
ベンダムスチンの固体投与剤
本発明では、経口投与に適した固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択される医薬的に許容できる糖類である少なくとも1種の医薬的に許容できる賦形剤とを含み、活性成分と糖類賦形剤の質量比が1:1〜5の範囲内である、医薬組成物を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物を含む経口投与用固体剤に関する。
【背景技術】
【0002】
(発明の背景)
ベンダムスチン(4-[5-[ビス(2-クロロエチル)アミノ]-1-メチルベンズイミダゾ-2-イル]ブタン酸、ナイトロジェンマスタード)は、二官能性アルキル化活性のあるアルキル化薬である。それは下記式(I)に相当する。
【0003】
【化1】

【0004】
ベンダムスチンは、他のアルキル化薬と如何なる交差耐性もないようであり、このことはアルキル化薬による治療を既に受けたことのある患者の化学療法の観点から利点を提供する。
ベンダムスチンは最初にドイツ民主共和国(GDR)で合成された。ベンダムスチンの塩酸は、1971〜1992年に商標名Cytostasan(登録商標)で入手可能な市販製品の活性成分だった。その時以来、それは独国で商標名Ribomustin(登録商標)にて市販され、慢性リンパ性白血病、非ホジキンリンパ腫及び多発性骨髄腫を治療するために広く使用されてきた。
市販製品は、ベンダムスチン塩酸塩の凍結乾燥粉末を含み、これを注射用水で再構成して濃縮物を得る。これを引き続き0.9%の塩化ナトリウム水溶液で希釈すると、注入用の最終溶液となる。この最終溶液を約30〜60分の時間をかけて静脈内注入で患者に投与する。
水中におけるベンダムスチンのビス-2-クロロエチルアミノ基の加水分解が効力の低下及び不純物形成をもたらす(B. Maas et al. (1994), Pharmazie 49: 775-777)。従って普通は病院内又は少なくとも医学的管理下で、凍結乾燥粉末の再構成後即座に投与を行なわなければならない。さらに、再構成は難しいと報告されている。再構成は30分より長い時間を要することがある。さらに、それは製品を2工程プロセスで再構成する責任がある医療専門家にとって厄介であり、時間がかかる。
【0005】
Preissら(1985)(Pharmazie 40:782-784)は、それぞれ4.2〜5.5mg/kgの範囲の用量で静脈内及び経口投与した後の7名の患者の血漿中のベンダムスチン塩酸塩の薬物動態を比較した。商業的に入手可能なCytostasan(登録商標)製品から調製した静脈内注射を3分かけて行ない、一方で等価用量の経口薬は25mgのベンダムスチン塩酸塩を含むカプセル剤の形を取った。患者が摂取すべきカプセル剤の数は10〜14で変動した。経口投与後1時間以内で最大血漿レベルが検出できた。平均経口バイオアベイラビリティーを計算すると57%だったが、25%〜94%まで変動し、個体間の大きい可変性を示唆している。
Weber(1991)(Pharmazie 46(8): 589-591)は、B6D2F1マウスにおけるベンダムスチン塩酸塩のバイオアベイラビリティーを調査し、胃腸管からの該薬物の吸収が不完全であり、たった約40%のバイオアベイラビリティーしかもたらさないことを見い出した。
US2006/0128777A1は、一般に死抵抗性細胞及びベンダムスチン含有組成物によって特徴づけられる癌の治療方法を記載している。これらの中で組成物は、カプセル剤、錠剤、丸剤、散剤又は顆粒剤である経口固体剤形であり、その中で活性化合物が少なくとも1種の不活性な賦形剤、例えばスクロース、ラクトース又はデンプンと混合され得る。しかしながら、具体的な組成物は例示されなかった。
【発明の概要】
【発明が解決しようとする課題】
【0006】
一度水で再構成した市販のi.v.製剤の安定性の問題を考慮して、かつ患者のコンプライアンスを改善するため、患者に投与しやすく、かつ既知の経口剤形に比べて可変性が少ないバイオアベイラビリティーの向上をもたらす、ベンダムスチンを含む安定な経口剤形が長年にわたって要望されている。
【課題を解決するための手段】
【0007】
(発明の概要)
上記課題を解決するため、本発明者らは詳細な調査を行なった。本発明者らは、最終的に本発明の安定医薬組成物を得ることに成功した。これらの組成物は、経口投与に適しており、かつ活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、少なくとも1種の医薬的に許容できる賦形剤とを含み、本組成物は、改善された溶解プロファイルを有する。
【図面の簡単な説明】
【0008】
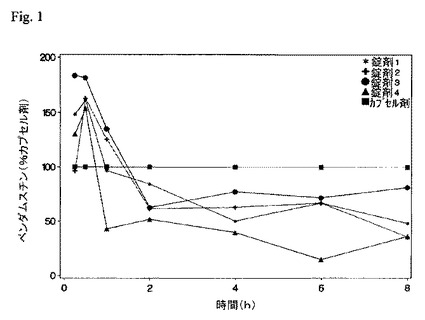
【図1】従来技術のカプセル剤並びに実施例6〜8の錠剤製剤(錠剤1〜3)及び実施例9の錠剤製剤(製剤3)(表4)の形態でベンダムスチン塩酸塩をイヌに投与した後に得られた平均血漿濃度(錠剤対カプセル剤)対時間曲線を示す。図1から、従来技術のカプセル剤に比し、本錠剤製剤がベンダムスチンのより高い最大濃度を提供することが明らかである。
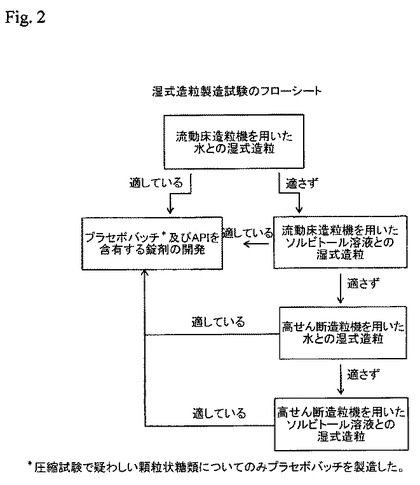
【図2】湿式造粒製造試験のフローシートを示す。
【発明を実施するための形態】
【0009】
(発明の詳細な説明)
本発明は、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、単糖類、二糖類、オリゴ糖類、環状オリゴ糖類、多糖及び糖アルコール類から選択される少なくとも1種の医薬的に許容できる賦形剤とを含む医薬組成物に関する。好ましくは、活性成分と賦形剤の質量比は、1対1〜5、好ましくは1対2〜5の範囲内、さらに好ましくは1:5〜1:2から選択される比である。
一実施形態では、本発明は、経口投与用固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択される医薬的に許容できる糖類である少なくとも1種の医薬的に許容できる賦形剤とを含み、前記活性成分と賦形剤の質量比が1:1の範囲内である、医薬組成物に関する。
さらなる実施形態では、本発明は、経口投与に適した固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択される医薬的に許容できる糖類である少なくとも1種の医薬的に許容できる賦形剤とを含み、前記活性成分と糖類賦形剤の質量比が1:2〜5の範囲内であり、かつ該組成物が、欧州薬局方(European Pharmacopoeia)に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す、医薬組成物に関する。
上記実施形態の範囲内のさらに好ましい実施形態は、前記医薬的に許容できる糖類が、1種以上の単糖、二糖及びオリゴ糖から成る群より選択され、前記活性成分と糖類賦形剤の質量比が1:2〜5の範囲内であり、かつ該組成物が、欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す、医薬組成物である。
【0010】
本発明は、医薬組成物に特定量の医薬的に許容できる糖類を組み入れることによって、特有の望ましい溶解プロファイルに到達できるという驚くべき発見に基づいている。
活性成分としてベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物を含む医薬組成物中の賦形剤として、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択され、好ましくは1種以上の単糖、二糖及びオリゴ糖から成る群より選択される医薬的に許容できる糖類を使用すると、安定性、打錠特性、溶解及び不純物形成に関して特に有利なプロファイルが達成されることが分かった。上記糖類は、欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す組成物をもたらす。
本発明の上記範囲内では、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールのいずれの組合せをも使用してよい。
特に、特定の糖類が安定性及び溶解に関して医薬組成物の特に有利なプロファイルと関係があることが分かった。本発明の組成物の好ましい糖類は、ブドウ糖無水物、ブドウ糖一水和物、ラクチトール一水和物、トレハロース、ソルビトール、エリスリトール、マルトース一水和物、マンニトール、ラクトース無水物、ラクトース一水和物、マルチトール、キシリトール、ショ糖、ショ糖97%+マルトデキストリン3%、β-シクロデキストリン、D-ラフィノース五水和物、D-メレジトース一水和物及び微結晶性セルロースである。本発明の医薬組成物は良い打錠特性、速い溶解及び医薬的に許容できる安定性を示す。
上記糖類は本発明の好ましい実施形態を構成し、そのいずれの組合せをも使用し得る。好ましくは、活性成分と上記糖類の比は、1:1〜5、好ましくは1:2〜5の範囲内、さらに好ましくは1:5〜1:2から選択される比である。
本発明のさらに好ましい実施形態は、経口投与用固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、ブドウ糖無水物、ブドウ糖一水和物、ラクチトール一水和物、トレハロース、ソルビトール、エリスリトール、マルトース一水和物、マンニトール、ラクトース無水物、ラクトース一水和物、マルチトール、キシリトール、ショ糖、ショ糖97%+マルトデキストリン3%、β-シクロデキストリン、D-ラフィノース五水和物、D-メレジトース一水和物及び微結晶性セルロースから選択される少なくとも1種の医薬的に許容できる賦形剤とを含み、かつ該組成物が、10分で少なくとも60%、20分で70%及び30分で80%のベンダムスチンの溶解を示す、医薬組成物である。
特に好ましい糖類は、マンニトール、マルチトール、エリスリトール、キシリトール、ラクトース、ショ糖、グルコース、ソルビトール、マルトース、トレハロース、ラクチトール及びブドウ糖(無水物又は一水和物)であり、活性成分と前記糖類の質量比は、好ましくは1:2〜5の範囲内である。上記糖類の範囲内の2種以上の組合せも本発明の範囲に包含される。
当業者は、上記糖類賦形剤の範囲内で適切な組合せを選択し、かつ欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す組成物を得るのに十分な立場にいる。
【0011】
好ましい実施形態では、本組成物は、錠剤、顆粒剤、又は丸剤の形態である。
好ましい剤形は錠剤である。錠剤という用語は、速崩壊錠剤をも含み、その中には分散性錠剤及び発泡性錠剤がある。
錠剤調製の最も一般的に用いられる方法は、直接圧縮、乾式造粒及び湿式造粒である。直接圧縮は、活性成分と賦形剤を含む混合物を錠剤プレス機で圧縮する工程を含む(L. Lachman et al., in: The Theory and Practice of Industrial Pharmacy, 3rd ed., 1986)。均一含量の活性成分を有する錠剤を作るためには、圧縮すべき混合物が良い流動及び圧縮の両特性を持たなければならない。混合物に適切な賦形剤、例えば潤沢剤、付着防止剤及び流動促進剤などを添加することによって常に良い流動特性を達成できるわけではない。従って、多くの場合、圧縮前に混合物を造粒する。
造粒は、粉末混合物から顆粒と呼ばれる球様又は規則的形状の凝集体を形成するプロセスである。これは、乾式造粒法及び湿式造粒法によって達成され得る。造粒は、凝集性が乏しい粉末混合物を、圧縮したときに良い粘着特性を有する錠剤をもたらす凝集体に変換するためにも利用される。
速崩壊錠剤の場合、必要に応じて1種以上の賦形剤との混合剤の活性成分は、該成分の味を隠すため並びに/或いは該成分を光及び/又は湿気によって起こり得る有害な作用に対して保護するため並びにベンダムスチンの場合は活性化合物によってもたらされる有害な作用に対して口内の粘膜を保護するため、有利にはコーティングを備えている。当該目的のため、好ましくはさらに以下に概要を示すように顆粒(granulate)を調製及び加工する。
【0012】
「顆粒(granulate)」という表現は、顆粒(granule)と呼ばれることもある粒子の凝集体を表す。顆粒は、一般的にコンパクション及び/若しくは圧縮法(乾式造粒)又は必要に応じて湿式造粒結合剤が溶解している液体を利用する湿式造粒法によって調製される(Remington's Pharmaceutical Sciences 18th ed. 1990, page 1641)。湿式造粒法には押出法も含まれる。従って、顆粒という用語は、ペレット、小球、及び押出品をも含み、顆粒の例として好ましくはペレットを用いる。
ペレットは直径が約1.0〜1.6mmで、特定の密度を有する小粒子として表され、この粒子は粉末混合物に押出し及び球形化の調剤プロセスを適用することで調製される。
必要に応じて1種以上の賦形剤との混合剤の活性成分は、該成分の味を隠すため並びに/或いは該成分を光及び/又は湿気によって起こり得る有害な作用に対して保護するため並びに/或いは活性化合物によってもたらされる有害な作用に対して口内の粘膜を保護するため、有利にはコーティングを備えていてよい。
丸剤は、活性成分をトリグリセリドの柔らかい混合物に添加することによって調製される小円形固体剤形である。混合物をローラーで伸ばして長いストリングにしてからバラバラにカットしてローラーで伸ばす(J.T. Carstensen: Pharmaceutical principles of solid dosage forms, 1993, Technomic Publishing Company, Inc. page 63)。
好ましくは本発明の剤形は乾式コンパクション法によって調製される。適切な技法は、例えばRemington's Pharmaceutical Science 18th. ed. 1990, page 1644に記載されている。それらの技法は乾式造粒、ローラーコンパクション及び直接圧縮を含む。錠剤をこれらの技法で調製する場合、直接圧縮を利用することがなおさらに有利である。
【0013】
本発明の剤形は好ましくはコーティングを備えている。コーティングには種々の目的がある。コーティングは、組成物中で使用される活性成分の味を隠すために役立ち得る。さらに同時にコーティングは、光及び/又は湿気によって起こり得る有害な作用、例えば酸化、分解などに対して活性成分を保護している。さらに、コーティング層は活性成分による口腔粘膜の損傷から対象を保護することができる。
技術上周知の技法、例えば噴霧コーティング及びマイクロカプセル化等によってコーティング層を剤形に適用することができる。錠剤では、それはフィルムコーティング、糖類コーティング又は圧縮コーティングの形態であり得る。好ましくはフィルムコーティングプロセスを使用する(Remington's Pharmaceutical Sciences 18th ed. 1990, page 1666)。活性成分が速崩壊錠剤用コーティングの適用を必要とする場合、錠剤への圧縮前に個々の顆粒が適宜コーティングを備えていてよい。
【0014】
「その医薬的に許容できるエステル」という表現は、ベンダムスチンのいずれの医薬的に許容できるエステル、例えばアルキルアルコール及び糖アルコールとのエステルをも表す。アルキルアルコールの例は、C1-6-アルキルアルコール、例えばメタノール、エタノール、プロパノール、イソプロパノール、ブタノール及びtert-ブタノールである。糖アルコールの例は、マンニトール、マルチトール、ソルビトール、エリスリトール、グリコール、グリセロール、アラビトール、キシリトール及びラクチトールである。ベンダムスチンエステルの好ましい例は、エチルエステル、イソプロピルエステル、マンニトールエステル及びソルビトールエステルであり、ベンダムスチンのエチルエステルが最も好ましい。
「その医薬的に許容できる塩」という表現は、患者に(直接又は間接的に)投与されるとベンダムスチンを与える、ベンダムスチンのいずれの医薬的に許容できる塩をも表す。この用語は、ベンダムスチンエステルの医薬的に許容できる塩をさらに含む。それにもかかわらず、医薬的に許容できない塩も、これらの化合物が医薬的に許容できる塩の調製に役立つことがあるので、本発明の範囲内に含まれる。例えば、ベンダムスチンの医薬的に許容できる塩は、従来の化学的方法によって、酸性又は塩基性基を含む対応化合物から合成される。一般的に、これらの塩は、例えば、水若しくは有機溶媒又は両者の混合物中における化学量論のこれらの化合物の遊離酸性形又は塩基性形と対応する塩基又は酸との反応を利用して調製される。一般的にエーテル、酢酸エチル、イソプロパノール又はアセトニトリル等の非水性媒体が好ましい。ベンダムスチンの医薬的に許容できる塩の塩形成に使用できる酸の例としては、無機酸、例えば塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、及びリン酸など、並びに有機酸、例えば酢酸、マレイン酸、フマル酸、クエン酸、シュウ酸、コハク酸、酒石酸、リンゴ酸、乳酸、メチルスルホン酸及びp-トルエンスルホン酸などが挙げられる。ベンダムスチンの医薬的に許容できる塩を無機又は有機塩基から誘導してアンモニウム塩;アルカリ金属塩(リチウム、ナトリウム、カリウム等)、カルシウム若しくはマグネシウム等のアルカリ土類金属の塩、アルミニウム塩、低級アルキルアミン塩、例えばメチルアミン若しくはエチルアミン塩、低級アルキルジアミン塩、例えばエチレンジアミン塩、エタノールアミン、N,N-ジアルキレンエタノールアミン、トリエタノールアミン、及びグルカミン塩、並びにアミノ酸の塩基性塩を得ることができる。塩酸、臭化水素酸、及びヨウ化水素酸から調製された酸性塩が特に好ましいが、塩酸塩がベンダムスチンの最も好まし医薬的に許容できる塩である。医薬的に許容できる塩は、技術上周知の従来法によって作られる。
【0015】
「その医薬的に許容できる溶媒和物」という表現は、患者に投与されると(直接又は間接的に)ベンダムスチンを与える、医薬的に許容できるいずれの溶媒和物をも表す。この用語は、ベンダムスチンエステルの医薬的に許容できる溶媒和物をさらに含む。好ましくは溶媒和物は水和物、メタノール、エタノール、プロパノール、若しくはイソプロパノール等のアルコールとの溶媒和物、酢酸エチル等のエステルとの溶媒和物、メチルエーテル、エチルエーテル若しくはTHF(テトラヒドロフラン)等のエーテルとの溶媒和物又はDMF(ジメチルホルムアミド)との溶媒和物であり、水和物又はエタノール等のアルコールとの溶媒和物がさらに好ましい。溶媒和物を構成するための溶媒は、好ましくは医薬的に許容できる溶媒である。
本発明の組成物中の活性成分がベンダムスチン又はその医薬的に許容できる塩であることが特に好ましい。活性成分がベンダムスチン塩酸塩であることが最も好ましい。
医薬組成物中の活性成分の用量は、患者の状態、性別、体重、体表面積、又は年齢に応じて、特に患者の体重及び体表面積に応じて当業者によって容易に決定され、10〜1000mgの範囲である。1日の投薬量が約50〜約1000mg、好ましくは約100〜約500mgの活性成分の範囲であることが好ましい。1日の投薬量を単回用量又は1日に2回若しくは3回などの複数回用量として摂取してよく、最も好ましくは単回1日用量として摂取する。1日用量を1週間に1回又は1週間に数回摂取してよい。剤形は、単回1日用量又はその一部の量を含有し得る。本発明の剤形が約10〜約1000mg、好ましくは約25〜約600mg、さらに好ましくは約50〜約200mg、最も好ましくは約100mgの活性成分を含むことが好ましい。
本発明の組成物には、実質的な量、好ましくは活性物質の質量の2〜5倍の範囲の量で糖類が存在する。糖類は、本発明の組成物に組み入れられると、活性化合物の安定性にプラス効果を及ぼすことが分かった。そのことに加えて、これらの賦形剤は驚くべきことに、参考カプセル剤と比較した場合、活性化合物、特にベンダムスチン塩酸塩のバイオアベイラビリティーの向上をもたらすことが分かった。
糖類の好ましい例としては、マンニトール、マルチトール、エリスリトール、キシリトール、ラクトース、ショ糖、グルコース、ソルビトール、マルトース、トレハロース、ラクチトール及びブドウ糖(無水物又は一水和物)が挙げられる。
【0016】
これらの糖類賦形剤に加えて、本発明の医薬組成物は、さらに以下に詳述するさらなる賦形剤、例えば潤沢剤、流動促進剤、充填剤(又は希釈剤)、結合剤及び崩壊剤を含んでよい。
潤沢剤は、医薬組成物において、特に錠剤製造において下記機能の1つ以上を有し得る物質である:錠剤機の部品(ホッパー、ダイ及びパンチ)の表面への錠剤材料の付着を防止すること、粒子間摩擦を低減させること、ダイからの錠剤の放出を促進すること及び(打錠すべき)混合物の流速を改善すること。前記潤沢剤は、典型的にステアリン酸、ステアリン酸の塩若しくはエステル、水素化植物油、酸化マグネシウム、ポリエチレングリコール、ラウリル硫酸ナトリウム及びタルク、並びにその混合物から成る群より選択される。好ましくは前記潤沢剤は、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、グリセリルパルミトステアラート及びステアリルフマル酸ナトリウム、並びにその混合物から選択される。ステアリン酸が最も好ましい選択肢である。
この出願では、流動促進剤という用語は、打錠すべき混合物の流動特性を改善する物質として解釈するものとする。流動促進剤については、タルク、二酸化ケイ素及びシリカゲル(Cab-O-Sil(登録商標)、Syloid(登録商標)、Aerosil(登録商標))、デンプン及びケイ酸カルシウム等のいずれの適切な流動促進剤をも使用し得る。典型的に、二酸化ケイ素を使用する。
一般的に、充填剤(又は希釈剤)という用語は、打錠すべき材料のバルクを増やすために使用する賦形剤を表す。このサイズの増加が固体組成物の取扱いを改善する。充填剤は通常、固体組成物毎の薬物の用量が低く、充填剤がなければ固体組成物が小さ過ぎる場合に必要である。適切な充填剤の例は、ラクトース、ショ糖、マンニトール、ソルビトール、サッカロース、デンプン、アルファ化デンプン、微結晶性セルロース、粉末セルロース、リン酸水素カルシウム、炭酸カルシウム及びそのいずれかの組合せである。好ましい実施形態では、充填剤は、ラクトース、デンプン、微結晶性セルロース、ミクロファインセルロース及びそのいずれかの組合せ、最も好ましくは無水ラクトース及び微結晶性セルロースから選択される。
【0017】
一般的に、結合剤という用語は、医薬製剤に凝集性を付与する薬剤のために使用され、凝集性は、特に圧縮後の錠剤の場合に組成が損なわれないままでいることを保証する。使用するコンパクション法(直接圧縮、乾式造粒又は湿式造粒)に応じて、異なる結合剤を使用する。乾式コンパクション法(直接圧縮及び乾式造粒)では、適切な結合剤はラクトース、ショ糖、マンニトール、ソルビトール、サッカロース、デンプン、アルファ化デンプン、微結晶性セルロース、粉末セルロース、リン酸水素カルシウム、炭酸カルシウム及びそのいずれかの組合せである。好ましい実施形態では、結合剤は、ラクトース、デンプン、微結晶性セルロース、ミクロファインセルロース及びそのいずれかの組合せ、最も好ましくは無水ラクトース及び微結晶性セルロースから成る群より選択される。湿式造粒プロセスでは、溶液として及び乾燥形態の両方で結合剤を使用することができる。適切な結合剤として、例えば、ポリビニルピロリドン、分散性セルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、メチルセルロース、デンプン、アルファ化デンプン、部分的アルファ化デンプン、アラビアゴム、デキストリン、プルラン等が挙げられる。これらの結合剤の中で、分散性セルロース、ポリビニルピロリドン、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチル セルロースがさらに好ましい。
【0018】
医薬組成物、特に錠剤組成物に崩壊剤を含めて、錠剤が生物学的水性液と接触した後のその分解又は崩壊を促進することができる。錠剤が嚥下されると、多くの場合、唾液、胃液及び腸液などの体液と錠剤が接触するときに錠剤の迅速な崩壊に崩壊剤が関与する。崩壊剤として役立つ物質は、化学的にデンプン、セルロース、架橋ポリマー等に分類されている。本発明の実施で使用すべき崩壊剤種及びその添加レベルに関する調査の結果として、デンプン、加工デンプン、例えばナトリウムデンプングリコラート(Primojel(登録商標))、ナトリウムカルボキシメチルセルロース、架橋カルボキシメチルセルロースナトリウム(Ac-Di-Sol(登録商標))、架橋ポリビニルピロリドン、ポラクリリン(polacrilin)カリウム(Amberlite(登録商標)IRP88)及び低置換ヒドロキシプロピルセルロースが非常に良い崩壊効果をもたらし得ることが分かった。
ベンダムスチンの水溶液の安定性は、pHによって強く影響を受ける。約5より高いpH値では、加水分解によるこの化合物の有意な分解が観察される。pH>5では、分解が急速に進行し、このpH範囲では結果として生じる副生物の含量が多い。主な加水分解生成物は、下記4-[5-[(2-クロロエチル)-(2-ヒドロキシ-エチル)アミノ]-1-メチル-ベンズイミダゾ-2-イル]-ブタン酸(HP1)、4-[5-[ビス(2-ヒドロキシエチル)アミノ]-1-メチル-ベンズイミダゾ-2-イル]-ブタン酸(HP2)及び4-(5-モルフォリノ-1-メチルベンズイミダゾール-2-イル)-ブタン酸(HP3)である。
【0019】
【化2】
【0020】
経口投与された薬物の吸収は、普通は胃、小腸及び/又は大腸内で起こる。胃内のpHは約1〜3.5であり、小腸内では約6.5〜7.6、大腸内では約7.5〜8.0である。従って、5より高いpHの水性環境内で分解しやすいベンダムスチンのような化合物では、分解を回避するため、それが胃内で吸収され、小腸或いは大腸をも通過しないことが非常に好ましい。従って、ベンダムスチンが胃内で完全に又は少なくとも高程度まで吸収され、それによって小腸又は大腸内でのベンダムスチンの分解を回避又は低減させる医薬組成物が要望されている。
出願人は、驚くべきことに本発明の医薬組成物、特に、上記好ましい糖類を有する医薬組成物を用いてこの問題を解決できることを見い出した。ベンダムスチンを含むこれらの組成物は、速い溶解、特に欧州薬局方に準拠して50rpmでパドル装置を用いて人工胃液中で測定した場合、20分、好ましくは10分で少なくとも60%、40分、好ましくは20分で70%、60分、好ましくは30分で80%、最も好ましくは10分で少なくとも75%、20分で85%、30分で90%のベンダムスチンの溶解を示す。本明細書で使用する場合、人工胃液は、1000mlの水に2gの塩化ナトリウムを溶解させてから5N塩酸でpHを1.5±0.05に調整することによって調製された溶液を表す。
薬物が胃を通って小腸に至るまでの全時間は約20分〜5時間、普通は約30分〜3時間である。従って、この発明の組成物は、胃内にある間にベンダムスチンが大部分の程度まで放出かつ溶解されるので、患者内でのベンダムスチンの分解を有利に低減させ、結果として本発明のベンダムスチン含有組成物のバイオアベイラビリティーを改善することとなるであろう。
【0021】
この発明のさらなる態様では、固体剤形の医薬組成物を、ヒト又は動物、好ましくはヒトにおける医学的状態の治療、導入、救命療法、幹細胞移植前の前処置、維持療法、残存疾患の治療のために使用し得る。この医学的状態は、慢性リンパ性白血病(CLL)、急性リンパ性白血病(ALL)、慢性骨髄性白血病(CML)、急性骨髄性白血病(AML)、ホジキン病、非ホジキンリンパ腫(NHL)、多発性骨髄腫、乳癌、卵巣癌、小細胞肺癌、非小細胞肺癌、及び自己免疫疾患から選択される。
本発明は、人体又は動物体における慢性リンパ性白血病、急性リンパ性白血病、慢性骨髄性白血病、急性骨髄性白血病、ホジキン病、非ホジキンリンパ腫、多発性骨髄腫、乳癌、卵巣癌、小細胞肺癌、非小細胞肺癌、及び自己免疫疾患から選択される医学的状態の治療方法であって、有効量のこの発明の医薬製剤を、治療が必要な人体又は動物体に投与する工程を含む方法をも含む。好ましくは医学的状態が非ホジキンリンパ腫である。
この発明の別の態様では、医薬組成物を少なくとも1種のさらなる活性薬と併用投与することができる。ここで、前記さらなる活性薬は、本医薬組成物の投与前、投与と同時、又は投与後に与えられる。この少なくとも1種のさらなる活性薬は、好ましくはCD20に特異的な抗体(例えばリツキシマブ又はオファツムマブ)、アントラサイクリン誘導体(例えばドキソルビシン又はダウノルビシン)、ビンカアルカロイド(例えばビンクリスチン)、プラチン誘導体(例えばシスプラチン又はカルボプラチン)、ダポリナド(daporinad)(FK866)、YM155、サリドマイド及びその類似体(例えばサリドマイド又はレナリドマイドである)、又はプロテアソーム阻害薬(例えばボルテズミブ(bortezumib))である。
この発明の医薬組成物を少なくとも1種のコルチコステロイドと併用投与してもよく、前記コルチコステロイドは、本医薬組成物の投与前、投与と同時、又は投与後に与えられる。コルチコステロイド例はプレドニゾン、プレドニゾロン及びデキサメタゾンである。
以下の実施例は、本発明をさらに説明する。当業者には当然のことながら、これらの実施例は単に説明の目的のためであり、本発明を限定するものと考えるべきでない。
【実施例】
【0022】
1.適合性試験
実施例1a
適合性試験のため、ベンダムスチン塩酸塩と賦形剤を1:1(m/m)の比で含む混合物を調製した。マンニトール及びラクトースから賦形剤を選択した。調製後、混合物をクリアガラスHPLC-バイアル(6ml)Agilentに入れ、下表1に示す種々の貯蔵条件で貯蔵した。規定時点でサンプルを貯蔵から取り出して純度(HPLC;カラム:Zorbax Bonus-RP、5μm;カラムオーブンの温度:30℃;オートサンプラーの温度:5℃;検出器:254nm)及び外観について試験した。
【0023】
表1:貯蔵条件
*50℃で1カ月貯蔵後、70℃で貯蔵
**25℃/60%r.h.で1カ月貯蔵後40℃/75%で貯蔵
【0024】
全てのこれらの混合物では、ベンダムスチン塩酸塩含量(HPLCで測定)はほとんど変化せず、全てのこれらの貯蔵条件で常に99%を超えたままだった。加水分解生成物HP1はこれらの全ての貯蔵条件でほとんど検出されなかった(面積%<0.2)。
指定ベンダムスチン塩酸塩混合物の外観試験を裸眼で行なった。調査した全ての混合物は仕様に従い、調製直後及び全てのこれらの貯蔵条件下で1カ月の貯蔵後の両方で白色乃至オフホワイトの粉末を与えた。
【0025】
実施例1b
実施例1aの方法に従うさらなる適合性試験のため、ベンダムスチン塩酸塩と賦形剤を1:1(m/m)の比で含む混合物を調製した。Opadry(登録商標)、Eudragit(登録商標)E PO、ナトリウムカルボキシメチルセルロース(Avicel(登録商標) RC 591)及び架橋ポリビニルピロリドン(Crospovidone)から賦形剤を選択した。
Eudragit(登録商標)E POの場合、不純物HP1(加水分解生成物)及びBM1DIMERの初期量が有意に増加したが(HP1:1.5%、BM1DIMER:1%)、貯蔵中に湿度の影響とは無関係に全ての貯蔵条件でこれらの不純物の減少を検出することができた。架橋ポリビニルピロリドンの場合、貯蔵条件40℃/75%R.H./開いたバイアルではHP1の0.1%から0.4%への有意な増加を検出できた。全ての他の貯蔵条件(閉じたバイアル)ではHP1の増加を検出できなかった。
Eudragit(登録商標)E PO及び架橋ポリビニルピロリドンを含む混合物の外観は、貯蔵条件70℃/閉じたバイアルで変化した。両混合物はわずかに粘着性になった。さらに架橋ポリビニルピロリドンとの混合物の色が白色からクリーム色に変化した。
Opadry(登録商標)及びAvicel(登録商標)RC591を含む混合物の場合も、貯蔵条件70℃/閉じたバイアルで色がクリーム色に変わった。
【0026】
2. 錠剤製剤
実施例2
主賦形剤としてマンニトールを含み、下表2aに示す相対量で微結晶性セルロース、Ac-Di-Sol(登録商標)、コロイド状二酸化ケイ素、タルク及びステアリン酸を含む混合物253gを1リットルのキューブブレンダー(Erweka)で15分間混合することによって調製した。その後、10.612gの混合物と3.0gのベンダムスチン塩酸塩を0.425mmの篩いを通して選別してから、50mlのガラスバイアルを備えたTurbulaミキサーT2Aに移し、引き続き60rpmで10分間混合した。
この混合物から、以下の特性を有する円形錠剤を圧縮成形した:
平均粒径:9.1mm;平均値質量:247.7mg;平均値硬度:81N。
【0027】
【0028】
錠剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩並びに分解生成物、合成の副生物のような関連物質の量をHPLCで測定した(カラム:Zorbax Bonus-RP、5μm;カラムオーブンの温度:30℃;オートサンプラーの温度:5℃;検出器:254nm)。結果を下表2bに示す。
【0029】
*1:NP1:4-[6-(2-クロロエチル)-3,6,7,8-テトラ-ヒドロ-3-メチル-イミダゾ[4,5-h]-[1,4]ベンゾチアジン-2-イル]ブタン酸
BM1Dimer:4-{5-[N-(2-クロロエチル)-N-(2-{4-[5-ビス(2-クロロエチル)アミノ-1-メチルベンズイミダゾール-2-イル]ブタノイルオキシ}エチル)アミノ]-1-メチルベンズイミダゾール-2-イル}ブタン酸
BM1EE:4-[5-[ビス(2-クロロエチル)アミノ]-1-メチル-ベンズイミダゾ-2-イル]ブタン酸エチルエステル
*2:n.d.:検出不能、すなわち検出限界超え(0.05%未満の面積パーセンテージ)
【0030】
実施例3
実施例2で述べたのと同様に混合物及び錠剤を調製したが、下表3aに示す化合物及び相対量を用いた。
錠剤は以下の特徴を有した:
平均値径:9.1mm;平均値質量:248.9mg。
【0031】
【0032】
錠剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表3bに示す。
【0033】
【0034】
実施例4
実施例2で述べたのと同様に錠剤を調製したが、下表4aに示した化合物及び相対量を用いた。
錠剤は以下の特徴を有した:
平均値径:9.1mm;平均値質量:247.8mg。
【0035】
【0036】
錠剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表4bに示す。
【0037】
【0038】
従来技術の参考例
20.0±1mgのベンダムスチン塩酸塩を空の硬ゼラチンカプセルのボディに量り入れ、AgilentのクリアガラスHPLCバイアル(6ml)に入れた。ボディの上部にキャップを置いてわずかに押すことによってカプセルを閉じた。このカプセル剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表5に示す。
【0039】
【0040】
すぐに分かるように、いずれのさらなる加工工程もなしで純粋なベンダムスチン塩酸塩からカプセル製剤を調製したが、カプセル製剤は、本発明の錠剤製剤より不安定なロットだった。40℃/75%RH(開いたガラスバイアル)及び50℃(閉じたバイアル)の両方で1カ月の貯蔵内に加水分解生成物が形成された。40℃及び75%RH(相対湿度)で開いたバイアルの場合、1カ月の貯蔵後に加水分解生成物HP1の量が4倍に増えた。閉じたバイアルではHP1含量がさらに高く、これはカプセルとの反応のためであろう。要約すると、錠剤はカプセル剤よりずっと安定な固体剤形を提供した。
【0041】
実施例5
8.0gのヒドロキシプロピルメチルセルロース及び1.5gのPEG6000を88.5gの精製水に溶解させる。その後2.0gの黄色酸化第二鉄及び0.5gの酸化チタンをその中に分散させてコーティング液を得る。実施例2で得た錠剤を、フィルムコーティング装置を用いて錠剤質量毎にこの溶液3%でコーティングする。
【0042】
実施例6
【0043】
(1000錠剤の製造方法)
コロイド状二酸化ケイ素及びステアリン酸以外の全ての錠剤コア成分をSomakon容器(5L)中に装填した。ベンダムスチンを加えて4分間1000rpm(ワイパー10rpm)でブレンディングを行なった。結果として生じたブレンドを0.5mmの篩いを通して選別した。容器にブレンドを装填し、コロイド状二酸化ケイ素を加えた。上記条件でブレンディングを2分間行なった。その後ステアリン酸を加え、ブレンディングを1分間続けた。引き続きブレンドを0.5mmの篩いを通して選別し、再び容器中に装填し、全て同一条件でさらに30秒間ブレンドした。
この混合物から下記特徴を有する円形錠剤を圧縮成形した:
平均値径:9.5mm;平均値質量:254.6mg(初め)〜257.2mg(終わり);脆砕性0.1%;平均値硬度:122N(初め)〜128N(終わり)。
引き続き錠剤をOpadry(登録商標)分散系で5%の質量増加が達成されるまでフィルムコーティングした。
フィルムコーティング錠の平均質量は268.4mgだった。
錠剤コアとフィルムコーティング錠を両方とも閉じた琥珀色ガラスバイアル内で40℃/75%RHにて貯蔵した。ベンダムスチン塩酸塩並びに分解生成物、合成の副生物のような関連物質の量を上述したようにHPLCで測定した。結果を下表6b.1及び6b.2に示す。
【0044】
*3:主ピークと比べて0.69の相対保持時間の未同定化合物ピーク
【0045】
【0046】
実施例7
【0047】
(1000錠剤の製造方法)
コロイド状二酸化ケイ素及びステアリン酸以外の全ての錠剤コア成分をSomakon容器(5L)中に装填した。ベンダムスチンを加えて4分間1000rpm(ワイパー10rpm)でブレンディングを行なった。結果として生じたブレンドを0.5mmの篩いを通して選別した。容器にブレンドを装填し、コロイド状二酸化ケイ素を加えた。上記条件でブレンディングを2分間行なった。その後ステアリン酸を加え、ブレンディングを1分間続けた。引き続きブレンドを0.5mmの篩いを通して選別し、再び容器中に装填し、全て同一条件でさらに30秒間ブレンドした。
この混合物から下記特徴を有する円形錠剤を圧縮成形した:
平均値径:9.5mm;平均値質量:262.4mg(初め)〜254.4mg(終わり);脆砕性0.1%(初め)〜0.2%(終わり);平均値硬度:98N(初め)〜91N(終わり)。
引き続き錠剤をEudragit(登録商標)分散系で3%の質量増加が達成されるまでフィルムコーティングした。
フィルムコーティング錠の平均質量は273.5mgだった。
錠剤コアとフィルムコーティング錠を両方とも閉じた琥珀色ガラスバイアル内で40℃/75%RHにて貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表7b.1及び7b.2に示す。
【0048】
【0049】
【0050】
実施例8
【0051】
(1000錠剤の製造方法)
コロイド状二酸化ケイ素及びステアリン酸以外の全ての錠剤コア成分をSomakon容器(2.5L)中に装填した。ベンダムスチンを加えて4分間1000rpm(ワイパー10rpm)でブレンディングを行なった。結果として生じたブレンドを0.5mmの篩いを通して選別した。容器にブレンドを装填し、コロイド状二酸化ケイ素を加えた。上記条件でブレンディングを2分間行なった。その後ステアリン酸を加え、ブレンディングを1分間続けた。引き続きブレンドを0.5mmの篩いを通して選別し、再び容器中に装填し、全て同一条件でさらに30秒間ブレンドした。
この混合物から下記特徴を有する円形錠剤を圧縮成形した:
平均値径:9.5mm;平均値質量:252.2mg(初め)〜250.7mg(終わり);脆砕性0.1%(初め)〜0.2%(終わり);平均値硬度:65N(初め)〜73N(終わり)。
引き続き錠剤をEudragit(登録商標)分散系で3%の質量増加が達成されるまでフィルムコーティングした。
フィルムコーティング錠の平均質量は253.6mgだった。
錠剤コアとフィルムコーティング錠を両方とも閉じた琥珀色ガラスバイアル内で40℃/75%RHにて貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表8b.1及び8b.2に示す。
【0052】
【0053】
【0054】
実施例9
【0055】
600錠剤のための製剤PF1の製造方法:
33.06gのベンダムスチン、111.60gのブドウ糖、40.92gのラクトース、11.22gの微結晶性セルロース及び1.20gのステアリン酸マグネシウムを秤量し、二重ポリエチレンバッグに移して5分間混合した。その後、該粉末ブレンドを偏心性錠剤機(Korsch EK0)のホッパーに移し、下記特徴を有する円形錠剤に圧縮成形した:平均値径:10.0mm;平均値質量:336.9mg(初め)〜335.98(終わり);脆砕性:0.15%;平均硬度値:69.25N(初め)〜68.60N(終わり)。
引き続き錠剤コアをコーティングパン(4M8 ForMate PanCoat)内で9%のOpadry(登録商標)TM White水性懸濁液を用いてコーティングして乾燥させた。錠剤の平均質量は342.42mgだった。その後、ねじ込みプラグで閉じた琥珀色ガラス瓶に錠剤を詰めて40℃/75%RHで貯蔵した。
600錠剤のための製剤PF2の製造方法:
33.06gのベンダムスチン、111.42gのラクトース、39.60gのトレハロース、12.60gの架橋ポリビニルピロリドン及び1.32gのステアリン酸マグネシウムを秤量し、二重ポリエチレンバッグに移して5分間混合した。その後、該粉末ブレンドを偏心性錠剤機(Korsch EK0)のホッパーに移し、下記特徴を有する円形錠剤に圧縮成形した:平均値径:10.0mm;平均値質量:332.95mg(初め)〜332.12(終わり);脆砕性:0.3%;平均硬度値:65.9N(初め)〜59.0N(終わり)。
引き続き錠剤コアをコーティングパン(4M8 ForMate PanCoat)内で9%のOpadry(登録商標)TM White水性懸濁液を用いてコーティングして乾燥させた。錠剤の平均質量は340.1mgだった。その後、ねじ込みプラグで閉じた琥珀色ガラス瓶に錠剤を詰めて40℃/75%RHで貯蔵した。
製剤PF3の製造方法:
ソルビトール及びブドウ糖無水物を秤量した。140.64gのソルビトールを105.48gの精製水に溶解させ、得られた溶液を引き続きFluid Bed Granulator(4M8ForMate FluidBed)内で用いて659.36gのブドウ糖を造粒した。その後、該顆粒を60℃で乾燥させ、850μmの篩いを通して選別した。
33.06gのベンダムスチン塩酸塩、149.82gのソルビトール/ブドウ糖顆粒、13.8gの微結晶性セルロース及び1.32gのステアリン酸マグネシウムを二重ポリエチレンバッグに量り入れて5分間混合した。その後、該粉末ブレンドを偏心性錠剤機(Korsch EK0O)のホッパーに移し、10.0mmの平均径を有する円形錠剤に圧縮成形した。錠剤は335.99mg(初め)〜339.50mg(終わり)の質量の平均値;脆砕性:0%;平均硬度値:125.60N(初め)〜129.7N(終わり)を有した。次に錠剤を下記2工程のコンディショニングプロセスに供した(選択したバッチについてのみ行なった):錠剤を25℃/60%R.Hに2時間置き、引き続き40℃に2時間置いた。
引き続き錠剤をコーティングパン(4M8 ForMate PanCoat)内で9%のOpadry(登録商標)TM White水性懸濁液を用いてコーティングした。錠剤の平均質量:341.43mg。その後、ねじ込みプラグで閉じた琥珀色ガラス瓶に錠剤を詰めて40℃/75%RHで貯蔵した。
貯蔵したフィルムコーティング錠中のベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表9b.1〜9b.3に示す。
【0056】
【0057】
【0058】
【0059】
3. 溶解試験
実施例10
実施例2及び3の錠剤製剤についての溶解試験を人工胃液内でT=0にて行なった。HPLC(カラム:Zorbax Bonus-RP、5μm;カラムオーブンの温度:30℃;オートサンプラーの温度:5℃;検出器:254nm)によるアッセイのため溶解サンプルを試験した。2gの塩化ナトリウムp.A.を1000mlの水に溶解させ、5N塩酸でpHを1.5±0.05に調整することによって、人工胃液pH1.5を調製した。欧州薬局方6.0の2.9.3章に準拠してApparatus 2(パドル装置)を用いて溶解試験を行なった。パドルの回転速度は50rpm、温度は37±0.5℃、溶解媒体の量は500mlだった。
実施例2の錠剤製剤(錠剤製剤1)及び実施例3の錠剤製剤(錠剤製剤2)の結果を表10aに示す。
【0060】
【0061】
実施例6、実施例7及び実施例8のコーティング錠製剤についてT=0で行なったのと同じ溶解試験の結果を下表10bに示す。
表10b
【0062】
実施例9の錠剤についての対応する溶解データは下表のとおりだった。
【0063】
【0064】
上記から得られるように、本発明の全ての錠剤製剤は、ベンダムスチンの速い溶解を示す。特に本発明の製剤は、本明細書で前述したベンダムスチンの溶解プロファイルを示す。
4. in vivo試験
ベンダムスチンの動物バイオアベイラビリティー研究をビーグル犬で行なった:PK研究概要
研究1
目的は、3種の錠剤製剤(T1〜3)及び1種のカプセル製剤(C)、全部で4種の経口製剤中の1用量(すなわち50mg)のベンダムスチンのバイオアベイラビリティーを決定することだった:AUC及びCmax。
要した動物の総数:16
基本設計:
交差設計、治療群(arm)毎に8匹の動物:
【0065】
表11a:期間1(単回用量の錠剤、又はカプセル剤、日1):
【0066】
1週間の休薬(wash-out)
【0067】
表11b:期間2(期間1の1週間後、以下のいずれかの単回用量、日8):
【0068】
1週間の休薬
【0069】
表11c:期間3(期間2の1週間後、以下のいずれかの単回用量、日15):
【0070】
研究2
目的は、1種の錠剤製剤T4、1種のカプセル製剤、全部で3種の経口製剤中の1用量(すなわち50mg)のベンダムスチンのバイオアベイラビリティーを決定することだった:AUC及びCmax。
要した動物の総数:16
基本設計:
交差設計、治療群毎に8匹の動物:
【0071】
表12a:期間1(単回用量のカプセル剤、日1):
【0072】
1週間の休薬
【0073】
表12b:期間2(期間1の1週間後、以下の製剤のどちらかの単回用量、日8):
【0074】
実施例11
50mgのベンダムスチンを含む実施例9のコーティング錠(製剤3、Opadry(登録商標)でコーティングした錠剤T4)をオスとメスのイヌに経口投与して参考例のカプセル剤と比較した。
カプセル製剤と実施例9のコーティング錠の両者について平均血漿プロファイル対時間を図1に示す。
実施例12
50mgのベンダムスチンを含む実施例6、7、又は8のコーティング錠(錠剤T1〜T3)をオスとメスのイヌに経口投与して参考例のカプセル剤と比較した。
カプセル製剤と実施例6〜8のコーティング錠の平均血漿プロファイル対時間を図1に示す。
実験は以下のために行なった:
−糖類又は糖類混合物が、速い溶解プロファイルとコーティングに適した硬度値とを有する化学的に安定した錠剤を得るのに適しているかを評価する;
−APIと賦形剤との適合性を評価する;
−異なる製造プロセス(乾式造粒、直接圧縮及び湿式造粒)を調査することによって、プラセボ及びAPI含有バッチを開発する;
−種々のベンダムスチン塩酸塩/糖類比を評価する;
−製造される錠剤の技術的特性及び安定性に及ぼす含水量の影響を調査する;
−商業的に入手可能な凍結乾燥ベンダムスチン塩酸塩製品(Ribomustin(登録商標))を用いて錠剤を製造し、かつこれらの錠剤を対応量のマンニトールとベンダムスチン塩酸塩を用いて作製した錠剤の特性と比較する。
本発明の錠剤、すなわち50mgのベンダムスチン(ベンダムスチン塩酸塩として55mg)を含む錠剤の製造のために以下の糖類を用いた。
【0075】
表13.
【0076】
作られたバッチの品質を物理的外観の観察、同定試験(HPLC)、溶解試験、含量及び関連物質アッセイ(HPLC)、含量均一性試験(HPLC)、硬度試験及び含水量(Karl Fischer法)によって評価した。下表に詳細に示す貯蔵条件下で琥珀色ガラス瓶に詰め込まれる加速安定性研究にバッチを委ねた。それぞれ製造されたAPI含有バッチについていくつかの錠剤をバックアップサンプルとして5℃で貯蔵した。
以下、それらの製造プロセスに関して種々の賦形剤を調査した。これらの賦形剤を使用することによって、乾式造粒でいくつかのプラセボ製造試験を行なって、良い品質の錠剤を得るのに適した製造方法についての予備情報を得た。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/002だけ架橋ポリビニルピロリドン(Crospovidone(登録商標))を用いた。バッチD001T/002(充填剤:無水ラクトース)に対するCrospovidone(登録商標)の選択は、この製剤と実施例9の原型製剤の間の類似性に基づいた。作製した全てのバッチ用の潤沢剤としてステアリン酸マグネシウムを使用した。プラセボ試験の乾式造粒製造プロセスは以下の工程に存した:
1. 糖類及び部分量の潤沢剤(総量の83.3%w/w)を正確に秤量してから2分間ポリエチレンバッグ内で混合した。
2. 得られた混合物を18mm径パンチを備えた錠剤機を用いて圧縮した。
3. 得られたスラグを850μmネットを用いて篩過した。
4. 顆粒を秤量し、2分間ポリエチレンバッグ内で崩壊剤及び残量の潤沢剤(16.7%w/w)と混合してから10mm径パンチを用いて打錠した。
【0077】
表14及び表15は、各プラセボ製剤の組成並びに最終混合物と錠剤の両方について行なった分析試験の結果を要約する。
表16には、プラセボバッチの製造プロセス中及び/又はそれらの分析特徴づけ中に行なった観察を報告する。
プラセボバッチD001T/001-D001T/002-D001T/004-D001T/013-D001T/015について行なった分析的及び/又は物理的試験結果は、これらの製剤が乾式造粒で製造し、かつAPIを添加してさらに調査するのに適していることを示した。全ての他の製剤は固めるのが難しく、得られても非常に脆砕性の錠剤によって特徴づけられる。
バッチD001T/005(充填剤:β-シクロデキストリン)は、乾式製造プロセスにおける良い挙動、高い硬度、低い脆砕性、長い崩壊時間を示した。超崩壊剤(Crospovidone(登録商標))を利用し、かつAPIを添加することによって、この製剤をさらに検討した(下記パラグラフ参照)。
【0078】
表14. 乾式造粒−プラセボバッチの組成及び分析結果(バッチD001T/001〜D001T/010)。
【0079】
(表14続き)
【0080】
(表14続き)
N.A. = 混合物が打錠プロセスに適していないので該当データなし(表5aに報告された観察参照)
【0081】
表15. 乾式造粒−プラセボバッチの組成及び分析結果(バッチD001T/011〜001T/025)。
【0082】
(表15続き)
【0083】
(表15続き)
N/A = 混合物が打錠プロセスに適さないので該当データなし(表5aに報告された観察参照)
【0084】
表16:それぞれ製造されたプラセボバッチの製造プロセス、製品の技術的特性及び分析試験についての所見
【0085】
(表16続き)
【0086】
(1:5のベンダムスチン塩酸塩/糖類質量比で乾式造粒によって製造したバッチ)
活性医薬成分(API)を含む錠剤を乾式造粒で製造するのに適していると評価されたプラセボ製剤を修正してAPIを含め、2つのAPI/糖類質量比1:5及び1:2を探究した。
このパラグラフでは、1:5のAPI/糖類質量比の製剤について述べる。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/022だけ架橋ポリビニルピロリドン(Crospovidone)を使用した。作製した全てのバッチの潤沢剤としてステアリン酸マグネシウムを用いた。
API含有バッチの乾式造粒による製造プロセスは以下の工程に存した:
1. 糖類、部分量の潤沢剤(総量の83.3%w/w)及びベンダムスチン塩酸塩を正確に秤量し、5分間二重ポリエチレンバッグ内で混合した。
2. 粉末ブレンドを18mm径パンチを備えた錠剤機を用いて圧縮した。
3. 顆粒を得るため、生成されたスラグを850μmネットを用いて篩過した。
4. 顆粒を秤量し、5分間二重ポリエチレンバッグ内で崩壊剤及び残量の潤沢剤(16.7%w/w)と混合した。
5. 得られた混合物を10mm径パンチを用いて打錠した。
表17は、製造された各API含有製剤の組成及びAPI含有最終混合物について行なった分析試験の結果を要約し;表18は、得られた製品について行なった分析試験の結果を要約する。
【0087】
表17. 乾式造粒−API/糖類質量比1:5。API含有バッチ最終混合の組成及び分析結果
【0088】
表18. 乾式造粒−API//糖類質量比1:5。API含有バッチ錠剤の分析結果
【0089】
最終混合物と最終製品の両者について行なった分析試験の結果は、主に含量均一性及び純度についてほとんどの場合に良かった。全てのAPI含有バッチは、満足のいく質量均一性、API含量の均質性、及び低い不純物含量を示した。全ての製剤のこの不純物プロファイルは、APIの仕様に従ったので(表中の仕様限界参照)、製造プロセス中に分解は起こらない。
2種のAPI含有バッチは、APIアッセイで低い値を示した;この結果は、小さいバッチサイズのため並びに製造プロセス中の損失及び最終混合物についてのIPC用サンプルのためであろう。
【0090】
(1:2のAPI/糖類質量比で乾式造粒によって製造したAPI含有バッチ)
1:5のAPI/糖類質量比を有する錠剤を製造するために乾式造粒で以前に調査した全ての糖類を1:2の比でも評価した。
製造プロセスについては上記を参照されたい。この場合、得られた混合物を8mm径パンチを用いて打錠した。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/105だけ架橋ポリビニルピロリドン(Ccrospovidone(登録商標))を使用した。このバッチでは、我々はAvicel(登録商標)PH 112及びCrospovidone(登録商標)の使用を検討した。1:5のAPI/糖類(以前の結果参照)で乾式造粒によって製造した前記シクロデキストリンベース製剤に従ってCrospovidone(登録商標)を選択した。
表19及び表20は、1:2のAPI/糖類質量比で乾式造粒によって製造した各API含有製剤の組成及び最終混合物と錠剤の両方について行なった分析試験の結果を要約する。全てのAPI含有バッチは、適切な質量均一性、API含量の均質性及び低不純物含量を示した。脆砕性及び硬度値は、ほとんどの場合に仕様に従っている。バッチD001T/093、D001T/095及びD001T/096の場合、6錠剤について行なった溶解試験の結果は、RSDが高く、仕様値の範囲外を示したので試験を12錠剤のサンプルに拡張した。
シクロデキストリンベース錠剤は、両崩壊剤(Avicel(登録商標)PH 112及びCrospovidone(登録商標))で良い特性を示す。
【0091】
表19. 乾式造粒−API/糖類質量比1:2。API含有バッチ最終混合物組成及び分析結果。
【0092】
表20. 乾式造粒−API/糖類質量比1:2。API含有バッチ錠剤分析結果。
【0093】
(表20続き)
n.d. = 検出されず
【0094】
(1:5のAPI/糖類質量比で直接圧縮によって製造したAPI含有バッチ)
乾式造粒で製造するのに適した特徴を有する糖類を直接圧縮を利用して1:5のAPI/糖類比を有する錠剤を開発することをも検討した。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/029だけ架橋ポリビニルピロリドン(Crospovidone(登録商標))を使用した。
この製造プロセスは以下の工程から成った。
1. API及び賦形剤を秤量する工程。
2. 原材料を二重ポリエチレンバッグに移して均質粉末ブレンドが得られるまで約5分間混合する工程。
3. 粉末ブレンドを錠剤機のホッパーに移す工程。
4. 10mm径パンチを備えた偏心性錠剤機を用いた粉末ブレンドの圧縮。
直接圧縮によって製造したAPI含有バッチの特性を下表に示す。
【0095】
表21. 直接圧縮−API/糖類質量比1:5。API含有バッチ最終混合物組成及び分析結果。
得られた分析試験結果を表22に掲載する。
【0096】
表22. 直接圧縮−API/糖類質量比1:5。API含有バッチ錠剤分析結果。
【0097】
上表で報告したように、直接圧縮で製造したAPI含有錠剤は、不均質なAPI含量と脆砕性のわずかな増加を示したバッチD001T/030(充填剤:ショ糖97%+マルトデキストリン3%)を除き、乾式造粒で作製した錠剤との重大な意味を持つ差異を示さなかった。
【0098】
湿式造粒:
(プラセボ探索的試験)
本プロジェクトの第一及び第二部で得られた結果に基づいて、乾式造粒又は直接圧縮に適さない糖類を湿式造粒によって調査した。
湿式造粒技術を調査するための本アプローチを以下に示す。
図2のフローシートに示す工程に従って各糖類を造粒した。各工程の最後に湿式造粒糖類を乾燥させ、圧縮試験を行なって該顆粒が打錠に適しているか評価した。圧縮試験の結果が疑わしい造粒糖類についてのみプラセボバッチを製造した。プラセボ試験の組成及び関連分析結果を表23で報告する。
以下の工程に従ってプラセボバッチを製造した。
1. 流動床又は高せん断造粒機を用いた水又はソルビトール溶液との糖類の湿式造粒(上記湿式造粒試験のフローシート、及び表23参照)。
2. 流動床造粒機又はオーブン内での湿式造粒糖類の乾燥工程。
3. 850及び710μmネットを用いて造粒糖類を篩過する工程。
4. 製剤の全成分の秤量及びポリエチレンバッグ内で2分間混合する工程。
5. 10mm径パンチを備えた偏心性錠剤機を用いた粉末ブレンドの圧縮。
作製した全てのバッチでは、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。
【0099】
表23. 湿式造粒。プラセボバッチ組成及びIPC結果。
【0100】
(表23続き)
【0101】
(表23続き)
【0102】
(表23続き)
【0103】
(1:5のAPI/糖類質量比で湿式造粒によって製造したAPI含有バッチ)
乾式造粒又は直接圧縮技術による錠剤製造に適さないことが分かった全ての糖類について湿式造粒プロセスを含む製造試験を行なった。
実験室規模で行なったこれらの試験の製造プロセスは以下のように要約される。
1. 流動床又は高せん断造粒機を用いた水又はソルビトール溶液との糖類の湿式造粒(湿式造粒製造試験の上記フローシート、及び表24参照)。
2. 流動床造粒機又はオーブン内での湿式造粒糖類の乾燥工程。
3. 850及び710μmネットを用いた篩過工程。
4. APIと賦形剤の秤量及びポリエチレンバッグ内で2分間混合する工程。
5. 10mm径パンチを備えた偏心性錠剤機を用いた粉末ブレンドの圧縮。
作製した全てのバッチでは、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。
【0104】
表24及び表25は、湿式造粒で製造した各API含有製剤の組成及び最終混合物と錠剤の両者について行なった分析試験の結果を掲載する。
最終混合物及び最終製品について行なった分析試験の結果は、ほとんどの場合、仕様に従う。製造プロセス中に分解は起こらない。
調査した糖類のうち、Fructose MS(Galam)だけが湿式造粒による加工に適さず:API含有バッチD001T/047は高い脆砕性を有し、バッチD001T/082は仕様の範囲外の脆砕性と硬度を示す。
バッチD001T/060、D001T/061、D001T/082、D001T/086はAPIアッセイで低い値を示し、バッチD001T/082及びD001T/086では、850μm及び710μmネットを用いて顆粒が篩過されたが、含量の均一性が従わない。この結果はおそらく不十分な粉末の混合のためであろう。
【0105】
表24. 湿式造粒−API/糖類質量比1:5。API含有バッチ最終混合物組成及び分析結果
【0106】
(表24続き)
【0107】
(表24続き)
【0108】
(表24続き)
(*) 好ましくはバッチは過剰のA.P.I(5.9%)を含む;(**) これらの糖類は空気流で流動化されないので、流動床を用いた糖類溶液による造粒工程を調査できなかった;(***) 最終混合物は打錠に適さない
【0109】
表25. 湿式造粒−API/糖類質量比1:5。API含有バッチ錠剤分析結果。
【0110】
(表25続き)
【0111】
(表25続き)
【0112】
(表25続き)
【0113】
(1:2のAPI/糖類質量比で湿式造粒によって製造したAPI含有バッチ)
1:5のAPI/糖類質量比の錠剤を製造するために湿式造粒で以前に調査した全ての糖類を1:2の比についても評価した。
フルクトースは得られた顆粒が打錠に適さないので、1:2の比では評価しなかった。
作製した全てのバッチでは、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。
API含量の均一性を改善するため、以下のアプローチを適用してこれらのAPI含有バッチを製造した。
1. 以前に最適化した手順を用いて糖類の湿式造粒
2. API含有混合物の調製
3. 混合物の乾式造粒(スラグ製造→スラグ篩過)
4. 得られた混合物を8mm径パンチを用いて打錠。
工程3(混合物の乾式造粒)については上記を参照されたい。
表26及び表27は、1:2のAPI/糖類質量比で湿式造粒した糖類を用いて製造したAPI含有バッチの組成及び分析結果を報告する。脆砕性は、ほとんどの場合、仕様の範囲外である。API/糖類の質量変化は、D001T/084バッチ(充填剤;顆粒化マンニトール)の技術的特性に影響を及ぼさない。
【0114】
表26. 湿式造粒−A.P.I./糖類質量比1:2。API含有バッチ最終混合物組成及び分析結果。
【0115】
(表26続き)
【0116】
(表26続き)
(*) 以前の製剤(API/糖類質量比1:5)を開発するために用いたラクチトールはもはや商業的に入手できないので、新しい製造業者によって購入したラクチトールを用いてこのバッチを製造した(DaniscoによるLactitol)。
【0117】
表27. 湿式造粒−A.P.I./糖類質量比1:2。API含有バッチ錠剤分析結果。
【0118】
(表27続き)
【0119】
(表27続き)
【0120】
(API/マンニトール質量比の影響)
マンニトールベース錠剤を製造して以下のAPI/マンニトール比(1:0.01、1:0.1、1:0.5、1:1.7、1:4、1:5、1:6及び1:10)を調査した。1:5のAPI/マンニトール質量比を有する製剤(標準的製剤)については上述した。
これらのバッチの製造では、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。製造プロセスに関し、1:1.7、1:4、及び1:6比では、湿式造粒したマンニトール、ベンダムスチン塩酸塩及び賦形剤を正確に秤量し、二重ポリエチレンバッグ内で5分間混合した。バッチD001T/110(1:10比)では、前混合を行なった。この場合、ベンダムスチン塩酸塩を5分間、半量の賦形剤混合物と混合した。次に、得られた混合物を残存量の賦形剤に加えてさらに5分間混合した。最終混合物を適切なパンチ(1:1、1:1.7及び1:2比には8mm径、1:4及び1:6比の場合は10mm、1:7比には12mm、1:10比には14mm)を備えた錠剤機を用いて打錠した。
1:0.01、1:0.1、1:0.5比については、我々は上述した製造プロセス(糖類の湿式造粒後に乾式造粒)を適用して、API含量の均一性を改善した。得られた混合物を6mm径パンチで打錠した。
下表(表28及び表29)は、異なるAPI/マンニトール比の影響を研究するために製造したAPI含有製剤の組成及び分析結果を要約する。バッチD001T/111、D001T/083及びD001T/106は高い脆砕性を示し、バッチD001T/106、D001T/108及びD001T/109では、含量の均一性が以前に得られたデータの傾向から逸脱して従わなかった。この結果は、これらのバッチが異なる物理的性質を有し得る新しいロット(ロット番号:F08-05873)のベンダムスチンHClを用いて作製したという事実のためであろう。
【0121】
表28. A.P.I./マンニトール質量比。API含有バッチ最終混合物組成
【0122】
(表28続き)
【0123】
(表28続き)
(*) 上記実験アプローチを用いて製造したバッチ
□ 標準製剤1:5API/糖類質量比
【0124】
表29. A.P.I./マンニトール質量比の研究。API含有バッチ錠剤分析結果。
【0125】
(表29続き)
【0126】
(表29続き)
□ 標準製剤1:5のAPI/糖類質量比
【0127】
(糖類の組合せの研究)
表30及び表31は、糖類の組合せの研究に関する結果を報告する。
以下の組合せを調査した。
−単糖/二糖1:1
(*)マンニトール(Pearlitol 200 SD)/ラクトース無水物(SuperTab 21 AN)
ソルビトール(Neosorb P60W)/マルトース(Sunmalt S)
−オリゴ糖/単糖1:1
(*)D-メレジトース一水和物/(*)ブドウ糖無水物ST 0.5
(*)ラフィノース五水和物(顆粒化)/(*)マンニトール(顆粒化)(Pearlitol 200 SD)
−オリゴ糖/二糖1:1
(*)ラフィノース五水和物(顆粒化)/ラクトース一水和物(Supertab 14SD)
β-シクロデキストリン(Kleptose DC)/ショ糖(EV Saccharide)
(*)これらの糖類は湿式造粒で顆粒化された(32ページ参照)。
製造プロセスは、未加工又は顆粒化糖類の直接圧縮に存した。
Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として用いて、以下の工程を実施してこれらのバッチを製造した。
1. 糖類(又は顆粒化糖類)、ベンダムスチン塩酸塩及び賦形剤を正確に秤量し、二重ポリエチレンバッグ内で5分間混合した。
2. 得られた混合物を10mm径パンチを用いて打錠した。
【0128】
表30. 糖類の組合せの研究。API含有バッチ最終混合物組成及び分析結果。
【0129】
表31. 糖類の組合せの研究。API含有バッチ錠剤分析結果。
【0130】
(表31続き)
【0131】
一般に、糖類の組合せの研究用に製造した錠剤は良い特性を示す。しかしながら、バッチD001T/102(ラフィノース五水和物/マンニトール(Pearlitol 200 SD))は高い脆砕性を示し、バッチD001T/100及びD001T/049はAPI含量の不均質性を示す。
【0132】
実施例14. 凍結乾燥ベンダムスチンHCl(Ribomustin)及びベンダムスチンHCl/マンニトール錠剤(Api/糖類質量比1:1.2)
静脈内適用のための市販製品(Ribomustin(登録商標))から得た凍結乾燥材料を使用するか又は湿式造粒したマンニトールとベンダムスチンHClを使用して、1:1.2の質量比でベンダムスチン塩酸塩/マンニトールを含む錠剤を調製した。
以下の実験操作に従って製造プロセスを行なった:Ribomustin(登録商標)バイアルから凍結乾燥粉末を取り出し、850μmネットを用いて篩過した。得られた粉末と潤沢剤(ステアリン酸マグネシウム)を正確に秤量し、ポリエチレンバッグ内で5分間混合した。混合物をゆっくり錠剤機のプレスチャンバーに移し、8mm径パンチを用いて手動でプレスして小スラグを得た。850μmネットを用いてスラグを篩過し、得られた顆粒を8mm径パンチを用いて手動でプレスした。
この実施例では、上述したのと同じ操作手順を施してベンダムスチン HCl/マンニトール錠剤を製造した。
製剤の組成を表32に示す。
【0133】
表32. Ribomustin及びベンダムスチン/マンニトール錠剤。API含有バッチ最終混合物組成
(*) 45.16%のベンダムスチンHCl及び54.20%のマンニトールに相当する
【0134】
表33は、凍結乾燥ベンダムスチン塩酸塩/マンニトール混合物及び非凍結乾燥ベンダムスチン塩酸塩/マンニトール混合物を用いて得た錠剤間の比較に関するデータを報告する。
【0135】
表33. Ribomustin及びベンダムスチン/マンニトール錠剤。API含有バッチ錠剤分析結果
【0136】
参考目標としてベンダムスチン塩酸塩APIの不純物プロファイルを例として挙げると(表中の仕様限界参照)、バッチD001T/125は、HP1不純物について仕様範囲外の値を示した。溶解試験の結果は、10分後には凍結乾燥ベンダムスチン塩酸塩/マンニトール混合物を含む錠剤の溶解プロファイルの方が速いが、30分後には両製剤で溶解が現仕様に従うことを明らかにしている。脆砕性は、バッチD001T/126では仕様の範囲外であり、バッチD001T/125については原料の十分な量を欠いたため試験を行なわなかった。
【0137】
(産業上の利用可能性)
本発明の組成物は多くの利点を示す。管理医療スタッフの補助なしで患者が容易に本組成物を使用することができる。従って、時間のかかる通院は時代遅れになり、それによって患者のコンプライアンスを向上させ得る。
剤形が固体なので、そのまま嚥下することができ、活性成分の溶解が達成されるまで患者が待つ必要がないことを意味する。さらに本剤形の良い安定性のため、室温で、かつ如何なる特殊な貯蔵条件をも必要とせずに本組成物を容易に貯蔵することができる。
本発明の剤形を利用することによって、剤形の体積のかなりの減少を果たすことができる。サイズの減少は、製造及び取扱いの観点からも患者のコンプライアンスの観点からも望ましい。
医薬組成物はin vitroで高い溶解を示し、in vivoでのベンダムスチンの分解を減少させ、それによってin vivoでのベンダムスチンのバイオアベイラビリティーを改善することになる。
【技術分野】
【0001】
本発明は、ベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物を含む経口投与用固体剤に関する。
【背景技術】
【0002】
(発明の背景)
ベンダムスチン(4-[5-[ビス(2-クロロエチル)アミノ]-1-メチルベンズイミダゾ-2-イル]ブタン酸、ナイトロジェンマスタード)は、二官能性アルキル化活性のあるアルキル化薬である。それは下記式(I)に相当する。
【0003】
【化1】
【0004】
ベンダムスチンは、他のアルキル化薬と如何なる交差耐性もないようであり、このことはアルキル化薬による治療を既に受けたことのある患者の化学療法の観点から利点を提供する。
ベンダムスチンは最初にドイツ民主共和国(GDR)で合成された。ベンダムスチンの塩酸は、1971〜1992年に商標名Cytostasan(登録商標)で入手可能な市販製品の活性成分だった。その時以来、それは独国で商標名Ribomustin(登録商標)にて市販され、慢性リンパ性白血病、非ホジキンリンパ腫及び多発性骨髄腫を治療するために広く使用されてきた。
市販製品は、ベンダムスチン塩酸塩の凍結乾燥粉末を含み、これを注射用水で再構成して濃縮物を得る。これを引き続き0.9%の塩化ナトリウム水溶液で希釈すると、注入用の最終溶液となる。この最終溶液を約30〜60分の時間をかけて静脈内注入で患者に投与する。
水中におけるベンダムスチンのビス-2-クロロエチルアミノ基の加水分解が効力の低下及び不純物形成をもたらす(B. Maas et al. (1994), Pharmazie 49: 775-777)。従って普通は病院内又は少なくとも医学的管理下で、凍結乾燥粉末の再構成後即座に投与を行なわなければならない。さらに、再構成は難しいと報告されている。再構成は30分より長い時間を要することがある。さらに、それは製品を2工程プロセスで再構成する責任がある医療専門家にとって厄介であり、時間がかかる。
【0005】
Preissら(1985)(Pharmazie 40:782-784)は、それぞれ4.2〜5.5mg/kgの範囲の用量で静脈内及び経口投与した後の7名の患者の血漿中のベンダムスチン塩酸塩の薬物動態を比較した。商業的に入手可能なCytostasan(登録商標)製品から調製した静脈内注射を3分かけて行ない、一方で等価用量の経口薬は25mgのベンダムスチン塩酸塩を含むカプセル剤の形を取った。患者が摂取すべきカプセル剤の数は10〜14で変動した。経口投与後1時間以内で最大血漿レベルが検出できた。平均経口バイオアベイラビリティーを計算すると57%だったが、25%〜94%まで変動し、個体間の大きい可変性を示唆している。
Weber(1991)(Pharmazie 46(8): 589-591)は、B6D2F1マウスにおけるベンダムスチン塩酸塩のバイオアベイラビリティーを調査し、胃腸管からの該薬物の吸収が不完全であり、たった約40%のバイオアベイラビリティーしかもたらさないことを見い出した。
US2006/0128777A1は、一般に死抵抗性細胞及びベンダムスチン含有組成物によって特徴づけられる癌の治療方法を記載している。これらの中で組成物は、カプセル剤、錠剤、丸剤、散剤又は顆粒剤である経口固体剤形であり、その中で活性化合物が少なくとも1種の不活性な賦形剤、例えばスクロース、ラクトース又はデンプンと混合され得る。しかしながら、具体的な組成物は例示されなかった。
【発明の概要】
【発明が解決しようとする課題】
【0006】
一度水で再構成した市販のi.v.製剤の安定性の問題を考慮して、かつ患者のコンプライアンスを改善するため、患者に投与しやすく、かつ既知の経口剤形に比べて可変性が少ないバイオアベイラビリティーの向上をもたらす、ベンダムスチンを含む安定な経口剤形が長年にわたって要望されている。
【課題を解決するための手段】
【0007】
(発明の概要)
上記課題を解決するため、本発明者らは詳細な調査を行なった。本発明者らは、最終的に本発明の安定医薬組成物を得ることに成功した。これらの組成物は、経口投与に適しており、かつ活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、少なくとも1種の医薬的に許容できる賦形剤とを含み、本組成物は、改善された溶解プロファイルを有する。
【図面の簡単な説明】
【0008】
【図1】従来技術のカプセル剤並びに実施例6〜8の錠剤製剤(錠剤1〜3)及び実施例9の錠剤製剤(製剤3)(表4)の形態でベンダムスチン塩酸塩をイヌに投与した後に得られた平均血漿濃度(錠剤対カプセル剤)対時間曲線を示す。図1から、従来技術のカプセル剤に比し、本錠剤製剤がベンダムスチンのより高い最大濃度を提供することが明らかである。
【図2】湿式造粒製造試験のフローシートを示す。
【発明を実施するための形態】
【0009】
(発明の詳細な説明)
本発明は、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、単糖類、二糖類、オリゴ糖類、環状オリゴ糖類、多糖及び糖アルコール類から選択される少なくとも1種の医薬的に許容できる賦形剤とを含む医薬組成物に関する。好ましくは、活性成分と賦形剤の質量比は、1対1〜5、好ましくは1対2〜5の範囲内、さらに好ましくは1:5〜1:2から選択される比である。
一実施形態では、本発明は、経口投与用固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択される医薬的に許容できる糖類である少なくとも1種の医薬的に許容できる賦形剤とを含み、前記活性成分と賦形剤の質量比が1:1の範囲内である、医薬組成物に関する。
さらなる実施形態では、本発明は、経口投与に適した固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択される医薬的に許容できる糖類である少なくとも1種の医薬的に許容できる賦形剤とを含み、前記活性成分と糖類賦形剤の質量比が1:2〜5の範囲内であり、かつ該組成物が、欧州薬局方(European Pharmacopoeia)に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す、医薬組成物に関する。
上記実施形態の範囲内のさらに好ましい実施形態は、前記医薬的に許容できる糖類が、1種以上の単糖、二糖及びオリゴ糖から成る群より選択され、前記活性成分と糖類賦形剤の質量比が1:2〜5の範囲内であり、かつ該組成物が、欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す、医薬組成物である。
【0010】
本発明は、医薬組成物に特定量の医薬的に許容できる糖類を組み入れることによって、特有の望ましい溶解プロファイルに到達できるという驚くべき発見に基づいている。
活性成分としてベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物を含む医薬組成物中の賦形剤として、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択され、好ましくは1種以上の単糖、二糖及びオリゴ糖から成る群より選択される医薬的に許容できる糖類を使用すると、安定性、打錠特性、溶解及び不純物形成に関して特に有利なプロファイルが達成されることが分かった。上記糖類は、欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す組成物をもたらす。
本発明の上記範囲内では、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールのいずれの組合せをも使用してよい。
特に、特定の糖類が安定性及び溶解に関して医薬組成物の特に有利なプロファイルと関係があることが分かった。本発明の組成物の好ましい糖類は、ブドウ糖無水物、ブドウ糖一水和物、ラクチトール一水和物、トレハロース、ソルビトール、エリスリトール、マルトース一水和物、マンニトール、ラクトース無水物、ラクトース一水和物、マルチトール、キシリトール、ショ糖、ショ糖97%+マルトデキストリン3%、β-シクロデキストリン、D-ラフィノース五水和物、D-メレジトース一水和物及び微結晶性セルロースである。本発明の医薬組成物は良い打錠特性、速い溶解及び医薬的に許容できる安定性を示す。
上記糖類は本発明の好ましい実施形態を構成し、そのいずれの組合せをも使用し得る。好ましくは、活性成分と上記糖類の比は、1:1〜5、好ましくは1:2〜5の範囲内、さらに好ましくは1:5〜1:2から選択される比である。
本発明のさらに好ましい実施形態は、経口投与用固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、ブドウ糖無水物、ブドウ糖一水和物、ラクチトール一水和物、トレハロース、ソルビトール、エリスリトール、マルトース一水和物、マンニトール、ラクトース無水物、ラクトース一水和物、マルチトール、キシリトール、ショ糖、ショ糖97%+マルトデキストリン3%、β-シクロデキストリン、D-ラフィノース五水和物、D-メレジトース一水和物及び微結晶性セルロースから選択される少なくとも1種の医薬的に許容できる賦形剤とを含み、かつ該組成物が、10分で少なくとも60%、20分で70%及び30分で80%のベンダムスチンの溶解を示す、医薬組成物である。
特に好ましい糖類は、マンニトール、マルチトール、エリスリトール、キシリトール、ラクトース、ショ糖、グルコース、ソルビトール、マルトース、トレハロース、ラクチトール及びブドウ糖(無水物又は一水和物)であり、活性成分と前記糖類の質量比は、好ましくは1:2〜5の範囲内である。上記糖類の範囲内の2種以上の組合せも本発明の範囲に包含される。
当業者は、上記糖類賦形剤の範囲内で適切な組合せを選択し、かつ欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、20分で少なくとも60%、40分で70%及び60分で80%のベンダムスチンの溶解を示す組成物を得るのに十分な立場にいる。
【0011】
好ましい実施形態では、本組成物は、錠剤、顆粒剤、又は丸剤の形態である。
好ましい剤形は錠剤である。錠剤という用語は、速崩壊錠剤をも含み、その中には分散性錠剤及び発泡性錠剤がある。
錠剤調製の最も一般的に用いられる方法は、直接圧縮、乾式造粒及び湿式造粒である。直接圧縮は、活性成分と賦形剤を含む混合物を錠剤プレス機で圧縮する工程を含む(L. Lachman et al., in: The Theory and Practice of Industrial Pharmacy, 3rd ed., 1986)。均一含量の活性成分を有する錠剤を作るためには、圧縮すべき混合物が良い流動及び圧縮の両特性を持たなければならない。混合物に適切な賦形剤、例えば潤沢剤、付着防止剤及び流動促進剤などを添加することによって常に良い流動特性を達成できるわけではない。従って、多くの場合、圧縮前に混合物を造粒する。
造粒は、粉末混合物から顆粒と呼ばれる球様又は規則的形状の凝集体を形成するプロセスである。これは、乾式造粒法及び湿式造粒法によって達成され得る。造粒は、凝集性が乏しい粉末混合物を、圧縮したときに良い粘着特性を有する錠剤をもたらす凝集体に変換するためにも利用される。
速崩壊錠剤の場合、必要に応じて1種以上の賦形剤との混合剤の活性成分は、該成分の味を隠すため並びに/或いは該成分を光及び/又は湿気によって起こり得る有害な作用に対して保護するため並びにベンダムスチンの場合は活性化合物によってもたらされる有害な作用に対して口内の粘膜を保護するため、有利にはコーティングを備えている。当該目的のため、好ましくはさらに以下に概要を示すように顆粒(granulate)を調製及び加工する。
【0012】
「顆粒(granulate)」という表現は、顆粒(granule)と呼ばれることもある粒子の凝集体を表す。顆粒は、一般的にコンパクション及び/若しくは圧縮法(乾式造粒)又は必要に応じて湿式造粒結合剤が溶解している液体を利用する湿式造粒法によって調製される(Remington's Pharmaceutical Sciences 18th ed. 1990, page 1641)。湿式造粒法には押出法も含まれる。従って、顆粒という用語は、ペレット、小球、及び押出品をも含み、顆粒の例として好ましくはペレットを用いる。
ペレットは直径が約1.0〜1.6mmで、特定の密度を有する小粒子として表され、この粒子は粉末混合物に押出し及び球形化の調剤プロセスを適用することで調製される。
必要に応じて1種以上の賦形剤との混合剤の活性成分は、該成分の味を隠すため並びに/或いは該成分を光及び/又は湿気によって起こり得る有害な作用に対して保護するため並びに/或いは活性化合物によってもたらされる有害な作用に対して口内の粘膜を保護するため、有利にはコーティングを備えていてよい。
丸剤は、活性成分をトリグリセリドの柔らかい混合物に添加することによって調製される小円形固体剤形である。混合物をローラーで伸ばして長いストリングにしてからバラバラにカットしてローラーで伸ばす(J.T. Carstensen: Pharmaceutical principles of solid dosage forms, 1993, Technomic Publishing Company, Inc. page 63)。
好ましくは本発明の剤形は乾式コンパクション法によって調製される。適切な技法は、例えばRemington's Pharmaceutical Science 18th. ed. 1990, page 1644に記載されている。それらの技法は乾式造粒、ローラーコンパクション及び直接圧縮を含む。錠剤をこれらの技法で調製する場合、直接圧縮を利用することがなおさらに有利である。
【0013】
本発明の剤形は好ましくはコーティングを備えている。コーティングには種々の目的がある。コーティングは、組成物中で使用される活性成分の味を隠すために役立ち得る。さらに同時にコーティングは、光及び/又は湿気によって起こり得る有害な作用、例えば酸化、分解などに対して活性成分を保護している。さらに、コーティング層は活性成分による口腔粘膜の損傷から対象を保護することができる。
技術上周知の技法、例えば噴霧コーティング及びマイクロカプセル化等によってコーティング層を剤形に適用することができる。錠剤では、それはフィルムコーティング、糖類コーティング又は圧縮コーティングの形態であり得る。好ましくはフィルムコーティングプロセスを使用する(Remington's Pharmaceutical Sciences 18th ed. 1990, page 1666)。活性成分が速崩壊錠剤用コーティングの適用を必要とする場合、錠剤への圧縮前に個々の顆粒が適宜コーティングを備えていてよい。
【0014】
「その医薬的に許容できるエステル」という表現は、ベンダムスチンのいずれの医薬的に許容できるエステル、例えばアルキルアルコール及び糖アルコールとのエステルをも表す。アルキルアルコールの例は、C1-6-アルキルアルコール、例えばメタノール、エタノール、プロパノール、イソプロパノール、ブタノール及びtert-ブタノールである。糖アルコールの例は、マンニトール、マルチトール、ソルビトール、エリスリトール、グリコール、グリセロール、アラビトール、キシリトール及びラクチトールである。ベンダムスチンエステルの好ましい例は、エチルエステル、イソプロピルエステル、マンニトールエステル及びソルビトールエステルであり、ベンダムスチンのエチルエステルが最も好ましい。
「その医薬的に許容できる塩」という表現は、患者に(直接又は間接的に)投与されるとベンダムスチンを与える、ベンダムスチンのいずれの医薬的に許容できる塩をも表す。この用語は、ベンダムスチンエステルの医薬的に許容できる塩をさらに含む。それにもかかわらず、医薬的に許容できない塩も、これらの化合物が医薬的に許容できる塩の調製に役立つことがあるので、本発明の範囲内に含まれる。例えば、ベンダムスチンの医薬的に許容できる塩は、従来の化学的方法によって、酸性又は塩基性基を含む対応化合物から合成される。一般的に、これらの塩は、例えば、水若しくは有機溶媒又は両者の混合物中における化学量論のこれらの化合物の遊離酸性形又は塩基性形と対応する塩基又は酸との反応を利用して調製される。一般的にエーテル、酢酸エチル、イソプロパノール又はアセトニトリル等の非水性媒体が好ましい。ベンダムスチンの医薬的に許容できる塩の塩形成に使用できる酸の例としては、無機酸、例えば塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、及びリン酸など、並びに有機酸、例えば酢酸、マレイン酸、フマル酸、クエン酸、シュウ酸、コハク酸、酒石酸、リンゴ酸、乳酸、メチルスルホン酸及びp-トルエンスルホン酸などが挙げられる。ベンダムスチンの医薬的に許容できる塩を無機又は有機塩基から誘導してアンモニウム塩;アルカリ金属塩(リチウム、ナトリウム、カリウム等)、カルシウム若しくはマグネシウム等のアルカリ土類金属の塩、アルミニウム塩、低級アルキルアミン塩、例えばメチルアミン若しくはエチルアミン塩、低級アルキルジアミン塩、例えばエチレンジアミン塩、エタノールアミン、N,N-ジアルキレンエタノールアミン、トリエタノールアミン、及びグルカミン塩、並びにアミノ酸の塩基性塩を得ることができる。塩酸、臭化水素酸、及びヨウ化水素酸から調製された酸性塩が特に好ましいが、塩酸塩がベンダムスチンの最も好まし医薬的に許容できる塩である。医薬的に許容できる塩は、技術上周知の従来法によって作られる。
【0015】
「その医薬的に許容できる溶媒和物」という表現は、患者に投与されると(直接又は間接的に)ベンダムスチンを与える、医薬的に許容できるいずれの溶媒和物をも表す。この用語は、ベンダムスチンエステルの医薬的に許容できる溶媒和物をさらに含む。好ましくは溶媒和物は水和物、メタノール、エタノール、プロパノール、若しくはイソプロパノール等のアルコールとの溶媒和物、酢酸エチル等のエステルとの溶媒和物、メチルエーテル、エチルエーテル若しくはTHF(テトラヒドロフラン)等のエーテルとの溶媒和物又はDMF(ジメチルホルムアミド)との溶媒和物であり、水和物又はエタノール等のアルコールとの溶媒和物がさらに好ましい。溶媒和物を構成するための溶媒は、好ましくは医薬的に許容できる溶媒である。
本発明の組成物中の活性成分がベンダムスチン又はその医薬的に許容できる塩であることが特に好ましい。活性成分がベンダムスチン塩酸塩であることが最も好ましい。
医薬組成物中の活性成分の用量は、患者の状態、性別、体重、体表面積、又は年齢に応じて、特に患者の体重及び体表面積に応じて当業者によって容易に決定され、10〜1000mgの範囲である。1日の投薬量が約50〜約1000mg、好ましくは約100〜約500mgの活性成分の範囲であることが好ましい。1日の投薬量を単回用量又は1日に2回若しくは3回などの複数回用量として摂取してよく、最も好ましくは単回1日用量として摂取する。1日用量を1週間に1回又は1週間に数回摂取してよい。剤形は、単回1日用量又はその一部の量を含有し得る。本発明の剤形が約10〜約1000mg、好ましくは約25〜約600mg、さらに好ましくは約50〜約200mg、最も好ましくは約100mgの活性成分を含むことが好ましい。
本発明の組成物には、実質的な量、好ましくは活性物質の質量の2〜5倍の範囲の量で糖類が存在する。糖類は、本発明の組成物に組み入れられると、活性化合物の安定性にプラス効果を及ぼすことが分かった。そのことに加えて、これらの賦形剤は驚くべきことに、参考カプセル剤と比較した場合、活性化合物、特にベンダムスチン塩酸塩のバイオアベイラビリティーの向上をもたらすことが分かった。
糖類の好ましい例としては、マンニトール、マルチトール、エリスリトール、キシリトール、ラクトース、ショ糖、グルコース、ソルビトール、マルトース、トレハロース、ラクチトール及びブドウ糖(無水物又は一水和物)が挙げられる。
【0016】
これらの糖類賦形剤に加えて、本発明の医薬組成物は、さらに以下に詳述するさらなる賦形剤、例えば潤沢剤、流動促進剤、充填剤(又は希釈剤)、結合剤及び崩壊剤を含んでよい。
潤沢剤は、医薬組成物において、特に錠剤製造において下記機能の1つ以上を有し得る物質である:錠剤機の部品(ホッパー、ダイ及びパンチ)の表面への錠剤材料の付着を防止すること、粒子間摩擦を低減させること、ダイからの錠剤の放出を促進すること及び(打錠すべき)混合物の流速を改善すること。前記潤沢剤は、典型的にステアリン酸、ステアリン酸の塩若しくはエステル、水素化植物油、酸化マグネシウム、ポリエチレングリコール、ラウリル硫酸ナトリウム及びタルク、並びにその混合物から成る群より選択される。好ましくは前記潤沢剤は、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、グリセリルパルミトステアラート及びステアリルフマル酸ナトリウム、並びにその混合物から選択される。ステアリン酸が最も好ましい選択肢である。
この出願では、流動促進剤という用語は、打錠すべき混合物の流動特性を改善する物質として解釈するものとする。流動促進剤については、タルク、二酸化ケイ素及びシリカゲル(Cab-O-Sil(登録商標)、Syloid(登録商標)、Aerosil(登録商標))、デンプン及びケイ酸カルシウム等のいずれの適切な流動促進剤をも使用し得る。典型的に、二酸化ケイ素を使用する。
一般的に、充填剤(又は希釈剤)という用語は、打錠すべき材料のバルクを増やすために使用する賦形剤を表す。このサイズの増加が固体組成物の取扱いを改善する。充填剤は通常、固体組成物毎の薬物の用量が低く、充填剤がなければ固体組成物が小さ過ぎる場合に必要である。適切な充填剤の例は、ラクトース、ショ糖、マンニトール、ソルビトール、サッカロース、デンプン、アルファ化デンプン、微結晶性セルロース、粉末セルロース、リン酸水素カルシウム、炭酸カルシウム及びそのいずれかの組合せである。好ましい実施形態では、充填剤は、ラクトース、デンプン、微結晶性セルロース、ミクロファインセルロース及びそのいずれかの組合せ、最も好ましくは無水ラクトース及び微結晶性セルロースから選択される。
【0017】
一般的に、結合剤という用語は、医薬製剤に凝集性を付与する薬剤のために使用され、凝集性は、特に圧縮後の錠剤の場合に組成が損なわれないままでいることを保証する。使用するコンパクション法(直接圧縮、乾式造粒又は湿式造粒)に応じて、異なる結合剤を使用する。乾式コンパクション法(直接圧縮及び乾式造粒)では、適切な結合剤はラクトース、ショ糖、マンニトール、ソルビトール、サッカロース、デンプン、アルファ化デンプン、微結晶性セルロース、粉末セルロース、リン酸水素カルシウム、炭酸カルシウム及びそのいずれかの組合せである。好ましい実施形態では、結合剤は、ラクトース、デンプン、微結晶性セルロース、ミクロファインセルロース及びそのいずれかの組合せ、最も好ましくは無水ラクトース及び微結晶性セルロースから成る群より選択される。湿式造粒プロセスでは、溶液として及び乾燥形態の両方で結合剤を使用することができる。適切な結合剤として、例えば、ポリビニルピロリドン、分散性セルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、メチルセルロース、デンプン、アルファ化デンプン、部分的アルファ化デンプン、アラビアゴム、デキストリン、プルラン等が挙げられる。これらの結合剤の中で、分散性セルロース、ポリビニルピロリドン、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチル セルロースがさらに好ましい。
【0018】
医薬組成物、特に錠剤組成物に崩壊剤を含めて、錠剤が生物学的水性液と接触した後のその分解又は崩壊を促進することができる。錠剤が嚥下されると、多くの場合、唾液、胃液及び腸液などの体液と錠剤が接触するときに錠剤の迅速な崩壊に崩壊剤が関与する。崩壊剤として役立つ物質は、化学的にデンプン、セルロース、架橋ポリマー等に分類されている。本発明の実施で使用すべき崩壊剤種及びその添加レベルに関する調査の結果として、デンプン、加工デンプン、例えばナトリウムデンプングリコラート(Primojel(登録商標))、ナトリウムカルボキシメチルセルロース、架橋カルボキシメチルセルロースナトリウム(Ac-Di-Sol(登録商標))、架橋ポリビニルピロリドン、ポラクリリン(polacrilin)カリウム(Amberlite(登録商標)IRP88)及び低置換ヒドロキシプロピルセルロースが非常に良い崩壊効果をもたらし得ることが分かった。
ベンダムスチンの水溶液の安定性は、pHによって強く影響を受ける。約5より高いpH値では、加水分解によるこの化合物の有意な分解が観察される。pH>5では、分解が急速に進行し、このpH範囲では結果として生じる副生物の含量が多い。主な加水分解生成物は、下記4-[5-[(2-クロロエチル)-(2-ヒドロキシ-エチル)アミノ]-1-メチル-ベンズイミダゾ-2-イル]-ブタン酸(HP1)、4-[5-[ビス(2-ヒドロキシエチル)アミノ]-1-メチル-ベンズイミダゾ-2-イル]-ブタン酸(HP2)及び4-(5-モルフォリノ-1-メチルベンズイミダゾール-2-イル)-ブタン酸(HP3)である。
【0019】
【化2】
【0020】
経口投与された薬物の吸収は、普通は胃、小腸及び/又は大腸内で起こる。胃内のpHは約1〜3.5であり、小腸内では約6.5〜7.6、大腸内では約7.5〜8.0である。従って、5より高いpHの水性環境内で分解しやすいベンダムスチンのような化合物では、分解を回避するため、それが胃内で吸収され、小腸或いは大腸をも通過しないことが非常に好ましい。従って、ベンダムスチンが胃内で完全に又は少なくとも高程度まで吸収され、それによって小腸又は大腸内でのベンダムスチンの分解を回避又は低減させる医薬組成物が要望されている。
出願人は、驚くべきことに本発明の医薬組成物、特に、上記好ましい糖類を有する医薬組成物を用いてこの問題を解決できることを見い出した。ベンダムスチンを含むこれらの組成物は、速い溶解、特に欧州薬局方に準拠して50rpmでパドル装置を用いて人工胃液中で測定した場合、20分、好ましくは10分で少なくとも60%、40分、好ましくは20分で70%、60分、好ましくは30分で80%、最も好ましくは10分で少なくとも75%、20分で85%、30分で90%のベンダムスチンの溶解を示す。本明細書で使用する場合、人工胃液は、1000mlの水に2gの塩化ナトリウムを溶解させてから5N塩酸でpHを1.5±0.05に調整することによって調製された溶液を表す。
薬物が胃を通って小腸に至るまでの全時間は約20分〜5時間、普通は約30分〜3時間である。従って、この発明の組成物は、胃内にある間にベンダムスチンが大部分の程度まで放出かつ溶解されるので、患者内でのベンダムスチンの分解を有利に低減させ、結果として本発明のベンダムスチン含有組成物のバイオアベイラビリティーを改善することとなるであろう。
【0021】
この発明のさらなる態様では、固体剤形の医薬組成物を、ヒト又は動物、好ましくはヒトにおける医学的状態の治療、導入、救命療法、幹細胞移植前の前処置、維持療法、残存疾患の治療のために使用し得る。この医学的状態は、慢性リンパ性白血病(CLL)、急性リンパ性白血病(ALL)、慢性骨髄性白血病(CML)、急性骨髄性白血病(AML)、ホジキン病、非ホジキンリンパ腫(NHL)、多発性骨髄腫、乳癌、卵巣癌、小細胞肺癌、非小細胞肺癌、及び自己免疫疾患から選択される。
本発明は、人体又は動物体における慢性リンパ性白血病、急性リンパ性白血病、慢性骨髄性白血病、急性骨髄性白血病、ホジキン病、非ホジキンリンパ腫、多発性骨髄腫、乳癌、卵巣癌、小細胞肺癌、非小細胞肺癌、及び自己免疫疾患から選択される医学的状態の治療方法であって、有効量のこの発明の医薬製剤を、治療が必要な人体又は動物体に投与する工程を含む方法をも含む。好ましくは医学的状態が非ホジキンリンパ腫である。
この発明の別の態様では、医薬組成物を少なくとも1種のさらなる活性薬と併用投与することができる。ここで、前記さらなる活性薬は、本医薬組成物の投与前、投与と同時、又は投与後に与えられる。この少なくとも1種のさらなる活性薬は、好ましくはCD20に特異的な抗体(例えばリツキシマブ又はオファツムマブ)、アントラサイクリン誘導体(例えばドキソルビシン又はダウノルビシン)、ビンカアルカロイド(例えばビンクリスチン)、プラチン誘導体(例えばシスプラチン又はカルボプラチン)、ダポリナド(daporinad)(FK866)、YM155、サリドマイド及びその類似体(例えばサリドマイド又はレナリドマイドである)、又はプロテアソーム阻害薬(例えばボルテズミブ(bortezumib))である。
この発明の医薬組成物を少なくとも1種のコルチコステロイドと併用投与してもよく、前記コルチコステロイドは、本医薬組成物の投与前、投与と同時、又は投与後に与えられる。コルチコステロイド例はプレドニゾン、プレドニゾロン及びデキサメタゾンである。
以下の実施例は、本発明をさらに説明する。当業者には当然のことながら、これらの実施例は単に説明の目的のためであり、本発明を限定するものと考えるべきでない。
【実施例】
【0022】
1.適合性試験
実施例1a
適合性試験のため、ベンダムスチン塩酸塩と賦形剤を1:1(m/m)の比で含む混合物を調製した。マンニトール及びラクトースから賦形剤を選択した。調製後、混合物をクリアガラスHPLC-バイアル(6ml)Agilentに入れ、下表1に示す種々の貯蔵条件で貯蔵した。規定時点でサンプルを貯蔵から取り出して純度(HPLC;カラム:Zorbax Bonus-RP、5μm;カラムオーブンの温度:30℃;オートサンプラーの温度:5℃;検出器:254nm)及び外観について試験した。
【0023】
表1:貯蔵条件
*50℃で1カ月貯蔵後、70℃で貯蔵
**25℃/60%r.h.で1カ月貯蔵後40℃/75%で貯蔵
【0024】
全てのこれらの混合物では、ベンダムスチン塩酸塩含量(HPLCで測定)はほとんど変化せず、全てのこれらの貯蔵条件で常に99%を超えたままだった。加水分解生成物HP1はこれらの全ての貯蔵条件でほとんど検出されなかった(面積%<0.2)。
指定ベンダムスチン塩酸塩混合物の外観試験を裸眼で行なった。調査した全ての混合物は仕様に従い、調製直後及び全てのこれらの貯蔵条件下で1カ月の貯蔵後の両方で白色乃至オフホワイトの粉末を与えた。
【0025】
実施例1b
実施例1aの方法に従うさらなる適合性試験のため、ベンダムスチン塩酸塩と賦形剤を1:1(m/m)の比で含む混合物を調製した。Opadry(登録商標)、Eudragit(登録商標)E PO、ナトリウムカルボキシメチルセルロース(Avicel(登録商標) RC 591)及び架橋ポリビニルピロリドン(Crospovidone)から賦形剤を選択した。
Eudragit(登録商標)E POの場合、不純物HP1(加水分解生成物)及びBM1DIMERの初期量が有意に増加したが(HP1:1.5%、BM1DIMER:1%)、貯蔵中に湿度の影響とは無関係に全ての貯蔵条件でこれらの不純物の減少を検出することができた。架橋ポリビニルピロリドンの場合、貯蔵条件40℃/75%R.H./開いたバイアルではHP1の0.1%から0.4%への有意な増加を検出できた。全ての他の貯蔵条件(閉じたバイアル)ではHP1の増加を検出できなかった。
Eudragit(登録商標)E PO及び架橋ポリビニルピロリドンを含む混合物の外観は、貯蔵条件70℃/閉じたバイアルで変化した。両混合物はわずかに粘着性になった。さらに架橋ポリビニルピロリドンとの混合物の色が白色からクリーム色に変化した。
Opadry(登録商標)及びAvicel(登録商標)RC591を含む混合物の場合も、貯蔵条件70℃/閉じたバイアルで色がクリーム色に変わった。
【0026】
2. 錠剤製剤
実施例2
主賦形剤としてマンニトールを含み、下表2aに示す相対量で微結晶性セルロース、Ac-Di-Sol(登録商標)、コロイド状二酸化ケイ素、タルク及びステアリン酸を含む混合物253gを1リットルのキューブブレンダー(Erweka)で15分間混合することによって調製した。その後、10.612gの混合物と3.0gのベンダムスチン塩酸塩を0.425mmの篩いを通して選別してから、50mlのガラスバイアルを備えたTurbulaミキサーT2Aに移し、引き続き60rpmで10分間混合した。
この混合物から、以下の特性を有する円形錠剤を圧縮成形した:
平均粒径:9.1mm;平均値質量:247.7mg;平均値硬度:81N。
【0027】
【0028】
錠剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩並びに分解生成物、合成の副生物のような関連物質の量をHPLCで測定した(カラム:Zorbax Bonus-RP、5μm;カラムオーブンの温度:30℃;オートサンプラーの温度:5℃;検出器:254nm)。結果を下表2bに示す。
【0029】
*1:NP1:4-[6-(2-クロロエチル)-3,6,7,8-テトラ-ヒドロ-3-メチル-イミダゾ[4,5-h]-[1,4]ベンゾチアジン-2-イル]ブタン酸
BM1Dimer:4-{5-[N-(2-クロロエチル)-N-(2-{4-[5-ビス(2-クロロエチル)アミノ-1-メチルベンズイミダゾール-2-イル]ブタノイルオキシ}エチル)アミノ]-1-メチルベンズイミダゾール-2-イル}ブタン酸
BM1EE:4-[5-[ビス(2-クロロエチル)アミノ]-1-メチル-ベンズイミダゾ-2-イル]ブタン酸エチルエステル
*2:n.d.:検出不能、すなわち検出限界超え(0.05%未満の面積パーセンテージ)
【0030】
実施例3
実施例2で述べたのと同様に混合物及び錠剤を調製したが、下表3aに示す化合物及び相対量を用いた。
錠剤は以下の特徴を有した:
平均値径:9.1mm;平均値質量:248.9mg。
【0031】
【0032】
錠剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表3bに示す。
【0033】
【0034】
実施例4
実施例2で述べたのと同様に錠剤を調製したが、下表4aに示した化合物及び相対量を用いた。
錠剤は以下の特徴を有した:
平均値径:9.1mm;平均値質量:247.8mg。
【0035】
【0036】
錠剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表4bに示す。
【0037】
【0038】
従来技術の参考例
20.0±1mgのベンダムスチン塩酸塩を空の硬ゼラチンカプセルのボディに量り入れ、AgilentのクリアガラスHPLCバイアル(6ml)に入れた。ボディの上部にキャップを置いてわずかに押すことによってカプセルを閉じた。このカプセル剤を40℃/75%RH(開いたガラスバイアル)又は50℃(閉じたガラスバイアル)で貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表5に示す。
【0039】
【0040】
すぐに分かるように、いずれのさらなる加工工程もなしで純粋なベンダムスチン塩酸塩からカプセル製剤を調製したが、カプセル製剤は、本発明の錠剤製剤より不安定なロットだった。40℃/75%RH(開いたガラスバイアル)及び50℃(閉じたバイアル)の両方で1カ月の貯蔵内に加水分解生成物が形成された。40℃及び75%RH(相対湿度)で開いたバイアルの場合、1カ月の貯蔵後に加水分解生成物HP1の量が4倍に増えた。閉じたバイアルではHP1含量がさらに高く、これはカプセルとの反応のためであろう。要約すると、錠剤はカプセル剤よりずっと安定な固体剤形を提供した。
【0041】
実施例5
8.0gのヒドロキシプロピルメチルセルロース及び1.5gのPEG6000を88.5gの精製水に溶解させる。その後2.0gの黄色酸化第二鉄及び0.5gの酸化チタンをその中に分散させてコーティング液を得る。実施例2で得た錠剤を、フィルムコーティング装置を用いて錠剤質量毎にこの溶液3%でコーティングする。
【0042】
実施例6
【0043】
(1000錠剤の製造方法)
コロイド状二酸化ケイ素及びステアリン酸以外の全ての錠剤コア成分をSomakon容器(5L)中に装填した。ベンダムスチンを加えて4分間1000rpm(ワイパー10rpm)でブレンディングを行なった。結果として生じたブレンドを0.5mmの篩いを通して選別した。容器にブレンドを装填し、コロイド状二酸化ケイ素を加えた。上記条件でブレンディングを2分間行なった。その後ステアリン酸を加え、ブレンディングを1分間続けた。引き続きブレンドを0.5mmの篩いを通して選別し、再び容器中に装填し、全て同一条件でさらに30秒間ブレンドした。
この混合物から下記特徴を有する円形錠剤を圧縮成形した:
平均値径:9.5mm;平均値質量:254.6mg(初め)〜257.2mg(終わり);脆砕性0.1%;平均値硬度:122N(初め)〜128N(終わり)。
引き続き錠剤をOpadry(登録商標)分散系で5%の質量増加が達成されるまでフィルムコーティングした。
フィルムコーティング錠の平均質量は268.4mgだった。
錠剤コアとフィルムコーティング錠を両方とも閉じた琥珀色ガラスバイアル内で40℃/75%RHにて貯蔵した。ベンダムスチン塩酸塩並びに分解生成物、合成の副生物のような関連物質の量を上述したようにHPLCで測定した。結果を下表6b.1及び6b.2に示す。
【0044】
*3:主ピークと比べて0.69の相対保持時間の未同定化合物ピーク
【0045】
【0046】
実施例7
【0047】
(1000錠剤の製造方法)
コロイド状二酸化ケイ素及びステアリン酸以外の全ての錠剤コア成分をSomakon容器(5L)中に装填した。ベンダムスチンを加えて4分間1000rpm(ワイパー10rpm)でブレンディングを行なった。結果として生じたブレンドを0.5mmの篩いを通して選別した。容器にブレンドを装填し、コロイド状二酸化ケイ素を加えた。上記条件でブレンディングを2分間行なった。その後ステアリン酸を加え、ブレンディングを1分間続けた。引き続きブレンドを0.5mmの篩いを通して選別し、再び容器中に装填し、全て同一条件でさらに30秒間ブレンドした。
この混合物から下記特徴を有する円形錠剤を圧縮成形した:
平均値径:9.5mm;平均値質量:262.4mg(初め)〜254.4mg(終わり);脆砕性0.1%(初め)〜0.2%(終わり);平均値硬度:98N(初め)〜91N(終わり)。
引き続き錠剤をEudragit(登録商標)分散系で3%の質量増加が達成されるまでフィルムコーティングした。
フィルムコーティング錠の平均質量は273.5mgだった。
錠剤コアとフィルムコーティング錠を両方とも閉じた琥珀色ガラスバイアル内で40℃/75%RHにて貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表7b.1及び7b.2に示す。
【0048】
【0049】
【0050】
実施例8
【0051】
(1000錠剤の製造方法)
コロイド状二酸化ケイ素及びステアリン酸以外の全ての錠剤コア成分をSomakon容器(2.5L)中に装填した。ベンダムスチンを加えて4分間1000rpm(ワイパー10rpm)でブレンディングを行なった。結果として生じたブレンドを0.5mmの篩いを通して選別した。容器にブレンドを装填し、コロイド状二酸化ケイ素を加えた。上記条件でブレンディングを2分間行なった。その後ステアリン酸を加え、ブレンディングを1分間続けた。引き続きブレンドを0.5mmの篩いを通して選別し、再び容器中に装填し、全て同一条件でさらに30秒間ブレンドした。
この混合物から下記特徴を有する円形錠剤を圧縮成形した:
平均値径:9.5mm;平均値質量:252.2mg(初め)〜250.7mg(終わり);脆砕性0.1%(初め)〜0.2%(終わり);平均値硬度:65N(初め)〜73N(終わり)。
引き続き錠剤をEudragit(登録商標)分散系で3%の質量増加が達成されるまでフィルムコーティングした。
フィルムコーティング錠の平均質量は253.6mgだった。
錠剤コアとフィルムコーティング錠を両方とも閉じた琥珀色ガラスバイアル内で40℃/75%RHにて貯蔵した。ベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表8b.1及び8b.2に示す。
【0052】
【0053】
【0054】
実施例9
【0055】
600錠剤のための製剤PF1の製造方法:
33.06gのベンダムスチン、111.60gのブドウ糖、40.92gのラクトース、11.22gの微結晶性セルロース及び1.20gのステアリン酸マグネシウムを秤量し、二重ポリエチレンバッグに移して5分間混合した。その後、該粉末ブレンドを偏心性錠剤機(Korsch EK0)のホッパーに移し、下記特徴を有する円形錠剤に圧縮成形した:平均値径:10.0mm;平均値質量:336.9mg(初め)〜335.98(終わり);脆砕性:0.15%;平均硬度値:69.25N(初め)〜68.60N(終わり)。
引き続き錠剤コアをコーティングパン(4M8 ForMate PanCoat)内で9%のOpadry(登録商標)TM White水性懸濁液を用いてコーティングして乾燥させた。錠剤の平均質量は342.42mgだった。その後、ねじ込みプラグで閉じた琥珀色ガラス瓶に錠剤を詰めて40℃/75%RHで貯蔵した。
600錠剤のための製剤PF2の製造方法:
33.06gのベンダムスチン、111.42gのラクトース、39.60gのトレハロース、12.60gの架橋ポリビニルピロリドン及び1.32gのステアリン酸マグネシウムを秤量し、二重ポリエチレンバッグに移して5分間混合した。その後、該粉末ブレンドを偏心性錠剤機(Korsch EK0)のホッパーに移し、下記特徴を有する円形錠剤に圧縮成形した:平均値径:10.0mm;平均値質量:332.95mg(初め)〜332.12(終わり);脆砕性:0.3%;平均硬度値:65.9N(初め)〜59.0N(終わり)。
引き続き錠剤コアをコーティングパン(4M8 ForMate PanCoat)内で9%のOpadry(登録商標)TM White水性懸濁液を用いてコーティングして乾燥させた。錠剤の平均質量は340.1mgだった。その後、ねじ込みプラグで閉じた琥珀色ガラス瓶に錠剤を詰めて40℃/75%RHで貯蔵した。
製剤PF3の製造方法:
ソルビトール及びブドウ糖無水物を秤量した。140.64gのソルビトールを105.48gの精製水に溶解させ、得られた溶液を引き続きFluid Bed Granulator(4M8ForMate FluidBed)内で用いて659.36gのブドウ糖を造粒した。その後、該顆粒を60℃で乾燥させ、850μmの篩いを通して選別した。
33.06gのベンダムスチン塩酸塩、149.82gのソルビトール/ブドウ糖顆粒、13.8gの微結晶性セルロース及び1.32gのステアリン酸マグネシウムを二重ポリエチレンバッグに量り入れて5分間混合した。その後、該粉末ブレンドを偏心性錠剤機(Korsch EK0O)のホッパーに移し、10.0mmの平均径を有する円形錠剤に圧縮成形した。錠剤は335.99mg(初め)〜339.50mg(終わり)の質量の平均値;脆砕性:0%;平均硬度値:125.60N(初め)〜129.7N(終わり)を有した。次に錠剤を下記2工程のコンディショニングプロセスに供した(選択したバッチについてのみ行なった):錠剤を25℃/60%R.Hに2時間置き、引き続き40℃に2時間置いた。
引き続き錠剤をコーティングパン(4M8 ForMate PanCoat)内で9%のOpadry(登録商標)TM White水性懸濁液を用いてコーティングした。錠剤の平均質量:341.43mg。その後、ねじ込みプラグで閉じた琥珀色ガラス瓶に錠剤を詰めて40℃/75%RHで貯蔵した。
貯蔵したフィルムコーティング錠中のベンダムスチン塩酸塩及び関連物質の量を上述したようにHPLCで測定した。結果を下表9b.1〜9b.3に示す。
【0056】
【0057】
【0058】
【0059】
3. 溶解試験
実施例10
実施例2及び3の錠剤製剤についての溶解試験を人工胃液内でT=0にて行なった。HPLC(カラム:Zorbax Bonus-RP、5μm;カラムオーブンの温度:30℃;オートサンプラーの温度:5℃;検出器:254nm)によるアッセイのため溶解サンプルを試験した。2gの塩化ナトリウムp.A.を1000mlの水に溶解させ、5N塩酸でpHを1.5±0.05に調整することによって、人工胃液pH1.5を調製した。欧州薬局方6.0の2.9.3章に準拠してApparatus 2(パドル装置)を用いて溶解試験を行なった。パドルの回転速度は50rpm、温度は37±0.5℃、溶解媒体の量は500mlだった。
実施例2の錠剤製剤(錠剤製剤1)及び実施例3の錠剤製剤(錠剤製剤2)の結果を表10aに示す。
【0060】
【0061】
実施例6、実施例7及び実施例8のコーティング錠製剤についてT=0で行なったのと同じ溶解試験の結果を下表10bに示す。
表10b
【0062】
実施例9の錠剤についての対応する溶解データは下表のとおりだった。
【0063】
【0064】
上記から得られるように、本発明の全ての錠剤製剤は、ベンダムスチンの速い溶解を示す。特に本発明の製剤は、本明細書で前述したベンダムスチンの溶解プロファイルを示す。
4. in vivo試験
ベンダムスチンの動物バイオアベイラビリティー研究をビーグル犬で行なった:PK研究概要
研究1
目的は、3種の錠剤製剤(T1〜3)及び1種のカプセル製剤(C)、全部で4種の経口製剤中の1用量(すなわち50mg)のベンダムスチンのバイオアベイラビリティーを決定することだった:AUC及びCmax。
要した動物の総数:16
基本設計:
交差設計、治療群(arm)毎に8匹の動物:
【0065】
表11a:期間1(単回用量の錠剤、又はカプセル剤、日1):
【0066】
1週間の休薬(wash-out)
【0067】
表11b:期間2(期間1の1週間後、以下のいずれかの単回用量、日8):
【0068】
1週間の休薬
【0069】
表11c:期間3(期間2の1週間後、以下のいずれかの単回用量、日15):
【0070】
研究2
目的は、1種の錠剤製剤T4、1種のカプセル製剤、全部で3種の経口製剤中の1用量(すなわち50mg)のベンダムスチンのバイオアベイラビリティーを決定することだった:AUC及びCmax。
要した動物の総数:16
基本設計:
交差設計、治療群毎に8匹の動物:
【0071】
表12a:期間1(単回用量のカプセル剤、日1):
【0072】
1週間の休薬
【0073】
表12b:期間2(期間1の1週間後、以下の製剤のどちらかの単回用量、日8):
【0074】
実施例11
50mgのベンダムスチンを含む実施例9のコーティング錠(製剤3、Opadry(登録商標)でコーティングした錠剤T4)をオスとメスのイヌに経口投与して参考例のカプセル剤と比較した。
カプセル製剤と実施例9のコーティング錠の両者について平均血漿プロファイル対時間を図1に示す。
実施例12
50mgのベンダムスチンを含む実施例6、7、又は8のコーティング錠(錠剤T1〜T3)をオスとメスのイヌに経口投与して参考例のカプセル剤と比較した。
カプセル製剤と実施例6〜8のコーティング錠の平均血漿プロファイル対時間を図1に示す。
実験は以下のために行なった:
−糖類又は糖類混合物が、速い溶解プロファイルとコーティングに適した硬度値とを有する化学的に安定した錠剤を得るのに適しているかを評価する;
−APIと賦形剤との適合性を評価する;
−異なる製造プロセス(乾式造粒、直接圧縮及び湿式造粒)を調査することによって、プラセボ及びAPI含有バッチを開発する;
−種々のベンダムスチン塩酸塩/糖類比を評価する;
−製造される錠剤の技術的特性及び安定性に及ぼす含水量の影響を調査する;
−商業的に入手可能な凍結乾燥ベンダムスチン塩酸塩製品(Ribomustin(登録商標))を用いて錠剤を製造し、かつこれらの錠剤を対応量のマンニトールとベンダムスチン塩酸塩を用いて作製した錠剤の特性と比較する。
本発明の錠剤、すなわち50mgのベンダムスチン(ベンダムスチン塩酸塩として55mg)を含む錠剤の製造のために以下の糖類を用いた。
【0075】
表13.
【0076】
作られたバッチの品質を物理的外観の観察、同定試験(HPLC)、溶解試験、含量及び関連物質アッセイ(HPLC)、含量均一性試験(HPLC)、硬度試験及び含水量(Karl Fischer法)によって評価した。下表に詳細に示す貯蔵条件下で琥珀色ガラス瓶に詰め込まれる加速安定性研究にバッチを委ねた。それぞれ製造されたAPI含有バッチについていくつかの錠剤をバックアップサンプルとして5℃で貯蔵した。
以下、それらの製造プロセスに関して種々の賦形剤を調査した。これらの賦形剤を使用することによって、乾式造粒でいくつかのプラセボ製造試験を行なって、良い品質の錠剤を得るのに適した製造方法についての予備情報を得た。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/002だけ架橋ポリビニルピロリドン(Crospovidone(登録商標))を用いた。バッチD001T/002(充填剤:無水ラクトース)に対するCrospovidone(登録商標)の選択は、この製剤と実施例9の原型製剤の間の類似性に基づいた。作製した全てのバッチ用の潤沢剤としてステアリン酸マグネシウムを使用した。プラセボ試験の乾式造粒製造プロセスは以下の工程に存した:
1. 糖類及び部分量の潤沢剤(総量の83.3%w/w)を正確に秤量してから2分間ポリエチレンバッグ内で混合した。
2. 得られた混合物を18mm径パンチを備えた錠剤機を用いて圧縮した。
3. 得られたスラグを850μmネットを用いて篩過した。
4. 顆粒を秤量し、2分間ポリエチレンバッグ内で崩壊剤及び残量の潤沢剤(16.7%w/w)と混合してから10mm径パンチを用いて打錠した。
【0077】
表14及び表15は、各プラセボ製剤の組成並びに最終混合物と錠剤の両方について行なった分析試験の結果を要約する。
表16には、プラセボバッチの製造プロセス中及び/又はそれらの分析特徴づけ中に行なった観察を報告する。
プラセボバッチD001T/001-D001T/002-D001T/004-D001T/013-D001T/015について行なった分析的及び/又は物理的試験結果は、これらの製剤が乾式造粒で製造し、かつAPIを添加してさらに調査するのに適していることを示した。全ての他の製剤は固めるのが難しく、得られても非常に脆砕性の錠剤によって特徴づけられる。
バッチD001T/005(充填剤:β-シクロデキストリン)は、乾式製造プロセスにおける良い挙動、高い硬度、低い脆砕性、長い崩壊時間を示した。超崩壊剤(Crospovidone(登録商標))を利用し、かつAPIを添加することによって、この製剤をさらに検討した(下記パラグラフ参照)。
【0078】
表14. 乾式造粒−プラセボバッチの組成及び分析結果(バッチD001T/001〜D001T/010)。
【0079】
(表14続き)
【0080】
(表14続き)
N.A. = 混合物が打錠プロセスに適していないので該当データなし(表5aに報告された観察参照)
【0081】
表15. 乾式造粒−プラセボバッチの組成及び分析結果(バッチD001T/011〜001T/025)。
【0082】
(表15続き)
【0083】
(表15続き)
N/A = 混合物が打錠プロセスに適さないので該当データなし(表5aに報告された観察参照)
【0084】
表16:それぞれ製造されたプラセボバッチの製造プロセス、製品の技術的特性及び分析試験についての所見
【0085】
(表16続き)
【0086】
(1:5のベンダムスチン塩酸塩/糖類質量比で乾式造粒によって製造したバッチ)
活性医薬成分(API)を含む錠剤を乾式造粒で製造するのに適していると評価されたプラセボ製剤を修正してAPIを含め、2つのAPI/糖類質量比1:5及び1:2を探究した。
このパラグラフでは、1:5のAPI/糖類質量比の製剤について述べる。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/022だけ架橋ポリビニルピロリドン(Crospovidone)を使用した。作製した全てのバッチの潤沢剤としてステアリン酸マグネシウムを用いた。
API含有バッチの乾式造粒による製造プロセスは以下の工程に存した:
1. 糖類、部分量の潤沢剤(総量の83.3%w/w)及びベンダムスチン塩酸塩を正確に秤量し、5分間二重ポリエチレンバッグ内で混合した。
2. 粉末ブレンドを18mm径パンチを備えた錠剤機を用いて圧縮した。
3. 顆粒を得るため、生成されたスラグを850μmネットを用いて篩過した。
4. 顆粒を秤量し、5分間二重ポリエチレンバッグ内で崩壊剤及び残量の潤沢剤(16.7%w/w)と混合した。
5. 得られた混合物を10mm径パンチを用いて打錠した。
表17は、製造された各API含有製剤の組成及びAPI含有最終混合物について行なった分析試験の結果を要約し;表18は、得られた製品について行なった分析試験の結果を要約する。
【0087】
表17. 乾式造粒−API/糖類質量比1:5。API含有バッチ最終混合の組成及び分析結果
【0088】
表18. 乾式造粒−API//糖類質量比1:5。API含有バッチ錠剤の分析結果
【0089】
最終混合物と最終製品の両者について行なった分析試験の結果は、主に含量均一性及び純度についてほとんどの場合に良かった。全てのAPI含有バッチは、満足のいく質量均一性、API含量の均質性、及び低い不純物含量を示した。全ての製剤のこの不純物プロファイルは、APIの仕様に従ったので(表中の仕様限界参照)、製造プロセス中に分解は起こらない。
2種のAPI含有バッチは、APIアッセイで低い値を示した;この結果は、小さいバッチサイズのため並びに製造プロセス中の損失及び最終混合物についてのIPC用サンプルのためであろう。
【0090】
(1:2のAPI/糖類質量比で乾式造粒によって製造したAPI含有バッチ)
1:5のAPI/糖類質量比を有する錠剤を製造するために乾式造粒で以前に調査した全ての糖類を1:2の比でも評価した。
製造プロセスについては上記を参照されたい。この場合、得られた混合物を8mm径パンチを用いて打錠した。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/105だけ架橋ポリビニルピロリドン(Ccrospovidone(登録商標))を使用した。このバッチでは、我々はAvicel(登録商標)PH 112及びCrospovidone(登録商標)の使用を検討した。1:5のAPI/糖類(以前の結果参照)で乾式造粒によって製造した前記シクロデキストリンベース製剤に従ってCrospovidone(登録商標)を選択した。
表19及び表20は、1:2のAPI/糖類質量比で乾式造粒によって製造した各API含有製剤の組成及び最終混合物と錠剤の両方について行なった分析試験の結果を要約する。全てのAPI含有バッチは、適切な質量均一性、API含量の均質性及び低不純物含量を示した。脆砕性及び硬度値は、ほとんどの場合に仕様に従っている。バッチD001T/093、D001T/095及びD001T/096の場合、6錠剤について行なった溶解試験の結果は、RSDが高く、仕様値の範囲外を示したので試験を12錠剤のサンプルに拡張した。
シクロデキストリンベース錠剤は、両崩壊剤(Avicel(登録商標)PH 112及びCrospovidone(登録商標))で良い特性を示す。
【0091】
表19. 乾式造粒−API/糖類質量比1:2。API含有バッチ最終混合物組成及び分析結果。
【0092】
表20. 乾式造粒−API/糖類質量比1:2。API含有バッチ錠剤分析結果。
【0093】
(表20続き)
n.d. = 検出されず
【0094】
(1:5のAPI/糖類質量比で直接圧縮によって製造したAPI含有バッチ)
乾式造粒で製造するのに適した特徴を有する糖類を直接圧縮を利用して1:5のAPI/糖類比を有する錠剤を開発することをも検討した。
2タイプの崩壊剤を使用した:標準的崩壊剤として微結晶性セルロース(Avicel(登録商標)PH 112)を用い、バッチD001T/029だけ架橋ポリビニルピロリドン(Crospovidone(登録商標))を使用した。
この製造プロセスは以下の工程から成った。
1. API及び賦形剤を秤量する工程。
2. 原材料を二重ポリエチレンバッグに移して均質粉末ブレンドが得られるまで約5分間混合する工程。
3. 粉末ブレンドを錠剤機のホッパーに移す工程。
4. 10mm径パンチを備えた偏心性錠剤機を用いた粉末ブレンドの圧縮。
直接圧縮によって製造したAPI含有バッチの特性を下表に示す。
【0095】
表21. 直接圧縮−API/糖類質量比1:5。API含有バッチ最終混合物組成及び分析結果。
得られた分析試験結果を表22に掲載する。
【0096】
表22. 直接圧縮−API/糖類質量比1:5。API含有バッチ錠剤分析結果。
【0097】
上表で報告したように、直接圧縮で製造したAPI含有錠剤は、不均質なAPI含量と脆砕性のわずかな増加を示したバッチD001T/030(充填剤:ショ糖97%+マルトデキストリン3%)を除き、乾式造粒で作製した錠剤との重大な意味を持つ差異を示さなかった。
【0098】
湿式造粒:
(プラセボ探索的試験)
本プロジェクトの第一及び第二部で得られた結果に基づいて、乾式造粒又は直接圧縮に適さない糖類を湿式造粒によって調査した。
湿式造粒技術を調査するための本アプローチを以下に示す。
図2のフローシートに示す工程に従って各糖類を造粒した。各工程の最後に湿式造粒糖類を乾燥させ、圧縮試験を行なって該顆粒が打錠に適しているか評価した。圧縮試験の結果が疑わしい造粒糖類についてのみプラセボバッチを製造した。プラセボ試験の組成及び関連分析結果を表23で報告する。
以下の工程に従ってプラセボバッチを製造した。
1. 流動床又は高せん断造粒機を用いた水又はソルビトール溶液との糖類の湿式造粒(上記湿式造粒試験のフローシート、及び表23参照)。
2. 流動床造粒機又はオーブン内での湿式造粒糖類の乾燥工程。
3. 850及び710μmネットを用いて造粒糖類を篩過する工程。
4. 製剤の全成分の秤量及びポリエチレンバッグ内で2分間混合する工程。
5. 10mm径パンチを備えた偏心性錠剤機を用いた粉末ブレンドの圧縮。
作製した全てのバッチでは、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。
【0099】
表23. 湿式造粒。プラセボバッチ組成及びIPC結果。
【0100】
(表23続き)
【0101】
(表23続き)
【0102】
(表23続き)
【0103】
(1:5のAPI/糖類質量比で湿式造粒によって製造したAPI含有バッチ)
乾式造粒又は直接圧縮技術による錠剤製造に適さないことが分かった全ての糖類について湿式造粒プロセスを含む製造試験を行なった。
実験室規模で行なったこれらの試験の製造プロセスは以下のように要約される。
1. 流動床又は高せん断造粒機を用いた水又はソルビトール溶液との糖類の湿式造粒(湿式造粒製造試験の上記フローシート、及び表24参照)。
2. 流動床造粒機又はオーブン内での湿式造粒糖類の乾燥工程。
3. 850及び710μmネットを用いた篩過工程。
4. APIと賦形剤の秤量及びポリエチレンバッグ内で2分間混合する工程。
5. 10mm径パンチを備えた偏心性錠剤機を用いた粉末ブレンドの圧縮。
作製した全てのバッチでは、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。
【0104】
表24及び表25は、湿式造粒で製造した各API含有製剤の組成及び最終混合物と錠剤の両者について行なった分析試験の結果を掲載する。
最終混合物及び最終製品について行なった分析試験の結果は、ほとんどの場合、仕様に従う。製造プロセス中に分解は起こらない。
調査した糖類のうち、Fructose MS(Galam)だけが湿式造粒による加工に適さず:API含有バッチD001T/047は高い脆砕性を有し、バッチD001T/082は仕様の範囲外の脆砕性と硬度を示す。
バッチD001T/060、D001T/061、D001T/082、D001T/086はAPIアッセイで低い値を示し、バッチD001T/082及びD001T/086では、850μm及び710μmネットを用いて顆粒が篩過されたが、含量の均一性が従わない。この結果はおそらく不十分な粉末の混合のためであろう。
【0105】
表24. 湿式造粒−API/糖類質量比1:5。API含有バッチ最終混合物組成及び分析結果
【0106】
(表24続き)
【0107】
(表24続き)
【0108】
(表24続き)
(*) 好ましくはバッチは過剰のA.P.I(5.9%)を含む;(**) これらの糖類は空気流で流動化されないので、流動床を用いた糖類溶液による造粒工程を調査できなかった;(***) 最終混合物は打錠に適さない
【0109】
表25. 湿式造粒−API/糖類質量比1:5。API含有バッチ錠剤分析結果。
【0110】
(表25続き)
【0111】
(表25続き)
【0112】
(表25続き)
【0113】
(1:2のAPI/糖類質量比で湿式造粒によって製造したAPI含有バッチ)
1:5のAPI/糖類質量比の錠剤を製造するために湿式造粒で以前に調査した全ての糖類を1:2の比についても評価した。
フルクトースは得られた顆粒が打錠に適さないので、1:2の比では評価しなかった。
作製した全てのバッチでは、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。
API含量の均一性を改善するため、以下のアプローチを適用してこれらのAPI含有バッチを製造した。
1. 以前に最適化した手順を用いて糖類の湿式造粒
2. API含有混合物の調製
3. 混合物の乾式造粒(スラグ製造→スラグ篩過)
4. 得られた混合物を8mm径パンチを用いて打錠。
工程3(混合物の乾式造粒)については上記を参照されたい。
表26及び表27は、1:2のAPI/糖類質量比で湿式造粒した糖類を用いて製造したAPI含有バッチの組成及び分析結果を報告する。脆砕性は、ほとんどの場合、仕様の範囲外である。API/糖類の質量変化は、D001T/084バッチ(充填剤;顆粒化マンニトール)の技術的特性に影響を及ぼさない。
【0114】
表26. 湿式造粒−A.P.I./糖類質量比1:2。API含有バッチ最終混合物組成及び分析結果。
【0115】
(表26続き)
【0116】
(表26続き)
(*) 以前の製剤(API/糖類質量比1:5)を開発するために用いたラクチトールはもはや商業的に入手できないので、新しい製造業者によって購入したラクチトールを用いてこのバッチを製造した(DaniscoによるLactitol)。
【0117】
表27. 湿式造粒−A.P.I./糖類質量比1:2。API含有バッチ錠剤分析結果。
【0118】
(表27続き)
【0119】
(表27続き)
【0120】
(API/マンニトール質量比の影響)
マンニトールベース錠剤を製造して以下のAPI/マンニトール比(1:0.01、1:0.1、1:0.5、1:1.7、1:4、1:5、1:6及び1:10)を調査した。1:5のAPI/マンニトール質量比を有する製剤(標準的製剤)については上述した。
これらのバッチの製造では、Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として使用した。製造プロセスに関し、1:1.7、1:4、及び1:6比では、湿式造粒したマンニトール、ベンダムスチン塩酸塩及び賦形剤を正確に秤量し、二重ポリエチレンバッグ内で5分間混合した。バッチD001T/110(1:10比)では、前混合を行なった。この場合、ベンダムスチン塩酸塩を5分間、半量の賦形剤混合物と混合した。次に、得られた混合物を残存量の賦形剤に加えてさらに5分間混合した。最終混合物を適切なパンチ(1:1、1:1.7及び1:2比には8mm径、1:4及び1:6比の場合は10mm、1:7比には12mm、1:10比には14mm)を備えた錠剤機を用いて打錠した。
1:0.01、1:0.1、1:0.5比については、我々は上述した製造プロセス(糖類の湿式造粒後に乾式造粒)を適用して、API含量の均一性を改善した。得られた混合物を6mm径パンチで打錠した。
下表(表28及び表29)は、異なるAPI/マンニトール比の影響を研究するために製造したAPI含有製剤の組成及び分析結果を要約する。バッチD001T/111、D001T/083及びD001T/106は高い脆砕性を示し、バッチD001T/106、D001T/108及びD001T/109では、含量の均一性が以前に得られたデータの傾向から逸脱して従わなかった。この結果は、これらのバッチが異なる物理的性質を有し得る新しいロット(ロット番号:F08-05873)のベンダムスチンHClを用いて作製したという事実のためであろう。
【0121】
表28. A.P.I./マンニトール質量比。API含有バッチ最終混合物組成
【0122】
(表28続き)
【0123】
(表28続き)
(*) 上記実験アプローチを用いて製造したバッチ
□ 標準製剤1:5API/糖類質量比
【0124】
表29. A.P.I./マンニトール質量比の研究。API含有バッチ錠剤分析結果。
【0125】
(表29続き)
【0126】
(表29続き)
□ 標準製剤1:5のAPI/糖類質量比
【0127】
(糖類の組合せの研究)
表30及び表31は、糖類の組合せの研究に関する結果を報告する。
以下の組合せを調査した。
−単糖/二糖1:1
(*)マンニトール(Pearlitol 200 SD)/ラクトース無水物(SuperTab 21 AN)
ソルビトール(Neosorb P60W)/マルトース(Sunmalt S)
−オリゴ糖/単糖1:1
(*)D-メレジトース一水和物/(*)ブドウ糖無水物ST 0.5
(*)ラフィノース五水和物(顆粒化)/(*)マンニトール(顆粒化)(Pearlitol 200 SD)
−オリゴ糖/二糖1:1
(*)ラフィノース五水和物(顆粒化)/ラクトース一水和物(Supertab 14SD)
β-シクロデキストリン(Kleptose DC)/ショ糖(EV Saccharide)
(*)これらの糖類は湿式造粒で顆粒化された(32ページ参照)。
製造プロセスは、未加工又は顆粒化糖類の直接圧縮に存した。
Avicel PH 112及びステアリン酸マグネシウムをそれぞれ崩壊剤及び潤沢剤として用いて、以下の工程を実施してこれらのバッチを製造した。
1. 糖類(又は顆粒化糖類)、ベンダムスチン塩酸塩及び賦形剤を正確に秤量し、二重ポリエチレンバッグ内で5分間混合した。
2. 得られた混合物を10mm径パンチを用いて打錠した。
【0128】
表30. 糖類の組合せの研究。API含有バッチ最終混合物組成及び分析結果。
【0129】
表31. 糖類の組合せの研究。API含有バッチ錠剤分析結果。
【0130】
(表31続き)
【0131】
一般に、糖類の組合せの研究用に製造した錠剤は良い特性を示す。しかしながら、バッチD001T/102(ラフィノース五水和物/マンニトール(Pearlitol 200 SD))は高い脆砕性を示し、バッチD001T/100及びD001T/049はAPI含量の不均質性を示す。
【0132】
実施例14. 凍結乾燥ベンダムスチンHCl(Ribomustin)及びベンダムスチンHCl/マンニトール錠剤(Api/糖類質量比1:1.2)
静脈内適用のための市販製品(Ribomustin(登録商標))から得た凍結乾燥材料を使用するか又は湿式造粒したマンニトールとベンダムスチンHClを使用して、1:1.2の質量比でベンダムスチン塩酸塩/マンニトールを含む錠剤を調製した。
以下の実験操作に従って製造プロセスを行なった:Ribomustin(登録商標)バイアルから凍結乾燥粉末を取り出し、850μmネットを用いて篩過した。得られた粉末と潤沢剤(ステアリン酸マグネシウム)を正確に秤量し、ポリエチレンバッグ内で5分間混合した。混合物をゆっくり錠剤機のプレスチャンバーに移し、8mm径パンチを用いて手動でプレスして小スラグを得た。850μmネットを用いてスラグを篩過し、得られた顆粒を8mm径パンチを用いて手動でプレスした。
この実施例では、上述したのと同じ操作手順を施してベンダムスチン HCl/マンニトール錠剤を製造した。
製剤の組成を表32に示す。
【0133】
表32. Ribomustin及びベンダムスチン/マンニトール錠剤。API含有バッチ最終混合物組成
(*) 45.16%のベンダムスチンHCl及び54.20%のマンニトールに相当する
【0134】
表33は、凍結乾燥ベンダムスチン塩酸塩/マンニトール混合物及び非凍結乾燥ベンダムスチン塩酸塩/マンニトール混合物を用いて得た錠剤間の比較に関するデータを報告する。
【0135】
表33. Ribomustin及びベンダムスチン/マンニトール錠剤。API含有バッチ錠剤分析結果
【0136】
参考目標としてベンダムスチン塩酸塩APIの不純物プロファイルを例として挙げると(表中の仕様限界参照)、バッチD001T/125は、HP1不純物について仕様範囲外の値を示した。溶解試験の結果は、10分後には凍結乾燥ベンダムスチン塩酸塩/マンニトール混合物を含む錠剤の溶解プロファイルの方が速いが、30分後には両製剤で溶解が現仕様に従うことを明らかにしている。脆砕性は、バッチD001T/126では仕様の範囲外であり、バッチD001T/125については原料の十分な量を欠いたため試験を行なわなかった。
【0137】
(産業上の利用可能性)
本発明の組成物は多くの利点を示す。管理医療スタッフの補助なしで患者が容易に本組成物を使用することができる。従って、時間のかかる通院は時代遅れになり、それによって患者のコンプライアンスを向上させ得る。
剤形が固体なので、そのまま嚥下することができ、活性成分の溶解が達成されるまで患者が待つ必要がないことを意味する。さらに本剤形の良い安定性のため、室温で、かつ如何なる特殊な貯蔵条件をも必要とせずに本組成物を容易に貯蔵することができる。
本発明の剤形を利用することによって、剤形の体積のかなりの減少を果たすことができる。サイズの減少は、製造及び取扱いの観点からも患者のコンプライアンスの観点からも望ましい。
医薬組成物はin vitroで高い溶解を示し、in vivoでのベンダムスチンの分解を減少させ、それによってin vivoでのベンダムスチンのバイオアベイラビリティーを改善することになる。
【特許請求の範囲】
【請求項1】
経口投与に適した固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択される医薬的に許容できる糖類である少なくとも1種の医薬的に許容できる賦形剤とを含み、前記活性成分と前記糖類賦形剤の質量比が1:1〜5の範囲内である、医薬組成物。
【請求項2】
前記活性成分と前記糖類の質量比が1:2〜5である、請求項1に記載の医薬組成物。
【請求項3】
錠剤、顆粒剤、又は丸剤の形態である、請求項1又は2に記載の医薬組成物。
【請求項4】
前記錠剤若しくは錠剤顆粒剤、顆粒剤又は丸剤がコーティングを備えている、請求項1〜3のいずれか1項に記載の医薬組成物。
【請求項5】
前記活性成分がベンダムスチン塩酸塩である、請求項1〜4のいずれか1項に記載の医薬組成物。
【請求項6】
10〜1000mgの前記活性成分と、30〜5000mgの前記糖類賦形剤とを含む、請求項1〜5のいずれか1項に記載の医薬組成物。
【請求項7】
前記糖類賦形剤が、マンニトール、マルチトール、エリスリトール、キシリトール、ラクトース、ショ糖、グルコース、ソルビトール、マルトース、トレハロース、ラクチトール、ブドウ糖及びフルクトースから選択される、請求項1〜6のいずれか1項に記載の医薬組成物。
【請求項8】
前記糖類賦形剤が、ブドウ糖無水物、ブドウ糖一水和物、ラクチトール一水和物、トレハロース、ソルビトール、エリスリトール、マルトース一水和物、マンニトール、ラクトース無水物、ラクトース一水和物、マルチトール、キシリトール、ショ糖、ショ糖 97%+マルトデキストリン3%、β-シクロデキストリン、D-ラフィノース五水和物、D-メレジトース一水和物及び微結晶性セルロースから選択される、請求項1〜6のいずれか1項に記載の医薬組成物。
【請求項9】
医薬的に許容できる潤沢剤、充填剤及び/又は崩壊剤をさらに含む、請求項1〜8のいずれか1項に記載の医薬組成物。
【請求項10】
前記医薬組成物が、欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、10分で少なくとも60%、20分で70%及び30分で80%のベンダムスチンの溶解を示す、請求項1に記載の医薬組成物。
【請求項11】
慢性リンパ性白血病、急性リンパ性白血病、慢性骨髄性白血病、急性骨髄性白血病、ホジキン病、非ホジキンリンパ腫、多発性骨髄腫、乳癌、卵巣癌、小細胞肺癌、非小細胞肺癌、及び自己免疫疾患から選択される医学的状態の治療用の、請求項1〜10のいずれか1項に記載の医薬組成物。
【請求項12】
前記医薬組成物が少なくとも1種のさらなる活性薬と併用投与されるものであり、前記さらなる活性薬が、前記医薬組成物の投与前、投与と同時、又は投与後に与えられる、請求項1〜11のいずれか1項に記載の医薬組成物。
【請求項13】
前記さらなる活性薬が、CD20に特異的な抗体、アントラサイクリン誘導体、ビンカアルカロイド又はプラチン誘導体である、請求項12に記載の医薬組成物。
【請求項14】
前記CD20に特異的な抗体がリツキシマブであり、前記アントラサイクリン誘導体がドキソルビシン又はダウノルビシンであり、前記ビンカアルカロイドがビンクリスチンであり、かつ前記プラチン誘導体がシスプラチン又はカルボプラチンである、請求項13に記載の医薬組成物。
【請求項15】
前記医薬組成物が少なくとも1種のコルチコステロイドと併用投与されるものであり、前記コルチコステロイドが前記医薬組成物の投与前、投与と同時、又は投与後に与えられる、請求項1〜14のいずれか1項に記載の医薬組成物。
【請求項16】
前記コルチコステロイドがプレドニゾン又はプレドニゾロンである、請求項15に記載の医薬組成物。
【請求項1】
経口投与に適した固体剤形の医薬組成物であって、活性成分としてのベンダムスチン又はその医薬的に許容できるエステル、塩若しくは溶媒和物と、1種以上の単糖、二糖、オリゴ糖、環状オリゴ糖、多糖及び糖アルコールから成る群より選択される医薬的に許容できる糖類である少なくとも1種の医薬的に許容できる賦形剤とを含み、前記活性成分と前記糖類賦形剤の質量比が1:1〜5の範囲内である、医薬組成物。
【請求項2】
前記活性成分と前記糖類の質量比が1:2〜5である、請求項1に記載の医薬組成物。
【請求項3】
錠剤、顆粒剤、又は丸剤の形態である、請求項1又は2に記載の医薬組成物。
【請求項4】
前記錠剤若しくは錠剤顆粒剤、顆粒剤又は丸剤がコーティングを備えている、請求項1〜3のいずれか1項に記載の医薬組成物。
【請求項5】
前記活性成分がベンダムスチン塩酸塩である、請求項1〜4のいずれか1項に記載の医薬組成物。
【請求項6】
10〜1000mgの前記活性成分と、30〜5000mgの前記糖類賦形剤とを含む、請求項1〜5のいずれか1項に記載の医薬組成物。
【請求項7】
前記糖類賦形剤が、マンニトール、マルチトール、エリスリトール、キシリトール、ラクトース、ショ糖、グルコース、ソルビトール、マルトース、トレハロース、ラクチトール、ブドウ糖及びフルクトースから選択される、請求項1〜6のいずれか1項に記載の医薬組成物。
【請求項8】
前記糖類賦形剤が、ブドウ糖無水物、ブドウ糖一水和物、ラクチトール一水和物、トレハロース、ソルビトール、エリスリトール、マルトース一水和物、マンニトール、ラクトース無水物、ラクトース一水和物、マルチトール、キシリトール、ショ糖、ショ糖 97%+マルトデキストリン3%、β-シクロデキストリン、D-ラフィノース五水和物、D-メレジトース一水和物及び微結晶性セルロースから選択される、請求項1〜6のいずれか1項に記載の医薬組成物。
【請求項9】
医薬的に許容できる潤沢剤、充填剤及び/又は崩壊剤をさらに含む、請求項1〜8のいずれか1項に記載の医薬組成物。
【請求項10】
前記医薬組成物が、欧州薬局方に準拠して50rpmでパドル装置を用いて500mlの溶解媒体中1.5のpHで測定した場合、10分で少なくとも60%、20分で70%及び30分で80%のベンダムスチンの溶解を示す、請求項1に記載の医薬組成物。
【請求項11】
慢性リンパ性白血病、急性リンパ性白血病、慢性骨髄性白血病、急性骨髄性白血病、ホジキン病、非ホジキンリンパ腫、多発性骨髄腫、乳癌、卵巣癌、小細胞肺癌、非小細胞肺癌、及び自己免疫疾患から選択される医学的状態の治療用の、請求項1〜10のいずれか1項に記載の医薬組成物。
【請求項12】
前記医薬組成物が少なくとも1種のさらなる活性薬と併用投与されるものであり、前記さらなる活性薬が、前記医薬組成物の投与前、投与と同時、又は投与後に与えられる、請求項1〜11のいずれか1項に記載の医薬組成物。
【請求項13】
前記さらなる活性薬が、CD20に特異的な抗体、アントラサイクリン誘導体、ビンカアルカロイド又はプラチン誘導体である、請求項12に記載の医薬組成物。
【請求項14】
前記CD20に特異的な抗体がリツキシマブであり、前記アントラサイクリン誘導体がドキソルビシン又はダウノルビシンであり、前記ビンカアルカロイドがビンクリスチンであり、かつ前記プラチン誘導体がシスプラチン又はカルボプラチンである、請求項13に記載の医薬組成物。
【請求項15】
前記医薬組成物が少なくとも1種のコルチコステロイドと併用投与されるものであり、前記コルチコステロイドが前記医薬組成物の投与前、投与と同時、又は投与後に与えられる、請求項1〜14のいずれか1項に記載の医薬組成物。
【請求項16】
前記コルチコステロイドがプレドニゾン又はプレドニゾロンである、請求項15に記載の医薬組成物。
【図1】

【図2】
【図2】
【公表番号】特表2012−510483(P2012−510483A)
【公表日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願番号】特願2011−538900(P2011−538900)
【出願日】平成21年12月3日(2009.12.3)
【国際出願番号】PCT/EP2009/008639
【国際公開番号】WO2010/063476
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(511134609)アステラス ドイチュランド ゲゼルシャフト ミット ベシュレンクテル ハフツング (2)
【Fターム(参考)】
【公表日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願日】平成21年12月3日(2009.12.3)
【国際出願番号】PCT/EP2009/008639
【国際公開番号】WO2010/063476
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(511134609)アステラス ドイチュランド ゲゼルシャフト ミット ベシュレンクテル ハフツング (2)
【Fターム(参考)】
[ Back to top ]