
ペグ化インスリンリスプロ化合物
本発明は糖尿病の分野に関する。本発明は特に、高分子量ポリエチレングリコールとペグ化し、生理的pHで高い可溶性を示し、作用時間が長く、薬物動態学的、薬力学的、および/または活性ピーク/トラフ比が2未満で特徴づけられるペグ化インスリンリスプロ化合物に関する。本発明は、当該分子を提供する方法、当該分子を含む医薬組成物、およびその治療学的使用にも関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は糖尿病の分野に関する。本発明は特に、非常に可溶性で拡張作用プロフィルを有するペグ化インスリンリスプロ化合物、当該分子の提供方法、当該分子を含む医薬組成物、および当該分子の治療的使用に関する。
【背景技術】
【0002】
正常血糖を達成するため、インスリン補充療法は、正常な個人における内因性インスリン分泌のパターンに出来る限り近づくようにデザインされている。インスリンの生理学的な要求は毎日変動し、(a)食事関連の血糖値の上昇に対応するためにインスリンを必要とする吸収段階、および(b)肝グルコース産生を調整して最適な空腹時血糖値を維持するため、一定量のインスリンを必要とする吸収後段階の2段階に分けられ得る。従って、糖尿病の効果的なインスリン療法では、ボーラス注射によって食事時に提供される速効性インスリンと、注射により1日に1度または2度投与して食間の血糖値を制御する長時間作用性インスリンとの2種類の外因性インスリン製剤が、一般的に組み合わされる。
【0003】
現在利用可能なインスリン補充療法は、1つ以上の重要な面が臨床的に欠けている。例えば、従来の中時間作用性および長時間作用性のインスリン製剤、例えばインスリンデテミルなどの基礎インスリン類似体を毎日投与する場合、基礎グルコース制御を一日中提供するには、その作用期間が不十分である。従って、基礎インスリンの作用期間は、多くの場合、1日に1回の投与によって高血糖を適切に制御するには不十分である。特に吸収後段階の要求を満たすには不十分である。更に、現在の療法では、単回注射を怠ると、「ピーク/トラフ」の薬物水準が有意に増大し、グルコース制御の不良に至る可能性がある。更に、インスリンの放出時間を長期化させるための不溶性ストラテジーの利用、例えば従来の中時間作用性および長時間作用性のインスリン製剤である、Neutral Protamine Hagedorn(NPH)インスリンおよびULTRALENTE(登録商標)の結晶懸濁液などの利用、またはインスリングラルギンの生体内沈降ストラテジーの利用は、注射内変動を増大させ、用量応答プロフィルの変動を増大させる結果となる。特に、NPHおよびULTRALENTE(登録商標)懸濁液は、製品の均一性を保証するため機械的な混合を必要とし、個体内変動を増大させ、最適な空腹時血糖を食間に長期間維持するのに必要な理想的に「平坦な」薬力プロフィルよりもむしろピークを与える傾向にある。従って、インスリン放出を長期化させるために不溶性の状態に依存するインスリン製剤は、可溶性製剤よりも本質的に予測性が低く、適切な血糖制御が難しく、致命的な低血糖エピソードに至る可能性が高い。更に、最近の基礎インスリン類似体は、速効性インスリン製剤または中時間作用性インスリン製剤とは混合しにくい。従って、現在のインスリン補充療法では、糖尿病患者は致命的な低血糖エピソードに罹りやすく、重篤で長期的な糖尿病の合併症を引き起こしやすく、および/または便利性や生活の質の面で患者にかなりの負担をかけやすい。
【0004】
「非免疫原性ポリペプチド」と題される特許文献1は、約500Daと約20,000Daの間の分子量を有する直鎖PEG分子に結合したインスリンを開示している。HindsとKimは、低分子量(600Da、750Daおよび2000Da)であるモノメトキシポリエチレングリコールと結合したインスリンを開示している(非特許文献1を参照)。この記事では、「より高分子のmPEG(5000Da)と結合すると、コンジュゲートの生物活性がかなり下がることが(以前に)見出されているので」、低分子量のmPEGインスリンコンジュゲートに研究を限定したと、著者達は述べている。特許文献2は、インスリンリスプロを含むインスリン誘導体と小分子の分岐鎖ポリマーとの結合を開示している。特許文献3は、特に、PEGの総分子量がそれぞれ約10kDaおよび約20kDaである直鎖および分岐鎖PEG分子と結合したインスリン分子を開示している。特許文献4(2008年2月7日に公開)および特許文献5(2008年7月17日に公開)は、特に、約200Daから約40,000Daの範囲の名目分子量を有するPEG分子に結合した種々のインスリン類似体を開示している。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第4,179,337号
【特許文献2】PCT特許出願国際公開公報第2006/079641号
【特許文献3】PCT特許出願国際公開公報第2004/091494号
【特許文献4】PCT特許出願国際公開公報第2008/015099号
【特許文献5】PCT特許出願国際公開公報第2008/084051号
【非特許文献】
【0006】
【非特許文献1】Hinds,K.D.およびKimら、Advanced Drug Delivery Reviews, 54:505−530(2002)
【発明の概要】
【発明が解決しようとする課題】
【0007】
基礎インスリン補充レジメンに更に適した長時間作用性のインスリンが今でも必要とされているのは明白である。特に、食事インスリン製剤と混合可能であり、長期間持続性の時間−作用プロフィル(すなわち1日1回またはそれ以下の頻度で注射することにより適切に血糖値を制御可能)を有し、より平坦な作用および薬力プロフィル(すなわち「ピーク/トラフ」比が低い)を有し、患者内変動が少なく(すなわち時間−作用プロフィルが予想可能なので、低血糖および/または体重増加の発生率が減少)、および/または注射部位刺激感または注射部位疼痛の少ない可溶性の基礎インスリンが必要とされている。
【課題を解決するための手段】
【0008】
我々はここに、インスリンリスプロが高分子量のポリエチレングリコール誘導体とペグ化することにより、治療上効果的な基礎インスリン活性を有し、長期間持続性の時間−作用プロフィルを有し、生理的pHで可溶性が高く、および/または一般的に使用される他の食事インスリン製剤と混合可能であるペグ化インスリンリスプロ化合物を提供できるという発見を報告する。
【0009】
本発明は式Iの化合物:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pは共有結合によって直接または間接に、Aの1位でグリシンのアルファアミノ基、Bの1位でフェニルアラニンのアルファアミノ基またはBの28位でリジンのイプシロンアミノ基に結合している)、またはその製薬上許容可能な塩を提供する。
【0010】
本発明は、複数のモノおよびジペグ化インスリンリスプロ化合物を含む組成物であって、組成物中のペグ化インスリンリスプロ化合物の約75%を超える量が、式Iのモノペグ化化合物である組成物も提供する。
【0011】
本発明は、式Iのモノペグ化インスリン化合物を含む組成物であって、組成物中のモノペグ化化合物の約50%を超える量が、B鎖の28位でリジンのイプシロンアミノ基に直接または間接に共有結合しているPEGを有する組成物も提供する。
【0012】
本発明は、式Iのペグ化インスリンリスプロおよび1つ以上の製薬上許容可能な防腐剤、等張剤、金属イオンまたは緩衝剤を含む医薬組成物も提供する。本発明の特定の実施形態では、式Iのペグ化インスリンリスプロおよび1つ以上の製薬上許容可能な防腐剤、等張剤、金属イオンまたは緩衝剤を含む医薬組成物が、治療上有効量のインスリン類似体を更に含む。
【0013】
本発明は、本発明のペグ化インスリンリスプロ化合物を含む治療上有効量の医薬組成物を患者に投与するステップを含む、高血糖症、真性糖尿病、または妊娠性糖尿病を治療する方法も提供する。
【0014】
本発明は、治療のために、本発明のペグ化インスリンリスプロ化合物を使用するステップも含む。
【0015】
本発明は、低血糖症、真性糖尿病、または妊娠性糖尿病を治療するための医薬品製造のために、本発明のペグ化インスリンリスプロ化合物を使用するステップも含む。
【図面の簡単な説明】
【0016】
【図1】ラットとイヌのPKパラメーターの相対成長スケーリングに基づき、20kDaのPEG−B28インスリンリスプロ、40kDaのPEG−B28インスリンリスプロ、およびインスリンデテミルに関してシミュレートした、ヒトPKプロフィルをグラフで表したものである。プロフィルは、1週間の投与後の1投与間隔を表したものである。数値は平均ピーク/トラフ比である。
【図2】PEG20kDaB28(約95%以上)/A1(約5%以下)インスリンリスプロ(LY、0.225mg/kg)またはインスリングラルギン(0.5U/kg)を皮下投与した後のヒトのグルコース注入速度(GIR)のグラフである。GIRプロフィルは観察データに基づいており、Lilly Resaerch Laboratories内で開発した「Loess平滑化」(Splus 2000、プロフェッショナル版、MathSoft社)関数を用いて観察データに適合させた。
【発明を実施するための形態】
【0017】
本明細書では以下の略語が使用される。ACN:アセトニトリル、Boc:tert−ブトキシカルボニル、BSA:ウシ血清アルブミン、DCM:ジクロロメタン、メチレンクロライド、DMF:N,N−ジメチルホルムアミド、DMSO:ジメチルスルホキシド、DTT:ジチオトレイトール、EDTA:エチレンジアミン四酢酸、Et:エチル、EtOH:エタノール、Fmoc:9−フルオレニルメチルオキシカルボニル、HCl:塩酸、Da:ダルトン、kDa:キロダルトン、Lilly:イーライリリー社(インディアナ州インディアナポリス)、mAb:モノクローナル抗体、Me:メチル、MeOH:メタノール、PBS:リン酸塩緩衝液生理食塩水、RP−HPLC:逆相高速液体クロマトグラフィ、SEC:粒径排除クロマトグラフィ、SEM:平均値の標準誤差、SPA:シンチレーション近接アッセイ、TFA:トリフルオロ酢酸。本開示に使用されるアミノ酸の略語は全て、37C.F.R§1.822(B)(2)の記載通り、米国特許商標局が認めたものである。
【0018】
「インスリン」と言う用語は、あらゆる種の野生型インスリンを含むことがここでは意図されており、限定されないが、ブタのインスリン、ウシのインスリン、ヒトのインスリンなどが含まれる。天然または野生型インスリンとは、天然に見出されるインスリンのアミノ酸配列に対応するアミノ酸配列を有するインスリンを意味する。様々な種から得られるインスリンをコードするポリヌクレオチドおよびアミノ酸配列は、当業者には周知である。例えば、ヒトインスリンは21のアミノ酸のA鎖と30のアミノ酸のB鎖(それぞれ配列番号1および2)を有している。インスリンは天然から得てもよい(つまり天然源から単離してもよい)し、生物合成または合成によって産生してもよい。「インスリン」と言う用語は、あらゆるインスリン誘導体および/またはインスリン類似体を含むことも意図されている。
【0019】
本明細書で使用する場合、「インスリン類似体」または「インスリン誘導体」と言う用語は、インスリン活性を有し、ヒトインスリンと実質的に同じアミノ酸配列を有してはいるが、1つ以上のアミノ酸の置換、欠失、反転または付加などのヒトインスリンの修飾によってヒトインスリンとは異なっている蛋白質であると定義される。かかる化合物は当業者には周知である。例えば、PCT特許出願国際公開公報第96/15804号および03/053339号、米国特許番号第3,528,960号、5,514,646号、5,618,913号、5,750,497号、6,011,007号、6,251,856号、欧州特許第254,516号および280,534号を参照の事。当業者に公知の典型的かつ非網羅的なインスリン類似体には、インスリンアスパルト、インスリンリスプロ、インスリングラルギン、インスリンデテミル、インスリングルリシンなどが含まれる。更に、本明細書で使用する場合、「インスリン」と言う用語には、インスリン誘導体とインスリン類似体の両方であると考えられる化合物も含まれる。かかる化合物の例は、米国特許第5,750,497号および6,011,007号に記載されている。当業者に公知のかかる化合物の具体的な例はインスリンデテミルである。
【0020】
種々のインスリン類似体および/または誘導体は、「速効性」または「迅速に作用する」インスリン類似体であることが知られている。本明細書で使用する場合、「速効性」および「迅速に作用する」と言う用語は、同じ意味で用いられる。「迅速に作用するインスリン類似体」によって生じる食事グルコース効果は、(a)ヒトインスリンよりも皮下注射後早く始まる、および/または(b)ヒトインスリンよりも皮下注射後の作用期間が短いことを示す。典型的な速効性インスリン類似体には、生理的条件下で一般的に2量体化または自己会合しにくいので速効性である、「単量体インスリン類似体」が含まれる。単量体インスリン類似体は当業者には公知である。例えば、米国特許第5,700,662号および欧州特許第214,826号を参照の事。インスリンリスプロは迅速に作用する単量体インスリン類似体であり、野生型インスリンB鎖(配列番号2)の28位にあるプロリンと、野生型インスリンB鎖(配列番号2)の29位にあるリジンが入れ替わっている。従って、インスリンリスプロは、限定されないが、LysB28ProB29ヒトインスリン、LysB28ProB29ヒトインスリン、B28LysB29Proヒトインスリンなど、様々な名称によって当業者に知られている。
【0021】
「架橋結合した」と言う用語は、システイン残基間に存在するジスルフィド結合を意味する。例えば、適切に架橋結合したヒトインスリンは、配列番号1の7位におけるシステインと配列番号2の7位におけるシステイン間のジスルフィド結合、配列番号1の20位におけるシステインと配列番号2の19位におけるシステイン間のジスルフィド結合、および配列番号1の6位におけるシステインと配列番号1の11位におけるシステイン間のジスルフィド結合を含む。同様に、適切に架橋結合したインスリンリスプロ化合物は、配列番号1の7位におけるシステインと配列番号3の7位におけるシステイン間のジスルフィド結合、配列番号1の20位におけるシステインと配列番号3の19位におけるシステイン間のジスルフィド結合、および配列番号1の6位におけるシステインと配列番号3の11位におけるシステイン間のジスルフィド結合を含む。
【0022】
本明細書で使用する場合、「PEG結合インスリンリスプロ」または「ペグ化インスリンリスプロ」と言う用語は、少なくとも1つのPEGと共有結合し、生体内でインスリン活性を有するヒトインスリンリスプロ、またはその誘導体を意味する。
【0023】
インスリンおよびインスリンリスプロの生物学的活性は既知である。本発明のペグ化インスリンリスプロ化合物に関して用いられる「インスリン活性」と言う用語は、限定されないが、それぞれ実施例5および実施例6で以下に記載される1型および2型糖尿病の動物モデルなど、一般に評価の定まった少なくとも1つの生体内動物モデルにおいて、血糖値を有意に低下させる能力を意味することが意図されている。従って、インスリン活性には、STZ処置のラットにおいて、用量568nmol/kgを単回皮下注射した後約4時間から少なくとも約36時間、血糖を100mg/dL以下のレベルにまで低下させるペグ化インスリンリスプロ化合物の能力が含まれる。
【0024】
「ポリエチレングリコール」または「PEG」は、ポリアルキレングリコール化合物またはその誘導体を意味し、カップリング剤が存在してもしなくても、あるいはカップリング部分または活性化部分との誘導化が存在してもしなくてもよい。典型的な例として、PEGは末端にヒドロキシ基を持つ線形ポリマーであり、HO−CH2CH2−(CH2CH2O)n−CH2CH2−OHの式を有している。PEG中の反復サブユニット「n」の数は、ダルトンで表される分子量に関して近似されたものである。ペグ化化合物を調製するのに使用されるPEG試薬は、一般的に、PEGポリマー中に異なる数(n)のエチレングリコール・サブユニットを有するPEGの異種混合物を含んでいる。PEGの1つのエチレングリコール・サブユニットである(−(CH2CH2O))の分子量は約44ダルトンである。従って、PEGポリマーの分子量は(n)数によって決定される。本発明のペグ化インスリンリスプロ化合物に結合するPEGは、約400から約1000の範囲のサブユニット数nを有している。本発明のペグ化インスリンリスプロ化合物に結合するPEGが、約400から約750の範囲のサブユニット数nを有しているのが好ましい。より好ましいのは、本発明のペグ化インスリンリスプロ化合物に結合するPEGが、約400から約550の範囲のサブユニット数nを有している場合である。最も好ましいのは、本発明のペグ化インスリンリスプロ化合物に結合するPEGが、約400から約500の範囲のサブユニット数nを有している場合である。
【0025】
PEGの数多くの誘導体、それを製造する方法およびインスリンやインスリンリスプロなどの蛋白質にそれを結合する方法は当業者には公知であり、本発明で使用するのに適している。例えば、PCT特許出願国際公開公報第01/62827号、2006/028745号、2006/096535号、2006/036825号、Zalipsky,S.Bioconjugate Chem.6:150−165,1995;Veroneseら,Applied Biochem.and Biotech.11:141−152,1985;およびRoberts,M.ら,Advanced Drug Delivery Reviews,54:459−476,2002を参照の事。本発明で使用される特に好ましい1つのPEGは、ポリマーの一端が下級C1−6アルコキシ基などの比較的不活性な基で終わっているPEGである。好ましくは、PEGはモノメトキシPEG(一般的にmPEGと呼ばれる)である。これは線形のPEGで、ポリマーの一端はメトキシ(−OCH3)基である。本発明で使用されるPEGは「活性化m−PEG」であるのが更に好ましい。この場合、線形PEGの一端はメトキシ基で終わり、他端は、好ましい活性化mPEGとのペグ化をインスリンリスプロ分子の特定の部位で促進させるため、インスリンリスプロまたはインスリンリスプロ誘導体の好ましい部位と結合するのに適した反応基で終わっている。
【0026】
PEGは一般的に分子量がある程度多様なPEG化合物の混合物として製造および使用されるので、当業者は通常、特定のペグ化化合物を生成するペグ化反応に使用されるPEG試薬の平均サイズによって、化合物に結合するPEGの分子量を表す。平均値を表すには数多くの方法が可能であるが、一般的には3つの方法が用いられている。すなわち、数平均分子量、重量平均分子量およびz平均分子量である。本明細書で使用する場合、「平均分子量」と言う用語は重量平均分子量を意味することが意図されるが、これは、限定されないが、マトリックス補助レーザー脱離イオン化時間(MALDI−TOF)質量分析法、ゲル浸透クロマトグラフィまたは他の液体クロマトグラフィ技術、光散乱技術、超遠心、粘度測定技術など、当業者に周知の技術を用いて測定できる。重量平均分子量を計算する式はΣ(Mi2Ni)/Σ(MiNi)で表されてもよく、式中、Niは混合物に分子量Miを有する分子のモル比率(または数比率)である。数平均分子量を計算する式はΣ(MiNi)/Σ(Ni)で表されてもよく、式中、Niは混合物に分子量Miを有する分子のモル比率(または数比率)である。重量平均分子量と数平均分子量の比は多重分散度指数(PDI)として知られており、分布の大体の幅を示すものである。本発明のペグ化インスリンリスプロ化合物を調製するのに適したPEG試薬は一般的に多重分散状態(すなわちポリマーの重量平均分子量と数平均分子量は等しくない)である。本発明の化合物または組成物を調製するのに使用されるPEG試薬のPDIは、約1.1未満であるのが好ましい。更に好ましくは、本発明の化合物または組成物を調製するのに使用されるPEG試薬のPDIは約1.05未満である。
【0027】
本発明のペグ化インスリンリスプロ化合物に関して、インスリンリスプロ分子に共有結合しているPEGの分子量は約17.5kDaから約40kDaの範囲(nは約400から約1000の範囲)、あるいはPEGの平均分子量は約17.5kDaから約40kDaである。好ましくは、インスリンリスプロ分子に共有結合しているPEGの分子量は約20kDaから約30kDaの範囲(nは約450から約750の範囲)、あるいはPEGの平均分子量は約20kDaから約30kDaである。更に好ましいのは、インスリンリスプロ分子に共有結合しているPEGの分子量は約17.5kDaから約25kDaの範囲(nは約400から約550の範囲)、あるいはPEGの平均分子量は約17.5kDaから約25kDaである。最も好ましいのは、インスリンリスプロ分子に共有結合しているPEGの分子量は約17.5kDaから約20kDaの範囲(nは約400から約500の範囲)、あるいはPEGの平均分子量は約17.5kDaから約20kDaである。
【0028】
特定の実施形態では、本発明のペグ化インスリンリスプロ化合物は、好ましい平均分子量を持った活性化mPEGをインスリンリスプロと共有結合させることにより調製される。ペグ化インスリンリスプロの反応条件は、使用される特定のPEG、インスリンリスプロの結合部位、インスリンリスプロ上で結合標的となる特定の種類の反応基、好ましいペグ化レベルなどによって異なり、当業者が容易に決定できる。特定のペグ化ストラテジーに最も適した実験条件は、一般的に、慣用の実験方法によって、当業者が容易に決定できる。
【0029】
好ましい実施形態では、本発明のペグ化インスリンリスプロ化合物は、mPEGマレイミド(mPEG−MAL)やmPEGチオール(mPEG−SH)など、比較的チオール選択的な活性化mPEGをインスリンリスプロに間接的に結合させることにより、すなわちN−スクシンイミジル−S−アセチルチオプロピオネート(SATP)やN−スクシンイミジル−S−アセチルチオアセテート(SATA)などの「アミン/チオール」調整剤を使ってインスリンリスプロに導入したチオール官能基に、チオール選択的活性化mPEGを結合させることにより調製される。更に好ましいのは、インスリンリスプロ上のチオール・スルフィドリル基にPEGを共有結合させるのに使用される活性化mPEGは、以下の(a)、(b)、(c)などのmPEGマレイミドである。
【化1】
【0030】
本発明のペグ化インスリンリスプロ化合物を調製する好ましい方法は、マイケル付加反応を利用して安定したチオエーテル結合を形成する方法である。反応は非常に特異的であり、他の官能基の存在下、穏やかな条件下で生じる。例えば、mPEGマレイミドは、活性化mPEGとして、本発明のペグ化インスリンリスプロコンジュゲートを調製するのに役立つ。ペグ化工程は、反応を完結させるため、mPEGマレイミドに対してチオール誘導インスリンリスプロをモル過剰使用するのが好ましい。反応は、pH4.0と9.0の間で、室温で1時間から40時間行うのが好ましい。チオールを含むペグ化されていない過剰のペプチドは、通常の分離方法によりペグ化生成物から容易に分離される。活性化mPEGマレイミドを用いるインスリンリスプロのペグ化に要求される典型的な条件は、実施例1に記載される。
【0031】
特定の実施形態では、本発明のペグ化インスリンリスプロ化合物は、アミンに比較的特異的な活性化mPEGを結合させることにより調製される。インスリンリスプロの主なアミン特異的ペグ化に適した活性化mPEGには、mPEGスクシンイミジルプロピオネート(mPEG−SPA)、mPEGスクシンイミジルブタノエート(mPEG−SBA)、mPEGスクシンイミジルアルファメチルブタノエート(mPEG−SMB)、mPEGスクシンイミジルカーボネート(mPEG−SC)、mPEGベンゾトリアゾールカーボネート、mPEG−p−ニトロフェニルカーボネート(mPEG−NPC)などが含まれる。
【0032】
本発明の好ましい実施形態では、インスリンリスプロのペグ化に使用される活性mPEGによって、加水分解的に安定した共有結合、例えば、アミド結合、ウレタン(カルバメートとしても知られる)結合、アミン結合、チオエーテル(スルフィドとしても知られる)結合、尿素(カルバミドとしても知られる)結合などにより、mPEGに共有結合的に結合するインスリンリスプロが生じる。更に好ましいのは、インスリンリスプロのペグ化に使用される活性化mPEGはmPEG−SCまたはmPEG−NPCである。いずれの場合も、ウレタン(すなわちカルバメート)結合によってPEGに共有結合するインスリンリスプロが生じる。種々の分子量のmPEG−NPCを用いるインスリンリスプロのペグ化に有効な典型的な条件は、実施例2に記載される。
【化2】
【0033】
本発明のペグ化インスリンリスプロ化合物は、一般的に、イオン交換クロマトグラフィ、粒径排除クロマトグラフィ、アフィニティクロマトグラフィ、疎水性相互作用クロマトグラフィ、逆相クロマトグラフィなど、1つ以上の精製技術を用いて精製される。試料中のペグ化インスリンリスプロ化合物の全体的な不均一性(ペグ化反応により生成されたペグ化インスリンリスプロ化合物の数および割合)は、クロマトグラフィ、電気泳動、質量分析、特にMALDI−MSおよびNMR分光技術のうち1つ以上を用いて評価できる。
【0034】
本発明のペグ化インスリンリスプロ化合物の調製に使用されるインスリンリスプロは、 液相方法、固相方法、半合成法、組み換えDNA技法など、一般に評価の定まった種々のペプチド合成技術のうちいずれかを用いて調製してよい。例えば、米国特許第5,700,662号(Chanceら)および欧州特許第214,826(Brangeら)は、種々のインスリン類似体の調製を開示している。インスリンリスプロのA鎖およびB鎖は、組み換えDNA技法を用いて、プロインスリン様前駆体分子から調製してもよい。好ましい実施形態では、本発明のペグ化インスリンリスプロを作るのに使用されるインスリンリスプロが、プロインスリン様前駆体を使って生成される。
【0035】
本発明は式Iの化合物:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約17.5kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pは共有結合によって直接または間接に、A鎖の1位でグリシンのアルファアミノ基、B鎖の1位でフェニルアラニンのアルファアミノ基またはB鎖の28位でリジンのイプシロンアミノ基に結合している)、またはその製薬上許容可能な塩を提供する。
【0036】
本発明の好ましい化合物は、(a)Pがウレタン結合またはチオエーテル結合によってインスリンリスプロと共有結合し、(b)STZ処置ラットにおいて、約568nmol/kgの用量で、ヒトインスリン受容体に対して約30nM、約20nM、約10nM、約5nMまたはそれ以下のKi値を有し、約6時間、約8時間、約10時間、約12時間または約14時間を超える除去半減期を有することによって特徴付けられる化合物、あるいはSTZ処置ラットにおいて、化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約36時間、約48時間、約60時間、約72時間、約84時間、約96時間、約108時間または約120時間、血糖値を約100mg/dL以下に低下させる活性によって特徴付けられる化合物である。更に好ましい化合物は、(a)Pがウレタン結合によってインスリンリスプロと共有結合し、(b)ヒトインスリン受容体に対して約10nM以下のKi値を有し、(c)STZ処置ラットにおいて、約568nmol/kgの用量で、6時間を超える除去半減期を有することによって特徴付けられ、(d)STZ処置ラットにおいて、化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約48時間、約60時間、約72時間、約84時間、約96時間、約108時間または約120時間、血糖値を約100mg/dL以下に低下させる活性によって特徴付けられる化合物である。更に好ましい化合物は、(a)Pがウレタン結合によってインスリンリスプロと共有結合し、(b)ヒトインスリン受容体に対して約10nM以下のKi値を有し、(c)STZ処置ラットにおいて、約568nmol/kgの用量で、6時間を超える除去半減期を有することによって特徴付けられ、(d)STZ処置ラットにおいて、化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約48時間、約60時間、約72時間、約84時間、約96時間、約108時間または約120時間、血糖値を約100mg/dL以下に低下させる活性によって特徴付けられ、(e)0.225mg/kgの用量で非経口的にヒトに単回投与した場合に、 約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することによって特徴付けられる化合物である。
【0037】
本発明に一貫する特徴および原理によれば、本発明の1つの実施形態は、インスリンリスプロのA鎖の1位におけるグリシンのアルファアミノ基(PEG−GlyA1インスリンリスプロ)、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基(PEG−PhB1インスリンリスプロ)、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基(PEG−LysB28インスリンリスプロ)に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含むモノペグ化インスリンリスプロ化合物を提供する。好ましくは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。更に好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。更に好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約17.5kDaまたは約20kDaを有するPEGを含む場合である(20kDa−PEG−LysB28インスリンリスプロ)。最も好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約20kDaを有するPEGを含む場合である(すなわち、PEG20kDa−LysB28インスリンリスプロ)。
【0038】
本発明の他の実施形態は、インスリンリスプロのA鎖の1位におけるグリシンのアルファアミノ基、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含むモノペグ化インスリンリスプロ化合物を提供する。好ましくは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。更に好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約17.5kDa、約20kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。最も好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約20kDaを有するPEGを含み、0.225mg/kgの化合物を単回皮下投与でヒトに投与した場合、PEG−LysB28インスリンリスプロが、約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することにより特徴づけられる場合である。
【0039】
別の実施形態では、本発明は、PEGの結合が異なる部位で生じるペグ化インスリンリスプロ化合物の混合物、および/またはモノペグ化インスリンリスプロ、ジペグ化インスリンリスプロおよびトリペグ化インスリンリスプロ化合物の混合物を含む組成物を提供する。本発明による具体的な組成物は、i)PEG−GlyA1インスリンリスプロ、ii)PEG−PheB1インスリンリスプロ、iii)PEG−LysB28インスリンリスプロ、iv)ジPEG−GlyA1PheB1インスリンリスプロ、v)ジPEG−GlyA1LysB28インスリンリスプロ、vi)ジPEG−PheB1LysB28インスリンリスプロおよびvii)ジPEG−GlyA1PheB1インスリンリスプロからなる群から選択される2つ以上のペグ化インスリンリスプロ化合物を含む。更に好ましいのは、本発明の組成物はペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量が式Iのモノペグ化インスリンリスプロ化合物である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がモノペグ化インスリンリスプロ化合物であり、全ペグ化インスリンリスプロ化合物の約20%、約15%、約10%、約5%または約3%未満がジペグ化されている場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がPEG−LysB28インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がPEG−LysB28インスリンリスプロであり、全ペグ化インスリンリスプロ化合物の約20%、約15%、約10%、約5%または約3%未満がPEG−GlyA1インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がPEG−LysB28インスリンリスプロであり、全ペグ化インスリンスプロ化合物の約20%、約15%、約10%、約5%または約3%未満がPEG−GlyA1インスリンリスプロであり、全ペグ化インスリンスプロ化合物の約10%、約5%または約3%未満がジペグ化インスリンリスプロ化合物の場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約80%がPEG−LysB28インスリンリスプロ、約10%がPEG−GlyA1インスリンリスプロ、約10%がジPEG−GlyA1LysB28インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約90%以上がPEG−LysB28インスリンリスプロ、約5%以下がPEG−GlyA1インスリンリスプロ、約5%以下がジPEG−GlyA1LysB28インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約90%以上がPEG−LysB28インスリンリスプロ、約5%以下がPEG−GlyA1インスリンリスプロ、約5%以下がジPEG−GlyA1LysB28インスリンリスプロであり、0.225mg/kgの化合物を単回皮下投与でヒトに投与した場合、PEG−LysB28インスリンリスプロが、約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することにより特徴づけられる場合である。最も好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約95%以上がPEG−LysB28インスリンリスプロ、約5%以下がPEG−GlyA1インスリンリスプロであり、0.225mg/kgの化合物を単回皮下投与でヒトに投与した場合、PEG−LysB28インスリンリスプロが、約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することにより特徴づけられる場合である。
【0040】
本明細書で使用する場合、「塩基条件」と言う用語は、ペグ化反応の塩基度を意味する。インスリンリスプロのB鎖の28位にリジンを有するペグ化インスリンリスプロに関して、反応は、より選択的に、実質的に脱プロトン化したインスリンリスプロのアルファアミノ基との間で行われるべきである。水性溶媒または共溶媒では、塩基条件とは7.0を超えるpHで反応が行われることを意味する。ペグ化反応が約8.5から約11.5のpHで行われるのが好ましい。有機溶媒では、反応は水中で10.75以上のpKaに等しい塩基度を有する塩基の存在下で行われる。
【0041】
本発明は、以下の式のペグ化インスリンリスプロ化合物、またはその製薬上許容可能な塩を製造する工程:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pはウレタン共有結合によってBの28位でリジンのイプシロンアミノ基に結合している)も含み、当該工程には、約25℃と約30℃の間の反応温度で、Bの28位におけるリジンのイプシロンアミノ基と、約20kDaと約40kDaの間の重量平均分子量を有するモノメトキシポリエチレングリコールp−ニトロフェニルカルボネート(mPEG−NPC)とをpH約8.5と約11.5の間の水性溶媒中で反応させるステップが含まれる。好ましくは、反応のpHは約10.5から約11.1の間に維持され、ペグ化反応は約25℃と約30℃の間の温度で約2時間から約12時間行われ、mPEG−NPC対インスリンリスプロの比率は約1.0から約5.0の範囲である。更に好ましいのは、mPEG−NPCの重量平均分子量が約20kDaで、反応のpHが約10.5と約11.1の間に維持され、ペグ化反応が約25℃と約30℃の間の温度で約2時間から約12時間行われ、mPEG−NPC対インスリンリスプロの比率が約1.0と約5.0の範囲にある場合である。更に好ましいのは、PEG対インスリンリスプロのモル比率が約2.5と約4.5の範囲にあり、mPEG−NPCの重量平均分子量が約20kDaで、ペグ化反応のpHが約10.5と約11.1の間に維持され、ペグ化反応が約25℃と約30℃の間の温度で約3時間から約6時間維持される場合である。最も好ましいのは、PEG対インスリンリスプロのモル比率が約2.6と約4.5の範囲にあり、mPEG−NPCの重量平均分子量が約20kDaで、ペグ化反応のpHが約10.5と約11.1の間に維持され、反応が約25℃と約30℃の間の温度で約3時間維持される場合である。
【0042】
異なる分子量を有する本発明のペグ化インスリンリスプロ化合物は、必要に応じて、当業者に公知の種々の技法を用いて単離できる。かかる技法には、限定されないが、ゲル濾過クロマトグラフィおよび/またはイオン交換クロマトグラフィが含まれる。すなわち、ゲル濾過クロマトグラフィを用いて、異なる分子量(本質的にペグ化反応に使用されるPEGの平均分子量に対応する差異)に基づいて、モノペグ化インスリンリスプロ化合物、ジペグ化インスリンリスプロ化合物およびトリペグ化インスリンリスプロ化合物の画分に分けてもよい。例えば、平均分子量約20kDaを有する活性化mPEGにインスリンリスプロを結合させる典型的な反応では、反応混合物は分子量約5,808ダルトンを有する非修飾インスリンリスプロ、平均分子量約25,808ダルトンを有するモノペグ化インスリンリスプロ、平均分子量約45,808ダルトンを有するジペグ化インスリンリスプロ、および平均分子量約65,808ダルトンを有するトリペグ化インスリンリスプロが含まれる場合がある。しかし、ゲル濾過技法は流体力学的サイズに基づいて化合物を分離するので、平均分子量約20kDaを有するmPEGと結合したモノペグ化インスリンリスプロは、平均分子量が約25,808ダルトンであるにもかかわらず、ゲル濾過中約82kDaの蛋白質として移動する。当業者は、精製および/または定量化に際し、平均分子量約20kDaのmPEGとペグ化したジペグ化種およびトリペグ化種が、非常に異なる移動または保持時間を有することを予期するであろう。
【0043】
「血漿半減期」と言う用語は、体内の薬物の血漿濃度が半分に低下する時間を意味する。「除去半減期」と言う用語は代替的に使用され、除去の最終対数線形率に相当する。当業者は、半減期がクリアランスと分配量の両方の関数として変化する誘導パラメーターであることを理解している。血漿半減期または除去半減期に関して用いられる「長期的」、「長期化」、または「増大」と言う用語は、本明細書では同じ意味で使用され、比較条件化で測定した場合、基準分子(例えばインスリンリスプロ)の半減期と比べて試験化合物(例えばペグ化インスリンリスプロ)の半減期が統計学的に有意に増大する意味であることが意図されている。
【0044】
クリアランスは薬物を排除する体の能力の尺度である。例えば薬物の修飾によってクリアランスが低下すれば、半減期は増大することが予想される。しかし、この相互関係は、分配量に変化がない場合のみ正確である。最終対数線形半減期(t1/2)、クリアランス(CL)および分配量(V)の間の有効な近似関係は、t1/2〜0.693(V/CL)の式で表される。クリアランスは、除去を説明するにあたり、薬物がどれほど除去されたかではなく、むしろ、薬物を完全に無くすのに必要な血液や血漿などの生物学的流体の量を示すものである。クリアランスは単位時間当たりの量として表わされる。
【0045】
本明細書で使用する場合、「治療」または「治療する」と言う用語は、糖尿病、高血糖症、あるいは症状や合併症と闘うまたはそれを軽減する目的でインスリン投与を必要とする他の病状に罹患している患者の管理およびケアを意味する。治療には、症状または合併症の発症、症状または合併症の軽減、または病気、病状または疾患の排除のために、本発明の化合物または組成物を投与するステップも含まれる。治療対象の患者は哺乳動物であり、好ましくはヒトである。
【0046】
式1のペグ化インスリンリスプロ化合物は、1つ以上の式Iの化合物をそれを必要とする患者に治療上有効量投与して、高血糖症を治療するのに有効である。本明細書で使用する場合、「治療上有効量」と言う用語は、患者の血糖を調節するのに十分な式Iのペグ化インスリンリスプロ化合物またはその組成物の量を意味する。式Iのペグ化インスリンリスプロの好ましい治療上有効量は、約0.01nmol/kgから約100nmol/kgである。更に好ましいのは、治療上有効量が約0.01nmol/kgから約50nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.01nmol/kgから約20nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.01nmol/kgから約10nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.1nmol/kgから約7.5nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.1nmol/kgから約5nmol/kgの場合である。もっとも好ましいのは、治療上有効量が約0.5nmol/kgから約5nmol/kgの場合である。しかし、ペグ化インスリンリスプロ化合物または実際に投与される1つ以上のペグ化インスリンリスプロ化合物を含む組成物の量は、治療対象の病状を含む様々な関連状況(すなわち高血糖症の原因)、投与されるペグ化インスリンリスプロの特定の種またはペグ化インスリンリスプロ化合物の特定の混合物、同時投与される他の薬物、インスリンまたはその他、選択された非経口的投与経路、個々の患者の年齢、体重および反応、並びに患者の症状の重篤性を考慮して、医師が決定するものであることは理解されるべきである。従って、上述の用量範囲は、如何なる意味でも、本発明の範囲を制限するものではない。
【0047】
「血糖を調節するのに十分」と言う用語は、化合物または組成物を患者に投与することにより、正常な空腹時血漿グルコースレベルになることを意味する。臨床的に正常な空腹時血漿グルコースレベルは70−110mg/dLである。臨床的に正常な食後血漿グルコースレベルは140mg/dL未満である。
【0048】
インスリン構造の共有結合的化学変化が貯蔵中に起きることが知られている。その結果、活性の低下した分子、脱アミド化生成物のように潜在的に免疫原性的な生成物、および高分子量の転換生成物(例えば、2量体、オリゴマー、ポリマー)が形成される場合がある。インスリンの化学的安定性に関する総合的な研究がJens Brange,“Stability of Insulin,” Kluwer Academic Publishers,1994に記載されている。インスリン組成物は一般的に約2−8℃以下という低温で貯蔵される(低温貯蔵は、室温で貯蔵する場合と比べて貯蔵寿命をかなり改善する)という事実があるにもかかわらず、インスリン製品の保存寿命は、可溶性の凝集体(2量体、オリゴマー、ポリマー)の形成によりやがて低下する。更に、インスリン製品は振動により(例えば、患者がポケットに入れて持ち運んだり、輸送したりすることにより)不溶性の凝集体(フィブリル)を形成する傾向にある。化学的または物理的影響により可溶性および不溶性の凝集体が形成される傾向を最小限にまで減らすことが、インスリン製品の質にとって非常に重要である。従って、インスリン組成物を治療に使用するためには、化学的および物理的な安定特性を証明しなければならない。
【0049】
本明細書で使用する場合、「安定性」と言う用語は、ペグ化インスリンリスプロ化合物の調剤の物理的および/または化学的安定性を意味する。ペグ化インスリンリスプロ調剤の物理的不安定は、蛋白質分子の凝集により高位のポリマーや更には沈殿物を形成することにより生じる場合がある。「安定した」調剤では、蛋白質の凝集度が受容可能なレベルに制御されており、時間とともに受容不可能なレベルにまで増大するということがない。本発明の特定の実施形態では、ペグ化インスリンリスプロ調剤は、凝集度が出発物質の凝集度の約1%、2%、3%、4%、5%、10%、15%、20%、25%または30%以内であれば、特定の期間安定であると考えられる。本発明の特定の実施形態では、ペグ化インスリンリスプロ調剤は、ポリペプチドの生物学的活性が出発物質の活性の少なくとも約99%、95%、90%、85%、80%、75%、70%、65%、60%、55%または50%であれば、特定の期間安定であると考えられる。
【0050】
本明細書で使用する場合、「化学的安定性」と言う用語は、ペグ化インスリンリスプロ組成物が、貯蔵中に、約2−8℃の低温貯蔵および約20−40℃の高温貯蔵を含む静止条件下で、可溶性の蛋白質凝集体を形成する傾向にあることを意味する。本発明のペグ化インスリンリスプロ化合物の化学的安定性は、特定の条件化で(例えば、特定の温度および湿度条件で特定の期間)、製剤の分析属性を決定することにより測定できる。測定可能な分析属性には、例えば、粒径排除HPLCを用いる高分子量種の形成などが含まれる。その結果は、次にモニターし、予め特定されたパラメーターと比較できる。
【0051】
本明細書で使用する場合、「物理的安定性」と言う用語は、ペグ化インスリンリスプロ組成物の振動など、物理的な行為の結果として不溶性の蛋白質凝集体が形成される傾向にあることを意味する。本発明のペグ化インスリンリスプロ化合物を種々の医薬調剤の形で、一定期間、種々の温度で貯蔵した場合の物理的安定性は、当業者に周知の方法により評価してよい。試料の見かけの光減衰(吸光度または光学密度)の測定もその中に含まれる。かかる光減衰の測定は、製剤の混濁に関係している。混濁は製剤中の蛋白質または複合体の凝集や沈殿により生じる。物理的安定性を評価する他の方法は当業者には周知であり、粒子の存在または不在の視覚的評価やチオフラビンT蛍光顕微鏡検査法によるフィブリル/ゲル形成の検出などもその中に含まれる。
【0052】
本発明の他の実施形態は、患者(特にヒト)に投与するのに適しており、式Iに示した少なくとも1つのペグ化インスリンリスプロ化合物の治療上有効量および1つ以上の製薬上許容可能な賦形剤、希釈剤、緩衝液、金属イオンまたは担体を含む医薬組成物を提供する。かかる医薬組成物はその性質上一般的に非経口的(そうでない場合もある)であり、当業者に周知の従来の非経口製品用賦形剤、緩衝液、希釈剤、金属イオンまたは担体を使用し、種々の技術のいずれかによって調製してもよい。
【0053】
本発明のペグ化インスリンリスプロ化合物はきわめて水溶性であるので、本発明の医薬組成物は水を主要溶媒として包含する組成物を含んでおり、ペグ化インスリンリスプロ化合物の全濃度は、少なくとも1mg/mL、少なくとも2mg/mL、少なくとも5mg/mL、少なくとも10mg/mL、少なくとも15mg/mL、少なくとも20mg/mL、少なくとも25mg/mL、少なくとも30mg/mL、少なくとも35mg/mL、少なくとも40mg/mL、少なくとも45mg/mL、少なくとも50mg/mLまたはそれ以上であり、製薬上許容可能な緩衝液を含み、医薬組成物のpHは約4.0から約8.5である。本発明の医薬組成物のpHは約6.0から約8.5であるのが好ましい。更に好ましいのは、本発明の医薬組成物が、全濃度が約2.5mg/mLから約60mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、組成物のpHが約6.0から約8.5の範囲の場合である。更に好ましいのは、本発明の医薬組成物が、全濃度が約5mg/mLから約50mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、医薬組成物のpHが約6.5から約7.5の範囲の場合である。更に好ましいのは、本発明の医薬組成物が、全濃度が約10mg/mLから約40mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、医薬組成物のpHが約6.5から約7.5の範囲の場合である。更に好ましいのは、本発明の医薬組成物が、全濃度が約15mg/mLから約40mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、医薬組成物のpHが約7.0から約7.5あるいは約7.0から約8.0の範囲の場合である。本発明の医薬組成物が更に治療上有効量のインスリンを含んでいるのが更に好ましい。更に好ましいのは、上記インスリンがインスリン類似体の場合である。更に好ましいのは、インスリン類似体が迅速に作用するインスリン類似体の場合である。最も好ましいのは、迅速に作用するインスリン類似体がインスリンリスプロの場合である。
【0054】
「緩衝液」と言う用語は、酸塩基共役成分の作用によりpHの変化に抵抗する溶液を意味する。使用される緩衝液は製薬上許容可能な緩衝液であるのが好ましい。「製薬上許容可能な緩衝液」と言う用語は、インスリン製剤に使用しても安全であり、医薬組成物のpHを目的のpHへ制御する効果のある溶液を意味する。好ましい実施形態では、緩衝液は約6.0から約8.5の範囲のpHを有している。更に好ましいのは、緩衝液が約7.0から約8.0の範囲のpHを有している場合である。本発明の組成物をこの範囲のpHに制御するのに用いられる製薬上許容可能な緩衝液は、限定されないが、リン酸塩緩衝液、酢酸塩緩衝液、クエン酸塩緩衝液、アルギニン緩衝液、トリス緩衝液、ヒスチジン緩衝液およびそれらの組み合わせを含む。「トリス」は2−アミノ−2−ヒドロキシメチル−1,3−プロパンジオールおよびその薬理学的に許容可能な塩を意味する。遊離塩基および塩酸塩(すなわちトリスHCl)が、トリスの一般的な2つの形態である。トリスはトリメチロールアミノメタン、トロメタミン、およびトリス(ヒドロキシメチル)アミノメタンとしても当業者に知られている。本発明の医薬組成物は、約2.5mMから約50mMのリン酸塩緩衝液またはトリス緩衝液を含むのが好ましい。更に好ましいのは、本発明の医薬組成物が約5mMから約20mMのリン酸塩緩衝液またはトリス緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約5mMから約10mMのリン酸塩緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約5mMのリン酸塩緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約7.5mMから約50mMのトリス緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約10mMから約25mMのトリス緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約15mMから約20mMのトリス緩衝液を含む場合である。最も好ましいのは、本発明の医薬組成物が約16mMのトリス緩衝液を含む場合である。
【0055】
本発明のペグ化インスリンリスプロ化合物および組成物は、非経口的に患者に投与される既知のインスリン製剤と同様の方法で製剤化してもよい。かかる製剤化は当業者には周知である。式Iのペグ化インスリンリスプロ化合物をHUMALOG(登録商標)インスリンリスプロまたはHumulin(登録商標)の製剤化と同様に製剤化するのが好ましい。従って、本発明の好ましい医薬組成物は、水、式Iのペグ化インスリンリスプロ化合物、等張剤および製薬上許容可能な緩衝液を含んでもよい。本発明の医薬組成物は製薬上許容可能な防腐剤を更に含むのが好ましい。更に好ましいのは、本発明の医薬組成物が、インスリンの6量体化を促進し得る二価の陽イオン、例えば亜鉛および/またはコバルトを更に含む場合である。更に好ましいのは、本発明の医薬組成物が、少なくとも1つの6量体安定化剤を更に含む場合である。更に、pHを調整するため、塩酸および/または水酸化ナトリウムを添加してもよい。
【0056】
「等張剤」は生理学的に寛容な化合物で、製剤に適切な等張性を与えることにより、投与された医薬組成物に接触する細胞膜に水の正味の流れが生じるのを防ぐ。グリセリンとしても知られるグリセロールは等張剤として一般的に使用される。他の等張剤としては、i)限定されないが、マンニトールやソルビトールなどの他の糖アルコール、ii)限定されないが、NaClなどの塩、iii)限定されないが、デキストロースなどの単糖類、iv)限定されないが、ラクトース、ショ糖、トレハロースなどの二糖類などが含まれる。本発明の医薬組成物には1つ以上の等張剤を含んでもよい。本発明の医薬組成物は、約270ミリオスモルから約330ミリオスモルの範囲の等張性を持つ製剤を生成することのできる1つ以上の等張剤を有するのが好ましい。等張剤がグリセロール、ソルビトール、ショ糖、NaCl、トレハロースおよび/またはマンニトールであれば更に好ましい。更に好ましいのは、等張剤がグリセロール、ソルビトール、ショ糖、NaClおよび/またはトレハロースの場合である。更に好ましいのは、グリセロール、ソルビトール、ショ糖、NaClまたはトレハロースが、約100mMから約200mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、グリセロールが、約100mMから約200mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、グリセロールが、約150mMから約180mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、グリセロールが、約130mMから約175mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、NaClが、約50mMから約300mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、NaClが、約100mMから約200mMの濃度で、本発明の医薬組成物の中に存在する場合である。最も好ましいのは、NaClが、約150mMの濃度で、本発明の医薬組成物の中に存在する場合である。
【0057】
本発明の医薬組成物は6量体安定化化合物を含んでもよい。「6量体安定化化合物」と言う用語は、6量体会合状態にある本発明のペグ化インスリンリスプロ化合物を安定化させる、非蛋白質様の小分子量の化合物である。インスリン分子用の既知の6量体安定化化合物は、カルシウムイオン、亜鉛、コバルト、銅、ニッケル、鉄、マグネシウム、マンガン、陰イオン、特に塩化物、臭化物、ヨウ化物、チオシアネート、およびフェノール系化合物、特にフェノール、フェノール系防腐剤、レゾルシノール、4’−ヒドロキシアセトアニリド、4−ヒドロキシベンズアミドおよび2,7−ジヒドロキシナフタレンである。好ましくは、6量体安定化化合物は、フェノール、m−クレゾール、o−クレゾール、p−クレゾール、クロロクレゾール、メチルパラベン、カルシウム、塩化物、または2つ以上の当該化合物の組み合わせである。6量体安定化化合物はフェノール、m−クレゾール、カルシウム、塩化物、またはそれらの組み合わせであるのが更に好ましい。好ましくは、本発明の医薬組成物は、約1mMと75mMの間のカルシウム、約1mMと約50mMの間のカルシウム、約1mMと約25mMの間のカルシウム、約5mMと約50mMの間のカルシウム、約2.5mMと約30mMの間のカルシウム、約2.5mMと約15mMの間のカルシウム、約2.5mMと約10mMの間のカルシウム、約5mMと約30mMの間のカルシウム、約5mMと約15mMの間のカルシウムを含む。より好ましくは、本発明の医薬組成物は約2.5mMと10mMの間のカルシウムを含む。
【0058】
本発明の医薬組成物の多用途製剤は防腐剤を含んでいてもよい。「防腐剤」と言う用語は、医薬製剤に添加され、抗菌薬として作用する化合物を意味する。非経口製剤として有効で許容可能な、当業者に公知の防腐剤には、塩化ベンザルコニウム、ベンゼトニウム、クロロヘキシジン、フェノール、m−クレゾール、ベンジルアルコール、メチルパラベン、プロピルパラベン、クロロブタノール、o−クレゾール、p−クレゾール、クロロクレゾール、硝酸フェニル水銀、チメロサル、安息香酸、およびそれらの種々の混合物が含まれる。特定のフェノール系防腐剤、例えばメチルパラベン、フェノールおよびm−クレゾールは、インスリンおよびインスリン様化合物と結合し、物理的または化学的安定性あるいは両方の安定性を増大させる立体配座的変化を誘起することが知られている(例えば、Birnbaum,D.T.ら,Pharmaceutical.Res.14:25−36(1997);Rahuel−Clermont,S.ら,Biochemistry 36:5837−5845(1997)を参照)。「フェノール系防腐剤」には、フェノール、m−クレゾール、o−クレゾール、p−クレゾール、クロロクレゾール、メチルパラベンの化合物、およびそれらの混合物が含まれる。本発明のペグ化インスリンリスプロ化合物の製剤に使用される防腐剤は、フェノール系防腐剤であってもよいし、6量体安定化化合物と同じであっても異なっていてもよい。好ましくは、フェノール系防腐剤はm−クレゾールまたはフェノールである。本発明の医薬組成物は、約0.1mMから約75mMの濃度のフェノールおよび/またはm−クレゾールを、防腐剤として、約7.0から約8.0のpHで含んでいるのが更に好ましい。更に好ましいのは、本発明の医薬組成物が、約1mMから約60mMの濃度のフェノールおよび/またはm−クレゾールを、防腐剤として、約7.0から約8.0のpHで含んでいる場合である。更に好ましいのは、本発明の医薬組成物が、約10mMから約40mMの濃度のフェノールおよび/またはm−クレゾールを、防腐剤として、約7.0から約8.0のpHで含んでいる場合である。更に好ましいのは、本発明の医薬組成物が、約30mMの濃度のフェノールおよび/またはm−クレゾールを、約7.0から約8.0のpHで含んでいる場合である。最も好ましいのは、本発明の医薬組成物が、約30mMの濃度のフェノールおよび/またはm−クレゾールを、約7.3から約7.5のpHで含んでいる場合である。
【0059】
上述したように、本発明の医薬組成物は、インスリンの6量体化を促進あるいはインスリン化合物を安定化する二価の金属陽イオン、例えば亜鉛またはコバルトを含んでいてもよい。「二価の金属陽イオン」と言う用語は、複数の蛋白質分子と複合体を形成するために共有するイオンまたはイオン群を意味する。遷移金属、アルカリ金属およびアルカリ土類金属は、インスリン化合物と複合体を形成することが知られている金属の例である。遷移金属が好ましい。二価の金属陽イオンは、亜鉛、銅、コバルト、ニッケル、マンガン、マグネシウム、カドミウムおよび鉄からなる群から選択される1つ以上の陽イオンであるのが好ましい。二価の金属陽イオンが亜鉛であるのが更に好ましい。亜鉛は、インスリンおよびインスリンリスプロを含む種々のインスリン類似体および/または誘導体の6量体形成を促進することが知られている。本発明の医薬組成物に含まれる亜鉛やコバルトなどの二価の陽イオンの主要な役割は、本発明のペグ化インスリンリスプロ化合物および/または本発明のペグ化インスリンリスプロ化合物を含む医薬組成物に含まれる他のインスリンまたはインスリン類似体の6量体形成を促進することである。フェノール系防腐剤が存在する場合、本発明の医薬組成物を含む溶液のpHを亜鉛(II)イオンの存在下で中性領域に調整することにより、あるいはpHを中性領域に調整した後に亜鉛(II)を添加することにより、6量体形成を促進できる場合がある。亜鉛対インスリン化合物、インスリン類似体および/またはペグ化インスリンリスプロ化合物の比率は、下限として約0.33に固定するのが好ましい。すなわち、2つの亜鉛原子に対して1つのインスリン6量体、インスリン類似体6量体および/またはペグ化インスリンリスプロ6量体が好ましい。更に好ましいのは、亜鉛対インスリン化合物、インスリン類似体および/またはペグ化インスリンリスプロ化合物の比率が約0.33から約0.67の場合である。亜鉛結合で蛋白質と競合する化合物、例えばクエン酸塩やリン酸塩が存在する場合は、処理中に追加の亜鉛を使用してもよい。6量体化に必要な量を超える過剰の亜鉛(例えば、亜鉛対インスリン化合物、インスリン類似体および/またはペグ化インスリンリスプロ化合物の比率が約0.33から約0.83の場合)が、6量体化を更に促進するのに好ましい場合がある。6量体化に必要な量を超える過剰の亜鉛を本発明の医薬組成物に存在させてもよく、それが化学的および物理的安定性の改善、「懸濁性」の改善、および時間−作用の更なる拡張にとって好ましい場合がある。一方、クエン酸塩緩衝液またはリン酸塩緩衝液中の過剰の亜鉛は、それぞれクエン酸亜鉛またはリン酸亜鉛並びにインスリンの沈殿の原因となる場合がある。
【0060】
本発明によれば、インスリン、インスリン類似体およびペグ化インスリンリスプロ6量体1モル当たり約0.3モルから約3モルの亜鉛を製剤中に存在させてもよい。全インスリン、インスリン類似体およびペグ化インスリンリスプロの6量体1モル当たり約0.3モルから約1モルの亜鉛を本発明の医薬組成物中に存在させるのが、更に好ましい。更に好ましいのは、全インスリン、インスリン類似体およびペグ化インスリンリスプロの6量体1モル当たり約0.3モルから約0.7モルの亜鉛を本発明の医薬組成物中に存在させる場合である。最も好ましいのは、インスリン、インスリン類似体およびペグ化インスリンリスプロの6量体1モル当たり約0.3モルから約0.55モルの亜鉛を本発明の医薬組成物中に存在させる場合である。本発明に亜鉛を提供する亜鉛化合物は、製薬上許容可能な亜鉛化合物であればどれでもよい。インスリン製剤への亜鉛の添加は、製薬上許容可能な亜鉛源同様、当業者には公知である。亜鉛は塩として供給されるのが好ましい。例えば、硫酸亜鉛、塩化亜鉛、酢酸亜鉛、クエン酸亜鉛、酸化亜鉛、硝酸亜鉛などである。
【0061】
本発明の更なる実施形態では、本発明の医薬組成物は更に1つ以上の表面活性剤を含む。本明細書で使用する場合、「表面活性剤」と言う用語は、気相液相界面の吸着により液相の表面張力を減少させる試剤を含む。表面活性剤の例には、限定されないが、非イオン性の表面活性剤、例えば、ポリソルベート(ポリソルベート80やポリソルベート20など)、ポロキサマー(ポロキサマー188など)、トリトン(商標)(トリトン(商標)X−100など)、ポリエチルグリコール、ポリプロピルグリコール、エチレンとプロピレングリコールのコポリマー(プルロニックスやPF68など)が含まれる。例えば、本発明の医薬組成物には、約0.001−0.5%(例えば、0.05−0.3%)の表面活性剤が存在してもよい。本発明の医薬組成物に使用される表面活性剤はポロキサマー188であるのが好ましい。更に好ましいのは、表面活性剤がポロキサマー188で、その濃度が約0.5mg/mLと約10mg/mLの間、約1mg/mLと約10mg/mLの間、約2mg/mLと約10mg/mLの間、約3mg/mLと約10mg/mLの間、約4mg/mLと約10mg/mLの間、約1mg/mLと約5mg/mLの間、約2mg/mLと約5mg/mLの間、約3mg/mLと約5mg/mLの間、および約4mg/mLと約5mg/mLの間の場合である。
【0062】
本発明は、好ましくはヒトにおいて、低血糖症および/または糖尿病の治療に使用される、式Iのペグ化インスリンリスプロ化合物またはその製薬上許容可能な塩も提供する。
【0063】
本発明は、好ましくはヒトにおいて、低血糖症および/または糖尿病の治療用の医薬品の製造に使用される、式Iのペグ化インスリンリスプロ化合物またはその製薬上許容可能な塩も提供する。
【0064】
本発明のペグ化インスリンリスプロを含む医薬組成物は、それを必要とする患者に非経口的に投与してもよい。非経口投与は、注射器、任意にペンタイプの注射器または機械駆動の注入器を用いて、皮下、筋肉内または静脈内注射によって行ってもよい。あるいは、非経口投与は輸液ポンプを用いて行ってもよい。
【実施例】
【0065】
(調製1:GlyA1−HSCH2CH2COインスリンリスプロ(3)およびLysB28−HSCH2CH2COインスリンリスプロ(4))
Trt−SCH2CH2CO−OH、N−ヒドロキシスクシンイミド(NHS)およびジイソプロピルカルボジイミド(DIC)を1mmolずつDMF2mL中で30分混合して、Trt−SCH2CH2CO−NHSエステルを調製する。1/10mmolのインスリンリスプロを5%トリエチルアミン(TEA)のDMSO溶液10mLに溶解する。その溶液に、活性化したTrt−SCH2CH2CO−NHSを0.2mmol加える。室温で2時間反応させた後、反応を終了させるため、エタノールアミン0.2mLを添加する。次に反応混合物を90mLの水で希釈し、RP−C18カラムで精製する。目的のLysB28−Trt−SCH2CH2COインスリンリスプロ(2)の画分をプールし凍結乾燥する。それとは別に、目的のGlyA1−Trt−SCH2CH2COインスリンリスプロ(1)の画分をプールし凍結乾燥する。0.2mLのチオアニソールと0.4mLのトリイソプロピルシランを含むTFA5mLに、1/10mmolの(1)または(2)を溶解する。30分後、TFAを蒸発によって除去し、残留性のペプチドを10%ACNの水溶液50mLに移す。その結果得られる溶液をRP−C18カラムで精製する。目的の(3)または(4)の画分をプールし、任意に凍結乾燥する。
【0066】
(調製2:PheB1−HSCH2CH2COインスリンリスプロ(7))
1/10mmolのインスリンリスプロを5%TEAのDMSO溶液10mLに溶解する。その溶液に、ジ−tert−ブチルカーボネートのDMSO溶液0.2mmolを添加する。室温で1時間反応させた後、反応を終了させるため、エタノールアミン0.2mLを添加する。次に反応混合物を90mLの水で希釈し、RP−C18カラムで精製する。目的のBoc−GlyAlおよびBoc−LysB28インスリンリスプロの画分をプールし、凍結乾燥して(5)を得る。1/10mmolの(5)を5%TEAのDMSO溶液10mLに溶解する。その溶液に、活性化したTrt−SCH2CH2CO−NHSを0.2mmol加える。室温で2時間反応させた後、反応を終了させるため、エタノールアミン0.2mLを添加する。次に反応混合物を90mLの水で希釈し、RP−C18カラムで精製する。目的の画分をプールし凍結乾燥すると、Trt−SCH2CH2CO−PheB1、Boc−GlyA1およびBoc−LysB28インスリンリスプロ(6)が得られる。0.2mLのチオアニソールと0.4mLのトリイソプロピルシランを含むTFA5mLに、1/10mmolの(6)を溶解する。30分後、TFAを蒸発によって除去し、残留性のペプチドを10%ACNの水溶液50mLに移す。その結果得られる溶液をRP−C18カラムで精製する。目的の画分をプールし凍結乾燥すると、(7)が得られる。
【0067】
(実施例1:チオール誘導化インスリンリスプロ中間体のペグ化)
平均分子量約20kDa(b)、30kDa(a)、40kDa(a)または60kDa(c)を有するモノメトキシPEG−MALを100mMのNH4Ac緩衝液(pH4.69)とACNの1対1混合液に溶解する。チオール誘導化インスリンリスプロ(例えば、化合物(3)、(4)または(7))の凍結乾燥粉末を上記溶液に添加する。反応は分析用RP−HPLCを使ってモニターしてもよい。反応が完結(通常約4時間後)したら、混合物は水で希釈し、RP−HPLCカラムを使って精製する。目的の画分をプールし凍結乾燥すると、ペグ化インスリンリスプロ化合物が得られる。実施例1に記載の方法で調製した典型的なペグ化インスリンリスプロ化合物は、8(a)、8(b)、9(a)、9(b)、10(a)、10(b)および15(c)として以下に示してある。これらのペグ化インスリンリスプロ化合物は、約400から約1000の範囲のnを有しているのが好ましい。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約400から約750の範囲のnを有している場合である。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約400から約550の範囲のnを有している場合である。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約400から約500の範囲のnを有している場合である。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約450から約500の範囲のnを有している場合である。最も好ましいのは、これらのペグ化インスリンリスプロ化合物が約450のnを有している場合である。
【化3−1】
【0068】
【化3−2】
【0069】
【化3−3】
【0070】
(実施例2:モノメトキシポリエチレングリコールp−ニトロフェニルカルボネート(mPEG−NPC)を用いたインスリンリスプロのペグ化)
1/10mmolのインスリンリスプロを20mLの0.2Mホウ酸塩緩衝液にpH10.5で溶解し、ACN20mL中の平均分子量約20kDaを有するmPEG−NPC1.98gを激しく攪拌しながら上記溶液に添加する。反応はRP−HPLCおよびSECでモニターする。約4時間後、反応混合物を酢酸を使ってpH5−7に酸性化し、RP−HPLCカラムを使って精製する。目的の画分をプールし凍結乾燥すると、モノペグ化PEG20Kインスリンリスプロが20%から45%の範囲の収率で得られる。RP−HPLC、SECおよびMALDI−MSで同定し、純度を確認する。A鎖またはB鎖に結合するmPEGの割合は、得られたコンジュゲートをトリス(2−カルボキシエチル)ホスフィン塩酸塩(TCEP)で処理した後に放出される遊離のA鎖およびB鎖の領域積分によって決定する。mPEG−NPC対インスリンリスプロの比率は、モノペグ化種とジペグ化種の生成物分布を決定する。反応pHはペグ化の部位特異性を支配する。pHが約8から約12に上がると、化合物(11)が主要生成物になる。pH10.5で、約20kDaの平均分子量(nは約450)を有するmPEG−NPCを用いて反応を行うと、(11)対(12)の比率は約85対15になる。
【0071】
上記のペグ化反応は、0.2MのNaOHを連続添加して反応混合物のpHを維持することにより、非緩衝水溶液中で行うこともできる。pHを約11.5に維持しながら、約20kDaの平均分子量を有するmPEG−NPCを用いて、非緩衝水溶液中で反応を行わせると、ペグ化反応生成物は、(11)と(12)を約92対8の割合で含む。化合物(11)は、約400から約1000の範囲のnを有するのが好ましい。更に好ましいのは、化合物(11)が約400から約750の範囲のnを有する場合である。更に好ましいのは、化合物(11)が約400から約550の範囲のnを有する場合である。更に好ましいのは、化合物(11)が約400から約500の範囲のnを有する場合である。更に好ましいのは、化合物(11)が約450から約500の範囲のnを有する場合である。最も好ましいのは、化合物(11)が約450のnを有する場合である。
【化4】
【0072】
(実施例3:インビトロ受容体親和性)
受容体結合アッセイは、ヒトインスリン受容体(hIR)またはヒトIGF−1受容体(hIGF−1R)を過剰発現している、安定的に形質移入された293EBNA HEK細胞から調製したP1膜上で行われる。結合親和性は、ヒト組み換え(3−[125I]ヨードチロシルA14)インスリン(2000Ci/mmol)またはヒト組み換え[125I]インスリン様成長因子(2000Ci/mmol)のいずれかを用いて、競合放射性リガンド結合アッセイで決定する。アッセイは、PVT PEI処置タイプA麦芽アグルチニン結合SPAビーズを用いてSPA法で行った。全ての試薬調製にSPAアッセイ緩衝液(50mMトリスHCl、pH7.5、150mM NaCl、0.1%BSA)を用いた。化合物の3倍段階希釈(100nMから2pM)はFreedom/Evoロボット(Tecan)を用いてアッセイ緩衝液で調製し、マルチメク(Beckman Coulter)を用いて96ウェル白色透明ボトムマイクロプレート(Corning/Costar)に添加した。放射性リガンド、膜およびSPAビーズをマルチドロップ装置(Titertek)を使って添加する。室温で10時間インキュベーションした後、マイクロベータ・トリラックス・シンチレーション・カウンターを用いて放射線を測定する。非標識インスリンリスプロと非標識IGF−Iをそれぞれ陽性対照および陰性対照として各実験に含める。IC50値は4パラメーター非線形ロジスティック回帰分析により決定する。親和定数(Ki)は、Ki=IC50/(1+D/Kd)の式に基づいてIC50値から計算する。式中、Dは実験に使用される放射性リガンドの濃度に等しく、Kdは飽和結合解析で決定される放射性リガンドの平衡結合親和定数に等しい(hIRとhIGF−1RのKdはそれぞれ0.124nMおよび0.130nMである)。以下に報告する幾何平均Kiは10^(平均対数Ki)であり、式中、平均対数Ki=平均(Ki1+Ki2+Ki3...Kin)であり、独立実験の数(n)は3以上である。しかし、ヒトIGF−1に関して以下に記載する場合、nは2である。
【0073】
実施例1に記載する方法で調製した以下のペグ化インスリンリスプロ化合物は、上述のhIR結合アッセイにおいて30nM未満の幾何平均Kiを有している。そのペグ化インスリンリスプロ化合物とは、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物10(a)、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物8(a)、平均分子量約60kDaを有する分岐mPEG−MALを用いて調製した化合物15(c)、平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、および平均分子量20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)である。hIRおよびhIGFR結合アッセイでは、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)の幾何平均Kiは、それぞれ3.07nM+/−0.32nM(+/−S.E.M、n=6)および84.3nM超(SEM=未決定、n=6)である。更に、40kDa、30kDaまたは20kDaの線形mPEG−NPCを用いて実施例2に記載の方法で調製した異種ペグ化インスリンリスプロ生成物も、上述のhIR結合アッセイにおいて30未満の幾何平均Kiを有している。上述のhIR結合アッセイでは、インスリンリスプロの幾何平均Kiは0.22+/−0.072nM(+/−SEM、n=4)である。上述のhIGFR結合アッセイにおいては、上記の全化合物が75nMを超える幾何平均Kiを有しており、ヒトIGF−1の幾何平均Kiは1.51+/−0.23nM(+/−SEM、n=2)である。
【0074】
ペグ化位置B28はhIR親和性を約10倍減少させ、これらインスリンリスプロのペグ化種をhIRの弱いアゴニストにしていることを、このデータは示している。更に、これらペグ化インスリンリスプロ種は、上述の条件下のアッセイにおいて、測定可能なIGFR−1結合特性を全く有していない。
【0075】
(実施例4:インスリン受容体リン酸化全細胞アッセイを用いたペグ化インスリンリスプロの効力評価)
本発明のペグ化インスリンリスプロ化合物は、商業的に入手可能な異種時間分解蛍光測定アッセイであるDELFIA(登録商標)(Perkin−Elmer)を用いて、機能活性を評価してもよい。簡単に言えば、ヒトインスリン受容体を過剰表現している293HEK細胞をトリプシン処理し、無血清培地(SFM)(脂肪酸のない0.1%BSAを含むDMEM)を用いて、ポリDリジンコートCostar96ウェルハーフエリア組織培養プレートの60,000細胞/ウェルで平板培養する。その細胞培養プレートをCO2インキュベーター中37℃で一晩インキュベーションする。抗インスリン受容体A鎖mAb8314キャプチャープレートも、Costar96ウェル黒色ハーフエリアマイクロタイタープレートを使って前の晩に調製し、30μLの抗インスリン受容体A鎖mAb8314(Soos,M.A.ら,Biochem J 235:199−208(1986)、マサチューセッツ州ケンブリッジのアブカム社などから商業的に入手可能)を用いて4℃で一晩処理し、10mM炭酸ナトリウムで1μg/mLに希釈する。結合していないmAb8314を除去するため、mAb8314キャプチャープレートを50mMトリス、pH7.5、150mMのNaClおよび0.1%ツイーン(TBST)で4度洗浄する。次にTBST中の1%BSAを用いて4℃で1時間を超える間、mAb8314キャプチャープレートをブロックする。ブロッキング後、キャプチャープレートをTBSTで2度洗浄して過剰のBSA溶液を除去する。キャプチャープレートをブロッキング緩衝液に浸漬したら、細胞培養プレートをインキュベーターから取り出し、温度を室温と同じにする。試験化合物をSFMで連続的に希釈する。インスリン受容体の自己リン酸化を刺激するため、希釈したテスト試薬50μLを細胞単層に加える。室温で30分反応させた後、試験化合物を吸引除去し、50μLの2x溶解緩衝液(2%NP40、100mMトリス、pH7.4、300mMのNaCl、EDTAを含むRoche Complete(商標)プロテアーゼインヒビターおよび4mMバナジン酸塩)を添加することにより、反応を停止させる。溶解緩衝液中、室温で30分反応させた後、30μLの溶解物を30μLのユウロピウムN1抗ホスホチロシンPY20抗体であるEu−N1−Py20(Perkin Elmer)を含むブロックされたキャプチャープレートに移し、10mMのヘペス緩衝液、140mMのNaClおよび0.1%ツイーンで50ng/mLに希釈する。この混合液を室温で1時間インキュベーションし、その後TBSTで6度洗浄して、結合しなかったEu−N1−Py20および細胞分解物を除去する。その後10分間、発生するシグナルに合わせて間歇的に振盪しながら、50μLのEnhancement Solution(Perkin Elmer)でインキュベーションする。リン酸化したインスリン受容体は、時間分解蛍光測定ユウロピウム設定でWallac Victorを用いて定量化する。リン酸化レベルはインスリン(100nM)の最大刺激用量に対する応答%として計算される。インスリン類似体の効力は、用量応答の4パラメーター適合を用いて、EC50用量として計算される。実施例4に記載されるアッセイにおいて、実施例1に記載される方法で調製した、15nM未満のEC50を有するペグ化インスリンリスプロ化合物には、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物10(a)、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)および平均分子量約20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)が含まれる。実施例4に記載されるアッセイでは、線形mPEG−MALを用いて調製した化合物9(a)のEC50は10.88nMである。約40kDa、約30kDaまたは約20kDaの平均分子量を有する線形mPEG−NPCを用いて実施例2に記載の方法で調製した異種ペグ化インスリンリスプロ生成物も、実施例4に記載のアッセイにおいて15nM未満のEC50を有している。約40kDaの平均分子量を有する線形mPEG−MALを使って調製した化合物8(a)と約40kDaの平均分子量を有する線形mPEG−MALを使って調製した化合物9(a)の50対50および70対30の混合物も、実施例4に記載のアッセイにおいて15nM未満のEC50を有している。同じアッセイにおいて、ヒトインスリンとインスリンリスプロのEC50はそれぞれ2.3nMおよび0.7nMである。
【0076】
ペグ化位置B28はhIRインビトロ活性を約10倍から20倍減少させ、これらインスリンリスプロのペグ化種をhIRの弱いアゴニストにしていることを、このデータは示している。更に、これらペグ化インスリンリスプロ種は、測定可能なIGFR−1結合特性を全く有していない。
【0077】
(実施例5:1型糖尿病ラットモデルを用いた、ペグ化インスリンリスプロのインビボ効力および薬物動態学的プロフィルの評価)
試験開始3日前に、10週目のHarlan Sprague−Dawley雄ラット(Harlan,Indianapolis)(体重250−280g)の尾静脈に0.5Mクエン酸(pH4.5)中のストレプトゾトシン(STZ)45mg/kgを静脈内投与する。試験開始時に、ラットを体重および血糖に基づいて群分けする。この研究には、血糖が400mg/dLと550mg/dLの間のラットだけを含める。試験開始日の朝、試験化合物をいくつかの所定用量の1つでラットに単回皮下注射する。尾静脈から定期的に血液の重複抜き取りを行い、EDTA二ナトリウムを含むチューブに集める。血糖値をグルコメーターで測定する。静脈血試料から血漿も集め、商業用のラットインスリン・ラジオイムノアッセイを用いて、投与された薬物の血漿濃度を測定する。経時的血糖(mg*h/dL)曲線の下の面積を各ラットについて計算し、それを4パラメーターロジスティック回帰分析に使用してED50を決定する。このアッセイでは、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)および平均分子量約20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)の効力(ED50)は、典型的な用量応答曲線に基づいて、それぞれ241nmol/kg、138nmol/kgおよび69nmol/kgである。更に、当該化合物は、568nmol/kgの単回皮下注射によって、STZ処置ラットの血糖値を少なくとも36時間、この種のラットにとっては正常なレベル(100mg/dL以下)にまで低下させることができた。一方、インスリンデテミルは、上記のエッセイにおいて、568nmol/kg単回投与によって血糖値を5−6時間正常化する。
【0078】
薬価パラメーターの評価に加えて、ラットにおける試験化合物の平均薬物動態パラメーター値も重複血液試料を用いて測定する。薬物動態パラメーターは、モデル非依存性の方法(WinNonlin Pro)を用いて計算する。得られる薬物動態パラメーター値は、用量の関数として非線形を示す。報告される値の範囲は、テストされる最高用量(568nmol/kg)と最低用量(5.6nmol/kg)の間の薬物動態パラメーター値に対応する。
【0079】
平均分子量約20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)の薬物動態的結果は、最高濃度に至るまでの時間(T最大)が6時間から12時間の範囲、見かけのクリアレンス速度(CL/F)が0.05L/h/kgから0.14L/h/kgの範囲、見かけの分配量(V/F)が0.6L/kgから7.2L/kgの範囲、除去半減期(t1/2)が8.5時間から34.5時間を示した。
【0080】
平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)の薬物動態的結果は、T最大が12時間、CL/Fが0.05L/h/kgから0.13L/h/kgの範囲、V/Fが0.6L/kgから2.0L/kgの範囲、t1/2が8.3時間から11.0時間を示した。
【0081】
平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)の薬物動態的結果は、T最大が12時間から24時間の範囲、CL/Fが0.06L/h/kgから0.2L/h/kgの範囲、V/Fが1.0L/kgから7.5L/kgの範囲、t1/2が11.1時間から48.5時間を示した。
【0082】
568nmol/kgのインスリンリスプロをSTZ処置雄ラットに同様にして投与すると、測定値は、t1/2が約1時間、CL/Fが約1.2L/h/kgである。
【0083】
18.9nmol/kgから568nmol/kgの範囲のインスリンデテミルをSTZ処置雄ラットに同様にして投与すると、t1/2が1.9時間から3.1時間の範囲、CL/Fが約0.8L/h/kgから1.7L/h/kgの範囲が観察される。
【0084】
デテミルと例示のペグ化インスリンリスプロ化合物の間の見かけのCL比は、薬物動態学的な複雑性の故に、測定に使用される用量によって結果が異なる。しかし、20kDaまたは40kDaのPEGと結合した例示のペグ化インスリンリスプロ化合物は、STZ誘発糖尿病ラットにおいて、見かけのクリアランスがデテミルよりも約5倍から30倍緩慢であることを、実施例5に記載の試験が示している。
【0085】
(実施例6:2型糖尿病ラットモデルを用いた、ペグ化インスリンリスプロのインビボ作用期間および薬物動態学的特性の評価)
40kDa線形PEGを有する化合物9(a)のグルコダイナミック(Glucodynamic)活性は、ビヒクル対照(PBS)または平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)517nmol/kgの単回皮下投与後、雄のZDF(fa/fa)ラット(n=4ラット/群)で評価する。薬物動態特性および薬価特性を評価するため、試料を連続して集める。平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)517nmol/kgを雄のZDF(fa/fa)ラットに単回皮下投与することにより、血糖値が統計学的に有意なレベルまで低下し、それが少なくとも7日間持続した(偽薬に対してp<0.05)。平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)への曝露も7日間検証可能である。
【0086】
(実施例7:コバルトイオンの存在下でフェノールを含むPEG−B28(92.4%)A1(7.6%)インスリンリスプロ、インスリンリスプロまたはその混合物(70%PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロ対30%インスリンリスプロ)のフェノール系防腐剤滴定)
PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロまたはインスリンリスプロを、20mMのKSCNおよび50mMのトリスClO4を含む溶液にpH8.0で溶解する。上記蛋白質の標的濃度は、蛋白質含有量(ε280=1.05(mg/mL)−1cm−1)に基づいて約4mg/mLである。塩化コバルト(41.9mg)を水1mLで溶解し、コバルトイオン濃度0.176Mの原液を得る。コバルトイオン対インスリン6量体の最終モル比が4になるように、コバルト原液のアリコート(蛋白質の濃度によって異なるが、約2μL)を0.8mLの蛋白質溶液に添加する。6量体化を評価するため、コバルト配位化学における歪みをフェノール系防腐剤濃度の関数として574nmでモニターした。具体的には、滴定終了時点での最終容量が0.84mLを超えないように、濃縮フェノール溶液(0.564M)をマイクロリットルアリコート用いて、蛋白質溶液0.8mLを滴定する。フェノールの各アリコートと400nmから800nmまでの可視スペクトルの溶液を集めた後、最終溶液を最低20分間攪拌する。574nmで記録した吸光度は、HISB10配位結合コバルトモル濃度、すなわちインスリン6量体のHISB10部分は二価の金属イオン2つと配位結合するという知識に基づいて6量体蛋白質濃度掛ける2でその吸光度を割ることによって、モル吸光係数に変換する。PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロとインスリンリスプロの70/30(モル対モル)混合製剤を調製するには、蛋白質をまず混合し、次にコバルトを添加し、それからフェノール系防腐剤で滴定する。
【0087】
インスリンリスプロでは、天然の配列であるB28の位置のプロリンとB29の位置のリジンが、野生型のヒトインスリンと比較して逆転している。この逆転によりB鎖のC末端で立体配座的な移動が生じ、その結果、2量体を形成するリスプロインスリン単量体の能力が立体的に妨害される。従って、2量体会合定数が野生型ヒトインスリンの2量体会合定数と比べて300倍減少する。驚いたことにかつ予想外にも、表1に示されるように、野生型のヒトインスリンと比べて既に脆弱化しているインスリンリスプロの2量体化ドメインの近くで6つの20kDaPEG部分が結合しているにもかかわらず、PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロは、Humalog(登録商標)で使用される調剤条件と類似の条件で、二価の金属イオンおよびフェノール系防腐剤の存在下、6量体の複合体として会合することを結果が示している。更に、PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロとインスリンリスプロの70/30混合物も6量体の複合体を形成する能力が証明されており、基礎インスリンおよび迅速に作用するインスリンの予備混合製剤を即座に、および/または安定して調製できることをこの事実は支持している。
【0088】
(表I)
【表1−1】
【0089】
【表1−2】
【0090】
(実施例8:亜鉛、フェノールおよび/またはカルシウムと共に製剤されたPEG20kDaB28(92.4%)/A1(7.6%)インスリンリスプロの6量体状態の分析)
インスリンおよび特定のインスリン類似体の化学的貯蔵安定性および使用時安定性は、溶液中で個別の6量体複合体を形成する能力によって促進される。二価の金属イオン(Zn+2またはCo+2)およびフェノール系防腐剤(フェノールまたはm−クレゾール)の存在下でインスリンまたはインスリンリスプロを6量体化する能力は、AsnA21の脱アミド化を遅らせ、その結果、高分子量の粒子(HMWP)の形成を最小限に抑える。
【0091】
PEG20kDa−LysB28インスリンリスプロが6量体を形成する能力を評価するため、A276nm=8.6mgインスリンリスプロ/mLまたは38.2mgペグ化インスリンリスプロコンジュゲート/mLに基づく最初の蛋白質濃度を有する、PEG20kDaB28(92.4%)/A1(7.6%)インスリンリスプロをpH6.7で一晩、水中で透析する。次に、透析した蛋白質を水で希釈して、蛋白質濃度を4.6mg/mLまたは20.6mgのペグ化蛋白質コンジュゲート/mLに調整する。4x緩衝液原液をpH7.0で調製する。リン酸塩緩衝液の最終濃度は40mM、m−クレゾールの最終濃度は12.8mg/mLとする。酸化亜鉛原液は酸化亜鉛を1NのHCl0.5mLに溶解し、次に水で希釈することにより調製し、最終亜鉛濃度を0.097Mとする。蛋白質濃度4.6mg/mLのPEG20kDaB28(92.4%)/A1(7.6%)インスリン450μLを亜鉛原液のアリコート(添加される亜鉛原液の最大全容量=2.5μL、またはPEG20kDaB28(92.4%)/A1(7.6%)インスリン6量体当たり4亜鉛イオンに相当する量)および4xリン酸塩緩衝液原液150μLと混合することにより、種々の亜鉛濃度(0μMから400μM)を有する、近UV円偏光二色性(CD)分析用の溶液試料を調製する。最終pHは、必要に応じてpH約7.0に調整する。PEG20kDaB28(96%)/A1(4%)インスリンリスプロまたはインスリンリスプロの6量体会合を、0.2cmセルを用いて、近UV円偏光二色性により、ジスルフィド変化に感応性の高い250nmでモニターする。平均残基楕円率をモル6量体当たりのモル亜鉛の比率に対してプロットする。
【0092】
驚いたことにかつ予想外にも、表IIに示されるように、野生型のヒトインスリンと比べて既に脆弱化しているインスリンリスプロの2量体化ドメインの近くで6つの20kDaPEG部分が結合しているにもかかわらず、PEG20kDaB28(92.4%)/A1(7.6%)インスリンは、Humalog(登録商標)の調剤条件と類似の条件で、二価の金属イオンおよびフェノール系防腐剤の存在下、6量体の複合体として会合することを結果が更に示している。
【0093】
6量体促進テスト製剤において、6量体化およびリガンド結合がPEG20kDaB28(92.4%)/A1(7.6%)インスリンの熱安定性に及ぼす影響についても、CD熱変性実験で調べた。全体的な信号変化はZn2+結合試験で使用した250nmよりも240nmの方が大きかったが、全溶液吸光度が高品質のCDデータが得られるほど十分低かったので、熱変性試験で用いた波長は240nmであった。1mmのキュベット中、240nmにおける熱スキャンデータを5℃から約95℃の間で集めた(最終温度は試料間でわずかな違いがあった)。スキャン速度およびデータピッチは1℃/分、周波数帯域幅は1.5nm、応答時間は8秒であった。熱変性データは、生信号(240nmにおける1000分の1度単位)およびFunf(T)=[Yobs(T)−Ynat(T)]/[Yunf(T)−Ynat(T)]の式で与えられる見かけの変性割合(Funf)の両方としてプロットしてよい。式中、Yobs(T)は温度の関数として観察される信号であり、Ynat(T)およびYunf(T)は、それぞれ未変性ベースラインおよび変性ベースラインの直線的外挿値である。変性開始温度は、Funfが未変性ベースライン(Funf=0)から増加し始める温度として定義され、中間温度はFunf=0.5の温度である。
【0094】
本質的に上述の方法で行われるCD熱変性実験が示すところによれば、PEG20kDaB28(92.4%)/A1(7.6%)インスリン6量体とインスリンリスプロ6量体を16mMトリス、pH7.2、3.1mg/mLのm−クレゾールおよび亜鉛で同様に製剤した場合、前者の熱安定性は後者の熱安定性よりも実質的に減少した。更に、上述のCD熱変性試験は、PEG20kDaB28(92.4%)/A1(7.6%)インスリンリスプロの融点がカルシウムと塩化物イオンの濃度に依存して有意に上昇することも見出した(データ省略)。具体的には、変性開始温度は16mMトリス、pH7.2、3.1mg/mLのm−クレゾールおよび亜鉛中では約30℃であるが、75mM塩化カルシウムを含む同じ製剤中では劇的に約50℃まで上昇する。製剤に塩化カルシウムの代わりにNaCl(25mMから150mMのNaCl)を加えた場合も同様の効果が観察された。従って、PEG20kDaB28インスリンリスプロ化合物を含む医薬組成物において、カルシウムおよび/または塩化物は非常に有効な6量体促進賦形剤であり、貯蔵中に当該医薬組成物の化学的および/または物理的安定性を増大する効果があろう。
【0095】
(表II:亜鉛/6量体比の関数としての平均残基楕円率(MRE)変動)
【表2】
【0096】
(実施例9:PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロの生成)
150mMの第二リン酸ナトリウムおよび50mMのEDTAを含む緩衝液を約4℃から約6℃で150mMの第三リン酸ナトリウムと混合し、10.85と11.10の間のpHを持つ緩衝液を得る。インスリンリスプロ結晶(30−60mg/mL)を約4℃から約6℃で緩衝液に徐々に添加し、結晶溶解中に凝集物の形成が生じるのを避けるため穏和に攪拌する。
【0097】
重量平均分子量約20kDa+/−約2kDaのPEGを有するモノメトキシポリエチレングリコールp−ニトロフェニルカーボネート(mPEG−NPC)を60mg/mLから120mg/mLの濃度で、冷水(4−6℃)に溶解する。その場合、必要量の冷水を容器に入れ、攪拌して渦流を作り、適切かつ急速な分散が起きるように、mPEG−NPCを渦流の眼に徐々に注ぐ。mPEG−NPC粉末は細かい粉末であり、分散させるとかなりの量の気泡が容器中に放出される。容器中のmPEG−NPC溶液は、容量に応じて30分間から60分間脱泡する。
【0098】
上記の方法で調製したインスリンリスプロ溶液は機械的攪拌用の冷却塔付き容器に移す。その容器には温度とpH測定用の装置が備わっている。攪拌は、レイノルズ数に基づいて乱流部分で操作される標準回転翼を用いて行う。mPEG−NPC(PEG)溶液は、全PEG添加時間が3時間から5時間になる速度で、容器中で測定する。冷却塔の温度は4℃から6℃に維持し、混合は継続して行う。反応液のpHは、必要量の150mM第三リン酸ナトリウムを添加することにより、10.85から11.10の間に維持する。PEG対インスリンリスプロの最終モル比が2.5から4.5の範囲になるまで、PEGを添加する。
【0099】
PEG添加の終了時点で、冷却塔の温度を60分以内に25℃から30℃に上昇させ、pHを約10.7から約11.0に維持しながら、反応混合物をその温度で約3時間から約6時間インキュベーションする。インキュベーション期間の終了時点で、2x緩衝液(100mM酢酸/酢酸ナトリウム、pH4.0)を添加することにより反応混合物をクエンチし、同じ2X緩衝液で希釈することにより伝導率(2.5mS/cm)および濃度(3−5mg/mL)を調節する。
【0100】
反応混合物は、適切な樹脂(高速流通SPセファロース樹脂など)を充填した陽イオン交換クロマトグラフィ(CEX)カラムを使って精製する。樹脂は層高約15cmから約30cmになるまでカラムに充填し、100mMの酢酸ナトリウム(緩衝液A)で平衡化し、希釈した反応混合物(生成物5−8g/樹脂L)を低いpH(約2.5から約4.0)かつ適切な流速(約50cm/時間から約90cm/時間)で充填する。モノペグ化生成物および未反応の蛋白質が樹脂上で選択的に結合するが、マルチペグ化した副生成物および過剰の試薬のほとんどはカラムを通過する。希釈した塩濃度(20−30mM)の緩衝液Aを用いて弱く吸着したマルチペグ化副生成物を洗い流し、次に塩濃度の高い(50−70mM)緩衝液(8−12CV)を使った勾配溶出によってペグ化生成物を樹脂から優先的に除去すると同時に、未反応の蛋白質はカラムに残ったままになる。生成物を集め(3−5CV)、カラムは塩濃度の高い(100mM)緩衝液Aで洗浄して未反応の蛋白質を除去する。
【0101】
3−5mg/mLのCEXカラム主流(3−5CV)は、接線流濾過により、標準のフラットシート膜(3−5kDa分子量カットオフ)を用いて40−80mg/mLまで濃縮する。この工程は、初期濃度を経て、緩衝液を交換し、目的の最終濃度まで行われる。操作フラックスは、工程中ずっと10−20L/m2(フィルター面積)/時間(LMH)に維持し、膜間差圧(TMP)は約15psiから約35psiに維持する。
【0102】
最終濃度のバルク活性医薬成分溶液は、適切な温度(−20℃から−70℃)で凍結させ、適切な温度(−20℃から−70℃)で保存する。
【0103】
(実施例10:イヌにおけるペグ化インスリンリスプロの薬物動態学的プロフィル)
2歳から4歳の雌のビーグル犬(体重7−10kg)に、例示の試験化合物18.9nmol/kgを皮下投与する。血液試料を定期的に腕頭静脈または伏在静脈から得て、それをEDTA二ナトリウムを含むチューブに集める。血漿を静脈血サンプリングで集め、商業用のインスリンラジオイムノアッセイを用いて血漿中の投与薬の濃度を測定する。本質的に実施例2に記載した方法で調製した例示化合物である、平均分子量約40kDa、約30kDaおよび約20kDaを有する線形mPEG−NPCを用いて調製した化合物11の各々について、薬物動態学的プロフィルおよびパラメーターを決定した。
【0104】
薬物動態学的パラメーターは、モデル非依存性の方法(WinNonlin Pro)を用いて計算した。平均分子量約20kDaを有する線形mPEG−NPCを用いて調製した化合物11は、最高濃度に至るまでの時間(T最大)が約12時間、見かけのクリアレンス速度(CL/F)が約0.046L/h/kg、最大濃度(C最大)が約14nM、除去半減期(t1/2)が約14時間を示した。
【0105】
平均分子量約30kDaを有する線形mPEG−NPCを用いて調製した化合物11は、T最大が約24時間、CL/Fが約0.027L/h/kg、C最大が約18nM、t1/2が約23時間を示した。
【0106】
平均分子量約40kDaを有する線形mPEG−NPCを用いて調製した化合物11は、T最大が約24時間、CL/Fが約0.026L/h/kg、C最大が約15nM、t1/2が約20時間を示した。
【0107】
インスリンデテミルも同様に18.9nmol/kgを雌のビーグル犬に投与し、T最大が約1.3時間、CL/Fが約0.12L/h/kg、C最大が約23nM、t1/2が約3.5時間を示した。
【0108】
表IIIはイヌにおける比較パラメーターを示す。この試験に使用したインスリン特異的なRIAは、ペグ化インスリンリスプロと内生的インスリンの両方を検出する。
【0109】
(表III:イヌにおけるペグ化インスリンリスプロのPKパラメーター)
【表3】
18.9nmol/kg皮下投与、n=3/群
【0110】
(実施例11:ヒトにおける平均「平坦化」および用量の推定)
改善された基礎インスリン療法の主要な基準は、患者への1日1回の投与に適した、真に平坦なプロフィルを達成する能力である。十分な平坦さはピーク/トラフ(PT)比が2未満の場合であると定義される。比較目的で述べると、公表されているPKプロフィルに基づいて計算したPT比は、デテミルで約4から9、グラルギンで約1.2から2.6である。18.9nmol/kgの例示化合物および同量のインスリンデテミルに関するラットとイヌのPKデータ(それぞれ実施例5および10を参照)は1−CMT PKモデルに適合させ、ka、CL/FおよびV/Fのパラメーターで表示した。次に各PKパラメーターを相対成長式P=aBWbに適合させる。式中、bはka、CL/FおよびV/Fに関して各々−0.25、0.75および1に固定し、aは適合パラメーターである。ヒト平均推定値を各PKパラメーターについて取得し、ヒトに1日量を投与後、シミュレーションは平均プロフィルを生成した。30kDaおよび40kDaペグ化インスリンリスプロコンジュゲートは類似しているので、シミュレーションは20kDaペグ化インスリンリスプロ、40kDaペグ化インスリンリスプロおよびインスリンデテミルだけを示してある。20kDaおよび40kDaペグ化インスリンリスプロ化合物のピーク/トラフ(PT)比は、インスリンデテミルのピーク/トラフ(PT)比よりも劇的に平坦であることを、生成されたシミュレーションが示している。図1はシミュレーションの結果を示す。
【0111】
効能に要求されるペグ化インスリンリスプロのヒト用量を推定するストラテジーは、ラットモデルにおけるペグ化インスリンリスプロとインスリンデテミルとの間の相対効力が臨床における相対効力と類似しているという推定の下に、既知の臨床コンパレーター(インスリンデテミル)をラット効能モデルの内部対照として用いることである。臨床におけるペグ化インスリンリスプロの1日必要量は、以下の式を用いて計算できる。
【数1】
表IVは各ペグ化インスリンリスプロコンジュゲートの相対効力比を示してある。表Vは、ラット、イヌおよびヒト(推定)における例示ペグ化インスリンリスプロ対デテミルの見かけの相対クリアランス比を示してある。
【0112】
(表IV:ラットにおけるペグ化インスリンリスプロコンジュゲートおよびインスリンデテミルの相対濃度ベース効力)
【表4】
【0113】
(表V:インスリンデテミル対ペグ化インスリンリスプロ化合物の相対CL/F比)
【表5】
【0114】
表IVの相対効力推定および表Vの相対CL/F推定を用いると、ヒトにおけるペグ化インスリンリスプロコンジュゲートの平均用量推定値は、20kDaペグ化インスリンリスプロ化合物と40kDaペグ化インスリンリスプロ化合物に関して、それぞれ4.2nmol/kgおよび6.9nmol/kgである。かかる推定値は、2型糖尿病患者に関してデテミルのラベルに記載されている臨床平均1日量18.5nmol/kgのインスリンデテミルに基づいている。時間作用および効力の両方を考慮した場合、臨床最大平均投与推定量はインスリンデテミルよりも約3倍低い。最善のケースでは、20kDaペグ化インスリンリスプロコンジュゲート、30kDaペグ化インスリンリスプロコンジュゲートおよび40kDaペグ化インスリンリスプロコンジュゲートの臨床平均投与推定量は、インスリンデテミルよりも約20倍および約45倍低い。
【0115】
(実施例12:PEG20kDaB28(約95%以上)/A1(約5%以下)インスリンリスプロおよびグラルギンを健康なボランティアに単回投与した後のグルコース注入速度)
本質的に実施例9に記載の方法で調製したPEG20kDaB28(約95%以上)/A1(約5%以下)インスリンリスプロ(LY)の単回用量を用いて、3部分からなる最初のヒト用量試験を行った。パートAは3つの試験期間からなり、第1期間で被験者にはLY用量が皮下(SC)注射され、第2期間でインスリングラルギン用量(0.5U/kg)が注射され、第3期間で別のLY用量が注射された。パートBは非盲検、単回用量、2回の反復期間試験で、被験者には24時間および36時間のグルコースクランプ法を用いて、両期間ともLYの単回用量が投与された。パートCは非盲検、2期間、単回用量、一定のシーケンス、コンパレーター制御試験で、第1期間で被験者にはLYのSC用量0.5mg/kgが単回投与され、第2期間ではインスリングラルギンのSC用量0.8U/kgが単回投与された。被験者にはパートAとパートCの各期間で24時間のグルコースクランプ法が施され、パートBの各期間ではより長い期間(36時間まで)グルコースクランプ法が施された。パートA、BおよびCにおいて、LYは以下のような単回用量が皮下投与された。
パートA用量:0.0025、0.0125、0.075、0.325mg/体重kg
パートB用量:0.15、0.225mg/kg
パートC用量:0.5mg/kg
【0116】
被験者には、ペグ化インスリンリスプロ化合物の単回用量またはコンパレーターとしてインスリングラルギンの単回用量(0.5U/kg)、そして第3期間にLYの別の単回用量が投与される。全ての治療期間で、各インスリン化合物の注射後、被験者にはオイグリセミッククランプ法を最高24/36時間施す。インスリン作用のグルコダイナミック(GD)尺度を提供する立証済みの経時的GIRを参考にして、グルコース注入速度(GIR)を正常血糖を維持するように調節する。オイグリセミックグルコースクランプ法の狙いは、インスリン化合物の用量を投与後、グルコース注入期間中ずっと、正常血糖が維持されることである。ここでは、内因性のインスリン分泌および肝グルコース産生が最少であり、グルコーススペースから輸送されるグルコース(すなわち代謝されたグルコース)は全て投与された外因性インスリンの直接の結果であることが想定されている。この場合のGIRは、経時的なインスリン作用のGD尺度となる。グルコースクランプ試験は全て、一晩(約8時間の)絶食後行われる。試験日の朝、容量ポンプの制御下で20%デキストロース溶液(緩衝液でpHを中性近くに調整)を投与する目的で、小さいカテーテルを1本の腕(理想的には肘窩)の静脈に挿入する。静脈グルコース採取のため、別のカテーテルを理想的には手首または手に挿入する。動脈血化した静脈血を採取するため、その部分を加熱装置で約55−60℃に加熱する。血液試料をベッドサイドで採取し、自動グルコースオキシダーゼ法を用いて直ちに全血中グルコース濃度を測定する。基礎血液採取および約30分間の安定期間の後、各被験者にインスリン化合物の用量が皮下投与される。インスリン化合物の皮下投与開始を時間ゼロと定義する。インスリン投与完了後、血糖を測定するために頻繁に血液を採取しながら、最高24時間または36時間正常血糖を維持するため、グルコースを種々の速度で静脈内に注入する。
【0117】
血液採取は、投与前約30分間約10分ごとに行い、投与後最初の2時間、5−10分ずつ継続(任意に、2.5分ずつの頻度で採取してもよい)し、その後クランプの最後まで10−30分間隔に頻度を下げる。
【0118】
グルコースクランプ中、グルコース注入速度を調節して、個々の被験者の所定標的血糖濃度を維持する。標的濃度は空腹時血糖値に近い値が好ましい。グルコースクランプの狙いは、投与前標的値(空腹時平均血糖値よりも5mg/dL下回る値として定義される)の+5%内に血糖濃度を維持することである。従って、GIRは変動しても、血糖濃度は一定に保たれる。だから、変動するグルコース注入速度は、試験インスリン化合物の活性を反映している。試料の血糖値および注入速度がクランプ中変動することは立証済みである。
【0119】
本質的に実施例12に記載の方法で行われた試験は、LYが理想的な「基礎」インスリンであり、作用期間が長く、見かけの半減期が24時間と44時間の範囲にあり、基礎特性すなわちピーク/トロフ比が2未満(図2)であるという特徴を有することを、ヒトにおいて実証した。更に、LYの作用期間はインスリングラルギンの作用期間よりも長い(図2)。グルコダイナミックスにおける被験者内変動は30%未満(データ省略)であり、これはグラルギンと同等またはそれよりも優れている。最後に、試験のパートC(0.5mg/kg)からのグルコダイナミック(GD)データは、LYのGIRプロフィルを「ピークなし」と示し、36時間を超える期間GDを維持し、グラルギンのピークGIR応答(0.5U/kg、データ省略)を凌駕した。
【0120】
(実施例13:PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ医薬組成物の化学的および物理的安定性)
実施例7および8で上述したように、PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロは、Humalog(登録商標)と類似の製剤条件において、二価の金属イオンおよびフェノール系防腐剤の存在下、6量体複合体として会合できる。従って、インスリンリスプロの商業用溶液製剤(すなわちHumalog(登録商標)溶液製剤)と類似のPEG20kDaB28インスリンリスプロ製剤に関して、化学的安定試験および物理的安定試験が行われた。試験対象の医薬製剤の化学的安定性は、指示された貯蔵期間中、様々な温度において、最初の時点と有意に異なる変化が種々の分析特性で検出されない場合、許容可能であると見なされる。試験対象の医薬製剤の物理的安定性は、視覚評価において粒子が全く観察されず、チオフラビンT蛍光顕微鏡検査法による検査でフィブリルやゲル形成が観察されない場合に、許容可能であると見なされる。
【0121】
PEG20kDaB28インスリンリスプロ1モル当たり0.5モルの亜鉛、16mg/mLのグリセリンおよび3.15mg/mLのm−クレゾール(リン酸塩緩衝液またはクエン酸塩緩衝液でそれぞれpH7.0またはpH6.5に調整)を含むPEG20kDaB28インスリンリスプロ製剤を調製し、化学的安定性および物理的安定性の両方を試験した。安定性への亜鉛の影響を評価するため、亜鉛を含まない類似の製剤も試験した。35℃の試料は約1ヶ月間貯蔵後ゲル粒子を形成し、25℃の試料は約2ヶ月の貯蔵後有意な数の粒子および気泡を形成した。視覚的評価ではクエン酸塩緩衝液の試料の方がリン酸塩緩衝液の試料よりもひどいように見えたが、クエン酸塩緩衝液とリン酸塩緩衝液の両方並びに亜鉛を含む製剤と亜鉛を含まない製剤の両方が、35℃の加速条件で類似の化学的/物理的安定性を示した。インスリンリスプロ対照製剤は透明であった。クエン酸塩緩衝液とリン酸塩緩衝液の両方が、5℃で少なくとも13ヶ月間、許容可能な化学的安定性を示した。
【0122】
その後の調査では、フェノール系防腐剤であるm−クレゾールは、高温(>25℃)に曝されたPEG20kDaB28インスリンリスプロと組み合わせた場合、ゲル化を促進することが示された。興味深いことに、m−クレゾールをmPEGだけ(活性化しているか否かに関係なく)に添加した場合、高温に曝しても、ゲル粒子は生じなかった。
【0123】
インスリンリスプロの基本的調剤方法では、高温でPEG20kDaB28インスリンリスプロ化合物に許容可能な安定性が与えられなかったので、PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ化合物を含み、改善された化学的および物理的安定性を示し、非経口的に投与される医薬組成物として商業化に適した医薬組成物を開発した。
【0124】
PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ(15mg/mL)の以下の製剤は、40℃で1週間、30℃で1ヶ月、25℃で3ヶ月および5℃で8ヶ月以上、化学的および物理的に許容可能な安定性を示した。
1)16mMのトリス緩衝液、pH7.0−8.0、10mMの塩化カルシウム、20mg/mLの砂糖(ショ糖またはトレハロース)、3mg/mL(28mM)のm−クレゾールおよびPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ1.0モル当たり0.5モルの亜鉛。
2)16mMのトリス緩衝液、pH7.0−8.0、10mMの塩化カルシウム、3mg/mLのポロキサマー、3mg/mL(28mM)のm−クレゾールおよびPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ1.0モル当たり0.5モルの亜鉛。
3)5mMのリン酸塩緩衝液、pH7.0、130mMのグリセリン、3mg/mL(28mM)のm−クレゾール、3mg/mLのポロキサマーおよびPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ1.0モル当たり0.3モルの亜鉛。
【0125】
亜鉛、m−クレゾール、カルシウムなどの6量体促進賦形剤を含むPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ製剤は、一般的に、物理的および化学的に高い安定性を示す。特に高温状態ではそうである。亜鉛およびm−クレゾールを含むPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ製剤にカルシウム、塩化物および/またはNaClを添加すると、40℃以上の温度に曝された製剤の物理的安定性が更に向上する。更に、リン酸塩緩衝液およびクエン酸塩緩衝液を用いた製剤は、一般的に、トリス緩衝液を用いた製剤よりも物理的安定性が低かった。
【技術分野】
【0001】
本発明は糖尿病の分野に関する。本発明は特に、非常に可溶性で拡張作用プロフィルを有するペグ化インスリンリスプロ化合物、当該分子の提供方法、当該分子を含む医薬組成物、および当該分子の治療的使用に関する。
【背景技術】
【0002】
正常血糖を達成するため、インスリン補充療法は、正常な個人における内因性インスリン分泌のパターンに出来る限り近づくようにデザインされている。インスリンの生理学的な要求は毎日変動し、(a)食事関連の血糖値の上昇に対応するためにインスリンを必要とする吸収段階、および(b)肝グルコース産生を調整して最適な空腹時血糖値を維持するため、一定量のインスリンを必要とする吸収後段階の2段階に分けられ得る。従って、糖尿病の効果的なインスリン療法では、ボーラス注射によって食事時に提供される速効性インスリンと、注射により1日に1度または2度投与して食間の血糖値を制御する長時間作用性インスリンとの2種類の外因性インスリン製剤が、一般的に組み合わされる。
【0003】
現在利用可能なインスリン補充療法は、1つ以上の重要な面が臨床的に欠けている。例えば、従来の中時間作用性および長時間作用性のインスリン製剤、例えばインスリンデテミルなどの基礎インスリン類似体を毎日投与する場合、基礎グルコース制御を一日中提供するには、その作用期間が不十分である。従って、基礎インスリンの作用期間は、多くの場合、1日に1回の投与によって高血糖を適切に制御するには不十分である。特に吸収後段階の要求を満たすには不十分である。更に、現在の療法では、単回注射を怠ると、「ピーク/トラフ」の薬物水準が有意に増大し、グルコース制御の不良に至る可能性がある。更に、インスリンの放出時間を長期化させるための不溶性ストラテジーの利用、例えば従来の中時間作用性および長時間作用性のインスリン製剤である、Neutral Protamine Hagedorn(NPH)インスリンおよびULTRALENTE(登録商標)の結晶懸濁液などの利用、またはインスリングラルギンの生体内沈降ストラテジーの利用は、注射内変動を増大させ、用量応答プロフィルの変動を増大させる結果となる。特に、NPHおよびULTRALENTE(登録商標)懸濁液は、製品の均一性を保証するため機械的な混合を必要とし、個体内変動を増大させ、最適な空腹時血糖を食間に長期間維持するのに必要な理想的に「平坦な」薬力プロフィルよりもむしろピークを与える傾向にある。従って、インスリン放出を長期化させるために不溶性の状態に依存するインスリン製剤は、可溶性製剤よりも本質的に予測性が低く、適切な血糖制御が難しく、致命的な低血糖エピソードに至る可能性が高い。更に、最近の基礎インスリン類似体は、速効性インスリン製剤または中時間作用性インスリン製剤とは混合しにくい。従って、現在のインスリン補充療法では、糖尿病患者は致命的な低血糖エピソードに罹りやすく、重篤で長期的な糖尿病の合併症を引き起こしやすく、および/または便利性や生活の質の面で患者にかなりの負担をかけやすい。
【0004】
「非免疫原性ポリペプチド」と題される特許文献1は、約500Daと約20,000Daの間の分子量を有する直鎖PEG分子に結合したインスリンを開示している。HindsとKimは、低分子量(600Da、750Daおよび2000Da)であるモノメトキシポリエチレングリコールと結合したインスリンを開示している(非特許文献1を参照)。この記事では、「より高分子のmPEG(5000Da)と結合すると、コンジュゲートの生物活性がかなり下がることが(以前に)見出されているので」、低分子量のmPEGインスリンコンジュゲートに研究を限定したと、著者達は述べている。特許文献2は、インスリンリスプロを含むインスリン誘導体と小分子の分岐鎖ポリマーとの結合を開示している。特許文献3は、特に、PEGの総分子量がそれぞれ約10kDaおよび約20kDaである直鎖および分岐鎖PEG分子と結合したインスリン分子を開示している。特許文献4(2008年2月7日に公開)および特許文献5(2008年7月17日に公開)は、特に、約200Daから約40,000Daの範囲の名目分子量を有するPEG分子に結合した種々のインスリン類似体を開示している。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第4,179,337号
【特許文献2】PCT特許出願国際公開公報第2006/079641号
【特許文献3】PCT特許出願国際公開公報第2004/091494号
【特許文献4】PCT特許出願国際公開公報第2008/015099号
【特許文献5】PCT特許出願国際公開公報第2008/084051号
【非特許文献】
【0006】
【非特許文献1】Hinds,K.D.およびKimら、Advanced Drug Delivery Reviews, 54:505−530(2002)
【発明の概要】
【発明が解決しようとする課題】
【0007】
基礎インスリン補充レジメンに更に適した長時間作用性のインスリンが今でも必要とされているのは明白である。特に、食事インスリン製剤と混合可能であり、長期間持続性の時間−作用プロフィル(すなわち1日1回またはそれ以下の頻度で注射することにより適切に血糖値を制御可能)を有し、より平坦な作用および薬力プロフィル(すなわち「ピーク/トラフ」比が低い)を有し、患者内変動が少なく(すなわち時間−作用プロフィルが予想可能なので、低血糖および/または体重増加の発生率が減少)、および/または注射部位刺激感または注射部位疼痛の少ない可溶性の基礎インスリンが必要とされている。
【課題を解決するための手段】
【0008】
我々はここに、インスリンリスプロが高分子量のポリエチレングリコール誘導体とペグ化することにより、治療上効果的な基礎インスリン活性を有し、長期間持続性の時間−作用プロフィルを有し、生理的pHで可溶性が高く、および/または一般的に使用される他の食事インスリン製剤と混合可能であるペグ化インスリンリスプロ化合物を提供できるという発見を報告する。
【0009】
本発明は式Iの化合物:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pは共有結合によって直接または間接に、Aの1位でグリシンのアルファアミノ基、Bの1位でフェニルアラニンのアルファアミノ基またはBの28位でリジンのイプシロンアミノ基に結合している)、またはその製薬上許容可能な塩を提供する。
【0010】
本発明は、複数のモノおよびジペグ化インスリンリスプロ化合物を含む組成物であって、組成物中のペグ化インスリンリスプロ化合物の約75%を超える量が、式Iのモノペグ化化合物である組成物も提供する。
【0011】
本発明は、式Iのモノペグ化インスリン化合物を含む組成物であって、組成物中のモノペグ化化合物の約50%を超える量が、B鎖の28位でリジンのイプシロンアミノ基に直接または間接に共有結合しているPEGを有する組成物も提供する。
【0012】
本発明は、式Iのペグ化インスリンリスプロおよび1つ以上の製薬上許容可能な防腐剤、等張剤、金属イオンまたは緩衝剤を含む医薬組成物も提供する。本発明の特定の実施形態では、式Iのペグ化インスリンリスプロおよび1つ以上の製薬上許容可能な防腐剤、等張剤、金属イオンまたは緩衝剤を含む医薬組成物が、治療上有効量のインスリン類似体を更に含む。
【0013】
本発明は、本発明のペグ化インスリンリスプロ化合物を含む治療上有効量の医薬組成物を患者に投与するステップを含む、高血糖症、真性糖尿病、または妊娠性糖尿病を治療する方法も提供する。
【0014】
本発明は、治療のために、本発明のペグ化インスリンリスプロ化合物を使用するステップも含む。
【0015】
本発明は、低血糖症、真性糖尿病、または妊娠性糖尿病を治療するための医薬品製造のために、本発明のペグ化インスリンリスプロ化合物を使用するステップも含む。
【図面の簡単な説明】
【0016】
【図1】ラットとイヌのPKパラメーターの相対成長スケーリングに基づき、20kDaのPEG−B28インスリンリスプロ、40kDaのPEG−B28インスリンリスプロ、およびインスリンデテミルに関してシミュレートした、ヒトPKプロフィルをグラフで表したものである。プロフィルは、1週間の投与後の1投与間隔を表したものである。数値は平均ピーク/トラフ比である。
【図2】PEG20kDaB28(約95%以上)/A1(約5%以下)インスリンリスプロ(LY、0.225mg/kg)またはインスリングラルギン(0.5U/kg)を皮下投与した後のヒトのグルコース注入速度(GIR)のグラフである。GIRプロフィルは観察データに基づいており、Lilly Resaerch Laboratories内で開発した「Loess平滑化」(Splus 2000、プロフェッショナル版、MathSoft社)関数を用いて観察データに適合させた。
【発明を実施するための形態】
【0017】
本明細書では以下の略語が使用される。ACN:アセトニトリル、Boc:tert−ブトキシカルボニル、BSA:ウシ血清アルブミン、DCM:ジクロロメタン、メチレンクロライド、DMF:N,N−ジメチルホルムアミド、DMSO:ジメチルスルホキシド、DTT:ジチオトレイトール、EDTA:エチレンジアミン四酢酸、Et:エチル、EtOH:エタノール、Fmoc:9−フルオレニルメチルオキシカルボニル、HCl:塩酸、Da:ダルトン、kDa:キロダルトン、Lilly:イーライリリー社(インディアナ州インディアナポリス)、mAb:モノクローナル抗体、Me:メチル、MeOH:メタノール、PBS:リン酸塩緩衝液生理食塩水、RP−HPLC:逆相高速液体クロマトグラフィ、SEC:粒径排除クロマトグラフィ、SEM:平均値の標準誤差、SPA:シンチレーション近接アッセイ、TFA:トリフルオロ酢酸。本開示に使用されるアミノ酸の略語は全て、37C.F.R§1.822(B)(2)の記載通り、米国特許商標局が認めたものである。
【0018】
「インスリン」と言う用語は、あらゆる種の野生型インスリンを含むことがここでは意図されており、限定されないが、ブタのインスリン、ウシのインスリン、ヒトのインスリンなどが含まれる。天然または野生型インスリンとは、天然に見出されるインスリンのアミノ酸配列に対応するアミノ酸配列を有するインスリンを意味する。様々な種から得られるインスリンをコードするポリヌクレオチドおよびアミノ酸配列は、当業者には周知である。例えば、ヒトインスリンは21のアミノ酸のA鎖と30のアミノ酸のB鎖(それぞれ配列番号1および2)を有している。インスリンは天然から得てもよい(つまり天然源から単離してもよい)し、生物合成または合成によって産生してもよい。「インスリン」と言う用語は、あらゆるインスリン誘導体および/またはインスリン類似体を含むことも意図されている。
【0019】
本明細書で使用する場合、「インスリン類似体」または「インスリン誘導体」と言う用語は、インスリン活性を有し、ヒトインスリンと実質的に同じアミノ酸配列を有してはいるが、1つ以上のアミノ酸の置換、欠失、反転または付加などのヒトインスリンの修飾によってヒトインスリンとは異なっている蛋白質であると定義される。かかる化合物は当業者には周知である。例えば、PCT特許出願国際公開公報第96/15804号および03/053339号、米国特許番号第3,528,960号、5,514,646号、5,618,913号、5,750,497号、6,011,007号、6,251,856号、欧州特許第254,516号および280,534号を参照の事。当業者に公知の典型的かつ非網羅的なインスリン類似体には、インスリンアスパルト、インスリンリスプロ、インスリングラルギン、インスリンデテミル、インスリングルリシンなどが含まれる。更に、本明細書で使用する場合、「インスリン」と言う用語には、インスリン誘導体とインスリン類似体の両方であると考えられる化合物も含まれる。かかる化合物の例は、米国特許第5,750,497号および6,011,007号に記載されている。当業者に公知のかかる化合物の具体的な例はインスリンデテミルである。
【0020】
種々のインスリン類似体および/または誘導体は、「速効性」または「迅速に作用する」インスリン類似体であることが知られている。本明細書で使用する場合、「速効性」および「迅速に作用する」と言う用語は、同じ意味で用いられる。「迅速に作用するインスリン類似体」によって生じる食事グルコース効果は、(a)ヒトインスリンよりも皮下注射後早く始まる、および/または(b)ヒトインスリンよりも皮下注射後の作用期間が短いことを示す。典型的な速効性インスリン類似体には、生理的条件下で一般的に2量体化または自己会合しにくいので速効性である、「単量体インスリン類似体」が含まれる。単量体インスリン類似体は当業者には公知である。例えば、米国特許第5,700,662号および欧州特許第214,826号を参照の事。インスリンリスプロは迅速に作用する単量体インスリン類似体であり、野生型インスリンB鎖(配列番号2)の28位にあるプロリンと、野生型インスリンB鎖(配列番号2)の29位にあるリジンが入れ替わっている。従って、インスリンリスプロは、限定されないが、LysB28ProB29ヒトインスリン、LysB28ProB29ヒトインスリン、B28LysB29Proヒトインスリンなど、様々な名称によって当業者に知られている。
【0021】
「架橋結合した」と言う用語は、システイン残基間に存在するジスルフィド結合を意味する。例えば、適切に架橋結合したヒトインスリンは、配列番号1の7位におけるシステインと配列番号2の7位におけるシステイン間のジスルフィド結合、配列番号1の20位におけるシステインと配列番号2の19位におけるシステイン間のジスルフィド結合、および配列番号1の6位におけるシステインと配列番号1の11位におけるシステイン間のジスルフィド結合を含む。同様に、適切に架橋結合したインスリンリスプロ化合物は、配列番号1の7位におけるシステインと配列番号3の7位におけるシステイン間のジスルフィド結合、配列番号1の20位におけるシステインと配列番号3の19位におけるシステイン間のジスルフィド結合、および配列番号1の6位におけるシステインと配列番号3の11位におけるシステイン間のジスルフィド結合を含む。
【0022】
本明細書で使用する場合、「PEG結合インスリンリスプロ」または「ペグ化インスリンリスプロ」と言う用語は、少なくとも1つのPEGと共有結合し、生体内でインスリン活性を有するヒトインスリンリスプロ、またはその誘導体を意味する。
【0023】
インスリンおよびインスリンリスプロの生物学的活性は既知である。本発明のペグ化インスリンリスプロ化合物に関して用いられる「インスリン活性」と言う用語は、限定されないが、それぞれ実施例5および実施例6で以下に記載される1型および2型糖尿病の動物モデルなど、一般に評価の定まった少なくとも1つの生体内動物モデルにおいて、血糖値を有意に低下させる能力を意味することが意図されている。従って、インスリン活性には、STZ処置のラットにおいて、用量568nmol/kgを単回皮下注射した後約4時間から少なくとも約36時間、血糖を100mg/dL以下のレベルにまで低下させるペグ化インスリンリスプロ化合物の能力が含まれる。
【0024】
「ポリエチレングリコール」または「PEG」は、ポリアルキレングリコール化合物またはその誘導体を意味し、カップリング剤が存在してもしなくても、あるいはカップリング部分または活性化部分との誘導化が存在してもしなくてもよい。典型的な例として、PEGは末端にヒドロキシ基を持つ線形ポリマーであり、HO−CH2CH2−(CH2CH2O)n−CH2CH2−OHの式を有している。PEG中の反復サブユニット「n」の数は、ダルトンで表される分子量に関して近似されたものである。ペグ化化合物を調製するのに使用されるPEG試薬は、一般的に、PEGポリマー中に異なる数(n)のエチレングリコール・サブユニットを有するPEGの異種混合物を含んでいる。PEGの1つのエチレングリコール・サブユニットである(−(CH2CH2O))の分子量は約44ダルトンである。従って、PEGポリマーの分子量は(n)数によって決定される。本発明のペグ化インスリンリスプロ化合物に結合するPEGは、約400から約1000の範囲のサブユニット数nを有している。本発明のペグ化インスリンリスプロ化合物に結合するPEGが、約400から約750の範囲のサブユニット数nを有しているのが好ましい。より好ましいのは、本発明のペグ化インスリンリスプロ化合物に結合するPEGが、約400から約550の範囲のサブユニット数nを有している場合である。最も好ましいのは、本発明のペグ化インスリンリスプロ化合物に結合するPEGが、約400から約500の範囲のサブユニット数nを有している場合である。
【0025】
PEGの数多くの誘導体、それを製造する方法およびインスリンやインスリンリスプロなどの蛋白質にそれを結合する方法は当業者には公知であり、本発明で使用するのに適している。例えば、PCT特許出願国際公開公報第01/62827号、2006/028745号、2006/096535号、2006/036825号、Zalipsky,S.Bioconjugate Chem.6:150−165,1995;Veroneseら,Applied Biochem.and Biotech.11:141−152,1985;およびRoberts,M.ら,Advanced Drug Delivery Reviews,54:459−476,2002を参照の事。本発明で使用される特に好ましい1つのPEGは、ポリマーの一端が下級C1−6アルコキシ基などの比較的不活性な基で終わっているPEGである。好ましくは、PEGはモノメトキシPEG(一般的にmPEGと呼ばれる)である。これは線形のPEGで、ポリマーの一端はメトキシ(−OCH3)基である。本発明で使用されるPEGは「活性化m−PEG」であるのが更に好ましい。この場合、線形PEGの一端はメトキシ基で終わり、他端は、好ましい活性化mPEGとのペグ化をインスリンリスプロ分子の特定の部位で促進させるため、インスリンリスプロまたはインスリンリスプロ誘導体の好ましい部位と結合するのに適した反応基で終わっている。
【0026】
PEGは一般的に分子量がある程度多様なPEG化合物の混合物として製造および使用されるので、当業者は通常、特定のペグ化化合物を生成するペグ化反応に使用されるPEG試薬の平均サイズによって、化合物に結合するPEGの分子量を表す。平均値を表すには数多くの方法が可能であるが、一般的には3つの方法が用いられている。すなわち、数平均分子量、重量平均分子量およびz平均分子量である。本明細書で使用する場合、「平均分子量」と言う用語は重量平均分子量を意味することが意図されるが、これは、限定されないが、マトリックス補助レーザー脱離イオン化時間(MALDI−TOF)質量分析法、ゲル浸透クロマトグラフィまたは他の液体クロマトグラフィ技術、光散乱技術、超遠心、粘度測定技術など、当業者に周知の技術を用いて測定できる。重量平均分子量を計算する式はΣ(Mi2Ni)/Σ(MiNi)で表されてもよく、式中、Niは混合物に分子量Miを有する分子のモル比率(または数比率)である。数平均分子量を計算する式はΣ(MiNi)/Σ(Ni)で表されてもよく、式中、Niは混合物に分子量Miを有する分子のモル比率(または数比率)である。重量平均分子量と数平均分子量の比は多重分散度指数(PDI)として知られており、分布の大体の幅を示すものである。本発明のペグ化インスリンリスプロ化合物を調製するのに適したPEG試薬は一般的に多重分散状態(すなわちポリマーの重量平均分子量と数平均分子量は等しくない)である。本発明の化合物または組成物を調製するのに使用されるPEG試薬のPDIは、約1.1未満であるのが好ましい。更に好ましくは、本発明の化合物または組成物を調製するのに使用されるPEG試薬のPDIは約1.05未満である。
【0027】
本発明のペグ化インスリンリスプロ化合物に関して、インスリンリスプロ分子に共有結合しているPEGの分子量は約17.5kDaから約40kDaの範囲(nは約400から約1000の範囲)、あるいはPEGの平均分子量は約17.5kDaから約40kDaである。好ましくは、インスリンリスプロ分子に共有結合しているPEGの分子量は約20kDaから約30kDaの範囲(nは約450から約750の範囲)、あるいはPEGの平均分子量は約20kDaから約30kDaである。更に好ましいのは、インスリンリスプロ分子に共有結合しているPEGの分子量は約17.5kDaから約25kDaの範囲(nは約400から約550の範囲)、あるいはPEGの平均分子量は約17.5kDaから約25kDaである。最も好ましいのは、インスリンリスプロ分子に共有結合しているPEGの分子量は約17.5kDaから約20kDaの範囲(nは約400から約500の範囲)、あるいはPEGの平均分子量は約17.5kDaから約20kDaである。
【0028】
特定の実施形態では、本発明のペグ化インスリンリスプロ化合物は、好ましい平均分子量を持った活性化mPEGをインスリンリスプロと共有結合させることにより調製される。ペグ化インスリンリスプロの反応条件は、使用される特定のPEG、インスリンリスプロの結合部位、インスリンリスプロ上で結合標的となる特定の種類の反応基、好ましいペグ化レベルなどによって異なり、当業者が容易に決定できる。特定のペグ化ストラテジーに最も適した実験条件は、一般的に、慣用の実験方法によって、当業者が容易に決定できる。
【0029】
好ましい実施形態では、本発明のペグ化インスリンリスプロ化合物は、mPEGマレイミド(mPEG−MAL)やmPEGチオール(mPEG−SH)など、比較的チオール選択的な活性化mPEGをインスリンリスプロに間接的に結合させることにより、すなわちN−スクシンイミジル−S−アセチルチオプロピオネート(SATP)やN−スクシンイミジル−S−アセチルチオアセテート(SATA)などの「アミン/チオール」調整剤を使ってインスリンリスプロに導入したチオール官能基に、チオール選択的活性化mPEGを結合させることにより調製される。更に好ましいのは、インスリンリスプロ上のチオール・スルフィドリル基にPEGを共有結合させるのに使用される活性化mPEGは、以下の(a)、(b)、(c)などのmPEGマレイミドである。
【化1】
【0030】
本発明のペグ化インスリンリスプロ化合物を調製する好ましい方法は、マイケル付加反応を利用して安定したチオエーテル結合を形成する方法である。反応は非常に特異的であり、他の官能基の存在下、穏やかな条件下で生じる。例えば、mPEGマレイミドは、活性化mPEGとして、本発明のペグ化インスリンリスプロコンジュゲートを調製するのに役立つ。ペグ化工程は、反応を完結させるため、mPEGマレイミドに対してチオール誘導インスリンリスプロをモル過剰使用するのが好ましい。反応は、pH4.0と9.0の間で、室温で1時間から40時間行うのが好ましい。チオールを含むペグ化されていない過剰のペプチドは、通常の分離方法によりペグ化生成物から容易に分離される。活性化mPEGマレイミドを用いるインスリンリスプロのペグ化に要求される典型的な条件は、実施例1に記載される。
【0031】
特定の実施形態では、本発明のペグ化インスリンリスプロ化合物は、アミンに比較的特異的な活性化mPEGを結合させることにより調製される。インスリンリスプロの主なアミン特異的ペグ化に適した活性化mPEGには、mPEGスクシンイミジルプロピオネート(mPEG−SPA)、mPEGスクシンイミジルブタノエート(mPEG−SBA)、mPEGスクシンイミジルアルファメチルブタノエート(mPEG−SMB)、mPEGスクシンイミジルカーボネート(mPEG−SC)、mPEGベンゾトリアゾールカーボネート、mPEG−p−ニトロフェニルカーボネート(mPEG−NPC)などが含まれる。
【0032】
本発明の好ましい実施形態では、インスリンリスプロのペグ化に使用される活性mPEGによって、加水分解的に安定した共有結合、例えば、アミド結合、ウレタン(カルバメートとしても知られる)結合、アミン結合、チオエーテル(スルフィドとしても知られる)結合、尿素(カルバミドとしても知られる)結合などにより、mPEGに共有結合的に結合するインスリンリスプロが生じる。更に好ましいのは、インスリンリスプロのペグ化に使用される活性化mPEGはmPEG−SCまたはmPEG−NPCである。いずれの場合も、ウレタン(すなわちカルバメート)結合によってPEGに共有結合するインスリンリスプロが生じる。種々の分子量のmPEG−NPCを用いるインスリンリスプロのペグ化に有効な典型的な条件は、実施例2に記載される。
【化2】
【0033】
本発明のペグ化インスリンリスプロ化合物は、一般的に、イオン交換クロマトグラフィ、粒径排除クロマトグラフィ、アフィニティクロマトグラフィ、疎水性相互作用クロマトグラフィ、逆相クロマトグラフィなど、1つ以上の精製技術を用いて精製される。試料中のペグ化インスリンリスプロ化合物の全体的な不均一性(ペグ化反応により生成されたペグ化インスリンリスプロ化合物の数および割合)は、クロマトグラフィ、電気泳動、質量分析、特にMALDI−MSおよびNMR分光技術のうち1つ以上を用いて評価できる。
【0034】
本発明のペグ化インスリンリスプロ化合物の調製に使用されるインスリンリスプロは、 液相方法、固相方法、半合成法、組み換えDNA技法など、一般に評価の定まった種々のペプチド合成技術のうちいずれかを用いて調製してよい。例えば、米国特許第5,700,662号(Chanceら)および欧州特許第214,826(Brangeら)は、種々のインスリン類似体の調製を開示している。インスリンリスプロのA鎖およびB鎖は、組み換えDNA技法を用いて、プロインスリン様前駆体分子から調製してもよい。好ましい実施形態では、本発明のペグ化インスリンリスプロを作るのに使用されるインスリンリスプロが、プロインスリン様前駆体を使って生成される。
【0035】
本発明は式Iの化合物:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約17.5kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pは共有結合によって直接または間接に、A鎖の1位でグリシンのアルファアミノ基、B鎖の1位でフェニルアラニンのアルファアミノ基またはB鎖の28位でリジンのイプシロンアミノ基に結合している)、またはその製薬上許容可能な塩を提供する。
【0036】
本発明の好ましい化合物は、(a)Pがウレタン結合またはチオエーテル結合によってインスリンリスプロと共有結合し、(b)STZ処置ラットにおいて、約568nmol/kgの用量で、ヒトインスリン受容体に対して約30nM、約20nM、約10nM、約5nMまたはそれ以下のKi値を有し、約6時間、約8時間、約10時間、約12時間または約14時間を超える除去半減期を有することによって特徴付けられる化合物、あるいはSTZ処置ラットにおいて、化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約36時間、約48時間、約60時間、約72時間、約84時間、約96時間、約108時間または約120時間、血糖値を約100mg/dL以下に低下させる活性によって特徴付けられる化合物である。更に好ましい化合物は、(a)Pがウレタン結合によってインスリンリスプロと共有結合し、(b)ヒトインスリン受容体に対して約10nM以下のKi値を有し、(c)STZ処置ラットにおいて、約568nmol/kgの用量で、6時間を超える除去半減期を有することによって特徴付けられ、(d)STZ処置ラットにおいて、化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約48時間、約60時間、約72時間、約84時間、約96時間、約108時間または約120時間、血糖値を約100mg/dL以下に低下させる活性によって特徴付けられる化合物である。更に好ましい化合物は、(a)Pがウレタン結合によってインスリンリスプロと共有結合し、(b)ヒトインスリン受容体に対して約10nM以下のKi値を有し、(c)STZ処置ラットにおいて、約568nmol/kgの用量で、6時間を超える除去半減期を有することによって特徴付けられ、(d)STZ処置ラットにおいて、化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約48時間、約60時間、約72時間、約84時間、約96時間、約108時間または約120時間、血糖値を約100mg/dL以下に低下させる活性によって特徴付けられ、(e)0.225mg/kgの用量で非経口的にヒトに単回投与した場合に、 約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することによって特徴付けられる化合物である。
【0037】
本発明に一貫する特徴および原理によれば、本発明の1つの実施形態は、インスリンリスプロのA鎖の1位におけるグリシンのアルファアミノ基(PEG−GlyA1インスリンリスプロ)、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基(PEG−PhB1インスリンリスプロ)、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基(PEG−LysB28インスリンリスプロ)に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含むモノペグ化インスリンリスプロ化合物を提供する。好ましくは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。更に好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。更に好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約17.5kDaまたは約20kDaを有するPEGを含む場合である(20kDa−PEG−LysB28インスリンリスプロ)。最も好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約20kDaを有するPEGを含む場合である(すなわち、PEG20kDa−LysB28インスリンリスプロ)。
【0038】
本発明の他の実施形態は、インスリンリスプロのA鎖の1位におけるグリシンのアルファアミノ基、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含むモノペグ化インスリンリスプロ化合物を提供する。好ましくは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の1位におけるフェニルアラニンのアルファアミノ基、またはインスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に直接または間接に共有結合する、平均分子量約17.5kDa、約20kDa、約25kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。更に好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約17.5kDa、約20kDa、約30kDaまたは約40kDaを有するPEGを含む場合である。最も好ましいのは、モノペグ化インスリンリスプロが、インスリンリスプロのB鎖の28位におけるリジンのイプシロンアミノ基に結合する、平均分子量約20kDaを有するPEGを含み、0.225mg/kgの化合物を単回皮下投与でヒトに投与した場合、PEG−LysB28インスリンリスプロが、約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することにより特徴づけられる場合である。
【0039】
別の実施形態では、本発明は、PEGの結合が異なる部位で生じるペグ化インスリンリスプロ化合物の混合物、および/またはモノペグ化インスリンリスプロ、ジペグ化インスリンリスプロおよびトリペグ化インスリンリスプロ化合物の混合物を含む組成物を提供する。本発明による具体的な組成物は、i)PEG−GlyA1インスリンリスプロ、ii)PEG−PheB1インスリンリスプロ、iii)PEG−LysB28インスリンリスプロ、iv)ジPEG−GlyA1PheB1インスリンリスプロ、v)ジPEG−GlyA1LysB28インスリンリスプロ、vi)ジPEG−PheB1LysB28インスリンリスプロおよびvii)ジPEG−GlyA1PheB1インスリンリスプロからなる群から選択される2つ以上のペグ化インスリンリスプロ化合物を含む。更に好ましいのは、本発明の組成物はペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量が式Iのモノペグ化インスリンリスプロ化合物である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がモノペグ化インスリンリスプロ化合物であり、全ペグ化インスリンリスプロ化合物の約20%、約15%、約10%、約5%または約3%未満がジペグ化されている場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がPEG−LysB28インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がPEG−LysB28インスリンリスプロであり、全ペグ化インスリンリスプロ化合物の約20%、約15%、約10%、約5%または約3%未満がPEG−GlyA1インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、そのペグ化インスリンリスプロ化合物の約80%、約85%、約90%、約95%または約97%を超える量がPEG−LysB28インスリンリスプロであり、全ペグ化インスリンスプロ化合物の約20%、約15%、約10%、約5%または約3%未満がPEG−GlyA1インスリンリスプロであり、全ペグ化インスリンスプロ化合物の約10%、約5%または約3%未満がジペグ化インスリンリスプロ化合物の場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約80%がPEG−LysB28インスリンリスプロ、約10%がPEG−GlyA1インスリンリスプロ、約10%がジPEG−GlyA1LysB28インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約90%以上がPEG−LysB28インスリンリスプロ、約5%以下がPEG−GlyA1インスリンリスプロ、約5%以下がジPEG−GlyA1LysB28インスリンリスプロの場合である。更に好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約90%以上がPEG−LysB28インスリンリスプロ、約5%以下がPEG−GlyA1インスリンリスプロ、約5%以下がジPEG−GlyA1LysB28インスリンリスプロであり、0.225mg/kgの化合物を単回皮下投与でヒトに投与した場合、PEG−LysB28インスリンリスプロが、約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することにより特徴づけられる場合である。最も好ましいのは、本発明の組成物がペグ化インスリンリスプロ化合物の混合物を含み、全ペグ化インスリンリスプロ化合物の約95%以上がPEG−LysB28インスリンリスプロ、約5%以下がPEG−GlyA1インスリンリスプロであり、0.225mg/kgの化合物を単回皮下投与でヒトに投与した場合、PEG−LysB28インスリンリスプロが、約24時間、約30時間、約32時間、約34時間、約36時間、約38時間、約40時間または約42時間を超える除去半減期を有することにより特徴づけられる場合である。
【0040】
本明細書で使用する場合、「塩基条件」と言う用語は、ペグ化反応の塩基度を意味する。インスリンリスプロのB鎖の28位にリジンを有するペグ化インスリンリスプロに関して、反応は、より選択的に、実質的に脱プロトン化したインスリンリスプロのアルファアミノ基との間で行われるべきである。水性溶媒または共溶媒では、塩基条件とは7.0を超えるpHで反応が行われることを意味する。ペグ化反応が約8.5から約11.5のpHで行われるのが好ましい。有機溶媒では、反応は水中で10.75以上のpKaに等しい塩基度を有する塩基の存在下で行われる。
【0041】
本発明は、以下の式のペグ化インスリンリスプロ化合物、またはその製薬上許容可能な塩を製造する工程:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pはウレタン共有結合によってBの28位でリジンのイプシロンアミノ基に結合している)も含み、当該工程には、約25℃と約30℃の間の反応温度で、Bの28位におけるリジンのイプシロンアミノ基と、約20kDaと約40kDaの間の重量平均分子量を有するモノメトキシポリエチレングリコールp−ニトロフェニルカルボネート(mPEG−NPC)とをpH約8.5と約11.5の間の水性溶媒中で反応させるステップが含まれる。好ましくは、反応のpHは約10.5から約11.1の間に維持され、ペグ化反応は約25℃と約30℃の間の温度で約2時間から約12時間行われ、mPEG−NPC対インスリンリスプロの比率は約1.0から約5.0の範囲である。更に好ましいのは、mPEG−NPCの重量平均分子量が約20kDaで、反応のpHが約10.5と約11.1の間に維持され、ペグ化反応が約25℃と約30℃の間の温度で約2時間から約12時間行われ、mPEG−NPC対インスリンリスプロの比率が約1.0と約5.0の範囲にある場合である。更に好ましいのは、PEG対インスリンリスプロのモル比率が約2.5と約4.5の範囲にあり、mPEG−NPCの重量平均分子量が約20kDaで、ペグ化反応のpHが約10.5と約11.1の間に維持され、ペグ化反応が約25℃と約30℃の間の温度で約3時間から約6時間維持される場合である。最も好ましいのは、PEG対インスリンリスプロのモル比率が約2.6と約4.5の範囲にあり、mPEG−NPCの重量平均分子量が約20kDaで、ペグ化反応のpHが約10.5と約11.1の間に維持され、反応が約25℃と約30℃の間の温度で約3時間維持される場合である。
【0042】
異なる分子量を有する本発明のペグ化インスリンリスプロ化合物は、必要に応じて、当業者に公知の種々の技法を用いて単離できる。かかる技法には、限定されないが、ゲル濾過クロマトグラフィおよび/またはイオン交換クロマトグラフィが含まれる。すなわち、ゲル濾過クロマトグラフィを用いて、異なる分子量(本質的にペグ化反応に使用されるPEGの平均分子量に対応する差異)に基づいて、モノペグ化インスリンリスプロ化合物、ジペグ化インスリンリスプロ化合物およびトリペグ化インスリンリスプロ化合物の画分に分けてもよい。例えば、平均分子量約20kDaを有する活性化mPEGにインスリンリスプロを結合させる典型的な反応では、反応混合物は分子量約5,808ダルトンを有する非修飾インスリンリスプロ、平均分子量約25,808ダルトンを有するモノペグ化インスリンリスプロ、平均分子量約45,808ダルトンを有するジペグ化インスリンリスプロ、および平均分子量約65,808ダルトンを有するトリペグ化インスリンリスプロが含まれる場合がある。しかし、ゲル濾過技法は流体力学的サイズに基づいて化合物を分離するので、平均分子量約20kDaを有するmPEGと結合したモノペグ化インスリンリスプロは、平均分子量が約25,808ダルトンであるにもかかわらず、ゲル濾過中約82kDaの蛋白質として移動する。当業者は、精製および/または定量化に際し、平均分子量約20kDaのmPEGとペグ化したジペグ化種およびトリペグ化種が、非常に異なる移動または保持時間を有することを予期するであろう。
【0043】
「血漿半減期」と言う用語は、体内の薬物の血漿濃度が半分に低下する時間を意味する。「除去半減期」と言う用語は代替的に使用され、除去の最終対数線形率に相当する。当業者は、半減期がクリアランスと分配量の両方の関数として変化する誘導パラメーターであることを理解している。血漿半減期または除去半減期に関して用いられる「長期的」、「長期化」、または「増大」と言う用語は、本明細書では同じ意味で使用され、比較条件化で測定した場合、基準分子(例えばインスリンリスプロ)の半減期と比べて試験化合物(例えばペグ化インスリンリスプロ)の半減期が統計学的に有意に増大する意味であることが意図されている。
【0044】
クリアランスは薬物を排除する体の能力の尺度である。例えば薬物の修飾によってクリアランスが低下すれば、半減期は増大することが予想される。しかし、この相互関係は、分配量に変化がない場合のみ正確である。最終対数線形半減期(t1/2)、クリアランス(CL)および分配量(V)の間の有効な近似関係は、t1/2〜0.693(V/CL)の式で表される。クリアランスは、除去を説明するにあたり、薬物がどれほど除去されたかではなく、むしろ、薬物を完全に無くすのに必要な血液や血漿などの生物学的流体の量を示すものである。クリアランスは単位時間当たりの量として表わされる。
【0045】
本明細書で使用する場合、「治療」または「治療する」と言う用語は、糖尿病、高血糖症、あるいは症状や合併症と闘うまたはそれを軽減する目的でインスリン投与を必要とする他の病状に罹患している患者の管理およびケアを意味する。治療には、症状または合併症の発症、症状または合併症の軽減、または病気、病状または疾患の排除のために、本発明の化合物または組成物を投与するステップも含まれる。治療対象の患者は哺乳動物であり、好ましくはヒトである。
【0046】
式1のペグ化インスリンリスプロ化合物は、1つ以上の式Iの化合物をそれを必要とする患者に治療上有効量投与して、高血糖症を治療するのに有効である。本明細書で使用する場合、「治療上有効量」と言う用語は、患者の血糖を調節するのに十分な式Iのペグ化インスリンリスプロ化合物またはその組成物の量を意味する。式Iのペグ化インスリンリスプロの好ましい治療上有効量は、約0.01nmol/kgから約100nmol/kgである。更に好ましいのは、治療上有効量が約0.01nmol/kgから約50nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.01nmol/kgから約20nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.01nmol/kgから約10nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.1nmol/kgから約7.5nmol/kgの場合である。更に好ましいのは、治療上有効量が約0.1nmol/kgから約5nmol/kgの場合である。もっとも好ましいのは、治療上有効量が約0.5nmol/kgから約5nmol/kgの場合である。しかし、ペグ化インスリンリスプロ化合物または実際に投与される1つ以上のペグ化インスリンリスプロ化合物を含む組成物の量は、治療対象の病状を含む様々な関連状況(すなわち高血糖症の原因)、投与されるペグ化インスリンリスプロの特定の種またはペグ化インスリンリスプロ化合物の特定の混合物、同時投与される他の薬物、インスリンまたはその他、選択された非経口的投与経路、個々の患者の年齢、体重および反応、並びに患者の症状の重篤性を考慮して、医師が決定するものであることは理解されるべきである。従って、上述の用量範囲は、如何なる意味でも、本発明の範囲を制限するものではない。
【0047】
「血糖を調節するのに十分」と言う用語は、化合物または組成物を患者に投与することにより、正常な空腹時血漿グルコースレベルになることを意味する。臨床的に正常な空腹時血漿グルコースレベルは70−110mg/dLである。臨床的に正常な食後血漿グルコースレベルは140mg/dL未満である。
【0048】
インスリン構造の共有結合的化学変化が貯蔵中に起きることが知られている。その結果、活性の低下した分子、脱アミド化生成物のように潜在的に免疫原性的な生成物、および高分子量の転換生成物(例えば、2量体、オリゴマー、ポリマー)が形成される場合がある。インスリンの化学的安定性に関する総合的な研究がJens Brange,“Stability of Insulin,” Kluwer Academic Publishers,1994に記載されている。インスリン組成物は一般的に約2−8℃以下という低温で貯蔵される(低温貯蔵は、室温で貯蔵する場合と比べて貯蔵寿命をかなり改善する)という事実があるにもかかわらず、インスリン製品の保存寿命は、可溶性の凝集体(2量体、オリゴマー、ポリマー)の形成によりやがて低下する。更に、インスリン製品は振動により(例えば、患者がポケットに入れて持ち運んだり、輸送したりすることにより)不溶性の凝集体(フィブリル)を形成する傾向にある。化学的または物理的影響により可溶性および不溶性の凝集体が形成される傾向を最小限にまで減らすことが、インスリン製品の質にとって非常に重要である。従って、インスリン組成物を治療に使用するためには、化学的および物理的な安定特性を証明しなければならない。
【0049】
本明細書で使用する場合、「安定性」と言う用語は、ペグ化インスリンリスプロ化合物の調剤の物理的および/または化学的安定性を意味する。ペグ化インスリンリスプロ調剤の物理的不安定は、蛋白質分子の凝集により高位のポリマーや更には沈殿物を形成することにより生じる場合がある。「安定した」調剤では、蛋白質の凝集度が受容可能なレベルに制御されており、時間とともに受容不可能なレベルにまで増大するということがない。本発明の特定の実施形態では、ペグ化インスリンリスプロ調剤は、凝集度が出発物質の凝集度の約1%、2%、3%、4%、5%、10%、15%、20%、25%または30%以内であれば、特定の期間安定であると考えられる。本発明の特定の実施形態では、ペグ化インスリンリスプロ調剤は、ポリペプチドの生物学的活性が出発物質の活性の少なくとも約99%、95%、90%、85%、80%、75%、70%、65%、60%、55%または50%であれば、特定の期間安定であると考えられる。
【0050】
本明細書で使用する場合、「化学的安定性」と言う用語は、ペグ化インスリンリスプロ組成物が、貯蔵中に、約2−8℃の低温貯蔵および約20−40℃の高温貯蔵を含む静止条件下で、可溶性の蛋白質凝集体を形成する傾向にあることを意味する。本発明のペグ化インスリンリスプロ化合物の化学的安定性は、特定の条件化で(例えば、特定の温度および湿度条件で特定の期間)、製剤の分析属性を決定することにより測定できる。測定可能な分析属性には、例えば、粒径排除HPLCを用いる高分子量種の形成などが含まれる。その結果は、次にモニターし、予め特定されたパラメーターと比較できる。
【0051】
本明細書で使用する場合、「物理的安定性」と言う用語は、ペグ化インスリンリスプロ組成物の振動など、物理的な行為の結果として不溶性の蛋白質凝集体が形成される傾向にあることを意味する。本発明のペグ化インスリンリスプロ化合物を種々の医薬調剤の形で、一定期間、種々の温度で貯蔵した場合の物理的安定性は、当業者に周知の方法により評価してよい。試料の見かけの光減衰(吸光度または光学密度)の測定もその中に含まれる。かかる光減衰の測定は、製剤の混濁に関係している。混濁は製剤中の蛋白質または複合体の凝集や沈殿により生じる。物理的安定性を評価する他の方法は当業者には周知であり、粒子の存在または不在の視覚的評価やチオフラビンT蛍光顕微鏡検査法によるフィブリル/ゲル形成の検出などもその中に含まれる。
【0052】
本発明の他の実施形態は、患者(特にヒト)に投与するのに適しており、式Iに示した少なくとも1つのペグ化インスリンリスプロ化合物の治療上有効量および1つ以上の製薬上許容可能な賦形剤、希釈剤、緩衝液、金属イオンまたは担体を含む医薬組成物を提供する。かかる医薬組成物はその性質上一般的に非経口的(そうでない場合もある)であり、当業者に周知の従来の非経口製品用賦形剤、緩衝液、希釈剤、金属イオンまたは担体を使用し、種々の技術のいずれかによって調製してもよい。
【0053】
本発明のペグ化インスリンリスプロ化合物はきわめて水溶性であるので、本発明の医薬組成物は水を主要溶媒として包含する組成物を含んでおり、ペグ化インスリンリスプロ化合物の全濃度は、少なくとも1mg/mL、少なくとも2mg/mL、少なくとも5mg/mL、少なくとも10mg/mL、少なくとも15mg/mL、少なくとも20mg/mL、少なくとも25mg/mL、少なくとも30mg/mL、少なくとも35mg/mL、少なくとも40mg/mL、少なくとも45mg/mL、少なくとも50mg/mLまたはそれ以上であり、製薬上許容可能な緩衝液を含み、医薬組成物のpHは約4.0から約8.5である。本発明の医薬組成物のpHは約6.0から約8.5であるのが好ましい。更に好ましいのは、本発明の医薬組成物が、全濃度が約2.5mg/mLから約60mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、組成物のpHが約6.0から約8.5の範囲の場合である。更に好ましいのは、本発明の医薬組成物が、全濃度が約5mg/mLから約50mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、医薬組成物のpHが約6.5から約7.5の範囲の場合である。更に好ましいのは、本発明の医薬組成物が、全濃度が約10mg/mLから約40mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、医薬組成物のpHが約6.5から約7.5の範囲の場合である。更に好ましいのは、本発明の医薬組成物が、全濃度が約15mg/mLから約40mg/mLの範囲のペグ化インスリンリスプロ化合物および緩衝液を含み、医薬組成物のpHが約7.0から約7.5あるいは約7.0から約8.0の範囲の場合である。本発明の医薬組成物が更に治療上有効量のインスリンを含んでいるのが更に好ましい。更に好ましいのは、上記インスリンがインスリン類似体の場合である。更に好ましいのは、インスリン類似体が迅速に作用するインスリン類似体の場合である。最も好ましいのは、迅速に作用するインスリン類似体がインスリンリスプロの場合である。
【0054】
「緩衝液」と言う用語は、酸塩基共役成分の作用によりpHの変化に抵抗する溶液を意味する。使用される緩衝液は製薬上許容可能な緩衝液であるのが好ましい。「製薬上許容可能な緩衝液」と言う用語は、インスリン製剤に使用しても安全であり、医薬組成物のpHを目的のpHへ制御する効果のある溶液を意味する。好ましい実施形態では、緩衝液は約6.0から約8.5の範囲のpHを有している。更に好ましいのは、緩衝液が約7.0から約8.0の範囲のpHを有している場合である。本発明の組成物をこの範囲のpHに制御するのに用いられる製薬上許容可能な緩衝液は、限定されないが、リン酸塩緩衝液、酢酸塩緩衝液、クエン酸塩緩衝液、アルギニン緩衝液、トリス緩衝液、ヒスチジン緩衝液およびそれらの組み合わせを含む。「トリス」は2−アミノ−2−ヒドロキシメチル−1,3−プロパンジオールおよびその薬理学的に許容可能な塩を意味する。遊離塩基および塩酸塩(すなわちトリスHCl)が、トリスの一般的な2つの形態である。トリスはトリメチロールアミノメタン、トロメタミン、およびトリス(ヒドロキシメチル)アミノメタンとしても当業者に知られている。本発明の医薬組成物は、約2.5mMから約50mMのリン酸塩緩衝液またはトリス緩衝液を含むのが好ましい。更に好ましいのは、本発明の医薬組成物が約5mMから約20mMのリン酸塩緩衝液またはトリス緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約5mMから約10mMのリン酸塩緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約5mMのリン酸塩緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約7.5mMから約50mMのトリス緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約10mMから約25mMのトリス緩衝液を含む場合である。更に好ましいのは、本発明の医薬組成物が約15mMから約20mMのトリス緩衝液を含む場合である。最も好ましいのは、本発明の医薬組成物が約16mMのトリス緩衝液を含む場合である。
【0055】
本発明のペグ化インスリンリスプロ化合物および組成物は、非経口的に患者に投与される既知のインスリン製剤と同様の方法で製剤化してもよい。かかる製剤化は当業者には周知である。式Iのペグ化インスリンリスプロ化合物をHUMALOG(登録商標)インスリンリスプロまたはHumulin(登録商標)の製剤化と同様に製剤化するのが好ましい。従って、本発明の好ましい医薬組成物は、水、式Iのペグ化インスリンリスプロ化合物、等張剤および製薬上許容可能な緩衝液を含んでもよい。本発明の医薬組成物は製薬上許容可能な防腐剤を更に含むのが好ましい。更に好ましいのは、本発明の医薬組成物が、インスリンの6量体化を促進し得る二価の陽イオン、例えば亜鉛および/またはコバルトを更に含む場合である。更に好ましいのは、本発明の医薬組成物が、少なくとも1つの6量体安定化剤を更に含む場合である。更に、pHを調整するため、塩酸および/または水酸化ナトリウムを添加してもよい。
【0056】
「等張剤」は生理学的に寛容な化合物で、製剤に適切な等張性を与えることにより、投与された医薬組成物に接触する細胞膜に水の正味の流れが生じるのを防ぐ。グリセリンとしても知られるグリセロールは等張剤として一般的に使用される。他の等張剤としては、i)限定されないが、マンニトールやソルビトールなどの他の糖アルコール、ii)限定されないが、NaClなどの塩、iii)限定されないが、デキストロースなどの単糖類、iv)限定されないが、ラクトース、ショ糖、トレハロースなどの二糖類などが含まれる。本発明の医薬組成物には1つ以上の等張剤を含んでもよい。本発明の医薬組成物は、約270ミリオスモルから約330ミリオスモルの範囲の等張性を持つ製剤を生成することのできる1つ以上の等張剤を有するのが好ましい。等張剤がグリセロール、ソルビトール、ショ糖、NaCl、トレハロースおよび/またはマンニトールであれば更に好ましい。更に好ましいのは、等張剤がグリセロール、ソルビトール、ショ糖、NaClおよび/またはトレハロースの場合である。更に好ましいのは、グリセロール、ソルビトール、ショ糖、NaClまたはトレハロースが、約100mMから約200mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、グリセロールが、約100mMから約200mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、グリセロールが、約150mMから約180mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、グリセロールが、約130mMから約175mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、NaClが、約50mMから約300mMの濃度で、本発明の医薬組成物の中に存在する場合である。更に好ましいのは、NaClが、約100mMから約200mMの濃度で、本発明の医薬組成物の中に存在する場合である。最も好ましいのは、NaClが、約150mMの濃度で、本発明の医薬組成物の中に存在する場合である。
【0057】
本発明の医薬組成物は6量体安定化化合物を含んでもよい。「6量体安定化化合物」と言う用語は、6量体会合状態にある本発明のペグ化インスリンリスプロ化合物を安定化させる、非蛋白質様の小分子量の化合物である。インスリン分子用の既知の6量体安定化化合物は、カルシウムイオン、亜鉛、コバルト、銅、ニッケル、鉄、マグネシウム、マンガン、陰イオン、特に塩化物、臭化物、ヨウ化物、チオシアネート、およびフェノール系化合物、特にフェノール、フェノール系防腐剤、レゾルシノール、4’−ヒドロキシアセトアニリド、4−ヒドロキシベンズアミドおよび2,7−ジヒドロキシナフタレンである。好ましくは、6量体安定化化合物は、フェノール、m−クレゾール、o−クレゾール、p−クレゾール、クロロクレゾール、メチルパラベン、カルシウム、塩化物、または2つ以上の当該化合物の組み合わせである。6量体安定化化合物はフェノール、m−クレゾール、カルシウム、塩化物、またはそれらの組み合わせであるのが更に好ましい。好ましくは、本発明の医薬組成物は、約1mMと75mMの間のカルシウム、約1mMと約50mMの間のカルシウム、約1mMと約25mMの間のカルシウム、約5mMと約50mMの間のカルシウム、約2.5mMと約30mMの間のカルシウム、約2.5mMと約15mMの間のカルシウム、約2.5mMと約10mMの間のカルシウム、約5mMと約30mMの間のカルシウム、約5mMと約15mMの間のカルシウムを含む。より好ましくは、本発明の医薬組成物は約2.5mMと10mMの間のカルシウムを含む。
【0058】
本発明の医薬組成物の多用途製剤は防腐剤を含んでいてもよい。「防腐剤」と言う用語は、医薬製剤に添加され、抗菌薬として作用する化合物を意味する。非経口製剤として有効で許容可能な、当業者に公知の防腐剤には、塩化ベンザルコニウム、ベンゼトニウム、クロロヘキシジン、フェノール、m−クレゾール、ベンジルアルコール、メチルパラベン、プロピルパラベン、クロロブタノール、o−クレゾール、p−クレゾール、クロロクレゾール、硝酸フェニル水銀、チメロサル、安息香酸、およびそれらの種々の混合物が含まれる。特定のフェノール系防腐剤、例えばメチルパラベン、フェノールおよびm−クレゾールは、インスリンおよびインスリン様化合物と結合し、物理的または化学的安定性あるいは両方の安定性を増大させる立体配座的変化を誘起することが知られている(例えば、Birnbaum,D.T.ら,Pharmaceutical.Res.14:25−36(1997);Rahuel−Clermont,S.ら,Biochemistry 36:5837−5845(1997)を参照)。「フェノール系防腐剤」には、フェノール、m−クレゾール、o−クレゾール、p−クレゾール、クロロクレゾール、メチルパラベンの化合物、およびそれらの混合物が含まれる。本発明のペグ化インスリンリスプロ化合物の製剤に使用される防腐剤は、フェノール系防腐剤であってもよいし、6量体安定化化合物と同じであっても異なっていてもよい。好ましくは、フェノール系防腐剤はm−クレゾールまたはフェノールである。本発明の医薬組成物は、約0.1mMから約75mMの濃度のフェノールおよび/またはm−クレゾールを、防腐剤として、約7.0から約8.0のpHで含んでいるのが更に好ましい。更に好ましいのは、本発明の医薬組成物が、約1mMから約60mMの濃度のフェノールおよび/またはm−クレゾールを、防腐剤として、約7.0から約8.0のpHで含んでいる場合である。更に好ましいのは、本発明の医薬組成物が、約10mMから約40mMの濃度のフェノールおよび/またはm−クレゾールを、防腐剤として、約7.0から約8.0のpHで含んでいる場合である。更に好ましいのは、本発明の医薬組成物が、約30mMの濃度のフェノールおよび/またはm−クレゾールを、約7.0から約8.0のpHで含んでいる場合である。最も好ましいのは、本発明の医薬組成物が、約30mMの濃度のフェノールおよび/またはm−クレゾールを、約7.3から約7.5のpHで含んでいる場合である。
【0059】
上述したように、本発明の医薬組成物は、インスリンの6量体化を促進あるいはインスリン化合物を安定化する二価の金属陽イオン、例えば亜鉛またはコバルトを含んでいてもよい。「二価の金属陽イオン」と言う用語は、複数の蛋白質分子と複合体を形成するために共有するイオンまたはイオン群を意味する。遷移金属、アルカリ金属およびアルカリ土類金属は、インスリン化合物と複合体を形成することが知られている金属の例である。遷移金属が好ましい。二価の金属陽イオンは、亜鉛、銅、コバルト、ニッケル、マンガン、マグネシウム、カドミウムおよび鉄からなる群から選択される1つ以上の陽イオンであるのが好ましい。二価の金属陽イオンが亜鉛であるのが更に好ましい。亜鉛は、インスリンおよびインスリンリスプロを含む種々のインスリン類似体および/または誘導体の6量体形成を促進することが知られている。本発明の医薬組成物に含まれる亜鉛やコバルトなどの二価の陽イオンの主要な役割は、本発明のペグ化インスリンリスプロ化合物および/または本発明のペグ化インスリンリスプロ化合物を含む医薬組成物に含まれる他のインスリンまたはインスリン類似体の6量体形成を促進することである。フェノール系防腐剤が存在する場合、本発明の医薬組成物を含む溶液のpHを亜鉛(II)イオンの存在下で中性領域に調整することにより、あるいはpHを中性領域に調整した後に亜鉛(II)を添加することにより、6量体形成を促進できる場合がある。亜鉛対インスリン化合物、インスリン類似体および/またはペグ化インスリンリスプロ化合物の比率は、下限として約0.33に固定するのが好ましい。すなわち、2つの亜鉛原子に対して1つのインスリン6量体、インスリン類似体6量体および/またはペグ化インスリンリスプロ6量体が好ましい。更に好ましいのは、亜鉛対インスリン化合物、インスリン類似体および/またはペグ化インスリンリスプロ化合物の比率が約0.33から約0.67の場合である。亜鉛結合で蛋白質と競合する化合物、例えばクエン酸塩やリン酸塩が存在する場合は、処理中に追加の亜鉛を使用してもよい。6量体化に必要な量を超える過剰の亜鉛(例えば、亜鉛対インスリン化合物、インスリン類似体および/またはペグ化インスリンリスプロ化合物の比率が約0.33から約0.83の場合)が、6量体化を更に促進するのに好ましい場合がある。6量体化に必要な量を超える過剰の亜鉛を本発明の医薬組成物に存在させてもよく、それが化学的および物理的安定性の改善、「懸濁性」の改善、および時間−作用の更なる拡張にとって好ましい場合がある。一方、クエン酸塩緩衝液またはリン酸塩緩衝液中の過剰の亜鉛は、それぞれクエン酸亜鉛またはリン酸亜鉛並びにインスリンの沈殿の原因となる場合がある。
【0060】
本発明によれば、インスリン、インスリン類似体およびペグ化インスリンリスプロ6量体1モル当たり約0.3モルから約3モルの亜鉛を製剤中に存在させてもよい。全インスリン、インスリン類似体およびペグ化インスリンリスプロの6量体1モル当たり約0.3モルから約1モルの亜鉛を本発明の医薬組成物中に存在させるのが、更に好ましい。更に好ましいのは、全インスリン、インスリン類似体およびペグ化インスリンリスプロの6量体1モル当たり約0.3モルから約0.7モルの亜鉛を本発明の医薬組成物中に存在させる場合である。最も好ましいのは、インスリン、インスリン類似体およびペグ化インスリンリスプロの6量体1モル当たり約0.3モルから約0.55モルの亜鉛を本発明の医薬組成物中に存在させる場合である。本発明に亜鉛を提供する亜鉛化合物は、製薬上許容可能な亜鉛化合物であればどれでもよい。インスリン製剤への亜鉛の添加は、製薬上許容可能な亜鉛源同様、当業者には公知である。亜鉛は塩として供給されるのが好ましい。例えば、硫酸亜鉛、塩化亜鉛、酢酸亜鉛、クエン酸亜鉛、酸化亜鉛、硝酸亜鉛などである。
【0061】
本発明の更なる実施形態では、本発明の医薬組成物は更に1つ以上の表面活性剤を含む。本明細書で使用する場合、「表面活性剤」と言う用語は、気相液相界面の吸着により液相の表面張力を減少させる試剤を含む。表面活性剤の例には、限定されないが、非イオン性の表面活性剤、例えば、ポリソルベート(ポリソルベート80やポリソルベート20など)、ポロキサマー(ポロキサマー188など)、トリトン(商標)(トリトン(商標)X−100など)、ポリエチルグリコール、ポリプロピルグリコール、エチレンとプロピレングリコールのコポリマー(プルロニックスやPF68など)が含まれる。例えば、本発明の医薬組成物には、約0.001−0.5%(例えば、0.05−0.3%)の表面活性剤が存在してもよい。本発明の医薬組成物に使用される表面活性剤はポロキサマー188であるのが好ましい。更に好ましいのは、表面活性剤がポロキサマー188で、その濃度が約0.5mg/mLと約10mg/mLの間、約1mg/mLと約10mg/mLの間、約2mg/mLと約10mg/mLの間、約3mg/mLと約10mg/mLの間、約4mg/mLと約10mg/mLの間、約1mg/mLと約5mg/mLの間、約2mg/mLと約5mg/mLの間、約3mg/mLと約5mg/mLの間、および約4mg/mLと約5mg/mLの間の場合である。
【0062】
本発明は、好ましくはヒトにおいて、低血糖症および/または糖尿病の治療に使用される、式Iのペグ化インスリンリスプロ化合物またはその製薬上許容可能な塩も提供する。
【0063】
本発明は、好ましくはヒトにおいて、低血糖症および/または糖尿病の治療用の医薬品の製造に使用される、式Iのペグ化インスリンリスプロ化合物またはその製薬上許容可能な塩も提供する。
【0064】
本発明のペグ化インスリンリスプロを含む医薬組成物は、それを必要とする患者に非経口的に投与してもよい。非経口投与は、注射器、任意にペンタイプの注射器または機械駆動の注入器を用いて、皮下、筋肉内または静脈内注射によって行ってもよい。あるいは、非経口投与は輸液ポンプを用いて行ってもよい。
【実施例】
【0065】
(調製1:GlyA1−HSCH2CH2COインスリンリスプロ(3)およびLysB28−HSCH2CH2COインスリンリスプロ(4))
Trt−SCH2CH2CO−OH、N−ヒドロキシスクシンイミド(NHS)およびジイソプロピルカルボジイミド(DIC)を1mmolずつDMF2mL中で30分混合して、Trt−SCH2CH2CO−NHSエステルを調製する。1/10mmolのインスリンリスプロを5%トリエチルアミン(TEA)のDMSO溶液10mLに溶解する。その溶液に、活性化したTrt−SCH2CH2CO−NHSを0.2mmol加える。室温で2時間反応させた後、反応を終了させるため、エタノールアミン0.2mLを添加する。次に反応混合物を90mLの水で希釈し、RP−C18カラムで精製する。目的のLysB28−Trt−SCH2CH2COインスリンリスプロ(2)の画分をプールし凍結乾燥する。それとは別に、目的のGlyA1−Trt−SCH2CH2COインスリンリスプロ(1)の画分をプールし凍結乾燥する。0.2mLのチオアニソールと0.4mLのトリイソプロピルシランを含むTFA5mLに、1/10mmolの(1)または(2)を溶解する。30分後、TFAを蒸発によって除去し、残留性のペプチドを10%ACNの水溶液50mLに移す。その結果得られる溶液をRP−C18カラムで精製する。目的の(3)または(4)の画分をプールし、任意に凍結乾燥する。
【0066】
(調製2:PheB1−HSCH2CH2COインスリンリスプロ(7))
1/10mmolのインスリンリスプロを5%TEAのDMSO溶液10mLに溶解する。その溶液に、ジ−tert−ブチルカーボネートのDMSO溶液0.2mmolを添加する。室温で1時間反応させた後、反応を終了させるため、エタノールアミン0.2mLを添加する。次に反応混合物を90mLの水で希釈し、RP−C18カラムで精製する。目的のBoc−GlyAlおよびBoc−LysB28インスリンリスプロの画分をプールし、凍結乾燥して(5)を得る。1/10mmolの(5)を5%TEAのDMSO溶液10mLに溶解する。その溶液に、活性化したTrt−SCH2CH2CO−NHSを0.2mmol加える。室温で2時間反応させた後、反応を終了させるため、エタノールアミン0.2mLを添加する。次に反応混合物を90mLの水で希釈し、RP−C18カラムで精製する。目的の画分をプールし凍結乾燥すると、Trt−SCH2CH2CO−PheB1、Boc−GlyA1およびBoc−LysB28インスリンリスプロ(6)が得られる。0.2mLのチオアニソールと0.4mLのトリイソプロピルシランを含むTFA5mLに、1/10mmolの(6)を溶解する。30分後、TFAを蒸発によって除去し、残留性のペプチドを10%ACNの水溶液50mLに移す。その結果得られる溶液をRP−C18カラムで精製する。目的の画分をプールし凍結乾燥すると、(7)が得られる。
【0067】
(実施例1:チオール誘導化インスリンリスプロ中間体のペグ化)
平均分子量約20kDa(b)、30kDa(a)、40kDa(a)または60kDa(c)を有するモノメトキシPEG−MALを100mMのNH4Ac緩衝液(pH4.69)とACNの1対1混合液に溶解する。チオール誘導化インスリンリスプロ(例えば、化合物(3)、(4)または(7))の凍結乾燥粉末を上記溶液に添加する。反応は分析用RP−HPLCを使ってモニターしてもよい。反応が完結(通常約4時間後)したら、混合物は水で希釈し、RP−HPLCカラムを使って精製する。目的の画分をプールし凍結乾燥すると、ペグ化インスリンリスプロ化合物が得られる。実施例1に記載の方法で調製した典型的なペグ化インスリンリスプロ化合物は、8(a)、8(b)、9(a)、9(b)、10(a)、10(b)および15(c)として以下に示してある。これらのペグ化インスリンリスプロ化合物は、約400から約1000の範囲のnを有しているのが好ましい。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約400から約750の範囲のnを有している場合である。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約400から約550の範囲のnを有している場合である。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約400から約500の範囲のnを有している場合である。更に好ましいのは、これらのペグ化インスリンリスプロ化合物が、約450から約500の範囲のnを有している場合である。最も好ましいのは、これらのペグ化インスリンリスプロ化合物が約450のnを有している場合である。
【化3−1】
【0068】
【化3−2】
【0069】
【化3−3】
【0070】
(実施例2:モノメトキシポリエチレングリコールp−ニトロフェニルカルボネート(mPEG−NPC)を用いたインスリンリスプロのペグ化)
1/10mmolのインスリンリスプロを20mLの0.2Mホウ酸塩緩衝液にpH10.5で溶解し、ACN20mL中の平均分子量約20kDaを有するmPEG−NPC1.98gを激しく攪拌しながら上記溶液に添加する。反応はRP−HPLCおよびSECでモニターする。約4時間後、反応混合物を酢酸を使ってpH5−7に酸性化し、RP−HPLCカラムを使って精製する。目的の画分をプールし凍結乾燥すると、モノペグ化PEG20Kインスリンリスプロが20%から45%の範囲の収率で得られる。RP−HPLC、SECおよびMALDI−MSで同定し、純度を確認する。A鎖またはB鎖に結合するmPEGの割合は、得られたコンジュゲートをトリス(2−カルボキシエチル)ホスフィン塩酸塩(TCEP)で処理した後に放出される遊離のA鎖およびB鎖の領域積分によって決定する。mPEG−NPC対インスリンリスプロの比率は、モノペグ化種とジペグ化種の生成物分布を決定する。反応pHはペグ化の部位特異性を支配する。pHが約8から約12に上がると、化合物(11)が主要生成物になる。pH10.5で、約20kDaの平均分子量(nは約450)を有するmPEG−NPCを用いて反応を行うと、(11)対(12)の比率は約85対15になる。
【0071】
上記のペグ化反応は、0.2MのNaOHを連続添加して反応混合物のpHを維持することにより、非緩衝水溶液中で行うこともできる。pHを約11.5に維持しながら、約20kDaの平均分子量を有するmPEG−NPCを用いて、非緩衝水溶液中で反応を行わせると、ペグ化反応生成物は、(11)と(12)を約92対8の割合で含む。化合物(11)は、約400から約1000の範囲のnを有するのが好ましい。更に好ましいのは、化合物(11)が約400から約750の範囲のnを有する場合である。更に好ましいのは、化合物(11)が約400から約550の範囲のnを有する場合である。更に好ましいのは、化合物(11)が約400から約500の範囲のnを有する場合である。更に好ましいのは、化合物(11)が約450から約500の範囲のnを有する場合である。最も好ましいのは、化合物(11)が約450のnを有する場合である。
【化4】
【0072】
(実施例3:インビトロ受容体親和性)
受容体結合アッセイは、ヒトインスリン受容体(hIR)またはヒトIGF−1受容体(hIGF−1R)を過剰発現している、安定的に形質移入された293EBNA HEK細胞から調製したP1膜上で行われる。結合親和性は、ヒト組み換え(3−[125I]ヨードチロシルA14)インスリン(2000Ci/mmol)またはヒト組み換え[125I]インスリン様成長因子(2000Ci/mmol)のいずれかを用いて、競合放射性リガンド結合アッセイで決定する。アッセイは、PVT PEI処置タイプA麦芽アグルチニン結合SPAビーズを用いてSPA法で行った。全ての試薬調製にSPAアッセイ緩衝液(50mMトリスHCl、pH7.5、150mM NaCl、0.1%BSA)を用いた。化合物の3倍段階希釈(100nMから2pM)はFreedom/Evoロボット(Tecan)を用いてアッセイ緩衝液で調製し、マルチメク(Beckman Coulter)を用いて96ウェル白色透明ボトムマイクロプレート(Corning/Costar)に添加した。放射性リガンド、膜およびSPAビーズをマルチドロップ装置(Titertek)を使って添加する。室温で10時間インキュベーションした後、マイクロベータ・トリラックス・シンチレーション・カウンターを用いて放射線を測定する。非標識インスリンリスプロと非標識IGF−Iをそれぞれ陽性対照および陰性対照として各実験に含める。IC50値は4パラメーター非線形ロジスティック回帰分析により決定する。親和定数(Ki)は、Ki=IC50/(1+D/Kd)の式に基づいてIC50値から計算する。式中、Dは実験に使用される放射性リガンドの濃度に等しく、Kdは飽和結合解析で決定される放射性リガンドの平衡結合親和定数に等しい(hIRとhIGF−1RのKdはそれぞれ0.124nMおよび0.130nMである)。以下に報告する幾何平均Kiは10^(平均対数Ki)であり、式中、平均対数Ki=平均(Ki1+Ki2+Ki3...Kin)であり、独立実験の数(n)は3以上である。しかし、ヒトIGF−1に関して以下に記載する場合、nは2である。
【0073】
実施例1に記載する方法で調製した以下のペグ化インスリンリスプロ化合物は、上述のhIR結合アッセイにおいて30nM未満の幾何平均Kiを有している。そのペグ化インスリンリスプロ化合物とは、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物10(a)、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物8(a)、平均分子量約60kDaを有する分岐mPEG−MALを用いて調製した化合物15(c)、平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、および平均分子量20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)である。hIRおよびhIGFR結合アッセイでは、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)の幾何平均Kiは、それぞれ3.07nM+/−0.32nM(+/−S.E.M、n=6)および84.3nM超(SEM=未決定、n=6)である。更に、40kDa、30kDaまたは20kDaの線形mPEG−NPCを用いて実施例2に記載の方法で調製した異種ペグ化インスリンリスプロ生成物も、上述のhIR結合アッセイにおいて30未満の幾何平均Kiを有している。上述のhIR結合アッセイでは、インスリンリスプロの幾何平均Kiは0.22+/−0.072nM(+/−SEM、n=4)である。上述のhIGFR結合アッセイにおいては、上記の全化合物が75nMを超える幾何平均Kiを有しており、ヒトIGF−1の幾何平均Kiは1.51+/−0.23nM(+/−SEM、n=2)である。
【0074】
ペグ化位置B28はhIR親和性を約10倍減少させ、これらインスリンリスプロのペグ化種をhIRの弱いアゴニストにしていることを、このデータは示している。更に、これらペグ化インスリンリスプロ種は、上述の条件下のアッセイにおいて、測定可能なIGFR−1結合特性を全く有していない。
【0075】
(実施例4:インスリン受容体リン酸化全細胞アッセイを用いたペグ化インスリンリスプロの効力評価)
本発明のペグ化インスリンリスプロ化合物は、商業的に入手可能な異種時間分解蛍光測定アッセイであるDELFIA(登録商標)(Perkin−Elmer)を用いて、機能活性を評価してもよい。簡単に言えば、ヒトインスリン受容体を過剰表現している293HEK細胞をトリプシン処理し、無血清培地(SFM)(脂肪酸のない0.1%BSAを含むDMEM)を用いて、ポリDリジンコートCostar96ウェルハーフエリア組織培養プレートの60,000細胞/ウェルで平板培養する。その細胞培養プレートをCO2インキュベーター中37℃で一晩インキュベーションする。抗インスリン受容体A鎖mAb8314キャプチャープレートも、Costar96ウェル黒色ハーフエリアマイクロタイタープレートを使って前の晩に調製し、30μLの抗インスリン受容体A鎖mAb8314(Soos,M.A.ら,Biochem J 235:199−208(1986)、マサチューセッツ州ケンブリッジのアブカム社などから商業的に入手可能)を用いて4℃で一晩処理し、10mM炭酸ナトリウムで1μg/mLに希釈する。結合していないmAb8314を除去するため、mAb8314キャプチャープレートを50mMトリス、pH7.5、150mMのNaClおよび0.1%ツイーン(TBST)で4度洗浄する。次にTBST中の1%BSAを用いて4℃で1時間を超える間、mAb8314キャプチャープレートをブロックする。ブロッキング後、キャプチャープレートをTBSTで2度洗浄して過剰のBSA溶液を除去する。キャプチャープレートをブロッキング緩衝液に浸漬したら、細胞培養プレートをインキュベーターから取り出し、温度を室温と同じにする。試験化合物をSFMで連続的に希釈する。インスリン受容体の自己リン酸化を刺激するため、希釈したテスト試薬50μLを細胞単層に加える。室温で30分反応させた後、試験化合物を吸引除去し、50μLの2x溶解緩衝液(2%NP40、100mMトリス、pH7.4、300mMのNaCl、EDTAを含むRoche Complete(商標)プロテアーゼインヒビターおよび4mMバナジン酸塩)を添加することにより、反応を停止させる。溶解緩衝液中、室温で30分反応させた後、30μLの溶解物を30μLのユウロピウムN1抗ホスホチロシンPY20抗体であるEu−N1−Py20(Perkin Elmer)を含むブロックされたキャプチャープレートに移し、10mMのヘペス緩衝液、140mMのNaClおよび0.1%ツイーンで50ng/mLに希釈する。この混合液を室温で1時間インキュベーションし、その後TBSTで6度洗浄して、結合しなかったEu−N1−Py20および細胞分解物を除去する。その後10分間、発生するシグナルに合わせて間歇的に振盪しながら、50μLのEnhancement Solution(Perkin Elmer)でインキュベーションする。リン酸化したインスリン受容体は、時間分解蛍光測定ユウロピウム設定でWallac Victorを用いて定量化する。リン酸化レベルはインスリン(100nM)の最大刺激用量に対する応答%として計算される。インスリン類似体の効力は、用量応答の4パラメーター適合を用いて、EC50用量として計算される。実施例4に記載されるアッセイにおいて、実施例1に記載される方法で調製した、15nM未満のEC50を有するペグ化インスリンリスプロ化合物には、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物10(a)、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)および平均分子量約20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)が含まれる。実施例4に記載されるアッセイでは、線形mPEG−MALを用いて調製した化合物9(a)のEC50は10.88nMである。約40kDa、約30kDaまたは約20kDaの平均分子量を有する線形mPEG−NPCを用いて実施例2に記載の方法で調製した異種ペグ化インスリンリスプロ生成物も、実施例4に記載のアッセイにおいて15nM未満のEC50を有している。約40kDaの平均分子量を有する線形mPEG−MALを使って調製した化合物8(a)と約40kDaの平均分子量を有する線形mPEG−MALを使って調製した化合物9(a)の50対50および70対30の混合物も、実施例4に記載のアッセイにおいて15nM未満のEC50を有している。同じアッセイにおいて、ヒトインスリンとインスリンリスプロのEC50はそれぞれ2.3nMおよび0.7nMである。
【0076】
ペグ化位置B28はhIRインビトロ活性を約10倍から20倍減少させ、これらインスリンリスプロのペグ化種をhIRの弱いアゴニストにしていることを、このデータは示している。更に、これらペグ化インスリンリスプロ種は、測定可能なIGFR−1結合特性を全く有していない。
【0077】
(実施例5:1型糖尿病ラットモデルを用いた、ペグ化インスリンリスプロのインビボ効力および薬物動態学的プロフィルの評価)
試験開始3日前に、10週目のHarlan Sprague−Dawley雄ラット(Harlan,Indianapolis)(体重250−280g)の尾静脈に0.5Mクエン酸(pH4.5)中のストレプトゾトシン(STZ)45mg/kgを静脈内投与する。試験開始時に、ラットを体重および血糖に基づいて群分けする。この研究には、血糖が400mg/dLと550mg/dLの間のラットだけを含める。試験開始日の朝、試験化合物をいくつかの所定用量の1つでラットに単回皮下注射する。尾静脈から定期的に血液の重複抜き取りを行い、EDTA二ナトリウムを含むチューブに集める。血糖値をグルコメーターで測定する。静脈血試料から血漿も集め、商業用のラットインスリン・ラジオイムノアッセイを用いて、投与された薬物の血漿濃度を測定する。経時的血糖(mg*h/dL)曲線の下の面積を各ラットについて計算し、それを4パラメーターロジスティック回帰分析に使用してED50を決定する。このアッセイでは、平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)、平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)および平均分子量約20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)の効力(ED50)は、典型的な用量応答曲線に基づいて、それぞれ241nmol/kg、138nmol/kgおよび69nmol/kgである。更に、当該化合物は、568nmol/kgの単回皮下注射によって、STZ処置ラットの血糖値を少なくとも36時間、この種のラットにとっては正常なレベル(100mg/dL以下)にまで低下させることができた。一方、インスリンデテミルは、上記のエッセイにおいて、568nmol/kg単回投与によって血糖値を5−6時間正常化する。
【0078】
薬価パラメーターの評価に加えて、ラットにおける試験化合物の平均薬物動態パラメーター値も重複血液試料を用いて測定する。薬物動態パラメーターは、モデル非依存性の方法(WinNonlin Pro)を用いて計算する。得られる薬物動態パラメーター値は、用量の関数として非線形を示す。報告される値の範囲は、テストされる最高用量(568nmol/kg)と最低用量(5.6nmol/kg)の間の薬物動態パラメーター値に対応する。
【0079】
平均分子量約20kDaを有する線形mPEG−MALを用いて調製した化合物9(b)の薬物動態的結果は、最高濃度に至るまでの時間(T最大)が6時間から12時間の範囲、見かけのクリアレンス速度(CL/F)が0.05L/h/kgから0.14L/h/kgの範囲、見かけの分配量(V/F)が0.6L/kgから7.2L/kgの範囲、除去半減期(t1/2)が8.5時間から34.5時間を示した。
【0080】
平均分子量約30kDaを有する線形mPEG−MALを用いて調製した化合物9(a)の薬物動態的結果は、T最大が12時間、CL/Fが0.05L/h/kgから0.13L/h/kgの範囲、V/Fが0.6L/kgから2.0L/kgの範囲、t1/2が8.3時間から11.0時間を示した。
【0081】
平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)の薬物動態的結果は、T最大が12時間から24時間の範囲、CL/Fが0.06L/h/kgから0.2L/h/kgの範囲、V/Fが1.0L/kgから7.5L/kgの範囲、t1/2が11.1時間から48.5時間を示した。
【0082】
568nmol/kgのインスリンリスプロをSTZ処置雄ラットに同様にして投与すると、測定値は、t1/2が約1時間、CL/Fが約1.2L/h/kgである。
【0083】
18.9nmol/kgから568nmol/kgの範囲のインスリンデテミルをSTZ処置雄ラットに同様にして投与すると、t1/2が1.9時間から3.1時間の範囲、CL/Fが約0.8L/h/kgから1.7L/h/kgの範囲が観察される。
【0084】
デテミルと例示のペグ化インスリンリスプロ化合物の間の見かけのCL比は、薬物動態学的な複雑性の故に、測定に使用される用量によって結果が異なる。しかし、20kDaまたは40kDaのPEGと結合した例示のペグ化インスリンリスプロ化合物は、STZ誘発糖尿病ラットにおいて、見かけのクリアランスがデテミルよりも約5倍から30倍緩慢であることを、実施例5に記載の試験が示している。
【0085】
(実施例6:2型糖尿病ラットモデルを用いた、ペグ化インスリンリスプロのインビボ作用期間および薬物動態学的特性の評価)
40kDa線形PEGを有する化合物9(a)のグルコダイナミック(Glucodynamic)活性は、ビヒクル対照(PBS)または平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)517nmol/kgの単回皮下投与後、雄のZDF(fa/fa)ラット(n=4ラット/群)で評価する。薬物動態特性および薬価特性を評価するため、試料を連続して集める。平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)517nmol/kgを雄のZDF(fa/fa)ラットに単回皮下投与することにより、血糖値が統計学的に有意なレベルまで低下し、それが少なくとも7日間持続した(偽薬に対してp<0.05)。平均分子量約40kDaを有する線形mPEG−MALを用いて調製した化合物9(a)への曝露も7日間検証可能である。
【0086】
(実施例7:コバルトイオンの存在下でフェノールを含むPEG−B28(92.4%)A1(7.6%)インスリンリスプロ、インスリンリスプロまたはその混合物(70%PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロ対30%インスリンリスプロ)のフェノール系防腐剤滴定)
PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロまたはインスリンリスプロを、20mMのKSCNおよび50mMのトリスClO4を含む溶液にpH8.0で溶解する。上記蛋白質の標的濃度は、蛋白質含有量(ε280=1.05(mg/mL)−1cm−1)に基づいて約4mg/mLである。塩化コバルト(41.9mg)を水1mLで溶解し、コバルトイオン濃度0.176Mの原液を得る。コバルトイオン対インスリン6量体の最終モル比が4になるように、コバルト原液のアリコート(蛋白質の濃度によって異なるが、約2μL)を0.8mLの蛋白質溶液に添加する。6量体化を評価するため、コバルト配位化学における歪みをフェノール系防腐剤濃度の関数として574nmでモニターした。具体的には、滴定終了時点での最終容量が0.84mLを超えないように、濃縮フェノール溶液(0.564M)をマイクロリットルアリコート用いて、蛋白質溶液0.8mLを滴定する。フェノールの各アリコートと400nmから800nmまでの可視スペクトルの溶液を集めた後、最終溶液を最低20分間攪拌する。574nmで記録した吸光度は、HISB10配位結合コバルトモル濃度、すなわちインスリン6量体のHISB10部分は二価の金属イオン2つと配位結合するという知識に基づいて6量体蛋白質濃度掛ける2でその吸光度を割ることによって、モル吸光係数に変換する。PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロとインスリンリスプロの70/30(モル対モル)混合製剤を調製するには、蛋白質をまず混合し、次にコバルトを添加し、それからフェノール系防腐剤で滴定する。
【0087】
インスリンリスプロでは、天然の配列であるB28の位置のプロリンとB29の位置のリジンが、野生型のヒトインスリンと比較して逆転している。この逆転によりB鎖のC末端で立体配座的な移動が生じ、その結果、2量体を形成するリスプロインスリン単量体の能力が立体的に妨害される。従って、2量体会合定数が野生型ヒトインスリンの2量体会合定数と比べて300倍減少する。驚いたことにかつ予想外にも、表1に示されるように、野生型のヒトインスリンと比べて既に脆弱化しているインスリンリスプロの2量体化ドメインの近くで6つの20kDaPEG部分が結合しているにもかかわらず、PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロは、Humalog(登録商標)で使用される調剤条件と類似の条件で、二価の金属イオンおよびフェノール系防腐剤の存在下、6量体の複合体として会合することを結果が示している。更に、PEG20kDaB28(92.4%)A1(7.6%)インスリンリスプロとインスリンリスプロの70/30混合物も6量体の複合体を形成する能力が証明されており、基礎インスリンおよび迅速に作用するインスリンの予備混合製剤を即座に、および/または安定して調製できることをこの事実は支持している。
【0088】
(表I)
【表1−1】
【0089】
【表1−2】
【0090】
(実施例8:亜鉛、フェノールおよび/またはカルシウムと共に製剤されたPEG20kDaB28(92.4%)/A1(7.6%)インスリンリスプロの6量体状態の分析)
インスリンおよび特定のインスリン類似体の化学的貯蔵安定性および使用時安定性は、溶液中で個別の6量体複合体を形成する能力によって促進される。二価の金属イオン(Zn+2またはCo+2)およびフェノール系防腐剤(フェノールまたはm−クレゾール)の存在下でインスリンまたはインスリンリスプロを6量体化する能力は、AsnA21の脱アミド化を遅らせ、その結果、高分子量の粒子(HMWP)の形成を最小限に抑える。
【0091】
PEG20kDa−LysB28インスリンリスプロが6量体を形成する能力を評価するため、A276nm=8.6mgインスリンリスプロ/mLまたは38.2mgペグ化インスリンリスプロコンジュゲート/mLに基づく最初の蛋白質濃度を有する、PEG20kDaB28(92.4%)/A1(7.6%)インスリンリスプロをpH6.7で一晩、水中で透析する。次に、透析した蛋白質を水で希釈して、蛋白質濃度を4.6mg/mLまたは20.6mgのペグ化蛋白質コンジュゲート/mLに調整する。4x緩衝液原液をpH7.0で調製する。リン酸塩緩衝液の最終濃度は40mM、m−クレゾールの最終濃度は12.8mg/mLとする。酸化亜鉛原液は酸化亜鉛を1NのHCl0.5mLに溶解し、次に水で希釈することにより調製し、最終亜鉛濃度を0.097Mとする。蛋白質濃度4.6mg/mLのPEG20kDaB28(92.4%)/A1(7.6%)インスリン450μLを亜鉛原液のアリコート(添加される亜鉛原液の最大全容量=2.5μL、またはPEG20kDaB28(92.4%)/A1(7.6%)インスリン6量体当たり4亜鉛イオンに相当する量)および4xリン酸塩緩衝液原液150μLと混合することにより、種々の亜鉛濃度(0μMから400μM)を有する、近UV円偏光二色性(CD)分析用の溶液試料を調製する。最終pHは、必要に応じてpH約7.0に調整する。PEG20kDaB28(96%)/A1(4%)インスリンリスプロまたはインスリンリスプロの6量体会合を、0.2cmセルを用いて、近UV円偏光二色性により、ジスルフィド変化に感応性の高い250nmでモニターする。平均残基楕円率をモル6量体当たりのモル亜鉛の比率に対してプロットする。
【0092】
驚いたことにかつ予想外にも、表IIに示されるように、野生型のヒトインスリンと比べて既に脆弱化しているインスリンリスプロの2量体化ドメインの近くで6つの20kDaPEG部分が結合しているにもかかわらず、PEG20kDaB28(92.4%)/A1(7.6%)インスリンは、Humalog(登録商標)の調剤条件と類似の条件で、二価の金属イオンおよびフェノール系防腐剤の存在下、6量体の複合体として会合することを結果が更に示している。
【0093】
6量体促進テスト製剤において、6量体化およびリガンド結合がPEG20kDaB28(92.4%)/A1(7.6%)インスリンの熱安定性に及ぼす影響についても、CD熱変性実験で調べた。全体的な信号変化はZn2+結合試験で使用した250nmよりも240nmの方が大きかったが、全溶液吸光度が高品質のCDデータが得られるほど十分低かったので、熱変性試験で用いた波長は240nmであった。1mmのキュベット中、240nmにおける熱スキャンデータを5℃から約95℃の間で集めた(最終温度は試料間でわずかな違いがあった)。スキャン速度およびデータピッチは1℃/分、周波数帯域幅は1.5nm、応答時間は8秒であった。熱変性データは、生信号(240nmにおける1000分の1度単位)およびFunf(T)=[Yobs(T)−Ynat(T)]/[Yunf(T)−Ynat(T)]の式で与えられる見かけの変性割合(Funf)の両方としてプロットしてよい。式中、Yobs(T)は温度の関数として観察される信号であり、Ynat(T)およびYunf(T)は、それぞれ未変性ベースラインおよび変性ベースラインの直線的外挿値である。変性開始温度は、Funfが未変性ベースライン(Funf=0)から増加し始める温度として定義され、中間温度はFunf=0.5の温度である。
【0094】
本質的に上述の方法で行われるCD熱変性実験が示すところによれば、PEG20kDaB28(92.4%)/A1(7.6%)インスリン6量体とインスリンリスプロ6量体を16mMトリス、pH7.2、3.1mg/mLのm−クレゾールおよび亜鉛で同様に製剤した場合、前者の熱安定性は後者の熱安定性よりも実質的に減少した。更に、上述のCD熱変性試験は、PEG20kDaB28(92.4%)/A1(7.6%)インスリンリスプロの融点がカルシウムと塩化物イオンの濃度に依存して有意に上昇することも見出した(データ省略)。具体的には、変性開始温度は16mMトリス、pH7.2、3.1mg/mLのm−クレゾールおよび亜鉛中では約30℃であるが、75mM塩化カルシウムを含む同じ製剤中では劇的に約50℃まで上昇する。製剤に塩化カルシウムの代わりにNaCl(25mMから150mMのNaCl)を加えた場合も同様の効果が観察された。従って、PEG20kDaB28インスリンリスプロ化合物を含む医薬組成物において、カルシウムおよび/または塩化物は非常に有効な6量体促進賦形剤であり、貯蔵中に当該医薬組成物の化学的および/または物理的安定性を増大する効果があろう。
【0095】
(表II:亜鉛/6量体比の関数としての平均残基楕円率(MRE)変動)
【表2】
【0096】
(実施例9:PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロの生成)
150mMの第二リン酸ナトリウムおよび50mMのEDTAを含む緩衝液を約4℃から約6℃で150mMの第三リン酸ナトリウムと混合し、10.85と11.10の間のpHを持つ緩衝液を得る。インスリンリスプロ結晶(30−60mg/mL)を約4℃から約6℃で緩衝液に徐々に添加し、結晶溶解中に凝集物の形成が生じるのを避けるため穏和に攪拌する。
【0097】
重量平均分子量約20kDa+/−約2kDaのPEGを有するモノメトキシポリエチレングリコールp−ニトロフェニルカーボネート(mPEG−NPC)を60mg/mLから120mg/mLの濃度で、冷水(4−6℃)に溶解する。その場合、必要量の冷水を容器に入れ、攪拌して渦流を作り、適切かつ急速な分散が起きるように、mPEG−NPCを渦流の眼に徐々に注ぐ。mPEG−NPC粉末は細かい粉末であり、分散させるとかなりの量の気泡が容器中に放出される。容器中のmPEG−NPC溶液は、容量に応じて30分間から60分間脱泡する。
【0098】
上記の方法で調製したインスリンリスプロ溶液は機械的攪拌用の冷却塔付き容器に移す。その容器には温度とpH測定用の装置が備わっている。攪拌は、レイノルズ数に基づいて乱流部分で操作される標準回転翼を用いて行う。mPEG−NPC(PEG)溶液は、全PEG添加時間が3時間から5時間になる速度で、容器中で測定する。冷却塔の温度は4℃から6℃に維持し、混合は継続して行う。反応液のpHは、必要量の150mM第三リン酸ナトリウムを添加することにより、10.85から11.10の間に維持する。PEG対インスリンリスプロの最終モル比が2.5から4.5の範囲になるまで、PEGを添加する。
【0099】
PEG添加の終了時点で、冷却塔の温度を60分以内に25℃から30℃に上昇させ、pHを約10.7から約11.0に維持しながら、反応混合物をその温度で約3時間から約6時間インキュベーションする。インキュベーション期間の終了時点で、2x緩衝液(100mM酢酸/酢酸ナトリウム、pH4.0)を添加することにより反応混合物をクエンチし、同じ2X緩衝液で希釈することにより伝導率(2.5mS/cm)および濃度(3−5mg/mL)を調節する。
【0100】
反応混合物は、適切な樹脂(高速流通SPセファロース樹脂など)を充填した陽イオン交換クロマトグラフィ(CEX)カラムを使って精製する。樹脂は層高約15cmから約30cmになるまでカラムに充填し、100mMの酢酸ナトリウム(緩衝液A)で平衡化し、希釈した反応混合物(生成物5−8g/樹脂L)を低いpH(約2.5から約4.0)かつ適切な流速(約50cm/時間から約90cm/時間)で充填する。モノペグ化生成物および未反応の蛋白質が樹脂上で選択的に結合するが、マルチペグ化した副生成物および過剰の試薬のほとんどはカラムを通過する。希釈した塩濃度(20−30mM)の緩衝液Aを用いて弱く吸着したマルチペグ化副生成物を洗い流し、次に塩濃度の高い(50−70mM)緩衝液(8−12CV)を使った勾配溶出によってペグ化生成物を樹脂から優先的に除去すると同時に、未反応の蛋白質はカラムに残ったままになる。生成物を集め(3−5CV)、カラムは塩濃度の高い(100mM)緩衝液Aで洗浄して未反応の蛋白質を除去する。
【0101】
3−5mg/mLのCEXカラム主流(3−5CV)は、接線流濾過により、標準のフラットシート膜(3−5kDa分子量カットオフ)を用いて40−80mg/mLまで濃縮する。この工程は、初期濃度を経て、緩衝液を交換し、目的の最終濃度まで行われる。操作フラックスは、工程中ずっと10−20L/m2(フィルター面積)/時間(LMH)に維持し、膜間差圧(TMP)は約15psiから約35psiに維持する。
【0102】
最終濃度のバルク活性医薬成分溶液は、適切な温度(−20℃から−70℃)で凍結させ、適切な温度(−20℃から−70℃)で保存する。
【0103】
(実施例10:イヌにおけるペグ化インスリンリスプロの薬物動態学的プロフィル)
2歳から4歳の雌のビーグル犬(体重7−10kg)に、例示の試験化合物18.9nmol/kgを皮下投与する。血液試料を定期的に腕頭静脈または伏在静脈から得て、それをEDTA二ナトリウムを含むチューブに集める。血漿を静脈血サンプリングで集め、商業用のインスリンラジオイムノアッセイを用いて血漿中の投与薬の濃度を測定する。本質的に実施例2に記載した方法で調製した例示化合物である、平均分子量約40kDa、約30kDaおよび約20kDaを有する線形mPEG−NPCを用いて調製した化合物11の各々について、薬物動態学的プロフィルおよびパラメーターを決定した。
【0104】
薬物動態学的パラメーターは、モデル非依存性の方法(WinNonlin Pro)を用いて計算した。平均分子量約20kDaを有する線形mPEG−NPCを用いて調製した化合物11は、最高濃度に至るまでの時間(T最大)が約12時間、見かけのクリアレンス速度(CL/F)が約0.046L/h/kg、最大濃度(C最大)が約14nM、除去半減期(t1/2)が約14時間を示した。
【0105】
平均分子量約30kDaを有する線形mPEG−NPCを用いて調製した化合物11は、T最大が約24時間、CL/Fが約0.027L/h/kg、C最大が約18nM、t1/2が約23時間を示した。
【0106】
平均分子量約40kDaを有する線形mPEG−NPCを用いて調製した化合物11は、T最大が約24時間、CL/Fが約0.026L/h/kg、C最大が約15nM、t1/2が約20時間を示した。
【0107】
インスリンデテミルも同様に18.9nmol/kgを雌のビーグル犬に投与し、T最大が約1.3時間、CL/Fが約0.12L/h/kg、C最大が約23nM、t1/2が約3.5時間を示した。
【0108】
表IIIはイヌにおける比較パラメーターを示す。この試験に使用したインスリン特異的なRIAは、ペグ化インスリンリスプロと内生的インスリンの両方を検出する。
【0109】
(表III:イヌにおけるペグ化インスリンリスプロのPKパラメーター)
【表3】
18.9nmol/kg皮下投与、n=3/群
【0110】
(実施例11:ヒトにおける平均「平坦化」および用量の推定)
改善された基礎インスリン療法の主要な基準は、患者への1日1回の投与に適した、真に平坦なプロフィルを達成する能力である。十分な平坦さはピーク/トラフ(PT)比が2未満の場合であると定義される。比較目的で述べると、公表されているPKプロフィルに基づいて計算したPT比は、デテミルで約4から9、グラルギンで約1.2から2.6である。18.9nmol/kgの例示化合物および同量のインスリンデテミルに関するラットとイヌのPKデータ(それぞれ実施例5および10を参照)は1−CMT PKモデルに適合させ、ka、CL/FおよびV/Fのパラメーターで表示した。次に各PKパラメーターを相対成長式P=aBWbに適合させる。式中、bはka、CL/FおよびV/Fに関して各々−0.25、0.75および1に固定し、aは適合パラメーターである。ヒト平均推定値を各PKパラメーターについて取得し、ヒトに1日量を投与後、シミュレーションは平均プロフィルを生成した。30kDaおよび40kDaペグ化インスリンリスプロコンジュゲートは類似しているので、シミュレーションは20kDaペグ化インスリンリスプロ、40kDaペグ化インスリンリスプロおよびインスリンデテミルだけを示してある。20kDaおよび40kDaペグ化インスリンリスプロ化合物のピーク/トラフ(PT)比は、インスリンデテミルのピーク/トラフ(PT)比よりも劇的に平坦であることを、生成されたシミュレーションが示している。図1はシミュレーションの結果を示す。
【0111】
効能に要求されるペグ化インスリンリスプロのヒト用量を推定するストラテジーは、ラットモデルにおけるペグ化インスリンリスプロとインスリンデテミルとの間の相対効力が臨床における相対効力と類似しているという推定の下に、既知の臨床コンパレーター(インスリンデテミル)をラット効能モデルの内部対照として用いることである。臨床におけるペグ化インスリンリスプロの1日必要量は、以下の式を用いて計算できる。
【数1】
表IVは各ペグ化インスリンリスプロコンジュゲートの相対効力比を示してある。表Vは、ラット、イヌおよびヒト(推定)における例示ペグ化インスリンリスプロ対デテミルの見かけの相対クリアランス比を示してある。
【0112】
(表IV:ラットにおけるペグ化インスリンリスプロコンジュゲートおよびインスリンデテミルの相対濃度ベース効力)
【表4】
【0113】
(表V:インスリンデテミル対ペグ化インスリンリスプロ化合物の相対CL/F比)
【表5】
【0114】
表IVの相対効力推定および表Vの相対CL/F推定を用いると、ヒトにおけるペグ化インスリンリスプロコンジュゲートの平均用量推定値は、20kDaペグ化インスリンリスプロ化合物と40kDaペグ化インスリンリスプロ化合物に関して、それぞれ4.2nmol/kgおよび6.9nmol/kgである。かかる推定値は、2型糖尿病患者に関してデテミルのラベルに記載されている臨床平均1日量18.5nmol/kgのインスリンデテミルに基づいている。時間作用および効力の両方を考慮した場合、臨床最大平均投与推定量はインスリンデテミルよりも約3倍低い。最善のケースでは、20kDaペグ化インスリンリスプロコンジュゲート、30kDaペグ化インスリンリスプロコンジュゲートおよび40kDaペグ化インスリンリスプロコンジュゲートの臨床平均投与推定量は、インスリンデテミルよりも約20倍および約45倍低い。
【0115】
(実施例12:PEG20kDaB28(約95%以上)/A1(約5%以下)インスリンリスプロおよびグラルギンを健康なボランティアに単回投与した後のグルコース注入速度)
本質的に実施例9に記載の方法で調製したPEG20kDaB28(約95%以上)/A1(約5%以下)インスリンリスプロ(LY)の単回用量を用いて、3部分からなる最初のヒト用量試験を行った。パートAは3つの試験期間からなり、第1期間で被験者にはLY用量が皮下(SC)注射され、第2期間でインスリングラルギン用量(0.5U/kg)が注射され、第3期間で別のLY用量が注射された。パートBは非盲検、単回用量、2回の反復期間試験で、被験者には24時間および36時間のグルコースクランプ法を用いて、両期間ともLYの単回用量が投与された。パートCは非盲検、2期間、単回用量、一定のシーケンス、コンパレーター制御試験で、第1期間で被験者にはLYのSC用量0.5mg/kgが単回投与され、第2期間ではインスリングラルギンのSC用量0.8U/kgが単回投与された。被験者にはパートAとパートCの各期間で24時間のグルコースクランプ法が施され、パートBの各期間ではより長い期間(36時間まで)グルコースクランプ法が施された。パートA、BおよびCにおいて、LYは以下のような単回用量が皮下投与された。
パートA用量:0.0025、0.0125、0.075、0.325mg/体重kg
パートB用量:0.15、0.225mg/kg
パートC用量:0.5mg/kg
【0116】
被験者には、ペグ化インスリンリスプロ化合物の単回用量またはコンパレーターとしてインスリングラルギンの単回用量(0.5U/kg)、そして第3期間にLYの別の単回用量が投与される。全ての治療期間で、各インスリン化合物の注射後、被験者にはオイグリセミッククランプ法を最高24/36時間施す。インスリン作用のグルコダイナミック(GD)尺度を提供する立証済みの経時的GIRを参考にして、グルコース注入速度(GIR)を正常血糖を維持するように調節する。オイグリセミックグルコースクランプ法の狙いは、インスリン化合物の用量を投与後、グルコース注入期間中ずっと、正常血糖が維持されることである。ここでは、内因性のインスリン分泌および肝グルコース産生が最少であり、グルコーススペースから輸送されるグルコース(すなわち代謝されたグルコース)は全て投与された外因性インスリンの直接の結果であることが想定されている。この場合のGIRは、経時的なインスリン作用のGD尺度となる。グルコースクランプ試験は全て、一晩(約8時間の)絶食後行われる。試験日の朝、容量ポンプの制御下で20%デキストロース溶液(緩衝液でpHを中性近くに調整)を投与する目的で、小さいカテーテルを1本の腕(理想的には肘窩)の静脈に挿入する。静脈グルコース採取のため、別のカテーテルを理想的には手首または手に挿入する。動脈血化した静脈血を採取するため、その部分を加熱装置で約55−60℃に加熱する。血液試料をベッドサイドで採取し、自動グルコースオキシダーゼ法を用いて直ちに全血中グルコース濃度を測定する。基礎血液採取および約30分間の安定期間の後、各被験者にインスリン化合物の用量が皮下投与される。インスリン化合物の皮下投与開始を時間ゼロと定義する。インスリン投与完了後、血糖を測定するために頻繁に血液を採取しながら、最高24時間または36時間正常血糖を維持するため、グルコースを種々の速度で静脈内に注入する。
【0117】
血液採取は、投与前約30分間約10分ごとに行い、投与後最初の2時間、5−10分ずつ継続(任意に、2.5分ずつの頻度で採取してもよい)し、その後クランプの最後まで10−30分間隔に頻度を下げる。
【0118】
グルコースクランプ中、グルコース注入速度を調節して、個々の被験者の所定標的血糖濃度を維持する。標的濃度は空腹時血糖値に近い値が好ましい。グルコースクランプの狙いは、投与前標的値(空腹時平均血糖値よりも5mg/dL下回る値として定義される)の+5%内に血糖濃度を維持することである。従って、GIRは変動しても、血糖濃度は一定に保たれる。だから、変動するグルコース注入速度は、試験インスリン化合物の活性を反映している。試料の血糖値および注入速度がクランプ中変動することは立証済みである。
【0119】
本質的に実施例12に記載の方法で行われた試験は、LYが理想的な「基礎」インスリンであり、作用期間が長く、見かけの半減期が24時間と44時間の範囲にあり、基礎特性すなわちピーク/トロフ比が2未満(図2)であるという特徴を有することを、ヒトにおいて実証した。更に、LYの作用期間はインスリングラルギンの作用期間よりも長い(図2)。グルコダイナミックスにおける被験者内変動は30%未満(データ省略)であり、これはグラルギンと同等またはそれよりも優れている。最後に、試験のパートC(0.5mg/kg)からのグルコダイナミック(GD)データは、LYのGIRプロフィルを「ピークなし」と示し、36時間を超える期間GDを維持し、グラルギンのピークGIR応答(0.5U/kg、データ省略)を凌駕した。
【0120】
(実施例13:PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ医薬組成物の化学的および物理的安定性)
実施例7および8で上述したように、PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロは、Humalog(登録商標)と類似の製剤条件において、二価の金属イオンおよびフェノール系防腐剤の存在下、6量体複合体として会合できる。従って、インスリンリスプロの商業用溶液製剤(すなわちHumalog(登録商標)溶液製剤)と類似のPEG20kDaB28インスリンリスプロ製剤に関して、化学的安定試験および物理的安定試験が行われた。試験対象の医薬製剤の化学的安定性は、指示された貯蔵期間中、様々な温度において、最初の時点と有意に異なる変化が種々の分析特性で検出されない場合、許容可能であると見なされる。試験対象の医薬製剤の物理的安定性は、視覚評価において粒子が全く観察されず、チオフラビンT蛍光顕微鏡検査法による検査でフィブリルやゲル形成が観察されない場合に、許容可能であると見なされる。
【0121】
PEG20kDaB28インスリンリスプロ1モル当たり0.5モルの亜鉛、16mg/mLのグリセリンおよび3.15mg/mLのm−クレゾール(リン酸塩緩衝液またはクエン酸塩緩衝液でそれぞれpH7.0またはpH6.5に調整)を含むPEG20kDaB28インスリンリスプロ製剤を調製し、化学的安定性および物理的安定性の両方を試験した。安定性への亜鉛の影響を評価するため、亜鉛を含まない類似の製剤も試験した。35℃の試料は約1ヶ月間貯蔵後ゲル粒子を形成し、25℃の試料は約2ヶ月の貯蔵後有意な数の粒子および気泡を形成した。視覚的評価ではクエン酸塩緩衝液の試料の方がリン酸塩緩衝液の試料よりもひどいように見えたが、クエン酸塩緩衝液とリン酸塩緩衝液の両方並びに亜鉛を含む製剤と亜鉛を含まない製剤の両方が、35℃の加速条件で類似の化学的/物理的安定性を示した。インスリンリスプロ対照製剤は透明であった。クエン酸塩緩衝液とリン酸塩緩衝液の両方が、5℃で少なくとも13ヶ月間、許容可能な化学的安定性を示した。
【0122】
その後の調査では、フェノール系防腐剤であるm−クレゾールは、高温(>25℃)に曝されたPEG20kDaB28インスリンリスプロと組み合わせた場合、ゲル化を促進することが示された。興味深いことに、m−クレゾールをmPEGだけ(活性化しているか否かに関係なく)に添加した場合、高温に曝しても、ゲル粒子は生じなかった。
【0123】
インスリンリスプロの基本的調剤方法では、高温でPEG20kDaB28インスリンリスプロ化合物に許容可能な安定性が与えられなかったので、PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ化合物を含み、改善された化学的および物理的安定性を示し、非経口的に投与される医薬組成物として商業化に適した医薬組成物を開発した。
【0124】
PEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ(15mg/mL)の以下の製剤は、40℃で1週間、30℃で1ヶ月、25℃で3ヶ月および5℃で8ヶ月以上、化学的および物理的に許容可能な安定性を示した。
1)16mMのトリス緩衝液、pH7.0−8.0、10mMの塩化カルシウム、20mg/mLの砂糖(ショ糖またはトレハロース)、3mg/mL(28mM)のm−クレゾールおよびPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ1.0モル当たり0.5モルの亜鉛。
2)16mMのトリス緩衝液、pH7.0−8.0、10mMの塩化カルシウム、3mg/mLのポロキサマー、3mg/mL(28mM)のm−クレゾールおよびPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ1.0モル当たり0.5モルの亜鉛。
3)5mMのリン酸塩緩衝液、pH7.0、130mMのグリセリン、3mg/mL(28mM)のm−クレゾール、3mg/mLのポロキサマーおよびPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ1.0モル当たり0.3モルの亜鉛。
【0125】
亜鉛、m−クレゾール、カルシウムなどの6量体促進賦形剤を含むPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ製剤は、一般的に、物理的および化学的に高い安定性を示す。特に高温状態ではそうである。亜鉛およびm−クレゾールを含むPEG20kDaB28(約95%)/A1(約5%)インスリンリスプロ製剤にカルシウム、塩化物および/またはNaClを添加すると、40℃以上の温度に曝された製剤の物理的安定性が更に向上する。更に、リン酸塩緩衝液およびクエン酸塩緩衝液を用いた製剤は、一般的に、トリス緩衝液を用いた製剤よりも物理的安定性が低かった。
【特許請求の範囲】
【請求項1】
以下の式のペグ化インスリンリスプロ化合物:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pはウレタンまたはチオエーテル共有結合によって、Aの1位でグリシンのアルファアミノ基、Bの1位でフェニルアラニンのアルファアミノ基またはBの28位でリジンのイプシロンアミノ基に結合している)、またはその製薬上許容可能な塩。
【請求項2】
前記PEGは、Bの28位におけるリジンのイプシロンアミノ基にウレタンまたはチオエーテル共有結合によって結合している、請求項1に記載の化合物。
【請求項3】
ヒトインスリン受容体に対して約30nM以下のKi値を持つことで更に特徴づけられる、請求項1または2に記載の化合物。
【請求項4】
ヒトインスリン受容体に対して約20nM以下のKi値を持つことで更に特徴づけられる、請求項1または2に記載の化合物。
【請求項5】
ヒトインスリン受容体に対して約10nM以下のKi値を持つことで更に特徴づけられる、請求項1または2に記載の化合物。
【請求項6】
約568nmol/kgを投与されたSTZ処置ラットにおいて、約6時間を超える除去半減期を有することで更に特徴づけられる、請求項1から5のいずれか1項に記載の化合物。
【請求項7】
STZ処置ラットにおいて、前記化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約36時間の範囲で、血糖を100mg/dL未満に低下させるのに十分である、請求項1から6のいずれか1項に記載の化合物。
【請求項8】
STZ処置ラットにおいて、前記化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約48時間の範囲で、血糖を100mg/dL未満に低下させるのに十分である、請求項1から7のいずれか1項に記載の化合物。
【請求項9】
ヒトにおいて、約0.225mg/kgの単回皮下投与により、約30時間を超える除去半減期を有することで更に特徴づけられる、請求項1から8のいずれか1項に記載の化合物。
【請求項10】
ヒトにおいて、約0.225mg/kgの単回皮下投与により、約36時間を超える除去半減期を有することで更に特徴づけられる、請求項1から9のいずれか1項に記載の化合物。
【請求項11】
前記PEGは約17.5kDaから約25kDaの範囲の分子量を有する、請求項1から10のいずれか1項に記載の化合物。
【請求項12】
前記PEGは約20kDaから約25kDaの範囲の分子量を有する、請求項1から11のいずれか1項に記載の化合物。
【請求項13】
前記共有結合はウレタン結合である、請求項1から12のいずれか1項に記載の化合物。
【請求項14】
患者に治療上有効量の前記化合物を非経口的に投与した場合、薬物動態学的、薬力学的、または活性ピーク/トラフ比が2未満であることにより更に特徴づけられる、請求項1から13のいずれか1項に記載の化合物。
【請求項15】
治療に使用するための、請求項1から14のいずれか1項に記載の化合物の使用。
【請求項16】
低血糖症または糖尿病の治療用医薬品を製造するための、請求項1から14のいずれか1項に記載の化合物の使用。
【請求項17】
請求項1から14のいずれか1項に記載の化合物と、1つ以上の製薬上許容可能な賦形剤、希釈剤または担体とを含む、医薬組成物。
【請求項18】
ペグ化インスリンリスプロ化合物のうち約95%を超える量がモノペグ化である、請求項17に記載の医薬組成物。
【請求項19】
ペグ化インスリンリスプロ化合物のうち約90%を超える量が、Bの28位でリジンのイプシロンアミノ基に共有結合するPEGを有する、請求項17から18のいずれか1項に記載の医薬組成物。
【請求項20】
約10mMトリスから約25mMトリスの範囲の濃度のトリス緩衝液をさらに含み、前記医薬組成物のpHが約7.0から約8.0であり、少なくとも1つの等張剤が約270ミリオスモルと約330ミリオスモルの間の等張性を持つ溶液を生成する、請求項17から19のいずれか1項に記載の医薬組成物。
【請求項21】
約5mMから約10mMの範囲の濃度のリン酸塩緩衝液をさらに含み、前記医薬組成物のpHが約7.0から約7.5であり、少なくとも1つの等張剤が約270ミリオスモルと約330ミリオスモルの間の等張性を持つ溶液を生成する、請求項17から20のいずれか1項に記載の医薬組成物。
【請求項22】
ペグ化インスリンリスプロ化合物の全濃度が約15mg/mLから約40mg/mLである、請求項17から21のいずれか1項に記載の医薬組成物。
【請求項23】
治療上有効量のインスリンリスプロを更に含む、請求項17から22のいずれか1項に記載の医薬組成物。
【請求項24】
約130mMのm−クレゾールと、インスリン、インスリン類似体およびペグ化インスリンリスプロの全6量体1モルにつき約2モルと約4モルの間の亜鉛と、約2.5mMと約25mMの間のカルシウムと、表面活性剤とを更に含む、請求項17から23のいずれか1項に記載の医薬組成物。
【請求項25】
製薬上許容可能なフェノール系防腐剤と、ペグ化インスリンリスプロ化合物を6量体化するのに十分な量の亜鉛とを更に含む、請求項17から24のいずれか1項に記載の医薬組成物。
【請求項26】
約100mMと約150mMの間のNaClと、約2.5mMと約25mMの間のカルシウムと、約0.5mg/mLと約5mg/mLの間のポロキサマー188とを更に含む、請求項17から25のいずれか1項に記載の医薬組成物。
【請求項27】
約1mg/mLと約5mg/mLの間のポロキサマー188を含む、請求項17から26のいずれか1項に記載の医薬組成物。
【請求項28】
ペグ化インスリンリスプロ1モルにつき約0.3モルと5モルの間の亜鉛を含む、請求項17から27のいずれか1項に記載の医薬組成物。
【請求項29】
治療を必要とする患者の高血糖症または糖尿病を治療する方法であって、治療上有効量の、請求項17から28のいずれか1項に記載の医薬組成物を患者に投与するステップを含む、方法。
【請求項30】
前記患者は真性糖尿病の治療を受ける、請求項29に記載の方法。
【請求項31】
前記患者は妊娠性糖尿病の治療を受ける、請求項29に記載の方法。
【請求項32】
以下の式のペグ化インスリンリスプロ化合物、またはその製薬上許容可能な塩を製造する工程であって:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pはウレタン共有結合によってBの28位でリジンのイプシロンアミノ基に結合している)、約25℃と約30℃の間の温度で、約3時間と約6時間の間、前記Bの28位におけるリジンのイプシロンアミノ基と、約20kDaと約40kDaの間の重量平均分子量を有するモノメトキシポリエチレングリコールp−ニトロフェニルカルボネート(mPEG−NPC)とをpH約8.5と約11.5の間の水性溶媒中で反応させるステップを含む、工程。
【請求項33】
PEGとインスリンリスプロのモル比の範囲は約2.5と約5.0の間である、請求項32に記載の工程。
【請求項34】
前記mPEG−NPCの重量平均分子量は約20kDaである、請求項32または33に記載の工程。
【請求項35】
前記反応のpHは約10.5と約11.5の間に維持される、請求項32から34のいずれか1項に記載の工程。
【請求項36】
前記反応は約25℃と約30℃の間の温度で約3時間行われる、請求項32から35のいずれか1項に記載の工程。
【請求項1】
以下の式のペグ化インスリンリスプロ化合物:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pはウレタンまたはチオエーテル共有結合によって、Aの1位でグリシンのアルファアミノ基、Bの1位でフェニルアラニンのアルファアミノ基またはBの28位でリジンのイプシロンアミノ基に結合している)、またはその製薬上許容可能な塩。
【請求項2】
前記PEGは、Bの28位におけるリジンのイプシロンアミノ基にウレタンまたはチオエーテル共有結合によって結合している、請求項1に記載の化合物。
【請求項3】
ヒトインスリン受容体に対して約30nM以下のKi値を持つことで更に特徴づけられる、請求項1または2に記載の化合物。
【請求項4】
ヒトインスリン受容体に対して約20nM以下のKi値を持つことで更に特徴づけられる、請求項1または2に記載の化合物。
【請求項5】
ヒトインスリン受容体に対して約10nM以下のKi値を持つことで更に特徴づけられる、請求項1または2に記載の化合物。
【請求項6】
約568nmol/kgを投与されたSTZ処置ラットにおいて、約6時間を超える除去半減期を有することで更に特徴づけられる、請求項1から5のいずれか1項に記載の化合物。
【請求項7】
STZ処置ラットにおいて、前記化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約36時間の範囲で、血糖を100mg/dL未満に低下させるのに十分である、請求項1から6のいずれか1項に記載の化合物。
【請求項8】
STZ処置ラットにおいて、前記化合物を約568nmol/kgの用量で単回皮下注射した後、約4時間から少なくとも約48時間の範囲で、血糖を100mg/dL未満に低下させるのに十分である、請求項1から7のいずれか1項に記載の化合物。
【請求項9】
ヒトにおいて、約0.225mg/kgの単回皮下投与により、約30時間を超える除去半減期を有することで更に特徴づけられる、請求項1から8のいずれか1項に記載の化合物。
【請求項10】
ヒトにおいて、約0.225mg/kgの単回皮下投与により、約36時間を超える除去半減期を有することで更に特徴づけられる、請求項1から9のいずれか1項に記載の化合物。
【請求項11】
前記PEGは約17.5kDaから約25kDaの範囲の分子量を有する、請求項1から10のいずれか1項に記載の化合物。
【請求項12】
前記PEGは約20kDaから約25kDaの範囲の分子量を有する、請求項1から11のいずれか1項に記載の化合物。
【請求項13】
前記共有結合はウレタン結合である、請求項1から12のいずれか1項に記載の化合物。
【請求項14】
患者に治療上有効量の前記化合物を非経口的に投与した場合、薬物動態学的、薬力学的、または活性ピーク/トラフ比が2未満であることにより更に特徴づけられる、請求項1から13のいずれか1項に記載の化合物。
【請求項15】
治療に使用するための、請求項1から14のいずれか1項に記載の化合物の使用。
【請求項16】
低血糖症または糖尿病の治療用医薬品を製造するための、請求項1から14のいずれか1項に記載の化合物の使用。
【請求項17】
請求項1から14のいずれか1項に記載の化合物と、1つ以上の製薬上許容可能な賦形剤、希釈剤または担体とを含む、医薬組成物。
【請求項18】
ペグ化インスリンリスプロ化合物のうち約95%を超える量がモノペグ化である、請求項17に記載の医薬組成物。
【請求項19】
ペグ化インスリンリスプロ化合物のうち約90%を超える量が、Bの28位でリジンのイプシロンアミノ基に共有結合するPEGを有する、請求項17から18のいずれか1項に記載の医薬組成物。
【請求項20】
約10mMトリスから約25mMトリスの範囲の濃度のトリス緩衝液をさらに含み、前記医薬組成物のpHが約7.0から約8.0であり、少なくとも1つの等張剤が約270ミリオスモルと約330ミリオスモルの間の等張性を持つ溶液を生成する、請求項17から19のいずれか1項に記載の医薬組成物。
【請求項21】
約5mMから約10mMの範囲の濃度のリン酸塩緩衝液をさらに含み、前記医薬組成物のpHが約7.0から約7.5であり、少なくとも1つの等張剤が約270ミリオスモルと約330ミリオスモルの間の等張性を持つ溶液を生成する、請求項17から20のいずれか1項に記載の医薬組成物。
【請求項22】
ペグ化インスリンリスプロ化合物の全濃度が約15mg/mLから約40mg/mLである、請求項17から21のいずれか1項に記載の医薬組成物。
【請求項23】
治療上有効量のインスリンリスプロを更に含む、請求項17から22のいずれか1項に記載の医薬組成物。
【請求項24】
約130mMのm−クレゾールと、インスリン、インスリン類似体およびペグ化インスリンリスプロの全6量体1モルにつき約2モルと約4モルの間の亜鉛と、約2.5mMと約25mMの間のカルシウムと、表面活性剤とを更に含む、請求項17から23のいずれか1項に記載の医薬組成物。
【請求項25】
製薬上許容可能なフェノール系防腐剤と、ペグ化インスリンリスプロ化合物を6量体化するのに十分な量の亜鉛とを更に含む、請求項17から24のいずれか1項に記載の医薬組成物。
【請求項26】
約100mMと約150mMの間のNaClと、約2.5mMと約25mMの間のカルシウムと、約0.5mg/mLと約5mg/mLの間のポロキサマー188とを更に含む、請求項17から25のいずれか1項に記載の医薬組成物。
【請求項27】
約1mg/mLと約5mg/mLの間のポロキサマー188を含む、請求項17から26のいずれか1項に記載の医薬組成物。
【請求項28】
ペグ化インスリンリスプロ1モルにつき約0.3モルと5モルの間の亜鉛を含む、請求項17から27のいずれか1項に記載の医薬組成物。
【請求項29】
治療を必要とする患者の高血糖症または糖尿病を治療する方法であって、治療上有効量の、請求項17から28のいずれか1項に記載の医薬組成物を患者に投与するステップを含む、方法。
【請求項30】
前記患者は真性糖尿病の治療を受ける、請求項29に記載の方法。
【請求項31】
前記患者は妊娠性糖尿病の治療を受ける、請求項29に記載の方法。
【請求項32】
以下の式のペグ化インスリンリスプロ化合物、またはその製薬上許容可能な塩を製造する工程であって:
P−〔(A)−(B)〕
(式中、
Aはインスリンリスプロ(配列番号1)のA鎖、
Bはインスリンリスプロ(配列番号3)のB鎖、
Pは約20kDaから約40kDaの範囲の分子量を有するPEGであり、AおよびBは適切に架橋結合しており、Pはウレタン共有結合によってBの28位でリジンのイプシロンアミノ基に結合している)、約25℃と約30℃の間の温度で、約3時間と約6時間の間、前記Bの28位におけるリジンのイプシロンアミノ基と、約20kDaと約40kDaの間の重量平均分子量を有するモノメトキシポリエチレングリコールp−ニトロフェニルカルボネート(mPEG−NPC)とをpH約8.5と約11.5の間の水性溶媒中で反応させるステップを含む、工程。
【請求項33】
PEGとインスリンリスプロのモル比の範囲は約2.5と約5.0の間である、請求項32に記載の工程。
【請求項34】
前記mPEG−NPCの重量平均分子量は約20kDaである、請求項32または33に記載の工程。
【請求項35】
前記反応のpHは約10.5と約11.5の間に維持される、請求項32から34のいずれか1項に記載の工程。
【請求項36】
前記反応は約25℃と約30℃の間の温度で約3時間行われる、請求項32から35のいずれか1項に記載の工程。
【図1】

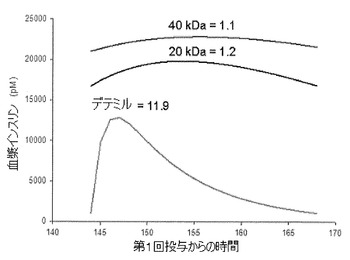
【図2】
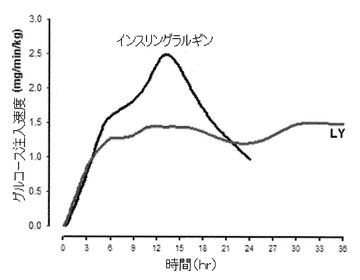
【図2】
【公表番号】特表2011−524357(P2011−524357A)
【公表日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願番号】特願2011−513625(P2011−513625)
【出願日】平成21年6月9日(2009.6.9)
【国際出願番号】PCT/US2009/046704
【国際公開番号】WO2009/152128
【国際公開日】平成21年12月17日(2009.12.17)
【出願人】(594197872)イーライ リリー アンド カンパニー (301)
【Fターム(参考)】
【公表日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願日】平成21年6月9日(2009.6.9)
【国際出願番号】PCT/US2009/046704
【国際公開番号】WO2009/152128
【国際公開日】平成21年12月17日(2009.12.17)
【出願人】(594197872)イーライ リリー アンド カンパニー (301)
【Fターム(参考)】
[ Back to top ]