
ベンジルピペリジン化合物
【課題】抗うつ薬として有用な新規薬剤の提供。
【解決手段】式(1):

[式中、R1は、水素原子またはメチル基を表し、R2は、メチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表し、Xは、メチレンまたは酸素原子を表し、nは、1〜3の整数を表す。]で表されるベンジルピペリジン化合物、またはその薬学上許容される塩。
【解決手段】式(1):
[式中、R1は、水素原子またはメチル基を表し、R2は、メチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表し、Xは、メチレンまたは酸素原子を表し、nは、1〜3の整数を表す。]で表されるベンジルピペリジン化合物、またはその薬学上許容される塩。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、セロトニン再取り込み阻害剤として有用な、新規なベンジルピペリジン化合物又はその薬学上許容される塩に関する。さらに詳しくは、本発明のベンジルピペリジン化合物は、ピペリジンを母骨格とする化合物であり、ピペリジンの4位に特定の置換ベンジル基を有し、さらにオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基(フェネチル基)をピペリジンの1位に有する。本発明のベンジルピペリジン化合物は、セロトニン再取り込み阻害作用を有することから、例えば抗うつ薬として有用である。
【背景技術】
【0002】
うつ病はあらゆる年令の人に影響を与える慢性病である。現在、使用されている各種の抗うつ薬のうち最も成功を収めているのは、選択的セロトニン再取り込み阻害剤(Selective serotonin reuptake inhibitor、以下SSRIと略すこともある)である。SSRIは、ドーパミン及びノルアドレナリン再取り込み阻害作用よりも高いセロトニン再取り込み阻害作用を有する。SSRIとして市販された最初の薬剤はジメリジン(zimelidine)であった。その後上市された又は開発下にある他のSSRIとしては、例えば、フルオキセチン(fluoxetine)、フルボキサミン(fluvoxamine)、シタロプラム(citalopram)、セルトラリン(sertraline)およびパロキセチン(paroxetine)が挙げられる。
【0003】
このようなSSRIはうつ病の治療薬として広く用いられているものの、まだいくつかの問題点を有することが指摘されている。全うつ病患者の約1/3を占める難治性の患者に対しては、SSRIでも十分な治療効果を上げられないことや、十分な抗うつ作用が発現するまでに3〜8週間もの長い期間を必要とすることが、代表的な問題として挙げられる。このようにSSRIの抗うつ作用の発現が緩慢である一方、その副作用は直ちに起こり得る。すなわち、患者が薬剤の治療効果を得ることなく副作用のみを経験する易損性期(vulnerable period)を招くという問題が生じる。このため、この期間中も同じ薬剤の服用を続けるように患者を説得することが治療医師にとってしばしば重い負担になる。更に、自殺を図る恐れのある患者にとっては、抗うつ作用の発現が緩慢であるため、十分なうつ症状の改善を経験する前に自発性(initiative)を回復することから、自殺の危険性やたびたびの入院の必要性などが生じる。従って、抗うつ作用が素早く発現するような抗うつ薬の開発が望まれている。
【0004】
SSRIが抗うつ作用を発現するまでに数週間もの長い期間を必要とする理由は、以下のように考えられている。
SSRIはセロトニン代謝回転の急性セロトニン再取り込みを阻害する。この阻害作用がセロトニンニューロンの神経終末において起こることにより、セロトニンによる神経伝達が強化され抗うつ作用が発現する。しかしながら、同阻害作用は縫線核に存在するセロトニンニューロン細胞体や樹状突起においても起こるため、縫線核ではセロトニン1A自己受容体を介するセロトニンニューロンの自己発火抑制(negative feedback反応)を強化してしまう。この結果、SSRI投与後の初期においては、セロトニンニューロンにおける神経伝達は全体として期待されるほど強化されないことになる。一方、数週間SSRIの服用を続けるうちに、縫線核のセロトニンニューロン細胞体や樹状突起上にあるセロトニン1A自己受容体は脱感作され、negative feedback反応が消失する。この結果、セロトニンニューロンの活動性の亢進と神経終末でのセロトニン取り込み阻害が協調して奏効し、セロトニン神経伝達が強化され、十分な抗うつ作用が発現する。
【0005】
従って、セロトニン1A受容体アンタゴニストの併用によりセロトニン1A自己受容体を遮断してセロトニンのnegative feedback反応を止めるか、あるいはセロトニン1A受容体アゴニストの併用によりセロトニン1A自己受容体を積極的に刺激し脱感作までの期間を短縮することで、SSRIの作用発現までの期間の短縮や、抗うつ作用の増強が可能となる。実際、セロトニン1A受容体に対して高い親和性を有するピンドロール(pindolol)をSSRIと併用すると、うつ病患者におけるセロトニン再取り込み阻害薬の作用を増強すること、また作用発現までの期間を短縮することが報告されている(Arch, Gen. Psychiatry, (1994),51,248−251)。
【0006】
患者が薬剤を服用する際、その薬剤の数や種類はより少ないことが望ましい。従って、上記知見に基づき、セロトニン再取り込み阻害作用とセロトニン1A受容体への親和性を併せ持つ化合物は、他の薬剤と併用することなく単剤で、抗うつ作用が強く、作用発現までの期間が短縮された新しい抗うつ薬となり得ると考えられ、このような化合物の薬剤としての開発が望まれている。
【0007】
セロトニン再取り込み阻害作用とセロトニン1A受容体への親和性を併せ持つ化合物としては、これまでに、4位に置換ベンジル基を、1位に置換フェニルエチル基を有するベンジルピペリジン誘導体が報告されている(例えば、特許文献1参照)。具体的には、式(A):
【0008】
【化1】
[式中、R0は水素原子、ハロゲン原子、アルキル基、置換アルコキシ基などを表し、R0は独立して複数存在し、R3は水素原子などを表し、nは整数2などを表し、mは整数2などを表し、R5およびR6は各々独立して水素原子などを表し、Zは置換アリール基などを表す。]
で表される環状アミンなどを有効成分とするセロトニン再取り込み阻害剤が開示されている。されにこれらのセロトニン再取り込み阻害剤がセロトニン1A拮抗作用を有することも開示されている。
【0009】
一方、ピペリジンの4位に置換ベンジル基を有する化合物は、複数の文献で報告されている。例えば、脳血管障害治療薬として作用する環状アミン誘導体を開示する文献(特許文献2参照)や、NMDA受容体アンタゴニストとして作用する4−置換ピペリジンを開示する文献(特許文献3参照)が挙げられる。
【0010】
さらに、ピペリジンの1位に置換フェニルエチル基を有する化合物も、いくつかの文献で報告されている。フェニルエチル基上の置換基として環状ケトン構造を持つピペリジン環を有するインドール誘導体が5−HT1Aアンタゴニストとして報告されている(例えば、特許文献4参照)。これらのインドール誘導体は、ピペリジンの4位に置換ベンジル基を有するベンジルピペリジン化合物とは骨格が異なる。また、同インドール誘導体がセロトニン再取り込み阻害作用を併せ持つとは報告されていない。
【0011】
これらの特許文献の何れにおいても、ピペリジンの4位に置換ベンジル基を有し、さらにオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基(フェネチル基)をピペリジンの1位に有するベンジルピペリジン化合物については具体的な開示や示唆はない。
【0012】
また三環系抗うつ薬(Tricyclic antidepressants、TCA)やSSRIなどの抗うつ薬の多くは薬の代謝に関与する酵素であってヒトチトクロームP450分子種の一つであるCYP2D6への阻害作用が強いことが知られている。一方、うつ病や不安症状の治療においてTCAやSSRIと併用され得る精神系疾患治療剤の多くがCYP2D6によって代謝されることも知られている。従って、これらの薬剤の併用においては、一方の薬剤によるCYP2D6の阻害作用に基づき他方の薬剤の代謝が阻害されることによって、後者の薬剤の血中濃度が上昇し、その結果重篤な副作用が発現する可能性がある。従って、抗うつ薬のCYP2D6の阻害作用がより弱い程、CYP2D6によって代謝される併用の精神系疾患治療剤との薬物相互作用がより小さくなることから、このような抗うつ薬は安全性が高い薬剤となり得ることが期待され、その開発が望まれている。
【0013】
更にCYP2D6は遺伝的多型による酵素活性の個体間変動が大きいことが知られている。CYP2D6によって代謝される割合の高い薬剤は、生体内薬物濃度に大きな個人差を生じ、通常代謝者(Extensive Metabolizer、EM)の場合と比較して代謝欠損者(Poor Metabolizer、PM)の場合、血中薬物濃度が大きく上昇する危険性が高い。またこのような薬剤は、CYP2D6を阻害する薬剤またはCYP2D6により代謝を受ける薬剤との薬物相互作用がより強く現れる危険性もある。従って、薬剤の代謝におけるCYP2D6の寄与率がより低いほど、CYP2D6の遺伝多型による薬物動態的影響がより小さくなることから、このような薬剤は安全性が高くなり得ることが期待され、その開発も望まれている。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】米国特許第6787560号
【特許文献2】国際公開第88/02365号パンフレット
【特許文献3】国際公開第97/23216号パンフレット
【特許文献4】国際公開第2005/108389号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明が解決しようとする課題は、セロトニン1A受容体に対する親和性を併せ持つ新しいセロトニン再取り込み阻害剤を提供することにある。このようなセロトニン再取り込み阻害剤は、例えばうつ病や不安(不安障害)などの治療薬になることが期待されることから、治療効果に優れ、さらに安全性の高い薬剤を提供することが本発明の課題である。具体的な課題としては、ヒトセロトニン再取り込み阻害活性が向上し、セロトニン1A受容体に対する親和性を有し、ヒトチトクロームP450分子種の一つであるCYP2D6に対する阻害作用が弱く、またはヒトにおける薬物代謝においてCYP2D6の寄与が小さい薬剤を提供することが挙げられる。
【課題を解決するための手段】
【0016】
本発明者らは上記課題を解決するために鋭意検討した結果、化学構造上の特徴として、ベンゼン環部分の3位が2−メトキシエトキシ基または2−ヒドロキシエトキシ基で置換されているジ置換ベンジル基を有し、且つオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基をピペリジンの1位に有するベンジルピペリジン化合物またはその薬学上許容される塩が、高いヒトセロトニン再取り込み阻害作用とヒト5−HT1A受容体に対する結合親和性を併せ持つのみならず、該化合物または該塩は、CYP2D6阻害が弱く、また代謝におけるCYP2D6の寄与が小さいことも見出した。これらの知見を基に、本発明を完成させるに至った。
本発明は、以下の〔1〕〜〔8〕で表される、セロトニン再取り込み阻害剤として有用な、ベンジルピペリジン化合物またはその薬学上許容される塩に関するものである。すなわち、本発明は、
〔1〕 式(1):
【0017】
【化2】
[式中、R1は、水素原子またはメチル基を表し、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表し、Xは、メチレン基または酸素原子を表し、nは、1〜3の整数を表す。]
で表される化合物、またはその薬学上許容される塩;
〔2〕 Xがメチレン基を表し、nが1〜2の整数を表すか、または、Xが酸素原子を表し、nが2〜3の整数を表す、〔1〕に記載の化合物、またはその薬学上許容される塩;
〔3〕 R1がメチル基を表す、〔1〕または〔2〕に記載の化合物、またはその薬学上許容される塩;
〔4〕 R2がp位に結合した臭素原子を表す、〔1〕〜〔3〕のいずれかに記載の化合物、またはその薬学上許容される塩;
〔5〕 Xが酸素原子を表し、nが2の整数を表す、〔1〕〜〔4〕のいずれかに記載の化合物、またはその薬学上許容される塩;
〔6〕 R2がp位に結合した臭素原子を表し、Xが酸素原子を表し、nが2の整数を表す、〔1〕または〔2〕に記載の化合物、またはその薬学上許容される塩;
〔7〕 式(1)で表される化合物が、以下の化合物(01)〜(15)からなる群:
(01)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(02)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(03)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(04)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(05)6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(06)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(07)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(08)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(09)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(10)6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(11)6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(12)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(13)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(14)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、および
(15)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン
から選択される、〔1〕に記載の化合物、またはその薬学上許容される塩;
〔8〕 薬学上許容される塩が塩酸塩、臭化水素酸塩、フマル酸塩、ベンゼンスルホン酸塩またはコハク酸塩である、〔1〕〜〔7〕に記載の化合物の薬学上許容される塩;
に関する。
また、本発明は、以下の〔9〕〜〔12〕で表される、医薬組成物または治療薬もしくは予防薬に関するものである。すなわち、本発明は、
〔9〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有する医薬組成物;
〔10〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有するセロトニン再取り込み阻害剤;
〔11〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有する抗うつ薬または抗不安薬;
〔12〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有する抗うつ薬;
に関する。
【0018】
また、本発明は、以下の〔13〕で表される、〔1〕〜〔7〕に記した本発明のベンジルピペリジン化合物の中間体に関するものである。すなわち、本発明は、
〔13〕 式(11):
【0019】
【化3】
[式中、R1は、水素原子またはメチル基を表し、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表す。]
で表される化合物に関する。
【0020】
また、本発明は、以下の〔14〕で表される、〔1〕〜〔7〕に記した本発明のベンジルピペリジン化合物の中間体に関するものである。すなわち、本発明は、
〔14〕 式(12):
【0021】
【化4】
[式中、Xは、メチレン基または酸素原子を表し、nは、1〜3の整数を表し、LG1は、ヨウ素原子、臭素原子、塩素原子、置換スルホニルオキシ基を表す。]
で表される化合物に関する。
【発明の効果】
【0022】
本発明により、うつ病等の治療薬となり得るセロトニン再取り込み阻害剤として有用な、ベンジルピペリジン化合物またはその薬学上許容される塩を提供することが可能になった。詳しくは、本発明により、高いヒトセロトニン再取り込み阻害活性とヒト5−HT1A受容体に対して結合親和性を有し、CYP2D6阻害が弱く、また代謝におけるCYP2D6の寄与が小さいベンジルピペリジン化合物またはその薬学上許容される塩を提供することが可能になった。
【発明を実施するための最良の形態】
【0023】
以下に、本発明をさらに具体的に説明する。
本発明の式(1)で表されるベンジルピペリジン化合物は、化学構造上の特徴として、ベンゼン環部分の3位が2−メトキシエトキシ基または2−ヒドロキシエトキシ基で置換されているジ置換ベンジル基を有し、且つオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基をピペリジンの1位に有するものである。
本発明において、「置換スルホニルオキシ基」は、アルキル基または置換されていてもよいフェニル基で置換されたスルホニルオキシ基を意味する。ここにおいて、アルキル基としては、炭素数1〜6の直鎖状又は分枝鎖状のアルキル基が挙げられ、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、ヘキシル基、トリフルオロメチル基などが挙げられる。置換されていてもよいフェニル基の置換基としては、ハロゲン原子(ここで、ハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられる。)、アルキル基(ここで、アルキル基は、炭素数1〜6の直鎖状又は分枝鎖状アルキル基を表し、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、ヘキシル基などが挙げられる。)、トリフルオロメチル基、シアノ基、ニトロ基、またはアルコキシ基(ここで、アルコキシ基は、炭素数1〜6の直鎖状又は分枝鎖状のアルコキシ基を表し、具体的には、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、ペンチルオキシ基、ヘキシルオキシ基などが挙げられる。)好ましい置換スルホニルオキシ基としては、メタンスルホニルオキシ基、ベンゼンスルホニルオキシ基およびp−トルエンスルホニルオキシ基が挙げられ、さらに好ましい置換スルホニルオキシ基としては、ベンゼンスルホニルオキシ基およびp−トルエンスルホニルオキシ基が挙げられる。
【0024】
式(1)において、R1として好ましくはメチル基が挙げられる。(ここへ移動しました)
式(1)において、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表す。例えば、R2がp位に結合した塩素原子、p位に結合した臭素原子またはp位に結合したメチル基を表す場合、式(1)の化合物は、式(1−p):
【0025】
【化5】
[式中、R1、Xおよびnは前記と同義であり、R2pは塩素原子、臭素原子またはメチル基を表す。]
で表される化合物を表す。
一方、R2がm位に結合した塩素原子またはm位に結合した臭素原子を表す場合、式(1)の化合物は、式(1−m):
【0026】
【化6】
[式中、R1、Xおよびnは前記と同義であり、R2mは塩素原子または臭素原子を表す。]
で表される化合物を表す。式(1)において、R2として好ましくはp位に結合した臭素原子が挙げられる。すなわち、式(1−p−Br):
【0027】
【化7】
[式中、R1、Xおよびnは、前記と同義である。]
で表される化合物が好ましい。
【0028】
式(1)において、Xがメチレン基を表し、nが1の整数を表す、式(1)の化合物としては、式(1−C−1):
【0029】
【化8】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xがメチレン基を表し、nが2の整数を表す、式(1)の化合物としては、式(1−C−2):
【0030】
【化9】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xがメチレン基を表し、nが3の整数を表す、式(1)の化合物としては、式(1−C−3):
【0031】
【化10】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xが酸素原子を表し、nが1の整数を表す、式(1)の化合物としては、式(1−O−1):
【0032】
【化11】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xが酸素原子を表し、nが2の整数を表す、式(1)の化合物としては、式(1−O−2):
【0033】
【化12】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xが酸素原子を表し、nが3の整数を表す、式(1)の化合物としては、式(1−O−3):
【0034】
【化13】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表す。
式(1)において、Xおよびnとしては、Xがメチレンを表しnが1〜2の整数を表すか、または、Xが酸素原子を表しnが2〜3の整数を表す場合が好ましい。すなわち、式(1−C−1)、式(1−C−2)、式(1−O−2)または式(1−O−3)で表される化合物が好ましい。Xおよびnとしてより好ましくは、Xが酸素原子を表しnが2の整数を表す場合があげられ、すなわち、式(1−O−2)で表される化合物がより好ましい。
より具体的には、以下の化合物(01)〜(15):
(01)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(02)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(03)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(04)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(05)6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(06)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(07)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(08)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(09)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(10)6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(11)6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(12)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(13)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(14)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、および
(15)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン
が好ましい。
【0035】
本発明のベンジルピペリジン化合物は、公知化合物から、以下に示す製造方法1〜5に示す方法、下記の製造方法に類似の方法、または当業者に周知の合成方法を適宜組み合わせて製造することができる。原料化合物(11)、(12)、(13)、(15)、(18)および(19)において、いくつかのものは新規であるが、後記の実施例に記載の方法、または実施例に類似の方法、または当業者に周知の合成方法を適宜組み合わせて製造することもできる。
また、本明細書において、記載の簡略化のために次の略号を使用する場合がある。
Boc:tert-ブトキシカルボニル基
Piv:tert-ブチルカルボニル基
Me:メチル基
Et:エチル基
Ph:フェニル基
Bn:ベンジル基
Ms:メタンスルホニル基
Bs:ベンゼンスルホニル基
Ts:p−トルエンスルホニル基
p:パラ(例えば、「p−Br」はパラ位に結合した臭素原子を意味する。)
m:メタ(例えば、「m−Br」はメタ位に結合した臭素原子を意味する。)
DMSO:ジメチルスルホキシド
【0036】
製造方法1:化合物(1)の製造方法
式(1)で表される化合物またはその塩は、例えば下記の方法によって製造できる。
【0037】
【化14】
[式中、R1、R2、X、nおよびLG1は、前記と同義である。]
【0038】
目的化合物(1)またはその塩は、化合物(11)またはその塩を化合物(12)と反応させることにより得ることができる。反応は、必要に応じ塩基の存在下、また、必要に応じ相間移動触媒の存在下、適当な不活性溶媒中で約−20℃〜用いた溶媒の沸点までの範囲の温度で、10分間〜48時間反応させることにより行うことができる。
塩基としては、例えばトリエチルアミン、ジイソプロピルエチルアミン、ピリジン等の有機塩基、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、炭酸水素カリウム、炭酸水素ナトリウム、りん酸二水素カリウム、りん酸水素二カリウム、りん酸カリウム、りん酸二水素ナトリウム、りん酸水素二ナトリウム、りん酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水素化ナトリウム等の無機塩基、ナトリウムメトキシド、カリウムtert-ブトキシド等の金属アルコキシド等が挙げられる。好ましくは炭酸カリウムおよびりん酸水素二カリウムが挙げられる。
相間移動触媒としては、例えば硫酸水素テトラブチルアンモニウムなどが挙げられる。
不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、アセトン、メチルエチルケトン、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。より好ましい溶媒としてはアセトニトリル、トルエン、ジメチルホルムアミドおよびN−メチル−2−ピロリジノンもしくはこれらの混合溶媒が挙げられる。
脱離基となるLG1としては臭素基や置換スルホニルオキシ基が好ましく、ベンゼンスルホニルオキシ基やp−トルエンスルホニルオキシ基がさらに好ましい。
【0039】
製造方法2:化合物(11)の製造方法
製造方法1の出発原料として用いられる化合物(11)またはその塩は、例えば米国特許第6787560号などの文献の方法を参考にして、下記の方法によって製造できる。
【0040】
【化15】
[式中、R1およびR2は、前記と同義である。PG1は窒素原子の保護基を、LG2は脱離基を表す。窒素原子の保護基PG1としては、例えばt−ブチルオキシカルボニル基、9−フルオレニルメチルオキシカルボニル基などのアルキルオキシカルボニル基が挙げられる。脱離基LG2としては、例えば塩素原子、臭素原子、ヨウ素原子などのハロゲン原子や、p−トルエンスルホニルオキシ基、メタンスルホニルオキシ基などの置換スルホニルオキシ基が挙げられる。]
【0041】
化合物(13)をホスホン酸エステル(14a)またはホスホニウム塩(14b)に変換する。この変換はホスホン酸エステル(14a)の場合、亜リン酸トリエチルを無溶媒もしくは不活性溶媒中、氷冷から用いた溶媒もしくは亜リン酸トリエチルの沸点までの間の温度で、1時間〜3日間反応させることにより行うことができる。ホスホニウム塩(14b)の場合、トリフェニルホスフィンを不活性溶媒中、氷冷から用いた溶媒の沸点までの間の温度で、1時間〜3日間反応させることにより行うことができる。
このホスホン酸エステル(14a)またはホスホニウム塩(14b)とケトン(15)を塩基の存在下、適当な不活性溶媒中で約−20℃から用いた溶媒の沸点までの温度で、10分間〜48時間反応させることにより、化合物(16)に変換することができる。
塩基としては、例えばトリエチルアミン、ピリジン等の有機塩基、炭酸カリウム、炭酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水素化ナトリウム等の無機塩基、ナトリウムメトキシド、カリウムtert-ブトキシド等の金属アルコキシドが挙げられる。
不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。
化合物(16)を接触還元することにより化合物(17)に変換することができる。R2がp位に結合した臭素原子またはm位に結合した臭素原子を表す場合には、この還元反応の触媒としてロジウム炭素等のロジウム系の触媒や白金炭素や酸化白金等の白金系の触媒、ルテニウム炭素等のルテニウム系の触媒や塩化パラジウムなどを用い、常圧もしくは加圧水素雰囲気下、適当な不活性溶媒中で0℃〜50℃にて反応させることにより行うことができる。適当な不活性溶媒としては酢酸エチルや、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。より好ましい触媒としてはロジウム炭素や白金炭素が挙げられる。またこの場合、より好ましい溶媒としては酢酸エチルが挙げられる。
化合物(17)を常法により脱保護することにより目的とする化合物(11)を得ることができる。保護基がt−ブチルオキシカルボニル基の場合は適当な不活性溶媒中で−20℃から用いた溶媒の沸点までの間の温度において塩酸や硫酸などの無機酸やトリフルオロ酢酸などの有機酸で処理することにより脱保護が行える。不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。保護基が9−フルオレニルメチルオキシカルボニル基の場合は適当な不活性溶媒中で−20℃から用いた溶媒の沸点までの間の温度において、ピロリジン、ピペリジン、モルホリン、トリエチルアミンやジイソプロピルエチルアミンなどの有機塩基で処理することにより脱保護が行える。不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。
【0042】
製造方法3:化合物(12)の製造方法
製造方法1の出発原料として用いられる化合物(12)は、例えば下記の方法によって製造できる。
【0043】
【化16】
[式中、Xおよびnは、前記と同義である。R3は水素原子またはアルキル基を表す。該アルキル基としては、直鎖状または分枝上の炭素原子数が1〜6のアルキル基が挙げられ、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基、1−メチルブチル基、ヘキシル基などを挙げることができる。]
【0044】
化合物(18)を適当な還元剤(例えば水素化リチウムアルミニウム、水素化ホウ素リチウム、水素化ホウ素ナトリウム、ジボランなど)を用いて、適当な不活性溶媒(例えばジエチルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン等のエーテル系溶媒など)中で、−20℃から用いた溶媒の沸点までの間の温度で、10分間〜48時間反応させることにより、化合物(19)を得ることができる。
化合物(19)を二酸化マンガンなどの酸化剤を用いて、適当な不活性溶媒中で酸化することにより化合物(20)を得ることができる。適当な不活性化溶媒としては、クロロホルムやジクロロメタンのようなハロゲン化溶媒、アセトニトリル、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリドン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。
化合物(20)の水酸基を常法により塩素原子、臭素原子もしくはヨウ素原子のハロゲン原子や、例えばp−トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基もしくはメタンスルホニルオキシ基などの置換スルホニルオキシ基に変換することにより化合物(12)を得ることができる。具体的には化合物(20)を例えばメタンスルホニルクロライド、ベンゼンスルホニルクロライドまたはp−トルエンスルホニルクロライドなどと不活性溶媒中で塩基の存在下、−20℃から用いた溶媒の沸点までの間の温度で、10分間〜48時間反応させることにより、化合物(12)を得ることができる。適当な不活性化溶媒としてはクロロホルムやジクロロメタンのようなハロゲン化溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリドン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。適当な塩基としてはトリエチルアミン、ピリジン等の有機塩基、炭酸カリウム、水酸化ナトリウムなどの無機塩基が挙げられる。またLG1が塩素原子、臭素原子などのハロゲンの場合は、LG1がp−トルエンスルホニルオキシ基、メタンスルホニルオキシ基などの置換スルホニルオキシ基である化合物(12)を、不活性溶媒中で例えば臭化リチウムなどと−20℃から用いた溶媒の沸点までの間の温度で10分間〜48時間反応させることにより得ることができる。適当な不活性化溶媒としてはクロロホルムやジクロロメタンのようなハロゲン化溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリドン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。また別法として化合物(20)を例えば適切な不活性溶媒中でトリフェニルホスフィン存在下、四塩化炭素や四臭化炭素と反応させることにより化合物(12)を得ることができる。
【0045】
製造方法4:化合物(12)の製造方法
原料化合物(12)は、例えば下記の方法によっても製造できる。
【0046】
【化17】
[上記式中、X、nおよびLG1は前記と同義である。]
【0047】
化合物(19)の一級水酸基を常法によりp−トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基、メタンスルホニルオキシ基などの置換スルホニルオキシ基に変換することにより化合物(21)を得ることができる。化合物(21)を適当な不活性溶媒中で、例えば二酸化マンガン酸化やジメチルスルホキシド(DMSO)酸化などの常法により、水酸基を酸化することにより、化合物(12)を得ることができる。
【0048】
製造方法5:化合物(20)の製造方法
製造方法3における中間体化合物(20)は、例えば下記の方法によって製造できる。
【0049】
【化18】
[式中、R3、Xおよびnは前記と同義である。PG2は、メトキシ基、メチルチオ基などを表すか、または2つのPG2が環を形成して、1,3−ジオキソラン基、1,3−ジオキサン基などの環状アセタール基を表してもよい。]
【0050】
化合物(18)のケトンを常法によりジアルキルアセタールやジアルキルチオアセタールに変換して化合物(22)とする。これを適当な不活性溶媒中、適当な還元剤(例えば水素化リチウムアルミニウム、水素化ホウ素リチウム、水素化ホウ素ナトリウムあるいはジボランなど)を用いて還元して化合物(23)とする。化合物(23)を適当な方法により脱保護することにより化合物(20)を得ることができる。
化合物(18)は例えばJournal of Medicinal Chemistry(1994), 37(21), 3482.、Journal of Medicinal Chemistry(1979), 22(12), 1464.、フランス特許第2672601号、日本特許 特開昭61−236774記載の方法などにより合成することができる。
【0051】
前記の製造方法において使用する原料や試薬などは、特に断らない限り、市販の化合物であるか、または公知の化合物から公知の方法を用いて製造することができる。また、前記式(1)の化合物において、官能基を適宜変換することによって、式(1)の別の化合物としてもよい。官能基の変換は、通常行われる一般的方法[例えば、コンプリヘンシブ・オーガニック・トランスフォーメーションズ(Comprehensive Organic Transformations)、アール.シー.ラロック(R.C.Larock)著(1989年)等参照]によって行うことができる。
【0052】
前記製造方法において、反応点以外の何れかの官能基が説明した反応条件下で変化するかまたは説明した方法を実施するのに不適切な場合は、当該官能基を予め適当な保護基で保護した上で、反応を実施し、その後、脱保護することにより、目的化合物を得ることができる。保護基としては、例えばプロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、ティー・ダブリュー・グリーン(T.W.Greene)著、ジョン・ワイリー・アンド・サンズ・インコーポレイテッド(John Wiley & Sons Inc.)(1981年)等に記載されているような通常の保護基を用いることができる。具体的には、アミンの保護基としてはエトキシカルボニル、tert-ブトキシカルボニル、ベンジルオキシカルボニル、アセチル、ベンゾイルまたはベンジル等を、また水酸基の保護基としてはトリアルキルシリル、アセチル、ベンゾイルまたはベンジル等を挙げることができる。ケトンの保護基としてはジメチルアセタール、1,3−ジオキサン、1,3−ジオキソラン、S,S’−ジメチルジチオアセタール、1,3−ジチアン、オキシム等をあげることができる。
保護基の導入および脱保護は、有機合成化学で常用される方法(例えば、上記のプロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis) 参照)またはそれらに準じた方法により行うことができる。
前記製造方法における中間体および目的化合物は、有機合成化学で常用される精製方法、例えば中和、濾過、抽出、洗浄、乾燥、濃縮、再結晶、各種クロマトグラフィー等により、単離精製することができる。また、中間体においては、特に精製することなく次の反応に供することも可能である。
【0053】
式(1)で表される本発明の化合物の中には、互変異性体が存在し得るものがある。互変異性の例としては、式(24)などが挙げられる。
【0054】
【化19】
本発明は、当該互変異性体を含め、全ての可能な異性体およびそれらの混合物を包含する。
【0055】
式(1)で表される化合物の薬学上許容される塩は、慣用の無毒性塩であり、有機酸塩(例えば酢酸塩、プロピオン酸塩、トリフルオロ酢酸塩、マレイン酸塩、フマル酸塩、クエン酸塩、コハク酸塩、酒石酸塩、メタンスルホン酸塩、ベンゼンスルホン酸塩、蟻酸塩、トルエンスルホン酸塩)もしくは無機酸塩(例えば塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、硝酸塩、リン酸塩)のような酸付加塩、アミノ酸(例えばアルギニン酸、アスパラギン酸、グルタミン酸)との塩、アルカリ金属塩(例えばナトリウム塩、カリウム塩)もしくはアルカリ土類金属塩(例えばカルシウム塩、マグネシウム塩)などの金属塩、アンモニウム塩、または有機塩基塩(例えばトリメチルアミン塩、トリエチルアミン塩、ピリジン塩、ピコリン塩、ジシクロヘキシルアミン塩、N,N’−ジベンジルエチレンジアミン塩)が挙げられる。
式(1)で表される化合物の薬学上許容される塩を取得するには、化合物(1)が薬学上許容される塩の形で得られる場合そのまま精製すればよく、一方、遊離の形で得られる場合、適当な有機溶媒に溶解もしくは懸濁させ、酸または塩基を加えて通常の方法により塩を形成させればよい。例えば水、メタノール、エタノール、アセトン等の溶媒中で、薬学上許容される酸又はアルカリと混合することで、塩にすることができる。
また、式(1)で表される化合物およびその薬学上許容される塩は、水との水和物またはエタノールなどの各種溶媒との溶媒和物として存在することもあるが、これら水和物や溶媒和物も本発明に包含される。
結晶として得られる式(1)で表される化合物およびその薬学上許容される塩には、結晶多形が存在する場合があり、その結晶多形も本発明に包含される。
【0056】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩は、ヒトセロトニン再取り込み阻害作用を有する。それゆえ、当該化合物および塩は、セロトニン神経系が介在する疾患の治療薬として有用である。セロトニン神経系が介在する疾患としては、例えば、うつ病や不安などが挙げられる。うつ病は、精神疾患の分類においては気分障害に含まれる。この気分障害の中には、主にはうつ病性障害と双極性障害がある。一般的なうつ病として、より詳細には、(i)大うつ病性障害、気分変調性障害もしくは特定不能なうつ病性障害を含むうつ病性障害、(ii)うつ病、または(iii)季節的情動障害などが挙げられる。これらの治療薬または再発予防薬として当該化合物および塩は有用である。さらに(iv)双極性障害における大うつ病エピソードの治療薬または再発予防薬としても当該化合物および塩は有用である。一方、不安(不安障害)の中には、主に不安障害と恐怖症がある。当該化合物および塩が治療薬または再発予防薬として有用な不安(不安障害)としては、(v)パニック障害、強迫性障害、外傷後ストレス障害、急性ストレス障害、全般性不安障害もしくは一般身体疾患による不安障害、(vi)物質誘発性不安障害を含む不安障害、(vii)広場恐怖症、(viii)社会的恐怖症、(ix)回避的人格異常、または(x)心身症などが挙げられる。また、他の疾患(統合失調症、認知症など)に伴ううつ症状または不安症状に対しても当該化合物および塩は有用である。さらに、当該化合物および塩は、痴呆、健忘症および加齢に関係した記憶障害を含む記憶障害;神経性食欲不良および神経性飢餓を含む摂食行動の障害;肥満症;睡眠障害;統合失調症;アルコール、たばこ、ニコチン、麻薬、覚せい剤、向精神薬等の薬物依存症;群発性頭痛;片頭痛;痛み;アルツハイマー病;慢性発作片頭痛;血管障害に関係した頭痛;パーキンソン病の痴呆、抑うつ、不安、神経弛緩薬誘導パーキンソン症候群および晩発性ジスキネジーを含むパーキンソン病;過プロラクチン血症などの内分泌異常;血管痙攣(特に、脳血管系の);高血圧症;運動性および分泌の変化が関与している胃腸管の障害;早発射精を含む性的機能不全などの治療または予防にも有用である。
【0057】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩の用量は患者の年齢および状態に応じて増減するが、化合物(1)の平均一回量約0.1mg、1mg、10mg、50mg、100mg、250mg、500mgおよび1000mgが、例えば前記のうつ病や不安などの疾患に対して有効である。一般には、ヒトに投与する場合、1日当り0.1mg/個体ないし約1,000mg/個体、好ましくは1日当り1mg/個体ないし約100mg/個体の量を投与することができる。1日の投与回数は、1回又は1日に数回、例えば各回1、2又は3用量を与える。
【0058】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩は、治療に使用する場合に、医薬組成物として、経口的または非経口的(例えば、静脈内、皮下、筋肉内、髄腔内、局所的、経直腸的、経皮的、経鼻的または経肺的)に投与することができる。経口投与のための投与形態としては、例えば、錠剤、カプセル剤、丸剤、顆粒剤、細粒剤、散剤、液剤、シロップ剤、懸濁剤などの剤形が挙げられ、非経口投与のための投与形態としては、例えば、注射用水性剤、注射用油性剤、坐剤、経鼻剤、経皮吸収剤[ローション剤、乳液剤、軟膏剤、クリーム剤、ゼリー剤、ゲル剤、貼付剤(テープ剤、経皮パッチ製剤、湿布剤等)、外用散剤等]等などの形態の製剤が挙げられる。これらの製剤は、従来公知の技術を用いて調製され、製剤分野において通常使用される無毒性かつ不活性な担体もしくは賦形剤を含有することができる。
【0059】
製剤用担体としては、製剤分野において常用され、かつ式(1)で表される化合物又はその薬学上許容される塩と反応しない物質が用いられる。すなわち、式(1)で表される化合物又はその薬学上許容される塩を含有する医薬組成物は、賦形剤、結合剤、滑沢剤、安定剤、崩壊剤、緩衝剤、溶解補助剤、等張化剤、溶解補助剤、pH調節剤、界面活性剤、乳化剤、懸濁化剤、分散剤、沈殿防止剤、増粘剤、粘度調節剤、ゲル化剤、無痛化剤、保存剤、可塑剤、経皮吸収促進剤、老化防止剤、保湿剤、防腐剤、香料等の製剤用担体を含有することができ、2種以上の製剤用担体添加物を適宜選択して用いることもできる。
【0060】
製剤用担体添加物として、具体的には、例えば乳糖、イノシトール、ブドウ糖、ショ糖、果糖、マンニトール(マンニット)、デキストラン、ソルビトール(ソルビット)、シクロデキストリン、デンプン(馬鈴薯デンプン、コーンスターチ、アミロペクチン等)、部分アルファー化デンプン、白糖、メタケイ酸アルミン酸マグネシウム、合成ケイ酸アルミニウム、アルギン酸ソーダ、結晶セルロース、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルデンプン、カルボキシメチルセルロースカルシウム、イオン交換樹脂、メチルセルロース、ゼラチン、アラビアゴム、プルラン、ヒドロキシプロピルセルロース、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロース、ヒドロキシエチルセルロース、ポリビニルピロリドン、ポリビニルアルコール、ゼラチン、アルギン酸、アルギン酸ナトリウム、軽質無水ケイ酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸アルミニウム、セトステアリルアルコール、ワックス、パラフィン、タルク、トラガント、ベントナイト、ビーガム、カルボキシビニルポリマー、酸化チタン、脂肪酸エステル、ソルビタン脂肪酸エステル、ラウリル硫酸ナトリウム、グリセリン、脂肪酸グリセリンエステル、精製ラノリン、グリセロゼラチン、ポリソルベート、マクロゴール、スクワラン、シリコーンオイル、植物油(ごま油、オリーブ油、大豆油、綿実油、ヒマシ油など)、液体パラフィン(流動パラフィン)、軟パラフィン、白色ワセリン、黄色ワセリン、パラフィン、羊毛脂、ロウ(蜜蝋、カルナウバロウ、サラシミツロウなど)、水、プロピレングリコール、ポリエチレングリコール、グリセロール、ラウリルアルコール、ミリスチルアルコール、オレイルアルコール、セチルアルコール、エタノール、塩化ナトリウム、水酸化ナトリウム、塩酸、クエン酸、ラウリル酸、ミリスチン酸、ステアリン酸、オレイン酸、ベンジルアルコール、グルタミン酸、グリシン、パラオキシ安息香酸メチル、パラオキシ安息香酸プロピル、p−ヒドロキシ安息香酸エステル類、コレステロールエステル、エチレングリコールモノアルキルエステル、プロピレングリコールモノアルキルエステル、モノステアリン酸グリセリン、ソルビタン脂肪酸エステル、ミリスチン酸イソプロピル、パルミチン酸イソプロピル、カルボキシポリメチレン、サッカリン、ストロベリーフレーバー、ペパーミントフレーバー、カカオ脂、ポリイソブチレン、酢酸ビニル共重合体、アクリル系共重合体、クエン酸トリエチル、クエン酸アセチルトリエチル、フタル酸ジエチル、セバシン酸ジエチル、セバシン酸ジブチル、アセチル化モノグリセリド、、ジエチレングリコール、ドデシルピロリドン、尿素、ラウリル酸エチル、エイゾン、カオリン、ベントナイト、酸化亜鉛、アガロース、カラギーナン、アカシアゴム、キサンタンガム、ラウリン酸カリウム、パルミチン酸カリウム、ミリスチン酸カリウム、セチル硫酸ナトリウム、ヒマシ油硫酸化物(ロート油)、Span(ステアリン酸ソルビタン、モノオレイン酸ソルビタン、セスキオレイン酸ソルビタン、トリオレイン酸ソルビタン等)、Tween(ポリソルベート20、ポリソルベート40、ポリソルベート60、ポリソルベート65、ポリソルベート80、ポリソルベート85、ポリオキシエチレンソルビタン脂肪酸エステル等)、ポリオキシエチレン硬化ヒマシ油(いわゆるHCO)、ポリオキシエチレンラウリルエーテル、ポリオキシエチレンセチルエーテル、ポリオキシエチレンオレイルエーテル、モノラウリン酸ポリエチレングリコール、モノステアリン酸ポリエチレングリコール、ポロキサマー(いわゆるプルロニック)、レシチン(ホスファチジルコリン、ホスファチジルセリンなどレシチンから単離された精製リン脂質をも含む)、レシチンの水素添加物等が挙げられる。
【0061】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩を、上記の如き医薬用途に使用する場合、通常、製剤用担体と混合して調製した製剤の形で投与され、製剤は通常の方法に従って調製される。例えば、本発明のベンジルピペリジン化合物およびその薬学上許容される塩を有効成分として0.051〜99重量%、好ましくは0.05〜80重量%、更に好ましくは0.1〜70%重量%、更に好ましくは0.1〜50重量%含有する医薬組成物とすることができる。これらの製剤はまた、治療上価値ある他の成分を含有していてもよい。
【0062】
本発明のベンジルピペリジル化合物およびその薬学上許容される塩は、その作用の増強を目的として、抗うつ薬、抗不安薬、統合失調症治療薬、ドパミン受容体作動薬、パーキンソン病治療薬、抗癲癇薬、抗痙攣薬、鎮痛薬、ホルモン製剤、偏頭痛治療薬、アドレナリンβ受容体拮抗薬、認知症治療薬、気分障害治療薬などの薬剤(併用薬剤)と組み合わせて用いることができる。また、その副作用抑制を目的として、制吐剤、睡眠導入剤、抗痙攣薬などの薬剤(併用薬剤)と組み合わせて用いることができる。本発明化合物及び併用薬剤の投与時期は限定されず、これらを投与対象に対し、同時に投与してもよいし、時間差をおいて投与してもよい。また、本発明化合物と併用薬剤の合剤としても良い。併用薬剤の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、本発明化合物と併用薬剤との配合比は、投与対象、投与ルート、対象疾患、症状、組み合わせなどにより適宜選択することができる。例えば投与対象がヒトである場合、本発明化合物1重量部に対し、併用薬剤を0.01〜1000重量部用いればよい。
【0063】
以下に本発明を、参考例、実施例及び試験例によりさらに詳細に説明するが、本発明の技術的範囲はこれら実施例に限定されるものではない。尚、以下の参考例及び実施例において示された化合物名は、必ずしもIUPAC命名法に従うものではない。
【0064】
化合物の同定には水素核磁気共鳴吸収スペクトル(1H−NMRスペクトル)等を用いた。いくつかの化合物については1H−NMRスペクトルスペクトルデータや融点を示した。また液体クロマトグラフィー分析により純度等の確認も行った。カラムにはSUMIPAX ODS C−212(5μm,6mmφ×15cm)を用い、測定波長を220 nm、移動層流速を1.0 ml/minに設定し、分析した。移動層としては0.05%トリフルオロ酢酸−アセトニトリル(A液)と0.05%トリフルオロ酢酸−水(B液)の混合溶媒を用いた。条件1ではA液とB液の混合比(A液:B液)を、測定開始時(0分)は25:75とし、測定開始40分後で50:50となるようにA液の比率を1分間に0.625%ずつ上昇させた。条件2ではA液とB液の混合比を常に40:60とした。化合物が検出された保持時間と、A液とB液の混合比(条件1または2)もいくつかの化合物について示した。
【0065】
参考例1
4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン 塩酸塩(4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidine hydrochloride,化合物(RE1))
下記の製造方法1または2に従い、合成した。
製造方法1
【0066】
【化20】
【0067】
化合物(1−1−1):1,3−ジブロモ−5−(2−メトキシエトキシ)ベンゼン(1,3-Dibromo-5-(2-methoxyethoxy)benzene)
3,5−ジブロモフェノール(37.7g,150mmol)、炭酸カリウム(41.4g,300mmol)のジメチルホルムアミド(150mmol)溶液に室温で2−ブロモエチルメチルエーテル(31.2g,224mmol)を加え、反応混合物を80℃で9時間攪拌した。室温に冷却後、水(300mL)、トルエン(150mL)と酢酸エチル(150mL)を加えて分液し、水層をトルエン(50mL)と酢酸エチル(50mL)の混合溶液で再抽出した。全有機層を水(50×2mL)、飽和食塩水(50mL)で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=20:1→15:1)で精製することで、表記化合物(1−1−1)(44.3g、95%)を褐色油状物として得た。
【0068】
化合物(1−1−2):3−ブロモ−5−(2−メトキシエトキシ)ベンズアルデヒド(3-Bromo-5-(2-methoxyethoxy)benzaldehyde)
n−ブチルリチウム(1.6M n−ヘキサン溶液,126 mL,198 mmol)のトルエン(120mL)溶液を氷浴で冷却して内温3〜5℃に保ちながらn−ブチルマグネシウムクロライド(0.89M テトラヒドロフラン溶液,116mL,100mmol)を25分間で滴下し、反応混合物をそのまま30分間攪拌した。化合物(1−1−1)(46.1g,149mmol)のトルエン(420mL)溶液を内温0〜3℃に保ちながら1時間で滴下し、反応混合物を2時間攪拌後、N,N−ジメチルホルムアミド(28.7mL,373mmol)を内温4〜5℃で40分間かけて滴下し、反応混合物を2時間攪拌した。2N−塩酸水(300mL)を加えて室温に昇温して分液した。水層をトルエン(100mL)で抽出し、全有機層を水、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=10:1→7:1)で精製することで、表記化合物(1−1−2)(28.8g、74%)を得た。
【0069】
化合物(1−1−3):[3−ブロモ−5−(2−メトキシエトキシ)フェニル]メタノール([3-Bromo-5-(2-methoxyethoxy)phenyl]methanol)
化合物(1−1−2)(28.9g,112mmol)のメタノール(112mL)溶液に水冷しながら室温で水素化ホウ素ナトリウム(4.22g,112mmol)を少しずつ加え、反応混合物を室温で3時間攪拌した。水(200 mL)を加え、メタノールを減圧留去し、酢酸エチル(200 mL+50 mL)で抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(1−1−3)(28.7g, 98%)を淡黄色油状物として得た。
【0070】
化合物(1−1−4):3−ブロモ−5−(2−メトキシエトキシ)ベンジル メタンスルホネート(3-Bromo-5-(2-methoxyethoxy)benzyl methanesulfonate)
化合物(1−1−3)(24.3g,93.1mmol)、トリメチルアミン塩酸塩(890mg,9.3mmol)とトリエチルアミン(25.9mL, 186mmol)のトルエン(186mL)溶液に、塩−氷浴で内温を10℃以下に保ちながらメタンスルホニルクロライド(16.0g,140mmol)のトルエン(15mL)溶液を50分間で滴下し、反応混合物を内温5℃以下で1時間攪拌した。内温5℃以下で2−ジエチルアミノエチルアミン(5.95g, 51.2mmol)を加えて20分間攪拌し、引き続き反応液に5%硫酸水素カリウム水溶液(250mL)と水(100mL)を加え、室温に昇温させて分液し、有機層を水洗後、トルエンを減圧留去することで、表記化合物(1−1−4)(33.1g)を得た。
【0071】
化合物(1−1−5):1−ブロモ−3−(ブロモメチル)−5−(2−メトキシエトキシ)ベンゼン(1-Bromo-3-(bromomethyl)-5-(2-methoxyethoxy)benzene)
化合物(1−1−4)(33.1g,97.6mmol相当)の無水テトラヒドロフラン(195mL)溶液に室温で臭化リチウム・1水和物(30.7g, 293mmol)を加え、反応混合物を1時間加熱還流した。室温に冷却後、トルエン(180mL)と水を加えて分液し、水層をトルエン(50mL)で再抽出した。全有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、トルエンを減圧留去することで、表記化合物(1−1−5)(29.3 g, 93%)を得た。
【0072】
化合物(1−1−6):[3−ブロモ−5−(2−メトキシエトキシ)ベンジル](トリフェニル)ホスホニウム ブロミド([3-Bromo-5-(2-methoxyethoxy)benzyl](triphenyl)phosphonium bromide)
化合物(1−1−5)(31.8g,98 mmol)とトリフェニルホスフィン(28.3g,108mmol)のトルエン(98mL)溶液を3時間加熱還流した。反応混合物をゆっくり室温まで冷却して生じた沈殿をろ取し、ろ上物をトルエン(40mL)で洗浄し、減圧乾燥することで、表記化合物(1−1−6)(50.1g,87%)を得た。
【0073】
化合物(1−1−7):tert−ブチル 4−{[3−ブロモ−5−(2−メトキシエトキシ)フェニル]メチリデン}ピペリジン−1−カルボキシレート(tert-Butyl 4-{[3-bromo-5-(2-methoxyethoxy)phenyl]methylidene}piperidine-1-carboxylate)
化合物(1−1−6)(50.1g、85mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(17.4g、87mmol)と炭酸カリウム(23.6g、171mmol)の2−プロパノール(250mL)懸濁溶液を40℃で4時間、50℃で2時間、60℃で3.5時間、80℃で7.5時間加熱還流した。反応混合物を室温まで冷却後、塩をろ去し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=10:1→7:1)で精製することで、表記化合物(1−1−7)(34.6g、95%)を無色油状物として得た。
【0074】
化合物(1−1−8):tert−ブチル 4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[3-bromo-5-(2-methoxyethoxy)benzyl]piperidine-1-carboxylate)
化合物(1−1−7)(34.6g、81mmol)を酢酸エチル(80mL)中、5%ロジウム炭素(9.74g)を用いて室温で26時間常圧水素添加反応を行った。触媒をろ去し、ろ液を減圧濃縮することで、表記化合物(1−1−8)(34.2g、98%)を得た。
【0075】
化合物(RE1):
化合物(1−1−8)(34.2g、80mmol)のメタノール(34mL)溶液に室温で10%塩酸メタノール溶液(103mL)を加え、反応混合物を室温で1日間攪拌した。溶媒を減圧留去後、得られた濃縮残渣にジエチルエーテルを加え、生じた沈殿をろ取し、ろ上物をジエチルエーテルで洗浄後、減圧乾燥することで白色固体(27.5g)を得た。これにアセトニトリル(132 mL)を加えて50℃に加温し、すべて溶解したことを確認後、ゆっくり冷却し、40℃で30分攪拌後、1時間で0℃まで冷却して、0℃で1時間攪拌した。析出物をろ取し、冷やしたアセトニトリル(20mL)で洗浄し、減圧乾燥することで、目的化合物(RE1)(22.6g,85%)を白色粉末として得た。
保持時間(条件1):15.56分
融点:107−108℃
1H-NMR (400 MHz, d6-DMSO) δ: 1.25-1.40 (2H, m), 1.67 (2H, d like, J = 14 Hz), 1.72-1.85 (1H, m), 2.49 (2H, d, J = 6.8 Hz), 2.77 (2H, dt, J = 2.4, 12 Hz), 3.20 (2H, br d,,J= 12.6 Hz), 3.34 (3H, s), 3.63 (2H, t like, J = 4.5 Hz), 4.09 (2H, t like, J = 4.5 Hz), 6.80 (1H, t, J = 1.7 Hz), 6.96-6.99 (2H, m).
【0076】
製造方法2
【0077】
【化21】
【0078】
化合物(1−2−1):ジエチル (3−ブロモ−5−メトキシベンジル)ホスホネート(Diethyl (3-Bromo-5-methoxybenzyl)phosphonate)
文献の方法(J.Med.Chem.2001,44,1866)により合成した1−ブロモ−3−メトキシ−5−メチルベンゼン(17.0g、91mmol)のモノクロロベンゼン(500mL)溶液に80℃で5,5−ジメチル−1,3−ジブロモヒダントイン(13.1g、46mmol)とアゾビスイソブチロニトリル(1.50g、9.1mmol)を同時に加え、反応混合物を30分間80℃で攪拌した。反応溶液を室温まで冷却後、10%チオ硫酸ナトリウム水溶液(100mL)に注ぎ30分間攪拌した。分液し、水層をトルエン(100mL)で抽出し、全有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、1−ブロモ−3−(ブロモメチル)−5−メトキシベンゼン(1-Bromo-3-(bromomethyl)-5-methoxybenzene)を得た。これを精製することなく、亜リン酸トリエチル(14.3mL、97mmol)とトルエン(50mL)に溶解させ、反応混合物を7時間加熱還流した。室温に冷却後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=1:2)で精製することで、表記化合物(1−2−1)(20.8g、64%)を得た。
【0079】
化合物(1−2−2):tert−ブチル 4−(3−ブロモ−5−メトキシベンジリデン)ピペリジン−1−カルボキシレート(tert-Butyl 4-(3-bromo-5-methoxybenzylidene)piperidine-1-carboxylate)
水素化ナトリウム(60%懸濁溶液、2.71g、68mmol)の無水テトラヒドロフラン(100mL)懸濁溶液に50℃で化合物(1−2−1) (19.0g、56mmol)の無水テトラヒドロフラン(100mL)溶液を15分間で滴下した。反応混合物を50℃で25分攪拌後、1−tert−ブトキシカルボニル−4−ピペリドン(14.5g、73mmol)の無水テトラヒドロフラン(100mL)溶液を50℃で30分間かけて滴下し、反応混合物を1時間攪拌した。引き続き、1−tert−ブトキシカルボニル−4−ピペリドン(5.00g、25mmol)と水素化ナトリウム(60%懸濁溶液、1.00g、25mmol)を加えて反応混合物を1.5時間攪拌した。室温に冷却後、水(100mL)を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去後、得られた濃縮残渣にn−ヘキサン(10mL)と酢酸エチル(10mL)を加え、生じた固体をろ取し、ろ上物をn−ヘキサン/酢酸エチル(1:1、5mL×3)で洗浄し、減圧乾燥することで、表記化合物(1−2−2)(11.3g、52%)を得た。
【0080】
化合物(1−2−3):tert−ブチル 4−(3−ブロモ−5−メトキシベンジル)ピペリジン−1−カルボキシレート(tert-Butyl 4-(3-bromo-5-methoxybenzyl)piperidine-1-carboxylate)
化合物(1−2−2)(19.0g、50mmol)を酢酸エチル(300mL)中、5%ロジウム炭素(10.3g、10mol%)を用いて室温で1.5時間常圧水素添加反応を行った。触媒をろ去し、ろ液を減圧濃縮することで、表記化合物(1−2−3)(20.6g、定量的)を得た。
【0081】
化合物(1−2−4):4−(3−ブロモ−5−メトキシベンジル)ピペリジン 塩酸塩(4-(3-Bromo-5-methoxybenzyl)piperidine hydrochloride)
化合物(1−2−3)(20.6g、50mmol)のメタノール(30mL)溶液に室温で10%塩酸メタノール溶液(150mL)を加え、反応混合物を50℃で45分間攪拌した。室温に冷却し、溶媒を減圧留去後、得られた濃縮残渣にジエチルエーテルを加え、生じた沈殿をろ取し、ろ上物をジエチルエーテルで洗浄後、減圧乾燥することで、表記化合物(1−2−4)(15.4g、97%)を得た。
保持時間(条件1):16.40分
【0082】
化合物(1−2−5):tert−ブチル 4−(3−ブロモ−5−ヒドロキシベンジル)ピペリジン−1−カルボキシレート(tert-Butyl 4-(3-bromo-5-hydroxybenzyl)piperidine-1-carboxylate)
化合物(1−2−4)(15.0g、47mmol)のジクロロメタン(200mL)溶液を氷浴で冷却し、1M−三臭化ホウ素−ジクロロメタン溶液(70mL、70mmol)を30分間で滴下した。反応混合物を氷冷下2時間攪拌し、さらに1M−三臭化ホウ素−ジクロロメタン溶液(50mL、50mmol)を氷冷しながら滴下した。反応混合物を2時間攪拌後、メタノール(50mL)を内温20℃以下に保ちながら滴下した。溶媒を減圧留去し、得られた濃縮残渣に2N−水酸化ナトリウム水溶液(200mL)と1,4−ジオキサン(400mL)を加えて溶解させ、室温でジ−tert−ブチルジカーボネート(10.3g、47mmol)の1,4−ジオキサン(50mL)溶液を30分間で滴下した。反応混合物を室温で1夜間攪拌後、1,4−ジオキサンを減圧留去し、濃縮残渣に水(300mL)を加えて酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(1−2−5)(15.8g、91%)を得た。
【0083】
化合物(1−1−8):
化合物(1−2−5)(15.8g、43mmol)、2−ブロモエチルメチルエーテル(6.0mL、64mmol)、ヨウ化カリウム(7.09g、43mmol)と炭酸カリウム(11.8g、85mmol)のジメチルホルムアミド(100mL)溶液を60〜70℃で8時間攪拌し、さらに2−ブロモエチルメチルエーテル(1.0mL、11mmol)を加えて、反応混合物を2時間攪拌した。室温に冷却後、水(500mL)に注ぎ、酢酸エチル/トルエン(1:1、200mL×3)で抽出し、有機層を水洗後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=5:1)で精製することで、表記化合物(1−1−8)(18.4g、定量的)を得た。
【0084】
化合物(RE1):
化合物(1−1−8)(18.4g、43mmol)のメタノール(30mL)溶液に室温で10%塩酸メタノール溶液(200mL)を加え、反応混合物を室温で1夜間攪拌した。メタノールを減圧留去し、得られた濃縮残渣にジエチルエーテル(100mL)を加えて生じた沈殿をろ取し、ろ上物をジエチルエーテル(50mL)で洗浄後、減圧乾燥することで、目的化合物(RE1)(14.7g、95%)を得た。
【0085】
参考例2
4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン(4-[3-Chloro-5-(2-methoxyethoxy)benzyl]piperidine,化合物(RE2))
【0086】
【化22】
化合物(1−1−8)(800mg、1.8mmol)と塩化銅(I)(537mg、5.4mmol)のジメチルホルムアミド(5.4mL)溶液を150℃で6時間攪拌した。室温に冷却後、塩をろ去し、ろ液を減圧留去して得られた濃縮残渣に2N−水酸化ナトリウム水溶液とクロロホルムを加えて生じた沈殿をろ去した。ろ液を酢酸エチルで抽出し、有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(RE2)(520mg、定量的)を得た。
1H-NMR (300 MHz, CDCl3) δ: 1.04-1.20 (2H, m), 1.52-1.83 (3H, m), 2.45 (2H, bd, J=6.8 Hz), 2.47-2.61 (2H, m), 3.04 (2H, bd, J=11.9 Hz), 3.45 (3H, s), 3.72-3.75 (2H, m), 4.07-4.10 (2H, m), 6.62 (1H, bt, J=1.8 Hz), 6.72-6.78 (2H, m).
【0087】
参考例3
4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン 塩酸塩(4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidine hydrochloride,化合物(RE3))
下記の製造方法1、2または3に従い、合成した。
製造方法1
【0088】
【化23】
【0089】
化合物(3−1−1):4−ブロモ−3−(2−メトキシエトキシ)安息香酸(4-Bromo-3-(2-methoxyethoxy)benzoic acid)
窒素雰囲気下、2−メトキシエタノール(16.48g,217mmol)の無水N−メチル−2−ピロリジノン(175mL)溶液に室温でカリウム t−ブトキシド(24.29g,217mmol)を加えた。目視にて溶解を確認後、4−ブロモ‐3‐フルオロ安息香酸(19.00g,86.8mmol)を少しずつ加えた。反応混合物を90℃で6時間攪拌した。室温まで冷却後、濃塩酸(36%,25mL)、水(500mL)の溶液に水冷しながら40分間で滴下し、内温20〜25℃で1時間攪拌後、生じた沈殿をろ取し、ろ上物を水(20 mL×2)、アセトニトリル(20mL×2)で洗浄し、ろ上物を減圧乾燥して白色固体(26.41g)を得た。これをアセトニトリル(380mL)に加え、還流温度付近まで加温し、目視にて溶解を確認後冷却し、75℃付近で結晶の析出が始まったら65〜70℃に保ちながら1時間攪拌後、再び2.5時間かけて30℃付近まで冷却し、引き続き水冷し内温20℃を保ちながら1時間攪拌した。生じた沈殿をろ取し、ろ上物をアセトニトリル(20mL×2)で洗浄することで、表記化合物(3−1−1)(20.09g,85%)を淡褐色針状晶として得た。
【0090】
化合物(3−1−2):[4−ブロモ−3−(2−メトキシエトキシ)フェニル]メタノール([4-Bromo-3-(2-methoxyethoxy)phenyl]methanol)
水素化ホウ素ナトリウム(8.08g,213.5mmol)の無水テトラヒドロフラン(100mL)懸濁溶液に水冷しながら三フッ化ホウ素・ジエチルエーテル錯体(35mL,285mmol)を滴下し、そのまま1時間攪拌した。化合物(3−1−1)(19.50g,71.2mmol)の無水テトラヒドロフラン(300mL)溶液を水冷により内温25℃以下に保ちながら30分間で滴下した。反応混合物を3時間攪拌後、氷冷して内温20℃以下に保ちながら水(200mL)を20分間で滴下した。トルエン(200mL)を加えて分液し、水層をトルエン(200mL)で再抽出した。全有機層を3%重曹水(200 mL)、水(200mL)で洗浄後、トルエンを減圧留去し、濃縮残渣にトルエン(200mL)を加えてトルエンを減圧留去することで、表記化合物(3−1−2)(18.18g)を得た。
【0091】
化合物(3−1−3):4−ブロモ−3−(2−メトキシエトキシ)ベンジル メタンスルホネート(4-Bromo-3-(2-methoxyethoxy)benzyl methanesulfonate)
化合物(3−1−2)(18.00g,71.17mmol相当)、トリメチルアミン塩酸塩(467mg,7.12mmol)とトリエチルアミン(19.8 mL,142mmol)のトルエン(90mL)溶液に、塩−氷浴で内温を5℃以下に保ちながらメタンスルホニルクロライド(8.56g,74.7mmol)のトルエン(18mL)溶液を30分間で滴下し、反応混合物を内温5℃以下で2時間攪拌した。反応液を5%硫酸水素カリウム水溶液(180mL)に氷浴で冷却して10℃以下に保ちながら注ぎ、30分間攪拌した。室温に昇温させて分液し、水層をトルエン(90mL)で再抽出した。全有機層を水(180 mL)で洗浄し、トルエンを減圧留去することで、表記化合物(3−1−3)(22.43g)を得た。
【0092】
化合物(3−1−4):1−ブロモ−4−(ブロモメチル)−2−(2−メトキシエトキシ)ベンゼン(1-Bromo-4-(bromomethyl)-2-(2-methoxyethoxy)benzene)
化合物(3−1−3)(22.43g,71.17mmol相当)の無水テトラヒドロフラン(100mL)溶液に室温で無水臭化リチウム(18.54g, 214mmol)を加え、反応混合物を1時間加熱還流した。室温に冷却後、水(100mL)およびトルエン(100mL)を加えて分液し、水層をトルエン(100mL)で再抽出した。全有機層を5%重曹水(100mL)、水(100mL)の順に洗浄し、トルエンを減圧留去することで、表記化合物(3−1−4)(19.54g)を白色固体として得た。
また、本表記化合物(3−1−4)は、化合物(3−1−3)を経由することなく、次に示すように、化合物(3−1−2)から直接合成することもできる。
化合物(3−1−2)(16.0g,61.3mmol)のトルエン(80g)と臭化水素水(47%,53g)の混合溶液を内温65−70℃で2時間攪拌した。室温に冷却後、水(16g)を加えて分液し、有機層を5%重曹水(48g)、水(48g)の順に洗浄した。有機層を減圧下濃縮することにより表記化合物(3−1−4)(17.9g,90%)を得た。
【0093】
化合物(3−1−5):[4−ブロモ−3−(2−メトキシエトキシ)ベンジル](トリフェニル)ホスホニウム ブロマイド([4-Bromo-3-(2-methoxyethoxy)benzyl](triphenyl)phosphonium bromide)
化合物(3−1−4)(19.54g,71.17mmol相当)のトルエン(100mL)溶液にトリフェニルホスフィン(18.67g,71.17mmol)を加え、反応混合物を3.5時間加熱還流した。室温まで冷却後、水冷しながら20℃に保って1時間攪拌後、生じた沈殿をろ取し、ろ上物をトルエン(40mL×3)で洗浄し、減圧乾燥することで、表記化合物(3−1−5)(32.44g,78%)を得た。
【0094】
化合物(3−1−6):tert−ブチル 4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジリデン]ピペリジン−1−カルボキシレート(tert-Butyl 4-[4-bromo-3-(2-methoxyethoxy)benzylidene]piperidine-1-carboxylate)
化合物(3−1−5)(32.00g,54.6mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(11.42g、57.3mmol)と炭酸カリウム(11.30g,81.9mmol)の2−プロパノール(160 mL)溶液を時間加熱還流した。室温に冷却後、塩をろ別し、ろ上の塩を2−プロパノール(30mL×2)で洗浄した。ろ液を減圧濃縮し、濃縮残渣(41.08g)を得た。トルエンを加えて減圧留去し(200mL×2)、引き続き濃縮残渣にトルエン(96mL)を加え、水冷して20−25℃に保ちながらn−ヘキサン(290mL)を30分間で滴下した。そのまま混合物を1時間攪拌後、氷冷で1時間攪拌し、生じた沈殿をろ別した。ろ上物をトルエン/n−ヘキサン(トルエン:n−ヘキサン〜1:3,20mL×2)で洗浄し、ろ液を減圧留去することで、表記化合物(3−1−6)(27.93g)を淡黄色油状物として得た。
【0095】
化合物(3−1−7):tert−ブチル 4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[4-bromo-3-(2-methoxyethoxy)benzyl]piperidine-1-carboxylate)
化合物(3−1−6)(27.93g,54.6mmol相当)を酢酸エチル(232mL)中で5%ロジウム炭素(5.80g)を用いて、内温15〜20℃で3時間常圧水素添加反応を行った。触媒をセライトろ過後、酢酸エチルを減圧留去することで、表記化合物(3−1−7)(25.73g)を白色固体として得た。
【0096】
化合物(RE3):
化合物(3−1−7)(25.73g,54.6mmol相当)の2−プロパノール(115mL)溶液を内温55〜60℃に加温し、濃塩酸(36%,23.2mL)を5分間で滴下し、反応混合物を内温55〜60℃で4時間攪拌した。室温まで冷却後、2−プロパノールを減圧留去して濃縮残渣(42.91g)を得た。これに水(115mL)とトルエン(115mL)を加えて分液し、有機層を水(50mL)で再抽出した。全水層を水酸化ナトリウムでpH10程度に調製し、トルエン(200+100+100mL)で抽出した。有機層を水(50mL)で洗浄し、トルエンを減圧留去して濃縮残渣(18.57g)を得た。これを2−プロパノールに溶解させ、室温で濃塩酸(36%,5.58g,54.6mmol)を加えて、2−プロパノールを減圧留去し、濃縮残渣に2−プロパノール(200mL×2)を加えて減圧留去することで濃縮残渣(18.61 g)を白色粉末として得た。これに2−プロパノール(115mL)を加え、内温65〜70℃付近で均一溶液になったことを目視にて確認後、徐冷した。60℃付近で結晶の析出を確認したところで、内温55〜60℃でn−ヘキサン(60mL)を20分間で滴下し、懸濁溶液を内温55〜60℃で1時間攪拌後、再び徐冷し、内温30℃以下になったところで水冷で内温15〜20℃に保ちながら1時間攪拌し、さらに氷冷して内温5℃以下で1時間攪拌した。析出物をろ取し、ろ上物を冷n−ヘキサン(23mL)−2−プロパノール(12mL)混合溶液で洗浄後、減圧乾燥することで、目的化合物(RE3)(17.30 g)を白色粉末として得た。
保持時間(条件1):15.55分
融点:171−172℃
1H-NMR (300 MHz, CDCl3) δ: 1.47-1.94 (5H, m), 2.55 (2H, d, J = 5.5 Hz), 2.79 (2H, t like, J = 12 Hz), 3.47 (2H, d like, J = 13 Hz), 3.51 (3H, s), 3.81 (2H, t, J = 4.8 Hz), 4.15 (2H, t, J = 4.8 Hz), 6.62 (1H, dd, J = 7.9, 1.8 Hz), 6.68 (1H, d, J = 1.7 Hz), 7.43 (1H, d, J = 8.1 Hz), 9.50 (2H, br s).
【0097】
製造方法2
【0098】
【化24】
【0099】
化合物(3−2−1):メチル 3−(2−メトキシエトキシ)−4−ニトロベンゾエート(Methyl 3-(2-methoxyethoxy)-4-nitrobenzoate)
メチル 3−ヒドロキシ−4−ニトロベンゾエート(15.0g、76mmol)、2−ブロモエチルメチルエーテル(14.5g、99mmol)、ヨウ化カリウム(12.6g、76mmol)と炭酸カリウム(21.4g、155mmol)のジメチルホルムアミド(250mL)溶液を60〜70℃で3時間攪拌した。さらに2−ブロモエチルメチルエーテル(5.00g、36mmol)を加えて、反応混合物を60〜70℃で2時間攪拌後、室温で1夜間攪拌した。反応混合物を水(600mL)に注ぎ、酢酸エチル/トルエン(1:1、500mL×2)で抽出し、有機層を5%炭酸カリウム水溶液、水の順に洗浄し、溶媒を減圧留去することで、表記化合物(3−2−1)(20.2g、定量的)を得た。
【0100】
化合物(3−2−2):メチル 4−アミノ−3−(2−メトキシエトキシ)ベンゾエート(Methyl 4-amino-3-(2-methoxyethoxy)benzoate)
化合物(3−2−1)(20.2g、76mmol)をメタノール(200mL)中で10%パラジウム炭素(8.06g、10mol%)を用いて、室温で2時間常圧水素添加反応を行った。水素の吸収が止まったことを確認して、触媒をろ去し、ろ液を減圧濃縮することで、表記化合物(3−2−2)(16.7g、98%)を得た。
【0101】
化合物(3−2−3):メチル 4−ブロモ−3−(2−メトキシエトキシ)ベンゾエート(Methyl 4-bromo-3-(2-methoxyethoxy)benzoate)
化合物(3−2−2)(10.0g、44mmol)の48%臭化水素酸水溶液(50mL)液を氷浴で冷却し、亜硝酸ナトリウム(3.07g、45mmol)の水(30mL)溶液を30分間で滴下し、反応混合物を1時間攪拌した。この溶液を5℃以下に保ち、60℃に加温した臭化銅(I)(4.17g、29mmol)の48%臭化水素酸水溶液(50mL)へ20分間で滴下し、反応混合物を60℃で1時間攪拌した。室温に冷却後、水(400mL)に注ぎ、ジエチルエーテルで抽出し、有機層を水、飽和食塩水の順で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(3−2−3)(9.77g、76%)を得た。
【0102】
化合物(3−2−4):[4−ブロモ−3−(2−メトキシエトキシ)フェニル]メタノール([4-Bromo-3-(2-methoxyethoxy)phenyl]methanol)
化合物(3−2−3)(10.0g、35mmol)の無水テトラヒドロフラン(50mL)溶液を穏やかに加熱還流させながら、1.0Mボラン・テトラヒドロフラン錯体テトラヒドロフラン溶液(140mL、140mmol)を滴下し、反応混合物を20時間加熱還流した。室温に冷却後、発泡が収まるまで水を加え、テトラヒドロフランを減圧留去した。濃縮残渣に飽和重曹水を加えてクロロホルムで抽出し、有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=1:1)で精製することで、表記化合物(3−2−4)(9.13g、定量的)を得た。
【0103】
化合物(3−1−4):
化合物(3−2−4)(9.00g、34.5mmol)とトリフェニルホスフィン(10.9g、41mmol)のジクロロメタン(200mL)溶液に、氷冷しながら内温20℃以下で四臭化炭素(15.5g、48mmol)を加え、反応混合物を氷冷下1.5時間攪拌した。溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(クロロホルム)で精製することで、表記化合物(3−1−4)(9.88g、88%)を得た。
【0104】
化合物(3−1−5):
化合物(3−1−4)(9.85g、30mmol)とトリフェニルホスフィン(9.57g、36.5mmol)のトルエン(200mL)溶液を16時間加熱還流した。反応混合物をゆっくり室温まで冷却し、引き続いて氷浴で30分間攪拌後、生じた沈殿をろ取し、ろ上物をトルエン(10mL×2)で洗浄し、減圧乾燥することで、表記化合物(3−1−5)(19.4g、定量的)を得た。
【0105】
化合物(3−1−6):
化合物(3−1−5)(19.0g、32mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(7.10g、36mmol)と炭酸カリウム(6.71g、49mmol)の2−プロパノール(200mL)懸濁溶液を11.5時間加熱還流した。反応混合物を室温まで冷却後、塩をろ去し、ろ液を減圧濃縮した。得られた濃縮残渣にn−ヘキサン/酢酸エチル(4:1、200mL)を加え、生じたトリフェニルホスフィンオキシドをろ去し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(3−1−6)(14.2g、定量的)を得た。
【0106】
化合物(3−1−7):
化合物(3−1−6)(4.00g、9.4mmol)を酢酸エチル(100mL)中で5%ロジウム炭素(1.91g、10mol%)を用いて室温で2.5時間常圧水素添加反応を行った。触媒をろ去し、ろ液を減圧濃縮し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(3−1−7)(3,88g、97%)を得た。
【0107】
化合物(RE3):
化合物(3−1−7)(3.88g、9.1mmol)のメタノール(5mL)溶液に室温で10%塩酸メタノール溶液(20mL)を加え、反応混合物を室温で5時間攪拌した。溶媒を減圧留去し、得られた濃縮残渣に少量の2−プロパノールを加えて固化させ、さらにジエチルエーテルを加えて懸濁させ、沈殿をろ取し、ジエチルエーテルで洗浄することで、目的化合物(RE3)(2.45g、74%)を得た。
【0108】
製造方法3
【0109】
【化25】
【0110】
化合物(3−3−1):(4−ブロモ−3−フルオロベンジル)(トリフェニル)ホスホニウム ブロマイド((4-Bromo-3-fluorobenzyl)(triphenyl)phosphonium bromide)
4−ブロモ−3−フルオロトルエン(25.0g,132 mmol)、5,5−ジメチル−1,3−ジブロモヒダントイン(18.9g,66.1mmol)、アゾビスイソブチロニトリル(1.09g,6.64mmol)のクロロベンゼン(400mL)溶液を内温80−90℃で1時間攪拌した。反応混合物を氷浴で冷却後、水(200mL)とチオ硫酸ナトリウム(33g,132mmol)を加えて攪拌した。分液し、有機層を水(200mL)で洗浄し、無水硫酸ナトリウムで乾燥後、全体が100mL程度の体積になるまで濃縮した。そこへトリフェニルホスフィン(34.69g,132mmol)とクロロベンゼン(30mL)を加えて3時間加熱還流した。反応混合物を室温まで冷却後、析出物をろ取し、ろ上物をトルエンで洗浄後、減圧乾燥することで、表記化合物(3−3−1)(47.0g)を得た。
【0111】
化合物(3−3−2):tert-ブチル 4−[(4−ブロモ−3−フルオロフェニル)メチリデン]ピペリジン−1−カルボキシレート(tert-Butyl 4-[(4-bromo-3-fluorophenyl)methylidene]piperidine-1-carboxylate)
化合物(3−3−1)(15.0g,28.3mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(3.76g,18.9mmol)および炭酸カリウム(5.21g,37,7mmol)の2−プロパノール(28mL)溶液を3時間加熱還流した。反応混合物を室温に冷却し、塩をろ別後、ろ液にトルエン(300mL)を加えて、水(100mL)、飽和食塩水(100mL)で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。濃縮残渣にn−ヘキサン(107mL)を加えて1時間加熱還流後、室温まで冷却して1時間攪拌後、氷浴で冷却しながら1時間攪拌した。析出したトリフェニルホスフィンオキシドをろ別し、n−ヘキサンで洗浄後、ろ液を減圧濃縮することで、表記化合物(3−3−2)(8.08g)を黄色固体として得た。
【0112】
化合物(3−3−3):tert-ブチル 4−(4−ブロモ−3−フルオロベンジル)ピペリジン−1−カルボキシレート(tert-Butyl 4-(4-bromo-3-fluorobenzyl)piperidine-1-carboxylate)
化合物(3−3−2)(8.08g)を酢酸エチル(57mL)中で5%白金炭素(800mg)を用いて時間常圧水素添加反応を行った。セライトろ過により触媒をろ別し、ろ液を濃縮することで、表記化合物(3−3−3)(8.61g)を淡黄色固体として得た。
【0113】
化合物(3−1−7):
化合物(3−3−3)(8.61g,18.9mmol相当)、2−メトキシエタノール(2.98mL,37.7mmol)のN−メチル−2−ピロリジノン(38mL)溶液に室温でカリウム t−ブトキシド(4.23g,37.7mmol)を加え、反応混合物を内温90℃で2.5時間攪拌後、カリウム t−ブトキシド(1.06g,9.43mmol)を加えて30分間90℃で攪拌し、さらにカリウム t−ブトキシド(1.06g,9.43mmol)を加えて30分間90℃で攪拌した。反応混合物を氷浴で冷却し、飽和塩化アンモニウム水溶液(80mL)を加え、トルエン(80mL×3)で抽出した。全有機層を水(40mL×2)、飽和食塩水(40mL)で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。濃縮残渣にn−ヘキサン(162mL)を加え、60℃に加温して溶解を確認後、ゆっくり室温まで冷却し、室温で1夜間攪拌後、氷浴で冷却して1時間攪拌し、析出物をろ取し、ろ上物をn−ヘキサンで洗浄後、減圧乾燥することで、表記化合物(3−1−7)(6.34g)を淡褐色粉末として得た。
【0114】
化合物(RE3):
化合物(3−1−7)(6.00g,14.0mmol)のメタノール(24 mL)溶液に室温で10%塩酸−メタノール溶液(24mL)を加え、反応混合物を50℃で5時間攪拌した。室温まで冷却後、溶媒を減圧留去し、濃縮残渣にアセトニトリル(6mL)を加えて減圧濃縮を4回繰り返した。濃縮残渣にアセトニトリル(38mL)を加えて80℃の油浴で加温し、固形物の溶解を確認後、1時間で室温まで冷却し、水浴で冷却して20℃で1時間、氷浴で1時間攪拌後、析出物をろ取し、ろ上物を冷アセトニトリル(30mL)で洗浄し、減圧乾燥することで、表記化合物(RE3)(4.56g,89%)を白色粉末として得た。
【0115】
参考例4
4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(4-[4-Chloro-3-(2-methoxyethoxy)benzyl]piperidine,化合物(RE4))
下記製造方法に従い、合成した。
製造方法
【0116】
【化26】
【0117】
化合物(4−1):メチル 4−クロロ−3−(2−メトキシエトキシ)ベンゾエート(Methyl 4-chloro-3-(2-methoxyethoxy)benzoate)
化合物(3−2−2)(5.20g、23mmol)の濃塩酸(36%,50mL)水溶液を氷浴で冷却し、亜硝酸ナトリウム(1.59g、23mmol)の水(20mL)溶液を10分間で滴下し、反応混合物を20分間攪拌した。この溶液を5℃以下に保ち、60℃に加温した塩化銅(I)(1.51g、15mmol)の濃塩酸(36%,50mL)水溶液へ10分間で滴下し、反応混合物を60℃で50分間攪拌した。室温に冷却後、水で希釈してジエチルエーテルで抽出した。有機層を飽和重曹水、飽和食塩水の順で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1→1:1)で精製することで、表記化合物(4−1)(4.43g、78%)を白色固体として得た。
【0118】
化合物(4−2):[4−クロロ−3−(2−メトキシエトキシ)フェニル]メタノール([4-Chloro-3-(2-methoxyethoxy)phenyl]methanol)
化合物(4−1)(4.33g、18mmol)の無水テトラヒドロフラン(50mL)溶液に室温で1.0Mボラン・テトラヒドロフラン錯体テトラヒドロフラン溶液(106mL、106mmol)を滴下し、引き続き反応混合物を27.5時間加熱還流した。室温に冷却後、発泡が収まるまでメタノールを加え、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=3:1→1:1)で精製することで、表記化合物(4−2)(3.62g、94%)を得た。
【0119】
化合物(4−3):4−(ブロモメチル)−1−クロロ−2−(2−メトキシエトキシ)ベンゼン(4-(Bromomethyl)-1-chloro-2-(2-methoxyethoxy)benzene)
化合物(4−2)(3.60g、16.6mmol)のジエチルエーテル(50mL)溶液に室温でトリフェニルホスフィン(6.54g、25mmol)、四臭化炭素(8.26g、25mmol)を一度に加え、反応混合物を16時間攪拌した。析出物をろ別し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン→n−ヘキサン:酢酸エチル=7:1)で精製することで、表記化合物(4−3)(3.93g、85%)を無色油状物として得た。
【0120】
化合物(4−4): [4−クロロ−3−(2−メトキシエトキシ)ベンジル](トリフェニル)ホスホニウム ブロマイド([4-Chloro-3-(2-methoxyethoxy)benzyl](triphenyl)phosphonium bromide)
化合物(4−3)(3.90g、14mmol)とトリフェニルホスフィン(4.41g、36.17mmol)のトルエン(100mL)溶液を5時間加熱還流した。反応混合物をゆっくり室温まで冷却し、引き続いて氷浴で1時間攪拌後、析出物をろ取し、ろ上物をトルエンで洗浄し、減圧乾燥することで、表記化合物(4−4)(7.24g、95%)を白色粉末として得た。
【0121】
化合物(4−5):tert−ブチル 4−{[4−クロロ−3−(2−メトキシエトキシ)フェニル]メチリデン}ピペリジン−1−カルボキシレート(tert-Butyl 4-{[4-chloro-3-(2-methoxyethoxy)phenyl]methylidene}piperidine-1-carboxylate)
化合物(4−4)(7.24g、13mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(2.40g、12mmol)と炭酸カリウム(2.78g、20mmol)の2−プロパノール(100mL)懸濁溶液を8時間加熱還流した。反応混合物を氷冷して1時間攪拌後、析出物をろ去し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=10:1→8:1)で精製することで、表記化合物(4−5)(4.52g、定量的)を無色油状物として得た。
【0122】
化合物(4−6):tert−ブチル 4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[4-chloro-3-(2-methoxyethoxy)benzyl]piperidine-1-carboxylate)
化合物(4−5)(4.52g、12mmol)を酢酸エチル(200mL)中で5%ロジウム炭素(1.53g)を用いて室温で1.5時間常圧水素添加反応を行った。触媒をセライトろ過でろ去し、ろ液を減圧濃縮後、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=8:1→4:1)で精製することで、表記化合物(4−6)(4.34g、96%)を得た。
【0123】
化合物(RE4):
化合物(4−6)(4.34g、11mmol)の1,4−ジオキサン(50mL)溶液を4N−塩酸−1,4−ジオキサン溶液(50mL)に加え、反応混合物を室温で15.5時間攪拌した。溶媒を減圧留去することで表記化合物の塩酸塩(3.24g,90%)を得た。このうち2gを2N−水酸化ナトリウム水溶液に加え、クロロホルムで抽出し、無水硫酸ナトリウムで乾燥後、溶媒を減圧濃縮することで、目的化合物(RE4)(1.91g)を淡黄色油状物として得た。
保持時間(条件1):13.71分
1H-NMR (300 MHz, CDCl3) δ: 1.03-1.26 (2H, m), 1.48-1.69 (3H, m), 2.48 (2H, t, J = 5.2 Hz), 2.54 (2H, td, J = 12.5, 2.6 Hz), 3.06 (2H, br d, J = 11.9 Hz), 3.49 (3H, s), 3.80 (2H, t, J = 4.8 Hz), 4.17 (2H, t, J = 4.8 Hz), 6.69 (1H, dd, J = 7.9, 1.8 Hz), 6.73 (1H, d, J = 1.8 Hz), 7.26 (1H, d, J = 8.1 Hz).
【0124】
参考例5
2−[2−ブロモ−5−(ピペリジン−4−イルメチル)フェノキシ]エタノール(2-[2-Bromo-5-(piperidin-4-ylmethyl)phenoxy]ethanol,化合物(RE5))
【0125】
【化27】
化合物(RE3)(3.00g、8.3mmol)のジクロロメタン(100mL)溶液を塩−氷浴で冷却し、0℃で1M−三臭化ホウ素ジクロロメタン溶液(7.8mL、7.8mmol)を30分間で滴下し、反応混合物を氷冷下1.5時間攪拌した。メタノール(20mL)を加え、溶媒を減圧留去した。得られた濃縮残渣に5%炭酸カリウム水溶液(50mL)を加え、クロロホルムで抽出し、有機層を無水硫酸ナトリウムで乾燥した。溶媒を減圧留去することで、表記化合物(RE5)(3.03g、定量的)を得た。
保持時間(条件1):7.56分
1H-NMR (400 MHz,CDCl3) δ:1.15-1.30 (2H, m), 1.40-1.55 (1H, m), 1.55-1.70 (2H, m), 2.49 (2H, d, J= 6.7 Hz), 2.58 (2H, dt, J= 2.4, 12 Hz), 3.11 (2H, d like, J= 12 Hz), 3.98 (2H, t, J= 4.6 Hz), 4.14 (2H, t, J= 4.6 Hz), 6.66 (1H, dd, J= 8.0, 1.8 Hz), 6.70 (1H, d, J= 1.8 Hz), 7.42 (1H, d, J= 8.0 Hz).
【0126】
参考例6
4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン(4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidine,化合物(RE6))
下記の製造方法に従い、合成した。
製造方法
【0127】
【化28】
【0128】
化合物(6−1):tert-ブチル 4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[3-(2-methoxyethoxy)-4-methylbenzyl]piperidine-1-carboxylate)
化合物(3−1−7) (5.00g、11.7g)、メチルボロニック アシッド(978mg、16mmol)とテトラキストリフェニルホスフィンパラジウム(674mg、5mol%)の1M−炭酸カリウム水溶液(35mL)、1,4−ジオキサン(80mL)溶液を4時間加熱還流した。室温まで冷却後、1,4−ジオキサンを減圧留去し、得られた濃縮残渣に水を加えて酢酸エチルで抽出し、全有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去後、濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(6−1)(3.46g、82%)を得た。
【0129】
化合物(RE6):
化合物(6−1)(3.46g、9.5mmol)に室温で10%塩酸メタノール溶液(40mL)を加え、反応混合物を1夜間攪拌した。メタノールを減圧留去し、得られた濃縮残渣に飽和重曹水を加え、塩析してクロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、目的化合物(RE6)(2.56g、定量的)を得た。
1H-NMR (300 MHz,CDCl3) δ: 1.39-1.58 (2H, m), 1.58-1.74 (1H, m), 1.77 (2H, d like, J = 13.9 Hz), 2.20 (3H, s), 2.52 (2H, d, J = 7.1 Hz), 2.70 (2H, t like, J = 12.6 Hz), 3.33 (2H, d like, J = 12.4 Hz), 3.47 (3H, s), 3.77 (2H, t, J = 4.8 Hz), 4.10 (2H, t, J = 4.8 Hz), 6.59 (1H, s), 6.63 (1H, d, J = 7.6 Hz), 7.03 (1H, d, J = 7.6 Hz).
【0130】
参考例7
2−(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)エチル 4−メチルベンゼンスルホネート(2-(4-Oxo-3,4-dihydro-2H-chromen-6-yl)ethyl 4-methylbenzenesulfonate,化合物(RE7))
下記の製造方法1、2または3に従い、合成した。
製造方法1
【0131】
【化29】
【0132】
化合物(7−1−1):メチル [4−(3−ヒドロキシプロポキシ)フェニル]アセテート(Methyl [4-(3-hydroxypropoxy)phenyl]acetate)
4−ヒドロキシフェニル酢酸 メチルエステル(50.0g,301mmol)のアセトニトリル(1000mL)、水(10mL)溶液に、室温で炭酸カリウム(91.5g,662mmol)、3−ブロモ−1−プロパノール(35.3mL,391mmol)の順で加え、反応混合物を3時間加熱還流した。室温に冷却後、塩をろ別し、ろ上物をアセトニトリル(50mL×2)で洗浄した。ろ液を減圧濃縮し、得られた濃縮残渣にトルエン(500mL)、水(250 mL)を加えて分液した。水層をトルエン(125mL×2)で再抽出し、全有機層を0.5N−水酸化ナトリウム水溶液(100mL)、1%硫酸水素カリウム水溶液(100mL)で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−1)(70.2 g)を黄色油状物として得た。
【0133】
化合物(7−1−2):3−[4−(2−メトキシ−2−オキソエチル)フェノキシ]プロパノイック アシッド(3-[4-(2-Methoxy-2-oxoethyl)phenoxy]propanoic acid)
2,2,6,6−テトラメチル−1−ピペリジニルオキシ、ラジカル(TEMPO)(4.71g,30.1mmol)のアセトニトリル(160mL)溶液に、0.25Mりん酸二水素カリウム水溶液(400mL)、0.25Mりん酸水素二ナトリウム水溶液(400mL)、80%亜塩素酸ナトリウム(54.5 g,482mmol)、5%次亜塩素酸ナトリウム水溶液(6.52mL,4.82mmol)を順に加え、引き続き水冷して内温20〜25℃に保ちながら化合物(7−1−1)(56.2g,241mmol相当)のアセトニトリル(800mL)溶液を約1時間で滴下した。反応混合物をそのまま2時間攪拌後、氷浴により内温15℃以下に保ちながら20%亜硫酸水素ナトリウム水溶液(400mL)を30分間で滴下した。溶液をゆっくり室温に昇温し、アセトニトリルを減圧留去した。水(400mL)を加えて室温で30分間攪拌し、生じた沈殿をろ取し、水(100mL×2)で洗浄した。ろ上物を酢酸エチル(400mL)に溶解させて、飽和食塩水(100mL)で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−2)(47.87 g)を得た。
【0134】
化合物(7−1−3):メチル [4−(3−クロロ−3−オキソプロポキシ)フェニル]アセテート(Methyl [4-(3-chloro-3-oxopropoxy)phenyl]acetate)
化合物(7−1−2)(47.0g,197mmol)のトルエン(470 mL)溶液に室温で塩化チオニル(42.7mL、592mmol)を加え、反応混合物を60℃で3時間攪拌した。室温に冷却後、トルエンを留去することで、表記化合物(7−1−3)(56.52g)を淡黄色油状物として得た。
【0135】
化合物(7−1−4):メチル (4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)アセテート(Methyl (4-oxo-3,4-dihydro-2H-chromen-6-yl)acetate)
塩化アルミニウム(52.5g,394mmol)のジクロロメタン(330 mL)溶液に化合物(7−1−3)(56.52g,197mmol相当)のジクロロメタン(140mL)溶液を水冷により内温20〜25℃に保ちながら30分間で滴下し、反応混合物をそのまま1.5時間攪拌した。反応混合物を冷却し、2N塩酸水溶液(470mL)を氷浴で冷却して内温15℃以下に保ちながら加えた。室温に昇温後、分液し、水層をクロロホルム(120mL)で再抽出した。全有機層を水(240mL)、飽和重曹水(240mL)の順で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−4)(38.91g)を褐色固体として得た。
【0136】
化合物(7−1−5):メチル (4,4−ジメトキシ−3,4−ジヒドロ−2H−クロメン−6−イル)アセテート(Methyl (4,4-dimethoxy-3,4-dihydro-2H-chromen-6-yl)acetate)
化合物(7−1−4)(35.0g,159mmol)のメタノール(105 mL)溶液にp−トルエンスルホン酸・1水和物(3.02g,15.9mmol)、オルトギ酸メチル(210mL)を加え、反応混合物を室温(15〜20℃)で20時間攪拌した。5%重曹水(175mL)を氷冷して内温15℃以下に保ちながら反応溶液を15分間で滴下し、トルエン(175mL)、水(88mL)を加えて分液した。水層をトルエン(88mL)で再抽出し、全有機層を水(44mL)で洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去することで、表記化合物(7−1−5)(46.49g)を黄色油状物として得た。
【0137】
化合物(7−1−6):6−(2−ヒドロキシエチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-Hydroxyethyl)-2,3-dihydro-4H-chromen-4-one)
水素化リチウムアルミニウム(9.05g,239mmol)のテトラヒドロフラン(600mL)懸濁溶液に化合物(7−1−5)(46.0g,159 mmol相当)のテトラヒドロフラン(90mL)溶液を水冷し内温30℃以下に保ちながら30分間で滴下し、反応混合物をそのまま1時間攪拌した。氷冷して内温15℃以下に保ちながらテトラヒドロフラン−水(1:2、12mL)を滴下した(途中で不溶物が析出し、攪拌が困難となった)。引き続き3N塩酸水(460mL)を内温15℃以下に保ちながら滴下した。そのまま1.5時間20〜25℃で攪拌し、トルエン(460mL)を加えて分液した。水層をトルエン(230mL)で再抽出した。全有機層を3N塩酸水(230mL×2)で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−6)(30 .0g)を無色油状物として得た。
【0138】
化合物(RE7):
化合物(7−1−6)(10.0g,52mmol相当)のアセトニトリル(150mL)溶液に、トリメチルアミン塩酸塩(497mg,5.20mmol)およびトリエチルアミン(14.4mL,104mmol)を加え、氷浴で冷却して内温15℃以下でp−トルエンスルホニルクロライド(11.9g, 62.4mmol)を少しずつ加え、反応混合物を内温5℃以下で1.5時間攪拌した。内温10℃以下で5%重曹水(75mL)を加え、室温に昇温してからトルエン(75mL)を加えて分液し、水層をトルエン(75mL)で再抽出した。全有機層を1%硫酸水素カリウム水溶液(38mL×2)で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで濃縮残渣(16.61 g)を得た。トルエン(50mL)を加えて50℃で1.5時間攪拌し、引き続き30分間で室温(20〜25℃)まで冷却した。水冷し内温20〜25℃で1時間攪拌後、析出物をろ取し、トルエン(10mL×2)で洗浄し、減圧乾燥することで、目的化合物(RE7)(10.65g)を淡黄色粉末として得た。
保持時間(条件2):15.50分
融点:121−122℃
1H-NMR (300 MHz,CDCl3) δ: 2.44 (3H, s), 2.79 (2H, t, J = 6.5 Hz), 2.91 (2H, t, J = 6.9 Hz), 4.18 (2H, t, J = 6.9 Hz), 4.51 (2H, t, J = 6.4 Hz), 6.88 (1H, d, J = 8.4 Hz), 7.26 (1H, dd, J = 8.5, 2.5 Hz), 7.30 (2H, d, J = 8.5 Hz), 7.60 (1H, d, J = 2.2 Hz), 7.71 (2H, d, J = 8.3 Hz).
【0139】
製造方法2
【0140】
【化30】
【0141】
化合物(7−2−1):3−[4−(2−メトキシエチル)フェノキシ]プロパン−1−オール(3-[4-(2-Methoxyethyl)phenoxy]propan-1-ol)
4−(2−メトキシエチル)フェノール(1.00g,6.57mmol)、3−ブロモ−1−プロパノール(771μL,8.54mmol)、炭酸カリウム(2.00g,14.5mmol)と水(200μL)のアセトニトリル(20mL)溶液を3時間加熱還流した。室温まで冷却後、塩をろ別し、ろ液を減圧濃縮した。得られた濃縮残渣に水(5mL)とトルエン(5mL)を加えて分液し、水層をトルエン(5mL×2)で再抽出した。全有機層を0.5N−水酸化ナトリウム水溶液(2mL)、1%硫酸水素カリウム(2mL)で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−2−1)(1.56g)を得た。
【0142】
化合物(7−2−2):3−[4−(2−メトキシエチル)フェノキシ]プロパノイック アシッド(3-[4-(2-Methoxyethyl)phenoxy]propanoic acid)
化合物(7−2−1)(1.56g,6.57mmol相当)を用いて実施例7−1−2)と同様の方法により、表記化合物(7−2−2)(1.30g, 88%)を白色粉末として得た。
【0143】
化合物(7−2−3):6−(2−メトキシエチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-Methoxyethyl)-2,3-dihydro-4H-chromen-4-one)
化合物(7−2−2)(1.30g,5.80mmol)のトルエン(13 mL)溶液に室温で塩化チオニル(1.25mL)を加え、反応混合物を60℃で3時間攪拌した。室温に冷却後、溶媒を減圧留去することで酸クロライド(1.46g)を得た。このうち292mg(1.16mmol相当)のジクロロメタン(1mL)溶液を室温で塩化アルミニウム(309mg,2.32mmol)のジクロロメタン(2mL)懸濁溶液に水冷しながら室温で5分間かけて滴下し、反応混合物を1時間攪拌した。2N塩酸水溶液(3mL)および氷に反応溶液を注ぎ、トルエンで抽出した。有機層を水、飽和重曹水、飽和食塩水の順で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−2−3)(235mg)を得た。
【0144】
化合物(7−2−4):2−(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)エチル 2,2−ジメチルプロパノエート(2-(4-Oxo-3,4-dihydro-2H-chromen-6-yl)ethyl 2,2-dimethylpropanoate)
化合物(7−2−3)(100mg,0.485mmol)、ヨウ化ナトリウム(436mg,2.91mmol)と水(3μL)のアセトニトリル(0.6 mL)溶液に40℃でピバロイルクロライド(298μL,2.43mmol)を加え、反応混合物を40℃で1.5時間攪拌した。室温に冷却後、トルエンで希釈し、水を加えて分液し、水層をトルエンで再抽出した。全有機層を飽和重曹水、飽和食塩水の順で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−2−4)(148mg)を褐色油状物として得た。
【0145】
化合物(7−1−6):
化合物(7−2−4)(105mg,0.354mmol)の濃塩酸(36%, 2mL)、メタノール(1mL)溶液を50℃で3時間攪拌した。室温に冷却後、反応混合物を氷に注ぎ、トルエンで抽出し、飽和重曹水、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−6)(62mg)を得た。
【0146】
化合物(RE7):
化合物(7−1−6)(62mg,0.354mmol)、トリメチルアミン塩酸塩(3.4mg,0.035mmol)とトリエチルアミン(98μL)のジクロロメタン(1mL)溶液に、氷浴で冷却しながらp−トルエンスルホニルクロライド(81mg,0.43mmol)を加え、反応混合物をそのまま1.5時間攪拌した。飽和重曹水を加えて室温に昇温し、トルエンで抽出し、全有機層を1%硫酸水素カリウム水溶液、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を減圧留去後、得られた濃縮残渣にトルエン(1mL)を加えて50℃で1時間攪拌し、ゆっくり室温まで冷却しながら攪拌後、室温で2時間攪拌した。析出物をろ取し、ろ上物をトルエンで洗浄後、減圧乾燥することで、目的化合物(RE7)(35mg,29%)を得た。
【0147】
製造方法3
【化31】
【0148】
化合物(7−3−1):6−(ブロモメチル)−2,3−ジヒドロ−4H−クロメン−4−オン (6-(Bromomethyl)-2,3-dihydro-4H-chromen-4-one)
6−メチル−2,3−ジヒドロ−4H−クロメン−4−オン(7.2g,44mmol)、5,5−ジメチル−1,3−ジブロモヒダントイン(7.7g,27mmol)およびアゾビスイソブチロニトリル(1.5g,9mmol)のモノクロロベンゼン(140mL)溶液を80℃で3時間攪拌した。反応溶液を氷水(100mL)に注ぎ、室温に昇温後分液した。水層をジクロロメタン(50mL×2)で抽出し、全有機層を飽和食塩水で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(7.8g,73%)を得た。
【0149】
化合物(7−3−2):(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)アセトニトリル((4-Oxo-3,4-dihydro-2H-chromen-6-yl)acetonitrile)
化合物(7−3−1)(500mg,2.1mmol)とシアン化カリウム(135mg,2.1mmol)の1,4−ジオキサン(7.5mL)および水(2.5mL)混合溶液を50℃で3時間攪拌した。反応溶液を室温まで冷却後、飽和食塩水(30mL)を加えてトルエン(30mL×2)で抽出した。全有機層を無水硫酸マグネシウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(311mg,70%)を得た。
【0150】
化合物(7−1−4):
化合物(7−3−2)(276mg,1.5mmol)の濃硫酸(2mL)、酢酸(2mL)および水(2mL)溶液を80℃で5時間攪拌した。反応溶液を室温に冷却後、10%水酸化ナトリウム水溶液でpH10−11に調製し、ジクロロメタン(30mL×2)で抽出した。全有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去することで化合物(7−3−3)(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)アセティックアシッド(4-Oxo-3,4-dihydro-2H-chromen-6-yl)acetic acidの粗生成物を得た。これを精製することなく濃硫酸(0.05mL)とともにメタノール(5mL)に溶解させ、反応混合物を2時間加熱還流した。室温に冷却後、水(10mL)を加えてメタノールを減圧留去し、濃縮残渣を酢酸エチル(30mL×2)で抽出した。全有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(251mg,88%)を得た。
【0151】
化合物(7−3−4):メチル 2,3−ジヒドロスピロ[クロメン−4,2’−[1,3]ジオキソラン]−6−イルアセテート(Methyl 2,3-dihydrospiro[chromene-4,2'-[1,3]dioxolan]-6-ylacetate)
化合物(7−1−4)(419mg,1.9mmol)、エチレングリコール(0.21mL,3.8mmol)、オルトギ酸メチル(0.42mL,3.8mmol)およびp−トルエンスルホン酸・1水和物(72mg,0.38mmol)のトルエン(8mL)溶液を3時間加熱還流した。室温に冷却後、トルエン(10mL)を加え、混合溶液を飽和重曹水(10mL)、水、飽和食塩水の順に洗浄した。有機層を無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで表記化合物の粗生成物(502mg)を得た。
【0152】
化合物(7−1−6):
水素化リチウムアルミニウム(72mg,1.9mmol)のテトラヒドロフラン(5mL)懸濁溶液に0℃で化合物(7−3−4)(502mg)のテトラヒドロフラン(3mL)溶液を滴下し、反応混合物をゆっくり室温に昇温させながら1時間攪拌した。ジエチルエーテル(8mL)および水を加え、生じた沈殿をろ別し、ろ液を減圧留去した。得られた濃縮残渣に10%塩酸水(8mL)およびテトラヒドロフラン(8mL)を加えて混合溶液を室温で30分間攪拌した。テトラヒドロフランを減圧留去後、酢酸エチルで抽出した。有機層を飽和食塩水で減圧留去後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(254mg,69%)を得た。
【0153】
化合物(RE7):
化合物(7−1−6)を用いて参考例7の製造法1と同様にして化合物(RE7)を得た。
【0154】
参考例8
2−(8−オキソ−5,6,7,8−テトラヒドロナフタレン−2−イル)エチル 4−メチルベンゼンスルホネート(2-(8-Oxo-5,6,7,8-tetrahydronaphthalen-2-yl)ethyl 4-methylbenzenesulfonate,化合物(RE8))
下記の製造方法に従い、合成した。
製造方法
【0155】
【化32】
【0156】
化合物(8−1):7−(2−ヒドロキシエチル)−1,2,3,4−テトラヒドロナフタレン−1−オール(7-(2-Hydroxyethyl)-1,2,3,4-tetrahydronaphthalen-1-ol)
水素化リチウムアルミニウム(33mg、0.86mmol)の無水テトラヒドロフラン(2mL)懸濁溶液に、加熱還流下で文献記載の方法(J. Med. Chem. 1994, 37(21), 3485)によって合成した(8−オキソ−5,6,7,8−テトラヒドロナフタレン−2−イル)アセティック アシッド(50mg、0.25mmol)の無水テトラヒドロフラン(1mL)溶液を加えて、反応混合物を1時間加熱還流した。室温に冷却後、反応溶液を氷浴で冷却し、水(32μL)を加え、引き続いて15%水酸化ナトリウム水溶液(32μL)と水(96μL)を加えて、溶液をそのまま30分間攪拌した。生じた沈殿をろ去し、ろ液を濃縮することで、表記化合物(8−1)(45mg、95%)を得た。
【0157】
化合物(8−2):7−(2−ヒドロキシエチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-Hydroxyethyl)-3,4-dihydronaphthalen-1(2H)-one)
化合物(8−1)(45mg、0.23mmol)と二酸化マンガン(20mg、2.3mmol)のジクロロメタン(2mL)懸濁溶液を室温で5日間攪拌した。二酸化マンガンをろ去し、ろ液を減圧濃縮して得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=2:1)で精製することで、表記化合物(8−2)(28mg、63%)を得た。
【0158】
化合物(RE8):
化合物(8−2)(28mg、0.15mmol)、トリエチルアミン(41μL、0.29mmol)とトリエチルアミン塩酸塩(1.4mg、0.015mmol)のジクロロメタン(2mL)溶液を氷浴で冷却し、p−トルエンスルホニルクロライド(42mg、0.22mmol)を加えて、反応混合物を氷冷下40分間攪拌した。水(20mL)を加えてクロロホルム(40mL×2)で抽出し、有機層を無水硫酸マグネシウムで乾燥した。溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=5:1→2:1)で精製することで、目的化合物(RE8)(49mg、96%)を得た。
保持時間(条件2):22.59分
1H-NMR(300 MHz, CDCl3):7.74 (d, J = 2.0 Hz, 1H, Ar), 7.71 (d, J = 8.4 Hz, 2H, Ar), 7.32-7.26(m, 3H, Ar), 7.17 (d, J = 7.7 Hz, 1H, Ar), 4.20 (t, J = 7.0 Hz, 2H, CH2), 3.00-2.90 (m, 4H), 2.64 (5, J = 6.5 Hz, 2H, CH2), 2.44 (s, 3H, CH3), 2.18-2.08 (m, 2H).
【0159】
参考例9
2−(3−オキソ−2,3−ジヒドロ−1H−インデン−5−イル)エチル 4−メチルベンゼンスルホネート(2-(3-Oxo-2,3-dihydro-1H-inden-5-yl)ethyl 4-methylbenzenesulfonate,化合物(RE9))
【0160】
【化33】
【0161】
文献記載の方法(J. Med. Chem. 1979, 22(12),1464)によって製造した(3−オキソ−2,3−ジヒドロ−1H−インデン−5−イル)アセティック アシッドを用いて、参考例8と同様にして、表記化合物(RE9)を製造した。
1H-NMR(300 MHz, CDCl3) δ:7.69 (d, J = 8.5 Hz, 2H, Ar), 7.44 (s, 1H, Ar), 7.41-7.38 (m, 2H, Ar), 7.28 (d, J = 8.5 Hz, 2H, Ar), 4.22 (t, J = 6.8 Hz, 2H, CH2), 3.11 (5, J = 6.0 Hz, 2H, CH2), 3.01 (t, J = 6.8 Hz, 2H, CH2), 2.69 (t, J = 6.0 Hz, 2H, CH2), 2.44 (s, 3H, CH3).
【0162】
参考例10
2−(5−オキソ−2,3,4,5−テトラヒドロ−1−ベンゾキセピン−7−イル)エチル 4−メチルベンゼンスルホネート(2-(5-Oxo-2,3,4,5-tetrahydro-1-benzoxepin-7-yl)ethyl 4-methylbenzenesulfonate,化合物(RE10))
【0163】
【化34】
【0164】
特許文献記載の方法(特開昭61-236774)によって製造した(5−オキソ−2,3,4,5−テトラヒドロ−1−ベンゾキセピン−7−イル)アセティック アシッドを用いて、参考例8と同様にして、表記化合物(RE10)を製造した。
保持時間(条件2):19.93分
1H-NMR (300 MHz, CDCl3) δ: 2.16-2.25 (2H, m), 2.44 (3H, s), 2.81-2.99 (4H, m), 4.13-4.27 (4H, m), 6.98 (1H, d, J = 8.3 Hz), 7.21 (1H, dd, J = 8.3, 2.3 Hz), 7.30 (2H, d, J = 8.1 Hz), 7.48 (1H, d, J = 2.4 Hz), 7.72 (2H, d, J = 8.3 Hz).
【0165】
参考例11
2−(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)エチル ベンゼンスルホネート(2-(4-Oxo-3,4-dihydro-2H-chromen-6-yl)ethyl benzenesulfonate),化合物(RE11))
【化35】
【0166】
化合物(7−1−6)とベンゼンスルホニルクロライドを用いて、参考例7の製造方法1と同様にして表記化合物を得た。
1H-NMR (300 MHz, CDCl3) δ: 2.85 (2H, t, J = 6.2 Hz), 2.90 (2H, t, J = 7.0 Hz), 4.19 (2H, t, J = 7.0 Hz), 4.22 (2H, t, J = 6.2 ), 6.79 (2H, d, J = 8.6 Hz), 7.01 (2H, d, J = 8.6 Hz), 7.45-7.52 (2H, m), 7.57-7.65 (1H, m), 7.75-7.82 (2H, m).
【0167】
実施例1
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
【0168】
【化36】
【0169】
化合物(RE3)(99mg,0.30mmol)、化合物(RE9)(100 mg,0.30mmol)と炭酸カリウム(0.39mmol)のアセトニトリル(3mL)溶液を60℃で27時間攪拌した。反応混合物を室温に冷却後、水(20mL)を加えて酢酸エチル(40mL)で抽出した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣を薄層シリカゲルクロマトグラフィー(クロロホルム:メタノール=10 : 1)で精製することで、表記化合物(137mg,93%)を淡黄色油状物として得た。
保持時間(条件1):27.44分
1H-NMR (300 MHz, CDCl3) δ: 1.33 (2H, m), 1.44-1.71 (3H, m), 1.97 (2H, bt, J = 9.9 Hz), 2.49 (2H, d, J = 6.8 Hz), 2.58 (2H, bt, J = 8.1 Hz), 2.67-2.73 (2H, m), 2.88 (2H, bt, J = 8.0 Hz), 2.99 (2 H, bd, J = 11.2 Hz), 3.11 (2H, t, J = 5.9 Hz), 3.50 (3H, s), 3.81 (2H, dd, J = 5.3, 4.2 Hz), 4.17 (2H, dd, J = 5.3, 4.2 Hz), 6.64 (1H, dd, J = 8.1, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 7.37-7.47 (3H, m), 7.58 (1H, bs).
【0170】
実施例2
7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
【0171】
【化37】
【0172】
化合物(RE9)の代わりに化合物(RE8)を用いて、実施例1と同様にして、表記化合物を合成した。
保持時間(条件1):31.95分
1H-NMR (400 MHz,CDCl3) δ: 1.26-1.37 (2H, m), 1.46-1.54 (1H, m), 1.64 (2H, bd, J = 12.2 Hz), 1.95 (2H, dt, J = 16.2, 5.8 Hz), 2.09-2.15 (2H, m), 2.49 (2H, d, J = 7.1 Hz), 2.55 (2H, bt, J = 8.4 Hz), 2.64 (2H, t, J = 6.6 Hz), 2.81 (2H, bt, J = 8.4 Hz), 2.93 (2H, t, J = 6.6 Hz), 2.96 (2H, d, J = 12.2 Hz), 3.50 (3H, s), 3.81 (2H, dd, J = 5.5, 4.3 Hz), 4.17 (2H, dd, J = 5.5, 4.3 Hz), 6.64 (1H, dd, J = 8.0, 2.0 Hz), 6.71 (1H, d, J = 2.0 Hz), 7.17 (1H, d, J = 7.8 Hz), 7.32 (1H, dd, J = 7.8, 1.8 Hz), 7.41 (1H, d, J = 8.0 Hz),7.86 (1H, d, J = 1.8 Hz).
【0173】
実施例3
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
【0174】
【化38】
【0175】
化合物(RE3)(52.0g,143mmol)を5%炭酸カリウム水溶液(350mL)に加え、トルエン(700mL×3)で抽出した。全有機層を無水硫酸ナトリウムで乾燥し、溶媒を減圧留去することで、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(48.1g)を得た。次に、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(2.00g,6.1mmol)、化合物(RE7)(2.01g,5.8mmol)および炭酸カリウム(1.66g,12mmol)のアセトニトリル(20mL)溶液を70〜80℃で7時間攪拌した。室温に冷却後、水(100mL)を加えて、クロロホルムで抽出し、クロロホルム層を無水硫酸ナトリウムで乾燥した。溶媒を減圧留去後、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=1:1→クロロホルム:メタノール=20:1)で精製することで、表記化合物(3.07g, 定量的)を得た。
保持時間(条件1):29.11分
1H-NMR (300 MHz,CDCl3) δ: 1.24-1.39 (2H, m), 1.40-1.73 (3H, m), 1.93 (2H, t, J = 10.6 Hz), 2.40-2.61 (2H, m), 2.48 (2H, d, J = 7.2 Hz), 2.66-2.87 (2H, m), 2.79 (2H, t, J = 6.4 Hz), 2.95 (2H, d, J = 11.7 Hz), 3.49 (3H, s), 3.81 (2H, t, J = 4.9 Hz), 4.17 (2H, t, J = 4.9 Hz), 4.51 (2H, t, J = 6.4 Hz), 6.64 (1H, dd, J = 8.1, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 6.89 (1H, d, J = 8.4 Hz), 7.32 (1H, dd, J = 8.4, 2.2 Hz), 7.41 (1H, d, J = 8.1 Hz), 7.70 (1H, d, J = 2.2 Hz).
【0176】
実施例4
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one hydrochloride)
【0177】
【化39】
【0178】
実施例3で得た化合物(3.07g , 5.8mmol)の2−プロパノール(20mL)溶液に室温で濃塩酸水溶液(36%,760μL,8.5mmol)を加え、溶液を室温で15.5時間攪拌した。析出物をろ取し、ろ上物を2−プロパノール(2mL×2)で洗浄し、減圧乾燥することで、表記化合物(2.26g,72%)を白色粉末として得た。
融点:156−157℃
【0179】
実施例5
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one benzenesulfonate)
【0180】
【化40】
【0181】
実施例3で得た化合物(1.00g,2.0mmol)とベンゼンスルホン酸1水和物(316mg,2.0mmol)の2−プロパノール(5mL)溶液を70℃に加温して固形物の溶解を確認後、室温まで4時間で冷却しながら攪拌した。生じた析出物をろ取し、2−プロパノール(1mL×2)で洗浄後、減圧乾燥することで表記化合物の粗生成物(1.13g)を得た。このうちの500mgをアセトン(10mL)と水(100μL)の混合溶液に加え、加温して溶解を確認後、徐々に冷却し、35−40℃で1時間攪拌後、さらに冷却して20−25℃で1時間攪拌した。析出物をろ取し、アセトン(1mL)で洗浄することで、表記化合物(323mg)を得た。
融点:143−144℃
【0182】
実施例1と同様の合成法で、以下に示す実施例6〜20の化合物を製造した。参考例に示した化合物(RE1)〜(RE10)の中から対応する化合物を原料として適宜用いた。
【0183】
実施例6
7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン(7-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydro-1-benzoxepin-5(2H)-one)
保持時間(条件1):30.84分
1H-NMR (300 MHz, CDCl3) δ: 1.25-1.42 (2H, m), 1.44-1.72 (3 H, m), 1.96 (2H, bt, J = 11.0 Hz), 2.15-2.24 (2H, m), 2.49 (2H, d, J = 7.0 Hz), 2.55 (2H, bdd, J = 9.9, 5.7 Hz), 2.79 (2H, bdd, J = 9.9, 5.7 Hz), 2.89 (2H, t, J = 6.9 Hz), 2.99 (2H, bd, J = 11.0 Hz), 3.50 (3H, s), 3.81 (2H, dd, J = 5.3, 4.2 Hz), 4.14-4.25 (4H, m), 6.64 (1H, dd, J = 8.0, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 6.99 (1H, d, J = 8.3 Hz), 7.27 (1H, dd, J = 8.3, 2.5 Hz), 7.41 (1 H, d, J = 8.1 Hz), 7.58 (1.0H, d, J = 2.5 Hz).
【0184】
実施例7
6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[4-Chloro-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):27.43分
1H-NMR (400 MHz,CDCl3) δ: 1.23-1.37 (2H, m), 1.43-1.56 (1H, m), 1.60-1.78 (2H, m), 1.94 (2H, t like, J = 12 Hz), 2.49 (2H, d, J = 7.2 Hz), 2.50-2.55 (2H, m), 2.72-2.80 (2H, m), 2.79 (2H, t, J = 6.4 Hz), 2.96 (2H, d like, J = 11.5 Hz), 3.49 (3H, s), 3.81 (2H, t, J = 4.8 Hz), 4.18 (2H, t, J = 4.8 Hz), 4.51 (2H, t, J = 6.4 Hz), 6.69 (1H, dd, J = 8.0, 1.8 Hz), 6.74 (1H, d, J = 1.8 Hz), 6.89 (1H, d, J = 8.5 Hz), 7.24(1H, d, J = 8.0 Hz), 7.31 (1H, dd, J = 8.5, 2.2 Hz), 7.70 (1H, d, J = 2.2 Hz).
【0185】
実施例8
6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル}−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):25.84分
1H-NMR(300 MHz,CDCl3) δ: 1.20-1.60(3H,m), 1.66(2H,d,J = 13.0Hz), 1.95(2H,t,J=11.7Hz), 2.21(3H,s), 2.49(2H,d,J=7.0Hz), 2.53-2.58(2H,m),
2.69(2H,t,J=5.9Hz), 2.86(2H,t,J=8.6Hz), 2.96(2H,d,J=11.4Hz),
3.10(2H,t,J=6.1Hz), 3.47(3H,s), 3.78(2H,t,J=4.6Hz), 4.12(2H,t,J=5.5Hz), 6.62(1H,s), 6.65(1H,d,J=7.9Hz), 7.03(1H,d,J=7.5Hz), 7.39(1H,d,J=7.9Hz), 7.44(1H,d,J=7.9Hz), 7.58(1H,s).
【0186】
実施例9
7−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
保持時間(条件1):30.36分
1H-NMR(300 MHz,CDCl3) δ: 1.30-1.80(5H,m), 1.99(2H,t,J=11.9Hz), 2.12(2H,quint,J=6.2Hz), 2.21(3H,s), 2.50(2H,d,J=6.6Hz), 2.54-2.70(4H,m), 2.80-2.90(2H,m), 2.93(2H,t,J=5.9Hz), 3.01(2H,d,J=10.8Hz), 3.47(3H,s), 3.78(2H,t,J=4.6Hz), 4.12(2H,t,J=5.5Hz), 6.61(1H,s), 6.65(1H,d,J=7.5Hz), 7.03(1H,d,J=7.5Hz), 7.18(1H,d,J=7.7Hz), 7.33(1H,dd,J=1.5,7.9Hz),
7.86(1H,d,J=1.7Hz).
【0187】
実施例10
6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):27.01分
1H-NMR (CDCl3) δ: 1.20-1.41 (2H, m), 1.43-1.73 (3H, m), 1.93 (2H, t like, J = 10.9 Hz), 2.20 (3H, s), 2.41-2.62 (2H, m), 2.49 (2H, d, J = 7.1 Hz), 2.67-2.87 (4H, m), 2.95 (2H, d, J = 10.7 Hz), 3.47 (3H, s), 3.77 (2H, t, J = 4.8 Hz), 4.12 (2H, t, J = 4.9 Hz), 4.51 (2H, t, J = 6.3 Hz), 6.61 (1H, s), 6.65 (1H, d, J = 7.6 Hz), 6.89 (1H, d, J = 8.5 Hz), 7.03 (1H, d, J = 7.6 Hz), 7.32 (1H, dd, J = 8.4, 2.3 Hz), 7.70 (1H, d, J = 2.2 Hz).
【0188】
実施例11
6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):17.50分
1H-NMR (400 MHz, CDCl3) δ: 1.38-1.26 (2H, m), 1.46-1.68 (3H, m), 1.96 (2H, bt, J = 11.1 Hz), 2.50 (2H, d, J = 7.1 Hz), 2.57 (2H, bt, J = 7.8 Hz), 2.67-2.71 (2H, m), 2.86 (2H, bt, J = 7.8 Hz), 2.98 (2H, bd, J = 11.1 Hz), 3.10 (2H, bt, J = 5.9 Hz), 3.99 (2H, bt, J = 4.5 Hz), 4.15 (2H, bt, J = 4.5 Hz), 6.67 (1H, dd, J = 8.0, 1.7 Hz), 6.71 (1H, d, J = 1.7 Hz), 7.39 (1H, d, J = 7.8 Hz), 7.42 (1H, d, J = 8.0 Hz), 7.44 (1H, dd, J = 7.8, 1.5 Hz), 7.58 (1H, bs).
【0189】
実施例12
7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
保持時間(条件1):21.91分
1H-NMR (400 MHz,CDCl3) δ: 1.24-1.39 (2H, m), 1.46-1.70 (3H, m), 1.95 (2H, bt, J = 12.2 Hz), 2.09-2.15 (2H, m), 2.50 (2H, d, J = 7.1 Hz), 2.51-2.59 (2H, m), 2.64 (2H, t, J = 6.6 Hz), 2.78-2.83 (2H, m), 2.93 (2H, t, J = 6.6 Hz), 2.96 (2H, d, J = 12.2 Hz), 3.99 (2H, bt, J = 4.4 Hz), 4.15 (2H, t, J = 4.5 Hz), 6.67 (1H, dd, J = 8.0, 2.0 Hz), 6.71 (1H, d, J = 2.0 Hz), 7.17 (1H, d, J = 7.8 Hz), 7.31 (1H, dd, J = 7.8, 2.0 Hz), 7.42 (1H, d, J = 8.0 Hz),7.86 (1H, d, J = 2.0 Hz).
【0190】
実施例13
6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):18.91分
1H NMR (300 MHz, CDCl3) δ: 1.28-1.42 (2H, m), 1.62-1.78 (1H, m), 1.66 (2H, bd, J=12.7 Hz), 2.00 (2H, bt, J=11.6 Hz), 2.50 (2H, d, J=7.0 Hz), 2.58 (2H, dd, J=11.0, 7.7 Hz), 2.76-2.81 (4H, m), 3.01 (2H, bd, J=11.6 Hz), 3.99 (2H, bt, J=4.5 Hz), 4.15 (2H, bd. J=4.5 Hz), 4.51 (2H, t, J=6.8 Hz), 6.66 (1H, dd, J = 8.1, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 6.90 (1H, d, J = 8.5 Hz), 7.33 (1H, dd, J =8.5, 2.4 Hz), 7.42 (1H, d, J = 8.1 Hz), 7.70 (1H, d, J = 2.4 Hz).
【0191】
実施例14
7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン(7-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydro-1-benzoxepin-5(2H)-one)
保持時間(条件1):20.89分
1H-NMR (300 MHz, CDCl3)δ: 1.25-1.45 (2H, m), 1.44-1.70 (3H, m), 1.96 (2H, bt, J = 11.2 Hz), 2.15-2.24 (2H, m), 2.50 (2H, d, J = 6.6 Hz), 2.54 (2H, bdd, J = 9.9, 6.1 Hz), 2.78 (2H, bdd, J = 9.9, 6.1 Hz), 2.89 (2H, t, J = 6.6 Hz), 2.98 (2H, bd, J = 11.2 Hz), 3.99 (2H, bt, J = 4.4 Hz), 4.15 (2H, t, J = 4.4 Hz), 4.21 (2H, t, J = 6.6 Hz), 6.67 (1H, dd, J = 8.1, 2.0 Hz), 6.71 (1H, d, J = 2.0 Hz), 6.99 (1H, d, J = 8.3 Hz), 7.26 (1H, dd, J =8.3, 2.4 Hz), 7.42 (1H, d, J = 8.1 Hz), 7.58 (1H, d, J = 2.4 Hz).
【0192】
実施例15
6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):27.89分
1H-NMR (300 MHz, CDCl3) δ: 1.24-1.42 (2H, m), 1.46-1.70 (3H, m), 1.97 (2H, bt, J = 10.3 Hz), 2.47 (2H, d, J = 7.0 Hz), 2.58 (2H, bt, J = 7.8 Hz), 2.67-2.71 (2H, m), 2.87 (2H, bt, J = 7.8 Hz), 2.98 (2H, bd, J = 10.3 Hz), 3.11 (2H, t, J = 5.9 Hz), 3.45 (3H, s), 3.72-3.75 (2H, m), 4.07-4.10 (2H, m), 6.66 (1H, td, J = 1.9, 0.3 Hz), 6.89-6.92 (2H, m), 7.39 (1H, d, J = 8.0 Hz), 7.45 (1H, dd, J = 8.0, 1.6 Hz), 7.58 (1H, d, J = 1.6 Hz).
【0193】
実施例16
7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
保持時間(条件1):32.39分
1H-NMR (400 MHz,CDCl3) δ: 1.28-1.40 (2H, m), 1.46-1.56 (1H, m), 1.64 (2H, bd, J = 12.6 Hz), 1.98 (2H, bt, J = 11.0 Hz), 2.08-2.15 (2H, m), 2.46 (2H, d, J = 7.0 Hz), 2.52-2.60 (2H, m), 2.64 (2H, t, J = 6.1 Hz), 2.80-2.87 (2H, m), 2.93 (2H, t, J = 6.1 Hz), 2.98 (2H, d, J = 11.0 Hz), 3.45 (3H, s), 3.73 (2H, t, J = 4.6 Hz), 4.08 (2H, t, J = 4.6 Hz), 6.66 (1H, bs), 6.88-6.92 (2H, m), 7.17 (1H, dt, J=7.8 Hz), 7.32 (1H, dd, J=7.8, 1.7 Hz), 7.85 (1H, d, J=1.7 Hz).
【0194】
実施例17
6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):29.33分
1H-NMR (400 MHz, CDCl3) δ: 1.26-1.37 (2H, m), 1.45-1.68 (3H, m), 1.94 (2H, bt, J=11.5 Hz), 2.46 (2H, d, J=7.1 Hz), 2.49-2.55 (2H, m), 2.73-2.81 (4H, m), 2.96 (2H, bd, J=11.5 Hz), 3.45 (3H, s), 3.72-3.75 (2H, m), 4.07-4.11 (2H, m), 4.51 (2H, t, J=6.3 Hz), 6.62 (1H, bt, J=1.8 Hz), 6.88-6.90 (3H, m), 7.32 (1H, dd, J=8.5, 2.3 Hz), 7.70 (1H, d, J=2.3 Hz).
【0195】
実施例18
7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン(7-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydro-1-benzoxepin-5(2H)-one)
保持時間(条件1):31.43分
1H-NMR (300 MHz, CDCl3)δ: 1.25-1.41 (2H, m), 1.44-1.69 (3H, m), 1.96 (2H, bt, J = 11.6 Hz), 2.15-2.24 (2H, m), 2.46 (2H, d, J = 7.0 Hz), 2.54 (2H, bdd, J = 9.9, 6.4 Hz), 2.79 (2H, bdd, J = 9.9, 6.4 Hz), 2.89 (2H, t, J = 7.0 Hz), 2.97 (2H, bd, J = 11.6 Hz), 3.72-3.75 (2H, m), 3.45 (3H, s), 4.07-4.10 (2H, m), 4.21 (2H, t, J = 6.7 Hz), 6.66 (1H, t, J = 1.8 Hz), 6.88-6.92 (2H, m), 6.99 (1H, d, J = 8.3 Hz), 7.27 (1H, dd, J = 8.3, 2.4 Hz), 7.58 (1H, d, J = 2.4 Hz).
【0196】
実施例19
6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[3-Chloro-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):26.77分
1H-NMR (300 MHz, CDCl3) δ: 1.25-1.39 (2H, m), 1.47-1.58 (1H, m), 1.65 (2H, bd, J=12.7 Hz), 1.96 (2H, bt, J=11.1 Hz), 2.47 (2H, d, J=7.1 Hz), 2.55-2.59 (2H, m), 2.68-2.71 (2H, m), 2.84-2.88 (2H, m), 2.98 (2H, bd, J=11.5 Hz), 3.10 (2H, t, J=5.7 Hz), 3.45 (3H, s), 3.73-3.75 (2H, m), 4.08-4.10 (2H, m), 6.62 (1H, bt, J=1.8 Hz), 6.73-6.77 (2H, m), 7.39 (1H, d, J=7.8 Hz), 7.44 (1H, dd, J=7.8, 1.7 Hz), 7.58 (1H, bs).
【0197】
実施例20
6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[3-Chloro-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):28.33分
1H-NMR (300 MHz, CDCl3) δ: 1.20-1.39 (2H, m), 1.45-1.58 (1H, m), 1.65 (2H, bd, J=13.4 Hz), 1.96 (2H, bt, J=10.6 Hz), 2.47 (2H, d, J=7.0 Hz), 2.50-2.59 (2H, m), 2.73-2.82 (4H, m), 2.98 (2H, bd, J=11.6 Hz), 3.45 (3H, s), 3.72-3.76 (2H, m), 4.07-4.10 (2H, m), 4.51 (2H, t, J=6.4 Hz), 6.62 (1H, bt, J=1.7 Hz), 6.73-6.78 (2H, m), 6.90 (1H, d, J=8.4 Hz), 7.32 (1H, dd, J=8.4, 2.4 Hz), 7.70 (1H, d, J=2.4 Hz).
【0198】
上記実施例6〜20の化合物の構造式を、下表1に示した。
【0199】
【表1】
【0200】
実施例21
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one fumarate)
【0201】
【化41】
【0202】
実施例3で得た化合物(503mg, 1.00mmol)とフマル酸(58mg, 0.50mmol)のエタノール(10mL)溶液を減圧濃縮することで濃縮残渣(557mg)を得た。このうちの100mgをアセトン(3mL)に加え、室温で1時間攪拌後、析出物をろ取し、減圧乾燥することで表記化合物(42mg)を得た。
融点:149−150℃
【0203】
実施例22
6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩(6-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one hydrochloride)
【0204】
【化42】
【0205】
実施例13で得た化合物(3.96g,8.1mmol)の2−プロパノール(40mL)とジクロロメタン(50mL)の混合溶液に、室温で濃塩酸水溶液(36%,860μL,10mmol)を加え、溶媒を減圧濃縮した。得られた濃縮残渣(3.61g)に2−プロパノール(72mL)を加えて2時間加熱還流し、3時間で20℃まで冷却しながら攪拌し、20℃で1時間攪拌後、氷冷しながら1時間攪拌した。析出物をろ取し、冷2−プロパノール(4mL×2)で洗浄することで白色固体(3.35g)を得た。これに2−プロパノール(100mL)と濃塩酸水溶液(36%,500μL)を加えて還流温度まで加温し、固形物の溶解を確認後、60℃まで冷却し、固体の析出を確認後、55〜60℃で1時間攪拌した。ひきつづいて2時間で20℃まで冷却しながら攪拌し、20℃で1時間攪拌した。析出物をろ取し、2−プロパノール(4mL×2)で洗浄することで表記化合物(3.10g,73%)を白色粉末として得た。
融点:177−178℃
【0206】
実施例23
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 臭化水素酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one hydrobromide)
【化43】
【0207】
実施例3で得た化合物(1.00 g, 2.00 mmol)の2−プロパノール(10mL)溶液に氷冷下48%臭化水素酸水(170μL, 2.4mmol)を加え、溶液を室温に昇温して1夜間攪拌した。生じた析出物をろ取し、2−プロパノール(1mL×2)で洗浄後、減圧乾燥することで表記化合物の粗結晶(797mg)を得た。粗結晶750mgを、2−プロパノール(30mL)に加え、加熱還流して固形物の溶解を確認後、室温までゆっくり冷却しながら19時間攪拌した。生じた析出物をろ取し、2−プロパノール(1mL×2)で洗浄後、減圧乾燥することで表記化合物(613mg)を白色結晶性固体として得た。
融点:148−150℃
【0208】
実施例24
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1コハク酸塩 6-(2-{4-[4-bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one butanedioate
【化44】
【0209】
実施例3で得た化合物(218 mg, 0.43 mmol)とコハク酸(26mg,0.22mmol)をエタノール(10mL)に加え、溶解を確認後、溶媒を減圧留去して濃縮残渣244mgを得た。濃縮残渣105mgを2−ブタノン/n−ヘキサン(1:1,1mL)中で1時間攪拌し、生じた析出物をろ取して室温で減圧乾燥することで表記化合物(36mg,62%)を白色粉末として得た。
融点:100−101℃
1H-NMR (DMSO-d6) δ: 1.10-1.25 (2H, m), 1.45-1.60 (3H, m), 1.94-2.07 (2H, m), 2.39 (4H, s), 2.47 (2H, d, J = 6.6 Hz), 2.53-2.58 (2H, m), 2.68-2.75 (2H, m), 2.76 (2H, t, J = 6.5 Hz), 2.92-3.01 (2H, m), 3.34 (3H, s), 3.68 (2H, t, J = 4.5 Hz), 4.16 (2H, t, J = 4.5 Hz), 4.50 (2H, t, J = 6.3 Hz), 6.69 (1H, dd, J = 8.0, 1.5 Hz), 6.94 (1H, d, J = 1.5 Hz), 6.95 (1H, d, J = 8.5 Hz), 7.42 (1H, dd, J = 8.5, 2.4 Hz), 7.44 (1H, d, J = 8.0 Hz), 7.58 (1H, d, J = 2.4 Hz).
【0210】
実施例25
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one benzenesulfonate)
化合物(RE3)(10.0g,27.4mmol)を水酸化カリウム水溶液(水酸化カリウム[2.15g,32.9mmol]と 水[50mL]から調製)に加え、トルエン(50mL×2)で抽出した。全有機層をあわせ、溶媒を減圧留去することで得た濃縮残渣1に、化合物(RE11)(9.57g,28.8mmol)、リン酸水素二カリウム(14.3g,82.3mmol)、N−メチル−2−ピロリジノン(3.0mL)、トルエン(100mL)を加え110〜120℃で6.5時間撹拌した。40〜50℃に冷却後、水(100mL)、テトラヒドロフラン(50mL)、トルエン(50mL)を加え、有機層を水(50mL)で洗浄した。溶媒を減圧留去して得た濃縮残渣2を精製することなく分割して次反応に用いた。濃縮残渣2の1/5量(5.5mmol相当)のアセトン(8mL)溶液をベンゼンスルホン酸アンモニウム(0.96g,5.48mmol)のアセトン(32mL)懸濁液に40〜50℃で加えた。50℃で1時間撹拌して固形物が全て溶解したことを確認後、溶媒を減圧留去した。濃縮残渣にアセトン(20mL)を加え、50℃に加熱して溶解後、40℃で種晶(5.0mg)を加えた。40℃で1時間撹拌してゆっくり室温に冷却後、氷浴で3時間撹拌し、析出物をろ取した。ろ上物を冷アセトン(4.0mL×2)で洗浄し、減圧乾燥することで、表記化合物(2.85g,79%)を得た。
【0211】
実施例26
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one benzenesulfonate)
化合物(RE3)(30.0 g, 82.3mmol)、化合物(RE7)(29.9g, 86,4 mmol)および炭酸カリウム(34.11 g, 247mmol)のアセトニトリル(236g)懸濁溶液を6.5時間加熱還流した。反応混合物を室温に冷却後、トルエン(519g)と水(600g)を加えて分液し、有機層を水(300g)で洗浄後、溶媒を減圧留去することで濃縮残渣(44.6g)を得た。濃縮残渣にアセトン(170g)を加えてろ過し、ろ上物をアセトン(40g)で洗浄した。全ろ液に水(3g)を加えて、40℃に加温し、ベンゼンスルホン酸1水和物(15.2g,96mmol)を加え、種晶(0.15g)を加えて40℃で1時間攪拌した。引き続き2℃までゆっくり冷やしながら攪拌し、内温2℃で1時間攪拌後、析出物をろ取し、冷アセトン(47.5g×2)で洗浄した。ろ上物を40℃で減圧乾燥することにより表記化合物(36.8g,68%)を白色結晶性固体として得た。
【0212】
実施例27
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩のForm1型結晶
化合物(RE3)(52.0g,143mmol)を5%炭酸カリウム水溶液(350mL)に加え、トルエン(700mL×3)で抽出した。全有機層を無水硫酸ナトリウムで乾燥し、溶媒を減圧留去することで4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(48.1g)を得た。これを化合物(RE7)(47.0g,143mmol)、炭酸カリウム(37.5 g,271mmol)とともにアセトニトリル(470mL)に加え、反応混合物を内温55−60℃で25時間攪拌した。室温に冷却後、水(940mL)、トルエン(940mL)を加えて分液し、水層をトルエン(470mL)で再抽出した。全有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣(76.6g)に2−プロパノール(1.1L)を加え、水冷しながら15−20℃で濃塩酸水(36%,14mL,171mmol)を5分間で滴下し、溶液を2時間15−20℃で22時間攪拌後、氷浴で冷却して4.5時間攪拌した。析出物をろ取し、冷却した2−プロパノール(55mL×2)で洗浄し、減圧乾燥することで濃縮残渣(61.6g)を得た。これに2−プロパノール(900mL)、36%濃塩酸水(36%,9.0mL)を加えて加温し、60℃付近ですべて溶解を確認後、溶液を20℃まで冷却して、15−20℃で14.5時間攪拌後、氷浴で冷却して5時間攪拌した。析出物をろ取し、冷2−プロパノール(70mL×2)で洗浄し、減圧乾燥することで表記化合物(51.1 g,70%)を無色結晶性固体として得た。
融点 157−159℃
元素分析
【0213】
【表2】
【0214】
実施例28
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormA型結晶
実施例3で得た化合物(1.00 g, 2.00 mmol)とベンゼンスルホン酸(316mg,2.0mmol)の2−プロパノール(5mL)溶液を70℃に加温して固形物の溶解を確認後,室温まで4時間で冷却しながら攪拌した。生じた析出物をろ取し,2−プロパノール(1mL×2)で洗浄後,減圧乾燥することで濃縮残渣(1.13 g)を得た。このうちの1.05gを2−プロパノール(32mL)に加えて,混合溶液を加熱還流して固形物が溶解したことを確認後,冷却しながら攪拌した。65℃で種晶(2mg)を加えて60−65℃で1時間攪拌した。引き続いて2時間で30℃まで冷却しながら攪拌し,水冷(25℃)で1時間攪拌後,析出物をろ取し,2−プロパノール(2mL×2)で洗浄後,減圧乾燥することで表記化合物(963mg)を無色結晶性固体として得た。
融点:142−143℃
元素分析
【0215】
【表3】
【0216】
実施例29
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormB型結晶
化合物(RE3)(2.00g,5.5mmol)を10%炭酸カリウム水溶液(20mL)に加え、トルエン(20mL×2)で抽出した。全有機層を溶媒を減圧留去することで、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(1.95g)を得た。次に、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(1.95g,5.5mmol)、化合物(RE7)(1.80g,5.2mmol)および炭酸カリウム(1.44g,10mmol)のアセトニトリル(10mL)溶液を55−60℃で19時間攪拌した。反応溶液を室温に冷却後、水(30mL)を加えて、トルエン(30mL×3)で抽出し、全有機層を5%重曹水(30mL)、水(30mL)の順で洗浄後、溶媒を減圧留去した。得られた濃縮残渣をアセトン(40mL)に溶解させてベンゼンスルホン酸1水和物(1.06g,6.0mmol)を加えて、固形物の溶解を確認後、溶媒を減圧留去した。得られた濃縮残渣にアセトン(40mL)および水(0.4mL)を加えて40℃に加温し、種晶(5mg)を加えて35−40℃で1.5時間攪拌した。引き続き1時間で20℃まで冷却後、1時間氷冷下攪拌した。析出物をろ取し、冷アセトン(5mL×2)で洗浄後、室温で減圧乾燥することで表記化合物(2.45g,71%)を無色結晶性固体として得た。
融点:143−144℃
元素分析
【0217】
【表4】
【0218】
実施例30
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormC型結晶
実施例3で得た化合物(7.76g,15.5mmol)を1%(v/v)含水アセトン(100mL)に溶解させ,溶液を40−45℃に加温してからベンゼンスルホン酸1水和物(2.93g、17mmol)を加えて攪拌した。固形物の溶解を確認後、種晶(50mg)を加えて1時間40−45℃で攪拌した。引き続き3時間で25℃まで冷却しながら攪拌し、その後室温で17.5時間攪拌した。引き続き1時間で5℃以下に冷却して、そのまま1時間攪拌し、析出物をろ取して、ろ上物を冷アセトン(10mL×2)で洗浄後、減圧乾燥することで表記化合物(8.83g,88%)を無色結晶性固体として得た。
融点:143−145℃(128℃でFormAに転移)
1H-NMR (400 MHz,DMSO-d6) δ: 1.30-1.46 (2H, m), 1.72-1.86 (3H, m), 2.52 (2H, d, J = 6.4 Hz), 2.78 (2H, t, J = 6.5 Hz), 2.82-3.00 (4H, m), 3.18-3.30 (2H, m), 3.34 (3H, s), 3.48-3.58 (2H, m), 3.66-3.72 (2H, m), 4.12-4.20 (2H, m), 4.51 (2H, t, J = 6.5 Hz), 6.72 (1H, dd, J = 8.1, 2.0 Hz), 6.96 (1H, d, J = 2.0 Hz), 7.01 (1H, d, J = 8.5 Hz), 7.27-7.34 (3H, m), 7.44 (1H, dd, J = 8.5, 2.2 Hz), 7.47 (1H, d, J = 8.1 Hz), 7.56-7.61 (2H, m), 7.67 (1H, d, J = 2.2 Hz), 9.02 (1H, br s).
元素分析
【0219】
【表5】
【0220】
実施例31
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA型結晶
実施例3で得た化合物(3.0g, 6.0mmol)をエタノール(75mL)に溶解させ、フマル酸(693mg, 6.0mmol)のエタノール(25mL)溶液を加え、さらにメタノール(100mL)を加えて溶解を確認後、溶媒を減圧留去して濃縮残渣を得た。これにアセトン(110mL)を加えて室温で2.5時間攪拌し、生じた析出物をろ取し、減圧乾燥(40℃、1mmHg、18時間)することで表記化合物(3.26g,88%)を無色結晶性固体として得た。
融点:149−151℃
1H-NMR (400 MHz,DMSO-d6) δ: 1.17-1.40 (2H, m), 1.47-1.73 (3.0H, m), 2.15-2.32 (2H, m), 2.48 (2H, d, J = 6.4 Hz), 2.63-2.85 (6H, m), 3.05-3.17 (2H, m), 3.34 (3H, s), 3.65-3.72 (2H, m), 4.13-4.20 (2H, m), 4.50 (2H, t, J = 6.4 Hz), 6.56 (2H, s), 6.70 (1H, dd, J = 8.0, 1.7 Hz), 6.95 (2H, d, J = 1.7 Hz), 6.97 (2H, d, J = 8.6 Hz), 7.42 (2H, dd, J = 8.6, 2.2 Hz), 7.45 (1H, d, J = 8.0 Hz), 7.60 (1H, d, J = 2.2 Hz).
【0221】
実施例32
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA+型結晶
実施例31で得たFormA型結晶(500mg)を1%含水アセトン(25mL)に加え65℃に加温して溶解を確認後、ゆっくり室温まで冷却しながら17時間攪拌した。生じた析出物をろ取し減圧乾燥(40℃、1mmHg、24時間)することで表記化合物(403mg,80%)を無色結晶性固体として得た。
融点:148−149℃
1H-NMR:実施例31と同じデータが得られた。
【0222】
実施例33
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩
4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン塩酸塩(2.0g,5.48mmol)を水酸化カリウム水溶液(水酸化カリウム[0.43g,6.58mmol]と 水[10ml]から調製)に加え、トルエン(10ml×2)で抽出した。全有機層をあわせ、溶媒を減圧留去した。濃縮残渣に、6−(2−p−ベンゼンスルホニルオキシエチル)−2,3−ジヒドロ−4H−クロメン−4−オン(1.91g,5.76mmol)、リン酸水素二カリウム(2.87g,16.5mmol)、N−メチル−2−ピペリドン(0.6ml)、トルエン(20ml)を加え110〜120℃で6時間攪拌した。40〜50℃に冷却後、水(20ml)、テトラヒドロフラン(10ml)、トルエン(10ml)を加え、有機層を水(10ml)で洗浄した。溶媒を減圧留去することで濃縮残渣を得た。得られた濃縮残渣を精製することなくアセトン(6.0ml)に溶解させて、50〜60℃でベンゼンスルホン酸(0.93 g,5.76mmol)のアセトン(4.0ml)溶液を滴下した。50〜60℃で30分攪拌後、酢酸n−ブチル(20.0ml)を滴下し、さらに1時間攪拌した。室温に冷却後、酢酸n−ブチル(10ml)を滴下し、氷浴で3.5時間攪拌した。析出物をろ取し、ろ上物を冷アセトン(4.0ml×2)で洗浄、減圧乾燥することで、表記化合物(3.21g,89%)を得た。
【0223】
実施例34
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormC型結晶
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(1.0g,1.51mmol)に0.5%含水アセトン(15.2mL)を加え、50℃以上で溶解した。溶液を40〜50℃に冷却し、種晶(実施例30で得られたFormC型結晶)(5mg)を加え、40〜50℃で1時間撹拌した。室温に冷却し、室温で1時間撹拌後、n−ヘプタン(8.8 mL)を加えた。氷浴で3時間撹拌後、析出物をろ取し、アセトン(2.5 mL)で洗浄後、50℃で減圧乾燥することで標記化合物(0.92g,92%)をFormC型結晶として得た。
【0224】
試験例1
ヒトセロトニン再取り込み阻害作用を評価するための[3H]シタロプラム結合 を用いたスクリーニング試験
【0225】
1−1 使用細胞および膜標品の調製
実験にはヒトセロトニントランスポーター(h−SERT)を発現させたCHO細胞(h−SERT/CHO)を用いた。細胞は5%CO2インキュベーター中で、10%FCS、500μg/ml Geneticinおよび100U/ml ペニシリン−100μg/ml ストレプトマイシンを含むF12(すべてSigma Aldrich製)にて培養し、SERT バッファー(120mM NaClおよび5mM KClを含む50mM Tris−HCl(pH=7.4))にて剥離・採取した細胞をテフロン(登録商標)製ホモジナイザーでホモジナイズした後、遠心操作(50,000xg、30min、4℃)を行なった。沈渣は適量のSERT バッファーに再懸濁し、使用まで−80℃で保存した。膜標品中のタンパク質量は、標準物質にウシ血清アルブミン(Sigma Aldrich製)を用いて、Dye Reagent Concentrate(BIO−RAD製)により定量した。
【0226】
1−2 受容体結合実験
[3H]シタロプラム結合の測定はOwensらの方法[Owens M. J. et al., J. Pharm. Exp. Ther., 283, 1305-1322(1997)]に準じて行った。すなわち、SERTバッファーで希釈した[3H]シタロプラム(最終濃度約2nM)50μl、h−SERT/CHO膜標品(蛋白量として40μg/well)149μl、およびジメチルスルホキシドに溶解した被験薬溶液1μlを加え全量を200μlとした。この液を室温で60分間反応させた後、0.05%ポリエチレンイミン水溶液でコーティングしたガラス繊維濾紙を用い速やかに低圧吸引ろ過した。ガラス繊維濾紙をSERT バッファー250μlで2回洗浄した後、ACS−II(Amersham製)4ml入りのガラスバイアルに移し、濾紙上に残存する放射活性を液体シンチレーションカウンターを用いて測定した。[3H]シタロプラムの非特異的結合は1μM クロミプラミン存在下での結合量とした。
IC50値をHill解析[Hill A. V., J. Physiol., 40, 190-200 (1910)参照]により算出し、h−SERT結合阻害定数(Ki)の算出は、式:
h−SERT結合阻害定数(Ki)=IC50/(1+S/Kd)
[Sは添加した[3H]シタロプラム濃度を示す。また、Kd値は[3H]シタロプラムの結合解離定数であり、別途同じ細胞膜を用いて実施した飽和結合実験より算出された値(2.16nM)を用いた。]
によって算出した。h−SERT結合阻害定数Ki値の数値が小さいほど、ヒトセロトニン再取り込み阻害作用が高いことを意味する。
【0227】
試験例2
ヒトセロトニン1A受容体に対する親和性を評価するための[3H]8−OH−DPAT結合試験
【0228】
2−1 使用細胞および膜標品の調製
実験にはヒトセロトニン1A受容体(h−5−HT1A)を発現させたCHO細胞(h−5−HT1A/CHO)を用いた。細胞は5%CO2インキュベーター中で、10%FCS、500μg/ml Geneticinおよび100U/ml ペニシリン−100μg/ml ストレプトマイシンを含むF12(すべてSigma Aldrich製)にて培養し、膜標品はYabuuchiらの方法3)に従って調製した。すなわち、50mM Tris−HCl(pH=7.4)にて剥離・採取した細胞を、テフロン(登録商標)製ホモジナイザーでホモジナイズした後、遠心操作(48,000xg、20min、4℃)を行なった。沈渣は適量の50mM Tris−HCl(pH=7.4)に再懸濁し、使用まで−80℃で保存した。膜標品中のタンパク質量は、標準物質にウシ血清アルブミン(Sigma Aldrich製)を用いて、Dye Reagent Concentrate(BIO−RAD製)により定量した。
【0229】
2−2 受容体結合実験
実験はYabuuchiらの方法[Yabuuchi K. et al., Biogenic Amines, 18, 319-328 (2004)]に準じて実施した。50mM Tris−HCl(pH=7.4)、4mM CaCl2を含む緩衝液中に、[3H]8−OH−DPAT(最終濃度0.5nM)を50μl、被検薬溶液を1μl、h−5−HT1A/CHO膜標品(蛋白質量として25μg/well)149μlを加え、全量200μlの反応液を用いて測定した。反応液を室温で30分間反応させた後、ガラス繊維濾紙上に速やかに低圧吸引濾過した。ガラス繊維濾紙を、50mM Tris−HCl(pH=7.4)250μlで2回洗浄した後、ACS−II(Amersham社製)4ml入りのカウンティングバイヤルに添加し、濾紙上に残存した受容体結合放射活性を液体シンチレーションカウンターで測定した。非特異的結合は10μM 8−OH−DPAT存在下での結合量とした。
IC50値をHill解析[Hill A. V., J. Physiol., 40, 190-200 (1910)参照]により算出し、h−5−HT1A結合阻害定数(Ki)の算出は、式:
h−5−HT1A結合阻害定数(Ki)=IC50/(1+S/Kd)
[Sは添加した[3H]8−OH−DPAT濃度を示す。また、Kd値は[3H]8−OH−DPATの結合解離定数であり、別途同じ細胞膜を用いて実施した飽和結合実験より算出された値(1.28nM)を用いた。]
によって算出した。h−5−HT1A結合阻害定数Ki値の数値が小さいほど、ヒトセロトニン1A受容体に対する親和性が高いことを意味する。
【0230】
実施例で得た本発明のベンジルピペリジン化合物について、上記の試験例1および2の試験を行い、結果を表6に示した。これらの試験結果から、本発明のベンジルピペリジン化合物またはその薬学上許容される塩が、ヒトセロトニン再取り込み阻害作用とヒト5−HT1A受容体に対する結合親和性を併せ持つだけでなく、ヒトセロトニン再取り込み阻害作用が高いことが明らかとなった。
【0231】
【表6】
【0232】
試験例3
CYP2D6阻害スクリーニング試験
【0233】
3−1 材料
ジクロフェナックはSigma社より、Pooled of Human Liver MicrosomesはXenotech,LLCより購入した。
【0234】
3−2−1 0.5Mリン酸カリウムBuffer(pH 7.4)の調製
0.5Mリン酸一カリウム溶液 150mLと0.5M リン酸二カリウム溶液 700mLを混合してpH7.4に調整した。
【0235】
3−2−2 165mM塩化マグネシウム溶液の調製
塩化マグネシウム六水和物、MgCl2・6H2O 3.35g/100mLになるよう純水に溶解した。
【0236】
3−2−3 ヒト肝ミクロソーム溶液の調製
Pooled of Human Liver Microsomes(20mg/ml)150μL、0.5M リン酸カリウムバッファー12mL、165 mM 塩化マグネシウム溶液1.2mLおよび純水34.65mLを混合して調製した。
【0237】
3−2−4 13mMβ−NADPH溶液の調製
β−NADPHを11.75mg/mLになるよう純水に溶解し調製した。
【0238】
3−2−5 基質溶液の調製
ジクロフェナックを2.0mMとなるようDMSOで溶解した後、純水で200倍に希釈した。
【0239】
3−3 実験方法
1. 被検薬物の10mM DMSO溶液をDMSOで5倍ずつ4段階希釈し、10、2、0.4、0.08mMのDMSO溶液を調製した。
2. 1.の被検薬物溶液およびDMSOをヒト肝ミクロソーム溶液で96倍に希釈し、80μLずつマイクロプレートに分注した。
3. 2.に基質溶液10μLおよびβ−NADPH溶液10μLを添加し、37℃で10minインキュベートした。
4. メタノール300μLを添加し、反応を停止させた。
5. 反応混合物をフィルターろ過し、LC−MSMSにて分析を行った。
【0240】
3−4 定量および計算
LC−MSMSにて4−ヒドロキシジクロフェナックの生成量を定量し、これを各ウェルにおけるCYP2D6の活性値とした。被検薬物としてDMSOを用いたウェルの活性と比較し、各サンプル添加群の残存活性を求め、被検薬物濃度よりCYP2D6阻害のIC50値を求めた。IC50値は残存活性50%をはさむ2点を結ぶ直線より算出した。CYP2D6阻害のIC50値の数値が大きいほど、CYP2D6阻害が弱いことを意味する。
【0241】
試験例4
ヒト肝ミクロソーム代謝におけるCYP2D6寄与率スクリーニング試験
【0242】
終濃度として3mMのNADPH(オリエンタル酵母工業製)、1mg/mLのヒト肝ミクロソーム(XENOTECH,LLC製)および1μMの被験物質を含む0.2mLの50mMリン酸カリウム緩衝液(pH7.4)を37℃の水浴上で加温することにより代謝反応を行った。15分または30分反応した後、反応液の3倍容のメタノールを添加して攪拌することにより反応を停止させた。この反応液を遠心分離して蛋白を沈殿させた後、上清を採取しLC−MS/MS分析に供した。結果の解析は以下の通り実施した。
被験物質の定量を行い、その残存量の経時変化を対数プロットしその傾きより代謝速度を算出した。
反応溶液に終濃度4μMのキニジンを添加しなかった場合の代謝速度に対する添加した場合の代謝速度の割合をCYP2D6以外の酵素の寄与率とし、その残りをCYP2D6の寄与率とした。すなわち、式:
寄与率(%)={1−(代謝速度[キニジン添加有]/代謝速度[キニジン添加なし])}×100
によって算出した。CYP2D6の寄与率の数値が小さいほど、CYP2D6の寄与が小さいことを意味する。
【0243】
実施例で得た本発明のベンジルピペリジン化合物について、試験例3および4の試験を行い、結果を表7に示した。これらの試験結果から、本発明のベンジルピペリジン化合物またはその薬学上許容される塩については、CYP2D6阻害が弱く、また代謝におけるCYP2D6の寄与が小さいことが明らかとなった。
【0244】
【表7】
*)ヒトミクロソーム代謝に対して安定であるため、
本試験ではCYP2D6の寄与率は判定不能であった。
【0245】
試験例5
[3H]5−HTを用いたセロトニントランスポーター機能阻害試験
1−1 使用細胞および細胞懸濁液の調製
実験にはヒトセロトニントランスポーター(h−SERT)を発現させたCHO細胞(h−SERT/CHO)を用いた。細胞は5%CO2インキュベーター中で、10% FCS、500μg/ml Geneticinおよび100U/ml ペニシリン−100μg/ml ストレプトマイシンを含むF12(すべてSigma Aldrich製)にて培養し、使用当日にCell Dissociation Buffer(Enzyme−free,PBS−based,GIBCO製)にて剥離・採取した。採取した細胞はPBS,0.1mM CaCl2および1mM MgCl2を含む緩衝液(以下PBSCM)に懸濁し、使用まで氷中にて保存した。
【0246】
1−2[3H]5−HT取り込み試験
[3H]5−HT取込の測定はRomanらの方法[Roman D.L. et al., J. Pharmacol. Exp. Ther., 308, 679-687 (2004)、Hill A. V., J. Physiol., 40, 190-200 (1910)]に準じて行った。すなわち、細胞懸濁液149μl(10×104cells/well)およびジメチルスルホキシドに溶解した被験薬溶液1μlを37℃で10分間プレインキュベーション後、PBSCMで希釈した[3H]5−HT(最終濃度約10nM)50μlを加え全量を200μlとした。この液を37℃で10分間反応させた後、0.3%ポリエチレンイミン水溶液でコーティングしたガラス繊維濾紙を用い速やかに低圧吸引ろ過した。ガラス繊維濾紙を氷冷整理食塩水250μlで2回洗浄した後、ACS−II(Amersham製)4ml入りのガラスバイアルに移し、濾紙上に残存する放射活性を液体シンチレーションカウンターを用いて測定した。[3H]5−HTの非特異的取込は13.3μM パロキセチン存在下での取込量とした。
IC50値の算出はHill解析[Hill A. V., J. Physiol., 40, 190-200 (1910)参照]により、取込阻害定数(Ki)の算出は次式によって算出した:
取込阻害定数(Ki)=IC50/(1+S/Km)
ただし、Sは添加した[3H]5−HT濃度を示す。また、Km値は[3H]5−HTのミカエリス定数であり、別途同じ細胞標品を用いて実施した飽和実験より算出された値(441nM)を用いた。得られた結果を、表8に示した。
【0247】
【表8】
【0248】
表8の試験結果から、本発明のベンジルピペリジン化合物またはその薬学上許容される塩については、セロトニンニューロンのセロトニントランスポーターに作用し、セロトニン再取り込みを強く阻害するすることが明らかとなった。
【0249】
試験例6
粉末エックス線回折(XRPD)分析
XRPD粉末エックス線回折装置としては、スペクトリス社製 X’Pert Pro(エキスパート プロ)を用いた。回折角度2シータ(2θ)4度〜40度の範囲でCu Kα1線(波長1.54060オングストローム)、X線管電流 40ミリアンペア、電圧45キロボルト、ステップ0.01700度、測定時間101.41770秒/ステップの条件にて測定した。測定はシリコン単結晶(Si単結晶)からなる無反射試料板にサンプル量約5ミリグラムを用いて測定を行った。
実施例27〜32で得られた各結晶について、本分析を行った。得られた粉末エックス線回折パターンを図1〜6に示す。結晶形の特定には、回折パターンを基に、各結晶の特徴となる回折ピークにより判断することができる。各結晶について、回折パターンから特定した特徴的な回折ピークは後述する。なお、回折角度2θにおけるピーク角度は、測定機器により、もしくは測定条件等により多少の誤差範囲を生じることがある。具体的には、測定誤差は±0.2、好ましくは±0.1の範囲であってよい。一方、相対強度については、試料調整の具合や測定条件等により、その値は変化しうる。
実施例27の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩のForm1型結晶の粉末エックス線回折のピーク角度、面間隔(d−spacing)およびその相対強度を表9に示す。
【0250】
【表9】
【0251】
表9に示したピークのうち、本塩酸塩のForm1型結晶の特徴的な回折ピークとしては、回折角度2θが、4.3゜、8.7゜、10.8゜、12.6゜、13.0゜、16.0゜、17.1゜、19.3゜、19.7゜、20.2゜であるピークが挙げられる。
【0252】
実施例28の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormA型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表10に示す。
【0253】
【表10】
【0254】
表10に示したピークのうち、本ベンゼンスルホン酸塩のFormA型結晶の特徴的な回折ピークとしては、回折角度2θが、8.7゜、11.9゜、13.0゜、14.9゜、15.3゜、17.4゜、17.7゜、18.2゜、19.3゜、20.4゜であるピークが挙げられる。
【0255】
実施例29の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormB型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表11に示す。
【0256】
【表11】
【0257】
表11に示したピークのうち、本ベンゼンスルホン酸塩のFormB型結晶の特徴的な回折ピークとしては、回折角度2θが、8.9゜、10.0゜、12.0゜、12.5゜、14.0゜、16.9゜、17.0゜、17.6゜、18.9゜、20.6゜であるピークが挙げられる。
【0258】
実施例30の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormC型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表12に示す。
【0259】
【表12−1】
【表12−2】
【0260】
表12に示したピークのうち、本ベンゼンスルホン酸塩のFormC型結晶の特徴的な回折ピークとしては、回折角度2θが、8.6゜、9.9゜、12.6゜、13.0゜、14.4゜、14.6゜、18.0゜、18.4゜、19.6゜、19.8゜であるピークが挙げられる。
【0261】
実施例31の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表13に示す。
【0262】
【表13】
【0263】
表13に示したピークのうち、本フマル酸塩のFormA型結晶の特徴的な回折ピークとしては、回折角度2θが、7.6゜、10.0゜、11.5゜、11.8゜、14.6゜、15.3゜、16.0゜、17.4゜、18.3゜、19.6゜、20.9゜、22.0゜、23.2゜であるピークが挙げられる。
【0264】
実施例32の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA+型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表14に示す。
【0265】
【表14】
【0266】
表14に示したピークのうち、本フマル酸塩のForm A+型結晶の特徴的な回折ピークとしては、回折角度2θが、7.4゜、10.1゜、11.1゜、14.7゜、16.0゜、18.4゜、19.8゜、22.2゜、23.4゜、25.9゜、26.7゜であるピークが挙げられる。
【0267】
製剤例1
錠剤の製造
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(5g)、乳糖(80g)、コーンスターチ(30g)、結晶セルロース(25g)、ヒドロキシプロピルセルロース(3g)、軽質無水ケイ酸(0.7g)及びステアリン酸マグネシウム(1.3g)を、常法により混合、造粒し、1錠あたり145mgで打錠し、1000錠を製する。
【0268】
製剤例2
散剤の製造
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(10g)、乳糖(960g)、ヒドロキシプロピルセルロース(25g)及び軽質無水ケイ酸(5g)を、常法により混合した後、散剤に製する。
【産業上の利用可能性】
【0269】
式(1)で表される本発明のベンジルピペリジン化合物及びその薬学上許容される塩は、ベンゼン環部分の3位が2−メトキシエトキシ基または2−ヒドロキシエトキシ基で置換されているジ置換ベンジル基を有し、且つオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基をピペリジンの1位に有する、という化学構造上の特徴を有し、セロトニン1A受容体に対する親和性を併せ持ち、ヒトセロトニン再取り込み阻害活性が向上し、ヒトチトクロームP450分子種の一つであるCYP2D6に対する阻害作用が弱く、もしくはヒトにおける薬物代謝においてCYP2D6の寄与が小さな、新しいセロトニン再取り込み阻害剤であることから、例えばうつ病や不安(不安障害)などの疾患に対し、治療効果に優れ安全性の高い治療薬または予防薬として使用しうる。
【図面の簡単な説明】
【0270】
【図1】試験例6の粉末エックス線回折分析で得られた、実施例27の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩のForm1型結晶の粉末エックス線回折パターンである。
【図2】試験例6の粉末エックス線回折分析で得られた、実施例28の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormA型結晶の粉末エックス線回折パターンである。
【図3】試験例6の粉末エックス線回折分析で得られた、実施例29の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormB型結晶の粉末エックス線回折パターンである。
【図4】試験例6の粉末エックス線回折分析で得られた、実施例30の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のForm C型結晶の粉末エックス線回折パターンである。
【図5】試験例6の粉末エックス線回折分析で得られた、実施例31の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のForm A型結晶の粉末エックス線回折パターンである。
【図6】試験例6の粉末エックス線回折分析で得られた、実施例32の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のForm A+型結晶の粉末エックス線回折パターンである。
【技術分野】
【0001】
本発明は、セロトニン再取り込み阻害剤として有用な、新規なベンジルピペリジン化合物又はその薬学上許容される塩に関する。さらに詳しくは、本発明のベンジルピペリジン化合物は、ピペリジンを母骨格とする化合物であり、ピペリジンの4位に特定の置換ベンジル基を有し、さらにオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基(フェネチル基)をピペリジンの1位に有する。本発明のベンジルピペリジン化合物は、セロトニン再取り込み阻害作用を有することから、例えば抗うつ薬として有用である。
【背景技術】
【0002】
うつ病はあらゆる年令の人に影響を与える慢性病である。現在、使用されている各種の抗うつ薬のうち最も成功を収めているのは、選択的セロトニン再取り込み阻害剤(Selective serotonin reuptake inhibitor、以下SSRIと略すこともある)である。SSRIは、ドーパミン及びノルアドレナリン再取り込み阻害作用よりも高いセロトニン再取り込み阻害作用を有する。SSRIとして市販された最初の薬剤はジメリジン(zimelidine)であった。その後上市された又は開発下にある他のSSRIとしては、例えば、フルオキセチン(fluoxetine)、フルボキサミン(fluvoxamine)、シタロプラム(citalopram)、セルトラリン(sertraline)およびパロキセチン(paroxetine)が挙げられる。
【0003】
このようなSSRIはうつ病の治療薬として広く用いられているものの、まだいくつかの問題点を有することが指摘されている。全うつ病患者の約1/3を占める難治性の患者に対しては、SSRIでも十分な治療効果を上げられないことや、十分な抗うつ作用が発現するまでに3〜8週間もの長い期間を必要とすることが、代表的な問題として挙げられる。このようにSSRIの抗うつ作用の発現が緩慢である一方、その副作用は直ちに起こり得る。すなわち、患者が薬剤の治療効果を得ることなく副作用のみを経験する易損性期(vulnerable period)を招くという問題が生じる。このため、この期間中も同じ薬剤の服用を続けるように患者を説得することが治療医師にとってしばしば重い負担になる。更に、自殺を図る恐れのある患者にとっては、抗うつ作用の発現が緩慢であるため、十分なうつ症状の改善を経験する前に自発性(initiative)を回復することから、自殺の危険性やたびたびの入院の必要性などが生じる。従って、抗うつ作用が素早く発現するような抗うつ薬の開発が望まれている。
【0004】
SSRIが抗うつ作用を発現するまでに数週間もの長い期間を必要とする理由は、以下のように考えられている。
SSRIはセロトニン代謝回転の急性セロトニン再取り込みを阻害する。この阻害作用がセロトニンニューロンの神経終末において起こることにより、セロトニンによる神経伝達が強化され抗うつ作用が発現する。しかしながら、同阻害作用は縫線核に存在するセロトニンニューロン細胞体や樹状突起においても起こるため、縫線核ではセロトニン1A自己受容体を介するセロトニンニューロンの自己発火抑制(negative feedback反応)を強化してしまう。この結果、SSRI投与後の初期においては、セロトニンニューロンにおける神経伝達は全体として期待されるほど強化されないことになる。一方、数週間SSRIの服用を続けるうちに、縫線核のセロトニンニューロン細胞体や樹状突起上にあるセロトニン1A自己受容体は脱感作され、negative feedback反応が消失する。この結果、セロトニンニューロンの活動性の亢進と神経終末でのセロトニン取り込み阻害が協調して奏効し、セロトニン神経伝達が強化され、十分な抗うつ作用が発現する。
【0005】
従って、セロトニン1A受容体アンタゴニストの併用によりセロトニン1A自己受容体を遮断してセロトニンのnegative feedback反応を止めるか、あるいはセロトニン1A受容体アゴニストの併用によりセロトニン1A自己受容体を積極的に刺激し脱感作までの期間を短縮することで、SSRIの作用発現までの期間の短縮や、抗うつ作用の増強が可能となる。実際、セロトニン1A受容体に対して高い親和性を有するピンドロール(pindolol)をSSRIと併用すると、うつ病患者におけるセロトニン再取り込み阻害薬の作用を増強すること、また作用発現までの期間を短縮することが報告されている(Arch, Gen. Psychiatry, (1994),51,248−251)。
【0006】
患者が薬剤を服用する際、その薬剤の数や種類はより少ないことが望ましい。従って、上記知見に基づき、セロトニン再取り込み阻害作用とセロトニン1A受容体への親和性を併せ持つ化合物は、他の薬剤と併用することなく単剤で、抗うつ作用が強く、作用発現までの期間が短縮された新しい抗うつ薬となり得ると考えられ、このような化合物の薬剤としての開発が望まれている。
【0007】
セロトニン再取り込み阻害作用とセロトニン1A受容体への親和性を併せ持つ化合物としては、これまでに、4位に置換ベンジル基を、1位に置換フェニルエチル基を有するベンジルピペリジン誘導体が報告されている(例えば、特許文献1参照)。具体的には、式(A):
【0008】
【化1】
[式中、R0は水素原子、ハロゲン原子、アルキル基、置換アルコキシ基などを表し、R0は独立して複数存在し、R3は水素原子などを表し、nは整数2などを表し、mは整数2などを表し、R5およびR6は各々独立して水素原子などを表し、Zは置換アリール基などを表す。]
で表される環状アミンなどを有効成分とするセロトニン再取り込み阻害剤が開示されている。されにこれらのセロトニン再取り込み阻害剤がセロトニン1A拮抗作用を有することも開示されている。
【0009】
一方、ピペリジンの4位に置換ベンジル基を有する化合物は、複数の文献で報告されている。例えば、脳血管障害治療薬として作用する環状アミン誘導体を開示する文献(特許文献2参照)や、NMDA受容体アンタゴニストとして作用する4−置換ピペリジンを開示する文献(特許文献3参照)が挙げられる。
【0010】
さらに、ピペリジンの1位に置換フェニルエチル基を有する化合物も、いくつかの文献で報告されている。フェニルエチル基上の置換基として環状ケトン構造を持つピペリジン環を有するインドール誘導体が5−HT1Aアンタゴニストとして報告されている(例えば、特許文献4参照)。これらのインドール誘導体は、ピペリジンの4位に置換ベンジル基を有するベンジルピペリジン化合物とは骨格が異なる。また、同インドール誘導体がセロトニン再取り込み阻害作用を併せ持つとは報告されていない。
【0011】
これらの特許文献の何れにおいても、ピペリジンの4位に置換ベンジル基を有し、さらにオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基(フェネチル基)をピペリジンの1位に有するベンジルピペリジン化合物については具体的な開示や示唆はない。
【0012】
また三環系抗うつ薬(Tricyclic antidepressants、TCA)やSSRIなどの抗うつ薬の多くは薬の代謝に関与する酵素であってヒトチトクロームP450分子種の一つであるCYP2D6への阻害作用が強いことが知られている。一方、うつ病や不安症状の治療においてTCAやSSRIと併用され得る精神系疾患治療剤の多くがCYP2D6によって代謝されることも知られている。従って、これらの薬剤の併用においては、一方の薬剤によるCYP2D6の阻害作用に基づき他方の薬剤の代謝が阻害されることによって、後者の薬剤の血中濃度が上昇し、その結果重篤な副作用が発現する可能性がある。従って、抗うつ薬のCYP2D6の阻害作用がより弱い程、CYP2D6によって代謝される併用の精神系疾患治療剤との薬物相互作用がより小さくなることから、このような抗うつ薬は安全性が高い薬剤となり得ることが期待され、その開発が望まれている。
【0013】
更にCYP2D6は遺伝的多型による酵素活性の個体間変動が大きいことが知られている。CYP2D6によって代謝される割合の高い薬剤は、生体内薬物濃度に大きな個人差を生じ、通常代謝者(Extensive Metabolizer、EM)の場合と比較して代謝欠損者(Poor Metabolizer、PM)の場合、血中薬物濃度が大きく上昇する危険性が高い。またこのような薬剤は、CYP2D6を阻害する薬剤またはCYP2D6により代謝を受ける薬剤との薬物相互作用がより強く現れる危険性もある。従って、薬剤の代謝におけるCYP2D6の寄与率がより低いほど、CYP2D6の遺伝多型による薬物動態的影響がより小さくなることから、このような薬剤は安全性が高くなり得ることが期待され、その開発も望まれている。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】米国特許第6787560号
【特許文献2】国際公開第88/02365号パンフレット
【特許文献3】国際公開第97/23216号パンフレット
【特許文献4】国際公開第2005/108389号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明が解決しようとする課題は、セロトニン1A受容体に対する親和性を併せ持つ新しいセロトニン再取り込み阻害剤を提供することにある。このようなセロトニン再取り込み阻害剤は、例えばうつ病や不安(不安障害)などの治療薬になることが期待されることから、治療効果に優れ、さらに安全性の高い薬剤を提供することが本発明の課題である。具体的な課題としては、ヒトセロトニン再取り込み阻害活性が向上し、セロトニン1A受容体に対する親和性を有し、ヒトチトクロームP450分子種の一つであるCYP2D6に対する阻害作用が弱く、またはヒトにおける薬物代謝においてCYP2D6の寄与が小さい薬剤を提供することが挙げられる。
【課題を解決するための手段】
【0016】
本発明者らは上記課題を解決するために鋭意検討した結果、化学構造上の特徴として、ベンゼン環部分の3位が2−メトキシエトキシ基または2−ヒドロキシエトキシ基で置換されているジ置換ベンジル基を有し、且つオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基をピペリジンの1位に有するベンジルピペリジン化合物またはその薬学上許容される塩が、高いヒトセロトニン再取り込み阻害作用とヒト5−HT1A受容体に対する結合親和性を併せ持つのみならず、該化合物または該塩は、CYP2D6阻害が弱く、また代謝におけるCYP2D6の寄与が小さいことも見出した。これらの知見を基に、本発明を完成させるに至った。
本発明は、以下の〔1〕〜〔8〕で表される、セロトニン再取り込み阻害剤として有用な、ベンジルピペリジン化合物またはその薬学上許容される塩に関するものである。すなわち、本発明は、
〔1〕 式(1):
【0017】
【化2】
[式中、R1は、水素原子またはメチル基を表し、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表し、Xは、メチレン基または酸素原子を表し、nは、1〜3の整数を表す。]
で表される化合物、またはその薬学上許容される塩;
〔2〕 Xがメチレン基を表し、nが1〜2の整数を表すか、または、Xが酸素原子を表し、nが2〜3の整数を表す、〔1〕に記載の化合物、またはその薬学上許容される塩;
〔3〕 R1がメチル基を表す、〔1〕または〔2〕に記載の化合物、またはその薬学上許容される塩;
〔4〕 R2がp位に結合した臭素原子を表す、〔1〕〜〔3〕のいずれかに記載の化合物、またはその薬学上許容される塩;
〔5〕 Xが酸素原子を表し、nが2の整数を表す、〔1〕〜〔4〕のいずれかに記載の化合物、またはその薬学上許容される塩;
〔6〕 R2がp位に結合した臭素原子を表し、Xが酸素原子を表し、nが2の整数を表す、〔1〕または〔2〕に記載の化合物、またはその薬学上許容される塩;
〔7〕 式(1)で表される化合物が、以下の化合物(01)〜(15)からなる群:
(01)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(02)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(03)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(04)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(05)6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(06)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(07)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(08)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(09)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(10)6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(11)6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(12)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(13)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(14)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、および
(15)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン
から選択される、〔1〕に記載の化合物、またはその薬学上許容される塩;
〔8〕 薬学上許容される塩が塩酸塩、臭化水素酸塩、フマル酸塩、ベンゼンスルホン酸塩またはコハク酸塩である、〔1〕〜〔7〕に記載の化合物の薬学上許容される塩;
に関する。
また、本発明は、以下の〔9〕〜〔12〕で表される、医薬組成物または治療薬もしくは予防薬に関するものである。すなわち、本発明は、
〔9〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有する医薬組成物;
〔10〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有するセロトニン再取り込み阻害剤;
〔11〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有する抗うつ薬または抗不安薬;
〔12〕 〔1〕〜〔7〕のいずれかに記載の化合物またはその薬学上許容される塩を有効成分として含有する抗うつ薬;
に関する。
【0018】
また、本発明は、以下の〔13〕で表される、〔1〕〜〔7〕に記した本発明のベンジルピペリジン化合物の中間体に関するものである。すなわち、本発明は、
〔13〕 式(11):
【0019】
【化3】
[式中、R1は、水素原子またはメチル基を表し、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表す。]
で表される化合物に関する。
【0020】
また、本発明は、以下の〔14〕で表される、〔1〕〜〔7〕に記した本発明のベンジルピペリジン化合物の中間体に関するものである。すなわち、本発明は、
〔14〕 式(12):
【0021】
【化4】
[式中、Xは、メチレン基または酸素原子を表し、nは、1〜3の整数を表し、LG1は、ヨウ素原子、臭素原子、塩素原子、置換スルホニルオキシ基を表す。]
で表される化合物に関する。
【発明の効果】
【0022】
本発明により、うつ病等の治療薬となり得るセロトニン再取り込み阻害剤として有用な、ベンジルピペリジン化合物またはその薬学上許容される塩を提供することが可能になった。詳しくは、本発明により、高いヒトセロトニン再取り込み阻害活性とヒト5−HT1A受容体に対して結合親和性を有し、CYP2D6阻害が弱く、また代謝におけるCYP2D6の寄与が小さいベンジルピペリジン化合物またはその薬学上許容される塩を提供することが可能になった。
【発明を実施するための最良の形態】
【0023】
以下に、本発明をさらに具体的に説明する。
本発明の式(1)で表されるベンジルピペリジン化合物は、化学構造上の特徴として、ベンゼン環部分の3位が2−メトキシエトキシ基または2−ヒドロキシエトキシ基で置換されているジ置換ベンジル基を有し、且つオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基をピペリジンの1位に有するものである。
本発明において、「置換スルホニルオキシ基」は、アルキル基または置換されていてもよいフェニル基で置換されたスルホニルオキシ基を意味する。ここにおいて、アルキル基としては、炭素数1〜6の直鎖状又は分枝鎖状のアルキル基が挙げられ、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、ヘキシル基、トリフルオロメチル基などが挙げられる。置換されていてもよいフェニル基の置換基としては、ハロゲン原子(ここで、ハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられる。)、アルキル基(ここで、アルキル基は、炭素数1〜6の直鎖状又は分枝鎖状アルキル基を表し、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、ヘキシル基などが挙げられる。)、トリフルオロメチル基、シアノ基、ニトロ基、またはアルコキシ基(ここで、アルコキシ基は、炭素数1〜6の直鎖状又は分枝鎖状のアルコキシ基を表し、具体的には、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、ペンチルオキシ基、ヘキシルオキシ基などが挙げられる。)好ましい置換スルホニルオキシ基としては、メタンスルホニルオキシ基、ベンゼンスルホニルオキシ基およびp−トルエンスルホニルオキシ基が挙げられ、さらに好ましい置換スルホニルオキシ基としては、ベンゼンスルホニルオキシ基およびp−トルエンスルホニルオキシ基が挙げられる。
【0024】
式(1)において、R1として好ましくはメチル基が挙げられる。(ここへ移動しました)
式(1)において、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表す。例えば、R2がp位に結合した塩素原子、p位に結合した臭素原子またはp位に結合したメチル基を表す場合、式(1)の化合物は、式(1−p):
【0025】
【化5】
[式中、R1、Xおよびnは前記と同義であり、R2pは塩素原子、臭素原子またはメチル基を表す。]
で表される化合物を表す。
一方、R2がm位に結合した塩素原子またはm位に結合した臭素原子を表す場合、式(1)の化合物は、式(1−m):
【0026】
【化6】
[式中、R1、Xおよびnは前記と同義であり、R2mは塩素原子または臭素原子を表す。]
で表される化合物を表す。式(1)において、R2として好ましくはp位に結合した臭素原子が挙げられる。すなわち、式(1−p−Br):
【0027】
【化7】
[式中、R1、Xおよびnは、前記と同義である。]
で表される化合物が好ましい。
【0028】
式(1)において、Xがメチレン基を表し、nが1の整数を表す、式(1)の化合物としては、式(1−C−1):
【0029】
【化8】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xがメチレン基を表し、nが2の整数を表す、式(1)の化合物としては、式(1−C−2):
【0030】
【化9】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xがメチレン基を表し、nが3の整数を表す、式(1)の化合物としては、式(1−C−3):
【0031】
【化10】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xが酸素原子を表し、nが1の整数を表す、式(1)の化合物としては、式(1−O−1):
【0032】
【化11】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xが酸素原子を表し、nが2の整数を表す、式(1)の化合物としては、式(1−O−2):
【0033】
【化12】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表し、
Xが酸素原子を表し、nが3の整数を表す、式(1)の化合物としては、式(1−O−3):
【0034】
【化13】
[式中、R1およびR2は、前記と同義である。]
で表される化合物を表す。
式(1)において、Xおよびnとしては、Xがメチレンを表しnが1〜2の整数を表すか、または、Xが酸素原子を表しnが2〜3の整数を表す場合が好ましい。すなわち、式(1−C−1)、式(1−C−2)、式(1−O−2)または式(1−O−3)で表される化合物が好ましい。Xおよびnとしてより好ましくは、Xが酸素原子を表しnが2の整数を表す場合があげられ、すなわち、式(1−O−2)で表される化合物がより好ましい。
より具体的には、以下の化合物(01)〜(15):
(01)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(02)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(03)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(04)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(05)6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(06)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(07)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(08)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(09)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(10)6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(11)6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(12)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(13)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(14)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、および
(15)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン
が好ましい。
【0035】
本発明のベンジルピペリジン化合物は、公知化合物から、以下に示す製造方法1〜5に示す方法、下記の製造方法に類似の方法、または当業者に周知の合成方法を適宜組み合わせて製造することができる。原料化合物(11)、(12)、(13)、(15)、(18)および(19)において、いくつかのものは新規であるが、後記の実施例に記載の方法、または実施例に類似の方法、または当業者に周知の合成方法を適宜組み合わせて製造することもできる。
また、本明細書において、記載の簡略化のために次の略号を使用する場合がある。
Boc:tert-ブトキシカルボニル基
Piv:tert-ブチルカルボニル基
Me:メチル基
Et:エチル基
Ph:フェニル基
Bn:ベンジル基
Ms:メタンスルホニル基
Bs:ベンゼンスルホニル基
Ts:p−トルエンスルホニル基
p:パラ(例えば、「p−Br」はパラ位に結合した臭素原子を意味する。)
m:メタ(例えば、「m−Br」はメタ位に結合した臭素原子を意味する。)
DMSO:ジメチルスルホキシド
【0036】
製造方法1:化合物(1)の製造方法
式(1)で表される化合物またはその塩は、例えば下記の方法によって製造できる。
【0037】
【化14】
[式中、R1、R2、X、nおよびLG1は、前記と同義である。]
【0038】
目的化合物(1)またはその塩は、化合物(11)またはその塩を化合物(12)と反応させることにより得ることができる。反応は、必要に応じ塩基の存在下、また、必要に応じ相間移動触媒の存在下、適当な不活性溶媒中で約−20℃〜用いた溶媒の沸点までの範囲の温度で、10分間〜48時間反応させることにより行うことができる。
塩基としては、例えばトリエチルアミン、ジイソプロピルエチルアミン、ピリジン等の有機塩基、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、炭酸水素カリウム、炭酸水素ナトリウム、りん酸二水素カリウム、りん酸水素二カリウム、りん酸カリウム、りん酸二水素ナトリウム、りん酸水素二ナトリウム、りん酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水素化ナトリウム等の無機塩基、ナトリウムメトキシド、カリウムtert-ブトキシド等の金属アルコキシド等が挙げられる。好ましくは炭酸カリウムおよびりん酸水素二カリウムが挙げられる。
相間移動触媒としては、例えば硫酸水素テトラブチルアンモニウムなどが挙げられる。
不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、アセトン、メチルエチルケトン、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。より好ましい溶媒としてはアセトニトリル、トルエン、ジメチルホルムアミドおよびN−メチル−2−ピロリジノンもしくはこれらの混合溶媒が挙げられる。
脱離基となるLG1としては臭素基や置換スルホニルオキシ基が好ましく、ベンゼンスルホニルオキシ基やp−トルエンスルホニルオキシ基がさらに好ましい。
【0039】
製造方法2:化合物(11)の製造方法
製造方法1の出発原料として用いられる化合物(11)またはその塩は、例えば米国特許第6787560号などの文献の方法を参考にして、下記の方法によって製造できる。
【0040】
【化15】
[式中、R1およびR2は、前記と同義である。PG1は窒素原子の保護基を、LG2は脱離基を表す。窒素原子の保護基PG1としては、例えばt−ブチルオキシカルボニル基、9−フルオレニルメチルオキシカルボニル基などのアルキルオキシカルボニル基が挙げられる。脱離基LG2としては、例えば塩素原子、臭素原子、ヨウ素原子などのハロゲン原子や、p−トルエンスルホニルオキシ基、メタンスルホニルオキシ基などの置換スルホニルオキシ基が挙げられる。]
【0041】
化合物(13)をホスホン酸エステル(14a)またはホスホニウム塩(14b)に変換する。この変換はホスホン酸エステル(14a)の場合、亜リン酸トリエチルを無溶媒もしくは不活性溶媒中、氷冷から用いた溶媒もしくは亜リン酸トリエチルの沸点までの間の温度で、1時間〜3日間反応させることにより行うことができる。ホスホニウム塩(14b)の場合、トリフェニルホスフィンを不活性溶媒中、氷冷から用いた溶媒の沸点までの間の温度で、1時間〜3日間反応させることにより行うことができる。
このホスホン酸エステル(14a)またはホスホニウム塩(14b)とケトン(15)を塩基の存在下、適当な不活性溶媒中で約−20℃から用いた溶媒の沸点までの温度で、10分間〜48時間反応させることにより、化合物(16)に変換することができる。
塩基としては、例えばトリエチルアミン、ピリジン等の有機塩基、炭酸カリウム、炭酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水素化ナトリウム等の無機塩基、ナトリウムメトキシド、カリウムtert-ブトキシド等の金属アルコキシドが挙げられる。
不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。
化合物(16)を接触還元することにより化合物(17)に変換することができる。R2がp位に結合した臭素原子またはm位に結合した臭素原子を表す場合には、この還元反応の触媒としてロジウム炭素等のロジウム系の触媒や白金炭素や酸化白金等の白金系の触媒、ルテニウム炭素等のルテニウム系の触媒や塩化パラジウムなどを用い、常圧もしくは加圧水素雰囲気下、適当な不活性溶媒中で0℃〜50℃にて反応させることにより行うことができる。適当な不活性溶媒としては酢酸エチルや、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。より好ましい触媒としてはロジウム炭素や白金炭素が挙げられる。またこの場合、より好ましい溶媒としては酢酸エチルが挙げられる。
化合物(17)を常法により脱保護することにより目的とする化合物(11)を得ることができる。保護基がt−ブチルオキシカルボニル基の場合は適当な不活性溶媒中で−20℃から用いた溶媒の沸点までの間の温度において塩酸や硫酸などの無機酸やトリフルオロ酢酸などの有機酸で処理することにより脱保護が行える。不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。保護基が9−フルオレニルメチルオキシカルボニル基の場合は適当な不活性溶媒中で−20℃から用いた溶媒の沸点までの間の温度において、ピロリジン、ピペリジン、モルホリン、トリエチルアミンやジイソプロピルエチルアミンなどの有機塩基で処理することにより脱保護が行える。不活性溶媒としては、例えばクロロホルム、ジクロロメタン等のハロゲン化炭化水素、ベンゼン、トルエン等の芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、2−プロパノール等の低級アルコール、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。
【0042】
製造方法3:化合物(12)の製造方法
製造方法1の出発原料として用いられる化合物(12)は、例えば下記の方法によって製造できる。
【0043】
【化16】
[式中、Xおよびnは、前記と同義である。R3は水素原子またはアルキル基を表す。該アルキル基としては、直鎖状または分枝上の炭素原子数が1〜6のアルキル基が挙げられ、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基、1−メチルブチル基、ヘキシル基などを挙げることができる。]
【0044】
化合物(18)を適当な還元剤(例えば水素化リチウムアルミニウム、水素化ホウ素リチウム、水素化ホウ素ナトリウム、ジボランなど)を用いて、適当な不活性溶媒(例えばジエチルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン等のエーテル系溶媒など)中で、−20℃から用いた溶媒の沸点までの間の温度で、10分間〜48時間反応させることにより、化合物(19)を得ることができる。
化合物(19)を二酸化マンガンなどの酸化剤を用いて、適当な不活性溶媒中で酸化することにより化合物(20)を得ることができる。適当な不活性化溶媒としては、クロロホルムやジクロロメタンのようなハロゲン化溶媒、アセトニトリル、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリドン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。
化合物(20)の水酸基を常法により塩素原子、臭素原子もしくはヨウ素原子のハロゲン原子や、例えばp−トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基もしくはメタンスルホニルオキシ基などの置換スルホニルオキシ基に変換することにより化合物(12)を得ることができる。具体的には化合物(20)を例えばメタンスルホニルクロライド、ベンゼンスルホニルクロライドまたはp−トルエンスルホニルクロライドなどと不活性溶媒中で塩基の存在下、−20℃から用いた溶媒の沸点までの間の温度で、10分間〜48時間反応させることにより、化合物(12)を得ることができる。適当な不活性化溶媒としてはクロロホルムやジクロロメタンのようなハロゲン化溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリドン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。適当な塩基としてはトリエチルアミン、ピリジン等の有機塩基、炭酸カリウム、水酸化ナトリウムなどの無機塩基が挙げられる。またLG1が塩素原子、臭素原子などのハロゲンの場合は、LG1がp−トルエンスルホニルオキシ基、メタンスルホニルオキシ基などの置換スルホニルオキシ基である化合物(12)を、不活性溶媒中で例えば臭化リチウムなどと−20℃から用いた溶媒の沸点までの間の温度で10分間〜48時間反応させることにより得ることができる。適当な不活性化溶媒としてはクロロホルムやジクロロメタンのようなハロゲン化溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン等のエーテル系溶媒、アセトニトリル、ジメチルホルムアミド、N−メチル−2−ピロリドン、ジメチルスルホキシド等の非プロトン性極性溶媒もしくはこれらの混合溶媒が挙げられる。また別法として化合物(20)を例えば適切な不活性溶媒中でトリフェニルホスフィン存在下、四塩化炭素や四臭化炭素と反応させることにより化合物(12)を得ることができる。
【0045】
製造方法4:化合物(12)の製造方法
原料化合物(12)は、例えば下記の方法によっても製造できる。
【0046】
【化17】
[上記式中、X、nおよびLG1は前記と同義である。]
【0047】
化合物(19)の一級水酸基を常法によりp−トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基、メタンスルホニルオキシ基などの置換スルホニルオキシ基に変換することにより化合物(21)を得ることができる。化合物(21)を適当な不活性溶媒中で、例えば二酸化マンガン酸化やジメチルスルホキシド(DMSO)酸化などの常法により、水酸基を酸化することにより、化合物(12)を得ることができる。
【0048】
製造方法5:化合物(20)の製造方法
製造方法3における中間体化合物(20)は、例えば下記の方法によって製造できる。
【0049】
【化18】
[式中、R3、Xおよびnは前記と同義である。PG2は、メトキシ基、メチルチオ基などを表すか、または2つのPG2が環を形成して、1,3−ジオキソラン基、1,3−ジオキサン基などの環状アセタール基を表してもよい。]
【0050】
化合物(18)のケトンを常法によりジアルキルアセタールやジアルキルチオアセタールに変換して化合物(22)とする。これを適当な不活性溶媒中、適当な還元剤(例えば水素化リチウムアルミニウム、水素化ホウ素リチウム、水素化ホウ素ナトリウムあるいはジボランなど)を用いて還元して化合物(23)とする。化合物(23)を適当な方法により脱保護することにより化合物(20)を得ることができる。
化合物(18)は例えばJournal of Medicinal Chemistry(1994), 37(21), 3482.、Journal of Medicinal Chemistry(1979), 22(12), 1464.、フランス特許第2672601号、日本特許 特開昭61−236774記載の方法などにより合成することができる。
【0051】
前記の製造方法において使用する原料や試薬などは、特に断らない限り、市販の化合物であるか、または公知の化合物から公知の方法を用いて製造することができる。また、前記式(1)の化合物において、官能基を適宜変換することによって、式(1)の別の化合物としてもよい。官能基の変換は、通常行われる一般的方法[例えば、コンプリヘンシブ・オーガニック・トランスフォーメーションズ(Comprehensive Organic Transformations)、アール.シー.ラロック(R.C.Larock)著(1989年)等参照]によって行うことができる。
【0052】
前記製造方法において、反応点以外の何れかの官能基が説明した反応条件下で変化するかまたは説明した方法を実施するのに不適切な場合は、当該官能基を予め適当な保護基で保護した上で、反応を実施し、その後、脱保護することにより、目的化合物を得ることができる。保護基としては、例えばプロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、ティー・ダブリュー・グリーン(T.W.Greene)著、ジョン・ワイリー・アンド・サンズ・インコーポレイテッド(John Wiley & Sons Inc.)(1981年)等に記載されているような通常の保護基を用いることができる。具体的には、アミンの保護基としてはエトキシカルボニル、tert-ブトキシカルボニル、ベンジルオキシカルボニル、アセチル、ベンゾイルまたはベンジル等を、また水酸基の保護基としてはトリアルキルシリル、アセチル、ベンゾイルまたはベンジル等を挙げることができる。ケトンの保護基としてはジメチルアセタール、1,3−ジオキサン、1,3−ジオキソラン、S,S’−ジメチルジチオアセタール、1,3−ジチアン、オキシム等をあげることができる。
保護基の導入および脱保護は、有機合成化学で常用される方法(例えば、上記のプロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis) 参照)またはそれらに準じた方法により行うことができる。
前記製造方法における中間体および目的化合物は、有機合成化学で常用される精製方法、例えば中和、濾過、抽出、洗浄、乾燥、濃縮、再結晶、各種クロマトグラフィー等により、単離精製することができる。また、中間体においては、特に精製することなく次の反応に供することも可能である。
【0053】
式(1)で表される本発明の化合物の中には、互変異性体が存在し得るものがある。互変異性の例としては、式(24)などが挙げられる。
【0054】
【化19】
本発明は、当該互変異性体を含め、全ての可能な異性体およびそれらの混合物を包含する。
【0055】
式(1)で表される化合物の薬学上許容される塩は、慣用の無毒性塩であり、有機酸塩(例えば酢酸塩、プロピオン酸塩、トリフルオロ酢酸塩、マレイン酸塩、フマル酸塩、クエン酸塩、コハク酸塩、酒石酸塩、メタンスルホン酸塩、ベンゼンスルホン酸塩、蟻酸塩、トルエンスルホン酸塩)もしくは無機酸塩(例えば塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、硝酸塩、リン酸塩)のような酸付加塩、アミノ酸(例えばアルギニン酸、アスパラギン酸、グルタミン酸)との塩、アルカリ金属塩(例えばナトリウム塩、カリウム塩)もしくはアルカリ土類金属塩(例えばカルシウム塩、マグネシウム塩)などの金属塩、アンモニウム塩、または有機塩基塩(例えばトリメチルアミン塩、トリエチルアミン塩、ピリジン塩、ピコリン塩、ジシクロヘキシルアミン塩、N,N’−ジベンジルエチレンジアミン塩)が挙げられる。
式(1)で表される化合物の薬学上許容される塩を取得するには、化合物(1)が薬学上許容される塩の形で得られる場合そのまま精製すればよく、一方、遊離の形で得られる場合、適当な有機溶媒に溶解もしくは懸濁させ、酸または塩基を加えて通常の方法により塩を形成させればよい。例えば水、メタノール、エタノール、アセトン等の溶媒中で、薬学上許容される酸又はアルカリと混合することで、塩にすることができる。
また、式(1)で表される化合物およびその薬学上許容される塩は、水との水和物またはエタノールなどの各種溶媒との溶媒和物として存在することもあるが、これら水和物や溶媒和物も本発明に包含される。
結晶として得られる式(1)で表される化合物およびその薬学上許容される塩には、結晶多形が存在する場合があり、その結晶多形も本発明に包含される。
【0056】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩は、ヒトセロトニン再取り込み阻害作用を有する。それゆえ、当該化合物および塩は、セロトニン神経系が介在する疾患の治療薬として有用である。セロトニン神経系が介在する疾患としては、例えば、うつ病や不安などが挙げられる。うつ病は、精神疾患の分類においては気分障害に含まれる。この気分障害の中には、主にはうつ病性障害と双極性障害がある。一般的なうつ病として、より詳細には、(i)大うつ病性障害、気分変調性障害もしくは特定不能なうつ病性障害を含むうつ病性障害、(ii)うつ病、または(iii)季節的情動障害などが挙げられる。これらの治療薬または再発予防薬として当該化合物および塩は有用である。さらに(iv)双極性障害における大うつ病エピソードの治療薬または再発予防薬としても当該化合物および塩は有用である。一方、不安(不安障害)の中には、主に不安障害と恐怖症がある。当該化合物および塩が治療薬または再発予防薬として有用な不安(不安障害)としては、(v)パニック障害、強迫性障害、外傷後ストレス障害、急性ストレス障害、全般性不安障害もしくは一般身体疾患による不安障害、(vi)物質誘発性不安障害を含む不安障害、(vii)広場恐怖症、(viii)社会的恐怖症、(ix)回避的人格異常、または(x)心身症などが挙げられる。また、他の疾患(統合失調症、認知症など)に伴ううつ症状または不安症状に対しても当該化合物および塩は有用である。さらに、当該化合物および塩は、痴呆、健忘症および加齢に関係した記憶障害を含む記憶障害;神経性食欲不良および神経性飢餓を含む摂食行動の障害;肥満症;睡眠障害;統合失調症;アルコール、たばこ、ニコチン、麻薬、覚せい剤、向精神薬等の薬物依存症;群発性頭痛;片頭痛;痛み;アルツハイマー病;慢性発作片頭痛;血管障害に関係した頭痛;パーキンソン病の痴呆、抑うつ、不安、神経弛緩薬誘導パーキンソン症候群および晩発性ジスキネジーを含むパーキンソン病;過プロラクチン血症などの内分泌異常;血管痙攣(特に、脳血管系の);高血圧症;運動性および分泌の変化が関与している胃腸管の障害;早発射精を含む性的機能不全などの治療または予防にも有用である。
【0057】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩の用量は患者の年齢および状態に応じて増減するが、化合物(1)の平均一回量約0.1mg、1mg、10mg、50mg、100mg、250mg、500mgおよび1000mgが、例えば前記のうつ病や不安などの疾患に対して有効である。一般には、ヒトに投与する場合、1日当り0.1mg/個体ないし約1,000mg/個体、好ましくは1日当り1mg/個体ないし約100mg/個体の量を投与することができる。1日の投与回数は、1回又は1日に数回、例えば各回1、2又は3用量を与える。
【0058】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩は、治療に使用する場合に、医薬組成物として、経口的または非経口的(例えば、静脈内、皮下、筋肉内、髄腔内、局所的、経直腸的、経皮的、経鼻的または経肺的)に投与することができる。経口投与のための投与形態としては、例えば、錠剤、カプセル剤、丸剤、顆粒剤、細粒剤、散剤、液剤、シロップ剤、懸濁剤などの剤形が挙げられ、非経口投与のための投与形態としては、例えば、注射用水性剤、注射用油性剤、坐剤、経鼻剤、経皮吸収剤[ローション剤、乳液剤、軟膏剤、クリーム剤、ゼリー剤、ゲル剤、貼付剤(テープ剤、経皮パッチ製剤、湿布剤等)、外用散剤等]等などの形態の製剤が挙げられる。これらの製剤は、従来公知の技術を用いて調製され、製剤分野において通常使用される無毒性かつ不活性な担体もしくは賦形剤を含有することができる。
【0059】
製剤用担体としては、製剤分野において常用され、かつ式(1)で表される化合物又はその薬学上許容される塩と反応しない物質が用いられる。すなわち、式(1)で表される化合物又はその薬学上許容される塩を含有する医薬組成物は、賦形剤、結合剤、滑沢剤、安定剤、崩壊剤、緩衝剤、溶解補助剤、等張化剤、溶解補助剤、pH調節剤、界面活性剤、乳化剤、懸濁化剤、分散剤、沈殿防止剤、増粘剤、粘度調節剤、ゲル化剤、無痛化剤、保存剤、可塑剤、経皮吸収促進剤、老化防止剤、保湿剤、防腐剤、香料等の製剤用担体を含有することができ、2種以上の製剤用担体添加物を適宜選択して用いることもできる。
【0060】
製剤用担体添加物として、具体的には、例えば乳糖、イノシトール、ブドウ糖、ショ糖、果糖、マンニトール(マンニット)、デキストラン、ソルビトール(ソルビット)、シクロデキストリン、デンプン(馬鈴薯デンプン、コーンスターチ、アミロペクチン等)、部分アルファー化デンプン、白糖、メタケイ酸アルミン酸マグネシウム、合成ケイ酸アルミニウム、アルギン酸ソーダ、結晶セルロース、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルデンプン、カルボキシメチルセルロースカルシウム、イオン交換樹脂、メチルセルロース、ゼラチン、アラビアゴム、プルラン、ヒドロキシプロピルセルロース、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロース、ヒドロキシエチルセルロース、ポリビニルピロリドン、ポリビニルアルコール、ゼラチン、アルギン酸、アルギン酸ナトリウム、軽質無水ケイ酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸アルミニウム、セトステアリルアルコール、ワックス、パラフィン、タルク、トラガント、ベントナイト、ビーガム、カルボキシビニルポリマー、酸化チタン、脂肪酸エステル、ソルビタン脂肪酸エステル、ラウリル硫酸ナトリウム、グリセリン、脂肪酸グリセリンエステル、精製ラノリン、グリセロゼラチン、ポリソルベート、マクロゴール、スクワラン、シリコーンオイル、植物油(ごま油、オリーブ油、大豆油、綿実油、ヒマシ油など)、液体パラフィン(流動パラフィン)、軟パラフィン、白色ワセリン、黄色ワセリン、パラフィン、羊毛脂、ロウ(蜜蝋、カルナウバロウ、サラシミツロウなど)、水、プロピレングリコール、ポリエチレングリコール、グリセロール、ラウリルアルコール、ミリスチルアルコール、オレイルアルコール、セチルアルコール、エタノール、塩化ナトリウム、水酸化ナトリウム、塩酸、クエン酸、ラウリル酸、ミリスチン酸、ステアリン酸、オレイン酸、ベンジルアルコール、グルタミン酸、グリシン、パラオキシ安息香酸メチル、パラオキシ安息香酸プロピル、p−ヒドロキシ安息香酸エステル類、コレステロールエステル、エチレングリコールモノアルキルエステル、プロピレングリコールモノアルキルエステル、モノステアリン酸グリセリン、ソルビタン脂肪酸エステル、ミリスチン酸イソプロピル、パルミチン酸イソプロピル、カルボキシポリメチレン、サッカリン、ストロベリーフレーバー、ペパーミントフレーバー、カカオ脂、ポリイソブチレン、酢酸ビニル共重合体、アクリル系共重合体、クエン酸トリエチル、クエン酸アセチルトリエチル、フタル酸ジエチル、セバシン酸ジエチル、セバシン酸ジブチル、アセチル化モノグリセリド、、ジエチレングリコール、ドデシルピロリドン、尿素、ラウリル酸エチル、エイゾン、カオリン、ベントナイト、酸化亜鉛、アガロース、カラギーナン、アカシアゴム、キサンタンガム、ラウリン酸カリウム、パルミチン酸カリウム、ミリスチン酸カリウム、セチル硫酸ナトリウム、ヒマシ油硫酸化物(ロート油)、Span(ステアリン酸ソルビタン、モノオレイン酸ソルビタン、セスキオレイン酸ソルビタン、トリオレイン酸ソルビタン等)、Tween(ポリソルベート20、ポリソルベート40、ポリソルベート60、ポリソルベート65、ポリソルベート80、ポリソルベート85、ポリオキシエチレンソルビタン脂肪酸エステル等)、ポリオキシエチレン硬化ヒマシ油(いわゆるHCO)、ポリオキシエチレンラウリルエーテル、ポリオキシエチレンセチルエーテル、ポリオキシエチレンオレイルエーテル、モノラウリン酸ポリエチレングリコール、モノステアリン酸ポリエチレングリコール、ポロキサマー(いわゆるプルロニック)、レシチン(ホスファチジルコリン、ホスファチジルセリンなどレシチンから単離された精製リン脂質をも含む)、レシチンの水素添加物等が挙げられる。
【0061】
本発明のベンジルピペリジン化合物およびその薬学上許容される塩を、上記の如き医薬用途に使用する場合、通常、製剤用担体と混合して調製した製剤の形で投与され、製剤は通常の方法に従って調製される。例えば、本発明のベンジルピペリジン化合物およびその薬学上許容される塩を有効成分として0.051〜99重量%、好ましくは0.05〜80重量%、更に好ましくは0.1〜70%重量%、更に好ましくは0.1〜50重量%含有する医薬組成物とすることができる。これらの製剤はまた、治療上価値ある他の成分を含有していてもよい。
【0062】
本発明のベンジルピペリジル化合物およびその薬学上許容される塩は、その作用の増強を目的として、抗うつ薬、抗不安薬、統合失調症治療薬、ドパミン受容体作動薬、パーキンソン病治療薬、抗癲癇薬、抗痙攣薬、鎮痛薬、ホルモン製剤、偏頭痛治療薬、アドレナリンβ受容体拮抗薬、認知症治療薬、気分障害治療薬などの薬剤(併用薬剤)と組み合わせて用いることができる。また、その副作用抑制を目的として、制吐剤、睡眠導入剤、抗痙攣薬などの薬剤(併用薬剤)と組み合わせて用いることができる。本発明化合物及び併用薬剤の投与時期は限定されず、これらを投与対象に対し、同時に投与してもよいし、時間差をおいて投与してもよい。また、本発明化合物と併用薬剤の合剤としても良い。併用薬剤の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、本発明化合物と併用薬剤との配合比は、投与対象、投与ルート、対象疾患、症状、組み合わせなどにより適宜選択することができる。例えば投与対象がヒトである場合、本発明化合物1重量部に対し、併用薬剤を0.01〜1000重量部用いればよい。
【0063】
以下に本発明を、参考例、実施例及び試験例によりさらに詳細に説明するが、本発明の技術的範囲はこれら実施例に限定されるものではない。尚、以下の参考例及び実施例において示された化合物名は、必ずしもIUPAC命名法に従うものではない。
【0064】
化合物の同定には水素核磁気共鳴吸収スペクトル(1H−NMRスペクトル)等を用いた。いくつかの化合物については1H−NMRスペクトルスペクトルデータや融点を示した。また液体クロマトグラフィー分析により純度等の確認も行った。カラムにはSUMIPAX ODS C−212(5μm,6mmφ×15cm)を用い、測定波長を220 nm、移動層流速を1.0 ml/minに設定し、分析した。移動層としては0.05%トリフルオロ酢酸−アセトニトリル(A液)と0.05%トリフルオロ酢酸−水(B液)の混合溶媒を用いた。条件1ではA液とB液の混合比(A液:B液)を、測定開始時(0分)は25:75とし、測定開始40分後で50:50となるようにA液の比率を1分間に0.625%ずつ上昇させた。条件2ではA液とB液の混合比を常に40:60とした。化合物が検出された保持時間と、A液とB液の混合比(条件1または2)もいくつかの化合物について示した。
【0065】
参考例1
4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン 塩酸塩(4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidine hydrochloride,化合物(RE1))
下記の製造方法1または2に従い、合成した。
製造方法1
【0066】
【化20】
【0067】
化合物(1−1−1):1,3−ジブロモ−5−(2−メトキシエトキシ)ベンゼン(1,3-Dibromo-5-(2-methoxyethoxy)benzene)
3,5−ジブロモフェノール(37.7g,150mmol)、炭酸カリウム(41.4g,300mmol)のジメチルホルムアミド(150mmol)溶液に室温で2−ブロモエチルメチルエーテル(31.2g,224mmol)を加え、反応混合物を80℃で9時間攪拌した。室温に冷却後、水(300mL)、トルエン(150mL)と酢酸エチル(150mL)を加えて分液し、水層をトルエン(50mL)と酢酸エチル(50mL)の混合溶液で再抽出した。全有機層を水(50×2mL)、飽和食塩水(50mL)で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=20:1→15:1)で精製することで、表記化合物(1−1−1)(44.3g、95%)を褐色油状物として得た。
【0068】
化合物(1−1−2):3−ブロモ−5−(2−メトキシエトキシ)ベンズアルデヒド(3-Bromo-5-(2-methoxyethoxy)benzaldehyde)
n−ブチルリチウム(1.6M n−ヘキサン溶液,126 mL,198 mmol)のトルエン(120mL)溶液を氷浴で冷却して内温3〜5℃に保ちながらn−ブチルマグネシウムクロライド(0.89M テトラヒドロフラン溶液,116mL,100mmol)を25分間で滴下し、反応混合物をそのまま30分間攪拌した。化合物(1−1−1)(46.1g,149mmol)のトルエン(420mL)溶液を内温0〜3℃に保ちながら1時間で滴下し、反応混合物を2時間攪拌後、N,N−ジメチルホルムアミド(28.7mL,373mmol)を内温4〜5℃で40分間かけて滴下し、反応混合物を2時間攪拌した。2N−塩酸水(300mL)を加えて室温に昇温して分液した。水層をトルエン(100mL)で抽出し、全有機層を水、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=10:1→7:1)で精製することで、表記化合物(1−1−2)(28.8g、74%)を得た。
【0069】
化合物(1−1−3):[3−ブロモ−5−(2−メトキシエトキシ)フェニル]メタノール([3-Bromo-5-(2-methoxyethoxy)phenyl]methanol)
化合物(1−1−2)(28.9g,112mmol)のメタノール(112mL)溶液に水冷しながら室温で水素化ホウ素ナトリウム(4.22g,112mmol)を少しずつ加え、反応混合物を室温で3時間攪拌した。水(200 mL)を加え、メタノールを減圧留去し、酢酸エチル(200 mL+50 mL)で抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(1−1−3)(28.7g, 98%)を淡黄色油状物として得た。
【0070】
化合物(1−1−4):3−ブロモ−5−(2−メトキシエトキシ)ベンジル メタンスルホネート(3-Bromo-5-(2-methoxyethoxy)benzyl methanesulfonate)
化合物(1−1−3)(24.3g,93.1mmol)、トリメチルアミン塩酸塩(890mg,9.3mmol)とトリエチルアミン(25.9mL, 186mmol)のトルエン(186mL)溶液に、塩−氷浴で内温を10℃以下に保ちながらメタンスルホニルクロライド(16.0g,140mmol)のトルエン(15mL)溶液を50分間で滴下し、反応混合物を内温5℃以下で1時間攪拌した。内温5℃以下で2−ジエチルアミノエチルアミン(5.95g, 51.2mmol)を加えて20分間攪拌し、引き続き反応液に5%硫酸水素カリウム水溶液(250mL)と水(100mL)を加え、室温に昇温させて分液し、有機層を水洗後、トルエンを減圧留去することで、表記化合物(1−1−4)(33.1g)を得た。
【0071】
化合物(1−1−5):1−ブロモ−3−(ブロモメチル)−5−(2−メトキシエトキシ)ベンゼン(1-Bromo-3-(bromomethyl)-5-(2-methoxyethoxy)benzene)
化合物(1−1−4)(33.1g,97.6mmol相当)の無水テトラヒドロフラン(195mL)溶液に室温で臭化リチウム・1水和物(30.7g, 293mmol)を加え、反応混合物を1時間加熱還流した。室温に冷却後、トルエン(180mL)と水を加えて分液し、水層をトルエン(50mL)で再抽出した。全有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、トルエンを減圧留去することで、表記化合物(1−1−5)(29.3 g, 93%)を得た。
【0072】
化合物(1−1−6):[3−ブロモ−5−(2−メトキシエトキシ)ベンジル](トリフェニル)ホスホニウム ブロミド([3-Bromo-5-(2-methoxyethoxy)benzyl](triphenyl)phosphonium bromide)
化合物(1−1−5)(31.8g,98 mmol)とトリフェニルホスフィン(28.3g,108mmol)のトルエン(98mL)溶液を3時間加熱還流した。反応混合物をゆっくり室温まで冷却して生じた沈殿をろ取し、ろ上物をトルエン(40mL)で洗浄し、減圧乾燥することで、表記化合物(1−1−6)(50.1g,87%)を得た。
【0073】
化合物(1−1−7):tert−ブチル 4−{[3−ブロモ−5−(2−メトキシエトキシ)フェニル]メチリデン}ピペリジン−1−カルボキシレート(tert-Butyl 4-{[3-bromo-5-(2-methoxyethoxy)phenyl]methylidene}piperidine-1-carboxylate)
化合物(1−1−6)(50.1g、85mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(17.4g、87mmol)と炭酸カリウム(23.6g、171mmol)の2−プロパノール(250mL)懸濁溶液を40℃で4時間、50℃で2時間、60℃で3.5時間、80℃で7.5時間加熱還流した。反応混合物を室温まで冷却後、塩をろ去し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=10:1→7:1)で精製することで、表記化合物(1−1−7)(34.6g、95%)を無色油状物として得た。
【0074】
化合物(1−1−8):tert−ブチル 4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[3-bromo-5-(2-methoxyethoxy)benzyl]piperidine-1-carboxylate)
化合物(1−1−7)(34.6g、81mmol)を酢酸エチル(80mL)中、5%ロジウム炭素(9.74g)を用いて室温で26時間常圧水素添加反応を行った。触媒をろ去し、ろ液を減圧濃縮することで、表記化合物(1−1−8)(34.2g、98%)を得た。
【0075】
化合物(RE1):
化合物(1−1−8)(34.2g、80mmol)のメタノール(34mL)溶液に室温で10%塩酸メタノール溶液(103mL)を加え、反応混合物を室温で1日間攪拌した。溶媒を減圧留去後、得られた濃縮残渣にジエチルエーテルを加え、生じた沈殿をろ取し、ろ上物をジエチルエーテルで洗浄後、減圧乾燥することで白色固体(27.5g)を得た。これにアセトニトリル(132 mL)を加えて50℃に加温し、すべて溶解したことを確認後、ゆっくり冷却し、40℃で30分攪拌後、1時間で0℃まで冷却して、0℃で1時間攪拌した。析出物をろ取し、冷やしたアセトニトリル(20mL)で洗浄し、減圧乾燥することで、目的化合物(RE1)(22.6g,85%)を白色粉末として得た。
保持時間(条件1):15.56分
融点:107−108℃
1H-NMR (400 MHz, d6-DMSO) δ: 1.25-1.40 (2H, m), 1.67 (2H, d like, J = 14 Hz), 1.72-1.85 (1H, m), 2.49 (2H, d, J = 6.8 Hz), 2.77 (2H, dt, J = 2.4, 12 Hz), 3.20 (2H, br d,,J= 12.6 Hz), 3.34 (3H, s), 3.63 (2H, t like, J = 4.5 Hz), 4.09 (2H, t like, J = 4.5 Hz), 6.80 (1H, t, J = 1.7 Hz), 6.96-6.99 (2H, m).
【0076】
製造方法2
【0077】
【化21】
【0078】
化合物(1−2−1):ジエチル (3−ブロモ−5−メトキシベンジル)ホスホネート(Diethyl (3-Bromo-5-methoxybenzyl)phosphonate)
文献の方法(J.Med.Chem.2001,44,1866)により合成した1−ブロモ−3−メトキシ−5−メチルベンゼン(17.0g、91mmol)のモノクロロベンゼン(500mL)溶液に80℃で5,5−ジメチル−1,3−ジブロモヒダントイン(13.1g、46mmol)とアゾビスイソブチロニトリル(1.50g、9.1mmol)を同時に加え、反応混合物を30分間80℃で攪拌した。反応溶液を室温まで冷却後、10%チオ硫酸ナトリウム水溶液(100mL)に注ぎ30分間攪拌した。分液し、水層をトルエン(100mL)で抽出し、全有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、1−ブロモ−3−(ブロモメチル)−5−メトキシベンゼン(1-Bromo-3-(bromomethyl)-5-methoxybenzene)を得た。これを精製することなく、亜リン酸トリエチル(14.3mL、97mmol)とトルエン(50mL)に溶解させ、反応混合物を7時間加熱還流した。室温に冷却後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=1:2)で精製することで、表記化合物(1−2−1)(20.8g、64%)を得た。
【0079】
化合物(1−2−2):tert−ブチル 4−(3−ブロモ−5−メトキシベンジリデン)ピペリジン−1−カルボキシレート(tert-Butyl 4-(3-bromo-5-methoxybenzylidene)piperidine-1-carboxylate)
水素化ナトリウム(60%懸濁溶液、2.71g、68mmol)の無水テトラヒドロフラン(100mL)懸濁溶液に50℃で化合物(1−2−1) (19.0g、56mmol)の無水テトラヒドロフラン(100mL)溶液を15分間で滴下した。反応混合物を50℃で25分攪拌後、1−tert−ブトキシカルボニル−4−ピペリドン(14.5g、73mmol)の無水テトラヒドロフラン(100mL)溶液を50℃で30分間かけて滴下し、反応混合物を1時間攪拌した。引き続き、1−tert−ブトキシカルボニル−4−ピペリドン(5.00g、25mmol)と水素化ナトリウム(60%懸濁溶液、1.00g、25mmol)を加えて反応混合物を1.5時間攪拌した。室温に冷却後、水(100mL)を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去後、得られた濃縮残渣にn−ヘキサン(10mL)と酢酸エチル(10mL)を加え、生じた固体をろ取し、ろ上物をn−ヘキサン/酢酸エチル(1:1、5mL×3)で洗浄し、減圧乾燥することで、表記化合物(1−2−2)(11.3g、52%)を得た。
【0080】
化合物(1−2−3):tert−ブチル 4−(3−ブロモ−5−メトキシベンジル)ピペリジン−1−カルボキシレート(tert-Butyl 4-(3-bromo-5-methoxybenzyl)piperidine-1-carboxylate)
化合物(1−2−2)(19.0g、50mmol)を酢酸エチル(300mL)中、5%ロジウム炭素(10.3g、10mol%)を用いて室温で1.5時間常圧水素添加反応を行った。触媒をろ去し、ろ液を減圧濃縮することで、表記化合物(1−2−3)(20.6g、定量的)を得た。
【0081】
化合物(1−2−4):4−(3−ブロモ−5−メトキシベンジル)ピペリジン 塩酸塩(4-(3-Bromo-5-methoxybenzyl)piperidine hydrochloride)
化合物(1−2−3)(20.6g、50mmol)のメタノール(30mL)溶液に室温で10%塩酸メタノール溶液(150mL)を加え、反応混合物を50℃で45分間攪拌した。室温に冷却し、溶媒を減圧留去後、得られた濃縮残渣にジエチルエーテルを加え、生じた沈殿をろ取し、ろ上物をジエチルエーテルで洗浄後、減圧乾燥することで、表記化合物(1−2−4)(15.4g、97%)を得た。
保持時間(条件1):16.40分
【0082】
化合物(1−2−5):tert−ブチル 4−(3−ブロモ−5−ヒドロキシベンジル)ピペリジン−1−カルボキシレート(tert-Butyl 4-(3-bromo-5-hydroxybenzyl)piperidine-1-carboxylate)
化合物(1−2−4)(15.0g、47mmol)のジクロロメタン(200mL)溶液を氷浴で冷却し、1M−三臭化ホウ素−ジクロロメタン溶液(70mL、70mmol)を30分間で滴下した。反応混合物を氷冷下2時間攪拌し、さらに1M−三臭化ホウ素−ジクロロメタン溶液(50mL、50mmol)を氷冷しながら滴下した。反応混合物を2時間攪拌後、メタノール(50mL)を内温20℃以下に保ちながら滴下した。溶媒を減圧留去し、得られた濃縮残渣に2N−水酸化ナトリウム水溶液(200mL)と1,4−ジオキサン(400mL)を加えて溶解させ、室温でジ−tert−ブチルジカーボネート(10.3g、47mmol)の1,4−ジオキサン(50mL)溶液を30分間で滴下した。反応混合物を室温で1夜間攪拌後、1,4−ジオキサンを減圧留去し、濃縮残渣に水(300mL)を加えて酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(1−2−5)(15.8g、91%)を得た。
【0083】
化合物(1−1−8):
化合物(1−2−5)(15.8g、43mmol)、2−ブロモエチルメチルエーテル(6.0mL、64mmol)、ヨウ化カリウム(7.09g、43mmol)と炭酸カリウム(11.8g、85mmol)のジメチルホルムアミド(100mL)溶液を60〜70℃で8時間攪拌し、さらに2−ブロモエチルメチルエーテル(1.0mL、11mmol)を加えて、反応混合物を2時間攪拌した。室温に冷却後、水(500mL)に注ぎ、酢酸エチル/トルエン(1:1、200mL×3)で抽出し、有機層を水洗後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=5:1)で精製することで、表記化合物(1−1−8)(18.4g、定量的)を得た。
【0084】
化合物(RE1):
化合物(1−1−8)(18.4g、43mmol)のメタノール(30mL)溶液に室温で10%塩酸メタノール溶液(200mL)を加え、反応混合物を室温で1夜間攪拌した。メタノールを減圧留去し、得られた濃縮残渣にジエチルエーテル(100mL)を加えて生じた沈殿をろ取し、ろ上物をジエチルエーテル(50mL)で洗浄後、減圧乾燥することで、目的化合物(RE1)(14.7g、95%)を得た。
【0085】
参考例2
4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン(4-[3-Chloro-5-(2-methoxyethoxy)benzyl]piperidine,化合物(RE2))
【0086】
【化22】
化合物(1−1−8)(800mg、1.8mmol)と塩化銅(I)(537mg、5.4mmol)のジメチルホルムアミド(5.4mL)溶液を150℃で6時間攪拌した。室温に冷却後、塩をろ去し、ろ液を減圧留去して得られた濃縮残渣に2N−水酸化ナトリウム水溶液とクロロホルムを加えて生じた沈殿をろ去した。ろ液を酢酸エチルで抽出し、有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(RE2)(520mg、定量的)を得た。
1H-NMR (300 MHz, CDCl3) δ: 1.04-1.20 (2H, m), 1.52-1.83 (3H, m), 2.45 (2H, bd, J=6.8 Hz), 2.47-2.61 (2H, m), 3.04 (2H, bd, J=11.9 Hz), 3.45 (3H, s), 3.72-3.75 (2H, m), 4.07-4.10 (2H, m), 6.62 (1H, bt, J=1.8 Hz), 6.72-6.78 (2H, m).
【0087】
参考例3
4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン 塩酸塩(4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidine hydrochloride,化合物(RE3))
下記の製造方法1、2または3に従い、合成した。
製造方法1
【0088】
【化23】
【0089】
化合物(3−1−1):4−ブロモ−3−(2−メトキシエトキシ)安息香酸(4-Bromo-3-(2-methoxyethoxy)benzoic acid)
窒素雰囲気下、2−メトキシエタノール(16.48g,217mmol)の無水N−メチル−2−ピロリジノン(175mL)溶液に室温でカリウム t−ブトキシド(24.29g,217mmol)を加えた。目視にて溶解を確認後、4−ブロモ‐3‐フルオロ安息香酸(19.00g,86.8mmol)を少しずつ加えた。反応混合物を90℃で6時間攪拌した。室温まで冷却後、濃塩酸(36%,25mL)、水(500mL)の溶液に水冷しながら40分間で滴下し、内温20〜25℃で1時間攪拌後、生じた沈殿をろ取し、ろ上物を水(20 mL×2)、アセトニトリル(20mL×2)で洗浄し、ろ上物を減圧乾燥して白色固体(26.41g)を得た。これをアセトニトリル(380mL)に加え、還流温度付近まで加温し、目視にて溶解を確認後冷却し、75℃付近で結晶の析出が始まったら65〜70℃に保ちながら1時間攪拌後、再び2.5時間かけて30℃付近まで冷却し、引き続き水冷し内温20℃を保ちながら1時間攪拌した。生じた沈殿をろ取し、ろ上物をアセトニトリル(20mL×2)で洗浄することで、表記化合物(3−1−1)(20.09g,85%)を淡褐色針状晶として得た。
【0090】
化合物(3−1−2):[4−ブロモ−3−(2−メトキシエトキシ)フェニル]メタノール([4-Bromo-3-(2-methoxyethoxy)phenyl]methanol)
水素化ホウ素ナトリウム(8.08g,213.5mmol)の無水テトラヒドロフラン(100mL)懸濁溶液に水冷しながら三フッ化ホウ素・ジエチルエーテル錯体(35mL,285mmol)を滴下し、そのまま1時間攪拌した。化合物(3−1−1)(19.50g,71.2mmol)の無水テトラヒドロフラン(300mL)溶液を水冷により内温25℃以下に保ちながら30分間で滴下した。反応混合物を3時間攪拌後、氷冷して内温20℃以下に保ちながら水(200mL)を20分間で滴下した。トルエン(200mL)を加えて分液し、水層をトルエン(200mL)で再抽出した。全有機層を3%重曹水(200 mL)、水(200mL)で洗浄後、トルエンを減圧留去し、濃縮残渣にトルエン(200mL)を加えてトルエンを減圧留去することで、表記化合物(3−1−2)(18.18g)を得た。
【0091】
化合物(3−1−3):4−ブロモ−3−(2−メトキシエトキシ)ベンジル メタンスルホネート(4-Bromo-3-(2-methoxyethoxy)benzyl methanesulfonate)
化合物(3−1−2)(18.00g,71.17mmol相当)、トリメチルアミン塩酸塩(467mg,7.12mmol)とトリエチルアミン(19.8 mL,142mmol)のトルエン(90mL)溶液に、塩−氷浴で内温を5℃以下に保ちながらメタンスルホニルクロライド(8.56g,74.7mmol)のトルエン(18mL)溶液を30分間で滴下し、反応混合物を内温5℃以下で2時間攪拌した。反応液を5%硫酸水素カリウム水溶液(180mL)に氷浴で冷却して10℃以下に保ちながら注ぎ、30分間攪拌した。室温に昇温させて分液し、水層をトルエン(90mL)で再抽出した。全有機層を水(180 mL)で洗浄し、トルエンを減圧留去することで、表記化合物(3−1−3)(22.43g)を得た。
【0092】
化合物(3−1−4):1−ブロモ−4−(ブロモメチル)−2−(2−メトキシエトキシ)ベンゼン(1-Bromo-4-(bromomethyl)-2-(2-methoxyethoxy)benzene)
化合物(3−1−3)(22.43g,71.17mmol相当)の無水テトラヒドロフラン(100mL)溶液に室温で無水臭化リチウム(18.54g, 214mmol)を加え、反応混合物を1時間加熱還流した。室温に冷却後、水(100mL)およびトルエン(100mL)を加えて分液し、水層をトルエン(100mL)で再抽出した。全有機層を5%重曹水(100mL)、水(100mL)の順に洗浄し、トルエンを減圧留去することで、表記化合物(3−1−4)(19.54g)を白色固体として得た。
また、本表記化合物(3−1−4)は、化合物(3−1−3)を経由することなく、次に示すように、化合物(3−1−2)から直接合成することもできる。
化合物(3−1−2)(16.0g,61.3mmol)のトルエン(80g)と臭化水素水(47%,53g)の混合溶液を内温65−70℃で2時間攪拌した。室温に冷却後、水(16g)を加えて分液し、有機層を5%重曹水(48g)、水(48g)の順に洗浄した。有機層を減圧下濃縮することにより表記化合物(3−1−4)(17.9g,90%)を得た。
【0093】
化合物(3−1−5):[4−ブロモ−3−(2−メトキシエトキシ)ベンジル](トリフェニル)ホスホニウム ブロマイド([4-Bromo-3-(2-methoxyethoxy)benzyl](triphenyl)phosphonium bromide)
化合物(3−1−4)(19.54g,71.17mmol相当)のトルエン(100mL)溶液にトリフェニルホスフィン(18.67g,71.17mmol)を加え、反応混合物を3.5時間加熱還流した。室温まで冷却後、水冷しながら20℃に保って1時間攪拌後、生じた沈殿をろ取し、ろ上物をトルエン(40mL×3)で洗浄し、減圧乾燥することで、表記化合物(3−1−5)(32.44g,78%)を得た。
【0094】
化合物(3−1−6):tert−ブチル 4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジリデン]ピペリジン−1−カルボキシレート(tert-Butyl 4-[4-bromo-3-(2-methoxyethoxy)benzylidene]piperidine-1-carboxylate)
化合物(3−1−5)(32.00g,54.6mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(11.42g、57.3mmol)と炭酸カリウム(11.30g,81.9mmol)の2−プロパノール(160 mL)溶液を時間加熱還流した。室温に冷却後、塩をろ別し、ろ上の塩を2−プロパノール(30mL×2)で洗浄した。ろ液を減圧濃縮し、濃縮残渣(41.08g)を得た。トルエンを加えて減圧留去し(200mL×2)、引き続き濃縮残渣にトルエン(96mL)を加え、水冷して20−25℃に保ちながらn−ヘキサン(290mL)を30分間で滴下した。そのまま混合物を1時間攪拌後、氷冷で1時間攪拌し、生じた沈殿をろ別した。ろ上物をトルエン/n−ヘキサン(トルエン:n−ヘキサン〜1:3,20mL×2)で洗浄し、ろ液を減圧留去することで、表記化合物(3−1−6)(27.93g)を淡黄色油状物として得た。
【0095】
化合物(3−1−7):tert−ブチル 4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[4-bromo-3-(2-methoxyethoxy)benzyl]piperidine-1-carboxylate)
化合物(3−1−6)(27.93g,54.6mmol相当)を酢酸エチル(232mL)中で5%ロジウム炭素(5.80g)を用いて、内温15〜20℃で3時間常圧水素添加反応を行った。触媒をセライトろ過後、酢酸エチルを減圧留去することで、表記化合物(3−1−7)(25.73g)を白色固体として得た。
【0096】
化合物(RE3):
化合物(3−1−7)(25.73g,54.6mmol相当)の2−プロパノール(115mL)溶液を内温55〜60℃に加温し、濃塩酸(36%,23.2mL)を5分間で滴下し、反応混合物を内温55〜60℃で4時間攪拌した。室温まで冷却後、2−プロパノールを減圧留去して濃縮残渣(42.91g)を得た。これに水(115mL)とトルエン(115mL)を加えて分液し、有機層を水(50mL)で再抽出した。全水層を水酸化ナトリウムでpH10程度に調製し、トルエン(200+100+100mL)で抽出した。有機層を水(50mL)で洗浄し、トルエンを減圧留去して濃縮残渣(18.57g)を得た。これを2−プロパノールに溶解させ、室温で濃塩酸(36%,5.58g,54.6mmol)を加えて、2−プロパノールを減圧留去し、濃縮残渣に2−プロパノール(200mL×2)を加えて減圧留去することで濃縮残渣(18.61 g)を白色粉末として得た。これに2−プロパノール(115mL)を加え、内温65〜70℃付近で均一溶液になったことを目視にて確認後、徐冷した。60℃付近で結晶の析出を確認したところで、内温55〜60℃でn−ヘキサン(60mL)を20分間で滴下し、懸濁溶液を内温55〜60℃で1時間攪拌後、再び徐冷し、内温30℃以下になったところで水冷で内温15〜20℃に保ちながら1時間攪拌し、さらに氷冷して内温5℃以下で1時間攪拌した。析出物をろ取し、ろ上物を冷n−ヘキサン(23mL)−2−プロパノール(12mL)混合溶液で洗浄後、減圧乾燥することで、目的化合物(RE3)(17.30 g)を白色粉末として得た。
保持時間(条件1):15.55分
融点:171−172℃
1H-NMR (300 MHz, CDCl3) δ: 1.47-1.94 (5H, m), 2.55 (2H, d, J = 5.5 Hz), 2.79 (2H, t like, J = 12 Hz), 3.47 (2H, d like, J = 13 Hz), 3.51 (3H, s), 3.81 (2H, t, J = 4.8 Hz), 4.15 (2H, t, J = 4.8 Hz), 6.62 (1H, dd, J = 7.9, 1.8 Hz), 6.68 (1H, d, J = 1.7 Hz), 7.43 (1H, d, J = 8.1 Hz), 9.50 (2H, br s).
【0097】
製造方法2
【0098】
【化24】
【0099】
化合物(3−2−1):メチル 3−(2−メトキシエトキシ)−4−ニトロベンゾエート(Methyl 3-(2-methoxyethoxy)-4-nitrobenzoate)
メチル 3−ヒドロキシ−4−ニトロベンゾエート(15.0g、76mmol)、2−ブロモエチルメチルエーテル(14.5g、99mmol)、ヨウ化カリウム(12.6g、76mmol)と炭酸カリウム(21.4g、155mmol)のジメチルホルムアミド(250mL)溶液を60〜70℃で3時間攪拌した。さらに2−ブロモエチルメチルエーテル(5.00g、36mmol)を加えて、反応混合物を60〜70℃で2時間攪拌後、室温で1夜間攪拌した。反応混合物を水(600mL)に注ぎ、酢酸エチル/トルエン(1:1、500mL×2)で抽出し、有機層を5%炭酸カリウム水溶液、水の順に洗浄し、溶媒を減圧留去することで、表記化合物(3−2−1)(20.2g、定量的)を得た。
【0100】
化合物(3−2−2):メチル 4−アミノ−3−(2−メトキシエトキシ)ベンゾエート(Methyl 4-amino-3-(2-methoxyethoxy)benzoate)
化合物(3−2−1)(20.2g、76mmol)をメタノール(200mL)中で10%パラジウム炭素(8.06g、10mol%)を用いて、室温で2時間常圧水素添加反応を行った。水素の吸収が止まったことを確認して、触媒をろ去し、ろ液を減圧濃縮することで、表記化合物(3−2−2)(16.7g、98%)を得た。
【0101】
化合物(3−2−3):メチル 4−ブロモ−3−(2−メトキシエトキシ)ベンゾエート(Methyl 4-bromo-3-(2-methoxyethoxy)benzoate)
化合物(3−2−2)(10.0g、44mmol)の48%臭化水素酸水溶液(50mL)液を氷浴で冷却し、亜硝酸ナトリウム(3.07g、45mmol)の水(30mL)溶液を30分間で滴下し、反応混合物を1時間攪拌した。この溶液を5℃以下に保ち、60℃に加温した臭化銅(I)(4.17g、29mmol)の48%臭化水素酸水溶液(50mL)へ20分間で滴下し、反応混合物を60℃で1時間攪拌した。室温に冷却後、水(400mL)に注ぎ、ジエチルエーテルで抽出し、有機層を水、飽和食塩水の順で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(3−2−3)(9.77g、76%)を得た。
【0102】
化合物(3−2−4):[4−ブロモ−3−(2−メトキシエトキシ)フェニル]メタノール([4-Bromo-3-(2-methoxyethoxy)phenyl]methanol)
化合物(3−2−3)(10.0g、35mmol)の無水テトラヒドロフラン(50mL)溶液を穏やかに加熱還流させながら、1.0Mボラン・テトラヒドロフラン錯体テトラヒドロフラン溶液(140mL、140mmol)を滴下し、反応混合物を20時間加熱還流した。室温に冷却後、発泡が収まるまで水を加え、テトラヒドロフランを減圧留去した。濃縮残渣に飽和重曹水を加えてクロロホルムで抽出し、有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=1:1)で精製することで、表記化合物(3−2−4)(9.13g、定量的)を得た。
【0103】
化合物(3−1−4):
化合物(3−2−4)(9.00g、34.5mmol)とトリフェニルホスフィン(10.9g、41mmol)のジクロロメタン(200mL)溶液に、氷冷しながら内温20℃以下で四臭化炭素(15.5g、48mmol)を加え、反応混合物を氷冷下1.5時間攪拌した。溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(クロロホルム)で精製することで、表記化合物(3−1−4)(9.88g、88%)を得た。
【0104】
化合物(3−1−5):
化合物(3−1−4)(9.85g、30mmol)とトリフェニルホスフィン(9.57g、36.5mmol)のトルエン(200mL)溶液を16時間加熱還流した。反応混合物をゆっくり室温まで冷却し、引き続いて氷浴で30分間攪拌後、生じた沈殿をろ取し、ろ上物をトルエン(10mL×2)で洗浄し、減圧乾燥することで、表記化合物(3−1−5)(19.4g、定量的)を得た。
【0105】
化合物(3−1−6):
化合物(3−1−5)(19.0g、32mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(7.10g、36mmol)と炭酸カリウム(6.71g、49mmol)の2−プロパノール(200mL)懸濁溶液を11.5時間加熱還流した。反応混合物を室温まで冷却後、塩をろ去し、ろ液を減圧濃縮した。得られた濃縮残渣にn−ヘキサン/酢酸エチル(4:1、200mL)を加え、生じたトリフェニルホスフィンオキシドをろ去し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(3−1−6)(14.2g、定量的)を得た。
【0106】
化合物(3−1−7):
化合物(3−1−6)(4.00g、9.4mmol)を酢酸エチル(100mL)中で5%ロジウム炭素(1.91g、10mol%)を用いて室温で2.5時間常圧水素添加反応を行った。触媒をろ去し、ろ液を減圧濃縮し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(3−1−7)(3,88g、97%)を得た。
【0107】
化合物(RE3):
化合物(3−1−7)(3.88g、9.1mmol)のメタノール(5mL)溶液に室温で10%塩酸メタノール溶液(20mL)を加え、反応混合物を室温で5時間攪拌した。溶媒を減圧留去し、得られた濃縮残渣に少量の2−プロパノールを加えて固化させ、さらにジエチルエーテルを加えて懸濁させ、沈殿をろ取し、ジエチルエーテルで洗浄することで、目的化合物(RE3)(2.45g、74%)を得た。
【0108】
製造方法3
【0109】
【化25】
【0110】
化合物(3−3−1):(4−ブロモ−3−フルオロベンジル)(トリフェニル)ホスホニウム ブロマイド((4-Bromo-3-fluorobenzyl)(triphenyl)phosphonium bromide)
4−ブロモ−3−フルオロトルエン(25.0g,132 mmol)、5,5−ジメチル−1,3−ジブロモヒダントイン(18.9g,66.1mmol)、アゾビスイソブチロニトリル(1.09g,6.64mmol)のクロロベンゼン(400mL)溶液を内温80−90℃で1時間攪拌した。反応混合物を氷浴で冷却後、水(200mL)とチオ硫酸ナトリウム(33g,132mmol)を加えて攪拌した。分液し、有機層を水(200mL)で洗浄し、無水硫酸ナトリウムで乾燥後、全体が100mL程度の体積になるまで濃縮した。そこへトリフェニルホスフィン(34.69g,132mmol)とクロロベンゼン(30mL)を加えて3時間加熱還流した。反応混合物を室温まで冷却後、析出物をろ取し、ろ上物をトルエンで洗浄後、減圧乾燥することで、表記化合物(3−3−1)(47.0g)を得た。
【0111】
化合物(3−3−2):tert-ブチル 4−[(4−ブロモ−3−フルオロフェニル)メチリデン]ピペリジン−1−カルボキシレート(tert-Butyl 4-[(4-bromo-3-fluorophenyl)methylidene]piperidine-1-carboxylate)
化合物(3−3−1)(15.0g,28.3mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(3.76g,18.9mmol)および炭酸カリウム(5.21g,37,7mmol)の2−プロパノール(28mL)溶液を3時間加熱還流した。反応混合物を室温に冷却し、塩をろ別後、ろ液にトルエン(300mL)を加えて、水(100mL)、飽和食塩水(100mL)で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。濃縮残渣にn−ヘキサン(107mL)を加えて1時間加熱還流後、室温まで冷却して1時間攪拌後、氷浴で冷却しながら1時間攪拌した。析出したトリフェニルホスフィンオキシドをろ別し、n−ヘキサンで洗浄後、ろ液を減圧濃縮することで、表記化合物(3−3−2)(8.08g)を黄色固体として得た。
【0112】
化合物(3−3−3):tert-ブチル 4−(4−ブロモ−3−フルオロベンジル)ピペリジン−1−カルボキシレート(tert-Butyl 4-(4-bromo-3-fluorobenzyl)piperidine-1-carboxylate)
化合物(3−3−2)(8.08g)を酢酸エチル(57mL)中で5%白金炭素(800mg)を用いて時間常圧水素添加反応を行った。セライトろ過により触媒をろ別し、ろ液を濃縮することで、表記化合物(3−3−3)(8.61g)を淡黄色固体として得た。
【0113】
化合物(3−1−7):
化合物(3−3−3)(8.61g,18.9mmol相当)、2−メトキシエタノール(2.98mL,37.7mmol)のN−メチル−2−ピロリジノン(38mL)溶液に室温でカリウム t−ブトキシド(4.23g,37.7mmol)を加え、反応混合物を内温90℃で2.5時間攪拌後、カリウム t−ブトキシド(1.06g,9.43mmol)を加えて30分間90℃で攪拌し、さらにカリウム t−ブトキシド(1.06g,9.43mmol)を加えて30分間90℃で攪拌した。反応混合物を氷浴で冷却し、飽和塩化アンモニウム水溶液(80mL)を加え、トルエン(80mL×3)で抽出した。全有機層を水(40mL×2)、飽和食塩水(40mL)で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。濃縮残渣にn−ヘキサン(162mL)を加え、60℃に加温して溶解を確認後、ゆっくり室温まで冷却し、室温で1夜間攪拌後、氷浴で冷却して1時間攪拌し、析出物をろ取し、ろ上物をn−ヘキサンで洗浄後、減圧乾燥することで、表記化合物(3−1−7)(6.34g)を淡褐色粉末として得た。
【0114】
化合物(RE3):
化合物(3−1−7)(6.00g,14.0mmol)のメタノール(24 mL)溶液に室温で10%塩酸−メタノール溶液(24mL)を加え、反応混合物を50℃で5時間攪拌した。室温まで冷却後、溶媒を減圧留去し、濃縮残渣にアセトニトリル(6mL)を加えて減圧濃縮を4回繰り返した。濃縮残渣にアセトニトリル(38mL)を加えて80℃の油浴で加温し、固形物の溶解を確認後、1時間で室温まで冷却し、水浴で冷却して20℃で1時間、氷浴で1時間攪拌後、析出物をろ取し、ろ上物を冷アセトニトリル(30mL)で洗浄し、減圧乾燥することで、表記化合物(RE3)(4.56g,89%)を白色粉末として得た。
【0115】
参考例4
4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(4-[4-Chloro-3-(2-methoxyethoxy)benzyl]piperidine,化合物(RE4))
下記製造方法に従い、合成した。
製造方法
【0116】
【化26】
【0117】
化合物(4−1):メチル 4−クロロ−3−(2−メトキシエトキシ)ベンゾエート(Methyl 4-chloro-3-(2-methoxyethoxy)benzoate)
化合物(3−2−2)(5.20g、23mmol)の濃塩酸(36%,50mL)水溶液を氷浴で冷却し、亜硝酸ナトリウム(1.59g、23mmol)の水(20mL)溶液を10分間で滴下し、反応混合物を20分間攪拌した。この溶液を5℃以下に保ち、60℃に加温した塩化銅(I)(1.51g、15mmol)の濃塩酸(36%,50mL)水溶液へ10分間で滴下し、反応混合物を60℃で50分間攪拌した。室温に冷却後、水で希釈してジエチルエーテルで抽出した。有機層を飽和重曹水、飽和食塩水の順で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1→1:1)で精製することで、表記化合物(4−1)(4.43g、78%)を白色固体として得た。
【0118】
化合物(4−2):[4−クロロ−3−(2−メトキシエトキシ)フェニル]メタノール([4-Chloro-3-(2-methoxyethoxy)phenyl]methanol)
化合物(4−1)(4.33g、18mmol)の無水テトラヒドロフラン(50mL)溶液に室温で1.0Mボラン・テトラヒドロフラン錯体テトラヒドロフラン溶液(106mL、106mmol)を滴下し、引き続き反応混合物を27.5時間加熱還流した。室温に冷却後、発泡が収まるまでメタノールを加え、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=3:1→1:1)で精製することで、表記化合物(4−2)(3.62g、94%)を得た。
【0119】
化合物(4−3):4−(ブロモメチル)−1−クロロ−2−(2−メトキシエトキシ)ベンゼン(4-(Bromomethyl)-1-chloro-2-(2-methoxyethoxy)benzene)
化合物(4−2)(3.60g、16.6mmol)のジエチルエーテル(50mL)溶液に室温でトリフェニルホスフィン(6.54g、25mmol)、四臭化炭素(8.26g、25mmol)を一度に加え、反応混合物を16時間攪拌した。析出物をろ別し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン→n−ヘキサン:酢酸エチル=7:1)で精製することで、表記化合物(4−3)(3.93g、85%)を無色油状物として得た。
【0120】
化合物(4−4): [4−クロロ−3−(2−メトキシエトキシ)ベンジル](トリフェニル)ホスホニウム ブロマイド([4-Chloro-3-(2-methoxyethoxy)benzyl](triphenyl)phosphonium bromide)
化合物(4−3)(3.90g、14mmol)とトリフェニルホスフィン(4.41g、36.17mmol)のトルエン(100mL)溶液を5時間加熱還流した。反応混合物をゆっくり室温まで冷却し、引き続いて氷浴で1時間攪拌後、析出物をろ取し、ろ上物をトルエンで洗浄し、減圧乾燥することで、表記化合物(4−4)(7.24g、95%)を白色粉末として得た。
【0121】
化合物(4−5):tert−ブチル 4−{[4−クロロ−3−(2−メトキシエトキシ)フェニル]メチリデン}ピペリジン−1−カルボキシレート(tert-Butyl 4-{[4-chloro-3-(2-methoxyethoxy)phenyl]methylidene}piperidine-1-carboxylate)
化合物(4−4)(7.24g、13mmol)、1−tert−ブトキシカルボニル−4−ピペリドン(2.40g、12mmol)と炭酸カリウム(2.78g、20mmol)の2−プロパノール(100mL)懸濁溶液を8時間加熱還流した。反応混合物を氷冷して1時間攪拌後、析出物をろ去し、ろ液を減圧濃縮した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=10:1→8:1)で精製することで、表記化合物(4−5)(4.52g、定量的)を無色油状物として得た。
【0122】
化合物(4−6):tert−ブチル 4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[4-chloro-3-(2-methoxyethoxy)benzyl]piperidine-1-carboxylate)
化合物(4−5)(4.52g、12mmol)を酢酸エチル(200mL)中で5%ロジウム炭素(1.53g)を用いて室温で1.5時間常圧水素添加反応を行った。触媒をセライトろ過でろ去し、ろ液を減圧濃縮後、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=8:1→4:1)で精製することで、表記化合物(4−6)(4.34g、96%)を得た。
【0123】
化合物(RE4):
化合物(4−6)(4.34g、11mmol)の1,4−ジオキサン(50mL)溶液を4N−塩酸−1,4−ジオキサン溶液(50mL)に加え、反応混合物を室温で15.5時間攪拌した。溶媒を減圧留去することで表記化合物の塩酸塩(3.24g,90%)を得た。このうち2gを2N−水酸化ナトリウム水溶液に加え、クロロホルムで抽出し、無水硫酸ナトリウムで乾燥後、溶媒を減圧濃縮することで、目的化合物(RE4)(1.91g)を淡黄色油状物として得た。
保持時間(条件1):13.71分
1H-NMR (300 MHz, CDCl3) δ: 1.03-1.26 (2H, m), 1.48-1.69 (3H, m), 2.48 (2H, t, J = 5.2 Hz), 2.54 (2H, td, J = 12.5, 2.6 Hz), 3.06 (2H, br d, J = 11.9 Hz), 3.49 (3H, s), 3.80 (2H, t, J = 4.8 Hz), 4.17 (2H, t, J = 4.8 Hz), 6.69 (1H, dd, J = 7.9, 1.8 Hz), 6.73 (1H, d, J = 1.8 Hz), 7.26 (1H, d, J = 8.1 Hz).
【0124】
参考例5
2−[2−ブロモ−5−(ピペリジン−4−イルメチル)フェノキシ]エタノール(2-[2-Bromo-5-(piperidin-4-ylmethyl)phenoxy]ethanol,化合物(RE5))
【0125】
【化27】
化合物(RE3)(3.00g、8.3mmol)のジクロロメタン(100mL)溶液を塩−氷浴で冷却し、0℃で1M−三臭化ホウ素ジクロロメタン溶液(7.8mL、7.8mmol)を30分間で滴下し、反応混合物を氷冷下1.5時間攪拌した。メタノール(20mL)を加え、溶媒を減圧留去した。得られた濃縮残渣に5%炭酸カリウム水溶液(50mL)を加え、クロロホルムで抽出し、有機層を無水硫酸ナトリウムで乾燥した。溶媒を減圧留去することで、表記化合物(RE5)(3.03g、定量的)を得た。
保持時間(条件1):7.56分
1H-NMR (400 MHz,CDCl3) δ:1.15-1.30 (2H, m), 1.40-1.55 (1H, m), 1.55-1.70 (2H, m), 2.49 (2H, d, J= 6.7 Hz), 2.58 (2H, dt, J= 2.4, 12 Hz), 3.11 (2H, d like, J= 12 Hz), 3.98 (2H, t, J= 4.6 Hz), 4.14 (2H, t, J= 4.6 Hz), 6.66 (1H, dd, J= 8.0, 1.8 Hz), 6.70 (1H, d, J= 1.8 Hz), 7.42 (1H, d, J= 8.0 Hz).
【0126】
参考例6
4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン(4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidine,化合物(RE6))
下記の製造方法に従い、合成した。
製造方法
【0127】
【化28】
【0128】
化合物(6−1):tert-ブチル 4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−カルボキシレート(tert-Butyl 4-[3-(2-methoxyethoxy)-4-methylbenzyl]piperidine-1-carboxylate)
化合物(3−1−7) (5.00g、11.7g)、メチルボロニック アシッド(978mg、16mmol)とテトラキストリフェニルホスフィンパラジウム(674mg、5mol%)の1M−炭酸カリウム水溶液(35mL)、1,4−ジオキサン(80mL)溶液を4時間加熱還流した。室温まで冷却後、1,4−ジオキサンを減圧留去し、得られた濃縮残渣に水を加えて酢酸エチルで抽出し、全有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去後、濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=4:1)で精製することで、表記化合物(6−1)(3.46g、82%)を得た。
【0129】
化合物(RE6):
化合物(6−1)(3.46g、9.5mmol)に室温で10%塩酸メタノール溶液(40mL)を加え、反応混合物を1夜間攪拌した。メタノールを減圧留去し、得られた濃縮残渣に飽和重曹水を加え、塩析してクロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、目的化合物(RE6)(2.56g、定量的)を得た。
1H-NMR (300 MHz,CDCl3) δ: 1.39-1.58 (2H, m), 1.58-1.74 (1H, m), 1.77 (2H, d like, J = 13.9 Hz), 2.20 (3H, s), 2.52 (2H, d, J = 7.1 Hz), 2.70 (2H, t like, J = 12.6 Hz), 3.33 (2H, d like, J = 12.4 Hz), 3.47 (3H, s), 3.77 (2H, t, J = 4.8 Hz), 4.10 (2H, t, J = 4.8 Hz), 6.59 (1H, s), 6.63 (1H, d, J = 7.6 Hz), 7.03 (1H, d, J = 7.6 Hz).
【0130】
参考例7
2−(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)エチル 4−メチルベンゼンスルホネート(2-(4-Oxo-3,4-dihydro-2H-chromen-6-yl)ethyl 4-methylbenzenesulfonate,化合物(RE7))
下記の製造方法1、2または3に従い、合成した。
製造方法1
【0131】
【化29】
【0132】
化合物(7−1−1):メチル [4−(3−ヒドロキシプロポキシ)フェニル]アセテート(Methyl [4-(3-hydroxypropoxy)phenyl]acetate)
4−ヒドロキシフェニル酢酸 メチルエステル(50.0g,301mmol)のアセトニトリル(1000mL)、水(10mL)溶液に、室温で炭酸カリウム(91.5g,662mmol)、3−ブロモ−1−プロパノール(35.3mL,391mmol)の順で加え、反応混合物を3時間加熱還流した。室温に冷却後、塩をろ別し、ろ上物をアセトニトリル(50mL×2)で洗浄した。ろ液を減圧濃縮し、得られた濃縮残渣にトルエン(500mL)、水(250 mL)を加えて分液した。水層をトルエン(125mL×2)で再抽出し、全有機層を0.5N−水酸化ナトリウム水溶液(100mL)、1%硫酸水素カリウム水溶液(100mL)で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−1)(70.2 g)を黄色油状物として得た。
【0133】
化合物(7−1−2):3−[4−(2−メトキシ−2−オキソエチル)フェノキシ]プロパノイック アシッド(3-[4-(2-Methoxy-2-oxoethyl)phenoxy]propanoic acid)
2,2,6,6−テトラメチル−1−ピペリジニルオキシ、ラジカル(TEMPO)(4.71g,30.1mmol)のアセトニトリル(160mL)溶液に、0.25Mりん酸二水素カリウム水溶液(400mL)、0.25Mりん酸水素二ナトリウム水溶液(400mL)、80%亜塩素酸ナトリウム(54.5 g,482mmol)、5%次亜塩素酸ナトリウム水溶液(6.52mL,4.82mmol)を順に加え、引き続き水冷して内温20〜25℃に保ちながら化合物(7−1−1)(56.2g,241mmol相当)のアセトニトリル(800mL)溶液を約1時間で滴下した。反応混合物をそのまま2時間攪拌後、氷浴により内温15℃以下に保ちながら20%亜硫酸水素ナトリウム水溶液(400mL)を30分間で滴下した。溶液をゆっくり室温に昇温し、アセトニトリルを減圧留去した。水(400mL)を加えて室温で30分間攪拌し、生じた沈殿をろ取し、水(100mL×2)で洗浄した。ろ上物を酢酸エチル(400mL)に溶解させて、飽和食塩水(100mL)で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−2)(47.87 g)を得た。
【0134】
化合物(7−1−3):メチル [4−(3−クロロ−3−オキソプロポキシ)フェニル]アセテート(Methyl [4-(3-chloro-3-oxopropoxy)phenyl]acetate)
化合物(7−1−2)(47.0g,197mmol)のトルエン(470 mL)溶液に室温で塩化チオニル(42.7mL、592mmol)を加え、反応混合物を60℃で3時間攪拌した。室温に冷却後、トルエンを留去することで、表記化合物(7−1−3)(56.52g)を淡黄色油状物として得た。
【0135】
化合物(7−1−4):メチル (4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)アセテート(Methyl (4-oxo-3,4-dihydro-2H-chromen-6-yl)acetate)
塩化アルミニウム(52.5g,394mmol)のジクロロメタン(330 mL)溶液に化合物(7−1−3)(56.52g,197mmol相当)のジクロロメタン(140mL)溶液を水冷により内温20〜25℃に保ちながら30分間で滴下し、反応混合物をそのまま1.5時間攪拌した。反応混合物を冷却し、2N塩酸水溶液(470mL)を氷浴で冷却して内温15℃以下に保ちながら加えた。室温に昇温後、分液し、水層をクロロホルム(120mL)で再抽出した。全有機層を水(240mL)、飽和重曹水(240mL)の順で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−4)(38.91g)を褐色固体として得た。
【0136】
化合物(7−1−5):メチル (4,4−ジメトキシ−3,4−ジヒドロ−2H−クロメン−6−イル)アセテート(Methyl (4,4-dimethoxy-3,4-dihydro-2H-chromen-6-yl)acetate)
化合物(7−1−4)(35.0g,159mmol)のメタノール(105 mL)溶液にp−トルエンスルホン酸・1水和物(3.02g,15.9mmol)、オルトギ酸メチル(210mL)を加え、反応混合物を室温(15〜20℃)で20時間攪拌した。5%重曹水(175mL)を氷冷して内温15℃以下に保ちながら反応溶液を15分間で滴下し、トルエン(175mL)、水(88mL)を加えて分液した。水層をトルエン(88mL)で再抽出し、全有機層を水(44mL)で洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去することで、表記化合物(7−1−5)(46.49g)を黄色油状物として得た。
【0137】
化合物(7−1−6):6−(2−ヒドロキシエチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-Hydroxyethyl)-2,3-dihydro-4H-chromen-4-one)
水素化リチウムアルミニウム(9.05g,239mmol)のテトラヒドロフラン(600mL)懸濁溶液に化合物(7−1−5)(46.0g,159 mmol相当)のテトラヒドロフラン(90mL)溶液を水冷し内温30℃以下に保ちながら30分間で滴下し、反応混合物をそのまま1時間攪拌した。氷冷して内温15℃以下に保ちながらテトラヒドロフラン−水(1:2、12mL)を滴下した(途中で不溶物が析出し、攪拌が困難となった)。引き続き3N塩酸水(460mL)を内温15℃以下に保ちながら滴下した。そのまま1.5時間20〜25℃で攪拌し、トルエン(460mL)を加えて分液した。水層をトルエン(230mL)で再抽出した。全有機層を3N塩酸水(230mL×2)で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−6)(30 .0g)を無色油状物として得た。
【0138】
化合物(RE7):
化合物(7−1−6)(10.0g,52mmol相当)のアセトニトリル(150mL)溶液に、トリメチルアミン塩酸塩(497mg,5.20mmol)およびトリエチルアミン(14.4mL,104mmol)を加え、氷浴で冷却して内温15℃以下でp−トルエンスルホニルクロライド(11.9g, 62.4mmol)を少しずつ加え、反応混合物を内温5℃以下で1.5時間攪拌した。内温10℃以下で5%重曹水(75mL)を加え、室温に昇温してからトルエン(75mL)を加えて分液し、水層をトルエン(75mL)で再抽出した。全有機層を1%硫酸水素カリウム水溶液(38mL×2)で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで濃縮残渣(16.61 g)を得た。トルエン(50mL)を加えて50℃で1.5時間攪拌し、引き続き30分間で室温(20〜25℃)まで冷却した。水冷し内温20〜25℃で1時間攪拌後、析出物をろ取し、トルエン(10mL×2)で洗浄し、減圧乾燥することで、目的化合物(RE7)(10.65g)を淡黄色粉末として得た。
保持時間(条件2):15.50分
融点:121−122℃
1H-NMR (300 MHz,CDCl3) δ: 2.44 (3H, s), 2.79 (2H, t, J = 6.5 Hz), 2.91 (2H, t, J = 6.9 Hz), 4.18 (2H, t, J = 6.9 Hz), 4.51 (2H, t, J = 6.4 Hz), 6.88 (1H, d, J = 8.4 Hz), 7.26 (1H, dd, J = 8.5, 2.5 Hz), 7.30 (2H, d, J = 8.5 Hz), 7.60 (1H, d, J = 2.2 Hz), 7.71 (2H, d, J = 8.3 Hz).
【0139】
製造方法2
【0140】
【化30】
【0141】
化合物(7−2−1):3−[4−(2−メトキシエチル)フェノキシ]プロパン−1−オール(3-[4-(2-Methoxyethyl)phenoxy]propan-1-ol)
4−(2−メトキシエチル)フェノール(1.00g,6.57mmol)、3−ブロモ−1−プロパノール(771μL,8.54mmol)、炭酸カリウム(2.00g,14.5mmol)と水(200μL)のアセトニトリル(20mL)溶液を3時間加熱還流した。室温まで冷却後、塩をろ別し、ろ液を減圧濃縮した。得られた濃縮残渣に水(5mL)とトルエン(5mL)を加えて分液し、水層をトルエン(5mL×2)で再抽出した。全有機層を0.5N−水酸化ナトリウム水溶液(2mL)、1%硫酸水素カリウム(2mL)で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−2−1)(1.56g)を得た。
【0142】
化合物(7−2−2):3−[4−(2−メトキシエチル)フェノキシ]プロパノイック アシッド(3-[4-(2-Methoxyethyl)phenoxy]propanoic acid)
化合物(7−2−1)(1.56g,6.57mmol相当)を用いて実施例7−1−2)と同様の方法により、表記化合物(7−2−2)(1.30g, 88%)を白色粉末として得た。
【0143】
化合物(7−2−3):6−(2−メトキシエチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-Methoxyethyl)-2,3-dihydro-4H-chromen-4-one)
化合物(7−2−2)(1.30g,5.80mmol)のトルエン(13 mL)溶液に室温で塩化チオニル(1.25mL)を加え、反応混合物を60℃で3時間攪拌した。室温に冷却後、溶媒を減圧留去することで酸クロライド(1.46g)を得た。このうち292mg(1.16mmol相当)のジクロロメタン(1mL)溶液を室温で塩化アルミニウム(309mg,2.32mmol)のジクロロメタン(2mL)懸濁溶液に水冷しながら室温で5分間かけて滴下し、反応混合物を1時間攪拌した。2N塩酸水溶液(3mL)および氷に反応溶液を注ぎ、トルエンで抽出した。有機層を水、飽和重曹水、飽和食塩水の順で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−2−3)(235mg)を得た。
【0144】
化合物(7−2−4):2−(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)エチル 2,2−ジメチルプロパノエート(2-(4-Oxo-3,4-dihydro-2H-chromen-6-yl)ethyl 2,2-dimethylpropanoate)
化合物(7−2−3)(100mg,0.485mmol)、ヨウ化ナトリウム(436mg,2.91mmol)と水(3μL)のアセトニトリル(0.6 mL)溶液に40℃でピバロイルクロライド(298μL,2.43mmol)を加え、反応混合物を40℃で1.5時間攪拌した。室温に冷却後、トルエンで希釈し、水を加えて分液し、水層をトルエンで再抽出した。全有機層を飽和重曹水、飽和食塩水の順で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−2−4)(148mg)を褐色油状物として得た。
【0145】
化合物(7−1−6):
化合物(7−2−4)(105mg,0.354mmol)の濃塩酸(36%, 2mL)、メタノール(1mL)溶液を50℃で3時間攪拌した。室温に冷却後、反応混合物を氷に注ぎ、トルエンで抽出し、飽和重曹水、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、表記化合物(7−1−6)(62mg)を得た。
【0146】
化合物(RE7):
化合物(7−1−6)(62mg,0.354mmol)、トリメチルアミン塩酸塩(3.4mg,0.035mmol)とトリエチルアミン(98μL)のジクロロメタン(1mL)溶液に、氷浴で冷却しながらp−トルエンスルホニルクロライド(81mg,0.43mmol)を加え、反応混合物をそのまま1.5時間攪拌した。飽和重曹水を加えて室温に昇温し、トルエンで抽出し、全有機層を1%硫酸水素カリウム水溶液、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を減圧留去後、得られた濃縮残渣にトルエン(1mL)を加えて50℃で1時間攪拌し、ゆっくり室温まで冷却しながら攪拌後、室温で2時間攪拌した。析出物をろ取し、ろ上物をトルエンで洗浄後、減圧乾燥することで、目的化合物(RE7)(35mg,29%)を得た。
【0147】
製造方法3
【化31】
【0148】
化合物(7−3−1):6−(ブロモメチル)−2,3−ジヒドロ−4H−クロメン−4−オン (6-(Bromomethyl)-2,3-dihydro-4H-chromen-4-one)
6−メチル−2,3−ジヒドロ−4H−クロメン−4−オン(7.2g,44mmol)、5,5−ジメチル−1,3−ジブロモヒダントイン(7.7g,27mmol)およびアゾビスイソブチロニトリル(1.5g,9mmol)のモノクロロベンゼン(140mL)溶液を80℃で3時間攪拌した。反応溶液を氷水(100mL)に注ぎ、室温に昇温後分液した。水層をジクロロメタン(50mL×2)で抽出し、全有機層を飽和食塩水で洗浄した。無水硫酸マグネシウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(7.8g,73%)を得た。
【0149】
化合物(7−3−2):(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)アセトニトリル((4-Oxo-3,4-dihydro-2H-chromen-6-yl)acetonitrile)
化合物(7−3−1)(500mg,2.1mmol)とシアン化カリウム(135mg,2.1mmol)の1,4−ジオキサン(7.5mL)および水(2.5mL)混合溶液を50℃で3時間攪拌した。反応溶液を室温まで冷却後、飽和食塩水(30mL)を加えてトルエン(30mL×2)で抽出した。全有機層を無水硫酸マグネシウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(311mg,70%)を得た。
【0150】
化合物(7−1−4):
化合物(7−3−2)(276mg,1.5mmol)の濃硫酸(2mL)、酢酸(2mL)および水(2mL)溶液を80℃で5時間攪拌した。反応溶液を室温に冷却後、10%水酸化ナトリウム水溶液でpH10−11に調製し、ジクロロメタン(30mL×2)で抽出した。全有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去することで化合物(7−3−3)(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)アセティックアシッド(4-Oxo-3,4-dihydro-2H-chromen-6-yl)acetic acidの粗生成物を得た。これを精製することなく濃硫酸(0.05mL)とともにメタノール(5mL)に溶解させ、反応混合物を2時間加熱還流した。室温に冷却後、水(10mL)を加えてメタノールを減圧留去し、濃縮残渣を酢酸エチル(30mL×2)で抽出した。全有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(251mg,88%)を得た。
【0151】
化合物(7−3−4):メチル 2,3−ジヒドロスピロ[クロメン−4,2’−[1,3]ジオキソラン]−6−イルアセテート(Methyl 2,3-dihydrospiro[chromene-4,2'-[1,3]dioxolan]-6-ylacetate)
化合物(7−1−4)(419mg,1.9mmol)、エチレングリコール(0.21mL,3.8mmol)、オルトギ酸メチル(0.42mL,3.8mmol)およびp−トルエンスルホン酸・1水和物(72mg,0.38mmol)のトルエン(8mL)溶液を3時間加熱還流した。室温に冷却後、トルエン(10mL)を加え、混合溶液を飽和重曹水(10mL)、水、飽和食塩水の順に洗浄した。有機層を無水硫酸マグネシウムで乾燥後、溶媒を減圧留去することで表記化合物の粗生成物(502mg)を得た。
【0152】
化合物(7−1−6):
水素化リチウムアルミニウム(72mg,1.9mmol)のテトラヒドロフラン(5mL)懸濁溶液に0℃で化合物(7−3−4)(502mg)のテトラヒドロフラン(3mL)溶液を滴下し、反応混合物をゆっくり室温に昇温させながら1時間攪拌した。ジエチルエーテル(8mL)および水を加え、生じた沈殿をろ別し、ろ液を減圧留去した。得られた濃縮残渣に10%塩酸水(8mL)およびテトラヒドロフラン(8mL)を加えて混合溶液を室温で30分間攪拌した。テトラヒドロフランを減圧留去後、酢酸エチルで抽出した。有機層を飽和食塩水で減圧留去後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン/酢酸エチル)で精製することで表記化合物(254mg,69%)を得た。
【0153】
化合物(RE7):
化合物(7−1−6)を用いて参考例7の製造法1と同様にして化合物(RE7)を得た。
【0154】
参考例8
2−(8−オキソ−5,6,7,8−テトラヒドロナフタレン−2−イル)エチル 4−メチルベンゼンスルホネート(2-(8-Oxo-5,6,7,8-tetrahydronaphthalen-2-yl)ethyl 4-methylbenzenesulfonate,化合物(RE8))
下記の製造方法に従い、合成した。
製造方法
【0155】
【化32】
【0156】
化合物(8−1):7−(2−ヒドロキシエチル)−1,2,3,4−テトラヒドロナフタレン−1−オール(7-(2-Hydroxyethyl)-1,2,3,4-tetrahydronaphthalen-1-ol)
水素化リチウムアルミニウム(33mg、0.86mmol)の無水テトラヒドロフラン(2mL)懸濁溶液に、加熱還流下で文献記載の方法(J. Med. Chem. 1994, 37(21), 3485)によって合成した(8−オキソ−5,6,7,8−テトラヒドロナフタレン−2−イル)アセティック アシッド(50mg、0.25mmol)の無水テトラヒドロフラン(1mL)溶液を加えて、反応混合物を1時間加熱還流した。室温に冷却後、反応溶液を氷浴で冷却し、水(32μL)を加え、引き続いて15%水酸化ナトリウム水溶液(32μL)と水(96μL)を加えて、溶液をそのまま30分間攪拌した。生じた沈殿をろ去し、ろ液を濃縮することで、表記化合物(8−1)(45mg、95%)を得た。
【0157】
化合物(8−2):7−(2−ヒドロキシエチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-Hydroxyethyl)-3,4-dihydronaphthalen-1(2H)-one)
化合物(8−1)(45mg、0.23mmol)と二酸化マンガン(20mg、2.3mmol)のジクロロメタン(2mL)懸濁溶液を室温で5日間攪拌した。二酸化マンガンをろ去し、ろ液を減圧濃縮して得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=2:1)で精製することで、表記化合物(8−2)(28mg、63%)を得た。
【0158】
化合物(RE8):
化合物(8−2)(28mg、0.15mmol)、トリエチルアミン(41μL、0.29mmol)とトリエチルアミン塩酸塩(1.4mg、0.015mmol)のジクロロメタン(2mL)溶液を氷浴で冷却し、p−トルエンスルホニルクロライド(42mg、0.22mmol)を加えて、反応混合物を氷冷下40分間攪拌した。水(20mL)を加えてクロロホルム(40mL×2)で抽出し、有機層を無水硫酸マグネシウムで乾燥した。溶媒を減圧留去し、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=5:1→2:1)で精製することで、目的化合物(RE8)(49mg、96%)を得た。
保持時間(条件2):22.59分
1H-NMR(300 MHz, CDCl3):7.74 (d, J = 2.0 Hz, 1H, Ar), 7.71 (d, J = 8.4 Hz, 2H, Ar), 7.32-7.26(m, 3H, Ar), 7.17 (d, J = 7.7 Hz, 1H, Ar), 4.20 (t, J = 7.0 Hz, 2H, CH2), 3.00-2.90 (m, 4H), 2.64 (5, J = 6.5 Hz, 2H, CH2), 2.44 (s, 3H, CH3), 2.18-2.08 (m, 2H).
【0159】
参考例9
2−(3−オキソ−2,3−ジヒドロ−1H−インデン−5−イル)エチル 4−メチルベンゼンスルホネート(2-(3-Oxo-2,3-dihydro-1H-inden-5-yl)ethyl 4-methylbenzenesulfonate,化合物(RE9))
【0160】
【化33】
【0161】
文献記載の方法(J. Med. Chem. 1979, 22(12),1464)によって製造した(3−オキソ−2,3−ジヒドロ−1H−インデン−5−イル)アセティック アシッドを用いて、参考例8と同様にして、表記化合物(RE9)を製造した。
1H-NMR(300 MHz, CDCl3) δ:7.69 (d, J = 8.5 Hz, 2H, Ar), 7.44 (s, 1H, Ar), 7.41-7.38 (m, 2H, Ar), 7.28 (d, J = 8.5 Hz, 2H, Ar), 4.22 (t, J = 6.8 Hz, 2H, CH2), 3.11 (5, J = 6.0 Hz, 2H, CH2), 3.01 (t, J = 6.8 Hz, 2H, CH2), 2.69 (t, J = 6.0 Hz, 2H, CH2), 2.44 (s, 3H, CH3).
【0162】
参考例10
2−(5−オキソ−2,3,4,5−テトラヒドロ−1−ベンゾキセピン−7−イル)エチル 4−メチルベンゼンスルホネート(2-(5-Oxo-2,3,4,5-tetrahydro-1-benzoxepin-7-yl)ethyl 4-methylbenzenesulfonate,化合物(RE10))
【0163】
【化34】
【0164】
特許文献記載の方法(特開昭61-236774)によって製造した(5−オキソ−2,3,4,5−テトラヒドロ−1−ベンゾキセピン−7−イル)アセティック アシッドを用いて、参考例8と同様にして、表記化合物(RE10)を製造した。
保持時間(条件2):19.93分
1H-NMR (300 MHz, CDCl3) δ: 2.16-2.25 (2H, m), 2.44 (3H, s), 2.81-2.99 (4H, m), 4.13-4.27 (4H, m), 6.98 (1H, d, J = 8.3 Hz), 7.21 (1H, dd, J = 8.3, 2.3 Hz), 7.30 (2H, d, J = 8.1 Hz), 7.48 (1H, d, J = 2.4 Hz), 7.72 (2H, d, J = 8.3 Hz).
【0165】
参考例11
2−(4−オキソ−3,4−ジヒドロ−2H−クロメン−6−イル)エチル ベンゼンスルホネート(2-(4-Oxo-3,4-dihydro-2H-chromen-6-yl)ethyl benzenesulfonate),化合物(RE11))
【化35】
【0166】
化合物(7−1−6)とベンゼンスルホニルクロライドを用いて、参考例7の製造方法1と同様にして表記化合物を得た。
1H-NMR (300 MHz, CDCl3) δ: 2.85 (2H, t, J = 6.2 Hz), 2.90 (2H, t, J = 7.0 Hz), 4.19 (2H, t, J = 7.0 Hz), 4.22 (2H, t, J = 6.2 ), 6.79 (2H, d, J = 8.6 Hz), 7.01 (2H, d, J = 8.6 Hz), 7.45-7.52 (2H, m), 7.57-7.65 (1H, m), 7.75-7.82 (2H, m).
【0167】
実施例1
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
【0168】
【化36】
【0169】
化合物(RE3)(99mg,0.30mmol)、化合物(RE9)(100 mg,0.30mmol)と炭酸カリウム(0.39mmol)のアセトニトリル(3mL)溶液を60℃で27時間攪拌した。反応混合物を室温に冷却後、水(20mL)を加えて酢酸エチル(40mL)で抽出した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた濃縮残渣を薄層シリカゲルクロマトグラフィー(クロロホルム:メタノール=10 : 1)で精製することで、表記化合物(137mg,93%)を淡黄色油状物として得た。
保持時間(条件1):27.44分
1H-NMR (300 MHz, CDCl3) δ: 1.33 (2H, m), 1.44-1.71 (3H, m), 1.97 (2H, bt, J = 9.9 Hz), 2.49 (2H, d, J = 6.8 Hz), 2.58 (2H, bt, J = 8.1 Hz), 2.67-2.73 (2H, m), 2.88 (2H, bt, J = 8.0 Hz), 2.99 (2 H, bd, J = 11.2 Hz), 3.11 (2H, t, J = 5.9 Hz), 3.50 (3H, s), 3.81 (2H, dd, J = 5.3, 4.2 Hz), 4.17 (2H, dd, J = 5.3, 4.2 Hz), 6.64 (1H, dd, J = 8.1, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 7.37-7.47 (3H, m), 7.58 (1H, bs).
【0170】
実施例2
7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
【0171】
【化37】
【0172】
化合物(RE9)の代わりに化合物(RE8)を用いて、実施例1と同様にして、表記化合物を合成した。
保持時間(条件1):31.95分
1H-NMR (400 MHz,CDCl3) δ: 1.26-1.37 (2H, m), 1.46-1.54 (1H, m), 1.64 (2H, bd, J = 12.2 Hz), 1.95 (2H, dt, J = 16.2, 5.8 Hz), 2.09-2.15 (2H, m), 2.49 (2H, d, J = 7.1 Hz), 2.55 (2H, bt, J = 8.4 Hz), 2.64 (2H, t, J = 6.6 Hz), 2.81 (2H, bt, J = 8.4 Hz), 2.93 (2H, t, J = 6.6 Hz), 2.96 (2H, d, J = 12.2 Hz), 3.50 (3H, s), 3.81 (2H, dd, J = 5.5, 4.3 Hz), 4.17 (2H, dd, J = 5.5, 4.3 Hz), 6.64 (1H, dd, J = 8.0, 2.0 Hz), 6.71 (1H, d, J = 2.0 Hz), 7.17 (1H, d, J = 7.8 Hz), 7.32 (1H, dd, J = 7.8, 1.8 Hz), 7.41 (1H, d, J = 8.0 Hz),7.86 (1H, d, J = 1.8 Hz).
【0173】
実施例3
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
【0174】
【化38】
【0175】
化合物(RE3)(52.0g,143mmol)を5%炭酸カリウム水溶液(350mL)に加え、トルエン(700mL×3)で抽出した。全有機層を無水硫酸ナトリウムで乾燥し、溶媒を減圧留去することで、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(48.1g)を得た。次に、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(2.00g,6.1mmol)、化合物(RE7)(2.01g,5.8mmol)および炭酸カリウム(1.66g,12mmol)のアセトニトリル(20mL)溶液を70〜80℃で7時間攪拌した。室温に冷却後、水(100mL)を加えて、クロロホルムで抽出し、クロロホルム層を無水硫酸ナトリウムで乾燥した。溶媒を減圧留去後、得られた濃縮残渣をシリカゲルカラムクロマトグラフィー(n−ヘキサン:酢酸エチル=1:1→クロロホルム:メタノール=20:1)で精製することで、表記化合物(3.07g, 定量的)を得た。
保持時間(条件1):29.11分
1H-NMR (300 MHz,CDCl3) δ: 1.24-1.39 (2H, m), 1.40-1.73 (3H, m), 1.93 (2H, t, J = 10.6 Hz), 2.40-2.61 (2H, m), 2.48 (2H, d, J = 7.2 Hz), 2.66-2.87 (2H, m), 2.79 (2H, t, J = 6.4 Hz), 2.95 (2H, d, J = 11.7 Hz), 3.49 (3H, s), 3.81 (2H, t, J = 4.9 Hz), 4.17 (2H, t, J = 4.9 Hz), 4.51 (2H, t, J = 6.4 Hz), 6.64 (1H, dd, J = 8.1, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 6.89 (1H, d, J = 8.4 Hz), 7.32 (1H, dd, J = 8.4, 2.2 Hz), 7.41 (1H, d, J = 8.1 Hz), 7.70 (1H, d, J = 2.2 Hz).
【0176】
実施例4
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one hydrochloride)
【0177】
【化39】
【0178】
実施例3で得た化合物(3.07g , 5.8mmol)の2−プロパノール(20mL)溶液に室温で濃塩酸水溶液(36%,760μL,8.5mmol)を加え、溶液を室温で15.5時間攪拌した。析出物をろ取し、ろ上物を2−プロパノール(2mL×2)で洗浄し、減圧乾燥することで、表記化合物(2.26g,72%)を白色粉末として得た。
融点:156−157℃
【0179】
実施例5
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one benzenesulfonate)
【0180】
【化40】
【0181】
実施例3で得た化合物(1.00g,2.0mmol)とベンゼンスルホン酸1水和物(316mg,2.0mmol)の2−プロパノール(5mL)溶液を70℃に加温して固形物の溶解を確認後、室温まで4時間で冷却しながら攪拌した。生じた析出物をろ取し、2−プロパノール(1mL×2)で洗浄後、減圧乾燥することで表記化合物の粗生成物(1.13g)を得た。このうちの500mgをアセトン(10mL)と水(100μL)の混合溶液に加え、加温して溶解を確認後、徐々に冷却し、35−40℃で1時間攪拌後、さらに冷却して20−25℃で1時間攪拌した。析出物をろ取し、アセトン(1mL)で洗浄することで、表記化合物(323mg)を得た。
融点:143−144℃
【0182】
実施例1と同様の合成法で、以下に示す実施例6〜20の化合物を製造した。参考例に示した化合物(RE1)〜(RE10)の中から対応する化合物を原料として適宜用いた。
【0183】
実施例6
7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン(7-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydro-1-benzoxepin-5(2H)-one)
保持時間(条件1):30.84分
1H-NMR (300 MHz, CDCl3) δ: 1.25-1.42 (2H, m), 1.44-1.72 (3 H, m), 1.96 (2H, bt, J = 11.0 Hz), 2.15-2.24 (2H, m), 2.49 (2H, d, J = 7.0 Hz), 2.55 (2H, bdd, J = 9.9, 5.7 Hz), 2.79 (2H, bdd, J = 9.9, 5.7 Hz), 2.89 (2H, t, J = 6.9 Hz), 2.99 (2H, bd, J = 11.0 Hz), 3.50 (3H, s), 3.81 (2H, dd, J = 5.3, 4.2 Hz), 4.14-4.25 (4H, m), 6.64 (1H, dd, J = 8.0, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 6.99 (1H, d, J = 8.3 Hz), 7.27 (1H, dd, J = 8.3, 2.5 Hz), 7.41 (1 H, d, J = 8.1 Hz), 7.58 (1.0H, d, J = 2.5 Hz).
【0184】
実施例7
6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[4-Chloro-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):27.43分
1H-NMR (400 MHz,CDCl3) δ: 1.23-1.37 (2H, m), 1.43-1.56 (1H, m), 1.60-1.78 (2H, m), 1.94 (2H, t like, J = 12 Hz), 2.49 (2H, d, J = 7.2 Hz), 2.50-2.55 (2H, m), 2.72-2.80 (2H, m), 2.79 (2H, t, J = 6.4 Hz), 2.96 (2H, d like, J = 11.5 Hz), 3.49 (3H, s), 3.81 (2H, t, J = 4.8 Hz), 4.18 (2H, t, J = 4.8 Hz), 4.51 (2H, t, J = 6.4 Hz), 6.69 (1H, dd, J = 8.0, 1.8 Hz), 6.74 (1H, d, J = 1.8 Hz), 6.89 (1H, d, J = 8.5 Hz), 7.24(1H, d, J = 8.0 Hz), 7.31 (1H, dd, J = 8.5, 2.2 Hz), 7.70 (1H, d, J = 2.2 Hz).
【0185】
実施例8
6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル}−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):25.84分
1H-NMR(300 MHz,CDCl3) δ: 1.20-1.60(3H,m), 1.66(2H,d,J = 13.0Hz), 1.95(2H,t,J=11.7Hz), 2.21(3H,s), 2.49(2H,d,J=7.0Hz), 2.53-2.58(2H,m),
2.69(2H,t,J=5.9Hz), 2.86(2H,t,J=8.6Hz), 2.96(2H,d,J=11.4Hz),
3.10(2H,t,J=6.1Hz), 3.47(3H,s), 3.78(2H,t,J=4.6Hz), 4.12(2H,t,J=5.5Hz), 6.62(1H,s), 6.65(1H,d,J=7.9Hz), 7.03(1H,d,J=7.5Hz), 7.39(1H,d,J=7.9Hz), 7.44(1H,d,J=7.9Hz), 7.58(1H,s).
【0186】
実施例9
7−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
保持時間(条件1):30.36分
1H-NMR(300 MHz,CDCl3) δ: 1.30-1.80(5H,m), 1.99(2H,t,J=11.9Hz), 2.12(2H,quint,J=6.2Hz), 2.21(3H,s), 2.50(2H,d,J=6.6Hz), 2.54-2.70(4H,m), 2.80-2.90(2H,m), 2.93(2H,t,J=5.9Hz), 3.01(2H,d,J=10.8Hz), 3.47(3H,s), 3.78(2H,t,J=4.6Hz), 4.12(2H,t,J=5.5Hz), 6.61(1H,s), 6.65(1H,d,J=7.5Hz), 7.03(1H,d,J=7.5Hz), 7.18(1H,d,J=7.7Hz), 7.33(1H,dd,J=1.5,7.9Hz),
7.86(1H,d,J=1.7Hz).
【0187】
実施例10
6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[3-(2-Methoxyethoxy)-4-methylbenzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):27.01分
1H-NMR (CDCl3) δ: 1.20-1.41 (2H, m), 1.43-1.73 (3H, m), 1.93 (2H, t like, J = 10.9 Hz), 2.20 (3H, s), 2.41-2.62 (2H, m), 2.49 (2H, d, J = 7.1 Hz), 2.67-2.87 (4H, m), 2.95 (2H, d, J = 10.7 Hz), 3.47 (3H, s), 3.77 (2H, t, J = 4.8 Hz), 4.12 (2H, t, J = 4.9 Hz), 4.51 (2H, t, J = 6.3 Hz), 6.61 (1H, s), 6.65 (1H, d, J = 7.6 Hz), 6.89 (1H, d, J = 8.5 Hz), 7.03 (1H, d, J = 7.6 Hz), 7.32 (1H, dd, J = 8.4, 2.3 Hz), 7.70 (1H, d, J = 2.2 Hz).
【0188】
実施例11
6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):17.50分
1H-NMR (400 MHz, CDCl3) δ: 1.38-1.26 (2H, m), 1.46-1.68 (3H, m), 1.96 (2H, bt, J = 11.1 Hz), 2.50 (2H, d, J = 7.1 Hz), 2.57 (2H, bt, J = 7.8 Hz), 2.67-2.71 (2H, m), 2.86 (2H, bt, J = 7.8 Hz), 2.98 (2H, bd, J = 11.1 Hz), 3.10 (2H, bt, J = 5.9 Hz), 3.99 (2H, bt, J = 4.5 Hz), 4.15 (2H, bt, J = 4.5 Hz), 6.67 (1H, dd, J = 8.0, 1.7 Hz), 6.71 (1H, d, J = 1.7 Hz), 7.39 (1H, d, J = 7.8 Hz), 7.42 (1H, d, J = 8.0 Hz), 7.44 (1H, dd, J = 7.8, 1.5 Hz), 7.58 (1H, bs).
【0189】
実施例12
7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
保持時間(条件1):21.91分
1H-NMR (400 MHz,CDCl3) δ: 1.24-1.39 (2H, m), 1.46-1.70 (3H, m), 1.95 (2H, bt, J = 12.2 Hz), 2.09-2.15 (2H, m), 2.50 (2H, d, J = 7.1 Hz), 2.51-2.59 (2H, m), 2.64 (2H, t, J = 6.6 Hz), 2.78-2.83 (2H, m), 2.93 (2H, t, J = 6.6 Hz), 2.96 (2H, d, J = 12.2 Hz), 3.99 (2H, bt, J = 4.4 Hz), 4.15 (2H, t, J = 4.5 Hz), 6.67 (1H, dd, J = 8.0, 2.0 Hz), 6.71 (1H, d, J = 2.0 Hz), 7.17 (1H, d, J = 7.8 Hz), 7.31 (1H, dd, J = 7.8, 2.0 Hz), 7.42 (1H, d, J = 8.0 Hz),7.86 (1H, d, J = 2.0 Hz).
【0190】
実施例13
6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):18.91分
1H NMR (300 MHz, CDCl3) δ: 1.28-1.42 (2H, m), 1.62-1.78 (1H, m), 1.66 (2H, bd, J=12.7 Hz), 2.00 (2H, bt, J=11.6 Hz), 2.50 (2H, d, J=7.0 Hz), 2.58 (2H, dd, J=11.0, 7.7 Hz), 2.76-2.81 (4H, m), 3.01 (2H, bd, J=11.6 Hz), 3.99 (2H, bt, J=4.5 Hz), 4.15 (2H, bd. J=4.5 Hz), 4.51 (2H, t, J=6.8 Hz), 6.66 (1H, dd, J = 8.1, 1.8 Hz), 6.71 (1H, d, J = 1.8 Hz), 6.90 (1H, d, J = 8.5 Hz), 7.33 (1H, dd, J =8.5, 2.4 Hz), 7.42 (1H, d, J = 8.1 Hz), 7.70 (1H, d, J = 2.4 Hz).
【0191】
実施例14
7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン(7-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydro-1-benzoxepin-5(2H)-one)
保持時間(条件1):20.89分
1H-NMR (300 MHz, CDCl3)δ: 1.25-1.45 (2H, m), 1.44-1.70 (3H, m), 1.96 (2H, bt, J = 11.2 Hz), 2.15-2.24 (2H, m), 2.50 (2H, d, J = 6.6 Hz), 2.54 (2H, bdd, J = 9.9, 6.1 Hz), 2.78 (2H, bdd, J = 9.9, 6.1 Hz), 2.89 (2H, t, J = 6.6 Hz), 2.98 (2H, bd, J = 11.2 Hz), 3.99 (2H, bt, J = 4.4 Hz), 4.15 (2H, t, J = 4.4 Hz), 4.21 (2H, t, J = 6.6 Hz), 6.67 (1H, dd, J = 8.1, 2.0 Hz), 6.71 (1H, d, J = 2.0 Hz), 6.99 (1H, d, J = 8.3 Hz), 7.26 (1H, dd, J =8.3, 2.4 Hz), 7.42 (1H, d, J = 8.1 Hz), 7.58 (1H, d, J = 2.4 Hz).
【0192】
実施例15
6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):27.89分
1H-NMR (300 MHz, CDCl3) δ: 1.24-1.42 (2H, m), 1.46-1.70 (3H, m), 1.97 (2H, bt, J = 10.3 Hz), 2.47 (2H, d, J = 7.0 Hz), 2.58 (2H, bt, J = 7.8 Hz), 2.67-2.71 (2H, m), 2.87 (2H, bt, J = 7.8 Hz), 2.98 (2H, bd, J = 10.3 Hz), 3.11 (2H, t, J = 5.9 Hz), 3.45 (3H, s), 3.72-3.75 (2H, m), 4.07-4.10 (2H, m), 6.66 (1H, td, J = 1.9, 0.3 Hz), 6.89-6.92 (2H, m), 7.39 (1H, d, J = 8.0 Hz), 7.45 (1H, dd, J = 8.0, 1.6 Hz), 7.58 (1H, d, J = 1.6 Hz).
【0193】
実施例16
7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン(7-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydronaphthalen-1(2H)-one)
保持時間(条件1):32.39分
1H-NMR (400 MHz,CDCl3) δ: 1.28-1.40 (2H, m), 1.46-1.56 (1H, m), 1.64 (2H, bd, J = 12.6 Hz), 1.98 (2H, bt, J = 11.0 Hz), 2.08-2.15 (2H, m), 2.46 (2H, d, J = 7.0 Hz), 2.52-2.60 (2H, m), 2.64 (2H, t, J = 6.1 Hz), 2.80-2.87 (2H, m), 2.93 (2H, t, J = 6.1 Hz), 2.98 (2H, d, J = 11.0 Hz), 3.45 (3H, s), 3.73 (2H, t, J = 4.6 Hz), 4.08 (2H, t, J = 4.6 Hz), 6.66 (1H, bs), 6.88-6.92 (2H, m), 7.17 (1H, dt, J=7.8 Hz), 7.32 (1H, dd, J=7.8, 1.7 Hz), 7.85 (1H, d, J=1.7 Hz).
【0194】
実施例17
6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):29.33分
1H-NMR (400 MHz, CDCl3) δ: 1.26-1.37 (2H, m), 1.45-1.68 (3H, m), 1.94 (2H, bt, J=11.5 Hz), 2.46 (2H, d, J=7.1 Hz), 2.49-2.55 (2H, m), 2.73-2.81 (4H, m), 2.96 (2H, bd, J=11.5 Hz), 3.45 (3H, s), 3.72-3.75 (2H, m), 4.07-4.11 (2H, m), 4.51 (2H, t, J=6.3 Hz), 6.62 (1H, bt, J=1.8 Hz), 6.88-6.90 (3H, m), 7.32 (1H, dd, J=8.5, 2.3 Hz), 7.70 (1H, d, J=2.3 Hz).
【0195】
実施例18
7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン(7-(2-{4-[3-Bromo-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-3,4-dihydro-1-benzoxepin-5(2H)-one)
保持時間(条件1):31.43分
1H-NMR (300 MHz, CDCl3)δ: 1.25-1.41 (2H, m), 1.44-1.69 (3H, m), 1.96 (2H, bt, J = 11.6 Hz), 2.15-2.24 (2H, m), 2.46 (2H, d, J = 7.0 Hz), 2.54 (2H, bdd, J = 9.9, 6.4 Hz), 2.79 (2H, bdd, J = 9.9, 6.4 Hz), 2.89 (2H, t, J = 7.0 Hz), 2.97 (2H, bd, J = 11.6 Hz), 3.72-3.75 (2H, m), 3.45 (3H, s), 4.07-4.10 (2H, m), 4.21 (2H, t, J = 6.7 Hz), 6.66 (1H, t, J = 1.8 Hz), 6.88-6.92 (2H, m), 6.99 (1H, d, J = 8.3 Hz), 7.27 (1H, dd, J = 8.3, 2.4 Hz), 7.58 (1H, d, J = 2.4 Hz).
【0196】
実施例19
6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(6-(2-{4-[3-Chloro-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-1H-inden-1-one)
保持時間(条件1):26.77分
1H-NMR (300 MHz, CDCl3) δ: 1.25-1.39 (2H, m), 1.47-1.58 (1H, m), 1.65 (2H, bd, J=12.7 Hz), 1.96 (2H, bt, J=11.1 Hz), 2.47 (2H, d, J=7.1 Hz), 2.55-2.59 (2H, m), 2.68-2.71 (2H, m), 2.84-2.88 (2H, m), 2.98 (2H, bd, J=11.5 Hz), 3.10 (2H, t, J=5.7 Hz), 3.45 (3H, s), 3.73-3.75 (2H, m), 4.08-4.10 (2H, m), 6.62 (1H, bt, J=1.8 Hz), 6.73-6.77 (2H, m), 7.39 (1H, d, J=7.8 Hz), 7.44 (1H, dd, J=7.8, 1.7 Hz), 7.58 (1H, bs).
【0197】
実施例20
6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン(6-(2-{4-[3-Chloro-5-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one)
保持時間(条件1):28.33分
1H-NMR (300 MHz, CDCl3) δ: 1.20-1.39 (2H, m), 1.45-1.58 (1H, m), 1.65 (2H, bd, J=13.4 Hz), 1.96 (2H, bt, J=10.6 Hz), 2.47 (2H, d, J=7.0 Hz), 2.50-2.59 (2H, m), 2.73-2.82 (4H, m), 2.98 (2H, bd, J=11.6 Hz), 3.45 (3H, s), 3.72-3.76 (2H, m), 4.07-4.10 (2H, m), 4.51 (2H, t, J=6.4 Hz), 6.62 (1H, bt, J=1.7 Hz), 6.73-6.78 (2H, m), 6.90 (1H, d, J=8.4 Hz), 7.32 (1H, dd, J=8.4, 2.4 Hz), 7.70 (1H, d, J=2.4 Hz).
【0198】
上記実施例6〜20の化合物の構造式を、下表1に示した。
【0199】
【表1】
【0200】
実施例21
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one fumarate)
【0201】
【化41】
【0202】
実施例3で得た化合物(503mg, 1.00mmol)とフマル酸(58mg, 0.50mmol)のエタノール(10mL)溶液を減圧濃縮することで濃縮残渣(557mg)を得た。このうちの100mgをアセトン(3mL)に加え、室温で1時間攪拌後、析出物をろ取し、減圧乾燥することで表記化合物(42mg)を得た。
融点:149−150℃
【0203】
実施例22
6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩(6-(2-{4-[4-Bromo-3-(2-hydroxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one hydrochloride)
【0204】
【化42】
【0205】
実施例13で得た化合物(3.96g,8.1mmol)の2−プロパノール(40mL)とジクロロメタン(50mL)の混合溶液に、室温で濃塩酸水溶液(36%,860μL,10mmol)を加え、溶媒を減圧濃縮した。得られた濃縮残渣(3.61g)に2−プロパノール(72mL)を加えて2時間加熱還流し、3時間で20℃まで冷却しながら攪拌し、20℃で1時間攪拌後、氷冷しながら1時間攪拌した。析出物をろ取し、冷2−プロパノール(4mL×2)で洗浄することで白色固体(3.35g)を得た。これに2−プロパノール(100mL)と濃塩酸水溶液(36%,500μL)を加えて還流温度まで加温し、固形物の溶解を確認後、60℃まで冷却し、固体の析出を確認後、55〜60℃で1時間攪拌した。ひきつづいて2時間で20℃まで冷却しながら攪拌し、20℃で1時間攪拌した。析出物をろ取し、2−プロパノール(4mL×2)で洗浄することで表記化合物(3.10g,73%)を白色粉末として得た。
融点:177−178℃
【0206】
実施例23
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 臭化水素酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one hydrobromide)
【化43】
【0207】
実施例3で得た化合物(1.00 g, 2.00 mmol)の2−プロパノール(10mL)溶液に氷冷下48%臭化水素酸水(170μL, 2.4mmol)を加え、溶液を室温に昇温して1夜間攪拌した。生じた析出物をろ取し、2−プロパノール(1mL×2)で洗浄後、減圧乾燥することで表記化合物の粗結晶(797mg)を得た。粗結晶750mgを、2−プロパノール(30mL)に加え、加熱還流して固形物の溶解を確認後、室温までゆっくり冷却しながら19時間攪拌した。生じた析出物をろ取し、2−プロパノール(1mL×2)で洗浄後、減圧乾燥することで表記化合物(613mg)を白色結晶性固体として得た。
融点:148−150℃
【0208】
実施例24
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1コハク酸塩 6-(2-{4-[4-bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one butanedioate
【化44】
【0209】
実施例3で得た化合物(218 mg, 0.43 mmol)とコハク酸(26mg,0.22mmol)をエタノール(10mL)に加え、溶解を確認後、溶媒を減圧留去して濃縮残渣244mgを得た。濃縮残渣105mgを2−ブタノン/n−ヘキサン(1:1,1mL)中で1時間攪拌し、生じた析出物をろ取して室温で減圧乾燥することで表記化合物(36mg,62%)を白色粉末として得た。
融点:100−101℃
1H-NMR (DMSO-d6) δ: 1.10-1.25 (2H, m), 1.45-1.60 (3H, m), 1.94-2.07 (2H, m), 2.39 (4H, s), 2.47 (2H, d, J = 6.6 Hz), 2.53-2.58 (2H, m), 2.68-2.75 (2H, m), 2.76 (2H, t, J = 6.5 Hz), 2.92-3.01 (2H, m), 3.34 (3H, s), 3.68 (2H, t, J = 4.5 Hz), 4.16 (2H, t, J = 4.5 Hz), 4.50 (2H, t, J = 6.3 Hz), 6.69 (1H, dd, J = 8.0, 1.5 Hz), 6.94 (1H, d, J = 1.5 Hz), 6.95 (1H, d, J = 8.5 Hz), 7.42 (1H, dd, J = 8.5, 2.4 Hz), 7.44 (1H, d, J = 8.0 Hz), 7.58 (1H, d, J = 2.4 Hz).
【0210】
実施例25
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one benzenesulfonate)
化合物(RE3)(10.0g,27.4mmol)を水酸化カリウム水溶液(水酸化カリウム[2.15g,32.9mmol]と 水[50mL]から調製)に加え、トルエン(50mL×2)で抽出した。全有機層をあわせ、溶媒を減圧留去することで得た濃縮残渣1に、化合物(RE11)(9.57g,28.8mmol)、リン酸水素二カリウム(14.3g,82.3mmol)、N−メチル−2−ピロリジノン(3.0mL)、トルエン(100mL)を加え110〜120℃で6.5時間撹拌した。40〜50℃に冷却後、水(100mL)、テトラヒドロフラン(50mL)、トルエン(50mL)を加え、有機層を水(50mL)で洗浄した。溶媒を減圧留去して得た濃縮残渣2を精製することなく分割して次反応に用いた。濃縮残渣2の1/5量(5.5mmol相当)のアセトン(8mL)溶液をベンゼンスルホン酸アンモニウム(0.96g,5.48mmol)のアセトン(32mL)懸濁液に40〜50℃で加えた。50℃で1時間撹拌して固形物が全て溶解したことを確認後、溶媒を減圧留去した。濃縮残渣にアセトン(20mL)を加え、50℃に加熱して溶解後、40℃で種晶(5.0mg)を加えた。40℃で1時間撹拌してゆっくり室温に冷却後、氷浴で3時間撹拌し、析出物をろ取した。ろ上物を冷アセトン(4.0mL×2)で洗浄し、減圧乾燥することで、表記化合物(2.85g,79%)を得た。
【0211】
実施例26
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(6-(2-{4-[4-Bromo-3-(2-methoxyethoxy)benzyl]piperidin-1-yl}ethyl)-2,3-dihydro-4H-chromen-4-one benzenesulfonate)
化合物(RE3)(30.0 g, 82.3mmol)、化合物(RE7)(29.9g, 86,4 mmol)および炭酸カリウム(34.11 g, 247mmol)のアセトニトリル(236g)懸濁溶液を6.5時間加熱還流した。反応混合物を室温に冷却後、トルエン(519g)と水(600g)を加えて分液し、有機層を水(300g)で洗浄後、溶媒を減圧留去することで濃縮残渣(44.6g)を得た。濃縮残渣にアセトン(170g)を加えてろ過し、ろ上物をアセトン(40g)で洗浄した。全ろ液に水(3g)を加えて、40℃に加温し、ベンゼンスルホン酸1水和物(15.2g,96mmol)を加え、種晶(0.15g)を加えて40℃で1時間攪拌した。引き続き2℃までゆっくり冷やしながら攪拌し、内温2℃で1時間攪拌後、析出物をろ取し、冷アセトン(47.5g×2)で洗浄した。ろ上物を40℃で減圧乾燥することにより表記化合物(36.8g,68%)を白色結晶性固体として得た。
【0212】
実施例27
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩のForm1型結晶
化合物(RE3)(52.0g,143mmol)を5%炭酸カリウム水溶液(350mL)に加え、トルエン(700mL×3)で抽出した。全有機層を無水硫酸ナトリウムで乾燥し、溶媒を減圧留去することで4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(48.1g)を得た。これを化合物(RE7)(47.0g,143mmol)、炭酸カリウム(37.5 g,271mmol)とともにアセトニトリル(470mL)に加え、反応混合物を内温55−60℃で25時間攪拌した。室温に冷却後、水(940mL)、トルエン(940mL)を加えて分液し、水層をトルエン(470mL)で再抽出した。全有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた濃縮残渣(76.6g)に2−プロパノール(1.1L)を加え、水冷しながら15−20℃で濃塩酸水(36%,14mL,171mmol)を5分間で滴下し、溶液を2時間15−20℃で22時間攪拌後、氷浴で冷却して4.5時間攪拌した。析出物をろ取し、冷却した2−プロパノール(55mL×2)で洗浄し、減圧乾燥することで濃縮残渣(61.6g)を得た。これに2−プロパノール(900mL)、36%濃塩酸水(36%,9.0mL)を加えて加温し、60℃付近ですべて溶解を確認後、溶液を20℃まで冷却して、15−20℃で14.5時間攪拌後、氷浴で冷却して5時間攪拌した。析出物をろ取し、冷2−プロパノール(70mL×2)で洗浄し、減圧乾燥することで表記化合物(51.1 g,70%)を無色結晶性固体として得た。
融点 157−159℃
元素分析
【0213】
【表2】
【0214】
実施例28
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormA型結晶
実施例3で得た化合物(1.00 g, 2.00 mmol)とベンゼンスルホン酸(316mg,2.0mmol)の2−プロパノール(5mL)溶液を70℃に加温して固形物の溶解を確認後,室温まで4時間で冷却しながら攪拌した。生じた析出物をろ取し,2−プロパノール(1mL×2)で洗浄後,減圧乾燥することで濃縮残渣(1.13 g)を得た。このうちの1.05gを2−プロパノール(32mL)に加えて,混合溶液を加熱還流して固形物が溶解したことを確認後,冷却しながら攪拌した。65℃で種晶(2mg)を加えて60−65℃で1時間攪拌した。引き続いて2時間で30℃まで冷却しながら攪拌し,水冷(25℃)で1時間攪拌後,析出物をろ取し,2−プロパノール(2mL×2)で洗浄後,減圧乾燥することで表記化合物(963mg)を無色結晶性固体として得た。
融点:142−143℃
元素分析
【0215】
【表3】
【0216】
実施例29
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormB型結晶
化合物(RE3)(2.00g,5.5mmol)を10%炭酸カリウム水溶液(20mL)に加え、トルエン(20mL×2)で抽出した。全有機層を溶媒を減圧留去することで、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(1.95g)を得た。次に、4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン(1.95g,5.5mmol)、化合物(RE7)(1.80g,5.2mmol)および炭酸カリウム(1.44g,10mmol)のアセトニトリル(10mL)溶液を55−60℃で19時間攪拌した。反応溶液を室温に冷却後、水(30mL)を加えて、トルエン(30mL×3)で抽出し、全有機層を5%重曹水(30mL)、水(30mL)の順で洗浄後、溶媒を減圧留去した。得られた濃縮残渣をアセトン(40mL)に溶解させてベンゼンスルホン酸1水和物(1.06g,6.0mmol)を加えて、固形物の溶解を確認後、溶媒を減圧留去した。得られた濃縮残渣にアセトン(40mL)および水(0.4mL)を加えて40℃に加温し、種晶(5mg)を加えて35−40℃で1.5時間攪拌した。引き続き1時間で20℃まで冷却後、1時間氷冷下攪拌した。析出物をろ取し、冷アセトン(5mL×2)で洗浄後、室温で減圧乾燥することで表記化合物(2.45g,71%)を無色結晶性固体として得た。
融点:143−144℃
元素分析
【0217】
【表4】
【0218】
実施例30
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormC型結晶
実施例3で得た化合物(7.76g,15.5mmol)を1%(v/v)含水アセトン(100mL)に溶解させ,溶液を40−45℃に加温してからベンゼンスルホン酸1水和物(2.93g、17mmol)を加えて攪拌した。固形物の溶解を確認後、種晶(50mg)を加えて1時間40−45℃で攪拌した。引き続き3時間で25℃まで冷却しながら攪拌し、その後室温で17.5時間攪拌した。引き続き1時間で5℃以下に冷却して、そのまま1時間攪拌し、析出物をろ取して、ろ上物を冷アセトン(10mL×2)で洗浄後、減圧乾燥することで表記化合物(8.83g,88%)を無色結晶性固体として得た。
融点:143−145℃(128℃でFormAに転移)
1H-NMR (400 MHz,DMSO-d6) δ: 1.30-1.46 (2H, m), 1.72-1.86 (3H, m), 2.52 (2H, d, J = 6.4 Hz), 2.78 (2H, t, J = 6.5 Hz), 2.82-3.00 (4H, m), 3.18-3.30 (2H, m), 3.34 (3H, s), 3.48-3.58 (2H, m), 3.66-3.72 (2H, m), 4.12-4.20 (2H, m), 4.51 (2H, t, J = 6.5 Hz), 6.72 (1H, dd, J = 8.1, 2.0 Hz), 6.96 (1H, d, J = 2.0 Hz), 7.01 (1H, d, J = 8.5 Hz), 7.27-7.34 (3H, m), 7.44 (1H, dd, J = 8.5, 2.2 Hz), 7.47 (1H, d, J = 8.1 Hz), 7.56-7.61 (2H, m), 7.67 (1H, d, J = 2.2 Hz), 9.02 (1H, br s).
元素分析
【0219】
【表5】
【0220】
実施例31
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA型結晶
実施例3で得た化合物(3.0g, 6.0mmol)をエタノール(75mL)に溶解させ、フマル酸(693mg, 6.0mmol)のエタノール(25mL)溶液を加え、さらにメタノール(100mL)を加えて溶解を確認後、溶媒を減圧留去して濃縮残渣を得た。これにアセトン(110mL)を加えて室温で2.5時間攪拌し、生じた析出物をろ取し、減圧乾燥(40℃、1mmHg、18時間)することで表記化合物(3.26g,88%)を無色結晶性固体として得た。
融点:149−151℃
1H-NMR (400 MHz,DMSO-d6) δ: 1.17-1.40 (2H, m), 1.47-1.73 (3.0H, m), 2.15-2.32 (2H, m), 2.48 (2H, d, J = 6.4 Hz), 2.63-2.85 (6H, m), 3.05-3.17 (2H, m), 3.34 (3H, s), 3.65-3.72 (2H, m), 4.13-4.20 (2H, m), 4.50 (2H, t, J = 6.4 Hz), 6.56 (2H, s), 6.70 (1H, dd, J = 8.0, 1.7 Hz), 6.95 (2H, d, J = 1.7 Hz), 6.97 (2H, d, J = 8.6 Hz), 7.42 (2H, dd, J = 8.6, 2.2 Hz), 7.45 (1H, d, J = 8.0 Hz), 7.60 (1H, d, J = 2.2 Hz).
【0221】
実施例32
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA+型結晶
実施例31で得たFormA型結晶(500mg)を1%含水アセトン(25mL)に加え65℃に加温して溶解を確認後、ゆっくり室温まで冷却しながら17時間攪拌した。生じた析出物をろ取し減圧乾燥(40℃、1mmHg、24時間)することで表記化合物(403mg,80%)を無色結晶性固体として得た。
融点:148−149℃
1H-NMR:実施例31と同じデータが得られた。
【0222】
実施例33
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩
4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン塩酸塩(2.0g,5.48mmol)を水酸化カリウム水溶液(水酸化カリウム[0.43g,6.58mmol]と 水[10ml]から調製)に加え、トルエン(10ml×2)で抽出した。全有機層をあわせ、溶媒を減圧留去した。濃縮残渣に、6−(2−p−ベンゼンスルホニルオキシエチル)−2,3−ジヒドロ−4H−クロメン−4−オン(1.91g,5.76mmol)、リン酸水素二カリウム(2.87g,16.5mmol)、N−メチル−2−ピペリドン(0.6ml)、トルエン(20ml)を加え110〜120℃で6時間攪拌した。40〜50℃に冷却後、水(20ml)、テトラヒドロフラン(10ml)、トルエン(10ml)を加え、有機層を水(10ml)で洗浄した。溶媒を減圧留去することで濃縮残渣を得た。得られた濃縮残渣を精製することなくアセトン(6.0ml)に溶解させて、50〜60℃でベンゼンスルホン酸(0.93 g,5.76mmol)のアセトン(4.0ml)溶液を滴下した。50〜60℃で30分攪拌後、酢酸n−ブチル(20.0ml)を滴下し、さらに1時間攪拌した。室温に冷却後、酢酸n−ブチル(10ml)を滴下し、氷浴で3.5時間攪拌した。析出物をろ取し、ろ上物を冷アセトン(4.0ml×2)で洗浄、減圧乾燥することで、表記化合物(3.21g,89%)を得た。
【0223】
実施例34
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormC型結晶
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(1.0g,1.51mmol)に0.5%含水アセトン(15.2mL)を加え、50℃以上で溶解した。溶液を40〜50℃に冷却し、種晶(実施例30で得られたFormC型結晶)(5mg)を加え、40〜50℃で1時間撹拌した。室温に冷却し、室温で1時間撹拌後、n−ヘプタン(8.8 mL)を加えた。氷浴で3時間撹拌後、析出物をろ取し、アセトン(2.5 mL)で洗浄後、50℃で減圧乾燥することで標記化合物(0.92g,92%)をFormC型結晶として得た。
【0224】
試験例1
ヒトセロトニン再取り込み阻害作用を評価するための[3H]シタロプラム結合 を用いたスクリーニング試験
【0225】
1−1 使用細胞および膜標品の調製
実験にはヒトセロトニントランスポーター(h−SERT)を発現させたCHO細胞(h−SERT/CHO)を用いた。細胞は5%CO2インキュベーター中で、10%FCS、500μg/ml Geneticinおよび100U/ml ペニシリン−100μg/ml ストレプトマイシンを含むF12(すべてSigma Aldrich製)にて培養し、SERT バッファー(120mM NaClおよび5mM KClを含む50mM Tris−HCl(pH=7.4))にて剥離・採取した細胞をテフロン(登録商標)製ホモジナイザーでホモジナイズした後、遠心操作(50,000xg、30min、4℃)を行なった。沈渣は適量のSERT バッファーに再懸濁し、使用まで−80℃で保存した。膜標品中のタンパク質量は、標準物質にウシ血清アルブミン(Sigma Aldrich製)を用いて、Dye Reagent Concentrate(BIO−RAD製)により定量した。
【0226】
1−2 受容体結合実験
[3H]シタロプラム結合の測定はOwensらの方法[Owens M. J. et al., J. Pharm. Exp. Ther., 283, 1305-1322(1997)]に準じて行った。すなわち、SERTバッファーで希釈した[3H]シタロプラム(最終濃度約2nM)50μl、h−SERT/CHO膜標品(蛋白量として40μg/well)149μl、およびジメチルスルホキシドに溶解した被験薬溶液1μlを加え全量を200μlとした。この液を室温で60分間反応させた後、0.05%ポリエチレンイミン水溶液でコーティングしたガラス繊維濾紙を用い速やかに低圧吸引ろ過した。ガラス繊維濾紙をSERT バッファー250μlで2回洗浄した後、ACS−II(Amersham製)4ml入りのガラスバイアルに移し、濾紙上に残存する放射活性を液体シンチレーションカウンターを用いて測定した。[3H]シタロプラムの非特異的結合は1μM クロミプラミン存在下での結合量とした。
IC50値をHill解析[Hill A. V., J. Physiol., 40, 190-200 (1910)参照]により算出し、h−SERT結合阻害定数(Ki)の算出は、式:
h−SERT結合阻害定数(Ki)=IC50/(1+S/Kd)
[Sは添加した[3H]シタロプラム濃度を示す。また、Kd値は[3H]シタロプラムの結合解離定数であり、別途同じ細胞膜を用いて実施した飽和結合実験より算出された値(2.16nM)を用いた。]
によって算出した。h−SERT結合阻害定数Ki値の数値が小さいほど、ヒトセロトニン再取り込み阻害作用が高いことを意味する。
【0227】
試験例2
ヒトセロトニン1A受容体に対する親和性を評価するための[3H]8−OH−DPAT結合試験
【0228】
2−1 使用細胞および膜標品の調製
実験にはヒトセロトニン1A受容体(h−5−HT1A)を発現させたCHO細胞(h−5−HT1A/CHO)を用いた。細胞は5%CO2インキュベーター中で、10%FCS、500μg/ml Geneticinおよび100U/ml ペニシリン−100μg/ml ストレプトマイシンを含むF12(すべてSigma Aldrich製)にて培養し、膜標品はYabuuchiらの方法3)に従って調製した。すなわち、50mM Tris−HCl(pH=7.4)にて剥離・採取した細胞を、テフロン(登録商標)製ホモジナイザーでホモジナイズした後、遠心操作(48,000xg、20min、4℃)を行なった。沈渣は適量の50mM Tris−HCl(pH=7.4)に再懸濁し、使用まで−80℃で保存した。膜標品中のタンパク質量は、標準物質にウシ血清アルブミン(Sigma Aldrich製)を用いて、Dye Reagent Concentrate(BIO−RAD製)により定量した。
【0229】
2−2 受容体結合実験
実験はYabuuchiらの方法[Yabuuchi K. et al., Biogenic Amines, 18, 319-328 (2004)]に準じて実施した。50mM Tris−HCl(pH=7.4)、4mM CaCl2を含む緩衝液中に、[3H]8−OH−DPAT(最終濃度0.5nM)を50μl、被検薬溶液を1μl、h−5−HT1A/CHO膜標品(蛋白質量として25μg/well)149μlを加え、全量200μlの反応液を用いて測定した。反応液を室温で30分間反応させた後、ガラス繊維濾紙上に速やかに低圧吸引濾過した。ガラス繊維濾紙を、50mM Tris−HCl(pH=7.4)250μlで2回洗浄した後、ACS−II(Amersham社製)4ml入りのカウンティングバイヤルに添加し、濾紙上に残存した受容体結合放射活性を液体シンチレーションカウンターで測定した。非特異的結合は10μM 8−OH−DPAT存在下での結合量とした。
IC50値をHill解析[Hill A. V., J. Physiol., 40, 190-200 (1910)参照]により算出し、h−5−HT1A結合阻害定数(Ki)の算出は、式:
h−5−HT1A結合阻害定数(Ki)=IC50/(1+S/Kd)
[Sは添加した[3H]8−OH−DPAT濃度を示す。また、Kd値は[3H]8−OH−DPATの結合解離定数であり、別途同じ細胞膜を用いて実施した飽和結合実験より算出された値(1.28nM)を用いた。]
によって算出した。h−5−HT1A結合阻害定数Ki値の数値が小さいほど、ヒトセロトニン1A受容体に対する親和性が高いことを意味する。
【0230】
実施例で得た本発明のベンジルピペリジン化合物について、上記の試験例1および2の試験を行い、結果を表6に示した。これらの試験結果から、本発明のベンジルピペリジン化合物またはその薬学上許容される塩が、ヒトセロトニン再取り込み阻害作用とヒト5−HT1A受容体に対する結合親和性を併せ持つだけでなく、ヒトセロトニン再取り込み阻害作用が高いことが明らかとなった。
【0231】
【表6】
【0232】
試験例3
CYP2D6阻害スクリーニング試験
【0233】
3−1 材料
ジクロフェナックはSigma社より、Pooled of Human Liver MicrosomesはXenotech,LLCより購入した。
【0234】
3−2−1 0.5Mリン酸カリウムBuffer(pH 7.4)の調製
0.5Mリン酸一カリウム溶液 150mLと0.5M リン酸二カリウム溶液 700mLを混合してpH7.4に調整した。
【0235】
3−2−2 165mM塩化マグネシウム溶液の調製
塩化マグネシウム六水和物、MgCl2・6H2O 3.35g/100mLになるよう純水に溶解した。
【0236】
3−2−3 ヒト肝ミクロソーム溶液の調製
Pooled of Human Liver Microsomes(20mg/ml)150μL、0.5M リン酸カリウムバッファー12mL、165 mM 塩化マグネシウム溶液1.2mLおよび純水34.65mLを混合して調製した。
【0237】
3−2−4 13mMβ−NADPH溶液の調製
β−NADPHを11.75mg/mLになるよう純水に溶解し調製した。
【0238】
3−2−5 基質溶液の調製
ジクロフェナックを2.0mMとなるようDMSOで溶解した後、純水で200倍に希釈した。
【0239】
3−3 実験方法
1. 被検薬物の10mM DMSO溶液をDMSOで5倍ずつ4段階希釈し、10、2、0.4、0.08mMのDMSO溶液を調製した。
2. 1.の被検薬物溶液およびDMSOをヒト肝ミクロソーム溶液で96倍に希釈し、80μLずつマイクロプレートに分注した。
3. 2.に基質溶液10μLおよびβ−NADPH溶液10μLを添加し、37℃で10minインキュベートした。
4. メタノール300μLを添加し、反応を停止させた。
5. 反応混合物をフィルターろ過し、LC−MSMSにて分析を行った。
【0240】
3−4 定量および計算
LC−MSMSにて4−ヒドロキシジクロフェナックの生成量を定量し、これを各ウェルにおけるCYP2D6の活性値とした。被検薬物としてDMSOを用いたウェルの活性と比較し、各サンプル添加群の残存活性を求め、被検薬物濃度よりCYP2D6阻害のIC50値を求めた。IC50値は残存活性50%をはさむ2点を結ぶ直線より算出した。CYP2D6阻害のIC50値の数値が大きいほど、CYP2D6阻害が弱いことを意味する。
【0241】
試験例4
ヒト肝ミクロソーム代謝におけるCYP2D6寄与率スクリーニング試験
【0242】
終濃度として3mMのNADPH(オリエンタル酵母工業製)、1mg/mLのヒト肝ミクロソーム(XENOTECH,LLC製)および1μMの被験物質を含む0.2mLの50mMリン酸カリウム緩衝液(pH7.4)を37℃の水浴上で加温することにより代謝反応を行った。15分または30分反応した後、反応液の3倍容のメタノールを添加して攪拌することにより反応を停止させた。この反応液を遠心分離して蛋白を沈殿させた後、上清を採取しLC−MS/MS分析に供した。結果の解析は以下の通り実施した。
被験物質の定量を行い、その残存量の経時変化を対数プロットしその傾きより代謝速度を算出した。
反応溶液に終濃度4μMのキニジンを添加しなかった場合の代謝速度に対する添加した場合の代謝速度の割合をCYP2D6以外の酵素の寄与率とし、その残りをCYP2D6の寄与率とした。すなわち、式:
寄与率(%)={1−(代謝速度[キニジン添加有]/代謝速度[キニジン添加なし])}×100
によって算出した。CYP2D6の寄与率の数値が小さいほど、CYP2D6の寄与が小さいことを意味する。
【0243】
実施例で得た本発明のベンジルピペリジン化合物について、試験例3および4の試験を行い、結果を表7に示した。これらの試験結果から、本発明のベンジルピペリジン化合物またはその薬学上許容される塩については、CYP2D6阻害が弱く、また代謝におけるCYP2D6の寄与が小さいことが明らかとなった。
【0244】
【表7】
*)ヒトミクロソーム代謝に対して安定であるため、
本試験ではCYP2D6の寄与率は判定不能であった。
【0245】
試験例5
[3H]5−HTを用いたセロトニントランスポーター機能阻害試験
1−1 使用細胞および細胞懸濁液の調製
実験にはヒトセロトニントランスポーター(h−SERT)を発現させたCHO細胞(h−SERT/CHO)を用いた。細胞は5%CO2インキュベーター中で、10% FCS、500μg/ml Geneticinおよび100U/ml ペニシリン−100μg/ml ストレプトマイシンを含むF12(すべてSigma Aldrich製)にて培養し、使用当日にCell Dissociation Buffer(Enzyme−free,PBS−based,GIBCO製)にて剥離・採取した。採取した細胞はPBS,0.1mM CaCl2および1mM MgCl2を含む緩衝液(以下PBSCM)に懸濁し、使用まで氷中にて保存した。
【0246】
1−2[3H]5−HT取り込み試験
[3H]5−HT取込の測定はRomanらの方法[Roman D.L. et al., J. Pharmacol. Exp. Ther., 308, 679-687 (2004)、Hill A. V., J. Physiol., 40, 190-200 (1910)]に準じて行った。すなわち、細胞懸濁液149μl(10×104cells/well)およびジメチルスルホキシドに溶解した被験薬溶液1μlを37℃で10分間プレインキュベーション後、PBSCMで希釈した[3H]5−HT(最終濃度約10nM)50μlを加え全量を200μlとした。この液を37℃で10分間反応させた後、0.3%ポリエチレンイミン水溶液でコーティングしたガラス繊維濾紙を用い速やかに低圧吸引ろ過した。ガラス繊維濾紙を氷冷整理食塩水250μlで2回洗浄した後、ACS−II(Amersham製)4ml入りのガラスバイアルに移し、濾紙上に残存する放射活性を液体シンチレーションカウンターを用いて測定した。[3H]5−HTの非特異的取込は13.3μM パロキセチン存在下での取込量とした。
IC50値の算出はHill解析[Hill A. V., J. Physiol., 40, 190-200 (1910)参照]により、取込阻害定数(Ki)の算出は次式によって算出した:
取込阻害定数(Ki)=IC50/(1+S/Km)
ただし、Sは添加した[3H]5−HT濃度を示す。また、Km値は[3H]5−HTのミカエリス定数であり、別途同じ細胞標品を用いて実施した飽和実験より算出された値(441nM)を用いた。得られた結果を、表8に示した。
【0247】
【表8】
【0248】
表8の試験結果から、本発明のベンジルピペリジン化合物またはその薬学上許容される塩については、セロトニンニューロンのセロトニントランスポーターに作用し、セロトニン再取り込みを強く阻害するすることが明らかとなった。
【0249】
試験例6
粉末エックス線回折(XRPD)分析
XRPD粉末エックス線回折装置としては、スペクトリス社製 X’Pert Pro(エキスパート プロ)を用いた。回折角度2シータ(2θ)4度〜40度の範囲でCu Kα1線(波長1.54060オングストローム)、X線管電流 40ミリアンペア、電圧45キロボルト、ステップ0.01700度、測定時間101.41770秒/ステップの条件にて測定した。測定はシリコン単結晶(Si単結晶)からなる無反射試料板にサンプル量約5ミリグラムを用いて測定を行った。
実施例27〜32で得られた各結晶について、本分析を行った。得られた粉末エックス線回折パターンを図1〜6に示す。結晶形の特定には、回折パターンを基に、各結晶の特徴となる回折ピークにより判断することができる。各結晶について、回折パターンから特定した特徴的な回折ピークは後述する。なお、回折角度2θにおけるピーク角度は、測定機器により、もしくは測定条件等により多少の誤差範囲を生じることがある。具体的には、測定誤差は±0.2、好ましくは±0.1の範囲であってよい。一方、相対強度については、試料調整の具合や測定条件等により、その値は変化しうる。
実施例27の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩のForm1型結晶の粉末エックス線回折のピーク角度、面間隔(d−spacing)およびその相対強度を表9に示す。
【0250】
【表9】
【0251】
表9に示したピークのうち、本塩酸塩のForm1型結晶の特徴的な回折ピークとしては、回折角度2θが、4.3゜、8.7゜、10.8゜、12.6゜、13.0゜、16.0゜、17.1゜、19.3゜、19.7゜、20.2゜であるピークが挙げられる。
【0252】
実施例28の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormA型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表10に示す。
【0253】
【表10】
【0254】
表10に示したピークのうち、本ベンゼンスルホン酸塩のFormA型結晶の特徴的な回折ピークとしては、回折角度2θが、8.7゜、11.9゜、13.0゜、14.9゜、15.3゜、17.4゜、17.7゜、18.2゜、19.3゜、20.4゜であるピークが挙げられる。
【0255】
実施例29の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormB型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表11に示す。
【0256】
【表11】
【0257】
表11に示したピークのうち、本ベンゼンスルホン酸塩のFormB型結晶の特徴的な回折ピークとしては、回折角度2θが、8.9゜、10.0゜、12.0゜、12.5゜、14.0゜、16.9゜、17.0゜、17.6゜、18.9゜、20.6゜であるピークが挙げられる。
【0258】
実施例30の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormC型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表12に示す。
【0259】
【表12−1】
【表12−2】
【0260】
表12に示したピークのうち、本ベンゼンスルホン酸塩のFormC型結晶の特徴的な回折ピークとしては、回折角度2θが、8.6゜、9.9゜、12.6゜、13.0゜、14.4゜、14.6゜、18.0゜、18.4゜、19.6゜、19.8゜であるピークが挙げられる。
【0261】
実施例31の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表13に示す。
【0262】
【表13】
【0263】
表13に示したピークのうち、本フマル酸塩のFormA型結晶の特徴的な回折ピークとしては、回折角度2θが、7.6゜、10.0゜、11.5゜、11.8゜、14.6゜、15.3゜、16.0゜、17.4゜、18.3゜、19.6゜、20.9゜、22.0゜、23.2゜であるピークが挙げられる。
【0264】
実施例32の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のFormA+型結晶の粉末エックス線回折のピーク角度、面間隔(d-spacing)およびその相対強度を表14に示す。
【0265】
【表14】
【0266】
表14に示したピークのうち、本フマル酸塩のForm A+型結晶の特徴的な回折ピークとしては、回折角度2θが、7.4゜、10.1゜、11.1゜、14.7゜、16.0゜、18.4゜、19.8゜、22.2゜、23.4゜、25.9゜、26.7゜であるピークが挙げられる。
【0267】
製剤例1
錠剤の製造
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン(5g)、乳糖(80g)、コーンスターチ(30g)、結晶セルロース(25g)、ヒドロキシプロピルセルロース(3g)、軽質無水ケイ酸(0.7g)及びステアリン酸マグネシウム(1.3g)を、常法により混合、造粒し、1錠あたり145mgで打錠し、1000錠を製する。
【0268】
製剤例2
散剤の製造
6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩(10g)、乳糖(960g)、ヒドロキシプロピルセルロース(25g)及び軽質無水ケイ酸(5g)を、常法により混合した後、散剤に製する。
【産業上の利用可能性】
【0269】
式(1)で表される本発明のベンジルピペリジン化合物及びその薬学上許容される塩は、ベンゼン環部分の3位が2−メトキシエトキシ基または2−ヒドロキシエトキシ基で置換されているジ置換ベンジル基を有し、且つオキソ基を持つ飽和環がベンゼン環部分で縮合したフェニルエチル基をピペリジンの1位に有する、という化学構造上の特徴を有し、セロトニン1A受容体に対する親和性を併せ持ち、ヒトセロトニン再取り込み阻害活性が向上し、ヒトチトクロームP450分子種の一つであるCYP2D6に対する阻害作用が弱く、もしくはヒトにおける薬物代謝においてCYP2D6の寄与が小さな、新しいセロトニン再取り込み阻害剤であることから、例えばうつ病や不安(不安障害)などの疾患に対し、治療効果に優れ安全性の高い治療薬または予防薬として使用しうる。
【図面の簡単な説明】
【0270】
【図1】試験例6の粉末エックス線回折分析で得られた、実施例27の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 塩酸塩のForm1型結晶の粉末エックス線回折パターンである。
【図2】試験例6の粉末エックス線回折分析で得られた、実施例28の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormA型結晶の粉末エックス線回折パターンである。
【図3】試験例6の粉末エックス線回折分析で得られた、実施例29の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のFormB型結晶の粉末エックス線回折パターンである。
【図4】試験例6の粉末エックス線回折分析で得られた、実施例30の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン ベンゼンスルホン酸塩のForm C型結晶の粉末エックス線回折パターンである。
【図5】試験例6の粉末エックス線回折分析で得られた、実施例31の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のForm A型結晶の粉末エックス線回折パターンである。
【図6】試験例6の粉末エックス線回折分析で得られた、実施例32の6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン 1フマル酸塩のForm A+型結晶の粉末エックス線回折パターンである。
【特許請求の範囲】
【請求項1】
式(1):
【化1】
[式中、R1は、水素原子またはメチル基を表し、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表し、Xは、メチレン基または酸素原子を表し、nは、1〜3の整数を表す。]
で表される化合物、またはその薬学上許容される塩。
【請求項2】
Xがメチレン基を表し、nが1〜2の整数を表すか、または、Xが酸素原子を表し、nが2〜3の整数を表す、請求項1に記載の化合物、またはその薬学上許容される塩。
【請求項3】
R1がメチル基を表す、請求項1または2に記載の化合物、またはその薬学上許容される塩。
【請求項4】
R2がp位に結合した臭素原子を表す、請求項1〜3のいずれか一項に記載の化合物、またはその薬学上許容される塩。
【請求項5】
Xが酸素原子を表し、nが2の整数を表す、請求項1〜4のいずれか一項に記載の化合物、またはその薬学上許容される塩。
【請求項6】
式(1)で表される化合物が、以下の化合物(01)〜(15)からなる群:
(01)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(02)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(04)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(05)6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(06)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(07)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(09)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(10)6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(11)6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(12)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(13)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(14)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、および
(15)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン
から選択される、請求項1に記載の化合物、またはその薬学上許容される塩。
【請求項1】
式(1):
【化1】
[式中、R1は、水素原子またはメチル基を表し、R2は、ピペリジン環に結合したメチレン基に対してp位またはm位に結合した基であって、p位に結合した塩素原子、p位に結合した臭素原子、p位に結合したメチル基、m位に結合した塩素原子またはm位に結合した臭素原子を表し、Xは、メチレン基または酸素原子を表し、nは、1〜3の整数を表す。]
で表される化合物、またはその薬学上許容される塩。
【請求項2】
Xがメチレン基を表し、nが1〜2の整数を表すか、または、Xが酸素原子を表し、nが2〜3の整数を表す、請求項1に記載の化合物、またはその薬学上許容される塩。
【請求項3】
R1がメチル基を表す、請求項1または2に記載の化合物、またはその薬学上許容される塩。
【請求項4】
R2がp位に結合した臭素原子を表す、請求項1〜3のいずれか一項に記載の化合物、またはその薬学上許容される塩。
【請求項5】
Xが酸素原子を表し、nが2の整数を表す、請求項1〜4のいずれか一項に記載の化合物、またはその薬学上許容される塩。
【請求項6】
式(1)で表される化合物が、以下の化合物(01)〜(15)からなる群:
(01)6−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(02)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(04)7−(2−{4−[4−ブロモ−3−(2−ヒドロキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(05)6−(2−{4−[4−クロロ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(06)6−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(07)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(09)7−(2−{4−[4−ブロモ−3−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン、
(10)6−(2−{4−[3−(2−メトキシエトキシ)−4−メチルベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(11)6−(2−{4−[3−クロロ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、
(12)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−1H−インデン−1−オン、
(13)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロナフタレン−1(2H)−オン、
(14)6−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−2,3−ジヒドロ−4H−クロメン−4−オン、および
(15)7−(2−{4−[3−ブロモ−5−(2−メトキシエトキシ)ベンジル]ピペリジン−1−イル}エチル)−3,4−ジヒドロ−1−ベンゾキセピン−5(2H)−オン
から選択される、請求項1に記載の化合物、またはその薬学上許容される塩。
【図1】

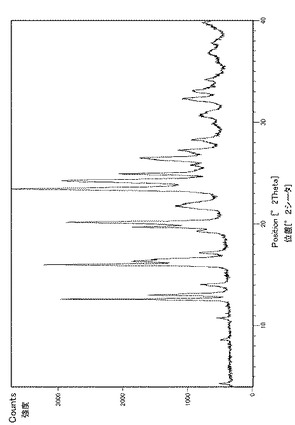
【図2】
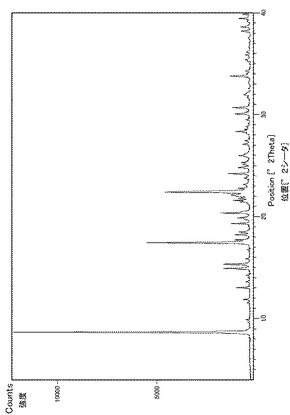
【図3】
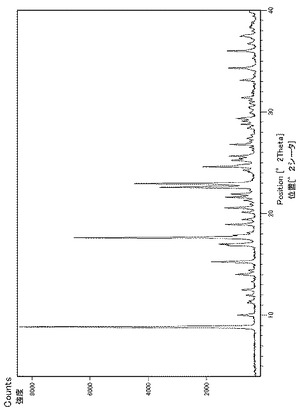
【図4】
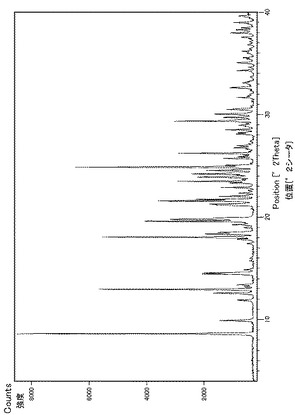
【図5】
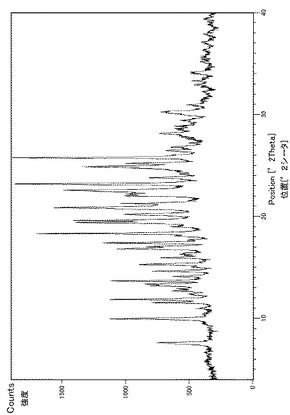
【図6】
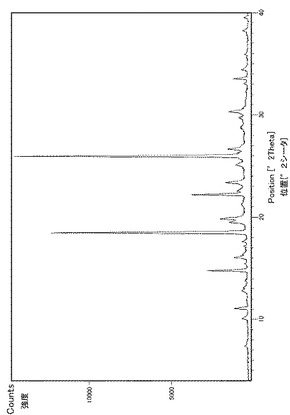
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−67111(P2012−67111A)
【公開日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願番号】特願2011−235127(P2011−235127)
【出願日】平成23年10月26日(2011.10.26)
【分割の表示】特願2009−537435(P2009−537435)の分割
【原出願日】平成21年2月4日(2009.2.4)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
【公開日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願日】平成23年10月26日(2011.10.26)
【分割の表示】特願2009−537435(P2009−537435)の分割
【原出願日】平成21年2月4日(2009.2.4)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
[ Back to top ]