
ペプチドデホルミラーゼ阻害物質、その使用、及びそれを含む医薬組成物
本発明は、a)次の一般式(I):

(式中:互いに独立して、R2及びR3は、H、炭素原子1〜5個のアルキル基、又は1〜5個のヘテロ原子を含む2〜20個の原子の金属キレート基を表し;互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;R1は、H、又は1〜50個の炭素原子を含む基を表し;但し、R2及びR3のうちの少なくとも1つが、上に定義した金属キレート基を表す)
を有し;並びに
b)大腸菌(Escherichia coli)ニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)ニッケル結合ペプチドデホルミラーゼ(配列番号:2)について約1μMよりも低いIC50を有する、
化合物又は薬学的に許容可能なその塩の、細菌感染症若しくは原生動物感染症の予防若しくは治療を対象とした医薬の製造のための使用、又は除草剤の製造のための使用に関する。
(式中:互いに独立して、R2及びR3は、H、炭素原子1〜5個のアルキル基、又は1〜5個のヘテロ原子を含む2〜20個の原子の金属キレート基を表し;互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;R1は、H、又は1〜50個の炭素原子を含む基を表し;但し、R2及びR3のうちの少なくとも1つが、上に定義した金属キレート基を表す)
を有し;並びに
b)大腸菌(Escherichia coli)ニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)ニッケル結合ペプチドデホルミラーゼ(配列番号:2)について約1μMよりも低いIC50を有する、
化合物又は薬学的に許容可能なその塩の、細菌感染症若しくは原生動物感染症の予防若しくは治療を対象とした医薬の製造のための使用、又は除草剤の製造のための使用に関する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
本発明は、ペプチドデホルミラーゼ阻害物質、それを含む医薬組成物、及び除草剤としてのその使用又は細菌感染症若しくは寄生虫感染症の治療のためのその使用に関する。
【0002】
ペプチドデホルミラーゼ(PDF;EC3.5.1.88)は、すべてのグラム陽性細菌及びグラム陰性細菌に見出された必須の酵素活性である。その細胞における役割は、すべての新生タンパク質からN−ホルミル基を除去することである。これが、N−末端メチオニン切出しを引き起こすもう一つの必須の細菌酵素であるメチオニンアミノペプチダーゼの作用を可能にする(総説についてはGiglione et al.,2004を参照のこと)。PDFは、ストレプトミセス種(Streptomyces sp.)が産生する天然抗生物質であるアクチノニンの天然標的である(Gordon et al.,1962;Chen et al.,2000)。効き目は少し低いが、ストレプトミセス種(Streptomyces sp.)が産生する、PDFを阻害する他の天然産物についても記述された(Chu et al.,2001)。
【化1】
【0003】
配列及び構造の両解析に基づき、2つの細菌性PDF型(PDF1及びPDF2)が区別されている(Giglione et al.,2000a;Guilloteau et al.,2002)。PDF2は、グラム陽性細菌にのみ見出される。高解像度の三次元(3D)構造及び精密酵素分析法が利用可能であるため、大腸菌(Escherichia coli)のPDF(PDF1B)及びバチルスステアロサーモフィラス(Bacillus stearothermophilus)のPDF2(PDF2)が、いずれかのPDFクラスの代表として選択された(Guilloteau et al.,2002;Ragusa et al.,1998)。細菌は、PDFをコード化する1つ又はいくつかの機能的遺伝子を有する可能性がある。例えば、大腸菌(Escherichia coli)は、defシストロンによりコード化される1つのPDF1遺伝子を有し、ブドウ球菌種及び連鎖球菌種(Staphylo- and Streptococci spp.)などの主要病原体は、ただ1つのPDF2を有し、並びにバチルス種(Bacillus spp.)は、2つのPDF遺伝子、すなわちPDF1をコード化するdef遺伝子及びPDF2をコード化するykrB遺伝子を有する。枯草菌(B.subilis)においては、PDF2が主要PDFである(Haas et al.,2001)。
【0004】
過去10年間、PDFは、最先端技術を結集した薬剤発見法を用いる新規抗生物質選択のための選択目標として選択されてきた(Giglione et al.,2000a;Meinnel,2000;Giglione & Meinnel,2001)。過去4年間、アクチノニンによく似た多数のPDF阻害物質(PDFI)が記述され、そのうちのいくつかが、最近、臨床試験に入った(総説については、Boularot et al.,2004を参照のこと)。従って、PDFIは、ゲノミクス戦略から生まれた抗菌性薬剤発見法の最初の成功である(Miesel et al.,2003)。
【0005】
しかしながら、機能的PDF配列が、当初は真正細菌に独特の形態であると考えられたけれども(Mazel et al.,1994)、ごく最近、多くの真核生物のゲノムに検出されている(Giglione et al.,2000b;Bracchi−Ricard et al.,2001)。これらのPDFは、細胞小器官、ミトコンドリア、及び植物色素体(葉緑体)、並びにマラリア病因である熱帯熱マラリア原虫(Plasmodium falciparum)などのアピコンプレキサン(apicomplexan)寄生虫(アピコプラスト)にターゲッティングされる。更に、PDFホモログは、キネトプラスチド原生動物でも証明されている(Meinnel,2000)。
【0006】
配列又は生化学的分析及び位置指定突然変異誘発データに基づき、すべてのミトコンドリアPDFsが、独特なサブクラス1型PDFs(PDF1A)に属することが示され、それらの活性部位が多数の特異性を示した(Giglione et al.,2000b;Serero et al.,2001;Serero et al.,2003)。色素体のPDFsは、細菌のPDF1sと強く関連しており、PDF1Bとして分類される(Giglione et al.,2004).
【0007】
アクチノニンがインビトロでもインビボでも、熱帯熱マラリア原虫(P.falciparum)に対しても植物のPDFに対しても阻害作用を示すので、この発見は先ず、細菌のPDFに対して活性なPDFIを、駆虫剤として、そして除草剤としてさえ使用できるということを示唆した(Meinnel,2000;Giglione & Meinnel,2001;Wiesner et al.,2001;Serero et al.,2001;Serero et al.,2001;Dirk et al.,2001)。
【0008】
それにもかかわらず、ヒトミトコンドリアにおけるPDF1A(HsPDF1)の存在が、前記PDFIの毒性に関して多数の問題を引き起こした。その理由は、特に、アクチノニンが、(i)インビトロで効果的にHsPDF1活性をブロックし、(ii)最も敏感なグラム陽性細菌に対する最良のPDFIについての最小阻止濃度(MIC)と類似した濃度範囲(2〜3μg/ml)において細胞毒性活性を及ぼすからである(Lee et al.,2003;Serero et al.,2003;Xu et al.,1998;Grujic et al.,2002,Lee et al 2004)。
【0009】
ミトコンドリアPDFの役割が不明瞭のままであり、ミトコンドリア膜が一般に多くの化合物に対して極めて不透過性であると考えられてはいるけれども、予防原則は、PDF1A及びPDF1B又はPDF2との間の、PDFIによるいかなる相互阻害をも回避するために、今や大きな注意をはらうべきであるということを強いる。換言すれば、このことが意味するのは、新世代のPDFIは、少なくともHsPDFを含むミトコンドリアPDF(PDF1A)に対する阻害作用を全く示してはならないということである。
【0010】
従って、本発明の目的は、細菌、植物色素体、並びにアピココンプレキサン(apicocomplexan)及びキネトプラスチド原生生物などの原生動物のペプチドデホルミラーゼを強く阻害しやすく、しかしミトコンドリア、特に哺乳類のミトコンドリアのペプチドデホルミラーゼを阻害しない新規な化合物を提供することである。
【0011】
本発明のもう一つの目的は、細菌若しくは寄生虫感染症を有する個体(individuals)を治療するための、又は除草剤としての前記化合物の使用に関する。
【0012】
本発明は、インドール誘導体が、細菌のペプチドデホルミラーゼを強く阻害することができるのに対して、ヒトミトコンドリアのペプチドデホルミラーゼに対して本質的に不活性であるという、本発明者らによる予想外の発見に基づくものである。
【0013】
本発明は、
a)下記一般式(I):
【化2】
(式中、
− 互いに独立して、R2及びR3は、H、炭素原子1〜5個のアルキル基、又は1〜5個のヘテロ原子を含む2〜20個の原子の金属キレート基を表し;
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し、R2及びR3のうちの少なくとも1つが、上に定義した金属キレート基を表す)
を有し;並びに
b)大腸菌(Escherichia coli)ニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)ニッケル結合ペプチドデホルミラーゼ(配列番号:2)について約1μMよりも低いIC50を有する;化合物又は薬学的に許容可能なその塩の、
細菌感染症若しくは原生動物感染症の予防若しくは治療を対象とした医薬の製造のための使用、又は除草剤の製造のための使用に関する。
【0014】
上に言及した「金属キレート基」は、金属原子又は金属イオンと配位錯体を形成しやすい基に関するものである(Whittaker et al.,1999;Boularot et al.,2004)。従って、「金属キレート基」は、特に、1つ又はいくつかの非共有電子対を有する基に関するものである。
【0015】
好ましい実施態様においては、本発明は、R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−(X)n1−Y
(式中、
− nは、整数0〜3であり、
− n1は、0又は1であり、
− Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
− Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド),−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
の金属キレート基で表される、上で定義した一般式(I)の化合物の使用に関する。
【0016】
その他の好ましい実施態様においては、本発明はまた、R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、上で定義した一般式(I)の化合物の使用に関する。
【0017】
その他の好ましい実施態様においては、本発明は、上に定義した一般式(I)で表される化合物の使用に関するものであり、前記式中、R4、R5及びR6が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、チオアルキル基、チオアリール基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるものである使用に関する。
【0018】
本発明は、より詳細には、R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、上で定義した一般式(I)で表される化合物の使用に関する。
【0019】
好ましい実施態様によれば、本発明はまた、R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるもので表される、上で定義した一般式(I)で表される化合物の使用に関する。
【0020】
その他の好ましい実施態様においては、本発明は、R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、上で定義した、一般式(I)の化合物の使用に関する。
【0021】
特に好ましい実施態様によれば、本発明は、次の一般式(II):
【化3】
(式中、Roは、請求項1〜7に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は上に定義したとおりである)
で表される、上に定義した化合物の使用に関する。
【0022】
その他の特に好ましい実施態様によれば、本発明は、次の一般式(III):
【化4】
(式中、Roは上に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは上に定義したとおりである)
で表される、上に定義した化合物の使用に関する。
【0023】
特に好ましい実施態様においては、本発明は、
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す
、上に定義した、式(III)で表される化合物の使用に関する。
【0024】
その他の特に好ましい実施態様においては、本発明は、前記化合物が、下記諸式:
【化5】
に相当することを特徴とする、上に定義した、一般式(I)で表される化合物の使用に関する。
【0025】
ペプチドデホルミラーゼ(PDF)は、金属酵素である(すなわち、その補欠分子族が金属イオンである)。従って、本明細書の目的どおり、表現「ニッケル結合ペプチドデホルミラーゼ」は、その金属イオンとして本質的にニッケルのみを含有する酵素に関する。例えば、そのような酵素は、Ragusa et al.(1998)に記述されたように、ニッケル存在下に精製することにより得ることができる。
【0026】
表現IC50は、大腸菌(Escherichia coli)のニッケル結合ペプチドデホルミラーゼ(配列番号:1)(EMBL/GenBank accession number P27251)活性及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)のニッケル結合ペプチドデホルミラーゼ(配列番号:2)(EMBL/GenBank accession number O31410)活性の50%を阻害するのに必要な一般式(I)で表される化合物の濃度に関するものである。
【0027】
ペプチドデホルミラーゼに対するIC50は、Serero et al.(2003)に記述された一般法に従うことにより測定することができる。
【0028】
手短に言えば、速度論的解析(Lazennec & Meinnel,1997に記述されている)の前に、阻害物質を、研究される酵素とともに、規定の最終濃度で25℃で15分間インキュベートする。速度論的アッセイは、小容量の基質を添加することにより開始される。基質は、1mMのホルミル−Met−Ala−Serである。速度論的解析は、阻害物質不在下に、脱ホルミル化速度0.5μM/sを与える酵素濃度存在下に行なわれる。IC50値は、50%阻害を与える濃度に相当する。
【0029】
有利には、一般式(I)で表される化合物は、細菌のPDF(すなわちPDF1及びPDF2)、並びに葉緑体、アピコプラスト及びキネトプラストのPDFを阻害しやすいが、ミトコンドリアのPDF(すなわちPDF1A)を阻害しにくいものである。
【0030】
その他の特に好ましい実施態様においては、本発明は、前記原生動物が、アピコプラスト又はキネトプラストを有する原生動物のうちから選択され、特に、下記属の原生動物、すなわちプラスモジウム属(Plasmodium)、トキソプラズマ属(Toxoplasma)、サルコシスティス属(Sarcocystis)、クリプトスポリジウム属(Cryptosporidium)、エイメリア属(Eimera)、タイレリア属(Theileria)、バベシア属(Babesia)、トリパノゾーマ属(Trypanosoma)、リーシュマニア属(Leishmania)を含む群から選択される、上で定義された、一般式(I)の化合物の使用に関する。
【0031】
有利には、式(I)で表される化合物は、アピコプラスト又はキネトプラストのPDFにターゲッティングして阻害し、こうしてそれぞれ、アピコプラストの機能及びキネトプラストの機能を障害し、それが原生動物寄生虫の成長を阻害し、及び/又は原生動物寄生虫を殺す。
【0032】
更にその他の特に好ましい実施態様においては、本発明は、前記細菌が、次の属の細菌、すなわち、ブドウ球菌属(Staphylococcus)、連鎖球菌属(Streptococcus)、バチルス属(Bacillus)、エンテロコッカス属(Enterococcus)、シュードモナス属(Pseudomonas)、アシネトバクター属(Acinetobacter)、エンテロバクター属(Enterobacter)、ヘモフィルス属(Haemophilus)を含む群から選択される、上に定義した一般式(I)の化合物の使用に関する。
【0033】
有利には、一般式(I)で表される化合物は、細菌のPDF1にもPDF2にもターゲッティングして阻害し、そのことが前記化合物を、グラム陰性細菌に対してもグラム陽性細菌に対しても使用することができる広域スペクトルの抗生物質にする。
【0034】
本発明はまた、活性物質として、上に定義した式(I)で表される少なくとも1つの化合物、又は薬学的に許容可能なその塩を、薬学的に許容可能な賦形剤とともに含むことを特徴とする医薬組成物に関する。
【0035】
好ましい実施態様においては、上に定義した医薬組成物は、一般式(I)で表される化合物を約250mg〜約5000mgの単位用量で個体に投与するのに適している。
【0036】
その他の実施態様においては、上に定義した医薬組成物は、一般式(I)で表される化合物を約250mg〜約5000mgの1日用量で個体に投与するのに適している。
【0037】
その他の実施態様においては、上に定義した医薬組成物は、経口、静脈内、又は腹腔内経路により投与される。
【0038】
本発明はまた、活性物質として、式(I)で表される少なくとも1つの化合物を含むことを特徴とする、除草剤組成物に関する。
【0039】
好ましい実施態様においては、上に定義した除草剤組成物は、式(I)で表される化合物を約1mg/l〜約1000mg/lの用量で、特に約10mg/l〜約100mg/lの用量で、植物に投与するのに適している。
【0040】
有利には、式(I)で表される化合物は、葉緑体のPDFにターゲッティングして阻害し、そのことが、例えば植物又は藻類などの、葉緑体を保有する生物の成長を絶つか又は重度に障害する。
【0041】
本発明はまた、上に定義したような一般式(I)で表される化合物に関する。
【0042】
本発明は、より詳細には、一般式(I):
【化6】
(式中、
− 互いに独立して、R2及びR3は、H、メチル基、エチル基、プロピル基、又は次の式:
−(CH2)n−(X)n1−Y
(式中、
*nは、整数0〜3であり、
*n1は、0又は1であり、
*Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
*Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド)、−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
で表される金属キレート基を表し、
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し:
*R2及びR3のうち少なくとも1つは、上に定義した金属キレート基を表し、及び
*R3が上に定義した金属キレート基を表すとき、−(CH2)n−(X)n1−Yは、−(CH2)2−NH2又は−(CH2)2−SHを表すことができない)
で表される化合物に関する。
【0043】
本発明は、より詳細には、R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、上で定義した、一般式(I)の化合物に関する。
【0044】
本発明の好ましい化合物は、R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、上で定義した、一般式(I)の化合物である。
【0045】
その他の好ましい実施態様においては、本発明は、R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、上で定義した、一般式(I)の化合物に関する。
【0046】
その他の特に好ましい実施態様においては、本発明は、R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、上で定義した一般式(I)で表される化合物に関する。
【0047】
本発明はまた、より詳細には、R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、請求項16〜20のいずれか一項に記載の一般式(I)の化合物に関する。
【0048】
本発明はまた、より詳細には、次の一般式(II):
【化7】
(式中、Roは、上に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は上に定義したとおりである)
で表される上に定義した化合物に関する。
【0049】
その他の好ましい実施態様においては、本発明は、次の一般式(III):
【化8】
(式中、Roは、上に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは上に定義したとおりである)
で表される上に定義した化合物に関する。
【0050】
その他の特に好ましい実施態様においては、本発明は、
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す、
上で定義された式(III)の化合物に関する。
【0051】
その他の特に好ましい実施態様においては、本発明は、上に定義した一般式(I)で表される化合物に関するものであり、前記化合物が、次の諸式:
【化9】
に相当することを特徴とする化合物に関する。
【0052】
本発明はまた、ペプチドデホルミラーゼ酵素をスクリーニングする化合物と接触させる工程、及び前記ペプチドデホルミラーゼ酵素に対する前記化合物の阻害活性を測定する工程を含むことを特徴とする、細菌又は原生動物感染症の予防若しくは治療、又は除草剤としての使用、を対象としたインドール構造を含む化合物をスクリーニングする方法に関する。
【0053】
本明細書の目的どおり、「インドール構造を含む化合物」は、次の構造:
【化10】
(構造中、あいた結合は、H又は任意の化学結合のいずれかに連結される)
を有する化合物である。
【0054】
上に定義したスクリーニング方法の好ましい実施態様においては、ペプチドデホルミラーゼ酵素に対するスクリーニング化合物の阻害活性は、前記ペプチドデホルミラーゼ酵素について前記化合物のIC50を測定することにより評価される。
【0055】
IC50は、前記の方法により測定することができる。
【0056】
上に定義したスクリーニング方法のその他の好ましい実施態様においては、IC50が約1μMより低いスクリーニング化合物が選択される。
【0057】
上に定義したスクリーニング方法のその他の好ましい実施態様においては、スクリーニング化合物は、上に定義した式(I)に相当する。
【0058】
上に定義したスクリーニング方法のその他の好ましい実施態様においては、ペプチドデホルミラーゼ酵素は、大腸菌(Escherichia coli)のニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)のニッケル結合ペプチドデホルミラーゼ(配列番号:2)である。
【0059】
上に定義したスクリーニング方法の特に好ましい実施態様においては、スクリーニング化合物が、ミトコンドリアのペプチドデホルミラーゼ、特にヒトミトコンドリアのペプチドデホルミラーゼに対する阻害特性を本質的に有しないということが更に試験される。
【0060】
一般式(I)を有する化合物の合成は、以下の手順に従い、又は実施例に記載するようにして行なうことができる。
【0061】
アミン、シアナミド及びヒドロキシグアニジンのインドール誘導体は、次の反応工程式1に示すようにして製造することができる。
【化11】
【0062】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基、チオアルキル基、又はチオアリール基を表す。
【0063】
手短に言えば、フィルスマイヤー−ハーク反応により相当するインドールから製造されるインドール−3−カルボキサルデヒドは、リン酸水素二アンモニウム、1−ニトロプロパン及び酢酸を用いて処理することにより、インドール−3−カルボニトリルに変換される(Jiang et al.,2000)。ニトリルは、CoCl2/NaBH4を用いてアミンに還元される(Leclerc et al.,1998)。シアナミドは、アミンのBrCNとの反応によって得られ、酢酸ナトリウム存在下に塩酸ヒドロキシルアミンとの反応によりヒドロキシグアニジンに変換される(Lefevre−Groboillot et al.,2001)。
【0064】
ヒドロキサム酸、ヒドロキシメチルケトン及びチオメチルケトンインドール誘導体は、次の反応工程式2に示すようにして製造することができる。
【化12】
【0065】
Wは、アルキル基、アリール基、アルコキシ基、アリールオキシ基又はチオアリール基を表す。
【0066】
手短に言えば、酢酸基は、亜鉛塩を2−ブロモ酢酸エチルエステルと反応させ、そのあとエステルを加水分解することにより、3−インドール位に導入される(Dillard et al.,1996)。ヒドロキシメチルケトンは、Wissnerにより記述された手順(Wissner et al.,1979)に従い、全くルイス酸触媒を用いないで、室温(r.t.)で、(インドール−3−イル)アセチルクロリドをトリス(トリメチルシリルオキシ)エチレンと反応させ、そのあと中間体を80℃で加水分解脱炭酸反応させることにより製造される。チオアセチル誘導体は、酸から出発して5ステップで製造され、塩化アシルのジアゾメタンとの反応によりα−ジアゾケトンに変換される(Salim & Capretta,2000)。HCl(g)エーテル溶液との処理で、クロロメチルケトンを得る。DMF中、室温で塩化物をチオ酢酸カリウムで置換することにより、対応するチオエステルを得、これを厳密な嫌気状態下にメタノール中のNa2CO3を用いて加水分解し、そのあとHCl(g)エーテル溶液で酸性化することにより脱保護することができる。
【0067】
対応する酸のインドール誘導体からヒドロキサム酸インドール誘導体を製造する一般手順は、次のとおりである。
【0068】
ヒドロキシルアミンをエステルと直接反応させることも可能ではあるが、(インドール−3−イル)酢酸誘導体の一部は市販品として入手可能であるので、酸から出発する一般合成手順を以下に記載する。
【0069】
手短に言えば、インドール酢酸誘導体(0.3mmol)のDMF溶液10mlに、1−ヒドロキシベンゾトリアゾール(1.1当量,m=44mg)、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(1.1当量,m=63mg)及びN−メチルモルホリン(1.1当量,36μL)を添加する。この溶液を室温で1時間撹拌する。塩酸ヒドロキシルアミン(1.1当量,m=23mg)を添加後、溶液を撹拌下に一晩放置する。次いでDMFを真空下に蒸発させ、酢酸エチルを残渣に添加する。この酢酸エチル溶液を、順次に、水、飽和NaHCO3水溶液及び飽和NaCl水溶液で洗浄する。Na2SO4で乾燥し、濾過後、溶媒を蒸発乾固して、ヒドロキサム酸誘導体を収率40〜60%で得る。
【0070】
インドール誘導体の2位置換は、反応工程式3に記載するようにして実施することができる。
【化13】
【0071】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基、又はチオアリール基を表す。
【0072】
手短に言えば、2−リチオ−N−ベンゼンスルホニルインドール(N−ベンゼンスルホニルインドールをBuLiと反応させて得る)をシュウ酸ジメチルと反応させ(Hasan et al.,1981)、そのあと引き続き塩基性加水分解とウォルフ−キシュナー還元を行ない、(インドール−2−イル)酢酸を得る(Weller & Ford,1984)。
【0073】
N−置換インドールは反応工程式4に記載するようにして製造することができる。
【化14】
【0074】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基又はチオアリール基を表す。
【0075】
手短に言えば、N−カルボキシメチル化は、リチオ誘導体(−78℃でTHF中2当量のリチウムビス(トリメチルシリル)アミドを用いて製造する)のクロロギ酸ベンジルとの反応により達成される(Horwell et al.,1997)。
【化15】
【0076】
本発明によるインドール窒素(R1)(反応工程式5)の様々な官能基化は、例えばDillard et al.,(1996)及びEissenstat et al.,(1995)に記述されたようにして製造することができる。
【0077】
リバースヒドロキサメートインドール誘導体は、反応工程式6に示すようにして製造することができる。
【化16】
【0078】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基、チオアルキル基、又はチオアリール基を表す。
【0079】
手短に言えば、ルートAに従い、アルデヒドのピリジン中塩酸ヒドロキシルアミンとの縮合で、ケトキシムを得、これを更にBorch条件下に還元する(Borch et al.,1971)。ルートBに従い、アルデヒドの還元後、ヒドロキシメチル誘導体を、Mitsunobu反応にかける(Kolossa et al.,1997)か又は臭素化するかいずれかしてから、N,O−ビス(tert−ブトキシカルボニル)ヒドロキシルアミンを用いる求核置換反応にかける。いったんトリフルオロ酢酸で脱保護すると、ヒドロキシルアミンはホルミル化され、リバースヒドロキサメートを与える。選択的N−ホルミル化反応は、特にHill et al.(2002)に記述されている。
【0080】
本発明による式(I)で表される化合物をすべて合成するためにこれらの手順にもたらし得る諸適合は、当業者の通常の技術の範囲内にある。
【0081】
《材料及び方法》
すべての溶媒及び化学薬品は、それぞれSDS及びAldrichから購入した。DMF、THF及びCH2Cl2は、標準的手順を用いて乾燥し、アルゴン雰囲気下に4Åモレキュラーシーブ上保存した。1HNMRスペクトルはBruker ARX−250スペクトロメーターで記録し、化学シフトはTMSから低磁場側でのppm単位で報告した。IRスペクトルは、MIRacleTM 単反射水平ATRユニット(ジルコニウム−セレン結晶)を備えた、Perkin−Elmer Spectrum One FT−IRスペクトロメーターを用いて得た。FAB及びCI質量スペクトルは、ParisのENSで記録した。元素分析は、Paris VI University(France)又はGif−sur−Yvette(CNRS、France)の微量分析サービスにより実施した。アルゴン下に行われる実験は、真空ラインで実施した。
【化17】
【0082】
《3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(1c)》
新しく蒸留したTHF(10mL)中の2−(インドール−3−イル)酢酸(500mg,M=175.19,2.85.mmol)及びリチウムビス(トリメチルシリル)アミド(1M/THF,6.27mmol,2.2当量,6,27mL)の溶液を、アルゴン下に、−78℃で1時間撹拌した。次いでクロロギ酸ベンジル(505μL,M=170.60,d=1.195,3.42.mmol,1.2当量)を添加した。反応混合物を、−78℃で2時間撹拌し、次いで溶媒を真空下に蒸発させた。残渣を水に溶解して、ジエチルエーテルで抽出した。水層をHCl水溶液(0.1N)でpH3まで酸性化して、結果として3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(1c)を白色沈殿として得、これを濾別して、ペンタンで洗浄した(795mg,収率90%)。Rf(シリカゲルMerckF254,CH2Cl2/CH3OH9/1v:v混合物)=0.4.Tf=152℃.IR(cm−1):1728及び1694(νCO).1HNMR(250MHz,[D6]DMSO):δ3.77(s,2H);5.53(s,2H);7.29−7.66(m,8H);7.74(s,1H);8,14(d,J=8.1,1H).CI−MS:m/z=327,[M+NH4+],100%.元素分析.C18H15NO4に対する計算値:C,69.89;H,4.89;N,4.53.実測値:C,69.78;H,5.00;N,4.55.
【0083】
《3−カルボキシメチル−5−ブロモ−インドール−1−カルボン酸ベンジルエステル(1d)》
2−(5−ブロモ−インドール−3−イル)酢酸(200mg,M=254.09,0.787mmol)に同じ手順を適用して、(1d)(280mg,収率:92%)を得た。Rf(シリカゲルCH2Cl2/CH3OH9/1v:v混合物)=0.5,Tf=88℃,IR(cm−1):1729及び1692(νCO).1HNMR(250MHz,[D6]アセトン):δ3.80(s,2H);5.51(s,2H);7.15(s,1H);7.24−7.82(m,7H);8.12(d,J=8,1,1H);11,16(s,1H).CI−MS:m/z=405,407,[M+NH4+],40%;388,390[M+],30%.元素分析.C18H14BrNO4.0.5H2Oに対する計算値:C,54.53;H,3.87;N,3.53.実測値:C,54.67;H,3.72;N,3.44.
【0084】
《1−(5−ブロモ−1H−インドール−3−イル)−3−ヒドロキシプロパン−2−オン(2)》
塩化オキサリル(104μl,M=126.93,d=1.455,1.18mmol,1.5当量)を、アルゴン下に0℃で、DMF数滴を含むTHF(10mL)中の2−(5−ブロモ−1H−インドール−3−イル)酢酸(200mg,M=254.09,0.788mmoL)の溶液に添加した。混合物を室温で2時間撹拌し、次いで真空下に蒸発させた。残渣をジオキサン10mlに溶解し、これに、アルゴン下に、トリス(トリメチルシリルオキシ)エチレン(675μL,M=292.59,d=0.885,1.94mmol,2.3当量)を添加した。室温で10時間撹拌後、HCl水溶液(0.1N)10mLを添加し、次いで溶液を80℃で30分間加熱した。NaClを添加後、溶液をジエチルエーテルで抽出した。有機層をブラインで洗浄し、MgSO4で乾燥して、真空下に蒸発乾固した。カラムクロマトグラフィー(シリカゲル,溶離CH2Cl2/CH3OH95/5v:v混合物)で精製後、1−(5−ブロモ−1H−インドール−3−イル)−3−ヒドロキシプロパン−2−オン(2)を得た(45mg,収率=21%)。Rf(シリカゲル,CH2Cl2/CH3OH9/1v:v混合物)=0.5.Tf=88℃.IR(cm−1):1702(νCO).1HNMR:(250MHz,CDCl3):δ3.00(s,1H);3.81(s,2H);4.31(s,2H);7.15(s,1H);7.19−7.32(m,2H);7.64(s,1H);8.21(s,1H).CI−MS:m/z=285,287,[M+NH4+],80%;268,270[M+],100%.元素分析.C11H10BrNO2に対する計算値:C,49.28;H,3.76;N,5.22.実測値:C,49.10;H,3.75;N,5.17.
【0085】
《チオ酢酸S−[3−(インドール−3−イル)−2−オキソ−プロピル]エステル(5a)》
塩化チオニル(150μl,M=118.97,d=1.63,2.06mmol,1.2当量)を、アルゴン下に、2−(インドール−3−イル)酢酸(300mg,M=175.19,1.72mmol)/CH2Cl2溶液(10mL)に添加した。混合物を40℃で3時間加熱し、次いで減圧下に蒸発させた。CH2Cl2(10mL)に溶解した残渣を、アルゴン下に、新しく調製したジアゾメタン/ジエチルエーテル(10.4mL,C=0.66M,6.88mmol,4当量)溶液中にカニューレで挿入した。0℃で4時間撹拌後、溶液を減圧下に蒸発させて、収率95%でジアゾ化合物(3a)を黄色油状物として得、これを更に精製することなく使用した。(3a):Rf(酢酸エチル/シクロヘキサン1/1v:v混合物)=0.65.IR(cm−1):2100(νCN);1726(νCO).1HNMR(250MHz,CDCl3):δ3.76(s,2H);5.28(s,1H);7.14−7.28(m,3H);7.39(d,J=7.8,1H);7.48(d,J=7.7,1H);8.12(s,1H).CI−MS:m/z=217,[M+NH4+],50%;200,([M+H]+),30%.HCl(g)エーテル溶液(630μL,C=6N,3.78mmol,2.2当量)を、(3a)/ジエチルエーテル(10mL)溶液に添加した。DMF5mLを添加して、ジエチルエーテルを注意深く蒸発させた。次いでKSAc(432mg,M=114.20,3.78mmol,2.2当量)/DMF溶液(5mL)を、アルゴン下に、カニューレにより移送した。室温で一晩撹拌後、溶媒を真空下に蒸発させて、(5a)を得、これを更にカラムクロマトグラフィー上、酢酸エチル/シクロヘキサン8/2v:v混合物で溶離して精製した(220mg,収率:52%)。Rf(シリカゲル酢酸エチル/シクロヘキサン1/1v:v混合物)=0.6.IR(cm−1):1788(νCO).1HNMR(250MHz,CDCl3):δ2,34(s,3H);3,78(s,2H);3,95(s,2H);7.09−7.57(m,5H);8.19(s,1H).CI−HRMS:C13H14NO2S+([M+H]+)に対する計算値,248.0445;実測値,248.0448.
【0086】
《チオ酢酸S−[3−(5−ブロモ−1H−インドール−3−イル)−2−オキソ−プロピル]エステル(5b)》
(5−ブロモ−インドール−3−イル)酢酸(300mg,M=254.09,1,18mmol)に同じ手順を適用して、チオ酢酸5−[3−(5−ブロモ−1H−インドール−3−イル)−2−オキソ−プロピル]エステル(5b)227mgを収率59%で得た。Rf(シリカゲル,酢酸エチル/シクロヘキサン1/1v:v混合物)=0.5.1HNMR(250MHz,CDCl3):δ2.36(s,3H);3.78(s,2H);3.92(s,2H);7.13−7.29(m,3H);7.72(s,1H);7.48(d,J=7.7,1H);8.18(s,1H).CI−HRMS:C13H13BrNO2S+([M+H]+)に対する計算値,325.9850,327.9830;実測値325.9843,327.9837.
ジアゾ化合物(3b)を収率95%で単離した:Rf(シリカゲル,シクロヘキサン/酢酸エチル3/7v:v混合物)=0.5.IR(cm−1):2101(νCN);1715(νCO).1HNMR(250MHz,CDCl3):δ3.75(s,2H);5.68(s,1H);7.22(dd,J3=8.5,4J=1.5,1H);7.34(s,1H);7,37(d,3J=8.5,1H);7,75(d,4J=1.5,1H).CI−MS:m/z=278,280,[M+],60%;250,252,[M−N2]+,100%.
【0087】
《3−(3−アセチルスルファニル−2−オキソ−プロピル)−インドール−1−カルボン酸ベンジルエステル(5c)》
3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(200mg,M=309.32,0.647mmol)に同じ手順を適用して、(5c)178mgを収率72%で得た。Rf(酢酸エチル/シクロヘキサン1/1v:v混合物)=0.6.1HNMR(250MHz,[D6]アセトン):δ2.34(s,3H);3.98(s,2H);4.07(s,2H);5,50(s,2H);7.15−7.80(m,9H);8.15(d,4J=1.5,1H).CI−HRMS:C21H23NO4S+[M+NH4+]に対する計算値,399.1379;実測値,399.1375.
ジアゾ化合物(3c)を収率91%で単離して、キャラクタリゼーションを行なった:Rf(酢酸エチル/シクロヘキサン1/1v:v混合物)=0.5.IR(cm−1):2102(νCN);1730及び1636(νCO).1HNMR(250MHz,[D6]アセトン):δ3.78(s,2H);5.50(s,2H);5,82(s,1H);7.26−7.69(m,8H);7.70(s,1H);8.17(d,J=8,1H).CI−MS:m/z=351,[M+NH4+],100%.
【0088】
《2−(インドール−3−イル)−N−ヒドロキシアセトアミド(6a).》
2−(インドール−3−イル)酢酸(500mg,M=175.19,2.85mmol)、1−ヒドロキシベンゾトリアゾール、1−ヒドロキシベンゾトリアゾール(HOBT)(425mg,M=135.13,3.14mmol,1.1当量)及び1−(3−ジメチルアミノプロピル)−3−エチルカルボキシルイミン塩酸塩、EDCI(602mg,M=191.71,3.14mmol,1.1当量)のDMF溶液(10mL)を、アルゴン下に5分間撹拌した。次いでN−メチルモルホリン(NMM)(345μl,M=101.15,d=0.92,3.14mmol,1.1当量)を添加した。反応混合物を室温で2時間撹拌して、塩酸ヒドロキシルアミン(218mg,M=69.5,3.14mmol,1,1当量)を添加した。溶液を撹拌下に一晩放置し、次いでDMFを真空下に蒸発させて、黄色油状物を得、これを酢酸エチルに溶解した。この溶液を、順次に、水及びNaHCO3水溶液で洗浄した。通常の処理後、溶媒を蒸発させて、(6a)を白色固体として得た(m=260mg,収率=60%)。IR(cm−1):1638(νCO).1HRMN(250MHz,[D6]DMSO):δ3.44(s,2H);7.02(t,J=7.1,1H);7.12(t,J=7.1,1H);7.19(s,1H);7.39(d,J=7.8,1H);7,62(d,J=7.6,1H);8,74(s,1H);10.65(s,1H);11.85(s,1H).元素分析.C10H10N2O2に対する計算値:C,63.15;H,5.30;N,14.73.実測値:C,62.96,H,5.31,N,14.63.
【0089】
《2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6b)》
(5−ブロモ−1H−インドール−3−イル)酢酸(200mg;M=254.09,0.787mmol)から出発して、(6b)116mgを収率55%で得た。Rf(C18シリカゲル,酢酸エチル/メタノール 1/1v:v混合物)=0.8.Tf=145℃.IR(cm−1):3417(νOH)3213(νNH),1633(νCO).1HNMR(250MHz,[D6]DMSO):δ3.49(s,1H);7.20−7.44(m,3H);7.87(s,1H);8.86(s,1H);10.59(s,1H);11.14(s,1H).CI−MS:m/z=270,272,[M+H]+,100%.元素分析.C10H9BrN2O2に対する計算値:C,44.63;H,3.37;N,10.41.実測値:C,44.52,H,3.52;N,10.45.
【0090】
《5−ブロモ−3−ヒドロキシカルバモイルメチル−インドール−1−カルボン酸ベンジルエステル(6d)》
Horwell et al.(1997)に記述されたカルボキシメチル化手順に従い、2−(5−ブロモ−インドール−3−イル)酢酸(200mg,M=254,09,0.787mmoL)から3−カルボキシメチル−5−ブロモ−インドール−1−カルボン酸ベンジルエステルを得た(280mg,収率:92%)。5−ブロモ−3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(120mg,M=388.21,0.309mmol)から出発して、(6d)50mgを収率40%で得た。IR(cm−1):1650及び1690(νCO).1HNMR(250MHz,[D6]DMSO):δ3.40(s,2H);5.56(s,2H);7.45−8.10(m,9H);8.95(s,1H);10.75(s,1H).元素分析.C18H15BrN2O4に対する計算値:C,53.62;H,3.75;N,6.95.実測値:C,53.73;H,3.54;N,7.11.
【0091】
《(4−フルオロ−1H−インドール−3−イル)−酢酸エチルエステル(8e)》
冷却した4−フルオロ−1H−インドール7e(M=135.05,300mg,2.22mmol)/THF3.3mL溶液に、溶液を氷浴で0℃未満に保持しながら、1.6Mのn−BuLi/ヘキサン 1.39mL(2.22mmol)を添加した。15分後、1NのZnCl2/Et2O 2.22mLを添加した。冷却浴を取り外し、混合物を24時間撹拌し、次いで真空下に蒸発させて、ロウ状物を得、これを更に無水トルエン(3.3mL)に溶解した。エチル−2−ブロモアセテート(246μL,2.22mmol)を添加後、溶液を24時間撹拌した。次いで混合物を1NHClで酸性にして、酢酸エチルに注ぎ入れた。有機層をブラインで洗浄して、MgSO4で乾燥した。エステルを、シリカゲル上10%AcOEt/シクロヘキサンで溶離するクロマトグラフを行ない、(8e)158mg(32%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.24(t,J=7,3H);3.88(s,2H);4.16(q,J=7,2H);6.71(dd,3JHF=11,3JHH=8,1H);7.06(td,3JHH=8,4JHF=5.4,1H);7.23(d,J=8,1H);7.28(d,5JHF=1.9,1H);10.34(s,1H).
【0092】
《2−(4−フルオロ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6e)》
NH2OH.HCl(497mg,7.15mmol)を粉末のまま、1MのEtONa/EtOH溶液6.4mLに添加した。この溶液を、エステル(8e)(158mg,0.715mmol,M=221.03)/エタノール溶液(10mL)に添加した。混合物をアルゴン下に80℃で24時間撹拌した。次いで冷却及び真空下に蒸発後、残渣をAcOEtに溶解した。この有機層をブライン、NaHCO3水溶液、0.1NHCl、ブラインで洗浄して、MgSO4で乾燥した。真空下に蒸発後、ヒドロキサム酸(6e)をアセトン/シクロヘキサンの1:1混合物に溶解した。減圧下にアセトンをゆっくり蒸発させて、(6e)75mgを白色固体として得た(51%)。IR(cm−1):3354(νNH),3280(νOH),1631(νCO).1HNMR(250MHz,[D6]アセトン):δ3.72(s,2H);6.70(dd,3JHF=11,3JHH=8,1H);7.05(td,3JHH=8,4JHF=5.4,1H);7.22(d,J=8,1H);7.28(s,1H);10.00(s,1H),10.43(s,1H).CI−HRMS:C10H13N3O2F([M+NH4+])に対する計算値,226.0992;実測値226.0991.
【0093】
《(5−フルオロ−1H−インドール−3−イル)−酢酸エチルエステル(8f)》
5−フルオロ−1H−インドール(7f)125mg(M=135.05,0.926mmol)からエステル(8f)91mg(収率=44%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.24(t,J=7.2,3H);3.74(s,2H);4.13(q,J=7.2,2H);6.92(t,J=9,1H);7.30(d,J=9,1H);7.38(m,2H);10.21(s,1H).
【0094】
《2−(5−フルオロ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6f)》
エステル(8f)91mg(M=221.23,0.41mmol)から出発して、ヒドロキサム酸(6f)(M=208.19)21mg(収率=25%)を得た。IR(cm−1):3360(νNH),3175(νOH),1625(νCO).1HNMR(250MHz,[D6]アセトン):δ3.57(s,2H);6.91(td,3JHF=3JHH=9.3,4JHH=2.3,1H);7.33−7.41(m,3H);8.00(s,1H);10.08(s,1H);10.22(s,1H).CI−MS:m/z=226.2,[M+NH4+],20%.
【0095】
《(5−クロロ−1H−インドール−3−イル)−酢酸エチルエステル(8g)》
5−クロロ−1H−インドール(7g)300mg(M=151.60,1.98mmol)からエステル(8g)198mg(収率=43%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.24(t,J=7,3H);3.77(s,2H);4.14(q,J=7,2H);7.24(d,J=8.5,1H);7.35−7.40(m,2H);7.79(s,1H);10.33(s,1H).
【0096】
《2−(5−クロロ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6g)》
エステル(8g)198mg(M=233.26,0.85mmol)から出発して、ヒドロキサム酸(6g)(M=224.64)194mg(収率=95%)を得た。IR(cm−1):3348(νNH),3190(νOH),1625(νCO).1HNMR(250MHz,[D6]アセトン):δ3.59(s,2H);7.10(d,J=8.5,1H);7.35(s,1H);7.41(d,J=8.5,1H);7.67(s,1H);10.00(s,1H);10.29(s,1H).CI−HRMS:に対する計算値C10H10N202Cl([M+NH4+]),225.04331(100%),227.0461(33%);実測値225.0432(100%),227.0413(32.4%).
【0097】
《(5−メトキシ−1H−インドール−3−イル)−酢酸エチルエステル(8h)》
5−メトキシ−1H−インドール(7h)300mg(M=147.18,2.04mmol)からエステル(8h)219mg(収率=46%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.23(t,J=7,3H);3.73(s,2H);3.82(s,3H);4.15(q,J=7,2H);6.79(d,J=8.5,1H);7.11(s,1H);7.26−7.32(m,2H);9.97(s,1H).
【0098】
《2−(5−メトキシ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6h)》
エステル(8g)219mg(M=233.26,0.94mmol)から出発して、ヒドロキサム酸(6h)(M=220.22)90mg(収率=43%)を得た。IR(cm−1):3307(νNH),3160(νOH),1620(νCO).1HNMR(250MHz,[D6]アセトン):δ3.57(s,2H);3.81(s,3H);6.77(dd,3J=8.8,4J=2.3,1H);7.15(d,J=2.3,1H);7.22(s,1H);7.28(d,J=8.8,1H);7.93(s,1H);10.04(s,1H).CI−MS:m/z=238,[M+NH4+],35%.
【0099】
《(6−ブロモ−1H−インドール−3−イル)−酢酸エチルエステル(8i)》
6−ブロモ−1H−インドール(7i)300mg(M=196.05,1.53mmol)からエステル(8i)192mg(収率=45%)を得た。1HNMR(250MHz,[D6]DMSO):δ1.23(t,J=7.2,3H);3.76(s,2H);4.13(q,J=7.2,2H);7.19(dd,3J=8.5,4J=1.5,1H);7.34(s,1H);7.55(d,J=8.5,1H);7.62(d,J=1.5,1H);10.28(s,1H).
【0100】
《2−(6−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6i)》
エステル(8i)192mg(M=282.16,0.68mmol)から出発して、ヒドロキサム酸(6i)(M=269.09)136mg(収率=74%)を得た。IR(cm−1):3335(νNH),3230(νOH),1615(νCO).1HNMR(250MHz,[D6]アセトン):δ3.60(s,2H);7.17(dd,3J=8.5,4J=1.5,1H);7.30(s,1H);7.6(m,2H);8.0(s,1H);10.0(s,1H);10.27(s,1H).CI−MS:m/z=270,272,[M+H]+,50%.
【0101】
《2−(5−ブロモ−2−メチル−1H−インドリル)−酢酸エチルエステル(8j)》
5−ブロモ−2−メチル−1H−インドール(7j)300mg(M=210.06,1.43mmol)からエステル(8j)201mg(収率=48%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.18(t,J=7,3H);2.33(S,3H);3.66(s,2H);4.06(q,J=7,2H);7.10(d,J=8,1H);7.22(d,J=8,1H);7.56(s,1H);10.11(s,1H).
【0102】
《2−(5−ブロモ−2−メチル−1H−インドリル)−N−ヒドロキシアセトアミド(6j)》
エステル(8j)201mg(M=296.16,0.68mmol)から出発して、ヒドロキサム酸(6j)(M=283.12)160mg(収率=83%)を得た。IR(cm−1):1690(νCO).1HNMR(250MHz,[D6]アセトン):δ2.44(s,3H);3.69(s,2H);7.14(d,J=8.4,1H);7.26(d,J=8.4,1H);7.67(s,1H);10.20(s,1H).
【化18】
【0103】
《5−ブロモ−2−メチル−1H−インドール−3−カルバルデヒド(9)》
POCl3(267μL,2.86mmoL,d=1.64)を、アルゴン下に0℃で、5−ブロモ−2−メチル−1H−インドール(7j)(500mg,2.38mmol)/DMF5mL溶液に添加した。室温で一晩撹拌後、2NNaOH水溶液2mLを添加し、溶液を更に2時間撹拌し、次いで酢酸エチルに注ぎ入れた。水で洗浄し、乾燥し、蒸発乾固した後、(9)526mgを収率95%で白色固体として単離した(収率=93%)。IR(cm−1):1628(νC=O).1HNMR(250MHz,[D6]アセトン):δ2.78(s,3H);7.33(d,3J=8.45,1H),7.36(d,3J=8.45,1H),8.33(s,1H),10.15(s,1H),11.04(s,1H).CI−MS:m/z=255,257[M+NH4+],85%;238−240,[M+],100%.元素分析.C10H8BrNOに対する計算値:C,50.45;H,3.39;N,5.88.実測値:C,50.42,H,3.49;N,5.87.
【0104】
《5−ブロモ−2−メチル−1H−インドール−3−カルバルデヒド−オキシム(10)》
NH2OH,HCl(70mg,1.01mmol)を、(9)(200mg,0.84mmoL)/ピリジン5mL溶液に添加した。室温で5時間撹拌後、溶液を蒸発乾固した。残渣を酢酸エチルに溶解後、溶液を順次、1NHCl、ブラインで洗浄して、MgSO4で乾燥した。溶媒を蒸発後、(10)210mgを黄色油状物として定量的収率で単離した。IR(cm−1):1622(νC=N).1HNMR(250MHz,[d6]アセトン):δ2.53(s,3H);7.2−7.35(m,2H);7.80(s,1H);8.32(s,1H);9.69(s,1H);10.49(s,1H).元素分析.C10H9BrN2O.0.25H2Oに対する計算値:C,46.63;H,3.72;N,10.87.実測値:C,46.69;H,3.66;N,10.58.
【0105】
《N−[2−(5−ブロモ−2−メチル−1H−インドール−3−イル)メチル]−ヒドロキシルアミン(11)》
(10)(110mg,0.43mmol)/メタノール10mL溶液に、メチルオレンジ結晶を数個添加し、次いでHCl(g)(2N)エタノール溶液を数滴添加した。溶液は赤紫色に変化した。HClを添加してそのあとも溶液の赤色を保持しながら、2当量のNaBH3CN(55mg,0.86mmol)/THF5mLを添加して、溶液を撹拌した。数時間後、赤色が安定してから、溶液を撹拌下に一晩放置した。蒸発後、残渣をメタノール5mLに溶解し、次いで水5mLを添加した。1NNaOH水溶液でpHを9まで調整した。この溶液をCH2Cl2で抽出した。通常の処理後、(11)(110mg)を黄色油状物として定量的収率で単離し、更に精製することなくキャラクタリゼーションを行なった。1HNMR(250MHz,[D6]アセトン):δ2.51(s,3H);5.16(s,2H);7.1−7.3(m,2H);8.10(s,1H);10.34(s,1H).CI−MS:m/z=255,257[M+],30%;224,226([M−NHOH]+),100%.元素分析.C10H11BrN2Oに対する計算値:C,47.08;H,4.35;N,10.98.実測値:C,46.97;H,4.51;N,11.13.
【0106】
《5−ブロモ−2−メチル−1−(フェニルスルホニル)−1H−インドール−3−カルバルデヒド(12)》
5−ブロモ−2−メチル−1H−インドール−3−カルバルデヒド(9)(500mg,2.10mmol)の溶液を、0℃でアルゴン下に、NaH(110mg,4.62mmol,2.2当量)/THF10mL懸濁液に添加した。1時間撹拌後、ベンゼンスルホニルクロリド(320μL,2.52mmol,1.2当量)の溶液を添加した。混合物を室温で一晩撹拌した。次いで水50mLを添加し、この溶液を酢酸エチルで抽出し、有機層を、順次、0.1NHCl、NaHCO3水溶液、ブラインで洗浄して、MgSO4で乾燥した。セライトを通して濾過し、蒸発させて、ペンタン/アセトン混合物から再結晶後、(12)700mg(95%収率)を得た。1HNMR(250MHz,[D6]アセトン):δ3.05(s,3H);7.57(dd,3J=8.9,4J=2.1,1H);7.6−8.0(m,5H),8.19(d,J=8.9,1H),8.41(d,J=2.1,1H),10.32(s,1H).CI−MS:m/z=378,380[M+],100%.
【0107】
《2−(5−ブロモ−2−メチル−1−(フェニルスルホニル)−1H−インドール−3−イル)エタノール(13)》
(12)(700mg,1.85mmol)/メタノール10mL溶液に、NaBH4(84mg,2.22mmol)/メタノール10mL溶液を添加した。室温で6時間撹拌後、溶液を酢酸エチルに注ぎ入れて、ブラインで洗浄した。有機層をMgSO4で乾燥して、真空下に蒸発させた。アルコールをシリカゲル上シクロヘキサン/酢酸エチル70/30で溶離するクロマトグラフを行ない、(13)700mgを定量的収率で淡黄色粉末として得た。1HNMR(250MHz,[D6]アセトン):δ2.64(s,3H);4.02(s,1H);4.72(s2H);7.45(dd,3J=8.9,4J=2,1H);7.83(d,J=2,1H);7.6−8.0(m,5H);8.13(d,J=8.9,1H).CI−MS:m/z=380,382[M+],100%.
【0108】
《Tert−ブチル−2−(5−ブロモ−2−メチル−1−(フェニルスルホニル)−1H−インドール−3−イル)メチル−(tert−ブトキシカルボニルオキシ)−カルバメート(14)》
(13)(220mg,0.58mmol)/THF10mL溶液に、室温でアルゴン下に、連続してPPh3(1.1当量,167mg,0.64mmol)を添加し、15分間撹拌後N−ブロモスクシンイミド(1.1当量,113mg,0.64mmol)を添加した。混合物を室温で一晩撹拌した。ブロモ誘導体を単離せず、更にこの溶液で使用した。N,O−ビス−(tert−ブトキシカルボニル)ヒドロキシルアミン(135mg,0.58mmol)/DMF5mL溶液をNaH(1.1当量,15mg)とともに予め15分間撹拌しておき、これを前記ブロモ誘導体に添加した。得られた溶液を2時間撹拌した。CH2CL2添加後、溶液を、水、NH4Cl水溶液、ブラインで洗浄して、MgSO4で乾燥した。溶媒蒸発後、残渣を、シリカゲル上CH2Cl2/C6H1270/30及び80/20で溶離するクロマトグラフを行ない、(14)150mgを得た(収率44%)。1HNMR(250MHz,[D6]アセトン):δ1.35(s,9H);1.49(s,9H);2.67(s,3H);4.87(s,2H,);7.47(dd,3J=8.8,4J=1.8,1H);7.63(m,3H);7.78(d,J=1.8,1H);7.9(d,J=7.5,2H);8.13(d,J=8.8,1H).CI−MS:m/z=612,614[M+],100%.
【0109】
《N−((5−ブロモ−2−メチル−1(フェニルスルホニル)−1H−インドール−3−イル)メチル)ヒドロキシルアミン(15)》
トリフルオロ酢酸775μL(40当量)を、(14)(150mg,0.25mmol)/CH2Cl210mL溶液に添加した。溶液を0℃でアルゴン下に2時間撹拌し、次いで水、NaHCO3水溶液で洗浄して、MgSO4で乾燥した。濾過し、減圧下に蒸発させて、(15)80mgを得た。シリカゲルによる精製において不安定であるため、これを更に精製することなく試験した。CI−HRMS:C16H1403N2BrS[M−H]+に対する計算値,392.9909(96%),394.9889(100%);実測値392.9911(24.7%),394.9895(24.2%).
【0110】
《N−(1−ベンゼンスルホニル−5−ブロモ−2−メチル−1H−インドール−3−イルメチル)−N−ヒドロキシホルムアミド(16).》
ギ酸(3.77mL,0.1mol)を、0℃で、無水酢酸(9.45mL,0.1mol)に滴下した。次いで混合物を50℃に2時間加熱し、室温まで放置した。CH2Cl210mLに溶解した(15)(80mg,0.2mmol)を、室温で前の溶液に滴下した。一晩撹拌後、真空下に蒸発させて、油状物を得、これを、シリカゲル上で数滴の酢酸存在下にCH2Cl2/C6H1299.5/0.5(vv混合物)で溶離するクロマトグラフを行なった。蒸発後、エーテル溶液からペンタン中に沈殿させて(16)50mg(収率60%)を得た。1HNMR(250MHz,[D6]アセトン):δ□272□3□;4.81(s,2H);7.47(dd,3J=8.8,4J=1.9,1H);7.63(t,J=7.5,2H);7.73(d,J=7.5,1H);7.8(d,J=1.9,1H);7.94(d,J=7.5,2H);8.13(d,J=8.8,1H);8.35(s,1H);9.10(s,1H).FAB+−MS:423,425,[M+H]+,10%;461,463,[M+NH4+],10%.CI−HRMS:C17H19O4N3BrS[M+NH4+]に対する計算値,440.0280(95.6%),442.0260(100%);実測値440.0292(86.4%),442.0247(92.9%).
【化19】
【0111】
《(1−ヒドロキシカルバモイルメチル−2−フェニル−エチル)−カルバミン酸tert−ブチルエステル(18)》
ヒドロキサム酸合成の通常の手順に従い、3−tert−ブトキシカルボニルアミノ−4−フェニル−酪酸(17)(0.308mmoL,M=279.33,m=86mg)から出発して、(18)50mgを収率55%で製造した。1HNMR(250MHz,[D6]DMSO):δ1.36(s,9H);2.16(m,2H);2.72(m,2H);3.99(m,1H),6.71(d,J=8.6,1H);7.19−7.35(m,5H);8.83(s,1H);10.40(s,1H).元素分析.C14H20N2O4.0.25H2Oに対する計算値:C,60.29;H,7.59;N,9.37.実測値:C,60.19;H,7.38;N,9.58.
【0112】
《(1−ヒドロキシカルバモイルメチル−2−フェニル−エチル)−カルバミン酸tert−ブチルエステル(20)》
Xiang et al.により記述されたようにして[Xiang,2002#1007]、(1−ホルミル−2−フェニル−エチル)−カルバミン酸tert−ブチルエステル(19)から化合物(20)を合成した。IR(cm−1):1682(νCO).1HNMR(250MHz,CDCl3):δ1.38(s,1H);2.83(d,J=6.7,2H);3.08−4.01(m,2H);4.19(m,1H);4.61(d,J=3.9,1H);7.15−7.80(m,5H);8.32(s,1H);8.85(s,1H).元素分析.C15H22N2O4.0.2H2Oに対する計算値:C,60.47;H,7.58;N,9.40.実測値:C,60.67;H,7.57;N,9.31.
【図面の簡単な説明】
【0113】
【図1A】15N−標識した大腸菌(E.coli)ペプチドデホルミラーゼ(1mM)上の2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(挿入図)のHSQC(異核種単量子相関(Heteronuclear Single Quantum Correlation))フットプリント。リファレンススペクトルは黒色輪郭線で示され、化学量論的量の化合物存在下のスペクトルは灰色でオーバーレイされる。著しく摂動されるアミド基がラベルされる。
【図1B】大腸菌(E.coli)ペプチドデホルミラーゼの3D構造。アミノ酸残基のアミド基NMRシフトが2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドの結合により摂動されるアミノ酸残基は、赤色で示される(図1Aでラベルされた残基)。触媒の金属イオンは球体として示される。
【図1C】大腸菌(E.coli)のPDF(100μM)から2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(1mM)への磁化移動を示すNMR飽和移動差スペクトル実験(STD)。上の図:混合物のリファレンススペクトル。星印でラベルしたピークは、緩衝液のシグナルに相当する。番号を付したピークは、図1Aでラベルした2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドに由来するシグナルに相当する。下の図:STDシグナル。矢印は、照射されたスペクトル領域を示す。正規化したSTD強度が示される。
【図2】本発明による2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド化合物が緑膿菌(Pseudomonas aeruginosa)ペプチドデホルミラーゼ酵素のS’1ポケットに結合している三次元モデルの三つの異なる視点から見た図であり、アクチノニンの構造及びベンザチオジオン−ヒドロキサム酸誘導体(benzathiozinone-hydroxamic derivative)の構造と重ね合わせてある。
【図3A】def遺伝子がPBADプロモーターの制御下にある大腸菌(Escherichiacoli)株について、アラビノース濃度(横軸,%)に関して、それぞれ、アクチノニン及び2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドのMIC(縦軸,μg/ml)を表す図である。
【図3B】def遺伝子がPBADプロモーターの制御下にある大腸菌(Escherichiacoli)株について、アラビノース濃度(横軸,%)に関して、それぞれ、アクチノニン及び2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドのMIC(縦軸,μg/ml)を表す図である。
【図3C】ykrB遺伝子もdef遺伝子も不活性化されてしまっており、且つ2つの遺伝子のうち1つがPxylAプロモーターの制御下にあった枯草菌(Bacillus subilis)株について、キシロース濃度(横軸,%)に関して、菌株に対するアクチノニン(黒丸)及び2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(黒四角)のMIC(縦軸,μg/ml)を表す図である。
【0114】
[実施例]
《使用した略語》
枯草菌(Bacillus subtilis),Bs;大腸菌(Escherichia coli),Ec;N−ホルミル,Fo;Met−tRNAfMetトランスホルミラーゼ,FMT;擬似結合定数,IC50;インドール誘導体,ID;ノックアウト,KO;最小阻止濃度,MIC;メタロプロテアーゼ,MP;阻害定数又は解離定数,Kd;マトリックスメタロプロテアーゼ,MMPs;ペプチドデホルミラーゼ,PDF;ペプチドデホルミラーゼ阻害物質,PDFI;プロテイン・データ・バンク,PDB;構造活性相関,SAR;飽和移動差スペクトル,STD;三次元,3D.
【実施例1】
【0115】
NMRによるメディアムスループットPDFIスクリーニングは、インドール誘導体をPDFの低親和性リガンドとして明らかにする
《材料及び方法》
15N−標識Ni−EcPDF1を、記述されたようにして得て(Dardelet al.,1998)、10mM HEPES−HCl(pH7.0)に溶解し、最終濃度を1mMとした。NMR実験は、3mm三重共鳴フローインジェクション・プローブを備えたBruker Avance 600MHz NMRスペクトロメーター上に記録された。プローブは、NMRコンソール(Bruker BEST(登録商標)システム)により制御されるGilsonリキッドハンドラーに連結される。インジェクション・プロトコルは、前に記述されたとおりである(Tisne et al.,2002)。化学シフト摂動実験のために、1mM 15N−標識EcPDF1(PDF1B)90μlを、96ウェルプレート中で同じ緩衝液に溶解した等容量の試験リガンド(3mM)と混合し、インジェクションの前に、Gilson 242 Peltierラック上、4℃で冷却した。飽和移動差スペクトル(STD,Mayer et al.,2001)実験のために、標識しないEcPDF1(PDF1B)又はBsPDF2(PDF2)を使用した。タンパク質及びリガンドの最終濃度をそれぞれ20μM及び1mMとした以外は、類似した緩衝液及びインジェクション容量を使用した。STD照射は、磁場の強さγB1/2π=100Hzを用いて、プロテイン・メチル・マッシーフ(0.5ppm)上のキャリヤセットで、1秒又は2秒間照射することにより行なった。
【0116】
《結果》
PDFに関して、ペプチドデホルミラーゼ阻害物質(PDFI)の結合能力は、本質的に2つの化学基により引き起こされる:
(i)金属結合基、及び
(ii)PDFのS1’ポケットに結合するP1’基(Boularotet al.,2004)。
いずれかの低親和性基の結合によるエントロピー増大は、ナノモル範囲の阻害定数を有する強力なPDFIを作り出す。
細菌PDFs(PDF1Bs及びPDF2s)のS1’ポケットは、n−ブチル基、n−ペンチル基、n−ヘキシル基、n−フェニル基(Ragusa et al.,1999;Molteni et al.,2004)及び他の環状側鎖(Boularot et al.,2004の引用文献を参照のこと)を、低選択性で受け入れることが知られている。以前の解析では、ミトコンドリアのPDF(PDF1A)は、例えばフェニル誘導体などの環状化合物を許容できない修正S1’ポケットを表示することが示された(Serero et al.,2003;Serero et al.,2001)。選択的PDFI(すなわちミトコンドリアのPDFをターゲッティングしないPDFI)を実証するために、環状P1’基を有する環状化合物を同定するという研究方法が、最も適当であると思われた。従って、本発明者らは、低結合定数でさえも細菌のPDFに結合するような環状化合物を同定するよう努力した。
従って、NMRによりそのような化学基を同定するために、そのような化合物のメディアムスケール・スクリーニングを設定した。フローインジェクションNMRプローブと連結されたピペッティング・ロボットを使用して、化合物ライブラリーを、連続して15N−標識EcPDFと混合し、インジェクションを行なった。化学シフト摂動は、HSQC−スペクトルを記録することによりモニターして、これを既に記述された非結合酵素で得られた対照データ(Meinnel et al.,1996;Meinnel et al.,1999)と比較した。化合物を添加して{1H−15N}HSQCタンパク質スペクトルの共鳴広がり及びシフトを引き起こすような化合物が同定された。影響されたアミド基を同定し、EcPDFの3D構造のバックボーンに位置決めした(例えば図1A参照)。
このスクリーニングから、インドール誘導体が酵素のS1’ポケットに対応する化学変化パターンを示したので、インドール誘導体が最も興味あるものとして浮上してきた(図1B)。相互に、これらのインドール誘導体のEcPDF1又はBsPDFいずれかに結合する様式の情報を得るために、リガンド観察飽和移動鎖スペクトル(STD)実験(Mayer et al.,2001)を行なった(図1C)。
例えば、化合物6bについて両方の場合に類似した結果が観察され(下の実施例2参照)、それは、インドール部分の4位について最も強いSTD効果が観察されたこと、及び3位に結合したメチレン基について最も弱いSTD効果が観察されたことを示した(図1C)。このことは、インドール基がPDF1B酵素にもPDF2酵素にも結合したことを確証したものであり、4位がタンパク質からの最も強い飽和移動を示したので、4位が最も深く埋め込まれていることを示唆したものである。
インドール及び5−ブロモインドールは、NMR滴定により調べられたようにミリモル範囲の結合定数を示した。この弱い結合定数は、細菌PDF(EcPDF1B及びBsPDF2)並びにミトコンドリアPDF(AtPDF1A)に対する阻害アッセイによって確認された(手順については実施例4を参照のこと)。
【0117】
インドール誘導体(ID)の効力を改良するために、本発明者らは、インドール誘導体の構造活性相関の研究に着手した。従って、下にラベルしたように位置1(R1)、2(R2)、3(R3)、4(R4)、5(R5)、6(R6)及び7(R7)のインドリル複素環に置換基を導入した。
【化20】
P1’(R5)側鎖に対応する4、5及び6位を、S1’ポケット中への適合性を改良するために、特にブロモ基などの親油基により置換した。2位及び3位においては、任意に短いCH2スペーサを介してインドール環に結合するいくつかの金属結合基を探索した。これには、カルボン酸、オキシム及びヒドロキシルアミンなどの単座官能基、並びにヒドロキシメチルケトン、チオメチルケトン及びヒドロキサム酸などの二座官能基が含まれた。最後に、1位は、あいたままにしたか又はベンジルオキシカルボニル残基などの諸残基により保護したが、これはP2’及び/又はP3’アミノ−酸基を模倣することを期待したものである(様々な酵素サブ部位の命名法についてはBoularot et al.,2004;Schechter et al.,1967を参照のこと)。
ヒドロキサム酸金属キレート基を3位に、臭素原子を5位に有する、そのようなインドール誘導体の2例を下に示し、それらのPDFに対する阻害作用及びそれらの抗菌特性も下に示した。
【実施例2】
【0118】
《インドール−ヒドロキサム酸誘導体のインビトロアッセイ》
様々な起源のPDFに対する本発明の化合物6b及び6dのインビトロ阻害プロファイルを評価して、アクチノニン並びに2つの合成PDF阻害物質であるRAS270及びRAS238のインビトロ阻害プロファイルと比較した。RAS358は、前に記述されたとおりに合成した(WO02/070653)。化合物RAS358は、リバースヒドロキサム酸化合物である。RAS270は、RAS358のヒドロキサメート誘導体に相当する。
3−tert−ブトキシカルボニルアミノ−4−フェニル−酪酸(市販品として入手可能)(0.308mmoL,M=279.33,m=86mg)から出発して、通常のヒドロキサム酸合成手順に従い、RAS270を50mg、収率55%で製造した。
【化21】
【0119】
《材料及び方法》
タンパク質分析のためのすべての化学薬品及び酵素はSigmaから購入した。ペプチドはBachem AGからのものであった。DNA操作のための酵素はNew England Biolabsから購入した。オリゴヌクレオチドはMWG−AG Biotechで合成された。
大腸菌(Escherichia coli)PDF(EcPDF1B)及びバチルスステアロサーモフィラス(Bacillus stearothermophilus)PDF2(BsPDF2)は、いずれかの細菌PDFクラスの代表として使用し、ニッケルイオン存在下に記述されたとおりに精製した(Ragusa et al.,1998)。シロイヌナズナ(Arabidospis thaliana)ミトコンドリアPDF(AtPDF1A)及びヒトミトコンドリアPDF(HsPDF1)は、記述されたとおりに精製した(Serero et al.,2001;Serero et al.,2003)。PDF活性を評価するために、PDF活性とギ酸脱水素酵素活性を共役させるアッセイを用いた。NADHの340nmでの吸光度(εM=6,300M−1.cm−1)を、37℃で、本質的に前に記述されたとおりにしてモニターした(Lazennec et al.,1997)。反応は、精製した酵素5〜15μlを添加して開始した。それぞれの場合、速度論的パラメーターは、前に記述されたとおりにして(Serero et al.,2001)、実験データを用いて、Michaelis−Menten式の逐次非線形最小二乗法から得た(Dardel et al.,1994)。阻害物質はすべてジメチルスルホキシドで希釈し、最終アッセイ緩衝液はこの溶媒10%を含有した。
【0120】
《結果》
結果を次の表1に示す:
【表1】
DF1Bに関する6b及び6dのIC50値は、アクチノニンの値より約1桁大きく、RAS270及びRAS358と同程度の大きさである。PDF2については、6b及び6dのIC50値は、アクチノニンの値と同程度であり、RAS270及びRAS358の値より1桁を超えて小さい。ミトコンドリアPDFsについては、アクチノニンが強力な阻害作用を示すのに対して、6b及び6dの阻害作用は特に低く、その結果前記化合物は全く阻害作用を欠いているということができる。
更に、6bが両方のPDF型に遅い結合様式を示すという事実は、酵素の阻害物質誘導適合コンホメーション変化を示していることに注意すべきである。
【実施例3】
【0121】
《インドール−ヒドロキサム酸誘導体のインビボアッセイ》
本発明の化合物6b及び6dのインビボ阻害プロファイルを評価し、アクチノニンのそれと比較した。
【0122】
《材料及び方法》
すべての細菌株は、Luria−Bertani(LB)培地(1%バクトトリプトン,0.5%酵母エキス,0.5%NaCl)で培養した。
株:2つの野生型又は擬似野生型枯草菌(B.subtilis)株を使用した。MO3482、すなわち、JH642の原栄養株skinプロファージ展覧誘導株(P.Stragier,IBPC,Franceの親切な供与)及び168(trpC2)(Anagnostopoulos et al.,1961)である。MHY101株(ΔykrB::nm,trpC2)及びMHD101株(Δdef::nm,trpC2)は、168株から得られ、前に記述されたものである(Haas et al.,2001)。更に2つの枯草菌(B.subtilis)株では、両方のデホルミラーゼの野生型遺伝子座(def及びykrB)が欠失しており、1つのデホルミラーゼバージョンがキシロース制御下に条件付で発現されるのであるが、そのような2つの枯草菌も使用した(Haas et al.,2001):MHY103(Δdef::nm;ΔykrB::erm;ΔthrC::xylR−PxylA−ykrB−spc,trpC2)及びMHD103(Δdef::erm;ΔykrB::nm;ΔthrC::xylR−PxylA−def−spc,trpC2)である。138株及びその誘導株は、C.Freiberg(Bayer AG,Germany)の親切な供与であった。大腸菌(E.coli)株JM101Tr(galK,rpsL,recA56,srl−300::Tn10)及びCAG1284(λ−,tolC210::Tn10,rph−)は、他のところで記述されている(Hirel et al.,1988;Singer et al.,1989).
大腸菌(E.coli)CAG1284感受性試験:tolC株CAG1284の薬剤感受性を測定するために感受性試験を計画した。EcPDF1のオープンリーディングフレームを、6−Hisタグ付きのフレームのC末端を有するpBADベクター(Invitrogen)中にクローニングした。このクローニングは、EcPDF1の合成をアラビノース濃度依存性にする(Guzman et al.,1995)。CAG1284株は、クロラムフェニコールを含むいくつかの抗生物質に対して感受性が高い(Sulavik et al.,2001)が、最初にpBADコンストラクトで形質転換されて、50μg/mlアンピシリン、3.4μg/mlクロラムフェニコール及び0.5%グルコースを補給したLB培地で選択された。細菌を、この培地中37℃で一晩培養し、次いで培養液を1:100に希釈して、培地3mlを接種するために使用した。OD600が0.9に達した時、懸濁液を適当に希釈して、100μl中2x104個の細菌を、ペトリ皿中、50μg/mlアンピシリン、アクチノニン(0.1〜3μM)及びグルコース(0.5%)又はアラビノース(0.0002〜0.2%)を補給した30mlの固体LB上に層状に重ねた。最小阻止濃度は、37℃で18時間インキュベーション後、全く増殖しなくなるアクチノニンの最低濃度として定義された。
【0123】
《結果》
1.グラム陰性細菌の阻害
2つの大腸菌(E.coli)誘導株(JM101Tr及びCAG1284)は、まず最初に化合物6bの最小阻止濃度(MIC)値への洞察を得るために並びにその値を天然の強力なPDFIアクチノニンの値(Chen et al.,2000)及びRAS270の値と比較するために使用した。
WT株(JM101Tr)については、アクチノニン及び6b両方のMICは類似しており、30〜40μg/mlの範囲にあったのに対して、RAS270のMICは300μg/mlを超えていた(表2)。
アクチノニンはAcrAB−tolC排出ポンプにより強く解毒されるので、tolC変異株(CAG1284株)を用いて、これが6bについても同じであるかどうかを調べた。アクチノニンの値が300倍低下したのに対して、6bのMICの値が唯の5.5倍だけわずかに低下したことが観察された(表2)。このことは、本発明のPDF阻害物質では細菌の排出系による解毒が不十分であることを示している。
【表2】
MICはまた、def遺伝子の発現がPBADプロモーターの制御下にあるというtolCコンテクストにおいても測定された。従って、PDF濃度増大のMICに対する影響力は、このプロモーターがアラビノース濃度増加により誘導されるおかげで、調べられた(Guzman et al.,1995;Chen et al.,2000)。アクチノニンの場合には、アクチノニン濃度についてほとんど直線的なMIC増加が存在する(図3A)。これは、(i)PDFが、アクチノニンの一次標的であること、及び(ii)PDF濃度が、排出ポンプ不在下にMICを限定する主要因子であることを示している。6bが同じ遺伝的背景において吟味されたとき、2種類のMIC曲線が観察された(図3B)。曲線の最初の部分は直線的であり、PDFが6bの標的であることを示す。65μg/mlを超えると飽和曲線が観察された。この値は、WTのMIC値と類似しており、膜透過性が6bの作用の律速段階であることを示す。
【0124】
2.グラム陽性細菌の阻害
第2系列の実験において、PDF1BもPDF2も発現するグラム陽性モデル生物である枯草菌(B.subtilis)を試験した。この場合もまたアクチノニンのMICを6b及び6dのMICと比較した。
WTコンテクスト(168株及びMO3482株)において、6b及び6dのMIC(20μg/ml)は、アクチノニンのMICよりも1桁高い大きさであった。これに対して、PDF2に対する低い結合能力から予想されるように、両フェニル誘導体RAS270及びRAS358は枯草菌に対して全く抗菌特性を有しなかった(表3)。
次いで、いずれかのPDFを発現する2つの枯草菌株誘導株(MHD101及びMHY101)に対する6b及び6dの影響力も研究した(表3)。
アクチノニン、6b及び6dのMICが、ykrB遺伝子不活性化コンテクストにおける減少値と、同じ傾向に従うことが見出された。これは、def遺伝子産物が、ykrBよりも少なく発現されたことを確証し(Haasetal.,2001)、6b及び6dが、実際にPDF1BをもPDF2をもターゲッティングしたことを示唆した。
【表3】
6b及び6dが枯草菌のPDFsを特異的にターゲッティングすることを証明するために、ykrB及びdefの両遺伝子が既に不活性化されており、且つ2つの遺伝子のうち1つがXylRプロモーターPxylAの制御下に置かれている株を使用した。これらの条件下に、PDF1B又はPDF2発現は、L−キシロースに依存している(Haas et al.,2001)。アクチノニンも6bもMICが、MHD103株かMYD103株かいずれかにおいて、L−キシロース濃度に依存することが観察された(図3C)。
【0125】
最後に、枯草菌における6b及びアクチノニンに対する耐性の出現を測定し、比較した。類似した値(5.10−7対6.10−7)が観察されたが、この結果は、他人の以前の測定値及びバチルス種(Bacillus spp)におけるトランスホルミラーゼ遺伝子不活性化により誘導されるホルミル化−脱ホルミル化経路のバイパス(Giglione et al.,2001に総説がある)と一致している。このデータは、6bの標的がPDFであることを示すもう1つの強い論拠であった。
【実施例4】
【0126】
ヒドロキサム酸インドール誘導体のPDFへの結合の三次元モデリング
《材料及び方法》
三次元(3D)モデリングは、InsightIIソフトウェア(Accelrys)を用いて行なった。ベンザチオジオン−ヒドロキサム酸誘導体(benzathiozinone-hydroxamic derivative)(BTH)に結合された緑膿菌(Pseudomonas aeruginosa)PDF(PaPDF1)の3D構造(PDB entry 1S17,Molteni et al.,2004)及びアクチノニンに結合されたPaPDF1の3D構造(PDB entry 1LRY,Guilloteau et al.,2002)を並べた。化合物6bをSketcherモジュールで組み立て、固定アンカーとしてヒドロキサメート基を用いて、アクチノニン及び化合物BTHの両方の構造上に並べ、そして3D構造において、いずれの化合物も置き換えるように使用した。PaPDF1にドッキングした6bの構造は、CharmM力場を用いて更に最小化し、最低エネルギー構造を選択した。
【0127】
《結果》
PDF1の活性部位における化合物6bの3Dモデリングは、それがS1’ポケットに完全に適合すること、そして臭化物基がn−ブチル基又はメチオニン側鎖、すなわち、S1’ポケットに受け入れられる最適側鎖、のC4メチル基を模倣することを明らかにした(図2)。更に、このモデルは、インドール部分の4位をS1’ポケットの内側深くに収納し、His132側鎖の下に埋め込まれた状態にした。これは実施例1で議論したSTDデータと一致している。
同時に、これらのデータは、インドール誘導体が抗菌活性を表すが、しかし動物のミトコンドリアPDFsを阻害しないことをも示している。
【実施例5】
【0128】
《シロイヌナズナの成長阻害》
シロイヌナズナ(生態型WS4)成長阻害は、6b又は6dについて50μMより高濃度で観察される。その上、AtPDF1BのIC50値は、化合物6b及び6dの場合にEcPDF1Bで観察される値と類似している。実験手法の詳細は、Serero et al.(2001)及びGiglione et al.(2003)に出ている。
【0129】
従って、本発明者らは、2つの実施例(6b及び6d)により、本発明のインドール化合物が:
(i)特にグラム陰性細菌及びグラム陽性細菌において、特異的にPDF1及びPDF2をターゲッティングすること、
(ii)ミトコンドリアPDFsに対して本質的に全く阻害活性を示さないこと
を証明した。
化合物6a、6g、6f、6i、6e、6h、6j、11、15、及び16で得た結果を、次の表4に示す。
【表4】
【0130】
《引用文献》
Anagnostopoulos,C.,and I.P.Crawford.1961.Transformation studies on the linkage of markers in the tryptophan pathway in Bacillus subtilis.Proc.Natl.Acad.Sci.U.S.A.47:378−90.
Borch,R.F.,M.D.Bernstein,and H.Dupont Durst.1971.The cyano hydridoborate anion as a selective reducing agent.J.Am.Chem.Soc.93:2897−2904.3.
Boularot,A.,C.Giglione,I.Artaud,andT.Meinnel.2004.Structure−activity relationship and therapeutic potential of peptide deformylase inhibitors.Curr.Opin.Invest.Drugs 5:809−822.4.
Bracchi−Ricard,V.,K.T.Nguyen,Y.Zhou,P.T.Rajagopalan,D.Chakrabarti,and D.Pei.2001.Characterization of an eukaryotic peptidedeformylase from Plasmodium falciparum.Arch.Biochem.Biophys.396:162−170.
Chen,D.Z.,D.V.Patel,C.J.Hackbarth,W.Wang,G.Dreyer,D.C.Young,P.S.Margolis,C.Wu,Z.J.Ni,J.Trias,R.J.White,and Z.Yuan.2000.Actinonin,a naturally occurring antibacterial agent,is a potent deformylase inhibitor.Biochemistry39:1256−1262.
Chu,M.,R.Mierza,L.He,L.Xu,F.Gentile,J.Taerracciano,M.Patel,L.Miesel,S.Bohanon,C.Kravec,C.Cramer,T.O.Fischman,A.Hruza,L.Ramanatan,P.Shipkova,and T.−M.Chan.2001.Isolation and structure elucidation of two novel deformylase inhibitors produced by Streptomyces sp.Tetrahedron Lett.42:3549−3451.
Dardel,F.1994.MC−Fit:Using Monte−Carlo methods to get accurate confidence limits on enzyme parameters.Comput.Appl.Biosci.10:273−275.
Dardel,F.,S.Ragusa,C.Lazennec,S.Blanquet,and T.Meinnel.1998.Solution structure of nickel−peptide deformylase.J.Mol.Biol.280:501−513.
Dillard,R.D.,N.J.Bach,S.E.Draheim,D.R.Berry,D.G.Carlson,N.Y.Chirgadze,D.K.Clawson,L.W.Hartley,L.M.Johnson,N.D.Jones,E.R.McKinney,E.D.Mihelich,J.L.Olkowski,R.W.Schevitz,A.C.Smith,D.W.Snyder,C.D.Sommers,and J.P.Wery.1996.Indole inhibitors of human nonpancreatic secretory phospholipase A2.1.Indole−3−acetamides.J.Med.Chem.39:5119−36.
Dirk,L.M.,M.A.Williams,and R.L.Houtz.2001.Eukaryotic peptidede formylases.Nuclear−encoded and chloroplast−targeted enzymes in Arabidopsis.Plant Physiol.127:97−107.
Eissenstat,M.A.,M.R.Bell,T.E.D’Ambra,E.J.Alexander,S.J.Daum,J.H.Ackerman,M.D.Gruett,V.Kumar,K.G.Estep,E.M.Olefirowicz,and etal.1995.Aminoalkylindoles:structure−activity relationships of novel cannabinoid mimetics.J.Med.Chem.38:3094−105.
Giglione,C.,and T.Meinnel.2001.Peptide deformylase as an emerging target for antiparasitic agents.Emerg.Therap.Targets 5:41−57.
Giglione,C.,M.Pierre,and T.Meinnel.2000a.Peptide deformylase as a target for new generation,broad spectrum antimicrobial agents.Mol.Microbiol.36:1197−1205.
Giglione,C.,A.Serero,M.Pierre,B.Boisson,and T.Meinnel.2000b.Identification of eukaryotic peptide deformylases reveals universality of N−terminal protein processing mechanisms.EMBOJ.19:5916−5929.
Giglione,C.,O.Vallon,and T.Meinnel.2003.Control of protein life−span by N−terminal methionine excision.EMBO J.22:13−23.
Giglione,C.,A.Boularot,and T.Meinnel.2004.Protein N−terminal methionine excision.Cell.Mol.Life Sci.61:1455−1474.
Gordon,J.J.,B.K.Kelly,and G.A.Miller.1962.Actinonin:an antibiotic substance produced by an actinomycete.Nature 195:701−702.
Grujic,M.,and M.Renko.2002.Aminopeptidase inhibitors bestatin and actinonin inhibit cell proliferation of myeloma cells predominantly by intracellular interactions.Cancer Lett182:113−9.
Guilloteau,J.P.,M.Mathieu,C.Giglione,V.Blanc,A.Dupuy,M.Chevrier,P.Gil,A.Famechon,T.Meinnel,and V.Mikol.2002.The crystal structures of four peptide deformylases bound to the antibiotic actinonin reveal two distinct types:a platform for the structure−based design of antibacterial agents.J.Mol.Biol.320:951−962.
Guzman,L.M.,D.Belin,M.J.Carson,and J.Beckwith.1995.Tight regulation,modulation,and high−level expression by vectors containing the arabinose PBAD promoter.J.Bacteriol.177:4121−4130.
Haas,M.,D.Beyer,R.Gahlmann,and C.Freiberg.2001.YkrB is the main peptide deformylase in Bacillus subtilis,a eubacterium containing two functional peptide deformylases.Microbiology147:1783−1791.
Hasan,I.,E.R.Marinelli,L.C.ChangLin,F.W.Fowler,and A.B.Levy.1981.Synthesis and reactions of N−protected 2−lithiated pyrroles and indoles.The tert−Butoxycarbonyl substituent as a protecting group.J.Org.Chem.46:157−164.
Hill,D.R.,C.N.Hsiao,R.Kurukulasuriya,and S.J.Wittenberger.2002.2,2,2−trifluoroethyl formate:a versatile and selective reagent for the formylation of alcohols,amines,and N−hydroxylamines.Org.Lett.4:111−3.
Hirel,P.−H.,F.Leveque,P.Mellot,F.Dardel,M.Panvert,Y.Mechulam,and G.Fayat.1988.Genetic engineering of methionyl−tRNA synthetase:in vitro regeneration of an active synthetase by proteolytic cleavage of a methionyl−tRNA synthetase−βgalactosidase chimeric protein.Biochimie 70:773−782.
Horwell,D.C.,D.Naylor,and H.M.G.Wilems.1997.2,3−disubstituted 2−azanorbornanes as polar β−turn mimetics.Bioorg.Med.Chem.Lett.7:31−36.
Jiang,B.,and X.H.Gu.2000.Syntheses and cytotoxicity evaluation of bis(indolyl)thiazole,bis(indolyl)pyrazinone and bis(indolyl)pyrazine:analogues of cytotoxic marine bis(indole)alkaloid.Bioorg.Med.Chem.Lett.8:363−71.
Kolossa,T.,Brooks C.D.W.,Rodrigues K.E.,Summers J.B.,Dellaria J.F.,Hulkower,K.I.,Bouska,J.,Bell,R.L.and Carter G.W.1997.Non−steroidal anti−inflammatory drugs as scaffolds for the dzsign of 5−lipoxygenase inhibitors.J.Med.Chem.40,819−824.
Lazennec,C.,and T.Meinnel.1997.Formate dehydrogenase−coupled spectrophotometric assay of peptide deformylase.Anal.Biochem.244:180−182.
Leclerc,V.,E.Fourmaintraux,P.Depreux,D.Lesieur,P.Morgan,H.E.Howell,P.Renard,D.H.Caignard,B.Pfeiffer,P.Delagrange,B.Guardiola−Lemaitre,and J.Andrieux.1998.Synthesis and structure−activity relationships of novel naphthalenic and bioisosteric related amidic derivatives as melatonin receptor ligands.Bioorg.Med.Chem.6:1875−87.
Lee,M.D.,C.Antczak,Y.Li,F.M.Sirotnak,W.G.Bornmann,and D.A.Scheinberg.2003.A new human peptide deformylase inhibitable by actinonin.Biochem.Biophys.Res.Commun.312:309−15.
Lee MD,She Y,Soskis MJ,Borella CP,Gardner JR,Hayes PA,Dy BM,Heaney ML,Philips MR,Bornmann WG,Sirotnak FM,Scheinberg DA(2004)Human mitochondrial peptide deformylase,a new anticancer target of actinonin−based antibiotics.J Clin Invest 114:1107−1116
Lefevre−Groboillot,D.,S.Dijols,J.L.Boucher,J.P.Mahy,R.Ricoux,A.Desbois,J.L.Zimmermann,and D.Mansuy.2001.N−hydroxyguanidines as new heme ligands:UV−visible,EPR,and resonance Raman studies of the interaction of various compounds bearing a C=NOHfunction with microperoxidase−8.Biochemistry 40:9909−17.
Mayer,M.,and B.Meyer.2001.Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor.J.Am.Chem.Soc.123:6108−17.
Mazel,D.,S.Pochet,and P.Marliere.1994.Genetic characterization of polypeptide deformylase,a distinctive enzyme of eubacterial translation.EMBO J.13:914−923.
Meinnel,T.2000.Peptide deformylase of eukaryotic protists:a target for new antiparasitic agents?Parasitol.Today16:165−168.
Meinnel,T.,S.Blanquet,and F.Dardel.1996.A new subclass of the zinc metalloproteases superfamily revealed by the solution structure of peptide deformylase.J.Mol.Biol.262:375−386.
Meinnel,T.,L.Patiny,S.Ragusa,and S.Blanquet.1999.Design and synthesis of substrate analogue inhibitors of peptide deformylase.Biochemistry 38:4287−4295.
Miesel,L.,J.Greene,and T.A.Black.2003.Genetic strategies for antibacterial drug discovery.Nat Rev Genet 4:442−56.
Molteni,V.,X.He,J.Nabakka,K.Yang,A.Kreusch,P.Gordon,B.Bursulaya,I.Warner,T.Shin,T.Biorac,N.S.Ryder,R.Goldberg,J.Doughty,and Y.He.2004.Identification of novel potent bicyclic peptide deformylase inhibitors.Bioorg.Med.Chem.Lett.14:1477−81.
Ragusa,S.,S.Blanquet,and T.Meinnel.1998.Control of peptide deformylase activity by metal cations.J.Mol.Biol.280:515−523.
Ragusa,S.,P.Mouchet,C.Lazennec,V.Dive,and T.Meinnel.1999.Substrate recognition and selectivity of peptide deformylase.Similarities and differences with metzincins and thermolysin.J.Mol.Biol.289:1445−1457.
Salim,M.,and A.Capretta.2000.Intramolecular carbenoid insertions:the reaction of α−diazoketones derived from pyrrolyl and indolyl carboxylic acids with rhodium(II) acetate.Tetrahedron56:8063−8069.41.
Schechter,I.,and A.Berger.1967.On the size of the active site in proteases.I.Papain.Bochem.Biophys.Res.Commun.27:157−162.
Serero,A.,C.Giglione,and T.Meinnel.2001.Distinctive features of the two classes of eukaryotic peptide deformylases.J.Mol.Biol 314:695−708.
Serero,A.,C.Giglione,A.Sardini,J.MartinezSanz,and T.Meinnel.2003.An unusual peptide deformylase features in the human mitochondrial N−terminal methionine excision pathway.J.Biol.Chem.278:52953−52963.
Singer,M.,T.A.Baker,G.Schnitzler,S.M.Deischel,M.Goel,W.Dove,K.J.Jaacks,A.D.Grossman,J.W.Erickson,and C.A.Gross.1989.A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli.Microbiol.Rev.53:1−24.
Sulavik,M.C.,C.Houseweart,C.Cramer,N.Jiwani,N.Murgolo,J.Greene,B.DiDomenico,K.J.Shaw,G.H.Miller,R.Hare,and G.Shimer.2001.Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes.Antimicrob.AgentsChemother.45:1126−36.
Tisne,C.,and F.Dardel.2002.Optimisation of a peptide library for screening specific RNA ligands by flow−injection NMR.Comb.Chem.High Throughput Screen.5:523−9.
Weller,D.D.,and D.W.Ford.1984.Synthesis of ellipticine.Tetrahedron Lett.25:2105−2108.
Whittaker,M.,C.D.Floyd,P.Brown,and A.J.H.Gearing.1999.Design and therapeutic application of matrix metalloproteinase inhibitors.Chem Rev.99:2735−2776.
Wiesner,J.,S.Sanderbrand,E.Beck,and H.Jomaa.2001.Seeking new targets for antiparasitic agents.Trends Parasitol.17:7.
Wissner,A.1979.2−hetero substituted silylated ketene acetals:reagents for the preparation of α−functionalized methyl ketones from carboxylic acid chlorides.J.Org.Chem.44:4717−4622.
Xu,Y.,L.T.Lai,J.L.Gabrilove,and D.A.Scheinberg.1998.Antitumor activity of actinonin in vitro and in vivo.Clin.Cancer Res.4:171−176.
【発明の詳細な説明】
【0001】
本発明は、ペプチドデホルミラーゼ阻害物質、それを含む医薬組成物、及び除草剤としてのその使用又は細菌感染症若しくは寄生虫感染症の治療のためのその使用に関する。
【0002】
ペプチドデホルミラーゼ(PDF;EC3.5.1.88)は、すべてのグラム陽性細菌及びグラム陰性細菌に見出された必須の酵素活性である。その細胞における役割は、すべての新生タンパク質からN−ホルミル基を除去することである。これが、N−末端メチオニン切出しを引き起こすもう一つの必須の細菌酵素であるメチオニンアミノペプチダーゼの作用を可能にする(総説についてはGiglione et al.,2004を参照のこと)。PDFは、ストレプトミセス種(Streptomyces sp.)が産生する天然抗生物質であるアクチノニンの天然標的である(Gordon et al.,1962;Chen et al.,2000)。効き目は少し低いが、ストレプトミセス種(Streptomyces sp.)が産生する、PDFを阻害する他の天然産物についても記述された(Chu et al.,2001)。
【化1】
【0003】
配列及び構造の両解析に基づき、2つの細菌性PDF型(PDF1及びPDF2)が区別されている(Giglione et al.,2000a;Guilloteau et al.,2002)。PDF2は、グラム陽性細菌にのみ見出される。高解像度の三次元(3D)構造及び精密酵素分析法が利用可能であるため、大腸菌(Escherichia coli)のPDF(PDF1B)及びバチルスステアロサーモフィラス(Bacillus stearothermophilus)のPDF2(PDF2)が、いずれかのPDFクラスの代表として選択された(Guilloteau et al.,2002;Ragusa et al.,1998)。細菌は、PDFをコード化する1つ又はいくつかの機能的遺伝子を有する可能性がある。例えば、大腸菌(Escherichia coli)は、defシストロンによりコード化される1つのPDF1遺伝子を有し、ブドウ球菌種及び連鎖球菌種(Staphylo- and Streptococci spp.)などの主要病原体は、ただ1つのPDF2を有し、並びにバチルス種(Bacillus spp.)は、2つのPDF遺伝子、すなわちPDF1をコード化するdef遺伝子及びPDF2をコード化するykrB遺伝子を有する。枯草菌(B.subilis)においては、PDF2が主要PDFである(Haas et al.,2001)。
【0004】
過去10年間、PDFは、最先端技術を結集した薬剤発見法を用いる新規抗生物質選択のための選択目標として選択されてきた(Giglione et al.,2000a;Meinnel,2000;Giglione & Meinnel,2001)。過去4年間、アクチノニンによく似た多数のPDF阻害物質(PDFI)が記述され、そのうちのいくつかが、最近、臨床試験に入った(総説については、Boularot et al.,2004を参照のこと)。従って、PDFIは、ゲノミクス戦略から生まれた抗菌性薬剤発見法の最初の成功である(Miesel et al.,2003)。
【0005】
しかしながら、機能的PDF配列が、当初は真正細菌に独特の形態であると考えられたけれども(Mazel et al.,1994)、ごく最近、多くの真核生物のゲノムに検出されている(Giglione et al.,2000b;Bracchi−Ricard et al.,2001)。これらのPDFは、細胞小器官、ミトコンドリア、及び植物色素体(葉緑体)、並びにマラリア病因である熱帯熱マラリア原虫(Plasmodium falciparum)などのアピコンプレキサン(apicomplexan)寄生虫(アピコプラスト)にターゲッティングされる。更に、PDFホモログは、キネトプラスチド原生動物でも証明されている(Meinnel,2000)。
【0006】
配列又は生化学的分析及び位置指定突然変異誘発データに基づき、すべてのミトコンドリアPDFsが、独特なサブクラス1型PDFs(PDF1A)に属することが示され、それらの活性部位が多数の特異性を示した(Giglione et al.,2000b;Serero et al.,2001;Serero et al.,2003)。色素体のPDFsは、細菌のPDF1sと強く関連しており、PDF1Bとして分類される(Giglione et al.,2004).
【0007】
アクチノニンがインビトロでもインビボでも、熱帯熱マラリア原虫(P.falciparum)に対しても植物のPDFに対しても阻害作用を示すので、この発見は先ず、細菌のPDFに対して活性なPDFIを、駆虫剤として、そして除草剤としてさえ使用できるということを示唆した(Meinnel,2000;Giglione & Meinnel,2001;Wiesner et al.,2001;Serero et al.,2001;Serero et al.,2001;Dirk et al.,2001)。
【0008】
それにもかかわらず、ヒトミトコンドリアにおけるPDF1A(HsPDF1)の存在が、前記PDFIの毒性に関して多数の問題を引き起こした。その理由は、特に、アクチノニンが、(i)インビトロで効果的にHsPDF1活性をブロックし、(ii)最も敏感なグラム陽性細菌に対する最良のPDFIについての最小阻止濃度(MIC)と類似した濃度範囲(2〜3μg/ml)において細胞毒性活性を及ぼすからである(Lee et al.,2003;Serero et al.,2003;Xu et al.,1998;Grujic et al.,2002,Lee et al 2004)。
【0009】
ミトコンドリアPDFの役割が不明瞭のままであり、ミトコンドリア膜が一般に多くの化合物に対して極めて不透過性であると考えられてはいるけれども、予防原則は、PDF1A及びPDF1B又はPDF2との間の、PDFIによるいかなる相互阻害をも回避するために、今や大きな注意をはらうべきであるということを強いる。換言すれば、このことが意味するのは、新世代のPDFIは、少なくともHsPDFを含むミトコンドリアPDF(PDF1A)に対する阻害作用を全く示してはならないということである。
【0010】
従って、本発明の目的は、細菌、植物色素体、並びにアピココンプレキサン(apicocomplexan)及びキネトプラスチド原生生物などの原生動物のペプチドデホルミラーゼを強く阻害しやすく、しかしミトコンドリア、特に哺乳類のミトコンドリアのペプチドデホルミラーゼを阻害しない新規な化合物を提供することである。
【0011】
本発明のもう一つの目的は、細菌若しくは寄生虫感染症を有する個体(individuals)を治療するための、又は除草剤としての前記化合物の使用に関する。
【0012】
本発明は、インドール誘導体が、細菌のペプチドデホルミラーゼを強く阻害することができるのに対して、ヒトミトコンドリアのペプチドデホルミラーゼに対して本質的に不活性であるという、本発明者らによる予想外の発見に基づくものである。
【0013】
本発明は、
a)下記一般式(I):
【化2】
(式中、
− 互いに独立して、R2及びR3は、H、炭素原子1〜5個のアルキル基、又は1〜5個のヘテロ原子を含む2〜20個の原子の金属キレート基を表し;
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し、R2及びR3のうちの少なくとも1つが、上に定義した金属キレート基を表す)
を有し;並びに
b)大腸菌(Escherichia coli)ニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)ニッケル結合ペプチドデホルミラーゼ(配列番号:2)について約1μMよりも低いIC50を有する;化合物又は薬学的に許容可能なその塩の、
細菌感染症若しくは原生動物感染症の予防若しくは治療を対象とした医薬の製造のための使用、又は除草剤の製造のための使用に関する。
【0014】
上に言及した「金属キレート基」は、金属原子又は金属イオンと配位錯体を形成しやすい基に関するものである(Whittaker et al.,1999;Boularot et al.,2004)。従って、「金属キレート基」は、特に、1つ又はいくつかの非共有電子対を有する基に関するものである。
【0015】
好ましい実施態様においては、本発明は、R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−(X)n1−Y
(式中、
− nは、整数0〜3であり、
− n1は、0又は1であり、
− Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
− Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド),−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
の金属キレート基で表される、上で定義した一般式(I)の化合物の使用に関する。
【0016】
その他の好ましい実施態様においては、本発明はまた、R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、上で定義した一般式(I)の化合物の使用に関する。
【0017】
その他の好ましい実施態様においては、本発明は、上に定義した一般式(I)で表される化合物の使用に関するものであり、前記式中、R4、R5及びR6が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、チオアルキル基、チオアリール基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるものである使用に関する。
【0018】
本発明は、より詳細には、R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、上で定義した一般式(I)で表される化合物の使用に関する。
【0019】
好ましい実施態様によれば、本発明はまた、R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるもので表される、上で定義した一般式(I)で表される化合物の使用に関する。
【0020】
その他の好ましい実施態様においては、本発明は、R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、上で定義した、一般式(I)の化合物の使用に関する。
【0021】
特に好ましい実施態様によれば、本発明は、次の一般式(II):
【化3】
(式中、Roは、請求項1〜7に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は上に定義したとおりである)
で表される、上に定義した化合物の使用に関する。
【0022】
その他の特に好ましい実施態様によれば、本発明は、次の一般式(III):
【化4】
(式中、Roは上に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは上に定義したとおりである)
で表される、上に定義した化合物の使用に関する。
【0023】
特に好ましい実施態様においては、本発明は、
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す
、上に定義した、式(III)で表される化合物の使用に関する。
【0024】
その他の特に好ましい実施態様においては、本発明は、前記化合物が、下記諸式:
【化5】
に相当することを特徴とする、上に定義した、一般式(I)で表される化合物の使用に関する。
【0025】
ペプチドデホルミラーゼ(PDF)は、金属酵素である(すなわち、その補欠分子族が金属イオンである)。従って、本明細書の目的どおり、表現「ニッケル結合ペプチドデホルミラーゼ」は、その金属イオンとして本質的にニッケルのみを含有する酵素に関する。例えば、そのような酵素は、Ragusa et al.(1998)に記述されたように、ニッケル存在下に精製することにより得ることができる。
【0026】
表現IC50は、大腸菌(Escherichia coli)のニッケル結合ペプチドデホルミラーゼ(配列番号:1)(EMBL/GenBank accession number P27251)活性及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)のニッケル結合ペプチドデホルミラーゼ(配列番号:2)(EMBL/GenBank accession number O31410)活性の50%を阻害するのに必要な一般式(I)で表される化合物の濃度に関するものである。
【0027】
ペプチドデホルミラーゼに対するIC50は、Serero et al.(2003)に記述された一般法に従うことにより測定することができる。
【0028】
手短に言えば、速度論的解析(Lazennec & Meinnel,1997に記述されている)の前に、阻害物質を、研究される酵素とともに、規定の最終濃度で25℃で15分間インキュベートする。速度論的アッセイは、小容量の基質を添加することにより開始される。基質は、1mMのホルミル−Met−Ala−Serである。速度論的解析は、阻害物質不在下に、脱ホルミル化速度0.5μM/sを与える酵素濃度存在下に行なわれる。IC50値は、50%阻害を与える濃度に相当する。
【0029】
有利には、一般式(I)で表される化合物は、細菌のPDF(すなわちPDF1及びPDF2)、並びに葉緑体、アピコプラスト及びキネトプラストのPDFを阻害しやすいが、ミトコンドリアのPDF(すなわちPDF1A)を阻害しにくいものである。
【0030】
その他の特に好ましい実施態様においては、本発明は、前記原生動物が、アピコプラスト又はキネトプラストを有する原生動物のうちから選択され、特に、下記属の原生動物、すなわちプラスモジウム属(Plasmodium)、トキソプラズマ属(Toxoplasma)、サルコシスティス属(Sarcocystis)、クリプトスポリジウム属(Cryptosporidium)、エイメリア属(Eimera)、タイレリア属(Theileria)、バベシア属(Babesia)、トリパノゾーマ属(Trypanosoma)、リーシュマニア属(Leishmania)を含む群から選択される、上で定義された、一般式(I)の化合物の使用に関する。
【0031】
有利には、式(I)で表される化合物は、アピコプラスト又はキネトプラストのPDFにターゲッティングして阻害し、こうしてそれぞれ、アピコプラストの機能及びキネトプラストの機能を障害し、それが原生動物寄生虫の成長を阻害し、及び/又は原生動物寄生虫を殺す。
【0032】
更にその他の特に好ましい実施態様においては、本発明は、前記細菌が、次の属の細菌、すなわち、ブドウ球菌属(Staphylococcus)、連鎖球菌属(Streptococcus)、バチルス属(Bacillus)、エンテロコッカス属(Enterococcus)、シュードモナス属(Pseudomonas)、アシネトバクター属(Acinetobacter)、エンテロバクター属(Enterobacter)、ヘモフィルス属(Haemophilus)を含む群から選択される、上に定義した一般式(I)の化合物の使用に関する。
【0033】
有利には、一般式(I)で表される化合物は、細菌のPDF1にもPDF2にもターゲッティングして阻害し、そのことが前記化合物を、グラム陰性細菌に対してもグラム陽性細菌に対しても使用することができる広域スペクトルの抗生物質にする。
【0034】
本発明はまた、活性物質として、上に定義した式(I)で表される少なくとも1つの化合物、又は薬学的に許容可能なその塩を、薬学的に許容可能な賦形剤とともに含むことを特徴とする医薬組成物に関する。
【0035】
好ましい実施態様においては、上に定義した医薬組成物は、一般式(I)で表される化合物を約250mg〜約5000mgの単位用量で個体に投与するのに適している。
【0036】
その他の実施態様においては、上に定義した医薬組成物は、一般式(I)で表される化合物を約250mg〜約5000mgの1日用量で個体に投与するのに適している。
【0037】
その他の実施態様においては、上に定義した医薬組成物は、経口、静脈内、又は腹腔内経路により投与される。
【0038】
本発明はまた、活性物質として、式(I)で表される少なくとも1つの化合物を含むことを特徴とする、除草剤組成物に関する。
【0039】
好ましい実施態様においては、上に定義した除草剤組成物は、式(I)で表される化合物を約1mg/l〜約1000mg/lの用量で、特に約10mg/l〜約100mg/lの用量で、植物に投与するのに適している。
【0040】
有利には、式(I)で表される化合物は、葉緑体のPDFにターゲッティングして阻害し、そのことが、例えば植物又は藻類などの、葉緑体を保有する生物の成長を絶つか又は重度に障害する。
【0041】
本発明はまた、上に定義したような一般式(I)で表される化合物に関する。
【0042】
本発明は、より詳細には、一般式(I):
【化6】
(式中、
− 互いに独立して、R2及びR3は、H、メチル基、エチル基、プロピル基、又は次の式:
−(CH2)n−(X)n1−Y
(式中、
*nは、整数0〜3であり、
*n1は、0又は1であり、
*Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
*Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド)、−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
で表される金属キレート基を表し、
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し:
*R2及びR3のうち少なくとも1つは、上に定義した金属キレート基を表し、及び
*R3が上に定義した金属キレート基を表すとき、−(CH2)n−(X)n1−Yは、−(CH2)2−NH2又は−(CH2)2−SHを表すことができない)
で表される化合物に関する。
【0043】
本発明は、より詳細には、R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、上で定義した、一般式(I)の化合物に関する。
【0044】
本発明の好ましい化合物は、R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、上で定義した、一般式(I)の化合物である。
【0045】
その他の好ましい実施態様においては、本発明は、R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、上で定義した、一般式(I)の化合物に関する。
【0046】
その他の特に好ましい実施態様においては、本発明は、R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、上で定義した一般式(I)で表される化合物に関する。
【0047】
本発明はまた、より詳細には、R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、請求項16〜20のいずれか一項に記載の一般式(I)の化合物に関する。
【0048】
本発明はまた、より詳細には、次の一般式(II):
【化7】
(式中、Roは、上に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は上に定義したとおりである)
で表される上に定義した化合物に関する。
【0049】
その他の好ましい実施態様においては、本発明は、次の一般式(III):
【化8】
(式中、Roは、上に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは上に定義したとおりである)
で表される上に定義した化合物に関する。
【0050】
その他の特に好ましい実施態様においては、本発明は、
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す、
上で定義された式(III)の化合物に関する。
【0051】
その他の特に好ましい実施態様においては、本発明は、上に定義した一般式(I)で表される化合物に関するものであり、前記化合物が、次の諸式:
【化9】
に相当することを特徴とする化合物に関する。
【0052】
本発明はまた、ペプチドデホルミラーゼ酵素をスクリーニングする化合物と接触させる工程、及び前記ペプチドデホルミラーゼ酵素に対する前記化合物の阻害活性を測定する工程を含むことを特徴とする、細菌又は原生動物感染症の予防若しくは治療、又は除草剤としての使用、を対象としたインドール構造を含む化合物をスクリーニングする方法に関する。
【0053】
本明細書の目的どおり、「インドール構造を含む化合物」は、次の構造:
【化10】
(構造中、あいた結合は、H又は任意の化学結合のいずれかに連結される)
を有する化合物である。
【0054】
上に定義したスクリーニング方法の好ましい実施態様においては、ペプチドデホルミラーゼ酵素に対するスクリーニング化合物の阻害活性は、前記ペプチドデホルミラーゼ酵素について前記化合物のIC50を測定することにより評価される。
【0055】
IC50は、前記の方法により測定することができる。
【0056】
上に定義したスクリーニング方法のその他の好ましい実施態様においては、IC50が約1μMより低いスクリーニング化合物が選択される。
【0057】
上に定義したスクリーニング方法のその他の好ましい実施態様においては、スクリーニング化合物は、上に定義した式(I)に相当する。
【0058】
上に定義したスクリーニング方法のその他の好ましい実施態様においては、ペプチドデホルミラーゼ酵素は、大腸菌(Escherichia coli)のニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラス(Bacillus stearothermophilus)のニッケル結合ペプチドデホルミラーゼ(配列番号:2)である。
【0059】
上に定義したスクリーニング方法の特に好ましい実施態様においては、スクリーニング化合物が、ミトコンドリアのペプチドデホルミラーゼ、特にヒトミトコンドリアのペプチドデホルミラーゼに対する阻害特性を本質的に有しないということが更に試験される。
【0060】
一般式(I)を有する化合物の合成は、以下の手順に従い、又は実施例に記載するようにして行なうことができる。
【0061】
アミン、シアナミド及びヒドロキシグアニジンのインドール誘導体は、次の反応工程式1に示すようにして製造することができる。
【化11】
【0062】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基、チオアルキル基、又はチオアリール基を表す。
【0063】
手短に言えば、フィルスマイヤー−ハーク反応により相当するインドールから製造されるインドール−3−カルボキサルデヒドは、リン酸水素二アンモニウム、1−ニトロプロパン及び酢酸を用いて処理することにより、インドール−3−カルボニトリルに変換される(Jiang et al.,2000)。ニトリルは、CoCl2/NaBH4を用いてアミンに還元される(Leclerc et al.,1998)。シアナミドは、アミンのBrCNとの反応によって得られ、酢酸ナトリウム存在下に塩酸ヒドロキシルアミンとの反応によりヒドロキシグアニジンに変換される(Lefevre−Groboillot et al.,2001)。
【0064】
ヒドロキサム酸、ヒドロキシメチルケトン及びチオメチルケトンインドール誘導体は、次の反応工程式2に示すようにして製造することができる。
【化12】
【0065】
Wは、アルキル基、アリール基、アルコキシ基、アリールオキシ基又はチオアリール基を表す。
【0066】
手短に言えば、酢酸基は、亜鉛塩を2−ブロモ酢酸エチルエステルと反応させ、そのあとエステルを加水分解することにより、3−インドール位に導入される(Dillard et al.,1996)。ヒドロキシメチルケトンは、Wissnerにより記述された手順(Wissner et al.,1979)に従い、全くルイス酸触媒を用いないで、室温(r.t.)で、(インドール−3−イル)アセチルクロリドをトリス(トリメチルシリルオキシ)エチレンと反応させ、そのあと中間体を80℃で加水分解脱炭酸反応させることにより製造される。チオアセチル誘導体は、酸から出発して5ステップで製造され、塩化アシルのジアゾメタンとの反応によりα−ジアゾケトンに変換される(Salim & Capretta,2000)。HCl(g)エーテル溶液との処理で、クロロメチルケトンを得る。DMF中、室温で塩化物をチオ酢酸カリウムで置換することにより、対応するチオエステルを得、これを厳密な嫌気状態下にメタノール中のNa2CO3を用いて加水分解し、そのあとHCl(g)エーテル溶液で酸性化することにより脱保護することができる。
【0067】
対応する酸のインドール誘導体からヒドロキサム酸インドール誘導体を製造する一般手順は、次のとおりである。
【0068】
ヒドロキシルアミンをエステルと直接反応させることも可能ではあるが、(インドール−3−イル)酢酸誘導体の一部は市販品として入手可能であるので、酸から出発する一般合成手順を以下に記載する。
【0069】
手短に言えば、インドール酢酸誘導体(0.3mmol)のDMF溶液10mlに、1−ヒドロキシベンゾトリアゾール(1.1当量,m=44mg)、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(1.1当量,m=63mg)及びN−メチルモルホリン(1.1当量,36μL)を添加する。この溶液を室温で1時間撹拌する。塩酸ヒドロキシルアミン(1.1当量,m=23mg)を添加後、溶液を撹拌下に一晩放置する。次いでDMFを真空下に蒸発させ、酢酸エチルを残渣に添加する。この酢酸エチル溶液を、順次に、水、飽和NaHCO3水溶液及び飽和NaCl水溶液で洗浄する。Na2SO4で乾燥し、濾過後、溶媒を蒸発乾固して、ヒドロキサム酸誘導体を収率40〜60%で得る。
【0070】
インドール誘導体の2位置換は、反応工程式3に記載するようにして実施することができる。
【化13】
【0071】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基、又はチオアリール基を表す。
【0072】
手短に言えば、2−リチオ−N−ベンゼンスルホニルインドール(N−ベンゼンスルホニルインドールをBuLiと反応させて得る)をシュウ酸ジメチルと反応させ(Hasan et al.,1981)、そのあと引き続き塩基性加水分解とウォルフ−キシュナー還元を行ない、(インドール−2−イル)酢酸を得る(Weller & Ford,1984)。
【0073】
N−置換インドールは反応工程式4に記載するようにして製造することができる。
【化14】
【0074】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基又はチオアリール基を表す。
【0075】
手短に言えば、N−カルボキシメチル化は、リチオ誘導体(−78℃でTHF中2当量のリチウムビス(トリメチルシリル)アミドを用いて製造する)のクロロギ酸ベンジルとの反応により達成される(Horwell et al.,1997)。
【化15】
【0076】
本発明によるインドール窒素(R1)(反応工程式5)の様々な官能基化は、例えばDillard et al.,(1996)及びEissenstat et al.,(1995)に記述されたようにして製造することができる。
【0077】
リバースヒドロキサメートインドール誘導体は、反応工程式6に示すようにして製造することができる。
【化16】
【0078】
Wは、ハロゲン原子、アルキル基、アリール基、アルコキシ基、アリールオキシ基、チオアルキル基、又はチオアリール基を表す。
【0079】
手短に言えば、ルートAに従い、アルデヒドのピリジン中塩酸ヒドロキシルアミンとの縮合で、ケトキシムを得、これを更にBorch条件下に還元する(Borch et al.,1971)。ルートBに従い、アルデヒドの還元後、ヒドロキシメチル誘導体を、Mitsunobu反応にかける(Kolossa et al.,1997)か又は臭素化するかいずれかしてから、N,O−ビス(tert−ブトキシカルボニル)ヒドロキシルアミンを用いる求核置換反応にかける。いったんトリフルオロ酢酸で脱保護すると、ヒドロキシルアミンはホルミル化され、リバースヒドロキサメートを与える。選択的N−ホルミル化反応は、特にHill et al.(2002)に記述されている。
【0080】
本発明による式(I)で表される化合物をすべて合成するためにこれらの手順にもたらし得る諸適合は、当業者の通常の技術の範囲内にある。
【0081】
《材料及び方法》
すべての溶媒及び化学薬品は、それぞれSDS及びAldrichから購入した。DMF、THF及びCH2Cl2は、標準的手順を用いて乾燥し、アルゴン雰囲気下に4Åモレキュラーシーブ上保存した。1HNMRスペクトルはBruker ARX−250スペクトロメーターで記録し、化学シフトはTMSから低磁場側でのppm単位で報告した。IRスペクトルは、MIRacleTM 単反射水平ATRユニット(ジルコニウム−セレン結晶)を備えた、Perkin−Elmer Spectrum One FT−IRスペクトロメーターを用いて得た。FAB及びCI質量スペクトルは、ParisのENSで記録した。元素分析は、Paris VI University(France)又はGif−sur−Yvette(CNRS、France)の微量分析サービスにより実施した。アルゴン下に行われる実験は、真空ラインで実施した。
【化17】
【0082】
《3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(1c)》
新しく蒸留したTHF(10mL)中の2−(インドール−3−イル)酢酸(500mg,M=175.19,2.85.mmol)及びリチウムビス(トリメチルシリル)アミド(1M/THF,6.27mmol,2.2当量,6,27mL)の溶液を、アルゴン下に、−78℃で1時間撹拌した。次いでクロロギ酸ベンジル(505μL,M=170.60,d=1.195,3.42.mmol,1.2当量)を添加した。反応混合物を、−78℃で2時間撹拌し、次いで溶媒を真空下に蒸発させた。残渣を水に溶解して、ジエチルエーテルで抽出した。水層をHCl水溶液(0.1N)でpH3まで酸性化して、結果として3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(1c)を白色沈殿として得、これを濾別して、ペンタンで洗浄した(795mg,収率90%)。Rf(シリカゲルMerckF254,CH2Cl2/CH3OH9/1v:v混合物)=0.4.Tf=152℃.IR(cm−1):1728及び1694(νCO).1HNMR(250MHz,[D6]DMSO):δ3.77(s,2H);5.53(s,2H);7.29−7.66(m,8H);7.74(s,1H);8,14(d,J=8.1,1H).CI−MS:m/z=327,[M+NH4+],100%.元素分析.C18H15NO4に対する計算値:C,69.89;H,4.89;N,4.53.実測値:C,69.78;H,5.00;N,4.55.
【0083】
《3−カルボキシメチル−5−ブロモ−インドール−1−カルボン酸ベンジルエステル(1d)》
2−(5−ブロモ−インドール−3−イル)酢酸(200mg,M=254.09,0.787mmol)に同じ手順を適用して、(1d)(280mg,収率:92%)を得た。Rf(シリカゲルCH2Cl2/CH3OH9/1v:v混合物)=0.5,Tf=88℃,IR(cm−1):1729及び1692(νCO).1HNMR(250MHz,[D6]アセトン):δ3.80(s,2H);5.51(s,2H);7.15(s,1H);7.24−7.82(m,7H);8.12(d,J=8,1,1H);11,16(s,1H).CI−MS:m/z=405,407,[M+NH4+],40%;388,390[M+],30%.元素分析.C18H14BrNO4.0.5H2Oに対する計算値:C,54.53;H,3.87;N,3.53.実測値:C,54.67;H,3.72;N,3.44.
【0084】
《1−(5−ブロモ−1H−インドール−3−イル)−3−ヒドロキシプロパン−2−オン(2)》
塩化オキサリル(104μl,M=126.93,d=1.455,1.18mmol,1.5当量)を、アルゴン下に0℃で、DMF数滴を含むTHF(10mL)中の2−(5−ブロモ−1H−インドール−3−イル)酢酸(200mg,M=254.09,0.788mmoL)の溶液に添加した。混合物を室温で2時間撹拌し、次いで真空下に蒸発させた。残渣をジオキサン10mlに溶解し、これに、アルゴン下に、トリス(トリメチルシリルオキシ)エチレン(675μL,M=292.59,d=0.885,1.94mmol,2.3当量)を添加した。室温で10時間撹拌後、HCl水溶液(0.1N)10mLを添加し、次いで溶液を80℃で30分間加熱した。NaClを添加後、溶液をジエチルエーテルで抽出した。有機層をブラインで洗浄し、MgSO4で乾燥して、真空下に蒸発乾固した。カラムクロマトグラフィー(シリカゲル,溶離CH2Cl2/CH3OH95/5v:v混合物)で精製後、1−(5−ブロモ−1H−インドール−3−イル)−3−ヒドロキシプロパン−2−オン(2)を得た(45mg,収率=21%)。Rf(シリカゲル,CH2Cl2/CH3OH9/1v:v混合物)=0.5.Tf=88℃.IR(cm−1):1702(νCO).1HNMR:(250MHz,CDCl3):δ3.00(s,1H);3.81(s,2H);4.31(s,2H);7.15(s,1H);7.19−7.32(m,2H);7.64(s,1H);8.21(s,1H).CI−MS:m/z=285,287,[M+NH4+],80%;268,270[M+],100%.元素分析.C11H10BrNO2に対する計算値:C,49.28;H,3.76;N,5.22.実測値:C,49.10;H,3.75;N,5.17.
【0085】
《チオ酢酸S−[3−(インドール−3−イル)−2−オキソ−プロピル]エステル(5a)》
塩化チオニル(150μl,M=118.97,d=1.63,2.06mmol,1.2当量)を、アルゴン下に、2−(インドール−3−イル)酢酸(300mg,M=175.19,1.72mmol)/CH2Cl2溶液(10mL)に添加した。混合物を40℃で3時間加熱し、次いで減圧下に蒸発させた。CH2Cl2(10mL)に溶解した残渣を、アルゴン下に、新しく調製したジアゾメタン/ジエチルエーテル(10.4mL,C=0.66M,6.88mmol,4当量)溶液中にカニューレで挿入した。0℃で4時間撹拌後、溶液を減圧下に蒸発させて、収率95%でジアゾ化合物(3a)を黄色油状物として得、これを更に精製することなく使用した。(3a):Rf(酢酸エチル/シクロヘキサン1/1v:v混合物)=0.65.IR(cm−1):2100(νCN);1726(νCO).1HNMR(250MHz,CDCl3):δ3.76(s,2H);5.28(s,1H);7.14−7.28(m,3H);7.39(d,J=7.8,1H);7.48(d,J=7.7,1H);8.12(s,1H).CI−MS:m/z=217,[M+NH4+],50%;200,([M+H]+),30%.HCl(g)エーテル溶液(630μL,C=6N,3.78mmol,2.2当量)を、(3a)/ジエチルエーテル(10mL)溶液に添加した。DMF5mLを添加して、ジエチルエーテルを注意深く蒸発させた。次いでKSAc(432mg,M=114.20,3.78mmol,2.2当量)/DMF溶液(5mL)を、アルゴン下に、カニューレにより移送した。室温で一晩撹拌後、溶媒を真空下に蒸発させて、(5a)を得、これを更にカラムクロマトグラフィー上、酢酸エチル/シクロヘキサン8/2v:v混合物で溶離して精製した(220mg,収率:52%)。Rf(シリカゲル酢酸エチル/シクロヘキサン1/1v:v混合物)=0.6.IR(cm−1):1788(νCO).1HNMR(250MHz,CDCl3):δ2,34(s,3H);3,78(s,2H);3,95(s,2H);7.09−7.57(m,5H);8.19(s,1H).CI−HRMS:C13H14NO2S+([M+H]+)に対する計算値,248.0445;実測値,248.0448.
【0086】
《チオ酢酸S−[3−(5−ブロモ−1H−インドール−3−イル)−2−オキソ−プロピル]エステル(5b)》
(5−ブロモ−インドール−3−イル)酢酸(300mg,M=254.09,1,18mmol)に同じ手順を適用して、チオ酢酸5−[3−(5−ブロモ−1H−インドール−3−イル)−2−オキソ−プロピル]エステル(5b)227mgを収率59%で得た。Rf(シリカゲル,酢酸エチル/シクロヘキサン1/1v:v混合物)=0.5.1HNMR(250MHz,CDCl3):δ2.36(s,3H);3.78(s,2H);3.92(s,2H);7.13−7.29(m,3H);7.72(s,1H);7.48(d,J=7.7,1H);8.18(s,1H).CI−HRMS:C13H13BrNO2S+([M+H]+)に対する計算値,325.9850,327.9830;実測値325.9843,327.9837.
ジアゾ化合物(3b)を収率95%で単離した:Rf(シリカゲル,シクロヘキサン/酢酸エチル3/7v:v混合物)=0.5.IR(cm−1):2101(νCN);1715(νCO).1HNMR(250MHz,CDCl3):δ3.75(s,2H);5.68(s,1H);7.22(dd,J3=8.5,4J=1.5,1H);7.34(s,1H);7,37(d,3J=8.5,1H);7,75(d,4J=1.5,1H).CI−MS:m/z=278,280,[M+],60%;250,252,[M−N2]+,100%.
【0087】
《3−(3−アセチルスルファニル−2−オキソ−プロピル)−インドール−1−カルボン酸ベンジルエステル(5c)》
3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(200mg,M=309.32,0.647mmol)に同じ手順を適用して、(5c)178mgを収率72%で得た。Rf(酢酸エチル/シクロヘキサン1/1v:v混合物)=0.6.1HNMR(250MHz,[D6]アセトン):δ2.34(s,3H);3.98(s,2H);4.07(s,2H);5,50(s,2H);7.15−7.80(m,9H);8.15(d,4J=1.5,1H).CI−HRMS:C21H23NO4S+[M+NH4+]に対する計算値,399.1379;実測値,399.1375.
ジアゾ化合物(3c)を収率91%で単離して、キャラクタリゼーションを行なった:Rf(酢酸エチル/シクロヘキサン1/1v:v混合物)=0.5.IR(cm−1):2102(νCN);1730及び1636(νCO).1HNMR(250MHz,[D6]アセトン):δ3.78(s,2H);5.50(s,2H);5,82(s,1H);7.26−7.69(m,8H);7.70(s,1H);8.17(d,J=8,1H).CI−MS:m/z=351,[M+NH4+],100%.
【0088】
《2−(インドール−3−イル)−N−ヒドロキシアセトアミド(6a).》
2−(インドール−3−イル)酢酸(500mg,M=175.19,2.85mmol)、1−ヒドロキシベンゾトリアゾール、1−ヒドロキシベンゾトリアゾール(HOBT)(425mg,M=135.13,3.14mmol,1.1当量)及び1−(3−ジメチルアミノプロピル)−3−エチルカルボキシルイミン塩酸塩、EDCI(602mg,M=191.71,3.14mmol,1.1当量)のDMF溶液(10mL)を、アルゴン下に5分間撹拌した。次いでN−メチルモルホリン(NMM)(345μl,M=101.15,d=0.92,3.14mmol,1.1当量)を添加した。反応混合物を室温で2時間撹拌して、塩酸ヒドロキシルアミン(218mg,M=69.5,3.14mmol,1,1当量)を添加した。溶液を撹拌下に一晩放置し、次いでDMFを真空下に蒸発させて、黄色油状物を得、これを酢酸エチルに溶解した。この溶液を、順次に、水及びNaHCO3水溶液で洗浄した。通常の処理後、溶媒を蒸発させて、(6a)を白色固体として得た(m=260mg,収率=60%)。IR(cm−1):1638(νCO).1HRMN(250MHz,[D6]DMSO):δ3.44(s,2H);7.02(t,J=7.1,1H);7.12(t,J=7.1,1H);7.19(s,1H);7.39(d,J=7.8,1H);7,62(d,J=7.6,1H);8,74(s,1H);10.65(s,1H);11.85(s,1H).元素分析.C10H10N2O2に対する計算値:C,63.15;H,5.30;N,14.73.実測値:C,62.96,H,5.31,N,14.63.
【0089】
《2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6b)》
(5−ブロモ−1H−インドール−3−イル)酢酸(200mg;M=254.09,0.787mmol)から出発して、(6b)116mgを収率55%で得た。Rf(C18シリカゲル,酢酸エチル/メタノール 1/1v:v混合物)=0.8.Tf=145℃.IR(cm−1):3417(νOH)3213(νNH),1633(νCO).1HNMR(250MHz,[D6]DMSO):δ3.49(s,1H);7.20−7.44(m,3H);7.87(s,1H);8.86(s,1H);10.59(s,1H);11.14(s,1H).CI−MS:m/z=270,272,[M+H]+,100%.元素分析.C10H9BrN2O2に対する計算値:C,44.63;H,3.37;N,10.41.実測値:C,44.52,H,3.52;N,10.45.
【0090】
《5−ブロモ−3−ヒドロキシカルバモイルメチル−インドール−1−カルボン酸ベンジルエステル(6d)》
Horwell et al.(1997)に記述されたカルボキシメチル化手順に従い、2−(5−ブロモ−インドール−3−イル)酢酸(200mg,M=254,09,0.787mmoL)から3−カルボキシメチル−5−ブロモ−インドール−1−カルボン酸ベンジルエステルを得た(280mg,収率:92%)。5−ブロモ−3−カルボキシメチル−インドール−1−カルボン酸ベンジルエステル(120mg,M=388.21,0.309mmol)から出発して、(6d)50mgを収率40%で得た。IR(cm−1):1650及び1690(νCO).1HNMR(250MHz,[D6]DMSO):δ3.40(s,2H);5.56(s,2H);7.45−8.10(m,9H);8.95(s,1H);10.75(s,1H).元素分析.C18H15BrN2O4に対する計算値:C,53.62;H,3.75;N,6.95.実測値:C,53.73;H,3.54;N,7.11.
【0091】
《(4−フルオロ−1H−インドール−3−イル)−酢酸エチルエステル(8e)》
冷却した4−フルオロ−1H−インドール7e(M=135.05,300mg,2.22mmol)/THF3.3mL溶液に、溶液を氷浴で0℃未満に保持しながら、1.6Mのn−BuLi/ヘキサン 1.39mL(2.22mmol)を添加した。15分後、1NのZnCl2/Et2O 2.22mLを添加した。冷却浴を取り外し、混合物を24時間撹拌し、次いで真空下に蒸発させて、ロウ状物を得、これを更に無水トルエン(3.3mL)に溶解した。エチル−2−ブロモアセテート(246μL,2.22mmol)を添加後、溶液を24時間撹拌した。次いで混合物を1NHClで酸性にして、酢酸エチルに注ぎ入れた。有機層をブラインで洗浄して、MgSO4で乾燥した。エステルを、シリカゲル上10%AcOEt/シクロヘキサンで溶離するクロマトグラフを行ない、(8e)158mg(32%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.24(t,J=7,3H);3.88(s,2H);4.16(q,J=7,2H);6.71(dd,3JHF=11,3JHH=8,1H);7.06(td,3JHH=8,4JHF=5.4,1H);7.23(d,J=8,1H);7.28(d,5JHF=1.9,1H);10.34(s,1H).
【0092】
《2−(4−フルオロ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6e)》
NH2OH.HCl(497mg,7.15mmol)を粉末のまま、1MのEtONa/EtOH溶液6.4mLに添加した。この溶液を、エステル(8e)(158mg,0.715mmol,M=221.03)/エタノール溶液(10mL)に添加した。混合物をアルゴン下に80℃で24時間撹拌した。次いで冷却及び真空下に蒸発後、残渣をAcOEtに溶解した。この有機層をブライン、NaHCO3水溶液、0.1NHCl、ブラインで洗浄して、MgSO4で乾燥した。真空下に蒸発後、ヒドロキサム酸(6e)をアセトン/シクロヘキサンの1:1混合物に溶解した。減圧下にアセトンをゆっくり蒸発させて、(6e)75mgを白色固体として得た(51%)。IR(cm−1):3354(νNH),3280(νOH),1631(νCO).1HNMR(250MHz,[D6]アセトン):δ3.72(s,2H);6.70(dd,3JHF=11,3JHH=8,1H);7.05(td,3JHH=8,4JHF=5.4,1H);7.22(d,J=8,1H);7.28(s,1H);10.00(s,1H),10.43(s,1H).CI−HRMS:C10H13N3O2F([M+NH4+])に対する計算値,226.0992;実測値226.0991.
【0093】
《(5−フルオロ−1H−インドール−3−イル)−酢酸エチルエステル(8f)》
5−フルオロ−1H−インドール(7f)125mg(M=135.05,0.926mmol)からエステル(8f)91mg(収率=44%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.24(t,J=7.2,3H);3.74(s,2H);4.13(q,J=7.2,2H);6.92(t,J=9,1H);7.30(d,J=9,1H);7.38(m,2H);10.21(s,1H).
【0094】
《2−(5−フルオロ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6f)》
エステル(8f)91mg(M=221.23,0.41mmol)から出発して、ヒドロキサム酸(6f)(M=208.19)21mg(収率=25%)を得た。IR(cm−1):3360(νNH),3175(νOH),1625(νCO).1HNMR(250MHz,[D6]アセトン):δ3.57(s,2H);6.91(td,3JHF=3JHH=9.3,4JHH=2.3,1H);7.33−7.41(m,3H);8.00(s,1H);10.08(s,1H);10.22(s,1H).CI−MS:m/z=226.2,[M+NH4+],20%.
【0095】
《(5−クロロ−1H−インドール−3−イル)−酢酸エチルエステル(8g)》
5−クロロ−1H−インドール(7g)300mg(M=151.60,1.98mmol)からエステル(8g)198mg(収率=43%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.24(t,J=7,3H);3.77(s,2H);4.14(q,J=7,2H);7.24(d,J=8.5,1H);7.35−7.40(m,2H);7.79(s,1H);10.33(s,1H).
【0096】
《2−(5−クロロ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6g)》
エステル(8g)198mg(M=233.26,0.85mmol)から出発して、ヒドロキサム酸(6g)(M=224.64)194mg(収率=95%)を得た。IR(cm−1):3348(νNH),3190(νOH),1625(νCO).1HNMR(250MHz,[D6]アセトン):δ3.59(s,2H);7.10(d,J=8.5,1H);7.35(s,1H);7.41(d,J=8.5,1H);7.67(s,1H);10.00(s,1H);10.29(s,1H).CI−HRMS:に対する計算値C10H10N202Cl([M+NH4+]),225.04331(100%),227.0461(33%);実測値225.0432(100%),227.0413(32.4%).
【0097】
《(5−メトキシ−1H−インドール−3−イル)−酢酸エチルエステル(8h)》
5−メトキシ−1H−インドール(7h)300mg(M=147.18,2.04mmol)からエステル(8h)219mg(収率=46%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.23(t,J=7,3H);3.73(s,2H);3.82(s,3H);4.15(q,J=7,2H);6.79(d,J=8.5,1H);7.11(s,1H);7.26−7.32(m,2H);9.97(s,1H).
【0098】
《2−(5−メトキシ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6h)》
エステル(8g)219mg(M=233.26,0.94mmol)から出発して、ヒドロキサム酸(6h)(M=220.22)90mg(収率=43%)を得た。IR(cm−1):3307(νNH),3160(νOH),1620(νCO).1HNMR(250MHz,[D6]アセトン):δ3.57(s,2H);3.81(s,3H);6.77(dd,3J=8.8,4J=2.3,1H);7.15(d,J=2.3,1H);7.22(s,1H);7.28(d,J=8.8,1H);7.93(s,1H);10.04(s,1H).CI−MS:m/z=238,[M+NH4+],35%.
【0099】
《(6−ブロモ−1H−インドール−3−イル)−酢酸エチルエステル(8i)》
6−ブロモ−1H−インドール(7i)300mg(M=196.05,1.53mmol)からエステル(8i)192mg(収率=45%)を得た。1HNMR(250MHz,[D6]DMSO):δ1.23(t,J=7.2,3H);3.76(s,2H);4.13(q,J=7.2,2H);7.19(dd,3J=8.5,4J=1.5,1H);7.34(s,1H);7.55(d,J=8.5,1H);7.62(d,J=1.5,1H);10.28(s,1H).
【0100】
《2−(6−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(6i)》
エステル(8i)192mg(M=282.16,0.68mmol)から出発して、ヒドロキサム酸(6i)(M=269.09)136mg(収率=74%)を得た。IR(cm−1):3335(νNH),3230(νOH),1615(νCO).1HNMR(250MHz,[D6]アセトン):δ3.60(s,2H);7.17(dd,3J=8.5,4J=1.5,1H);7.30(s,1H);7.6(m,2H);8.0(s,1H);10.0(s,1H);10.27(s,1H).CI−MS:m/z=270,272,[M+H]+,50%.
【0101】
《2−(5−ブロモ−2−メチル−1H−インドリル)−酢酸エチルエステル(8j)》
5−ブロモ−2−メチル−1H−インドール(7j)300mg(M=210.06,1.43mmol)からエステル(8j)201mg(収率=48%)を得た。1HNMR(250MHz,[D6]アセトン):δ1.18(t,J=7,3H);2.33(S,3H);3.66(s,2H);4.06(q,J=7,2H);7.10(d,J=8,1H);7.22(d,J=8,1H);7.56(s,1H);10.11(s,1H).
【0102】
《2−(5−ブロモ−2−メチル−1H−インドリル)−N−ヒドロキシアセトアミド(6j)》
エステル(8j)201mg(M=296.16,0.68mmol)から出発して、ヒドロキサム酸(6j)(M=283.12)160mg(収率=83%)を得た。IR(cm−1):1690(νCO).1HNMR(250MHz,[D6]アセトン):δ2.44(s,3H);3.69(s,2H);7.14(d,J=8.4,1H);7.26(d,J=8.4,1H);7.67(s,1H);10.20(s,1H).
【化18】
【0103】
《5−ブロモ−2−メチル−1H−インドール−3−カルバルデヒド(9)》
POCl3(267μL,2.86mmoL,d=1.64)を、アルゴン下に0℃で、5−ブロモ−2−メチル−1H−インドール(7j)(500mg,2.38mmol)/DMF5mL溶液に添加した。室温で一晩撹拌後、2NNaOH水溶液2mLを添加し、溶液を更に2時間撹拌し、次いで酢酸エチルに注ぎ入れた。水で洗浄し、乾燥し、蒸発乾固した後、(9)526mgを収率95%で白色固体として単離した(収率=93%)。IR(cm−1):1628(νC=O).1HNMR(250MHz,[D6]アセトン):δ2.78(s,3H);7.33(d,3J=8.45,1H),7.36(d,3J=8.45,1H),8.33(s,1H),10.15(s,1H),11.04(s,1H).CI−MS:m/z=255,257[M+NH4+],85%;238−240,[M+],100%.元素分析.C10H8BrNOに対する計算値:C,50.45;H,3.39;N,5.88.実測値:C,50.42,H,3.49;N,5.87.
【0104】
《5−ブロモ−2−メチル−1H−インドール−3−カルバルデヒド−オキシム(10)》
NH2OH,HCl(70mg,1.01mmol)を、(9)(200mg,0.84mmoL)/ピリジン5mL溶液に添加した。室温で5時間撹拌後、溶液を蒸発乾固した。残渣を酢酸エチルに溶解後、溶液を順次、1NHCl、ブラインで洗浄して、MgSO4で乾燥した。溶媒を蒸発後、(10)210mgを黄色油状物として定量的収率で単離した。IR(cm−1):1622(νC=N).1HNMR(250MHz,[d6]アセトン):δ2.53(s,3H);7.2−7.35(m,2H);7.80(s,1H);8.32(s,1H);9.69(s,1H);10.49(s,1H).元素分析.C10H9BrN2O.0.25H2Oに対する計算値:C,46.63;H,3.72;N,10.87.実測値:C,46.69;H,3.66;N,10.58.
【0105】
《N−[2−(5−ブロモ−2−メチル−1H−インドール−3−イル)メチル]−ヒドロキシルアミン(11)》
(10)(110mg,0.43mmol)/メタノール10mL溶液に、メチルオレンジ結晶を数個添加し、次いでHCl(g)(2N)エタノール溶液を数滴添加した。溶液は赤紫色に変化した。HClを添加してそのあとも溶液の赤色を保持しながら、2当量のNaBH3CN(55mg,0.86mmol)/THF5mLを添加して、溶液を撹拌した。数時間後、赤色が安定してから、溶液を撹拌下に一晩放置した。蒸発後、残渣をメタノール5mLに溶解し、次いで水5mLを添加した。1NNaOH水溶液でpHを9まで調整した。この溶液をCH2Cl2で抽出した。通常の処理後、(11)(110mg)を黄色油状物として定量的収率で単離し、更に精製することなくキャラクタリゼーションを行なった。1HNMR(250MHz,[D6]アセトン):δ2.51(s,3H);5.16(s,2H);7.1−7.3(m,2H);8.10(s,1H);10.34(s,1H).CI−MS:m/z=255,257[M+],30%;224,226([M−NHOH]+),100%.元素分析.C10H11BrN2Oに対する計算値:C,47.08;H,4.35;N,10.98.実測値:C,46.97;H,4.51;N,11.13.
【0106】
《5−ブロモ−2−メチル−1−(フェニルスルホニル)−1H−インドール−3−カルバルデヒド(12)》
5−ブロモ−2−メチル−1H−インドール−3−カルバルデヒド(9)(500mg,2.10mmol)の溶液を、0℃でアルゴン下に、NaH(110mg,4.62mmol,2.2当量)/THF10mL懸濁液に添加した。1時間撹拌後、ベンゼンスルホニルクロリド(320μL,2.52mmol,1.2当量)の溶液を添加した。混合物を室温で一晩撹拌した。次いで水50mLを添加し、この溶液を酢酸エチルで抽出し、有機層を、順次、0.1NHCl、NaHCO3水溶液、ブラインで洗浄して、MgSO4で乾燥した。セライトを通して濾過し、蒸発させて、ペンタン/アセトン混合物から再結晶後、(12)700mg(95%収率)を得た。1HNMR(250MHz,[D6]アセトン):δ3.05(s,3H);7.57(dd,3J=8.9,4J=2.1,1H);7.6−8.0(m,5H),8.19(d,J=8.9,1H),8.41(d,J=2.1,1H),10.32(s,1H).CI−MS:m/z=378,380[M+],100%.
【0107】
《2−(5−ブロモ−2−メチル−1−(フェニルスルホニル)−1H−インドール−3−イル)エタノール(13)》
(12)(700mg,1.85mmol)/メタノール10mL溶液に、NaBH4(84mg,2.22mmol)/メタノール10mL溶液を添加した。室温で6時間撹拌後、溶液を酢酸エチルに注ぎ入れて、ブラインで洗浄した。有機層をMgSO4で乾燥して、真空下に蒸発させた。アルコールをシリカゲル上シクロヘキサン/酢酸エチル70/30で溶離するクロマトグラフを行ない、(13)700mgを定量的収率で淡黄色粉末として得た。1HNMR(250MHz,[D6]アセトン):δ2.64(s,3H);4.02(s,1H);4.72(s2H);7.45(dd,3J=8.9,4J=2,1H);7.83(d,J=2,1H);7.6−8.0(m,5H);8.13(d,J=8.9,1H).CI−MS:m/z=380,382[M+],100%.
【0108】
《Tert−ブチル−2−(5−ブロモ−2−メチル−1−(フェニルスルホニル)−1H−インドール−3−イル)メチル−(tert−ブトキシカルボニルオキシ)−カルバメート(14)》
(13)(220mg,0.58mmol)/THF10mL溶液に、室温でアルゴン下に、連続してPPh3(1.1当量,167mg,0.64mmol)を添加し、15分間撹拌後N−ブロモスクシンイミド(1.1当量,113mg,0.64mmol)を添加した。混合物を室温で一晩撹拌した。ブロモ誘導体を単離せず、更にこの溶液で使用した。N,O−ビス−(tert−ブトキシカルボニル)ヒドロキシルアミン(135mg,0.58mmol)/DMF5mL溶液をNaH(1.1当量,15mg)とともに予め15分間撹拌しておき、これを前記ブロモ誘導体に添加した。得られた溶液を2時間撹拌した。CH2CL2添加後、溶液を、水、NH4Cl水溶液、ブラインで洗浄して、MgSO4で乾燥した。溶媒蒸発後、残渣を、シリカゲル上CH2Cl2/C6H1270/30及び80/20で溶離するクロマトグラフを行ない、(14)150mgを得た(収率44%)。1HNMR(250MHz,[D6]アセトン):δ1.35(s,9H);1.49(s,9H);2.67(s,3H);4.87(s,2H,);7.47(dd,3J=8.8,4J=1.8,1H);7.63(m,3H);7.78(d,J=1.8,1H);7.9(d,J=7.5,2H);8.13(d,J=8.8,1H).CI−MS:m/z=612,614[M+],100%.
【0109】
《N−((5−ブロモ−2−メチル−1(フェニルスルホニル)−1H−インドール−3−イル)メチル)ヒドロキシルアミン(15)》
トリフルオロ酢酸775μL(40当量)を、(14)(150mg,0.25mmol)/CH2Cl210mL溶液に添加した。溶液を0℃でアルゴン下に2時間撹拌し、次いで水、NaHCO3水溶液で洗浄して、MgSO4で乾燥した。濾過し、減圧下に蒸発させて、(15)80mgを得た。シリカゲルによる精製において不安定であるため、これを更に精製することなく試験した。CI−HRMS:C16H1403N2BrS[M−H]+に対する計算値,392.9909(96%),394.9889(100%);実測値392.9911(24.7%),394.9895(24.2%).
【0110】
《N−(1−ベンゼンスルホニル−5−ブロモ−2−メチル−1H−インドール−3−イルメチル)−N−ヒドロキシホルムアミド(16).》
ギ酸(3.77mL,0.1mol)を、0℃で、無水酢酸(9.45mL,0.1mol)に滴下した。次いで混合物を50℃に2時間加熱し、室温まで放置した。CH2Cl210mLに溶解した(15)(80mg,0.2mmol)を、室温で前の溶液に滴下した。一晩撹拌後、真空下に蒸発させて、油状物を得、これを、シリカゲル上で数滴の酢酸存在下にCH2Cl2/C6H1299.5/0.5(vv混合物)で溶離するクロマトグラフを行なった。蒸発後、エーテル溶液からペンタン中に沈殿させて(16)50mg(収率60%)を得た。1HNMR(250MHz,[D6]アセトン):δ□272□3□;4.81(s,2H);7.47(dd,3J=8.8,4J=1.9,1H);7.63(t,J=7.5,2H);7.73(d,J=7.5,1H);7.8(d,J=1.9,1H);7.94(d,J=7.5,2H);8.13(d,J=8.8,1H);8.35(s,1H);9.10(s,1H).FAB+−MS:423,425,[M+H]+,10%;461,463,[M+NH4+],10%.CI−HRMS:C17H19O4N3BrS[M+NH4+]に対する計算値,440.0280(95.6%),442.0260(100%);実測値440.0292(86.4%),442.0247(92.9%).
【化19】
【0111】
《(1−ヒドロキシカルバモイルメチル−2−フェニル−エチル)−カルバミン酸tert−ブチルエステル(18)》
ヒドロキサム酸合成の通常の手順に従い、3−tert−ブトキシカルボニルアミノ−4−フェニル−酪酸(17)(0.308mmoL,M=279.33,m=86mg)から出発して、(18)50mgを収率55%で製造した。1HNMR(250MHz,[D6]DMSO):δ1.36(s,9H);2.16(m,2H);2.72(m,2H);3.99(m,1H),6.71(d,J=8.6,1H);7.19−7.35(m,5H);8.83(s,1H);10.40(s,1H).元素分析.C14H20N2O4.0.25H2Oに対する計算値:C,60.29;H,7.59;N,9.37.実測値:C,60.19;H,7.38;N,9.58.
【0112】
《(1−ヒドロキシカルバモイルメチル−2−フェニル−エチル)−カルバミン酸tert−ブチルエステル(20)》
Xiang et al.により記述されたようにして[Xiang,2002#1007]、(1−ホルミル−2−フェニル−エチル)−カルバミン酸tert−ブチルエステル(19)から化合物(20)を合成した。IR(cm−1):1682(νCO).1HNMR(250MHz,CDCl3):δ1.38(s,1H);2.83(d,J=6.7,2H);3.08−4.01(m,2H);4.19(m,1H);4.61(d,J=3.9,1H);7.15−7.80(m,5H);8.32(s,1H);8.85(s,1H).元素分析.C15H22N2O4.0.2H2Oに対する計算値:C,60.47;H,7.58;N,9.40.実測値:C,60.67;H,7.57;N,9.31.
【図面の簡単な説明】
【0113】
【図1A】15N−標識した大腸菌(E.coli)ペプチドデホルミラーゼ(1mM)上の2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(挿入図)のHSQC(異核種単量子相関(Heteronuclear Single Quantum Correlation))フットプリント。リファレンススペクトルは黒色輪郭線で示され、化学量論的量の化合物存在下のスペクトルは灰色でオーバーレイされる。著しく摂動されるアミド基がラベルされる。
【図1B】大腸菌(E.coli)ペプチドデホルミラーゼの3D構造。アミノ酸残基のアミド基NMRシフトが2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドの結合により摂動されるアミノ酸残基は、赤色で示される(図1Aでラベルされた残基)。触媒の金属イオンは球体として示される。
【図1C】大腸菌(E.coli)のPDF(100μM)から2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(1mM)への磁化移動を示すNMR飽和移動差スペクトル実験(STD)。上の図:混合物のリファレンススペクトル。星印でラベルしたピークは、緩衝液のシグナルに相当する。番号を付したピークは、図1Aでラベルした2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドに由来するシグナルに相当する。下の図:STDシグナル。矢印は、照射されたスペクトル領域を示す。正規化したSTD強度が示される。
【図2】本発明による2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド化合物が緑膿菌(Pseudomonas aeruginosa)ペプチドデホルミラーゼ酵素のS’1ポケットに結合している三次元モデルの三つの異なる視点から見た図であり、アクチノニンの構造及びベンザチオジオン−ヒドロキサム酸誘導体(benzathiozinone-hydroxamic derivative)の構造と重ね合わせてある。
【図3A】def遺伝子がPBADプロモーターの制御下にある大腸菌(Escherichiacoli)株について、アラビノース濃度(横軸,%)に関して、それぞれ、アクチノニン及び2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドのMIC(縦軸,μg/ml)を表す図である。
【図3B】def遺伝子がPBADプロモーターの制御下にある大腸菌(Escherichiacoli)株について、アラビノース濃度(横軸,%)に関して、それぞれ、アクチノニン及び2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミドのMIC(縦軸,μg/ml)を表す図である。
【図3C】ykrB遺伝子もdef遺伝子も不活性化されてしまっており、且つ2つの遺伝子のうち1つがPxylAプロモーターの制御下にあった枯草菌(Bacillus subilis)株について、キシロース濃度(横軸,%)に関して、菌株に対するアクチノニン(黒丸)及び2−(5−ブロモ−1H−インドール−3−イル)−N−ヒドロキシアセトアミド(黒四角)のMIC(縦軸,μg/ml)を表す図である。
【0114】
[実施例]
《使用した略語》
枯草菌(Bacillus subtilis),Bs;大腸菌(Escherichia coli),Ec;N−ホルミル,Fo;Met−tRNAfMetトランスホルミラーゼ,FMT;擬似結合定数,IC50;インドール誘導体,ID;ノックアウト,KO;最小阻止濃度,MIC;メタロプロテアーゼ,MP;阻害定数又は解離定数,Kd;マトリックスメタロプロテアーゼ,MMPs;ペプチドデホルミラーゼ,PDF;ペプチドデホルミラーゼ阻害物質,PDFI;プロテイン・データ・バンク,PDB;構造活性相関,SAR;飽和移動差スペクトル,STD;三次元,3D.
【実施例1】
【0115】
NMRによるメディアムスループットPDFIスクリーニングは、インドール誘導体をPDFの低親和性リガンドとして明らかにする
《材料及び方法》
15N−標識Ni−EcPDF1を、記述されたようにして得て(Dardelet al.,1998)、10mM HEPES−HCl(pH7.0)に溶解し、最終濃度を1mMとした。NMR実験は、3mm三重共鳴フローインジェクション・プローブを備えたBruker Avance 600MHz NMRスペクトロメーター上に記録された。プローブは、NMRコンソール(Bruker BEST(登録商標)システム)により制御されるGilsonリキッドハンドラーに連結される。インジェクション・プロトコルは、前に記述されたとおりである(Tisne et al.,2002)。化学シフト摂動実験のために、1mM 15N−標識EcPDF1(PDF1B)90μlを、96ウェルプレート中で同じ緩衝液に溶解した等容量の試験リガンド(3mM)と混合し、インジェクションの前に、Gilson 242 Peltierラック上、4℃で冷却した。飽和移動差スペクトル(STD,Mayer et al.,2001)実験のために、標識しないEcPDF1(PDF1B)又はBsPDF2(PDF2)を使用した。タンパク質及びリガンドの最終濃度をそれぞれ20μM及び1mMとした以外は、類似した緩衝液及びインジェクション容量を使用した。STD照射は、磁場の強さγB1/2π=100Hzを用いて、プロテイン・メチル・マッシーフ(0.5ppm)上のキャリヤセットで、1秒又は2秒間照射することにより行なった。
【0116】
《結果》
PDFに関して、ペプチドデホルミラーゼ阻害物質(PDFI)の結合能力は、本質的に2つの化学基により引き起こされる:
(i)金属結合基、及び
(ii)PDFのS1’ポケットに結合するP1’基(Boularotet al.,2004)。
いずれかの低親和性基の結合によるエントロピー増大は、ナノモル範囲の阻害定数を有する強力なPDFIを作り出す。
細菌PDFs(PDF1Bs及びPDF2s)のS1’ポケットは、n−ブチル基、n−ペンチル基、n−ヘキシル基、n−フェニル基(Ragusa et al.,1999;Molteni et al.,2004)及び他の環状側鎖(Boularot et al.,2004の引用文献を参照のこと)を、低選択性で受け入れることが知られている。以前の解析では、ミトコンドリアのPDF(PDF1A)は、例えばフェニル誘導体などの環状化合物を許容できない修正S1’ポケットを表示することが示された(Serero et al.,2003;Serero et al.,2001)。選択的PDFI(すなわちミトコンドリアのPDFをターゲッティングしないPDFI)を実証するために、環状P1’基を有する環状化合物を同定するという研究方法が、最も適当であると思われた。従って、本発明者らは、低結合定数でさえも細菌のPDFに結合するような環状化合物を同定するよう努力した。
従って、NMRによりそのような化学基を同定するために、そのような化合物のメディアムスケール・スクリーニングを設定した。フローインジェクションNMRプローブと連結されたピペッティング・ロボットを使用して、化合物ライブラリーを、連続して15N−標識EcPDFと混合し、インジェクションを行なった。化学シフト摂動は、HSQC−スペクトルを記録することによりモニターして、これを既に記述された非結合酵素で得られた対照データ(Meinnel et al.,1996;Meinnel et al.,1999)と比較した。化合物を添加して{1H−15N}HSQCタンパク質スペクトルの共鳴広がり及びシフトを引き起こすような化合物が同定された。影響されたアミド基を同定し、EcPDFの3D構造のバックボーンに位置決めした(例えば図1A参照)。
このスクリーニングから、インドール誘導体が酵素のS1’ポケットに対応する化学変化パターンを示したので、インドール誘導体が最も興味あるものとして浮上してきた(図1B)。相互に、これらのインドール誘導体のEcPDF1又はBsPDFいずれかに結合する様式の情報を得るために、リガンド観察飽和移動鎖スペクトル(STD)実験(Mayer et al.,2001)を行なった(図1C)。
例えば、化合物6bについて両方の場合に類似した結果が観察され(下の実施例2参照)、それは、インドール部分の4位について最も強いSTD効果が観察されたこと、及び3位に結合したメチレン基について最も弱いSTD効果が観察されたことを示した(図1C)。このことは、インドール基がPDF1B酵素にもPDF2酵素にも結合したことを確証したものであり、4位がタンパク質からの最も強い飽和移動を示したので、4位が最も深く埋め込まれていることを示唆したものである。
インドール及び5−ブロモインドールは、NMR滴定により調べられたようにミリモル範囲の結合定数を示した。この弱い結合定数は、細菌PDF(EcPDF1B及びBsPDF2)並びにミトコンドリアPDF(AtPDF1A)に対する阻害アッセイによって確認された(手順については実施例4を参照のこと)。
【0117】
インドール誘導体(ID)の効力を改良するために、本発明者らは、インドール誘導体の構造活性相関の研究に着手した。従って、下にラベルしたように位置1(R1)、2(R2)、3(R3)、4(R4)、5(R5)、6(R6)及び7(R7)のインドリル複素環に置換基を導入した。
【化20】
P1’(R5)側鎖に対応する4、5及び6位を、S1’ポケット中への適合性を改良するために、特にブロモ基などの親油基により置換した。2位及び3位においては、任意に短いCH2スペーサを介してインドール環に結合するいくつかの金属結合基を探索した。これには、カルボン酸、オキシム及びヒドロキシルアミンなどの単座官能基、並びにヒドロキシメチルケトン、チオメチルケトン及びヒドロキサム酸などの二座官能基が含まれた。最後に、1位は、あいたままにしたか又はベンジルオキシカルボニル残基などの諸残基により保護したが、これはP2’及び/又はP3’アミノ−酸基を模倣することを期待したものである(様々な酵素サブ部位の命名法についてはBoularot et al.,2004;Schechter et al.,1967を参照のこと)。
ヒドロキサム酸金属キレート基を3位に、臭素原子を5位に有する、そのようなインドール誘導体の2例を下に示し、それらのPDFに対する阻害作用及びそれらの抗菌特性も下に示した。
【実施例2】
【0118】
《インドール−ヒドロキサム酸誘導体のインビトロアッセイ》
様々な起源のPDFに対する本発明の化合物6b及び6dのインビトロ阻害プロファイルを評価して、アクチノニン並びに2つの合成PDF阻害物質であるRAS270及びRAS238のインビトロ阻害プロファイルと比較した。RAS358は、前に記述されたとおりに合成した(WO02/070653)。化合物RAS358は、リバースヒドロキサム酸化合物である。RAS270は、RAS358のヒドロキサメート誘導体に相当する。
3−tert−ブトキシカルボニルアミノ−4−フェニル−酪酸(市販品として入手可能)(0.308mmoL,M=279.33,m=86mg)から出発して、通常のヒドロキサム酸合成手順に従い、RAS270を50mg、収率55%で製造した。
【化21】
【0119】
《材料及び方法》
タンパク質分析のためのすべての化学薬品及び酵素はSigmaから購入した。ペプチドはBachem AGからのものであった。DNA操作のための酵素はNew England Biolabsから購入した。オリゴヌクレオチドはMWG−AG Biotechで合成された。
大腸菌(Escherichia coli)PDF(EcPDF1B)及びバチルスステアロサーモフィラス(Bacillus stearothermophilus)PDF2(BsPDF2)は、いずれかの細菌PDFクラスの代表として使用し、ニッケルイオン存在下に記述されたとおりに精製した(Ragusa et al.,1998)。シロイヌナズナ(Arabidospis thaliana)ミトコンドリアPDF(AtPDF1A)及びヒトミトコンドリアPDF(HsPDF1)は、記述されたとおりに精製した(Serero et al.,2001;Serero et al.,2003)。PDF活性を評価するために、PDF活性とギ酸脱水素酵素活性を共役させるアッセイを用いた。NADHの340nmでの吸光度(εM=6,300M−1.cm−1)を、37℃で、本質的に前に記述されたとおりにしてモニターした(Lazennec et al.,1997)。反応は、精製した酵素5〜15μlを添加して開始した。それぞれの場合、速度論的パラメーターは、前に記述されたとおりにして(Serero et al.,2001)、実験データを用いて、Michaelis−Menten式の逐次非線形最小二乗法から得た(Dardel et al.,1994)。阻害物質はすべてジメチルスルホキシドで希釈し、最終アッセイ緩衝液はこの溶媒10%を含有した。
【0120】
《結果》
結果を次の表1に示す:
【表1】
DF1Bに関する6b及び6dのIC50値は、アクチノニンの値より約1桁大きく、RAS270及びRAS358と同程度の大きさである。PDF2については、6b及び6dのIC50値は、アクチノニンの値と同程度であり、RAS270及びRAS358の値より1桁を超えて小さい。ミトコンドリアPDFsについては、アクチノニンが強力な阻害作用を示すのに対して、6b及び6dの阻害作用は特に低く、その結果前記化合物は全く阻害作用を欠いているということができる。
更に、6bが両方のPDF型に遅い結合様式を示すという事実は、酵素の阻害物質誘導適合コンホメーション変化を示していることに注意すべきである。
【実施例3】
【0121】
《インドール−ヒドロキサム酸誘導体のインビボアッセイ》
本発明の化合物6b及び6dのインビボ阻害プロファイルを評価し、アクチノニンのそれと比較した。
【0122】
《材料及び方法》
すべての細菌株は、Luria−Bertani(LB)培地(1%バクトトリプトン,0.5%酵母エキス,0.5%NaCl)で培養した。
株:2つの野生型又は擬似野生型枯草菌(B.subtilis)株を使用した。MO3482、すなわち、JH642の原栄養株skinプロファージ展覧誘導株(P.Stragier,IBPC,Franceの親切な供与)及び168(trpC2)(Anagnostopoulos et al.,1961)である。MHY101株(ΔykrB::nm,trpC2)及びMHD101株(Δdef::nm,trpC2)は、168株から得られ、前に記述されたものである(Haas et al.,2001)。更に2つの枯草菌(B.subtilis)株では、両方のデホルミラーゼの野生型遺伝子座(def及びykrB)が欠失しており、1つのデホルミラーゼバージョンがキシロース制御下に条件付で発現されるのであるが、そのような2つの枯草菌も使用した(Haas et al.,2001):MHY103(Δdef::nm;ΔykrB::erm;ΔthrC::xylR−PxylA−ykrB−spc,trpC2)及びMHD103(Δdef::erm;ΔykrB::nm;ΔthrC::xylR−PxylA−def−spc,trpC2)である。138株及びその誘導株は、C.Freiberg(Bayer AG,Germany)の親切な供与であった。大腸菌(E.coli)株JM101Tr(galK,rpsL,recA56,srl−300::Tn10)及びCAG1284(λ−,tolC210::Tn10,rph−)は、他のところで記述されている(Hirel et al.,1988;Singer et al.,1989).
大腸菌(E.coli)CAG1284感受性試験:tolC株CAG1284の薬剤感受性を測定するために感受性試験を計画した。EcPDF1のオープンリーディングフレームを、6−Hisタグ付きのフレームのC末端を有するpBADベクター(Invitrogen)中にクローニングした。このクローニングは、EcPDF1の合成をアラビノース濃度依存性にする(Guzman et al.,1995)。CAG1284株は、クロラムフェニコールを含むいくつかの抗生物質に対して感受性が高い(Sulavik et al.,2001)が、最初にpBADコンストラクトで形質転換されて、50μg/mlアンピシリン、3.4μg/mlクロラムフェニコール及び0.5%グルコースを補給したLB培地で選択された。細菌を、この培地中37℃で一晩培養し、次いで培養液を1:100に希釈して、培地3mlを接種するために使用した。OD600が0.9に達した時、懸濁液を適当に希釈して、100μl中2x104個の細菌を、ペトリ皿中、50μg/mlアンピシリン、アクチノニン(0.1〜3μM)及びグルコース(0.5%)又はアラビノース(0.0002〜0.2%)を補給した30mlの固体LB上に層状に重ねた。最小阻止濃度は、37℃で18時間インキュベーション後、全く増殖しなくなるアクチノニンの最低濃度として定義された。
【0123】
《結果》
1.グラム陰性細菌の阻害
2つの大腸菌(E.coli)誘導株(JM101Tr及びCAG1284)は、まず最初に化合物6bの最小阻止濃度(MIC)値への洞察を得るために並びにその値を天然の強力なPDFIアクチノニンの値(Chen et al.,2000)及びRAS270の値と比較するために使用した。
WT株(JM101Tr)については、アクチノニン及び6b両方のMICは類似しており、30〜40μg/mlの範囲にあったのに対して、RAS270のMICは300μg/mlを超えていた(表2)。
アクチノニンはAcrAB−tolC排出ポンプにより強く解毒されるので、tolC変異株(CAG1284株)を用いて、これが6bについても同じであるかどうかを調べた。アクチノニンの値が300倍低下したのに対して、6bのMICの値が唯の5.5倍だけわずかに低下したことが観察された(表2)。このことは、本発明のPDF阻害物質では細菌の排出系による解毒が不十分であることを示している。
【表2】
MICはまた、def遺伝子の発現がPBADプロモーターの制御下にあるというtolCコンテクストにおいても測定された。従って、PDF濃度増大のMICに対する影響力は、このプロモーターがアラビノース濃度増加により誘導されるおかげで、調べられた(Guzman et al.,1995;Chen et al.,2000)。アクチノニンの場合には、アクチノニン濃度についてほとんど直線的なMIC増加が存在する(図3A)。これは、(i)PDFが、アクチノニンの一次標的であること、及び(ii)PDF濃度が、排出ポンプ不在下にMICを限定する主要因子であることを示している。6bが同じ遺伝的背景において吟味されたとき、2種類のMIC曲線が観察された(図3B)。曲線の最初の部分は直線的であり、PDFが6bの標的であることを示す。65μg/mlを超えると飽和曲線が観察された。この値は、WTのMIC値と類似しており、膜透過性が6bの作用の律速段階であることを示す。
【0124】
2.グラム陽性細菌の阻害
第2系列の実験において、PDF1BもPDF2も発現するグラム陽性モデル生物である枯草菌(B.subtilis)を試験した。この場合もまたアクチノニンのMICを6b及び6dのMICと比較した。
WTコンテクスト(168株及びMO3482株)において、6b及び6dのMIC(20μg/ml)は、アクチノニンのMICよりも1桁高い大きさであった。これに対して、PDF2に対する低い結合能力から予想されるように、両フェニル誘導体RAS270及びRAS358は枯草菌に対して全く抗菌特性を有しなかった(表3)。
次いで、いずれかのPDFを発現する2つの枯草菌株誘導株(MHD101及びMHY101)に対する6b及び6dの影響力も研究した(表3)。
アクチノニン、6b及び6dのMICが、ykrB遺伝子不活性化コンテクストにおける減少値と、同じ傾向に従うことが見出された。これは、def遺伝子産物が、ykrBよりも少なく発現されたことを確証し(Haasetal.,2001)、6b及び6dが、実際にPDF1BをもPDF2をもターゲッティングしたことを示唆した。
【表3】
6b及び6dが枯草菌のPDFsを特異的にターゲッティングすることを証明するために、ykrB及びdefの両遺伝子が既に不活性化されており、且つ2つの遺伝子のうち1つがXylRプロモーターPxylAの制御下に置かれている株を使用した。これらの条件下に、PDF1B又はPDF2発現は、L−キシロースに依存している(Haas et al.,2001)。アクチノニンも6bもMICが、MHD103株かMYD103株かいずれかにおいて、L−キシロース濃度に依存することが観察された(図3C)。
【0125】
最後に、枯草菌における6b及びアクチノニンに対する耐性の出現を測定し、比較した。類似した値(5.10−7対6.10−7)が観察されたが、この結果は、他人の以前の測定値及びバチルス種(Bacillus spp)におけるトランスホルミラーゼ遺伝子不活性化により誘導されるホルミル化−脱ホルミル化経路のバイパス(Giglione et al.,2001に総説がある)と一致している。このデータは、6bの標的がPDFであることを示すもう1つの強い論拠であった。
【実施例4】
【0126】
ヒドロキサム酸インドール誘導体のPDFへの結合の三次元モデリング
《材料及び方法》
三次元(3D)モデリングは、InsightIIソフトウェア(Accelrys)を用いて行なった。ベンザチオジオン−ヒドロキサム酸誘導体(benzathiozinone-hydroxamic derivative)(BTH)に結合された緑膿菌(Pseudomonas aeruginosa)PDF(PaPDF1)の3D構造(PDB entry 1S17,Molteni et al.,2004)及びアクチノニンに結合されたPaPDF1の3D構造(PDB entry 1LRY,Guilloteau et al.,2002)を並べた。化合物6bをSketcherモジュールで組み立て、固定アンカーとしてヒドロキサメート基を用いて、アクチノニン及び化合物BTHの両方の構造上に並べ、そして3D構造において、いずれの化合物も置き換えるように使用した。PaPDF1にドッキングした6bの構造は、CharmM力場を用いて更に最小化し、最低エネルギー構造を選択した。
【0127】
《結果》
PDF1の活性部位における化合物6bの3Dモデリングは、それがS1’ポケットに完全に適合すること、そして臭化物基がn−ブチル基又はメチオニン側鎖、すなわち、S1’ポケットに受け入れられる最適側鎖、のC4メチル基を模倣することを明らかにした(図2)。更に、このモデルは、インドール部分の4位をS1’ポケットの内側深くに収納し、His132側鎖の下に埋め込まれた状態にした。これは実施例1で議論したSTDデータと一致している。
同時に、これらのデータは、インドール誘導体が抗菌活性を表すが、しかし動物のミトコンドリアPDFsを阻害しないことをも示している。
【実施例5】
【0128】
《シロイヌナズナの成長阻害》
シロイヌナズナ(生態型WS4)成長阻害は、6b又は6dについて50μMより高濃度で観察される。その上、AtPDF1BのIC50値は、化合物6b及び6dの場合にEcPDF1Bで観察される値と類似している。実験手法の詳細は、Serero et al.(2001)及びGiglione et al.(2003)に出ている。
【0129】
従って、本発明者らは、2つの実施例(6b及び6d)により、本発明のインドール化合物が:
(i)特にグラム陰性細菌及びグラム陽性細菌において、特異的にPDF1及びPDF2をターゲッティングすること、
(ii)ミトコンドリアPDFsに対して本質的に全く阻害活性を示さないこと
を証明した。
化合物6a、6g、6f、6i、6e、6h、6j、11、15、及び16で得た結果を、次の表4に示す。
【表4】
【0130】
《引用文献》
Anagnostopoulos,C.,and I.P.Crawford.1961.Transformation studies on the linkage of markers in the tryptophan pathway in Bacillus subtilis.Proc.Natl.Acad.Sci.U.S.A.47:378−90.
Borch,R.F.,M.D.Bernstein,and H.Dupont Durst.1971.The cyano hydridoborate anion as a selective reducing agent.J.Am.Chem.Soc.93:2897−2904.3.
Boularot,A.,C.Giglione,I.Artaud,andT.Meinnel.2004.Structure−activity relationship and therapeutic potential of peptide deformylase inhibitors.Curr.Opin.Invest.Drugs 5:809−822.4.
Bracchi−Ricard,V.,K.T.Nguyen,Y.Zhou,P.T.Rajagopalan,D.Chakrabarti,and D.Pei.2001.Characterization of an eukaryotic peptidedeformylase from Plasmodium falciparum.Arch.Biochem.Biophys.396:162−170.
Chen,D.Z.,D.V.Patel,C.J.Hackbarth,W.Wang,G.Dreyer,D.C.Young,P.S.Margolis,C.Wu,Z.J.Ni,J.Trias,R.J.White,and Z.Yuan.2000.Actinonin,a naturally occurring antibacterial agent,is a potent deformylase inhibitor.Biochemistry39:1256−1262.
Chu,M.,R.Mierza,L.He,L.Xu,F.Gentile,J.Taerracciano,M.Patel,L.Miesel,S.Bohanon,C.Kravec,C.Cramer,T.O.Fischman,A.Hruza,L.Ramanatan,P.Shipkova,and T.−M.Chan.2001.Isolation and structure elucidation of two novel deformylase inhibitors produced by Streptomyces sp.Tetrahedron Lett.42:3549−3451.
Dardel,F.1994.MC−Fit:Using Monte−Carlo methods to get accurate confidence limits on enzyme parameters.Comput.Appl.Biosci.10:273−275.
Dardel,F.,S.Ragusa,C.Lazennec,S.Blanquet,and T.Meinnel.1998.Solution structure of nickel−peptide deformylase.J.Mol.Biol.280:501−513.
Dillard,R.D.,N.J.Bach,S.E.Draheim,D.R.Berry,D.G.Carlson,N.Y.Chirgadze,D.K.Clawson,L.W.Hartley,L.M.Johnson,N.D.Jones,E.R.McKinney,E.D.Mihelich,J.L.Olkowski,R.W.Schevitz,A.C.Smith,D.W.Snyder,C.D.Sommers,and J.P.Wery.1996.Indole inhibitors of human nonpancreatic secretory phospholipase A2.1.Indole−3−acetamides.J.Med.Chem.39:5119−36.
Dirk,L.M.,M.A.Williams,and R.L.Houtz.2001.Eukaryotic peptidede formylases.Nuclear−encoded and chloroplast−targeted enzymes in Arabidopsis.Plant Physiol.127:97−107.
Eissenstat,M.A.,M.R.Bell,T.E.D’Ambra,E.J.Alexander,S.J.Daum,J.H.Ackerman,M.D.Gruett,V.Kumar,K.G.Estep,E.M.Olefirowicz,and etal.1995.Aminoalkylindoles:structure−activity relationships of novel cannabinoid mimetics.J.Med.Chem.38:3094−105.
Giglione,C.,and T.Meinnel.2001.Peptide deformylase as an emerging target for antiparasitic agents.Emerg.Therap.Targets 5:41−57.
Giglione,C.,M.Pierre,and T.Meinnel.2000a.Peptide deformylase as a target for new generation,broad spectrum antimicrobial agents.Mol.Microbiol.36:1197−1205.
Giglione,C.,A.Serero,M.Pierre,B.Boisson,and T.Meinnel.2000b.Identification of eukaryotic peptide deformylases reveals universality of N−terminal protein processing mechanisms.EMBOJ.19:5916−5929.
Giglione,C.,O.Vallon,and T.Meinnel.2003.Control of protein life−span by N−terminal methionine excision.EMBO J.22:13−23.
Giglione,C.,A.Boularot,and T.Meinnel.2004.Protein N−terminal methionine excision.Cell.Mol.Life Sci.61:1455−1474.
Gordon,J.J.,B.K.Kelly,and G.A.Miller.1962.Actinonin:an antibiotic substance produced by an actinomycete.Nature 195:701−702.
Grujic,M.,and M.Renko.2002.Aminopeptidase inhibitors bestatin and actinonin inhibit cell proliferation of myeloma cells predominantly by intracellular interactions.Cancer Lett182:113−9.
Guilloteau,J.P.,M.Mathieu,C.Giglione,V.Blanc,A.Dupuy,M.Chevrier,P.Gil,A.Famechon,T.Meinnel,and V.Mikol.2002.The crystal structures of four peptide deformylases bound to the antibiotic actinonin reveal two distinct types:a platform for the structure−based design of antibacterial agents.J.Mol.Biol.320:951−962.
Guzman,L.M.,D.Belin,M.J.Carson,and J.Beckwith.1995.Tight regulation,modulation,and high−level expression by vectors containing the arabinose PBAD promoter.J.Bacteriol.177:4121−4130.
Haas,M.,D.Beyer,R.Gahlmann,and C.Freiberg.2001.YkrB is the main peptide deformylase in Bacillus subtilis,a eubacterium containing two functional peptide deformylases.Microbiology147:1783−1791.
Hasan,I.,E.R.Marinelli,L.C.ChangLin,F.W.Fowler,and A.B.Levy.1981.Synthesis and reactions of N−protected 2−lithiated pyrroles and indoles.The tert−Butoxycarbonyl substituent as a protecting group.J.Org.Chem.46:157−164.
Hill,D.R.,C.N.Hsiao,R.Kurukulasuriya,and S.J.Wittenberger.2002.2,2,2−trifluoroethyl formate:a versatile and selective reagent for the formylation of alcohols,amines,and N−hydroxylamines.Org.Lett.4:111−3.
Hirel,P.−H.,F.Leveque,P.Mellot,F.Dardel,M.Panvert,Y.Mechulam,and G.Fayat.1988.Genetic engineering of methionyl−tRNA synthetase:in vitro regeneration of an active synthetase by proteolytic cleavage of a methionyl−tRNA synthetase−βgalactosidase chimeric protein.Biochimie 70:773−782.
Horwell,D.C.,D.Naylor,and H.M.G.Wilems.1997.2,3−disubstituted 2−azanorbornanes as polar β−turn mimetics.Bioorg.Med.Chem.Lett.7:31−36.
Jiang,B.,and X.H.Gu.2000.Syntheses and cytotoxicity evaluation of bis(indolyl)thiazole,bis(indolyl)pyrazinone and bis(indolyl)pyrazine:analogues of cytotoxic marine bis(indole)alkaloid.Bioorg.Med.Chem.Lett.8:363−71.
Kolossa,T.,Brooks C.D.W.,Rodrigues K.E.,Summers J.B.,Dellaria J.F.,Hulkower,K.I.,Bouska,J.,Bell,R.L.and Carter G.W.1997.Non−steroidal anti−inflammatory drugs as scaffolds for the dzsign of 5−lipoxygenase inhibitors.J.Med.Chem.40,819−824.
Lazennec,C.,and T.Meinnel.1997.Formate dehydrogenase−coupled spectrophotometric assay of peptide deformylase.Anal.Biochem.244:180−182.
Leclerc,V.,E.Fourmaintraux,P.Depreux,D.Lesieur,P.Morgan,H.E.Howell,P.Renard,D.H.Caignard,B.Pfeiffer,P.Delagrange,B.Guardiola−Lemaitre,and J.Andrieux.1998.Synthesis and structure−activity relationships of novel naphthalenic and bioisosteric related amidic derivatives as melatonin receptor ligands.Bioorg.Med.Chem.6:1875−87.
Lee,M.D.,C.Antczak,Y.Li,F.M.Sirotnak,W.G.Bornmann,and D.A.Scheinberg.2003.A new human peptide deformylase inhibitable by actinonin.Biochem.Biophys.Res.Commun.312:309−15.
Lee MD,She Y,Soskis MJ,Borella CP,Gardner JR,Hayes PA,Dy BM,Heaney ML,Philips MR,Bornmann WG,Sirotnak FM,Scheinberg DA(2004)Human mitochondrial peptide deformylase,a new anticancer target of actinonin−based antibiotics.J Clin Invest 114:1107−1116
Lefevre−Groboillot,D.,S.Dijols,J.L.Boucher,J.P.Mahy,R.Ricoux,A.Desbois,J.L.Zimmermann,and D.Mansuy.2001.N−hydroxyguanidines as new heme ligands:UV−visible,EPR,and resonance Raman studies of the interaction of various compounds bearing a C=NOHfunction with microperoxidase−8.Biochemistry 40:9909−17.
Mayer,M.,and B.Meyer.2001.Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor.J.Am.Chem.Soc.123:6108−17.
Mazel,D.,S.Pochet,and P.Marliere.1994.Genetic characterization of polypeptide deformylase,a distinctive enzyme of eubacterial translation.EMBO J.13:914−923.
Meinnel,T.2000.Peptide deformylase of eukaryotic protists:a target for new antiparasitic agents?Parasitol.Today16:165−168.
Meinnel,T.,S.Blanquet,and F.Dardel.1996.A new subclass of the zinc metalloproteases superfamily revealed by the solution structure of peptide deformylase.J.Mol.Biol.262:375−386.
Meinnel,T.,L.Patiny,S.Ragusa,and S.Blanquet.1999.Design and synthesis of substrate analogue inhibitors of peptide deformylase.Biochemistry 38:4287−4295.
Miesel,L.,J.Greene,and T.A.Black.2003.Genetic strategies for antibacterial drug discovery.Nat Rev Genet 4:442−56.
Molteni,V.,X.He,J.Nabakka,K.Yang,A.Kreusch,P.Gordon,B.Bursulaya,I.Warner,T.Shin,T.Biorac,N.S.Ryder,R.Goldberg,J.Doughty,and Y.He.2004.Identification of novel potent bicyclic peptide deformylase inhibitors.Bioorg.Med.Chem.Lett.14:1477−81.
Ragusa,S.,S.Blanquet,and T.Meinnel.1998.Control of peptide deformylase activity by metal cations.J.Mol.Biol.280:515−523.
Ragusa,S.,P.Mouchet,C.Lazennec,V.Dive,and T.Meinnel.1999.Substrate recognition and selectivity of peptide deformylase.Similarities and differences with metzincins and thermolysin.J.Mol.Biol.289:1445−1457.
Salim,M.,and A.Capretta.2000.Intramolecular carbenoid insertions:the reaction of α−diazoketones derived from pyrrolyl and indolyl carboxylic acids with rhodium(II) acetate.Tetrahedron56:8063−8069.41.
Schechter,I.,and A.Berger.1967.On the size of the active site in proteases.I.Papain.Bochem.Biophys.Res.Commun.27:157−162.
Serero,A.,C.Giglione,and T.Meinnel.2001.Distinctive features of the two classes of eukaryotic peptide deformylases.J.Mol.Biol 314:695−708.
Serero,A.,C.Giglione,A.Sardini,J.MartinezSanz,and T.Meinnel.2003.An unusual peptide deformylase features in the human mitochondrial N−terminal methionine excision pathway.J.Biol.Chem.278:52953−52963.
Singer,M.,T.A.Baker,G.Schnitzler,S.M.Deischel,M.Goel,W.Dove,K.J.Jaacks,A.D.Grossman,J.W.Erickson,and C.A.Gross.1989.A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli.Microbiol.Rev.53:1−24.
Sulavik,M.C.,C.Houseweart,C.Cramer,N.Jiwani,N.Murgolo,J.Greene,B.DiDomenico,K.J.Shaw,G.H.Miller,R.Hare,and G.Shimer.2001.Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes.Antimicrob.AgentsChemother.45:1126−36.
Tisne,C.,and F.Dardel.2002.Optimisation of a peptide library for screening specific RNA ligands by flow−injection NMR.Comb.Chem.High Throughput Screen.5:523−9.
Weller,D.D.,and D.W.Ford.1984.Synthesis of ellipticine.Tetrahedron Lett.25:2105−2108.
Whittaker,M.,C.D.Floyd,P.Brown,and A.J.H.Gearing.1999.Design and therapeutic application of matrix metalloproteinase inhibitors.Chem Rev.99:2735−2776.
Wiesner,J.,S.Sanderbrand,E.Beck,and H.Jomaa.2001.Seeking new targets for antiparasitic agents.Trends Parasitol.17:7.
Wissner,A.1979.2−hetero substituted silylated ketene acetals:reagents for the preparation of α−functionalized methyl ketones from carboxylic acid chlorides.J.Org.Chem.44:4717−4622.
Xu,Y.,L.T.Lai,J.L.Gabrilove,and D.A.Scheinberg.1998.Antitumor activity of actinonin in vitro and in vivo.Clin.Cancer Res.4:171−176.
【特許請求の範囲】
【請求項1】
a)下記一般式(I):
【化1】
(式中、
− 互いに独立して、R2及びR3は、H、炭素原子1〜5個のアルキル基、又は1〜5個のヘテロ原子を含む2〜20個の原子の金属キレート基を表し;
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し、R2及びR3のうちの少なくとも1つが、上に定義した金属キレート基を表す)
を有し;並びに
b)大腸菌ニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラスニッケル結合ペプチドデホルミラーゼ(配列番号:2)について約1μMよりも低いIC50を有する;化合物又は薬学的に許容可能なその塩の、
細菌感染症若しくは原生動物感染症の予防若しくは治療を対象とした医薬の製造のための使用、又は除草剤の製造のための使用。
【請求項2】
R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−(X)n1−Y
(式中、
− nは、整数0〜3であり、
− n1は、0又は1であり、
− Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
− Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド),−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
の金属キレート基で表される、請求項1に記載の一般式(I)の化合物の使用。
【請求項3】
R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、請求項1又は2に記載の一般式(I)の化合物の使用。
【請求項4】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるもので表される、請求項1〜3のいずれか一項に記載の、一般式(I)で表される化合物の使用。
【請求項5】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、請求項1〜4のいずれか一項に記載の一般式(I)で表される化合物の使用。
【請求項6】
R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるもので表される、請求項1〜5のいずれか一項に記載の、一般式(I)で表される化合物の使用。
【請求項7】
R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、請求項1〜6のいずれか一項に記載の、一般式(I)の化合物の使用。
【請求項8】
下記一般式(II):
【化2】
(式中、Roは、請求項1〜7に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は、請求項1〜7に定義したとおりである)
で表される、請求項1〜7のいずれか一項に記載の化合物の使用。
【請求項9】
下記一般式(III):
【化3】
(式中、Roは、請求項1〜7に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは、請求項1〜7に定義したとおりである)
で表される、請求項1〜8のいずれか一項に記載の化合物の使用。
【請求項10】
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す
、請求項9に記載の式(III)で表される化合物の使用。
【請求項11】
前記化合物が、下記諸式:
【化4】
に相当することを特徴とする、請求項1〜10のいずれか一項に記載の、一般式(I)で表される化合物の使用。
【請求項12】
前記原生動物が、アピコプラスト又はキネトプラストを有する原生動物のうちから選択され、特に、下記属の原生動物、すなわちプラスモジウム属(Plasmodium)、トキソプラズマ属(Toxoplasma)、サルコシスティス属(Sarcocystis)、クリプトスポリジウム属(Cryptosporidium)、エイメリア属(Eimera)、タイレリア属(Theileria)、バベシア属(Babesia)、トリパノゾーマ属(Trypanosoma)、リーシュマニア属(Leishmania)を含む群から選択される、請求項1〜11のいずれか一項に記載の一般式(I)の化合物の使用。
【請求項13】
前記細菌が、下記属の細菌、すなわちブドウ球菌属(Staphylococcus)、連鎖球菌属(Streptococcus)、バチルス属(Bacillus)、エンテロコッカス属(Enterococcus)、シュードモナス属(Pseudomonas)、アシネトバクター属(Acinetobacter)、ヘモフィルス属(Haemophilus)、エンテロバクター属(Enterobacter)を含む群から選択される、請求項1〜11のいずれか一項に記載の、一般式(I)の化合物の使用。
【請求項14】
活性物質として、請求項1〜11のいずれか一項に記載の少なくとも1つの化合物、又は薬学的に許容可能なその塩を、薬学的に許容可能な賦形剤とともに含むことを特徴とする医薬組成物。
【請求項15】
活性物質として、請求項1〜11のいずれか一項に記載の少なくとも1つの化合物を含むことを特徴とする除草剤組成物。
【請求項16】
一般式(I):
【化5】
(式中、
−R2及びR3は、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−(X)n1−Y
(式中、
*nは、整数0〜3であり、
*n1は、0又は1であり、
*Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
*Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド)、−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
で表される金属キレート基を表し、
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し:
*R2及びR3のうち少なくとも1つは、上に定義した金属キレート基を表し、及び
*R3が上に定義した金属キレート基を表すとき、−(CH2)n−(X)n1−Yは、−(CH2)2−NH2又は−(CH2)2−SHを表すことができない)
で表される化合物。
【請求項17】
R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、請求項16に記載の一般式(I)の化合物。
【請求項18】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、請求項16又は17に記載の一般式(I)の化合物。
【請求項19】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、請求項16〜18のいずれか一項に記載の一般式(I)の化合物。
【請求項20】
R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、請求項16〜19のいずれか一項に記載の一般式(I)で表される化合物。
【請求項21】
R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、請求項16〜20のいずれか一項に記載の一般式(I)の化合物。
【請求項22】
下記一般式(II):
【化6】
(式中、Roは、請求項16〜21に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は、請求項16〜21に定義したとおりである)
で表される請求項16〜21のいずれか一項に記載の化合物。
【請求項23】
下記一般式(III):
【化7】
(式中、Roは、請求項16〜21に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは請求項16〜21に定義したとおりである)
で表される請求項16〜22のいずれか一項に記載の化合物。
【請求項24】
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す、
請求項23に記載の式(III)の化合物。
【請求項25】
前記化合物が、下記諸式:
【化8】
に相当することを特徴とする、請求項16〜24のいずれか一項に記載の、一般式(I)の化合物。
【請求項26】
ペプチドデホルミラーゼ酵素をスクリーニングする化合物と接触させる工程、及び前記ペプチドデホルミラーゼ酵素に対する前記化合物の阻害活性を測定する工程を含むことを特徴とする、細菌又は原生動物感染症の予防若しくは治療、又は除草剤としての使用、を対象としたインドール構造を含む化合物をスクリーニングする方法。
【請求項1】
a)下記一般式(I):
【化1】
(式中、
− 互いに独立して、R2及びR3は、H、炭素原子1〜5個のアルキル基、又は1〜5個のヘテロ原子を含む2〜20個の原子の金属キレート基を表し;
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し、R2及びR3のうちの少なくとも1つが、上に定義した金属キレート基を表す)
を有し;並びに
b)大腸菌ニッケル結合ペプチドデホルミラーゼ(配列番号:1)及び/又はバチルスステアロサーモフィラスニッケル結合ペプチドデホルミラーゼ(配列番号:2)について約1μMよりも低いIC50を有する;化合物又は薬学的に許容可能なその塩の、
細菌感染症若しくは原生動物感染症の予防若しくは治療を対象とした医薬の製造のための使用、又は除草剤の製造のための使用。
【請求項2】
R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−(X)n1−Y
(式中、
− nは、整数0〜3であり、
− n1は、0又は1であり、
− Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
− Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド),−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
の金属キレート基で表される、請求項1に記載の一般式(I)の化合物の使用。
【請求項3】
R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、請求項1又は2に記載の一般式(I)の化合物の使用。
【請求項4】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるもので表される、請求項1〜3のいずれか一項に記載の、一般式(I)で表される化合物の使用。
【請求項5】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、請求項1〜4のいずれか一項に記載の一般式(I)で表される化合物の使用。
【請求項6】
R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基で置換されるもので表される、請求項1〜5のいずれか一項に記載の、一般式(I)で表される化合物の使用。
【請求項7】
R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、請求項1〜6のいずれか一項に記載の、一般式(I)の化合物の使用。
【請求項8】
下記一般式(II):
【化2】
(式中、Roは、請求項1〜7に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は、請求項1〜7に定義したとおりである)
で表される、請求項1〜7のいずれか一項に記載の化合物の使用。
【請求項9】
下記一般式(III):
【化3】
(式中、Roは、請求項1〜7に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは、請求項1〜7に定義したとおりである)
で表される、請求項1〜8のいずれか一項に記載の化合物の使用。
【請求項10】
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す
、請求項9に記載の式(III)で表される化合物の使用。
【請求項11】
前記化合物が、下記諸式:
【化4】
に相当することを特徴とする、請求項1〜10のいずれか一項に記載の、一般式(I)で表される化合物の使用。
【請求項12】
前記原生動物が、アピコプラスト又はキネトプラストを有する原生動物のうちから選択され、特に、下記属の原生動物、すなわちプラスモジウム属(Plasmodium)、トキソプラズマ属(Toxoplasma)、サルコシスティス属(Sarcocystis)、クリプトスポリジウム属(Cryptosporidium)、エイメリア属(Eimera)、タイレリア属(Theileria)、バベシア属(Babesia)、トリパノゾーマ属(Trypanosoma)、リーシュマニア属(Leishmania)を含む群から選択される、請求項1〜11のいずれか一項に記載の一般式(I)の化合物の使用。
【請求項13】
前記細菌が、下記属の細菌、すなわちブドウ球菌属(Staphylococcus)、連鎖球菌属(Streptococcus)、バチルス属(Bacillus)、エンテロコッカス属(Enterococcus)、シュードモナス属(Pseudomonas)、アシネトバクター属(Acinetobacter)、ヘモフィルス属(Haemophilus)、エンテロバクター属(Enterobacter)を含む群から選択される、請求項1〜11のいずれか一項に記載の、一般式(I)の化合物の使用。
【請求項14】
活性物質として、請求項1〜11のいずれか一項に記載の少なくとも1つの化合物、又は薬学的に許容可能なその塩を、薬学的に許容可能な賦形剤とともに含むことを特徴とする医薬組成物。
【請求項15】
活性物質として、請求項1〜11のいずれか一項に記載の少なくとも1つの化合物を含むことを特徴とする除草剤組成物。
【請求項16】
一般式(I):
【化5】
(式中、
−R2及びR3は、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−(X)n1−Y
(式中、
*nは、整数0〜3であり、
*n1は、0又は1であり、
*Xは、−O−、−NH−、−CHOH−、−CHF−、又は−CO−から選択される基を表し、及び
*Yは、−NH2(アミン)、−NHOH(ヒドロキシルアミン)、−CH=NOH(オキシム)、−NHCN(シアナミド)、−NH−C(NOH)NH2(ヒドロキシグアニジン)、−SCN(チオシアネート)、−SH(チオール)、−B(OH)2(ホウ酸)、−COOH(カルボン酸)、−COCH2OH(ヒドロキシメチルケトン)、−COCH2SH(チオメチルケトン)、−CONHOH(ヒドロキサム酸)、−SO2NHOH(スルホヒドロキサム酸)、−NO−N=O(NONOエート)、−NOH−COH(リバースヒドロキサム酸)を含むリストから選択される基を表す)
で表される金属キレート基を表し、
− 互いに独立して、R4、R5、R6、及びR7は、H、ハロゲン原子、又は1〜10個の炭素原子を含む基を表し;
− R1は、H、又は1〜50個の炭素原子を含む基を表し;
但し:
*R2及びR3のうち少なくとも1つは、上に定義した金属キレート基を表し、及び
*R3が上に定義した金属キレート基を表すとき、−(CH2)n−(X)n1−Yは、−(CH2)2−NH2又は−(CH2)2−SHを表すことができない)
で表される化合物。
【請求項17】
R2及びR3が、互いに独立して、H、メチル基、エチル基、プロピル基、又は下記式:
−(CH2)n−Y
(式中、
− nは、整数0〜3であり、及び
− Yは、−NHOH、−CONHOH、又は−NOH−COHを含むリストから選択される基を表す)
の金属キレート基で表される、請求項16に記載の一般式(I)の化合物。
【請求項18】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基又はアリールオキシカルボニル基を表し、適切な場合には、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、請求項16又は17に記載の一般式(I)の化合物。
【請求項19】
R4、R5、R6、及びR7が、互いに独立して、H、ハロゲン原子、アルキル基、又は炭素原子1〜10個のアルコキシ基で表される、請求項16〜18のいずれか一項に記載の一般式(I)の化合物。
【請求項20】
R1が、
− H、又は
− ペプチド配列、又は
− アルキル基、アルコキシ基、アセチル基、アルコキシカルボニル基、アリール基、アリールオキシ基、チオアルキル基、チオアリール基、アリールスルホニル基、アリールオキシカルボニル基、アルコキシカルボニルアルキル基、又はアリールオキシカルボニルアルキル基を表し、適切な場合には、ピリジン基、モルホリン基、チオモルホリン基、ピペリジン基、ピペラジン基、ヒドロキシル基、チオール基、チオアルキル基、チオアリール基、又はアミノ基により置換されるもので表される、請求項16〜19のいずれか一項に記載の一般式(I)で表される化合物。
【請求項21】
R1が、H、式−SO2−C6H5で表されるフェニルスルホニル基、又は式−CO−O−CH2−C6H5で表されるベンジルオキシカルボニル基で表される、請求項16〜20のいずれか一項に記載の一般式(I)の化合物。
【請求項22】
下記一般式(II):
【化6】
(式中、Roは、請求項16〜21に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びR3は、請求項16〜21に定義したとおりである)
で表される請求項16〜21のいずれか一項に記載の化合物。
【請求項23】
下記一般式(III):
【化7】
(式中、Roは、請求項16〜21に定義したR4、R5、R6、又はR7に相当し、並びにR1、R2、及びYは請求項16〜21に定義したとおりである)
で表される請求項16〜22のいずれか一項に記載の化合物。
【請求項24】
− Roが、H、ハロゲン原子(例えば、Cl、F又はBr)、炭素原子1〜10個のアルコキシ基(例えば、メトキシ基OCH3)を表し、
− R2が、H又はCH3を表し、
− Yが、−NHOH、−CONHOH、又は−NOH−COHを表し、及び
− R1が、H、−SO2−C6H5、又は−CO−O−CH2−C6H5を表す、
請求項23に記載の式(III)の化合物。
【請求項25】
前記化合物が、下記諸式:
【化8】
に相当することを特徴とする、請求項16〜24のいずれか一項に記載の、一般式(I)の化合物。
【請求項26】
ペプチドデホルミラーゼ酵素をスクリーニングする化合物と接触させる工程、及び前記ペプチドデホルミラーゼ酵素に対する前記化合物の阻害活性を測定する工程を含むことを特徴とする、細菌又は原生動物感染症の予防若しくは治療、又は除草剤としての使用、を対象としたインドール構造を含む化合物をスクリーニングする方法。
【図1A】

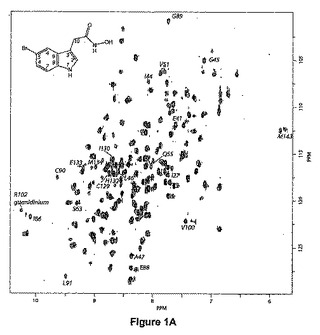
【図1B】
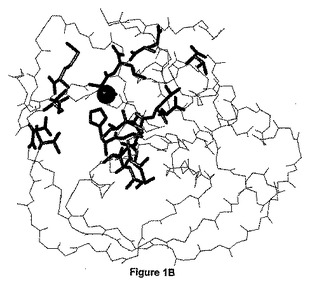
【図1C】
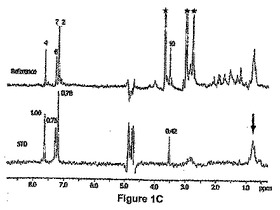
【図2】
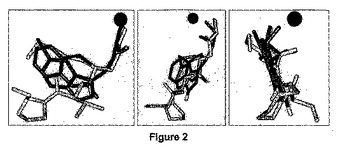
【図3A】
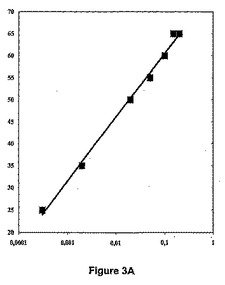
【図3B】
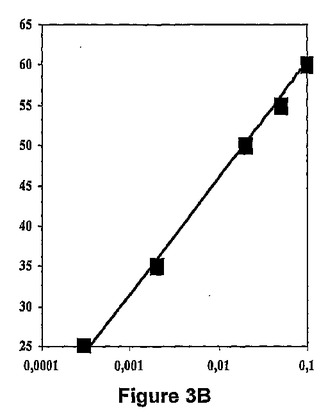
【図3C】
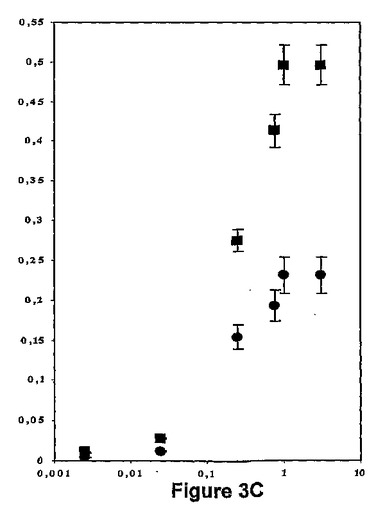
【図1B】
【図1C】
【図2】
【図3A】
【図3B】
【図3C】
【公表番号】特表2008−528455(P2008−528455A)
【公表日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願番号】特願2007−551621(P2007−551621)
【出願日】平成18年1月20日(2006.1.20)
【国際出願番号】PCT/EP2006/000508
【国際公開番号】WO2006/077139
【国際公開日】平成18年7月27日(2006.7.27)
【出願人】(505409247)サントル ナシオナル ドゥ ラ ルシェルシェ シアンティフィク (16)
【氏名又は名称原語表記】CENTRE NATIONAL DE LA RECHERCHE SCIENTIFIQUE
【出願人】(506413856)ユニヴェルシテ・ルネ・デカルト・パリ・サンク (6)
【Fターム(参考)】
【公表日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願日】平成18年1月20日(2006.1.20)
【国際出願番号】PCT/EP2006/000508
【国際公開番号】WO2006/077139
【国際公開日】平成18年7月27日(2006.7.27)
【出願人】(505409247)サントル ナシオナル ドゥ ラ ルシェルシェ シアンティフィク (16)
【氏名又は名称原語表記】CENTRE NATIONAL DE LA RECHERCHE SCIENTIFIQUE
【出願人】(506413856)ユニヴェルシテ・ルネ・デカルト・パリ・サンク (6)
【Fターム(参考)】
[ Back to top ]