
ベンゾチアジン化合物ジマレイン酸塩の新規結晶
【課題】本発明は、安定性に優れ、取り扱いが容易である、アレルギー性疾患及び炎症性疾患の予防及び/又は治療剤に有用な新規結晶形を提供することを目的とする。
【解決手段】X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(β型結晶)、及びX線回折スペクトルにおいて、回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(γ型結晶)。
【解決手段】X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(β型結晶)、及びX線回折スペクトルにおいて、回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(γ型結晶)。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ベンゾチアジン化合物のジマレイン酸塩の新規結晶及びその製造方法、並びに該結晶を含有するアレルギー疾患又は炎症性疾患の予防及び/又は治療剤に関する。
【背景技術】
【0002】
次式(1):
【0003】
【化1】

【0004】
(式中、lは1又は2の整数を示し、mは1又は2の整数を示し、nは1〜4の整数を示す)
で表されるベンゾチアジン化合物は、抗ヒスタミン作用及び抗ロイコトリエン作用を有し、アレルギー性鼻炎、アレルギー性結膜炎等のアレルギー性疾患、喘息、乾癬、リウマチ及び炎症性大腸炎等の炎症性疾患の予防又は治療薬として有用である(特許文献1)。
【0005】
また、該ベンゾチアジン化合物は、その構造中に塩基性窒素原子を3個有することから、種々の酸付加塩の形態になり、特許文献1においてはジマレイン酸塩として単離されている(特許文献1の実施例28)。
【0006】
一方、医薬品原体は、医薬品製造の容易性に加えて、製造スケール等が変化した場合であっても、常に同程度の収率と純度をもって製造できるという一定の再現性を有することが望まれる。そして、それに加えて、医薬品原体の結晶性、非吸湿性、水溶性、可視光及び紫外線に対する安定性、温度及び湿度に対する分解度の低さなどの様々な条件を満たすことが必要とされる。医薬として活性な化合物がこの様な医薬品原体として適当な物理化学的性質を有さない場合、通常種々の改善検討がなされる。すなわち、結晶化、水溶性、バイオアベイラビリティ、光安定性、保存安定性、非吸湿性等の改善を目的として、化合物の塩、水和物、結晶形の検討がしばしば行われており、化合物は改善された結果、適当な物理化学的性質を有する医薬品原体として医薬品製造に用いられている。
【0007】
例えば、油状物や不定形の化合物を結晶化することで、取り扱いやすくなるので、品質管理が容易となる一方で、その化合物の溶解度やバイオアベイラビリティに差が出ることが報告されている(非特許文献1参照)。また、化合物の結晶形が安定形であれば、製剤工程において結晶形変化のリスクが少なくなる。また、化合物の結晶化が容易であれば、再結晶による目的化合物の精製収率を向上させることができ、生産過程での品質管理も有利に進めることが可能である(非特許文献2)。化合物の水溶性の改善は経口剤のバイオアベイラビリティを向上させ、注射剤や点眼剤の製造工程を容易にすることができる。化合物の熱及び光安定性の改善は医薬の品質を保証する上で有用であり、化合物の非吸湿性は原体の保管や製剤化工程において有利である。このようなことから、医薬品製造にあたっては最適な物理化学的性質を有する医薬原体を見出すことが非常に重要であり、そのために様々な医薬原体の形態を徹底的に検討することが望まれている。しかしながら、新たに生成した塩、水和物、結晶形がいかなる物理化学的性状を有するかを事前に予測することは困難である。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特許第3588126号公報
【非特許文献】
【0009】
【非特許文献1】Rharmaceutical Research,945,12(7),1995
【非特許文献2】International Journal of Pharmaceutics,209,105,1994
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、前記式(1)で表されるベンゾチアジン化合物のジマレイン酸塩の新たな結晶形を提供することを目的とする。
【課題を解決するための手段】
【0011】
そこで、本発明者らは、前記ベンゾチアジン化合物のジマレイン酸塩について種々検討を行った結果、既に前記特許文献1で報告されている結晶形(α型結晶)とは異なる新規結晶形β型及びγ型が存在することを見出し、本発明を完成するに至った。さらに、本発明者らは、該新規結晶体がアレルギー性疾患(特に、アレルギー性結膜炎)の治療又は予防に有効に用いることができることを見出し、本発明を完成するに至った。
【0012】
すなわち、本発明は、下記の通りである。
[1]X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(β型結晶)。
[2]融点が約112℃±1.0℃である、前記[1]に記載の結晶。
[3]X線回折スペクトルにおいて、回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(γ型結晶)。
[4]融点が約126.2±0.5℃である、前記[3]に記載の結晶。
[5]前記[1]〜[4]のいずれか一つに記載の結晶を含有する医薬組成物。
[6]前記[1]〜[4]のいずれか一つに記載の結晶を含有するアレルギー性疾患の予防及び/又は治療剤。
[7]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[6]に記載の予防及び/又は治療剤。
[8]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[7]に記載の予防及び/又は治療剤。
【0013】
[9]プロパノールを溶媒として用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(β型結晶)の製造方法。
[10]前記プロパノールが、1−プロパノール又は2−プロパノールである、前記[9]に記載の製造方法。
[11]さらに、メタノールを溶媒として用いて結晶化を行う、前記[9]又は[10]に記載の製造方法。
[12]アルコール系溶媒とジアルキルケトン溶媒との混合溶媒を用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(γ型結晶)の製造方法。
[13]前記アルコール系溶媒が、メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール又はヘプタノールである、前記[12]に記載の製造方法。
[14]前記ジアルキルケトン溶媒が、次式(4):
【0014】
【化2】
【0015】
〔式中、R1及びR2は、互いに同一又は異なっていてもよいC1-6アルキル基を示す。〕
である、前記[12]又は[13]に記載の製造方法。
[15]前記ジアルキルケトン溶媒が、メチルイソブチルケトンである、前記[12]又は[13]に記載の製造方法。
[16]アレルギー性疾患の予防及び/又は治療用の、前記[1]〜[4]のいずれか一つに記載の結晶。
[17]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[16]に記載の結晶。
[18]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[17]に記載の結晶。
【0016】
[19]前記[1]〜[4]のいずれか一つに記載の結晶の、アレルギー性疾患の予防及び/又は治療剤製造のための使用。
[20]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[19]に記載の使用。
[21]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[20]に記載の使用。
【0017】
[22]前記[1]〜[4]のいずれか一つに記載の結晶を投与することを特徴とするアレルギー性疾患の予防及び/又は治療方法。
[23]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[22]に記載の方法。
[24]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[23]に記載の方法。
【発明の効果】
【0018】
本発明に係るベンゾチアジン化合物のジマレイン酸塩新規結晶形β型結晶及びγ型結晶は、安定性に優れているため、取り扱いが容易であり、アレルギー疾患又は炎症性疾患の予防及び/又は治療剤の製造に適している。また、該新規結晶形β型結晶及びγ型結晶は、アレルギー性疾患又は炎症性疾患に対して優れた治療及び予防効果を示す。
【図面の簡単な説明】
【0019】
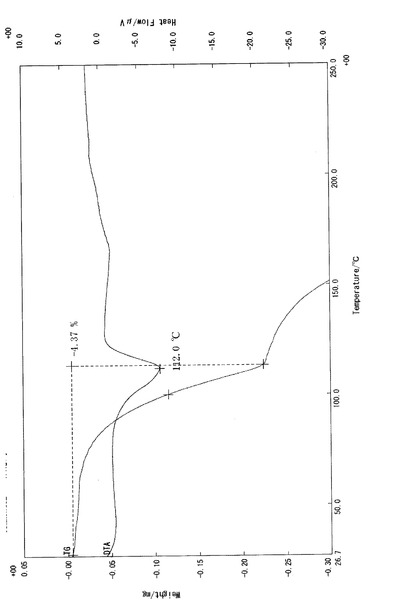
【図1】実施例3−(i)で得られたβ型結晶(3b)の熱分析データ(TG−DTA測定)を示すチャートである。
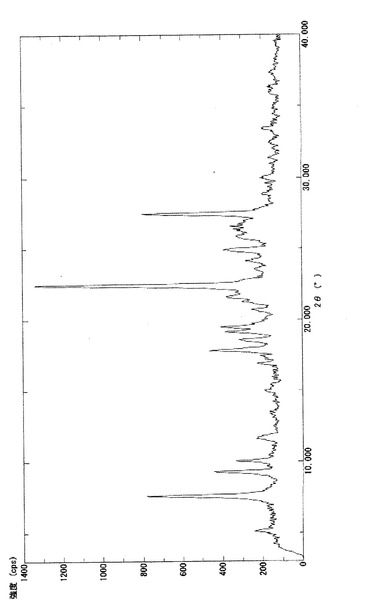
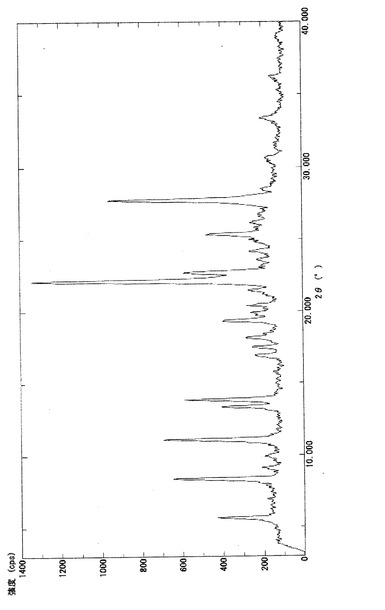
【図2】実施例3−(i)で得られたβ型結晶(3b)の粉末X線回折パターンを示す図である。
【図3】実施例3−(ii)で得られたγ型結晶(3c)の熱分析データ(TG−DTA測定)を示すチャートである。
【図4】実施例3−(ii)で得られたγ型結晶(3c)の粉末X線回折パターンを示す図である。
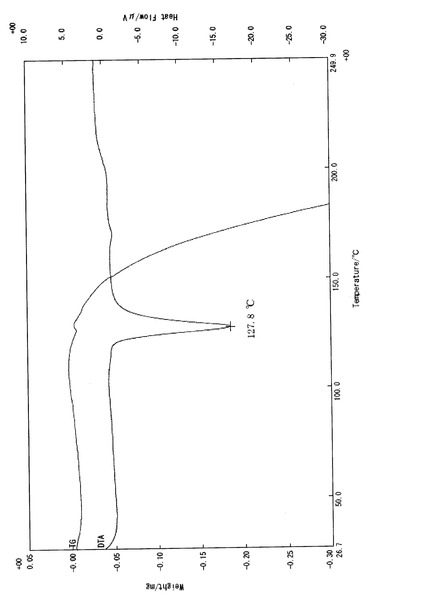
【図5】比較例1で得られたα型結晶(3a)の熱分析データ(TG−DTA測定)を示すチャートである。
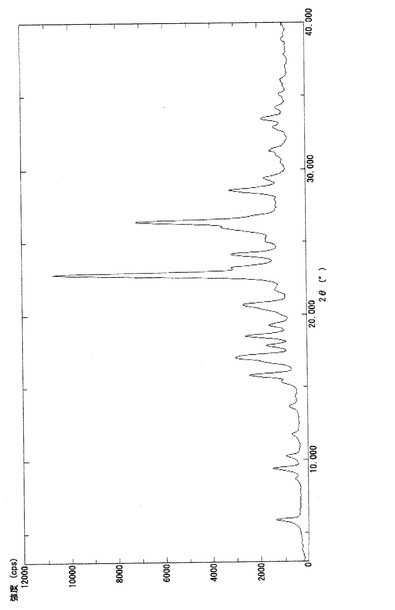
【図6】比較例1で得られたα型結晶(3a)の粉末X線回折パターンを示す図である。
【発明を実施するための形態】
【0020】
本発明に係る7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形β型結晶及びγ型結晶は、次に示す方法により製造することができる。
【0021】
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2a)(以下、単にジマレイン酸化合物(2a)と略称する場合がある。)は、例えば、日本特許登録公報第3588126号に記載の方法に従って製造することができる。また、該化合物の製造を行う際において、反応部位以外の官能基については必要に応じて予め保護しておき、適当な段階においてこれを脱保護してもよい。保護、脱保護条件としては一般に用いられる方法(例えば、Protective Groups in Organic Synthesis Third Edition,John Wiley & Sons,Inc.,1999 に記載された方法)を参考にして行うことができる。さらに、各工程において、反応は通常行われる方法(例えば、Comprehensive Organic Transformations Second Edition,John Wiley & Sons,Inc;1999に記載された方法)で行えばよく、単離精製は結晶化、再結晶化又はクロマトグラフィー等の通常の方法を適宜選択し、又は組み合わせて行えばよい。
【0022】
ジマレイン酸化合物(2a)のβ型結晶又はγ型結晶は、非晶形のジマレイン酸化合物(2a)又はジマレイン酸化合物(2a)の他の結晶形を、加熱又は未加熱の状態で所定の溶媒に溶かした後、該溶液を冷却することによって該化合物を結晶析出させることで製造できる。
【0023】
(i)β型結晶の製造
β型結晶を製造するために用いることができる溶媒は、プロパノール、又はプロパノールとメタノールとの混合溶媒である。該プロパノールとして、1−プロパノール又は2−プロパノールを用いることができるが、2−プロパノールを用いることが好ましい。混合溶媒を用いる場合には、2種の溶媒を混合した後にジマレイン酸化合物(2a)を溶解させてもよく、あるいは1種の溶媒にジマレイン酸化合物(2a)を溶解させた後に、残りの溶媒を加えてもよい。また、これら溶媒にさらに異なる種類の溶媒を加えることもできる。
【0024】
溶媒の使用量は、特に限定されないが、例えば、ジマレイン酸化合物(2a)1gに対して、溶媒全体として、通常約2〜10mLであり、好ましくは約4〜6mLである。混合溶媒を用いる場合の混合比は、特に限定されないが、容量換算で、メタノール:プロパノール=1:1〜1:6が好ましく、メタノール:プロパノール=1:4がより好ましい。
【0025】
ジマレイン酸化合物(2a)を溶媒に溶かす際の加熱温度は、特に限定されないが、例えば、通常40℃〜100℃である。また、ジマレイン酸化合物(2a)の結晶を析出させるための冷却温度は、特に限定されないが、例えば、通常約10℃〜約35℃、好ましくは約20℃〜約30℃である。冷却下では溶液撹拌を行ってもよく、その際の撹拌時間は、通常約1時間〜約5時間、好ましくは約3時間である。析出した結晶は、濾過などの自体公知の方法で単離することができる。濾過温度は、通常約15℃〜約30℃である。
【0026】
上記操作により得られた結晶を自体公知の方法で乾燥させることにより、本発明の結晶を得ることができる。乾燥手段は、特に限定されないが、減圧乾燥、あるいは通風による乾燥でもよい。乾燥温度は40℃以下が好ましく、より好ましくは約25℃〜約35℃である。乾燥時間は、通常約3時間〜約15時間、好ましくは約6時間〜約9時間である。
【0027】
(ii)γ型結晶の製造
γ型結晶を製造するために用いることができる溶媒は、アルコール系溶媒とジアルキルケトン溶媒との混合溶媒である。アルコール系溶媒としては、特に限定されないが、例えば、メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール又はヘプタノール等を用いることができ、メタノールが特に好ましい。ジアルキルケトン溶媒としては、特に限定されないが、例えば、次式(4):
【0028】
【化3】
【0029】
〔式中、R1及びR2は、互いに同一又は異なっていてもよいC1-6アルキル基を示す。〕で示される溶媒を用いることができ、メチルイソブチルケトン(MIBK)が特に好ましい。ジマレイン酸化合物(2a)を溶解させる際には、2種の溶媒を混合した後にジマレイン酸化合物(2a)を溶解させてもよく、あるいは1種の溶媒にジマレイン酸化合物(2a)を溶解させた後に残りの溶媒を加えてもよい。また、これら混合溶媒にさらに異なる種類の溶媒を加えることもできる。
【0030】
溶媒の使用量は、特に限定されないが、例えば、ジマレイン酸化合物(2a)1gに対して、溶媒全体として、通常約2〜10mLであり、好ましくは約4〜6mLである。混合溶媒の混合比は、特に限定されないが、容量換算で、アルコール系溶媒:ジアルキルケトン溶媒=1:1〜1:6が好ましく、アルコール系溶媒:ジアルキルケトン溶媒=1:4がより好ましい。
【0031】
ジマレイン酸化合物(2a)を溶媒に溶かす際の加熱温度は、特に限定されないが、例えば、通常40℃〜100℃である。また、ジマレイン酸化合物(2a)の結晶を析出させるための冷却温度は、特に限定されないが、例えば、通常約10℃〜約35℃、好ましくは約20℃〜約30℃である。冷却下では溶液撹拌を行ってもよく、その際の撹拌時間は、通常約1時間〜約5時間、好ましくは約3時間である。析出した結晶は、濾過などの自体公知の方法で単離することができる。濾過温度は、通常約15℃〜約30℃である。
【0032】
上記操作により得られた結晶を自体公知の方法で乾燥させることにより、本発明の結晶を得ることができる。乾燥手段は、特に限定されないが、減圧乾燥、あるいは通風による乾燥でもよい。乾燥温度は40℃以下が好ましく、より好ましくは約25℃〜約35℃である。乾燥時間は、通常約3時間〜約15時間、好ましくは約6時間〜約9時間である。
【0033】
得られた結晶の解析方法としては、X線回折による結晶解析の方法が一般的である。さらに、結晶の方位を決定する方法としては、機械的な方法または光学的な方法(例えば、FT−ラマンスペクトル、固体NMRスペクトル)なども挙げられる。上記解析方法により得られるスペクトルのピークは、その性質上一定の測定誤差が必然的に生じる。スペクトルのピークの数値が当該誤差範囲のものも本発明の結晶に含まれる。
【0034】
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶としては、X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する結晶(β型結晶:融点112℃±1.0℃)、あるいは回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する結晶(γ型結晶:融点約126.2±0.5℃)が挙げられる。
【0035】
本発明のジマレイン酸化合物(2a)の結晶は、水和物などの溶媒和物であってもよく、非溶媒和物であってもよい。該「水和物」としては、0.5水和物ないし5.0水和物が挙げられる。このうち、0.5水和物、1.0水和物、1.5水和物、2.0水和物、2.5水和物が好ましい。また、本発明のジマレイン酸化合物(2a)の結晶は、水和物以外の溶媒和物であってもよい。
【0036】
本発明のジマレイン酸化合物(2a)の溶媒和物結晶としては、例えばメタノール和物結晶、エタノール和物結晶等のアルコール和物結晶(好ましくはC1-6アルコール和物結晶)や水および有機溶媒が付加した有機溶媒・水和物結晶(例えば、アルコール・水和物結晶、好ましくはメタノール・水和物、エタノール・水和物などのC1-6アルコール・水和物結晶)などが挙げられる。
【0037】
本発明のジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶を含有する医薬を製造する際には、この結晶を単独で用いてよいが、通常は医薬として許容される担体、添加物等を配合して使用してもよい。医薬組成物の投与形態は、特に限定されず、治療目的に応じて適宜選択できる。例えば、経口剤、注射剤、坐剤、軟膏剤、吸入剤、点眼剤、点鼻剤、貼付剤等のいずれでもよい。これらの投与形態に適した医薬組成物は、公知の製剤方法により製造できる。また、本発明のジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶は、他の疾患治療に有効な化合物、例えばアレルギー性疾患の治療に有効な他の化合物と併用することもできる。
【0038】
経口用固形製剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に賦形剤、さらに必要に応じて結合剤、崩壊剤、滑沢剤、着色剤、矯味剤、矯臭剤等を加えた後、常法により錠剤、被覆錠剤、顆粒剤、散剤、カプセル剤等を製造することができる。添加剤は、当該分野で一般的に使用されているものでよい。
【0039】
経口用液体製剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に矯味剤、緩衝剤、安定化剤、矯臭剤等を加えて常法により内服液剤、シロップ剤、エリキシル剤等を製造することができる。
【0040】
点眼剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶にpH調節剤、緩衝剤、安定化剤、等張化剤、防腐剤、抗酸化剤、粘稠化剤、キレート剤等を用いて、常法により製造することができる。
【0041】
注射剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶にpH調節剤、緩衝剤、安定化剤、等張化剤、局所麻酔剤等を添加し、常法により皮下、筋肉及び静脈内注射剤を製造することができる。
【0042】
坐剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に公知の坐剤用担体、例えば、ポリエチレングリコール、ラノリン、カカオ脂、脂肪酸トリグリセライド等、さらに必要に応じてツイーン(登録商標)等の界面活性剤等を加えた後、常法により製造することができる。
【0043】
軟膏剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に通常使用される基剤、安定化剤、湿潤剤、保存剤等を必要に応じて配合し、常法により混合、製剤化することができる。
【0044】
ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶は、上記以外に常法を利用して適宜好ましい製剤とすることができ、常法により吸入剤、点眼剤、点鼻剤とすることもできる。
【0045】
本発明の医薬の投与量は、患者の体重、年齢、性別、症状等によって異なるが、通常成人の場合、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶として、約0.01〜1000mgを1日1〜4回に分けて投与することができる。好ましくは約0.1〜100mgを1日1〜4回に分けて投与することができる。
【実施例】
【0046】
以下、実施例、比較例及び試験例により本発明をより具体的に説明するが、本発明はこれらにより何ら限定されるものではない。なお、下記実施例中で用いられている略号は下記の意味を示す。
s:シングレット(singlet)
d:ダブレット(doublet)
t:トリプレット(triplet)
q:クアルテット(quartet)
m:マルチプレット(multiplet)
br:ブロード(broad)
J:カップリング定数(coupling constant)
Hz:ヘルツ(Hertz)
CDCl3:重クロロホルム
1H-NMR:プロトン核磁気共鳴
【0047】
実施例1:7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン(1a)の製造(フリー体の製造)
【0048】
【化4】
【0049】
a)特開昭60−4176号、特開昭59−70675号に記載の方法によって得られた7−ヒドロキシ−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン65 g(359mmol)をアルゴン雰囲気下、テトラヒドロフラン(194mL)に懸濁し、トリフェニルホスフィン104 g(397mmol)及び3−クロロプロパノール32mL(379mmol)を加え、0℃に冷却した。次いで、得られた反応液に、アゾジカルボン酸ジイソプロピルエステル78mL(396mmol)を30℃以下で滴下した後、室温で1時間攪拌した。得られた溶液から溶媒を減圧留去した後、メタノール(390mL)を加えて室温で1時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−(3−クロロプロポキシ)−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン59g(収率64%)を青白色結晶として得た。
【0050】
【化5】
【0051】
1H−NMR(400MHz,DMSO−d6)δ:2.12(2H,quint,J=6.2Hz),3.28(2H,s),3.76(2H,t,J=6.2Hz),4.03(2H,t,J=5.8Hz),6.78(1H,dd,J=2.8,8.8Hz),6.88(1H,d,J=8.8Hz),6.90(1H,d,J=2.8Hz),10.38(1H,s)
【0052】
b)7−(3−クロロプロポキシ)−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン57g(221mmol)をジメチルホルムアミド(172mL)に懸濁し、炭酸カリウム49g(355mmol)、ヨウ化カリウム40g(241mmol)、N−t−ブトキシカルボニルピペラジン43g(231mmol)を加え、100℃に加熱して4時間攪拌した。反応液に水(344mL)を加えた後、0℃に冷却し、さらに同温で1時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−〔3−(N−t−ブトキシカルボニルピペラジニル)プロポキシ〕−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン89g(収率99%)を青白色結晶として得た。
【0053】
【化6】
【0054】
1H−NMR(400MHz,DMSO−d6)δ:1.39(9H,s),1.83(2H,quint,J=6.8Hz),2.31(4H,t,J=4.8Hz),2.39(2H,t,J=7.0Hz),3.30(4H,t,J=4.6Hz),3.41(2H,s),3.95(2H,t,J=6.4Hz),6.78(1H,dd,J=2.8,8.8Hz),6.88(1H,d,J=8.8Hz),6.89(1H,s),10.38(1H,s)
【0055】
c)7−{3−(N−t−ブトキシカルボニルピペラジニル)プロポキシ}−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン87g(214mmol)をエタノール(174mL)に懸濁し、6N塩酸水溶液(174mL)を50℃で滴下し、同温で1時間攪拌した。反応液にエタノール(522mL)を加えた後、0℃に冷却し、さらに同温で1時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−{3−(ピペラジン−1−イル)プロポキシ}−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・2塩酸塩75g(収率92%)を青白色結晶として得た。
【0056】
【化7】
【0057】
1H−NMR(400MHz,D2O)δ:2.13(2H,td,J=5.9,15.6Hz),3.34(2H,s),3.35(2H,t,J=8.0Hz),3.44−3.64(8H,m),4.02(2H,t,J=5.6Hz),6.74(1H,dd,J=2.4,8.8Hz),6.85(1H,d,J=8.8Hz),6.90(1H,d,J=2.4Hz)
【0058】
d)Journal of Heterocyclic Chemistry(1987),24(1),31−37に記載の方法によって得られた1−(2−エトキシエチル)−2−クロロメチル−1H−ベンゾイミダゾールをテトラヒドロフラン(293mL)と水(147mL)の混合液に溶解し、実施例1−1の工程c)で製造した7−{3−(N−t−ブトキシカルボニルピペラジニル)プロポキシ}−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン73g(192mmol)を加えた。次いで、ジイソプロピルエチルアミン117mL(673mmol)、ヨウ化カリウム35g(211mmol)を加え、室温で15時間攪拌した。反応液に酢酸エチル(293mL)及び水(147mL)を加えて抽出し、有機層を20%食塩水(147mL)で洗浄した。有機層を減圧濃縮し、表題化合物(1a)115g(2工程、定量的)を褐色油状物として得た。
【0059】
1H−NMR(400MHz,CDCl3)δ:1.13(3H,t,J=7.0Hz),1.93(2H,quint,J=6.9Hz),2.40−2.70(8H,m),2.51(2H,t,J=7.2Hz),3.41(2H,s),3.42(2H,q,J=6.8Hz),3.76(2H,t,J=6.0Hz),3.88(2H,s),3.97(2H,t,J=6.2Hz),4.51(2H,t,J=5.8Hz),6.71(1H,dd,J=2.6,8.6Hz),6.77(1H,d,J=8.8Hz),6.85(1H,d,J=2.4Hz),7.24−7.28(2H,m),7.39(1H,ddd,J=1.2,6.0,6.8Hz),7.73(1H,ddd,J=1.2,6.0,6.8Hz),8.35(1H,s)
【0060】
実施例2:7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の製造
【0061】
【化8】
【0062】
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン15.9g(31.1mmol)をエタノール70mLに溶解し、溶液を60℃に加温した後、マレイン酸8.0g(68.9mmol)を加え、室温で15時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩を13.3g得た。得られた化合物をメタノール(13mL)に溶解し、60℃に加温した後、THF(52mL)を加え、室温で20時間攪拌した。得られた結晶をろ取した後、50℃で5時間減圧乾燥し、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2a)を10.3g(収率45%)を青白色結晶として得た。
【0063】
1H−NMR(400MHz,DMSO−d6)δ:1.01(3H,t,J=7.0Hz),2.00−2.07(2H,m),3.00(4H,m),3.20(2H,m),3.37(2H,q,J=6.9Hz),3.41−3.47(4H,m),3.70(2H,t,J=5.2Hz),3.95(2H,s),3.99(2H,t,J=5.8Hz),4.50(2H,t,J=5.0Hz),6.14(4H,s),6.76(1H,dd,J=2.4,8.8Hz),6.88(1H,s),6.90(1H,m),7.19−7.27(2H,m),7.60(2H,d,J=7.6Hz),10.40(1H,s)
【0064】
実施例3:7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形の製造
【0065】
(i)β型結晶の製造
(a)7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(1g)を取り、2−プロパノール(20mL)を加えて100℃に加熱し、その後自然放冷しながら3時間撹拌した。析出した結晶をろ取し、減圧下室温にて9時間乾燥して、β型結晶(3b)を得た(収率89%)。
【0066】
(b)7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2g)を取り、メタノール(2mL)を加えて60℃まで加熱し、2−プロパノール(8mL)を加えた。その後室温まで自然放冷しながら15時間撹拌し、析出した結晶をろ取した。ろ取した結晶を減圧下室温にて8時間乾燥し、β型結晶(3b)を得た(収率104%)。
【0067】
実施例3−(i)で得られたβ型結晶(3b)について熱分析測定を行った。熱分析測定は、サンプル約5mgを熱分析用アルミパンに精密に秤量し、基準物質としてAl2O3を使用して、雰囲気N2ガス(150mL/min)存在下、昇温速度10℃/分とし、熱分析装置Thermo Plus 2 システム(リガク社製)を用いて、示差熱分析法(DTA)及び熱質量測定法(TG)によって行った。熱分析測定の結果を図1に示す。図1で見られるように、β型結晶(3b)の融点は112℃であった(BUCHI社製、B−545)。
【0068】
実施例3−(i)で得られたβ型結晶(3b)について粉末X線回折の測定を行った。粉末X線回折の測定は、サンプルをX線回折用シリコン無反射試料板の試料ホルダー部分に充填し、デスクトップX線回折装置:MiniFlex(リガク社製)により、回折角2θの走査範囲;3.00°〜40.00°、サンプリング幅;0.02°、スキャン速度;2.00°/分の条件下で行った。得られた回折パターンは図2に示す。β型結晶(3b)は、下記表1に示す特有の回折角度及び相対強度を有した。
【0069】
【表1】
【0070】
(ii)γ型結晶の製造
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2g)を取り、メタノール(2mL)を加えて60℃まで加熱し、メチルイソブチルケトン(MIBK)(8mL)を加えた。その後室温まで自然放冷しながら15時間撹拌し、析出した結晶をろ取した。ろ取した結晶を減圧下50℃で8時間乾燥し、γ型結晶(3c)を得た(収率94%)。
【0071】
実施例3−(i)と同じ条件下にて、γ型結晶(3c)について熱分析測定及び粉末X線回折測定を行った。熱分析測定の結果を図3に示し、得られた回折パターンを図4に示す。図3で見られるように、γ型結晶(3c)の融点は126.2℃であった(BUCHI社製、B−545)。また、γ型結晶(3c)は、下記表2に示す特有の回折角度及び相対強度を有した。
【0072】
【表2】
【0073】
比較例1:α型結晶の製造
(a)7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(0.5g)を取り、メタノール(0.5mL)を加えて60℃まで加熱し、エタノール(2mL)を加えた。その後室温まで自然放冷しながら18時間撹拌し、析出した結晶をろ取した。ろ取した結晶を減圧下室温にて乾燥し、α型結晶(3a)を得た(収率82%)。
【0074】
(b)前記溶媒であるエタノールの代わりに、アセトン、テトラヒドロフラン、アセトニトリルのいずれかを用いる点を除いて、上記比較例1(a)に記載の方法と同じ手法によって結晶化工程を行った。結果、アセトン、テトラヒドロフラン、アセトニトリルのいずれの溶媒を用いた場合にも、α型結晶(3a)が得られた(収率アセトン:79%、テトラヒドロフラン:85%、アセトニトリル:62%)。
【0075】
実施例3−(i)と同じ条件下にて、α型結晶(3a)について熱分析測定及び粉末X線回折測定を行った。熱分析測定の結果を図5に示し、得られた回折パターンを図6に示す。図5で見られるように、α型結晶(3a)の融点は127.8℃であった(BUCHI社製、B−545)。また、α型結晶(3a)は、下記表3に示す特有の回折角度及び相対強度を有した。
【0076】
【表3】
【0077】
比較例2
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(0.5g)を取り、メタノール(0.5mL)を加えて60℃まで加熱し、トルエン又は酢酸エチル(2mL)を加えた。その後室温まで自然放冷しながら撹拌したが、結晶を析出させることはできなかった。
【0078】
以上、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形の検討結果を下記表に纏める(表4参照)。
【0079】
【表4】
【0080】
以上の検討結果から、特定の溶媒または特定溶媒の組み合わせを用いた場合に、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形であるβ型結晶又はγ型結晶を得られることが分かった。
【産業上の利用可能性】
【0081】
本発明に係るベンゾチアジン化合物のジマレイン酸塩新規結晶形β型結晶及びγ型結晶は、安定性に優れているため、取り扱いが容易であり、アレルギー疾患又は炎症性疾患の予防及び/又は治療剤の製造に適している。また、該新規結晶形β型結晶及びγ型結晶は、アレルギー性疾患又は炎症性疾患に対して優れた治療及び予防効果を示す。
【技術分野】
【0001】
本発明は、ベンゾチアジン化合物のジマレイン酸塩の新規結晶及びその製造方法、並びに該結晶を含有するアレルギー疾患又は炎症性疾患の予防及び/又は治療剤に関する。
【背景技術】
【0002】
次式(1):
【0003】
【化1】
【0004】
(式中、lは1又は2の整数を示し、mは1又は2の整数を示し、nは1〜4の整数を示す)
で表されるベンゾチアジン化合物は、抗ヒスタミン作用及び抗ロイコトリエン作用を有し、アレルギー性鼻炎、アレルギー性結膜炎等のアレルギー性疾患、喘息、乾癬、リウマチ及び炎症性大腸炎等の炎症性疾患の予防又は治療薬として有用である(特許文献1)。
【0005】
また、該ベンゾチアジン化合物は、その構造中に塩基性窒素原子を3個有することから、種々の酸付加塩の形態になり、特許文献1においてはジマレイン酸塩として単離されている(特許文献1の実施例28)。
【0006】
一方、医薬品原体は、医薬品製造の容易性に加えて、製造スケール等が変化した場合であっても、常に同程度の収率と純度をもって製造できるという一定の再現性を有することが望まれる。そして、それに加えて、医薬品原体の結晶性、非吸湿性、水溶性、可視光及び紫外線に対する安定性、温度及び湿度に対する分解度の低さなどの様々な条件を満たすことが必要とされる。医薬として活性な化合物がこの様な医薬品原体として適当な物理化学的性質を有さない場合、通常種々の改善検討がなされる。すなわち、結晶化、水溶性、バイオアベイラビリティ、光安定性、保存安定性、非吸湿性等の改善を目的として、化合物の塩、水和物、結晶形の検討がしばしば行われており、化合物は改善された結果、適当な物理化学的性質を有する医薬品原体として医薬品製造に用いられている。
【0007】
例えば、油状物や不定形の化合物を結晶化することで、取り扱いやすくなるので、品質管理が容易となる一方で、その化合物の溶解度やバイオアベイラビリティに差が出ることが報告されている(非特許文献1参照)。また、化合物の結晶形が安定形であれば、製剤工程において結晶形変化のリスクが少なくなる。また、化合物の結晶化が容易であれば、再結晶による目的化合物の精製収率を向上させることができ、生産過程での品質管理も有利に進めることが可能である(非特許文献2)。化合物の水溶性の改善は経口剤のバイオアベイラビリティを向上させ、注射剤や点眼剤の製造工程を容易にすることができる。化合物の熱及び光安定性の改善は医薬の品質を保証する上で有用であり、化合物の非吸湿性は原体の保管や製剤化工程において有利である。このようなことから、医薬品製造にあたっては最適な物理化学的性質を有する医薬原体を見出すことが非常に重要であり、そのために様々な医薬原体の形態を徹底的に検討することが望まれている。しかしながら、新たに生成した塩、水和物、結晶形がいかなる物理化学的性状を有するかを事前に予測することは困難である。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特許第3588126号公報
【非特許文献】
【0009】
【非特許文献1】Rharmaceutical Research,945,12(7),1995
【非特許文献2】International Journal of Pharmaceutics,209,105,1994
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、前記式(1)で表されるベンゾチアジン化合物のジマレイン酸塩の新たな結晶形を提供することを目的とする。
【課題を解決するための手段】
【0011】
そこで、本発明者らは、前記ベンゾチアジン化合物のジマレイン酸塩について種々検討を行った結果、既に前記特許文献1で報告されている結晶形(α型結晶)とは異なる新規結晶形β型及びγ型が存在することを見出し、本発明を完成するに至った。さらに、本発明者らは、該新規結晶体がアレルギー性疾患(特に、アレルギー性結膜炎)の治療又は予防に有効に用いることができることを見出し、本発明を完成するに至った。
【0012】
すなわち、本発明は、下記の通りである。
[1]X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(β型結晶)。
[2]融点が約112℃±1.0℃である、前記[1]に記載の結晶。
[3]X線回折スペクトルにおいて、回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(γ型結晶)。
[4]融点が約126.2±0.5℃である、前記[3]に記載の結晶。
[5]前記[1]〜[4]のいずれか一つに記載の結晶を含有する医薬組成物。
[6]前記[1]〜[4]のいずれか一つに記載の結晶を含有するアレルギー性疾患の予防及び/又は治療剤。
[7]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[6]に記載の予防及び/又は治療剤。
[8]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[7]に記載の予防及び/又は治療剤。
【0013】
[9]プロパノールを溶媒として用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(β型結晶)の製造方法。
[10]前記プロパノールが、1−プロパノール又は2−プロパノールである、前記[9]に記載の製造方法。
[11]さらに、メタノールを溶媒として用いて結晶化を行う、前記[9]又は[10]に記載の製造方法。
[12]アルコール系溶媒とジアルキルケトン溶媒との混合溶媒を用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶(γ型結晶)の製造方法。
[13]前記アルコール系溶媒が、メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール又はヘプタノールである、前記[12]に記載の製造方法。
[14]前記ジアルキルケトン溶媒が、次式(4):
【0014】
【化2】
【0015】
〔式中、R1及びR2は、互いに同一又は異なっていてもよいC1-6アルキル基を示す。〕
である、前記[12]又は[13]に記載の製造方法。
[15]前記ジアルキルケトン溶媒が、メチルイソブチルケトンである、前記[12]又は[13]に記載の製造方法。
[16]アレルギー性疾患の予防及び/又は治療用の、前記[1]〜[4]のいずれか一つに記載の結晶。
[17]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[16]に記載の結晶。
[18]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[17]に記載の結晶。
【0016】
[19]前記[1]〜[4]のいずれか一つに記載の結晶の、アレルギー性疾患の予防及び/又は治療剤製造のための使用。
[20]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[19]に記載の使用。
[21]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[20]に記載の使用。
【0017】
[22]前記[1]〜[4]のいずれか一つに記載の結晶を投与することを特徴とするアレルギー性疾患の予防及び/又は治療方法。
[23]前記アレルギー性疾患が、アレルギー性眼疾患である、前記[22]に記載の方法。
[24]前記アレルギー性眼疾患が、アレルギー性結膜炎である、前記[23]に記載の方法。
【発明の効果】
【0018】
本発明に係るベンゾチアジン化合物のジマレイン酸塩新規結晶形β型結晶及びγ型結晶は、安定性に優れているため、取り扱いが容易であり、アレルギー疾患又は炎症性疾患の予防及び/又は治療剤の製造に適している。また、該新規結晶形β型結晶及びγ型結晶は、アレルギー性疾患又は炎症性疾患に対して優れた治療及び予防効果を示す。
【図面の簡単な説明】
【0019】
【図1】実施例3−(i)で得られたβ型結晶(3b)の熱分析データ(TG−DTA測定)を示すチャートである。
【図2】実施例3−(i)で得られたβ型結晶(3b)の粉末X線回折パターンを示す図である。
【図3】実施例3−(ii)で得られたγ型結晶(3c)の熱分析データ(TG−DTA測定)を示すチャートである。
【図4】実施例3−(ii)で得られたγ型結晶(3c)の粉末X線回折パターンを示す図である。
【図5】比較例1で得られたα型結晶(3a)の熱分析データ(TG−DTA測定)を示すチャートである。
【図6】比較例1で得られたα型結晶(3a)の粉末X線回折パターンを示す図である。
【発明を実施するための形態】
【0020】
本発明に係る7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形β型結晶及びγ型結晶は、次に示す方法により製造することができる。
【0021】
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2a)(以下、単にジマレイン酸化合物(2a)と略称する場合がある。)は、例えば、日本特許登録公報第3588126号に記載の方法に従って製造することができる。また、該化合物の製造を行う際において、反応部位以外の官能基については必要に応じて予め保護しておき、適当な段階においてこれを脱保護してもよい。保護、脱保護条件としては一般に用いられる方法(例えば、Protective Groups in Organic Synthesis Third Edition,John Wiley & Sons,Inc.,1999 に記載された方法)を参考にして行うことができる。さらに、各工程において、反応は通常行われる方法(例えば、Comprehensive Organic Transformations Second Edition,John Wiley & Sons,Inc;1999に記載された方法)で行えばよく、単離精製は結晶化、再結晶化又はクロマトグラフィー等の通常の方法を適宜選択し、又は組み合わせて行えばよい。
【0022】
ジマレイン酸化合物(2a)のβ型結晶又はγ型結晶は、非晶形のジマレイン酸化合物(2a)又はジマレイン酸化合物(2a)の他の結晶形を、加熱又は未加熱の状態で所定の溶媒に溶かした後、該溶液を冷却することによって該化合物を結晶析出させることで製造できる。
【0023】
(i)β型結晶の製造
β型結晶を製造するために用いることができる溶媒は、プロパノール、又はプロパノールとメタノールとの混合溶媒である。該プロパノールとして、1−プロパノール又は2−プロパノールを用いることができるが、2−プロパノールを用いることが好ましい。混合溶媒を用いる場合には、2種の溶媒を混合した後にジマレイン酸化合物(2a)を溶解させてもよく、あるいは1種の溶媒にジマレイン酸化合物(2a)を溶解させた後に、残りの溶媒を加えてもよい。また、これら溶媒にさらに異なる種類の溶媒を加えることもできる。
【0024】
溶媒の使用量は、特に限定されないが、例えば、ジマレイン酸化合物(2a)1gに対して、溶媒全体として、通常約2〜10mLであり、好ましくは約4〜6mLである。混合溶媒を用いる場合の混合比は、特に限定されないが、容量換算で、メタノール:プロパノール=1:1〜1:6が好ましく、メタノール:プロパノール=1:4がより好ましい。
【0025】
ジマレイン酸化合物(2a)を溶媒に溶かす際の加熱温度は、特に限定されないが、例えば、通常40℃〜100℃である。また、ジマレイン酸化合物(2a)の結晶を析出させるための冷却温度は、特に限定されないが、例えば、通常約10℃〜約35℃、好ましくは約20℃〜約30℃である。冷却下では溶液撹拌を行ってもよく、その際の撹拌時間は、通常約1時間〜約5時間、好ましくは約3時間である。析出した結晶は、濾過などの自体公知の方法で単離することができる。濾過温度は、通常約15℃〜約30℃である。
【0026】
上記操作により得られた結晶を自体公知の方法で乾燥させることにより、本発明の結晶を得ることができる。乾燥手段は、特に限定されないが、減圧乾燥、あるいは通風による乾燥でもよい。乾燥温度は40℃以下が好ましく、より好ましくは約25℃〜約35℃である。乾燥時間は、通常約3時間〜約15時間、好ましくは約6時間〜約9時間である。
【0027】
(ii)γ型結晶の製造
γ型結晶を製造するために用いることができる溶媒は、アルコール系溶媒とジアルキルケトン溶媒との混合溶媒である。アルコール系溶媒としては、特に限定されないが、例えば、メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール又はヘプタノール等を用いることができ、メタノールが特に好ましい。ジアルキルケトン溶媒としては、特に限定されないが、例えば、次式(4):
【0028】
【化3】
【0029】
〔式中、R1及びR2は、互いに同一又は異なっていてもよいC1-6アルキル基を示す。〕で示される溶媒を用いることができ、メチルイソブチルケトン(MIBK)が特に好ましい。ジマレイン酸化合物(2a)を溶解させる際には、2種の溶媒を混合した後にジマレイン酸化合物(2a)を溶解させてもよく、あるいは1種の溶媒にジマレイン酸化合物(2a)を溶解させた後に残りの溶媒を加えてもよい。また、これら混合溶媒にさらに異なる種類の溶媒を加えることもできる。
【0030】
溶媒の使用量は、特に限定されないが、例えば、ジマレイン酸化合物(2a)1gに対して、溶媒全体として、通常約2〜10mLであり、好ましくは約4〜6mLである。混合溶媒の混合比は、特に限定されないが、容量換算で、アルコール系溶媒:ジアルキルケトン溶媒=1:1〜1:6が好ましく、アルコール系溶媒:ジアルキルケトン溶媒=1:4がより好ましい。
【0031】
ジマレイン酸化合物(2a)を溶媒に溶かす際の加熱温度は、特に限定されないが、例えば、通常40℃〜100℃である。また、ジマレイン酸化合物(2a)の結晶を析出させるための冷却温度は、特に限定されないが、例えば、通常約10℃〜約35℃、好ましくは約20℃〜約30℃である。冷却下では溶液撹拌を行ってもよく、その際の撹拌時間は、通常約1時間〜約5時間、好ましくは約3時間である。析出した結晶は、濾過などの自体公知の方法で単離することができる。濾過温度は、通常約15℃〜約30℃である。
【0032】
上記操作により得られた結晶を自体公知の方法で乾燥させることにより、本発明の結晶を得ることができる。乾燥手段は、特に限定されないが、減圧乾燥、あるいは通風による乾燥でもよい。乾燥温度は40℃以下が好ましく、より好ましくは約25℃〜約35℃である。乾燥時間は、通常約3時間〜約15時間、好ましくは約6時間〜約9時間である。
【0033】
得られた結晶の解析方法としては、X線回折による結晶解析の方法が一般的である。さらに、結晶の方位を決定する方法としては、機械的な方法または光学的な方法(例えば、FT−ラマンスペクトル、固体NMRスペクトル)なども挙げられる。上記解析方法により得られるスペクトルのピークは、その性質上一定の測定誤差が必然的に生じる。スペクトルのピークの数値が当該誤差範囲のものも本発明の結晶に含まれる。
【0034】
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶としては、X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する結晶(β型結晶:融点112℃±1.0℃)、あるいは回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する結晶(γ型結晶:融点約126.2±0.5℃)が挙げられる。
【0035】
本発明のジマレイン酸化合物(2a)の結晶は、水和物などの溶媒和物であってもよく、非溶媒和物であってもよい。該「水和物」としては、0.5水和物ないし5.0水和物が挙げられる。このうち、0.5水和物、1.0水和物、1.5水和物、2.0水和物、2.5水和物が好ましい。また、本発明のジマレイン酸化合物(2a)の結晶は、水和物以外の溶媒和物であってもよい。
【0036】
本発明のジマレイン酸化合物(2a)の溶媒和物結晶としては、例えばメタノール和物結晶、エタノール和物結晶等のアルコール和物結晶(好ましくはC1-6アルコール和物結晶)や水および有機溶媒が付加した有機溶媒・水和物結晶(例えば、アルコール・水和物結晶、好ましくはメタノール・水和物、エタノール・水和物などのC1-6アルコール・水和物結晶)などが挙げられる。
【0037】
本発明のジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶を含有する医薬を製造する際には、この結晶を単独で用いてよいが、通常は医薬として許容される担体、添加物等を配合して使用してもよい。医薬組成物の投与形態は、特に限定されず、治療目的に応じて適宜選択できる。例えば、経口剤、注射剤、坐剤、軟膏剤、吸入剤、点眼剤、点鼻剤、貼付剤等のいずれでもよい。これらの投与形態に適した医薬組成物は、公知の製剤方法により製造できる。また、本発明のジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶は、他の疾患治療に有効な化合物、例えばアレルギー性疾患の治療に有効な他の化合物と併用することもできる。
【0038】
経口用固形製剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に賦形剤、さらに必要に応じて結合剤、崩壊剤、滑沢剤、着色剤、矯味剤、矯臭剤等を加えた後、常法により錠剤、被覆錠剤、顆粒剤、散剤、カプセル剤等を製造することができる。添加剤は、当該分野で一般的に使用されているものでよい。
【0039】
経口用液体製剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に矯味剤、緩衝剤、安定化剤、矯臭剤等を加えて常法により内服液剤、シロップ剤、エリキシル剤等を製造することができる。
【0040】
点眼剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶にpH調節剤、緩衝剤、安定化剤、等張化剤、防腐剤、抗酸化剤、粘稠化剤、キレート剤等を用いて、常法により製造することができる。
【0041】
注射剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶にpH調節剤、緩衝剤、安定化剤、等張化剤、局所麻酔剤等を添加し、常法により皮下、筋肉及び静脈内注射剤を製造することができる。
【0042】
坐剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に公知の坐剤用担体、例えば、ポリエチレングリコール、ラノリン、カカオ脂、脂肪酸トリグリセライド等、さらに必要に応じてツイーン(登録商標)等の界面活性剤等を加えた後、常法により製造することができる。
【0043】
軟膏剤を調製する場合は、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶に通常使用される基剤、安定化剤、湿潤剤、保存剤等を必要に応じて配合し、常法により混合、製剤化することができる。
【0044】
ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶は、上記以外に常法を利用して適宜好ましい製剤とすることができ、常法により吸入剤、点眼剤、点鼻剤とすることもできる。
【0045】
本発明の医薬の投与量は、患者の体重、年齢、性別、症状等によって異なるが、通常成人の場合、ジマレイン酸化合物(2a)のβ型結晶及び/又はγ型結晶として、約0.01〜1000mgを1日1〜4回に分けて投与することができる。好ましくは約0.1〜100mgを1日1〜4回に分けて投与することができる。
【実施例】
【0046】
以下、実施例、比較例及び試験例により本発明をより具体的に説明するが、本発明はこれらにより何ら限定されるものではない。なお、下記実施例中で用いられている略号は下記の意味を示す。
s:シングレット(singlet)
d:ダブレット(doublet)
t:トリプレット(triplet)
q:クアルテット(quartet)
m:マルチプレット(multiplet)
br:ブロード(broad)
J:カップリング定数(coupling constant)
Hz:ヘルツ(Hertz)
CDCl3:重クロロホルム
1H-NMR:プロトン核磁気共鳴
【0047】
実施例1:7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン(1a)の製造(フリー体の製造)
【0048】
【化4】
【0049】
a)特開昭60−4176号、特開昭59−70675号に記載の方法によって得られた7−ヒドロキシ−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン65 g(359mmol)をアルゴン雰囲気下、テトラヒドロフラン(194mL)に懸濁し、トリフェニルホスフィン104 g(397mmol)及び3−クロロプロパノール32mL(379mmol)を加え、0℃に冷却した。次いで、得られた反応液に、アゾジカルボン酸ジイソプロピルエステル78mL(396mmol)を30℃以下で滴下した後、室温で1時間攪拌した。得られた溶液から溶媒を減圧留去した後、メタノール(390mL)を加えて室温で1時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−(3−クロロプロポキシ)−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン59g(収率64%)を青白色結晶として得た。
【0050】
【化5】
【0051】
1H−NMR(400MHz,DMSO−d6)δ:2.12(2H,quint,J=6.2Hz),3.28(2H,s),3.76(2H,t,J=6.2Hz),4.03(2H,t,J=5.8Hz),6.78(1H,dd,J=2.8,8.8Hz),6.88(1H,d,J=8.8Hz),6.90(1H,d,J=2.8Hz),10.38(1H,s)
【0052】
b)7−(3−クロロプロポキシ)−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン57g(221mmol)をジメチルホルムアミド(172mL)に懸濁し、炭酸カリウム49g(355mmol)、ヨウ化カリウム40g(241mmol)、N−t−ブトキシカルボニルピペラジン43g(231mmol)を加え、100℃に加熱して4時間攪拌した。反応液に水(344mL)を加えた後、0℃に冷却し、さらに同温で1時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−〔3−(N−t−ブトキシカルボニルピペラジニル)プロポキシ〕−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン89g(収率99%)を青白色結晶として得た。
【0053】
【化6】
【0054】
1H−NMR(400MHz,DMSO−d6)δ:1.39(9H,s),1.83(2H,quint,J=6.8Hz),2.31(4H,t,J=4.8Hz),2.39(2H,t,J=7.0Hz),3.30(4H,t,J=4.6Hz),3.41(2H,s),3.95(2H,t,J=6.4Hz),6.78(1H,dd,J=2.8,8.8Hz),6.88(1H,d,J=8.8Hz),6.89(1H,s),10.38(1H,s)
【0055】
c)7−{3−(N−t−ブトキシカルボニルピペラジニル)プロポキシ}−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン87g(214mmol)をエタノール(174mL)に懸濁し、6N塩酸水溶液(174mL)を50℃で滴下し、同温で1時間攪拌した。反応液にエタノール(522mL)を加えた後、0℃に冷却し、さらに同温で1時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−{3−(ピペラジン−1−イル)プロポキシ}−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・2塩酸塩75g(収率92%)を青白色結晶として得た。
【0056】
【化7】
【0057】
1H−NMR(400MHz,D2O)δ:2.13(2H,td,J=5.9,15.6Hz),3.34(2H,s),3.35(2H,t,J=8.0Hz),3.44−3.64(8H,m),4.02(2H,t,J=5.6Hz),6.74(1H,dd,J=2.4,8.8Hz),6.85(1H,d,J=8.8Hz),6.90(1H,d,J=2.4Hz)
【0058】
d)Journal of Heterocyclic Chemistry(1987),24(1),31−37に記載の方法によって得られた1−(2−エトキシエチル)−2−クロロメチル−1H−ベンゾイミダゾールをテトラヒドロフラン(293mL)と水(147mL)の混合液に溶解し、実施例1−1の工程c)で製造した7−{3−(N−t−ブトキシカルボニルピペラジニル)プロポキシ}−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン73g(192mmol)を加えた。次いで、ジイソプロピルエチルアミン117mL(673mmol)、ヨウ化カリウム35g(211mmol)を加え、室温で15時間攪拌した。反応液に酢酸エチル(293mL)及び水(147mL)を加えて抽出し、有機層を20%食塩水(147mL)で洗浄した。有機層を減圧濃縮し、表題化合物(1a)115g(2工程、定量的)を褐色油状物として得た。
【0059】
1H−NMR(400MHz,CDCl3)δ:1.13(3H,t,J=7.0Hz),1.93(2H,quint,J=6.9Hz),2.40−2.70(8H,m),2.51(2H,t,J=7.2Hz),3.41(2H,s),3.42(2H,q,J=6.8Hz),3.76(2H,t,J=6.0Hz),3.88(2H,s),3.97(2H,t,J=6.2Hz),4.51(2H,t,J=5.8Hz),6.71(1H,dd,J=2.6,8.6Hz),6.77(1H,d,J=8.8Hz),6.85(1H,d,J=2.4Hz),7.24−7.28(2H,m),7.39(1H,ddd,J=1.2,6.0,6.8Hz),7.73(1H,ddd,J=1.2,6.0,6.8Hz),8.35(1H,s)
【0060】
実施例2:7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の製造
【0061】
【化8】
【0062】
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン15.9g(31.1mmol)をエタノール70mLに溶解し、溶液を60℃に加温した後、マレイン酸8.0g(68.9mmol)を加え、室温で15時間攪拌した。析出した結晶をろ取した後、50℃で5時間減圧乾燥し、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩を13.3g得た。得られた化合物をメタノール(13mL)に溶解し、60℃に加温した後、THF(52mL)を加え、室温で20時間攪拌した。得られた結晶をろ取した後、50℃で5時間減圧乾燥し、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2a)を10.3g(収率45%)を青白色結晶として得た。
【0063】
1H−NMR(400MHz,DMSO−d6)δ:1.01(3H,t,J=7.0Hz),2.00−2.07(2H,m),3.00(4H,m),3.20(2H,m),3.37(2H,q,J=6.9Hz),3.41−3.47(4H,m),3.70(2H,t,J=5.2Hz),3.95(2H,s),3.99(2H,t,J=5.8Hz),4.50(2H,t,J=5.0Hz),6.14(4H,s),6.76(1H,dd,J=2.4,8.8Hz),6.88(1H,s),6.90(1H,m),7.19−7.27(2H,m),7.60(2H,d,J=7.6Hz),10.40(1H,s)
【0064】
実施例3:7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形の製造
【0065】
(i)β型結晶の製造
(a)7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(1g)を取り、2−プロパノール(20mL)を加えて100℃に加熱し、その後自然放冷しながら3時間撹拌した。析出した結晶をろ取し、減圧下室温にて9時間乾燥して、β型結晶(3b)を得た(収率89%)。
【0066】
(b)7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2g)を取り、メタノール(2mL)を加えて60℃まで加熱し、2−プロパノール(8mL)を加えた。その後室温まで自然放冷しながら15時間撹拌し、析出した結晶をろ取した。ろ取した結晶を減圧下室温にて8時間乾燥し、β型結晶(3b)を得た(収率104%)。
【0067】
実施例3−(i)で得られたβ型結晶(3b)について熱分析測定を行った。熱分析測定は、サンプル約5mgを熱分析用アルミパンに精密に秤量し、基準物質としてAl2O3を使用して、雰囲気N2ガス(150mL/min)存在下、昇温速度10℃/分とし、熱分析装置Thermo Plus 2 システム(リガク社製)を用いて、示差熱分析法(DTA)及び熱質量測定法(TG)によって行った。熱分析測定の結果を図1に示す。図1で見られるように、β型結晶(3b)の融点は112℃であった(BUCHI社製、B−545)。
【0068】
実施例3−(i)で得られたβ型結晶(3b)について粉末X線回折の測定を行った。粉末X線回折の測定は、サンプルをX線回折用シリコン無反射試料板の試料ホルダー部分に充填し、デスクトップX線回折装置:MiniFlex(リガク社製)により、回折角2θの走査範囲;3.00°〜40.00°、サンプリング幅;0.02°、スキャン速度;2.00°/分の条件下で行った。得られた回折パターンは図2に示す。β型結晶(3b)は、下記表1に示す特有の回折角度及び相対強度を有した。
【0069】
【表1】
【0070】
(ii)γ型結晶の製造
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(2g)を取り、メタノール(2mL)を加えて60℃まで加熱し、メチルイソブチルケトン(MIBK)(8mL)を加えた。その後室温まで自然放冷しながら15時間撹拌し、析出した結晶をろ取した。ろ取した結晶を減圧下50℃で8時間乾燥し、γ型結晶(3c)を得た(収率94%)。
【0071】
実施例3−(i)と同じ条件下にて、γ型結晶(3c)について熱分析測定及び粉末X線回折測定を行った。熱分析測定の結果を図3に示し、得られた回折パターンを図4に示す。図3で見られるように、γ型結晶(3c)の融点は126.2℃であった(BUCHI社製、B−545)。また、γ型結晶(3c)は、下記表2に示す特有の回折角度及び相対強度を有した。
【0072】
【表2】
【0073】
比較例1:α型結晶の製造
(a)7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(0.5g)を取り、メタノール(0.5mL)を加えて60℃まで加熱し、エタノール(2mL)を加えた。その後室温まで自然放冷しながら18時間撹拌し、析出した結晶をろ取した。ろ取した結晶を減圧下室温にて乾燥し、α型結晶(3a)を得た(収率82%)。
【0074】
(b)前記溶媒であるエタノールの代わりに、アセトン、テトラヒドロフラン、アセトニトリルのいずれかを用いる点を除いて、上記比較例1(a)に記載の方法と同じ手法によって結晶化工程を行った。結果、アセトン、テトラヒドロフラン、アセトニトリルのいずれの溶媒を用いた場合にも、α型結晶(3a)が得られた(収率アセトン:79%、テトラヒドロフラン:85%、アセトニトリル:62%)。
【0075】
実施例3−(i)と同じ条件下にて、α型結晶(3a)について熱分析測定及び粉末X線回折測定を行った。熱分析測定の結果を図5に示し、得られた回折パターンを図6に示す。図5で見られるように、α型結晶(3a)の融点は127.8℃であった(BUCHI社製、B−545)。また、α型結晶(3a)は、下記表3に示す特有の回折角度及び相対強度を有した。
【0076】
【表3】
【0077】
比較例2
7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩(0.5g)を取り、メタノール(0.5mL)を加えて60℃まで加熱し、トルエン又は酢酸エチル(2mL)を加えた。その後室温まで自然放冷しながら撹拌したが、結晶を析出させることはできなかった。
【0078】
以上、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形の検討結果を下記表に纏める(表4参照)。
【0079】
【表4】
【0080】
以上の検討結果から、特定の溶媒または特定溶媒の組み合わせを用いた場合に、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の新規結晶形であるβ型結晶又はγ型結晶を得られることが分かった。
【産業上の利用可能性】
【0081】
本発明に係るベンゾチアジン化合物のジマレイン酸塩新規結晶形β型結晶及びγ型結晶は、安定性に優れているため、取り扱いが容易であり、アレルギー疾患又は炎症性疾患の予防及び/又は治療剤の製造に適している。また、該新規結晶形β型結晶及びγ型結晶は、アレルギー性疾患又は炎症性疾患に対して優れた治療及び予防効果を示す。
【特許請求の範囲】
【請求項1】
X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶。
【請求項2】
融点が約112℃±1.0℃である、請求項1に記載の結晶。
【請求項3】
X線回折スペクトルにおいて、回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶。
【請求項4】
融点が約126.2±0.5℃である、請求項3に記載の結晶。
【請求項5】
請求項1〜4のいずれか一項に記載の結晶を含有する医薬組成物。
【請求項6】
請求項1〜4のいずれか一項に記載の結晶を含有するアレルギー性疾患の予防及び/又は治療剤。
【請求項7】
前記アレルギー性疾患が、アレルギー性眼疾患である、請求項6に記載の予防及び/又は治療剤。
【請求項8】
前記アレルギー性眼疾患が、アレルギー性結膜炎である、請求項7に記載の予防及び/又は治療剤。
【請求項9】
プロパノールを溶媒として用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶の製造方法。
【請求項10】
前記プロパノールが、1−プロパノール又は2−プロパノールである、請求項9に記載の製造方法。
【請求項11】
さらに、メタノールを溶媒として用いて結晶化を行う、請求項9又は10に記載の製造方法。
【請求項12】
アルコール系溶媒とジアルキルケトン溶媒との混合溶媒を用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶の製造方法。
【請求項13】
前記アルコール系溶媒が、メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール又はヘプタノールである、請求項12に記載の製造方法。
【請求項14】
前記ジアルキルケトン溶媒が、次式(4):
【化1】
〔式中、R1及びR2は、互いに同一又は異なっていてもよいC1-6アルキル基を示す。〕
である、請求項12又は13に記載の製造方法。
【請求項15】
前記ジアルキルケトン溶媒が、メチルイソブチルケトンである、請求項12又は13に記載の製造方法。
【請求項1】
X線回折スペクトルにおいて、回折角(2θ)=7.60付近、9.28付近、17.84付近、22.42付近及び27.48付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶。
【請求項2】
融点が約112℃±1.0℃である、請求項1に記載の結晶。
【請求項3】
X線回折スペクトルにおいて、回折角(2θ)=17.10付近、22.78付近、24.14付近、25.96付近、26.40付近及び28.54付近に特徴的なピークを有する、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶。
【請求項4】
融点が約126.2±0.5℃である、請求項3に記載の結晶。
【請求項5】
請求項1〜4のいずれか一項に記載の結晶を含有する医薬組成物。
【請求項6】
請求項1〜4のいずれか一項に記載の結晶を含有するアレルギー性疾患の予防及び/又は治療剤。
【請求項7】
前記アレルギー性疾患が、アレルギー性眼疾患である、請求項6に記載の予防及び/又は治療剤。
【請求項8】
前記アレルギー性眼疾患が、アレルギー性結膜炎である、請求項7に記載の予防及び/又は治療剤。
【請求項9】
プロパノールを溶媒として用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶の製造方法。
【請求項10】
前記プロパノールが、1−プロパノール又は2−プロパノールである、請求項9に記載の製造方法。
【請求項11】
さらに、メタノールを溶媒として用いて結晶化を行う、請求項9又は10に記載の製造方法。
【請求項12】
アルコール系溶媒とジアルキルケトン溶媒との混合溶媒を用いて結晶化を行う、7−[3−{4−(N−エトキシエチルベンゾイミダゾール−2−イルメチル)−1−ピペラジニル}プロポキシ]−3,4−ジヒドロ−2H−1,4−ベンゾチアジン−3−オン・ジマレイン酸塩の結晶の製造方法。
【請求項13】
前記アルコール系溶媒が、メタノール、エタノール、プロパノール、ブタノール、ペンタノール、ヘキサノール又はヘプタノールである、請求項12に記載の製造方法。
【請求項14】
前記ジアルキルケトン溶媒が、次式(4):
【化1】
〔式中、R1及びR2は、互いに同一又は異なっていてもよいC1-6アルキル基を示す。〕
である、請求項12又は13に記載の製造方法。
【請求項15】
前記ジアルキルケトン溶媒が、メチルイソブチルケトンである、請求項12又は13に記載の製造方法。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2013−35773(P2013−35773A)
【公開日】平成25年2月21日(2013.2.21)
【国際特許分類】
【出願番号】特願2011−172256(P2011−172256)
【出願日】平成23年8月5日(2011.8.5)
【出願人】(000163006)興和株式会社 (618)
【Fターム(参考)】
【公開日】平成25年2月21日(2013.2.21)
【国際特許分類】
【出願日】平成23年8月5日(2011.8.5)
【出願人】(000163006)興和株式会社 (618)
【Fターム(参考)】
[ Back to top ]