
ペプチド及び関連化合物の経皮送達システム
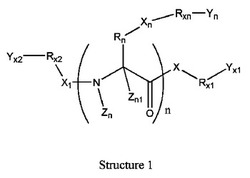
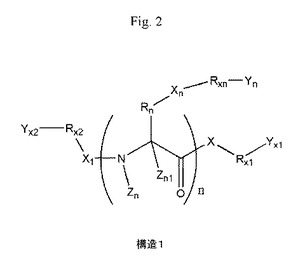
構造1で表されるペプチド及び関連化合物の新規な正荷電プロドラッグを合成した。構造1で表される化合物は、標準的なペプチド合成プロトコルにより合成され得る。ペプチドのプロドラッグの正に荷電したアミノ基は、薬物を水中に溶解させるだけでなく、膜のホスファート頭基上の負電荷と結合し、プロドラッグを細胞質ゾルの中に押し込む。プロドラッグは、ペプチド及び関連化合物よりも〜100〜1000倍速く人の皮膚を通して拡散する。プロドラッグは、人又は動物におけるペプチド及び関連化合物治療可能な状態の治療に使用され得る。プロドラッグは、どんな種類の薬物治療のためにも経皮投与され得、消化管におけるタンパク質分解酵素によるタンパク質分解を回避し得る。プロドラッグの制御経皮投与システムは、オキシカム及び関連化合物が絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、その副作用を減少させることを可能にする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ペプチドを正に荷電した水溶性プロドラッグに移行させることによるペプチド及び関連化合物の経皮送達システム、及び人又は動物において、ペプチド及び関連化合物により治療可能な状態を治療する際のその医薬用途に関する。より具体的に言うと、本発明は、ペプチド及び関連化合物の速い浸透を可能にし、それらを経皮投与可能にするためにある。
【背景技術】
【0002】
全てのペプチドはポリマーであり、結合してポリマーを作るモノマーはアミノ酸である。2〜50アミノ酸残基を含有する鎖は、集合的にペプチドと称される。もし鎖が50アミノ酸残基より多いならば、それらはタンパク質と呼ばれる。ペプチドは、全ての生物において莫大な種類の役割を果たしている。ペプチドホルモンは、ホルモンのうちの最も大きなグループである。特定され、合成されるペプチドホルモンの数は増え続けている。それらは、生命をコントロールするプロセスにおいて興味深い役割を有している。1ナノグラムの甲状腺刺激ホルモン放出ホルモン(hyrotropin-releasing hormone)をマウスに注入すると、血液から甲状腺へのヨウ化物の取り込みを増大させる (R.L. Kisliuk, Principles of Medicinal Chemistry, 4th Ed., W.O. Foyeなど、Eds., Williams & Wilkins, 4th Ed. 1995, p.606)。タフトシン (Thr-Lys-Pro-Arg) は、食作用を刺激し、抗体依存性の細胞傷害性を促進し (V .A. Najjar, Mol. Cell. Biochem. 41. 1, 1981)、脳及び小腸から単離されたMet-エンケファリン (Tyr-Gly-Gly-Phe-Met) は、同じ受容体と結合し、鎮痛作用を有するという点で、モルヒネが作用するように作用する (J.R.Jaffe及びW.R. Martin, in Pharmacological Basis of Therapeutics, A.G. Gilmanなど、Eds., New York, Pergamon Press, 1990, p.481)。オキシトシン (Pierceなど、J. Biol. Chem. 199, 929, 1952)、バソプレシン (Kammなど、J. Am. Chem. Soc. 50, 573, 1928), アンジオテンシン (J.C. Garrison及びM.J. Peach, in Pharmacological Basis of Therapeutics, A.G. Gilmanなど、Eds., New York, Pergamon Press, 1990, p.749)、ガストリン (P.C. Emson及びB.E.B. Sandberg, Annu, Rep. Med. Chem., 18, 31, 1983)、ソマトスタチン (A.V. Schallyなど、Annu. Rev. Biochem., 47, 89, 1978), ダイノルフィン (M.G. Weisskopfなど、Nature, 362, 423, 1993)、エンドセリン (A.M. Doherty, J. Med. Chem., 35, 1493, 1992)、セクレチン (E. Jorper, Gastroenterology, 55, 157, 1968)、カルシトニン (M.V.L. Rayなど、Biotechnology, 11, 64, 1993)、インスリン (F. Sanger, Br. Med. Bull., 16. 183, 1960)、及びその他多くは、構造が推定されたペプチドであり、それらは多くの病気の治療のために使用される。
【0003】
残念ながら、ペプチド及び関連化合物は、タンパク質分解酵素により急速にタンパク質分解される。ペプチドを経口的に服用すると、それらは数分で破壊される。注射の場合、ペプチドの投与は痛みを伴い、多くの場合、慢性症状を治療するために頻繁で金のかかる来院が必要とされる。
【0004】
薬物投与の一代替方法は、局所送達である。局所薬物送達は、いくつかの利点を有する。この方法は、肝臓及び消化管での初回通過代謝により引き起こされる薬物の不活性化を回避するのに役立つ。それは、全身に曝露されることなく、意図した作用部位に、適切な濃度の薬物の局所的な送達を供給し得る。Fishman(Fishman; Robert, 米国特許第7,052,715号明細書)は、経口薬物治療と関連した追加的な課題を指摘した。それは、疼痛又は炎症のある末端部を効果的に治療するためには、血流中で達成されなければならない濃度レベルが、有意でなければならないということである。これらのレベルはしばしば、疼痛又は損傷のある特定部位を正確に標的とすることが可能である場合に必要となるレベルよりもずっと高い。Yeagerは、男性の勃起不全の治療を目的にPGE1を送達するため、浸透エンハンサーを使用することを試みた(Yeager, James L. 米国特許第6,693,135号明細書)。Susan Milosovichらは、テストステロニル−4−ジメチルアミノブチラート塩酸塩(testosteronyl-4-dimethylaminobutyrate.HCl)(TSBH)を設計し、調製した。これは、親油性部分及び生理学的pHにおいてプロトン化された形態で存在する第3級アミン基を有する。彼らは、プロドラッグ(TSBH)は、薬物(TS)それ自体よりも〜60倍速く人の皮膚を通して拡散することを見出した[Susan Milosovichなど、J. Pharm. Sci., 82,227(1993)]。
【発明の概要】
【発明が解決しようとする課題】
【0005】
ペプチドは、全ての生物において莫大な種類の役割を果たしており、それらは多くの病気の治療のために使用される。
残念ながら、ペプチド及び関連化合物は、タンパク質分解酵素により急速にタンパク質分解される。ペプチドを経口的に服用すると、それらは数分で破壊される。注射の場合、ペプチドの投与は痛みを伴い、多くの場合、慢性症状を治療するために頻繁で金のかかる来院が必要とされる。
【課題を解決するための手段】
【0006】
本発明は、ペプチド及び関連化合物の新規な正荷電プロドラッグの調製、並びにそれらの医薬用途に関する。ペプチド及び関連化合物のプロドラッグは、次式の構造1を有する。
【化1】

【0007】
式中、XはO、S、又はNHを表し、X1又はXnはCO、SO、SO2、PO(OR)、NO、又は何もないことを表し、Zn又はZn1はH、CH3、C2H5、C3H7、CF3、C2F5、又はC3F7を表し、Rnはアミノ酸の脂肪族側鎖、アミノ酸のヒドロキシル−若しくは硫黄含有側鎖、アミノ酸の芳香族側鎖、アミノ酸のアミノ、イミダゾリル、若しくはクアニジノ(quanidino)基含有側鎖、又はアミノ酸のカルボキシル若しくはカルボキサミド基含有側鎖のいずれか一つ、H、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、Yx1、Yx2、又はYnはH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は以下の基、
【化2】
又は何もないことを表し、Rx1、Rx2、Rx3、Rx4、Rx5、又はRxnはH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10……であり、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよい。いずれのCH2基はO、S又はNHと置換されてもよい。配列中の1種以上のアミノ酸残基は非天然アミノ酸、例えば、β−アミノ酸、2−ナフチルアラニン、及びアルキル、アルキルオキシ、アルケニル若しくはアルキニルのいずれか一つ、アリール又はヘテロアリール残基により置換され得る。ペプチド鎖上のアミノ酸のアミノ及びカルボキシル官能基は、ラクタムブリッジを形成しホモデティック(homodetic)な環状ペプチドを形成してもよい。システイン、ホメシステイン(homecysteine)、又は他のアミノ酸のチオール基は、ジスルフィドブリッジを形成しヘテロデティックな(heterodetic)環状ペプチドを形成してもよい。
【0008】
ペプチド又は関連化合物のこれらのプロドラッグをデザインするための原則は、以下の通りである。1.プロドラッグは、親油性部分及び生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護された(monoprotected)クアニジノ基(親水性部分)を有していなければならない。2.ペプチドの全てのプロドラッグは1つ又は2つだけ(好ましくは1つ)の、生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有するべきである。3.第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基は、N末端、C末端、又はペプチドの側鎖であり得る。N末端又はC末端部分が好ましい。4.ペプチドを親油性にするために、カルボキシル基、アミノ基、グアニジン基又は他の親水性基は、アルキル、アリール、若しくはヘテロアリールエステル又はアミド基で保護され得る。
【0009】
以下のものはこれらのプロドラッグの例である。
【化3】
【化4】
【化5】
【0010】
式中、RはH、分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10……、アリール又はヘテロアリール残基を表し、X4、X5、X6、X7、X8、又はX9はCO、SO2、PO(OR)、NO、又は何もないことを表し、R1、R2、R3、R4、R5、R6、R7、R8、又はR9はH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基を表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10であり、Arはフェニル、2’−ナフチル、4−ヨードフェニル、又は他のアリール若しくはヘテロアリール残基を表し、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、いずれのCH2基はO、S又はNHと置換されてもよい。
【0011】
【化6】
【化7】
【0012】
薬物吸収は、胃腸管からであろうと他の部位からであろうと、薬物が分子形で障壁膜を横断して通過することを必要とする。薬物は初めに溶解しなければならず、もし薬物が望ましい生物薬剤的性質を有しているならば、高濃度の領域から低濃度の領域へと膜を横断して通過し、血液循環又は体循環に入るだろう。全ての生物学的膜は、主要な構成成分として脂質を含有している。膜形成について主要な役割を果たしている分子は、全てホスファート含有高極性頭基(head group)を有し、たいていの場合、2つの高疎水性炭化水素尾部(tail)を有している。膜は2分子膜であり、その2つの親水性頭基は、両側にある水性領域へと外側を向いている。非常に親水性な薬物(ほとんどのペプチド)は膜の疎水性層を通過することができず、非常に疎水性な薬物はそれらの類似性のため膜の一部として疎水性層に留まり、それ故に内側にある細胞質ゾルに能率的に入ることができないだろう。
【0013】
本発明の目的は、膜及び皮膚障壁を通してのそれらの浸透率を増大させることによって、ペプチド及び関連化合物を経皮投与(局所適用)可能にすることである。ペプチド及び関連化合物のこれらの新規なプロドラッグは、共通して2つの構造的特徴を有している。すなわち、これらは、(親油性のアルコールを使用してカルボキシル基を保護し、親油性の酸を使用してアミノ、ヒドロキシル、若しくはグアニジン基、又はペプチドの他の親水性基を保護することにより形成され得る)親油性部分、及び生理学的pHにおいてプロトン化された形態で存在する第1級、第2級又は第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有している。膜障壁を通して能率的に通過するためには、このような親水性親油性バランスが必要とされる[Susan Milosovichなど、J. Pharm. Sci., 82,227(1993)]。正に荷電されたアミノ基は薬物の溶解性を大きく増大させ、親油性部分は、プロドラッグが親油性の膜及び皮膚障壁に入るのに役立つだろう。これらの新規なプロドラッグは、溶液、スプレー、ローション、軟膏、エマルジョン又はゲルのような投与形態で経皮投与されると、皮膚表面の水分中に直ちに溶解するだろう。これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は、半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中にプロドラッグを押し込むだろう。
【0014】
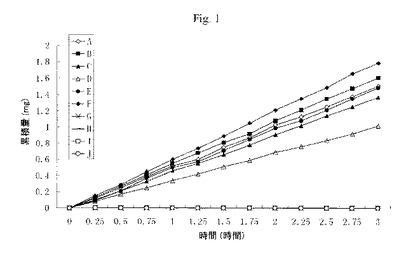
いくつかのプロドラッグの人の皮膚を通しての浸透率を、修正フランツ(Franz)細胞を使用することにより試験管内で測定した。修正フランツ細胞は前大腿部及び後大腿部のヒト皮膚組織(厚さ360〜400μm)から単離した。受容液(receiving fluid)は生理食塩水中2%ウシ血清アルブミン2mlからなり、600rpmで撹拌した。時間に対する、皮膚に浸透するこれらのプロドラッグ及びそれらの親薬物の累積量を特異的な高速液体クロマトグラフィー法により測定した。pH7.4ホスファート緩衝液(0.2M)2mL中、いつくかのプロドラッグ及びペプチドの10%溶液からなるドナーを使用して得た結果を図1に示す。人の皮膚を通して拡散する、Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、HC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg(Ac)-Trp-Lys-NH2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Ala(2-ナフチル)-Arg-Trp-Lys-NH2.HCl、Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HCl、Ac-Tyr-Gly-Gly-Phe-Met-OH、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg-Trp-Lys-NH2、及びH-Val-Pro-Gly-Pro-Arg-OHに対して、0.52 mg、0.55 mg、0.46 mg、0.34 mg、0.50 mg、0.60 mg、0.001 mg、0.001 mg、0.001 mg、及び0.001 mg/cm2/hという明白な流速値を計算した。本結果は、プロドラッグは、ペプチド及び関連化合物よりも340〜600倍速く人の皮膚を通して拡散するということを示す。本結果は、ジアルキルアミノエチル基上の正電荷は、薬物が膜又は皮膚障壁を横断して通過することにおいて非常に重要な役割を有していることを示す。
【0015】
良いプロドラッグは、血漿中ですぐに変化して薬物それ自体に戻るべきである。本発明
におけるペプチドのプロドラッグは、ヒト血漿中、良い収率で非常に速く変化して親ペプチドに戻り得る。全血1ml中、Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC120mgを37℃で30分間インキュベートした。混合物をHPLCにより解析した。3%のAc-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、2%のAc-Tyr-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、8%のAc-Tyr-Gly-Gly-Phe-Met-OH、60%のH-Tyr-Gly-Gly-Phe-Met-OH、27%の他の副生成物(アミノ酸、ジペプチド、トリペプチド、テトラペプチド)を観測した。HC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3に対して、5%のHC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、6%の(CH3)2NCH2CH2CH2CO-Tyr-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、10%の(CH3)2NCH2CH2CH2CO-Tyr-Gly-Gly-Phe-Met-OH、55%のH-Tyr-Gly-Gly-Phe-Met-OH、及び24%の他の副生成物を観測した。シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HClに対して、4%のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl、8%のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(Ac)-Trp-Lys-OH、10%のシクロ(1,6)-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、45%のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、33%の他の副生成物を観測した。本結果は、ペプチドのプロドラッグのほとんどが変化して親ペプチドに戻り、従って、ペプチドの経皮送達システムは成功であるということを示す。
【0016】
エンテロスタチン[Val-Pro-Asp-Pro-Arg(VPDPR)、Val-Pro-Gly-Pro-Arg(VPGPR)及びAla-Pro-Gly-Pro-Arg(APGPR)]は、トリプシン開裂(tryptic cleavage)後のプロコリパーゼのNH2末端に由来するペンタペプチドであり、腸−脳(gut-brain)ペプチドのファミリーに属する。それらは脂肪摂取を調整し、肥満治療のために使用されてもよい (Erlanson-Albertsson C, York D, Obes. Rev. 1997 Jul; 5(4): 360-72及びSorhede M, Mei J, Erlanson-Albertsson C. , J Physiol. 87:273-275,1993)。H-Val-Pro-Asp-Pro-Arg-OHを一晩中飢えさせたOsborne-Mendelラットに腹腔内投与した場合、食物摂取の用量依存的な減少をもたらした。この摂食抑制は、ラットが高脂肪食を与えられた場合に観察されたが、高炭水化物、低脂肪食を与えられたラットにおいては観察されなかった (Okada S.など、Physiol Behav., 1991 Jun; 49(6): 1185-9)。暗いときに始まる(dark-onset)給餌時間の初めにおいて、ラットの背中に適用される水0.5ml中、5mg/kgのAc-Val-Pro-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClを、ラット(n=5)に経皮投与した (n=5 それぞれ、食物を提示されて一晩中絶食したラットと随時食物を与えられたラットの両方)。脂肪摂取のこの選択的な抑制が観察された。
【0017】
メラノコルチンIIは環状ラクタムペプチド、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OHである。それは、男性及び女性の性機能障害を治療するための、Palatinの(AMEX:PTN)新規な薬剤候補である。まず第一に、メラノコルチンアゴニストと呼ばれる治療の新規な分類において、メラノコルチンIIは、現在利用できるED薬において見られる心臓血管系への作用なしに、勃起不全(ED)及び女性の性機能障害を治療するのに有効である見込みを示した。メラノコルチンIIは、直接的に血管系に関係するよりむしろ、中枢神経系に関係するメカニズムを通して作用する。結果として、それは、現在利用できる製品を超える顕著な安全性及び有効性という利点を提供し得る。本発明の新規なプロドラッグは、非常に高い割合(〜0.3〜0.5mg/h/cm2)で人の皮膚を通して拡散し得、ほとんど副作用のない、勃起不全を治療する又は女性の性的刺激を増強する方法を提供し得る。pH7.0ホスファート緩衝液(0.1M)0.2ml中、2mg/kgのシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドA)及びシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(NO2)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドB)を、1日に1回、5日間、雄ラット(30匹)の背中に適用した。結果、ペプチドA又はペプチドBが与えられたラットでは、それが与えられなかったラットに比べて、勧誘(solicitation)において、ペプチドAでは5倍の増加、ペプチドBでは6倍の増加を示し、交尾において、ペプチドAとペプチドBの両方とも3倍の増加を示した。pH7.0ホスファート緩衝液(0.1M)0.2ml中、同量のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドA)及びシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(NO2)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドB)を、1日に1回、5日間、雄ラット(30匹)と雌ラット(30匹)の両方の背中に適用した場合、その結果、ペプチドA又はペプチドBが与えられたラットでは、それが与えられなかったラットに比べて、勧誘において6倍の増加を示し、交尾において5倍の増加を示した。
【0018】
オピオイドペプチド、例えば、Met-エンケファリン(H-Tyr-Gly-Gly-Phe-Met-OH)、Leu-エンケファリン(H-Tyr-Gly-Gly-Phe-Leu-OH)、H-Tyr-D-Ala-Gly-N-Me-Phe-Met(O)-OL、H-Tyr-D-Ala-Gly-Phe-Leu-OHなどは、モルヒネ様鎮痛作用を発揮する。酢酸溶液をマウスの腹腔内に投与したときに起こるライジング(writhing)数を数え、対照群に基づく抑制率を計算した。HCl.H-Tyr(Ac)-D-Ala-Gly-Phe-Leu-OCH2(CH2)4CH3(10mg/kg、B)、Ac-Tyr(Ac)-D-Ala-Gly-Phe-Leu-OCH2CH2N(CH2CH3).HCl(10mg/kg、C)、及びHCl.H-Tyr(Ac)-D-Ala-Gly-Phe-Met(O)-OL(10mg/kg、D)を、酢酸溶液を投与する30分前にマウスの頸部に経皮投与した。グループAは対照群である。結果を表1に示す。
表1.エンケファリン及び関連化合物のプロドラッグによるライジング抑制率
本結果は、ペプチドプロドラッグを使用する経皮送達システムは、肥満及び疼痛の治療、並びに男性及び女性の性機能障害の治療に対して非常によく作用する、ということを示す。
【0019】
ペプチド及び関連化合物は非常に親水性であり、皮膚膜障壁に浸透することができない。ペプチドを経口的に服用すると、ペプチド及び関連化合物は、消化管内で、数分でタンパク質分解酵素により急速にタンパク質分解される。注射の場合、ペプチドの投与は痛みを伴い、多くの場合、慢性症状を治療するために頻繁で金のかかる来院が必要とされる。ペプチドのプロドラッグが皮膚に局所適用されると、それらは皮膚の水分中に直ちに溶解するだろう。これらのプロドラッグのアミノ基上にある正電荷は、皮膚膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は細胞質ゾルの中にプロドラッグを押し込むだろう。
【0020】
構造1で表される化合物は、標準的なペプチド合成プロトコルにより調製され得る。構造4−Cで表されるシクロペプチド及び関連化合物の調製では、標準的なペプチド合成プロトコルによりペプチド鎖は合成され得、リシンの側鎖は2−、又は4−Pyoc保護基により保護され、ペプチドの環化は樹脂上で行われた。
【発明の効果】
【0021】
本発明におけるこれらのペプチド及び関連化合物のプロドラッグは、親油性部分及び親水性部分(生理学的pHにおいてプロトン化された形態で存在するアミン基)を有する。これらのプロドラッグの正に荷電したアミノ基は2つの優れた利点を有する。第一に、プロドラッグを水中に溶解させる。すなわち、これらの新規なプロドラッグが、液体、スプレー、ローション、軟膏、エマルジョン又はゲルのような投与形態で経皮投与されるとき、これらは直ちに、皮膚、眼、生殖器部、口、鼻、又は身体の他の部分における水分と混合するだろう。第二に、これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。プロドラッグの(親油性アルキル基で極性基を修飾することによる)親油性部分は、薬物が皮膚膜に入るのに役立つだろう。これらのプロドラッグが膜に入るとき、親水性部分は、半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中にプロドラッグを押し込むだろう。皮膚、眼、生殖器部、口、鼻、又は身体の他の部分での短い残留のため、プロドラッグは、皮膚、眼、生殖器部、口、鼻、又は身体の他の部分に痒み、ヒリヒリ感又は痛みを引き起こさないだろう。実験結果は、40%を超えるプロドラッグが、数分で変化して親ペプチドに戻ったということを示す。プロドラッグは、容易に血液脳関門に浸透しさえし得る。ペプチド及び関連化合物の経皮送達システムは、ペプチドホルモンを医薬的に使用できるようにし得る。これらのプロドラッグを経皮投与することのもう一つの大きな利点は、特に子供への、薬物投与がいっそう容易になるだろうということである。
【図面の簡単な説明】
【0022】
【図1】フランツ細胞(n=5)での、単離されたヒト皮膚組織を横断する、Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、HC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg(Ac)-Trp-Lys-NH2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Ala(2-ナフチル)-Arg-Trp-Lys-NH2.HCl、Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HCl、Ac-Tyr-Gly-Gly-Phe-Met-OH、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg-Trp-Lys-NH2、及びH-Val-Pro-Gly-Pro-Arg-OHの累積量。それぞれの場合において、媒体はpH7.4ホスファート緩衝液(0.2M)であった。
【図2】構造1。式中、XはO、S、又はNHを表し、X1又はXnはCO、SO、SO2、PO(OR)、NO、又は何もないことを表し、Zn又はZn1はH、CH3、C2H5、C3H7、CF3、C2F5、又はC3F7を表し、Rnはアミノ酸の脂肪族側鎖、アミノ酸のヒドロキシル−若しくは硫黄含有側鎖、アミノ酸の芳香族側鎖、アミノ酸のアミノ、イミダゾリル、若しくはクアニジノ基含有側鎖、又はアミノ酸のカルボキシル若しくはカルボキサミド基含有側鎖のいずれか一つ、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、Yx1、Yx2、又はYnはH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、Rx3Rx4Rx5N+A-又は何もないことを表し、Rx1、Rx2、Rx3、Rx4、Rx5、又はRxnはH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよい。いずれのCH2基はO、S又はNHと置換されてもよい。配列中の1種以上のアミノ酸残基は非天然アミノ酸、例えば、β−アミノ酸、2−ナフチルアラニン、及びアルキル、アルキルオキシ、アルケニル若しくはアルキニルのいずれか一つ、アリール又はヘテロアリール残基により置換され得る。ペプチド鎖上のアミノ酸のアミノ及びカルボキシル官能基は、ラクタムブリッジを形成しホモデティックな環状ペプチドを形成してもよい。システイン、ホメシステイン、又は他のアミノ酸のチオール基は、ジスルフィドブリッジを形成しヘテロデティックな環状ペプチドを形成してもよい。
【発明を実施するための形態】
【0023】
(最良の形態)
Ac-Val-Pro-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
1.H-Arg(diAc)-OCH2CH2N(CH2CH3)2の調製
Z-Arg-OH30.8gをアセトン500ml中に溶解した。20%NaOH200mlを反応混合物に加えた。無水酢酸40gを反応混合物に一滴ずつ加えた。混合物を室温で2時間撹拌した。溶媒を蒸発させた。残留物を酢酸エチル500mlで抽出した。酢酸エチル溶液を水(3×100ml)で洗浄した。酢酸エチル層を硫酸ナトリウム上で乾燥させた。酢酸エチル溶液を蒸発させて乾燥した。残留物(Z-Arg(diAc)-OH、30g)をアセトニトリル300ml中に溶解した。混合物を氷水浴で0℃にまで冷却した。N,N−ジエチルアミノエタノール12g、4−ジメチルアミノピリジン2g、及び1,3−ジシクロヘキシルカルボジイミド22gを反応混合物に加えた。反応混合物を0℃で1時間、及び室温で一晩中撹拌する。固形物を濾過により除去し、溶液を蒸発させて乾燥した。残留物を酢酸エチル(2×250ml)で抽出した。酢酸エチル溶液を5%重炭酸ナトリウム(1×500ml)及び水(3×100ml)で洗浄した。酢酸エチル溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥する。残留物[Z-Arg(diAc)-OCH2CH2N(CH2CH3)2、28g]をメタノール300ml中に溶解した。10%Pd/C2gを溶液に加えた。混合物を水素下、室温で10時間撹拌した。Pd/Cを濾過により除去した。溶液を蒸発させて乾燥した。H-Arg(diAc)-OCH2CH2N(CH2CH3)222gを得た。
【0024】
2.Boc-Asp(OEt)-Pro-OSuの調製
L-プロリン15gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びBoc-Asp(OEt)-OSu36gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Boc-Asp(OEt)-Pro-OH)25g及びN−ヒドロキシスクシンイミド11gをジクロロメチレン(dichloromethlene)300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド16gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Boc-Asp(OEt)-Pro-OSu28gを得た。
【0025】
3.H-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFAの調製
H-Arg(diAc)-OCH2CH2N(CH2CH3)222gを5%NaHCO3300ml中に溶解した。アセトン150ml中のBoc-Asp(OEt)-Pro-OSu24gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。酢酸エチル溶液を水(3×100ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸250mlを混合物に加え、混合物を30分間撹拌した。混合物を蒸発させて乾燥した。H-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA32gを得た。
【0026】
4.Ac-Val-Pro-OSuの調製
L-プロリン15gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-VaI-OSu26gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Ac-Val-Pro-OH)20g及びN−ヒドロキシスクシンイミド11gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド16gを反応混合物に加えた。混合物を0℃で1時間、及び室温で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Ac-Val-Pro-OSu20gを得た。
【0027】
5.Ac-Val-Pro-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
H-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA31gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Val-Pro-OSu15gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。有機層を水(3×100ml)で洗浄する。酢酸エチル層を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。ジオキサン(50ml)中HClガス3.5gを溶液に加える。固形物を集め、エーテル(3×50ml)で洗浄する。乾燥後、所望の吸湿性生成物20gを得た。水中での溶解性:150mg/ml、元素分析:C39H66ClN9O11、分子量:872.45、計算% C: 53.69; H: 7.62; Cl: 4.06; N: 14.45; O: 20.17、実測% C: 53.61; H: 7.67; Cl: 4.10; N: 14.40, O: 20.22、MS: m/e: 836.4; m/e+1: 836.4。
【0028】
(発明の形態)
Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HClの調製
1.H-Met-OCH2CH2N(CH2CH3)2.TFAの調製
Boc-Met-OH25gをジクロロメチレン300ml中に溶解した。混合物を氷水浴で0℃にまで冷却した。N,N−ジエチルアミノエタノール12g、4−ジメチルアミノピリジン2g、及び1,3−ジシクロヘキシルカルボジイミド22gを反応混合物に加えた。反応混合物を0℃で1時間、及び室温で一晩中撹拌する。固形物を濾過により除去し、ジクロロメチレン溶液を5%重炭酸ナトリウム(1×500ml)及び水(3×100ml)で洗浄した。酢酸エチル溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥する。残留物[Boc-Met-OCH2CH2N(CH2CH3)2、30g]をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸250mlを混合物に加え、混合物を30分間撹拌する。溶液を蒸発させて乾燥した。H-Met-OCH2CH2N(CH2CH3)2.TFA26gを得た。
【0029】
2.Boc-Gly-Phe-OSuの調製
L-フェニルアラニン20gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びBoc-Gly-OSu28gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Boc-Gly-Phe-OH)22g及びN−ヒドロキシスクシンイミド10gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド15gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄する。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Boc-Gly-Phe-OSu25gを得た。
【0030】
3.H-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.TFAの調製
H-Met-OCH2CH2N(CH2CH3)2.TFA25gを5%NaHCO3300ml中に溶解した。アセトン150ml中のBoc-Gly-Phe-OSu22gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。酢酸エチル溶液を水(3×100ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸200mlを混合物に加え、混合物を30分間撹拌した。混合物を蒸発させて乾燥した。H-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.TFA25gを得た。
【0031】
4.Ac-Tyr(Ac)-Gly-OSuの調製
L-グリシン11gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Tyr(Ac)-OSu36gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Ac-Tyr(Ac)-Gly-OH)28g及びN−ヒドロキシスクシンイミド13gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド18gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄する。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Ac-Tyr(Ac)-Gly-OSu20gを得た。
【0032】
5.Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HClの調製
H-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.TFA24gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Tyr(Ac)-Gly-OSu15gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。有機層を水(3×100ml)で洗浄する。酢酸エチル層を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。ジオキサン(50ml)中HClガス3.5gを溶液に加える。固形物を集め、エーテル(3×50ml)で洗浄する。乾燥後、所望の吸湿性生成物18gを得た。水中での溶解性:200mg/ml、元素分析:C37H53ClN6O9S、分子量:793.37、計算% C: 56.01; H: 6.73; Cl: 4.47; N: 10.59; O: 18.15; S: 4.04、実測% C: 55.96; H: 6.76; Cl: 4.52; N: 10.54, O: 18.19; S: 4.03、MS: m/e: 757.4; m/e+1: 758.4。
【0033】
Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
1.Boc-Gly-Pro-OSuの調製
L-プロリン15gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びBoc-Gly-OSu27.2gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Boc-Gly-Pro-OH)21g及びN−ヒドロキシスクシンイミド11gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド17gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄する。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Boc-Gly-Pro-OSu23gを得た。
【0034】
2.H-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFAの調製
H-Arg(diAc)-OCH2CH2N(CH2CH3)222gを5%NaHCO3300ml中に溶解した。アセトン150ml中のBoc-Gly-Pro-OSu20gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。酢酸エチル溶液を水(3×100ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。溶液を蒸発させて乾燥した。残留物をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸250mlを混合物に加え、混合物を30分間撹拌した。混合物を蒸発させて乾燥した。H-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA28gを得た。
【0035】
3.Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
H-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA26gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Val-Pro-OSu15gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。有機層を水(3×100ml)で洗浄する。酢酸エチル層を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。ジオキサン(50ml)中HClガス3.5gを溶液に加える。固形物を集め、エーテル(3×50ml)で洗浄した。乾燥後、所望の吸湿性生成物18gを得た。水中での溶解性:150mg/ml、元素分析:C35H60ClN9O9、分子量:786.36、計算% C: 53.46; H: 7.69; Cl: 4.51; N: 16.03; O: 18.31、実測% C: 53.43; H: 7.73; Cl: 4.55; N: 16.01, O: 18.29、MS: m/e: 750.4; m/e+1: 751.4。
【0036】
シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HClの調製
1.Ac-Nle-Asp(OFm)-OHの調製
H-Asp(OFm)-OH.TFA43g及びAc-Nle-OSu27gをアセトン300ml中に懸濁した。5%NaHCO3300mlを反応混合物に加えた。混合物を室温で一晩中撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Ac-Nle-Asp(OFm)-OH42gを得た。
【0037】
2.Fmoc-Trp-Lys(4-Pyoc)-OHの調製
H-Lys(4-Pyoc)-OHを参考文献(H. Kunz及びS. Birnbach, Tetrahedron Lett., 25, 3567, 1984; H. Kunz及びR. Barthels, Angew. Chem., Int. Ed. Engl., 22, 783, 1983)に従って調製した。H-Lys(4-Pyoc)-OH33gを5%NaHCO3300ml中に懸濁した。アセトン300ml及びFmoc-Trp-OSu52gを反応混合物に加えた。混合物を室温で一晩中撹拌した。混合物をエーテル(1×500ml)で洗浄した。酢酸エチル500mlを混合物に加え、混合物のpHを氷冷した3NHClで2.2〜2.3に調整する。酢酸エチル層を集め、水で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。有機溶液を蒸発させて乾燥した。Fmoc-Trp-Lys(4-Pyoc)-OH55gを得た。
【0038】
3.シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OHの調製
Wang樹脂100gをDMF700 ml中に懸濁した。Fmoc-Trp-Lys(4-Pyoc)-OH50g、1−ヒドロキシベンゾトリアゾール13g、4−ジメチルアミノピリジン2g、及びN,N’−ジイソプロピルカルボジイミド12g。混合物を室温で一晩中撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Fmoc-Arg(diAc)-OH48g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Fmoc-Phe-OH39g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Fmoc-His(Fmoc)-OH60g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Ac-Nle-Asp(OFm)-OH60g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。ペプチドの(peptided)樹脂をDMF700 ml中に懸濁した。MeI50gを反応混合物に加えた。混合物を室温で1時間、及び50℃で1時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。30%ピペリジン700 mlを樹脂に加えた。混合物を60分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で10時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。トリフルオロ酢酸500mlを樹脂に加え、混合物を室温で1時間撹拌した。樹脂を濾過により除去し、溶液を蒸発させて乾燥する。残留物をエーテル(3×100 ml)で洗浄した。
【0039】
4.シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HClの調製
シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OH10gをDMF300ml中に溶解した。混合物を氷水浴で0℃にまで冷却した。N,N−ジエチルアミノエタノール12g、4−ジメチルアミノピリジン2g、及び1,3−ジシクロヘキシルカルボジイミド22gを反応混合物に加えた。反応混合物を0℃で1時間、及び室温で一晩中撹拌する。固形物を濾過により除去し、ジクロロメチレン溶液を5%重炭酸ナトリウム(1×500ml)及び水(3×100ml)で洗浄した。酢酸エチル溶液を硫酸ナトリウム上で乾燥させた。ジオキサン(20ml)中2gのHClを溶液に加えた。固形物を集め、エーテル(3×30ml)で洗浄した。生成物8gを得た。
【産業上の利用可能性】
【0040】
構造1で表されるプロドラッグは、容易に皮膚膜に浸透し得る。ペプチド及び関連化合物の経皮送達システムは、人又は動物における多くの疾患、例えば、関節リウマチ及び変形性関節症に由来する疼痛、熱、男性の勃起不全、女性の性機能障害、全身血圧、降圧コントロール(hypotensive control)、血小板凝集の抑制、肺疾患、消化器疾患、炎症、ショック、生殖能(reproduction)、受精能(fertility)などの治療において、ペプチドホルモンを有用にし得る。
【技術分野】
【0001】
本発明は、ペプチドを正に荷電した水溶性プロドラッグに移行させることによるペプチド及び関連化合物の経皮送達システム、及び人又は動物において、ペプチド及び関連化合物により治療可能な状態を治療する際のその医薬用途に関する。より具体的に言うと、本発明は、ペプチド及び関連化合物の速い浸透を可能にし、それらを経皮投与可能にするためにある。
【背景技術】
【0002】
全てのペプチドはポリマーであり、結合してポリマーを作るモノマーはアミノ酸である。2〜50アミノ酸残基を含有する鎖は、集合的にペプチドと称される。もし鎖が50アミノ酸残基より多いならば、それらはタンパク質と呼ばれる。ペプチドは、全ての生物において莫大な種類の役割を果たしている。ペプチドホルモンは、ホルモンのうちの最も大きなグループである。特定され、合成されるペプチドホルモンの数は増え続けている。それらは、生命をコントロールするプロセスにおいて興味深い役割を有している。1ナノグラムの甲状腺刺激ホルモン放出ホルモン(hyrotropin-releasing hormone)をマウスに注入すると、血液から甲状腺へのヨウ化物の取り込みを増大させる (R.L. Kisliuk, Principles of Medicinal Chemistry, 4th Ed., W.O. Foyeなど、Eds., Williams & Wilkins, 4th Ed. 1995, p.606)。タフトシン (Thr-Lys-Pro-Arg) は、食作用を刺激し、抗体依存性の細胞傷害性を促進し (V .A. Najjar, Mol. Cell. Biochem. 41. 1, 1981)、脳及び小腸から単離されたMet-エンケファリン (Tyr-Gly-Gly-Phe-Met) は、同じ受容体と結合し、鎮痛作用を有するという点で、モルヒネが作用するように作用する (J.R.Jaffe及びW.R. Martin, in Pharmacological Basis of Therapeutics, A.G. Gilmanなど、Eds., New York, Pergamon Press, 1990, p.481)。オキシトシン (Pierceなど、J. Biol. Chem. 199, 929, 1952)、バソプレシン (Kammなど、J. Am. Chem. Soc. 50, 573, 1928), アンジオテンシン (J.C. Garrison及びM.J. Peach, in Pharmacological Basis of Therapeutics, A.G. Gilmanなど、Eds., New York, Pergamon Press, 1990, p.749)、ガストリン (P.C. Emson及びB.E.B. Sandberg, Annu, Rep. Med. Chem., 18, 31, 1983)、ソマトスタチン (A.V. Schallyなど、Annu. Rev. Biochem., 47, 89, 1978), ダイノルフィン (M.G. Weisskopfなど、Nature, 362, 423, 1993)、エンドセリン (A.M. Doherty, J. Med. Chem., 35, 1493, 1992)、セクレチン (E. Jorper, Gastroenterology, 55, 157, 1968)、カルシトニン (M.V.L. Rayなど、Biotechnology, 11, 64, 1993)、インスリン (F. Sanger, Br. Med. Bull., 16. 183, 1960)、及びその他多くは、構造が推定されたペプチドであり、それらは多くの病気の治療のために使用される。
【0003】
残念ながら、ペプチド及び関連化合物は、タンパク質分解酵素により急速にタンパク質分解される。ペプチドを経口的に服用すると、それらは数分で破壊される。注射の場合、ペプチドの投与は痛みを伴い、多くの場合、慢性症状を治療するために頻繁で金のかかる来院が必要とされる。
【0004】
薬物投与の一代替方法は、局所送達である。局所薬物送達は、いくつかの利点を有する。この方法は、肝臓及び消化管での初回通過代謝により引き起こされる薬物の不活性化を回避するのに役立つ。それは、全身に曝露されることなく、意図した作用部位に、適切な濃度の薬物の局所的な送達を供給し得る。Fishman(Fishman; Robert, 米国特許第7,052,715号明細書)は、経口薬物治療と関連した追加的な課題を指摘した。それは、疼痛又は炎症のある末端部を効果的に治療するためには、血流中で達成されなければならない濃度レベルが、有意でなければならないということである。これらのレベルはしばしば、疼痛又は損傷のある特定部位を正確に標的とすることが可能である場合に必要となるレベルよりもずっと高い。Yeagerは、男性の勃起不全の治療を目的にPGE1を送達するため、浸透エンハンサーを使用することを試みた(Yeager, James L. 米国特許第6,693,135号明細書)。Susan Milosovichらは、テストステロニル−4−ジメチルアミノブチラート塩酸塩(testosteronyl-4-dimethylaminobutyrate.HCl)(TSBH)を設計し、調製した。これは、親油性部分及び生理学的pHにおいてプロトン化された形態で存在する第3級アミン基を有する。彼らは、プロドラッグ(TSBH)は、薬物(TS)それ自体よりも〜60倍速く人の皮膚を通して拡散することを見出した[Susan Milosovichなど、J. Pharm. Sci., 82,227(1993)]。
【発明の概要】
【発明が解決しようとする課題】
【0005】
ペプチドは、全ての生物において莫大な種類の役割を果たしており、それらは多くの病気の治療のために使用される。
残念ながら、ペプチド及び関連化合物は、タンパク質分解酵素により急速にタンパク質分解される。ペプチドを経口的に服用すると、それらは数分で破壊される。注射の場合、ペプチドの投与は痛みを伴い、多くの場合、慢性症状を治療するために頻繁で金のかかる来院が必要とされる。
【課題を解決するための手段】
【0006】
本発明は、ペプチド及び関連化合物の新規な正荷電プロドラッグの調製、並びにそれらの医薬用途に関する。ペプチド及び関連化合物のプロドラッグは、次式の構造1を有する。
【化1】
【0007】
式中、XはO、S、又はNHを表し、X1又はXnはCO、SO、SO2、PO(OR)、NO、又は何もないことを表し、Zn又はZn1はH、CH3、C2H5、C3H7、CF3、C2F5、又はC3F7を表し、Rnはアミノ酸の脂肪族側鎖、アミノ酸のヒドロキシル−若しくは硫黄含有側鎖、アミノ酸の芳香族側鎖、アミノ酸のアミノ、イミダゾリル、若しくはクアニジノ(quanidino)基含有側鎖、又はアミノ酸のカルボキシル若しくはカルボキサミド基含有側鎖のいずれか一つ、H、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、Yx1、Yx2、又はYnはH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は以下の基、
【化2】
又は何もないことを表し、Rx1、Rx2、Rx3、Rx4、Rx5、又はRxnはH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10……であり、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよい。いずれのCH2基はO、S又はNHと置換されてもよい。配列中の1種以上のアミノ酸残基は非天然アミノ酸、例えば、β−アミノ酸、2−ナフチルアラニン、及びアルキル、アルキルオキシ、アルケニル若しくはアルキニルのいずれか一つ、アリール又はヘテロアリール残基により置換され得る。ペプチド鎖上のアミノ酸のアミノ及びカルボキシル官能基は、ラクタムブリッジを形成しホモデティック(homodetic)な環状ペプチドを形成してもよい。システイン、ホメシステイン(homecysteine)、又は他のアミノ酸のチオール基は、ジスルフィドブリッジを形成しヘテロデティックな(heterodetic)環状ペプチドを形成してもよい。
【0008】
ペプチド又は関連化合物のこれらのプロドラッグをデザインするための原則は、以下の通りである。1.プロドラッグは、親油性部分及び生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護された(monoprotected)クアニジノ基(親水性部分)を有していなければならない。2.ペプチドの全てのプロドラッグは1つ又は2つだけ(好ましくは1つ)の、生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有するべきである。3.第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基は、N末端、C末端、又はペプチドの側鎖であり得る。N末端又はC末端部分が好ましい。4.ペプチドを親油性にするために、カルボキシル基、アミノ基、グアニジン基又は他の親水性基は、アルキル、アリール、若しくはヘテロアリールエステル又はアミド基で保護され得る。
【0009】
以下のものはこれらのプロドラッグの例である。
【化3】
【化4】
【化5】
【0010】
式中、RはH、分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10……、アリール又はヘテロアリール残基を表し、X4、X5、X6、X7、X8、又はX9はCO、SO2、PO(OR)、NO、又は何もないことを表し、R1、R2、R3、R4、R5、R6、R7、R8、又はR9はH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基を表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10であり、Arはフェニル、2’−ナフチル、4−ヨードフェニル、又は他のアリール若しくはヘテロアリール残基を表し、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、いずれのCH2基はO、S又はNHと置換されてもよい。
【0011】
【化6】
【化7】
【0012】
薬物吸収は、胃腸管からであろうと他の部位からであろうと、薬物が分子形で障壁膜を横断して通過することを必要とする。薬物は初めに溶解しなければならず、もし薬物が望ましい生物薬剤的性質を有しているならば、高濃度の領域から低濃度の領域へと膜を横断して通過し、血液循環又は体循環に入るだろう。全ての生物学的膜は、主要な構成成分として脂質を含有している。膜形成について主要な役割を果たしている分子は、全てホスファート含有高極性頭基(head group)を有し、たいていの場合、2つの高疎水性炭化水素尾部(tail)を有している。膜は2分子膜であり、その2つの親水性頭基は、両側にある水性領域へと外側を向いている。非常に親水性な薬物(ほとんどのペプチド)は膜の疎水性層を通過することができず、非常に疎水性な薬物はそれらの類似性のため膜の一部として疎水性層に留まり、それ故に内側にある細胞質ゾルに能率的に入ることができないだろう。
【0013】
本発明の目的は、膜及び皮膚障壁を通してのそれらの浸透率を増大させることによって、ペプチド及び関連化合物を経皮投与(局所適用)可能にすることである。ペプチド及び関連化合物のこれらの新規なプロドラッグは、共通して2つの構造的特徴を有している。すなわち、これらは、(親油性のアルコールを使用してカルボキシル基を保護し、親油性の酸を使用してアミノ、ヒドロキシル、若しくはグアニジン基、又はペプチドの他の親水性基を保護することにより形成され得る)親油性部分、及び生理学的pHにおいてプロトン化された形態で存在する第1級、第2級又は第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有している。膜障壁を通して能率的に通過するためには、このような親水性親油性バランスが必要とされる[Susan Milosovichなど、J. Pharm. Sci., 82,227(1993)]。正に荷電されたアミノ基は薬物の溶解性を大きく増大させ、親油性部分は、プロドラッグが親油性の膜及び皮膚障壁に入るのに役立つだろう。これらの新規なプロドラッグは、溶液、スプレー、ローション、軟膏、エマルジョン又はゲルのような投与形態で経皮投与されると、皮膚表面の水分中に直ちに溶解するだろう。これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は、半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中にプロドラッグを押し込むだろう。
【0014】
いくつかのプロドラッグの人の皮膚を通しての浸透率を、修正フランツ(Franz)細胞を使用することにより試験管内で測定した。修正フランツ細胞は前大腿部及び後大腿部のヒト皮膚組織(厚さ360〜400μm)から単離した。受容液(receiving fluid)は生理食塩水中2%ウシ血清アルブミン2mlからなり、600rpmで撹拌した。時間に対する、皮膚に浸透するこれらのプロドラッグ及びそれらの親薬物の累積量を特異的な高速液体クロマトグラフィー法により測定した。pH7.4ホスファート緩衝液(0.2M)2mL中、いつくかのプロドラッグ及びペプチドの10%溶液からなるドナーを使用して得た結果を図1に示す。人の皮膚を通して拡散する、Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、HC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg(Ac)-Trp-Lys-NH2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Ala(2-ナフチル)-Arg-Trp-Lys-NH2.HCl、Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HCl、Ac-Tyr-Gly-Gly-Phe-Met-OH、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg-Trp-Lys-NH2、及びH-Val-Pro-Gly-Pro-Arg-OHに対して、0.52 mg、0.55 mg、0.46 mg、0.34 mg、0.50 mg、0.60 mg、0.001 mg、0.001 mg、0.001 mg、及び0.001 mg/cm2/hという明白な流速値を計算した。本結果は、プロドラッグは、ペプチド及び関連化合物よりも340〜600倍速く人の皮膚を通して拡散するということを示す。本結果は、ジアルキルアミノエチル基上の正電荷は、薬物が膜又は皮膚障壁を横断して通過することにおいて非常に重要な役割を有していることを示す。
【0015】
良いプロドラッグは、血漿中ですぐに変化して薬物それ自体に戻るべきである。本発明
におけるペプチドのプロドラッグは、ヒト血漿中、良い収率で非常に速く変化して親ペプチドに戻り得る。全血1ml中、Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC120mgを37℃で30分間インキュベートした。混合物をHPLCにより解析した。3%のAc-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、2%のAc-Tyr-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、8%のAc-Tyr-Gly-Gly-Phe-Met-OH、60%のH-Tyr-Gly-Gly-Phe-Met-OH、27%の他の副生成物(アミノ酸、ジペプチド、トリペプチド、テトラペプチド)を観測した。HC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3に対して、5%のHC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、6%の(CH3)2NCH2CH2CH2CO-Tyr-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、10%の(CH3)2NCH2CH2CH2CO-Tyr-Gly-Gly-Phe-Met-OH、55%のH-Tyr-Gly-Gly-Phe-Met-OH、及び24%の他の副生成物を観測した。シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HClに対して、4%のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl、8%のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(Ac)-Trp-Lys-OH、10%のシクロ(1,6)-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、45%のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、33%の他の副生成物を観測した。本結果は、ペプチドのプロドラッグのほとんどが変化して親ペプチドに戻り、従って、ペプチドの経皮送達システムは成功であるということを示す。
【0016】
エンテロスタチン[Val-Pro-Asp-Pro-Arg(VPDPR)、Val-Pro-Gly-Pro-Arg(VPGPR)及びAla-Pro-Gly-Pro-Arg(APGPR)]は、トリプシン開裂(tryptic cleavage)後のプロコリパーゼのNH2末端に由来するペンタペプチドであり、腸−脳(gut-brain)ペプチドのファミリーに属する。それらは脂肪摂取を調整し、肥満治療のために使用されてもよい (Erlanson-Albertsson C, York D, Obes. Rev. 1997 Jul; 5(4): 360-72及びSorhede M, Mei J, Erlanson-Albertsson C. , J Physiol. 87:273-275,1993)。H-Val-Pro-Asp-Pro-Arg-OHを一晩中飢えさせたOsborne-Mendelラットに腹腔内投与した場合、食物摂取の用量依存的な減少をもたらした。この摂食抑制は、ラットが高脂肪食を与えられた場合に観察されたが、高炭水化物、低脂肪食を与えられたラットにおいては観察されなかった (Okada S.など、Physiol Behav., 1991 Jun; 49(6): 1185-9)。暗いときに始まる(dark-onset)給餌時間の初めにおいて、ラットの背中に適用される水0.5ml中、5mg/kgのAc-Val-Pro-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClを、ラット(n=5)に経皮投与した (n=5 それぞれ、食物を提示されて一晩中絶食したラットと随時食物を与えられたラットの両方)。脂肪摂取のこの選択的な抑制が観察された。
【0017】
メラノコルチンIIは環状ラクタムペプチド、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OHである。それは、男性及び女性の性機能障害を治療するための、Palatinの(AMEX:PTN)新規な薬剤候補である。まず第一に、メラノコルチンアゴニストと呼ばれる治療の新規な分類において、メラノコルチンIIは、現在利用できるED薬において見られる心臓血管系への作用なしに、勃起不全(ED)及び女性の性機能障害を治療するのに有効である見込みを示した。メラノコルチンIIは、直接的に血管系に関係するよりむしろ、中枢神経系に関係するメカニズムを通して作用する。結果として、それは、現在利用できる製品を超える顕著な安全性及び有効性という利点を提供し得る。本発明の新規なプロドラッグは、非常に高い割合(〜0.3〜0.5mg/h/cm2)で人の皮膚を通して拡散し得、ほとんど副作用のない、勃起不全を治療する又は女性の性的刺激を増強する方法を提供し得る。pH7.0ホスファート緩衝液(0.1M)0.2ml中、2mg/kgのシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドA)及びシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(NO2)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドB)を、1日に1回、5日間、雄ラット(30匹)の背中に適用した。結果、ペプチドA又はペプチドBが与えられたラットでは、それが与えられなかったラットに比べて、勧誘(solicitation)において、ペプチドAでは5倍の増加、ペプチドBでは6倍の増加を示し、交尾において、ペプチドAとペプチドBの両方とも3倍の増加を示した。pH7.0ホスファート緩衝液(0.1M)0.2ml中、同量のシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドA)及びシクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(NO2)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl(ペプチドB)を、1日に1回、5日間、雄ラット(30匹)と雌ラット(30匹)の両方の背中に適用した場合、その結果、ペプチドA又はペプチドBが与えられたラットでは、それが与えられなかったラットに比べて、勧誘において6倍の増加を示し、交尾において5倍の増加を示した。
【0018】
オピオイドペプチド、例えば、Met-エンケファリン(H-Tyr-Gly-Gly-Phe-Met-OH)、Leu-エンケファリン(H-Tyr-Gly-Gly-Phe-Leu-OH)、H-Tyr-D-Ala-Gly-N-Me-Phe-Met(O)-OL、H-Tyr-D-Ala-Gly-Phe-Leu-OHなどは、モルヒネ様鎮痛作用を発揮する。酢酸溶液をマウスの腹腔内に投与したときに起こるライジング(writhing)数を数え、対照群に基づく抑制率を計算した。HCl.H-Tyr(Ac)-D-Ala-Gly-Phe-Leu-OCH2(CH2)4CH3(10mg/kg、B)、Ac-Tyr(Ac)-D-Ala-Gly-Phe-Leu-OCH2CH2N(CH2CH3).HCl(10mg/kg、C)、及びHCl.H-Tyr(Ac)-D-Ala-Gly-Phe-Met(O)-OL(10mg/kg、D)を、酢酸溶液を投与する30分前にマウスの頸部に経皮投与した。グループAは対照群である。結果を表1に示す。
表1.エンケファリン及び関連化合物のプロドラッグによるライジング抑制率
本結果は、ペプチドプロドラッグを使用する経皮送達システムは、肥満及び疼痛の治療、並びに男性及び女性の性機能障害の治療に対して非常によく作用する、ということを示す。
【0019】
ペプチド及び関連化合物は非常に親水性であり、皮膚膜障壁に浸透することができない。ペプチドを経口的に服用すると、ペプチド及び関連化合物は、消化管内で、数分でタンパク質分解酵素により急速にタンパク質分解される。注射の場合、ペプチドの投与は痛みを伴い、多くの場合、慢性症状を治療するために頻繁で金のかかる来院が必要とされる。ペプチドのプロドラッグが皮膚に局所適用されると、それらは皮膚の水分中に直ちに溶解するだろう。これらのプロドラッグのアミノ基上にある正電荷は、皮膚膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は細胞質ゾルの中にプロドラッグを押し込むだろう。
【0020】
構造1で表される化合物は、標準的なペプチド合成プロトコルにより調製され得る。構造4−Cで表されるシクロペプチド及び関連化合物の調製では、標準的なペプチド合成プロトコルによりペプチド鎖は合成され得、リシンの側鎖は2−、又は4−Pyoc保護基により保護され、ペプチドの環化は樹脂上で行われた。
【発明の効果】
【0021】
本発明におけるこれらのペプチド及び関連化合物のプロドラッグは、親油性部分及び親水性部分(生理学的pHにおいてプロトン化された形態で存在するアミン基)を有する。これらのプロドラッグの正に荷電したアミノ基は2つの優れた利点を有する。第一に、プロドラッグを水中に溶解させる。すなわち、これらの新規なプロドラッグが、液体、スプレー、ローション、軟膏、エマルジョン又はゲルのような投与形態で経皮投与されるとき、これらは直ちに、皮膚、眼、生殖器部、口、鼻、又は身体の他の部分における水分と混合するだろう。第二に、これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。プロドラッグの(親油性アルキル基で極性基を修飾することによる)親油性部分は、薬物が皮膚膜に入るのに役立つだろう。これらのプロドラッグが膜に入るとき、親水性部分は、半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中にプロドラッグを押し込むだろう。皮膚、眼、生殖器部、口、鼻、又は身体の他の部分での短い残留のため、プロドラッグは、皮膚、眼、生殖器部、口、鼻、又は身体の他の部分に痒み、ヒリヒリ感又は痛みを引き起こさないだろう。実験結果は、40%を超えるプロドラッグが、数分で変化して親ペプチドに戻ったということを示す。プロドラッグは、容易に血液脳関門に浸透しさえし得る。ペプチド及び関連化合物の経皮送達システムは、ペプチドホルモンを医薬的に使用できるようにし得る。これらのプロドラッグを経皮投与することのもう一つの大きな利点は、特に子供への、薬物投与がいっそう容易になるだろうということである。
【図面の簡単な説明】
【0022】
【図1】フランツ細胞(n=5)での、単離されたヒト皮膚組織を横断する、Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HC1、HC1.(CH3)2NCH2CH2CH2CO-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2CH2CH3、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg(Ac)-Trp-Lys-NH2.HCl、シクロ(1,6)-Ac-Nle-Asp-His-D-Ala(2-ナフチル)-Arg-Trp-Lys-NH2.HCl、Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HCl、Ac-Tyr-Gly-Gly-Phe-Met-OH、シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg-Trp-Lys-OH、シクロ(1,6)-Ac-Nle-Asp-His-D-Phe(4-I)-Arg-Trp-Lys-NH2、及びH-Val-Pro-Gly-Pro-Arg-OHの累積量。それぞれの場合において、媒体はpH7.4ホスファート緩衝液(0.2M)であった。
【図2】構造1。式中、XはO、S、又はNHを表し、X1又はXnはCO、SO、SO2、PO(OR)、NO、又は何もないことを表し、Zn又はZn1はH、CH3、C2H5、C3H7、CF3、C2F5、又はC3F7を表し、Rnはアミノ酸の脂肪族側鎖、アミノ酸のヒドロキシル−若しくは硫黄含有側鎖、アミノ酸の芳香族側鎖、アミノ酸のアミノ、イミダゾリル、若しくはクアニジノ基含有側鎖、又はアミノ酸のカルボキシル若しくはカルボキサミド基含有側鎖のいずれか一つ、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、Yx1、Yx2、又はYnはH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、Rx3Rx4Rx5N+A-又は何もないことを表し、Rx1、Rx2、Rx3、Rx4、Rx5、又はRxnはH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよい。いずれのCH2基はO、S又はNHと置換されてもよい。配列中の1種以上のアミノ酸残基は非天然アミノ酸、例えば、β−アミノ酸、2−ナフチルアラニン、及びアルキル、アルキルオキシ、アルケニル若しくはアルキニルのいずれか一つ、アリール又はヘテロアリール残基により置換され得る。ペプチド鎖上のアミノ酸のアミノ及びカルボキシル官能基は、ラクタムブリッジを形成しホモデティックな環状ペプチドを形成してもよい。システイン、ホメシステイン、又は他のアミノ酸のチオール基は、ジスルフィドブリッジを形成しヘテロデティックな環状ペプチドを形成してもよい。
【発明を実施するための形態】
【0023】
(最良の形態)
Ac-Val-Pro-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
1.H-Arg(diAc)-OCH2CH2N(CH2CH3)2の調製
Z-Arg-OH30.8gをアセトン500ml中に溶解した。20%NaOH200mlを反応混合物に加えた。無水酢酸40gを反応混合物に一滴ずつ加えた。混合物を室温で2時間撹拌した。溶媒を蒸発させた。残留物を酢酸エチル500mlで抽出した。酢酸エチル溶液を水(3×100ml)で洗浄した。酢酸エチル層を硫酸ナトリウム上で乾燥させた。酢酸エチル溶液を蒸発させて乾燥した。残留物(Z-Arg(diAc)-OH、30g)をアセトニトリル300ml中に溶解した。混合物を氷水浴で0℃にまで冷却した。N,N−ジエチルアミノエタノール12g、4−ジメチルアミノピリジン2g、及び1,3−ジシクロヘキシルカルボジイミド22gを反応混合物に加えた。反応混合物を0℃で1時間、及び室温で一晩中撹拌する。固形物を濾過により除去し、溶液を蒸発させて乾燥した。残留物を酢酸エチル(2×250ml)で抽出した。酢酸エチル溶液を5%重炭酸ナトリウム(1×500ml)及び水(3×100ml)で洗浄した。酢酸エチル溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥する。残留物[Z-Arg(diAc)-OCH2CH2N(CH2CH3)2、28g]をメタノール300ml中に溶解した。10%Pd/C2gを溶液に加えた。混合物を水素下、室温で10時間撹拌した。Pd/Cを濾過により除去した。溶液を蒸発させて乾燥した。H-Arg(diAc)-OCH2CH2N(CH2CH3)222gを得た。
【0024】
2.Boc-Asp(OEt)-Pro-OSuの調製
L-プロリン15gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びBoc-Asp(OEt)-OSu36gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Boc-Asp(OEt)-Pro-OH)25g及びN−ヒドロキシスクシンイミド11gをジクロロメチレン(dichloromethlene)300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド16gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Boc-Asp(OEt)-Pro-OSu28gを得た。
【0025】
3.H-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFAの調製
H-Arg(diAc)-OCH2CH2N(CH2CH3)222gを5%NaHCO3300ml中に溶解した。アセトン150ml中のBoc-Asp(OEt)-Pro-OSu24gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。酢酸エチル溶液を水(3×100ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸250mlを混合物に加え、混合物を30分間撹拌した。混合物を蒸発させて乾燥した。H-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA32gを得た。
【0026】
4.Ac-Val-Pro-OSuの調製
L-プロリン15gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-VaI-OSu26gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Ac-Val-Pro-OH)20g及びN−ヒドロキシスクシンイミド11gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド16gを反応混合物に加えた。混合物を0℃で1時間、及び室温で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Ac-Val-Pro-OSu20gを得た。
【0027】
5.Ac-Val-Pro-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
H-Asp(OEt)-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA31gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Val-Pro-OSu15gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。有機層を水(3×100ml)で洗浄する。酢酸エチル層を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。ジオキサン(50ml)中HClガス3.5gを溶液に加える。固形物を集め、エーテル(3×50ml)で洗浄する。乾燥後、所望の吸湿性生成物20gを得た。水中での溶解性:150mg/ml、元素分析:C39H66ClN9O11、分子量:872.45、計算% C: 53.69; H: 7.62; Cl: 4.06; N: 14.45; O: 20.17、実測% C: 53.61; H: 7.67; Cl: 4.10; N: 14.40, O: 20.22、MS: m/e: 836.4; m/e+1: 836.4。
【0028】
(発明の形態)
Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HClの調製
1.H-Met-OCH2CH2N(CH2CH3)2.TFAの調製
Boc-Met-OH25gをジクロロメチレン300ml中に溶解した。混合物を氷水浴で0℃にまで冷却した。N,N−ジエチルアミノエタノール12g、4−ジメチルアミノピリジン2g、及び1,3−ジシクロヘキシルカルボジイミド22gを反応混合物に加えた。反応混合物を0℃で1時間、及び室温で一晩中撹拌する。固形物を濾過により除去し、ジクロロメチレン溶液を5%重炭酸ナトリウム(1×500ml)及び水(3×100ml)で洗浄した。酢酸エチル溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥する。残留物[Boc-Met-OCH2CH2N(CH2CH3)2、30g]をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸250mlを混合物に加え、混合物を30分間撹拌する。溶液を蒸発させて乾燥した。H-Met-OCH2CH2N(CH2CH3)2.TFA26gを得た。
【0029】
2.Boc-Gly-Phe-OSuの調製
L-フェニルアラニン20gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びBoc-Gly-OSu28gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Boc-Gly-Phe-OH)22g及びN−ヒドロキシスクシンイミド10gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド15gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄する。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Boc-Gly-Phe-OSu25gを得た。
【0030】
3.H-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.TFAの調製
H-Met-OCH2CH2N(CH2CH3)2.TFA25gを5%NaHCO3300ml中に溶解した。アセトン150ml中のBoc-Gly-Phe-OSu22gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。酢酸エチル溶液を水(3×100ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸200mlを混合物に加え、混合物を30分間撹拌した。混合物を蒸発させて乾燥した。H-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.TFA25gを得た。
【0031】
4.Ac-Tyr(Ac)-Gly-OSuの調製
L-グリシン11gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Tyr(Ac)-OSu36gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Ac-Tyr(Ac)-Gly-OH)28g及びN−ヒドロキシスクシンイミド13gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド18gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄する。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Ac-Tyr(Ac)-Gly-OSu20gを得た。
【0032】
5.Ac-Tyr(Ac)-Gly-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.HClの調製
H-Gly-Phe-Met-OCH2CH2N(CH2CH3)2.TFA24gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Tyr(Ac)-Gly-OSu15gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。有機層を水(3×100ml)で洗浄する。酢酸エチル層を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。ジオキサン(50ml)中HClガス3.5gを溶液に加える。固形物を集め、エーテル(3×50ml)で洗浄する。乾燥後、所望の吸湿性生成物18gを得た。水中での溶解性:200mg/ml、元素分析:C37H53ClN6O9S、分子量:793.37、計算% C: 56.01; H: 6.73; Cl: 4.47; N: 10.59; O: 18.15; S: 4.04、実測% C: 55.96; H: 6.76; Cl: 4.52; N: 10.54, O: 18.19; S: 4.03、MS: m/e: 757.4; m/e+1: 758.4。
【0033】
Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
1.Boc-Gly-Pro-OSuの調製
L-プロリン15gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びBoc-Gly-OSu27.2gを反応混合物に加えた。混合物を室温で5時間撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。残留物(Boc-Gly-Pro-OH)21g及びN−ヒドロキシスクシンイミド11gをジクロロメチレン300ml中に溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド17gを反応混合物に加えた。混合物を0℃で1時間撹拌した。固形物を濾過により除去した。ジクロロメチレン溶液を5%重炭酸ナトリウム(1×200ml)及び水(3×200ml)で洗浄する。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Boc-Gly-Pro-OSu23gを得た。
【0034】
2.H-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFAの調製
H-Arg(diAc)-OCH2CH2N(CH2CH3)222gを5%NaHCO3300ml中に溶解した。アセトン150ml中のBoc-Gly-Pro-OSu20gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。酢酸エチル溶液を水(3×100ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。溶液を蒸発させて乾燥した。残留物をジクロロメチレン250ml中に溶解した。トリフルオロ酢酸250mlを混合物に加え、混合物を30分間撹拌した。混合物を蒸発させて乾燥した。H-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA28gを得た。
【0035】
3.Ac-Val-Pro-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.HClの調製
H-Gly-Pro-Arg(diAc)-OCH2CH2N(CH2CH3)2.2TFA26gを10%重炭酸ナトリウム300ml中に溶解した。アセトン150ml及びAc-Val-Pro-OSu15gを反応混合物に加えた。混合物を室温で5時間撹拌した。酢酸エチル500mlを混合物に加えた。有機層を水(3×100ml)で洗浄する。酢酸エチル層を硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去する。ジオキサン(50ml)中HClガス3.5gを溶液に加える。固形物を集め、エーテル(3×50ml)で洗浄した。乾燥後、所望の吸湿性生成物18gを得た。水中での溶解性:150mg/ml、元素分析:C35H60ClN9O9、分子量:786.36、計算% C: 53.46; H: 7.69; Cl: 4.51; N: 16.03; O: 18.31、実測% C: 53.43; H: 7.73; Cl: 4.55; N: 16.01, O: 18.29、MS: m/e: 750.4; m/e+1: 751.4。
【0036】
シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HClの調製
1.Ac-Nle-Asp(OFm)-OHの調製
H-Asp(OFm)-OH.TFA43g及びAc-Nle-OSu27gをアセトン300ml中に懸濁した。5%NaHCO3300mlを反応混合物に加えた。混合物を室温で一晩中撹拌した。混合物をエーテル(1×300ml)で洗浄した。酢酸エチル500mlを水層に加えた。混合物のpHを氷冷した3NHClで2.4〜2.5に調整した。酢酸エチル層を集め、水(3×300ml)で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。溶液を蒸発させて乾燥した。Ac-Nle-Asp(OFm)-OH42gを得た。
【0037】
2.Fmoc-Trp-Lys(4-Pyoc)-OHの調製
H-Lys(4-Pyoc)-OHを参考文献(H. Kunz及びS. Birnbach, Tetrahedron Lett., 25, 3567, 1984; H. Kunz及びR. Barthels, Angew. Chem., Int. Ed. Engl., 22, 783, 1983)に従って調製した。H-Lys(4-Pyoc)-OH33gを5%NaHCO3300ml中に懸濁した。アセトン300ml及びFmoc-Trp-OSu52gを反応混合物に加えた。混合物を室温で一晩中撹拌した。混合物をエーテル(1×500ml)で洗浄した。酢酸エチル500mlを混合物に加え、混合物のpHを氷冷した3NHClで2.2〜2.3に調整する。酢酸エチル層を集め、水で洗浄した。有機溶液を硫酸ナトリウム上で乾燥させた。有機溶液を蒸発させて乾燥した。Fmoc-Trp-Lys(4-Pyoc)-OH55gを得た。
【0038】
3.シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OHの調製
Wang樹脂100gをDMF700 ml中に懸濁した。Fmoc-Trp-Lys(4-Pyoc)-OH50g、1−ヒドロキシベンゾトリアゾール13g、4−ジメチルアミノピリジン2g、及びN,N’−ジイソプロピルカルボジイミド12g。混合物を室温で一晩中撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Fmoc-Arg(diAc)-OH48g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Fmoc-Phe-OH39g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Fmoc-His(Fmoc)-OH60g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。20%ピペリジン700 mlを樹脂に加えた。混合物を30分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、Ac-Nle-Asp(OFm)-OH60g、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で2時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。ペプチドの(peptided)樹脂をDMF700 ml中に懸濁した。MeI50gを反応混合物に加えた。混合物を室温で1時間、及び50℃で1時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。30%ピペリジン700 mlを樹脂に加えた。混合物を60分間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。DMF700 ml、1−ヒドロキシベンゾトリアゾール13g、トリエチルアミン35 ml、及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウム38gを樹脂に加えた。混合物を室温で10時間撹拌した。樹脂を濾過により集め、DMF(3×400 ml)、メタノール(3×400 ml)、及びジクロロメチレン(3×400 ml)で洗浄した。トリフルオロ酢酸500mlを樹脂に加え、混合物を室温で1時間撹拌した。樹脂を濾過により除去し、溶液を蒸発させて乾燥する。残留物をエーテル(3×100 ml)で洗浄した。
【0039】
4.シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OCH2CH2N(CH2CH3)2.HClの調製
シクロ(1,6)-Ac-Nle-Asp-His-Phe-Arg(diAc)-Trp-Lys-OH10gをDMF300ml中に溶解した。混合物を氷水浴で0℃にまで冷却した。N,N−ジエチルアミノエタノール12g、4−ジメチルアミノピリジン2g、及び1,3−ジシクロヘキシルカルボジイミド22gを反応混合物に加えた。反応混合物を0℃で1時間、及び室温で一晩中撹拌する。固形物を濾過により除去し、ジクロロメチレン溶液を5%重炭酸ナトリウム(1×500ml)及び水(3×100ml)で洗浄した。酢酸エチル溶液を硫酸ナトリウム上で乾燥させた。ジオキサン(20ml)中2gのHClを溶液に加えた。固形物を集め、エーテル(3×30ml)で洗浄した。生成物8gを得た。
【産業上の利用可能性】
【0040】
構造1で表されるプロドラッグは、容易に皮膚膜に浸透し得る。ペプチド及び関連化合物の経皮送達システムは、人又は動物における多くの疾患、例えば、関節リウマチ及び変形性関節症に由来する疼痛、熱、男性の勃起不全、女性の性機能障害、全身血圧、降圧コントロール(hypotensive control)、血小板凝集の抑制、肺疾患、消化器疾患、炎症、ショック、生殖能(reproduction)、受精能(fertility)などの治療において、ペプチドホルモンを有用にし得る。
【特許請求の範囲】
【請求項1】
次式の構造1で表される化合物であって、
【化1】
式中、XはO、S、又はNHを表し、X1又はXnはCO、SO、SO2、PO(OR)、NO、又は何もないことを表し、Zn又はZn1はH、CH3、C2H5、C3H7、CF3、C2F5、又はC3F7を表し、Rnはアミノ酸の脂肪族側鎖、アミノ酸のヒドロキシル−若しくは硫黄含有側鎖、アミノ酸の芳香族側鎖、アミノ酸のアミノ、イミダゾリル、若しくはクアニジノ基含有側鎖、又はアミノ酸のカルボキシル若しくはカルボキサミド基含有側鎖のいずれか一つ、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、Yx1、Yx2、又はYnはH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は以下の基、
【化2】
又は何もないことを表し、Rx1、Rx2、Rx3、Rx4、Rx5、又はRxnはH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよく、アミノ酸はL−型でもD−型でもよく、配列中の1種以上のアミノ酸残基は非天然アミノ酸、例えば、β−アミノ酸、2−ナフチルアラニン、及びアルキル、アルキルオキシ、アルケニル若しくはアルキニルのいずれか一つ、アリール又はヘテロアリール残基により置換されてもよく、ペプチド鎖上のアミノ酸のアミノ及びカルボキシル官能基は、ラクタムブリッジを形成しホモデティックな環状ペプチドを形成してもよく、システイン、ホメシステイン、又は他のアミノ酸のチオール基は、ジスルフィドブリッジを形成しヘテロデティックな環状ペプチドを形成してもよい。
【請求項2】
ペプチド及び関連化合物の経皮送達システムのデザインであって、ペプチド又は関連化合物のこれらのプロドラッグをデザインするための原則は、1.プロドラッグは、親油性部分及び生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有していなければならず、2.ペプチドの全てプロドラッグは1つ又は2つだけ(好ましくは1つ)の、生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有するべきである。3.第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基は、N末端、C末端、又はペプチドの側鎖であり得、N末端又はC末端部分が好ましく、4.ペプチドを親油性にするために、カルボキシル基、アミノ基、グアニジン基又は他の親水性基は、アルキル、アリール、若しくはヘテロアリールエステル又はアミド基で保護され得る。
【請求項3】
次式の構造2−Cで表される化合物、及び歯痛、頭痛、関節炎、他の炎症性、熱、癌、月経困難症、及び急性片頭痛に由来する疼痛を治療するためのそれらの使用。
【化3】
式中、Rは分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10…、アリール又はヘテロアリール残基を表し、XはCO、SO、SO2、又は何もないことを表し、X1はCH3SCH2CH2-、CH3SOCH2CH2-、又は(CH3)2CHCH2-を表し、X2はH、CH3又はCF3を表し、X3はH、CH3、C2H5、又はCF3を表し、R1、R2、R3、R4、又はR5はH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、アミノ酸はL−型又はD−型であり得、全てのR、Rn、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項4】
次式の構造3−Cで表される化合物、及び歯痛、頭痛、関節炎、他の炎症性、熱、癌、月経困難症、及び急性片頭痛に由来する疼痛を治療するためのそれらの使用。
【化4】
式中、R1又はR2はH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、R3はH又はCH3を表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、アミノ酸はL−型又はD−型であり得、全てのR、Rn、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項5】
次式の構造4−Cで表される化合物、及び男性の勃起不全、女性の性機能障害、白皮症、及び他の皮膚病を治療するためのそれらの使用。
【化5】
式中、Rは分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10…、アリール又はヘテロアリール残基を表し、X4、X5、X6、X7、X8、又はX9はCO、SO2、PO(OR)、NO、又は何もないことを表し、R1、R2、R3、R4、R5、R6、R7、R8、又はR9はH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、Arはフェニル、2’−ナフチル、4−ヨードフェニル、又は他のアリール若しくはヘテロアリール残基を表し、アミノ酸はL−型又はD−型であり得、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項6】
次式の構造5−Cで表される化合物、及び肥満を治療するためのそれらの使用。
【化6】
式中、Rは分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10…、アリール又はヘテロアリール残基、又は何もないことを表し、X1はH又はR1OCOCH2−を表し、X2はH又は(CH3)2CH−を表し、R1、R2、R3、R4、R5、又はR6はH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基を表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、全てのR、Rn、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項7】
請求項5に記載の構造4−Cで表されるシクロペプチド及び関連化合物を調製する方法であって、ペプチド鎖が標準的なペプチド合成プロトコルにより合成され得、リシンの側鎖が2−,又は4−Pyoc保護基により保護され、ペプチドの環化が樹脂上で行われる方法。
【請求項8】
請求項1に記載の、構造1で表される化合物、又は少なくとも構造1で表される化合物を活性成分として含む組成物であって、人又は動物において、ペプチド及び関連化合物により治療可能な状態を治療するために、経口投与又は経皮投与可能である化合物又は組成物であって、前記状態が、緑内障又は高眼圧症、男性の勃起不全及び女性の性機能障害、全身血圧、堕胎、降圧コントロール、血小板凝集の抑制、肺疾患、消化器疾患、炎症、ショック、生殖能、受精能、肥満などを含むが、これに限定されるものではない化合物又は組成物。
【請求項9】
人又は動物において、ペプチド及び関連化合物治療可能な状態を治療する方法であって、身体の一部に(溶液、スプレー、ローション、軟膏、エマルジョン又はゲルの形態で)経皮投与することにより、請求項1に記載の、構造1で表される化合物、又は少なくとも構造1で表される化合物を活性成分として含む組成物の治療有効血漿レベルを送達することを特徴とする方法。
【請求項10】
人又は動物において、ペプチド及び関連化合物治療可能な状態を治療するための、請求項1に記載の、構造1で表される化合物、又は少なくとも構造1で表される化合物を活性成分として含む組成物の経皮治療応用システムであって、活性物質含有マトリックス層及び非透過性支持層からなる包帯又はパッチであり得、最も好ましくは、活性物質貯蔵庫であり、これが皮膚に面する透過性底面を有し、放出の割合を制御することによって、ペプチド及び関連化合物が絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、ペプチド及び関連化合物の副作用を減少させることを可能にすることを特徴とするシステム。
【請求項1】
次式の構造1で表される化合物であって、
【化1】
式中、XはO、S、又はNHを表し、X1又はXnはCO、SO、SO2、PO(OR)、NO、又は何もないことを表し、Zn又はZn1はH、CH3、C2H5、C3H7、CF3、C2F5、又はC3F7を表し、Rnはアミノ酸の脂肪族側鎖、アミノ酸のヒドロキシル−若しくは硫黄含有側鎖、アミノ酸の芳香族側鎖、アミノ酸のアミノ、イミダゾリル、若しくはクアニジノ基含有側鎖、又はアミノ酸のカルボキシル若しくはカルボキサミド基含有側鎖のいずれか一つ、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、Yx1、Yx2、又はYnはH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は以下の基、
【化2】
又は何もないことを表し、Rx1、Rx2、Rx3、Rx4、Rx5、又はRxnはH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよく、アミノ酸はL−型でもD−型でもよく、配列中の1種以上のアミノ酸残基は非天然アミノ酸、例えば、β−アミノ酸、2−ナフチルアラニン、及びアルキル、アルキルオキシ、アルケニル若しくはアルキニルのいずれか一つ、アリール又はヘテロアリール残基により置換されてもよく、ペプチド鎖上のアミノ酸のアミノ及びカルボキシル官能基は、ラクタムブリッジを形成しホモデティックな環状ペプチドを形成してもよく、システイン、ホメシステイン、又は他のアミノ酸のチオール基は、ジスルフィドブリッジを形成しヘテロデティックな環状ペプチドを形成してもよい。
【請求項2】
ペプチド及び関連化合物の経皮送達システムのデザインであって、ペプチド又は関連化合物のこれらのプロドラッグをデザインするための原則は、1.プロドラッグは、親油性部分及び生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有していなければならず、2.ペプチドの全てプロドラッグは1つ又は2つだけ(好ましくは1つ)の、生理学的pHにおいてプロトン化された形態で存在する第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基(親水性部分)を有するべきである。3.第1級、第2級若しくは第3級アミン基、クアニジノ基、又は1つが保護されたクアニジノ基は、N末端、C末端、又はペプチドの側鎖であり得、N末端又はC末端部分が好ましく、4.ペプチドを親油性にするために、カルボキシル基、アミノ基、グアニジン基又は他の親水性基は、アルキル、アリール、若しくはヘテロアリールエステル又はアミド基で保護され得る。
【請求項3】
次式の構造2−Cで表される化合物、及び歯痛、頭痛、関節炎、他の炎症性、熱、癌、月経困難症、及び急性片頭痛に由来する疼痛を治療するためのそれらの使用。
【化3】
式中、Rは分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10…、アリール又はヘテロアリール残基を表し、XはCO、SO、SO2、又は何もないことを表し、X1はCH3SCH2CH2-、CH3SOCH2CH2-、又は(CH3)2CHCH2-を表し、X2はH、CH3又はCF3を表し、X3はH、CH3、C2H5、又はCF3を表し、R1、R2、R3、R4、又はR5はH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、アミノ酸はL−型又はD−型であり得、全てのR、Rn、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項4】
次式の構造3−Cで表される化合物、及び歯痛、頭痛、関節炎、他の炎症性、熱、癌、月経困難症、及び急性片頭痛に由来する疼痛を治療するためのそれらの使用。
【化4】
式中、R1又はR2はH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、R3はH又はCH3を表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、アミノ酸はL−型又はD−型であり得、全てのR、Rn、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項5】
次式の構造4−Cで表される化合物、及び男性の勃起不全、女性の性機能障害、白皮症、及び他の皮膚病を治療するためのそれらの使用。
【化5】
式中、Rは分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10…、アリール又はヘテロアリール残基を表し、X4、X5、X6、X7、X8、又はX9はCO、SO2、PO(OR)、NO、又は何もないことを表し、R1、R2、R3、R4、R5、R6、R7、R8、又はR9はH、O、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基、又は何もないことを表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、Arはフェニル、2’−ナフチル、4−ヨードフェニル、又は他のアリール若しくはヘテロアリール残基を表し、アミノ酸はL−型又はD−型であり得、全てのR、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項6】
次式の構造5−Cで表される化合物、及び肥満を治療するためのそれらの使用。
【化6】
式中、Rは分枝鎖若しくは直鎖、-(CH2)n-であってn=0、1、2、3、4、5、6、7、8、9、10…、アリール又はヘテロアリール残基、又は何もないことを表し、X1はH又はR1OCOCH2−を表し、X2はH又は(CH3)2CH−を表し、R1、R2、R3、R4、R5、又はR6はH、1〜12炭素原子を有するアルキル、アルキルオキシ、アルケニル若しくはアルキニル残基のいずれか一つ、アリール又はヘテロアリール残基を表し、A-はCl-、Br-、F-、I-、AcO-、シトラート、又はいずれの陰イオンを表し、n=0、1、2、3、4、5、6、7、8、9、10…であり、全てのR、Rn、-(CH2)n-又は-(CH2)m-基は分枝鎖若しくは直鎖であり、C、H、O、S、又はN原子を含んでもよく、単結合、二重結合、及び三重結合を有してもよく、CH2基はO、S又はNHと置換されてもよい。
【請求項7】
請求項5に記載の構造4−Cで表されるシクロペプチド及び関連化合物を調製する方法であって、ペプチド鎖が標準的なペプチド合成プロトコルにより合成され得、リシンの側鎖が2−,又は4−Pyoc保護基により保護され、ペプチドの環化が樹脂上で行われる方法。
【請求項8】
請求項1に記載の、構造1で表される化合物、又は少なくとも構造1で表される化合物を活性成分として含む組成物であって、人又は動物において、ペプチド及び関連化合物により治療可能な状態を治療するために、経口投与又は経皮投与可能である化合物又は組成物であって、前記状態が、緑内障又は高眼圧症、男性の勃起不全及び女性の性機能障害、全身血圧、堕胎、降圧コントロール、血小板凝集の抑制、肺疾患、消化器疾患、炎症、ショック、生殖能、受精能、肥満などを含むが、これに限定されるものではない化合物又は組成物。
【請求項9】
人又は動物において、ペプチド及び関連化合物治療可能な状態を治療する方法であって、身体の一部に(溶液、スプレー、ローション、軟膏、エマルジョン又はゲルの形態で)経皮投与することにより、請求項1に記載の、構造1で表される化合物、又は少なくとも構造1で表される化合物を活性成分として含む組成物の治療有効血漿レベルを送達することを特徴とする方法。
【請求項10】
人又は動物において、ペプチド及び関連化合物治療可能な状態を治療するための、請求項1に記載の、構造1で表される化合物、又は少なくとも構造1で表される化合物を活性成分として含む組成物の経皮治療応用システムであって、活性物質含有マトリックス層及び非透過性支持層からなる包帯又はパッチであり得、最も好ましくは、活性物質貯蔵庫であり、これが皮膚に面する透過性底面を有し、放出の割合を制御することによって、ペプチド及び関連化合物が絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、ペプチド及び関連化合物の副作用を減少させることを可能にすることを特徴とするシステム。
【図1】

【図2】
【図2】
【公表番号】特表2010−509308(P2010−509308A)
【公表日】平成22年3月25日(2010.3.25)
【国際特許分類】
【出願番号】特願2009−535822(P2009−535822)
【出願日】平成18年11月8日(2006.11.8)
【国際出願番号】PCT/IB2006/054170
【国際公開番号】WO2008/056207
【国際公開日】平成20年5月15日(2008.5.15)
【出願人】(509023539)
【Fターム(参考)】
【公表日】平成22年3月25日(2010.3.25)
【国際特許分類】
【出願日】平成18年11月8日(2006.11.8)
【国際出願番号】PCT/IB2006/054170
【国際公開番号】WO2008/056207
【国際公開日】平成20年5月15日(2008.5.15)
【出願人】(509023539)
【Fターム(参考)】
[ Back to top ]