
ヘモグロビン類の測定方法
【課題】陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定方法に関し、特に安定型ヘモグロビンA1cを高精度で測定することが可能なヘモグロビン類の測定方法を提供する。
【解決手段】陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する方法であって、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出するヘモグロビン類の測定方法。
【解決手段】陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する方法であって、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出するヘモグロビン類の測定方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定方法に関し、特に安定型ヘモグロビンA1cを高精度で測定することが可能なヘモグロビン類の測定方法に関する。
【背景技術】
【0002】
有機化学、生化学、医学等の分野では、試料中の成分の分離や分取等の操作を行う機器として液体クロマトグラフィーが汎用されている。液体クロマトグラフィーは、通常、図13に示すような構成となっており、移動相用タンク31a及び31bから送液された溶離液は、脱気装置(デガッサー)32により脱気された後、マニホールド(液切換装置)34を通り、送液ポンプ35により圧力が掛けられ、検体用タンク38から試料導入装置37を介して検体が導入された後、フィルター39を経由して分離カラム40に導入される。そして、この分離カラム40により試料中の各成分が分離される。分離された各成分は検出器41によって、例えば、吸光度を測定する等によって検出され、その結果がデータ処理装置42により処理されてクロマトグラムとして表される。なお、移動相の定流量、定圧を目的として必要に応じてダンパー36が設置されてもよい。
【0003】
このような液体クロマトグラフィーを用いた測定方法は、血液中の糖化ヘモグロビンの測定にも用いられており、特にヘモグロビンA1c(以下、HbA1cという)は、過去1〜2カ月間の血液中の平均的な糖濃度を反映しているため、糖尿病のスクリーニング検査や糖尿病患者の血糖管理状態を把握するための検査項目として広く利用されている。なお、HbA1cは、血液中のグルコースとヘモグロビンA(以下、HbAという)とが反応して生成された糖化ヘモグロビン(以下、GHbという)であり、可逆的に反応したものは、不安定型HbA1c(unstable HbA1c)と呼ばれ、不安定型HbA1cを経て不可逆的に反応したものは、安定型HbA1c(stable HbA1c)と呼ばれている。
【0004】
通常、ヘモグロビンは2種類のサブユニット2つずつから構成される4量体のタンパク質である。HbAのサブユニットはα鎖とβ鎖であり、このβ鎖のN末端アミノ酸にグルコースが結合したものがHbA1cである。このうち過去1〜2カ月間の平均的な血糖値を良く反映しているものは、安定型HbA1cであり、臨床検査分野では、安定型HbA1c値(%)を高精度に得ることができる測定法の開発が望まれている。なお、安定型HbA1cは、全ヘモグロビンピークの面積(主に、ヘモグロビンA0ピークの面積)に対する安定型HbA1cピークの面積の比率(%)を求めることにより測定できる。即ち、精度良く安定型HbA1c値を測定するためには、精度良く安定型HbA1cピークの面積と全ヘモグロビンピークの面積(主に、ヘモグロビンA0ピークの面積)を求める必要がある。
【0005】
従来、HbA1cの主な測定法としては、例えば、特許文献1に開示されているように、血液検体を溶血希釈して調製した試料中のヘモグロビン類を、陽イオン交換法により、ヘモグロビン成分毎に異なるプラス荷電状態の違いを利用して分離する液体クロマトグラフィーを用いた方法が行われてきた。
【0006】
通常、陽イオン交換液体クロマトグラフィーを用い、充分な時間を掛けて溶血試料中のヘモグロビン類を分離すると、ヘモグロビンA1a(以下、HbA1aという)及びヘモグロビンA1b(以下、HbA1bという)、ヘモグロビンF(以下、HbFという)、不安定型HbA1c、安定型HbA1c及びヘモグロビンA0(以下、HbA0という)の順に溶離されてくる。ここで、HbA1a、HbA1b及びHbA1cはHbAが糖化されたGHbであり、HbFはα鎖とγ鎖から成る胎児性ヘモグロビンであり、HbA0はHbAを主成分とする一群のヘモグロビン成分であって、HbA1cより強くカラムに保持されたものである。
【0007】
しかしながら、従来の陽イオン交換液体クロマトグラフィーを用いた方法では、安定型HbA1cから不安定型HbA1cを充分に分離することができないばかりでなく、アセチル化ヘモグロビン(以下、アセチル化Hbという)やカルバミル化ヘモグロビン(以下、カルバミル化Hbという)等の「修飾ヘモグロビン」が安定型HbA1cと重なって溶離してくるという問題があった。
即ち、陽イオン交換液体クロマトグラフィーによる安定型HbA1c値(%)の測定を目的として、血液検体のヘモグロビン類を測定する際、安定型HbA1cの測定値に影響を与えないように、安定型HbA1cと溶出挙動が近似している不安定型HbA1cやアセチル化Hb及びカルバミル化Hbピークを、安定型HbA1cピークから分離することが困難であった。
【0008】
これに対して、近年、カルバミル化Hb、アセチル化Hb等の修飾ヘモグロビンの分離性能を向上させ、安定型HbA1cを測定できる技術が開発されており、例えば、特許文献2には、陽イオン交換液体クロマトグラフィーによるヘモグロビン類の測定方法であって、カオトロピックイオンを含有し、かつ、pH4.0〜6.8で緩衝能を持つ無機酸、有機酸及び/又はこれらの塩を含む溶離液を用いることで安定型HbA1cを分離する方法が開示されている。
しかしながら、このような方法では、高度に修飾されたアセチル化Hb、カルバミル化Hbを充分に分離できないという欠点があった。
【0009】
一方で、カラムを経て分離されたヘモグロビン類のピークを検出するための測定器(図13の検出器41)についても、近年研究がなされており、例えば、特許文献3、4には、GHbを検出するための測定器として、波長415nmまたはその付近の青色単色光を発するレーザーを光源とした吸光度計が開示されている。
このような吸光度計では、GHbの最大吸収波長である415nm付近の波長を測定波長とすることで、理論上はGHbを精度よく測定することが可能となるが、実際の臨床検査においては、血液中に含まれるビリルビン類、乳ビ等の不純物(干渉物質)が測定結果に悪影響を及ぼすことによって、特に安定型HbA1cの測定精度が低いものとなっていた。また、このような干渉物質の血液中の濃度は、個人によって異なることから、干渉物質の存在下で正確に安定型HbA1c測定することは非常に困難であった。
【特許文献1】特公平8−7198号公報
【特許文献2】特開2000−111539号公報
【特許文献3】特開平8−159954号公報
【特許文献4】特開平11−142326号公報
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明は、上記現状に鑑み、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定方法であって、特に安定型HbA1cを高精度で測定することが可能なヘモグロビン類の測定方法を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明は、陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する方法であって、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出するヘモグロビン類の測定方法である。
以下に本発明を詳述する。
【0012】
本発明者らは、鋭意検討した結果、ヘモグロビン類の測定において、所定範囲の波長を有する2種類の光を用い、得られた吸光度の差からクロマトグラムを作成し、ピーク比率を算出することにより、干渉物質による測定値の影響を大幅に低減させ、高精度なヘモグロビン類の測定、特に安定型HbA1cの高度な測定が可能となることを見出し、本発明を完成させるに至った。
【0013】
本発明では、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出することにより、ヘモグロビン類の測定を行う。
なお、本明細書において、「ピーク比率」とは、ピーク面積比率、ピーク高さ比率等のことをいい、測定するヘモグロビン類の種類によって、適宜選択することができる。ピーク面積比率を算出する方法としては、例えば、安定型HbA1c値を測定する場合における、全ヘモグロビンピークの面積(主に、HbA0ピークの面積)に対する安定型HbA1cピークの面積の比率(%)を算出する方法等を用いることができる。
【0014】
本発明の方法を用いた場合に、測定精度が向上する理由は以下の通りであると考えられる。まず、ヘモグロビン類の吸収特性を図1に示す。図1において、縦軸は吸光度、横軸は波長を表す。図1に示すように、ヘモグロビン類は最大吸収波長が415nm付近であるため、検出光として波長が415±10nmの範囲にある光を用いれば、ヘモグロビン類は充分検出できるが、ビリルビン類、乳ビ等の不純物(即ち干渉物質)による干渉によって、精度が低下する。これに対して、本発明では、ヘモグロビン類を検出するための第一波長光(波長405〜425nm)に加えて、上記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出することにより、ヘモグロビン類の測定を行う。
上記第二波長光は、上記干渉物質を検出するためのものであり、第一波長光における吸光度と、第二波長光における吸光度との差を求めることで、得られるクロマトグラムは、干渉物質由来のピーク強度が低減され、安定型HbA1cのピークが明確化したものとなる。これにより、干渉物質による影響を極力抑制して、安定型HbA1cを高精度で測定することが可能となる。また、本発明の方法は、干渉物質の含有量の大小に関わらず、幅広い血液検体に適応することができる。
【0015】
上記第一波長光は、ヘモグロビン類を検出するためのものであり、ヘモグロビン類を検出可能な波長を有する光であれば、特に限定されず、測定対象となるヘモグロビン類の種類により適宜変更することができる。
上記第一波長光は、波長の好ましい下限が405nm、好ましい上限が425nmである。上記範囲外であると、ヘモグロビン類を感度良く検出できないことがある。より好ましい下限は410nm、より好ましい上限は420nmである。
【0016】
上記第二波長光は、上記第一波長光よりも波長が大きい光である。このような第二波長光を用いて、吸光度を測定することで、主に上記干渉物質を検出することができる。
また、上記第二波長光は、波長の下限が450nm、上限が480nmである。上記範囲外であると、干渉物質がヘモグロビン類の測定値に与える影響が大きくなる。好ましい下限は460nm、好ましい上限は470nmである。
【0017】
本発明のヘモグロビン類の測定方法において、測定対象となるヘモグロビン類としては、特に限定されず、例えば、公知のヘモグロビン類が挙げられ、具体的には例えば、HbA1a、HbA1b、HbF、不安定型HbA1c、安定型HbA1c、HbA0、HbA2等が挙げられる。また、アセチル化Hb、カルバミル化Hb等の修飾ヘモグロビン、ヘモグロビンS(以下、HbSという)、ヘモグロビンC(以下、HbCという)等の異常ヘモグロビンについても測定することができる。
【0018】
本発明において、陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する際に使用する溶離液としては、酸解離定数(pKa)が、2.15〜6.39及び6.40〜10.50の範囲内である緩衝剤を含有することが好ましい。
上記pKaが上記範囲外であると、溶離液のpHを、測定目的のピークを分離するのに適切な範囲とすることができず、結果として、ヘモグロビン類が変性したり、ヘモグロビン類の分離が困難となったりすることがある。
なお、上記緩衝剤としては、pKaを2.15〜6.39及び6.40〜10.50の範囲に少なくとも1つずつもつ単一の物質を用いてもよく、2.15〜6.39の範囲内に少なくとも1つのpKaをもつ物質と6.40〜10.50の範囲内に少なくとも1つのpKaをもつ物質とを組み合わせて緩衝剤として用いてもよい。また、上記緩衝剤を複数組み合わせて用いてもよい。
【0019】
上記緩衝剤のpKaの範囲は、測定目的のピークを分離するのに適切な溶離液のpH付近において、より優れた緩衝能を発揮できるように、2.61〜6.39及び6.40〜10.50の範囲内であることが好ましく、より好ましくは、2.80〜6.35及び6.80〜10.00の範囲内である。更に好ましくは、3.50〜6.25及び7.00〜9.50の範囲内である。
【0020】
上記緩衝剤としては、例えば、リン酸、ホウ酸、炭酸等の無機物のほか、カルボン酸、ジカルボン酸、カルボン酸誘導体、ヒドロキシカルボン酸、アニリン又はアニリン誘導体、アミノ酸、アミン類、イミダゾール類、アルコール類等の有機物が挙げられる。また、エチレンジアミン四酢酸、ピロリン酸、ピリジン、カコジル酸、グリセロールリン酸、2,4,6−コリジン、N−エチルモルホリン、モルホリン、4−アミノピリジン、アンモニア、エフェドリン、ヒドロキシプロリン、ペリジン、トリス(ヒドロキシメチル)アミノメタン、グリシルグリシン等の有機物でもよい。
【0021】
上記カルボン酸としては、例えば、ギ酸、酢酸、プロピオン酸、安息香酸等が挙げられる。上記ジカルボン酸としては、例えば、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、マレイン酸、フタル酸、フマル酸等が挙げられる。
【0022】
上記カルボン酸誘導体としては、例えば、β,β’−ジメチルグルタル酸、バルビツール酸、5,5−ジエチルバルビツール酸、γ−アミノ酪酸、ピルビン酸、フランカルボン酸、ε−アミノカプロン酸等が挙げられる。
【0023】
上記ヒドロキシカルボン酸としては、例えば、酒石酸、クエン酸、乳酸、リンゴ酸等が挙げられる。上記アニリン又はアニリン誘導体としては、例えば、アニリン、ジメチルアニリン等が挙げられる。上記アミノ酸としては、例えば、アスパラギン酸、アスパラギン、グリシン、α−アラニン、β−アラニン、ヒスチジン、セリン、ロイシン等が挙げられる。
【0024】
上記アミン類としては、例えば、エチレンジアミン、エタノールアミン、トリメチルアミン、ジエタノールアミン等が挙げられる。上記イミダゾール類としては、例えば、イミダゾール、5(4)−ヒドロキシイミダゾール、5(4)−メチルイミダゾール、2,5(4)−ジメチルイミダゾール等が挙げられる。
【0025】
上記アルコール類としては、例えば、2−アミノ−2−メチル−1,3−プロパンジオール、2−アミノ−2−エチル−1,3−プロパンジオール、2−アミノ−2−メチル−1−プロパノール等が挙げられる。
【0026】
また、上記緩衝剤としては、2−(N−モリホリノ)エタンスルホン酸(MES)、ビス(2−ヒドロキシエチル)イミノトリス−(ヒドロキシメチル)メタン(Bistris)、N−(2−アセトアミド)イミドジ酢酸(ADA)、ピペラジン−N,N’−ビス(2−エタンスルホン酸)(PIPES)、1,3−ビス(トリス(ヒドロキシメチル)−メチルアミノ)プロパン(Bistrispropane)、N−(アセトアミド)−2−アミノエタンスルホン酸(ACES)、3−(N−モルフォリン)プロパンスルホン酸(MOPS)、N,N’−ビス(2−ヒドロキシエチル)−2−アミノエタンスルホン酸(BES)、N−トリス(ヒドロキシメチル)メチル−2−アミノエタンスルホン酸(TES)、N−2−ヒドロキシエチルピペラジン−N’−エタンスルホン酸(HEPES)、N−2−ヒドロキシエチルピペラジン−N’−プロパンスルホン酸(HEPPS)、N−トリス(ヒドロキシメチル)メチルグリシン(Tricine)、トリス(ヒドロキシメチル)アミノエタン(Tris)、N,N’−ビス(2−ヒドロキシエチル)グリシン(Bicine)、グリシルグリシン、N−トリス(ヒドロキシメチル)メチル−3−アミノプロパンスルホン酸(TAPS)、グリシン、シクロヘキシルアミノプロパンスルホン酸(CAPS)等の一般にグッド(Good)の緩衝液といわれるものを組成する物質も使用できる。これらの物質のpKaを表1、2に示す。(引用文献:堀尾武一、山下仁平著、蛋白質・酵素の基礎実験法、南江堂)
【0027】
【表1】

【0028】
【表2】
【0029】
上記溶離液中の緩衝剤の含有量は、緩衝作用がある範囲であればよく、好ましい下限は1mM、好ましい上限は1000mMである。より好ましい下限は10mM、より好ましい上限は500mMである。また、上記緩衝剤は、単独でも複数混合して用いてもよく、例えば、有機物と無機物を混合して用いてもよい。
【0030】
上記溶離液は、HbA1cピーク溶出を最適化することを目的として、塩化ナトリウム、塩化カリウム、硫酸ナトリウム、硫酸カリウム、リン酸ナトリウム等の無機塩類を含有していてもよい。なお、これらの無機塩類の濃度は、特に限定されないが、好ましい下限が1mM、好ましい上限が1500mMである。
【0031】
上記溶離液は、pH調節剤として、公知の酸、塩基を含有していてもよい。上記酸としては、例えば、塩酸、リン酸、硝酸、硫酸等が挙げられ、上記塩基としては、例えば、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化マグネシウム、水酸化バリウム、水酸化カルシウム等が挙げられる。これらの酸、塩基の含有量は、特に限定されないが、好ましい下限は0.001mM、好ましい上限は500mMである。
【0032】
上記溶離液は、メタノール、エタノール、アセトニトリル、アセトン等の水溶性有機溶媒を含有していてもよい。上記有機溶媒の濃度は、塩等が析出しない程度で用いることが好ましく、好ましい上限は80%(v/v)である。
【0033】
また、上記溶離液は、アジ化ナトリウム、チモール等の防腐剤を含有していてもよく、ヘモグロビンの安定剤として、公知の安定剤、例えば、エチレンジアミン四酢酸(EDTA)等のキレート剤、グルタチオン、アジ化ナトリウム等の還元剤・酸化防止剤等を含有していてもよい。
【0034】
本発明において用いる溶離液は、カオトロピックイオンを含有することが好ましい。上記カオトロピックイオンとは、化合物が水溶液に溶解したときに解離により生じたイオンであり、水の構造を破壊し、疎水性物質と水が接触したときに起こる水のエントロピー減少を抑制するものである。
【0035】
上記カオトロピックイオンには、陰イオン及び陽イオンのカオトロピックイオンがあり、上記陰イオンのカオトロピックイオンとしては、トリブロモ酢酸イオン、トリクロロ酢酸イオン、チオシアン酸イオン、ヨウ化物イオン、過塩素酸イオン、ジクロロ酢酸イオン、硝酸イオン、臭化物イオン、塩化物イオン、酢酸イオン等が挙げられ、陽イオンのカオトロピックイオンとしては、バリウムイオン、カルシウムイオン、リチウムイオン、セシウムイオン、カリウムイオン、マグネシウムイオン、グアニジンイオン等が挙げられる。
【0036】
上記カオトロピックイオンの中でも、陰イオンのカオトロピックイオンとしては、トリブロモ酢酸イオン、トリクロロ酢酸イオン、チオシアン酸イオン、ヨウ化物イオン、過塩素酸イオン、ジクロロ酢酸イオン、硝酸イオン、臭化物イオン等を用いることが好ましく、陽イオンのカオトロピックイオンとしては、バリウムイオン、カルシウムイオン、マグネシウムイオン、リチウムイオン、セシウムイオン、グアニジンイオン等を用いることが好ましい。より好ましくは、チオシアン酸イオン、過塩素酸イオン、硝酸イオン、グアニジンイオン等が用いられる。
【0037】
上記溶離液中のカオトロピックイオンの含有量の好ましい下限は0.1mM、好ましい上限は3000mMである。0.1mM未満であると、ヘモグロビン類の測定において、分離効果が低下するおそれがあり、3000mMを超えて添加しても、ヘモグロビン類の分離効果はそれ以上向上しない。より好ましい下限は1mM、より好ましい上限は1000mMであり、更に好ましい下限は10mM、更に好ましい上限は500mMである。
【0038】
本発明において、ヘモグロビン類の測定を行う場合は、pHの異なる少なくとも2種類以上の溶離液を用いることが好ましい。その場合、測定目的のピークを分離するにあたって用いる溶離液は、同一の緩衝剤を含有するものを用いるのが好ましいが、溶離液を切り替える際の、検出器出力のベースライン変動が、測定値に悪影響を与えなければ、必ずしもこれに限定されない。なお、溶離液のpHは、例えば、pH調節剤の添加量によって調節することができる。
更に、ベースライン変動をより小さくするために、上記測定目的のピークを分離するにあたって用いる溶離液は、緩衝剤の濃度も同一であるものを用いるのがより好ましい。
【0039】
本発明において、上記溶離液を送液する方法としては特に限定されないが、pHの異なる2種類以上の溶離液を用いる場合は、少なくとも溶出力の異なる3種の溶離液を用い、HbA0を溶出するための溶離液を送液する前に、HbA0より前に溶出されてくる各ヘモグロビン成分を分離することを目的として、該HbA0を溶出するための溶離液以外の溶離液を送液する方法を行うことが好ましい。
なお、通常のヘモグロビン類の測定において、測定目的となるヘモグロビン類としては、HbA1a、HbA1b、HbF、不安定型HbA1c、安定型HbA1c、HbA0等が挙げられるが、このうちHbA0より前に溶出されてくる各ヘモグロビン成分とは、HbA1a、HbA1b、HbF、不安定型HbA1c、安定型HbA1cのことをいう。
【0040】
また、安定型HbA1cの測定に悪影響を与える可能性のあるHbA2、HbS、HbC等のヘモグロビン成分を含む血液検体を測定する場合は、HbA0成分(ピーク)として主にHbAを溶出させた後、HbA2、HbS、HbC等を溶離させることが好ましい。これにより、HbA0ピークからHbA以外のヘモグロビン成分を除けるため、より正確な安定型HbA1c(%)を算出できる。
なお、HbA0を溶出するための溶離液を送液した後、HbA2、HbS、HbC等を溶離するために使用する溶離液としては、より溶出力の強い溶離液を使用する必要がある。
【0041】
また、本発明のヘモグロビン類の測定方法では、溶離液を勾配溶出法又は段階溶出法によって送液し、その途中において溶出力の低い溶離液を送液することが好ましい。
【0042】
従来のヘモグロビン類の測定方法では、分離対象成分のピークをシャープにしたり、隣り合って溶出する2つ以上のピークの分離度を向上させたりするために、複数の溶離液を用いた勾配溶出法や段階溶出法が利用されていた。
【0043】
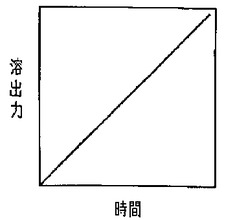
上記勾配溶出法は「グラジエント溶出」と呼ばれる方法であり、例えば、複数台の送液ポンプを用い、溶出力が異なる複数の溶離液の送液比率を連続的に変化させることにより送液する方法である。これにより、図10に示すように、時間と共に溶出力が連続的に上昇するように溶出が行われる。
【0044】
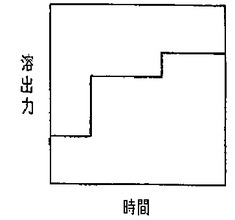
また、上記段階溶出法は「ステップワイズ溶出」と呼ばれる方法であり、例えば、1台の送液ポンプを、電磁弁等を介して複数の溶離液に連結し、電磁弁を切り替えることにより、溶出力の低い溶離液から、溶出力の高い溶離液に切り替えて送液する方法である。従って、図11に示すように、溶出力は段階的に上昇する。
【0045】
しかしながら、従来の勾配溶出法や段階溶出法では、溶出される各成分の性質が類似していたり、短時間で溶出することが要求されている場合、類似した性質の成分間でピークが重なり、分離度が低下するおそれがあった。
【0046】
これに対して、勾配溶出法又は段階溶出法によって溶離液を送液するに際し、その途中、即ち、勾配溶出法又は段階溶出法により複数の溶離液を順に切り替えて送液していく途中において、溶出力が低い溶離液を送液することにより、分離対象のピーク又はピーク間の分離状態を良くすることができる。具体的には、段階溶出法の場合、溶出力の弱い溶離液から溶出力の強い溶離液に切り替えて送液した後、溶出力の弱い溶離液に切り替え、しばらくしてから溶出力の強い溶離液に切り替えて送液する方法等が挙げられる。
【0047】
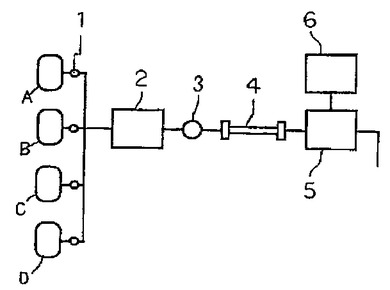
本発明の方法を段階溶出法によって行う場合の、装置の構成例を図12に示した。A、B、C、Dは、各々溶出力の異なる(例えば、塩濃度、pH、極性等において異なる)溶離液であり、電磁弁1によって設定時間に各溶離液に切り替えられるように構成されている。溶離液は、送液ポンプ2により、試料注入部3から導入された試料とともにカラム4に導かれ、各成分が検出器5により検出される。各ピークの面積、高さ等はインテグレータ6により算出される。
【0048】
本発明では、ヘモグロビン類の測定において、陽イオン交換液体クロマトグラフィーを用いる。上記陽イオン交換液体クロマトグラフィーの充填剤は、少なくとも1種以上のカチオン交換基を有している粒子よりなるものであり、例えば、高分子粒子にカチオン交換基を導入することにより得ることができる。
【0049】
上記カチオン交換基としては、公知のものを用いることができ、例えば、カルボキシル基、スルホン酸基、リン酸基等のカチオン交換基等が挙げられる。なお、上記カチオン交換基は、複数種導入してもよい。
【0050】
充填剤として用いる粒子の直径の好ましい下限は0.5μm、好ましい上限は20μmであり、より好ましい下限は1μm、より好ましい上限は10μmである。
また、上記粒子の粒度分布を示す変動係数値(CV値)の好ましい上限は40%、より好ましい上限は30%である。なお、粒子径の標準偏差÷平均直径×100から求めることができる。
【0051】
上記高分子粒子としては、例えば、シリカ、ジルコニア等の無機系粒子;セルロース、ポリアミノ酸、キトサン等の天然高分子粒子;ポリスチレン、ポリアクリル酸エステル等の合成高分子粒子等が挙げられる。
上記高分子粒子において、導入されるカチオン交換基以外の構成成分は、より親水性であることが好ましい。また耐圧性・耐膨潤性の点から架橋度の高いものが好ましい。
【0052】
上記高分子粒子へのカチオン交換基の導入は、公知の方法により行うことができるが、例えば、高分子粒子を調製後、粒子が有する官能基(水酸基、アミノ基、カルボキシル基、エポキシ基等)に、化学反応でカチオン交換基を粒子に導入させる方法により行うことができる。
【0053】
また、カチオン交換基を有する単量体を重合して高分子粒子を調製する方法によってもカチオン交換基を有する充填剤粒子を調製することができる。例えば、カチオン交換基含有単量体と架橋性単量体等とを混合し、重合開始剤の存在下に重合する方法等が挙げられる。
【0054】
更に、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル等の重合性カチオン交換基含有エステルを架橋性単量体等と混合し、重合開始剤存在下で重合した後、得られた粒子を加水分解処理し、エステルをカチオン交換基に変換させる方法により、カチオン交換基を有する充填剤粒子を調製してもよい。
また、特公平8−7197号公報に記載のように、架橋重合体粒子を調製した後、カチオン交換基を有する単量体を添加して、重合体粒子の表面付近に、該単量体を重合させる方法により、カチオン交換基を有する充填剤粒子を調製してもよい。
【0055】
本発明では、上記充填剤をカラムに充填して使用する。上記カラムの材質としては特に限定されず、例えば、公知のステンレス製、ガラス製、樹脂製等が挙げられる。
上記カラムのサイズとしては、内径0.1〜50mm、長さ1〜300mmのものが好ましく、内径0.2〜30mm、長さ5〜200mmのものがより好ましい。
【0056】
上記充填剤のカラムへの充填方法としては、特に限定されず、公知の方法を使用できるが、特にスラリー充填法が好ましい。具体的には、例えば、充填剤粒子を溶離液等の緩衝液に分散させたスラリーを送液ポンプ等により、充填剤をカラムに圧入する方法等が挙げられる。
【0057】
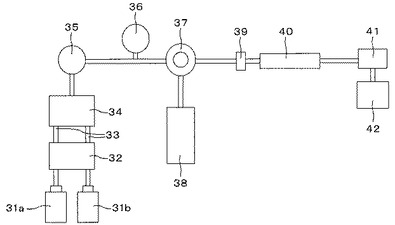
本発明に使用される陽イオン交換液体クロマトグラフィー装置としては、公知のものを使用することができ、例えば、図13に示すように、溶離液用タンク31a及び31b、脱気装置32、液切換電磁弁34、送液ポンプ35、ダンパー36、試料導入装置37、検体用タンク38、フィルター39、カラム40、検出器41及びデータ処理装置42から構成されるものを用いることができる。また、カラム恒温槽等の他の付属装置が適宜付加されてもよい。なお、液切換電磁弁34は、電磁弁を用いたマニホールド方式としてもよく、ロータリーバルブ方式等の公知の液切換装置としてもよい。
【0058】
フィルター39としては、公知のフィルター素材で、測定成分の測定値に影響を与えないものが好ましい。具体的には例えば、不織布とろ紙を積層したフィルター、不織布とメンブレンフィルターを積層したフィルター、焼結フィルターを表面処理剤(シリコーン処理、ブロッキング剤)で処理したフィルター等を用いることができる。
【0059】
上記フィルターの材質としては、測定成分の吸着が少ない素材が好ましく、例えば、不織布の素材としては、ポリエチレン、ポリプロピレン、ポリエステル、ポリエチレンテレフタレート、ナイロン、レーヨン、アクリル、ココナッツファイバー、塩化ビニリデン、綿、ウール、麻、ガラス繊維等が挙げられる。
また、必要に応じて、生体試料成分の吸着を小さくするために、上記不織布の表面を少なくとも、セルロース系樹脂、フッ素系樹脂、スルホン系樹脂、ポリエチレン、エチレンのアルキル誘導体の重合体、アクリル系樹脂、ナイロン、炭化物セラミック、窒化物セラミック、珪化物セラミック、硼化物セラミック、表面がシリル化処理された二酸化珪素、ガラス及びチタンからなる群より選ばれる少なくとも1種からなるもの、又は、これらを複数組み合わせた素材で処理しても良い。
【0060】
上記不織布の厚みの好ましい下限は0.1mm、好ましい上限は10mmであり、上記不織布の空隙率の好ましい下限は40%、好ましい上限は90%である。
【0061】
上記ろ紙としては、公知のものを用いることができ、素材は特に限定されないが、セルロース、ガラス繊維、フッ素樹脂、シリカ繊維等からなるものを用いることができる。また、ろ紙の強度向上、生体試料の吸着抑制等を実現するため、公知のろ紙に特殊処理を施したものを用いることができる。具体的には例えば、湿潤強度を高めたろ紙(アドバンテック東洋社製、ウェットストレングスろ紙)等が挙げられる。上記ろ紙の保留粒子径は、生体試料中の異物を除去するため、3μm以下であることが好ましい。
【0062】
上記フィルターは、不織布及びろ紙の有効ろ過面積を大きくするために、カラムから離れた側から「支持体、不織布、ろ紙、支持体」又は「不織布、ろ紙、支持体」の順の構成にすることができる。上記支持体の形状は、不織布及びろ紙の有効ろ過面積を大きくできるものであれば特に限定されないが、メッシュ状のものが好ましい。上記支持体の素材としては、例えば、ポリプロピレン、ポリエチレン、ナイロン、ポリエチレンテレフタレート、金属メッシュ(ステンレス、チタン)等が挙げられる。
【0063】
上記フィルターは、必要に応じて、シリコーンコーティングされてもよく、更に、ブロッキング試薬でブロッキング処理されていることが好ましい。上記ブロッキング試薬としては、例えば、ウシ血清アルブミン、カゼイン、ゼラチン、ヘモグロビン、ミオグロビンなどの蛋白質;例えば、リン脂質等の極性脂質;例えば、SDS、ポリエチレングリコールモノ−4−オクチルフェニルエーテル(トリトンX−100)などの界面活性剤などが挙げられる。
【0064】
本発明の測定方法における、他の測定条件は、使用する測定試料、カラム等の種類によって適宜選択できるが、溶離液の流速は、好ましくは0.05〜5mL/分、より好ましくは0.2〜3mL/分である。測定試料は、通常、界面活性剤等溶血活性を有する物質を含む溶液により溶血された溶血液を希釈したものを用いる。試料注入量は、血液検体の希釈倍率により異なるが、好ましくは0.1〜100μL程度である。
【発明の効果】
【0065】
本発明によれば、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定において、ヘモグロビン類を検出するための第一波長光と、主に干渉物質を検出する第二波長光という2種類の光を用いて吸光度測定を行った後、得られた吸光度の差を求めることにより、干渉物質による測定値の影響を大幅に低減させ、高精度なヘモグロビン類の測定、特に安定型HbA1cの高度な測定が可能なヘモグロビン類の測定方法を提供することができる。
【発明を実施するための最良の形態】
【0066】
以下に実施例を掲げて本発明を更に詳しく説明するが、本発明はこれら実施例のみに限定されるものではない。
【0067】
(実施例1)
(1)充填剤の調製
テトラエチレングリコールジメタクリレート(新中村化学社製)400g及び2−アクリルアミド−2−メチルプロパンスルホン酸150gの混合物に過酸化ベンゾイル(和光純薬社製)1.5gを溶解した。これを4重量%ポリビニルアルコール(日本合成化学社製)水溶液2500mLに分散させ、撹拌しながら窒素雰囲気下で75℃に昇温し、8時間重合した。重合後、洗浄し乾燥した後、分級して平均粒子径6μmの粒子を得た。
【0068】
(2)充填剤のカラムへの充填
得られた粒子0.7gを、50mMリン酸緩衝液(pH5.8)30mLに分散し、5分間超音波処理した後、よく撹拌した。全量をステンレス製の空カラム(内径4.6×30mm)を接続したパッカー(梅谷精機社製)に注入した。次いで、パッカーに送液ポンプ(サヌキ工業社製)を接続し、圧力30MPaで定圧充填した。
【0069】
(3)溶離液A、Bの調製
50mMリン酸緩衝液に、過塩素酸ナトリウムを60mM添加し、pHを5.3に調整して溶離液Aとした。また、80mMリン酸緩衝液に、過塩素酸ナトリウムを300mM添加し、pHを8.0に調整して溶離液Bとした。
【0070】
(4)測定試料の調製
(測定試料Aの調製)
健常人血をフッ化ナトリウム採血した全血検体から以下の試料を調製した。
市販のグリコHbコントロール(国際試薬社製)を、グリコHbコントロール「レベル2」1バイアルに精製水0.2mL添加溶解した後、溶血希釈液(0.1重量%トリトンX−100(東京化成社製)のリン酸緩衝液溶液、pH7.0)で101倍に希釈して測定試料Aとした。
【0071】
(測定試料Bの調製)
市販品である干渉物質検討用の「干渉チェック・Aプラス(国際試薬社製)」のビリルビンF(遊離型:サンプル)、ビリルビンF(ブランク)を取扱説明書「標準操作法」に従って、調製した測定試料Aに添加し、混和することにより測定試料Bとした。
具体的には、溶解したビリルビンF(遊離型:サンプル)1.0mLと、測定試料A9.0mLとを混和し、試料b1(サンプル)とした。次いで、溶解したビリルビンF(ブランク)1.0mLと、測定試料A9.0mLとを混和し、試料b2(ブランク)とした後、試料b1(サンプル)0.5mLと試料b2(ブランク)0.5mLとを混和することにより、測定試料Bとした。
【0072】
(測定試料Cの調製)
市販品である干渉物質検討用の「干渉チェック・Aプラス(国際試薬社製)」のビリルビンC(抱合型:サンプル)、ビリルビンC(ブランク)を取扱説明書「標準操作法」に従って、調製した測定試料Aに添加し、混和することにより測定試料Cとした。
具体的には、溶解したビリルビンC(抱合型:サンプル)1.0mLと、測定試料A9.0mLとを混和し、試料c1(サンプル)とした。次いで、溶解したビリルビンC(ブランク)1.0mLと、測定試料A9.0mLとを混和し、試料c2(ブランク)とした後、試料c1(サンプル)0.5mLと試料c2(ブランク)0.5mLとを混和することにより、測定試料Cとした。
【0073】
(測定試料Dの調製)
市販品である干渉物質検討用の「干渉チェック・Aプラス(国際試薬社製)」の乳ビ(サンプル)、乳ビ(ブランク)を取扱説明書「標準操作法」に従って、調製した測定試料Aに添加し、混和することにより測定試料Dとした。
具体的には、溶解した乳ビ(サンプル)1.0mLと、測定試料A9.0mLとを混和し、試料d1(サンプル)とした。次いで、溶解した乳ビ(ブランク)1.0mLと、測定試料A9.0mLとを混和し、試料d2(ブランク)とした後、試料d1(サンプル)0.5mLと試料d2(ブランク)0.5mLとを混和することにより、測定試料Dとした。
【0074】
(5)ヘモグロビン類の測定
得られたカラム、溶離液及び測定試料A〜Dを用いて、以下の条件でヘモグロビン類の測定を行い、クロマトグラムを得た後、HbA0ピークの面積に対する安定型HbA1cピークの面積の比率(%)を求めることにより安定型HbA1c値を算出した。なお、安定型HbA1cの算出には、第一波長光(波長;415nm)により測定した吸光度から、第二波長光(波長;460nm)により測定した吸光度を差し引いたものを示すクロマトグラムを用いた。
測定は、室温(25℃)で行った。また、溶離液は25℃に保温したものを用い、測定開始より0〜2分の間は溶離液Aを送液し、2〜3分の間は溶離液Bを送液し、3〜5分の間は溶離液Aを送液した。
【0075】
測定装置としては、図13に示す構成のものを用い、送液ポンプはLC−9A(島津製作所社製)、オートサンプラはASU−420(積水化学工業社製)、検出器はSPD−6AV(島津製作所社製)を使用した。なお、測定条件は以下の通りとした。
測定条件:流速:2.0mL/分
検出波長:415nm(第一波長)、460nm(第二波長)
試料注入量:10μL
【0076】
(実施例2)
実施例1の(5)において、検出波長を415nm(第一波長)、470nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0077】
(実施例3)
実施例1の(5)において、検出波長を415nm(第一波長)、480nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0078】
(実施例4)
実施例1の(5)において、検出波長を420nm(第一波長)、470nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0079】
(比較例1)
実施例1の(5)において、検出波長を415nm(第一波長)のみとした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0080】
(比較例2)
実施例1の(5)において、検出波長を420nm(第一波長)のみとした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0081】
(比較例3)
実施例1の(5)において、検出波長を415nm(第一波長)、500nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0082】
(比較例4)
実施例1の(5)において、検出波長を415nm(第一波長)、600nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0083】
(測定結果)
実施例1〜4、比較例1〜4により、干渉物質を添加していない測定試料A、及び、干渉物質として、ビリルビンF、ビリルビンC、乳ビをそれぞれ添加した測定試料B〜Dを測定し、安定型HbA1c値を求めた結果を表3に示した。
その結果、実施例1〜4では、干渉物質を添加していない測定試料Aと、干渉物質を添加して調製した測定試料B〜Dの安定型HbA1c値は同じ値であった。これにより、実施例では、干渉物質濃度が高くても、安定型HbA1c値を精度良く測定できることがわかる。
一方、比較例1〜4では、干渉物質を添加していない測定試料Aの安定型HbA1c値に比べて、干渉物質を添加して調製した測定試料B〜Dの安定型HbA1c値がより小さくなった。これにより、比較例では、干渉物質の影響を受けて、安定型HbA1c値を精度良く測定できないことがわかる。
【0084】
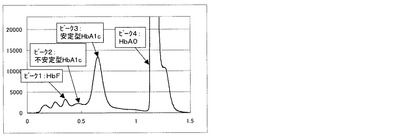
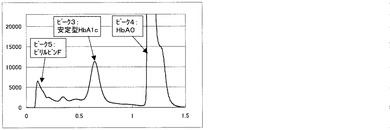
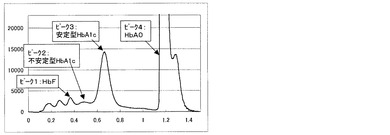
また、実施例2の測定方法にて得られるクロマトグラムを図2〜5に示す。図2は測定試料A(干渉物質無し)、図3は測定試料B(ビリルビンF)、図4は測定試料C(ビリルビンC)、図5は測定試料D(乳ビ)を測定した結果である。なお、図2〜5のクロマトグラムは、第一波長光により測定した吸光度から、第二波長光により測定した吸光度を差し引いたものを表すものである。ピーク1はHbF、ピーク2は不安定型HbA1c、ピーク3は安定型HbA1c、ピーク4はHbA0(主にHbA)、ピーク5はビリルビンF、ピーク6はビリルビンC、ピーク7は乳ビを示す。図4に示すように、測定試料C(ビリルビンC)を測定した場合は、ビリルビンC由来のピークが比較例3の場合(図8)に比べて非常に小さく、安定型HbA1cピークの溶出位置まで影響していないことが明らかで、そのために、表3に示すように、安定型HbA1c値を精度良く求めることができる。
【0085】
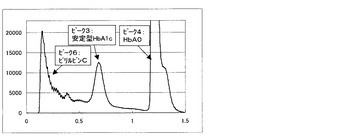
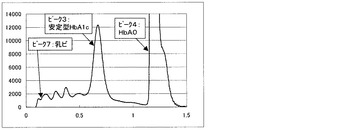
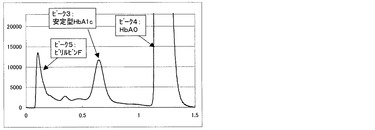
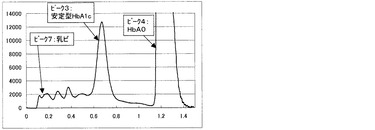
一方、比較例3の測定方法にて得られるクロマトグラムを図6〜9に示す。図6は測定試料A(干渉物質無し)、図7は測定試料B(ビリルビンF)、図8は測定試料C(ビリルビンC)、図9は測定試料D(乳ビ)を測定して得られた結果である。なお、図6〜9のクロマトグラムは、第一波長光により測定した吸光度から、第二波長光により測定した吸光度を差し引いたものを表すものである。ピーク1はHbF、ピーク2は不安定型HbA1c、ピーク3は安定型HbA1c、ピーク4はHbA0(主にHbA)、ピーク5はビリルビンF、ピーク6はビリルビンC、ピーク7は乳ビを示す。図8に示すように、測定試料C(ビリルビンC)では、ビリルビンC由来のピークが非常に大きく、しかも、安定型HbA1cピークの溶出位置まで影響していることが明らかで、そのために、安定型HbA1c値を精度良く求めることが困難であることがわかる。
【0086】
【表3】
【産業上の利用可能性】
【0087】
本発明によれば、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定方法に関し、特に安定型ヘモグロビンA1cを高精度で測定することが可能なヘモグロビン類の測定方法を提供することができる。
【図面の簡単な説明】
【0088】
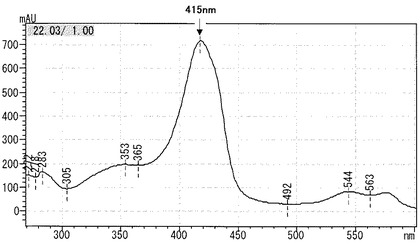
【図1】ヘモグロビン類の吸光特性を示す図である。
【図2】実施例2の測定条件により、測定試料Aの測定を行った際に得られたクロマトグラムを示す図である。
【図3】実施例2の測定条件により、測定試料Bの測定を行った際に得られたクロマトグラムを示す図である。
【図4】実施例2の測定条件により、測定試料Cの測定を行った際に得られたクロマトグラムを示す図である。
【図5】実施例2の測定条件により、測定試料Dの測定を行った際に得られたクロマトグラムを示す図である。
【図6】比較例3の測定条件により、測定試料Aの測定を行った際に得られたクロマトグラムを示す図である。
【図7】比較例3の測定条件により、測定試料Bの測定を行った際に得られたクロマトグラムを示す図である。
【図8】比較例3の測定条件により、測定試料Cの測定を行った際に得られたクロマトグラムを示す図である。
【図9】比較例3の測定条件により、測定試料Dの測定を行った際に得られたクロマトグラムを示す図である。
【図10】勾配溶出法における溶出力と時間との関係を示すグラフである。
【図11】段階溶出法における溶出力と時間との関係を示すグラフである。
【図12】段階溶出法を行う場合の装置の構成例である。
【図13】一般的に使用されている液体クロマトグラフィーの概略図である。
【符号の説明】
【0089】
1 電磁弁
2 送液ポンプ
3 試料注入部
4 カラム
5 検出器
6 インテグレータ
【技術分野】
【0001】
本発明は、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定方法に関し、特に安定型ヘモグロビンA1cを高精度で測定することが可能なヘモグロビン類の測定方法に関する。
【背景技術】
【0002】
有機化学、生化学、医学等の分野では、試料中の成分の分離や分取等の操作を行う機器として液体クロマトグラフィーが汎用されている。液体クロマトグラフィーは、通常、図13に示すような構成となっており、移動相用タンク31a及び31bから送液された溶離液は、脱気装置(デガッサー)32により脱気された後、マニホールド(液切換装置)34を通り、送液ポンプ35により圧力が掛けられ、検体用タンク38から試料導入装置37を介して検体が導入された後、フィルター39を経由して分離カラム40に導入される。そして、この分離カラム40により試料中の各成分が分離される。分離された各成分は検出器41によって、例えば、吸光度を測定する等によって検出され、その結果がデータ処理装置42により処理されてクロマトグラムとして表される。なお、移動相の定流量、定圧を目的として必要に応じてダンパー36が設置されてもよい。
【0003】
このような液体クロマトグラフィーを用いた測定方法は、血液中の糖化ヘモグロビンの測定にも用いられており、特にヘモグロビンA1c(以下、HbA1cという)は、過去1〜2カ月間の血液中の平均的な糖濃度を反映しているため、糖尿病のスクリーニング検査や糖尿病患者の血糖管理状態を把握するための検査項目として広く利用されている。なお、HbA1cは、血液中のグルコースとヘモグロビンA(以下、HbAという)とが反応して生成された糖化ヘモグロビン(以下、GHbという)であり、可逆的に反応したものは、不安定型HbA1c(unstable HbA1c)と呼ばれ、不安定型HbA1cを経て不可逆的に反応したものは、安定型HbA1c(stable HbA1c)と呼ばれている。
【0004】
通常、ヘモグロビンは2種類のサブユニット2つずつから構成される4量体のタンパク質である。HbAのサブユニットはα鎖とβ鎖であり、このβ鎖のN末端アミノ酸にグルコースが結合したものがHbA1cである。このうち過去1〜2カ月間の平均的な血糖値を良く反映しているものは、安定型HbA1cであり、臨床検査分野では、安定型HbA1c値(%)を高精度に得ることができる測定法の開発が望まれている。なお、安定型HbA1cは、全ヘモグロビンピークの面積(主に、ヘモグロビンA0ピークの面積)に対する安定型HbA1cピークの面積の比率(%)を求めることにより測定できる。即ち、精度良く安定型HbA1c値を測定するためには、精度良く安定型HbA1cピークの面積と全ヘモグロビンピークの面積(主に、ヘモグロビンA0ピークの面積)を求める必要がある。
【0005】
従来、HbA1cの主な測定法としては、例えば、特許文献1に開示されているように、血液検体を溶血希釈して調製した試料中のヘモグロビン類を、陽イオン交換法により、ヘモグロビン成分毎に異なるプラス荷電状態の違いを利用して分離する液体クロマトグラフィーを用いた方法が行われてきた。
【0006】
通常、陽イオン交換液体クロマトグラフィーを用い、充分な時間を掛けて溶血試料中のヘモグロビン類を分離すると、ヘモグロビンA1a(以下、HbA1aという)及びヘモグロビンA1b(以下、HbA1bという)、ヘモグロビンF(以下、HbFという)、不安定型HbA1c、安定型HbA1c及びヘモグロビンA0(以下、HbA0という)の順に溶離されてくる。ここで、HbA1a、HbA1b及びHbA1cはHbAが糖化されたGHbであり、HbFはα鎖とγ鎖から成る胎児性ヘモグロビンであり、HbA0はHbAを主成分とする一群のヘモグロビン成分であって、HbA1cより強くカラムに保持されたものである。
【0007】
しかしながら、従来の陽イオン交換液体クロマトグラフィーを用いた方法では、安定型HbA1cから不安定型HbA1cを充分に分離することができないばかりでなく、アセチル化ヘモグロビン(以下、アセチル化Hbという)やカルバミル化ヘモグロビン(以下、カルバミル化Hbという)等の「修飾ヘモグロビン」が安定型HbA1cと重なって溶離してくるという問題があった。
即ち、陽イオン交換液体クロマトグラフィーによる安定型HbA1c値(%)の測定を目的として、血液検体のヘモグロビン類を測定する際、安定型HbA1cの測定値に影響を与えないように、安定型HbA1cと溶出挙動が近似している不安定型HbA1cやアセチル化Hb及びカルバミル化Hbピークを、安定型HbA1cピークから分離することが困難であった。
【0008】
これに対して、近年、カルバミル化Hb、アセチル化Hb等の修飾ヘモグロビンの分離性能を向上させ、安定型HbA1cを測定できる技術が開発されており、例えば、特許文献2には、陽イオン交換液体クロマトグラフィーによるヘモグロビン類の測定方法であって、カオトロピックイオンを含有し、かつ、pH4.0〜6.8で緩衝能を持つ無機酸、有機酸及び/又はこれらの塩を含む溶離液を用いることで安定型HbA1cを分離する方法が開示されている。
しかしながら、このような方法では、高度に修飾されたアセチル化Hb、カルバミル化Hbを充分に分離できないという欠点があった。
【0009】
一方で、カラムを経て分離されたヘモグロビン類のピークを検出するための測定器(図13の検出器41)についても、近年研究がなされており、例えば、特許文献3、4には、GHbを検出するための測定器として、波長415nmまたはその付近の青色単色光を発するレーザーを光源とした吸光度計が開示されている。
このような吸光度計では、GHbの最大吸収波長である415nm付近の波長を測定波長とすることで、理論上はGHbを精度よく測定することが可能となるが、実際の臨床検査においては、血液中に含まれるビリルビン類、乳ビ等の不純物(干渉物質)が測定結果に悪影響を及ぼすことによって、特に安定型HbA1cの測定精度が低いものとなっていた。また、このような干渉物質の血液中の濃度は、個人によって異なることから、干渉物質の存在下で正確に安定型HbA1c測定することは非常に困難であった。
【特許文献1】特公平8−7198号公報
【特許文献2】特開2000−111539号公報
【特許文献3】特開平8−159954号公報
【特許文献4】特開平11−142326号公報
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明は、上記現状に鑑み、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定方法であって、特に安定型HbA1cを高精度で測定することが可能なヘモグロビン類の測定方法を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明は、陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する方法であって、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出するヘモグロビン類の測定方法である。
以下に本発明を詳述する。
【0012】
本発明者らは、鋭意検討した結果、ヘモグロビン類の測定において、所定範囲の波長を有する2種類の光を用い、得られた吸光度の差からクロマトグラムを作成し、ピーク比率を算出することにより、干渉物質による測定値の影響を大幅に低減させ、高精度なヘモグロビン類の測定、特に安定型HbA1cの高度な測定が可能となることを見出し、本発明を完成させるに至った。
【0013】
本発明では、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出することにより、ヘモグロビン類の測定を行う。
なお、本明細書において、「ピーク比率」とは、ピーク面積比率、ピーク高さ比率等のことをいい、測定するヘモグロビン類の種類によって、適宜選択することができる。ピーク面積比率を算出する方法としては、例えば、安定型HbA1c値を測定する場合における、全ヘモグロビンピークの面積(主に、HbA0ピークの面積)に対する安定型HbA1cピークの面積の比率(%)を算出する方法等を用いることができる。
【0014】
本発明の方法を用いた場合に、測定精度が向上する理由は以下の通りであると考えられる。まず、ヘモグロビン類の吸収特性を図1に示す。図1において、縦軸は吸光度、横軸は波長を表す。図1に示すように、ヘモグロビン類は最大吸収波長が415nm付近であるため、検出光として波長が415±10nmの範囲にある光を用いれば、ヘモグロビン類は充分検出できるが、ビリルビン類、乳ビ等の不純物(即ち干渉物質)による干渉によって、精度が低下する。これに対して、本発明では、ヘモグロビン類を検出するための第一波長光(波長405〜425nm)に加えて、上記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出することにより、ヘモグロビン類の測定を行う。
上記第二波長光は、上記干渉物質を検出するためのものであり、第一波長光における吸光度と、第二波長光における吸光度との差を求めることで、得られるクロマトグラムは、干渉物質由来のピーク強度が低減され、安定型HbA1cのピークが明確化したものとなる。これにより、干渉物質による影響を極力抑制して、安定型HbA1cを高精度で測定することが可能となる。また、本発明の方法は、干渉物質の含有量の大小に関わらず、幅広い血液検体に適応することができる。
【0015】
上記第一波長光は、ヘモグロビン類を検出するためのものであり、ヘモグロビン類を検出可能な波長を有する光であれば、特に限定されず、測定対象となるヘモグロビン類の種類により適宜変更することができる。
上記第一波長光は、波長の好ましい下限が405nm、好ましい上限が425nmである。上記範囲外であると、ヘモグロビン類を感度良く検出できないことがある。より好ましい下限は410nm、より好ましい上限は420nmである。
【0016】
上記第二波長光は、上記第一波長光よりも波長が大きい光である。このような第二波長光を用いて、吸光度を測定することで、主に上記干渉物質を検出することができる。
また、上記第二波長光は、波長の下限が450nm、上限が480nmである。上記範囲外であると、干渉物質がヘモグロビン類の測定値に与える影響が大きくなる。好ましい下限は460nm、好ましい上限は470nmである。
【0017】
本発明のヘモグロビン類の測定方法において、測定対象となるヘモグロビン類としては、特に限定されず、例えば、公知のヘモグロビン類が挙げられ、具体的には例えば、HbA1a、HbA1b、HbF、不安定型HbA1c、安定型HbA1c、HbA0、HbA2等が挙げられる。また、アセチル化Hb、カルバミル化Hb等の修飾ヘモグロビン、ヘモグロビンS(以下、HbSという)、ヘモグロビンC(以下、HbCという)等の異常ヘモグロビンについても測定することができる。
【0018】
本発明において、陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する際に使用する溶離液としては、酸解離定数(pKa)が、2.15〜6.39及び6.40〜10.50の範囲内である緩衝剤を含有することが好ましい。
上記pKaが上記範囲外であると、溶離液のpHを、測定目的のピークを分離するのに適切な範囲とすることができず、結果として、ヘモグロビン類が変性したり、ヘモグロビン類の分離が困難となったりすることがある。
なお、上記緩衝剤としては、pKaを2.15〜6.39及び6.40〜10.50の範囲に少なくとも1つずつもつ単一の物質を用いてもよく、2.15〜6.39の範囲内に少なくとも1つのpKaをもつ物質と6.40〜10.50の範囲内に少なくとも1つのpKaをもつ物質とを組み合わせて緩衝剤として用いてもよい。また、上記緩衝剤を複数組み合わせて用いてもよい。
【0019】
上記緩衝剤のpKaの範囲は、測定目的のピークを分離するのに適切な溶離液のpH付近において、より優れた緩衝能を発揮できるように、2.61〜6.39及び6.40〜10.50の範囲内であることが好ましく、より好ましくは、2.80〜6.35及び6.80〜10.00の範囲内である。更に好ましくは、3.50〜6.25及び7.00〜9.50の範囲内である。
【0020】
上記緩衝剤としては、例えば、リン酸、ホウ酸、炭酸等の無機物のほか、カルボン酸、ジカルボン酸、カルボン酸誘導体、ヒドロキシカルボン酸、アニリン又はアニリン誘導体、アミノ酸、アミン類、イミダゾール類、アルコール類等の有機物が挙げられる。また、エチレンジアミン四酢酸、ピロリン酸、ピリジン、カコジル酸、グリセロールリン酸、2,4,6−コリジン、N−エチルモルホリン、モルホリン、4−アミノピリジン、アンモニア、エフェドリン、ヒドロキシプロリン、ペリジン、トリス(ヒドロキシメチル)アミノメタン、グリシルグリシン等の有機物でもよい。
【0021】
上記カルボン酸としては、例えば、ギ酸、酢酸、プロピオン酸、安息香酸等が挙げられる。上記ジカルボン酸としては、例えば、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、マレイン酸、フタル酸、フマル酸等が挙げられる。
【0022】
上記カルボン酸誘導体としては、例えば、β,β’−ジメチルグルタル酸、バルビツール酸、5,5−ジエチルバルビツール酸、γ−アミノ酪酸、ピルビン酸、フランカルボン酸、ε−アミノカプロン酸等が挙げられる。
【0023】
上記ヒドロキシカルボン酸としては、例えば、酒石酸、クエン酸、乳酸、リンゴ酸等が挙げられる。上記アニリン又はアニリン誘導体としては、例えば、アニリン、ジメチルアニリン等が挙げられる。上記アミノ酸としては、例えば、アスパラギン酸、アスパラギン、グリシン、α−アラニン、β−アラニン、ヒスチジン、セリン、ロイシン等が挙げられる。
【0024】
上記アミン類としては、例えば、エチレンジアミン、エタノールアミン、トリメチルアミン、ジエタノールアミン等が挙げられる。上記イミダゾール類としては、例えば、イミダゾール、5(4)−ヒドロキシイミダゾール、5(4)−メチルイミダゾール、2,5(4)−ジメチルイミダゾール等が挙げられる。
【0025】
上記アルコール類としては、例えば、2−アミノ−2−メチル−1,3−プロパンジオール、2−アミノ−2−エチル−1,3−プロパンジオール、2−アミノ−2−メチル−1−プロパノール等が挙げられる。
【0026】
また、上記緩衝剤としては、2−(N−モリホリノ)エタンスルホン酸(MES)、ビス(2−ヒドロキシエチル)イミノトリス−(ヒドロキシメチル)メタン(Bistris)、N−(2−アセトアミド)イミドジ酢酸(ADA)、ピペラジン−N,N’−ビス(2−エタンスルホン酸)(PIPES)、1,3−ビス(トリス(ヒドロキシメチル)−メチルアミノ)プロパン(Bistrispropane)、N−(アセトアミド)−2−アミノエタンスルホン酸(ACES)、3−(N−モルフォリン)プロパンスルホン酸(MOPS)、N,N’−ビス(2−ヒドロキシエチル)−2−アミノエタンスルホン酸(BES)、N−トリス(ヒドロキシメチル)メチル−2−アミノエタンスルホン酸(TES)、N−2−ヒドロキシエチルピペラジン−N’−エタンスルホン酸(HEPES)、N−2−ヒドロキシエチルピペラジン−N’−プロパンスルホン酸(HEPPS)、N−トリス(ヒドロキシメチル)メチルグリシン(Tricine)、トリス(ヒドロキシメチル)アミノエタン(Tris)、N,N’−ビス(2−ヒドロキシエチル)グリシン(Bicine)、グリシルグリシン、N−トリス(ヒドロキシメチル)メチル−3−アミノプロパンスルホン酸(TAPS)、グリシン、シクロヘキシルアミノプロパンスルホン酸(CAPS)等の一般にグッド(Good)の緩衝液といわれるものを組成する物質も使用できる。これらの物質のpKaを表1、2に示す。(引用文献:堀尾武一、山下仁平著、蛋白質・酵素の基礎実験法、南江堂)
【0027】
【表1】
【0028】
【表2】
【0029】
上記溶離液中の緩衝剤の含有量は、緩衝作用がある範囲であればよく、好ましい下限は1mM、好ましい上限は1000mMである。より好ましい下限は10mM、より好ましい上限は500mMである。また、上記緩衝剤は、単独でも複数混合して用いてもよく、例えば、有機物と無機物を混合して用いてもよい。
【0030】
上記溶離液は、HbA1cピーク溶出を最適化することを目的として、塩化ナトリウム、塩化カリウム、硫酸ナトリウム、硫酸カリウム、リン酸ナトリウム等の無機塩類を含有していてもよい。なお、これらの無機塩類の濃度は、特に限定されないが、好ましい下限が1mM、好ましい上限が1500mMである。
【0031】
上記溶離液は、pH調節剤として、公知の酸、塩基を含有していてもよい。上記酸としては、例えば、塩酸、リン酸、硝酸、硫酸等が挙げられ、上記塩基としては、例えば、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化マグネシウム、水酸化バリウム、水酸化カルシウム等が挙げられる。これらの酸、塩基の含有量は、特に限定されないが、好ましい下限は0.001mM、好ましい上限は500mMである。
【0032】
上記溶離液は、メタノール、エタノール、アセトニトリル、アセトン等の水溶性有機溶媒を含有していてもよい。上記有機溶媒の濃度は、塩等が析出しない程度で用いることが好ましく、好ましい上限は80%(v/v)である。
【0033】
また、上記溶離液は、アジ化ナトリウム、チモール等の防腐剤を含有していてもよく、ヘモグロビンの安定剤として、公知の安定剤、例えば、エチレンジアミン四酢酸(EDTA)等のキレート剤、グルタチオン、アジ化ナトリウム等の還元剤・酸化防止剤等を含有していてもよい。
【0034】
本発明において用いる溶離液は、カオトロピックイオンを含有することが好ましい。上記カオトロピックイオンとは、化合物が水溶液に溶解したときに解離により生じたイオンであり、水の構造を破壊し、疎水性物質と水が接触したときに起こる水のエントロピー減少を抑制するものである。
【0035】
上記カオトロピックイオンには、陰イオン及び陽イオンのカオトロピックイオンがあり、上記陰イオンのカオトロピックイオンとしては、トリブロモ酢酸イオン、トリクロロ酢酸イオン、チオシアン酸イオン、ヨウ化物イオン、過塩素酸イオン、ジクロロ酢酸イオン、硝酸イオン、臭化物イオン、塩化物イオン、酢酸イオン等が挙げられ、陽イオンのカオトロピックイオンとしては、バリウムイオン、カルシウムイオン、リチウムイオン、セシウムイオン、カリウムイオン、マグネシウムイオン、グアニジンイオン等が挙げられる。
【0036】
上記カオトロピックイオンの中でも、陰イオンのカオトロピックイオンとしては、トリブロモ酢酸イオン、トリクロロ酢酸イオン、チオシアン酸イオン、ヨウ化物イオン、過塩素酸イオン、ジクロロ酢酸イオン、硝酸イオン、臭化物イオン等を用いることが好ましく、陽イオンのカオトロピックイオンとしては、バリウムイオン、カルシウムイオン、マグネシウムイオン、リチウムイオン、セシウムイオン、グアニジンイオン等を用いることが好ましい。より好ましくは、チオシアン酸イオン、過塩素酸イオン、硝酸イオン、グアニジンイオン等が用いられる。
【0037】
上記溶離液中のカオトロピックイオンの含有量の好ましい下限は0.1mM、好ましい上限は3000mMである。0.1mM未満であると、ヘモグロビン類の測定において、分離効果が低下するおそれがあり、3000mMを超えて添加しても、ヘモグロビン類の分離効果はそれ以上向上しない。より好ましい下限は1mM、より好ましい上限は1000mMであり、更に好ましい下限は10mM、更に好ましい上限は500mMである。
【0038】
本発明において、ヘモグロビン類の測定を行う場合は、pHの異なる少なくとも2種類以上の溶離液を用いることが好ましい。その場合、測定目的のピークを分離するにあたって用いる溶離液は、同一の緩衝剤を含有するものを用いるのが好ましいが、溶離液を切り替える際の、検出器出力のベースライン変動が、測定値に悪影響を与えなければ、必ずしもこれに限定されない。なお、溶離液のpHは、例えば、pH調節剤の添加量によって調節することができる。
更に、ベースライン変動をより小さくするために、上記測定目的のピークを分離するにあたって用いる溶離液は、緩衝剤の濃度も同一であるものを用いるのがより好ましい。
【0039】
本発明において、上記溶離液を送液する方法としては特に限定されないが、pHの異なる2種類以上の溶離液を用いる場合は、少なくとも溶出力の異なる3種の溶離液を用い、HbA0を溶出するための溶離液を送液する前に、HbA0より前に溶出されてくる各ヘモグロビン成分を分離することを目的として、該HbA0を溶出するための溶離液以外の溶離液を送液する方法を行うことが好ましい。
なお、通常のヘモグロビン類の測定において、測定目的となるヘモグロビン類としては、HbA1a、HbA1b、HbF、不安定型HbA1c、安定型HbA1c、HbA0等が挙げられるが、このうちHbA0より前に溶出されてくる各ヘモグロビン成分とは、HbA1a、HbA1b、HbF、不安定型HbA1c、安定型HbA1cのことをいう。
【0040】
また、安定型HbA1cの測定に悪影響を与える可能性のあるHbA2、HbS、HbC等のヘモグロビン成分を含む血液検体を測定する場合は、HbA0成分(ピーク)として主にHbAを溶出させた後、HbA2、HbS、HbC等を溶離させることが好ましい。これにより、HbA0ピークからHbA以外のヘモグロビン成分を除けるため、より正確な安定型HbA1c(%)を算出できる。
なお、HbA0を溶出するための溶離液を送液した後、HbA2、HbS、HbC等を溶離するために使用する溶離液としては、より溶出力の強い溶離液を使用する必要がある。
【0041】
また、本発明のヘモグロビン類の測定方法では、溶離液を勾配溶出法又は段階溶出法によって送液し、その途中において溶出力の低い溶離液を送液することが好ましい。
【0042】
従来のヘモグロビン類の測定方法では、分離対象成分のピークをシャープにしたり、隣り合って溶出する2つ以上のピークの分離度を向上させたりするために、複数の溶離液を用いた勾配溶出法や段階溶出法が利用されていた。
【0043】
上記勾配溶出法は「グラジエント溶出」と呼ばれる方法であり、例えば、複数台の送液ポンプを用い、溶出力が異なる複数の溶離液の送液比率を連続的に変化させることにより送液する方法である。これにより、図10に示すように、時間と共に溶出力が連続的に上昇するように溶出が行われる。
【0044】
また、上記段階溶出法は「ステップワイズ溶出」と呼ばれる方法であり、例えば、1台の送液ポンプを、電磁弁等を介して複数の溶離液に連結し、電磁弁を切り替えることにより、溶出力の低い溶離液から、溶出力の高い溶離液に切り替えて送液する方法である。従って、図11に示すように、溶出力は段階的に上昇する。
【0045】
しかしながら、従来の勾配溶出法や段階溶出法では、溶出される各成分の性質が類似していたり、短時間で溶出することが要求されている場合、類似した性質の成分間でピークが重なり、分離度が低下するおそれがあった。
【0046】
これに対して、勾配溶出法又は段階溶出法によって溶離液を送液するに際し、その途中、即ち、勾配溶出法又は段階溶出法により複数の溶離液を順に切り替えて送液していく途中において、溶出力が低い溶離液を送液することにより、分離対象のピーク又はピーク間の分離状態を良くすることができる。具体的には、段階溶出法の場合、溶出力の弱い溶離液から溶出力の強い溶離液に切り替えて送液した後、溶出力の弱い溶離液に切り替え、しばらくしてから溶出力の強い溶離液に切り替えて送液する方法等が挙げられる。
【0047】
本発明の方法を段階溶出法によって行う場合の、装置の構成例を図12に示した。A、B、C、Dは、各々溶出力の異なる(例えば、塩濃度、pH、極性等において異なる)溶離液であり、電磁弁1によって設定時間に各溶離液に切り替えられるように構成されている。溶離液は、送液ポンプ2により、試料注入部3から導入された試料とともにカラム4に導かれ、各成分が検出器5により検出される。各ピークの面積、高さ等はインテグレータ6により算出される。
【0048】
本発明では、ヘモグロビン類の測定において、陽イオン交換液体クロマトグラフィーを用いる。上記陽イオン交換液体クロマトグラフィーの充填剤は、少なくとも1種以上のカチオン交換基を有している粒子よりなるものであり、例えば、高分子粒子にカチオン交換基を導入することにより得ることができる。
【0049】
上記カチオン交換基としては、公知のものを用いることができ、例えば、カルボキシル基、スルホン酸基、リン酸基等のカチオン交換基等が挙げられる。なお、上記カチオン交換基は、複数種導入してもよい。
【0050】
充填剤として用いる粒子の直径の好ましい下限は0.5μm、好ましい上限は20μmであり、より好ましい下限は1μm、より好ましい上限は10μmである。
また、上記粒子の粒度分布を示す変動係数値(CV値)の好ましい上限は40%、より好ましい上限は30%である。なお、粒子径の標準偏差÷平均直径×100から求めることができる。
【0051】
上記高分子粒子としては、例えば、シリカ、ジルコニア等の無機系粒子;セルロース、ポリアミノ酸、キトサン等の天然高分子粒子;ポリスチレン、ポリアクリル酸エステル等の合成高分子粒子等が挙げられる。
上記高分子粒子において、導入されるカチオン交換基以外の構成成分は、より親水性であることが好ましい。また耐圧性・耐膨潤性の点から架橋度の高いものが好ましい。
【0052】
上記高分子粒子へのカチオン交換基の導入は、公知の方法により行うことができるが、例えば、高分子粒子を調製後、粒子が有する官能基(水酸基、アミノ基、カルボキシル基、エポキシ基等)に、化学反応でカチオン交換基を粒子に導入させる方法により行うことができる。
【0053】
また、カチオン交換基を有する単量体を重合して高分子粒子を調製する方法によってもカチオン交換基を有する充填剤粒子を調製することができる。例えば、カチオン交換基含有単量体と架橋性単量体等とを混合し、重合開始剤の存在下に重合する方法等が挙げられる。
【0054】
更に、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル等の重合性カチオン交換基含有エステルを架橋性単量体等と混合し、重合開始剤存在下で重合した後、得られた粒子を加水分解処理し、エステルをカチオン交換基に変換させる方法により、カチオン交換基を有する充填剤粒子を調製してもよい。
また、特公平8−7197号公報に記載のように、架橋重合体粒子を調製した後、カチオン交換基を有する単量体を添加して、重合体粒子の表面付近に、該単量体を重合させる方法により、カチオン交換基を有する充填剤粒子を調製してもよい。
【0055】
本発明では、上記充填剤をカラムに充填して使用する。上記カラムの材質としては特に限定されず、例えば、公知のステンレス製、ガラス製、樹脂製等が挙げられる。
上記カラムのサイズとしては、内径0.1〜50mm、長さ1〜300mmのものが好ましく、内径0.2〜30mm、長さ5〜200mmのものがより好ましい。
【0056】
上記充填剤のカラムへの充填方法としては、特に限定されず、公知の方法を使用できるが、特にスラリー充填法が好ましい。具体的には、例えば、充填剤粒子を溶離液等の緩衝液に分散させたスラリーを送液ポンプ等により、充填剤をカラムに圧入する方法等が挙げられる。
【0057】
本発明に使用される陽イオン交換液体クロマトグラフィー装置としては、公知のものを使用することができ、例えば、図13に示すように、溶離液用タンク31a及び31b、脱気装置32、液切換電磁弁34、送液ポンプ35、ダンパー36、試料導入装置37、検体用タンク38、フィルター39、カラム40、検出器41及びデータ処理装置42から構成されるものを用いることができる。また、カラム恒温槽等の他の付属装置が適宜付加されてもよい。なお、液切換電磁弁34は、電磁弁を用いたマニホールド方式としてもよく、ロータリーバルブ方式等の公知の液切換装置としてもよい。
【0058】
フィルター39としては、公知のフィルター素材で、測定成分の測定値に影響を与えないものが好ましい。具体的には例えば、不織布とろ紙を積層したフィルター、不織布とメンブレンフィルターを積層したフィルター、焼結フィルターを表面処理剤(シリコーン処理、ブロッキング剤)で処理したフィルター等を用いることができる。
【0059】
上記フィルターの材質としては、測定成分の吸着が少ない素材が好ましく、例えば、不織布の素材としては、ポリエチレン、ポリプロピレン、ポリエステル、ポリエチレンテレフタレート、ナイロン、レーヨン、アクリル、ココナッツファイバー、塩化ビニリデン、綿、ウール、麻、ガラス繊維等が挙げられる。
また、必要に応じて、生体試料成分の吸着を小さくするために、上記不織布の表面を少なくとも、セルロース系樹脂、フッ素系樹脂、スルホン系樹脂、ポリエチレン、エチレンのアルキル誘導体の重合体、アクリル系樹脂、ナイロン、炭化物セラミック、窒化物セラミック、珪化物セラミック、硼化物セラミック、表面がシリル化処理された二酸化珪素、ガラス及びチタンからなる群より選ばれる少なくとも1種からなるもの、又は、これらを複数組み合わせた素材で処理しても良い。
【0060】
上記不織布の厚みの好ましい下限は0.1mm、好ましい上限は10mmであり、上記不織布の空隙率の好ましい下限は40%、好ましい上限は90%である。
【0061】
上記ろ紙としては、公知のものを用いることができ、素材は特に限定されないが、セルロース、ガラス繊維、フッ素樹脂、シリカ繊維等からなるものを用いることができる。また、ろ紙の強度向上、生体試料の吸着抑制等を実現するため、公知のろ紙に特殊処理を施したものを用いることができる。具体的には例えば、湿潤強度を高めたろ紙(アドバンテック東洋社製、ウェットストレングスろ紙)等が挙げられる。上記ろ紙の保留粒子径は、生体試料中の異物を除去するため、3μm以下であることが好ましい。
【0062】
上記フィルターは、不織布及びろ紙の有効ろ過面積を大きくするために、カラムから離れた側から「支持体、不織布、ろ紙、支持体」又は「不織布、ろ紙、支持体」の順の構成にすることができる。上記支持体の形状は、不織布及びろ紙の有効ろ過面積を大きくできるものであれば特に限定されないが、メッシュ状のものが好ましい。上記支持体の素材としては、例えば、ポリプロピレン、ポリエチレン、ナイロン、ポリエチレンテレフタレート、金属メッシュ(ステンレス、チタン)等が挙げられる。
【0063】
上記フィルターは、必要に応じて、シリコーンコーティングされてもよく、更に、ブロッキング試薬でブロッキング処理されていることが好ましい。上記ブロッキング試薬としては、例えば、ウシ血清アルブミン、カゼイン、ゼラチン、ヘモグロビン、ミオグロビンなどの蛋白質;例えば、リン脂質等の極性脂質;例えば、SDS、ポリエチレングリコールモノ−4−オクチルフェニルエーテル(トリトンX−100)などの界面活性剤などが挙げられる。
【0064】
本発明の測定方法における、他の測定条件は、使用する測定試料、カラム等の種類によって適宜選択できるが、溶離液の流速は、好ましくは0.05〜5mL/分、より好ましくは0.2〜3mL/分である。測定試料は、通常、界面活性剤等溶血活性を有する物質を含む溶液により溶血された溶血液を希釈したものを用いる。試料注入量は、血液検体の希釈倍率により異なるが、好ましくは0.1〜100μL程度である。
【発明の効果】
【0065】
本発明によれば、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定において、ヘモグロビン類を検出するための第一波長光と、主に干渉物質を検出する第二波長光という2種類の光を用いて吸光度測定を行った後、得られた吸光度の差を求めることにより、干渉物質による測定値の影響を大幅に低減させ、高精度なヘモグロビン類の測定、特に安定型HbA1cの高度な測定が可能なヘモグロビン類の測定方法を提供することができる。
【発明を実施するための最良の形態】
【0066】
以下に実施例を掲げて本発明を更に詳しく説明するが、本発明はこれら実施例のみに限定されるものではない。
【0067】
(実施例1)
(1)充填剤の調製
テトラエチレングリコールジメタクリレート(新中村化学社製)400g及び2−アクリルアミド−2−メチルプロパンスルホン酸150gの混合物に過酸化ベンゾイル(和光純薬社製)1.5gを溶解した。これを4重量%ポリビニルアルコール(日本合成化学社製)水溶液2500mLに分散させ、撹拌しながら窒素雰囲気下で75℃に昇温し、8時間重合した。重合後、洗浄し乾燥した後、分級して平均粒子径6μmの粒子を得た。
【0068】
(2)充填剤のカラムへの充填
得られた粒子0.7gを、50mMリン酸緩衝液(pH5.8)30mLに分散し、5分間超音波処理した後、よく撹拌した。全量をステンレス製の空カラム(内径4.6×30mm)を接続したパッカー(梅谷精機社製)に注入した。次いで、パッカーに送液ポンプ(サヌキ工業社製)を接続し、圧力30MPaで定圧充填した。
【0069】
(3)溶離液A、Bの調製
50mMリン酸緩衝液に、過塩素酸ナトリウムを60mM添加し、pHを5.3に調整して溶離液Aとした。また、80mMリン酸緩衝液に、過塩素酸ナトリウムを300mM添加し、pHを8.0に調整して溶離液Bとした。
【0070】
(4)測定試料の調製
(測定試料Aの調製)
健常人血をフッ化ナトリウム採血した全血検体から以下の試料を調製した。
市販のグリコHbコントロール(国際試薬社製)を、グリコHbコントロール「レベル2」1バイアルに精製水0.2mL添加溶解した後、溶血希釈液(0.1重量%トリトンX−100(東京化成社製)のリン酸緩衝液溶液、pH7.0)で101倍に希釈して測定試料Aとした。
【0071】
(測定試料Bの調製)
市販品である干渉物質検討用の「干渉チェック・Aプラス(国際試薬社製)」のビリルビンF(遊離型:サンプル)、ビリルビンF(ブランク)を取扱説明書「標準操作法」に従って、調製した測定試料Aに添加し、混和することにより測定試料Bとした。
具体的には、溶解したビリルビンF(遊離型:サンプル)1.0mLと、測定試料A9.0mLとを混和し、試料b1(サンプル)とした。次いで、溶解したビリルビンF(ブランク)1.0mLと、測定試料A9.0mLとを混和し、試料b2(ブランク)とした後、試料b1(サンプル)0.5mLと試料b2(ブランク)0.5mLとを混和することにより、測定試料Bとした。
【0072】
(測定試料Cの調製)
市販品である干渉物質検討用の「干渉チェック・Aプラス(国際試薬社製)」のビリルビンC(抱合型:サンプル)、ビリルビンC(ブランク)を取扱説明書「標準操作法」に従って、調製した測定試料Aに添加し、混和することにより測定試料Cとした。
具体的には、溶解したビリルビンC(抱合型:サンプル)1.0mLと、測定試料A9.0mLとを混和し、試料c1(サンプル)とした。次いで、溶解したビリルビンC(ブランク)1.0mLと、測定試料A9.0mLとを混和し、試料c2(ブランク)とした後、試料c1(サンプル)0.5mLと試料c2(ブランク)0.5mLとを混和することにより、測定試料Cとした。
【0073】
(測定試料Dの調製)
市販品である干渉物質検討用の「干渉チェック・Aプラス(国際試薬社製)」の乳ビ(サンプル)、乳ビ(ブランク)を取扱説明書「標準操作法」に従って、調製した測定試料Aに添加し、混和することにより測定試料Dとした。
具体的には、溶解した乳ビ(サンプル)1.0mLと、測定試料A9.0mLとを混和し、試料d1(サンプル)とした。次いで、溶解した乳ビ(ブランク)1.0mLと、測定試料A9.0mLとを混和し、試料d2(ブランク)とした後、試料d1(サンプル)0.5mLと試料d2(ブランク)0.5mLとを混和することにより、測定試料Dとした。
【0074】
(5)ヘモグロビン類の測定
得られたカラム、溶離液及び測定試料A〜Dを用いて、以下の条件でヘモグロビン類の測定を行い、クロマトグラムを得た後、HbA0ピークの面積に対する安定型HbA1cピークの面積の比率(%)を求めることにより安定型HbA1c値を算出した。なお、安定型HbA1cの算出には、第一波長光(波長;415nm)により測定した吸光度から、第二波長光(波長;460nm)により測定した吸光度を差し引いたものを示すクロマトグラムを用いた。
測定は、室温(25℃)で行った。また、溶離液は25℃に保温したものを用い、測定開始より0〜2分の間は溶離液Aを送液し、2〜3分の間は溶離液Bを送液し、3〜5分の間は溶離液Aを送液した。
【0075】
測定装置としては、図13に示す構成のものを用い、送液ポンプはLC−9A(島津製作所社製)、オートサンプラはASU−420(積水化学工業社製)、検出器はSPD−6AV(島津製作所社製)を使用した。なお、測定条件は以下の通りとした。
測定条件:流速:2.0mL/分
検出波長:415nm(第一波長)、460nm(第二波長)
試料注入量:10μL
【0076】
(実施例2)
実施例1の(5)において、検出波長を415nm(第一波長)、470nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0077】
(実施例3)
実施例1の(5)において、検出波長を415nm(第一波長)、480nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0078】
(実施例4)
実施例1の(5)において、検出波長を420nm(第一波長)、470nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0079】
(比較例1)
実施例1の(5)において、検出波長を415nm(第一波長)のみとした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0080】
(比較例2)
実施例1の(5)において、検出波長を420nm(第一波長)のみとした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0081】
(比較例3)
実施例1の(5)において、検出波長を415nm(第一波長)、500nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0082】
(比較例4)
実施例1の(5)において、検出波長を415nm(第一波長)、600nm(第二波長)とした以外は、実施例1と同様にして、安定型HbA1c値を測定した。
【0083】
(測定結果)
実施例1〜4、比較例1〜4により、干渉物質を添加していない測定試料A、及び、干渉物質として、ビリルビンF、ビリルビンC、乳ビをそれぞれ添加した測定試料B〜Dを測定し、安定型HbA1c値を求めた結果を表3に示した。
その結果、実施例1〜4では、干渉物質を添加していない測定試料Aと、干渉物質を添加して調製した測定試料B〜Dの安定型HbA1c値は同じ値であった。これにより、実施例では、干渉物質濃度が高くても、安定型HbA1c値を精度良く測定できることがわかる。
一方、比較例1〜4では、干渉物質を添加していない測定試料Aの安定型HbA1c値に比べて、干渉物質を添加して調製した測定試料B〜Dの安定型HbA1c値がより小さくなった。これにより、比較例では、干渉物質の影響を受けて、安定型HbA1c値を精度良く測定できないことがわかる。
【0084】
また、実施例2の測定方法にて得られるクロマトグラムを図2〜5に示す。図2は測定試料A(干渉物質無し)、図3は測定試料B(ビリルビンF)、図4は測定試料C(ビリルビンC)、図5は測定試料D(乳ビ)を測定した結果である。なお、図2〜5のクロマトグラムは、第一波長光により測定した吸光度から、第二波長光により測定した吸光度を差し引いたものを表すものである。ピーク1はHbF、ピーク2は不安定型HbA1c、ピーク3は安定型HbA1c、ピーク4はHbA0(主にHbA)、ピーク5はビリルビンF、ピーク6はビリルビンC、ピーク7は乳ビを示す。図4に示すように、測定試料C(ビリルビンC)を測定した場合は、ビリルビンC由来のピークが比較例3の場合(図8)に比べて非常に小さく、安定型HbA1cピークの溶出位置まで影響していないことが明らかで、そのために、表3に示すように、安定型HbA1c値を精度良く求めることができる。
【0085】
一方、比較例3の測定方法にて得られるクロマトグラムを図6〜9に示す。図6は測定試料A(干渉物質無し)、図7は測定試料B(ビリルビンF)、図8は測定試料C(ビリルビンC)、図9は測定試料D(乳ビ)を測定して得られた結果である。なお、図6〜9のクロマトグラムは、第一波長光により測定した吸光度から、第二波長光により測定した吸光度を差し引いたものを表すものである。ピーク1はHbF、ピーク2は不安定型HbA1c、ピーク3は安定型HbA1c、ピーク4はHbA0(主にHbA)、ピーク5はビリルビンF、ピーク6はビリルビンC、ピーク7は乳ビを示す。図8に示すように、測定試料C(ビリルビンC)では、ビリルビンC由来のピークが非常に大きく、しかも、安定型HbA1cピークの溶出位置まで影響していることが明らかで、そのために、安定型HbA1c値を精度良く求めることが困難であることがわかる。
【0086】
【表3】
【産業上の利用可能性】
【0087】
本発明によれば、陽イオン交換液体クロマトグラフィーを用いたヘモグロビン類の測定方法に関し、特に安定型ヘモグロビンA1cを高精度で測定することが可能なヘモグロビン類の測定方法を提供することができる。
【図面の簡単な説明】
【0088】
【図1】ヘモグロビン類の吸光特性を示す図である。
【図2】実施例2の測定条件により、測定試料Aの測定を行った際に得られたクロマトグラムを示す図である。
【図3】実施例2の測定条件により、測定試料Bの測定を行った際に得られたクロマトグラムを示す図である。
【図4】実施例2の測定条件により、測定試料Cの測定を行った際に得られたクロマトグラムを示す図である。
【図5】実施例2の測定条件により、測定試料Dの測定を行った際に得られたクロマトグラムを示す図である。
【図6】比較例3の測定条件により、測定試料Aの測定を行った際に得られたクロマトグラムを示す図である。
【図7】比較例3の測定条件により、測定試料Bの測定を行った際に得られたクロマトグラムを示す図である。
【図8】比較例3の測定条件により、測定試料Cの測定を行った際に得られたクロマトグラムを示す図である。
【図9】比較例3の測定条件により、測定試料Dの測定を行った際に得られたクロマトグラムを示す図である。
【図10】勾配溶出法における溶出力と時間との関係を示すグラフである。
【図11】段階溶出法における溶出力と時間との関係を示すグラフである。
【図12】段階溶出法を行う場合の装置の構成例である。
【図13】一般的に使用されている液体クロマトグラフィーの概略図である。
【符号の説明】
【0089】
1 電磁弁
2 送液ポンプ
3 試料注入部
4 カラム
5 検出器
6 インテグレータ
【特許請求の範囲】
【請求項1】
陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する方法であって、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出することを特徴とするヘモグロビン類の測定方法。
【請求項1】
陽イオン交換液体クロマトグラフィーを用いてヘモグロビン類を測定する方法であって、ヘモグロビン類を検出するための第一波長光、及び、前記第一波長光よりも波長が大きく、かつ、波長が450〜480nmの第二波長光を用いて吸光度測定を行った後、第一波長光における吸光度と、第二波長光における吸光度との差からクロマトグラムを作成し、ピーク比率を算出することを特徴とするヘモグロビン類の測定方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】

【図9】
【図10】
【図11】
【図12】
【図13】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2007−315942(P2007−315942A)
【公開日】平成19年12月6日(2007.12.6)
【国際特許分類】
【出願番号】特願2006−146369(P2006−146369)
【出願日】平成18年5月26日(2006.5.26)
【出願人】(000002174)積水化学工業株式会社 (5,781)
【公開日】平成19年12月6日(2007.12.6)
【国際特許分類】
【出願日】平成18年5月26日(2006.5.26)
【出願人】(000002174)積水化学工業株式会社 (5,781)
[ Back to top ]