
ペニシリン結合タンパク質(PBP)2bの結晶
【課題】 X線結晶構造解析および阻害剤の設計等を可能とする高品質のペニシリン結合タンパク質(PBP)2bの結晶を提供する。
【解決手段】 肺炎連鎖球菌(Streptococcus pneumoniae)由来のPBP2b可溶性部分およびトランスペプチダーゼドメインにアミノ酸変異を導入し、高品質で再現性良く得られるPBP2bの結晶を作製した。得られた結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功した。
【解決手段】 肺炎連鎖球菌(Streptococcus pneumoniae)由来のPBP2b可溶性部分およびトランスペプチダーゼドメインにアミノ酸変異を導入し、高品質で再現性良く得られるPBP2bの結晶を作製した。得られた結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、X線結晶構造解析および阻害剤の設計等に利用するためのペニシリン結合タンパク質(PBP)2bの結晶に関する。
【背景技術】
【0002】
ペニシリン結合タンパク質(PBP)は細菌における細胞壁を構成する構造ユニットであるムレインモノマーを基質として細胞壁ペプチドグリカンを組み立てる一連の酵素群であり、細菌のペプチドグリカンの重合および架橋反応に働く膜結合型のタンパク質である。肺炎連鎖球菌(Streptococcus pneumoniae)には6種類のPBPが確認されているが、ペニシリン耐性肺炎連鎖球菌では主に3種類のPBP(PBP1a、PBP2x、PBP2b)における変異がβ−ラクタム薬の感受性低下に関与している。特にペニシリン系薬剤およびカルバペネム系薬剤においては、耐性とPBP2b変異との密接な関連が示唆されている。また、配列番号3に示されるR6株(ATCC49619)由来PBP2bにおいては、アミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換された変異が薬剤耐性に重要であることが示唆されている(非特許文献1および特許文献1参照)。従って、PBP2bはβ−ラクタム薬の耐性に重要な役割を果たしており、PBP2bの立体構造をX線結晶構造解析により解明することは、耐性のメカニズムを説明するだけでなく、耐性菌に有効な新薬を創出する上でも非常に重要である。
【0003】
肺炎連鎖球菌(Streptococcus pneumoniae)由来PBP2bは、細胞内ドメイン、膜貫通領域、N末端ドメインおよびトランスペプチダーゼドメインから構成される膜結合型タンパク質であり、細胞内ドメインおよび膜貫通領域を除去し、膜結合機能を喪失させたPBP2b可溶性部分の調製方法が明らかになっている(特許文献1および非特許文献2参照)。しかしながら、PBP2b可溶性部分からさらにN末端ドメインを除去したトランスペプチダーゼドメインの調製方法は未だ明らかとなっていない。また、膜結合型PBP2b、PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインのいずれについても、結晶および結晶構造解析の報告はない。従って、X線結晶構造解析によりPBP2bの立体構造を決定し、効率的な阻害剤の設計を可能とする高品質のPBP2b結晶の創出が望まれている。
【0004】
一般にタンパク質のN末端、C末端やループ領域は構造が揺らいでいる場合があり、その際には複数のコンフォメーションを取り得ることから、タンパク質の揺らいでいる領域の除去やドメインの切り出しは均一のコンフォメーションから構成される高分解能のX線回折能を有する結晶を作製するために有効な手段である(非特許文献3参照)。その一つの方法としてプロテアーゼであるトリプシンを利用してタンパク質を限定分解する方法が知られている。トリプシンは表面に露出している塩基性アミノ酸であるリジンやアルギニン残基の直後を選択的に切断することにより、タンパク質の揺らいでいる領域を除去可能な場合がある。肺炎連鎖球菌PBP1aではトリプシン限定分解物において分解能2.6ÅのX線回折能を示す結晶が得られており、その際、トリプシン限定分解物の純度を確保しマイナー部位での切断を避けるため1箇所点変異を導入している(非特許文献4参照)。また、肺炎連鎖球菌PBP1bではトリプシン限定分解物において分解能1.9ÅのX線回折能を示す結晶が得られており、その際、トリプシン限定分解物の純度を確保しマイナー部位での切断を避けるため3箇所点変異を導入している(非特許文献5参照)。しかしながら、トリプシン限定分解のパターンは個々のタンパク質に依存し、PBP2bでは未だ明らかになっていないことから、点変異導入の必要性も含め結晶化に適したトリプシン限定分解物の作製には試行錯誤が必要である上、PBP2bに対してトリプシン限定分解が有効な手段であるかも明らかではない。結晶化に適したPBP2bトランスペプチダーゼドメインをトリプシン限定分解することにより新規に見出すことができれば、高品質の結晶を作製し、PBP2bの立体構造を解明することに多いに貢献できると考えられる。
【0005】
また、タンパク質の表面に露出しているグルタミン酸やリジン等の特定のアミノ酸残基に変異を導入し、タンパク質表面の性状を変化させることにより高品質の結晶を作製した報告も知られている(非特許文献6参照)。しかしながら、この方法は立体構造が既知のタンパク質においては有効であるが、PBP2bのように立体構造が未知のタンパク質の場合には表面に露出している残基を正確に同定することができないため、理論に基づいて変異を導入することは困難である。立体構造が未知のタンパク質に対して表面の性状あるいはコンフォメーションを変化させるために変異を導入することは可能であるが、結晶化に適した変異であるかどうかについては、実際にタンパク質を調製し、結晶化スクリーニングを実施した後、X線回折実験で検証する必要があり、多大な試行錯誤を要する。さらに、作製した変異体の結晶化において、特定の試薬の添加が結晶化に有効であるかどうかを予想することも困難である。従って、PBP2bにおいて、結晶化に適したアミノ酸変異の同定と結晶化に有効な試薬の組み合わせを見出すことができれば、高品質の結晶を作製し、PBP2bの立体構造を解明することに多いに貢献できると考えられる。
【特許文献1】特許公開2004−290070号広報
【非特許文献1】Antimicrob.Agents Chemother. 48(6)2244−2250(2004)
【非特許文献2】Antimicrob.Agents Chemother. 48(5)1848−1855(2004)
【非特許文献3】BIOベンチャー 3(5)、36−39(2003)
【非特許文献4】J.Mol.Biol.355(4)684−696(2006)
【非特許文献5】Proc.Natl.Acad.Sci.USA.102(3)577−582(2005)
【非特許文献6】Structure 12(4)529−535(2004)
【発明の開示】
【発明が解決しようとする課題】
【0006】
これまでPBP2bの結晶および結晶構造解析の報告はない。そのため、創薬において重要であるPBP2bの立体構造情報が存在せず効率的な阻害剤の設計が困難である。また、一般にトリプシン限定分解および適切なアミノ酸変異導入等により良質の結晶が得られる可能性があるが、PBP2bへの適用はなく、X線結晶構造解析に適したPBP2b結晶の製造方法は不明である。
【課題を解決するための手段】
【0007】
本発明は、結晶化に適したPBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインを調製し、さらにアミノ酸変異を導入することにより、X線結晶構造解析を実施し効率的な阻害剤の設計等を可能とする高品質のPBP2b結晶を提供することで上記課題を解決する。
【0008】
本発明者は、肺炎連鎖球菌(Streptococcus pneumoniae)由来PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインを調製し、膨大な数の結晶化条件探索の試行錯誤により再現性よく得られる結晶を取得した。さらに、薬剤耐性に関与するアミノ酸変異を導入することにより高品質で再現性よく得られるPBP2bの結晶を取得した。この結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功し、創薬研究に重要なPBP2bの立体構造を決定した。すなわち、本発明はX線結晶構造解析および阻害剤の設計等に利用するためのPBP2b結晶に関する。ここで、PBP2b変異体可溶性部分の結晶において、空間群はF222であり、格子定数はa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°である。また、PBP2b変異体トランスペプチダーゼドメインの結晶において、空間群はP43212であり、格子定数はa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°である。さらに、PBP2b変異体トランスペプチダーゼドメインの結晶構造から、配列番号3におけるアミノ酸残基番号431に対応するスレオニンをリジンに置換する変異により導入したリジン残基の側鎖が結晶化剤として使用した硫酸塩と相互作用することで高品質の結晶が製造できることを見出した。
【0009】
すなわち、本発明は以下の発明を含有する。
(1)連鎖球菌科(Streptococcaceae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
(2)ストレプトコッカス属(Streptococcus)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
(3)肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
(4)ペニシリン結合タンパク質(PBP)2b可溶性部分から構成される(1)〜(3)のいずれか一項に記載の結晶。
(5)配列番号1で示されるアミノ酸配列からなるタンパク質から構成される(1)〜(4)のいずれか一項に記載の結晶。
(6)配列番号1で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される(1)〜(4)のいずれか一項に記載の結晶。
(7)配列番号1で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される(1)〜(4)のいずれか一項に記載の結晶。
(8)空間群がF222である(1)〜(7)のいずれか一項に記載の結晶。
(9)空間群がF222であり、格子定数がa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°である(1)〜(7)のいずれか一項に記載の結晶。
(10)ペニシリン結合タンパク質(PBP)2bトランスペプチダーゼドメインから構成される(1)〜(3)のいずれか一項に記載の結晶。
(11)配列番号2で示されるアミノ酸配列からなるタンパク質から構成される(1)〜(4)、(10)のいずれか一項に記載の結晶。
(12)配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される(1)〜(4)、(10)のいずれか一項に記載の結晶。
(13)配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される(1)〜(4)、(10)のいずれか一項に記載の結晶。
(14)空間群がP43212である(1)〜(4)、(10)〜(13)のいずれか一項に記載の結晶。
(15)空間群がP43212であり、格子定数がa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°である(1)〜(4)、(10)〜(13)のいずれか一項に記載の結晶。
(16)(1)〜(15)のいずれか一項に記載の結晶であって、該結晶を構成するタンパク質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
(17)硫酸塩またはリン酸塩の存在下で成長する(1)〜(16)のいずれか一項に記載の結晶。
(18)硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムの存在下で成長する(1)〜(16)のいずれか一項に記載の結晶。
(19)配列番号2で示されるアミノ酸配列からなるタンパク質。
(20)配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質。
(21)配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質。
(22)肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶の製造方法であって、結晶化剤として硫酸塩またはリン酸塩を用いて結晶化することを特徴とする、(1)〜(18)のいずれか一項に記載の結晶の製造方法。
(23)結晶化剤として硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムを用いることを特徴とする(22)記載の製造方法。
(24)配列番号3におけるアミノ酸残基番号431のスレオニンがリジンに置換されているアミノ酸変異を有することを特徴とする、肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの可溶性部分またはトランスペプチダーゼドメインを調製する工程を含む、(22)または(23)記載の製造方法。
(25)配列番号1または2で示されるアミノ酸配列からなるタンパク質を調製する工程を含む、(22)〜(24)のいずれか一項に記載の製造方法。
【発明の効果】
【0010】
本発明のPBP2b結晶は高品質で再現性良く調製可能であるため、X線結晶構造解析に適しており、これまでに全く報告の無かったPBP2bの立体構造解析が初めて可能となった。本発明のPBP2b結晶を利用することにより、PBP2bを阻害する化合物の効率的な設計等に役立つ立体構造情報を取得できるため、創薬において極めて有用である。
【発明を実施するための最良の形態】
【0011】
以下、本発明を詳細に説明する。
<<PBP2bアミノ酸変異と可溶性部分およびトランスペプチダーゼドメイン>>
本発明の結晶を構成するタンパク質は、連鎖球菌科(Streptococcaceae)、好ましくはストレプトコッカス属(Streptococcus)、より好ましくは肺炎連鎖球菌(Streptococcus pneumoniae)、さらに好ましくは肺炎連鎖球菌R6株(ATCC49619)由来のタンパク質である。特に好ましくは、配列番号3に示されるアミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換された変異体タンパク質であり、これらの変異は薬剤耐性菌の一部に存在する変異である。また、本発明において、「トランスペプチダーゼ」とは、細菌細胞壁の一つの成分であるペプチドグリカンの生合成の最終段階に関与し、ペプチドグリカン連鎖を構成しているN−アセチルムラミン酸から伸びるポリペプチド鎖の架橋を触媒する酵素を意味する。さらに、「トランスペプチダーゼドメイン」とは、上記ポリペプチド鎖を架橋する能力を有し、β−ラクタム薬との結合が可能であるタンパク質の構造単位である。本発明における「相同タンパク質」とは、配列番号1または2に示されるアミノ酸配列を有するタンパク質と少なくとも85%、好ましくは90%、より好ましくは95%、さらに好ましくは98%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼ活性を有するタンパク質である。ここで示した相同性の数値は、当業者に公知の相同性検索プログラムであるFASTA(Science,227,1435−1441(1985);Proc.Natl.Acad.Sci.USA,85,2444−2448(1988);http://www.ddbj.nig.ac.jp/search/fasta−j.html)においてデフォルト(初期設定)のパラメータを用いて算出される数値を示す。本発明における「改変タンパク質」とは、配列番号1または2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼ活性を有するタンパク質である。ここで、「置換、欠失、付加もしくは挿入」などの改変に係るアミノ酸の数は、好ましくは1〜40個、より好ましくは1〜20個、さらに好ましくは1〜10個、特に好ましくは1〜6個である。
【0012】
PBP2bは、細胞内ドメイン、膜貫通領域、N末端ドメインおよびトランスペプチダーゼドメインから構成される膜結合型タンパク質である。PBP2b可溶性部分とは、細胞内ドメインおよび膜貫通領域を除去し、膜結合機能を喪失させたPBP2bである。PBP2b可溶性部分は由来となる株およびN末端が異なる種々のものが存在可能であるが、肺炎連鎖球菌R6株においては、好ましくは、配列番号3に示されるアミノ酸配列のアミノ酸残基番号43から685で構成される。PBP2bトランスペプチダーゼドメインは、PBP2b可溶性部分からさらにN末端ドメインを除去することにより調製可能であり、除去する領域の違いにより種々のものが存在可能であるが、肺炎連鎖球菌R6株においては、配列番号3に示されるアミノ酸残基番号315から685で構成されるのが好ましいことをプロテアーゼであるトリプシンを用いた限定分解実験から見出せる。トランスペプチダーゼドメインの同定方法としては精製したPBP2b可溶性部分を種々の濃度のトリプシン存在下で限定分解した試料の質量分析および生じた各断片のN末端アミノ酸配列分析により決定することができる。PBP2bトランスペプチダーゼドメインはその領域のみを発現させることも可能であるが、好ましくは、PBP2b可溶性部分をトリプシン限定分解することにより調製することが望ましい。その際にマイナー部位での切断を回避するために、配列番号3に示されるアミノ酸残基番号229および561のリジン残基をグルタミンに置換する方法が有用であり、特に残基番号561のリジン残基をグルタミンに置換することが好ましい。なお、広義の解釈においては、PBP2bトランスペプチダーゼドメインは膜結合機能を喪失させた可溶性タンパク質であるため、PBP2b可溶性部分に含有される。
【0013】
<<PBP2b可溶性部分の発現>>
PBP2b可溶性部分は、それをコードするDNA断片を、宿主細胞内で複製可能でかつ同遺伝子が発現可能な状態で含むDNA分子、特にDNA発現ベクターの形態とし、それによって宿主細胞の形質転換を行い、その形質転換体を培養することによって得られる。このDNA分子は、ベクター分子にPBP2b可溶性部分をコードするDNA断片を組み込むことによって得ることができる。本発明の好ましい態様によれば、このベクターはプラスミドである。本発明において利用されるベクターは、使用する宿主細胞の種類を勘案して、ウィルス、プラスミド、コスミドベクターなどから適宜選択することができる。例えば宿主細胞が大腸菌の場合はpUC、pBR系のプラスミド、枯草菌の場合はpUB系のプラスミド、酵母の場合はYEp、YRp、YCp系のプラスミドベクターが挙げられる。宿主細胞としては、宿主−ベクター系が確立されているものであれば利用可能であり、好ましくは大腸菌が挙げられる。宿主細胞の形質転換により得られた形質転換体は、適当な条件で培養し、得られた形質転換体の細胞抽出液を慣用の方法に従い回収することができる。ここで、PBP2b可溶性部分に付加的ポリペプチド、例えばグルタチオン−S−トランスフェラーゼ(GST)またはヒスチジン残基に富むポリペプチド(His−tag)を融合タンパク質として発現することも慣用の技術により可能である。
【0014】
<<PBP2b可溶性部分およびトランスペプチダーゼドメインの精製>>
GSTを融合して大腸菌で発現させたPBP2b可溶性部分は、グルタチオンを固定化したアフィニティーカラムを用いた慣用の技術により容易に精製が可能である。精製したPBP2b可溶性部分を20mM トリス塩酸 pH 7.9、1mM EDTA溶液に対して透析した後、適当量のトリプシンを加え、室温で1〜2時間反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう加えることでPBP2bトランスペプチダーゼドメインが得られる。添加するトリプシンの終濃度は典型的には0.1〜1mg/mLであるが、PBP2b可溶性部分のタンパク質濃度に依存するため、好ましくは、毎回、少量での確認実験により決定する必要がある。PBP2b可溶性部分およびトランスペプチダーゼドメインは陰イオン交換カラム、ハイドロキシアパタイトカラム、陽イオン交換カラム、ゲルろ過カラムを用いた一般的なクロマトグラフィーの手法を組み合わせて精製することが可能であり、配列番号3に示されるアミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換された変異体においても同様に精製が可能である。
【0015】
<<PBP2b可溶性部分およびトランスペプチダーゼドメインの結晶化>>
一般的に蛋白質の結晶化は容易ではない。結晶化できる条件が比較的容易に見出される場合もあるが、通常は種々の結晶化条件をスクリーニングしなければならない。さらに結晶化剤の濃度、pH、塩の種類と濃度、添加剤などの条件を最適化することによって、X線結晶構造解析に供することができる良質の結晶が得られる。また、いかなる条件でも結晶化しない蛋白質も存在すると考えられている。蛋白質の結晶が得られなければX線結晶構造解析を行うことができない。また、X線結晶構造解析に用いる蛋白質の結晶は、結晶であればいかなるものでも解析可能であるわけではない。実際にX線を照射した際に、高分解能の回折強度データが得られることが必要である。本発明におけるPBP2b結晶化の方法は、ハンギングドロップ法、シッティングドロップ法、透析法などのいかなる方法を用いてもよいが、好ましくは蒸気拡散法であるハンギングドロップ法またはシッティングドロップ法が用いられる。また、シーディング法またはリザーバー溶液にオイルを重層することにより結晶核の形成および結晶成長をコントロールすることもできる。結晶化の温度は4℃〜20℃が一般的であるが、本発明では好ましくは20℃である。結晶化に用いるタンパク質としては、(1)配列番号4に示されるR6株由来PBP2b可溶性部分、(2)配列番号5に示されるR6株由来PBP2bトランスペプチダーゼドメイン、(3)配列番号1に示されるR6株由来PBP2b変異体可溶性部分および(4)配列番号2に示されるR6株由来PBP2b変異体トランスペプチダーゼドメインのいずれを用いても良いが、好ましくは(3)R6株由来PBP2b変異体可溶性部分、より好ましくは(4)R6株由来PBP2b変異体トランスペプチダーゼドメインが用いられる。また、前記タンパク質中のメチオニン残基がセレノメチオニンに置換されたセレノメチオニン置換体も同様に結晶化に用いられる。結晶化に用いる精製タンパク質の濃度は、4〜20mg/mL程度であることが好ましい。そして、蛋白質溶液に結晶化剤、塩類、緩衝液、添加剤などを適当量加えて、結晶化を行う。本発明のPBP2bの結晶化に用いられる結晶化剤としては、硫酸アンモニウムなどの無機塩類、ポリエチレングリコールなどの水溶性高分子、イソプロパノールやエタノールなどの有機溶媒などが挙げられる。本発明においては、無機塩類を好ましく用いることができ、硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウム等の硫酸塩もしくはリン酸水素二カリウム等のリン酸塩を特に好ましく用いることができる。また、必要に応じてポリエチレングリコールを組み合わせることも可能である。また、緩衝液としては、公知の緩衝液のいずれも用いることができる。さらに添加剤としては、種々の金属イオン、有機溶媒、還元剤等を用いることができる。セレノメチオニン置換体の場合には未置換体の場合における周辺の条件で結晶を作製できるが、セレン原子の酸化を抑制するためにジチオスレイトール等の還元剤を添加することが好ましい。各PBP2bと好ましい結晶化剤の組み合わせについては次に示す通りである。
【0016】
(1)R6株由来PBP2b可溶性部分:
0.8−1.6M 硫酸アンモニウム、0.1M 酢酸ナトリウム緩衝液 pH 4.6
(2)R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体:
1.1−1.6M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9
(3−1)R6株由来PBP2b変異体可溶性部分:
10−20% ポリエチレングリコール8000、0.2M 硫酸アンモニウム、0.1M トリス塩酸緩衝液 pH 8.0
(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体:
18−20% ポリエチレングリコール8000、0.3M 硫酸リチウム、0.1M トリス塩酸緩衝液 pH 8.5、10mM ジチオスレイトール
(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体:
1.1−1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9−8.5、または、0.7−1.1M 硫酸ナトリウム、10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.0−6.5
上記の条件で得られる結晶のうち、好ましいものは(3−1)R6株由来PBP2b変異体可溶性部分の結晶および(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶であり、特に好ましいものは(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶である。
【0017】
<<PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメイン結晶のX線回折強度データの収集>>
上記のようにして得られる結晶の外観、単位格子の種類・大きさなどはその結晶に固有のものであり、良質の結晶を用いれば、X線回折実験により高分解能の回折強度データを取得することができる。結晶を多価アルコールを含有する抗凍結溶液中に浸した後凍結させ、凍結状態でX線回折データを収集し、X線回折像を得る。ここで、多価アルコールとしては、スクロース、トレハロース、グリセロールなどが挙げられる。これらの中で特にグリセロールが好ましい。抗凍結溶液中の多価アルコールの濃度は、好ましくは10%〜30%である。X線を照射して回折強度データを取得するには、いかなるX線発生装置を用いることができるが、好ましくは、(財)高輝度光科学研究センターの大型放射光施設SPring−8などの第3世代の放射光施設を利用することができる。得られたX線回折強度データを処理することにより、結晶学的パラメータを特定することが可能である。前段落(3−1)R6株由来PBP2b変異体可溶性部分の結晶および(3−2)セレノメチオニン置換体結晶は、空間群がF222である。また、格子定数はa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°であり、より好ましくはa=175±9Å、b=186±9Å、c=369±19Å、α=β=γ=90°であり、さらに好ましくはa=175±2Å、b=186±2Å、c=369±4Å、α=β=γ=90°であり、特に好ましくはa=175±1Å、b=186±1Å、c=369±1Å、α=β=γ=90°である。また、(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶は空間群がP43212であり、格子定数がa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°であり、より好ましくはa=86±4Å、b=86±4Å、c=143±7Å、α=β=γ=90°であり、さらに好ましくはa=86±1Å、b=86±1Å、c=143±2Å、α=β=γ=90°であり、特に好ましくはa=86±1Å、b=86±1Å、c=143±1Å、α=β=γ=90°である。
【0018】
<<PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインの結晶構造の決定>>
上記のように得られたデータを用いてPBP2bの結晶構造の決定が可能となる。一般に位相決定には、分子置換法、重原子同型置換法、多波長異常分散法(MAD法)などが用いられる。目的蛋白質に対する類縁蛋白質の立体構造が未知あるいは相同性が低い場合には、その立体構造を利用した分子置換法による構造解析が不可能であるため、PBP2b結晶構造の決定には、重原子同型置換法またはMAD法などが好ましく、より好ましくはセレノメチオニン置換体結晶を用いたMAD法が用いられる。特に好ましくは、前段落に記載した(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶を用いたMAD法であり、新規結晶構造決定が初めて可能となる。前段落(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶を用いた場合には、分解能がやや劣ることおよび非対称単位中の分子数が多いことによりMAD法による構造決定は困難であるが、(3−1)R6株由来PBP2b変異体可溶性部分の結晶を用いた場合も含め、構造決定したトランスペプチダーゼドメインの結晶構造をサーチモデルに利用する分子置換法により構造決定が可能となる。
【0019】
以下、実施例により本発明をさらに詳細に説明するが、本発明の技術的範囲は実施例によって限定されるものではない。また、各種ベクターの作製、蛋白質の発現等は、特に記載のない限り、Molecular Cloning,A Laboratory Manual,3rd edition(Sambrook and Russell著,Cold Spring Harbor laboratory Press刊(2001))などに記載の公知の手法に従って実施できる。
【実施例1】
【0020】
<<肺炎連鎖球菌R6株由来PBP2bおよび変異体の可溶性部分の大腸菌発現ベクター構築>>
配列番号4で示されるアミノ酸配列を有する肺炎連鎖球菌R6株由来PBP2b可溶性部分のグルタチオン−S−トランスフェラーゼ(GST)融合タンパク質の大腸菌発現ベクター(pGEX/SpPBP2b−wt)は特許公開2004−290070号広報に記載の方法により作製した。また、配列番号3に示されるアミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換した変異体である、配列番号1で示されるアミノ酸配列を有する肺炎連鎖球菌R6株由来PBP2b変異体可溶性部分のGST融合タンパク質の大腸菌発現ベクター(pGEX/SpPBP2b−mt7)についても同様に特許公開2004−290070号広報に記載の方法により作製した。
【実施例2】
【0021】
<<R6株由来PBP2bおよび変異体の可溶性部分の発現>>
pGEX/SpPBP2b−wtを大腸菌BL21(Novagen社製)に、また、pGEX/SpPBP2b−mt7を大腸菌DH5α(TOYOBO社製)に形質転換し得られた形質転換体を100μg/mLのアンピシリンを含むSB培地(1.2%(w/v)Bacto Tryptone、2.4%(w/v)Yeast Extract、0.5%(v/v)グリセロール、0.072M リン酸水素二カリウム、0.028M リン酸二水素カリウム)5L中で600nmにおけるO.D.が1.0に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、28℃で2時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)400mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体を菌体破砕バッファー(1mM EDTA、1mM フッ化フェニルメチルスルホニル、1μM ロイペプチン、1μM ペプスタチンA、0.1mg/mL リゾチーム含有PBS)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を2回施し、細胞を破砕した。遠心分離(60分間、12000rpm)とそれに続く0.2μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【実施例3】
【0022】
<<R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体の発現>>
MAD法によるX線結晶構造解析に使用する重原子誘導体結晶を作製するために、メチオニンの硫黄原子が重原子であるセレンに置換されたR6株由来PBP2b変異体のセレノメチオニン置換体の発現を試みた。実施例1記載の組み換えプラスミドpGEX/SpPBP2b−mt7をメチオニン要求株である大腸菌B834(DE3)(Novagen社製)に形質転換し、100μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。得られた形質転換体は100μg/mLのアンピシリンを含むLB培地100mLにて、37℃で一晩培養し、100μg/mLのアンピシリンを含む4LのLeMaster培地(組成は下記の表1に示す)に40mL接種し、600nmにおけるO.D.が0.5−0.8に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、30℃で一晩誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)225mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体を菌体破砕バッファー(1mM EDTA、5mM β−メルカプトエタノール、1mM フッ化フェニルメチルスルホニル、1μM ロイペプチン、1μM ペプスタチンA、0.1mg/mL リゾチーム含有PBS)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を3回施し、細胞を破砕した。遠心分離(60分間、12000rpm)とそれに続く0.2μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【0023】
【表1】

【実施例4】
【0024】
<<R6株由来PBP2bおよび変異体の可溶性部分の精製>>
(1)アフィニティー精製
以下の精製操作は4℃で実施した。実施例2で調製した細胞抽出液を予めPBSで平衡化したGlutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)担体80mLにバッチ法で結合させた。これを担体の10倍容量のPBSで5回洗浄した後、担体容量と等量の溶出液(10mM 還元型グルタチオン、50mM トリス塩酸 pH 7.9)で3回溶出した。
【0025】
(2)GSTタグの切断
上記(1)で精製したサンプルを1mg/mLに希釈し、終濃度2.5unit/mLのトロンビン(シグマ社製)を添加し、4℃で一晩反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。
【0026】
(3)陰イオン交換カラムによる精製
上記(2)で得られたサンプルを陰イオン交換カラムであるPOROS HQ/20(PerSeptive Biosystems社製)を用いて精製した。流速は10mL/minであり、サンプルを3分割し、精製は3度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9中の0−600mM 塩化ナトリウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめ、Glutathione Sepharose 4 Fast Flow担体にアプライし、バッチ法によりGSTタグを除去した。
【0027】
(4)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(3)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8)に対して透析しバッファー置換を実施した。サンプルを3分割し、精製は3度実施した。平衡化バッファーで平衡化したカラムに流速3mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、5−250mM リン酸カリウムバッファー pH 6.8の直線勾配(20カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0028】
(5)ゲルろ過カラムによる精製
上記(4)で得られたサンプルを保持分子量30000以上の限外ろ過膜であるAmicon Ultra−15 30kDa NMWL(ミリポア社製)を用いて2mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH7.5、150mM 塩化ナトリウム)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。精製はサンプルを2等分し、2度に分けて実施した。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて10mg/mLまで濃縮後、液体窒素中で凍結し、−80℃で保存した。
【実施例5】
【0029】
<<R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体の精製>>
(1)アフィニティー精製
以下の精製操作は4℃で実施した。アフィニティーカラムは、Glutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量50mLのカラムを作製し、平衡化バッファー(1mM EDTA、5mM β−メルカプトエタノール含有PBS)で平衡化した。そこへ実施例3で調製した細胞抽出液を流速2mL/minでアプライし、5カラム容量の平衡化バッファーで洗浄した後、溶出バッファー(50mM トリス塩酸 pH 7.9、10mM 還元型グルタチオン、1mM EDTA、5mM β−メルカプトエタノール)で溶出した。SDS−PAGEを実施し、溶出画分を確認し、主要なフラクションをまとめて回収した。
【0030】
(2)GSTタグの切断
上記(1)で精製したサンプルを透析バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)に対して透析した後、2倍量の透析バッファーを加えて希釈し、終濃度5unit/mLのトロンビン(シグマ社製)を添加し、4℃で一晩反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。
【0031】
(3)陰イオン交換カラムによる精製
上記(2)で得られたサンプルを陰イオン交換カラムであるPOROS HQ/20(PerSeptive Biosystems社製)を用いて精製した。流速は10mL/minであり、サンプルを3分割し、精製は3度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、10mM ジチオスレイトール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9、1mM EDTA、10mM ジチオスレイトール中の0−500mM 塩化ナトリウムの直線勾配(16カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめ、Glutathione Sepharose 4 Fast Flow担体2mLにアプライし、バッチ法によりGSTタグを除去した。
【0032】
(4)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(3)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8、1mM ジチオスレイトール)で平衡化した脱塩カラムHiPrep 26/10 Desalting(アマシャムバイオサイエンス社製)に流速7.5mL/minでアプライし、バッファー置換を実施した。サンプルを3分割し、精製は3度実施した。平衡化バッファーで平衡化したハイドロキシアパタイトカラムに流速3mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、1mM ジチオスレイトール中の5−250mM リン酸カリウムバッファー pH 6.8の直線勾配(25カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0033】
(5)ゲルろ過カラムによる精製
上記(4)で得られたサンプルを保持分子量50000以上の限外ろ過膜であるAmicon Ultra−15 50kDa NMWL(ミリポア社製)を用いて1.7mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH7.5、150mM 塩化ナトリウム、10mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。精製はサンプルを2等分し、2度に分けて実施した。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて20mg/mLまで濃縮後、終濃度0.05mMのEDTAを添加後、液体窒素中で凍結し、−80℃で保存した。
【実施例6】
【0034】
<<トランスペプチダーゼドメイン調製のための大腸菌発現系の構築>>
(1)トリプシン限定分解によるトランスペプチダーゼドメインの同定
実施例5で精製したR6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体を20mM Tris塩酸 pH 7.9、1mM EDTA、1mM ジチオスレイトールでタンパク質終濃度0.4mg/mLに希釈した。次に、希釈液10μL、5mg/mL トリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)溶液1.6μL、10mM EDTA溶液2μL、MilliQ水6.4μLを混合し、26℃、1時間反応後、サンプルを2等分した。一方は終濃度1mM PMSFおよび還元剤含有のSDS−PAGEサンプルバッファーと等量混合し、3分間煮沸後、SDS−PAGEにアプライし泳動した後、PVDF膜に転写し、主要なバンドを切り出しプロテインシークエンサーによりN末端アミノ酸配列を分析した。残りの一方は10分の1量の10%酢酸を加えた後、ZipTipC4(ミリポア社製)を用いて精製しMALDI−TOF型質量分析装置にて分子量を測定した。その結果、SDS−PAGEにおいて分子量41k、25k、19k付近の主要なバンドおよび12k付近のマイナーなバンドが確認された。N末端アミノ酸配列の結果では、41k付近のバンドのN末端アミノ酸配列はLTI、19k付近のバンドはGEIYD、12k付近のバンドはGGLGD、TGTGEおよびSAYPGの混合物であり、この内SAYPGの配列はトリプシン由来であった。質量分析では多数のピークが観測されたが、その中に分子量18284、18158、13156のピークが存在しており、また、23300のピークはトリプシン由来であることが判明した。以上の結果から、SDS−PAGEでの25kのバンドはトリプシン由来、41kのバンドは配列番号3に示されるR6株由来PBP2bにおいてはアミノ酸番号315〜685、19kのバンドは66〜228、66〜229の混合物、12kのバンドは562〜680、621〜685の混合物に対応することが判明した。トランスペプチダーゼドメインに存在する保存モチーフはアミノ酸番号391から始まるSVVK、448から始まるSSN、620から始まるKTGであることから、トランスペプチダーゼドメインとしてアミノ酸番号315〜685からなる領域を選択した。
【0035】
(2)トリプシン限定分解時におけるマイナー部位での切断を回避するための点変異の導入
上記(1)においてトリプシン限定分解でのマイナー部位での切断箇所として配列番号3に示されるR6株由来PBP2bにおいてはアミノ酸番号561および620のリジン残基の直後が対応する。この内、アミノ酸番号620のリジンはトランスペプチダーゼドメインの保存モチーフに存在するため、変異を導入するのは好ましくないが、アミノ酸番号561のリジンは変異を導入することでトリプシン限定分解時におけるマイナー部位での切断を回避することができる。また、アミノ酸番号66〜228、66〜229の混合物からなる部分についてはトランスペプチダーゼドメインと相互作用している可能性が否定できなかったため、アミノ酸番号229のリジン残基についても変異を導入し、混合物生成を回避する方針とした。ここで、アミノ酸番号561および229のいずれについてもグルタミンへの変異を試みた。実施例1に記載のpGEX/SpPBP2b−wtおよびpGEX/SpPBP2b−mt7を鋳型として、下記のプライマーを用い、QuickChange Site−Directed Mutagenesis Kit(Stratagene社製)で、添付の説明書の方法に従い、アミノ酸番号561の部位への変異を導入した。
【0036】
2b_R6_K561Q_5:5’−GGCATTTATGGTAATAATGATCAGGGAGGACTGGGTGACTTG−3’(配列番号6)
2b_R6_K561Q_3:5’−CAAGTCACCCAGTCCTCCCTGATCATTATTACCATAAATGCC−3’(配列番号7)
【0037】
次に下記のプライマーを用いて同様の手順に従い、さらにアミノ酸番号229の部位への変異を導入し、pGEX/SpPBP2b−wt−229Q−561QおよびpGEX/SpPBP2b−mt7−229Q−561Qを作製した。これらのプラスミドのDNA配列を確認し、各々配列番号4、配列番号1に記載のアミノ酸配列に目的の2箇所の点変異が導入されたR6株由来および変異体のPBP2b可溶性部分をコードすることを確認した。
【0038】
2b_R6_K229Q_5:5’−CTGGCATTAGTATTTCTACTTCTTGGGATAGACAGGTTTTGGA−3’(配列番号8)
2b_R6_K229Q_3:5’−TCCAAAACCTGTCTATCCCAAGAAGTAGAAATACTAATGCCAG−3’(配列番号9)
【実施例7】
【0039】
<<R6株由来PBP2bおよび変異体トランスペプチダーゼドメイン取得のための可溶性部分セレノメチオニン置換体の発現>>
実施例6で得られたpGEX/SpPBP2b−wt−229Q−561QおよびpGEX/SpPBP2b−mt7−229Q−561Qをメチオニン要求株である大腸菌B834(DE3)(Novagen社製)に形質転換し、100μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。得られた形質転換体は100μg/mLのアンピシリンを含むLB培地100mLにて、37℃で一晩培養し、遠心分離機で集菌後、100μg/mLのアンピシリンを含む100mlのLeMaster培地(組成は実施例3の表1に示す)で懸濁し、再度、遠心分離機によって集菌し、100μg/mLのアンピシリンを含むLeMaster培地100mlに再懸濁した。この懸濁液を100μg/mLのアンピシリンを含む4.8LのLeMaster培地に48mL接種し、600nmにおけるO.D.が0.5−0.8に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、30℃で4時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)200mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体を菌体破砕バッファー(1mM EDTA、5mM β−メルカプトエタノール、1mM フッ化フェニルメチルスルホニル、1μM ロイペプチン、1μM ペプスタチンA、0.1mg/mL リゾチーム含有PBS)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を4回施し、細胞を破砕した。遠心分離(60分間、12000rpm)とそれに続く0.2μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【実施例8】
【0040】
<<R6株由来PBP2bおよび変異体トランスペプチダーゼドメインのセレノメチオニン置換体の精製>>
(1)R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体の精製
(1−1)アフィニティー精製
以下の精製操作は4℃で実施した。アフィニティーカラムは、Glutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量50mLのカラムを作製し、平衡化バッファー(1mM EDTA、5mM β−メルカプトエタノール含有PBS)で平衡化した。そこへ実施例7においてpGEX/SpPBP2b−wt−229Q−561Qを用いて調製した細胞抽出液を流速2mL/minでアプライし、5カラム容量の平衡化バッファーで洗浄した後、溶出バッファー(5unit/mLトロンビン(シグマ社製)含有PBS)を50mLアプライした段階でポンプを止めて10℃で一晩反応させGSTタグを切断した。平衡化バッファーを送り込んで溶出した後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。SDS−PAGEにより溶出画分を確認し、主要なフラクションをまとめて回収した。
【0041】
(1−2)陰イオン交換カラムによる精製
上記(1−1)で得られたサンプルを透析バッファー(20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール)に対して透析した後、陰イオン交換カラムであるPOROS HQ/20(PerSeptive Biosystems社製)を用いて精製した。流速は10mL/minであり、サンプルを4分割し、精製は4度実施した。平衡化バッファー(20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール中の0−300mM 塩化ナトリウムの直線勾配(12カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0042】
(1−3)トリプシン限定分解
上記(1−2)で精製したR6株由来PBP2b可溶性部分のセレノメチオニン置換体を20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール溶液に対して透析した後回収し、はじめに小スケールでのトリプシン限定分解の条件検討を実施した。回収液5μLを0.125〜5mg/mLのトリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)溶液と等量混合し、24℃、2時間反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。SDS−PAGEにより各濃度におけるトリプシン消化のパターンを確認し、トリプシンの添加量を終濃度0.125mg/mlと決定し、残りの全サンプルのトリプシン限定分解を実施した。
【0043】
(1−4)陰イオン交換カラムによる精製
上記(1−3)で得られたサンプルを陰イオン交換カラムであるMonoQ HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。流速は4mL/minであり、サンプルを4分割し、精製は4度実施した。平衡化バッファー(20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール中の0−300mM 塩化ナトリウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0044】
(1−5)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(1−4)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8、1mM ジチオスレイトール)に対して透析し、バッファー置換を実施した。サンプルを2分割し、精製は2度実施した。平衡化バッファーで平衡化したハイドロキシアパタイトカラムに流速3.5mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、1mM ジチオスレイトール中の5−250mM リン酸カリウムバッファー pH 6.8の直線勾配(25カラム容量)で溶出した。SDS−PAGEを実施し、非吸着画分をまとめた。
【0045】
(1−6)ゲルろ過カラムによる精製
上記(1−5)で得られたサンプルを保持分子量10000以上の限外ろ過膜であるAmicon Ultra−15 10kDa NMWL(ミリポア社製)を用いて0.75mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH8.0、150mM 塩化ナトリウム、1mM EDTA、10mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて10mg/mLまで濃縮後、液体窒素中で凍結し、−80℃で保存した。SDS−PAGEの結果から、分子量41k付近のバンドのみが確認され、19k付近のバンドについては除去されたことから、配列番号5に示されるアミノ酸配列からなるR6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体であることが示唆された。
【0046】
(2)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体の精製
(2−1)アフィニティー精製
以下の精製操作は4℃で実施した。アフィニティーカラムは、Glutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量50mLのカラムを作製し、平衡化バッファー(1mM EDTA、5mM β−メルカプトエタノール含有PBS)で平衡化した。そこへ実施例7においてpGEX/SpPBP2b−mt7−229Q−561Qを用いて調製した細胞抽出液を流速2mL/minでアプライし、5カラム容量の平衡化バッファーで洗浄した後、溶出バッファー(50mM トリス塩酸 pH 7.9、10mM 還元型グルタチオン、1mM EDTA、5mM β−メルカプトエタノール)で溶出した。SDS−PAGEにより溶出画分を確認し、主要なフラクションをまとめて透析バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)に対して透析することでバッファー置換をした。次に、トリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)を終濃度2.3μg/mLの条件で添加し、25℃で1時間反応させることでGSTタグのみを切断し、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。
【0047】
(2−2)陰イオン交換カラムによる精製
上記(2−1)で得られたサンプルを陰イオン交換カラムであるMonoQ HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。流速は4mL/minであり、サンプルを5分割し、精製は5度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール中の0−250mM 塩化ナトリウムの直線勾配(8カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0048】
(2−3)トリプシン限定分解
上記(2−2)で精製したR6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体を20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール溶液に対して透析した後回収し、はじめに小スケールでのトリプシン限定分解の条件検討を実施した。回収液9μLに0.039〜10mg/mLのトリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)溶液1μLを添加し、25℃、1時間反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。SDS−PAGEにより各濃度におけるトリプシン消化のパターンを確認し、トリプシンの添加量を終濃度0.125mg/mlと決定し、残りの全サンプルのトリプシン限定分解を実施した。
【0049】
(2−4)陰イオン交換カラムによる精製
上記(2−3)で得られたサンプルを陰イオン交換カラムであるMonoQ HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。流速は4mL/minであり、サンプルを4分割し、精製は4度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール中の0−250mM 塩化ナトリウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0050】
(2−5)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(2−4)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8、1mM ジチオスレイトール)に対して透析し、バッファー置換を実施した。サンプルを2分割し、精製は2度実施した。平衡化バッファーで平衡化したハイドロキシアパタイトカラムに流速3.5mL/minでサンプルをアプライし、非吸着画分を回収した。
【0051】
(2−6)陽イオン交換カラムによる精製
上記(2−5)で得られたサンプルを平衡化バッファー(50mM 酢酸ナトリウムバッファー pH 5.0、1mM EDTA、1mM ジチオスレイトール)に対して透析し、バッファー置換を実施した後、陽イオン交換カラムであるMonoS HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。サンプルを2分割し、精製は2度実施した。平衡化バッファーで平衡化したカラムに流速4mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、50mM 酢酸ナトリウムバッファー pH 5.0、1mM EDTA、1mM ジチオスレイトール中の0−200mM 塩化ナトリウムの直線勾配(16カラム容量)で溶出した。SDS−PAGEにより主要な溶出画分をまとめた。
【0052】
(2−7)ゲルろ過カラムによる精製
上記(2−6)で得られたサンプルを保持分子量10000以上の限外ろ過膜であるAmicon Ultra−15 10kDa NMWL(ミリポア社製)を用いて1.2mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH7.9、150mM 塩化ナトリウム、1mM EDTA、1mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて4.7mg/mLまで濃縮後、液体窒素中で凍結し、−80℃で保存した。SDS−PAGEでは分子量41k付近のバンドのみが確認された。また、エレクトロスプレイイオン化法による質量分析装置により解析したところ、分子量39851に主要なピークが確認されたことから、計算値で39843の分子量を持つ、配列番号2に示されるアミノ酸配列からなるR6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体であることが確認された。
【実施例9】
【0053】
<<PBP2bの結晶化>>
(1)R6株由来PBP2b可溶性部分の結晶化
R6株由来PBP2b可溶性部分の結晶は、シッティングドロップ蒸気拡散法により得た。実施例4で精製したタンパク質溶液(10mg/mL)0.5μLと結晶化剤(0.8−1.6M 硫酸アンモニウム、0.1M 酢酸ナトリウム緩衝液 pH 4.6)0.5μLを混合し結晶化ドロップとし、リザーバー溶液として上記結晶化剤100μL分注し密閉した。プレートを20℃の恒温槽内に静置すると数日で針状のクラスター結晶が得られた。0.8M 硫酸アンモニウム、0.1M 酢酸ナトリウム緩衝液 pH 4.6からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図1に示す。
【0054】
(2)R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体の結晶化
R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体の結晶は、ハンギングドロップ蒸気拡散法により得た。実施例8(1)で精製したタンパク質溶液(10mg/mL)2.0μLと結晶化剤(1.1−1.6M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9)2.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注し、Al’sオイル(ハンプトンリサーチ社製)250μLをリザーバー溶液の上に重層した。ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを20℃の恒温槽内に静置すると数日で針状のクラスター結晶が得られた。1.1M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図2に示す。
【0055】
(3)R6株由来PBP2b変異体の可溶性部分およびセレノメチオニン置換体の結晶化
(3−1)R6株由来PBP2b変異体の可溶性部分の結晶化
R6株由来PBP2b変異体の可溶性部分の結晶は、ハンギングドロップ蒸気拡散法により得た。実施例4で精製したタンパク質溶液(10mg/mL)1.0μLと結晶化剤(10−20% ポリエチレングリコール8000、0.2M 硫酸アンモニウム、0.1M トリス塩酸緩衝液 pH 8.0)1.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注した。ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを20℃の恒温槽内に静置すると数日で結晶が得られた。また、硫酸アンモニウムの代わりに硫酸リチウム、硫酸ナトリウムを用いた場合も同様に結晶が得られた。10% ポリエチレングリコール8000、0.2M 硫酸アンモニウム、0.1M トリス塩酸緩衝液 pH 8.0からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図3に示す。
【0056】
(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体の結晶化
R6株由来PBP2b変異体可溶性部分の結晶は、ハンギングドロップ蒸気拡散法により得た。実施例5で精製したタンパク質溶液(10mg/mL)1.0μLと結晶化剤(18−20% ポリエチレングリコール8000、0.3M 硫酸リチウム、0.1M トリス塩酸緩衝液 pH 8.5、10mM ジチオスレイトール)1.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注し、Al’sオイル(ハンプトンリサーチ社製)200μLをリザーバー溶液の上に重層した。ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを20℃の恒温槽内に静置すると数日で結晶が得られた。また、硫酸リチウムの代わりに硫酸アンモニウム、硫酸ナトリウムを用いた場合も同様に結晶が得られた。19% ポリエチレングリコール8000、0.3M 硫酸リチウム、0.1M トリス塩酸緩衝液 pH 8.5、10mM ジチオスレイトールからなる結晶化剤を用いて得られた結晶の顕微鏡写真を図4に示す。
【0057】
(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体の結晶化
R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体の結晶は、シッティングドロップ蒸気拡散法により得た。実施例8(2)で精製したタンパク質溶液(4.7mg/mL)0.4μLと結晶化剤(1.1−1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9−8.5)0.4μLを混合し結晶化ドロップとし、リザーバー溶液として上記結晶化剤100μL分注し密閉した。プレートを20℃の恒温槽内に静置すると数日で結晶が得られた。1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 8.5からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図5に示す。また、硫酸アンモニウムの代わりに硫酸ナトリウムを用いた条件(0.7−1.1M 硫酸ナトリウム、10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.0−6.5または0.1M クエン酸ナトリウム緩衝液 pH 5.5または0.1M HEPES緩衝液 pH 7.0−7.5またはトリス塩酸緩衝液 pH 7.9−8.5)および硫酸マグネシウムを用いた条件(0.9−1.1M 硫酸マグネシウム、1−10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.5)でも同様の結晶が得られた。また、N−Benzoyl−D−alanylthioglycolic acidを添加した条件でも同様の結晶が得られ、ハンギングドロップ蒸気拡散法においても同様の結晶が得られた。さらに、マイクロ・フリューディック技術を応用したTOPAZ 4.96スクリーニングチップ(フリューダイム社製)を用いた場合には結晶化剤として(i)1M 硫酸アンモニウム、0.1M 酢酸ナトリウム pH 4.6および(ii)1.5M リン酸水素二カリウムを用いた条件において外見上同様の結晶が得られた。
【実施例10】
【0058】
<<PBP2b結晶のX線回折強度データの収集>>
(1)R6株由来PBP2b変異体の可溶性部分の結晶
実施例9(3−1)において得られた結晶を35%グリセロール含有の抗凍結剤溶液に移した後、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しイメージングプレートを装備したR−AXIS V(リガク社製)を検出装置としてX線回折データを振動法にて測定した。X線の波長は1Å、振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、回折強度データを分解能3.5Åで処理した。その結果、空間群がF222であり、格子定数がa=175.47Å、b=186.16Å、c=368.53Å、α=β=γ=90°であった。得られたデータのCompletenessは99.9%、Rmergeは9.4%であった。データ収集の結果を下記の表2にまとめた。
【0059】
【表2】
【0060】
(2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶
実施例9(3−2)において得られた結晶を20%グリセロール含有の抗凍結剤溶液に移した後、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しイメージングプレートを装備したR−AXIS V(リガク社製)を検出装置としてX線回折データを振動法にて測定した。このとき、セレンの異常散乱効果を用いて構造解析を行うために、波長は0.97947、0.97879および0.90000Åの3波長、即ち、吸収端のエッジ、ピーク、リモートの3カ所でMADデータを収集した。振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、回折強度データを分解能3.5Åで処理した。その結果、空間群がF222であり、格子定数がa=175.75Å、b=186.21Å、c=369.21Å、α=β=γ=90°であった。データ収集の結果を下記の表3にまとめた。
【0061】
【表3】
【0062】
(3)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶
実施例9(4)において、1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 8.5からなる結晶化剤を用いて得られた結晶を25%グリセロール含有の抗凍結剤溶液に移した後、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しイメージングプレートを装備したR−AXIS V(リガク社製)を検出装置としてX線回折データを振動法にて測定した。このとき、セレンの異常散乱効果を用いて構造解析を行うために、波長は0.97948、0.97907および0.90000Åの3波長、即ち、吸収端のエッジ、ピーク、リモートの3カ所でMADデータを収集した。振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、回折強度データを分解能2.4Åで処理した。その結果、空間群の候補がP43212またはP41212となったが、下記に示す実施例11の解析結果からP43212と判明した。格子定数はa=86.39Å、b=86.39Å、c=143.27Å、α=β=γ=90°であり、データ収集の結果を下記の表4にまとめた。また、同様の手順で調製した結晶を(財)高輝度光科学研究センターの大型放射光施設SPring−8の構造生物学IビームラインBL41XUを利用しQuantum315CCDカメラ(ADSC社製)を検出器としてX線回折データを振動法にて測定した。X線の波長は1Å、振動角は1°/フレームであり、回折強度データを2.2Åで処理した結果、空間群はP43212、格子定数はa=86.34Å、b=86.34Å、c=143.40Å、α=β=γ=90°であった。データ収集の結果を下記の表5にまとめた。また、実施例9(4)において、0.8M 硫酸ナトリウム、10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.5からなる結晶化剤を用いてN−Benzoyl−D−alanylthioglycolic acid存在下で得られた結晶を用いて、同様の条件でX線回折測定を実施したところ、空間群はP43212、格子定数はa=86.41Å、b=86.41Å、c=143.96Å、α=β=γ=90°であった。
【0063】
【表4】
【0064】
【表5】
【実施例11】
【0065】
<<PBP2bの結晶構造決定>>
次に、プログラムSOLVE(Terwillinger, T.C. & Berendzen,J. Acta Crystallogr. D 55,849−861(1999))を用いて、実施例10の表4に示したMADデータを処理し、セレン原子の位置を決定し初期位相を計算した。位相の改良及び初期モデルの構築はプログラムRESOLVE(Terwilliger, T.C. Acta Crystallogr. D56,965−972(2000))を用いた。この際、空間群P41212で処理した場合には妥当な電子密度が得られず、空間群P43212で処理した場合においてタンパク質の明瞭の電子密度が得られたため、空間群をP43212と決定した。次いで、プログラムLafire(YAO M.他、Acta.Cryst.D62(2)、189−196(2006))、プログラムTurbo Frodo(AFMB−CNRS製)、プログラムRefmac5(Collaborative Computational Project Number 4(ccp4))およびプログラムCNX(アクセルリス社製)を利用したモデルの構築および修正を繰り返し実施し、R6株由来PBP2b変異体トランスペプチダーゼドメインの結晶構造を決定し、最終的には実施例10の表5に示した高分解能のデータを用いて分解能2.2Åで構造精密化を実施した。本結晶の非対称単位中にはR6株由来PBP2b変異体トランスペプチダーゼドメインが1分子存在していた。決定した結晶構造の正確さの指標であるR因子は、0.209であった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.230であった。また、決定したR6株由来PBP2b変異体トランスペプチダーゼドメインの構造を図6に示した。さらに、結晶中において、隣り合うPBP2b分子の境界に存在する2回軸上に硫酸イオンが存在し、各PBP2b分子のリジン、アスパラギン残基の側鎖および水分子と水素結合を形成していた。結晶中において、分子間のパッキングに関与している硫酸イオン周辺の電子密度図とモデルを図7に示した。ここで、リジンは配列番号3に示されるR6株由来PBP2bの配列における431番目のスレオニンをリジンへ変異させた残基に対応しており、薬剤耐性に関与しているアミノ酸変異の導入と硫酸アンモニウム等の硫酸塩を結晶化剤として使用することを組み合わせることにより高品質の結晶が製造できることを見出した。
【産業上の利用可能性】
【0066】
本発明のPBP2b結晶は、X線結晶構造解析可能な新規な結晶であり、解析に成功した立体構造情報を利用することにより阻害剤の効率的な設計等が可能となり、新薬創出に活用できるため、産業上極めて有用である。
【図面の簡単な説明】
【0067】
【図1】本発明のR6株由来PBP2b可溶性部分の結晶の顕微鏡写真である。
【図2】本発明のR6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体結晶の顕微鏡写真である。
【図3】本発明のR6株由来PBP2b変異体可溶性部分の結晶の顕微鏡写真である。
【図4】本発明のR6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶の顕微鏡写真である。
【図5】本発明のR6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶の顕微鏡写真である。
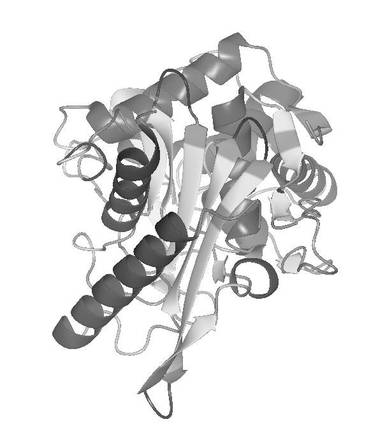
【図6】決定したR6株由来PBP2b変異体トランスペプチダーゼドメインの構造をリボン図で示す。
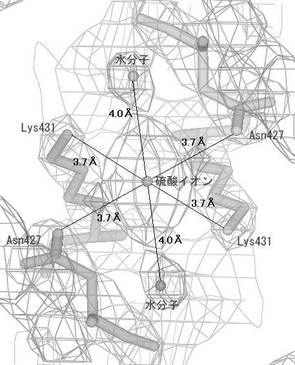
【図7】本発明のR6株由来PBP2b変異体トランスペプチダーゼドメイン結晶中において、分子間のパッキングに関与している硫酸イオン周辺の電子密度図とモデルを示す。
【技術分野】
【0001】
本発明は、X線結晶構造解析および阻害剤の設計等に利用するためのペニシリン結合タンパク質(PBP)2bの結晶に関する。
【背景技術】
【0002】
ペニシリン結合タンパク質(PBP)は細菌における細胞壁を構成する構造ユニットであるムレインモノマーを基質として細胞壁ペプチドグリカンを組み立てる一連の酵素群であり、細菌のペプチドグリカンの重合および架橋反応に働く膜結合型のタンパク質である。肺炎連鎖球菌(Streptococcus pneumoniae)には6種類のPBPが確認されているが、ペニシリン耐性肺炎連鎖球菌では主に3種類のPBP(PBP1a、PBP2x、PBP2b)における変異がβ−ラクタム薬の感受性低下に関与している。特にペニシリン系薬剤およびカルバペネム系薬剤においては、耐性とPBP2b変異との密接な関連が示唆されている。また、配列番号3に示されるR6株(ATCC49619)由来PBP2bにおいては、アミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換された変異が薬剤耐性に重要であることが示唆されている(非特許文献1および特許文献1参照)。従って、PBP2bはβ−ラクタム薬の耐性に重要な役割を果たしており、PBP2bの立体構造をX線結晶構造解析により解明することは、耐性のメカニズムを説明するだけでなく、耐性菌に有効な新薬を創出する上でも非常に重要である。
【0003】
肺炎連鎖球菌(Streptococcus pneumoniae)由来PBP2bは、細胞内ドメイン、膜貫通領域、N末端ドメインおよびトランスペプチダーゼドメインから構成される膜結合型タンパク質であり、細胞内ドメインおよび膜貫通領域を除去し、膜結合機能を喪失させたPBP2b可溶性部分の調製方法が明らかになっている(特許文献1および非特許文献2参照)。しかしながら、PBP2b可溶性部分からさらにN末端ドメインを除去したトランスペプチダーゼドメインの調製方法は未だ明らかとなっていない。また、膜結合型PBP2b、PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインのいずれについても、結晶および結晶構造解析の報告はない。従って、X線結晶構造解析によりPBP2bの立体構造を決定し、効率的な阻害剤の設計を可能とする高品質のPBP2b結晶の創出が望まれている。
【0004】
一般にタンパク質のN末端、C末端やループ領域は構造が揺らいでいる場合があり、その際には複数のコンフォメーションを取り得ることから、タンパク質の揺らいでいる領域の除去やドメインの切り出しは均一のコンフォメーションから構成される高分解能のX線回折能を有する結晶を作製するために有効な手段である(非特許文献3参照)。その一つの方法としてプロテアーゼであるトリプシンを利用してタンパク質を限定分解する方法が知られている。トリプシンは表面に露出している塩基性アミノ酸であるリジンやアルギニン残基の直後を選択的に切断することにより、タンパク質の揺らいでいる領域を除去可能な場合がある。肺炎連鎖球菌PBP1aではトリプシン限定分解物において分解能2.6ÅのX線回折能を示す結晶が得られており、その際、トリプシン限定分解物の純度を確保しマイナー部位での切断を避けるため1箇所点変異を導入している(非特許文献4参照)。また、肺炎連鎖球菌PBP1bではトリプシン限定分解物において分解能1.9ÅのX線回折能を示す結晶が得られており、その際、トリプシン限定分解物の純度を確保しマイナー部位での切断を避けるため3箇所点変異を導入している(非特許文献5参照)。しかしながら、トリプシン限定分解のパターンは個々のタンパク質に依存し、PBP2bでは未だ明らかになっていないことから、点変異導入の必要性も含め結晶化に適したトリプシン限定分解物の作製には試行錯誤が必要である上、PBP2bに対してトリプシン限定分解が有効な手段であるかも明らかではない。結晶化に適したPBP2bトランスペプチダーゼドメインをトリプシン限定分解することにより新規に見出すことができれば、高品質の結晶を作製し、PBP2bの立体構造を解明することに多いに貢献できると考えられる。
【0005】
また、タンパク質の表面に露出しているグルタミン酸やリジン等の特定のアミノ酸残基に変異を導入し、タンパク質表面の性状を変化させることにより高品質の結晶を作製した報告も知られている(非特許文献6参照)。しかしながら、この方法は立体構造が既知のタンパク質においては有効であるが、PBP2bのように立体構造が未知のタンパク質の場合には表面に露出している残基を正確に同定することができないため、理論に基づいて変異を導入することは困難である。立体構造が未知のタンパク質に対して表面の性状あるいはコンフォメーションを変化させるために変異を導入することは可能であるが、結晶化に適した変異であるかどうかについては、実際にタンパク質を調製し、結晶化スクリーニングを実施した後、X線回折実験で検証する必要があり、多大な試行錯誤を要する。さらに、作製した変異体の結晶化において、特定の試薬の添加が結晶化に有効であるかどうかを予想することも困難である。従って、PBP2bにおいて、結晶化に適したアミノ酸変異の同定と結晶化に有効な試薬の組み合わせを見出すことができれば、高品質の結晶を作製し、PBP2bの立体構造を解明することに多いに貢献できると考えられる。
【特許文献1】特許公開2004−290070号広報
【非特許文献1】Antimicrob.Agents Chemother. 48(6)2244−2250(2004)
【非特許文献2】Antimicrob.Agents Chemother. 48(5)1848−1855(2004)
【非特許文献3】BIOベンチャー 3(5)、36−39(2003)
【非特許文献4】J.Mol.Biol.355(4)684−696(2006)
【非特許文献5】Proc.Natl.Acad.Sci.USA.102(3)577−582(2005)
【非特許文献6】Structure 12(4)529−535(2004)
【発明の開示】
【発明が解決しようとする課題】
【0006】
これまでPBP2bの結晶および結晶構造解析の報告はない。そのため、創薬において重要であるPBP2bの立体構造情報が存在せず効率的な阻害剤の設計が困難である。また、一般にトリプシン限定分解および適切なアミノ酸変異導入等により良質の結晶が得られる可能性があるが、PBP2bへの適用はなく、X線結晶構造解析に適したPBP2b結晶の製造方法は不明である。
【課題を解決するための手段】
【0007】
本発明は、結晶化に適したPBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインを調製し、さらにアミノ酸変異を導入することにより、X線結晶構造解析を実施し効率的な阻害剤の設計等を可能とする高品質のPBP2b結晶を提供することで上記課題を解決する。
【0008】
本発明者は、肺炎連鎖球菌(Streptococcus pneumoniae)由来PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインを調製し、膨大な数の結晶化条件探索の試行錯誤により再現性よく得られる結晶を取得した。さらに、薬剤耐性に関与するアミノ酸変異を導入することにより高品質で再現性よく得られるPBP2bの結晶を取得した。この結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功し、創薬研究に重要なPBP2bの立体構造を決定した。すなわち、本発明はX線結晶構造解析および阻害剤の設計等に利用するためのPBP2b結晶に関する。ここで、PBP2b変異体可溶性部分の結晶において、空間群はF222であり、格子定数はa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°である。また、PBP2b変異体トランスペプチダーゼドメインの結晶において、空間群はP43212であり、格子定数はa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°である。さらに、PBP2b変異体トランスペプチダーゼドメインの結晶構造から、配列番号3におけるアミノ酸残基番号431に対応するスレオニンをリジンに置換する変異により導入したリジン残基の側鎖が結晶化剤として使用した硫酸塩と相互作用することで高品質の結晶が製造できることを見出した。
【0009】
すなわち、本発明は以下の発明を含有する。
(1)連鎖球菌科(Streptococcaceae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
(2)ストレプトコッカス属(Streptococcus)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
(3)肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
(4)ペニシリン結合タンパク質(PBP)2b可溶性部分から構成される(1)〜(3)のいずれか一項に記載の結晶。
(5)配列番号1で示されるアミノ酸配列からなるタンパク質から構成される(1)〜(4)のいずれか一項に記載の結晶。
(6)配列番号1で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される(1)〜(4)のいずれか一項に記載の結晶。
(7)配列番号1で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される(1)〜(4)のいずれか一項に記載の結晶。
(8)空間群がF222である(1)〜(7)のいずれか一項に記載の結晶。
(9)空間群がF222であり、格子定数がa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°である(1)〜(7)のいずれか一項に記載の結晶。
(10)ペニシリン結合タンパク質(PBP)2bトランスペプチダーゼドメインから構成される(1)〜(3)のいずれか一項に記載の結晶。
(11)配列番号2で示されるアミノ酸配列からなるタンパク質から構成される(1)〜(4)、(10)のいずれか一項に記載の結晶。
(12)配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される(1)〜(4)、(10)のいずれか一項に記載の結晶。
(13)配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される(1)〜(4)、(10)のいずれか一項に記載の結晶。
(14)空間群がP43212である(1)〜(4)、(10)〜(13)のいずれか一項に記載の結晶。
(15)空間群がP43212であり、格子定数がa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°である(1)〜(4)、(10)〜(13)のいずれか一項に記載の結晶。
(16)(1)〜(15)のいずれか一項に記載の結晶であって、該結晶を構成するタンパク質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
(17)硫酸塩またはリン酸塩の存在下で成長する(1)〜(16)のいずれか一項に記載の結晶。
(18)硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムの存在下で成長する(1)〜(16)のいずれか一項に記載の結晶。
(19)配列番号2で示されるアミノ酸配列からなるタンパク質。
(20)配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質。
(21)配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質。
(22)肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶の製造方法であって、結晶化剤として硫酸塩またはリン酸塩を用いて結晶化することを特徴とする、(1)〜(18)のいずれか一項に記載の結晶の製造方法。
(23)結晶化剤として硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムを用いることを特徴とする(22)記載の製造方法。
(24)配列番号3におけるアミノ酸残基番号431のスレオニンがリジンに置換されているアミノ酸変異を有することを特徴とする、肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの可溶性部分またはトランスペプチダーゼドメインを調製する工程を含む、(22)または(23)記載の製造方法。
(25)配列番号1または2で示されるアミノ酸配列からなるタンパク質を調製する工程を含む、(22)〜(24)のいずれか一項に記載の製造方法。
【発明の効果】
【0010】
本発明のPBP2b結晶は高品質で再現性良く調製可能であるため、X線結晶構造解析に適しており、これまでに全く報告の無かったPBP2bの立体構造解析が初めて可能となった。本発明のPBP2b結晶を利用することにより、PBP2bを阻害する化合物の効率的な設計等に役立つ立体構造情報を取得できるため、創薬において極めて有用である。
【発明を実施するための最良の形態】
【0011】
以下、本発明を詳細に説明する。
<<PBP2bアミノ酸変異と可溶性部分およびトランスペプチダーゼドメイン>>
本発明の結晶を構成するタンパク質は、連鎖球菌科(Streptococcaceae)、好ましくはストレプトコッカス属(Streptococcus)、より好ましくは肺炎連鎖球菌(Streptococcus pneumoniae)、さらに好ましくは肺炎連鎖球菌R6株(ATCC49619)由来のタンパク質である。特に好ましくは、配列番号3に示されるアミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換された変異体タンパク質であり、これらの変異は薬剤耐性菌の一部に存在する変異である。また、本発明において、「トランスペプチダーゼ」とは、細菌細胞壁の一つの成分であるペプチドグリカンの生合成の最終段階に関与し、ペプチドグリカン連鎖を構成しているN−アセチルムラミン酸から伸びるポリペプチド鎖の架橋を触媒する酵素を意味する。さらに、「トランスペプチダーゼドメイン」とは、上記ポリペプチド鎖を架橋する能力を有し、β−ラクタム薬との結合が可能であるタンパク質の構造単位である。本発明における「相同タンパク質」とは、配列番号1または2に示されるアミノ酸配列を有するタンパク質と少なくとも85%、好ましくは90%、より好ましくは95%、さらに好ましくは98%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼ活性を有するタンパク質である。ここで示した相同性の数値は、当業者に公知の相同性検索プログラムであるFASTA(Science,227,1435−1441(1985);Proc.Natl.Acad.Sci.USA,85,2444−2448(1988);http://www.ddbj.nig.ac.jp/search/fasta−j.html)においてデフォルト(初期設定)のパラメータを用いて算出される数値を示す。本発明における「改変タンパク質」とは、配列番号1または2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼ活性を有するタンパク質である。ここで、「置換、欠失、付加もしくは挿入」などの改変に係るアミノ酸の数は、好ましくは1〜40個、より好ましくは1〜20個、さらに好ましくは1〜10個、特に好ましくは1〜6個である。
【0012】
PBP2bは、細胞内ドメイン、膜貫通領域、N末端ドメインおよびトランスペプチダーゼドメインから構成される膜結合型タンパク質である。PBP2b可溶性部分とは、細胞内ドメインおよび膜貫通領域を除去し、膜結合機能を喪失させたPBP2bである。PBP2b可溶性部分は由来となる株およびN末端が異なる種々のものが存在可能であるが、肺炎連鎖球菌R6株においては、好ましくは、配列番号3に示されるアミノ酸配列のアミノ酸残基番号43から685で構成される。PBP2bトランスペプチダーゼドメインは、PBP2b可溶性部分からさらにN末端ドメインを除去することにより調製可能であり、除去する領域の違いにより種々のものが存在可能であるが、肺炎連鎖球菌R6株においては、配列番号3に示されるアミノ酸残基番号315から685で構成されるのが好ましいことをプロテアーゼであるトリプシンを用いた限定分解実験から見出せる。トランスペプチダーゼドメインの同定方法としては精製したPBP2b可溶性部分を種々の濃度のトリプシン存在下で限定分解した試料の質量分析および生じた各断片のN末端アミノ酸配列分析により決定することができる。PBP2bトランスペプチダーゼドメインはその領域のみを発現させることも可能であるが、好ましくは、PBP2b可溶性部分をトリプシン限定分解することにより調製することが望ましい。その際にマイナー部位での切断を回避するために、配列番号3に示されるアミノ酸残基番号229および561のリジン残基をグルタミンに置換する方法が有用であり、特に残基番号561のリジン残基をグルタミンに置換することが好ましい。なお、広義の解釈においては、PBP2bトランスペプチダーゼドメインは膜結合機能を喪失させた可溶性タンパク質であるため、PBP2b可溶性部分に含有される。
【0013】
<<PBP2b可溶性部分の発現>>
PBP2b可溶性部分は、それをコードするDNA断片を、宿主細胞内で複製可能でかつ同遺伝子が発現可能な状態で含むDNA分子、特にDNA発現ベクターの形態とし、それによって宿主細胞の形質転換を行い、その形質転換体を培養することによって得られる。このDNA分子は、ベクター分子にPBP2b可溶性部分をコードするDNA断片を組み込むことによって得ることができる。本発明の好ましい態様によれば、このベクターはプラスミドである。本発明において利用されるベクターは、使用する宿主細胞の種類を勘案して、ウィルス、プラスミド、コスミドベクターなどから適宜選択することができる。例えば宿主細胞が大腸菌の場合はpUC、pBR系のプラスミド、枯草菌の場合はpUB系のプラスミド、酵母の場合はYEp、YRp、YCp系のプラスミドベクターが挙げられる。宿主細胞としては、宿主−ベクター系が確立されているものであれば利用可能であり、好ましくは大腸菌が挙げられる。宿主細胞の形質転換により得られた形質転換体は、適当な条件で培養し、得られた形質転換体の細胞抽出液を慣用の方法に従い回収することができる。ここで、PBP2b可溶性部分に付加的ポリペプチド、例えばグルタチオン−S−トランスフェラーゼ(GST)またはヒスチジン残基に富むポリペプチド(His−tag)を融合タンパク質として発現することも慣用の技術により可能である。
【0014】
<<PBP2b可溶性部分およびトランスペプチダーゼドメインの精製>>
GSTを融合して大腸菌で発現させたPBP2b可溶性部分は、グルタチオンを固定化したアフィニティーカラムを用いた慣用の技術により容易に精製が可能である。精製したPBP2b可溶性部分を20mM トリス塩酸 pH 7.9、1mM EDTA溶液に対して透析した後、適当量のトリプシンを加え、室温で1〜2時間反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう加えることでPBP2bトランスペプチダーゼドメインが得られる。添加するトリプシンの終濃度は典型的には0.1〜1mg/mLであるが、PBP2b可溶性部分のタンパク質濃度に依存するため、好ましくは、毎回、少量での確認実験により決定する必要がある。PBP2b可溶性部分およびトランスペプチダーゼドメインは陰イオン交換カラム、ハイドロキシアパタイトカラム、陽イオン交換カラム、ゲルろ過カラムを用いた一般的なクロマトグラフィーの手法を組み合わせて精製することが可能であり、配列番号3に示されるアミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換された変異体においても同様に精製が可能である。
【0015】
<<PBP2b可溶性部分およびトランスペプチダーゼドメインの結晶化>>
一般的に蛋白質の結晶化は容易ではない。結晶化できる条件が比較的容易に見出される場合もあるが、通常は種々の結晶化条件をスクリーニングしなければならない。さらに結晶化剤の濃度、pH、塩の種類と濃度、添加剤などの条件を最適化することによって、X線結晶構造解析に供することができる良質の結晶が得られる。また、いかなる条件でも結晶化しない蛋白質も存在すると考えられている。蛋白質の結晶が得られなければX線結晶構造解析を行うことができない。また、X線結晶構造解析に用いる蛋白質の結晶は、結晶であればいかなるものでも解析可能であるわけではない。実際にX線を照射した際に、高分解能の回折強度データが得られることが必要である。本発明におけるPBP2b結晶化の方法は、ハンギングドロップ法、シッティングドロップ法、透析法などのいかなる方法を用いてもよいが、好ましくは蒸気拡散法であるハンギングドロップ法またはシッティングドロップ法が用いられる。また、シーディング法またはリザーバー溶液にオイルを重層することにより結晶核の形成および結晶成長をコントロールすることもできる。結晶化の温度は4℃〜20℃が一般的であるが、本発明では好ましくは20℃である。結晶化に用いるタンパク質としては、(1)配列番号4に示されるR6株由来PBP2b可溶性部分、(2)配列番号5に示されるR6株由来PBP2bトランスペプチダーゼドメイン、(3)配列番号1に示されるR6株由来PBP2b変異体可溶性部分および(4)配列番号2に示されるR6株由来PBP2b変異体トランスペプチダーゼドメインのいずれを用いても良いが、好ましくは(3)R6株由来PBP2b変異体可溶性部分、より好ましくは(4)R6株由来PBP2b変異体トランスペプチダーゼドメインが用いられる。また、前記タンパク質中のメチオニン残基がセレノメチオニンに置換されたセレノメチオニン置換体も同様に結晶化に用いられる。結晶化に用いる精製タンパク質の濃度は、4〜20mg/mL程度であることが好ましい。そして、蛋白質溶液に結晶化剤、塩類、緩衝液、添加剤などを適当量加えて、結晶化を行う。本発明のPBP2bの結晶化に用いられる結晶化剤としては、硫酸アンモニウムなどの無機塩類、ポリエチレングリコールなどの水溶性高分子、イソプロパノールやエタノールなどの有機溶媒などが挙げられる。本発明においては、無機塩類を好ましく用いることができ、硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウム等の硫酸塩もしくはリン酸水素二カリウム等のリン酸塩を特に好ましく用いることができる。また、必要に応じてポリエチレングリコールを組み合わせることも可能である。また、緩衝液としては、公知の緩衝液のいずれも用いることができる。さらに添加剤としては、種々の金属イオン、有機溶媒、還元剤等を用いることができる。セレノメチオニン置換体の場合には未置換体の場合における周辺の条件で結晶を作製できるが、セレン原子の酸化を抑制するためにジチオスレイトール等の還元剤を添加することが好ましい。各PBP2bと好ましい結晶化剤の組み合わせについては次に示す通りである。
【0016】
(1)R6株由来PBP2b可溶性部分:
0.8−1.6M 硫酸アンモニウム、0.1M 酢酸ナトリウム緩衝液 pH 4.6
(2)R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体:
1.1−1.6M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9
(3−1)R6株由来PBP2b変異体可溶性部分:
10−20% ポリエチレングリコール8000、0.2M 硫酸アンモニウム、0.1M トリス塩酸緩衝液 pH 8.0
(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体:
18−20% ポリエチレングリコール8000、0.3M 硫酸リチウム、0.1M トリス塩酸緩衝液 pH 8.5、10mM ジチオスレイトール
(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体:
1.1−1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9−8.5、または、0.7−1.1M 硫酸ナトリウム、10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.0−6.5
上記の条件で得られる結晶のうち、好ましいものは(3−1)R6株由来PBP2b変異体可溶性部分の結晶および(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶であり、特に好ましいものは(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶である。
【0017】
<<PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメイン結晶のX線回折強度データの収集>>
上記のようにして得られる結晶の外観、単位格子の種類・大きさなどはその結晶に固有のものであり、良質の結晶を用いれば、X線回折実験により高分解能の回折強度データを取得することができる。結晶を多価アルコールを含有する抗凍結溶液中に浸した後凍結させ、凍結状態でX線回折データを収集し、X線回折像を得る。ここで、多価アルコールとしては、スクロース、トレハロース、グリセロールなどが挙げられる。これらの中で特にグリセロールが好ましい。抗凍結溶液中の多価アルコールの濃度は、好ましくは10%〜30%である。X線を照射して回折強度データを取得するには、いかなるX線発生装置を用いることができるが、好ましくは、(財)高輝度光科学研究センターの大型放射光施設SPring−8などの第3世代の放射光施設を利用することができる。得られたX線回折強度データを処理することにより、結晶学的パラメータを特定することが可能である。前段落(3−1)R6株由来PBP2b変異体可溶性部分の結晶および(3−2)セレノメチオニン置換体結晶は、空間群がF222である。また、格子定数はa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°であり、より好ましくはa=175±9Å、b=186±9Å、c=369±19Å、α=β=γ=90°であり、さらに好ましくはa=175±2Å、b=186±2Å、c=369±4Å、α=β=γ=90°であり、特に好ましくはa=175±1Å、b=186±1Å、c=369±1Å、α=β=γ=90°である。また、(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶は空間群がP43212であり、格子定数がa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°であり、より好ましくはa=86±4Å、b=86±4Å、c=143±7Å、α=β=γ=90°であり、さらに好ましくはa=86±1Å、b=86±1Å、c=143±2Å、α=β=γ=90°であり、特に好ましくはa=86±1Å、b=86±1Å、c=143±1Å、α=β=γ=90°である。
【0018】
<<PBP2b可溶性部分およびPBP2bトランスペプチダーゼドメインの結晶構造の決定>>
上記のように得られたデータを用いてPBP2bの結晶構造の決定が可能となる。一般に位相決定には、分子置換法、重原子同型置換法、多波長異常分散法(MAD法)などが用いられる。目的蛋白質に対する類縁蛋白質の立体構造が未知あるいは相同性が低い場合には、その立体構造を利用した分子置換法による構造解析が不可能であるため、PBP2b結晶構造の決定には、重原子同型置換法またはMAD法などが好ましく、より好ましくはセレノメチオニン置換体結晶を用いたMAD法が用いられる。特に好ましくは、前段落に記載した(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶を用いたMAD法であり、新規結晶構造決定が初めて可能となる。前段落(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶を用いた場合には、分解能がやや劣ることおよび非対称単位中の分子数が多いことによりMAD法による構造決定は困難であるが、(3−1)R6株由来PBP2b変異体可溶性部分の結晶を用いた場合も含め、構造決定したトランスペプチダーゼドメインの結晶構造をサーチモデルに利用する分子置換法により構造決定が可能となる。
【0019】
以下、実施例により本発明をさらに詳細に説明するが、本発明の技術的範囲は実施例によって限定されるものではない。また、各種ベクターの作製、蛋白質の発現等は、特に記載のない限り、Molecular Cloning,A Laboratory Manual,3rd edition(Sambrook and Russell著,Cold Spring Harbor laboratory Press刊(2001))などに記載の公知の手法に従って実施できる。
【実施例1】
【0020】
<<肺炎連鎖球菌R6株由来PBP2bおよび変異体の可溶性部分の大腸菌発現ベクター構築>>
配列番号4で示されるアミノ酸配列を有する肺炎連鎖球菌R6株由来PBP2b可溶性部分のグルタチオン−S−トランスフェラーゼ(GST)融合タンパク質の大腸菌発現ベクター(pGEX/SpPBP2b−wt)は特許公開2004−290070号広報に記載の方法により作製した。また、配列番号3に示されるアミノ酸残基番号431のスレオニン、432のグルタミン、451のスレオニン、624のアラニンが各々リジン、ロイシン、アラニン、グリシンに置換した変異体である、配列番号1で示されるアミノ酸配列を有する肺炎連鎖球菌R6株由来PBP2b変異体可溶性部分のGST融合タンパク質の大腸菌発現ベクター(pGEX/SpPBP2b−mt7)についても同様に特許公開2004−290070号広報に記載の方法により作製した。
【実施例2】
【0021】
<<R6株由来PBP2bおよび変異体の可溶性部分の発現>>
pGEX/SpPBP2b−wtを大腸菌BL21(Novagen社製)に、また、pGEX/SpPBP2b−mt7を大腸菌DH5α(TOYOBO社製)に形質転換し得られた形質転換体を100μg/mLのアンピシリンを含むSB培地(1.2%(w/v)Bacto Tryptone、2.4%(w/v)Yeast Extract、0.5%(v/v)グリセロール、0.072M リン酸水素二カリウム、0.028M リン酸二水素カリウム)5L中で600nmにおけるO.D.が1.0に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、28℃で2時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)400mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体を菌体破砕バッファー(1mM EDTA、1mM フッ化フェニルメチルスルホニル、1μM ロイペプチン、1μM ペプスタチンA、0.1mg/mL リゾチーム含有PBS)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を2回施し、細胞を破砕した。遠心分離(60分間、12000rpm)とそれに続く0.2μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【実施例3】
【0022】
<<R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体の発現>>
MAD法によるX線結晶構造解析に使用する重原子誘導体結晶を作製するために、メチオニンの硫黄原子が重原子であるセレンに置換されたR6株由来PBP2b変異体のセレノメチオニン置換体の発現を試みた。実施例1記載の組み換えプラスミドpGEX/SpPBP2b−mt7をメチオニン要求株である大腸菌B834(DE3)(Novagen社製)に形質転換し、100μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。得られた形質転換体は100μg/mLのアンピシリンを含むLB培地100mLにて、37℃で一晩培養し、100μg/mLのアンピシリンを含む4LのLeMaster培地(組成は下記の表1に示す)に40mL接種し、600nmにおけるO.D.が0.5−0.8に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、30℃で一晩誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)225mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体を菌体破砕バッファー(1mM EDTA、5mM β−メルカプトエタノール、1mM フッ化フェニルメチルスルホニル、1μM ロイペプチン、1μM ペプスタチンA、0.1mg/mL リゾチーム含有PBS)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を3回施し、細胞を破砕した。遠心分離(60分間、12000rpm)とそれに続く0.2μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【0023】
【表1】
【実施例4】
【0024】
<<R6株由来PBP2bおよび変異体の可溶性部分の精製>>
(1)アフィニティー精製
以下の精製操作は4℃で実施した。実施例2で調製した細胞抽出液を予めPBSで平衡化したGlutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)担体80mLにバッチ法で結合させた。これを担体の10倍容量のPBSで5回洗浄した後、担体容量と等量の溶出液(10mM 還元型グルタチオン、50mM トリス塩酸 pH 7.9)で3回溶出した。
【0025】
(2)GSTタグの切断
上記(1)で精製したサンプルを1mg/mLに希釈し、終濃度2.5unit/mLのトロンビン(シグマ社製)を添加し、4℃で一晩反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。
【0026】
(3)陰イオン交換カラムによる精製
上記(2)で得られたサンプルを陰イオン交換カラムであるPOROS HQ/20(PerSeptive Biosystems社製)を用いて精製した。流速は10mL/minであり、サンプルを3分割し、精製は3度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9中の0−600mM 塩化ナトリウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめ、Glutathione Sepharose 4 Fast Flow担体にアプライし、バッチ法によりGSTタグを除去した。
【0027】
(4)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(3)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8)に対して透析しバッファー置換を実施した。サンプルを3分割し、精製は3度実施した。平衡化バッファーで平衡化したカラムに流速3mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、5−250mM リン酸カリウムバッファー pH 6.8の直線勾配(20カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0028】
(5)ゲルろ過カラムによる精製
上記(4)で得られたサンプルを保持分子量30000以上の限外ろ過膜であるAmicon Ultra−15 30kDa NMWL(ミリポア社製)を用いて2mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH7.5、150mM 塩化ナトリウム)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。精製はサンプルを2等分し、2度に分けて実施した。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて10mg/mLまで濃縮後、液体窒素中で凍結し、−80℃で保存した。
【実施例5】
【0029】
<<R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体の精製>>
(1)アフィニティー精製
以下の精製操作は4℃で実施した。アフィニティーカラムは、Glutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量50mLのカラムを作製し、平衡化バッファー(1mM EDTA、5mM β−メルカプトエタノール含有PBS)で平衡化した。そこへ実施例3で調製した細胞抽出液を流速2mL/minでアプライし、5カラム容量の平衡化バッファーで洗浄した後、溶出バッファー(50mM トリス塩酸 pH 7.9、10mM 還元型グルタチオン、1mM EDTA、5mM β−メルカプトエタノール)で溶出した。SDS−PAGEを実施し、溶出画分を確認し、主要なフラクションをまとめて回収した。
【0030】
(2)GSTタグの切断
上記(1)で精製したサンプルを透析バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)に対して透析した後、2倍量の透析バッファーを加えて希釈し、終濃度5unit/mLのトロンビン(シグマ社製)を添加し、4℃で一晩反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。
【0031】
(3)陰イオン交換カラムによる精製
上記(2)で得られたサンプルを陰イオン交換カラムであるPOROS HQ/20(PerSeptive Biosystems社製)を用いて精製した。流速は10mL/minであり、サンプルを3分割し、精製は3度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、10mM ジチオスレイトール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9、1mM EDTA、10mM ジチオスレイトール中の0−500mM 塩化ナトリウムの直線勾配(16カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめ、Glutathione Sepharose 4 Fast Flow担体2mLにアプライし、バッチ法によりGSTタグを除去した。
【0032】
(4)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(3)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8、1mM ジチオスレイトール)で平衡化した脱塩カラムHiPrep 26/10 Desalting(アマシャムバイオサイエンス社製)に流速7.5mL/minでアプライし、バッファー置換を実施した。サンプルを3分割し、精製は3度実施した。平衡化バッファーで平衡化したハイドロキシアパタイトカラムに流速3mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、1mM ジチオスレイトール中の5−250mM リン酸カリウムバッファー pH 6.8の直線勾配(25カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0033】
(5)ゲルろ過カラムによる精製
上記(4)で得られたサンプルを保持分子量50000以上の限外ろ過膜であるAmicon Ultra−15 50kDa NMWL(ミリポア社製)を用いて1.7mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH7.5、150mM 塩化ナトリウム、10mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。精製はサンプルを2等分し、2度に分けて実施した。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて20mg/mLまで濃縮後、終濃度0.05mMのEDTAを添加後、液体窒素中で凍結し、−80℃で保存した。
【実施例6】
【0034】
<<トランスペプチダーゼドメイン調製のための大腸菌発現系の構築>>
(1)トリプシン限定分解によるトランスペプチダーゼドメインの同定
実施例5で精製したR6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体を20mM Tris塩酸 pH 7.9、1mM EDTA、1mM ジチオスレイトールでタンパク質終濃度0.4mg/mLに希釈した。次に、希釈液10μL、5mg/mL トリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)溶液1.6μL、10mM EDTA溶液2μL、MilliQ水6.4μLを混合し、26℃、1時間反応後、サンプルを2等分した。一方は終濃度1mM PMSFおよび還元剤含有のSDS−PAGEサンプルバッファーと等量混合し、3分間煮沸後、SDS−PAGEにアプライし泳動した後、PVDF膜に転写し、主要なバンドを切り出しプロテインシークエンサーによりN末端アミノ酸配列を分析した。残りの一方は10分の1量の10%酢酸を加えた後、ZipTipC4(ミリポア社製)を用いて精製しMALDI−TOF型質量分析装置にて分子量を測定した。その結果、SDS−PAGEにおいて分子量41k、25k、19k付近の主要なバンドおよび12k付近のマイナーなバンドが確認された。N末端アミノ酸配列の結果では、41k付近のバンドのN末端アミノ酸配列はLTI、19k付近のバンドはGEIYD、12k付近のバンドはGGLGD、TGTGEおよびSAYPGの混合物であり、この内SAYPGの配列はトリプシン由来であった。質量分析では多数のピークが観測されたが、その中に分子量18284、18158、13156のピークが存在しており、また、23300のピークはトリプシン由来であることが判明した。以上の結果から、SDS−PAGEでの25kのバンドはトリプシン由来、41kのバンドは配列番号3に示されるR6株由来PBP2bにおいてはアミノ酸番号315〜685、19kのバンドは66〜228、66〜229の混合物、12kのバンドは562〜680、621〜685の混合物に対応することが判明した。トランスペプチダーゼドメインに存在する保存モチーフはアミノ酸番号391から始まるSVVK、448から始まるSSN、620から始まるKTGであることから、トランスペプチダーゼドメインとしてアミノ酸番号315〜685からなる領域を選択した。
【0035】
(2)トリプシン限定分解時におけるマイナー部位での切断を回避するための点変異の導入
上記(1)においてトリプシン限定分解でのマイナー部位での切断箇所として配列番号3に示されるR6株由来PBP2bにおいてはアミノ酸番号561および620のリジン残基の直後が対応する。この内、アミノ酸番号620のリジンはトランスペプチダーゼドメインの保存モチーフに存在するため、変異を導入するのは好ましくないが、アミノ酸番号561のリジンは変異を導入することでトリプシン限定分解時におけるマイナー部位での切断を回避することができる。また、アミノ酸番号66〜228、66〜229の混合物からなる部分についてはトランスペプチダーゼドメインと相互作用している可能性が否定できなかったため、アミノ酸番号229のリジン残基についても変異を導入し、混合物生成を回避する方針とした。ここで、アミノ酸番号561および229のいずれについてもグルタミンへの変異を試みた。実施例1に記載のpGEX/SpPBP2b−wtおよびpGEX/SpPBP2b−mt7を鋳型として、下記のプライマーを用い、QuickChange Site−Directed Mutagenesis Kit(Stratagene社製)で、添付の説明書の方法に従い、アミノ酸番号561の部位への変異を導入した。
【0036】
2b_R6_K561Q_5:5’−GGCATTTATGGTAATAATGATCAGGGAGGACTGGGTGACTTG−3’(配列番号6)
2b_R6_K561Q_3:5’−CAAGTCACCCAGTCCTCCCTGATCATTATTACCATAAATGCC−3’(配列番号7)
【0037】
次に下記のプライマーを用いて同様の手順に従い、さらにアミノ酸番号229の部位への変異を導入し、pGEX/SpPBP2b−wt−229Q−561QおよびpGEX/SpPBP2b−mt7−229Q−561Qを作製した。これらのプラスミドのDNA配列を確認し、各々配列番号4、配列番号1に記載のアミノ酸配列に目的の2箇所の点変異が導入されたR6株由来および変異体のPBP2b可溶性部分をコードすることを確認した。
【0038】
2b_R6_K229Q_5:5’−CTGGCATTAGTATTTCTACTTCTTGGGATAGACAGGTTTTGGA−3’(配列番号8)
2b_R6_K229Q_3:5’−TCCAAAACCTGTCTATCCCAAGAAGTAGAAATACTAATGCCAG−3’(配列番号9)
【実施例7】
【0039】
<<R6株由来PBP2bおよび変異体トランスペプチダーゼドメイン取得のための可溶性部分セレノメチオニン置換体の発現>>
実施例6で得られたpGEX/SpPBP2b−wt−229Q−561QおよびpGEX/SpPBP2b−mt7−229Q−561Qをメチオニン要求株である大腸菌B834(DE3)(Novagen社製)に形質転換し、100μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。得られた形質転換体は100μg/mLのアンピシリンを含むLB培地100mLにて、37℃で一晩培養し、遠心分離機で集菌後、100μg/mLのアンピシリンを含む100mlのLeMaster培地(組成は実施例3の表1に示す)で懸濁し、再度、遠心分離機によって集菌し、100μg/mLのアンピシリンを含むLeMaster培地100mlに再懸濁した。この懸濁液を100μg/mLのアンピシリンを含む4.8LのLeMaster培地に48mL接種し、600nmにおけるO.D.が0.5−0.8に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、30℃で4時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)200mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体を菌体破砕バッファー(1mM EDTA、5mM β−メルカプトエタノール、1mM フッ化フェニルメチルスルホニル、1μM ロイペプチン、1μM ペプスタチンA、0.1mg/mL リゾチーム含有PBS)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を4回施し、細胞を破砕した。遠心分離(60分間、12000rpm)とそれに続く0.2μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【実施例8】
【0040】
<<R6株由来PBP2bおよび変異体トランスペプチダーゼドメインのセレノメチオニン置換体の精製>>
(1)R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体の精製
(1−1)アフィニティー精製
以下の精製操作は4℃で実施した。アフィニティーカラムは、Glutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量50mLのカラムを作製し、平衡化バッファー(1mM EDTA、5mM β−メルカプトエタノール含有PBS)で平衡化した。そこへ実施例7においてpGEX/SpPBP2b−wt−229Q−561Qを用いて調製した細胞抽出液を流速2mL/minでアプライし、5カラム容量の平衡化バッファーで洗浄した後、溶出バッファー(5unit/mLトロンビン(シグマ社製)含有PBS)を50mLアプライした段階でポンプを止めて10℃で一晩反応させGSTタグを切断した。平衡化バッファーを送り込んで溶出した後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。SDS−PAGEにより溶出画分を確認し、主要なフラクションをまとめて回収した。
【0041】
(1−2)陰イオン交換カラムによる精製
上記(1−1)で得られたサンプルを透析バッファー(20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール)に対して透析した後、陰イオン交換カラムであるPOROS HQ/20(PerSeptive Biosystems社製)を用いて精製した。流速は10mL/minであり、サンプルを4分割し、精製は4度実施した。平衡化バッファー(20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール中の0−300mM 塩化ナトリウムの直線勾配(12カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0042】
(1−3)トリプシン限定分解
上記(1−2)で精製したR6株由来PBP2b可溶性部分のセレノメチオニン置換体を20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール溶液に対して透析した後回収し、はじめに小スケールでのトリプシン限定分解の条件検討を実施した。回収液5μLを0.125〜5mg/mLのトリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)溶液と等量混合し、24℃、2時間反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。SDS−PAGEにより各濃度におけるトリプシン消化のパターンを確認し、トリプシンの添加量を終濃度0.125mg/mlと決定し、残りの全サンプルのトリプシン限定分解を実施した。
【0043】
(1−4)陰イオン交換カラムによる精製
上記(1−3)で得られたサンプルを陰イオン交換カラムであるMonoQ HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。流速は4mL/minであり、サンプルを4分割し、精製は4度実施した。平衡化バッファー(20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 8.0、1mM EDTA、5mM β−メルカプトエタノール中の0−300mM 塩化ナトリウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0044】
(1−5)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(1−4)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8、1mM ジチオスレイトール)に対して透析し、バッファー置換を実施した。サンプルを2分割し、精製は2度実施した。平衡化バッファーで平衡化したハイドロキシアパタイトカラムに流速3.5mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、1mM ジチオスレイトール中の5−250mM リン酸カリウムバッファー pH 6.8の直線勾配(25カラム容量)で溶出した。SDS−PAGEを実施し、非吸着画分をまとめた。
【0045】
(1−6)ゲルろ過カラムによる精製
上記(1−5)で得られたサンプルを保持分子量10000以上の限外ろ過膜であるAmicon Ultra−15 10kDa NMWL(ミリポア社製)を用いて0.75mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH8.0、150mM 塩化ナトリウム、1mM EDTA、10mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて10mg/mLまで濃縮後、液体窒素中で凍結し、−80℃で保存した。SDS−PAGEの結果から、分子量41k付近のバンドのみが確認され、19k付近のバンドについては除去されたことから、配列番号5に示されるアミノ酸配列からなるR6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体であることが示唆された。
【0046】
(2)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体の精製
(2−1)アフィニティー精製
以下の精製操作は4℃で実施した。アフィニティーカラムは、Glutathione Sepharose 4 Fast Flow(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量50mLのカラムを作製し、平衡化バッファー(1mM EDTA、5mM β−メルカプトエタノール含有PBS)で平衡化した。そこへ実施例7においてpGEX/SpPBP2b−mt7−229Q−561Qを用いて調製した細胞抽出液を流速2mL/minでアプライし、5カラム容量の平衡化バッファーで洗浄した後、溶出バッファー(50mM トリス塩酸 pH 7.9、10mM 還元型グルタチオン、1mM EDTA、5mM β−メルカプトエタノール)で溶出した。SDS−PAGEにより溶出画分を確認し、主要なフラクションをまとめて透析バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)に対して透析することでバッファー置換をした。次に、トリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)を終濃度2.3μg/mLの条件で添加し、25℃で1時間反応させることでGSTタグのみを切断し、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。
【0047】
(2−2)陰イオン交換カラムによる精製
上記(2−1)で得られたサンプルを陰イオン交換カラムであるMonoQ HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。流速は4mL/minであり、サンプルを5分割し、精製は5度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール中の0−250mM 塩化ナトリウムの直線勾配(8カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0048】
(2−3)トリプシン限定分解
上記(2−2)で精製したR6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体を20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール溶液に対して透析した後回収し、はじめに小スケールでのトリプシン限定分解の条件検討を実施した。回収液9μLに0.039〜10mg/mLのトリプシン(Trypsin TPCK treated From Bovine Pancreas、Sigma社製)溶液1μLを添加し、25℃、1時間反応させた後、フッ化フェニルメチルスルホニルを終濃度1mMになるよう添加し反応を止めた。SDS−PAGEにより各濃度におけるトリプシン消化のパターンを確認し、トリプシンの添加量を終濃度0.125mg/mlと決定し、残りの全サンプルのトリプシン限定分解を実施した。
【0049】
(2−4)陰イオン交換カラムによる精製
上記(2−3)で得られたサンプルを陰イオン交換カラムであるMonoQ HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。流速は4mL/minであり、サンプルを4分割し、精製は4度実施した。平衡化バッファー(20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール)で平衡化したカラムにサンプルをアプライし、3カラム容量の平衡化バッファーで洗浄した後、20mM トリス塩酸 pH 7.9、1mM EDTA、5mM β−メルカプトエタノール中の0−250mM 塩化ナトリウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEを実施し、主要な溶出画分をまとめた。
【0050】
(2−5)ハイドロキシアパタイトカラムによる精製
ハイドロキシアパタイトカラムは、Macro−Prep Ceramic Hydroxyapatite TYPE I 20μm(バイオラッド社製)を担体として用い、マニュアル記載の方法でカラム容量8mLのカラムを作製した。次に、(2−4)で得られたサンプルを平衡化バッファー(5mM リン酸カリウムバッファー pH 6.8、1mM ジチオスレイトール)に対して透析し、バッファー置換を実施した。サンプルを2分割し、精製は2度実施した。平衡化バッファーで平衡化したハイドロキシアパタイトカラムに流速3.5mL/minでサンプルをアプライし、非吸着画分を回収した。
【0051】
(2−6)陽イオン交換カラムによる精製
上記(2−5)で得られたサンプルを平衡化バッファー(50mM 酢酸ナトリウムバッファー pH 5.0、1mM EDTA、1mM ジチオスレイトール)に対して透析し、バッファー置換を実施した後、陽イオン交換カラムであるMonoS HR10/10(アマシャムバイオサイエンス社製)を用いて精製した。サンプルを2分割し、精製は2度実施した。平衡化バッファーで平衡化したカラムに流速4mL/minでサンプルをアプライし、2カラム容量の平衡化バッファーで洗浄した後、50mM 酢酸ナトリウムバッファー pH 5.0、1mM EDTA、1mM ジチオスレイトール中の0−200mM 塩化ナトリウムの直線勾配(16カラム容量)で溶出した。SDS−PAGEにより主要な溶出画分をまとめた。
【0052】
(2−7)ゲルろ過カラムによる精製
上記(2−6)で得られたサンプルを保持分子量10000以上の限外ろ過膜であるAmicon Ultra−15 10kDa NMWL(ミリポア社製)を用いて1.2mLまで濃縮し、ゲルろ過バッファー(20mM トリス塩酸 pH7.9、150mM 塩化ナトリウム、1mM EDTA、1mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1mL/minでアプライした。SDS−PAGEを実施し、主要な画分をまとめ、上記記載の限外ろ過膜を用いて4.7mg/mLまで濃縮後、液体窒素中で凍結し、−80℃で保存した。SDS−PAGEでは分子量41k付近のバンドのみが確認された。また、エレクトロスプレイイオン化法による質量分析装置により解析したところ、分子量39851に主要なピークが確認されたことから、計算値で39843の分子量を持つ、配列番号2に示されるアミノ酸配列からなるR6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体であることが確認された。
【実施例9】
【0053】
<<PBP2bの結晶化>>
(1)R6株由来PBP2b可溶性部分の結晶化
R6株由来PBP2b可溶性部分の結晶は、シッティングドロップ蒸気拡散法により得た。実施例4で精製したタンパク質溶液(10mg/mL)0.5μLと結晶化剤(0.8−1.6M 硫酸アンモニウム、0.1M 酢酸ナトリウム緩衝液 pH 4.6)0.5μLを混合し結晶化ドロップとし、リザーバー溶液として上記結晶化剤100μL分注し密閉した。プレートを20℃の恒温槽内に静置すると数日で針状のクラスター結晶が得られた。0.8M 硫酸アンモニウム、0.1M 酢酸ナトリウム緩衝液 pH 4.6からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図1に示す。
【0054】
(2)R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体の結晶化
R6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体の結晶は、ハンギングドロップ蒸気拡散法により得た。実施例8(1)で精製したタンパク質溶液(10mg/mL)2.0μLと結晶化剤(1.1−1.6M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9)2.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注し、Al’sオイル(ハンプトンリサーチ社製)250μLをリザーバー溶液の上に重層した。ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを20℃の恒温槽内に静置すると数日で針状のクラスター結晶が得られた。1.1M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図2に示す。
【0055】
(3)R6株由来PBP2b変異体の可溶性部分およびセレノメチオニン置換体の結晶化
(3−1)R6株由来PBP2b変異体の可溶性部分の結晶化
R6株由来PBP2b変異体の可溶性部分の結晶は、ハンギングドロップ蒸気拡散法により得た。実施例4で精製したタンパク質溶液(10mg/mL)1.0μLと結晶化剤(10−20% ポリエチレングリコール8000、0.2M 硫酸アンモニウム、0.1M トリス塩酸緩衝液 pH 8.0)1.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注した。ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを20℃の恒温槽内に静置すると数日で結晶が得られた。また、硫酸アンモニウムの代わりに硫酸リチウム、硫酸ナトリウムを用いた場合も同様に結晶が得られた。10% ポリエチレングリコール8000、0.2M 硫酸アンモニウム、0.1M トリス塩酸緩衝液 pH 8.0からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図3に示す。
【0056】
(3−2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体の結晶化
R6株由来PBP2b変異体可溶性部分の結晶は、ハンギングドロップ蒸気拡散法により得た。実施例5で精製したタンパク質溶液(10mg/mL)1.0μLと結晶化剤(18−20% ポリエチレングリコール8000、0.3M 硫酸リチウム、0.1M トリス塩酸緩衝液 pH 8.5、10mM ジチオスレイトール)1.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注し、Al’sオイル(ハンプトンリサーチ社製)200μLをリザーバー溶液の上に重層した。ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを20℃の恒温槽内に静置すると数日で結晶が得られた。また、硫酸リチウムの代わりに硫酸アンモニウム、硫酸ナトリウムを用いた場合も同様に結晶が得られた。19% ポリエチレングリコール8000、0.3M 硫酸リチウム、0.1M トリス塩酸緩衝液 pH 8.5、10mM ジチオスレイトールからなる結晶化剤を用いて得られた結晶の顕微鏡写真を図4に示す。
【0057】
(4)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体の結晶化
R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体の結晶は、シッティングドロップ蒸気拡散法により得た。実施例8(2)で精製したタンパク質溶液(4.7mg/mL)0.4μLと結晶化剤(1.1−1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 7.9−8.5)0.4μLを混合し結晶化ドロップとし、リザーバー溶液として上記結晶化剤100μL分注し密閉した。プレートを20℃の恒温槽内に静置すると数日で結晶が得られた。1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 8.5からなる結晶化剤を用いて得られた結晶の顕微鏡写真を図5に示す。また、硫酸アンモニウムの代わりに硫酸ナトリウムを用いた条件(0.7−1.1M 硫酸ナトリウム、10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.0−6.5または0.1M クエン酸ナトリウム緩衝液 pH 5.5または0.1M HEPES緩衝液 pH 7.0−7.5またはトリス塩酸緩衝液 pH 7.9−8.5)および硫酸マグネシウムを用いた条件(0.9−1.1M 硫酸マグネシウム、1−10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.5)でも同様の結晶が得られた。また、N−Benzoyl−D−alanylthioglycolic acidを添加した条件でも同様の結晶が得られ、ハンギングドロップ蒸気拡散法においても同様の結晶が得られた。さらに、マイクロ・フリューディック技術を応用したTOPAZ 4.96スクリーニングチップ(フリューダイム社製)を用いた場合には結晶化剤として(i)1M 硫酸アンモニウム、0.1M 酢酸ナトリウム pH 4.6および(ii)1.5M リン酸水素二カリウムを用いた条件において外見上同様の結晶が得られた。
【実施例10】
【0058】
<<PBP2b結晶のX線回折強度データの収集>>
(1)R6株由来PBP2b変異体の可溶性部分の結晶
実施例9(3−1)において得られた結晶を35%グリセロール含有の抗凍結剤溶液に移した後、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しイメージングプレートを装備したR−AXIS V(リガク社製)を検出装置としてX線回折データを振動法にて測定した。X線の波長は1Å、振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、回折強度データを分解能3.5Åで処理した。その結果、空間群がF222であり、格子定数がa=175.47Å、b=186.16Å、c=368.53Å、α=β=γ=90°であった。得られたデータのCompletenessは99.9%、Rmergeは9.4%であった。データ収集の結果を下記の表2にまとめた。
【0059】
【表2】
【0060】
(2)R6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶
実施例9(3−2)において得られた結晶を20%グリセロール含有の抗凍結剤溶液に移した後、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しイメージングプレートを装備したR−AXIS V(リガク社製)を検出装置としてX線回折データを振動法にて測定した。このとき、セレンの異常散乱効果を用いて構造解析を行うために、波長は0.97947、0.97879および0.90000Åの3波長、即ち、吸収端のエッジ、ピーク、リモートの3カ所でMADデータを収集した。振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、回折強度データを分解能3.5Åで処理した。その結果、空間群がF222であり、格子定数がa=175.75Å、b=186.21Å、c=369.21Å、α=β=γ=90°であった。データ収集の結果を下記の表3にまとめた。
【0061】
【表3】
【0062】
(3)R6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶
実施例9(4)において、1.5M 硫酸アンモニウム、15% グリセロール、10mM ジチオスレイトール、0.1M トリス塩酸緩衝液 pH 8.5からなる結晶化剤を用いて得られた結晶を25%グリセロール含有の抗凍結剤溶液に移した後、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しイメージングプレートを装備したR−AXIS V(リガク社製)を検出装置としてX線回折データを振動法にて測定した。このとき、セレンの異常散乱効果を用いて構造解析を行うために、波長は0.97948、0.97907および0.90000Åの3波長、即ち、吸収端のエッジ、ピーク、リモートの3カ所でMADデータを収集した。振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、回折強度データを分解能2.4Åで処理した。その結果、空間群の候補がP43212またはP41212となったが、下記に示す実施例11の解析結果からP43212と判明した。格子定数はa=86.39Å、b=86.39Å、c=143.27Å、α=β=γ=90°であり、データ収集の結果を下記の表4にまとめた。また、同様の手順で調製した結晶を(財)高輝度光科学研究センターの大型放射光施設SPring−8の構造生物学IビームラインBL41XUを利用しQuantum315CCDカメラ(ADSC社製)を検出器としてX線回折データを振動法にて測定した。X線の波長は1Å、振動角は1°/フレームであり、回折強度データを2.2Åで処理した結果、空間群はP43212、格子定数はa=86.34Å、b=86.34Å、c=143.40Å、α=β=γ=90°であった。データ収集の結果を下記の表5にまとめた。また、実施例9(4)において、0.8M 硫酸ナトリウム、10mM ジチオスレイトール、0.1M MES緩衝液 pH 6.5からなる結晶化剤を用いてN−Benzoyl−D−alanylthioglycolic acid存在下で得られた結晶を用いて、同様の条件でX線回折測定を実施したところ、空間群はP43212、格子定数はa=86.41Å、b=86.41Å、c=143.96Å、α=β=γ=90°であった。
【0063】
【表4】
【0064】
【表5】
【実施例11】
【0065】
<<PBP2bの結晶構造決定>>
次に、プログラムSOLVE(Terwillinger, T.C. & Berendzen,J. Acta Crystallogr. D 55,849−861(1999))を用いて、実施例10の表4に示したMADデータを処理し、セレン原子の位置を決定し初期位相を計算した。位相の改良及び初期モデルの構築はプログラムRESOLVE(Terwilliger, T.C. Acta Crystallogr. D56,965−972(2000))を用いた。この際、空間群P41212で処理した場合には妥当な電子密度が得られず、空間群P43212で処理した場合においてタンパク質の明瞭の電子密度が得られたため、空間群をP43212と決定した。次いで、プログラムLafire(YAO M.他、Acta.Cryst.D62(2)、189−196(2006))、プログラムTurbo Frodo(AFMB−CNRS製)、プログラムRefmac5(Collaborative Computational Project Number 4(ccp4))およびプログラムCNX(アクセルリス社製)を利用したモデルの構築および修正を繰り返し実施し、R6株由来PBP2b変異体トランスペプチダーゼドメインの結晶構造を決定し、最終的には実施例10の表5に示した高分解能のデータを用いて分解能2.2Åで構造精密化を実施した。本結晶の非対称単位中にはR6株由来PBP2b変異体トランスペプチダーゼドメインが1分子存在していた。決定した結晶構造の正確さの指標であるR因子は、0.209であった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.230であった。また、決定したR6株由来PBP2b変異体トランスペプチダーゼドメインの構造を図6に示した。さらに、結晶中において、隣り合うPBP2b分子の境界に存在する2回軸上に硫酸イオンが存在し、各PBP2b分子のリジン、アスパラギン残基の側鎖および水分子と水素結合を形成していた。結晶中において、分子間のパッキングに関与している硫酸イオン周辺の電子密度図とモデルを図7に示した。ここで、リジンは配列番号3に示されるR6株由来PBP2bの配列における431番目のスレオニンをリジンへ変異させた残基に対応しており、薬剤耐性に関与しているアミノ酸変異の導入と硫酸アンモニウム等の硫酸塩を結晶化剤として使用することを組み合わせることにより高品質の結晶が製造できることを見出した。
【産業上の利用可能性】
【0066】
本発明のPBP2b結晶は、X線結晶構造解析可能な新規な結晶であり、解析に成功した立体構造情報を利用することにより阻害剤の効率的な設計等が可能となり、新薬創出に活用できるため、産業上極めて有用である。
【図面の簡単な説明】
【0067】
【図1】本発明のR6株由来PBP2b可溶性部分の結晶の顕微鏡写真である。
【図2】本発明のR6株由来PBP2bトランスペプチダーゼドメインのセレノメチオニン置換体結晶の顕微鏡写真である。
【図3】本発明のR6株由来PBP2b変異体可溶性部分の結晶の顕微鏡写真である。
【図4】本発明のR6株由来PBP2b変異体可溶性部分のセレノメチオニン置換体結晶の顕微鏡写真である。
【図5】本発明のR6株由来PBP2b変異体トランスペプチダーゼドメインのセレノメチオニン置換体結晶の顕微鏡写真である。
【図6】決定したR6株由来PBP2b変異体トランスペプチダーゼドメインの構造をリボン図で示す。
【図7】本発明のR6株由来PBP2b変異体トランスペプチダーゼドメイン結晶中において、分子間のパッキングに関与している硫酸イオン周辺の電子密度図とモデルを示す。
【特許請求の範囲】
【請求項1】
連鎖球菌科(Streptococcaceae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
【請求項2】
ストレプトコッカス属(Streptococcus)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
【請求項3】
肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
【請求項4】
ペニシリン結合タンパク質(PBP)2b可溶性部分から構成される請求項1〜3のいずれか一項に記載の結晶。
【請求項5】
配列番号1で示されるアミノ酸配列からなるタンパク質から構成される請求項1〜4のいずれか一項に記載の結晶。
【請求項6】
配列番号1で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される請求項1〜4のいずれか一項に記載の結晶。
【請求項7】
配列番号1で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される請求項1〜4のいずれか一項に記載の結晶。
【請求項8】
空間群がF222である請求項1〜7のいずれか一項に記載の結晶。
【請求項9】
空間群がF222であり、格子定数がa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°である請求項1〜7のいずれか一項に記載の結晶。
【請求項10】
ペニシリン結合タンパク質(PBP)2bトランスペプチダーゼドメインから構成される請求項1〜3のいずれか一項に記載の結晶。
【請求項11】
配列番号2で示されるアミノ酸配列からなるタンパク質から構成される請求項1〜4、10のいずれか一項に記載の結晶。
【請求項12】
配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される請求項1〜4、10のいずれか一項に記載の結晶。
【請求項13】
配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される請求項1〜4、10のいずれか一項に記載の結晶。
【請求項14】
空間群がP43212である請求項1〜4、10〜13のいずれか一項に記載の結晶。
【請求項15】
空間群がP43212であり、格子定数がa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°である請求項1〜4、10〜13のいずれか一項に記載の結晶。
【請求項16】
請求項1〜15のいずれか一項に記載の結晶であって、該結晶を構成するタンパク質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
【請求項17】
硫酸塩またはリン酸塩の存在下で成長する請求項1〜16のいずれか一項に記載の結晶。
【請求項18】
硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムの存在下で成長する請求項1〜16のいずれか一項に記載の結晶。
【請求項19】
配列番号2で示されるアミノ酸配列からなるタンパク質。
【請求項20】
配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質。
【請求項21】
配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質。
【請求項22】
肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶の製造方法であって、結晶化剤として硫酸塩またはリン酸塩を用いて結晶化することを特徴とする、請求項1〜18のいずれか一項に記載の結晶の製造方法。
【請求項23】
結晶化剤として硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムを用いることを特徴とする、請求項22記載の製造方法。
【請求項24】
配列番号3におけるアミノ酸残基番号431のスレオニンがリジンに置換されているアミノ酸変異を有することを特徴とする、肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの可溶性部分またはトランスペプチダーゼドメインを調製する工程を含む、請求項22または23記載の製造方法。
【請求項25】
配列番号1または2で示されるアミノ酸配列からなるタンパク質を調製する工程を含む、請求項22〜24のいずれか一項に記載の製造方法。
【請求項1】
連鎖球菌科(Streptococcaceae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
【請求項2】
ストレプトコッカス属(Streptococcus)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
【請求項3】
肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶。
【請求項4】
ペニシリン結合タンパク質(PBP)2b可溶性部分から構成される請求項1〜3のいずれか一項に記載の結晶。
【請求項5】
配列番号1で示されるアミノ酸配列からなるタンパク質から構成される請求項1〜4のいずれか一項に記載の結晶。
【請求項6】
配列番号1で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される請求項1〜4のいずれか一項に記載の結晶。
【請求項7】
配列番号1で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される請求項1〜4のいずれか一項に記載の結晶。
【請求項8】
空間群がF222である請求項1〜7のいずれか一項に記載の結晶。
【請求項9】
空間群がF222であり、格子定数がa=175±18Å、b=186±19Å、c=369±37Å、α=β=γ=90°である請求項1〜7のいずれか一項に記載の結晶。
【請求項10】
ペニシリン結合タンパク質(PBP)2bトランスペプチダーゼドメインから構成される請求項1〜3のいずれか一項に記載の結晶。
【請求項11】
配列番号2で示されるアミノ酸配列からなるタンパク質から構成される請求項1〜4、10のいずれか一項に記載の結晶。
【請求項12】
配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質から構成される請求項1〜4、10のいずれか一項に記載の結晶。
【請求項13】
配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質から構成される請求項1〜4、10のいずれか一項に記載の結晶。
【請求項14】
空間群がP43212である請求項1〜4、10〜13のいずれか一項に記載の結晶。
【請求項15】
空間群がP43212であり、格子定数がa=86±9Å、b=86±9Å、c=143±14Å、α=β=γ=90°である請求項1〜4、10〜13のいずれか一項に記載の結晶。
【請求項16】
請求項1〜15のいずれか一項に記載の結晶であって、該結晶を構成するタンパク質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
【請求項17】
硫酸塩またはリン酸塩の存在下で成長する請求項1〜16のいずれか一項に記載の結晶。
【請求項18】
硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムの存在下で成長する請求項1〜16のいずれか一項に記載の結晶。
【請求項19】
配列番号2で示されるアミノ酸配列からなるタンパク質。
【請求項20】
配列番号2で示されるアミノ酸配列からなるタンパク質と少なくとも85%の相同性を有するアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する相同タンパク質。
【請求項21】
配列番号2で示されるアミノ酸配列において、1個もしくは複数個のアミノ酸が置換、欠失、付加もしくは挿入されたアミノ酸配列を含んでおり、かつトランスペプチダーゼドメインを有する改変タンパク質。
【請求項22】
肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの結晶の製造方法であって、結晶化剤として硫酸塩またはリン酸塩を用いて結晶化することを特徴とする、請求項1〜18のいずれか一項に記載の結晶の製造方法。
【請求項23】
結晶化剤として硫酸アンモニウム、硫酸マグネシウム、硫酸ナトリウム、硫酸リチウムまたはリン酸水素二カリウムを用いることを特徴とする、請求項22記載の製造方法。
【請求項24】
配列番号3におけるアミノ酸残基番号431のスレオニンがリジンに置換されているアミノ酸変異を有することを特徴とする、肺炎連鎖球菌(Streptococcus pneumoniae)由来であるペニシリン結合タンパク質(PBP)2bの可溶性部分またはトランスペプチダーゼドメインを調製する工程を含む、請求項22または23記載の製造方法。
【請求項25】
配列番号1または2で示されるアミノ酸配列からなるタンパク質を調製する工程を含む、請求項22〜24のいずれか一項に記載の製造方法。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2009−137840(P2009−137840A)
【公開日】平成21年6月25日(2009.6.25)
【国際特許分類】
【出願番号】特願2006−91462(P2006−91462)
【出願日】平成18年3月29日(2006.3.29)
【出願人】(000006091)明治製菓株式会社 (180)
【Fターム(参考)】
【公開日】平成21年6月25日(2009.6.25)
【国際特許分類】
【出願日】平成18年3月29日(2006.3.29)
【出願人】(000006091)明治製菓株式会社 (180)
【Fターム(参考)】
[ Back to top ]