
ペグ修飾ハイドロキシアパタイト及びそれを基材とする医薬とその製造方法
【課題】ハイドロキシアパタイト粒子表面をポリエチレングリコール誘導体で修飾することにより、安全性の高い、新たな機能を有するPEG修飾HAPと、それを用いる用途とその製造方法を提供する。
【解決手段】粒径が50μm〜10nmのハイドロキシアパタイトが、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体と−O(CO)で結合されていて、炭素含量が10〜0.1%の物質であり、また、該物質と医薬品有効成分又は医薬品添加物とからなり、該医薬品有効成分の重量比が1〜30%の物質であり、該物質は、粒径50μm〜10nmのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体の活性エステルとを無水有機溶媒中で処理して得られる。
【解決手段】粒径が50μm〜10nmのハイドロキシアパタイトが、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体と−O(CO)で結合されていて、炭素含量が10〜0.1%の物質であり、また、該物質と医薬品有効成分又は医薬品添加物とからなり、該医薬品有効成分の重量比が1〜30%の物質であり、該物質は、粒径50μm〜10nmのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体の活性エステルとを無水有機溶媒中で処理して得られる。
【発明の詳細な説明】
【技術分野】
【0001】
薬物を担持させる微粒子は、そのサイズを調製することで、又は微粒子を適当に修飾することで、剤形的には、経口、静脈注、皮下注、経肺、経鼻に有効に利用し得る。また、機能的には、薬物の肝臓、肺又は炎症部位等への選択的デリバリー、放出制御、いやな味のマスキング又は腸管吸収の改善等に有効に利用し得る。
このような微粒子としては、リポゾーム、高分子ミセル、プロトスフィア(登録商標)、樹脂又はシリカゲル、ゼオライト、ハイドロキシアパタイト等の無機パーテイクル(無機マイクロスフィア、ナノスフィア)が知られている。本発明は、ハイドロキシアパタイト表面をポリエチレングリコール(以下、ペグ又はPEGと略記する)で修飾した新規ペグ修飾ハイドロキシアパタイト(以下、PEG修飾HAPと略記する)及びその用途と製造方法に関する。
【背景技術】
【0002】
ハイドロキシアパタイト(以下、HAPと略記する)は、骨や歯の基本成分であり、生体親和性が高く、糖やタンパク質を吸着しやすい性質がある。そのため、HAPは、骨や歯の補填材料等の医療用基材、カラム充填材、薬物輸送担体、あるいは細胞培養基盤として幅広く利用されている。こうしたHAPの特性をさらに生かすために、その表面を機能性高分子や生理活性物質で化学修飾させることが求められている。しかしながら、その足がかりとなるHAP表面の水酸基は、反応性が低く、機能性高分子や生理活性物質等の有機化合物を均一に結合させることが難しい。
【0003】
アパタイト粒子の化学修飾については、ナノアパタイト粒子にヘキサメチレンジイソシアネートを用いてポリエチレングリコールを結合させた、Liu Qiugらの報告がある(Biomaterials (1998), 19(11-12), 1067-1072)。また、古薗らは、アパタイト粒子にアミノ基を持つシランカップリング剤を結合させ、カルボン酸とアミノ基との縮合反応によりポリアクリル酸を介してシリコーンシートと結合した経皮デバイスの開発に成功している(J Biomed Mater Res. (2001), 56(1),9-16)。さらに、田中らは、2種類以上の官能基を有するシランカップリング剤やイソシアネート化合物を用いて、HAP多孔体表面に反応性の高い有機官能基を導入し、該HAP多孔体表面に有機物質を共有結合させることに成功し、これがクロマトグラフ用カラム充填剤、DDS担体、イオン交換媒体、細胞培養基盤、インプラント等として幅広く利用できることを示した(特開2003−342011号公報)。
【0004】
しかしこれらは、いずれもその調製過程でバイファンクショナルなリンカー試薬を用いるため、ハイドロキシアパタイト粒子同士の架橋結合が必然的に避けられず、この架橋ハイドロキシアパタイト粒子が、副生成物として混在する問題点がある。また、反応性の高いシランカップリング剤やイソシアネート化合物を使用しているため、残存するこれら反応性官能基の安全性も問題と考えられる。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Biomaterials (1998), 19(11-12), 1067-1072
【非特許文献2】J Biomed Mater Res. (2001), 56(1),9-16
【特許文献】
【0006】
【特許文献1】特開2003−342011号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、バイファンクショナルリンカーを用いず、ハイドロキシアパタイト粒子表面をポリエチレングリコール誘導体で修飾することにより、安全性の高い、新たな機能を有するPEG修飾HAPを提供し、加えてそれを利用する用途とその製造方法を提供することを課題とする。
【課題を解決するための手段】
【0008】
上記課題を解決するため、本発明者らは、鋭意検討した結果、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体を用いてHAPの表面にポリエチレングリコールを−O(CO)で結合導入することに成功した。さらに、該新規PEG修飾HAPに、各種医薬をローデイングさせたドラッグデリバリーシステム(DDS)は、剤形的には、経口、静脈注、皮下注、経肺、経鼻に、有効に利用し得ること、及び薬物の肝臓、肺又は炎症部位等への選択的デリバリー、放出制御、いやな味のマスキング又は腸管吸収の改善等に有効に応用できることを見出した。
【0009】
このPEG修飾HAPは、アパタイト固有の高い機械的強度と各種物質吸着能を維持すると共に、血中滞留性等の特性を併せ持つことが期待できる。そのため、DDS担体を始め、クロマトグラフ用カラム充填剤、イオン交換媒体、細胞培養基盤、インプラント等として幅広く利用し得る。
【0010】
すなわち、本発明は、以下の(1)〜(23)を提供するものである。
(1)
粒径が50μm〜10nmのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体からなる混合物であって、炭素含量が10〜0.1%の物質。
(2)
粒径が50μm〜10nmのハイドロキシアパタイトが、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体と−O(CO)で結合されていて、炭素含量が10〜0.1%の物質。
【0011】
(3)
請求項1記載の物質及び医薬品有効成分からなり、医薬品有効成分の重量比が1〜30%の物質、又は、請求項1記載の物質及び医薬品有効成分および医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
(4)
請求項2記載の物質及び医薬品有効成分からなり、医薬品有効成分の重量比が1〜30%の物質、又は、請求項2記載の物質及び医薬品有効成分および医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
【0012】
(5)
請求項3又4記載の物質から調製される医薬品。
(6)
請求項3又は4記載の医薬品有効成分がsiRNA、アプタマー、RNA、DNA、ペプチド又は蛋白質であるところの請求項3又は4記載の物質。
(7)
請求項3又は4記載の医薬品有効成分がクラリスロマイシンであるところの請求項3又は4記載の物質。
(8)
請求項3又は4記載の医薬品有効成分がイトラコナゾールであるところの請求項3又は4記載の物質。
(9)
請求項3又は4記載の医薬品有効成分がsiRNAであるところの請求項3又は4記載の物質。
【0013】
(10)
サブミクロンサイズのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体の活性エステルを無水有機溶媒中で処理してサブミクロンサイズの請求項1又は2記載の物質を得る方法。
(11)
サブミクロンサイズの請求項1又は2記載の物質と医薬有効成分又は医薬添加物を有機溶媒中で処理して請求項3又は4記載の物質を得る方法。
【0014】
(12)
請求項3又は4記載の医薬品有効成分がカンデサルタンであるところの請求項3又は4記載の物質。
(13)
請求項3又は4記載の医薬品有効成分がカンデサルタンシレキセチルであるところの請求項3又は4記載の物質。
(14)
請求項3又は4記載の医薬品有効成分が、配列が
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
である二重螺旋のsiRNAであるところの請求項3又は4記載の物質。
(15)
請求項3又は4記載の医薬品有効成分が、配列が
5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
である二重螺旋のsiRNAであるところの請求項3又は4記載の物質。
【0015】
(16)
請求項3又は4記載の医薬品有効成分がエトポシドであるところの請求項3又は4記載の物質。
(17)
請求項3又は4記載の医薬品有効成分がメシル酸ネルフィナビルであるところの請求項3又は4記載の物質。
(18)
請求項3又は4記載の医薬品有効成分がシンバスタチンであるところの請求項3又は4記載の物質。
(19)
請求項3又は4記載の医薬品有効成分が7−エチル−10−ヒドロキシ−カンプトテシンであるところの請求項3又は4記載の物質。
【0016】
(20)
請求項3又は4記載の医薬品有効成分がパクリタキセルであるところの請求項3又は4記載の物質。
(21)
請求項3又は4記載の医薬品有効成分がメシル酸サキナビルであるところの請求項3又は4記載の物質。
(22)
請求項3又は4記載の医薬品有効成分がインスリンであるところの請求項3又は4記載の物質。
(23)
請求項3又は4記載の医薬品有効成分がブロモクリプチンメシル酸塩であるところの請求項3又は4記載の物質。
【発明の効果】
【0017】
本発明によれば、次のような効果を奏することができる。
(1) 難溶性の医薬物質であっても、本発明のPEG修飾HAPを基材に用いることで、可溶性物質のように取り扱うことができ、体内への薬物投与が容易となり、さらに体内血中滞留性の向上が図られる
(2) HAP表面をPEG化することで、HAP粒子同士の凝集を防止することができる。
(3) 表面をPEG化したHAPを基材に用いることで、有効成分をローディングしたHAP粒子についても、粒子同士の凝集を防止することができる。
【図面の簡単な説明】
【0018】
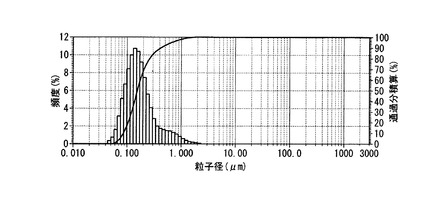
【図1】実施例1のPEG修飾HAPの粒度分布を示すグラフ。
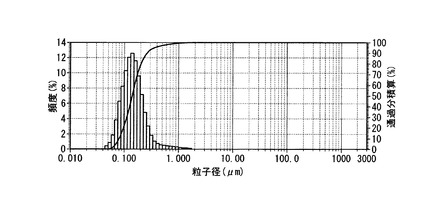
【図2】実施例2のPEG修飾HAPとクラリスロマイシンとからなる医薬の粒度分布を示すグラフ。
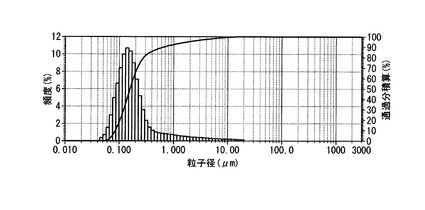
【図3】実施例3のPEG修飾HAPとイトラコナゾールとからなる医薬の粒度分布を示すグラフ。
【図4】実施例4のsiRNAコーティングPEG修飾HAPの蛍光顕微鏡写真(×1000)で、(a)フルオレセイン標識siRNAコーティング品、(b)ローダミン標識siRNAコーティング品。
【図5】実施例18の結果のカンデサルタンの時間(分)による溶出濃度を示すグラフ。
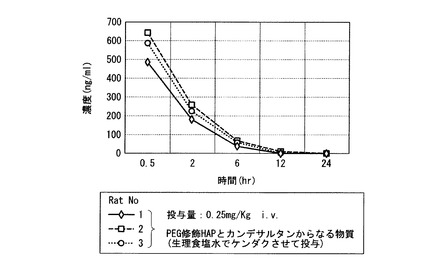
【図6】実施例19の結果のラットに静脈注射後のプラズマ中の時間(時)による濃度変化を示すグラフ。
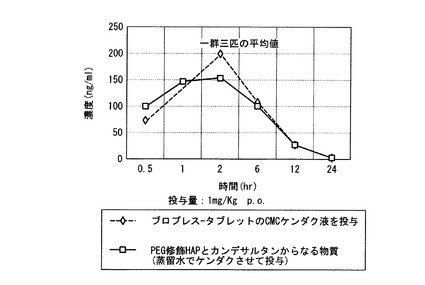
【図7】実施例20の結果のラットに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフ。
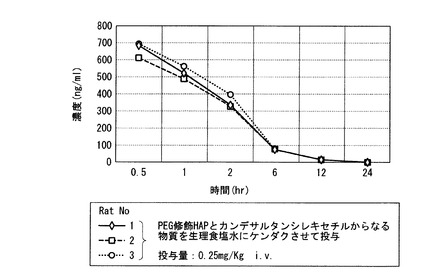
【図8】実施例21の結果のラットに静脈注射後のプラズマ中の時間(時)による濃度変化を示すグラフ。
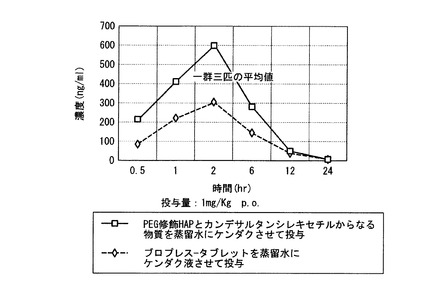
【図9】実施例22の結果のラットに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフ。
【図10】実施例23の結果のA549細胞に対する細胞毒性を示すグラフ。
【図11】実施例24の結果のA549細胞へのトランスフェクション試験の4時間経過後の細胞の蛍光顕微鏡写真。
【図12】実施例26の結果のレーザー共焦点顕微鏡写真。
【図13】実施例27の結果のレーザー共焦点顕微鏡写真。
【発明を実施するための形態】
【0019】
以下、本発明について詳細に説明する。
本発明は、バイファンクショナルリンカーを用いず、モノファンクショナルポリエチレングリコール誘導体でハイドロキシアパタイト粒子表面にポリエチレングリコール誘導体を結合させて得られた、安全性の高い、新たな機能を有するPEG修飾HAP及びその用途と製造方法に関する。
PEG修飾に付されるHAPは、多数の細孔(気孔)を有するHAP固体であっても良いし、気孔率のあまり高くないHAPでも良い。
【0020】
HAPは、一般組成をCa5(PO4)3OH、とする化合物であり、その反応の非化学量論性によって、CaHPO4 、Ca3(PO4)2、Ca4O(PO4)2、Ca10(PO4)6(OH)2、CaP4O11、Ca(PO3)2、Ca2P2O7、Ca(H2PO4)2H2Oなどリン酸カルシウムと称される1群の化合物を含む。また、HAPは、Ca5(PO4)3OH、又はCa10(PO4)6(OH)2の組成式で示される化合物を基本成分とするもので、Ca成分の一部分は、Sr、Ba、Mg、Fe、Al、Y、La、Na、K、Hなどから選ばれる1種以上で置換されてもよい。また、(PO4)成分の一部分が、VO4、BO3、SO4、CO3、SiO4等から選ばれる1種以上で置換されてもよい。更に、(OH)成分の一部分が、F、Cl、O、CO3等から選ばれる1種以上で置換されてもよい。また、これらの各成分の一部が欠陥となっていてもよい。生体骨中のアパタイトのPO4及びOH成分の一部は、通常CO3に置換されているため、本複合生体材料の製造中、大気中からのCO3の混入と各成分への一部置換(0〜10質量%程度)があってもよい。
【0021】
なお、HAPは、通常の微結晶・非晶質並びに結晶体の他に、同型固溶体、置換型固溶体、侵入型固溶体であってもよく、非量子論的欠陥を含むものであってもよい。また、この「HAP」中、カルシウム及びリンの原子比(Ca/P)は1.3〜1.8の範囲内にあることが好ましく、特に1.5〜1.7がより好ましい。原子比が1.3〜1.8の範囲内にあると、生成物中のアパタイト(リン酸カルシウム化合物)の組成と結晶構造が、脊椎動物の骨の中に存在するアパタイトと類似の組成と構造をとりうるため、生体親和性がより高くなるからである。
PEG修飾に付されるHAPは、公知の方法で調製することが出来るが、市販のもの、例えばAldrich社製Hydroxyapatite, nanopowderを利用しても良い。
【0022】
本発明で用いられる「モノファンクショナルポリエチレングリコール誘導体」としては、市販の高純度一官能性活性化PEG修飾剤を用いたがこれに限定するものではない。PEG部分を直鎖にするか分枝にするか、又はPEG部分の分子量等は、目的に応じて任意に選択調節できる。
修飾時の反応溶媒は、無水有機溶媒、特にジメチルホルムアミド(DMF)、ジメチルスルホキサイド(DMSO)、酢酸、アセトン、テトラヒドロフラン(THF)、酢酸エチル、ジクロロメタンが使用できるが、特に医薬品の残留溶媒ガイドラインに記すクラス2〜3のジメチルスルホキサイド(DMSO)、アセトンが好ましい。反応温度は、氷冷下から100℃、反応時間は2〜72時間、「モノファンクショナルポリエチレングリコール誘導体」は、HAP1gあたり大過剰(1〜0.1g)を用いた。反応終了後、残存する過剰の「モノファンクショナルポリエチレングリコール誘導体」及び副生成物のN‐ヒドロキシスクシンイミドを反応に使用した有機溶媒で洗浄ろ過して除き、不溶物を減圧下乾燥して、PEG修飾HAPを得た。PEG修飾HAPの炭素含量は、PEG修飾試薬量を調節することで0.1〜10%で調節できるが、特に1〜3%前後が好ましい。
【0023】
本発明のPEG修飾HAPは、医薬品有効成分を吸着させることで、DDS担体として利用することができる。HAP又はPEGは生体適合性が高いため、生体内への薬物送達にも安心して利用できる(J Mater Sci. (2000), 11(2),67‐72)。
標的器官に特異的なリガンドを結合させれば、より確実に目的とする器官に薬剤を送達することも可能となる。また、医薬品有効成分が難水溶性で注射製剤が不可能又は腸管吸収の悪い場合、サブミクロンサイズのPEG修飾HAPに医薬品有効成分を吸着させることで、間接的に医薬品有効成分をサブミクロンサイズに調整でき、注射製剤の開発及び経口吸収改善に広く応用できる。さらに、サブミクロンサイズのPEG修飾HAPに医薬品有効成分としてRNA、DNA、蛋白質等を吸着させた物質は、それら医薬品有効成分の有望なDDSとして応用可能である。
【0024】
PEG修飾HAPと医薬品有効成分又は医薬品添加物とからなる物質は以下の方法で調製した。医薬品有効成分又は医薬品添加物を医薬品の残留溶媒ガイドラインに記すクラス2〜3のDMSO、エタノール(EtOH)、アセトン等の溶媒に溶解させ、これに重量比で90%比のPEG修飾HAPを加え、室温下超音波処理後、この懸濁液全量を凍結乾燥又は減圧下溶媒留去して、請求項記載の物質を得た。医薬品有効成分又は医薬品添加物のPEG修飾HAPに対する搭載率は、医薬品有効成分又は医薬品添加物にも依存するが1〜30%で調製できる。特に10%前後が好ましい。
以下、実施例によって本発明をさらに詳細に説明するが本発明はこれらの実施例によって限定されるものではない。
【実施例1】
【0025】
〔PEG修飾HAPの調製〕
(1)PEG修飾HAPの調製
ハイドロキシアパタイト−ナノパウダー〔Aldrich 677418〕200mgのアセトン20ml懸濁液に、ポリエチレングリコール(PEG)修飾剤(NOF社製 SUNBRIGHT ME‐020CS)200mgを加え、30分間超音波(周波数28kHz、出力100W)を照射した。この懸濁液を室温にて18時間攪拌後、遠心分離(9000×g, 20℃,30分間)し、上清をデカンテーションにて除去した。沈殿物をアセトンにて2回洗浄(20ml×2)した後、減圧下、50℃にて18時間乾燥し、PEG修飾HAP 158mgを白色粉末として得た。
【0026】
(2)残留溶媒、及びPEG修飾率の測定
調製したPEG修飾HAP中の残留溶媒をガスクロマトグラフ(GC)法で、PEG修飾率としてCHN元素分析法で炭素含量を定量した結果を以下に示す。
残留溶媒(アセトン)濃度:< 100μg/g
炭素含量:2.01%
<GC分析条件>
装置:HP‐5890IIシステム (Hewlett Packard製)
カラム:DB‐624 75mm×0.53 mm 膜厚0.3μm
カラム温度:40℃ → 260℃
キャリアガス:ヘリウム 7 psi
検出器:水素炎イオン検出器(FID) 250℃
<CHN元素分析条件>
機器:vario EL III (Elementar AnalysensystemeGmbH)
燃焼炉温度:950℃
還元炉温度:500℃
ヘリウム流量:200 ml/min
酸素流量:30 ml/min
燃焼時間:90 sec
【0027】
(3)粒子径分布測定
調製したPEG修飾HAP 1mgをミリQ水 (15ml)に懸濁し、超音波(周波数28 kHz、出力100W)を5分間照射した後、粒子経測定を行った。[測定機器:HORIBA レーザー回折/散乱式粒子径分布測定装置 LA‐950] 結果を図1に示す。
【実施例2】
【0028】
〔PEG修飾HAPとクラリスロマイシンからなる物質の調製〕
(1)PEG修飾HAPとクラリスロマイシンからなる物質の調製
PEG修飾HAP(100mg)にクラリスロマイシン(8mg)のDMSO(2ml)溶液を加えた後、超音波(周波数28kHz、出力100W)を2分間照射した。この懸濁液を凍結乾燥し、白色粉末 108.6 mgを得た。これを更に減圧下、50℃にて36時間乾燥し目的物108.1mgを白色粉末として得た。
【0029】
(2)薬物吸着率の測定
本品10mgにアセトニトリル 1mlを加え、超音波(周波数28 kHz、出力100W)を5分間照射した。この懸濁液を遠心分離(9000×g, 20℃, 3分間)し、上清を更に0.22μmのフィルターにてろ過しHPLC用サンプルとした。HPLC分析より、本品10mg中にクラリスロマイシン0.74mgが含有されていることを確認した。吸着率:7.4%(w/w)
<HPLC分析条件>
機器:Waters Alliance 2695 Separations Module, Waters 2487 Dual λAbsorbance Detector
カラム:Atlantis dC18, particle size 3.0μm, 3.9 mm×100mm(Waters)
移動層:A : 0.67 mol/Lリン酸二水素カリウム試液 B : アセトニトリル。
A:B=65:35(v/v)
流速:1.0 ml/min
検出波長:210 nm
Retention time:7.1 min
【0030】
(3)粒子径分布測定
本品1mgをミリQ水 (15ml)に懸濁し、超音波(周波数28kHz、出力100W)を5分間照射した後、粒子計測定を行った。結果を図2に示す。
【実施例3】
【0031】
〔PEG修飾HAPとイトラコナゾールからなる物質の調製〕
(1)組成物の調製
PEG修飾HAP(300mg)にイトラコナゾール(24mg)のDMSO(4.8ml)溶液を加えた後、超音波(周波数28kHz、出力100W)を2分間照射した。この懸濁液を凍結乾燥し、白色粉末324.3mgを得た。これをミリQ水(15ml)に懸濁後、コンドロイチン硫酸ナトリウム水溶液 [10mg/ml] (0.3ml)を添加し、超音波(周波数28kHz、出力100W)を2分間照射した。この懸濁液を凍結乾燥し白色粉末322.6mg得た。
【0032】
(2)薬物吸着率の測定
本品10mgにアセトニトリル1mlを加え、超音波(周波数28kHz、出力100W)を5分間照射した。この懸濁液を遠心分離(9000×g, 20℃, 3分間)し、上清を更に0.22μmのフィルターでろ過し、HPLC用サンプルとした。HPLC分析
より、本品10mg中にイトラコナゾール0.74mgが含有されていることを確認した。吸着率:7.4%(w/w)
<HPLC分析条件>
機器:Waters Alliance 2695 Separations Module, Waters 2487 Dual λAbsorbance Detector
カラム:XBridge C18, particle size 3.5μm, 4.6 mm×100mm(Waters)
移動層:A:0.2% ジイソプロピルアミン-メタノール溶液 B:0.5% 酢酸アンモニウム水溶液。 A:B=4:1(v/v)
流速:0.9 ml/min、検出波長:263 nm
Retention time:3.0 min
【0033】
(3)粒子径分布測定
本品1mgをミリQ水 (15ml)に懸濁し、超音波(周波数28kHz、出力100W)を5分間照射した後、粒子計測定を行った。結果を図3に示す。
【実施例4】
【0034】
〔PEG修飾HAPとsiRNAからなる物質の調製〕
(1) PEG修飾HAPと蛍光標識siRNAからなる物質の調製
PEG修飾HAP6mgを量り取り、純水10mlを加えた。混合液を乳化・分散機へ移し、16000rpmで1分間処理し、均一な懸濁液を得た。懸濁液3mlに対し、10mg/ml 蛍光標識siRNA水溶液18μlを加え、十分混合した。
(2) 蛍光顕微鏡観察
本品3mlにグリセリン6mlを加えた。蛍光顕微鏡にてサンプルの観察を行った。蛍光励起波長は、フルオレセイン標識品は490nm、ローダミン標識品は550nmに設定した。結果を図4に示す。
(3) 結果
蛍光顕微鏡観察より、siRNAはPEG修飾HAP表面にコーティングされていることがわかった。
【実施例5】
【0035】
〔PEG修飾HAPとイトラコナゾールからなる物質をラットに投与した際の血中動態試験〕
(1) 試験目的
PEG修飾HAPとイトラコナゾールからなる物質を、ラットに静脈内及び経口投与し血中濃度の推移を確認し、バイオアベイラビリテイーを算出した。
(2) 被験物質
(1)静脈内投与用
PEG修飾HAPとイトラコナゾールからなる物質の水性懸濁液
保存条件:遮光・室温保存
(2)経口投与用
PEG修飾HAPとイトラコナゾールからなる物質の水性懸濁液
保存条件:遮光・室温保存
(3) 動物
・ 動物種:ラット,系統:SD,性別:雄
・ 例数:n=3
投与時週齢:7週齢
・ 投与時摂餌条件:非絶食
・ 投与量:静脈内投与; 5 mg/2 ml/kg(27G注射針
を使用して静注)
経口投与; 12 mg/4.8 ml/kg
【0036】
(4) 採血
血漿採取時点:投与後0.5,2,6,18,24,48,168時間
血漿採取:尾静脈からヘパリンナトリウム処理した毛細管を用いて血液約0.5 mlを採取する。
血液を遠心分離(12000 rpm,4℃,3分間)して得られた血漿は、測定まで−20℃で凍結保存した。
(5) 症状観察:一般状態の観察のみ実施し、特定部位及び特定組織の観察は実施していない。
(6) 血漿中濃度の測定
分析方法
・ 測定対象:イトラコナゾール
・ 標準物質:イトラコナゾール
保存条件:冷暗所
・ 内標準物質:ロラタジン
保存条件:冷暗所
・ 分析条件:LC/MS/MS
【0037】
(7) LC/MS/MS条件
カラム:CAPCELL PAK C18 MG II, 50mm×4.6mm id, 5mm(資生堂)
移動層:A : 2mmol/L酢酸アンモニウム B : アセトニトリル。
A:B=35:65(v/v)
流速0.5ml/min
イオン源:APCI
極性:正イオン
検出イオン:m/z705.1, 392.1(イトラコナゾール)
m/z383.5, 337.2[ I.S.(インターナルスタンダード)]
(8) 前処理
・ 検量線試料及び測定用試料にI.S.溶液100μlを加え、攪拌した。ブランク試料にはアセトニトリル100μlを加え、攪拌した。
・ アセトニトリル700μlを加え、攪拌した。
・ 約12000×gで10分間(4℃)遠心分離した。
・ 上清5μlをLC/MS/MSに注入した。
【0038】
(9) 結果
PEG修飾HAPとイトラコナゾールからなる物質の0.5〜24時間でのバイオアベイラビリテイーは、約57%と高い腸管吸収改善効果がみられた。(表1)
同時に行った市販イトリゾール注及びイトリゾールカプセル50の血中動態インビボ試験結果から、経口イトリゾールカプセル50のイトリゾール注に対する0.5〜48時間でのバイオアベイラビリテイーは約39%であった。
この結果から、サブミクロンサイズのPEG修飾HAPと難水溶性医薬とからなる物質が、新規注射製剤として及び高い経口吸収を示す優れた経口製剤とし有用であることが示された。
【0039】
【表1】

【実施例6】
【0040】
〔PEG修飾HAPとクラリスロマイシンからなる物質をラットに投与した際の血中動態試験〕
(1) 試験目的
PEG修飾HAPとクラリスロマイシンからなる物質を、ラットに静脈内及び皮下投与し血中濃度の推移を確認した。
(2) 被験物質
(1)静脈内投与用
PEG修飾HAPとクラリスロマイシンからなる物質の水性懸濁液
保存条件:遮光・室温保存
(2)皮下投与用
PEG修飾HAPとクラリスロマイシンからなる物質の水性懸濁液
保存条件:遮光・室温保存
(3)動物
・ 動物種:ラット,系統:SD,性別:雄
・ 例数:n=3
投与時週齢:7週齢
・ 投与時摂餌条件:非絶食
・ 投与量:静脈内投与; 1 mg/ml/kg(27G注射針を使用して静注)
皮下投与; 2 mg/2 mL/kg(27G注射針を使用して皮下注)
【0041】
(4) 採血
血漿採取時点:投与後0.5,2,6,12,24,48,168時間
血漿採取:尾静脈からヘパリンナトリウム処理したパスツールピペットを用いて血液約0.5 mlを採取する。
血液を遠心分離(8000×g,4℃,3分間)して得られた血漿は、測定まで−20℃で凍結保存した。
(5) 症状観察:一般状態の観察のみ実施し、特定部位及び特定組織の観察は実施していない。
(6) 血漿中濃度の測定
分析方法
・ 測定対象:クラリスロマイシン
・ 標準物質:クラリスロマイシン
保存条件:冷暗所
・ 内標準物質:エリスロマイシンB
保存条件:冷暗所
・ 分析条件:LC/MS/MS
【0042】
(7) LC/MS/MS条件
カラム:Atlantis dC18 3mm, 4.6mm ID x 75mm(ウオーターズ)
ガードカラム:Atlantis dC18 3mm, 4.6mm ID x 20mm(ウオーターズ)
移動層:A: 20mmol/Lギ酸アンモニウム B : アセトニトリル。
A:B=55:45(v/v),流速0.5ml/min
イオン源:ESI
極性:正イオン
検出イオン:m/z748.6, 158.2(クラリスロマイシン)
m/z718.6, 158.3[ I.S.(インターナルスタンダード)]
(8) 前処理
1. 検量線試料、ブランク試料及び測定用試料に5%(w/v)炭酸ナトリウム200μl及び酢酸エチル4μlを添加した。
2. 室温で約15分間振とうした後、約1800 x g、室温、10分間で遠心分離した。
3. 上清(有機層)を13mlポリプロピレン製(p.p.)チューブに移した。
4. 上清を窒素気流下濃縮乾固(40度C、約30分)した。
5. 残渣に再溶解溶液1mlを添加後、攪拌した。
6. 10μlをLC/MS/MSに注入した。
【0043】
(9) 結果
表2に示すように、サブミクロンサイズのPEG修飾HAPと難水溶性医薬クラリスロマイシンとからなる物質が、新規注射製剤(27G注射針を使用して静注及び皮下注可能)として有用であることが示された。
【表2】
【実施例7】
【0044】
〔PEG修飾HAPとカンデサルタンからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.4 %(w/w)
【実施例8】
【0045】
〔PEG修飾HAPとカンデサルタンシレキセチルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:6.3 %(w/w)
【実施例9】
【0046】
〔PEG修飾HAPと
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
からなる物質の調製〕
(1) 組成物の調製
実施例4と類似の方法により調製した。
(2) 薬物吸着率の測定
蛍光分析法により確認した。吸着率:20 %(w/w)
【実施例10】
【0047】
[PEG修飾HAPと
5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
からなる物質の調製]
(1) 組成物の調製
実施例4と類似の方法により調製した。
(2) 薬物吸着率の測定
蛍光分析法により確認した。吸着率:20 %(w/w)
【実施例11】
【0048】
[PEG修飾HAPとエトポシドからなる物質の調製]
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:8.0 %(w/w)
【実施例12】
【0049】
〔PEG修飾HAPとメシル酸ネルフィナビルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.4 %(w/w)
【実施例13】
【0050】
〔PEG修飾HAPとシンバスタチンからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.9 %(w/w)
【実施例14】
【0051】
〔PEG修飾HAPと7-エチル-10-ヒドロキシ-カンプトテシンからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:6.8 %(w/w)
【実施例15】
【0052】
〔PEG修飾HAPとパクリタキセルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.4 %(w/w)
【実施例16】
【0053】
〔PEG修飾HAPとメシル酸サキナビルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:8.0 %(w/w)
【実施例17】
【0054】
〔PEG修飾HAPとインスリンからなる物質の調製〕
(1) 組成物の調製
実施例4と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:12.7 %(w/w)
【実施例18】
【0055】
〔PEG修飾HAPとカンデサルタンからなる物質のパドル法による溶出試験〕
(結果)
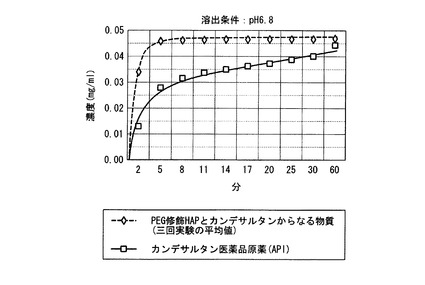
図5は、カンデサルタンの時間(分)による溶出濃度を示すグラフである。
図5に示すように、カンデサルタン医薬品原薬からのカンデサルタンの溶出は、およそ一時間を要するのに比べ、PEG修飾HAPとカンデサルタンからなる物質からのカンデサルタンの溶出は早く、およそ5分で完了した。
【実施例19】
【0056】
〔PEG修飾HAPとカンデサルタンからなる物質をラットにi.v.(静脈注射)投与した際の血中動態試験〕
(結果)
図6は、ラットに静脈注射後のプラズマ(血漿)中の時間(時)による濃度変化を示すグラフである。
図6に示すように、サブミクロンサイズのPEG修飾HAPと難水溶性医薬カンデサルタンからなる物質が、新規注射製剤(27G注射針を使用して静注及び皮下注可能)として有用であることが示された。
【実施例20】
【0057】
〔PEG修飾HAPとカンデサルタンからなる物質をラットにpo.(経口)投与した際の血中動態試験〕
(結果)
図7は、ラッドに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフである。
図7に示すように、PEG修飾HAPとカンデサルタンからなる物質は、市販のブロプレス(登録商標)-タブレット錠に匹敵する経口吸収性を示した。
【実施例21】
【0058】
〔PEG修飾HAPとカンデサルタンシレキセチルからなる物質をラットにi.v.投与した際の血中動態試験〕
(結果)
図8は、ラットに静脈注射後のプラズマ中の時間(時)による濃度変化を示すグラフである。
図8に示すように、サブミクロンサイズのPEG修飾HAPと難水溶性医薬カンデサルタンシレキセチルからなる物質が、新規注射製剤(27G注射針を使用して静注及び皮下注可能)として有用であることが示された。
【実施例22】
【0059】
〔PEG修飾HAPとカンデサルタンシレキセチルからなる物質をラットにpo.投与した際の血中動態試験〕
(結果)
図9は、ラットに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフである。
図9に示すように、PEG修飾HAPとカンデサルタンシレキセチルからなる物質は、市販のブロプレス-タブレット錠のおよそ1.5倍の経口吸収性を示した。
【実施例23】
【0060】
〔PEG修飾HAPと
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
からなる物質のin vitro細胞毒性評価試験〕
(結果)
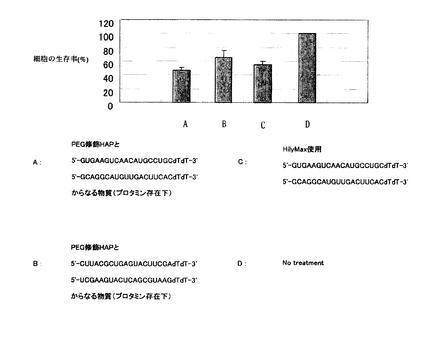
図10は、A549細胞に対する細胞毒性を示すグラフである。
図10中、Aに示すように、PEG修飾HAPと
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
からなる物質は、A549ヒト肺癌細胞に対しおよそ50%細胞生存率を示した。図10中Cに示すポジテイブコントロール(株式会社同仁化学研究所製HilyMaxを使用)に匹敵する強い効果を示した。一方図10中、Bに示すネガテイブコントロールでは、およそ70%細胞生存率を示した。
【実施例24】
【0061】
〔PEG修飾HAPと
蛍光ラベル化5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
からなる物質のA549細胞へのトランスフェクション試験と蛍光顕微鏡観察〕
(結果)
A549ヒト肺癌細胞に、PEG修飾HAPと
蛍光ラベル化5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
からなる物質を添加後、4時間経過後の細胞の蛍光顕微鏡写真を図11に示す。
【実施例25】
【0062】
〔PEG修飾HAPとエトポシドからなる物質をラットにi.v.投与した際の動態試験〕
(結果)
表3に示すようにサブミクロンサイズのPEG修飾HAPと難水溶性医薬エトポシドからなる物質は市販のエトポシド注射薬(ベプシド注)と比較し、エトポシドが肝臓に高く集積することが認められた。
【0063】
【表3】
【実施例26】
【0064】
〔PEG修飾HAPとシンバスタチンからなる物質の顕微鏡観察〕
(結果)
PEG修飾HAPとシンバスタチンからなる物質のレーザー共焦点顕微鏡写真を図12に示す。粒径のそろったサブミクロンサイズのPEG修飾HAPとシンバスタチンからなる物質粒子がブラウン運動している様子が観察された。
【実施例27】
【0065】
〔PEG修飾HAPとメシル酸ネルフィナビルからなる物質の顕微鏡観察〕
(結果)
PEG修飾HAPとメシル酸ネルフィナビルからなる物質のレーザー共焦点顕微鏡写真を図13に示す。粒径のそろったサブミクロンサイズのPEG修飾HAPとメシル酸ネルフィナビルからなる物質粒子がブラウン運動している様子が観察された。
【実施例28】
【0066】
〔PEG修飾HAPとブロモクリプチンメシル酸塩からなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:4.2 %(w/w)
【技術分野】
【0001】
薬物を担持させる微粒子は、そのサイズを調製することで、又は微粒子を適当に修飾することで、剤形的には、経口、静脈注、皮下注、経肺、経鼻に有効に利用し得る。また、機能的には、薬物の肝臓、肺又は炎症部位等への選択的デリバリー、放出制御、いやな味のマスキング又は腸管吸収の改善等に有効に利用し得る。
このような微粒子としては、リポゾーム、高分子ミセル、プロトスフィア(登録商標)、樹脂又はシリカゲル、ゼオライト、ハイドロキシアパタイト等の無機パーテイクル(無機マイクロスフィア、ナノスフィア)が知られている。本発明は、ハイドロキシアパタイト表面をポリエチレングリコール(以下、ペグ又はPEGと略記する)で修飾した新規ペグ修飾ハイドロキシアパタイト(以下、PEG修飾HAPと略記する)及びその用途と製造方法に関する。
【背景技術】
【0002】
ハイドロキシアパタイト(以下、HAPと略記する)は、骨や歯の基本成分であり、生体親和性が高く、糖やタンパク質を吸着しやすい性質がある。そのため、HAPは、骨や歯の補填材料等の医療用基材、カラム充填材、薬物輸送担体、あるいは細胞培養基盤として幅広く利用されている。こうしたHAPの特性をさらに生かすために、その表面を機能性高分子や生理活性物質で化学修飾させることが求められている。しかしながら、その足がかりとなるHAP表面の水酸基は、反応性が低く、機能性高分子や生理活性物質等の有機化合物を均一に結合させることが難しい。
【0003】
アパタイト粒子の化学修飾については、ナノアパタイト粒子にヘキサメチレンジイソシアネートを用いてポリエチレングリコールを結合させた、Liu Qiugらの報告がある(Biomaterials (1998), 19(11-12), 1067-1072)。また、古薗らは、アパタイト粒子にアミノ基を持つシランカップリング剤を結合させ、カルボン酸とアミノ基との縮合反応によりポリアクリル酸を介してシリコーンシートと結合した経皮デバイスの開発に成功している(J Biomed Mater Res. (2001), 56(1),9-16)。さらに、田中らは、2種類以上の官能基を有するシランカップリング剤やイソシアネート化合物を用いて、HAP多孔体表面に反応性の高い有機官能基を導入し、該HAP多孔体表面に有機物質を共有結合させることに成功し、これがクロマトグラフ用カラム充填剤、DDS担体、イオン交換媒体、細胞培養基盤、インプラント等として幅広く利用できることを示した(特開2003−342011号公報)。
【0004】
しかしこれらは、いずれもその調製過程でバイファンクショナルなリンカー試薬を用いるため、ハイドロキシアパタイト粒子同士の架橋結合が必然的に避けられず、この架橋ハイドロキシアパタイト粒子が、副生成物として混在する問題点がある。また、反応性の高いシランカップリング剤やイソシアネート化合物を使用しているため、残存するこれら反応性官能基の安全性も問題と考えられる。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Biomaterials (1998), 19(11-12), 1067-1072
【非特許文献2】J Biomed Mater Res. (2001), 56(1),9-16
【特許文献】
【0006】
【特許文献1】特開2003−342011号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、バイファンクショナルリンカーを用いず、ハイドロキシアパタイト粒子表面をポリエチレングリコール誘導体で修飾することにより、安全性の高い、新たな機能を有するPEG修飾HAPを提供し、加えてそれを利用する用途とその製造方法を提供することを課題とする。
【課題を解決するための手段】
【0008】
上記課題を解決するため、本発明者らは、鋭意検討した結果、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体を用いてHAPの表面にポリエチレングリコールを−O(CO)で結合導入することに成功した。さらに、該新規PEG修飾HAPに、各種医薬をローデイングさせたドラッグデリバリーシステム(DDS)は、剤形的には、経口、静脈注、皮下注、経肺、経鼻に、有効に利用し得ること、及び薬物の肝臓、肺又は炎症部位等への選択的デリバリー、放出制御、いやな味のマスキング又は腸管吸収の改善等に有効に応用できることを見出した。
【0009】
このPEG修飾HAPは、アパタイト固有の高い機械的強度と各種物質吸着能を維持すると共に、血中滞留性等の特性を併せ持つことが期待できる。そのため、DDS担体を始め、クロマトグラフ用カラム充填剤、イオン交換媒体、細胞培養基盤、インプラント等として幅広く利用し得る。
【0010】
すなわち、本発明は、以下の(1)〜(23)を提供するものである。
(1)
粒径が50μm〜10nmのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体からなる混合物であって、炭素含量が10〜0.1%の物質。
(2)
粒径が50μm〜10nmのハイドロキシアパタイトが、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体と−O(CO)で結合されていて、炭素含量が10〜0.1%の物質。
【0011】
(3)
請求項1記載の物質及び医薬品有効成分からなり、医薬品有効成分の重量比が1〜30%の物質、又は、請求項1記載の物質及び医薬品有効成分および医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
(4)
請求項2記載の物質及び医薬品有効成分からなり、医薬品有効成分の重量比が1〜30%の物質、又は、請求項2記載の物質及び医薬品有効成分および医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
【0012】
(5)
請求項3又4記載の物質から調製される医薬品。
(6)
請求項3又は4記載の医薬品有効成分がsiRNA、アプタマー、RNA、DNA、ペプチド又は蛋白質であるところの請求項3又は4記載の物質。
(7)
請求項3又は4記載の医薬品有効成分がクラリスロマイシンであるところの請求項3又は4記載の物質。
(8)
請求項3又は4記載の医薬品有効成分がイトラコナゾールであるところの請求項3又は4記載の物質。
(9)
請求項3又は4記載の医薬品有効成分がsiRNAであるところの請求項3又は4記載の物質。
【0013】
(10)
サブミクロンサイズのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体の活性エステルを無水有機溶媒中で処理してサブミクロンサイズの請求項1又は2記載の物質を得る方法。
(11)
サブミクロンサイズの請求項1又は2記載の物質と医薬有効成分又は医薬添加物を有機溶媒中で処理して請求項3又は4記載の物質を得る方法。
【0014】
(12)
請求項3又は4記載の医薬品有効成分がカンデサルタンであるところの請求項3又は4記載の物質。
(13)
請求項3又は4記載の医薬品有効成分がカンデサルタンシレキセチルであるところの請求項3又は4記載の物質。
(14)
請求項3又は4記載の医薬品有効成分が、配列が
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
である二重螺旋のsiRNAであるところの請求項3又は4記載の物質。
(15)
請求項3又は4記載の医薬品有効成分が、配列が
5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
である二重螺旋のsiRNAであるところの請求項3又は4記載の物質。
【0015】
(16)
請求項3又は4記載の医薬品有効成分がエトポシドであるところの請求項3又は4記載の物質。
(17)
請求項3又は4記載の医薬品有効成分がメシル酸ネルフィナビルであるところの請求項3又は4記載の物質。
(18)
請求項3又は4記載の医薬品有効成分がシンバスタチンであるところの請求項3又は4記載の物質。
(19)
請求項3又は4記載の医薬品有効成分が7−エチル−10−ヒドロキシ−カンプトテシンであるところの請求項3又は4記載の物質。
【0016】
(20)
請求項3又は4記載の医薬品有効成分がパクリタキセルであるところの請求項3又は4記載の物質。
(21)
請求項3又は4記載の医薬品有効成分がメシル酸サキナビルであるところの請求項3又は4記載の物質。
(22)
請求項3又は4記載の医薬品有効成分がインスリンであるところの請求項3又は4記載の物質。
(23)
請求項3又は4記載の医薬品有効成分がブロモクリプチンメシル酸塩であるところの請求項3又は4記載の物質。
【発明の効果】
【0017】
本発明によれば、次のような効果を奏することができる。
(1) 難溶性の医薬物質であっても、本発明のPEG修飾HAPを基材に用いることで、可溶性物質のように取り扱うことができ、体内への薬物投与が容易となり、さらに体内血中滞留性の向上が図られる
(2) HAP表面をPEG化することで、HAP粒子同士の凝集を防止することができる。
(3) 表面をPEG化したHAPを基材に用いることで、有効成分をローディングしたHAP粒子についても、粒子同士の凝集を防止することができる。
【図面の簡単な説明】
【0018】
【図1】実施例1のPEG修飾HAPの粒度分布を示すグラフ。
【図2】実施例2のPEG修飾HAPとクラリスロマイシンとからなる医薬の粒度分布を示すグラフ。
【図3】実施例3のPEG修飾HAPとイトラコナゾールとからなる医薬の粒度分布を示すグラフ。
【図4】実施例4のsiRNAコーティングPEG修飾HAPの蛍光顕微鏡写真(×1000)で、(a)フルオレセイン標識siRNAコーティング品、(b)ローダミン標識siRNAコーティング品。
【図5】実施例18の結果のカンデサルタンの時間(分)による溶出濃度を示すグラフ。
【図6】実施例19の結果のラットに静脈注射後のプラズマ中の時間(時)による濃度変化を示すグラフ。
【図7】実施例20の結果のラットに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフ。
【図8】実施例21の結果のラットに静脈注射後のプラズマ中の時間(時)による濃度変化を示すグラフ。
【図9】実施例22の結果のラットに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフ。
【図10】実施例23の結果のA549細胞に対する細胞毒性を示すグラフ。
【図11】実施例24の結果のA549細胞へのトランスフェクション試験の4時間経過後の細胞の蛍光顕微鏡写真。
【図12】実施例26の結果のレーザー共焦点顕微鏡写真。
【図13】実施例27の結果のレーザー共焦点顕微鏡写真。
【発明を実施するための形態】
【0019】
以下、本発明について詳細に説明する。
本発明は、バイファンクショナルリンカーを用いず、モノファンクショナルポリエチレングリコール誘導体でハイドロキシアパタイト粒子表面にポリエチレングリコール誘導体を結合させて得られた、安全性の高い、新たな機能を有するPEG修飾HAP及びその用途と製造方法に関する。
PEG修飾に付されるHAPは、多数の細孔(気孔)を有するHAP固体であっても良いし、気孔率のあまり高くないHAPでも良い。
【0020】
HAPは、一般組成をCa5(PO4)3OH、とする化合物であり、その反応の非化学量論性によって、CaHPO4 、Ca3(PO4)2、Ca4O(PO4)2、Ca10(PO4)6(OH)2、CaP4O11、Ca(PO3)2、Ca2P2O7、Ca(H2PO4)2H2Oなどリン酸カルシウムと称される1群の化合物を含む。また、HAPは、Ca5(PO4)3OH、又はCa10(PO4)6(OH)2の組成式で示される化合物を基本成分とするもので、Ca成分の一部分は、Sr、Ba、Mg、Fe、Al、Y、La、Na、K、Hなどから選ばれる1種以上で置換されてもよい。また、(PO4)成分の一部分が、VO4、BO3、SO4、CO3、SiO4等から選ばれる1種以上で置換されてもよい。更に、(OH)成分の一部分が、F、Cl、O、CO3等から選ばれる1種以上で置換されてもよい。また、これらの各成分の一部が欠陥となっていてもよい。生体骨中のアパタイトのPO4及びOH成分の一部は、通常CO3に置換されているため、本複合生体材料の製造中、大気中からのCO3の混入と各成分への一部置換(0〜10質量%程度)があってもよい。
【0021】
なお、HAPは、通常の微結晶・非晶質並びに結晶体の他に、同型固溶体、置換型固溶体、侵入型固溶体であってもよく、非量子論的欠陥を含むものであってもよい。また、この「HAP」中、カルシウム及びリンの原子比(Ca/P)は1.3〜1.8の範囲内にあることが好ましく、特に1.5〜1.7がより好ましい。原子比が1.3〜1.8の範囲内にあると、生成物中のアパタイト(リン酸カルシウム化合物)の組成と結晶構造が、脊椎動物の骨の中に存在するアパタイトと類似の組成と構造をとりうるため、生体親和性がより高くなるからである。
PEG修飾に付されるHAPは、公知の方法で調製することが出来るが、市販のもの、例えばAldrich社製Hydroxyapatite, nanopowderを利用しても良い。
【0022】
本発明で用いられる「モノファンクショナルポリエチレングリコール誘導体」としては、市販の高純度一官能性活性化PEG修飾剤を用いたがこれに限定するものではない。PEG部分を直鎖にするか分枝にするか、又はPEG部分の分子量等は、目的に応じて任意に選択調節できる。
修飾時の反応溶媒は、無水有機溶媒、特にジメチルホルムアミド(DMF)、ジメチルスルホキサイド(DMSO)、酢酸、アセトン、テトラヒドロフラン(THF)、酢酸エチル、ジクロロメタンが使用できるが、特に医薬品の残留溶媒ガイドラインに記すクラス2〜3のジメチルスルホキサイド(DMSO)、アセトンが好ましい。反応温度は、氷冷下から100℃、反応時間は2〜72時間、「モノファンクショナルポリエチレングリコール誘導体」は、HAP1gあたり大過剰(1〜0.1g)を用いた。反応終了後、残存する過剰の「モノファンクショナルポリエチレングリコール誘導体」及び副生成物のN‐ヒドロキシスクシンイミドを反応に使用した有機溶媒で洗浄ろ過して除き、不溶物を減圧下乾燥して、PEG修飾HAPを得た。PEG修飾HAPの炭素含量は、PEG修飾試薬量を調節することで0.1〜10%で調節できるが、特に1〜3%前後が好ましい。
【0023】
本発明のPEG修飾HAPは、医薬品有効成分を吸着させることで、DDS担体として利用することができる。HAP又はPEGは生体適合性が高いため、生体内への薬物送達にも安心して利用できる(J Mater Sci. (2000), 11(2),67‐72)。
標的器官に特異的なリガンドを結合させれば、より確実に目的とする器官に薬剤を送達することも可能となる。また、医薬品有効成分が難水溶性で注射製剤が不可能又は腸管吸収の悪い場合、サブミクロンサイズのPEG修飾HAPに医薬品有効成分を吸着させることで、間接的に医薬品有効成分をサブミクロンサイズに調整でき、注射製剤の開発及び経口吸収改善に広く応用できる。さらに、サブミクロンサイズのPEG修飾HAPに医薬品有効成分としてRNA、DNA、蛋白質等を吸着させた物質は、それら医薬品有効成分の有望なDDSとして応用可能である。
【0024】
PEG修飾HAPと医薬品有効成分又は医薬品添加物とからなる物質は以下の方法で調製した。医薬品有効成分又は医薬品添加物を医薬品の残留溶媒ガイドラインに記すクラス2〜3のDMSO、エタノール(EtOH)、アセトン等の溶媒に溶解させ、これに重量比で90%比のPEG修飾HAPを加え、室温下超音波処理後、この懸濁液全量を凍結乾燥又は減圧下溶媒留去して、請求項記載の物質を得た。医薬品有効成分又は医薬品添加物のPEG修飾HAPに対する搭載率は、医薬品有効成分又は医薬品添加物にも依存するが1〜30%で調製できる。特に10%前後が好ましい。
以下、実施例によって本発明をさらに詳細に説明するが本発明はこれらの実施例によって限定されるものではない。
【実施例1】
【0025】
〔PEG修飾HAPの調製〕
(1)PEG修飾HAPの調製
ハイドロキシアパタイト−ナノパウダー〔Aldrich 677418〕200mgのアセトン20ml懸濁液に、ポリエチレングリコール(PEG)修飾剤(NOF社製 SUNBRIGHT ME‐020CS)200mgを加え、30分間超音波(周波数28kHz、出力100W)を照射した。この懸濁液を室温にて18時間攪拌後、遠心分離(9000×g, 20℃,30分間)し、上清をデカンテーションにて除去した。沈殿物をアセトンにて2回洗浄(20ml×2)した後、減圧下、50℃にて18時間乾燥し、PEG修飾HAP 158mgを白色粉末として得た。
【0026】
(2)残留溶媒、及びPEG修飾率の測定
調製したPEG修飾HAP中の残留溶媒をガスクロマトグラフ(GC)法で、PEG修飾率としてCHN元素分析法で炭素含量を定量した結果を以下に示す。
残留溶媒(アセトン)濃度:< 100μg/g
炭素含量:2.01%
<GC分析条件>
装置:HP‐5890IIシステム (Hewlett Packard製)
カラム:DB‐624 75mm×0.53 mm 膜厚0.3μm
カラム温度:40℃ → 260℃
キャリアガス:ヘリウム 7 psi
検出器:水素炎イオン検出器(FID) 250℃
<CHN元素分析条件>
機器:vario EL III (Elementar AnalysensystemeGmbH)
燃焼炉温度:950℃
還元炉温度:500℃
ヘリウム流量:200 ml/min
酸素流量:30 ml/min
燃焼時間:90 sec
【0027】
(3)粒子径分布測定
調製したPEG修飾HAP 1mgをミリQ水 (15ml)に懸濁し、超音波(周波数28 kHz、出力100W)を5分間照射した後、粒子経測定を行った。[測定機器:HORIBA レーザー回折/散乱式粒子径分布測定装置 LA‐950] 結果を図1に示す。
【実施例2】
【0028】
〔PEG修飾HAPとクラリスロマイシンからなる物質の調製〕
(1)PEG修飾HAPとクラリスロマイシンからなる物質の調製
PEG修飾HAP(100mg)にクラリスロマイシン(8mg)のDMSO(2ml)溶液を加えた後、超音波(周波数28kHz、出力100W)を2分間照射した。この懸濁液を凍結乾燥し、白色粉末 108.6 mgを得た。これを更に減圧下、50℃にて36時間乾燥し目的物108.1mgを白色粉末として得た。
【0029】
(2)薬物吸着率の測定
本品10mgにアセトニトリル 1mlを加え、超音波(周波数28 kHz、出力100W)を5分間照射した。この懸濁液を遠心分離(9000×g, 20℃, 3分間)し、上清を更に0.22μmのフィルターにてろ過しHPLC用サンプルとした。HPLC分析より、本品10mg中にクラリスロマイシン0.74mgが含有されていることを確認した。吸着率:7.4%(w/w)
<HPLC分析条件>
機器:Waters Alliance 2695 Separations Module, Waters 2487 Dual λAbsorbance Detector
カラム:Atlantis dC18, particle size 3.0μm, 3.9 mm×100mm(Waters)
移動層:A : 0.67 mol/Lリン酸二水素カリウム試液 B : アセトニトリル。
A:B=65:35(v/v)
流速:1.0 ml/min
検出波長:210 nm
Retention time:7.1 min
【0030】
(3)粒子径分布測定
本品1mgをミリQ水 (15ml)に懸濁し、超音波(周波数28kHz、出力100W)を5分間照射した後、粒子計測定を行った。結果を図2に示す。
【実施例3】
【0031】
〔PEG修飾HAPとイトラコナゾールからなる物質の調製〕
(1)組成物の調製
PEG修飾HAP(300mg)にイトラコナゾール(24mg)のDMSO(4.8ml)溶液を加えた後、超音波(周波数28kHz、出力100W)を2分間照射した。この懸濁液を凍結乾燥し、白色粉末324.3mgを得た。これをミリQ水(15ml)に懸濁後、コンドロイチン硫酸ナトリウム水溶液 [10mg/ml] (0.3ml)を添加し、超音波(周波数28kHz、出力100W)を2分間照射した。この懸濁液を凍結乾燥し白色粉末322.6mg得た。
【0032】
(2)薬物吸着率の測定
本品10mgにアセトニトリル1mlを加え、超音波(周波数28kHz、出力100W)を5分間照射した。この懸濁液を遠心分離(9000×g, 20℃, 3分間)し、上清を更に0.22μmのフィルターでろ過し、HPLC用サンプルとした。HPLC分析
より、本品10mg中にイトラコナゾール0.74mgが含有されていることを確認した。吸着率:7.4%(w/w)
<HPLC分析条件>
機器:Waters Alliance 2695 Separations Module, Waters 2487 Dual λAbsorbance Detector
カラム:XBridge C18, particle size 3.5μm, 4.6 mm×100mm(Waters)
移動層:A:0.2% ジイソプロピルアミン-メタノール溶液 B:0.5% 酢酸アンモニウム水溶液。 A:B=4:1(v/v)
流速:0.9 ml/min、検出波長:263 nm
Retention time:3.0 min
【0033】
(3)粒子径分布測定
本品1mgをミリQ水 (15ml)に懸濁し、超音波(周波数28kHz、出力100W)を5分間照射した後、粒子計測定を行った。結果を図3に示す。
【実施例4】
【0034】
〔PEG修飾HAPとsiRNAからなる物質の調製〕
(1) PEG修飾HAPと蛍光標識siRNAからなる物質の調製
PEG修飾HAP6mgを量り取り、純水10mlを加えた。混合液を乳化・分散機へ移し、16000rpmで1分間処理し、均一な懸濁液を得た。懸濁液3mlに対し、10mg/ml 蛍光標識siRNA水溶液18μlを加え、十分混合した。
(2) 蛍光顕微鏡観察
本品3mlにグリセリン6mlを加えた。蛍光顕微鏡にてサンプルの観察を行った。蛍光励起波長は、フルオレセイン標識品は490nm、ローダミン標識品は550nmに設定した。結果を図4に示す。
(3) 結果
蛍光顕微鏡観察より、siRNAはPEG修飾HAP表面にコーティングされていることがわかった。
【実施例5】
【0035】
〔PEG修飾HAPとイトラコナゾールからなる物質をラットに投与した際の血中動態試験〕
(1) 試験目的
PEG修飾HAPとイトラコナゾールからなる物質を、ラットに静脈内及び経口投与し血中濃度の推移を確認し、バイオアベイラビリテイーを算出した。
(2) 被験物質
(1)静脈内投与用
PEG修飾HAPとイトラコナゾールからなる物質の水性懸濁液
保存条件:遮光・室温保存
(2)経口投与用
PEG修飾HAPとイトラコナゾールからなる物質の水性懸濁液
保存条件:遮光・室温保存
(3) 動物
・ 動物種:ラット,系統:SD,性別:雄
・ 例数:n=3
投与時週齢:7週齢
・ 投与時摂餌条件:非絶食
・ 投与量:静脈内投与; 5 mg/2 ml/kg(27G注射針
を使用して静注)
経口投与; 12 mg/4.8 ml/kg
【0036】
(4) 採血
血漿採取時点:投与後0.5,2,6,18,24,48,168時間
血漿採取:尾静脈からヘパリンナトリウム処理した毛細管を用いて血液約0.5 mlを採取する。
血液を遠心分離(12000 rpm,4℃,3分間)して得られた血漿は、測定まで−20℃で凍結保存した。
(5) 症状観察:一般状態の観察のみ実施し、特定部位及び特定組織の観察は実施していない。
(6) 血漿中濃度の測定
分析方法
・ 測定対象:イトラコナゾール
・ 標準物質:イトラコナゾール
保存条件:冷暗所
・ 内標準物質:ロラタジン
保存条件:冷暗所
・ 分析条件:LC/MS/MS
【0037】
(7) LC/MS/MS条件
カラム:CAPCELL PAK C18 MG II, 50mm×4.6mm id, 5mm(資生堂)
移動層:A : 2mmol/L酢酸アンモニウム B : アセトニトリル。
A:B=35:65(v/v)
流速0.5ml/min
イオン源:APCI
極性:正イオン
検出イオン:m/z705.1, 392.1(イトラコナゾール)
m/z383.5, 337.2[ I.S.(インターナルスタンダード)]
(8) 前処理
・ 検量線試料及び測定用試料にI.S.溶液100μlを加え、攪拌した。ブランク試料にはアセトニトリル100μlを加え、攪拌した。
・ アセトニトリル700μlを加え、攪拌した。
・ 約12000×gで10分間(4℃)遠心分離した。
・ 上清5μlをLC/MS/MSに注入した。
【0038】
(9) 結果
PEG修飾HAPとイトラコナゾールからなる物質の0.5〜24時間でのバイオアベイラビリテイーは、約57%と高い腸管吸収改善効果がみられた。(表1)
同時に行った市販イトリゾール注及びイトリゾールカプセル50の血中動態インビボ試験結果から、経口イトリゾールカプセル50のイトリゾール注に対する0.5〜48時間でのバイオアベイラビリテイーは約39%であった。
この結果から、サブミクロンサイズのPEG修飾HAPと難水溶性医薬とからなる物質が、新規注射製剤として及び高い経口吸収を示す優れた経口製剤とし有用であることが示された。
【0039】
【表1】
【実施例6】
【0040】
〔PEG修飾HAPとクラリスロマイシンからなる物質をラットに投与した際の血中動態試験〕
(1) 試験目的
PEG修飾HAPとクラリスロマイシンからなる物質を、ラットに静脈内及び皮下投与し血中濃度の推移を確認した。
(2) 被験物質
(1)静脈内投与用
PEG修飾HAPとクラリスロマイシンからなる物質の水性懸濁液
保存条件:遮光・室温保存
(2)皮下投与用
PEG修飾HAPとクラリスロマイシンからなる物質の水性懸濁液
保存条件:遮光・室温保存
(3)動物
・ 動物種:ラット,系統:SD,性別:雄
・ 例数:n=3
投与時週齢:7週齢
・ 投与時摂餌条件:非絶食
・ 投与量:静脈内投与; 1 mg/ml/kg(27G注射針を使用して静注)
皮下投与; 2 mg/2 mL/kg(27G注射針を使用して皮下注)
【0041】
(4) 採血
血漿採取時点:投与後0.5,2,6,12,24,48,168時間
血漿採取:尾静脈からヘパリンナトリウム処理したパスツールピペットを用いて血液約0.5 mlを採取する。
血液を遠心分離(8000×g,4℃,3分間)して得られた血漿は、測定まで−20℃で凍結保存した。
(5) 症状観察:一般状態の観察のみ実施し、特定部位及び特定組織の観察は実施していない。
(6) 血漿中濃度の測定
分析方法
・ 測定対象:クラリスロマイシン
・ 標準物質:クラリスロマイシン
保存条件:冷暗所
・ 内標準物質:エリスロマイシンB
保存条件:冷暗所
・ 分析条件:LC/MS/MS
【0042】
(7) LC/MS/MS条件
カラム:Atlantis dC18 3mm, 4.6mm ID x 75mm(ウオーターズ)
ガードカラム:Atlantis dC18 3mm, 4.6mm ID x 20mm(ウオーターズ)
移動層:A: 20mmol/Lギ酸アンモニウム B : アセトニトリル。
A:B=55:45(v/v),流速0.5ml/min
イオン源:ESI
極性:正イオン
検出イオン:m/z748.6, 158.2(クラリスロマイシン)
m/z718.6, 158.3[ I.S.(インターナルスタンダード)]
(8) 前処理
1. 検量線試料、ブランク試料及び測定用試料に5%(w/v)炭酸ナトリウム200μl及び酢酸エチル4μlを添加した。
2. 室温で約15分間振とうした後、約1800 x g、室温、10分間で遠心分離した。
3. 上清(有機層)を13mlポリプロピレン製(p.p.)チューブに移した。
4. 上清を窒素気流下濃縮乾固(40度C、約30分)した。
5. 残渣に再溶解溶液1mlを添加後、攪拌した。
6. 10μlをLC/MS/MSに注入した。
【0043】
(9) 結果
表2に示すように、サブミクロンサイズのPEG修飾HAPと難水溶性医薬クラリスロマイシンとからなる物質が、新規注射製剤(27G注射針を使用して静注及び皮下注可能)として有用であることが示された。
【表2】
【実施例7】
【0044】
〔PEG修飾HAPとカンデサルタンからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.4 %(w/w)
【実施例8】
【0045】
〔PEG修飾HAPとカンデサルタンシレキセチルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:6.3 %(w/w)
【実施例9】
【0046】
〔PEG修飾HAPと
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
からなる物質の調製〕
(1) 組成物の調製
実施例4と類似の方法により調製した。
(2) 薬物吸着率の測定
蛍光分析法により確認した。吸着率:20 %(w/w)
【実施例10】
【0047】
[PEG修飾HAPと
5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
からなる物質の調製]
(1) 組成物の調製
実施例4と類似の方法により調製した。
(2) 薬物吸着率の測定
蛍光分析法により確認した。吸着率:20 %(w/w)
【実施例11】
【0048】
[PEG修飾HAPとエトポシドからなる物質の調製]
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:8.0 %(w/w)
【実施例12】
【0049】
〔PEG修飾HAPとメシル酸ネルフィナビルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.4 %(w/w)
【実施例13】
【0050】
〔PEG修飾HAPとシンバスタチンからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.9 %(w/w)
【実施例14】
【0051】
〔PEG修飾HAPと7-エチル-10-ヒドロキシ-カンプトテシンからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:6.8 %(w/w)
【実施例15】
【0052】
〔PEG修飾HAPとパクリタキセルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:7.4 %(w/w)
【実施例16】
【0053】
〔PEG修飾HAPとメシル酸サキナビルからなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:8.0 %(w/w)
【実施例17】
【0054】
〔PEG修飾HAPとインスリンからなる物質の調製〕
(1) 組成物の調製
実施例4と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:12.7 %(w/w)
【実施例18】
【0055】
〔PEG修飾HAPとカンデサルタンからなる物質のパドル法による溶出試験〕
(結果)
図5は、カンデサルタンの時間(分)による溶出濃度を示すグラフである。
図5に示すように、カンデサルタン医薬品原薬からのカンデサルタンの溶出は、およそ一時間を要するのに比べ、PEG修飾HAPとカンデサルタンからなる物質からのカンデサルタンの溶出は早く、およそ5分で完了した。
【実施例19】
【0056】
〔PEG修飾HAPとカンデサルタンからなる物質をラットにi.v.(静脈注射)投与した際の血中動態試験〕
(結果)
図6は、ラットに静脈注射後のプラズマ(血漿)中の時間(時)による濃度変化を示すグラフである。
図6に示すように、サブミクロンサイズのPEG修飾HAPと難水溶性医薬カンデサルタンからなる物質が、新規注射製剤(27G注射針を使用して静注及び皮下注可能)として有用であることが示された。
【実施例20】
【0057】
〔PEG修飾HAPとカンデサルタンからなる物質をラットにpo.(経口)投与した際の血中動態試験〕
(結果)
図7は、ラッドに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフである。
図7に示すように、PEG修飾HAPとカンデサルタンからなる物質は、市販のブロプレス(登録商標)-タブレット錠に匹敵する経口吸収性を示した。
【実施例21】
【0058】
〔PEG修飾HAPとカンデサルタンシレキセチルからなる物質をラットにi.v.投与した際の血中動態試験〕
(結果)
図8は、ラットに静脈注射後のプラズマ中の時間(時)による濃度変化を示すグラフである。
図8に示すように、サブミクロンサイズのPEG修飾HAPと難水溶性医薬カンデサルタンシレキセチルからなる物質が、新規注射製剤(27G注射針を使用して静注及び皮下注可能)として有用であることが示された。
【実施例22】
【0059】
〔PEG修飾HAPとカンデサルタンシレキセチルからなる物質をラットにpo.投与した際の血中動態試験〕
(結果)
図9は、ラットに経口投与後のプラズマ中の時間(時)による濃度変化を示すグラフである。
図9に示すように、PEG修飾HAPとカンデサルタンシレキセチルからなる物質は、市販のブロプレス-タブレット錠のおよそ1.5倍の経口吸収性を示した。
【実施例23】
【0060】
〔PEG修飾HAPと
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
からなる物質のin vitro細胞毒性評価試験〕
(結果)
図10は、A549細胞に対する細胞毒性を示すグラフである。
図10中、Aに示すように、PEG修飾HAPと
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
からなる物質は、A549ヒト肺癌細胞に対しおよそ50%細胞生存率を示した。図10中Cに示すポジテイブコントロール(株式会社同仁化学研究所製HilyMaxを使用)に匹敵する強い効果を示した。一方図10中、Bに示すネガテイブコントロールでは、およそ70%細胞生存率を示した。
【実施例24】
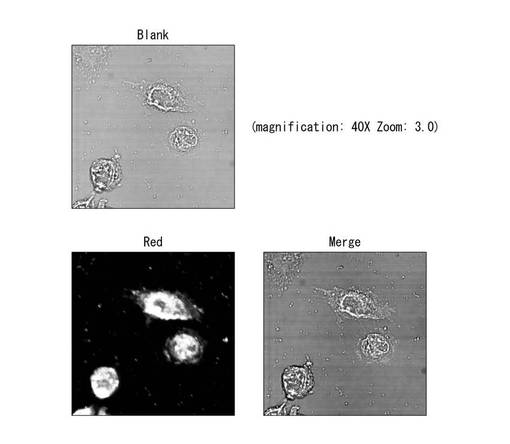
【0061】
〔PEG修飾HAPと
蛍光ラベル化5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
からなる物質のA549細胞へのトランスフェクション試験と蛍光顕微鏡観察〕
(結果)
A549ヒト肺癌細胞に、PEG修飾HAPと
蛍光ラベル化5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
からなる物質を添加後、4時間経過後の細胞の蛍光顕微鏡写真を図11に示す。
【実施例25】
【0062】
〔PEG修飾HAPとエトポシドからなる物質をラットにi.v.投与した際の動態試験〕
(結果)
表3に示すようにサブミクロンサイズのPEG修飾HAPと難水溶性医薬エトポシドからなる物質は市販のエトポシド注射薬(ベプシド注)と比較し、エトポシドが肝臓に高く集積することが認められた。
【0063】
【表3】
【実施例26】
【0064】
〔PEG修飾HAPとシンバスタチンからなる物質の顕微鏡観察〕
(結果)
PEG修飾HAPとシンバスタチンからなる物質のレーザー共焦点顕微鏡写真を図12に示す。粒径のそろったサブミクロンサイズのPEG修飾HAPとシンバスタチンからなる物質粒子がブラウン運動している様子が観察された。
【実施例27】
【0065】
〔PEG修飾HAPとメシル酸ネルフィナビルからなる物質の顕微鏡観察〕
(結果)
PEG修飾HAPとメシル酸ネルフィナビルからなる物質のレーザー共焦点顕微鏡写真を図13に示す。粒径のそろったサブミクロンサイズのPEG修飾HAPとメシル酸ネルフィナビルからなる物質粒子がブラウン運動している様子が観察された。
【実施例28】
【0066】
〔PEG修飾HAPとブロモクリプチンメシル酸塩からなる物質の調製〕
(1) 組成物の調製
実施例2又は実施例3と類似の方法により調製した。
(2) 薬物吸着率の測定
実施例2又は実施例3と類似の方法により確認した。吸着率:4.2 %(w/w)
【特許請求の範囲】
【請求項1】
粒径が50μm〜10nmのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体からなり、炭素含量が10〜0.1%の物質。
【請求項2】
粒径が50μm〜10nmのハイドロキシアパタイトが、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体と−O(CO)で結合されていて、炭素含量が10〜0.1%の物質。
【請求項3】
請求項1記載の物質及び医薬品有効成分又は医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
【請求項4】
請求項2記載の物質及び医薬品有効成分又は医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
【請求項5】
請求項3又4記載の物質から調製される医薬品。
【請求項6】
請求項3又は4記載の医薬品有効成分がsiRNA、アプタマー、RNA、DNA、ペプチド又は蛋白質であるところの請求項3又は4記載の物質。
【請求項7】
請求項3又は4記載の医薬品有効成分がクラリスロマイシンであるところの請求項3又は4記載の物質。
【請求項8】
請求項3又は4記載の医薬品有効成分がイトラコナゾールであるところの請求項3又は4記載の物質。
【請求項9】
請求項3又は4記載の医薬品有効成分がsiRNAであるところの請求項3又は4記載の物質。
【請求項10】
サブミクロンサイズのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体の活性エステルを無水有機溶媒中で処理してサブミクロンサイズの請求項1又は2記載の物質を得る方法。
【請求項11】
サブミクロンサイズの請求項1又は2記載の物質と医薬有効成分又は医薬添加物を有機溶媒中で処理して請求項3又は4記載の物質を得る方法。
【請求項12】
請求項3又は4記載の医薬品有効成分がカンデサルタンであるところの請求項3又は4記載の物質。
【請求項13】
請求項3又は4記載の医薬品有効成分がカンデサルタンシレキセチルであるところの請求項3又は4記載の物質。
【請求項14】
請求項3又は4記載の医薬品有効成分が
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
であるところの請求項3又は4記載の物質。
【請求項15】
請求項3又は4記載の医薬品有効成分が
5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
であるところの請求項3又は4記載の物質。
【請求項16】
請求項3又は4記載の医薬品有効成分がエトポシドであるところの請求項3又は4記載の物質。
【請求項17】
請求項3又は4記載の医薬品有効成分がメシル酸ネルフィナビルであるところの請求項3又は4記載の物質。
【請求項18】
請求項3又は4記載の医薬品有効成分がシンバスタチンであるところの請求項3又は4記載の物質。
【請求項19】
請求項3又は4記載の医薬品有効成分が7−エチル−10−ヒドロキシ−カンプトテシンであるところの請求項3又は4記載の物質。
【請求項20】
請求項3又は4記載の医薬品有効成分がパクリタキセルであるところの請求項3又は4記載の物質。
【請求項21】
請求項3又は4記載の医薬品有効成分がメシル酸サキナビルであるところの請求項3又は4記載の物質。
【請求項22】
請求項3又は4記載の医薬品有効成分がインスリンであるところの請求項3又は4記載の物質。
【請求項23】
請求項3又は4記載の医薬品有効成分がブロモクリプチンメシル酸塩であるところの請求項3又は4記載の物質。
【請求項1】
粒径が50μm〜10nmのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体からなり、炭素含量が10〜0.1%の物質。
【請求項2】
粒径が50μm〜10nmのハイドロキシアパタイトが、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体と−O(CO)で結合されていて、炭素含量が10〜0.1%の物質。
【請求項3】
請求項1記載の物質及び医薬品有効成分又は医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
【請求項4】
請求項2記載の物質及び医薬品有効成分又は医薬品添加物からなり、医薬品有効成分の重量比が1〜30%の物質。
【請求項5】
請求項3又4記載の物質から調製される医薬品。
【請求項6】
請求項3又は4記載の医薬品有効成分がsiRNA、アプタマー、RNA、DNA、ペプチド又は蛋白質であるところの請求項3又は4記載の物質。
【請求項7】
請求項3又は4記載の医薬品有効成分がクラリスロマイシンであるところの請求項3又は4記載の物質。
【請求項8】
請求項3又は4記載の医薬品有効成分がイトラコナゾールであるところの請求項3又は4記載の物質。
【請求項9】
請求項3又は4記載の医薬品有効成分がsiRNAであるところの請求項3又は4記載の物質。
【請求項10】
サブミクロンサイズのハイドロキシアパタイトと、末端官能基としてカルボキシル基を有するポリエチレングリコール誘導体の活性エステルを無水有機溶媒中で処理してサブミクロンサイズの請求項1又は2記載の物質を得る方法。
【請求項11】
サブミクロンサイズの請求項1又は2記載の物質と医薬有効成分又は医薬添加物を有機溶媒中で処理して請求項3又は4記載の物質を得る方法。
【請求項12】
請求項3又は4記載の医薬品有効成分がカンデサルタンであるところの請求項3又は4記載の物質。
【請求項13】
請求項3又は4記載の医薬品有効成分がカンデサルタンシレキセチルであるところの請求項3又は4記載の物質。
【請求項14】
請求項3又は4記載の医薬品有効成分が
5'-GUGAAGUCAACAUGCCUGCTT-3'(配列番号1)
5'-GCAGGCAUGUUGACUUCACTT-3'(配列番号2)
であるところの請求項3又は4記載の物質。
【請求項15】
請求項3又は4記載の医薬品有効成分が
5'-CUUACGCUGAGUACUUCGATT-3'(配列番号3)
5'-UCGAAGUACUCAGCGUAAGTT-3'(配列番号4)
であるところの請求項3又は4記載の物質。
【請求項16】
請求項3又は4記載の医薬品有効成分がエトポシドであるところの請求項3又は4記載の物質。
【請求項17】
請求項3又は4記載の医薬品有効成分がメシル酸ネルフィナビルであるところの請求項3又は4記載の物質。
【請求項18】
請求項3又は4記載の医薬品有効成分がシンバスタチンであるところの請求項3又は4記載の物質。
【請求項19】
請求項3又は4記載の医薬品有効成分が7−エチル−10−ヒドロキシ−カンプトテシンであるところの請求項3又は4記載の物質。
【請求項20】
請求項3又は4記載の医薬品有効成分がパクリタキセルであるところの請求項3又は4記載の物質。
【請求項21】
請求項3又は4記載の医薬品有効成分がメシル酸サキナビルであるところの請求項3又は4記載の物質。
【請求項22】
請求項3又は4記載の医薬品有効成分がインスリンであるところの請求項3又は4記載の物質。
【請求項23】
請求項3又は4記載の医薬品有効成分がブロモクリプチンメシル酸塩であるところの請求項3又は4記載の物質。
【図1】

【図2】
【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図4】

【図11】
【図12】

【図13】

【図2】
【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図4】
【図11】
【図12】
【図13】
【公開番号】特開2010−18772(P2010−18772A)
【公開日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願番号】特願2009−13585(P2009−13585)
【出願日】平成21年1月23日(2009.1.23)
【出願人】(000000239)株式会社荏原製作所 (1,477)
【Fターム(参考)】
【公開日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願日】平成21年1月23日(2009.1.23)
【出願人】(000000239)株式会社荏原製作所 (1,477)
【Fターム(参考)】
[ Back to top ]