
ベツリンの誘導体
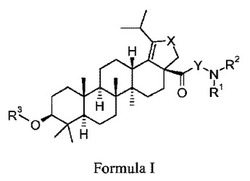
本発明は、以下の式Iにより特徴付けられる化合物またはその薬学上許容される塩:

(式中、R1、R2、R3、XおよびYは本明細書に定義される通りである)に関する。本発明の化合物はHIV−1の治療に有用である。
(式中、R1、R2、R3、XおよびYは本明細書に定義される通りである)に関する。本発明の化合物はHIV−1の治療に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はベツリンの誘導体に関する。
【背景技術】
【0002】
現在、抗レトロウイルス薬を用いるウイルス複製の長期間の抑制は、HIV−1感染を治療するための唯一の選択である。今までに、多くの承認された薬物が患者の生存期間を非常に延長させることが示されている。しかしながら、非常に活性のある抗レトロウイルス治療(HAART)として知られている治療レジメンはしばしば複雑である。なぜなら、薬物耐性HIV−1変異体の急速出現を回避するために、異なる薬物の組合せが患者に投与されなければならないからである。患者の生存に対するHAARTの効果のある作用にも関わらず、薬物耐性が生じる可能性もある。
【0003】
多剤耐性(MDR)HIV−1単離株の出現は重大な臨床的帰結を有し、サルベージ療法として知られている、新規投薬レジメンを用いて抑制されなければならない。現在の指針は、サルベージ療法が少なくとも2つ、好ましくは3つの完全に活性な薬物を含むことを推奨している。典型的に、第1の治療は、ウイルス酵素RTおよびプロテアーゼ(PR)を標的とする3〜4個の薬物を組合わせる。サルベージ療法の1つの選択は、耐性分離株に対して活性を維持する同じ機構的クラス由来の薬物の異なる組合せを投与することである。しかしながら、このアプローチの選択はしばしば、同じクラスにおける異なる薬物に広範な交差耐性を頻繁に与える耐性変異として制限される。現在、融合、侵入、およびインテグラーゼ(IN)阻害剤の開発による代替の治療ストラテジーが利用可能となっている。しかしながら、3つ全ての新規薬物クラスに対する耐性がインビトロおよびインビボの両方において既に報告されている。したがって、抗レトロウイルス薬物を用いるHIV−1感染患者の持続した首尾良い治療は、新しい標的および作用機構を有する新規および改良された薬物の継続した開発を必要とする。
【0004】
4つのタンパク質ドメイン−マトリクス(MA)、カプシド(CA)、ヌクレオカプシド(NC)およびp6−ならびに2つのスペーサーペプチド、SP1およびSP2から構成される、HIV Gagポリプロテイン前駆体(Pr55Gag)が新規の治療標的として表されている。Gagポリプロテインの開裂は、感染性ウイルス粒子産生の進行において重要な役割を果たすが、今まで、抗レトロウイルス薬はこの機構について認められていなかった。
【0005】
ほとんどの細胞種類において、集合は細胞膜で起こり、GagのMAドメインが膜結合を媒介する。集合は細胞由来の未成熟粒子の出芽により達成される。粒子放出と同時に、ウイルス的にコードされるPRが、4つの成熟タンパク質、MA、CA、NCおよびp6、ならびに2つのスペーサーペプチド、SP1およびSP2にGagを開裂する。Gag−PolはまたPRにより開裂され、ウイルス酵素PR、RTおよびINを遊離させる。Gagタンパク質分解処理は、成熟として知られている、粒子内の形態学的再配列を含む。成熟により、未成熟なドーナツ型の粒子が成熟ビリオンに変換され、その成熟ビリオンは、NCとウイルス酵素RTとINとの複合体においてウイルスRNAゲノムを囲むCAシェルから構成される凝縮した円錐形コアを含有する。成熟は新規細胞の感染のためのウイルスを作製し、粒子感染性に必要不可欠である。
【0006】
ベビリマット(bevirimat)(PA−457)は、感染性ウイルス粒子の形成に必要なGagの処理である、カプシド−SP1(p25)のカプシドへの変換において最終段階を阻害する成熟阻害剤である。ベビリマットは、ART耐性株および野生型HIVに対して活性を有し、全てのクラス由来の抗レトロウイルスとの相乗効果を示す。ベビリマットは、患者においてHIVウイルス負荷を平均1.3log10/mL減少させ、その患者は>=20μg/mLのトラフ濃度を達成し、Q369、V370またはT371において重要なベースラインのGag多形をいずれも有さなかった。しかしながら、Q369、V370またはT371においてGag多形を有するベビリマットの使用者は、それらの部位においてGag多形を有さない患者より顕著に低い負荷軽減を実証した。
【0007】
したがって、成熟阻害剤である代替の化合物を発見することは当該技術分野における進歩であろう。
【発明の概要】
【課題を解決するための手段】
【0008】
第1の態様において、本発明は、以下の式により特徴付けられる化合物またはその薬学上許容される塩である:
【化1】
【0009】
(式中、
R1およびR2は各々独立して、H、C1−C6−アルキル、t−ブチルオキシカルボニル、Me−SO2−、HOOCC(CH3)2CH2C(O)−、CH3C(O)、(R4)2N−(CH2)m−、(R5)n−フェニル−Q−、(R6)q−Hetアリール−(CH2)p−、(R6)q−Hetalk−(CH2)r−、または(R6)q−シクロalk−(CH2)p−であるか、あるいはR1およびR2は、それらが結合する窒素原子と一緒に、メチルスルホニル基または2個以下のC1−C4−アルキル基で置換されてもよい3〜7員のヘテロシクロアルキル環を形成し、
R3は、HOOCC(CH3)2CH2C(O)−またはHOOCCH2C(CH3)2CH2C(O)−であり、
各R4は独立してHまたはC1−C6−アルキルであり、
各R5は独立してハロ、C1−C6−アルキル、C1−C6−アルコキシ、CF3、OCF3、N(CH3)2、またはNO2であり、
各R6は独立してハロ、C1−C6−アルキル、−COOH、−C(O)NH2、ジメチルアミノメチル、または1−メチル−4−ピペラジニルメチルであり、
XおよびYは各々独立してメチレンまたはカルボニルであり、
Qは、−(CH2)p−、−C(O)−、−NH−C(O)−、−CH(CH3)−、−C(CH3)2−、1,1−シクロプロピルジイル、または1,1−シクロペンチルジイルであり、
Hetアリールは5〜6員のヘテロアリール基であり、
Hetalkは3〜7員のヘテロシクロアルキル基であり、
シクロalkは3〜6員のシクロアルキル基であり、
各mは独立して2または3であり、
各nは独立して0、1、または2であり、
各pは独立して0または1であり、
各qは独立して0、1、または2であり、かつ、
各rは独立して0、1、2、3、または4である)。
【0010】
第2の態様において、本発明は、a)式Iの化合物またはその薬学上許容される塩と、b)薬学上許容される賦形剤とを含んでなる、組成物に関する。
【0011】
第3の態様において、本発明は、HIV−1を治療する方法であって、それを患っている患者に、有効量の式Iの化合物、またはその薬学上許容される塩を投与することを含んでなる、方法である。
【0012】
本発明の化合物はHIV−1を有する患者の治療に有用である。
【発明を実施するための形態】
【0013】
本発明は、以下の式の化合物またはその薬学上許容される塩:
【化2】
【0014】
(式中、R1、R2、R3、XおよびYは上記に定義される通りである)
に関する。
【0015】
本明細書で使用する場合、C1−C6−アルキルとは、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、t−ブチル、n−ペンチル、およびn−ヘキシルを含む、直鎖または分枝アルキル基を指す。
【0016】
本明細書で使用するハロとは、フルオロ、クロロ、またはブロモを指す。
【0017】
ヘテロアリールとは、5〜6員のヘテロアリール基を指し、その例には、限定されないが、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、オキサゾリル、チアゾリル、イミダゾリル、チエニル、フリル、およびピロリルが含まれる。
【0018】
Hetalkとは、3〜7員のヘテロシクロアルキル基を指し、その例には、アゼチジニル、ピロリジニル、ピペリジニル、N−メチルピペリジン−4−イル、4−メチルピペラジニル、モルホリノ、およびチオモルホリノが含まれる。
【0019】
本明細書で使用するシクロalkとは、シクロプロピル、シクロブチル、シクロペンチル、およびシクロヘキシルを意味する。
【0020】
R1およびR2は、それらが結合する窒素原子と一緒に、ヘテロシクロアルキル基を形成でき、そのような基の例には、限定されないが、アゼチジニル、ピペリジニル、モルホリノ、チオモルホリノ、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、1−メチル−2−ピペラジノン−4−イル、チオモルホリン−1,1ジオキシド−4−イル、およびピロリジニル基が含まれる。
【0021】
本発明の別の態様において、Hetアリールは、ピリジル、チエニル、またはフリルである。
【0022】
別の態様において、R1は、H、メチル、ジメチルアミノエチル、t−ブチルオキシカルボニル、Me−SO2−、またはHOOCC(CH3)2CH2C(O)−であり、別の態様において、R1は、H、CH3、またはジメチルアミノエチルである。
【0023】
別の態様において、R2は、H、(R5)n−フェニル−Q−、(R6)q−フラニル−(CH2)p−、(R6)q−ピリジル−(CH2)p−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、Hetalk−(CH2)r−、またはC3−C6−シクロアルキル−(CH2)p−であり、別の態様において、R2は、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−である。
【0024】
別の態様において、R3は、HOOCC(CH3)2CH2C(O)−であり、
別の態様において、各R4はメチルであり、mは2であり、
別の態様において、各R5は独立してメチル、メトキシ、ハロ、CF3、またはOCF3であり、
別の態様において、各R6は独立してメチル、F、またはClであり、
別の態様において、XおよびYは両方カルボニルであり、
別の態様において、XおよびYは両方メチレンであり、
別の態様において、Xはカルボニルであり、Yはメチレンであり、
別の態様において、Xはメチレンであり、Xはカルボニルであり、
別の態様において、qは0または1である。
【0025】
本発明は、化合物およびそれらの薬学上許容される塩を含む。したがって、「化合物またはその薬学上許容される塩」の文脈において「または」という用語は、化合物またはその薬学上許容される塩(代替)、あるいは化合物およびその薬学上許容される塩(組合せ)のいずれかを指すものと理解される。
【0026】
本発明の化合物は、中間体1と本明細書に称される、ベツリンの誘導体である:
【化3】
【0027】
ベツリン(ルペ−20(29)−エン−3β,28−ジオール)は、樺の木の樹皮から一般的に単離される豊富に天然に存在するトリテルペンであり、ベツリンはその抽出物の30乾燥重量%までを形成する。Green,Brian;Bentley,Michael D.;Chung,Bong Y.;Lynch,Nicholas G.;Jensen,Bruce L.(1985),「Isolation of Betulin and Rearrangement to Allobetulin A Biomimetic Natural Product Synthesis」,J.Chem.Educ.200:7を参照のこと。
【0028】
本明細書で使用する場合、「薬学上許容される塩」という用語は、これらの化合物、物質、組成物、および剤形を指し、それらは、過剰な毒性、炎症、または他の問題もしくは合併症を生じずに、ヒトおよび動物の組織と接触させる使用に適切である十分な医学的判断の範囲内である。当業者は、式(I)に係る化合物の薬学上許容される塩が調製され得ることを理解するだろう。それらの薬学上許容される塩は、化合物の最終単離および精製の間にインサイチュで調製され得るか、またはその遊離酸もしくは遊離塩基形態で精製化合物をそれぞれの好適な塩基もしくは酸と別々に反応させることにより調製され得る。
【0029】
本発明の化合物は、好適な酸または塩基との反応により薬学上許容される塩を形成できる。好適な酸には無機酸および有機酸が含まれ、好適な無機酸の例には、塩酸、臭化水素酸、リン酸、メタリン酸、硝酸、および硫酸が含まれ、好適な有機酸の例には、酒石酸、酢酸、トリフルオロ酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、ギ酸、プロピオン酸、グリコール酸、グルコン酸、マレイン酸、コハク酸、メタンスルホン酸、エタンスルホン酸、ステアリン酸、ベンゼンスルホン酸、ブロモベンゼンスルホン酸、およびp−トルエンスルホン酸が含まれる。好適な塩基には、例えば、NaOH、KOH、Na2CO3、K2CO3、NaH、およびカリウム−t−ブトキシドを含む、水酸化物、炭酸塩、水素化物、およびアルコキシドが含まれる。
【実施例】
【0030】
上記の本発明の実施形態は単に例示であることを意図し、多数の変更および修飾が、当業者には明らかであろう。全てのこのような変更および修飾は、添付の特許請求の範囲に定義されるように本発明の範囲内であることが意図される。
【0031】
装置の説明
1H NMRスペクトルを、Bruker Avance−III400分光計で記録した。化学シフトは100万分の1(ppm、δ単位)で表す。結合定数はヘルツ(Hz)の単位である。分割パターンは見かけの多重度を表し、s(一重線)、d(二重線)、t(三重線)、q(四重線)、quint(五重線)、m(多重線)、br(広幅)と指定する。
【0032】
分析的低分解能質量分析(MS)は、勾配溶離法を用いるSunFire C18、4.6×50mm、3.5μmを用いてAgilent1200HPLC/6110またはAgilent1200HPLC/6130で記録した。
【0033】
溶媒A:水中の0.01%トリフルオロ酢酸(TFA);
溶媒B:アセトニトリル中の0.01%TFA;
1.2分間、一定のA、続いて4分にわたって5%〜95%または20%〜95%のB。
【0034】
生物学的アッセイ
試験化合物の抗ウイルス活性を、2つの細胞同時培養HIVライフサイクルアッセイにおいて決定した。このアッセイにおいて、HIV−1 HxB2に慢性的に感性させたJurkat T−リンパ球を、修飾されたHIV LTR−ルシフェラーゼレポーターを有するインジケーターHOS細胞と同時培養した。感染したJurkat細胞により産生されたウイルスは、ルシフェラーゼレポーターのLTR指令発現を導くHOS細胞に感染し得る。Jurkat細胞におけるウイルス産生、ウイルスの成熟、HIVライフサイクルにおける侵入または侵入後段階を妨げる化合物は、ルシフェラーゼシグナルを減少させる。
【0035】
アッセイを開始する前に、感染したJurkat、J4HxB2、細胞の凍結ストックバイアルを37℃の水槽中で急速に解凍させ、穏やかに攪拌しながら細胞培地(10%ウシ胎仔血清およびゲンタマイシンを含有するRPMI 1640培地)と共に15mLにゆっくりと希釈する。次いで細胞を、37℃、5%CO2にて培地中に入れ、細胞培地を添加することによって4日目に30mL、および7日目に60mLに拡張する。
【0036】
1日目に、アッセイを開始し、HOS細胞を急速に解凍させ、細胞培地中に15mLまでゆっくりと希釈し、1400rpmにて5分間遠心分離し、10mLの細胞培地中で再懸濁する。2×107HOS細胞を、冷却した(4℃)細胞培地中で1112mLに希釈し、攪拌プレート上に置いた。培地中の6.7×107J4HxB2細胞を、1400rpmにて5分間、遠心分離によりペレットにし、冷却したHOS細胞懸濁液中に再懸濁した。HOSおよびJ4HxB2細胞を、96または384ウェルプレート中に播種する前に少なくとも5分間、攪拌プレート上で混合した。マルチドロップ(Multidrop)(または同様の機器)を使用して、細胞を、試験化合物を含有するアッセイプレートに分配する。96ウェルアッセフォーマットにおいて、0.2mLのJ4HxB2/HOS細胞懸濁液を、アッセイプレート中の2μLの試験化合物に加えた。384ウェルアッセイフォーマットに関して、0.05mLの細胞懸濁液を、アッセイプレート中の0.5μLの試験化合物に加えた。化合物は10点または11点連続希釈として試験した。細胞を化合物のプレートに添加した後、プレートを室温にて30分〜1時間攪拌させ、次いで5日間、加湿した5%CO2 37℃のインキュベーターに移した。5日の終わりに、プレートをインキュベーターから取り除き、室温で30分〜1時間、平衡化した。製造業社の指示書に従ってPromega Steady−Glo試薬を調製し、マルチドロップ(または同様の機器)を使用してプレートに加えた。0.02mLのSteady−Gloを384ウェルプレートのウェルに加えた。96ウェルプレートに関して、0.1mLの培地を各ウェルから取り除き、0.06mLのSteady−Gloを添加した。次いで発光を、EnvisionもしくはTopcount Microplate Readerまたは同様の機器を用いて検出した。データ分析のために、DMSOまたはHIV−1阻害剤(エファビレンツ、ブレカナビルまたは同様の抗ウイルス薬)を含有するウェルに対応する高および低シグナル対照を使用してデータを正規化し、次いでそれを、適切なデータ解析パッケージを用いて非線形回帰モデルにフィットさせて、IC50値を求めた。
【0037】
スキームおよび実験手順
以下のスキームおよび手順は本発明の化合物がどのように調製できるかを例示する。参照されている特定の溶媒および反応条件もまた例示であり、限定することを意図するものではない。説明されていない化合物は、市販されているか、または入手可能な出発物質を用いて当業者により容易に調製できる。本明細書に開示される実施例は、例示のみの目的のためであり、本発明の範囲を限定することを意図するものではない。全ての実施例は、本明細書に開示されたアッセイを用いると、1μMから1nMのLHIV IC50値を示した。
【0038】
式Iの化合物についての中間体10の合成:
【化4】
【化5】
【0039】
工程A:中間体2
室温にて、CH2Cl2(DCM、100mL)中の中間体1(20g、45.2mmol)、4−ジメチルアミノピリジン(DMAP、1.66g、13.6mmol)、およびEt3N(63mL、136mmol)の溶液に、無水酢酸(Ac2O、17.1mL、113mmol)を加えた。それを一晩還流で加熱した後、室温まで冷却し、反応物を水(50mL)でクエンチした。次いで有機相を水(50mL×2)で洗浄し、硫酸ナトリウムで乾燥させた。減圧下でほとんどの有機溶媒を除去した後、無水エタノール(50mL)を加え、得られた沈殿物を、白色の固体(中間体2、20g、84%)として濾過により回収した。1H NMR(400MHz,CDCl3)δppm4.69(1H,m),4.59(1H,m),4.51−4.43(1H,m),4.25(1H,d,J=11.2Hz),3.85(1H,d,J=10.8Hz),2.49−2.40(1H,m),2.07(3H,s),2.04(3H,s),1.98−0.77(42H,m).LC/MS:m/z計算値526.4,実測値527.7(M+1)+。
【0040】
工程B:中間体3
酢酸(40mL、33%)中のHBrを、105℃にて予め加熱したトルエン(40mL)、Ac2O(40mL)、および酢酸(AcOH、40mL)中の中間体2(20g、38mmol)の懸濁液に加えた。反応混合物を攪拌し、この温度で1.5時間加熱した。冷却後、酢酸ナトリウム(24g)を加え、得られた反応混合物を蒸発乾固した。薄茶色の残渣をDCM(200mL)中に取り、有機相を水(100mL×3)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発乾固させて残渣を得て、それを95%エタノールおよびDCMから再結晶させて、白色の固体として中間体3(13.8g、69%)を得た。1H NMR(400MHz,CDCl3)δppm4.50−4.46(1H,m),4.02(1H,d,J=10.8Hz),3.98(1H,d,J=10.8Hz),3.18−3.10(1H,m),2.43−2.40(1H,m),2.26−2.22(2H,m),2.04(3H,s),2.05(3H,s),2.00−1.95(1H,m),1.90−1.85(1H,m),1.77−0.83(39H,m).LC/MS:m/z計算値526.4,実測値549.2(M+Na)+。
【0041】
工程C:中間体4
無水トルエン(90mL)、AcOH(119mL)、およびAc2O(29mL)中の中間体3(7g、13.29mmol)、酢酸ナトリウム(NaOAc、6.21g、76mmol)および二クロム酸ナトリウム二水和物(4.75g、15.95mmol)の混合物を60℃にて一晩攪拌した。冷却後、反応混合物を水(150mL)と酢酸エチル(EtOAc、250mL)との間に分配した。有機相を、水(100mL)、飽和炭酸ナトリウム溶液(100mL×2)およびブライン(100mL×2)で連続して洗浄し、硫酸ナトリウムで乾燥させ、減圧下で濃縮して、粘性のある油を得た。粘性のある油をメタノール(MeOH、250mL)で粉砕し、沈殿物を白色の固体として中間体4(6g、11.1mmol、83%収率)として回収した。1H NMR(400MHz,CDCl3)δppm4.52−4.46(1H,m),4.33(1H,d,J=10.8Hz),4.06(1H,d,J=11.2Hz),3.21−3.16(1H,m),2.86(1H,dd,J=12.8,3.2Hz),2.42−2.36(1H,m),2.05(3H,s),2.00(3H,s),1.94−0.84(40H,m).LC/MS:m/z計算値540.4,実測値563.3(M+Na)+。
【0042】
工程D:中間体5
1:1のエタノール(EtOH)およびトルエン(400mL)の混合物中の中間体4(7g、12.94mmol)および水酸化カリウム(KOH、0.872g、15.5mmol)の混合物を、室温にて1時間激しく攪拌した。反応混合物をHCl水溶液(1N)でpH7に中和し、蒸発乾固した。得た残渣を水および少量のアセトン中に取った。沈殿物を回収し、次いで水で洗浄し、真空中で乾燥させて、中間体5(6.0g、93%)を白色の固体として得た。LC/MS:m/z 計算値498.4、実測値499.3(M+1)+。
【0043】
工程E:中間体6
室温にてDCM(300mL)中の中間体5(5.1g、10.23mmol)の溶液に、クロロクロム酸ピリジニウム(PCC、6.61g、30.7mmol)、およびシリカゲル(6.6g)を加えた。反応混合物を室温にて1時間攪拌した。反応物を水でクエンチした後、有機相を飽和重炭酸ナトリウム溶液(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、粗生成物を得て、それをシリカゲル上でのカラムクロマトグラフィー(EtOAc:PE=1:10〜1:5)により精製して、白色の固体として中間体6(4.2g、83%)を得た。LC/MS:m/z 計算値496.4,実測値497.2(M+1)+。
【0044】
工程F:中間体7
−40℃にて窒素雰囲気下で、無水テトラヒドロフラン(THF、60mL)中の1,3−ジチアン(5.7g、47.4mmol)の溶液に、n−BuLi(27mL、67.5mmol)の溶液をゆっくりと加えた。反応混合物をさらに2時間、−20℃にて攪拌した後、無水THF(40mL)中の中間体6(4.2g、8.46mmol)の溶液を、−70℃にて窒素雰囲気下でゆっくりと加えた。次いで反応物を1時間、−78℃にて攪拌し、その後、それを飽和NaHCO3溶液でクエンチした。抽出をEtOAcで実施し、有機相を、水(50mL)、飽和ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、粗生成物を得て、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=8:1〜4:1)により精製して、中間体7(3.0g、5.22mmol、61.7%)を得た。LC/MS:m/z 計算値574.4,実測値575.0(M+1)+。
【0045】
工程G:中間体8
DCM(40mL)中の中間体7(3.5g、6.09mmol)、Et3N(2.55mL、18.26mmol)、およびDMAP(0.149g、1.218mmol)の溶液に、室温にてAc2O(3.45mL、36.5mmol)を加えた。50℃にて2時間、攪拌した後、反応物を水でクエンチした。有機相を水(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体8(3.41g、85%)を得た。LC/MS:m/z 計算値658.4,実測値659.1(M+1)+。
【0046】
工程H:中間体9
アセトニトリル(160mL)および水(40mL)中の中間体8(4.5g、6.83mmol)の溶液に、室温にてN−ブロモスクシンイミド(NBS、7.29g、41.0mmol)を加えた。室温にて10分間攪拌した後、反応物を水でクエンチした。有機層を飽和亜硫酸ナトリウム(200mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体9(3.2g、82%)を得た。LC/MS:m/z 計算値568.4,実測値569.3(M+1)+。
【0047】
工程I:中間体10
t−ブタノール(300mL)、THF(60mL)、および2−メチル−2−ブテン(2mL)中の中間体9(3.2g、5.63mmol)の氷冷した溶液に、水(60mL)中のNaClO2(7.19g、67.5mmol)およびNaH2PO4(6.75g、56.3mmol)の溶液を15分にわたってゆっくりと加えた。0℃にて10分間攪拌した後、反応混合物を室温まで加温し、さらに30分間攪拌し、続いてEtOAcで希釈した。有機相を水(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮乾固した。得られた残渣をシリカゲル上でのカラムクロマトグラフィー(DCM:MeOH=50:1〜10:1)により精製して、白色の固体として中間体10(2.8g、81%)を得た。LC/MS:m/z 計算値584.4,実測値585.3(M+1)+。
【0048】
以下の実施例は例示のみの目的であり、本発明の範囲を限定することを意図しない。以下の実施例1のスキームに関して、R1NHR2はアゼチジニル基を表し、R3はHOOCC(CH3)2CH2C(O)O−である。
【0049】
実施例1:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[1−アゼチジニル(オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化6】
【化7】
【0050】
工程A:中間体11
DCM(5mL)中の中間体10(680mg、1.163mmol)の溶液に、塩化オキサリル(3mL、35.5mmol)および数滴のDMFを加えた。反応混合物を室温にて3時間攪拌し、減圧下で蒸発させて、淡黄色の固体として中間体11を得た。
【0051】
工程B:中間体12−1
DCM(10mL)中の中間体11(430mg、0.713mmol)、DMAP(100.0mg、0.819mmol)およびEt3N(2mL、14.35mmol)の溶液に、室温にてアゼチジン(200mg、3.5mmol)を加えた。室温にて4時間攪拌した後、反応物をDCM(50mL)で希釈した。有機相を、水(50mL)、塩酸水溶液(2N、50mL)で洗浄し、次いで硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、粗生成物を得て、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=5:1〜1:1)により精製して、白色の固体として中間体12−1(300mg、67.5%)を得た。LC/MS:m/z 計算値623.4,実測値624.3(M+1)+。
【0052】
工程C:中間体13−1
1:1のEtOHおよびトルエン(12mL)の混合物中の中間体12−1(350mg、0.561mmol)およびKOH(19mg、0.353mmol)の混合物を、室温にて15分間激しく攪拌した。反応混合物をHCl水溶液(1N)でpH7に中和した。水を加えた後、反応物をEtOAcで抽出した。有機相を硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮して、中間体13−1(320mg、97%)を淡黄色の固体として得た。LC/MS:m/z 計算値581.4,実測値582.3(M+1)+。
【0053】
工程D:中間体14−1
ジメチルスルホキシド(DMSO、8mL)中の中間体13−1(430mg、0.74mmol)の溶液に、室温にて2−ヨードキシ安息香酸(IBX、1.1g、3.93mmol)を加えた。室温にて一晩攪拌した後、反応物を水でクエンチし、EtOAcで抽出した。有機相を硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮して残渣を得、それをシリカゲル上でのカラムクロマトグラフィー(PE:EA=5:1〜3:1)により精製して、白色の固体として中間体14−1(300mg、70%)を得た。LC/MS:m/z 計算値579.4,実測値580.3(M+1)+。
【0054】
工程E:中間体18−1
1,4−ジオキサン(15mL)および濃HCl(5mL)中の中間体14−1(300mg、0.52mmol)の溶液を室温にて一晩攪拌した。反応物を水(30mL)でクエンチし、EtOAc(60mL)で抽出した。有機相を水で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて残渣を得、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=5:1〜3:1)により精製して、白色の固体として中間体18−1(70mg、25%)を得た。LC/MS:m/z 計算値537.4,実測値538.3(M+1)+。
【0055】
工程F:化合物19−1
ピリミジン(3mL)中の中間体18−1(70mg、0.13mmol)の溶液に、3,3−ジメチルジヒドロ−2,5−フランジオン(159mg、1.302mmol)およびDMAP(334mg、2.6mmol)を加えた。それを90℃にて一晩加熱した後、反応混合物をDCMで抽出した。有機相をHCl(2N、25mL)、水(50mL×2)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて残渣を得、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAC=3:1〜2:1)により精製して、白色の固体の生成物として化合物19−1(30mg、34.6%)を得た。1H NMR(400MHz,CDCl3)δppm4.54−4.42(2H,m),4.28−4.35(1H,m),4.05−4.15(2H,m),3.17−3.25(1H,m),2.75−2.62(4H,m),2.60−2.52(2H,m),2.35(1H,quint,J=8.0Hz),2.17(1H,d,J=19.2Hz),2.10−2.00(1H,m),1.96−1.82(1H,m),1.78−0.78(42H,m).LC/MS:m/z 計算値665.9,実測値664.4(M−1)−。
【0056】
実施例2:2,2−ジメチル−4−オキソ−4−({(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−3a−[[4−(メチルスルホニル)−1−ピペラジニル](オキソ)アセチル]−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル}オキシ)ブタン酸
【化8】
【化9】
【0057】
中間体12−2
DMF(8mL)中の中間体10(600mg、1.026mmol)、ジイソプロピルエチルアミン(DIPEA、2g、15.47mmol)、および2−(1H−7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェートメタンアミニウム(HATU、1.4g、3.68mmol)の混合物に、室温にて1−(メチルスルホニル)ピペラジン(700mg、4.26mmol)を加えた。室温にて4時間攪拌した後、反応物を水(100mL)でクエンチし、EtOAc(200mL)で抽出した。有機相を硫酸ナトリウムで乾燥させ、減圧下で蒸発させて残渣を得て、それをシリカゲル上でのカラムクロマトグラフィー(DCM:MeOH=100:1〜30:1)により精製して、白色の固体として中間体12−2(650mg、87%)を得た。LC/MS:m/z 計算値730.4,実測値731.3(M+1)+。
【0058】
化合物19−2
化合物19−2を、実施例1(式中、R1NHR2はメチルスルホニルピペラジンである)に使用したものと同様の手順を用いて中間体12−2から白色の固体として調製した。1H NMR(400MHz,CDCl3)δppm4.54−4.46(1H,m),3.82−3.73(1H,m),3.68−3.53(2H,m),3.50−3.42(1H,m),3.36−3.18(5H,m),2.82(3H,s),2.78−2.50(5H,m),2.20(1H,d,J=18.4Hz),2.10−2.06(1H,m),1.90−0.82(43H,m).LC/MS:m/z 計算値772.4,実測値773.3(M+1)+。
【0059】
実施例3:2,2−ジメチル−4−オキソ−4−({(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−3a−[(4−メチル−1−ピペラジニル)(オキソ)アセチル]−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル}オキシ)ブタン酸
【化10】
【0060】
実施例2の化合物を調製するために使用したものと同様の手順に従って、淡黄色の固体として標題化合物を調製した。この場合、R1NHR2は1−メチルピペラジンである。1H NMR(400MHz,DMSO−d6)δppm4.38−4.32(1H,m),3.52−3.17(6H,m),2.60−2.20(9H,m),2.19(3H,s),1.98−0.79(44H,m).LC/MS:m/z 計算値709.0,実測値707.5(M−1)−。
【0061】
実施例4:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[[(4−フルオロフェニル)アミノ](オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化11】
【化12】
【0062】
工程A:中間体15
EtOH(2mL)およびトルエン(2mL)中の中間体10(500mg、0.855mmol)の溶液に室温にてKOH(192mg、3.42mmol)を加えた。室温にて30分攪拌後、反応物をDCM(100mL)で希釈した。有機層をNH4Cl水溶液(50mL×2)およびブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体15(400mg、86%)を得た。LC/MS:m/z計算値542.4,実測値543.1(M+1)+。
【0063】
工程B:中間体16
DMSO(6mL)中の中間体15(290mg、0.534mmol)の溶液に、室温にてIBX(1496mg、5.34mmol)を加えた。それを50℃にて3時間攪拌後、次いで反応混合物を室温まで冷却し、DCM(100mL)で希釈した。有機相を水(50mL×4)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体16(234mg、81%)を黄色固体として得た。LC/MS:m/z計算値540.4,実測値541.1(M+1)+。
【0064】
工程C:中間体17
DCM(8mL)中の中間体16(350mg、0.664mmol)の溶液に、窒素雰囲気下で0℃にて、塩化オキサリル(1.4mL、15.99mmol)を5分にわたってゆっくり加えた。それを室温にて1時間攪拌後、反応混合物を蒸発乾固して、中間体17を淡黄色の固体として得た。
【0065】
工程D:中間体14−4
DCM(7mL)中の4−フルオロアニリン(221mg、1.992mmol)およびEt3N(0.463mL、3.32mmol)の混合物に、中間体17(371mg、0.664mmol)を室温にて加えた。室温にて1時間攪拌後、反応混合物をDCM(50mL)で希釈した。有機相を水(50mL)、ブライン(25mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=5:1〜1:1)により精製することで、中間体14−4(150mg、36%)を白色の固体として得た。LC/MS:m/z計算値634.4,実測値635.3(M+1)+。
【0066】
化合物19−4
化合物19−4を、実施例1において使用したものと同様の手順で中間体14−4から黄色の固体として調製した。1HNMR(400MHz,CDCl3)δppm8.72(1H,s),7.61−7.57(2H,m),7.08−7.03(2H,m),4.52−4.48(1H,m),3.28−3.20(1H,m),2.78−2.53(5H,m),2.25(1H,d,J=18.8Hz),2.08−1.87(2H,m),1.18−0.81(42H,m).LC/MS:m/z計算値719.4,実測値720.3(M+1)+。
【0067】
実施例5:2,2−ジメチル−4−オキソ−4−[((3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3a−{オキソ[(2−チエニルメチル)アミノ]アセチル}−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル)オキシ]ブタン酸
【化13】
【化14】
【0068】
中間体14−5
DMF(10mL)中の中間体16(220mg、0.407mmol)、HATU(309mg、0.814mmol)、およびDIPEA(0.36mL、2.1mmol)の溶液に、(2−チエニルメチル)アミン(46mg、0.407mmol)を室温にて加えた。反応混合物を室温にて一晩攪拌し、DCMで希釈した。有機相を水(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=20:1〜4:1)により精製することで、中間体14−5(60mg、23%)を黄色の固体として得た。LC/MS:m/z計算値636.4,実測値637.3(M+1)+。
【0069】
化合物19−5
化合物19−5を、実施例1において使用したものと同様の手順で中間体14−5から黄色がかった固体として調製した。1HNMR(400MHz,CDCl3):δppm7.25(1H,dd,J=4.8Hz,1.6Hz),7.19(1H,t,J=5.6Hz),7.00−6.94(2H,m),4.66(1H,dd,J=15.2Hz,6.0Hz),4.57(1H,dd,J=15.2Hz,6.0Hz),4.54−4.47(1H,m),3.25−3.20(1H,m),2.76−2.52(5H,m),2.21(1H,d,J=18.8Hz),2.08−1.86(2H,m),1.78−0.81(42H,m).LC/MS:m/z計算値722.4,実測値723.3(M+1)+。
【0070】
実施例6:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[{[(4−クロロフェニル)メチル]アミノ}(オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化15】
【0071】
実施例2に記載されたものと同様の手順(ここで、R1NHR2は[(4−クロロフェニル)メチル]アミンである)を使用して、標題化合物を淡黄色の固体として得た。1HNMR(400MHz,CDCl3)δppm7.32−7.17(5H,m),4.54−4.32(3H,m),3.26−3.16(1H,m),2.76−2.48(5H,m),2.20(1H,d,J=18.8Hz),2.03−1.86(2H,m),1.77−0.81(43H,m).LC/MS:m/z計算値749.4,実測値748.4(M−1)−。
【0072】
実施例7:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[{[(2−クロロフェニル)メチル]アミノ}(オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化16】
【0073】
実施例2に記載されたものと同様の手順(ここで、R1NHR2は[(2−クロロフェニル)メチル]アミンである)を使用して、標題化合物を淡黄色の固体として得た。1HNMR(400MHz,CDCl3):δppm7.41−7.21(5H,m),4.61−4.44(3H,m),3.22−3.12(1H,m),2.76−2.46(5H,m),2.18(1H,d,J=19.2Hz),1.96−1.84(2H,m),1.78−0.81(43H,m).LC/MS:m/z計算値749.4,実測値748.4(M−1)−。
【0074】
表1に列挙された以下の化合物を、上記で列挙した方法と同様の方法を用いて調製した。各場合において、XおよびYは、それぞれカルボニルである。
【化17】
【表1−1】
【表1−2】
【表1−3】
【0075】
実施例8:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−({[(4−クロロフェニル)メチル]アミノ}アセチル)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化18】
【化19】
【0076】
工程A:中間体20
中間体6(300mg、0.604mmol)およびMeNO2(7.5mL、139mmol)の混合物に、Et3N(0.6mL,4.30mmol)を室温にて加えた。一晩攪拌後、反応物を水(1.0mL)でクエンチし、EtOAc(50mL)と水(25mL)との間に分配した。有機相を飽和ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固した。得られた残渣をシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=4:1)により精製することで、中間体20(335mg、99%)を白色の固体として得た。
【0077】
工程B:中間体21
MeOH(125mL)中のNiCl2・6H2O(0.929g、7.17mmol)の溶液に、水素化ホウ素ナトリウム(0.271g、7.17mmol)を0℃にて少量ずつ加えた。30分攪拌後、中間体20(2g,3.59mmol)を加え、その後追加の水素化ホウ素ナトリウム(2.44g、64.53mmol)を少量加えた。反応混合物を0℃でさらに30分攪拌し、濾過し、不溶性物質を除去した。濾液を真空内で濃縮乾固した。得られた残渣をEtOAc(100mL)中に溶解し、有機相を水(40mL)、ブライン(40mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固して、中間体21(1.79g、95%)をオフホワイトの固体として得た。LC/MS:m/z計算値527.3,実測値528.2(M+1)+。
【0078】
工程C:中間体22−1および23−1
MeOH(30mL)中の中間体21(450mg、0.853mmol)および4−クロロベンズアルデヒド(120mg、0.853mmol)の懸濁液に、ZnCl2(69.7mg、0.083mmol)を室温にて加えた。室温にて2時間攪拌後、シアノ水素化ホウ素ナトリウム(107mg、0.25mmol)を加え、得られた混合物をさらに2時間攪拌して、中間体22−1を得た。
【0079】
上述のように得た反応混合物を、Boc2O(0.297mL、1.279mmol)およびEt3N(0.238mL、1.705mmol)に加えた。室温にて一晩攪拌後、不溶性物質を濾過により除去し、濾液を減圧下で濃縮乾固した。得られた残渣をEtOAc(50mL)中に溶解し、有機相を水(25mL)、ブライン(25m)で洗浄し、硫酸マグネシウムで乾燥させ、真空内で蒸発乾固して、残渣を得て、これを分取TLCにより精製することで、中間体23−1(164mg,25.6%)を白色の固体として得た。
【0080】
工程D:中間体24−1
DCM(5mL)中の中間体23−1(130mg、0.173mmol)の溶液に、PCC(372mg、1.728mmol)およびシリカゲル(120mg)を室温にて加えた。室温にて8時間攪拌後、不溶性物質を濾過により除去し、濾液を減圧下で濃縮乾固した。得られた残渣をシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=1:6)により精製することで、中間体24−1(92mg、71%)を白色の固体として得た。LC/MS:m/z計算値785.4,実測値686.0(M−Boc+1)+。
【0081】
工程E:中間体25−1
1,4−ジオキサン(2.5mL)中の中間体24−1(28mg、0.037mmol)の溶液に、濃HCl(1.0mL、32.9mmol)を加えた。室温にて一晩攪拌後、反応混合物をEtOAc(50mL)で希釈し、飽和重炭酸ナトリウム溶液で中和し、EtOAc(50mL)と飽和重炭酸ナトリウム(25mL)との間に分配した。有機相を飽和重炭酸ナトリウム(25mL)、ブライン(25mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体25−1(22.3mg,98%)を無色の油として得た。LC/MS:m/z計算値607.3,実測値608.1(M+1)+。
【0082】
工程F:中間体26−1
DCM(5mL)中の中間体25−1(79mg、0.13mmol)およびBoc2O(28.3mg、0.13mmol)の溶液に、Et3N(18μL、0.13mmol)を加えた。室温にて1時間攪拌後、反応物を水(1.0mL)でクエンチし、EtOAc(25mL)と水(10mL)との間に分配した。有機相を飽和重炭酸ナトリウム(10mL)、ブライン(10mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=5:1)により精製して、中間体26−1(57mg、62%)を白色の泡状物として得た。
【0083】
工程G:中間体27−1
無水ピリジン(2mL)中の中間体26−1(26mg、0.037mmol)の溶液に、DMAP(22.42mg、0.184mmol)および3,3−ジメチルジヒドロ−2,5−フランジオン(47mg、0.367mmol)を加えた。それを80℃にて一晩攪拌後、反応混合物をEtOAc(30mL)で希釈した。有機相をHCl水溶液(2N、10mL)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc4:1)により精製して、中間体27−1(15mg、48.9%)を白色の泡状物として得た。LC/MS:m/z計算値835.4,実測値858.4(M+Na)+。
【0084】
工程H:化合物28−1
DCM(1.0mL)中の中間体27−1(27mg、0.032mmol)の溶液に、TFA(0.5mL、6.49mmol)を滴下して加えた。室温にて1時間攪拌後、反応混合物を重炭酸ナトリウム水溶液(25mL)で中和した。反応混合物をEtOAc(50mL)と重炭酸ナトリウム水溶液(25mL)との間に分配し、有機相をブライン(25mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、標題化合物28−1(9mg、29.5%)を得た。LC/MS:m/z計算値735.3,実測値736.3(M+1)+。
【0085】
実施例9:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−{[(2−フラニルメチル)アミノ]アセチル}−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化20】
【化21】
【0086】
中間体24−2
実施例8における記載に従って調製した、DMSO(10mL)中の中間体23−2(1.46g、1.24mmol)の溶液に、IBX(2g、7.14mmol)を加えた。それを60℃にて2時間攪拌後、反応混合物をEtOAc(50mL)で希釈した。有機相を水(30mL)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得た。これをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=100:0〜85:15)により精製して、中間体24−2(180mg、20.6%)を白色の固体として得た。LC/MS:m/z計算値727.3,実測値728.3(M+1)+。
【0087】
化合物28−2
化合物28−2を、実施例8において使用したものと同様の手順により、中間体24−2から白色の固体として調製した。1HNMR(400MHz,CD3OD)δppm7.54(1H,d,J=1.2Hz),6.52(1H,d,J=3.2Hz),6.41(1H,dd,J=3.2,2.0Hz),4.39(1H,dd,J=10.8,5.6Hz),4.21(2H,s),3.99(1H,d,J=18.0Hz),3.89(1H,d,J=18.0Hz),3.27−3.19(1H,m),2.54(1H,d,J=16.0Hz),2.46(1H,d,J=16.0Hz),2.49−2.39(2H,m),2.34(1H,d,J=19.2Hz),2.13(1H,d,J=19.2Hz),1.99−1.81(2H,m),1.73−1.18(13H,m),1.17(3H,s),1.16(3H,s),1.13(3H,s),1.11(3H,s),1.04−0.92(3H,m),0.96(3H,s),0.90(3H,s),0.85(3H,s),0.77(6H,s).LC/MS:m/z計算値691.3,実測値692.1(M+1)+。
【0088】
実施例10:2,2−ジメチル−4−オキソ−4−[((3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3a−{[(3−ピリジニルメチル)アミノ]アセチル}−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル)オキシ]ブタン酸
【化22】
【0089】
実施例8および9Aに記載されたものと同様の手順を用いて、標題化合物を黄色の固体として調製した。1HNMR(400MHz,CDCl3)δppm8.51−8.48(2H,m),7.68(1H,d,J=8.0H),7.22(1H,br),4.49(1H,dd,J=11.2,4.8Hz),3.71(2H,s),3.41(1H,d,J=18.4Hz),3.30(1H,d,J=18.4Hz),3.17−3.11(1H,m),2.68−0.74(52H,m).LC/MS:m/z計算値702.4,実測値703.3(M+1)+。
【0090】
実施例11:2−2,ジメチル−4−オキソ−4−({(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3a−[({[3−(トリフルオロメチル)フェニル]メチル}アミノ)アセチル]−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル}オキシ)ブタン酸
【化23】
【0091】
実施例8および9に記載されたものと同様の手順を用いて、標題化合物を調製した。1HNMR(400MHz,CD3OD)δppm7.86−7.66(4H,m),4.47(1H,dd,J=11.2,5.2Hz),4.31(2H,s),4.22(1H,d,J=17.6Hz),4.11(1H,d,J=17.6Hz),3.38−3.27(1H,m),2.64−0.87(52H,m).LC/MS:m/z計算値769.4,実測値770.3(M+1)+。
【0092】
実施例12:4−((3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル(isopstickyl)−5a,5b,8,8,11a−ペンタメチル−3a−(2−(4−メチルピペラジン−1−イル)アセチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化24】
【化25】
【0093】
工程A:中間体29
1:1のトルエン(20mL)およびEtOH(20mL)の混合物中の中間体8(863mg、1.31mmol)およびKOH(367mg、6.55mmol)の懸濁液を、室温にて1時間激しく攪拌した。それをHCl水溶液(1N)で中和した後、反応混合物を蒸発乾固した。得られた残渣をDCM(200mL)中に取り、有機相を水(200mL)、飽和重炭酸ナトリウム溶液(100mL×2)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(EtOAc:PE=1:30〜1:15)により精製して、中間体29(464mg、57.4%)を白色の固体として得た。LC/MS:m/z計算値616.3,実測値617.0(M+1)+。
【0094】
工程B:中間体30
アセトニトリル(20mL)および水(5mL)中のNBS(803mg、4.51mmol)の溶液に、中間体29(464mg、0.752mmol)を室温にて加えた。室温にて0.5時間攪拌後、反応物を亜硫酸ナトリウム水溶液でクエンチし、濃縮した。得られた残渣をエーテル(30mL)中に溶解し、有機相をブライン(15mL)で洗浄し、硫酸ナトリウムで乾燥させ、蒸発乾固して、中間体30(541mg、96%)を白色の固体として得た。LC/MS:m/z計算値526.3,実測値527.1(M+1)+。
【0095】
工程C:中間体31−1
MeOH(20mL)中の中間体30(450mg、0.598mmol)、1−メチルピペラジン(0.133mL、1.196mmol)、および塩化亜鉛(48.9mg、0.359mmol)の懸濁液を、70℃にて0.5時間加熱した。室温まで冷却後、シアノ水素化ホウ素ナトリウム(225mg、3.59mmol)を少量加えた。反応混合物を室温にてさらに17時間攪拌し、その後、それをEtOAc(50mL)で希釈した。有機相を飽和重炭酸ナトリウム水溶液(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて残渣を得て、これを分取−HPLCにより精製することで、中間体31−1(291mg、80%)を白色の固体として得た。LC/MS:m/z計算値610.3,実測値611.4(M+1)+。
【0096】
工程D:中間体32−1
DMSO(5mL)中の中間体31−1(50mg、0.082mmol)、およびIBX(68.8mg、0.246mmol)の溶液を、60℃にて4時間攪拌した。その後、反応混合物をEtOAc(30mL)で希釈し、有機相を飽和重炭酸ナトリウム(15mL)、水(15mL)、ブライン(15mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体32−1(42mg、84%)を白色の泡状物として得た。LC/MS:m/z計算値608.3,実測値609.3(M+1)+。
【0097】
工程E:中間体33−1
1,4−ジオキサン(4mL)および濃HCl(2mL、65.8mmol)中の中間体32−1(42mg、0.069mmol)の溶液を、室温にて一晩攪拌した。その後、反応混合物を蒸発乾固した。得られた残渣をEtOAc(30mL)中に溶解し、有機相を飽和重炭酸ナトリウム(15mL)、ブライン(15mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体33−1(42mg,97%)を白色の泡状物として得た。LC/MS:m/z計算値566.3,実測値567.4(M+1)+。
【0098】
工程F:化合物28−5
ピリジン(0.8mL)中の中間体33−1(80mg、0.141mmol)、3,3−ジメチル−ジヒドロフラン−2,5−ジオン(362mg、2.82mmol)およびDMAP(86mg、0.706mmol)の溶液を、窒素雰囲気下で100℃にて3時間攪拌した。室温まで冷却後、反応混合物をEtOAc(30mL)で希釈した。有機相をブラインで洗浄し、硫酸ナトリウムで乾燥させ、蒸発乾固した。得られた残渣を分取−HPLCにより精製することで、化合物28−5(50mg、38%)を白色の泡状物として得た。1HNMR(400MHz,CD3OD)δppm4.50(1H,dd,J=10.8,5.6Hz),3.62−3.40(2H,m),3.38−3.20(4H,m),2.99−2.81(6H,m),2.68−2.53(4H,m),2.40(1H,d,J=19.2Hz),2.15(1H,d,J=19.2Hz),2.10−1.93(2H,m),1.83−0.87(44H,m).LC/MS:m/z計算値694.3,実測値695.4(M+1)+。
【0099】
表2中の化合物を、上記で示した手順と同様の方法で調製した。各場合において、Xはカルボニルであり、YはCH2である。
【化26】
【表2−1】
【表2−2】
【表2−3】
【表2−4】
【0100】
実施例13:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(4−クロロベンジルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化27】
【化28】
【0101】
工程A:中間体34
EtOH(100mL)およびトルエン(100mL)中の中間体3(5g、7.59mmol)の溶液に、KOH(0.51g、9.11mmol)を加えた。室温にて4時間攪拌後、次いで反応混合物を水(500mL)とEtOAc(500mL)との間に分配した。有機相を水(200mL×3)、ブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=6:1〜4:1)により精製して、中間体34(2.5g,67.9%)を白色の固体として得た。1HNMR(400Hz,CDCl3)δppm4.50−4.67(1H,m),3.68(1H,d,J=10.4Hz),3.32(1H,d,J=10.4Hz),3.23−3.15(1H,m),2.42−2.28(3H,m),2.05(3H,s),2.02−1.89(2H,m),1.77−0.83(40H,m)。
【0102】
工程B:中間体35
DCM(75mL)中の中間体34(3g、6.19mmol)の溶液に、室温にてPCC(4g、18.57mmol)およびシリカゲル(3.0g)を加えた。室温にて2時間攪拌後、反応物を水(100mL)でクエンチした。有機相を飽和重炭酸ナトリウム(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で濃縮して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=10:1)により精製して、中間体35(3g、100%)を白色の固体として得た。1HNMR(400Hz,CDCl3)δppm9.43(1H,s),4.50−4.46(1H,m),3.25−3.21(1H,m),2.43−2.02(5H,m),2.04(3H,m),2.00−1.93(1H,m),1.75−0.81(38H,m)。
【0103】
工程C:中間体36
MeNO2(128mL、2382mmol)中の中間体36(5g、10.36mmol)の溶液に、Et3N(10.11mL、72.5mmol)を加えた。それを60℃にて一晩攪拌後、反応混合物を水とEtOAc(各100mL)との間に分配した。有機相を水(20mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(ヘキサン:EtOAc=10:1〜6:1)により精製して、中間体36(2.8g、49.7%)を白色の粉末として得た。LC/MS:m/z計算値543.4,実測値566.3(M+Na+)+。
【0104】
工程D:中間体37
MeOH(166mL)中の中間体36(2.8g、5.15mmol)の溶液に、塩化ニッケル(II)(1.67g、12.87mmol)および水素化ホウ素ナトリウム(4.87g、129mmol)を0℃にて加えた。0℃にて10分攪拌後、反応混合物を水とEtOAc(各200mL)との間に分配し、有機相を水(100mL×3)、ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、中間体37(2.65g、100%)を固体として得た。LC/MS:m/z計算値513.4,実測値514.3(M+1)+。
【0105】
工程E:中間体39
MeOH(15mL)およびジクロロエタン(DCE、15mL)中の中間体37(350mg、0.613mmol)および4−クロロベンズアルデヒド(86mg、0.613mmol)の溶液に、塩化亜鉛(50.1mg、0.368mmol)を加えた。反応混合物を80℃にて1時間攪拌後、室温まで冷却し、シアノ水素化ホウ素ナトリウム(57.8mg、0.92mmol)を加えた。得られた混合物を室温にてさらに1時間攪拌して、中間体38を得た。
【0106】
上記の得られた反応混合物に、Et3N(0.18mL、1.38mmol)およびジ−tert−ブチルジカーボネート(0.157mL、0.674mmol)を加えた。室温にて30分攪拌後、反応混合物を水(20mL)とEtOAc(100mL)との間に分配した。有機相を水(30mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=15:1)により精製して、中間体39(125mg,27.6%)を白色の固体として得た。
【0107】
工程F:中間体40
DCM(10mL)中の中間体39(120mg、0.162mmol)の溶液に、PCC(35mg、0.162mmol)およびシリカゲル(100mg)を加えた。室温にて2時間攪拌後、不溶性物質を濾過により除去し、濾液を濃縮して、中間体40(110mg、92%)を白色の固体として得た。
【0108】
工程G:中間体41
MeOH(1mL)、THF(1mL)、および水(0.5mL)中のNaOH(597mg、14.94mmol)の溶液に、中間体40(110mg、0.149mmol)を加えた。室温にて1時間攪拌後、反応物を水(20mL)で希釈し、EtOAc(50mL)で抽出した。有機相をブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体41(100mg、96%)を白色の固体として得た。
【0109】
工程H:中間体42
無水ピリジン(2mL)中の中間体41(100mg、0.144mmol)の溶液に、DMAP(106mg、0.864mmol)および3,3−ジメチルジヒドロ−2,5−フランジオン(369mg、2.88mmol)を加えた。反応混合物を80℃にて一晩加熱後、溶媒を真空内で除去し、残渣をDCM(50mL)中に取った。有機相をHCl水溶液(0.5N,20mL)、水(2×30mL)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させて、真空内で蒸発させて、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=15:1)により精製することで、中間体42(56mg、44.9%)を白色の泡状物として得た。
【0110】
工程I:化合物43−1
DCM(1mL)中の中間体42(45mg、0.055mmol)の溶液に、TFA(0.5mL、6.49mmol)を加えた。室温にて1時間攪拌後、溶媒を除去して、残渣を得て、これを分取−HPLCにより精製することで、化合物43−1(38mg、77%)を白色の固体として得た。1HNMR(400Hz,CD3OD)δppm7.42−7.36(4H,m),4.40−4.36(1H,m),4.15(2H,dd,J=16.8,13.2Hz),3.93(1H,d,J=17.6Hz),3.85(1H,d,J=17.6Hz),3.19−3.14(m,1H),2.57−1.98(5H,m),1.97−0.77(48H,m).LC/MS:m/z計算値721.4,実測値722.1(M+1)+。
【0111】
実施例14:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(4−フルオロベンジルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化29】
【0112】
実施例13に記載されたものと同様の方法を用いて標題化合物を白色の固体として調製した。1HNMR(400Hz,MeOD)δppm7.55−7.53(2H,m),7.24−7.20(2H,m),4.50−4.40(1H,m),4.27−4.24(2H,dd,J=11.6,11.2Hz),4.03−3.98(2H,dd,J=18.8,14.4Hz),3.31−3.25(1H,m),2.67−2.46(4H,m),2.34−2.31(1H,m),2.12−0.88(48H,m).LC/MS:m/z計算値705.4,実測値706.4(M+1)+。
【0113】
実施例15:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(フラン−2−イルメチルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化30】
【0114】
実施例13に記載されたものと同様の方法を用いて標題化合物を白色の固体として調製した。1HNMR(400Hz,MeOD)δppm7.55(1H,br),6.54(1H,d,J=3.2Hz),6.43−6.42(1H,m),4.40−4.36(1H,m),4.23(2H,s),3.87(2H,s),3.19−3.16(1H,m),2.57−2.22(5H,m),2.05−0.77(48H,m).LC/MS:m/z計算値677.4,実測値678.4(M+1)+。
【0115】
実施例16:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(シクロペンチルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化31】
【0116】
実施例13に記載されたものと同様の方法を用いて標題化合物を白色の固体として調製した。1HNMR(400Hz,MeOD)δppm4.41−4.37(1H,m),3.88(2H,br),3.47(1H,br),3.22−3.17(1H,m),2.57−2.45(4H,m),2.22(1H,d,J=13.2Hz),2.09(1H,d,J=13.6Hz),1.99−0.77(55H,m).LC/MS:m/z計算値665.5,実測値666.4(M+1)+。
【0117】
実施例17:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3a−(2−(4−メチルピペラジン−1−イル)アセチル)−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化32】
【化33】
【0118】
工程A:中間体44
無水THF(50mL)中の水素化ナトリウム(60%、250mg、6.25mmol)の懸濁液に、窒素雰囲気下でヨウ化トリメチルスルホキソニウム(900mg、4.09mmol)を室温にて加えた。反応混合物を2時間還流後、60℃まで冷却し、無水THF(2mL)中の中間体35(750mg、1.554mmol)を滴下して加えた。得られた混合物を60℃にて3時間攪拌し、次いで室温にて1時間攪拌し、その後、反応物を飽和重炭酸ナトリウム水溶液(50mL)でクエンチした。反応混合物をDCM(200mL)で希釈し、有機相を水(50mL×3)、飽和ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=10:1)により精製して、中間体44(300mg、42.5%)を白色の固体として得た。
【0119】
工程B:中間体45
DCM(10mL)中の中間体44(300mg、0.66mmol)の混合物に、DMAP(7.25mg、0.059mmol)、無水酢酸(0.182mL、1.781mmol)およびEt3N(0.494mL、3.56mmol)を加えた。室温にて1時間攪拌後、反応混合物を氷水(20mL)で希釈し、DCM(100mL)で抽出した。有機相を水(40mL×3)、ブライン(40mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、淡黄色の固体を得た。その固体をMeOH(5mL)中に取り、沈殿物を回収し、冷MeOH(5mL)でリンスして、中間体45(250mg、85%)を白色の固体として得た。
【0120】
工程C:化合物46
1−メチルピペラジン(5mL)中の中間体45(250mg、0.503mmol)の溶液に、酢酸(0.014mL、0.252mmol)を加えた。それを150℃にて一晩攪拌後、室温まで冷却し、反応混合物を水(50mL)とDCM(100mL)との間に分配した。次いで有機相を分け、飽和塩化アンモニウム(40mL)、飽和重炭酸ナトリウム(40mL)、水(100mL×3)、ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して中間体46(270mg、84%)を淡黄色の固体として得た。LC/MS:m/z計算値596.5,実測値597.4(M+1)+。
【0121】
工程D:中間体47
DMSO(20mL)中のIBX(1.178g、4.21mmol)の溶液に、中間体46(270mg、0.421mmol)を加えた。それを50℃にて1時間攪拌後、反応混合物を水(150mL)とDCM(50mL)との間に分配した。有機相を分け、水(20mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、中間体47(200mg、80%)を淡黄色の固体として得た。LC/MS:m/z計算値594.5,実測値595.4(M+1)+。
【0122】
工程E:中間体48
MeOH(3mL)、THF(3mL)、および水(1.5mL)中の中間体47(200mg、0.336mmol)の溶液に、NaOH(1076mg、26.9mmol)を加えた。室温にて1時間攪拌後、反応混合物を水(150mL)とDCM(100mL)との間に分配した。有機相を分け、水(20mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、中間体48(77.7mg、37.7%)をオフホワイトの固体として得た。LC/MS:m/z計算値552.5,実測値553.2(M+1)+。
【0123】
工程F:化合物43−5
無水ピリジン(3mL)中の中間体48(100mg、0.181mmol)の溶液に、3,3−ジメチル−ジヒドロフラン−2,5−ジオン(232mg、1.809mmol)およびDMAP(66.3mg,0.543mmol)を加えた。反応混合物を120℃にてマイクロ波下で1.5時間加熱し、溶媒を真空内で除去し、残渣をEtOAc(50mL)中に取った。有機相をHCl水溶液(0.5N、20mL)、水(30mL×2)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、化合物43−5(32mg、18.74%)を白色の固体として得た。1HNMR(400Hz,MeOD)δppm4.51−4.47(1H,m),3.75−3.65(2H,t),3.32(4H,br),3.31−3.27(1H,m),3.05(4H,br),2.91(3H,s),2.67−2.55(4H,m),2.32−2.28(1H,m),2.20−2.17(1H,m),2.06−2.03(1H,m),1.95−0.88(45H,m).LC/MS:m/z計算値680.5,実測値681.4(M+1)+。
【0124】
表3中の化合物を、上記で示した手順と同様の方法で調製した。各場合において、XおよびYは、それぞれCH2である。
【化34】
【表3−1】
【表3−2】
【表3−3】
【0125】
実施例18:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3a−(2−(4−メチルピペラジン−1−イル)−2−オキソアセチル)−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化35】
【化36】
【0126】
工程A:中間体49
無水THF(50mL)中の1,3−ジアチン(4g、33.2mmol)の溶液に、n−BuLi(THF中2.5M)(18mL、45mmol)を−20℃にてアルゴン雰囲気下でゆっくりと加えた。得られた混合物を−20℃にて2時間攪拌した。−78℃まで冷却後、無水THF(5mL)中の中間体35(3g、6.21mmol)を反応混合物に加え、得られた混合物を−70℃にてさらに1時間攪拌し、その後反応物、重炭酸ナトリウム(10mL)の10%溶液を添加することでクエンチした。反応混合物をDCM(100mL)で希釈し、有機相を分け、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロトマグラフィー(Hex:EtOAc=10:1)により精製することで、中間体49(1.7g、48.8%)を白色の固体として得た。LC/MS:m/z計算値560.3,実測値583.3(M+Na)+。
【0127】
工程B:中間体50
DCM(100mL)中の中間体49(1g、1.78mmol)、Et3N(0.743mL、5.35mmol)およびDMAP(65.3mg、0.535mmol)の溶液に、無水酢酸(0.184mL、1.961mmol)を加えた。それを0℃にて4時間攪拌後、反応混合物を水(50mL)とDCM(100mL)との間に分配した。有機相を分け、水、ブラインで洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して残渣を得て、これをEtOHおよびDCMから再結晶化して、中間体50(510mg、47.4%)を白色の固体として得た。LC/MS:m/z計算値602.9,実測値625.3(M+Na)+。
【0128】
工程C:中間体51
EtOH(45mL)および水(5mL)中の中間体50(2g、3.32mmol)の溶液に、硝酸銀(5.63g、33.2mmol)を加えた。反応混合物を60℃にて一晩攪拌後、不溶性物質を濾過で除去し、DCM(100mL×3)で洗浄した。有機相を水(100mL×3)、ブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させた。減圧下で蒸発させて、中間体51(1.5g、88%)を黄色の固体として得た。LC/MS:m/z計算値512.8,実測値535.2(M+Na)+。
【0129】
工程D:中間体52
DMSO(25mL)中の中間体51(700mg、1.37mmol)の溶液に、IBX(3823mg、13.65mmol)を加えた。それを30℃にて1時間攪拌後、反応混合物を水(20mL)とDCM(300mL)との間に分配した。次いで有機相を分け、水、ブラインで洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、中間体52(370mg、51.5%)を黄色の固体として得た。LC/MS:m/z計算値526.3,実測値525.3(M−1)−。
【0130】
工程E:中間体53
DCM(10mL)中の中間体52(260mg、0.494mmol)の溶液に、シュウ酸ジクロライド(0.418mL、4.94mmol)を加えた。室温にて1時間攪拌後、反応混合物を真空内で蒸発させて、中間体53(269mg、100%)を黄色の固体として得た。
【0131】
工程F:中間体54
DCM(10mL)中の中間体53(270mg、0.495mmol)、およびEt3N(551mg、5.45mmol)の溶液に、1−メチルピペラジン(496mg、4.95mmol)を加えた。室温にて1時間攪拌後、反応混合物を水(20mL)とDCM(150mL)との間に分配した。有機相を分け、水(20mL)、ブライン(10mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固させて、中間体54(220mg、73%)を黄色の固体として得た。LC/MS:m/z計算値608.4,実測値609.4(M+1)+。
【0132】
工程G:中間体55
THF(2mL)、MeOH(2mL)、および水(1mL)中の中間体54(220mg、0.361mmol)の溶液に、NaOH(1.1g、28.9mmol)を加えた。室温にて1時間攪拌後、反応混合物を水(20mL)とDCM(150mL)との間に分配した。有機相を分け、水(20mL)、ブライン(10mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体55(150mg、73.2%)を黄色の固体として得た。LC/MS:m/z計算値566.4,実測値567.3(M+1)+。
【0133】
工程H:化合物56−1
ピリジン(2mL)中の中間体55(678mg、5.29mmol)およびDMAP(97mg、0.794mmol)の溶液に、3,3−ジメチル−ジヒドロフラン−2,5−ジオン(678mg、5.29mmol)を加えた。反応混合物を120℃にて3時間攪拌後、溶媒を真空内で除去し、残渣をEtOAc(50mL)中に取った。有機相をHCl水溶液(0.5N、20mL)、水(30mL×2)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、標題化合物56−1(30mg、14.01%)を白色の固体として得た。1HNMR(400MHz,MeOD)δppm:4.51−4.47(1H,m),3.72−3.35(8H,m),3.29−3.22(1H,m),2.98(3H,s),2.61(2H,q,J=16Hz),2.50−0.88(50H,m).LC/MS:計算値694.5,実測値695.4(M+1)+。
【0134】
実施例19:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3a−(2−オキソ−2−(2−フェニルプロパン−2−イルアミノ)アセチル)−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化37】
【0135】
実施例18に記載されたものと同様の手順を用いて標題化合物を調製した。1HNMR(400MHz,MeOD)δppm:8.23(1H,s),7.29−7.28(2H,m),7.21−7.17(2H,m),7.11−7.07(1H,m),4.40−4.36(1H,m),3.65−3.44(1H,m),3.11−3.04(1H,m),2.50(2H,q,J=16.4Hz),2.40−2.05(6H,m),1.91−0.77(49H,m).LC/MS:計算値729.5,実測値m/z730.3(M+1)+。
【0136】
実施例20:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−((4−クロロベンジル)(メチル)アミノ)−2−オキソアセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化38】
【0137】
実施例18に記載されたものと同様の手順を用いて標題化合物を調製した。1HNMR(400MHz,CDCl3)δppm:7.32−7.25(3H,m),7.18(1H,m),4.67−4.24(3H,m),3.23−3.19(1H,m),2.86(2H,2/3CH3,s),2.79(1H,1/3CH3,s),2.63(1H,d,J=15.6Hz),2.58(1H,d,J=15.6Hz),2.44−0.79(50H,m).LC/MS:計算値749.44,実測値772.5(M+Na)+。
【技術分野】
【0001】
本発明はベツリンの誘導体に関する。
【背景技術】
【0002】
現在、抗レトロウイルス薬を用いるウイルス複製の長期間の抑制は、HIV−1感染を治療するための唯一の選択である。今までに、多くの承認された薬物が患者の生存期間を非常に延長させることが示されている。しかしながら、非常に活性のある抗レトロウイルス治療(HAART)として知られている治療レジメンはしばしば複雑である。なぜなら、薬物耐性HIV−1変異体の急速出現を回避するために、異なる薬物の組合せが患者に投与されなければならないからである。患者の生存に対するHAARTの効果のある作用にも関わらず、薬物耐性が生じる可能性もある。
【0003】
多剤耐性(MDR)HIV−1単離株の出現は重大な臨床的帰結を有し、サルベージ療法として知られている、新規投薬レジメンを用いて抑制されなければならない。現在の指針は、サルベージ療法が少なくとも2つ、好ましくは3つの完全に活性な薬物を含むことを推奨している。典型的に、第1の治療は、ウイルス酵素RTおよびプロテアーゼ(PR)を標的とする3〜4個の薬物を組合わせる。サルベージ療法の1つの選択は、耐性分離株に対して活性を維持する同じ機構的クラス由来の薬物の異なる組合せを投与することである。しかしながら、このアプローチの選択はしばしば、同じクラスにおける異なる薬物に広範な交差耐性を頻繁に与える耐性変異として制限される。現在、融合、侵入、およびインテグラーゼ(IN)阻害剤の開発による代替の治療ストラテジーが利用可能となっている。しかしながら、3つ全ての新規薬物クラスに対する耐性がインビトロおよびインビボの両方において既に報告されている。したがって、抗レトロウイルス薬物を用いるHIV−1感染患者の持続した首尾良い治療は、新しい標的および作用機構を有する新規および改良された薬物の継続した開発を必要とする。
【0004】
4つのタンパク質ドメイン−マトリクス(MA)、カプシド(CA)、ヌクレオカプシド(NC)およびp6−ならびに2つのスペーサーペプチド、SP1およびSP2から構成される、HIV Gagポリプロテイン前駆体(Pr55Gag)が新規の治療標的として表されている。Gagポリプロテインの開裂は、感染性ウイルス粒子産生の進行において重要な役割を果たすが、今まで、抗レトロウイルス薬はこの機構について認められていなかった。
【0005】
ほとんどの細胞種類において、集合は細胞膜で起こり、GagのMAドメインが膜結合を媒介する。集合は細胞由来の未成熟粒子の出芽により達成される。粒子放出と同時に、ウイルス的にコードされるPRが、4つの成熟タンパク質、MA、CA、NCおよびp6、ならびに2つのスペーサーペプチド、SP1およびSP2にGagを開裂する。Gag−PolはまたPRにより開裂され、ウイルス酵素PR、RTおよびINを遊離させる。Gagタンパク質分解処理は、成熟として知られている、粒子内の形態学的再配列を含む。成熟により、未成熟なドーナツ型の粒子が成熟ビリオンに変換され、その成熟ビリオンは、NCとウイルス酵素RTとINとの複合体においてウイルスRNAゲノムを囲むCAシェルから構成される凝縮した円錐形コアを含有する。成熟は新規細胞の感染のためのウイルスを作製し、粒子感染性に必要不可欠である。
【0006】
ベビリマット(bevirimat)(PA−457)は、感染性ウイルス粒子の形成に必要なGagの処理である、カプシド−SP1(p25)のカプシドへの変換において最終段階を阻害する成熟阻害剤である。ベビリマットは、ART耐性株および野生型HIVに対して活性を有し、全てのクラス由来の抗レトロウイルスとの相乗効果を示す。ベビリマットは、患者においてHIVウイルス負荷を平均1.3log10/mL減少させ、その患者は>=20μg/mLのトラフ濃度を達成し、Q369、V370またはT371において重要なベースラインのGag多形をいずれも有さなかった。しかしながら、Q369、V370またはT371においてGag多形を有するベビリマットの使用者は、それらの部位においてGag多形を有さない患者より顕著に低い負荷軽減を実証した。
【0007】
したがって、成熟阻害剤である代替の化合物を発見することは当該技術分野における進歩であろう。
【発明の概要】
【課題を解決するための手段】
【0008】
第1の態様において、本発明は、以下の式により特徴付けられる化合物またはその薬学上許容される塩である:
【化1】
【0009】
(式中、
R1およびR2は各々独立して、H、C1−C6−アルキル、t−ブチルオキシカルボニル、Me−SO2−、HOOCC(CH3)2CH2C(O)−、CH3C(O)、(R4)2N−(CH2)m−、(R5)n−フェニル−Q−、(R6)q−Hetアリール−(CH2)p−、(R6)q−Hetalk−(CH2)r−、または(R6)q−シクロalk−(CH2)p−であるか、あるいはR1およびR2は、それらが結合する窒素原子と一緒に、メチルスルホニル基または2個以下のC1−C4−アルキル基で置換されてもよい3〜7員のヘテロシクロアルキル環を形成し、
R3は、HOOCC(CH3)2CH2C(O)−またはHOOCCH2C(CH3)2CH2C(O)−であり、
各R4は独立してHまたはC1−C6−アルキルであり、
各R5は独立してハロ、C1−C6−アルキル、C1−C6−アルコキシ、CF3、OCF3、N(CH3)2、またはNO2であり、
各R6は独立してハロ、C1−C6−アルキル、−COOH、−C(O)NH2、ジメチルアミノメチル、または1−メチル−4−ピペラジニルメチルであり、
XおよびYは各々独立してメチレンまたはカルボニルであり、
Qは、−(CH2)p−、−C(O)−、−NH−C(O)−、−CH(CH3)−、−C(CH3)2−、1,1−シクロプロピルジイル、または1,1−シクロペンチルジイルであり、
Hetアリールは5〜6員のヘテロアリール基であり、
Hetalkは3〜7員のヘテロシクロアルキル基であり、
シクロalkは3〜6員のシクロアルキル基であり、
各mは独立して2または3であり、
各nは独立して0、1、または2であり、
各pは独立して0または1であり、
各qは独立して0、1、または2であり、かつ、
各rは独立して0、1、2、3、または4である)。
【0010】
第2の態様において、本発明は、a)式Iの化合物またはその薬学上許容される塩と、b)薬学上許容される賦形剤とを含んでなる、組成物に関する。
【0011】
第3の態様において、本発明は、HIV−1を治療する方法であって、それを患っている患者に、有効量の式Iの化合物、またはその薬学上許容される塩を投与することを含んでなる、方法である。
【0012】
本発明の化合物はHIV−1を有する患者の治療に有用である。
【発明を実施するための形態】
【0013】
本発明は、以下の式の化合物またはその薬学上許容される塩:
【化2】
【0014】
(式中、R1、R2、R3、XおよびYは上記に定義される通りである)
に関する。
【0015】
本明細書で使用する場合、C1−C6−アルキルとは、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、t−ブチル、n−ペンチル、およびn−ヘキシルを含む、直鎖または分枝アルキル基を指す。
【0016】
本明細書で使用するハロとは、フルオロ、クロロ、またはブロモを指す。
【0017】
ヘテロアリールとは、5〜6員のヘテロアリール基を指し、その例には、限定されないが、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、オキサゾリル、チアゾリル、イミダゾリル、チエニル、フリル、およびピロリルが含まれる。
【0018】
Hetalkとは、3〜7員のヘテロシクロアルキル基を指し、その例には、アゼチジニル、ピロリジニル、ピペリジニル、N−メチルピペリジン−4−イル、4−メチルピペラジニル、モルホリノ、およびチオモルホリノが含まれる。
【0019】
本明細書で使用するシクロalkとは、シクロプロピル、シクロブチル、シクロペンチル、およびシクロヘキシルを意味する。
【0020】
R1およびR2は、それらが結合する窒素原子と一緒に、ヘテロシクロアルキル基を形成でき、そのような基の例には、限定されないが、アゼチジニル、ピペリジニル、モルホリノ、チオモルホリノ、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、1−メチル−2−ピペラジノン−4−イル、チオモルホリン−1,1ジオキシド−4−イル、およびピロリジニル基が含まれる。
【0021】
本発明の別の態様において、Hetアリールは、ピリジル、チエニル、またはフリルである。
【0022】
別の態様において、R1は、H、メチル、ジメチルアミノエチル、t−ブチルオキシカルボニル、Me−SO2−、またはHOOCC(CH3)2CH2C(O)−であり、別の態様において、R1は、H、CH3、またはジメチルアミノエチルである。
【0023】
別の態様において、R2は、H、(R5)n−フェニル−Q−、(R6)q−フラニル−(CH2)p−、(R6)q−ピリジル−(CH2)p−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、Hetalk−(CH2)r−、またはC3−C6−シクロアルキル−(CH2)p−であり、別の態様において、R2は、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−である。
【0024】
別の態様において、R3は、HOOCC(CH3)2CH2C(O)−であり、
別の態様において、各R4はメチルであり、mは2であり、
別の態様において、各R5は独立してメチル、メトキシ、ハロ、CF3、またはOCF3であり、
別の態様において、各R6は独立してメチル、F、またはClであり、
別の態様において、XおよびYは両方カルボニルであり、
別の態様において、XおよびYは両方メチレンであり、
別の態様において、Xはカルボニルであり、Yはメチレンであり、
別の態様において、Xはメチレンであり、Xはカルボニルであり、
別の態様において、qは0または1である。
【0025】
本発明は、化合物およびそれらの薬学上許容される塩を含む。したがって、「化合物またはその薬学上許容される塩」の文脈において「または」という用語は、化合物またはその薬学上許容される塩(代替)、あるいは化合物およびその薬学上許容される塩(組合せ)のいずれかを指すものと理解される。
【0026】
本発明の化合物は、中間体1と本明細書に称される、ベツリンの誘導体である:
【化3】
【0027】
ベツリン(ルペ−20(29)−エン−3β,28−ジオール)は、樺の木の樹皮から一般的に単離される豊富に天然に存在するトリテルペンであり、ベツリンはその抽出物の30乾燥重量%までを形成する。Green,Brian;Bentley,Michael D.;Chung,Bong Y.;Lynch,Nicholas G.;Jensen,Bruce L.(1985),「Isolation of Betulin and Rearrangement to Allobetulin A Biomimetic Natural Product Synthesis」,J.Chem.Educ.200:7を参照のこと。
【0028】
本明細書で使用する場合、「薬学上許容される塩」という用語は、これらの化合物、物質、組成物、および剤形を指し、それらは、過剰な毒性、炎症、または他の問題もしくは合併症を生じずに、ヒトおよび動物の組織と接触させる使用に適切である十分な医学的判断の範囲内である。当業者は、式(I)に係る化合物の薬学上許容される塩が調製され得ることを理解するだろう。それらの薬学上許容される塩は、化合物の最終単離および精製の間にインサイチュで調製され得るか、またはその遊離酸もしくは遊離塩基形態で精製化合物をそれぞれの好適な塩基もしくは酸と別々に反応させることにより調製され得る。
【0029】
本発明の化合物は、好適な酸または塩基との反応により薬学上許容される塩を形成できる。好適な酸には無機酸および有機酸が含まれ、好適な無機酸の例には、塩酸、臭化水素酸、リン酸、メタリン酸、硝酸、および硫酸が含まれ、好適な有機酸の例には、酒石酸、酢酸、トリフルオロ酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、ギ酸、プロピオン酸、グリコール酸、グルコン酸、マレイン酸、コハク酸、メタンスルホン酸、エタンスルホン酸、ステアリン酸、ベンゼンスルホン酸、ブロモベンゼンスルホン酸、およびp−トルエンスルホン酸が含まれる。好適な塩基には、例えば、NaOH、KOH、Na2CO3、K2CO3、NaH、およびカリウム−t−ブトキシドを含む、水酸化物、炭酸塩、水素化物、およびアルコキシドが含まれる。
【実施例】
【0030】
上記の本発明の実施形態は単に例示であることを意図し、多数の変更および修飾が、当業者には明らかであろう。全てのこのような変更および修飾は、添付の特許請求の範囲に定義されるように本発明の範囲内であることが意図される。
【0031】
装置の説明
1H NMRスペクトルを、Bruker Avance−III400分光計で記録した。化学シフトは100万分の1(ppm、δ単位)で表す。結合定数はヘルツ(Hz)の単位である。分割パターンは見かけの多重度を表し、s(一重線)、d(二重線)、t(三重線)、q(四重線)、quint(五重線)、m(多重線)、br(広幅)と指定する。
【0032】
分析的低分解能質量分析(MS)は、勾配溶離法を用いるSunFire C18、4.6×50mm、3.5μmを用いてAgilent1200HPLC/6110またはAgilent1200HPLC/6130で記録した。
【0033】
溶媒A:水中の0.01%トリフルオロ酢酸(TFA);
溶媒B:アセトニトリル中の0.01%TFA;
1.2分間、一定のA、続いて4分にわたって5%〜95%または20%〜95%のB。
【0034】
生物学的アッセイ
試験化合物の抗ウイルス活性を、2つの細胞同時培養HIVライフサイクルアッセイにおいて決定した。このアッセイにおいて、HIV−1 HxB2に慢性的に感性させたJurkat T−リンパ球を、修飾されたHIV LTR−ルシフェラーゼレポーターを有するインジケーターHOS細胞と同時培養した。感染したJurkat細胞により産生されたウイルスは、ルシフェラーゼレポーターのLTR指令発現を導くHOS細胞に感染し得る。Jurkat細胞におけるウイルス産生、ウイルスの成熟、HIVライフサイクルにおける侵入または侵入後段階を妨げる化合物は、ルシフェラーゼシグナルを減少させる。
【0035】
アッセイを開始する前に、感染したJurkat、J4HxB2、細胞の凍結ストックバイアルを37℃の水槽中で急速に解凍させ、穏やかに攪拌しながら細胞培地(10%ウシ胎仔血清およびゲンタマイシンを含有するRPMI 1640培地)と共に15mLにゆっくりと希釈する。次いで細胞を、37℃、5%CO2にて培地中に入れ、細胞培地を添加することによって4日目に30mL、および7日目に60mLに拡張する。
【0036】
1日目に、アッセイを開始し、HOS細胞を急速に解凍させ、細胞培地中に15mLまでゆっくりと希釈し、1400rpmにて5分間遠心分離し、10mLの細胞培地中で再懸濁する。2×107HOS細胞を、冷却した(4℃)細胞培地中で1112mLに希釈し、攪拌プレート上に置いた。培地中の6.7×107J4HxB2細胞を、1400rpmにて5分間、遠心分離によりペレットにし、冷却したHOS細胞懸濁液中に再懸濁した。HOSおよびJ4HxB2細胞を、96または384ウェルプレート中に播種する前に少なくとも5分間、攪拌プレート上で混合した。マルチドロップ(Multidrop)(または同様の機器)を使用して、細胞を、試験化合物を含有するアッセイプレートに分配する。96ウェルアッセフォーマットにおいて、0.2mLのJ4HxB2/HOS細胞懸濁液を、アッセイプレート中の2μLの試験化合物に加えた。384ウェルアッセイフォーマットに関して、0.05mLの細胞懸濁液を、アッセイプレート中の0.5μLの試験化合物に加えた。化合物は10点または11点連続希釈として試験した。細胞を化合物のプレートに添加した後、プレートを室温にて30分〜1時間攪拌させ、次いで5日間、加湿した5%CO2 37℃のインキュベーターに移した。5日の終わりに、プレートをインキュベーターから取り除き、室温で30分〜1時間、平衡化した。製造業社の指示書に従ってPromega Steady−Glo試薬を調製し、マルチドロップ(または同様の機器)を使用してプレートに加えた。0.02mLのSteady−Gloを384ウェルプレートのウェルに加えた。96ウェルプレートに関して、0.1mLの培地を各ウェルから取り除き、0.06mLのSteady−Gloを添加した。次いで発光を、EnvisionもしくはTopcount Microplate Readerまたは同様の機器を用いて検出した。データ分析のために、DMSOまたはHIV−1阻害剤(エファビレンツ、ブレカナビルまたは同様の抗ウイルス薬)を含有するウェルに対応する高および低シグナル対照を使用してデータを正規化し、次いでそれを、適切なデータ解析パッケージを用いて非線形回帰モデルにフィットさせて、IC50値を求めた。
【0037】
スキームおよび実験手順
以下のスキームおよび手順は本発明の化合物がどのように調製できるかを例示する。参照されている特定の溶媒および反応条件もまた例示であり、限定することを意図するものではない。説明されていない化合物は、市販されているか、または入手可能な出発物質を用いて当業者により容易に調製できる。本明細書に開示される実施例は、例示のみの目的のためであり、本発明の範囲を限定することを意図するものではない。全ての実施例は、本明細書に開示されたアッセイを用いると、1μMから1nMのLHIV IC50値を示した。
【0038】
式Iの化合物についての中間体10の合成:
【化4】
【化5】
【0039】
工程A:中間体2
室温にて、CH2Cl2(DCM、100mL)中の中間体1(20g、45.2mmol)、4−ジメチルアミノピリジン(DMAP、1.66g、13.6mmol)、およびEt3N(63mL、136mmol)の溶液に、無水酢酸(Ac2O、17.1mL、113mmol)を加えた。それを一晩還流で加熱した後、室温まで冷却し、反応物を水(50mL)でクエンチした。次いで有機相を水(50mL×2)で洗浄し、硫酸ナトリウムで乾燥させた。減圧下でほとんどの有機溶媒を除去した後、無水エタノール(50mL)を加え、得られた沈殿物を、白色の固体(中間体2、20g、84%)として濾過により回収した。1H NMR(400MHz,CDCl3)δppm4.69(1H,m),4.59(1H,m),4.51−4.43(1H,m),4.25(1H,d,J=11.2Hz),3.85(1H,d,J=10.8Hz),2.49−2.40(1H,m),2.07(3H,s),2.04(3H,s),1.98−0.77(42H,m).LC/MS:m/z計算値526.4,実測値527.7(M+1)+。
【0040】
工程B:中間体3
酢酸(40mL、33%)中のHBrを、105℃にて予め加熱したトルエン(40mL)、Ac2O(40mL)、および酢酸(AcOH、40mL)中の中間体2(20g、38mmol)の懸濁液に加えた。反応混合物を攪拌し、この温度で1.5時間加熱した。冷却後、酢酸ナトリウム(24g)を加え、得られた反応混合物を蒸発乾固した。薄茶色の残渣をDCM(200mL)中に取り、有機相を水(100mL×3)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発乾固させて残渣を得て、それを95%エタノールおよびDCMから再結晶させて、白色の固体として中間体3(13.8g、69%)を得た。1H NMR(400MHz,CDCl3)δppm4.50−4.46(1H,m),4.02(1H,d,J=10.8Hz),3.98(1H,d,J=10.8Hz),3.18−3.10(1H,m),2.43−2.40(1H,m),2.26−2.22(2H,m),2.04(3H,s),2.05(3H,s),2.00−1.95(1H,m),1.90−1.85(1H,m),1.77−0.83(39H,m).LC/MS:m/z計算値526.4,実測値549.2(M+Na)+。
【0041】
工程C:中間体4
無水トルエン(90mL)、AcOH(119mL)、およびAc2O(29mL)中の中間体3(7g、13.29mmol)、酢酸ナトリウム(NaOAc、6.21g、76mmol)および二クロム酸ナトリウム二水和物(4.75g、15.95mmol)の混合物を60℃にて一晩攪拌した。冷却後、反応混合物を水(150mL)と酢酸エチル(EtOAc、250mL)との間に分配した。有機相を、水(100mL)、飽和炭酸ナトリウム溶液(100mL×2)およびブライン(100mL×2)で連続して洗浄し、硫酸ナトリウムで乾燥させ、減圧下で濃縮して、粘性のある油を得た。粘性のある油をメタノール(MeOH、250mL)で粉砕し、沈殿物を白色の固体として中間体4(6g、11.1mmol、83%収率)として回収した。1H NMR(400MHz,CDCl3)δppm4.52−4.46(1H,m),4.33(1H,d,J=10.8Hz),4.06(1H,d,J=11.2Hz),3.21−3.16(1H,m),2.86(1H,dd,J=12.8,3.2Hz),2.42−2.36(1H,m),2.05(3H,s),2.00(3H,s),1.94−0.84(40H,m).LC/MS:m/z計算値540.4,実測値563.3(M+Na)+。
【0042】
工程D:中間体5
1:1のエタノール(EtOH)およびトルエン(400mL)の混合物中の中間体4(7g、12.94mmol)および水酸化カリウム(KOH、0.872g、15.5mmol)の混合物を、室温にて1時間激しく攪拌した。反応混合物をHCl水溶液(1N)でpH7に中和し、蒸発乾固した。得た残渣を水および少量のアセトン中に取った。沈殿物を回収し、次いで水で洗浄し、真空中で乾燥させて、中間体5(6.0g、93%)を白色の固体として得た。LC/MS:m/z 計算値498.4、実測値499.3(M+1)+。
【0043】
工程E:中間体6
室温にてDCM(300mL)中の中間体5(5.1g、10.23mmol)の溶液に、クロロクロム酸ピリジニウム(PCC、6.61g、30.7mmol)、およびシリカゲル(6.6g)を加えた。反応混合物を室温にて1時間攪拌した。反応物を水でクエンチした後、有機相を飽和重炭酸ナトリウム溶液(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、粗生成物を得て、それをシリカゲル上でのカラムクロマトグラフィー(EtOAc:PE=1:10〜1:5)により精製して、白色の固体として中間体6(4.2g、83%)を得た。LC/MS:m/z 計算値496.4,実測値497.2(M+1)+。
【0044】
工程F:中間体7
−40℃にて窒素雰囲気下で、無水テトラヒドロフラン(THF、60mL)中の1,3−ジチアン(5.7g、47.4mmol)の溶液に、n−BuLi(27mL、67.5mmol)の溶液をゆっくりと加えた。反応混合物をさらに2時間、−20℃にて攪拌した後、無水THF(40mL)中の中間体6(4.2g、8.46mmol)の溶液を、−70℃にて窒素雰囲気下でゆっくりと加えた。次いで反応物を1時間、−78℃にて攪拌し、その後、それを飽和NaHCO3溶液でクエンチした。抽出をEtOAcで実施し、有機相を、水(50mL)、飽和ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、粗生成物を得て、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=8:1〜4:1)により精製して、中間体7(3.0g、5.22mmol、61.7%)を得た。LC/MS:m/z 計算値574.4,実測値575.0(M+1)+。
【0045】
工程G:中間体8
DCM(40mL)中の中間体7(3.5g、6.09mmol)、Et3N(2.55mL、18.26mmol)、およびDMAP(0.149g、1.218mmol)の溶液に、室温にてAc2O(3.45mL、36.5mmol)を加えた。50℃にて2時間、攪拌した後、反応物を水でクエンチした。有機相を水(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体8(3.41g、85%)を得た。LC/MS:m/z 計算値658.4,実測値659.1(M+1)+。
【0046】
工程H:中間体9
アセトニトリル(160mL)および水(40mL)中の中間体8(4.5g、6.83mmol)の溶液に、室温にてN−ブロモスクシンイミド(NBS、7.29g、41.0mmol)を加えた。室温にて10分間攪拌した後、反応物を水でクエンチした。有機層を飽和亜硫酸ナトリウム(200mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体9(3.2g、82%)を得た。LC/MS:m/z 計算値568.4,実測値569.3(M+1)+。
【0047】
工程I:中間体10
t−ブタノール(300mL)、THF(60mL)、および2−メチル−2−ブテン(2mL)中の中間体9(3.2g、5.63mmol)の氷冷した溶液に、水(60mL)中のNaClO2(7.19g、67.5mmol)およびNaH2PO4(6.75g、56.3mmol)の溶液を15分にわたってゆっくりと加えた。0℃にて10分間攪拌した後、反応混合物を室温まで加温し、さらに30分間攪拌し、続いてEtOAcで希釈した。有機相を水(100mL)で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮乾固した。得られた残渣をシリカゲル上でのカラムクロマトグラフィー(DCM:MeOH=50:1〜10:1)により精製して、白色の固体として中間体10(2.8g、81%)を得た。LC/MS:m/z 計算値584.4,実測値585.3(M+1)+。
【0048】
以下の実施例は例示のみの目的であり、本発明の範囲を限定することを意図しない。以下の実施例1のスキームに関して、R1NHR2はアゼチジニル基を表し、R3はHOOCC(CH3)2CH2C(O)O−である。
【0049】
実施例1:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[1−アゼチジニル(オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化6】
【化7】
【0050】
工程A:中間体11
DCM(5mL)中の中間体10(680mg、1.163mmol)の溶液に、塩化オキサリル(3mL、35.5mmol)および数滴のDMFを加えた。反応混合物を室温にて3時間攪拌し、減圧下で蒸発させて、淡黄色の固体として中間体11を得た。
【0051】
工程B:中間体12−1
DCM(10mL)中の中間体11(430mg、0.713mmol)、DMAP(100.0mg、0.819mmol)およびEt3N(2mL、14.35mmol)の溶液に、室温にてアゼチジン(200mg、3.5mmol)を加えた。室温にて4時間攪拌した後、反応物をDCM(50mL)で希釈した。有機相を、水(50mL)、塩酸水溶液(2N、50mL)で洗浄し、次いで硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、粗生成物を得て、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=5:1〜1:1)により精製して、白色の固体として中間体12−1(300mg、67.5%)を得た。LC/MS:m/z 計算値623.4,実測値624.3(M+1)+。
【0052】
工程C:中間体13−1
1:1のEtOHおよびトルエン(12mL)の混合物中の中間体12−1(350mg、0.561mmol)およびKOH(19mg、0.353mmol)の混合物を、室温にて15分間激しく攪拌した。反応混合物をHCl水溶液(1N)でpH7に中和した。水を加えた後、反応物をEtOAcで抽出した。有機相を硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮して、中間体13−1(320mg、97%)を淡黄色の固体として得た。LC/MS:m/z 計算値581.4,実測値582.3(M+1)+。
【0053】
工程D:中間体14−1
ジメチルスルホキシド(DMSO、8mL)中の中間体13−1(430mg、0.74mmol)の溶液に、室温にて2−ヨードキシ安息香酸(IBX、1.1g、3.93mmol)を加えた。室温にて一晩攪拌した後、反応物を水でクエンチし、EtOAcで抽出した。有機相を硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮して残渣を得、それをシリカゲル上でのカラムクロマトグラフィー(PE:EA=5:1〜3:1)により精製して、白色の固体として中間体14−1(300mg、70%)を得た。LC/MS:m/z 計算値579.4,実測値580.3(M+1)+。
【0054】
工程E:中間体18−1
1,4−ジオキサン(15mL)および濃HCl(5mL)中の中間体14−1(300mg、0.52mmol)の溶液を室温にて一晩攪拌した。反応物を水(30mL)でクエンチし、EtOAc(60mL)で抽出した。有機相を水で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて残渣を得、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=5:1〜3:1)により精製して、白色の固体として中間体18−1(70mg、25%)を得た。LC/MS:m/z 計算値537.4,実測値538.3(M+1)+。
【0055】
工程F:化合物19−1
ピリミジン(3mL)中の中間体18−1(70mg、0.13mmol)の溶液に、3,3−ジメチルジヒドロ−2,5−フランジオン(159mg、1.302mmol)およびDMAP(334mg、2.6mmol)を加えた。それを90℃にて一晩加熱した後、反応混合物をDCMで抽出した。有機相をHCl(2N、25mL)、水(50mL×2)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて残渣を得、それをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAC=3:1〜2:1)により精製して、白色の固体の生成物として化合物19−1(30mg、34.6%)を得た。1H NMR(400MHz,CDCl3)δppm4.54−4.42(2H,m),4.28−4.35(1H,m),4.05−4.15(2H,m),3.17−3.25(1H,m),2.75−2.62(4H,m),2.60−2.52(2H,m),2.35(1H,quint,J=8.0Hz),2.17(1H,d,J=19.2Hz),2.10−2.00(1H,m),1.96−1.82(1H,m),1.78−0.78(42H,m).LC/MS:m/z 計算値665.9,実測値664.4(M−1)−。
【0056】
実施例2:2,2−ジメチル−4−オキソ−4−({(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−3a−[[4−(メチルスルホニル)−1−ピペラジニル](オキソ)アセチル]−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル}オキシ)ブタン酸
【化8】
【化9】
【0057】
中間体12−2
DMF(8mL)中の中間体10(600mg、1.026mmol)、ジイソプロピルエチルアミン(DIPEA、2g、15.47mmol)、および2−(1H−7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェートメタンアミニウム(HATU、1.4g、3.68mmol)の混合物に、室温にて1−(メチルスルホニル)ピペラジン(700mg、4.26mmol)を加えた。室温にて4時間攪拌した後、反応物を水(100mL)でクエンチし、EtOAc(200mL)で抽出した。有機相を硫酸ナトリウムで乾燥させ、減圧下で蒸発させて残渣を得て、それをシリカゲル上でのカラムクロマトグラフィー(DCM:MeOH=100:1〜30:1)により精製して、白色の固体として中間体12−2(650mg、87%)を得た。LC/MS:m/z 計算値730.4,実測値731.3(M+1)+。
【0058】
化合物19−2
化合物19−2を、実施例1(式中、R1NHR2はメチルスルホニルピペラジンである)に使用したものと同様の手順を用いて中間体12−2から白色の固体として調製した。1H NMR(400MHz,CDCl3)δppm4.54−4.46(1H,m),3.82−3.73(1H,m),3.68−3.53(2H,m),3.50−3.42(1H,m),3.36−3.18(5H,m),2.82(3H,s),2.78−2.50(5H,m),2.20(1H,d,J=18.4Hz),2.10−2.06(1H,m),1.90−0.82(43H,m).LC/MS:m/z 計算値772.4,実測値773.3(M+1)+。
【0059】
実施例3:2,2−ジメチル−4−オキソ−4−({(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−3a−[(4−メチル−1−ピペラジニル)(オキソ)アセチル]−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル}オキシ)ブタン酸
【化10】
【0060】
実施例2の化合物を調製するために使用したものと同様の手順に従って、淡黄色の固体として標題化合物を調製した。この場合、R1NHR2は1−メチルピペラジンである。1H NMR(400MHz,DMSO−d6)δppm4.38−4.32(1H,m),3.52−3.17(6H,m),2.60−2.20(9H,m),2.19(3H,s),1.98−0.79(44H,m).LC/MS:m/z 計算値709.0,実測値707.5(M−1)−。
【0061】
実施例4:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[[(4−フルオロフェニル)アミノ](オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化11】
【化12】
【0062】
工程A:中間体15
EtOH(2mL)およびトルエン(2mL)中の中間体10(500mg、0.855mmol)の溶液に室温にてKOH(192mg、3.42mmol)を加えた。室温にて30分攪拌後、反応物をDCM(100mL)で希釈した。有機層をNH4Cl水溶液(50mL×2)およびブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体15(400mg、86%)を得た。LC/MS:m/z計算値542.4,実測値543.1(M+1)+。
【0063】
工程B:中間体16
DMSO(6mL)中の中間体15(290mg、0.534mmol)の溶液に、室温にてIBX(1496mg、5.34mmol)を加えた。それを50℃にて3時間攪拌後、次いで反応混合物を室温まで冷却し、DCM(100mL)で希釈した。有機相を水(50mL×4)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、中間体16(234mg、81%)を黄色固体として得た。LC/MS:m/z計算値540.4,実測値541.1(M+1)+。
【0064】
工程C:中間体17
DCM(8mL)中の中間体16(350mg、0.664mmol)の溶液に、窒素雰囲気下で0℃にて、塩化オキサリル(1.4mL、15.99mmol)を5分にわたってゆっくり加えた。それを室温にて1時間攪拌後、反応混合物を蒸発乾固して、中間体17を淡黄色の固体として得た。
【0065】
工程D:中間体14−4
DCM(7mL)中の4−フルオロアニリン(221mg、1.992mmol)およびEt3N(0.463mL、3.32mmol)の混合物に、中間体17(371mg、0.664mmol)を室温にて加えた。室温にて1時間攪拌後、反応混合物をDCM(50mL)で希釈した。有機相を水(50mL)、ブライン(25mL)で洗浄し、硫酸ナトリウムで乾燥させ、減圧下で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=5:1〜1:1)により精製することで、中間体14−4(150mg、36%)を白色の固体として得た。LC/MS:m/z計算値634.4,実測値635.3(M+1)+。
【0066】
化合物19−4
化合物19−4を、実施例1において使用したものと同様の手順で中間体14−4から黄色の固体として調製した。1HNMR(400MHz,CDCl3)δppm8.72(1H,s),7.61−7.57(2H,m),7.08−7.03(2H,m),4.52−4.48(1H,m),3.28−3.20(1H,m),2.78−2.53(5H,m),2.25(1H,d,J=18.8Hz),2.08−1.87(2H,m),1.18−0.81(42H,m).LC/MS:m/z計算値719.4,実測値720.3(M+1)+。
【0067】
実施例5:2,2−ジメチル−4−オキソ−4−[((3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3a−{オキソ[(2−チエニルメチル)アミノ]アセチル}−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル)オキシ]ブタン酸
【化13】
【化14】
【0068】
中間体14−5
DMF(10mL)中の中間体16(220mg、0.407mmol)、HATU(309mg、0.814mmol)、およびDIPEA(0.36mL、2.1mmol)の溶液に、(2−チエニルメチル)アミン(46mg、0.407mmol)を室温にて加えた。反応混合物を室温にて一晩攪拌し、DCMで希釈した。有機相を水(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=20:1〜4:1)により精製することで、中間体14−5(60mg、23%)を黄色の固体として得た。LC/MS:m/z計算値636.4,実測値637.3(M+1)+。
【0069】
化合物19−5
化合物19−5を、実施例1において使用したものと同様の手順で中間体14−5から黄色がかった固体として調製した。1HNMR(400MHz,CDCl3):δppm7.25(1H,dd,J=4.8Hz,1.6Hz),7.19(1H,t,J=5.6Hz),7.00−6.94(2H,m),4.66(1H,dd,J=15.2Hz,6.0Hz),4.57(1H,dd,J=15.2Hz,6.0Hz),4.54−4.47(1H,m),3.25−3.20(1H,m),2.76−2.52(5H,m),2.21(1H,d,J=18.8Hz),2.08−1.86(2H,m),1.78−0.81(42H,m).LC/MS:m/z計算値722.4,実測値723.3(M+1)+。
【0070】
実施例6:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[{[(4−クロロフェニル)メチル]アミノ}(オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化15】
【0071】
実施例2に記載されたものと同様の手順(ここで、R1NHR2は[(4−クロロフェニル)メチル]アミンである)を使用して、標題化合物を淡黄色の固体として得た。1HNMR(400MHz,CDCl3)δppm7.32−7.17(5H,m),4.54−4.32(3H,m),3.26−3.16(1H,m),2.76−2.48(5H,m),2.20(1H,d,J=18.8Hz),2.03−1.86(2H,m),1.77−0.81(43H,m).LC/MS:m/z計算値749.4,実測値748.4(M−1)−。
【0072】
実施例7:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−[{[(2−クロロフェニル)メチル]アミノ}(オキソ)アセチル]−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化16】
【0073】
実施例2に記載されたものと同様の手順(ここで、R1NHR2は[(2−クロロフェニル)メチル]アミンである)を使用して、標題化合物を淡黄色の固体として得た。1HNMR(400MHz,CDCl3):δppm7.41−7.21(5H,m),4.61−4.44(3H,m),3.22−3.12(1H,m),2.76−2.46(5H,m),2.18(1H,d,J=19.2Hz),1.96−1.84(2H,m),1.78−0.81(43H,m).LC/MS:m/z計算値749.4,実測値748.4(M−1)−。
【0074】
表1に列挙された以下の化合物を、上記で列挙した方法と同様の方法を用いて調製した。各場合において、XおよびYは、それぞれカルボニルである。
【化17】
【表1−1】
【表1−2】
【表1−3】
【0075】
実施例8:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−({[(4−クロロフェニル)メチル]アミノ}アセチル)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化18】
【化19】
【0076】
工程A:中間体20
中間体6(300mg、0.604mmol)およびMeNO2(7.5mL、139mmol)の混合物に、Et3N(0.6mL,4.30mmol)を室温にて加えた。一晩攪拌後、反応物を水(1.0mL)でクエンチし、EtOAc(50mL)と水(25mL)との間に分配した。有機相を飽和ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固した。得られた残渣をシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=4:1)により精製することで、中間体20(335mg、99%)を白色の固体として得た。
【0077】
工程B:中間体21
MeOH(125mL)中のNiCl2・6H2O(0.929g、7.17mmol)の溶液に、水素化ホウ素ナトリウム(0.271g、7.17mmol)を0℃にて少量ずつ加えた。30分攪拌後、中間体20(2g,3.59mmol)を加え、その後追加の水素化ホウ素ナトリウム(2.44g、64.53mmol)を少量加えた。反応混合物を0℃でさらに30分攪拌し、濾過し、不溶性物質を除去した。濾液を真空内で濃縮乾固した。得られた残渣をEtOAc(100mL)中に溶解し、有機相を水(40mL)、ブライン(40mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固して、中間体21(1.79g、95%)をオフホワイトの固体として得た。LC/MS:m/z計算値527.3,実測値528.2(M+1)+。
【0078】
工程C:中間体22−1および23−1
MeOH(30mL)中の中間体21(450mg、0.853mmol)および4−クロロベンズアルデヒド(120mg、0.853mmol)の懸濁液に、ZnCl2(69.7mg、0.083mmol)を室温にて加えた。室温にて2時間攪拌後、シアノ水素化ホウ素ナトリウム(107mg、0.25mmol)を加え、得られた混合物をさらに2時間攪拌して、中間体22−1を得た。
【0079】
上述のように得た反応混合物を、Boc2O(0.297mL、1.279mmol)およびEt3N(0.238mL、1.705mmol)に加えた。室温にて一晩攪拌後、不溶性物質を濾過により除去し、濾液を減圧下で濃縮乾固した。得られた残渣をEtOAc(50mL)中に溶解し、有機相を水(25mL)、ブライン(25m)で洗浄し、硫酸マグネシウムで乾燥させ、真空内で蒸発乾固して、残渣を得て、これを分取TLCにより精製することで、中間体23−1(164mg,25.6%)を白色の固体として得た。
【0080】
工程D:中間体24−1
DCM(5mL)中の中間体23−1(130mg、0.173mmol)の溶液に、PCC(372mg、1.728mmol)およびシリカゲル(120mg)を室温にて加えた。室温にて8時間攪拌後、不溶性物質を濾過により除去し、濾液を減圧下で濃縮乾固した。得られた残渣をシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=1:6)により精製することで、中間体24−1(92mg、71%)を白色の固体として得た。LC/MS:m/z計算値785.4,実測値686.0(M−Boc+1)+。
【0081】
工程E:中間体25−1
1,4−ジオキサン(2.5mL)中の中間体24−1(28mg、0.037mmol)の溶液に、濃HCl(1.0mL、32.9mmol)を加えた。室温にて一晩攪拌後、反応混合物をEtOAc(50mL)で希釈し、飽和重炭酸ナトリウム溶液で中和し、EtOAc(50mL)と飽和重炭酸ナトリウム(25mL)との間に分配した。有機相を飽和重炭酸ナトリウム(25mL)、ブライン(25mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体25−1(22.3mg,98%)を無色の油として得た。LC/MS:m/z計算値607.3,実測値608.1(M+1)+。
【0082】
工程F:中間体26−1
DCM(5mL)中の中間体25−1(79mg、0.13mmol)およびBoc2O(28.3mg、0.13mmol)の溶液に、Et3N(18μL、0.13mmol)を加えた。室温にて1時間攪拌後、反応物を水(1.0mL)でクエンチし、EtOAc(25mL)と水(10mL)との間に分配した。有機相を飽和重炭酸ナトリウム(10mL)、ブライン(10mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=5:1)により精製して、中間体26−1(57mg、62%)を白色の泡状物として得た。
【0083】
工程G:中間体27−1
無水ピリジン(2mL)中の中間体26−1(26mg、0.037mmol)の溶液に、DMAP(22.42mg、0.184mmol)および3,3−ジメチルジヒドロ−2,5−フランジオン(47mg、0.367mmol)を加えた。それを80℃にて一晩攪拌後、反応混合物をEtOAc(30mL)で希釈した。有機相をHCl水溶液(2N、10mL)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc4:1)により精製して、中間体27−1(15mg、48.9%)を白色の泡状物として得た。LC/MS:m/z計算値835.4,実測値858.4(M+Na)+。
【0084】
工程H:化合物28−1
DCM(1.0mL)中の中間体27−1(27mg、0.032mmol)の溶液に、TFA(0.5mL、6.49mmol)を滴下して加えた。室温にて1時間攪拌後、反応混合物を重炭酸ナトリウム水溶液(25mL)で中和した。反応混合物をEtOAc(50mL)と重炭酸ナトリウム水溶液(25mL)との間に分配し、有機相をブライン(25mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、標題化合物28−1(9mg、29.5%)を得た。LC/MS:m/z計算値735.3,実測値736.3(M+1)+。
【0085】
実施例9:4−{[(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−{[(2−フラニルメチル)アミノ]アセチル}−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル]オキシ}−2,2−ジメチル−4−オキソブタン酸
【化20】
【化21】
【0086】
中間体24−2
実施例8における記載に従って調製した、DMSO(10mL)中の中間体23−2(1.46g、1.24mmol)の溶液に、IBX(2g、7.14mmol)を加えた。それを60℃にて2時間攪拌後、反応混合物をEtOAc(50mL)で希釈した。有機相を水(30mL)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得た。これをシリカゲル上でのカラムクロマトグラフィー(PE:EtOAc=100:0〜85:15)により精製して、中間体24−2(180mg、20.6%)を白色の固体として得た。LC/MS:m/z計算値727.3,実測値728.3(M+1)+。
【0087】
化合物28−2
化合物28−2を、実施例8において使用したものと同様の手順により、中間体24−2から白色の固体として調製した。1HNMR(400MHz,CD3OD)δppm7.54(1H,d,J=1.2Hz),6.52(1H,d,J=3.2Hz),6.41(1H,dd,J=3.2,2.0Hz),4.39(1H,dd,J=10.8,5.6Hz),4.21(2H,s),3.99(1H,d,J=18.0Hz),3.89(1H,d,J=18.0Hz),3.27−3.19(1H,m),2.54(1H,d,J=16.0Hz),2.46(1H,d,J=16.0Hz),2.49−2.39(2H,m),2.34(1H,d,J=19.2Hz),2.13(1H,d,J=19.2Hz),1.99−1.81(2H,m),1.73−1.18(13H,m),1.17(3H,s),1.16(3H,s),1.13(3H,s),1.11(3H,s),1.04−0.92(3H,m),0.96(3H,s),0.90(3H,s),0.85(3H,s),0.77(6H,s).LC/MS:m/z計算値691.3,実測値692.1(M+1)+。
【0088】
実施例10:2,2−ジメチル−4−オキソ−4−[((3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3a−{[(3−ピリジニルメチル)アミノ]アセチル}−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル)オキシ]ブタン酸
【化22】
【0089】
実施例8および9Aに記載されたものと同様の手順を用いて、標題化合物を黄色の固体として調製した。1HNMR(400MHz,CDCl3)δppm8.51−8.48(2H,m),7.68(1H,d,J=8.0H),7.22(1H,br),4.49(1H,dd,J=11.2,4.8Hz),3.71(2H,s),3.41(1H,d,J=18.4Hz),3.30(1H,d,J=18.4Hz),3.17−3.11(1H,m),2.68−0.74(52H,m).LC/MS:m/z計算値702.4,実測値703.3(M+1)+。
【0090】
実施例11:2−2,ジメチル−4−オキソ−4−({(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−5a,5b,8,8,11a−ペンタメチル−1−(1−メチルエチル)−2−オキソ−3a−[({[3−(トリフルオロメチル)フェニル]メチル}アミノ)アセチル]−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イル}オキシ)ブタン酸
【化23】
【0091】
実施例8および9に記載されたものと同様の手順を用いて、標題化合物を調製した。1HNMR(400MHz,CD3OD)δppm7.86−7.66(4H,m),4.47(1H,dd,J=11.2,5.2Hz),4.31(2H,s),4.22(1H,d,J=17.6Hz),4.11(1H,d,J=17.6Hz),3.38−3.27(1H,m),2.64−0.87(52H,m).LC/MS:m/z計算値769.4,実測値770.3(M+1)+。
【0092】
実施例12:4−((3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル(isopstickyl)−5a,5b,8,8,11a−ペンタメチル−3a−(2−(4−メチルピペラジン−1−イル)アセチル)−2−オキソ−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化24】
【化25】
【0093】
工程A:中間体29
1:1のトルエン(20mL)およびEtOH(20mL)の混合物中の中間体8(863mg、1.31mmol)およびKOH(367mg、6.55mmol)の懸濁液を、室温にて1時間激しく攪拌した。それをHCl水溶液(1N)で中和した後、反応混合物を蒸発乾固した。得られた残渣をDCM(200mL)中に取り、有機相を水(200mL)、飽和重炭酸ナトリウム溶液(100mL×2)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(EtOAc:PE=1:30〜1:15)により精製して、中間体29(464mg、57.4%)を白色の固体として得た。LC/MS:m/z計算値616.3,実測値617.0(M+1)+。
【0094】
工程B:中間体30
アセトニトリル(20mL)および水(5mL)中のNBS(803mg、4.51mmol)の溶液に、中間体29(464mg、0.752mmol)を室温にて加えた。室温にて0.5時間攪拌後、反応物を亜硫酸ナトリウム水溶液でクエンチし、濃縮した。得られた残渣をエーテル(30mL)中に溶解し、有機相をブライン(15mL)で洗浄し、硫酸ナトリウムで乾燥させ、蒸発乾固して、中間体30(541mg、96%)を白色の固体として得た。LC/MS:m/z計算値526.3,実測値527.1(M+1)+。
【0095】
工程C:中間体31−1
MeOH(20mL)中の中間体30(450mg、0.598mmol)、1−メチルピペラジン(0.133mL、1.196mmol)、および塩化亜鉛(48.9mg、0.359mmol)の懸濁液を、70℃にて0.5時間加熱した。室温まで冷却後、シアノ水素化ホウ素ナトリウム(225mg、3.59mmol)を少量加えた。反応混合物を室温にてさらに17時間攪拌し、その後、それをEtOAc(50mL)で希釈した。有機相を飽和重炭酸ナトリウム水溶液(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて残渣を得て、これを分取−HPLCにより精製することで、中間体31−1(291mg、80%)を白色の固体として得た。LC/MS:m/z計算値610.3,実測値611.4(M+1)+。
【0096】
工程D:中間体32−1
DMSO(5mL)中の中間体31−1(50mg、0.082mmol)、およびIBX(68.8mg、0.246mmol)の溶液を、60℃にて4時間攪拌した。その後、反応混合物をEtOAc(30mL)で希釈し、有機相を飽和重炭酸ナトリウム(15mL)、水(15mL)、ブライン(15mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体32−1(42mg、84%)を白色の泡状物として得た。LC/MS:m/z計算値608.3,実測値609.3(M+1)+。
【0097】
工程E:中間体33−1
1,4−ジオキサン(4mL)および濃HCl(2mL、65.8mmol)中の中間体32−1(42mg、0.069mmol)の溶液を、室温にて一晩攪拌した。その後、反応混合物を蒸発乾固した。得られた残渣をEtOAc(30mL)中に溶解し、有機相を飽和重炭酸ナトリウム(15mL)、ブライン(15mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体33−1(42mg,97%)を白色の泡状物として得た。LC/MS:m/z計算値566.3,実測値567.4(M+1)+。
【0098】
工程F:化合物28−5
ピリジン(0.8mL)中の中間体33−1(80mg、0.141mmol)、3,3−ジメチル−ジヒドロフラン−2,5−ジオン(362mg、2.82mmol)およびDMAP(86mg、0.706mmol)の溶液を、窒素雰囲気下で100℃にて3時間攪拌した。室温まで冷却後、反応混合物をEtOAc(30mL)で希釈した。有機相をブラインで洗浄し、硫酸ナトリウムで乾燥させ、蒸発乾固した。得られた残渣を分取−HPLCにより精製することで、化合物28−5(50mg、38%)を白色の泡状物として得た。1HNMR(400MHz,CD3OD)δppm4.50(1H,dd,J=10.8,5.6Hz),3.62−3.40(2H,m),3.38−3.20(4H,m),2.99−2.81(6H,m),2.68−2.53(4H,m),2.40(1H,d,J=19.2Hz),2.15(1H,d,J=19.2Hz),2.10−1.93(2H,m),1.83−0.87(44H,m).LC/MS:m/z計算値694.3,実測値695.4(M+1)+。
【0099】
表2中の化合物を、上記で示した手順と同様の方法で調製した。各場合において、Xはカルボニルであり、YはCH2である。
【化26】
【表2−1】
【表2−2】
【表2−3】
【表2−4】
【0100】
実施例13:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(4−クロロベンジルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化27】
【化28】
【0101】
工程A:中間体34
EtOH(100mL)およびトルエン(100mL)中の中間体3(5g、7.59mmol)の溶液に、KOH(0.51g、9.11mmol)を加えた。室温にて4時間攪拌後、次いで反応混合物を水(500mL)とEtOAc(500mL)との間に分配した。有機相を水(200mL×3)、ブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=6:1〜4:1)により精製して、中間体34(2.5g,67.9%)を白色の固体として得た。1HNMR(400Hz,CDCl3)δppm4.50−4.67(1H,m),3.68(1H,d,J=10.4Hz),3.32(1H,d,J=10.4Hz),3.23−3.15(1H,m),2.42−2.28(3H,m),2.05(3H,s),2.02−1.89(2H,m),1.77−0.83(40H,m)。
【0102】
工程B:中間体35
DCM(75mL)中の中間体34(3g、6.19mmol)の溶液に、室温にてPCC(4g、18.57mmol)およびシリカゲル(3.0g)を加えた。室温にて2時間攪拌後、反応物を水(100mL)でクエンチした。有機相を飽和重炭酸ナトリウム(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で濃縮して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=10:1)により精製して、中間体35(3g、100%)を白色の固体として得た。1HNMR(400Hz,CDCl3)δppm9.43(1H,s),4.50−4.46(1H,m),3.25−3.21(1H,m),2.43−2.02(5H,m),2.04(3H,m),2.00−1.93(1H,m),1.75−0.81(38H,m)。
【0103】
工程C:中間体36
MeNO2(128mL、2382mmol)中の中間体36(5g、10.36mmol)の溶液に、Et3N(10.11mL、72.5mmol)を加えた。それを60℃にて一晩攪拌後、反応混合物を水とEtOAc(各100mL)との間に分配した。有機相を水(20mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(ヘキサン:EtOAc=10:1〜6:1)により精製して、中間体36(2.8g、49.7%)を白色の粉末として得た。LC/MS:m/z計算値543.4,実測値566.3(M+Na+)+。
【0104】
工程D:中間体37
MeOH(166mL)中の中間体36(2.8g、5.15mmol)の溶液に、塩化ニッケル(II)(1.67g、12.87mmol)および水素化ホウ素ナトリウム(4.87g、129mmol)を0℃にて加えた。0℃にて10分攪拌後、反応混合物を水とEtOAc(各200mL)との間に分配し、有機相を水(100mL×3)、ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、中間体37(2.65g、100%)を固体として得た。LC/MS:m/z計算値513.4,実測値514.3(M+1)+。
【0105】
工程E:中間体39
MeOH(15mL)およびジクロロエタン(DCE、15mL)中の中間体37(350mg、0.613mmol)および4−クロロベンズアルデヒド(86mg、0.613mmol)の溶液に、塩化亜鉛(50.1mg、0.368mmol)を加えた。反応混合物を80℃にて1時間攪拌後、室温まで冷却し、シアノ水素化ホウ素ナトリウム(57.8mg、0.92mmol)を加えた。得られた混合物を室温にてさらに1時間攪拌して、中間体38を得た。
【0106】
上記の得られた反応混合物に、Et3N(0.18mL、1.38mmol)およびジ−tert−ブチルジカーボネート(0.157mL、0.674mmol)を加えた。室温にて30分攪拌後、反応混合物を水(20mL)とEtOAc(100mL)との間に分配した。有機相を水(30mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=15:1)により精製して、中間体39(125mg,27.6%)を白色の固体として得た。
【0107】
工程F:中間体40
DCM(10mL)中の中間体39(120mg、0.162mmol)の溶液に、PCC(35mg、0.162mmol)およびシリカゲル(100mg)を加えた。室温にて2時間攪拌後、不溶性物質を濾過により除去し、濾液を濃縮して、中間体40(110mg、92%)を白色の固体として得た。
【0108】
工程G:中間体41
MeOH(1mL)、THF(1mL)、および水(0.5mL)中のNaOH(597mg、14.94mmol)の溶液に、中間体40(110mg、0.149mmol)を加えた。室温にて1時間攪拌後、反応物を水(20mL)で希釈し、EtOAc(50mL)で抽出した。有機相をブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体41(100mg、96%)を白色の固体として得た。
【0109】
工程H:中間体42
無水ピリジン(2mL)中の中間体41(100mg、0.144mmol)の溶液に、DMAP(106mg、0.864mmol)および3,3−ジメチルジヒドロ−2,5−フランジオン(369mg、2.88mmol)を加えた。反応混合物を80℃にて一晩加熱後、溶媒を真空内で除去し、残渣をDCM(50mL)中に取った。有機相をHCl水溶液(0.5N,20mL)、水(2×30mL)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させて、真空内で蒸発させて、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=15:1)により精製することで、中間体42(56mg、44.9%)を白色の泡状物として得た。
【0110】
工程I:化合物43−1
DCM(1mL)中の中間体42(45mg、0.055mmol)の溶液に、TFA(0.5mL、6.49mmol)を加えた。室温にて1時間攪拌後、溶媒を除去して、残渣を得て、これを分取−HPLCにより精製することで、化合物43−1(38mg、77%)を白色の固体として得た。1HNMR(400Hz,CD3OD)δppm7.42−7.36(4H,m),4.40−4.36(1H,m),4.15(2H,dd,J=16.8,13.2Hz),3.93(1H,d,J=17.6Hz),3.85(1H,d,J=17.6Hz),3.19−3.14(m,1H),2.57−1.98(5H,m),1.97−0.77(48H,m).LC/MS:m/z計算値721.4,実測値722.1(M+1)+。
【0111】
実施例14:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(4−フルオロベンジルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化29】
【0112】
実施例13に記載されたものと同様の方法を用いて標題化合物を白色の固体として調製した。1HNMR(400Hz,MeOD)δppm7.55−7.53(2H,m),7.24−7.20(2H,m),4.50−4.40(1H,m),4.27−4.24(2H,dd,J=11.6,11.2Hz),4.03−3.98(2H,dd,J=18.8,14.4Hz),3.31−3.25(1H,m),2.67−2.46(4H,m),2.34−2.31(1H,m),2.12−0.88(48H,m).LC/MS:m/z計算値705.4,実測値706.4(M+1)+。
【0113】
実施例15:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(フラン−2−イルメチルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化30】
【0114】
実施例13に記載されたものと同様の方法を用いて標題化合物を白色の固体として調製した。1HNMR(400Hz,MeOD)δppm7.55(1H,br),6.54(1H,d,J=3.2Hz),6.43−6.42(1H,m),4.40−4.36(1H,m),4.23(2H,s),3.87(2H,s),3.19−3.16(1H,m),2.57−2.22(5H,m),2.05−0.77(48H,m).LC/MS:m/z計算値677.4,実測値678.4(M+1)+。
【0115】
実施例16:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−(シクロペンチルアミノ)アセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化31】
【0116】
実施例13に記載されたものと同様の方法を用いて標題化合物を白色の固体として調製した。1HNMR(400Hz,MeOD)δppm4.41−4.37(1H,m),3.88(2H,br),3.47(1H,br),3.22−3.17(1H,m),2.57−2.45(4H,m),2.22(1H,d,J=13.2Hz),2.09(1H,d,J=13.6Hz),1.99−0.77(55H,m).LC/MS:m/z計算値665.5,実測値666.4(M+1)+。
【0117】
実施例17:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3a−(2−(4−メチルピペラジン−1−イル)アセチル)−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化32】
【化33】
【0118】
工程A:中間体44
無水THF(50mL)中の水素化ナトリウム(60%、250mg、6.25mmol)の懸濁液に、窒素雰囲気下でヨウ化トリメチルスルホキソニウム(900mg、4.09mmol)を室温にて加えた。反応混合物を2時間還流後、60℃まで冷却し、無水THF(2mL)中の中間体35(750mg、1.554mmol)を滴下して加えた。得られた混合物を60℃にて3時間攪拌し、次いで室温にて1時間攪拌し、その後、反応物を飽和重炭酸ナトリウム水溶液(50mL)でクエンチした。反応混合物をDCM(200mL)で希釈し、有機相を水(50mL×3)、飽和ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて残渣を得て、これをシリカゲル上でのカラムクロマトグラフィー(Hex:EtOAc=10:1)により精製して、中間体44(300mg、42.5%)を白色の固体として得た。
【0119】
工程B:中間体45
DCM(10mL)中の中間体44(300mg、0.66mmol)の混合物に、DMAP(7.25mg、0.059mmol)、無水酢酸(0.182mL、1.781mmol)およびEt3N(0.494mL、3.56mmol)を加えた。室温にて1時間攪拌後、反応混合物を氷水(20mL)で希釈し、DCM(100mL)で抽出した。有機相を水(40mL×3)、ブライン(40mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、淡黄色の固体を得た。その固体をMeOH(5mL)中に取り、沈殿物を回収し、冷MeOH(5mL)でリンスして、中間体45(250mg、85%)を白色の固体として得た。
【0120】
工程C:化合物46
1−メチルピペラジン(5mL)中の中間体45(250mg、0.503mmol)の溶液に、酢酸(0.014mL、0.252mmol)を加えた。それを150℃にて一晩攪拌後、室温まで冷却し、反応混合物を水(50mL)とDCM(100mL)との間に分配した。次いで有機相を分け、飽和塩化アンモニウム(40mL)、飽和重炭酸ナトリウム(40mL)、水(100mL×3)、ブライン(50mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して中間体46(270mg、84%)を淡黄色の固体として得た。LC/MS:m/z計算値596.5,実測値597.4(M+1)+。
【0121】
工程D:中間体47
DMSO(20mL)中のIBX(1.178g、4.21mmol)の溶液に、中間体46(270mg、0.421mmol)を加えた。それを50℃にて1時間攪拌後、反応混合物を水(150mL)とDCM(50mL)との間に分配した。有機相を分け、水(20mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、中間体47(200mg、80%)を淡黄色の固体として得た。LC/MS:m/z計算値594.5,実測値595.4(M+1)+。
【0122】
工程E:中間体48
MeOH(3mL)、THF(3mL)、および水(1.5mL)中の中間体47(200mg、0.336mmol)の溶液に、NaOH(1076mg、26.9mmol)を加えた。室温にて1時間攪拌後、反応混合物を水(150mL)とDCM(100mL)との間に分配した。有機相を分け、水(20mL×3)、ブライン(20mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、中間体48(77.7mg、37.7%)をオフホワイトの固体として得た。LC/MS:m/z計算値552.5,実測値553.2(M+1)+。
【0123】
工程F:化合物43−5
無水ピリジン(3mL)中の中間体48(100mg、0.181mmol)の溶液に、3,3−ジメチル−ジヒドロフラン−2,5−ジオン(232mg、1.809mmol)およびDMAP(66.3mg,0.543mmol)を加えた。反応混合物を120℃にてマイクロ波下で1.5時間加熱し、溶媒を真空内で除去し、残渣をEtOAc(50mL)中に取った。有機相をHCl水溶液(0.5N、20mL)、水(30mL×2)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、化合物43−5(32mg、18.74%)を白色の固体として得た。1HNMR(400Hz,MeOD)δppm4.51−4.47(1H,m),3.75−3.65(2H,t),3.32(4H,br),3.31−3.27(1H,m),3.05(4H,br),2.91(3H,s),2.67−2.55(4H,m),2.32−2.28(1H,m),2.20−2.17(1H,m),2.06−2.03(1H,m),1.95−0.88(45H,m).LC/MS:m/z計算値680.5,実測値681.4(M+1)+。
【0124】
表3中の化合物を、上記で示した手順と同様の方法で調製した。各場合において、XおよびYは、それぞれCH2である。
【化34】
【表3−1】
【表3−2】
【表3−3】
【0125】
実施例18:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3a−(2−(4−メチルピペラジン−1−イル)−2−オキソアセチル)−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化35】
【化36】
【0126】
工程A:中間体49
無水THF(50mL)中の1,3−ジアチン(4g、33.2mmol)の溶液に、n−BuLi(THF中2.5M)(18mL、45mmol)を−20℃にてアルゴン雰囲気下でゆっくりと加えた。得られた混合物を−20℃にて2時間攪拌した。−78℃まで冷却後、無水THF(5mL)中の中間体35(3g、6.21mmol)を反応混合物に加え、得られた混合物を−70℃にてさらに1時間攪拌し、その後反応物、重炭酸ナトリウム(10mL)の10%溶液を添加することでクエンチした。反応混合物をDCM(100mL)で希釈し、有機相を分け、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これをシリカゲル上でのカラムクロトマグラフィー(Hex:EtOAc=10:1)により精製することで、中間体49(1.7g、48.8%)を白色の固体として得た。LC/MS:m/z計算値560.3,実測値583.3(M+Na)+。
【0127】
工程B:中間体50
DCM(100mL)中の中間体49(1g、1.78mmol)、Et3N(0.743mL、5.35mmol)およびDMAP(65.3mg、0.535mmol)の溶液に、無水酢酸(0.184mL、1.961mmol)を加えた。それを0℃にて4時間攪拌後、反応混合物を水(50mL)とDCM(100mL)との間に分配した。有機相を分け、水、ブラインで洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して残渣を得て、これをEtOHおよびDCMから再結晶化して、中間体50(510mg、47.4%)を白色の固体として得た。LC/MS:m/z計算値602.9,実測値625.3(M+Na)+。
【0128】
工程C:中間体51
EtOH(45mL)および水(5mL)中の中間体50(2g、3.32mmol)の溶液に、硝酸銀(5.63g、33.2mmol)を加えた。反応混合物を60℃にて一晩攪拌後、不溶性物質を濾過で除去し、DCM(100mL×3)で洗浄した。有機相を水(100mL×3)、ブライン(100mL)で洗浄し、硫酸ナトリウムで乾燥させた。減圧下で蒸発させて、中間体51(1.5g、88%)を黄色の固体として得た。LC/MS:m/z計算値512.8,実測値535.2(M+Na)+。
【0129】
工程D:中間体52
DMSO(25mL)中の中間体51(700mg、1.37mmol)の溶液に、IBX(3823mg、13.65mmol)を加えた。それを30℃にて1時間攪拌後、反応混合物を水(20mL)とDCM(300mL)との間に分配した。次いで有機相を分け、水、ブラインで洗浄し、硫酸ナトリウムで乾燥させた。溶媒を除去して、中間体52(370mg、51.5%)を黄色の固体として得た。LC/MS:m/z計算値526.3,実測値525.3(M−1)−。
【0130】
工程E:中間体53
DCM(10mL)中の中間体52(260mg、0.494mmol)の溶液に、シュウ酸ジクロライド(0.418mL、4.94mmol)を加えた。室温にて1時間攪拌後、反応混合物を真空内で蒸発させて、中間体53(269mg、100%)を黄色の固体として得た。
【0131】
工程F:中間体54
DCM(10mL)中の中間体53(270mg、0.495mmol)、およびEt3N(551mg、5.45mmol)の溶液に、1−メチルピペラジン(496mg、4.95mmol)を加えた。室温にて1時間攪拌後、反応混合物を水(20mL)とDCM(150mL)との間に分配した。有機相を分け、水(20mL)、ブライン(10mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発乾固させて、中間体54(220mg、73%)を黄色の固体として得た。LC/MS:m/z計算値608.4,実測値609.4(M+1)+。
【0132】
工程G:中間体55
THF(2mL)、MeOH(2mL)、および水(1mL)中の中間体54(220mg、0.361mmol)の溶液に、NaOH(1.1g、28.9mmol)を加えた。室温にて1時間攪拌後、反応混合物を水(20mL)とDCM(150mL)との間に分配した。有機相を分け、水(20mL)、ブライン(10mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、中間体55(150mg、73.2%)を黄色の固体として得た。LC/MS:m/z計算値566.4,実測値567.3(M+1)+。
【0133】
工程H:化合物56−1
ピリジン(2mL)中の中間体55(678mg、5.29mmol)およびDMAP(97mg、0.794mmol)の溶液に、3,3−ジメチル−ジヒドロフラン−2,5−ジオン(678mg、5.29mmol)を加えた。反応混合物を120℃にて3時間攪拌後、溶媒を真空内で除去し、残渣をEtOAc(50mL)中に取った。有機相をHCl水溶液(0.5N、20mL)、水(30mL×2)、ブライン(30mL)で洗浄し、硫酸ナトリウムで乾燥させ、真空内で蒸発させて、残渣を得て、これを分取−HPLCにより精製して、標題化合物56−1(30mg、14.01%)を白色の固体として得た。1HNMR(400MHz,MeOD)δppm:4.51−4.47(1H,m),3.72−3.35(8H,m),3.29−3.22(1H,m),2.98(3H,s),2.61(2H,q,J=16Hz),2.50−0.88(50H,m).LC/MS:計算値694.5,実測値695.4(M+1)+。
【0134】
実施例19:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3a−(2−オキソ−2−(2−フェニルプロパン−2−イルアミノ)アセチル)−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化37】
【0135】
実施例18に記載されたものと同様の手順を用いて標題化合物を調製した。1HNMR(400MHz,MeOD)δppm:8.23(1H,s),7.29−7.28(2H,m),7.21−7.17(2H,m),7.11−7.07(1H,m),4.40−4.36(1H,m),3.65−3.44(1H,m),3.11−3.04(1H,m),2.50(2H,q,J=16.4Hz),2.40−2.05(6H,m),1.91−0.77(49H,m).LC/MS:計算値729.5,実測値m/z730.3(M+1)+。
【0136】
実施例20:4−((3aS,5aR,5bR,7aR,9S,11aR,11bR,13aS)−3a−(2−((4−クロロベンジル)(メチル)アミノ)−2−オキソアセチル)−1−イソプスティキル−5a,5b,8,8,11a−ペンタメチル−3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a−オクタデカヒドロ−2H−シクロペンタ[a]クリセン−9−イルオキシ)−2,2−ジメチル−4−オキソブタン酸
【化38】
【0137】
実施例18に記載されたものと同様の手順を用いて標題化合物を調製した。1HNMR(400MHz,CDCl3)δppm:7.32−7.25(3H,m),7.18(1H,m),4.67−4.24(3H,m),3.23−3.19(1H,m),2.86(2H,2/3CH3,s),2.79(1H,1/3CH3,s),2.63(1H,d,J=15.6Hz),2.58(1H,d,J=15.6Hz),2.44−0.79(50H,m).LC/MS:計算値749.44,実測値772.5(M+Na)+。
【特許請求の範囲】
【請求項1】
以下の式により特徴付けられる化合物またはその薬学上許容される塩:
【化1】
(式中、
R1およびR2は各々独立して、H、C1−C6−アルキル、t−ブチルオキシカルボニル、Me−SO2−、HOOCC(CH3)2CH2C(O)−、CH3C(O)、(R4)2N−(CH2)m−、(R5)n−フェニル−Q−、(R6)q−Hetアリール−(CH2)p−、(R6)q−Hetalk−(CH2)r−、または(R6)q−シクロalk−(CH2)p−であるか、あるいはR1およびR2は、それらが結合する窒素原子と一緒に、メチルスルホニル基または2個以下のC1−C4−アルキル基で置換されてもよい3〜7員のヘテロシクロアルキル環を形成し、
R3は、
【化2】
であり、
各R4は独立してHまたはC1−C6−アルキルであり、
各R5は独立してハロ、C1−C6−アルキル、C1−C6−アルコキシ、CF3、OCF3、N(CH3)2、またはNO2であり、
各R6は独立してハロ、C1−C6−アルキル、−COOH、−C(O)NH2、ジメチルアミノメチル、または1−メチル−4−ピペラジニルメチルであり、
XおよびYは各々独立してメチレンまたはカルボニルであり、
Qは、−(CH2)p−、−C(O)−、−NH−C(O)−、−CH(CH3)−、−C(CH3)2−、1,1−シクロプロピルジイル、または1,1−シクロペンチルジイルであり、
Hetアリールは5〜6員のヘテロアリール基であり、
Hetalkは3〜7員のヘテロシクロアルキル基であり、
シクロalkは3〜6員のシクロアルキル基であり、
各mは独立して2または3であり、
各nは独立して0、1、または2であり、
各pは独立して0または1であり、
各qは独立して0、1、または2であり、かつ、
各rは独立して0、1、2、3、または4である)。
【請求項2】
R1が、H、メチル、ジメチルアミノエチル、t−ブチルオキシカルボニル、Me−SO2−、またはHOOCC(CH3)2CH2C(O)−であり、
R2が、H、(R5)n−フェニル−Q−、(R6)q−フラニル−(CH2)p−、(R6)q−ピリジル−(CH2)p−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、Hetalk−(CH2)r−、またはC3−C6−シクロアルキル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペリジニル、モルホリノ、チオモルホリノ、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、1−メチル−2−ピペラジノン−4−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、かつ、
各R5が独立してメチル、メトキシ、ハロ、CF3、またはOCF3であり、かつ、
各R6が独立してメチル、F、またはClである、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項3】
Xがメチレンであり、かつYがメチレンであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペリジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項4】
Xがメチレンであり、かつYがカルボニルであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペラジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項5】
Xがカルボニルであり、かつYがカルボニルであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペラジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項6】
Xがカルボニルであり、かつYがメチレンであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペラジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項7】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化3】
である、
請求項3に記載の化合物またはその薬学上許容される塩。
【請求項8】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化4】
である、
請求項4に記載の化合物またはその薬学上許容される塩。
【請求項9】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化5】
である、
請求項5に記載の化合物またはその薬学上許容される塩。
【請求項10】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化6】
である、
請求項6に記載の化合物またはその薬学上許容される塩。
【請求項11】
a)請求項1に記載の化合物またはその薬学上許容される塩と、2)薬学上許容される賦形剤とを含んでなる、組成物。
【請求項12】
HIV−1を治療する方法であって、それを患っている患者に、有効量の請求項1に記載の化合物またはその薬学上許容される塩を投与することを含んでなる、方法。
【請求項1】
以下の式により特徴付けられる化合物またはその薬学上許容される塩:
【化1】
(式中、
R1およびR2は各々独立して、H、C1−C6−アルキル、t−ブチルオキシカルボニル、Me−SO2−、HOOCC(CH3)2CH2C(O)−、CH3C(O)、(R4)2N−(CH2)m−、(R5)n−フェニル−Q−、(R6)q−Hetアリール−(CH2)p−、(R6)q−Hetalk−(CH2)r−、または(R6)q−シクロalk−(CH2)p−であるか、あるいはR1およびR2は、それらが結合する窒素原子と一緒に、メチルスルホニル基または2個以下のC1−C4−アルキル基で置換されてもよい3〜7員のヘテロシクロアルキル環を形成し、
R3は、
【化2】
であり、
各R4は独立してHまたはC1−C6−アルキルであり、
各R5は独立してハロ、C1−C6−アルキル、C1−C6−アルコキシ、CF3、OCF3、N(CH3)2、またはNO2であり、
各R6は独立してハロ、C1−C6−アルキル、−COOH、−C(O)NH2、ジメチルアミノメチル、または1−メチル−4−ピペラジニルメチルであり、
XおよびYは各々独立してメチレンまたはカルボニルであり、
Qは、−(CH2)p−、−C(O)−、−NH−C(O)−、−CH(CH3)−、−C(CH3)2−、1,1−シクロプロピルジイル、または1,1−シクロペンチルジイルであり、
Hetアリールは5〜6員のヘテロアリール基であり、
Hetalkは3〜7員のヘテロシクロアルキル基であり、
シクロalkは3〜6員のシクロアルキル基であり、
各mは独立して2または3であり、
各nは独立して0、1、または2であり、
各pは独立して0または1であり、
各qは独立して0、1、または2であり、かつ、
各rは独立して0、1、2、3、または4である)。
【請求項2】
R1が、H、メチル、ジメチルアミノエチル、t−ブチルオキシカルボニル、Me−SO2−、またはHOOCC(CH3)2CH2C(O)−であり、
R2が、H、(R5)n−フェニル−Q−、(R6)q−フラニル−(CH2)p−、(R6)q−ピリジル−(CH2)p−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、Hetalk−(CH2)r−、またはC3−C6−シクロアルキル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペリジニル、モルホリノ、チオモルホリノ、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、1−メチル−2−ピペラジノン−4−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、かつ、
各R5が独立してメチル、メトキシ、ハロ、CF3、またはOCF3であり、かつ、
各R6が独立してメチル、F、またはClである、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項3】
Xがメチレンであり、かつYがメチレンであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペリジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項4】
Xがメチレンであり、かつYがカルボニルであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペラジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項5】
Xがカルボニルであり、かつYがカルボニルであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペラジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項6】
Xがカルボニルであり、かつYがメチレンであり、
R1が、H、メチル、t−ブチルオキシカルボニル、Me−SO2−、またはジメチルアミノエチルであり、
R2が、H、(R5)n−フェニル−(CH2)p−、(R6)n−フラニル−(CH2)q−、(R6)n−ピリジル−(CH2)q−、(R6)q−チエニル−(CH2)p−、1−メチルピラゾール−3−イル、ピロリジニル−(CH2)r−、4−メチルピペラジニル、N−メチルピペラジン−4−イル、シクロプロピル−(CH2)p−、シクロヘキシル−(CH2)p−、またはシクロペンチル−(CH2)p−であるか、あるいは
R1およびR2が、それらが結合する窒素原子と一緒に、アゼチジニル、ピペラジニル、4−メチル−ピペラジン−1−イル、4−メチルスルホニル−ピペラジン−1−イル、2,4−ジメチル−ピペラジン−1−イル、4−メチル−ジアゼパン−1−イル、チオモルホリン−1,1ジオキシド−4−イル、またはピロリジニルを形成し、
R5が、メチル、メトキシ、F、Cl、CF3、またはOCF3であり、かつ、
qが0または1である、
請求項1に記載の化合物またはその薬学上許容される塩。
【請求項7】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化3】
である、
請求項3に記載の化合物またはその薬学上許容される塩。
【請求項8】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化4】
である、
請求項4に記載の化合物またはその薬学上許容される塩。
【請求項9】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化5】
である、
請求項5に記載の化合物またはその薬学上許容される塩。
【請求項10】
R1が、H、CH3、またはジメチルアミノエチルであり、
R2が、(R5)n−フェニル−CH2、チエニル−CH2−、フラニル−CH2、ピリジニル−CH2−であり、かつ、
R3が、
【化6】
である、
請求項6に記載の化合物またはその薬学上許容される塩。
【請求項11】
a)請求項1に記載の化合物またはその薬学上許容される塩と、2)薬学上許容される賦形剤とを含んでなる、組成物。
【請求項12】
HIV−1を治療する方法であって、それを患っている患者に、有効量の請求項1に記載の化合物またはその薬学上許容される塩を投与することを含んでなる、方法。
【公表番号】特表2013−519674(P2013−519674A)
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願番号】特願2012−552944(P2012−552944)
【出願日】平成23年2月9日(2011.2.9)
【国際出願番号】PCT/US2011/024174
【国際公開番号】WO2011/100308
【国際公開日】平成23年8月18日(2011.8.18)
【出願人】(509329523)グラクソスミスクライン エルエルシー (38)
【Fターム(参考)】
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願日】平成23年2月9日(2011.2.9)
【国際出願番号】PCT/US2011/024174
【国際公開番号】WO2011/100308
【国際公開日】平成23年8月18日(2011.8.18)
【出願人】(509329523)グラクソスミスクライン エルエルシー (38)
【Fターム(参考)】
[ Back to top ]