
ベンゾオキサゾール化合物および使用方法
本発明は、ベンゾオキサゾール化合物およびその医薬上許容される塩ならびにそれを含む医薬組成物を提供する。本発明は、本明細書に記載される使用方法をさらに提供する。
【発明の詳細な説明】
【背景技術】
【0001】
損傷または感染に対する多くの初期の免疫応答は、自然免疫Toll様受容体(TLR)のファミリーによって開始される。TLRファミリーは、幅広い特異性を有し、病原生物の間で高度に保存された分子構造または異なる種類の物理的損傷を認識する。特異的TLRのリガンドとしては、エンドトキシン、一本鎖もしくは二本鎖RNA、ペプチドグリカン、フラジェリン、熱ショックタンパク質が挙げられ、TLR9の場合には、DNA中の非メチル化CpG配列が挙げられる(Bell et al.,Trends Immunol 24:528−533(2003)およびWagner,Trends Immunol 25:381−6(2004))。この疾患または損傷の初期のセンシングは、続いて起こる免疫応答に絶対不可欠であるが、同時にそれは不適切な応答または損傷性の応答の原因でもあり得る。例えば、TLR9は、狼瘡を含む自己免疫障害に結び付けられているが、それは長い間抗DNA抗体反応性、および多発性硬化症に関連付けられている(Prinz et al.,J Clin Invest 116:456−464(2006))。また、TLR9は、敗血性ショックによる死に関連する、制御されない炎症応答の根底にあると思われる(Plitas et al.,J Exp Med 205:1277−83(2008))。
【0002】
TLR9は、非メチル化CpG配列に結合し、細胞活性化およびサイトカイン分泌をもたらす、ヒトB細胞および形質細胞様樹状細胞(PDC)に発現される自然免疫受容体として最初に同定された(Hemmi et al.,Nature 408:740−745(2000))。これらの配列は、真核生物のDNAと比較して、細菌のDNAで過剰に発現され、そのため、細菌感染の指標としての機能を果たすことができる。しかし、真核生物のDNAは、いくらかの非メチル化CpGを含み、同様にTLR9を刺激する能力がある(Vallin et al.,J Immunol.163:6306−13(1999)およびLeadbetter et al.(2002))。瀕死の細胞のDNAは、一般に摂取され系から隔離されるが、狼瘡は、過剰な細胞片のクリアランス不良および蓄積を引き起こす遺伝的欠損に関連し(Krishnan et al.,Seminars in Immunology 18:240−243(2006))、免疫系を異常に高レベルのリガンドに曝露する。この状態は、B細胞上の複合体特異的免疫グロブリンか、または樹状細胞もしくは抗原提示細胞上のFc受容体のいずれかによるDNA含有複合体の標的化取り込みと相まって、該刺激複合体にTLR9リガンドが存在することにより後押しされた応答である自己抗原の提示および反応をもたらし得る。この知見は、PDCが、全身性紅斑性狼瘡(SLE)血清中に見出される複合体にα−インターフェロンを分泌することにより応答し、これがFc受容体であってDNA依存性であり、TLR9に媒介されるという事実により例証される(Leadbetter et al.,(2002)およびMeans et al.,J Clin Invest 1152:407−17(2005))。得られるα−インターフェロンは、樹状細胞およびB細胞の成熟をさらに駆動することができる。それに対応して、アレイデータにより、重篤な疾患の患者におけるα−インターフェロン誘導性遺伝子の活性化が示される(Bennett et al.,J Exp Med.197:711−23(2003))。PDCは、身体においてα−インターフェロンの主な供給源であるので、この知見は疾患をもつこの細胞集団にさらに関連する。
【0003】
B細胞も狼瘡自己免疫の中心であり、合成オリゴヌクレオチドを用いるTLR9刺激によりIL−6および抗体を増殖および産生するように駆動される。自己反応性細胞表面免疫グロブリンを発現しているB細胞との選択的結合のために、自己抗原特異的細胞は、この応答において優勢となる。従って、DNAまたはRNAを含有する細胞片がTLR9により刺激された自己抗原特異的B細胞からの抗体と結合して、Fc受容体を介して形質細胞様樹状細胞を刺激する、自己強化サイクルが働いている。
【発明の概要】
【0004】
本発明の第1の実施形態は、式(I):
【化1】

の化合物またはその医薬上許容される塩を提供し、
式中、R5、R6、またはR7のうちの1つが、式(a):
【化2】
の基であり、かつ、R5が(a)である場合、R6およびR7は双方ともHであり、R6が(a)である場合、R5およびR7は双方ともHであり、R7が(a)である場合、R5およびR6は双方ともHであり、
iおよびjは同一であって、かつ0、1、2、3または4であり、
R1およびR4は同一であって、かつH、CH3およびCH2CH3からなる群から選択され、
R2はCH3であり、R3は(CH2)hN(CH3)2(hが2、3または4である)および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは、
R2およびR3は同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
(CH2)nN(CH3)2(式中、nは2、3または4である)、
(CH2)pO(CH2)qN(CH3)2(式中、pおよびqは同一であって、かつ2または3である)、
式(b):
【化3】
(式中、uは0または1である)の基、または
式(c):
【化4】
(式中、vは0または1であり、ZはNまたはCHであり、XはOまたはNCH3である)の基からなる群から選択されるか、又は
R1−N−R2およびR3−N−R4は同一であって、かつ
式(d):
【化5】
の基、および
式(e):
【化6】
(式中、rは1、2または3であり、
YはCHまたはNであり、
R8はH、CH3、CH(CH3)2、N(CH3)2、CH2OCH3または式(f):
【化7】
(式中、tは、0または1である)の基であり、
R9はH、CH2OCH3または式(f)の基である)
の基からなる群から選択される。
【0005】
本発明の第2の実施形態は、本明細書に記載される化合物を含む医薬組成物を提供する。
【0006】
本発明の第3の実施形態は、被験体において免疫学的障害、例えば、敗血症、狼瘡、関節リウマチ、または多発性硬化症などを治療する方法を提供し、この方法は、本明細書に記載される化合物を被験体に有効量で投与することを含む。
【0007】
本発明の第4の実施形態は、被験体において狼瘡を治療する方法を提供し、この方法は、本明細書に記載される化合物を被験体に有効量で投与することを含む。
【0008】
本発明の第5の実施形態は、免疫学的障害、例えば、敗血症、狼瘡、関節リウマチ、または多発性硬化症などを治療するための薬物の製造のために、本明細書に記載される化合物を使用することを提供する。
【0009】
本発明のその他の実施形態は、本明細書において開示され、以下でさらに詳細に考察される。
【図面の簡単な説明】
【0010】
【図1】実施例11に記載されるように、インビボで20mg/kgまたは60mg/kgの化合物2、化合物23、またはヒドロキシクロロキン(HCQ)の2週間の投与、そしてエキソビボでオリゴCpG1668を用いるリンパ節の刺激後の、マウスから取り出したリンパ節において検出されたインターロイキン6(IL−6)の濃度を示す棒グラフである。
【図2】実施例12Aに記載されるように、MRL/MpJ−faslpr/J(「MRL/lpr」)マウスの20mg/kgまたは60mg/kgの化合物23を用いる投薬の前後7週間の、マウス抗dsDNAのELISAアッセイにおける、450ナノメートル(OD450)での光学濃度測定の結果を示すプロット図である。
【図3】実施例12Aに記載されるように、投薬の4、10または12週間後に、20mg/kgまたは60mg/kgの化合物23を投与されたマウスのMRL/lpr自発的狼瘡モデルにおける抗核抗体(ANA)試験の結果を示す一連の棒グラフを示す図である。結果は、ANA結果の5つの考えられるカテゴリー(−、−/+、0、+、または++)の各々にスコアされたマウスの数として示す。
【図4】実施例12Bに記載されるように、化合物23(20mg/kgまたは60mg/kg)、HCQ、またはシクロホスファミドを用いる12週間の投薬前および投薬後の、MRL/lprマウス抗dsDNAのOD450でのELISAアッセイの結果を示すグラフである。
【図5】実施例13に記載されるように、20mg/kgまたは60mg/kgの化合物23または化合物30を用いる16週間のNZB/WF1/Jマウスの投薬前および投薬後の、マウス抗dsDNAのOD450でのELISAアッセイの結果を示すプロット図である。
【図6】実施例13に記載されるように、20mg/kgまたは60mg/kgの化合物23または化合物30を用いる投薬の16週間後の、5つの考えられるANAスコアの各々のNZB/WF1/Jマウスの数を示す一連の棒グラフを示す図である。
【図7】実施例14に記載されるように、合成ペプチドMOGp35−55で免疫化して、多発性硬化症に似た症状の実験的自己免疫性脳炎(EAE)を誘導したC57BL/6Jマウスに、20mg/kgまたは60mg/kgの化合物23を投与した結果を示すグラフである。
【図8】実施例15に記載されるように、完全フロイントアジュバントで乳化したウシII型コラーゲンで免疫化して、関節炎を誘導した雄DBA/1Jマウスに、6mg/kg、20mg/kgまたは60mg/kgの化合物2を投与した結果を示すグラフである。
【発明を実施するための形態】
【0011】
(A.定義)
「治療」、「治療する」、および「治療すること」とは、本明細書に記載される障害または疾患の進行を後退させること、軽減すること、または阻止することをさす。さらに、本明細書において用いられるように、被験体の「治療」とは、免疫学的障害を有するか、かかる免疫学的障害の症状を有するか、あるいはかかる免疫学的障害の危険性がある(または免疫学的障害に罹患しやすい)被験体への、前記障害、前記障害の症状、あるいは前記障害の危険性(または前記障害に対する易罹患性)を治す、癒す、軽減する、緩和する、変化させる、除去する、改善する、好転させる、または作用する目的での、本明細書に記載される本発明の化合物の被験体への適用または投与、あるいは被験体の細胞または組織への前記化合物の適用または投与が含まれる。用語「治療すること」とは、損傷、病変または状態の治療または改善の成功のいずれかの徴候をさし、それには、任意の客観的または主観的パラメーター、例えば、軽減、緩解、症状の減少あるいは損傷、病変または状態を被験体にとってより許容できるものにすること、変性または減退の速度を遅らせること、変性の最終状態の衰弱を抑えること、被験体の身体的または精神的健康を向上させること、あるいは、場合によっては、前記障害のより重症な型または症状の発症を予防することなど、が含まれる。症状の治療または改善は、身体検査、精神鑑定、または当分野で公知の診断検査の結果を含む客観的または主観的パラメーターに基づくことができる。
【0012】
一実施形態において、用語「治療すること」には、免疫学的障害によって引き起こされるかまたは免疫学的障害に関連する炎症、感染、発熱、異常な精神状態、臓器機能不全、貧血、関節痛、疲労、認知的困難、痙攣および/または振戦からの好転またはその除去などの免疫学的障害の症状を好転させることが含まれる。
【0013】
「予防」、「予防する」、および「予防すること」とは、本明細書に記載される障害または疾患の発生または発症を、処置を取らない場合に起こると思われるその発生または発症と比べてなくすまたは低減させることをさす。
【0014】
「有効な量」とは、臨床検査および評価、患者の観察、および/または同類のものによって示される障害または疾患の症状の除去をもたらす量をさす。「有効な量」は、生物学的または化学的活性において検出可能な変化をもたらす用量をさらに表すことができる。検出可能な変化は、当該機構またはプロセスについて当業者によって検出し、かつ/またはさらに定量することができる。さらに、「有効な量」は、所望の生理学的状態を維持する、すなわち、目的の状態の著しい減退を低減もしくは予防し、かつ/または好転を促す量を表すことができる。「有効な量」は、治療上有効な量をさらにさすことができる。
【0015】
本明細書において用いられる「被験体」とは、哺乳類被験体(例えば、イヌ、ネコ、ウマ、ウシ、ヒツジ、ヤギ、サルなど)、特にヒト被験体(男性および女性の両方の被験体を含み、新生児、乳児、小児、青年期、成人および高齢者の被験体を含み、様々な人種および民族(限定されるものではないが、白人、黒人、アジア系、アメリカインディアンおよびヒスパニック系を含む)をさらに含む)を意味する。
【0016】
本明細書において用いられるように、用語「医薬上許容される塩」とは、本発明の化合物の比較的毒性のない無機酸塩および有機酸塩をさす。これらの塩は、前記化合物の最終的な単離および精製中にインサイチューで調製することができ、あるいは生成した化合物をその遊離形態で適した有機酸または無機酸と別々に反応させ、そのようにして形成された塩を単離することにより調製することもできる。代表的な酸性塩としては、酢酸塩、アジピン酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、重炭酸塩/炭酸塩、重硫酸塩/硫酸塩、ホウ酸塩、カンシル酸塩、クエン酸塩、シクラミン酸塩、エジシル酸塩、エシレート、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンズ酸塩、塩酸塩/塩化物、臭化水素酸塩/臭化物、ヨウ化水素酸塩/ヨウ化物、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシル酸塩、メチル硫酸塩、ナフチル酸塩、2−ナプシル酸塩、ニコチン酸塩、硝酸塩、オロチン酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素/リン酸二水素、ピログルタミン酸塩、サッカリン酸塩、ステアリン酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、トシル酸塩、トリフルオロ酢酸塩およびキシナホ酸塩が挙げられる。一実施形態において、医薬上許容される塩は塩酸塩/塩化物塩である。
【0017】
(B.化合物)
本明細書に記載される一部の化合物は、1以上の不斉中心を含んでよく、従ってそれらの化合物は、様々な異性体形、例えば立体異性体および/またはジアステレオマーとして存在することができる。キラル中心の周囲の結合の配向が式に明記されていない場合には、その式は、本発明の化合物の構造において起こり得る、不斉炭素に基づく幾何異性体、光学異性体、立体異性体および互変異性体などの考え得るあらゆる異性体を包含すると理解すべきである。
【0018】
上に述べたように、本発明は、式(I):
【化8】
の化合物またはその医薬上許容される塩(式中、i、R1、R2、R5、R6、およびR7は、本明細書において上述した記載の通りに定義される)を提供する。
【0019】
式(I)の化合物のいくつかの実施形態では、iおよびjは、双方とも1である。
【0020】
さらなる実施形態では、R2およびR3は同一であって、以下の:
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
本明細書で上述した式(b)の基、および
本明細書で上述した式(c)の基
からなる群から選択される。
【0021】
他の実施形態によれば、R1−N−R2およびR3−N−R4は同一であって、かつ
式(g):
【化9】
の基、式(h):
【化10】
の基、 式(i):
【化11】
の基、 式(j):
【化12】
の基、 式(k):
【化13】
の基または式(m):
【化14】
の基から選択される。
【0022】
式(I)の化合物のいくつかの実施形態では、
iおよびjは双方とも1であり、
R1およびR4は同一であって、かつHおよびCH3からなる群から選択され、
R2はCH3であり、R3は(CH2)2N(CH3)2および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは
R2およびR3は同一であって、かつ
a)(CH2)kCH3(式中、kは0、1または2である)、
b)(CH2)mN(CH2CH3)2(式中、mは2または3である)、
c)本明細書で上述した式(b)の基、および
d)本明細書で上述した式(c)の基
からなる群から選択されるか、又は、
R1−N−R2およびR3−N−R4は同一であって、かつ
式(g)の基、式(h)の基、式(i)の基、式(j)の基、式(k)の基、および式(m)の基からなる群から選択される(全ては本明細書で上述した記載の通りである)。
【0023】
さらなる実施形態では、R5は、本明細書で上述した式(a)の基であり、R6およびR7は各々Hである。
【0024】
式(I)の化合物の特定の実施形態では、この化合物は以下から選択される構造を有する。
【0025】
【表1A】
【表1B】
【表1C】
【表1D】
【表1E】
【表1F】
【表1G】
【0026】
もう1つの実施形態では、式Iの化合物は、化合物1、化合物2、化合物10、化合物11、化合物12、化合物13、化合物14、化合物16、化合物17、化合物26、化合物29、化合物30、化合物31、化合物32、化合物34、および化合物35またはその医薬上許容される塩からなる群から選択される。もう1つの実施形態では、前記化合物は、化合物13、化合物16、化合物22、化合物23、化合物26、化合物27、化合物30、化合物34、および化合物35またはその医薬上許容される塩からなる群から選択される。もう1つの実施形態では、前記化合物は、化合物23、化合物26、および化合物30、またはその医薬上許容される塩からなる群から選択される。さらにもう1つの実施形態では、前記化合物は、化合物23またはその医薬上許容される塩である。
【0027】
(C.化合物の合成)
本明細書に記載される化合物は、一連の反応によって調製することができる。本発明の化合物を調製するための一般的方法を下に示す。実施例を記載するセクションにおいて下にさらに述べるように、ある特定の場合において例として特定の化合物を記載する。下に記述されている場合を除き、以下の方法では、R1、R2、R3、R4、i、およびjは、上の式Iにおいて定義されるものと同一である。
【0028】
方法1−アルキル化、アミノ化またはアルキル化、アミノ化およびN−メチル化
【化15】
(R1aおよびR4aがHである場合、またはR2aおよびR3aがHである場合、N−メチル化)
上に示した方法1の反応スキームにおいて、
n、i、およびjは同一であって、かつ0、1、2、3または4であり、
R1aおよびR4aは双方ともRxと同一であり、
R2aおよびR3aは双方ともRyと同一であり、
R1bおよびR4bは同一であり、
R2bおよびR3bは同一であり、
R1aおよびR4aが双方ともHである場合には、R1bおよびR4bは双方ともメチルであり、
R2aおよびR3aが双方ともHである場合には、R2bおよびR3bは双方ともメチルである。
【0029】
本発明の実施形態による対称の化合物を合成する一方法では、例えば、2−(4−ヒドロキシフェニル)ベンゾオキサゾール−6−オール(1A)を、ジメチルスルホキシド(DMSO)中炭酸カリウムの存在下、ブロモクロロアルカン(1B)と反応させてジクロロエーテル誘導体(1C)を形成することができる。また、化合物1Aの代わりに2−(3−ヒドロキシフェニル)ベンゾオキサゾール−6−オールおよび2−(2−ヒドロキシフェニル)ベンゾオキサゾール−6−オールを用いてもよい。次に、ジクロロエーテル誘導体(1C)をアミン(HNRxRy)と反応させてジアミン(1D)を形成することができる。方法1の合成スキームにおいて、単一アミン反応物を用いてジアミン(1D)中にアミン基−NR1aR2aおよび−NR3aR4aを形成することができることを示すために、前記アミンはHNRxRyと総称的に表現される。そのようなものとして、RxはR1aおよびR4aと同一であり、RyはR2aおよびR3aと同じである。その上、R1aおよびR4aが水素である場合、またはR2aおよびR3aが水素である場合には、前記化合物は、所望によりN−メチル化を経てN−メチル誘導体(1E)を形成することができる。化合物1Dおよび1Eは、双方とも本発明の実施形態に従う化合物であり得る。
【0030】
方法2−光延反応または光延反応およびN−メチル化
【化16】
(光延)(R1aおよびR4aがHである場合、またはR2aおよび/もしくはR3aがHである場合、N−メチル化)
上に示した方法2の反応スキームにおいて、
n、i、およびjは同一であって、かつ0、1、2、3または4であり、
R1aおよびR4aは双方ともRxと同一であり、
R2aおよびR3aは双方ともRyと同一であり、
R1bおよびR4bは同一であり、
R2bおよびR3bは同一であり、
R1aおよびR4aが双方ともHである場合には、R1bおよびR4bは双方ともメチルであり、
R2aおよびR3aが双方ともHである場合には、R2bおよびR3bは双方ともメチルである。
【0031】
本発明の実施形態による対称の化合物を合成するもう1つの方法では、光延反応によって、2−(4−ヒドロキシフェニル)ベンゾオキサゾール−6−オール(2A)をヒドロキシアルキルアミン(2B)と反応させてジアミン(2C)を作出することができる。また、化合物2Aの代わりに2−(3−ヒドロキシフェニル)ベンゾオキサゾール−6−オールおよび2−(2−ヒドロキシフェニル)ベンゾオキサゾール−6−オールを用いてもよい。その上、R1aおよびR4aが水素である場合、またはR2aおよびR3aが水素である場合には、前記化合物は、所望によりN−メチル化を経てN−メチル誘導体(2D)を形成することができる。化合物2Cおよび2Dは、双方とも本発明の実施形態による化合物であり得る。
【0032】
方法3−不斉側鎖類似体について
【化17】
(方法1または方法2)(方法1または方法2)
方法3の反応スキームにおいて、
R1およびR4は同一であって、かつHおよびCH3からなる群から選択され、
R2はCH3であり、R3は(CH2)2N(CHs)2および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択される。
【0033】
本発明のいくつかの実施形態による不斉化合物は方法3によって合成することができる。かかる場合では、4−アミノベンゼン−1,3−ジオール(3A)を、ヘキサフルオロリン酸ベンゾトリアゾール−1−イル−オキシ−トリス−(ジメチルアミノ)−ホスホニウム(PyBop)の存在下、保護されたヒドロキシル基を有する安息香酸、例えば、4−(ベンジルオキシ)安息香酸(3B)、3−(ベンジルオキシ)安息香酸または2−(ベンジルオキシ)安息香酸などと反応させてベンズアミド誘導体(3C)を形成することができる。次いで、そのベンズアミドを環化してベンゾオキサゾール環(3D)を形成することができる。この時点において、上に記載される、方法1または方法2のいずれかを用いてヒドロキシ官能性ベンゾオキサゾール(3D)をアミン(3E)へと変換してよい。方法1および2に関し、R1はR1aまたはR1bであってよく、R2はR2aまたはR2bであってよい。その後、例えば、パラジウムを用いる水素化によって、ベンジルエーテル基を脱保護してヒドロキシ官能性アミン(3F)を形成することができる。その後、方法1または方法2のいずれかを用いてヒドロキシル官能性アミンをジアミン(3G)へと変えてよい。方法1および2に関し、R4はR4aまたはR4bであってよく、R3はR3aまたはR3bであってよい。
【0034】
(D.医薬組成物)
一実施形態では、本発明は、化合物を含む医薬組成物である。もう1つの実施形態では、前記医薬組成物は医薬上許容される担体をさらに含む。本明細書において用いられる用語「医薬上許容される担体」とは、被験体への治療薬の送達のためのビヒクルとして用いられる、それ自体は治療薬でない、任意の物質をさす。
【0035】
本発明の組成物は、経口投与、非経口投与、吸入スプレー投与、局所投与、直腸投与、鼻腔投与、舌下投与、口内投与、膣投与または植え込み型リザーバー投与などに用いる処方物に適し得る。好ましくは、前記組成物は、経口的に、局所に、腹膜内にまたは静脈内に投与される。本発明の組成物の滅菌注射剤型は水性懸濁液であっても油性懸濁液であってもよい。これらの懸濁液は、適した分散剤または湿潤剤および懸濁化剤を用いて当分野で公知の技術によって処方され得る。滅菌注射用調製物は、毒性のない非経口的に許容される希釈剤または溶媒中の滅菌注射用溶液または懸濁液(例えば、1,3−ブタンジオールの溶液として)であってよい。使用することのできる許容されるビヒクルおよび溶媒には、水、リンゲル液および等張塩化ナトリウム溶液がある。その上、滅菌固定油が溶媒または懸濁媒として通常使用される。
【0036】
医薬上許容される油は、本発明の組成物中の溶媒または懸濁媒として使用され得る。脂肪酸、例えば、オレイン酸およびそのグリセリド誘導体などは、天然の医薬上許容される油、例えば、オリーブ油またはヒマシ油、特にそれらのポリオキシエチル化型などと同様に、注射用処方物中に含められることが適している。本発明の油含有組成物はまた、長鎖アルコール希釈剤または分散剤、例えば、カルボキシメチルセルロースまたはエマルジョンおよび懸濁液を含む医薬上許容される投与形の処方物において一般的に用いられる類似の分散剤なども含有することができる。前記組成物は、界面活性剤(Tween(登録商標)またはSpan(登録商標)を含む非イオン性洗剤など)その他の乳化剤、またはバイオアベイラビリティー向上剤をさらに含むことが適している。
【0037】
本発明の組成物は、限定されるものではないが、カプセル剤、錠剤、懸濁液または溶液を含む経口的に許容される投与形の形であってよい。経口投与形は、少なくとも1種類の賦形剤を含むことができる。本経口処方物に用いられる賦形剤には、希釈剤、不快な味または臭いを遮断するまたは中和するために加えられる物質、香味料、色素、香料、および前記組成物の外観を良くするために加えられる物質を含めることができる。本発明の一部の経口投与形は、前記組成物の用量単位を、追加的経口投与に適したカプセル剤または錠剤などの個別物品の形にすることを可能にするまたは容易にする賦形剤、例えば、崩壊剤、結合剤、接着剤、湿潤剤、ポリマー、滑沢剤、または磨砕剤などを含むことが適している。本発明の賦形剤含有錠剤組成物は、溶解型、懸濁型、ナノ粒子型、マイクロ粒子型またはその制御放出型、遅延放出型、プログラム放出型、時限放出型、パルス放出型、徐放出型もしくは長期放出型の組合せで1種類以上の賦形剤を本発明の化合物と混合する段階を含む任意の適した調剤方法によって調製することができる。
【0038】
あるいは、本発明の医薬上許容される組成物は直腸投与用の坐剤の形であってよい。それらの坐剤は、室温では固体であるが直腸温では液体であるために直腸内で溶けて薬物を放出する好適な非刺激性賦形剤と前記薬剤を混合することによって調製することができる。かかる材料には、ココア乳脂、蜜蝋およびポリエチレングリコールが含まれる。
【0039】
本発明の医薬上許容される組成物は、活性成分が1種類以上の担体中に懸濁されているまたは溶解されている局所用溶液、軟膏、またはクリームの形であってよい。本発明の化合物の局所投与に用いる担体としては、限定されるものではないが、鉱油、流動ワセリン、白色ワセリン、プロピレングリコール、ポリオキシエチレン、ポリオキシプロピレン化合物、乳化蝋および水が挙げられる。局所処方物が軟膏またはクリームの形である場合には、適した担体としては、限定されるものではないが、鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステルワックス、セテアリルアルコール、2 オクチルドデカノール、ベンジルアルコールおよび水が挙げられる。
【0040】
医薬上許容される組成物が眼用処方物である場合には、その組成物は、塩化ベンジルアルコニウムなどの防腐剤を含むまたは含まない、等張のpH調整された滅菌水溶液中の微粒子懸濁液であってよく、または等張のpH調整された滅菌生理食塩水中の溶液としてであってもよい。あるいは、眼に用いるために、医薬上許容される組成物は軟膏の形で処方してもよい。
【0041】
本発明の医薬上許容される組成物はまた、鼻腔エアゾールによっても吸入投与経路によっても投与することができる。かかる組成物は、医薬処方技術分野で周知の技術によって調製され、ベンジルアルコールまたは他の適した防腐剤、バイオアベイラビリティーを高めるための吸収促進剤、フッ化炭素、および/または他の通常の可溶化剤または分散剤を使用して、生理食塩水中の溶液として調製することができる。
【0042】
その上、本発明の化合物を含む医薬処方物は非経口処方物の形でもあり得る。
【0043】
ある特定の実施形態では、本発明の医薬上許容される組成物は経口投与用に処方されることが好ましい。
【0044】
(E.使用方法)
もう1つの実施形態では、本発明は、被験体において免疫学的障害を予防または治療する方法であり、その方法は、本発明の化合物の治療上有効な量を被験体に投与することを含む。特定の実施形態では、本発明の方法による、免疫学的障害を予防または治療するために用いられる本発明の化合物は、化合物13、化合物16、化合物22、化合物23、化合物26、化合物27、化合物30、化合物34、および化合物35またはその医薬上許容される塩、からなる群から選択される式Iの化合物である。もう1つの実施形態では、前記化合物は、化合物23、化合物26、および化合物30またはその医薬上許容される塩からなる群から選択される。もう1つの実施形態では、前記化合物は、化合物23またはその医薬上許容される塩である。
【0045】
一実施形態では、本発明の方法を用いて予防または治療される免疫学的障害は、自己免疫疾患である。本明細書において用いられるように、用語「自己免疫疾患」とは、自己成分と非自己成分とを識別する免疫系の障害によって一般的に引き起こされる疾患をさす。本発明の化合物により予防または治療することのできる自己免疫疾患には、限定されるものではないが、狼瘡(SLEなど)、尋常性天疱瘡、重症筋無力症、溶血性貧血、血小板減少性紫斑病、グレーブス病、シェーグレン病、皮膚筋炎、橋本病、多発性筋炎、炎症性腸疾患、多発性硬化症、真性糖尿病、潰瘍性大腸炎、関節リウマチ、強皮症および乾癬が含まれる。本発明の特定の実施形態では、前記自己免疫疾患は狼瘡または多発性硬化症である。一部の実施形態では、前記自己免疫疾患はSLEである。本明細書において用いられるように、狼瘡とは、DNA、RNA、ヒストン、およびヌクレオソームを含む自己細胞成分に対する抗体を特徴とする自己免疫疾患をさす。身体症状には、自己抗体または糸球体での免疫複合体の沈着によって生じる腎臓疾患、皮膚疹、光感受性、関節炎ならびに痙攣および精神病を含む神経学的障害が含まれる。この疾患は主に女性が罹患し、通常若年成人を襲う。直接命にかかわることはまれであるが、この疾患は死亡率の増加と関連し、生活の質に深刻な影響をもたらし、疲労の継続、体重減少、アテローム性動脈硬化の危険性、および発熱を伴う。
【0046】
もう1つの実施形態では、前記免疫学的障害は敗血症である。本明細書において用いられるように、敗血症とは、感染によって引き起こされる全身性炎症応答をさす。本発明の化合物は、敗血症によって生じる可能性がある、重症敗血症および敗血性ショックを治療するために投与することができる。特定の実施形態では、本発明の化合物は、重症敗血症を治療するために投与することができる。他の実施形態では、本発明の化合物は、敗血性ショックを治療するために用いることができる。
【0047】
本発明のさらなる実施形態によれば、本明細書に記載される化合物は、疾患の症状が強度および/または種類において増加するように疾患が再発した場合に、比較的短期間に、狼瘡、多発性硬化症および/または関節リウマチの治療のために用いることができる。一部の実施形態では、本明細書に記載される化合物は、長期治療、例えば、数日、数ヶ月または数年続く治療として用いることができる。
【0048】
本発明の一部の実施形態では、本明細書に記載される化合物は、狼瘡の治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。狼瘡の治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、ステロイド(例えば、グルココルチコイドおよびコルチコステロイド)、免疫抑制薬、非ステロイド系抗炎症薬(NSAID)、抗体療法、インターフェロン、細胞毒性薬および抗マラリア薬。一部の実施形態では、狼瘡の治療に有用な追加の薬剤は以下の少なくとも1種類である、デヒドロエピアンドロステロン(DHEA)、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、クロベタゾール、ハロベタゾール、トリアムシノロン、ベタメタゾン、フルオシノロン、フルオシノニド、レフルノミド、アザチオプリン、メトトレキサート、ミトキサントロン、クラドリビン、クロラムブシル、シクロホスファミド、シクロスポリン、ミコフェノレート(ミコフェノール酸、ミコフェノール酸モフェチルおよびミコフェノール酸ナトリウムを含む)、セレコキシブ、ジクロフェナク、フェノプロフェン、フルルビプロフェン、イブプロフェン、ケトプロフェン、フェンブフェン、インドメタシン、メクロファメート(meclofamate)、メロキシカム、ナブメトン、ナプロキセン、オキサプロジン、ロフェコキシブ、サリチレート、アセチルサリチル酸、アセトアミノフェン、スリンダク、トルメチン、リツキシマブ、ベリムマブ、インフリキシマブ、アダリムマブ、エプラツズマブ、ナタリズマブ、α−インターフェロンならびに/またはキニーネおよびヒドロキシクロロキン、クロロキン、キニジンおよびキナクリンなどのキニーネ誘導体。
【0049】
一部の実施形態では、本明細書に記載される化合物は、多発性硬化症の治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。多発性硬化症の治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、ステロイド(例えば、グルココルチコイドおよびコルチコステロイド)、免疫抑制薬、抗体療法およびインターフェロン。一部の実施形態では、多発性硬化症の治療に有用な追加の薬剤は以下の少なくとも1種類である、デヒドロエピアンドロステロン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、クロベタゾール、ハロベタゾール、トリアムシノロン、ベタメタゾン、フルオシノロン、フルオシノニド、アザチオプリン、メトトレキサート、ノバントロン、コパクソン、クラドリビン、クロラムブシル、シクロホスファミド、シクロスポリン、ミコフェノレート(ミコフェノール酸、ミコフェノール酸モフェチルおよびミコフェノール酸ナトリウムを含む)、リツキシマブ、ベリムマブ、エプラツズマブ、ナタリズマブおよび/またはβ−インターフェロン。
【0050】
さらに他の実施形態では、本明細書に記載される化合物は、関節リウマチの治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。関節リウマチの治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、シクロオキシゲナーゼ−2(COX−2)阻害薬、NSAID、疾患修飾性抗リウマチ薬(DMARD)、ステロイド、免疫抑制薬および抗体療法。一部の実施形態では、関節リウマチの治療に有用な追加の薬剤は以下の少なくとも1種類である、セレコキシブ、ジクロフェナク、フェノプロフェン、フルルビプロフェン、イブプロフェン、ケトプロフェン、フェンブフェン、インドメタシン、メクロファメート、メロキシカム、ナブメトン、ナプロキセン、オキサプロジン、ロフェコキシブ、アセチルサリチル酸、アセトアミノフェン、スリンダク、トルメチン、デヒドロエピアンドロステロン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、クロベタゾール、ハロベタゾール、トリアムシノロン、ベタメタゾン、フルオシノロン、フルオシノニド、レフルノミド、アザチオプリン、メトトレキサート、ミトキサントロン、クラドリビン、クロラムブシル、シクロホスファミド、シクロスポリン、ミコフェノール酸モフェチル、リツキシマブ、ベリムマブ、インフリキシマブ、アダリムマブ、エプラツズマブ、ナタリズマブ、アナキンラ、アバタセプト、アウラノフィン、オーロチオグルコース、オーロチオマレート、スルファサラジン、ミノサイクリンならびに/またはキニーネおよびヒドロキシクロロキン、クロロキン、キニジンおよびキナクリンなどのキニーネ誘導体。
【0051】
一部の実施形態では、本明細書に記載される化合物は、敗血症の治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。敗血症の治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、抗生物質、アミノグリコシド、コロイド溶液または晶質液(すなわち、輸液療法)、昇圧薬、変力薬、ステロイドおよび抗凝固薬。一部の実施形態では、敗血症の治療に有用な追加の薬剤は以下の少なくとも1種類である、アルブミン、デンプン、ノルエピネフリン、ドーパミン、バソプレシン、ドブタミン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、および、限定されるものではないが、ヒト組換え活性化プロテインC(rhAPC)を含む活性化プロテインC。
【0052】
さらなる実施形態では、前記化合物は、本明細書に記載される障害を治療するために併用する他の治療の用量を減らすために用いることができる(例えば、ステロイド節約型レジメンにおいて)。
【0053】
一部の実施形態では、本明細書に記載される化合物は、狼瘡、多発性硬化症、関節リウマチおよび/または敗血症の治療に有用な追加の薬剤の投与前に、その投与と同時にまたはその投与の後に投与される。
【0054】
追加の薬剤の投与「前の」本明細書に記載される化合物の投与とは、追加の薬剤による初期治療の前に、すなわち、狼瘡、多発性硬化症、関節リウマチおよび/または敗血症の危険性があるまたは患っている被験体集団に本明細書に記載される化合物と追加の薬剤とを投与することを含む治療プロトコール中の追加薬剤の投与の前に、本明細書に記載される化合物を被験体に投与することをさす。
【0055】
同時投与では、本明細書に記載される化合物と追加の薬剤は、同一時点にまたは一方の直後に投与される。一般的には、本明細書に記載される化合物と追加の薬剤は、観察される結果が、それらが同一時点において投与されたときに得られる結果と比較的変わらない十分近い時点において投与される。
【0056】
化合物は、経口的に(口腔を介する投与を含み、経口経管栄養チューブを介する投与をさらに含む)、非経口的に、吸入スプレーにより、局所に、経皮的に、直腸に、鼻腔に(経鼻経管栄養チューブを含む)、舌下に、口内に、膣にまたは埋め込み型リザーバーを介してなど、任意の適した経路によって被験体に投与してよい。本明細書において用いられる用語「非経口の」には、皮下、静脈内、筋肉内、関節内、滑液嚢内、胸骨内、くも膜下腔内、肝内、病巣内および頭蓋内の注射または注入技法が含まれる。前記組成物は、経口的に、非経口的に、経皮的にまたは吸入スプレーにより投与されることが好ましい。
【0057】
化合物は有効な量で被験体に投与される。有効な量は、一般的に1日あたり体重1kgあたり0.01mg〜500mgである。一部の実施形態では、医薬上許容される組成物は、1日あたり体重1kgあたり前記化合物0.01mg〜100mgの間の投薬量を、これらの組成物を受ける患者に投与することができるように処方され得る。ある特定の実施形態では、本発明の組成物は、0.01mg〜70mgの間の投薬量を与えるように処方される。その他の実施形態では、本発明の組成物は、0.1mg〜25mgの間または5mg〜60mgの間の投薬量を与えるように処方される。
【0058】
一部の実施形態では、有効な用量は、約5〜250mg/kgの間、約10〜200mg/kgの間、または約20〜120mg/kgの間である。一部の実施形態では、有効な用量には、5mg/kg、10mg/kg、20mg/kg、25mg/kg、40mg/kg、50mg/kg、60mg/kg、75mg/kg、100mg/kg、120mg/kgおよび150mg/kgが含まれる。投与形は、例えば、錠剤またはカプセル剤の形であってよく、有効な用量は1以上の錠剤、カプセル剤または同類のもので与えることができ、1日1回または1日を通して、例えば、4時間、8時間または12時間の間隔で与えることができる。例えば、錠剤またはカプセル剤は、例えば、10mg、25mg、50mg、75mg、100mg、150mg、200mgの化合物を含有することができる。あらゆる用量が容易にかつ便宜に分配され得るように、液体処方物も調製されてよい。
【0059】
一部の実施形態では、単一投与形で組成物を作製するために賦形剤材料と組み合わせることのできる本発明の化合物の量は、治療する宿主、および特定の投与経路によって変動することになる。
【0060】
また、特定患者についての具体的な投薬量および治療計画が、使用される具体的な化合物の活性、年齢、体重、全般的健康、性別、食生活、投与時期、排出速度、薬物組合せ、および治療する医師の判断および治療される特定の疾患の重篤度を含む、様々な因子によって決まることは当然理解される。組成物中の本発明の化合物の量もまた、組成物中の特定の化合物によって決まる。
【0061】
本明細書に記載される本発明がさらに十分に理解されるように、以下の実施例を示す。これらの実施例は例示を目的としたものでしかなく、本発明を限定するものと解釈してはならないことは当然理解される。比較化合物が下のいくつかの実施例で使用される。比較化合物の式は以下の通りである。
【化18】
【0062】
本発明の例となる化合物は、下に記載されるスキームおよび実験計画に従って調製された。
【実施例】
【0063】
(実施例1)
(化合物40の合成)
【化19】
化合物40。上に表されるように、1−ブロモ−3−クロロプロパン(7.3mL、0.074モル)を、化合物39(5.62g、0.0247モル)および炭酸カリウム(10g、0.074モル)のDMSO(50mL)中の攪拌混合物に室温にて添加し、得られる混合物を4時間攪拌し、薄層クロマトグラフィー(TLC)により反応が完了したことが示された。次に、混合物を水(200mL)に注入し、150mL酢酸エチル(EtOAc)で3回抽出し、Na2SO4で乾燥させ、濾過し、濃縮させた。残渣の真空クロマトグラフィー(10%〜30%EtOAc/ヘキサン)により、白色の固体生成物である化合物40を得た(8.10g、86%)。ヘキサン/EtOAc中で結晶化させて白色の純粋な固体生成物(6.40g)を得、母液は少量の不純物(1.60g)を含んだ。1H NMR(400MHz,CD3OD)δ 8.11(d,J=9.1Hz,2H)、7.55(d,J=8.8Hz,1H)、7.27(d,J=2.3Hz,1H)、7.11(d,J=9.1Hz,2H)、7.00(dd,J=2.3,8.8Hz,1H)、4.22(t,J=5.9Hz,2H)、4.19(t,J=6.15,2H)、3.78(tt,J=6.4Hz,4H)、2.30(dt,J=1.76,1.76Hz,4H)。出発化合物の化合物39の調製については、J.Med.Chem.2004,47,5021−5040を参照されたい。
【0064】
同じ反応を化合物39の異性体を用いて行った。異性体化合物47および化合物48を、J.Med.Chem.2004,47,5021−5040に記載される合成手順によって調製した。
【化20】
【0065】
(実施例2)
(化合物49の合成)
【化21】
化合物49。実施例1の1−ブロモ−3−クロロプロパンを1−ブロモ−2−クロロエタンに置き換えて、化合物49を得た。
【0066】
(実施例3)
(化合物50の合成)
【化22】
(化合物39)(化合物50)
化合物50。実施例1の1−ブロモ−3−クロロプロパンを1−ブロモ−5−クロロペンタンで置き換えて、化合物50を得た。
【0067】
(実施例4)
(化合物23の合成)
【化23】
化合物23。上のスキームを参照して、実施例1に記載される通り製造された化合物40(8.40g、0.0221モル)とピロリジン(7.4mL、0.088モル)のエタノール(50mL)中の混合物を、封管下、100℃にて12時間加熱した。TLC(メタノール(Et3N/MeOH)中20%トリエチルアミン)により、単一の新しいスポットが示され、質量分析により、450(M+H)の単一の所望のピークが示された。混合物を室温まで冷却し、濃縮させ、トルエンとの共沸濃縮により過剰なピロリジンを除去した(50mL中3回)。残渣をエタノール(EtOH)に溶解させ、濾過し、濃縮させた。残留固体をMeOH/EtOAc中で結晶化させて、純粋な生成物である化合物23(7.88g、68%)を得た。1H NMR(400MHz,CD3OD)δ 8.12(d,J=8.9Hz,2H)、7.57(d,J=8.8Hz,1H)、7.28(d,J=2.2Hz,1H)、7.12(d,J=8.9Hz,2H)、7.01(dd,J=2.2.8.8Hz,1H)、4.18(t,J=5.86Hz,2H)、4.15(t,J=6.15Hz,2H)、3.07−2.94(m,12H)、2.15(m,4H)、1.96(m,8H):MS(ES+)m/z 450.4。
【0068】
(実施例5)
(化合物30の合成)
【化24】
化合物30。上のスキームを参照して、実施例1に記載される通り製造された化合物40(13.4g、0.0352モル)とピペリジン(14mL、0.14モル)のエタノール(100mL、2モル)中の混合物を、封管下、100℃にて18時間加熱した。TLC(10%Et3N/MeOH)により、単一のスポットが示され、MSにより、478(M+H)の1つの単一の所望のピークが示された。混合物を、濃縮させ、トルエンとの共沸濃縮により過剰なピペリジンを除去した(50mL中3回)。残渣を、MeOHを添加した水に溶解させ、次いで1NのNaOHを、適度に攪拌しながら、さらなる沈殿が形成されなくなるまで添加した。混合物を濃縮して残留MeOHを除去し、濾過し、水で洗浄した。収集した遊離形態の固体(真空乾燥後16.55g、34.7ミリモル)を、暖かいMeOH(100mL)に溶解させ、70mLの1NのHCl(70ミリモル)で処理して二塩酸塩を生じ、混合物を濃縮乾固した。残留固体をMeOH/EtOAc中で結晶化させて、純粋な生成物である化合物30(16.0g、82%)を得た。1H NMR(400MHz,CD3OD)δ 8.13(d,J=8.8Hz,2H)、7.58(d,J=8.8Hz,1H)、7.29(d,J=2.3Hz,1H)、7.14(d,J=8.8Hz,2H)、7.02(dd,J=2.3,8.8Hz,1H)、4.21(t,J=5.57Hz,2H)、4.19(t,J=5.57Hz,2H)、3.59(m,4H)、3.34(m,4)、2.99(m,4h)、2.28(tt,J=2.3、5.6Hz,4H)、2.00−1.50(m,12H):MS(ES+)m/z 478.5。
【0069】
(実施例6)
(化合物51の合成)
【化25】
化合物51。化合物51などの類似体は、次の手順を用いて半自動化合成(SAS)を用いて調製することができる。化合物40(27.0mg、0.071ミリモル)とシクロペンチルアミン(98.6mg、1.16ミリモル)のN−メチルピロリジノン(0.5mL)中の混合物を、180℃にて300秒間マイクロ波により加熱した。粗反応混合物を、酸性条件下、LC/MSにより精製した。回収した、目的生成物を含有する画分を、分析的LC/MSにより純度についてチェックした。この化合物の全体的な純度は、>95%であり、m/z=478.5であった。純粋な生成物を含有する2種類の画分を合し、蒸発させて生成物である化合物51(22.5mg、66.4%)を得た。
【0070】
(実施例7)
(例となる化合物)
下の表1中の以下の類似体が調製された。これらの化合物は、表中に特定される対応するジクロロアルカンおよび側鎖アミンを用いて、化合物23(実施例4)および/または化合物30(実施例5)の調製のための手順に従って調製することができる。これらの化合物は、化合物51(実施例6)の調製のための手順に従っても調製することができる。
【0071】
表1.例となる化合物
【表2A】
【表2B】
【表2C】
【表2D】
【表2E】
【表2F】
【表2G】
【0072】
(上の表の化合物選択のためのNMRデータ)
化合物1:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.58(d,J=8.8Hz,1H)、7.33(d,J=2.4Hz,1H)、7.16(d,J=8.8Hz,2H)、7.06(dd,J=2.2,8.8Hz,1H)、4.28(m,4H)、3.10(m,4H)、2.68(s,6H)、2.57(s,6H)。
【0073】
化合物2:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.59(d,J=8.8Hz,1H)、7.30(d,J=2.4Hz,1H)、7.15(d,J=8.8Hz,2H)、7.04(dd,J=2.4,8.8Hz,1H)、4.21(m,4H)、3.34(m,4H)、2.93(s,6H)、2.92(s,6H)、2.68(s,6H)、2.26(m,4H)。
【0074】
化合物3:1H NMR(400MHz,CD3OD)δ 8.15(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.24(d,J=2.4Hz,1H)、7.10(d,J=8.8Hz,2H)、6.98(dd,J=2.4,8.8Hz,1H)、4.10(m,4H)、3.25(m,4H)、1.90(m,4H)、1.78(m,4H)、1.59(m,4H)。
【0075】
化合物4:1H NMR(400MHz,CD3OD)δ 8.18(dd,J=1.6,8.0Hz,1H)、7.72(d,J=9.0Hz,1H)、7.60(m,1H)、7.37(d,J=2.4Hz,1H)、7.30(d,J=8.4Hz,1H)、7.21(dd,J=7.2,8.0Hz,1H)、7.09(dd,J=2.6,9.0Hz,1H)4.39(t,J=5.6Hz,2H)、4.20(t,J=5.8,2H)、3.22(dd,J=7.6,8.4Hz,2H)、3.05(s,6H)、2.82(s,6H)、2.38(m,2H)、2.23(m,2H)。
【0076】
化合物5:1H NMR(400MHz,CD3OD)δ 7.79(d,J=8.0Hz,1H)、7.75(dd,J=1.6,2.4Hz,1H)、7.63(d,J=8.8Hz,1H)、7.50(dd,J=8.0,8.0Hz,1H)、7.32(d,J=2.4Hz,1H)、7.18(dd,J=2.6,8.6Hz,1H)、7.06(dd,J=2.4,8.8Hz,1H)4.21(m,4H)、3.32(m,4H)、2.91(s,6H)、2.90(s,6H)、2.25(m,4H)。
【0077】
化合物20:1H NMR(400MHz,CD3OD)δ 8.10(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.25(d,J=2.4Hz,1H)、7.10(d,J=8.8Hz,2H)、6.99(dd,J=2.4、8.8Hz,1H)、4.12(m,4H)、3.04(bd,J=10.8Hz,4H)、2.66(bs,8H)、2.57(m,4H)、2.15−1.90(m,14H)、1.82(m,8H)、1.60(m,4H)。
【0078】
化合物27:1H NMR(400MHz,CD3OD)δ 8.13(d,J=9.2Hz,2H)、7.58(d,J=8.8Hz,1H)、7.29(d,J=2.4Hz,1H)、7.13(d,J=9.2Hz,2H)、7.03(dd,J=2.2,8.6Hz,1H)、4.20(m,4H)、3.60−3.10(m,12H)、2.98(s,6H)、2.93(s,12H)、2.25(m,8H)。
【0079】
化合物35:1H NMR(400MHz,CD3OD)δ 8.10(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.25(d,J=2.4Hz,1H)、7.10(d,J=8.8Hz,2H)、6.99(dd,J=2.4,8.8Hz,1H)、4.10(m,4H)、3.60(m,8H)、2.65(m,8H)、2.35(m,18H)、2.02(m,4H)。
【0080】
化合物36:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.30(d、J=2.4Hz,1H)、7.13(d,J=8.8Hz,2H)、7.03(dd,J=2.4,8.8Hz,1H)、4.22(m,4H)、3.92(bs,4H)、3.87(m,4H)、3.60−3.20(m,12H)、3.00(s,6H)、2.92(s,12H)、2.30(m,4H)。
【0081】
化合物37:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.59(d,J=8.8Hz,1H)、7.31(d,J=2.4Hz,1H)、7.15(d,J=8.8Hz,2H)、7.04(dd,J=2.4,8.8Hz,1H)、4.23(m,4H)、3.58(m,8H)、3.55−3.35(m,6H)、m 3.23(m,6H)、2.96(s,6H)、2.90(m,12H)、2.30(m,4H)、2.05(m,8H)。
【0082】
化合物38:1H NMR(400MHz,CD3OD)δ 8.13(d,J=8.8Hz,2H)、7.58(d,J=8.8Hz,1H)、7.29(d,J=2.0Hz,1H)、7.13(d,J=8.8Hz,2H)、7.02(dd,J=2.4,8.8Hz,1H)、4.20(m,4H)、3.30−3.50(m,4H)、3.18(m,6H)、2.85−2.96(m,18H)、2.28(m,4H)、1.82(m,6H)。
【0083】
(実施例8)
(化合物34の合成)
【化26】
化合物34。上のスキームを参照して、4−アミノレソルシノールヒドロクロリド(5.3g、33ミリモル)、4−ベンジルオキシ安息香酸(5.0g、22ミリモル)、1−ヒドロキシベンゾトリアゾール(5.9g、44ミリモル)、ベンゾトリアゾール−1−イル−オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(23g、44ミリモル)およびN,N−ジイソプロピルエチルアミン(19mL,110ミリモル)のジメチルホルムアミド(DMF)(50mL)中の混合物を、0℃で室温まで一晩攪拌した。濃縮によってDMFを除去した後、反応混合物をシリカゲルクロマトグラフィー(20%〜80%EtOAc/ヘキサン、EtOAcおよび30%MeOH/EtOAc)により精製して、所望のアミド化合物41(または/およびエステル、7.3g、95%)を得た。
【0084】
化合物41(7.3g、22ミリモル)の酢酸(90mL)中溶液を、120℃にて3時間加熱した。反応混合物を室温まで冷却し、濃縮した。残渣をシリカゲルクロマトグラフィー(10%〜30%EtOAc/ヘキサン)により精製して、目的生成物である化合物42(2.0g、29%)を淡黄色の固体として得た。1H NMR(400MHz,CD3OD)δ 8.09(d,J=9.1Hz,2H)、7.47−7.4(m,6H)、7.16(d,J=9.1Hz,2H)、7.02(d,J=2.4Hz,1H)、7.02(dd,J=2.4,8.5Hz,1H)、5.18(s,2H):MS(ES−)m/z 316.4。
【0085】
化合物42(1.77g、5.58ミリモル)、1−ブロモ−3−クロロプロパン(1.6mL、17ミリモル)および炭酸カリウム(2.7g、20ミリモル)の混合物を10mLのDMSO中で室温にて2.5時間、TLCにより反応が完了するまで攪拌した。反応を水の添加によってクエンチし、次いでEtOAcで5、3回(five three times)抽出した。合した有機相をNa2SO4で乾燥させ、濾過し、濃縮させた。残渣を酢酸エチル−へキサンから結晶化させて、化合物43を淡色の固体(1.93g、88%)として得た。
【0086】
化合物43(1.10g、2.8ミリモル)の40mLのエタノール中の懸濁液に、5mLのジメチルアミンを−78℃で添加した。混合物を封管に入れて封をし、100℃にて10時間加熱した。混合物を濃縮して化合物44(1.1g、98%)を得た。生成物を次の段階の反応に進めた。
【0087】
化合物44(1.1g、2.7ミリモル)およびパラジウムヒドロキシド(7mg)の32mLのメタノールおよび5mLのジクロロメタン中の混合物を、水素バルーン下、室温にて一晩攪拌した。混合物をセライトを通じて濾過し、濾液を濃縮して粗生成物である化合物45を得た。理論収率を0.85gと仮定し、生成物をさらなる精製をせずに次の段階に直接進めた。
【0088】
化合物45(200mg、0.64ミリモル)、1−ブロモ−3−クロロプロパン(0.32mL、3.2ミリモル)および炭酸カリウム(470mg、3.4ミリモル)の混合物を4mLのDMSO中で室温にて2時間、TLCにより反応が完了するまで攪拌した。反応を水の添加によってクエンチし、次いでEtOAcで4回抽出した。合した有機相をNa2SO4で乾燥させ、濾過し、濃縮させた。残渣を分取TLC(5%MeOH/THF中、4%Et3N)により精製して、化合物46(170mg、68%)を得た。
【0089】
化合物46(30mg、0.08ミリモル)、N,N,N’−トリメチル−1,3−プロパン−ジアミン(50mg、0.43ミリモル)の混合物を2mLのエタノール中で、封管下、100℃にて10時間加熱した。混合物を濃縮させ、残渣をHPLCにより精製して、8mgの化合物34を白色固体(8mg、20%)として得た。1H NMR(400MHz,CD3OD)δ 8.10(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz 1H)、7.25(d,J=2.3Hz,1H)、7.10(d,J=8.8Hz,2H)、7.00(dd,J=2.3,8.8Hz,1H)、4.14(t,J=5.9Hz,2H)、4.10(t,J=6.2Hz,2H)、2.69(m,4H)、2.56(m,4H)、2.39(s,12H)、2.36(s,3H)、2.04(m,4H)、1.77(m,2H)、MS(ES−)m/z+H 469.55。
【0090】
(実施例9)
(インビトロ生物活性)
(A.一次アッセイ)
一次アッセイは、ヒト胚腎臓上皮(HEK)細胞にTLR9をトランスフェクトすることを伴う。より具体的には、pGL3−エンハンサーベクター(Promega Corp.Madison,WI)にクローニングされた、E−セレクチン−1プロモーターに含められた3つのNF−kB結合部位の制御下、HEK293線維芽細胞(ATCC番号 CRP−1573、American Type Culture Collection,Manassas,VA)を、ヒトTLR9をコードするpcDNA3.1D/V5−His−TOPO(登録商標)プラスミド(Invitrogen,Carlsbad,CA)およびホタルルシフェラーゼを用いてトランスフェクトした(Taqポリメラーゼ増幅PCR産物として直接挿入した)。このアッセイで試験された化合物を下の表1に記載する。各々の化合物は、30分細胞に添加された後、ヌクレオチド配列5’−TCG TCG TTT TGT CGT TTT GTC GTT−3’(配列番号1)とともに、合成ホスホロチオエートオリゴヌクレオチドであるオリゴCpG2006(Hartmann et al.,J Immunol 164:1617−24(2000))で刺激した。細胞を、37℃にて一晩インキュベートした。ルシフェラーゼ基質Steady−Glo(登録商標)(Promega)をウェルに添加し、発光を測定してTLR9に駆動される(TLR9−driven)遺伝子活性化の程度を決定した。発光データを用いて各々の試料についてのIC50を算出した。IC50は、化合物の不在下で、言い換えれば完全な刺激を伴って観察される発光の50%まで発光を抑制する化合物の濃度として定義される。このアッセイの結果を下の表2に示す。比較化合物のIC50は、0.16マイクロモルであることが見出された。化合物1のIC50は、0.06マイクロモルであることが見出され、残りの化合物のIC50は、0.04マイクロモル未満であった。
【0091】
(B.二次アッセイ)
(1)HEK/TLR7活性化アッセイ:一次アッセイと同じ化合物を、以下の違いを伴って同じように試験した。HEK細胞株をトランスフェクトするために使用したプラスミドは、TLR9の代わりにヒトTLR7をコードし、オリゴCpG2006の代わりにR848を用いて細胞を刺激した。このアッセイの結果も表2に記載する。試験した各々の化合物について、上のHEK/TLR9活性化アッセイから得られるIC50は、HEK/TLR7アッセイから得られるIC50よりも有意に低く、TLR7に対するよりもTLR9に対する特異性が大きいことが示される。
【0092】
(2)脾細胞CpG1668アッセイ:一次アッセイで試験した一部の化合物は、ヌクレオチド配列5’−TCC ATG ACG TTC CTG ATG CT−3’(配列番号2)とともに、合成ホスホロチオエートオリゴヌクレオチドであるオリゴCpG1668(Krieg et al.Nature.374:546−9(1995))による刺激に応答するマウス脾臓IL−6産生の抑制についてもアッセイした。各々の化合物を分離された脾細胞に添加してから、刺激性オリゴヌクレオチドを添加した。細胞を培養中で72時間刺激し、IL−6レベルのELISA分析用に上清を取り出した。ここで、IC50は、化合物の不在下で、言い換えれば完全な刺激を伴って観察されるサイトカイン産生の50%までサイトカイン産生を抑制する化合物の濃度として定義される。比較化合物を除いて、試験した全ての化合物は、1μMより低いIC50でマウス脾細胞の刺激を阻害した。このアッセイの結果を下の表2に示す。
【0093】
(3)脾臓CT Gloアッセイ:化合物が生細胞への有害作用を有するかどうかを決定するため、分離されたマウス脾細胞を一部の化合物とともに72時間インキュベートした。インキュベーションの終わりにCellTiter−Glo(商標)試薬(Promega Corporation.Madison.WI)を添加し、製造業者の指図書によって発光を読み取った。この試薬は、ATPを細胞代謝の指標として測定する。死細胞は、シグナルを生じないものである。IC50は、化合物の不在下で観察される生存力の50%まで生存力を抑制する化合物の濃度として定義される。生存力に測定可能な程度に影響を及ぼした少数の化合物は、TLR9刺激に応答するサイトカイン放出に影響を及ぼす濃度よりもはるかに高い濃度で影響を及ぼした。結果を表2に示す。これらの結果は、TLR9抑制が細胞の死または脆弱性に起因するものでないことを示す。
【0094】
(4)PBMC CpG2216アッセイ:上記のように試験した化合物の一部は、ヌクレオチド配列5’−ggG GGA CGA TCG TCg ggg gG−3’(小文字はホスホロチオエート結合を示す、その他は全てホスホジエステル)(配列番号3)とともに、合成オリゴホスホロチオエート/ホスホジエステルオリゴヌクレオチドであるオリゴCpG2216(Vollmer et al.Eur J Immunol.34:251−62(2004))による刺激に対するヒト末梢血単核細胞(PBMC)α−インターフェロン応答の抑制についてもアッセイした。各々の化合物をFicoll−Paque(商標)(GE Healthcare,UK)に添加して、健常なボランティアドナーから単核細胞を分離した後、刺激性オリゴヌクレオチドを添加した。細胞を培養中で72時間刺激し、α−インターフェロンのELISA分析用に上清を取り出し、ELISA分析をVeriKine(商標)ヒトIFN−αELISAキット(Pestka Biomedical Laboratories,NJ)を用いて行った。このアッセイで試験した全ての化合物は、1μMより低いIC50でヒトPBMCの刺激を阻害した。
【0095】
下の表2は、試験した化合物、および上に記載され考察されるアッセイの各々から得られる結果を示す。
【0096】
【表3A】
【表3B】
【0097】
(実施例10)
(インビボ生物活性:短期CpG刺激)
短期CpG刺激アッセイ:上の実施例9に記載されるHEK/TLR9および脾細胞CpG1668アッセイにおいて効力を有すると特定された化合物を、インビボでの短期CpG刺激アッセイにおいて試験した。マウスに水中の化合物を経口投与し、90分以内に、30μgのオリゴCpG1668の皮下(s.c.)注射を投与した。2時間後、血清を採取し、ELISA分析を用いてIL−6についてアッセイした。このアッセイの結果を下の表3に示す。異なる日付に反復されたアッセイから得られる結果は、コンマで隔てられる複数の数字として示される。
【0098】
【表4】
【0099】
表3は、20mg/kgの化合物23、化合物30、または化合物26の経口投与の1.5時間後に少なくとも69%のIL−6の抑制が観察されたことを示す。少なくとも75%のIL−6の抑制が化合物23および化合物30の経口投与後に観察された。上記のインビトロアッセイで高レベルの抑制を提示したにもかかわらず、試験した2種類の化合物、化合物14および化合物20の投与後には抑制は観察されなかった。残りの化合物は様々なレベルのIL−6抑制を提示した。
【0100】
(実施例11)
(インビボ生物活性:リンパ節応答)
上の実施例10に記載されるアッセイされた2種類の化合物、化合物23および化合物30は、特に高レベルの抑制を示した。マウスにそれらの2種類の化合物の1種類またはヒドロキシクロロキン(「HCQ」)を、毎日20mg/kgかまたは60mg/kgで2週間経口投与した。インビボ投薬の後、リンパ節を取り出してインビトロで1μg/mlのCpG1668を用いて72時間刺激し、その後上清を取り出してELISAによりIL−6についてアッセイした。このアッセイの結果を図1に示す。投薬のレベルは図中に表示される各々の化合物名の後に示される。
【0101】
図1の結果は、化合物2と化合物23の両方がリンパ節においてIL−6放出を抑制したことを示し、化合物23はわずか20mg/kgの投与によっても特に高レベルの抑制を示した。
【0102】
(実施例12)
(インビボ生物活性:自発的狼瘡モデル)
(A.MRL/MpJ−faslpr/J 実験1)
この実験では、Jackson Laboratoriesより入手したMRL/MpJ−faslpr/Jマウス(「MRL/lpr」)に、水中20または60mg/kgの化合物23を経口的に投薬した。MRL/lprマウスは、リンパ球死に欠陥を有し、ヒト全身性紅斑性狼瘡(SLE)に見出される抗核抗体を含む自己抗体を自発的に発生させる。投薬は、5週齢に開始して週に5回毎日行った。血液および尿試料を1日目の投薬前に採取し、以降3〜4週毎に採取した。ELISAアッセイを用いて投薬の7週前および7週後の血清抗dsDNAを測定した。マウス抗dsDNA全Ig定性ELISAキット(Alpha Diagnostic International.San Antonio,TX)を用いてアッセイを行った。結果を図2に示す。図2は、この自己抗体への用量設定効果を示し、ビヒクルを投与された動物に対して450nmでの平均光学濃度(Optical Density)(OD450)が統計上有意に抑制された。統計的有意差は投薬10週でなお観察されたが、その後化合物治療群は12週およびそれ以降に、経口によってdH2Oを投薬されたビヒクル治療動物に一層類似するようになった。全体的な様子または体重測定で判断して、投薬量に関連する毒性の根拠は見られなかった。
【0103】
抗核抗体(「ANA」)も、この実験において、Antibodies Inc.製のスライド上の固定された試料として得た固定されたHepG2細胞を、上記のように化合物23を20または60mg/kgで投薬したマウスから得た希釈血清に曝露し、Antibodies Inc.により供給されるキット試薬をキットの説明書に従って使用してマウス免疫グロブリンについて染色することにより調査された。次に、蛍光染色の強度を顕微鏡で読み取り、スコアに−から+++の間の値を割り当てた。試料の識別表示はデータ収集のために隠した(blinded)。
【0104】
図3は、ANA試験の結果をグラフによって図示する。図3は、ビヒクル処理マウスが5〜12週齢の間に徐々にANA陽性となったが、一方、化合物処理マウスから得た血清が陽性ANA染色に対してはるかに遅い進行を示したという点で、ANA発生の用量依存性の抑制を示す。
【0105】
(B.MRL/lpr 実験2)
このSLE実験において、投薬群は、下の表4に記載される通りであった。化合物23の2種類の用量レベルをHCQおよびシクロホスファミド(Cytoxan(登録商標))と比較した。HCQおよびシクロホスファミドは、それぞれ、皮膚狼瘡および全身性狼瘡の治療に現在使用されている。シクロホスファミドは、一貫した血清抗dsDNAレベルの抑制を示した。この差は、ANOVAによりビヒクルに対して試験した場合に8週および12週の投薬時点で統計上有意であった。12週間の投薬前および投薬後の個々の動物血清について450ナノメートルでの光学濃度(OD450)のELISAアッセイから得られる結果を図4に示す。血清は、PBS中1:600希釈でアッセイした。(マウス抗dsDNA全Ig定性ELISAキット、Alpha Diagnostic International)。投薬は、経口(p.o.)かまたは腹腔内(i.p.)投与のいずれかにより行った。このアッセイから得られる個々のデータポイントを図4に図示する。図4は、12週で、ビヒクルと高用量の化合物23との間に平均dsDNAレベルの統計上の有意差があったことも示す。
【0106】
タンパク尿(BCAタンパク質アッセイにより測定)または皮膚病変への影響は、この実験でいずれの化合物によっても見られなかった。
【0107】
【表5】
【0108】
(実施例13)
(インビボ生物活性:NZBWF1/J自発的狼瘡モデル)
化合物23および化合物30のインビボ研究を、NZBWF1/Jマウスモデルで行った。マウスはJackson Laboratoriesより入手し、12の群に分け、水中の化合物を5ヶ月齢で開始し週に5日、経口的に投薬した。投薬の詳細を下の表5に提供する。
【0109】
【表6】
【0110】
この研究で試験した化合物の化合物23または化合物30はいずれも、20mg/kgまたは60mg/kgの化合物の投薬後の16週の治療の間、抗dsDNAへの効果を示さなかった。この研究の結果を図5に図示する。短期アッセイで化合物23よりも多少高い効力を示した化合物30は、60mg/kgの高用量で抗dsDNAをいくらか抑制したが、これは統計上有意でなかった。ANAを同じ実験で測定し、12頭のビヒクル治療動物のうち5頭しか+/−またはそれよりも高いスコアに達しなかったものの、12週の治療後のANAスコアの分布に化合物の効果はなかった。ANAスコアリングの結果を図6に図示する。
【0111】
化合物23および化合物30は、3〜4ヶ月の投薬後にNZBWF1/Jマウス系統において用量に関連する体重減少を一貫して生じた。筋肉、脳、腎臓および脾臓の組織学的検査は、ヘマトキシリンおよびエオシン染色において明らかな病態を示さなかった。
【0112】
60および20mg/kgの化合物23を用いる毎日経口投薬の結果として、SLEのMLR/lprモデルにおける抗dsDNAの抑制がもたらされた。化合物23はまた、抗核抗体発生の用量依存性の阻害も示した。シクロホスファミドはMRL/lprモデルにおいて抗dsDNAを抑制したが、一方ヒドロキシクロロキンは検出可能な効果がなかった。さらに、NZB/W系での化合物23の試験により、抗dsDNA、ANA、またはタンパク尿への効果は示されなかった。用量依存性の体重減少は、NZB/Wマウスにおいて数ヶ月の投薬の後に観察された。
【0113】
(実施例14)
(インビボ生物活性:実験的自己免疫性脳炎 多発性硬化症モデル)
実験的自己免疫性脳炎(EAE)は、ヒト多発性硬化症を模倣する、マウスにおいて誘導される自己免疫であり、自己免疫には、自己抗体、および中枢神経系抗原に対して標的とされる細胞応答が含まれる。EAEを誘導するため、8頭の雄C57BL/6Jマウス(Jackson Laboratories)の群を、完全フロイントアジュバント(CFA)中200μg/マウスの用量で、アミノ酸配列MEVGWYRSPFSRVVHLYRNGK(配列番号4)とともに、合成ペプチドMOGp35−55(Becher et al.J.Clin.Invest.112:1186−1191(2003))で0日目に皮下に免疫化した。同時に、動物に百日咳毒素を腹腔内に30ng/マウスで投与した。2日目に、百日咳注射を繰り返した。7日目に、マウスを不完全フロイントアジュバント中200μg/マウスのMOGp35−55で追加免疫した。9日目から、20mg/kgまたは60mg/kgの水中の化合物23を用いて経口胃管栄養法により毎日投与した。尾および四肢の麻痺の程度および位置を観察することにより症状をスコア化し、各々の8頭のマウス群の平均スコアを示す。
【0114】
この研究の結果を図7に示す。MOGに誘導されるEAEは、対照マウスと比較して、20mg/kgの化合物23を投与したマウスにおいて有意に抑制され、60mg/kgの同じ化合物を投与したマウスにおいてさらに抑制されたことを観察することができる。
【0115】
(実施例15)
(インビボ生物活性:コラーゲン誘発関節炎)
II型コラーゲン誘発関節炎モデルを、関節炎の治療のために化合物2をインビボで評価するために使用した。このモデルでは、関節炎を、完全フロイントアジュバントを用いて雄DBA/1J(Jackson Labs)マウスにおいて誘導した。最初のプライミング/免疫化用量の300μgの、等容積の完全フロイントアジュバントで乳化したウシII型コラーゲンを、尾の付け根から少なくとも5mmの皮下に(s.c.)注射する。
【0116】
免疫化後9日目に開始して、以降毎日週に5日、各々のマウスに、6mg/kg、20mg/kg、または60mg/kgの化合物2あるいはプラセボを1週間あたり5日、経口的に投与した。
【0117】
2回目の免疫化用量は、最初の免疫化の21日後に、等容積の不完全フロイントアジュバントで乳化した、200μgのウシII型コラーゲンの最初の免疫化部位よりも上に皮下注射により投与した。関節炎の進行をさらに促進するため、2回目の免疫化の3日後に、動物にリポ多糖(LPS)(PBS中1μg/マウス)の腹腔内注射を投与した。
【0118】
最初の免疫化用量の21、25、28、および31日後に、足腫脹のエビデンスについて動物を調べ、各々の時点の関節炎スコアを動物に割り当てた。このアッセイの結果を図8に示す。足腫脹の抑制は、6および20mg/kgを用いる投薬後に観察されたが、60mg/kgを用いる投薬後の結果は不明であった。異例の60mg/kgの結果から、この特定の動物モデルについての適切な上限投薬レベルを決定するさらなる研究の必要性が示唆される。
【0119】
(実施例16)
(インビボ生物活性:盲腸結紮穿刺術)
盲腸結紮穿刺(CLP)モデルは、腹部切開を行って盲腸(消化管の容易に単離される部分)を露出させ、盲腸の一部分を結紮する外科手技を伴う。次に、盲腸を穿刺し、少量の腸内容物を押し出す。切開を閉じ、動物に蘇生のための生理食塩水および抗生物質を与える。敗血症は、主に押し出された腸内容物からの殺菌に起因して発症し、疾患の発症は、盲腸の穿刺のサイズおよび数に依存する迅速性および重篤度とともに起こる。
【0120】
(A.盲腸結紮穿刺術)
ケタミン、キシラジンおよびアセプロマジンの組合せの腹腔内投与によりマウスにおいて麻酔を誘導する。動物の腹部領域は短く刈ってよく、体温を測定するためのトランスポンダを皮下に埋め込む。この手順は無菌生存手術に適した領域で行う。この手順の間中、動物は加温パッドの上に置く。腹部領域は、ポビドン/ヨードおよびアルコールで最低3回交互に拭いて準備する。
【0121】
正中切開を行って腹部を開き、盲腸を露出する。生理食塩水で湿らせた綿棒(cotton-tipped applicators)を用いて盲腸を腹腔から摘出する。回盲弁のすぐ遠位で4−0縫合糸(sutures)を用いて盲腸の先端を結紮する。盲腸の内容物を一端に移動させて、次に盲腸を滅菌針で穿刺する。圧力を加えて少量の物質を盲腸から腹膜腔に押し出す。結紮は所定の位置に残して動かさない。筋肉および筋膜を連続的な4−0縫合糸で閉じ、皮膚を創傷ステープル(wound staples)で閉じる。
【0122】
手術直後に、体重100gあたり5mlの暖かい蘇生のための生理食塩水を皮下注射する。温度および体重のベースライン読みを記録する。手術1時間後、20mg/kg用量の抗生物質モキシフロキサシンを試験されているいずれかの抗敗血症薬剤候補とともに腹腔内に投与する。研究の持続のために抗生物質を24時間間隔で投与する。投与する抗生物質の用量は、CLPにより放出される細菌を根絶することを目的としているのではなく、その代わりにヒトが抗生物質を投与されるがそれらが効果のない場合の臨床シナリオをモデル化するために使用される。モキシフロキサシンはQT間隔の延長に関係しているが、好ましくない薬物相互作用が回避される限り、臨床使用に安全であると考えられ(Torres et al.,J Surg Res.125:88−93(2005))、該抗生物質は別のマウス敗血症モデルにおいて安全かつ効果的であると既に証明されている(Alkorta et al.,Int J.Antimicrob.Agents 25(2):163−7,(2005))。
【0123】
(B.試験化合物の投与)
化合物は、手術の約1時間後に抗生物質注射の時に投与されるが、このタイミングは変動する可能性がある。化合物は、候補化合物の薬物動態特性に応じて1日に2回投与される。試験化合物の投与は、投与のために確立されたガイドラインに従って、次の経路:尾静脈からの経口、腹腔内、皮下、または静脈注射のいずれかによって行われる。
【0124】
10頭前後の動物の群が使用される。薬物評価実験は、盲腸結紮穿刺を経験し、ビヒクルのみを投与される1つの群(対照群)、およびCLPを経験し、試験治療化合物を投与される群を有する。化合物は手術後1時間に投与され、動物をその日中2時間おきに生存についてモニターする。トランスポンダを介して2時間おきに体温をとり、動物の健康を各時点でモニターする。体重は4時間間隔で量る。毎日少なくとも8時間の期間動物をモニターする。サイトカインレベルの測定のために血清試料を採取する。死亡数をモニターし、薬物治療の有効性の尺度として使用する。
【背景技術】
【0001】
損傷または感染に対する多くの初期の免疫応答は、自然免疫Toll様受容体(TLR)のファミリーによって開始される。TLRファミリーは、幅広い特異性を有し、病原生物の間で高度に保存された分子構造または異なる種類の物理的損傷を認識する。特異的TLRのリガンドとしては、エンドトキシン、一本鎖もしくは二本鎖RNA、ペプチドグリカン、フラジェリン、熱ショックタンパク質が挙げられ、TLR9の場合には、DNA中の非メチル化CpG配列が挙げられる(Bell et al.,Trends Immunol 24:528−533(2003)およびWagner,Trends Immunol 25:381−6(2004))。この疾患または損傷の初期のセンシングは、続いて起こる免疫応答に絶対不可欠であるが、同時にそれは不適切な応答または損傷性の応答の原因でもあり得る。例えば、TLR9は、狼瘡を含む自己免疫障害に結び付けられているが、それは長い間抗DNA抗体反応性、および多発性硬化症に関連付けられている(Prinz et al.,J Clin Invest 116:456−464(2006))。また、TLR9は、敗血性ショックによる死に関連する、制御されない炎症応答の根底にあると思われる(Plitas et al.,J Exp Med 205:1277−83(2008))。
【0002】
TLR9は、非メチル化CpG配列に結合し、細胞活性化およびサイトカイン分泌をもたらす、ヒトB細胞および形質細胞様樹状細胞(PDC)に発現される自然免疫受容体として最初に同定された(Hemmi et al.,Nature 408:740−745(2000))。これらの配列は、真核生物のDNAと比較して、細菌のDNAで過剰に発現され、そのため、細菌感染の指標としての機能を果たすことができる。しかし、真核生物のDNAは、いくらかの非メチル化CpGを含み、同様にTLR9を刺激する能力がある(Vallin et al.,J Immunol.163:6306−13(1999)およびLeadbetter et al.(2002))。瀕死の細胞のDNAは、一般に摂取され系から隔離されるが、狼瘡は、過剰な細胞片のクリアランス不良および蓄積を引き起こす遺伝的欠損に関連し(Krishnan et al.,Seminars in Immunology 18:240−243(2006))、免疫系を異常に高レベルのリガンドに曝露する。この状態は、B細胞上の複合体特異的免疫グロブリンか、または樹状細胞もしくは抗原提示細胞上のFc受容体のいずれかによるDNA含有複合体の標的化取り込みと相まって、該刺激複合体にTLR9リガンドが存在することにより後押しされた応答である自己抗原の提示および反応をもたらし得る。この知見は、PDCが、全身性紅斑性狼瘡(SLE)血清中に見出される複合体にα−インターフェロンを分泌することにより応答し、これがFc受容体であってDNA依存性であり、TLR9に媒介されるという事実により例証される(Leadbetter et al.,(2002)およびMeans et al.,J Clin Invest 1152:407−17(2005))。得られるα−インターフェロンは、樹状細胞およびB細胞の成熟をさらに駆動することができる。それに対応して、アレイデータにより、重篤な疾患の患者におけるα−インターフェロン誘導性遺伝子の活性化が示される(Bennett et al.,J Exp Med.197:711−23(2003))。PDCは、身体においてα−インターフェロンの主な供給源であるので、この知見は疾患をもつこの細胞集団にさらに関連する。
【0003】
B細胞も狼瘡自己免疫の中心であり、合成オリゴヌクレオチドを用いるTLR9刺激によりIL−6および抗体を増殖および産生するように駆動される。自己反応性細胞表面免疫グロブリンを発現しているB細胞との選択的結合のために、自己抗原特異的細胞は、この応答において優勢となる。従って、DNAまたはRNAを含有する細胞片がTLR9により刺激された自己抗原特異的B細胞からの抗体と結合して、Fc受容体を介して形質細胞様樹状細胞を刺激する、自己強化サイクルが働いている。
【発明の概要】
【0004】
本発明の第1の実施形態は、式(I):
【化1】
の化合物またはその医薬上許容される塩を提供し、
式中、R5、R6、またはR7のうちの1つが、式(a):
【化2】
の基であり、かつ、R5が(a)である場合、R6およびR7は双方ともHであり、R6が(a)である場合、R5およびR7は双方ともHであり、R7が(a)である場合、R5およびR6は双方ともHであり、
iおよびjは同一であって、かつ0、1、2、3または4であり、
R1およびR4は同一であって、かつH、CH3およびCH2CH3からなる群から選択され、
R2はCH3であり、R3は(CH2)hN(CH3)2(hが2、3または4である)および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは、
R2およびR3は同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
(CH2)nN(CH3)2(式中、nは2、3または4である)、
(CH2)pO(CH2)qN(CH3)2(式中、pおよびqは同一であって、かつ2または3である)、
式(b):
【化3】
(式中、uは0または1である)の基、または
式(c):
【化4】
(式中、vは0または1であり、ZはNまたはCHであり、XはOまたはNCH3である)の基からなる群から選択されるか、又は
R1−N−R2およびR3−N−R4は同一であって、かつ
式(d):
【化5】
の基、および
式(e):
【化6】
(式中、rは1、2または3であり、
YはCHまたはNであり、
R8はH、CH3、CH(CH3)2、N(CH3)2、CH2OCH3または式(f):
【化7】
(式中、tは、0または1である)の基であり、
R9はH、CH2OCH3または式(f)の基である)
の基からなる群から選択される。
【0005】
本発明の第2の実施形態は、本明細書に記載される化合物を含む医薬組成物を提供する。
【0006】
本発明の第3の実施形態は、被験体において免疫学的障害、例えば、敗血症、狼瘡、関節リウマチ、または多発性硬化症などを治療する方法を提供し、この方法は、本明細書に記載される化合物を被験体に有効量で投与することを含む。
【0007】
本発明の第4の実施形態は、被験体において狼瘡を治療する方法を提供し、この方法は、本明細書に記載される化合物を被験体に有効量で投与することを含む。
【0008】
本発明の第5の実施形態は、免疫学的障害、例えば、敗血症、狼瘡、関節リウマチ、または多発性硬化症などを治療するための薬物の製造のために、本明細書に記載される化合物を使用することを提供する。
【0009】
本発明のその他の実施形態は、本明細書において開示され、以下でさらに詳細に考察される。
【図面の簡単な説明】
【0010】
【図1】実施例11に記載されるように、インビボで20mg/kgまたは60mg/kgの化合物2、化合物23、またはヒドロキシクロロキン(HCQ)の2週間の投与、そしてエキソビボでオリゴCpG1668を用いるリンパ節の刺激後の、マウスから取り出したリンパ節において検出されたインターロイキン6(IL−6)の濃度を示す棒グラフである。
【図2】実施例12Aに記載されるように、MRL/MpJ−faslpr/J(「MRL/lpr」)マウスの20mg/kgまたは60mg/kgの化合物23を用いる投薬の前後7週間の、マウス抗dsDNAのELISAアッセイにおける、450ナノメートル(OD450)での光学濃度測定の結果を示すプロット図である。
【図3】実施例12Aに記載されるように、投薬の4、10または12週間後に、20mg/kgまたは60mg/kgの化合物23を投与されたマウスのMRL/lpr自発的狼瘡モデルにおける抗核抗体(ANA)試験の結果を示す一連の棒グラフを示す図である。結果は、ANA結果の5つの考えられるカテゴリー(−、−/+、0、+、または++)の各々にスコアされたマウスの数として示す。
【図4】実施例12Bに記載されるように、化合物23(20mg/kgまたは60mg/kg)、HCQ、またはシクロホスファミドを用いる12週間の投薬前および投薬後の、MRL/lprマウス抗dsDNAのOD450でのELISAアッセイの結果を示すグラフである。
【図5】実施例13に記載されるように、20mg/kgまたは60mg/kgの化合物23または化合物30を用いる16週間のNZB/WF1/Jマウスの投薬前および投薬後の、マウス抗dsDNAのOD450でのELISAアッセイの結果を示すプロット図である。
【図6】実施例13に記載されるように、20mg/kgまたは60mg/kgの化合物23または化合物30を用いる投薬の16週間後の、5つの考えられるANAスコアの各々のNZB/WF1/Jマウスの数を示す一連の棒グラフを示す図である。
【図7】実施例14に記載されるように、合成ペプチドMOGp35−55で免疫化して、多発性硬化症に似た症状の実験的自己免疫性脳炎(EAE)を誘導したC57BL/6Jマウスに、20mg/kgまたは60mg/kgの化合物23を投与した結果を示すグラフである。
【図8】実施例15に記載されるように、完全フロイントアジュバントで乳化したウシII型コラーゲンで免疫化して、関節炎を誘導した雄DBA/1Jマウスに、6mg/kg、20mg/kgまたは60mg/kgの化合物2を投与した結果を示すグラフである。
【発明を実施するための形態】
【0011】
(A.定義)
「治療」、「治療する」、および「治療すること」とは、本明細書に記載される障害または疾患の進行を後退させること、軽減すること、または阻止することをさす。さらに、本明細書において用いられるように、被験体の「治療」とは、免疫学的障害を有するか、かかる免疫学的障害の症状を有するか、あるいはかかる免疫学的障害の危険性がある(または免疫学的障害に罹患しやすい)被験体への、前記障害、前記障害の症状、あるいは前記障害の危険性(または前記障害に対する易罹患性)を治す、癒す、軽減する、緩和する、変化させる、除去する、改善する、好転させる、または作用する目的での、本明細書に記載される本発明の化合物の被験体への適用または投与、あるいは被験体の細胞または組織への前記化合物の適用または投与が含まれる。用語「治療すること」とは、損傷、病変または状態の治療または改善の成功のいずれかの徴候をさし、それには、任意の客観的または主観的パラメーター、例えば、軽減、緩解、症状の減少あるいは損傷、病変または状態を被験体にとってより許容できるものにすること、変性または減退の速度を遅らせること、変性の最終状態の衰弱を抑えること、被験体の身体的または精神的健康を向上させること、あるいは、場合によっては、前記障害のより重症な型または症状の発症を予防することなど、が含まれる。症状の治療または改善は、身体検査、精神鑑定、または当分野で公知の診断検査の結果を含む客観的または主観的パラメーターに基づくことができる。
【0012】
一実施形態において、用語「治療すること」には、免疫学的障害によって引き起こされるかまたは免疫学的障害に関連する炎症、感染、発熱、異常な精神状態、臓器機能不全、貧血、関節痛、疲労、認知的困難、痙攣および/または振戦からの好転またはその除去などの免疫学的障害の症状を好転させることが含まれる。
【0013】
「予防」、「予防する」、および「予防すること」とは、本明細書に記載される障害または疾患の発生または発症を、処置を取らない場合に起こると思われるその発生または発症と比べてなくすまたは低減させることをさす。
【0014】
「有効な量」とは、臨床検査および評価、患者の観察、および/または同類のものによって示される障害または疾患の症状の除去をもたらす量をさす。「有効な量」は、生物学的または化学的活性において検出可能な変化をもたらす用量をさらに表すことができる。検出可能な変化は、当該機構またはプロセスについて当業者によって検出し、かつ/またはさらに定量することができる。さらに、「有効な量」は、所望の生理学的状態を維持する、すなわち、目的の状態の著しい減退を低減もしくは予防し、かつ/または好転を促す量を表すことができる。「有効な量」は、治療上有効な量をさらにさすことができる。
【0015】
本明細書において用いられる「被験体」とは、哺乳類被験体(例えば、イヌ、ネコ、ウマ、ウシ、ヒツジ、ヤギ、サルなど)、特にヒト被験体(男性および女性の両方の被験体を含み、新生児、乳児、小児、青年期、成人および高齢者の被験体を含み、様々な人種および民族(限定されるものではないが、白人、黒人、アジア系、アメリカインディアンおよびヒスパニック系を含む)をさらに含む)を意味する。
【0016】
本明細書において用いられるように、用語「医薬上許容される塩」とは、本発明の化合物の比較的毒性のない無機酸塩および有機酸塩をさす。これらの塩は、前記化合物の最終的な単離および精製中にインサイチューで調製することができ、あるいは生成した化合物をその遊離形態で適した有機酸または無機酸と別々に反応させ、そのようにして形成された塩を単離することにより調製することもできる。代表的な酸性塩としては、酢酸塩、アジピン酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、重炭酸塩/炭酸塩、重硫酸塩/硫酸塩、ホウ酸塩、カンシル酸塩、クエン酸塩、シクラミン酸塩、エジシル酸塩、エシレート、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンズ酸塩、塩酸塩/塩化物、臭化水素酸塩/臭化物、ヨウ化水素酸塩/ヨウ化物、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシル酸塩、メチル硫酸塩、ナフチル酸塩、2−ナプシル酸塩、ニコチン酸塩、硝酸塩、オロチン酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素/リン酸二水素、ピログルタミン酸塩、サッカリン酸塩、ステアリン酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、トシル酸塩、トリフルオロ酢酸塩およびキシナホ酸塩が挙げられる。一実施形態において、医薬上許容される塩は塩酸塩/塩化物塩である。
【0017】
(B.化合物)
本明細書に記載される一部の化合物は、1以上の不斉中心を含んでよく、従ってそれらの化合物は、様々な異性体形、例えば立体異性体および/またはジアステレオマーとして存在することができる。キラル中心の周囲の結合の配向が式に明記されていない場合には、その式は、本発明の化合物の構造において起こり得る、不斉炭素に基づく幾何異性体、光学異性体、立体異性体および互変異性体などの考え得るあらゆる異性体を包含すると理解すべきである。
【0018】
上に述べたように、本発明は、式(I):
【化8】
の化合物またはその医薬上許容される塩(式中、i、R1、R2、R5、R6、およびR7は、本明細書において上述した記載の通りに定義される)を提供する。
【0019】
式(I)の化合物のいくつかの実施形態では、iおよびjは、双方とも1である。
【0020】
さらなる実施形態では、R2およびR3は同一であって、以下の:
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
本明細書で上述した式(b)の基、および
本明細書で上述した式(c)の基
からなる群から選択される。
【0021】
他の実施形態によれば、R1−N−R2およびR3−N−R4は同一であって、かつ
式(g):
【化9】
の基、式(h):
【化10】
の基、 式(i):
【化11】
の基、 式(j):
【化12】
の基、 式(k):
【化13】
の基または式(m):
【化14】
の基から選択される。
【0022】
式(I)の化合物のいくつかの実施形態では、
iおよびjは双方とも1であり、
R1およびR4は同一であって、かつHおよびCH3からなる群から選択され、
R2はCH3であり、R3は(CH2)2N(CH3)2および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは
R2およびR3は同一であって、かつ
a)(CH2)kCH3(式中、kは0、1または2である)、
b)(CH2)mN(CH2CH3)2(式中、mは2または3である)、
c)本明細書で上述した式(b)の基、および
d)本明細書で上述した式(c)の基
からなる群から選択されるか、又は、
R1−N−R2およびR3−N−R4は同一であって、かつ
式(g)の基、式(h)の基、式(i)の基、式(j)の基、式(k)の基、および式(m)の基からなる群から選択される(全ては本明細書で上述した記載の通りである)。
【0023】
さらなる実施形態では、R5は、本明細書で上述した式(a)の基であり、R6およびR7は各々Hである。
【0024】
式(I)の化合物の特定の実施形態では、この化合物は以下から選択される構造を有する。
【0025】
【表1A】
【表1B】
【表1C】
【表1D】
【表1E】
【表1F】
【表1G】
【0026】
もう1つの実施形態では、式Iの化合物は、化合物1、化合物2、化合物10、化合物11、化合物12、化合物13、化合物14、化合物16、化合物17、化合物26、化合物29、化合物30、化合物31、化合物32、化合物34、および化合物35またはその医薬上許容される塩からなる群から選択される。もう1つの実施形態では、前記化合物は、化合物13、化合物16、化合物22、化合物23、化合物26、化合物27、化合物30、化合物34、および化合物35またはその医薬上許容される塩からなる群から選択される。もう1つの実施形態では、前記化合物は、化合物23、化合物26、および化合物30、またはその医薬上許容される塩からなる群から選択される。さらにもう1つの実施形態では、前記化合物は、化合物23またはその医薬上許容される塩である。
【0027】
(C.化合物の合成)
本明細書に記載される化合物は、一連の反応によって調製することができる。本発明の化合物を調製するための一般的方法を下に示す。実施例を記載するセクションにおいて下にさらに述べるように、ある特定の場合において例として特定の化合物を記載する。下に記述されている場合を除き、以下の方法では、R1、R2、R3、R4、i、およびjは、上の式Iにおいて定義されるものと同一である。
【0028】
方法1−アルキル化、アミノ化またはアルキル化、アミノ化およびN−メチル化
【化15】
(R1aおよびR4aがHである場合、またはR2aおよびR3aがHである場合、N−メチル化)
上に示した方法1の反応スキームにおいて、
n、i、およびjは同一であって、かつ0、1、2、3または4であり、
R1aおよびR4aは双方ともRxと同一であり、
R2aおよびR3aは双方ともRyと同一であり、
R1bおよびR4bは同一であり、
R2bおよびR3bは同一であり、
R1aおよびR4aが双方ともHである場合には、R1bおよびR4bは双方ともメチルであり、
R2aおよびR3aが双方ともHである場合には、R2bおよびR3bは双方ともメチルである。
【0029】
本発明の実施形態による対称の化合物を合成する一方法では、例えば、2−(4−ヒドロキシフェニル)ベンゾオキサゾール−6−オール(1A)を、ジメチルスルホキシド(DMSO)中炭酸カリウムの存在下、ブロモクロロアルカン(1B)と反応させてジクロロエーテル誘導体(1C)を形成することができる。また、化合物1Aの代わりに2−(3−ヒドロキシフェニル)ベンゾオキサゾール−6−オールおよび2−(2−ヒドロキシフェニル)ベンゾオキサゾール−6−オールを用いてもよい。次に、ジクロロエーテル誘導体(1C)をアミン(HNRxRy)と反応させてジアミン(1D)を形成することができる。方法1の合成スキームにおいて、単一アミン反応物を用いてジアミン(1D)中にアミン基−NR1aR2aおよび−NR3aR4aを形成することができることを示すために、前記アミンはHNRxRyと総称的に表現される。そのようなものとして、RxはR1aおよびR4aと同一であり、RyはR2aおよびR3aと同じである。その上、R1aおよびR4aが水素である場合、またはR2aおよびR3aが水素である場合には、前記化合物は、所望によりN−メチル化を経てN−メチル誘導体(1E)を形成することができる。化合物1Dおよび1Eは、双方とも本発明の実施形態に従う化合物であり得る。
【0030】
方法2−光延反応または光延反応およびN−メチル化
【化16】
(光延)(R1aおよびR4aがHである場合、またはR2aおよび/もしくはR3aがHである場合、N−メチル化)
上に示した方法2の反応スキームにおいて、
n、i、およびjは同一であって、かつ0、1、2、3または4であり、
R1aおよびR4aは双方ともRxと同一であり、
R2aおよびR3aは双方ともRyと同一であり、
R1bおよびR4bは同一であり、
R2bおよびR3bは同一であり、
R1aおよびR4aが双方ともHである場合には、R1bおよびR4bは双方ともメチルであり、
R2aおよびR3aが双方ともHである場合には、R2bおよびR3bは双方ともメチルである。
【0031】
本発明の実施形態による対称の化合物を合成するもう1つの方法では、光延反応によって、2−(4−ヒドロキシフェニル)ベンゾオキサゾール−6−オール(2A)をヒドロキシアルキルアミン(2B)と反応させてジアミン(2C)を作出することができる。また、化合物2Aの代わりに2−(3−ヒドロキシフェニル)ベンゾオキサゾール−6−オールおよび2−(2−ヒドロキシフェニル)ベンゾオキサゾール−6−オールを用いてもよい。その上、R1aおよびR4aが水素である場合、またはR2aおよびR3aが水素である場合には、前記化合物は、所望によりN−メチル化を経てN−メチル誘導体(2D)を形成することができる。化合物2Cおよび2Dは、双方とも本発明の実施形態による化合物であり得る。
【0032】
方法3−不斉側鎖類似体について
【化17】
(方法1または方法2)(方法1または方法2)
方法3の反応スキームにおいて、
R1およびR4は同一であって、かつHおよびCH3からなる群から選択され、
R2はCH3であり、R3は(CH2)2N(CHs)2および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択される。
【0033】
本発明のいくつかの実施形態による不斉化合物は方法3によって合成することができる。かかる場合では、4−アミノベンゼン−1,3−ジオール(3A)を、ヘキサフルオロリン酸ベンゾトリアゾール−1−イル−オキシ−トリス−(ジメチルアミノ)−ホスホニウム(PyBop)の存在下、保護されたヒドロキシル基を有する安息香酸、例えば、4−(ベンジルオキシ)安息香酸(3B)、3−(ベンジルオキシ)安息香酸または2−(ベンジルオキシ)安息香酸などと反応させてベンズアミド誘導体(3C)を形成することができる。次いで、そのベンズアミドを環化してベンゾオキサゾール環(3D)を形成することができる。この時点において、上に記載される、方法1または方法2のいずれかを用いてヒドロキシ官能性ベンゾオキサゾール(3D)をアミン(3E)へと変換してよい。方法1および2に関し、R1はR1aまたはR1bであってよく、R2はR2aまたはR2bであってよい。その後、例えば、パラジウムを用いる水素化によって、ベンジルエーテル基を脱保護してヒドロキシ官能性アミン(3F)を形成することができる。その後、方法1または方法2のいずれかを用いてヒドロキシル官能性アミンをジアミン(3G)へと変えてよい。方法1および2に関し、R4はR4aまたはR4bであってよく、R3はR3aまたはR3bであってよい。
【0034】
(D.医薬組成物)
一実施形態では、本発明は、化合物を含む医薬組成物である。もう1つの実施形態では、前記医薬組成物は医薬上許容される担体をさらに含む。本明細書において用いられる用語「医薬上許容される担体」とは、被験体への治療薬の送達のためのビヒクルとして用いられる、それ自体は治療薬でない、任意の物質をさす。
【0035】
本発明の組成物は、経口投与、非経口投与、吸入スプレー投与、局所投与、直腸投与、鼻腔投与、舌下投与、口内投与、膣投与または植え込み型リザーバー投与などに用いる処方物に適し得る。好ましくは、前記組成物は、経口的に、局所に、腹膜内にまたは静脈内に投与される。本発明の組成物の滅菌注射剤型は水性懸濁液であっても油性懸濁液であってもよい。これらの懸濁液は、適した分散剤または湿潤剤および懸濁化剤を用いて当分野で公知の技術によって処方され得る。滅菌注射用調製物は、毒性のない非経口的に許容される希釈剤または溶媒中の滅菌注射用溶液または懸濁液(例えば、1,3−ブタンジオールの溶液として)であってよい。使用することのできる許容されるビヒクルおよび溶媒には、水、リンゲル液および等張塩化ナトリウム溶液がある。その上、滅菌固定油が溶媒または懸濁媒として通常使用される。
【0036】
医薬上許容される油は、本発明の組成物中の溶媒または懸濁媒として使用され得る。脂肪酸、例えば、オレイン酸およびそのグリセリド誘導体などは、天然の医薬上許容される油、例えば、オリーブ油またはヒマシ油、特にそれらのポリオキシエチル化型などと同様に、注射用処方物中に含められることが適している。本発明の油含有組成物はまた、長鎖アルコール希釈剤または分散剤、例えば、カルボキシメチルセルロースまたはエマルジョンおよび懸濁液を含む医薬上許容される投与形の処方物において一般的に用いられる類似の分散剤なども含有することができる。前記組成物は、界面活性剤(Tween(登録商標)またはSpan(登録商標)を含む非イオン性洗剤など)その他の乳化剤、またはバイオアベイラビリティー向上剤をさらに含むことが適している。
【0037】
本発明の組成物は、限定されるものではないが、カプセル剤、錠剤、懸濁液または溶液を含む経口的に許容される投与形の形であってよい。経口投与形は、少なくとも1種類の賦形剤を含むことができる。本経口処方物に用いられる賦形剤には、希釈剤、不快な味または臭いを遮断するまたは中和するために加えられる物質、香味料、色素、香料、および前記組成物の外観を良くするために加えられる物質を含めることができる。本発明の一部の経口投与形は、前記組成物の用量単位を、追加的経口投与に適したカプセル剤または錠剤などの個別物品の形にすることを可能にするまたは容易にする賦形剤、例えば、崩壊剤、結合剤、接着剤、湿潤剤、ポリマー、滑沢剤、または磨砕剤などを含むことが適している。本発明の賦形剤含有錠剤組成物は、溶解型、懸濁型、ナノ粒子型、マイクロ粒子型またはその制御放出型、遅延放出型、プログラム放出型、時限放出型、パルス放出型、徐放出型もしくは長期放出型の組合せで1種類以上の賦形剤を本発明の化合物と混合する段階を含む任意の適した調剤方法によって調製することができる。
【0038】
あるいは、本発明の医薬上許容される組成物は直腸投与用の坐剤の形であってよい。それらの坐剤は、室温では固体であるが直腸温では液体であるために直腸内で溶けて薬物を放出する好適な非刺激性賦形剤と前記薬剤を混合することによって調製することができる。かかる材料には、ココア乳脂、蜜蝋およびポリエチレングリコールが含まれる。
【0039】
本発明の医薬上許容される組成物は、活性成分が1種類以上の担体中に懸濁されているまたは溶解されている局所用溶液、軟膏、またはクリームの形であってよい。本発明の化合物の局所投与に用いる担体としては、限定されるものではないが、鉱油、流動ワセリン、白色ワセリン、プロピレングリコール、ポリオキシエチレン、ポリオキシプロピレン化合物、乳化蝋および水が挙げられる。局所処方物が軟膏またはクリームの形である場合には、適した担体としては、限定されるものではないが、鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステルワックス、セテアリルアルコール、2 オクチルドデカノール、ベンジルアルコールおよび水が挙げられる。
【0040】
医薬上許容される組成物が眼用処方物である場合には、その組成物は、塩化ベンジルアルコニウムなどの防腐剤を含むまたは含まない、等張のpH調整された滅菌水溶液中の微粒子懸濁液であってよく、または等張のpH調整された滅菌生理食塩水中の溶液としてであってもよい。あるいは、眼に用いるために、医薬上許容される組成物は軟膏の形で処方してもよい。
【0041】
本発明の医薬上許容される組成物はまた、鼻腔エアゾールによっても吸入投与経路によっても投与することができる。かかる組成物は、医薬処方技術分野で周知の技術によって調製され、ベンジルアルコールまたは他の適した防腐剤、バイオアベイラビリティーを高めるための吸収促進剤、フッ化炭素、および/または他の通常の可溶化剤または分散剤を使用して、生理食塩水中の溶液として調製することができる。
【0042】
その上、本発明の化合物を含む医薬処方物は非経口処方物の形でもあり得る。
【0043】
ある特定の実施形態では、本発明の医薬上許容される組成物は経口投与用に処方されることが好ましい。
【0044】
(E.使用方法)
もう1つの実施形態では、本発明は、被験体において免疫学的障害を予防または治療する方法であり、その方法は、本発明の化合物の治療上有効な量を被験体に投与することを含む。特定の実施形態では、本発明の方法による、免疫学的障害を予防または治療するために用いられる本発明の化合物は、化合物13、化合物16、化合物22、化合物23、化合物26、化合物27、化合物30、化合物34、および化合物35またはその医薬上許容される塩、からなる群から選択される式Iの化合物である。もう1つの実施形態では、前記化合物は、化合物23、化合物26、および化合物30またはその医薬上許容される塩からなる群から選択される。もう1つの実施形態では、前記化合物は、化合物23またはその医薬上許容される塩である。
【0045】
一実施形態では、本発明の方法を用いて予防または治療される免疫学的障害は、自己免疫疾患である。本明細書において用いられるように、用語「自己免疫疾患」とは、自己成分と非自己成分とを識別する免疫系の障害によって一般的に引き起こされる疾患をさす。本発明の化合物により予防または治療することのできる自己免疫疾患には、限定されるものではないが、狼瘡(SLEなど)、尋常性天疱瘡、重症筋無力症、溶血性貧血、血小板減少性紫斑病、グレーブス病、シェーグレン病、皮膚筋炎、橋本病、多発性筋炎、炎症性腸疾患、多発性硬化症、真性糖尿病、潰瘍性大腸炎、関節リウマチ、強皮症および乾癬が含まれる。本発明の特定の実施形態では、前記自己免疫疾患は狼瘡または多発性硬化症である。一部の実施形態では、前記自己免疫疾患はSLEである。本明細書において用いられるように、狼瘡とは、DNA、RNA、ヒストン、およびヌクレオソームを含む自己細胞成分に対する抗体を特徴とする自己免疫疾患をさす。身体症状には、自己抗体または糸球体での免疫複合体の沈着によって生じる腎臓疾患、皮膚疹、光感受性、関節炎ならびに痙攣および精神病を含む神経学的障害が含まれる。この疾患は主に女性が罹患し、通常若年成人を襲う。直接命にかかわることはまれであるが、この疾患は死亡率の増加と関連し、生活の質に深刻な影響をもたらし、疲労の継続、体重減少、アテローム性動脈硬化の危険性、および発熱を伴う。
【0046】
もう1つの実施形態では、前記免疫学的障害は敗血症である。本明細書において用いられるように、敗血症とは、感染によって引き起こされる全身性炎症応答をさす。本発明の化合物は、敗血症によって生じる可能性がある、重症敗血症および敗血性ショックを治療するために投与することができる。特定の実施形態では、本発明の化合物は、重症敗血症を治療するために投与することができる。他の実施形態では、本発明の化合物は、敗血性ショックを治療するために用いることができる。
【0047】
本発明のさらなる実施形態によれば、本明細書に記載される化合物は、疾患の症状が強度および/または種類において増加するように疾患が再発した場合に、比較的短期間に、狼瘡、多発性硬化症および/または関節リウマチの治療のために用いることができる。一部の実施形態では、本明細書に記載される化合物は、長期治療、例えば、数日、数ヶ月または数年続く治療として用いることができる。
【0048】
本発明の一部の実施形態では、本明細書に記載される化合物は、狼瘡の治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。狼瘡の治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、ステロイド(例えば、グルココルチコイドおよびコルチコステロイド)、免疫抑制薬、非ステロイド系抗炎症薬(NSAID)、抗体療法、インターフェロン、細胞毒性薬および抗マラリア薬。一部の実施形態では、狼瘡の治療に有用な追加の薬剤は以下の少なくとも1種類である、デヒドロエピアンドロステロン(DHEA)、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、クロベタゾール、ハロベタゾール、トリアムシノロン、ベタメタゾン、フルオシノロン、フルオシノニド、レフルノミド、アザチオプリン、メトトレキサート、ミトキサントロン、クラドリビン、クロラムブシル、シクロホスファミド、シクロスポリン、ミコフェノレート(ミコフェノール酸、ミコフェノール酸モフェチルおよびミコフェノール酸ナトリウムを含む)、セレコキシブ、ジクロフェナク、フェノプロフェン、フルルビプロフェン、イブプロフェン、ケトプロフェン、フェンブフェン、インドメタシン、メクロファメート(meclofamate)、メロキシカム、ナブメトン、ナプロキセン、オキサプロジン、ロフェコキシブ、サリチレート、アセチルサリチル酸、アセトアミノフェン、スリンダク、トルメチン、リツキシマブ、ベリムマブ、インフリキシマブ、アダリムマブ、エプラツズマブ、ナタリズマブ、α−インターフェロンならびに/またはキニーネおよびヒドロキシクロロキン、クロロキン、キニジンおよびキナクリンなどのキニーネ誘導体。
【0049】
一部の実施形態では、本明細書に記載される化合物は、多発性硬化症の治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。多発性硬化症の治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、ステロイド(例えば、グルココルチコイドおよびコルチコステロイド)、免疫抑制薬、抗体療法およびインターフェロン。一部の実施形態では、多発性硬化症の治療に有用な追加の薬剤は以下の少なくとも1種類である、デヒドロエピアンドロステロン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、クロベタゾール、ハロベタゾール、トリアムシノロン、ベタメタゾン、フルオシノロン、フルオシノニド、アザチオプリン、メトトレキサート、ノバントロン、コパクソン、クラドリビン、クロラムブシル、シクロホスファミド、シクロスポリン、ミコフェノレート(ミコフェノール酸、ミコフェノール酸モフェチルおよびミコフェノール酸ナトリウムを含む)、リツキシマブ、ベリムマブ、エプラツズマブ、ナタリズマブおよび/またはβ−インターフェロン。
【0050】
さらに他の実施形態では、本明細書に記載される化合物は、関節リウマチの治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。関節リウマチの治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、シクロオキシゲナーゼ−2(COX−2)阻害薬、NSAID、疾患修飾性抗リウマチ薬(DMARD)、ステロイド、免疫抑制薬および抗体療法。一部の実施形態では、関節リウマチの治療に有用な追加の薬剤は以下の少なくとも1種類である、セレコキシブ、ジクロフェナク、フェノプロフェン、フルルビプロフェン、イブプロフェン、ケトプロフェン、フェンブフェン、インドメタシン、メクロファメート、メロキシカム、ナブメトン、ナプロキセン、オキサプロジン、ロフェコキシブ、アセチルサリチル酸、アセトアミノフェン、スリンダク、トルメチン、デヒドロエピアンドロステロン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、クロベタゾール、ハロベタゾール、トリアムシノロン、ベタメタゾン、フルオシノロン、フルオシノニド、レフルノミド、アザチオプリン、メトトレキサート、ミトキサントロン、クラドリビン、クロラムブシル、シクロホスファミド、シクロスポリン、ミコフェノール酸モフェチル、リツキシマブ、ベリムマブ、インフリキシマブ、アダリムマブ、エプラツズマブ、ナタリズマブ、アナキンラ、アバタセプト、アウラノフィン、オーロチオグルコース、オーロチオマレート、スルファサラジン、ミノサイクリンならびに/またはキニーネおよびヒドロキシクロロキン、クロロキン、キニジンおよびキナクリンなどのキニーネ誘導体。
【0051】
一部の実施形態では、本明細書に記載される化合物は、敗血症の治療に有用な追加の薬剤と共投与することができる(すなわち、併用療法)。敗血症の治療に有用な追加の薬剤は以下の種類の薬物の1種類以上であり得る、抗生物質、アミノグリコシド、コロイド溶液または晶質液(すなわち、輸液療法)、昇圧薬、変力薬、ステロイドおよび抗凝固薬。一部の実施形態では、敗血症の治療に有用な追加の薬剤は以下の少なくとも1種類である、アルブミン、デンプン、ノルエピネフリン、ドーパミン、バソプレシン、ドブタミン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、ヒドロコルチゾン、フルドロコルチゾン、および、限定されるものではないが、ヒト組換え活性化プロテインC(rhAPC)を含む活性化プロテインC。
【0052】
さらなる実施形態では、前記化合物は、本明細書に記載される障害を治療するために併用する他の治療の用量を減らすために用いることができる(例えば、ステロイド節約型レジメンにおいて)。
【0053】
一部の実施形態では、本明細書に記載される化合物は、狼瘡、多発性硬化症、関節リウマチおよび/または敗血症の治療に有用な追加の薬剤の投与前に、その投与と同時にまたはその投与の後に投与される。
【0054】
追加の薬剤の投与「前の」本明細書に記載される化合物の投与とは、追加の薬剤による初期治療の前に、すなわち、狼瘡、多発性硬化症、関節リウマチおよび/または敗血症の危険性があるまたは患っている被験体集団に本明細書に記載される化合物と追加の薬剤とを投与することを含む治療プロトコール中の追加薬剤の投与の前に、本明細書に記載される化合物を被験体に投与することをさす。
【0055】
同時投与では、本明細書に記載される化合物と追加の薬剤は、同一時点にまたは一方の直後に投与される。一般的には、本明細書に記載される化合物と追加の薬剤は、観察される結果が、それらが同一時点において投与されたときに得られる結果と比較的変わらない十分近い時点において投与される。
【0056】
化合物は、経口的に(口腔を介する投与を含み、経口経管栄養チューブを介する投与をさらに含む)、非経口的に、吸入スプレーにより、局所に、経皮的に、直腸に、鼻腔に(経鼻経管栄養チューブを含む)、舌下に、口内に、膣にまたは埋め込み型リザーバーを介してなど、任意の適した経路によって被験体に投与してよい。本明細書において用いられる用語「非経口の」には、皮下、静脈内、筋肉内、関節内、滑液嚢内、胸骨内、くも膜下腔内、肝内、病巣内および頭蓋内の注射または注入技法が含まれる。前記組成物は、経口的に、非経口的に、経皮的にまたは吸入スプレーにより投与されることが好ましい。
【0057】
化合物は有効な量で被験体に投与される。有効な量は、一般的に1日あたり体重1kgあたり0.01mg〜500mgである。一部の実施形態では、医薬上許容される組成物は、1日あたり体重1kgあたり前記化合物0.01mg〜100mgの間の投薬量を、これらの組成物を受ける患者に投与することができるように処方され得る。ある特定の実施形態では、本発明の組成物は、0.01mg〜70mgの間の投薬量を与えるように処方される。その他の実施形態では、本発明の組成物は、0.1mg〜25mgの間または5mg〜60mgの間の投薬量を与えるように処方される。
【0058】
一部の実施形態では、有効な用量は、約5〜250mg/kgの間、約10〜200mg/kgの間、または約20〜120mg/kgの間である。一部の実施形態では、有効な用量には、5mg/kg、10mg/kg、20mg/kg、25mg/kg、40mg/kg、50mg/kg、60mg/kg、75mg/kg、100mg/kg、120mg/kgおよび150mg/kgが含まれる。投与形は、例えば、錠剤またはカプセル剤の形であってよく、有効な用量は1以上の錠剤、カプセル剤または同類のもので与えることができ、1日1回または1日を通して、例えば、4時間、8時間または12時間の間隔で与えることができる。例えば、錠剤またはカプセル剤は、例えば、10mg、25mg、50mg、75mg、100mg、150mg、200mgの化合物を含有することができる。あらゆる用量が容易にかつ便宜に分配され得るように、液体処方物も調製されてよい。
【0059】
一部の実施形態では、単一投与形で組成物を作製するために賦形剤材料と組み合わせることのできる本発明の化合物の量は、治療する宿主、および特定の投与経路によって変動することになる。
【0060】
また、特定患者についての具体的な投薬量および治療計画が、使用される具体的な化合物の活性、年齢、体重、全般的健康、性別、食生活、投与時期、排出速度、薬物組合せ、および治療する医師の判断および治療される特定の疾患の重篤度を含む、様々な因子によって決まることは当然理解される。組成物中の本発明の化合物の量もまた、組成物中の特定の化合物によって決まる。
【0061】
本明細書に記載される本発明がさらに十分に理解されるように、以下の実施例を示す。これらの実施例は例示を目的としたものでしかなく、本発明を限定するものと解釈してはならないことは当然理解される。比較化合物が下のいくつかの実施例で使用される。比較化合物の式は以下の通りである。
【化18】
【0062】
本発明の例となる化合物は、下に記載されるスキームおよび実験計画に従って調製された。
【実施例】
【0063】
(実施例1)
(化合物40の合成)
【化19】
化合物40。上に表されるように、1−ブロモ−3−クロロプロパン(7.3mL、0.074モル)を、化合物39(5.62g、0.0247モル)および炭酸カリウム(10g、0.074モル)のDMSO(50mL)中の攪拌混合物に室温にて添加し、得られる混合物を4時間攪拌し、薄層クロマトグラフィー(TLC)により反応が完了したことが示された。次に、混合物を水(200mL)に注入し、150mL酢酸エチル(EtOAc)で3回抽出し、Na2SO4で乾燥させ、濾過し、濃縮させた。残渣の真空クロマトグラフィー(10%〜30%EtOAc/ヘキサン)により、白色の固体生成物である化合物40を得た(8.10g、86%)。ヘキサン/EtOAc中で結晶化させて白色の純粋な固体生成物(6.40g)を得、母液は少量の不純物(1.60g)を含んだ。1H NMR(400MHz,CD3OD)δ 8.11(d,J=9.1Hz,2H)、7.55(d,J=8.8Hz,1H)、7.27(d,J=2.3Hz,1H)、7.11(d,J=9.1Hz,2H)、7.00(dd,J=2.3,8.8Hz,1H)、4.22(t,J=5.9Hz,2H)、4.19(t,J=6.15,2H)、3.78(tt,J=6.4Hz,4H)、2.30(dt,J=1.76,1.76Hz,4H)。出発化合物の化合物39の調製については、J.Med.Chem.2004,47,5021−5040を参照されたい。
【0064】
同じ反応を化合物39の異性体を用いて行った。異性体化合物47および化合物48を、J.Med.Chem.2004,47,5021−5040に記載される合成手順によって調製した。
【化20】
【0065】
(実施例2)
(化合物49の合成)
【化21】
化合物49。実施例1の1−ブロモ−3−クロロプロパンを1−ブロモ−2−クロロエタンに置き換えて、化合物49を得た。
【0066】
(実施例3)
(化合物50の合成)
【化22】
(化合物39)(化合物50)
化合物50。実施例1の1−ブロモ−3−クロロプロパンを1−ブロモ−5−クロロペンタンで置き換えて、化合物50を得た。
【0067】
(実施例4)
(化合物23の合成)
【化23】
化合物23。上のスキームを参照して、実施例1に記載される通り製造された化合物40(8.40g、0.0221モル)とピロリジン(7.4mL、0.088モル)のエタノール(50mL)中の混合物を、封管下、100℃にて12時間加熱した。TLC(メタノール(Et3N/MeOH)中20%トリエチルアミン)により、単一の新しいスポットが示され、質量分析により、450(M+H)の単一の所望のピークが示された。混合物を室温まで冷却し、濃縮させ、トルエンとの共沸濃縮により過剰なピロリジンを除去した(50mL中3回)。残渣をエタノール(EtOH)に溶解させ、濾過し、濃縮させた。残留固体をMeOH/EtOAc中で結晶化させて、純粋な生成物である化合物23(7.88g、68%)を得た。1H NMR(400MHz,CD3OD)δ 8.12(d,J=8.9Hz,2H)、7.57(d,J=8.8Hz,1H)、7.28(d,J=2.2Hz,1H)、7.12(d,J=8.9Hz,2H)、7.01(dd,J=2.2.8.8Hz,1H)、4.18(t,J=5.86Hz,2H)、4.15(t,J=6.15Hz,2H)、3.07−2.94(m,12H)、2.15(m,4H)、1.96(m,8H):MS(ES+)m/z 450.4。
【0068】
(実施例5)
(化合物30の合成)
【化24】
化合物30。上のスキームを参照して、実施例1に記載される通り製造された化合物40(13.4g、0.0352モル)とピペリジン(14mL、0.14モル)のエタノール(100mL、2モル)中の混合物を、封管下、100℃にて18時間加熱した。TLC(10%Et3N/MeOH)により、単一のスポットが示され、MSにより、478(M+H)の1つの単一の所望のピークが示された。混合物を、濃縮させ、トルエンとの共沸濃縮により過剰なピペリジンを除去した(50mL中3回)。残渣を、MeOHを添加した水に溶解させ、次いで1NのNaOHを、適度に攪拌しながら、さらなる沈殿が形成されなくなるまで添加した。混合物を濃縮して残留MeOHを除去し、濾過し、水で洗浄した。収集した遊離形態の固体(真空乾燥後16.55g、34.7ミリモル)を、暖かいMeOH(100mL)に溶解させ、70mLの1NのHCl(70ミリモル)で処理して二塩酸塩を生じ、混合物を濃縮乾固した。残留固体をMeOH/EtOAc中で結晶化させて、純粋な生成物である化合物30(16.0g、82%)を得た。1H NMR(400MHz,CD3OD)δ 8.13(d,J=8.8Hz,2H)、7.58(d,J=8.8Hz,1H)、7.29(d,J=2.3Hz,1H)、7.14(d,J=8.8Hz,2H)、7.02(dd,J=2.3,8.8Hz,1H)、4.21(t,J=5.57Hz,2H)、4.19(t,J=5.57Hz,2H)、3.59(m,4H)、3.34(m,4)、2.99(m,4h)、2.28(tt,J=2.3、5.6Hz,4H)、2.00−1.50(m,12H):MS(ES+)m/z 478.5。
【0069】
(実施例6)
(化合物51の合成)
【化25】
化合物51。化合物51などの類似体は、次の手順を用いて半自動化合成(SAS)を用いて調製することができる。化合物40(27.0mg、0.071ミリモル)とシクロペンチルアミン(98.6mg、1.16ミリモル)のN−メチルピロリジノン(0.5mL)中の混合物を、180℃にて300秒間マイクロ波により加熱した。粗反応混合物を、酸性条件下、LC/MSにより精製した。回収した、目的生成物を含有する画分を、分析的LC/MSにより純度についてチェックした。この化合物の全体的な純度は、>95%であり、m/z=478.5であった。純粋な生成物を含有する2種類の画分を合し、蒸発させて生成物である化合物51(22.5mg、66.4%)を得た。
【0070】
(実施例7)
(例となる化合物)
下の表1中の以下の類似体が調製された。これらの化合物は、表中に特定される対応するジクロロアルカンおよび側鎖アミンを用いて、化合物23(実施例4)および/または化合物30(実施例5)の調製のための手順に従って調製することができる。これらの化合物は、化合物51(実施例6)の調製のための手順に従っても調製することができる。
【0071】
表1.例となる化合物
【表2A】
【表2B】
【表2C】
【表2D】
【表2E】
【表2F】
【表2G】
【0072】
(上の表の化合物選択のためのNMRデータ)
化合物1:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.58(d,J=8.8Hz,1H)、7.33(d,J=2.4Hz,1H)、7.16(d,J=8.8Hz,2H)、7.06(dd,J=2.2,8.8Hz,1H)、4.28(m,4H)、3.10(m,4H)、2.68(s,6H)、2.57(s,6H)。
【0073】
化合物2:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.59(d,J=8.8Hz,1H)、7.30(d,J=2.4Hz,1H)、7.15(d,J=8.8Hz,2H)、7.04(dd,J=2.4,8.8Hz,1H)、4.21(m,4H)、3.34(m,4H)、2.93(s,6H)、2.92(s,6H)、2.68(s,6H)、2.26(m,4H)。
【0074】
化合物3:1H NMR(400MHz,CD3OD)δ 8.15(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.24(d,J=2.4Hz,1H)、7.10(d,J=8.8Hz,2H)、6.98(dd,J=2.4,8.8Hz,1H)、4.10(m,4H)、3.25(m,4H)、1.90(m,4H)、1.78(m,4H)、1.59(m,4H)。
【0075】
化合物4:1H NMR(400MHz,CD3OD)δ 8.18(dd,J=1.6,8.0Hz,1H)、7.72(d,J=9.0Hz,1H)、7.60(m,1H)、7.37(d,J=2.4Hz,1H)、7.30(d,J=8.4Hz,1H)、7.21(dd,J=7.2,8.0Hz,1H)、7.09(dd,J=2.6,9.0Hz,1H)4.39(t,J=5.6Hz,2H)、4.20(t,J=5.8,2H)、3.22(dd,J=7.6,8.4Hz,2H)、3.05(s,6H)、2.82(s,6H)、2.38(m,2H)、2.23(m,2H)。
【0076】
化合物5:1H NMR(400MHz,CD3OD)δ 7.79(d,J=8.0Hz,1H)、7.75(dd,J=1.6,2.4Hz,1H)、7.63(d,J=8.8Hz,1H)、7.50(dd,J=8.0,8.0Hz,1H)、7.32(d,J=2.4Hz,1H)、7.18(dd,J=2.6,8.6Hz,1H)、7.06(dd,J=2.4,8.8Hz,1H)4.21(m,4H)、3.32(m,4H)、2.91(s,6H)、2.90(s,6H)、2.25(m,4H)。
【0077】
化合物20:1H NMR(400MHz,CD3OD)δ 8.10(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.25(d,J=2.4Hz,1H)、7.10(d,J=8.8Hz,2H)、6.99(dd,J=2.4、8.8Hz,1H)、4.12(m,4H)、3.04(bd,J=10.8Hz,4H)、2.66(bs,8H)、2.57(m,4H)、2.15−1.90(m,14H)、1.82(m,8H)、1.60(m,4H)。
【0078】
化合物27:1H NMR(400MHz,CD3OD)δ 8.13(d,J=9.2Hz,2H)、7.58(d,J=8.8Hz,1H)、7.29(d,J=2.4Hz,1H)、7.13(d,J=9.2Hz,2H)、7.03(dd,J=2.2,8.6Hz,1H)、4.20(m,4H)、3.60−3.10(m,12H)、2.98(s,6H)、2.93(s,12H)、2.25(m,8H)。
【0079】
化合物35:1H NMR(400MHz,CD3OD)δ 8.10(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.25(d,J=2.4Hz,1H)、7.10(d,J=8.8Hz,2H)、6.99(dd,J=2.4,8.8Hz,1H)、4.10(m,4H)、3.60(m,8H)、2.65(m,8H)、2.35(m,18H)、2.02(m,4H)。
【0080】
化合物36:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz,1H)、7.30(d、J=2.4Hz,1H)、7.13(d,J=8.8Hz,2H)、7.03(dd,J=2.4,8.8Hz,1H)、4.22(m,4H)、3.92(bs,4H)、3.87(m,4H)、3.60−3.20(m,12H)、3.00(s,6H)、2.92(s,12H)、2.30(m,4H)。
【0081】
化合物37:1H NMR(400MHz,CD3OD)δ 8.14(d,J=8.8Hz,2H)、7.59(d,J=8.8Hz,1H)、7.31(d,J=2.4Hz,1H)、7.15(d,J=8.8Hz,2H)、7.04(dd,J=2.4,8.8Hz,1H)、4.23(m,4H)、3.58(m,8H)、3.55−3.35(m,6H)、m 3.23(m,6H)、2.96(s,6H)、2.90(m,12H)、2.30(m,4H)、2.05(m,8H)。
【0082】
化合物38:1H NMR(400MHz,CD3OD)δ 8.13(d,J=8.8Hz,2H)、7.58(d,J=8.8Hz,1H)、7.29(d,J=2.0Hz,1H)、7.13(d,J=8.8Hz,2H)、7.02(dd,J=2.4,8.8Hz,1H)、4.20(m,4H)、3.30−3.50(m,4H)、3.18(m,6H)、2.85−2.96(m,18H)、2.28(m,4H)、1.82(m,6H)。
【0083】
(実施例8)
(化合物34の合成)
【化26】
化合物34。上のスキームを参照して、4−アミノレソルシノールヒドロクロリド(5.3g、33ミリモル)、4−ベンジルオキシ安息香酸(5.0g、22ミリモル)、1−ヒドロキシベンゾトリアゾール(5.9g、44ミリモル)、ベンゾトリアゾール−1−イル−オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(23g、44ミリモル)およびN,N−ジイソプロピルエチルアミン(19mL,110ミリモル)のジメチルホルムアミド(DMF)(50mL)中の混合物を、0℃で室温まで一晩攪拌した。濃縮によってDMFを除去した後、反応混合物をシリカゲルクロマトグラフィー(20%〜80%EtOAc/ヘキサン、EtOAcおよび30%MeOH/EtOAc)により精製して、所望のアミド化合物41(または/およびエステル、7.3g、95%)を得た。
【0084】
化合物41(7.3g、22ミリモル)の酢酸(90mL)中溶液を、120℃にて3時間加熱した。反応混合物を室温まで冷却し、濃縮した。残渣をシリカゲルクロマトグラフィー(10%〜30%EtOAc/ヘキサン)により精製して、目的生成物である化合物42(2.0g、29%)を淡黄色の固体として得た。1H NMR(400MHz,CD3OD)δ 8.09(d,J=9.1Hz,2H)、7.47−7.4(m,6H)、7.16(d,J=9.1Hz,2H)、7.02(d,J=2.4Hz,1H)、7.02(dd,J=2.4,8.5Hz,1H)、5.18(s,2H):MS(ES−)m/z 316.4。
【0085】
化合物42(1.77g、5.58ミリモル)、1−ブロモ−3−クロロプロパン(1.6mL、17ミリモル)および炭酸カリウム(2.7g、20ミリモル)の混合物を10mLのDMSO中で室温にて2.5時間、TLCにより反応が完了するまで攪拌した。反応を水の添加によってクエンチし、次いでEtOAcで5、3回(five three times)抽出した。合した有機相をNa2SO4で乾燥させ、濾過し、濃縮させた。残渣を酢酸エチル−へキサンから結晶化させて、化合物43を淡色の固体(1.93g、88%)として得た。
【0086】
化合物43(1.10g、2.8ミリモル)の40mLのエタノール中の懸濁液に、5mLのジメチルアミンを−78℃で添加した。混合物を封管に入れて封をし、100℃にて10時間加熱した。混合物を濃縮して化合物44(1.1g、98%)を得た。生成物を次の段階の反応に進めた。
【0087】
化合物44(1.1g、2.7ミリモル)およびパラジウムヒドロキシド(7mg)の32mLのメタノールおよび5mLのジクロロメタン中の混合物を、水素バルーン下、室温にて一晩攪拌した。混合物をセライトを通じて濾過し、濾液を濃縮して粗生成物である化合物45を得た。理論収率を0.85gと仮定し、生成物をさらなる精製をせずに次の段階に直接進めた。
【0088】
化合物45(200mg、0.64ミリモル)、1−ブロモ−3−クロロプロパン(0.32mL、3.2ミリモル)および炭酸カリウム(470mg、3.4ミリモル)の混合物を4mLのDMSO中で室温にて2時間、TLCにより反応が完了するまで攪拌した。反応を水の添加によってクエンチし、次いでEtOAcで4回抽出した。合した有機相をNa2SO4で乾燥させ、濾過し、濃縮させた。残渣を分取TLC(5%MeOH/THF中、4%Et3N)により精製して、化合物46(170mg、68%)を得た。
【0089】
化合物46(30mg、0.08ミリモル)、N,N,N’−トリメチル−1,3−プロパン−ジアミン(50mg、0.43ミリモル)の混合物を2mLのエタノール中で、封管下、100℃にて10時間加熱した。混合物を濃縮させ、残渣をHPLCにより精製して、8mgの化合物34を白色固体(8mg、20%)として得た。1H NMR(400MHz,CD3OD)δ 8.10(d,J=8.8Hz,2H)、7.55(d,J=8.8Hz 1H)、7.25(d,J=2.3Hz,1H)、7.10(d,J=8.8Hz,2H)、7.00(dd,J=2.3,8.8Hz,1H)、4.14(t,J=5.9Hz,2H)、4.10(t,J=6.2Hz,2H)、2.69(m,4H)、2.56(m,4H)、2.39(s,12H)、2.36(s,3H)、2.04(m,4H)、1.77(m,2H)、MS(ES−)m/z+H 469.55。
【0090】
(実施例9)
(インビトロ生物活性)
(A.一次アッセイ)
一次アッセイは、ヒト胚腎臓上皮(HEK)細胞にTLR9をトランスフェクトすることを伴う。より具体的には、pGL3−エンハンサーベクター(Promega Corp.Madison,WI)にクローニングされた、E−セレクチン−1プロモーターに含められた3つのNF−kB結合部位の制御下、HEK293線維芽細胞(ATCC番号 CRP−1573、American Type Culture Collection,Manassas,VA)を、ヒトTLR9をコードするpcDNA3.1D/V5−His−TOPO(登録商標)プラスミド(Invitrogen,Carlsbad,CA)およびホタルルシフェラーゼを用いてトランスフェクトした(Taqポリメラーゼ増幅PCR産物として直接挿入した)。このアッセイで試験された化合物を下の表1に記載する。各々の化合物は、30分細胞に添加された後、ヌクレオチド配列5’−TCG TCG TTT TGT CGT TTT GTC GTT−3’(配列番号1)とともに、合成ホスホロチオエートオリゴヌクレオチドであるオリゴCpG2006(Hartmann et al.,J Immunol 164:1617−24(2000))で刺激した。細胞を、37℃にて一晩インキュベートした。ルシフェラーゼ基質Steady−Glo(登録商標)(Promega)をウェルに添加し、発光を測定してTLR9に駆動される(TLR9−driven)遺伝子活性化の程度を決定した。発光データを用いて各々の試料についてのIC50を算出した。IC50は、化合物の不在下で、言い換えれば完全な刺激を伴って観察される発光の50%まで発光を抑制する化合物の濃度として定義される。このアッセイの結果を下の表2に示す。比較化合物のIC50は、0.16マイクロモルであることが見出された。化合物1のIC50は、0.06マイクロモルであることが見出され、残りの化合物のIC50は、0.04マイクロモル未満であった。
【0091】
(B.二次アッセイ)
(1)HEK/TLR7活性化アッセイ:一次アッセイと同じ化合物を、以下の違いを伴って同じように試験した。HEK細胞株をトランスフェクトするために使用したプラスミドは、TLR9の代わりにヒトTLR7をコードし、オリゴCpG2006の代わりにR848を用いて細胞を刺激した。このアッセイの結果も表2に記載する。試験した各々の化合物について、上のHEK/TLR9活性化アッセイから得られるIC50は、HEK/TLR7アッセイから得られるIC50よりも有意に低く、TLR7に対するよりもTLR9に対する特異性が大きいことが示される。
【0092】
(2)脾細胞CpG1668アッセイ:一次アッセイで試験した一部の化合物は、ヌクレオチド配列5’−TCC ATG ACG TTC CTG ATG CT−3’(配列番号2)とともに、合成ホスホロチオエートオリゴヌクレオチドであるオリゴCpG1668(Krieg et al.Nature.374:546−9(1995))による刺激に応答するマウス脾臓IL−6産生の抑制についてもアッセイした。各々の化合物を分離された脾細胞に添加してから、刺激性オリゴヌクレオチドを添加した。細胞を培養中で72時間刺激し、IL−6レベルのELISA分析用に上清を取り出した。ここで、IC50は、化合物の不在下で、言い換えれば完全な刺激を伴って観察されるサイトカイン産生の50%までサイトカイン産生を抑制する化合物の濃度として定義される。比較化合物を除いて、試験した全ての化合物は、1μMより低いIC50でマウス脾細胞の刺激を阻害した。このアッセイの結果を下の表2に示す。
【0093】
(3)脾臓CT Gloアッセイ:化合物が生細胞への有害作用を有するかどうかを決定するため、分離されたマウス脾細胞を一部の化合物とともに72時間インキュベートした。インキュベーションの終わりにCellTiter−Glo(商標)試薬(Promega Corporation.Madison.WI)を添加し、製造業者の指図書によって発光を読み取った。この試薬は、ATPを細胞代謝の指標として測定する。死細胞は、シグナルを生じないものである。IC50は、化合物の不在下で観察される生存力の50%まで生存力を抑制する化合物の濃度として定義される。生存力に測定可能な程度に影響を及ぼした少数の化合物は、TLR9刺激に応答するサイトカイン放出に影響を及ぼす濃度よりもはるかに高い濃度で影響を及ぼした。結果を表2に示す。これらの結果は、TLR9抑制が細胞の死または脆弱性に起因するものでないことを示す。
【0094】
(4)PBMC CpG2216アッセイ:上記のように試験した化合物の一部は、ヌクレオチド配列5’−ggG GGA CGA TCG TCg ggg gG−3’(小文字はホスホロチオエート結合を示す、その他は全てホスホジエステル)(配列番号3)とともに、合成オリゴホスホロチオエート/ホスホジエステルオリゴヌクレオチドであるオリゴCpG2216(Vollmer et al.Eur J Immunol.34:251−62(2004))による刺激に対するヒト末梢血単核細胞(PBMC)α−インターフェロン応答の抑制についてもアッセイした。各々の化合物をFicoll−Paque(商標)(GE Healthcare,UK)に添加して、健常なボランティアドナーから単核細胞を分離した後、刺激性オリゴヌクレオチドを添加した。細胞を培養中で72時間刺激し、α−インターフェロンのELISA分析用に上清を取り出し、ELISA分析をVeriKine(商標)ヒトIFN−αELISAキット(Pestka Biomedical Laboratories,NJ)を用いて行った。このアッセイで試験した全ての化合物は、1μMより低いIC50でヒトPBMCの刺激を阻害した。
【0095】
下の表2は、試験した化合物、および上に記載され考察されるアッセイの各々から得られる結果を示す。
【0096】
【表3A】
【表3B】
【0097】
(実施例10)
(インビボ生物活性:短期CpG刺激)
短期CpG刺激アッセイ:上の実施例9に記載されるHEK/TLR9および脾細胞CpG1668アッセイにおいて効力を有すると特定された化合物を、インビボでの短期CpG刺激アッセイにおいて試験した。マウスに水中の化合物を経口投与し、90分以内に、30μgのオリゴCpG1668の皮下(s.c.)注射を投与した。2時間後、血清を採取し、ELISA分析を用いてIL−6についてアッセイした。このアッセイの結果を下の表3に示す。異なる日付に反復されたアッセイから得られる結果は、コンマで隔てられる複数の数字として示される。
【0098】
【表4】
【0099】
表3は、20mg/kgの化合物23、化合物30、または化合物26の経口投与の1.5時間後に少なくとも69%のIL−6の抑制が観察されたことを示す。少なくとも75%のIL−6の抑制が化合物23および化合物30の経口投与後に観察された。上記のインビトロアッセイで高レベルの抑制を提示したにもかかわらず、試験した2種類の化合物、化合物14および化合物20の投与後には抑制は観察されなかった。残りの化合物は様々なレベルのIL−6抑制を提示した。
【0100】
(実施例11)
(インビボ生物活性:リンパ節応答)
上の実施例10に記載されるアッセイされた2種類の化合物、化合物23および化合物30は、特に高レベルの抑制を示した。マウスにそれらの2種類の化合物の1種類またはヒドロキシクロロキン(「HCQ」)を、毎日20mg/kgかまたは60mg/kgで2週間経口投与した。インビボ投薬の後、リンパ節を取り出してインビトロで1μg/mlのCpG1668を用いて72時間刺激し、その後上清を取り出してELISAによりIL−6についてアッセイした。このアッセイの結果を図1に示す。投薬のレベルは図中に表示される各々の化合物名の後に示される。
【0101】
図1の結果は、化合物2と化合物23の両方がリンパ節においてIL−6放出を抑制したことを示し、化合物23はわずか20mg/kgの投与によっても特に高レベルの抑制を示した。
【0102】
(実施例12)
(インビボ生物活性:自発的狼瘡モデル)
(A.MRL/MpJ−faslpr/J 実験1)
この実験では、Jackson Laboratoriesより入手したMRL/MpJ−faslpr/Jマウス(「MRL/lpr」)に、水中20または60mg/kgの化合物23を経口的に投薬した。MRL/lprマウスは、リンパ球死に欠陥を有し、ヒト全身性紅斑性狼瘡(SLE)に見出される抗核抗体を含む自己抗体を自発的に発生させる。投薬は、5週齢に開始して週に5回毎日行った。血液および尿試料を1日目の投薬前に採取し、以降3〜4週毎に採取した。ELISAアッセイを用いて投薬の7週前および7週後の血清抗dsDNAを測定した。マウス抗dsDNA全Ig定性ELISAキット(Alpha Diagnostic International.San Antonio,TX)を用いてアッセイを行った。結果を図2に示す。図2は、この自己抗体への用量設定効果を示し、ビヒクルを投与された動物に対して450nmでの平均光学濃度(Optical Density)(OD450)が統計上有意に抑制された。統計的有意差は投薬10週でなお観察されたが、その後化合物治療群は12週およびそれ以降に、経口によってdH2Oを投薬されたビヒクル治療動物に一層類似するようになった。全体的な様子または体重測定で判断して、投薬量に関連する毒性の根拠は見られなかった。
【0103】
抗核抗体(「ANA」)も、この実験において、Antibodies Inc.製のスライド上の固定された試料として得た固定されたHepG2細胞を、上記のように化合物23を20または60mg/kgで投薬したマウスから得た希釈血清に曝露し、Antibodies Inc.により供給されるキット試薬をキットの説明書に従って使用してマウス免疫グロブリンについて染色することにより調査された。次に、蛍光染色の強度を顕微鏡で読み取り、スコアに−から+++の間の値を割り当てた。試料の識別表示はデータ収集のために隠した(blinded)。
【0104】
図3は、ANA試験の結果をグラフによって図示する。図3は、ビヒクル処理マウスが5〜12週齢の間に徐々にANA陽性となったが、一方、化合物処理マウスから得た血清が陽性ANA染色に対してはるかに遅い進行を示したという点で、ANA発生の用量依存性の抑制を示す。
【0105】
(B.MRL/lpr 実験2)
このSLE実験において、投薬群は、下の表4に記載される通りであった。化合物23の2種類の用量レベルをHCQおよびシクロホスファミド(Cytoxan(登録商標))と比較した。HCQおよびシクロホスファミドは、それぞれ、皮膚狼瘡および全身性狼瘡の治療に現在使用されている。シクロホスファミドは、一貫した血清抗dsDNAレベルの抑制を示した。この差は、ANOVAによりビヒクルに対して試験した場合に8週および12週の投薬時点で統計上有意であった。12週間の投薬前および投薬後の個々の動物血清について450ナノメートルでの光学濃度(OD450)のELISAアッセイから得られる結果を図4に示す。血清は、PBS中1:600希釈でアッセイした。(マウス抗dsDNA全Ig定性ELISAキット、Alpha Diagnostic International)。投薬は、経口(p.o.)かまたは腹腔内(i.p.)投与のいずれかにより行った。このアッセイから得られる個々のデータポイントを図4に図示する。図4は、12週で、ビヒクルと高用量の化合物23との間に平均dsDNAレベルの統計上の有意差があったことも示す。
【0106】
タンパク尿(BCAタンパク質アッセイにより測定)または皮膚病変への影響は、この実験でいずれの化合物によっても見られなかった。
【0107】
【表5】
【0108】
(実施例13)
(インビボ生物活性:NZBWF1/J自発的狼瘡モデル)
化合物23および化合物30のインビボ研究を、NZBWF1/Jマウスモデルで行った。マウスはJackson Laboratoriesより入手し、12の群に分け、水中の化合物を5ヶ月齢で開始し週に5日、経口的に投薬した。投薬の詳細を下の表5に提供する。
【0109】
【表6】
【0110】
この研究で試験した化合物の化合物23または化合物30はいずれも、20mg/kgまたは60mg/kgの化合物の投薬後の16週の治療の間、抗dsDNAへの効果を示さなかった。この研究の結果を図5に図示する。短期アッセイで化合物23よりも多少高い効力を示した化合物30は、60mg/kgの高用量で抗dsDNAをいくらか抑制したが、これは統計上有意でなかった。ANAを同じ実験で測定し、12頭のビヒクル治療動物のうち5頭しか+/−またはそれよりも高いスコアに達しなかったものの、12週の治療後のANAスコアの分布に化合物の効果はなかった。ANAスコアリングの結果を図6に図示する。
【0111】
化合物23および化合物30は、3〜4ヶ月の投薬後にNZBWF1/Jマウス系統において用量に関連する体重減少を一貫して生じた。筋肉、脳、腎臓および脾臓の組織学的検査は、ヘマトキシリンおよびエオシン染色において明らかな病態を示さなかった。
【0112】
60および20mg/kgの化合物23を用いる毎日経口投薬の結果として、SLEのMLR/lprモデルにおける抗dsDNAの抑制がもたらされた。化合物23はまた、抗核抗体発生の用量依存性の阻害も示した。シクロホスファミドはMRL/lprモデルにおいて抗dsDNAを抑制したが、一方ヒドロキシクロロキンは検出可能な効果がなかった。さらに、NZB/W系での化合物23の試験により、抗dsDNA、ANA、またはタンパク尿への効果は示されなかった。用量依存性の体重減少は、NZB/Wマウスにおいて数ヶ月の投薬の後に観察された。
【0113】
(実施例14)
(インビボ生物活性:実験的自己免疫性脳炎 多発性硬化症モデル)
実験的自己免疫性脳炎(EAE)は、ヒト多発性硬化症を模倣する、マウスにおいて誘導される自己免疫であり、自己免疫には、自己抗体、および中枢神経系抗原に対して標的とされる細胞応答が含まれる。EAEを誘導するため、8頭の雄C57BL/6Jマウス(Jackson Laboratories)の群を、完全フロイントアジュバント(CFA)中200μg/マウスの用量で、アミノ酸配列MEVGWYRSPFSRVVHLYRNGK(配列番号4)とともに、合成ペプチドMOGp35−55(Becher et al.J.Clin.Invest.112:1186−1191(2003))で0日目に皮下に免疫化した。同時に、動物に百日咳毒素を腹腔内に30ng/マウスで投与した。2日目に、百日咳注射を繰り返した。7日目に、マウスを不完全フロイントアジュバント中200μg/マウスのMOGp35−55で追加免疫した。9日目から、20mg/kgまたは60mg/kgの水中の化合物23を用いて経口胃管栄養法により毎日投与した。尾および四肢の麻痺の程度および位置を観察することにより症状をスコア化し、各々の8頭のマウス群の平均スコアを示す。
【0114】
この研究の結果を図7に示す。MOGに誘導されるEAEは、対照マウスと比較して、20mg/kgの化合物23を投与したマウスにおいて有意に抑制され、60mg/kgの同じ化合物を投与したマウスにおいてさらに抑制されたことを観察することができる。
【0115】
(実施例15)
(インビボ生物活性:コラーゲン誘発関節炎)
II型コラーゲン誘発関節炎モデルを、関節炎の治療のために化合物2をインビボで評価するために使用した。このモデルでは、関節炎を、完全フロイントアジュバントを用いて雄DBA/1J(Jackson Labs)マウスにおいて誘導した。最初のプライミング/免疫化用量の300μgの、等容積の完全フロイントアジュバントで乳化したウシII型コラーゲンを、尾の付け根から少なくとも5mmの皮下に(s.c.)注射する。
【0116】
免疫化後9日目に開始して、以降毎日週に5日、各々のマウスに、6mg/kg、20mg/kg、または60mg/kgの化合物2あるいはプラセボを1週間あたり5日、経口的に投与した。
【0117】
2回目の免疫化用量は、最初の免疫化の21日後に、等容積の不完全フロイントアジュバントで乳化した、200μgのウシII型コラーゲンの最初の免疫化部位よりも上に皮下注射により投与した。関節炎の進行をさらに促進するため、2回目の免疫化の3日後に、動物にリポ多糖(LPS)(PBS中1μg/マウス)の腹腔内注射を投与した。
【0118】
最初の免疫化用量の21、25、28、および31日後に、足腫脹のエビデンスについて動物を調べ、各々の時点の関節炎スコアを動物に割り当てた。このアッセイの結果を図8に示す。足腫脹の抑制は、6および20mg/kgを用いる投薬後に観察されたが、60mg/kgを用いる投薬後の結果は不明であった。異例の60mg/kgの結果から、この特定の動物モデルについての適切な上限投薬レベルを決定するさらなる研究の必要性が示唆される。
【0119】
(実施例16)
(インビボ生物活性:盲腸結紮穿刺術)
盲腸結紮穿刺(CLP)モデルは、腹部切開を行って盲腸(消化管の容易に単離される部分)を露出させ、盲腸の一部分を結紮する外科手技を伴う。次に、盲腸を穿刺し、少量の腸内容物を押し出す。切開を閉じ、動物に蘇生のための生理食塩水および抗生物質を与える。敗血症は、主に押し出された腸内容物からの殺菌に起因して発症し、疾患の発症は、盲腸の穿刺のサイズおよび数に依存する迅速性および重篤度とともに起こる。
【0120】
(A.盲腸結紮穿刺術)
ケタミン、キシラジンおよびアセプロマジンの組合せの腹腔内投与によりマウスにおいて麻酔を誘導する。動物の腹部領域は短く刈ってよく、体温を測定するためのトランスポンダを皮下に埋め込む。この手順は無菌生存手術に適した領域で行う。この手順の間中、動物は加温パッドの上に置く。腹部領域は、ポビドン/ヨードおよびアルコールで最低3回交互に拭いて準備する。
【0121】
正中切開を行って腹部を開き、盲腸を露出する。生理食塩水で湿らせた綿棒(cotton-tipped applicators)を用いて盲腸を腹腔から摘出する。回盲弁のすぐ遠位で4−0縫合糸(sutures)を用いて盲腸の先端を結紮する。盲腸の内容物を一端に移動させて、次に盲腸を滅菌針で穿刺する。圧力を加えて少量の物質を盲腸から腹膜腔に押し出す。結紮は所定の位置に残して動かさない。筋肉および筋膜を連続的な4−0縫合糸で閉じ、皮膚を創傷ステープル(wound staples)で閉じる。
【0122】
手術直後に、体重100gあたり5mlの暖かい蘇生のための生理食塩水を皮下注射する。温度および体重のベースライン読みを記録する。手術1時間後、20mg/kg用量の抗生物質モキシフロキサシンを試験されているいずれかの抗敗血症薬剤候補とともに腹腔内に投与する。研究の持続のために抗生物質を24時間間隔で投与する。投与する抗生物質の用量は、CLPにより放出される細菌を根絶することを目的としているのではなく、その代わりにヒトが抗生物質を投与されるがそれらが効果のない場合の臨床シナリオをモデル化するために使用される。モキシフロキサシンはQT間隔の延長に関係しているが、好ましくない薬物相互作用が回避される限り、臨床使用に安全であると考えられ(Torres et al.,J Surg Res.125:88−93(2005))、該抗生物質は別のマウス敗血症モデルにおいて安全かつ効果的であると既に証明されている(Alkorta et al.,Int J.Antimicrob.Agents 25(2):163−7,(2005))。
【0123】
(B.試験化合物の投与)
化合物は、手術の約1時間後に抗生物質注射の時に投与されるが、このタイミングは変動する可能性がある。化合物は、候補化合物の薬物動態特性に応じて1日に2回投与される。試験化合物の投与は、投与のために確立されたガイドラインに従って、次の経路:尾静脈からの経口、腹腔内、皮下、または静脈注射のいずれかによって行われる。
【0124】
10頭前後の動物の群が使用される。薬物評価実験は、盲腸結紮穿刺を経験し、ビヒクルのみを投与される1つの群(対照群)、およびCLPを経験し、試験治療化合物を投与される群を有する。化合物は手術後1時間に投与され、動物をその日中2時間おきに生存についてモニターする。トランスポンダを介して2時間おきに体温をとり、動物の健康を各時点でモニターする。体重は4時間間隔で量る。毎日少なくとも8時間の期間動物をモニターする。サイトカインレベルの測定のために血清試料を採取する。死亡数をモニターし、薬物治療の有効性の尺度として使用する。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
(式中、R5、R6またはR7のうちの1つが式(a):
【化2】
の基であり、かつ、R5が(a)である場合、R6およびR7は双方ともHであり、R6が(a)である場合、R5およびR7は双方ともHであり、R7が(a)である場合、R5およびR6は双方ともHであり、
iおよびjは同一であって、かつ0、1、2、3または4であり、
R1およびR4は同一であって、かつH、CH3およびCH2CH3からなる群から選択され、
R2はCH3であり、R3は(CH2)hN(CH3)2(式中、hが2、3または4である)および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは、
R2およびR3は同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
(CH2)nN(CH3)2(式中、nは2、3または4である)、
(CH2)pO(CH2)qN(CH3)2(式中、pおよびqは同一であって、かつ2または3である)、
式(b):
【化3】
(式中、uは0または1である)の基、および
式(c):
【化4】
(式中、vは0または1であり、ZはNまたはCHであり、かつXはOまたはNCH3である)の基からなる群から選択されるか、又は、
R1−N−R2およびR3−N−R4は同一であって、かつ
式(d):
【化5】
の基および
式(e):
【化6】
(式中、rは1、2または3であり、
YはCHまたはNであり、
R8はH、CH3、CH(CH3)2、N(CH3)2、CH2OCH3または式(f):
【化7】
(式中、tは、0または1である)の基であり、
R9はH、CH2OCH3、または式(f)の基である)
の基からなる群から選択される)
の化合物またはその医薬上許容される塩。
【請求項2】
iおよびjが双方とも1である請求項1に記載の化合物。
【請求項3】
R2およびR3が同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
式(b)の基、および
式(c)の基からなる群から選択される請求項1に記載の化合物。
【請求項4】
R1−N−R2およびR3−N−R4が同一であって、かつ
式(g):
【化8】
の基、式(h):
【化9】
の基、式(i):
【化10】
の基、式(j):
【化11】
の基、式(k):
【化12】
の基および式(m):
【化13】
の基からなる群から選択される請求項1に記載の化合物。
【請求項5】
iおよびjが双方とも1であり、
R1およびR4が同一であって、かつHおよびCH3からなる群から選択され、
R2がCH3であり、R3が(CH2)2N(CH3)2および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは
R2およびR3が、
a)(CH2)kCH3(式中、kは0、1または2である)、
b)(CH2)mN(CH2CH3)2(式中、mは2または3である)、
c)式(b)の基、および
d)式(c)の基からなる群から選択されるか、または
R1−N−R2およびR3−N−R4が同一であって、かつ式(g)の基、式(h)の基、式(i)の基、式(j)の基、式(k)の基および式(m)の基からなる群から選択される請求項1に記載の化合物。
【請求項6】
R5が式(a)の基であり、R6およびR7が各々Hである請求項1に記載の化合物。
【請求項7】
本化合物が、
【化14】
(化合物1)、
【化15】
(化合物2)、
【化16】
(化合物3)、
【化17】
(化合物4)、
【化18】
(化合物5)、
【化19】
(化合物6)、
【化20】
(化合物7)、
【化21】
(化合物8)、
【化22】
(化合物9)、
【化23】
(化合物10)、
【化24】
(化合物11)、
【化25】
(化合物12)、
【化26】
(化合物13)、
【化27】
(化合物14)、
【化28】
(化合物15)、
【化29】
(化合物16)、
【化30】
(化合物17)、
【化31】
(化合物18)、
【化32】
(化合物19)、
【化33】
(化合物20)、
【化34】
(化合物21)、
【化35】
(化合物22)、
【化36】
(化合物23)、
【化37】
(化合物24)、
【化38】
(化合物25)、
【化39】
(化合物26)、
【化40】
(化合物27)、
【化41】
(化合物28)、
【化42】
(化合物29)、
【化43】
(化合物30)、
【化44】
(化合物31)、
【化45】
(化合物32)、
【化46】
(化合物33)、
【化47】
(化合物34)、
【化48】
(化合物35)、
【化49】
(化合物36)、
【化50】
(化合物37)、および
【化51】
(化合物38)
またはその医薬上許容される塩からなる群から選択される請求項1に記載の化合物。
【請求項8】
本化合物が、
【化52】
(化合物13)、
【化53】
(化合物16)、
【化54】
(化合物22)、
【化55】
(化合物26)、
【化56】
(化合物27)、
【化57】
(化合物30)、
【化58】
(化合物23)、
【化59】
(化合物34)、および
【化60】
(化合物35)
またはその医薬上許容される塩からなる群から選択される請求項1に記載の化合物。
【請求項9】
式:
【化61】
(化合物23)
を有する化合物またはその医薬上許容される塩。
【請求項10】
式(I):
【化62】
(式中、R5、R6またはR7のうちの1つが、式(a):
【化63】
の基であり、かつ、R5が(a)である場合、R6およびR7は双方ともHであり、R6が(a)である場合、R5およびR7は双方ともHであり、R7が(a)である場合、R5およびR6は双方ともHであり、
iおよびjは同一であって、かつ0、1、2、3または4であり、
R1およびR4は同一であって、かつH、CH3およびCH2CH3からなる群から選択され、
R2はCH3であり、R3は(CH2)hN(CH3)2(hが2、3、または4である)および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは
R2およびR3は同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
(CH2)nN(CH3)2(式中、nは2、3または4である)、
(CH2)pO(CH2)qN(CH3)2(式中、pおよびqは同一であって、かつ2または3である)、
式(b):
【化64】
(式中、uは0または1である)の基、および
式(c):
【化65】
(式中、vは0または1であり、ZはNまたはCHであり、XはOまたはNCH3である)の基からなる群から選択されるか、又は
R1−N−R2およびR3−N−R4は同一であって、かつ
式(d):
【化66】
の基および
式(e):
【化67】
(式中、rは1、2または3であり、
YはCHまたはNであり、
R8はH、CH3、CH(CH3)2、N(CH3)2、CH2OCH3または式(f):
【化68】
(式中、tは0または1である)の基であり、
R9はH、CH2OCH3または式(f)の基である)
の基からなる群から選択される)
の化合物またはその医薬上許容される塩を含む医薬組成物。
【請求項11】
前記化合物が、
【化69】
(化合物1)、
【化70】
(化合物2)、
【化71】
(化合物3)、
【化72】
(化合物4)、
【化73】
(化合物5)、
【化74】
(化合物6)、
【化75】
(化合物7)、
【化76】
(化合物8)、
【化77】
(化合物9)、
【化78】
(化合物10)、
【化79】
(化合物11)、
【化80】
(化合物12)、
【化81】
(化合物13)、
【化82】
(化合物14)、
【化83】
(化合物15)、
【化84】
(化合物16)、
【化85】
(化合物17)、
【化86】
(化合物18)、
【化87】
(化合物19)、
【化88】
(化合物20)、
【化89】
(化合物21)、
【化90】
(化合物22)、
【化91】
(化合物23)、
【化92】
(化合物24)、
【化93】
(化合物25)、
【化94】
(化合物26)、
【化95】
(化合物27)、
【化96】
(化合物28)、
【化97】
(化合物29)、
【化98】
(化合物30)、
【化99】
(化合物31)、
【化100】
(化合物32)、
【化101】
(化合物33)、
【化102】
(化合物34)、
【化103】
(化合物35)、
【化104】
(化合物36)、
【化105】
(化合物37)、および
【化106】
(化合物38)
からなる群から選択される化合物またはその医薬上許容される塩である請求項10に記載の医薬組成物。
【請求項12】
前記化合物が、
【化107】
(化合物13)、
【化108】
(化合物16)、
【化109】
(化合物22)、
【化110】
(化合物26)、
【化111】
(化合物27)、
【化112】
(化合物30)、
【化113】
(化合物23)、
【化114】
(化合物34)、および
【化115】
(化合物35)
なる群から選択される化合物またはその医薬上許容される塩である請求項10に記載の医薬組成物。
【請求項13】
前記化合物が、式:
【化116】
(化合物23)
を有する化合物またはその医薬上許容される塩である請求項10に記載の医薬組成物。
【請求項14】
被験体において、敗血症、狼瘡、関節リウマチおよび多発性硬化症からなる群から選択される免疫障害を予防または治療する方法であって、前記被験体に、
【化117】
(化合物13)、
【化118】
(化合物16)、
【化119】
(化合物22)、
【化120】
(化合物23)、
【化121】
(化合物26)、
【化122】
(化合物27)、
【化123】
(化合物30)、
【化124】
(化合物34)、および
【化125】
(化合物35)
からなる群から選択される化合物またはその医薬上許容される塩を有効量で投与することを含む方法。
【請求項15】
前記被験体がヒトである請求項14に記載の方法。
【請求項16】
前記化合物が、
【化126】
(化合物30)、
【化127】
(化合物23)、および
【化128】
(化合物26)
からなる群から選択される化合物またはその医薬上許容される塩である請求項14に記載の方法。
【請求項17】
前記化合物が、
【化129】
(化合物23)
の化合物またはその医薬上許容される塩である請求項14に記載の方法。
【請求項18】
前記免疫障害が敗血症である請求項14に記載の方法。
【請求項19】
前記免疫障害が、狼瘡、関節リウマチおよび多発性硬化症からなる群から選択される請求項14に記載の方法。
【請求項20】
被験体において狼瘡を予防または治療する方法であって、被験体に、
【化130】
(化合物13)、
【化131】
(化合物16)、
【化132】
(化合物22)、
【化133】
(化合物23)、
【化134】
(化合物26)、
【化135】
(化合物27)、
【化136】
(化合物30)、
【化137】
(化合物34)、および
【化138】
(化合物35)
からなる群から選択される化合物またはその医薬上許容される塩を有効量で投与することを含む方法。
【請求項21】
前記化合物が、
【化139】
(化合物23)
の化合物またはその医薬上許容される塩である請求項20に記載の方法。
【請求項22】
敗血症、狼瘡、関節リウマチおよび多発性硬化症からなる群から選択される免疫障害を治療するための薬物の製造のための請求項8に記載の化合物の使用。
【請求項23】
前記免疫障害が敗血症である請求項22に記載の化合物の使用。
【請求項1】
式(I):
【化1】
(式中、R5、R6またはR7のうちの1つが式(a):
【化2】
の基であり、かつ、R5が(a)である場合、R6およびR7は双方ともHであり、R6が(a)である場合、R5およびR7は双方ともHであり、R7が(a)である場合、R5およびR6は双方ともHであり、
iおよびjは同一であって、かつ0、1、2、3または4であり、
R1およびR4は同一であって、かつH、CH3およびCH2CH3からなる群から選択され、
R2はCH3であり、R3は(CH2)hN(CH3)2(式中、hが2、3または4である)および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは、
R2およびR3は同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
(CH2)nN(CH3)2(式中、nは2、3または4である)、
(CH2)pO(CH2)qN(CH3)2(式中、pおよびqは同一であって、かつ2または3である)、
式(b):
【化3】
(式中、uは0または1である)の基、および
式(c):
【化4】
(式中、vは0または1であり、ZはNまたはCHであり、かつXはOまたはNCH3である)の基からなる群から選択されるか、又は、
R1−N−R2およびR3−N−R4は同一であって、かつ
式(d):
【化5】
の基および
式(e):
【化6】
(式中、rは1、2または3であり、
YはCHまたはNであり、
R8はH、CH3、CH(CH3)2、N(CH3)2、CH2OCH3または式(f):
【化7】
(式中、tは、0または1である)の基であり、
R9はH、CH2OCH3、または式(f)の基である)
の基からなる群から選択される)
の化合物またはその医薬上許容される塩。
【請求項2】
iおよびjが双方とも1である請求項1に記載の化合物。
【請求項3】
R2およびR3が同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
式(b)の基、および
式(c)の基からなる群から選択される請求項1に記載の化合物。
【請求項4】
R1−N−R2およびR3−N−R4が同一であって、かつ
式(g):
【化8】
の基、式(h):
【化9】
の基、式(i):
【化10】
の基、式(j):
【化11】
の基、式(k):
【化12】
の基および式(m):
【化13】
の基からなる群から選択される請求項1に記載の化合物。
【請求項5】
iおよびjが双方とも1であり、
R1およびR4が同一であって、かつHおよびCH3からなる群から選択され、
R2がCH3であり、R3が(CH2)2N(CH3)2および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは
R2およびR3が、
a)(CH2)kCH3(式中、kは0、1または2である)、
b)(CH2)mN(CH2CH3)2(式中、mは2または3である)、
c)式(b)の基、および
d)式(c)の基からなる群から選択されるか、または
R1−N−R2およびR3−N−R4が同一であって、かつ式(g)の基、式(h)の基、式(i)の基、式(j)の基、式(k)の基および式(m)の基からなる群から選択される請求項1に記載の化合物。
【請求項6】
R5が式(a)の基であり、R6およびR7が各々Hである請求項1に記載の化合物。
【請求項7】
本化合物が、
【化14】
(化合物1)、
【化15】
(化合物2)、
【化16】
(化合物3)、
【化17】
(化合物4)、
【化18】
(化合物5)、
【化19】
(化合物6)、
【化20】
(化合物7)、
【化21】
(化合物8)、
【化22】
(化合物9)、
【化23】
(化合物10)、
【化24】
(化合物11)、
【化25】
(化合物12)、
【化26】
(化合物13)、
【化27】
(化合物14)、
【化28】
(化合物15)、
【化29】
(化合物16)、
【化30】
(化合物17)、
【化31】
(化合物18)、
【化32】
(化合物19)、
【化33】
(化合物20)、
【化34】
(化合物21)、
【化35】
(化合物22)、
【化36】
(化合物23)、
【化37】
(化合物24)、
【化38】
(化合物25)、
【化39】
(化合物26)、
【化40】
(化合物27)、
【化41】
(化合物28)、
【化42】
(化合物29)、
【化43】
(化合物30)、
【化44】
(化合物31)、
【化45】
(化合物32)、
【化46】
(化合物33)、
【化47】
(化合物34)、
【化48】
(化合物35)、
【化49】
(化合物36)、
【化50】
(化合物37)、および
【化51】
(化合物38)
またはその医薬上許容される塩からなる群から選択される請求項1に記載の化合物。
【請求項8】
本化合物が、
【化52】
(化合物13)、
【化53】
(化合物16)、
【化54】
(化合物22)、
【化55】
(化合物26)、
【化56】
(化合物27)、
【化57】
(化合物30)、
【化58】
(化合物23)、
【化59】
(化合物34)、および
【化60】
(化合物35)
またはその医薬上許容される塩からなる群から選択される請求項1に記載の化合物。
【請求項9】
式:
【化61】
(化合物23)
を有する化合物またはその医薬上許容される塩。
【請求項10】
式(I):
【化62】
(式中、R5、R6またはR7のうちの1つが、式(a):
【化63】
の基であり、かつ、R5が(a)である場合、R6およびR7は双方ともHであり、R6が(a)である場合、R5およびR7は双方ともHであり、R7が(a)である場合、R5およびR6は双方ともHであり、
iおよびjは同一であって、かつ0、1、2、3または4であり、
R1およびR4は同一であって、かつH、CH3およびCH2CH3からなる群から選択され、
R2はCH3であり、R3は(CH2)hN(CH3)2(hが2、3、または4である)および(CH2)2O(CH2)2O(CH2)2N(CH3)2からなる群から選択されるか、若しくは
R2およびR3は同一であって、かつ
(CH2)kCH3(式中、kは0、1または2である)、
(CH2)mN(CH2CH3)2(式中、mは2または3である)、
(CH2)nN(CH3)2(式中、nは2、3または4である)、
(CH2)pO(CH2)qN(CH3)2(式中、pおよびqは同一であって、かつ2または3である)、
式(b):
【化64】
(式中、uは0または1である)の基、および
式(c):
【化65】
(式中、vは0または1であり、ZはNまたはCHであり、XはOまたはNCH3である)の基からなる群から選択されるか、又は
R1−N−R2およびR3−N−R4は同一であって、かつ
式(d):
【化66】
の基および
式(e):
【化67】
(式中、rは1、2または3であり、
YはCHまたはNであり、
R8はH、CH3、CH(CH3)2、N(CH3)2、CH2OCH3または式(f):
【化68】
(式中、tは0または1である)の基であり、
R9はH、CH2OCH3または式(f)の基である)
の基からなる群から選択される)
の化合物またはその医薬上許容される塩を含む医薬組成物。
【請求項11】
前記化合物が、
【化69】
(化合物1)、
【化70】
(化合物2)、
【化71】
(化合物3)、
【化72】
(化合物4)、
【化73】
(化合物5)、
【化74】
(化合物6)、
【化75】
(化合物7)、
【化76】
(化合物8)、
【化77】
(化合物9)、
【化78】
(化合物10)、
【化79】
(化合物11)、
【化80】
(化合物12)、
【化81】
(化合物13)、
【化82】
(化合物14)、
【化83】
(化合物15)、
【化84】
(化合物16)、
【化85】
(化合物17)、
【化86】
(化合物18)、
【化87】
(化合物19)、
【化88】
(化合物20)、
【化89】
(化合物21)、
【化90】
(化合物22)、
【化91】
(化合物23)、
【化92】
(化合物24)、
【化93】
(化合物25)、
【化94】
(化合物26)、
【化95】
(化合物27)、
【化96】
(化合物28)、
【化97】
(化合物29)、
【化98】
(化合物30)、
【化99】
(化合物31)、
【化100】
(化合物32)、
【化101】
(化合物33)、
【化102】
(化合物34)、
【化103】
(化合物35)、
【化104】
(化合物36)、
【化105】
(化合物37)、および
【化106】
(化合物38)
からなる群から選択される化合物またはその医薬上許容される塩である請求項10に記載の医薬組成物。
【請求項12】
前記化合物が、
【化107】
(化合物13)、
【化108】
(化合物16)、
【化109】
(化合物22)、
【化110】
(化合物26)、
【化111】
(化合物27)、
【化112】
(化合物30)、
【化113】
(化合物23)、
【化114】
(化合物34)、および
【化115】
(化合物35)
なる群から選択される化合物またはその医薬上許容される塩である請求項10に記載の医薬組成物。
【請求項13】
前記化合物が、式:
【化116】
(化合物23)
を有する化合物またはその医薬上許容される塩である請求項10に記載の医薬組成物。
【請求項14】
被験体において、敗血症、狼瘡、関節リウマチおよび多発性硬化症からなる群から選択される免疫障害を予防または治療する方法であって、前記被験体に、
【化117】
(化合物13)、
【化118】
(化合物16)、
【化119】
(化合物22)、
【化120】
(化合物23)、
【化121】
(化合物26)、
【化122】
(化合物27)、
【化123】
(化合物30)、
【化124】
(化合物34)、および
【化125】
(化合物35)
からなる群から選択される化合物またはその医薬上許容される塩を有効量で投与することを含む方法。
【請求項15】
前記被験体がヒトである請求項14に記載の方法。
【請求項16】
前記化合物が、
【化126】
(化合物30)、
【化127】
(化合物23)、および
【化128】
(化合物26)
からなる群から選択される化合物またはその医薬上許容される塩である請求項14に記載の方法。
【請求項17】
前記化合物が、
【化129】
(化合物23)
の化合物またはその医薬上許容される塩である請求項14に記載の方法。
【請求項18】
前記免疫障害が敗血症である請求項14に記載の方法。
【請求項19】
前記免疫障害が、狼瘡、関節リウマチおよび多発性硬化症からなる群から選択される請求項14に記載の方法。
【請求項20】
被験体において狼瘡を予防または治療する方法であって、被験体に、
【化130】
(化合物13)、
【化131】
(化合物16)、
【化132】
(化合物22)、
【化133】
(化合物23)、
【化134】
(化合物26)、
【化135】
(化合物27)、
【化136】
(化合物30)、
【化137】
(化合物34)、および
【化138】
(化合物35)
からなる群から選択される化合物またはその医薬上許容される塩を有効量で投与することを含む方法。
【請求項21】
前記化合物が、
【化139】
(化合物23)
の化合物またはその医薬上許容される塩である請求項20に記載の方法。
【請求項22】
敗血症、狼瘡、関節リウマチおよび多発性硬化症からなる群から選択される免疫障害を治療するための薬物の製造のための請求項8に記載の化合物の使用。
【請求項23】
前記免疫障害が敗血症である請求項22に記載の化合物の使用。
【図1】

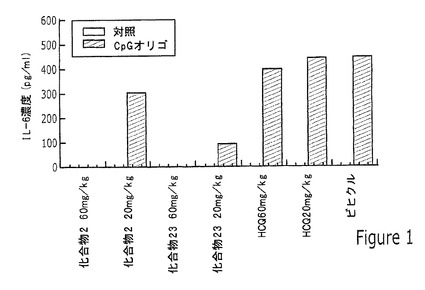
【図2】

【図3A−3C】

【図3D−3F】
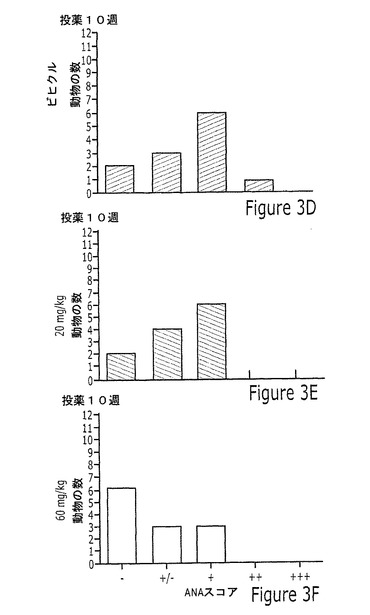
【図3G−3I】
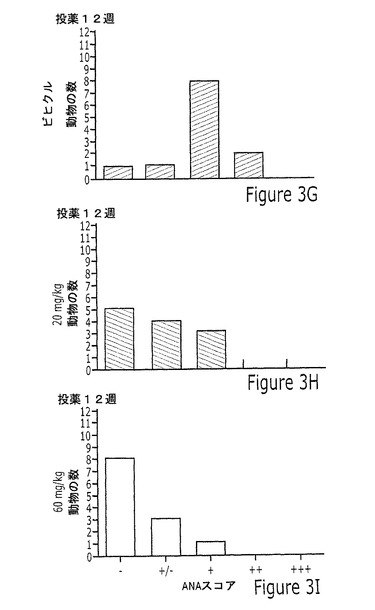
【図4】
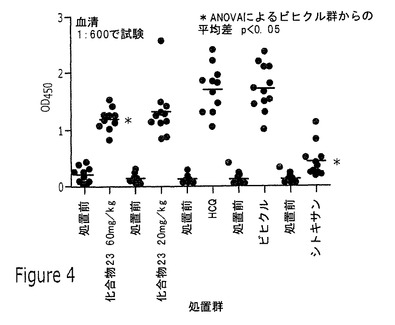
【図5】
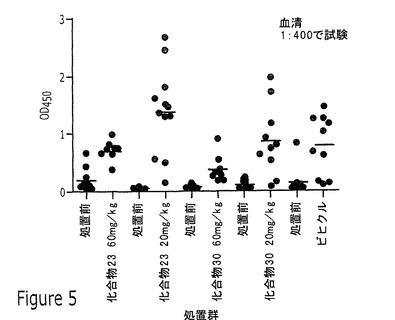
【図6A−6C】
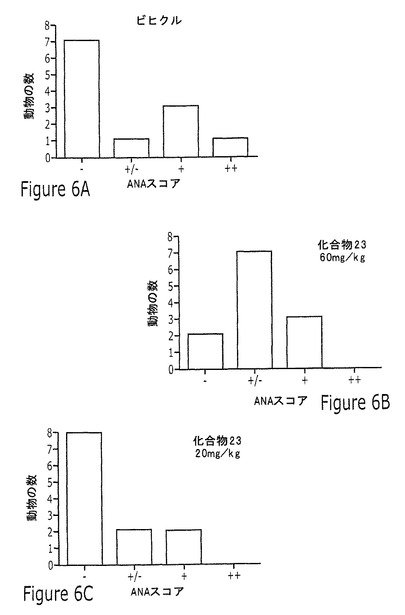
【図6D.6E】
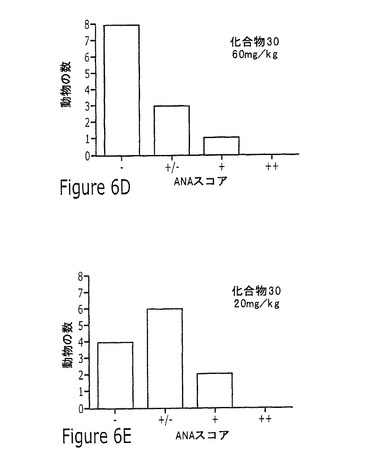
【図7】
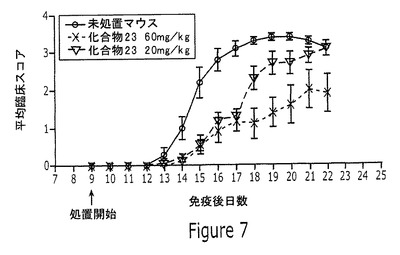
【図8】
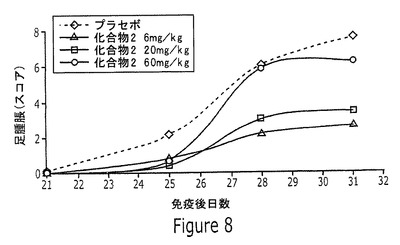
【図2】
【図3A−3C】
【図3D−3F】
【図3G−3I】
【図4】
【図5】
【図6A−6C】
【図6D.6E】
【図7】
【図8】
【公表番号】特表2012−504133(P2012−504133A)
【公表日】平成24年2月16日(2012.2.16)
【国際特許分類】
【出願番号】特願2011−529267(P2011−529267)
【出願日】平成21年9月25日(2009.9.25)
【国際出願番号】PCT/US2009/058398
【国際公開番号】WO2010/036905
【国際公開日】平成22年4月1日(2010.4.1)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
【公表日】平成24年2月16日(2012.2.16)
【国際特許分類】
【出願日】平成21年9月25日(2009.9.25)
【国際出願番号】PCT/US2009/058398
【国際公開番号】WO2010/036905
【国際公開日】平成22年4月1日(2010.4.1)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
[ Back to top ]