
ペニシリン結合タンパク質の発現及びその抗生物質のハイスループット検出への応用
【課題】細菌から完全長クラスA型ペニシリン結合タンパク質を発現及び精製するための方法、及び該タンパク質を用いた抗生物質のハイスループットスクリーニング方法を提供する。
【解決手段】細菌のゲノムDNAから完全長ペニシリン結合タンパク質のDNA配列を増幅させ、DNA配列をベクターにクローニングし、ホスト細胞内でDNA配列を発現させて完全長ペニシリン結合タンパク質を得て、タンパク質を界面活性剤で可溶化し、タンパク質を精製する、完全長クラスA型ペニシリン結合タンパク質を精製する方法。
【解決手段】細菌のゲノムDNAから完全長ペニシリン結合タンパク質のDNA配列を増幅させ、DNA配列をベクターにクローニングし、ホスト細胞内でDNA配列を発現させて完全長ペニシリン結合タンパク質を得て、タンパク質を界面活性剤で可溶化し、タンパク質を精製する、完全長クラスA型ペニシリン結合タンパク質を精製する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、細菌から完全長クラスA型ペニシリン結合タンパク質を発現及び精製するための方法に関する。本発明は抗生物質のハイスループットスクリーニング法にも関する。
【背景技術】
【0002】
Staphylococcus aureus、Streptococcus pneumoniae及びEnterococcus faecalis等の多くの一般的な病原菌は今や多剤耐性となって公衆衛生問題として浮上してきており、新しい抗生物質が医療において求められているが、いまだもってそのニーズは満たされていない。
【0003】
抗菌剤の開発においては、細菌細胞壁又はペプチドグリカン合成経路がターゲットであった(Silver LL (2003) Curr Opin Microbiol 6:431-438)。ペプチドグリカンの合成は幾つかの段階から成り、脂質I及び脂質IIの合成、それに続く脂質IIの最終トランスグリコシル化とペプチド転移によるペプチドグリカンの形成を含む(Ostash B, Walker S (2005) Curr Opin Chem Biol 9:459-466)。現在の抗生物質の多くがペプチド転移をターゲットとしたβ−ラクタム誘導体である。トランスグリコシル化プロセスを阻害する医薬品は未だ開発されていない。トランスグリコシラーゼ(TG)について唯一知られている強力な阻害剤はモエノマイシン複合体(フラボマイシン)であり、モエノマイシンA(図1A、Moe A、1)、A12、Cl、C3及びC4が含まれる(Zehl M, Pittenauer E, Rizzi A, Allmaier G (2006) Amer Soc Mass Spec 17:1081-1090)。中でも、Moe Aがそのファミリーで最も豊富な薬剤である(Eichorn P, Aga DS (2005) Rapid Commun Mass Spectrom 19:2179-2186)。その特異な抗菌特性は化学者にモエノマイシンフラグメントと誘導体の合成を促すこととなった(Adachi M, Zhang Y, Leimkuhler C, Sun B, LaTour JV, Kahne DE (2006) J Am Chem Soc 128:14012-14013; Welzel P (2005) Chem Rev 105:4610-4660; Welzel P (2007) Angew Chem Int Ed Engl 46:4825-4829)。モエノマイシンAの最初の全合成はつい最近Kahneとその同僚によって成された(Taylor TG, Li X, Oberthur M, Zhu W, Kahne DE (2006) J Am Chem Soc 128:15084-15085)。その生合成経路もまた解明されている(Ostash B, Saghatelian A, Walker S (2007) Chem Biol 14:257-267)。しかしながら、その生物学的利用能の低さから、フラボマイシンだけが動物飼料における成長促進抗生物質として利用されている(Butaye P, Devriese LA, Haesebrouck F (2003) Clin Microbiol Rev 16:175-188)。
【0004】
トランスグリコシル化は主に二機能性クラスA型ペニシリン結合タンパク質(PBP)によって触媒されている(Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A (2006) FEMS Microbiol Rev 30:673-691)。PBPはN−アセチルグルコサミン−N−アセチルムラミル−ペンタペプチドの重合及びペンタペプチド間の架橋による細胞壁の最終形成段階に関わる主要酵素であることから、抗生物質の発見にとって重要なターゲットの1つであると考えられている。これらの膜に固定された酵素は3つの別個のタンパク質ドメイン、つまりそのアミノ末端からカルボキシル末端に向かって膜貫通(Transmembrane:TM)ドメイン、トランスグリコシラーゼドメイン(Transglycosilyase:TG)、トランスペプチダーゼ(Transpeptidase:TP)ドメインから成る。S.aureusから得たPBP2細胞外ドメインと(Lovering AL, de Castro LH, D. Lim D, Strynadka NC (2007) Science 315:1402-1405)及びA.aeolicusから得たPBP1aのトランスグリコシラーゼドメイン(Yuan Y, Barrett D, Zhang Y, Kahne D, Sliz P, Walker S (2007) Proc Natl Acad Sci 104:5348-5353)の近年のX線結晶構造は、触媒作用におけるPBPの膜貫通ドメインの役割についての情報は欠けているものの、細胞壁ペプチドグリカン重合の説得力のあるメカニズムについて非常に貴重な知見をもたらした。PBPの膜貫通ドメインは、モエノマイシンの脂質部分又は脂質IIと相互作用するのではないかと推測されている(Ye XY, Lo MC, Brunner L, Walker D, Kahne D, Walker S (2001) J Am Chem Soc 123:3155-3156; Anikin A, Buchynskyy A, Kempin U, Stembera K, Welzel P, Lantzsch G (1999) Angew Chem Int Ed 38:3703-3707)。
【0005】
クラスA型PBPの特性解析及びTG阻害剤の同定には機能性PBP及び重合用の基質としての脂質IIを必要とする。脂質IIの入手には限界があることから、阻害剤の開発のみならず酵素の研究が阻まれていた。脂質IIの不足を解消するために、TG阻害剤を探すためのスクリーニング方法の多くは主にモエノマイシンに頼っている(Di Guilmi AM. Dessen A, Dideberg O, Vernet T (2002) Curr Pharm Biotechnol 3:63-75)。モエノマイシン活性に基づいて、SPRアッセイ(Stembera K, Vogel S, Buchynskyy A, Ayala JA, Welzel P (2002) Chembiochem 3:559-565)又は放射性アッセイ(Vollmer W, Holtje J-V (2000) Antimicrob Agents Chemother 44:1181-1185; Jha RK, de Sousa SM (2006) J Biomol Screen 11:1005-1014; Ravishankar S, Kumar VP, Chandrakala B, Jha RK, Solapure SM, de Sousa SM (2005) Antimicrob Agents Chemother 49:1410-1418; Chandrakala B, Shandil RK, Mehra U, Ravishankar S, Kaur P, Usha V, Joe B, deSousa SM (2004) Antimicrob Agents Chemother 48:30-40)を用いたロースループット阻害剤スクリーニング法が開発された。そのため、ハイスループットスクリーニングに適したTG活性アッセイプラットフォームが阻害剤の同定に望ましい。
【発明を実施するための最良の形態】
【0006】
本発明は細菌から完全長クラスA型ペニシリン結合タンパク質を発現及び精製するための方法を提供するものであり、
(a)細菌のゲノムDNAから完全長ペニシリン結合タンパク質のDNA配列を増幅させ、
(b)DNA配列をベクターにクローニングし、
(c)ホスト細胞内でDNA配列を発現させて完全長ペニシリン結合タンパク質を得て、
(d)タンパク質を界面活性剤で可溶化し、
(e)タンパク質を精製することを含み、
界面活性剤はn−デシル−β−D−マルトピラノシド、n−ウンデシル−β−D−マルトピラノシド、n−ドデシル−β−D−マルトピラノシド、n−オクチル−β−D−グルコピラノシド、n−ノニル−β−D−グルコピラノシド、n−テトラデシルホスホクロリン、n−ドデシル−N,N−ジメチルアミン−N−オキシド、3−[(3−コラミドプロピル)−ジメチルアンモニオ]−1−プロパンスルホネート又はα−[4−(1,1,3,3−テトラメチルブチル)フェニル]−w−ヒドロキシ−ポリ(オキシ−1,2−エタンジイル)である。
【0007】
ある好ましい実施形態において、細菌はEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである。
【0008】
好ましくは、完全長クラスA型ペニシリン結合タンパク質の精製に使用する界面活性剤はn−ドデシル−β−D−マルトピラノシドである。
【0009】
加えて、完全長タンパク質は3つのドメインを含み、N末端からC末端に向かって膜貫通ドメイン、トランスグリコシラーゼドメイン、トランスペプチダーゼドメインである。
【0010】
本発明は抗生物質のハイスループットスクリーニング法を更に提供し、この方法は
(a)スクリーニングの候補を用意し、
(b)蛍光モエノマイシンと膜貫通ドメインとトランスグリコシラーゼドメインを含むクラスA型ペニシリン結合タンパク質で蛍光異方性測定を実行し、
(c)蛍光異方性値とKD値を計算し、
(d)トランスグリコシラーゼ阻害剤のKI及びIC50値を計算し、
(e)阻害剤の最低阻害濃度を求めることを含み、
KDはモエノマイシンとタンパク質との間の解離定数であり、KIは阻害剤とタンパク質との間の解離定数であり、IC50は細菌の増殖を半分に阻害するのに必要な阻害濃度であり、最低阻害濃度とは細胞の増殖を防止する阻害剤の最低濃度である。
【0011】
ある実施形態において、抗生物質はトランスグリコシラーゼ阻害剤であり、好ましくは(Z)−5−(4−ブロモフェニル)−3−((5−ニトロフラン−2−イル)メチレン)フラン−2(3H)−オン、(Z)−1,3−ジフェニル−4−(2−(チアゾール−2−イル)ヒドラゾノ−1H−ピラゾール−5(4H)−オン、(E)−4−(2−(5,5,8,8−テトラメチル−5,6,7,8−テトラヒドロナフタレン−2−イル)プロプ−1−エニル)安息香酸である。
【0012】
ある好ましい実施形態において、タンパク質は3つのドメインを含み、N末端からC末端に向かって膜貫通ドメイン、トランスグリコシラーゼドメイン、トランスペプチダーゼドメインである。
【0013】
本発明において、蛍光プローブとして使用するモエノマイシンは、蛍光異方性に基づいたアッセイの実行時にペニシリン結合タンパク質に結合する。
【0014】
その他の好ましい実施形態において、本発明の方法でスクリーニングされたヒット化合物であるところの抗生物質はEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである細菌の増殖を阻害する。
【0015】
好ましい実施形態において、細菌はE.faecalis又はS.aureusである。
【0016】
以下の実施例は非限定的であり、本発明の多様な態様と特徴を表しているにすぎない。
【実施例1】
【0017】
完全長ペニシリン結合タンパク質の発現
E.coliのPBP1b変異体の発現と精製
E.coli(Swiss−Prot accession number:P02919)からの完全長PBP1bをゲノムDNAから増幅し、pETベクター(EMD Sciences社。サンディエゴ、カリフォルニア州)にクローニングし、N末端(His)6タグで発現させた。BL21(DE3)E.coliホスト細胞を37℃で600nmでのODが0.6に達するまで増殖させ、タンパク質発現を1mMのIPTGで3時間に亘って誘発させた。細胞ペレットをpH8.0の20mMのトリス、300mMのNaClに再懸濁させ、マイクロフルイダイザ(Microfluidics社、ニュートン、マサチューセッツ州、米国)で粉砕した。組み換え完全長膜タンパク質の可溶化と精製を様々な界面活性剤を用いてテストし(Anatrace社、マウミー、オハイオ州、米国)、n−デシル−β−D−マルトピラノシド、n−ウンデシル−β−D−マルトピラノシド、n−ドデシル−β−D−マルトピラノシド、n−オクチル−β−D−グルコピラノシド、n−ノニル−β−D−グルコピラノシド、n−テトラデシルホスホクロリン、n−ドデシル−N,N−ジメチルアミン−N−オキシド、CHAPS(3−[(3−コラミドプロピル)−ジメチルアンモニオ]−1−プロパンスルホネート)及びTriton X−100(α−[4−(1,1,3,3−テトラメチルブチル)フェニル]−w−ヒドロキシ−ポリ(オキシ−1,2−エタンジイル)が含まれる。中でも、DDM(n−ドデシル−β−D−マルトピラノシド)は完全長PBP1bを可溶化し、かつそのモエノマイシン結合活性を維持させることが判明した。従って、完全長PBP1bを20mMのDDMで可溶化させ、1mMのDDMの存在下で製造業者の指示に従ってニッケルキレート化クロマトグラフィで精製した。
【0018】
PBP1bのドメインは膜貫通ドメイン(TM、残基64−87)、トランスグリコシラーゼドメイン(TG、残基195−409)、トランスペプチダーゼ(TP、残基444−736)として定義した。ヌクレオチドプライマはTG+TP(残基88−844)、TM+TG(残基1−409)、TG(残基195−409)、及びTP(残基444−736)を含有するそれぞれの変異体を増幅するように設計された。増幅物は発現と精製のためにpETベクターへとクローニングした。変異体TM+TGを得るための手順は完全長タンパク質の調製について記載したとおりである。TG+TP、TG及びTPの変異体については、13mMのn−テトラデシルホスホクロリン(Anatrace社)をタンパク質の抽出に、0.25mMのものをニッケルキレート化クロマトグラフィに用いた。界面活性剤を1mMのDDMに交換し、アミコンウルトラフィルターユニット(Millipore社、ビレリカ、マサチューセッツ州、米国)を用いて濃縮した。
【0019】
16の細菌株からの組み換え完全長二機能性PBPの発現及び精製
完全長二機能性PBPのホモログを、E.coliからのPBP1bタンパク質配列(Swiss−Prot accession number:P02919)を用いてBLASTでNCBIデータベースでの同定を行った(http://www.ncbi.nlm.nih.gov/BLAST/)。トランスグリコシラーゼモチーフと活性部位の存在を確認するため、Jalviewプログラム(4)をMUSCLE多重アラインメントアルゴリズム(5)と共に用いて配列をアラインし、アラインされた出力(図2)をESPript(6)を用いて作成した。明確にするため、以下の残基はアライメントから除外した:Bpe(B. pertussis):354−443、Ngo(N. gonorrhoeae):331−416、Bsu(B. subtilis):615−643、735−787、797−835、及び897−914、及びCdi(C. difficile):267−322及び804−842。
【0020】
個々のPBPの発現のために、ゲノムDNAをAmerican Type Culture Collection(ATCC、マナサス、ヴァージニア州、米国)から購入した。Escherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica及びNeisseria gonorrhoeaeを含む16の細菌からの完全長遺伝子の組み換えタンパク質を完全長E.coliのPBP1bについて記載したものと同じプロトコルを用いて調製した。その殆どは80%より高い純度で得ることができ(図3)、結合実験のために使用可能である。
【実施例2】
【0021】
抗生物質のハイスループット検出
材料及び方法
表面プラズモン共鳴検出
精製したPBP及びその変異体をCM3チップ(GE healthcare社、ウプサラ、スウェーデン)上に約1500〜2000RUまでアミン結合法を用いて固定化した。次にチップを異なる濃度のモエノマイシンA(0−2000nM)に通した。固定化及びデータ収集をBIAcoreT100(GE Healthcare社、ウプサラ、スウェーデン)を用いて25℃で行った。
【0022】
蛍光異方性測定
蛍光異方性測定を488nmレーザを備えたレーザ蛍光分析装置(IsoCyte、Blueshift Biotech社、サニーヴェール、カリフォルニア州、米国)を用いて384ウェルプレートのウェルで3回行った。各種バッファ、塩、pH値、及び二価陽イオン(Ca++、Mg++、Co++)を蛍光異方性測定にむけて最適化した。KD及びKIは100mMのNaCl、pH8.0の10mMのトリスにおいて求めた。走査の焦点は、検出の障害とならないようにプレート底部の上方であった。データ分析はプロプライエタリ・ソフトウェアであるBlueImage(Blueshift Biotech社)を用いて行った。蛍光異方性値(A)は方程式:A= (I平行G*I垂直)/(I平行+2G*I垂直)を用いて計算し、I平行は励起光に平行な放射光の蛍光強度であり、I垂直は励起光に対して垂直な放射光の蛍光強度であり、Gは機器バイアスを修正するためのゲーティングファクタである。Gファクタは各回ごとにプローブだけのウェルを基準異方性として用いて実験的に求めた。
【0023】
直接結合アッセイからのKD値の測定
直接結合アッセイにおいて、初期条件にはpH8.0、10mMのトリス、100mMのNaCl中の40μlの100nMのF−Moeが含まれた。少量のPBP原液を添加した後、5分間の平衡後に異方性の変化を測定した。直接滴定はリガンド(L)とレセプタ(R)との単純な結合平衡に沿うものとみなす(R+LS⇔RLSでKD=R・LS/RLS)。データをPrism(GraphPad Software社、サンディエゴ、カリフォルニア州)を用いて[方程式1KD]にフィットさせ、KD(3)を求めた。
(A測定−A最低)/(A最高−A最低)=(KD+LST+RT)−[KD+LST+RT]2−4・LST・RT]1/2 [方程式1]
であり、A測定は観察された異方性、A最低は最低異方性、A最高は最高異方性、LSTはF−Moeの総濃度、RTはPBPの総濃度、KDはリガンド(F−Moe)とレセプタ(PBP)との間の解離定数である。報告値は4回の平均である。
【0024】
KI及びIC50値の競合置換アッセイによる測定
競合置換において、初期条件にはpH8.0、10mMのトリス、100mMのNaCll中の40μlの100nMのF−Moe、10μg/mlのH.pyloriのPBP1aが含まれた。化合物原液のアリコートを添加し、5分間の平衡の後、異方性をモニタした。置換アッセイによって得られたデータを用いて、完全競合結合モデル(RL+LS⇔KLR+LS+L⇔KDRLS+L)(3)を用いて阻害剤の阻害定数(KI)とIC50値を計算した。KI値は総阻害剤濃度(C)を結合/遊離F−Moeのモル比に対してプロットし、得られた曲線を[方程式2]にフィットさせることで求めた。
C=[(KI/KD*X)+1][RT−(LST*X)/(X+1)−KD*X] [方程式2]
X=結合/遊離=(A測定−A最低)/(A最高−A測定)
ここでA測定は化合物の特定濃度(C)で観察された異方性、A最低は最低異方性、A最高は最高異方性、LSTはF−Moeの総濃度、RTはPBP1aの総濃度、KDはリガンド(F−Moe)とレセプタ(PBP)との間の解離定数、KIは阻害剤とPBP1aとの間の解離定数である。報告値は4回の平均である。
【0025】
IC50を求めるために、結合蛍光プローブの割合を競合物濃度(C)の対数値に対してプロットする。得られたS次型曲線を[方程式3]を用いてフィットさせると、IC50値が得られる。
(A測定−A最低)/(A最高−A最低)=1/(1+10^((C−log(IC50))*傾き) [方程式3]
ここでA測定は化合物の特定濃度(C)で観察された異方性であり、A最低は最低異方性、A最高は最高異方性である。
【0026】
TG阻害剤のハイスループットスクリーニング
FAアッセイを購入した50000個の小分子(ChemBridge社、サンディエゴ、カリフォルニア州、米国)と我々が所有するコレクションのうちの7000個に対してのスクリーニングに使用した。化合物を96ウェルプレート(Freedom Evo、Tecan Schweiz AG社、マネドルフ、スイス)に、次に384ウェルプレートへとマルチディスペンサ(Labcyte社、サニーヴェール、カリフォルニア州、米国)を用いて移し、スクリーニング用の化合物のプレートを準備した。最終容量40μl、pH8.0の100nMのF−Moe、10mMのトリス、100mMのNaCl中のH.pyloriのPBP1a(10μg/ml)を384ウェルプレートに加えた(Freedom Evo150、Tecan社)。2mM化合物原液の1μlをマルチディスペンサ(Labcyte社)を用いてウェルに加えた。各プレートの最後2つのカラムはコントロールである10μMのモエノマイシンと2.5%のDMSOにそれぞれ用いた。30分の培養後、蛍光異方性における変化をIsocyte(Blueshift Biotech社)を用いて求めた。コントロールの異方性値と比較して75%より高い低下を示したヒット化合物を更に調べるために選択した。
【0027】
最低阻害濃度(minimal inhibitory concentration:MIC)の測定
試験化合物の最低阻害濃度(MIC)をNCCLS基準に従って求めた。実験は血液を加えた(Streptococcus pneumonia)又は加えていない(Bacillus subtilis、Enterococcus faecalis、Staphylococcus aureus)ミュラー・ヒルトン培養液での二倍希釈を用いて96ウェルマイクロタイタープレートで行った。5x105細胞/mlで指数関数的に増殖する細胞を様々な濃度の試験化合物で培養した。18〜24時間に亘る37℃での培養後、細菌の増殖を防止する化合物の最低濃度としてMICを求めた。
【0028】
結果
クラスA型ペニシリン結合タンパク質へのモエノマイシン結合のドメイン要件(二機能性トランスグリコシラーゼ)
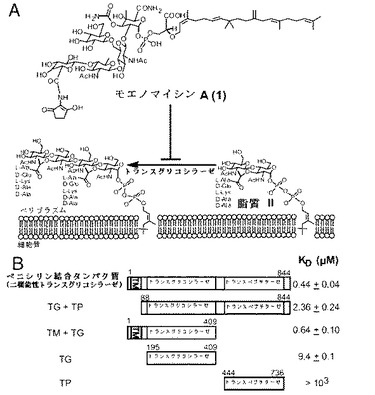
E.coliからのPBP1bの異なるドメインを含むタンパク質構築物を、表面プラズモン共鳴(SPR)を用いたモエノマイシン結合活性測定用に発現、適した界面活性剤を用いて精製した(実施例1)。5つのE.coliのPBP1b変異体を発現、精製した:膜貫通ドメイン、トランスグリコシラーゼドメイン及びトランスペプチダーゼドメインを含む完全長タンパク質(TM+TG+TP)、(2)TG+TP、(3)TM+TG、(4)TG及び(5)TP(図1B)。ターゲットタンパク質をセンサーチップ上に固定化した後、異なる濃度のモエノマイシンを表面に通し、結合親和性を求めた。図1Bに図示されるように、膜貫通ドメインを有していないPBP(TG+TP)のモエノマイシン結合活性は完全長PBP1bの結合活性と比較すると約5倍低かった。その一方で、TM+TGの結合活性は完全長タンパク質の結合活性と同様である。結果は、膜貫通ドメインがモエノマイシン結合において重要な役割を果たす可能性があることを示唆している。完全長タンパク質のモエノマイシン結合親和性は4.4x10−7Mと求められており、TG反応についての阻害能と同様であった(Stembera K, Vogel S, Buchynskyy A, Ayala JA, Welzel P (2002) Chembiochem 3:559-565)。我々は、SPR分析でのモエノマイシンAのC25脂質の非特異的な結合を憂慮していたが、TP変異体をコントロール用のタンパク質として用い、求められたモエノマイシンのPBP1b変異体に対しての結合親和性が顕著であり特異的なものであることが確認された。切断PBP1b変異体を使用した結合実験は、TM+PG変異体の結合親和性が完全長タンパク質の結合親和性と同様であることを更に示した。Moe A結合におけるTMドメインの役割が、TG+TP変異体の結合能が完全長PBP1bのものと比較して5倍も低下したという観察結果から示された(図1B)。
【0029】
モエノマイシンのPBP結合活性と最低阻害濃度(MIC)の相関関係
膜貫通ドメインはモエノマイシン結合に大きく関与することから、モエノマイシン結合の研究のために16のグラム陰性及びグラム陽性細菌(表1)からの完全長PBPの準備に着手した。クラスA型PBPの遺伝子はNCBIデータベース(http://www.ncbi.nlm.nih.gov/BLAST/)を用いて同定し、この全てがTG及びTPモチーフを有していることが確認された(Barrett DS, Chen L, Litterman NK, Walker S (2004) Biochemistry 43:12375; Di Guilmi AM, Dessen A, Dideberg O, Vernet T (2003) J Bacteriol 185:1650-1658)(図2)。ターゲット遺伝子を個々の細菌種のそれぞれのゲノムDNAから増幅して発現ベクターにクローニングし、E.coliホストを用いて組み換えタンパク質を産生させた。精製したタンパク質の酵素活性は脂質II重合によって確認され(データ提示せず)、モエノマイシン結合はSPRで測定した。表1に示されるように、異なる種から得たPBP間には様々な定常状態親和性(KD)値が見えられたが、測定されたKD値は全て10−7Mの範囲内に収まり、トランスグリコシル化プロセスについて報告された阻害濃度に近い(Stembera K, Vogel S, Buchynskyy A, Ayala JA, Welzel P (2002) Chembiochem 3:559-565)。興味深いことに、テストしたグラム陽性細菌の中でも、E.faecalisとS.aureusのPBPのKD値はMIC値と相関関係にあり、PBPのモエノマイシン結合部位がこれらの種に対しての抗生物質の開発にとってよいターゲットとなり得ることを示唆している。
【0030】
表1
16の細菌株についての抗菌活性とモエノマイシンのPBP結合親和性との相関関係

*1 異なる細菌種に対してのモエノマイシンのMIC。
*2 文献から得たモエノマイシンのMIC値。
*3 個々の細菌株から得たクラスA型PBP遺伝子の配列ID。クラスA型PBPのホモログをBLASTを用いてNCBIデータベースで同定した。
*4 SPRを用いてのPBPホモログとモエノマイシンのKD。平均値を示す。
*5 本発明のFAに基づいたアッセイを用いて得たPBPホモログとモエノマイシンとのKD。
【0031】
トランスグリコシラーゼ阻害剤についての蛍光異方性に基づいたアッセイの開発
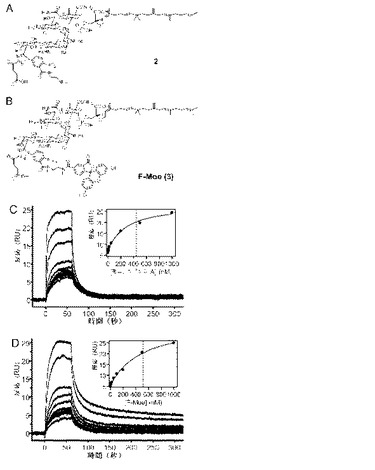
完全長タンパク質をモエノマイシン結合研究に使用できるとの結論に基づいて、蛍光異方性(FA)に基づいたアッセイを設計し、TGに対しての小分子の結合親和性をモニタした。複雑な結晶構造(Lovering AL, de Castro LH, D. Lim D, Strynadka NC (2007) Science 315:1402-1405)及び構造・活性関係(Welzel P (2005) Chem Rev 105:4610-4660)についての従来の研究は、化合物1から2への変性(図4A)は結合と抗菌性を劇的に低下させることはないことを示唆した。更に、2のアミノ部分により、結合研究用のモエノマイシン型のプローブとしてのいずれのフルオロフォアとの共役が可能となる(Buchynskyy A, Kempin U, Vogel S, Hennig L, Findeisen M, Muller D, Giesa S, Knoll H, Welzel P (2002) Eur J Org Chem 7:1149-1162; Donnerstag A, Marzian S, Muller D, Welzel P (1995) Tetrahedron 51:1931-1940)。実際、化合物2は基本条件下で即座にフルオレセイン(6−カルボキシフルオレセイン N−ヒドロキシスクシンイミドエステル)に結合して蛍光プローブ3を生成した(F−Moe、図4B)。FAアッセイで使用の蛍光プローブについての主な懸念の1つはプローブそれ自体、つまりフルオロフォア又は構造変性のいずれかがターゲットタンパク質と小分子との結合を阻害することである。従って、Moe AのPBP結合親和性及び蛍光プローブをSPRを用いて比較した。求めた定常状態親和性(KD)値はMoe A及びF−Moeについて同様であった(4.4x10−7対5.2x10−7M)(図4C及び4D)。この結果によりF−MoeがFAに基づいたPBP結合アッセイに有用な有効蛍光プローブであることが確認された。
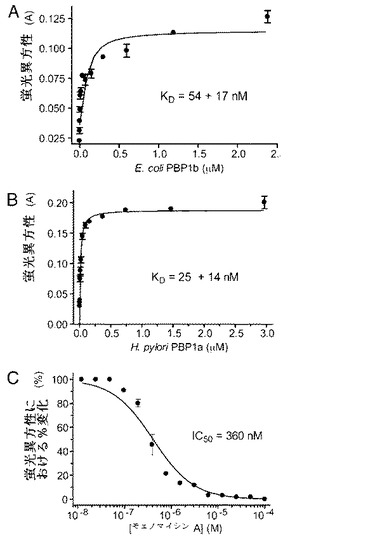
【0032】
F−Moeの異方性はE.coliのPBP1bとの培養により著しく上昇し、これはF−Moe−PBP1b複合体の形成によるものと思われる(図5A)。対照的に、F−Moeの異方性は、100μMまで、ウシ血清アルブミンとの培養では変化しなかった(データ提示せず)。タンパク質濃縮物を、異方性変化の用量依存性を確立するために更に滴定に供した。F−Moeについての推定解離定数(KD)はE.coliのPBP1bにおいてはサブミクロモル範囲にあった(図5A)。しかしながら、E.coliのPBP1bを用いたFAアッセイの信号対雑音比は不十分であり、最高異方性は0.120である。従って、異なるクラスA型PBPをスクリーニングしてアッセイを改良した。試験したホモログ全てにおいて、Helicobacter pyloriのPBP1aを用いたFAアッセイが25+/−14nMの最適な信号対雑音比を出し、異方性はF−Moeへの結合時に0.018から0.2へと上昇した(図5B)。阻害剤スクリーニング用のアッセイを開発するために、F−MoeをH.pyloriのPBP1aと予備培養し、次に様々な濃度の無標識のMoe Aとの競合に供した。Moe Aの濃度が上昇するにつれて異方性の低下が観察され、KI(阻害定数)とIC50値はそれぞれ0.47+/−0.10μMと0.36μMとなった(図5C)。この結果により、FAアッセイがPBPのモエノマイシン結合ポケットからプローブを競合的に置換する阻害剤をスクリーニングすることが実証された。
【0033】
トランスグリコシラーゼ阻害剤のハイスループットスクリーニング
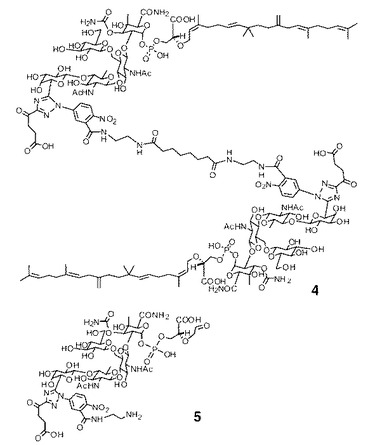
幾つかのモエノマイシン類似体(2、4、5)を調製してFAに基づいたアッセイの信頼性を評価した。表2に示されるように、これらのモエノマイシン類似体のFAに基づいたアッセイで求めた異なる結合親和性(IC50値)は、発表された論文のものに類似している(Marzian S, Happel M, Wagner U, Muller D, Welzel P, Fehlhaber HW, Stark A, Schutz HJ, Markus A, Limbert M, van Heijenoort Y, van Heijenoort J (1994) Tetrahedron 50:5299-5308)。モエノマイシンAから2への変性によりTG結合親和性は若干低下した。8炭素スペーサを介した2の二量体化により、PBP結合能と抗菌性の双方が2より2倍上昇した4が得られたが、4はそれでもMoe A(1)よりも強度が低い。より顕著には、C25脂質部分を有していない化合物5はF−Moeプローブ3を置換することがほとんどできず、これはモエノマイシンの疎水性部分が完全長PBP1bへの結合に不可欠であることを示している。阻害能の順序がMIC値の順序と一致していることは注目に値する(表2)。
【0034】
表2
選択されたヒット化合物のTG活性の阻害と抗菌性測定
*1 化合物2、4、5はモエノマイシン誘導体である。化合物6〜8はハイスループットスクリーニングでスクリーニングされたヒット化合物である。
*2 IC50値は図5Aで図示の蛍光異方性アッセイを用いて求められた。
*3 異なる細菌種に対してのモエノマイシンのMICを「材料と方法」で記載したように求めた。ATCCはアメリカンタイプカルチャーコレクションの略。
【0035】
モエノマイシン及びその類似体はグラム陽性細菌に対してよく効くものの、その薬物動態特性の悪さによりTG阻害剤開発において新たなリード化合物が必要とされている。HTSフォーマットでの効率的かつ経済的なTGアッセイは、新規のTG阻害剤の同定に役立つと思われる。
【0036】
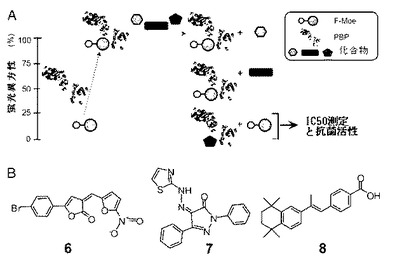
FAアッセイを用いて50μMでMoeA誘導体(図6)と共に57000個の小分子のコレクションをスクリーニングした。モエノマイシン1μMでのアッセイをハイコントロール、2.5%DMSOでのアッセイをローコントロールとした。ウシ血清アルブミン等のその他のタンパク質もコントロールとして含めることで、異方性の上昇が非特異的な結合によるものでないことを確認した。ハイスループットスクリーニングのロバスト性を評価するための0から1の範囲の統計パラメータであるZ´値(Zhang JH, Chung TD, Oldenburg KR (1999) J Biomol Screen 4:67-73)を100を越える個々の実験から0.895と求めた。スクリーニングでヒットした11の少なくとも75%の阻害を示した化合物を、PBP結合に関して抗菌性アッセイとIC50値の測定を含め更に研究するため選び出した(図7A)。中でも、2つのモエノマイシン類似体(2及び4)及び3つの小分子(化合物6、7及び8)が抗菌性とTG結合阻害活性の双方を有していると確認された(表2)。化合物6、7、8はそれぞれ(Z)−5−(4−ブロモフェニル)−3−((5−ニトロフラン−2−イル)メチレン)フラン−2(3H)−オン(Z)−1,3−ジフェニル−4−(2−(チアゾール−2−イル)ヒドラゾノ−1H−ピラゾール−5−(4H)−オン;(E)−4−(2−(5,5,8,8−テトラメチル−5,6,7,8−テトラヒドロナフタレン―2―イル)プロプ−1−エニル)安息香酸である。
【0037】
本発明のFAに基づいたアッセイを用いて、0.895のZ´値で0.02%のヒット率が得られ、これはこのアッセイを膨大な化合物ライブラリから可能性のあるヒット化合物を迅速に同定するためのロバストな一次スクリーニングとして使用可能なことを示唆している。選択したヒット化合物を抗菌性アッセイ又は脂質II重合活性分析を用いて更にスクリーニングし、リード化合物を同定する。全ての蛍光を利用したアッセイと同様に、本発明のFAに基づいたアッセイは蛍光化合物についてTG阻害活性を求めるのに使用することはできない。しかしながら、FAに基づいたPBP結合アッセイは新規の抗菌剤の発見に非常に強力なHTSアッセイであることが実証された。
【0038】
本発明を当業者がそれを実践するのに十分な詳細さでもって説明、例示してきたが、本発明の精神と範囲から逸脱することなく様々な代替、改変、改良が可能であることは明白である。
【0039】
本発明が目的を達成し、本来備えているもののみならず、記載の結果と利点を得るに十分に適合されていることを当業者は容易に理解するものである。その改変及びその他の使用は他の当業者の想定内である。これらの改変は本発明の精神の範囲に含まれ、請求項の範囲によって定められる。
【図面の簡単な説明】
【0040】
【図1】モエノマイシンによるトランスグリコシラーゼの阻害と切断されたPBP変異体の結合親和性を示す。(A)モエノマイシンA(1)が細菌細胞壁の合成におけるトランスグリコシル化工程を阻害している。(B)モエノマイシンの結合の研究に使用したPBP変異体の概略図である。完全長PBP(TM+TG+TP)はE.coliのPBP1b(P02919)のアミノ酸残基1−844を含有している。TG+TP(残基88−844)、TM+TG(残基1−409)、TG(残基195−409)及びTP(残基444−736)は完全長PBP1bの一部を含む切断変異体である。これらの変異体のモエノマイシン結合定数(KD)はSPRを用いて求めた。
【図2】16の細菌株から得た完全長二機能性PBPの配列アラインメントを示す。本研究で使用した16の完全長PBP配列をJalviewログラムを用いてアラインし、図はESPriptを用いて作成した。保存した残基について、同一の残基の場合は背景を濃い灰色、似た残基は灰色でそれぞれ囲んだ。灰色の星印はTGの場合モチーフ:EDxxFxxHxxG、GxSTxTQQ、RKxxE、KxxILxxYxN、及びRxxVL、TPの場合はモチーフSxxK、SxN及びKTGを示す。下線のついた星印は触媒残基を記している。アラインされた配列の順番は表1と図3の順番と同一である。
【図3】16の細菌種から得たクラスA型PBPの精製タンパク質のSDS−PAGE分析を示す。各レーンは5μgのタンパク質を含有し、MはkDa単位での分子量マーカーを示す。1:Bordetella pertussisからのPBP1a、2:Citrobacter freundiiからのPBP1b、3:Escherichia coliからのPBP1b、4:Haemophilus influenzaeからのPBP1b、5:Helicobacter pyloriからのPBP1a、6:Klebsiella pneumoniaeからのPBP1b、7:Neisseria gonorrhoeaeからのPBP1、8:Pseudomonas aeruginosaからのPBP1b、9:Salmonella entericaからのPBP1b、10:Shigella flexneriからのPBP2、11:Bacillus subtilisからのPBP1a/1b、12:Clostridium difficileからのPBP、13:Enterococcus faecalisからのPBP2a、14:Enterococcus faeciumからのPBP1、15:Staphylococcus aureusからのPBP2、16:Streptococcus pneumoniaeからのPBP1b。
【図4】トランスグリコシラーゼについての新規の蛍光異方性アッセイの設計。(A、B)変性モエノマイシンA(2)及びフルオレセインで標識したモエノマイシンF−Moe(3)の化学構造。(C、D)モエノマイシンA(C)とF−Moe(D)のE.coliのPBP1bに対しての結合活性のSPR分析。固定化したE.coliのPBP1bへのモエノマイシン結合についての反応を示している。データは、挿入したグラフで図示されるように、定常状態での親和性を用いて分析し、1:1の相互作用モデルへとフィットさせた。X軸と点線の切片から推定したKD値はMoe A及びF−Moeについてそれぞれ4.4x10−7M及び5.161x10−7Mであった。
【図5】トランスグリコシラーゼについてのハイスループットFAアッセイの開発。(A)E.coliのPBP1bがF−Moeに結合すると、蛍光異方性において濃度依存性の変化が観察された。(B)Helicobacter pyloriのPBP1aでの改善されたFAアッセイ。H.pyloriのPBP1aを用いて同様に濃度依存性の蛍光異方性変化を行った。最大異方性値は0.2であった。(C)様々な濃度の無標識モエノマイシンによるPBP1a結合F−Moe複合体の置換を示す。蛍光異方性における変化は[(A測定−A最低)/(A最高−A最低)x100%]と定義する。KD及びIC50値を実施例2に記載したように計算した。
【図6】モエノマイシン誘導体の化学構造を図示する(4及び5)。
【図7】クラスA型PBPを用いたTG阻害剤としての小分子のスクリーニングを示す。(A)FAアッセイを用いたTG阻害剤のハイスループットスクリーニングのスキーム。タンパク質構造の画像はPDB ID 2OLV(12)から作成した。(B)ハイスループットスクリーニングでヒットした化合物の化学構造。
【技術分野】
【0001】
本発明は、細菌から完全長クラスA型ペニシリン結合タンパク質を発現及び精製するための方法に関する。本発明は抗生物質のハイスループットスクリーニング法にも関する。
【背景技術】
【0002】
Staphylococcus aureus、Streptococcus pneumoniae及びEnterococcus faecalis等の多くの一般的な病原菌は今や多剤耐性となって公衆衛生問題として浮上してきており、新しい抗生物質が医療において求められているが、いまだもってそのニーズは満たされていない。
【0003】
抗菌剤の開発においては、細菌細胞壁又はペプチドグリカン合成経路がターゲットであった(Silver LL (2003) Curr Opin Microbiol 6:431-438)。ペプチドグリカンの合成は幾つかの段階から成り、脂質I及び脂質IIの合成、それに続く脂質IIの最終トランスグリコシル化とペプチド転移によるペプチドグリカンの形成を含む(Ostash B, Walker S (2005) Curr Opin Chem Biol 9:459-466)。現在の抗生物質の多くがペプチド転移をターゲットとしたβ−ラクタム誘導体である。トランスグリコシル化プロセスを阻害する医薬品は未だ開発されていない。トランスグリコシラーゼ(TG)について唯一知られている強力な阻害剤はモエノマイシン複合体(フラボマイシン)であり、モエノマイシンA(図1A、Moe A、1)、A12、Cl、C3及びC4が含まれる(Zehl M, Pittenauer E, Rizzi A, Allmaier G (2006) Amer Soc Mass Spec 17:1081-1090)。中でも、Moe Aがそのファミリーで最も豊富な薬剤である(Eichorn P, Aga DS (2005) Rapid Commun Mass Spectrom 19:2179-2186)。その特異な抗菌特性は化学者にモエノマイシンフラグメントと誘導体の合成を促すこととなった(Adachi M, Zhang Y, Leimkuhler C, Sun B, LaTour JV, Kahne DE (2006) J Am Chem Soc 128:14012-14013; Welzel P (2005) Chem Rev 105:4610-4660; Welzel P (2007) Angew Chem Int Ed Engl 46:4825-4829)。モエノマイシンAの最初の全合成はつい最近Kahneとその同僚によって成された(Taylor TG, Li X, Oberthur M, Zhu W, Kahne DE (2006) J Am Chem Soc 128:15084-15085)。その生合成経路もまた解明されている(Ostash B, Saghatelian A, Walker S (2007) Chem Biol 14:257-267)。しかしながら、その生物学的利用能の低さから、フラボマイシンだけが動物飼料における成長促進抗生物質として利用されている(Butaye P, Devriese LA, Haesebrouck F (2003) Clin Microbiol Rev 16:175-188)。
【0004】
トランスグリコシル化は主に二機能性クラスA型ペニシリン結合タンパク質(PBP)によって触媒されている(Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A (2006) FEMS Microbiol Rev 30:673-691)。PBPはN−アセチルグルコサミン−N−アセチルムラミル−ペンタペプチドの重合及びペンタペプチド間の架橋による細胞壁の最終形成段階に関わる主要酵素であることから、抗生物質の発見にとって重要なターゲットの1つであると考えられている。これらの膜に固定された酵素は3つの別個のタンパク質ドメイン、つまりそのアミノ末端からカルボキシル末端に向かって膜貫通(Transmembrane:TM)ドメイン、トランスグリコシラーゼドメイン(Transglycosilyase:TG)、トランスペプチダーゼ(Transpeptidase:TP)ドメインから成る。S.aureusから得たPBP2細胞外ドメインと(Lovering AL, de Castro LH, D. Lim D, Strynadka NC (2007) Science 315:1402-1405)及びA.aeolicusから得たPBP1aのトランスグリコシラーゼドメイン(Yuan Y, Barrett D, Zhang Y, Kahne D, Sliz P, Walker S (2007) Proc Natl Acad Sci 104:5348-5353)の近年のX線結晶構造は、触媒作用におけるPBPの膜貫通ドメインの役割についての情報は欠けているものの、細胞壁ペプチドグリカン重合の説得力のあるメカニズムについて非常に貴重な知見をもたらした。PBPの膜貫通ドメインは、モエノマイシンの脂質部分又は脂質IIと相互作用するのではないかと推測されている(Ye XY, Lo MC, Brunner L, Walker D, Kahne D, Walker S (2001) J Am Chem Soc 123:3155-3156; Anikin A, Buchynskyy A, Kempin U, Stembera K, Welzel P, Lantzsch G (1999) Angew Chem Int Ed 38:3703-3707)。
【0005】
クラスA型PBPの特性解析及びTG阻害剤の同定には機能性PBP及び重合用の基質としての脂質IIを必要とする。脂質IIの入手には限界があることから、阻害剤の開発のみならず酵素の研究が阻まれていた。脂質IIの不足を解消するために、TG阻害剤を探すためのスクリーニング方法の多くは主にモエノマイシンに頼っている(Di Guilmi AM. Dessen A, Dideberg O, Vernet T (2002) Curr Pharm Biotechnol 3:63-75)。モエノマイシン活性に基づいて、SPRアッセイ(Stembera K, Vogel S, Buchynskyy A, Ayala JA, Welzel P (2002) Chembiochem 3:559-565)又は放射性アッセイ(Vollmer W, Holtje J-V (2000) Antimicrob Agents Chemother 44:1181-1185; Jha RK, de Sousa SM (2006) J Biomol Screen 11:1005-1014; Ravishankar S, Kumar VP, Chandrakala B, Jha RK, Solapure SM, de Sousa SM (2005) Antimicrob Agents Chemother 49:1410-1418; Chandrakala B, Shandil RK, Mehra U, Ravishankar S, Kaur P, Usha V, Joe B, deSousa SM (2004) Antimicrob Agents Chemother 48:30-40)を用いたロースループット阻害剤スクリーニング法が開発された。そのため、ハイスループットスクリーニングに適したTG活性アッセイプラットフォームが阻害剤の同定に望ましい。
【発明を実施するための最良の形態】
【0006】
本発明は細菌から完全長クラスA型ペニシリン結合タンパク質を発現及び精製するための方法を提供するものであり、
(a)細菌のゲノムDNAから完全長ペニシリン結合タンパク質のDNA配列を増幅させ、
(b)DNA配列をベクターにクローニングし、
(c)ホスト細胞内でDNA配列を発現させて完全長ペニシリン結合タンパク質を得て、
(d)タンパク質を界面活性剤で可溶化し、
(e)タンパク質を精製することを含み、
界面活性剤はn−デシル−β−D−マルトピラノシド、n−ウンデシル−β−D−マルトピラノシド、n−ドデシル−β−D−マルトピラノシド、n−オクチル−β−D−グルコピラノシド、n−ノニル−β−D−グルコピラノシド、n−テトラデシルホスホクロリン、n−ドデシル−N,N−ジメチルアミン−N−オキシド、3−[(3−コラミドプロピル)−ジメチルアンモニオ]−1−プロパンスルホネート又はα−[4−(1,1,3,3−テトラメチルブチル)フェニル]−w−ヒドロキシ−ポリ(オキシ−1,2−エタンジイル)である。
【0007】
ある好ましい実施形態において、細菌はEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである。
【0008】
好ましくは、完全長クラスA型ペニシリン結合タンパク質の精製に使用する界面活性剤はn−ドデシル−β−D−マルトピラノシドである。
【0009】
加えて、完全長タンパク質は3つのドメインを含み、N末端からC末端に向かって膜貫通ドメイン、トランスグリコシラーゼドメイン、トランスペプチダーゼドメインである。
【0010】
本発明は抗生物質のハイスループットスクリーニング法を更に提供し、この方法は
(a)スクリーニングの候補を用意し、
(b)蛍光モエノマイシンと膜貫通ドメインとトランスグリコシラーゼドメインを含むクラスA型ペニシリン結合タンパク質で蛍光異方性測定を実行し、
(c)蛍光異方性値とKD値を計算し、
(d)トランスグリコシラーゼ阻害剤のKI及びIC50値を計算し、
(e)阻害剤の最低阻害濃度を求めることを含み、
KDはモエノマイシンとタンパク質との間の解離定数であり、KIは阻害剤とタンパク質との間の解離定数であり、IC50は細菌の増殖を半分に阻害するのに必要な阻害濃度であり、最低阻害濃度とは細胞の増殖を防止する阻害剤の最低濃度である。
【0011】
ある実施形態において、抗生物質はトランスグリコシラーゼ阻害剤であり、好ましくは(Z)−5−(4−ブロモフェニル)−3−((5−ニトロフラン−2−イル)メチレン)フラン−2(3H)−オン、(Z)−1,3−ジフェニル−4−(2−(チアゾール−2−イル)ヒドラゾノ−1H−ピラゾール−5(4H)−オン、(E)−4−(2−(5,5,8,8−テトラメチル−5,6,7,8−テトラヒドロナフタレン−2−イル)プロプ−1−エニル)安息香酸である。
【0012】
ある好ましい実施形態において、タンパク質は3つのドメインを含み、N末端からC末端に向かって膜貫通ドメイン、トランスグリコシラーゼドメイン、トランスペプチダーゼドメインである。
【0013】
本発明において、蛍光プローブとして使用するモエノマイシンは、蛍光異方性に基づいたアッセイの実行時にペニシリン結合タンパク質に結合する。
【0014】
その他の好ましい実施形態において、本発明の方法でスクリーニングされたヒット化合物であるところの抗生物質はEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである細菌の増殖を阻害する。
【0015】
好ましい実施形態において、細菌はE.faecalis又はS.aureusである。
【0016】
以下の実施例は非限定的であり、本発明の多様な態様と特徴を表しているにすぎない。
【実施例1】
【0017】
完全長ペニシリン結合タンパク質の発現
E.coliのPBP1b変異体の発現と精製
E.coli(Swiss−Prot accession number:P02919)からの完全長PBP1bをゲノムDNAから増幅し、pETベクター(EMD Sciences社。サンディエゴ、カリフォルニア州)にクローニングし、N末端(His)6タグで発現させた。BL21(DE3)E.coliホスト細胞を37℃で600nmでのODが0.6に達するまで増殖させ、タンパク質発現を1mMのIPTGで3時間に亘って誘発させた。細胞ペレットをpH8.0の20mMのトリス、300mMのNaClに再懸濁させ、マイクロフルイダイザ(Microfluidics社、ニュートン、マサチューセッツ州、米国)で粉砕した。組み換え完全長膜タンパク質の可溶化と精製を様々な界面活性剤を用いてテストし(Anatrace社、マウミー、オハイオ州、米国)、n−デシル−β−D−マルトピラノシド、n−ウンデシル−β−D−マルトピラノシド、n−ドデシル−β−D−マルトピラノシド、n−オクチル−β−D−グルコピラノシド、n−ノニル−β−D−グルコピラノシド、n−テトラデシルホスホクロリン、n−ドデシル−N,N−ジメチルアミン−N−オキシド、CHAPS(3−[(3−コラミドプロピル)−ジメチルアンモニオ]−1−プロパンスルホネート)及びTriton X−100(α−[4−(1,1,3,3−テトラメチルブチル)フェニル]−w−ヒドロキシ−ポリ(オキシ−1,2−エタンジイル)が含まれる。中でも、DDM(n−ドデシル−β−D−マルトピラノシド)は完全長PBP1bを可溶化し、かつそのモエノマイシン結合活性を維持させることが判明した。従って、完全長PBP1bを20mMのDDMで可溶化させ、1mMのDDMの存在下で製造業者の指示に従ってニッケルキレート化クロマトグラフィで精製した。
【0018】
PBP1bのドメインは膜貫通ドメイン(TM、残基64−87)、トランスグリコシラーゼドメイン(TG、残基195−409)、トランスペプチダーゼ(TP、残基444−736)として定義した。ヌクレオチドプライマはTG+TP(残基88−844)、TM+TG(残基1−409)、TG(残基195−409)、及びTP(残基444−736)を含有するそれぞれの変異体を増幅するように設計された。増幅物は発現と精製のためにpETベクターへとクローニングした。変異体TM+TGを得るための手順は完全長タンパク質の調製について記載したとおりである。TG+TP、TG及びTPの変異体については、13mMのn−テトラデシルホスホクロリン(Anatrace社)をタンパク質の抽出に、0.25mMのものをニッケルキレート化クロマトグラフィに用いた。界面活性剤を1mMのDDMに交換し、アミコンウルトラフィルターユニット(Millipore社、ビレリカ、マサチューセッツ州、米国)を用いて濃縮した。
【0019】
16の細菌株からの組み換え完全長二機能性PBPの発現及び精製
完全長二機能性PBPのホモログを、E.coliからのPBP1bタンパク質配列(Swiss−Prot accession number:P02919)を用いてBLASTでNCBIデータベースでの同定を行った(http://www.ncbi.nlm.nih.gov/BLAST/)。トランスグリコシラーゼモチーフと活性部位の存在を確認するため、Jalviewプログラム(4)をMUSCLE多重アラインメントアルゴリズム(5)と共に用いて配列をアラインし、アラインされた出力(図2)をESPript(6)を用いて作成した。明確にするため、以下の残基はアライメントから除外した:Bpe(B. pertussis):354−443、Ngo(N. gonorrhoeae):331−416、Bsu(B. subtilis):615−643、735−787、797−835、及び897−914、及びCdi(C. difficile):267−322及び804−842。
【0020】
個々のPBPの発現のために、ゲノムDNAをAmerican Type Culture Collection(ATCC、マナサス、ヴァージニア州、米国)から購入した。Escherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica及びNeisseria gonorrhoeaeを含む16の細菌からの完全長遺伝子の組み換えタンパク質を完全長E.coliのPBP1bについて記載したものと同じプロトコルを用いて調製した。その殆どは80%より高い純度で得ることができ(図3)、結合実験のために使用可能である。
【実施例2】
【0021】
抗生物質のハイスループット検出
材料及び方法
表面プラズモン共鳴検出
精製したPBP及びその変異体をCM3チップ(GE healthcare社、ウプサラ、スウェーデン)上に約1500〜2000RUまでアミン結合法を用いて固定化した。次にチップを異なる濃度のモエノマイシンA(0−2000nM)に通した。固定化及びデータ収集をBIAcoreT100(GE Healthcare社、ウプサラ、スウェーデン)を用いて25℃で行った。
【0022】
蛍光異方性測定
蛍光異方性測定を488nmレーザを備えたレーザ蛍光分析装置(IsoCyte、Blueshift Biotech社、サニーヴェール、カリフォルニア州、米国)を用いて384ウェルプレートのウェルで3回行った。各種バッファ、塩、pH値、及び二価陽イオン(Ca++、Mg++、Co++)を蛍光異方性測定にむけて最適化した。KD及びKIは100mMのNaCl、pH8.0の10mMのトリスにおいて求めた。走査の焦点は、検出の障害とならないようにプレート底部の上方であった。データ分析はプロプライエタリ・ソフトウェアであるBlueImage(Blueshift Biotech社)を用いて行った。蛍光異方性値(A)は方程式:A= (I平行G*I垂直)/(I平行+2G*I垂直)を用いて計算し、I平行は励起光に平行な放射光の蛍光強度であり、I垂直は励起光に対して垂直な放射光の蛍光強度であり、Gは機器バイアスを修正するためのゲーティングファクタである。Gファクタは各回ごとにプローブだけのウェルを基準異方性として用いて実験的に求めた。
【0023】
直接結合アッセイからのKD値の測定
直接結合アッセイにおいて、初期条件にはpH8.0、10mMのトリス、100mMのNaCl中の40μlの100nMのF−Moeが含まれた。少量のPBP原液を添加した後、5分間の平衡後に異方性の変化を測定した。直接滴定はリガンド(L)とレセプタ(R)との単純な結合平衡に沿うものとみなす(R+LS⇔RLSでKD=R・LS/RLS)。データをPrism(GraphPad Software社、サンディエゴ、カリフォルニア州)を用いて[方程式1KD]にフィットさせ、KD(3)を求めた。
(A測定−A最低)/(A最高−A最低)=(KD+LST+RT)−[KD+LST+RT]2−4・LST・RT]1/2 [方程式1]
であり、A測定は観察された異方性、A最低は最低異方性、A最高は最高異方性、LSTはF−Moeの総濃度、RTはPBPの総濃度、KDはリガンド(F−Moe)とレセプタ(PBP)との間の解離定数である。報告値は4回の平均である。
【0024】
KI及びIC50値の競合置換アッセイによる測定
競合置換において、初期条件にはpH8.0、10mMのトリス、100mMのNaCll中の40μlの100nMのF−Moe、10μg/mlのH.pyloriのPBP1aが含まれた。化合物原液のアリコートを添加し、5分間の平衡の後、異方性をモニタした。置換アッセイによって得られたデータを用いて、完全競合結合モデル(RL+LS⇔KLR+LS+L⇔KDRLS+L)(3)を用いて阻害剤の阻害定数(KI)とIC50値を計算した。KI値は総阻害剤濃度(C)を結合/遊離F−Moeのモル比に対してプロットし、得られた曲線を[方程式2]にフィットさせることで求めた。
C=[(KI/KD*X)+1][RT−(LST*X)/(X+1)−KD*X] [方程式2]
X=結合/遊離=(A測定−A最低)/(A最高−A測定)
ここでA測定は化合物の特定濃度(C)で観察された異方性、A最低は最低異方性、A最高は最高異方性、LSTはF−Moeの総濃度、RTはPBP1aの総濃度、KDはリガンド(F−Moe)とレセプタ(PBP)との間の解離定数、KIは阻害剤とPBP1aとの間の解離定数である。報告値は4回の平均である。
【0025】
IC50を求めるために、結合蛍光プローブの割合を競合物濃度(C)の対数値に対してプロットする。得られたS次型曲線を[方程式3]を用いてフィットさせると、IC50値が得られる。
(A測定−A最低)/(A最高−A最低)=1/(1+10^((C−log(IC50))*傾き) [方程式3]
ここでA測定は化合物の特定濃度(C)で観察された異方性であり、A最低は最低異方性、A最高は最高異方性である。
【0026】
TG阻害剤のハイスループットスクリーニング
FAアッセイを購入した50000個の小分子(ChemBridge社、サンディエゴ、カリフォルニア州、米国)と我々が所有するコレクションのうちの7000個に対してのスクリーニングに使用した。化合物を96ウェルプレート(Freedom Evo、Tecan Schweiz AG社、マネドルフ、スイス)に、次に384ウェルプレートへとマルチディスペンサ(Labcyte社、サニーヴェール、カリフォルニア州、米国)を用いて移し、スクリーニング用の化合物のプレートを準備した。最終容量40μl、pH8.0の100nMのF−Moe、10mMのトリス、100mMのNaCl中のH.pyloriのPBP1a(10μg/ml)を384ウェルプレートに加えた(Freedom Evo150、Tecan社)。2mM化合物原液の1μlをマルチディスペンサ(Labcyte社)を用いてウェルに加えた。各プレートの最後2つのカラムはコントロールである10μMのモエノマイシンと2.5%のDMSOにそれぞれ用いた。30分の培養後、蛍光異方性における変化をIsocyte(Blueshift Biotech社)を用いて求めた。コントロールの異方性値と比較して75%より高い低下を示したヒット化合物を更に調べるために選択した。
【0027】
最低阻害濃度(minimal inhibitory concentration:MIC)の測定
試験化合物の最低阻害濃度(MIC)をNCCLS基準に従って求めた。実験は血液を加えた(Streptococcus pneumonia)又は加えていない(Bacillus subtilis、Enterococcus faecalis、Staphylococcus aureus)ミュラー・ヒルトン培養液での二倍希釈を用いて96ウェルマイクロタイタープレートで行った。5x105細胞/mlで指数関数的に増殖する細胞を様々な濃度の試験化合物で培養した。18〜24時間に亘る37℃での培養後、細菌の増殖を防止する化合物の最低濃度としてMICを求めた。
【0028】
結果
クラスA型ペニシリン結合タンパク質へのモエノマイシン結合のドメイン要件(二機能性トランスグリコシラーゼ)
E.coliからのPBP1bの異なるドメインを含むタンパク質構築物を、表面プラズモン共鳴(SPR)を用いたモエノマイシン結合活性測定用に発現、適した界面活性剤を用いて精製した(実施例1)。5つのE.coliのPBP1b変異体を発現、精製した:膜貫通ドメイン、トランスグリコシラーゼドメイン及びトランスペプチダーゼドメインを含む完全長タンパク質(TM+TG+TP)、(2)TG+TP、(3)TM+TG、(4)TG及び(5)TP(図1B)。ターゲットタンパク質をセンサーチップ上に固定化した後、異なる濃度のモエノマイシンを表面に通し、結合親和性を求めた。図1Bに図示されるように、膜貫通ドメインを有していないPBP(TG+TP)のモエノマイシン結合活性は完全長PBP1bの結合活性と比較すると約5倍低かった。その一方で、TM+TGの結合活性は完全長タンパク質の結合活性と同様である。結果は、膜貫通ドメインがモエノマイシン結合において重要な役割を果たす可能性があることを示唆している。完全長タンパク質のモエノマイシン結合親和性は4.4x10−7Mと求められており、TG反応についての阻害能と同様であった(Stembera K, Vogel S, Buchynskyy A, Ayala JA, Welzel P (2002) Chembiochem 3:559-565)。我々は、SPR分析でのモエノマイシンAのC25脂質の非特異的な結合を憂慮していたが、TP変異体をコントロール用のタンパク質として用い、求められたモエノマイシンのPBP1b変異体に対しての結合親和性が顕著であり特異的なものであることが確認された。切断PBP1b変異体を使用した結合実験は、TM+PG変異体の結合親和性が完全長タンパク質の結合親和性と同様であることを更に示した。Moe A結合におけるTMドメインの役割が、TG+TP変異体の結合能が完全長PBP1bのものと比較して5倍も低下したという観察結果から示された(図1B)。
【0029】
モエノマイシンのPBP結合活性と最低阻害濃度(MIC)の相関関係
膜貫通ドメインはモエノマイシン結合に大きく関与することから、モエノマイシン結合の研究のために16のグラム陰性及びグラム陽性細菌(表1)からの完全長PBPの準備に着手した。クラスA型PBPの遺伝子はNCBIデータベース(http://www.ncbi.nlm.nih.gov/BLAST/)を用いて同定し、この全てがTG及びTPモチーフを有していることが確認された(Barrett DS, Chen L, Litterman NK, Walker S (2004) Biochemistry 43:12375; Di Guilmi AM, Dessen A, Dideberg O, Vernet T (2003) J Bacteriol 185:1650-1658)(図2)。ターゲット遺伝子を個々の細菌種のそれぞれのゲノムDNAから増幅して発現ベクターにクローニングし、E.coliホストを用いて組み換えタンパク質を産生させた。精製したタンパク質の酵素活性は脂質II重合によって確認され(データ提示せず)、モエノマイシン結合はSPRで測定した。表1に示されるように、異なる種から得たPBP間には様々な定常状態親和性(KD)値が見えられたが、測定されたKD値は全て10−7Mの範囲内に収まり、トランスグリコシル化プロセスについて報告された阻害濃度に近い(Stembera K, Vogel S, Buchynskyy A, Ayala JA, Welzel P (2002) Chembiochem 3:559-565)。興味深いことに、テストしたグラム陽性細菌の中でも、E.faecalisとS.aureusのPBPのKD値はMIC値と相関関係にあり、PBPのモエノマイシン結合部位がこれらの種に対しての抗生物質の開発にとってよいターゲットとなり得ることを示唆している。
【0030】
表1
16の細菌株についての抗菌活性とモエノマイシンのPBP結合親和性との相関関係
*1 異なる細菌種に対してのモエノマイシンのMIC。
*2 文献から得たモエノマイシンのMIC値。
*3 個々の細菌株から得たクラスA型PBP遺伝子の配列ID。クラスA型PBPのホモログをBLASTを用いてNCBIデータベースで同定した。
*4 SPRを用いてのPBPホモログとモエノマイシンのKD。平均値を示す。
*5 本発明のFAに基づいたアッセイを用いて得たPBPホモログとモエノマイシンとのKD。
【0031】
トランスグリコシラーゼ阻害剤についての蛍光異方性に基づいたアッセイの開発
完全長タンパク質をモエノマイシン結合研究に使用できるとの結論に基づいて、蛍光異方性(FA)に基づいたアッセイを設計し、TGに対しての小分子の結合親和性をモニタした。複雑な結晶構造(Lovering AL, de Castro LH, D. Lim D, Strynadka NC (2007) Science 315:1402-1405)及び構造・活性関係(Welzel P (2005) Chem Rev 105:4610-4660)についての従来の研究は、化合物1から2への変性(図4A)は結合と抗菌性を劇的に低下させることはないことを示唆した。更に、2のアミノ部分により、結合研究用のモエノマイシン型のプローブとしてのいずれのフルオロフォアとの共役が可能となる(Buchynskyy A, Kempin U, Vogel S, Hennig L, Findeisen M, Muller D, Giesa S, Knoll H, Welzel P (2002) Eur J Org Chem 7:1149-1162; Donnerstag A, Marzian S, Muller D, Welzel P (1995) Tetrahedron 51:1931-1940)。実際、化合物2は基本条件下で即座にフルオレセイン(6−カルボキシフルオレセイン N−ヒドロキシスクシンイミドエステル)に結合して蛍光プローブ3を生成した(F−Moe、図4B)。FAアッセイで使用の蛍光プローブについての主な懸念の1つはプローブそれ自体、つまりフルオロフォア又は構造変性のいずれかがターゲットタンパク質と小分子との結合を阻害することである。従って、Moe AのPBP結合親和性及び蛍光プローブをSPRを用いて比較した。求めた定常状態親和性(KD)値はMoe A及びF−Moeについて同様であった(4.4x10−7対5.2x10−7M)(図4C及び4D)。この結果によりF−MoeがFAに基づいたPBP結合アッセイに有用な有効蛍光プローブであることが確認された。
【0032】
F−Moeの異方性はE.coliのPBP1bとの培養により著しく上昇し、これはF−Moe−PBP1b複合体の形成によるものと思われる(図5A)。対照的に、F−Moeの異方性は、100μMまで、ウシ血清アルブミンとの培養では変化しなかった(データ提示せず)。タンパク質濃縮物を、異方性変化の用量依存性を確立するために更に滴定に供した。F−Moeについての推定解離定数(KD)はE.coliのPBP1bにおいてはサブミクロモル範囲にあった(図5A)。しかしながら、E.coliのPBP1bを用いたFAアッセイの信号対雑音比は不十分であり、最高異方性は0.120である。従って、異なるクラスA型PBPをスクリーニングしてアッセイを改良した。試験したホモログ全てにおいて、Helicobacter pyloriのPBP1aを用いたFAアッセイが25+/−14nMの最適な信号対雑音比を出し、異方性はF−Moeへの結合時に0.018から0.2へと上昇した(図5B)。阻害剤スクリーニング用のアッセイを開発するために、F−MoeをH.pyloriのPBP1aと予備培養し、次に様々な濃度の無標識のMoe Aとの競合に供した。Moe Aの濃度が上昇するにつれて異方性の低下が観察され、KI(阻害定数)とIC50値はそれぞれ0.47+/−0.10μMと0.36μMとなった(図5C)。この結果により、FAアッセイがPBPのモエノマイシン結合ポケットからプローブを競合的に置換する阻害剤をスクリーニングすることが実証された。
【0033】
トランスグリコシラーゼ阻害剤のハイスループットスクリーニング
幾つかのモエノマイシン類似体(2、4、5)を調製してFAに基づいたアッセイの信頼性を評価した。表2に示されるように、これらのモエノマイシン類似体のFAに基づいたアッセイで求めた異なる結合親和性(IC50値)は、発表された論文のものに類似している(Marzian S, Happel M, Wagner U, Muller D, Welzel P, Fehlhaber HW, Stark A, Schutz HJ, Markus A, Limbert M, van Heijenoort Y, van Heijenoort J (1994) Tetrahedron 50:5299-5308)。モエノマイシンAから2への変性によりTG結合親和性は若干低下した。8炭素スペーサを介した2の二量体化により、PBP結合能と抗菌性の双方が2より2倍上昇した4が得られたが、4はそれでもMoe A(1)よりも強度が低い。より顕著には、C25脂質部分を有していない化合物5はF−Moeプローブ3を置換することがほとんどできず、これはモエノマイシンの疎水性部分が完全長PBP1bへの結合に不可欠であることを示している。阻害能の順序がMIC値の順序と一致していることは注目に値する(表2)。
【0034】
表2
選択されたヒット化合物のTG活性の阻害と抗菌性測定
*1 化合物2、4、5はモエノマイシン誘導体である。化合物6〜8はハイスループットスクリーニングでスクリーニングされたヒット化合物である。
*2 IC50値は図5Aで図示の蛍光異方性アッセイを用いて求められた。
*3 異なる細菌種に対してのモエノマイシンのMICを「材料と方法」で記載したように求めた。ATCCはアメリカンタイプカルチャーコレクションの略。
【0035】
モエノマイシン及びその類似体はグラム陽性細菌に対してよく効くものの、その薬物動態特性の悪さによりTG阻害剤開発において新たなリード化合物が必要とされている。HTSフォーマットでの効率的かつ経済的なTGアッセイは、新規のTG阻害剤の同定に役立つと思われる。
【0036】
FAアッセイを用いて50μMでMoeA誘導体(図6)と共に57000個の小分子のコレクションをスクリーニングした。モエノマイシン1μMでのアッセイをハイコントロール、2.5%DMSOでのアッセイをローコントロールとした。ウシ血清アルブミン等のその他のタンパク質もコントロールとして含めることで、異方性の上昇が非特異的な結合によるものでないことを確認した。ハイスループットスクリーニングのロバスト性を評価するための0から1の範囲の統計パラメータであるZ´値(Zhang JH, Chung TD, Oldenburg KR (1999) J Biomol Screen 4:67-73)を100を越える個々の実験から0.895と求めた。スクリーニングでヒットした11の少なくとも75%の阻害を示した化合物を、PBP結合に関して抗菌性アッセイとIC50値の測定を含め更に研究するため選び出した(図7A)。中でも、2つのモエノマイシン類似体(2及び4)及び3つの小分子(化合物6、7及び8)が抗菌性とTG結合阻害活性の双方を有していると確認された(表2)。化合物6、7、8はそれぞれ(Z)−5−(4−ブロモフェニル)−3−((5−ニトロフラン−2−イル)メチレン)フラン−2(3H)−オン(Z)−1,3−ジフェニル−4−(2−(チアゾール−2−イル)ヒドラゾノ−1H−ピラゾール−5−(4H)−オン;(E)−4−(2−(5,5,8,8−テトラメチル−5,6,7,8−テトラヒドロナフタレン―2―イル)プロプ−1−エニル)安息香酸である。
【0037】
本発明のFAに基づいたアッセイを用いて、0.895のZ´値で0.02%のヒット率が得られ、これはこのアッセイを膨大な化合物ライブラリから可能性のあるヒット化合物を迅速に同定するためのロバストな一次スクリーニングとして使用可能なことを示唆している。選択したヒット化合物を抗菌性アッセイ又は脂質II重合活性分析を用いて更にスクリーニングし、リード化合物を同定する。全ての蛍光を利用したアッセイと同様に、本発明のFAに基づいたアッセイは蛍光化合物についてTG阻害活性を求めるのに使用することはできない。しかしながら、FAに基づいたPBP結合アッセイは新規の抗菌剤の発見に非常に強力なHTSアッセイであることが実証された。
【0038】
本発明を当業者がそれを実践するのに十分な詳細さでもって説明、例示してきたが、本発明の精神と範囲から逸脱することなく様々な代替、改変、改良が可能であることは明白である。
【0039】
本発明が目的を達成し、本来備えているもののみならず、記載の結果と利点を得るに十分に適合されていることを当業者は容易に理解するものである。その改変及びその他の使用は他の当業者の想定内である。これらの改変は本発明の精神の範囲に含まれ、請求項の範囲によって定められる。
【図面の簡単な説明】
【0040】
【図1】モエノマイシンによるトランスグリコシラーゼの阻害と切断されたPBP変異体の結合親和性を示す。(A)モエノマイシンA(1)が細菌細胞壁の合成におけるトランスグリコシル化工程を阻害している。(B)モエノマイシンの結合の研究に使用したPBP変異体の概略図である。完全長PBP(TM+TG+TP)はE.coliのPBP1b(P02919)のアミノ酸残基1−844を含有している。TG+TP(残基88−844)、TM+TG(残基1−409)、TG(残基195−409)及びTP(残基444−736)は完全長PBP1bの一部を含む切断変異体である。これらの変異体のモエノマイシン結合定数(KD)はSPRを用いて求めた。
【図2】16の細菌株から得た完全長二機能性PBPの配列アラインメントを示す。本研究で使用した16の完全長PBP配列をJalviewログラムを用いてアラインし、図はESPriptを用いて作成した。保存した残基について、同一の残基の場合は背景を濃い灰色、似た残基は灰色でそれぞれ囲んだ。灰色の星印はTGの場合モチーフ:EDxxFxxHxxG、GxSTxTQQ、RKxxE、KxxILxxYxN、及びRxxVL、TPの場合はモチーフSxxK、SxN及びKTGを示す。下線のついた星印は触媒残基を記している。アラインされた配列の順番は表1と図3の順番と同一である。
【図3】16の細菌種から得たクラスA型PBPの精製タンパク質のSDS−PAGE分析を示す。各レーンは5μgのタンパク質を含有し、MはkDa単位での分子量マーカーを示す。1:Bordetella pertussisからのPBP1a、2:Citrobacter freundiiからのPBP1b、3:Escherichia coliからのPBP1b、4:Haemophilus influenzaeからのPBP1b、5:Helicobacter pyloriからのPBP1a、6:Klebsiella pneumoniaeからのPBP1b、7:Neisseria gonorrhoeaeからのPBP1、8:Pseudomonas aeruginosaからのPBP1b、9:Salmonella entericaからのPBP1b、10:Shigella flexneriからのPBP2、11:Bacillus subtilisからのPBP1a/1b、12:Clostridium difficileからのPBP、13:Enterococcus faecalisからのPBP2a、14:Enterococcus faeciumからのPBP1、15:Staphylococcus aureusからのPBP2、16:Streptococcus pneumoniaeからのPBP1b。
【図4】トランスグリコシラーゼについての新規の蛍光異方性アッセイの設計。(A、B)変性モエノマイシンA(2)及びフルオレセインで標識したモエノマイシンF−Moe(3)の化学構造。(C、D)モエノマイシンA(C)とF−Moe(D)のE.coliのPBP1bに対しての結合活性のSPR分析。固定化したE.coliのPBP1bへのモエノマイシン結合についての反応を示している。データは、挿入したグラフで図示されるように、定常状態での親和性を用いて分析し、1:1の相互作用モデルへとフィットさせた。X軸と点線の切片から推定したKD値はMoe A及びF−Moeについてそれぞれ4.4x10−7M及び5.161x10−7Mであった。
【図5】トランスグリコシラーゼについてのハイスループットFAアッセイの開発。(A)E.coliのPBP1bがF−Moeに結合すると、蛍光異方性において濃度依存性の変化が観察された。(B)Helicobacter pyloriのPBP1aでの改善されたFAアッセイ。H.pyloriのPBP1aを用いて同様に濃度依存性の蛍光異方性変化を行った。最大異方性値は0.2であった。(C)様々な濃度の無標識モエノマイシンによるPBP1a結合F−Moe複合体の置換を示す。蛍光異方性における変化は[(A測定−A最低)/(A最高−A最低)x100%]と定義する。KD及びIC50値を実施例2に記載したように計算した。
【図6】モエノマイシン誘導体の化学構造を図示する(4及び5)。
【図7】クラスA型PBPを用いたTG阻害剤としての小分子のスクリーニングを示す。(A)FAアッセイを用いたTG阻害剤のハイスループットスクリーニングのスキーム。タンパク質構造の画像はPDB ID 2OLV(12)から作成した。(B)ハイスループットスクリーニングでヒットした化合物の化学構造。
【特許請求の範囲】
【請求項1】
抗生物質のハイスループットスクリーニング方法であり、
(a)スクリーニングの候補を用意し、
(b)蛍光モエノマイシンと膜貫通ドメインとトランスグリコシラーゼドメインを含むクラスA型ペニシリン結合タンパク質で蛍光異方性測定を実行し、
(c)蛍光異方性値とKD値を計算し、
(d)トランスグリコシラーゼ阻害剤のKI及びIC50値を計算し、
(e)阻害剤の最低阻害濃度を求めることを含み、
KDはモエノマイシンとタンパク質との間の解離定数であり、KIは阻害剤とタンパク質との間の解離定数であり、IC50は細菌の増殖を半分に阻害するのに必要な阻害濃度であり、最低阻害濃度とは細胞の増殖を防止する阻害剤の最低濃度である方法。
【請求項2】
抗生物質がトランスグリコシラーゼ阻害剤である、請求項1に記載の方法。
【請求項3】
トランスグリコシラーゼ阻害剤が(Z)−5−(4−ブロモフェニル)−3−((5−ニトロフラン−2−イル)メチレン)フラン−2(3H)−オン、(Z)−1,3−ジフェニル−4−(2−(チアゾール−2−イル)ヒドラゾノ−1H−ピラゾール−5(4H)−オン、(E)−4−(2−(5,5,8,8−テトラメチル−5,6,7,8−テトラヒドロナフタレン−2−イル)プロプ−1−エニル)安息香酸である、請求項2に記載の方法。
【請求項4】
タンパク質がN末端からC末端に向かって膜貫通、トランスグリコシラーゼ、トランスペプチダーゼの3つのドメインを含む、請求項1に記載の方法。
【請求項5】
モエノマイシンがペニシリン結合タンパク質に結合する、請求項1に記載の方法。
【請求項6】
抗生物質が細菌の増殖を阻害し、細菌がEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである、請求項1に記載の方法。
【請求項7】
細菌がE.faecalis又はS.aureusである、請求項6に記載の方法。
【請求項8】
前記クラスA型ペニシリン結合タンパク質が以下の手段によって得られることを特徴とする、請求項1記載の方法。
(a)細菌のゲノムDNAから完全長ペニシリン結合タンパク質のDNA配列を増幅させ、
(b)DNA配列をベクターにクローニングし、
(c)ホスト細胞内でDNA配列を発現させて完全長ペニシリン結合タンパク質を得て、
(d)タンパク質を界面活性剤で可溶化し、
(e)タンパク質を精製することを含み、
界面活性剤がn−デシル−β−D−マルトピラノシド、n−ウンデシル−β−D−マルトピラノシド、n−ドデシル−β−D−マルトピラノシド、n−オクチル−β−D−グルコピラノシド、n−ノニル−β−D−グルコピラノシド、n−テトラデシルホスホクロリン、n−ドデシル−N,N−ジメチルアミン−N−オキシド、3−[(3−コラミドプロピル)−ジメチルアンモニオ]−1−プロパンスルホネート又はα−[4−(1,1,3,3−テトラメチルブチル)フェニル]−w−ヒドロキシ−ポリ(オキシ−1,2−エタンジイル)である。
【請求項9】
細菌がEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである、請求項8に記載の方法。
【請求項10】
界面活性剤がn−ドデシル−β−D−マルトピラノシドである、請求項8に記載の方法。
【請求項11】
タンパク質がN末端からC末端に向かって膜貫通、トランスグリコシラーゼ、トランスペプチダーゼの3つのドメインを含む、請求項8に記載の方法。
【請求項1】
抗生物質のハイスループットスクリーニング方法であり、
(a)スクリーニングの候補を用意し、
(b)蛍光モエノマイシンと膜貫通ドメインとトランスグリコシラーゼドメインを含むクラスA型ペニシリン結合タンパク質で蛍光異方性測定を実行し、
(c)蛍光異方性値とKD値を計算し、
(d)トランスグリコシラーゼ阻害剤のKI及びIC50値を計算し、
(e)阻害剤の最低阻害濃度を求めることを含み、
KDはモエノマイシンとタンパク質との間の解離定数であり、KIは阻害剤とタンパク質との間の解離定数であり、IC50は細菌の増殖を半分に阻害するのに必要な阻害濃度であり、最低阻害濃度とは細胞の増殖を防止する阻害剤の最低濃度である方法。
【請求項2】
抗生物質がトランスグリコシラーゼ阻害剤である、請求項1に記載の方法。
【請求項3】
トランスグリコシラーゼ阻害剤が(Z)−5−(4−ブロモフェニル)−3−((5−ニトロフラン−2−イル)メチレン)フラン−2(3H)−オン、(Z)−1,3−ジフェニル−4−(2−(チアゾール−2−イル)ヒドラゾノ−1H−ピラゾール−5(4H)−オン、(E)−4−(2−(5,5,8,8−テトラメチル−5,6,7,8−テトラヒドロナフタレン−2−イル)プロプ−1−エニル)安息香酸である、請求項2に記載の方法。
【請求項4】
タンパク質がN末端からC末端に向かって膜貫通、トランスグリコシラーゼ、トランスペプチダーゼの3つのドメインを含む、請求項1に記載の方法。
【請求項5】
モエノマイシンがペニシリン結合タンパク質に結合する、請求項1に記載の方法。
【請求項6】
抗生物質が細菌の増殖を阻害し、細菌がEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである、請求項1に記載の方法。
【請求項7】
細菌がE.faecalis又はS.aureusである、請求項6に記載の方法。
【請求項8】
前記クラスA型ペニシリン結合タンパク質が以下の手段によって得られることを特徴とする、請求項1記載の方法。
(a)細菌のゲノムDNAから完全長ペニシリン結合タンパク質のDNA配列を増幅させ、
(b)DNA配列をベクターにクローニングし、
(c)ホスト細胞内でDNA配列を発現させて完全長ペニシリン結合タンパク質を得て、
(d)タンパク質を界面活性剤で可溶化し、
(e)タンパク質を精製することを含み、
界面活性剤がn−デシル−β−D−マルトピラノシド、n−ウンデシル−β−D−マルトピラノシド、n−ドデシル−β−D−マルトピラノシド、n−オクチル−β−D−グルコピラノシド、n−ノニル−β−D−グルコピラノシド、n−テトラデシルホスホクロリン、n−ドデシル−N,N−ジメチルアミン−N−オキシド、3−[(3−コラミドプロピル)−ジメチルアンモニオ]−1−プロパンスルホネート又はα−[4−(1,1,3,3−テトラメチルブチル)フェニル]−w−ヒドロキシ−ポリ(オキシ−1,2−エタンジイル)である。
【請求項9】
細菌がEscherichia coli、Klebsiella pneumoniae、Streptococcus pneumoniae、Shigella flexneri、Haemophilus influenzae、Helicobacter pylori、Citrobacter freundii、Bordetella pertussi、Staphylococcus aureus (MRSA Mu50)、Bacillus subtilis、Pseudomonas aeruginosa、Clostridium difficile、Enterococcus faecium、Enterococcus faecalis、Salmonella enterica又はNeisseria gonorrhoeaeである、請求項8に記載の方法。
【請求項10】
界面活性剤がn−ドデシル−β−D−マルトピラノシドである、請求項8に記載の方法。
【請求項11】
タンパク質がN末端からC末端に向かって膜貫通、トランスグリコシラーゼ、トランスペプチダーゼの3つのドメインを含む、請求項8に記載の方法。
【図1】

【図2】

【図3】

【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−229528(P2011−229528A)
【公開日】平成23年11月17日(2011.11.17)
【国際特許分類】
【出願番号】特願2011−102306(P2011−102306)
【出願日】平成23年4月28日(2011.4.28)
【分割の表示】特願2008−178187(P2008−178187)の分割
【原出願日】平成20年7月8日(2008.7.8)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 発行者名 National Academy of Sciences 刊行物名 Proceeding of the National Academy of Science of the United States of America 巻数 105 第2 発行年月日 2008年1月15日
【出願人】(505008028)中央研究院 (4)
【Fターム(参考)】
【公開日】平成23年11月17日(2011.11.17)
【国際特許分類】
【出願日】平成23年4月28日(2011.4.28)
【分割の表示】特願2008−178187(P2008−178187)の分割
【原出願日】平成20年7月8日(2008.7.8)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 発行者名 National Academy of Sciences 刊行物名 Proceeding of the National Academy of Science of the United States of America 巻数 105 第2 発行年月日 2008年1月15日
【出願人】(505008028)中央研究院 (4)
【Fターム(参考)】
[ Back to top ]