
ペミロラストの新しい結晶形
ペミロラストナトリウム塩半水和物形を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬物の新しい固体状態の形態、それらを含有する医薬組成物、およびそれらを得る方法に関する。
【背景技術】
【0002】
薬物組成物の製剤化において、原薬は、好都合に取扱いおよび加工を行うことができる形態であることが重要である。これは、商業的に実現可能な製造方法を得るという点からだけでなく、活性化合物を含む医薬製剤のその後の製造という点から見ても重要である。
【0003】
活性成分の化学的安定性、固体状態安定性、および「貯蔵寿命」も極めて重要な因子である。原薬、およびそれを含有する組成物は、活性成分の物理化学的特性(例えば、その化学組成、密度、吸湿性および溶解性)に大きな変化を示すことなく、かなりの期間にわたって有効に貯蔵することができるべきである。
【0004】
さらに、化学的にできるだけ純粋な形態の薬物を提供することも重要である。
【0005】
この点で、非晶質または半非晶質材料は重大な問題を提起する恐れがある。例えば、このような材料は、典型的には取扱いおよび製剤化が困難であり、信頼性のない溶解性をもたらし、不安定でありかつ化学的に不純であることがしばしば認められている。
【0006】
上記の問題は、薬物を安定した結晶形で容易に得ることができる場合に解決可能であることを当業者は理解するであろう。
【0007】
さらに、結晶性薬物化合物は、患者に投与された後、信頼性および再現性のより高い血漿中濃度プロファイルを実現することがわかっている。
【0008】
したがって、商業的に実現可能でかつ薬学的に許容される薬物組成物の製造において、可能な場合はいつでも、実質的に結晶性でかつ安定な形態の薬物を実現することが重要である。
【0009】
しかし、この目的は、必ずしも実現可能ではないことに留意されたい。実際、典型的には、化合物の結晶化挙動がどうなるかを分子構造だけから予想することは可能でない。これは、通常実験に基づいてしか決定することができない。
【0010】
ペミロラストは、喘息、アレルギー性鼻炎、および結膜炎などの病態の治療で使用される、経口投与で活性な抗アレルギー薬である。例えば、米国特許第4,122,274号、欧州特許出願公開第316 174号および同第1 285 921号、Yanagiharaら、Japanese Journal of Pharmacology、51、93(1989年)およびDrugs of Today、28、29(1992年)を参照のこと。この薬物は、例えば日本で、ALEGYSAL(商標)という商標でカリウム塩として現在市販されている。
【0011】
市販のペミロラストカリウムは、ヒトにおいて鋭い血漿中濃度ピークを生じることが知られているという欠点がある(例えば、Kinbaraら、「Plasma Level and Urinary Excretion of TBX in Humans」、Japanese Pharmacology & Therapeutics、18(3)(1990年)、および「Antiallergic agent - ALEGYSAL tablet 5mg - ALEGYSAL tablet 10mg - ALEGYSAL dry syrup」、Pharmaceutical Interview Form(IF)、2007年10月改訂(第7版)、Standard Commodity Classification No.:87449を参照のこと)。後者の文献には、ペミロラストのカリウム塩は吸湿性であることも報告されており、これにより、化学的不安定性がもたらされ、苦味をもつと考えられる。
【0012】
米国特許第4,122,274号には、カリウム塩および(実施例14における)ナトリウム塩を含む、ペミロラストの塩の生成方法が記載されている。本明細書に記載されているように、この技法により、物理的に不安定なナトリウム塩が生成される。ペミロラストのナトリウム塩は、国際公開第2008/074975号および同第2008/075028号にも記載されている(しかし、その合成法は記載されていない)。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】米国特許第4,122,274号
【特許文献2】欧州特許出願公開第316 174号
【特許文献3】欧州特許出願公開第1 285 921号
【特許文献4】国際公開第2008/074975号
【特許文献5】国際公開第2008/075028号
【非特許文献】
【0014】
【非特許文献1】Yanagiharaら、Japanese Journal of Pharmacology、51、93(1989年)
【非特許文献2】Yanagiharaら、Drugs of Today、28、29(1992年)
【非特許文献3】Kinbaraら、「Plasma Level and Urinary Excretion of TBX in Humans」、Japanese Pharmacology & Therapeutics、18(3)(1990年)、
【非特許文献4】Kinbaraら、「Antiallergic agent - ALEGYSAL tablet 5mg - ALEGYSAL tablet 10mg - ALEGYSAL dry syrup」、Pharmaceutical Interview Form(IF)、2007年10月改訂(第7版)、Standard Commodity Classification No.:87449
【非特許文献5】Marzら、Circulation、110(2004年)
【非特許文献6】Nicklasら、CMAJ、172、1199(2005年)
【非特許文献7】Ridkerら、N. Engl. J. Med., 352, 20(2005年)
【非特許文献8】RobbinsおよびCotran、Pathologic Basis of Disease、第8版、Saunders Elsevier
【非特許文献9】Remington The Science and Practice of Pharmacy、第19版、Mack Printing Company, Easton, Pennsylvania(1995年)
【非特許文献10】Martindale - The Complete Drug Reference(第35版)およびその中の引用文献
【非特許文献11】Sanoおよびlshihara、Heterocycles、48、775(1998年)
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明者らは、今回、ペミロラストの安定な結晶性ナトリウム塩を生成することが可能であること、また予想外なことに、このような塩が、対応する先行技術のペミロラストカリウム塩より、水性媒体に可溶でなく、かつ吸湿性でないことも見出した。したがって、このような塩は、ペミロラストカリウムについて上述した鋭い血漿中濃度ピークも、芳しくない味および吸湿性の問題も生じないものと予想される。
【課題を解決するための手段】
【0016】
本発明の第1の態様によれば、ペミロラストナトリウム塩の半水和物形(以下に、「本発明の化合物」と呼ばれる)が提供される。本発明の化合物は、別の場合には「ペミロラストナトリウム半水和物」と呼ばれることがある。
【0017】
ペミロラストナトリウム塩、ならびに/あるいはペミロラスト1モル当たり少なくとも約0.3モル、好ましくは少なくとも約0.35モル、より好ましくは少なくとも約0.4モル、および/または約0.7モル以下、好ましくは約0.65モル以下、より好ましくは約0.6モル以下の水(約0.5モルの水など)をこのような水が密接に結合していようと緩く結合していようと(すなわち、結晶水またはその他の形)含有する本発明の化合物もさらに提供される。
【0018】
本発明者らは、本発明の化合物を自然状態において実質的に結晶性である形態で容易に得ることができることを見出した。
【0019】
結晶化度が約98%を超える、例えば約95%を超える形態の本発明の化合物を生成することが可能であるが、「実質的に結晶性」について、本発明者らは、結晶化度が約60%を超える、好ましくは約75%を超える、より好ましくは約80%を超える(約90%など)形態を含める。結晶化度(%)は、粉末X線回折(PXRD)を用いて当業者が決定することができる。固体NMR法、FT-IR法、ラマン分光法、示差走査熱量測定法(DSC)、マイクロカロリメトリー法、および真密度の算出など、他の技法も使用することができる。
【0020】
好ましい本発明の化合物は、2θ値(単位:度)およそ(すなわち、約またはほぼ)26.6の特有の結晶性ピーク(図1/表2において、およそ26.5である)を含み、好ましくは2θ値(単位:度)およそ(すなわち、約またはほぼ)25.3の別の強い結晶性ピーク(図1/表2において、およそ25.2である)も含む粉末X線回折パターンで特徴付けることができる。より好ましくは、本発明の化合物は、2θ値(単位:度)およそ(すなわち、約またはほぼ)13.0(図1/表2において、およそ12.9)、15.3(図1/表2において、およそ15.2)、18.2(図1/表2において、およそ18.1)および/または28.4(図1/表2において、およそ28.3)の追加の強い結晶性ピークを含み; より好ましくは、2θ値(単位:度)およそ(すなわち、約またはほぼ)16.8(図1/表2において、およそ16.7)、19.2(図1/表2において、およそ19.1)、および/または27.0(図1/表2において、およそ26.9)の別の強い結晶性ピークを含み; より好ましくは、2θ値(単位:度)およそ(すなわち、約またはほぼ)14.9および/または29.5の別の強い結晶性ピークを含む、粉末X線回折パターンで特徴付けることができる。
【0021】
好ましい本発明の化合物は、本明細書に添付されている図5(および/または図1)に示され、かつ/または下記の表4(および/または表2)にまとめられているものに本質的に一致している粉末X線回折パターンで特徴付けることもできる。当業者は、ペミロラストナトリウム半水和物の形態が他の粉末X線回折パターンと「本質的に」同じ粉末X線回折パターンを示すことをそれぞれのパターン(すなわち、試料の好ましい配向や各機器の設定(例えば、装置のタイプ、標準化、および/または較正)などの実験誤差を考慮して、それぞれのピーク間隔)から同じ結晶形が形成されていること(それぞれ図5および表4で特徴付けられる形態と図1および表2で特徴付けられる形態との間ではよくあることである)がその当業者に明らかであったときに理解するであろう。したがって、本明細書で指定されているd値の実験誤差の限界は、最後の小数位で±2ぐらいの範囲とすることができる。
【0022】
本発明の化合物は、好ましくは実質的に結晶学的に純粋である。「実質的に結晶学的に純粋」によって、本発明者らは、PXRD測定によって判断できる範囲で、ペミロラストナトリウムの他の結晶形を約5%未満、より好ましくは約3%未満、特に約1%未満しか含有しないペミロラストナトリウム半水和物の結晶形を含める(半水和物の形それともその他の形かどうか、このような他の結晶形に由来するPXRDピークの存在によって判断される)。
【0023】
本発明者らは、本発明の化合物が、米国特許第4,122,274号に記載されているように調製されたものを含む、ペミロラストナトリウム塩の他の形態と比べると、驚くほど改善された物理的および/または化学的安定性を有することを見出した。
【0024】
本明細書に定義する「安定な」という用語は、化学的安定性および固体状態安定性を包含する。
【0025】
「化学的安定性」によって、本発明者らは、化合物が、単独の固体形、または薬学的に許容される担体、希釈剤、もしくは助剤と混合して提供され得る固体製剤の形で、標準貯蔵条件下で、化学的崩壊または分解をほとんど伴うことなく貯蔵できることを含める。
【0026】
「固体状態安定性」によって、本発明者らは、化合物が、単独の固体形、または薬学的に許容される担体、希釈剤、もしくは助剤と混合して提供され得る固体製剤の形で、標準貯蔵条件下で、固体状態変態(例えば、結晶化、再結晶、結晶性損失(loss of crystallinity)、固体状態相転移、水和、脱水、溶媒和、または脱溶媒和)をほとんど伴うことなく貯蔵できることを含める。
【0027】
「標準貯蔵条件」の例としては、長期間(すなわち、6か月以上)にわたる、温度-80〜+50℃(好ましくは0〜40℃、より好ましくは15〜30℃などの周囲温度)、圧力0.1〜2バール(好ましくは大気圧)、および/または460ルクスのUV/可視光への曝露が挙げられる。このような条件下では、本発明の化合物は、適宜に化学的崩壊/分解または固体状態変態を約15%未満、より好ましくは約10%未満、特に約5%未満しか生じないことがわかり得る。温度および圧力の上記の上限および下限は、標準貯蔵条件の両端点を表すものであり、これら端点のある種の組合せ(例えば、温度50℃と圧力0.1バール)は通常の貯蔵時にはないことを当業者は理解するであろう。
【0028】
「標準貯蔵条件」という用語は、相対湿度5〜95%(好ましくは10〜60%)も包含することができる。しかし、本発明によるいくつかの結晶形の場合、水和および/または脱水による立体配座または結晶構造の変化が、標準温度/圧力における相対湿度の特定の端点への長期曝露の結果として生じる可能性がある。
【0029】
本発明者らは、有利にはペミロラストナトリウム塩の少なくとも部分溶解および/または懸濁に続いて結晶化を経て、本発明の化合物を得ることが可能であることを見出した。
【0030】
この点に関して、有利にはペミロラストとナトリウム含有塩基を反応させ、続いて適切な溶媒系から結晶化を行うことによって、本発明の化合物を得ることができる。前記結晶化に先立って、ペミロラストナトリウムを単離することができる。
【0031】
ナトリウム含有塩基は、ペミロラスト分子のテトラゾール部分からプロトンを除去するのに十分な塩基性である必要がある。したがって、好ましい塩基としては、水酸化ナトリウム、ならびに水素化ナトリウム、ナトリウムアミドおよびナトリウムアルコキシドが挙げられる。記載され得るナトリウムアルコキシドとしては、ナトリウムのC1〜6アルコキシド、具体的にはC1〜4アルコキシド、例えばC1〜3アルコキシド、例えばナトリウムエトキシドまたは例えばナトリウムメトキシドが挙げられる。
【0032】
本発明の化合物の適切な溶媒系からの結晶化は、ペミロラストナトリウム塩を含む溶媒系において過飽和を(例えば、冷却、溶媒蒸発、および/または好適な貧溶媒の添加により)達成することによって実現することができる。結晶化は、ナトリウム塩を加えて、溶液のイオン強度を高めること(NaClまたはNaSO4など; いわゆる「塩析」)により物質の溶解性を低下させ、かつ/または種結晶を過飽和溶液に加えること(一度利用可能)によって行うこともできる。
【0033】
溶媒系としては、酢酸アルキル(例えば、酢酸エチル、酢酸iso-プロピル、酢酸ブチルなどの酢酸直鎖状または分枝状C1〜6アルキル)、低級(例えば、直鎖状または分枝状C1〜6、好ましくはC1〜4)アルキルアルコール(例えば、メタノール、エタノール、iso-プロパノール)、脂肪族および芳香族炭化水素(例えば、iso-オクタン、n-ヘプタン、およびトルエン)、ジアルキルケトン(例えば、メチルエチルケトン、アセトン、およびメチルiso-ブチルケトン)、ジアルキルエーテル(例えば、ジ-iso-プロピルエーテルおよびtert-ブチルメチルエーテル)、環状エーテル(例えば、テトラヒドロフランおよびジオキサン)、アセトニトリル、ジメチルホルムアミドなどの1つまたは複数の有機溶媒を挙げることができる。上記溶媒のうちのいずれかの混合物を使用することができる。有機溶媒は水を含有していてもよい。
【0034】
結晶形が異なれば、所与の温度での有機溶媒における溶解性が異なることがある。この点に関して、上記または他の溶媒を「貧溶媒」(すなわち、メタノール、エタノール、またはイソプロパノールなど、本発明の化合物が難溶性であるが、本発明の化合物がより可溶性である別の溶媒(水など)と混和可能である溶媒)として使用することができ、したがって結晶化プロセスを助けることができる。
【0035】
特に好適な溶媒としては、少量の水と混合することができる低級アルキルアルコール(例えば、イソプロパノールなどのC1〜4アルコール、好ましくはメタノール、またはより好ましくはエタノール)が挙げられる。
【0036】
特に、本発明者らは、有機溶媒(例えば、エタノールなどの低級アルキルアルコール)の存在下、さらに約12%以下(w/w、有機溶媒の割合として)、特に約11%以下、特に約10%以下(例えば、約8%未満、具体的には約5%未満、例えば約3%未満)の水の存在下、ペミロラストナトリウムの部分溶解(平衡および/またはスラリー形成とも呼ばれる)に続いて結晶化を経由して、本発明の化合物を得ることができることを見出した。このような水は、結晶化混合物に加えてもよく、あるいは有機溶媒または結晶化させる対象のペミロラストナトリウムのうちの一方または両方にすでに存在してもよい。この結晶化は、半水和物を形成するためのいくらかの水を系中に存在させる必要があるので、完全に無水の条件下で行うことはできないことを当業者は理解するであろう。部分溶解は、(例えば、飽和溶液を生成するための)完全溶解ではなく、したがってこのプロセスは、標準再結晶を含まないことも、当業者は理解するであろう。このプロセスは、約75℃未満、具体的には約72℃、例えば約70℃の温度で実施されることも好ましい。
【0037】
本発明者らは、本発明の化合物を下記によって得ることができることも見出した。
(a) 約70℃超、例えば約72℃、具体的には約75℃、例えば約80℃以上の水性溶媒(すなわち、純水など、少なくとも約95%(w/w)の水を含む)におけるペミロラストナトリウムの部分溶解(平衡および/またはスラリー形成)に続いて、結晶化。このプロセスは、再結晶ではなく、好ましくは上記の温度で濾過して、生成された結晶を単離することを含む; および
(b) 水性溶媒(すなわち、純水など、少なくとも約95%(w/w)の水を含む)におけるペミロラストナトリウムの完全(例えば、少なくとも約95%)溶解と、その後に続く過剰量の貧溶媒(例えば、エタノール)の添加。このプロセスは、好ましくは高温(例えば、およそ溶媒の沸点)における貧溶媒の添加を含む。エタノールの場合、これは、約75℃など、約70℃(例えば、約72℃)〜約80℃であり、結晶の単離前に、より低い温度(例えば、約20℃などの室温)に冷却する。
【0038】
結晶化させる対象の化合物の溶液(および/または部分溶液)中の濃度、および使用される溶媒系は、結晶化温度および結晶化時間に影響を及ぼす可能性があることを当業者は理解するであろう。
【0039】
当業者によって理解され得るように、得られる結晶形は、結晶化プロセスの動力学と熱力学とに依存する。特定の熱力学条件下(溶媒系、温度、圧力、および本発明の化合物の濃度)で、ある結晶形は、別の結晶形(または、実際には他のいかなる結晶形)より安定であり得る。しかし、他の結晶形が、比較して相対的に低い熱力学的安定性を有することができると、動力学的に好まれる。したがって、さらに、時間、不純物プロファイル、撹拌、種結晶の存在などの動力学的因子も、どの形態が出現するかに影響を及ぼすことがある。したがって、ペミロラストナトリウムの半水和物、および実際には(適切な場合は)ペミロラストナトリウム塩の半水和物形の異なる結晶形を得るために、本明細書に述べられる手順を当業者が適宜適応させることができる。
【0040】
本明細書に記載される結晶形が他の結晶形の非存在下で調製されるようにするために、他の結晶形の核および/または種結晶の非存在下で、所望の結晶形の核および/または種結晶を加えることによって、結晶化を実施することができる。
【0041】
さらに、乾燥温度および乾燥時間は、本発明の化合物の固体状態特性および/または固体状態の形態に影響を及ぼすことがある。例えば、脱水は、低湿度および/または高温および/または減圧で行われることがある。例えば、結晶性半水和物が生成した後、臨界湿度レベルが存在し得る。そのレベル未満で、乾燥が行われることがあり、それによって、結晶水が失われ、無水物への少なくとも部分的な固体状態変態が起こり得る。
【0042】
これにも関わらず、例えば本明細書の以下に記載されるペミロラストナトリウムの高水和物(例えば、七水和物)を脱水することによって、本発明の化合物を生成することもできる。
【0043】
本発明の化合物の調製およびキャラクタリゼーションを本明細書の以下に記載する。本発明の化合物の様々な結晶形のキャラクタリゼーションは、例えば本明細書の以下に記載される粉末X線回折(PXRD)法を用いて容易に行うことができる。
【0044】
本発明の化合物は、当業者に周知である技法、例えばデカンテーション、濾過および/または遠心分離を行って単離することができる。
【0045】
本発明者らは、本明細書に記載される結晶化プロセスを使用することによって、化学的純度の高い本発明の化合物を生成することが可能であることを見出した。
【0046】
本発明の化合物が本明細書に記載されたとおりに結晶化されると、得られた化合物は、ペミロラストの他の塩、および/またはペミロラストナトリウム塩の他の形態に比べて、本明細書の以上に記載された改善された化学的および固体状態安定性、ならびに改善された溶解性および吸湿性プロファイルを有する形態をとる。
【0047】
(医薬品製剤および医学的使用)
本発明の化合物は、薬理活性を有しているので有用である。したがって、これらは、医薬品として示される。
【0048】
特に、本発明の化合物は、炎症性病態の治療において有用である。
【0049】
炎症性病態は、典型的には、宿主にとって有益であるより有害である効果をもたらす、免疫防御機構の活性化で特徴付けられる。このような病態は、一般に様々な程度の組織の発赤もしくは充血、腫脹、高熱、疼痛、そう痒、細胞の死および組織の破壊、細胞増殖、ならびに/または機能喪失に伴う。記載され得る炎症性病態としては、動脈炎、糖尿病(1型糖尿病、および好ましくは2型糖尿病を含む)、肥満、メタボリックシンドローム、子宮内膜症、アレルギー(アレルギー性結膜炎およびアレルギー性鼻炎を含む)、強直性脊椎炎、喘息、アトピー性皮膚炎、ざ瘡、皮膚熱傷、酒さ、脂漏性皮膚炎、皮膚潰瘍、慢性閉塞性肺疾患、接触性皮膚炎、膀胱炎、痛風性関節炎、炎症性腸疾患(クローン病や潰瘍性大腸炎など)、多発性硬化症、変形性関節症、膵炎、前立腺炎、乾癬、乾癬性関節炎、関節リウマチ、腱炎、滑液包炎、シェーグレン症候群、全身性エリテマトーデス、ブドウ膜炎、蕁麻疹、血管炎、糖尿病性血管合併症、偏頭痛、アテローム性動脈硬化症およびそれに伴う心血管障害が挙げられる。記載され得る病態としては、アトピー性皮膚炎、子宮内膜症、偏頭痛、喘息、慢性閉塞性肺疾患、クローン病、多発性硬化症、乾癬、関節リウマチ、全身性エリテマトーデス、潰瘍性大腸炎、さらに詳細にはアテローム性動脈硬化症およびそれに伴う心血管障害が挙げられる。記載され得る他の病態としては、メタボリックシンドローム、肥満、糖尿病および/または糖尿病性血管合併症が挙げられる。
【0050】
「アテローム性動脈硬化症」という用語は、当業者によって、血管、特に動脈壁におけるコレステロール蓄積、泡沫細胞形成、炎症および細胞増殖で特徴付けられる任意の疾患を包含するものと理解される。アテローム性動脈硬化症「に伴う」心血管障害は、大動脈瘤(腹部大動脈瘤および/またはアテローム動脈硬化性大動脈瘤を含む)、動脈硬化症、末梢動脈閉塞性疾患、冠動脈疾患(例えば、狭心症、心筋梗塞、心臓発作など)、冠疾患(心臓病および心疾患を含む、虚血性心疾患など)を包含し、プラークもしくはアテローム破綻および/または不安定性、血管もしくは動脈疾患、虚血性疾患/虚血、ならびに脳卒中(脳血管障害および一過性脳虚血発作を含む)も包含することができる。
【0051】
記載され得る患者群としては、急性冠症候群患者が挙げられる。「急性冠症候群」という用語は、胸痛および/または心電図(ECG)異常に伴うことが多いがこれに限らない任意の心筋虚血の異常状態(abnormal myocardial ischaemic state)を包含すると、当業者によって理解されよう。このような症候群は、心筋梗塞(心臓発作)の最も一般的な症状である。この用語は、「安定狭心症」(すなわち、労作時に発症し、安静時に消失する狭心症)とは対照的に「不安定狭心症」という用語とおおむね同義であることを当業者は理解するであろう。発生するペースがますます悪化する労作性狭心症(「漸増性狭心症」)は同様に、当業者によって「不安定」の定義の範囲内であるとみなされるであろう。
【0052】
本発明の別の態様によれば、炎症性障害、特にアテローム性動脈硬化症および/またはそれに伴う心血管障害、具体的には大動脈瘤の治療方法であって、このような治療を必要とする患者への本発明の化合物の投与を含む方法が提供される。
【0053】
記載され得る他の炎症性病態としては、文献中で様々に「低グレード全身性炎症」、「無症候性全身性炎症」、「慢性低グレード炎症」、「持続性低グレード炎症」、または文脈に応じて、単に「低グレード炎症」もしくは「全身性炎症」とも呼ばれる病態を包含するように理解される全身性低グレード炎症(SLGI)が挙げられる(例えば、Marzら、Circulation、110(2004年)およびNicklasら、CMAJ、172、1199(2005年)を参照のこと)。他の炎症マーカー(例えば、血中サイトカイン、接着分子、および白血球)がSLGIを示すことが知られており、本発明によれば測定および低減され得るが、SLGIは常に、とりわけ血漿C反応性タンパク質(CRP)値を特徴とし、その値は(例えば、他の点では外見上健常でかつ/または非アレルギー/非喘息哺乳類対象において)約10mg/L未満であるが、約7mg/L超、例えば約5mg/L超、好ましくは約3mg/L超、より好ましくは約2mg/L超、特に約1mg/L超、さらに特に約0.9mg/L超である。このような血漿CRP値は、薬理学的に有効量の本発明の化合物を投与することによって低減することができる。
【0054】
したがって、患者における血漿CRP値を(本明細書の以上に記載される値のいずれか1つの値未満に)低減するための本発明の化合物、および本発明の化合物を患者に投与することを含む、患者における血漿CRP値を(本明細書の以上に記載される値のいずれか1つの値未満に)低減する方法もさらに提供される。
【0055】
SLGIは、例えばメタボリックシンドローム、糖尿病(例えば、2型糖尿病)、インスリン抵抗性症候群、肥満、心血管疾患(例えば、アテローム性動脈硬化症、腹部大動脈瘤、および他の心血管イベント)、および一部の癌(例えば、大腸癌)に関連していることが知られている。CRP値の僅かな上昇が、その他の点では外見上健常な対象における疾患の唯一の徴候にもなり得る。
【0056】
CRPの僅かな上昇によって、様々な医学的病態における望ましくない転帰もしくは合併症、または様々な疾患において死に至る可能性も予測することができる。特に、CRPの上昇によって、心血管病的状態および死亡、ならびに/または2型糖尿病の発症を予測することができ、それらの両方のリスクは、本発明によれば、本発明の化合物を使用することによって低減することができる。
【0057】
したがって、患者において心血管病的状態および死亡のリスクを低減(すなわち、予防)し、かつ/または2型糖尿病の発症を低減(すなわち、予防)する方法が提供され、その方法は:
(a) その患者における血漿CRP値を測定すること;
(b) その血漿CRP値が、本明細書の以上に記載される値の1つの値を超える、特に約0.9mg/Lを超えるかどうか決定すること; および
(c) 超えている場合、本発明の化合物をその患者にしばらく適切な投与量で投与して、CRP値を例えば本明細書の以上に記載される関連性のある値まで低減すること
を含む。
【0058】
アメリカ心臓協会(American Heart Association(AHA))および米国疾病管理予防センター(CDC)は、CRPをリスクアセスメントツールとして評価し、1mg/L未満、1〜3mg/L、および3mg/L超の切点を使用して、それぞれ心血管病的状態または死亡を発症する相対リスクのより小さい、平均的な、および大きい対象を特定することを提案した。0.9mg/Lを超える血漿CRPが、心血管イベントのリスク増大の切点としても使用されている(Ridkerら、N. Engl. J. Med., 352, 20(2005年))。
【0059】
「病的状態」という用語は、一般に任意の病的状態(diseased state)、障害(disability)、病気(illness)、および/または健康不良(poor health)を包含すると、当業者によって理解されよう。したがって、「心血管」病的状態は、潜在的な(underlying)心血管合併症の結果として示されるような状態を包含するが、その心血管合併症は、それ自体が肥満、メタボリックシンドローム、(例えば、2型)糖尿病など本明細書の以上に記載される他の病態のうちの1つまたは複数の結果であり得る(下記参照)。
【0060】
2型糖尿病は、末梢組織のインスリンに対する応答性低下(インスリン抵抗性)、ならびにインスリン抵抗性および高血糖に直面してインスリン分泌不足として顕在化するβ細胞機能不全で特徴付けられる障害である(例えば、RobbinsおよびCotran、Pathologic Basis of Disease、第8版、Saunders Elsevierを参照のこと)。2型糖尿病の症状としては、慢性疲労、過剰の尿生成、過度の口渇、および水分摂取の増加が挙げられる。糖尿病の現行の世界保健機関(World Health Organisation)診断基準は、(a) 空腹時血漿ブドウ糖値:少なくとも7.0mmol/L、または(b) 経口ブドウ糖負荷試験(OGTT)での血漿グルコース値:少なくとも11.1mmol/Lである。「2型糖尿病の発症を低減する」によって、本発明者らは、先在病態の進展(例えば、悪化)を予防するためのSLGIの治療に加えて、2型糖尿病の発症の予防を含める。
【0061】
本発明者らは、CRPが0.9mg/Lを超える対象において、ペミロラストが血漿トリプターゼ値を同時に低減することはなく、さらに対象において、血漿CRP値と肥満細胞トリプターゼ値の間に相互関係がないことも見出した。
【0062】
したがって、本明細書に記載されるSLGIと特に関係がある使用および方法は、非アレルギー患者におけるまたは非アレルギー患者に関するものであることが好ましい。「非アレルギー」によって、本発明者らは、免疫系のアトピー性障害の外に現れる徴候を(このような治療を受ける時点では)患者が示さないことを意味する。この点に関して、このような患者は、IgEを介した肥満細胞および/または好塩基球の活性化を含む免疫学的応答で特徴付けられるアレルゲンに対する過敏症の徴候を示さないことがある。非アレルギー患者であるかどうかの決定は、例えば既知のアレルゲンに対する応答について(例えば、皮膚を)検査し、またはアレルゲン特異的IgEの有無およびレベルについて血液を分析することによってルーチンで行うことができる。
【0063】
本明細書に記載されるSLGIと特に関係がある使用および方法は、非喘息患者におけるまたは非喘息患者に関するものであることがさらに好ましい。「非喘息」によって、本発明者らは、気管支がその平滑筋細胞の収縮、気道炎症および呼吸困難によって可逆的狭窄を起こしている、両肺の慢性炎症に対する素因の外に現れる徴候を(このような治療を受ける時点では)患者が示さないことを意味する。喘息は、アレルギー性でも、非アレルギー性でもよい。
【0064】
SLGI治療の好ましい使用および方法としては、患者が喫煙者または元喫煙者であり、その対象者は糖尿病および/もしくはメタボリックシンドロームを患い、または肥満度指数が25を超える、使用および方法が挙げられる。
【0065】
記載され得る他の炎症性病態としては、うっ血性心不全、心房細動、高血圧(本態性肺動脈高血圧および/または門脈高圧を含む); 照射、手術、および/または外傷の結果(炎症、線維症、瘢痕、および癒着を含む); 炎症によって引き起こされた線維症、瘢痕、および/または癒着; 癌、骨粗鬆症、サルコイドーシス、過敏性腸症候群、網膜症(糖尿病網膜症を含む)、加齢黄斑変性、腎症(糖尿病性腎症を含む)、糸球体腎炎(IgA腎炎/腎症を含む)が挙げられる。
【0066】
誤解を避けるために、本発明の文脈においては、「治療(treatment)」「療法(therapy)」および「治療法(therapy method)」という用語は、治療的または姑息的処置を必要とする患者の治療的または姑息的処置、ならびにアテローム性動脈硬化症やそれに伴う心血管障害などの炎症性障害の影響を受け易い患者の予防的処置および/または診断を包含する。
【0067】
「患者」は、哺乳類(ヒトを含む)患者を包含する。
【0068】
本発明の化合物は、薬学的に許容される剤形の化合物を含む製剤の形で、好ましくは、局所(locally)投与もしくは全身投与、例えば経口投与、静脈内もしくは動脈内投与(血管内または他の血管周囲装置/剤形(例えば、ステント)によることを含む)、筋肉内投与、皮膚適用、皮下投与、経粘膜(例えば、舌下または頬側)投与、直腸投与、経皮投与、経鼻投与、経肺(例えば、気管または気管支)投与、局所(topically)投与され、または他の何らかの非経口経路で投与される。好ましい送達様式としては、経口(特に)、静脈内、皮膚、または皮下、経鼻、筋肉内、または腹腔内送達が挙げられる。
【0069】
本発明の化合物は一般に、所期の投与経路および標準的製剤慣行を十分に考慮して選択され得る薬学的に許容される助剤、希釈剤、または担体と混合して1つまたは複数の医薬製剤の形で投与されることになる。このような薬学的に許容される担体は、好ましくは無菌、純粋、非発熱性であり、活性化合物に対して化学的に不活性であり、使用条件下で有害な副作用または毒性を示すことがない。このような薬学的に許容される担体は、本発明の化合物の即放性製剤または放出調節製剤をもたらすこともできる。
【0070】
本発明の化合物は、好適な担体、希釈剤、または助剤と混合する前にさらに処理することができる。例えば、結晶形をミルまたはグラインダーでより小さい粒子に粉砕することができる。
【0071】
好適な医薬製剤は市販のものとすることができ、または他には、文献、例えばRemington The Science and Practice of Pharmacy、第19版、Mack Printing Company, Easton, Pennsylvania(1995年)ならびにMartindale - The Complete Drug Reference(第35版)およびその中の引用文献に記載されており、それらの文献すべての中の関連する開示内容は参照により本明細書に組み込まれる。別の場合では、好適な製剤の調製は、当業者が工夫を凝らすことなくルーチンの技法を用いて実現することができる。
【0072】
製剤中の本発明の化合物の量は、治療対象の病態の重症度および患者、ならびに使用される化合物に依存することになるが、当業者が工夫を凝らすことなく決定することができる。
【0073】
治療対象の障害および患者、ならびに投与経路に応じて、本発明の化合物を、様々な治療上有効量で、それを必要とする患者に投与することができる。
【0074】
しかし、本発明の文脈において、哺乳類、特にヒトに投与される量は、哺乳類における治療効果を合理的な時間枠にわたってもたらすのに十分な量とすべきである。的確な投与量および組成物ならびに最適な送達レジメンの選択は、とりわけ、製剤の薬理学的特性、治療対象の病態の性質および重症度、レシピエントの健康状態および精神的鋭敏さ(mental acuity)、ならびに治療対象の患者の年齢、状態、体重、性別、および応答、疾患の病期/重症度、ならびに患者間の遺伝子の差の影響も受けることになることを当業者は認識するであろう。
【0075】
本発明の化合物は、継続投与または間欠投与することができる(例えば、ボーラス注射による投与)。用量は、投与のタイミングおよび頻度によっても決定され得る。
【0076】
好適な投与量としては、Martindale-The Complete Drug Reference(第35版)およびその中の引用文献などの医学文献に記載されているものが挙げられ、それらの文献すべての中の関連する開示内容は参照により本明細書に組み込まれる。したがって、本発明の化合物の(遊離酸として算出された)好適な投与量は、約0.01mg/体重kg〜約1,000mg/体重kgの範囲である。より好ましい範囲は、経口投与の場合、1日ごとに約0.1mg/kg〜約20mg/kgである。
【0077】
しかし、ペミロラストの好適な投与量は当業者に周知である。例えば、(遊離酸として算出された)経口投与量は、1日当たり約0.5mg〜約900mgなど、約0.1mg〜約1.2gの範囲とすることができる。例えば、1日量の範囲の好適な下限は、約1mg、具体的には約2mg、例えば約5mg、具体的には約10mg、より好ましくは約20mgであり、1日量の範囲の好適な上限は、約200mg、例えば約100mg、具体的には約80mg、より好ましくは約60mgである。したがって、経口1日量は、約2mg〜約60(例えば、約50)mg、具体的には約5mg〜約45(例えば、約40)mg、好ましくは約10mg〜約35(例えば約30)mgとすることができる。好適な個々の投与量は、1日当たり約10mg〜約100mg、具体的には約20mg〜約90mg、例えば約30mg〜約80mgとすることができる。好ましい投与量は、1日約40mg〜約80mgの範囲、特に約60mgである。投与量は、1日1回の投与に基づくものとすることができ、または1日2回もしくは3回(好ましくは、2回)の投与量に(例えば、等量)分割し得ることを当業者は理解するであろう。
【0078】
いずれにしても、医師または他の当業者は、個々の患者に最も適した実際の用量をルーチンで決定することができるであろう。上記の用量は、平均的な場合の例である。もちろん、より高いまたはより低い用量範囲が相応である個々の例が存在することがあり、このような場合も、本発明の範囲内である。
【0079】
本発明の化合物は、本明細書に定義する炎症性障害の治療に有用である1つまたは複数の活性成分と組み合わせることもできる。
【0080】
したがって、本発明の別の態様によれば、
(a) 本発明の化合物; および
(b) 炎症性障害の治療に有用である1つもしくは複数の活性成分、またはその薬学的に許容される塩もしくは溶媒和物
を含む配合剤も提供される。
【0081】
このような配合剤は、本発明の化合物を炎症性障害の治療に有用である活性成分と併用投与することを実現し、したがって別個の製剤(それらの製剤の少なくとも1つが本発明の化合物を含み、少なくとも1つがもう一方の活性成分を含む)として提供することができ、あるいは複合成分製剤として提供する(すなわち、製剤化する)ことができる(すなわち、本発明の化合物および別の活性成分を含む単一の製剤として提供することができる)。
【0082】
したがって、
(1) 本発明の化合物; 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物; および薬学的に許容される助剤、希釈剤、または担体を含む医薬製剤(その製剤は、以下に「複合成分製剤」と呼ばれる); ならびに
(2)
成分(A) 本発明の化合物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤; および
成分(B) 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤を含み、それらの成分(A)および(B)はそれぞれ、他方と併用投与するのに適した形態で提供されている
パーツキットをさらに提供する。
【0083】
本発明の別の態様によれば、上記で定義されたパーツキットを作製する方法が提供され、その方法は、上記で定義された成分(A)を上記で定義された成分(B)と結合させ、したがって2つの成分を互いに併用投与するのに適したものとする工程を含む。
【0084】
2つの成分を互いに「結合」させることについて、本発明者らは、パーツキットの成分(A)および(B)が、
(i) 別個の製剤として(すなわち、互いに関わりなく)提供され得るものであり、その後一緒にまとめられ、併用療法において互いに併用され; または
(ii) 併用療法において互いに併用されるための「配合パック」の別個の成分として一緒に包装および提供され得るものであることを含める。
【0085】
したがって、
(I) 本明細書に定義する成分(A)および(B)の一方と、
(II) その成分を2つの成分の他方と併用するための指示書
を一緒に含むパーツキットもさらに提供される。
【0086】
本明細書に記載されるパーツキットは、反復投与を実現するために、適切な量/投与量の本発明の化合物を含む1つを超える製剤、および/または適切な量/投与量のもう一方の活性成分/塩/溶媒和物を含む1つを超える製剤を含んでもよい。1つを超える製剤(どちらかの活性化合物を含む)が存在する場合、このような製剤は、どちらかの化合物の投与量、化学組成、および/または物理的形態の点から同じでもよく、または異なってもよい。
【0087】
本明細書に記載されるパーツキットに関して、「併用投与」によって、本発明者らは、本発明の化合物および他の活性成分(またはその塩/溶媒和物)を含む各製剤を、関連する病態の治療の過程において順次に、別々に、かつ/または同時に投与することを含める。
【0088】
したがって、本発明による配合剤に関して、「併用投与」という用語は、配合剤の2成分(本発明の化合物および他の活性成分)を一緒にまたは時間的に十分に接近して投与(場合によっては、反復投与)して、本発明の化合物を含む製剤またはもう一方の活性成分を含む製剤が、同じ治療の過程において他方の成分の非存在下に単独で投与(場合によっては、反復投与)される場合より高い薬効が、関連する病態の治療の過程において患者にもたらされることを包含する。配合剤が特定の病態についてより高い薬効を治療の過程においてもたらすかどうかの決定は、治療または予防対象の病態に依存するが、当業者がルーチンで実現することができる。
【0089】
さらに、本発明によるパーツキットに関連して、「併用」という用語は、2つの製剤のうちのどちらかが、もう一方の成分を投与する前、後、および/または同時に投与(場合によっては、反復投与)され得ることを包含する。本文脈で使用される場合、「同時に投与(administered simultaneously)」および「同時に投与(administered at the same time as)」という用語は、個々の投与量の本発明の化合物および他方の活性成分が、互いに48時間(例えば、24時間)以内に投与されることを包含する。
【0090】
本明細書に定義される炎症性障害の治療に有用である活性成分としては、トロンボキサンA2アンタゴニスト、P2Y12アンタゴニスト、PPARγアゴニスト、アンジオテンシンIIの生成および/または作用を抑制する化合物、他の血小板凝集抑制薬、ならびにより好ましくはスタチンが挙げられる。
【0091】
「トロンボキサンA2アンタゴニスト」という用語は、インビトロおよび/またはインビボ試験で、(i) トロンボキサンTP受容体の遮断、(ii) 酵素トロンボキサンシンターゼの抑制、または(iii) 血小板シクロオキシゲナーゼ-1の(例えば、選択的)抑制、それによる例えば血小板凝集の抑制のうちの1つまたは複数によって、トロンボキサンA2の効果を実験に基づいて決定することができる程度に抑制することができる化合物であればいずれも包含する。
【0092】
好ましいトロンボキサンA2アンタゴニストとしては、アスピリン/アセチルサリチル酸、より好ましくはエグアレン、特にオザグレル、さらに特にピコタミド、およびテルトロバン、特にセラトロダスト、さらに特にラマトロバンが挙げられる。
【0093】
「P2Y12アンタゴニスト」という用語は、インビトロおよび/またはインビボ試験で、ADPの血小板受容体P2Y12との結合を実験に基づいて決定することができる程度に(例えば、選択的に)抑制し、それによって血小板凝集を抑制することができる化合物であればいずれも包含する。
【0094】
好ましいP2Y12アンタゴニストとしては、プラスグレル、チカグレロル、特にクロピドグレルが挙げられる。
【0095】
「PPARγ」アゴニストという用語は、インビトロおよび/またはインビボ試験で、実験に基づいて決定することができる程度にペルオキシソーム増殖剤活性化受容体γと結合し、かつ/またはその機能に影響を及ぼすことができる化合物であればいずれも包含する。
【0096】
したがって、好ましいPPARγアゴニストとしては、リボグリタゾン、ナベグリタザル、バラグリタゾン、またはより好ましくはロシグリタゾン、特にピオグリタゾンを含む、チアゾリジンジオンと総称される化合物が挙げられる。記載され得る他のPPARγアゴニストとしては、チグリタザル(chiglitazar)、エタロシブ、ファルグリタザル、ロベグリタゾン(lobeglitazone)、ネトグリタゾン、ソデルグリタザル、ならびに次の医薬品開発コード(developmental drug code)によって文献に定義されているもの:THR-0921(Theracos Inc.)、またはより好ましくはAVE-0847およびAVE-0897(両方ともSanofi-Aventis)、CLX-0921(Calyx Therapeutics)、CS-7017(Daiichi Sankyo Co Ltd)、DRF-11605(Dr Reddy's Laboratories Ltd)、GFT-505(Genfit SA)、GSK-376501(GlaxoSmithKline plc)、INT-131(Amgen Inc; InteKrin Therapeutics)、(LBM-642; セボグリタザル; Novartis AG)、ONO-5129(Ono Pharmaceutical Co Ltd)、(PLX-204; インデグリタザル; Plexxikon Inc)、およびSDX-101が挙げられる。
【0097】
「アンジオテンシンIIの生成および/または作用を抑制する化合物」という用語は、インビトロおよび/またはインビボ試験で、アンジオテンシンIIの生成および/または作用を実験に基づいて決定することができる程度に(例えば、選択的に)抑制することができる化合物であればいずれも包含し、アンジオテンシン変換酵素(ACE)阻害物質、アンジオテンシン受容体遮断薬(ARB)、およびレニン阻害物質を包含すると理解されよう。
【0098】
「アンジオテンシン変換酵素(ACE)阻害物質」という用語は、インビトロおよび/またはインビボ試験で、アンジオテンシンIのアンジオテンシンIIへの変換を実験に基づいて決定することができる程度に(例えば、選択的に)抑制することができる化合物であればいずれも包含する。
【0099】
記載され得るACE阻害物質としては、アラセプリル、ベナゼプリル、カプトプリル、セロナプリル、シラザプリル、デラプリル、エナラプリル、ホシノプリル、ゲモパトリラト、グリコプリル(glycopril)、イドラプリル、イレパトリル(ilepatril)、イミダプリル、リベンザプリル、リシノプリル、ミクロギニン-FR1、ミキサンプリル、モエキシプリル、モエキシプリラート、モベルチプリル、オマパトリラト、プレンチル(Prentyl)、ペリンドプリル、キナプリル、ラミプリル、サンパトリラット、スピラプリル、シネコル(Synecor)、テモカプリル、トランドラプリル、ウチバプリル、ゾフェノプリルおよびザビシプリラトが挙げられる。より好ましいACE阻害物質としては、ベナゼプリル、シラザプリル、イレパトリル、イミダプリル、モエキシプリル、スピラプリル、テモカプリル、およびゾフェノプリル、より好ましくはホシノプリルおよびトランドラプリル、さらに特にエナラプリル、リシノプリル、およびキナプリル、特にカプトプリル、ペリンドプリル、およびラミプリルが挙げられる。
【0100】
「アンジオテンシン受容体遮断薬(ARB)」は、「アンジオテンシンII AT1受容体アンタゴニスト」という用語とおおむね同義であると当業者が理解するものであり、したがってインビトロおよび/またはインビボ試験で、アンジオテンシンII AT1受容体の活性化を実験に基づいて決定することができる程度に(例えば、選択的に)遮断することができる物質であればいずれも包含する。
【0101】
記載され得るARBとしては、アジルサルタン、アジルサルタンメドキソミル、カンデサルタン、カンデサルタンシレキセチル、アンジオカイン(angiokine)であるDival、エリサルタン、エリサルタンカリウム(elisartan potassium)、エプロサルタン、エンブサルタン、フィマサルタン(fimasartan)、フォンサルタン(fonsartan)、イルベサルタン、ロサルタン、ミルファサルタン、オルメサルタン、ポミサルタン、プラトサルタン、リピサルタン、サプリサルタン、サララシン、タソサルタン、テルミサルタン、バルサルタン、およびゾラサルタンが挙げられる。より好ましいARBとしては、アジルサルタン、エプロサルタン、フィマサルタン(fimasartan)、およびプラトサルタン、より好ましくはテルミサルタン、さらに特にイルベサルタン、およびオルメサルタン、特にカンデサルタン、ロサルタン、およびバルサルタンが挙げられる。
【0102】
「レニン阻害物質」という用語は、インビトロおよび/またはインビボ試験で、レニン-アンジオテンシン系におけるレニンの機能を実験に基づいて決定することができる程度に(例えば、選択的に)遮断することができる物質であればいずれも包含すると、当業者によって理解されよう。
【0103】
記載され得るレニン阻害物質としては、シクロチアゾマイシン、アリスキレン、シプロキレン、ジテキレン、エナルキレン、レミキレン、テルラキレン、およびザンキレンが挙げられる。好ましいレニン阻害物質としては、アリスキレンが挙げられる。
【0104】
アンジオテンシンIIの生成および/または作用を抑制する化合物としては、次の医薬品開発コードによって文献に定義されているものも挙げられる:100240、606A、A-65317、A-68064、A-74273、A-81282、A-81988、A-82186、AB-47、BIBR-363、BIBS-222、BIBS-39、BILA-2157BS、BL-2040、BMS-180560、BMS-181688、BMS-182657、BMS-183920、BMS-184698、BRL-36378、CGP-38560、CGP-38560a、CGP-42112-A、CGP-42112、CGP-421132-B、CGP-48369、CGP-49870、CGP-55128A、CGP-56346A、CGS-26670、CGS-26582、CGS-27025、CGS-28106、CGS-30440、CHF-1521、CI-996、CL-329167、CL-331049、CL-332877、CP-191166、CP-71362、CV-11194、CV-11974、DMP-581、DMP-811、DU-1777、DuP-167、DuP-532、E-4030、E-4177、EC-33、EK-112、EMD-56133、EMD-58265、EMD-66684、ER-32897、ER-32935、ER-32945、ES-1005、ES-305、ES-RWJ-38970、RWJ-46458、RWJ-47639、RXP-407、S-2864、S-5590、SB-203220、SC-50560、SC-51316、SC-51895、SC-52458、SC-54629、SC-565254、Sch-47896、Sch-54470、SK-1080、SKF-107328、SL-910102、SQ-30774、SQ-31844、SQ-33800、SR-43845、TA-606、TH-142177、U-97018、UK-63831、UK-77568、UK-79942、UP-275-22、WAY-121604、WAY-126227、VNP-489、XH-148、XR-510、YM-21095、YM-26365、YM-31472、YM-358、およびZD-7155。
【0105】
記載され得る他の血小板凝集抑制薬としては、アスピリン/アセチルサリチル酸の一酸化窒素供与性誘導体(例えば、NCX-4016、NicOx S.A.)、またはより好ましくはアナグレリド、アルガトロバン、ベラプロスト、カングレロール、シロスタゾール、ジピリダモール、リマプロスト、パログレリル、プロカインアミド、サルポグレラート(例えば、サルポグレラート塩酸塩)、チクロピジン、チロフィバンおよびトリフルサール、ならびに次の医薬品開発コードによって文献に定義されているものが挙げられる:DA-697b(国際公開第2007/032498号を参照のこと; Daiichi Seiyaku Co Ltd)、DG-041(deCODE Genetics Inc)、K-134(CAS RN 189362-06-9)、PL-2200(CAS RN 50-78-2)、PRT-60128(Portola Pharmaceuticals Inc)、SH-529(イロプロスト/β-シクロデキストリンクラスレート; Bayer Schering Pharma AG)、およびYY-280(チクロピジンとEGb-761(タナミン(tanamin); イチョウエキス; Yuyu Inc.)の併用療法)。
【0106】
「スタチン」という用語は、HMG-CoA還元酵素の阻害物質であればいずれも包含し、フルバスタチン、シンバスタチン、ロバスタチン、ロスバスタチン、ピタバスタチン、グレンバスタチン、セリバスタチン、プラバスタチン、メバスタチン、ベルバスタチン、ダルバスタチン、およびアトルバスタチンが含まれる。
【0107】
記載され得る他のスタチンとしては、アシテマート、ベンフルオレクス、クレスチン(Clestin)、コレストロン、ジヒドロメビノリン、メグルトール、ラウソノール、および次のコード名の化合物が挙げられる:ATI-16000、BAY-10-2987、BAY-x-2678、BB-476、BIO-002、BIO-003、BIO-2、BMS-180431、CP-83101、DMP-565、FR-901512、GR-95030、HBS-107、KS-01-019、L-659699、L-669262、NR-300、P-882222、PTX-023595、RP 61969、S-2468、SC-32561、sc-45355、SDZ-265859、SQ-33600、U-20685、およびNCX-6550(ニトロプラバスタチン)やNCX-6560(ニトロアトルバスタチン)などのNO-増強性/放出性スタチン。
【0108】
より好ましいスタチンとしては、ピタバスタチン(例えば、Livalo(登録商標)、Pitava(登録商標))、フルバスタチン(例えば、Lescol(登録商標))、シンバスタチン(例えば、Zocor(登録商標)、Lipex(登録商標))、ロバスタチン(例えば、Mevacor(登録商標)、Altocor(登録商標))、ロスバスタチン(例えば、Crestor(登録商標))、プラバスタチン(例えば、Pravachol(登録商標)、Selektine(登録商標)、Lipostat(登録商標))、およびアトルバスタチン(例えば、Lipitor(登録商標)、Torvast(登録商標))が挙げられる。特に好ましいスタチンとしては、ピタバスタチン、より好ましくはシンバスタチン、さらに特にアトルバスタチン、特にロスバスタチンが挙げられる。
【0109】
記載され得る炎症の治療に有用である他の活性成分の薬学的に許容される塩としては、酸付加塩および塩基付加塩が挙げられる。このような塩は、通常の手段、例えば遊離酸または遊離塩基の形の活性成分と1当量または複数当量の適切な酸または塩基の、場合によっては溶媒中または塩が不溶性である媒体中での反応、続いて標準技法(例えば、真空中で凍結乾燥または濾過)を用いた前記溶媒または前記媒体の除去によって生成され得る。塩は、例えば好適なイオン交換樹脂を用いて、塩の形の活性成分の対イオンを別の対イオンと交換することによっても調製され得る。
【0110】
記載され得るピコタミドの塩としては、塩酸塩、重硫酸塩、マレイン酸塩およびトシル酸塩が挙げられる。記載され得るオザグレル、テルトロバン、エグアレン、およびアスピリンの塩としては、リチウム塩、ナトリウム塩、カリウム塩などのアルカリ金属塩が挙げられる。オザグレルおよびエグアレンの好ましい塩としては、ナトリウム塩が挙げられる。
【0111】
クロピドグレルの好ましい塩としては、重硫酸塩が挙げられるが、記載され得る他の塩および記載され得るチカグレロルの塩としては、塩酸塩、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。記載され得るプラスグレルの好ましい塩としては、塩酸塩が挙げられるが、記載され得る他の塩としては、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。
【0112】
記載され得るピオグリタゾンの好ましい塩としては、塩酸塩が挙げられるが、記載され得る他の塩としては、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。記載され得るロシグリタゾンの好ましい塩としては、マレイン酸塩が挙げられるが、記載され得る他の塩としては、塩酸塩、重硫酸塩、およびトシル酸塩が挙げられる。記載され得るリボグリタゾンの塩としては、塩酸塩、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。ナベグリタザルの好ましい塩としては、ナトリウム塩が挙げられるが、記載され得る他の塩としては、リチウム塩およびカリウム塩が挙げられる。記載され得るバラグリタゾンの好ましい塩としては、ナトリウム塩、カリウム塩、およびカルシウム塩が挙げられる。
【0113】
アンジオテンシンIIの生成および/または作用を抑制する化合物の好ましい塩としては、例えば塩酸塩、重硫酸塩、マレイン酸塩、メシル酸塩、トシル酸塩、カルシウムやマグネシウムなどのアルカリ土類金属塩、またはナトリウム塩やカリウム塩などのアルカリ金属塩が挙げられる。このような塩は、ペリンドプリル、エナラプリル、リシノプリル、キナプリル、イルベサルタン、オルメサルタン、トランドラプリル、テルミサルタン、ベナゼプリル、シラザプリル、モエキシプリル、スピラプリル、エプロサルタン、およびフィマサルタン(fimasartan)を含めて、化合物について、ルーチンの技法を用いて調製され得る。塩酸塩、重硫酸塩、マレイン酸塩、メシル酸塩、およびトシル酸塩が、ラミプリルやアリスキレンなどの化合物については好ましい。アルカリ土類金属塩、さらに特にアルカリ金属塩が、カンデサルタン、バルサルタン、カプトプリル、ロサルタン、特にホシノプリルなどの化合物については好ましく、それらの好ましい塩としては、カルシウム塩、マグネシウム塩、カリウム塩、特にナトリウム塩が挙げられる。記載され得るベナゼプリルおよびモエキシプリルの好ましい塩としては、塩酸塩が挙げられるが、記載され得る他の塩としては、重硫酸塩、マレイン酸塩、メシル酸塩、およびトシル酸塩が挙げられる。記載され得るエプロサルタンの好ましい塩としては、メシル酸塩が挙げられるが、記載され得る他の塩としては、塩酸塩、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。
【0114】
スタチンの好ましい塩としては、ピタバスタチンカルシウム、フルバスタチンナトリウム、プラバスタチンナトリウム、ロスバスタチンカルシウム、アトルバスタチンカルシウムなどのナトリウム塩、カリウム塩、およびカルシウム塩が挙げられる。
【0115】
炎症の治療に有用である他の活性成分の好適な投与量は、当業者に周知であり、Martindale - The Complete Drug Reference(第35版)およびその中の引用文献などの医学文献中で該当する薬物について列挙されているものが含まれ、それらの文献すべての中の関連する開示内容は参照により本明細書に組み込まれる。
【0116】
「約」という単語が、本明細書で、例えば量(例えば、値、重量、体積、モル)、温度、結晶化度、崩壊度(degree of degradation)、純度、溶解度、および活性成分の投与量に関連して使用されるときはいつでも、このような変数は近似的なものであり、そういうものとして本明細書に特定される数と±10%、例えば±5%、好ましくは±2%(例えば±1%)異なる可能性があることを理解されたい。
【0117】
本発明の化合物は、取り扱いやすさが改善される形態であり、以前に調製されたペミロラストの形態に比べると改善された化学的および固体状態安定性を有する形態で生成され得るという利点を有する。したがって、化合物は、長期間にわたって貯蔵される場合に安定であり得る。
【0118】
本発明の化合物は、公知および/または市販のペミロラストの形態に比べて改善された溶解性および吸湿性プロファイルも有する。本発明の化合物は、公知および/または市販のペミロラストの形態に比べて改善された味覚プロファイルも有することができる。
【0119】
本発明の化合物は、好収量で、以前に調製されたペミロラストの形態より高純度で、より短時間で、より好都合に、より低コストで調製され得るという利点も有することができる。
【0120】
本発明の化合物は、本明細書の以上に記載される病態の治療において、炎症性障害(アテローム性動脈硬化症およびそれに伴う心血管病態など)またはその他の治療で使用する先行技術において公知の同様な化合物に比べて、医師および/もしくは患者にとってより好都合で、より有効で、より低毒性で、活性範囲がより広く、より強力で、副作用がより少なく、または他の有用な薬理学的特性を有することができるという利点も有することができる。
【0121】
含まれている図面を参照しながら、以下の実施例によって、本発明を説明するが、決して限定するものではない。
【図面の簡単な説明】
【0122】
【図1】実施例1によって得られたペミロラストナトリウム半水和物の結晶形の粉末X線回折図(diffractogram)を示す図である。
【図2】実施例1によって得られたペミロラストナトリウム半水和物の結晶形のFT-ラマンスペクトルを示す図である。
【図3】比較例5によって得られたペミロラストナトリウムの結晶形の粉末X線回折図を示す図である。
【図4】比較例5によって得られたペミロラストナトリウムの結晶形の異なる時点(調製後(下側のトレース)および約1か月後(上側のトレース))におけるFT-ラマンスペクトルを示す図である。
【図5】実施例10によって得られたペミロラストナトリウム半水和物の結晶形の粉末X線回折図を示す図である。
【図6】実施例11によって得られたペミロラストナトリウム七水和物の結晶形の粉末X線回折図を示す図である。
【図7】実施例14によって得られたペミロラストナトリウムの追加の結晶形の粉末X線回折図を示す図である。
【図8】ペミロラストナトリウム半水和物(四角)とペミロラストカリウム(三角)の溶解プロファイル(溶解対時間の百分率)比較を示す図である; 平均値±SD(1群当たりn=3)。
【発明を実施するための形態】
【0123】
実施例1〜5の一般手順
FT-ラマンスペクトルを1064nmで操作される近赤外Nd:YAGレーザーおよび液体窒素冷却ゲルマニウム検出器を備えたBruker RFS 100 FT-ラマンシステムで記録した。3500〜50cm-1の範囲を2cm-1の分解能で64回スキャンし、集積した。一般に、100mWのレーザー出力を使用した。
【0124】
Bruker D8を使用して、粉末X線回折を実施した; 銅Kα線、40kV/40mA; LynxEye検出器、ステップサイズ0.02°(2θ)、ステップ時間37秒。試料調製: 僅かな圧力を加えて、平面にしたこと以外は、一般に何ら特別な処理を行うことなく、試料を測定した。シリコン単結晶サンプルホルダーのタイプ: a) 標準ホルダー深さ0.1mm、b) 深さ0.5mm、空洞直径12mm、c) 深さ1.0mm、空洞直径12mm。Bruker D8で測定した試料はすべて、測定時に回転させた。別段の指定のない限り、周囲空気雰囲気を使用した。選択された試料をPhilips X'pert PW 3040またはPhilips PW1710(銅Kα線、ステップサイズ0.02°(2θ)、2秒/ステップ、2〜50°(2θ)で測定した。僅かな圧力を加えて、平面にしたこと以外は、何ら特別な処理を行うことなく、試料を測定した。別段の指定のない限り、周囲空気雰囲気を使用した。
(下記の実施例に従って調製された形態は、関連するパターンから(実験誤差を考慮して)、同じ結晶形が生成されていたことが明らかであったとき、下記に開示される他の実施例と「本質的に」同じPXRD回折パターンを示した。したがって、PXRD間隔値の実験誤差の限界は、本明細書のいずれかの部分で使用されているように、最後の小数位で±2ぐらいの範囲とすることができる。)
【0125】
C、H、およびNの元素分析は、Leco CHN 800またはLeco CHNS 932装置を使用して、燃焼によって行った。Oの元素分析は、Leco RO-478装置を使用して、熱分解によって行った。Naの元素分析は、原子吸収分光法で行った。
【0126】
1H/13C NMRスペクトルをBruker DPX300装置で記録した。別の方法では、1H NMRスペクトルをVarian MERCURY+400分光計(400MHz)で記録した。スペクトルは周囲温度で記録した。化学シフトは、TMSを基準にして、溶媒シグナルδ7.26ppm CHCl3、δ2.50ppm DMSO、およびδ4.79ppm H2Oを経て、δ値(ppm)として記載される。
【0127】
質量分析スペクトルは、Onyx Monolithic C18 50mm×4.6mmカラム(Phenomenex)を装備したFinnigan ThermoQuest AQA四重極型質量分析計を有するGilson HPLCシステムを使用するLC-MSシステム(流速4mL/分、アセトニトリル/水勾配および0.05%ギ酸)、またはVarianクロムパックキャピラリーカラムCP-SIL 8 CB Low Bleed/MS(30 m_0.22mm、0.25mm)を装備し、イオン生成電位70eVを利用するGC-MS装置で記録した。
【0128】
示差走査熱量測定をPerkin Elmer DSC 7で記録した。密閉型金るつぼ、加熱速度:10K/分、範囲:50℃〜350℃。DSC分析の過程において記録された、熱イベントと関係付けられた温度は、それぞれの熱イベントのピーク温度(最低/最高)である。
【0129】
TG-FTIRは、Bruker FT-IR Spectrometer Vector 22を備えたNetzsch Thermo-Microbalance TG 209で記録した。Alるつぼ(開放型または微細孔付き)、N2雰囲気、加熱速度:10K/分、範囲:25〜250℃。
【0130】
HPLCをTSP HPLC(UV3000、AS3000、P4000、SCM1000ソフトウェアバージョン4.1)で行った; カラム:Waters、X Terra MS C18 4.6×100mm、5μ(CC01); 移動相A:H2O+0.1%TFA; 移動相B:アセトニトリル+0.1%TFA; 標準濃度:約0.09mg/mL; 保持時間:6.6分; 勾配:0.0分:A:95%/B:5%; 20.0分:A:5%/B:95%; 21.0分:A:95%/B:5%; 30.0分:A:95%/B:5%; 流速:1.0mL/分; 注入量:10μL; 波長:254nm。
【0131】
動的水蒸気収着(Dynamic Vapor Sorption)(DVS)測定を収着測定システムSPS11-100nで実施した。試料をアルミニウムるつぼに入れ、事前定義湿度プログラムを開始する前に、その試料を所与のRHで平衡化させておいた。(1) 50%RHで2時間; (2) 50→0%RH(5%/時); (3) 0%RHで5時間; (4) 0→95%RH(5%/h); (5) 95%RHで5時間; (6) 95→50%RH(5%/時); (7) 50%RHで2時間。吸湿性をヨーロッパ薬局方に従って分類した(80%RH/25℃で24時間貯蔵)。(周囲条件での)出発材料に比べて、85%RHにおける質量変化率を分類に使用した-非常に吸湿性:質量の増加≧15%; 吸湿性:質量の増加は15%未満であり、2%以上である; 若干吸湿性:質量の増加は2%未満であり、0.2%以上である; 潮解性:十分な水が吸収されて、液体になる。
【実施例】
【0132】
(実施例1)
ペミロラストナトリウム半水和物
ペミロラストカリウムを水に溶解し、6M HClを用いてpH 1まで酸性化し、遊離酸を溶液から沈澱させることによって、ペミロラスト遊離酸を調製した。生成した結晶を濾過し、水で洗浄し、真空乾燥した。次に、2M NaOHの代わりに2M KOHを使用し、水:イソプロパノール(2:1の比)の代わりに水:イソプロパノール(1:2の比)から再結晶をして、下記の比較例4(ステップIaおよびIb)に記載の方法と同様にして、ペミロラストカリウムを調製した。
【0133】
ペミロラスト遊離酸(89mg)を783μLのメタノール中0.5Mナトリウムメトキシド(Fluka)に懸濁した。懸濁液を1日撹拌した。沈殿物が生成した。濾過し、真空下で約2時間乾燥した後、固体物質が得られた(収量:81mg)。
【0134】
元素組成分析を下記の表1にまとめる。そのデータから、1:1の化学量論でペミロラストナトリウム塩の半水和物が示唆される。式:Na(C10H7N6O)×0.5H2Oの理論データを算出した。
【0135】
【表1】
【0136】
実施例1によって得られた形態のPXRDパターンを図1に示し、下記の表2にまとめる。
【0137】
【表2A】
【0138】
【表2B】
【0139】
塩は高結晶性であった。
【0140】
実施例1によって得られた形態のFT-ラマンスペクトルを図2に示す。
【0141】
NMR、DSC、TG-FTIR、およびDVS分析も実施した。DSCは、280.3℃で1つの吸熱を示した。DVS分析によって、タイトル化合物は、上記の分類体系によれば吸湿性である(80%超RHで、著しい吸水が開始する)ことが明らかである。(対照的に、DVSによって、ペミロラストカリウムは、上記の分類体系によれば非常に吸湿性である(70%超RHで、著しい吸水が開始する)ことが明らかである。)
【0142】
(実施例2)
ペミロラストナトリウム半水和物
ペミロラスト遊離酸(上記の実施例1に記載のとおりに調製した; 90mg)を791μLのメタノール中0.5Mナトリウムメトキシド(Fluka)に懸濁した。懸濁液を1日撹拌した。沈殿物が生成した。濾過し、真空下で約1.5時間乾燥した後、固体物質が得られた(収量:59mg)。
【0143】
結晶をFT-ラマンで分析した。関連するスペクトルは、上記の実施例1に従って得られた形態によって示されたものと本質的に同じであった。
【0144】
(実施例3)
溶解性の比較
約40mgの試料(上記の実施例2に記載された手順を経て得られた)を0.25mLの再蒸留水に分散した。懸濁液を22℃で24時間振盪した。その後、Eppendorf Thermomixer Comfort(400rpm)を使用して、迅速固/液分離を行った。Centrifuge Hettich EBA 12 R(15,000g、1分、22℃)で、Millipore Centrifugal Filter Devices(PTFEフィルター; 0.2μm)を用いて、懸濁液を濾過した。濾液中の試料の濃度をHPLCで分析し、固相をFT-ラマン分光法で分析した。
【0145】
実施例2の化合物は、検討された条件下で水溶解度23.64mg/mLを示した。飽和溶液はpH 8.0であった。
【0146】
ペミロラストカリウム塩(メタノール中0.5Mナトリウムメトキシドの代わりにメタノール中3.4Mカリウムメトキシド(Fluka)を使用して、実施例1の第2段落、および上記の実施例2に記載のように調製された)の水溶解度を同様に決定し、検討された条件下で192.38mg/mLであることがわかった。飽和溶液はpH 9.0であった。(市販のペミロラストカリウムの水溶解度は、179〜182mg/mLであると報告されている; 出典:上記Pharmaceutical Interview Form)
【0147】
(比較例4)
ペミロラストナトリウムの合成
ペミロラストナトリウム塩を次の2つの方法によって調製した:
(I)
(Ia) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1.2-a]ピリミジン-4-オン(ペミロラスト)
これは、マロノニトリル(1.64g、24.8mmol; Acros Organics)、2-アミノ-3-ピコリン(2.51mL、24.8mmol; Acros Organics)、オルトギ酸エチル(4.55mL、27.3mmol; Sigma-Aldrich)、およびアジ化ナトリウム(1.78g、27.4mmol; Sigma-Aldrich)を出発物として、SanoおよびIshiharaによって記載された方法(Heterocycles、48、775(1998年))に従って合成され、サブタイトルの化合物が得られた(2.64g; 46.7%)。
1H NMR (DMSO-d6) δ: 9.21 (s, 1H, CH), 9.16〜9.11 (m, 1H, CH), 8.13〜8.07 (m, 1H, CH), 7.58〜7.51 (m, 1H, CH), 2.62 (s, 3H, CH3)。
【0148】
(Ib) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(ペミロラスト)ナトリウム塩
9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(2g、8.76mmol; 上記の工程Iaによるもの)を2-プロパノール(9mL)に懸濁し、2M NaOH(8.8mL、17.6mmol)を加えた。反応混合物を50℃に1時間加熱した。さらに17mLの2-プロパノールで処理した後、粗タイトル化合物が沈殿した。氷浴中で冷却した後、固体物質を濾過で回収し、100mLの水に再溶解した。未溶解の物質を濾過で除去し、濾液を蒸発させた。残渣を水および2-プロパノール(2:1の比)から再結晶し、真空乾燥して、純粋なペミロラストNa塩を得た(1.26g、57.5%)。
1H NMR (D2O) δ: 8.86〜8.80 (m, 1H, CH), 8.57 (s, 1H, CH), 7.68〜7.59 (m, 1H, CH), 7.22〜7.13 (m, 1H, CH), 2.39 (s, 3H, CH3)。
【0149】
(II) (米国特許第4,122,274号に記載の方法)
(IIa) 2-シアノ-3-(3-メチル-2-ピリジルアミノ)アクリル酸エチル
エトキシメチレンシアノ酢酸エチル(7.82g、46.2mmol; Sigma-Aldrich)および2-アミノ-3-ピコリン(4.67mL、46.2mmol; Acros Organics)のトルエン(4mL)溶液を100℃で15分間加熱した。反応混合物を冷却し、サブタイトルの生成物(10.45g、97.8%)を濾過で回収した。
GC-MS(70eV) m/z(相対強度)231(M+、15)、158(100)
1H NMR (CDCl3) δ: 11.20〜11.07 (m, 1H, NH), 8.82 (d, J = 12.4 Hz, 1H, CH), 8.22〜8.17 (m, 1H, CH), 7.53〜7.47 (m, 1H, CH), 7.07〜6.96 (m, 1H, CH), 4.31 (q, J = 7.2 Hz, 2H, CH2), 2.33 (s, 3H, CH3), 1.37 (t, J = 7.2 Hz, 3H, CH3)。
【0150】
(IIb) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(ペミロラスト)
THF(375mL)を-30℃に冷却し、塩化アルミニウム(7.30g、54.7mmol)と、続いてNaN3(10.65g、163.8mmol)を加えた。反応混合物を30分間加熱還流し、その後、5℃に冷却した。2-シアノ-3-(3-メチル-2-ピリジルアミノ)アクリル酸エチル(10.40、45.0mmol; 上記のステップIIaによるもの)を加え、反応混合物を18時間加熱還流した。反応混合物を放冷し、THFを減圧下で除去した。残渣を氷水(210mL)で処理し、6M HClを用いて、pH 3に酸性化した。固体物質を濾過で回収し、DMFから再結晶して、サブタイトルの生成物(4.61g、44.9%)を得た。
LC-MS (M + H+) 229.1
1H NMR (DMSO-d6) δ: 9.21 (s, 1H, CH), 9.16〜9.11 (m, 1H, CH), 8.13〜8.07 (m, 1H, CH), 7.58〜7.51 (m, 1H, CH), 2.62 (s, 3H, CH3)。
【0151】
(IIc) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(ペミロラスト)ナトリウム塩
1M NaOH(20.30mL、20.3mmol)を9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(4.60g、20.1mmol; 上記の工程IIbによるもの)の水(115mL)懸濁液に滴下した。反応混合物を100mLの水で希釈し、50℃に2分間加熱した。溶液を濾過し、水を凍結乾燥で除去した。粗生成物(6.13g)を小分けし、様々な比の水:エタノールから再結晶して、純粋なタイトル化合物を得た。
【0152】
(比較例5)
米国特許第4,122,274号の方法に従ったペミロラストナトリウムの再結晶
米国特許第4,122,274号には、粗タイトル生成物(ペミロラストナトリウム)を水:エタノールから再結晶して、純粋なタイトル生成物を得たことが記載されている。このレベルの詳細からでは、使用された水:エタノールの比がどんなものであったか明らかでなく、したがって先行技術の技法を再現する目的で、いくつかの実験を行った。
【0153】
(i) 粗ペミロラストナトリウム塩(480mg; 上記の比較例4、方法(I)によるもの)を1:1の比の水およびエタノール(95%)から再結晶した。ペミロラストNa塩(480mg、1.92mmol)を70℃でH2O(8mL)に溶解し、EtOH95%(8mL)を加えた。透明な溶液を室温に到達するまで放置し、生成された固体物質を濾取し、少量のエタノールで洗浄し、真空乾燥して、316mgの純粋なナトリウム塩を得た。
【0154】
(ii) 粗ペミロラストナトリウム塩(500mg; 上記の比較例4、方法(II)によるもの)を70℃で水(4.9mL)に溶解した。その後、EtOH 95%(約4.0mL)を固体が生成し始めるまで70℃で加えた。さらに0.1mLの水を加えて、すべてを溶液にした。冷却すると生成した固体物質を濾過で回収し、真空乾燥して、348mgの純粋なナトリウム塩を得た。
【0155】
(iii) 粗ペミロラストナトリウム塩(300mg; 上記の比較例4、方法(II)によるもの)を70℃で水:エタノール(1:1の比; 10mL)から再結晶した。冷却すると生成した固体物質を濾過で回収し、真空乾燥して、174mgの純粋なナトリウム塩を得た。
【0156】
(iv) 粗ペミロラストナトリウム塩(300mg; 上記の比較例4、方法(II)によるもの)を70℃で水:エタノール(9:1の比、4mL)から再結晶した。冷却すると生成した固体物質を濾過で回収し、真空乾燥して、219mgの純粋なナトリウム塩を得た。
【0157】
純粋なペミロラスナトリウム塩の4つの試料はすべて、同じ物理化学的特性(ラマンスペクトルおよびNMR)を有するものであった:
1H NMR (D2O) δ: 8.86〜8.80 (m, 1H, CH), 8.57 (s, 1H, CH), 7.68〜7.59 (m, 1H, CH), 7.22〜7.13 (m, 1H, CH), 2.39 (s, 3H, CH3)。
【0158】
PXRDパターン(上記の比較例5(i)について測定された)を図3に示す。このことから、ナトリウム塩のこの形態は、結晶性部分と混合された非晶質物質であるというのが結論であった。
【0159】
ラマンスペクトルを再結晶直後に記録した。次いで、すべての試料をドラフトの棚に周囲条件下で貯蔵した。約1か月後に、ラマンスペクトルを記録し、先に記録されたものと有意差があった。これは、図4に示されており、下側のスペクトルは先の測定と一致し、上側のスペクトルは、後の測定と一致する。これらの結果を考慮に入れて、先行技術のペミロラストナトリウムの非晶質形は、物理的に不安定であるというのが結論であった。
【0160】
非晶質物質は、下記の実施例11に従って得られた形態を40℃で40時間減圧乾燥して、12gの淡黄色綿様非晶質固体を得ることによっても調製された。
【0161】
実施例6〜16の一般手順
Thermal Advantage TGA Q5000IR(TA装置)モジュールを使用して、TGAを測定した。試料(約10〜16mg)を白金パン(100 HI)に入れ込み、窒素パージ下に、加熱速度10℃/分で25から350℃に加熱した。
【0162】
平面偏光を装備したNikon SMZ800顕微鏡を用いて、顕微鏡検査を行った。
【0163】
(290℃に設定された)乾燥炉を装備した装置で、カールフィッシャー電量滴定を実施した。
【0164】
冷蔵冷却システムを装備したThermal Advantage DSC Q1000(TA装置)を使用して、DSCを試験した。装置は、インジウムを使用して温度およびエンタルピーについて較正した。非気密性アルミニウムパンに、約2〜3mgの試料を正確に秤量し、クリンプした。試料を連続窒素パージ(50mL/分)下に、加熱速度10℃/分で25から275℃までスキャンした。
【0165】
試料のPXRDパターンは、Siemens D5000粉末回折計でCuK線(1.540 56Å)を使用して収集した。管電圧およびアンペア数を40kVおよび40mAにそれぞれ設定した。発散スリットおよび散乱線除去スリットの設定は、20mmの試料領域の照明のため、可変である。各試料を2θにおいて5〜40°の範囲をステップサイズ0.02°でスキャンした。1ステップ当たりの測定時間は1秒であり、分析中、試料台は30rpmで回転し(粉末試料)、または回転しなかった(錠剤)。小容積のゼロバックグラウンドホルダーも使用した。シリコン標準物質を使用して、装置を事前に較正した。(下記の実施例に従って調製された形態は、関連するパターンから(実験誤差を考慮して)、同じ結晶形が生成されていたことが明らかであったとき、下記に開示される他の実施例と「本質的に」同じPXRD回折パターンを示した。したがって、PXRD間隔値の実験誤差の限界は、本明細書のいずれかの部分で使用されているように、最後の小数位で±2ぐらいの範囲とすることができる。)
【0166】
Agilent XDB C18 50×4.6、1.8μmカラムを使用して、HPLC-UV(実施例16)を行った。カラムオーブンの温度は40.0℃であり、流速は1mL/分であり、検出はUV/VISにより370.0 nmで行った。注入量は10μLであった。Chromolith Performance RP-18 100×4.6mmカラム(Merck)を使用して、HPLC-UV(実施例10)を行った。カラムオーブンを周囲温度に設定し、流速は3mL/分であり、検出はUVにより254nmで行った。HPLC法は両方とも、移動相Aの0.1%TFA(水溶液)および移動相Bのアセトニトリルを使用した。
【0167】
(実施例6)
ペミロラストナトリウム半水和物
ペミロラスト遊離酸(1g; 対応するカリウム塩(Chemtronica AB、Stockholm、Sweden)から、水に溶解し、酢酸で酸性化し、その結果沈殿した遊離酸を濾取し、乾燥することによって調製された)を下記の表3に記載される一連の選択された有機溶媒または溶媒混合物(15mL)に懸濁した。その後、得られたスラリーを50〜60℃に加熱し、塩形成剤(1当量の水酸化ナトリウム(50%水溶液もしくは8%メタノール溶液)またはナトリウムエトキシド(21%エタノール溶液))を加えた。固体の部分溶解が認められた。しかし、数秒以内に、新しい沈殿物が得られ、スラリーがより濃厚になった。スラリーを50〜60℃で1〜2時間平衡化し、次いで室温に冷却し、濾過した。固体を室温および大気圧で、1〜2日で乾燥した。
【0168】
【表3】
【0169】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0170】
上記のメタノールおよびイソプロパノール結晶化によって得られた結晶を、DSCおよびTGAで分析し、すべて半水和物であることが確認された。
【0171】
(比較例7)
ペミロラストナトリウム七水和物
ペミロラスト遊離酸(3g; 上記の実施例6に記載のとおりに調製された)を水(30mL)に懸濁し、スラリーを約50℃に加熱した。1当量の50%NaOH(水溶液)を加え、その結果透明な溶液が得られた。溶液を冷却した。溶液温度約40℃で、塩は結晶化し始め、非常に濃厚なスラリーが得られた。スラリーを希釈するために、水(80mL)を追加した。最後に、スラリーを0℃に冷却し、1時間平衡化し、続いて濾過した。固体を室温および大気圧で1〜2日間乾燥して、4.04gのタイトル化合物を得た。
【0172】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例11に従って得られた形態によって示されたものと本質的に同じであった。
【0173】
(実施例8)
ペミロラストナトリウム半水和物
ペミロラストナトリウム七水和物の試料(3.5g、上記の実施例7に記載された方法に従って得られた)を水(3.0mL)に懸濁し、約80℃に加熱した。スラリーをこの温度で濾過し、次いで室温および大気圧で乾燥した。収量は1.3gであった。
【0174】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0175】
(実施例9)
ペミロラストナトリウム半水和物
(a) ペミロラスト
遊離酸を、ペミロラストカリウム(15.8g; Chemtronica AB)から、水(100mL)とTHF(80mL)の混合物に室温で溶解し、酢酸(1当量; 17.5g; 20%水溶液)で酸性化することによって調製した。まず、約1mLの酢酸を添加し、得られた希薄なスラリーを約30分間平衡化した。その後、残りの酢酸をゆっくり加えた。得られた濃厚なスラリーを水(50mL)で希釈し、2時間平衡化し、次いで濾過し、水で洗浄した。得られた固体を40℃および減圧下で5〜10時間乾燥して、7.5gのサブタイトル化合物を得た。
【0176】
(b) ペミロラストナトリウム半水和物
遊離酸(1g; 上記のステップ(a)によるもの)をエタノール(13.5mL)と水(1.5mL)の混合物に懸濁し、懸濁液を60℃に加熱した。ナトリウムエトキシド(1当量; 1.43g; 21%エタノール溶液)を加え、その結果固体の部分溶解によって、スラリーが生成した。ナトリウム塩は直ちに結晶化し、新しいスラリーが生成した。スラリーを60℃で約1時間平衡化し、その後20〜25℃に冷却し、濾過した。濾過ケーキをエタノールで洗浄し、室温および大気圧で1〜2日間乾燥して、0.99gのタイトル化合物を得た。
【0177】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0178】
(実施例10)
ペミロラストナトリウム半水和物-大規模生産
(a) ペミロラスト
水(2,500mL)とTHF(1,400mL)の混合物に、室温で1時間撹拌しながらペミロラストカリウム(190g; 0.71mol; Chemtronica AB)を溶解した。次いで、混合物のクリア濾過を行い、水(400mL)中酢酸(43g、0.72mol)を2つに分けて加えた。まず、約50mLの混合物を加え、得られたスラリーを30分間撹拌した。次いで、残りの酢酸溶液をゆっくり加えた。得られたスラリーを2時間にわたって平衡化し、濾過し、水で洗浄した。サブタイトル化合物を終夜風乾し、次いで真空オーブン中、40℃で24時間乾燥した。収量は164g(白色固体、硬質塊)であった。
【0179】
(b) ペミロラストナトリウム半水和物
遊離酸(45.5g、0.2mol; 上記のステップ(a)によるもの)をエタノール(630mL)に懸濁し、混合物を57℃に加熱した(内部温度)。水(72mL)中水酸化ナトリウム(8.0g、0.2mol)を加えた。固体の部分溶解によって、ほとんど直ちにスラリーが生成し、続いてその結晶化が起こった。スラリーを57℃で1時間にわたって平衡化し、次いで20〜25℃に冷却し、濾過した。濾過ケーキをエタノールで洗浄し、真空中、室温で48時間乾燥した。タイトル化合物は淡黄色粉末として得られ、その収量は45gであった。純度は、HPLC-UVにより>99%であると判定された。
【0180】
結晶をPXRDで分析した。関連性のあるスペクトルを図5に示し、主ピークを下記の表4にまとめる。(この分析から、実施例10に従って得られた形態は、実施例1および2に従って得られたものと同じであるが、前者の試料は、出発材料(ペミロラスト遊離酸)、不純物、および/または副生物を含み得るものであったことが明らかである。)
【0181】
【表4】
【0182】
(比較例11)
ペミロラストナトリウム七水和物-大規模生産
ペミロラスト遊離酸(80g、0.35mol; 上記の実施例10(a)を参照のこと)を水(3L)に懸濁し、50℃に加熱した。水(14g)中水酸化ナトリウム(14g、0.35mol)を加え、得られた溶液のクリア濾過を行った。溶液を20℃に冷却した。生成物が、約25〜28℃で自発的に結晶化し始めた。スラリーを20℃で30分間平衡化し、次いで0℃に冷却した。スラリーを0℃で2時間平衡化し、濾過し、氷-冷水(400mL)で洗浄した。残っている湿った物質を大気圧、45℃、および75%相対湿度で乾燥した。結晶は一緒に溶融し、飴様物質が得られた。この物質を2.5Lの水に50℃で溶解し、結晶化手順を繰り返した。得られた沈殿物を濾過し、乾燥し、約40分間それを通して吸気し、次いで大気圧、25℃、および相対湿度60%で乾燥した。収量は80gの淡黄色固体であった。
【0183】
実施例11によって得られた形態のPXRDパターンを図7に示し、主ピークを下記の表5にまとめる。
【0184】
【表5】
【0185】
DSC(実施例11によってではなく、以前に調製された試料に関する)によって、2つの吸熱(1つは71.1℃、1つは90.2℃)を示した。
【0186】
(実施例12)
ペミロラストナトリウム半水和物
ペミロラストナトリウム半水和物(0.35g; 実施例10に記載された方法に従って得られた)を水(0.8g)に加えた。試料を85〜90℃に加熱した。得られた透明な溶液を75℃に冷却し、熱エタノール(75℃)を2mLずつ5分ごとに加えた。6mLを加えたところで、結晶化が始まった。さらに6mLを10分かけて加えた。試料を20℃に冷却し、濾過し、室温でおよび大気圧で終夜乾燥して、0.19gのタイトル化合物を得た。単離された沈殿物を平面偏光を装備した光学顕微鏡で検査し、PXRDで分析した。関連するスペクトルは、実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0187】
(実施例13)
結晶化実験
ペミロラストナトリウム半水和物(0.2〜0.4g; 実施例10に記載された方法に従って得られた)を、10mLのエタノールおよび水、ならびにそれらの比80:20、60:40、40:60、および20:80の混合物に加えた。試料を50〜70℃に加熱した。透明な溶液が、0:100、20:80、40:60、および60:40(エタノール:水の比)で得られた。80:20および100:0(エタノール:水の比)では、未溶解の結晶を沈降させ、透明な溶液をデカンテーションした。次いで、透明な溶液を放置して、20℃までゆっくり冷却させた。
【0188】
得られた沈殿物の目視検査を、平面偏光を装備した光学顕微鏡で実施した。上記の実施例10(ペミロラストナトリウム半水和物; キューブ結晶として出現する)または上記の実施例11(ペミロラストナトリウム七水和物; 針様結晶として出現する)に記載された方法に従って得られ、完全に特徴付けられた結晶形の公知の物理的外見に基づいて、2つの結晶形のうちのどちらが得られたかについて判定を行った。
【0189】
その検査が実施された後、再び試料を(すべての場合に)完全な溶解が認められるまで加熱した。溶液を再び放置して、約35℃までゆっくり冷却させ、(上記の実施例10に記載された方法に従って得られた)ペミロラストナトリウム半水和物結晶と(上記の実施例11に記載された方法に従って得られた)ペミロラストナトリウム七水和物結晶との1:1混合物を種結晶として加えた。最後に、試料を20℃に冷却し、得られた沈殿物を上述された平面偏光を装備した光学顕微鏡で検査した。
【0190】
その結果を下記の表6にまとめる。
【0191】
【表6】
【0192】
この後、ペミロラストナトリウム半水和物(0.2〜0.4g; 実施例10に記載された方法に従って得られた)を10mLのエタノール:水の80:20、85:15、90:10および95:5の比混合物に加えた。試料を50〜70℃に加熱した。未溶解の結晶を沈降させ、透明な溶液をデカンテーションした。次いで、透明な溶液を放置して、20℃までゆっくり冷却させ、得られた沈殿物を上述された平面偏光を装備した光学顕微鏡で検査した。
【0193】
その検査が実施された後、再び試料を(すべての場合に)完全な溶解が認められるまで加熱した。溶液を放置して、約50℃の温度にゆっくり到達させ、次いで種結晶を添加することなく行われた実験で得られたものとは反対の形態の結晶を種結晶として添加した。最後に、試料を20℃に冷却し、得られた沈殿物を上述された平面偏光を装備した光学顕微鏡で検査した。
【0194】
その結果を下記の表7にまとめる。
【0195】
【表7】
【0196】
この実験の結論は、有機溶媒中、約10%以下の水の存在下で部分溶解することによって、ペミロラストナトリウム半水和物を調製できるということである。
【0197】
(実施例14)
安定性試験
実施例10(半水和物)、実施例11(七水和物)、および実施例5(非晶質形)に記載された方法に従って得られたペミロラストナトリウムの相対安定性を様々な水分レベルで決定するために、安定性試験を行った。
【0198】
試験は、4つの異なる貯蔵条件下、40℃/75%RH、25℃/60%RH、室温/10〜20%RH、および室温/>90%RHという異なる水分レベルおよび温度で行った。物質をガラスのビーカーに開放して貯蔵し、分析のため試料を定期的(時間0、1週間、および4週間)に抜き取った。
【0199】
記載された時間間隔で、(a) スパーテル1杯の物質(物質のスミアリングまたは疎砕を行わなかった)を白地のmunktellフィルターに置き、(b) 平面偏光を装備した顕微鏡で検査することによって外観を検査した。含水率も、カールフィッシャー滴定で分析した。PXRDを0時点および4週目の時点で実施した。
【0200】
試験によって、半水和物形は、相対湿度60%以下(10〜20%RHに至るまで調査)で4週まで貯蔵したとき、安定である(固相)ことが実証された。光学顕微鏡検査を含めて、外観は、これらの貯蔵条件下で不変のままであった。水の分析(カールフィッシャー)は、これらの条件下で不変の結果を示した(2.8%+/-0.2)。
【0201】
75%RH以上において、半水和物が今まで知られていなかった固体状態の形態に変換した(図7に示されるPXRDパターンによって実証されるように、その主ピークを下記の表8にまとめる)。
【0202】
【表8A】
【0203】
【表8B】
【0204】
この形態は、七水和物とほぼ同じ量の水を含有する(ほぼ30%(w/w))。
【0205】
一方、七水和物形は、1つの調査条件の60%RHでしか4週間まで安定でなかった。乾燥条件(10〜20%RH)下で、七水和物形は、半水和物形に変換された。より高い湿度レベル(75%RH)では、試料は、図7に特徴付けられる上記の新しい固体状態の形態に部分的に変換した。しかし、90%RHでは、相転移の徴候は認められなかった。
【0206】
非晶質形は、60%RH以上の貯蔵条件下で結晶化したが、10〜20%RHの乾燥条件下では、非晶質のままである。60%RH以上では、七水和物と、図7および上記の表8(表)に特徴付けられる上記の新しい固体状態の形態の2つの固体形が、貯蔵後4週間出現した。
【0207】
(実施例15)
錠剤の妥当性試験
(A) 5mg錠剤
まず、プラセボバッチ81002-1002-16を生成し、ロータリー式打錠機(Korsch PH106)の調整に使用した。
【0208】
次いで、表9にまとめた医薬品添加物を含む半水和物含有錠剤1000個を1バッチ生成した。試験中、温度および相対湿度をモニターした。この場合、RHは12.7%であった。上記の実施例10に記載されている方法に従って、半水和物が得られ、乾式混合する前に、500μmの篩に通すことによって篩過した。下記に列挙された医薬品添加物(ステアリン酸マグネシウムを除く)との混合は、Turbulaミキサーで10分間実施した。この後、ステアリン酸マグネシウムを500μmの篩に通し、混合物に加え、続いてさらに2分間混合する。ロータリー式打錠機(Korsch PH 106)で、25rpmで直径7mmの丸形凹型パンチを使用して、錠剤を生成した。低、中、および高(それぞれ、ほぼ3、4、および7kN)の3つの圧縮力を評価した。
【0209】
【表9】
【0210】
同じ方法を使用して、12.2%RHで次の医薬品添加物(下記の表10を参照のこと)を含む7水和物含有錠剤1000個を1バッチ生成した。上記の実施例11に記載された方法に従って、七水和物を得た。
【0211】
【表10】
【0212】
(B) 0.2mgミニ錠剤
上記の(A)に記載されたのと同じ方法を使用して、11.6%RH(半水和物)および12.2%RH(七水和物)で、次の医薬品添加物(下記の表11および表12)を含む半水和物含有錠剤および七水和物含有錠剤5000個を別個の2バッチ生成した。上述のように、まずプラセボを生成し、打錠機調整に使用した。直径3mmの丸形凹型パンチを、打錠機において1つの圧縮力(ほぼ400N)で使用した。
【0213】
【表11】
【0214】
【表12】
【0215】
(C) 30mg錠剤
上記の(A)に記載されたものと同様の方法を使用して、8.0%RH(半水和物)および10.2%RH(七水和物)で、次の医薬品添加物(下記の表13および表14)を含む半水和物含有錠剤および七水和物含有錠剤250個を別個の2バッチ生成した。
【0216】
含量均一性を改善する試みとして、まず活性物質を500μmの篩の代わりに250μmの篩で篩過した。次いで、活性物質を手でMCCと前もって混合した。直径6mmの円板パンチを打錠機で使用した。約31%の活性物質を含有する半水和物製剤では(活性物質の不足のため、計画されたものから変更した)、2つの圧縮力(5kNおよび14kN)を使用した。約47%の活性物質を含有する七水和物製剤では、粉体流の不足というだけで、評価され得る圧縮力が1つになった(約2kN)。この圧縮力は、七水和物製剤のフロー特性のため、可変性の高いものであった。
【0217】
【表13】
【0218】
【表14】
【0219】
すべての錠剤について、脆弱性および破砕抵抗性試験を標準方法に従って実施した(それぞれPh. Eur. 6.0, 2.9.7および2.9.8、破砕抵抗性の結果をPh. Eur.によるNではなくkpで表すことを除く)。30mg錠剤についても、破砕した錠剤と無傷な錠剤とを打錠した後PXRDを行った。
【0220】
結果
七水和物は、粘性/石鹸様フレークに似ていることが認められ、500μmの篩を通過するのは本当に困難であることがわかった。篩過した後、何らかの物質は篩に残ったままであった。これは、半水和物の場合には経験されなかったことである。ほとんどの場合、適度な重量バラツキ、脆弱性、および含量均一性と同様、許容できる圧縮力/破砕強度曲線が認められた。
【0221】
しかし、30mg錠剤では、主に混合粉体(固体)のフロー特性が不良であるため、七水和物製剤の打錠プロセスが簡単ではなく、結果として錠剤重量変動性が高くなった。このバラツキは、また圧縮力のバラツキを生じた(これは、下側パンチと上側パンチの間の一定の距離に調整されているので、パンチ間の距離が一定のダイ中の粉体が少なくなると、圧縮力が低下する)。また、パンチ表面への接着が、錠剤表面の損傷に伴って起こり、その結果僅かな脆弱性が生じる。
【0222】
七水和物についてDSCで観察(および上記の実施例11に記載)された吸熱を鑑み、これは、(内部温度70℃が、圧縮プロセス中に容易に到達することができることを考えれば)その多形体が圧縮によって錠剤になる能力を限定することができるものと予想された。したがって、30mg錠剤を上述の通りPXRDで分析するよう決定された。PXRD分析から、半水和物製剤についても七水和物製剤についても、打錠時に相転移は起こらなかったことがわかった。
【0223】
(実施例16)
溶解試験
インビトロ溶解試験をペミロラストカリウム(入手先Chemtronica AB、Stockholm、Sweden)およびペミロラストナトリウム半水和物(上記の実施例10に記載された方法に従って調製された)について、標準Ph. Eur. Methodologyを使用して行った(Apparatus 2(paddle)、Ph. Eur., 2.9.3、50mMホスファートバッファーpH 6.8を溶解媒体として使用した。撹拌速度50rpmを用いた。温度は37℃であった)。
【0224】
試験は、3回繰り返して行った。各物質30mgを別個の容器に加え、タイマーを開始した。目視検査サンプリングは、1mLのプラスチック注射器を使用して、(目視検査によって)物質がいかに迅速に溶解するように見えるかに応じて、一定間隔で行った。小さい直径のフィルターを使用して、試料をHPLCバイアルに濾過した。試料を逆相HPLCで分析した。ペミロラスト定量をUV検出によって行った。
【0225】
結果
カリウム塩は溶解媒体に加えられるとすぐに溶解した一方、ナトリウム塩の半水和物は大きな凝集体を形成し、溶解がより困難であるように思われた。2つの物質について得られた3つの溶解曲線の平均(±SD)(溶解対時間の百分率)を図8に示す。
【技術分野】
【0001】
本発明は、薬物の新しい固体状態の形態、それらを含有する医薬組成物、およびそれらを得る方法に関する。
【背景技術】
【0002】
薬物組成物の製剤化において、原薬は、好都合に取扱いおよび加工を行うことができる形態であることが重要である。これは、商業的に実現可能な製造方法を得るという点からだけでなく、活性化合物を含む医薬製剤のその後の製造という点から見ても重要である。
【0003】
活性成分の化学的安定性、固体状態安定性、および「貯蔵寿命」も極めて重要な因子である。原薬、およびそれを含有する組成物は、活性成分の物理化学的特性(例えば、その化学組成、密度、吸湿性および溶解性)に大きな変化を示すことなく、かなりの期間にわたって有効に貯蔵することができるべきである。
【0004】
さらに、化学的にできるだけ純粋な形態の薬物を提供することも重要である。
【0005】
この点で、非晶質または半非晶質材料は重大な問題を提起する恐れがある。例えば、このような材料は、典型的には取扱いおよび製剤化が困難であり、信頼性のない溶解性をもたらし、不安定でありかつ化学的に不純であることがしばしば認められている。
【0006】
上記の問題は、薬物を安定した結晶形で容易に得ることができる場合に解決可能であることを当業者は理解するであろう。
【0007】
さらに、結晶性薬物化合物は、患者に投与された後、信頼性および再現性のより高い血漿中濃度プロファイルを実現することがわかっている。
【0008】
したがって、商業的に実現可能でかつ薬学的に許容される薬物組成物の製造において、可能な場合はいつでも、実質的に結晶性でかつ安定な形態の薬物を実現することが重要である。
【0009】
しかし、この目的は、必ずしも実現可能ではないことに留意されたい。実際、典型的には、化合物の結晶化挙動がどうなるかを分子構造だけから予想することは可能でない。これは、通常実験に基づいてしか決定することができない。
【0010】
ペミロラストは、喘息、アレルギー性鼻炎、および結膜炎などの病態の治療で使用される、経口投与で活性な抗アレルギー薬である。例えば、米国特許第4,122,274号、欧州特許出願公開第316 174号および同第1 285 921号、Yanagiharaら、Japanese Journal of Pharmacology、51、93(1989年)およびDrugs of Today、28、29(1992年)を参照のこと。この薬物は、例えば日本で、ALEGYSAL(商標)という商標でカリウム塩として現在市販されている。
【0011】
市販のペミロラストカリウムは、ヒトにおいて鋭い血漿中濃度ピークを生じることが知られているという欠点がある(例えば、Kinbaraら、「Plasma Level and Urinary Excretion of TBX in Humans」、Japanese Pharmacology & Therapeutics、18(3)(1990年)、および「Antiallergic agent - ALEGYSAL tablet 5mg - ALEGYSAL tablet 10mg - ALEGYSAL dry syrup」、Pharmaceutical Interview Form(IF)、2007年10月改訂(第7版)、Standard Commodity Classification No.:87449を参照のこと)。後者の文献には、ペミロラストのカリウム塩は吸湿性であることも報告されており、これにより、化学的不安定性がもたらされ、苦味をもつと考えられる。
【0012】
米国特許第4,122,274号には、カリウム塩および(実施例14における)ナトリウム塩を含む、ペミロラストの塩の生成方法が記載されている。本明細書に記載されているように、この技法により、物理的に不安定なナトリウム塩が生成される。ペミロラストのナトリウム塩は、国際公開第2008/074975号および同第2008/075028号にも記載されている(しかし、その合成法は記載されていない)。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】米国特許第4,122,274号
【特許文献2】欧州特許出願公開第316 174号
【特許文献3】欧州特許出願公開第1 285 921号
【特許文献4】国際公開第2008/074975号
【特許文献5】国際公開第2008/075028号
【非特許文献】
【0014】
【非特許文献1】Yanagiharaら、Japanese Journal of Pharmacology、51、93(1989年)
【非特許文献2】Yanagiharaら、Drugs of Today、28、29(1992年)
【非特許文献3】Kinbaraら、「Plasma Level and Urinary Excretion of TBX in Humans」、Japanese Pharmacology & Therapeutics、18(3)(1990年)、
【非特許文献4】Kinbaraら、「Antiallergic agent - ALEGYSAL tablet 5mg - ALEGYSAL tablet 10mg - ALEGYSAL dry syrup」、Pharmaceutical Interview Form(IF)、2007年10月改訂(第7版)、Standard Commodity Classification No.:87449
【非特許文献5】Marzら、Circulation、110(2004年)
【非特許文献6】Nicklasら、CMAJ、172、1199(2005年)
【非特許文献7】Ridkerら、N. Engl. J. Med., 352, 20(2005年)
【非特許文献8】RobbinsおよびCotran、Pathologic Basis of Disease、第8版、Saunders Elsevier
【非特許文献9】Remington The Science and Practice of Pharmacy、第19版、Mack Printing Company, Easton, Pennsylvania(1995年)
【非特許文献10】Martindale - The Complete Drug Reference(第35版)およびその中の引用文献
【非特許文献11】Sanoおよびlshihara、Heterocycles、48、775(1998年)
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明者らは、今回、ペミロラストの安定な結晶性ナトリウム塩を生成することが可能であること、また予想外なことに、このような塩が、対応する先行技術のペミロラストカリウム塩より、水性媒体に可溶でなく、かつ吸湿性でないことも見出した。したがって、このような塩は、ペミロラストカリウムについて上述した鋭い血漿中濃度ピークも、芳しくない味および吸湿性の問題も生じないものと予想される。
【課題を解決するための手段】
【0016】
本発明の第1の態様によれば、ペミロラストナトリウム塩の半水和物形(以下に、「本発明の化合物」と呼ばれる)が提供される。本発明の化合物は、別の場合には「ペミロラストナトリウム半水和物」と呼ばれることがある。
【0017】
ペミロラストナトリウム塩、ならびに/あるいはペミロラスト1モル当たり少なくとも約0.3モル、好ましくは少なくとも約0.35モル、より好ましくは少なくとも約0.4モル、および/または約0.7モル以下、好ましくは約0.65モル以下、より好ましくは約0.6モル以下の水(約0.5モルの水など)をこのような水が密接に結合していようと緩く結合していようと(すなわち、結晶水またはその他の形)含有する本発明の化合物もさらに提供される。
【0018】
本発明者らは、本発明の化合物を自然状態において実質的に結晶性である形態で容易に得ることができることを見出した。
【0019】
結晶化度が約98%を超える、例えば約95%を超える形態の本発明の化合物を生成することが可能であるが、「実質的に結晶性」について、本発明者らは、結晶化度が約60%を超える、好ましくは約75%を超える、より好ましくは約80%を超える(約90%など)形態を含める。結晶化度(%)は、粉末X線回折(PXRD)を用いて当業者が決定することができる。固体NMR法、FT-IR法、ラマン分光法、示差走査熱量測定法(DSC)、マイクロカロリメトリー法、および真密度の算出など、他の技法も使用することができる。
【0020】
好ましい本発明の化合物は、2θ値(単位:度)およそ(すなわち、約またはほぼ)26.6の特有の結晶性ピーク(図1/表2において、およそ26.5である)を含み、好ましくは2θ値(単位:度)およそ(すなわち、約またはほぼ)25.3の別の強い結晶性ピーク(図1/表2において、およそ25.2である)も含む粉末X線回折パターンで特徴付けることができる。より好ましくは、本発明の化合物は、2θ値(単位:度)およそ(すなわち、約またはほぼ)13.0(図1/表2において、およそ12.9)、15.3(図1/表2において、およそ15.2)、18.2(図1/表2において、およそ18.1)および/または28.4(図1/表2において、およそ28.3)の追加の強い結晶性ピークを含み; より好ましくは、2θ値(単位:度)およそ(すなわち、約またはほぼ)16.8(図1/表2において、およそ16.7)、19.2(図1/表2において、およそ19.1)、および/または27.0(図1/表2において、およそ26.9)の別の強い結晶性ピークを含み; より好ましくは、2θ値(単位:度)およそ(すなわち、約またはほぼ)14.9および/または29.5の別の強い結晶性ピークを含む、粉末X線回折パターンで特徴付けることができる。
【0021】
好ましい本発明の化合物は、本明細書に添付されている図5(および/または図1)に示され、かつ/または下記の表4(および/または表2)にまとめられているものに本質的に一致している粉末X線回折パターンで特徴付けることもできる。当業者は、ペミロラストナトリウム半水和物の形態が他の粉末X線回折パターンと「本質的に」同じ粉末X線回折パターンを示すことをそれぞれのパターン(すなわち、試料の好ましい配向や各機器の設定(例えば、装置のタイプ、標準化、および/または較正)などの実験誤差を考慮して、それぞれのピーク間隔)から同じ結晶形が形成されていること(それぞれ図5および表4で特徴付けられる形態と図1および表2で特徴付けられる形態との間ではよくあることである)がその当業者に明らかであったときに理解するであろう。したがって、本明細書で指定されているd値の実験誤差の限界は、最後の小数位で±2ぐらいの範囲とすることができる。
【0022】
本発明の化合物は、好ましくは実質的に結晶学的に純粋である。「実質的に結晶学的に純粋」によって、本発明者らは、PXRD測定によって判断できる範囲で、ペミロラストナトリウムの他の結晶形を約5%未満、より好ましくは約3%未満、特に約1%未満しか含有しないペミロラストナトリウム半水和物の結晶形を含める(半水和物の形それともその他の形かどうか、このような他の結晶形に由来するPXRDピークの存在によって判断される)。
【0023】
本発明者らは、本発明の化合物が、米国特許第4,122,274号に記載されているように調製されたものを含む、ペミロラストナトリウム塩の他の形態と比べると、驚くほど改善された物理的および/または化学的安定性を有することを見出した。
【0024】
本明細書に定義する「安定な」という用語は、化学的安定性および固体状態安定性を包含する。
【0025】
「化学的安定性」によって、本発明者らは、化合物が、単独の固体形、または薬学的に許容される担体、希釈剤、もしくは助剤と混合して提供され得る固体製剤の形で、標準貯蔵条件下で、化学的崩壊または分解をほとんど伴うことなく貯蔵できることを含める。
【0026】
「固体状態安定性」によって、本発明者らは、化合物が、単独の固体形、または薬学的に許容される担体、希釈剤、もしくは助剤と混合して提供され得る固体製剤の形で、標準貯蔵条件下で、固体状態変態(例えば、結晶化、再結晶、結晶性損失(loss of crystallinity)、固体状態相転移、水和、脱水、溶媒和、または脱溶媒和)をほとんど伴うことなく貯蔵できることを含める。
【0027】
「標準貯蔵条件」の例としては、長期間(すなわち、6か月以上)にわたる、温度-80〜+50℃(好ましくは0〜40℃、より好ましくは15〜30℃などの周囲温度)、圧力0.1〜2バール(好ましくは大気圧)、および/または460ルクスのUV/可視光への曝露が挙げられる。このような条件下では、本発明の化合物は、適宜に化学的崩壊/分解または固体状態変態を約15%未満、より好ましくは約10%未満、特に約5%未満しか生じないことがわかり得る。温度および圧力の上記の上限および下限は、標準貯蔵条件の両端点を表すものであり、これら端点のある種の組合せ(例えば、温度50℃と圧力0.1バール)は通常の貯蔵時にはないことを当業者は理解するであろう。
【0028】
「標準貯蔵条件」という用語は、相対湿度5〜95%(好ましくは10〜60%)も包含することができる。しかし、本発明によるいくつかの結晶形の場合、水和および/または脱水による立体配座または結晶構造の変化が、標準温度/圧力における相対湿度の特定の端点への長期曝露の結果として生じる可能性がある。
【0029】
本発明者らは、有利にはペミロラストナトリウム塩の少なくとも部分溶解および/または懸濁に続いて結晶化を経て、本発明の化合物を得ることが可能であることを見出した。
【0030】
この点に関して、有利にはペミロラストとナトリウム含有塩基を反応させ、続いて適切な溶媒系から結晶化を行うことによって、本発明の化合物を得ることができる。前記結晶化に先立って、ペミロラストナトリウムを単離することができる。
【0031】
ナトリウム含有塩基は、ペミロラスト分子のテトラゾール部分からプロトンを除去するのに十分な塩基性である必要がある。したがって、好ましい塩基としては、水酸化ナトリウム、ならびに水素化ナトリウム、ナトリウムアミドおよびナトリウムアルコキシドが挙げられる。記載され得るナトリウムアルコキシドとしては、ナトリウムのC1〜6アルコキシド、具体的にはC1〜4アルコキシド、例えばC1〜3アルコキシド、例えばナトリウムエトキシドまたは例えばナトリウムメトキシドが挙げられる。
【0032】
本発明の化合物の適切な溶媒系からの結晶化は、ペミロラストナトリウム塩を含む溶媒系において過飽和を(例えば、冷却、溶媒蒸発、および/または好適な貧溶媒の添加により)達成することによって実現することができる。結晶化は、ナトリウム塩を加えて、溶液のイオン強度を高めること(NaClまたはNaSO4など; いわゆる「塩析」)により物質の溶解性を低下させ、かつ/または種結晶を過飽和溶液に加えること(一度利用可能)によって行うこともできる。
【0033】
溶媒系としては、酢酸アルキル(例えば、酢酸エチル、酢酸iso-プロピル、酢酸ブチルなどの酢酸直鎖状または分枝状C1〜6アルキル)、低級(例えば、直鎖状または分枝状C1〜6、好ましくはC1〜4)アルキルアルコール(例えば、メタノール、エタノール、iso-プロパノール)、脂肪族および芳香族炭化水素(例えば、iso-オクタン、n-ヘプタン、およびトルエン)、ジアルキルケトン(例えば、メチルエチルケトン、アセトン、およびメチルiso-ブチルケトン)、ジアルキルエーテル(例えば、ジ-iso-プロピルエーテルおよびtert-ブチルメチルエーテル)、環状エーテル(例えば、テトラヒドロフランおよびジオキサン)、アセトニトリル、ジメチルホルムアミドなどの1つまたは複数の有機溶媒を挙げることができる。上記溶媒のうちのいずれかの混合物を使用することができる。有機溶媒は水を含有していてもよい。
【0034】
結晶形が異なれば、所与の温度での有機溶媒における溶解性が異なることがある。この点に関して、上記または他の溶媒を「貧溶媒」(すなわち、メタノール、エタノール、またはイソプロパノールなど、本発明の化合物が難溶性であるが、本発明の化合物がより可溶性である別の溶媒(水など)と混和可能である溶媒)として使用することができ、したがって結晶化プロセスを助けることができる。
【0035】
特に好適な溶媒としては、少量の水と混合することができる低級アルキルアルコール(例えば、イソプロパノールなどのC1〜4アルコール、好ましくはメタノール、またはより好ましくはエタノール)が挙げられる。
【0036】
特に、本発明者らは、有機溶媒(例えば、エタノールなどの低級アルキルアルコール)の存在下、さらに約12%以下(w/w、有機溶媒の割合として)、特に約11%以下、特に約10%以下(例えば、約8%未満、具体的には約5%未満、例えば約3%未満)の水の存在下、ペミロラストナトリウムの部分溶解(平衡および/またはスラリー形成とも呼ばれる)に続いて結晶化を経由して、本発明の化合物を得ることができることを見出した。このような水は、結晶化混合物に加えてもよく、あるいは有機溶媒または結晶化させる対象のペミロラストナトリウムのうちの一方または両方にすでに存在してもよい。この結晶化は、半水和物を形成するためのいくらかの水を系中に存在させる必要があるので、完全に無水の条件下で行うことはできないことを当業者は理解するであろう。部分溶解は、(例えば、飽和溶液を生成するための)完全溶解ではなく、したがってこのプロセスは、標準再結晶を含まないことも、当業者は理解するであろう。このプロセスは、約75℃未満、具体的には約72℃、例えば約70℃の温度で実施されることも好ましい。
【0037】
本発明者らは、本発明の化合物を下記によって得ることができることも見出した。
(a) 約70℃超、例えば約72℃、具体的には約75℃、例えば約80℃以上の水性溶媒(すなわち、純水など、少なくとも約95%(w/w)の水を含む)におけるペミロラストナトリウムの部分溶解(平衡および/またはスラリー形成)に続いて、結晶化。このプロセスは、再結晶ではなく、好ましくは上記の温度で濾過して、生成された結晶を単離することを含む; および
(b) 水性溶媒(すなわち、純水など、少なくとも約95%(w/w)の水を含む)におけるペミロラストナトリウムの完全(例えば、少なくとも約95%)溶解と、その後に続く過剰量の貧溶媒(例えば、エタノール)の添加。このプロセスは、好ましくは高温(例えば、およそ溶媒の沸点)における貧溶媒の添加を含む。エタノールの場合、これは、約75℃など、約70℃(例えば、約72℃)〜約80℃であり、結晶の単離前に、より低い温度(例えば、約20℃などの室温)に冷却する。
【0038】
結晶化させる対象の化合物の溶液(および/または部分溶液)中の濃度、および使用される溶媒系は、結晶化温度および結晶化時間に影響を及ぼす可能性があることを当業者は理解するであろう。
【0039】
当業者によって理解され得るように、得られる結晶形は、結晶化プロセスの動力学と熱力学とに依存する。特定の熱力学条件下(溶媒系、温度、圧力、および本発明の化合物の濃度)で、ある結晶形は、別の結晶形(または、実際には他のいかなる結晶形)より安定であり得る。しかし、他の結晶形が、比較して相対的に低い熱力学的安定性を有することができると、動力学的に好まれる。したがって、さらに、時間、不純物プロファイル、撹拌、種結晶の存在などの動力学的因子も、どの形態が出現するかに影響を及ぼすことがある。したがって、ペミロラストナトリウムの半水和物、および実際には(適切な場合は)ペミロラストナトリウム塩の半水和物形の異なる結晶形を得るために、本明細書に述べられる手順を当業者が適宜適応させることができる。
【0040】
本明細書に記載される結晶形が他の結晶形の非存在下で調製されるようにするために、他の結晶形の核および/または種結晶の非存在下で、所望の結晶形の核および/または種結晶を加えることによって、結晶化を実施することができる。
【0041】
さらに、乾燥温度および乾燥時間は、本発明の化合物の固体状態特性および/または固体状態の形態に影響を及ぼすことがある。例えば、脱水は、低湿度および/または高温および/または減圧で行われることがある。例えば、結晶性半水和物が生成した後、臨界湿度レベルが存在し得る。そのレベル未満で、乾燥が行われることがあり、それによって、結晶水が失われ、無水物への少なくとも部分的な固体状態変態が起こり得る。
【0042】
これにも関わらず、例えば本明細書の以下に記載されるペミロラストナトリウムの高水和物(例えば、七水和物)を脱水することによって、本発明の化合物を生成することもできる。
【0043】
本発明の化合物の調製およびキャラクタリゼーションを本明細書の以下に記載する。本発明の化合物の様々な結晶形のキャラクタリゼーションは、例えば本明細書の以下に記載される粉末X線回折(PXRD)法を用いて容易に行うことができる。
【0044】
本発明の化合物は、当業者に周知である技法、例えばデカンテーション、濾過および/または遠心分離を行って単離することができる。
【0045】
本発明者らは、本明細書に記載される結晶化プロセスを使用することによって、化学的純度の高い本発明の化合物を生成することが可能であることを見出した。
【0046】
本発明の化合物が本明細書に記載されたとおりに結晶化されると、得られた化合物は、ペミロラストの他の塩、および/またはペミロラストナトリウム塩の他の形態に比べて、本明細書の以上に記載された改善された化学的および固体状態安定性、ならびに改善された溶解性および吸湿性プロファイルを有する形態をとる。
【0047】
(医薬品製剤および医学的使用)
本発明の化合物は、薬理活性を有しているので有用である。したがって、これらは、医薬品として示される。
【0048】
特に、本発明の化合物は、炎症性病態の治療において有用である。
【0049】
炎症性病態は、典型的には、宿主にとって有益であるより有害である効果をもたらす、免疫防御機構の活性化で特徴付けられる。このような病態は、一般に様々な程度の組織の発赤もしくは充血、腫脹、高熱、疼痛、そう痒、細胞の死および組織の破壊、細胞増殖、ならびに/または機能喪失に伴う。記載され得る炎症性病態としては、動脈炎、糖尿病(1型糖尿病、および好ましくは2型糖尿病を含む)、肥満、メタボリックシンドローム、子宮内膜症、アレルギー(アレルギー性結膜炎およびアレルギー性鼻炎を含む)、強直性脊椎炎、喘息、アトピー性皮膚炎、ざ瘡、皮膚熱傷、酒さ、脂漏性皮膚炎、皮膚潰瘍、慢性閉塞性肺疾患、接触性皮膚炎、膀胱炎、痛風性関節炎、炎症性腸疾患(クローン病や潰瘍性大腸炎など)、多発性硬化症、変形性関節症、膵炎、前立腺炎、乾癬、乾癬性関節炎、関節リウマチ、腱炎、滑液包炎、シェーグレン症候群、全身性エリテマトーデス、ブドウ膜炎、蕁麻疹、血管炎、糖尿病性血管合併症、偏頭痛、アテローム性動脈硬化症およびそれに伴う心血管障害が挙げられる。記載され得る病態としては、アトピー性皮膚炎、子宮内膜症、偏頭痛、喘息、慢性閉塞性肺疾患、クローン病、多発性硬化症、乾癬、関節リウマチ、全身性エリテマトーデス、潰瘍性大腸炎、さらに詳細にはアテローム性動脈硬化症およびそれに伴う心血管障害が挙げられる。記載され得る他の病態としては、メタボリックシンドローム、肥満、糖尿病および/または糖尿病性血管合併症が挙げられる。
【0050】
「アテローム性動脈硬化症」という用語は、当業者によって、血管、特に動脈壁におけるコレステロール蓄積、泡沫細胞形成、炎症および細胞増殖で特徴付けられる任意の疾患を包含するものと理解される。アテローム性動脈硬化症「に伴う」心血管障害は、大動脈瘤(腹部大動脈瘤および/またはアテローム動脈硬化性大動脈瘤を含む)、動脈硬化症、末梢動脈閉塞性疾患、冠動脈疾患(例えば、狭心症、心筋梗塞、心臓発作など)、冠疾患(心臓病および心疾患を含む、虚血性心疾患など)を包含し、プラークもしくはアテローム破綻および/または不安定性、血管もしくは動脈疾患、虚血性疾患/虚血、ならびに脳卒中(脳血管障害および一過性脳虚血発作を含む)も包含することができる。
【0051】
記載され得る患者群としては、急性冠症候群患者が挙げられる。「急性冠症候群」という用語は、胸痛および/または心電図(ECG)異常に伴うことが多いがこれに限らない任意の心筋虚血の異常状態(abnormal myocardial ischaemic state)を包含すると、当業者によって理解されよう。このような症候群は、心筋梗塞(心臓発作)の最も一般的な症状である。この用語は、「安定狭心症」(すなわち、労作時に発症し、安静時に消失する狭心症)とは対照的に「不安定狭心症」という用語とおおむね同義であることを当業者は理解するであろう。発生するペースがますます悪化する労作性狭心症(「漸増性狭心症」)は同様に、当業者によって「不安定」の定義の範囲内であるとみなされるであろう。
【0052】
本発明の別の態様によれば、炎症性障害、特にアテローム性動脈硬化症および/またはそれに伴う心血管障害、具体的には大動脈瘤の治療方法であって、このような治療を必要とする患者への本発明の化合物の投与を含む方法が提供される。
【0053】
記載され得る他の炎症性病態としては、文献中で様々に「低グレード全身性炎症」、「無症候性全身性炎症」、「慢性低グレード炎症」、「持続性低グレード炎症」、または文脈に応じて、単に「低グレード炎症」もしくは「全身性炎症」とも呼ばれる病態を包含するように理解される全身性低グレード炎症(SLGI)が挙げられる(例えば、Marzら、Circulation、110(2004年)およびNicklasら、CMAJ、172、1199(2005年)を参照のこと)。他の炎症マーカー(例えば、血中サイトカイン、接着分子、および白血球)がSLGIを示すことが知られており、本発明によれば測定および低減され得るが、SLGIは常に、とりわけ血漿C反応性タンパク質(CRP)値を特徴とし、その値は(例えば、他の点では外見上健常でかつ/または非アレルギー/非喘息哺乳類対象において)約10mg/L未満であるが、約7mg/L超、例えば約5mg/L超、好ましくは約3mg/L超、より好ましくは約2mg/L超、特に約1mg/L超、さらに特に約0.9mg/L超である。このような血漿CRP値は、薬理学的に有効量の本発明の化合物を投与することによって低減することができる。
【0054】
したがって、患者における血漿CRP値を(本明細書の以上に記載される値のいずれか1つの値未満に)低減するための本発明の化合物、および本発明の化合物を患者に投与することを含む、患者における血漿CRP値を(本明細書の以上に記載される値のいずれか1つの値未満に)低減する方法もさらに提供される。
【0055】
SLGIは、例えばメタボリックシンドローム、糖尿病(例えば、2型糖尿病)、インスリン抵抗性症候群、肥満、心血管疾患(例えば、アテローム性動脈硬化症、腹部大動脈瘤、および他の心血管イベント)、および一部の癌(例えば、大腸癌)に関連していることが知られている。CRP値の僅かな上昇が、その他の点では外見上健常な対象における疾患の唯一の徴候にもなり得る。
【0056】
CRPの僅かな上昇によって、様々な医学的病態における望ましくない転帰もしくは合併症、または様々な疾患において死に至る可能性も予測することができる。特に、CRPの上昇によって、心血管病的状態および死亡、ならびに/または2型糖尿病の発症を予測することができ、それらの両方のリスクは、本発明によれば、本発明の化合物を使用することによって低減することができる。
【0057】
したがって、患者において心血管病的状態および死亡のリスクを低減(すなわち、予防)し、かつ/または2型糖尿病の発症を低減(すなわち、予防)する方法が提供され、その方法は:
(a) その患者における血漿CRP値を測定すること;
(b) その血漿CRP値が、本明細書の以上に記載される値の1つの値を超える、特に約0.9mg/Lを超えるかどうか決定すること; および
(c) 超えている場合、本発明の化合物をその患者にしばらく適切な投与量で投与して、CRP値を例えば本明細書の以上に記載される関連性のある値まで低減すること
を含む。
【0058】
アメリカ心臓協会(American Heart Association(AHA))および米国疾病管理予防センター(CDC)は、CRPをリスクアセスメントツールとして評価し、1mg/L未満、1〜3mg/L、および3mg/L超の切点を使用して、それぞれ心血管病的状態または死亡を発症する相対リスクのより小さい、平均的な、および大きい対象を特定することを提案した。0.9mg/Lを超える血漿CRPが、心血管イベントのリスク増大の切点としても使用されている(Ridkerら、N. Engl. J. Med., 352, 20(2005年))。
【0059】
「病的状態」という用語は、一般に任意の病的状態(diseased state)、障害(disability)、病気(illness)、および/または健康不良(poor health)を包含すると、当業者によって理解されよう。したがって、「心血管」病的状態は、潜在的な(underlying)心血管合併症の結果として示されるような状態を包含するが、その心血管合併症は、それ自体が肥満、メタボリックシンドローム、(例えば、2型)糖尿病など本明細書の以上に記載される他の病態のうちの1つまたは複数の結果であり得る(下記参照)。
【0060】
2型糖尿病は、末梢組織のインスリンに対する応答性低下(インスリン抵抗性)、ならびにインスリン抵抗性および高血糖に直面してインスリン分泌不足として顕在化するβ細胞機能不全で特徴付けられる障害である(例えば、RobbinsおよびCotran、Pathologic Basis of Disease、第8版、Saunders Elsevierを参照のこと)。2型糖尿病の症状としては、慢性疲労、過剰の尿生成、過度の口渇、および水分摂取の増加が挙げられる。糖尿病の現行の世界保健機関(World Health Organisation)診断基準は、(a) 空腹時血漿ブドウ糖値:少なくとも7.0mmol/L、または(b) 経口ブドウ糖負荷試験(OGTT)での血漿グルコース値:少なくとも11.1mmol/Lである。「2型糖尿病の発症を低減する」によって、本発明者らは、先在病態の進展(例えば、悪化)を予防するためのSLGIの治療に加えて、2型糖尿病の発症の予防を含める。
【0061】
本発明者らは、CRPが0.9mg/Lを超える対象において、ペミロラストが血漿トリプターゼ値を同時に低減することはなく、さらに対象において、血漿CRP値と肥満細胞トリプターゼ値の間に相互関係がないことも見出した。
【0062】
したがって、本明細書に記載されるSLGIと特に関係がある使用および方法は、非アレルギー患者におけるまたは非アレルギー患者に関するものであることが好ましい。「非アレルギー」によって、本発明者らは、免疫系のアトピー性障害の外に現れる徴候を(このような治療を受ける時点では)患者が示さないことを意味する。この点に関して、このような患者は、IgEを介した肥満細胞および/または好塩基球の活性化を含む免疫学的応答で特徴付けられるアレルゲンに対する過敏症の徴候を示さないことがある。非アレルギー患者であるかどうかの決定は、例えば既知のアレルゲンに対する応答について(例えば、皮膚を)検査し、またはアレルゲン特異的IgEの有無およびレベルについて血液を分析することによってルーチンで行うことができる。
【0063】
本明細書に記載されるSLGIと特に関係がある使用および方法は、非喘息患者におけるまたは非喘息患者に関するものであることがさらに好ましい。「非喘息」によって、本発明者らは、気管支がその平滑筋細胞の収縮、気道炎症および呼吸困難によって可逆的狭窄を起こしている、両肺の慢性炎症に対する素因の外に現れる徴候を(このような治療を受ける時点では)患者が示さないことを意味する。喘息は、アレルギー性でも、非アレルギー性でもよい。
【0064】
SLGI治療の好ましい使用および方法としては、患者が喫煙者または元喫煙者であり、その対象者は糖尿病および/もしくはメタボリックシンドロームを患い、または肥満度指数が25を超える、使用および方法が挙げられる。
【0065】
記載され得る他の炎症性病態としては、うっ血性心不全、心房細動、高血圧(本態性肺動脈高血圧および/または門脈高圧を含む); 照射、手術、および/または外傷の結果(炎症、線維症、瘢痕、および癒着を含む); 炎症によって引き起こされた線維症、瘢痕、および/または癒着; 癌、骨粗鬆症、サルコイドーシス、過敏性腸症候群、網膜症(糖尿病網膜症を含む)、加齢黄斑変性、腎症(糖尿病性腎症を含む)、糸球体腎炎(IgA腎炎/腎症を含む)が挙げられる。
【0066】
誤解を避けるために、本発明の文脈においては、「治療(treatment)」「療法(therapy)」および「治療法(therapy method)」という用語は、治療的または姑息的処置を必要とする患者の治療的または姑息的処置、ならびにアテローム性動脈硬化症やそれに伴う心血管障害などの炎症性障害の影響を受け易い患者の予防的処置および/または診断を包含する。
【0067】
「患者」は、哺乳類(ヒトを含む)患者を包含する。
【0068】
本発明の化合物は、薬学的に許容される剤形の化合物を含む製剤の形で、好ましくは、局所(locally)投与もしくは全身投与、例えば経口投与、静脈内もしくは動脈内投与(血管内または他の血管周囲装置/剤形(例えば、ステント)によることを含む)、筋肉内投与、皮膚適用、皮下投与、経粘膜(例えば、舌下または頬側)投与、直腸投与、経皮投与、経鼻投与、経肺(例えば、気管または気管支)投与、局所(topically)投与され、または他の何らかの非経口経路で投与される。好ましい送達様式としては、経口(特に)、静脈内、皮膚、または皮下、経鼻、筋肉内、または腹腔内送達が挙げられる。
【0069】
本発明の化合物は一般に、所期の投与経路および標準的製剤慣行を十分に考慮して選択され得る薬学的に許容される助剤、希釈剤、または担体と混合して1つまたは複数の医薬製剤の形で投与されることになる。このような薬学的に許容される担体は、好ましくは無菌、純粋、非発熱性であり、活性化合物に対して化学的に不活性であり、使用条件下で有害な副作用または毒性を示すことがない。このような薬学的に許容される担体は、本発明の化合物の即放性製剤または放出調節製剤をもたらすこともできる。
【0070】
本発明の化合物は、好適な担体、希釈剤、または助剤と混合する前にさらに処理することができる。例えば、結晶形をミルまたはグラインダーでより小さい粒子に粉砕することができる。
【0071】
好適な医薬製剤は市販のものとすることができ、または他には、文献、例えばRemington The Science and Practice of Pharmacy、第19版、Mack Printing Company, Easton, Pennsylvania(1995年)ならびにMartindale - The Complete Drug Reference(第35版)およびその中の引用文献に記載されており、それらの文献すべての中の関連する開示内容は参照により本明細書に組み込まれる。別の場合では、好適な製剤の調製は、当業者が工夫を凝らすことなくルーチンの技法を用いて実現することができる。
【0072】
製剤中の本発明の化合物の量は、治療対象の病態の重症度および患者、ならびに使用される化合物に依存することになるが、当業者が工夫を凝らすことなく決定することができる。
【0073】
治療対象の障害および患者、ならびに投与経路に応じて、本発明の化合物を、様々な治療上有効量で、それを必要とする患者に投与することができる。
【0074】
しかし、本発明の文脈において、哺乳類、特にヒトに投与される量は、哺乳類における治療効果を合理的な時間枠にわたってもたらすのに十分な量とすべきである。的確な投与量および組成物ならびに最適な送達レジメンの選択は、とりわけ、製剤の薬理学的特性、治療対象の病態の性質および重症度、レシピエントの健康状態および精神的鋭敏さ(mental acuity)、ならびに治療対象の患者の年齢、状態、体重、性別、および応答、疾患の病期/重症度、ならびに患者間の遺伝子の差の影響も受けることになることを当業者は認識するであろう。
【0075】
本発明の化合物は、継続投与または間欠投与することができる(例えば、ボーラス注射による投与)。用量は、投与のタイミングおよび頻度によっても決定され得る。
【0076】
好適な投与量としては、Martindale-The Complete Drug Reference(第35版)およびその中の引用文献などの医学文献に記載されているものが挙げられ、それらの文献すべての中の関連する開示内容は参照により本明細書に組み込まれる。したがって、本発明の化合物の(遊離酸として算出された)好適な投与量は、約0.01mg/体重kg〜約1,000mg/体重kgの範囲である。より好ましい範囲は、経口投与の場合、1日ごとに約0.1mg/kg〜約20mg/kgである。
【0077】
しかし、ペミロラストの好適な投与量は当業者に周知である。例えば、(遊離酸として算出された)経口投与量は、1日当たり約0.5mg〜約900mgなど、約0.1mg〜約1.2gの範囲とすることができる。例えば、1日量の範囲の好適な下限は、約1mg、具体的には約2mg、例えば約5mg、具体的には約10mg、より好ましくは約20mgであり、1日量の範囲の好適な上限は、約200mg、例えば約100mg、具体的には約80mg、より好ましくは約60mgである。したがって、経口1日量は、約2mg〜約60(例えば、約50)mg、具体的には約5mg〜約45(例えば、約40)mg、好ましくは約10mg〜約35(例えば約30)mgとすることができる。好適な個々の投与量は、1日当たり約10mg〜約100mg、具体的には約20mg〜約90mg、例えば約30mg〜約80mgとすることができる。好ましい投与量は、1日約40mg〜約80mgの範囲、特に約60mgである。投与量は、1日1回の投与に基づくものとすることができ、または1日2回もしくは3回(好ましくは、2回)の投与量に(例えば、等量)分割し得ることを当業者は理解するであろう。
【0078】
いずれにしても、医師または他の当業者は、個々の患者に最も適した実際の用量をルーチンで決定することができるであろう。上記の用量は、平均的な場合の例である。もちろん、より高いまたはより低い用量範囲が相応である個々の例が存在することがあり、このような場合も、本発明の範囲内である。
【0079】
本発明の化合物は、本明細書に定義する炎症性障害の治療に有用である1つまたは複数の活性成分と組み合わせることもできる。
【0080】
したがって、本発明の別の態様によれば、
(a) 本発明の化合物; および
(b) 炎症性障害の治療に有用である1つもしくは複数の活性成分、またはその薬学的に許容される塩もしくは溶媒和物
を含む配合剤も提供される。
【0081】
このような配合剤は、本発明の化合物を炎症性障害の治療に有用である活性成分と併用投与することを実現し、したがって別個の製剤(それらの製剤の少なくとも1つが本発明の化合物を含み、少なくとも1つがもう一方の活性成分を含む)として提供することができ、あるいは複合成分製剤として提供する(すなわち、製剤化する)ことができる(すなわち、本発明の化合物および別の活性成分を含む単一の製剤として提供することができる)。
【0082】
したがって、
(1) 本発明の化合物; 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物; および薬学的に許容される助剤、希釈剤、または担体を含む医薬製剤(その製剤は、以下に「複合成分製剤」と呼ばれる); ならびに
(2)
成分(A) 本発明の化合物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤; および
成分(B) 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤を含み、それらの成分(A)および(B)はそれぞれ、他方と併用投与するのに適した形態で提供されている
パーツキットをさらに提供する。
【0083】
本発明の別の態様によれば、上記で定義されたパーツキットを作製する方法が提供され、その方法は、上記で定義された成分(A)を上記で定義された成分(B)と結合させ、したがって2つの成分を互いに併用投与するのに適したものとする工程を含む。
【0084】
2つの成分を互いに「結合」させることについて、本発明者らは、パーツキットの成分(A)および(B)が、
(i) 別個の製剤として(すなわち、互いに関わりなく)提供され得るものであり、その後一緒にまとめられ、併用療法において互いに併用され; または
(ii) 併用療法において互いに併用されるための「配合パック」の別個の成分として一緒に包装および提供され得るものであることを含める。
【0085】
したがって、
(I) 本明細書に定義する成分(A)および(B)の一方と、
(II) その成分を2つの成分の他方と併用するための指示書
を一緒に含むパーツキットもさらに提供される。
【0086】
本明細書に記載されるパーツキットは、反復投与を実現するために、適切な量/投与量の本発明の化合物を含む1つを超える製剤、および/または適切な量/投与量のもう一方の活性成分/塩/溶媒和物を含む1つを超える製剤を含んでもよい。1つを超える製剤(どちらかの活性化合物を含む)が存在する場合、このような製剤は、どちらかの化合物の投与量、化学組成、および/または物理的形態の点から同じでもよく、または異なってもよい。
【0087】
本明細書に記載されるパーツキットに関して、「併用投与」によって、本発明者らは、本発明の化合物および他の活性成分(またはその塩/溶媒和物)を含む各製剤を、関連する病態の治療の過程において順次に、別々に、かつ/または同時に投与することを含める。
【0088】
したがって、本発明による配合剤に関して、「併用投与」という用語は、配合剤の2成分(本発明の化合物および他の活性成分)を一緒にまたは時間的に十分に接近して投与(場合によっては、反復投与)して、本発明の化合物を含む製剤またはもう一方の活性成分を含む製剤が、同じ治療の過程において他方の成分の非存在下に単独で投与(場合によっては、反復投与)される場合より高い薬効が、関連する病態の治療の過程において患者にもたらされることを包含する。配合剤が特定の病態についてより高い薬効を治療の過程においてもたらすかどうかの決定は、治療または予防対象の病態に依存するが、当業者がルーチンで実現することができる。
【0089】
さらに、本発明によるパーツキットに関連して、「併用」という用語は、2つの製剤のうちのどちらかが、もう一方の成分を投与する前、後、および/または同時に投与(場合によっては、反復投与)され得ることを包含する。本文脈で使用される場合、「同時に投与(administered simultaneously)」および「同時に投与(administered at the same time as)」という用語は、個々の投与量の本発明の化合物および他方の活性成分が、互いに48時間(例えば、24時間)以内に投与されることを包含する。
【0090】
本明細書に定義される炎症性障害の治療に有用である活性成分としては、トロンボキサンA2アンタゴニスト、P2Y12アンタゴニスト、PPARγアゴニスト、アンジオテンシンIIの生成および/または作用を抑制する化合物、他の血小板凝集抑制薬、ならびにより好ましくはスタチンが挙げられる。
【0091】
「トロンボキサンA2アンタゴニスト」という用語は、インビトロおよび/またはインビボ試験で、(i) トロンボキサンTP受容体の遮断、(ii) 酵素トロンボキサンシンターゼの抑制、または(iii) 血小板シクロオキシゲナーゼ-1の(例えば、選択的)抑制、それによる例えば血小板凝集の抑制のうちの1つまたは複数によって、トロンボキサンA2の効果を実験に基づいて決定することができる程度に抑制することができる化合物であればいずれも包含する。
【0092】
好ましいトロンボキサンA2アンタゴニストとしては、アスピリン/アセチルサリチル酸、より好ましくはエグアレン、特にオザグレル、さらに特にピコタミド、およびテルトロバン、特にセラトロダスト、さらに特にラマトロバンが挙げられる。
【0093】
「P2Y12アンタゴニスト」という用語は、インビトロおよび/またはインビボ試験で、ADPの血小板受容体P2Y12との結合を実験に基づいて決定することができる程度に(例えば、選択的に)抑制し、それによって血小板凝集を抑制することができる化合物であればいずれも包含する。
【0094】
好ましいP2Y12アンタゴニストとしては、プラスグレル、チカグレロル、特にクロピドグレルが挙げられる。
【0095】
「PPARγ」アゴニストという用語は、インビトロおよび/またはインビボ試験で、実験に基づいて決定することができる程度にペルオキシソーム増殖剤活性化受容体γと結合し、かつ/またはその機能に影響を及ぼすことができる化合物であればいずれも包含する。
【0096】
したがって、好ましいPPARγアゴニストとしては、リボグリタゾン、ナベグリタザル、バラグリタゾン、またはより好ましくはロシグリタゾン、特にピオグリタゾンを含む、チアゾリジンジオンと総称される化合物が挙げられる。記載され得る他のPPARγアゴニストとしては、チグリタザル(chiglitazar)、エタロシブ、ファルグリタザル、ロベグリタゾン(lobeglitazone)、ネトグリタゾン、ソデルグリタザル、ならびに次の医薬品開発コード(developmental drug code)によって文献に定義されているもの:THR-0921(Theracos Inc.)、またはより好ましくはAVE-0847およびAVE-0897(両方ともSanofi-Aventis)、CLX-0921(Calyx Therapeutics)、CS-7017(Daiichi Sankyo Co Ltd)、DRF-11605(Dr Reddy's Laboratories Ltd)、GFT-505(Genfit SA)、GSK-376501(GlaxoSmithKline plc)、INT-131(Amgen Inc; InteKrin Therapeutics)、(LBM-642; セボグリタザル; Novartis AG)、ONO-5129(Ono Pharmaceutical Co Ltd)、(PLX-204; インデグリタザル; Plexxikon Inc)、およびSDX-101が挙げられる。
【0097】
「アンジオテンシンIIの生成および/または作用を抑制する化合物」という用語は、インビトロおよび/またはインビボ試験で、アンジオテンシンIIの生成および/または作用を実験に基づいて決定することができる程度に(例えば、選択的に)抑制することができる化合物であればいずれも包含し、アンジオテンシン変換酵素(ACE)阻害物質、アンジオテンシン受容体遮断薬(ARB)、およびレニン阻害物質を包含すると理解されよう。
【0098】
「アンジオテンシン変換酵素(ACE)阻害物質」という用語は、インビトロおよび/またはインビボ試験で、アンジオテンシンIのアンジオテンシンIIへの変換を実験に基づいて決定することができる程度に(例えば、選択的に)抑制することができる化合物であればいずれも包含する。
【0099】
記載され得るACE阻害物質としては、アラセプリル、ベナゼプリル、カプトプリル、セロナプリル、シラザプリル、デラプリル、エナラプリル、ホシノプリル、ゲモパトリラト、グリコプリル(glycopril)、イドラプリル、イレパトリル(ilepatril)、イミダプリル、リベンザプリル、リシノプリル、ミクロギニン-FR1、ミキサンプリル、モエキシプリル、モエキシプリラート、モベルチプリル、オマパトリラト、プレンチル(Prentyl)、ペリンドプリル、キナプリル、ラミプリル、サンパトリラット、スピラプリル、シネコル(Synecor)、テモカプリル、トランドラプリル、ウチバプリル、ゾフェノプリルおよびザビシプリラトが挙げられる。より好ましいACE阻害物質としては、ベナゼプリル、シラザプリル、イレパトリル、イミダプリル、モエキシプリル、スピラプリル、テモカプリル、およびゾフェノプリル、より好ましくはホシノプリルおよびトランドラプリル、さらに特にエナラプリル、リシノプリル、およびキナプリル、特にカプトプリル、ペリンドプリル、およびラミプリルが挙げられる。
【0100】
「アンジオテンシン受容体遮断薬(ARB)」は、「アンジオテンシンII AT1受容体アンタゴニスト」という用語とおおむね同義であると当業者が理解するものであり、したがってインビトロおよび/またはインビボ試験で、アンジオテンシンII AT1受容体の活性化を実験に基づいて決定することができる程度に(例えば、選択的に)遮断することができる物質であればいずれも包含する。
【0101】
記載され得るARBとしては、アジルサルタン、アジルサルタンメドキソミル、カンデサルタン、カンデサルタンシレキセチル、アンジオカイン(angiokine)であるDival、エリサルタン、エリサルタンカリウム(elisartan potassium)、エプロサルタン、エンブサルタン、フィマサルタン(fimasartan)、フォンサルタン(fonsartan)、イルベサルタン、ロサルタン、ミルファサルタン、オルメサルタン、ポミサルタン、プラトサルタン、リピサルタン、サプリサルタン、サララシン、タソサルタン、テルミサルタン、バルサルタン、およびゾラサルタンが挙げられる。より好ましいARBとしては、アジルサルタン、エプロサルタン、フィマサルタン(fimasartan)、およびプラトサルタン、より好ましくはテルミサルタン、さらに特にイルベサルタン、およびオルメサルタン、特にカンデサルタン、ロサルタン、およびバルサルタンが挙げられる。
【0102】
「レニン阻害物質」という用語は、インビトロおよび/またはインビボ試験で、レニン-アンジオテンシン系におけるレニンの機能を実験に基づいて決定することができる程度に(例えば、選択的に)遮断することができる物質であればいずれも包含すると、当業者によって理解されよう。
【0103】
記載され得るレニン阻害物質としては、シクロチアゾマイシン、アリスキレン、シプロキレン、ジテキレン、エナルキレン、レミキレン、テルラキレン、およびザンキレンが挙げられる。好ましいレニン阻害物質としては、アリスキレンが挙げられる。
【0104】
アンジオテンシンIIの生成および/または作用を抑制する化合物としては、次の医薬品開発コードによって文献に定義されているものも挙げられる:100240、606A、A-65317、A-68064、A-74273、A-81282、A-81988、A-82186、AB-47、BIBR-363、BIBS-222、BIBS-39、BILA-2157BS、BL-2040、BMS-180560、BMS-181688、BMS-182657、BMS-183920、BMS-184698、BRL-36378、CGP-38560、CGP-38560a、CGP-42112-A、CGP-42112、CGP-421132-B、CGP-48369、CGP-49870、CGP-55128A、CGP-56346A、CGS-26670、CGS-26582、CGS-27025、CGS-28106、CGS-30440、CHF-1521、CI-996、CL-329167、CL-331049、CL-332877、CP-191166、CP-71362、CV-11194、CV-11974、DMP-581、DMP-811、DU-1777、DuP-167、DuP-532、E-4030、E-4177、EC-33、EK-112、EMD-56133、EMD-58265、EMD-66684、ER-32897、ER-32935、ER-32945、ES-1005、ES-305、ES-RWJ-38970、RWJ-46458、RWJ-47639、RXP-407、S-2864、S-5590、SB-203220、SC-50560、SC-51316、SC-51895、SC-52458、SC-54629、SC-565254、Sch-47896、Sch-54470、SK-1080、SKF-107328、SL-910102、SQ-30774、SQ-31844、SQ-33800、SR-43845、TA-606、TH-142177、U-97018、UK-63831、UK-77568、UK-79942、UP-275-22、WAY-121604、WAY-126227、VNP-489、XH-148、XR-510、YM-21095、YM-26365、YM-31472、YM-358、およびZD-7155。
【0105】
記載され得る他の血小板凝集抑制薬としては、アスピリン/アセチルサリチル酸の一酸化窒素供与性誘導体(例えば、NCX-4016、NicOx S.A.)、またはより好ましくはアナグレリド、アルガトロバン、ベラプロスト、カングレロール、シロスタゾール、ジピリダモール、リマプロスト、パログレリル、プロカインアミド、サルポグレラート(例えば、サルポグレラート塩酸塩)、チクロピジン、チロフィバンおよびトリフルサール、ならびに次の医薬品開発コードによって文献に定義されているものが挙げられる:DA-697b(国際公開第2007/032498号を参照のこと; Daiichi Seiyaku Co Ltd)、DG-041(deCODE Genetics Inc)、K-134(CAS RN 189362-06-9)、PL-2200(CAS RN 50-78-2)、PRT-60128(Portola Pharmaceuticals Inc)、SH-529(イロプロスト/β-シクロデキストリンクラスレート; Bayer Schering Pharma AG)、およびYY-280(チクロピジンとEGb-761(タナミン(tanamin); イチョウエキス; Yuyu Inc.)の併用療法)。
【0106】
「スタチン」という用語は、HMG-CoA還元酵素の阻害物質であればいずれも包含し、フルバスタチン、シンバスタチン、ロバスタチン、ロスバスタチン、ピタバスタチン、グレンバスタチン、セリバスタチン、プラバスタチン、メバスタチン、ベルバスタチン、ダルバスタチン、およびアトルバスタチンが含まれる。
【0107】
記載され得る他のスタチンとしては、アシテマート、ベンフルオレクス、クレスチン(Clestin)、コレストロン、ジヒドロメビノリン、メグルトール、ラウソノール、および次のコード名の化合物が挙げられる:ATI-16000、BAY-10-2987、BAY-x-2678、BB-476、BIO-002、BIO-003、BIO-2、BMS-180431、CP-83101、DMP-565、FR-901512、GR-95030、HBS-107、KS-01-019、L-659699、L-669262、NR-300、P-882222、PTX-023595、RP 61969、S-2468、SC-32561、sc-45355、SDZ-265859、SQ-33600、U-20685、およびNCX-6550(ニトロプラバスタチン)やNCX-6560(ニトロアトルバスタチン)などのNO-増強性/放出性スタチン。
【0108】
より好ましいスタチンとしては、ピタバスタチン(例えば、Livalo(登録商標)、Pitava(登録商標))、フルバスタチン(例えば、Lescol(登録商標))、シンバスタチン(例えば、Zocor(登録商標)、Lipex(登録商標))、ロバスタチン(例えば、Mevacor(登録商標)、Altocor(登録商標))、ロスバスタチン(例えば、Crestor(登録商標))、プラバスタチン(例えば、Pravachol(登録商標)、Selektine(登録商標)、Lipostat(登録商標))、およびアトルバスタチン(例えば、Lipitor(登録商標)、Torvast(登録商標))が挙げられる。特に好ましいスタチンとしては、ピタバスタチン、より好ましくはシンバスタチン、さらに特にアトルバスタチン、特にロスバスタチンが挙げられる。
【0109】
記載され得る炎症の治療に有用である他の活性成分の薬学的に許容される塩としては、酸付加塩および塩基付加塩が挙げられる。このような塩は、通常の手段、例えば遊離酸または遊離塩基の形の活性成分と1当量または複数当量の適切な酸または塩基の、場合によっては溶媒中または塩が不溶性である媒体中での反応、続いて標準技法(例えば、真空中で凍結乾燥または濾過)を用いた前記溶媒または前記媒体の除去によって生成され得る。塩は、例えば好適なイオン交換樹脂を用いて、塩の形の活性成分の対イオンを別の対イオンと交換することによっても調製され得る。
【0110】
記載され得るピコタミドの塩としては、塩酸塩、重硫酸塩、マレイン酸塩およびトシル酸塩が挙げられる。記載され得るオザグレル、テルトロバン、エグアレン、およびアスピリンの塩としては、リチウム塩、ナトリウム塩、カリウム塩などのアルカリ金属塩が挙げられる。オザグレルおよびエグアレンの好ましい塩としては、ナトリウム塩が挙げられる。
【0111】
クロピドグレルの好ましい塩としては、重硫酸塩が挙げられるが、記載され得る他の塩および記載され得るチカグレロルの塩としては、塩酸塩、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。記載され得るプラスグレルの好ましい塩としては、塩酸塩が挙げられるが、記載され得る他の塩としては、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。
【0112】
記載され得るピオグリタゾンの好ましい塩としては、塩酸塩が挙げられるが、記載され得る他の塩としては、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。記載され得るロシグリタゾンの好ましい塩としては、マレイン酸塩が挙げられるが、記載され得る他の塩としては、塩酸塩、重硫酸塩、およびトシル酸塩が挙げられる。記載され得るリボグリタゾンの塩としては、塩酸塩、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。ナベグリタザルの好ましい塩としては、ナトリウム塩が挙げられるが、記載され得る他の塩としては、リチウム塩およびカリウム塩が挙げられる。記載され得るバラグリタゾンの好ましい塩としては、ナトリウム塩、カリウム塩、およびカルシウム塩が挙げられる。
【0113】
アンジオテンシンIIの生成および/または作用を抑制する化合物の好ましい塩としては、例えば塩酸塩、重硫酸塩、マレイン酸塩、メシル酸塩、トシル酸塩、カルシウムやマグネシウムなどのアルカリ土類金属塩、またはナトリウム塩やカリウム塩などのアルカリ金属塩が挙げられる。このような塩は、ペリンドプリル、エナラプリル、リシノプリル、キナプリル、イルベサルタン、オルメサルタン、トランドラプリル、テルミサルタン、ベナゼプリル、シラザプリル、モエキシプリル、スピラプリル、エプロサルタン、およびフィマサルタン(fimasartan)を含めて、化合物について、ルーチンの技法を用いて調製され得る。塩酸塩、重硫酸塩、マレイン酸塩、メシル酸塩、およびトシル酸塩が、ラミプリルやアリスキレンなどの化合物については好ましい。アルカリ土類金属塩、さらに特にアルカリ金属塩が、カンデサルタン、バルサルタン、カプトプリル、ロサルタン、特にホシノプリルなどの化合物については好ましく、それらの好ましい塩としては、カルシウム塩、マグネシウム塩、カリウム塩、特にナトリウム塩が挙げられる。記載され得るベナゼプリルおよびモエキシプリルの好ましい塩としては、塩酸塩が挙げられるが、記載され得る他の塩としては、重硫酸塩、マレイン酸塩、メシル酸塩、およびトシル酸塩が挙げられる。記載され得るエプロサルタンの好ましい塩としては、メシル酸塩が挙げられるが、記載され得る他の塩としては、塩酸塩、重硫酸塩、マレイン酸塩、およびトシル酸塩が挙げられる。
【0114】
スタチンの好ましい塩としては、ピタバスタチンカルシウム、フルバスタチンナトリウム、プラバスタチンナトリウム、ロスバスタチンカルシウム、アトルバスタチンカルシウムなどのナトリウム塩、カリウム塩、およびカルシウム塩が挙げられる。
【0115】
炎症の治療に有用である他の活性成分の好適な投与量は、当業者に周知であり、Martindale - The Complete Drug Reference(第35版)およびその中の引用文献などの医学文献中で該当する薬物について列挙されているものが含まれ、それらの文献すべての中の関連する開示内容は参照により本明細書に組み込まれる。
【0116】
「約」という単語が、本明細書で、例えば量(例えば、値、重量、体積、モル)、温度、結晶化度、崩壊度(degree of degradation)、純度、溶解度、および活性成分の投与量に関連して使用されるときはいつでも、このような変数は近似的なものであり、そういうものとして本明細書に特定される数と±10%、例えば±5%、好ましくは±2%(例えば±1%)異なる可能性があることを理解されたい。
【0117】
本発明の化合物は、取り扱いやすさが改善される形態であり、以前に調製されたペミロラストの形態に比べると改善された化学的および固体状態安定性を有する形態で生成され得るという利点を有する。したがって、化合物は、長期間にわたって貯蔵される場合に安定であり得る。
【0118】
本発明の化合物は、公知および/または市販のペミロラストの形態に比べて改善された溶解性および吸湿性プロファイルも有する。本発明の化合物は、公知および/または市販のペミロラストの形態に比べて改善された味覚プロファイルも有することができる。
【0119】
本発明の化合物は、好収量で、以前に調製されたペミロラストの形態より高純度で、より短時間で、より好都合に、より低コストで調製され得るという利点も有することができる。
【0120】
本発明の化合物は、本明細書の以上に記載される病態の治療において、炎症性障害(アテローム性動脈硬化症およびそれに伴う心血管病態など)またはその他の治療で使用する先行技術において公知の同様な化合物に比べて、医師および/もしくは患者にとってより好都合で、より有効で、より低毒性で、活性範囲がより広く、より強力で、副作用がより少なく、または他の有用な薬理学的特性を有することができるという利点も有することができる。
【0121】
含まれている図面を参照しながら、以下の実施例によって、本発明を説明するが、決して限定するものではない。
【図面の簡単な説明】
【0122】
【図1】実施例1によって得られたペミロラストナトリウム半水和物の結晶形の粉末X線回折図(diffractogram)を示す図である。
【図2】実施例1によって得られたペミロラストナトリウム半水和物の結晶形のFT-ラマンスペクトルを示す図である。
【図3】比較例5によって得られたペミロラストナトリウムの結晶形の粉末X線回折図を示す図である。
【図4】比較例5によって得られたペミロラストナトリウムの結晶形の異なる時点(調製後(下側のトレース)および約1か月後(上側のトレース))におけるFT-ラマンスペクトルを示す図である。
【図5】実施例10によって得られたペミロラストナトリウム半水和物の結晶形の粉末X線回折図を示す図である。
【図6】実施例11によって得られたペミロラストナトリウム七水和物の結晶形の粉末X線回折図を示す図である。
【図7】実施例14によって得られたペミロラストナトリウムの追加の結晶形の粉末X線回折図を示す図である。
【図8】ペミロラストナトリウム半水和物(四角)とペミロラストカリウム(三角)の溶解プロファイル(溶解対時間の百分率)比較を示す図である; 平均値±SD(1群当たりn=3)。
【発明を実施するための形態】
【0123】
実施例1〜5の一般手順
FT-ラマンスペクトルを1064nmで操作される近赤外Nd:YAGレーザーおよび液体窒素冷却ゲルマニウム検出器を備えたBruker RFS 100 FT-ラマンシステムで記録した。3500〜50cm-1の範囲を2cm-1の分解能で64回スキャンし、集積した。一般に、100mWのレーザー出力を使用した。
【0124】
Bruker D8を使用して、粉末X線回折を実施した; 銅Kα線、40kV/40mA; LynxEye検出器、ステップサイズ0.02°(2θ)、ステップ時間37秒。試料調製: 僅かな圧力を加えて、平面にしたこと以外は、一般に何ら特別な処理を行うことなく、試料を測定した。シリコン単結晶サンプルホルダーのタイプ: a) 標準ホルダー深さ0.1mm、b) 深さ0.5mm、空洞直径12mm、c) 深さ1.0mm、空洞直径12mm。Bruker D8で測定した試料はすべて、測定時に回転させた。別段の指定のない限り、周囲空気雰囲気を使用した。選択された試料をPhilips X'pert PW 3040またはPhilips PW1710(銅Kα線、ステップサイズ0.02°(2θ)、2秒/ステップ、2〜50°(2θ)で測定した。僅かな圧力を加えて、平面にしたこと以外は、何ら特別な処理を行うことなく、試料を測定した。別段の指定のない限り、周囲空気雰囲気を使用した。
(下記の実施例に従って調製された形態は、関連するパターンから(実験誤差を考慮して)、同じ結晶形が生成されていたことが明らかであったとき、下記に開示される他の実施例と「本質的に」同じPXRD回折パターンを示した。したがって、PXRD間隔値の実験誤差の限界は、本明細書のいずれかの部分で使用されているように、最後の小数位で±2ぐらいの範囲とすることができる。)
【0125】
C、H、およびNの元素分析は、Leco CHN 800またはLeco CHNS 932装置を使用して、燃焼によって行った。Oの元素分析は、Leco RO-478装置を使用して、熱分解によって行った。Naの元素分析は、原子吸収分光法で行った。
【0126】
1H/13C NMRスペクトルをBruker DPX300装置で記録した。別の方法では、1H NMRスペクトルをVarian MERCURY+400分光計(400MHz)で記録した。スペクトルは周囲温度で記録した。化学シフトは、TMSを基準にして、溶媒シグナルδ7.26ppm CHCl3、δ2.50ppm DMSO、およびδ4.79ppm H2Oを経て、δ値(ppm)として記載される。
【0127】
質量分析スペクトルは、Onyx Monolithic C18 50mm×4.6mmカラム(Phenomenex)を装備したFinnigan ThermoQuest AQA四重極型質量分析計を有するGilson HPLCシステムを使用するLC-MSシステム(流速4mL/分、アセトニトリル/水勾配および0.05%ギ酸)、またはVarianクロムパックキャピラリーカラムCP-SIL 8 CB Low Bleed/MS(30 m_0.22mm、0.25mm)を装備し、イオン生成電位70eVを利用するGC-MS装置で記録した。
【0128】
示差走査熱量測定をPerkin Elmer DSC 7で記録した。密閉型金るつぼ、加熱速度:10K/分、範囲:50℃〜350℃。DSC分析の過程において記録された、熱イベントと関係付けられた温度は、それぞれの熱イベントのピーク温度(最低/最高)である。
【0129】
TG-FTIRは、Bruker FT-IR Spectrometer Vector 22を備えたNetzsch Thermo-Microbalance TG 209で記録した。Alるつぼ(開放型または微細孔付き)、N2雰囲気、加熱速度:10K/分、範囲:25〜250℃。
【0130】
HPLCをTSP HPLC(UV3000、AS3000、P4000、SCM1000ソフトウェアバージョン4.1)で行った; カラム:Waters、X Terra MS C18 4.6×100mm、5μ(CC01); 移動相A:H2O+0.1%TFA; 移動相B:アセトニトリル+0.1%TFA; 標準濃度:約0.09mg/mL; 保持時間:6.6分; 勾配:0.0分:A:95%/B:5%; 20.0分:A:5%/B:95%; 21.0分:A:95%/B:5%; 30.0分:A:95%/B:5%; 流速:1.0mL/分; 注入量:10μL; 波長:254nm。
【0131】
動的水蒸気収着(Dynamic Vapor Sorption)(DVS)測定を収着測定システムSPS11-100nで実施した。試料をアルミニウムるつぼに入れ、事前定義湿度プログラムを開始する前に、その試料を所与のRHで平衡化させておいた。(1) 50%RHで2時間; (2) 50→0%RH(5%/時); (3) 0%RHで5時間; (4) 0→95%RH(5%/h); (5) 95%RHで5時間; (6) 95→50%RH(5%/時); (7) 50%RHで2時間。吸湿性をヨーロッパ薬局方に従って分類した(80%RH/25℃で24時間貯蔵)。(周囲条件での)出発材料に比べて、85%RHにおける質量変化率を分類に使用した-非常に吸湿性:質量の増加≧15%; 吸湿性:質量の増加は15%未満であり、2%以上である; 若干吸湿性:質量の増加は2%未満であり、0.2%以上である; 潮解性:十分な水が吸収されて、液体になる。
【実施例】
【0132】
(実施例1)
ペミロラストナトリウム半水和物
ペミロラストカリウムを水に溶解し、6M HClを用いてpH 1まで酸性化し、遊離酸を溶液から沈澱させることによって、ペミロラスト遊離酸を調製した。生成した結晶を濾過し、水で洗浄し、真空乾燥した。次に、2M NaOHの代わりに2M KOHを使用し、水:イソプロパノール(2:1の比)の代わりに水:イソプロパノール(1:2の比)から再結晶をして、下記の比較例4(ステップIaおよびIb)に記載の方法と同様にして、ペミロラストカリウムを調製した。
【0133】
ペミロラスト遊離酸(89mg)を783μLのメタノール中0.5Mナトリウムメトキシド(Fluka)に懸濁した。懸濁液を1日撹拌した。沈殿物が生成した。濾過し、真空下で約2時間乾燥した後、固体物質が得られた(収量:81mg)。
【0134】
元素組成分析を下記の表1にまとめる。そのデータから、1:1の化学量論でペミロラストナトリウム塩の半水和物が示唆される。式:Na(C10H7N6O)×0.5H2Oの理論データを算出した。
【0135】
【表1】
【0136】
実施例1によって得られた形態のPXRDパターンを図1に示し、下記の表2にまとめる。
【0137】
【表2A】
【0138】
【表2B】
【0139】
塩は高結晶性であった。
【0140】
実施例1によって得られた形態のFT-ラマンスペクトルを図2に示す。
【0141】
NMR、DSC、TG-FTIR、およびDVS分析も実施した。DSCは、280.3℃で1つの吸熱を示した。DVS分析によって、タイトル化合物は、上記の分類体系によれば吸湿性である(80%超RHで、著しい吸水が開始する)ことが明らかである。(対照的に、DVSによって、ペミロラストカリウムは、上記の分類体系によれば非常に吸湿性である(70%超RHで、著しい吸水が開始する)ことが明らかである。)
【0142】
(実施例2)
ペミロラストナトリウム半水和物
ペミロラスト遊離酸(上記の実施例1に記載のとおりに調製した; 90mg)を791μLのメタノール中0.5Mナトリウムメトキシド(Fluka)に懸濁した。懸濁液を1日撹拌した。沈殿物が生成した。濾過し、真空下で約1.5時間乾燥した後、固体物質が得られた(収量:59mg)。
【0143】
結晶をFT-ラマンで分析した。関連するスペクトルは、上記の実施例1に従って得られた形態によって示されたものと本質的に同じであった。
【0144】
(実施例3)
溶解性の比較
約40mgの試料(上記の実施例2に記載された手順を経て得られた)を0.25mLの再蒸留水に分散した。懸濁液を22℃で24時間振盪した。その後、Eppendorf Thermomixer Comfort(400rpm)を使用して、迅速固/液分離を行った。Centrifuge Hettich EBA 12 R(15,000g、1分、22℃)で、Millipore Centrifugal Filter Devices(PTFEフィルター; 0.2μm)を用いて、懸濁液を濾過した。濾液中の試料の濃度をHPLCで分析し、固相をFT-ラマン分光法で分析した。
【0145】
実施例2の化合物は、検討された条件下で水溶解度23.64mg/mLを示した。飽和溶液はpH 8.0であった。
【0146】
ペミロラストカリウム塩(メタノール中0.5Mナトリウムメトキシドの代わりにメタノール中3.4Mカリウムメトキシド(Fluka)を使用して、実施例1の第2段落、および上記の実施例2に記載のように調製された)の水溶解度を同様に決定し、検討された条件下で192.38mg/mLであることがわかった。飽和溶液はpH 9.0であった。(市販のペミロラストカリウムの水溶解度は、179〜182mg/mLであると報告されている; 出典:上記Pharmaceutical Interview Form)
【0147】
(比較例4)
ペミロラストナトリウムの合成
ペミロラストナトリウム塩を次の2つの方法によって調製した:
(I)
(Ia) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1.2-a]ピリミジン-4-オン(ペミロラスト)
これは、マロノニトリル(1.64g、24.8mmol; Acros Organics)、2-アミノ-3-ピコリン(2.51mL、24.8mmol; Acros Organics)、オルトギ酸エチル(4.55mL、27.3mmol; Sigma-Aldrich)、およびアジ化ナトリウム(1.78g、27.4mmol; Sigma-Aldrich)を出発物として、SanoおよびIshiharaによって記載された方法(Heterocycles、48、775(1998年))に従って合成され、サブタイトルの化合物が得られた(2.64g; 46.7%)。
1H NMR (DMSO-d6) δ: 9.21 (s, 1H, CH), 9.16〜9.11 (m, 1H, CH), 8.13〜8.07 (m, 1H, CH), 7.58〜7.51 (m, 1H, CH), 2.62 (s, 3H, CH3)。
【0148】
(Ib) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(ペミロラスト)ナトリウム塩
9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(2g、8.76mmol; 上記の工程Iaによるもの)を2-プロパノール(9mL)に懸濁し、2M NaOH(8.8mL、17.6mmol)を加えた。反応混合物を50℃に1時間加熱した。さらに17mLの2-プロパノールで処理した後、粗タイトル化合物が沈殿した。氷浴中で冷却した後、固体物質を濾過で回収し、100mLの水に再溶解した。未溶解の物質を濾過で除去し、濾液を蒸発させた。残渣を水および2-プロパノール(2:1の比)から再結晶し、真空乾燥して、純粋なペミロラストNa塩を得た(1.26g、57.5%)。
1H NMR (D2O) δ: 8.86〜8.80 (m, 1H, CH), 8.57 (s, 1H, CH), 7.68〜7.59 (m, 1H, CH), 7.22〜7.13 (m, 1H, CH), 2.39 (s, 3H, CH3)。
【0149】
(II) (米国特許第4,122,274号に記載の方法)
(IIa) 2-シアノ-3-(3-メチル-2-ピリジルアミノ)アクリル酸エチル
エトキシメチレンシアノ酢酸エチル(7.82g、46.2mmol; Sigma-Aldrich)および2-アミノ-3-ピコリン(4.67mL、46.2mmol; Acros Organics)のトルエン(4mL)溶液を100℃で15分間加熱した。反応混合物を冷却し、サブタイトルの生成物(10.45g、97.8%)を濾過で回収した。
GC-MS(70eV) m/z(相対強度)231(M+、15)、158(100)
1H NMR (CDCl3) δ: 11.20〜11.07 (m, 1H, NH), 8.82 (d, J = 12.4 Hz, 1H, CH), 8.22〜8.17 (m, 1H, CH), 7.53〜7.47 (m, 1H, CH), 7.07〜6.96 (m, 1H, CH), 4.31 (q, J = 7.2 Hz, 2H, CH2), 2.33 (s, 3H, CH3), 1.37 (t, J = 7.2 Hz, 3H, CH3)。
【0150】
(IIb) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(ペミロラスト)
THF(375mL)を-30℃に冷却し、塩化アルミニウム(7.30g、54.7mmol)と、続いてNaN3(10.65g、163.8mmol)を加えた。反応混合物を30分間加熱還流し、その後、5℃に冷却した。2-シアノ-3-(3-メチル-2-ピリジルアミノ)アクリル酸エチル(10.40、45.0mmol; 上記のステップIIaによるもの)を加え、反応混合物を18時間加熱還流した。反応混合物を放冷し、THFを減圧下で除去した。残渣を氷水(210mL)で処理し、6M HClを用いて、pH 3に酸性化した。固体物質を濾過で回収し、DMFから再結晶して、サブタイトルの生成物(4.61g、44.9%)を得た。
LC-MS (M + H+) 229.1
1H NMR (DMSO-d6) δ: 9.21 (s, 1H, CH), 9.16〜9.11 (m, 1H, CH), 8.13〜8.07 (m, 1H, CH), 7.58〜7.51 (m, 1H, CH), 2.62 (s, 3H, CH3)。
【0151】
(IIc) 9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(ペミロラスト)ナトリウム塩
1M NaOH(20.30mL、20.3mmol)を9-メチル-3-(1H-テトラゾル-5-イル)-4H-ピリド[1,2-a]ピリミジン-4-オン(4.60g、20.1mmol; 上記の工程IIbによるもの)の水(115mL)懸濁液に滴下した。反応混合物を100mLの水で希釈し、50℃に2分間加熱した。溶液を濾過し、水を凍結乾燥で除去した。粗生成物(6.13g)を小分けし、様々な比の水:エタノールから再結晶して、純粋なタイトル化合物を得た。
【0152】
(比較例5)
米国特許第4,122,274号の方法に従ったペミロラストナトリウムの再結晶
米国特許第4,122,274号には、粗タイトル生成物(ペミロラストナトリウム)を水:エタノールから再結晶して、純粋なタイトル生成物を得たことが記載されている。このレベルの詳細からでは、使用された水:エタノールの比がどんなものであったか明らかでなく、したがって先行技術の技法を再現する目的で、いくつかの実験を行った。
【0153】
(i) 粗ペミロラストナトリウム塩(480mg; 上記の比較例4、方法(I)によるもの)を1:1の比の水およびエタノール(95%)から再結晶した。ペミロラストNa塩(480mg、1.92mmol)を70℃でH2O(8mL)に溶解し、EtOH95%(8mL)を加えた。透明な溶液を室温に到達するまで放置し、生成された固体物質を濾取し、少量のエタノールで洗浄し、真空乾燥して、316mgの純粋なナトリウム塩を得た。
【0154】
(ii) 粗ペミロラストナトリウム塩(500mg; 上記の比較例4、方法(II)によるもの)を70℃で水(4.9mL)に溶解した。その後、EtOH 95%(約4.0mL)を固体が生成し始めるまで70℃で加えた。さらに0.1mLの水を加えて、すべてを溶液にした。冷却すると生成した固体物質を濾過で回収し、真空乾燥して、348mgの純粋なナトリウム塩を得た。
【0155】
(iii) 粗ペミロラストナトリウム塩(300mg; 上記の比較例4、方法(II)によるもの)を70℃で水:エタノール(1:1の比; 10mL)から再結晶した。冷却すると生成した固体物質を濾過で回収し、真空乾燥して、174mgの純粋なナトリウム塩を得た。
【0156】
(iv) 粗ペミロラストナトリウム塩(300mg; 上記の比較例4、方法(II)によるもの)を70℃で水:エタノール(9:1の比、4mL)から再結晶した。冷却すると生成した固体物質を濾過で回収し、真空乾燥して、219mgの純粋なナトリウム塩を得た。
【0157】
純粋なペミロラスナトリウム塩の4つの試料はすべて、同じ物理化学的特性(ラマンスペクトルおよびNMR)を有するものであった:
1H NMR (D2O) δ: 8.86〜8.80 (m, 1H, CH), 8.57 (s, 1H, CH), 7.68〜7.59 (m, 1H, CH), 7.22〜7.13 (m, 1H, CH), 2.39 (s, 3H, CH3)。
【0158】
PXRDパターン(上記の比較例5(i)について測定された)を図3に示す。このことから、ナトリウム塩のこの形態は、結晶性部分と混合された非晶質物質であるというのが結論であった。
【0159】
ラマンスペクトルを再結晶直後に記録した。次いで、すべての試料をドラフトの棚に周囲条件下で貯蔵した。約1か月後に、ラマンスペクトルを記録し、先に記録されたものと有意差があった。これは、図4に示されており、下側のスペクトルは先の測定と一致し、上側のスペクトルは、後の測定と一致する。これらの結果を考慮に入れて、先行技術のペミロラストナトリウムの非晶質形は、物理的に不安定であるというのが結論であった。
【0160】
非晶質物質は、下記の実施例11に従って得られた形態を40℃で40時間減圧乾燥して、12gの淡黄色綿様非晶質固体を得ることによっても調製された。
【0161】
実施例6〜16の一般手順
Thermal Advantage TGA Q5000IR(TA装置)モジュールを使用して、TGAを測定した。試料(約10〜16mg)を白金パン(100 HI)に入れ込み、窒素パージ下に、加熱速度10℃/分で25から350℃に加熱した。
【0162】
平面偏光を装備したNikon SMZ800顕微鏡を用いて、顕微鏡検査を行った。
【0163】
(290℃に設定された)乾燥炉を装備した装置で、カールフィッシャー電量滴定を実施した。
【0164】
冷蔵冷却システムを装備したThermal Advantage DSC Q1000(TA装置)を使用して、DSCを試験した。装置は、インジウムを使用して温度およびエンタルピーについて較正した。非気密性アルミニウムパンに、約2〜3mgの試料を正確に秤量し、クリンプした。試料を連続窒素パージ(50mL/分)下に、加熱速度10℃/分で25から275℃までスキャンした。
【0165】
試料のPXRDパターンは、Siemens D5000粉末回折計でCuK線(1.540 56Å)を使用して収集した。管電圧およびアンペア数を40kVおよび40mAにそれぞれ設定した。発散スリットおよび散乱線除去スリットの設定は、20mmの試料領域の照明のため、可変である。各試料を2θにおいて5〜40°の範囲をステップサイズ0.02°でスキャンした。1ステップ当たりの測定時間は1秒であり、分析中、試料台は30rpmで回転し(粉末試料)、または回転しなかった(錠剤)。小容積のゼロバックグラウンドホルダーも使用した。シリコン標準物質を使用して、装置を事前に較正した。(下記の実施例に従って調製された形態は、関連するパターンから(実験誤差を考慮して)、同じ結晶形が生成されていたことが明らかであったとき、下記に開示される他の実施例と「本質的に」同じPXRD回折パターンを示した。したがって、PXRD間隔値の実験誤差の限界は、本明細書のいずれかの部分で使用されているように、最後の小数位で±2ぐらいの範囲とすることができる。)
【0166】
Agilent XDB C18 50×4.6、1.8μmカラムを使用して、HPLC-UV(実施例16)を行った。カラムオーブンの温度は40.0℃であり、流速は1mL/分であり、検出はUV/VISにより370.0 nmで行った。注入量は10μLであった。Chromolith Performance RP-18 100×4.6mmカラム(Merck)を使用して、HPLC-UV(実施例10)を行った。カラムオーブンを周囲温度に設定し、流速は3mL/分であり、検出はUVにより254nmで行った。HPLC法は両方とも、移動相Aの0.1%TFA(水溶液)および移動相Bのアセトニトリルを使用した。
【0167】
(実施例6)
ペミロラストナトリウム半水和物
ペミロラスト遊離酸(1g; 対応するカリウム塩(Chemtronica AB、Stockholm、Sweden)から、水に溶解し、酢酸で酸性化し、その結果沈殿した遊離酸を濾取し、乾燥することによって調製された)を下記の表3に記載される一連の選択された有機溶媒または溶媒混合物(15mL)に懸濁した。その後、得られたスラリーを50〜60℃に加熱し、塩形成剤(1当量の水酸化ナトリウム(50%水溶液もしくは8%メタノール溶液)またはナトリウムエトキシド(21%エタノール溶液))を加えた。固体の部分溶解が認められた。しかし、数秒以内に、新しい沈殿物が得られ、スラリーがより濃厚になった。スラリーを50〜60℃で1〜2時間平衡化し、次いで室温に冷却し、濾過した。固体を室温および大気圧で、1〜2日で乾燥した。
【0168】
【表3】
【0169】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0170】
上記のメタノールおよびイソプロパノール結晶化によって得られた結晶を、DSCおよびTGAで分析し、すべて半水和物であることが確認された。
【0171】
(比較例7)
ペミロラストナトリウム七水和物
ペミロラスト遊離酸(3g; 上記の実施例6に記載のとおりに調製された)を水(30mL)に懸濁し、スラリーを約50℃に加熱した。1当量の50%NaOH(水溶液)を加え、その結果透明な溶液が得られた。溶液を冷却した。溶液温度約40℃で、塩は結晶化し始め、非常に濃厚なスラリーが得られた。スラリーを希釈するために、水(80mL)を追加した。最後に、スラリーを0℃に冷却し、1時間平衡化し、続いて濾過した。固体を室温および大気圧で1〜2日間乾燥して、4.04gのタイトル化合物を得た。
【0172】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例11に従って得られた形態によって示されたものと本質的に同じであった。
【0173】
(実施例8)
ペミロラストナトリウム半水和物
ペミロラストナトリウム七水和物の試料(3.5g、上記の実施例7に記載された方法に従って得られた)を水(3.0mL)に懸濁し、約80℃に加熱した。スラリーをこの温度で濾過し、次いで室温および大気圧で乾燥した。収量は1.3gであった。
【0174】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0175】
(実施例9)
ペミロラストナトリウム半水和物
(a) ペミロラスト
遊離酸を、ペミロラストカリウム(15.8g; Chemtronica AB)から、水(100mL)とTHF(80mL)の混合物に室温で溶解し、酢酸(1当量; 17.5g; 20%水溶液)で酸性化することによって調製した。まず、約1mLの酢酸を添加し、得られた希薄なスラリーを約30分間平衡化した。その後、残りの酢酸をゆっくり加えた。得られた濃厚なスラリーを水(50mL)で希釈し、2時間平衡化し、次いで濾過し、水で洗浄した。得られた固体を40℃および減圧下で5〜10時間乾燥して、7.5gのサブタイトル化合物を得た。
【0176】
(b) ペミロラストナトリウム半水和物
遊離酸(1g; 上記のステップ(a)によるもの)をエタノール(13.5mL)と水(1.5mL)の混合物に懸濁し、懸濁液を60℃に加熱した。ナトリウムエトキシド(1当量; 1.43g; 21%エタノール溶液)を加え、その結果固体の部分溶解によって、スラリーが生成した。ナトリウム塩は直ちに結晶化し、新しいスラリーが生成した。スラリーを60℃で約1時間平衡化し、その後20〜25℃に冷却し、濾過した。濾過ケーキをエタノールで洗浄し、室温および大気圧で1〜2日間乾燥して、0.99gのタイトル化合物を得た。
【0177】
結晶をPXRDで分析した。関連するスペクトルは、下記の実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0178】
(実施例10)
ペミロラストナトリウム半水和物-大規模生産
(a) ペミロラスト
水(2,500mL)とTHF(1,400mL)の混合物に、室温で1時間撹拌しながらペミロラストカリウム(190g; 0.71mol; Chemtronica AB)を溶解した。次いで、混合物のクリア濾過を行い、水(400mL)中酢酸(43g、0.72mol)を2つに分けて加えた。まず、約50mLの混合物を加え、得られたスラリーを30分間撹拌した。次いで、残りの酢酸溶液をゆっくり加えた。得られたスラリーを2時間にわたって平衡化し、濾過し、水で洗浄した。サブタイトル化合物を終夜風乾し、次いで真空オーブン中、40℃で24時間乾燥した。収量は164g(白色固体、硬質塊)であった。
【0179】
(b) ペミロラストナトリウム半水和物
遊離酸(45.5g、0.2mol; 上記のステップ(a)によるもの)をエタノール(630mL)に懸濁し、混合物を57℃に加熱した(内部温度)。水(72mL)中水酸化ナトリウム(8.0g、0.2mol)を加えた。固体の部分溶解によって、ほとんど直ちにスラリーが生成し、続いてその結晶化が起こった。スラリーを57℃で1時間にわたって平衡化し、次いで20〜25℃に冷却し、濾過した。濾過ケーキをエタノールで洗浄し、真空中、室温で48時間乾燥した。タイトル化合物は淡黄色粉末として得られ、その収量は45gであった。純度は、HPLC-UVにより>99%であると判定された。
【0180】
結晶をPXRDで分析した。関連性のあるスペクトルを図5に示し、主ピークを下記の表4にまとめる。(この分析から、実施例10に従って得られた形態は、実施例1および2に従って得られたものと同じであるが、前者の試料は、出発材料(ペミロラスト遊離酸)、不純物、および/または副生物を含み得るものであったことが明らかである。)
【0181】
【表4】
【0182】
(比較例11)
ペミロラストナトリウム七水和物-大規模生産
ペミロラスト遊離酸(80g、0.35mol; 上記の実施例10(a)を参照のこと)を水(3L)に懸濁し、50℃に加熱した。水(14g)中水酸化ナトリウム(14g、0.35mol)を加え、得られた溶液のクリア濾過を行った。溶液を20℃に冷却した。生成物が、約25〜28℃で自発的に結晶化し始めた。スラリーを20℃で30分間平衡化し、次いで0℃に冷却した。スラリーを0℃で2時間平衡化し、濾過し、氷-冷水(400mL)で洗浄した。残っている湿った物質を大気圧、45℃、および75%相対湿度で乾燥した。結晶は一緒に溶融し、飴様物質が得られた。この物質を2.5Lの水に50℃で溶解し、結晶化手順を繰り返した。得られた沈殿物を濾過し、乾燥し、約40分間それを通して吸気し、次いで大気圧、25℃、および相対湿度60%で乾燥した。収量は80gの淡黄色固体であった。
【0183】
実施例11によって得られた形態のPXRDパターンを図7に示し、主ピークを下記の表5にまとめる。
【0184】
【表5】
【0185】
DSC(実施例11によってではなく、以前に調製された試料に関する)によって、2つの吸熱(1つは71.1℃、1つは90.2℃)を示した。
【0186】
(実施例12)
ペミロラストナトリウム半水和物
ペミロラストナトリウム半水和物(0.35g; 実施例10に記載された方法に従って得られた)を水(0.8g)に加えた。試料を85〜90℃に加熱した。得られた透明な溶液を75℃に冷却し、熱エタノール(75℃)を2mLずつ5分ごとに加えた。6mLを加えたところで、結晶化が始まった。さらに6mLを10分かけて加えた。試料を20℃に冷却し、濾過し、室温でおよび大気圧で終夜乾燥して、0.19gのタイトル化合物を得た。単離された沈殿物を平面偏光を装備した光学顕微鏡で検査し、PXRDで分析した。関連するスペクトルは、実施例10(および上記の実施例1)に従って得られた形態によって示されたものと本質的に同じであった。
【0187】
(実施例13)
結晶化実験
ペミロラストナトリウム半水和物(0.2〜0.4g; 実施例10に記載された方法に従って得られた)を、10mLのエタノールおよび水、ならびにそれらの比80:20、60:40、40:60、および20:80の混合物に加えた。試料を50〜70℃に加熱した。透明な溶液が、0:100、20:80、40:60、および60:40(エタノール:水の比)で得られた。80:20および100:0(エタノール:水の比)では、未溶解の結晶を沈降させ、透明な溶液をデカンテーションした。次いで、透明な溶液を放置して、20℃までゆっくり冷却させた。
【0188】
得られた沈殿物の目視検査を、平面偏光を装備した光学顕微鏡で実施した。上記の実施例10(ペミロラストナトリウム半水和物; キューブ結晶として出現する)または上記の実施例11(ペミロラストナトリウム七水和物; 針様結晶として出現する)に記載された方法に従って得られ、完全に特徴付けられた結晶形の公知の物理的外見に基づいて、2つの結晶形のうちのどちらが得られたかについて判定を行った。
【0189】
その検査が実施された後、再び試料を(すべての場合に)完全な溶解が認められるまで加熱した。溶液を再び放置して、約35℃までゆっくり冷却させ、(上記の実施例10に記載された方法に従って得られた)ペミロラストナトリウム半水和物結晶と(上記の実施例11に記載された方法に従って得られた)ペミロラストナトリウム七水和物結晶との1:1混合物を種結晶として加えた。最後に、試料を20℃に冷却し、得られた沈殿物を上述された平面偏光を装備した光学顕微鏡で検査した。
【0190】
その結果を下記の表6にまとめる。
【0191】
【表6】
【0192】
この後、ペミロラストナトリウム半水和物(0.2〜0.4g; 実施例10に記載された方法に従って得られた)を10mLのエタノール:水の80:20、85:15、90:10および95:5の比混合物に加えた。試料を50〜70℃に加熱した。未溶解の結晶を沈降させ、透明な溶液をデカンテーションした。次いで、透明な溶液を放置して、20℃までゆっくり冷却させ、得られた沈殿物を上述された平面偏光を装備した光学顕微鏡で検査した。
【0193】
その検査が実施された後、再び試料を(すべての場合に)完全な溶解が認められるまで加熱した。溶液を放置して、約50℃の温度にゆっくり到達させ、次いで種結晶を添加することなく行われた実験で得られたものとは反対の形態の結晶を種結晶として添加した。最後に、試料を20℃に冷却し、得られた沈殿物を上述された平面偏光を装備した光学顕微鏡で検査した。
【0194】
その結果を下記の表7にまとめる。
【0195】
【表7】
【0196】
この実験の結論は、有機溶媒中、約10%以下の水の存在下で部分溶解することによって、ペミロラストナトリウム半水和物を調製できるということである。
【0197】
(実施例14)
安定性試験
実施例10(半水和物)、実施例11(七水和物)、および実施例5(非晶質形)に記載された方法に従って得られたペミロラストナトリウムの相対安定性を様々な水分レベルで決定するために、安定性試験を行った。
【0198】
試験は、4つの異なる貯蔵条件下、40℃/75%RH、25℃/60%RH、室温/10〜20%RH、および室温/>90%RHという異なる水分レベルおよび温度で行った。物質をガラスのビーカーに開放して貯蔵し、分析のため試料を定期的(時間0、1週間、および4週間)に抜き取った。
【0199】
記載された時間間隔で、(a) スパーテル1杯の物質(物質のスミアリングまたは疎砕を行わなかった)を白地のmunktellフィルターに置き、(b) 平面偏光を装備した顕微鏡で検査することによって外観を検査した。含水率も、カールフィッシャー滴定で分析した。PXRDを0時点および4週目の時点で実施した。
【0200】
試験によって、半水和物形は、相対湿度60%以下(10〜20%RHに至るまで調査)で4週まで貯蔵したとき、安定である(固相)ことが実証された。光学顕微鏡検査を含めて、外観は、これらの貯蔵条件下で不変のままであった。水の分析(カールフィッシャー)は、これらの条件下で不変の結果を示した(2.8%+/-0.2)。
【0201】
75%RH以上において、半水和物が今まで知られていなかった固体状態の形態に変換した(図7に示されるPXRDパターンによって実証されるように、その主ピークを下記の表8にまとめる)。
【0202】
【表8A】
【0203】
【表8B】
【0204】
この形態は、七水和物とほぼ同じ量の水を含有する(ほぼ30%(w/w))。
【0205】
一方、七水和物形は、1つの調査条件の60%RHでしか4週間まで安定でなかった。乾燥条件(10〜20%RH)下で、七水和物形は、半水和物形に変換された。より高い湿度レベル(75%RH)では、試料は、図7に特徴付けられる上記の新しい固体状態の形態に部分的に変換した。しかし、90%RHでは、相転移の徴候は認められなかった。
【0206】
非晶質形は、60%RH以上の貯蔵条件下で結晶化したが、10〜20%RHの乾燥条件下では、非晶質のままである。60%RH以上では、七水和物と、図7および上記の表8(表)に特徴付けられる上記の新しい固体状態の形態の2つの固体形が、貯蔵後4週間出現した。
【0207】
(実施例15)
錠剤の妥当性試験
(A) 5mg錠剤
まず、プラセボバッチ81002-1002-16を生成し、ロータリー式打錠機(Korsch PH106)の調整に使用した。
【0208】
次いで、表9にまとめた医薬品添加物を含む半水和物含有錠剤1000個を1バッチ生成した。試験中、温度および相対湿度をモニターした。この場合、RHは12.7%であった。上記の実施例10に記載されている方法に従って、半水和物が得られ、乾式混合する前に、500μmの篩に通すことによって篩過した。下記に列挙された医薬品添加物(ステアリン酸マグネシウムを除く)との混合は、Turbulaミキサーで10分間実施した。この後、ステアリン酸マグネシウムを500μmの篩に通し、混合物に加え、続いてさらに2分間混合する。ロータリー式打錠機(Korsch PH 106)で、25rpmで直径7mmの丸形凹型パンチを使用して、錠剤を生成した。低、中、および高(それぞれ、ほぼ3、4、および7kN)の3つの圧縮力を評価した。
【0209】
【表9】
【0210】
同じ方法を使用して、12.2%RHで次の医薬品添加物(下記の表10を参照のこと)を含む7水和物含有錠剤1000個を1バッチ生成した。上記の実施例11に記載された方法に従って、七水和物を得た。
【0211】
【表10】
【0212】
(B) 0.2mgミニ錠剤
上記の(A)に記載されたのと同じ方法を使用して、11.6%RH(半水和物)および12.2%RH(七水和物)で、次の医薬品添加物(下記の表11および表12)を含む半水和物含有錠剤および七水和物含有錠剤5000個を別個の2バッチ生成した。上述のように、まずプラセボを生成し、打錠機調整に使用した。直径3mmの丸形凹型パンチを、打錠機において1つの圧縮力(ほぼ400N)で使用した。
【0213】
【表11】
【0214】
【表12】
【0215】
(C) 30mg錠剤
上記の(A)に記載されたものと同様の方法を使用して、8.0%RH(半水和物)および10.2%RH(七水和物)で、次の医薬品添加物(下記の表13および表14)を含む半水和物含有錠剤および七水和物含有錠剤250個を別個の2バッチ生成した。
【0216】
含量均一性を改善する試みとして、まず活性物質を500μmの篩の代わりに250μmの篩で篩過した。次いで、活性物質を手でMCCと前もって混合した。直径6mmの円板パンチを打錠機で使用した。約31%の活性物質を含有する半水和物製剤では(活性物質の不足のため、計画されたものから変更した)、2つの圧縮力(5kNおよび14kN)を使用した。約47%の活性物質を含有する七水和物製剤では、粉体流の不足というだけで、評価され得る圧縮力が1つになった(約2kN)。この圧縮力は、七水和物製剤のフロー特性のため、可変性の高いものであった。
【0217】
【表13】
【0218】
【表14】
【0219】
すべての錠剤について、脆弱性および破砕抵抗性試験を標準方法に従って実施した(それぞれPh. Eur. 6.0, 2.9.7および2.9.8、破砕抵抗性の結果をPh. Eur.によるNではなくkpで表すことを除く)。30mg錠剤についても、破砕した錠剤と無傷な錠剤とを打錠した後PXRDを行った。
【0220】
結果
七水和物は、粘性/石鹸様フレークに似ていることが認められ、500μmの篩を通過するのは本当に困難であることがわかった。篩過した後、何らかの物質は篩に残ったままであった。これは、半水和物の場合には経験されなかったことである。ほとんどの場合、適度な重量バラツキ、脆弱性、および含量均一性と同様、許容できる圧縮力/破砕強度曲線が認められた。
【0221】
しかし、30mg錠剤では、主に混合粉体(固体)のフロー特性が不良であるため、七水和物製剤の打錠プロセスが簡単ではなく、結果として錠剤重量変動性が高くなった。このバラツキは、また圧縮力のバラツキを生じた(これは、下側パンチと上側パンチの間の一定の距離に調整されているので、パンチ間の距離が一定のダイ中の粉体が少なくなると、圧縮力が低下する)。また、パンチ表面への接着が、錠剤表面の損傷に伴って起こり、その結果僅かな脆弱性が生じる。
【0222】
七水和物についてDSCで観察(および上記の実施例11に記載)された吸熱を鑑み、これは、(内部温度70℃が、圧縮プロセス中に容易に到達することができることを考えれば)その多形体が圧縮によって錠剤になる能力を限定することができるものと予想された。したがって、30mg錠剤を上述の通りPXRDで分析するよう決定された。PXRD分析から、半水和物製剤についても七水和物製剤についても、打錠時に相転移は起こらなかったことがわかった。
【0223】
(実施例16)
溶解試験
インビトロ溶解試験をペミロラストカリウム(入手先Chemtronica AB、Stockholm、Sweden)およびペミロラストナトリウム半水和物(上記の実施例10に記載された方法に従って調製された)について、標準Ph. Eur. Methodologyを使用して行った(Apparatus 2(paddle)、Ph. Eur., 2.9.3、50mMホスファートバッファーpH 6.8を溶解媒体として使用した。撹拌速度50rpmを用いた。温度は37℃であった)。
【0224】
試験は、3回繰り返して行った。各物質30mgを別個の容器に加え、タイマーを開始した。目視検査サンプリングは、1mLのプラスチック注射器を使用して、(目視検査によって)物質がいかに迅速に溶解するように見えるかに応じて、一定間隔で行った。小さい直径のフィルターを使用して、試料をHPLCバイアルに濾過した。試料を逆相HPLCで分析した。ペミロラスト定量をUV検出によって行った。
【0225】
結果
カリウム塩は溶解媒体に加えられるとすぐに溶解した一方、ナトリウム塩の半水和物は大きな凝集体を形成し、溶解がより困難であるように思われた。2つの物質について得られた3つの溶解曲線の平均(±SD)(溶解対時間の百分率)を図8に示す。
【特許請求の範囲】
【請求項1】
ペミロラストナトリウム塩の半水和物形。
【請求項2】
ペミロラスト1モル当たり約0.3〜約0.7モルの水を含有する、請求項1に記載の化合物。
【請求項3】
実質的に結晶性である、請求項1または請求項2に記載の化合物。
【請求項4】
およそ26.6の2θ値(単位:度)を有する特徴的な結晶性ピークを含む粉末X線回折パターンを特徴とする、請求項1から3のいずれか一項に記載の化合物。
【請求項5】
およそ25.3の2θ値を有する別の強い結晶性ピークを含む粉末X線回折パターンを特徴とする、請求項4に記載の化合物。
【請求項6】
およそ13.0、15.3、18.2、および/または28.4の2θ値を有する別の強い結晶性ピークを含む粉末X線回折パターンを特徴とする、請求項4または請求項5に記載の化合物。
【請求項7】
溶媒から結晶化させる工程を含む、請求項1から6のいずれか一項に記載の化合物の調製方法。
【請求項8】
前記結晶化の前に、ペミロラストとナトリウム含有塩基を反応させる工程をさらに含む、請求項7に記載の方法。
【請求項9】
前記塩基が水酸化ナトリウムまたはナトリウムアルコキシドである、請求項8に記載の方法。
【請求項10】
前記溶媒が低級アルキルアルコールを含む、請求項7から9のいずれか一項に記載の方法。
【請求項11】
前記溶媒がメタノールまたはエタノールである、請求項10に記載の方法。
【請求項12】
前記結晶化が、ペミロラストナトリウムを、約10%以下(w/w、溶媒の割合として)の水の存在下で溶媒に部分溶解する工程を含む、請求項7から11のいずれか一項に記載の方法。
【請求項13】
約70℃未満の温度で実施される、請求項12に記載の方法。
【請求項14】
前記結晶化が、ペミロラストナトリウムを水性溶媒に約72℃以上で部分溶解する工程を含む、請求項7から11のいずれか一項に記載の方法。
【請求項15】
約72℃以上で濾過して、化合物を単離する工程をさらに含む、請求項14に記載の方法。
【請求項16】
前記結晶化が、ペミロラストナトリウムを水性溶媒に溶解し、続いて過剰量の貧溶媒を加える工程を含む、請求項7から11のいずれか一項に記載の方法。
【請求項17】
前記貧溶媒がエタノールを含む、請求項16に記載の方法。
【請求項18】
前記貧溶媒を溶媒のおよそ沸点で沈殿が起おこるまで加え、得られたものを約室温まで冷却する、請求項16または請求項17に記載の方法。
【請求項19】
ペミロラストナトリウムの高水和物の脱水を含む、請求項1から6のいずれか一項に記載の化合物の調製方法。
【請求項20】
請求項7から19のいずれか一項に記載の方法で得ることができる化合物。
【請求項21】
薬剤として使用するための、請求項1から6または20のいずれか一項に記載の化合物。
【請求項22】
請求項1から6または20のいずれか一項に記載の化合物を、薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤。
【請求項23】
炎症性障害の治療で使用するための、請求項1から6または20のいずれか一項に記載の化合物。
【請求項24】
炎症性障害を治療するための医薬を製造するための、請求項1から6または20のいずれか一項に記載の化合物の使用。
【請求項25】
炎症性障害の治療方法であって、請求項1から6または20のいずれか一項に記載の化合物のこのような治療を必要とする患者への投与を含む方法。
【請求項26】
前記障害がアテローム性動脈硬化症またはそれに伴う心血管障害である、請求項23に記載の化合物、請求項24に記載の使用、または請求項25に記載の方法。
【請求項27】
前記障害がアテローム性動脈硬化症である、請求項26に記載の化合物、使用、または方法。
【請求項28】
前記アテローム性動脈硬化症に伴う心血管障害が、大動脈瘤、動脈硬化症、末梢動脈閉塞性疾患、冠動脈疾患、冠疾患、プラーク破綻および/または不安定性、アテローム破綻および/または不安定性、血管疾患、動脈疾患、虚血性疾患、虚血、ならびに脳卒中から選択される、請求項26に記載の化合物、使用、または方法。
【請求項29】
前記冠動脈疾患が、狭心症、心筋梗塞および心臓発作から選択され、冠疾患が、心臓病および心疾患から選択され、脳卒中が、脳血管障害および一過性脳虚血発作から選択され、かつ/または障害が、プラーク破綻および/もしくは不安定性、またはアテローム破綻および/もしくは不安定性である、請求項28に記載の化合物、使用、または方法。
【請求項30】
前記障害が大動脈瘤である、請求項28に記載の化合物、使用、または方法。
【請求項31】
患者が急性冠症候群患者である、請求項26から30のいずれか一項に記載の化合物、使用、または方法。
【請求項32】
前記障害が全身性低グレード炎症である、請求項23に記載の化合物、請求項24に記載の使用、または請求項25に記載の方法。
【請求項33】
前記障害が、メタボリックシンドローム、肥満、糖尿病、および/または糖尿病性血管合併症から選択される、請求項23に記載の化合物、請求項24に記載の使用、または請求項25に記載の方法。
【請求項34】
(a) 請求項1から6または20のいずれか一項に記載の化合物; および
(b) 炎症性障害の治療に有用である1種もしくは複数の活性成分、またはその薬学的に許容される塩もしくは溶媒和物
を含む配合剤。
【請求項35】
請求項1から6または20のいずれか一項に記載の化合物を含む医薬製剤; 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物; および薬学的に許容される助剤、希釈剤、または担体を含む、請求項34に記載の配合剤。
【請求項36】
成分(A) 請求項1から6または20のいずれか一項に記載の化合物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤;および
成分(B) 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤
を含むパーツキットを含み、構成要素(A)および(B)はそれぞれ、他方と併用投与するのに適した形で提供されている、請求項34に記載の配合剤。
【請求項37】
炎症性障害の治療に有用である前記活性成分が、トロンボキサンA2アンタゴニスト、P2Y12アンタゴニスト、PPARγアゴニスト、アンジオテンシンIIの生成および/もしくは作用を抑制する化合物、血小板凝集抑制薬、またはスタチンである、請求項34から36のいずれか一項に記載の配合剤。
【請求項38】
前記活性成分がスタチンである、請求項37に記載の配合剤。
【請求項39】
前記活性成分が、アトルバスタチンまたはロスバスタチンである、請求項38に記載の配合剤。
【請求項40】
前記活性成分が、アスピリン/アセチルサリチル酸、エグアレン、オザグレル、ピコタミド、テルトロバン、セラトロダスト、ラマトロバン、プラスグレル、チカグレロル、クロピドグレル、リボグリタゾン、ナベグリタザル、バラグリタゾン、ロシグリタゾン、ピオグリタゾン、カプトプリル、ペリンドプリル、ラミプリル、カンデサルタン、ロサルタン、バルサルタン、またはアリスキレンである、請求項37に記載の配合剤。
【請求項1】
ペミロラストナトリウム塩の半水和物形。
【請求項2】
ペミロラスト1モル当たり約0.3〜約0.7モルの水を含有する、請求項1に記載の化合物。
【請求項3】
実質的に結晶性である、請求項1または請求項2に記載の化合物。
【請求項4】
およそ26.6の2θ値(単位:度)を有する特徴的な結晶性ピークを含む粉末X線回折パターンを特徴とする、請求項1から3のいずれか一項に記載の化合物。
【請求項5】
およそ25.3の2θ値を有する別の強い結晶性ピークを含む粉末X線回折パターンを特徴とする、請求項4に記載の化合物。
【請求項6】
およそ13.0、15.3、18.2、および/または28.4の2θ値を有する別の強い結晶性ピークを含む粉末X線回折パターンを特徴とする、請求項4または請求項5に記載の化合物。
【請求項7】
溶媒から結晶化させる工程を含む、請求項1から6のいずれか一項に記載の化合物の調製方法。
【請求項8】
前記結晶化の前に、ペミロラストとナトリウム含有塩基を反応させる工程をさらに含む、請求項7に記載の方法。
【請求項9】
前記塩基が水酸化ナトリウムまたはナトリウムアルコキシドである、請求項8に記載の方法。
【請求項10】
前記溶媒が低級アルキルアルコールを含む、請求項7から9のいずれか一項に記載の方法。
【請求項11】
前記溶媒がメタノールまたはエタノールである、請求項10に記載の方法。
【請求項12】
前記結晶化が、ペミロラストナトリウムを、約10%以下(w/w、溶媒の割合として)の水の存在下で溶媒に部分溶解する工程を含む、請求項7から11のいずれか一項に記載の方法。
【請求項13】
約70℃未満の温度で実施される、請求項12に記載の方法。
【請求項14】
前記結晶化が、ペミロラストナトリウムを水性溶媒に約72℃以上で部分溶解する工程を含む、請求項7から11のいずれか一項に記載の方法。
【請求項15】
約72℃以上で濾過して、化合物を単離する工程をさらに含む、請求項14に記載の方法。
【請求項16】
前記結晶化が、ペミロラストナトリウムを水性溶媒に溶解し、続いて過剰量の貧溶媒を加える工程を含む、請求項7から11のいずれか一項に記載の方法。
【請求項17】
前記貧溶媒がエタノールを含む、請求項16に記載の方法。
【請求項18】
前記貧溶媒を溶媒のおよそ沸点で沈殿が起おこるまで加え、得られたものを約室温まで冷却する、請求項16または請求項17に記載の方法。
【請求項19】
ペミロラストナトリウムの高水和物の脱水を含む、請求項1から6のいずれか一項に記載の化合物の調製方法。
【請求項20】
請求項7から19のいずれか一項に記載の方法で得ることができる化合物。
【請求項21】
薬剤として使用するための、請求項1から6または20のいずれか一項に記載の化合物。
【請求項22】
請求項1から6または20のいずれか一項に記載の化合物を、薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤。
【請求項23】
炎症性障害の治療で使用するための、請求項1から6または20のいずれか一項に記載の化合物。
【請求項24】
炎症性障害を治療するための医薬を製造するための、請求項1から6または20のいずれか一項に記載の化合物の使用。
【請求項25】
炎症性障害の治療方法であって、請求項1から6または20のいずれか一項に記載の化合物のこのような治療を必要とする患者への投与を含む方法。
【請求項26】
前記障害がアテローム性動脈硬化症またはそれに伴う心血管障害である、請求項23に記載の化合物、請求項24に記載の使用、または請求項25に記載の方法。
【請求項27】
前記障害がアテローム性動脈硬化症である、請求項26に記載の化合物、使用、または方法。
【請求項28】
前記アテローム性動脈硬化症に伴う心血管障害が、大動脈瘤、動脈硬化症、末梢動脈閉塞性疾患、冠動脈疾患、冠疾患、プラーク破綻および/または不安定性、アテローム破綻および/または不安定性、血管疾患、動脈疾患、虚血性疾患、虚血、ならびに脳卒中から選択される、請求項26に記載の化合物、使用、または方法。
【請求項29】
前記冠動脈疾患が、狭心症、心筋梗塞および心臓発作から選択され、冠疾患が、心臓病および心疾患から選択され、脳卒中が、脳血管障害および一過性脳虚血発作から選択され、かつ/または障害が、プラーク破綻および/もしくは不安定性、またはアテローム破綻および/もしくは不安定性である、請求項28に記載の化合物、使用、または方法。
【請求項30】
前記障害が大動脈瘤である、請求項28に記載の化合物、使用、または方法。
【請求項31】
患者が急性冠症候群患者である、請求項26から30のいずれか一項に記載の化合物、使用、または方法。
【請求項32】
前記障害が全身性低グレード炎症である、請求項23に記載の化合物、請求項24に記載の使用、または請求項25に記載の方法。
【請求項33】
前記障害が、メタボリックシンドローム、肥満、糖尿病、および/または糖尿病性血管合併症から選択される、請求項23に記載の化合物、請求項24に記載の使用、または請求項25に記載の方法。
【請求項34】
(a) 請求項1から6または20のいずれか一項に記載の化合物; および
(b) 炎症性障害の治療に有用である1種もしくは複数の活性成分、またはその薬学的に許容される塩もしくは溶媒和物
を含む配合剤。
【請求項35】
請求項1から6または20のいずれか一項に記載の化合物を含む医薬製剤; 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物; および薬学的に許容される助剤、希釈剤、または担体を含む、請求項34に記載の配合剤。
【請求項36】
成分(A) 請求項1から6または20のいずれか一項に記載の化合物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤;および
成分(B) 炎症性障害の治療に有用である活性成分、またはその薬学的に許容される塩もしくは溶媒和物を薬学的に許容される助剤、希釈剤、または担体と混合して含む医薬製剤
を含むパーツキットを含み、構成要素(A)および(B)はそれぞれ、他方と併用投与するのに適した形で提供されている、請求項34に記載の配合剤。
【請求項37】
炎症性障害の治療に有用である前記活性成分が、トロンボキサンA2アンタゴニスト、P2Y12アンタゴニスト、PPARγアゴニスト、アンジオテンシンIIの生成および/もしくは作用を抑制する化合物、血小板凝集抑制薬、またはスタチンである、請求項34から36のいずれか一項に記載の配合剤。
【請求項38】
前記活性成分がスタチンである、請求項37に記載の配合剤。
【請求項39】
前記活性成分が、アトルバスタチンまたはロスバスタチンである、請求項38に記載の配合剤。
【請求項40】
前記活性成分が、アスピリン/アセチルサリチル酸、エグアレン、オザグレル、ピコタミド、テルトロバン、セラトロダスト、ラマトロバン、プラスグレル、チカグレロル、クロピドグレル、リボグリタゾン、ナベグリタザル、バラグリタゾン、ロシグリタゾン、ピオグリタゾン、カプトプリル、ペリンドプリル、ラミプリル、カンデサルタン、ロサルタン、バルサルタン、またはアリスキレンである、請求項37に記載の配合剤。
【図1】


【図2】

【図3】
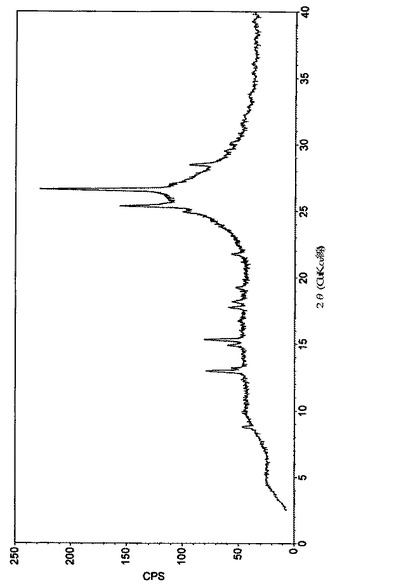
【図4】
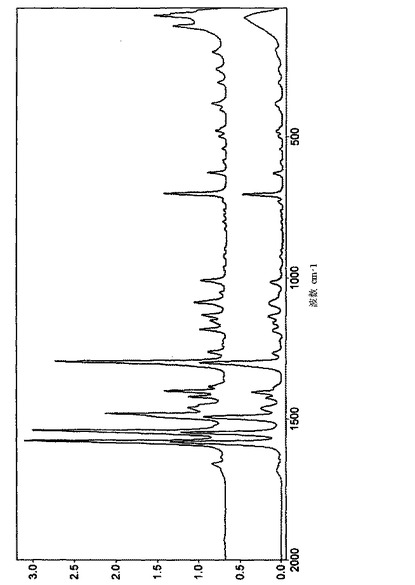
【図5】

【図6】
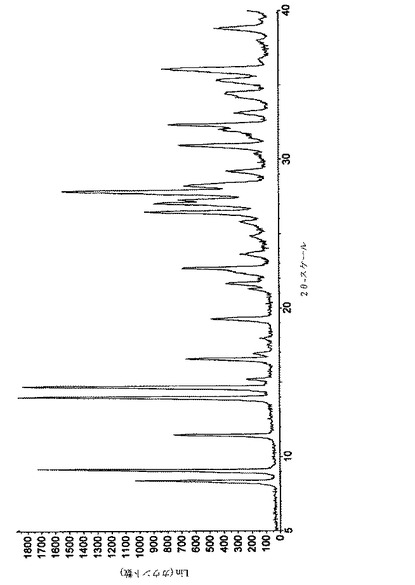
【図7】
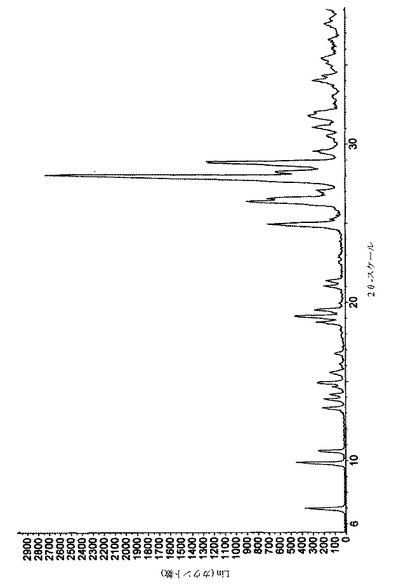
【図8】
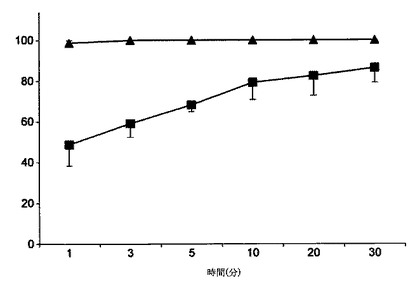
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2012−530118(P2012−530118A)
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願番号】特願2012−515552(P2012−515552)
【出願日】平成22年6月15日(2010.6.15)
【国際出願番号】PCT/GB2010/001168
【国際公開番号】WO2010/146348
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(509171232)カルドズ・アーベー (4)
【Fターム(参考)】
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願日】平成22年6月15日(2010.6.15)
【国際出願番号】PCT/GB2010/001168
【国際公開番号】WO2010/146348
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(509171232)カルドズ・アーベー (4)
【Fターム(参考)】
[ Back to top ]