
ペプチダーゼに対する耐性が向上しているGPL−1(グルカゴン様ペプチド−1)融合ペプチド
【課題】生物活性を有し、タンパク分解に対して耐性のあるペプチド分子に基づいたGLP−1を提供すること。
【解決手段】本発明は、GLP−1活性を有しており、インビボにおける安定性が向上している(特にジペプチジルペプチダーゼIVに対して耐性がある)融合ペプチドを提供する。前記融合ペプチドは、構成要素(I)として、N末端にGLP−1(7−35、7−36または7−37)の配列、および構成要素(II)として、C末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体もしくは誘導体を含んでいる。構成要素(II)は、完全または部分的なIP2(介在性ペプチド2)であることが好ましい。好ましい実施の形態は、GLP−1(7−35、36または37)/IP2/GLP−1(7−35、36または37)またはGLP−2の配列を含んでいる。融合ペプチドは、組み換え細胞において、または合成的に生産されていてもよいし、様々な病気または疾患(例えば、1型糖尿病または2型糖尿病、アポトーシスが関連する病気、または、神経変性疾患など)を治療するための薬物を調製するために使用していてもよい。
【解決手段】本発明は、GLP−1活性を有しており、インビボにおける安定性が向上している(特にジペプチジルペプチダーゼIVに対して耐性がある)融合ペプチドを提供する。前記融合ペプチドは、構成要素(I)として、N末端にGLP−1(7−35、7−36または7−37)の配列、および構成要素(II)として、C末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体もしくは誘導体を含んでいる。構成要素(II)は、完全または部分的なIP2(介在性ペプチド2)であることが好ましい。好ましい実施の形態は、GLP−1(7−35、36または37)/IP2/GLP−1(7−35、36または37)またはGLP−2の配列を含んでいる。融合ペプチドは、組み換え細胞において、または合成的に生産されていてもよいし、様々な病気または疾患(例えば、1型糖尿病または2型糖尿病、アポトーシスが関連する病気、または、神経変性疾患など)を治療するための薬物を調製するために使用していてもよい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、C末端が伸長した新規GLP−1融合ペプチドに関する。このGLP−1ペプチドは、(i)エンドペプチダーゼIVによる不活性化に耐性があり、(ii)形質転換された動物細胞中において高レベルで発現されていてもよく、(iii)例えば2型糖尿病の治療に有効である。
【背景技術】
【0002】
グルカゴン遺伝子は十分に研究されている(例えば、「White, J.W.ら, 1986 Nucleic Acid Res. 14(12) 4719-4730」を参照)。高分子量の前躯体分子としてのプレプログルカゴン分子は、膵臓のα細胞、および、空腸および大腸のL細胞において合成される。プレプログルカゴンは180アミノ酸長のプロホルモンであり、その配列は、グルカゴンに加えて、グルカゴン様ペプチド−1(GLP−1)およびグルカゴン様ペプチド−2(GLP−2)という関連した構造の2つの配列を含んでいる。プレプログルカゴン分子において、GLP−1およびGLP−2の間には、17アミノ酸ペプチド配列(厳密に言えば、15アミノ酸配列とC末端RRの切断部位とである)、介在性ペプチド2(IP2)が存在している。IP2配列(前躯体分子においてGLP−1およびGLP−2の間に位置している)は、通常は、GLP−1のaa37の後でタンパク質分解により切断される。したがって、プレプログルカゴンモジュールは、細胞および環境に応じて切断されて、GLP−1(1−37)(37アミノ酸であるGLP−1の未加工の形態)を含む様々なペプチドが生じる。一般的に、膵臓および腸において、この加工が行われる。さらにGLP(1−37)配列はタンパク質分解されて、31アミノ酸の加工された形態である活性型GLP−1(7−37)、または、GLP−1(7−36)アミドが生じる。したがって、GLP−1(7−37)の記載は、もとのペプチドであるGLP−1のN末端から数えたときに、該当する断片が7番から37番までのアミノ酸残基を含んでいる(なお、7番、37番のアミノ酸残基も含んでいる)、ということを示している。GLP−1(7−36)アミドおよびGLP−1(7−37)のアミノ酸配列は、一般式I(配列番号43):His−Ala−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Ser−Tyr−Leu−Glu−Gly−Gln−Ala−Ala−Lys−Glu−Phe−Ile−Ala−Trp−Leu−Val−Lys−Gly−Arg−X(I)(なお、XがNH2であるときにGLP−1(7−36)アミドを表し、XがGly−OHであるときにGLP−1(7−37)を表す)で表されている。
【0003】
GLP−1は、消化管ホルモンであり、β細胞においてアデニル酸シクラーゼ活性およびタンパク質キナーゼ活性を刺激するなどの作用がある効き目の強い内因性のインスリン分泌性の作用因子である。該GLP−1は、生理学的に、上部消化管からの胃抑制ポリペプチドと共に、血糖値を下げる内分泌物ホルモンとして機能する。したがって、食物の摂取に反応して分泌されるGLP−1は、例えば血液中に含まれる糖質を制限するために強調して働く胃、肝臓、膵臓および脳に対して複数の効果を与える。その結果、糖質代謝に対するGLP−1の強力な作用、および、糖尿病(2型糖尿病など)を治療するための潜在的な適用可能性のために、グルカゴン様ペプチドGLP−1(7−36)アミド、およびアミド化されていない類似体のGLP−1(7−37)は、興味を非常に引き寄せていた。インスリンが存在しているときに細胞が適切に反応しないので、2型糖尿病はインスリンへの耐性を示すという特徴がある。これは、1型糖尿病よりも複雑な問題である。症状は比較的に軽度(ケトアシドーシスではない)で、かつ散発性であったりするので、患者は、数年間、2型糖尿病であると診断されるまで気づかないことがある。しかしながら、2型糖尿病に気づかないために、腎不全、冠状動脈性心臓病を含む重症合併症が発症することになり、死亡率がますます増加する。
【0004】
GLP−1(7−36)アミドまたはGLP−1(7−37)は、血清において短命なペプチドである。該ペプチドは、ジペプチジルペプチダーゼIV(DPP−IV)により残基と残基9との間において切断される。切断されたペプチドは不活性である。したがって、体外から投与されたGLP−1は、非常に短命であり、治療への応用における実用性が限られている。
【0005】
天然に生じるGLP−1(GLP−1(7−37))の(DPP−IVに対して)安定な類似体を、合成できるような、様々な試みが行われている。特に、インビボにおいてAlaである残基8は、他の残基、例えばGly、Ser、またはThrに置き換えられている(Burcelin, R., ら (1999) Metabolism 48, 252-258)。Gly8またはG8の類似体を、両方とも合成された分子として広範囲にわたって検証し、突然変異体のポリペプチドを分泌するための遺伝子組み換え細胞株により生産した(Burcelin, R., ら (1999) Annals of the New York Academy of Sciences 875: 277-285)。GLP−1(7−37)の生物活性が失われることなく、インビボにおけるGLP−1(7−37)の安定性を高めるためにGLP−1(7−37)に、様々な他の修飾が導入されている。しかしながら、これらの全てのアプローチの結果、関係する多くの問題が原因で、顕著な治療上の有意性が少しも見られなかった。
【0006】
ソレセン,B(Thorens, B.)は、多量体のGLP−1を発現するカセットを作製するための方法を国際公開公報第9953064号パンフレットに公開した。該多量体のGLP−1を発現するカセットを、様々な種類の細胞の中に組み込むが可能である。前記様々なタイプの細胞は、一般的に利用できる不死化された細胞株および分裂性の初代培養細胞である。例えば、哺乳類のCNS(中枢神経系)に由来するEGF−応答性ニューロスフィア、bFGF−応答性神経前躯体幹細胞が挙げられる。また、上手くいった例においては、仔ハムスター腎臓由来のBHK細胞が用いられている。埋め込まれたトランスフェクションされた細胞は、グルコース調節を糖尿病ではないときのグルコース調節と実質的に同等になるように、糖尿病マウスを上手く治療するために使用されると考えられた。しかしながら、この技術は、糖尿病患者に対して定期的な投与が行われる治療の必要条件を満たさない。
【0007】
外因的にグルコース量を安定させるるための他のアプローチは、2型糖尿病の治療に関する研究のもとに、内分泌物模倣剤として公知である新しい分類の医薬に基づいている。エキセナチド(exenatide)(バイエッタ(Byetta)(登録商標))は、アメリカドクトカゲ(Gila monster lizard)の唾液において発見された天然化合物を合成したものである。臨床試験において、内分泌物模倣剤(エキセチナド)は、血液中に含まれる糖質を減少すること、および、β細胞の機能マーカーを向上させることを示した。しかしながら、エキセナチドは、ヒト内分泌物ホルモンのグルカゴン様ペプチド−1(GLP−1)のある程度の効果だけを示す。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開公報第9953064号パンフレット
【非特許文献】
【0009】
【非特許文献1】White, J.W.ら, 1986 Nucleic Acid Res. 14(12) 4719-4730
【非特許文献2】Burcelin, R., ら (1999) Metabolism 48, 252-258
【非特許文献3】Burcelin, R., ら (1999) Annals of the New York Academy of Sciences 875: 277-285
【発明の概要】
【発明が解決しようとする課題】
【0010】
要約すると、GLP−1に基づいて血糖値を下げることができる、2型糖尿病に対する利用可能な有効な治療が、現在のところ存在していない。言い換えれば、GLP−1に関して公知の有益な効果(例えば、(i)肥満被検体における胃内容物排出速度を短縮することにより、血液の循環の中に入る栄養物の浸入速度を強力に減らすような生理的濃度におけるGLP−1の活性、または、(ii)インスリンを刺激するGPL−1の活性)を全面にわたって反映する治療は提供されていない。従って、生物活性を有し、タンパク分解に対して耐性のあるペプチド分子に基づいたGLP−1を提供することが本発明の目的である。
【課題を解決するための手段】
【0011】
1.構成要素(I)として、N末端に一般式II
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Xaa16−Ser−Xaa18−Xaa19−Xaa20−Glu−Xaa22−Xaa23−Ala−Xaa25−Xaa26−Xaa27−Phe−Ile−Xaa3o−Trp−Leu−Xaa33−Xaa34−Xaa35−Xaa36−Xaa37、
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、3−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し(ここで、Xaa8はGlyを表すことが特に好ましい);
Xaa16は、ValまたはLeuを表し;
Xaaは、Ser、LysまたはArgを表し;
Xaa19は、TyrまたはGlnを表し;
Xaa20は、LeuまたはMetを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa25は、AlaまたはValを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa27は、GluまたはLeuを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa33は、ValまたはLysを表し;
Xaa34は、Lys、Glu、AsnまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、Gly、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、Pro、もしくはLys、またはアミドを表すか、あるいは存在していない)、あるいは、
一般式III
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Xaa8−Tyr−Leu−Glu−Xaa22−Xaa23−Ala−Ala−Xaa26−Glu−Phe−lle−Xaa30−Trp−Leu−Val−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し;
Xaa18は、Ser、LysまたはArgを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa34は、Lys、GluまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、もしくはLys、またはアミドを表すか、あるいは存在していない)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる融合ペプチド。
2.構成要素(I)として、N末端にGLP−1(7−35、7−36、または7−37)配列、および、
構成要素(II)として、C末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、1の融合ペプチド。
3.構成要素(I)として、N末端に一般式I
His−Ala−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Ser−Tyr−Leu−Glu−Gly−Gln−Ala−Ala−Lys−Glu−Phe−Ile−Ala−Trp−Leu−Val−Lys−Gly−Arg−X(I)
(式中、Xは、NH2またはGly−OHを表す)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、1の融合ペプチド。
4.構成要素(I)が、配列番号1と少なくとも80%の配列相同性を有する配列を含んでいる、2または3の融合ペプチド。
5.構成要素(II)が、βターン様構造を形成するペプチド配列である、1〜4の何れか1つの融合ペプチド。
6.構成要素(II)が、少なくとも1つのアラニン残基およびプロリン残基を含んでいるペプチド配列である、1〜5の何れか1つの融合ペプチド。
7.構成要素(II)が、βターンを形成する性質を有している4量体を、含んでいるペプチド配列であって、例えば該4量体の2位にプロリン残基を有している、1〜6の何れか1つの融合ペプチド。
8.構成要素(II)が、VAIA、IAEE、PEEV、AEEV、EELG、AAAA、AAVA、AALG、DFPE、AADX、AXDX、およびXADXから成る群から選ばれる配列モチーフを含んでいるペプチド配列であって、Xは任意のアミノ酸を表す、1〜7の何れか1つの融合ペプチド。
9.構成要素(II)が、AA、XA、AX、RR、RXおよびXRから成る群から選ばれる構成要素(II)のN末端配列モチーフにより、構成要素(I)のC末端と連結しているペプチド配列である、1〜8の何れか1つの融合ペプチド。
10.構成要素(II)が、配列番号25(DFPEEVA)の配列モチーフを含んでいるペプチド配列であるか、または、配列番号25と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜9の何れか1つの融合ペプチド。
11.構成要素(II)が、配列番号22(RRDFPEEVAI)および配列番号26(AADFPEEVAI)から成る群から選ばれる配列を含んでいるペプチド配列であるか、あるいは、配列番号22または配列番号26と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜10の何れか1つの融合ペプチド。
12.構成要素(II)が、配列番号23(RRDFPEEVAIVEEL)、配列番号24(RRDFPEEVAIAEEL)、配列番号27(AADFPEEVAIVEEL)、および配列番号28(AADFPEEVAIAEEL)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号23、24、27または28の何れかと少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜11の何れか1つの融合ペプチド。
13.構成要素(II)が、配列番号2(RRDFPEEVAIVEELG)、配列番号3(RRDFPEEVAIAEELG)、配列番号29(AADFPEEVAIVEELG)、および配列番号30(AADFPEEVAIAEELG)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号2、3、29または30と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜12の何れか1つの融合ペプチド。
14.構成要素(II)が、9−30アミノ酸、好ましくは9−20アミノ酸、および最も好ましくは9−15アミノ酸を有しているペプチド配列である、1〜13の何れか1つの融合ペプチド。
15.構成要素(I)および構成要素(II)が、直接連結されているか、または、リンカー配列を介して連結されている、1〜14の何れか1つの融合ペプチド。
16.前記リンカー配列が、1−10アミノ酸の長さを有している、15の融合ペプチド。
17.前記融合ペプチドが、
配列番号8(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELG)、
配列番号12(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELG)、
配列番号31(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIAEELG)、
配列番号32(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIAEELG)、
配列番号33(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIAEELG)、
配列番号34(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIAAALG)、
配列番号35(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFP)、
配列番号36(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVA)、
配列番号37(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELGRRHAC)、
配列番号38(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIVEELG)、
配列番号39(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIVEELG)、
配列番号40(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIVEELG)、
配列番号41(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIVAALG)、および
配列番号42(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELGRRHAC)
から成る群から選ばれる配列を含んでいるか、または、配列番号8、12と少なくとも80%の配列相同性を有する配列を含んでいる、1〜16の何れか1つの融合ペプチド。
18.前記融合ペプチドが、構成要素(II)のC末端、および/または、構成要素(I)のN末端と連結した他の構成要素(III)を含んでいる、1〜17の何れか1つの融合ペプチド。
19.構成要素(III)が、少なくとも4のアミノ酸残基、好ましくは少なくとも10の付加的なアミノ酸残基、より好ましくは、少なくとも20または少なくとも30の付加的なアミノ酸残基を含んでいる、18の融合ペプチド。
20.構成要素(III)が、プログルカゴンにおけるようなGLP−2のN末端配列、またはGLP−1(7−37)のN末端配列の、少なくとも4、好ましくは10、より好ましくは少なくとも20の付加的なアミノ酸残基を含んでいる、17または19の融合ペプチド。
21.構成要素(III)が、配列番号4または5の配列を含んでいるか、もしくは、配列番号4または5と少なくとも80%の配列相同性を有する配列を含んでいる、18〜20の何れか1つの融合ペプチド。
22.前記融合ペプチドが、配列番号6、7、10および11から成る群から選ばれるペプチド配列を含んでいるか、あるいは、配列番号6、7、10または11と少なくとも80%の配列相同性を有する配列を含んでいる、18〜21の何れか1つの融合ペプチド。
23.前記アミノ酸の少なくとも1つが、(i)天然に生じるアミノ酸の側鎖を共有結合修飾することにより誘導体化される、(ii)ペプチド骨格を修飾することにより誘導体化される、(iii)NH2末端基またはカルボキシ末端基を修飾することにより誘導体化される、1〜22の何れか1つの融合ペプチド。
24.前記アミノ酸の少なくとも1つが、リピル(lipyl)基または炭水化物基により誘導体化される、23の融合ペプチド。
25.GLP−1(GLP−1(7))のN末端のHis残基が、該GLP−1のNH2末端および/または該GLP−1のヒスチジル側鎖において、特に疎水性の部分によって化学的に修飾される、23または24の融合ペプチド。
26.前記融合ペプチドが、構成要素(IV)として、担体タンパク質、特にトランスフェリンまたはアルブミンを含んでいる、1〜25の何れか1つの融合ペプチド。
27.構成要素(I)、(II)および/または(III)のアミノ酸配列が反転されており、前記アミノ酸配列は、少なくとも部分的にDアミノ酸アイソマーから構成されている、1〜26の何れか1つの融合ペプチド。
28.固体状態のペプチド合成により、1〜27の何れか1つの融合ペプチドの製造方法。
29.1〜22または26の何れか1つの融合ペプチドをコードする核酸。
30.29の核酸を含んでいるベクター。
31.前記融合ペプチドを発現することができる、外因的に導入された29のDNA(特に30のベクター)を含んでいる宿主細胞。
32.融合ペプチドをコードする核酸を含有させるために形質転換した微生物を発酵させて、タンパク質を回収する、1〜22または26の何れか1つの融合ペプチドの製造方法。
33.動物細胞を、上記タンパク質が上記細胞から排出される条件下において成長させる、32のタンパク質の製造方法。
34.ヒトまたは動物の治療の場合に使用するための薬物としての、1〜27の何れか1つの融合ペプチド。
35.1型または2型糖尿病、インスリン耐性、体重異常および疾病、またはそれに関連する症状を治療するための薬物を製造するための、(i)1〜27の何れか1つの融合ペプチド、(ii)29の核酸、(iii)30のベクター、あるいは(iv)31の宿主細胞の使用。
36.神経変性疾患および神経変性病、またはそれらに関連する状態を治療するための薬物を製造するための、(i)1〜27の何れか1つの融合ペプチド、(ii)29の核酸、(iii)30のベクター、あるいは(iv)31の宿主細胞の使用。
37.アポトーシスに関係する疾患および病気、または状態を治療するための薬物を製造するための、(i)1〜27の何れか1つの融合ペプチド、(ii)29の核酸、(iii)30のベクター、あるいは(iv)31の宿主細胞の使用。
【図面の簡単な説明】
【0012】
【図1】GLP−1の各コンストラクトを示す図である。
【図2】hTERT−MSC細胞およびHEK293細胞に一過的にトランスフェクションした後の活性型GLP1を示す図である。
【図3】哺乳類細胞から分泌されたGLP−1ペプチドのウェスタンブロット解析を示す図である。
【図4】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図5】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図6】GLP−1ペプチドのウェスタンブロット解析を示す図である。
【図7】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図8】インビトロにおけるバイオアッセイ(環状AMPの産生)を示す図である。
【図9】db/dbマウスの絶食時の相対的グルコース量を示す図である。
【図10】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図11】AはT細胞の想起反応アッセイの結果を示す図であり、Bは動物の免疫血清における抗原特異的IgG抗体の力価を示す図である。
【図12】AはGLP−1の用量反応曲線を示す図であり、BはCM1の用量反応曲線を示す図であり、CはCM3の用量反応曲線を示す図であり、DはCM3−ANA01の用量反応曲線を示す図である。
【図13】Aはdb/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図であり、Bは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
【図14】122日に及ぶ処置の間における、db/dbマウスの1週間の飼料消費に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
【図15】Aは非絶食時の血糖値を示す図であり、Bは注射から1時間後の対照に対する血糖の減少を示す図であり、Cは絶食時の血糖値を示す図である。
【図16】Aは腹腔内注射によるグルコース負荷テストを示す図であり、Bは腹腔内注射によるグルコース負荷テスト(相対値)を示す図である。
【図17】0.9%生理食塩水(グループA)、CM1ペプチド(グループB)、CM3−ANA01ペプチド(グループC)、またはエキセンディン−4(グループD)を用いて18週間処置した後の、マウスの血清中インスリン量に関するデータを示す図である。
【図18】Aは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて6、12および18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図であり、Bは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。
【図19】0.9%生理食塩水(A、n=11)、CM1(B、n=12)、CM3−ANA01(C、n=12)、またはエキセンディン(D、n=12)を用いて、18週間処置した後の動物を乱切した日に決定された膵臓の重量示す図である。
【図20】異なる濃度のテスト物質であるCM1(A)およびCM3−ANA01(B)、ならびに参照物質であるエキセンディン−4(C)に対する、インビトロにおける脾臓細胞の想起反応を示す図である。
【発明を実施するための形態】
【0013】
本発明は、構成要素(I)として、N末端にGLP−1(7−35、7−36または7−37)配列、ならびに構成要素(II)としてC末端に、少なくとも9つのアミノ酸のペプチド配列、またはその機能的な断片、変異体もしくは誘導体を含んでいる融合ペプチドに関する。
【0014】
本発明は、生じた本発明のペプチドが、タンパク質分解性のエンドペプチダーゼIVの活性が主に原因である、生体内におけるタンパク質分解から保護されるという知見に基づく。少なくとも2つの構成要素(I)および(II)を有する本発明のペプチドは、GLP−1の生物活性を示し、かつC末端の延長によって構成要素(I)としてのGPL−1に安定性を付与する。
【0015】
本明細書において用いられる「本発明のペプチド」という用語は、本明細書において定義されるような融合ペプチド、その変異体、類似体、断片または誘導体(例えば、融合ペプチドの誘導体化された断片、類似体、または変異体の組み合わせが含まれる)のことである。本明細書において用いられる「GLP−1ペプチド」という用語は、GLP−1(7−35、36または37)を意味しており、一方において、「修飾されたGLP−1ペプチド」は、本発明のペプチドの構成要素(I)および(III)のいずれか一方に生じ得る、GLP−1(7−35、36または37)の誘導体化された断片、類似体、変異体を含む、任意のGLP−1類似体、GLP−1誘導体、GLP−1変異体またはGLP−1断片を意味することが意図される。本明細書において用いられる「GLP−2ペプチド」という用語は、GLP−2(1−33、34または35)を意味しており、その一方で、「修飾されたGLP−2ペプチド」は、GLP−2(1−33、34または35)の誘導体化された断片、類似体または変異体を含む、任意のGLP−2類似体、断片、変異体、GLP−2誘導体またはGLP−2類似体の誘導体を意味することが意図される。変異体、類似体、断片および誘導体は、修飾されていない配列(例えば、GLP−1(7−35、36または37)またはGLP−2(1−33、34または35))の修飾物として分類される。本発明の目的の範囲内において、任意の変異体、類似体、断片または誘導体は、機能的でなければならない(例えば、修飾されていないGLP−1と同じまたは類似の生物学的効果を及ぼさなければならない)。
【0016】
本発明のペプチドは、融合ペプチド、またはその変異体、類似体、断片もしくは誘導体であることが好ましい。ここで、構成要素(I)は、配列番号1と少なくとも80%、より好ましくは少なくとも85%、さらに好ましくは少なくとも90%の配列相同性を有している配列を含んでいる。配列番号1は、哺乳類において厳密に保存されている、GLP−1(7−37)の天然アミノ酸配列(31アミノ酸の長さ)を表している。
【0017】
本発明の融合ペプチド(またはより広く、融合ペプチドの類似体、断片、変異体もしくは誘導体を含む任意の本発明のペプチド)の第2の構成要素(構成要素(II))は、典型的に、βターン様構造を形成してもよりし、またはしていなくてもよい、少なくとも9アミノ酸の長さの配列を含んでいる。βターン様構造は、タンパク質またはペプチドの典型的な2次構造要素である。βターン様構造は、典型的に、ペプチドまたはタンパク質の中心鎖の向かう方向が逆戻りしている、4アミノ酸によって形成されている。構成要素(II)のアミノ酸配列は、少なくとも9つのアミノ酸を含んでおり、かつ好ましくはその配列の中に少なくとも1つのプロリンまたはアラニンを含んでいる。プロリン残基は、βターンを形成する4量体のアミノ酸配列に共通して含まれるアミノ酸である。プロリン残基は、融合ペプチドの構成要素(II)に存在する4量体のβターン配列の2または3位、好ましくは2位に代表的に配置されていることが共通している。
【0018】
本発明の融合物は、通常、構成要素(II)において、9〜30、好ましくは9〜20、さらに最も好ましくは9〜15アミノ酸の配列長を有している。一般的にいえば、構成要素(II)における配列が短いほど好ましい。というのも、より長い配列と比べてGLP受容体に対する結合活性がより優れているためである。必須の条件ではないが、構成要素(II)の配列は、中性であることが好ましく、またはpH7において負電荷を有していてもよい。
【0019】
本発明の融合タンパク質は、構成要素(II)がVAIA、IAEE、PEEV、AEEV、EELG、AAAA、AAVA、AALG、DFPE、AADX、AXDXおよびXADX(ここで、Xは、任意のアミノ酸(天然に生じるまたは修飾された天然ではないアミノ酸)を表す)から成る群から選択された配列モチーフを含んでいれば、好ましい。これらの4量体のモチーフは、構成要素(II)におけるあらゆる位置に配置されていてもよい。特に好ましい実施形態において、本発明の融合ペプチドの構成要素(II)は、AA、XA、AX、RR、RXおよびXR(ここで、Xは、任意のアミノ酸(天然に生じるまたは修飾された天然ではないアミノ酸)を表す)から成る群から選択された構成要素(II)のN末端配列モチーフにより、構成要素(I)のC末端と連結しているペプチド配列である。
【0020】
本発明の融合ペプチドにおける構成要素(II)の好ましいモチーフは、ヒトまたはマウスのIP−2の部分配列に対応する配列番号25の配列モチーフ(DFPEEVA)含んでいるか、または配列モチーフDFPEEVAと少なくとも80%の配列相同性を有する配列を含んでいる。
【0021】
融合ペプチドとしては、構成要素(II)が、配列番号22(RRDFPEEVAI)もしくは配列番号26(AADFPEEVAI)に記載の配列(すべてのペプチド配列は、一文字表記によって示されている)、または配列番号22もしくは26と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列であることが特に好ましい。配列番号22は、長さ15アミノ酸である全長IP−2配列におけるN末端側の10アミノ酸を含む、全長(ヒトまたはマウスの)IP−2配列(介在性ペプチド2)の部分配列である。配列番号26は、N末端の残基(RR)を(AA)に置換することによって配列番号22から誘導されている。IP−2の好ましい例は、βターンを含んでいるペプチド配列である。従って、構成要素(II)に含まれる他の特に好ましい配列は、ヒトに存在するN末端の14アミノ酸配列(配列番号23(RRDEPEEVAIVEEL))、もしくはマウスにおける等価物(配列番号24(RRDFPEEVAIAEEL))、または配列番号23もしくは24と少なくとも80%の配列相同性を有する配列のような、より長いIP−2の部分アミノ酸配列である。融合ペプチドの構成要素(II)に含まれる最も好ましい要素は、天然に生じるIP−2配列の15アミノ酸をすべて有する全長IP−2配列(配列番号2(RRDFPEEVAIVEELG)のヒト、または配列番号3(RRDFPEEVAIAEELG)のマウス)、または配列番号2もしくは3と少なくとも80%の配列相同性を有する配列である。IP2の哺乳類におけるアイソフォームのすべて(哺乳類の間におけるIP2の天然変異体)もまた本発明の範囲に含まれる。この段落において述べた配列のすべては、天然に生じるN末端のモチーフ(RR)の代わりに、モチーフ(AA)、(AX)または(XA)を備えていてもよい(例えば、配列番号27(AADFPEEVAIVEEL)、配列番号28(AADFPEEVAIAEEL)、配列番号29(AADFPEEVAIVEELG)および配列番号30(AADFPEEVAIAEELG))。構成要素(II)に含まれる配列の1よりも多い複製物(コピー)が含まれていてもよい(例えば、IP2、またはIP2断片、IP2変異体もしくは類似体、または誘導体の2、3またはそれ以上の複製物)。
【0022】
従って、本発明のペプチドは、配列番号8に記載の配列(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELG)(すなわち、マウスIP2に対してC末端を介して任意のリンカー配列なしで連結されたGLP−1(7−37))、または、配列番号12に記載の配列(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELG)(すなわち、ヒトIP2に対して任意のリンカー配列なしでC末端を介して連結されたGLP−1(7−37))を含んでいることが好ましい。本発明のペプチドとしてさらに好ましい配列番号8および12の発明の変異体は、配列番号31(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIAEELG)、配列番号32(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIAEELG)、配列番号33(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIAEELG)、配列番号34(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIAAALG)、配列番号35(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFP)、配列番号36(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVA)、配列番号37(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELGRRHAC)、配列番号38(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIVEELG)、配列番号39(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIVEELG)、配列番号40(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIVEELG)、配列番号41(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIVAALG)および配列番42(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELGRRHAC)(すなわち、IP2配列の特定の類似体または変異体に対してC末端を介して任意のリンカー配列なしで連結されたGLP−1(7−37))である。配列番号8および12と少なくとも80%の配列相同性を有するこれらの変異体、類似体もしくは断片、またはこれらの誘導体が同様に含まれ、またそれらは好ましい。
【0023】
あらゆる理論に制限されることなく、これらを必要とするあらゆる患者に投与されるときに、GLP−1(7−35、36または37)の不安定性は非保護の3次元構造に起因していることが、本発明の発明者らによって結論されている。プロテアーゼは、インビボにおいてGLP−1(7−35、36または37)ペプチドを切断し、かつ急速にその生理活性を消失させることがある。GLP−1(7−35、36または37)のC末端にペプチド配列を連結することによって、その構造物は、酵素による分解に対する安定性を増す。構成要素(II)への強剛性の提供、および1次構造によって形成されるβターンの存在に起因して、付加的なC末端のペプチド配列(本発明の融合ペプチドの構成要素(II)に含まれている)が折り返されるならば、安定性の増強は向上すると思われる。しかし、本発明のペプチドの構成要素(II)におけるβターン構造は、酵素による分解に対する構成要素(I)のGLP−1配列の安定化に必須であるとは、思われない。C末端のペプチドの伸長部分(例えば、βターン構造要素を含む)の長所によって本発明のペプチドは、DPP−IVの不活性化に対する耐性の向上が見られる。C末端のペプチドは、標的細胞にある受容体に作用する前にGLP−1(7−35、36または37)配列から切断されない、またはインビボにおいて酵素的に切断されてGLP−1(7−35、36または37)を形成する、の内のいずれか一方であってもよい。GLP−1受容体の部位と結合した本発明のペプチドの正確な形成に関係なく、本発明のペプチドは、活性なインシュリン分泌性の化合物としてその機能を発揮する。
【0024】
βターン要素を形成する1次構造のおかげで、構成要素(II)に含まれることが適すると思われるペプチド配列は、当該分野における当業者によって公知の適切な(例えば、(例えば、円偏光2色性の)分光法)、または他の方法によって容易に決定され得る。
【0025】
構成要素(II)および構成要素(I)は、直接に連結されていても、またはリンカー配列を介して連結されていてもよい。両方の構成要素は、互いに直接に連結されていることが好ましい。両方の構成要素がリンカー(またはスペーサ)を介して連結されている場合に、当該リンカーは、ペプチドリンカーまたは有機リンカーであることが好ましい。ペプチドリンカーは、典型的に1〜10アミノ酸、好ましくは1〜5、より好ましくは1〜3のアミノ酸の長さを有しており、ある場合において、当該ペプチドリンカーは、11〜50アミノ酸を含むより長いものであってもよい。ペプチドリンカーは、種々のアミノ酸配列から構成されていてもよい。ペプチドリンカーにより、連結される構成要素の間に構造的な自由度がもたらされる。構造的な自由度は、例えば、リンカー配列内に種々のグリシンまたはプロリン残基、好ましくは少なくとも30%、より好ましくは少なくとも40%、さらに好ましくは60%のプロリンおよびグリシン残基を含むペプチドリンカーを有することによって達成される。特定の配列とは関係なく、当該ペプチドリンカーは、免疫学的に不活性であることが好ましい。
【0026】
本発明の好ましい実施形態において、本発明のペプチド(例えば、融合ペプチド、またはその類似体、断片、変異体もしくは誘導体)は、構成要素(II)のC末端、および/または構成要素(I)のN末端のいずれかと連結されている、第3の構成要素(III)を含んでいる。構成要素(III)は、構成要素(II)のC末端に配置されていることが好ましい。構成要素(III)が(そのC末端によって)構成要素(I)のN末端または(そのN末端によって)構成要素(II)のC末端と連結されているか否かに関わらず、その結合は、直接的またはリンカー配列を介して間接的であってもよい。上記リンカー配列に関して、構成要素(I)および構成要素(II)と接続しているリンカーとして上述において開示されているものが好ましい。通常、構成要素(III)は、少なくとも4アミノ酸残基、好ましくは少なくとも10アミノ酸残基、より好ましくは少なくとも20または少なくとも30アミノ酸残基を含んでいる。機能的な観点から、構成要素(III)が備えられることによって、本発明のペプチドの安定性がさらに増強する。構成要素(III)は、GLP−1(7−37)の生物活性にほぼ類似する、本発明のペプチドの生物学的な機能を損なわないことが期待される。
【0027】
本発明のペプチドの構成要素(III)は、任意の哺乳生物のGLP−2のアイソフォーム((哺乳類において他の天然に生じるGLP−2のアイソフォーム)、例えば、配列番号4および5に示されているようなマウスまたはヒトのアイソフォーム)のN末端配列における、少なくとも4、好ましくは少なくとも10、より好ましくは少なくとも20の付加的なアミノ酸残基を包含することが好ましい。GLP−2は、プログルカゴンにおいて生じ、かつまた炭水化物代謝に関与する。構成要素(I)(GLP−1ペプチド)に含まれる生物学的に活性な配列と同様に、構成要素(III)もまた、GLP−2の天然に生じる形態の類似体、変異体または誘導体を含んでいてもよい。また、構成要素(III)は、GLP−1(7−37)(なお、これに対応する、すべての哺乳類のアイソフォーム、または本明細書に開示されているような、それらのすべての機能的な変異体、類似体もしくは誘導体も含まれる)のN末端配列の少なくとも4、好ましくは10、より好ましくは20の付加的なアミノ酸残基を含んでいてもよい。一般的に言えば、構成要素(III)は、本発明のペプチドの構成要素(I)に好適なものとして本明細書に開示されている、GLP−1ペプチド、または修飾されたGLP−1ペプチドの任意の形態を含んでいてもよい。さらに、構成要素(III)はまた、GLP−1(7−37)とGLP−2とのキメラ形態を含んでいてもよい。キメラ形態は、GLP−1(7−37)とGLP−2と(または両方の断片、類似体、変異体もしくは誘導体と)を互いに連結させて、構成要素(III)としてのキメラ形態を本発明のペプチドに導入することによって、生成されていてもよい。上記キメラ形態は、互いに連結されたGLP−1(7−37)の部分配列およびGLP−2の部分配列から構成されることが好ましい。例えば、上記キメラ形態は、GLP−1のN末端における5〜30アミノ酸とGLP−2のC末端における5〜30アミノ酸とを含んでいてもよいし、その逆であってもよい(例えば、GLP−1(7−37)の7または8〜22、23、24、25、26、27または28アミノ酸と、GLP−2の15、16、17、18、19、20、21、22、23または24位から例えば、C末端までのアミノ酸)。
【0028】
GLP−2またはGLP−1(7−37)の天然に生じる形態の修飾物のそれぞれが、構成要素(III)として使用されるならば、構成要素(III)は、配列番号4もしくは5、または配列番号1の配列のそれぞれ、あるいは配列番号4もしくは5、または配列番号1と少なくとも80%の配列相同性を有する配列を含むことが好ましい。また、例えば、側鎖の修飾またはペプチド骨格の修飾などの結果である(ここで開示されている「誘導体」に属するような)、これらの好ましい配列の誘導体は、本発明による構成要素(III)として包含される。
【0029】
他の実施形態において、構成要素(III)は、上述のような配列を複数含んでいてもよい。例えば、構成要素(III)は、GLP−1(7−37)および/またはGLP−2の少なくとも2、好ましくは2、3、または4の複製物、または配列番号1、4または5と少なくとも80%の配列相同性を有する配列の少なくとも2つの複製物を含んでいてもよい。また、構成要素(III)は、例えば、GLP−1(7−37)および/またはGLP−2あるいは少なくとも80%の配列相同性を有するその修飾物とともにキメラ型の組み合わせを最終的に形成する、上述したような、GLP−1(7−37)またはGLP−2のキメラ型の複製物以外も含んでいてもよい。また、例えば、(1)そのN末端によって構成要素(II)のC末端と、および(2)そのC末端によって構成要素(I)のN末端と、リンカーを介してまたは直接に連結されている2つ以上の、好ましくは2つの構成要素(III)は、本発明の範囲内にある。2つの構成要素(III)が備えられている場合には、これらは、同一であっても、異なっていてもよい。
【0030】
従って、3つの構成要素(I)、(II)および(III)を含む発明の融合ペプチドは、特に好ましい。これらの構成要素のすべてを含んでいる4つの特定の実施形態は、配列番号6(N−GLP−1(7−37)−IP2(マウス)−RR−GLP−1(7−37)−C、またここではマウスCM1を表している)、配列番号7(N−GLP−1(7−37)−IP2(マウス)−RR−GLP−2−C、またここではマウスCM2を表している)、配列番号10(N−GLP−1(7−37)−IP2(ヒト)−RR−GLP−1(7−37)−C、またヒトCM1を表している)および配列番号11(N−GLP−1(7−37)−IP2(ヒト)−RR−GLP−2−C、またヒトCM2を表している)、または配列番号6、7、10もしくは11と少なくとも80%の配列相同性を有する配列、またはこれらの誘導体から成る群から選択される。配列6、7、10および11のすべては、IP2(構成要素(II))のC末端にRR−リンカー(2つのアルギニン残基)を含んでいる。なお、このRR−リンカーは、代替案においてはなくてもよい。配列番号6、7、10または11の各実施形態における構成要素(I)は、GLP−1(7−37)であり、(構成要素(II)のC末端に連結されたこれらの各実施形態において)構成要素(III)は、GLP−1(7−37)またはGLP−2である。
【0031】
本発明のペプチドは、種々の修飾された形態を生じてもよい。これらの修飾された形態は、以下に開示され、より詳細に説明されている。
【0032】
本明細書において、「塩」という用語は、上述した融合ペプチド、またはその類似体、断片、誘導体もしくは変異体のカルボキシル基の塩およびアミノ基の酸付加塩の両方を意味する。カルボキシル基の塩は、当業者に公知の方法によって形成されてもよく、例えば、ナトリウム、カルシウム、アンモニウム、鉄、亜鉛の塩などの無機塩、ならびに例えば、エタノールアミン、アルギニンもしくはリジンのようなアミン、ピペリジンおよびプロカインなどを用いて形成されるような有機塩基を有する塩を含む。酸付加塩は、例えば、塩酸または硫酸のような例えば、無機酸を有する塩、ならびに例えば、酢酸またはシュウ酸のような有機酸を有する塩を含む。もちろん、該当するあらゆる塩は、本発明に適した本発明のペプチドの生物活性(例えば、血液の循環に入る栄養物の割合を減少させる能力)を保持する必要がある。ペプチド塩の形態は、薬学的製剤に含まれていてもよい。
【0033】
本発明の融合ペプチドの「断片」は、所望の生物活性を保持しているより短いペプチドである、分子の任意の部分集合を意味する。断片は、上記分子のいずれかの末端からアミノ酸を取り除き、生じたものをインクレチンのようなその特性に関して試験することによって容易に調製され得る。ポリペプチドのN末端および/またはC末端のいずれかから同時に1つのアミノ酸を取り除くプロテアーゼは、公知であり、このため、所望の生物活性を保持する断片の決定は、決まりきった実験のみを含んでいる。結論として、断片は、ペプチド末端におけるアミノ酸および/またはペプチド配列内にあるアミノ酸の欠損に原因し得る。
【0034】
さらに、自身が融合ペプチド、類似体、変異体、機能的な誘導体および/またはこれらの断片である、抗2型糖尿病活性を有する本発明のペプチドは、本発明のペプチドに隣接する付加的なアミノ酸残基をまた含むことができる。生じた分子がプロテアーゼに対する耐性または安定性を保持し、かつ当該分子がインクレチンとして作用する能力を保持する限りにおいて、任意の当該隣接する残基が、核になるペプチドの基本的なおよび新規な性質に影響するか否かを、例えば、決まりきった実験を用いた膵臓細胞に対する影響によって、決定することができる。「本質的に成る」という用語は、特定の配列に言及するとき、付加的な隣接する残基が特定の本発明のペプチドが有する基本的なおよび新規の性質に影響せずに存在し得ることを意味する。この用語は、特定の配列内における置換、欠損または付加を包含していない。
【0035】
本発明の「変異体」という用語は、上述した本発明のペプチドの全体またはその断片のいずれかと実質的に類似している分子を意味している。変異体ペプチドは、当該分野において公知の方法を用いた、変異体ペプチドの直接的な化学合成によって簡便に調製され得る。もちろん、本発明のペプチドの当該変異体は、類似の糖尿病性(例えば、対応する天然に生じるGLP−1ペプチドのようなインシュリン刺激活性)を有している。
【0036】
また、上述したペプチドのアミノ酸配列変異体は、合成された誘導体をコードするDNAにおける変異によって調製され得る。当該変異体は、例えば、アミノ酸残基からの欠損体、アミノ酸残基への挿入体、またはアミノ酸残基の置換体を含む。また、欠損、挿入および置換のあらゆる組み合わせにより、最終的なコンストラクトを完成させてもよいが、この場合、最終的なコンストラクトは所望の活性を保有している。当然、変異体ペプチドをコードするDNAに施される変異は、読み枠を変更する必要はなく、かつmRNAの2次構造の生成を可能にする相補的な領域を作り出さないことが好ましい。
【0037】
本発明の上述したペプチドの「類似体」は、分子の全体またはこれらの活性な断片のいずれかと実質的に類似の非天然分子を意味する。当該類似体は、対応する天然に生じるGLP−1ペプチドと同じ活性を示す。
【0038】
本発明に係る、本発明のペプチドに属して作製される置換の種類は、異なる種の相同なタンパク質/ペプチドの間におけるアミノ酸変化の頻度の解析に基づいていてもよい。当該解析に基づくと、保存された置換は、以下の5つのグループ内の1つの中における交換として本明細書では定義され得る:
I.小さな、脂肪族の、非極性のまたはわずかに極性の残基(Ala、Ser、Thr、ProおよびGly);
II.極性の、負電荷の残基(Asp、Asn、GluおよびGln)およびそれらのアミド;
III.極性の、正電荷の残基(His、ArgおよびLys);
IV.大きい、脂肪族の非極性の残基(Met、Leu、Ile、ValおよびCys)、ならびに;
V.大きな芳香族の残基(Phe、TryおよびTrp)。
【0039】
上記グループにおいて、下記の置換が、「保存性が高い」と考えられる:Asp/Gul;His/Arg/Lys;Phe/Tyr/Trp;Met/Leu/Ile/Val。保存性が中程度である置換は、上記(I)、(II)、および(III)を含んでいるスーパーグループ(A)または、上記(IV)および(V)を含んでいるスーパーグループ(B)に限定される、グループ(I)−(IV)のうちの2つの間において、交換されるように定義される。置換は、遺伝的にコードされるアミノ酸、または天然に生じるアミノ酸に限定されない。
【0040】
通常、本発明のペプチドの類似体または変異体はまた、例えば、可溶性の向上(親水性アミノ酸による疎水性アミノ酸の置換)を目的として作製されたアミノ酸置換体を含む。本発明のペプチドであるGLP−1ペプチドの(本発明のペプチドの構成要素(I)および/または(III)において生じる)変異体/類似体の一実施形態において、(修飾された)GLP−1ペプチドは、GLP−1ペプチドの7、8、11、12、16、22、23、24、25、27、30、33、34、35、36または37位における1つ以上の置換を特徴としている。例えば、以下の名称のように、[Arg34−GLP−1(7−37)]は、天然に生じる34位のリジンがアルギニンにより置換されているGLP−1類似体を示している。
【0041】
特に、本発明のペプチドの構成要素(I)および/または(III)は、例えば、Gln9−GLP−1(7−37)、D−Gln9−GLP−1(7−37)、アセチル−Lys9−GLP−1(7−37)、Thr16−Lys18−GLP−1(7−37)、ならびにLys18−GLP−1(7−37)、Arg34−GLP−1(7−37)、Lys38−Arg26−GLP−1(7−38)−OH、Lys36−Arg26−GLP−1(7−36)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26−Lys38−GLP−1(7−38)、Arg26−Lys38−GLP−1(7−38)、Arg26−Lys38−GLP−1(7−38)、Arg34−Lys38−GLP−1(7−38)、Ala37−Lys38−GLP−1(7−38)、ならびにLys37−GLP−1(7−37)を含むGLP−1(7−35、36または37)の変異体および類似体を含んでいてもよい。
【0042】
本発明の他の実施形態において、本発明のペプチドは、構成要素(I)または(III)として、以下の一般式II(配列番号44):
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Xaa16−Ser−Xaa18−Xaa19−Xaa20−Glu−Xaa22−Xaa23−Ala−Xaa25−Xaa26−Xaa27−Phe−Ile−Xaa3o−Trp−Leu−Xaa33−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノヒスチジン、2−アミノヒスチジン、3−ヒドロキシヒスチジン、ホモヒスチジン、N−アセチルヒスチジン、a−フルオロメチルヒスチジン、a−メチルヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸または(1−アミノシクロオクチル)カルボン酸を表し(ここで、Xaa8はGlyを表すことが特に好ましい);Xaa16は、ValまたはLeuを表し;Xaaは、Ser、LysまたはArgを表し;Xaa19は、TyrまたはGlnを表し;Xaa20は、LeuまたはMetを表し;Xaa22は、Gly、GluまたはAibを表し;Xaa23は、Gln、Glu、LysまたはArgを表し;Xaa25は、AlaまたはValを表し;Xaa26は、Lys、GluまたはArgを表し;Xaa27は、GluまたはLeuを表し;Xaa30は、Ala、GluまたはArgを表し;Xaa33は、ValまたはLysを表し;Xaa34は、Lys、Glu、AsnまたはArgを表し;Xaa35は、GlyまたはAibを表し;Xaa36は、Arg、GlyもしくはLys、またはアミドを表すか、あるいは存在せず;ならびにXaa37は、Gly、Ala、Glu、Pro、もしくはLys、またはアミドを表すか、あるいは存在しない。)
のアミノ酸配列を含んでいる修飾されたGLP−1を含んでいる。
【0043】
本発明の他の実施形態において、本発明のペプチドの構成要素(I)および/または(III)は、以下の一般式式III(配列番号45):
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Xaa8−Tyr−Leu−Glu−Xaa22−Xaa23−Ala−Ala−Xaa26−Glu−Phe−lle−Xaa30−Trp−Leu−Val−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノヒスチジン、2−アミノヒスチジン、−ヒドロキシヒスチジン、ホモヒスチジン、N−アセチルヒスチジン、a−フルオロメチルヒスチジン、a−メチルヒスチジン、3−ピリジルアラニン、2−ピリジルアラニンまたは4−ピリジルアラニンを表し; Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸または(1−アミノシクロオクチル)カルボン酸を表し; Xaa18は、Ser、LysまたはArgを表し; Xaa22は、Gly、GluまたはAibを表し; Xaa23は、Gln、Glu、LysまたはArgを表し; Xaa26は、Lys、GluまたはArgを表し;Xaa30は、Ala、GluまたはArgを表し; Xaa34は、Lys、GluまたはArgを表し;Xaa35は、GlyまたはAibを表し; Xaa36は、ArgもしくはLys、またはアミドを表すか、あるいは存在せず; Xaa37は、Gly、Ala、GluもしくはLys、またはアミドを表すか、あるいは存在していない。)
のアミノ酸配列を含んでいる修飾されたGLP−1ペプチドを含んでいる。
【0044】
本発明の特に好ましい実施形態において、本発明のペプチドの構成要素(I)および/または(III)は、GLP−1(7−35)、GLP−1(7−36)、GLP−1(7−36)−アミド、GLP−1(7−37)またはこれらの変異体、類似体もしくは誘導体から選択される(修飾された)GLP−1ペプチドを含んでいる。また、本発明のペプチドは、構成要素(I)および/または(III)に、GLP−1ペプチドの8位にAib残基を、あるいはGLP−1ペプチドの7位にD−ヒスチジン、脱アミノヒスチジン、2−アミノヒスチジン、ヒドロキシヒスチジン、ホモヒスチジン、N−アセチルヒスチジン、a−フルオロメチルヒスチジン、a−メチルヒスチジン、3−ピリジルアラニン、2−ピリジルアラニンおよび4−ピリジルアラニンから成る群から選択される1つのアミノ酸残基を有する修飾されたGLP−1ペプチドを含んでいることが好ましい。
【0045】
本発明に使用する類似体を得るために使用され得るタンパク質におけるアミノ酸の置換体の生成例は、Markらによる米国再発行特許発明第33.653号明細書、米国特許第4,959,314号明細書、米国特許第4,588,585号明細書および米国特許第4,737,462号明細書、Kothsらによる米国特許第5.116.943号明細書、Namenらによる米国特許第4.965.195号明細書ならびにLeeらによる米国特許第5.017.691号明細書に示されているような、あらゆる周知の方法段階を含み、かつ米国特許第4.904.584号明細書(Shawら)に示されているリジンに置換されたタンパク質を含む。
【0046】
好ましくは、上述の、かつ構成要素 (I)、(II)および/または(III)に含まれる変異体または類似体は、「天然の」配列(例えば、天然アミノ酸配列と少なくとも70%の相同性を有し、かつこれらの生物活性を維持する、GLP−1(7−37)、GLP−2もしくはこれらの生物学的に活性な断片またはあらゆるIP2アイソフォーム)と同じである核となる配列を有する。さらに好ましくは、当該配列は、天然の配列と少なくとも80%の相同性、少なくとも90%の相同性、最も好ましくは少なくとも95%の相同性を有する。ここで、特定のペプチドは、決まった長さの参照ポリペプチドと特定の割合の相同性を有し、その相同性の割合は、参照ペプチドに対する比である。従って、100アミノ酸の長さである参照ポリペプチドと50%の相同性であるペプチドは、参照ポリペプチドにおける50アミノ酸の長さの部分と完全に一致している。また、その全長に渡って参照ポリペプチドと50%の相同性であるペプチドは、100アミノ酸の長さのポリペプチドである。当然、他のポリペプチドは、同じ判断基準に合致する。本明細書で使用されるような、「配列相同性」という用語は、当該配列が以下のように比較されることを意味している。当該配列は、遺伝的な演算群のGAP(Genetic Computing Group’s global alignment program)の様式9を用いて整列される。このGAPの様式9では、間隔が空く(間隔の初めの空白に対する)罰則(−12)、および間隔が伸びる(間隔における付加的な連続する空白につき)罰則(−4)を用いる、初期(BLOSUM62)のマトリックス(−4〜+11の値)が使用される。整列の後に、要求した配列におけるアミノ酸数の百分率として合致した数を表すことによって、相同性の百分率が演算される。
【0047】
また、融合ペプチドまたはその類似体、断片もしくは変異体の誘導体は、本発明に包含される。本発明のペプチドの「誘導体」という用語は、1つのアミノ酸が別の20の一般的に生じる天然アミノ酸に変化されていない、上記修飾された本発明のペプチドのみを意味している。その結果、遺伝的にコードされているアミノ酸は、(i)選択された残基の側鎖、または末端残基のアミノ基またはカルボキシル基と(好ましくは共有結合修飾することによって)反応可能な有機誘導試薬と、遺伝的にコードされているアミノ酸を反応させることによって、あるいは、(ii)非天然アミノ酸または遺伝暗号にコードされていない天然アミノ酸(例えば、ヒドロキシプロリン、γ−カルボキシグルタミン酸、オルチニン、ホスホセリン、D−アラニン、およびD−グルタミン)の導入によって、あるいは、(iii)別のペプチド骨格配列によるペプチド骨格の修飾によって修飾されていてもよい。上記非天然アミノ酸は、化学合成によって作製されたものであり、すなわち、遺伝暗号によりコードされるアミノ酸のD−アイソマー、Aib(a−アミノイソブチル酸)、Abu(a−アミノブチル酸)、Tie(tert−ブチルグリシン)、p―アイアニン、3−アミノメチル安息香酸、およびアントラニル酸である。
【0048】
以下において、例えば、構成要素 (I)、(II)および/または(III)に配置されている本発明のペプチドの任意の部位(任意のアミノ酸)に生じ得る、本発明のペプチド(上述のように、融合ペプチドの変異体、類似体、または断片を含む)におけるアミノ酸の好ましい修飾が、開示されている。
【0049】
本発明のペプチドの任意の形態(例えば、本発明のペプチドの類似体)においてシステイニル残基が存在するならば、システイン残基は、クロロ酢酸またはクロロ酢酸アミドのようなアルファハロ酢酸塩(および対応するアミン)とほとんど普遍的に反応し、カルボキシルメチルまたはカルボキシアミドメチル誘導体が得られる。また、システイニル残基は、ブロモトリフルオロアセトン、アルファ−ブロモ−ベータ−(5−イミダゾイル)プロピオン酸、クロロ酢酸リン酸塩、N−アルキルマレイミド、3−ニトロ−2−ピリジル二硫化物、メチル−2−ピリジル二硫化物、p−塩化水銀安息香酸、2−塩化水銀−4−ニトロフェノール、またはクロロ−7−ニトロベンゾ−2−オキサ−1,3−ジアゾール用いた反応によって誘導体化される。
【0050】
本発明におけるヒスチジン残基は、pH5.5−7.0におけるジエチルプロカーボネート(この試薬は、ヒスチジン側鎖に比較的に特異的であるので、)を用いた反応によって誘導体化される。パラブロモフェナシルブロマイドもまた有効であり、この反応は、pH6.0の0.1Mのナトリウムカコジル酸において好ましく実施される。特に、本発明のペプチドの構成要素 (I)および/または(III)に含まれるようなGLP−1(7−37)のN末端ヒスチジン残基は、Suzukiら(Diabetes Res.; Clinical Practice 5 (Supp. 1): S30 (1988))によって示されているように、GLP−1ペプチドのインシュリン分泌性の活性に非常に重要である。これに対応して、本発明のペプチドは、アルキルもしくはアシル(C1−6C)基により、構成要素 (I)および/または(III)の一部としてのGLP−1のHis7を修飾してもよいし、機能的に等価なC5−C6の環構造によりHisを置換することにより、構成要素 (I)および/または(III)の一部としてのGLP−1のHis7を修飾してもよい。好ましい修飾は、His7のアミノ末端またはそのヒスチジン側鎖に疎水性の部分を導入することである。
【0051】
本発明のペプチドにおけるリジニル残基およびアミノ末端残基は、例えば、琥珀酸または他のカルボン酸の無水物と反応されてもよい。これらの試薬を用いた誘導は、リジニル残基の電荷を逆転させる効果を有する。本発明のペプチドにおけるリジン残基のイプシロンアミノ基のアシル(C12−C18)修飾によって、該ペプチドの血液循環における半減期が増大する。アルギニン残基は、従来の試薬の1つまたはいくつか(フェニルグリオキサールおよびニンヒドリンなど)を用いた反応によって修飾されてもよい。アルギニン残基の誘導は、グアジニン官能基の高いpKaのために、その反応がアルカリ条件において行われる必要がある。さらに、これらの試薬は、アルギニンイプシロンアミノ基と同様に、リジン基と反応し得る。
【0052】
チロシン残基の特異的修飾は、それ自体、広範囲に亘って研究されている。通常、N−アセチルイミダゾールはO−アセチルチロシル種を、またテトラニトロメタンはe−ニトロ誘導体を形成するために用いることができる。
【0053】
カルボキシルの側基(アスパルチルまたはグルタミル)は、1−シクロヘキシル−3−〔2−モルフォリニル−(4−エチル)〕カルボジイミド、または1−エチル−3−(4−アゾニア−4,4−ジメチルフェニル)カルボジイミドのようなカルボジイミド(R’N−C−N−R’)との反応によって選択的に修飾されてもよい。
【0054】
さらに、アスパルチルおよびグルタミル残基は、アンモニウムイオンとの反応によって、アスパラギニルおよびグルタミニル残基に転換されてもよい。
【0055】
グルタミニルおよびアスパラギニル残基は、対応するグルタミルおよびアスパルチル残基に脱アミド化されてもよい。また、これらの残基は、弱酸性の環境において脱アミド化されてもよい。これら残基の形態のうち、どちらかは本発明の範囲に含まれる。本発明のペプチドを脱アミド化すると、プロテアーゼ酵素またはペプチダーゼ酵素によるタンパク質分解に対する感受性が変化することがある。このことから、脱アミド化は、本発明のペプチドのタンパク質分解による切断に向かわせる、生理学的な重要性を有する可能性があることが示唆される。なお、本発明の生合成ペプチドは、或る保存条件において分解することがあり、これにより本発明のペプチドにおける別の位置において脱アミド化が起こるということに注意されたい。本発明のペプチド中のメチオニン残基は、酸化の影響を受けやすく、それは主にスルホキシドによる影響である。上述のその他の誘導体として、本発明の脱アミドペプチドおよび/または、本発明のスルホキシドペプチドは、十分な生物活性を示すために用いられてもよい。
【0056】
アルファアミノ酸含有残基を誘導体化する、その他の好ましい試薬には、イミドエステルが含まれる。上記イミドエステルとしては、例えば、メチルピコリニミデート;ピリドキサルリン酸;ピリドキサル;クロロボロヒドリド;トリニトロベンゼンスルホン酸;0−メチルリオスレア;2,4−ペンタンジオン;およびグリオキシレートと反応するトランスアミナーゼ触媒等が挙げられる。
【0057】
適切なアミノまたはカルボキシル保護基を用いることによって、カルボキシ基(C−末端)およびアミノ基(N−末端)(上述したカルボキシまたはアミドアミノ酸の側鎖基と同様の)を有する本発明のペプチドの末端アミノ酸残基は、当該残基を保護された(例えば、アミド基によるC末端の保護のような)および/または保護されていない形状で存在し得る。また、本発明のペプチドの酸添加塩が提供されてもよい。代表的な酸添加塩は、ハロゲン化水素酸塩である。そのようなハロゲン化水素酸塩としては、例えば、HBrまたはHIが挙げられるが、HClであることがより好ましい。
【0058】
本発明のペプチドに存在するリジンの末端、側鎖カルボキシル基、またはイプシロン−アミノ基におけるPEG付加は、酸化に対する耐性を与えるものであり、またそれは本発明の範囲に含まれる。
【0059】
本発明のペプチドの誘導体に起こるその他の修飾としては、本発明のペプチドと共有結合され得る炭水化物および/または脂質に基づくものである。脂質および/または炭水化物は、セリン、スレオニン、アスパラギン、グルタミン、またはチロシンと結合するか、あるいは、グルタミン酸塩、またはアスパラギン酸塩とそれらの反応性側鎖の構成成分を経由して結合することが好ましい。また、炭水化物および/または脂質は、本発明のペプチドの末端構成成分と結合されてもよい。さらに、本発明のペプチドは、機能的に異なるペプチドまたはタンパク質構成成分と結合されてもよい。これにより、本発明のペプチドが安定化されて、および/または体液中、特に血液中における本発明のペプチドの輸送性を改善する働きがもたらされてもよい。ペプチドまたはタンパク質としては、例えば、アルブミン、およびトランスフェリン等から選択されることが好ましい。これらは、本発明のペプチドと直接結合されるか(構成要素IVとして)、またはペプチドもしくは有機連結配列を経由して結合される。これらのペプチドまたはタンパク質は、本発明のペプチドの末端の1つと結合されることがより好ましい。
【0060】
本発明のペプチドにおける分解という問題を解決するために、本発明の別の実施形態では、Dアミノ酸または、少なくとも一部分はDアミノ酸から成る本発明のペプチドのレトロインバーソ異性体が提供される。「レトロインバーソ異性体」とは、配列の方向性が逆であり、各アミノ酸残基のキラリティーが反転した直鎖ペプチドの異性体を意味する(Jamesonら, Nature, 368, 744-746 (1994); Bradyら, Nature, 368, 692-693 (1994)を参照)。もとのペプチドに関しては、当該レトロインバーソペプチドは、アミノ酸、特にF−mocアミノ酸誘導体を用いて、アミノ酸を逆の順序で結合される。特に、粗精製のペプチドは、逆相HPLCによって精製されてもよい。
【0061】
本発明のペプチドに導入されてもよいその他の修飾は、ペプチド骨格の修飾に関する。修飾された当該ペプチドは、骨格模倣物質であることが好ましい。当該骨格の側鎖構造は、本発明のペプチド、その断片、変異体、誘導体、または類似体と同一である。しかしながら当該骨格は、天然に生じる骨格とは異なっている。一般的に、骨格模倣物質では、1つ以上の当該骨格の鎖部分(NH,CH,CO)が修飾される。この修飾としては、挿入、または置換のどちらかがあるが、置換がより好ましい。置換基としては、例えば、(I)―NH−と置換される−O−,−S−,および−CH2−;(II)−CHR−と置換される−N−,−C−アルキル−,および−BH−;および(III)−CO−と置換される―CS−,−CH2−,−SOn−,−P=O(OH)―および−B(OH)−が挙げられる。本発明のペプチドの模倣ペプチドは、上記修飾を組み合わせてもよい。特に、I、II、およびIIIの各基の修飾は、組み合わせることができる。模倣ペプチドにおいて、各骨格の鎖部分は修飾されてもよく、また、鎖部分のある程度が人工的に生じる部分と交換されてもよい。本発明のペプチドにおけるすべての骨格の鎖部分における各―NH−,−CHR−またはCOは、別の人工的に生じる基と交換されることが好ましい。本発明のペプチド骨格のアミド結合(―NH−CO−)が置換される場合(分子全体であるか、または少なくとも1つ位置において)には、置換部分は、生物学的に等価であるもの、例えば例えば、レトロインバースアミド結合(―CO−NH−)、ヒドロキシルエチレン(−CH(OH)−CH2−)、アルケン(CH2=CH−)、カルバ(CH2−CH2−)および/または−P=O(OH−CH2−)が挙げられる。また、挿入による当該骨格の鎖伸張は、本発明のペプチドの骨格模倣物質において(例えばC−アルファ原子に隣接する部分の近くで)起こってもよい。C−アルファ原子の片側の部分において、例えば−O−、−S−、−CH−、―NH−が挿入されてもよい。
【0062】
さらに、本発明のペプチドは、オリゴカルバメートペプチド骨格構造であることがより好ましい。上記アミド結合は、カルバメート部分において置換される。N−保護アミノアルキル炭酸のモノマーは、対応するアミノ酸またはアミノアルコールを経由して利用可能であり、活性エステルに転換される。そのような活性エステルとしては、例えば、F−moc部分を用いてp−ニトロフェニルエステルに転換されてもよいし、また、は固相合成を用いて光感受性ニトロアトリルオキシカルボニル基に転換されてもよい。
【0063】
本発明のペプチドは、上述のようなタンパク質分解的切断から保護されるものであり、特に、ジペプチジルアミノペプチターゼ−4(DPP−IV)に対して保護される。本明細書において、「DPP−IVからの保護」とは、請求項1に係るペプチドに関して用いられる。本発明のペプチド、ならびに、当該ペプチドの誘導体、類似体、断片、および変異体により、本発明のペプチドの構成要素(I)および/または(III)の一部としてのGLP(7−35、36または37)は、血しょうペプチダーゼ(DPP−IV)対する耐性を獲得する。
【0064】
ジペプチジルアミノペプチダーゼIVによる分解に対するペプチドの耐性は、例えば、以下の分解分析によって測定される。すなわち、当該ペプチドのアリコートは、pH7〜8の最適な緩衝液(アルブミンは含まれていない)において、4〜22時間精製されたジペプチジルアミノペプチダーゼIVのアリコートと共に、37℃でインキュベートされる。酵素反応は、トリフルオロ酢酸の添加によって終結され、その後、ペプチドの分解生成物はHPLCまたはLC−MS分析によって分離および定量される。この分析を実施するための方法の1つとして、次の方法が挙げられる。すなわち、混合物は、Zorbax300SB−C18(30nmの微細孔、5μm粒子)150×2.1mmカラムの上に乗せられ、0.1%トリフルオロ酢酸中にアセトニトリルが直線勾配で、30分間で0%〜100%になるようにしながら、0.5ml/分の流速で溶解される。ペプチドおよびその分解生成物を、214nm(ペプチド結合)または280nm(芳香族アミノ酸)におけるそれらの吸光度によって観察することが可能であり、それらのピーク領域の積分によって定量される。分解傾向は、分離されたピークのMSスペクトルを測定することが可能であるLC−MSを用いて測定することができる。一定時間における、原型を保持した/分解された化合物の百分率は、ペプチドDPP−IV安定性の評価に用いられる。
【0065】
一定時間における原型を保持した化合物の百分率に基づくGLP−1(7−37)よりも10倍安定であるとき、本発明のペプチドはDPP−IVに対して安定であると考えられる。このように、DPP−IVに対して安定である本発明のペプチドは、上記のようなGLP−1(7−37)よりも少なくとも10倍安定であることが好ましく、少なくとも20倍安定であることがより好ましい。安定性は、当業者に公知である如何なる方法によって評価されてもよく、例えば、本発明のペプチド溶液にDPP−IVを加えることによって試験されてもよいし、例えば、当該ペプチドの経時的な分解を測定してもよく、分光法、ウェスタンブロット分析、抗体スクリーニング等によって測定されてもよい。同時に、本発明のペプチドは、例えば、GLP−1(7−37)の天然のレセプター(GLP−1レセプター)と結合することによって、GLP−1(7−37)の効果を与える化合物として規定される。本発明のペプチドとGLP−1レセプターとの間の結合親和性は、天然に生じるGLP−1ペプチドとGLP−1レセプターとの結合親和性の少なくとも10%、より好ましくは少なくとも50%に相当するGLP−1レセプターとの結合親和性を有することが好ましい。上記結合親和性は、如何なる適切な方法によって測定されてもよく、例えば、表面プラズモン共鳴等が挙げられる。さらに、例えば、本発明のペプチドが、細胞にシグナルを伝達する細胞外液レセプターと結合することによって、細胞内液cAMPの形成を誘引することが好ましい。
【0066】
本発明のペプチドは、技術的にGLP−1(7−36)アミドおよびGLP−1(7−37)の製造方法と類似した固相ペプチド合成技術を用いて、合成的に製造することができる。また、その後、例えば、逆相HPLCカラム上の単純精製方法、または好ましいクロマトグラフィー法によって、実験室規模で精製され得る。
【0067】
しかしながら、本発明のペプチドは、当該ペプチドを産生する微生物細胞または動物細胞系である組み換え細胞において形成されることが好ましい。本発明のペプチドは、当該ペプチドが発現された細胞から、例えば、従来の分離技術を用いて単離されてもよい。このように単離された細胞を、インビボにおいて、例えば支持体および栄養素を含む適切な条件で飼育することが可能であり、細胞外に分泌されたタンパク質(すなわち本発明のペプチド)を、細胞外の培地から回収することができる。したがって、細胞内に導入されるように設計された配列は、本発明のペプチドの分泌を導く、リーダー配列およびシグナルペプチド配列を含むことがより好ましい。上記細胞は、内在的に、または組み換え遺伝子配列によって、リーダー配列およびシグナル配列を分解することができるプロテアーゼを発現することが好ましい。別の方法において、本発明のペプチドをコードしている組み換え遺伝子配列は、上記リーダー配列およびシグナルペプチド配列を含まない。そのため、細胞内に発現された本発明のペプチドは分泌されず、細胞を溶解する段階を含む工程によって、細胞から回収される。上記方法において、コード化している配列は、培地から産生ペプチドの効率的な抽出を可能にする精製タグを含んでもよい。なお、当該タグとは、本発明のペプチドを放出するために切断されてもよい。
【0068】
さらに、本発明は、本発明のペプチド、あるいは融合ペプチド、またはその断片、類似体、もしくは変異体をコードする核酸を提供する本発明のペプチドをコードしている核酸であれば、任意のものが本発明に包含される。遺伝コードの縮重のため、多数の核酸配列は本発明のペプチドをコードしてもよい。本発明の範囲に含まれる核酸分子は、本発明のペプチドをコードする核酸を含んでおり、さらに、(機能的な)ヌクレオチド配列を含んでもよい。本発明の好ましい実施形態において、上記核酸分子は、(a)すべてのGLP−1 a a 配列(GLP−1(7−37)、または機能的GLP−1(7−35,36,および37)配列、また(b)任意のプロテアーゼのための(a)のGLP−1配列のN−末端における切断配列、をコードしてもよく、(b)から上流は、リーダー配列をコードすることができる。別の好ましい実施形態において、(b)の核酸分子をコードしている核酸配列から上流は、さらに(c)シグナルペプチドをコードしている配列を含んでもよい。また、本発明の核酸分子は、配列(c)を有していてもよく、該配列(c)は、(a)の上流と、リーダー配列をコードしている任意の配列を含まない(b)との間に融合されている。なお、上記導入配列およびシグナルペプチド配列は、プレプログルカゴンとは異なる種類であることが好ましい。
【0069】
さらに、本発明は、本発明の核酸(分子)および本発明の核酸(分子)の発現のためのその他の機能的な構成要素を含んでいるベクターを提供する。特に、本発明の核酸(分子)は、プロモーター配列と融合され、最終的には、例えば、エンハンサー配列のようなその他の調節配列と結合される。複製のために、上記プラスミドは複製開始点を含んでもよい。当該ベクターをトランスフェクションされる細胞を選択するために、1つ以上の抗生物質耐性遺伝子(例えば、カナマイシン、アンピシリン)がベクターに備えられてもよい。当該ベクターは、哺乳類細胞中において複製および発現するための、バクテリア由来プロモーター、抗生物質耐性遺伝子および複製起点プロモーター、および抗生物質耐性遺伝子を含むプラスミドであってもよい。さらに、本発明は、上述の前躯体タンパク質を翻訳することが可能な外因的に導入された本発明のDNAを含む宿主細胞を備えていてもよい。当該宿主細胞は、原核宿主細胞であるか、または、例えば哺乳類細胞のような真核宿主細胞であってもよい。
【0070】
さらに、本発明の態様によれば、成分(I)、(II)、ひいては成分(III)を含む本発明のペプチドの投与による動物(その中でもヒトであることが好ましい)の治療方法を提供する。また、糖代謝に関連する疾病または病気の治療および予防のための製品を製造するための、上記本発明のペプチドの対応する使用も提供する。糖疾患の例としては、これに限定されるものではないが、次のものを含む。すなわち、I型およびII型糖尿病(NIDDM)、インスリン耐性、体重異常および疾病、またはそれに関連する症状が挙げられる。なお、上記体重異常または関連する症状には、肥満、太りすぎに関連する症状、満腹中枢の調節不調、血しょうインスリン量の減少、血糖値上昇、または膵臓β細胞量の減少が含まれる。望ましくは、2型糖尿病(NIDDM)を治療するための薬物を製造するための本発明のペプチドの使用が、本明細書に開示されている。結果として、本発明は、例えば、被験者の体重を減少させるため、被験者の飽満を低下させるため、被験者の食後の血漿インスリン値を増加させるため、被験者の空腹時の血糖値を減少させるため、被験者の膵臓β細胞量を増加させるため、または被験者のI型またはII型糖尿病を治療するための本発明のペプチドの使用に関する。
【0071】
その他の病気または疾患のある患者は、本発明のペプチドによって治療されてもよく、例えば、融合ペプチド、または当該ペプチドの類似体、断片、変異体、誘導体によって治療されてもよい。本発明のペプチドは、神経変性疾患、および神経変性病、およびそれらに関連する状態を治療するための薬物、ならびに、アポトーシスに関連する疾患、病気および状態を治療するための薬物を調製するために用いられてもよい。以下のことから、これら障害を治療するために本発明のペプチドが使用される。すなわち、サイクリックAMP第2のメッセンジャー経路に関連するGLP−1レセプターが、げっ歯類およびヒトの脳の至るところに発現している。脳におけるレセプター分布の化学構造は、食事摂取量の調節および嫌悪ストレスへの応答におけるGLP−1の中心的な役割と関連しているだけではない。GLP−1レセプターにおいて結合しているGLP−1は、神経栄養性の性質を発揮し、培養された神経細胞において、グルタミン酸に誘導されるアポトーシスと酸化的損傷とに対して保護を与えることが示された。さらに、GLP−1は、細胞培養物においてアミロイドβ−プロテイン前躯体の修飾プロセシングを修飾し、インビボでの脳におけるアミロイドβ−ペプチド量の用量依存的な減少を示す。そのため、GLP−1は、中枢神経系の調節因子として知られている。生理活性型GLP−1の生物活性を模倣している本発明のペプチドは、例えば、アルツハイマー疾患(AD)、その他中枢および抹消神経変性障害(例えば、筋萎縮性側索硬化症(ALS)、アレキサンダー症、アルパース症、毛細血管拡張性運動失調症、カナバン症、コケーン症候群、クロイツフェルト・ヤコブ病、多発性硬化症、サンドホフ病、ピックス病、脊髄小脳失調、シルダー病、およびパーキンソン病)の治療に、治療上の関連性を有する。
【0072】
さらに、生理活性型GLP−1は、様々な細胞に抗アポトーシス作用を及ぼすことが示されている。例えば、GLP−1は、新たに単.離されたヒトの島細胞または他の種類の細胞の質量および機能の保存のために有用である。その範囲において、本発明の生物活性型ペプチドは、細胞または組織のアポトーシスによってもたらされる障害を治療するために用いられる。
【0073】
本発明のペプチドは、体外から投与される組成物の製造のために使用してもよく、その組成物は、単.離された本発明のペプチドを含んでいてもよい。得られた組成物は、上述した障害の治療として用いられてもよい。また、本明細書中に開示されている障害は、本発明の宿主細胞、核酸(分子)、およびベクター、によって治療されてもよい。さらに本発明の宿主細胞、核酸(分子)、およびベクターを、当該障害を治療するための薬物の調製に用いてもよい。
【0074】
活性成分として本発明のペプチドを含む製剤の調整は、米国特許第4,608,251号明細書;米国特許第4,601,903号明細書;米国特許第4,599,231号明細書;米国特許第4,599,230号明細書;米国特許第4,596,792号明細書;および米国特許第4,578,770号明細書において実証されているように、技術的に広く知られているところであり、これら全ては、本明細書に参考によって援用される。特に、上記製剤は溶液または懸濁液のように注入可能物質(水を含有していることが好ましい(水性製剤))として調製されるか、または乳化されてもよい。「水性製剤」とは、少なくとも50% w / w の割合で水を含んでいる溶液を意味する。同様に、「水性溶液」とは、少なくとも50% w / w の割合で水を含んでいる溶液を意味するものであり、「水性懸濁液」とは、少なくとも50% w / w の割合で水を含んでいる懸濁液を意味するものである。
【0075】
静脈注射、皮膚もしくは皮下注射、または病巣部位での注射のために、活性成分は非経口投与が可能な水性溶液の形状にされる。なお、当該水性溶液は、発熱物質がなく、適切なpH、等張性、および安定性を有する溶液である。一般的に液体の薬学的組成物は、水のような液体溶剤を含む。液体溶剤は、生理食塩水、D型グルコースエタノールを含んでもよく、その他の糖類溶液または、例えばエチレングリコール、プロピレングリコール、およびポリエチレングリコールのようなグリコール、またはその混合物が含まれてもよい。さらに、実施例は、リンガー液または乳酸加リンガー液のようなその他の液体溶媒である。
【0076】
本発明が、本発明の化合物の水性溶液および緩衝液を含んでいる薬学的製剤に関する場合には、上記化合物は0.1mg/ml以上の濃度において存在し、また上記製剤は約2.0から約10.0のpHを有することが好ましく、さらに約7.0から約8.5であることがより好ましい。上記製剤のpHは、本発明の化合物の等電点から少なくとも1pHユニットであることが好ましく、当該等電点から少なくとも2pHユニットであることがさらに好ましい。
【0077】
また、注入前の液体である溶液または懸濁液に入れるのに適した固形形態も調整され得る。薬学的製剤は、医者または患者が使用前に溶媒または希釈剤を加えるために、フリーズドライ製剤であってもよい。すなわち、一度調製された製剤は、患者へ直ちに投与されるわけではない。どちらかといえば、調製の後、溶液形態または患者へ投与するのに適した他の形態へと後で再構成するために、氷結状態または乾燥状態において貯蔵するように包装される。別の実施形態において、薬学的製剤は事前に溶解することなく使用する状態の乾燥製剤(例えば、フリーズドライまたはスプレードライ)である。「乾燥形態」は、液体の薬学的組成物または薬学的製剤が、(i)フリーズドライ(例えば、凍結乾燥;「Williams and Polli (1984) J. Parenteral Sci. Technol. 38: 48-59」を参照)、(ii)スプレードライ(「Masters (1991) in Spray-Drying Handbook (5th ed; Longman Scientific and Technical, Essez, U. K. ), pp. 491-676」; 「Broadheadら (1992) Drug Devel. Ind. Pharm. 18: 1169-1206」;および「Mumenthalerら (1994) Pharm. Res. 11: 12-20」を参照)、または、(iii)空気乾燥(「Carpenter and Crowe (1988) Cryobiology 25: 459-470; and Roser (1991) Biopharm. 4: 47-53」を参照)のいずれかによって乾燥されたものをいう。液体の薬学的組成物の貯蔵中に、ポリペプチドによる凝集が形成されると当該ポリペプチドの生物活性に、悪影響が及ぼされることがある。そして、これにより薬学的組成物の治療効力を損失するという結果が生じる。さらに、当該ポリペプチドを含んでいる薬学的組成物が輸液システムを用いて投与されるとき、形成された凝集は、管類、膜、またはポンプを閉塞するというようなその他の問題をもたらす可能性もある。
【0078】
その他の成分は、本発明のペプチド薬学的製剤において存在していてもよい。追加の上記成分には、湿潤剤、乳化剤、酸化防止剤、充填剤、pH緩衝剤(例えば、リン酸塩、クエン酸塩、マレイン酸塩の緩衝剤)、保存料、界面活性剤、安定剤、等張性調整剤、キレート剤、金属イオン、油性賦形剤、タンパク質(例えば、ヒト血清アルブミン、ゼラチン、またはタンパク質)および/または両性イオン(例えば、ベタイン、タウリン、アルギニン、グリシン、リジン、およびヒスチジンのようなアミノ酸)を含んでもよい。
【0079】
本発明の製剤のための安定剤としては、高分子量ポリマーまたは低分子量化合物の群から選択されることが好ましい。さらに、本実施の形態の安定剤は、ポリエチレングリコール(例えば、PEG3350)、ポリビニルアルコール(PVA)、ポリビニルピロリドン、カルボキシ−ヒドロキシセルロース、またはその誘導体(例えば、HPC、HPC−SL、HPC−L、およびHPMC)、シクロデキストリン、また、モノチオグリセロール、チオグリコール酸、および2−メチルチオエタノールのような含硫物質、および種々の塩(例えば、塩化ナトリウム)から選択されてもよい。これらの具体的な安定剤は、各自本発明の別の実施形態を構成する。また、薬学的組成物は、薬物活性ポリペプチドの安定性をさらに増強する分解防止剤をさらに含んでもよい。本発明に特に影響がある分解防止剤としては、特に限定されるものではないが、メチオニンおよびEDTA(これらはメチオニン酸化に対してポリペプチドを保護する)、ならびに、非イオン性界面活性剤(これは凍結融解または機械的剪断に関連のある凝集に対してポリペプチドを保護する)が含まれる。
【0080】
本発明の製剤の界面活性剤としては、清浄剤、エトキシル化されたキャスターオイル、ポリグリコール化されたグリセリド、アセチル化されたモノグリセリド、ソルビタン脂肪酸エステル、ポリオキシプロピレン−ポリオキシエチレンブロック重合体(例えば、PluronieF68、poloxamer188および407、またはトリトンX−100のようなpoloxamers)、ポリオキシエチレンソルビタン脂肪酸エステル、星形PEO、アルキル化およびアルコキシル化された誘導体(Tween−20、Tween−40、Tween−80、およびBrij−35)のようなポリオキシエチレンおよびポリエチレン誘導体、ポリオキシエチレンヒドロキシステアレート、モノグリセリド、またはエトキシル化されたその誘導体、ジグリセリド、またはそのポリオキシエチレン誘導体、アルコール、グリセロール、レクチンおよびリン脂質(例えば、ホスファチジルセリン、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルイノシトール、ジホスファチジルグリセロール、およびスフィンゴミエリン)、また、(例えば、ジパルミトイルホスファチジン酸のような)リン脂質の誘導体、(例えば、パルミトイルリゾホスファチジル−L−セリンおよび、エタノールアミン、コリン、セリン、またはスレオニンの1−アシル−sn−グリセロ−3−リン酸エステルのような)リゾリン脂質、および、リゾホスファチジルおよびホスファチジルコリンのアルキルアルコキシル(アルキルエステル)、アルコキシ(アルキルエーテル)誘導体(例えば、リゾホスファチジルコリンおよびジパルミトイルホスファチジルコリンのラウロイルおよびミリストイル誘導体、および極性頭部基(コリン、エタノールアミン、ホスファチジン酸、セリン、スレオニン、グリセロール、およびイノシトール)が修飾されたもの、正電荷を帯びたDODAC、DOTMA、DCP、BISHOP、リゾホスファチジルセリン、およびリゾホスファチジルスレオニン、ならびに、グリセロリン脂質(例えば、ケファリン)、グリセロ糖脂質(例えば、ガラクトピラノシド)、スフィンゴ糖脂質(例えば、セラミド、ガングリオシド)、ドデシルホスホコリン、鶏卵リゾレシチン、フシジン酸誘導体(例えば、タウロジヒドロフシジン酸ナトリウム等)、長鎖脂肪酸および脂肪酸のC6−C12を有する塩(例えば、オレイン酸およびカプリン酸)、アシルカルニチンおよび誘導体、また、リジン、アルギニン、またはヒスチジンのN'X−アシル化誘導体、側鎖がアセチル化されたリジンもしくはアルギニンの誘導体、または、リジン、アルギニン、またはヒスチジンおよび中性もしくは酸性アミノ酸の任意の組み合わせを含んでいる、N−アシル化されたジペプチドの誘導体、中性アミノ酸および2つの電荷を帯びたアミノ酸の如何なる組み合わせを含んでいるN−アシル化されたトリペプチドの誘導体、DSS(ドキュセートナトリウム、CAS登録番号〔577−11−7〕、ドキュセートカルシウム、CAS登録番号〔128−49−4〕ドキュセートカリウム、CAS登録番号〔7491−09−0〕)、SDS(ドデシル硫酸ナトリウムまたはラウリル硫酸ナトリウム)、カプリル酸ナトリウム、コール酸またはその誘導体、胆汁酸およびその塩、グリシンまたはタウリンの複合体(conjugates)、ウルソデオキシコール酸、コール酸ナトリウム、デオキシコール酸ナトリウム、タウロコール酸ナトリウム、グリコール酸ナトリウム、N−ヘキサデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホン酸、一価のアニオン(アルキル−アリル−スルホン酸塩)界面活性剤、(例えば、N−アルキル−N、N−ジメチルアンモニオ−1−プロパンスルホン酸、3−胆汁アミド−1−プロピルジメチルアンモニオ−1−プロパン−スルホン酸のような)両性イオン界面活性剤、(例えば、セチル−トリメチルアンモニウム臭化物、セチルピリジニウム塩化物のような)4級アンモニウム塩基である陽イオン界面活性剤、(例えば、ドデシル−D−グルコピラノシドのような)非イオン性の界面活性剤、例えばテトロニクスのような、エチレンジアミンへのプロピレン酸化、およびエチレン酸化の選択的付加に由来する四官能性ブロック共重合体であるポロキサミン、イミダゾリン誘導体類の群から選択されてもよい界面活性剤、あるいは、それらの混合物から選択されてもよい。これらの具体的な界面活性剤は、それぞれ本発明の別の実施形態の構成要素である。薬学的組成物における界面活性剤の使用は、当業者に知られるところである。便宜上、レミントン:薬学の科学と演習、19編、1995が参照される。
【0081】
薬学的に利用可能な保存料としては、フェノール、O−クレゾール、m−クレゾール、p−クレゾール、メチルp−ヒドロキシ安息香酸、プロピルp−ヒドロキシ安息香酸、2−フェノキシエタノール、ブチルp−ヒドロキシ安息香酸、2−フェニルエタノール、ベンジルアルコール、エタノール、およびクロロブタノールならびに、チオメロサル、ブロノポル、安息香酸、イミド尿素、クロロヘキシジン、デヒドロ酢酸ナトリウム、クロロクレゾール、エチルp−ヒドロキシ安息香酸、塩化ベンゼトニウム、およびクロルフェネシン(3p−クロルフェネキシプロパン−1,2−ジオール)、またはそれらの混合物から成る群から選択されることが好ましい。
【0082】
等張剤としては、(例えば塩化ナトリウムのような)塩、糖または糖アルコール、(例えば、L−グリシン、L−ヒスチジン、アルギニン、リジン、イソロイシン、アスパラギン酸、トリプトファン、スレオニンのような)アミノ酸、(例えば、グリセロール(グリセリン)、1,2−プロパンジオール(プロピレングリコール)、1,3−プロパンジオール、1,3−ブタンジオールのような)アルジトール、および(例えばPEG400のような)ポリエチレングリコール、またはそれらの混合物から成る群から選択されることが好ましい。なお、糖としては、モノ、ジ、または多糖、あるいは水溶性グルカンのような如何なる糖を用いてもよく、これらにはフルクトース、グルコース、マンノース、ソルボース、キシロース、マルトース、ラクトース、スクロース、トレハロース、デキストラン、プルラン、デキストリン、シクロデキストリン、水溶性デンプン、ヒドロキシエチルデンプン、およびカルボキシメチルセルロースナトリウムが含まれる。また、一実施形態において、糖添加物はスクロースである。糖アルコールとしては、少なくとも1つの水酸基を有するC4−C8炭化水素として規定され、例えば、マンニトール、ソルビトール、イノシトール、ガラシチトール、ズルシトール、キシリトール、およびアラビトールが含まれる。一実施形態において、糖アルコール添加物はマンニトールである。また、糖アルコールは、上述の糖または糖アルコールは、単独で用いてもよく、組み合わせて用いてもよい。なお、糖または糖アルコールの使用量は、液体の調製において可溶であり、本発明の方法を用いて達成される効果の安定化に悪影響を及ぼさない限り限定されるものではない。
【0083】
キレート剤としては、エチレンジアミンテトラ酢酸(EDTA)、クエン酸、およびアスパラギン酸の塩、またそれらの混合物から選択されることが好ましい。
【0084】
緩衝剤としては、酢酸ナトリウム、炭酸ナトリウム、クエン酸、グリシルグリシン、ヒスチジン、グリシン、リジン、アルギニン、リン酸二水素ナトリウム、リン酸一水素二ナトリウム、およびリン酸ナトリウム、トリス(ヒドロキシメチル)−アミノメタン、へペス、バイシン、トリシン、リンゴ酸、コハク酸、フマル酸、酒石酸、アスパラギン酸、またはそれらの混合物から成る群から選択されることが好ましい。これらの具体的な緩衝剤は、それぞれ本発明の別の実施形態の構成要素である。
【0085】
本発明の治療ペプチドを含んでいる薬学的組成物において、すべての上記添加剤の使用、特にそのの濃縮範囲に関しては、当業者に知られるところである。便宜上、レミントン:薬学の科学と演習、19編、1995が参照される。
【0086】
本発明のペプチドを含んでいる製剤は、例えば、皮下、皮内、または筋内への注射によって、従来の非経口的な投与がされる。本発明のペプチドの非経口的投与の組成は、例えば、国際公開第03/002136号パンフレットにおいて記載されているように調製されてもよい。
【0087】
さらに、その他の投与形態に適している製剤は坐薬を含んでおり、場合によっては経口、口腔、舌下、腹腔内、膣内、肛門、および頭蓋内処方であってもよい。坐薬には、従来の結合剤および担体である、例えば、ポリアルカレングリコール、またはトリグリセリドが含まれてよい。;上記坐薬は、0.5%から10%の範囲で活性成分を含んでいる混合物から形成されてもよく、上記範囲は0.5%から12%であることがさらに好ましい。経口製剤は、例えば、医薬品等級のマンニトール、ラクトース、デンプン、ステアリン酸マグネシウム、サッカリンナトリウム、セルロース、および炭酸マグネシウム等のような、一般的に使用される賦形剤を含む。これらの組成物は、溶液、懸濁液、タブレット、錠剤、カプセル、持続放出性製剤、または粉末の形態をとり、また、活性成分を10%−95%含んでおり、より好ましくは25%−70%含む。
【0088】
上述のように、添加された薬剤の用途は、作用の持続時間を制御するためであってもよい。放出を制御して調製することは、本発明のペプチドを複合または取り込むためのポリマーを用いることによって達成され得る。活性成分(ペプチド)の制御された運搬は、(例えば、ポリエステル、ポリアミノ酸、ポリビニルピロリドン、エチレンビニル酢酸共重合体、メチルセルロース、カルボキシメチルセルロース、および硫酸プロタミンのような)適切なマクロ分子、マクロ分子の濃度、ならびに混和方法を選択することによって実施され得る。上述の技術は、レミントンの薬学(上述を参照)において開示されている。放出を制御して調製することによって作用の持続時間を制御するために可能な方法としては、その他に本発明のペプチドを、ポリエステル、ポリアミノ酸、ヒドロゲル、ポリ(乳酸)、またはエチレンビニル酢酸共重合体のような高分子材料の粒子に混和する方法がある。
【0089】
本発明のペプチドの形態としては、中性または塩であってもよい。本発明のペプチドは、多くの有機塩基および無機塩基、ならびに有機酸および無機酸の何れかとの反応して、例えば、ペプチドの遊離アミノ基と共に形成されるような(付加)塩を形成するために十分に酸性もしくは塩基性であってもよい。酸付加塩を形成するために一般的に使用される酸としては、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸等のような無機酸、または酒石酸、アスパラギン−トルエンスルホン酸、メタンスルホン酸、シュウ酸、マンデル酸、p−ブロモフェニル−スルホン酸、炭酸、コハク酸、クエン酸、および安息香酸のような有機酸が挙げられる。上記塩としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、亜硫酸水素塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸、カプリン酸塩、アクリル酸塩、ギ酸塩、イソブチル酸塩、カプロン酸塩、ヘプタン酸塩、プロピオン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオエート、へキシン−1,6−ジオエート、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、ガンマ−ヒドロキシ酪酸塩、グリコール酸塩、酒石酸塩、メタンスルホン酸塩、およびプロパンスルホン酸塩が例示できる。また、遊離カルボキシル基から成る塩は、例えば、ナトリウム、カリウム、アンモニウム、カルシウム、または水酸化第2鉄のような無機塩基、またイソプロピルアミン、トリメチルアミン、2−エチルアミノエタノール、ヒスチジン、プロカイン等のような有機塩基由来であってもよい。本発明のペプチドの酸付加塩、カルボン酸塩、低アルキルエステル、およびアミドは、国際公開第91/11457号パンフレット(1991);欧州特許出願公開第0733644号明細書(1996);および米国特許第5,512,549号明細書(1996)にしたがって作製されてもよい。
【0090】
本発明のペプチド配列を含んでいる製剤は、投与製剤と適合するようにして投与され、その投与量は、治療効果が期待できる量とする。治療される患者によって決まる投与量は、例えば、患者の症状の重症度によっても決まる。適切な投与量の範囲は、1回の治療的投与につき、活性成分が数百マイクログラムであるが、好ましくは、該活性成分は、(1−10mgの範囲という高い投与量が検討されるが)約0.1μgから2000μgの範囲であり、例えば、約0.5μgから1000μgであってもよく、また、好ましくは1μgから500μgの範囲であり、特に約10μgから100μgの範囲である。
【0091】
本発明のペプチドを含み、さらに、例えば付加賦形剤としてグリシン、マンニトール、またはその他の添加剤を加えたような製剤は、凍結乾燥された形態で薬瓶として市販されてもよい。希釈剤を有する対の薬瓶が提供され、患者は、投与前に製剤を望ましい濃度に調整することができる。また、本発明の製剤は、注射器に充填される形態のような、その他公知形態において市販されてもよい。
【0092】
本発明は、さらに下記の実施例において実証される。
【0093】
〔実施例〕
<実施例1>
−遺伝子コンストラクトの作製−
GLP−1(7−37)cDNAのコード配列を、図1aに示されたようなHincIIおよびEcoRIの制限酵素認識部位を含む配列として人工的に合成した。図1bに示されたようなGLP−1(7−37)のコード配列、IP2ならびにSfoI、EcoRIおよびXbaIの制限酵素認識部位を含むcDNA(図1b)を、別に合成した。GLP−1を分泌経路に向かわせるために、ストロメライシン3(stromelysin3、(登録番号NM_005940)の異種シグナル配列を用いた。つまり、ストロメライシンのシグナル配列およびリーダー配列をコードするcDNAを、逆転写PCRを用いてヒトRNAから増幅した。そして、図1aまたは図1bのコンストラクトを用いて、図1cおよび図1dに示されたコンストラクトをそれぞれ作製した。
【0094】
図1aのコンストラクトのHincll/EcoRI断片を、図1dの配列のSfoI部位に挿入してクローニングすることにより、図1eのコンストラクトを作製した。同様に、図1dのコンストラクトのEcoRI断片を、真核生物発現プラスミドのEcoRI部位に導入してクローニングすることにより、図1fに示されたコンストラクトを作製した。図1bに示されたコンストラクトのHincII/XbaI断片を繰り返して、図1dに示されたコンストラクトのSfoI/XbaI部位に挿入してクローニングすることにより、図1gに示されたコンストラクトを作製した。図1hは、ヒトGLP−1(7−37)、IP2およびGLP−2(1−35)をコードする配列に融合している、ストロメライシンのリーダー配列およびシグナル配列をコードする、コドンが最適化された合成さ配列を示している。また、ストロメライシンのリーダー配列およびシグナル配列は、内因性のイントロン配列が短くなったことによって中断されている。図1hのコンストラクトのDNA配列は、配列番号16に示されており、翻訳されたペプチドの配列は、配列番号15に示されている。
【0095】
また、図1iおよび1jに示された配列を合成する。その後、これらの配列を用いて、図1jのNaeI/BssHII断片を、図1hの直鎖状のNaeI/BssHII配列に挿入してクローニングすることによって、図1kに示されたコンストラクトを作製する。図1kのコンストラクトのDNA配列は配列番号14に示されており、翻訳されたペプチドの配列は配列番号13に示されている。図1hの配列をBssHIIにより消化して、再度ライゲーションすることにより図1lのコンストラクトを作製する。図1lのコンストラクトのDNA配列は配列番号18に示されており、翻訳されたペプチドの配列は配列番号17に示されている。図1iの配列のAfeI/BssHII断片を、図1hの直鎖状のAfeI/BssHII配列に挿入してクローニングすることにより、図1mのコンストラクトを作製した。図1mのコンストラクトのDNA配列は配列番号20に示されており、翻訳されたペプチドの配列は配列番号19に示されている。
【0096】
上記コンストラクトは、通常の技術を用いて当業者により作製されてもよい。
【0097】
<実施例2>
−哺乳類細胞に対するトランスフェクション、クローン選抜およびGLP−1の発現−
細胞源:HEK293(ヒト胎児腎臓細胞株、カタログ番号ACC305、DSMZ Cell Culture Collection、ドイツ)、AtT20(マウスLAF1下垂体腫瘍細胞株、カタログ番号87021902、European Cell Culture Collection、英国)、hTERT−MSC細胞は、カセム(Kassem)教授(オーデンセ大学病院(University Hospital of Odense)、デンマーク)から生み出された。
【0098】
106の細胞にトランスフェクションするために、異なるGLP−1のコンストラクトを有している0.5〜2μgのプラスミドDNAを用いた。該コンストラクトを、実施例1に記載したように作製した。Current Protocols in Molecular Biology (Ausubelら 1994ff Harvard Medical School Vol2., Unit 9.1)に記載のような標準のリン酸カルシウム共沈法により、HEK293細胞にトランスフェクションした。Current Protocols in Molecular Biology (Ausubelら 1994ff, Harvard Medical School Vol 2., Unit 9.4に記載されているようなフュージーン(FuGene(Roche))を用いて、AtT20細胞にトランスフェクションした。電気パラメーターと細胞種に特異的な溶液との組み合わせに基づいた非ウィルス性の方法である、ヌクレオフェクター技術(Nucleofector technology (Amaxa))を用いて、hTERT−MSC細胞にトランスフェクションした。ヌクレオフェクター装置(プログラムC17)およびヌクレオフェクター溶液(VPE−1001)を用いることにより、60%よりも大きいトランスフェクション効率が達成された。トランスフェクションから48時間後に、選抜剤であるブラストサイジン(2μg/ml)を細胞培養液に添加することによって、DNAが染色体に安定に組み込まれた細胞のクローンの選抜を行った。12〜15日後、トランスフェクションされた細胞の安定クローンを単離することができ、該クローンの特徴を調べるために増殖させた。
【0099】
異なるGLP−1コンストラクトの一過性発現を、hTERT−MSC細胞およびHEK293細胞において測定した。hTERT−MSC細胞およびHEK293細胞の両方において測定することができた、活性型GLP−1の発現量は、単量体GLP−1のコンストラクトである♯103および♯317(GLP−1(7−37)を1コピーだけ有している)では、ごくわずかであったが、2量体GLP−1のコンストラクトである♯217(GLP−1(7−37)を、構成要素(I)および構成要素(III)として有している)では、非常に多かった。この結果をまとめて図2に示す。コンストラクトをGLP−1コンストラクト♯159にまで伸ばした(構成要素(II)としてIP2を4コピー有している)が、さらに顕著に増加することはなかった(示さず)。hTERT−MSC細胞に、異なるコンストラクトをトランスフェクションした後、GLP−1を安定発現するクローンを選抜した。発現量を表1に示す。
【0100】
【表1】

【0101】
<実施例3>
−哺乳類細胞から分泌されたGLP−1ペプチドのウェスタンブロット解析−
GLP−1を分泌する細胞の細胞培養上清を、10%−20%の勾配のSDS PAGE(120V、90分)により分離して、PVDFメンブレン(Immobilon-P Membrane 0.45 μm ミリポアIPVH 00010)にセミドライブロッティング(2.0mA/cm2、60分)によりトランスファーした。メタノール固定と、ブロッキング(3%(w:v)BSAおよび0.1%(v:v)Tween−20を含むTBS)とを行った後、上記メンブレンを、1μg/mlの抗GLP−1抗体(HYB 147−12、Antibodyshop)を用いて、4℃で一晩、免疫ブロットした。洗浄して、0.02μg/mlの検出抗体(Anti Mouse IgG, HRP conjugated, パーキンエルマー PC 2855-1197)と一緒に、室温で4時間インキュベーションした後、化学発光検出を行いタンパク質の位置を明らかにした。
【0102】
ウェスタンブロット解析を図3に示す。なお、1は、モック(mock)をトランスフェクションしたhTERT−MSC細胞の上清に溶解した、100ngの合成GLP−1(7−37)を表し、2は、コンストラクト♯217に由来する2量体GLP−1を分泌するhTERT−MSC細胞(クローン79TM217/13)の上清を表し、3は、コンストラクト♯217に由来する2量体GLP−1を分泌するAtT20細胞(クローン81−A−217/3)の上清を表し、Mは、前もって着色しているタンパク質マーカー(kDa)を表す。この結果によれば、GLP−1(7−37)およびC末端付加物を含んでいる本発明のペプチド(図3における2および3)が、トランスフェクションされた細胞株から分泌されること、そして該本発明のペプチドを、GLP−1(7−37)の中央にある分子エピトープに結合する抗GLP−1抗体を用いて検出できることが示された。
【0103】
<実施例4>
−ヒト細胞から分泌されたGLP−1ペプチドの血しょうにおけるインビトロでの安定性−
HEK293細胞およびhTERT−MSC細胞に、ストロメライシンの異種シグナル配列をコードするコンストラクトを一過的にトランスフェクションした。なお、上記異種シグナル配列は、以下のペプチドをコードする各GLP−1変異体と連結している。
【0104】
1:GLP−1(7−37)、
2:GLP−1(7−37)−IP2(11AAだけ伸長)
3:GLP1(7−37)−IP2−GLP1(7−37)。
【0105】
細胞から分泌されたGLP−1ペプチドまたは合成GLP−1(7−37)(Bachem)を含んでいる細胞培養上清を、ヒトのリンパ球が豊富な血しょう(ジペプチジルペプチダーゼ活性を含む)と一緒に、37℃および5%のCO2の環境下において、3または4時間インキュベーションした。モックをトランスフェクションした細胞の上清に含まれる合成GLP−1(7−37)を、DPP−IV活性の陽性対照として用いた(このDPP−IV活性は、DPP−IV阻害剤((#DPP4, Biotrend)の添加によって阻害されることが示された)。活性型GLPを、GLP(活性)ELISA(#EGLP-35K, Biotrend)を用いて測定した。このGLP(活性)ELISAでは、GLP−1(7−37)のN末端のエピトープに結合する抗体が用いられるので、DPP−IVにより分解された不活性型GLP−1(9−37)ペプチドが識別される。
【0106】
この結果を図4(HEK293細胞)および図5(hTERT−MSC細胞)に示す。HEK293細胞およびhTERT−MSC細胞の両方は、遺伝子コンストラクトに対する有効な宿主である。実施例3と同様に、1〜3の種類のトランスフェクションされた細胞に関する結果に番号をつけた。1は、モックをトランスフェクションしたhTERT−MSC細胞の上清に溶解した、100ngの合成GLP−1(7−37)を表し、2は、コンストラクト♯217に由来する2量体GLP−1を分泌するhTERT−MSC細胞(クローン79TM217/13)の上清を表し、3は、コンストラクト♯217に由来する2量体GLP−1を分泌するAtT20細胞(クローン81−A−217/3)の上清を表す。コンストラクト1は、合成GLP−1と同様の方法でDPP−IVによって不活性化される野生型のGLP−1を製造するのに対し、本発明のC−末端が伸長したGLP−1の形態(図4の2および3、ならびに図5の3)は、分解に対する耐性がより高く、少なくとも40%の活性を維持している。C末端が伸長したGLP−1は、インビトロで、ヒトの血しょうにおいて顕著に安定化されている。2量体のGLP−1配列(3)を有するペプチドは、インビトロでのDPP−IVによる分解に対してほぼ完全に安定化されている。
【0107】
<実施例5>
−GLP−1ペプチドのウェスタンブロット解析−
様々なGLP−1ペプチドを、固相により(syn)、または、E.coliを用いた組み換えにより(rec)合成的に製造した。GLP−1ペプチド(配列番号1(31ng)、ならびに、配列番号6、7および8(各10ng))を、10%−20%の勾配のSDS PAGE(120V、90分)により分離して、PVDFメンブレン(Immobilon-P Membrane 0,45μmミリポア IPVH 00010)にセミドライブロッティング(2.0mA/cm2、60分)によりトランスファーした。メタノール固定と、ブロッキング(3%(w:v)BSAおよび0.1%(v:v)Tween−20を含むTBS)とを行った後、上記メンブレンを、1μg/mlの抗GLP−1抗体(HYB 147−12, Antibodyshop)を用いて、4℃で一晩、免疫ブロットした。洗浄して、0.02μg/mlの検出抗体(Anti Mouse IgG, HRP conjugated, パーキンエルマー PC 2855-1197)と一緒に、室温で4時間インキュベーションした後、化学発光検出を行いタンパク質の位置を明らかにした。示されたペプチドのウェスタンブロットを図6に示す。以下の値を与えることができる。配列番号1(ID1syn)は、GLP−1(7−37)に対応し、31aaおよび3.3kDである;配列番号8(ID8syn、CM3)は、GLP−1(7−37)−IP2に対応し、46aaおよび5.1kDである;配列番号7(ID7rec、CM2)は、GLP−1(7−37)−IP2−RR−GLP2に対応し、83aaおよび9.4kDである;配列番号6(ID6syn、CM1)は、GLP−1(7−37)−IP2−RR−GLP1(7−37)に対応し、79aaおよび8.7kDである。
【0108】
<実施例6>
−GLP−1CMペプチドのインビトロでのヒト血しょうにおける安定性−
濃度20ng/mlの合成GLP−1ペプチド(配列番号1syn、配列番号6syn、配列番号7rec、配列番号8syn)を、ヒト血しょうと一緒に、37℃および5%CO2の環境下において3時間インキュベートした。血しょうのジペプチジルペプチダーゼ活性を、DPP−IV阻害剤(#DPP4, Biotrend)を用いて阻害した。活性型GLPを、GLP(活性)ELISA(#EGLP-35K、Biotrend)を用いて測定した。
【0109】
天然GLP−1(7−37)(配列番号1)と比べて、本発明のC−末端が伸長したGLP−1ペプチド(配列番号6、配列番号7および配列番号8)は、インビトロでのヒト血しょうにおいて顕著に安定化している(図7)。右側の対照のように、DPP−IVを添加した実験に関する結果を示す。GLP−1の活性が、これらの対照実験において、完全に維持されている。
【0110】
<実施例7>
−インビトロにおけるバイオアッセイ(環状AMPの産生)−
RIN−5細胞(ラットの島細胞種、ECACC 番号95090402)を、24ウェルプレートにおいて、4日間、70%の集密度に達するまで育てた。細胞をDMEM(E15−009、PAA)で2回洗浄してから、1%HSA(Aventis)、0.2mM IBMX(858455、シグマ)およびテストペプチドが添加されたDMEM(E15−009、PAA)を0.5ml加えた。25℃で20分間インキュベートした後、細胞を氷冷のPBSを用いて2回洗浄した。0.5%TritonX−100を含む0.1N HCLを添加して、細胞のcAMPを抽出した。cAMP(低pH)EIA(カタログ番号DE0355、R&D)を用いて、環状AMPを定量した。3×10−8Mの刺激については、配列番号1、配列番号6syn、配列番号6rec、配列番号7rec、配列番号8synを用いた。
【0111】
この結果を、図8に示す。100%のcAMP産生は、GLP−1が無いときの基本産生に対応している。GLP−1は、Gタンパク質共役型受容体に結合し、cAMPの産生を刺激する。テストした全ての分子により、細胞のcAMP産生が増加する。
【0112】
<実施例8>
−インビボでの生物活性−
11週齢のII型糖尿病のマウス(C57BL/Ks−Leprdb/db、Harlan)に、5μgのペプチドを、1日に二回(午前9時および午後5時)皮下注射した(1グループあたりn=5である)。一晩絶食させてから午前10時における血糖を、GLPCMペプチドを用いて処置する前(0日)および処置した後(2日、4日、7日、10日後)について、測定した。データを、0日における血糖値に対する相対値として示した。
【0113】
テストされた全ての本発明のペプチド((合成または組み換えの)配列番号6および(合成または組み換えの)配列番号7)は、抗高血糖症効果を有している。組み換えの配列番号6(CM1)および合成の配列番号8(CM3)を用いたときに、最良の結果が得られた。図9のy軸に、上記処置の相対的効果を示す。0日における血糖を1にした。未処置の動物では、血糖値が経時的に連続して増加しているが、本発明のペプチドを用いて処置された動物では、血糖値が経時的に、概ね連続して減少している。
【0114】
<実施例9>
−GLP1CMペプチドのインビトロでの安定性の測定(速度論的テスト方法)−
インキュベーション緩衝液(50mMトリエタノールアミン−HCl(pH7.8)、0.2%HSA)中のCM3、アラニンが置換されたCM3類似体(CM3−ANA01、CM3−ANA02、CM3−ANA03、CM3−ANA04)、C末端が短くなったCM3類似体(CM3−ANA06、CM3−ANA07)、またはC末端が伸長したCM3類似体(CM3−ANA09)からの1μMのペプチドのアリコートを、10%ヒト血しょうと一緒に、37℃および5%CO2の環境下において0、3、6および9時間インキュベートした。DPPIV阻害剤(#DPP、Biotrend)を添加することにより、ジペプチジルペプチダーゼ活性を停止させて、GLP−1(活性)ELISA(#EGLP-35K, Linco)を用いて、活性型GLP−1量を測定した。2以上の独立な実験のそれぞれにおいて、これを3回行い、結果を得た。
【0115】
GLP−1のC末端に付加された少なくとも9個のアミノ酸を有している本発明のペプチドは、CM3(マウス、GLP−1(7−37)−IP2)および、構成要素(II)(IP2)の配列が修飾されているその誘導体(すなわちCM3−ANA01、CM3−ANA01、CM3−ANA02、CM3−ANA03、CM3−ANA04、CM3−ANA05、CM3−ANA07、CM3−ANA07)である。GLP−1ペプチドおよびCM3−ANA06ペプチドは、対照物質または参照物質を表している。
【0116】
図10は、活性型GLP−1のうちで活性を有しているものを、0時間における値の%として示す(すなわち0時間=100%)図である。GLP−1は、血しょうに晒されてから9時間後に9%だけ残っていることから、血しょう安定性が最も悪いことが明確に示されている。CM3−ANA01は、9時間後に84%が残っているので、安定性が最も良好であることが示されている。本発明のペプチドは、9時間後に少なくとも30%が残っており、伸長部分が短いペプチドはこの値に達していない。速度論から、テストした物質に関して、活性型GLP−1を0時間の値の80%にまで分解するのに必要な時間(DT80:80%になるまでの分解時間)を計算することができる。
【0117】
【表2】
【0118】
本発明の物質は、参照物質と比べて、インビトロでの血しょう安定性が大幅に高いことが示されている。任意の理論に束縛されることなく、GLP−1のC末端を伸長させたことにより、立体障害が生じ、上記類似体がDPPIVの活性部位に入り込むことが阻止される。そして、このため上記類似体は分解から免れる。これに対して、より短い参照物質は、GLP−1の安定性に関するこの保護効果を示さない。この仮説は、CM3−ANA06(36aa)、CM3−ANA07(40aa)およびCM3(46aa)のペプチドに見られるように、ペプチドのC末端が長くなればなるほど、安定性が増加することに、支持されている。それにもかかわらず、この現象は、CM3のIP2領域におけるアラニン置換によって示されたように、アミノ酸の数に依存するだけではない。CM3とアミノ酸の数が同じであるCM3−ANA03ペプチドは、CM3と比べて、安定性が非常に減少している。一方、46アミノ酸からなるCM3−ANA01は、CM3と比べて、安定性がより高くなっている。このことは、アミノ酸の数とは関係なく、別の立体効果が安定性に影響するかもしれないということを示している可能性がある。特に好ましいペプチドは、GLP−1のC末端においてIP2を含んでいる伸長部分を有しているペプチド、またはIP2とある程度の相同性を有する伸長部分を有しているペプチドである。これら伸長部分により、DPPIVの活性部位にN末端領域が侵入することを防ぐ特定の立体構造が生じてもよい。
【0119】
<実施例10>
インビボでの研究−(免疫原性)
テスト物質であるCM1に免疫原性があるかどうかを明らかにするために、パラバイオサイエンス(Parabioscience(グローニンゲン、ドイツ))において、マウスの研究を実施した。22日間、5回繰り返して静脈注射された(70μgのペプチド/用量)BALB/cマウス(n=5)において、試験を行った。
【0120】
CM1synにより毒性は誘導されず、以下の図に示されるように誘導されたT細胞抗体の反応と、抗体力価とは最小であった。ペプチドの純度は95%だけであるので、副産物の免疫原性効果を、排除することができない。CM1synに免疫原性効果があるかどうかを評価する研究を、図11に示す。左側(図11A)に、T細胞の想起反応アッセイの結果を示す。処置されたマウスおよび対照のマウスの脾臓を体外培養した。それからT細胞を単離し、抗原の量を増やしながら刺激した。増殖性の想起反応が測定される。右側(図11B)において、動物の免疫血清における抗原特異的IgG抗体の力価を決定した。
【0121】
<実施例11>
−糖尿病マウスにおける用量と有効性とに関する研究−
2時間の絶食のあと、5つの異なる濃度のGLP−1、CM1、CM3、CM3−ANA01およびエキセンディン−4を用いて、糖尿病のC57BL/KsJ@Rj−db(db/db)マウスを処置した。6番目のグループを、生理食塩水のみを用いて処置した。1グループにつき7匹の動物を処置した。注射から1時間および4時間後に尾より採血し、この血液から血糖を直接決定した。
【0122】
図12(図10A(GLP−1)、図12B(CM1)、図12C(CM3)および図12D(CM3−ANA01))に示された用量反応曲線に、その結果を示す。全てのテスト物質に関して、異なる濃度の初期値に対する、血糖の差の百分率を示すグラフを作成した。1時間における測定値を用いて、基準値と比較して百分率が減少するような用量反応曲線を作成することにより、異なるテスト物質に関するED50値を決定した。モーガン−メルセル−フローディン(Morgan-Mercer-Flodin(MMF))モデルを用いて、GLP−1、CM3−ANA01、CM3recおよびエキセンディン−4に関するED50値を評価し、リチャーズモデルを用いて、CM1に関するED50値を評価した。両者のモデルは、用量反応を評価するために適切なシグモイド適合モデルである。全てのペプチドに関して、血しょうグルコースのED50値(一回の皮下注射の用量(μg/マウス))を、血糖値の百分率の減少から決定した。上記ED50値を表1にまとめて示す。
【0123】
【表3】
【0124】
上記ED50値によれば、GLP−1類似体であるCM1、CM3およびCM3−ANA01のインビボにおける生物活性は、20倍よりも大きいと推定される。また、このことは、血しょうにおける安定性が増強されたことによるという可能性が非常に高いといえる。
【0125】
テストした全てのペプチドにより、高血糖のdb/dbマウスにおける血糖値が、少なくとも最高の濃度に関して大幅に減少した。1.1−3.0nmolのペプチドを1回皮下注射した後の、対照に対する血しょうグルコースの低下を、表2にまとめて示す。表2は、対照に対するテスト物質のグルコース低下効果を示している。ペプチドの注射から1時間後(1h)および4時間後(4h)における血しょうグルコースの低下および対応する有意差を求めた。ペプチドの長期有効性について、差異が見られた。エキセンディン−4およびCM3のみにおいて、4時間後に血糖値が有意に減少していた。
【0126】
【表4】
【0127】
<実施例12>
−db/dbマウスの長期間処置−
以下の4つのグループ(n=12)のC57BL/KsJ@Rj−db(db/db)マウスを調査した:
グループA;1日に1回、溶剤(0.9%生理食塩水)を用いて処置された、
グループB;1日に1回、24nmol/kgのテスト物質1(CM1rec)を用いて処置された、
グループC;1日に1回、24nmol/kgのテスト物質2(CM3−ANA01)を用いて処置された、
グループD;1日に1回、24nmol/kgの参照物質(エキセンディン−4)を用いて処置された。なお、エキセンディン−4は技術的に知られており、承認されている。しかしエキセンディン−4はGLP−1類似体ではない。
【0128】
ペプチドまたは溶剤を、1日に1回、2○○−3○○p.m.の間に、グループA−Dにおいて背中の皮膚のひだに皮下投与した。この投与計画を、18週間続けた。12週目から18週目までにおいて、1グループあたり12匹いるうちの6匹に、1日に2回処置を行った。
【0129】
マウスの様々なパラメーター、すなわち、健康状態(1)、体重(2)、飼料消費(3)、血糖(4)、グルコース負荷テスト(5)、インスリンのデータ(6)、グリコシル化ホモグロビン(7)、病状(8)、およびT細胞の再刺激(9)を調査した。
【0130】
健康状態(1)
全てのグループの健康状態は良好であり、処置の副作用は見られなかった。
【0131】
体重(2)
18週に及ぶ処置の後において、処置された全てのグループにおける体重は減少していた(図13)。相対的体重増加は、CM3−ANA01のグループにおいて有意に少ない。図13Aは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。1グループあたり12匹の動物の平均値をプロットされている。図13Bは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。122日に及ぶ処置の後に関する1グループあたり12匹の動物の相対的体重増加をプロットされている。テスト物質としてCM3−ANA01を用いて処置したときのみに(グループB)、体重増加量が有意に少なくなっていた(p<0.001)。
【0132】
飼料消費(3)
対照と比較して、CM1およびCM3−ANA01のグループにおいて、飼料消費が有意に低くなっていた。図14は、122日に及ぶ処置の間における、db/dbマウスの1週間の飼料消費に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。マウス1匹あたりの相対的飼料消費は、グループBおよびCにおいて有意に減少している(p<0.001)。
【0133】
血糖値(4)
血糖値を、様々な状況において決定した。非絶食時の血糖値(ペプチドの注射から1時間後)を図15Aに示す。すなわち、図15Aには、非絶食時の血糖値に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果が示されている。生理食塩水(A)、または、24nmol/kgの濃度のCM1(B)、CM3−ANA01(C)、もしくはエキセンディン(D)ペプチドを皮下注射した。それから1時間後に尾より採血し、この血液から血糖を測定した。対照と比較して、ペプチドの皮下注射から1時間後の、非絶食時の血糖値は、グループBでは55%、グループでは51%、グループDでは60%に減少している(図15B)。処置されたグループにおける血糖は、極めて有意に減少している(p<0.0001)。
【0134】
絶食時の血糖値(ペプチドの注射から18時間後)を図15Cに示す。すなわち、図15Cには、絶食時の血糖値に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果が示されている。テスト物質を皮下注射してから18時間後に、一晩(12時間)絶食させた。それから、尾より採血し、この血液から血糖値を測定した。ペプチドを注射してから18時間後の絶食時の、血糖値は、CM1およびCM3−ANA01のグループにおいて減少している。
【0135】
グルコース負荷テスト(5)(IPGTT)
グルコース負荷テストを、グループA−Dのマウスに対する処置を8週行った後に、実施した。尾より採血することによって、12時間絶食させた動物の基準血糖値(0分)を決定した。その後、テスト物質を皮下注射し、20%のグルコース溶液を腹腔内注射した(1kgあたり1gのグルコース)。そして、グルコース注射から15、30、45、60、90、および120分後の血糖値を続けて決定した。全ての処置されたグループにおいて、グルコース負荷の有意な正規化が示された(図16A)。図16Aでは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて8週間処置された後に、腹腔内注射によりグルコース負荷テスト(IPGTT)を実施している間の血糖値が絶対値で示されている。なお、上記絶対値は、各グループの平均値(n=12)である。
【0136】
図16Aにより提示されるデータを別の表現にて、図16Bに示す。図16Bでは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて8週間処置された後に、腹腔内注射によりグルコース負荷テスト(IPGTT)を実施している間の血糖値が、初期血糖値(100%)に対して標準化された相対値で示されている。
【0137】
インスリン量(6)
一晩(12時間)絶食させた後のマウスのインスリン量を決定した。18週間の処置の後、処置されたグループの全てのマウスは、処置されていない対照よりも有意に多くのインスリンを産生している。0.9%生理食塩水(グループA)、CM1ペプチド(グループB)、CM3−ANA01ペプチド(グループC)、またはエキセンディン−4(グループD)を用いて18週間処置した後の、マウスの血清中インスリン量に関するデータを、図17に示す。血液を採取した直後、プロテアーゼ阻害剤カクテルを用いて試料を処置することにより、インスリンが分解することを防いだ。インスリンマウスウルトラセンシティブELISA(Insulin Mouse Ultrasensitive ELISA(カタログ番号EIA-3440、DRG))を用いて、血清をインスリンについて分析した。スチューデントt−検定により、有意差を決定した。
【0138】
グリコシル化ヘモグロビン(7)
赤血球(RBC)におけるヘモグロビンに非常に多くの血しょうグルコースが結合することによって、グリコシル化ヘモグロビンが形成される。RBCの寿命は120日であるため、グリコシル化ヘモグロビンは、グルコース制御の長期指標を与え、血しょうグルコース量よりも良好な指標として見られる。後眼窩洞(retro-orbital sinus)から全血液を回収した。そして、酵素HbA1cテストキット(Enzymatic HbA1c Test Kit (#DZ121A, Diazyme))を用いて分析して、グリコシル化ヘモグロビン量を決定した。グリコシル化ヘモグロビンをパラメーターとして用いた場合に、増加が有意に減少していることが、全ての処置されたグループにおいて見られた。
【0139】
図18Aは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて6、12および18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。図18Bは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。1グループあたりの全動物の平均値(n=12)で表されている。処置された全グループのグリコシル化ヘモグロビンの増加は、対照よりも有意に低い。
【0140】
病状(8)
処置されたマウスの病状を調査した。検視の結果、処置されたグループと処置されていないグループAとの間の臓器については、膵臓の体積のこと以外は違いがないことが明らかになった。膵臓の重量を決定すると、処置された全グループの脾臓の重量が、極めて有意に重いことが明らかになった。0.9%生理食塩水(A、n=11)、CM1(B、n=12)、CM3−ANA01(C、n=12)、またはエキセンディン(D、n=12)を用いて、18週間処置した後の動物を乱切した日に決定された膵臓の重量を、図19に示す。
【0141】
IV型免疫原性(9)
T細胞の再刺激によりIV型免疫原性を調べて、テストされた物質の可能性がある免疫原性効果を評価した。したがって、1グループあたり8匹の動物の脾臓を体外培養して、T細胞を単離し、異なる濃度の対応するテスト物質を用いて再刺激した。処置されていない対照と比べて、再刺激されたT細胞の増殖は、有意に増加していなかった。図20は、異なる濃度のテスト物質であるCM1(図20A)およびCM3−ANA01(図20B)、ならびに参照物質であるエキセンディン−4(図20C)に対する、インビトロにおける脾臓細胞の想起反応を示す図である。非放射性細胞増殖分析(細胞増殖のためのELISA(Cell Proliferation ELISA)、BrdU、化学発光法)を用いて、テスト物質であるCM1およびCM3−ANA01、ならびに参照物質であるエキセンディン−4を含んでいる溶液に対する、グループA−Dのマウスの脾臓細胞のインビトロにおける想起反応を、別々のマウスにおいて3回測定した。なお、上記溶液における各テスト物質の濃度を、初めは50μg/mlに調製し、その後3倍に希釈していった。刺激指数(SI)を、それぞれのペプチドが存在するときの反応、およびペプチドがないときの反応の指数として計算した。それぞれの処置されたグループおよび対照のグループの平均値±標準偏差を示す(グループA:n=3、グループB:n=6、グループC:n=7、グループD:n=6)。平均値を計算するために、SIが6.5よりも大きいマウスだけを分析した。なお、この6.5は5μg/mlのConAにより刺激した陽性対照培養物におけるSIである。
【0142】
糖尿病マウスにおける長期研究によって、本発明のC末端が伸長したペプチドの有効性が、さらに支持された。テストされた全てのパラメーター(体重、飼料消費、血糖値、グリコシル化ヘモグロビン、グルコース負荷、インスリン分泌、膵臓の重量)において、C末端が伸長したペプチドの有意な治療効果が明らかになった。内因的に生じた配列を有しているC末端が伸長したCellMedペプチドの相同性を考慮すると、上述した本発明のペプチドは、非哺乳類のエキセンディン−4と比較して、免疫原性の点において有利であることが示された。マウスの免疫原性研究から得られたデータは、任意の臨床的に関連するCM1ペプチドの免疫原性がないということを支持している。
【0143】
〔配列表〕
【0144】
【化1】
【0145】
【化2】
【0146】
【化3】
【0147】
【化4】
【0148】
【化5】
【0149】
【化6】
【0150】
【化7】
【0151】
【化8】
【0152】
〔図面の簡単な説明〕
〔図1〕
GLP−1の各コンストラクトを示す図である。
〔図2〕
hTERT−MSC細胞およびHEK293細胞に一過的にトランスフェクションした後の活性型GLP1を示す図である。
〔図3〕
哺乳類細胞から分泌されたGLP−1ペプチドのウェスタンブロット解析を示す図である。
〔図4〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図5〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図6〕
GLP−1ペプチドのウェスタンブロット解析を示す図である。
〔図7〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図8〕
インビトロにおけるバイオアッセイ(環状AMPの産生)を示す図である。
〔図9〕
db/dbマウスの絶食時の相対的グルコース量を示す図である。
〔図10〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図11〕
AはT細胞の想起反応アッセイの結果を示す図であり、Bは動物の免疫血清における抗原特異的IgG抗体の力価を示す図である。
〔図12〕
AはGLP−1の用量反応曲線を示す図であり、BはCM1の用量反応曲線を示す図であり、CはCM3の用量反応曲線を示す図であり、DはCM3−ANA01の用量反応曲線を示す図である。
〔図13〕
Aはdb/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図であり、Bは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
〔図14〕
122日に及ぶ処置の間における、db/dbマウスの1週間の飼料消費に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
〔図15〕
Aは非絶食時の血糖値を示す図であり、Bは注射から1時間後の対照に対する血糖の減少を示す図であり、Cは絶食時の血糖値を示す図である。
〔図16〕
Aは腹腔内注射によるグルコース負荷テストを示す図であり、Bは腹腔内注射によるグルコース負荷テスト(相対値)を示す図である。
〔図17〕
0.9%生理食塩水(グループA)、CM1ペプチド(グループB)、CM3−ANA01ペプチド(グループC)、またはエキセンディン−4(グループD)を用いて18週間処置した後の、マウスの血清中インスリン量に関するデータを示す図である。
〔図18〕
Aは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて6、12および18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図であり、Bは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。
〔図19〕
0.9%生理食塩水(A、n=11)、CM1(B、n=12)、CM3−ANA01(C、n=12)、またはエキセンディン(D、n=12)を用いて、18週間処置した後の動物を乱切した日に決定された膵臓の重量示す図である。
〔図20〕
異なる濃度のテスト物質であるCM1(A)およびCM3−ANA01(B)、ならびに参照物質であるエキセンディン−4(C)に対する、インビトロにおける脾臓細胞の想起反応を示す図である。
【技術分野】
【0001】
本発明は、C末端が伸長した新規GLP−1融合ペプチドに関する。このGLP−1ペプチドは、(i)エンドペプチダーゼIVによる不活性化に耐性があり、(ii)形質転換された動物細胞中において高レベルで発現されていてもよく、(iii)例えば2型糖尿病の治療に有効である。
【背景技術】
【0002】
グルカゴン遺伝子は十分に研究されている(例えば、「White, J.W.ら, 1986 Nucleic Acid Res. 14(12) 4719-4730」を参照)。高分子量の前躯体分子としてのプレプログルカゴン分子は、膵臓のα細胞、および、空腸および大腸のL細胞において合成される。プレプログルカゴンは180アミノ酸長のプロホルモンであり、その配列は、グルカゴンに加えて、グルカゴン様ペプチド−1(GLP−1)およびグルカゴン様ペプチド−2(GLP−2)という関連した構造の2つの配列を含んでいる。プレプログルカゴン分子において、GLP−1およびGLP−2の間には、17アミノ酸ペプチド配列(厳密に言えば、15アミノ酸配列とC末端RRの切断部位とである)、介在性ペプチド2(IP2)が存在している。IP2配列(前躯体分子においてGLP−1およびGLP−2の間に位置している)は、通常は、GLP−1のaa37の後でタンパク質分解により切断される。したがって、プレプログルカゴンモジュールは、細胞および環境に応じて切断されて、GLP−1(1−37)(37アミノ酸であるGLP−1の未加工の形態)を含む様々なペプチドが生じる。一般的に、膵臓および腸において、この加工が行われる。さらにGLP(1−37)配列はタンパク質分解されて、31アミノ酸の加工された形態である活性型GLP−1(7−37)、または、GLP−1(7−36)アミドが生じる。したがって、GLP−1(7−37)の記載は、もとのペプチドであるGLP−1のN末端から数えたときに、該当する断片が7番から37番までのアミノ酸残基を含んでいる(なお、7番、37番のアミノ酸残基も含んでいる)、ということを示している。GLP−1(7−36)アミドおよびGLP−1(7−37)のアミノ酸配列は、一般式I(配列番号43):His−Ala−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Ser−Tyr−Leu−Glu−Gly−Gln−Ala−Ala−Lys−Glu−Phe−Ile−Ala−Trp−Leu−Val−Lys−Gly−Arg−X(I)(なお、XがNH2であるときにGLP−1(7−36)アミドを表し、XがGly−OHであるときにGLP−1(7−37)を表す)で表されている。
【0003】
GLP−1は、消化管ホルモンであり、β細胞においてアデニル酸シクラーゼ活性およびタンパク質キナーゼ活性を刺激するなどの作用がある効き目の強い内因性のインスリン分泌性の作用因子である。該GLP−1は、生理学的に、上部消化管からの胃抑制ポリペプチドと共に、血糖値を下げる内分泌物ホルモンとして機能する。したがって、食物の摂取に反応して分泌されるGLP−1は、例えば血液中に含まれる糖質を制限するために強調して働く胃、肝臓、膵臓および脳に対して複数の効果を与える。その結果、糖質代謝に対するGLP−1の強力な作用、および、糖尿病(2型糖尿病など)を治療するための潜在的な適用可能性のために、グルカゴン様ペプチドGLP−1(7−36)アミド、およびアミド化されていない類似体のGLP−1(7−37)は、興味を非常に引き寄せていた。インスリンが存在しているときに細胞が適切に反応しないので、2型糖尿病はインスリンへの耐性を示すという特徴がある。これは、1型糖尿病よりも複雑な問題である。症状は比較的に軽度(ケトアシドーシスではない)で、かつ散発性であったりするので、患者は、数年間、2型糖尿病であると診断されるまで気づかないことがある。しかしながら、2型糖尿病に気づかないために、腎不全、冠状動脈性心臓病を含む重症合併症が発症することになり、死亡率がますます増加する。
【0004】
GLP−1(7−36)アミドまたはGLP−1(7−37)は、血清において短命なペプチドである。該ペプチドは、ジペプチジルペプチダーゼIV(DPP−IV)により残基と残基9との間において切断される。切断されたペプチドは不活性である。したがって、体外から投与されたGLP−1は、非常に短命であり、治療への応用における実用性が限られている。
【0005】
天然に生じるGLP−1(GLP−1(7−37))の(DPP−IVに対して)安定な類似体を、合成できるような、様々な試みが行われている。特に、インビボにおいてAlaである残基8は、他の残基、例えばGly、Ser、またはThrに置き換えられている(Burcelin, R., ら (1999) Metabolism 48, 252-258)。Gly8またはG8の類似体を、両方とも合成された分子として広範囲にわたって検証し、突然変異体のポリペプチドを分泌するための遺伝子組み換え細胞株により生産した(Burcelin, R., ら (1999) Annals of the New York Academy of Sciences 875: 277-285)。GLP−1(7−37)の生物活性が失われることなく、インビボにおけるGLP−1(7−37)の安定性を高めるためにGLP−1(7−37)に、様々な他の修飾が導入されている。しかしながら、これらの全てのアプローチの結果、関係する多くの問題が原因で、顕著な治療上の有意性が少しも見られなかった。
【0006】
ソレセン,B(Thorens, B.)は、多量体のGLP−1を発現するカセットを作製するための方法を国際公開公報第9953064号パンフレットに公開した。該多量体のGLP−1を発現するカセットを、様々な種類の細胞の中に組み込むが可能である。前記様々なタイプの細胞は、一般的に利用できる不死化された細胞株および分裂性の初代培養細胞である。例えば、哺乳類のCNS(中枢神経系)に由来するEGF−応答性ニューロスフィア、bFGF−応答性神経前躯体幹細胞が挙げられる。また、上手くいった例においては、仔ハムスター腎臓由来のBHK細胞が用いられている。埋め込まれたトランスフェクションされた細胞は、グルコース調節を糖尿病ではないときのグルコース調節と実質的に同等になるように、糖尿病マウスを上手く治療するために使用されると考えられた。しかしながら、この技術は、糖尿病患者に対して定期的な投与が行われる治療の必要条件を満たさない。
【0007】
外因的にグルコース量を安定させるるための他のアプローチは、2型糖尿病の治療に関する研究のもとに、内分泌物模倣剤として公知である新しい分類の医薬に基づいている。エキセナチド(exenatide)(バイエッタ(Byetta)(登録商標))は、アメリカドクトカゲ(Gila monster lizard)の唾液において発見された天然化合物を合成したものである。臨床試験において、内分泌物模倣剤(エキセチナド)は、血液中に含まれる糖質を減少すること、および、β細胞の機能マーカーを向上させることを示した。しかしながら、エキセナチドは、ヒト内分泌物ホルモンのグルカゴン様ペプチド−1(GLP−1)のある程度の効果だけを示す。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開公報第9953064号パンフレット
【非特許文献】
【0009】
【非特許文献1】White, J.W.ら, 1986 Nucleic Acid Res. 14(12) 4719-4730
【非特許文献2】Burcelin, R., ら (1999) Metabolism 48, 252-258
【非特許文献3】Burcelin, R., ら (1999) Annals of the New York Academy of Sciences 875: 277-285
【発明の概要】
【発明が解決しようとする課題】
【0010】
要約すると、GLP−1に基づいて血糖値を下げることができる、2型糖尿病に対する利用可能な有効な治療が、現在のところ存在していない。言い換えれば、GLP−1に関して公知の有益な効果(例えば、(i)肥満被検体における胃内容物排出速度を短縮することにより、血液の循環の中に入る栄養物の浸入速度を強力に減らすような生理的濃度におけるGLP−1の活性、または、(ii)インスリンを刺激するGPL−1の活性)を全面にわたって反映する治療は提供されていない。従って、生物活性を有し、タンパク分解に対して耐性のあるペプチド分子に基づいたGLP−1を提供することが本発明の目的である。
【課題を解決するための手段】
【0011】
1.構成要素(I)として、N末端に一般式II
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Xaa16−Ser−Xaa18−Xaa19−Xaa20−Glu−Xaa22−Xaa23−Ala−Xaa25−Xaa26−Xaa27−Phe−Ile−Xaa3o−Trp−Leu−Xaa33−Xaa34−Xaa35−Xaa36−Xaa37、
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、3−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し(ここで、Xaa8はGlyを表すことが特に好ましい);
Xaa16は、ValまたはLeuを表し;
Xaaは、Ser、LysまたはArgを表し;
Xaa19は、TyrまたはGlnを表し;
Xaa20は、LeuまたはMetを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa25は、AlaまたはValを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa27は、GluまたはLeuを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa33は、ValまたはLysを表し;
Xaa34は、Lys、Glu、AsnまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、Gly、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、Pro、もしくはLys、またはアミドを表すか、あるいは存在していない)、あるいは、
一般式III
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Xaa8−Tyr−Leu−Glu−Xaa22−Xaa23−Ala−Ala−Xaa26−Glu−Phe−lle−Xaa30−Trp−Leu−Val−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し;
Xaa18は、Ser、LysまたはArgを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa34は、Lys、GluまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、もしくはLys、またはアミドを表すか、あるいは存在していない)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる融合ペプチド。
2.構成要素(I)として、N末端にGLP−1(7−35、7−36、または7−37)配列、および、
構成要素(II)として、C末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、1の融合ペプチド。
3.構成要素(I)として、N末端に一般式I
His−Ala−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Ser−Tyr−Leu−Glu−Gly−Gln−Ala−Ala−Lys−Glu−Phe−Ile−Ala−Trp−Leu−Val−Lys−Gly−Arg−X(I)
(式中、Xは、NH2またはGly−OHを表す)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、1の融合ペプチド。
4.構成要素(I)が、配列番号1と少なくとも80%の配列相同性を有する配列を含んでいる、2または3の融合ペプチド。
5.構成要素(II)が、βターン様構造を形成するペプチド配列である、1〜4の何れか1つの融合ペプチド。
6.構成要素(II)が、少なくとも1つのアラニン残基およびプロリン残基を含んでいるペプチド配列である、1〜5の何れか1つの融合ペプチド。
7.構成要素(II)が、βターンを形成する性質を有している4量体を、含んでいるペプチド配列であって、例えば該4量体の2位にプロリン残基を有している、1〜6の何れか1つの融合ペプチド。
8.構成要素(II)が、VAIA、IAEE、PEEV、AEEV、EELG、AAAA、AAVA、AALG、DFPE、AADX、AXDX、およびXADXから成る群から選ばれる配列モチーフを含んでいるペプチド配列であって、Xは任意のアミノ酸を表す、1〜7の何れか1つの融合ペプチド。
9.構成要素(II)が、AA、XA、AX、RR、RXおよびXRから成る群から選ばれる構成要素(II)のN末端配列モチーフにより、構成要素(I)のC末端と連結しているペプチド配列である、1〜8の何れか1つの融合ペプチド。
10.構成要素(II)が、配列番号25(DFPEEVA)の配列モチーフを含んでいるペプチド配列であるか、または、配列番号25と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜9の何れか1つの融合ペプチド。
11.構成要素(II)が、配列番号22(RRDFPEEVAI)および配列番号26(AADFPEEVAI)から成る群から選ばれる配列を含んでいるペプチド配列であるか、あるいは、配列番号22または配列番号26と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜10の何れか1つの融合ペプチド。
12.構成要素(II)が、配列番号23(RRDFPEEVAIVEEL)、配列番号24(RRDFPEEVAIAEEL)、配列番号27(AADFPEEVAIVEEL)、および配列番号28(AADFPEEVAIAEEL)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号23、24、27または28の何れかと少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜11の何れか1つの融合ペプチド。
13.構成要素(II)が、配列番号2(RRDFPEEVAIVEELG)、配列番号3(RRDFPEEVAIAEELG)、配列番号29(AADFPEEVAIVEELG)、および配列番号30(AADFPEEVAIAEELG)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号2、3、29または30と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、1〜12の何れか1つの融合ペプチド。
14.構成要素(II)が、9−30アミノ酸、好ましくは9−20アミノ酸、および最も好ましくは9−15アミノ酸を有しているペプチド配列である、1〜13の何れか1つの融合ペプチド。
15.構成要素(I)および構成要素(II)が、直接連結されているか、または、リンカー配列を介して連結されている、1〜14の何れか1つの融合ペプチド。
16.前記リンカー配列が、1−10アミノ酸の長さを有している、15の融合ペプチド。
17.前記融合ペプチドが、
配列番号8(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELG)、
配列番号12(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELG)、
配列番号31(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIAEELG)、
配列番号32(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIAEELG)、
配列番号33(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIAEELG)、
配列番号34(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIAAALG)、
配列番号35(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFP)、
配列番号36(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVA)、
配列番号37(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELGRRHAC)、
配列番号38(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIVEELG)、
配列番号39(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIVEELG)、
配列番号40(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIVEELG)、
配列番号41(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIVAALG)、および
配列番号42(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELGRRHAC)
から成る群から選ばれる配列を含んでいるか、または、配列番号8、12と少なくとも80%の配列相同性を有する配列を含んでいる、1〜16の何れか1つの融合ペプチド。
18.前記融合ペプチドが、構成要素(II)のC末端、および/または、構成要素(I)のN末端と連結した他の構成要素(III)を含んでいる、1〜17の何れか1つの融合ペプチド。
19.構成要素(III)が、少なくとも4のアミノ酸残基、好ましくは少なくとも10の付加的なアミノ酸残基、より好ましくは、少なくとも20または少なくとも30の付加的なアミノ酸残基を含んでいる、18の融合ペプチド。
20.構成要素(III)が、プログルカゴンにおけるようなGLP−2のN末端配列、またはGLP−1(7−37)のN末端配列の、少なくとも4、好ましくは10、より好ましくは少なくとも20の付加的なアミノ酸残基を含んでいる、17または19の融合ペプチド。
21.構成要素(III)が、配列番号4または5の配列を含んでいるか、もしくは、配列番号4または5と少なくとも80%の配列相同性を有する配列を含んでいる、18〜20の何れか1つの融合ペプチド。
22.前記融合ペプチドが、配列番号6、7、10および11から成る群から選ばれるペプチド配列を含んでいるか、あるいは、配列番号6、7、10または11と少なくとも80%の配列相同性を有する配列を含んでいる、18〜21の何れか1つの融合ペプチド。
23.前記アミノ酸の少なくとも1つが、(i)天然に生じるアミノ酸の側鎖を共有結合修飾することにより誘導体化される、(ii)ペプチド骨格を修飾することにより誘導体化される、(iii)NH2末端基またはカルボキシ末端基を修飾することにより誘導体化される、1〜22の何れか1つの融合ペプチド。
24.前記アミノ酸の少なくとも1つが、リピル(lipyl)基または炭水化物基により誘導体化される、23の融合ペプチド。
25.GLP−1(GLP−1(7))のN末端のHis残基が、該GLP−1のNH2末端および/または該GLP−1のヒスチジル側鎖において、特に疎水性の部分によって化学的に修飾される、23または24の融合ペプチド。
26.前記融合ペプチドが、構成要素(IV)として、担体タンパク質、特にトランスフェリンまたはアルブミンを含んでいる、1〜25の何れか1つの融合ペプチド。
27.構成要素(I)、(II)および/または(III)のアミノ酸配列が反転されており、前記アミノ酸配列は、少なくとも部分的にDアミノ酸アイソマーから構成されている、1〜26の何れか1つの融合ペプチド。
28.固体状態のペプチド合成により、1〜27の何れか1つの融合ペプチドの製造方法。
29.1〜22または26の何れか1つの融合ペプチドをコードする核酸。
30.29の核酸を含んでいるベクター。
31.前記融合ペプチドを発現することができる、外因的に導入された29のDNA(特に30のベクター)を含んでいる宿主細胞。
32.融合ペプチドをコードする核酸を含有させるために形質転換した微生物を発酵させて、タンパク質を回収する、1〜22または26の何れか1つの融合ペプチドの製造方法。
33.動物細胞を、上記タンパク質が上記細胞から排出される条件下において成長させる、32のタンパク質の製造方法。
34.ヒトまたは動物の治療の場合に使用するための薬物としての、1〜27の何れか1つの融合ペプチド。
35.1型または2型糖尿病、インスリン耐性、体重異常および疾病、またはそれに関連する症状を治療するための薬物を製造するための、(i)1〜27の何れか1つの融合ペプチド、(ii)29の核酸、(iii)30のベクター、あるいは(iv)31の宿主細胞の使用。
36.神経変性疾患および神経変性病、またはそれらに関連する状態を治療するための薬物を製造するための、(i)1〜27の何れか1つの融合ペプチド、(ii)29の核酸、(iii)30のベクター、あるいは(iv)31の宿主細胞の使用。
37.アポトーシスに関係する疾患および病気、または状態を治療するための薬物を製造するための、(i)1〜27の何れか1つの融合ペプチド、(ii)29の核酸、(iii)30のベクター、あるいは(iv)31の宿主細胞の使用。
【図面の簡単な説明】
【0012】
【図1】GLP−1の各コンストラクトを示す図である。
【図2】hTERT−MSC細胞およびHEK293細胞に一過的にトランスフェクションした後の活性型GLP1を示す図である。
【図3】哺乳類細胞から分泌されたGLP−1ペプチドのウェスタンブロット解析を示す図である。
【図4】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図5】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図6】GLP−1ペプチドのウェスタンブロット解析を示す図である。
【図7】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図8】インビトロにおけるバイオアッセイ(環状AMPの産生)を示す図である。
【図9】db/dbマウスの絶食時の相対的グルコース量を示す図である。
【図10】GLP−1ペプチドの血しょうにおける安定性を示す図である。
【図11】AはT細胞の想起反応アッセイの結果を示す図であり、Bは動物の免疫血清における抗原特異的IgG抗体の力価を示す図である。
【図12】AはGLP−1の用量反応曲線を示す図であり、BはCM1の用量反応曲線を示す図であり、CはCM3の用量反応曲線を示す図であり、DはCM3−ANA01の用量反応曲線を示す図である。
【図13】Aはdb/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図であり、Bは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
【図14】122日に及ぶ処置の間における、db/dbマウスの1週間の飼料消費に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
【図15】Aは非絶食時の血糖値を示す図であり、Bは注射から1時間後の対照に対する血糖の減少を示す図であり、Cは絶食時の血糖値を示す図である。
【図16】Aは腹腔内注射によるグルコース負荷テストを示す図であり、Bは腹腔内注射によるグルコース負荷テスト(相対値)を示す図である。
【図17】0.9%生理食塩水(グループA)、CM1ペプチド(グループB)、CM3−ANA01ペプチド(グループC)、またはエキセンディン−4(グループD)を用いて18週間処置した後の、マウスの血清中インスリン量に関するデータを示す図である。
【図18】Aは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて6、12および18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図であり、Bは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。
【図19】0.9%生理食塩水(A、n=11)、CM1(B、n=12)、CM3−ANA01(C、n=12)、またはエキセンディン(D、n=12)を用いて、18週間処置した後の動物を乱切した日に決定された膵臓の重量示す図である。
【図20】異なる濃度のテスト物質であるCM1(A)およびCM3−ANA01(B)、ならびに参照物質であるエキセンディン−4(C)に対する、インビトロにおける脾臓細胞の想起反応を示す図である。
【発明を実施するための形態】
【0013】
本発明は、構成要素(I)として、N末端にGLP−1(7−35、7−36または7−37)配列、ならびに構成要素(II)としてC末端に、少なくとも9つのアミノ酸のペプチド配列、またはその機能的な断片、変異体もしくは誘導体を含んでいる融合ペプチドに関する。
【0014】
本発明は、生じた本発明のペプチドが、タンパク質分解性のエンドペプチダーゼIVの活性が主に原因である、生体内におけるタンパク質分解から保護されるという知見に基づく。少なくとも2つの構成要素(I)および(II)を有する本発明のペプチドは、GLP−1の生物活性を示し、かつC末端の延長によって構成要素(I)としてのGPL−1に安定性を付与する。
【0015】
本明細書において用いられる「本発明のペプチド」という用語は、本明細書において定義されるような融合ペプチド、その変異体、類似体、断片または誘導体(例えば、融合ペプチドの誘導体化された断片、類似体、または変異体の組み合わせが含まれる)のことである。本明細書において用いられる「GLP−1ペプチド」という用語は、GLP−1(7−35、36または37)を意味しており、一方において、「修飾されたGLP−1ペプチド」は、本発明のペプチドの構成要素(I)および(III)のいずれか一方に生じ得る、GLP−1(7−35、36または37)の誘導体化された断片、類似体、変異体を含む、任意のGLP−1類似体、GLP−1誘導体、GLP−1変異体またはGLP−1断片を意味することが意図される。本明細書において用いられる「GLP−2ペプチド」という用語は、GLP−2(1−33、34または35)を意味しており、その一方で、「修飾されたGLP−2ペプチド」は、GLP−2(1−33、34または35)の誘導体化された断片、類似体または変異体を含む、任意のGLP−2類似体、断片、変異体、GLP−2誘導体またはGLP−2類似体の誘導体を意味することが意図される。変異体、類似体、断片および誘導体は、修飾されていない配列(例えば、GLP−1(7−35、36または37)またはGLP−2(1−33、34または35))の修飾物として分類される。本発明の目的の範囲内において、任意の変異体、類似体、断片または誘導体は、機能的でなければならない(例えば、修飾されていないGLP−1と同じまたは類似の生物学的効果を及ぼさなければならない)。
【0016】
本発明のペプチドは、融合ペプチド、またはその変異体、類似体、断片もしくは誘導体であることが好ましい。ここで、構成要素(I)は、配列番号1と少なくとも80%、より好ましくは少なくとも85%、さらに好ましくは少なくとも90%の配列相同性を有している配列を含んでいる。配列番号1は、哺乳類において厳密に保存されている、GLP−1(7−37)の天然アミノ酸配列(31アミノ酸の長さ)を表している。
【0017】
本発明の融合ペプチド(またはより広く、融合ペプチドの類似体、断片、変異体もしくは誘導体を含む任意の本発明のペプチド)の第2の構成要素(構成要素(II))は、典型的に、βターン様構造を形成してもよりし、またはしていなくてもよい、少なくとも9アミノ酸の長さの配列を含んでいる。βターン様構造は、タンパク質またはペプチドの典型的な2次構造要素である。βターン様構造は、典型的に、ペプチドまたはタンパク質の中心鎖の向かう方向が逆戻りしている、4アミノ酸によって形成されている。構成要素(II)のアミノ酸配列は、少なくとも9つのアミノ酸を含んでおり、かつ好ましくはその配列の中に少なくとも1つのプロリンまたはアラニンを含んでいる。プロリン残基は、βターンを形成する4量体のアミノ酸配列に共通して含まれるアミノ酸である。プロリン残基は、融合ペプチドの構成要素(II)に存在する4量体のβターン配列の2または3位、好ましくは2位に代表的に配置されていることが共通している。
【0018】
本発明の融合物は、通常、構成要素(II)において、9〜30、好ましくは9〜20、さらに最も好ましくは9〜15アミノ酸の配列長を有している。一般的にいえば、構成要素(II)における配列が短いほど好ましい。というのも、より長い配列と比べてGLP受容体に対する結合活性がより優れているためである。必須の条件ではないが、構成要素(II)の配列は、中性であることが好ましく、またはpH7において負電荷を有していてもよい。
【0019】
本発明の融合タンパク質は、構成要素(II)がVAIA、IAEE、PEEV、AEEV、EELG、AAAA、AAVA、AALG、DFPE、AADX、AXDXおよびXADX(ここで、Xは、任意のアミノ酸(天然に生じるまたは修飾された天然ではないアミノ酸)を表す)から成る群から選択された配列モチーフを含んでいれば、好ましい。これらの4量体のモチーフは、構成要素(II)におけるあらゆる位置に配置されていてもよい。特に好ましい実施形態において、本発明の融合ペプチドの構成要素(II)は、AA、XA、AX、RR、RXおよびXR(ここで、Xは、任意のアミノ酸(天然に生じるまたは修飾された天然ではないアミノ酸)を表す)から成る群から選択された構成要素(II)のN末端配列モチーフにより、構成要素(I)のC末端と連結しているペプチド配列である。
【0020】
本発明の融合ペプチドにおける構成要素(II)の好ましいモチーフは、ヒトまたはマウスのIP−2の部分配列に対応する配列番号25の配列モチーフ(DFPEEVA)含んでいるか、または配列モチーフDFPEEVAと少なくとも80%の配列相同性を有する配列を含んでいる。
【0021】
融合ペプチドとしては、構成要素(II)が、配列番号22(RRDFPEEVAI)もしくは配列番号26(AADFPEEVAI)に記載の配列(すべてのペプチド配列は、一文字表記によって示されている)、または配列番号22もしくは26と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列であることが特に好ましい。配列番号22は、長さ15アミノ酸である全長IP−2配列におけるN末端側の10アミノ酸を含む、全長(ヒトまたはマウスの)IP−2配列(介在性ペプチド2)の部分配列である。配列番号26は、N末端の残基(RR)を(AA)に置換することによって配列番号22から誘導されている。IP−2の好ましい例は、βターンを含んでいるペプチド配列である。従って、構成要素(II)に含まれる他の特に好ましい配列は、ヒトに存在するN末端の14アミノ酸配列(配列番号23(RRDEPEEVAIVEEL))、もしくはマウスにおける等価物(配列番号24(RRDFPEEVAIAEEL))、または配列番号23もしくは24と少なくとも80%の配列相同性を有する配列のような、より長いIP−2の部分アミノ酸配列である。融合ペプチドの構成要素(II)に含まれる最も好ましい要素は、天然に生じるIP−2配列の15アミノ酸をすべて有する全長IP−2配列(配列番号2(RRDFPEEVAIVEELG)のヒト、または配列番号3(RRDFPEEVAIAEELG)のマウス)、または配列番号2もしくは3と少なくとも80%の配列相同性を有する配列である。IP2の哺乳類におけるアイソフォームのすべて(哺乳類の間におけるIP2の天然変異体)もまた本発明の範囲に含まれる。この段落において述べた配列のすべては、天然に生じるN末端のモチーフ(RR)の代わりに、モチーフ(AA)、(AX)または(XA)を備えていてもよい(例えば、配列番号27(AADFPEEVAIVEEL)、配列番号28(AADFPEEVAIAEEL)、配列番号29(AADFPEEVAIVEELG)および配列番号30(AADFPEEVAIAEELG))。構成要素(II)に含まれる配列の1よりも多い複製物(コピー)が含まれていてもよい(例えば、IP2、またはIP2断片、IP2変異体もしくは類似体、または誘導体の2、3またはそれ以上の複製物)。
【0022】
従って、本発明のペプチドは、配列番号8に記載の配列(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELG)(すなわち、マウスIP2に対してC末端を介して任意のリンカー配列なしで連結されたGLP−1(7−37))、または、配列番号12に記載の配列(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELG)(すなわち、ヒトIP2に対して任意のリンカー配列なしでC末端を介して連結されたGLP−1(7−37))を含んでいることが好ましい。本発明のペプチドとしてさらに好ましい配列番号8および12の発明の変異体は、配列番号31(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIAEELG)、配列番号32(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIAEELG)、配列番号33(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIAEELG)、配列番号34(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIAAALG)、配列番号35(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFP)、配列番号36(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVA)、配列番号37(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELGRRHAC)、配列番号38(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIVEELG)、配列番号39(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIVEELG)、配列番号40(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIVEELG)、配列番号41(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIVAALG)および配列番42(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELGRRHAC)(すなわち、IP2配列の特定の類似体または変異体に対してC末端を介して任意のリンカー配列なしで連結されたGLP−1(7−37))である。配列番号8および12と少なくとも80%の配列相同性を有するこれらの変異体、類似体もしくは断片、またはこれらの誘導体が同様に含まれ、またそれらは好ましい。
【0023】
あらゆる理論に制限されることなく、これらを必要とするあらゆる患者に投与されるときに、GLP−1(7−35、36または37)の不安定性は非保護の3次元構造に起因していることが、本発明の発明者らによって結論されている。プロテアーゼは、インビボにおいてGLP−1(7−35、36または37)ペプチドを切断し、かつ急速にその生理活性を消失させることがある。GLP−1(7−35、36または37)のC末端にペプチド配列を連結することによって、その構造物は、酵素による分解に対する安定性を増す。構成要素(II)への強剛性の提供、および1次構造によって形成されるβターンの存在に起因して、付加的なC末端のペプチド配列(本発明の融合ペプチドの構成要素(II)に含まれている)が折り返されるならば、安定性の増強は向上すると思われる。しかし、本発明のペプチドの構成要素(II)におけるβターン構造は、酵素による分解に対する構成要素(I)のGLP−1配列の安定化に必須であるとは、思われない。C末端のペプチドの伸長部分(例えば、βターン構造要素を含む)の長所によって本発明のペプチドは、DPP−IVの不活性化に対する耐性の向上が見られる。C末端のペプチドは、標的細胞にある受容体に作用する前にGLP−1(7−35、36または37)配列から切断されない、またはインビボにおいて酵素的に切断されてGLP−1(7−35、36または37)を形成する、の内のいずれか一方であってもよい。GLP−1受容体の部位と結合した本発明のペプチドの正確な形成に関係なく、本発明のペプチドは、活性なインシュリン分泌性の化合物としてその機能を発揮する。
【0024】
βターン要素を形成する1次構造のおかげで、構成要素(II)に含まれることが適すると思われるペプチド配列は、当該分野における当業者によって公知の適切な(例えば、(例えば、円偏光2色性の)分光法)、または他の方法によって容易に決定され得る。
【0025】
構成要素(II)および構成要素(I)は、直接に連結されていても、またはリンカー配列を介して連結されていてもよい。両方の構成要素は、互いに直接に連結されていることが好ましい。両方の構成要素がリンカー(またはスペーサ)を介して連結されている場合に、当該リンカーは、ペプチドリンカーまたは有機リンカーであることが好ましい。ペプチドリンカーは、典型的に1〜10アミノ酸、好ましくは1〜5、より好ましくは1〜3のアミノ酸の長さを有しており、ある場合において、当該ペプチドリンカーは、11〜50アミノ酸を含むより長いものであってもよい。ペプチドリンカーは、種々のアミノ酸配列から構成されていてもよい。ペプチドリンカーにより、連結される構成要素の間に構造的な自由度がもたらされる。構造的な自由度は、例えば、リンカー配列内に種々のグリシンまたはプロリン残基、好ましくは少なくとも30%、より好ましくは少なくとも40%、さらに好ましくは60%のプロリンおよびグリシン残基を含むペプチドリンカーを有することによって達成される。特定の配列とは関係なく、当該ペプチドリンカーは、免疫学的に不活性であることが好ましい。
【0026】
本発明の好ましい実施形態において、本発明のペプチド(例えば、融合ペプチド、またはその類似体、断片、変異体もしくは誘導体)は、構成要素(II)のC末端、および/または構成要素(I)のN末端のいずれかと連結されている、第3の構成要素(III)を含んでいる。構成要素(III)は、構成要素(II)のC末端に配置されていることが好ましい。構成要素(III)が(そのC末端によって)構成要素(I)のN末端または(そのN末端によって)構成要素(II)のC末端と連結されているか否かに関わらず、その結合は、直接的またはリンカー配列を介して間接的であってもよい。上記リンカー配列に関して、構成要素(I)および構成要素(II)と接続しているリンカーとして上述において開示されているものが好ましい。通常、構成要素(III)は、少なくとも4アミノ酸残基、好ましくは少なくとも10アミノ酸残基、より好ましくは少なくとも20または少なくとも30アミノ酸残基を含んでいる。機能的な観点から、構成要素(III)が備えられることによって、本発明のペプチドの安定性がさらに増強する。構成要素(III)は、GLP−1(7−37)の生物活性にほぼ類似する、本発明のペプチドの生物学的な機能を損なわないことが期待される。
【0027】
本発明のペプチドの構成要素(III)は、任意の哺乳生物のGLP−2のアイソフォーム((哺乳類において他の天然に生じるGLP−2のアイソフォーム)、例えば、配列番号4および5に示されているようなマウスまたはヒトのアイソフォーム)のN末端配列における、少なくとも4、好ましくは少なくとも10、より好ましくは少なくとも20の付加的なアミノ酸残基を包含することが好ましい。GLP−2は、プログルカゴンにおいて生じ、かつまた炭水化物代謝に関与する。構成要素(I)(GLP−1ペプチド)に含まれる生物学的に活性な配列と同様に、構成要素(III)もまた、GLP−2の天然に生じる形態の類似体、変異体または誘導体を含んでいてもよい。また、構成要素(III)は、GLP−1(7−37)(なお、これに対応する、すべての哺乳類のアイソフォーム、または本明細書に開示されているような、それらのすべての機能的な変異体、類似体もしくは誘導体も含まれる)のN末端配列の少なくとも4、好ましくは10、より好ましくは20の付加的なアミノ酸残基を含んでいてもよい。一般的に言えば、構成要素(III)は、本発明のペプチドの構成要素(I)に好適なものとして本明細書に開示されている、GLP−1ペプチド、または修飾されたGLP−1ペプチドの任意の形態を含んでいてもよい。さらに、構成要素(III)はまた、GLP−1(7−37)とGLP−2とのキメラ形態を含んでいてもよい。キメラ形態は、GLP−1(7−37)とGLP−2と(または両方の断片、類似体、変異体もしくは誘導体と)を互いに連結させて、構成要素(III)としてのキメラ形態を本発明のペプチドに導入することによって、生成されていてもよい。上記キメラ形態は、互いに連結されたGLP−1(7−37)の部分配列およびGLP−2の部分配列から構成されることが好ましい。例えば、上記キメラ形態は、GLP−1のN末端における5〜30アミノ酸とGLP−2のC末端における5〜30アミノ酸とを含んでいてもよいし、その逆であってもよい(例えば、GLP−1(7−37)の7または8〜22、23、24、25、26、27または28アミノ酸と、GLP−2の15、16、17、18、19、20、21、22、23または24位から例えば、C末端までのアミノ酸)。
【0028】
GLP−2またはGLP−1(7−37)の天然に生じる形態の修飾物のそれぞれが、構成要素(III)として使用されるならば、構成要素(III)は、配列番号4もしくは5、または配列番号1の配列のそれぞれ、あるいは配列番号4もしくは5、または配列番号1と少なくとも80%の配列相同性を有する配列を含むことが好ましい。また、例えば、側鎖の修飾またはペプチド骨格の修飾などの結果である(ここで開示されている「誘導体」に属するような)、これらの好ましい配列の誘導体は、本発明による構成要素(III)として包含される。
【0029】
他の実施形態において、構成要素(III)は、上述のような配列を複数含んでいてもよい。例えば、構成要素(III)は、GLP−1(7−37)および/またはGLP−2の少なくとも2、好ましくは2、3、または4の複製物、または配列番号1、4または5と少なくとも80%の配列相同性を有する配列の少なくとも2つの複製物を含んでいてもよい。また、構成要素(III)は、例えば、GLP−1(7−37)および/またはGLP−2あるいは少なくとも80%の配列相同性を有するその修飾物とともにキメラ型の組み合わせを最終的に形成する、上述したような、GLP−1(7−37)またはGLP−2のキメラ型の複製物以外も含んでいてもよい。また、例えば、(1)そのN末端によって構成要素(II)のC末端と、および(2)そのC末端によって構成要素(I)のN末端と、リンカーを介してまたは直接に連結されている2つ以上の、好ましくは2つの構成要素(III)は、本発明の範囲内にある。2つの構成要素(III)が備えられている場合には、これらは、同一であっても、異なっていてもよい。
【0030】
従って、3つの構成要素(I)、(II)および(III)を含む発明の融合ペプチドは、特に好ましい。これらの構成要素のすべてを含んでいる4つの特定の実施形態は、配列番号6(N−GLP−1(7−37)−IP2(マウス)−RR−GLP−1(7−37)−C、またここではマウスCM1を表している)、配列番号7(N−GLP−1(7−37)−IP2(マウス)−RR−GLP−2−C、またここではマウスCM2を表している)、配列番号10(N−GLP−1(7−37)−IP2(ヒト)−RR−GLP−1(7−37)−C、またヒトCM1を表している)および配列番号11(N−GLP−1(7−37)−IP2(ヒト)−RR−GLP−2−C、またヒトCM2を表している)、または配列番号6、7、10もしくは11と少なくとも80%の配列相同性を有する配列、またはこれらの誘導体から成る群から選択される。配列6、7、10および11のすべては、IP2(構成要素(II))のC末端にRR−リンカー(2つのアルギニン残基)を含んでいる。なお、このRR−リンカーは、代替案においてはなくてもよい。配列番号6、7、10または11の各実施形態における構成要素(I)は、GLP−1(7−37)であり、(構成要素(II)のC末端に連結されたこれらの各実施形態において)構成要素(III)は、GLP−1(7−37)またはGLP−2である。
【0031】
本発明のペプチドは、種々の修飾された形態を生じてもよい。これらの修飾された形態は、以下に開示され、より詳細に説明されている。
【0032】
本明細書において、「塩」という用語は、上述した融合ペプチド、またはその類似体、断片、誘導体もしくは変異体のカルボキシル基の塩およびアミノ基の酸付加塩の両方を意味する。カルボキシル基の塩は、当業者に公知の方法によって形成されてもよく、例えば、ナトリウム、カルシウム、アンモニウム、鉄、亜鉛の塩などの無機塩、ならびに例えば、エタノールアミン、アルギニンもしくはリジンのようなアミン、ピペリジンおよびプロカインなどを用いて形成されるような有機塩基を有する塩を含む。酸付加塩は、例えば、塩酸または硫酸のような例えば、無機酸を有する塩、ならびに例えば、酢酸またはシュウ酸のような有機酸を有する塩を含む。もちろん、該当するあらゆる塩は、本発明に適した本発明のペプチドの生物活性(例えば、血液の循環に入る栄養物の割合を減少させる能力)を保持する必要がある。ペプチド塩の形態は、薬学的製剤に含まれていてもよい。
【0033】
本発明の融合ペプチドの「断片」は、所望の生物活性を保持しているより短いペプチドである、分子の任意の部分集合を意味する。断片は、上記分子のいずれかの末端からアミノ酸を取り除き、生じたものをインクレチンのようなその特性に関して試験することによって容易に調製され得る。ポリペプチドのN末端および/またはC末端のいずれかから同時に1つのアミノ酸を取り除くプロテアーゼは、公知であり、このため、所望の生物活性を保持する断片の決定は、決まりきった実験のみを含んでいる。結論として、断片は、ペプチド末端におけるアミノ酸および/またはペプチド配列内にあるアミノ酸の欠損に原因し得る。
【0034】
さらに、自身が融合ペプチド、類似体、変異体、機能的な誘導体および/またはこれらの断片である、抗2型糖尿病活性を有する本発明のペプチドは、本発明のペプチドに隣接する付加的なアミノ酸残基をまた含むことができる。生じた分子がプロテアーゼに対する耐性または安定性を保持し、かつ当該分子がインクレチンとして作用する能力を保持する限りにおいて、任意の当該隣接する残基が、核になるペプチドの基本的なおよび新規な性質に影響するか否かを、例えば、決まりきった実験を用いた膵臓細胞に対する影響によって、決定することができる。「本質的に成る」という用語は、特定の配列に言及するとき、付加的な隣接する残基が特定の本発明のペプチドが有する基本的なおよび新規の性質に影響せずに存在し得ることを意味する。この用語は、特定の配列内における置換、欠損または付加を包含していない。
【0035】
本発明の「変異体」という用語は、上述した本発明のペプチドの全体またはその断片のいずれかと実質的に類似している分子を意味している。変異体ペプチドは、当該分野において公知の方法を用いた、変異体ペプチドの直接的な化学合成によって簡便に調製され得る。もちろん、本発明のペプチドの当該変異体は、類似の糖尿病性(例えば、対応する天然に生じるGLP−1ペプチドのようなインシュリン刺激活性)を有している。
【0036】
また、上述したペプチドのアミノ酸配列変異体は、合成された誘導体をコードするDNAにおける変異によって調製され得る。当該変異体は、例えば、アミノ酸残基からの欠損体、アミノ酸残基への挿入体、またはアミノ酸残基の置換体を含む。また、欠損、挿入および置換のあらゆる組み合わせにより、最終的なコンストラクトを完成させてもよいが、この場合、最終的なコンストラクトは所望の活性を保有している。当然、変異体ペプチドをコードするDNAに施される変異は、読み枠を変更する必要はなく、かつmRNAの2次構造の生成を可能にする相補的な領域を作り出さないことが好ましい。
【0037】
本発明の上述したペプチドの「類似体」は、分子の全体またはこれらの活性な断片のいずれかと実質的に類似の非天然分子を意味する。当該類似体は、対応する天然に生じるGLP−1ペプチドと同じ活性を示す。
【0038】
本発明に係る、本発明のペプチドに属して作製される置換の種類は、異なる種の相同なタンパク質/ペプチドの間におけるアミノ酸変化の頻度の解析に基づいていてもよい。当該解析に基づくと、保存された置換は、以下の5つのグループ内の1つの中における交換として本明細書では定義され得る:
I.小さな、脂肪族の、非極性のまたはわずかに極性の残基(Ala、Ser、Thr、ProおよびGly);
II.極性の、負電荷の残基(Asp、Asn、GluおよびGln)およびそれらのアミド;
III.極性の、正電荷の残基(His、ArgおよびLys);
IV.大きい、脂肪族の非極性の残基(Met、Leu、Ile、ValおよびCys)、ならびに;
V.大きな芳香族の残基(Phe、TryおよびTrp)。
【0039】
上記グループにおいて、下記の置換が、「保存性が高い」と考えられる:Asp/Gul;His/Arg/Lys;Phe/Tyr/Trp;Met/Leu/Ile/Val。保存性が中程度である置換は、上記(I)、(II)、および(III)を含んでいるスーパーグループ(A)または、上記(IV)および(V)を含んでいるスーパーグループ(B)に限定される、グループ(I)−(IV)のうちの2つの間において、交換されるように定義される。置換は、遺伝的にコードされるアミノ酸、または天然に生じるアミノ酸に限定されない。
【0040】
通常、本発明のペプチドの類似体または変異体はまた、例えば、可溶性の向上(親水性アミノ酸による疎水性アミノ酸の置換)を目的として作製されたアミノ酸置換体を含む。本発明のペプチドであるGLP−1ペプチドの(本発明のペプチドの構成要素(I)および/または(III)において生じる)変異体/類似体の一実施形態において、(修飾された)GLP−1ペプチドは、GLP−1ペプチドの7、8、11、12、16、22、23、24、25、27、30、33、34、35、36または37位における1つ以上の置換を特徴としている。例えば、以下の名称のように、[Arg34−GLP−1(7−37)]は、天然に生じる34位のリジンがアルギニンにより置換されているGLP−1類似体を示している。
【0041】
特に、本発明のペプチドの構成要素(I)および/または(III)は、例えば、Gln9−GLP−1(7−37)、D−Gln9−GLP−1(7−37)、アセチル−Lys9−GLP−1(7−37)、Thr16−Lys18−GLP−1(7−37)、ならびにLys18−GLP−1(7−37)、Arg34−GLP−1(7−37)、Lys38−Arg26−GLP−1(7−38)−OH、Lys36−Arg26−GLP−1(7−36)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26および34−Lys38−GLP−1(7−38)、Arg26−Lys38−GLP−1(7−38)、Arg26−Lys38−GLP−1(7−38)、Arg26−Lys38−GLP−1(7−38)、Arg34−Lys38−GLP−1(7−38)、Ala37−Lys38−GLP−1(7−38)、ならびにLys37−GLP−1(7−37)を含むGLP−1(7−35、36または37)の変異体および類似体を含んでいてもよい。
【0042】
本発明の他の実施形態において、本発明のペプチドは、構成要素(I)または(III)として、以下の一般式II(配列番号44):
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Xaa16−Ser−Xaa18−Xaa19−Xaa20−Glu−Xaa22−Xaa23−Ala−Xaa25−Xaa26−Xaa27−Phe−Ile−Xaa3o−Trp−Leu−Xaa33−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノヒスチジン、2−アミノヒスチジン、3−ヒドロキシヒスチジン、ホモヒスチジン、N−アセチルヒスチジン、a−フルオロメチルヒスチジン、a−メチルヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸または(1−アミノシクロオクチル)カルボン酸を表し(ここで、Xaa8はGlyを表すことが特に好ましい);Xaa16は、ValまたはLeuを表し;Xaaは、Ser、LysまたはArgを表し;Xaa19は、TyrまたはGlnを表し;Xaa20は、LeuまたはMetを表し;Xaa22は、Gly、GluまたはAibを表し;Xaa23は、Gln、Glu、LysまたはArgを表し;Xaa25は、AlaまたはValを表し;Xaa26は、Lys、GluまたはArgを表し;Xaa27は、GluまたはLeuを表し;Xaa30は、Ala、GluまたはArgを表し;Xaa33は、ValまたはLysを表し;Xaa34は、Lys、Glu、AsnまたはArgを表し;Xaa35は、GlyまたはAibを表し;Xaa36は、Arg、GlyもしくはLys、またはアミドを表すか、あるいは存在せず;ならびにXaa37は、Gly、Ala、Glu、Pro、もしくはLys、またはアミドを表すか、あるいは存在しない。)
のアミノ酸配列を含んでいる修飾されたGLP−1を含んでいる。
【0043】
本発明の他の実施形態において、本発明のペプチドの構成要素(I)および/または(III)は、以下の一般式式III(配列番号45):
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Xaa8−Tyr−Leu−Glu−Xaa22−Xaa23−Ala−Ala−Xaa26−Glu−Phe−lle−Xaa30−Trp−Leu−Val−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノヒスチジン、2−アミノヒスチジン、−ヒドロキシヒスチジン、ホモヒスチジン、N−アセチルヒスチジン、a−フルオロメチルヒスチジン、a−メチルヒスチジン、3−ピリジルアラニン、2−ピリジルアラニンまたは4−ピリジルアラニンを表し; Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸または(1−アミノシクロオクチル)カルボン酸を表し; Xaa18は、Ser、LysまたはArgを表し; Xaa22は、Gly、GluまたはAibを表し; Xaa23は、Gln、Glu、LysまたはArgを表し; Xaa26は、Lys、GluまたはArgを表し;Xaa30は、Ala、GluまたはArgを表し; Xaa34は、Lys、GluまたはArgを表し;Xaa35は、GlyまたはAibを表し; Xaa36は、ArgもしくはLys、またはアミドを表すか、あるいは存在せず; Xaa37は、Gly、Ala、GluもしくはLys、またはアミドを表すか、あるいは存在していない。)
のアミノ酸配列を含んでいる修飾されたGLP−1ペプチドを含んでいる。
【0044】
本発明の特に好ましい実施形態において、本発明のペプチドの構成要素(I)および/または(III)は、GLP−1(7−35)、GLP−1(7−36)、GLP−1(7−36)−アミド、GLP−1(7−37)またはこれらの変異体、類似体もしくは誘導体から選択される(修飾された)GLP−1ペプチドを含んでいる。また、本発明のペプチドは、構成要素(I)および/または(III)に、GLP−1ペプチドの8位にAib残基を、あるいはGLP−1ペプチドの7位にD−ヒスチジン、脱アミノヒスチジン、2−アミノヒスチジン、ヒドロキシヒスチジン、ホモヒスチジン、N−アセチルヒスチジン、a−フルオロメチルヒスチジン、a−メチルヒスチジン、3−ピリジルアラニン、2−ピリジルアラニンおよび4−ピリジルアラニンから成る群から選択される1つのアミノ酸残基を有する修飾されたGLP−1ペプチドを含んでいることが好ましい。
【0045】
本発明に使用する類似体を得るために使用され得るタンパク質におけるアミノ酸の置換体の生成例は、Markらによる米国再発行特許発明第33.653号明細書、米国特許第4,959,314号明細書、米国特許第4,588,585号明細書および米国特許第4,737,462号明細書、Kothsらによる米国特許第5.116.943号明細書、Namenらによる米国特許第4.965.195号明細書ならびにLeeらによる米国特許第5.017.691号明細書に示されているような、あらゆる周知の方法段階を含み、かつ米国特許第4.904.584号明細書(Shawら)に示されているリジンに置換されたタンパク質を含む。
【0046】
好ましくは、上述の、かつ構成要素 (I)、(II)および/または(III)に含まれる変異体または類似体は、「天然の」配列(例えば、天然アミノ酸配列と少なくとも70%の相同性を有し、かつこれらの生物活性を維持する、GLP−1(7−37)、GLP−2もしくはこれらの生物学的に活性な断片またはあらゆるIP2アイソフォーム)と同じである核となる配列を有する。さらに好ましくは、当該配列は、天然の配列と少なくとも80%の相同性、少なくとも90%の相同性、最も好ましくは少なくとも95%の相同性を有する。ここで、特定のペプチドは、決まった長さの参照ポリペプチドと特定の割合の相同性を有し、その相同性の割合は、参照ペプチドに対する比である。従って、100アミノ酸の長さである参照ポリペプチドと50%の相同性であるペプチドは、参照ポリペプチドにおける50アミノ酸の長さの部分と完全に一致している。また、その全長に渡って参照ポリペプチドと50%の相同性であるペプチドは、100アミノ酸の長さのポリペプチドである。当然、他のポリペプチドは、同じ判断基準に合致する。本明細書で使用されるような、「配列相同性」という用語は、当該配列が以下のように比較されることを意味している。当該配列は、遺伝的な演算群のGAP(Genetic Computing Group’s global alignment program)の様式9を用いて整列される。このGAPの様式9では、間隔が空く(間隔の初めの空白に対する)罰則(−12)、および間隔が伸びる(間隔における付加的な連続する空白につき)罰則(−4)を用いる、初期(BLOSUM62)のマトリックス(−4〜+11の値)が使用される。整列の後に、要求した配列におけるアミノ酸数の百分率として合致した数を表すことによって、相同性の百分率が演算される。
【0047】
また、融合ペプチドまたはその類似体、断片もしくは変異体の誘導体は、本発明に包含される。本発明のペプチドの「誘導体」という用語は、1つのアミノ酸が別の20の一般的に生じる天然アミノ酸に変化されていない、上記修飾された本発明のペプチドのみを意味している。その結果、遺伝的にコードされているアミノ酸は、(i)選択された残基の側鎖、または末端残基のアミノ基またはカルボキシル基と(好ましくは共有結合修飾することによって)反応可能な有機誘導試薬と、遺伝的にコードされているアミノ酸を反応させることによって、あるいは、(ii)非天然アミノ酸または遺伝暗号にコードされていない天然アミノ酸(例えば、ヒドロキシプロリン、γ−カルボキシグルタミン酸、オルチニン、ホスホセリン、D−アラニン、およびD−グルタミン)の導入によって、あるいは、(iii)別のペプチド骨格配列によるペプチド骨格の修飾によって修飾されていてもよい。上記非天然アミノ酸は、化学合成によって作製されたものであり、すなわち、遺伝暗号によりコードされるアミノ酸のD−アイソマー、Aib(a−アミノイソブチル酸)、Abu(a−アミノブチル酸)、Tie(tert−ブチルグリシン)、p―アイアニン、3−アミノメチル安息香酸、およびアントラニル酸である。
【0048】
以下において、例えば、構成要素 (I)、(II)および/または(III)に配置されている本発明のペプチドの任意の部位(任意のアミノ酸)に生じ得る、本発明のペプチド(上述のように、融合ペプチドの変異体、類似体、または断片を含む)におけるアミノ酸の好ましい修飾が、開示されている。
【0049】
本発明のペプチドの任意の形態(例えば、本発明のペプチドの類似体)においてシステイニル残基が存在するならば、システイン残基は、クロロ酢酸またはクロロ酢酸アミドのようなアルファハロ酢酸塩(および対応するアミン)とほとんど普遍的に反応し、カルボキシルメチルまたはカルボキシアミドメチル誘導体が得られる。また、システイニル残基は、ブロモトリフルオロアセトン、アルファ−ブロモ−ベータ−(5−イミダゾイル)プロピオン酸、クロロ酢酸リン酸塩、N−アルキルマレイミド、3−ニトロ−2−ピリジル二硫化物、メチル−2−ピリジル二硫化物、p−塩化水銀安息香酸、2−塩化水銀−4−ニトロフェノール、またはクロロ−7−ニトロベンゾ−2−オキサ−1,3−ジアゾール用いた反応によって誘導体化される。
【0050】
本発明におけるヒスチジン残基は、pH5.5−7.0におけるジエチルプロカーボネート(この試薬は、ヒスチジン側鎖に比較的に特異的であるので、)を用いた反応によって誘導体化される。パラブロモフェナシルブロマイドもまた有効であり、この反応は、pH6.0の0.1Mのナトリウムカコジル酸において好ましく実施される。特に、本発明のペプチドの構成要素 (I)および/または(III)に含まれるようなGLP−1(7−37)のN末端ヒスチジン残基は、Suzukiら(Diabetes Res.; Clinical Practice 5 (Supp. 1): S30 (1988))によって示されているように、GLP−1ペプチドのインシュリン分泌性の活性に非常に重要である。これに対応して、本発明のペプチドは、アルキルもしくはアシル(C1−6C)基により、構成要素 (I)および/または(III)の一部としてのGLP−1のHis7を修飾してもよいし、機能的に等価なC5−C6の環構造によりHisを置換することにより、構成要素 (I)および/または(III)の一部としてのGLP−1のHis7を修飾してもよい。好ましい修飾は、His7のアミノ末端またはそのヒスチジン側鎖に疎水性の部分を導入することである。
【0051】
本発明のペプチドにおけるリジニル残基およびアミノ末端残基は、例えば、琥珀酸または他のカルボン酸の無水物と反応されてもよい。これらの試薬を用いた誘導は、リジニル残基の電荷を逆転させる効果を有する。本発明のペプチドにおけるリジン残基のイプシロンアミノ基のアシル(C12−C18)修飾によって、該ペプチドの血液循環における半減期が増大する。アルギニン残基は、従来の試薬の1つまたはいくつか(フェニルグリオキサールおよびニンヒドリンなど)を用いた反応によって修飾されてもよい。アルギニン残基の誘導は、グアジニン官能基の高いpKaのために、その反応がアルカリ条件において行われる必要がある。さらに、これらの試薬は、アルギニンイプシロンアミノ基と同様に、リジン基と反応し得る。
【0052】
チロシン残基の特異的修飾は、それ自体、広範囲に亘って研究されている。通常、N−アセチルイミダゾールはO−アセチルチロシル種を、またテトラニトロメタンはe−ニトロ誘導体を形成するために用いることができる。
【0053】
カルボキシルの側基(アスパルチルまたはグルタミル)は、1−シクロヘキシル−3−〔2−モルフォリニル−(4−エチル)〕カルボジイミド、または1−エチル−3−(4−アゾニア−4,4−ジメチルフェニル)カルボジイミドのようなカルボジイミド(R’N−C−N−R’)との反応によって選択的に修飾されてもよい。
【0054】
さらに、アスパルチルおよびグルタミル残基は、アンモニウムイオンとの反応によって、アスパラギニルおよびグルタミニル残基に転換されてもよい。
【0055】
グルタミニルおよびアスパラギニル残基は、対応するグルタミルおよびアスパルチル残基に脱アミド化されてもよい。また、これらの残基は、弱酸性の環境において脱アミド化されてもよい。これら残基の形態のうち、どちらかは本発明の範囲に含まれる。本発明のペプチドを脱アミド化すると、プロテアーゼ酵素またはペプチダーゼ酵素によるタンパク質分解に対する感受性が変化することがある。このことから、脱アミド化は、本発明のペプチドのタンパク質分解による切断に向かわせる、生理学的な重要性を有する可能性があることが示唆される。なお、本発明の生合成ペプチドは、或る保存条件において分解することがあり、これにより本発明のペプチドにおける別の位置において脱アミド化が起こるということに注意されたい。本発明のペプチド中のメチオニン残基は、酸化の影響を受けやすく、それは主にスルホキシドによる影響である。上述のその他の誘導体として、本発明の脱アミドペプチドおよび/または、本発明のスルホキシドペプチドは、十分な生物活性を示すために用いられてもよい。
【0056】
アルファアミノ酸含有残基を誘導体化する、その他の好ましい試薬には、イミドエステルが含まれる。上記イミドエステルとしては、例えば、メチルピコリニミデート;ピリドキサルリン酸;ピリドキサル;クロロボロヒドリド;トリニトロベンゼンスルホン酸;0−メチルリオスレア;2,4−ペンタンジオン;およびグリオキシレートと反応するトランスアミナーゼ触媒等が挙げられる。
【0057】
適切なアミノまたはカルボキシル保護基を用いることによって、カルボキシ基(C−末端)およびアミノ基(N−末端)(上述したカルボキシまたはアミドアミノ酸の側鎖基と同様の)を有する本発明のペプチドの末端アミノ酸残基は、当該残基を保護された(例えば、アミド基によるC末端の保護のような)および/または保護されていない形状で存在し得る。また、本発明のペプチドの酸添加塩が提供されてもよい。代表的な酸添加塩は、ハロゲン化水素酸塩である。そのようなハロゲン化水素酸塩としては、例えば、HBrまたはHIが挙げられるが、HClであることがより好ましい。
【0058】
本発明のペプチドに存在するリジンの末端、側鎖カルボキシル基、またはイプシロン−アミノ基におけるPEG付加は、酸化に対する耐性を与えるものであり、またそれは本発明の範囲に含まれる。
【0059】
本発明のペプチドの誘導体に起こるその他の修飾としては、本発明のペプチドと共有結合され得る炭水化物および/または脂質に基づくものである。脂質および/または炭水化物は、セリン、スレオニン、アスパラギン、グルタミン、またはチロシンと結合するか、あるいは、グルタミン酸塩、またはアスパラギン酸塩とそれらの反応性側鎖の構成成分を経由して結合することが好ましい。また、炭水化物および/または脂質は、本発明のペプチドの末端構成成分と結合されてもよい。さらに、本発明のペプチドは、機能的に異なるペプチドまたはタンパク質構成成分と結合されてもよい。これにより、本発明のペプチドが安定化されて、および/または体液中、特に血液中における本発明のペプチドの輸送性を改善する働きがもたらされてもよい。ペプチドまたはタンパク質としては、例えば、アルブミン、およびトランスフェリン等から選択されることが好ましい。これらは、本発明のペプチドと直接結合されるか(構成要素IVとして)、またはペプチドもしくは有機連結配列を経由して結合される。これらのペプチドまたはタンパク質は、本発明のペプチドの末端の1つと結合されることがより好ましい。
【0060】
本発明のペプチドにおける分解という問題を解決するために、本発明の別の実施形態では、Dアミノ酸または、少なくとも一部分はDアミノ酸から成る本発明のペプチドのレトロインバーソ異性体が提供される。「レトロインバーソ異性体」とは、配列の方向性が逆であり、各アミノ酸残基のキラリティーが反転した直鎖ペプチドの異性体を意味する(Jamesonら, Nature, 368, 744-746 (1994); Bradyら, Nature, 368, 692-693 (1994)を参照)。もとのペプチドに関しては、当該レトロインバーソペプチドは、アミノ酸、特にF−mocアミノ酸誘導体を用いて、アミノ酸を逆の順序で結合される。特に、粗精製のペプチドは、逆相HPLCによって精製されてもよい。
【0061】
本発明のペプチドに導入されてもよいその他の修飾は、ペプチド骨格の修飾に関する。修飾された当該ペプチドは、骨格模倣物質であることが好ましい。当該骨格の側鎖構造は、本発明のペプチド、その断片、変異体、誘導体、または類似体と同一である。しかしながら当該骨格は、天然に生じる骨格とは異なっている。一般的に、骨格模倣物質では、1つ以上の当該骨格の鎖部分(NH,CH,CO)が修飾される。この修飾としては、挿入、または置換のどちらかがあるが、置換がより好ましい。置換基としては、例えば、(I)―NH−と置換される−O−,−S−,および−CH2−;(II)−CHR−と置換される−N−,−C−アルキル−,および−BH−;および(III)−CO−と置換される―CS−,−CH2−,−SOn−,−P=O(OH)―および−B(OH)−が挙げられる。本発明のペプチドの模倣ペプチドは、上記修飾を組み合わせてもよい。特に、I、II、およびIIIの各基の修飾は、組み合わせることができる。模倣ペプチドにおいて、各骨格の鎖部分は修飾されてもよく、また、鎖部分のある程度が人工的に生じる部分と交換されてもよい。本発明のペプチドにおけるすべての骨格の鎖部分における各―NH−,−CHR−またはCOは、別の人工的に生じる基と交換されることが好ましい。本発明のペプチド骨格のアミド結合(―NH−CO−)が置換される場合(分子全体であるか、または少なくとも1つ位置において)には、置換部分は、生物学的に等価であるもの、例えば例えば、レトロインバースアミド結合(―CO−NH−)、ヒドロキシルエチレン(−CH(OH)−CH2−)、アルケン(CH2=CH−)、カルバ(CH2−CH2−)および/または−P=O(OH−CH2−)が挙げられる。また、挿入による当該骨格の鎖伸張は、本発明のペプチドの骨格模倣物質において(例えばC−アルファ原子に隣接する部分の近くで)起こってもよい。C−アルファ原子の片側の部分において、例えば−O−、−S−、−CH−、―NH−が挿入されてもよい。
【0062】
さらに、本発明のペプチドは、オリゴカルバメートペプチド骨格構造であることがより好ましい。上記アミド結合は、カルバメート部分において置換される。N−保護アミノアルキル炭酸のモノマーは、対応するアミノ酸またはアミノアルコールを経由して利用可能であり、活性エステルに転換される。そのような活性エステルとしては、例えば、F−moc部分を用いてp−ニトロフェニルエステルに転換されてもよいし、また、は固相合成を用いて光感受性ニトロアトリルオキシカルボニル基に転換されてもよい。
【0063】
本発明のペプチドは、上述のようなタンパク質分解的切断から保護されるものであり、特に、ジペプチジルアミノペプチターゼ−4(DPP−IV)に対して保護される。本明細書において、「DPP−IVからの保護」とは、請求項1に係るペプチドに関して用いられる。本発明のペプチド、ならびに、当該ペプチドの誘導体、類似体、断片、および変異体により、本発明のペプチドの構成要素(I)および/または(III)の一部としてのGLP(7−35、36または37)は、血しょうペプチダーゼ(DPP−IV)対する耐性を獲得する。
【0064】
ジペプチジルアミノペプチダーゼIVによる分解に対するペプチドの耐性は、例えば、以下の分解分析によって測定される。すなわち、当該ペプチドのアリコートは、pH7〜8の最適な緩衝液(アルブミンは含まれていない)において、4〜22時間精製されたジペプチジルアミノペプチダーゼIVのアリコートと共に、37℃でインキュベートされる。酵素反応は、トリフルオロ酢酸の添加によって終結され、その後、ペプチドの分解生成物はHPLCまたはLC−MS分析によって分離および定量される。この分析を実施するための方法の1つとして、次の方法が挙げられる。すなわち、混合物は、Zorbax300SB−C18(30nmの微細孔、5μm粒子)150×2.1mmカラムの上に乗せられ、0.1%トリフルオロ酢酸中にアセトニトリルが直線勾配で、30分間で0%〜100%になるようにしながら、0.5ml/分の流速で溶解される。ペプチドおよびその分解生成物を、214nm(ペプチド結合)または280nm(芳香族アミノ酸)におけるそれらの吸光度によって観察することが可能であり、それらのピーク領域の積分によって定量される。分解傾向は、分離されたピークのMSスペクトルを測定することが可能であるLC−MSを用いて測定することができる。一定時間における、原型を保持した/分解された化合物の百分率は、ペプチドDPP−IV安定性の評価に用いられる。
【0065】
一定時間における原型を保持した化合物の百分率に基づくGLP−1(7−37)よりも10倍安定であるとき、本発明のペプチドはDPP−IVに対して安定であると考えられる。このように、DPP−IVに対して安定である本発明のペプチドは、上記のようなGLP−1(7−37)よりも少なくとも10倍安定であることが好ましく、少なくとも20倍安定であることがより好ましい。安定性は、当業者に公知である如何なる方法によって評価されてもよく、例えば、本発明のペプチド溶液にDPP−IVを加えることによって試験されてもよいし、例えば、当該ペプチドの経時的な分解を測定してもよく、分光法、ウェスタンブロット分析、抗体スクリーニング等によって測定されてもよい。同時に、本発明のペプチドは、例えば、GLP−1(7−37)の天然のレセプター(GLP−1レセプター)と結合することによって、GLP−1(7−37)の効果を与える化合物として規定される。本発明のペプチドとGLP−1レセプターとの間の結合親和性は、天然に生じるGLP−1ペプチドとGLP−1レセプターとの結合親和性の少なくとも10%、より好ましくは少なくとも50%に相当するGLP−1レセプターとの結合親和性を有することが好ましい。上記結合親和性は、如何なる適切な方法によって測定されてもよく、例えば、表面プラズモン共鳴等が挙げられる。さらに、例えば、本発明のペプチドが、細胞にシグナルを伝達する細胞外液レセプターと結合することによって、細胞内液cAMPの形成を誘引することが好ましい。
【0066】
本発明のペプチドは、技術的にGLP−1(7−36)アミドおよびGLP−1(7−37)の製造方法と類似した固相ペプチド合成技術を用いて、合成的に製造することができる。また、その後、例えば、逆相HPLCカラム上の単純精製方法、または好ましいクロマトグラフィー法によって、実験室規模で精製され得る。
【0067】
しかしながら、本発明のペプチドは、当該ペプチドを産生する微生物細胞または動物細胞系である組み換え細胞において形成されることが好ましい。本発明のペプチドは、当該ペプチドが発現された細胞から、例えば、従来の分離技術を用いて単離されてもよい。このように単離された細胞を、インビボにおいて、例えば支持体および栄養素を含む適切な条件で飼育することが可能であり、細胞外に分泌されたタンパク質(すなわち本発明のペプチド)を、細胞外の培地から回収することができる。したがって、細胞内に導入されるように設計された配列は、本発明のペプチドの分泌を導く、リーダー配列およびシグナルペプチド配列を含むことがより好ましい。上記細胞は、内在的に、または組み換え遺伝子配列によって、リーダー配列およびシグナル配列を分解することができるプロテアーゼを発現することが好ましい。別の方法において、本発明のペプチドをコードしている組み換え遺伝子配列は、上記リーダー配列およびシグナルペプチド配列を含まない。そのため、細胞内に発現された本発明のペプチドは分泌されず、細胞を溶解する段階を含む工程によって、細胞から回収される。上記方法において、コード化している配列は、培地から産生ペプチドの効率的な抽出を可能にする精製タグを含んでもよい。なお、当該タグとは、本発明のペプチドを放出するために切断されてもよい。
【0068】
さらに、本発明は、本発明のペプチド、あるいは融合ペプチド、またはその断片、類似体、もしくは変異体をコードする核酸を提供する本発明のペプチドをコードしている核酸であれば、任意のものが本発明に包含される。遺伝コードの縮重のため、多数の核酸配列は本発明のペプチドをコードしてもよい。本発明の範囲に含まれる核酸分子は、本発明のペプチドをコードする核酸を含んでおり、さらに、(機能的な)ヌクレオチド配列を含んでもよい。本発明の好ましい実施形態において、上記核酸分子は、(a)すべてのGLP−1 a a 配列(GLP−1(7−37)、または機能的GLP−1(7−35,36,および37)配列、また(b)任意のプロテアーゼのための(a)のGLP−1配列のN−末端における切断配列、をコードしてもよく、(b)から上流は、リーダー配列をコードすることができる。別の好ましい実施形態において、(b)の核酸分子をコードしている核酸配列から上流は、さらに(c)シグナルペプチドをコードしている配列を含んでもよい。また、本発明の核酸分子は、配列(c)を有していてもよく、該配列(c)は、(a)の上流と、リーダー配列をコードしている任意の配列を含まない(b)との間に融合されている。なお、上記導入配列およびシグナルペプチド配列は、プレプログルカゴンとは異なる種類であることが好ましい。
【0069】
さらに、本発明は、本発明の核酸(分子)および本発明の核酸(分子)の発現のためのその他の機能的な構成要素を含んでいるベクターを提供する。特に、本発明の核酸(分子)は、プロモーター配列と融合され、最終的には、例えば、エンハンサー配列のようなその他の調節配列と結合される。複製のために、上記プラスミドは複製開始点を含んでもよい。当該ベクターをトランスフェクションされる細胞を選択するために、1つ以上の抗生物質耐性遺伝子(例えば、カナマイシン、アンピシリン)がベクターに備えられてもよい。当該ベクターは、哺乳類細胞中において複製および発現するための、バクテリア由来プロモーター、抗生物質耐性遺伝子および複製起点プロモーター、および抗生物質耐性遺伝子を含むプラスミドであってもよい。さらに、本発明は、上述の前躯体タンパク質を翻訳することが可能な外因的に導入された本発明のDNAを含む宿主細胞を備えていてもよい。当該宿主細胞は、原核宿主細胞であるか、または、例えば哺乳類細胞のような真核宿主細胞であってもよい。
【0070】
さらに、本発明の態様によれば、成分(I)、(II)、ひいては成分(III)を含む本発明のペプチドの投与による動物(その中でもヒトであることが好ましい)の治療方法を提供する。また、糖代謝に関連する疾病または病気の治療および予防のための製品を製造するための、上記本発明のペプチドの対応する使用も提供する。糖疾患の例としては、これに限定されるものではないが、次のものを含む。すなわち、I型およびII型糖尿病(NIDDM)、インスリン耐性、体重異常および疾病、またはそれに関連する症状が挙げられる。なお、上記体重異常または関連する症状には、肥満、太りすぎに関連する症状、満腹中枢の調節不調、血しょうインスリン量の減少、血糖値上昇、または膵臓β細胞量の減少が含まれる。望ましくは、2型糖尿病(NIDDM)を治療するための薬物を製造するための本発明のペプチドの使用が、本明細書に開示されている。結果として、本発明は、例えば、被験者の体重を減少させるため、被験者の飽満を低下させるため、被験者の食後の血漿インスリン値を増加させるため、被験者の空腹時の血糖値を減少させるため、被験者の膵臓β細胞量を増加させるため、または被験者のI型またはII型糖尿病を治療するための本発明のペプチドの使用に関する。
【0071】
その他の病気または疾患のある患者は、本発明のペプチドによって治療されてもよく、例えば、融合ペプチド、または当該ペプチドの類似体、断片、変異体、誘導体によって治療されてもよい。本発明のペプチドは、神経変性疾患、および神経変性病、およびそれらに関連する状態を治療するための薬物、ならびに、アポトーシスに関連する疾患、病気および状態を治療するための薬物を調製するために用いられてもよい。以下のことから、これら障害を治療するために本発明のペプチドが使用される。すなわち、サイクリックAMP第2のメッセンジャー経路に関連するGLP−1レセプターが、げっ歯類およびヒトの脳の至るところに発現している。脳におけるレセプター分布の化学構造は、食事摂取量の調節および嫌悪ストレスへの応答におけるGLP−1の中心的な役割と関連しているだけではない。GLP−1レセプターにおいて結合しているGLP−1は、神経栄養性の性質を発揮し、培養された神経細胞において、グルタミン酸に誘導されるアポトーシスと酸化的損傷とに対して保護を与えることが示された。さらに、GLP−1は、細胞培養物においてアミロイドβ−プロテイン前躯体の修飾プロセシングを修飾し、インビボでの脳におけるアミロイドβ−ペプチド量の用量依存的な減少を示す。そのため、GLP−1は、中枢神経系の調節因子として知られている。生理活性型GLP−1の生物活性を模倣している本発明のペプチドは、例えば、アルツハイマー疾患(AD)、その他中枢および抹消神経変性障害(例えば、筋萎縮性側索硬化症(ALS)、アレキサンダー症、アルパース症、毛細血管拡張性運動失調症、カナバン症、コケーン症候群、クロイツフェルト・ヤコブ病、多発性硬化症、サンドホフ病、ピックス病、脊髄小脳失調、シルダー病、およびパーキンソン病)の治療に、治療上の関連性を有する。
【0072】
さらに、生理活性型GLP−1は、様々な細胞に抗アポトーシス作用を及ぼすことが示されている。例えば、GLP−1は、新たに単.離されたヒトの島細胞または他の種類の細胞の質量および機能の保存のために有用である。その範囲において、本発明の生物活性型ペプチドは、細胞または組織のアポトーシスによってもたらされる障害を治療するために用いられる。
【0073】
本発明のペプチドは、体外から投与される組成物の製造のために使用してもよく、その組成物は、単.離された本発明のペプチドを含んでいてもよい。得られた組成物は、上述した障害の治療として用いられてもよい。また、本明細書中に開示されている障害は、本発明の宿主細胞、核酸(分子)、およびベクター、によって治療されてもよい。さらに本発明の宿主細胞、核酸(分子)、およびベクターを、当該障害を治療するための薬物の調製に用いてもよい。
【0074】
活性成分として本発明のペプチドを含む製剤の調整は、米国特許第4,608,251号明細書;米国特許第4,601,903号明細書;米国特許第4,599,231号明細書;米国特許第4,599,230号明細書;米国特許第4,596,792号明細書;および米国特許第4,578,770号明細書において実証されているように、技術的に広く知られているところであり、これら全ては、本明細書に参考によって援用される。特に、上記製剤は溶液または懸濁液のように注入可能物質(水を含有していることが好ましい(水性製剤))として調製されるか、または乳化されてもよい。「水性製剤」とは、少なくとも50% w / w の割合で水を含んでいる溶液を意味する。同様に、「水性溶液」とは、少なくとも50% w / w の割合で水を含んでいる溶液を意味するものであり、「水性懸濁液」とは、少なくとも50% w / w の割合で水を含んでいる懸濁液を意味するものである。
【0075】
静脈注射、皮膚もしくは皮下注射、または病巣部位での注射のために、活性成分は非経口投与が可能な水性溶液の形状にされる。なお、当該水性溶液は、発熱物質がなく、適切なpH、等張性、および安定性を有する溶液である。一般的に液体の薬学的組成物は、水のような液体溶剤を含む。液体溶剤は、生理食塩水、D型グルコースエタノールを含んでもよく、その他の糖類溶液または、例えばエチレングリコール、プロピレングリコール、およびポリエチレングリコールのようなグリコール、またはその混合物が含まれてもよい。さらに、実施例は、リンガー液または乳酸加リンガー液のようなその他の液体溶媒である。
【0076】
本発明が、本発明の化合物の水性溶液および緩衝液を含んでいる薬学的製剤に関する場合には、上記化合物は0.1mg/ml以上の濃度において存在し、また上記製剤は約2.0から約10.0のpHを有することが好ましく、さらに約7.0から約8.5であることがより好ましい。上記製剤のpHは、本発明の化合物の等電点から少なくとも1pHユニットであることが好ましく、当該等電点から少なくとも2pHユニットであることがさらに好ましい。
【0077】
また、注入前の液体である溶液または懸濁液に入れるのに適した固形形態も調整され得る。薬学的製剤は、医者または患者が使用前に溶媒または希釈剤を加えるために、フリーズドライ製剤であってもよい。すなわち、一度調製された製剤は、患者へ直ちに投与されるわけではない。どちらかといえば、調製の後、溶液形態または患者へ投与するのに適した他の形態へと後で再構成するために、氷結状態または乾燥状態において貯蔵するように包装される。別の実施形態において、薬学的製剤は事前に溶解することなく使用する状態の乾燥製剤(例えば、フリーズドライまたはスプレードライ)である。「乾燥形態」は、液体の薬学的組成物または薬学的製剤が、(i)フリーズドライ(例えば、凍結乾燥;「Williams and Polli (1984) J. Parenteral Sci. Technol. 38: 48-59」を参照)、(ii)スプレードライ(「Masters (1991) in Spray-Drying Handbook (5th ed; Longman Scientific and Technical, Essez, U. K. ), pp. 491-676」; 「Broadheadら (1992) Drug Devel. Ind. Pharm. 18: 1169-1206」;および「Mumenthalerら (1994) Pharm. Res. 11: 12-20」を参照)、または、(iii)空気乾燥(「Carpenter and Crowe (1988) Cryobiology 25: 459-470; and Roser (1991) Biopharm. 4: 47-53」を参照)のいずれかによって乾燥されたものをいう。液体の薬学的組成物の貯蔵中に、ポリペプチドによる凝集が形成されると当該ポリペプチドの生物活性に、悪影響が及ぼされることがある。そして、これにより薬学的組成物の治療効力を損失するという結果が生じる。さらに、当該ポリペプチドを含んでいる薬学的組成物が輸液システムを用いて投与されるとき、形成された凝集は、管類、膜、またはポンプを閉塞するというようなその他の問題をもたらす可能性もある。
【0078】
その他の成分は、本発明のペプチド薬学的製剤において存在していてもよい。追加の上記成分には、湿潤剤、乳化剤、酸化防止剤、充填剤、pH緩衝剤(例えば、リン酸塩、クエン酸塩、マレイン酸塩の緩衝剤)、保存料、界面活性剤、安定剤、等張性調整剤、キレート剤、金属イオン、油性賦形剤、タンパク質(例えば、ヒト血清アルブミン、ゼラチン、またはタンパク質)および/または両性イオン(例えば、ベタイン、タウリン、アルギニン、グリシン、リジン、およびヒスチジンのようなアミノ酸)を含んでもよい。
【0079】
本発明の製剤のための安定剤としては、高分子量ポリマーまたは低分子量化合物の群から選択されることが好ましい。さらに、本実施の形態の安定剤は、ポリエチレングリコール(例えば、PEG3350)、ポリビニルアルコール(PVA)、ポリビニルピロリドン、カルボキシ−ヒドロキシセルロース、またはその誘導体(例えば、HPC、HPC−SL、HPC−L、およびHPMC)、シクロデキストリン、また、モノチオグリセロール、チオグリコール酸、および2−メチルチオエタノールのような含硫物質、および種々の塩(例えば、塩化ナトリウム)から選択されてもよい。これらの具体的な安定剤は、各自本発明の別の実施形態を構成する。また、薬学的組成物は、薬物活性ポリペプチドの安定性をさらに増強する分解防止剤をさらに含んでもよい。本発明に特に影響がある分解防止剤としては、特に限定されるものではないが、メチオニンおよびEDTA(これらはメチオニン酸化に対してポリペプチドを保護する)、ならびに、非イオン性界面活性剤(これは凍結融解または機械的剪断に関連のある凝集に対してポリペプチドを保護する)が含まれる。
【0080】
本発明の製剤の界面活性剤としては、清浄剤、エトキシル化されたキャスターオイル、ポリグリコール化されたグリセリド、アセチル化されたモノグリセリド、ソルビタン脂肪酸エステル、ポリオキシプロピレン−ポリオキシエチレンブロック重合体(例えば、PluronieF68、poloxamer188および407、またはトリトンX−100のようなpoloxamers)、ポリオキシエチレンソルビタン脂肪酸エステル、星形PEO、アルキル化およびアルコキシル化された誘導体(Tween−20、Tween−40、Tween−80、およびBrij−35)のようなポリオキシエチレンおよびポリエチレン誘導体、ポリオキシエチレンヒドロキシステアレート、モノグリセリド、またはエトキシル化されたその誘導体、ジグリセリド、またはそのポリオキシエチレン誘導体、アルコール、グリセロール、レクチンおよびリン脂質(例えば、ホスファチジルセリン、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルイノシトール、ジホスファチジルグリセロール、およびスフィンゴミエリン)、また、(例えば、ジパルミトイルホスファチジン酸のような)リン脂質の誘導体、(例えば、パルミトイルリゾホスファチジル−L−セリンおよび、エタノールアミン、コリン、セリン、またはスレオニンの1−アシル−sn−グリセロ−3−リン酸エステルのような)リゾリン脂質、および、リゾホスファチジルおよびホスファチジルコリンのアルキルアルコキシル(アルキルエステル)、アルコキシ(アルキルエーテル)誘導体(例えば、リゾホスファチジルコリンおよびジパルミトイルホスファチジルコリンのラウロイルおよびミリストイル誘導体、および極性頭部基(コリン、エタノールアミン、ホスファチジン酸、セリン、スレオニン、グリセロール、およびイノシトール)が修飾されたもの、正電荷を帯びたDODAC、DOTMA、DCP、BISHOP、リゾホスファチジルセリン、およびリゾホスファチジルスレオニン、ならびに、グリセロリン脂質(例えば、ケファリン)、グリセロ糖脂質(例えば、ガラクトピラノシド)、スフィンゴ糖脂質(例えば、セラミド、ガングリオシド)、ドデシルホスホコリン、鶏卵リゾレシチン、フシジン酸誘導体(例えば、タウロジヒドロフシジン酸ナトリウム等)、長鎖脂肪酸および脂肪酸のC6−C12を有する塩(例えば、オレイン酸およびカプリン酸)、アシルカルニチンおよび誘導体、また、リジン、アルギニン、またはヒスチジンのN'X−アシル化誘導体、側鎖がアセチル化されたリジンもしくはアルギニンの誘導体、または、リジン、アルギニン、またはヒスチジンおよび中性もしくは酸性アミノ酸の任意の組み合わせを含んでいる、N−アシル化されたジペプチドの誘導体、中性アミノ酸および2つの電荷を帯びたアミノ酸の如何なる組み合わせを含んでいるN−アシル化されたトリペプチドの誘導体、DSS(ドキュセートナトリウム、CAS登録番号〔577−11−7〕、ドキュセートカルシウム、CAS登録番号〔128−49−4〕ドキュセートカリウム、CAS登録番号〔7491−09−0〕)、SDS(ドデシル硫酸ナトリウムまたはラウリル硫酸ナトリウム)、カプリル酸ナトリウム、コール酸またはその誘導体、胆汁酸およびその塩、グリシンまたはタウリンの複合体(conjugates)、ウルソデオキシコール酸、コール酸ナトリウム、デオキシコール酸ナトリウム、タウロコール酸ナトリウム、グリコール酸ナトリウム、N−ヘキサデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホン酸、一価のアニオン(アルキル−アリル−スルホン酸塩)界面活性剤、(例えば、N−アルキル−N、N−ジメチルアンモニオ−1−プロパンスルホン酸、3−胆汁アミド−1−プロピルジメチルアンモニオ−1−プロパン−スルホン酸のような)両性イオン界面活性剤、(例えば、セチル−トリメチルアンモニウム臭化物、セチルピリジニウム塩化物のような)4級アンモニウム塩基である陽イオン界面活性剤、(例えば、ドデシル−D−グルコピラノシドのような)非イオン性の界面活性剤、例えばテトロニクスのような、エチレンジアミンへのプロピレン酸化、およびエチレン酸化の選択的付加に由来する四官能性ブロック共重合体であるポロキサミン、イミダゾリン誘導体類の群から選択されてもよい界面活性剤、あるいは、それらの混合物から選択されてもよい。これらの具体的な界面活性剤は、それぞれ本発明の別の実施形態の構成要素である。薬学的組成物における界面活性剤の使用は、当業者に知られるところである。便宜上、レミントン:薬学の科学と演習、19編、1995が参照される。
【0081】
薬学的に利用可能な保存料としては、フェノール、O−クレゾール、m−クレゾール、p−クレゾール、メチルp−ヒドロキシ安息香酸、プロピルp−ヒドロキシ安息香酸、2−フェノキシエタノール、ブチルp−ヒドロキシ安息香酸、2−フェニルエタノール、ベンジルアルコール、エタノール、およびクロロブタノールならびに、チオメロサル、ブロノポル、安息香酸、イミド尿素、クロロヘキシジン、デヒドロ酢酸ナトリウム、クロロクレゾール、エチルp−ヒドロキシ安息香酸、塩化ベンゼトニウム、およびクロルフェネシン(3p−クロルフェネキシプロパン−1,2−ジオール)、またはそれらの混合物から成る群から選択されることが好ましい。
【0082】
等張剤としては、(例えば塩化ナトリウムのような)塩、糖または糖アルコール、(例えば、L−グリシン、L−ヒスチジン、アルギニン、リジン、イソロイシン、アスパラギン酸、トリプトファン、スレオニンのような)アミノ酸、(例えば、グリセロール(グリセリン)、1,2−プロパンジオール(プロピレングリコール)、1,3−プロパンジオール、1,3−ブタンジオールのような)アルジトール、および(例えばPEG400のような)ポリエチレングリコール、またはそれらの混合物から成る群から選択されることが好ましい。なお、糖としては、モノ、ジ、または多糖、あるいは水溶性グルカンのような如何なる糖を用いてもよく、これらにはフルクトース、グルコース、マンノース、ソルボース、キシロース、マルトース、ラクトース、スクロース、トレハロース、デキストラン、プルラン、デキストリン、シクロデキストリン、水溶性デンプン、ヒドロキシエチルデンプン、およびカルボキシメチルセルロースナトリウムが含まれる。また、一実施形態において、糖添加物はスクロースである。糖アルコールとしては、少なくとも1つの水酸基を有するC4−C8炭化水素として規定され、例えば、マンニトール、ソルビトール、イノシトール、ガラシチトール、ズルシトール、キシリトール、およびアラビトールが含まれる。一実施形態において、糖アルコール添加物はマンニトールである。また、糖アルコールは、上述の糖または糖アルコールは、単独で用いてもよく、組み合わせて用いてもよい。なお、糖または糖アルコールの使用量は、液体の調製において可溶であり、本発明の方法を用いて達成される効果の安定化に悪影響を及ぼさない限り限定されるものではない。
【0083】
キレート剤としては、エチレンジアミンテトラ酢酸(EDTA)、クエン酸、およびアスパラギン酸の塩、またそれらの混合物から選択されることが好ましい。
【0084】
緩衝剤としては、酢酸ナトリウム、炭酸ナトリウム、クエン酸、グリシルグリシン、ヒスチジン、グリシン、リジン、アルギニン、リン酸二水素ナトリウム、リン酸一水素二ナトリウム、およびリン酸ナトリウム、トリス(ヒドロキシメチル)−アミノメタン、へペス、バイシン、トリシン、リンゴ酸、コハク酸、フマル酸、酒石酸、アスパラギン酸、またはそれらの混合物から成る群から選択されることが好ましい。これらの具体的な緩衝剤は、それぞれ本発明の別の実施形態の構成要素である。
【0085】
本発明の治療ペプチドを含んでいる薬学的組成物において、すべての上記添加剤の使用、特にそのの濃縮範囲に関しては、当業者に知られるところである。便宜上、レミントン:薬学の科学と演習、19編、1995が参照される。
【0086】
本発明のペプチドを含んでいる製剤は、例えば、皮下、皮内、または筋内への注射によって、従来の非経口的な投与がされる。本発明のペプチドの非経口的投与の組成は、例えば、国際公開第03/002136号パンフレットにおいて記載されているように調製されてもよい。
【0087】
さらに、その他の投与形態に適している製剤は坐薬を含んでおり、場合によっては経口、口腔、舌下、腹腔内、膣内、肛門、および頭蓋内処方であってもよい。坐薬には、従来の結合剤および担体である、例えば、ポリアルカレングリコール、またはトリグリセリドが含まれてよい。;上記坐薬は、0.5%から10%の範囲で活性成分を含んでいる混合物から形成されてもよく、上記範囲は0.5%から12%であることがさらに好ましい。経口製剤は、例えば、医薬品等級のマンニトール、ラクトース、デンプン、ステアリン酸マグネシウム、サッカリンナトリウム、セルロース、および炭酸マグネシウム等のような、一般的に使用される賦形剤を含む。これらの組成物は、溶液、懸濁液、タブレット、錠剤、カプセル、持続放出性製剤、または粉末の形態をとり、また、活性成分を10%−95%含んでおり、より好ましくは25%−70%含む。
【0088】
上述のように、添加された薬剤の用途は、作用の持続時間を制御するためであってもよい。放出を制御して調製することは、本発明のペプチドを複合または取り込むためのポリマーを用いることによって達成され得る。活性成分(ペプチド)の制御された運搬は、(例えば、ポリエステル、ポリアミノ酸、ポリビニルピロリドン、エチレンビニル酢酸共重合体、メチルセルロース、カルボキシメチルセルロース、および硫酸プロタミンのような)適切なマクロ分子、マクロ分子の濃度、ならびに混和方法を選択することによって実施され得る。上述の技術は、レミントンの薬学(上述を参照)において開示されている。放出を制御して調製することによって作用の持続時間を制御するために可能な方法としては、その他に本発明のペプチドを、ポリエステル、ポリアミノ酸、ヒドロゲル、ポリ(乳酸)、またはエチレンビニル酢酸共重合体のような高分子材料の粒子に混和する方法がある。
【0089】
本発明のペプチドの形態としては、中性または塩であってもよい。本発明のペプチドは、多くの有機塩基および無機塩基、ならびに有機酸および無機酸の何れかとの反応して、例えば、ペプチドの遊離アミノ基と共に形成されるような(付加)塩を形成するために十分に酸性もしくは塩基性であってもよい。酸付加塩を形成するために一般的に使用される酸としては、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸等のような無機酸、または酒石酸、アスパラギン−トルエンスルホン酸、メタンスルホン酸、シュウ酸、マンデル酸、p−ブロモフェニル−スルホン酸、炭酸、コハク酸、クエン酸、および安息香酸のような有機酸が挙げられる。上記塩としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、亜硫酸水素塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸、カプリン酸塩、アクリル酸塩、ギ酸塩、イソブチル酸塩、カプロン酸塩、ヘプタン酸塩、プロピオン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオエート、へキシン−1,6−ジオエート、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、ガンマ−ヒドロキシ酪酸塩、グリコール酸塩、酒石酸塩、メタンスルホン酸塩、およびプロパンスルホン酸塩が例示できる。また、遊離カルボキシル基から成る塩は、例えば、ナトリウム、カリウム、アンモニウム、カルシウム、または水酸化第2鉄のような無機塩基、またイソプロピルアミン、トリメチルアミン、2−エチルアミノエタノール、ヒスチジン、プロカイン等のような有機塩基由来であってもよい。本発明のペプチドの酸付加塩、カルボン酸塩、低アルキルエステル、およびアミドは、国際公開第91/11457号パンフレット(1991);欧州特許出願公開第0733644号明細書(1996);および米国特許第5,512,549号明細書(1996)にしたがって作製されてもよい。
【0090】
本発明のペプチド配列を含んでいる製剤は、投与製剤と適合するようにして投与され、その投与量は、治療効果が期待できる量とする。治療される患者によって決まる投与量は、例えば、患者の症状の重症度によっても決まる。適切な投与量の範囲は、1回の治療的投与につき、活性成分が数百マイクログラムであるが、好ましくは、該活性成分は、(1−10mgの範囲という高い投与量が検討されるが)約0.1μgから2000μgの範囲であり、例えば、約0.5μgから1000μgであってもよく、また、好ましくは1μgから500μgの範囲であり、特に約10μgから100μgの範囲である。
【0091】
本発明のペプチドを含み、さらに、例えば付加賦形剤としてグリシン、マンニトール、またはその他の添加剤を加えたような製剤は、凍結乾燥された形態で薬瓶として市販されてもよい。希釈剤を有する対の薬瓶が提供され、患者は、投与前に製剤を望ましい濃度に調整することができる。また、本発明の製剤は、注射器に充填される形態のような、その他公知形態において市販されてもよい。
【0092】
本発明は、さらに下記の実施例において実証される。
【0093】
〔実施例〕
<実施例1>
−遺伝子コンストラクトの作製−
GLP−1(7−37)cDNAのコード配列を、図1aに示されたようなHincIIおよびEcoRIの制限酵素認識部位を含む配列として人工的に合成した。図1bに示されたようなGLP−1(7−37)のコード配列、IP2ならびにSfoI、EcoRIおよびXbaIの制限酵素認識部位を含むcDNA(図1b)を、別に合成した。GLP−1を分泌経路に向かわせるために、ストロメライシン3(stromelysin3、(登録番号NM_005940)の異種シグナル配列を用いた。つまり、ストロメライシンのシグナル配列およびリーダー配列をコードするcDNAを、逆転写PCRを用いてヒトRNAから増幅した。そして、図1aまたは図1bのコンストラクトを用いて、図1cおよび図1dに示されたコンストラクトをそれぞれ作製した。
【0094】
図1aのコンストラクトのHincll/EcoRI断片を、図1dの配列のSfoI部位に挿入してクローニングすることにより、図1eのコンストラクトを作製した。同様に、図1dのコンストラクトのEcoRI断片を、真核生物発現プラスミドのEcoRI部位に導入してクローニングすることにより、図1fに示されたコンストラクトを作製した。図1bに示されたコンストラクトのHincII/XbaI断片を繰り返して、図1dに示されたコンストラクトのSfoI/XbaI部位に挿入してクローニングすることにより、図1gに示されたコンストラクトを作製した。図1hは、ヒトGLP−1(7−37)、IP2およびGLP−2(1−35)をコードする配列に融合している、ストロメライシンのリーダー配列およびシグナル配列をコードする、コドンが最適化された合成さ配列を示している。また、ストロメライシンのリーダー配列およびシグナル配列は、内因性のイントロン配列が短くなったことによって中断されている。図1hのコンストラクトのDNA配列は、配列番号16に示されており、翻訳されたペプチドの配列は、配列番号15に示されている。
【0095】
また、図1iおよび1jに示された配列を合成する。その後、これらの配列を用いて、図1jのNaeI/BssHII断片を、図1hの直鎖状のNaeI/BssHII配列に挿入してクローニングすることによって、図1kに示されたコンストラクトを作製する。図1kのコンストラクトのDNA配列は配列番号14に示されており、翻訳されたペプチドの配列は配列番号13に示されている。図1hの配列をBssHIIにより消化して、再度ライゲーションすることにより図1lのコンストラクトを作製する。図1lのコンストラクトのDNA配列は配列番号18に示されており、翻訳されたペプチドの配列は配列番号17に示されている。図1iの配列のAfeI/BssHII断片を、図1hの直鎖状のAfeI/BssHII配列に挿入してクローニングすることにより、図1mのコンストラクトを作製した。図1mのコンストラクトのDNA配列は配列番号20に示されており、翻訳されたペプチドの配列は配列番号19に示されている。
【0096】
上記コンストラクトは、通常の技術を用いて当業者により作製されてもよい。
【0097】
<実施例2>
−哺乳類細胞に対するトランスフェクション、クローン選抜およびGLP−1の発現−
細胞源:HEK293(ヒト胎児腎臓細胞株、カタログ番号ACC305、DSMZ Cell Culture Collection、ドイツ)、AtT20(マウスLAF1下垂体腫瘍細胞株、カタログ番号87021902、European Cell Culture Collection、英国)、hTERT−MSC細胞は、カセム(Kassem)教授(オーデンセ大学病院(University Hospital of Odense)、デンマーク)から生み出された。
【0098】
106の細胞にトランスフェクションするために、異なるGLP−1のコンストラクトを有している0.5〜2μgのプラスミドDNAを用いた。該コンストラクトを、実施例1に記載したように作製した。Current Protocols in Molecular Biology (Ausubelら 1994ff Harvard Medical School Vol2., Unit 9.1)に記載のような標準のリン酸カルシウム共沈法により、HEK293細胞にトランスフェクションした。Current Protocols in Molecular Biology (Ausubelら 1994ff, Harvard Medical School Vol 2., Unit 9.4に記載されているようなフュージーン(FuGene(Roche))を用いて、AtT20細胞にトランスフェクションした。電気パラメーターと細胞種に特異的な溶液との組み合わせに基づいた非ウィルス性の方法である、ヌクレオフェクター技術(Nucleofector technology (Amaxa))を用いて、hTERT−MSC細胞にトランスフェクションした。ヌクレオフェクター装置(プログラムC17)およびヌクレオフェクター溶液(VPE−1001)を用いることにより、60%よりも大きいトランスフェクション効率が達成された。トランスフェクションから48時間後に、選抜剤であるブラストサイジン(2μg/ml)を細胞培養液に添加することによって、DNAが染色体に安定に組み込まれた細胞のクローンの選抜を行った。12〜15日後、トランスフェクションされた細胞の安定クローンを単離することができ、該クローンの特徴を調べるために増殖させた。
【0099】
異なるGLP−1コンストラクトの一過性発現を、hTERT−MSC細胞およびHEK293細胞において測定した。hTERT−MSC細胞およびHEK293細胞の両方において測定することができた、活性型GLP−1の発現量は、単量体GLP−1のコンストラクトである♯103および♯317(GLP−1(7−37)を1コピーだけ有している)では、ごくわずかであったが、2量体GLP−1のコンストラクトである♯217(GLP−1(7−37)を、構成要素(I)および構成要素(III)として有している)では、非常に多かった。この結果をまとめて図2に示す。コンストラクトをGLP−1コンストラクト♯159にまで伸ばした(構成要素(II)としてIP2を4コピー有している)が、さらに顕著に増加することはなかった(示さず)。hTERT−MSC細胞に、異なるコンストラクトをトランスフェクションした後、GLP−1を安定発現するクローンを選抜した。発現量を表1に示す。
【0100】
【表1】
【0101】
<実施例3>
−哺乳類細胞から分泌されたGLP−1ペプチドのウェスタンブロット解析−
GLP−1を分泌する細胞の細胞培養上清を、10%−20%の勾配のSDS PAGE(120V、90分)により分離して、PVDFメンブレン(Immobilon-P Membrane 0.45 μm ミリポアIPVH 00010)にセミドライブロッティング(2.0mA/cm2、60分)によりトランスファーした。メタノール固定と、ブロッキング(3%(w:v)BSAおよび0.1%(v:v)Tween−20を含むTBS)とを行った後、上記メンブレンを、1μg/mlの抗GLP−1抗体(HYB 147−12、Antibodyshop)を用いて、4℃で一晩、免疫ブロットした。洗浄して、0.02μg/mlの検出抗体(Anti Mouse IgG, HRP conjugated, パーキンエルマー PC 2855-1197)と一緒に、室温で4時間インキュベーションした後、化学発光検出を行いタンパク質の位置を明らかにした。
【0102】
ウェスタンブロット解析を図3に示す。なお、1は、モック(mock)をトランスフェクションしたhTERT−MSC細胞の上清に溶解した、100ngの合成GLP−1(7−37)を表し、2は、コンストラクト♯217に由来する2量体GLP−1を分泌するhTERT−MSC細胞(クローン79TM217/13)の上清を表し、3は、コンストラクト♯217に由来する2量体GLP−1を分泌するAtT20細胞(クローン81−A−217/3)の上清を表し、Mは、前もって着色しているタンパク質マーカー(kDa)を表す。この結果によれば、GLP−1(7−37)およびC末端付加物を含んでいる本発明のペプチド(図3における2および3)が、トランスフェクションされた細胞株から分泌されること、そして該本発明のペプチドを、GLP−1(7−37)の中央にある分子エピトープに結合する抗GLP−1抗体を用いて検出できることが示された。
【0103】
<実施例4>
−ヒト細胞から分泌されたGLP−1ペプチドの血しょうにおけるインビトロでの安定性−
HEK293細胞およびhTERT−MSC細胞に、ストロメライシンの異種シグナル配列をコードするコンストラクトを一過的にトランスフェクションした。なお、上記異種シグナル配列は、以下のペプチドをコードする各GLP−1変異体と連結している。
【0104】
1:GLP−1(7−37)、
2:GLP−1(7−37)−IP2(11AAだけ伸長)
3:GLP1(7−37)−IP2−GLP1(7−37)。
【0105】
細胞から分泌されたGLP−1ペプチドまたは合成GLP−1(7−37)(Bachem)を含んでいる細胞培養上清を、ヒトのリンパ球が豊富な血しょう(ジペプチジルペプチダーゼ活性を含む)と一緒に、37℃および5%のCO2の環境下において、3または4時間インキュベーションした。モックをトランスフェクションした細胞の上清に含まれる合成GLP−1(7−37)を、DPP−IV活性の陽性対照として用いた(このDPP−IV活性は、DPP−IV阻害剤((#DPP4, Biotrend)の添加によって阻害されることが示された)。活性型GLPを、GLP(活性)ELISA(#EGLP-35K, Biotrend)を用いて測定した。このGLP(活性)ELISAでは、GLP−1(7−37)のN末端のエピトープに結合する抗体が用いられるので、DPP−IVにより分解された不活性型GLP−1(9−37)ペプチドが識別される。
【0106】
この結果を図4(HEK293細胞)および図5(hTERT−MSC細胞)に示す。HEK293細胞およびhTERT−MSC細胞の両方は、遺伝子コンストラクトに対する有効な宿主である。実施例3と同様に、1〜3の種類のトランスフェクションされた細胞に関する結果に番号をつけた。1は、モックをトランスフェクションしたhTERT−MSC細胞の上清に溶解した、100ngの合成GLP−1(7−37)を表し、2は、コンストラクト♯217に由来する2量体GLP−1を分泌するhTERT−MSC細胞(クローン79TM217/13)の上清を表し、3は、コンストラクト♯217に由来する2量体GLP−1を分泌するAtT20細胞(クローン81−A−217/3)の上清を表す。コンストラクト1は、合成GLP−1と同様の方法でDPP−IVによって不活性化される野生型のGLP−1を製造するのに対し、本発明のC−末端が伸長したGLP−1の形態(図4の2および3、ならびに図5の3)は、分解に対する耐性がより高く、少なくとも40%の活性を維持している。C末端が伸長したGLP−1は、インビトロで、ヒトの血しょうにおいて顕著に安定化されている。2量体のGLP−1配列(3)を有するペプチドは、インビトロでのDPP−IVによる分解に対してほぼ完全に安定化されている。
【0107】
<実施例5>
−GLP−1ペプチドのウェスタンブロット解析−
様々なGLP−1ペプチドを、固相により(syn)、または、E.coliを用いた組み換えにより(rec)合成的に製造した。GLP−1ペプチド(配列番号1(31ng)、ならびに、配列番号6、7および8(各10ng))を、10%−20%の勾配のSDS PAGE(120V、90分)により分離して、PVDFメンブレン(Immobilon-P Membrane 0,45μmミリポア IPVH 00010)にセミドライブロッティング(2.0mA/cm2、60分)によりトランスファーした。メタノール固定と、ブロッキング(3%(w:v)BSAおよび0.1%(v:v)Tween−20を含むTBS)とを行った後、上記メンブレンを、1μg/mlの抗GLP−1抗体(HYB 147−12, Antibodyshop)を用いて、4℃で一晩、免疫ブロットした。洗浄して、0.02μg/mlの検出抗体(Anti Mouse IgG, HRP conjugated, パーキンエルマー PC 2855-1197)と一緒に、室温で4時間インキュベーションした後、化学発光検出を行いタンパク質の位置を明らかにした。示されたペプチドのウェスタンブロットを図6に示す。以下の値を与えることができる。配列番号1(ID1syn)は、GLP−1(7−37)に対応し、31aaおよび3.3kDである;配列番号8(ID8syn、CM3)は、GLP−1(7−37)−IP2に対応し、46aaおよび5.1kDである;配列番号7(ID7rec、CM2)は、GLP−1(7−37)−IP2−RR−GLP2に対応し、83aaおよび9.4kDである;配列番号6(ID6syn、CM1)は、GLP−1(7−37)−IP2−RR−GLP1(7−37)に対応し、79aaおよび8.7kDである。
【0108】
<実施例6>
−GLP−1CMペプチドのインビトロでのヒト血しょうにおける安定性−
濃度20ng/mlの合成GLP−1ペプチド(配列番号1syn、配列番号6syn、配列番号7rec、配列番号8syn)を、ヒト血しょうと一緒に、37℃および5%CO2の環境下において3時間インキュベートした。血しょうのジペプチジルペプチダーゼ活性を、DPP−IV阻害剤(#DPP4, Biotrend)を用いて阻害した。活性型GLPを、GLP(活性)ELISA(#EGLP-35K、Biotrend)を用いて測定した。
【0109】
天然GLP−1(7−37)(配列番号1)と比べて、本発明のC−末端が伸長したGLP−1ペプチド(配列番号6、配列番号7および配列番号8)は、インビトロでのヒト血しょうにおいて顕著に安定化している(図7)。右側の対照のように、DPP−IVを添加した実験に関する結果を示す。GLP−1の活性が、これらの対照実験において、完全に維持されている。
【0110】
<実施例7>
−インビトロにおけるバイオアッセイ(環状AMPの産生)−
RIN−5細胞(ラットの島細胞種、ECACC 番号95090402)を、24ウェルプレートにおいて、4日間、70%の集密度に達するまで育てた。細胞をDMEM(E15−009、PAA)で2回洗浄してから、1%HSA(Aventis)、0.2mM IBMX(858455、シグマ)およびテストペプチドが添加されたDMEM(E15−009、PAA)を0.5ml加えた。25℃で20分間インキュベートした後、細胞を氷冷のPBSを用いて2回洗浄した。0.5%TritonX−100を含む0.1N HCLを添加して、細胞のcAMPを抽出した。cAMP(低pH)EIA(カタログ番号DE0355、R&D)を用いて、環状AMPを定量した。3×10−8Mの刺激については、配列番号1、配列番号6syn、配列番号6rec、配列番号7rec、配列番号8synを用いた。
【0111】
この結果を、図8に示す。100%のcAMP産生は、GLP−1が無いときの基本産生に対応している。GLP−1は、Gタンパク質共役型受容体に結合し、cAMPの産生を刺激する。テストした全ての分子により、細胞のcAMP産生が増加する。
【0112】
<実施例8>
−インビボでの生物活性−
11週齢のII型糖尿病のマウス(C57BL/Ks−Leprdb/db、Harlan)に、5μgのペプチドを、1日に二回(午前9時および午後5時)皮下注射した(1グループあたりn=5である)。一晩絶食させてから午前10時における血糖を、GLPCMペプチドを用いて処置する前(0日)および処置した後(2日、4日、7日、10日後)について、測定した。データを、0日における血糖値に対する相対値として示した。
【0113】
テストされた全ての本発明のペプチド((合成または組み換えの)配列番号6および(合成または組み換えの)配列番号7)は、抗高血糖症効果を有している。組み換えの配列番号6(CM1)および合成の配列番号8(CM3)を用いたときに、最良の結果が得られた。図9のy軸に、上記処置の相対的効果を示す。0日における血糖を1にした。未処置の動物では、血糖値が経時的に連続して増加しているが、本発明のペプチドを用いて処置された動物では、血糖値が経時的に、概ね連続して減少している。
【0114】
<実施例9>
−GLP1CMペプチドのインビトロでの安定性の測定(速度論的テスト方法)−
インキュベーション緩衝液(50mMトリエタノールアミン−HCl(pH7.8)、0.2%HSA)中のCM3、アラニンが置換されたCM3類似体(CM3−ANA01、CM3−ANA02、CM3−ANA03、CM3−ANA04)、C末端が短くなったCM3類似体(CM3−ANA06、CM3−ANA07)、またはC末端が伸長したCM3類似体(CM3−ANA09)からの1μMのペプチドのアリコートを、10%ヒト血しょうと一緒に、37℃および5%CO2の環境下において0、3、6および9時間インキュベートした。DPPIV阻害剤(#DPP、Biotrend)を添加することにより、ジペプチジルペプチダーゼ活性を停止させて、GLP−1(活性)ELISA(#EGLP-35K, Linco)を用いて、活性型GLP−1量を測定した。2以上の独立な実験のそれぞれにおいて、これを3回行い、結果を得た。
【0115】
GLP−1のC末端に付加された少なくとも9個のアミノ酸を有している本発明のペプチドは、CM3(マウス、GLP−1(7−37)−IP2)および、構成要素(II)(IP2)の配列が修飾されているその誘導体(すなわちCM3−ANA01、CM3−ANA01、CM3−ANA02、CM3−ANA03、CM3−ANA04、CM3−ANA05、CM3−ANA07、CM3−ANA07)である。GLP−1ペプチドおよびCM3−ANA06ペプチドは、対照物質または参照物質を表している。
【0116】
図10は、活性型GLP−1のうちで活性を有しているものを、0時間における値の%として示す(すなわち0時間=100%)図である。GLP−1は、血しょうに晒されてから9時間後に9%だけ残っていることから、血しょう安定性が最も悪いことが明確に示されている。CM3−ANA01は、9時間後に84%が残っているので、安定性が最も良好であることが示されている。本発明のペプチドは、9時間後に少なくとも30%が残っており、伸長部分が短いペプチドはこの値に達していない。速度論から、テストした物質に関して、活性型GLP−1を0時間の値の80%にまで分解するのに必要な時間(DT80:80%になるまでの分解時間)を計算することができる。
【0117】
【表2】
【0118】
本発明の物質は、参照物質と比べて、インビトロでの血しょう安定性が大幅に高いことが示されている。任意の理論に束縛されることなく、GLP−1のC末端を伸長させたことにより、立体障害が生じ、上記類似体がDPPIVの活性部位に入り込むことが阻止される。そして、このため上記類似体は分解から免れる。これに対して、より短い参照物質は、GLP−1の安定性に関するこの保護効果を示さない。この仮説は、CM3−ANA06(36aa)、CM3−ANA07(40aa)およびCM3(46aa)のペプチドに見られるように、ペプチドのC末端が長くなればなるほど、安定性が増加することに、支持されている。それにもかかわらず、この現象は、CM3のIP2領域におけるアラニン置換によって示されたように、アミノ酸の数に依存するだけではない。CM3とアミノ酸の数が同じであるCM3−ANA03ペプチドは、CM3と比べて、安定性が非常に減少している。一方、46アミノ酸からなるCM3−ANA01は、CM3と比べて、安定性がより高くなっている。このことは、アミノ酸の数とは関係なく、別の立体効果が安定性に影響するかもしれないということを示している可能性がある。特に好ましいペプチドは、GLP−1のC末端においてIP2を含んでいる伸長部分を有しているペプチド、またはIP2とある程度の相同性を有する伸長部分を有しているペプチドである。これら伸長部分により、DPPIVの活性部位にN末端領域が侵入することを防ぐ特定の立体構造が生じてもよい。
【0119】
<実施例10>
インビボでの研究−(免疫原性)
テスト物質であるCM1に免疫原性があるかどうかを明らかにするために、パラバイオサイエンス(Parabioscience(グローニンゲン、ドイツ))において、マウスの研究を実施した。22日間、5回繰り返して静脈注射された(70μgのペプチド/用量)BALB/cマウス(n=5)において、試験を行った。
【0120】
CM1synにより毒性は誘導されず、以下の図に示されるように誘導されたT細胞抗体の反応と、抗体力価とは最小であった。ペプチドの純度は95%だけであるので、副産物の免疫原性効果を、排除することができない。CM1synに免疫原性効果があるかどうかを評価する研究を、図11に示す。左側(図11A)に、T細胞の想起反応アッセイの結果を示す。処置されたマウスおよび対照のマウスの脾臓を体外培養した。それからT細胞を単離し、抗原の量を増やしながら刺激した。増殖性の想起反応が測定される。右側(図11B)において、動物の免疫血清における抗原特異的IgG抗体の力価を決定した。
【0121】
<実施例11>
−糖尿病マウスにおける用量と有効性とに関する研究−
2時間の絶食のあと、5つの異なる濃度のGLP−1、CM1、CM3、CM3−ANA01およびエキセンディン−4を用いて、糖尿病のC57BL/KsJ@Rj−db(db/db)マウスを処置した。6番目のグループを、生理食塩水のみを用いて処置した。1グループにつき7匹の動物を処置した。注射から1時間および4時間後に尾より採血し、この血液から血糖を直接決定した。
【0122】
図12(図10A(GLP−1)、図12B(CM1)、図12C(CM3)および図12D(CM3−ANA01))に示された用量反応曲線に、その結果を示す。全てのテスト物質に関して、異なる濃度の初期値に対する、血糖の差の百分率を示すグラフを作成した。1時間における測定値を用いて、基準値と比較して百分率が減少するような用量反応曲線を作成することにより、異なるテスト物質に関するED50値を決定した。モーガン−メルセル−フローディン(Morgan-Mercer-Flodin(MMF))モデルを用いて、GLP−1、CM3−ANA01、CM3recおよびエキセンディン−4に関するED50値を評価し、リチャーズモデルを用いて、CM1に関するED50値を評価した。両者のモデルは、用量反応を評価するために適切なシグモイド適合モデルである。全てのペプチドに関して、血しょうグルコースのED50値(一回の皮下注射の用量(μg/マウス))を、血糖値の百分率の減少から決定した。上記ED50値を表1にまとめて示す。
【0123】
【表3】
【0124】
上記ED50値によれば、GLP−1類似体であるCM1、CM3およびCM3−ANA01のインビボにおける生物活性は、20倍よりも大きいと推定される。また、このことは、血しょうにおける安定性が増強されたことによるという可能性が非常に高いといえる。
【0125】
テストした全てのペプチドにより、高血糖のdb/dbマウスにおける血糖値が、少なくとも最高の濃度に関して大幅に減少した。1.1−3.0nmolのペプチドを1回皮下注射した後の、対照に対する血しょうグルコースの低下を、表2にまとめて示す。表2は、対照に対するテスト物質のグルコース低下効果を示している。ペプチドの注射から1時間後(1h)および4時間後(4h)における血しょうグルコースの低下および対応する有意差を求めた。ペプチドの長期有効性について、差異が見られた。エキセンディン−4およびCM3のみにおいて、4時間後に血糖値が有意に減少していた。
【0126】
【表4】
【0127】
<実施例12>
−db/dbマウスの長期間処置−
以下の4つのグループ(n=12)のC57BL/KsJ@Rj−db(db/db)マウスを調査した:
グループA;1日に1回、溶剤(0.9%生理食塩水)を用いて処置された、
グループB;1日に1回、24nmol/kgのテスト物質1(CM1rec)を用いて処置された、
グループC;1日に1回、24nmol/kgのテスト物質2(CM3−ANA01)を用いて処置された、
グループD;1日に1回、24nmol/kgの参照物質(エキセンディン−4)を用いて処置された。なお、エキセンディン−4は技術的に知られており、承認されている。しかしエキセンディン−4はGLP−1類似体ではない。
【0128】
ペプチドまたは溶剤を、1日に1回、2○○−3○○p.m.の間に、グループA−Dにおいて背中の皮膚のひだに皮下投与した。この投与計画を、18週間続けた。12週目から18週目までにおいて、1グループあたり12匹いるうちの6匹に、1日に2回処置を行った。
【0129】
マウスの様々なパラメーター、すなわち、健康状態(1)、体重(2)、飼料消費(3)、血糖(4)、グルコース負荷テスト(5)、インスリンのデータ(6)、グリコシル化ホモグロビン(7)、病状(8)、およびT細胞の再刺激(9)を調査した。
【0130】
健康状態(1)
全てのグループの健康状態は良好であり、処置の副作用は見られなかった。
【0131】
体重(2)
18週に及ぶ処置の後において、処置された全てのグループにおける体重は減少していた(図13)。相対的体重増加は、CM3−ANA01のグループにおいて有意に少ない。図13Aは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。1グループあたり12匹の動物の平均値をプロットされている。図13Bは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。122日に及ぶ処置の後に関する1グループあたり12匹の動物の相対的体重増加をプロットされている。テスト物質としてCM3−ANA01を用いて処置したときのみに(グループB)、体重増加量が有意に少なくなっていた(p<0.001)。
【0132】
飼料消費(3)
対照と比較して、CM1およびCM3−ANA01のグループにおいて、飼料消費が有意に低くなっていた。図14は、122日に及ぶ処置の間における、db/dbマウスの1週間の飼料消費に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。マウス1匹あたりの相対的飼料消費は、グループBおよびCにおいて有意に減少している(p<0.001)。
【0133】
血糖値(4)
血糖値を、様々な状況において決定した。非絶食時の血糖値(ペプチドの注射から1時間後)を図15Aに示す。すなわち、図15Aには、非絶食時の血糖値に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果が示されている。生理食塩水(A)、または、24nmol/kgの濃度のCM1(B)、CM3−ANA01(C)、もしくはエキセンディン(D)ペプチドを皮下注射した。それから1時間後に尾より採血し、この血液から血糖を測定した。対照と比較して、ペプチドの皮下注射から1時間後の、非絶食時の血糖値は、グループBでは55%、グループでは51%、グループDでは60%に減少している(図15B)。処置されたグループにおける血糖は、極めて有意に減少している(p<0.0001)。
【0134】
絶食時の血糖値(ペプチドの注射から18時間後)を図15Cに示す。すなわち、図15Cには、絶食時の血糖値に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果が示されている。テスト物質を皮下注射してから18時間後に、一晩(12時間)絶食させた。それから、尾より採血し、この血液から血糖値を測定した。ペプチドを注射してから18時間後の絶食時の、血糖値は、CM1およびCM3−ANA01のグループにおいて減少している。
【0135】
グルコース負荷テスト(5)(IPGTT)
グルコース負荷テストを、グループA−Dのマウスに対する処置を8週行った後に、実施した。尾より採血することによって、12時間絶食させた動物の基準血糖値(0分)を決定した。その後、テスト物質を皮下注射し、20%のグルコース溶液を腹腔内注射した(1kgあたり1gのグルコース)。そして、グルコース注射から15、30、45、60、90、および120分後の血糖値を続けて決定した。全ての処置されたグループにおいて、グルコース負荷の有意な正規化が示された(図16A)。図16Aでは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて8週間処置された後に、腹腔内注射によりグルコース負荷テスト(IPGTT)を実施している間の血糖値が絶対値で示されている。なお、上記絶対値は、各グループの平均値(n=12)である。
【0136】
図16Aにより提示されるデータを別の表現にて、図16Bに示す。図16Bでは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて8週間処置された後に、腹腔内注射によりグルコース負荷テスト(IPGTT)を実施している間の血糖値が、初期血糖値(100%)に対して標準化された相対値で示されている。
【0137】
インスリン量(6)
一晩(12時間)絶食させた後のマウスのインスリン量を決定した。18週間の処置の後、処置されたグループの全てのマウスは、処置されていない対照よりも有意に多くのインスリンを産生している。0.9%生理食塩水(グループA)、CM1ペプチド(グループB)、CM3−ANA01ペプチド(グループC)、またはエキセンディン−4(グループD)を用いて18週間処置した後の、マウスの血清中インスリン量に関するデータを、図17に示す。血液を採取した直後、プロテアーゼ阻害剤カクテルを用いて試料を処置することにより、インスリンが分解することを防いだ。インスリンマウスウルトラセンシティブELISA(Insulin Mouse Ultrasensitive ELISA(カタログ番号EIA-3440、DRG))を用いて、血清をインスリンについて分析した。スチューデントt−検定により、有意差を決定した。
【0138】
グリコシル化ヘモグロビン(7)
赤血球(RBC)におけるヘモグロビンに非常に多くの血しょうグルコースが結合することによって、グリコシル化ヘモグロビンが形成される。RBCの寿命は120日であるため、グリコシル化ヘモグロビンは、グルコース制御の長期指標を与え、血しょうグルコース量よりも良好な指標として見られる。後眼窩洞(retro-orbital sinus)から全血液を回収した。そして、酵素HbA1cテストキット(Enzymatic HbA1c Test Kit (#DZ121A, Diazyme))を用いて分析して、グリコシル化ヘモグロビン量を決定した。グリコシル化ヘモグロビンをパラメーターとして用いた場合に、増加が有意に減少していることが、全ての処置されたグループにおいて見られた。
【0139】
図18Aは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて6、12および18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。図18Bは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。1グループあたりの全動物の平均値(n=12)で表されている。処置された全グループのグリコシル化ヘモグロビンの増加は、対照よりも有意に低い。
【0140】
病状(8)
処置されたマウスの病状を調査した。検視の結果、処置されたグループと処置されていないグループAとの間の臓器については、膵臓の体積のこと以外は違いがないことが明らかになった。膵臓の重量を決定すると、処置された全グループの脾臓の重量が、極めて有意に重いことが明らかになった。0.9%生理食塩水(A、n=11)、CM1(B、n=12)、CM3−ANA01(C、n=12)、またはエキセンディン(D、n=12)を用いて、18週間処置した後の動物を乱切した日に決定された膵臓の重量を、図19に示す。
【0141】
IV型免疫原性(9)
T細胞の再刺激によりIV型免疫原性を調べて、テストされた物質の可能性がある免疫原性効果を評価した。したがって、1グループあたり8匹の動物の脾臓を体外培養して、T細胞を単離し、異なる濃度の対応するテスト物質を用いて再刺激した。処置されていない対照と比べて、再刺激されたT細胞の増殖は、有意に増加していなかった。図20は、異なる濃度のテスト物質であるCM1(図20A)およびCM3−ANA01(図20B)、ならびに参照物質であるエキセンディン−4(図20C)に対する、インビトロにおける脾臓細胞の想起反応を示す図である。非放射性細胞増殖分析(細胞増殖のためのELISA(Cell Proliferation ELISA)、BrdU、化学発光法)を用いて、テスト物質であるCM1およびCM3−ANA01、ならびに参照物質であるエキセンディン−4を含んでいる溶液に対する、グループA−Dのマウスの脾臓細胞のインビトロにおける想起反応を、別々のマウスにおいて3回測定した。なお、上記溶液における各テスト物質の濃度を、初めは50μg/mlに調製し、その後3倍に希釈していった。刺激指数(SI)を、それぞれのペプチドが存在するときの反応、およびペプチドがないときの反応の指数として計算した。それぞれの処置されたグループおよび対照のグループの平均値±標準偏差を示す(グループA:n=3、グループB:n=6、グループC:n=7、グループD:n=6)。平均値を計算するために、SIが6.5よりも大きいマウスだけを分析した。なお、この6.5は5μg/mlのConAにより刺激した陽性対照培養物におけるSIである。
【0142】
糖尿病マウスにおける長期研究によって、本発明のC末端が伸長したペプチドの有効性が、さらに支持された。テストされた全てのパラメーター(体重、飼料消費、血糖値、グリコシル化ヘモグロビン、グルコース負荷、インスリン分泌、膵臓の重量)において、C末端が伸長したペプチドの有意な治療効果が明らかになった。内因的に生じた配列を有しているC末端が伸長したCellMedペプチドの相同性を考慮すると、上述した本発明のペプチドは、非哺乳類のエキセンディン−4と比較して、免疫原性の点において有利であることが示された。マウスの免疫原性研究から得られたデータは、任意の臨床的に関連するCM1ペプチドの免疫原性がないということを支持している。
【0143】
〔配列表〕
【0144】
【化1】
【0145】
【化2】
【0146】
【化3】
【0147】
【化4】
【0148】
【化5】
【0149】
【化6】
【0150】
【化7】
【0151】
【化8】
【0152】
〔図面の簡単な説明〕
〔図1〕
GLP−1の各コンストラクトを示す図である。
〔図2〕
hTERT−MSC細胞およびHEK293細胞に一過的にトランスフェクションした後の活性型GLP1を示す図である。
〔図3〕
哺乳類細胞から分泌されたGLP−1ペプチドのウェスタンブロット解析を示す図である。
〔図4〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図5〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図6〕
GLP−1ペプチドのウェスタンブロット解析を示す図である。
〔図7〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図8〕
インビトロにおけるバイオアッセイ(環状AMPの産生)を示す図である。
〔図9〕
db/dbマウスの絶食時の相対的グルコース量を示す図である。
〔図10〕
GLP−1ペプチドの血しょうにおける安定性を示す図である。
〔図11〕
AはT細胞の想起反応アッセイの結果を示す図であり、Bは動物の免疫血清における抗原特異的IgG抗体の力価を示す図である。
〔図12〕
AはGLP−1の用量反応曲線を示す図であり、BはCM1の用量反応曲線を示す図であり、CはCM3の用量反応曲線を示す図であり、DはCM3−ANA01の用量反応曲線を示す図である。
〔図13〕
Aはdb/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図であり、Bは、db/dbマウスの体重に対する、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
〔図14〕
122日に及ぶ処置の間における、db/dbマウスの1週間の飼料消費に対する生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いた処置の効果を示す図である。
〔図15〕
Aは非絶食時の血糖値を示す図であり、Bは注射から1時間後の対照に対する血糖の減少を示す図であり、Cは絶食時の血糖値を示す図である。
〔図16〕
Aは腹腔内注射によるグルコース負荷テストを示す図であり、Bは腹腔内注射によるグルコース負荷テスト(相対値)を示す図である。
〔図17〕
0.9%生理食塩水(グループA)、CM1ペプチド(グループB)、CM3−ANA01ペプチド(グループC)、またはエキセンディン−4(グループD)を用いて18週間処置した後の、マウスの血清中インスリン量に関するデータを示す図である。
〔図18〕
Aは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて6、12および18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図であり、Bは、生理食塩水(A)、CM1(B)、CM3−ANA01(C)、またはエキセンディン(D)を用いて18週間処置した後の、全血液試料中のグリコシル化ヘモグロビン(GHb)の相対的増加を示す図である。
〔図19〕
0.9%生理食塩水(A、n=11)、CM1(B、n=12)、CM3−ANA01(C、n=12)、またはエキセンディン(D、n=12)を用いて、18週間処置した後の動物を乱切した日に決定された膵臓の重量示す図である。
〔図20〕
異なる濃度のテスト物質であるCM1(A)およびCM3−ANA01(B)、ならびに参照物質であるエキセンディン−4(C)に対する、インビトロにおける脾臓細胞の想起反応を示す図である。
【特許請求の範囲】
【請求項1】
構成要素(I)として、N末端に一般式II
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Xaa16−Ser−Xaa18−Xaa19−Xaa20−Glu−Xaa22−Xaa23−Ala−Xaa25−Xaa26−Xaa27−Phe−Ile−Xaa3o−Trp−Leu−Xaa33−Xaa34−Xaa35−Xaa36−Xaa37、
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、3−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し(ここで、Xaa8はGlyを表すことが特に好ましい);
Xaa16は、ValまたはLeuを表し;
Xaaは、Ser、LysまたはArgを表し;
Xaa19は、TyrまたはGlnを表し;
Xaa20は、LeuまたはMetを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa25は、AlaまたはValを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa27は、GluまたはLeuを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa33は、ValまたはLysを表し;
Xaa34は、Lys、Glu、AsnまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、Gly、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、Pro、もしくはLys、またはアミドを表すか、あるいは存在していない)、あるいは、
一般式III
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Xaa8−Tyr−Leu−Glu−Xaa22−Xaa23−Ala−Ala−Xaa26−Glu−Phe−lle−Xaa30−Trp−Leu−Val−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し;
Xaa18は、Ser、LysまたはArgを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa34は、Lys、GluまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、もしくはLys、またはアミドを表すか、あるいは存在していない)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる融合ペプチド。
【請求項2】
構成要素(I)として、N末端にGLP−1(7−35、7−36、または7−37)配列、および、
構成要素(II)として、C末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、請求項1に記載の融合ペプチド。
【請求項3】
構成要素(I)として、N末端に一般式I
His−Ala−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Ser−Tyr−Leu−Glu−Gly−Gln−Ala−Ala−Lys−Glu−Phe−Ile−Ala−Trp−Leu−Val−Lys−Gly−Arg−X(I)
(式中、Xは、NH2またはGly−OHを表す)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、請求項1に記載の融合ペプチド。
【請求項4】
構成要素(I)が、配列番号1と少なくとも80%の配列相同性を有する配列を含んでいる、請求項2または3に記載の融合ペプチド。
【請求項5】
構成要素(II)が、βターン様構造を形成するペプチド配列である、請求項1〜4の何れか1項に記載の融合ペプチド。
【請求項6】
構成要素(II)が、少なくとも1つのアラニン残基およびプロリン残基を含んでいるペプチド配列である、請求項1〜5の何れか1項に記載の融合ペプチド。
【請求項7】
構成要素(II)が、βターンを形成する性質を有している4量体を、含んでいるペプチド配列であって、例えば該4量体の2位にプロリン残基を有している、請求項1〜6の何れか1項に記載の融合ペプチド。
【請求項8】
構成要素(II)が、VAIA、IAEE、PEEV、AEEV、EELG、AAAA、AAVA、AALG、DFPE、AADX、AXDX、およびXADXから成る群から選ばれる配列モチーフを含んでいるペプチド配列であって、Xは任意のアミノ酸を表す、請求項1〜7の何れか1項に記載の融合ペプチド。
【請求項9】
構成要素(II)が、AA、XA、AX、RR、RXおよびXRから成る群から選ばれる構成要素(II)のN末端配列モチーフにより、構成要素(I)のC末端と連結しているペプチド配列である、請求項1〜8の何れか1項に記載の融合ペプチド。
【請求項10】
構成要素(II)が、配列番号25(DFPEEVA)の配列モチーフを含んでいるペプチド配列であるか、または、配列番号25と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜9の何れか1項に記載の融合ペプチド。
【請求項11】
構成要素(II)が、配列番号22(RRDFPEEVAI)および配列番号26(AADFPEEVAI)から成る群から選ばれる配列を含んでいるペプチド配列であるか、あるいは、配列番号22または配列番号26と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜10の何れか1項に記載の融合ペプチド。
【請求項12】
構成要素(II)が、配列番号23(RRDFPEEVAIVEEL)、配列番号24(RRDFPEEVAIAEEL)、配列番号27(AADFPEEVAIVEEL)、および配列番号28(AADFPEEVAIAEEL)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号23、24、27または28の何れかと少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜11の何れか1項に記載の融合ペプチド。
【請求項13】
構成要素(II)が、配列番号2(RRDFPEEVAIVEELG)、配列番号3(RRDFPEEVAIAEELG)、配列番号29(AADFPEEVAIVEELG)、および配列番号30(AADFPEEVAIAEELG)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号2、3、29または30と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜12の何れか1項に記載の融合ペプチド。
【請求項14】
構成要素(II)が、9−30アミノ酸、好ましくは9−20アミノ酸、および最も好ましくは9−15アミノ酸を有しているペプチド配列である、請求項1〜13の何れか1項に記載の融合ペプチド。
【請求項15】
構成要素(I)および構成要素(II)が、直接連結されているか、または、リンカー配列を介して連結されている、請求項1〜14の何れか1項に記載の融合ペプチド。
【請求項16】
前記リンカー配列が、1−10アミノ酸の長さを有している、請求項15に記載の融合ペプチド。
【請求項17】
前記融合ペプチドが、
配列番号8(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELG)、
配列番号12(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELG)、
配列番号31(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIAEELG)、
配列番号32(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIAEELG)、
配列番号33(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIAEELG)、
配列番号34(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIAAALG)、
配列番号35(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFP)、
配列番号36(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVA)、
配列番号37(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELGRRHAC)、
配列番号38(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIVEELG)、
配列番号39(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIVEELG)、
配列番号40(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIVEELG)、
配列番号41(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIVAALG)、および
配列番号42(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELGRRHAC)
から成る群から選ばれる配列を含んでいるか、または、配列番号8、12と少なくとも80%の配列相同性を有する配列を含んでいる、請求項1〜16の何れか1項に記載の融合ペプチド。
【請求項18】
前記融合ペプチドが、構成要素(II)のC末端、および/または、構成要素(I)のN末端と連結した他の構成要素(III)を含んでいる、請求項1〜17の何れか1項に記載の融合ペプチド。
【請求項19】
構成要素(III)が、少なくとも4のアミノ酸残基、好ましくは少なくとも10の付加的なアミノ酸残基、より好ましくは、少なくとも20または少なくとも30の付加的なアミノ酸残基を含んでいる、請求項18に記載の融合ペプチド。
【請求項20】
構成要素(III)が、プログルカゴンにおけるようなGLP−2のN末端配列、またはGLP−1(7−37)のN末端配列の、少なくとも4、好ましくは10、より好ましくは少なくとも20の付加的なアミノ酸残基を含んでいる、請求項17または19に記載の融合ペプチド。
【請求項21】
構成要素(III)が、配列番号4または5の配列を含んでいるか、もしくは、配列番号4または5と少なくとも80%の配列相同性を有する配列を含んでいる、請求項18〜20の何れか1項に記載の融合ペプチド。
【請求項22】
前記融合ペプチドが、配列番号6、7、10および11から成る群から選ばれるペプチド配列を含んでいるか、あるいは、配列番号6、7、10または11と少なくとも80%の配列相同性を有する配列を含んでいる、請求項18〜21の何れか1項に記載の融合ペプチド。
【請求項23】
前記アミノ酸の少なくとも1つが、(i)天然に生じるアミノ酸の側鎖を共有結合修飾することにより誘導体化される、(ii)ペプチド骨格を修飾することにより誘導体化される、(iii)NH2末端基またはカルボキシ末端基を修飾することにより誘導体化される、請求項1〜22の何れか1項に記載の融合ペプチド。
【請求項24】
前記アミノ酸の少なくとも1つが、リピル(lipyl)基または炭水化物基により誘導体化される、請求項23に記載の融合ペプチド。
【請求項25】
GLP−1(GLP−1(7))のN末端のHis残基が、該GLP−1のNH2末端および/または該GLP−1のヒスチジル側鎖において、特に疎水性の部分によって化学的に修飾される、請求項23または24に記載の融合ペプチド。
【請求項26】
前記融合ペプチドが、構成要素(IV)として、担体タンパク質、特にトランスフェリンまたはアルブミンを含んでいる、請求項1〜25の何れか1項に記載の融合ペプチド。
【請求項27】
構成要素(I)、(II)および/または(III)のアミノ酸配列が反転されており、前記アミノ酸配列は、少なくとも部分的にDアミノ酸アイソマーから構成されている、請求項1〜26の何れか1項に記載の融合ペプチド。
【請求項28】
固体状態のペプチド合成により、請求項1〜27の何れか1項に記載の融合ペプチドの製造方法。
【請求項29】
請求項1〜22または26の何れか1項に記載の融合ペプチドをコードする核酸。
【請求項30】
請求項29に記載の核酸を含んでいるベクター。
【請求項31】
前記融合ペプチドを発現することができる、外因的に導入された請求項29に記載のDNA(特に請求項30に記載のベクター)を含んでいる宿主細胞。
【請求項32】
融合ペプチドをコードする核酸を含有させるために形質転換した微生物を発酵させて、タンパク質を回収する、請求項1〜22または26の何れか1項に記載の融合ペプチドの製造方法。
【請求項33】
動物細胞を、上記タンパク質が上記細胞から排出される条件下において成長させる、請求項32に記載のタンパク質の製造方法。
【請求項34】
ヒトまたは動物の治療の場合に使用するための薬物としての、請求項1〜27の何れか1項に記載の融合ペプチド。
【請求項35】
1型または2型糖尿病、インスリン耐性、体重異常および疾病、またはそれに関連する症状を治療するための薬物を製造するための、(i)請求項1〜27の何れか1項に記載の融合ペプチド、(ii)請求項29に記載の核酸、(iii)請求項30に記載のベクター、あるいは(iv)請求項31に記載の宿主細胞の使用。
【請求項36】
神経変性疾患および神経変性病、またはそれらに関連する状態を治療するための薬物を製造するための、(i)請求項1〜27の何れか1項に記載の融合ペプチド、(ii)請求項29に記載の核酸、(iii)請求項30に記載のベクター、あるいは(iv)請求項31に記載の宿主細胞の使用。
【請求項37】
アポトーシスに関係する疾患および病気、または状態を治療するための薬物を製造するための、(i)請求項1〜27の何れか1項に記載の融合ペプチド、(ii)請求項29に記載の核酸、(iii)請求項30に記載のベクター、あるいは(iv)請求項31に記載の宿主細胞の使用。
【請求項1】
構成要素(I)として、N末端に一般式II
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Xaa16−Ser−Xaa18−Xaa19−Xaa20−Glu−Xaa22−Xaa23−Ala−Xaa25−Xaa26−Xaa27−Phe−Ile−Xaa3o−Trp−Leu−Xaa33−Xaa34−Xaa35−Xaa36−Xaa37、
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、3−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し(ここで、Xaa8はGlyを表すことが特に好ましい);
Xaa16は、ValまたはLeuを表し;
Xaaは、Ser、LysまたはArgを表し;
Xaa19は、TyrまたはGlnを表し;
Xaa20は、LeuまたはMetを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa25は、AlaまたはValを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa27は、GluまたはLeuを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa33は、ValまたはLysを表し;
Xaa34は、Lys、Glu、AsnまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、Gly、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、Pro、もしくはLys、またはアミドを表すか、あるいは存在していない)、あるいは、
一般式III
Xaa7−Xaa8−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Xaa8−Tyr−Leu−Glu−Xaa22−Xaa23−Ala−Ala−Xaa26−Glu−Phe−lle−Xaa30−Trp−Leu−Val−Xaa34−Xaa35−Xaa36−Xaa37
(式中、Xaa7は、L−ヒスチジン、D−ヒスチジン、脱アミノ−ヒスチジン、2−アミノ−ヒスチジン、−ヒドロキシ−ヒスチジン、ホモヒスチジン、N−アセチル−ヒスチジン、a−フルオロメチル−ヒスチジン、a−メチル−ヒスチジン、3−ピリジルアラニン、2−ピリジルアラニン、または4−ピリジルアラニンを表し;
Xaa8は、Ala、Gly、Val、Leu、Ile、Lys、Aib、(1−アミノシクロプロピル)カルボン酸、(1−アミノシクロブチル)カルボン酸、(1−アミノシクロペンチル)カルボン酸、(1−アミノシクロヘキシル)カルボン酸、(1−アミノシクロヘプチル)カルボン酸、または(1−アミノシクロオクチル)カルボン酸を表し;
Xaa18は、Ser、LysまたはArgを表し;
Xaa22は、Gly、GluまたはAibを表し;
Xaa23は、Gln、Glu、LysまたはArgを表し;
Xaa26は、Lys、GluまたはArgを表し;
Xaa30は、Ala、GluまたはArgを表し;
Xaa34は、Lys、GluまたはArgを表し;
Xaa35は、GlyまたはAibを表し;
Xaa36は、Arg、もしくはLys、またはアミドを表すか、あるいは存在せず;
Xaa37は、Gly、Ala、Glu、もしくはLys、またはアミドを表すか、あるいは存在していない)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる融合ペプチド。
【請求項2】
構成要素(I)として、N末端にGLP−1(7−35、7−36、または7−37)配列、および、
構成要素(II)として、C末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、請求項1に記載の融合ペプチド。
【請求項3】
構成要素(I)として、N末端に一般式I
His−Ala−Glu−Gly−Thr−Phe−Thr−Ser−Asp−Val−Ser−Ser−Tyr−Leu−Glu−Gly−Gln−Ala−Ala−Lys−Glu−Phe−Ile−Ala−Trp−Leu−Val−Lys−Gly−Arg−X(I)
(式中、Xは、NH2またはGly−OHを表す)
に記載の配列、および、
構成要素(II)としてC末端に少なくとも9アミノ酸のペプチド配列、またはその機能的な断片、変異体、もしくは誘導体を含んでいる、請求項1に記載の融合ペプチド。
【請求項4】
構成要素(I)が、配列番号1と少なくとも80%の配列相同性を有する配列を含んでいる、請求項2または3に記載の融合ペプチド。
【請求項5】
構成要素(II)が、βターン様構造を形成するペプチド配列である、請求項1〜4の何れか1項に記載の融合ペプチド。
【請求項6】
構成要素(II)が、少なくとも1つのアラニン残基およびプロリン残基を含んでいるペプチド配列である、請求項1〜5の何れか1項に記載の融合ペプチド。
【請求項7】
構成要素(II)が、βターンを形成する性質を有している4量体を、含んでいるペプチド配列であって、例えば該4量体の2位にプロリン残基を有している、請求項1〜6の何れか1項に記載の融合ペプチド。
【請求項8】
構成要素(II)が、VAIA、IAEE、PEEV、AEEV、EELG、AAAA、AAVA、AALG、DFPE、AADX、AXDX、およびXADXから成る群から選ばれる配列モチーフを含んでいるペプチド配列であって、Xは任意のアミノ酸を表す、請求項1〜7の何れか1項に記載の融合ペプチド。
【請求項9】
構成要素(II)が、AA、XA、AX、RR、RXおよびXRから成る群から選ばれる構成要素(II)のN末端配列モチーフにより、構成要素(I)のC末端と連結しているペプチド配列である、請求項1〜8の何れか1項に記載の融合ペプチド。
【請求項10】
構成要素(II)が、配列番号25(DFPEEVA)の配列モチーフを含んでいるペプチド配列であるか、または、配列番号25と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜9の何れか1項に記載の融合ペプチド。
【請求項11】
構成要素(II)が、配列番号22(RRDFPEEVAI)および配列番号26(AADFPEEVAI)から成る群から選ばれる配列を含んでいるペプチド配列であるか、あるいは、配列番号22または配列番号26と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜10の何れか1項に記載の融合ペプチド。
【請求項12】
構成要素(II)が、配列番号23(RRDFPEEVAIVEEL)、配列番号24(RRDFPEEVAIAEEL)、配列番号27(AADFPEEVAIVEEL)、および配列番号28(AADFPEEVAIAEEL)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号23、24、27または28の何れかと少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜11の何れか1項に記載の融合ペプチド。
【請求項13】
構成要素(II)が、配列番号2(RRDFPEEVAIVEELG)、配列番号3(RRDFPEEVAIAEELG)、配列番号29(AADFPEEVAIVEELG)、および配列番号30(AADFPEEVAIAEELG)から成る群から選ばれる配列を含んでいるペプチド配列であるか、もしくは、配列番号2、3、29または30と少なくとも80%の配列相同性を有する配列を含んでいるペプチド配列である、請求項1〜12の何れか1項に記載の融合ペプチド。
【請求項14】
構成要素(II)が、9−30アミノ酸、好ましくは9−20アミノ酸、および最も好ましくは9−15アミノ酸を有しているペプチド配列である、請求項1〜13の何れか1項に記載の融合ペプチド。
【請求項15】
構成要素(I)および構成要素(II)が、直接連結されているか、または、リンカー配列を介して連結されている、請求項1〜14の何れか1項に記載の融合ペプチド。
【請求項16】
前記リンカー配列が、1−10アミノ酸の長さを有している、請求項15に記載の融合ペプチド。
【請求項17】
前記融合ペプチドが、
配列番号8(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELG)、
配列番号12(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELG)、
配列番号31(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIAEELG)、
配列番号32(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIAEELG)、
配列番号33(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIAEELG)、
配列番号34(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIAAALG)、
配列番号35(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFP)、
配列番号36(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVA)、
配列番号37(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIAEELGRRHAC)、
配列番号38(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADFPEEVAIVEELG)、
配列番号39(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFAEEVAIVEELG)、
配列番号40(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDAAAAVAIVEELG)、
配列番号41(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGAADAAAAVAIVAALG)、および
配列番号42(HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRGRRDFPEEVAIVEELGRRHAC)
から成る群から選ばれる配列を含んでいるか、または、配列番号8、12と少なくとも80%の配列相同性を有する配列を含んでいる、請求項1〜16の何れか1項に記載の融合ペプチド。
【請求項18】
前記融合ペプチドが、構成要素(II)のC末端、および/または、構成要素(I)のN末端と連結した他の構成要素(III)を含んでいる、請求項1〜17の何れか1項に記載の融合ペプチド。
【請求項19】
構成要素(III)が、少なくとも4のアミノ酸残基、好ましくは少なくとも10の付加的なアミノ酸残基、より好ましくは、少なくとも20または少なくとも30の付加的なアミノ酸残基を含んでいる、請求項18に記載の融合ペプチド。
【請求項20】
構成要素(III)が、プログルカゴンにおけるようなGLP−2のN末端配列、またはGLP−1(7−37)のN末端配列の、少なくとも4、好ましくは10、より好ましくは少なくとも20の付加的なアミノ酸残基を含んでいる、請求項17または19に記載の融合ペプチド。
【請求項21】
構成要素(III)が、配列番号4または5の配列を含んでいるか、もしくは、配列番号4または5と少なくとも80%の配列相同性を有する配列を含んでいる、請求項18〜20の何れか1項に記載の融合ペプチド。
【請求項22】
前記融合ペプチドが、配列番号6、7、10および11から成る群から選ばれるペプチド配列を含んでいるか、あるいは、配列番号6、7、10または11と少なくとも80%の配列相同性を有する配列を含んでいる、請求項18〜21の何れか1項に記載の融合ペプチド。
【請求項23】
前記アミノ酸の少なくとも1つが、(i)天然に生じるアミノ酸の側鎖を共有結合修飾することにより誘導体化される、(ii)ペプチド骨格を修飾することにより誘導体化される、(iii)NH2末端基またはカルボキシ末端基を修飾することにより誘導体化される、請求項1〜22の何れか1項に記載の融合ペプチド。
【請求項24】
前記アミノ酸の少なくとも1つが、リピル(lipyl)基または炭水化物基により誘導体化される、請求項23に記載の融合ペプチド。
【請求項25】
GLP−1(GLP−1(7))のN末端のHis残基が、該GLP−1のNH2末端および/または該GLP−1のヒスチジル側鎖において、特に疎水性の部分によって化学的に修飾される、請求項23または24に記載の融合ペプチド。
【請求項26】
前記融合ペプチドが、構成要素(IV)として、担体タンパク質、特にトランスフェリンまたはアルブミンを含んでいる、請求項1〜25の何れか1項に記載の融合ペプチド。
【請求項27】
構成要素(I)、(II)および/または(III)のアミノ酸配列が反転されており、前記アミノ酸配列は、少なくとも部分的にDアミノ酸アイソマーから構成されている、請求項1〜26の何れか1項に記載の融合ペプチド。
【請求項28】
固体状態のペプチド合成により、請求項1〜27の何れか1項に記載の融合ペプチドの製造方法。
【請求項29】
請求項1〜22または26の何れか1項に記載の融合ペプチドをコードする核酸。
【請求項30】
請求項29に記載の核酸を含んでいるベクター。
【請求項31】
前記融合ペプチドを発現することができる、外因的に導入された請求項29に記載のDNA(特に請求項30に記載のベクター)を含んでいる宿主細胞。
【請求項32】
融合ペプチドをコードする核酸を含有させるために形質転換した微生物を発酵させて、タンパク質を回収する、請求項1〜22または26の何れか1項に記載の融合ペプチドの製造方法。
【請求項33】
動物細胞を、上記タンパク質が上記細胞から排出される条件下において成長させる、請求項32に記載のタンパク質の製造方法。
【請求項34】
ヒトまたは動物の治療の場合に使用するための薬物としての、請求項1〜27の何れか1項に記載の融合ペプチド。
【請求項35】
1型または2型糖尿病、インスリン耐性、体重異常および疾病、またはそれに関連する症状を治療するための薬物を製造するための、(i)請求項1〜27の何れか1項に記載の融合ペプチド、(ii)請求項29に記載の核酸、(iii)請求項30に記載のベクター、あるいは(iv)請求項31に記載の宿主細胞の使用。
【請求項36】
神経変性疾患および神経変性病、またはそれらに関連する状態を治療するための薬物を製造するための、(i)請求項1〜27の何れか1項に記載の融合ペプチド、(ii)請求項29に記載の核酸、(iii)請求項30に記載のベクター、あるいは(iv)請求項31に記載の宿主細胞の使用。
【請求項37】
アポトーシスに関係する疾患および病気、または状態を治療するための薬物を製造するための、(i)請求項1〜27の何れか1項に記載の融合ペプチド、(ii)請求項29に記載の核酸、(iii)請求項30に記載のベクター、あるいは(iv)請求項31に記載の宿主細胞の使用。
【図1】


【図2】
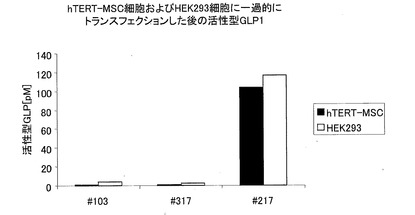
【図3】
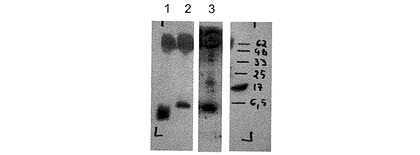
【図4】
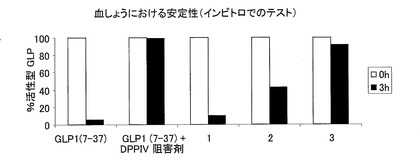
【図5】
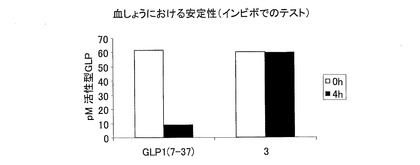
【図6】
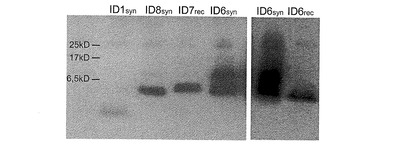
【図7】
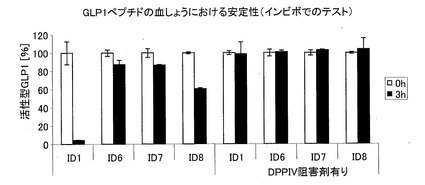
【図8】
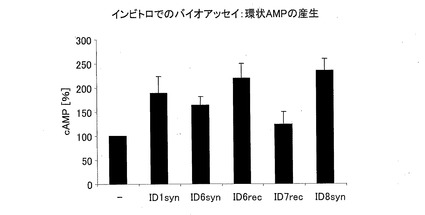
【図9】
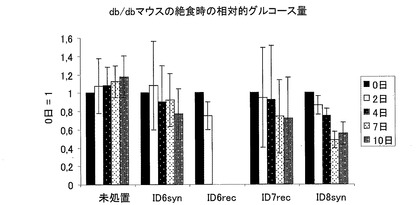
【図10】
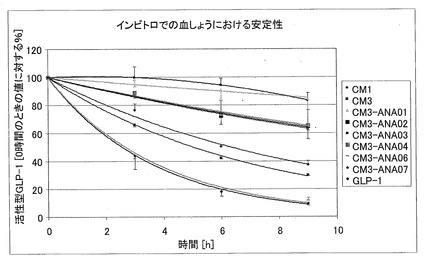
【図11】
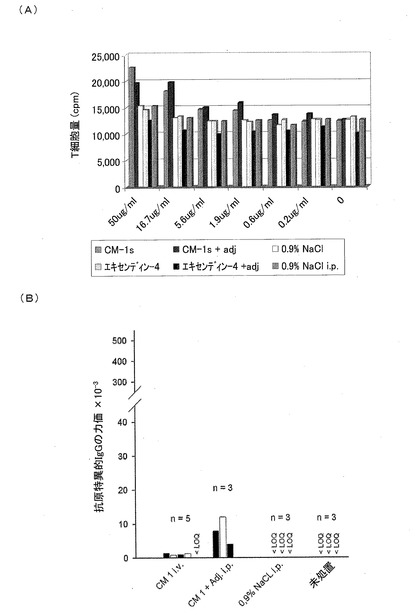
【図12】
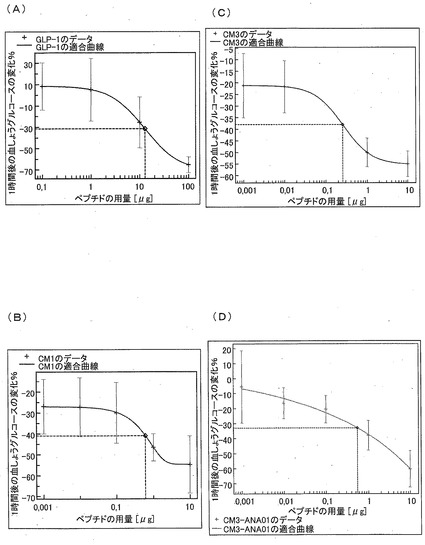
【図13】
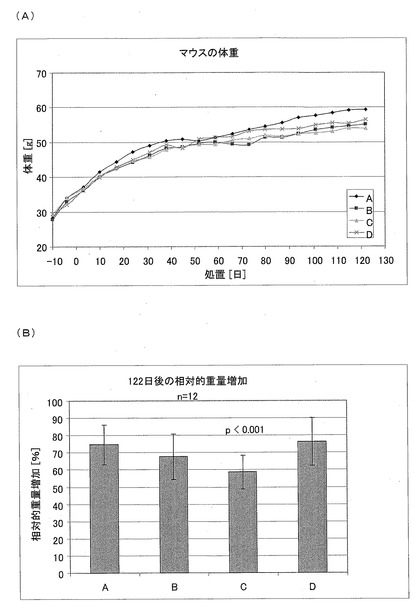
【図14】
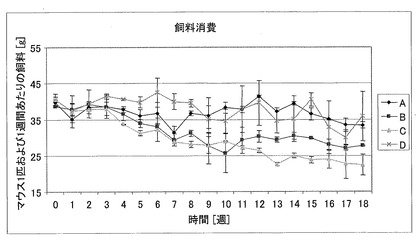
【図15】
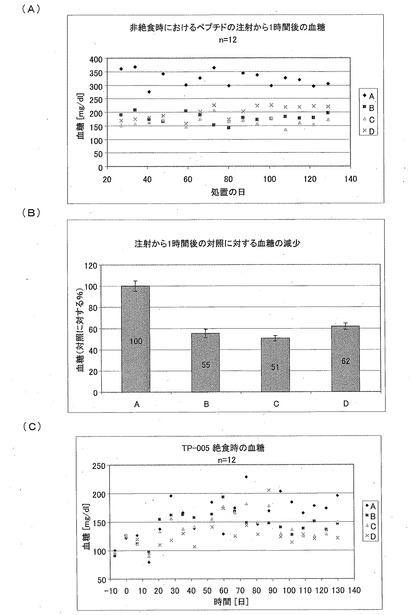
【図16】
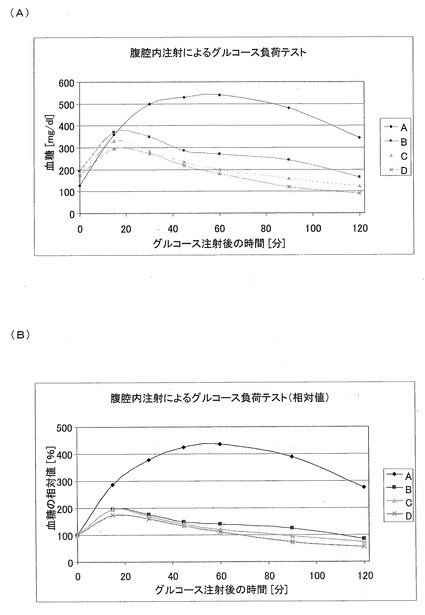
【図17】
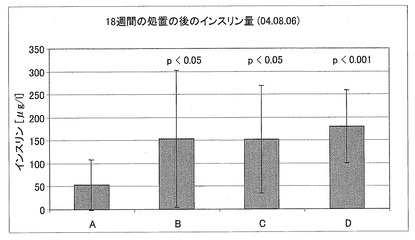
【図18】
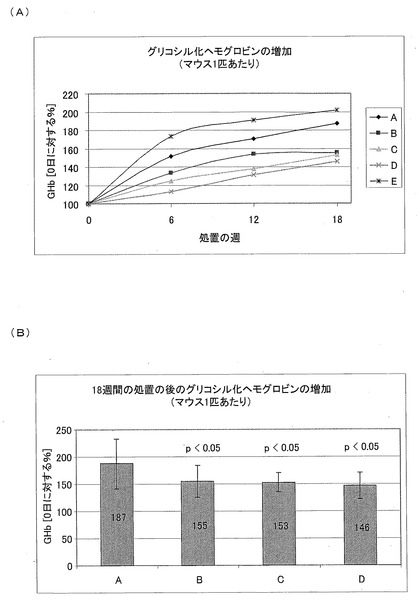
【図19】
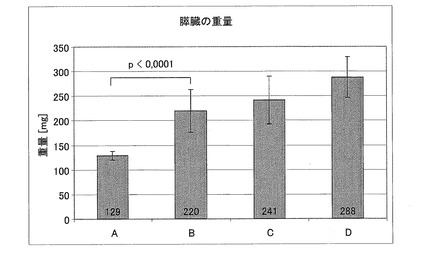
【図20】
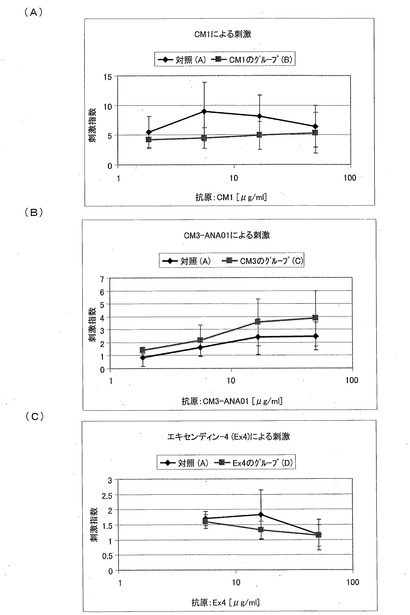
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2013−99352(P2013−99352A)
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願番号】特願2013−12558(P2013−12558)
【出願日】平成25年1月25日(2013.1.25)
【分割の表示】特願2008−531616(P2008−531616)の分割
【原出願日】平成18年9月22日(2006.9.22)
【出願人】(303039785)バイオコンパティブルズ ユーケー リミテッド (23)
【Fターム(参考)】
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願日】平成25年1月25日(2013.1.25)
【分割の表示】特願2008−531616(P2008−531616)の分割
【原出願日】平成18年9月22日(2006.9.22)
【出願人】(303039785)バイオコンパティブルズ ユーケー リミテッド (23)
【Fターム(参考)】
[ Back to top ]