
ベンズイミダゾールカンナビノイドアゴニスト
【化1】

本発明は、カンナビノイド受容体作動性を有する新規な式(I)のベンズイミダゾール化合物、これらの化合物を含んでなる製薬学的組成物、これらの化合物の化学的製造方法ならびに動物、特にヒトにおけるカンナビノイド受容体の媒介に結び付けられる疾患の処置におけるそれらの使用に関する。
本発明は、カンナビノイド受容体作動性を有する新規な式(I)のベンズイミダゾール化合物、これらの化合物を含んでなる製薬学的組成物、これらの化合物の化学的製造方法ならびに動物、特にヒトにおけるカンナビノイド受容体の媒介に結び付けられる疾患の処置におけるそれらの使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、選択的カンナビノイド受容体2作動性を有する新規な式(I)のベンズイミダゾール化合物、これらの化合物を含んでなる製薬学的組成物、これらの化合物の化学的製造方法ならびに動物、特にヒトにおけるカンナビノイド受容体の媒介に結び付けられる疾患の処置におけるそれらの使用に関する。
【背景技術】
【0002】
マリファナ由来カンナビノイドΔ9−テトラヒドロ−カンナビノイド(Δ9−THC)のような古典的なカンナビノイドは、体内の特異的なカンナビノイド受容体との相互作用を介してそれらの薬理学的効果を生む。これまでに2つのカンナビノイド受容体:哺乳類脳及び末梢組織において見出された受容体CB1及び末梢組織において主に見出された受容体CB2が特性化された。これらの受容体の1つ又は両方に関するアゴニスト又はアンタゴニストである化合物は、多様な薬理学的効果を与えることが見出された。CB2受容体に関する高い選択性は、カンナビノイド構造に伴って見られる中心的な不利な事柄を避けながらCB受容体アゴニストの有益な効果を利用するための手段を与えることができると思われるので、選択的CB2作動活性を有するカンナビノイド類似物の開発に有意な興味が持たれる(例えば非特許文献1を参照されたい)。
【0003】
特許文献1は、アルツハイマー病、不安障害、抑うつ性障害、骨粗しょう症、心臓血管病、慢性関節リウマチ又は前立腺ガンのようなエストロゲン受容体−βに関連する疾患の処置における使用のための、エストロゲン受容体−βリガンドとしてのベンズイミダゾール化合物を開示している。特許文献2は、CB2受容体活性により媒介される状態の処置において有用な、CB2作動活性を有するスルホニルベンズイミダゾール誘導体を開示している。
【0004】
本発明の化合物は、複素原子が常に置換されている1−ピペリジン−4−イルメチル又は1−テトラヒドロチオピラン−4−イルメチル基の存在により、挙げられた当該技術分野において既知の化合物と構造的に異なる。
【0005】
本発明の化合物は、予期に反し、当該技術分野において既知の特許文献1の化合物と比較して、体温の低下及び平伏体姿勢(flat body posture)のようなCB1関連副作用のない選択的CB2アゴニストであることが見出された。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第2002/46168号パンフレット
【特許文献2】国際公開第2006/048754号パンフレット
【非特許文献】
【0007】
【非特許文献1】Expert Opinion on Investigational Drugs,14(6),2005年,695−703
【発明の概要】
【0008】
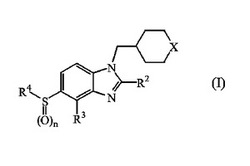
本発明は、立体化学的異性体を含んで、式(I)
【0009】
【化1】
【0010】
[式中、
nは整数0、1又は2であり;
XはSO、SO2又はN−(CO)−R1であり;
R1は水素;
C1−6アルキル;
C1−6アルキルオキシ;
C1−4アルキルオキシC1−4アルキル;又は
ポリハロC1−6アルキル
であり;
R2はC1−6アルキルであり;
R3は水素、ハロ、C1−4アルキル、C1−4アルキルオキシ、トリフルオロメチル又はシアノであり;
R4はC1−8アルキル;
C3−8シクロアルキルで置換されたC1−8アルキル;
ポリハロC1−8アルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC1−8アルキル;
C3−8シクロアルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC3−8シクロアルキル;
テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、
アリール;あるいは
ヘテロアリール
であり;
アリールはフェニル;あるいはハロ、ヒドロキシ、C1−4アルキル、ポリハロC1−4アルキル、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、NR5R6、R7−カルボニル、R7−SO2−又はヒドロキシ、NR5R6、R7−カルボニルもしくはR7−SO2−で置換されたC1−4アルキルからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたフェニルであり;
ヘテロアリールはフラニル、チオフェニル、ピロリル、ピラゾリル、イミダゾリル、イソオキサゾリル、チアゾリル、トリアゾリル、テトラゾリル、イソチアゾリル、チアジアゾリル、オキサジアゾリル、ピリジニル、ピリダジニル、ピリミジニル又はピラジニルから選ばれ;
ここでR5及びR6は互いに独立して水素、C1−4アルキル、ポリハロC1−4アルキル、アミノスルホニル又はC1−8アルキルスルホニル;あるいはR7−カルボニルから選ばれ;
ここでR5及びR6は、R5及びR6を有する窒素原子と一緒になって、ピロリジニル、ピペリジニル、ピペラジニル又はモルホリニル環を形成することができ;そして
ここでR7はC1−4アルキル、ヒドロキシ、アミノ、モノ−もしくはジ−(C1−4アルキル)アミノ、(ヒドロキシC1−4アルキル)アミノ、(C1−4アルキルオキシC1−4アルキル)アミノ、ジ(C1−4アルキル)アミノC1−4アルキル、ピロリジニル、ピペリジニル、モルホリニル又はN−メチル−ピペラジニルである]
の化合物あるいはその製薬学的に許容され得る酸付加塩又はその溶媒和物に関する。
【0011】
前記の定義において用いられる場合:
−ハロはフルオロ、クロロ、ブロモ及びヨードの総称であり;
−C1−4アルキルは、1〜4個の炭素原子を有する直鎖状及び分枝鎖状飽和炭化水素基、例えばメチル、エチル、プロピル、ブチル、1−メチルエチル、2−メチルプロピルなどを定義し;
−C1−6アルキルは、C1−4アルキル及び5もしくは6個の炭素原子を有するその高級同族体、例えば2−メチルブチル、ペンチル、ヘキシルなどを含むものとし;
−C1−8アルキルは、C1−6アルキル及び7〜8個の炭素原子を有するその高級同族体、例えばヘプチル、エチルヘキシル、オクチルなどを含むものとし;
−ポリハロC1−4アルキルは、ポリハロ置換されたC1−4アルキル、特に2〜6個のハロゲン原子で置換された(上記で定義されたような)C1−4アルキル、例えばジフルオロメチル、トリフルオロメチル、トリフルオロエチルなどとして定義され;
−C3−6シクロアルキルは、シクロプロピル、シクロブチル、シクロペンチル及びシクロヘキシルの総称であり;
−C3−8シクロアルキルは、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル及びシクロオクチルの総称であり;
−C6−8シクロアルキルは、シクロヘキシル、シクロヘプチル及びシクロオクチルの総称である。
【0012】
前記で用いられた「立体化学的異性体」という用語は、式(I)の化合物が有することができるすべての可能な異性体を定義する。他にことわるか又は指示しなければ、化合物の化学的名称はすべての可能な立体化学的異性体の混合物を示し、該混合物は基本の分子構造のすべてのジアステレオマー及びエナンチオマーを含有する。さらに特定的に、ステレオジェン中心はR−又はS−立体配置を有することができ;二価環状(部分的)飽和基上の置換基は、シス−又はトランス−立体配置のいずれかを有することができる。
【0013】
式(I)の化合物の立体化学的異性体は、明らかに本発明の範囲内に包含されることが意図されている。
【0014】
式(I)の化合物及びそれらの製造において用いられる中間体の絶対立体化学的配置は、例えばX−線回折のような周知の方法を用いて当該技術分野における熟練者が容易に決定することができる。
【0015】
さらに、式(I)のいくつかの化合物及びそれらの製造において用いられる中間体のいくつかは、多形を示し得る。本発明は、上記に記載した状態の処置において有用な性質を有するいずれの多形相も包含することが理解されるべきである。
【0016】
上記で言及した製薬学的に許容され得る酸付加塩は、式(I)の化合物が形成することができる治療的に活性な無毒性の酸付加塩の形態を含むものとする。これらの製薬学的に許容され得る酸付加塩は、塩基形態をそのような適した酸で処理することにより簡単に得ることができる。適した酸は、例えば無機酸、例えばハロゲン化水素酸、例えば塩酸又は臭化水素酸、硫酸、硝酸、リン酸などの酸;あるいは有機酸、例えば酢酸、プロパン酸、
ヒドロキシ酢酸、乳酸、ピルビン酸、シュウ酸(すなわちエタン二酸)、マロン酸、コハク酸(すなわちブタン二酸)、マレイン酸、フマル酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、シクラミン酸、サリチル酸、p−アミノサリチル酸、パモ酸などの酸を含む。
【0017】
逆に、適した塩基を用いる処理により、該塩形態を遊離の塩基形態に転換することができる。
【0018】
式(I)の化合物は、溶媒和されない形態及び溶媒和された形態の両方において存在することができる。「溶媒和物」という用語は本明細書で、本発明の化合物及び1種もしくはそれより多い製薬学的に許容され得る溶媒分子、例えば水又はエタノールを含んでなる分子会合を記述するために用いられる。「水和物」という用語は、該溶媒が水である場合に用いられる。
【0019】
興味深い式(I)の化合物は、以下の制限の1つもしくはそれより多くが適用される式(I)の化合物である:
a)nが整数0であるか、又はnが整数2であるか;あるいは
b)XがSO2であるか;あるいは
c)XがN−(CO)−R1であり、ここでR1は水素であるか;あるいは
d)XがN−(CO)−R1であり、ここでR1はC1−6アルキル、好ましくはメチル又はエチルであるか;あるいは
e)XがN−(CO)−R1であり、ここでR1はC1−6アルキルオキシ、好ましくはメチルオキシであるか;あるいは
f)XがN−(CO)−R1であり、ここでR1はC1−4アルキルオキシC1−4アルキル、好ましくはメチルオキシメチルであるか;あるいは
g)XがN−(CO)−R1であり、ここでR1はポリハロC1−6アルキル、好ましくはトリフルオロメチルであるか;あるいは
h)R2がC1−6アルキルである、特にR2がtert−ブチル又は−CH2−tert−ブチルであるか;あるいは
i)R3が水素であるか;あるいは
j)R4がC1−8アルキル、C3−8シクロアルキルで置換されたC1−8アルキル、ポリハロC1−8アルキルであるか;あるいは
k)R4がヒドロキシ、C1−4アルキルオキシ、シアノで置換されたC1−8アルキルであるか;あるいは
l)R4がアリールであるか;あるいは
m)R4がヘテロアリールである。
【0020】
1つの態様において本発明は、nが整数0、1又は2であり;XがSO2又はN−(CO)−R1であり;R1が水素、C1−6アルキル、C1−6アルキルオキシ、C1−4アルキルオキシC1−4アルキル;あるいはポリハロC1−6アルキルであり;R2がC1−6アルキルであり;R3が水素であり;R4がC1−8アルキル;C3−8シクロアルキルで置換されたC1−8アルキル;ポリハロC1−8アルキル;C3−8シクロアルキル;ヒドロキシ、C1−4アルキルオキシ、シアノ又はアリールで置換されたC1−8アルキル;アリール;又はヘテロアリールであり;ここでアリールはハロ、C1−4アルキルオキシ又はシアノで置換されたフェニルであり;そしてヘテロアリールはチアゾリル又はピリジニルである、立体化学的異性体を含む式(I)の化合物;あるいは製薬学的に許容され得るその酸付加塩又はその溶媒和物に関する。
【0021】
nが0である式(I)の化合物として定義される式(I−a)の化合物は、Cs2CO3のような適した塩基の存在下に、例えば2−プロパノン、1,4−ジオキサン又はTH
Fのような反応に不活性な溶媒中で中間体(II)を中間体(III)と反応させることにより製造することができ、ここでLは離脱基、例えばハロ、メタンスルホニルオキシ、ベンゼンスルホニルオキシ、トリフルオロメタンスルホニルオキシなどの反応性離脱基である。中間体(III)中に存在する置換基の型に依存して、カップリング反応後に除去することができる保護基を、中間体(III)中に導入することが必要であり得る。
【0022】
【化2】
【0023】
nが0である式(I)の化合物として定義される式(I−a)の化合物を、Cs2CO3のような適した塩基、Pd2(dba)3のような触媒及びキサントフォス(Xantphos)のような適したリガンドの存在下に、例えば2−プロパノン、1,4−ジオキサン又はTHFのような反応に不活性な溶媒中で、通常の条件又はマイクロ波条件下で加熱しながら、中間体(II)を中間体(IV)と反応させることによって製造することもできる。
【0024】
【化3】
【0025】
当該技術分野において既知のS−酸化反応により、式(I−a)の化合物を、nが1を示す式(I)の化合物として定義される式(I−b)の化合物に、あるいはnが2を示す式(I)の化合物として定義される式(I−c)の化合物に転換することができる。
【0026】
【化4】
【0027】
過酸化水素の30%水溶液を用いて、又は他の酸化剤、例えばNaIO4、tert−ブチルオキシクロリド、亜硝酸アシル、過ホウ酸ナトリウム及び過酸、例えばmCPBA(メタ−クロロ過安息香酸)により、S−酸化反応を行なうことができる。スルフィドをスルホキシドに酸化することができ、それをさらに1当量の過酸化水素、KMnO4、過ホウ酸ナトリウム、過硫酸水素カリウム、mCPBAなどの試薬の添加によりスルホンにさらに酸化することができる。十分な酸化剤が存在すれば、スルホキシドを単離せずに直接、スルフィドをスルホンに転換することができる。
【0028】
XがN−(CO)−R1を示す式(I)の化合物として定義される式(I−d)の化合物は、中間体(VI)を用いる中間体(V)のN−アルキル化により製造することができ、ここでWは適した離脱基、例えばハロ、例えばフルオロ、クロロ、ブロモ、ヨードであるか、あるいはいくつかの場合、Wはスルホニルオキシ基、例えばメタンスルホニルオキシ、ベンゼンスルホニルオキシ、トリフルオロメタンスルホニルオキシなどの反応性離脱基であることもできる。例えばアセトニトリル又はジクロロメタンのような反応に不活性な溶媒中で、且つ場合により例えば炭酸ナトリウム、炭酸カリウム又はトリエチルアミンのような適した塩基の存在下で反応を行なうことができる。攪拌は反応の速度を増すことができる。室温〜反応混合物の還流温度の範囲の温度で反応を簡単に行なうことができる。
【0029】
【化5】
【0030】
nが2であり、R1が水素以外と定義される式(I)の化合物として定義される式(I−e)の化合物は、スキーム1に記載されるとおりに製造することができる。
【0031】
【化6】
【0032】
式(I−e)の化合物を得るための縮合反応は、酸性又は塩基性条件下で行なうことができる。酸性条件下で、縮合は、酢酸のような有機酸あるいはHCl又はH2SO4のような無機酸あるいはそれらの組み合わせの存在下に、酢酸、H2O、メタノール、エタノール、ジオキサン、トルエン又はジクロロエタンのような溶媒中で行なわれる。塩基性条件下で、縮合反応は、例えばK2CO3のような無機塩基の存在下に、DMSOのような反応に不活性な溶媒中で、あるいはアルコール性NaOH溶液中で行なわれる。室温〜反応混合物の還流温度の範囲の温度で反応を簡単に行なうことができる。溶媒としてのジクロロエタン中で、例えば190℃におけるマイクロ波補助加熱により反応速度及び収率を増すことができ、おそらくさらに酸又は塩基を加える必要が取り除かれる。
【0033】
上記の方法において製造される式(I)の化合物は、エナンチオマーのラセミ混合物の形態で合成され得、それを当該技術分野において既知の分割法に従って互いから分離する
ことができる。ラセミ形態で得られる式(I)の化合物を、適したキラル酸との反応により対応するジアステレオマー塩の形態に転換することができる。該ジアステレオマー塩の形態を、続いて例えば選択的又は分別結晶化により分離し、アルカリによりそこからエナンチオマーを遊離させる。式(I)の化合物のエナンチオマーの形態を分離する代わりの方法は、キラル固定相を用いる液体クロマトグラフィーを含む。該純粋な立体化学的異性体を、適した出発材料の対応する純粋な立体化学的異性体から誘導することもでき、但し、反応は立体特異的に起こる。好ましくは、特定の立体異性体が望まれる場合、該化合物は立体特異的製造方法により合成されるであろう。これらの方法は、有利にはエナンチオマー的に純粋な出発材料を用いるであろう。
【0034】
式(I)の化合物、その製薬学的に許容され得る塩及び立体異性体は、薬理学的実施例において示されるように、選択的カンナビノイド受容体2(CB2)作動性を有する。薬理学的実施例C.1は、CB2アゴニズムを測定する方法を記載しており、結果は表C.1に挙げられている。
【0035】
従って本式(I)の化合物は、特にカンナビノイド2受容体、特にCB2作動活性により媒介される状態又は疾患の処置における薬剤として有用である。続いて、本化合物をCB2受容体活性、特にCB2作動活性により媒介される状態又は疾患の処置用の薬剤の製造のために用いることができる。
【0036】
好ましくは、本発明は、CB2状態又は疾患から選ばれる状態又は疾患の処置用の薬剤の製造のための式(I)の化合物又はその製薬学的に許容され得る塩の使用も提供する。
【0037】
さらに本発明は、哺乳類の患者におけるCB2受容体活性により媒介される状態の処置方法を提供し、それは、そのような処置の必要な哺乳類に式(I)の化合物又はその製薬学的に許容され得る塩の治療的に有効な量を投与することを含む。
【0038】
カンナビノイド受容体2が媒介する状態又は障害は、例えば、心臓血管病、例えばアテローム性動脈硬化症、高血圧、心筋虚血;慢性疼痛障害(chronic pain disorders)、例えば痛覚過敏症、神経障害疼痛(neuropathic pain)、末梢痛、内臓痛、炎症痛、熱性痛覚過敏症、侵害受容疼痛(nociceptive pain)、線維筋痛、慢性背下部痛及び歯痛;炎症、浮腫、膀胱炎、神経炎症性疾患、免疫系障害、自己免疫疾患、多発性硬化症、慢性関節リウマチ、胃腸障害、腸運動障害、過敏性腸症候群(IBS)、炎症性腸疾患(IBD)、クローン病、慢性肝臓損傷(肝硬変)、ガン、前立腺ガン、ガン痛、神経膠腫、アレルギー、吐気及び嘔吐、喘息、慢性閉塞性肺疾患、乾癬、てんかん及び骨喪失障害(bone loss disorders)、例えば骨粗しょう症である(下記で「CB2障害又は疾患」と言及する)。
【0039】
本明細書で用いられる場合、「処置する」及び「処置」という用語は、そのような用語が適用される疾患、障害又は状態あるいはそのような疾患、障害又は状態の1つもしくはそれより多い症状を逆転させるか、緩和するか、その進行を妨げるか、又は予防することを含む、治癒的、緩和的及び予防的処置を指す。
【0040】
さらに本発明は、少なくとも1種の製薬学的に許容され得る担体及び式(I)の化合物の治療的に有効な量を含んでなる製薬学的組成物を提供する。
【0041】
本発明の製薬学的組成物の調製のために、塩基又は酸付加塩の形態にある特定の化合物の活性成分として有効な量を、少なくとも1種の製薬学的に許容され得る担体と緊密な混合物において合わせ、その担体は、投与のために望ましい調製物の形態に依存して多様な形態をとることができる。望ましくはこれらの製薬学的組成物は、好ましくは経口的投与
、直腸的投与、経皮的投与又は非経口的注入に適した単位投薬形態にある。
【0042】
例えば経口的投薬形態における組成物の調製において、懸濁剤、シロップ、エリキシル剤及び溶液のような経口用液体調製物の場合、通常の液体の製薬学的担体のいずれか、例えば水、グリコール、油、アルコールなど;あるいは粉剤、丸薬、カプセル及び錠剤の場合、澱粉、糖類、カオリン、滑沢剤、結合剤、崩壊剤などのような固体の製薬学的担体を用いることができる。それらの容易な投与のために、錠剤及びカプセルは最も有利な経口的投薬単位形態物を与え、その場合には固体の製薬学的担体が用いられるのは明らかである。非経口的注入組成物の場合、製薬学的担体は主に無菌水を含んでなるであろうが、活性成分の溶解性を向上させるための他の成分が含まれることができる。例えば食塩水、グルコース溶液又は両者の混合物を含んでなる製薬学的担体を用いることにより、注入可能な溶液を調製することができる。適した液体担体、懸濁化剤などを用いることにより、注入可能な懸濁剤を調製することもできる。経皮的投与に適した組成物において、製薬学的担体は場合により浸透促進剤及び/又は適した湿潤剤を含んでなることができ、それらは場合により皮膚に有意な悪影響を引き起こさない小さい割合の適した添加剤と組み合わされていることができる。該添加剤は、皮膚への活性成分の投与を促進するように及び/又は所望の組成物の調製の助けとなるように選ばれることができる。これらの局所用組成物を種々の方法で、例えば経皮パッチ、スポット−オン又は軟膏として投与することができる。式(I)の化合物の付加塩は、対応する塩基形態より向上したそれらの水溶性のために、水性組成物の調製において明らかにより適している。
【0043】
投与の容易さ及び投薬量の均一性のために、本発明の製薬学的組成物を投薬単位形態物において調製するのが特に有利である。本明細書で用いられる「投薬単位形態物」は、1回の投薬量として適した物理的に分離された単位を指し、各単位は所望の治療効果を生むために計算されたあらかじめ決められた量の活性成分を、必要な製薬学的担体と一緒に含有する。そのような投薬単位形態物の例は錠剤(刻み付き又はコーティング錠を含む)、カプセル、丸薬、粉剤小包、ウェハース、注入可能な溶液又は懸濁剤、小さじ一杯、大さじ一杯など、ならびに分離されたそれらの複数である。
【0044】
経口的投与のために、本発明の製薬学的組成物は、製薬学的に許容され得る賦形剤及び担体、例えば結合剤(例えば予備ゼラチン化トウモロコシ澱粉、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロースなど)、充填剤(例えばラクトース、微結晶性セルロース、リン酸カルシウムなど)、滑沢剤(例えばステアリン酸マグネシウム、タルク、シリカなど)、崩壊剤(例えばポテト澱粉、ナトリウム澱粉グリコレートなど)、湿潤剤(例えばラウリル硫酸ナトリウム)などを用いて通常の手段により調製される固体の投薬形態物、例えば錠剤(嚥下可能及びチュワブル形態の両方)、カプセル又はゲルカップの形態をとることができる。そのような錠剤を当該技術分野において周知の方法によりコーティングすることもできる。
【0045】
経口的投与のための液体調製物は、例えば溶液、シロップ又は懸濁剤の形態をとることができるか、あるいはそれらを使用前に水及び/又は他の適した液体担体と混合するための乾燥生成物として調製することができる。そのような液体調製物は通常の方法により、場合により懸濁化剤(例えばソルビトールシロップ、メチルセルロース、ヒドロキシプロピルメチルセルロース又は水素化食用脂肪)、乳化剤(例えばレシチン又はアラビアゴム)、非−水性担体(例えばアーモンド油、油性エステル又はエチルアルコール)、甘味料、風味料、隠蔽剤及び防腐剤(例えばp−ヒドロキシ安息香酸メチルもしくはプロピル又はソルビン酸)のような他の製薬学的に許容され得る添加剤を用いて調製することができる。
【0046】
本発明の製薬学的組成物において有用な製薬学的に許容され得る甘味料は、好ましくは
少なくとも1種の強力甘味料、例えばアスパルテーム、アセスルフェームカリウム、シクラミン酸ナトリウム、アリテーム、ジヒドロカルコン甘味料、モネリン、ステビオシドスクラロース(4,1’,6’−トリクロロ−4,1’,6’−トリデオキシガラクトスクロース)又は、好ましくはサッカリン、サッカリンナトリウムもしくはカルシウム及び場合により少なくとも1種のバルク甘味料(bulk sweetener)、例えばソルビトール、マンニトール、フルクトース、スクロース、マルトース、イソマルト、グルコース、水素化グルコースシロップ、キシリトール、カラメル又はハチミツを含む。強力甘味料は、簡便に低濃度で用いられる。例えばサッカリンナトリウムの場合、該濃度は、最終的な調剤の約0.04%〜0.1%(重量/容量)の範囲であることができる。バルク甘味料は、約10%〜約35%、好ましくは約10%〜15%(重量/容量)の範囲の比較的高い濃度で有効に用いられ得る。
【0047】
低−投薬量調剤において苦い味の成分を隠蔽することができる製薬学的に許容され得る風味料は、好ましくはフルーツ風味料、例えばチェリー、ラズベリー、黒スグリ又はストロベリー風味料である。2種の風味料の組み合わせは、非常に良い結果を与えることができる。高−投薬量の調剤においては、より強い製薬学的に許容され得る風味料、例えばカラメルチョコレート(Caramel Chocolate)、ミントクール(Mint
Cool)、ファンタジー(Fantasy)などが必要であり得る。各風味料は、約0.05%〜1%(重量/容量)の範囲の濃度で、最終的な組成物中に存在することができる。該強い風味料の組み合わせは、有利に用いられる。好ましくは、調剤の環境下で味及び/又は色の変化又は喪失を経ない風味料が用いられる。
【0048】
式(I)の化合物を、注入、簡便には静脈内、筋肉内又は皮下注入による、例えば大量注入又は継続的静脈内注入による非経口的投与用に調製することができる。注入用の調剤は、単位投薬形態物において、例えばアンプル中で、又は加えられた防腐剤を含む多投薬量容器中で存在することができる。それらは、油性又は水性ビヒクル中の懸濁剤、溶液又は乳剤のような形態をとることができ、等張化剤、懸濁化剤、安定剤及び/又は分散剤のような調製剤を含有することができる。あるいはまた、活性成分は、使用前に適したビヒクル、例えば発熱物質を含まない無菌水と混合するための粉末形態で存在することができる。
【0049】
式(I)の化合物を、例えば通常の座薬基剤、例えばココアバター及び/又は他のグリセリドを含有する座薬又は保持浣腸(retention enema)のような直腸用組成物において調製することもできる。
【0050】
カンナビノイド受容体の媒介に結び付けられる疾患の処置における熟練者は、下記に示される試験結果から、式(I)の化合物の治療的に有効な量を容易に決定することができるであろう。一般に治療的に有効な投薬量は、処置されるべき患者の体重のkg当たり約0.001mg〜kg当たり約50mg、より好ましくは体重のkg当たり約0.01mg〜kg当たり約10mgであろうと思われる。治療的に有効な投薬量を、1日を通じて適した間隔における2回かもしくはそれより多い細分−投薬量の形態で投与するのが適しているかも知れない。該細分−投薬量を、例えばそれぞれ単位投薬形態物当たりに約0.1mg〜約1000mg、さらに特定的に約1〜約500mgの活性成分を含有する単位投薬形態物として調製することができる。
【0051】
本明細書で用いられる場合、化合物の「治療的に有効な量」は、患者又は動物に投与されると、その患者又は動物において、カンナビノイド受容体の刺激における識別可能な増加又は減少を引き起こすのに十分に高いレベルのその化合物を与える化合物の量である。
【0052】
投与の正確な用量及び頻度は、当該技術分野における熟練者に周知のとおり、用いられ
る特定の式(I)の化合物、処置されている特定の状態、処置されいてる状態の重度、特定の患者の年令、体重及び一般的な身体条件ならびに患者が摂取しているかも知れない他の投薬に依存する。さらに、処置される患者の反応に依存して及び/又は本発明の化合物を処方する医師の評価に依存して、該「治療的に有効な量」を減少させるか又は増加させることができる。従って上記で挙げた有効な1日の量の範囲は、単に指針である。
【発明を実施するための形態】
【0053】
実験部分
下記に記載する方法において、以下の略語を用いた:「DCM」はジクロロメタンを示し、「MeOH」はメタノールを示し、「NH3」はアンモニアを示し、「CH3CN」はアセトニトリルを示し、「THF」はテトラヒドロフランを示し、「DIPE」はジイソプロピルエーテルを示し、「NaBH3(CN)」はナトリウムシアノトリヒドロボレートを示し、「Cs2CO3」は炭酸セシウムを意味し、「MgSO4」は硫酸マグネシウムを意味し、「NaHCO3」は炭酸一ナトリウム塩を意味し、「NaOH」は水酸化ナトリウムを意味し、「Pd2(dba)3」はトリス[μ−[(1,2−η:4,5−η)−(1E,4E)−1,5−ジフェニル−1,4−ペンタジエン−3−オン]ジパラジウムを意味し、そして「キサントフォス」は(9,9−ジメチル−9H−キサンテン−4,5−ジイル)ビス[ジフェニルホスフィン]を意味し、「DMSO」はジメチルスルホキシドを意味し;「DMAP」は4−(ジメチルアミノ)ピリジンを意味し、「HBTU」は1−[ビス(ジメチルアミノ)メチレン]−1H−ベンゾトリアゾリウムヘキサフルオロホスフェート(1−)3−オキシドを意味する。
【0054】
Isolute HM−NTMフィルターは、Argonaut,Foster City,CA 94404,USAの製品であり、改質された形態(modified form)の珪藻土を含んでなる短カラムであり、コンビナトリアルケミストリー用途において試料から水を除去することができる。
【0055】
高−性能液体クロマトグラフィー精製法:
−精製法A
生成物を逆相高−性能液体クロマトグラフィー(Shandon Hyperprep(R) C18 BDS(Base Deactivated Silica)8μm,250g,内径5cm)により精製した。2つの移動相を用いた(相A:水中の0.25%NH4HCO3溶液;相B:CH3CN)。最初に40ml/分の流量で85%A及び15%Bを0.5分間保持した。次いで80ml/分の流量で勾配を適用し、41分内に10%A及び90%Bとした。次いで80ml/分の流量で勾配を適用し、20分内に100%Cとし、4分間保持した。
−精製法B
生成物を逆相高−性能液体クロマトグラフィー(Shandon Hyperprep(R) C18 BDS(Base Deactivated Silica)8μm,250g,内径5cm)により精製した。3つの移動相を用いる勾配を適用した(相A:水中の0.25%NH4HCO3溶液;相B:CH3OH;相C:CH3CN)。所望の画分を集め、仕上げた。
【0056】
A.中間体の合成
実施例A.1
a)
【化7】
ジオキサン(120ml)中の1−クロロ−4−(エチルスルホニル)−2−ニトロベンゼン(7.5g,0.03モル)及び4−アミノメチル−ピペリジン−1−カルボン酸tert−ブチルエステル(7.72g,0.036モル)の混合物に、炭酸カリウム(4.55g,0.033モル)を加えた。反応混合物を75〜80℃で3時間及び次いで100℃で3時間攪拌した。固体を濾過し、反応混合物を蒸発させた。残留物をDCM(200ml)中に溶解した。有機層を水(200ml)で洗浄した。水層をDCM(150ml)で抽出した。合わせた有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、12.1gの中間体(1)を与えた。
b)
【化8】
メタノール(150ml)中の中間体(1)(12.1g)の混合物を、チオフェン溶液(1ml)及び酸化バナジウム(0.2g)の存在下で、触媒として活性炭上のパラジウム(10%)(2g)を用いて水素化した。水素(3当量)の吸収後、反応混合物をジカライト上で濾過し、濾液を蒸発させ、中間体(2)を与えた。
c)
【化9】
DCM(90ml)中の中間体(2)(0.014モル)、2,2−ジメチルプロパノイルクロリド(2.1ml,0.017モル)及びピリジン(2ml)の混合物を室温で終夜攪拌した。溶媒を蒸発させ、酢酸(80ml)及び塩酸(8ml)を残留物に加えた。この混合物を3時間還流させ、次いで混合物を蒸発させた。残留物をDCM(300ml)と水(250ml)に分配した。NH3水を用いて混合物を塩基性とし、層を分離した。分離された有機層を水で洗浄し、乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、3.4gの残留物を与えた。この残留物をシリカゲル上のカラムクロマトグラフィー(Biotage;溶離剤:DCM/(MeOH/NH3) 99/1から92/8まで)により精製した。所望の画分を集め、溶媒を蒸発させ、2.7gの中間体(3)を与えた。
【0057】
実施例A.2
a)
【化10】
2−プロパノン(1000ml)中の4−クロロベンゼンチオール(0.2モル)及び1,1,1−トリフルオロ−4−ヨードブタン(0.21モル)の混合物を氷−浴上で冷却した。Cs2CO3(0.215モル)を加え、反応混合物を窒素流下に室温で終夜攪拌した。次いでDIPE(1000ml)を加え、固体を濾過し、濾液を蒸発させ、中間体(4)を与えた。
b)
【化11】
トリクロロメタン(1200ml)中の中間体(4)(0.2モル)の混合物を、氷−浴上で冷却した。3−クロロベンゼンカルボペルオキソ酸(100g;70〜75%)を、20分かけて分けて加え、反応混合物を室温で210分間攪拌した。混合物を氷−浴上で冷却し、NaOH水溶液(1000ml,5%)を加えた。分離された有機層をNaOH水溶液(1000ml,5%)で2回洗浄し、次いで水で洗浄した。合わせた有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、57gの中間体(5)を与えた。
c)
【化12】
濃硫酸(500ml)中の中間体(5)(0.2モル)の混合物に、冷水を用いて反応混合物を冷却しながら、濃硫酸と硝酸の混合物(50/50)(100ml)を1時間かけて滴下した。混合物を室温で2時間攪拌し、次いで氷(2000ml)上に注いだ。沈殿を濾過し、水で洗浄し、乾燥し(真空)、66gの中間体(6)を与えた。
d)
【化13】
DMSO(4ml)中の中間体(6)(0.002モル)、1−アセチル−4−ピペリジンメタナミン(0.00235モル)及びトリエチルアミン(0.003モル)の混合物を100℃で終夜攪拌した。この混合物を氷−水上に注ぎ、DCMで抽出した。有機層を水で2回洗浄し、乾燥し(MgSO4)、溶媒を蒸発させた。残留物をシリカゲル上のコンビフラッシュカラムクロマトグラフィー(溶離剤:DCM/(CH3OH/NH3)
100/0から97/3)により精製した。生成物画分を集め、溶媒を蒸発させ、0.4gの中間体(7)を与えた。
e)
【化14】
メタノール(40ml)中の中間体(7)(0.0009モル)の混合物を、チオフェン溶液(0.1ml)の存在下に触媒として活性炭上のパラジウム(10%)(0.1g)を用いて水素化した。水素(3当量)の吸収後、触媒をセライト上で濾過し、濾液を蒸発させ、中間体(8)を与えた(そのまま次の段階で使用した)。
【0058】
実施例A.3
a)
【化15】
DMSO(15ml)中の中間体(6)(0.008モル)、4−アミノメチル−ピペリジン−1−カルボン酸tert−ブチルエステル(0.0093モル)及びトリエチルアミン(0.012モル)の混合物を、密閉された容器中で100℃において終夜攪拌した。反応混合物を60℃に冷まし、次いで氷−水(200ml)上に注いだ。黄色の混合物を室温で30分間攪拌し、黄色の沈殿を濾過し、多量の水で洗浄し、乾燥し(真空)、3.95gの中間体(10)を与えた。
b)
【化16】
メタノール(100ml)中の中間体(10)(0.00738モル)の混合物を、チオフェン溶液(0.5ml)の存在下に触媒として活性炭上のパラジウム(10%)(1g)を用いて水素化した。水素(3当量)の吸収後、触媒をセライト上で濾過し、濾液を蒸発させ、中間体(11)を与えた。
c)
【化17】
DCM(50ml)中の中間体(11)(最大で0.00768モル;粗)、2,2−ジメチルプロパノイルクロリド(0.0096モル)及びピリジン(2ml)の混合物を室温で2時間攪拌した。溶媒を蒸発させ、酢酸(50ml)及び塩酸(濃)(5ml)を
残留物に加えた。この混合物を120℃で3時間攪拌した。混合物を冷却し、溶媒を蒸発させた。残留物をDCMとNH3水に分配した。分離された有機層をブラインで洗浄し、乾燥し(MgSO4)、溶媒を蒸発させた。粗残留物をシリカゲル上のコンビフラッシュカラムクロマトグラフィー(溶離剤:DCM/(CH3OH/NH3) 92/8)により精製した。生成物画分を集め、溶媒を蒸発させ、2.65gの中間体(12)を与えた。
【0059】
実施例A.4
a)
【化18】
アセトン(500ml)中の4−クロロベンゼンチオール(0.1モル)及び4−ブロモブタンニトリル(0.15モル)の混合物を攪拌した。Cs2CO3(0.11モル)を加え、反応混合物を窒素下で攪拌した。DIPE(500ml)を加え、沈殿を濾過した。濾液を蒸発させ、中間体(13)を与えた。
b)
【化19】
トリクロロメタン(500ml)中の中間体(13)(0.1モル)の混合物を氷上で冷却しながら、3−クロロベンゼンカルボペルオキソ酸(0.22モル)を分けて加えた。反応混合物を室温で3時間攪拌した。有機層をNaOH−溶液(500ml,1N NaOH,3x)及び水で洗浄した。有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、35gの中間体(14)を与えた。
c)
【化20】
硫酸(120ml)を塩/氷浴(−10℃)上で冷却した。中間体(14)(0.05モル)を混合物に加え、続いて硫酸と硝酸の混合物(1:1)(20ml)を30分間かけて滴下した。混合物を室温に加熱し、混合物は黄オレンジ色の溶液になった。室温で1時間攪拌した後、反応混合物を氷−水上に注ぎ、明黄色の固体を濾過し、水で洗浄した。沈殿を乾燥し(真空)、13.5gの残留物を与えた。室温でDCMを用いて残留物を磨砕し、残る固体を濾過した。濾液を蒸発させ、乾燥し、7.1gの中間体(15)を与えた。
d)
【化21】
DMSO(5ml)中の中間体(15)(0.002モル)、1−アセチル−4−ピペリジンメタナミン(0.00235モル)及びトリエチルアミン(約0.003モル)の混合物を100℃で終夜攪拌した。混合物を氷水上に注ぎ、DCMで抽出した。有機層を水で2回洗浄し、乾燥し(MgSO4)、溶媒を蒸発させた。残留物をシリカゲル上のカラムクロマトグラフィー(溶離剤:DCM/(CH3OH/NH3) 100/0から96/4)により精製した。生成物画分を集め、溶媒を蒸発させ、0.41gの中間体(16)を与えた。
e)
【化22】
メタノール(40ml)中の中間体(16)(約0.001モル)の混合物を、チオフェン溶液(0.1ml)の存在下に触媒として活性炭上のパラジウム(10%)(0.1g)を用いて水素化した。水素(3当量)の吸収後、触媒をセライト上で濾過し、濾液を蒸発させた。残留物をそのまま次の段階において用い、中間体(17)を与えた。
【0060】
実施例A.5
a)
【化23】
エタノール(500ml)中の5−クロロ−2−ニトロベンゼンアミン(0.16モル)、4−メトキシベンゼンメタンチオール(0.16モル)及び水酸化カリウム(0.30モル)の混合物を2時間攪拌し、且つ還流させた。反応混合物を冷却した。沈殿を濾過し、エタノールで洗浄し、乾燥し、48.5gの中間体(18)を与えた。
b)
【化24】
氷浴上で冷却されたDCM(180ml)中の中間体(18)(0.03モル)及びピリジン(0.06モル)の混合物に、DCM(20ml)中の2,2−ジメチルプロパノイルクロリド(0.032モル)の溶液を滴下した。反応混合物が室温に達するのを許し
た。DMAPを加え、混合物を20時間攪拌し、且つ還流させた。追加の中間体(18)(0.01モル)、2,2−ジメチルプロパノイルクロリド(0.048モル)及びピリジン(1.2モル)を加えた。混合物を2時間還流させた。溶媒を蒸発させた。残留物をDCM中に取り上げ、水で洗浄した。有機層を分離し、乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物をDIPEから結晶化させた。沈殿を濾過し、洗浄し、乾燥し、9.1gの中間体(19)を与えた。
c)
【化25】
水(500ml)中の中間体(19)(0.0748モル)、鉄(56g)及び酢酸(10ml)の混合物を4時間攪拌し、且つ還流させた。混合物を冷却した。溶媒をデカンテーションした。残留物をメタノール及びTHF中に取り上げた。混合物をジカライト上で濾過した。溶媒を蒸発させた。残留物をDCM中に取り上げた。有機層を分離し、MgSO4及びジカライト上で濾過した。溶媒を蒸発させた。残留物をDIPEから結晶化させた。沈殿を濾過し、乾燥し、21gの中間体(20)を与えた。
d)
【化26】
DCM(250ml)中の中間体(20)に、4−ホルミル−1−ピペリジンカルボン酸tert−ブチルエステル、次いで酢酸及びチタン(IV)イソプロポキシドを加えた。反応混合物を20分間攪拌した。次いでNaBH3(CN)を加え、反応混合物を2時間攪拌した。反応混合物に水を加え、有機層を分離し、乾燥し(MgSO4)、濾過し、蒸発させ、20gの中間体(21)を与えた。
e)
【化27】
中間体(21)、酢酸及び塩酸(濃)を、還流温度で終夜攪拌した。反応混合物を濃縮した。残留物を水(500ml)中に取り上げ、NaHCO3を用いて塩基性とし、300mlのDCMを用いて3回抽出した。合わせた有機層をブラインで洗浄し、MgSO4上で乾燥し、濃縮して、11.4gの中間体(22)を与えた。
【0061】
実施例A.6
【化28】
化合物(11)及びトリフルオロ酢酸を、マイクロ波中で120℃において60分間攪拌した。反応混合物を冷却した。溶媒を蒸発させた。残留物を酢酸エチル中に取り上げ、次いでH2O/NaHCO3溶液で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させ、2.3gの中間体(23)を与えた。
【0062】
以下の中間体を、化合物(44)から類似して製造した:
【化29】
【0063】
実施例A.7
a)
【化30】
窒素流下の反応。メチルテトラヒドロ−2H−チオピラン−4−イル−ケトン(0.039モル;エタノール中の50%溶液)を、DCM(32ml)及び酢酸(4ml)中の中間体(20)(0.03モル)の混合物に室温で加え、次いで5分間攪拌した。反応混合物にNaBH3(CN)(0.04モル)を加え、次いで室温で1時間攪拌した。反応混合物を水で洗浄した。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物をDIPE中に懸濁させた。沈殿を濾過し、乾燥し(真空,室温)、11.2gの中間体(25)を与えた。
b)
【化31】
中間体(25)と酢酸の混合物を、マイクロ波中で150℃において100分間加熱した。溶媒を蒸発させた。残留物をDCM中に取り上げ、水/NaHCO3で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させた。シリカゲル及び溶離剤としてDCM:MeOH/NH3(100から97:3)を用い、短フィルター上で残留物を精製した。生成物画分を集め、蒸発させ、1.3gの中間体(26)を与えた。
c)
【化32】
中間体(26)及びトリフルオロ酢酸を、マイクロ波中で120℃において30分間攪拌した。反応混合物を冷却した。溶媒を蒸発させた。残留物を酢酸エチル中に取り上げ、次いで水/NaHCO3溶液で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させた。粗残留物を次の段階に用い、1.3gの中間体(27)を与えた。
【0064】
実施例A.9
【化33】
DCM(20ml)中の中間体(2)(0.0047モル)、tert−ブチルアセチルクロリド(0.81g,0.006モル)及びピリジン(2ml)の混合物を、室温で終夜攪拌した。溶媒を蒸発させた。酢酸(25ml)及びHCl(2ml)を残留物に加えた。この混合物をマイクロ波オーブン中で190℃において75分間攪拌した(2つに分けて(in 2 portions))。溶媒を蒸発させた。残留物をDCM(250ml)とNH3水に分配した。分離された有機層を水で洗浄し、乾燥し(MgSO4)、濾過し、溶媒を蒸発させて残留物を与え、それをシリカゲル上のカラムクロマトグラフィー(Biotage;溶離剤:CH2Cl2/(CH3OH/NH3) 100/0から96)により精製し、0.85gの中間体(28)を与えた。
【0065】
実施例A.10
【化34】
アセトン(10ml)中の3−ヨードフェニル(10ミリモル)、1−ブロモ−3−メトキシプロパン(14.7ミリモル)及びK2CO3(20.26ミリモル)の混合物を、50℃で40時間攪拌した。塩を濾過し、洗浄した。濾液を濃縮し、残留物をカラムクロマトグラフィーにより、溶離剤として酢酸エチル/ヘプタン(0:100から20:80)を用いて精製した。生成物画分を集め、蒸発させ、中間体(29)を与えた。
【0066】
実施例A.11
【化35】
アセトン(10ml)中の3−ヨードフェニル(10ミリモル)、1−ブロモ−3−シアノプロパン(14.7ミリモル)及びK2CO3(20.26ミリモル)の混合物を、50℃で40時間攪拌した。塩を濾過し、洗浄した。濾液を濃縮し、残留物をカラムクロマトグラフィーにより、溶離剤として酢酸エチル/ヘプタン(0:100から20:80)を用いて精製した。生成物画分を集め、蒸発させ、中間体(30)を与えた。
【0067】
B.最終的化合物の合成
実施例B.1
【化36】
中間体(3)(0.7g,0.00193モル)、アセチルアセテート(0.26g,0.0025モル)及びDCM(20ml)の混合物を、室温で終夜反応させた。混合物を最初に水(15ml)で洗浄し、次いでNH3水(2x15ml)で2回及び最後にブライン(15ml)で洗浄した。混合物をIsolute HM−NTM上で濾過し、次いで窒素流下で溶媒を蒸発させた。残留物をDIPEから結晶化させ、0.035gの化合物(1)を与えた。油性の生成物を2−プロパノール(8ml)中に溶解し、HCl/2−プロパノール(6N,0.5ml)溶液を加えた。溶媒を蒸発させた。ジエチルエーテルを用いて残留物を磨砕した。沈殿を濾過し、乾燥し(真空)、0.53gの化合物(2)を与えた。
【0068】
中間体(28)を無水酢酸と反応させることにより、化合物(46)を類似して製造した。
実施例B.2
【化37】
DCM(20ml)中の中間体(8)(最大で0.0009モル)、2,2−ジメチルプロパノイルクロリド(0.170ml)及びピリジン(0.25ml)の混合物を、室温で2時間攪拌した。溶媒を蒸発させ、粗残留物に1,2−ジクロロエタン(4ml)を加えた。混合物をマイクロ波中で190℃において1時間加熱した。混合物を冷まし、DCM(4ml)及びNaOH溶液(1ml,1N)を加えた。反応混合物を攪拌し、Isolute HM−NTMフィルター上で濾過し、溶媒を蒸発させた。生成物を逆相高−性能液体クロマトグラフィーにより精製した。生成物画分を集め、溶媒を蒸発させ、0.123gの化合物(3)を与えた。
【0069】
2,2−ジメチルプロパノイルクロリドを中間体(17)と反応させることにより、化合物(10)を類似して製造し、中間体(2)を2,2−ジメチルアセチルクロリドと反応させることにより、化合物(47)を類似して製造した。
【0070】
実施例B.3
【化38】
DCM(10ml)中の中間体(12)(0.00061モル)、無水酢酸(0.0008モル)及びトリエチルアミン(0.17ml)の混合物を、室温で終夜攪拌した。水を加え、混合物を攪拌し、層を分離した。Isolute HM−NTMフィルターに通過させることにより有機層を乾燥した。溶媒を蒸発させた。ジエチルエーテル中で塩酸を用いて残留物を結晶化させ、0.260gの化合物(4)を与えた。
【0071】
中間体(12)をプロパノイルクロリド、メトキシカルボニルクロリド、ビス(トリフルオロ酢酸)無水物又は2−メトキシアセチルクロリドと反応させることにより、それぞれ化合物(6)、(7)、(8)及び(9)を類似して製造した。溶媒としてピリジン及びTHFの存在下で中間体(22)を無水酢酸と反応させることにより、化合物(11)を類似して製造した。
【0072】
実施例B.4
【化39】
DCM(5ml)中の中間体(12)(0.000675モル)、ギ酸(0.00087モル)、HBTU(0.00087モル)及びトリエチルアミン(0.15ml)の混合物を50℃で攪拌した。2時間後、追加分のギ酸(0.00087モル)を加え、混合物を室温で終夜攪拌した。次いでDCM(5ml)及び水(1ml)を加え、混合物をIsolute HM−NTMフィルター上で濾過した。溶媒を蒸発させ、残留物を逆相高−性能液体クロマトグラフィーにより精製した。生成物画分を集め、溶媒を蒸発させ、0.205gの化合物(5)を与えた。
【0073】
実施例B.5
【化40】
トリクロロメタン(10ml)中の化合物(11)(0.0003モル)の混合物に、3−クロロベンゼンカルボペルオキソ酸(0.0007モル;77%)を室温で加えた。反応混合物を室温で30分間攪拌し、次いで水及びNaOH(1N)を加えた。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物を逆相カラム上で精製した。生成物画分を集め、溶媒を蒸発させ、乾燥するまで共−蒸発させ、0.075gの化合物(12)を与えた。
【0074】
中間体(29)から出発し、類似して化合物(49)を製造した。化合物(50)から出発し、類似して化合物(53)を製造した。化合物(54)から出発し、類似して化合物(62)及び(63)を製造した。化合物(72)から出発し、類似して化合物(73)を製造した。化合物(52)から出発し、類似して化合物(56)を製造した。化合物(51)から出発し、類似して化合物(61)を製造した。化合物(57)から出発し、類似して化合物(60)を製造した。化合物(58)から出発し、類似して化合物(59)を製造した。化合物(65)から出発し、類似して化合物(67)を製造した。化合物(66)から出発し、類似して化合物(68)を製造した。化合物(74)から出発し、類似して化合物(64)を製造した。化合物(75)から出発し、類似して化合物(69)を製造した。化合物(70)から出発し、類似して化合物(71)を製造した。化合物(81)から出発し、類似して化合物(82)を製造した。化合物(83)から出発し、類似して化合物(84)を製造した。化合物(85)から出発し、類似して化合物(86)を製造した。化合物(87)から出発し、類似して化合物(88)を製造した。化合物(89)から出発し、類似して化合物(90)を製造した。化合物(91)から出発し、類似して化合物(92)を製造した。化合物(93)から出発し、類似して化合物(94)を製造した。化合物(95)から出発し、類似して化合物(96)を製造した。化合物(97)から出発し、類似して化合物(98)を製造した。化合物(99)から出発し、類似して化合物(100)を製造した。化合物(101)から出発し、類似して化合物(102)を製造した。
【0075】
実施例B.6
【化41】
THF(10ml)中の中間体(23)(0.001モル)、3−ブロモ−1−プロパノール(0.003モル)及びCs2CO3(0.002モル)の混合物を、60℃で1時間攪拌した。反応混合物を冷却し、ジカライト上で濾過し、濾液の溶媒を蒸発させた。残留物を逆相HPLCにより精製した。所望の画分を集め、溶媒を蒸発させ、完全に乾燥するまで共−蒸発させ、0.170gの化合物(13)を与えた。
【0076】
中間体(23)を1−ブロモ−2−メトキシ−エタン、3−ブロモプロパンニトリル、1−(ブロモメチル)−4−フルオロ−ベンゼン、(ブロモメチル)−シクロブタン又は2−ブロモプロパン−ニトリルと反応させることにより、それぞれ化合物(14)、(15)、(17)、(41)及び(45)を類似して製造した。中間体(24)を1−ブロモ−3−ヒドロキシ−プロパン、1−ブロモ−2−メトキシ−エタン又は(ブロモメチル)−シクロプロパンと反応させることにより、それぞれ化合物(21)、(22)及び(37)を類似して製造した。K2CO3の存在下に、溶媒としてのDMF中で中間体(23)を3−ペンチルブロミド、2−(4−フルオロフェニル)エチルブロミド、4−ヘプチルブロミド、ベンジルブロミド、2−プロピルヨーダイド又は4−ニトロベンジルブロミドと反応させることにより、それぞれ化合物(51)、(52)、(65)、(72)、(74)及び(75)を類似して製造した。
【0077】
実施例B.7
【化42】
ジオキサン(80ml)中の中間体(23)(最大で0.005モル)及び4−ヨードベンゾニトリル(0.010モル)の混合物を脱ガスし、反応混合物上に窒素流を導入した(3回)。次いでCs2CO3(4g)を加え、混合物を脱ガスし、再び窒素を反応混合物上に導入した。次いでPd2(dba)3(0.200g)及びキサントフォス(0.150g)を加え、脱ガス及び窒素作用を行なった。反応混合物上に窒素バルーンを残し、混合物を100℃で終夜攪拌した。混合物を冷却し、濾過し、濾液を蒸発させた。残留物をDCM中に取り上げ、水で洗浄した。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物をHPLCにより精製した。生成物画分を集め、溶媒を蒸発させ、1.100gの化合物(18)を与えた。
【0078】
中間体(23)を1−ヨードシクロペンタン、4−クロロピリジン又は2−ブロモチアゾールと反応させることにより、それぞれ化合物(32)、(33)及び(38)を類似して製造した。中間体(23)を3−ブロモピリジン、2,6−ジクロロヨードベンゼン、1−フルオロ−4−ヨードベンゼン、2−ヨードチオフェン及び2−クロロヨードベンゼンと反応させることにより、それぞれ化合物(54)、(57)、(58)、(66)及び(70)を類似して製造した。中間体(24)を2−ブロモ−チアゾールと反応させることにより、化合物(77)を類似して製造した。中間体(23)を1−ヨード−3−メトキシベンゼン、3−ヨード−ベンゾニトリル、中間体(29)、1−ヨード−4−(トリフルオロメチル)ベンゼン、1−クロロ−3−ヨードベンゼン、1−ヨード−3−(トリフルオロメチル)ベンゼン及び2−ヨードベンゾニトリルと反応させることにより、それぞれ化合物(81)、(83)、(85)、(87)、(89)、(91)及び(93)を類似して製造した。中間体(23)を中間体(30)、3−ブロモ−N,N−ジメチル−ベンゼンアミン、1−ブロモ−3−(1−メチルエトキシ)ベンゼン及び2−ヨード−1,3−ジメトキシベンゼンと反応させることにより、それぞれ化合物(95)、(97)、(99)及び(101)を類似して製造した。
【0079】
実施例B.8
【化43】
中間体(3)(1.7g,0.0047モル)及びギ酸メチル(25ml)の混合物を40℃で終夜反応させた。混合物を窒素流下に60℃において濃縮した。残留物をDIPEから、1滴の2−プロパノールを用いて結晶化させ、1.55gの化合物(28)を与えた。
【0080】
中間体(22)から出発して、化合物(44)を類似して製造した。
【0081】
中間体(28)から出発して、化合物(48)を類似して製造した。
【0082】
実施例B.9
【化44】
ジオキサン(15ml)中のヨードエタン(0.01モル)の混合物に、Cs2CO3(0.006モル)を加えた。ジオキサン(10ml)中の中間体(27)(0.003モル)を加え、混合物を90℃で2時間攪拌した。反応混合物を冷却し、溶媒を蒸発させた。残留物をトリクロロメタン(50ml)中に取り上げ、次いで3−クロロベンゼンカルボペルオキソ酸(0.018モル)を加えた。この反応混合物を室温で1時間攪拌した。反応混合物をNaOH 1M水溶液で2回洗浄した。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物を高−性能液体クロマトグラフィー(NH4HCO3緩衝液を用いる標準的な勾配溶離)により精製した。生成物画分を集め、溶媒を蒸発させた。この残留物をDIPE及び少量のCH3CN中に懸濁させた。沈殿を濾過し、乾燥し(真空,50℃)、0.064gの化合物(29)を与えた(融点194℃)。
【0083】
中間体(27)を2−ヨード−プロパン又は(ブロモメチル)−シクロプロパンと反応させることにより、それぞれ化合物(30)及び(31)を類似して製造した。
【0084】
実施例B.10
【化45】
酢酸中の1−{4−[2−tert−ブチル−5−(1−オキシ−ピリジン−4−スルホニル)−ベンゾイミダゾール−1−イルメチル]−ピペリジン−1−イル}−エタノン(B.5の方法に従って化合物(33)を酸化することにより製造される)及び鉄の混合物を、密閉された容器中で60℃において2時間振盪させた。反応混合物を冷却した。生成物を含有する溶媒をデカンテーションすることにより、過剰の鉄を除去した。5mlの酢酸を用いて鉄の残留物を濯いだ(再びデカンテーション)。合わせた溶媒層を蒸発させた。残留物をDCM中に取り上げ、水で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させた。残留物をDIPE及びいくらかの2−プロパノールから結晶化させた。固体を濾過し、洗浄し、乾燥して0.133gの化合物(34)を与えた。
【0085】
実施例B.11
【化46】
エタノール中の化合物(39)及びナトリウムエトキシドを脱ガスし、反応物上に窒素を導入した(密閉された容器中で)。この混合物を室温で15分間攪拌した。室温でヨー
ドメタンを加え、反応混合物を3時間攪拌した。溶媒を濃縮した。残留物をDCM中に取り上げ、水で洗浄した。MgSO4を用いて有機層を乾燥し、濾過し、蒸発させた。シリカゲルを有するカラム上で、溶離剤として100/0から98/2のDCM/CH3OH(7N NH3)を用い、残留物を精製した。生成物画分を集め、蒸発させた。エーテル中のHCl(1M)及びアセトニトリルを用い、残留物をHCl塩として結晶化させた。固体を濾過し、洗浄し、乾燥し、0.09gの化合物(42)を与えた。
【0086】
実施例B.12
【化47】
ジイソプロピルエチルアミン(0.003814モル)及び2−ブロモピリジンをトルエン(5ml)に加えた。真空にすることによりこの溶液を脱ガスし、その上に窒素雰囲気を導入した。最初に調製された溶液に、次いでシリンジを用いてキサントフォス(0.041g)、Pd2(dba)3(0.0165g)及びジオキサン(5ml)の新しい溶液を加えた。反応物を軽真空下に置いた。ジオキサン(5ml)中の中間体(23)の溶液も、シリンジを用いて加えた。反応混合物を週末に及んで84℃で振盪させた。溶媒を蒸発させ、DCM(40ml)及び水(10ml)を用いて生成物を仕上げた。次いで溶媒を蒸発させた。40gのシリカゲルカラムを用いて生成物を精製した(溶離剤:DCM:CH3OH/NH3(7N) 100/0から98/2)。生成物画分を一緒にし、完全に乾燥するまで溶媒を蒸発させ、0.743gの化合物(50)を与えた。
【0087】
実施例B.13
【化48】
化合物(73)(0.001326モル)をTHF(10ml)中に溶解した。溶液を脱ガスし、次いで溶液を窒素雰囲気下とした。溶液を0℃で冷却した。次いでシリンジを用い、ナトリウムビス(トリメチルシリル)アミドを加えた。反応混合物を0℃で1時間攪拌した。シリンジを用いてヨードメタン(0.947ml)を反応溶液に加え、反応混合物を0℃で1時間攪拌した。ジクロロメタン及び水を用いて生成物を仕上げた。有機層を乾燥した(MgSO4)。溶媒を蒸発させた。RP HPLC,方法Bを用いて残留物を精製した。画分を一緒にし、溶媒を蒸発させた。DIPEを用いて生成物を固化させた。固体を濾過し、洗浄し、オーブン中で乾燥し、0.165gの化合物(55)を与えた。
【0088】
実施例B.14
【化49】
THF(10ml)中の中間体(23)(0.0043モル)、ブロモメチルシクロプロパン(0.01モル)及びCs2CO3(0.008モル)の混合物を脱ガスした。反応混合物を窒素下に65℃において20時間攪拌した。いくらかのジスルフィドが生成したので、NaBH4を加え、反応混合物を65℃でさらに24時間攪拌した。反応混合物を濾過して塩を除去した。濾液をDCMで希釈し、水で洗浄した。有機層を乾燥し(MgSO4)、濾過し、完全に乾燥するまで蒸発させ、2gの化合物(16)を与えた。
【0089】
実施例B.15
【化50】
炭素上の白金(5%)+0.5%V(0.3g)を、窒素流下でTHF(50ml)中に懸濁させ、次いで化合物(69)(0.00157モル)を加え、3当量の水素が吸収されるまで反応混合物を水素雰囲気下で攪拌した。ジカライト上の濾過により触媒を除去し、次いで溶媒を蒸発させ、0.839gの化合物(78)を与えた。
【0090】
実施例B.16
【化51】
化合物(78)(0.001521モル)をTHF(100ml)中に溶解し、DIPE(0.001521モル)を加えた。アセチリクロリド(0.001521モル)を加え、反応混合物を室温で30分間攪拌した。いくらかの1M NaOH溶液を加え、THFを用いて生成物を仕上げた。有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。方法Aに従って残留物を精製した。画分を一緒にし、溶媒を蒸発させ、0.375gの化合物(79)を与えた。
【0091】
実施例B.17
【化52】
化合物(79)(0.000286モル)をDMF(10nl)中に溶解した。反応混合物をN2雰囲気下に0℃において冷却した。次いで水素化ナトリウム(0.002001モル;鉱油中の60%分散液)を加えた。反応混合物を30分間攪拌し、次いでヨードメタン(0.002573モル)を加えた。反応混合物を1時間攪拌し、ジクロロメタン(50ml)及びH2O(20ml)を用いて生成物を仕上げた。有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。DIPEを用いて生成物を固化させた。固体を濾過し、洗浄し、オーブン中で乾燥し、0.132gの化合物(80)を与えた。
【0092】
表F−1は、上記の実施例の1つに従って製造された化合物を挙げている。
【0093】
【表1】
【0094】
【表2】
【0095】
【表3】
【0096】
【表4】
【0097】
【表5】
【0098】
【表6】
【0099】
【表7】
【0100】
【表8】
【0101】
【表9】
【0102】
表F−2は、実施例B.5及びB.7に記載された方法を用いて、且つ化合物(116)の場合にはさらに実施例B.10に記載されたFe/酢酸を用いる処理を用いて製造された化合物を挙げている。
【0103】
【表10】
【0104】
C.化合物の同定
C1.LCMS
本発明の化合物のLCMS−特性化のために、以下の方法を用いた。
【0105】
一般的方法A
HPLC測定は、脱ガス器を有するクウォーターナリーポンプ、オートサンプラー、カラムオーブン(他に指示しなければ40℃に設定)、ダイオード−アレー検出器(DAD)及び下記のそれぞれの方法において特定されるカラムを含んでなるAlliance HT 2790(Waters)システムを用いて行なわれた。カラムからの流れはMS分光計に分けられた。MS検出器は、エレクトロスプレーイオン化源を用いて形成された。0.1秒の滞留時間を用いて1秒内に100から1000まで走査することにより、質量スペクトルを取得した。毛管針電圧は3kVであり、源温度は140℃に保たれた。ネブライザーガスとして窒素を用いた。Waters−Micromass MassLynx−Openlynxデータシステムを用いてデータ取得を行なった。
【0106】
一般的方法B
LC測定は、バイナリーポンプ、サンプルオルガナイザー、カラムヒーター(55℃に設定)、ダイオードアレー検出器(DAD)及び下記のそれぞれの方法において特定されるカラムを含んでなるAcquity UPLC(Waters)システムを用いて行なわれた。カラムからの流れはMS分光計に分けられた。MS検出器は、エレクトロスプレーイオン化源を用いて形成された。0.02秒の滞留時間を用いて0.18秒内に100から1000まで走査することにより、質量スペクトルを取得した。毛管針電圧は3.5kVであり、源温度は140℃に保たれた。ネブライザーガスとして窒素を用いた。Waters−Micromass MassLynx−Openlynxデータシステムを用いてデータ取得を行なった。
【0107】
LCMS法1
一般的方法Aに加え:逆相HPLCをXterra MS C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。3種の移動相(移動相A:95% 25mM酢酸アンモニウム+5%アセトニトリル;移動相B:アセトニトリル;移動相C:メタノール)を用い、6.5分内に100%Aから1%A、49%B及び50%Cにし、1分内に1%A及び99%Bにし、これらの条件を1分間保持し、100%Aを用いて1.5分間再平衡化する勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0108】
LCMS法2
一般的方法Bに加え:架橋エチルシロキサン/シリカハイブリッド(BEH) C18カラム(1.7μm,2.1x50mm;WatersAcquity)上で、0.8ml/分の流量を用い、逆相UPLC(超性能液体クロマトグラフィー(Ultra Performance Liquid Chromatography))を行なった。2種の移動相(移動相A:H2O中の0.1%ギ酸/メタノール 95/5;移動相B:メタノール)を用い、1.3分内に95%A及び5%Bから5%A及び95%Bにし、0.2分間保持する勾配条件を実施した。0.5μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0109】
LCMS法3
一般的方法Aに加え:カラムヒーターを45℃に設定した。逆相HPLCをXterra MS C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。3種の移動相(移動相A:H2O中の0.1%ギ酸/メタノール 95/5;移動相B:アセトニトリル;移動相C:メタノール)を用い、7分内に100%Aから1%A、49%B及び50%Cにし、これらの条件を1分間保持する勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に1
0Vであった。
【0110】
LCMS法4
一般的方法Aに加え:カラムヒーターを60℃に設定した。逆相HPLCをXterra MS C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。3種の移動相(移動相A:95% 25mM酢酸アンモニウム+5%アセトニトリル;移動相B:アセトニトリル;移動相C:メタノール)を用い、6.5分内に100%Aから50%B及び50%Cにし、0.5分内に100%Bにし、これらの条件を1分間保持し、100%Aを用いて1.5分間再平衡化する勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0111】
LCMS法5
一般的方法Aに加え:逆相HPLCをAtlantis C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。2種の移動相(移動相A:70%メタノール+30% H2O;移動相B:H2O中の0.1%ギ酸/メタノール 95/5)を用い、12分内に100%Bから5%B+95%Aにする勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0112】
C2.融点
複数の化合物に関し、DSC823e(Mettler−Toledo)を用いて融点を決定した。30℃/分の温度勾配を用いて融点を測定した。報告される値はピーク値である。最高温度は400℃であった。
【0113】
複数の化合物に関し、直線状温度勾配を有する加熱板、スライディングポインター(sliding pointer)及び度摂氏における温度目盛からなるKoflerホットベンチ(hot bench)を用いて融点を得た。
【0114】
【表11】
【0115】
【表12】
【0116】
【表13】
【0117】
D.薬理学的実施例
D.1 ヒトCB2受容体の活性化に反応するcAMPの阻害
均一時間分解蛍光(HTRF)アッセイを介し、ヒトCB2(hCB2)受容体が活性化される時のホルスコリン−活性化cAMP生産を阻害する能力を測定することにより、試験化合物の機能的活性を評価した。
【0118】
hCB2が安定にトランスフェクションされたCHO−K1細胞を、T175 Falconフラスコ中で、2%の溶液A(5.106IU/lのペニシリンG,5g/lの硫酸ストレプトマイシン,5.5g/lのピルベート,14.6g/lのL−グルタミン,1MのNaOH)及び10%の胎児ウシ血清が補足されたDMEM/NUT MIX F−12倍地中において80〜90%密集まで生育させた。実験の前に倍地を除去し、細胞をPBS/EDTA(140mM NaCl,1mM Na2−EDTA,8mM Na2HPO4.2H2O,8.5mM KH2PO4,2.7mM KCl,21mM グルコース)で洗浄し、刺激緩衝液(HBSS 1x,IBMX 1mM,Hepes 5mM,MgCl2 10mM,BSA 0.1%,pH7.4)中に再懸濁させた。hCB2実験のために、細胞をml当たり106個の細胞の濃度まで希釈した。cAMP Dynamic HTRFキット(CIS bio international,France)を用い、製造者の推薦に従ってアッセイを行なった。
【0119】
CB2に関し、384平底黒ポリスチレンアッセイプレート(Costar)の各ウェルを、15μMのホルスコリン及び試験化合物(3%DMSO中)、3%DMSO又は10μM Win55212−2(3%DMSO中)を含有する10μlの刺激緩衝液で満たした。次いで20μlの希釈されたhCB2−CHO−K1細胞を加えた(ウェル当たり20,000個の細胞)。室温における暗所中での30分間のインキュベーションの後、10μlのcAMP−XL665及び10μlの抗−cAMPクリプテート(両方とも1/100の最終的な希釈において)を細胞に加えた。
【0120】
室温における暗所中で反応混合物を1〜24時間平衡化した後、Discoveryミクロプレート蛍光カウンター(Perkin Elmer)を用いて665nm及び620nmにおいて蛍光を測定し、665nm/620nmのシグナル比を計算した。試験化合物のシグナル比を、それぞれDMSO標準(最大シグナル比,cAMPの阻害なし)及びhCB2に関するWIN55212−2(最小シグナル比,cAMPの最大阻害)のシグナル比に対して相対的に表した。各試験化合物に関して作られる用量反応曲線から、cAMPレベルの最大阻害の50%が観察される用量(EC50,表中でpEC50=−log(EC50)値として表される)及びWIN55212−2(hCB2に関する)と比較して10μMの試験化合物を用いて達成される阻害のレベルを計算した。
【0121】
【表14】
【0122】
【表15】
【0123】
D.2 比較データ
体温の低下、平伏体姿勢及び散瞳のようなCB1に関連する望ましくない副作用を、本発明の複数の化合物及び引用文献国際公開第2006/048754号パンフレットにより包含される複数の化合物に関して測定した。両方の組の化合物に関し、処置された動物の半分より多くで体温への効果、すなわち低下が観察されるLAD(最低許容用量)を決定した。データを表D−3に挙げる。
【0124】
【表16】
【0125】
【表17】
【技術分野】
【0001】
本発明は、選択的カンナビノイド受容体2作動性を有する新規な式(I)のベンズイミダゾール化合物、これらの化合物を含んでなる製薬学的組成物、これらの化合物の化学的製造方法ならびに動物、特にヒトにおけるカンナビノイド受容体の媒介に結び付けられる疾患の処置におけるそれらの使用に関する。
【背景技術】
【0002】
マリファナ由来カンナビノイドΔ9−テトラヒドロ−カンナビノイド(Δ9−THC)のような古典的なカンナビノイドは、体内の特異的なカンナビノイド受容体との相互作用を介してそれらの薬理学的効果を生む。これまでに2つのカンナビノイド受容体:哺乳類脳及び末梢組織において見出された受容体CB1及び末梢組織において主に見出された受容体CB2が特性化された。これらの受容体の1つ又は両方に関するアゴニスト又はアンタゴニストである化合物は、多様な薬理学的効果を与えることが見出された。CB2受容体に関する高い選択性は、カンナビノイド構造に伴って見られる中心的な不利な事柄を避けながらCB受容体アゴニストの有益な効果を利用するための手段を与えることができると思われるので、選択的CB2作動活性を有するカンナビノイド類似物の開発に有意な興味が持たれる(例えば非特許文献1を参照されたい)。
【0003】
特許文献1は、アルツハイマー病、不安障害、抑うつ性障害、骨粗しょう症、心臓血管病、慢性関節リウマチ又は前立腺ガンのようなエストロゲン受容体−βに関連する疾患の処置における使用のための、エストロゲン受容体−βリガンドとしてのベンズイミダゾール化合物を開示している。特許文献2は、CB2受容体活性により媒介される状態の処置において有用な、CB2作動活性を有するスルホニルベンズイミダゾール誘導体を開示している。
【0004】
本発明の化合物は、複素原子が常に置換されている1−ピペリジン−4−イルメチル又は1−テトラヒドロチオピラン−4−イルメチル基の存在により、挙げられた当該技術分野において既知の化合物と構造的に異なる。
【0005】
本発明の化合物は、予期に反し、当該技術分野において既知の特許文献1の化合物と比較して、体温の低下及び平伏体姿勢(flat body posture)のようなCB1関連副作用のない選択的CB2アゴニストであることが見出された。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第2002/46168号パンフレット
【特許文献2】国際公開第2006/048754号パンフレット
【非特許文献】
【0007】
【非特許文献1】Expert Opinion on Investigational Drugs,14(6),2005年,695−703
【発明の概要】
【0008】
本発明は、立体化学的異性体を含んで、式(I)
【0009】
【化1】
【0010】
[式中、
nは整数0、1又は2であり;
XはSO、SO2又はN−(CO)−R1であり;
R1は水素;
C1−6アルキル;
C1−6アルキルオキシ;
C1−4アルキルオキシC1−4アルキル;又は
ポリハロC1−6アルキル
であり;
R2はC1−6アルキルであり;
R3は水素、ハロ、C1−4アルキル、C1−4アルキルオキシ、トリフルオロメチル又はシアノであり;
R4はC1−8アルキル;
C3−8シクロアルキルで置換されたC1−8アルキル;
ポリハロC1−8アルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC1−8アルキル;
C3−8シクロアルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC3−8シクロアルキル;
テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、
アリール;あるいは
ヘテロアリール
であり;
アリールはフェニル;あるいはハロ、ヒドロキシ、C1−4アルキル、ポリハロC1−4アルキル、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、NR5R6、R7−カルボニル、R7−SO2−又はヒドロキシ、NR5R6、R7−カルボニルもしくはR7−SO2−で置換されたC1−4アルキルからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたフェニルであり;
ヘテロアリールはフラニル、チオフェニル、ピロリル、ピラゾリル、イミダゾリル、イソオキサゾリル、チアゾリル、トリアゾリル、テトラゾリル、イソチアゾリル、チアジアゾリル、オキサジアゾリル、ピリジニル、ピリダジニル、ピリミジニル又はピラジニルから選ばれ;
ここでR5及びR6は互いに独立して水素、C1−4アルキル、ポリハロC1−4アルキル、アミノスルホニル又はC1−8アルキルスルホニル;あるいはR7−カルボニルから選ばれ;
ここでR5及びR6は、R5及びR6を有する窒素原子と一緒になって、ピロリジニル、ピペリジニル、ピペラジニル又はモルホリニル環を形成することができ;そして
ここでR7はC1−4アルキル、ヒドロキシ、アミノ、モノ−もしくはジ−(C1−4アルキル)アミノ、(ヒドロキシC1−4アルキル)アミノ、(C1−4アルキルオキシC1−4アルキル)アミノ、ジ(C1−4アルキル)アミノC1−4アルキル、ピロリジニル、ピペリジニル、モルホリニル又はN−メチル−ピペラジニルである]
の化合物あるいはその製薬学的に許容され得る酸付加塩又はその溶媒和物に関する。
【0011】
前記の定義において用いられる場合:
−ハロはフルオロ、クロロ、ブロモ及びヨードの総称であり;
−C1−4アルキルは、1〜4個の炭素原子を有する直鎖状及び分枝鎖状飽和炭化水素基、例えばメチル、エチル、プロピル、ブチル、1−メチルエチル、2−メチルプロピルなどを定義し;
−C1−6アルキルは、C1−4アルキル及び5もしくは6個の炭素原子を有するその高級同族体、例えば2−メチルブチル、ペンチル、ヘキシルなどを含むものとし;
−C1−8アルキルは、C1−6アルキル及び7〜8個の炭素原子を有するその高級同族体、例えばヘプチル、エチルヘキシル、オクチルなどを含むものとし;
−ポリハロC1−4アルキルは、ポリハロ置換されたC1−4アルキル、特に2〜6個のハロゲン原子で置換された(上記で定義されたような)C1−4アルキル、例えばジフルオロメチル、トリフルオロメチル、トリフルオロエチルなどとして定義され;
−C3−6シクロアルキルは、シクロプロピル、シクロブチル、シクロペンチル及びシクロヘキシルの総称であり;
−C3−8シクロアルキルは、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル及びシクロオクチルの総称であり;
−C6−8シクロアルキルは、シクロヘキシル、シクロヘプチル及びシクロオクチルの総称である。
【0012】
前記で用いられた「立体化学的異性体」という用語は、式(I)の化合物が有することができるすべての可能な異性体を定義する。他にことわるか又は指示しなければ、化合物の化学的名称はすべての可能な立体化学的異性体の混合物を示し、該混合物は基本の分子構造のすべてのジアステレオマー及びエナンチオマーを含有する。さらに特定的に、ステレオジェン中心はR−又はS−立体配置を有することができ;二価環状(部分的)飽和基上の置換基は、シス−又はトランス−立体配置のいずれかを有することができる。
【0013】
式(I)の化合物の立体化学的異性体は、明らかに本発明の範囲内に包含されることが意図されている。
【0014】
式(I)の化合物及びそれらの製造において用いられる中間体の絶対立体化学的配置は、例えばX−線回折のような周知の方法を用いて当該技術分野における熟練者が容易に決定することができる。
【0015】
さらに、式(I)のいくつかの化合物及びそれらの製造において用いられる中間体のいくつかは、多形を示し得る。本発明は、上記に記載した状態の処置において有用な性質を有するいずれの多形相も包含することが理解されるべきである。
【0016】
上記で言及した製薬学的に許容され得る酸付加塩は、式(I)の化合物が形成することができる治療的に活性な無毒性の酸付加塩の形態を含むものとする。これらの製薬学的に許容され得る酸付加塩は、塩基形態をそのような適した酸で処理することにより簡単に得ることができる。適した酸は、例えば無機酸、例えばハロゲン化水素酸、例えば塩酸又は臭化水素酸、硫酸、硝酸、リン酸などの酸;あるいは有機酸、例えば酢酸、プロパン酸、
ヒドロキシ酢酸、乳酸、ピルビン酸、シュウ酸(すなわちエタン二酸)、マロン酸、コハク酸(すなわちブタン二酸)、マレイン酸、フマル酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、シクラミン酸、サリチル酸、p−アミノサリチル酸、パモ酸などの酸を含む。
【0017】
逆に、適した塩基を用いる処理により、該塩形態を遊離の塩基形態に転換することができる。
【0018】
式(I)の化合物は、溶媒和されない形態及び溶媒和された形態の両方において存在することができる。「溶媒和物」という用語は本明細書で、本発明の化合物及び1種もしくはそれより多い製薬学的に許容され得る溶媒分子、例えば水又はエタノールを含んでなる分子会合を記述するために用いられる。「水和物」という用語は、該溶媒が水である場合に用いられる。
【0019】
興味深い式(I)の化合物は、以下の制限の1つもしくはそれより多くが適用される式(I)の化合物である:
a)nが整数0であるか、又はnが整数2であるか;あるいは
b)XがSO2であるか;あるいは
c)XがN−(CO)−R1であり、ここでR1は水素であるか;あるいは
d)XがN−(CO)−R1であり、ここでR1はC1−6アルキル、好ましくはメチル又はエチルであるか;あるいは
e)XがN−(CO)−R1であり、ここでR1はC1−6アルキルオキシ、好ましくはメチルオキシであるか;あるいは
f)XがN−(CO)−R1であり、ここでR1はC1−4アルキルオキシC1−4アルキル、好ましくはメチルオキシメチルであるか;あるいは
g)XがN−(CO)−R1であり、ここでR1はポリハロC1−6アルキル、好ましくはトリフルオロメチルであるか;あるいは
h)R2がC1−6アルキルである、特にR2がtert−ブチル又は−CH2−tert−ブチルであるか;あるいは
i)R3が水素であるか;あるいは
j)R4がC1−8アルキル、C3−8シクロアルキルで置換されたC1−8アルキル、ポリハロC1−8アルキルであるか;あるいは
k)R4がヒドロキシ、C1−4アルキルオキシ、シアノで置換されたC1−8アルキルであるか;あるいは
l)R4がアリールであるか;あるいは
m)R4がヘテロアリールである。
【0020】
1つの態様において本発明は、nが整数0、1又は2であり;XがSO2又はN−(CO)−R1であり;R1が水素、C1−6アルキル、C1−6アルキルオキシ、C1−4アルキルオキシC1−4アルキル;あるいはポリハロC1−6アルキルであり;R2がC1−6アルキルであり;R3が水素であり;R4がC1−8アルキル;C3−8シクロアルキルで置換されたC1−8アルキル;ポリハロC1−8アルキル;C3−8シクロアルキル;ヒドロキシ、C1−4アルキルオキシ、シアノ又はアリールで置換されたC1−8アルキル;アリール;又はヘテロアリールであり;ここでアリールはハロ、C1−4アルキルオキシ又はシアノで置換されたフェニルであり;そしてヘテロアリールはチアゾリル又はピリジニルである、立体化学的異性体を含む式(I)の化合物;あるいは製薬学的に許容され得るその酸付加塩又はその溶媒和物に関する。
【0021】
nが0である式(I)の化合物として定義される式(I−a)の化合物は、Cs2CO3のような適した塩基の存在下に、例えば2−プロパノン、1,4−ジオキサン又はTH
Fのような反応に不活性な溶媒中で中間体(II)を中間体(III)と反応させることにより製造することができ、ここでLは離脱基、例えばハロ、メタンスルホニルオキシ、ベンゼンスルホニルオキシ、トリフルオロメタンスルホニルオキシなどの反応性離脱基である。中間体(III)中に存在する置換基の型に依存して、カップリング反応後に除去することができる保護基を、中間体(III)中に導入することが必要であり得る。
【0022】
【化2】
【0023】
nが0である式(I)の化合物として定義される式(I−a)の化合物を、Cs2CO3のような適した塩基、Pd2(dba)3のような触媒及びキサントフォス(Xantphos)のような適したリガンドの存在下に、例えば2−プロパノン、1,4−ジオキサン又はTHFのような反応に不活性な溶媒中で、通常の条件又はマイクロ波条件下で加熱しながら、中間体(II)を中間体(IV)と反応させることによって製造することもできる。
【0024】
【化3】
【0025】
当該技術分野において既知のS−酸化反応により、式(I−a)の化合物を、nが1を示す式(I)の化合物として定義される式(I−b)の化合物に、あるいはnが2を示す式(I)の化合物として定義される式(I−c)の化合物に転換することができる。
【0026】
【化4】
【0027】
過酸化水素の30%水溶液を用いて、又は他の酸化剤、例えばNaIO4、tert−ブチルオキシクロリド、亜硝酸アシル、過ホウ酸ナトリウム及び過酸、例えばmCPBA(メタ−クロロ過安息香酸)により、S−酸化反応を行なうことができる。スルフィドをスルホキシドに酸化することができ、それをさらに1当量の過酸化水素、KMnO4、過ホウ酸ナトリウム、過硫酸水素カリウム、mCPBAなどの試薬の添加によりスルホンにさらに酸化することができる。十分な酸化剤が存在すれば、スルホキシドを単離せずに直接、スルフィドをスルホンに転換することができる。
【0028】
XがN−(CO)−R1を示す式(I)の化合物として定義される式(I−d)の化合物は、中間体(VI)を用いる中間体(V)のN−アルキル化により製造することができ、ここでWは適した離脱基、例えばハロ、例えばフルオロ、クロロ、ブロモ、ヨードであるか、あるいはいくつかの場合、Wはスルホニルオキシ基、例えばメタンスルホニルオキシ、ベンゼンスルホニルオキシ、トリフルオロメタンスルホニルオキシなどの反応性離脱基であることもできる。例えばアセトニトリル又はジクロロメタンのような反応に不活性な溶媒中で、且つ場合により例えば炭酸ナトリウム、炭酸カリウム又はトリエチルアミンのような適した塩基の存在下で反応を行なうことができる。攪拌は反応の速度を増すことができる。室温〜反応混合物の還流温度の範囲の温度で反応を簡単に行なうことができる。
【0029】
【化5】
【0030】
nが2であり、R1が水素以外と定義される式(I)の化合物として定義される式(I−e)の化合物は、スキーム1に記載されるとおりに製造することができる。
【0031】
【化6】
【0032】
式(I−e)の化合物を得るための縮合反応は、酸性又は塩基性条件下で行なうことができる。酸性条件下で、縮合は、酢酸のような有機酸あるいはHCl又はH2SO4のような無機酸あるいはそれらの組み合わせの存在下に、酢酸、H2O、メタノール、エタノール、ジオキサン、トルエン又はジクロロエタンのような溶媒中で行なわれる。塩基性条件下で、縮合反応は、例えばK2CO3のような無機塩基の存在下に、DMSOのような反応に不活性な溶媒中で、あるいはアルコール性NaOH溶液中で行なわれる。室温〜反応混合物の還流温度の範囲の温度で反応を簡単に行なうことができる。溶媒としてのジクロロエタン中で、例えば190℃におけるマイクロ波補助加熱により反応速度及び収率を増すことができ、おそらくさらに酸又は塩基を加える必要が取り除かれる。
【0033】
上記の方法において製造される式(I)の化合物は、エナンチオマーのラセミ混合物の形態で合成され得、それを当該技術分野において既知の分割法に従って互いから分離する
ことができる。ラセミ形態で得られる式(I)の化合物を、適したキラル酸との反応により対応するジアステレオマー塩の形態に転換することができる。該ジアステレオマー塩の形態を、続いて例えば選択的又は分別結晶化により分離し、アルカリによりそこからエナンチオマーを遊離させる。式(I)の化合物のエナンチオマーの形態を分離する代わりの方法は、キラル固定相を用いる液体クロマトグラフィーを含む。該純粋な立体化学的異性体を、適した出発材料の対応する純粋な立体化学的異性体から誘導することもでき、但し、反応は立体特異的に起こる。好ましくは、特定の立体異性体が望まれる場合、該化合物は立体特異的製造方法により合成されるであろう。これらの方法は、有利にはエナンチオマー的に純粋な出発材料を用いるであろう。
【0034】
式(I)の化合物、その製薬学的に許容され得る塩及び立体異性体は、薬理学的実施例において示されるように、選択的カンナビノイド受容体2(CB2)作動性を有する。薬理学的実施例C.1は、CB2アゴニズムを測定する方法を記載しており、結果は表C.1に挙げられている。
【0035】
従って本式(I)の化合物は、特にカンナビノイド2受容体、特にCB2作動活性により媒介される状態又は疾患の処置における薬剤として有用である。続いて、本化合物をCB2受容体活性、特にCB2作動活性により媒介される状態又は疾患の処置用の薬剤の製造のために用いることができる。
【0036】
好ましくは、本発明は、CB2状態又は疾患から選ばれる状態又は疾患の処置用の薬剤の製造のための式(I)の化合物又はその製薬学的に許容され得る塩の使用も提供する。
【0037】
さらに本発明は、哺乳類の患者におけるCB2受容体活性により媒介される状態の処置方法を提供し、それは、そのような処置の必要な哺乳類に式(I)の化合物又はその製薬学的に許容され得る塩の治療的に有効な量を投与することを含む。
【0038】
カンナビノイド受容体2が媒介する状態又は障害は、例えば、心臓血管病、例えばアテローム性動脈硬化症、高血圧、心筋虚血;慢性疼痛障害(chronic pain disorders)、例えば痛覚過敏症、神経障害疼痛(neuropathic pain)、末梢痛、内臓痛、炎症痛、熱性痛覚過敏症、侵害受容疼痛(nociceptive pain)、線維筋痛、慢性背下部痛及び歯痛;炎症、浮腫、膀胱炎、神経炎症性疾患、免疫系障害、自己免疫疾患、多発性硬化症、慢性関節リウマチ、胃腸障害、腸運動障害、過敏性腸症候群(IBS)、炎症性腸疾患(IBD)、クローン病、慢性肝臓損傷(肝硬変)、ガン、前立腺ガン、ガン痛、神経膠腫、アレルギー、吐気及び嘔吐、喘息、慢性閉塞性肺疾患、乾癬、てんかん及び骨喪失障害(bone loss disorders)、例えば骨粗しょう症である(下記で「CB2障害又は疾患」と言及する)。
【0039】
本明細書で用いられる場合、「処置する」及び「処置」という用語は、そのような用語が適用される疾患、障害又は状態あるいはそのような疾患、障害又は状態の1つもしくはそれより多い症状を逆転させるか、緩和するか、その進行を妨げるか、又は予防することを含む、治癒的、緩和的及び予防的処置を指す。
【0040】
さらに本発明は、少なくとも1種の製薬学的に許容され得る担体及び式(I)の化合物の治療的に有効な量を含んでなる製薬学的組成物を提供する。
【0041】
本発明の製薬学的組成物の調製のために、塩基又は酸付加塩の形態にある特定の化合物の活性成分として有効な量を、少なくとも1種の製薬学的に許容され得る担体と緊密な混合物において合わせ、その担体は、投与のために望ましい調製物の形態に依存して多様な形態をとることができる。望ましくはこれらの製薬学的組成物は、好ましくは経口的投与
、直腸的投与、経皮的投与又は非経口的注入に適した単位投薬形態にある。
【0042】
例えば経口的投薬形態における組成物の調製において、懸濁剤、シロップ、エリキシル剤及び溶液のような経口用液体調製物の場合、通常の液体の製薬学的担体のいずれか、例えば水、グリコール、油、アルコールなど;あるいは粉剤、丸薬、カプセル及び錠剤の場合、澱粉、糖類、カオリン、滑沢剤、結合剤、崩壊剤などのような固体の製薬学的担体を用いることができる。それらの容易な投与のために、錠剤及びカプセルは最も有利な経口的投薬単位形態物を与え、その場合には固体の製薬学的担体が用いられるのは明らかである。非経口的注入組成物の場合、製薬学的担体は主に無菌水を含んでなるであろうが、活性成分の溶解性を向上させるための他の成分が含まれることができる。例えば食塩水、グルコース溶液又は両者の混合物を含んでなる製薬学的担体を用いることにより、注入可能な溶液を調製することができる。適した液体担体、懸濁化剤などを用いることにより、注入可能な懸濁剤を調製することもできる。経皮的投与に適した組成物において、製薬学的担体は場合により浸透促進剤及び/又は適した湿潤剤を含んでなることができ、それらは場合により皮膚に有意な悪影響を引き起こさない小さい割合の適した添加剤と組み合わされていることができる。該添加剤は、皮膚への活性成分の投与を促進するように及び/又は所望の組成物の調製の助けとなるように選ばれることができる。これらの局所用組成物を種々の方法で、例えば経皮パッチ、スポット−オン又は軟膏として投与することができる。式(I)の化合物の付加塩は、対応する塩基形態より向上したそれらの水溶性のために、水性組成物の調製において明らかにより適している。
【0043】
投与の容易さ及び投薬量の均一性のために、本発明の製薬学的組成物を投薬単位形態物において調製するのが特に有利である。本明細書で用いられる「投薬単位形態物」は、1回の投薬量として適した物理的に分離された単位を指し、各単位は所望の治療効果を生むために計算されたあらかじめ決められた量の活性成分を、必要な製薬学的担体と一緒に含有する。そのような投薬単位形態物の例は錠剤(刻み付き又はコーティング錠を含む)、カプセル、丸薬、粉剤小包、ウェハース、注入可能な溶液又は懸濁剤、小さじ一杯、大さじ一杯など、ならびに分離されたそれらの複数である。
【0044】
経口的投与のために、本発明の製薬学的組成物は、製薬学的に許容され得る賦形剤及び担体、例えば結合剤(例えば予備ゼラチン化トウモロコシ澱粉、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロースなど)、充填剤(例えばラクトース、微結晶性セルロース、リン酸カルシウムなど)、滑沢剤(例えばステアリン酸マグネシウム、タルク、シリカなど)、崩壊剤(例えばポテト澱粉、ナトリウム澱粉グリコレートなど)、湿潤剤(例えばラウリル硫酸ナトリウム)などを用いて通常の手段により調製される固体の投薬形態物、例えば錠剤(嚥下可能及びチュワブル形態の両方)、カプセル又はゲルカップの形態をとることができる。そのような錠剤を当該技術分野において周知の方法によりコーティングすることもできる。
【0045】
経口的投与のための液体調製物は、例えば溶液、シロップ又は懸濁剤の形態をとることができるか、あるいはそれらを使用前に水及び/又は他の適した液体担体と混合するための乾燥生成物として調製することができる。そのような液体調製物は通常の方法により、場合により懸濁化剤(例えばソルビトールシロップ、メチルセルロース、ヒドロキシプロピルメチルセルロース又は水素化食用脂肪)、乳化剤(例えばレシチン又はアラビアゴム)、非−水性担体(例えばアーモンド油、油性エステル又はエチルアルコール)、甘味料、風味料、隠蔽剤及び防腐剤(例えばp−ヒドロキシ安息香酸メチルもしくはプロピル又はソルビン酸)のような他の製薬学的に許容され得る添加剤を用いて調製することができる。
【0046】
本発明の製薬学的組成物において有用な製薬学的に許容され得る甘味料は、好ましくは
少なくとも1種の強力甘味料、例えばアスパルテーム、アセスルフェームカリウム、シクラミン酸ナトリウム、アリテーム、ジヒドロカルコン甘味料、モネリン、ステビオシドスクラロース(4,1’,6’−トリクロロ−4,1’,6’−トリデオキシガラクトスクロース)又は、好ましくはサッカリン、サッカリンナトリウムもしくはカルシウム及び場合により少なくとも1種のバルク甘味料(bulk sweetener)、例えばソルビトール、マンニトール、フルクトース、スクロース、マルトース、イソマルト、グルコース、水素化グルコースシロップ、キシリトール、カラメル又はハチミツを含む。強力甘味料は、簡便に低濃度で用いられる。例えばサッカリンナトリウムの場合、該濃度は、最終的な調剤の約0.04%〜0.1%(重量/容量)の範囲であることができる。バルク甘味料は、約10%〜約35%、好ましくは約10%〜15%(重量/容量)の範囲の比較的高い濃度で有効に用いられ得る。
【0047】
低−投薬量調剤において苦い味の成分を隠蔽することができる製薬学的に許容され得る風味料は、好ましくはフルーツ風味料、例えばチェリー、ラズベリー、黒スグリ又はストロベリー風味料である。2種の風味料の組み合わせは、非常に良い結果を与えることができる。高−投薬量の調剤においては、より強い製薬学的に許容され得る風味料、例えばカラメルチョコレート(Caramel Chocolate)、ミントクール(Mint
Cool)、ファンタジー(Fantasy)などが必要であり得る。各風味料は、約0.05%〜1%(重量/容量)の範囲の濃度で、最終的な組成物中に存在することができる。該強い風味料の組み合わせは、有利に用いられる。好ましくは、調剤の環境下で味及び/又は色の変化又は喪失を経ない風味料が用いられる。
【0048】
式(I)の化合物を、注入、簡便には静脈内、筋肉内又は皮下注入による、例えば大量注入又は継続的静脈内注入による非経口的投与用に調製することができる。注入用の調剤は、単位投薬形態物において、例えばアンプル中で、又は加えられた防腐剤を含む多投薬量容器中で存在することができる。それらは、油性又は水性ビヒクル中の懸濁剤、溶液又は乳剤のような形態をとることができ、等張化剤、懸濁化剤、安定剤及び/又は分散剤のような調製剤を含有することができる。あるいはまた、活性成分は、使用前に適したビヒクル、例えば発熱物質を含まない無菌水と混合するための粉末形態で存在することができる。
【0049】
式(I)の化合物を、例えば通常の座薬基剤、例えばココアバター及び/又は他のグリセリドを含有する座薬又は保持浣腸(retention enema)のような直腸用組成物において調製することもできる。
【0050】
カンナビノイド受容体の媒介に結び付けられる疾患の処置における熟練者は、下記に示される試験結果から、式(I)の化合物の治療的に有効な量を容易に決定することができるであろう。一般に治療的に有効な投薬量は、処置されるべき患者の体重のkg当たり約0.001mg〜kg当たり約50mg、より好ましくは体重のkg当たり約0.01mg〜kg当たり約10mgであろうと思われる。治療的に有効な投薬量を、1日を通じて適した間隔における2回かもしくはそれより多い細分−投薬量の形態で投与するのが適しているかも知れない。該細分−投薬量を、例えばそれぞれ単位投薬形態物当たりに約0.1mg〜約1000mg、さらに特定的に約1〜約500mgの活性成分を含有する単位投薬形態物として調製することができる。
【0051】
本明細書で用いられる場合、化合物の「治療的に有効な量」は、患者又は動物に投与されると、その患者又は動物において、カンナビノイド受容体の刺激における識別可能な増加又は減少を引き起こすのに十分に高いレベルのその化合物を与える化合物の量である。
【0052】
投与の正確な用量及び頻度は、当該技術分野における熟練者に周知のとおり、用いられ
る特定の式(I)の化合物、処置されている特定の状態、処置されいてる状態の重度、特定の患者の年令、体重及び一般的な身体条件ならびに患者が摂取しているかも知れない他の投薬に依存する。さらに、処置される患者の反応に依存して及び/又は本発明の化合物を処方する医師の評価に依存して、該「治療的に有効な量」を減少させるか又は増加させることができる。従って上記で挙げた有効な1日の量の範囲は、単に指針である。
【発明を実施するための形態】
【0053】
実験部分
下記に記載する方法において、以下の略語を用いた:「DCM」はジクロロメタンを示し、「MeOH」はメタノールを示し、「NH3」はアンモニアを示し、「CH3CN」はアセトニトリルを示し、「THF」はテトラヒドロフランを示し、「DIPE」はジイソプロピルエーテルを示し、「NaBH3(CN)」はナトリウムシアノトリヒドロボレートを示し、「Cs2CO3」は炭酸セシウムを意味し、「MgSO4」は硫酸マグネシウムを意味し、「NaHCO3」は炭酸一ナトリウム塩を意味し、「NaOH」は水酸化ナトリウムを意味し、「Pd2(dba)3」はトリス[μ−[(1,2−η:4,5−η)−(1E,4E)−1,5−ジフェニル−1,4−ペンタジエン−3−オン]ジパラジウムを意味し、そして「キサントフォス」は(9,9−ジメチル−9H−キサンテン−4,5−ジイル)ビス[ジフェニルホスフィン]を意味し、「DMSO」はジメチルスルホキシドを意味し;「DMAP」は4−(ジメチルアミノ)ピリジンを意味し、「HBTU」は1−[ビス(ジメチルアミノ)メチレン]−1H−ベンゾトリアゾリウムヘキサフルオロホスフェート(1−)3−オキシドを意味する。
【0054】
Isolute HM−NTMフィルターは、Argonaut,Foster City,CA 94404,USAの製品であり、改質された形態(modified form)の珪藻土を含んでなる短カラムであり、コンビナトリアルケミストリー用途において試料から水を除去することができる。
【0055】
高−性能液体クロマトグラフィー精製法:
−精製法A
生成物を逆相高−性能液体クロマトグラフィー(Shandon Hyperprep(R) C18 BDS(Base Deactivated Silica)8μm,250g,内径5cm)により精製した。2つの移動相を用いた(相A:水中の0.25%NH4HCO3溶液;相B:CH3CN)。最初に40ml/分の流量で85%A及び15%Bを0.5分間保持した。次いで80ml/分の流量で勾配を適用し、41分内に10%A及び90%Bとした。次いで80ml/分の流量で勾配を適用し、20分内に100%Cとし、4分間保持した。
−精製法B
生成物を逆相高−性能液体クロマトグラフィー(Shandon Hyperprep(R) C18 BDS(Base Deactivated Silica)8μm,250g,内径5cm)により精製した。3つの移動相を用いる勾配を適用した(相A:水中の0.25%NH4HCO3溶液;相B:CH3OH;相C:CH3CN)。所望の画分を集め、仕上げた。
【0056】
A.中間体の合成
実施例A.1
a)
【化7】
ジオキサン(120ml)中の1−クロロ−4−(エチルスルホニル)−2−ニトロベンゼン(7.5g,0.03モル)及び4−アミノメチル−ピペリジン−1−カルボン酸tert−ブチルエステル(7.72g,0.036モル)の混合物に、炭酸カリウム(4.55g,0.033モル)を加えた。反応混合物を75〜80℃で3時間及び次いで100℃で3時間攪拌した。固体を濾過し、反応混合物を蒸発させた。残留物をDCM(200ml)中に溶解した。有機層を水(200ml)で洗浄した。水層をDCM(150ml)で抽出した。合わせた有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、12.1gの中間体(1)を与えた。
b)
【化8】
メタノール(150ml)中の中間体(1)(12.1g)の混合物を、チオフェン溶液(1ml)及び酸化バナジウム(0.2g)の存在下で、触媒として活性炭上のパラジウム(10%)(2g)を用いて水素化した。水素(3当量)の吸収後、反応混合物をジカライト上で濾過し、濾液を蒸発させ、中間体(2)を与えた。
c)
【化9】
DCM(90ml)中の中間体(2)(0.014モル)、2,2−ジメチルプロパノイルクロリド(2.1ml,0.017モル)及びピリジン(2ml)の混合物を室温で終夜攪拌した。溶媒を蒸発させ、酢酸(80ml)及び塩酸(8ml)を残留物に加えた。この混合物を3時間還流させ、次いで混合物を蒸発させた。残留物をDCM(300ml)と水(250ml)に分配した。NH3水を用いて混合物を塩基性とし、層を分離した。分離された有機層を水で洗浄し、乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、3.4gの残留物を与えた。この残留物をシリカゲル上のカラムクロマトグラフィー(Biotage;溶離剤:DCM/(MeOH/NH3) 99/1から92/8まで)により精製した。所望の画分を集め、溶媒を蒸発させ、2.7gの中間体(3)を与えた。
【0057】
実施例A.2
a)
【化10】
2−プロパノン(1000ml)中の4−クロロベンゼンチオール(0.2モル)及び1,1,1−トリフルオロ−4−ヨードブタン(0.21モル)の混合物を氷−浴上で冷却した。Cs2CO3(0.215モル)を加え、反応混合物を窒素流下に室温で終夜攪拌した。次いでDIPE(1000ml)を加え、固体を濾過し、濾液を蒸発させ、中間体(4)を与えた。
b)
【化11】
トリクロロメタン(1200ml)中の中間体(4)(0.2モル)の混合物を、氷−浴上で冷却した。3−クロロベンゼンカルボペルオキソ酸(100g;70〜75%)を、20分かけて分けて加え、反応混合物を室温で210分間攪拌した。混合物を氷−浴上で冷却し、NaOH水溶液(1000ml,5%)を加えた。分離された有機層をNaOH水溶液(1000ml,5%)で2回洗浄し、次いで水で洗浄した。合わせた有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、57gの中間体(5)を与えた。
c)
【化12】
濃硫酸(500ml)中の中間体(5)(0.2モル)の混合物に、冷水を用いて反応混合物を冷却しながら、濃硫酸と硝酸の混合物(50/50)(100ml)を1時間かけて滴下した。混合物を室温で2時間攪拌し、次いで氷(2000ml)上に注いだ。沈殿を濾過し、水で洗浄し、乾燥し(真空)、66gの中間体(6)を与えた。
d)
【化13】
DMSO(4ml)中の中間体(6)(0.002モル)、1−アセチル−4−ピペリジンメタナミン(0.00235モル)及びトリエチルアミン(0.003モル)の混合物を100℃で終夜攪拌した。この混合物を氷−水上に注ぎ、DCMで抽出した。有機層を水で2回洗浄し、乾燥し(MgSO4)、溶媒を蒸発させた。残留物をシリカゲル上のコンビフラッシュカラムクロマトグラフィー(溶離剤:DCM/(CH3OH/NH3)
100/0から97/3)により精製した。生成物画分を集め、溶媒を蒸発させ、0.4gの中間体(7)を与えた。
e)
【化14】
メタノール(40ml)中の中間体(7)(0.0009モル)の混合物を、チオフェン溶液(0.1ml)の存在下に触媒として活性炭上のパラジウム(10%)(0.1g)を用いて水素化した。水素(3当量)の吸収後、触媒をセライト上で濾過し、濾液を蒸発させ、中間体(8)を与えた(そのまま次の段階で使用した)。
【0058】
実施例A.3
a)
【化15】
DMSO(15ml)中の中間体(6)(0.008モル)、4−アミノメチル−ピペリジン−1−カルボン酸tert−ブチルエステル(0.0093モル)及びトリエチルアミン(0.012モル)の混合物を、密閉された容器中で100℃において終夜攪拌した。反応混合物を60℃に冷まし、次いで氷−水(200ml)上に注いだ。黄色の混合物を室温で30分間攪拌し、黄色の沈殿を濾過し、多量の水で洗浄し、乾燥し(真空)、3.95gの中間体(10)を与えた。
b)
【化16】
メタノール(100ml)中の中間体(10)(0.00738モル)の混合物を、チオフェン溶液(0.5ml)の存在下に触媒として活性炭上のパラジウム(10%)(1g)を用いて水素化した。水素(3当量)の吸収後、触媒をセライト上で濾過し、濾液を蒸発させ、中間体(11)を与えた。
c)
【化17】
DCM(50ml)中の中間体(11)(最大で0.00768モル;粗)、2,2−ジメチルプロパノイルクロリド(0.0096モル)及びピリジン(2ml)の混合物を室温で2時間攪拌した。溶媒を蒸発させ、酢酸(50ml)及び塩酸(濃)(5ml)を
残留物に加えた。この混合物を120℃で3時間攪拌した。混合物を冷却し、溶媒を蒸発させた。残留物をDCMとNH3水に分配した。分離された有機層をブラインで洗浄し、乾燥し(MgSO4)、溶媒を蒸発させた。粗残留物をシリカゲル上のコンビフラッシュカラムクロマトグラフィー(溶離剤:DCM/(CH3OH/NH3) 92/8)により精製した。生成物画分を集め、溶媒を蒸発させ、2.65gの中間体(12)を与えた。
【0059】
実施例A.4
a)
【化18】
アセトン(500ml)中の4−クロロベンゼンチオール(0.1モル)及び4−ブロモブタンニトリル(0.15モル)の混合物を攪拌した。Cs2CO3(0.11モル)を加え、反応混合物を窒素下で攪拌した。DIPE(500ml)を加え、沈殿を濾過した。濾液を蒸発させ、中間体(13)を与えた。
b)
【化19】
トリクロロメタン(500ml)中の中間体(13)(0.1モル)の混合物を氷上で冷却しながら、3−クロロベンゼンカルボペルオキソ酸(0.22モル)を分けて加えた。反応混合物を室温で3時間攪拌した。有機層をNaOH−溶液(500ml,1N NaOH,3x)及び水で洗浄した。有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させ、35gの中間体(14)を与えた。
c)
【化20】
硫酸(120ml)を塩/氷浴(−10℃)上で冷却した。中間体(14)(0.05モル)を混合物に加え、続いて硫酸と硝酸の混合物(1:1)(20ml)を30分間かけて滴下した。混合物を室温に加熱し、混合物は黄オレンジ色の溶液になった。室温で1時間攪拌した後、反応混合物を氷−水上に注ぎ、明黄色の固体を濾過し、水で洗浄した。沈殿を乾燥し(真空)、13.5gの残留物を与えた。室温でDCMを用いて残留物を磨砕し、残る固体を濾過した。濾液を蒸発させ、乾燥し、7.1gの中間体(15)を与えた。
d)
【化21】
DMSO(5ml)中の中間体(15)(0.002モル)、1−アセチル−4−ピペリジンメタナミン(0.00235モル)及びトリエチルアミン(約0.003モル)の混合物を100℃で終夜攪拌した。混合物を氷水上に注ぎ、DCMで抽出した。有機層を水で2回洗浄し、乾燥し(MgSO4)、溶媒を蒸発させた。残留物をシリカゲル上のカラムクロマトグラフィー(溶離剤:DCM/(CH3OH/NH3) 100/0から96/4)により精製した。生成物画分を集め、溶媒を蒸発させ、0.41gの中間体(16)を与えた。
e)
【化22】
メタノール(40ml)中の中間体(16)(約0.001モル)の混合物を、チオフェン溶液(0.1ml)の存在下に触媒として活性炭上のパラジウム(10%)(0.1g)を用いて水素化した。水素(3当量)の吸収後、触媒をセライト上で濾過し、濾液を蒸発させた。残留物をそのまま次の段階において用い、中間体(17)を与えた。
【0060】
実施例A.5
a)
【化23】
エタノール(500ml)中の5−クロロ−2−ニトロベンゼンアミン(0.16モル)、4−メトキシベンゼンメタンチオール(0.16モル)及び水酸化カリウム(0.30モル)の混合物を2時間攪拌し、且つ還流させた。反応混合物を冷却した。沈殿を濾過し、エタノールで洗浄し、乾燥し、48.5gの中間体(18)を与えた。
b)
【化24】
氷浴上で冷却されたDCM(180ml)中の中間体(18)(0.03モル)及びピリジン(0.06モル)の混合物に、DCM(20ml)中の2,2−ジメチルプロパノイルクロリド(0.032モル)の溶液を滴下した。反応混合物が室温に達するのを許し
た。DMAPを加え、混合物を20時間攪拌し、且つ還流させた。追加の中間体(18)(0.01モル)、2,2−ジメチルプロパノイルクロリド(0.048モル)及びピリジン(1.2モル)を加えた。混合物を2時間還流させた。溶媒を蒸発させた。残留物をDCM中に取り上げ、水で洗浄した。有機層を分離し、乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物をDIPEから結晶化させた。沈殿を濾過し、洗浄し、乾燥し、9.1gの中間体(19)を与えた。
c)
【化25】
水(500ml)中の中間体(19)(0.0748モル)、鉄(56g)及び酢酸(10ml)の混合物を4時間攪拌し、且つ還流させた。混合物を冷却した。溶媒をデカンテーションした。残留物をメタノール及びTHF中に取り上げた。混合物をジカライト上で濾過した。溶媒を蒸発させた。残留物をDCM中に取り上げた。有機層を分離し、MgSO4及びジカライト上で濾過した。溶媒を蒸発させた。残留物をDIPEから結晶化させた。沈殿を濾過し、乾燥し、21gの中間体(20)を与えた。
d)
【化26】
DCM(250ml)中の中間体(20)に、4−ホルミル−1−ピペリジンカルボン酸tert−ブチルエステル、次いで酢酸及びチタン(IV)イソプロポキシドを加えた。反応混合物を20分間攪拌した。次いでNaBH3(CN)を加え、反応混合物を2時間攪拌した。反応混合物に水を加え、有機層を分離し、乾燥し(MgSO4)、濾過し、蒸発させ、20gの中間体(21)を与えた。
e)
【化27】
中間体(21)、酢酸及び塩酸(濃)を、還流温度で終夜攪拌した。反応混合物を濃縮した。残留物を水(500ml)中に取り上げ、NaHCO3を用いて塩基性とし、300mlのDCMを用いて3回抽出した。合わせた有機層をブラインで洗浄し、MgSO4上で乾燥し、濃縮して、11.4gの中間体(22)を与えた。
【0061】
実施例A.6
【化28】
化合物(11)及びトリフルオロ酢酸を、マイクロ波中で120℃において60分間攪拌した。反応混合物を冷却した。溶媒を蒸発させた。残留物を酢酸エチル中に取り上げ、次いでH2O/NaHCO3溶液で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させ、2.3gの中間体(23)を与えた。
【0062】
以下の中間体を、化合物(44)から類似して製造した:
【化29】
【0063】
実施例A.7
a)
【化30】
窒素流下の反応。メチルテトラヒドロ−2H−チオピラン−4−イル−ケトン(0.039モル;エタノール中の50%溶液)を、DCM(32ml)及び酢酸(4ml)中の中間体(20)(0.03モル)の混合物に室温で加え、次いで5分間攪拌した。反応混合物にNaBH3(CN)(0.04モル)を加え、次いで室温で1時間攪拌した。反応混合物を水で洗浄した。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物をDIPE中に懸濁させた。沈殿を濾過し、乾燥し(真空,室温)、11.2gの中間体(25)を与えた。
b)
【化31】
中間体(25)と酢酸の混合物を、マイクロ波中で150℃において100分間加熱した。溶媒を蒸発させた。残留物をDCM中に取り上げ、水/NaHCO3で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させた。シリカゲル及び溶離剤としてDCM:MeOH/NH3(100から97:3)を用い、短フィルター上で残留物を精製した。生成物画分を集め、蒸発させ、1.3gの中間体(26)を与えた。
c)
【化32】
中間体(26)及びトリフルオロ酢酸を、マイクロ波中で120℃において30分間攪拌した。反応混合物を冷却した。溶媒を蒸発させた。残留物を酢酸エチル中に取り上げ、次いで水/NaHCO3溶液で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させた。粗残留物を次の段階に用い、1.3gの中間体(27)を与えた。
【0064】
実施例A.9
【化33】
DCM(20ml)中の中間体(2)(0.0047モル)、tert−ブチルアセチルクロリド(0.81g,0.006モル)及びピリジン(2ml)の混合物を、室温で終夜攪拌した。溶媒を蒸発させた。酢酸(25ml)及びHCl(2ml)を残留物に加えた。この混合物をマイクロ波オーブン中で190℃において75分間攪拌した(2つに分けて(in 2 portions))。溶媒を蒸発させた。残留物をDCM(250ml)とNH3水に分配した。分離された有機層を水で洗浄し、乾燥し(MgSO4)、濾過し、溶媒を蒸発させて残留物を与え、それをシリカゲル上のカラムクロマトグラフィー(Biotage;溶離剤:CH2Cl2/(CH3OH/NH3) 100/0から96)により精製し、0.85gの中間体(28)を与えた。
【0065】
実施例A.10
【化34】
アセトン(10ml)中の3−ヨードフェニル(10ミリモル)、1−ブロモ−3−メトキシプロパン(14.7ミリモル)及びK2CO3(20.26ミリモル)の混合物を、50℃で40時間攪拌した。塩を濾過し、洗浄した。濾液を濃縮し、残留物をカラムクロマトグラフィーにより、溶離剤として酢酸エチル/ヘプタン(0:100から20:80)を用いて精製した。生成物画分を集め、蒸発させ、中間体(29)を与えた。
【0066】
実施例A.11
【化35】
アセトン(10ml)中の3−ヨードフェニル(10ミリモル)、1−ブロモ−3−シアノプロパン(14.7ミリモル)及びK2CO3(20.26ミリモル)の混合物を、50℃で40時間攪拌した。塩を濾過し、洗浄した。濾液を濃縮し、残留物をカラムクロマトグラフィーにより、溶離剤として酢酸エチル/ヘプタン(0:100から20:80)を用いて精製した。生成物画分を集め、蒸発させ、中間体(30)を与えた。
【0067】
B.最終的化合物の合成
実施例B.1
【化36】
中間体(3)(0.7g,0.00193モル)、アセチルアセテート(0.26g,0.0025モル)及びDCM(20ml)の混合物を、室温で終夜反応させた。混合物を最初に水(15ml)で洗浄し、次いでNH3水(2x15ml)で2回及び最後にブライン(15ml)で洗浄した。混合物をIsolute HM−NTM上で濾過し、次いで窒素流下で溶媒を蒸発させた。残留物をDIPEから結晶化させ、0.035gの化合物(1)を与えた。油性の生成物を2−プロパノール(8ml)中に溶解し、HCl/2−プロパノール(6N,0.5ml)溶液を加えた。溶媒を蒸発させた。ジエチルエーテルを用いて残留物を磨砕した。沈殿を濾過し、乾燥し(真空)、0.53gの化合物(2)を与えた。
【0068】
中間体(28)を無水酢酸と反応させることにより、化合物(46)を類似して製造した。
実施例B.2
【化37】
DCM(20ml)中の中間体(8)(最大で0.0009モル)、2,2−ジメチルプロパノイルクロリド(0.170ml)及びピリジン(0.25ml)の混合物を、室温で2時間攪拌した。溶媒を蒸発させ、粗残留物に1,2−ジクロロエタン(4ml)を加えた。混合物をマイクロ波中で190℃において1時間加熱した。混合物を冷まし、DCM(4ml)及びNaOH溶液(1ml,1N)を加えた。反応混合物を攪拌し、Isolute HM−NTMフィルター上で濾過し、溶媒を蒸発させた。生成物を逆相高−性能液体クロマトグラフィーにより精製した。生成物画分を集め、溶媒を蒸発させ、0.123gの化合物(3)を与えた。
【0069】
2,2−ジメチルプロパノイルクロリドを中間体(17)と反応させることにより、化合物(10)を類似して製造し、中間体(2)を2,2−ジメチルアセチルクロリドと反応させることにより、化合物(47)を類似して製造した。
【0070】
実施例B.3
【化38】
DCM(10ml)中の中間体(12)(0.00061モル)、無水酢酸(0.0008モル)及びトリエチルアミン(0.17ml)の混合物を、室温で終夜攪拌した。水を加え、混合物を攪拌し、層を分離した。Isolute HM−NTMフィルターに通過させることにより有機層を乾燥した。溶媒を蒸発させた。ジエチルエーテル中で塩酸を用いて残留物を結晶化させ、0.260gの化合物(4)を与えた。
【0071】
中間体(12)をプロパノイルクロリド、メトキシカルボニルクロリド、ビス(トリフルオロ酢酸)無水物又は2−メトキシアセチルクロリドと反応させることにより、それぞれ化合物(6)、(7)、(8)及び(9)を類似して製造した。溶媒としてピリジン及びTHFの存在下で中間体(22)を無水酢酸と反応させることにより、化合物(11)を類似して製造した。
【0072】
実施例B.4
【化39】
DCM(5ml)中の中間体(12)(0.000675モル)、ギ酸(0.00087モル)、HBTU(0.00087モル)及びトリエチルアミン(0.15ml)の混合物を50℃で攪拌した。2時間後、追加分のギ酸(0.00087モル)を加え、混合物を室温で終夜攪拌した。次いでDCM(5ml)及び水(1ml)を加え、混合物をIsolute HM−NTMフィルター上で濾過した。溶媒を蒸発させ、残留物を逆相高−性能液体クロマトグラフィーにより精製した。生成物画分を集め、溶媒を蒸発させ、0.205gの化合物(5)を与えた。
【0073】
実施例B.5
【化40】
トリクロロメタン(10ml)中の化合物(11)(0.0003モル)の混合物に、3−クロロベンゼンカルボペルオキソ酸(0.0007モル;77%)を室温で加えた。反応混合物を室温で30分間攪拌し、次いで水及びNaOH(1N)を加えた。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物を逆相カラム上で精製した。生成物画分を集め、溶媒を蒸発させ、乾燥するまで共−蒸発させ、0.075gの化合物(12)を与えた。
【0074】
中間体(29)から出発し、類似して化合物(49)を製造した。化合物(50)から出発し、類似して化合物(53)を製造した。化合物(54)から出発し、類似して化合物(62)及び(63)を製造した。化合物(72)から出発し、類似して化合物(73)を製造した。化合物(52)から出発し、類似して化合物(56)を製造した。化合物(51)から出発し、類似して化合物(61)を製造した。化合物(57)から出発し、類似して化合物(60)を製造した。化合物(58)から出発し、類似して化合物(59)を製造した。化合物(65)から出発し、類似して化合物(67)を製造した。化合物(66)から出発し、類似して化合物(68)を製造した。化合物(74)から出発し、類似して化合物(64)を製造した。化合物(75)から出発し、類似して化合物(69)を製造した。化合物(70)から出発し、類似して化合物(71)を製造した。化合物(81)から出発し、類似して化合物(82)を製造した。化合物(83)から出発し、類似して化合物(84)を製造した。化合物(85)から出発し、類似して化合物(86)を製造した。化合物(87)から出発し、類似して化合物(88)を製造した。化合物(89)から出発し、類似して化合物(90)を製造した。化合物(91)から出発し、類似して化合物(92)を製造した。化合物(93)から出発し、類似して化合物(94)を製造した。化合物(95)から出発し、類似して化合物(96)を製造した。化合物(97)から出発し、類似して化合物(98)を製造した。化合物(99)から出発し、類似して化合物(100)を製造した。化合物(101)から出発し、類似して化合物(102)を製造した。
【0075】
実施例B.6
【化41】
THF(10ml)中の中間体(23)(0.001モル)、3−ブロモ−1−プロパノール(0.003モル)及びCs2CO3(0.002モル)の混合物を、60℃で1時間攪拌した。反応混合物を冷却し、ジカライト上で濾過し、濾液の溶媒を蒸発させた。残留物を逆相HPLCにより精製した。所望の画分を集め、溶媒を蒸発させ、完全に乾燥するまで共−蒸発させ、0.170gの化合物(13)を与えた。
【0076】
中間体(23)を1−ブロモ−2−メトキシ−エタン、3−ブロモプロパンニトリル、1−(ブロモメチル)−4−フルオロ−ベンゼン、(ブロモメチル)−シクロブタン又は2−ブロモプロパン−ニトリルと反応させることにより、それぞれ化合物(14)、(15)、(17)、(41)及び(45)を類似して製造した。中間体(24)を1−ブロモ−3−ヒドロキシ−プロパン、1−ブロモ−2−メトキシ−エタン又は(ブロモメチル)−シクロプロパンと反応させることにより、それぞれ化合物(21)、(22)及び(37)を類似して製造した。K2CO3の存在下に、溶媒としてのDMF中で中間体(23)を3−ペンチルブロミド、2−(4−フルオロフェニル)エチルブロミド、4−ヘプチルブロミド、ベンジルブロミド、2−プロピルヨーダイド又は4−ニトロベンジルブロミドと反応させることにより、それぞれ化合物(51)、(52)、(65)、(72)、(74)及び(75)を類似して製造した。
【0077】
実施例B.7
【化42】
ジオキサン(80ml)中の中間体(23)(最大で0.005モル)及び4−ヨードベンゾニトリル(0.010モル)の混合物を脱ガスし、反応混合物上に窒素流を導入した(3回)。次いでCs2CO3(4g)を加え、混合物を脱ガスし、再び窒素を反応混合物上に導入した。次いでPd2(dba)3(0.200g)及びキサントフォス(0.150g)を加え、脱ガス及び窒素作用を行なった。反応混合物上に窒素バルーンを残し、混合物を100℃で終夜攪拌した。混合物を冷却し、濾過し、濾液を蒸発させた。残留物をDCM中に取り上げ、水で洗浄した。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物をHPLCにより精製した。生成物画分を集め、溶媒を蒸発させ、1.100gの化合物(18)を与えた。
【0078】
中間体(23)を1−ヨードシクロペンタン、4−クロロピリジン又は2−ブロモチアゾールと反応させることにより、それぞれ化合物(32)、(33)及び(38)を類似して製造した。中間体(23)を3−ブロモピリジン、2,6−ジクロロヨードベンゼン、1−フルオロ−4−ヨードベンゼン、2−ヨードチオフェン及び2−クロロヨードベンゼンと反応させることにより、それぞれ化合物(54)、(57)、(58)、(66)及び(70)を類似して製造した。中間体(24)を2−ブロモ−チアゾールと反応させることにより、化合物(77)を類似して製造した。中間体(23)を1−ヨード−3−メトキシベンゼン、3−ヨード−ベンゾニトリル、中間体(29)、1−ヨード−4−(トリフルオロメチル)ベンゼン、1−クロロ−3−ヨードベンゼン、1−ヨード−3−(トリフルオロメチル)ベンゼン及び2−ヨードベンゾニトリルと反応させることにより、それぞれ化合物(81)、(83)、(85)、(87)、(89)、(91)及び(93)を類似して製造した。中間体(23)を中間体(30)、3−ブロモ−N,N−ジメチル−ベンゼンアミン、1−ブロモ−3−(1−メチルエトキシ)ベンゼン及び2−ヨード−1,3−ジメトキシベンゼンと反応させることにより、それぞれ化合物(95)、(97)、(99)及び(101)を類似して製造した。
【0079】
実施例B.8
【化43】
中間体(3)(1.7g,0.0047モル)及びギ酸メチル(25ml)の混合物を40℃で終夜反応させた。混合物を窒素流下に60℃において濃縮した。残留物をDIPEから、1滴の2−プロパノールを用いて結晶化させ、1.55gの化合物(28)を与えた。
【0080】
中間体(22)から出発して、化合物(44)を類似して製造した。
【0081】
中間体(28)から出発して、化合物(48)を類似して製造した。
【0082】
実施例B.9
【化44】
ジオキサン(15ml)中のヨードエタン(0.01モル)の混合物に、Cs2CO3(0.006モル)を加えた。ジオキサン(10ml)中の中間体(27)(0.003モル)を加え、混合物を90℃で2時間攪拌した。反応混合物を冷却し、溶媒を蒸発させた。残留物をトリクロロメタン(50ml)中に取り上げ、次いで3−クロロベンゼンカルボペルオキソ酸(0.018モル)を加えた。この反応混合物を室温で1時間攪拌した。反応混合物をNaOH 1M水溶液で2回洗浄した。分離された有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。残留物を高−性能液体クロマトグラフィー(NH4HCO3緩衝液を用いる標準的な勾配溶離)により精製した。生成物画分を集め、溶媒を蒸発させた。この残留物をDIPE及び少量のCH3CN中に懸濁させた。沈殿を濾過し、乾燥し(真空,50℃)、0.064gの化合物(29)を与えた(融点194℃)。
【0083】
中間体(27)を2−ヨード−プロパン又は(ブロモメチル)−シクロプロパンと反応させることにより、それぞれ化合物(30)及び(31)を類似して製造した。
【0084】
実施例B.10
【化45】
酢酸中の1−{4−[2−tert−ブチル−5−(1−オキシ−ピリジン−4−スルホニル)−ベンゾイミダゾール−1−イルメチル]−ピペリジン−1−イル}−エタノン(B.5の方法に従って化合物(33)を酸化することにより製造される)及び鉄の混合物を、密閉された容器中で60℃において2時間振盪させた。反応混合物を冷却した。生成物を含有する溶媒をデカンテーションすることにより、過剰の鉄を除去した。5mlの酢酸を用いて鉄の残留物を濯いだ(再びデカンテーション)。合わせた溶媒層を蒸発させた。残留物をDCM中に取り上げ、水で洗浄した。有機層を乾燥し(MgSO4)、濾過し、蒸発させた。残留物をDIPE及びいくらかの2−プロパノールから結晶化させた。固体を濾過し、洗浄し、乾燥して0.133gの化合物(34)を与えた。
【0085】
実施例B.11
【化46】
エタノール中の化合物(39)及びナトリウムエトキシドを脱ガスし、反応物上に窒素を導入した(密閉された容器中で)。この混合物を室温で15分間攪拌した。室温でヨー
ドメタンを加え、反応混合物を3時間攪拌した。溶媒を濃縮した。残留物をDCM中に取り上げ、水で洗浄した。MgSO4を用いて有機層を乾燥し、濾過し、蒸発させた。シリカゲルを有するカラム上で、溶離剤として100/0から98/2のDCM/CH3OH(7N NH3)を用い、残留物を精製した。生成物画分を集め、蒸発させた。エーテル中のHCl(1M)及びアセトニトリルを用い、残留物をHCl塩として結晶化させた。固体を濾過し、洗浄し、乾燥し、0.09gの化合物(42)を与えた。
【0086】
実施例B.12
【化47】
ジイソプロピルエチルアミン(0.003814モル)及び2−ブロモピリジンをトルエン(5ml)に加えた。真空にすることによりこの溶液を脱ガスし、その上に窒素雰囲気を導入した。最初に調製された溶液に、次いでシリンジを用いてキサントフォス(0.041g)、Pd2(dba)3(0.0165g)及びジオキサン(5ml)の新しい溶液を加えた。反応物を軽真空下に置いた。ジオキサン(5ml)中の中間体(23)の溶液も、シリンジを用いて加えた。反応混合物を週末に及んで84℃で振盪させた。溶媒を蒸発させ、DCM(40ml)及び水(10ml)を用いて生成物を仕上げた。次いで溶媒を蒸発させた。40gのシリカゲルカラムを用いて生成物を精製した(溶離剤:DCM:CH3OH/NH3(7N) 100/0から98/2)。生成物画分を一緒にし、完全に乾燥するまで溶媒を蒸発させ、0.743gの化合物(50)を与えた。
【0087】
実施例B.13
【化48】
化合物(73)(0.001326モル)をTHF(10ml)中に溶解した。溶液を脱ガスし、次いで溶液を窒素雰囲気下とした。溶液を0℃で冷却した。次いでシリンジを用い、ナトリウムビス(トリメチルシリル)アミドを加えた。反応混合物を0℃で1時間攪拌した。シリンジを用いてヨードメタン(0.947ml)を反応溶液に加え、反応混合物を0℃で1時間攪拌した。ジクロロメタン及び水を用いて生成物を仕上げた。有機層を乾燥した(MgSO4)。溶媒を蒸発させた。RP HPLC,方法Bを用いて残留物を精製した。画分を一緒にし、溶媒を蒸発させた。DIPEを用いて生成物を固化させた。固体を濾過し、洗浄し、オーブン中で乾燥し、0.165gの化合物(55)を与えた。
【0088】
実施例B.14
【化49】
THF(10ml)中の中間体(23)(0.0043モル)、ブロモメチルシクロプロパン(0.01モル)及びCs2CO3(0.008モル)の混合物を脱ガスした。反応混合物を窒素下に65℃において20時間攪拌した。いくらかのジスルフィドが生成したので、NaBH4を加え、反応混合物を65℃でさらに24時間攪拌した。反応混合物を濾過して塩を除去した。濾液をDCMで希釈し、水で洗浄した。有機層を乾燥し(MgSO4)、濾過し、完全に乾燥するまで蒸発させ、2gの化合物(16)を与えた。
【0089】
実施例B.15
【化50】
炭素上の白金(5%)+0.5%V(0.3g)を、窒素流下でTHF(50ml)中に懸濁させ、次いで化合物(69)(0.00157モル)を加え、3当量の水素が吸収されるまで反応混合物を水素雰囲気下で攪拌した。ジカライト上の濾過により触媒を除去し、次いで溶媒を蒸発させ、0.839gの化合物(78)を与えた。
【0090】
実施例B.16
【化51】
化合物(78)(0.001521モル)をTHF(100ml)中に溶解し、DIPE(0.001521モル)を加えた。アセチリクロリド(0.001521モル)を加え、反応混合物を室温で30分間攪拌した。いくらかの1M NaOH溶液を加え、THFを用いて生成物を仕上げた。有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。方法Aに従って残留物を精製した。画分を一緒にし、溶媒を蒸発させ、0.375gの化合物(79)を与えた。
【0091】
実施例B.17
【化52】
化合物(79)(0.000286モル)をDMF(10nl)中に溶解した。反応混合物をN2雰囲気下に0℃において冷却した。次いで水素化ナトリウム(0.002001モル;鉱油中の60%分散液)を加えた。反応混合物を30分間攪拌し、次いでヨードメタン(0.002573モル)を加えた。反応混合物を1時間攪拌し、ジクロロメタン(50ml)及びH2O(20ml)を用いて生成物を仕上げた。有機層を乾燥し(MgSO4)、濾過し、溶媒を蒸発させた。DIPEを用いて生成物を固化させた。固体を濾過し、洗浄し、オーブン中で乾燥し、0.132gの化合物(80)を与えた。
【0092】
表F−1は、上記の実施例の1つに従って製造された化合物を挙げている。
【0093】
【表1】
【0094】
【表2】
【0095】
【表3】
【0096】
【表4】
【0097】
【表5】
【0098】
【表6】
【0099】
【表7】
【0100】
【表8】
【0101】
【表9】
【0102】
表F−2は、実施例B.5及びB.7に記載された方法を用いて、且つ化合物(116)の場合にはさらに実施例B.10に記載されたFe/酢酸を用いる処理を用いて製造された化合物を挙げている。
【0103】
【表10】
【0104】
C.化合物の同定
C1.LCMS
本発明の化合物のLCMS−特性化のために、以下の方法を用いた。
【0105】
一般的方法A
HPLC測定は、脱ガス器を有するクウォーターナリーポンプ、オートサンプラー、カラムオーブン(他に指示しなければ40℃に設定)、ダイオード−アレー検出器(DAD)及び下記のそれぞれの方法において特定されるカラムを含んでなるAlliance HT 2790(Waters)システムを用いて行なわれた。カラムからの流れはMS分光計に分けられた。MS検出器は、エレクトロスプレーイオン化源を用いて形成された。0.1秒の滞留時間を用いて1秒内に100から1000まで走査することにより、質量スペクトルを取得した。毛管針電圧は3kVであり、源温度は140℃に保たれた。ネブライザーガスとして窒素を用いた。Waters−Micromass MassLynx−Openlynxデータシステムを用いてデータ取得を行なった。
【0106】
一般的方法B
LC測定は、バイナリーポンプ、サンプルオルガナイザー、カラムヒーター(55℃に設定)、ダイオードアレー検出器(DAD)及び下記のそれぞれの方法において特定されるカラムを含んでなるAcquity UPLC(Waters)システムを用いて行なわれた。カラムからの流れはMS分光計に分けられた。MS検出器は、エレクトロスプレーイオン化源を用いて形成された。0.02秒の滞留時間を用いて0.18秒内に100から1000まで走査することにより、質量スペクトルを取得した。毛管針電圧は3.5kVであり、源温度は140℃に保たれた。ネブライザーガスとして窒素を用いた。Waters−Micromass MassLynx−Openlynxデータシステムを用いてデータ取得を行なった。
【0107】
LCMS法1
一般的方法Aに加え:逆相HPLCをXterra MS C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。3種の移動相(移動相A:95% 25mM酢酸アンモニウム+5%アセトニトリル;移動相B:アセトニトリル;移動相C:メタノール)を用い、6.5分内に100%Aから1%A、49%B及び50%Cにし、1分内に1%A及び99%Bにし、これらの条件を1分間保持し、100%Aを用いて1.5分間再平衡化する勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0108】
LCMS法2
一般的方法Bに加え:架橋エチルシロキサン/シリカハイブリッド(BEH) C18カラム(1.7μm,2.1x50mm;WatersAcquity)上で、0.8ml/分の流量を用い、逆相UPLC(超性能液体クロマトグラフィー(Ultra Performance Liquid Chromatography))を行なった。2種の移動相(移動相A:H2O中の0.1%ギ酸/メタノール 95/5;移動相B:メタノール)を用い、1.3分内に95%A及び5%Bから5%A及び95%Bにし、0.2分間保持する勾配条件を実施した。0.5μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0109】
LCMS法3
一般的方法Aに加え:カラムヒーターを45℃に設定した。逆相HPLCをXterra MS C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。3種の移動相(移動相A:H2O中の0.1%ギ酸/メタノール 95/5;移動相B:アセトニトリル;移動相C:メタノール)を用い、7分内に100%Aから1%A、49%B及び50%Cにし、これらの条件を1分間保持する勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に1
0Vであった。
【0110】
LCMS法4
一般的方法Aに加え:カラムヒーターを60℃に設定した。逆相HPLCをXterra MS C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。3種の移動相(移動相A:95% 25mM酢酸アンモニウム+5%アセトニトリル;移動相B:アセトニトリル;移動相C:メタノール)を用い、6.5分内に100%Aから50%B及び50%Cにし、0.5分内に100%Bにし、これらの条件を1分間保持し、100%Aを用いて1.5分間再平衡化する勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0111】
LCMS法5
一般的方法Aに加え:逆相HPLCをAtlantis C18カラム(3.5μm,4.6x100mm)上で、1.6ml/分の流量を用いて行なった。2種の移動相(移動相A:70%メタノール+30% H2O;移動相B:H2O中の0.1%ギ酸/メタノール 95/5)を用い、12分内に100%Bから5%B+95%Aにする勾配条件を実施した。10μlの注入容積を用いた。コーン電圧は、正のイオン化モードの場合に10Vであり、負のイオン化モードの場合に20Vであった。
【0112】
C2.融点
複数の化合物に関し、DSC823e(Mettler−Toledo)を用いて融点を決定した。30℃/分の温度勾配を用いて融点を測定した。報告される値はピーク値である。最高温度は400℃であった。
【0113】
複数の化合物に関し、直線状温度勾配を有する加熱板、スライディングポインター(sliding pointer)及び度摂氏における温度目盛からなるKoflerホットベンチ(hot bench)を用いて融点を得た。
【0114】
【表11】
【0115】
【表12】
【0116】
【表13】
【0117】
D.薬理学的実施例
D.1 ヒトCB2受容体の活性化に反応するcAMPの阻害
均一時間分解蛍光(HTRF)アッセイを介し、ヒトCB2(hCB2)受容体が活性化される時のホルスコリン−活性化cAMP生産を阻害する能力を測定することにより、試験化合物の機能的活性を評価した。
【0118】
hCB2が安定にトランスフェクションされたCHO−K1細胞を、T175 Falconフラスコ中で、2%の溶液A(5.106IU/lのペニシリンG,5g/lの硫酸ストレプトマイシン,5.5g/lのピルベート,14.6g/lのL−グルタミン,1MのNaOH)及び10%の胎児ウシ血清が補足されたDMEM/NUT MIX F−12倍地中において80〜90%密集まで生育させた。実験の前に倍地を除去し、細胞をPBS/EDTA(140mM NaCl,1mM Na2−EDTA,8mM Na2HPO4.2H2O,8.5mM KH2PO4,2.7mM KCl,21mM グルコース)で洗浄し、刺激緩衝液(HBSS 1x,IBMX 1mM,Hepes 5mM,MgCl2 10mM,BSA 0.1%,pH7.4)中に再懸濁させた。hCB2実験のために、細胞をml当たり106個の細胞の濃度まで希釈した。cAMP Dynamic HTRFキット(CIS bio international,France)を用い、製造者の推薦に従ってアッセイを行なった。
【0119】
CB2に関し、384平底黒ポリスチレンアッセイプレート(Costar)の各ウェルを、15μMのホルスコリン及び試験化合物(3%DMSO中)、3%DMSO又は10μM Win55212−2(3%DMSO中)を含有する10μlの刺激緩衝液で満たした。次いで20μlの希釈されたhCB2−CHO−K1細胞を加えた(ウェル当たり20,000個の細胞)。室温における暗所中での30分間のインキュベーションの後、10μlのcAMP−XL665及び10μlの抗−cAMPクリプテート(両方とも1/100の最終的な希釈において)を細胞に加えた。
【0120】
室温における暗所中で反応混合物を1〜24時間平衡化した後、Discoveryミクロプレート蛍光カウンター(Perkin Elmer)を用いて665nm及び620nmにおいて蛍光を測定し、665nm/620nmのシグナル比を計算した。試験化合物のシグナル比を、それぞれDMSO標準(最大シグナル比,cAMPの阻害なし)及びhCB2に関するWIN55212−2(最小シグナル比,cAMPの最大阻害)のシグナル比に対して相対的に表した。各試験化合物に関して作られる用量反応曲線から、cAMPレベルの最大阻害の50%が観察される用量(EC50,表中でpEC50=−log(EC50)値として表される)及びWIN55212−2(hCB2に関する)と比較して10μMの試験化合物を用いて達成される阻害のレベルを計算した。
【0121】
【表14】
【0122】
【表15】
【0123】
D.2 比較データ
体温の低下、平伏体姿勢及び散瞳のようなCB1に関連する望ましくない副作用を、本発明の複数の化合物及び引用文献国際公開第2006/048754号パンフレットにより包含される複数の化合物に関して測定した。両方の組の化合物に関し、処置された動物の半分より多くで体温への効果、すなわち低下が観察されるLAD(最低許容用量)を決定した。データを表D−3に挙げる。
【0124】
【表16】
【0125】
【表17】
【特許請求の範囲】
【請求項1】
立体化学的異性体を含む式(I)
【化1】
[式中、
nは整数0、1又は2であり;
XはSO、SO2又はN−(CO)−R1であり;
R1は水素;
C1−6アルキル;
C1−6アルキルオキシ;
C1−4アルキルオキシC1−4アルキル;又は
ポリハロC1−6アルキル
であり;
R2はC1−6アルキルであり;
R3は水素、ハロ、C1−4アルキル、C1−4アルキルオキシ、トリフルオロメチル又はシアノであり;
R4はC1−8アルキル;
C3−8シクロアルキルで置換されたC1−8アルキル;
ポリハロC1−8アルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC1−8アルキル;
C3−8シクロアルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC3−8シクロアルキル;
テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、
アリール;あるいは
ヘテロアリール
であり;
アリールはフェニル;あるいはハロ、ヒドロキシ、C1−4アルキル、ポリハロC1−4アルキル、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、NR5R6、R7−カルボニル、R7−SO2−、又はヒドロキシ、NR5R6、R7−カルボニルもしくはR7−SO2−で置換されたC1−4アルキルからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたフェニルであり;
ヘテロアリールはフラニル、チオフェニル、ピロリル、ピラゾリル、イミダゾリル、イソオキサゾリル、チアゾリル、トリアゾリル、テトラゾリル、イソチアゾリル、チアジアゾリル、オキサジアゾリル、ピリジニル、ピリダジニル、ピリミジニル又はピラジニルから選ばれ;
ここでR5及びR6は互いに独立して水素、C1−4アルキル、ポリハロC1−4アルキル、アミノスルホニル又はC1−8アルキルスルホニル;あるいはR7−カルボニルから
選ばれ;
ここでR5及びR6は、R5及びR6を有する窒素原子と一緒になって、ピロリジニル、ピペリジニル、ピペラジニル又はモルホリニル環を形成することができ;そして
ここでR7はC1−4アルキル、ヒドロキシ、アミノ、モノ−もしくはジ−(C1−4アルキル)アミノ、(ヒドロキシC1−4アルキル)アミノ、(C1−4アルキルオキシC1−4アルキル)アミノ、ジ(C1−4アルキル)アミノC1−4アルキル、ピロリジニル、ピペリジニル、モルホリニル又はN−メチル−ピペラジニルである]
の化合物あるいはその製薬学的に許容され得る酸付加塩又はその溶媒和物。
【請求項2】
XがSO2である請求項1に記載の化合物。
【請求項3】
XがN−(CO)−R1である請求項1に記載の化合物。
【請求項4】
R2がC1−6アルキルである請求項1〜3のいずれかに記載の化合物。
【請求項5】
製薬学的に許容され得る担体及び請求項1〜4のいずれかに記載の化合物の治療的に活性な量を含んでなる製薬学的組成物。
【請求項6】
請求項1〜4のいずれかに記載の化合物の治療的に活性な量を製薬学的に許容され得る担体と緊密に混合する、請求項5に記載の製薬学的組成物の調製方法。
【請求項7】
薬剤としての使用のための請求項1〜4のいずれかに記載の化合物。
【請求項8】
カンナビノイド受容体2活性、特にCB2作動活性により媒介される状態又は疾患の処置用の薬剤の製造のための請求項1〜4のいずれかに記載の化合物。
【請求項9】
nが0である請求項1に記載の式(I)の化合物として定義される式(I−a)の化合物を、適した塩基の存在下に、反応に不活性な溶媒中で、中間体(II)を、Lが離脱基である中間体(III)と反応させることにより;
【化2】
[式中、R2、R3及びR4は請求項1におけるとおりに定義される];
あるいは;必要に応じて;式(I−a)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−a)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することにより製造する方法。
【請求項10】
nが1である請求項1に記載の式(I)の化合物として定義される式(I−b)の化合物を、X、R2、R3及びR4が請求項1において定義されたとおりである式(I−a)の化合物を、酸化剤を用いてS−酸化することにより;
【化3】
あるいは;必要に応じて;式(I−b)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−b)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することにより製造する方法。
【請求項11】
nが1である請求項1に記載の式(I)の化合物として定義される式(I−c)の化合物を、X、R2、R3及びR4が請求項1において定義されたとおりである式(I−a)の化合物を、酸化剤を用いてS−酸化することにより;
【化4】
あるいは;必要に応じて;式(I−c)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−c)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することにより製造する方法。
【請求項12】
XがN−(CO)−R1を示す式(I)の化合物として定義される式(I−d)の化合物を、反応に不活性な溶媒中で、Wが適した離脱基である中間体(VI)を用いて中間体(V)をN−アルキル化することにより、
【化5】
あるいは;必要に応じて;式(I−d)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−d)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することによる製造方法
。
【請求項1】
立体化学的異性体を含む式(I)
【化1】
[式中、
nは整数0、1又は2であり;
XはSO、SO2又はN−(CO)−R1であり;
R1は水素;
C1−6アルキル;
C1−6アルキルオキシ;
C1−4アルキルオキシC1−4アルキル;又は
ポリハロC1−6アルキル
であり;
R2はC1−6アルキルであり;
R3は水素、ハロ、C1−4アルキル、C1−4アルキルオキシ、トリフルオロメチル又はシアノであり;
R4はC1−8アルキル;
C3−8シクロアルキルで置換されたC1−8アルキル;
ポリハロC1−8アルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC1−8アルキル;
C3−8シクロアルキル;
ヒドロキシ、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、アリール又はヘテロアリールからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたC3−8シクロアルキル;
テトラヒドロピラニル、テトラヒドロフラニル、オキセタニル、
アリール;あるいは
ヘテロアリール
であり;
アリールはフェニル;あるいはハロ、ヒドロキシ、C1−4アルキル、ポリハロC1−4アルキル、C1−4アルキルオキシ、ポリハロC1−4アルキルオキシ、シアノ、ニトロ、NR5R6、R7−カルボニル、R7−SO2−、又はヒドロキシ、NR5R6、R7−カルボニルもしくはR7−SO2−で置換されたC1−4アルキルからそれぞれ独立して選ばれる1、2又は3個の置換基で置換されたフェニルであり;
ヘテロアリールはフラニル、チオフェニル、ピロリル、ピラゾリル、イミダゾリル、イソオキサゾリル、チアゾリル、トリアゾリル、テトラゾリル、イソチアゾリル、チアジアゾリル、オキサジアゾリル、ピリジニル、ピリダジニル、ピリミジニル又はピラジニルから選ばれ;
ここでR5及びR6は互いに独立して水素、C1−4アルキル、ポリハロC1−4アルキル、アミノスルホニル又はC1−8アルキルスルホニル;あるいはR7−カルボニルから
選ばれ;
ここでR5及びR6は、R5及びR6を有する窒素原子と一緒になって、ピロリジニル、ピペリジニル、ピペラジニル又はモルホリニル環を形成することができ;そして
ここでR7はC1−4アルキル、ヒドロキシ、アミノ、モノ−もしくはジ−(C1−4アルキル)アミノ、(ヒドロキシC1−4アルキル)アミノ、(C1−4アルキルオキシC1−4アルキル)アミノ、ジ(C1−4アルキル)アミノC1−4アルキル、ピロリジニル、ピペリジニル、モルホリニル又はN−メチル−ピペラジニルである]
の化合物あるいはその製薬学的に許容され得る酸付加塩又はその溶媒和物。
【請求項2】
XがSO2である請求項1に記載の化合物。
【請求項3】
XがN−(CO)−R1である請求項1に記載の化合物。
【請求項4】
R2がC1−6アルキルである請求項1〜3のいずれかに記載の化合物。
【請求項5】
製薬学的に許容され得る担体及び請求項1〜4のいずれかに記載の化合物の治療的に活性な量を含んでなる製薬学的組成物。
【請求項6】
請求項1〜4のいずれかに記載の化合物の治療的に活性な量を製薬学的に許容され得る担体と緊密に混合する、請求項5に記載の製薬学的組成物の調製方法。
【請求項7】
薬剤としての使用のための請求項1〜4のいずれかに記載の化合物。
【請求項8】
カンナビノイド受容体2活性、特にCB2作動活性により媒介される状態又は疾患の処置用の薬剤の製造のための請求項1〜4のいずれかに記載の化合物。
【請求項9】
nが0である請求項1に記載の式(I)の化合物として定義される式(I−a)の化合物を、適した塩基の存在下に、反応に不活性な溶媒中で、中間体(II)を、Lが離脱基である中間体(III)と反応させることにより;
【化2】
[式中、R2、R3及びR4は請求項1におけるとおりに定義される];
あるいは;必要に応じて;式(I−a)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−a)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することにより製造する方法。
【請求項10】
nが1である請求項1に記載の式(I)の化合物として定義される式(I−b)の化合物を、X、R2、R3及びR4が請求項1において定義されたとおりである式(I−a)の化合物を、酸化剤を用いてS−酸化することにより;
【化3】
あるいは;必要に応じて;式(I−b)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−b)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することにより製造する方法。
【請求項11】
nが1である請求項1に記載の式(I)の化合物として定義される式(I−c)の化合物を、X、R2、R3及びR4が請求項1において定義されたとおりである式(I−a)の化合物を、酸化剤を用いてS−酸化することにより;
【化4】
あるいは;必要に応じて;式(I−c)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−c)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することにより製造する方法。
【請求項12】
XがN−(CO)−R1を示す式(I)の化合物として定義される式(I−d)の化合物を、反応に不活性な溶媒中で、Wが適した離脱基である中間体(VI)を用いて中間体(V)をN−アルキル化することにより、
【化5】
あるいは;必要に応じて;式(I−d)の化合物を製薬学的に許容され得る酸付加塩に転換するか、又は逆に、アルカリを用いて式(I−d)の化合物の酸付加塩を遊離の塩基形態に転換し;そして必要に応じて、その立体化学的異性体を製造することによる製造方法
。
【公表番号】特表2010−523482(P2010−523482A)
【公表日】平成22年7月15日(2010.7.15)
【国際特許分類】
【出願番号】特願2010−500244(P2010−500244)
【出願日】平成20年3月25日(2008.3.25)
【国際出願番号】PCT/EP2008/053480
【国際公開番号】WO2008/119694
【国際公開日】平成20年10月9日(2008.10.9)
【出願人】(390033008)ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ (616)
【氏名又は名称原語表記】JANSSEN PHARMACEUTICA NAAMLOZE VENNOOTSCHAP
【Fターム(参考)】
【公表日】平成22年7月15日(2010.7.15)
【国際特許分類】
【出願日】平成20年3月25日(2008.3.25)
【国際出願番号】PCT/EP2008/053480
【国際公開番号】WO2008/119694
【国際公開日】平成20年10月9日(2008.10.9)
【出願人】(390033008)ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ (616)
【氏名又は名称原語表記】JANSSEN PHARMACEUTICA NAAMLOZE VENNOOTSCHAP
【Fターム(参考)】
[ Back to top ]