にて15時間攪拌する。反応物を後処理及びカラム精製(100%のジクロロメタン)した後、4(5.89g)を得る。生成物をHPLCにより精製し(純度90%)、1H−NMRによって同定する。
【0115】
THF/H2O(300mL、1:1)中の4(5.89g)の懸濁液に、室温にてLiOH・H2O(1.74g)を加え、得られた混合物を14時間攪拌する。酸/塩基による後処理の後に、白色固体の生成物を得る。生成物を高真空下で乾燥させて、NMR及び質量分析により同定する。
【0116】
(実施例4)
糖模倣物(図3)の合成
中間体IIの合成:メタノール(200mL)及び硫酸(2mL、98%)中の(−)シキミ酸(20g)を、室温にて50時間攪拌する。反応混合物を2Nの冷NaOH水溶液によって中和する。蒸発乾固後、その残留物をシリカゲルクロマトグラフィーにより精製し、II(19.2g)を得る。
【0117】
中間体(III)の合成:シキミ酸メチル(II、10g)、2,2−ジメトキシプロパン(10mL)、及びp−TsOH(0.8g)をアセトニトリル(125mL)に溶解させ、室温にて1時間攪拌する。次に、反応混合物をトリエチルアミン(2mL)により中和し、蒸発乾固する。その残留物をシリカゲルクロマトグラフィーで処理して、III(11g)を得る。
【0118】
中間体IVの合成:MeOH(40mL)中のシキミ酸誘導体III(10g)及びPtO2/C(10%、250mg)を、室温にて激しく攪拌しながら水素化する。16時間後、反応混合物をセライトによって濾過し、蒸発乾固する。その残留物をシリカゲルクロマトグラフィーで処理して、IVを得る。
【0119】
中間体Vの合成:DCM(100mL)中のIV(8g)の溶液に、0℃にてピリジン(12mL)、無水酢酸(7mL)、及びDMAP(25mg)を加える。反応混合物を室温にて1時間攪拌し、EtOAc(250mL)で希釈する。0.5Mの塩酸(50mLで3回)、KHCO3の飽和溶液(50mLで3回)、及び塩水(50mLで3回)で洗浄後、収集した有機層を乾燥させ(Na2SO4)、蒸発乾固する。残留物をシリカゲルクロマトグラフィーにより精製し、V(6.8g)を得る。
【0120】
中間体VIの合成:酢酸(30mL、80%)中のV(6.0g)の溶液を80℃にて1時間攪拌する。溶媒を蒸発除去し、残留物をシリカゲルクロマトグラフィー(DCM/メタノール14:1)により精製し、VI(3.6g)を得る。
【0121】
中間体VIIの合成:ピリジン(30mL)中のVI(3g)及びp−TsCl(3.5g)の溶液を、室温にて6時間攪拌する。MeOH(5mL)を加え、溶媒を減圧蒸発させて、その残留物をEtOAcに溶解させ(150mLで3回)、その有機層を0.5Mの塩酸水溶液(0℃)、冷水、及び冷塩水で洗浄する。収集した有機層を乾燥させ(Na2SO4により)、セライトで濾過して、蒸発乾固する。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=4:1)により精製し、VII(3.7g)を得る。
【0122】
化合物VIIIの合成:DMF(20mL)中のVII(3g)及びNaN3(2.5g)の溶液を80℃にて攪拌する。反応混合物を室温に冷却し、EtOAc(200mL)及び水(50mL)で希釈する。有機層を更に水で2回(50mLで2回)、塩水で1回(50mL)洗浄する。全ての水層をEtOAcで2回抽出する(50mLで2回)。収集した有機層をNa2SO4で乾燥させ、濾過して、その溶媒を蒸発除去する。残留物をシリカゲルクロマトグラフィー(石油エーテル/EtOAc=5:2)により精製し、VIII(2.2g)を得る。
【0123】
化合物Xの合成:DCM(3mL)中のエチル−2,3,4−トリ−o−ベンジル−α−L−フコチオピラノシドIX(1.5g)の溶液に、0℃にてアルゴン雰囲気下で臭素(150μL)を加える。5分後、冷却槽を取り外して、反応混合物を更に室温にて25分間攪拌する。シクロヘキセン(200μL)を加え、その反応混合物をDCM(10mL)及びDMF(5mL)中のVIII(400mg)、(Et)4NBr(750mg)、及び粉末状の4Åモレキュラーシーブの溶液に加える。16時間後、トリエチルアミン(1.5mL)を加え、更に10分間攪拌し、EtOAc(50mL)で希釈して、飽和NaHCO3水溶液、水、塩水で洗浄する。水層をEtOAcで2回抽出する(50mLで2回)。収集した有機層を、乾燥させ(Na2SO4により)、濾過して蒸発乾固する。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、X(700mg)を得る。
【0124】
化合物XIの合成:メタノール(20mL)中のX(1.5g)の溶液に、新たに調製したNaOMe(80mg)を加え、その反応混合物を圧力管内にて80℃にて20時間攪拌する。反応混合物を室温に冷却し、酢酸によって中和する。溶媒を蒸発乾固して、残留物をエーテルに溶解させる。新たに調製したジアゾメタンを加え、過剰なジアゾメタンを酢酸によって中和する。溶媒を蒸発除去し、XI(1.25g)を得る。
【0125】
構築ブロックXVの合成:この合成は、既述のものと同じ方法で実施する(「Helvetica Chemica Acta 83:2893−2907 (2000))。
【0126】
化合物XVIの合成:DCM(17mL)中のXI(1.6g)、XV(3g)、及び粉末状の活性化モレキュラーシーブ4Å(1g)の混合物を、アルゴン雰囲気下にて室温にて2時間攪拌する。次いで、DMTST(2g)を等量ずつ4回に分けて1.5時間かけて加える。24時間後、反応混合物をセライトによって濾過し、その濾液をDCM(100mL)で希釈する。有機層を飽和NaHCO3水溶液及び塩水で洗浄し、その水層をDCMで2回抽出する。収集した有機層を乾燥させて(Na2SO4により)、濾過して蒸発乾固する。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=8:1)により精製し、XVI(1.5g)を得る。
【0127】
化合物XVIIの合成:ジクロロメタン(10mL)中のXVI(500mg)及びオロチン酸塩化物(500mL)の溶液に、トリフェニルホスフィンの溶液(5mLのジクロロメタン中に500mg)を10分間かけて滴下する。反応混合物を室温にて25時間攪拌し、溶媒を蒸発除去する。残留物を精製し(シリカゲルクロマトグラフィー、DCM/メタノール=19:1)、XVII(250mg)を得る。
【0128】
化合物XVIIIの合成:ジオキサン−水(5:1、12mL)中のXVII(200mg)の溶液に、10%Pd−C(100mg)を加え、その反応混合物を水素下(55psi)で24時間激しく攪拌する。触媒をセライトのベッドを通して濾過し、溶媒を蒸発除去する。残留物をシリカゲルクロマトグラフィーにより精製し、化合物XVIII(150mg)を得る。
【0129】
XIXの合成:メタノール(5mL)中の化合物XVIII(145mg)の溶液に、NaOMeのメタノール溶液(25%、0.025mL)を加え、反応混合物を室温にて4時間攪拌し、酢酸で中和して、溶媒を蒸発除去する。残留物を水に溶解させ、ダウエックス50wX−8(Na形態)樹脂のベッドを通して濾過する。水洗浄物を蒸発除去し、化合物XIX(100mg)を得る。
【0130】
EDA−XIXの合成:XIX(80mg)をエチレンジアミン(EDA)(1mL)と共に攪拌しながら70℃にて5時間加熱する。溶媒を蒸発除去し、セファデックスG−25カラムにより精製して、EDA−XIX(82mg)を得る。
【0131】
(実施例5)
糖模倣物(図4)の合成
化合物XXIの合成:ピリジン(60mL)中の化合物XX(1.5g、以前に公開された手順(Carbohydrate Chemistry and Biochemistry 2000, vol.1, page 345−365)に従って合成)の溶液に、無水安息香酸(0.73g)及びジメチルアミノピリジン(0.02g)を加える。反応混合物を室温にて20時間攪拌する。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させる。溶液を1Mの冷塩酸及び水で連続して洗浄する。溶液を乾燥させ(酢酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXI(1g)を得る。
【0132】
化合物XXIIの合成:ジクロロメタン(20mL)中の化合物XXIの溶液に、トリフルオロ酢酸(20mL)を0℃にて加え、反応混合物を同じ温度で1時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカゲル)により精製して、化合物XXII(0.6g)を得る。
【0133】
化合物XXIIIの合成:ジクロロメタン(40mL)中の化合物XXII(1g)の溶液に、DBU(0.05mL)及びトリクロロアセトニトリル(0.4g)を0℃にて加える。溶液を同じ温度で1.5時間攪拌する。溶媒を蒸発除去し、カラムクロマトグラフィー(シリカゲル)により精製して、化合物XXIII(0.6g)を得る。
【0134】
化合物XXIVの合成:化合物XI(0.7g)、ジクロロメタン(40mL)中の化合物XXIII(0.5g)、及びモレキュラーシーブ(4Å、5g)の混合物に、TMSOTf溶液(ジクロロメタン(5mL)中に0.15mL)を0℃にて攪拌しながら滴下する。同じ温度で2時間攪拌を継続する。トリエチルアミン(0.2mL)を加え、反応混合物をセライトベッドを通して濾過する。反応混合物を、冷飽和重炭酸ナトリウム液によって洗浄する。乾燥させて(酢酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXIV(0.7g)を得る。
【0135】
化合物XXVの合成:DMF(30mL)中の化合物XXIV(0.6g)の溶液に、DBU(30滴)及びdl−ジチオトイトール(DTT、0.28g)を加える。反応混合物を室温にて1時間攪拌する。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させて、水で洗浄する。有機層を乾燥させ(酢酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXV(0.45g)を得る。
【0136】
化合物XXVIの合成:DMF(10mL)中の化合物XXV(0.4g)の溶液に、オロチン酸(0.14g)、EEDQ(0.19g)、及び4−メチル−モルフォリン(0.09g)を加え、反応混合物を70℃にて20時間攪拌する。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させる。溶液を冷飽和重炭酸ナトリウム液及び水で洗浄する。有機層を乾燥させ(硫酸ソーダにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XXVI(0.2g)を得る。
【0137】
化合物XXVIIの合成:化合物XXVI(0.2g)を、記載と同じ条件下で水素化し、記載の通りMeOH中のNaOMeを使用して中間体を部分的に脱ベンゾイル化し、クロマトグラフィーにより精製して、化合物XXVII(0.05Og)を得る。
【0138】
化合物XXVIIIの合成:化合物XXVIIを、記載の通りにエチレンジアミンで処理し、カラムクロマトグラフィー(シリカゲル及びゲル濾過セファデックスG−25)により精製して、化合物XXVIII(25mg)を得る。
【0139】
(実施例6)
PEG化BASA(図7B)の合成
DMF(1mL)中の3,6−ジオキサオクタン二酸(PEG、200mg、市販)の溶液にヒューニッヒ塩基(0.4mL)を加え、5分後にHATU(0.35g)を加える。溶液を室温にて10分間攪拌した後、DMF(0.1mL)中に溶解させた実施例2(50mg)のBASAの溶液を加える。反応混合物を室温にて4時間攪拌し、溶媒を蒸発除去する。残留物をHPLC(逆相C18カラム)により精製し、XXXIII(40mg)を得る。
【0140】
(実施例7)
ペグ化BASA(図7A)の合成
この合成は、実施例1のBASAを使用することを除き実施例6で説明したのと同じ方法で実施し、XXXII(50mg)を得る。
【0141】
(実施例8)
ペグ化BACA(図7C)の合成
中間体XXXIV(方法1)の合成:実施例3のBACA(0.5g)をメタノール−水(1mL、9:1)に懸濁させ、Cs2CO3の水溶液を加えてpHを8.2に調整する。溶媒をトルエンで共蒸発させて除去する。残留物をDMF(1mL)に溶解させる。臭化ベンジル(0.5mL)を加え、室温にて20時間攪拌する。ジクロロメタン(15mL)を加え、冷水で洗浄する。有機層を乾燥させ(無水硫酸ナトリウムにより)、溶媒を蒸発除去する。残留物をカラムクロマトグラフィー(シリカ)により精製し、XXXIV(0.48g)を得る。
【0142】
中間体XXXIV(方法2)の合成:DMF中に溶解させた実施例3のBACA(1g)の溶液にN,N−ジイソプロピルエチルアミン(1.5g)及び臭化ベンジル(1.5g)を加える。反応混合物を50℃にて20時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカ)により精製して、XXXIVを得る。
【0143】
中間体XXXVの合成:メタノール(10mL)中のXXXIV(0.2g)の溶液に、0℃にて水素化ホウ素ナトリウム(0.070g)を加え、反応混合物を0℃にて1時間攪拌する。酢酸を加えて反応物を急冷し、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカ)により精製し、XXXV(0.16g)を得る。
【0144】
XXXVbの中間体の合成:ジクロロメタン中のXXXV(0.36g)の溶液にトリエチルアミン(0.6mL)及びMeSO2Cl(0.29mL)を加える。反応混合物を室温にて21時間攪拌する。反応混合物をジクロロメタンで希釈し、水、1Mの塩酸、及び塩水で洗浄する。有機層を乾燥させ(Na2SO4により)、乾燥状態まで濃縮して、粗生成物XXXVaを得る。DMF(5mL)中の粗XXXVaの溶液に、NaN3(0.18g)を加える。反応混合物を100℃にて4時間攪拌し、溶媒を蒸発除去する。残留物をCH2Cl2に溶解させ、冷塩水、1Mの冷塩酸、冷炭酸水素ナトリウム溶液、及び冷水で洗浄する。有機層を乾燥させ(Na2SO4により)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカ)により精製して、XXXVb(0.14g)を得る。
【0145】
中間体XXXVcの合成:DMF(3mL)中のXXXVb(0.08g)の溶液にDTT(0.04g)及びDBU(0.02mL)を加え、室温にて1時間攪拌する。溶媒を蒸発除去し、残留物をEtOAcに溶解させ、H2Oで洗浄して、有機層を乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカ)により精製し、中間体XXXVc(0.061g)を得る。
【0146】
中間体XXXVdの合成:DMF(3mL)中のモノ保護PEGジカルボン酸(0.6g)の溶液に、ジイソプロピルエチルアミン(0.24mL)及びHATU(0.513g)を攪拌しながら加える。上記の溶液にDMF(3mL)中の中間体XXXVc(0.185g)の溶液を加える。反応混合物を室温にて1時間攪拌する。溶媒を蒸発除去し、残留物をEtOAcに溶解させる。EtOAc層を水で洗浄し、カラムクロマトグラフィーにより精製して、中間体XXXVdを得る。
【0147】
XXXVIの合成:氷酢酸(3mL)中のXXXVd(0.185g)の溶液にZn粉末(0.1g)を加え、反応物を40℃にて30分攪拌する。反応混合物をセライトベッドを通して濾過し、Zn固形物をメタノールによって洗浄する。濾液を乾固状態まで濃縮し、セップパックC18カラムにより精製し、XXXVI(0.050g)を得る。
【0148】
中間体XXXIVaの合成:THF(25mL)中のTiCl4−テトラヒドロフラン錯体(0.705g)及びZn粉末(0.28g)の懸濁液を、不活性雰囲気下で攪拌しながら75℃にて2時間還流する。この混合物に実施例3のBACA(0.3g)及びN−フルオレニルメトキシカルボニル−3−アミノプロパノール(0.312g、参考文献Casimiro−Garcia, et al., Bioorg. Med. Chem. 1979 (2001) 2827に記載の通りに調製)の溶液を加える。反応混合物を不活性雰囲気下で75℃にて2.5時間攪拌する(McMurrayカップリング)。反応物を室温に冷却し、H2O(30mL)を加え、セライトベッドを通して濾過し、濾液をEtOAcで3回洗浄する(各30mL)。有機層を収集し、乾燥させ(Na2SO4によって)、濾過して、乾固状態まで濃縮する。残留物をカラムクロマトグラフィーにより精製し、XXXIVaを得る。
【0149】
中間体XXXIVbの合成:無水THF(21mL)中のXXXIVa(0.444g)の溶液に、ピペリジン(6.25mL)を加え、反応混合物を室温にて3時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカ)により精製し、中間体XXXIVb(0.26g)を得る。
【0150】
中間体XXXIVcの固相合成:PS−カルボジイミド樹脂(0.20Og)、HOBt(0.03Og)、及びモノ保護PEG−COOH(0.08Og)のCH2Cl2(3mL)中での混合物を、シリンジ反応器中にて室温にて5分間攪拌する。その混合物に、CH2Cl2(3mL)中の中間体XXXIVb(0.08Og)の溶液を加え、反応混合物を室温にて3時間攪拌する。MP炭酸樹脂(0.216g)を加え、室温にて2時間攪拌する。樹脂を濾別した後、その樹脂をCH2Cl2で5回洗浄する。濾液を収集し、乾固状態まで濃縮して、中間体XXXIVcを得る。
【0151】
中間体XXXVIaの合成:中間体XXXVIの合成について説明したのと全く同じ方法で、XXXVIc(0.12g)をZn/酢酸で処理して、中間体XXXVIa(0.104g)を得る。
【0152】
(実施例9)
ペグ化BACA(図7C)の合成
中間体XXXIVaの合成:THF(25mL)中のTiCl4−テトラヒドロフラン錯体(0.705g)及びZn−粉末(0.28g)の懸濁液を、不活性雰囲気下で攪拌しながら75℃にて2時間還流する。この混合物に実施例3のBACA(0.3g)及びN−フルオレニルメトキシカルボニル3アミノプロパノール(0.312g、参考文献Casimiro−Garcia, et al., Bioorg. Med. hem. 1979 (2001) 2827に記載の通り調製)の溶液を加える。反応混合物を不活性雰囲気下で75℃にて2.5時間攪拌する(McMurrayカップリング)。室温まで反応物を冷却し、H2O(30mL)を加え、セライトベッドを通して濾過し、濾液をEtOAcで3回洗浄する(各30mL)。有機層を収集し、乾燥させて(Na2SO4により)、濾過して乾固状態まで濃縮する。残留物をカラムクロマトグラフィーにより精製し、XXXIVaを得る。
【0153】
中間体XXXIVbの合成:無水THF(21mL)中のXXXIVa(0.444g)の溶液に、ピペリジン(6.25mL)を加え、反応混合物を室温にて3時間攪拌する。溶媒を蒸発除去し、残留物をカラムクロマトグラフィー(シリカ)により精製し、中間体XXXIVb(0.26g)を得る。
【0154】
中間体XXXIVcの固相合成:PSカルボジイミド樹脂(0.200g)、HOBt(0.030g)及びモノ保護PEG−COOH(0.080g)のCH2Cl2(3mL)中での混合物を、シリンジ反応器中にて室温にて5分攪拌する。上記の混合物に、CH2Cl2(3mL)中の中間体XXXIVb(0.080g)の溶液を加え、反応混合物を室温にて3時間攪拌する。MP−炭酸樹脂(0.216g)を加え、室温にて2時間攪拌する。樹脂を濾別した後、その樹脂をCH2Cl2で5回洗浄する。濾液を収集し、乾固状態まで濃縮して、中間体XXXIVcを得る。
【0155】
XXXVIaの合成:中間体XXXVIの合成について説明したのと同じ方法で、XXXIVc(0.12g)をZn/酢酸で処理して、中間体XXXVIa(0.104g)を得る。
【0156】
(実施例10)
糖模倣BASA(図8A)の合成
実施例7(0.015g)のXXXIIのDMF(0.1mL)中の溶液に、ヒューニッヒ塩基(0.015mL)を加えた後、HATU(0.007g)を加える。反応混合物を室温にて10分攪拌する。実施例4のEDA−XIX(DMFmL中に0.010g)の溶液を加え、反応混合物を室温にて8時間攪拌する。溶媒を蒸発除去し、残留物をセファデックスG−25クロマトグラフィーにより精製し、図8Aの糖模倣BASA(0.008g)を得る。
【0157】
(実施例11)
糖模倣BACA(図8B)の合成
EDA−XIX及びXXXVI間のカップリング:実施例8のXXXVIのDMF中の溶液にジイソプロピルエチルアミンを加えた後、HATUを加える。溶液を室温にて3分間攪拌する。その後、円錐形のバイアル瓶中で攪拌しながら上記の溶液をEDA−XIXに加える。反応混合物を室温にて2時間攪拌する。溶媒を蒸発除去し、粗中間体XXXVIbを得る。これは精製せずに次の段階で使用する。
【0158】
粗XXXVIbをNaOMe−メタノール−H2Oで2時間処理した後、ゲル濾過により精製して、糖模倣BACAを得る。
【0159】
EDA−XIX及び実施例9のXXXVIa間のカップリング:このカップリング反応は、XXXVIbの合成について説明したのと全く同じ方法で実施し、粗生成物XXXVIcを得る。
【0160】
XXXVIcを、記載と同じ方法によりNaOMe−メタノール−H2Oで処理し、糖模倣BACAを得る。
【0161】
(実施例12)
糖模倣BASA(図8C)の合成
この合成は、実施例6のXXXIII及び実施例4のEDA−XIXを使用して、実施例10で説明したのと同じ方法で実施し、図8Cの糖模倣BASAを得る。或いは、EDA−XIXとの反応に、XXXIIIを実施例18のXLVに置き換えることも可能である。
【0162】
(実施例13)
糖模倣BASA(図9A)の合成
この合成は、実施例7のXXXII及び実施例5のEDA−XXVIIIを使用して、実施例10で説明したのと同じ方法で実施し、図9Aの糖模倣BASAを得る。
【0163】
(実施例14)
糖模倣BACA(図9B)の合成
EDA−XXVIII及びXXXVI間のカップリング:このカップリング反応は、XXXVIbの合成について説明したのと全く同じ方法で実施し、粗生成物XXXVIdを得る。
【0164】
記載と全く同じ方法でXXXVIdをNaOMe−メタノール−H2Oで処理し、糖模倣BACAを得る。
【0165】
EDA−XXVIII及びXXXVIa間のカップリング:このカップリング反応は、XXXVIbの合成について説明したのと全く同じ方法で実施し、粗生成物XXXVIeを得る。
【0166】
記載と全く同じ方法によりXXXVIeをNaOMe−メタノール−H2Oで処理し、糖模倣BACAを得る。
【0167】
(実施例15)
糖模倣BASA(図9C)の合成
この合成は、実施例6のXXXIII及び実施例5のEDA−XXVIIIを使用して、実施例10で説明したのと同じ方法で実施し、図9Cの糖模倣BASAを得る。或いは、EDA−XXVIIIとの反応に、XXXIIIを実施例18のXLVに置き換えることも可能である。
【0168】
(実施例16)
糖模倣物(図12)の合成
中間体XXXVIIの合成:中間体XXVの合成について実施例5で説明したのと同じ方法により、実施例5の中間体XXIV(0.1g)のピリジン溶液を、ピリジン中の塩化ニコチニル(0.08mL)及びジメチルアミノピリジン(0.04g)で処理して、中間体XXXVII(0.075g)を得る。
【0169】
中間体XXXVIIIの合成:化合物XVIIの合成について実施例4で説明したのと同じ方法により、中間体XXXVII(0.07g)をトリフェニルホスフィン及びオロチン酸塩化物で処理して、XXXVIII(0.048g)を得る。
【0170】
中間体XXXIXの合成:記載の通り、Pd/Cによる中間体XXXVIII(0.04g)の水素化により、化合物XXXIX(0.02g)を得る。
【0171】
化合物XLの合成:中間体XXXIX(0.015g)を60℃にて10分間0.5NのNaOHで処理し、セファデックスG−25ゲル濾過により精製して、化合物XL(0.010g)を得る。
【0172】
(実施例17)
糖模倣物(図13)の合成
XLIの合成:t−BuOH−水(4mL、1:1)中の化合物XVI(0.1g)の懸濁液に、1−エチニル−3−フルオロベンゼン(0.9g)、1%のCuSO4(0.1mL)、及びL−アスコルビン酸Na(4mg)を加える。混合物を20時間攪拌しながら加熱する(70℃)。溶媒を蒸発除去し、残留物をジクロロメタンに溶解させる。有機層を水で洗浄し、乾燥させ(無水硫酸ナトリウムにより)、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XLI(0.08g)を得る。
【0173】
XLIIの合成:化合物XLI(0.25g)をジオキサン−水(4:1、7.5mL)に溶解させる。10%のPd/C(0.25g)を加え、続いて酢酸(7滴)を加える。混合物を40psiにて15時間水素化する。反応混合物をセライトベッドを通して濾過し、乾固状態まで濃縮して、化合物XLII(0.2g)を得る。
【0174】
XLIIIの合成:メタノール(5mL)中の化合物XLII(0.2g)の溶液に、メタノール(0.05mL)中のNaOMeの溶液を加え、反応混合物を室温にて2時間攪拌する。反応混合物を、酢酸を数滴加えて中和し、乾固状態まで濃縮する。残留物をカラムクロマトグラフィー(シリカゲル)により精製し、化合物XLIII(0.15g)を得る。
【0175】
XLIVの合成:化合物XLIII(0.15g)をエチレンジアミン(7mL)に溶解させ、反応混合物を70℃にて9時間攪拌する。溶媒を蒸発除去し、残留物を、最初にカラムクロマトグラフィー(シリカゲル)により精製した後、逆相C18により精製し、化合物XLIV(0.11g)を得る。
【0176】
糖模倣BASA(化合物XLV)の合成:この合成は、実施例6のXXXIIIとXLIVを使用して、実施例10で説明したのと同じ方法で実施し、化合物XLVを得る。
【0177】
(実施例18)
糖模倣BASA(図14)の合成
化合物XLVの合成:DMF(1mL)中の3,6−ジオキサオクタン二酸(PEG、200mg、市販)の溶液にヒューニッヒ塩基(0.4mL)を加え、5分後にHATU(0.35g)を加える。溶液を室温にて10分間攪拌した後、8−アミノナフタレン−1,3,6トリスルホン酸(50mg、市販)のDMF溶液を加える。反応混合物を室温にて4時間攪拌して、溶媒を蒸発除去する。残留物をHPLC(逆相C18カラム)により精製し、XLV(25mg)を得る。
【0178】
化合物XLVIの合成:この合成は、実施例4のXLV及びEDA−XIXを使用して、実施例10で説明したのと同じ方法で実施し、化合物XLVI(4mg)を得る。
【0179】
(実施例19)
糖模倣BASA(図15)の合成
XLVIIの合成:この合成は、化合物XXIVを出発物質として、XLIの合成について説明したのと同じ方法で実施し、化合物XLVIを得る。
【0180】
XLVIIIの合成:この合成は、化合物XLVIIを出発物質として、化合物XLIII(XLIから)で説明したのと同じ方法で実施し、化合物XLVIIIを得る。
【0181】
XLIXの合成:この合成は、化合物XLVIIIを出発物質として、XLIVの合成について説明したのと同じ方法で実施し、化合物XLIXを得る。
【0182】
糖模倣BASA(化合物L)の合成:この合成は、実施例6のXXXIII及びXLIXを使用して、実施例10で説明したのと同じ方法で実施し、化合物Lを得る。或いは、XLIXとの反応のために、XXXIIIを実施例18のXLVに置き換えることも可能である。
【0183】
(実施例20)
糖模倣物(図16)の合成
化合物Iの合成:参考文献[J. Org. Chem. 54, 3738−3740 (1989);Liebigs Annalen der Chemie 575,
1 (1952)]に記載の通り合成する。
【0184】
中間体IIの合成:I(2.8g、15.04mmol)、ピリジン(4.8mL、60.15mmol)、塩化ベンゾイル(3.5mL、30.07mmol)、及び触媒量のジメチルアミノピリジンを、ジクロロメタン(6mL)中にて室温にて攪拌する。2時間後、TLC対照が反応の完了を示す。次に、反応混合物を酢酸エチル(200mL)で希釈し、水、1NのHCI水溶液(氷冷)、飽和炭酸水素ナトリウム水溶液、及び塩水で洗浄する(各50mL)。水層を、酢酸エチルで2回洗浄し(150mLで2回)、収集してNa2SO4で乾燥させる。溶媒を濾過し蒸発させた後、残留物をシリカゲルクロマトグラフィー(PE/EtOAc=4:1)により精製し、化合物II(3.78g、86%)を得る。
【0185】
中間体IIIの合成:II(3.78g、13.02mmol)及びNaBH4を、メタノール(35mL)中にて0℃にて攪拌する。30分後、反応混合物を水(15mL)で急冷し、1NのAcOH水溶液で中和する。再び水(10mL)を加え、混合物をジクロロメタンで3回抽出する(150mLで3回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾燥させる。残留物をシリカゲルクロマトグラフィー(PE/EtOAc=3:2)により処理し、化合物III(3.7g、97%)を得る。[α]D+79.78°(c=0.940、CH2Cl2)。
【0186】
中間体IVの合成:CHCl3(50mL、標準的アロックスで濾過)中のIII(3.4mg、11.64mmol)の溶液に、1−クロローN,N,2−トリメチルプロペニルアミン(4.94mL、34.93mmol)をアルゴン雰囲気下で室温にてシリンジによって加える。TLC対照が反応の完了を示すまで、反応混合物を還流しながら攪拌する(30分)。反応混合物を室温に冷却した後、トリエチルアミン(6mL)によって急冷して、蒸発乾固する(浴温30℃)。残留物をシリカゲルクロマトグラフィー(PE/EtOAc=9:1)により精製し、化合物IV(3.3g、92%)を得る。
【0187】
中間体Vの合成:乾燥トルエン中(40mL)におけるIV(3.25g、10.47mmol)の溶液に、新たに蒸留されたBu3SnH(30.58mL、115.14mmol)及びAIBN(1.7g、10.47mmol)をアルゴン雰囲気下で加える。反応物を還流する。75分後、TLC対照が反応の完了を示したら、反応混合物を室温に冷却した後、アセトニトリル(50mL)で希釈する。溶液をヘキサン(50mL)によって洗浄し、そのヘキサン層をアセトニトリル(50mL)によって再び抽出する。収集したアセトニトリル層を蒸発させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=14:1)で処理し、化合物V(2.66g、92%)を得る。[α]D−117.79°(c=1.810、CH2Cl2)。
【0188】
中間体VIの合成:酢酸80%水溶液中のV(2.63g、9.54mmol)の溶液を80℃にて攪拌する。TLC対照が反応の完了を示したら(30分)、反応混合物を室温に冷却する。NaOH水溶液で中和した後、混合物をジクロロメタンで3回抽出する(200mLで3回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾固する。残留物をシリカゲルクロマトグラフィー(PE/EtOAc=3:2)により精製して、化合物Vl(2.01g、89%)を得る。[α]D−54.86°(c=1.420、CH2Cl2)。
【0189】
中間体VIIの合成:乾燥ジクロロメタン(40mL)中の、Vl(1.98g、8.39mmol)、新たに再結晶したトルエン−4−スルホニルクロリド(1.9g、10.07mmol)、Bu2SnO(2.09g、8.39mmol)、及びトリエチルアミン(1.8mL、16.78mmol)を、アルゴン雰囲気下で室温にて攪拌する。20時間後、TLC対照が反応の完了を示す。その後、反応混合物をメタノール(15mL)で急冷し、蒸発させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=8:1)で処理し、化合物VII(2.65g、85%)を得る。[α]D−68.08°(c=0.448、CH2Cl2)
中間体VIIIの合成:DMF(30mL)中のVII(640mg、1.71mmol)及びNaN3(555mg、8.55mmol)の混合物を、アルゴン雰囲気下で80℃にて攪拌する。TLCにより反応の完了を示したら(1時間後)、反応混合物を室温に冷却し、ジクロロメタン(50mL)で希釈して、水(50mL)で洗浄する。次に、水層をジクロロメタンで2回抽出し(50mLで2回)、収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=6:1)により精製して、化合物VIII(391mg、87%)を得る。[α]D−69.39°(c=2.330、CH2Cl2)。
【0190】
中間体IXの合成:乾燥ジクロロメタン(1mL)中のエチル2,3,4−トリ−O−ベンジル−α−L−フコチオピラノシド(223mg、0.47mmol)の攪拌溶液に、アルゴン雰囲気下で0℃にて臭素(48μ、0.54mmol)を加える。5分後、冷却浴を取り外し、反応混合物を更に室温にて40分攪拌する。過剰の臭素を除去するために、反応混合物にシクロヘキセン(50μL)を加えると、反応混合物の色が消える。次に、この反応混合物を、VIII(61mg、0.23mmol)、(Et)4NBr(98mg、0.47mmol)、及び粉末状の4Åモレキュラーシーブ(100mg)をジクロロメタン(1.6mL)及びDMF(1mL)中にてあらかじめ攪拌しておいた溶液(室温にて1時間)に加える。18時間後、反応をピリジン(2mL)によって急冷し、更に15分間攪拌し、EtOAc(50mL)で希釈して、飽和KHCO3水溶液、水、及び塩水で洗浄する(各50mL)。水層をEtOAcで2回抽出する(50mLで2回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾固させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、IX(116mg、73%)を得る。[α]D−95.96°(c=1.040、CH2Cl2)。
【0191】
中間体Xの合成:メタノール(10mL)中のIX(530mg、0.78mmol)、及び新たに調製した触媒量のNaOMeを、アルゴン雰囲気下で室温にて攪拌する。反応混合物を、粉末状のアンバーリスト−15で中和し、セライトで濾過する。濾液を蒸発乾固させ、シリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、化合物X(380mg、85%)を得る。[α]D−76.425°(c=4.00、CH2Cl2)。
【0192】
中間体XIの合成:ジクロロメタン(3mL)中の、X(184mg、0.32mmol)、ガラクトース構築ブロック[375mg、0.4811mmol、HeIv. Chim. Acta 83:2893−2907 (2000)に既述の通り合成]、及び活性化粉末状のモレキュラーシーブ4Å(200mg)の混合物を、アルゴン雰囲気下で室温にて4時間攪拌する。その後、DMTST[B](165mg、0.64mmol)を4等分して1.5時間かけて、ジクロロメタン(3mL)中にて活性化粉末状のモレキュラーシーブ4Å(100mg)を予め攪拌しておいた混合物(室温にて4時間)に加える。92時間後、TLC対照が反応の完了を示したら、反応混合物をセライトで濾過し、濾液をジクロロメタン(50mL)で希釈する。有機層を飽和炭酸水素ナトリウム水溶液及び塩水(各20mL)で洗浄し、水層をジクロロメタンで2回抽出する(50mLで2回)。収集した有機層をNa2SO4で乾燥させ、濾過して、蒸発乾固させる。残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=9:1)により精製し、化合物XI(310mg、75%)を得る。[α]D−36.98°(c=2.32、CH2Cl2)。
基本手順A
中間体XIIの合成:乾燥ジクロロメタン(3mL)中のXl(100mg、0.077mol)の攪拌溶液に、塩化酢酸(27μL、0.387mmol)を加える。5分後、PPh3(101mg、0.387mmol)を加え、溶液を室温にて攪拌する。22時間後、TLC対照が反応の完了を示したら、溶媒を除去し、残留物をシリカゲルクロマトグラフィー(トルエン/EtOAc=2:1)で処理して、化合物XII(43mg、43%)を得る。[α]D−55.33°(c=2.185、CH2Cl2)。
【0193】
中間体XIIIの合成:手順Aに従って:Xl(80mg、0.062mol)及びオロチン酸塩化物(59mg、0.310mmol)、及びPPh3(81mg、0.310mmol)を使用して、4時間後、(ジクロロメタン/メタノール25:1)で処理して、化合物XIII(35mg、40%)を得る。[α]D−26.08°(c=1.48、CH2Cl2)。
【0194】
中間体XIVの合成:手順Aに従って:Xl(70mg、0.054mmol)及びビフェニル−4−カルボン酸塩化物(58mg、0.271mmol)、及びPPh3(71mg、0.271mmol)を使用して、4時間後、(トルエン/EtOAc=6:1)で処理して、XIV(22mg、28%)を得る。[α]D−32.11°(c=1.065、CH2Cl2)
中間体XVの合成:手順Aに従って:Xl(45mg、0.035mmol)及びビフェニル−2−カルボン酸塩化物(125mg0.577mmol)、及びPPh3(151mg、0.577mmol)を使用して、5時間後、(トルエン/EtOAc6:1)で処理して、化合物XV(22mg、44%)を得る。[α]D−19.54°(c=1.10、CH2Cl2)。
基本手順B
中間体XVIの合成:CH2Cl2(2mL)及び水(100μL)中のXl(120mg、0.093mmol)及びPPh3(30mg、0.116mmol)の混合物を、室温にて44時間攪拌する。次に、溶媒を除去し、残留物をCH2Cl2(2mL)に溶解させ、DIC(11.8mg、0.094mmol)及びバニリン酸(24mg、0.139mmol)を加える。混合物を室温にて更に62時間攪拌し、溶媒を除去して、粗生成物をシリカゲルクロマトグラフィー(トルエン/EtOAc=2.5:1)により精製し、化合物XVI(62mg、47%)を得る。[α]D−29.58°(c=2.86、CH2Cl2)。
基本手順C
中間体XVIIの合成:XII(40mg、0.0306mmol)、Pd(OH)2(30mg)、ジオキサン(2mL)、及び水(0.4mL)の混合物を、室温にて5barにてParrシェーカーで水素化する。20時間後、TLC対照が反応の完了を示す。反応混合物をセライトで濾過し、蒸発乾固する。クロマトグラフィー(CH2Cl2/メタノール=9:1)によって粗生成物を精製し、XVII(20mg、69%)を得る。[α]D−43.50°(c=1.00、MeOH)。
【0195】
中間体XVIIIの合成:手順Cに従って:XIV(26mg、0.018mmol)、Pd(OH)2(11mg)、ジオキサン(1.2mL)、及び水(0.25mL)を使用して、50時間後、(CH2Cl2/メタノール=2:1)で処理して、XVIII(10mg、50%)を得る。
【0196】
中間体XIXの合成:手順Cに従って:XV(22mg、0.015mmol)、Pd(OH)2(20mg)、ジオキサン(1mL)、及び水(0.25mL)を使用して、22時間後、(CH2Cl2/メタノール=15:1)で処理して、化合物XIX(13mg、82%)得る。[α]D−9.50°(c=1.21、MeOH)。
【0197】
中間体XXIの合成:手順Cに従って:XIII(21mg、0.015mmol)、Pd(OH)2(20mg)、ジオキサン(1mL)、及び水(0.25mL)を使用して、22時間後、(CH2Cl2/メタノール=15:1)で処理して、化合物XXI(13mg、86%)を得る。
【0198】
中間体XXの合成:XVI(20mg、0.0141mmol)、10%Pd/C(20mg)、メタノール(2mL)、及び酢酸(50μL)の混合物を、水素雰囲気下で攪拌する。TLC対照が反応の完了を示したら(22時間後)、混合物をセライトで濾過し、蒸発乾固する。残留物をシリカゲルクロマトグラフィー(CH2Cl2/メタノール=2:1)で処理し、XX(15mg、定量)を得る。[α]D−29.32°(c=1.105、MeOH)。
基本手順D
生成物XXIIの合成:乾燥メタノール(1mL)中のXVII(27mg、0.0278mmol)の溶液、及び新たに調製した触媒量のNaOMeを、アルゴン雰囲気下で室温にて攪拌する。1時間後、TLCにより反応の完了を確認する。反応混合物を粉末状のAmberlyst−15で中和し、セライトで濾過して、濾液を蒸発乾固する。残留物をメタノール(20mL)に溶解させて、イオン交換ダウエックスNa(登録商標)で濾過する。濾液を蒸発乾固して、粗生成物をクロマトグラフィー(CH2Cl2/メタノール/水=10:4:0.8)で処理し、XXII(12mg、56%)を得る。[α]D−97.49°(c=0.546、MeOH)。
【0199】
生成物XXIIIの合成:手順Dに従って:XVIII(9.0mg、0.0083mmol)を使用して、2時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理して、化合物XXIII(7.5mg、定量)を得る。[α]D−79.50°(c=0.400、MeOH)。
【0200】
生成物XXIVの合成:手順Dに従って:XIX(23mg、0.0212mmol)及びMeOH(2mL)を使用して、2.5時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理して、化合物XXIV(7.0mg、39%)を得る。[α]D−54.54°(c=0.586、MeOH)。
【0201】
生成物XXVの合成:手順Dに従って:XX(19mg、0.0180mmol)及びMeOH(2mL)を使用して、4時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理し、化合物XXV(10.5mg、70%)を得る。[α]D−69.96°(c=0.793、MeOH)。
【0202】
生成物XXVIの合成:手順Dに従って:XXI(19mg、0.0180mmol)及びメタノール(2mL)を使用して、4時間後、(CH2Cl2/メタノール/水=10:3:0.5)で処理して、化合物XXVI(10mg、65%)を得る。
【0203】
(実施例21)
E−セレクチン拮抗薬活性の検定(図17A)
マイクロタイタープレート(プレート1)のウェルをE−セレクチン/hIgキメラ(GlycoTech Corp.[米国メリーランド州ロックビル])により、37℃にて2時間インキュベートすることによってコーティングする。50mMのTris HCl、150mMのNaCl、2mMのCaCl2、pH7.4(Tris−Ca)でプレートを5回洗浄した後、Tris−Ca/Stabilcoat(SurModics[米国ミネソタ州エデン・プレーリー])(1:1,v/v)中の1%BSA(100μL)を、各ウェルに添加して、非特異的結合をブロッキングする。試験化合物を、第2の低結合丸底プレート(プレート2)において、Tris−Ca(60μL/ウェル)中、順次希釈する。ストレプトアビジン−HRP(Sigma[米国ミズーリ州セントルイス])をSLea−PAA−ビオチン(GlycoTech Corp.[米国メリーランド州ロックビル])と混合させて予め形成しておいた結合体を、プレート2の各ウェルに添加する(60μL/ウェルの1μg/mL)。プレート1を、Tris−Caで数回洗浄し、100μL/ウェルをプレート2からプレート1に移す。正確に2時間室温にてインキュベートした後、プレートを洗浄し、100μL/ウェルのTMB試薬(KPL labs[米国メリーランド州ゲイザースバーグ])を各ウェルに加える。3分間室温にてインキュベートした後、100μL/ウェルの1MのH3PO4を加えることによってこの反応を停止させ、450nmでの吸光度をマイクロタイタープレートリーダーによって測定する。
【0204】
(実施例22)
P−セレクチン拮抗薬活性の検定(図17B)
ネオ糖タンパク質であるシアリルLea−HSA(Isosep AB[スウェーデン])を、マイクロタイタープレート(プレート1)のウェル上にコーティングした後、そのウェルをダルベッコのリン酸緩衝化生理食塩水(DPBS)中で希釈された2%のウシ血清アルブミン(BSA)を添加することによりブロックする。第2のマイクロタイタープレート(プレート2)において、試験拮抗薬をDPBS中の1%BSAにおいて順次希釈する。ブロック後、プレート1を洗浄し、プレート2の内容物をプレート1に移す。P−セレクチン/hIg組換えキメラタンパク質(GlycoTech Corp.[米国メリーランド州ロックビル])をプレート1における各ウェルに更に加え、この結合プロセスを室温にて2時間インキュベートする。次いで、プレート1をDPBSで洗浄し、ペルオキシダーゼ標識ヤギ抗ヒトIg(γ)(KPL Labs[米国メリーランド州ゲイザースバーグ])の1μg/mLを各ウェルに加える。室温にて1時間インキュベーションした後、そのプレートをDBPSで洗浄した後、TMB基質(KPL Labs)を各ウェルに加える。5分後、1MのH3PO4を添加して反応を停止させる。その後、450nmでの吸光度をマイクロタイタープレートリーダーを使用して測定する。
【0205】
(実施例23)
抗炎症性マウスモデル。in vivoでの空気嚢に対するIL−1β−誘発性好中球遊走についての試験化合物の効果(図19)
動物
雄の非近交系スイスアルビノマウス(15〜18g体重)を、Bantin and Kingmanから購入し(T.O. stain;ハル、ハンバーサイド)、標準の固形飼料を与え、水道水を自由に摂取できるようにして、12時間の明/暗サイクルで飼育する。全ての動物は、実験前の7日間飼育し、実験日(6日目;以下を参照)には体重が約25gに達する。
実験設計
0日目及び3日目に空気注入(2.5mL、皮下注射)により、マウスの背部に空気嚢を形成する(Perretti & Flower, 1993)。カルボキシメチルセルロース(CMC)の均質な懸濁液を、PBSにおいて0.5%w/vで調製し、その懸濁液にマウス組換え型IL−1βを20ng/mLの濃度で加える。
【0206】
IL−1β投与の直前の0時間の時点で試験化合物を投与する。又、基底の陰性対照を提供するために、CMCだけ(IL−1βなし)を投与した比較用のマウスの群にも投与する。
【0207】
全ての場合において、IL−1β後4時間で3mMのEDTAを含むPBS(2mL)によって空気嚢を洗浄し、肺胞洗浄液の定比容量(100μL)を採取し、チュルク溶液(3%酢酸中の0.01%クリスタルバイオレット)で1:10に希釈して、遊走した白血球(>=90% 多形核白血球、PMN)の数を測定する。次に、サンプルをボルテックスで攪拌し、10μLの染色した細胞溶液をノイバウエル血液波長計にセットして光学顕微鏡(Olympus B061)を使用して好中球数を計数する。
化合物投与
実験当日、試験化合物の新鮮な溶液を、1mMのCaCl2及びMgCl2を補ったPBSのダルベッコ緩衝剤において調製する。マウスのP−セレクチン又はE−セレクチンに対するモノクローナル抗体(mAb)を、BD Pharmingenから購入し、一方、抗L−セレクチンmAbはSerotecから購入する。
【0208】
ラット抗マウスL−セレクチン(クローンMEL−14):1mg
ラット抗マウスP−セレクチン(クローンRB40.34):0.5mg/mL
ラット抗マウスE−セレクチン(クローン10E9.6):0.5mg/mL
図19
6日後のマウス空気嚢は、0時間の時点でIL−1β(10ng)によって炎症を起こしている。実施例17の糖模倣BASA(図13)及び実施例12の糖模倣BASA(図8C)を、0時間及び4時間の時点で静脈内に投与する。抗セレクチンmAbの混合物を0時間の時点で静脈内に投与する。
【0209】
8時間の時点で空気嚢を洗浄し、染色して光学顕微鏡検査により遊走したPMNの数を測定する。
【0210】
図19において、n数は、群A、B、C及びD当たり、それぞれマウス9、8、7及び8匹である。
【0211】
本明細書で引用し及び/又は出願データシートに列挙した上記の米国特許、米国特許出願公開、米国特許出願、外国特許、外国特許出願及び特許以外の刊行物は全て、全体が参考として本明細書で援用される。
【0212】
以上において本発明の特定の実施形態を例示のために本明細書で説明してきたが、本発明の趣旨及び適用範囲から逸脱することなく、種々の改変が行われる場合があることが、以上のことから理解されるであろう。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】

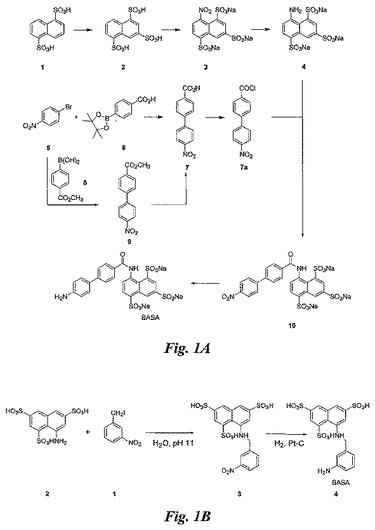
【図2】
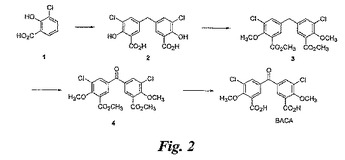
【図3】
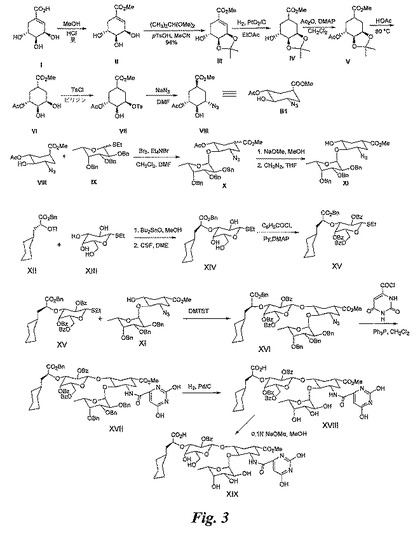
【図4】
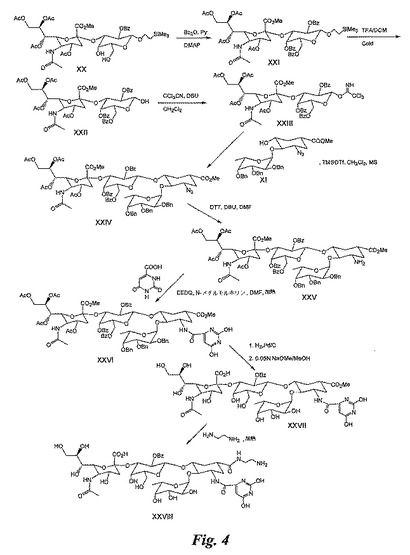
【図5】
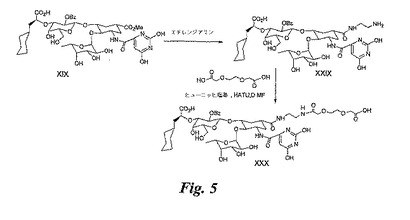
【図6】
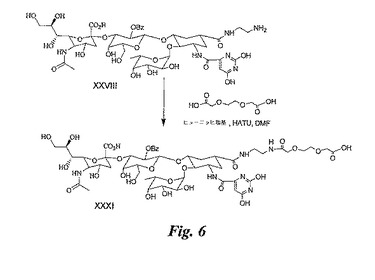
【図7A】
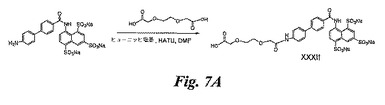
【図7B】
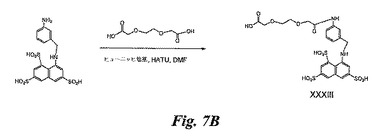
【図7C】
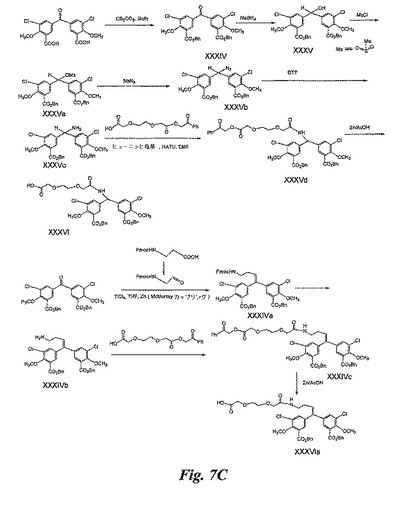
【図8A】
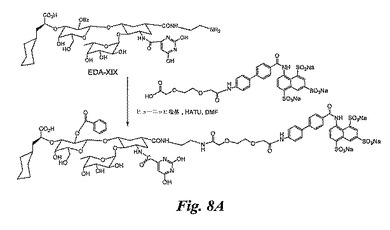
【図8B】
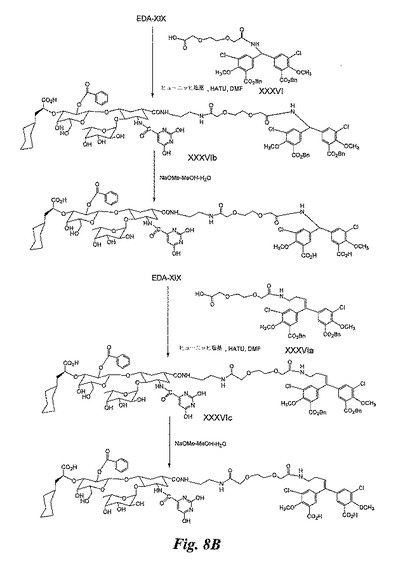
【図8C】
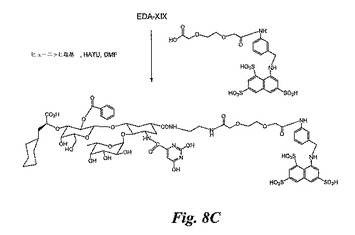
【図9A】
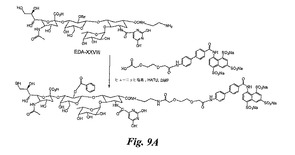
【図9B】
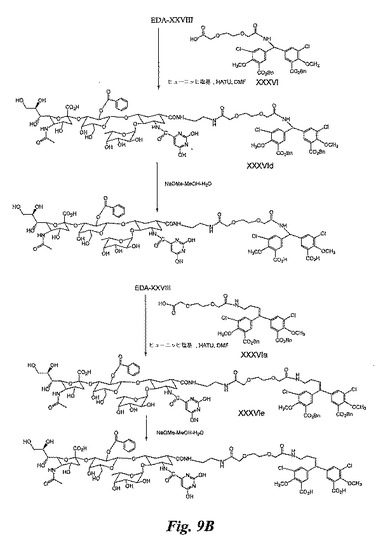
【図9C】
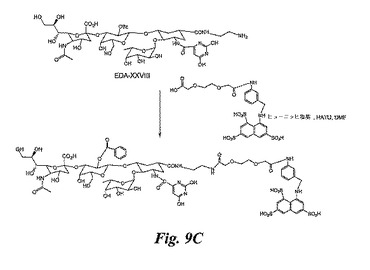
【図10】
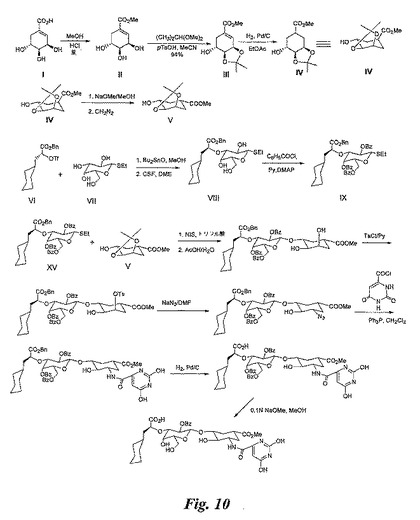
【図11】
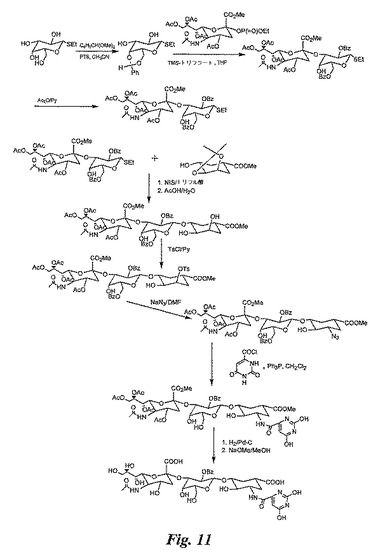
【図12】
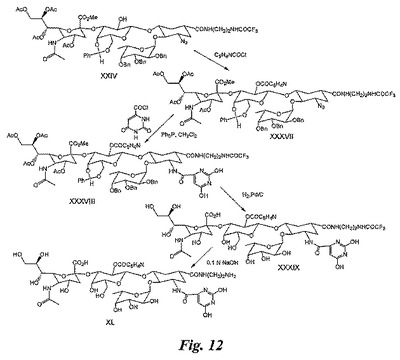
【図13】
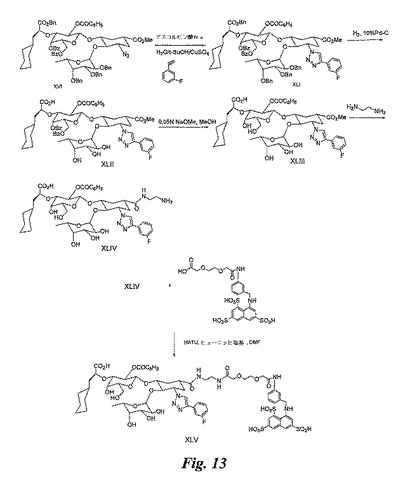
【図14】
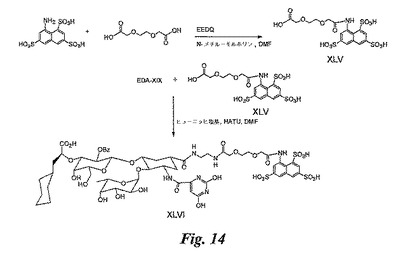
【図15】
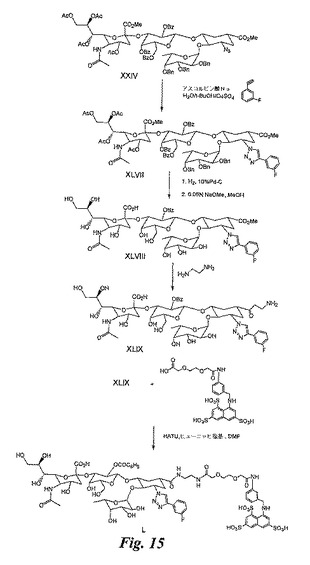
【図16】
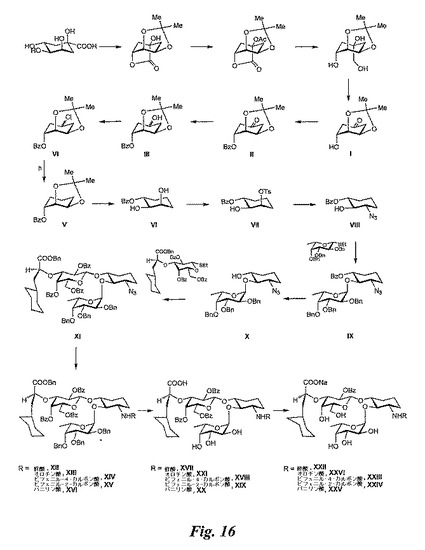
【図17】

【図18】
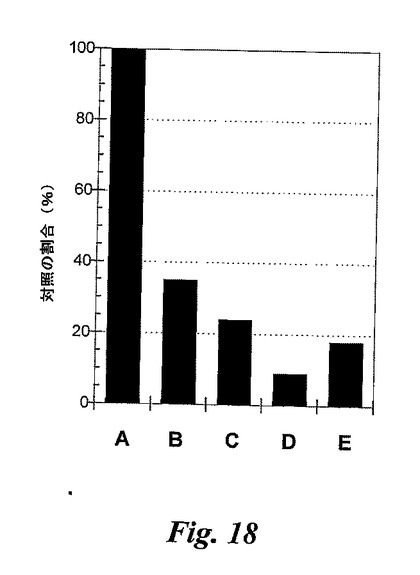
【図19】
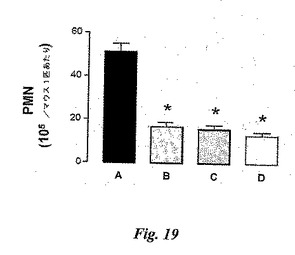
【図2】
【図3】
【図4】
【図5】
【図6】
【図7A】
【図7B】
【図7C】
【図8A】
【図8B】
【図8C】
【図9A】
【図9B】
【図9C】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2012−184268(P2012−184268A)
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−149401(P2012−149401)
【出願日】平成24年7月3日(2012.7.3)
【分割の表示】特願2008−529320(P2008−529320)の分割
【原出願日】平成18年9月1日(2006.9.1)
【出願人】(506167421)グリコミメティクス, インコーポレイテッド (7)
【Fターム(参考)】
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願番号】特願2012−149401(P2012−149401)
【出願日】平成24年7月3日(2012.7.3)
【分割の表示】特願2008−529320(P2008−529320)の分割
【原出願日】平成18年9月1日(2006.9.1)
【出願人】(506167421)グリコミメティクス, インコーポレイテッド (7)
【Fターム(参考)】
[ Back to top ]