
ペプチドリガンドによる指向性ドラッグデリバリー
EGFRの3D結晶構造に基づいて得られた、EGFRの表面ポケットに結合する新規のペプチドリガンド(Leu−Ala−Arg−Leu−Leu−Thr)を提供する。上記ペプチドリガンドがリポソーム表面のPEG部分の末端にコンジュゲートした場合、EGFR高発現癌細胞(H1299及びSPCA1)によるリポソームの結合及び取込みは特異的且つ効率的に誘導される。リポソームに含まれる抗癌剤ドキソルビシンの標的指向性デリバリーによって、in vitroの細胞に対するより優れた治療効果が得られる。In vivoでは、上記標的指向性リポソームを尾静脈から注射し、異種移植された腫瘍組織中のそれらの分布及び蓄積を、生体蛍光イメージングシステムを用いて経時的に観察する。LARLLTによる指向性リポソームは腫瘍部分に次第に濃縮され、注射後80時間以上、選択的に保持されることが示された。
【発明の詳細な説明】
【背景技術】
【0001】
癌治療において、化学療法は広く用いられているが、その有効性は、正常な組織に対する薬剤毒性に起因する重篤な副作用によって損なわれることも多い。これまでに多くの研究が、化学療法剤の治療指数を改善することを目的とした、正常組織を回避して腫瘍組織に特異的に薬剤を送達するための戦略に焦点を当ててきた。
【0002】
いくつかのリポソーム化抗癌製剤が開発されており、それらは既に、リポソーム化されていない製剤より治療効果が高く、副作用の少ない製剤として患者に用いられている。最も治療効果の高いリポソーム化抗癌製剤の1つは、ドキシル(カエリックスとしても知られる)である(非特許文献1)。ドキシルは、PEGでコートされたリポソーム(ステルスリポソーム)を含んでおり、血清半減期が長く、透過性の高い血管を通じて徐々に漏出して腫瘍内部に蓄積することができる。このような受動的な標的指向メカニズムに加えて、能動的な標的指向戦略も提案されている。能動的な標的指向戦略においては、薬剤を封入したリポソームと腫瘍との相互作用をより強めるために、抗体や標的指向性リガンドが用いられる。抗体による標的指向性免疫リポソームは非常に活発に研究されており、動物実験と臨床試験の両方において有望視されている(非特許文献2〜6)。また、低分子リガンド(非特許文献7及び8)やペプチド(非特許文献9及び10)も同様に、癌治療用の能動的標的指向性リポソームを構築するために研究されている。
【0003】
in silico又はin vitroでの結合試験と比較して、in vivoでの結合を取り巻く環境は、多くの解剖学的障壁や細網内皮系(RES)等の自然の排出機構、さらに、他の非特異的な相互作用が関与しているために、はるかに複雑なものとなっている。免疫リポソーム及び低分子による標的指向性リポソームを用いて、結合に影響を及ぼす多くの要因が同定されている(非特許文献11及び12)。また、様々な能動的標的指向性リポソームの、薬物動態特性や血管外漏出挙動に影響を及ぼすいくつかの因子が同定されている(非特許文献11及び12)。リポソーム表面にコンジュゲートされたリガンドに対する免疫除去は大きな問題であり(非特許文献13及び14)、IgGを含む免疫リポソームは、Fcレセプターと相互作用して速やかに除去されることが明らかにされている(非特許文献15〜17)。また、低分子リガンドである葉酸をコンジュゲートした場合にも、in vivoでのRESによるリポソームの除去が促進された(非特許文献8)。さらに、リポソーム表面のリガンド密度(非特許文献18)及びPEGリンカーの長さ(非特許文献8及び19)についても、ともに重要であることが明らかにされている。
【0004】
上皮成長因子受容体(EGFR)は、肺癌、乳癌、膀胱癌、及び卵巣癌を含む(ただしこれらに限定されるわけではない)、幅広い種類のヒトの癌において過剰発現している。また、EGFRは、進行疾患の様々な特性や予後不良とも関連していることが明らかにされている(非特許文献20)。EGFRを標的とする様々なベクターやコンジュゲートは、癌細胞への細胞毒性薬、毒素、又は放射性核種の送達を増加させること、また、動物モデルでは腫瘍の増殖を抑制する作用を持つことが報告されている(非特許文献21〜25)。Mamotらは、EGFRに特異的な免疫リポソームを開発し、一連の研究により、in vitro及びin vivoにおいて、この免疫リポソームが優れた送達特性と高い抗癌作用を持つことを示した(非特許文献19、26、及び27)。
【0005】
EGFRを標的とした、いくつかの効果的な薬剤が開発されており、その中には、チロシンキナーゼ阻害剤であるタルセバ(登録商標)及びイレッサ(登録商標)、及び、EGFRのブロッキング抗体であるアービタックス(登録商標)が含まれる。このようなEGFR自体を標的とする薬剤を設計することに加えて、他の戦略として、従来の治療剤をEGFRに向けて送達するシステムの開発がある。多くの研究では、EGFRの天然のリガンドである上皮成長因子(EGF)を使用することによって、標的指向性の細胞毒性薬剤、毒素、リポソーム、及び、他の薬剤/遺伝子のデリバリーシステムの開発や、標的指向性の放射性核種システムの開発が行われてきた(非特許文献21〜24)。しかし、前記の戦略では、EGFのもつ内在性増殖促進作用が、癌細胞に及ぼす影響が常に問題となる。このため、他に2つの戦略が提案されている。1つは、EGFRに結合する抗体又は抗体断片を用いる方法であり(非特許文献26〜28)、もう1つは低分子ペプチド又はEGFの断片を用いる方法である(非特許文献21、29、及び30)。このような方法においては、EGFRに強固且つ特異的に結合することが可能で、免疫原性及び細胞増殖促進作用が低い低分子ペプチドは特に望ましい。
【0006】
当技術分野においては、例えば、EGFRを過剰発現する細胞等の標的細胞に、治療用薬剤を効果的に送達するための材料及び方法は依然として必要とされている。本発明は、このような必要性に応えるものである。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res 1994;54(4):987-92
【非特許文献2】Maruyama K, Takizawa T, Yuda T, Kennel SJ, Huang L, Iwatsuru M. Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies. Biochim Biophys Acta 1995;1234(1):74-80
【非特許文献3】Park JW, Hong K, Carter P, et al. Development of anti-p185HER2 immunoliposomes for cancer therapy. Proc Natl Acad Sci U S A 1995;92(5):1327-31
【非特許文献4】Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: in vitro and in vivo studies. Br J Cancer 1996;74:1749-1756
【非特許文献5】Lopes de Menezes DE, Pilarski LM, Allen TM. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res 1998;58(15):3320-30
【非特許文献6】Matsumura Y, Gotoh M, Muro K, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol 2004;15(3):517-25
【非特許文献7】Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta 1995;1233(2):134-44
【非特許文献8】Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9
【非特許文献9】Terada T, Mizobata M, Kawakami S, Yabe Y, Yamashita F, Hashida M. Basic fibroblast growth factor-binding peptide as a novel targeting ligand of drug carrier to tumor cells. J Drug Target 2006;14(8):536-45
【非特許文献10】Schiffelers RM, Koning GA, ten Hagen TL, et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release 2003;91(1-2):115-22
【非特許文献11】Mastrobattista E, Koning GA, Storm G. Immunoliposomes for the targeted delivery of antitumor drugs. Adv Drug Deliv Rev 1999;40(1-2):103-127
【非特許文献12】Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res 2006;16(3):175-83
【非特許文献13】Koning GA, Morselt HW, Gorter A, et al. Pharmacokinetics of differently designed immunoliposome formulations in rats with or without hepatic colon cancer metastases. Pharm Res 2001;18(9):1291-8
【非特許文献14】Koning GA, Morselt HW, Gorter A, et al. Interaction of differently designed immunoliposomes with colon cancer cells and Kupffer cells. An in vitro comparison. Pharm Res 2003;20(8):1249-57
【非特許文献15】Derksen JT, Morselt HW, Scherphof GL. Uptake and processing of immunoglobulin-coated liposomes by subpopulations of rat liver macrophages. Biochim Biophys Acta 1988;971(2):127-36
【非特許文献16】Maruyama K, Takahashi N, Tagawa T, Nagaike K, Iwatsuru M. Immunoliposomes bearing poloethylene glycol-coupled Fab' fragment show prolonged circulation time and high extravasation into targeted solid tumors in vivo. FEBS Lett 1997;413(1):177-80
【非特許文献17】Huwyler J, Yang J, Pardridge WM. Receptor mediated delivery of daunomycin using immunoliposomes: pharmacokinetics and tissue distribution in the rat. J Pharmacol Exp Ther 1997; 282(3):1541-6
【非特許文献18】Tardi P, Bally MB, Harasym TO. Clearance properties of liposomes involving conjugated proteins for targeting. Adv Drug Deliv Rev 1998;32(1-2):99-118
【非特許文献19】Mamot C, Ritschard R, Kung W, Park JW, Herrmann R, Rochlitz CF. EGFR-targeted immunoliposomes derived from the monoclonal antibody EMD72000 mediate specific and efficient drug delivery to a variety of colorectal cancer cells. J Drug Target 2006; 14(4):215-23
【非特許文献20】Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995;19:183-232
【非特許文献21】Lutsenko SV, Feldman NB, Severin SE. Cytotoxic and antitumor activities of doxorubicin conjugates with the epidermal growth factor and its receptor-binding fragment. J Drug Target 2002;10(7):567-71
【非特許文献22】Jinno H, Ueda M, Ozawa S, et al. Epidermal growth factor receptor-dependent cytotoxic effect by an EGF-ribonuclease conjugate on human cancer cell lines--a trial for less immunogenic chimeric toxin. Cancer Chemother Pharmacol 1996;38(4):303-8
【非特許文献23】Kullberg EB, Nestor M, Gedda L. Tumor-cell targeted epiderimal growth factor liposomes loaded with boronated acridine: uptake and processing. Pharm Res 2003; 20(2):229-36
【非特許文献24】Chen P, Mrkobrada M, Vallis KA, et al. Comparative antiproliferative effects of (111) In-DTPA-hEGF, chemotherapeutic agents and gamma-radiation on EGFR-positive breast cancer cells. Nucl Med Biol 2002;29(6):693-9
【非特許文献25】Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different strategies for formation of pegylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjug Chem 2001;12(4):529-37
【非特許文献26】Mamot C, Drummond DC, Greiser U, et al. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR-and EGFRvIII-overexpressing tumor cells. Cancer Res 2003;63(12):3154-61
【非特許文献27】Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res 2005; 65(24):11631-8
【非特許文献28】Schmidt M, Vakalopoulou E, Schneider DW, Wels W. Construction and functional characterization of scFv(14E1)-ETA -a novel, highly potent antibody-toxin specific for the EGF receptor. Br J Cancer 1997; 75(11):1575-84
【非特許文献29】Liu X, Tian P, Yu Y, Yao M, Cao X, Gu J. Enhanced antitumor effect of EGF R-targeted p21WAF-1 and GM-CSF gene transfer in the established murine hepatoma by peritumoral injection. Cancer Gene Ther 2002;9(1):100-8
【非特許文献30】Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J 2005;19(14):1978-85
【発明の概要】
【発明の効果】
【0008】
本発明は、目的とする標的細胞に物質を送達するために用いることができるペプチドを提供する。前記物質としては、例えば、治療剤及び/又は造影剤を挙げることができる。1つの実施形態において、本発明は、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドを提供する。また、1つの実施形態においては、前記ペプチドは、LARLLT配列(配列番号1)を含んでいてもよい。本発明のペプチドはどのような長さであってもよく、前記ペプチドの長さとしては、例えば、約6アミノ酸〜約15アミノ酸、又は約6アミノ酸〜約10アミノ酸の長さを挙げることができる。本発明のペプチドは、6、7、8、9、10、又は11アミノ酸以上の長さであってもよい。いくつかの実施形態においては、本発明のペプチドは、長さが6アミノ酸のペプチドであって、且つ、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列からなるペプチドである。いくつかの実施形態においては、本発明のペプチドは、アミノ酸配列LARLLTからなるペプチドであってもよい。さらに、本発明は、本発明のペプチドのEGFR結合活性を模倣したペプチド模倣物や、本発明のペプチドの機能を保持したアミノ酸置換ペプチドを提供する。
【0009】
他の実施形態においては、本発明のペプチド及びペプチド模倣物は、1又は2以上の化合物に直接的又は間接的に結合(attached)していてもよい。いくつかの実施形態においては、本発明のペプチドは、治療剤及び/又は造影剤に結合している。1つの実施形態においては、本発明は、脂質と、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドとを含む分子を提供する。いくつかの実施形態においては、前記ペプチドは、LARLLT配列を含んでいてもよい。また、本発明のペプチドは、リンカーを使用することによって間接的に脂質に結合していてもよく、リンカーとしては、当業者に公知の任意のリンカーを用いることができる。本発明のペプチド及びペプチド模倣物と結合する脂質の好適な例としては、特に制限されないが、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)等を挙げることができる。いくつかの実施形態においては、リンカーはポリエチレングリコール(PEG)を含んでいてもよい。リンカーと結合した脂質の例としては、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)を挙げることができる。前記のようなリンカーと結合した脂質を含む分子は、さらに、1又は数個の本発明のペプチド及びペプチド模倣物と結合していてもよい。
【0010】
本発明のペプチド及びペプチド模倣物は、活性物質と結合していてもよく、活性物質としては、例えば、治療剤及び/又は造影剤を挙げることができる。また、本発明のペプチド及びペプチド模倣物と活性物質との結合は、直接的であっても、間接的であってもよい。本発明のペプチド及びペプチド模倣物は、活性物質と、本発明のペプチド及びペプチド模倣物と前記活性物質の官能基とが直接的に反応することよって結合していてもよい。また、本発明のペプチド及びペプチド模倣物は、活性物質と、前記ペプチドと前記活性物質とがリンカーを介して間接的に反応することよって結合していてもよく、この場合、リンカーは通常、前記ペプチド及び前記活性物質の両方と共有結合を形成することが可能な二官能性の分子である。
【0011】
他の実施形態においては、本発明はリポソームを包含する。本発明のリポソームの例としては、脂質及び本発明のペプチドを含む分子を含むリポソームを挙げることができ、本発明のペプチドとしては、例えば、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドを挙げることができる。また、いくつかの実施形態においては、前記ペプチドはLARLLT配列を含んでいてもよい。上述のように、本発明のリポソームにおいて用いられる分子は、脂質部分とペプチド部分との間にリンカーを含んでいてもよい。1つの実施形態においては、前記脂質は、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)であってもよい。また、1つの実施形態においては、前記リンカーはポリエチレングリコール(PEG)であってもよい。1つの実施形態においては、脂質−リンカー−ペプチドの組合せは、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTであってもよい。また、本発明のリポソームは、当業者に公知の任意の脂質又は脂質の組合せを用いて作製してもよい。リポソームの作製に用いる好適な脂質の例としては、特に制限されないが、リン脂質(例えば、ホスファチジルコリン、ホスファチジルグリセロール、及びホスファチジルエタノールアミン)、リゾ脂質、及びPEG化リン脂質等を挙げることができ、このような好適な脂質はどのような比率で組み合わされていてもよい。本発明に用いるリポソームは、任意選択で、ゲル相から液相への相転移温度が約39.0℃〜約45℃の脂質を含む熱感受性リポソームであってもよい。好適なリポソームの例の1つとして、DPPC及びMSPCを含むリポソームであって、DPPC:MSPCのモルパーセント比が、99:1、98:2、97:3、96:4、95:5、90:10から約80:20、75:25、70:30、65:35、60:40のリポソームや、又は、DPPC:MSPCのモルパーセント比が51:49のリポソームを挙げることができる。また、熱感受性リポソームには、さらに1又は数種の脂質(例えば、前記ペプチド−リンカー−及び/又はPEG化脂質)が組み込まれていてもよい。
【0012】
本発明のリポソームは、治療用化合物及び造影化合物からなる群より選択される1又は数種の化合物を、さらに含んでいてもよい。本発明のリポソームは通常、二重膜を有して内部空間を画定し、さらに、化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる。また、いくつかの実施形態においては、化合物は前記リポソームの外側に会合(associated)していてもよい。本発明のリポソームに封入するための、好適な種類の化合物の例の1つとして、抗癌化合物を挙げることができる。好適な抗癌化合物の例としては、特に制限されないが、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物等を挙げることができる。いくつかの実施形態においては、本発明のリポソームは、ドセタキセルを含んでいてもよい。また、いくつかの実施形態においては、本発明のリポソームは、ドキソルビシンを含んでいてもよい。さらに、いくつかの実施形態においては、本発明のリポソームは、プラチナ含有化合物を含んでいてもよく、プラチナ含有化合物としては、例えば、カルボプラチン又はシスプラチンを挙げることができる。
【0013】
さらに本発明は、化合物を、その化合物を必要とする対象に投与する方法を提供する。この方法は通常、前記化合物と、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドとを会合させる工程、及び、この工程によって得られたペプチドと会合した化合物を対象に接触させる工程を含む。いくつかの実施形態において、前記ペプチドはLARLLT配列を含んでいてもよい。また、ペプチドと化合物との会合は、共有結合性であっても、非共有結合性であってもよい。いくつかの実施形態において、前記ペプチドは、直接的又は間接的に活性物質と結合していてもよい。いくつかの実施形態においては、前記ペプチドは脂質に結合していてもよく、さらに、前記ペプチドが結合した脂質は、リポソームを形成していてもよい。送達される化合物はリポソームの一部であってもよく、例えば、リポソームの二重膜中にあっても、リポソームの内部空間に封入されていてもよい。また、前記ペプチドは化合物に結合していてもよく、さらに、前記化合物がリポソームに含まれていてもよい。ここで、化合物は、例えば、前記リポソームの二重膜中に含まれていても、前記リポソームの内部空間に封入されていてもよい。前記ペプチドが脂質に結合する場合には、前記ペプチドが結合した脂質は、前記脂質と前記ペプチドの間にリンカーを含んでいてもよい。本発明に用いる好適な脂質の例としては、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)を挙げることができ、好適なリンカーは、ポリエチレングリコール(PEG)を含んでいてもよい。従って、リンカーを介して本発明のペプチドと結合した脂質の例の1つとしては、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを挙げることができる。さらに、前記リンカーを介して本発明のペプチドと結合した脂質は、リポソームに組み込んで本発明の方法に用いてもよい。本発明の脂質−リンカー−ペプチドを含むリポソームは、当業者に公知の他のどのような脂質を含んでいてもよい。いくつかの本発明の方法においては、上述のような脂質−リンカー−ペプチドを含むリポソームに化合物が会合していてもよく、さらに、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃であってもよい。このようなリポソームは、それを必要とする対象に投与されてもよく、さらに、かかる対象の一部は、前記リポソームのゲル相から液相への相転移温度よりも高い温度まで加温されてもよい。
【0014】
標的組織を加温する方法としては、適切な方法であればどのような方法を用いてもよいが、例えば、無線周波数照射の使用、高密度焦点式超音波等の超音波の使用、マイクロ波放射の使用、温浴槽や光などの赤外線放射源の使用を挙げることができ、また同様に、他の形態として、放射性同位体等による外部又は内部からの放射線照射、又は電場及び磁場を挙げることができる。
【0015】
本発明の方法を用いることによって、任意の化合物を投与することができる。投与する化合物の好適な例としては、特に制限されないが、治療用化合物及び造影化合物等を挙げることができる。これら化合物は、本発明のペプチドに直接的に又は間接的に結合していても、又は、本発明のペプチドと結合した脂質を含むリポソームに含まれていてもよい。本発明の方法の1つにおいては、リポソームは二重膜を有して内部空間を画定していてもよく、さらに、前記化合物は、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれていてもよい。また、本発明の方法は、治療用化合物を送達するために用いることができ、治療用化合物としては、例えば、抗癌化合物を挙げることができる。抗癌化合物としては、特に制限されないが、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物等を挙げることができる。いくつかの実施形態においては、本発明の方法は、ドセタキセルを送達するために用いてもよい。また、いくつかの実施形態においては、本発明の方法は、ドキソルビシンを送達するために用いてもよい。さらに、いくつかの実施形態においては、本発明の方法は、プラチナ含有化合物を送達するために用いてもよく、プラチナ含有化合物としては、例えば、カルボプラチン又はシスプラチンを挙げることができる。
【0016】
1つの実施形態においては、本発明は癌を治療する方法を提供する。この方法は、本発明のペプチドと会合した抗癌化合物を、それを必要とする対象に投与する工程を含む。前記会合の例の1つとしては、本発明のペプチドと抗癌化合物との直接的又は間接的な結合を挙げることができる。また、他の例としては、前記抗癌化合物がリポソームに封入されており、ここで、リポソームは、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドと会合した脂質を含む。いくつかの実施形態においては、前記ペプチドはLARLLT配列を含んでいてもよい。前記ペプチドは通常、前記脂質と直接又はリンカーを介して結合する。好適な脂質は、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)であり、好適なリンカーは、ポリエチレングリコール(PEG)である。従って、抗癌化合物を送達するために用いられるリポソームに組み込まれる好適なペプチド−リンカー−脂質の例の1つとして、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを挙げることができる。リポソームの作製に用いることのできる脂質の好適な例としては、特に制限されないが、リン脂質(例えば、ホスファチジルコリン、ホスファチジルグリセロール、及びホスファチジルエタノールアミン)、リゾ脂質、及びPEG化リン脂質等を挙げることができる。本発明の癌を治療する方法に用いるリポソームは、任意選択で、ゲル相から液相への相転移温度が約39.0℃〜約45℃のリポソームであってもよい。好適なリポソームの例の1つとして、DPPC及びMSPCを含むリポソームであって、DPPC:MSPCのモルパーセント比が、99:1、98:2、97:3、96:4、95:5、90:10から約80:20、75:25、70:30、65:35、60:40のリポソームや、又は、DPPC:MSPCのモルパーセント比が51:49のリポソームを挙げることができる。前記ペプチド−リンカー−脂質は、このようなリポソームに組み込むことができる。本発明の癌を治療する方法には、公知の任意の抗癌剤を用いることができる。かかる抗癌剤としては、特に制限されないが、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物等を挙げることができる。いくつかの実施形態において抗癌化合物は、ドセタキセルであってもよい。また、いくつかの実施形態において抗癌化合物は、ドキソルビシンであってもよい。さらに、いくつかの実施形態において抗癌化合物は、プラチナ含有化合物であってもよく、プラチナ含有化合物としては、例えば、カルボプラチンを挙げることができる。いくつかの本発明の癌を治療する方法においては、抗癌化合物は、上述のペプチド−リンカー−脂質を含むリポソームと会合していてもよく、さらに、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃であってもよい。前記方法は、対象の一部を加温する工程をさらに含んでいてもよい。対象を加温する方法としては、どのような方法を用いてもよいが、例えば、マイクロ波エネルギーの使用、他の電磁波エネルギー、高周波焼灼、高密度焦点式超音波、超音波、温水の使用等を挙げることができる。
【図面の簡単な説明】
【0017】
【図1】EGFRにおける、LARLLT及びコントロールペプチドのドッキング構造。A.PDBによるEGFR構造モデル。アスタリスクで示した領域は、EGF結合部位である。円内の領域は、本発明者らがドッキング解析に選んだ結合ポケットである。B.EGFRのポケット内における、LARLLTの最小エネルギードッキング構造。LARLLTペプチドを棒球モデルで示す。また、水素結合の可能性のある部位を円柱で示す。C.EGFRのポケット内における、コントロールペプチドの最小エネルギードッキング構造。
【図2】EGFRを高発現する細胞に対する、リガンドによる標的指向性リポソームの結合に関する蛍光顕微鏡実験。パネルA:LARLLT、コントロールペプチド、及びEGFによる指向性リポソームのH1299細胞への結合。(i)LARLLTリポソーム、(iii)EGFリポソーム、(v)コントロールペプチドリポソーム。(ii)、(iv)、(vi)は、(i)、(ii)、(v)と同視野の位相差顕微鏡写真。パネルB:4℃又は37℃における、50倍モル過剰量のフリーのリガンドの存在下での、LARLLTによる指向性リポソームのH1299細胞への結合。(i)37℃における、過剰量のフリーのLARLLT存在下での結合、(ii)37℃における、フリーのLARLLT非存在下での結合、(iii)4℃における、過剰量のフリーのLARLLT存在下での結合、(iv)4℃における、フリーのLARLLT非存在下での結合、(v)37℃における、過剰量のフリーのEGF存在下での結合、(vi)37℃における、フリーのEGF非存在下での結合。パネルC:LARLLT、コントロールペプチドリポソームのSPC−A1細胞への結合。(i)LARLLTリポソーム、(iii)コントロールペプチドリポソーム。(ii)及び(iv)は、(i)及び(iii)と同視野の位相差顕微鏡写真。
【図3】H1299細胞における、LARLLTがコンジュゲートしたリポソームの内在化。A.エンドサイトーシスによって細胞に取り込まれたLARLLTリポソームの位相差画像及び蛍光画像のオーバーレイ画像。B.共焦点蛍光顕微鏡のZ軸スキャンモードを用いて得られた細胞の上部から底部までの11の断面画像。
【図4】ドキソルビシンを含む標的指向性リポソームの殺細胞効果。A.H1299細胞。算出したIC50の値は、LARLLTリポソーム(黒色ひし形)では〜13μg/ml、コントロールペプチドリポソーム(白抜き四角)では85μg/ml、フリーのドキソルビシン単独(黒色三角)では5.7μg/mlであった。B.SPCA1細胞。算出したIC50の値は、LARLLTリポソーム(黒色ひし形)では〜5μg/ml、コントロールペプチドリポソーム(白抜き四角)では15μg/ml、フリーのドキソルビシン単独(黒色三角)では2μg/mlであった。
【図5】担癌マウス体内のCy5.5及びCy5.5標識ペプチドの蛍光画像。ここで示す蛍光画像は、フリーのCy5.5色素、LARLLT−Cy5.5、及びコントロールペプチド−Cy5.5を注射して1及び6時間後に撮影した。
【図6】腫瘍組織における、ペプチドによる指向性リポソームの分布及び蓄積を示す蛍光画像。ここで示す画像(左から右に向かって)は実験に用いたマウスの可視光画像、LARLLTリポソーム(上段)及びコントロールペプチドリポソーム(下段)を注射して1時間、6時間、12時間、24時間、48時間、80時間後の蛍光画像である。
【図7】図7は、薬剤を注射した後の様々な時点における、ヌードマウスに異種移植されたH460腫瘍塊に蓄積したドキソルビシンの濃度を示す棒グラフである。黒色バーは、ドキソルビシンの溶液を注射後の腫瘍中のドキソルビシン濃度を示す。また、点線付き黒色バーは、従来のドキソルビシンを含むリポソームを注射した後の腫瘍中のドキソルビシン濃度を、波線付き黒色バーは、ドキソルビシンを含むLARLLT修飾リポソームを注射した後の腫瘍中のドキソルビシン濃度を示す。
【図8】図8は、0.01〜10μg/mlでの様々な濃度のドキソルビシンを含む、ドキソルビシン含有製剤により処理したH460細胞の蛍光強度を示す線グラフである。x=フリーのドキソルビシン、◇=D4*リポソーム、▲=D4 4+4リポソーム、○=D4 2+2リポソーム、●=サーモドックスリポソーム。D4は配列番号1及を含むリポソームであり、D4*は配列番号2を含むリポソームである。D4−サーモドックス2+2は、DPPC:MSPC:DSPE−MPEG2000:D4−DSPE−MPEG2000を90:10:2:2のモル比で含み、D4−サーモドックス4+4は、DPPC:MSPC:DSPE−MPEG2000:D4−DSPE−MPEG2000を90:10:4:4のモル比で含む。D4*−サーモドックスはDPPC:MSPC:DSPE−MPEG2000:D4*−DSPE−MPEG2000を90:10:2:2のモル比で含む。
【発明を実施するための形態】
【0018】
ペプチド
【0019】
本発明は、EGFRと強固且つ特異的に結合し得る低分子ペプチドを提供する。本発明のペプチド及びペプチド模倣物は通常、免疫原性及び/又は増殖促進作用を伴うことなく、EGFRと結合することができる。
【0020】
本発明の典型的なペプチドとしては、アミノ酸配列LeuAlaArgLeuLeuThr(配列番号1)を含むペプチドを挙げることができる。これに加えて、本発明のペプチドの例としては、特に制限されないが、配列番号1に示されるアミノ酸配列のうち、1又は数個のアミノ酸が異なるアミノ酸によって置換されたペプチド等を挙げることができる。また、いくつかの実施形態においては、配列番号1アミノ酸位置のうち、1箇所のみが置換されていてもよく、さらに、いくつかの実施形態においては、配列番号1のアミノ酸位置のうち、2箇所以上が置換されていてもよい。これらの置換は、配列番号1の任意のアミノ酸位置において行うことができる。また、いくつかの実施形態においては、配列番号1のアミノ酸位置のうち、1又は数箇所のアミノ酸位置が、1又は数個の天然アミノ酸によって置換されていてもよく、さらに、いくつかの実施形態においては、配列番号1のアミノ酸位置のうち、1又は数箇所のアミノ酸位置が、1又は数個の非天然アミノ酸によって置換されていてもよい。いくつかの実施形態においては、本発明のペプチドは、1又は数個のD−アミノ酸を含んでいてもよい。
【0021】
本発明のペプチドに含まれるアミノ酸の置換は通常、同類置換であってもよい。本明細書において、同類置換とは、1つのアミノ酸を、物理的及び/又は化学的な性質が類似した他の1つのアミノ酸によって置き換えることを指す。遺伝学的にコードされた標準的なアミノ酸は、その性質、特に極性と電荷に従って分類することができる。便宜的な分類の1つを以下に示す:グリシン及びアラニン;セリン、トレオニン、アスパラギン、グルタミン及びシステイン;リシン、アルギニン及びヒスチジン;アスパラギン酸及びグルタミン酸;バリン、ロイシン、イソロイシン、及びメチオニン;及びフェニルアラニン、トリプトファン及びチロシン。ある同類グループのアミノ酸を、同じ同類グループの他のアミノ酸によって置き換えることは「同類置換」であると見なされる。本発明のペプチドの性質は通常、このような置換によって実質的に影響を受けることはなく、ここで本発明のペプチドの性質としては、例えば、EGFRに対する結合能を挙げることができる。このように、本発明のペプチドは、配列番号1に示されるアミノ酸配列から、同類置換のみによって1又は数個のアミノ酸が置換された、配列番号1とは異なるペプチドを含む。
【0022】
本発明のペプチドの例としては、特に制限されないが、以下に示すアミノ酸配列等を挙げることができる:
【0023】
Leu Ala Arg Leu Leu Thr(配列番号1)
【0024】
Xaa1 Ala Arg Leu Leu Thr(配列番号3)
【0025】
Leu Xaa2 Arg Leu Leu Thr(配列番号4)
【0026】
Leu Ala Xaa3 Leu Leu Thr(配列番号5)
【0027】
Leu Ala Arg Xaa4 Leu Thr(配列番号6)
【0028】
Leu Ala Arg Leu Xaa5 Thr(配列番号7)
【0029】
Leu Ala Arg Leu Leu Xaa6(配列番号8)
【0030】
ここで、Xaa1はバリン、イソロイシン、及びメチオニンからなる群より選択され、Xaa2はグリシンであり、Xaa3はリシン及びヒスチジンからなる群より選択され、Xaa4はバリン、イソロイシン、及びメチオニンからなる群より選択され、Xaa5はバリン、イソロイシン、及びメチオニンからなる群より選択され、Xaa6はセリン、アスパラギン、グルタミン及びシステインからなる群より選択される。また、本発明には前記の置換を2以上含むペプチドも包含される。
【0031】
さらに本発明は、前記本発明のペプチドと同様のEGFRに対する結合能を示す非ペプチド性化合物にも関する。本発明の実施に際しては、このような本発明のペプチドを模倣した能力を有するペプチド模倣物又は「低分子」を用いてもよい。当業者であれば、Fauchere J., Adv. Drug Res. 15: 29 (1986)、及びEvans et al., J. Med. Chem. 30: 1229 (1987)に述べられているような周知の手法を用いて、ペプチド模倣物を設計することができる。
【0032】
EGFRに対する結合能を有するペプチド模倣物は、本発明のペプチドと同じように用いることができる。かかるペプチド模倣物は通常、in vivoでの化学分解に対する耐性等の望ましい性質を付与するような結合によって、任意に置換された1又は数個のペプチド結合を有している。このような結合としては、特に制限されないが、−−CH2NH−−、−−CH2S−−、−−CH2CH2−−、−−CH=CH−−、−−COCH2−−、−−CH(OH)CH2−−、及び、−−CH2SO−−等を挙げることができる。
【0033】
本発明のペプチド及びペプチド模倣物を、任意の所望の物質と会合させることによって、前記物質を目的とする標的細胞に送達することができる。前記物質の好適な例としては、特に制限されないが、化合物(例えば、活性物質、治療剤、造影剤等)、リポソーム(例えば、熱感受性リポソーム及びステルスリポソーム)、及びナノ粒子(例えば、タンパク質ナノ粒子、金属ナノ粒子、及び結晶化低分子ナノ粒子等)等を挙げることができる。
【0034】
本発明のペプチド及びペプチド模倣物と、送達される物質との会合は、共有結合性であっても、非共有結合性であってもよく、また、直接的であっても、間接的であってもよい。いくつかの実施形態において、本発明のペプチドは、送達される物質と共有結合によって結合していてもよく、かかる結合は、例えば、リンカーを介したものであってもよい。
【0035】
リンカーの例としては、本発明のペプチドと、送達される物質とに、共有結合によって結合することができる架橋剤を挙げることができる。このような架橋剤は通常、本発明のペプチド及び結合相手の物質に存在する官能基(例えば、−OH基、NH2基、COOH基、及び−SH基)と反応する。また、様々な官能基との、架橋剤の異なる反応性に基づいて、様々な架橋剤を選択することができる。
【0036】
現在当業者に公知の、活性化した官能基を介してポリペプチドをコンジュゲート又はカップリングさせる手法が特に適用可能である。これについては、例えば、Aurameas, et al., Scand. J. Immunol., Vol. 8, Suppl. 7:7-23 (1978)、米国特許第4493795号明細書、米国特許第3791932号明細書、及び米国特許第3839153号明細書を参照することができる。これらの文献は、ペプチドのカップリングの開示に関して特に本明細書に援用される。また、部位特異的なカップリング反応を行うことによって、カップリング後のペプチドの配向による活性の喪失を最小限に抑えることができる。これについては、例えば、Rodwell et al., Biotech., 3:889-894 (1985)、及び米国特許第4671958号明細書の記載を参照することができる。前記の手法の他、ペプチドを架橋するための手法として、Klipstein, et al., J. Infect. Dis., 147:318-326 (1983)等に記載の、マイケル付加反応体、グルタルアルデヒド等のジアルデヒドを用いる手法や、アミド結合の形成のために水溶性カルボジイミドを用いるカルボジイミド法を例示することができる。また、N−又はC−末端にシステインが導入されたペプチドをコンジュゲートさせるために、ヘテロ二官能性架橋剤であるSPDP(N−サクシニミジル−3−(2−ピリジルジチオ)プロピオナート)を用いることもできる。
【0037】
本明細書に記載のように、ペプチド結合を形成させる方法や、縮合剤を使用する方法や、さらに、公知の二官能性架橋剤を用いる方法などの、当業者に公知の化学的なコンジュゲート方法によって、前記ペプチドは活性物質とコンジュゲートさせることができる。コンジュゲートにおいては、ペプチドと活性物質とが、その間にどのような基も含まずに直接的に結合していてもよく、このような直接的な結合の例としては、直接的なペプチド結合を挙げることができる。また、コンジュゲートにおいては、ペプチドと活性物質とが、仲介する基を挟んで間接的に結合していてもよく、仲介する基としては、例えば、血漿アルブミン等のタンパク質やペプチド、又は他のスペーサー分子等を挙げることができる。また、例えば、前記間接的な結合はヘテロ二官能性又はホモ二官能性架橋剤を介したものであってもよく、架橋剤としては、例えば、カルボジイミド、グルタルアルデヒド、N−サクシニミジル 3−(2−ピリジルジチオ)プロピオナート(SPDP)及びその誘導体、ビスマレイミド、4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシラート等を挙げることができる。また、架橋反応は、外来の架橋剤を用いることなく、コンジュゲートする分子上の反応基を用いて行ってもよい。ペプチド分子を化学的に架橋する方法は一般的に当業者に公知であり、多数のヘテロ及びホモ二官能性架橋剤が、例えば、米国特許第4355023号明細書、米国特許第4657853号明細書、米国特許第4676980号明細書、米国特許第4925921号明細書、米国特許第4970156号明細書に記載されている。また、これらに加えて、市販の架橋剤を用いることもできる。市販の架橋剤としては、例えば、Pierce Chemical Co.,(Rockford, IL. 61105)社製の架橋剤を挙げることができる。前記各文献は参照することにより本明細書に援用される。生理的な条件下で前記コンジュゲートから治療剤を切り離すために、開裂可能な架橋剤、特にジスルフィド結合の開裂が可能な架橋剤を用いてもよい。このような開裂可能な架橋剤の例としては、4−サクシニミジルオキシカルボニル−a−(2−ピリジルジチオ)−トルエンを挙げることができる。以上のような架橋を含むコンジュゲーションは、コンジュゲートされた後のペプチド及び活性物質の望ましい機能を損なわないように行う必要がある。
【0038】
本発明の1つの実施形態においては、本発明のペプチド及びペプチド模倣物はナノ粒子に結合していてもよく、ナノ粒子としては種々の重合体や金属ナノ粒子、リポソーム、ニオソーム、固体脂質粒子、ミセル、量子ドット、デンドリマー、マイクロカプセル、細胞、ゴースト細胞、リポタンパク質、及び、様々なナノ集合体等の様々なナノ粒子を用いることができる。タンパク質ナノ粒子は、当技術分野に公知の方法を用いて作製することができる。タンパクナノ粒子の作製方法としては、例えば、修正した脱溶媒和−架橋法を挙げることできる。タンパク質を含む水溶液(2%、w/v)を、活性物質とともに緩衝液中でインキュベートした後に、エタノールに滴下して加えることによって、活性物質が組み込まれたタンパク質ナノ粒子を形成させることができる。ナノ粒子は、本発明のペプチドと架橋されていてもよく、例えば、グルタルアルデヒドを用いて本発明のペプチドと架橋されていてもよい。金ナノ粒子は、還元剤として水素化ホウ素ナトリウムを用いる、標準的な湿式化学合成法に従って合成することができる。
【0039】
1つの実施形態においては、本発明のペプチド及びペプチド模倣物は脂質に結合して、リポソームに組み込まれていてもよい。前記結合としては、特に制限されないが、例えば、本発明のペプチド及びペプチド模倣物が脂質の頭部基部分に直接結合していても、又は、脂質の頭部基部分にポリマー(例えば、PEG)等のリンカーを介して結合していてもよい。リポソーム(熱感受性リポソーム、すなわち、ゲル相から液相への相転移温度が約39.0℃〜約45℃であるリポソーム)は通常、活性物質(例えば、治療剤及び/又は造影剤)を含む。
【0040】
本発明のリポソームは通常、1又は数種類のホスファチジルコリンを含む。本発明の実施に用いることのできるホスファチジルコリンの好適な例としては、特に制限されないが、1,2−ジラウロイル−sn−グリセロ−3−ホスホコリン(DLPC)、1,2−ジミリストイル−sn−グリセロ−3−ホスホコリン(DMPC)、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)、1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン(DSPC)、1,2−ジオレオイル−sn−グリセロ−3−ホスホコリン(DOPC)、及び1−パルミトイル−2−オレオイル−sn−グリセロ−3−ホスホコリン(POPC)等を挙げることができる。
【0041】
本発明のリポソームは通常、1又は数種類のホスファチジルグリセロールを含む。ホスファチジルグリセロールの好適な例としては、特に制限されないが、1,2−ジミリストイル−sn−グリセロ−3−ホスホグリセロール(DMPG)、1,2−ジパルミトイル−sn−グリセロ−3−ホスホグリセロール(DPPG)、1,2−ジステアロイル−sn−グリセロ−3−ホスホグリセロール(DSPG)、及び1−パルミトイル−2−オレオイル−sn−グリセロ−3−ホスホグリセロール(POPG)等を挙げることができる。
【0042】
本発明のリポソームは通常、1又は数種類のリゾ脂質を含む。本明細書において、「リゾ脂質」は、ホスファチジン酸(1,2−ジアシル−snグリセロ−3−リン酸)の任意の誘導体であって、グリセロール部分に共有結合した1つのアシル鎖を含むものを示す。前記ホスファチジン酸の任意の誘導体としては、特に制限されないが、ホスファチジルコリン、ホスファチジルグリセロール、及びホスファチジルエタノールアミン等を挙げることができる。また、本発明の実施には、当業者に公知の任意のリゾ脂質を用いることができ、ここで、リゾ脂質としては、特に制限されないが、モノパルミトイルホスファチジルコリン(MPPC)、モノラウリルホスファチジルコリン(MLPC)、モノミリストイルホスファチジルコリン(MMPC)、モノステアロイルホスファチジルコリン(MSPC)、及びそれらの混合物等を挙げることができる。
【0043】
さらに、本発明のリポソームは、親水性ポリマーに結合した脂質を含んでいてもよい。親水性ポリマーに結合した脂質としては、例えば、PEGに共有結合した脂質を挙げることができる。
【0044】
活性物質
【0045】
本発明のペプチド及びペプチド模倣物は、活性物質を送達するために用いることができる。本明細書において、「活性物質」は、対象中の特異的な部位に送達されることが望まれる化合物であればどのような化合物であってもよい。本発明の実施には、どのような活性物質を用いてもよい。
【0046】
本発明の熱感受性リポソームにおいては、活性物質として抗癌剤を用いることができる。好適な抗癌剤の例を以下に示す:
【0047】
アルキル化剤、例えば、ナイトロジェンマスタード(例えば、クロラムブシル、クロルメチン、シクロホスファミド、イホスファミド、メルファラン、ニトロソ尿素(例えば、カルムスチン、フォテムスチン、ロムスチン、ストレプトゾシン)、プラチナ含有化合物(例えば、カルボプラチン、シスプラチン、オキサリプラチン、BBR3464)、ブスルファン、ダカルバジン、メクロレタミン、プロカルバジン、テモゾロマイド、チオテパ、及びウラムスチン;
【0048】
代謝拮抗剤、例えば、葉酸を標的とする代謝拮抗剤(例えば、アミノプテリン、メトトレキサート、ペメトレキセド、ラルチトレキセド)、プリン代謝を標的とする代謝拮抗剤(例えば、クラドリビン、クロファラビン、フルダラビン、メルカプトプリン、ペントスタチン、チオグアニン)、ピリミジン代謝を標的とする代謝拮抗剤(例えば、カペシタビン、シタラビン、フルオロウラシル、フロクスウリジン、ゲムシタビン);
【0049】
紡錘体毒植物アルカロイド、例えば、タキサン(例えば、ドセタキセル、パクリタキセル)、及びビンカ(例えば、ビンブラスチン、ビンクリスチン、ビンデシンビノレルビン);
【0050】
細胞障害性/抗腫瘍抗生物質、例えば、アントラサイクリン抗生物質(例えば、ダウノルビシン、ドキソルビシン、エピルビシン、イダルビシン、ミトキサントロン、バルルビシン、カルミノマイシン、nアセチルアドリアマイシン、ルビダゾン、5−イミドダウノマイシン、N30アセチルダウノマイシン、及びエピルビシン)、ブレオマイシン、マイトマイシン、及びアクチノマイシン;
【0051】
トポイソメラーゼ阻害剤、例えば、カンプトテシン(例えば、カンプトテシン、トポテカン、イリノテカン)、ポドフィルム(例えば、エトポシド、テニポシド);
【0052】
モノクローナル抗体又はその断片、例えば、アレムツズマブ、ベバシズマブ、セツキシマブ、ゲムツズマブ、パニツムマブ、リツキシマブ、トシツモマブ、及びトラスツズマブ;
【0053】
光感作性物質、例えば、アミノレブリン酸、アミノレブリン酸メチル、ポルフィマーナトリウム、及びベルテポルフィン;
【0054】
キナーゼ阻害剤、例えば、ダサチニブ、エルロチニブ、ゲフィチニブ、イマチニブ、ラパチニブ、ニロチニブ、ソラフェニブ、スニチニブ、及びバンデタニブ;
【0055】
酵素、例えば、アスパラギナーゼ、ペガスパルガーゼ、及び、酵素阻害剤、例えば、ヒドロキシ尿素;
【0056】
アポトーシス誘導剤、例えば、亜ヒ酸、ベルケイド、及びジェナセンス;
【0057】
生物学的反応修飾物質、例えば、デニロイキンジフチトクス;
【0058】
抗ホルモン剤、例えば、ゴセレリン酢酸塩、ロイプロリド酢酸塩、トリプトレリンパモ酸塩、酢酸メゲストロール、タモキシフェン、トレミフェン、フルベストラント、テストラクトン、アナストロゾール、エキセメスタン、及びレトロゾール;
【0059】
レチノイド、例えば、9−cis−レチノイン酸及びオールトランスレチノイン酸。
【0060】
他の実施形態においては、本発明の熱感受性リポソームは2以上の抗腫瘍薬を含むか、又は、それぞれが異なる種類の活性物質を含む2以上の熱感受性リポソームを本発明の方法に用いることができる。前記異なる種類の活性物質としては、例えば、異なる種類の抗癌剤を挙げることができる。
【0061】
本発明の実施においては、さらに他の活性物質を用いることができる。前記活性物質としては、特に制限されないが、例えば、抗生物質、抗真菌剤、抗炎症薬、免疫抑制薬、抗感染症薬、抗ウイルス薬、駆虫薬、及び抗寄生虫性化合物等を挙げることができる。
【0062】
前記活性物質は造影剤であってもよい。本発明のリポソーム製剤に好適に用いることのできる造影剤としては、超音波造影剤、X線造影剤(例えば、金及びバリウムを含む造影剤、イオヘキソール、ビジパーク、及び、他のヨードを含む造影剤や他の微粒子造影剤)、磁気共鳴造影剤(例えば、マンガンを含む常磁性体、鉄を含む常磁性体、ガドリニウムを含む常磁性体、及び、他のランタニドを含む常磁性体)、放射性医薬品(放射性同位体、又は放射性同位体を含む化合物)等を挙げることができる。
【0063】
使用する方法
【0064】
本発明のペプチドと会合した活性物質は、任意の適切な投与経路によって対象に投与することができる。ここで、好適な投与経路としては、静脈内、動脈内、筋肉内腹腔内、皮下、皮内、関節内、髄腔内、脳室内を例示することができ、さらに、例えば吸入、経口、又は粘膜(舌下)上に直接投与する他の投与経路も同様に挙げることができる。また、本発明の材料及び方法を用いて、任意の組織を治療することができる。前記本発明の材料及び方法を用いて治療可能な組織の例としては、特に制限されないが、肝臓、腎臓、骨、軟組織、頭部及び頸部の組織、筋肉、副腎組織、肺、胸部、甲状腺、膵臓、子宮内膜及び子宮頸部の組織を含む子宮組織、卵巣、及び前立腺等を挙げることができる。また、治療可能な組織は、癌組織であっても、罹患した組織又は損傷した組織であってもよく、さらに、必要であれば健康な組織であってもよい。
【0065】
本発明の方法を用いて対象に投与される活性物質の量は、当業者が決定することができ、好適には前記投与量の活性物質を長時間にわたって静脈内に投与する。ここでの投与時間としては、例えば、およそ1分間〜およそ24時間を挙げることができる。
【0066】
前記活性物質の投与量は、当技術分野で公知のように、キャリアに含まれる活性物質に応じて調整してもよい。
【0067】
活性物質が、本発明のペプチドを含む熱感受性リポソームに組み込まれている場合には、対象中の標的組織を、前記熱感受性リポソームの投与前、及び/又は投与中、及び/又は投与後に加温してもよい。1つの実施形態においては、前記標的組織を最初に加温し(例えば、10〜30分間)、加温後、可能な限り早く対象に前記熱感受性リポソームを送達する。他の実施形態においては、熱感受性リポソームを対象に投与し、投与後、可能な限り早く標的組織を加温する。このような加温処置は最長で3時間継続して行ってもよい。
【0068】
標的組織を加温する方法としては、好適な方法であればどのような方法を用いてもよい。好適な方法としては、例えば、無線周波数照射の使用、高密度焦点式超音波等の超音波の使用、マイクロ波放射の使用、温浴槽や光などの任意の赤外線放射源の使用を挙げることができ、また、他の形態として、放射性同位体等による外部又は内部からの放射線照射、又は電場及び磁場も同様に挙げることができる。
【0069】
当業者であれば、本発明の範囲又は本発明のいかなる実施形態からも逸脱することなく、本明細書に開示されている方法及び用途に対して、他の適切な改変及び修正を容易に行うことができる。ここまでに詳細に述べられた本発明は、以下の実施例を参照することにより、より明確に理解される。以下の実施例は、本発明を説明する目的で本明細書に組み込まれており、本発明を限定することを目的とするものではない。
[実施例]
【実施例1】
【0070】
卵ホスファチジルコリン、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[マレイミド(ポリエチレングリコール)2000](DSPE−PEG2000−Mal)及びコレステロールは、全てAvanti Polar Lipids社(米国アラバマ州)より購入した。Lissamine(登録商標)ローダミンB 1,2−ジヘキサデカノイル−sn−グリセロ−3−ホスホエタノールアミン(ローダミンDHPE)及びN−(フルオロセイン−5−チオカルバモイル)−1,2−ジヘキサデカノイル−sn−グリセロ−3−ホスホエタノールアミン(フルオロセインDHPE)は、Invitrogen Corporation社より購入した。N−サクシニミジル 3−(2−ピリジルジチオ)プロピオナート(SPDP)及びトリス(2−カルボキシエチル)ホスフィン(TCEP)はPierce Biotechnology, Inc.社より購入した。Cy5.5モノNHSエステルはGE Healthcare社より提供された。他の全ての分析グレードの化学薬品はSinopharm Chemical Reagent Co. Ltd社(中国上海)より入手した。
【0071】
不活性状態のEGFRの結晶構造データは、PDB(http://www.rcsb.org/pdb/、code:1NQL)よりダウンロードした。Sheng-Hung Wang博士より供与されたPSCAN2.2.2プログラム(http://home.pchome.com.tw/team/gentamicin/mol/mol.htm)を用いてEGFRの3D構造モデルを構築し、結合ポケットの候補を探索した。この検索により、EGFRの上端付近の表面上にある1つのポケットを見い出し、図1に示した。前記ポケット内部のアミノ酸残基構造を詳細に分析し、Mekler-Idlisのアミノ酸ペアリング理論(Heal JR, Roberts GW, Raynes JG, Bhakoo A, Miller AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochem 2002;3(2-3):136-51)に基づいて、132個のヘキサペプチドを含むフォーカストライブラリを設計した。
【0072】
前記のEGFR表面ポケットに対するペプチドの結合性に関して、Autodock3.0.を用いて前記ライブラリをスクリーニングし、ペプチドの順位付けを行った。計算を高速化し、複数のプロセッサ上でプログラムを実行するために、本発明者らは前記ソフトウェアをパラレルコンピューティングが可能になるように修正した。前記の全てのコンピュータ処理は、SJTUスーパーコンピューティングセンター内のSGI Onyx3800機を用いて行った。最初に大まかなスクリーニングを行い、前記ライブラリに含まれる132個のペプチドの中から、より低い結合エネルギーを持つ50個のペプチドを選び出した。ラマルク型遺伝アルゴリズム局所検索(GALS)アルゴリズム(Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639-62)を用いて、1回あたり25万回のエネルギー計算からなるドッキング試行を、各ペプチドに対して独立して100回行った。また、その他の全てのドッキングパラメータは、Csaba及びDavid(Hetenyi C. van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci 2002;11(7):1729-37)の記載に基づき決定した。次に、前記スクリーニングにより選び出された50個のペプチドについて、同様のアルゴリズムで、ドッキングあたり1000万回のエネルギー計算からなる精密な解析段階により、さらに詳細な検討を行った。各ペプチドの最小ドッキングエネルギーを比較して最も低いドッキングエネルギーを持つ20個のペプチドを選び、合成及びウエットスクリーニングを行った。BIACore結合試験により、これら20個のペプチドのうちの2つは濃度依存的にEGFRに結合することが示された。中でも、LARLLT配列からなるペプチド(配列番号1)(ドッキング試験における順位は4位)はEGFRに結合する可能性が明確に示された。
【0073】
図1A(円内)にPSCANプログラムを用いて見い出された結合ポケットを示す。この結合ポケットは、ペプチドが外部から容易に結合できるEGFR表面上に存在し、また、EGF結合部位からは少し離れた場所に位置する。これらのことから本発明者らは、前記ポケット内部の立体構造が、EGF結合及びEGFRの二量体化の有無によって変化する可能性は低いと推測した。前記ポケットは、底部にいくつかの疎水性残基を、開口部周辺に親水性残基を有する(帯電残基:Asp51、Glu73、Glu110、Arg48、Lys56、Arg74;極性残基:Thr106、Ser53、Ser62)。
【0074】
我々が、本実験に使用するために選択した2つのペプチド(LARLLT及びコントロールペプチド)を前記ポケットにドッキングさせ、最適なドッキング構造を探索した。LARLLTの最小ドッキングエネルギー及び結合自由エネルギーは、それぞれ−17.9kcal/mol及び−8.54kcal/molであり、コントロールペプチドの最小ドッキングエネルギー及び結合自由エネルギーは、それぞれ−12.52kcal/mol及び−3.85kcal/molであった。
【0075】
図1Bに示すように、ペプチドLARLLTは前記ポケットの中にすっぽりと収まり、LARLLTのほぼ全ての残基がEGFRの残基と相互作用した。LARLLTの両端(LEU1、ALA2及びTHR6)と中央(ARG3)の残基については、6つの水素結合が形成されることが示唆された。前記ペプチドのN末端においては、LEU1のアミノ基の水素は、EGFRのGLU73のカルボキシル基の酸素と相互作用し、ALA2の主鎖の酸素は、EGFRのGLU73の主鎖のアミド基の水素と相互作用した。また、前記ペプチドのC末端においては、THR6は水素結合の供与体と受容体の両方の役割を持ち、THR6のC末端の酸素はEGFRのSER53のヒドロキシル基の水素と相互作用し、THR6の側鎖のヒドロキシル基の水素はEGFRのASP51のカルボキシル基の酸素と相互作用した。さらに、前記ペプチドの中央部のARG3は、静電的相互作用(EGFRのGLU73及びGLU110との)と水素結合の両方を介してEGFRと相互作用した。以上の3つの鍵となる残基(LEU1、THR6、ARG3)は、空間的にちょうど良い位置にあり、EGFRのポケット内で錨のように固定されるとともに、LARLLTの疎水性残基ALA2、LEU4及びLEU5と、ポケット内の疎水性底部(hydrophobic bottom)とのより強い相互作用を容易にした。ドッキング構造(図1)に示すように、LARLLTの残基とEGFRの残基との詳細な相互作用解析は、M−Iのペアリング理論(Heal JR, Roberts GW, Raynes JG, Bhakoo A, Miller AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochem 2002;3(2-3):136-51)によく一致していた。
【0076】
これに対して、LARLLTと同じアミノ酸組成を持つが、配列をランダムに並べ替えたコントロールペプチド配列番号2は、図1Cに示すように、ポケット内に収まることすらできなかった。コントロールペプチドの帯電したARG1は、EGFRのASP51と相互作用したが、他の相互作用が弱過ぎるためにコントロールペプチドをポケット内に固定することができなかった。
【実施例2】
【0077】
ペプチドLARLLTは、GL Biochem Ltd.社(上海)において合成及び精製され、その構造と純度はHPLC及びMSにより全て確認された。また、比較のために、D4*と名付けた、LARLLTと同じアミノ酸組成を持つが、配列をランダムに並べ替えたペプチド(RTALLL、コントロールペプチド(配列番号2))を同様に合成した。前記の2つのペプチドを蛍光標識するために、Cy5.5−NHSと、LARLLT又はコントロールペプチドとを1:2のモル比で混合し、遮光下、25℃で一晩インキュベートした。これらの蛍光ペプチドは、その後さらに精製することなく実験に用いた。
【実施例3】
【0078】
EGF又はLARLLT/コントロールペプチドをPBS−EDTAに溶解し、DMSOに溶解したN−サクシニミジル 3−(2−ピリジルジチオ)プロピオナート(SPDP)と1:1.2のモル比で、室温で1時間混合した後、凍結乾燥した。また、クロロホルムに溶解したDSPE−PEG2000−Mal脂質を乾燥させて薄膜を作製し、HEPES緩衝液(pH7.4)で約0.4mMの濃度となるように水和させた。コンジュゲーションを行うために、チオール化したタンパク質又ペプチドをトリス(2−カルボキシエチル)ホスフィン(TCEP)溶液に加え、窒素雰囲気下、室温で1時間インキュベートした。さらに、MAL−PEG2000−DSPEミセル溶液とモル比5:1で素早く混合し、窒素雰囲気下、攪拌しながら10℃で一晩反応させた。HPLC分析を行ったところ、前記の反応によってほぼ全てのMal−PEG−DSPEが改変していた(modified)ことが示された。ここで、余剰のタンパク質/ペプチドを、例えば、ゲルろ過カラムや透析等の通常の方法により除去することもできる。
【実施例4】
【0079】
実験に用いたリポソームは全て、乾燥薄膜水和法を行った後、100nmの膜を数回通過させることより作製した。本実験に用いたほとんどのリポソームの脂質組成は、EPC:CHOL:DSPE−PEG=10:5:0.5から開始した。ローダミン又はフルオロセイン標識リポソームを除いて、ローダミン−DHPE又はフルオロセイン−DHPEのいずれかを全脂質の約0.6%モルとなるように前記脂質組成に加えた。
【0080】
Cy5.5標識リポソームについては、Cy5.5−NHS、DSPE、及びトリエチルアミンを3:1:3.5のモル比となるようにクロロホルム中で混合し、25℃で一晩インキュベート(遮光下)することにより、Cy5.5−DSPEを合成した。得られたCy5.5−DSPEを、全脂質の約0.5%モルとなるように前記脂質組成に組み込んだ。リポソーム形成後に透析を行って、余剰のCy5.5を除去した。
【実施例5】
【0081】
ドキソルビシンを封入したリポソームの作製は膜通過勾配法に従って行った(Mayer LD, Tai LC, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta 1990;1025(2):143-51、Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta 1993;1151(2):201-15. Erratum in: Biochim Biophys Acta 1994;1190(1):197)。簡単に説明すると、脂質をpH4.0の125mM硫酸アンモニウム中で水和させ、膜通過処理により〜100nmのリポソームを作製した。得られたリポソームをHEPES緩衝液(pH7.5)中で透析し、20mMのHEPES、150mMのNaCl(pH7.5)を用いて必要な濃度に希釈した。そして、ドキソルビシンを加え、前記リポソームとともに60℃で10分間インキュベートした。
【0082】
前記リガンドをコンジュゲートさせた脂質を、Ishida et al.(Ishida T, Iden DL, Allen TM. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett 1999;460 (1):129-33)の開発した方法を若干改変した方法によって、事前に作製したリポソームに挿入した。簡単に説明すると、DSPE−PEG2000−リガンドのミセル溶液を、作製済みのリポソーム溶液に9:100のモル脂質比で加え、60℃で1時間インキュベートした(ドキソルビシンを含むリポソームの場合には55℃で30分間)。SnakeSkin(登録商標)Pleated Dialysis Tubing 10,000 MWCO(Pierce Chemical Company社製)を用いて、前記溶液をPBSで4時間透析した。また、コントロールとして、mPEG2000−DSPEのミセルを同じ割合で混合してインキュベートし、標的指向性を有さない(non-targeted)リポソームを作製した。
【0083】
前記ペプチドリガンドをDSPE−PEG2000に、スルフヒドリル−マレイミドカップリング反応によってコンジュゲートさせた。逆相HPLC分析によりこの反応をモニターして確認した。前記反応により得られたペプチド−PEG−DSPE、又はmPEG−DSPEコントロールを、広く用いられているインキュベーション方法(Ishida T, Iden DL, Allen TM. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett 1999;460 (1):129-33)によって、事前に作製したリポソームに組み込んだ。このインキュベート処理において、ペプチド−PEG−脂質(又はmPEG−脂質)の、脂質に対する割合を様々に変えて検討し(3:100、6:100、及び9:100モル)、ほとんどの細胞に対してリポソームの非特異的な結合を抑制する比として、9:100の比率を決定した。また、前記手法によってリポソームの安定性(integrity)が損なわれないことを確認した。前記工程中又は工程後に漏れ出した封入ドキソルビシンはわずか5%だった。リポソームの最終的なサイズ分布は、Zetasizer3000H(Malvern Instruments社製)を用いた光子相関分光法(PCS)により決定した。膜通過処理直後に得られたリポソーム(〜110nm)と比較すると、ペプチド脂質を挿入した後のリポソームの粒子径はわずかに大きかったが(130〜150nm)、安定性は保たれていた。
【実施例6】
【0084】
本実験においては、EGFR発現レベルの高い2つの細胞株、ヒト非小細胞肺癌細胞株H1299及びヒト肺腺癌細胞株SPCA1を用いた。前記細胞は、10%のウシ胎児血清を含むRPMI1640培地にて、5%CO2を含む加湿気相条件下、37℃で培養を行った。
【0085】
H1299及びSPCA1の異種移植マウスモデルは、上海がん研究所の動物実験センターで作製した。それらのマウスモデルは、腫瘍細胞を移植した後約3〜4週間後(腫瘍サイズは直径約5〜7mm)にin vivoイメージング実験に用い、その後、人道的に安楽死させた。前記動物実験のプロトコールは、上海交通大学薬学部(Shanghai Jiaotong University, School of Pharmacy)の動物実験委員会によって承認された。
【実施例7】
【0086】
EGFRを高発現しているH1299又はSPCA1細胞を35mm径の培養ディッシュに播種し(1×106細胞/ディッシュ)、RPMI1640培地中で24〜48時間培養した。細胞培養がおよそ80%コンフルエントに達した後、リガンドとコンジュゲートさせたローダミンリポソームをRPMI1640で希釈し、4℃又は37℃で前記培養ディッシュに添加した。競合結合実験のために、LARLLTを組み込んだリポソームを添加する2時間前に、50倍モル濃度過剰量のフリーのLARLLT又はEGFを培地に加えた。前記特定の温度で、細胞を4時間インキュベートした後に、PBS(pH7.4)で6回洗浄した。細胞に結合し、内在化した蛍光脂質を、共焦点レーザー顕微鏡(CLSM、Zeiss LSM社製、ドイツ)を用いて可視化した。また、フローサイトメトリー実験のために、H1299又はSPCA1細胞を35mm径のウェルに播種して約80%コンフルエントに達するまで増殖させた。細胞を、リガンドで修飾した(modified)フルオロセイン標識リポソームとともに37℃で3時間インキュベートした後に、PBSで洗浄し、トリプシンで処理して懸濁した。その後、フローサイトメーター(BD FACSCalibur、Becton Dickinson社製)により解析した。
【0087】
EGF、LARLLT、又はコントロールペプチドで修飾したローダミン標識リポソームの、EGFR高発現細胞に対する結合能をin vitroで調べた。図2Aに示すように、EGF及びLARLLTで修飾したリポソームは、ともに効率よくH1299細胞に結合した。EGFで修飾したリポソームによる蛍光は、LARLLT修飾リポソームよりもわずかに強いことから、EGFによるより強い結合が示唆された。しかし、コントロールペプチドを用いた場合には、蛍光はほとんど観察されなかった。同様の結果は、EGFRを発現する別の細胞株SPC−A1を用いた場合にも観察された(図2C)。
【0088】
さらに詳細な結合特性に関する評価を、H1299細胞を用いて様々な温度条件で行った。4℃の温度条件では、蛍光は主に細胞表面上で認められ、過剰量の標識されていないフリーのLARLLTと競合させることによって、大部分の蛍光は認められなくなった(図2B−iii及びiv)。これに対し、37℃の温度条件では、フリーのLARLLTによる競合作用はそれほど明確には認められなかった(図2B−I及びii)。このことは、37℃では、LARLLTがEGFRに結合した後に、エンドサイトーシス及びレセプターの再利用(turnaround)が活発に起こっていることを示唆している。また、興味深いことに、37℃の温度条件では、フリーのEGFの存在はLARLLTリポソームの蛍光強度に、完全ではないが、実際にある程度の影響を及ぼした(図2B−v及びvi)。LARLLTが結合した後のエンドサイトーシス経路を、EGFが阻害した可能性がある。
【0089】
図3に、LARLLTで修飾したリポソームの、H1299細胞への結合及び取込みについて高倍率で示し(図3A)、さらに、Z軸連続スキャン画像を示した(図3B)。細胞内の多数のエンドサイトーシス小胞において脂質の蛍光が認められたことから、EGF結合部位から遠く離れた、EGFR表面上のわずかな窪みにLARLLTが結合することによって、活発なエンドサイトーシス活性が誘導され得ることが示された。
【0090】
フローサイトメトリーを用いて、より大きな細胞集団に対する、LARLLT及びコントロールペプチドが組み込まれたリポソームの結合能の違いを比較検討した。リポソームをフルオロセイン−DSPEで標識し、H1299及びSPCA1細胞にそれぞれ加えた。3時間のインキュベート後、H1299細胞の75%でLARLLTリポソームの結合による高い蛍光シグナルが認められた(101〜103 FL1値)。一方、コントロールペプチドリポソームの結合による蛍光は、H1299細胞の27%でしか認められなかった。SPCA1細胞の場合には、LARLLTリポソームによって68%が蛍光を発したが(35〜103 FL1値ゲート)、コントロールペプチドリポソームでは17%であった。
【実施例8】
【0091】
H1299及びSPCA1細胞を104細胞/ウェルの濃度で96ウェルプレートに播種し、一晩増殖させた。フリーのドキソルビシン、LARLLTによって修飾したドキソルビシン封入リポソーム、及びコントロールペプチドによって修飾したドキソルビシン封入リポソームを培地に添加し、2時間インキュベートした。その後、細胞を新しい培地により洗浄し、さらに48時間増殖させた。異なる処理後の細胞の生存率はMTTアッセイ(Scudiero DA, Shoemaker RH, Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988;48(17):4827-33)を用いて決定した。
【0092】
脂質とコンジュゲートしたLARLLT又はコントロールペプチドによって、ドキソルビシン封入リポソームを修飾してEGFR高発現細胞に添加し、これらのリポソームの殺細胞効果をMTTアッセイにより評価した。H1299及びSPCA1のいずれの細胞においても、LARLLTによって修飾したリポソームはコントロールペプチドによって修飾したリポソームよりも効率的に薬物を送達した(図4A及びB)。両方の細胞におけるLARLLTリポソームのIC50を算出したところ、コントロールペプチドリポソームと比較しておよそ3〜5倍細胞毒性が強かった。
【実施例9】
【0093】
Cy5.5、LARLLT−Cy5.5、コントロールペプチド−Cy5.5、及び、LARLLT又はコントロールペプチドを導入したCy5.5標識リポソームを、H1299又はSPCA1を異種移植した担癌マウスの尾静脈から注射した。注射後の様々な時点において、前記マウスに麻酔をかけて、Optix in vivo蛍光イメージングシステム(GE Healthcare社製)を用いてイメージングを行った。Cy5.5色素のみのコントロール群を除く、他の全ての投与群では少なくとも3匹のマウスを含む。各投与群において最も典型的な1匹のマウスから得られた、代表的な画像を示した。これらの画像は、自家蛍光による障害を除去するために、Cy5.5(0.8〜1.1ns)の蛍光寿命の時間分解を行った。
【0094】
LARLLT及びコントロールペプチドを近赤外色素Cy5.5により標識し、H1299を移植した担癌マウスの尾静脈に注射した。注射後の様々な時点での、前記マウス体内におけるCy5.5の蛍光の分布を画像化した。フリーの色素コントロールを含む全ての投与群において、蛍光は最初体全体で認められ、その後、すぐに腎臓と膀胱に蓄積した。このような腎臓と膀胱における強い蛍光による障害を防ぐために、皮下に腫瘍が存在する部分のみに絞った画像を示した(図5)。Cy5.5色素を注射したマウスにおいては、腫瘍部分における蛍光シグナルはバックグラウンドと同じレベルであった。しかし、LARLLT−Cy5.5を注射したマウスにおいては、注射後1〜6時間の間、腫瘍組織中の蛍光シグナルは周囲の組織と比較して非常に強かった。これに対して、Cy5.5−コントロールペプチドを注射したマウスでは、腫瘍部分における蛍光の蓄積は全く認められなかった。
【0095】
LARLLT及びコントロールペプチド修飾Cy5.5標識リポソームを、H1299を移植した担癌マウスの尾静脈に注射した。注射後の様々な時点において、これらのマウスの体全体をスキャンし、異なる時点での腫瘍部分での蛍光強度を定量化した。図6に示すように、LARLLT修飾リポソームは、注射6時間後から、次第に腫瘍部分の内部に蓄積された。注射後80時間経過した後も、蛍光シグナル及びその対比が有意に認められた。しかし、コントロールペプチド修飾リポソームの場合には、腫瘍部分における蛍光強度は、常にバックグラウンドと同レベルであった。我々は、マウスの体全体における蛍光強度に対する、腫瘍部分の蛍光強度の相対比率を算出した(表1)。腫瘍部分及び体全体の蛍光強度(NC)は、eXplore OptiView 1.0.0.0ソフトウェアを用いて算出した。また、体全体の蛍光強度に対する腫瘍部分の蛍光強度のパーセンテージを表にして示した。
【0096】
表1 蛍光標識リポソームを注射した後の様々な時点における腫瘍部分の蛍光強度。
【0097】
【表1】

【0098】
マウスの体全体の蛍光強度に対する腫瘍部分の蛍光強度の比率は、LARLLT修飾リポソームの注射1時間後から、プラトーに達したと考えられる76〜80時間後まで次第に上昇していった。以上のことから、LARLLT標的指向性リポソームは徐々に腫瘍部分に蓄積され、その後は強力に保持されることが示唆された。
【0099】
いくつかの実施形態においては、本発明のペプチド及びペプチド模倣物は、EGF結合部位から離れた表面ポケットに結合するように設計されていてもよい。このように設計された本発明のペプチド及びペプチド模倣物(及び本発明のペプチドと結合した治療薬)は、EGFRに結合する際にEGFと直接競合することがないため、EGFRに対する結合親和性が、EGFの親和性よりも低かったとしても、EGFR発現細胞に結合することができる。
【0100】
本発明のペプチド及びペプチド模倣物を、例えば、PEG2000−DSPEの末端のような、様々な化合物とコンジュゲートさせてリポソームに導入した場合にも、本発明のペプチド及びペプチド製剤のEGFRへの結合能は維持される。そして、本発明のペプチド及びペプチド模倣物は、化合物(例えば、リポソーム)をEGFR高発現細胞に特異的に接着させてEGFR高発現細胞による取込みを可能とする。以上に示したように、本発明のペプチド及びペプチド模倣物とコンジュゲートした化合物(例えば、前記のような本発明のペプチドを組み込んだリポソーム)をin vivoに投与した場合、その化合物は標的細胞(例えば、腫瘍細胞)に対して指向性を有することから、結果的に、本発明のペプチドとコンジュゲートした化合物(例えば、ペプチドがコンジュゲートしたリポソーム)は前記標的(例えば、腫瘍内)に蓄積される。
【0101】
本研究ではPEG2000リンカーのみを用いた。また、我々の研究においては、リガンドと結合した脂質を2〜4:100の脂質比率の条件で組み込んだ場合に、in vitroでの細胞への結合が最も有意に認められた(バックグラウンドの非特異的な結合が最も低かった)。しかし、当業者に公知のリンカーであれば、他のどのようなリンカーを用いてもよく、また同様に、他の脂質比率を用いてもよい。
【0102】
前記と同様のリポソーム製剤をin vivoで用いた。このリポソームは、その表面上に比較的高いリガンド−PEG密度を持ち、腫瘍にゆっくりと蓄積された。実際、これまでのいくつかの研究において、循環中及び浸潤中の能動的標的指向性リポソームは、ステルスリポソームと同様の振舞いをするに過ぎないことが示唆されている(Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: in vitro and in vivo studies. Br J Cancer 1996;74:1749-1756、Matsumura Y, Gotoh M, Muro K, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol 2004;15(3):517-25、Sapra P, Moase EH, Ma J, Allen TM. Improved therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab' fragments. Clin Cancer Res 2004;10(3):1100-11)。このように、能動的標的指向性リポソームの機能は、腫瘍組織に浸潤した後にのみ意味を持つ。能動的標的指向性リポソームは、その標的指向性によって癌細胞と強固に結合し、容易に細胞に取り込まれることができる(Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9、Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 2006;66(13):6732-40)。in vivo蛍光イメージングシステムを用いて観察した結果(図6)は、前記の作用メカニズムに一致していた。リガンドを組み込んだLARLLTリポソームは、非常に長時間(>80時間)にわたって腫瘍組織に留まることが明らかにされた。一方、我々が標的指向性を持たないステルスリポソームであると予測したとおり、コントロール群を用いた場合には、12〜24時間経過した時点で、腫瘍組織中に有意な蓄積は認められなかった。
【0103】
小動物用in vivo蛍光イメージングシステムは、標識された薬剤及びデリバリーシステムのin vivoでの分布を追跡するために最近開発された装置である。Cy5.5は、組織透過性に優れており、バックグラウンドが低いことから、最も一般的に使用される色素である。in vivo蛍光イメージング技術の主な利点は、非侵襲的であること、さらに、実験の全ての経過を通じて、同一の生きた動物を継続的に観察することが可能なことである。これまでの多くの研究では、in vivoで薬品又は脂質の分布を追跡するために、放射標識した薬品又は脂質が用いられてきたが、平均的な測定結果を得るために、多くの動物を異なる時点で犠牲にする必要があった。本発明者らのin vivo蛍光イメージングシステムを用いた経験では、同じ実験群中の異なる動物個体において、一般的な健康状態、生理機能、代謝機能、腫瘍のサイズ及び位置等がばらついていても問題が無い。また、本実験により得られた能動的標的指向性リポソームの時間連続分布プロファイルは、他の方法を用いたこれまでの報告とよく一致していた(Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9、Sapra P, Moase EH, Ma J, Allen TM. Improved therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab' fragments. Clin Cancer Res 2004;10(3):1100-11)。
【実施例10】
【0104】
本発明のペプチドを用いた標的指向性を有するリポソームが、標的腫瘍における薬剤の蓄積を増加させることができるかについて評価した。5mg/kgのドキソルビシンを、H460腫瘍細胞を移植したヌードマウスの静脈に注射した。また、フリーのドキソルビシン(図7、Dox)と、PEG化したリポソームに封入されたドキソルビシン(図7、PEG)、及び本発明のペプチドを含むリポソームに封入されたドキソルビシン(図7、D4)とを比較した。これらをマウスに注射して0.5時間、6時間、及び48時間後にマウスを安楽死させ、腫瘍を摘出して腫瘍に含まれるドキソルビシンを測定した。各試験群あたりN=3の実験動物を用いた。
【0105】
図7に示すように、フリーのドキソルビシン及びPEG化したリポソームに封入されたドキソルビシンと比較して、本発明のペプチドを含むリポソームを用いた結果、有意に多量のドキソルビシンが腫瘍に蓄積された。
【実施例11】
【0106】
FACS解析による本発明のペプチドを含むリポソームの結合解析
【0107】
H−460細胞を無菌条件下、培養プレート上で増殖させ、スクレーピングによって回収した。回収したH−460細胞に、リポソームサンプル(空のサーモドックスのリポソーム、サーモドックス、D4−サーモドックス、D4*−サーモドックス、ここで、D4−サーモドックスは配列番号1で示されるペプチドを含み、D4*−サーモドックスは配列番号2で示されるペプチドを含む)及びドキソルビシン塩酸塩を加えて、37℃で4時間インキュベートした。本発明のペプチドをそれぞれ異なる量で含む2種類のリポソーム(D4−サーモドックス)の評価を行った。前記の2種類のリポソームをそれぞれD4−サーモドックス2+2及びD4−サーモドックス4+4と名付けた。D4−サーモドックス2+2は、DPPC、MSPC、DSPE−MPEG2000及びD4−DSPE−MPEG2000を90:10:2:2のモル比で含み、D4−サーモドックス4+4は、DPPC、MSPC、DSPE−MPEG2000及びD4−DSPE−MPEG2000を、90:10:4:4のモル比で含んでいた。また、D4*−サーモドックスは、DPPC、MSPC、DSPE−MPEG2000、及びD4*−DSPE−MPEG2000を90:10:2:2のモル比で含んでいた。インキュベーション後、細胞をPBSで4回洗浄し、その後、ドキソルビシンの発する蛍光を用いてFACSによって細胞をカウントした(励起波長488nm、及び発光波長575nm)。
【0108】
以下の群について実験を行った:a.無処理コントロール;b.約0.01〜10μg(ドキソルビシン)/mLのサーモドックス;c.約0.01〜10μg(ドキソルビシン)/mLのD4−サーモドックス2+2;d.約0.01〜10μg(ドキソルビシン)/mLのD4−サーモドックス4+4;e.約0.01〜10μg(ドキソルビシン)/mLのD4*−サーモドックス、及びf.0.01〜10μgのドキソルビシン塩酸塩。
【0109】
実験は少なくとも3回繰り返して行った。同一条件の実験の中から、代表的なヒストグラムを示す。厳密には、細胞数のばらつきやFACSのパラメータによって、実験から得られた数値は変化するかもしれないが、大まかなパターンや傾向の違いは常に一定であった。
【0110】
濃度が10μg/mlの場合、in vivoでのD4−サーモドックス(2+2)のドキソルビシンによる蛍光強度は、サーモドックス及びD4*−サーモドックスよりも強かったが、フリーのドキソルビシン塩酸塩よりは弱かった。フリーの薬剤は細胞膜の透過性が比較的高いので、以上の結果から、D4−リポソームは、コントロールリポソームよりも多くH460細胞に結合することが確認された。濃度が1μg/mlの場合、D4−サーモドックス(2+2、4+4)により処理された細胞は、サーモドックス又はD4*−サーモドックスにより処理された細胞と比較して、より強いドキソルビシン蛍光シグナルを発していた。濃度が0.1μg/ml及び0.01μg/mlの場合、組成の異なるリポソーム間での明確な違いは認められなかった。
【0111】
図8は、実験に用いた各リポソームに含まれるドキソルビシンの投与量に対して、FACS解析により得られたヒストグラムの平均値をプロットした線グラフを示す。ドキソルビシンの細胞への取込み(平均蛍光強度)は、薬剤溶液を用いた場合に最も高かった。これは、フリーのドキソルビシンが非常に効率的に細胞膜を透過することができるためである。異なる組成のリポソームを比較した場合、2つのD4リポソーム製剤は、サーモドックスコントロール及びD4*リポソームコントロールよりも細胞への取込み効率が非常に優れていた。
【0112】
本明細書において言及された全ての刊行物、特許、及び特許出願は、本発明が関連する技術分野における当業者の技術レベルの指標となる。また、本明細書において言及された全ての刊行物、特許、及び特許出願は、それぞれを参照することによって具体的かつ個々に示されるのと同じ程度の範囲が、参照することによって本明細書に援用される。
【0113】
参考文献
1. Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res 1994;54(4):987-92.
2. Maruyama K, Takizawa T, Yuda T, Kennel SJ, Huang L, Iwatsuru M. Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies. Biochim Biophys Acta 1995;1234(1):74-80.
3. Park JW, Hong K, Carter P, et al. Development of anti-p185HER2 immunoliposomes for cancer therapy. Proc Natl Acad Sci U S A 1995;92(5):1327-31.
4. Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: in vitro and in vivo studies. Br J Cancer 1996;74:1749-1756.
5. Lopes de Menezes DE, Pilarski LM, Allen TM. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res 1998;58(15):3320-30.
6. Matsumura Y, Gotoh M, Muro K, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol 2004;15(3):517-25.
7. Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta 1995;1233(2):134-44.
8. Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9.
9. Terada T, Mizobata M, Kawakami S, Yabe Y, Yamashita F, Hashida M. Basic fibroblast growth factor-binding peptide as a novel targeting ligand of drug carrier to tumor cells. J Drug Target 2006;14(8):536-45.
10. Schiffelers RM, Koning GA, ten Hagen TL, et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release 2003;91(1-2):115-22.
11. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995;19:183-232.
12. Lutsenko SV, Feldman NB, Severin SE. Cytotoxic and antitumor activities of doxorubicinconjugates with the epidermal growth factor and its receptor-binding fragment. J Drug Target 2002;10(7):567-71.
13. Jinno H, Ueda M, Ozawa S, et al. Epidermal growth factor receptor-dependent cytotoxic effect by an EGF-ribonuclease conjugate on human cancer cell lines--a trial for less immunogenic chimeric toxin. Cancer Chemother Pharmacol 1996;38(4):303-8.
14. Kullberg EB, Nestor M, Gedda L. Tumor-cell targeted epiderimal growth factor liposomes loaded with boronated acridine: uptake and processing. Pharm Res 2003; 20(2):229-36.
15. Chen P, Mrkobrada M, Vallis KA, et al. Comparative antiproliferative effects of (111) In-DTPA-hEGF, chemotherapeutic agents and gamma-radiation on EGFR-positive breast cancer cells. Nucl Med Biol 2002;29(6):693-9.
16. Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different strategies for formation of pegylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjug Chem 2001;12(4):529-37.
17. Mamot C, Drummond DC, Greiser U, et al. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR-and EGFRvIII-overexpressing tumor cells. Cancer Res 2003;63(12):3154-61.
18. Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res 2005; 65(24):11631-8.
19. Mamot C, Ritschard R, Kung W, Park JW, Herrmann R, Rochlitz CF. EGFR-targeted immunoliposomes derived from the monoclonal antibody EMD72000 mediate specific and efficient drug delivery to a variety of colorectal cancer cells. J Drug Target 2006; 14(4):215-23.
20. Heal JR, Roberts GW, Raynes JG, Bhakoo A, Miller AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochem 2002;3(2-3):136-51.
21. Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639-62.
22. Hetenyi C. van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci 2002;11(7):1729-37.
23. Mayer LD, Tai LC, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta 1990;1025(2):143-51.
24. Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta 1993;1151(2):201-15. Erratum in: Biochim Biophys Acta 1994;1190(1):197.
25. Ishida T, Iden DL, Allen TM. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett 1999;460 (1):129-33.
26. Scudiero DA, Shoemaker RH, Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988;48(17):4827-33.
27. Schmidt M, Vakalopoulou E, Schneider DW, Wels W. Construction and functional characterization of scFv(14E1)-ETA -a novel, highly potent antibody-toxin specific for the EGF receptor. Br J Cancer 1997; 75(11):1575-84.
28. Liu X, Tian P, Yu Y, Yao M, Cao X, Gu J. Enhanced antitumor effect of EGF R-targeted p21WAF-1 and GM-CSF gene transfer in the established murine hepatoma by peritumoral injection. Cancer Gene Ther 2002;9(1):100-8.
29. Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J 2005;19(14):1978-85.
30. de Graaf C, Pospisil P, Pos W, Folkers G, Vermeulen NP. Binding mode prediction of cytochrome p450 and thymidine kinase protein-ligand complexes by consideration of water and rescoring in automated docking. J Med Chem 2005;48(7):2308-18.
31. Rogers JP, Beuscher AE 4th, Flajolet M, et al.. Discovery of protein phosphatase 2C inhibitors by virtual screening. J Med Chem 2006; 49(5):1658-67.
32. Bhakoo A, Raynes JG, Heal JR, Keller M, Miller AD. De-novo design of complementary (antisense) peptide mini-receptor inhibitor of interleukin 18 (IL-18). Mol Immunol 2004;41(12):1217-24.
33. Mastrobattista E, Koning GA, Storm G. Immunoliposomes for the targeted delivery of antitumor drugs. Adv Drug Deliv Rev 1999;40(1-2):103-127.
34. Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res 2006;16(3):175-83.
35. Koning GA, Morselt HW, Gorter A, et al. Pharmacokinetics of differently designed immunoliposome formulations in rats with or without hepatic colon cancer metastases. Pharm Res 2001;18(9):1291-8.
36. Koning GA, Morselt HW, Gorter A, et al. Interaction of differently designed immunoliposomes with colon cancer cells and Kupffer cells. An in vitro comparison. Pharm Res 2003;20(8):1249-57.
37. Derksen JT, Morselt HW, Scherphof GL. Uptake and processing of immunoglobulin-coated liposomes by subpopulations of rat liver macrophages. Biochim Biophys Acta 1988;971(2):127-36.
38. Maruyama K, Takahashi N, Tagawa T, Nagaike K, Iwatsuru M. Immunoliposomes bearing poloethylene glycol-coupled Fab' fragment show prolonged circulation time and high extravasation into targeted solid tumors in vivo. FEBS Lett 1997;413(1):177-80.
39. Huwyler J, Yang J, Pardridge WM. Receptor mediated delivery of daunomycin using immunoliposomes: pharmacokinetics and tissue distribution in the rat. J Pharmacol Exp Ther 1997; 282(3):1541-6.
40. Tardi P, Bally MB, Harasym TO. Clearance properties of liposomes involving conjugated proteins for targeting. Adv Drug Deliv Rev 1998;32(1-2):99-118.
41. Sapra P, Moase EH, Ma J, Allen TM. Improved therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab' fragments. Clin Cancer Res 2004;10(3):1100-11.
42. Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 2006;66(13):6732-40.
【背景技術】
【0001】
癌治療において、化学療法は広く用いられているが、その有効性は、正常な組織に対する薬剤毒性に起因する重篤な副作用によって損なわれることも多い。これまでに多くの研究が、化学療法剤の治療指数を改善することを目的とした、正常組織を回避して腫瘍組織に特異的に薬剤を送達するための戦略に焦点を当ててきた。
【0002】
いくつかのリポソーム化抗癌製剤が開発されており、それらは既に、リポソーム化されていない製剤より治療効果が高く、副作用の少ない製剤として患者に用いられている。最も治療効果の高いリポソーム化抗癌製剤の1つは、ドキシル(カエリックスとしても知られる)である(非特許文献1)。ドキシルは、PEGでコートされたリポソーム(ステルスリポソーム)を含んでおり、血清半減期が長く、透過性の高い血管を通じて徐々に漏出して腫瘍内部に蓄積することができる。このような受動的な標的指向メカニズムに加えて、能動的な標的指向戦略も提案されている。能動的な標的指向戦略においては、薬剤を封入したリポソームと腫瘍との相互作用をより強めるために、抗体や標的指向性リガンドが用いられる。抗体による標的指向性免疫リポソームは非常に活発に研究されており、動物実験と臨床試験の両方において有望視されている(非特許文献2〜6)。また、低分子リガンド(非特許文献7及び8)やペプチド(非特許文献9及び10)も同様に、癌治療用の能動的標的指向性リポソームを構築するために研究されている。
【0003】
in silico又はin vitroでの結合試験と比較して、in vivoでの結合を取り巻く環境は、多くの解剖学的障壁や細網内皮系(RES)等の自然の排出機構、さらに、他の非特異的な相互作用が関与しているために、はるかに複雑なものとなっている。免疫リポソーム及び低分子による標的指向性リポソームを用いて、結合に影響を及ぼす多くの要因が同定されている(非特許文献11及び12)。また、様々な能動的標的指向性リポソームの、薬物動態特性や血管外漏出挙動に影響を及ぼすいくつかの因子が同定されている(非特許文献11及び12)。リポソーム表面にコンジュゲートされたリガンドに対する免疫除去は大きな問題であり(非特許文献13及び14)、IgGを含む免疫リポソームは、Fcレセプターと相互作用して速やかに除去されることが明らかにされている(非特許文献15〜17)。また、低分子リガンドである葉酸をコンジュゲートした場合にも、in vivoでのRESによるリポソームの除去が促進された(非特許文献8)。さらに、リポソーム表面のリガンド密度(非特許文献18)及びPEGリンカーの長さ(非特許文献8及び19)についても、ともに重要であることが明らかにされている。
【0004】
上皮成長因子受容体(EGFR)は、肺癌、乳癌、膀胱癌、及び卵巣癌を含む(ただしこれらに限定されるわけではない)、幅広い種類のヒトの癌において過剰発現している。また、EGFRは、進行疾患の様々な特性や予後不良とも関連していることが明らかにされている(非特許文献20)。EGFRを標的とする様々なベクターやコンジュゲートは、癌細胞への細胞毒性薬、毒素、又は放射性核種の送達を増加させること、また、動物モデルでは腫瘍の増殖を抑制する作用を持つことが報告されている(非特許文献21〜25)。Mamotらは、EGFRに特異的な免疫リポソームを開発し、一連の研究により、in vitro及びin vivoにおいて、この免疫リポソームが優れた送達特性と高い抗癌作用を持つことを示した(非特許文献19、26、及び27)。
【0005】
EGFRを標的とした、いくつかの効果的な薬剤が開発されており、その中には、チロシンキナーゼ阻害剤であるタルセバ(登録商標)及びイレッサ(登録商標)、及び、EGFRのブロッキング抗体であるアービタックス(登録商標)が含まれる。このようなEGFR自体を標的とする薬剤を設計することに加えて、他の戦略として、従来の治療剤をEGFRに向けて送達するシステムの開発がある。多くの研究では、EGFRの天然のリガンドである上皮成長因子(EGF)を使用することによって、標的指向性の細胞毒性薬剤、毒素、リポソーム、及び、他の薬剤/遺伝子のデリバリーシステムの開発や、標的指向性の放射性核種システムの開発が行われてきた(非特許文献21〜24)。しかし、前記の戦略では、EGFのもつ内在性増殖促進作用が、癌細胞に及ぼす影響が常に問題となる。このため、他に2つの戦略が提案されている。1つは、EGFRに結合する抗体又は抗体断片を用いる方法であり(非特許文献26〜28)、もう1つは低分子ペプチド又はEGFの断片を用いる方法である(非特許文献21、29、及び30)。このような方法においては、EGFRに強固且つ特異的に結合することが可能で、免疫原性及び細胞増殖促進作用が低い低分子ペプチドは特に望ましい。
【0006】
当技術分野においては、例えば、EGFRを過剰発現する細胞等の標的細胞に、治療用薬剤を効果的に送達するための材料及び方法は依然として必要とされている。本発明は、このような必要性に応えるものである。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res 1994;54(4):987-92
【非特許文献2】Maruyama K, Takizawa T, Yuda T, Kennel SJ, Huang L, Iwatsuru M. Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies. Biochim Biophys Acta 1995;1234(1):74-80
【非特許文献3】Park JW, Hong K, Carter P, et al. Development of anti-p185HER2 immunoliposomes for cancer therapy. Proc Natl Acad Sci U S A 1995;92(5):1327-31
【非特許文献4】Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: in vitro and in vivo studies. Br J Cancer 1996;74:1749-1756
【非特許文献5】Lopes de Menezes DE, Pilarski LM, Allen TM. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res 1998;58(15):3320-30
【非特許文献6】Matsumura Y, Gotoh M, Muro K, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol 2004;15(3):517-25
【非特許文献7】Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta 1995;1233(2):134-44
【非特許文献8】Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9
【非特許文献9】Terada T, Mizobata M, Kawakami S, Yabe Y, Yamashita F, Hashida M. Basic fibroblast growth factor-binding peptide as a novel targeting ligand of drug carrier to tumor cells. J Drug Target 2006;14(8):536-45
【非特許文献10】Schiffelers RM, Koning GA, ten Hagen TL, et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release 2003;91(1-2):115-22
【非特許文献11】Mastrobattista E, Koning GA, Storm G. Immunoliposomes for the targeted delivery of antitumor drugs. Adv Drug Deliv Rev 1999;40(1-2):103-127
【非特許文献12】Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res 2006;16(3):175-83
【非特許文献13】Koning GA, Morselt HW, Gorter A, et al. Pharmacokinetics of differently designed immunoliposome formulations in rats with or without hepatic colon cancer metastases. Pharm Res 2001;18(9):1291-8
【非特許文献14】Koning GA, Morselt HW, Gorter A, et al. Interaction of differently designed immunoliposomes with colon cancer cells and Kupffer cells. An in vitro comparison. Pharm Res 2003;20(8):1249-57
【非特許文献15】Derksen JT, Morselt HW, Scherphof GL. Uptake and processing of immunoglobulin-coated liposomes by subpopulations of rat liver macrophages. Biochim Biophys Acta 1988;971(2):127-36
【非特許文献16】Maruyama K, Takahashi N, Tagawa T, Nagaike K, Iwatsuru M. Immunoliposomes bearing poloethylene glycol-coupled Fab' fragment show prolonged circulation time and high extravasation into targeted solid tumors in vivo. FEBS Lett 1997;413(1):177-80
【非特許文献17】Huwyler J, Yang J, Pardridge WM. Receptor mediated delivery of daunomycin using immunoliposomes: pharmacokinetics and tissue distribution in the rat. J Pharmacol Exp Ther 1997; 282(3):1541-6
【非特許文献18】Tardi P, Bally MB, Harasym TO. Clearance properties of liposomes involving conjugated proteins for targeting. Adv Drug Deliv Rev 1998;32(1-2):99-118
【非特許文献19】Mamot C, Ritschard R, Kung W, Park JW, Herrmann R, Rochlitz CF. EGFR-targeted immunoliposomes derived from the monoclonal antibody EMD72000 mediate specific and efficient drug delivery to a variety of colorectal cancer cells. J Drug Target 2006; 14(4):215-23
【非特許文献20】Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995;19:183-232
【非特許文献21】Lutsenko SV, Feldman NB, Severin SE. Cytotoxic and antitumor activities of doxorubicin conjugates with the epidermal growth factor and its receptor-binding fragment. J Drug Target 2002;10(7):567-71
【非特許文献22】Jinno H, Ueda M, Ozawa S, et al. Epidermal growth factor receptor-dependent cytotoxic effect by an EGF-ribonuclease conjugate on human cancer cell lines--a trial for less immunogenic chimeric toxin. Cancer Chemother Pharmacol 1996;38(4):303-8
【非特許文献23】Kullberg EB, Nestor M, Gedda L. Tumor-cell targeted epiderimal growth factor liposomes loaded with boronated acridine: uptake and processing. Pharm Res 2003; 20(2):229-36
【非特許文献24】Chen P, Mrkobrada M, Vallis KA, et al. Comparative antiproliferative effects of (111) In-DTPA-hEGF, chemotherapeutic agents and gamma-radiation on EGFR-positive breast cancer cells. Nucl Med Biol 2002;29(6):693-9
【非特許文献25】Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different strategies for formation of pegylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjug Chem 2001;12(4):529-37
【非特許文献26】Mamot C, Drummond DC, Greiser U, et al. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR-and EGFRvIII-overexpressing tumor cells. Cancer Res 2003;63(12):3154-61
【非特許文献27】Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res 2005; 65(24):11631-8
【非特許文献28】Schmidt M, Vakalopoulou E, Schneider DW, Wels W. Construction and functional characterization of scFv(14E1)-ETA -a novel, highly potent antibody-toxin specific for the EGF receptor. Br J Cancer 1997; 75(11):1575-84
【非特許文献29】Liu X, Tian P, Yu Y, Yao M, Cao X, Gu J. Enhanced antitumor effect of EGF R-targeted p21WAF-1 and GM-CSF gene transfer in the established murine hepatoma by peritumoral injection. Cancer Gene Ther 2002;9(1):100-8
【非特許文献30】Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J 2005;19(14):1978-85
【発明の概要】
【発明の効果】
【0008】
本発明は、目的とする標的細胞に物質を送達するために用いることができるペプチドを提供する。前記物質としては、例えば、治療剤及び/又は造影剤を挙げることができる。1つの実施形態において、本発明は、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドを提供する。また、1つの実施形態においては、前記ペプチドは、LARLLT配列(配列番号1)を含んでいてもよい。本発明のペプチドはどのような長さであってもよく、前記ペプチドの長さとしては、例えば、約6アミノ酸〜約15アミノ酸、又は約6アミノ酸〜約10アミノ酸の長さを挙げることができる。本発明のペプチドは、6、7、8、9、10、又は11アミノ酸以上の長さであってもよい。いくつかの実施形態においては、本発明のペプチドは、長さが6アミノ酸のペプチドであって、且つ、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列からなるペプチドである。いくつかの実施形態においては、本発明のペプチドは、アミノ酸配列LARLLTからなるペプチドであってもよい。さらに、本発明は、本発明のペプチドのEGFR結合活性を模倣したペプチド模倣物や、本発明のペプチドの機能を保持したアミノ酸置換ペプチドを提供する。
【0009】
他の実施形態においては、本発明のペプチド及びペプチド模倣物は、1又は2以上の化合物に直接的又は間接的に結合(attached)していてもよい。いくつかの実施形態においては、本発明のペプチドは、治療剤及び/又は造影剤に結合している。1つの実施形態においては、本発明は、脂質と、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドとを含む分子を提供する。いくつかの実施形態においては、前記ペプチドは、LARLLT配列を含んでいてもよい。また、本発明のペプチドは、リンカーを使用することによって間接的に脂質に結合していてもよく、リンカーとしては、当業者に公知の任意のリンカーを用いることができる。本発明のペプチド及びペプチド模倣物と結合する脂質の好適な例としては、特に制限されないが、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)等を挙げることができる。いくつかの実施形態においては、リンカーはポリエチレングリコール(PEG)を含んでいてもよい。リンカーと結合した脂質の例としては、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)を挙げることができる。前記のようなリンカーと結合した脂質を含む分子は、さらに、1又は数個の本発明のペプチド及びペプチド模倣物と結合していてもよい。
【0010】
本発明のペプチド及びペプチド模倣物は、活性物質と結合していてもよく、活性物質としては、例えば、治療剤及び/又は造影剤を挙げることができる。また、本発明のペプチド及びペプチド模倣物と活性物質との結合は、直接的であっても、間接的であってもよい。本発明のペプチド及びペプチド模倣物は、活性物質と、本発明のペプチド及びペプチド模倣物と前記活性物質の官能基とが直接的に反応することよって結合していてもよい。また、本発明のペプチド及びペプチド模倣物は、活性物質と、前記ペプチドと前記活性物質とがリンカーを介して間接的に反応することよって結合していてもよく、この場合、リンカーは通常、前記ペプチド及び前記活性物質の両方と共有結合を形成することが可能な二官能性の分子である。
【0011】
他の実施形態においては、本発明はリポソームを包含する。本発明のリポソームの例としては、脂質及び本発明のペプチドを含む分子を含むリポソームを挙げることができ、本発明のペプチドとしては、例えば、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドを挙げることができる。また、いくつかの実施形態においては、前記ペプチドはLARLLT配列を含んでいてもよい。上述のように、本発明のリポソームにおいて用いられる分子は、脂質部分とペプチド部分との間にリンカーを含んでいてもよい。1つの実施形態においては、前記脂質は、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)であってもよい。また、1つの実施形態においては、前記リンカーはポリエチレングリコール(PEG)であってもよい。1つの実施形態においては、脂質−リンカー−ペプチドの組合せは、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTであってもよい。また、本発明のリポソームは、当業者に公知の任意の脂質又は脂質の組合せを用いて作製してもよい。リポソームの作製に用いる好適な脂質の例としては、特に制限されないが、リン脂質(例えば、ホスファチジルコリン、ホスファチジルグリセロール、及びホスファチジルエタノールアミン)、リゾ脂質、及びPEG化リン脂質等を挙げることができ、このような好適な脂質はどのような比率で組み合わされていてもよい。本発明に用いるリポソームは、任意選択で、ゲル相から液相への相転移温度が約39.0℃〜約45℃の脂質を含む熱感受性リポソームであってもよい。好適なリポソームの例の1つとして、DPPC及びMSPCを含むリポソームであって、DPPC:MSPCのモルパーセント比が、99:1、98:2、97:3、96:4、95:5、90:10から約80:20、75:25、70:30、65:35、60:40のリポソームや、又は、DPPC:MSPCのモルパーセント比が51:49のリポソームを挙げることができる。また、熱感受性リポソームには、さらに1又は数種の脂質(例えば、前記ペプチド−リンカー−及び/又はPEG化脂質)が組み込まれていてもよい。
【0012】
本発明のリポソームは、治療用化合物及び造影化合物からなる群より選択される1又は数種の化合物を、さらに含んでいてもよい。本発明のリポソームは通常、二重膜を有して内部空間を画定し、さらに、化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる。また、いくつかの実施形態においては、化合物は前記リポソームの外側に会合(associated)していてもよい。本発明のリポソームに封入するための、好適な種類の化合物の例の1つとして、抗癌化合物を挙げることができる。好適な抗癌化合物の例としては、特に制限されないが、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物等を挙げることができる。いくつかの実施形態においては、本発明のリポソームは、ドセタキセルを含んでいてもよい。また、いくつかの実施形態においては、本発明のリポソームは、ドキソルビシンを含んでいてもよい。さらに、いくつかの実施形態においては、本発明のリポソームは、プラチナ含有化合物を含んでいてもよく、プラチナ含有化合物としては、例えば、カルボプラチン又はシスプラチンを挙げることができる。
【0013】
さらに本発明は、化合物を、その化合物を必要とする対象に投与する方法を提供する。この方法は通常、前記化合物と、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドとを会合させる工程、及び、この工程によって得られたペプチドと会合した化合物を対象に接触させる工程を含む。いくつかの実施形態において、前記ペプチドはLARLLT配列を含んでいてもよい。また、ペプチドと化合物との会合は、共有結合性であっても、非共有結合性であってもよい。いくつかの実施形態において、前記ペプチドは、直接的又は間接的に活性物質と結合していてもよい。いくつかの実施形態においては、前記ペプチドは脂質に結合していてもよく、さらに、前記ペプチドが結合した脂質は、リポソームを形成していてもよい。送達される化合物はリポソームの一部であってもよく、例えば、リポソームの二重膜中にあっても、リポソームの内部空間に封入されていてもよい。また、前記ペプチドは化合物に結合していてもよく、さらに、前記化合物がリポソームに含まれていてもよい。ここで、化合物は、例えば、前記リポソームの二重膜中に含まれていても、前記リポソームの内部空間に封入されていてもよい。前記ペプチドが脂質に結合する場合には、前記ペプチドが結合した脂質は、前記脂質と前記ペプチドの間にリンカーを含んでいてもよい。本発明に用いる好適な脂質の例としては、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)を挙げることができ、好適なリンカーは、ポリエチレングリコール(PEG)を含んでいてもよい。従って、リンカーを介して本発明のペプチドと結合した脂質の例の1つとしては、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを挙げることができる。さらに、前記リンカーを介して本発明のペプチドと結合した脂質は、リポソームに組み込んで本発明の方法に用いてもよい。本発明の脂質−リンカー−ペプチドを含むリポソームは、当業者に公知の他のどのような脂質を含んでいてもよい。いくつかの本発明の方法においては、上述のような脂質−リンカー−ペプチドを含むリポソームに化合物が会合していてもよく、さらに、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃であってもよい。このようなリポソームは、それを必要とする対象に投与されてもよく、さらに、かかる対象の一部は、前記リポソームのゲル相から液相への相転移温度よりも高い温度まで加温されてもよい。
【0014】
標的組織を加温する方法としては、適切な方法であればどのような方法を用いてもよいが、例えば、無線周波数照射の使用、高密度焦点式超音波等の超音波の使用、マイクロ波放射の使用、温浴槽や光などの赤外線放射源の使用を挙げることができ、また同様に、他の形態として、放射性同位体等による外部又は内部からの放射線照射、又は電場及び磁場を挙げることができる。
【0015】
本発明の方法を用いることによって、任意の化合物を投与することができる。投与する化合物の好適な例としては、特に制限されないが、治療用化合物及び造影化合物等を挙げることができる。これら化合物は、本発明のペプチドに直接的に又は間接的に結合していても、又は、本発明のペプチドと結合した脂質を含むリポソームに含まれていてもよい。本発明の方法の1つにおいては、リポソームは二重膜を有して内部空間を画定していてもよく、さらに、前記化合物は、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれていてもよい。また、本発明の方法は、治療用化合物を送達するために用いることができ、治療用化合物としては、例えば、抗癌化合物を挙げることができる。抗癌化合物としては、特に制限されないが、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物等を挙げることができる。いくつかの実施形態においては、本発明の方法は、ドセタキセルを送達するために用いてもよい。また、いくつかの実施形態においては、本発明の方法は、ドキソルビシンを送達するために用いてもよい。さらに、いくつかの実施形態においては、本発明の方法は、プラチナ含有化合物を送達するために用いてもよく、プラチナ含有化合物としては、例えば、カルボプラチン又はシスプラチンを挙げることができる。
【0016】
1つの実施形態においては、本発明は癌を治療する方法を提供する。この方法は、本発明のペプチドと会合した抗癌化合物を、それを必要とする対象に投与する工程を含む。前記会合の例の1つとしては、本発明のペプチドと抗癌化合物との直接的又は間接的な結合を挙げることができる。また、他の例としては、前記抗癌化合物がリポソームに封入されており、ここで、リポソームは、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドと会合した脂質を含む。いくつかの実施形態においては、前記ペプチドはLARLLT配列を含んでいてもよい。前記ペプチドは通常、前記脂質と直接又はリンカーを介して結合する。好適な脂質は、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)であり、好適なリンカーは、ポリエチレングリコール(PEG)である。従って、抗癌化合物を送達するために用いられるリポソームに組み込まれる好適なペプチド−リンカー−脂質の例の1つとして、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを挙げることができる。リポソームの作製に用いることのできる脂質の好適な例としては、特に制限されないが、リン脂質(例えば、ホスファチジルコリン、ホスファチジルグリセロール、及びホスファチジルエタノールアミン)、リゾ脂質、及びPEG化リン脂質等を挙げることができる。本発明の癌を治療する方法に用いるリポソームは、任意選択で、ゲル相から液相への相転移温度が約39.0℃〜約45℃のリポソームであってもよい。好適なリポソームの例の1つとして、DPPC及びMSPCを含むリポソームであって、DPPC:MSPCのモルパーセント比が、99:1、98:2、97:3、96:4、95:5、90:10から約80:20、75:25、70:30、65:35、60:40のリポソームや、又は、DPPC:MSPCのモルパーセント比が51:49のリポソームを挙げることができる。前記ペプチド−リンカー−脂質は、このようなリポソームに組み込むことができる。本発明の癌を治療する方法には、公知の任意の抗癌剤を用いることができる。かかる抗癌剤としては、特に制限されないが、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物等を挙げることができる。いくつかの実施形態において抗癌化合物は、ドセタキセルであってもよい。また、いくつかの実施形態において抗癌化合物は、ドキソルビシンであってもよい。さらに、いくつかの実施形態において抗癌化合物は、プラチナ含有化合物であってもよく、プラチナ含有化合物としては、例えば、カルボプラチンを挙げることができる。いくつかの本発明の癌を治療する方法においては、抗癌化合物は、上述のペプチド−リンカー−脂質を含むリポソームと会合していてもよく、さらに、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃であってもよい。前記方法は、対象の一部を加温する工程をさらに含んでいてもよい。対象を加温する方法としては、どのような方法を用いてもよいが、例えば、マイクロ波エネルギーの使用、他の電磁波エネルギー、高周波焼灼、高密度焦点式超音波、超音波、温水の使用等を挙げることができる。
【図面の簡単な説明】
【0017】
【図1】EGFRにおける、LARLLT及びコントロールペプチドのドッキング構造。A.PDBによるEGFR構造モデル。アスタリスクで示した領域は、EGF結合部位である。円内の領域は、本発明者らがドッキング解析に選んだ結合ポケットである。B.EGFRのポケット内における、LARLLTの最小エネルギードッキング構造。LARLLTペプチドを棒球モデルで示す。また、水素結合の可能性のある部位を円柱で示す。C.EGFRのポケット内における、コントロールペプチドの最小エネルギードッキング構造。
【図2】EGFRを高発現する細胞に対する、リガンドによる標的指向性リポソームの結合に関する蛍光顕微鏡実験。パネルA:LARLLT、コントロールペプチド、及びEGFによる指向性リポソームのH1299細胞への結合。(i)LARLLTリポソーム、(iii)EGFリポソーム、(v)コントロールペプチドリポソーム。(ii)、(iv)、(vi)は、(i)、(ii)、(v)と同視野の位相差顕微鏡写真。パネルB:4℃又は37℃における、50倍モル過剰量のフリーのリガンドの存在下での、LARLLTによる指向性リポソームのH1299細胞への結合。(i)37℃における、過剰量のフリーのLARLLT存在下での結合、(ii)37℃における、フリーのLARLLT非存在下での結合、(iii)4℃における、過剰量のフリーのLARLLT存在下での結合、(iv)4℃における、フリーのLARLLT非存在下での結合、(v)37℃における、過剰量のフリーのEGF存在下での結合、(vi)37℃における、フリーのEGF非存在下での結合。パネルC:LARLLT、コントロールペプチドリポソームのSPC−A1細胞への結合。(i)LARLLTリポソーム、(iii)コントロールペプチドリポソーム。(ii)及び(iv)は、(i)及び(iii)と同視野の位相差顕微鏡写真。
【図3】H1299細胞における、LARLLTがコンジュゲートしたリポソームの内在化。A.エンドサイトーシスによって細胞に取り込まれたLARLLTリポソームの位相差画像及び蛍光画像のオーバーレイ画像。B.共焦点蛍光顕微鏡のZ軸スキャンモードを用いて得られた細胞の上部から底部までの11の断面画像。
【図4】ドキソルビシンを含む標的指向性リポソームの殺細胞効果。A.H1299細胞。算出したIC50の値は、LARLLTリポソーム(黒色ひし形)では〜13μg/ml、コントロールペプチドリポソーム(白抜き四角)では85μg/ml、フリーのドキソルビシン単独(黒色三角)では5.7μg/mlであった。B.SPCA1細胞。算出したIC50の値は、LARLLTリポソーム(黒色ひし形)では〜5μg/ml、コントロールペプチドリポソーム(白抜き四角)では15μg/ml、フリーのドキソルビシン単独(黒色三角)では2μg/mlであった。
【図5】担癌マウス体内のCy5.5及びCy5.5標識ペプチドの蛍光画像。ここで示す蛍光画像は、フリーのCy5.5色素、LARLLT−Cy5.5、及びコントロールペプチド−Cy5.5を注射して1及び6時間後に撮影した。
【図6】腫瘍組織における、ペプチドによる指向性リポソームの分布及び蓄積を示す蛍光画像。ここで示す画像(左から右に向かって)は実験に用いたマウスの可視光画像、LARLLTリポソーム(上段)及びコントロールペプチドリポソーム(下段)を注射して1時間、6時間、12時間、24時間、48時間、80時間後の蛍光画像である。
【図7】図7は、薬剤を注射した後の様々な時点における、ヌードマウスに異種移植されたH460腫瘍塊に蓄積したドキソルビシンの濃度を示す棒グラフである。黒色バーは、ドキソルビシンの溶液を注射後の腫瘍中のドキソルビシン濃度を示す。また、点線付き黒色バーは、従来のドキソルビシンを含むリポソームを注射した後の腫瘍中のドキソルビシン濃度を、波線付き黒色バーは、ドキソルビシンを含むLARLLT修飾リポソームを注射した後の腫瘍中のドキソルビシン濃度を示す。
【図8】図8は、0.01〜10μg/mlでの様々な濃度のドキソルビシンを含む、ドキソルビシン含有製剤により処理したH460細胞の蛍光強度を示す線グラフである。x=フリーのドキソルビシン、◇=D4*リポソーム、▲=D4 4+4リポソーム、○=D4 2+2リポソーム、●=サーモドックスリポソーム。D4は配列番号1及を含むリポソームであり、D4*は配列番号2を含むリポソームである。D4−サーモドックス2+2は、DPPC:MSPC:DSPE−MPEG2000:D4−DSPE−MPEG2000を90:10:2:2のモル比で含み、D4−サーモドックス4+4は、DPPC:MSPC:DSPE−MPEG2000:D4−DSPE−MPEG2000を90:10:4:4のモル比で含む。D4*−サーモドックスはDPPC:MSPC:DSPE−MPEG2000:D4*−DSPE−MPEG2000を90:10:2:2のモル比で含む。
【発明を実施するための形態】
【0018】
ペプチド
【0019】
本発明は、EGFRと強固且つ特異的に結合し得る低分子ペプチドを提供する。本発明のペプチド及びペプチド模倣物は通常、免疫原性及び/又は増殖促進作用を伴うことなく、EGFRと結合することができる。
【0020】
本発明の典型的なペプチドとしては、アミノ酸配列LeuAlaArgLeuLeuThr(配列番号1)を含むペプチドを挙げることができる。これに加えて、本発明のペプチドの例としては、特に制限されないが、配列番号1に示されるアミノ酸配列のうち、1又は数個のアミノ酸が異なるアミノ酸によって置換されたペプチド等を挙げることができる。また、いくつかの実施形態においては、配列番号1アミノ酸位置のうち、1箇所のみが置換されていてもよく、さらに、いくつかの実施形態においては、配列番号1のアミノ酸位置のうち、2箇所以上が置換されていてもよい。これらの置換は、配列番号1の任意のアミノ酸位置において行うことができる。また、いくつかの実施形態においては、配列番号1のアミノ酸位置のうち、1又は数箇所のアミノ酸位置が、1又は数個の天然アミノ酸によって置換されていてもよく、さらに、いくつかの実施形態においては、配列番号1のアミノ酸位置のうち、1又は数箇所のアミノ酸位置が、1又は数個の非天然アミノ酸によって置換されていてもよい。いくつかの実施形態においては、本発明のペプチドは、1又は数個のD−アミノ酸を含んでいてもよい。
【0021】
本発明のペプチドに含まれるアミノ酸の置換は通常、同類置換であってもよい。本明細書において、同類置換とは、1つのアミノ酸を、物理的及び/又は化学的な性質が類似した他の1つのアミノ酸によって置き換えることを指す。遺伝学的にコードされた標準的なアミノ酸は、その性質、特に極性と電荷に従って分類することができる。便宜的な分類の1つを以下に示す:グリシン及びアラニン;セリン、トレオニン、アスパラギン、グルタミン及びシステイン;リシン、アルギニン及びヒスチジン;アスパラギン酸及びグルタミン酸;バリン、ロイシン、イソロイシン、及びメチオニン;及びフェニルアラニン、トリプトファン及びチロシン。ある同類グループのアミノ酸を、同じ同類グループの他のアミノ酸によって置き換えることは「同類置換」であると見なされる。本発明のペプチドの性質は通常、このような置換によって実質的に影響を受けることはなく、ここで本発明のペプチドの性質としては、例えば、EGFRに対する結合能を挙げることができる。このように、本発明のペプチドは、配列番号1に示されるアミノ酸配列から、同類置換のみによって1又は数個のアミノ酸が置換された、配列番号1とは異なるペプチドを含む。
【0022】
本発明のペプチドの例としては、特に制限されないが、以下に示すアミノ酸配列等を挙げることができる:
【0023】
Leu Ala Arg Leu Leu Thr(配列番号1)
【0024】
Xaa1 Ala Arg Leu Leu Thr(配列番号3)
【0025】
Leu Xaa2 Arg Leu Leu Thr(配列番号4)
【0026】
Leu Ala Xaa3 Leu Leu Thr(配列番号5)
【0027】
Leu Ala Arg Xaa4 Leu Thr(配列番号6)
【0028】
Leu Ala Arg Leu Xaa5 Thr(配列番号7)
【0029】
Leu Ala Arg Leu Leu Xaa6(配列番号8)
【0030】
ここで、Xaa1はバリン、イソロイシン、及びメチオニンからなる群より選択され、Xaa2はグリシンであり、Xaa3はリシン及びヒスチジンからなる群より選択され、Xaa4はバリン、イソロイシン、及びメチオニンからなる群より選択され、Xaa5はバリン、イソロイシン、及びメチオニンからなる群より選択され、Xaa6はセリン、アスパラギン、グルタミン及びシステインからなる群より選択される。また、本発明には前記の置換を2以上含むペプチドも包含される。
【0031】
さらに本発明は、前記本発明のペプチドと同様のEGFRに対する結合能を示す非ペプチド性化合物にも関する。本発明の実施に際しては、このような本発明のペプチドを模倣した能力を有するペプチド模倣物又は「低分子」を用いてもよい。当業者であれば、Fauchere J., Adv. Drug Res. 15: 29 (1986)、及びEvans et al., J. Med. Chem. 30: 1229 (1987)に述べられているような周知の手法を用いて、ペプチド模倣物を設計することができる。
【0032】
EGFRに対する結合能を有するペプチド模倣物は、本発明のペプチドと同じように用いることができる。かかるペプチド模倣物は通常、in vivoでの化学分解に対する耐性等の望ましい性質を付与するような結合によって、任意に置換された1又は数個のペプチド結合を有している。このような結合としては、特に制限されないが、−−CH2NH−−、−−CH2S−−、−−CH2CH2−−、−−CH=CH−−、−−COCH2−−、−−CH(OH)CH2−−、及び、−−CH2SO−−等を挙げることができる。
【0033】
本発明のペプチド及びペプチド模倣物を、任意の所望の物質と会合させることによって、前記物質を目的とする標的細胞に送達することができる。前記物質の好適な例としては、特に制限されないが、化合物(例えば、活性物質、治療剤、造影剤等)、リポソーム(例えば、熱感受性リポソーム及びステルスリポソーム)、及びナノ粒子(例えば、タンパク質ナノ粒子、金属ナノ粒子、及び結晶化低分子ナノ粒子等)等を挙げることができる。
【0034】
本発明のペプチド及びペプチド模倣物と、送達される物質との会合は、共有結合性であっても、非共有結合性であってもよく、また、直接的であっても、間接的であってもよい。いくつかの実施形態において、本発明のペプチドは、送達される物質と共有結合によって結合していてもよく、かかる結合は、例えば、リンカーを介したものであってもよい。
【0035】
リンカーの例としては、本発明のペプチドと、送達される物質とに、共有結合によって結合することができる架橋剤を挙げることができる。このような架橋剤は通常、本発明のペプチド及び結合相手の物質に存在する官能基(例えば、−OH基、NH2基、COOH基、及び−SH基)と反応する。また、様々な官能基との、架橋剤の異なる反応性に基づいて、様々な架橋剤を選択することができる。
【0036】
現在当業者に公知の、活性化した官能基を介してポリペプチドをコンジュゲート又はカップリングさせる手法が特に適用可能である。これについては、例えば、Aurameas, et al., Scand. J. Immunol., Vol. 8, Suppl. 7:7-23 (1978)、米国特許第4493795号明細書、米国特許第3791932号明細書、及び米国特許第3839153号明細書を参照することができる。これらの文献は、ペプチドのカップリングの開示に関して特に本明細書に援用される。また、部位特異的なカップリング反応を行うことによって、カップリング後のペプチドの配向による活性の喪失を最小限に抑えることができる。これについては、例えば、Rodwell et al., Biotech., 3:889-894 (1985)、及び米国特許第4671958号明細書の記載を参照することができる。前記の手法の他、ペプチドを架橋するための手法として、Klipstein, et al., J. Infect. Dis., 147:318-326 (1983)等に記載の、マイケル付加反応体、グルタルアルデヒド等のジアルデヒドを用いる手法や、アミド結合の形成のために水溶性カルボジイミドを用いるカルボジイミド法を例示することができる。また、N−又はC−末端にシステインが導入されたペプチドをコンジュゲートさせるために、ヘテロ二官能性架橋剤であるSPDP(N−サクシニミジル−3−(2−ピリジルジチオ)プロピオナート)を用いることもできる。
【0037】
本明細書に記載のように、ペプチド結合を形成させる方法や、縮合剤を使用する方法や、さらに、公知の二官能性架橋剤を用いる方法などの、当業者に公知の化学的なコンジュゲート方法によって、前記ペプチドは活性物質とコンジュゲートさせることができる。コンジュゲートにおいては、ペプチドと活性物質とが、その間にどのような基も含まずに直接的に結合していてもよく、このような直接的な結合の例としては、直接的なペプチド結合を挙げることができる。また、コンジュゲートにおいては、ペプチドと活性物質とが、仲介する基を挟んで間接的に結合していてもよく、仲介する基としては、例えば、血漿アルブミン等のタンパク質やペプチド、又は他のスペーサー分子等を挙げることができる。また、例えば、前記間接的な結合はヘテロ二官能性又はホモ二官能性架橋剤を介したものであってもよく、架橋剤としては、例えば、カルボジイミド、グルタルアルデヒド、N−サクシニミジル 3−(2−ピリジルジチオ)プロピオナート(SPDP)及びその誘導体、ビスマレイミド、4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシラート等を挙げることができる。また、架橋反応は、外来の架橋剤を用いることなく、コンジュゲートする分子上の反応基を用いて行ってもよい。ペプチド分子を化学的に架橋する方法は一般的に当業者に公知であり、多数のヘテロ及びホモ二官能性架橋剤が、例えば、米国特許第4355023号明細書、米国特許第4657853号明細書、米国特許第4676980号明細書、米国特許第4925921号明細書、米国特許第4970156号明細書に記載されている。また、これらに加えて、市販の架橋剤を用いることもできる。市販の架橋剤としては、例えば、Pierce Chemical Co.,(Rockford, IL. 61105)社製の架橋剤を挙げることができる。前記各文献は参照することにより本明細書に援用される。生理的な条件下で前記コンジュゲートから治療剤を切り離すために、開裂可能な架橋剤、特にジスルフィド結合の開裂が可能な架橋剤を用いてもよい。このような開裂可能な架橋剤の例としては、4−サクシニミジルオキシカルボニル−a−(2−ピリジルジチオ)−トルエンを挙げることができる。以上のような架橋を含むコンジュゲーションは、コンジュゲートされた後のペプチド及び活性物質の望ましい機能を損なわないように行う必要がある。
【0038】
本発明の1つの実施形態においては、本発明のペプチド及びペプチド模倣物はナノ粒子に結合していてもよく、ナノ粒子としては種々の重合体や金属ナノ粒子、リポソーム、ニオソーム、固体脂質粒子、ミセル、量子ドット、デンドリマー、マイクロカプセル、細胞、ゴースト細胞、リポタンパク質、及び、様々なナノ集合体等の様々なナノ粒子を用いることができる。タンパク質ナノ粒子は、当技術分野に公知の方法を用いて作製することができる。タンパクナノ粒子の作製方法としては、例えば、修正した脱溶媒和−架橋法を挙げることできる。タンパク質を含む水溶液(2%、w/v)を、活性物質とともに緩衝液中でインキュベートした後に、エタノールに滴下して加えることによって、活性物質が組み込まれたタンパク質ナノ粒子を形成させることができる。ナノ粒子は、本発明のペプチドと架橋されていてもよく、例えば、グルタルアルデヒドを用いて本発明のペプチドと架橋されていてもよい。金ナノ粒子は、還元剤として水素化ホウ素ナトリウムを用いる、標準的な湿式化学合成法に従って合成することができる。
【0039】
1つの実施形態においては、本発明のペプチド及びペプチド模倣物は脂質に結合して、リポソームに組み込まれていてもよい。前記結合としては、特に制限されないが、例えば、本発明のペプチド及びペプチド模倣物が脂質の頭部基部分に直接結合していても、又は、脂質の頭部基部分にポリマー(例えば、PEG)等のリンカーを介して結合していてもよい。リポソーム(熱感受性リポソーム、すなわち、ゲル相から液相への相転移温度が約39.0℃〜約45℃であるリポソーム)は通常、活性物質(例えば、治療剤及び/又は造影剤)を含む。
【0040】
本発明のリポソームは通常、1又は数種類のホスファチジルコリンを含む。本発明の実施に用いることのできるホスファチジルコリンの好適な例としては、特に制限されないが、1,2−ジラウロイル−sn−グリセロ−3−ホスホコリン(DLPC)、1,2−ジミリストイル−sn−グリセロ−3−ホスホコリン(DMPC)、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)、1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン(DSPC)、1,2−ジオレオイル−sn−グリセロ−3−ホスホコリン(DOPC)、及び1−パルミトイル−2−オレオイル−sn−グリセロ−3−ホスホコリン(POPC)等を挙げることができる。
【0041】
本発明のリポソームは通常、1又は数種類のホスファチジルグリセロールを含む。ホスファチジルグリセロールの好適な例としては、特に制限されないが、1,2−ジミリストイル−sn−グリセロ−3−ホスホグリセロール(DMPG)、1,2−ジパルミトイル−sn−グリセロ−3−ホスホグリセロール(DPPG)、1,2−ジステアロイル−sn−グリセロ−3−ホスホグリセロール(DSPG)、及び1−パルミトイル−2−オレオイル−sn−グリセロ−3−ホスホグリセロール(POPG)等を挙げることができる。
【0042】
本発明のリポソームは通常、1又は数種類のリゾ脂質を含む。本明細書において、「リゾ脂質」は、ホスファチジン酸(1,2−ジアシル−snグリセロ−3−リン酸)の任意の誘導体であって、グリセロール部分に共有結合した1つのアシル鎖を含むものを示す。前記ホスファチジン酸の任意の誘導体としては、特に制限されないが、ホスファチジルコリン、ホスファチジルグリセロール、及びホスファチジルエタノールアミン等を挙げることができる。また、本発明の実施には、当業者に公知の任意のリゾ脂質を用いることができ、ここで、リゾ脂質としては、特に制限されないが、モノパルミトイルホスファチジルコリン(MPPC)、モノラウリルホスファチジルコリン(MLPC)、モノミリストイルホスファチジルコリン(MMPC)、モノステアロイルホスファチジルコリン(MSPC)、及びそれらの混合物等を挙げることができる。
【0043】
さらに、本発明のリポソームは、親水性ポリマーに結合した脂質を含んでいてもよい。親水性ポリマーに結合した脂質としては、例えば、PEGに共有結合した脂質を挙げることができる。
【0044】
活性物質
【0045】
本発明のペプチド及びペプチド模倣物は、活性物質を送達するために用いることができる。本明細書において、「活性物質」は、対象中の特異的な部位に送達されることが望まれる化合物であればどのような化合物であってもよい。本発明の実施には、どのような活性物質を用いてもよい。
【0046】
本発明の熱感受性リポソームにおいては、活性物質として抗癌剤を用いることができる。好適な抗癌剤の例を以下に示す:
【0047】
アルキル化剤、例えば、ナイトロジェンマスタード(例えば、クロラムブシル、クロルメチン、シクロホスファミド、イホスファミド、メルファラン、ニトロソ尿素(例えば、カルムスチン、フォテムスチン、ロムスチン、ストレプトゾシン)、プラチナ含有化合物(例えば、カルボプラチン、シスプラチン、オキサリプラチン、BBR3464)、ブスルファン、ダカルバジン、メクロレタミン、プロカルバジン、テモゾロマイド、チオテパ、及びウラムスチン;
【0048】
代謝拮抗剤、例えば、葉酸を標的とする代謝拮抗剤(例えば、アミノプテリン、メトトレキサート、ペメトレキセド、ラルチトレキセド)、プリン代謝を標的とする代謝拮抗剤(例えば、クラドリビン、クロファラビン、フルダラビン、メルカプトプリン、ペントスタチン、チオグアニン)、ピリミジン代謝を標的とする代謝拮抗剤(例えば、カペシタビン、シタラビン、フルオロウラシル、フロクスウリジン、ゲムシタビン);
【0049】
紡錘体毒植物アルカロイド、例えば、タキサン(例えば、ドセタキセル、パクリタキセル)、及びビンカ(例えば、ビンブラスチン、ビンクリスチン、ビンデシンビノレルビン);
【0050】
細胞障害性/抗腫瘍抗生物質、例えば、アントラサイクリン抗生物質(例えば、ダウノルビシン、ドキソルビシン、エピルビシン、イダルビシン、ミトキサントロン、バルルビシン、カルミノマイシン、nアセチルアドリアマイシン、ルビダゾン、5−イミドダウノマイシン、N30アセチルダウノマイシン、及びエピルビシン)、ブレオマイシン、マイトマイシン、及びアクチノマイシン;
【0051】
トポイソメラーゼ阻害剤、例えば、カンプトテシン(例えば、カンプトテシン、トポテカン、イリノテカン)、ポドフィルム(例えば、エトポシド、テニポシド);
【0052】
モノクローナル抗体又はその断片、例えば、アレムツズマブ、ベバシズマブ、セツキシマブ、ゲムツズマブ、パニツムマブ、リツキシマブ、トシツモマブ、及びトラスツズマブ;
【0053】
光感作性物質、例えば、アミノレブリン酸、アミノレブリン酸メチル、ポルフィマーナトリウム、及びベルテポルフィン;
【0054】
キナーゼ阻害剤、例えば、ダサチニブ、エルロチニブ、ゲフィチニブ、イマチニブ、ラパチニブ、ニロチニブ、ソラフェニブ、スニチニブ、及びバンデタニブ;
【0055】
酵素、例えば、アスパラギナーゼ、ペガスパルガーゼ、及び、酵素阻害剤、例えば、ヒドロキシ尿素;
【0056】
アポトーシス誘導剤、例えば、亜ヒ酸、ベルケイド、及びジェナセンス;
【0057】
生物学的反応修飾物質、例えば、デニロイキンジフチトクス;
【0058】
抗ホルモン剤、例えば、ゴセレリン酢酸塩、ロイプロリド酢酸塩、トリプトレリンパモ酸塩、酢酸メゲストロール、タモキシフェン、トレミフェン、フルベストラント、テストラクトン、アナストロゾール、エキセメスタン、及びレトロゾール;
【0059】
レチノイド、例えば、9−cis−レチノイン酸及びオールトランスレチノイン酸。
【0060】
他の実施形態においては、本発明の熱感受性リポソームは2以上の抗腫瘍薬を含むか、又は、それぞれが異なる種類の活性物質を含む2以上の熱感受性リポソームを本発明の方法に用いることができる。前記異なる種類の活性物質としては、例えば、異なる種類の抗癌剤を挙げることができる。
【0061】
本発明の実施においては、さらに他の活性物質を用いることができる。前記活性物質としては、特に制限されないが、例えば、抗生物質、抗真菌剤、抗炎症薬、免疫抑制薬、抗感染症薬、抗ウイルス薬、駆虫薬、及び抗寄生虫性化合物等を挙げることができる。
【0062】
前記活性物質は造影剤であってもよい。本発明のリポソーム製剤に好適に用いることのできる造影剤としては、超音波造影剤、X線造影剤(例えば、金及びバリウムを含む造影剤、イオヘキソール、ビジパーク、及び、他のヨードを含む造影剤や他の微粒子造影剤)、磁気共鳴造影剤(例えば、マンガンを含む常磁性体、鉄を含む常磁性体、ガドリニウムを含む常磁性体、及び、他のランタニドを含む常磁性体)、放射性医薬品(放射性同位体、又は放射性同位体を含む化合物)等を挙げることができる。
【0063】
使用する方法
【0064】
本発明のペプチドと会合した活性物質は、任意の適切な投与経路によって対象に投与することができる。ここで、好適な投与経路としては、静脈内、動脈内、筋肉内腹腔内、皮下、皮内、関節内、髄腔内、脳室内を例示することができ、さらに、例えば吸入、経口、又は粘膜(舌下)上に直接投与する他の投与経路も同様に挙げることができる。また、本発明の材料及び方法を用いて、任意の組織を治療することができる。前記本発明の材料及び方法を用いて治療可能な組織の例としては、特に制限されないが、肝臓、腎臓、骨、軟組織、頭部及び頸部の組織、筋肉、副腎組織、肺、胸部、甲状腺、膵臓、子宮内膜及び子宮頸部の組織を含む子宮組織、卵巣、及び前立腺等を挙げることができる。また、治療可能な組織は、癌組織であっても、罹患した組織又は損傷した組織であってもよく、さらに、必要であれば健康な組織であってもよい。
【0065】
本発明の方法を用いて対象に投与される活性物質の量は、当業者が決定することができ、好適には前記投与量の活性物質を長時間にわたって静脈内に投与する。ここでの投与時間としては、例えば、およそ1分間〜およそ24時間を挙げることができる。
【0066】
前記活性物質の投与量は、当技術分野で公知のように、キャリアに含まれる活性物質に応じて調整してもよい。
【0067】
活性物質が、本発明のペプチドを含む熱感受性リポソームに組み込まれている場合には、対象中の標的組織を、前記熱感受性リポソームの投与前、及び/又は投与中、及び/又は投与後に加温してもよい。1つの実施形態においては、前記標的組織を最初に加温し(例えば、10〜30分間)、加温後、可能な限り早く対象に前記熱感受性リポソームを送達する。他の実施形態においては、熱感受性リポソームを対象に投与し、投与後、可能な限り早く標的組織を加温する。このような加温処置は最長で3時間継続して行ってもよい。
【0068】
標的組織を加温する方法としては、好適な方法であればどのような方法を用いてもよい。好適な方法としては、例えば、無線周波数照射の使用、高密度焦点式超音波等の超音波の使用、マイクロ波放射の使用、温浴槽や光などの任意の赤外線放射源の使用を挙げることができ、また、他の形態として、放射性同位体等による外部又は内部からの放射線照射、又は電場及び磁場も同様に挙げることができる。
【0069】
当業者であれば、本発明の範囲又は本発明のいかなる実施形態からも逸脱することなく、本明細書に開示されている方法及び用途に対して、他の適切な改変及び修正を容易に行うことができる。ここまでに詳細に述べられた本発明は、以下の実施例を参照することにより、より明確に理解される。以下の実施例は、本発明を説明する目的で本明細書に組み込まれており、本発明を限定することを目的とするものではない。
[実施例]
【実施例1】
【0070】
卵ホスファチジルコリン、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[マレイミド(ポリエチレングリコール)2000](DSPE−PEG2000−Mal)及びコレステロールは、全てAvanti Polar Lipids社(米国アラバマ州)より購入した。Lissamine(登録商標)ローダミンB 1,2−ジヘキサデカノイル−sn−グリセロ−3−ホスホエタノールアミン(ローダミンDHPE)及びN−(フルオロセイン−5−チオカルバモイル)−1,2−ジヘキサデカノイル−sn−グリセロ−3−ホスホエタノールアミン(フルオロセインDHPE)は、Invitrogen Corporation社より購入した。N−サクシニミジル 3−(2−ピリジルジチオ)プロピオナート(SPDP)及びトリス(2−カルボキシエチル)ホスフィン(TCEP)はPierce Biotechnology, Inc.社より購入した。Cy5.5モノNHSエステルはGE Healthcare社より提供された。他の全ての分析グレードの化学薬品はSinopharm Chemical Reagent Co. Ltd社(中国上海)より入手した。
【0071】
不活性状態のEGFRの結晶構造データは、PDB(http://www.rcsb.org/pdb/、code:1NQL)よりダウンロードした。Sheng-Hung Wang博士より供与されたPSCAN2.2.2プログラム(http://home.pchome.com.tw/team/gentamicin/mol/mol.htm)を用いてEGFRの3D構造モデルを構築し、結合ポケットの候補を探索した。この検索により、EGFRの上端付近の表面上にある1つのポケットを見い出し、図1に示した。前記ポケット内部のアミノ酸残基構造を詳細に分析し、Mekler-Idlisのアミノ酸ペアリング理論(Heal JR, Roberts GW, Raynes JG, Bhakoo A, Miller AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochem 2002;3(2-3):136-51)に基づいて、132個のヘキサペプチドを含むフォーカストライブラリを設計した。
【0072】
前記のEGFR表面ポケットに対するペプチドの結合性に関して、Autodock3.0.を用いて前記ライブラリをスクリーニングし、ペプチドの順位付けを行った。計算を高速化し、複数のプロセッサ上でプログラムを実行するために、本発明者らは前記ソフトウェアをパラレルコンピューティングが可能になるように修正した。前記の全てのコンピュータ処理は、SJTUスーパーコンピューティングセンター内のSGI Onyx3800機を用いて行った。最初に大まかなスクリーニングを行い、前記ライブラリに含まれる132個のペプチドの中から、より低い結合エネルギーを持つ50個のペプチドを選び出した。ラマルク型遺伝アルゴリズム局所検索(GALS)アルゴリズム(Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639-62)を用いて、1回あたり25万回のエネルギー計算からなるドッキング試行を、各ペプチドに対して独立して100回行った。また、その他の全てのドッキングパラメータは、Csaba及びDavid(Hetenyi C. van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci 2002;11(7):1729-37)の記載に基づき決定した。次に、前記スクリーニングにより選び出された50個のペプチドについて、同様のアルゴリズムで、ドッキングあたり1000万回のエネルギー計算からなる精密な解析段階により、さらに詳細な検討を行った。各ペプチドの最小ドッキングエネルギーを比較して最も低いドッキングエネルギーを持つ20個のペプチドを選び、合成及びウエットスクリーニングを行った。BIACore結合試験により、これら20個のペプチドのうちの2つは濃度依存的にEGFRに結合することが示された。中でも、LARLLT配列からなるペプチド(配列番号1)(ドッキング試験における順位は4位)はEGFRに結合する可能性が明確に示された。
【0073】
図1A(円内)にPSCANプログラムを用いて見い出された結合ポケットを示す。この結合ポケットは、ペプチドが外部から容易に結合できるEGFR表面上に存在し、また、EGF結合部位からは少し離れた場所に位置する。これらのことから本発明者らは、前記ポケット内部の立体構造が、EGF結合及びEGFRの二量体化の有無によって変化する可能性は低いと推測した。前記ポケットは、底部にいくつかの疎水性残基を、開口部周辺に親水性残基を有する(帯電残基:Asp51、Glu73、Glu110、Arg48、Lys56、Arg74;極性残基:Thr106、Ser53、Ser62)。
【0074】
我々が、本実験に使用するために選択した2つのペプチド(LARLLT及びコントロールペプチド)を前記ポケットにドッキングさせ、最適なドッキング構造を探索した。LARLLTの最小ドッキングエネルギー及び結合自由エネルギーは、それぞれ−17.9kcal/mol及び−8.54kcal/molであり、コントロールペプチドの最小ドッキングエネルギー及び結合自由エネルギーは、それぞれ−12.52kcal/mol及び−3.85kcal/molであった。
【0075】
図1Bに示すように、ペプチドLARLLTは前記ポケットの中にすっぽりと収まり、LARLLTのほぼ全ての残基がEGFRの残基と相互作用した。LARLLTの両端(LEU1、ALA2及びTHR6)と中央(ARG3)の残基については、6つの水素結合が形成されることが示唆された。前記ペプチドのN末端においては、LEU1のアミノ基の水素は、EGFRのGLU73のカルボキシル基の酸素と相互作用し、ALA2の主鎖の酸素は、EGFRのGLU73の主鎖のアミド基の水素と相互作用した。また、前記ペプチドのC末端においては、THR6は水素結合の供与体と受容体の両方の役割を持ち、THR6のC末端の酸素はEGFRのSER53のヒドロキシル基の水素と相互作用し、THR6の側鎖のヒドロキシル基の水素はEGFRのASP51のカルボキシル基の酸素と相互作用した。さらに、前記ペプチドの中央部のARG3は、静電的相互作用(EGFRのGLU73及びGLU110との)と水素結合の両方を介してEGFRと相互作用した。以上の3つの鍵となる残基(LEU1、THR6、ARG3)は、空間的にちょうど良い位置にあり、EGFRのポケット内で錨のように固定されるとともに、LARLLTの疎水性残基ALA2、LEU4及びLEU5と、ポケット内の疎水性底部(hydrophobic bottom)とのより強い相互作用を容易にした。ドッキング構造(図1)に示すように、LARLLTの残基とEGFRの残基との詳細な相互作用解析は、M−Iのペアリング理論(Heal JR, Roberts GW, Raynes JG, Bhakoo A, Miller AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochem 2002;3(2-3):136-51)によく一致していた。
【0076】
これに対して、LARLLTと同じアミノ酸組成を持つが、配列をランダムに並べ替えたコントロールペプチド配列番号2は、図1Cに示すように、ポケット内に収まることすらできなかった。コントロールペプチドの帯電したARG1は、EGFRのASP51と相互作用したが、他の相互作用が弱過ぎるためにコントロールペプチドをポケット内に固定することができなかった。
【実施例2】
【0077】
ペプチドLARLLTは、GL Biochem Ltd.社(上海)において合成及び精製され、その構造と純度はHPLC及びMSにより全て確認された。また、比較のために、D4*と名付けた、LARLLTと同じアミノ酸組成を持つが、配列をランダムに並べ替えたペプチド(RTALLL、コントロールペプチド(配列番号2))を同様に合成した。前記の2つのペプチドを蛍光標識するために、Cy5.5−NHSと、LARLLT又はコントロールペプチドとを1:2のモル比で混合し、遮光下、25℃で一晩インキュベートした。これらの蛍光ペプチドは、その後さらに精製することなく実験に用いた。
【実施例3】
【0078】
EGF又はLARLLT/コントロールペプチドをPBS−EDTAに溶解し、DMSOに溶解したN−サクシニミジル 3−(2−ピリジルジチオ)プロピオナート(SPDP)と1:1.2のモル比で、室温で1時間混合した後、凍結乾燥した。また、クロロホルムに溶解したDSPE−PEG2000−Mal脂質を乾燥させて薄膜を作製し、HEPES緩衝液(pH7.4)で約0.4mMの濃度となるように水和させた。コンジュゲーションを行うために、チオール化したタンパク質又ペプチドをトリス(2−カルボキシエチル)ホスフィン(TCEP)溶液に加え、窒素雰囲気下、室温で1時間インキュベートした。さらに、MAL−PEG2000−DSPEミセル溶液とモル比5:1で素早く混合し、窒素雰囲気下、攪拌しながら10℃で一晩反応させた。HPLC分析を行ったところ、前記の反応によってほぼ全てのMal−PEG−DSPEが改変していた(modified)ことが示された。ここで、余剰のタンパク質/ペプチドを、例えば、ゲルろ過カラムや透析等の通常の方法により除去することもできる。
【実施例4】
【0079】
実験に用いたリポソームは全て、乾燥薄膜水和法を行った後、100nmの膜を数回通過させることより作製した。本実験に用いたほとんどのリポソームの脂質組成は、EPC:CHOL:DSPE−PEG=10:5:0.5から開始した。ローダミン又はフルオロセイン標識リポソームを除いて、ローダミン−DHPE又はフルオロセイン−DHPEのいずれかを全脂質の約0.6%モルとなるように前記脂質組成に加えた。
【0080】
Cy5.5標識リポソームについては、Cy5.5−NHS、DSPE、及びトリエチルアミンを3:1:3.5のモル比となるようにクロロホルム中で混合し、25℃で一晩インキュベート(遮光下)することにより、Cy5.5−DSPEを合成した。得られたCy5.5−DSPEを、全脂質の約0.5%モルとなるように前記脂質組成に組み込んだ。リポソーム形成後に透析を行って、余剰のCy5.5を除去した。
【実施例5】
【0081】
ドキソルビシンを封入したリポソームの作製は膜通過勾配法に従って行った(Mayer LD, Tai LC, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta 1990;1025(2):143-51、Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta 1993;1151(2):201-15. Erratum in: Biochim Biophys Acta 1994;1190(1):197)。簡単に説明すると、脂質をpH4.0の125mM硫酸アンモニウム中で水和させ、膜通過処理により〜100nmのリポソームを作製した。得られたリポソームをHEPES緩衝液(pH7.5)中で透析し、20mMのHEPES、150mMのNaCl(pH7.5)を用いて必要な濃度に希釈した。そして、ドキソルビシンを加え、前記リポソームとともに60℃で10分間インキュベートした。
【0082】
前記リガンドをコンジュゲートさせた脂質を、Ishida et al.(Ishida T, Iden DL, Allen TM. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett 1999;460 (1):129-33)の開発した方法を若干改変した方法によって、事前に作製したリポソームに挿入した。簡単に説明すると、DSPE−PEG2000−リガンドのミセル溶液を、作製済みのリポソーム溶液に9:100のモル脂質比で加え、60℃で1時間インキュベートした(ドキソルビシンを含むリポソームの場合には55℃で30分間)。SnakeSkin(登録商標)Pleated Dialysis Tubing 10,000 MWCO(Pierce Chemical Company社製)を用いて、前記溶液をPBSで4時間透析した。また、コントロールとして、mPEG2000−DSPEのミセルを同じ割合で混合してインキュベートし、標的指向性を有さない(non-targeted)リポソームを作製した。
【0083】
前記ペプチドリガンドをDSPE−PEG2000に、スルフヒドリル−マレイミドカップリング反応によってコンジュゲートさせた。逆相HPLC分析によりこの反応をモニターして確認した。前記反応により得られたペプチド−PEG−DSPE、又はmPEG−DSPEコントロールを、広く用いられているインキュベーション方法(Ishida T, Iden DL, Allen TM. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett 1999;460 (1):129-33)によって、事前に作製したリポソームに組み込んだ。このインキュベート処理において、ペプチド−PEG−脂質(又はmPEG−脂質)の、脂質に対する割合を様々に変えて検討し(3:100、6:100、及び9:100モル)、ほとんどの細胞に対してリポソームの非特異的な結合を抑制する比として、9:100の比率を決定した。また、前記手法によってリポソームの安定性(integrity)が損なわれないことを確認した。前記工程中又は工程後に漏れ出した封入ドキソルビシンはわずか5%だった。リポソームの最終的なサイズ分布は、Zetasizer3000H(Malvern Instruments社製)を用いた光子相関分光法(PCS)により決定した。膜通過処理直後に得られたリポソーム(〜110nm)と比較すると、ペプチド脂質を挿入した後のリポソームの粒子径はわずかに大きかったが(130〜150nm)、安定性は保たれていた。
【実施例6】
【0084】
本実験においては、EGFR発現レベルの高い2つの細胞株、ヒト非小細胞肺癌細胞株H1299及びヒト肺腺癌細胞株SPCA1を用いた。前記細胞は、10%のウシ胎児血清を含むRPMI1640培地にて、5%CO2を含む加湿気相条件下、37℃で培養を行った。
【0085】
H1299及びSPCA1の異種移植マウスモデルは、上海がん研究所の動物実験センターで作製した。それらのマウスモデルは、腫瘍細胞を移植した後約3〜4週間後(腫瘍サイズは直径約5〜7mm)にin vivoイメージング実験に用い、その後、人道的に安楽死させた。前記動物実験のプロトコールは、上海交通大学薬学部(Shanghai Jiaotong University, School of Pharmacy)の動物実験委員会によって承認された。
【実施例7】
【0086】
EGFRを高発現しているH1299又はSPCA1細胞を35mm径の培養ディッシュに播種し(1×106細胞/ディッシュ)、RPMI1640培地中で24〜48時間培養した。細胞培養がおよそ80%コンフルエントに達した後、リガンドとコンジュゲートさせたローダミンリポソームをRPMI1640で希釈し、4℃又は37℃で前記培養ディッシュに添加した。競合結合実験のために、LARLLTを組み込んだリポソームを添加する2時間前に、50倍モル濃度過剰量のフリーのLARLLT又はEGFを培地に加えた。前記特定の温度で、細胞を4時間インキュベートした後に、PBS(pH7.4)で6回洗浄した。細胞に結合し、内在化した蛍光脂質を、共焦点レーザー顕微鏡(CLSM、Zeiss LSM社製、ドイツ)を用いて可視化した。また、フローサイトメトリー実験のために、H1299又はSPCA1細胞を35mm径のウェルに播種して約80%コンフルエントに達するまで増殖させた。細胞を、リガンドで修飾した(modified)フルオロセイン標識リポソームとともに37℃で3時間インキュベートした後に、PBSで洗浄し、トリプシンで処理して懸濁した。その後、フローサイトメーター(BD FACSCalibur、Becton Dickinson社製)により解析した。
【0087】
EGF、LARLLT、又はコントロールペプチドで修飾したローダミン標識リポソームの、EGFR高発現細胞に対する結合能をin vitroで調べた。図2Aに示すように、EGF及びLARLLTで修飾したリポソームは、ともに効率よくH1299細胞に結合した。EGFで修飾したリポソームによる蛍光は、LARLLT修飾リポソームよりもわずかに強いことから、EGFによるより強い結合が示唆された。しかし、コントロールペプチドを用いた場合には、蛍光はほとんど観察されなかった。同様の結果は、EGFRを発現する別の細胞株SPC−A1を用いた場合にも観察された(図2C)。
【0088】
さらに詳細な結合特性に関する評価を、H1299細胞を用いて様々な温度条件で行った。4℃の温度条件では、蛍光は主に細胞表面上で認められ、過剰量の標識されていないフリーのLARLLTと競合させることによって、大部分の蛍光は認められなくなった(図2B−iii及びiv)。これに対し、37℃の温度条件では、フリーのLARLLTによる競合作用はそれほど明確には認められなかった(図2B−I及びii)。このことは、37℃では、LARLLTがEGFRに結合した後に、エンドサイトーシス及びレセプターの再利用(turnaround)が活発に起こっていることを示唆している。また、興味深いことに、37℃の温度条件では、フリーのEGFの存在はLARLLTリポソームの蛍光強度に、完全ではないが、実際にある程度の影響を及ぼした(図2B−v及びvi)。LARLLTが結合した後のエンドサイトーシス経路を、EGFが阻害した可能性がある。
【0089】
図3に、LARLLTで修飾したリポソームの、H1299細胞への結合及び取込みについて高倍率で示し(図3A)、さらに、Z軸連続スキャン画像を示した(図3B)。細胞内の多数のエンドサイトーシス小胞において脂質の蛍光が認められたことから、EGF結合部位から遠く離れた、EGFR表面上のわずかな窪みにLARLLTが結合することによって、活発なエンドサイトーシス活性が誘導され得ることが示された。
【0090】
フローサイトメトリーを用いて、より大きな細胞集団に対する、LARLLT及びコントロールペプチドが組み込まれたリポソームの結合能の違いを比較検討した。リポソームをフルオロセイン−DSPEで標識し、H1299及びSPCA1細胞にそれぞれ加えた。3時間のインキュベート後、H1299細胞の75%でLARLLTリポソームの結合による高い蛍光シグナルが認められた(101〜103 FL1値)。一方、コントロールペプチドリポソームの結合による蛍光は、H1299細胞の27%でしか認められなかった。SPCA1細胞の場合には、LARLLTリポソームによって68%が蛍光を発したが(35〜103 FL1値ゲート)、コントロールペプチドリポソームでは17%であった。
【実施例8】
【0091】
H1299及びSPCA1細胞を104細胞/ウェルの濃度で96ウェルプレートに播種し、一晩増殖させた。フリーのドキソルビシン、LARLLTによって修飾したドキソルビシン封入リポソーム、及びコントロールペプチドによって修飾したドキソルビシン封入リポソームを培地に添加し、2時間インキュベートした。その後、細胞を新しい培地により洗浄し、さらに48時間増殖させた。異なる処理後の細胞の生存率はMTTアッセイ(Scudiero DA, Shoemaker RH, Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988;48(17):4827-33)を用いて決定した。
【0092】
脂質とコンジュゲートしたLARLLT又はコントロールペプチドによって、ドキソルビシン封入リポソームを修飾してEGFR高発現細胞に添加し、これらのリポソームの殺細胞効果をMTTアッセイにより評価した。H1299及びSPCA1のいずれの細胞においても、LARLLTによって修飾したリポソームはコントロールペプチドによって修飾したリポソームよりも効率的に薬物を送達した(図4A及びB)。両方の細胞におけるLARLLTリポソームのIC50を算出したところ、コントロールペプチドリポソームと比較しておよそ3〜5倍細胞毒性が強かった。
【実施例9】
【0093】
Cy5.5、LARLLT−Cy5.5、コントロールペプチド−Cy5.5、及び、LARLLT又はコントロールペプチドを導入したCy5.5標識リポソームを、H1299又はSPCA1を異種移植した担癌マウスの尾静脈から注射した。注射後の様々な時点において、前記マウスに麻酔をかけて、Optix in vivo蛍光イメージングシステム(GE Healthcare社製)を用いてイメージングを行った。Cy5.5色素のみのコントロール群を除く、他の全ての投与群では少なくとも3匹のマウスを含む。各投与群において最も典型的な1匹のマウスから得られた、代表的な画像を示した。これらの画像は、自家蛍光による障害を除去するために、Cy5.5(0.8〜1.1ns)の蛍光寿命の時間分解を行った。
【0094】
LARLLT及びコントロールペプチドを近赤外色素Cy5.5により標識し、H1299を移植した担癌マウスの尾静脈に注射した。注射後の様々な時点での、前記マウス体内におけるCy5.5の蛍光の分布を画像化した。フリーの色素コントロールを含む全ての投与群において、蛍光は最初体全体で認められ、その後、すぐに腎臓と膀胱に蓄積した。このような腎臓と膀胱における強い蛍光による障害を防ぐために、皮下に腫瘍が存在する部分のみに絞った画像を示した(図5)。Cy5.5色素を注射したマウスにおいては、腫瘍部分における蛍光シグナルはバックグラウンドと同じレベルであった。しかし、LARLLT−Cy5.5を注射したマウスにおいては、注射後1〜6時間の間、腫瘍組織中の蛍光シグナルは周囲の組織と比較して非常に強かった。これに対して、Cy5.5−コントロールペプチドを注射したマウスでは、腫瘍部分における蛍光の蓄積は全く認められなかった。
【0095】
LARLLT及びコントロールペプチド修飾Cy5.5標識リポソームを、H1299を移植した担癌マウスの尾静脈に注射した。注射後の様々な時点において、これらのマウスの体全体をスキャンし、異なる時点での腫瘍部分での蛍光強度を定量化した。図6に示すように、LARLLT修飾リポソームは、注射6時間後から、次第に腫瘍部分の内部に蓄積された。注射後80時間経過した後も、蛍光シグナル及びその対比が有意に認められた。しかし、コントロールペプチド修飾リポソームの場合には、腫瘍部分における蛍光強度は、常にバックグラウンドと同レベルであった。我々は、マウスの体全体における蛍光強度に対する、腫瘍部分の蛍光強度の相対比率を算出した(表1)。腫瘍部分及び体全体の蛍光強度(NC)は、eXplore OptiView 1.0.0.0ソフトウェアを用いて算出した。また、体全体の蛍光強度に対する腫瘍部分の蛍光強度のパーセンテージを表にして示した。
【0096】
表1 蛍光標識リポソームを注射した後の様々な時点における腫瘍部分の蛍光強度。
【0097】
【表1】
【0098】
マウスの体全体の蛍光強度に対する腫瘍部分の蛍光強度の比率は、LARLLT修飾リポソームの注射1時間後から、プラトーに達したと考えられる76〜80時間後まで次第に上昇していった。以上のことから、LARLLT標的指向性リポソームは徐々に腫瘍部分に蓄積され、その後は強力に保持されることが示唆された。
【0099】
いくつかの実施形態においては、本発明のペプチド及びペプチド模倣物は、EGF結合部位から離れた表面ポケットに結合するように設計されていてもよい。このように設計された本発明のペプチド及びペプチド模倣物(及び本発明のペプチドと結合した治療薬)は、EGFRに結合する際にEGFと直接競合することがないため、EGFRに対する結合親和性が、EGFの親和性よりも低かったとしても、EGFR発現細胞に結合することができる。
【0100】
本発明のペプチド及びペプチド模倣物を、例えば、PEG2000−DSPEの末端のような、様々な化合物とコンジュゲートさせてリポソームに導入した場合にも、本発明のペプチド及びペプチド製剤のEGFRへの結合能は維持される。そして、本発明のペプチド及びペプチド模倣物は、化合物(例えば、リポソーム)をEGFR高発現細胞に特異的に接着させてEGFR高発現細胞による取込みを可能とする。以上に示したように、本発明のペプチド及びペプチド模倣物とコンジュゲートした化合物(例えば、前記のような本発明のペプチドを組み込んだリポソーム)をin vivoに投与した場合、その化合物は標的細胞(例えば、腫瘍細胞)に対して指向性を有することから、結果的に、本発明のペプチドとコンジュゲートした化合物(例えば、ペプチドがコンジュゲートしたリポソーム)は前記標的(例えば、腫瘍内)に蓄積される。
【0101】
本研究ではPEG2000リンカーのみを用いた。また、我々の研究においては、リガンドと結合した脂質を2〜4:100の脂質比率の条件で組み込んだ場合に、in vitroでの細胞への結合が最も有意に認められた(バックグラウンドの非特異的な結合が最も低かった)。しかし、当業者に公知のリンカーであれば、他のどのようなリンカーを用いてもよく、また同様に、他の脂質比率を用いてもよい。
【0102】
前記と同様のリポソーム製剤をin vivoで用いた。このリポソームは、その表面上に比較的高いリガンド−PEG密度を持ち、腫瘍にゆっくりと蓄積された。実際、これまでのいくつかの研究において、循環中及び浸潤中の能動的標的指向性リポソームは、ステルスリポソームと同様の振舞いをするに過ぎないことが示唆されている(Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: in vitro and in vivo studies. Br J Cancer 1996;74:1749-1756、Matsumura Y, Gotoh M, Muro K, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol 2004;15(3):517-25、Sapra P, Moase EH, Ma J, Allen TM. Improved therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab' fragments. Clin Cancer Res 2004;10(3):1100-11)。このように、能動的標的指向性リポソームの機能は、腫瘍組織に浸潤した後にのみ意味を持つ。能動的標的指向性リポソームは、その標的指向性によって癌細胞と強固に結合し、容易に細胞に取り込まれることができる(Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9、Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 2006;66(13):6732-40)。in vivo蛍光イメージングシステムを用いて観察した結果(図6)は、前記の作用メカニズムに一致していた。リガンドを組み込んだLARLLTリポソームは、非常に長時間(>80時間)にわたって腫瘍組織に留まることが明らかにされた。一方、我々が標的指向性を持たないステルスリポソームであると予測したとおり、コントロール群を用いた場合には、12〜24時間経過した時点で、腫瘍組織中に有意な蓄積は認められなかった。
【0103】
小動物用in vivo蛍光イメージングシステムは、標識された薬剤及びデリバリーシステムのin vivoでの分布を追跡するために最近開発された装置である。Cy5.5は、組織透過性に優れており、バックグラウンドが低いことから、最も一般的に使用される色素である。in vivo蛍光イメージング技術の主な利点は、非侵襲的であること、さらに、実験の全ての経過を通じて、同一の生きた動物を継続的に観察することが可能なことである。これまでの多くの研究では、in vivoで薬品又は脂質の分布を追跡するために、放射標識した薬品又は脂質が用いられてきたが、平均的な測定結果を得るために、多くの動物を異なる時点で犠牲にする必要があった。本発明者らのin vivo蛍光イメージングシステムを用いた経験では、同じ実験群中の異なる動物個体において、一般的な健康状態、生理機能、代謝機能、腫瘍のサイズ及び位置等がばらついていても問題が無い。また、本実験により得られた能動的標的指向性リポソームの時間連続分布プロファイルは、他の方法を用いたこれまでの報告とよく一致していた(Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9、Sapra P, Moase EH, Ma J, Allen TM. Improved therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab' fragments. Clin Cancer Res 2004;10(3):1100-11)。
【実施例10】
【0104】
本発明のペプチドを用いた標的指向性を有するリポソームが、標的腫瘍における薬剤の蓄積を増加させることができるかについて評価した。5mg/kgのドキソルビシンを、H460腫瘍細胞を移植したヌードマウスの静脈に注射した。また、フリーのドキソルビシン(図7、Dox)と、PEG化したリポソームに封入されたドキソルビシン(図7、PEG)、及び本発明のペプチドを含むリポソームに封入されたドキソルビシン(図7、D4)とを比較した。これらをマウスに注射して0.5時間、6時間、及び48時間後にマウスを安楽死させ、腫瘍を摘出して腫瘍に含まれるドキソルビシンを測定した。各試験群あたりN=3の実験動物を用いた。
【0105】
図7に示すように、フリーのドキソルビシン及びPEG化したリポソームに封入されたドキソルビシンと比較して、本発明のペプチドを含むリポソームを用いた結果、有意に多量のドキソルビシンが腫瘍に蓄積された。
【実施例11】
【0106】
FACS解析による本発明のペプチドを含むリポソームの結合解析
【0107】
H−460細胞を無菌条件下、培養プレート上で増殖させ、スクレーピングによって回収した。回収したH−460細胞に、リポソームサンプル(空のサーモドックスのリポソーム、サーモドックス、D4−サーモドックス、D4*−サーモドックス、ここで、D4−サーモドックスは配列番号1で示されるペプチドを含み、D4*−サーモドックスは配列番号2で示されるペプチドを含む)及びドキソルビシン塩酸塩を加えて、37℃で4時間インキュベートした。本発明のペプチドをそれぞれ異なる量で含む2種類のリポソーム(D4−サーモドックス)の評価を行った。前記の2種類のリポソームをそれぞれD4−サーモドックス2+2及びD4−サーモドックス4+4と名付けた。D4−サーモドックス2+2は、DPPC、MSPC、DSPE−MPEG2000及びD4−DSPE−MPEG2000を90:10:2:2のモル比で含み、D4−サーモドックス4+4は、DPPC、MSPC、DSPE−MPEG2000及びD4−DSPE−MPEG2000を、90:10:4:4のモル比で含んでいた。また、D4*−サーモドックスは、DPPC、MSPC、DSPE−MPEG2000、及びD4*−DSPE−MPEG2000を90:10:2:2のモル比で含んでいた。インキュベーション後、細胞をPBSで4回洗浄し、その後、ドキソルビシンの発する蛍光を用いてFACSによって細胞をカウントした(励起波長488nm、及び発光波長575nm)。
【0108】
以下の群について実験を行った:a.無処理コントロール;b.約0.01〜10μg(ドキソルビシン)/mLのサーモドックス;c.約0.01〜10μg(ドキソルビシン)/mLのD4−サーモドックス2+2;d.約0.01〜10μg(ドキソルビシン)/mLのD4−サーモドックス4+4;e.約0.01〜10μg(ドキソルビシン)/mLのD4*−サーモドックス、及びf.0.01〜10μgのドキソルビシン塩酸塩。
【0109】
実験は少なくとも3回繰り返して行った。同一条件の実験の中から、代表的なヒストグラムを示す。厳密には、細胞数のばらつきやFACSのパラメータによって、実験から得られた数値は変化するかもしれないが、大まかなパターンや傾向の違いは常に一定であった。
【0110】
濃度が10μg/mlの場合、in vivoでのD4−サーモドックス(2+2)のドキソルビシンによる蛍光強度は、サーモドックス及びD4*−サーモドックスよりも強かったが、フリーのドキソルビシン塩酸塩よりは弱かった。フリーの薬剤は細胞膜の透過性が比較的高いので、以上の結果から、D4−リポソームは、コントロールリポソームよりも多くH460細胞に結合することが確認された。濃度が1μg/mlの場合、D4−サーモドックス(2+2、4+4)により処理された細胞は、サーモドックス又はD4*−サーモドックスにより処理された細胞と比較して、より強いドキソルビシン蛍光シグナルを発していた。濃度が0.1μg/ml及び0.01μg/mlの場合、組成の異なるリポソーム間での明確な違いは認められなかった。
【0111】
図8は、実験に用いた各リポソームに含まれるドキソルビシンの投与量に対して、FACS解析により得られたヒストグラムの平均値をプロットした線グラフを示す。ドキソルビシンの細胞への取込み(平均蛍光強度)は、薬剤溶液を用いた場合に最も高かった。これは、フリーのドキソルビシンが非常に効率的に細胞膜を透過することができるためである。異なる組成のリポソームを比較した場合、2つのD4リポソーム製剤は、サーモドックスコントロール及びD4*リポソームコントロールよりも細胞への取込み効率が非常に優れていた。
【0112】
本明細書において言及された全ての刊行物、特許、及び特許出願は、本発明が関連する技術分野における当業者の技術レベルの指標となる。また、本明細書において言及された全ての刊行物、特許、及び特許出願は、それぞれを参照することによって具体的かつ個々に示されるのと同じ程度の範囲が、参照することによって本明細書に援用される。
【0113】
参考文献
1. Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res 1994;54(4):987-92.
2. Maruyama K, Takizawa T, Yuda T, Kennel SJ, Huang L, Iwatsuru M. Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies. Biochim Biophys Acta 1995;1234(1):74-80.
3. Park JW, Hong K, Carter P, et al. Development of anti-p185HER2 immunoliposomes for cancer therapy. Proc Natl Acad Sci U S A 1995;92(5):1327-31.
4. Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: in vitro and in vivo studies. Br J Cancer 1996;74:1749-1756.
5. Lopes de Menezes DE, Pilarski LM, Allen TM. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res 1998;58(15):3320-30.
6. Matsumura Y, Gotoh M, Muro K, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol 2004;15(3):517-25.
7. Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta 1995;1233(2):134-44.
8. Gabizon A, Horowitz AT, Goren D, TzemachD, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin cancer Res 2003;9:6551-9.
9. Terada T, Mizobata M, Kawakami S, Yabe Y, Yamashita F, Hashida M. Basic fibroblast growth factor-binding peptide as a novel targeting ligand of drug carrier to tumor cells. J Drug Target 2006;14(8):536-45.
10. Schiffelers RM, Koning GA, ten Hagen TL, et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release 2003;91(1-2):115-22.
11. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995;19:183-232.
12. Lutsenko SV, Feldman NB, Severin SE. Cytotoxic and antitumor activities of doxorubicinconjugates with the epidermal growth factor and its receptor-binding fragment. J Drug Target 2002;10(7):567-71.
13. Jinno H, Ueda M, Ozawa S, et al. Epidermal growth factor receptor-dependent cytotoxic effect by an EGF-ribonuclease conjugate on human cancer cell lines--a trial for less immunogenic chimeric toxin. Cancer Chemother Pharmacol 1996;38(4):303-8.
14. Kullberg EB, Nestor M, Gedda L. Tumor-cell targeted epiderimal growth factor liposomes loaded with boronated acridine: uptake and processing. Pharm Res 2003; 20(2):229-36.
15. Chen P, Mrkobrada M, Vallis KA, et al. Comparative antiproliferative effects of (111) In-DTPA-hEGF, chemotherapeutic agents and gamma-radiation on EGFR-positive breast cancer cells. Nucl Med Biol 2002;29(6):693-9.
16. Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different strategies for formation of pegylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjug Chem 2001;12(4):529-37.
17. Mamot C, Drummond DC, Greiser U, et al. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR-and EGFRvIII-overexpressing tumor cells. Cancer Res 2003;63(12):3154-61.
18. Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res 2005; 65(24):11631-8.
19. Mamot C, Ritschard R, Kung W, Park JW, Herrmann R, Rochlitz CF. EGFR-targeted immunoliposomes derived from the monoclonal antibody EMD72000 mediate specific and efficient drug delivery to a variety of colorectal cancer cells. J Drug Target 2006; 14(4):215-23.
20. Heal JR, Roberts GW, Raynes JG, Bhakoo A, Miller AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochem 2002;3(2-3):136-51.
21. Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639-62.
22. Hetenyi C. van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci 2002;11(7):1729-37.
23. Mayer LD, Tai LC, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta 1990;1025(2):143-51.
24. Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta 1993;1151(2):201-15. Erratum in: Biochim Biophys Acta 1994;1190(1):197.
25. Ishida T, Iden DL, Allen TM. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett 1999;460 (1):129-33.
26. Scudiero DA, Shoemaker RH, Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988;48(17):4827-33.
27. Schmidt M, Vakalopoulou E, Schneider DW, Wels W. Construction and functional characterization of scFv(14E1)-ETA -a novel, highly potent antibody-toxin specific for the EGF receptor. Br J Cancer 1997; 75(11):1575-84.
28. Liu X, Tian P, Yu Y, Yao M, Cao X, Gu J. Enhanced antitumor effect of EGF R-targeted p21WAF-1 and GM-CSF gene transfer in the established murine hepatoma by peritumoral injection. Cancer Gene Ther 2002;9(1):100-8.
29. Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J 2005;19(14):1978-85.
30. de Graaf C, Pospisil P, Pos W, Folkers G, Vermeulen NP. Binding mode prediction of cytochrome p450 and thymidine kinase protein-ligand complexes by consideration of water and rescoring in automated docking. J Med Chem 2005;48(7):2308-18.
31. Rogers JP, Beuscher AE 4th, Flajolet M, et al.. Discovery of protein phosphatase 2C inhibitors by virtual screening. J Med Chem 2006; 49(5):1658-67.
32. Bhakoo A, Raynes JG, Heal JR, Keller M, Miller AD. De-novo design of complementary (antisense) peptide mini-receptor inhibitor of interleukin 18 (IL-18). Mol Immunol 2004;41(12):1217-24.
33. Mastrobattista E, Koning GA, Storm G. Immunoliposomes for the targeted delivery of antitumor drugs. Adv Drug Deliv Rev 1999;40(1-2):103-127.
34. Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res 2006;16(3):175-83.
35. Koning GA, Morselt HW, Gorter A, et al. Pharmacokinetics of differently designed immunoliposome formulations in rats with or without hepatic colon cancer metastases. Pharm Res 2001;18(9):1291-8.
36. Koning GA, Morselt HW, Gorter A, et al. Interaction of differently designed immunoliposomes with colon cancer cells and Kupffer cells. An in vitro comparison. Pharm Res 2003;20(8):1249-57.
37. Derksen JT, Morselt HW, Scherphof GL. Uptake and processing of immunoglobulin-coated liposomes by subpopulations of rat liver macrophages. Biochim Biophys Acta 1988;971(2):127-36.
38. Maruyama K, Takahashi N, Tagawa T, Nagaike K, Iwatsuru M. Immunoliposomes bearing poloethylene glycol-coupled Fab' fragment show prolonged circulation time and high extravasation into targeted solid tumors in vivo. FEBS Lett 1997;413(1):177-80.
39. Huwyler J, Yang J, Pardridge WM. Receptor mediated delivery of daunomycin using immunoliposomes: pharmacokinetics and tissue distribution in the rat. J Pharmacol Exp Ther 1997; 282(3):1541-6.
40. Tardi P, Bally MB, Harasym TO. Clearance properties of liposomes involving conjugated proteins for targeting. Adv Drug Deliv Rev 1998;32(1-2):99-118.
41. Sapra P, Moase EH, Ma J, Allen TM. Improved therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab' fragments. Clin Cancer Res 2004;10(3):1100-11.
42. Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 2006;66(13):6732-40.
【特許請求の範囲】
【請求項1】
配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドであって、長さが約6〜約15アミノ酸であるペプチド。
【請求項2】
配列番号1を含む、請求項1記載のペプチド。
【請求項3】
配列番号1からなる、請求項1記載のペプチド。
【請求項4】
配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドと会合した脂質を含む分子。
【請求項5】
脂質とペプチドとの間にリンカーを含む、請求項4記載の分子。
【請求項6】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項5記載の分子。
【請求項7】
リンカーが、ポリエチレングリコール(PEG)を含む請求項5記載の分子。
【請求項8】
1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)を含む、請求項4記載の分子。
【請求項9】
配列番号1を含む、請求項4記載の分子。
【請求項10】
ペプチドと会合した脂質を含む分子を含むリポソームであって、
前記ペプチドが、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドであるリポソーム。
【請求項11】
分子が、脂質とペプチドとの間にリンカーを含む請求項10記載のリポソーム。
【請求項12】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項10記載のリポソーム。
【請求項13】
リンカーが、ポリエチレングリコール(PEG)を含む請求項12記載のリポソーム。
【請求項14】
1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含む、請求項12記載のリポソーム。
【請求項15】
リポソームが、ホスファチジルコリン、ホスファチジルグリセロール、及びリゾ脂質からなる群より選択される脂質をさらに含む請求項10記載のリポソーム。
【請求項16】
リポソームが、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)及びモノステアロイルホスファチジルコリン(MSPC)を含む請求項10記載のリポソーム。
【請求項17】
治療用化合物及び造影化合物からなる群より選択される化合物をさらに含む、請求項10記載のリポソーム。
【請求項18】
リポソームが、二重膜を有して内部空間を画定し、さらに、化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる、請求項17記載のリポソーム。
【請求項19】
治療用化合物が、抗癌化合物である請求項17記載のリポソーム。
【請求項20】
抗癌化合物が、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物からなる群より選択される請求項19記載のリポソーム。
【請求項21】
抗癌化合物が、ドセタキセル、ドキソルビシン、及びカルボプラチンからなる群より選択される請求項19記載のリポソーム。
【請求項22】
造影剤を含む請求項17記載のリポソーム。
【請求項23】
1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含み、治療剤がカルボプラチンである請求項17記載のリポソーム。
【請求項24】
以下の工程を含む、化合物を、それを必要とする対象に投与する方法:
前記化合物と、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドとを会合させる工程;及び
前記工程によって得られたペプチドと会合した化合物を、前記対象に接触させる工程。
【請求項25】
ペプチドが、脂質と結合しており、且つ、前記ペプチドと結合した脂質がリポソームを形成している、請求項24記載の方法。
【請求項26】
ペプチドと結合した脂質が、前記脂質と前記ペプチドの間にリンカーを含む請求項24記載の方法。
【請求項27】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項25記載の方法。
【請求項28】
リンカーが、ポリエチレングリコール(PEG)を含む請求項26記載の方法。
【請求項29】
ペプチドと結合した脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含む、請求項25記載の方法。
【請求項30】
リポソームが、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)及びモノステアロイルホスファチジルコリン(MSPC)をさらに含み、且つ、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃である、請求項24記載の方法。
【請求項31】
リポソームが、PEG化脂質をさらに含む請求項24記載の方法。
【請求項32】
化合物が、治療用化合物及び造影化合物からなる群より選択される請求項24記載の方法。
【請求項33】
リポソームが、二重膜を有して内部空間を画定し、さらに、化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる、請求項24記載の方法。
【請求項34】
治療用化合物が、抗癌化合物である請求項32記載の方法。
【請求項35】
抗癌化合物が、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物からなる群より選択される請求項34記載の方法。
【請求項36】
抗癌化合物が、ドセタキセル、ドキソルビシン、及びカルボプラチンからなる群より選択される請求項34記載の方法。
【請求項37】
化合物が、造影剤である請求項32記載の方法。
【請求項38】
対象の一部を加温する工程をさらに含む請求項24記載の方法。
【請求項39】
加温する工程が、電磁エネルギー、赤外線放射、又は超音波の使用を含む請求項38記載の方法。
【請求項40】
以下の工程を含む、癌の治療を必要とする対象において、癌を治療する方法:
前記対象にリポソームを投与する工程であって、前記リポソームが、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドと会合した脂質を含み、且つ、前記リポソームが抗癌化合物を含む工程。
【請求項41】
ペプチドが、脂質と結合している請求項40記載の方法。
【請求項42】
ペプチドと結合した脂質が、前記脂質と前記ペプチドの間にリンカーを含む請求項41記載の方法。
【請求項43】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項41記載の方法。
【請求項44】
リンカーが、ポリエチレングリコール(PEG)を含む請求項41記載の方法。
【請求項45】
ペプチドと結合した脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含む、請求項41記載の方法。
【請求項46】
リポソームが、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)及びモノステアロイルホスファチジルコリン(MSPC)をさらに含み、且つ、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃である、請求項40記載の方法。
【請求項47】
リポソームが、PEG化脂質を含む請求項40記載の方法。
【請求項48】
リポソームが、二重膜を有して内部空間を画定し、さらに、抗癌化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる、請求項40記載の方法。
【請求項49】
抗癌化合物が、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物からなる群より選択される請求項40記載の方法。
【請求項50】
抗癌化合物が、ドセタキセル、ドキソルビシン、及びカルボプラチンからなる群より選択される請求項40記載の方法。
【請求項51】
対象の一部を加温する工程をさらに含む請求項40記載の方法。
【請求項52】
加温する工程が、電磁エネルギー、赤外線放射、又は超音波の使用を含む請求項51記載の方法。
【請求項1】
配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドであって、長さが約6〜約15アミノ酸であるペプチド。
【請求項2】
配列番号1を含む、請求項1記載のペプチド。
【請求項3】
配列番号1からなる、請求項1記載のペプチド。
【請求項4】
配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドと会合した脂質を含む分子。
【請求項5】
脂質とペプチドとの間にリンカーを含む、請求項4記載の分子。
【請求項6】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項5記載の分子。
【請求項7】
リンカーが、ポリエチレングリコール(PEG)を含む請求項5記載の分子。
【請求項8】
1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)を含む、請求項4記載の分子。
【請求項9】
配列番号1を含む、請求項4記載の分子。
【請求項10】
ペプチドと会合した脂質を含む分子を含むリポソームであって、
前記ペプチドが、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドであるリポソーム。
【請求項11】
分子が、脂質とペプチドとの間にリンカーを含む請求項10記載のリポソーム。
【請求項12】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項10記載のリポソーム。
【請求項13】
リンカーが、ポリエチレングリコール(PEG)を含む請求項12記載のリポソーム。
【請求項14】
1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含む、請求項12記載のリポソーム。
【請求項15】
リポソームが、ホスファチジルコリン、ホスファチジルグリセロール、及びリゾ脂質からなる群より選択される脂質をさらに含む請求項10記載のリポソーム。
【請求項16】
リポソームが、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)及びモノステアロイルホスファチジルコリン(MSPC)を含む請求項10記載のリポソーム。
【請求項17】
治療用化合物及び造影化合物からなる群より選択される化合物をさらに含む、請求項10記載のリポソーム。
【請求項18】
リポソームが、二重膜を有して内部空間を画定し、さらに、化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる、請求項17記載のリポソーム。
【請求項19】
治療用化合物が、抗癌化合物である請求項17記載のリポソーム。
【請求項20】
抗癌化合物が、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物からなる群より選択される請求項19記載のリポソーム。
【請求項21】
抗癌化合物が、ドセタキセル、ドキソルビシン、及びカルボプラチンからなる群より選択される請求項19記載のリポソーム。
【請求項22】
造影剤を含む請求項17記載のリポソーム。
【請求項23】
1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含み、治療剤がカルボプラチンである請求項17記載のリポソーム。
【請求項24】
以下の工程を含む、化合物を、それを必要とする対象に投与する方法:
前記化合物と、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドとを会合させる工程;及び
前記工程によって得られたペプチドと会合した化合物を、前記対象に接触させる工程。
【請求項25】
ペプチドが、脂質と結合しており、且つ、前記ペプチドと結合した脂質がリポソームを形成している、請求項24記載の方法。
【請求項26】
ペプチドと結合した脂質が、前記脂質と前記ペプチドの間にリンカーを含む請求項24記載の方法。
【請求項27】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項25記載の方法。
【請求項28】
リンカーが、ポリエチレングリコール(PEG)を含む請求項26記載の方法。
【請求項29】
ペプチドと結合した脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含む、請求項25記載の方法。
【請求項30】
リポソームが、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)及びモノステアロイルホスファチジルコリン(MSPC)をさらに含み、且つ、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃である、請求項24記載の方法。
【請求項31】
リポソームが、PEG化脂質をさらに含む請求項24記載の方法。
【請求項32】
化合物が、治療用化合物及び造影化合物からなる群より選択される請求項24記載の方法。
【請求項33】
リポソームが、二重膜を有して内部空間を画定し、さらに、化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる、請求項24記載の方法。
【請求項34】
治療用化合物が、抗癌化合物である請求項32記載の方法。
【請求項35】
抗癌化合物が、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物からなる群より選択される請求項34記載の方法。
【請求項36】
抗癌化合物が、ドセタキセル、ドキソルビシン、及びカルボプラチンからなる群より選択される請求項34記載の方法。
【請求項37】
化合物が、造影剤である請求項32記載の方法。
【請求項38】
対象の一部を加温する工程をさらに含む請求項24記載の方法。
【請求項39】
加温する工程が、電磁エネルギー、赤外線放射、又は超音波の使用を含む請求項38記載の方法。
【請求項40】
以下の工程を含む、癌の治療を必要とする対象において、癌を治療する方法:
前記対象にリポソームを投与する工程であって、前記リポソームが、配列番号1、配列番号3、配列番号4、配列番号5、配列番号6、配列番号7、及び配列番号8からなる群より選択される配列を含むペプチドと会合した脂質を含み、且つ、前記リポソームが抗癌化合物を含む工程。
【請求項41】
ペプチドが、脂質と結合している請求項40記載の方法。
【請求項42】
ペプチドと結合した脂質が、前記脂質と前記ペプチドの間にリンカーを含む請求項41記載の方法。
【請求項43】
脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン(DSPE)である請求項41記載の方法。
【請求項44】
リンカーが、ポリエチレングリコール(PEG)を含む請求項41記載の方法。
【請求項45】
ペプチドと結合した脂質が、1,2−ジステアロイル−sn−グリセロ−3−ホスホエタノールアミン−N−[メトキシ(ポリエチレングリコール)−2000](DSPE−PEG2000)−LARLLTを含む、請求項41記載の方法。
【請求項46】
リポソームが、1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン(DPPC)及びモノステアロイルホスファチジルコリン(MSPC)をさらに含み、且つ、前記リポソームのゲル相から液相への相転移温度が約39.0℃〜約45℃である、請求項40記載の方法。
【請求項47】
リポソームが、PEG化脂質を含む請求項40記載の方法。
【請求項48】
リポソームが、二重膜を有して内部空間を画定し、さらに、抗癌化合物が、前記リポソームの内部空間、前記リポソームの二重膜、又は、前記内部空間及び前記二重膜の両方に含まれる、請求項40記載の方法。
【請求項49】
抗癌化合物が、アルキル化剤、代謝拮抗剤、紡錘体毒植物アルカロイド、細胞障害性抗腫瘍抗生物質、トポイソメラーゼ阻害剤、モノクローナル抗体又はその断片、光感作性物質、キナーゼ阻害剤、抗腫瘍性酵素及び酵素阻害剤、アポトーシス誘導剤、生物学的反応修飾物質、抗ホルモン剤、レチノイド、及びプラチナ含有化合物からなる群より選択される請求項40記載の方法。
【請求項50】
抗癌化合物が、ドセタキセル、ドキソルビシン、及びカルボプラチンからなる群より選択される請求項40記載の方法。
【請求項51】
対象の一部を加温する工程をさらに含む請求項40記載の方法。
【請求項52】
加温する工程が、電磁エネルギー、赤外線放射、又は超音波の使用を含む請求項51記載の方法。
【図1】

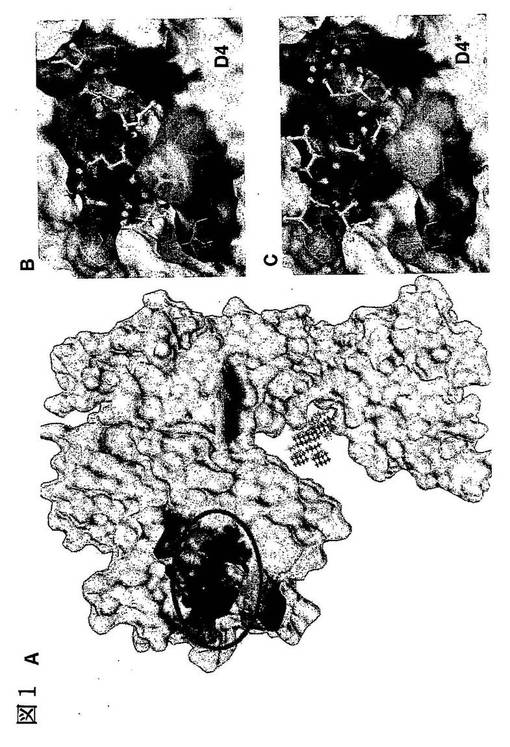
【図2】

【図3】
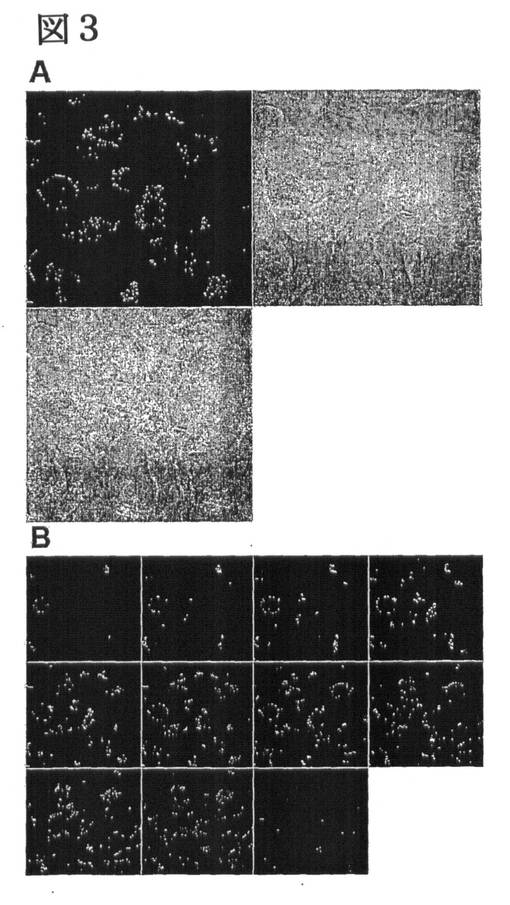
【図4】
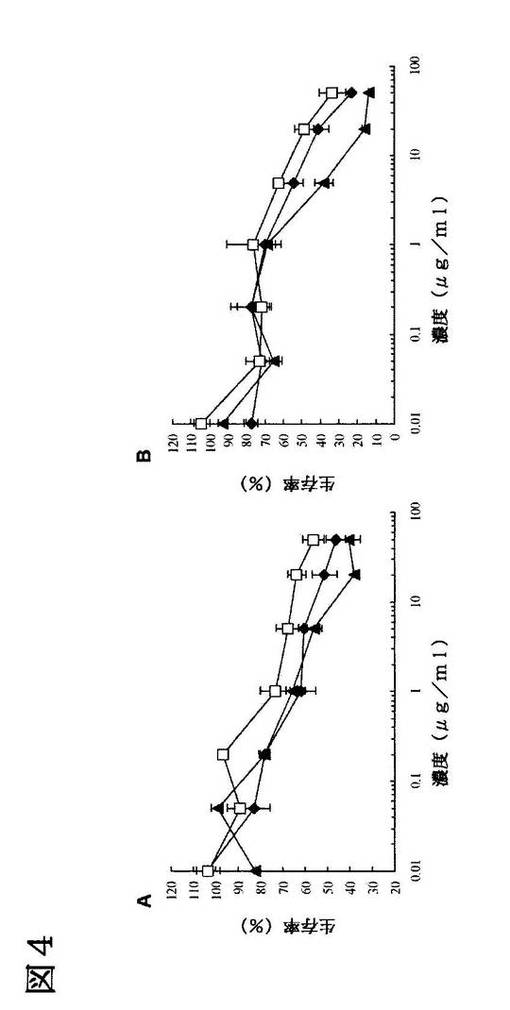
【図5】
【図6】
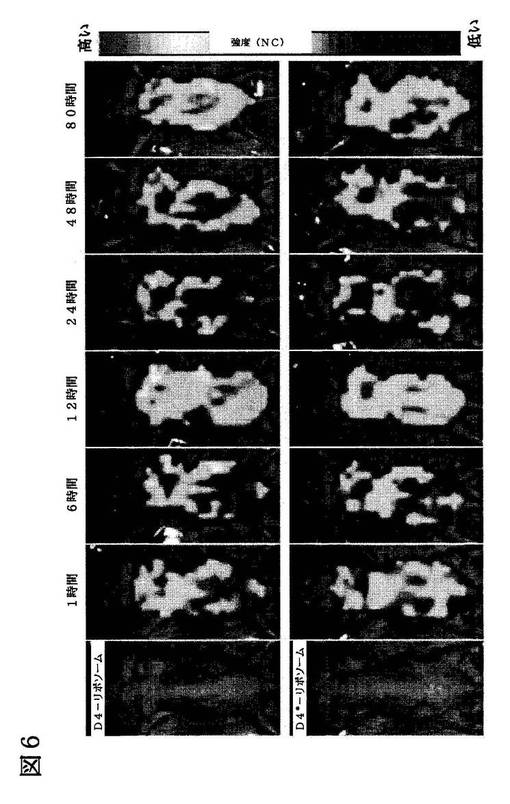
【図7】
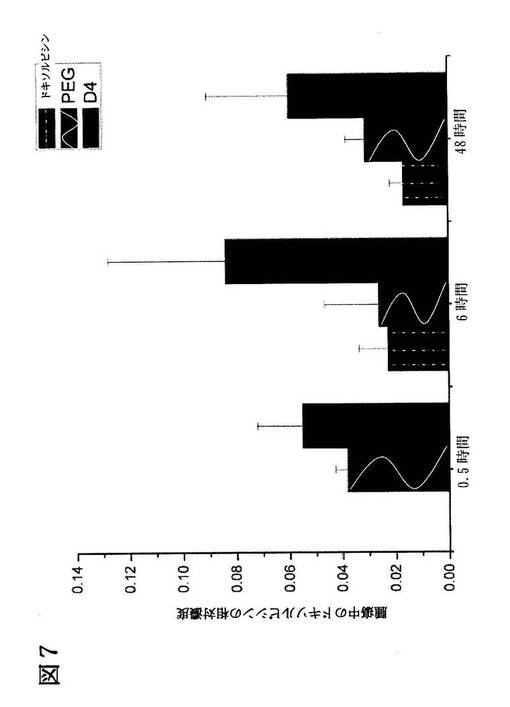
【図8】
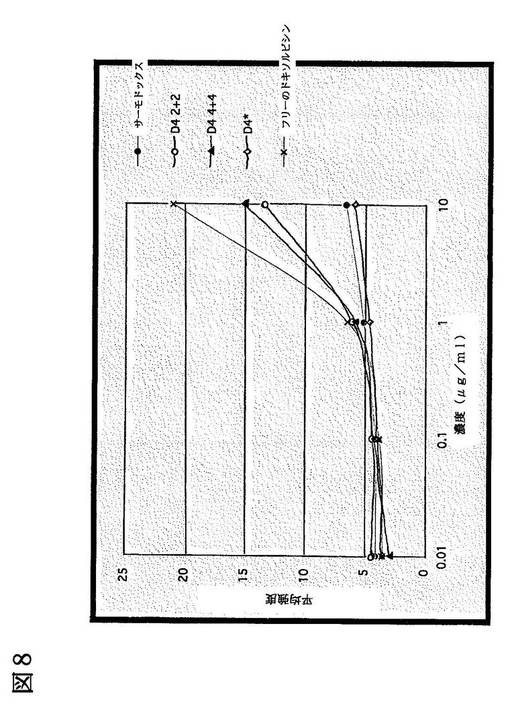
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2011−502177(P2011−502177A)
【公表日】平成23年1月20日(2011.1.20)
【国際特許分類】
【出願番号】特願2010−532406(P2010−532406)
【出願日】平成20年11月5日(2008.11.5)
【国際出願番号】PCT/CN2008/001847
【国際公開番号】WO2009/062399
【国際公開日】平成21年5月22日(2009.5.22)
【出願人】(510124180)シャンハイ ジャオトン ユニバーシティ (1)
【氏名又は名称原語表記】SHANGHAI JIAOTONG UNIVERSITY
【Fターム(参考)】
【公表日】平成23年1月20日(2011.1.20)
【国際特許分類】
【出願日】平成20年11月5日(2008.11.5)
【国際出願番号】PCT/CN2008/001847
【国際公開番号】WO2009/062399
【国際公開日】平成21年5月22日(2009.5.22)
【出願人】(510124180)シャンハイ ジャオトン ユニバーシティ (1)
【氏名又は名称原語表記】SHANGHAI JIAOTONG UNIVERSITY
【Fターム(参考)】
[ Back to top ]