
ペプチドの製造方法
本発明は、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド、環状(1→6)−ジスルフィドの改良された製造方法に関し、それは、固相樹脂上に適切な保護基を有するアミノ酸残基およびチオアルキルカルボン酸を組み上げ、このようにして得られたペプチドを樹脂から切断し、同時に、チオール部分のAcm保護基以外の側鎖保護基を除去して式(3)のペプチドアミドを得、保護されたチオール基を有する式(3)のペプチドアミドのリジン残基をグアニル化によりホモアルギニン残基に変換して式(4)のペプチドアミドを得、式(5)の銀ペプチドを製造し、続いて、その銀ペプチドを同時に脱保護および酸化して、式(1)の化合物を含む粗ペプチドアミドを得、最終的にクロマトグラフで精製することを含む。記載の方法は、簡単で、容易であり、環境に優しく、且つ費用効率が高い。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、固相Fmoc化学を用いる、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドの改良された製造方法に関する。
【0002】
〔発明の背景および従来技術〕
米国特許第5318899号明細書には、血小板関連虚血疾患の治療および予防用の治療剤としての式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドが記載されている。それは、ヒト血小板の血小板受容体糖タンパク質(GP)に結合して、血小板凝集を阻害する。血小板凝集には、血小板膜の表面上のGP複合体が介在する。それは、非活性形態においては、刺激されていない血小板の表面に存在する。血小板が付着および生理学的アゴニストにより活性化されたとき、GPはまた、フィブリノーゲン、フォン・ヴィレブランド因子(vWF)、およびフィブロネクチンの受容体となるように活性化される。しかしながら、生体内での血小板凝集および血栓生成の主な原因であると考えられているものは、フィブリノーゲンおよび/またはvWFの結合である。このことは、フィブリノーゲンまたはvWFのGPへの結合を特異的に阻害する物質は血小板凝集を阻害し、生体内での血栓生成を阻害する候補物質となりうることを教示している(Eric J. Topol, Tatiana V. Byzova, Edward F. Plow and The Lancet; Vol 353; January 16, 1999; pg 227-231)。この論文には、血小板凝集阻害活性を有する化合物が記載されているが、その分子を合成する方法は教示されていない。
【0003】
血小板糖タンパク質IIb/IIIaのアンタゴニストは、経皮的冠動脈形成術(PCI)中の心筋損傷をもたらす血栓合併症の程度を軽減する上で、認められた役割を果たす。
【0004】
式(1)の化合物は、6個のアミノ酸と1個のメルカプトプロピオニル(デスアミノシステイニル)残基を含む、ジスルフィドで輪になった環状ヘプタペプチドである。ジスルフィド架橋は、システインアミドとメルカプトプロピオニル部位の間で形成される。溶液相ペプチド合成により製造され、分取用逆相液体クロマトグラフィーにより精製されることが知られており、凍結乾燥される(www.fda.gov/cder/foi/label/1998/20718lbl.pdf)。
【0005】
ペプチド合成法に関して、2つの主な合成手法が、現在の実施で優位を占めている。これらは、溶液(均一相)中での合成および固相(不均一相)上での合成である。しかしながら、溶液相ルートは、各々のカップリングの後、生成したペプチドを単離しなくてはならないので、固相ルートに比べて、より煩雑であり、一方、固相合成では、過剰の試薬および副生物は、簡単な濾過により洗い去られる。両者において、所望のペプチド化合物は、構築しているペプチド鎖にアミノ酸部分を段階的に付加することにより製造される。
【0006】
米国特許第5958732号および第5318899号明細書には、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドのようなペプチドを合成する組み換え技術がクレームされている。この組み換え法により得られるペプチドは、リジン残基をホモアルギニン残基に変換するために、溶液相合成により修飾される。これらの特許文献は、Boc化学を用いる固相合成をもクレームしており、これらの特許の主題は、根本的に本発明とは異なるものである。
【0007】
Boc化学と比較して、Fmoc化学に基づく合成は、温和な手順を使用し、Fmoc基は塩基に不安定性であるため、酸に不安定な側鎖保護基が使用されて、直交保護を与える。保護基の使用原理は、保護基の結合を切断するエネルギーは、他のいかなる基よりも低いということにある。
【0008】
特許US5686566、US5686567、US5686569、US5686570およびUS5756451は、式(1)の化合物のそれらの塩または他の形態における異なるPAIを取り扱っているが、Fmoc固相合成を使用するその製造方法を教示していない。
【0009】
同様に、特許US5759999、US5786333、US5770564、US5807825、US5807828、US5843897、US5968902およびUS5935926には、血小板関連疾患の処置方法およびboc化学を用いる式(1)のペプチドアミドの製造方法が記載されている。
【0010】
特許US5344783およびUS5851839は、血小板凝集阻害剤(PAI)を選択し、同定する方法に関し、そして式(1)のペプチドアミドの製造用のboc化学を開示している。
【0011】
US特許5780595は、PAIに特異的な抗体をクレームしており、また、式(1)のペプチドアミドの製造用のboc化学を開示している。
【0012】
種々の他のペプチドの合成のFmocルートは、従来技術で周知であり、いくつかの文献が、それらの製造に利用できる。しかしながら、経済的であり、最少の工程を含み、且つ環境に優しい、式(1)の化合物の製造方法を開発することが明らかに必要である。
【0013】
先に説明したように、Fmoc化学をベースとする合成は、温和な手順を利用し、Fmoc基は塩基に不安定性であるため、酸に不安定な側鎖保護基が使用されて、直交保護を与える。Fmoc化学で使用される保護基は、tert−ブチル部分に基づく:Ser、Thrに対してはtert−ブチルエーテル、Asp、Gluに対してはtert−ブチルエステル、およびLys、Hisに対してはBoc。trtおよびacm基は、Cysの保護に使用される。ArgおよびHarのグアニジン基は、Mtr、PmcまたはPbfで保護される。Fmoc−アミノ酸誘導体の多くは、市販されている。しかしながら、最終生成物を精製する前に、経費が増加し、最終生成物の純度および収率に影響を与える、別個の操作が必要であるので、ホモアルギニンを含有するペプチドおよびジスルフィド結合に基づく環状ペプチド化合物のようないくつかのアミノ酸類縁体の製造に対して、ある問題がこの分野に存在している。鎖の組み立てに使用するために商業的に購入した場合、Fmoc−ホモアルギニン残基は、高価なものとなる。あるいは、ペプチドの組み立てにおいて、Har単位は、α−NH2でリジン残基のグアニル化により構築され、それは、その生理学的活性の評価用のバソプレシン類縁体を得るために実証されている(Lindebergら、Int. J. Peptide Protein Res. 10, 1977, 240-244)。
【0014】
WO03/093302には、Fmoc−α−窒素保護Cα−カルボキサミドシステインを使用する、式(1)のペプチドの合成が開示されている。そのチオール側鎖を介しての、析出した形態での第一のアミノ酸システインの固体支持体4−メトキシトリチルポリスチレン樹脂への付着と、引き続くα−窒素保護基の除去およびその窒素上でのペプチドの組み立てが記載されている。しかしながら、この方法では、一般的な市販の態様ではない固体支持体4−メトキシトリチルポリスチレン樹脂が使用されており、また、Fmoc−α−窒素保護Cα−カルボキサミドシステインは、市販されていない。これは、本発明の方法に対して、増大した数の工程を有する方法とし、また高価なものにする。切断条件は、エタンジオールを使用し、それは、その方法を、毒性が高く、環境に優しくないものとし、高価なスクラバーの使用を必要とする。鎖の組み立てにおいてFmoc−ホモアルギニン残基の使用はまた、商業的に購入した場合、この方法を、非常に高価なものとする。全体として、この文献でクレームされている方法は、本発明でクレームされている方法とは異なる。さらに、WO03/093302の方法には、ある制限が伴い、それは、本発明のプロセス工程において好適な修飾をもたらすことにより、克服される。
【0015】
したがって、本発明の方法は、以下に記載のように、WO03/093302−A2特許公報中に記載のものに対して、改良された、有効な方法である。
1 不純物の生成をもたらす空気酸化に感受性で、最終生成物の精製および収率を妨げる、−SHペプチドの製造を含まない。
2 ペプチド鎖を構築するための、アミノ酸に対する保護基の的確な選択。
3 アミノ酸のラセミ化を妨げる、適切な活性化反応剤を使用する、アミノ酸のカルボキシル官能の活性化。
4 −SHペプチドを単離せずに、銀ペプチド塩中間体から式(1)のペプチドアミド中のジスルフィドループを得るための効率的な方法。
【0016】
かなりの数の公知で天然由来の小型のおよび中型の環状ペプチドならびに所望の薬理学的特性を有するそれらの合成誘導体および類縁体のいくつかが合成されている。しかしながら、より広範囲な医学的な使用は、しばしば、それらの合成および精製が複雑であるために、妨げられている。したがって、これらの化合物を簡単に、より少ない工程で、しかもより少ないコストで製造する改良方法が望まれており、また、それは産業および人類の要求である。
【0017】
ペプチドの純度および収率は、合成の全てのルートでの重要な観点である。最終生成物中における薬理学的に活性な化合物の相対的含有量で表される収率は、可能な限り高い必要がある。純度は、薬理学的に活性な不純物の存在の程度により表され、その不純物は、痕跡量しか存在しないが、ペプチドが治療剤として使用される場合にその有益な作用を妨害するかあるいは無効にさえしてしまうものである。薬理学的な面では、双方の観点が考慮される必要がある。一般には、精製は、より大きなペプチド分子であるほど、より困難になる。均一(溶液)相合成(これが、より大量のペプチドの工業生産で選択される現在の方法である)においては、個々の工程の間で必要とされる精製の繰り返しは、より純粋な生成物をもたらすが、収率は低い。そこで、合成の最終段階での収率および精製法の改良が必要である。本発明は、工業的に実施可能な固相合成であり、向上した収率で高純度の生成物を与える新規な方法である。
【0018】
従来技術には、アミノ酸を活性化するためにHOBtおよびDICを使用することが記載されており、それは、OBtエステルの生成をもたらす。しかしながら、この手法を使用することの主な欠点は、OBtエステルそれ自体の製造は、約20分を要し、また、反応を0℃で行う必要があることである。SPPS中でのNα保護アミノ酸の段階的な導入は、通常、新たに入るアミノ酸の系中でのカルボキシル基の活性化または予め形成された活性化アミノ酸誘導体の使用を含む。近年、アミニウムおよびホスホニウム系誘導体(HBTU、TBTU、Py Boc、およびHATU)が、系中でのカルボキシル活性化用の好ましいツールとなっている。それらは、カップリング効率および鏡像異性化抑制の双方の点で、優れた結果を与えることが示されている(Fmoc Solid Phase Peptide Synthesis by Chan W.C. and White P. D., Oxford University Press, 2000, p. 41-74)。HBTUの使用は、高収率および高純度をもたらす。それは、冷却を必要とせずに、活性化工程において時間を節約する。二重カップリングもまた、Mpr(Acm)−OHについて必要としない。
【0019】
Fmoc−アミノ酸誘導体の多くは市販されている。しかしながら、最終生成物を精製する前に、経費が増加し、最終生成物の純度および収率に影響を与える、別個の操作が必要であるので、ホモアルギニンを含有するペプチドおよびジスルフィド結合に基づく環状ペプチド化合物のようなある種のアミノ酸類縁体の製造に対して、ある問題がこの分野に存在している。ペプチド鎖の組み立てに使用するために商業的に購入した場合、Fmoc−ホモアルギニン残基は、非常に高価なものとなる。あるいは、ペプチド組み立ては、リジンを使用して構築され、続いて、α−NH2でリジン残基がグアニル化される(Lindebergら、Int. J. Peptide Protein Res. 10, 1977, 240-244)。
【0020】
ジスルフィド構造の形成を伴う保護または非保護スルフヒドリル基の酸化的環化は、通常、最終合成工程として行われるが、その理由は、ジスルフィド結合がかなり熱的に及び化学的に不安定であるからである。数少ないケースでは、それはまた、固体支持体からペプチド分子を切断する前に行われる。例えばメタノールまたは酢酸中での、遊離のおよび/またはある種の保護されたスルフヒドリル基を含有する鎖状ペプチドのヨウ素での酸化は、CRC Handbook of Neurohypophyseal Hormone Analogs, Vol. 1, Part 1; Jost, K.,ら編、CRC Press, Boca Raton, Fla. 1987, p. 31にさらに説明されている。しかしながら、ヨウ素は、環化剤としては、欠点がないわけではない。例えば、ペプチド基質中に存在するトリプトファン部位は、ヨウ素化される危険があり、原料を完全に変換させることと副反応を最少にすることとのバランスはデリケートなものとなり、ひいては生成物の純度に影響を与える。Tam(Tam J. P.ら、1990, J. Am. Chem. Soc., Vol. 113, p. 6657)は、種々の緩衝液系中でのDMSOの20〜50%溶液の使用が、他の方法、例えば空気酸化に比べて、ジスルフィド結合の生成を非常に促進することを論証している。DMSOはまた、他の酸化手法を使用すると生じるペプチドの凝集および析出を大きく減少させ、ある場合には、完全に抑止することが見出されている。そこで、DMSOで酸化された、ジスルフィドで輪になったペプチドの収率および純度は、他の公知の方法に比べて、さらにより高い。本発明において、この点は、酸化的環化にヨウ素ルートを選択しないことにより、正しく対処されている。また、本発明においては、チオール基を含むペプチドアミドの代わりにペプチドアミドの銀塩を、SH−ペプチドを単離せずに酸化に付し、酸化反応中にオフサイド生成物の生成を回避している。したがって、本発明において採用される脱保護プロセス工程および引き続くグアニル化ペプチドアミドの酸化は、純度および収率が向上した式(1)の化合物を含む粗ペプチドアミドを与える。最終的に、粗ペプチドの精製は、最終純粋ペプチドの向上した収率をもたらす。
【0021】
合成の公知のルートにおける他の複雑化の因子は、所望の環化ジスルフィドと、反応の終わりに過剰のヨウ素を還元するために使用される無機イオウ化合物、例えば亜ジチオン酸ナトリウムまたはチオ硫酸ナトリウムとの間の相互作用の可能性である。そのような還元性イオウ含有化合物は、一般に求核的攻撃に感受性であるジスルフィド結合と相互作用しうる。本発明の方法では、ヨウ素の使用が回避されるので、得られる生成物は、純度が高く、関連する不純物は検出されない。
【0022】
本方法は、2,3の容易で単純な工程で達成される。固相合成の使用は、プロセスをより単純なものとし、Fmoc化学の使用は、HFのようなきつい化学薬品の使用を排除し、これにより、生成物の安定性に影響を与えない。その方法は、中間体の精製をしなくてもすむようにし、それにより、収率が増大し、コストが減少する。工程(b)および(e)におけるスカベンジャーとしてのチオールの置換は、チオール用のスクラバーの使用が不要となることにより、方法をより環境に優しく、経済的なものとする。
【0023】
方法の選択は、しばしば、治療用ペプチドの安定性に影響する。従来技術の方法に付随する制限を回避しうる、式(1)のペプチドを得るために長く待ち望まれていた要件がある。したがって、トリプトファン、ジスルフィドループ、ε−NH2側鎖等を含むペプチド合成の工業的方法は、ペプチド鎖を構築するための保護基および反応条件の適切な選択を要する。この目的は、ここに、出願人により、本出願に完全に記載の方法を開発することにより、成功裡に達成された。
【0024】
明細書中で使用されている用語解説
AA アミノ酸
Acm アセトアミドメチル
ACT 活性化剤
ADP アデノシン二リン酸
AgOTf トリフルオロメタンスルホン酸銀
Arg アルギニン
Asp アスパラギン酸
Boc/boc tert−ブチルオキシカルボニル
Cys システイン
DCM ジクロロメタン
DEP 脱保護反応剤
DMF ジメチルホルムアミド
DMSO ジメチルスルホキシド
DTT ジチオトレイトール
EDT エタンジチオール
Fmoc 9−フルオレニルメチルオキシカルボニル
Glu グルタミン酸
Gly グリシン
HBTU ヘキサフルオロリン酸2−(1H−ベンゾトリアゾール1−イル)−
1,1,3,3−テトラメチルウロニウム
HF フッ化水素
HIC 疎水性相互作用クロマトグラフィー
His ヒスチジン
IEC イオン交換クロマトグラフィー
LC−MS 液体クロマトグラフィー−質量分析
Lys リジン
Mpr メルカプトプロピオン酸
Mtr 4−メトキシ−2,3,6−トリメチルベンゼンスルホニル
NMM N−メチルモルホリン
Obut O−t−ブチル
Pbf 2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−
スルホニル
Pmc 2,2,5,7,8−ペンタメチルクロマン−6−スルホニル
PPP 乏血小板血漿
Pro プロリン
PRP 多血小板血漿
RP−HPLC 逆相高速液体クロマトグラフィー
RV 反応容器
Ser セリン
SOLV 溶媒
SP 合成ペプチド
TEA トリエチルアミン
TFA トリフルオロ酢酸
Thr スレオニン
TIS トリイソプロピルシラン
Trp トリプトファン
Trt トリチル
【0025】
〔発明の目的〕
本発明の主たる目的は、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド、環状(1→6)−ジスルフィドを得るための改良方法を提供することである。
【0026】
本発明の他の目的は、高収率且つ高純度の式(1)のペプチドアミドを得るための方法を開示することである。
【0027】
本発明のさらに他の目的は、Fmoc化学を用いることによる、式(1)のペプチドアミドの固相合成の方法を開示することである。
【0028】
本発明のさらなる他の目的は、溶液相合成に比べて、より少ない工程数の式(1)のペプチドの製造方法を開示することである。
【0029】
本発明のさらに他の目的は、式(1)の化合物の従来技術の固相合成に付随する制限がない、式(1)のペプチドアミドの製造方法を設計することである。
【0030】
本発明のさらなる他の目的は、向上した純度を有する、ジスルフィド部位を含む小型および中型ペプチドの製造方法を提供することである。
【0031】
本発明のさらなる他の目的は、プロセス工程における随伴不純物の生成を最少にするために適切な保護基および反応剤を選択して、それにより収率を向上させ且つコストを軽減させることである。
【0032】
〔発明の要約〕
本発明は、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド、環状(1→6)−ジスルフィドの改良された製造方法に関し、それは、固相樹脂上に適切な保護基を有するアミノ酸残基およびチオアルキルカルボン酸を組み上げ、このようにして得られたペプチドを樹脂から切断し、同時にチオール部分のAcm保護基以外の側鎖保護基を除去して式(3)のペプチドアミドを得、保護されたチオール基を有する式(3)のペプチドアミドのリジン残基をグアニル化によりホモアルギニン残基に変換し、続いて、同時に脱保護して式(5)の銀ペプチドを得、そして銀ペプチドを酸化して式(1)の粗ペプチドアミドを得、最終的にクロマトグラフで精製することを含む。記載の方法は、簡単で、容易であり、環境にやさしく、且つ費用効率が高い。
【0033】
〔発明の詳細な説明〕
したがって、本発明は、固相上での式(1):
【化12】

のペプチドN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド、環状(1→6)−ジスルフィドの製造方法であって、
a)カップリングしてペプチド結合により互いに直接連結させることにより、固体支持体樹脂上に6個のアミノ酸およびチオアルキルカルボン酸からなるペプチド鎖を必要な配列に組み上げて、下記の式(2):
【化13】
のペプチドを得る工程、
b)工程(a)の各々のカップリングの後に、遊離アミノ基を無水酢酸でキャッピングする工程、
c)工程(b)のペプチドを固体支持体樹脂から切断し且つAcm基以外の全ての基を脱保護して、下記の式(3):
【化14】
のペプチドアミドを得る工程、
d)有機溶媒中、ε−リジン−NH2で工程(c)のペプチドをグアニル化し、続いて、他の溶媒で析出させて、下記の式(4):
【化15】
のペプチドアミドを得る工程、
e)適切な溶媒中で工程(d)の式(4)のペプチドアミドを重金属塩で処理し、続いて、有機溶媒を用いて析出させて、式(5):
【化16】
の重金属−ペプチド塩を得る工程、
f)適切な求核剤で、工程(e)の重金属−ペプチド塩を処理し、式(1)の粗ペプチドアミドを得る工程、そして
g)工程(f)の粗ペプチドアミドを、クロマトグラフィー法で精製する工程、を含むその製造方法を提供する。
【0034】
本発明の実施態様は、そのペプチド結合を形成するための、化合物のアミノおよびカルボキシル等価体の反応を含む。
【0035】
本発明の他の実施態様は、固相結合アミノ酸を得るための、リンカーを介して固相樹脂に結合された、保護された第一のアミノ酸のC−末端を提供する。
【0036】
本発明のさらに他の実施態様は、任意のアミド樹脂、好ましくはRinkアミド樹脂を有する固体支持体を使用する。
【0037】
本発明のさらなる他の実施態様は、チオール保護Fmocシステインとして第一の保護されたアミノ酸を使用する。
【0038】
本発明のさらに他の実施態様は、カップリング剤としてHBTUを使用する。
【0039】
本発明のさらなる他の実施態様は、組み上げられたペプチドアミドの放出をもたらす、リンカーを有する樹脂の切断を提供する。
【0040】
本発明のさらに他の実施態様は、アミノもしくはカルボン酸基またはそれらの誘導体である、末端官能性の各々に連結されることにより得られる式(1)のペプチドアミド化合物を提供する。
【0041】
本発明のさらなる他の実施態様は、Cys、Pro、Trp、Asp、Lys、Gly、Arg、Har、LeuおよびGluよりなる群から選択されるアミノ酸を使用する。
【0042】
本発明の実施態様は、チオアルキルカルボン酸、2−チオプロピオン酸を使用する。
【0043】
本発明の他の実施態様は、FmocまたはBocである、アミノ酸のアミノ官能の保護基を使用することを提供する。
【0044】
本発明のさらに他の実施態様は、非保護または保護O−tBuエステルである、カルボキシ官能を使用することを提供する。
【0045】
本発明のさらなる他の実施態様は、Acm基である、チオール官能の保護基を使用することを提供する。
【0046】
本発明のさらに他の実施態様は、ある規定の比率での試薬TFA、TIS、EDT、DCM、フェノールおよび水、好ましくはTFA(85〜98%):TIS(0〜5%):H2O(0〜5%):EDT(0〜5%):フェノール(0〜5%)、より好ましくはTFA(94.5〜95.5%):TIS(0〜2.5%):H2O(0〜3%):EDT(0〜2.5%)を用いての固体支持体樹脂からのペプチドの切断を提供する。
【0047】
本発明の他の実施態様は、DMF、エタノールおよびメタノールよりなる群から選択されるグアニル化のための有機溶媒を使用する。
【0048】
本発明のさらに他の実施態様では、ペプチドのグアニル化が、好ましくは溶媒DMFを用いることにより行われる。
【0049】
本発明のさらなる他の実施態様では、式(4)のペプチドアミドの析出が、アセトン、アセトニトリル、メタノール、エーテル類、ペンタン、ヘキサンおよびこれらの混合物よりなる群から選択される溶媒を使用して行われる。
【0050】
本発明のさらなる他の実施態様では、析出が、好ましくはアセトニトリルを使用して行われる。
【0051】
本発明の他の実施態様では、式(4)のペプチドの精製が、RP−HPLCにより達成することができる。
【0052】
本発明のさらに他の実施態様では、得られた式(1)のペプチドが、99%より高い純度を有する。
【0053】
本発明のさらなる他の実施態様では、固相合成法による式(1)のペプチドの製造が、Fmoc化学を使用して行われる。
【0054】
本発明のさらなる実施態様では、トリフルオロメタンスルホン酸タリウム、酢酸水銀またはトリフルオロメタンスルホン酸銀から選択されるacm除去用の重金属塩、好ましくはトリフルオロメタンスルホン酸銀を使用する。
【0055】
本発明の他の実施態様では、重金属ペプチド塩が、式(4)のペプチドを、TFA中のトリフルオロメタンスルホン酸銀で処理することにより得られる。
【0056】
本発明のさらに他の実施態様では、式(5)の重金属ペプチド塩の析出が、好ましくはエーテル系溶媒、より好ましくはジイソプロピルエーテルを使用して行われる。
【0057】
本発明のさらなる他の実施態様では、重金属ペプチド塩が、HClとDMSOで処理されて、同時に重金属を除去し、且つ得られたペプチドを酸化して、式(1)の粗ペプチドアミドを生成する。
【0058】
本発明のさらなる他の実施態様では、式(1)の粗ペプチドアミドが、RP−HPLCで精製される。
【0059】
本発明の他の実施態様では、式(1)の粗ペプチドアミドの精製が、好ましくは、移動相として、メタノールおよび/またはアセトニトリルを水性TFA(0〜0.5%)と組み合わせて使用される、C−4、C−8またはC−18シリカまたはポリマー逆相カラムを使用するRP−HPLCで行われる。
【0060】
本発明のさらなる他の実施態様は、方法を高価ではないものにできる、粗ペプチド精製用メタノール(ARグレード)を用いる。
【0061】
本発明のさらに他の実施態様は、以下の式(2):
【化17】
の中間体ペプチドアミドの製造方法を提供する。
【0062】
本発明のさらなる他の実施態様は、以下の式(3):
【化18】
の中間体ペプチドアミドの製造方法を提供する。
【0063】
本発明のさらなる他の実施態様は、以下の式(4):
【化19】
のペプチドアミドの製造方法を提供する。
【0064】
本発明のさらに他の実施態様は、以下の式(5):
【化20】
の中間体ペプチドアミド銀塩の製造方法を提供する。
【0065】
以下の実施例は、本発明を例証するものであり、本発明の範囲を限定するものではない。
【0066】
〔実施例〕
実施例(1):鎖状ペプチドの化学合成
(Acm)Mpr-Lys(Boc)-Gly-Asp(OBut)-Trp-Pro-Cys(Acm)-樹脂
式(2)
一般的手順:
ペプチド鎖の組み上げは、以下の方法で行う。樹脂を、ペプチド合成装置[CS936、CS BIO, Calif. Peputide Synthesizer]のRVに移し、ペプチド合成装置上で、鎖状ペプチドを、1.5〜4.0倍モル過剰のアミノ酸誘導体を用いてその上で組み上げる。樹脂上のFmoc基を脱保護し、続いて、NMMの存在下にHBTUで第一のアミノ酸、Fmoc−Cys(C)を活性化することにより、Fmoc−Cys(C)を樹脂にカップリングする。次のアミノ酸、プロリンをカップリングするために、第一のアミノ酸、すなわちFmoc−Cys(C)のα窒素を脱保護し、続いて、NMMの存在下にHBTUでFmoc−Proを活性化する。完全な鎖状ペプチド鎖が固体支持体上に組み上げられるまで、このプロセスを、全てのアミノ酸で繰り返す。Mprが、その最後に組み上げられる。各々のカップリングは、45〜90分の時間の範囲で行われる。カップリング工程には、30〜60分間の無水酢酸でのキャッピングが続く。カップリングが完了した後、樹脂を、DMF、N−メチルピロリドンまたはDCMの範囲から選択しうる有機溶媒、好ましくはDMFと引き続くDCMで洗浄し、次いで、真空下に乾燥させる。式(2)の鎖状ペプチドが得られる。
【0067】
ペプチドは、Fmoc化学を用いるrinkアミド樹脂上での固相ペプチド合成技術により、ペプチドアミドとして合成された。
【0068】
【表1】
【0069】
樹脂(15.38g−rinkアミド、10mmol)をCS936のRVに移し、DMF中で膨潤させた。
【0070】
(i)Fmoc−Cys(Acm)/HBTUの樹脂へのカップリングによるFmoc Cys(Acm)−樹脂の合成。予め膨潤させた樹脂(10mmol)をDMFで2回洗浄し、続いて、20%ピペリジンで2回処理してFmocを除去した。樹脂を6回DMFで洗浄した。FmocCys(Acm)(20mmol)およびHBTU(アミノ酸と等モル)を0.4M NMM中に溶解し、樹脂に加えた。カップリングを、60分間、最適撹拌下に行った。樹脂を、再度、DMFで3回洗浄した。カップリングの後、遊離アミノ基を無水酢酸(2.5M)で45分間キャッピングし、続いて、DMFで3回洗浄した。このHBTU法は、エステルを単離しない一工程法である。
【0071】
合成サイクルは、以下のようにプログラムされた:
【表2】
【0072】
(ii)Fmoc−Pro/HBTUをFmoc−Cys(Acm)−樹脂にカップリングすることによるFmoc−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0073】
【表3】
【0074】
(iii)Fmoc−Trp/HBTUをFmoc−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0075】
【表4】
【0076】
(iv)Fmoc−Asp(Obut)/HBTUをFmoc−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0077】
【表5】
【0078】
(v)Fmoc−Gly/HBTUをFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0079】
【表6】
【0080】
(vi)Fmoc−Lys(Boc)/HBTUをFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0081】
【表7】
【0082】
(vii)Mpr(Acm)/HBTUをFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるMpr(Acm)−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0083】
【表8】
【0084】
実施例(2):鎖状ペプチドの化学合成
【0085】
【化21】
【0086】
一般的手順:
ペプチド鎖の組み上げは、以下の方法で行う。樹脂を、ペプチド合成装置[CS936、CS BIO, Calif. Peputide Synthesizer]のRVに移し、ペプチド合成装置上で、鎖状ペプチドを、1.5〜4.0倍モル過剰のアミノ酸誘導体を用いてその上で組み上げる。樹脂上のFmoc基を脱保護し、続いて、Fmoc−Cys(C)を活性化することにより、第一のアミノ酸、Fmoc−Cys(C)を樹脂にカップリングする。Fmoc−Cys(C)(1.3mmol)およびHOBt(2.6mmol)をDMF(5.0ml)に溶解し、氷浴中で10℃未満に冷却した。DIC(1.74mmol)を、単一アリコートとして、反応混合物に加えた。次いで、混合物を、反応容器中の湿潤樹脂に加える前に、6分間撹拌した。カップリング反応は、60分間行われる。
【0087】
次のアミノ酸、プロリンをカップリングするために、第一のアミノ酸、すなわちFmoc−Cys(C)のα窒素を脱保護する。これに続いて、上記のように冷条件でDIC/HOBtでFmoc−Proを活性化し、次いで、この混合物を反応容器に移す。完全な鎖状ペプチド鎖が固体支持体上に組み上げられるまで、このプロセスを、全てのアミノ酸で繰り返す。Mprが、その最後に組み上げられる。各々のカップリングは、45〜90分の時間の範囲で行われる。Mprのカップリングを繰り返す。カップリング工程には、30〜60分間の無水酢酸でのキャッピングが続く。カップリングが完了した後、樹脂を、DMF、N−メチルピロリドンまたはDCMの範囲から選択しうる有機溶媒、好ましくはDMFと引き続くDCMで洗浄し、次いで、真空下に乾燥させる。式(2)の鎖状ペプチドが得られる。
【0088】
ペプチドは、Fmoc化学を用いるrinkアミド樹脂上での固相ペプチド合成技術により、ペプチドアミドとして合成された。
【0089】
【表9】
【0090】
樹脂(1g−rinkアミド、0.65mmol)をPS3のRVに移し、DMF中で膨潤させた。
【0091】
活性化Fmoc−Cys(Acm)の樹脂へのカップリングによるFmoc Cys(Acm)−樹脂の合成。予め膨潤させた樹脂(0.65mmol)をDMFで2回洗浄し、続いて、20%ピペリジンで2回処理してFmocを除去した。樹脂をDMFで6回洗浄した。Fmoc Cys(Acm)(1.3mmol)およびHOBt(2.6mmol)をDMF(5.0ml)中に溶解し、氷浴中で10℃未満に冷却した。DIC(1.74mmol)を、単一アリコートとして、反応混合物に加えた。次いで、混合物を、湿潤樹脂に加える前に、6分間撹拌した。カップリングを、60分間、最適撹拌下に行った。樹脂を、再度、DMFで3回洗浄した。カップリングの後、遊離アミノ基を無水酢酸(2.5M)で45分間キャッピングし、続いて、DMFで3回洗浄した。このDIC/HOBt法は、手動方式で、多工程法である。
【0092】
合成サイクルは、以下のようにプログラムされた:
【表10】
【0093】
(ii)活性化Fmoc−ProをFmoc−Cys(Acm)−樹脂にカップリングすることによるFmoc−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0094】
【表11】
【0095】
(iii)活性化Fmoc−TrpをFmoc−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0096】
【表12】
【0097】
(iv)Fmoc−AspをFmoc−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0098】
【表13】
【0099】
(v)Fmoc−GlyをFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0100】
【表14】
【0101】
(vi)Fmoc−Lys(Boc)をFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0102】
【表15】
【0103】
(vii)Mpr(Acm)をFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるMpr(Acm)−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0104】
【表16】
【0105】
合成において、Mpr(Acm)のカップリングは、カップリング反応を完結させるために、2回行う必要があった。
【0106】
実施例(3):樹脂からのペプチドの切断によるペプチドアミド(Acm)Mpr−Lys−Gly−Asp−Trp−Pro−Cys(Acm)−CONH2(式(3))の生成
組み上げられたペプチド樹脂(実施例1または2よりのもの)を、CS936中で、TFA(95%):TIS(2.5%):H2O(2.5%):EDT(0%):フェノール(0%)からなる切断用混合物500mlでR.T.にて2時間処理した。反応混合物を、RVを通して濾過し、TFAをRotavap上でエバポレートした。ペプチドの析出を、−20℃で、一定の撹拌をしながら、冷ジイソプロピルエーテル300mlを加えることにより、行った。溶媒中の粗ペプチド析出物を、−20℃で10時間放置する。ワットマン濾紙5番を通して濾過して単離し、続いて、冷溶媒で洗浄(100mlx3)して、切断用混合物中で使用したスカベンジャーを除去した。粗ペプチド析出物を、P2O5で真空乾燥し、RP−HPLCで特徴付けした(図1および図2)。
実施例1 実施例2
収率:58.73 収率:48.73
ペプチドの%純度:90% ペプチドの%純度:79.68%
【0107】
実施例(4):粗ペプチドのグアニル化による(Acm)Mpr−Homoarg−Gly−Asp−Trp−Pro−Cys(Acm)−CONH2(式(4))の生成
ペプチド(1g、1.157mmol)をDMF15mlに溶解し、pHをTEAで9.0に調整した。DMF(15ml)中の反応剤、硝酸3,5−ジメチルピラゾール−1−カルボキサミジン(931.5mg)を、ペプチドに加えた。反応混合物を、1倍過剰量の反応剤、硝酸3,5−ジメチルピラゾール−1−カルボキサミジンを多数回で添加しながら、30℃で4日間撹拌した。
アセトニトリル(TEAでpHを8.0に調整)280mlを加えることにより、ペプチドを反応混合物から析出させた。混合物を、さらに−20℃で10時間保持した。それを、ワットマン5番濾紙を通して濾過し、アセトニトリル(pH8.0)で3回洗浄し、続いて、手を加えていないアセトニトリルでpHを中和した。析出物を、一晩、高真空下に乾燥した。収率:85%。ペプチドを、RP−HPLCで特徴付けした(図2)。
【0108】
実施例(5):グアニル化ペプチドの脱ACMと引き続く酸化による、式(1):
【化22】
の生成
【0109】
TFA(134.9ml)およびアニソール(2.7ml)を混合し、氷中で冷却し、実施例3からの予め冷却したペプチド658mgに加え、窒素で飽和させた。続いて、AgOTf(3.47g)を添加し、氷浴中で2時間撹拌した。TFAを高真空下に除去し、ペプチドの銀塩を、ジイソプロピルエーテル(〜400ml)を加えて析出させた。反応混合物をG−4焼結ロートを通して濾過し、析出物(銀−ペプチド)をジイソプロピルエーテル(60mlx3)中に再懸濁し、上記と同様に洗浄し、真空下にP2O5で乾燥させた。
銀ペプチドの酸化は、氷冷条件で、銀−ペプチド塩10mgを50%DMSO/IM HCl15.6ml中に溶解することにより行った。反応混合物を25℃で3時間撹拌した。析出物を、G−4焼結ロートまたはHyfloベッドを通して濾過して、塩化銀を除去した。濾液を、酸化の完結についてチェックした(図4)。反応が完結すると、式(1)の粗ペプチドが得られた。純度百分率:85%。
【0110】
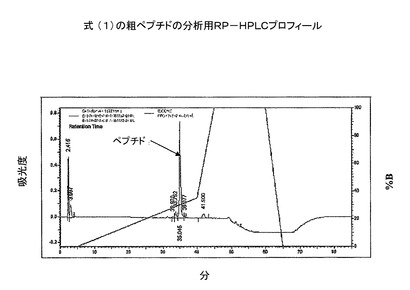
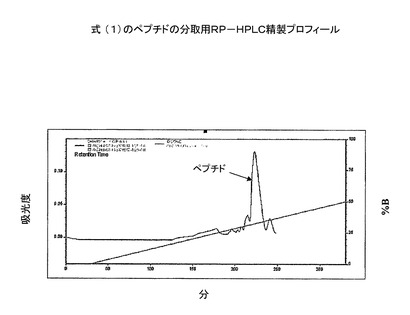
実施例(6):S−Sペプチドの精製
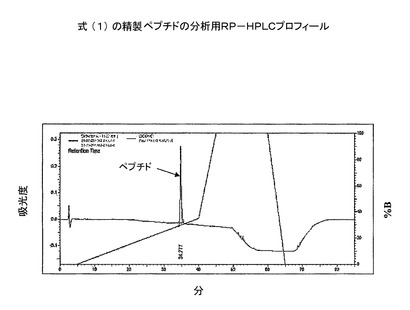
式(1)の粗ジスルフィドで輪になったペプチドを、prepC−18カラム(50x250mm、100Å)に入れた。ペプチドを、グラジエントプログラムでTFA(0.1%)水溶液とメタノールを用いることにより精製した(図5)。続いて、コントローラー、2個のLC8Aポンプ、UV−Vis検出器よりなる島津分取用HPLCシステム上で、上記の溶媒系を用いて定組成運転を行った。式(1)の精製ペプチドアミドを分析用RP−HPLCで分析した(図6)。質量は、質量分析機で決定した(図7)。
【0111】
実施例(7):S−Sペプチドの精製
メタノールの代わりにアセトニトリルを用いたこと以外は、実施例5と同様にして、精製を行い、式(1)のペプチドアミドを得た。
【0112】
実施例(8):酢酸水銀(II)を用いるグアニル化ペプチドの脱ACM
システインのAcm基保護を酢酸水銀(II)での処理によりグアニル化ペプチドから除去した以外は、実施例(4)と同様。
ペプチド(13.4mg、ローリー法で確認、Cys−Acmの)を酢酸(10%)400μl中に溶解した。10倍過剰量の酢酸水銀(II)(82.96mg)をそれに加え、反応物を渦撹拌し、R.T.で5時間保持した。100倍過剰量のβ−メルカプトエタノール(181.37μl)を加え、溶液を渦撹拌し、一晩、室温で放置した。反応混合物を4分間遠心分離し、上清を採集した。析出物を、遠心により、400μlx3の10%酢酸で抽出した。濾液をプールし、RP−HPLCで決定した純度百分率は55%であった(図3)。
【0113】
実施例(9):ヨウ素を用いるグアニル化ペプチドの脱ACM
システインのAcm基保護をヨウ素での処理によりグアニル化ペプチドから除去した以外は、実施例(4)と同様。
ペプチド(9.18mg、ローリー法で確認、Cys−Acmの)を酢酸(80%)17.8ml中に溶解し、N2で15分間パージした。I2の1mM溶液(80%酢酸中、〜4ml)を1時間かけて、黄色が持続するまでペプチド溶液に加えた。混合物をさらに30分間撹拌し、続いて、1N Na2S2O3で、黄色が消失するまで、中和し、凍結乾燥した。‘SH’の確認をエルマン(ellman)試験で行ったが、ACMの除去が達成されなかったとを示す、陰性であった。
【0114】
実施例(10):脱ACMペプチドの精製
酢酸水銀(II)処理ペプチド試料およびヨウ素処理ペプチド試料を、hyperprep(250x10mm、12μ、C−18カラム)を用いて、RP−HPLCにより脱塩した。
【0115】
実施例(11):式(1)の生物活性を調べるための血小板凝集阻害アッセイ
式(1)のペプチドの生物活性を、4Xレーザー血小板凝集計(EMA)を使用して、血小板凝集阻害アッセイを用いて調べた。承諾を得たヒトの提供者からの新鮮な静脈血を取り、クエン酸緩衝液に入れた。多血小板血漿(PRP)および乏血小板血漿を遠心分離により分離した。PRP中の血小板数は、2〜3x108個/mlに調整した。基準凝集量をPPPで調整した後、PRPを10〜20mM ADPで処理し、総凝集百分率を調べた。次いで、PRPをまず、種々の濃度の参照標準および式(1)の合成ペプチドと共にインキュベートした。その後、ADPを加えて、凝集の阻害を調べた。式1の合成ペプチドの生物活性の再現性を、数回確認し、参照標準と比較した。表1は、多くの実験のうちの一つを示す(12回の実験から)。合成ペプチド(SP)についてのIC50投与量は、市販の参照標準に比べて、140nM未満であった。SPでのADP誘起血小板凝集の50%より多い阻害が、大多数の試料において見られ、結果は、市販の参照標準に匹敵するものであった。
【0116】
【表17】
【図面の簡単な説明】
【0117】
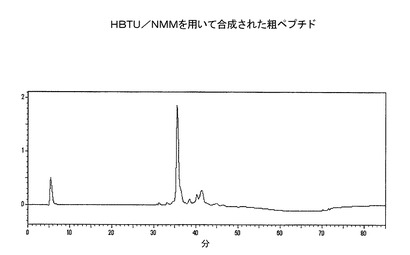
【図1】樹脂からのHBTU−粗ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。
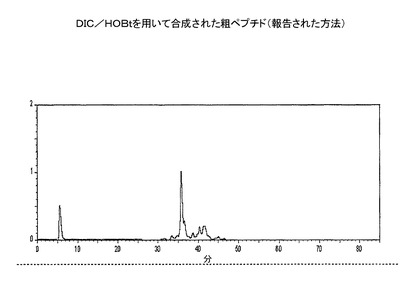
【図2】樹脂からのDIC−粗ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。
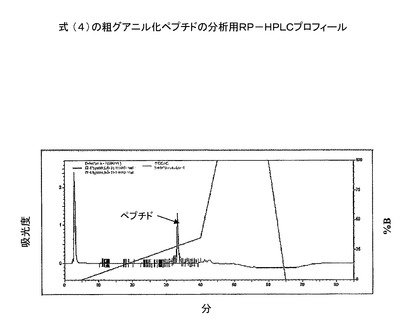
【図3】粗グアニル化ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。
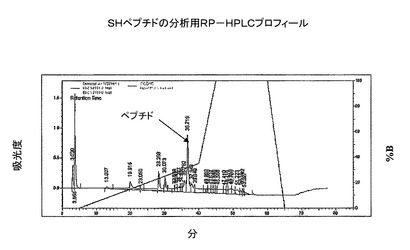
【図4】SHペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。ピークA−粗SHペプチド。
【図5】粗環状ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。ピークA−粗環状ペプチド。
【図6】粗環状ペプチドの分取用RP−HPLC溶出プロフィール(カラム:Phenomenex Luna;C−18(2);10μ;250X50mm;流速:50ml/分;溶媒系:A:0.1%TFA、B:100%メタノール)。
【図7】精製した環状ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。ピークA−純粋ペプチド。
【図8】質量が832であり、不純物が903であることを示す純粋ペプチドのMS分析。
【技術分野】
【0001】
本発明は、固相Fmoc化学を用いる、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドの改良された製造方法に関する。
【0002】
〔発明の背景および従来技術〕
米国特許第5318899号明細書には、血小板関連虚血疾患の治療および予防用の治療剤としての式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドが記載されている。それは、ヒト血小板の血小板受容体糖タンパク質(GP)に結合して、血小板凝集を阻害する。血小板凝集には、血小板膜の表面上のGP複合体が介在する。それは、非活性形態においては、刺激されていない血小板の表面に存在する。血小板が付着および生理学的アゴニストにより活性化されたとき、GPはまた、フィブリノーゲン、フォン・ヴィレブランド因子(vWF)、およびフィブロネクチンの受容体となるように活性化される。しかしながら、生体内での血小板凝集および血栓生成の主な原因であると考えられているものは、フィブリノーゲンおよび/またはvWFの結合である。このことは、フィブリノーゲンまたはvWFのGPへの結合を特異的に阻害する物質は血小板凝集を阻害し、生体内での血栓生成を阻害する候補物質となりうることを教示している(Eric J. Topol, Tatiana V. Byzova, Edward F. Plow and The Lancet; Vol 353; January 16, 1999; pg 227-231)。この論文には、血小板凝集阻害活性を有する化合物が記載されているが、その分子を合成する方法は教示されていない。
【0003】
血小板糖タンパク質IIb/IIIaのアンタゴニストは、経皮的冠動脈形成術(PCI)中の心筋損傷をもたらす血栓合併症の程度を軽減する上で、認められた役割を果たす。
【0004】
式(1)の化合物は、6個のアミノ酸と1個のメルカプトプロピオニル(デスアミノシステイニル)残基を含む、ジスルフィドで輪になった環状ヘプタペプチドである。ジスルフィド架橋は、システインアミドとメルカプトプロピオニル部位の間で形成される。溶液相ペプチド合成により製造され、分取用逆相液体クロマトグラフィーにより精製されることが知られており、凍結乾燥される(www.fda.gov/cder/foi/label/1998/20718lbl.pdf)。
【0005】
ペプチド合成法に関して、2つの主な合成手法が、現在の実施で優位を占めている。これらは、溶液(均一相)中での合成および固相(不均一相)上での合成である。しかしながら、溶液相ルートは、各々のカップリングの後、生成したペプチドを単離しなくてはならないので、固相ルートに比べて、より煩雑であり、一方、固相合成では、過剰の試薬および副生物は、簡単な濾過により洗い去られる。両者において、所望のペプチド化合物は、構築しているペプチド鎖にアミノ酸部分を段階的に付加することにより製造される。
【0006】
米国特許第5958732号および第5318899号明細書には、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドのようなペプチドを合成する組み換え技術がクレームされている。この組み換え法により得られるペプチドは、リジン残基をホモアルギニン残基に変換するために、溶液相合成により修飾される。これらの特許文献は、Boc化学を用いる固相合成をもクレームしており、これらの特許の主題は、根本的に本発明とは異なるものである。
【0007】
Boc化学と比較して、Fmoc化学に基づく合成は、温和な手順を使用し、Fmoc基は塩基に不安定性であるため、酸に不安定な側鎖保護基が使用されて、直交保護を与える。保護基の使用原理は、保護基の結合を切断するエネルギーは、他のいかなる基よりも低いということにある。
【0008】
特許US5686566、US5686567、US5686569、US5686570およびUS5756451は、式(1)の化合物のそれらの塩または他の形態における異なるPAIを取り扱っているが、Fmoc固相合成を使用するその製造方法を教示していない。
【0009】
同様に、特許US5759999、US5786333、US5770564、US5807825、US5807828、US5843897、US5968902およびUS5935926には、血小板関連疾患の処置方法およびboc化学を用いる式(1)のペプチドアミドの製造方法が記載されている。
【0010】
特許US5344783およびUS5851839は、血小板凝集阻害剤(PAI)を選択し、同定する方法に関し、そして式(1)のペプチドアミドの製造用のboc化学を開示している。
【0011】
US特許5780595は、PAIに特異的な抗体をクレームしており、また、式(1)のペプチドアミドの製造用のboc化学を開示している。
【0012】
種々の他のペプチドの合成のFmocルートは、従来技術で周知であり、いくつかの文献が、それらの製造に利用できる。しかしながら、経済的であり、最少の工程を含み、且つ環境に優しい、式(1)の化合物の製造方法を開発することが明らかに必要である。
【0013】
先に説明したように、Fmoc化学をベースとする合成は、温和な手順を利用し、Fmoc基は塩基に不安定性であるため、酸に不安定な側鎖保護基が使用されて、直交保護を与える。Fmoc化学で使用される保護基は、tert−ブチル部分に基づく:Ser、Thrに対してはtert−ブチルエーテル、Asp、Gluに対してはtert−ブチルエステル、およびLys、Hisに対してはBoc。trtおよびacm基は、Cysの保護に使用される。ArgおよびHarのグアニジン基は、Mtr、PmcまたはPbfで保護される。Fmoc−アミノ酸誘導体の多くは、市販されている。しかしながら、最終生成物を精製する前に、経費が増加し、最終生成物の純度および収率に影響を与える、別個の操作が必要であるので、ホモアルギニンを含有するペプチドおよびジスルフィド結合に基づく環状ペプチド化合物のようないくつかのアミノ酸類縁体の製造に対して、ある問題がこの分野に存在している。鎖の組み立てに使用するために商業的に購入した場合、Fmoc−ホモアルギニン残基は、高価なものとなる。あるいは、ペプチドの組み立てにおいて、Har単位は、α−NH2でリジン残基のグアニル化により構築され、それは、その生理学的活性の評価用のバソプレシン類縁体を得るために実証されている(Lindebergら、Int. J. Peptide Protein Res. 10, 1977, 240-244)。
【0014】
WO03/093302には、Fmoc−α−窒素保護Cα−カルボキサミドシステインを使用する、式(1)のペプチドの合成が開示されている。そのチオール側鎖を介しての、析出した形態での第一のアミノ酸システインの固体支持体4−メトキシトリチルポリスチレン樹脂への付着と、引き続くα−窒素保護基の除去およびその窒素上でのペプチドの組み立てが記載されている。しかしながら、この方法では、一般的な市販の態様ではない固体支持体4−メトキシトリチルポリスチレン樹脂が使用されており、また、Fmoc−α−窒素保護Cα−カルボキサミドシステインは、市販されていない。これは、本発明の方法に対して、増大した数の工程を有する方法とし、また高価なものにする。切断条件は、エタンジオールを使用し、それは、その方法を、毒性が高く、環境に優しくないものとし、高価なスクラバーの使用を必要とする。鎖の組み立てにおいてFmoc−ホモアルギニン残基の使用はまた、商業的に購入した場合、この方法を、非常に高価なものとする。全体として、この文献でクレームされている方法は、本発明でクレームされている方法とは異なる。さらに、WO03/093302の方法には、ある制限が伴い、それは、本発明のプロセス工程において好適な修飾をもたらすことにより、克服される。
【0015】
したがって、本発明の方法は、以下に記載のように、WO03/093302−A2特許公報中に記載のものに対して、改良された、有効な方法である。
1 不純物の生成をもたらす空気酸化に感受性で、最終生成物の精製および収率を妨げる、−SHペプチドの製造を含まない。
2 ペプチド鎖を構築するための、アミノ酸に対する保護基の的確な選択。
3 アミノ酸のラセミ化を妨げる、適切な活性化反応剤を使用する、アミノ酸のカルボキシル官能の活性化。
4 −SHペプチドを単離せずに、銀ペプチド塩中間体から式(1)のペプチドアミド中のジスルフィドループを得るための効率的な方法。
【0016】
かなりの数の公知で天然由来の小型のおよび中型の環状ペプチドならびに所望の薬理学的特性を有するそれらの合成誘導体および類縁体のいくつかが合成されている。しかしながら、より広範囲な医学的な使用は、しばしば、それらの合成および精製が複雑であるために、妨げられている。したがって、これらの化合物を簡単に、より少ない工程で、しかもより少ないコストで製造する改良方法が望まれており、また、それは産業および人類の要求である。
【0017】
ペプチドの純度および収率は、合成の全てのルートでの重要な観点である。最終生成物中における薬理学的に活性な化合物の相対的含有量で表される収率は、可能な限り高い必要がある。純度は、薬理学的に活性な不純物の存在の程度により表され、その不純物は、痕跡量しか存在しないが、ペプチドが治療剤として使用される場合にその有益な作用を妨害するかあるいは無効にさえしてしまうものである。薬理学的な面では、双方の観点が考慮される必要がある。一般には、精製は、より大きなペプチド分子であるほど、より困難になる。均一(溶液)相合成(これが、より大量のペプチドの工業生産で選択される現在の方法である)においては、個々の工程の間で必要とされる精製の繰り返しは、より純粋な生成物をもたらすが、収率は低い。そこで、合成の最終段階での収率および精製法の改良が必要である。本発明は、工業的に実施可能な固相合成であり、向上した収率で高純度の生成物を与える新規な方法である。
【0018】
従来技術には、アミノ酸を活性化するためにHOBtおよびDICを使用することが記載されており、それは、OBtエステルの生成をもたらす。しかしながら、この手法を使用することの主な欠点は、OBtエステルそれ自体の製造は、約20分を要し、また、反応を0℃で行う必要があることである。SPPS中でのNα保護アミノ酸の段階的な導入は、通常、新たに入るアミノ酸の系中でのカルボキシル基の活性化または予め形成された活性化アミノ酸誘導体の使用を含む。近年、アミニウムおよびホスホニウム系誘導体(HBTU、TBTU、Py Boc、およびHATU)が、系中でのカルボキシル活性化用の好ましいツールとなっている。それらは、カップリング効率および鏡像異性化抑制の双方の点で、優れた結果を与えることが示されている(Fmoc Solid Phase Peptide Synthesis by Chan W.C. and White P. D., Oxford University Press, 2000, p. 41-74)。HBTUの使用は、高収率および高純度をもたらす。それは、冷却を必要とせずに、活性化工程において時間を節約する。二重カップリングもまた、Mpr(Acm)−OHについて必要としない。
【0019】
Fmoc−アミノ酸誘導体の多くは市販されている。しかしながら、最終生成物を精製する前に、経費が増加し、最終生成物の純度および収率に影響を与える、別個の操作が必要であるので、ホモアルギニンを含有するペプチドおよびジスルフィド結合に基づく環状ペプチド化合物のようなある種のアミノ酸類縁体の製造に対して、ある問題がこの分野に存在している。ペプチド鎖の組み立てに使用するために商業的に購入した場合、Fmoc−ホモアルギニン残基は、非常に高価なものとなる。あるいは、ペプチド組み立ては、リジンを使用して構築され、続いて、α−NH2でリジン残基がグアニル化される(Lindebergら、Int. J. Peptide Protein Res. 10, 1977, 240-244)。
【0020】
ジスルフィド構造の形成を伴う保護または非保護スルフヒドリル基の酸化的環化は、通常、最終合成工程として行われるが、その理由は、ジスルフィド結合がかなり熱的に及び化学的に不安定であるからである。数少ないケースでは、それはまた、固体支持体からペプチド分子を切断する前に行われる。例えばメタノールまたは酢酸中での、遊離のおよび/またはある種の保護されたスルフヒドリル基を含有する鎖状ペプチドのヨウ素での酸化は、CRC Handbook of Neurohypophyseal Hormone Analogs, Vol. 1, Part 1; Jost, K.,ら編、CRC Press, Boca Raton, Fla. 1987, p. 31にさらに説明されている。しかしながら、ヨウ素は、環化剤としては、欠点がないわけではない。例えば、ペプチド基質中に存在するトリプトファン部位は、ヨウ素化される危険があり、原料を完全に変換させることと副反応を最少にすることとのバランスはデリケートなものとなり、ひいては生成物の純度に影響を与える。Tam(Tam J. P.ら、1990, J. Am. Chem. Soc., Vol. 113, p. 6657)は、種々の緩衝液系中でのDMSOの20〜50%溶液の使用が、他の方法、例えば空気酸化に比べて、ジスルフィド結合の生成を非常に促進することを論証している。DMSOはまた、他の酸化手法を使用すると生じるペプチドの凝集および析出を大きく減少させ、ある場合には、完全に抑止することが見出されている。そこで、DMSOで酸化された、ジスルフィドで輪になったペプチドの収率および純度は、他の公知の方法に比べて、さらにより高い。本発明において、この点は、酸化的環化にヨウ素ルートを選択しないことにより、正しく対処されている。また、本発明においては、チオール基を含むペプチドアミドの代わりにペプチドアミドの銀塩を、SH−ペプチドを単離せずに酸化に付し、酸化反応中にオフサイド生成物の生成を回避している。したがって、本発明において採用される脱保護プロセス工程および引き続くグアニル化ペプチドアミドの酸化は、純度および収率が向上した式(1)の化合物を含む粗ペプチドアミドを与える。最終的に、粗ペプチドの精製は、最終純粋ペプチドの向上した収率をもたらす。
【0021】
合成の公知のルートにおける他の複雑化の因子は、所望の環化ジスルフィドと、反応の終わりに過剰のヨウ素を還元するために使用される無機イオウ化合物、例えば亜ジチオン酸ナトリウムまたはチオ硫酸ナトリウムとの間の相互作用の可能性である。そのような還元性イオウ含有化合物は、一般に求核的攻撃に感受性であるジスルフィド結合と相互作用しうる。本発明の方法では、ヨウ素の使用が回避されるので、得られる生成物は、純度が高く、関連する不純物は検出されない。
【0022】
本方法は、2,3の容易で単純な工程で達成される。固相合成の使用は、プロセスをより単純なものとし、Fmoc化学の使用は、HFのようなきつい化学薬品の使用を排除し、これにより、生成物の安定性に影響を与えない。その方法は、中間体の精製をしなくてもすむようにし、それにより、収率が増大し、コストが減少する。工程(b)および(e)におけるスカベンジャーとしてのチオールの置換は、チオール用のスクラバーの使用が不要となることにより、方法をより環境に優しく、経済的なものとする。
【0023】
方法の選択は、しばしば、治療用ペプチドの安定性に影響する。従来技術の方法に付随する制限を回避しうる、式(1)のペプチドを得るために長く待ち望まれていた要件がある。したがって、トリプトファン、ジスルフィドループ、ε−NH2側鎖等を含むペプチド合成の工業的方法は、ペプチド鎖を構築するための保護基および反応条件の適切な選択を要する。この目的は、ここに、出願人により、本出願に完全に記載の方法を開発することにより、成功裡に達成された。
【0024】
明細書中で使用されている用語解説
AA アミノ酸
Acm アセトアミドメチル
ACT 活性化剤
ADP アデノシン二リン酸
AgOTf トリフルオロメタンスルホン酸銀
Arg アルギニン
Asp アスパラギン酸
Boc/boc tert−ブチルオキシカルボニル
Cys システイン
DCM ジクロロメタン
DEP 脱保護反応剤
DMF ジメチルホルムアミド
DMSO ジメチルスルホキシド
DTT ジチオトレイトール
EDT エタンジチオール
Fmoc 9−フルオレニルメチルオキシカルボニル
Glu グルタミン酸
Gly グリシン
HBTU ヘキサフルオロリン酸2−(1H−ベンゾトリアゾール1−イル)−
1,1,3,3−テトラメチルウロニウム
HF フッ化水素
HIC 疎水性相互作用クロマトグラフィー
His ヒスチジン
IEC イオン交換クロマトグラフィー
LC−MS 液体クロマトグラフィー−質量分析
Lys リジン
Mpr メルカプトプロピオン酸
Mtr 4−メトキシ−2,3,6−トリメチルベンゼンスルホニル
NMM N−メチルモルホリン
Obut O−t−ブチル
Pbf 2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−
スルホニル
Pmc 2,2,5,7,8−ペンタメチルクロマン−6−スルホニル
PPP 乏血小板血漿
Pro プロリン
PRP 多血小板血漿
RP−HPLC 逆相高速液体クロマトグラフィー
RV 反応容器
Ser セリン
SOLV 溶媒
SP 合成ペプチド
TEA トリエチルアミン
TFA トリフルオロ酢酸
Thr スレオニン
TIS トリイソプロピルシラン
Trp トリプトファン
Trt トリチル
【0025】
〔発明の目的〕
本発明の主たる目的は、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド、環状(1→6)−ジスルフィドを得るための改良方法を提供することである。
【0026】
本発明の他の目的は、高収率且つ高純度の式(1)のペプチドアミドを得るための方法を開示することである。
【0027】
本発明のさらに他の目的は、Fmoc化学を用いることによる、式(1)のペプチドアミドの固相合成の方法を開示することである。
【0028】
本発明のさらなる他の目的は、溶液相合成に比べて、より少ない工程数の式(1)のペプチドの製造方法を開示することである。
【0029】
本発明のさらに他の目的は、式(1)の化合物の従来技術の固相合成に付随する制限がない、式(1)のペプチドアミドの製造方法を設計することである。
【0030】
本発明のさらなる他の目的は、向上した純度を有する、ジスルフィド部位を含む小型および中型ペプチドの製造方法を提供することである。
【0031】
本発明のさらなる他の目的は、プロセス工程における随伴不純物の生成を最少にするために適切な保護基および反応剤を選択して、それにより収率を向上させ且つコストを軽減させることである。
【0032】
〔発明の要約〕
本発明は、式(1)のN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド、環状(1→6)−ジスルフィドの改良された製造方法に関し、それは、固相樹脂上に適切な保護基を有するアミノ酸残基およびチオアルキルカルボン酸を組み上げ、このようにして得られたペプチドを樹脂から切断し、同時にチオール部分のAcm保護基以外の側鎖保護基を除去して式(3)のペプチドアミドを得、保護されたチオール基を有する式(3)のペプチドアミドのリジン残基をグアニル化によりホモアルギニン残基に変換し、続いて、同時に脱保護して式(5)の銀ペプチドを得、そして銀ペプチドを酸化して式(1)の粗ペプチドアミドを得、最終的にクロマトグラフで精製することを含む。記載の方法は、簡単で、容易であり、環境にやさしく、且つ費用効率が高い。
【0033】
〔発明の詳細な説明〕
したがって、本発明は、固相上での式(1):
【化12】
のペプチドN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド、環状(1→6)−ジスルフィドの製造方法であって、
a)カップリングしてペプチド結合により互いに直接連結させることにより、固体支持体樹脂上に6個のアミノ酸およびチオアルキルカルボン酸からなるペプチド鎖を必要な配列に組み上げて、下記の式(2):
【化13】
のペプチドを得る工程、
b)工程(a)の各々のカップリングの後に、遊離アミノ基を無水酢酸でキャッピングする工程、
c)工程(b)のペプチドを固体支持体樹脂から切断し且つAcm基以外の全ての基を脱保護して、下記の式(3):
【化14】
のペプチドアミドを得る工程、
d)有機溶媒中、ε−リジン−NH2で工程(c)のペプチドをグアニル化し、続いて、他の溶媒で析出させて、下記の式(4):
【化15】
のペプチドアミドを得る工程、
e)適切な溶媒中で工程(d)の式(4)のペプチドアミドを重金属塩で処理し、続いて、有機溶媒を用いて析出させて、式(5):
【化16】
の重金属−ペプチド塩を得る工程、
f)適切な求核剤で、工程(e)の重金属−ペプチド塩を処理し、式(1)の粗ペプチドアミドを得る工程、そして
g)工程(f)の粗ペプチドアミドを、クロマトグラフィー法で精製する工程、を含むその製造方法を提供する。
【0034】
本発明の実施態様は、そのペプチド結合を形成するための、化合物のアミノおよびカルボキシル等価体の反応を含む。
【0035】
本発明の他の実施態様は、固相結合アミノ酸を得るための、リンカーを介して固相樹脂に結合された、保護された第一のアミノ酸のC−末端を提供する。
【0036】
本発明のさらに他の実施態様は、任意のアミド樹脂、好ましくはRinkアミド樹脂を有する固体支持体を使用する。
【0037】
本発明のさらなる他の実施態様は、チオール保護Fmocシステインとして第一の保護されたアミノ酸を使用する。
【0038】
本発明のさらに他の実施態様は、カップリング剤としてHBTUを使用する。
【0039】
本発明のさらなる他の実施態様は、組み上げられたペプチドアミドの放出をもたらす、リンカーを有する樹脂の切断を提供する。
【0040】
本発明のさらに他の実施態様は、アミノもしくはカルボン酸基またはそれらの誘導体である、末端官能性の各々に連結されることにより得られる式(1)のペプチドアミド化合物を提供する。
【0041】
本発明のさらなる他の実施態様は、Cys、Pro、Trp、Asp、Lys、Gly、Arg、Har、LeuおよびGluよりなる群から選択されるアミノ酸を使用する。
【0042】
本発明の実施態様は、チオアルキルカルボン酸、2−チオプロピオン酸を使用する。
【0043】
本発明の他の実施態様は、FmocまたはBocである、アミノ酸のアミノ官能の保護基を使用することを提供する。
【0044】
本発明のさらに他の実施態様は、非保護または保護O−tBuエステルである、カルボキシ官能を使用することを提供する。
【0045】
本発明のさらなる他の実施態様は、Acm基である、チオール官能の保護基を使用することを提供する。
【0046】
本発明のさらに他の実施態様は、ある規定の比率での試薬TFA、TIS、EDT、DCM、フェノールおよび水、好ましくはTFA(85〜98%):TIS(0〜5%):H2O(0〜5%):EDT(0〜5%):フェノール(0〜5%)、より好ましくはTFA(94.5〜95.5%):TIS(0〜2.5%):H2O(0〜3%):EDT(0〜2.5%)を用いての固体支持体樹脂からのペプチドの切断を提供する。
【0047】
本発明の他の実施態様は、DMF、エタノールおよびメタノールよりなる群から選択されるグアニル化のための有機溶媒を使用する。
【0048】
本発明のさらに他の実施態様では、ペプチドのグアニル化が、好ましくは溶媒DMFを用いることにより行われる。
【0049】
本発明のさらなる他の実施態様では、式(4)のペプチドアミドの析出が、アセトン、アセトニトリル、メタノール、エーテル類、ペンタン、ヘキサンおよびこれらの混合物よりなる群から選択される溶媒を使用して行われる。
【0050】
本発明のさらなる他の実施態様では、析出が、好ましくはアセトニトリルを使用して行われる。
【0051】
本発明の他の実施態様では、式(4)のペプチドの精製が、RP−HPLCにより達成することができる。
【0052】
本発明のさらに他の実施態様では、得られた式(1)のペプチドが、99%より高い純度を有する。
【0053】
本発明のさらなる他の実施態様では、固相合成法による式(1)のペプチドの製造が、Fmoc化学を使用して行われる。
【0054】
本発明のさらなる実施態様では、トリフルオロメタンスルホン酸タリウム、酢酸水銀またはトリフルオロメタンスルホン酸銀から選択されるacm除去用の重金属塩、好ましくはトリフルオロメタンスルホン酸銀を使用する。
【0055】
本発明の他の実施態様では、重金属ペプチド塩が、式(4)のペプチドを、TFA中のトリフルオロメタンスルホン酸銀で処理することにより得られる。
【0056】
本発明のさらに他の実施態様では、式(5)の重金属ペプチド塩の析出が、好ましくはエーテル系溶媒、より好ましくはジイソプロピルエーテルを使用して行われる。
【0057】
本発明のさらなる他の実施態様では、重金属ペプチド塩が、HClとDMSOで処理されて、同時に重金属を除去し、且つ得られたペプチドを酸化して、式(1)の粗ペプチドアミドを生成する。
【0058】
本発明のさらなる他の実施態様では、式(1)の粗ペプチドアミドが、RP−HPLCで精製される。
【0059】
本発明の他の実施態様では、式(1)の粗ペプチドアミドの精製が、好ましくは、移動相として、メタノールおよび/またはアセトニトリルを水性TFA(0〜0.5%)と組み合わせて使用される、C−4、C−8またはC−18シリカまたはポリマー逆相カラムを使用するRP−HPLCで行われる。
【0060】
本発明のさらなる他の実施態様は、方法を高価ではないものにできる、粗ペプチド精製用メタノール(ARグレード)を用いる。
【0061】
本発明のさらに他の実施態様は、以下の式(2):
【化17】
の中間体ペプチドアミドの製造方法を提供する。
【0062】
本発明のさらなる他の実施態様は、以下の式(3):
【化18】
の中間体ペプチドアミドの製造方法を提供する。
【0063】
本発明のさらなる他の実施態様は、以下の式(4):
【化19】
のペプチドアミドの製造方法を提供する。
【0064】
本発明のさらに他の実施態様は、以下の式(5):
【化20】
の中間体ペプチドアミド銀塩の製造方法を提供する。
【0065】
以下の実施例は、本発明を例証するものであり、本発明の範囲を限定するものではない。
【0066】
〔実施例〕
実施例(1):鎖状ペプチドの化学合成
(Acm)Mpr-Lys(Boc)-Gly-Asp(OBut)-Trp-Pro-Cys(Acm)-樹脂
式(2)
一般的手順:
ペプチド鎖の組み上げは、以下の方法で行う。樹脂を、ペプチド合成装置[CS936、CS BIO, Calif. Peputide Synthesizer]のRVに移し、ペプチド合成装置上で、鎖状ペプチドを、1.5〜4.0倍モル過剰のアミノ酸誘導体を用いてその上で組み上げる。樹脂上のFmoc基を脱保護し、続いて、NMMの存在下にHBTUで第一のアミノ酸、Fmoc−Cys(C)を活性化することにより、Fmoc−Cys(C)を樹脂にカップリングする。次のアミノ酸、プロリンをカップリングするために、第一のアミノ酸、すなわちFmoc−Cys(C)のα窒素を脱保護し、続いて、NMMの存在下にHBTUでFmoc−Proを活性化する。完全な鎖状ペプチド鎖が固体支持体上に組み上げられるまで、このプロセスを、全てのアミノ酸で繰り返す。Mprが、その最後に組み上げられる。各々のカップリングは、45〜90分の時間の範囲で行われる。カップリング工程には、30〜60分間の無水酢酸でのキャッピングが続く。カップリングが完了した後、樹脂を、DMF、N−メチルピロリドンまたはDCMの範囲から選択しうる有機溶媒、好ましくはDMFと引き続くDCMで洗浄し、次いで、真空下に乾燥させる。式(2)の鎖状ペプチドが得られる。
【0067】
ペプチドは、Fmoc化学を用いるrinkアミド樹脂上での固相ペプチド合成技術により、ペプチドアミドとして合成された。
【0068】
【表1】
【0069】
樹脂(15.38g−rinkアミド、10mmol)をCS936のRVに移し、DMF中で膨潤させた。
【0070】
(i)Fmoc−Cys(Acm)/HBTUの樹脂へのカップリングによるFmoc Cys(Acm)−樹脂の合成。予め膨潤させた樹脂(10mmol)をDMFで2回洗浄し、続いて、20%ピペリジンで2回処理してFmocを除去した。樹脂を6回DMFで洗浄した。FmocCys(Acm)(20mmol)およびHBTU(アミノ酸と等モル)を0.4M NMM中に溶解し、樹脂に加えた。カップリングを、60分間、最適撹拌下に行った。樹脂を、再度、DMFで3回洗浄した。カップリングの後、遊離アミノ基を無水酢酸(2.5M)で45分間キャッピングし、続いて、DMFで3回洗浄した。このHBTU法は、エステルを単離しない一工程法である。
【0071】
合成サイクルは、以下のようにプログラムされた:
【表2】
【0072】
(ii)Fmoc−Pro/HBTUをFmoc−Cys(Acm)−樹脂にカップリングすることによるFmoc−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0073】
【表3】
【0074】
(iii)Fmoc−Trp/HBTUをFmoc−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0075】
【表4】
【0076】
(iv)Fmoc−Asp(Obut)/HBTUをFmoc−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0077】
【表5】
【0078】
(v)Fmoc−Gly/HBTUをFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0079】
【表6】
【0080】
(vi)Fmoc−Lys(Boc)/HBTUをFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0081】
【表7】
【0082】
(vii)Mpr(Acm)/HBTUをFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるMpr(Acm)−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0083】
【表8】
【0084】
実施例(2):鎖状ペプチドの化学合成
【0085】
【化21】
【0086】
一般的手順:
ペプチド鎖の組み上げは、以下の方法で行う。樹脂を、ペプチド合成装置[CS936、CS BIO, Calif. Peputide Synthesizer]のRVに移し、ペプチド合成装置上で、鎖状ペプチドを、1.5〜4.0倍モル過剰のアミノ酸誘導体を用いてその上で組み上げる。樹脂上のFmoc基を脱保護し、続いて、Fmoc−Cys(C)を活性化することにより、第一のアミノ酸、Fmoc−Cys(C)を樹脂にカップリングする。Fmoc−Cys(C)(1.3mmol)およびHOBt(2.6mmol)をDMF(5.0ml)に溶解し、氷浴中で10℃未満に冷却した。DIC(1.74mmol)を、単一アリコートとして、反応混合物に加えた。次いで、混合物を、反応容器中の湿潤樹脂に加える前に、6分間撹拌した。カップリング反応は、60分間行われる。
【0087】
次のアミノ酸、プロリンをカップリングするために、第一のアミノ酸、すなわちFmoc−Cys(C)のα窒素を脱保護する。これに続いて、上記のように冷条件でDIC/HOBtでFmoc−Proを活性化し、次いで、この混合物を反応容器に移す。完全な鎖状ペプチド鎖が固体支持体上に組み上げられるまで、このプロセスを、全てのアミノ酸で繰り返す。Mprが、その最後に組み上げられる。各々のカップリングは、45〜90分の時間の範囲で行われる。Mprのカップリングを繰り返す。カップリング工程には、30〜60分間の無水酢酸でのキャッピングが続く。カップリングが完了した後、樹脂を、DMF、N−メチルピロリドンまたはDCMの範囲から選択しうる有機溶媒、好ましくはDMFと引き続くDCMで洗浄し、次いで、真空下に乾燥させる。式(2)の鎖状ペプチドが得られる。
【0088】
ペプチドは、Fmoc化学を用いるrinkアミド樹脂上での固相ペプチド合成技術により、ペプチドアミドとして合成された。
【0089】
【表9】
【0090】
樹脂(1g−rinkアミド、0.65mmol)をPS3のRVに移し、DMF中で膨潤させた。
【0091】
活性化Fmoc−Cys(Acm)の樹脂へのカップリングによるFmoc Cys(Acm)−樹脂の合成。予め膨潤させた樹脂(0.65mmol)をDMFで2回洗浄し、続いて、20%ピペリジンで2回処理してFmocを除去した。樹脂をDMFで6回洗浄した。Fmoc Cys(Acm)(1.3mmol)およびHOBt(2.6mmol)をDMF(5.0ml)中に溶解し、氷浴中で10℃未満に冷却した。DIC(1.74mmol)を、単一アリコートとして、反応混合物に加えた。次いで、混合物を、湿潤樹脂に加える前に、6分間撹拌した。カップリングを、60分間、最適撹拌下に行った。樹脂を、再度、DMFで3回洗浄した。カップリングの後、遊離アミノ基を無水酢酸(2.5M)で45分間キャッピングし、続いて、DMFで3回洗浄した。このDIC/HOBt法は、手動方式で、多工程法である。
【0092】
合成サイクルは、以下のようにプログラムされた:
【表10】
【0093】
(ii)活性化Fmoc−ProをFmoc−Cys(Acm)−樹脂にカップリングすることによるFmoc−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0094】
【表11】
【0095】
(iii)活性化Fmoc−TrpをFmoc−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0096】
【表12】
【0097】
(iv)Fmoc−AspをFmoc−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0098】
【表13】
【0099】
(v)Fmoc−GlyをFmoc−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0100】
【表14】
【0101】
(vi)Fmoc−Lys(Boc)をFmoc−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0102】
【表15】
【0103】
(vii)Mpr(Acm)をFmoc−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂にカップリングすることによるMpr(Acm)−Lys(Boc)−Gly−Asp(Obut)−Trp−Pro−Cys(Acm)−樹脂の合成。反応を、工程1と同様に行った。合成サイクルは、以下のようにプログラムされた:
【0104】
【表16】
【0105】
合成において、Mpr(Acm)のカップリングは、カップリング反応を完結させるために、2回行う必要があった。
【0106】
実施例(3):樹脂からのペプチドの切断によるペプチドアミド(Acm)Mpr−Lys−Gly−Asp−Trp−Pro−Cys(Acm)−CONH2(式(3))の生成
組み上げられたペプチド樹脂(実施例1または2よりのもの)を、CS936中で、TFA(95%):TIS(2.5%):H2O(2.5%):EDT(0%):フェノール(0%)からなる切断用混合物500mlでR.T.にて2時間処理した。反応混合物を、RVを通して濾過し、TFAをRotavap上でエバポレートした。ペプチドの析出を、−20℃で、一定の撹拌をしながら、冷ジイソプロピルエーテル300mlを加えることにより、行った。溶媒中の粗ペプチド析出物を、−20℃で10時間放置する。ワットマン濾紙5番を通して濾過して単離し、続いて、冷溶媒で洗浄(100mlx3)して、切断用混合物中で使用したスカベンジャーを除去した。粗ペプチド析出物を、P2O5で真空乾燥し、RP−HPLCで特徴付けした(図1および図2)。
実施例1 実施例2
収率:58.73 収率:48.73
ペプチドの%純度:90% ペプチドの%純度:79.68%
【0107】
実施例(4):粗ペプチドのグアニル化による(Acm)Mpr−Homoarg−Gly−Asp−Trp−Pro−Cys(Acm)−CONH2(式(4))の生成
ペプチド(1g、1.157mmol)をDMF15mlに溶解し、pHをTEAで9.0に調整した。DMF(15ml)中の反応剤、硝酸3,5−ジメチルピラゾール−1−カルボキサミジン(931.5mg)を、ペプチドに加えた。反応混合物を、1倍過剰量の反応剤、硝酸3,5−ジメチルピラゾール−1−カルボキサミジンを多数回で添加しながら、30℃で4日間撹拌した。
アセトニトリル(TEAでpHを8.0に調整)280mlを加えることにより、ペプチドを反応混合物から析出させた。混合物を、さらに−20℃で10時間保持した。それを、ワットマン5番濾紙を通して濾過し、アセトニトリル(pH8.0)で3回洗浄し、続いて、手を加えていないアセトニトリルでpHを中和した。析出物を、一晩、高真空下に乾燥した。収率:85%。ペプチドを、RP−HPLCで特徴付けした(図2)。
【0108】
実施例(5):グアニル化ペプチドの脱ACMと引き続く酸化による、式(1):
【化22】
の生成
【0109】
TFA(134.9ml)およびアニソール(2.7ml)を混合し、氷中で冷却し、実施例3からの予め冷却したペプチド658mgに加え、窒素で飽和させた。続いて、AgOTf(3.47g)を添加し、氷浴中で2時間撹拌した。TFAを高真空下に除去し、ペプチドの銀塩を、ジイソプロピルエーテル(〜400ml)を加えて析出させた。反応混合物をG−4焼結ロートを通して濾過し、析出物(銀−ペプチド)をジイソプロピルエーテル(60mlx3)中に再懸濁し、上記と同様に洗浄し、真空下にP2O5で乾燥させた。
銀ペプチドの酸化は、氷冷条件で、銀−ペプチド塩10mgを50%DMSO/IM HCl15.6ml中に溶解することにより行った。反応混合物を25℃で3時間撹拌した。析出物を、G−4焼結ロートまたはHyfloベッドを通して濾過して、塩化銀を除去した。濾液を、酸化の完結についてチェックした(図4)。反応が完結すると、式(1)の粗ペプチドが得られた。純度百分率:85%。
【0110】
実施例(6):S−Sペプチドの精製
式(1)の粗ジスルフィドで輪になったペプチドを、prepC−18カラム(50x250mm、100Å)に入れた。ペプチドを、グラジエントプログラムでTFA(0.1%)水溶液とメタノールを用いることにより精製した(図5)。続いて、コントローラー、2個のLC8Aポンプ、UV−Vis検出器よりなる島津分取用HPLCシステム上で、上記の溶媒系を用いて定組成運転を行った。式(1)の精製ペプチドアミドを分析用RP−HPLCで分析した(図6)。質量は、質量分析機で決定した(図7)。
【0111】
実施例(7):S−Sペプチドの精製
メタノールの代わりにアセトニトリルを用いたこと以外は、実施例5と同様にして、精製を行い、式(1)のペプチドアミドを得た。
【0112】
実施例(8):酢酸水銀(II)を用いるグアニル化ペプチドの脱ACM
システインのAcm基保護を酢酸水銀(II)での処理によりグアニル化ペプチドから除去した以外は、実施例(4)と同様。
ペプチド(13.4mg、ローリー法で確認、Cys−Acmの)を酢酸(10%)400μl中に溶解した。10倍過剰量の酢酸水銀(II)(82.96mg)をそれに加え、反応物を渦撹拌し、R.T.で5時間保持した。100倍過剰量のβ−メルカプトエタノール(181.37μl)を加え、溶液を渦撹拌し、一晩、室温で放置した。反応混合物を4分間遠心分離し、上清を採集した。析出物を、遠心により、400μlx3の10%酢酸で抽出した。濾液をプールし、RP−HPLCで決定した純度百分率は55%であった(図3)。
【0113】
実施例(9):ヨウ素を用いるグアニル化ペプチドの脱ACM
システインのAcm基保護をヨウ素での処理によりグアニル化ペプチドから除去した以外は、実施例(4)と同様。
ペプチド(9.18mg、ローリー法で確認、Cys−Acmの)を酢酸(80%)17.8ml中に溶解し、N2で15分間パージした。I2の1mM溶液(80%酢酸中、〜4ml)を1時間かけて、黄色が持続するまでペプチド溶液に加えた。混合物をさらに30分間撹拌し、続いて、1N Na2S2O3で、黄色が消失するまで、中和し、凍結乾燥した。‘SH’の確認をエルマン(ellman)試験で行ったが、ACMの除去が達成されなかったとを示す、陰性であった。
【0114】
実施例(10):脱ACMペプチドの精製
酢酸水銀(II)処理ペプチド試料およびヨウ素処理ペプチド試料を、hyperprep(250x10mm、12μ、C−18カラム)を用いて、RP−HPLCにより脱塩した。
【0115】
実施例(11):式(1)の生物活性を調べるための血小板凝集阻害アッセイ
式(1)のペプチドの生物活性を、4Xレーザー血小板凝集計(EMA)を使用して、血小板凝集阻害アッセイを用いて調べた。承諾を得たヒトの提供者からの新鮮な静脈血を取り、クエン酸緩衝液に入れた。多血小板血漿(PRP)および乏血小板血漿を遠心分離により分離した。PRP中の血小板数は、2〜3x108個/mlに調整した。基準凝集量をPPPで調整した後、PRPを10〜20mM ADPで処理し、総凝集百分率を調べた。次いで、PRPをまず、種々の濃度の参照標準および式(1)の合成ペプチドと共にインキュベートした。その後、ADPを加えて、凝集の阻害を調べた。式1の合成ペプチドの生物活性の再現性を、数回確認し、参照標準と比較した。表1は、多くの実験のうちの一つを示す(12回の実験から)。合成ペプチド(SP)についてのIC50投与量は、市販の参照標準に比べて、140nM未満であった。SPでのADP誘起血小板凝集の50%より多い阻害が、大多数の試料において見られ、結果は、市販の参照標準に匹敵するものであった。
【0116】
【表17】
【図面の簡単な説明】
【0117】
【図1】樹脂からのHBTU−粗ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。
【図2】樹脂からのDIC−粗ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。
【図3】粗グアニル化ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。
【図4】SHペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。ピークA−粗SHペプチド。
【図5】粗環状ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;注入量:20μl;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。ピークA−粗環状ペプチド。
【図6】粗環状ペプチドの分取用RP−HPLC溶出プロフィール(カラム:Phenomenex Luna;C−18(2);10μ;250X50mm;流速:50ml/分;溶媒系:A:0.1%TFA、B:100%メタノール)。
【図7】精製した環状ペプチドの分析用RP−HPLC溶出プロフィール(カラム:PEP300;C−18;5μ;150X3mm;流速:0.5ml/分;溶媒系:A:0.1%TFA、B:100%アセトニトリル)。ピークA−純粋ペプチド。
【図8】質量が832であり、不純物が903であることを示す純粋ペプチドのMS分析。
【特許請求の範囲】
【請求項1】
固相上での式(1):
【化1】
のペプチドN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドの製造方法であって、
a.カップリングしてペプチド結合により互いに直接連結させることにより、固体支持体樹脂上に6個のアミノ酸およびチオアルキルカルボン酸からなるペプチド鎖を必要な配列に組み上げて、下記の式(2):
【化2】
のペプチド結合樹脂を得、
b.工程(a)の各々のカップリングの後に、遊離アミノ基を無水酢酸でキャッピングし、
c.工程(a)のペプチドを樹脂から切断し且つAcm基以外の全ての基を脱保護して、下記の式(3):
【化3】
のペプチドアミドを得、
d.有機溶媒中、ε−リジン−NH2で式(3)のペプチドをグアニル化し、続いて、他の溶媒で析出させて、下記の式(4):
【化4】
のペプチドを得、
e.適切な溶媒中で式(4)のペプチドを重金属塩で処理し、続いて、有機溶媒を用いて重金属−ペプチド塩を析出させて、式(5):
【化5】
の重金属−ペプチド塩を得、
f.適切な求核剤で、工程(e)の重金属−ペプチドを酸化し且つ脱塩して、式(1)のペプチドを得、そして
g.工程(f)の粗ペプチドを、RPHPLCで精製することを含む、その製造方法。
【請求項2】
化合物のアミノおよびカルボキシル等価体の反応が、そのペプチド結合を形成する、請求項1に記載の方法。
【請求項3】
保護された第一のアミノ酸のC−末端を、リンカーを介して固相に結合し、固相結合アミノ酸を得る、請求項1に記載の方法。
【請求項4】
使用される固体支持体が任意のアミド樹脂、好ましくはRinkアミド樹脂である、請求項1に記載の方法。
【請求項5】
第一の保護されたアミノ酸が、チオール保護Fmocシステインである、請求項1に記載の方法。
【請求項6】
リンカーを有する樹脂の切断が、組み上げられたペプチドアミドの放出をもたらすものである、請求項1に記載の方法。
【請求項7】
ペプチドアミド化合物が、ペプチド結合により末端官能性の各々に連結された化合物であり、各々の末端官能性がアミノもしくはカルボン酸基またはそれらの誘導体である、請求項1に記載の方法。
【請求項8】
使用されるアミノ酸が、Cys、Pro、Trp、Asp、Lys、Gly、Arg、Har、LeuおよびGluよりなる群から選択される、請求項1に記載の方法。
【請求項9】
使用されるチオアルキルカルボン酸が、2−チオプロピオン酸である、請求項1に記載の方法。
【請求項10】
アミノ酸の−NH2官能基の保護基がFmocまたはBocである、請求項1に記載の方法。
【請求項11】
−COOH官能基の保護基が非保護または保護O−tBuエステルである、請求項1に記載の方法。
【請求項12】
SH−官能の保護基がAcm基である、請求項1に記載の方法。
【請求項13】
工程(c)において、ペプチドが、ある規定の比率での反応剤TFA、TIS、EDT、DCM、フェノールおよび水、好ましくはTFA(85〜98%):TIS(0〜5%):H2O(0〜5%):EDT(0〜5%):フェノール(0〜5%)、より好ましくはTFA(94.5〜95.5%):TIS(0〜2.5%):H2O(0〜3%):EDT(0〜2.5%)を用いて固体支持体樹脂から切断される、請求項1に記載の方法。
【請求項14】
工程(d)において、グアニル化に使用される有機溶媒が、DMF、エタノールおよびメタノールよりなる群から選択される、請求項1に記載の方法。
【請求項15】
工程(d)において、式(4)のペプチドの析出が、アセトン、アセトニトリル、メタノール、エーテル類、ペンタン、ヘキサンおよびこれらの混合物よりなる群から選択される溶媒を使用して行われる、請求項1に記載の方法。
【請求項16】
析出が、好ましくはアセトニトリルを使用して行われる、請求項15に記載の方法。
【請求項17】
工程(g)において、得られた式(1)のペプチドが、99%より高い純度を有する、請求項1に記載の方法。
【請求項18】
固相合成による式(1)のペプチドの製造が、Fmoc化学を使用して行われる、請求項1に記載の方法。
【請求項19】
アミノ酸の組み上げが、式(2):
【化6】
のペプチド結合樹脂を与える、請求項1に記載の方法。
【請求項20】
工程(d)において、式(3)のペプチドのグアニル化が、好ましくは、DMF中の溶媒を使用することにより行われる、請求項1に記載の方法。
【請求項21】
式(4)のペプチドの精製が、RP−HPLCにより行われ、達成される、請求項1に記載の方法。
【請求項22】
式(4)のペプチドの処理に使用される重金属塩が、TFA中のトリフルオロメタンスルホン酸銀である、請求項1に記載の方法。
【請求項23】
式(5):
【化7】
の重金属ペプチド塩の析出が、好ましくはエーテル系溶媒、より好ましくはジイソプロピルエーテルを使用して行われる、請求項1に記載の方法。
【請求項24】
工程(f)において、重金属ペプチド塩が、HClとDMSOで処理されて、同時に重金属を除去し、且つ得られたペプチドを酸化して、式(1)の粗ペプチドアミドを生成する、請求項1に記載の方法。
【請求項25】
式(1)の粗ペプチドアミドが、RP−HPLCで精製される、請求項1に記載の方法。
【請求項26】
式(1)の粗ペプチドアミドの精製が、好ましくは、移動相として、メタノールおよび/またはアセトニトリルが単独でまたは水性TFA(0〜0.5%)と組み合わせて使用される、C−4、C−8またはC−18シリカまたはポリマー逆相カラムを使用するRP−HPLCで行われる、請求項1に記載の方法。
【請求項27】
式(2):
【化8】
の中間体ペプチド。
【請求項28】
式(3):
【化9】
の中間体ペプチド。
【請求項29】
式(4):
【化10】
の中間体ペプチド。
【請求項30】
式(5):
【化11】
の中間体ペプチド塩。
【請求項1】
固相上での式(1):
【化1】
のペプチドN6−(アミノイミノメチル)−N2−(3−メルカプト−1−オキソプロピル−L−リジルグリシル−L−α−アスパルチル−L−トリプトフィル−L−プロリル−L−システインアミド,環状(1→6)−ジスルフィドの製造方法であって、
a.カップリングしてペプチド結合により互いに直接連結させることにより、固体支持体樹脂上に6個のアミノ酸およびチオアルキルカルボン酸からなるペプチド鎖を必要な配列に組み上げて、下記の式(2):
【化2】
のペプチド結合樹脂を得、
b.工程(a)の各々のカップリングの後に、遊離アミノ基を無水酢酸でキャッピングし、
c.工程(a)のペプチドを樹脂から切断し且つAcm基以外の全ての基を脱保護して、下記の式(3):
【化3】
のペプチドアミドを得、
d.有機溶媒中、ε−リジン−NH2で式(3)のペプチドをグアニル化し、続いて、他の溶媒で析出させて、下記の式(4):
【化4】
のペプチドを得、
e.適切な溶媒中で式(4)のペプチドを重金属塩で処理し、続いて、有機溶媒を用いて重金属−ペプチド塩を析出させて、式(5):
【化5】
の重金属−ペプチド塩を得、
f.適切な求核剤で、工程(e)の重金属−ペプチドを酸化し且つ脱塩して、式(1)のペプチドを得、そして
g.工程(f)の粗ペプチドを、RPHPLCで精製することを含む、その製造方法。
【請求項2】
化合物のアミノおよびカルボキシル等価体の反応が、そのペプチド結合を形成する、請求項1に記載の方法。
【請求項3】
保護された第一のアミノ酸のC−末端を、リンカーを介して固相に結合し、固相結合アミノ酸を得る、請求項1に記載の方法。
【請求項4】
使用される固体支持体が任意のアミド樹脂、好ましくはRinkアミド樹脂である、請求項1に記載の方法。
【請求項5】
第一の保護されたアミノ酸が、チオール保護Fmocシステインである、請求項1に記載の方法。
【請求項6】
リンカーを有する樹脂の切断が、組み上げられたペプチドアミドの放出をもたらすものである、請求項1に記載の方法。
【請求項7】
ペプチドアミド化合物が、ペプチド結合により末端官能性の各々に連結された化合物であり、各々の末端官能性がアミノもしくはカルボン酸基またはそれらの誘導体である、請求項1に記載の方法。
【請求項8】
使用されるアミノ酸が、Cys、Pro、Trp、Asp、Lys、Gly、Arg、Har、LeuおよびGluよりなる群から選択される、請求項1に記載の方法。
【請求項9】
使用されるチオアルキルカルボン酸が、2−チオプロピオン酸である、請求項1に記載の方法。
【請求項10】
アミノ酸の−NH2官能基の保護基がFmocまたはBocである、請求項1に記載の方法。
【請求項11】
−COOH官能基の保護基が非保護または保護O−tBuエステルである、請求項1に記載の方法。
【請求項12】
SH−官能の保護基がAcm基である、請求項1に記載の方法。
【請求項13】
工程(c)において、ペプチドが、ある規定の比率での反応剤TFA、TIS、EDT、DCM、フェノールおよび水、好ましくはTFA(85〜98%):TIS(0〜5%):H2O(0〜5%):EDT(0〜5%):フェノール(0〜5%)、より好ましくはTFA(94.5〜95.5%):TIS(0〜2.5%):H2O(0〜3%):EDT(0〜2.5%)を用いて固体支持体樹脂から切断される、請求項1に記載の方法。
【請求項14】
工程(d)において、グアニル化に使用される有機溶媒が、DMF、エタノールおよびメタノールよりなる群から選択される、請求項1に記載の方法。
【請求項15】
工程(d)において、式(4)のペプチドの析出が、アセトン、アセトニトリル、メタノール、エーテル類、ペンタン、ヘキサンおよびこれらの混合物よりなる群から選択される溶媒を使用して行われる、請求項1に記載の方法。
【請求項16】
析出が、好ましくはアセトニトリルを使用して行われる、請求項15に記載の方法。
【請求項17】
工程(g)において、得られた式(1)のペプチドが、99%より高い純度を有する、請求項1に記載の方法。
【請求項18】
固相合成による式(1)のペプチドの製造が、Fmoc化学を使用して行われる、請求項1に記載の方法。
【請求項19】
アミノ酸の組み上げが、式(2):
【化6】
のペプチド結合樹脂を与える、請求項1に記載の方法。
【請求項20】
工程(d)において、式(3)のペプチドのグアニル化が、好ましくは、DMF中の溶媒を使用することにより行われる、請求項1に記載の方法。
【請求項21】
式(4)のペプチドの精製が、RP−HPLCにより行われ、達成される、請求項1に記載の方法。
【請求項22】
式(4)のペプチドの処理に使用される重金属塩が、TFA中のトリフルオロメタンスルホン酸銀である、請求項1に記載の方法。
【請求項23】
式(5):
【化7】
の重金属ペプチド塩の析出が、好ましくはエーテル系溶媒、より好ましくはジイソプロピルエーテルを使用して行われる、請求項1に記載の方法。
【請求項24】
工程(f)において、重金属ペプチド塩が、HClとDMSOで処理されて、同時に重金属を除去し、且つ得られたペプチドを酸化して、式(1)の粗ペプチドアミドを生成する、請求項1に記載の方法。
【請求項25】
式(1)の粗ペプチドアミドが、RP−HPLCで精製される、請求項1に記載の方法。
【請求項26】
式(1)の粗ペプチドアミドの精製が、好ましくは、移動相として、メタノールおよび/またはアセトニトリルが単独でまたは水性TFA(0〜0.5%)と組み合わせて使用される、C−4、C−8またはC−18シリカまたはポリマー逆相カラムを使用するRP−HPLCで行われる、請求項1に記載の方法。
【請求項27】
式(2):
【化8】
の中間体ペプチド。
【請求項28】
式(3):
【化9】
の中間体ペプチド。
【請求項29】
式(4):
【化10】
の中間体ペプチド。
【請求項30】
式(5):
【化11】
の中間体ペプチド塩。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2008−503597(P2008−503597A)
【公表日】平成20年2月7日(2008.2.7)
【国際特許分類】
【出願番号】特願2007−526703(P2007−526703)
【出願日】平成16年10月10日(2004.10.10)
【国際出願番号】PCT/IN2004/000315
【国際公開番号】WO2005/121164
【国際公開日】平成17年12月22日(2005.12.22)
【出願人】(506249303)ユーエスブイ・リミテッド (4)
【氏名又は名称原語表記】USV LIMITED
【Fターム(参考)】
【公表日】平成20年2月7日(2008.2.7)
【国際特許分類】
【出願日】平成16年10月10日(2004.10.10)
【国際出願番号】PCT/IN2004/000315
【国際公開番号】WO2005/121164
【国際公開日】平成17年12月22日(2005.12.22)
【出願人】(506249303)ユーエスブイ・リミテッド (4)
【氏名又は名称原語表記】USV LIMITED
【Fターム(参考)】
[ Back to top ]