
ペリプラズム発現によるコンビナトリアルタンパク質ライブラリーのスクリーニング
本発明は、低分子およびペプチドと結合できる結合タンパク質を単離するための迅速なアプローチを提供することにより、先行技術の欠点を克服する。本技法では、抗体配列等の候補結合タンパク質のライブラリーをグラム陰性菌のペリプラズム内で発現させ、標識リガンドと混合する。リガンドに対する親和性を有する組換えポリペプチドを発現するクローンにおいて、結合タンパク質に結合する標識リガンドの濃度が増加し、これにより残りのライブラリーからの細胞の単離が可能となる。標的リガンドを蛍光標識する場合、蛍光活性化細胞分類法(FACS)により細胞を単離できる。本アプローチは先行技術の方法よりも迅速であり、ファージディスプレイで用いられるリガンド融合タンパク質の外表面発現に関連する問題を回避するものである。本発明者らはまた、開発したスクリーニング法を用いて最初に調製した改良抗体を提供した。
【発明の詳細な説明】
【背景技術】
【0001】
発明の背景
本出願は2002年7月15日に提出した米国特許仮出願第60/396,058号の優先権を主張し、また本出願は2000年10月27日に提出した米国特許出願第09/699,023号の一部継続出願でもある。前述の出願の全開示は、参照として本明細書に組み入れられる。政府は、米陸軍ARO MURIプログラムおよび生物センサー開発のテキサス連合(Texas Consortium)に従って、かつ米陸軍研究所との契約番号DADD17-01-D-0001に関連して、本発明において権利を所有すると考えられる。
【技術分野】
【0002】
1. 発明の分野
本発明は、一般にタンパク質工学の分野に関する。より詳細には、リガンド結合ポリペプチドの単離を可能にするための、ポリペプチドのコンビナトリアルライブラリーのスクリーニングの改良法に関する。
【0003】
2. 関連技術の説明
高い親和性および特異性でリガンドと結合するか、または反応物(基質)の所望の生成物への酵素転換を触媒するポリペプチドの単離は、生物工学における重要な過程である。適切なスクリーニング法が利用できるのであれば、変異体の大きなライブラリーから、所望の基質特異性を有するタンパク質および酵素を含むリガンド結合ポリペプチドを単離することが可能である。103〜105個の異なる変異体から構成される小さなタンパク質ライブラリーは、まず各クローンを個別に培養し、次に特異的結合を示すクローンを検出する従来のアッセイ法により、スクリーニングすることができる。例えば、異なるタンパク質変異体を発現する個々のクローンをマイクロタイターウェルプレートで増殖することも、または個々のコロニーを寒天プレート等の半固形培地で培養することもできる。結合を検出するには、細胞を溶解してタンパク質を放出し、溶解物をナイロンフィルターに転写し、次いでこれを放射標識または蛍光標識したリガンドを用いてプロービングする(DeWildtら、2000)。しかし、たとえロボットによる自動化および高密度アレイにおいて結合を検出するためのデジタル画像システムを用いたとしても、数千万ないし数十億個のクローンからなる大きなライブラリーをスクリーニングすることは不可能である。そのような大きさのライブラリーのスクリーニングは、ナノモル以下の範囲の親和性を有する酵素またはタンパク質結合物の新たな単離に必要とされる。
【0004】
非常に大きなタンパク質ライブラリーのスクリーニングは、ウイルスまたは細胞の表面上でのタンパク質の提示に依存する様々な技法によって達成された(Ladnerら、1993)。提示技術の根底にある前提は、生物粒子(すなわち、細胞またはウイルス)の外部表面に固定されるように操作されたタンパク質が、細胞の溶解を必要とせずにリガンドへの結合に直接利用できることである。リガンドに対する親和性を有するタンパク質を提示するウイルスまたは細胞は、磁気分離またはフローサイトメトリーによる、固定化リガンドに対する連続的な吸着/脱離を含む様々な方法によって単離され得る(Ladnerら、1993、米国特許第5,223,409号、Ladnerら、1998、米国特許第5,837,500号、Georgiouら、1997、Shustaら、1999)。
【0005】
タンパク質ライブラリーのスクリーニング用途のために最も広く使用される提示技術は、ファージディスプレイである。ファージディスプレイは、特定のリガンドに結合するタンパク質を発見するため、ならびに結合親和性および特異性を操作するための、十分に確立された強力な技法である(RodiおよびMakowski、1999)。ファージディスプレイでは、関心対象の遺伝子を、表面に露出されるタンパク質、最も一般的にはpIIIをコードするファージ遺伝子にインフレームで融合させる。遺伝子融合物は、2つのドメインが独立して折りたたまれるキメラタンパク質に翻訳される。リガンドに対する結合親和性を有するタンパク質を提示するファージは、固定化リガンドに対する選択的吸着、「パニング」として周知の過程により、容易に濃縮され得る。結合したファージを、通常は酸性溶離によって表面から脱着し、大腸菌細胞の感染を介して増幅する。通常、最大で1010個までの多様性を有する非常に大きなライブラリーからでさえ、特異的ポリペプチドを提示するファージを選択するのに、4〜6ラウンドのパニングおよび増幅で十分である。密に結合するポリペプチドを提示するクローンを迅速に濃縮するための、ファージディスプレイのいくつかの変法が開発されている(DuenasおよびBorrebaeck、1994;Malmborgら、1996;Kjaerら、1998;Burioniら、1998;Levitan、1998;Mutuberriaら、1999;Johnsら、2000)。
【0006】
ファージディスプレイ技術の最も重要な用途の1つは、高親和性抗体の単離である(Dall'AcquaおよびCarter、1998;Hudsonら、1998;Hoogenboomら、1998;MaynardおよびGeorgiou、2000)。scFvまたはFAB断片の非常に大きくかつ構造的に多様なライブラリーが構築されており、多数の合成抗原および天然抗原に対する抗体のインビトロ単離のための使用に成功している(Griffithsら、1994;Vaughanら、1996;Sheetsら、1998;Piniら、1998;de Haardら、1999;Knappikら、2000;SblatteroおよびBradbury、2000)。選択した抗体をCDRまたは全遺伝子CDRの突然変異誘発に供したライブラリーから、改善された親和性または特異性を有する抗体断片を単離することができる(Hawkinsら、1992;Lowら、1996;Thompsonら、1996;ChowdhuryおよびPastan、1999)。最終的に、溶解度が低いことで知られているscFvの発現特性も、変異体ライブラリーのファージディスプレイによって改善された(Dengら、1994;Coiaら、1997)。
【0007】
しかし、いくつかの目覚しい成功にもかかわらず、ファージに提示されるライブラリーのスクリーニングは多くの要素によって複雑になり得る。第1に、ファージディスプレイでは、ファージの会合を可能にし依然として細胞増殖を維持するために必要である抗体断片遺伝子III融合物の低発現レベルが理由で、細菌における適切な発現に対して最小限の選択が課される(Krebberら、1996、1997)。結果として、数ラウンドのパニング後に単離されたクローンは、大腸菌で調製規模で産生することが困難である場合が多い。第2に、ファージの提示したタンパク質がリガンドに結合し得ても、場合によってはそれらの非融合可溶性対応物は結合しない(Griepら、1999)。第3に、高い結合親和性を有するリガンド結合タンパク質およびより特異的な抗体の単離は、ファージの会合を完了するために遺伝子IIIタンパク質がビリオン当たり約5コピーで存在する必要性による結合活性効果によって複雑になり得る。主に一価のタンパク質提示を生じる系でさえ、ほぼ常にタンパク質の複数コピーを含むクローンの小さな画分が存在する。そのようなクローンは固定化表面により密接に結合し、一価のファージと比較してより高い親和性で濃縮される(Dengら、1995;MacKenzieら、1996、1998)。第4に、理論的な分析は別として(Levitan、1998)、パニングは、実験条件の影響、例えば弱くまたは非特異的に結合したファージを除去するための洗浄段階のストリンジェンシーが、実験の最終結果に基づいて試行錯誤によってのみ決定され得る点で、今だに「ブラックボックス」過程である。最後に、たとえpIIIおよびより少ない程度でファージコートの他のタンパク質が、異種性ポリペプチドの融合に対して一般に耐性があるとしても、ファージ生合成過程に組み込まれる必要性により、ライブラリーの多様性を制限し得る生物学的制約が課される。したがって、当技術分野において、これらの制限を克服し得る技法の大きな必要性が存在する。
【0008】
タンパク質ライブラリーはまた、細菌細胞、菌類細胞、または高等生物の細胞の表面上にも提示されている。細胞が提示するライブラリーは、典型的にフローサイトメトリーによってスクリーニングされる(Georgiouら、1997、Daughertyら、2000)。しかし、ファージディスプレイと同様に、タンパク質を細胞表面上で発現するように操作する必要がある。これにより、いくつかの潜在的な制限が課される。例えば、細胞表面上にタンパク質を提示する必要性により、スクリーニングされ得るタンパク質およびタンパク質変異体の多様性を制限する生物学的制約が課される。また、いくつかのポリペプチド鎖からなる複合タンパク質は、細菌、繊維状ファージ、または酵母の表面上に容易に提示され得ない。このように、当技術分野において、上記の制限をすべて回避し、非常に大きなポリペプチドライブラリーをスクリーニングするための完全に新規な手段を提供する技術の大きな必要性が存在する。
【発明の開示】
【0009】
発明の概要
1つの局面において、本発明は、以下の段階を含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を含む細菌を取得する方法を提供する:(a) 内膜および外膜およびペリプラズムを含むグラム陰性菌であって、細菌のペリプラズム内に候補結合ポリペプチドをコードする核酸配列を発現するグラム陰性菌を提供する段階;(b) 細菌をペリプラズム内に拡散し得る標識リガンドと接触させる段階;および(c) 候補結合ポリペプチドに結合した標識リガンドの存在に基づき、細菌を選択する段階。本発明の1つの態様において、本方法は、(a) 候補結合ポリペプチドと細菌の内膜の外側に固定されるポリペプチドとを含む融合タンパク質をコードする核酸配列を発現するグラム陰性菌を提供する段階; (b) 細菌を細菌内に拡散し得る標識リガンドと接触させる段階;および(c) 候補結合ポリペプチドに結合した標識リガンドの存在に基づき、細菌を選択する段階を含む。
【0010】
本発明の特定の態様において、本方法は、(d) 細菌から候補結合ポリペプチドをコードする核酸配列をクローニングする段階をさらに含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を取得する方法としてさらに定義され得る。本方法において、核酸配列は、融合ポリペプチドの発現を内膜の外側に方向づけ得るリーダー配列に機能的に結合しているとしてさらに定義され得る。
【0011】
本発明の1つの態様では、グラム陰性菌は大腸菌である。本発明の特定のさらなる態様では、本方法は、グラム陰性菌の集団の使用を含むとしてさらに定義され得る。そのような集団は、複数の候補結合ポリペプチドを含む複数の融合ポリペプチドをまとめて発現し得る。集団は、以下の段階を含む方法によって取得し得る:(a) 候補結合ポリペプチドおよび内膜アンカーポリペプチドを含む複数の融合ポリペプチドをコードする複数の核酸配列を調製する段階;および(b) このDNA挿入物でグラム陰性菌の集団を形質転換する段階。本方法において、グラム陰性菌の集団を標識リガンドと接触させてもよい。
【0012】
本発明の1つの態様では、候補結合ポリペプチドは抗体またはその断片としてさらに定義され、または候補結合ポリペプチドは抗体以外の結合タンパク質であってもよい。候補結合ポリペプチドはまた、酵素としてさらに定義され得る。標識リガンドには、ペプチド、ポリペプチド、酵素、核酸、および/または合成分子が含まれ得る。標識リガンドは、蛍光標識を含む任意の適切な手段によって標識され得る。本発明の特定の態様において、候補結合ポリペプチドをコードする核酸が既知の核酸配列に隣接しているとしてさらに定義される場合、それによって選択後に核酸が増幅され得る。
【0013】
本発明の特定の態様において、結合ポリペプチドをコードする核酸配列を含む細菌を取得する方法は、標識リガンドに対する細菌の外膜の透過性を増大させるために細菌を処理する段階を含む。本発明の1つの態様において、処理段階には、高浸透圧条件による細菌の処理、物理的ストレスによる細菌の処理、および/またはファージによる細菌の処理が含まれ得る。本方法は、細菌の外膜を除去する段階、または別法として様々な分子量のポリペプチドの拡散を可能にする欠陥外膜を有する突然変異細菌を使用する段階を含んでもよい。本方法はまた、約25℃を含む準生理的温度で細菌を培養する段階を含んでよい。本方法は、候補結合ポリペプチドに結合していない標識リガンドを除去する段階をさらに含んでよい。
【0014】
本発明による選択は、任意の適切な方法を含み得る。本発明の1つの態様では、選択はフローサイトメトリー(例えば、蛍光活性化細胞分類法(FACS))を含む。別の態様では、選択は磁気選別を含む。リガンドと候補結合ポリペプチドは、可逆的に結合し得る。ポリペプチドは、天然大腸菌リポタンパク質NlpAに由来する6残基配列へのN末端融合、任意の膜貫通タンパク質またはその断片、および繊維状ファージの遺伝子IIIタンパク質またはその断片を含む、任意の適切なアンカーにより内膜の外側に固定され得る。
【0015】
さらに別の局面において、本発明は、表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合する、単離された抗体またはその断片を提供する。そのような抗体またはその断片は、約96 pMから約21 pMの間および/または約35 pMから約21 pMの間の結合親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義され得る。単離された抗体またはその断片は、IgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義され得る。抗体はヒト化抗体であってよく、ヒト抗体であってもよい。特定の態様において、単離された抗体またはその断片は、少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む。
【0016】
さらに別の局面において、本発明は、炭疽菌防御抗原に免疫学的に結合し、かつ、I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、L94P、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lからなる群より選択される改変(I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、およびL94Pは可変軽鎖内に存在し、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lは可変重鎖内に存在する)を含む可変軽鎖および可変重鎖を除く、配列番号:21の可変軽鎖および可変重鎖を含む、単離された抗体またはその断片を提供する。本発明の特定の態様では、単離された抗体またはその断片は、上記改変の可能なすべての組み合わせを含む、約3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18個、またはすべての改変を含む、約2個からすべてのそのような改変を含むと定義され得る。
【0017】
本発明の特定の局面では、単離された抗体またはその断片は、表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される。本発明のさらなる態様において、抗体またはその断片はQ55LおよびS56Pを含む。単離された抗体またはその断片は、配列番号:22または24の可変軽鎖および/または可変重鎖を含み得る。1つの態様において、単離された抗体またはその断片は、配列番号:22または24を含む。単離された抗体またはその断片は、scAb、Fab、またはSFvとしてさらに定義され得り、またIgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義され得る。単離された抗体またはその断片はヒト化抗体であってよく、ヒト型であってもよい。特定の態様において、単離された抗体またはその断片は、少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む。
【0018】
さらに別の局面において、本発明は、本発明が提供する抗体またはその断片をコードする単離された核酸を提供する。1つの態様において、核酸は、配列番号:23および/または25の可変軽鎖をコードする。別の態様において、核酸は、配列番号:23および/または25の可変重鎖をコードする。さらに別の態様において、核酸は配列番号:23のポリペプチドをコードし、別の態様においては、配列番号:25のポリペプチドをコードする。
【0019】
例示的態様の説明
本発明は、抗体または抗体断片を含む、特定の分子標的を認識する結合ポリペプチドを単離する新規方法を提供することにより、先行技術の制限を克服する。この技法では、ポリペプチド(例えば、抗体または他の結合ポリペプチド)変異体のライブラリーを作製し、グラム陰性菌で発現させることができる。変異体ポリペプチドは、ペリプラズムに面した細菌の内(細胞質)膜上に固定された融合タンパク質として発現させることができる。融合タンパク質とは、連続したポリペプチドを形成するために連結された2つまたはそれ以上の開始ポリペプチドからなるポリペプチドである。連結されたポリペプチドは、典型的に異なる供給源に由来する。次に、様々な化学的、物理的、もしくは他の処理により、または透過性の増加をもたらす変異体を使用することにより、細菌のペリプラズム(外)膜を透過性にする。細菌外膜の透過処理により、膜に固定されたポリペプチドが外部溶液に添加した標的高分子に接近できるようになる。
【0020】
微生物骨格上への異種性タンパク質の提示は、ワクチン開発、バイオレメディエーション、およびタンパク質工学を含む多くの異なる分野において魅力的な用途を有する。グラム陰性菌では、N末端またはC末端キメラ融合によりタンパク質が細胞表面に提示されるように設計されたディスプレイシステムが存在する。外膜タンパク質、リポタンパク質、表面構造タンパク質、およびリーダーペプチドへの融合を含む、タンパク質の局在を方向づけるために用いられる多くの異なる戦略が存在するが、多くが同じ制限を共有する。1つの制限は、提示できるタンパク質の大きさである。多くの提示骨格は数百アミノ酸を許容するにとどまり、このことが提示できるタンパク質の範囲を著しく限定する。また、提示とは関心対象のタンパク質がその環境と相互作用し得るように位置づけられることを意味するが、これらの系の多くの主要な制限は、グラム陰性菌の外表面の構造、特にリポ多糖(LPS)分子の存在が、外部から添加したリガンドの結合を阻害する立体的制限を有する点である。別の制限は、提示されたタンパク質が外膜の外部表面に局在化されるという必要条件に起因する。このため、ポリペプチドはまず細胞質膜を越えて分泌されなければならず、次にペリプラズム空間を横断しなければならず、最後に外膜中に適切に構築されなければならない。結合ポリペプチドは、所与のリガンドに結合し得る、少なくとも2つのアミノ酸残基の任意の種類の分子であってよい。結合によって、免疫学的相互作用が起こることを意味する。生合成の制限により、この様式で提示され得るタンパク質の種類が制限される。例えば、大きなポリペプチド(例えばアルカリホスファターゼ)は、大腸菌表面上に提示することができない(Stathopoulosら、1999)。
【0021】
本発明に従って、タンパク質を内膜の外表面に固定して提示させることにより、先行技術の制限を克服することができる。外膜を透過性にする条件を利用することにより、内膜に固定されるscFv抗体(約30 kDaの大きさ)を発現する大腸菌を、例えばフルオロフォアに複合化した標的抗原で標識することができ、続いてこの大腸菌を用いて、機能獲得変異体を単離するために、フローサイトメトリーを利用してタンパク質ライブラリーを選別することができることが、この技法を用いて実証された。
【0022】
当業者に周知であり、例えばTris-EDTA-リゾチームの使用を含む細菌外膜の破壊の後では、少なくとも240 kDaまでの大きさの標識抗原を検出することができる。蛍光標識を用いると、細胞をフローサイトメトリーにより単離することができ、単離されたクローンのDNAをPCRによりレスキューすることができる。本発明者らは、2ラウンドのAPExにより、炭疽菌防御抗原(PA)に対する中和抗体の親和性が120倍を超えて改善され、最終KD=35 pMを示したことを実証する。
【0023】
本発明の1つの態様では、標的分子を蛍光色素で標識する。したがって、標的分子を認識するポリペプチドを発現する細菌クローンは蛍光標識した標的に結合し、次には蛍光を発するようになる。続いて、所望の結合タンパク質を発現する蛍光細菌を、フローサイトメトリー等の自動化技法を用いて集団から濃縮することができる。
【0024】
ポリペプチドライブラリーを、内膜アンカーポリペプチドへの融合を介して、大腸菌または他のグラム陰性菌の内膜のペリプラズム面に結合することができる。使用できる1つのアンカーには、大腸菌のNlpA(新規リポタンパク質A)遺伝子の最初の6アミノ酸が含まれる。しかし、他の単一膜貫通または多指向性(polytropic)膜タンパク質またはペプチド配列を、固定する目的で使用することも可能である。
【0025】
本技法の1つの恩典は、候補結合ポリペプチドを内膜のペリプラズム面に固定することにより、通常ポリペプチドの標識標的分子への到達性を制限する細菌外膜の透過処理が可能になる点である。結合ポリペプチドを膜のペリプラズム面に固定することにより、外膜が損なわれた場合に細胞からの結合ポリペプチドの遊離が妨げられる。したがって、コンビナトリアルライブラリーから抗体および他の結合タンパク質を含む大きな結合ポリペプチドおよびリガンドを単離するために、本技法を用いることができる。本技法は、高いシグナル対ノイズ比を提供するばかりでなく、非常に大きな抗原分子に対するポリペプチドまたは抗体結合物の単離を可能にする。本方法によって、より大きな標的の選択が可能になるため、例えばメラノーマまたは他の特定の種類の腫瘍細胞等の腫瘍細胞を含む細胞上に発現する特定の抗原マーカー等の標的の選択において使用できる可能性が存在する。腫瘍特異的抗体は、癌の治療において大きな展望を示している。
【0026】
ペリプラズムは、グラム陰性菌の内膜および外膜によって規定される空間である。野生型大腸菌および他のグラム陰性菌では、外膜は、600 Daよりも大きい分子のペリプラズム空間への拡散を厳密に制限する透過性障壁として役立つ(DecadおよびNikado、1976)。より大きい分子のペリプラズム内への拡散を可能にする外膜の透過性を増大させる条件は、ライブラリーをスクリーニングする能力の点から見て2つの悪影響を有する:(a) 細胞生存度がかなりの程度で影響を受ける、および(b) 細胞内への分子の拡散は、タンパク質および他の高分子の拡散を伴う。
【0027】
本発明者らは、候補結合ポリペプチドを内膜の外(ペリプラズム)側に固定することにより、または可溶型の候補結合ポリペプチドをペリプラズム中に発現させることにより、リガンドおよびポリペプチドの蛍光複合体が外膜を通過し、候補結合タンパク質に結合し、内膜に結合してとどまることを可能にする技法を同定した。したがって、リガンドに対する親和性を有する組換えポリペプチドを発現する細菌細胞において、結合タンパク質に結合している標識リガンドを検出することができ、それによって残りのライブラリーからその細菌を単離することができる。標的リガンドの蛍光標識を用いる場合には、フローサイトメトリー(蛍光活性化細胞分類法(FACS))により細胞を効率よく単離することができる。このアプローチを用いると、細菌において融合タンパク質を発現する既存のライブラリーを、ファージディスプレイシステムまたは細胞表面ディスプレイシステムにサブクローニングする必要なく、リガンド結合に関して容易に試験することができる。
【0028】
ペリプラズム発現はまた、本発明に従って可溶型での発現によっても実施することが可能である。本発明に従って使用可能である、ペリプラズムでの可溶性発現、および候補結合タンパク質のスクリーニングに関する技法は、2000年10月27日に提出した米国特許出願第09/699,023号に詳細に記載されており、この開示全体は本明細書に参照として明確に組み入れられる。
【0029】
I. 固定化ペリプラズム発現
ファージディスプレイおよび細菌細胞表面ディスプレイという先行技術の方法は、定義により、標的への限定されない接近を可能にするために、タンパク質が用いるビヒクルの外表面上に物理的に提示される必要があるという制限に遭う(ファージへの固定化、またはフローサイトメトリーのため蛍光複合化リガンド)(米国特許第5,223,409号、その開示はその全体が参照として本明細書に明確に組み入れられる)。しかし、ある種のタンパク質はファージ上に十分に提示されないことが知られており(Maenakaら、1996;Coreyら、1993)、細胞表面提示の毒性効果が詳細に論じられている(Daughertyら、1999)。さらに、内膜上への結合を妨げるリポ多糖が存在しない。
【0030】
本発明者らは本明細書において、まだ依然として比較的大きなリガンドに接近しやすい融合タンパク質として、結合タンパク質を内膜のペリプラズム面上に発現できる技法について説明した。本明細書で用いる「結合ポリペプチド」という用語には、抗体だけでなく、抗体の断片、および所与の標的分子に潜在的に結合し得るタンパク質を含む任意の他のペプチドも含まれる。抗体または他の結合ペプチドは、本発明により、内膜のペリプラズム面へのアンカーとして機能し得るポリペプチドとの融合ポリペプチドとして発現され得る。そのような技法を、「固定化ペリプラズム発現」または「APEx」と命名できる。
【0031】
ペリプラズム区画は、グラム陰性細胞の内膜と外膜との間に含まれる(例えば、Oliver、1996を参照のこと)。ペリプラズム区画は、細胞区画として、細胞の増殖および分裂に伴って大きさ、形状、および内容物が変動する場合がある。ペプチドグリカンの枠組み内で、ヘテロポリマーはペリプラズムタンパク質およびわずかな水の高密度環境であり、この区画にゲル様の粘稠度をもたらす(Hobotら、1984;van WielinkおよびDuine、1990)。ペプチドグリカンは、外膜への近接性に依存して様々な程度に重合され、細胞形状および浸透圧溶解に対する耐性を与えるムレイン袋状構造を形成する。
【0032】
外膜(Nikaido、1996を参照のこと)は、リン脂質、ポーリンタンパク質、
および、培地内へ伸長するリポ多糖(LPS)から構成される。外膜の完全性の分子基盤は、2価カチオン(Mg2+およびCa2+)を結合し、互いを静電気的に連結して表面上に高度に規則正しい準結晶配向した「瓦屋根」を形成するLPSの能力により備わっている(Labischinskiら、1985)。膜は、ポーリンを介して、約650 Da以下の分子の通過を可能にする非常に厳密な透過性障壁を形成する(Burmanら、1972;DecadおよびNikaido、1976)。大量の水で満たされたポーリンチャネルは、単糖および二糖、イオン、ならびにアミノ酸のペリプラズム区画への自由な通過の許容を主に担う(Naeke、1976;NikaidoおよびNakae、1979;NikaidoおよびVaara、1985)。分子のペリプラズムへの接近の、そのような厳密な生理的制御により、使用するリガンドが650 Da以下の排除限界でなくとも、または正常に浸透性である化合物の類似体でなくとも、APExが作用するはずであることは、一見して不可解に見えるかもしれない。しかし本発明者らは、2000 Daよりも大きいリガンドがペリプラズム膜を破壊することなくペリプラズム内に拡散し得ることを示した。このような拡散は、本明細書において以下に記載するように、外膜をより透過性にする、細菌細胞の1つまたは複数の処理によって支援され得る。
【0033】
II. アンカーなしのディスプレイライブラリーのスクリーニング
ファージディスプレイおよび細菌細胞表面ディスプレイという先行技術の方法は、定義により、標的への限定されない接近を可能にするために、タンパク質が用いるビヒクルの表面上に物理的に提示される必要があるという制限に遭う(ファージへの固定化、またはフローサイトメトリーのため蛍光複合化リガンド)(米国特許第5,223,409号、その開示はその全体が参照として本明細書に明確に組み入れられる)。ある種のタンパク質はファージ上に十分に提示されないことが知られており(Maenakaら、1996;Coreyら、1993)、細胞表面提示の毒性効果が詳細に論じられている(Daughertyら、1999)。提示すべきタンパク質はまた融合タンパク質として発現される必要があり、融合タンパク質はその機能を変化させる可能性がある。したがって、任意のディスプレイシステムによって課される選択の制約により、その適用が比較的小さくかつ「単純な」タンパク質に限定され、多くの大きくかつ複雑な多サブユニット種の接近が拒絶される可能性がある。後者は、宿主細胞の生存度に非常に重大な影響を及ぼすことなく、ファージ会合の終結または外膜の移行の複雑な過程に効率よく参加することができない可能性が非常に高い。
【0034】
本明細書において、発現した結合タンパク質、例えば抗体が、大腸菌のペリプラズム区画に標的され得り、なお結合リガンドおよびペプチドを受け入れられる条件を記載する。本明細書で用いる「結合タンパク質」という用語には、抗体だけでなく、抗体の断片、および所与の標的分子に潜在的に結合し得る任意の他のポリペプチドまたはタンパク質も含まれる。固定されるのと同様に、抗体または他の結合タンパク質は、本発明により、融合タンパク質としてではなく直接発現させることができる。このような技法を「アンカーなしの提示」(ALD)と命名できる。ALDがどのように機能し得るかを理解するため、それが機能する位置を認識する必要がある。
【0035】
ペリプラズム区画は、グラム陰性細胞の内膜と外膜との間に含まれる(例えば、Oliver、1996を参照のこと)。ペリプラズム区画は、細胞区画として、細胞の増殖および分裂に伴って大きさ、形状、および内容物が変動する場合がある。ペプチドグリカンの枠組み内で、ヘテロポリマーはペリプラズムタンパク質およびわずかな水の高密度環境であり、この区画にゲル様の粘稠度をもたらす(Hobotら、1984;van WielinkおよびDuine、1990)。ペプチドグリカンは、外膜への近接性に依存して様々な程度に重合され、細胞形状および浸透圧溶解に対する耐性を与えるムレイン袋状構造を形成する。
【0036】
外膜(Nikaido、1996を参照のこと)は、リン脂質、ポーリンタンパク質、
および、培地内へ伸長するリポ多糖(LPS)から構成される。外膜の完全性の分子基盤は、2価カチオン(Mg2+およびCa2+)を結合し、互いを静電気的に連結して表面上に高度に規則正しい準結晶配向した「瓦屋根」を形成するLPSの能力により備わっている(Labischinskiら、1985)。膜は、ポーリンを介して、約650 Da以下の分子の通過を可能にする非常に厳密な透過性障壁を形成する(Burmanら、1972;DecadおよびNikaido、1976)。大量の水で満たされたポーリンチャネルは、単糖および二糖、イオン、ならびにアミノ酸のペリプラズム区画への自由な通過の許容を主に担う(Naeke、1976;NikaidoおよびNakae、1979;NikaidoおよびVaara、1985)。分子のペリプラズムへの接近の、そのような厳密な生理的制御により、使用するリガンドが650 Da以下の排除限界でなくとも、または正常に浸透性である化合物の類似体でなくとも、ALDが作用するはずであることは、一見して不可解に見えるかもしれない。しかし本発明者らは、ALDに関して、リガンドがペリプラズム内に拡散し得ることを示した。このような拡散は、本明細書において以下に記載するように、外膜をより透過性にする、細菌細胞の1つまたは複数の処理によって支援され得る。
【0037】
III. 外膜の透過処理
本発明の1つの態様では、1つまたは複数の標識リガンドに対する外膜の透過性を増大させる方法を使用する。これにより、通常なら外膜を通過できない標識リガンドにスクリーニングを利用することが可能になる。しかし、ある種類の分子、例えば650 Da排除限界よりも大きい疎水性抗生物質は、膜ポーリンとは無関係にそれ自体で細菌外膜を通して拡散し得る(Farmerら、1999)。その過程は、その際に膜を実際に透過処理し得る(JouenneおよびJunter、1990)。このような機構は、ポリミキシンBノナペプチドを用いてインビボで細胞質膜タンパク質のペリプラズムループを選択的に標識するために採用された(Wadaら、1999)。また、ある種の長鎖リン酸ポリマー(100 Pi)も、外膜の正常な分子ふるい活性を完全に回避するようである(RaoおよびTorriani、1988)。
【0038】
生存度を喪失することなくまたは発現したタンパク質が細胞から放出されることなく、リガンドのペリプラズム内への透過をもたらす条件を同定したが、本発明は外膜の維持をせずに行うことが可能である。融合ポリペプチドを用いて候補結合ポリペプチドを内(細胞質)膜の外側に固定することにより、結合した標識リガンドを検出するために(細胞からの結合タンパク質の漏出を妨げる障壁としての)外膜を維持する必要がなくなる。結果として、細胞質膜の外(ペリプラズム)面に固定される結合タンパク質を発現する細胞は、ある程度透過処理した膜を有するまたはほぼ完全に外膜を除去した細胞において、単に蛍光標識リガンドの溶液と共にインキュベートすることにより、蛍光標識され得る。
【0039】
異なる細菌宿主株の外膜の透過性は、大きく異なり得る。OmpFの過剰発現に起因する透過性の増大が、OmpFの翻訳に関する負の調節性mRNAの量の減少をもたらすヒストン様タンパク質の欠除によって起こることが以前に示されている(Painbeniら、1997)。また、DMA複製および染色体分離も、レプリソームと内膜との密接した接触に依存することが周知であり、内膜自体は多くの箇所で外膜と接触している。ライブラリースクリーニング用途に関して好ましい宿主は、プラスミドコピー数を減少させる変異をさらに有する大腸菌ABLEC株である。
【0040】
本発明者らはまた、高浸透圧ショック等の処理により標識が有意に改善され得ることを認めた。カルシウムイオン(Bukauら、1985)、およびさらにTris緩衝液(Irvinら、1981)を含む多く薬剤により、外膜の透過性が変化することは周知である。さらに、本発明者らは、ファージ感染により標識過程が促進されることを見出した。繊維状ファージの内膜タンパク質pIIIおよび大きな多量体外膜タンパク質pIVによって膜透過性は変化し得り(Boekeら、1982)、pIVの変異体では通常なら排除されるマルトデキストリンの接近が改善されたことが知られている(Marcianoら、1999)。株、塩、およびファージの賢明な組み合わせを含む本発明の技法を用いて、高度な透過性が達成され得た(Daughertyら、1999)。次に、フローサイトメトリーまたは他の関連技法を用いて、蛍光標識リガンドに結合している固定化結合ポリペプチドを含む細胞を、標識リガンドに対して親和性のない結合タンパク質を発現する細胞から容易に単離することができる。しかし、細胞の生存度を維持するために、破壊的な技法の使用を控えることが典型的に望ましいと考えられる。
【0041】
IV. 固定化ペリプラズム発現
本発明の1つの態様では、内膜の外面上に融合ポリペプチドを発現する細菌細胞を提供する。そのような融合ポリペプチドは、候補結合ポリペプチドと内膜の外面へのアンカーとして機能するポリペプチドとの間の融合を含み得る。本発明の範囲から逸脱することなく、さらなるポリペプチド配列を融合ポリペプチドに付加することができることは、当業者に理解されよう。そのようなポリペプチドの一例は、アンカーポリペプチドと候補結合ポリペプチドを連結する働きをするリンカーポリペプチドである。本発明の背景にある一般的図式は、候補結合ポリペプチドの不均一な集合の有利な発現を含む。
【0042】
内膜への固定は、内膜リポタンパク質のリーダーペプチドおよび最初の6アミノ酸を用いて達成され得る。内膜リポタンパク質の一例はNlpA(新規リポタンパク質A)である。NlpAの最初の6アミノ酸を、内膜に発現させるべきタンパク質のN末端アンカーとして使用することができる。NlpAは、内膜に独占的に局在する非必須リポタンパク質として、大腸菌において同定され特徴づけされた(Yu、1986;Yamaguchi、1988)。
【0043】
すべての原核生物リポタンパク質と同様に、NlpAは、それをSec経路を介する内膜を越えた移行に標的するリーダー配列と共に合成される。前駆タンパク質が内膜の外側に現れたならば、成熟リポタンパク質のシステイン残基がジアシルグリセリドとチオエーテル結合を形成する。その後シグナルペプチドがシグナルペプチダーゼIIによって切断され、システイン残基はアミノアシル化される(Pugsley、1993)。N末端のシステインが脂質で修飾された生じたタンパク質は、続いて内膜または外膜に局在し得る。この局在化は、成熟リポタンパク質の第2アミノ酸によって決まることが実証されている(Yamaguchi、1988)。この位置がアスパラギン酸であると、タンパク質はN末端脂質成分を介して内膜に固定されたままになるのに対し、2番目の位置に任意の他のアミノ酸がくると、一般にリポタンパク質は外膜に配向される(GennityおよびInouye、1992)。これは、タンパク質LolA、LolB、およびATP依存性ABCトランスポーター複合体LolCDEによって達成される(Yakushi、2000、Masuda、2002)。NlpAは第2アミノ酸残基としてアスパラギン酸を有し、よって内膜内に固定されてとどまる。
【0044】
リポタンパク質の2番目のアミノ酸をアルギニン(R)に変換することにより、リポタンパク質が内膜に存在するよう標的されるであろうことが報告されている(Yakushi、1997)。したがって、大腸菌(および場合によっては他のグラム陰性菌)のすべてのリポタンパク質が、アンカー配列となり得る。必要なのは、シグナル配列およびアミノ酸2位のアルギニンのみである。その後にsec切断領域が続く人工的secシグナル配列を用いて、および成熟タンパク質のアミノ酸1としてシステインをアミノ酸2としてアルギニンをコードすることにより、この構築物を人工的に設計することが可能である。大きな融合構築物を必要とするものの、膜貫通タンパク質もまた、場合によってはアンカー配列として使用できる。
【0045】
本発明で使用し得るアンカーの例には、リポタンパク質、CDNSSS成熟リポタンパク質アンカーを有する肺炎桿菌のプルラナーゼ、ファージにコードされるcelB、および大腸菌acrE(envC)が含まれる。タンパク質アンカーとして使用し得る内膜タンパク質の例には、

が含まれる。さらに、任意の細胞質タンパク質の単一の膜貫通ループを膜アンカーとして用いることも可能である。
【0046】
ファージディスプレイとの関連において、融合タンパク質の多様な集団の調製は周知である(例えば、米国特許第5,571,698号を参照のこと)。例えば繊維状ファージM13の遺伝子IIIコートタンパク質のアミノ末端ドメイン、または別の表面関連分子の代わりに、細胞質膜のペリプラズム面へのアンカーに関心対象のタンパク質を連結することにより、本発明において同様の技法を使用することができる。本発明に従って、そのような融合物を変異させ、細胞質膜のペリプラズム面上に低量で発現する、構造的に関連した融合タンパク質のライブラリーを形成することができる。そのように、ファージディスプレイとの関連において当業者に周知である候補分子の不均一な集合を作製する技法を、本発明で使用するために適合化することができる。当業者は、そのような適合化には、プロモーター、エンハンサー、またはリーダー配列を含む、内膜のペリプラズム面に固定される融合タンパク質を発現させるための細菌エレメントの使用が含まれることを認識するであろう。本発明は、ファージの使用または細胞表面上への分子の発現を必要としない、十分に発現されない場合もあり、または宿主細胞にとって有害である可能性のあるファージディスプレイに対する恩典を提供する。
【0047】
多様な候補結合タンパク質および/または抗体を作製するために本発明に関連して使用できる技法の例には、米国特許第5,824,520号に記載されている免疫グロブリン重鎖ライブラリーを発現させるための技法が含まれる。この技法では、高度に多様な合成超可変領域を作製することにより、一本鎖抗体ライブラリーを作製する。抗体提示のための同様の技法は、米国特許第5,922,545号によっても示される。次に、固定化融合ポリペプチドを発現させるため、これらの配列を、グラム陰性菌の内膜のペリプラズム面に対するアンカー配列をコードする核酸に融合することができる。
【0048】
融合タンパク質を作製する方法は、当業者には周知である(例えば、米国特許第5,780,279号を参照のこと)。そうするための1つの手段には、候補結合ポリペプチドとアンカー配列との間に遺伝子融合を構築する段階、および1つまたは複数のコドンにおいて結合タンパク質をコードする核酸を変異させ、それにより変異体のファミリーを作製する段階が含まれる。次に、変異させた融合タンパク質を、細菌の大きな集団において発現させることができる。その後、標的リガンドが結合した細菌を単離することができ、結合タンパク質をコードする対応する核酸をクローニングすることができる。
【0049】
V. 候補分子のスクリーニング
本発明は、標的リガンドを結合し得る分子を同定する方法を提供する。スクリーニングする結合ポリペプチドには、多様な候補物質の大きなライブラリーが含まれ得り、または別法として標的リガンドを結合する可能性が高いと考えられる構造特性を目的として選択される特定の種類の化合物が含まれ得る。本発明の1つの態様において、候補結合ポリペプチドは、抗体またはその断片もしくは一部である。本発明の他の態様では、候補分子は別の結合タンパク質であってよい。
【0050】
本発明に従って標的リガンドを結合し得る候補分子を同定するため、以下の段階を実施することができる:候補結合ポリペプチドと内膜のペリプラズム面に固定される配列との融合タンパク質を含むグラム陰性菌の集団を提供する段階;細菌と、候補結合ポリペプチドと接触し得る少なくとも1つの第1標識標的リガンドを混合する段階;および標的リガンドを結合し得る分子を発現する少なくとも1つの第1細菌を同定する段階。
【0051】
上記の方法では、固定された候補結合タンパク質と標識リガンドとが結合することにより、細胞の外への拡散が妨げられることになる。このように、標識リガンドの分子が細菌のペリプラズム内に保持され得る。または、ペリプラズムを除去することもでき、したがって固定化により結合した候補分子の保持が起こることになる。次いで、標識を用いて、標的リガンドを結合し得る結合ポリペプチドを発現する細胞を単離することができ、このようにして結合ポリペプチドをコードする遺伝子が単離される。その後、インビボまたはエクスビボ発現方法により、標的リガンドを結合し得る分子を大量に産生することができ、次いで、例えば以下に記載するような診断または治療用途等の任意の所望の用途に用いることができる。
【0052】
本明細書で用いる「候補分子」または「候補ポリペプチド」という用語は、標的分子に対する親和性を潜在的に有する可能性のある任意の分子またはポリペプチドを指す。候補物質は、合成分子等の低分子を含む、タンパク質またはその断片であってよい。本発明の1つの態様において、候補分子は抗体配列またはその断片を含んでよい。そのような配列は、特に、標的リガンドに結合する可能性に合わせて設計することができる。
【0053】
本発明に従って単離する結合ポリペプチドまたは抗体はまた、標的リガンドの構造を解明する助けとなり得る。原理上は、このアプローチにより、次の薬物設計の基礎となり得るファーマコアが得られる。機能的な、薬理学的に活性のある抗体に対する抗イディオタイプ抗体を作製することにより、タンパク質結晶学を完全に回避できる可能性がある。鏡像の鏡像として、抗イディオタイプ抗体の結合部位は、本来の抗原の類似体であることが期待される。次いで、抗イディオタイプ抗体を用いて、化学的または生物学的に産生したペプチドのバンクからペプチドを同定および単離することができる。選択したペプチドはファーマコアとして役立つと考えられる。抗イディオタイプは、抗原として抗体を使用し、抗体の産生に関して本明細書に記載した方法により作製することができる。一方、様々な市販業者から、標的リガンドの結合に関して基本的な基準を満たすと考えられる低分子ライブラリーを単純に取得してもよい。そのようなライブラリーは、低分子をコードする核酸または分子を発現する細菌として提供され得る
【0054】
A. 結合タンパク質コード配列のクローニング
抗体または他の結合タンパク質の結合親和性は、例えば、MunsonおよびPollardのスキャッチャード分析(1980)によって決定することができる。所望の特異性、親和性、および/または活性の分子を産生する細菌細胞を同定した後、対応するコード配列をクローニングすることが可能である。この方法では、従来の手順により(例えば、抗体または結合タンパク質をコードする遺伝子に特異的に結合し得るオリゴヌクレオチドプローブを用いて)、分子をコードするDNAを単離し配列決定することができる。
【0055】
単離したならば、抗体または結合タンパク質DNAを発現ベクター内に配置し、次にこのベクターを、通常であれば免疫グロブリンタンパク質を産生しないサルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞、またはミエローマ細胞等の宿主細胞にトランスフェクションし、組換え宿主細胞において結合タンパク質を合成させることができる。例えば、相同的なマウス配列の代わりにヒト重鎖および軽鎖定常ドメインのコード配列を用いることより(Morrisonら、1984)、または免疫グロブリンコード配列に非免疫グロブリンポリペプチドのコード配列のすべてまたは一部を共有結合することにより、DNAを修飾してもよい。この様式で、所望の結合特異性を有する「キメラ」または「ハイブリッド」結合タンパク質が作製される。
【0056】
典型的に、そのような非免疫グロブリンポリペプチドは抗体の定常ドメインと置換するか、または標的リガンドに対する特異性を有する1つの抗原結合部位と異なる抗体に対する特異性を有する別の抗原結合部位を含むキメラ二価抗体を作製するために、抗体の1つの抗原結合部位の可変領域と置換する。
【0057】
キメラ抗体またはハイブリッド抗体はまた、架橋剤が関与する方法を含む合成タンパク質化学における既知の方法を用いて、インビトロで作製され得る。例えば免疫毒素は、ジスルフィド交換反応を用いるかまたはチオエーテル結合の形成により、構築され得る。この方法に適した試薬の例には、イミノチオラートおよびメチル-4-メルカプトブチルイミダートが含まれる。
【0058】
生細胞または非生細胞から核酸をクローニングし得ることは、当業者に理解されよう。非生細胞の場合には、例えば、PCRを用いるなどしてクローン化DNAの増幅を利用することが望ましいと考えられる。これは、生細胞を用いて、細胞をさらに増殖させるかまたはさせずに行うことも可能である。
【0059】
B. リガンドに対するタンパク質親和性の最大化
天然の免疫反応において、抗体遺伝子は高率で変異を蓄積する(体細胞超変異)。導入される変化のいくつかによって、より高い親和性および高親和性表面免疫グロブリンを提示するB細胞が付与されることになる。「チェーンシャフリング」(Marksら、1992)として知られる技法を用いて、この天然の過程を模倣することができる。この方法では、重鎖および軽鎖V領域遺伝子を、非免疫化ドナーから得られたVドメイン遺伝子の天然変種(レパートリー)と順次置換することにより、本発明に従って得られた「一次」ヒト抗体の親和性を改善することができる。この技法によって、nM範囲の親和性を有する抗体または抗体断片の産生が可能になる。非常に大きな抗体レパートリーを作製するための戦略はWaterhouseら(1993)によって記載されており、そのような大きなファージライブラリーからの高親和性ヒト抗体の直接的な単離はGriffithら(1994)によって報告されている。遺伝子シャッフリング法を用いて、げっ歯類抗体からヒト抗体を導出することも可能であり、この場合、ヒト抗体は初めのげっ歯類抗体と類似した親和性および特異性を有する。「エピトープ刷り込み」とも称されるこの方法により、ファージディスプレイ技法によって得られたげっ歯類抗体の重鎖または軽鎖VドメインをヒトVドメイン遺伝子のレパートリーと置換し、げっ歯類-ヒトキメラが作製される。抗原を選択することにより、機能的な抗原結合部位を回復し得るヒト可変領域が単離される、すなわち、エピトープによってパートナーの選択が支配される(刷り込まれる)。残りのげっ歯類Vドメインを置換するためにこの過程を繰り返すと、ヒト抗体が得られる(1993年4月1日に公開された国際公開公報第93/06213号を参照のこと)。CDR移植によるげっ歯類抗体の従来のヒト化とは異なり、この技法により、げっ歯類起源のフレームワークまたはCDR残基も有さない、完全なヒト抗体が提供される。
【0060】
C. 標識リガンド
本発明の1つの態様では、標識リガンドに対する親和性を有する抗体または結合タンパク質を単離する。本発明に従ってグラム陰性菌のペリプラズム膜を透過処理および/または除去することにより、潜在的に任意の大きさの標識リガンドをスクリーニングすることができる。ペリプラズム膜の除去を行わない場合には、細菌ペリプラズム膜を超えてリガンドが効率的に拡散できるように、標識リガンドは典型的に50,000 Da未満の大きさであることが好ましい。
【0061】
上記のように、本発明に従って、1つまたは複数の検出可能な薬剤で標識したリガンドを提供することが典型的に望ましい。これは、例えば、リガンドを少なくとも1つの検出可能な薬剤に連結して複合体を形成させることによって、実施することができる。例えば、少なくとも1つの検出可能な分子または成分を連結または共有結合または複合体化することは、従来から行われている。「標識」または「検出可能な標識」とは、特定の機能的特性および/または化学的特性によって検出され得る化合物および/または成分であり、これを用いることによって、これが結合したリガンドの検出、および/または必要に応じてさらなる定量が可能になる。本発明に使用できる標識の例には、酵素、放射標識、ハプテン、蛍光標識、リン光性分子、化学発光性分子、発色団、発光性分子、光親和性分子、ビオチン等の着色粒子またはリガンドが含まれるが、これらに限定されない。
【0062】
本発明の1つの態様では、標識に対する細胞の自動化スクリーニングが実施できるように、視覚的に検出可能なマーカーを用いる。特に、所望の結合タンパク質または抗体を発現する細胞を単離するためにフローサイトメトリーの使用を可能にするという点で、蛍光標識が有利である。適切な機器を用いて可視化することにより検出できる薬剤の例は、当技術分野において周知であり、所望のリガンドに結合する方法も同様に周知である(例えば、それぞれ参照として本明細書に組み入れられる米国特許第5,021,236号;第4,938,948号;および第4,472,509号を参照のこと)。そのような薬剤には、常磁性イオン;放射性同位元素;蛍光色素;NMR検出可能物質、およびX線像用の物質が含まれる。本発明で使用可能な蛍光標識の種類は当業者に周知であり、例えば、Alexa350、Alexa430、AMCA、BODIPY 630/650、BODIPY 650/655、BODIPY-FL、BODIPY-R6G、BODIPY-TMR、BODIPY-TRX、カスケードブルー、Cy3、Cy5、6-FAM、フルオレセインイソチオシアネート、HEX、6-JOE、オレゴングリーン488、オレゴングリーン500、オレゴングリーン514、パシフィックブルー、REG、ローダミングリーン、ローダミンレッド、レノグラフィン、ROX、TAMRA、TET、テトラメチルローダミン、および/またはテキサスレッドが含まれる。
【0063】
磁気スクリーニング法は当業者には周知である(例えば、米国特許第4,988,618号、米国特許第5,567,326号、および米国特許第5,779,907号を参照のこと)。そのような技法に従って標識として使用し得る常磁性イオンの例には、クロム(III)、マンガン(II)、鉄(III)、鉄(II)、コバルト(II)、ニッケル(II)、銅(II)、ネオジム(III)、サマリウム(III)、イッテルビウム(III)、ガドリニウム(III)、バナジウム(II)、テルビウム(III)、ジスプロシウム(III)、ホルミウム(III)、および/またはエルビウム(III)が含まれる。他の状況において有用なイオンには、ランタン(III)、金(III)、鉛(II)、および特にビスマス(III)が含まれるが、これらに限定されない。
【0064】
本発明において意図する別の種類のリガンド複合体は、リガンドが、二次結合分子および/または発色基質と接触した際に有色産物を生じる酵素(酵素タグ)に結合しているリガンド複合体である、そのような酵素の例には、ウレアーゼ、アルカリホスファターゼ、(西洋ワサビ)過酸化水素ペルオキシダーゼ、およびグルコースオキシダーゼが含まれる。このような例では、選択した細胞が生存したままであることが望ましい。好ましい二次結合リガンドは、ビオチンおよび/またはアビジンおよびストレプトアビジン化合物である。そのような標識の使用は当業者に周知であり、例えば米国特許第3,817,837号;第3,850,752号;第3,939,350号;第3,996,345号;第4,277,437号;第4,275,149号;および第4,366,241号に記載されており、それぞれ参照として本明細書に組み入れられる。
【0065】
低強度の紫外光によって生成される反応性ナイトレン中間体を介してタンパク質への共有結合を形成するために、アジド基を含む分子を用いることも可能である(PotterおよびHaley、1983)。特に、プリンヌクレオチドの2-アジド類似体および8-アジド類似体が、粗製細胞抽出物中のヌクレオチド結合タンパク質を同定するための部位特異的光プローブとして用いられた(OwenおよびHaley、1987;Athertonら、1985)。2-アジド類似体および8-アジド類似体はまた、精製したタンパク質のヌクレオチド結合ドメインをマッピングするためにも使用され(Khatoonら、1989;Kingら、1989;およびDholakiaら、1989)、リガンド結合剤としても使用することができる。
【0066】
当業者に周知の任意の技法により、標識を行うことができる。例えば、リガンドを、所望の標識および次亜塩素酸ナトリウム等の化学酸化剤またはラクトペルオキシダーゼ等の酵素的酸化剤と接触させることにより、リガンドを標識することができる。同様に、リガンド交換過程を用いることも可能である。または、例えば、標識、SNCl2等の還元剤、ナトリウム-カリウムフタル酸溶液等の緩衝液 、およびリガンドをインキュベートすることにより、直接的標識技法を用いてもよい。例えば、ジエチレントリアミン五酢酸(DTPA)またはエチレンジアミン四酢酸(EDTA)の存在下で標識をリガンドに結合するために、リガンド上の媒介性官能基を用いることも可能である。
【0067】
複合体成分へのリガンドの結合または複合体化に関して、他の方法もまた当技術分野において周知である。いくつかの結合法は、リガンドに結合させた無水ジエチレントリアミン五酢酸(DTPA);エチレントリアミン四酢酸;N-クロロ-p-トルエンスルホンアミド;および/またはテトラクロロ-3α-6α-ジフェニルグリコウリル(diphenylglycouril)-3等の有機キレート剤の使用を含む(それぞれ参照として本明細書に組み入れられる米国特許第4,472,509号および第4,938,948号)。グルタルアルデヒドまたは過ヨウ素酸等のカップリング剤の存在下で、リガンドを酵素と反応させてもよい。蛍光マーカーとの複合体は、これらのカップリング剤の存在下で、またはイソチオシアネートと反応させることにより、調製することができる。米国特許第4,938,948号では、モノクローナル抗体を用いて乳房腫瘍の画像診断が達成されるが、メチル-p-ヒドロキシベンズイミデートまたはN-スクシニミジル-3-(4-ヒドロキシフェニル)プロピオン酸等のリンカーを用いて、検出可能な撮像成分を抗体に結合する。
【0068】
ペリプラズムで発現させたタンパク質を適切な蛍光リガンドで特異的に標識する能力は、ライブラリースクリーニング以外の用途も有する。タンパク質生産の過程において産生をモニタリングするために、蛍光リガンドでの特異的標識およびフローサイトメトリーを用いることができる。フローサイトメトリーはこれまで細菌細胞の解析のために用いられてきたが、ペリプラズムタンパク質の特異的標識および定量のためには用いられていなかった。しかし、IGF-1、いくつかのインターロイキン、ウロキナーゼ型プラスミノーゲン活性化因子等の酵素、抗体断片、阻害剤(例えば、ウシ膵臓トリプシンインヒビター)を含む多数の商業的に重要なタンパク質は、ペリプラズム空間に分泌される形態で、組換え大腸菌において発現される。本発明による技法に従い、適切な蛍光リガンドおよびフローサイトメトリー解析を利用することにより、培養物中の各細胞内のそのようなタンパク質の産生レベルをモニタリングすることができる。
【0069】
一般に、タンパク質発現のモニタリングは、細胞の溶解および免疫学的技法または次のクロマトグラフィー分離によるタンパク質の検出を必要とする。しかし、ELISAまたはウェスタンブロット解析は時間を要し、これらの解析からは細胞集団内での発現の分布に関する情報が提供されず、さらにオンラインモニタリングに用いることができない(Thorstensonら、1997;Berrierら、2000)。対照的に、FACS標識は迅速かつ単純であり、グラム陰性菌で発現した組換えタンパク質の工業的規模での発酵をオンラインモニタリングするためにうまく適合化することができる。同様に、本発明を用いて、細菌においてインビボで発現したまたはエクスビボで提供された酵素の相対濃度または比活性を測定することも可能である。
【0070】
本発明に従って抗体等のリガンド結合タンパク質が単離されたならば、その分子の有用性を増すために、少なくとも1つの薬剤に分子を連結して複合体を形成することが望ましいと考えられる。例えば、診断薬または治療薬として抗体分子の有効性を高めるために、少なくとも1つの所望の分子または成分を連結または共有結合または複合体化することは、従来から行われている。そのような分子または成分は、少なくとも1つのエフェクターまたはレポーター分子であるが、これらに限定されない。エフェクター分子には、細胞障害性等の所望の活性を有する分子が含まれる。抗体に結合されているエフェクター分子の限定されない例には、毒素、抗腫瘍薬、治療的酵素、放射標識ヌクレオチド、抗ウイルス薬、キレート薬、サイトカイン、増殖因子、およびオリゴヌクレオチドまたはポリヌクレオチドが含まれる。これに反して、レポーター分子は、アッセイにより検出され得る任意の成分と定義される。そのような分子を標識する技法は、当業者には周知であり、本明細書において先に記載した。
【0071】
次に、例えばタンパク質、ポリペプチド、またはペプチド等の生物成分を結合、精製、除去、定量、および/またはそうでない場合は一般に検出するための免疫検出法において、本明細書に従って調製した抗体等の標識結合タンパク質を使用してもよい。いくつかの免疫検出法には、少し記述するだけでも酵素結合免疫吸着測定法(ELISA)、放射性免疫測定法(RIA)、免疫放射定量測定法、蛍光免疫測定法、化学発光測定法、生物発光測定法、およびウェスタンブロット法が含まれる。様々な有用な免疫検出法の段階は、例えば、Doolittle MHおよびBen-Zeev O、1999;Gulbis BおよびGaland P、1933;およびDe Jager Rら、1993等の科学文献に記載されており、これらはそれぞれ参照として本明細書に組み入れられる。そのような技法には、当技術分野で周知である様々な種類の酵素結合免疫吸着測定法(ELISA)および/または放射性免疫測定法(RIA)等の結合測定法が含まれる。
【0072】
本発明に従って調製した、抗体を含むリガンド結合分子は、例えば、免疫組織化学法(IHC)によって研究するために調製した、新鮮凍結した組織ブロックおよび/またはホルマリン固定、パラフィン包埋した組織ブロックと共に用いることも可能である。これらの粒子状試料から組織ブロックを調製する方法は、様々な予後因子のこれまでのIHC研究において用いられて成功しており、および/または当業者に周知である(Abbondanzoら、1990)。
【0073】
VI. フローサイトメトリーを用いた自動化スクリーニング
本発明の1つの態様において、候補分子に結合し、細菌の細胞質膜の外面に結合している標識リガンドを含む細菌細胞を効率よく単離するために、蛍光活性化細胞分類 (FACS)スクリーニングまたは他の自動化フローサイトメトリー技法を用いることができる。フローサイトメトリーを行うための機器は当業者に周知であり、市販されている。そのような機器の例には、Becton Dickinson(カリフォルニア州、フォスターシティー)のFACS Star Plus、FACSCan、FACSort機器、Coulter Epics Division(フロリダ州、ハイアリーア)のEpics C、およびCytomation(コロラド州、コロラドスプリングス)のMoFloが含まれる。
【0074】
フローサイトメトリー技法は、一般に、液体試料中の細胞または他の粒子の分離を伴う。典型的に、フローサイトメトリーの目的は、分離された粒子をその1つまたは複数の特徴、例えば標識リガンドまたは他の分子の存在に関して分析することである。フローサイトメトリーの基本的な段階は、液体流動が検出領域を通過するような、装置内の液体試料の方向づけを含む。粒子は一つずつセンサーを通過すべきであり、大きさ、屈折、光散乱、不透明度、粗雑度、形状、蛍光等に基づき分類される。
【0075】
細胞の迅速な定量解析は、生物医学研究および医学において有用となる。装置により、1秒当たり数千個の細胞の速度での、細胞特性の定量的多パラメータ解析が可能になる。これらの機器により、細胞種を区別する能力が提供される。データは、測定した変数の1次元(ヒストグラム)または2次元(等高線プロット、散布プロット)度数分布で示される場合が多い。多パラメータデータファイルの分割は、双方向性の1次元または2次元画像プログラムの連続した使用を必要とする。
【0076】
迅速に細胞を検出するため、多パラメータフローサイトメトリーデータの定量的解析は、以下の2段階からなる:細胞種の特徴づけおよび試料の処理。一般に、細胞種特徴づけの過程により、細胞特性が関心対象の細胞と関心対象でない細胞に分別される。次いで、試料の処理において、収まる領域に従って各細胞が2つのカテゴリーのうちの一方に分類される。高度な検出性能は細胞の適切な特性が得られる場合にのみ期待され得るため、細胞の種類の解析は非常に重要である。
【0077】
フローサイトメトリーによって細胞分析が行われるだけでなく、細胞の分別も行われる。米国特許第3,826,364号において、機能的に異なる細胞種等の粒子を物理的に分離する装置が開示されている。この機械では、レーザーにより、適切なレンズまたはレンズシステムにより粒子の流れに焦点を合わせた照明が提供され、その結果その中の粒子から高度に限局化した散乱が得られる。さらに、流動中の蛍光粒子を励起するために、高輝度源照明が粒子の流れに向けられる。流動中の特定の粒子は選択的に荷電され、次いで、指定の容器に向けて偏向させることによって分離され得る。この分離の古典的な形態は蛍光タグ化抗体によるものであり、この抗体は、分離のために1つまたは複数の細胞種を標識するために使用される。
【0078】
フローサイトメトリー法の他の例には、米国特許第4,284,412号;第4,989,977号;第4,498,766号;第5,478,722号;4,857,451号;第4,774,189号;4,767,206号;第4,714,682号;第5,160,974号;および第4,661,913号に記載されている方法が含まれるが、これらに限定されず、これらの開示はそれぞれ参照として本明細書に明確に組み入れられる。
【0079】
本発明に関して、フローサイトメトリーの重要な局面は、複数ラウンドのスクリーニングを連続して行うことができる点である。選別の最初のラウンドから細胞を単離し、直ちにフローサイトメーターに再導入し、スクリーニングのストリンジェンシーを改善するために再度スクリーニングすることが可能である。当業者に周知である別の恩典は、フローサイトメトリーを用いて非生細胞を回収できる点である。フローサイトメトリーは基本的に粒子選別技術であるため、細胞が成長または増殖する能力は必要ではない。そのような非生細胞から核酸を回収する技法は当技術分野において周知であり、これには、例えばPCRを含む鋳型依存的増幅技法の使用が含まれる。
【0080】
VII. 核酸に基づいた発現系
本発明の特定の態様においては、組換えタンパク質を発現させるために核酸に基づいた発現系を使用できる。例えば、本発明の1つの態様は、選択したリガンドに対する親和性を有する候補抗体または他の結合タンパク質を含む融合ポリペプチドのコード配列によるグラム陰性菌の形質転換、およびグラム陰性菌の細胞質膜上でのそのような分子の発現を含む。本発明の他の態様では、標的リガンドに対する特異性を有する単離された結合タンパク質を調製するために、そのようなコード配列の発現を例えば真核生物宿主細胞において行うことができる。次に、単離したタンパク質を1つまたは複数の治療用途または診断用途に用いることができる。
【0081】
A. 核酸送達の方法
本発明の特定の局面は、標的細胞への核酸の送達を含み得る。例えば、細菌宿主細胞を、標的リガンドに潜在的に結合し得る候補分子をコードする核酸で形質転換することができる。本発明の特定の態様においては、発現を細菌の細胞質膜に標的することが望ましい場合がある。真核生物宿主細胞の形質転換も同様に、標的リガンドを結合し得ると同定された様々な候補分子の発現に使用できる。
【0082】
細胞を形質転換するための核酸送達に適した方法には、核酸(例えばDNA)をそのような細胞またはその細胞小器官にさえ導入し得る実質的に任意の方法が含まれると考えられる。そのような方法には、マイクロインジェクション(HarlanおよびWeintraub、1985;米国特許第5,789,215号、参照として本明細書に組み入れられる)を含む注入(米国特許第5,994,624号、第5,981,274号、第5,945,100号、第5,780,448号、第5,736,524号、第5,702,932号、第5,656,610号。第5,589,466号、および第5,580,859号、それぞれ参照として本明細書に組み入れられる);エレクトロポレーション(米国特許第5,384,253号、参照として本明細書に組み入れられる);リン酸カルシウム沈殿(GrahamおよびVan Der Eb、1973;ChenおよびOkayama、1987;Rippeら、1990);DEAE-デキストランとそれに続くポリエチレングリコールの使用(Gopal、1985);直接的超音波負荷(Fechheimerら、1987);リポソームを介するトランスフェクション(NicolauおよびSene、1982;Fraleyら、1979;Nicolauら、1987;Wongら、1980;Kanedaら、1989;Katoら、1991);微粒子銃(国際公開公報第94/09699号および第95/06128号;米国特許第5,610,042号;第5,322,783号、第5,563,055号、第5,550,318号、第5,538,877号、および第5,538,880号、それぞれ参照として本明細書に組み入れられる);炭化ケイ素繊維との撹拌(Kaepplerら、1990;米国特許第5,302,523号および第5,464,765号、それぞれ参照として本明細書に組み入れられる);アグロバクテリウムを介する形質転換(米国特許第5,591,616号および第5,563,055号、それぞれ参照として本明細書に組み入れられる); またはプロトプラストのPEGを介する形質転換(Omirullehら、1993;米国特許第4,684,611号および第4,952,500号、それぞれ参照として本明細書に組み入れられる);乾燥/抑制を介するDNA取り込み(Potrykusら、1985)等のDNAの直接的送達が含まれるが、これらに限定されない。これらのような技法を適用し、細胞小器官、細胞、組織、または生物体を安定にまたは一過性に形質転換することができる。
【0083】
1. エレクトロポレーション法
本発明の特定の態様では、エレクトロポレーションにより核酸を細胞に導入する。エレクトロポレーションは、細胞懸濁液およびDNAの高圧放電への曝露を含む。この方法のいくつかの変法では、ペクチン分解酵素等のある種の細胞壁分解酵素を用いて、標的レシピエント細胞を未処理の細胞よりもエレクトロポレーションによってより形質転換されやすくする(米国特許第5,384,253号、参照として本明細書に組み入れられる)。または、機械的損傷によって、レシピエント細胞をより形質転換されやすくできる。
【0084】
2. リン酸カルシウム法
本発明の他の態様では、リン酸カルシウム沈殿法により核酸を細胞に導入する。この技法を用いて、ヒトKB細胞にアデノウイルス5 DNAがトランスフェクションされた(GrahamおよびVen Der Eb、1973)。同様にこの方法により、マウスL(A9)、マウスC127、CHO、CV-1、BHK、NIH3T3、およびHeLa細胞に、ネオマイシンマーカー遺伝子がトランスフェクションされ(ChenおよびOkayama、1987)、ラット肝細胞に様々なマーカー遺伝子がトランスフェクションされた(Rippeら、1990)。
【0085】
B. ベクター
例えば、標的リガンドに結合する能力についてスクリーニングすることが望まれる候補ポリペプチドをコードする核酸配列によるグラム陰性菌の形質転換において、本発明によりベクターを使用できる。本発明の1つの態様において、標的ポリペプチドをコードする核酸配列の不均一性「ライブラリー」全体を細菌集団に導入することができ、それにより全ライブラリーのスクリーニングが可能になる。「ベクター」という用語を使用して、細胞内に導入されそこで複製され得るための、核酸配列を挿入し得る担体核酸分子を指す。核酸配列は「外因性」または「異種性」であってよく、これはその配列がベクターを導入する細胞にとって外来性であること、またはその配列が細胞内の配列と相同的であるが、通常はその配列が見出されない宿主細胞核酸内の位置にあることを意味する。ベクターには、プラスミド、コスミド、ウイルス(バクテリオファージ、動物ウイルス、および植物ウイルス)、および人工染色体(例えばYAC)が含まれる。当業者は標準的な組換え技法によりベクターを構築することができ、この技法は、どちらも参照として本明細書に組み入れられるManiatisら、1988、およびAusubelら、1994に記載されている。
【0086】
「発現ベクター」という用語は、転写され得る遺伝子産物の少なくとも一部をコードする核酸配列を含むベクターを指す。場合によっては、次にRNA分子がタンパク質、ポリペプチド、またはペプチドに翻訳される。別の場合には、例えばアンチセンス分子またはリボザイムの産生において、これらの配列は翻訳されない。発現ベクターは様々な「制御配列」を含み得り、「制御配列」とは、特定の宿主生物体において機能的に結合したコード配列の転写および場合によっては翻訳に必要な核酸配列を指す。ベクターおよび発現ベクターは、転写および翻訳を支配する制御配列に加えて、同様に他の機能に役立ち、以下に記載する核酸配列を含んでよい。
【0087】
1. プロモーターおよびエンハンサー
「プロモーター」とは、そこで転写の開始および速度が制御される核酸配列の領域である制御配列である。プロモーターは、RNAポリメラーゼおよび他の転写因子等の制御タンパク質および分子が結合し得る遺伝子エレメントを含み得る。「機能的に位置する」、「機能的に結合した」、「制御下で」、および「転写制御下で」という語句は、配列の転写開始およびまたは発現を制御すべき核酸配列に関して、プロモーターが正しく機能的な位置および/または方向にあることを意味する。プロモーターは「エンハンサー」と共に使用してもしなくてもよく、「エンハンサー」とは、核酸配列の転写活性化に関与するシス作動性の制御配列を指す。
【0088】
プロモーターは、コード部分および/またはエキソンの上流に位置する5'非コード配列を単離することによって得られ得るように、遺伝子または配列と天然で付随しているものであってよい。そのようなプロモーターは「内因性」と称すことができる。同様にエンハンサーも、核酸配列の下流または上流に位置して、核酸配列と天然に付随しているものであってよい。または、組換えまたは異種性プロモーターの制御下にコード核酸部分を置くことにより、ある種の恩典が得られることになるが、そのようなプロモーターとは、その天然環境において核酸配列と通常は付随していないプロモーターを指す。組換えまたは異種性エンハンサーもまた、その天然環境において核酸配列と通常は付随していないエンハンサーを指す。そのようなプロモーターまたはエンハンサーには、他の遺伝子のプロモーターまたはエンハンサー、任意の他の原核生物、ウイルス、または真核生物細胞から単離されたプロモーターまたはエンハンサー、ならびに「天然に存在」しない、すなわち異なる転写制御領域の異なるエレメントおよび/または発現を変化させる変異を含むプロモーターまたはエンハンサーが含まれ得る。合成によりプロモーターおよびエンハンサーの核酸配列を作製することに加えて、本明細書で開示する組成物と関連して、組換えクローニング技法および/またはPCR(商標)を含む核酸増幅技法を用いて配列を産生してもよい(それぞれ参照として本明細書に組み入れられる米国特許第4,683,202号、米国特許第5,928,906号を参照のこと)。さらに、ミトコンドリア、葉緑体等の非核細胞小器官内で配列の転写および/または発現を指示する制御配列も同様に使用し得ることを意図する。
【0089】
当然のことながら、発現させるために選択した細胞種、細胞小器官、および生物体において、DNA部分の発現を効率的に指示するプロモーターおよび/またはエンハンサーを使用することが重要であろう。本発明で使用し得るそのようなプロモーターの一例は、大腸菌アラビノースプロモーターである。分子生物学の分野における当業者は、一般に、タンパク質発現のためのプロモーター、エンハンサー、および細胞種の組み合わせの使用に詳しく、例えば、参照として本明細書に組み入れられるSambrookら(1989)を参照されたい。使用するプロモーターは、構成的、組織特異的、誘導性、および/または組換えタンパク質および/またはペプチドの大量発現において有利であるように、導入したDNA部分の高レベル発現を指示する適切な条件下で有用であってよい。プロモーターは、異種性であっても内因性であってもよい。
【0090】
2. 開始シグナルおよび内部リボソーム結合部位
コード配列の効率的な翻訳には、特異的な開始シグナルも必要とされ得る。これらのシグナルには、ATG開始コドンまたは隣接配列が含まれる。ATG開始コドンを含む外因性翻訳制御シグナルが提供される必要がある場合もある。当業者は、容易にこれを決定し必要なシグナルを提供し得ると考えられる。挿入物全体の翻訳を確実にするため、開始コドンは所望のコード配列のフレームと「インフレーム」でなければならないことは周知である。外因性の翻訳制御シグナルおよび開始コドンは、天然であっても合成であってもよい。発現の効率は、適切な転写エンハンサーエレメントを含めることによって増強され得る。
。
【0091】
3. マルチクローニング部位
ベクターはマルチクローニング部位(MCS)を含み得るが、MCSとは、そのいずれかがベクターを消化するための標準的な組換え技術と併せて使用され得る、複数の制限酵素部位を含む核酸領域である(参照として本明細書に組み入れられるCarbonelliら、1999、Lavensonら、1998、およびCocea、1997を参照のこと)。「制限酵素消化」とは、核酸分子内の特定の位置でのみ機能する酵素による核酸分子の触媒作用的切断を指す。これらの制限酵素の多くは市販されている。そのような酵素の使用は、当業者に理解されている。外因性配列をベクター内に連結できるように、MCS内で切断する制限酵素を用いてベクターを線状化または断片化する場合が多い。「連結」とは、2つの核酸断片間でリン酸ジエステル結合を形成する過程を指し、これらの断片は相互に隣接してもしなくてもよい。制限酵素および連結反応を含む技法は、組換え技術の分野の当業者に周知である。
【0092】
4. 終結シグナル
本発明に従って調製するベクターまたは構築物は、一般に、少なくとも1つの終結シグナルを含むことになる。「終結シグナル」または「ターミネーター」とは、RNAポリメラーゼによるRNA転写の特異的終結に関与するDNA配列からなる。したがって、特定の態様においては、RNA転写産物の産生を終了させる終結シグナルを意図する。ターミネーターは、所望のメッセージレベルを達成するためにインビボで必要とされる場合がある。
【0093】
本発明における使用が意図されるターミネーターには、本明細書に記載するかまたは当業者に周知である任意の周知である転写ターミネーターが含まれ、これには、例えばrhp依存性ターミネーターまたはrho依存性ターミネーターが含まれるが、これらに限定されない。特定の態様において、終結シグナルは、配列切断に起因するなどして、転写可能なまたは翻訳可能な配列を欠いている可能性がある。
【0094】
5. 複製起点
宿主細胞においてベクターを増幅するため、ベクターは、そこで複製が開始される特異的核酸配列である1つまたは複数の複製起点(多くの場合「ori」と称される)を含み得る。または、宿主細胞が酵母である場合は、自己複製配列(ARS)が使用され得る。
【0095】
6. 選択可能なおよびスクリーニング可能なマーカー
本発明の特定の態様では、発現ベクター内にマーカーを含めることによって、本発明の核酸構築物を含む細胞をインビトロまたはインビボで同定することができる。そのようなマーカーによって同定可能な変化が細胞に付与され、発現ベクターを含む細胞の容易な同定が可能となる。一般に、選択可能なマーカーは、選択を可能にする特性を付与するものである。陽性選択可能なマーカーは、マーカーの存在によって選択が可能になるマーカーであるのに対して、陰性選択可能なマーカーは、その存在によって選択が妨げられるマーカーである。陽性選択可能なマーカーの例は、薬剤耐性マーカーである。
【0096】
通常、薬物選択マーカーを含めることは形質転換体のクローニングおよび同定に役立ち、例えば、ネオマイシン、ピューロマイシン、ハイグロマイシン、DHFR、GPT、ゼオシン、およびヒスチジノールに対する耐性を付与する遺伝子は有用な選択可能マーカーである。条件の実行に基づいて形質転換体の識別を可能にする表現型を付与するマーカーに加えて、その基本が比色分析であるGFP等のスクリーニング可能なマーカーを含む、他の種類のマーカーもまた意図する。または、単純ヘルペスウイルスチミジンキナーゼ(tk)またはクロラムフェニコールアセチルトランスフェラーゼ(CAT)等のスクリーニング可能な酵素を使用してもよい。当業者はまた、おそらくFACS解析に関連して、免疫学的マーカーをいかにして使用するかを理解すると考えられる。遺伝子産物をコードする核酸が同時に発現され得る限り、使用するマーカーは重要でないと考えられる。選択可能なまたはスクリーニング可能なマーカーのさらなる例は、当業者に周知である。
【0097】
C. 宿主細胞
本明細書では、「細胞」、「細胞株」、および「細胞培養物」という用語は互換的に用いられ得る。これらの用語すべてには、任意のおよびすべての次の世代であるそれらの子孫も含まれる。すべての子孫は、意図的変異または偶発的変異により同一でない可能性があることが理解される。異種性核酸配列の発現との関連において、「宿主細胞」とは原核細胞を指し、これには、ベクターを複製し得るおよび/またはベクターによりコードされる異種性遺伝子を発現し得る形質転換可能な任意の生物が含まれる。宿主細胞は、ベクターのレシピエントとして使用することができ、現に使用されている。宿主細胞は「トランスフェクション」または「形質転換」することができ、これらの用語は、外因性核酸を宿主細胞内に伝達または導入する過程を指す。形質転換細胞には、最初の被験細胞およびその子孫が含まれる。
【0098】
本発明の特定の態様において、宿主細胞はグラム陰性菌である。この細菌は、内膜と外膜との間にペリプラズム膜を有し、特にペリプラズムと細胞質との間に細胞質膜としても知られている上記の内膜を有するという点で、本発明での使用に適している。そのようなものとして、そのようなペリプラズム空間を有する任意の他の細胞も本発明に従って使用することができる。本発明で使用し得るグラム陰性菌の例には、大腸菌、緑膿菌、コレラ菌、ネズミチフス菌、フレクスナー赤痢菌、インフルエンザ菌、百日咳菌、火傷病菌、根粒菌種が含まれるが、これらに限定されない。グラム陰性菌細胞は、選択したリガンドを結合し得る候補結合ポリペプチドを含む融合ポリペプチドをコードする配列で形質転換した細菌としてさらに定義され得る。ポリペプチドはペリプラズム空間に面した細胞質膜の外面に固定されるが、これは抗体コード配列または別の配列を含み得る。ポリペプチドを発現させるための1つの手段は、そのような方向づけを起こし得るリーダー配列をポリペプチドに結合させることによる。
【0099】
多数の原核生物細胞株および培養物が宿主細胞としての使用に利用でき、これらは、生きた培養物および遺伝物質の保管所としての機能を果たす機関であるアメリカン・タイプ・カルチャー・コレクション(ATCC)(www.atcc.org)を通して入手できる。適切な宿主は、ベクター骨格および所望の結果に基づき、当業者により決定され得る。プラスミドまたはコスミドは、例えば、多くのベクターを複製するために原核宿主細胞内に導入され得る。ベクターを複製および/または発現させるために宿主細胞として使用される細菌細胞には、DH5α、JM109、およびKC8、ならびSURE(登録商標)コンピテント細胞およびSOLOPACK(商標)ゴールド細胞(STRATAGENE(登録商標)、ラ・ホーヤ)等の多くの市販の細菌宿主が含まれる。または、大腸菌LE392等の細菌細胞をバクテリオファージの宿主細胞として使用することも可能である。
【0100】
様々な細胞種および生物に由来する多くの宿主細胞が入手可能であり、当業者に周知であると考えられる。同様に、ウイルスベクターを、原核生物宿主細胞、特にベクターの複製または発現に関して許容状態である原核生物宿主細胞とともに用いてもよい。いくつかのベクターは、原核細胞および真核細胞の両方においてベクターの複製および/または発現を可能にする制御配列を利用できる。当業者はさらに、上記の宿主細胞を維持するためおよびベクターの複製を可能にするためにそれらすべてをインキュベートする条件を理解するものと考えられる。ベクターの大量生産、ならびにベクターによってコードされる核酸、およびその同族ポリペプチド、タンパク質、またはペプチドの産生を可能にする技法および条件もまた理解され、周知である。
【0101】
D. 発現系
上記の組成物の少なくとも一部またはすべてを含む多数の発現系が存在する。そのような系は、例えば、本発明により特定のリガンドに結合し得ると同定されるポリペプチド産物を産生するために使用できる。原核生物に基づく系は、本発明での、核酸配列、またはその同族ポリヌクレオチド、タンパク質、およびペプチドを産生させるための使用に利用することができる。多くのそのような系は広く市販されている。発現系の他の例には、T7、Tac、Trc、BAD、λpL、テトラサイクリン、またはLacプロモーター等の強力な原核生物プロモーターを含むベクター、pET発現系、および大腸菌発現系が含まれる。
【0102】
E. 候補の結合タンパク質および抗体
本発明の特定の局面において、標的リガンドに潜在的に結合し得るタンパク質および短いペプチドを含む候補の抗体または他の組換えポリペプチドは、宿主細菌細胞の細胞質膜上に発現する。そのような抗体または他の結合ポリペプチドの異種性集団を発現させることにより、標的リガンドに対して高い親和性を有する抗体が同定され得る。同定された抗体は、次に、本明細書に記載するように様々な診断用途または治療用途に使用することが可能である。
【0103】
本明細書で使用する「抗体」という用語は、IgG、IgM、IgA、IgD、およびIgE等の任意の免疫学的結合因子を広く指すよう意図される。「抗体」という用語はまた、抗原結合領域を有する任意の抗体様分子を指すためにも使用され、これにはFab'、Fab, F(ab')2、一本鎖ドメイン抗体(DAB)、Fv、scFv(一本鎖Fv)等の抗体断片、ならびに二価抗体、三価抗体、および多価抗体等の操作された多価抗体断片が含まれる。抗体に基づいた様々な構築物および断片を調製および使用するための技法は、当技術分野において周知である。抗体を調製および特徴づけするための手段もまた、当技術分野において周知である(例えば、Antibodies: A Laboratory Manual、Cold Spring Harbor Laboratory、1988;参照として本明細書に組み入れられる、を参照のこと)。
【0104】
標的リガンドに対する親和性を有する抗体が同定されたならば、必要に応じて、ろ過、遠心分離、およびHPLCまたはアフィニティークロマトグラフィー等の様々なクロマトグラフィー法を用いて、抗体またはリガンド結合ポリペプチドを精製することができる。抗体を含むそのようなポリペプチドの断片は、ペプシンまたはパパイン等の酵素による消化を含む方法によるか、または化学的還元によるジスルフィド結合の切断によって、そのように産生された抗体から得ることができる。または、本発明の包含する、タンパク質断片を含む抗体または他のポリペプチドは、自動ペプチド合成機を使用して合成することも可能である。
【0105】
分子クローニングアプローチは、本発明に従ってその後標的リガンドに対する親和性に関してスクリーニングされ得る候補抗体の異種性集団を作製するための1つの適切な方法を含む。本発明の1つの態様では、動物の脾臓から単離されたRNAから、コンビナトリアル免疫グロブリンファージミドを調製することができる。スクリーニングされるべきリガンドで動物を免疫することにより、アッセイを特定の抗原に標的することが可能である。従来技術に勝る本アプローチの恩典は、1ラウンドで約104倍の抗体を産生させスクリーニングすることができる点、およびH鎖およびL鎖の組み合わせによって、適切な抗体の発見の機会をさらに増す新たな特異性が産生される点である。
【0106】
VIII. 核酸の操作および検出
本発明の特定の態様では、核酸を操作、単離、および/または検出するための1つまたは複数の技法を使用することが所望され得る。そのような技法には、例えば、宿主細胞を形質転換するためのベクターの調製、およびトランスジェニック細胞から選択された核酸部分をクローニングする方法が含まれ得る。そのような操作を実施するための方法は、本開示を考慮して当業者に周知であろう。
【0107】
増幅用の鋳型として使用する核酸は、標準的な方法に従って細胞、組織、または他の試料から単離することができる(Sambrookら、1989)。特定の態様においては、鋳型核酸を実質的に精製せずに、細胞全体、または組織ホモジネートまたは生体液試料で解析を行うことが可能である。核酸は、ゲノムDNAまたは分画したRNAもしくは全細胞RNAであってよい。RNAを使用する場合、まずRNAを相補DNAに変換することが望ましい場合がある。
【0108】
本明細書で用いる「プライマー」という用語は、鋳型依存的過程において、新生核酸の合成をプライミングし得る任意の核酸を包含することが意図される。典型的には、プライマーは10〜20および/または30塩基対長のオリゴヌクレオチドであるが、より長い配列も使用できる。プライマーは、二本鎖および/または一本鎖の形態で提供され得るが、一本鎖の形態が好ましい。
【0109】
選択した核酸配列に相当する核酸に選択的にハイブリダイズするように設計したプライマー対を、選択的ハイブリダイゼーションを可能にする条件下で、鋳型核酸と接触させる。所望の用途に応じて、プライマーに完全に相補的な配列へのハイブリダイゼーションのみを可能にする高ストリンジェンシー条件を選択してもよい。他の態様では、プライマー配列との1つまたは複数のミスマッチを含む核酸の増幅を可能にするため、ストリンジェンシーを下げてハイブリダイゼーションを行ってもよい。ハイブリダイズしたならば、鋳型-プライマー複合体を、鋳型依存的核酸合成を促進する1つまたは複数の酵素と接触させる。十分量の増幅産物が産生されるまで、「サイクル」とも称される複数ラウンドの増幅を行う。
【0110】
増幅産物を検出および定量することができる。特定の適用では、検出は視覚的手段によって行われ得る。または、検出は、取り込まれた放射標識または蛍光標識の化学発光、放射性シンチグラフィーを介した、またはさらに電気インパルス信号および/または熱インパルス信号を用いる系を介した、産物の間接的同定を含み得る(Affymax技術;Bellus、1994)。
【0111】
所与の鋳型試料中に存在するオリゴヌクレオチド配列を増幅するために、多くの鋳型依存的過程が利用可能である。最もよく知られている増幅法はポリメラーゼ連鎖反応法(PCR(商標)と称される)であり、これは米国特許第4,683,195号、第4,683,202号、および第4,800,159号、ならびにInnisら、1988に詳細に記載されており、これらはそれぞれその全体が参照として本明細書に組み入れられる。
【0112】
逆転写PCR(商標)増幅手順は、増幅したmRNAの量を定量化するために実施され得る。RNAをcDNAに逆転写する方法は周知である(Sambrookら、1989を参照のこと)。逆転写の別法では、熱安定性DNAポリメラーゼを利用する。これらの方法は、国際公開公報第90/07641号に記載されている。ポリメラーゼ連鎖反応法は、当技術分野で周知である。RT-PCRの代表的な方法は、米国特許第5,882,864号に記載されている。
【0113】
増幅の別の方法はリガーゼ連鎖反応法(「LCR」)であり、これは、その全体が参照として本明細書に組み入れられる欧州出願第320 308号に開示されている。米国特許第4,883,750号では、標的配列にプローブ対を結合させるための、LCRに類似した方法について記載している。米国特許第5,912,148号に開示されている、PCR(商標)およびオリゴヌクレオチドリガーゼアッセイ(OLA)に基づく方法も用いることが可能である。
【0114】
本発明を実施する上で使用できる、標的核酸配列を増幅するための別法は、米国特許第5,843,650号、第5,846,709号、第5,846,783号、第5,849,546号、第5,849,497号、第5,849,547号、第5,858,652号、第5,866,366号、第5,916,776号、第5,922,574号、第5,928,905号、第5,928,906号、第5,932,451号、第5,935,825号、第5,939,291号、および第5,942,391号、英国出願第2202 328号、ならびにPCT出願PCT/US89/01025に開示されており、これらはそれぞれその全体が参照として本明細書に組み入れられる。
【0115】
PCT出願PCT/US87/00880に記載されているQbetaレプリカーゼもまた、本発明における増幅法として使用することが可能である。この方法では、標的の配列に相補的な領域を有するRNAの複製配列を、RNAポリメラーゼの存在下で試料に添加する。ポリメラーゼが複製配列をコピーすることになり、次にこれを検出することができる。
【0116】
制限エンドヌクレアーゼおよびリガーゼを用いて、制限酵素部位の一方の鎖内にヌクレオチド5'-[α-チオ]-三リン酸を含む標的分子の増幅を達成する等温増幅法もまた、本発明における核酸の増幅に有用である場合がある(Walkerら、1992)。米国特許第5,916,779号に開示されている鎖置換増幅(SDA)は、複数ラウンドの鎖置換および合成、すなわちニックトランスレーションを含む、核酸の等温増幅を実施する別の方法である。
【0117】
他の核酸増幅手順には、核酸配列に基づく増幅(NASBA)および3SR(Kwohら、1989;Gingerasら、国際公開公報第88/10315号、その全体が参照として本明細書に組み入れられる)を含む、転写に基づく増幅系(TAS)が含まれる。欧州出願第329 822号では、一本鎖RNA(「ssRNA」)、ssDNA、および二本鎖DNA(dsDNA)を繰り返して合成する段階を含む核酸増幅過程を開示しており、本発明に基づいてこの過程を使用することが可能である。
【0118】
国際公開公報第89/06700号(その全体が参照として本明細書に組み入れられる)では、標的一本鎖DNA(「ssDNA」)に対するプロモーター領域/プライマー配列のハイブリダイゼーション、それに続くその配列の多くのRNAコピーの転写に基づいた核酸配列増幅の図式を開示している。この図式は周期的ではない、すなわち、生じたRNA転写産物から新たな鋳型が産生されない。他の増幅法には、「race」および「片側PCR」が含まれる(Frohman、1990;Oharaら、1989)。
【0119】
IX. 実施例
以下の実施例は、本発明の好ましい態様を実証するために含めるものである。以下の実施例において開示する技法により、本発明者らが発見した技法が本発明の実施に際して十分に機能することが示され、したがって以下の実施例において開示する技法がその実施の好ましい様式を構成すると見なされ得ることが、当業者によって理解されるべきである。しかし当業者は、本開示を踏まえて、開示する特定の態様において多くの変更がなされ得り、本発明の精神および範囲から逸脱することなくなお同様または類似の結果が得られることを理解すべきである。
【0120】
実施例1
固定化ペリプラズム発現が低分子およびペプチドを標的することの実証
APExにより提示されたscFvが低分子およびペプチドを標的する能力を図1A〜図1Bおよび図1Cにそれぞれ示す。NlpAの最初の6アミノ酸(APEx解析のため内膜標的配列として機能する)の、抗メタンフェタミン、抗ジゴキシン、または抗ペプチドscfvのいずれかへの融合体を含む3つの大腸菌培養物を培養し、以下に記載するようにタンパク質発現を誘導した。次に、各構築物の細胞を、5×PBS緩衝液中で200 nM濃度のメタンフェタミン-FL(図1A)、ジゴキシゲニン-bodipy(図1B)、または200 nMペプチド(18 mer)-BodipyFL(図1C)で標識した。提示したデータは、各標識細胞培養物による10,000イベントのヒストグラム表示を示す。結果から、APExによって提示されたscfvが、非特異的リガンドに対する交差反応性を最小限に抑えつつ、その特異的抗原複合化フルオロフォアに結合する能力が実証される。
【0121】
実施例2
固定化ペリプラズム発現によるAb断片の認識の実証
scFvがより大きなタンパク質に接近できることを実証するため、まず、ヒトAb断片またはマウスAb断片に対するポリクローナル抗体血清が、固定化ペリプラズム発現によって大腸菌内膜上に提示されたそれぞれに由来するscFvを認識することを実証した。固定化ペリプラズム発現によりマウス由来scFvを発現する大腸菌(図2A)または固定化ペリプラズム発現によりヒト由来scFvを発現する大腸菌(図2B)を、以下に記載するように、抗マウスポリクローナルIgG(H+L)-Alexa-FLまたは抗ヒトポリクローナルIgG(Fab)-FITCのいずれかで標識した。それぞれ10000イベントのヒストグラム表示形態における結果から(図2A、図2B)、抗ヒトポリクローナル(約150 kDaの大きさ)がヒト由来scFvを特異的に認識し、抗マウスポリクローナル(150 kDa)がマウス由来scFvを認識することが実証された。
【0122】
実施例3
固定化ペリプラズム発現によって提示されるscFvが、大きな抗原複合化フルオロフォアに特異的に結合する能力の実証
固定化ペリプラズム発現によって提示されるscFvが、大きな抗原複合化フルオロフォアに特異的に結合する能力を実証するため、固定化ペリプラズム発現によって抗防御抗原(PA) scFv(PAは炭疽菌毒素の一成分である:83 kDaタンパク質)または抗ジゴキシゲニンscFvを発現する大腸菌を、以下に示すように誘導し標識した。10,000イベントのヒストグラムデータから、抗ジゴキシゲニンscFvを発現する細胞と比較して、PA-Cy5抗原複合化フルオロフォアへの特異的な結合が実証された(図3A)。この点をさらに明らかにするため、ジゴキシゲニンを240 kDa蛍光タンパク質であるフィコエリトリン(PE)に結合した。以下に示すように、細胞をこの複合体で標識した。固定化ペリプラズム発現により抗ジゴキシゲニンscFvを発現する大腸菌(10,000イベント)が大きなPE-ジゴキシゲニン複合体で標識されるのに対し、固定化ペリプラズム発現によって非特異的scFvを発現する大腸菌は蛍光をほとんど示さないことが認められた(図3B)。
【0123】
実施例4
フローサイトメトリー選択によるscFvライブラリーからの改良scFv変種の選択の実証
固定化ペリプラズム発現によってメタンフェタミンに対する親和性を有するscFvの変異ライブラリーを発現するポリクローナル大腸菌のスキャンを行った。メタンフェタミン複合化フルオロフォアを用いた選別および再選別を2ラウンド行うことにより、ライブラリーの亜集団が単離された(図4)。
【0124】
このライブラリーに由来する個々のクローンを同じメタンフェタミンフルオロフォアで標識し、以下のように解析した。親の抗メタンフェタミンscFvよりも高い平均FLシグナルを有する、変異体9と命名したクローンの例を図5に示す。
【0125】
実施例5
材料および方法:
A. ベクターの構築
リーダーペプチドおよび成熟NlpAタンパク質の最初の6アミノ酸は、プライマー
、VENTポリメラーゼ(New England Biolabs)、およびdNTP(Roche)を用いて、XL1-ブルー大腸菌(Stratagene)において全細胞PCR(Perken Elmer)を行うことにより作製した。次いで、これをNde1およびSfi1制限酵素エンドヌクレアーゼで切断し、マルチクローニングサイト(MCS)の下流に、エレメント(mycおよびhisタグ、Cm耐性マーカー、colE1起点、ならびにlac I)を有する大腸菌発現ベクターのlacプロモーターとMCSとの間にクローニングした。次に、関心対象のscFvをMCS内にクローニングし、このベクターをAbleC大腸菌(Stratagene)に形質転換した。
【0126】
B. 発現
TB培地+2%グルコースおよび30 mg/lクロラムフェニコール中に、大腸菌をOD600が0.1になるまで播種する。細胞を37℃で2時間培養した後、30分間で温度を25℃に下げる。次に、1 mM IPTGにより細胞を25℃で4時間誘導する。
【0127】
元のscFv配列を鋳型として利用し、Fromant Mら(1995)によって記載されているように、変異原性PCR法によりscFv配列の変異ライブラリーを構築した。次に、これらの変異産物を上記のAPEx発現ベクターにクローニングし、ABLEC大腸菌に形質転換し、2%グルコースおよび30 ug/mlクロラムフェニコールを含むSOC培地で寒天プレートにプレーティングした。30℃で一晩インキュベートした後、プレートから大腸菌を掻き取り、15%グリセロールアリコート中で凍結し、後にフローサイトメトリー選別するために-80℃で保存した。
【0128】
C. 標識手順
誘導した後、細胞を200 nMプローブを添加した5×PBS中で45分間インキュベートするか、または350μlの0.75 Mショ糖、100 mM Tris中に再懸濁する。次に10 mg/mlリゾチーム35μlを添加し、続いて穏やかに振盪しながら1 mM EDTA 700μlを1滴ずつ添加する。氷上で10分間静置させた後、0.5 M MgCl2を50μl添加する。さらに10分間氷上で静置させた後、懸濁液を13,200gで1分間遠心分離し、上清を除去し、1×PBS 500μl中に再懸濁する。次に細胞を200 nMプローブで45分間標識し、その後フローサイトメトリーにより解析し、改善された蛍光を選択する。
【0129】
D. 株およびプラスミド
APExによるスクリーニングには、株ABLE(商標)C(Stratagene)を使用した。大腸菌株TG1およびHB2151は、Griffinライブラリーと共に提供された。ABLE(商標)CおよびABLE(商標)KはStratageneから購入し、ファージM13K07はPharmaciaから購入した。ファージディスプレイビヒクルのFACS解析の陽性対照を、pHEN2内の既存のscFvを26.10 scFvに置換することにより構築し、pHEN2.digを作製した。陰性対照は、Griffin.1ライブラリーと共に提供された、抗サイログロブリンscFvを有するpHEN.thyである。PtacベクターはpIMS120の誘導体である(Hayhurst、2000)。
【0130】
E. ファージのパニング
Griffin.1ライブラリーは、CDR3ループの一部またはすべてがランダムに変異され、インビボで組換えられたヒト重鎖および軽鎖の大きなレパートリーに由来する半合成scFvライブラリーである(Griffithsら、1994)。このライブラリーは、本発明に基づく固定化ペリプラズム発現によるスクリーニングのための、候補結合ポリペプチドの1つの可能な供給源を示す。以下の実施例9に概説するように、ウェブサイトの取り扱い説明(www.mrc-cpe.cam.ac.uk/~phage/glp.html)に従って、ライブラリーをレスキューし、5ラウンドのパニングに供した。10μgml-1ジゴキシン-BSA複合体でイムノチューブをコーティングし、中和した溶出液を等分し、次のラウンドのファージパニング用にTG-1を、またはFAC解析用にABLE(商標)Cを感染させるために使用した。
【0131】
抗原結合ファージの濃縮を示すため、溶出液の力価をモニタリングした。反応性を確認するため、精製して力価を標準化した、各ラウンドで生じるファージ保存液のポリクローナルファージELISAを、ジゴキシン-オボアルブミン複合体で実施した。第3、第4、および第5ラウンドで生じた陽性クローンの割合は、各ラウンド後の96個の単離体のモノクローナルファージELISAにより決定した。陽性を、バックグラウンドシグナルが0.01を超えることはほとんどない、0.5よりも大きい吸光度と任意に定義した。第3、第4、および第5ラウンドによる24個の陽性クローンに対して、MvaIフィンガープリントを実施した。
【0132】
F. FACSスクリーニング
APEx発現をスキャンするため、B項およびC項に記載したように、APEx構築物を有する大腸菌のグリセロール保存液を培養して標識した。標識した後、細胞をPBSで一度洗浄し、スキャンした。bodipyまたはFL標識抗原を用いた上記の研究では、励起するために488 nmレーザーを使用し、Cy5には633 nmレーザーを使用した。機器を以下のように設定し、FACSCalibur(BD)でスキャンを達成した:側方散乱トリガーV 400、閾値250、前方散乱E01、FL1 V 400 FL2 V400(488 nm ex)、FL4 V 700(633 nm ex)。
【0133】
APEx発現の選別は以下の通りであった。選別はすべて、MoFlo FC(Cytomation)を用いて行った。B項およびC項に記載したように、前記ライブラリーを培養して標識し、PBSで一度洗浄し、FL強度が増加したものを選別した。集団のポリクローナルスキャンが濃縮を示すまで、次のラウンドの選別を行った(図4を参照のこと)。次に、個々のクローンを拾い上げ、FL活性を解析した。
【0134】
他の研究のため、ファージミド含有ABLE(商標)Cグリセロール保存液のアリコートを、600 nmにおけるODが約0.1 cm-1になるように1 mlの2×TY(2%グルコース、100μgml-1アンピシリン)中に掻き取った。37℃で2時間激しく振盪した後、1 mMになるようにIPTGを添加し、培養物を25℃で4時間振盪した。培養物50μlを、室温で1時間、5×PBS 1 ml中で適度に撹拌しながら、100 nM BODIPY(商標)-ジゴキシゲニンで標識した(Daughertyら、1999)。標識の最後の10分間、ヨウ化プロピジウムを2μg/ml-1になるように添加した。細胞をペレット化し、標識混合液100μl中に再懸濁した。Becton-Dickinson FACSortでスキャンを行い、1500s-1に104イベントを回収した。
【0135】
FACSライブラリーを選別するため、テリフィックブロス中で細胞を培養し、0.1 mM IPTGで誘導した。1000 s-1の排除モードで、106イベントに対して(第2ラウンドでは107)選別を実施した。回収された選別溶液を0.7μm膜フィルターに通して、適切な抗生物質を添加したSOC寒天の表面上にフィルターを乗せた後、コロニーを30℃で24時間培養した。
【0136】
E. ファージクローンの解析
ELISAによるファージ粒子のスクリーニングは、以下のように要約される。ELISAにおけるファージの結合を、一次ヒツジ抗M13抗血清(CP laboratoriesまたは5 prime ? 3 prime)、その後の西洋ワサビペルオキシダーゼ(HRP)複合化抗ヒツジ抗体(Sigma)により検出する。または、HRP-抗M13複合体を用いることも可能である(Pharmacia)。2% MPBSまたは3% BSA-PBSでプレートをブロッキングできる。ポリクローナルファージELISAに関しては、その技法は一般に以下の通りである:MicroTest III可塑性アッセイプレート(Falcon)を、ウェルあたり100μlのタンパク質抗原でコーティングする。抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし4℃で一晩コーティングする。ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、37℃で2時間、ウェルを2% MPBSまたは3% BSA-PBSで満たす。PBSでウェルを3回濯ぐ。選択の各ラウンドの終了時に保存したファージのアリコートから、PEG沈殿したファージ10μlを添加する(約1010 tfu)。2% MPBSまたは3% BSA-PBSで100μlに調整する。室温で90分間インキュベートする。被験溶液を除去し、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。2% MPBSまたは3% BSA-PBS中に適切に希釈したHRP-抗M13またはヒツジ抗M13抗血清を添加する。室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。ヒツジ抗M13抗血清を用いる場合には、2% MPBSまたは3% BSA中に適切に希釈したHRP-抗ヒツジ抗血清と共に室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液、pH 6.0、使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0137】
モノクローナルファージELISAは、以下のように要約され得る。モノクローナルファージ抗体を同定するため、pHENファージ粒子をレスキューする必要がある:C10のプレートから(各ラウンドの選択後)、96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを播種し、30℃で振盪しながら(300 rpm)一晩培養する。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび1%グルコースを含む2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。37℃で振盪しながら1時間培養する。最終濃度が15%になるようにグリセロールを添加し、次いでこのプレートを-70℃で保存することによって、元の96ウェルプレートのグリセロール保存液を作製する。(2枚目のプレートの)各ウェルに、感染効率(moi)10となるようにVCS-M13またはM13KO7ヘルパーファージを添加する。37℃で30分間静置する。1,800gで10分間遠心分離した後、上清を吸引除去する。100μg/mlアンピシリンおよび50μg/mlカナマイシンを含む2×TY 200μl中に、ペレットを再懸濁する。30℃で振盪しながら一晩培養する。1,800gで10分間遠心分離し、上清100μlを先に詳述したファージELISAにおいて使用する。
【0138】
抗体断片の産生は以下のように要約される。選択されたpHENは、HB2151に感染させ、次いでELISA用に抗体断片の可溶性発現が起こるように誘導する必要がある。各選択から溶出されたファージ10μl(約105 t.u.)を採取し、対数増殖中のHB2151細菌200μlに37℃で30分間感染させる(湯浴)。100μg/mlアンピシリンおよび1%グルコースを含むTYE上に、1μl、10μl、100μl、および1:10希釈物をプレーティングする。これらのプレートを37℃で一晩インキュベートする。96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを釣菌し、37℃で振盪しながら(300 rpm)一晩培養する。別のプレートに接種するために使用したならば、最終濃度が15%となるようにグリセロールを添加し、-70℃で保存することによって、このプレートのグリセロール保存液を作製できる。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび0.1%グルコースを含む新鮮2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。600 nmにおけるODが約0.9になるまで(約3時間)、37℃で振盪しながら培養する。所要のODに達したならば、100μg/mlアンピシリンおよび9 mM IPTG(最終濃度1 mM IPTG)を含む2×TY 25μlを添加する。30℃でさらに16時間〜24時間振盪し続ける。ウェル当たり100μlのタンパク質抗原で、MicroTest III可塑性アッセイプレート(Falcon)をコーティングする。
【0139】
抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし、室温で一晩コーティングする。翌日、ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、ウェル当たり200μlの3% BSA-PBSで37℃で2時間ブロッキングする。細菌プレートを1,800gで10分間遠心分離し、上清(可溶性scFvを含む)100μlをELISAプレートに添加し、室温で1時間インキュベートする。被験溶液を除去し、PBSで3回洗浄する。1% BSA-PBS中で濃度4μg/mlの精製9E10抗体(mycタグ化抗体断片を検出する)50μl、および1% BSA-PBS中で1:500希釈したHRP-抗マウス抗体50μlを添加する。室温で60分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液(pH 6.0)。使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する。)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0140】
ライブラリー中の挿入物は、
と命名したプライマーを用いてPCRスクリーニングすることにより、スクリーニングできる。VHおよびVLの配列決定に関しては、プライマー
の使用を推奨する。
【0141】
実施例6
抗体親和性成熟
Shortら(1995)は、表面プラズモン共鳴により測定される、ジゴキシンに対する平衡解離定数(KD) が300 pMである、A4-19と命名された26-10変異体を単離した。A4-19は、重鎖CDR1内に3つのアミノ酸置換を含む(VH:T30→P、VH:D31→S、およびVH:T34→Y)。既に非常に堅固な結合を示す抗体から開始する場合でさえも、可溶性ペリプラズム発現/FACSスクリーニングによって、結合親和性の増大した変異体が得られ得るかどうかを試験した。ジゴキシンハプテンに接触するか(VL:T91、VL:P96)または近接する(VL:V94)3つの軽鎖CDR3残基を(Jeffreyら、1993)、NNS(S=GまたはC)戦略を用いてランダム化した(Daughertyら、1998)。pelBリーダーによりペリプラズム中に発現する2.5×106個の形質転換体のライブラリーを作製し、2ラウンドのFACSによりスクリーニングした。1ラウンド目のスクリーニングでは、100 nMの蛍光プローブで標識した細胞をPBSで1度洗浄し、非蛍光粒子が蛍光粒子と同じ液体エレメント内に検出される場合にも、機器がすべての蛍光イベントを回収するという回収様式を用いて選別した。回収様式での操作により、純度は犠牲になるものの、非常に稀な細胞が回収されることが十分に保証された。
【0142】
回収した細胞を再度培養して標識し、洗浄し、次いで50倍過剰(50μM)の遊離ジゴキシンと共に様々な時間(15分〜90分)インキュベートした。同時発生的蛍光イベントおよび非蛍光イベントを拒絶し、それにより高度の純度が得られる排除様式を用いた選別により、所望のレベルの蛍光を保持する細胞を単離した。非蛍光競合物質と様々な時間インキュベートした後に得られた細胞プールの蛍光減衰速度を測定した。初期の時点で(競合物質とのインキュベーション<60分)、開始のA4-19抗体と比較してわずかに速い速度が認められたが、60分および90分の集団では速度が減少した。競合60分後に得られた細胞集団由来の5個のランダムなクローンおよび90分プールに由来する13クローンを無作為に拾い、配列決定した(表1)。強い配列共通性が明らかに明白であった。精製抗体のハプテン結合反応速度をSPRにより決定し、その結果を表1に示す。対応するアミノ酸配列を、配列番号:8〜19に示す。ゲル濾過FPLCによる精製および解析の際に、どの変異体も二量体化することが認められなかったことに留意されたい。試験した変異体はすべて、開始のA4-19抗体の結合速度定数(0.9±0.2x106 M-1)とは識別できない結合速度定数(kon)を示した。競合60分後に単離されたクローンのkdissは、A4-19のkdissと同じか、またはそれよりも速かった。競合90分後に単離されたクローンは、溶液中でより遅いkdissを示した。1つのクローン90.3は、150 pMのKDを生じる2倍遅い解離速度定数を示した。したがって、本発明のライブラリースクリーニング方法によって、既にナノモル以下のKDを示す抗体から開始する場合でも、特異的標識によりさらに優れた変異体の単離が可能となった。驚くほどのことではないが、興味深いことに、4-19に存在する3つの重鎖CDR1変異および軽鎖の残基94および96における2つの変異の効果は、相加的であった。
【0143】
(表1)60分(クローン60.1〜60.4)および90分(クローン90.1〜90.6)オフレート選択により単離された、重鎖および軽鎖CDR3アミノ酸配列(配列番号:8〜20)。括弧内に同一クローン数を示す。ND:未検。
【0144】
実施例7
蛍光シグナルの最大化
ペリプラズム中に可溶型のscFv抗体を発現する細胞の蛍光強度は、用いた大腸菌株および培養条件に強く依存した。26-10抗体を用いると、細胞を25℃で培養した場合に最大蛍光強度が得られた。準生理的温度での培養は、いくつかの有益な効果を有する。低い温度(すなわち25℃)でscFvを発現させることにより、折りたたみ経路を遅くすることによって直接的に、および発現の負荷を低減するためにプラスミドのコピー数を減少することによって間接的に、scFvの適切な折りたたみが促進される。実際に、37℃でのscFvの直接発現では、一般に可溶性タンパク質はほとんどまたは全く産生されない(例えば、Goughら、1999を参照のこと)。外膜の組成もまた非生理的温度で変わり、透過性の増大が生じる(Martinezら、1999)。それどころかむしろ、様々な大腸菌株間の著しい相違に注目した。試験した様々な株の中で、ABLE(商標)Cにおいて最も高い蛍光強度が得られた。この株におけるタンパク質発現および外膜タンパク質特性の予備的な解析から、高い蛍光シグナルはColE1起点プラスミドのコピー数を減少させるpcnB変異に起因せず、むしろ細胞外被のタンパク質組成における相違に起因することが示された。実際に、ABLE(商標)Cのより強力な染色は、ELISAおよびウェスタンブロッティングによって推定されるように、他の株に対する高レベルのタンパク質発現とは関連していなかった。
【0145】
高浸透圧条件下で蛍光標識することにより、有意に高い蛍光が生じた。標識中に細胞を5×PBS中でインキュベートした場合、5〜7倍の蛍光の増加が得られた(通常のPBS中でインキュベートした細胞の平均FL1が20〜30であるのに対し、平均FL1>150)。しかし、細胞の生存度がかなり減少したため、増大したシグナルは損なわれた。タンパク質の高度に多様化したライブラリーをスクリーニングする場合、そのような生存度の減少は所望されない場合があり、その発現は宿主細胞に対する有害な効果を既に有している可能性がある。同様に、M13KO7等の繊維状ファージとの同時感染によってファージショック応答が誘導され、特に外膜透過性の増大が生じる。M13KO7の感染により、集団の平均蛍光において3倍の増大が生じた。しかし、高浸透圧ショックを用いた場合と同様に、ヨウ化プロピジウム染色によって決定される培養物の生存度は若干減少した。
【0146】
蛍光リガンドによる細胞の標識、それに続く大過剰の遊離リガンドとのインキュベーションにより、平均蛍光強度が時間依存的に減少する。蛍光減衰の速度は、抗体-抗原複合体の解離速度を反映する(Daughertyら、2000)。ジゴキシンに関して、蛍光減衰の速度が、BIACOREを使用して精製抗体で測定した解離速度と比較して約3〜4倍遅いことが認められた。インビトロにおける抗体/抗原複合体の解離速度と比較して蛍光減衰の速度が低いことは、リガンドと細胞との間の衝突頻度、ペリプラズムにおける抗体の濃度、および当然、外膜を介する拡散の速度を含む様々な効果に由来する(ペリプラズム空間における反応速度論の解析に関しては、Martinezら、(1996)を参照のこと)。予測され得るように、インビトロで決定したkoff速度に対するペリプラズムにおける蛍光減衰の速度の比は、抗原に依存する。
【0147】
実施例8
ペリプラズム中に可溶型のscFv抗体を発現する細胞の蛍光検出および濃縮
26-10 scFv抗体は、ジゴキシンおよびジゴキシゲニン等の強心配糖体に高い親和性で結合する(ジゴキシンおよびジゴキシゲニンに対する精製抗体のKDは、それぞれ0.9±0.2×10-9 M-1および2.4±0.4×10-9 M-1である。Chenら、1999)。26-10 scFvおよびその変種は、ハプテン結合に及ぼすCDRおよびフレームワーク領域における変異の影響を理解するためのモデル系として広く用いられている(Schilbachら、1992;Shortら、1995;Daughertyら、1998、2000;Chenら、1999)。26-10 scFvの誘導体を、大腸菌アラビノースプロモーター下で、大腸菌ペリプラズムへの分泌を可能にするpelBリーダーペプチドと共に可溶型で発現させた。生じたプラスミドベクター(pBAD30pelB-Dig)をara-大腸菌株LMG194に形質転換し、0.2% w/vアラビノースでタンパク質合成を誘導した。200 nMのジゴキシゲニン-BODIPY(商標)と共にインキュベートした場合、25℃で培養した細胞が強く蛍光を発し、非特異的に結合したリガンドを除去するために十分に洗浄した後でさえも蛍光シグナルが保持されることが認められた。ペリプラズムにおける親水性溶質の透過に関して一般に受容される大きさの限界である約600 Da(DecadおよびNikaido、1976)よりも有意に高い分子量を有するプローブで細胞が標識されることから、蛍光シグナルが主に生存不能な透過可能となった細胞に起因する可能性が生じた。しかし、通常隔絶された核酸にインターカレートすることにより、膜損傷細胞に特異的に結合する生存度染色ヨウ化プロピジウムでの染色から、>90%の細胞がこの色素に透過性でないことが明らかになった。これは、対数期後期に回収した対照大腸菌培養物における無傷の細胞の割合と類似している。
【0148】
ペリプラズム中に可溶型の26-10抗体を発現する細胞を、1ラウンドの選別のみで、ベクターで形質転換した大過剰の大腸菌から濃縮し得た。具体的には、LMG194(pBAD30pelB-Dig)を、空ベクター(pBAD30)を含む10,000倍過剰の大腸菌と混合した。前者の細胞はアンピシリンおよびクロラムフェニコールの両方に耐性であるのに対して(ampr、Cmr)、後者はアンピシリンのみに耐性である(ampr)。0.2% w/vアラビノースで4時間誘導した後、細胞を100 nMのジゴキシゲニン-BODIPY(商標)で1時間標識し、FACSにより蛍光性の細胞を単離した。選別された細胞を再培養し上記のように再標識した後に、この集団は5〜8倍の平均蛍光強度の増加を示した(選別前の細胞混合物についてFL1=4であるのに対してFL1=20)。濃縮された集団におけるscFv発現クローンの画分を、Cmrでもあるamprクローンの数によって測定した。amprコロニーの80%はCmrでもあり、このことから、蛍光標識および細胞選別により1ラウンドで1,000倍をはるかに上回る濃縮が起こることが示された。
【0149】
実施例9
準最適温度における細胞透過性の増加
ペリプラズム中に可溶型のscFv抗体を発現する細胞の蛍光強度は、用いた大腸菌株および培養条件に強く依存した。26-10抗体を用いた場合、細胞を25℃で培養した場合に最大蛍光強度が得られた。準生理的温度での培養は、いくつかの有益な効果を有する。低い温度(すなわち25℃)でscFvを発現させることにより、折りたたみ経路を遅くすることによって直接的に、および発現の負荷を低減するためにプラスミドのコピー数を減少することによって間接的に、scFvの適切な折りたたみが促進される。実際に、37℃でのscFvの直接発現では、一般に可溶性タンパク質はほとんどまたは全く産生されない(例えば、Goughら、1999を参照のこと)。外膜の組成もまた非生理的温度で変わり、透過性の増大が生じる(Martinezら、1999)。それどころかむしろ、様々な大腸菌株間の著しい相違に注目した。試験した様々な株の中で、ABLE(商標)Cにおいて最も高い蛍光強度が得られた。この株におけるタンパク質発現および外膜タンパク質特性の予備的な解析から、高い蛍光シグナルはColE1起点プラスミドのコピー数を減少させるpcnB変異に起因せず、むしろ細胞外被のタンパク質組成における相違に起因することが示された。実際に、ABLE(商標)Cのより強力な染色は、ELISAおよびウェスタンブロッティングによって推定されるように、他の株に対する高レベルのタンパク質発現とは関連していなかった。
【0150】
高浸透圧条件下で蛍光標識することにより、有意に高い蛍光が生じた。標識中に細胞を5×PBS中でインキュベートした場合、5〜7倍の蛍光の増加が得られた(通常のPBS中でインキュベートした細胞の平均FL1が20〜30であるのに対し、平均FL1>150)。しかし、細胞の生存度がかなり減少したため、増大したシグナルは損なわれた。タンパク質の高度に多様化したライブラリーをスクリーニングする場合、そのような生存度の減少は所望されない場合があり、その発現は宿主細胞に対する有害な効果を既に有している可能性がある。同様に、M13KO7等の繊維状ファージとの同時感染によってファージショック応答が誘導され、特に外膜透過性の増大が生じる。M13KO7の感染により、集団の平均蛍光において3倍の増大が生じた。しかし、高浸透圧ショックを用いた場合と同様に、ヨウ化プロピジウム染色によって決定される培養物の生存度は若干減少した。
【0151】
蛍光リガンドによる細胞の標識、それに続く大過剰の遊離リガンドとのインキュベーションにより、平均蛍光強度が時間依存的に減少する。蛍光減衰の速度は、抗体-抗原複合体の解離速度を反映する(Daughertyら、2000)。ジゴキシンに関して、蛍光減衰の速度が、BIACOREを使用して精製抗体で測定した解離速度と比較して約3〜4倍遅いことが認められた。インビトロにおける抗体/抗原複合体の解離速度と比較して蛍光減衰の速度が低いことは、リガンドと細胞との間の衝突頻度、ペリプラズムにおける抗体の濃度、および当然、外膜を介する拡散の速度を含む様々な効果に由来する(ペリプラズム空間における反応速度論の解析に関しては、Martinezら、(1996)を参照のこと)。予測され得るように、インビトロで決定したkoff速度に対するペリプラズムにおける蛍光減衰の速度の比は、抗原に依存する。
【0152】
実施例10
FACSによるレパートリー抗体ライブラリーの解析およびスクリーニング
多種多様な抗体配列を含む大きなレパートリーライブラリーをスクリーニングすることにより、新規に、すなわち動物を免疫することなく、抗体を単離することができる。そのような大きなライブラリーのスクリーニングは、十分に確立されている(Nissimら、1994、Winterら、1994、Griffithら、1994、Knappikら、2000)。これまで、利用可能な大きな抗体レパートリーライブラリーはすべて、ファージディスプレイによる使用のために構築されてきた。しかし、ファージディスプレイ用に構築されたライブラリーは、内膜への固定化または可溶型での、細菌のペリプラズム空間内でのタンパク質発現のためにも使用することが可能である。特に、繊維状バクテリオファージにおける低タンパク質コピー数提示では、組換えポリペプチドをpIIIへのN末端融合物として発現させる。ファージ生合成過程において、pIII融合物はまずペリプラズムに標的され、pIIIの小さなC末端部分によって内膜に固定される。ファージが放出される際に、scFv-pIII融合物はファージの末端で野生型pIIIと同時に組み込まれ、これにより会合過程が完了する(RakonjacおよびModel、1998;Rakonjacら、1999)。ファージディスプレイに関して最も広く用いられるベクターでは、N末端scFvとpIII遺伝子との間にアンバーコドンが配置されている。したがって、適切な大腸菌サプレッサー株では、scFvを提示するために完全長のscFv-pIII融合タンパク質が産生されるが、非サプレッサー株では、可溶性scFvのみが発現する。または、融合物に内膜固定化ペプチドを含めることにより、固定化発現が達成され得る。
【0153】
ファージディスプレイでの抑制の程度はベクターおよび株によって変化するが、10%の読み過ごししか可能にしない傾向がある。したがって、ファージディスプレイの生物学の結果として、アンバーコドンを含むすべてのライブラリーは、宿主に関係なくある程度のペリプラズムの発現を生じる。よって、FACSが、既存の高度に多様化した天然ライブラリーからのリガンド結合タンパク質の単離に役立ち得るかどうかを検討することは、非常に興味深いことであった(Griffithsら、1994;Vaughanら、1996;Sheetsら、1998;Piniら、1998;de Haardら、1999;Knappikら、2000;SblatteroおよびBradbury、2000)。
【0154】
ファージパニングによるファージライブラリーの従来のスクリーニングにより、天然抗体レパートリーライブラリーから、強心配糖体ジゴキシンに特異的なscFvを発現するファージが濃縮された。BSA複合体に対してパニング過程を行い、オボアルブミン複合体に対してスクリーニングを行い、タンパク質およびハプテン-タンパク質界面の結合体の出現を減少させた。パン4由来の24個の陽性単離体は同じフィンガープリントを共有し、6クローンのDNA配列決定から、同じ重鎖配列および軽鎖配列が確認され(「dig1」)、6つのうち1つ(「dig2」)は特有のHCDR3とLCDR3との組み合わせを有した。同一条件下および異なる条件下でファージライブラリーを再度スクリーニングしたところ、同じDNAフィンガープリントを有するクローンの単離のみが生じた。
【0155】
パニングの各ラウンド後に大腸菌ABLE(商標)CでレスキューしたファージのFACS解析から、ファージELISAシグナルに酷似した、3ラウンドにおける平均蛍光の増加が示された。1ラウンドのFACSによる結合クローンの有意な濃縮は、3ラウンド目のファージパニングから得られた集団を用いて開始することにより得られた。この結果は、パニング実験の過程で得られた濃縮特性と一致する。第3、第4、および第5ラウンド由来の106個の細胞をFACSスクリーニングおよび選別することによって、それぞれ30%、80%、および100%の頻度で陽性クローンが単離された。
【0156】
第3ラウンドの集団からFACSによって単離された14クローンのうちの5つは、ジゴキシンに対する結合に関して陽性であることが認められた。重要なことには、このクローンのうちの3つは、ファージパニングで見逃された別の抗体に相当していた(本明細書において「dig3」と称する)。残りの2つは、dig1クローンであった。この結果から、ペリプラズム空間に発現し、蛍光リガンドによって標識されたライブラリーのFACSスクリーニングにより、他のライブラリースクリーニング法によって単離され得ないクローンが単離されることが実証される。
【0157】
実施例11
Griffin.1ライブラリーを使用するための方法の要約
Griffin.1ライブラリーを使用するための方法は、以下のように要約され得る。Griffin.1ライブラリーは、合成V遺伝子断片から作製されたscFvファージミドライブラリーである。このライブラリーは、重鎖および軽鎖可変領域をloxライブラリーベクター(Griffithsら、1994)からファージミドベクターpHEN2に再クローニングすることにより作製された。このライブラリーを使用するためのキットは、合成scFvライブラリーのチューブ(1 ml)、陽性対照(抗サイログロブリンクローンを含むTG1)のグリセロール保存液、陰性対照(pHEN2を含むTG1)のグリセロール保存液、ファージ粒子を増殖させるための大腸菌TG1(Gibson、1984)サプレッサー株(K12、del(lac-pro)、supE、thi、hsdD5/F'traD36、proA+B+、lacIq、lacZdelM15)のグリセロール保存液(供給される株は、このT-ファージ耐性変種である)、抗体断片を発現させるための大腸菌HB2151(Carterら、1985)および非サプレッサー株(K12、ara、del(lac-pro)、thi/F'proA+B+、lacIq、lacZdelM15)のグリセロール保存液を含む。このライブラリーは、必要時まで-70℃で凍結保存する。
【0158】
株をプレーティングし、次に100μg/mlアンピシリンおよび1%グルコースを含む2×TY中のそれぞれの一晩培養物として(37℃で振盪しながら)培養する。100μg/mlアンピシリンおよび1%グルコースを含む2×TY(2×TYは、1リットルあたり、トリプトン16 g、酵母エキス10 g、およびNaCl 5 g)で培養物を1:100に希釈し、以下に記載する手順に従ってファージミドをレスキューする。サイログロブリンでコーティングしたイムノチューブにおける1ラウンドの選択には、陽性対照および陰性対照の1:100混合物を共に用いる。
【0159】
ライブラリーを使用するための手順は、以下のように要約される。ファージ/ファージミドは、性繊毛を介してF+大腸菌に感染する。性繊毛の生成および効率的な感染のためには、大腸菌を37℃で培養し、これが対数期(600 nmにおけるODが0.4〜0.6)になければならない。以下の手順全体にわたり、そのような培養物が必要である。それは以下のように調製し得る:最小培地プレートから2×TY培地5 ml中に細菌コロニーを移し、37℃で振盪しながら一晩培養する。翌日、新鮮な2×TY培地中に1:100希釈することにより継代培養し、OD 0.4〜0.6まで37℃で振盪しながら培養し、次いでファージに感染させる。様々なヘルパーファージが、ファージミドライブラリーのレスキューに利用可能である。VCS-M13(Stratagene)およびM13KO7(Pharmacia)は小アリコートで購入することができ、ファージミドライブラリーをレスキューするために以下のように大量に調製することができる:OD 0.2の大腸菌TG1(または他の適切な株)200μlに、(十分に分離したプラークを得るために)ヘルパーファージの段階希釈物10μlを、37℃(温浴)で振盪せずに30分間感染させる。融解したH-トップアガー(42℃)3 mlを添加し、温めたTYE(注7)プレート上に注ぐ。静置した後、37℃で一晩インキュベートする。TG1の対数増殖培養物(上記参照)3〜4 ml中に、小プラークを採取する。37℃で振盪しながら約2時間培養する。2リットルフラスコ中の2×TY 500 ml中に接種し、上記のように1時間培養し、次いで最終濃度が50〜70μg/mlになるようにカナマイシン(25μg/ml水溶液)を添加する。さらに8〜16時間培養する。細菌を10,800 gで15分間遠心分離する。ファージの上清に1/5量のPEG/NaCl(20%ポリエチレングリコール6000-2.5 M NaCl)を添加し、氷上で最低30分間インキュベートする。10,800 gで15分間遠心分離する。ペレットをTE 2 ml中に再懸濁し、0.45μnフィルター(Minisart NML;Sartorius)を通して保存液をろ過滅菌する。保存液の力価を測定した後、約1×1012 p.f.u./mlになるように希釈する。-20℃でアリコートを保存する。特記されない限り、遠心はすべて4℃で行う。
【0160】
ライブラリーの増殖に関して、その手順は以下のように要約される:細菌ライブラリー保存液のすべて(約1×1010クローン)を、100μg/mlアンピシリンおよび1%グルコースを含む2×TY 500 mlに接種する。600 nmにおけるODが0.5になるまで、37℃で振盪しながら培養するが、これには約1.5〜2時間を要するはずである。1:20の比率でヘルパーファージを添加することにより(600 nmにおけるODが1の細菌=約8x108細菌/mlであることを考慮した、細菌細胞数:ヘルパーファージ粒子数)、この培養物25 ml(1x1010細菌)にVCS-M13またはM13KO7ヘルパーファージを感染させる。
【0161】
感染させた細胞を3,300 gで10分間遠心分離する。100μg/mlアンピシリンおよび25μg/mlカナマイシンを含む2×TY 30 ml中に、ペレットを穏やかに再懸濁する。100μg/mlアンピシリンおよび25μg/mlカナマイシンを含む予め温めた2×TY 470 mlを添加し、30℃で振盪しながら一晩インキュベートする。ポリエチレングリコール(PEG)6000で沈殿させることにより、ファージが濃縮され、あらゆる可溶性抗体が除去され得る(TG1において、抗体遺伝子とgIIIとの連結部においてコードされるアンバー終止コドンの抑制が決して完全ではないので)。A6からの培養物を10,800 gで10分間(または3,300 gで30分間)遠心分離する。上清に1/5量のPEG/NaCl(20%ポリエチレングリコール6000、2.5 M NaCl)を添加する。十分に混合し、4℃で1時間以上静置する。10,800 gで30分間遠心分離する。ペレットを水40 ml中に再懸濁し、PEG/NaCl 8 mlを添加する。混合し、4℃で20分以上静置する。10,800 gで10分間、または3,300 gで30分間遠心分離した後、上清を吸引除去する。短時間再遠心分離した後、残存するすべてのPEG/NaClを吸引除去する。ペレットをPBS 5 ml中に再懸濁し、微量遠心機で11,600 gで10分間遠心分離し、残りの細菌残屑のほとんどを除去する。短期保存の場合は4℃で、または長期保存の場合はPBS、15% グリセロール中で-70℃で、ファージ上清を保存する。ファージ保存液の力価を測定するため、PBS 1 ml中にファージ1μlを希釈し、このうちの1μlを用いて、OD600で0.4〜0.6のTG1 1 mlを感染させる。このうちの50μl、1:102希釈物の50μl、および1:104希釈物の50μlを、100μg/mlアンピシリンおよび1%グルコースを含むTYEプレート上にプレーティングし、37℃で一晩培養する。ファージ保存液は、1012〜1013/mlであるはずである。
【0162】
イムノチューブにおける選択は、以下のように要約される。Nuncイムノチューブ(Maxisorp カタログ番号4-44202)を、所要の抗原4 mlで一晩コーティングする。コーティングの効率は、抗原濃度、緩衝液、および温度に依存できる。通常、PBS中または50 mM炭酸水素ナトリウム、pH 9.6中の抗原10μg/ml〜100μg/mlを室温(rt)で用いる。翌日、PBSでチューブを3回洗浄する(単にチューブにPBSを注ぎ込み、次いでさらに直ちにそのPBSを捨てる)。2% MPBSでいっぱいになるまでチューブを満たす。カバーをし、37℃で(または抗原の安定性に応じて室温で)2時間インキュベートしてブロッキングする。PBSでチューブを3回洗浄する。2% MPBS 4 ml中に、A13からのファージ1012〜1013 cfuを添加する。上下回転台上で連続して回転させながら室温で30分間インキュベートし、次いで室温で少なくともさらに90分間静置する。上清中の非結合ファージを捨てる。1ラウンド目の選択では、0.1% Tween-20を含むPBSでチューブを10回洗浄し、次にPBSで10回洗浄して界面活性剤を除去する。各洗浄段階は、緩衝液を注ぎ込み、直ちに捨てることによって行う。2ラウンド目およびそれに続くラウンドでの選択では、0.1% Tween-20を含むPBSでチューブを20回洗浄し、次にPBSで20回洗浄する。チューブから過剰のPBSを振り出し、100 mMトリエチルアミン1 ml(水50 ml中のトリエチルアミン(7.18 M) 700μl、使用する日に希釈する)を添加することによってファージを溶出し、上下回転台上で10分間連続して回転させる。インキュベーションを行っている間に、1M Tris(pH 7.4)0.5 mlを含むチューブを予め調製しておき、即座に中和するために、7から溶出したファージ1 mlに添加する。ファージは4℃で保存することもできるし、または上記のようにTG1を感染させるために用いることもできる。溶出後、1M Tris(pH 7.4)200μlをイムノチューブにさらに添加し、チューブ内の残ったファージを中和する。TG1の対数増殖培養物を9.25 ml採取し、溶出したファージを0.75 ml添加する。また、TG1培養物4 mlをイムノチューブに添加する。両培養物を37℃(湯浴)で30分間、感染させるために振盪せずにインキュベートする。感染させたTG1細菌10 mlおよび4 mlをプールし、100μl採取して4つ〜5つの100倍段階希釈物を作製する。100μg/mlアンピシリンおよび1%グルコースを含むTYE上に、これらの希釈物をプレーティングする。37℃で一晩培養する。残りの感染TG1培養物を採取し、3,300 gで10分間遠心分離する。ペレット化した細菌を2×TY 1 ml中に再懸濁し、100μg/mlアンピシリンおよび1%グルコースを含むTYEの大きなNuncバイオアッセイディッシュ(Gibco-BRL(注8))上にプレーティングする。一晩またはコロニーが見えるまで、30℃で培養する。
【0163】
さらなる選択ラウンドのため、細胞のバイオアッセイディッシュに5 ml〜6 mlの2×TY、15%グリセロールを添加し、ガラススプレッダーで細胞をほぐす。100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100 mlに、掻き取った細菌50〜100μlを接種した後、残りの細菌を-70℃で保存する。ここでも、600 nmでの開始OD=<0.1であることを確認することが望ましい。600 nmでのODが0.5になるまで(約2時間)、37℃で振盪しながら細菌を培養する。1:20の比率でヘルパーファージを添加することにより(600 nmにおける1 OD細菌=約8×108細菌/mlであることを考慮した、細菌細胞数:ヘルパーファージ粒子数)、この培養物10 mlにVCS-M13またはM13KO7ヘルパーファージを感染させる。37℃の湯浴中、振盪せずに30分間インキュベートする。感染細胞を3,300 gで10分間遠心分離する。100μg/mlアンピシリンおよび25μg/mlカナマイシンを含む2×TY 50 ml中に、ペレットを穏やかに再懸濁し、30℃で振盪しながら一晩インキュベートする。一晩培養物40 mlを採取し、10,800 gで10分間、または3,300 gで30分間遠心分離する。上清に1/5量(8 ml)のPEG/NaCl(20%ポリエチレングリコール6000、2.5 M NaCl)を添加する。十分に混合し、4℃で1時間以上静置する。10,800 gで10分間、または3,300 gで30分間遠心分離した後、上清を吸引除去する。短時間再遠心分離した後、残存するすべてのPEG/NaCl屑を吸引除去する。ペレットをPBS 2 ml中に再懸濁し、微量遠心機で11,600 gで10分間遠心分離し、残りの細菌残屑のほとんどを除去する。このファージ1 mlを4℃で保存し、残りの1 mlアリコートを次の選択ラウンドに使用できる。この選択をさらに2〜3ラウンド繰り返す。
【0164】
ELISAによるファージ粒子のスクリーニングは、以下のように要約される。ELISAにおけるファージの結合を、一次ヒツジ抗M13抗血清(CP laboratoriesまたは5 prime ? 3 prime)、その後の西洋ワサビペルオキシダーゼ(HRP)複合化抗ヒツジ抗体(Sigma)により検出する。または、HRP-抗M13複合体を用いることも可能である(Pharmacia)。2% MPBSまたは3% BSA-PBSでプレートをブロッキングできる。ポリクローナルファージELISAに関しては、その技法は一般に以下の通りである:MicroTest III可塑性アッセイプレート(Falcon)を、ウェルあたり100μlのタンパク質抗原でコーティングする。抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし室温で一晩コーティングする。ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、ウェル当たり200μlの2% MPBSまたは3% BSA-PBSで37℃で2時間ブロッキングする。PBSでウェルを3回濯ぐ。選択の各ラウンドの終了時に保存したファージのアリコートから、PEG沈殿したファージ10μlを添加する(約1010 cfu)。2% MPBSまたは3% BSA-PBSで100μlに調整する。室温で90分間インキュベートする。被験溶液を除去し、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。2% MPBSまたは3% BSA-PBS中に適切に希釈したHRP-抗M13またはヒツジ抗M13抗血清を添加する。室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。ヒツジ抗M13抗血清を用いる場合には、2% MPBSまたは3% BSA中に適切に希釈したHRP-抗ヒツジ抗血清と共に室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液(pH 6.0)。使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0165】
モノクローナルファージELISAは、以下のように要約され得る。モノクローナルファージ抗体を同定するため、pHENファージ粒子をレスキューする必要がある。C10のプレートから(各ラウンドの選択後)、96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを播種し、37℃で振盪しながら(300 rpm)一晩培養する。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび1%グルコースを含む2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。37℃で振盪しながら1時間培養する。最終濃度が15%になるようにグリセロールを添加し、次いでこのプレートを-70℃で保存することによって、元の96ウェルプレートのグリセロール保存液を作製する。(2枚目のプレートの)各ウェルに、100μg/mlアンピシリン、1%グルコース、および109 pfu VCS-M13またはM13KO7ヘルパーファージを含む2×TY 25μlを添加する。37℃で30分間静置した後、37℃で1時間振盪する。1,800gで10分間遠心分離した後、上清を吸引除去する。100μg/mlアンピシリンおよび50μg/mlカナマイシンを含む2×TY 200μl中に、ペレットを再懸濁する。30℃で振盪しながら一晩培養する。1,800gで10分間遠心分離し、上清100μlを先に詳述したファージELISAにおいて使用する。
【0166】
可溶性抗体断片の産生は以下のように要約される。選択されたpHENは、HB2151に感染させ、次いでELISA用に抗体断片の可溶性発現が起こるように誘導する必要がある。各選択から溶出されたファージ10μl(約105 t.u.)を採取し、対数増殖中のHB2151細菌200μlに37℃で30分間感染させる(湯浴)。100μg/mlアンピシリンおよび1%グルコースを含むTYE上に、1μl、10μl、100μl、および1:10希釈物をプレーティングする。これらのプレートを37℃で一晩インキュベートする。96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを釣菌し、37℃で振盪しながら(300 rpm)一晩培養する。別のプレートに接種するために使用したならば、最終濃度が15%となるようにグリセロールを添加し、-70℃で保存することによって、このプレートのグリセロール保存液を作製できる。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび0.1%グルコースを含む新鮮2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。600 nmにおけるODが約0.9になるまで(約3時間)、37℃で振盪しながら培養する。所要のODに達したならば、100μg/mlアンピシリンおよび9 mM IPTG(最終濃度1 mM IPTG)を含む2×TY 25μlを添加する。30℃でさらに16時間〜24時間振盪し続ける。ウェル当たり100μlのタンパク質抗原で、MicroTest III可塑性アッセイプレート(Falcon)をコーティングする。抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし、室温で一晩コーティングする。翌日、ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、ウェル当たり200μlの3% BSA-PBSで37℃で2時間ブロッキングする。細菌プレートを1,800gで10分間遠心分離し、上清(可溶性scFvを含む)100μlをELISAプレートに添加し、室温で1時間インキュベートする。被験溶液を除去し、PBSで3回洗浄する。1% BSA-PBS中で濃度4μg/mlの精製9E10抗体(mycタグ化抗体断片を検出する)50μl、および1% BSA-PBS中に1:500希釈したHRP-抗マウス抗体50μlを添加する。室温で60分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液(pH 6.0)。使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する。)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0167】
ライブラリー中の挿入物は、
と命名したプライマーを用いてPCRスクリーニングすることにより、スクリーニングできる。VHおよびVLの配列決定に関しては、プライマー
の使用を推奨する。
【0168】
実施例12
レパートリーライブラリーからのTNBに特異的なscFv抗体の単離
本実施例では、リガンドTNB(トリニトロベンゼン)に対するレパートリー抗体ライブラリーのスクリーニングについて要約する。標準的な手順により(実施例9を参照、またwww.mrc-cpe.cam.ac.uk/~phage/glp.htmlにも記載されている)、レパートリーライブラリー(Griffin-1ライブラリー)のファージパニングをまず3ラウンド行うことにより、ライブラリースクリーニングを開始した。様々なラウンドのパニングからレスキューしたファージを用いて、大腸菌ABLE Cを感染させた。この細胞を対数期中期まで培養し、上記のように可溶型のscFv抗体の発現を誘導し、蛍光色素Cy5に複合化した100 nM TNBSで標識した。標識した細胞を、633 nmで放射する5 mMダイオードレーザーを装備したCytomation MoFlo装置を用いて、フローサイトメトリーにより解析した。膜フィルター上で高い蛍光を発するクローンを単離し、さらに解析した。FACSにより単離された10クローンのうち3クローンをさらに解析し、それらがTNBS-BSA複合体への強い結合を示すことが認められた。配列解析により、TNBS特異的クローンのうちの1つは、ファージディスプレイによっても見出されていたことが確認された。しかし、ライブラリーの可溶性ペリプラズム発現およびFACSスクリーニングによって単離された他の2つのクローンは、ファージパニングによって単離されたクローンのいずれにも相当しなかった。
【0169】
実施例13
大腸菌ペリプラズム中に可溶型で発現された抗体によるオリゴヌクレオチドプローブの検出
本実施例は、修飾したオリゴヌクレオチドが細菌の外膜を通して拡散し得ることを示す。5つののA残基間に4つのヌクレアーゼ耐性ホスホロチオエート結合を含む配列5'-ジゴキシゲニン-AAAAA-フルオレセイン-3'(dig-5A-FLと命名、分子量2,384 Da、配列番号:5)を有するオリゴヌクレオチドは、Integrated DNA Technologies、アイオワ州により合成され、精製された(RP HPLC)。このオリゴヌクレオチドのジゴキシゲニン部分は、ジゴキシンに特異的なscFv抗体(抗ジゴキシンscFv)によって認識され得る。プローブ分子が外膜を通して拡散し得るならば、ペリプラズム内に抗ジゴキシンscFvを発現する細胞は5A-Flを結合し得り、次にはこれによって細胞が蛍光性になるはずである。
【0170】
陰性対照としてのアトラジン(Hayhurst 2000)またはジゴキシゲニンに特異的なペリプラズムscFvを発現するABLE(商標)C細胞を、5×強度のPBS中で、100 nMジゴキシゲニン-BODIPY(商標)または100 nM dig-5A-FLのいずれかと共にインキュベートした。生存度染色のため、ヨウ化プロピジウムも添加した。ヨウ化プロピジウム排除(無傷の膜を有する細胞を同定するため)および側方散乱に基づいて、生細胞にゲートをかけた。1秒当たり1,000イベントの速度で、約10,000個の細胞を解析した。得られたデータを図3に示す。プローブに結合しない非関連の抗アトラジン抗体を発現する細胞は、バックグラウンドの蛍光のみを示した。対照的に、抗ジゴキシンscFv抗体を提示する細胞は、ジゴキシゲニン-BODIPY(商標)および5-A-FLの両方で明らかに標識された。後者のプローブは、対照細胞で観察されたシグナルよりも明らかに高いシグナルを生じた。5-A-FLは、より小さく非電荷であるジゴキシゲニン-BODIPYと比較して低い蛍光強度を生じるものの、前者プローブで得られたシグナルはFACSによるscFvライブラリーのスクリーニングに十分であった。
【0171】
実施例14
市販の蛍光基質を用いた、フザリウム・ソラニ(Fusarium solani)のリパーゼクチナーゼを発現する大腸菌の、フローサイトメトリーによる識別
本実施例では、市販の蛍光基質を用いて、ペリプラズム内に関連酵素を提示する大腸菌を特異的に標識し得ることを実証する。驚くべきことに、これらの反応の可溶性蛍光生成物は細胞内に十分に保持され、これにより非酵素発現細菌からの酵素発現大腸菌の識別および選択が可能になる。
【0172】
全遺伝子合成によりフザリウム・ソラニのリパーゼクチナーゼをコードする遺伝子を構築し、プラスミドpBAD18Cm内の強力な誘導性プロモーターpBADの下流に配置した。pBADプロモーターからのタンパク質発現は、FACSによるタンパク質ライブラリーのスクリーニングに有益である(Daughertyら、1999)。クチナーゼ遺伝子をコードする得られたプラスミドを、pKG3-53-1と命名した。pKG3-53-1および対照としてのpBAD18Cmを、DH5αに形質転換した。本実施例では、対照細胞からクチナーゼ発現細胞(DH5α(pKG3-53-1))を識別する能力を、2つの異なる市販の基質:フルオレセインジブチレートまたはLysoSensorグリーンDND-189(LSG)(どちらも、Molecular Probes、オレゴン州による)を用いて判定した。後者は、酵素によるエステル加水分解に起因して起こる細胞内のpH変化を検出する、正に荷電した蛍光プローブである。
【0173】
テリフィックブロス/クロラムフェニコール50:g/ml(TB/Cm)中、37℃で激しく振盪しながら細胞を一晩培養した。TB/Cm(50:g/ml)10 ml中に、一晩培養物100:lから継代培養物を作製した。OD600=0.6になるまで、37℃で激しく振盪しながらこれらの継代培養物を培養した。継代培養物4 mlアリコートを、Beckman Allegra 6R遠心機で3650 rpmで20分間ペレット化した。上清を除去し、0.2%グルコースおよび50:g/mlのクロラムフェニコール(Cm)を含むM9最小培地4 ml中に、ペレットを再懸濁した。最終濃度が0.2%になるように、20%保存液のアラビノースを添加した。この培養物を、25℃で激しく振盪しながら4時間誘導した。続いて、誘導した培養物の2 mlアリコートを、Eppendorf 5415C遠心機で8000 rpmで10分間ペレット化し、新鮮培地で洗浄し、再度8000 rpmで10分間ペレット化した。洗浄したペレットを、吸光度OD600=1.0になるように、グルコースを含まないM9塩培地中に再懸濁した。この保存溶液を1:10希釈し、希釈した細胞懸濁液1 mlを、ジメチルスルホキシド(DMSO)中の0.1 mMフルオレセインジブチレート(FDB)保存溶液0.1 mlと混合した。最終FDP濃度は10:Mであった。37℃で30分間、反応を進行させた。標識した細胞を、Ar 488 nmレーザーを装備したBecton Dickinson FACSortで直ちに解析した。クチナーゼ発現細胞および対照細胞の蛍光分布を図9Aに示す。
【0174】
陽性(酵素発現)細胞と対照細胞との識別に対する第2のプローブの有用性もまた試験した。上記のように、pKG3-53-4プラスミドからクチナーゼを発現する大腸菌および陰性細胞(未改変のpBAD18Cmプラスミドを発現する)を培養して誘導し、洗浄した。ペレットを1%ショ糖で洗浄し、再度ペレット化し、OD600=1.0になるように新鮮な1%ショ糖中に再懸濁した。この細胞の保存溶液を、氷上で保存した。
【0175】
標識用に、LysoSensorグリーンDND-189(LSG、Molecular Probes)保存溶液を、DMSO中で1 mMになるように調製した。1 M 4-ニトロフェニルブチレート保存溶液もまた、DMSO中で調製した。まず細胞保存溶液を希釈し、最終濃度が1μMになるようにLSGを添加し、最終濃度が1μMになるように4-ニトロフェニルブチレートを1:1000希釈することにより、細胞の標識を開始した。細胞による4-ニトロフェニルブチレートの酵素的加水分解を25℃で5分間進行させ、次いで上記のようにBecton Dickinson FACSortで直ちに細胞を解析した。LysoSensorグリーンDND-189プローブで染色したクチナーゼ発現細胞および対照細胞の蛍光分布を、図9Bに示す。
【0176】
実施例15
炭疽菌防御抗原に対する親和性が120倍を上回って改善された抗体を単離するための、固定化ペリプラズム発現の使用
大きなライブラリーのスクリーニングには、遺伝子、それがコードするタンパク質、および所望の機能の間の物理的関連性が必要である。そのような関連性は、機構的研究、生物工学的目的、およびプロテオミクス研究に有益であると判明した様々なインビボ提示技術を用いて確立され得る(Hoess、2001;HayhurstおよびGeorgiou、2001;Wittrup、2000)。
【0177】
APExは、フローサイトメトリー(FC)によるスクリーニングを可能にする別のアプローチである。FCは、ハイスループットと、各ライブラリーメンバーのリアルタイム、定量的、多パラメータ解析を兼ね備える。時間当たり4億個細胞を超える次数の選別速度で市販のFC機械を使用して、微生物形質転換効率の制約内で利用可能な大きさのライブラリーをスクリーニングすることができる。さらに、多パラメータFCにより、リアルタイムでそれぞれのおよびすべてのクローンの機能に関して有益な情報が提供され得り、よってこれはライブラリー構築過程を導き選別条件を最適化するのに役立つ(BoderおよびWittrup、2000;Daughertyら、2000)。
【0178】
細菌および酵母のタンパク質提示はFCと組み合わせて、様々なリガンドに対する高親和性抗体を操作するために用いられている(Daughertyら、1999;Boderら、2000)。しかし、タンパク質を細胞表面に提示するための必要条件から、ライブラリースクリーニングの適用に影響し得る生物学的制約が課される。真核生物における折りたたまれないタンパク質反応またはグラム陰性菌の外膜に対するタンパク質選別のストリンジェンシー等の過程により、実際に表面提示と適合するポリペプチドの多様性が制限される(Sagtら、2002;Sathopoulosら、1996)。さらに、微生物表面は、その巨大分子組成がタンパク質:リガンド認識を妨げ得る化学的に複雑な構造をしている。外膜上のリポ多糖の存在がタンパク質:リガンド認識に対する立体的障壁を示すため、この問題はグラム陰性菌において特に著しく、実際にこれが線毛等の特殊化した付属器の進化に寄与したと思われる(Hultgrenら、1996)。
【0179】
APExは、以前の提示戦略の生物学的制約および抗原の接近限界を克服し、これにより実質的に任意の大きさの抗原に対する抗体の効率的な単離が可能になる。APExでは、タンパク質はN末端またはC末端融合物として大腸菌細胞質膜の外(ペリプラズム)面に係留され、よって細胞表面におけるタンパク質の提示に関連した生物学的制約が除かれる。細菌外膜を化学的/酵素的に透過処理した後、固定化scFv抗体を発現する大腸菌を、少なくとも240 kDaの蛍光抗原で特異的に標識し、FCによって解析することができる。本発明者らは、APExを用いることによって、120倍を超えて親和性が増加した、炭疽菌の防御抗原に対する抗体断片を含む、著しく改善されたリガンド親和性を有する抗体の効率的単離を実証した。
【0180】
A. 固定化ペリプラズム発現およびリガンド結合の検出
スクリーニング用途のためには、理想的な発現系は、異種性ポリペプチドの合成に起因し得る細胞毒性または増殖異常を最小限に抑えるべきである(Daughertyら、2000)。APExを使用することにより、膜貫通タンパク質融合物に関連する複雑な問題が回避される(MirouxおよびWalker、1996;Mingarroら、1997)。細菌リポタンパク質は、膜タンパク質とは異なり、膜固定化のためにSRPまたはYidC経路を必要としないことが知られている(Samuelsonら、2000)。リポタンパク質はSec経路を介して膜を超えて分泌され、ペリプラズムに入ると、シグナル配列のC末端側に存在するシステイン残基にジアシルグリセリド基がチオエーテル結合を介して付着する。その後、シグナルペプチダーゼIIによってシグナルペプチドが切断され、修飾されたシステイン残基においてタンパク質が脂肪アシル化され、最終的に親油性脂肪酸が膜内に挿入し、これによりタンパク質が固定される(Pugsley、1993;Seydelら、1999;Yajushiら、2000)。
【0181】
リーダーペプチドおよび成熟NlpAタンパク質の最初の6アミノ酸をコードする配列(推定上の脂肪アシル化部位および内膜標的部位を含む)を、内膜のペリプラズム面へのscFv抗体の固定化に使用した。NlpAは、内膜に独占的に局在する非必須大腸菌リポタンパク質である(Yuら、1986;Yamaguchiら、1988)。特に注目すべきは、内膜標的のための共通残基と考えられている、脂肪アシル化システイン残基に隣接したアスパラギン酸残基である(Yamaguchiら、1988)。26-10抗ジゴキシン/ジゴキシゲニン(Dig) scFvおよび抗炭疽菌防御抗原(PA)14B7 scFvへのNlpA融合物を構築し、大腸菌においてlacプロモーターから発現させた。IPTGによりNlpA-[scFv]合成を誘導した後、細胞をEDTAおよびリゾチームと共にインキュベートして外膜および細胞壁を破壊した。透過処理した細胞を、蛍光色素BODIPY(商標)(200 nM)に複合化したそれぞれの抗原と混合し、フローサイトメトリーにより細胞の蛍光を測定した。NlpA-[14B7 scFv]およびNlpA-[Dig scFv]を発現する処理細胞は、対照と比較して、それぞれ9倍および16倍高い平均蛍光強度を示した(図7A)。細胞を非関連の蛍光抗原と混合した場合にはバックグラウンドの蛍光のみが検出され、このことから、本研究の条件下ではバックグラウンドの結合が無視できることが示された。
【0182】
細胞質膜上に固定された抗体断片が大きな抗原に結合する能力をさらに評価するため、NlpA-[Dig scFv]が、240 kDA蛍光タンパク質フィコエリトリン(PE)に複合化したジゴキシゲニンを認識する能力について試験した。複合体をNlpA-[Dig scFv]を発現する細胞と混合し、EDTA-リゾチームで処理した。高い細胞蛍光が観察されたことから、膜に固定化された抗体によるジゴキシゲニン-PEの結合が示された(図7B)。全体として、蓄積されたデータから、Tris-EDTA-リゾチームで処理した細胞において、細胞質膜上に固定されたscFvは、低分子から少なくとも最大で分子量240 kDaまでのタンパク質に及ぶリガンドに容易に結合し得ることが実証された。重要なことには、ジゴキシゲニン-PEで標識し、その後フローサイトメトリーを1ラウンド行うことにより、非関連の抗原特異性を有するscFvとの同様の融合物を発現する細胞から、NlpA-[Dig scFv]を発現する細菌が500倍を超えて濃縮された。
【0183】
B. APExによるライブラリースクリーニング
抗PA 14B7 scFvの遺伝子のエラープローンPCRにより1×107個のメンバーのライブラリーを構築し、NlpA膜固定化配列に融合した。無作為に選択した12個のライブラリークローンをDNA配列決定したところ、遺伝子当たり平均して2%のヌクレオチド置換が明らかになった。IPTGによりNlpA-[14B7変異体scFv]合成を融合した後、細胞をTris-EDTA-リゾチームで処理し、洗浄し、200 nM PA-BODIPY(商標)で標識した。内膜の完全性を、ヨウ化プロピジウム(PI)で染色することによりモニタリングした。超ハイスループットCytomation Inc. MoFlo液滴偏向フローサイトメーターを用いて、低いPI蛍光(630 nm放射)および高いBODIPY(商標)蛍光に選択的にゲートをかけて、全部で2×108個の細菌を選別した。最も高い530 nm蛍光(FL1)を有して選別された約5%の細胞を回収し、直ちにPIのみで再度染色し、上記のように再度選別した。この2回目の選別サイクルには抗原を添加しなかったため、解離反応速度の低い抗体を発現する細胞のみが、依然として蛍光性のままである。おそらくは潜在的scFv毒性(Somervilleら、1994;HayhurstおよびHarris、1999)、Tris-EDTA-リゾチーム処理、および高剪断フローサイトメトリー環境への曝露の組み合わせに起因して、この集団のプレーティング効率は低かった。したがって、潜在的高親和性クローンの喪失を回避するため、選別によって回収された約1×104個の蛍光イベントをPCR(商標)増幅することにより、scFvをコードするDNAをレスキューした。PCR(商標)増幅に用いた条件により、標識過程におけるTris-EDTA-リゾチーム処理に起因する、部分的に加水分解された細胞壁を有する細胞から、細胞DNAが量的に放出されたことに留意されたい。30ラウンドのPCR(商標)増幅を行った後、DNAをpAPEx1に連結し、新鮮な大腸菌に形質転換した。選別の2ラウンド目は、この場合には集団のうちの最も蛍光の高い2%のみを回収することを除いては先の通りに行い、次いで直ちに再選別して約5,000蛍光イベントを得た。
【0184】
2ラウンド目によるscFv DNAをPCR(商標)により増幅し、scAb型で可溶型の抗体断片を発現させるためにpMoPac16(Hayhurstら、2003)に連結した。scAb抗体断片は、軽鎖がヒトκ定常領域に融合したscFvである。この抗体断片型は、scFvと比較して優れたペリプラズム溶解性を示す(Maynardら、2002;Hayhurst、2000)。scAb型の20クローンを無作為に拾い、液体培地中で培養した。IPTGで誘導した後、ペリプラズムタンパク質を単離し、表面プラズモン共鳴(SPR)を用いて、相対抗原解離反応速度に関してscAbタンパク質を順位づけした。20クローンのうち11クローンが、14B7親抗体と比較してより低い抗原解離反応速度を示した。抗原解離反応速度が最も低い3つのscAbを大量に産生し、Niクロマトグラフィー、それに続くゲル濾過FPLCによりこれを精製した。興味深いことに、ライブラリーから選択されたクローンはすべて、優れた発現特性を示し、振盪フラスコ培養においてL当たり4 mg〜8 mgの精製タンパク質が得られた。詳細なBIACore解析により、3クローンはすべて親の14B7抗体と比較して、PAに対して実質的に低いKDを示すことが実証された(図8Aおよび図8B)。KDの改善は主に、抗原解離の低下(すなわち、koffの低下)に起因した。最も親和性の高いクローンM18は、(koffが4.2×10-5 M-1 秒-1で)KD 35 pMを示し、これはM18-PA半減期6.6時間に相当する。これは、親抗体14B7(BIACore 3000により決定されたKD=4.3 nM)と比較して、親和性が120倍を超えて改善されたことを表す。同定された変異体を図8Bに示し、1H、M5、M6、およびM18抗体の高次構造を示す略図を図10に示す。M5の変異は以下の通りであった:軽鎖においてQ38R、Q55L、S56P、T74A、Q78L、および重鎖においてK62R。M6に関して変異は以下の通りであった:軽鎖においてS22G、L33S、Q55L、S56P、Q78L、およびL94P、ならびに重鎖においてS7P、K19R、S30N、T68I、およびM80L。M18に関して変異は以下の通りであった:軽鎖においてI21V、L46F、S56P、S76N、Q78L、およびL94P、ならびに重鎖においてS30N、T57S、K64E、およびT68。図11は、可変重鎖および可変軽鎖ならびに形成された変異を示す、14B7 scFv配列(配列番号:21)とM18 scFv配列(配列番号:23)とのアラインメントを示す。これらの配列をコードする核酸を、それぞれ配列番号:20および22に示す。
【0185】
変異体抗体へのNlpA融合物を発現する、Tris-EDTA-リゾチームで透過処理した細胞の蛍光強度は、抗原結合親和性に応じて変化した(図8C)。例えば、バックグラウンド蛍光が約5であるのと比較し、NlpA-[M18 scFv]タンパク質を発現する細胞は平均蛍光強度250を示すのに対して、親の14B7 scFvを発現する細胞は平均蛍光強度30を示した(図8B)。中程度の親和性を有する抗体は、その相対親和性順位と一致して中程度の蛍光強度を示した。35 pM程度の低い解離定数を示す抗体を発現する細胞を分離する能力により、何故3つの独特な非常に高い親和性変種が単離され得たかに関する道理にかなった説明が提供され、フローサイトメトリー解析を用いて得られた優れた分解能が示される。
【0186】
詳細に解析した3つのクローン、M5、M6、およびM18は、それぞれ7個、12個、および11個のアミノ酸置換を含んでいた。ファージディスプレイを用いた以前の研究において(Maynardら、2002)、本発明者らは、1) 変異誘発エラープローンPCR(商標)、2) 5ラウンドのファージパニング、および3) パニング後のクローンのDNAシャッフリングからそれぞれなる3サイクルにより、14B7 scFvの変種を単離した。その研究で単離された最も優れたクローンは、Q55LおよびS56P置換を含み、(BIACore 3000により決定された)KD 150 pMを示す1Hであった。これらの2つの変異は結合ポケットの疎水性を増すようであり、疎水性相互作用の増大が抗体の親和性成熟において優性効果であるという山のような証拠を拡充するものである(Liら、2003)。同じアミノ酸置換は、APExによって単離されたM5およびM6クローンにおいても認められる。しかし、これらの2つのクローンにおけるさらなる変異により、親和性のさらなる増大が付与された。M5、M6、およびM18が単一ラウンドのアセクシャル(asexual)PCR(商標)後に単離され、なおどれも、ファージディスプレイによって実に複数ラウンドのセクシャル(sexual)変異誘発および選択の後に単離され得た最も優れた抗体と比較して、より高い親和性を有していたことは注目に値する。
【0187】
APExによって単離された最も親和性の高いクローンM18は、S56P変異を含んでいたが、1H、M5、およびM6に見出されたQ55L置換を欠いていた。部位特異的変異誘発によりQ55L置換をM18に導入すると、生じたScAbは、kon 1.1×106 M-1 秒-1およびkoff 2.4×10-5 秒-1の抗原結合(KD=21 pM)のさらなる改善を示し、これは複合体半減期11.6時間に相当する。しかし、この変異の導入により、精製タンパク質の収量が5倍を超えて減少し、振盪フラスコ培養で1.2 mg/Lになった。修飾したM18配列を配列番号:25に示し、この配列をコードする核酸を配列番号:24に示す。
【0188】
C. ファージディスプレイされたscFv抗体のAPEx
重要な治療標的および診断標的に対する多くの抗体断片が、ファージディスプレイによりスクリーニングしてレパートリーライブラリーから単離されている。時間を要するサブクローニング段階を必要としない、迅速な抗原結合解析およびそのような抗体の親和性成熟のための手段を開発することが望ましい。抗体は最も一般的には、ファージ遺伝子3マイナーコートタンパク質(g3p)のN末端への融合を介して、繊維状ファージ上に提示される(Barbasら、1991)。ファージの形態形成過程において、g3pはその最C末端を介して内膜に一時的に付着し、その後成長していくビリオンに取り込まれ得る(BoekeおよびModel、1982)。したがって、抗体断片はペリプラズム区画内に固定されかつ提示もされる。よって本発明者らは、APEx形式を用いて、抗体ライブラリーのスクリーニング目的で、g3p融合タンパク質が利用できるかどうかを評価した。上記の高親和性抗PA M18 scFv、抗ジゴキシン/ジゴキシゲニン26-10 scFv、および抗メタンフェタミン scFv(Meth)を、ファージディスプレイの目的で広く用いられ、g3p提示のために遺伝子IIIの短い変種を利用するファージミドpAK200内のlacプロモーターの下流のg3pのN末端にインフレームでクローニングした(Krebberら、1997)。IPTGで誘導した後、scFv-g3p融合物を発現する細胞をTris-EDTA-リゾチームにより透過処理し、それぞれの蛍光抗原で標識した(図9)。3つのscFvいずれに関しても、それぞれの抗原とインキュベートした場合にのみ高い蛍光が得られた。重要なことには、g3pのN末端に融合したscFvの平均蛍光強度が、NlpAアンカーのC末端への融合により得られた平均蛍光強度に匹敵した。図9の結果から、 (i) 大きな可溶性ドメインがN末端で膜アンカーに係留され得ること;(ii) 繊維状ファージ上に提示するためにファージミド内にクローニングした抗体断片が、APEx形式を用いてフローサイトメトリーにより容易に解析され得ること;および(iii) scFv抗体が、N末端またはC末端の融合物として、抗原結合を喪失することなく細胞質膜上に固定され得ることが実証される。
【0189】
D. 考察
本発明者らは、コンビナトリアルライブラリーから高親和性リガンド結合タンパク質、特にscFv抗体の効率的な選択を可能にする方法を開発した。1つの局面において、APExは、内膜の外面へのタンパク質の固定化、次の外膜の破壊、その後の蛍光標識抗原とのインキュベーションおよびFC選別に基づく。この戦略により、以前の細菌ペリプラズムおよび表面提示アプローチを超えるいくつかの恩典が提供される:1) 内膜中にタンパク質を保持するために脂肪アシル化アンカーを利用することにより、提示の成功のために必要とされるのは6アミノ酸程度の短いアミノ酸の融合のみであり、これによって場合によっては大きな融合物が課し得る悪影響が低減される;2) 内膜は、提示された抗体断片への大きな抗原の結合を立体的に妨げ得るLPSまたは他の複雑な糖質等の分子を欠いている;3) 融合物質は、提示される前に膜1枚を横切らなくてはならないだけである;4) N末端およびC末端融合戦略の両方を使用することができる;および5) 一般のファージディスプレイベクターから発現されるタンパク質に対して、APExを直接使用することができる。この最後の点が特に重要であるが、それはこの点によって、サブクローニングする段階を全く必要とせずに、ファージパニング実験によるクローンを量的に解析しまたはフローサイトメトリーによりさらに選別し得るハイブリッドライブラリースクリーニング戦略が可能となるからである。
【0190】
低分子(例えば、ジゴキシゲニンおよびメタンフェタミン <1 kDa)からフィコエリトリン複合体(240 kDa)に及ぶ抗原を検出するために、APExを使用することができる。実際には、より大きなタンパク質を同様に使用し得ることを意図するように、図3Bで使用したフィコエリトリン複合体は、抗原検出の上限を規定することを意図していない。
【0191】
実施例において、蛍光標識抗原に結合するscFvをコードする遺伝子は、PCR(商標)により選別した細胞からレスキューした。このアプローチの恩典は、潜在的scFv毒性、Tris-EDTA-リゾチーム破壊、およびFC剪断力の組み合わせに起因する、もはや生存できないクローンの単離が可能になる点である。このような方法で、単離されたクローンの多様性が最大化される。PCR(商標)レスキューのさらに別の恩典は、サブクローニングする前に、プールした細胞からのDNA増幅を変異誘発条件下で実施し得る点である。したがって、選択の各ラウンド後に、単離した遺伝子にランダム変異を導入することができ、これにより定向進化のさらなるラウンドが簡略化される(HanesおよびPluckthun、1997)。さらに、増幅段階において、プール内のタンパク質コード遺伝子間の鋳型交換に有利に働くPCR(商標)条件を使用することにより、選択されたクローン間の組換えが可能になる。PCR(商標)レスキューは、他のライブラリースクリーニング形式においても同様に有利であると考えられる。
【0192】
いずれのライブラリースクリーニングにも関わる重要な問題は、単離されたクローンを高レベルで発現する能力である。既存の提示形式は、提示されたタンパク質の発現特性に影響し得る大きな固定化配列への融合を含む。したがって、十分に提示されるscFvは、非融合タンパク質としての可溶型での高発現に必ずしも従い得ない(Hayhurstら、2003)。一方、本発明において細胞質膜へのタンパク質のN末端係留に使用し得る短い(6アミノ酸)尾部は、融合物の発現特性に影響しないようである。この仮説と一致して、APExにより単離された炭疽菌PAに対して親和性が増大した3つのクローンはすべて、多くのアミノ酸置換を有するにもかかわらず、優れた可溶性発現特性を示した。同様に、メタンフェタミン抗体の親和性成熟においても十分に発現するクローンが得られたことから、細菌において可溶型で大量に容易に産生され得るクローンの単離は、本発明を用いた選択の固有の特性であることが示唆される。
【0193】
本実施例において、本発明者らは親和性成熟のためにAPExを使用し、21 pM程度に低いKD値を示す、炭疽菌防御抗原に対するscFvを操作した。PAに対して最も高い親和性を示すscFv結合部位をヒト化し、全長IgGに変換し、前臨床試験において、炭疽菌中毒に対するその中和の可能性を評価している。親和性成熟に加えて、ペリプラズムループ内に固定されたscFv抗体が蛍光抗原に結合し得り、蛍光レポーターとして機能する膜タンパク質トポロジーの解析、および亢進した機能を有する酵素変種の選択を含むいくつかの他のタンパク質工学用途に、APExを利用することが可能である。特に、細胞外被によって酵素触媒による産物を保持する部位が提供されるため、酵素ライブラリー選別にAPExを容易に適合化することができ、これにより触媒による代謝回転に直接基づいた選択が可能になる(Olsenら、2000)。本発明者らはまた、膜タンパク質に対するリガンドのスクリーニングへのAPExの利用に関して評価している。要するに、固定化ペリプラズム発現が、コンビナトリアルライブラリースクリーニングおよび他のタンパク質工学用途を促進する可能性を有することが実証された。
【0194】
E. 材料および方法
1. 組換えDNA技法
NdeI部位およびSfiI部位に隣接したリーダーペプチドおよび成熟NlpAタンパク質の最初の6アミノ酸を、プライマー
を用いてXL1-ブルー(Stratagene、カリフォルニア州)の全細胞PCRを行うことにより増幅した。生じたNlpA断片を用いて、NdeIおよびSfiIを介してpMoPac1(Hayhurstら、2003)のpelBリーダー配列を置換し、pAPEx1を作製した。ジゴキシン(Chenら、1999)、炭疽菌防御抗原PA(Maynardら、2002)、およびメタンフェタミンに特異的なscFvを、非適合性SfiI部位を介してpAPEx1内のNlpA断片の下流に挿入した。同じ遺伝子をファージディスプレイベクターpAK200(Krebberら、1997)にクローニングすることにより、scFvの対応するg3p融合物を作製した。
【0195】
2. 培養条件
全体にわたって使用した宿主株は、大腸菌ABLE C(商標)(Stratagene)であった。pAPEx1またはpAK200誘導体で形質転換した大腸菌を、2%グルコースおよび30ug/mlクロラムフェニコールを添加したテリフィックブロス(TB)中に、OD600が0.1になるまで播種した。細胞の培養および誘導は、以前に記載されている通りに行った(Chenら、2001)。誘導した後、細胞の外膜を記載されている通りに透過処理した(NeuおよびHeppel、1965)。手短に説明すると、細胞(OD600=20で約1 mlに相当する)をペレット化し、0.75 Mショ糖、0.1 M Tris-HCl、pH 8.0、100μg/mlニワトリ卵白リゾチームの氷冷溶液350μl中に再懸濁した。氷冷1 mM EDTA 700μlを穏やかに添加し、懸濁液を氷上に10分間置いた。0.5 M MgCl2 50μlを添加し、混合物を氷上にさらに10分間置いた。得られた細胞を穏やかにペレット化し、200 nMプローブを添加したリン酸緩衝食塩水(1×PBS)中に再懸濁し室温で45分間インキュベートしてから、FCにより評価した。
【0196】
3. 蛍光プローブ
ジゴキシゲニン-BODIPYの合成は、以前に記載されている(Daughertyら、1999)。メタンフェタミン-フルオレセイン複合体は、Roche Diagnosticsより分与されたものであった。S. Leppla NIHにより分与された精製PAタンパク質は、製造業者の説明に従って、bodipy FL SE D-2184とのモル比1対7でBODIPY(商標)と複合体化した。複合化されなかったBODIPY(商標)は、透析により除去した。
【0197】
ジゴキシゲニン-フィコエリトリンを合成するため、製造業者の説明に従って、R-フィコエリトリンおよび3-アミノ-3-ジオキシジゴキシゲニンヘミスクシンアミド、スクシニミジルエステル(Molecular Probe)をモル比1対5で複合化した。過剰のPBS中で透析することにより、遊離のジゴキシゲニンを除去した。
【0198】
4. FCによるscFvライブラリーの親和性成熟
標準的な手順によりエラープローンPCRを用いて14B7親scFvからライブラリーを作製し(Fromantら、1995)、pAPEx1発現ベクターにクローニングした。形質転換、誘導、および標識し、次いで内膜の完全性をモニタリングするために細胞をヨウ化プロピジウムで染色した(PI放射 617 nm)。励起に488 nmアルゴンレーザーを用いて、MoFlo(Cytomation)液滴偏向フローサイトメーターで細胞を解析した。530/40バンドパスフィルターを通して検出されるフルオレセイン/Bodipy FL放射スペクトラムの蛍光の改善、および630/40バンドパスフィルターを通して検出されるPI放射における標識の欠除に基づいて、細胞を選択した。
【0199】
1回目の選別後に捕獲された大腸菌を、フローサイトメーターで直ちに再選別した。続いて、選別された細胞懸濁液中のscFv遺伝子をPCR(商標)により増幅した。増幅されたならば、次に変異体scFv遺伝子をpAPEx1ベクターに再クローニングし、細胞に再形質転換し、次いで寒天プレート上で30℃で一晩培養した。得られたクローンを上記のように2ラウンド目の選別および再選別に供し、その後scAbタンパク質を発現させるためにscFv遺伝子をpMoPac16(Hayhurstら、2003)にサブクローニングした。
【0200】
5. 表面プラズモン共鳴解析
以前に記載されている通りに、IMAC/サイズ排除FPLCにより、単量体scAbタンパク質を精製した(Hayhurstら、2003)。BIACore3000機器を用いて、SPRにより親和性測定値を取得した。EDC/NHS化学により、約500RUのPAをCM5チップにカップリングした。同様にBSAもカップリングし、線の減算に使用した。BIA HBS-EP緩衝液中25℃で流速100μl/分にて、反応速度解析を実施した。20 nMから開始した各抗体の2倍希釈物を5種類、3つ組で解析した。
【0201】
本明細書で開示され請求される方法はすべて、本開示の観点から、過度に実験することなく作製および実行することができる。本発明の組成物および方法を好ましい態様に関して記載したが、本発明の概念、精神、および範囲から逸脱することなく、本明細書に記載した方法および方法の段階または方法の段階の順序に変更がなされ得ることは、当業者に明白であろう。さらに具体的には、本明細書に記載した物質の代わりに、化学的および生理学的に関連する特定の物質を代用し得て、同じもしくは類似した結果が得られることは明白であろう。当業者にとって明白であるこのような類似の代用品および変更はすべて、添付の特許請求の範囲によって定義される本発明の精神、範囲、および概念の範囲内であると見なされる。
【0202】
参考文献
以下の参考文献は、例示的な手順の補足または他の詳細な補足を、本明細書に記載したものに提供する範囲内において、参照として本明細書に明確に組み入れられる。
【図面の簡単な説明】
【0203】
添付の図面は本明細書の一部を形成し、本発明の特定の局面をさらに実証するために含めるものである。本明細書に示した特定の態様の詳細な説明と組み合わせてこれらの図面の1つまたは複数を参照することにより、本発明をより良く理解できる。
【図1】図1A〜図1Cは、APExによる抗原標的の選択的同定を示す。APExは、大腸菌において表示されるscFvを発現した。低分子の(A) ジゴキシゲニン-Bodipy FLおよび(B) メタンフェタミン-FLと結合する発現したscFv;または(C) 例えばペプチド18aaといったペプチドに結合する発現したScFvを示す。
【図2】図2Aおよび図2Bは、スフェロプラストの表面上でのScFvの検出を示す。APExは、大腸菌において表示されるscFvを発現した。発現したScFvは、大きな抗原、例えばPA-Cy5(83 kD)、フィコエリトリン-ジゴキシゲニン(240 kD)を結合し得た。APExによって発現したscFvが大きなタンパク質に接近し得るという証拠が提供される。
【図3】図3Aおよび図3Bは、大きな標的抗原を複合化したフルオロフォアについてのScFvの検出を示す。
【図4】Meth-FLプローブに対するメタンフェタミン結合scFvの成熟を示す。
【図5】親の抗メタンフェタミンscFvと比較して高い平均FLシグナルを有する、変異体9と命名したクローンの解析。固定化ペリプラズム発現により発現したscFvを示す。
【図6】高親和性抗体断片をフローサイトメトリーに基づいて単離するための、固定化ペリプラズム発現(APEx)の原理を示す略図である。
【図7】APExにより描出される標的の例。(A) 200 nM Bodipy(商標)複合化蛍光抗原で標識した、PA特異的(14B7)scFvおよびジゴキシゲニン特異的(Dig)scFvを発現するABLEC(商標)の蛍光分布。ヒストグラムは、大腸菌10,000イベントの平均蛍光強度を示す。(B) 200 nMの240 kDaジゴキシゲニン-フィコエリトリン複合体で標識した、14B7 scFvまたはDig scFvを発現する細胞のヒストグラム。
【図8】APExにより選択された抗PA抗体断片の解析を示す。(A) 抗PA scAbとPAとの結合のシグナルプラズモン共鳴(SPR)解析。(B) SPRによって得られた親和性データの表。(C) 大腸菌で発現させ、200 nM PA-Bodipy(商標)複合体で標識した、pAPEx1中の抗PA scFvの、抗メタンフェタミン(Meth)scFv陰性対照と比較してのFCヒストグラム。
【図9】N末端とC末端固定化戦略の比較を示す。(A) 大腸菌においてpAPEx1ベクター中のN末端融合物として発現する抗ジゴキシゲニンDig scfv、抗PA M18 scFv、および抗メタンフェタミンMeth scFvは、200 nMのそれぞれの抗原で特異的に標識される。(B) pAK200ベクター中の同じscFvのC末端融合物も、200 nMのそれぞれの抗原で特異的に標識される。
【図10】1H、M5、M6、およびM18配列の高次構造およびアミノ酸置換を示す、抗体結合ポケットを上から見た図を示す。
【図11】可変重鎖および可変軽鎖ならびに結合親和性を改善するために形成された変異を示す、14B7 scFv配列(配列番号:21)とM18 scFv配列(配列番号:23)とのアラインメントを示す。
【背景技術】
【0001】
発明の背景
本出願は2002年7月15日に提出した米国特許仮出願第60/396,058号の優先権を主張し、また本出願は2000年10月27日に提出した米国特許出願第09/699,023号の一部継続出願でもある。前述の出願の全開示は、参照として本明細書に組み入れられる。政府は、米陸軍ARO MURIプログラムおよび生物センサー開発のテキサス連合(Texas Consortium)に従って、かつ米陸軍研究所との契約番号DADD17-01-D-0001に関連して、本発明において権利を所有すると考えられる。
【技術分野】
【0002】
1. 発明の分野
本発明は、一般にタンパク質工学の分野に関する。より詳細には、リガンド結合ポリペプチドの単離を可能にするための、ポリペプチドのコンビナトリアルライブラリーのスクリーニングの改良法に関する。
【0003】
2. 関連技術の説明
高い親和性および特異性でリガンドと結合するか、または反応物(基質)の所望の生成物への酵素転換を触媒するポリペプチドの単離は、生物工学における重要な過程である。適切なスクリーニング法が利用できるのであれば、変異体の大きなライブラリーから、所望の基質特異性を有するタンパク質および酵素を含むリガンド結合ポリペプチドを単離することが可能である。103〜105個の異なる変異体から構成される小さなタンパク質ライブラリーは、まず各クローンを個別に培養し、次に特異的結合を示すクローンを検出する従来のアッセイ法により、スクリーニングすることができる。例えば、異なるタンパク質変異体を発現する個々のクローンをマイクロタイターウェルプレートで増殖することも、または個々のコロニーを寒天プレート等の半固形培地で培養することもできる。結合を検出するには、細胞を溶解してタンパク質を放出し、溶解物をナイロンフィルターに転写し、次いでこれを放射標識または蛍光標識したリガンドを用いてプロービングする(DeWildtら、2000)。しかし、たとえロボットによる自動化および高密度アレイにおいて結合を検出するためのデジタル画像システムを用いたとしても、数千万ないし数十億個のクローンからなる大きなライブラリーをスクリーニングすることは不可能である。そのような大きさのライブラリーのスクリーニングは、ナノモル以下の範囲の親和性を有する酵素またはタンパク質結合物の新たな単離に必要とされる。
【0004】
非常に大きなタンパク質ライブラリーのスクリーニングは、ウイルスまたは細胞の表面上でのタンパク質の提示に依存する様々な技法によって達成された(Ladnerら、1993)。提示技術の根底にある前提は、生物粒子(すなわち、細胞またはウイルス)の外部表面に固定されるように操作されたタンパク質が、細胞の溶解を必要とせずにリガンドへの結合に直接利用できることである。リガンドに対する親和性を有するタンパク質を提示するウイルスまたは細胞は、磁気分離またはフローサイトメトリーによる、固定化リガンドに対する連続的な吸着/脱離を含む様々な方法によって単離され得る(Ladnerら、1993、米国特許第5,223,409号、Ladnerら、1998、米国特許第5,837,500号、Georgiouら、1997、Shustaら、1999)。
【0005】
タンパク質ライブラリーのスクリーニング用途のために最も広く使用される提示技術は、ファージディスプレイである。ファージディスプレイは、特定のリガンドに結合するタンパク質を発見するため、ならびに結合親和性および特異性を操作するための、十分に確立された強力な技法である(RodiおよびMakowski、1999)。ファージディスプレイでは、関心対象の遺伝子を、表面に露出されるタンパク質、最も一般的にはpIIIをコードするファージ遺伝子にインフレームで融合させる。遺伝子融合物は、2つのドメインが独立して折りたたまれるキメラタンパク質に翻訳される。リガンドに対する結合親和性を有するタンパク質を提示するファージは、固定化リガンドに対する選択的吸着、「パニング」として周知の過程により、容易に濃縮され得る。結合したファージを、通常は酸性溶離によって表面から脱着し、大腸菌細胞の感染を介して増幅する。通常、最大で1010個までの多様性を有する非常に大きなライブラリーからでさえ、特異的ポリペプチドを提示するファージを選択するのに、4〜6ラウンドのパニングおよび増幅で十分である。密に結合するポリペプチドを提示するクローンを迅速に濃縮するための、ファージディスプレイのいくつかの変法が開発されている(DuenasおよびBorrebaeck、1994;Malmborgら、1996;Kjaerら、1998;Burioniら、1998;Levitan、1998;Mutuberriaら、1999;Johnsら、2000)。
【0006】
ファージディスプレイ技術の最も重要な用途の1つは、高親和性抗体の単離である(Dall'AcquaおよびCarter、1998;Hudsonら、1998;Hoogenboomら、1998;MaynardおよびGeorgiou、2000)。scFvまたはFAB断片の非常に大きくかつ構造的に多様なライブラリーが構築されており、多数の合成抗原および天然抗原に対する抗体のインビトロ単離のための使用に成功している(Griffithsら、1994;Vaughanら、1996;Sheetsら、1998;Piniら、1998;de Haardら、1999;Knappikら、2000;SblatteroおよびBradbury、2000)。選択した抗体をCDRまたは全遺伝子CDRの突然変異誘発に供したライブラリーから、改善された親和性または特異性を有する抗体断片を単離することができる(Hawkinsら、1992;Lowら、1996;Thompsonら、1996;ChowdhuryおよびPastan、1999)。最終的に、溶解度が低いことで知られているscFvの発現特性も、変異体ライブラリーのファージディスプレイによって改善された(Dengら、1994;Coiaら、1997)。
【0007】
しかし、いくつかの目覚しい成功にもかかわらず、ファージに提示されるライブラリーのスクリーニングは多くの要素によって複雑になり得る。第1に、ファージディスプレイでは、ファージの会合を可能にし依然として細胞増殖を維持するために必要である抗体断片遺伝子III融合物の低発現レベルが理由で、細菌における適切な発現に対して最小限の選択が課される(Krebberら、1996、1997)。結果として、数ラウンドのパニング後に単離されたクローンは、大腸菌で調製規模で産生することが困難である場合が多い。第2に、ファージの提示したタンパク質がリガンドに結合し得ても、場合によってはそれらの非融合可溶性対応物は結合しない(Griepら、1999)。第3に、高い結合親和性を有するリガンド結合タンパク質およびより特異的な抗体の単離は、ファージの会合を完了するために遺伝子IIIタンパク質がビリオン当たり約5コピーで存在する必要性による結合活性効果によって複雑になり得る。主に一価のタンパク質提示を生じる系でさえ、ほぼ常にタンパク質の複数コピーを含むクローンの小さな画分が存在する。そのようなクローンは固定化表面により密接に結合し、一価のファージと比較してより高い親和性で濃縮される(Dengら、1995;MacKenzieら、1996、1998)。第4に、理論的な分析は別として(Levitan、1998)、パニングは、実験条件の影響、例えば弱くまたは非特異的に結合したファージを除去するための洗浄段階のストリンジェンシーが、実験の最終結果に基づいて試行錯誤によってのみ決定され得る点で、今だに「ブラックボックス」過程である。最後に、たとえpIIIおよびより少ない程度でファージコートの他のタンパク質が、異種性ポリペプチドの融合に対して一般に耐性があるとしても、ファージ生合成過程に組み込まれる必要性により、ライブラリーの多様性を制限し得る生物学的制約が課される。したがって、当技術分野において、これらの制限を克服し得る技法の大きな必要性が存在する。
【0008】
タンパク質ライブラリーはまた、細菌細胞、菌類細胞、または高等生物の細胞の表面上にも提示されている。細胞が提示するライブラリーは、典型的にフローサイトメトリーによってスクリーニングされる(Georgiouら、1997、Daughertyら、2000)。しかし、ファージディスプレイと同様に、タンパク質を細胞表面上で発現するように操作する必要がある。これにより、いくつかの潜在的な制限が課される。例えば、細胞表面上にタンパク質を提示する必要性により、スクリーニングされ得るタンパク質およびタンパク質変異体の多様性を制限する生物学的制約が課される。また、いくつかのポリペプチド鎖からなる複合タンパク質は、細菌、繊維状ファージ、または酵母の表面上に容易に提示され得ない。このように、当技術分野において、上記の制限をすべて回避し、非常に大きなポリペプチドライブラリーをスクリーニングするための完全に新規な手段を提供する技術の大きな必要性が存在する。
【発明の開示】
【0009】
発明の概要
1つの局面において、本発明は、以下の段階を含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を含む細菌を取得する方法を提供する:(a) 内膜および外膜およびペリプラズムを含むグラム陰性菌であって、細菌のペリプラズム内に候補結合ポリペプチドをコードする核酸配列を発現するグラム陰性菌を提供する段階;(b) 細菌をペリプラズム内に拡散し得る標識リガンドと接触させる段階;および(c) 候補結合ポリペプチドに結合した標識リガンドの存在に基づき、細菌を選択する段階。本発明の1つの態様において、本方法は、(a) 候補結合ポリペプチドと細菌の内膜の外側に固定されるポリペプチドとを含む融合タンパク質をコードする核酸配列を発現するグラム陰性菌を提供する段階; (b) 細菌を細菌内に拡散し得る標識リガンドと接触させる段階;および(c) 候補結合ポリペプチドに結合した標識リガンドの存在に基づき、細菌を選択する段階を含む。
【0010】
本発明の特定の態様において、本方法は、(d) 細菌から候補結合ポリペプチドをコードする核酸配列をクローニングする段階をさらに含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を取得する方法としてさらに定義され得る。本方法において、核酸配列は、融合ポリペプチドの発現を内膜の外側に方向づけ得るリーダー配列に機能的に結合しているとしてさらに定義され得る。
【0011】
本発明の1つの態様では、グラム陰性菌は大腸菌である。本発明の特定のさらなる態様では、本方法は、グラム陰性菌の集団の使用を含むとしてさらに定義され得る。そのような集団は、複数の候補結合ポリペプチドを含む複数の融合ポリペプチドをまとめて発現し得る。集団は、以下の段階を含む方法によって取得し得る:(a) 候補結合ポリペプチドおよび内膜アンカーポリペプチドを含む複数の融合ポリペプチドをコードする複数の核酸配列を調製する段階;および(b) このDNA挿入物でグラム陰性菌の集団を形質転換する段階。本方法において、グラム陰性菌の集団を標識リガンドと接触させてもよい。
【0012】
本発明の1つの態様では、候補結合ポリペプチドは抗体またはその断片としてさらに定義され、または候補結合ポリペプチドは抗体以外の結合タンパク質であってもよい。候補結合ポリペプチドはまた、酵素としてさらに定義され得る。標識リガンドには、ペプチド、ポリペプチド、酵素、核酸、および/または合成分子が含まれ得る。標識リガンドは、蛍光標識を含む任意の適切な手段によって標識され得る。本発明の特定の態様において、候補結合ポリペプチドをコードする核酸が既知の核酸配列に隣接しているとしてさらに定義される場合、それによって選択後に核酸が増幅され得る。
【0013】
本発明の特定の態様において、結合ポリペプチドをコードする核酸配列を含む細菌を取得する方法は、標識リガンドに対する細菌の外膜の透過性を増大させるために細菌を処理する段階を含む。本発明の1つの態様において、処理段階には、高浸透圧条件による細菌の処理、物理的ストレスによる細菌の処理、および/またはファージによる細菌の処理が含まれ得る。本方法は、細菌の外膜を除去する段階、または別法として様々な分子量のポリペプチドの拡散を可能にする欠陥外膜を有する突然変異細菌を使用する段階を含んでもよい。本方法はまた、約25℃を含む準生理的温度で細菌を培養する段階を含んでよい。本方法は、候補結合ポリペプチドに結合していない標識リガンドを除去する段階をさらに含んでよい。
【0014】
本発明による選択は、任意の適切な方法を含み得る。本発明の1つの態様では、選択はフローサイトメトリー(例えば、蛍光活性化細胞分類法(FACS))を含む。別の態様では、選択は磁気選別を含む。リガンドと候補結合ポリペプチドは、可逆的に結合し得る。ポリペプチドは、天然大腸菌リポタンパク質NlpAに由来する6残基配列へのN末端融合、任意の膜貫通タンパク質またはその断片、および繊維状ファージの遺伝子IIIタンパク質またはその断片を含む、任意の適切なアンカーにより内膜の外側に固定され得る。
【0015】
さらに別の局面において、本発明は、表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合する、単離された抗体またはその断片を提供する。そのような抗体またはその断片は、約96 pMから約21 pMの間および/または約35 pMから約21 pMの間の結合親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義され得る。単離された抗体またはその断片は、IgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義され得る。抗体はヒト化抗体であってよく、ヒト抗体であってもよい。特定の態様において、単離された抗体またはその断片は、少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む。
【0016】
さらに別の局面において、本発明は、炭疽菌防御抗原に免疫学的に結合し、かつ、I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、L94P、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lからなる群より選択される改変(I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、およびL94Pは可変軽鎖内に存在し、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lは可変重鎖内に存在する)を含む可変軽鎖および可変重鎖を除く、配列番号:21の可変軽鎖および可変重鎖を含む、単離された抗体またはその断片を提供する。本発明の特定の態様では、単離された抗体またはその断片は、上記改変の可能なすべての組み合わせを含む、約3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18個、またはすべての改変を含む、約2個からすべてのそのような改変を含むと定義され得る。
【0017】
本発明の特定の局面では、単離された抗体またはその断片は、表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される。本発明のさらなる態様において、抗体またはその断片はQ55LおよびS56Pを含む。単離された抗体またはその断片は、配列番号:22または24の可変軽鎖および/または可変重鎖を含み得る。1つの態様において、単離された抗体またはその断片は、配列番号:22または24を含む。単離された抗体またはその断片は、scAb、Fab、またはSFvとしてさらに定義され得り、またIgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義され得る。単離された抗体またはその断片はヒト化抗体であってよく、ヒト型であってもよい。特定の態様において、単離された抗体またはその断片は、少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む。
【0018】
さらに別の局面において、本発明は、本発明が提供する抗体またはその断片をコードする単離された核酸を提供する。1つの態様において、核酸は、配列番号:23および/または25の可変軽鎖をコードする。別の態様において、核酸は、配列番号:23および/または25の可変重鎖をコードする。さらに別の態様において、核酸は配列番号:23のポリペプチドをコードし、別の態様においては、配列番号:25のポリペプチドをコードする。
【0019】
例示的態様の説明
本発明は、抗体または抗体断片を含む、特定の分子標的を認識する結合ポリペプチドを単離する新規方法を提供することにより、先行技術の制限を克服する。この技法では、ポリペプチド(例えば、抗体または他の結合ポリペプチド)変異体のライブラリーを作製し、グラム陰性菌で発現させることができる。変異体ポリペプチドは、ペリプラズムに面した細菌の内(細胞質)膜上に固定された融合タンパク質として発現させることができる。融合タンパク質とは、連続したポリペプチドを形成するために連結された2つまたはそれ以上の開始ポリペプチドからなるポリペプチドである。連結されたポリペプチドは、典型的に異なる供給源に由来する。次に、様々な化学的、物理的、もしくは他の処理により、または透過性の増加をもたらす変異体を使用することにより、細菌のペリプラズム(外)膜を透過性にする。細菌外膜の透過処理により、膜に固定されたポリペプチドが外部溶液に添加した標的高分子に接近できるようになる。
【0020】
微生物骨格上への異種性タンパク質の提示は、ワクチン開発、バイオレメディエーション、およびタンパク質工学を含む多くの異なる分野において魅力的な用途を有する。グラム陰性菌では、N末端またはC末端キメラ融合によりタンパク質が細胞表面に提示されるように設計されたディスプレイシステムが存在する。外膜タンパク質、リポタンパク質、表面構造タンパク質、およびリーダーペプチドへの融合を含む、タンパク質の局在を方向づけるために用いられる多くの異なる戦略が存在するが、多くが同じ制限を共有する。1つの制限は、提示できるタンパク質の大きさである。多くの提示骨格は数百アミノ酸を許容するにとどまり、このことが提示できるタンパク質の範囲を著しく限定する。また、提示とは関心対象のタンパク質がその環境と相互作用し得るように位置づけられることを意味するが、これらの系の多くの主要な制限は、グラム陰性菌の外表面の構造、特にリポ多糖(LPS)分子の存在が、外部から添加したリガンドの結合を阻害する立体的制限を有する点である。別の制限は、提示されたタンパク質が外膜の外部表面に局在化されるという必要条件に起因する。このため、ポリペプチドはまず細胞質膜を越えて分泌されなければならず、次にペリプラズム空間を横断しなければならず、最後に外膜中に適切に構築されなければならない。結合ポリペプチドは、所与のリガンドに結合し得る、少なくとも2つのアミノ酸残基の任意の種類の分子であってよい。結合によって、免疫学的相互作用が起こることを意味する。生合成の制限により、この様式で提示され得るタンパク質の種類が制限される。例えば、大きなポリペプチド(例えばアルカリホスファターゼ)は、大腸菌表面上に提示することができない(Stathopoulosら、1999)。
【0021】
本発明に従って、タンパク質を内膜の外表面に固定して提示させることにより、先行技術の制限を克服することができる。外膜を透過性にする条件を利用することにより、内膜に固定されるscFv抗体(約30 kDaの大きさ)を発現する大腸菌を、例えばフルオロフォアに複合化した標的抗原で標識することができ、続いてこの大腸菌を用いて、機能獲得変異体を単離するために、フローサイトメトリーを利用してタンパク質ライブラリーを選別することができることが、この技法を用いて実証された。
【0022】
当業者に周知であり、例えばTris-EDTA-リゾチームの使用を含む細菌外膜の破壊の後では、少なくとも240 kDaまでの大きさの標識抗原を検出することができる。蛍光標識を用いると、細胞をフローサイトメトリーにより単離することができ、単離されたクローンのDNAをPCRによりレスキューすることができる。本発明者らは、2ラウンドのAPExにより、炭疽菌防御抗原(PA)に対する中和抗体の親和性が120倍を超えて改善され、最終KD=35 pMを示したことを実証する。
【0023】
本発明の1つの態様では、標的分子を蛍光色素で標識する。したがって、標的分子を認識するポリペプチドを発現する細菌クローンは蛍光標識した標的に結合し、次には蛍光を発するようになる。続いて、所望の結合タンパク質を発現する蛍光細菌を、フローサイトメトリー等の自動化技法を用いて集団から濃縮することができる。
【0024】
ポリペプチドライブラリーを、内膜アンカーポリペプチドへの融合を介して、大腸菌または他のグラム陰性菌の内膜のペリプラズム面に結合することができる。使用できる1つのアンカーには、大腸菌のNlpA(新規リポタンパク質A)遺伝子の最初の6アミノ酸が含まれる。しかし、他の単一膜貫通または多指向性(polytropic)膜タンパク質またはペプチド配列を、固定する目的で使用することも可能である。
【0025】
本技法の1つの恩典は、候補結合ポリペプチドを内膜のペリプラズム面に固定することにより、通常ポリペプチドの標識標的分子への到達性を制限する細菌外膜の透過処理が可能になる点である。結合ポリペプチドを膜のペリプラズム面に固定することにより、外膜が損なわれた場合に細胞からの結合ポリペプチドの遊離が妨げられる。したがって、コンビナトリアルライブラリーから抗体および他の結合タンパク質を含む大きな結合ポリペプチドおよびリガンドを単離するために、本技法を用いることができる。本技法は、高いシグナル対ノイズ比を提供するばかりでなく、非常に大きな抗原分子に対するポリペプチドまたは抗体結合物の単離を可能にする。本方法によって、より大きな標的の選択が可能になるため、例えばメラノーマまたは他の特定の種類の腫瘍細胞等の腫瘍細胞を含む細胞上に発現する特定の抗原マーカー等の標的の選択において使用できる可能性が存在する。腫瘍特異的抗体は、癌の治療において大きな展望を示している。
【0026】
ペリプラズムは、グラム陰性菌の内膜および外膜によって規定される空間である。野生型大腸菌および他のグラム陰性菌では、外膜は、600 Daよりも大きい分子のペリプラズム空間への拡散を厳密に制限する透過性障壁として役立つ(DecadおよびNikado、1976)。より大きい分子のペリプラズム内への拡散を可能にする外膜の透過性を増大させる条件は、ライブラリーをスクリーニングする能力の点から見て2つの悪影響を有する:(a) 細胞生存度がかなりの程度で影響を受ける、および(b) 細胞内への分子の拡散は、タンパク質および他の高分子の拡散を伴う。
【0027】
本発明者らは、候補結合ポリペプチドを内膜の外(ペリプラズム)側に固定することにより、または可溶型の候補結合ポリペプチドをペリプラズム中に発現させることにより、リガンドおよびポリペプチドの蛍光複合体が外膜を通過し、候補結合タンパク質に結合し、内膜に結合してとどまることを可能にする技法を同定した。したがって、リガンドに対する親和性を有する組換えポリペプチドを発現する細菌細胞において、結合タンパク質に結合している標識リガンドを検出することができ、それによって残りのライブラリーからその細菌を単離することができる。標的リガンドの蛍光標識を用いる場合には、フローサイトメトリー(蛍光活性化細胞分類法(FACS))により細胞を効率よく単離することができる。このアプローチを用いると、細菌において融合タンパク質を発現する既存のライブラリーを、ファージディスプレイシステムまたは細胞表面ディスプレイシステムにサブクローニングする必要なく、リガンド結合に関して容易に試験することができる。
【0028】
ペリプラズム発現はまた、本発明に従って可溶型での発現によっても実施することが可能である。本発明に従って使用可能である、ペリプラズムでの可溶性発現、および候補結合タンパク質のスクリーニングに関する技法は、2000年10月27日に提出した米国特許出願第09/699,023号に詳細に記載されており、この開示全体は本明細書に参照として明確に組み入れられる。
【0029】
I. 固定化ペリプラズム発現
ファージディスプレイおよび細菌細胞表面ディスプレイという先行技術の方法は、定義により、標的への限定されない接近を可能にするために、タンパク質が用いるビヒクルの外表面上に物理的に提示される必要があるという制限に遭う(ファージへの固定化、またはフローサイトメトリーのため蛍光複合化リガンド)(米国特許第5,223,409号、その開示はその全体が参照として本明細書に明確に組み入れられる)。しかし、ある種のタンパク質はファージ上に十分に提示されないことが知られており(Maenakaら、1996;Coreyら、1993)、細胞表面提示の毒性効果が詳細に論じられている(Daughertyら、1999)。さらに、内膜上への結合を妨げるリポ多糖が存在しない。
【0030】
本発明者らは本明細書において、まだ依然として比較的大きなリガンドに接近しやすい融合タンパク質として、結合タンパク質を内膜のペリプラズム面上に発現できる技法について説明した。本明細書で用いる「結合ポリペプチド」という用語には、抗体だけでなく、抗体の断片、および所与の標的分子に潜在的に結合し得るタンパク質を含む任意の他のペプチドも含まれる。抗体または他の結合ペプチドは、本発明により、内膜のペリプラズム面へのアンカーとして機能し得るポリペプチドとの融合ポリペプチドとして発現され得る。そのような技法を、「固定化ペリプラズム発現」または「APEx」と命名できる。
【0031】
ペリプラズム区画は、グラム陰性細胞の内膜と外膜との間に含まれる(例えば、Oliver、1996を参照のこと)。ペリプラズム区画は、細胞区画として、細胞の増殖および分裂に伴って大きさ、形状、および内容物が変動する場合がある。ペプチドグリカンの枠組み内で、ヘテロポリマーはペリプラズムタンパク質およびわずかな水の高密度環境であり、この区画にゲル様の粘稠度をもたらす(Hobotら、1984;van WielinkおよびDuine、1990)。ペプチドグリカンは、外膜への近接性に依存して様々な程度に重合され、細胞形状および浸透圧溶解に対する耐性を与えるムレイン袋状構造を形成する。
【0032】
外膜(Nikaido、1996を参照のこと)は、リン脂質、ポーリンタンパク質、
および、培地内へ伸長するリポ多糖(LPS)から構成される。外膜の完全性の分子基盤は、2価カチオン(Mg2+およびCa2+)を結合し、互いを静電気的に連結して表面上に高度に規則正しい準結晶配向した「瓦屋根」を形成するLPSの能力により備わっている(Labischinskiら、1985)。膜は、ポーリンを介して、約650 Da以下の分子の通過を可能にする非常に厳密な透過性障壁を形成する(Burmanら、1972;DecadおよびNikaido、1976)。大量の水で満たされたポーリンチャネルは、単糖および二糖、イオン、ならびにアミノ酸のペリプラズム区画への自由な通過の許容を主に担う(Naeke、1976;NikaidoおよびNakae、1979;NikaidoおよびVaara、1985)。分子のペリプラズムへの接近の、そのような厳密な生理的制御により、使用するリガンドが650 Da以下の排除限界でなくとも、または正常に浸透性である化合物の類似体でなくとも、APExが作用するはずであることは、一見して不可解に見えるかもしれない。しかし本発明者らは、2000 Daよりも大きいリガンドがペリプラズム膜を破壊することなくペリプラズム内に拡散し得ることを示した。このような拡散は、本明細書において以下に記載するように、外膜をより透過性にする、細菌細胞の1つまたは複数の処理によって支援され得る。
【0033】
II. アンカーなしのディスプレイライブラリーのスクリーニング
ファージディスプレイおよび細菌細胞表面ディスプレイという先行技術の方法は、定義により、標的への限定されない接近を可能にするために、タンパク質が用いるビヒクルの表面上に物理的に提示される必要があるという制限に遭う(ファージへの固定化、またはフローサイトメトリーのため蛍光複合化リガンド)(米国特許第5,223,409号、その開示はその全体が参照として本明細書に明確に組み入れられる)。ある種のタンパク質はファージ上に十分に提示されないことが知られており(Maenakaら、1996;Coreyら、1993)、細胞表面提示の毒性効果が詳細に論じられている(Daughertyら、1999)。提示すべきタンパク質はまた融合タンパク質として発現される必要があり、融合タンパク質はその機能を変化させる可能性がある。したがって、任意のディスプレイシステムによって課される選択の制約により、その適用が比較的小さくかつ「単純な」タンパク質に限定され、多くの大きくかつ複雑な多サブユニット種の接近が拒絶される可能性がある。後者は、宿主細胞の生存度に非常に重大な影響を及ぼすことなく、ファージ会合の終結または外膜の移行の複雑な過程に効率よく参加することができない可能性が非常に高い。
【0034】
本明細書において、発現した結合タンパク質、例えば抗体が、大腸菌のペリプラズム区画に標的され得り、なお結合リガンドおよびペプチドを受け入れられる条件を記載する。本明細書で用いる「結合タンパク質」という用語には、抗体だけでなく、抗体の断片、および所与の標的分子に潜在的に結合し得る任意の他のポリペプチドまたはタンパク質も含まれる。固定されるのと同様に、抗体または他の結合タンパク質は、本発明により、融合タンパク質としてではなく直接発現させることができる。このような技法を「アンカーなしの提示」(ALD)と命名できる。ALDがどのように機能し得るかを理解するため、それが機能する位置を認識する必要がある。
【0035】
ペリプラズム区画は、グラム陰性細胞の内膜と外膜との間に含まれる(例えば、Oliver、1996を参照のこと)。ペリプラズム区画は、細胞区画として、細胞の増殖および分裂に伴って大きさ、形状、および内容物が変動する場合がある。ペプチドグリカンの枠組み内で、ヘテロポリマーはペリプラズムタンパク質およびわずかな水の高密度環境であり、この区画にゲル様の粘稠度をもたらす(Hobotら、1984;van WielinkおよびDuine、1990)。ペプチドグリカンは、外膜への近接性に依存して様々な程度に重合され、細胞形状および浸透圧溶解に対する耐性を与えるムレイン袋状構造を形成する。
【0036】
外膜(Nikaido、1996を参照のこと)は、リン脂質、ポーリンタンパク質、
および、培地内へ伸長するリポ多糖(LPS)から構成される。外膜の完全性の分子基盤は、2価カチオン(Mg2+およびCa2+)を結合し、互いを静電気的に連結して表面上に高度に規則正しい準結晶配向した「瓦屋根」を形成するLPSの能力により備わっている(Labischinskiら、1985)。膜は、ポーリンを介して、約650 Da以下の分子の通過を可能にする非常に厳密な透過性障壁を形成する(Burmanら、1972;DecadおよびNikaido、1976)。大量の水で満たされたポーリンチャネルは、単糖および二糖、イオン、ならびにアミノ酸のペリプラズム区画への自由な通過の許容を主に担う(Naeke、1976;NikaidoおよびNakae、1979;NikaidoおよびVaara、1985)。分子のペリプラズムへの接近の、そのような厳密な生理的制御により、使用するリガンドが650 Da以下の排除限界でなくとも、または正常に浸透性である化合物の類似体でなくとも、ALDが作用するはずであることは、一見して不可解に見えるかもしれない。しかし本発明者らは、ALDに関して、リガンドがペリプラズム内に拡散し得ることを示した。このような拡散は、本明細書において以下に記載するように、外膜をより透過性にする、細菌細胞の1つまたは複数の処理によって支援され得る。
【0037】
III. 外膜の透過処理
本発明の1つの態様では、1つまたは複数の標識リガンドに対する外膜の透過性を増大させる方法を使用する。これにより、通常なら外膜を通過できない標識リガンドにスクリーニングを利用することが可能になる。しかし、ある種類の分子、例えば650 Da排除限界よりも大きい疎水性抗生物質は、膜ポーリンとは無関係にそれ自体で細菌外膜を通して拡散し得る(Farmerら、1999)。その過程は、その際に膜を実際に透過処理し得る(JouenneおよびJunter、1990)。このような機構は、ポリミキシンBノナペプチドを用いてインビボで細胞質膜タンパク質のペリプラズムループを選択的に標識するために採用された(Wadaら、1999)。また、ある種の長鎖リン酸ポリマー(100 Pi)も、外膜の正常な分子ふるい活性を完全に回避するようである(RaoおよびTorriani、1988)。
【0038】
生存度を喪失することなくまたは発現したタンパク質が細胞から放出されることなく、リガンドのペリプラズム内への透過をもたらす条件を同定したが、本発明は外膜の維持をせずに行うことが可能である。融合ポリペプチドを用いて候補結合ポリペプチドを内(細胞質)膜の外側に固定することにより、結合した標識リガンドを検出するために(細胞からの結合タンパク質の漏出を妨げる障壁としての)外膜を維持する必要がなくなる。結果として、細胞質膜の外(ペリプラズム)面に固定される結合タンパク質を発現する細胞は、ある程度透過処理した膜を有するまたはほぼ完全に外膜を除去した細胞において、単に蛍光標識リガンドの溶液と共にインキュベートすることにより、蛍光標識され得る。
【0039】
異なる細菌宿主株の外膜の透過性は、大きく異なり得る。OmpFの過剰発現に起因する透過性の増大が、OmpFの翻訳に関する負の調節性mRNAの量の減少をもたらすヒストン様タンパク質の欠除によって起こることが以前に示されている(Painbeniら、1997)。また、DMA複製および染色体分離も、レプリソームと内膜との密接した接触に依存することが周知であり、内膜自体は多くの箇所で外膜と接触している。ライブラリースクリーニング用途に関して好ましい宿主は、プラスミドコピー数を減少させる変異をさらに有する大腸菌ABLEC株である。
【0040】
本発明者らはまた、高浸透圧ショック等の処理により標識が有意に改善され得ることを認めた。カルシウムイオン(Bukauら、1985)、およびさらにTris緩衝液(Irvinら、1981)を含む多く薬剤により、外膜の透過性が変化することは周知である。さらに、本発明者らは、ファージ感染により標識過程が促進されることを見出した。繊維状ファージの内膜タンパク質pIIIおよび大きな多量体外膜タンパク質pIVによって膜透過性は変化し得り(Boekeら、1982)、pIVの変異体では通常なら排除されるマルトデキストリンの接近が改善されたことが知られている(Marcianoら、1999)。株、塩、およびファージの賢明な組み合わせを含む本発明の技法を用いて、高度な透過性が達成され得た(Daughertyら、1999)。次に、フローサイトメトリーまたは他の関連技法を用いて、蛍光標識リガンドに結合している固定化結合ポリペプチドを含む細胞を、標識リガンドに対して親和性のない結合タンパク質を発現する細胞から容易に単離することができる。しかし、細胞の生存度を維持するために、破壊的な技法の使用を控えることが典型的に望ましいと考えられる。
【0041】
IV. 固定化ペリプラズム発現
本発明の1つの態様では、内膜の外面上に融合ポリペプチドを発現する細菌細胞を提供する。そのような融合ポリペプチドは、候補結合ポリペプチドと内膜の外面へのアンカーとして機能するポリペプチドとの間の融合を含み得る。本発明の範囲から逸脱することなく、さらなるポリペプチド配列を融合ポリペプチドに付加することができることは、当業者に理解されよう。そのようなポリペプチドの一例は、アンカーポリペプチドと候補結合ポリペプチドを連結する働きをするリンカーポリペプチドである。本発明の背景にある一般的図式は、候補結合ポリペプチドの不均一な集合の有利な発現を含む。
【0042】
内膜への固定は、内膜リポタンパク質のリーダーペプチドおよび最初の6アミノ酸を用いて達成され得る。内膜リポタンパク質の一例はNlpA(新規リポタンパク質A)である。NlpAの最初の6アミノ酸を、内膜に発現させるべきタンパク質のN末端アンカーとして使用することができる。NlpAは、内膜に独占的に局在する非必須リポタンパク質として、大腸菌において同定され特徴づけされた(Yu、1986;Yamaguchi、1988)。
【0043】
すべての原核生物リポタンパク質と同様に、NlpAは、それをSec経路を介する内膜を越えた移行に標的するリーダー配列と共に合成される。前駆タンパク質が内膜の外側に現れたならば、成熟リポタンパク質のシステイン残基がジアシルグリセリドとチオエーテル結合を形成する。その後シグナルペプチドがシグナルペプチダーゼIIによって切断され、システイン残基はアミノアシル化される(Pugsley、1993)。N末端のシステインが脂質で修飾された生じたタンパク質は、続いて内膜または外膜に局在し得る。この局在化は、成熟リポタンパク質の第2アミノ酸によって決まることが実証されている(Yamaguchi、1988)。この位置がアスパラギン酸であると、タンパク質はN末端脂質成分を介して内膜に固定されたままになるのに対し、2番目の位置に任意の他のアミノ酸がくると、一般にリポタンパク質は外膜に配向される(GennityおよびInouye、1992)。これは、タンパク質LolA、LolB、およびATP依存性ABCトランスポーター複合体LolCDEによって達成される(Yakushi、2000、Masuda、2002)。NlpAは第2アミノ酸残基としてアスパラギン酸を有し、よって内膜内に固定されてとどまる。
【0044】
リポタンパク質の2番目のアミノ酸をアルギニン(R)に変換することにより、リポタンパク質が内膜に存在するよう標的されるであろうことが報告されている(Yakushi、1997)。したがって、大腸菌(および場合によっては他のグラム陰性菌)のすべてのリポタンパク質が、アンカー配列となり得る。必要なのは、シグナル配列およびアミノ酸2位のアルギニンのみである。その後にsec切断領域が続く人工的secシグナル配列を用いて、および成熟タンパク質のアミノ酸1としてシステインをアミノ酸2としてアルギニンをコードすることにより、この構築物を人工的に設計することが可能である。大きな融合構築物を必要とするものの、膜貫通タンパク質もまた、場合によってはアンカー配列として使用できる。
【0045】
本発明で使用し得るアンカーの例には、リポタンパク質、CDNSSS成熟リポタンパク質アンカーを有する肺炎桿菌のプルラナーゼ、ファージにコードされるcelB、および大腸菌acrE(envC)が含まれる。タンパク質アンカーとして使用し得る内膜タンパク質の例には、
が含まれる。さらに、任意の細胞質タンパク質の単一の膜貫通ループを膜アンカーとして用いることも可能である。
【0046】
ファージディスプレイとの関連において、融合タンパク質の多様な集団の調製は周知である(例えば、米国特許第5,571,698号を参照のこと)。例えば繊維状ファージM13の遺伝子IIIコートタンパク質のアミノ末端ドメイン、または別の表面関連分子の代わりに、細胞質膜のペリプラズム面へのアンカーに関心対象のタンパク質を連結することにより、本発明において同様の技法を使用することができる。本発明に従って、そのような融合物を変異させ、細胞質膜のペリプラズム面上に低量で発現する、構造的に関連した融合タンパク質のライブラリーを形成することができる。そのように、ファージディスプレイとの関連において当業者に周知である候補分子の不均一な集合を作製する技法を、本発明で使用するために適合化することができる。当業者は、そのような適合化には、プロモーター、エンハンサー、またはリーダー配列を含む、内膜のペリプラズム面に固定される融合タンパク質を発現させるための細菌エレメントの使用が含まれることを認識するであろう。本発明は、ファージの使用または細胞表面上への分子の発現を必要としない、十分に発現されない場合もあり、または宿主細胞にとって有害である可能性のあるファージディスプレイに対する恩典を提供する。
【0047】
多様な候補結合タンパク質および/または抗体を作製するために本発明に関連して使用できる技法の例には、米国特許第5,824,520号に記載されている免疫グロブリン重鎖ライブラリーを発現させるための技法が含まれる。この技法では、高度に多様な合成超可変領域を作製することにより、一本鎖抗体ライブラリーを作製する。抗体提示のための同様の技法は、米国特許第5,922,545号によっても示される。次に、固定化融合ポリペプチドを発現させるため、これらの配列を、グラム陰性菌の内膜のペリプラズム面に対するアンカー配列をコードする核酸に融合することができる。
【0048】
融合タンパク質を作製する方法は、当業者には周知である(例えば、米国特許第5,780,279号を参照のこと)。そうするための1つの手段には、候補結合ポリペプチドとアンカー配列との間に遺伝子融合を構築する段階、および1つまたは複数のコドンにおいて結合タンパク質をコードする核酸を変異させ、それにより変異体のファミリーを作製する段階が含まれる。次に、変異させた融合タンパク質を、細菌の大きな集団において発現させることができる。その後、標的リガンドが結合した細菌を単離することができ、結合タンパク質をコードする対応する核酸をクローニングすることができる。
【0049】
V. 候補分子のスクリーニング
本発明は、標的リガンドを結合し得る分子を同定する方法を提供する。スクリーニングする結合ポリペプチドには、多様な候補物質の大きなライブラリーが含まれ得り、または別法として標的リガンドを結合する可能性が高いと考えられる構造特性を目的として選択される特定の種類の化合物が含まれ得る。本発明の1つの態様において、候補結合ポリペプチドは、抗体またはその断片もしくは一部である。本発明の他の態様では、候補分子は別の結合タンパク質であってよい。
【0050】
本発明に従って標的リガンドを結合し得る候補分子を同定するため、以下の段階を実施することができる:候補結合ポリペプチドと内膜のペリプラズム面に固定される配列との融合タンパク質を含むグラム陰性菌の集団を提供する段階;細菌と、候補結合ポリペプチドと接触し得る少なくとも1つの第1標識標的リガンドを混合する段階;および標的リガンドを結合し得る分子を発現する少なくとも1つの第1細菌を同定する段階。
【0051】
上記の方法では、固定された候補結合タンパク質と標識リガンドとが結合することにより、細胞の外への拡散が妨げられることになる。このように、標識リガンドの分子が細菌のペリプラズム内に保持され得る。または、ペリプラズムを除去することもでき、したがって固定化により結合した候補分子の保持が起こることになる。次いで、標識を用いて、標的リガンドを結合し得る結合ポリペプチドを発現する細胞を単離することができ、このようにして結合ポリペプチドをコードする遺伝子が単離される。その後、インビボまたはエクスビボ発現方法により、標的リガンドを結合し得る分子を大量に産生することができ、次いで、例えば以下に記載するような診断または治療用途等の任意の所望の用途に用いることができる。
【0052】
本明細書で用いる「候補分子」または「候補ポリペプチド」という用語は、標的分子に対する親和性を潜在的に有する可能性のある任意の分子またはポリペプチドを指す。候補物質は、合成分子等の低分子を含む、タンパク質またはその断片であってよい。本発明の1つの態様において、候補分子は抗体配列またはその断片を含んでよい。そのような配列は、特に、標的リガンドに結合する可能性に合わせて設計することができる。
【0053】
本発明に従って単離する結合ポリペプチドまたは抗体はまた、標的リガンドの構造を解明する助けとなり得る。原理上は、このアプローチにより、次の薬物設計の基礎となり得るファーマコアが得られる。機能的な、薬理学的に活性のある抗体に対する抗イディオタイプ抗体を作製することにより、タンパク質結晶学を完全に回避できる可能性がある。鏡像の鏡像として、抗イディオタイプ抗体の結合部位は、本来の抗原の類似体であることが期待される。次いで、抗イディオタイプ抗体を用いて、化学的または生物学的に産生したペプチドのバンクからペプチドを同定および単離することができる。選択したペプチドはファーマコアとして役立つと考えられる。抗イディオタイプは、抗原として抗体を使用し、抗体の産生に関して本明細書に記載した方法により作製することができる。一方、様々な市販業者から、標的リガンドの結合に関して基本的な基準を満たすと考えられる低分子ライブラリーを単純に取得してもよい。そのようなライブラリーは、低分子をコードする核酸または分子を発現する細菌として提供され得る
【0054】
A. 結合タンパク質コード配列のクローニング
抗体または他の結合タンパク質の結合親和性は、例えば、MunsonおよびPollardのスキャッチャード分析(1980)によって決定することができる。所望の特異性、親和性、および/または活性の分子を産生する細菌細胞を同定した後、対応するコード配列をクローニングすることが可能である。この方法では、従来の手順により(例えば、抗体または結合タンパク質をコードする遺伝子に特異的に結合し得るオリゴヌクレオチドプローブを用いて)、分子をコードするDNAを単離し配列決定することができる。
【0055】
単離したならば、抗体または結合タンパク質DNAを発現ベクター内に配置し、次にこのベクターを、通常であれば免疫グロブリンタンパク質を産生しないサルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞、またはミエローマ細胞等の宿主細胞にトランスフェクションし、組換え宿主細胞において結合タンパク質を合成させることができる。例えば、相同的なマウス配列の代わりにヒト重鎖および軽鎖定常ドメインのコード配列を用いることより(Morrisonら、1984)、または免疫グロブリンコード配列に非免疫グロブリンポリペプチドのコード配列のすべてまたは一部を共有結合することにより、DNAを修飾してもよい。この様式で、所望の結合特異性を有する「キメラ」または「ハイブリッド」結合タンパク質が作製される。
【0056】
典型的に、そのような非免疫グロブリンポリペプチドは抗体の定常ドメインと置換するか、または標的リガンドに対する特異性を有する1つの抗原結合部位と異なる抗体に対する特異性を有する別の抗原結合部位を含むキメラ二価抗体を作製するために、抗体の1つの抗原結合部位の可変領域と置換する。
【0057】
キメラ抗体またはハイブリッド抗体はまた、架橋剤が関与する方法を含む合成タンパク質化学における既知の方法を用いて、インビトロで作製され得る。例えば免疫毒素は、ジスルフィド交換反応を用いるかまたはチオエーテル結合の形成により、構築され得る。この方法に適した試薬の例には、イミノチオラートおよびメチル-4-メルカプトブチルイミダートが含まれる。
【0058】
生細胞または非生細胞から核酸をクローニングし得ることは、当業者に理解されよう。非生細胞の場合には、例えば、PCRを用いるなどしてクローン化DNAの増幅を利用することが望ましいと考えられる。これは、生細胞を用いて、細胞をさらに増殖させるかまたはさせずに行うことも可能である。
【0059】
B. リガンドに対するタンパク質親和性の最大化
天然の免疫反応において、抗体遺伝子は高率で変異を蓄積する(体細胞超変異)。導入される変化のいくつかによって、より高い親和性および高親和性表面免疫グロブリンを提示するB細胞が付与されることになる。「チェーンシャフリング」(Marksら、1992)として知られる技法を用いて、この天然の過程を模倣することができる。この方法では、重鎖および軽鎖V領域遺伝子を、非免疫化ドナーから得られたVドメイン遺伝子の天然変種(レパートリー)と順次置換することにより、本発明に従って得られた「一次」ヒト抗体の親和性を改善することができる。この技法によって、nM範囲の親和性を有する抗体または抗体断片の産生が可能になる。非常に大きな抗体レパートリーを作製するための戦略はWaterhouseら(1993)によって記載されており、そのような大きなファージライブラリーからの高親和性ヒト抗体の直接的な単離はGriffithら(1994)によって報告されている。遺伝子シャッフリング法を用いて、げっ歯類抗体からヒト抗体を導出することも可能であり、この場合、ヒト抗体は初めのげっ歯類抗体と類似した親和性および特異性を有する。「エピトープ刷り込み」とも称されるこの方法により、ファージディスプレイ技法によって得られたげっ歯類抗体の重鎖または軽鎖VドメインをヒトVドメイン遺伝子のレパートリーと置換し、げっ歯類-ヒトキメラが作製される。抗原を選択することにより、機能的な抗原結合部位を回復し得るヒト可変領域が単離される、すなわち、エピトープによってパートナーの選択が支配される(刷り込まれる)。残りのげっ歯類Vドメインを置換するためにこの過程を繰り返すと、ヒト抗体が得られる(1993年4月1日に公開された国際公開公報第93/06213号を参照のこと)。CDR移植によるげっ歯類抗体の従来のヒト化とは異なり、この技法により、げっ歯類起源のフレームワークまたはCDR残基も有さない、完全なヒト抗体が提供される。
【0060】
C. 標識リガンド
本発明の1つの態様では、標識リガンドに対する親和性を有する抗体または結合タンパク質を単離する。本発明に従ってグラム陰性菌のペリプラズム膜を透過処理および/または除去することにより、潜在的に任意の大きさの標識リガンドをスクリーニングすることができる。ペリプラズム膜の除去を行わない場合には、細菌ペリプラズム膜を超えてリガンドが効率的に拡散できるように、標識リガンドは典型的に50,000 Da未満の大きさであることが好ましい。
【0061】
上記のように、本発明に従って、1つまたは複数の検出可能な薬剤で標識したリガンドを提供することが典型的に望ましい。これは、例えば、リガンドを少なくとも1つの検出可能な薬剤に連結して複合体を形成させることによって、実施することができる。例えば、少なくとも1つの検出可能な分子または成分を連結または共有結合または複合体化することは、従来から行われている。「標識」または「検出可能な標識」とは、特定の機能的特性および/または化学的特性によって検出され得る化合物および/または成分であり、これを用いることによって、これが結合したリガンドの検出、および/または必要に応じてさらなる定量が可能になる。本発明に使用できる標識の例には、酵素、放射標識、ハプテン、蛍光標識、リン光性分子、化学発光性分子、発色団、発光性分子、光親和性分子、ビオチン等の着色粒子またはリガンドが含まれるが、これらに限定されない。
【0062】
本発明の1つの態様では、標識に対する細胞の自動化スクリーニングが実施できるように、視覚的に検出可能なマーカーを用いる。特に、所望の結合タンパク質または抗体を発現する細胞を単離するためにフローサイトメトリーの使用を可能にするという点で、蛍光標識が有利である。適切な機器を用いて可視化することにより検出できる薬剤の例は、当技術分野において周知であり、所望のリガンドに結合する方法も同様に周知である(例えば、それぞれ参照として本明細書に組み入れられる米国特許第5,021,236号;第4,938,948号;および第4,472,509号を参照のこと)。そのような薬剤には、常磁性イオン;放射性同位元素;蛍光色素;NMR検出可能物質、およびX線像用の物質が含まれる。本発明で使用可能な蛍光標識の種類は当業者に周知であり、例えば、Alexa350、Alexa430、AMCA、BODIPY 630/650、BODIPY 650/655、BODIPY-FL、BODIPY-R6G、BODIPY-TMR、BODIPY-TRX、カスケードブルー、Cy3、Cy5、6-FAM、フルオレセインイソチオシアネート、HEX、6-JOE、オレゴングリーン488、オレゴングリーン500、オレゴングリーン514、パシフィックブルー、REG、ローダミングリーン、ローダミンレッド、レノグラフィン、ROX、TAMRA、TET、テトラメチルローダミン、および/またはテキサスレッドが含まれる。
【0063】
磁気スクリーニング法は当業者には周知である(例えば、米国特許第4,988,618号、米国特許第5,567,326号、および米国特許第5,779,907号を参照のこと)。そのような技法に従って標識として使用し得る常磁性イオンの例には、クロム(III)、マンガン(II)、鉄(III)、鉄(II)、コバルト(II)、ニッケル(II)、銅(II)、ネオジム(III)、サマリウム(III)、イッテルビウム(III)、ガドリニウム(III)、バナジウム(II)、テルビウム(III)、ジスプロシウム(III)、ホルミウム(III)、および/またはエルビウム(III)が含まれる。他の状況において有用なイオンには、ランタン(III)、金(III)、鉛(II)、および特にビスマス(III)が含まれるが、これらに限定されない。
【0064】
本発明において意図する別の種類のリガンド複合体は、リガンドが、二次結合分子および/または発色基質と接触した際に有色産物を生じる酵素(酵素タグ)に結合しているリガンド複合体である、そのような酵素の例には、ウレアーゼ、アルカリホスファターゼ、(西洋ワサビ)過酸化水素ペルオキシダーゼ、およびグルコースオキシダーゼが含まれる。このような例では、選択した細胞が生存したままであることが望ましい。好ましい二次結合リガンドは、ビオチンおよび/またはアビジンおよびストレプトアビジン化合物である。そのような標識の使用は当業者に周知であり、例えば米国特許第3,817,837号;第3,850,752号;第3,939,350号;第3,996,345号;第4,277,437号;第4,275,149号;および第4,366,241号に記載されており、それぞれ参照として本明細書に組み入れられる。
【0065】
低強度の紫外光によって生成される反応性ナイトレン中間体を介してタンパク質への共有結合を形成するために、アジド基を含む分子を用いることも可能である(PotterおよびHaley、1983)。特に、プリンヌクレオチドの2-アジド類似体および8-アジド類似体が、粗製細胞抽出物中のヌクレオチド結合タンパク質を同定するための部位特異的光プローブとして用いられた(OwenおよびHaley、1987;Athertonら、1985)。2-アジド類似体および8-アジド類似体はまた、精製したタンパク質のヌクレオチド結合ドメインをマッピングするためにも使用され(Khatoonら、1989;Kingら、1989;およびDholakiaら、1989)、リガンド結合剤としても使用することができる。
【0066】
当業者に周知の任意の技法により、標識を行うことができる。例えば、リガンドを、所望の標識および次亜塩素酸ナトリウム等の化学酸化剤またはラクトペルオキシダーゼ等の酵素的酸化剤と接触させることにより、リガンドを標識することができる。同様に、リガンド交換過程を用いることも可能である。または、例えば、標識、SNCl2等の還元剤、ナトリウム-カリウムフタル酸溶液等の緩衝液 、およびリガンドをインキュベートすることにより、直接的標識技法を用いてもよい。例えば、ジエチレントリアミン五酢酸(DTPA)またはエチレンジアミン四酢酸(EDTA)の存在下で標識をリガンドに結合するために、リガンド上の媒介性官能基を用いることも可能である。
【0067】
複合体成分へのリガンドの結合または複合体化に関して、他の方法もまた当技術分野において周知である。いくつかの結合法は、リガンドに結合させた無水ジエチレントリアミン五酢酸(DTPA);エチレントリアミン四酢酸;N-クロロ-p-トルエンスルホンアミド;および/またはテトラクロロ-3α-6α-ジフェニルグリコウリル(diphenylglycouril)-3等の有機キレート剤の使用を含む(それぞれ参照として本明細書に組み入れられる米国特許第4,472,509号および第4,938,948号)。グルタルアルデヒドまたは過ヨウ素酸等のカップリング剤の存在下で、リガンドを酵素と反応させてもよい。蛍光マーカーとの複合体は、これらのカップリング剤の存在下で、またはイソチオシアネートと反応させることにより、調製することができる。米国特許第4,938,948号では、モノクローナル抗体を用いて乳房腫瘍の画像診断が達成されるが、メチル-p-ヒドロキシベンズイミデートまたはN-スクシニミジル-3-(4-ヒドロキシフェニル)プロピオン酸等のリンカーを用いて、検出可能な撮像成分を抗体に結合する。
【0068】
ペリプラズムで発現させたタンパク質を適切な蛍光リガンドで特異的に標識する能力は、ライブラリースクリーニング以外の用途も有する。タンパク質生産の過程において産生をモニタリングするために、蛍光リガンドでの特異的標識およびフローサイトメトリーを用いることができる。フローサイトメトリーはこれまで細菌細胞の解析のために用いられてきたが、ペリプラズムタンパク質の特異的標識および定量のためには用いられていなかった。しかし、IGF-1、いくつかのインターロイキン、ウロキナーゼ型プラスミノーゲン活性化因子等の酵素、抗体断片、阻害剤(例えば、ウシ膵臓トリプシンインヒビター)を含む多数の商業的に重要なタンパク質は、ペリプラズム空間に分泌される形態で、組換え大腸菌において発現される。本発明による技法に従い、適切な蛍光リガンドおよびフローサイトメトリー解析を利用することにより、培養物中の各細胞内のそのようなタンパク質の産生レベルをモニタリングすることができる。
【0069】
一般に、タンパク質発現のモニタリングは、細胞の溶解および免疫学的技法または次のクロマトグラフィー分離によるタンパク質の検出を必要とする。しかし、ELISAまたはウェスタンブロット解析は時間を要し、これらの解析からは細胞集団内での発現の分布に関する情報が提供されず、さらにオンラインモニタリングに用いることができない(Thorstensonら、1997;Berrierら、2000)。対照的に、FACS標識は迅速かつ単純であり、グラム陰性菌で発現した組換えタンパク質の工業的規模での発酵をオンラインモニタリングするためにうまく適合化することができる。同様に、本発明を用いて、細菌においてインビボで発現したまたはエクスビボで提供された酵素の相対濃度または比活性を測定することも可能である。
【0070】
本発明に従って抗体等のリガンド結合タンパク質が単離されたならば、その分子の有用性を増すために、少なくとも1つの薬剤に分子を連結して複合体を形成することが望ましいと考えられる。例えば、診断薬または治療薬として抗体分子の有効性を高めるために、少なくとも1つの所望の分子または成分を連結または共有結合または複合体化することは、従来から行われている。そのような分子または成分は、少なくとも1つのエフェクターまたはレポーター分子であるが、これらに限定されない。エフェクター分子には、細胞障害性等の所望の活性を有する分子が含まれる。抗体に結合されているエフェクター分子の限定されない例には、毒素、抗腫瘍薬、治療的酵素、放射標識ヌクレオチド、抗ウイルス薬、キレート薬、サイトカイン、増殖因子、およびオリゴヌクレオチドまたはポリヌクレオチドが含まれる。これに反して、レポーター分子は、アッセイにより検出され得る任意の成分と定義される。そのような分子を標識する技法は、当業者には周知であり、本明細書において先に記載した。
【0071】
次に、例えばタンパク質、ポリペプチド、またはペプチド等の生物成分を結合、精製、除去、定量、および/またはそうでない場合は一般に検出するための免疫検出法において、本明細書に従って調製した抗体等の標識結合タンパク質を使用してもよい。いくつかの免疫検出法には、少し記述するだけでも酵素結合免疫吸着測定法(ELISA)、放射性免疫測定法(RIA)、免疫放射定量測定法、蛍光免疫測定法、化学発光測定法、生物発光測定法、およびウェスタンブロット法が含まれる。様々な有用な免疫検出法の段階は、例えば、Doolittle MHおよびBen-Zeev O、1999;Gulbis BおよびGaland P、1933;およびDe Jager Rら、1993等の科学文献に記載されており、これらはそれぞれ参照として本明細書に組み入れられる。そのような技法には、当技術分野で周知である様々な種類の酵素結合免疫吸着測定法(ELISA)および/または放射性免疫測定法(RIA)等の結合測定法が含まれる。
【0072】
本発明に従って調製した、抗体を含むリガンド結合分子は、例えば、免疫組織化学法(IHC)によって研究するために調製した、新鮮凍結した組織ブロックおよび/またはホルマリン固定、パラフィン包埋した組織ブロックと共に用いることも可能である。これらの粒子状試料から組織ブロックを調製する方法は、様々な予後因子のこれまでのIHC研究において用いられて成功しており、および/または当業者に周知である(Abbondanzoら、1990)。
【0073】
VI. フローサイトメトリーを用いた自動化スクリーニング
本発明の1つの態様において、候補分子に結合し、細菌の細胞質膜の外面に結合している標識リガンドを含む細菌細胞を効率よく単離するために、蛍光活性化細胞分類 (FACS)スクリーニングまたは他の自動化フローサイトメトリー技法を用いることができる。フローサイトメトリーを行うための機器は当業者に周知であり、市販されている。そのような機器の例には、Becton Dickinson(カリフォルニア州、フォスターシティー)のFACS Star Plus、FACSCan、FACSort機器、Coulter Epics Division(フロリダ州、ハイアリーア)のEpics C、およびCytomation(コロラド州、コロラドスプリングス)のMoFloが含まれる。
【0074】
フローサイトメトリー技法は、一般に、液体試料中の細胞または他の粒子の分離を伴う。典型的に、フローサイトメトリーの目的は、分離された粒子をその1つまたは複数の特徴、例えば標識リガンドまたは他の分子の存在に関して分析することである。フローサイトメトリーの基本的な段階は、液体流動が検出領域を通過するような、装置内の液体試料の方向づけを含む。粒子は一つずつセンサーを通過すべきであり、大きさ、屈折、光散乱、不透明度、粗雑度、形状、蛍光等に基づき分類される。
【0075】
細胞の迅速な定量解析は、生物医学研究および医学において有用となる。装置により、1秒当たり数千個の細胞の速度での、細胞特性の定量的多パラメータ解析が可能になる。これらの機器により、細胞種を区別する能力が提供される。データは、測定した変数の1次元(ヒストグラム)または2次元(等高線プロット、散布プロット)度数分布で示される場合が多い。多パラメータデータファイルの分割は、双方向性の1次元または2次元画像プログラムの連続した使用を必要とする。
【0076】
迅速に細胞を検出するため、多パラメータフローサイトメトリーデータの定量的解析は、以下の2段階からなる:細胞種の特徴づけおよび試料の処理。一般に、細胞種特徴づけの過程により、細胞特性が関心対象の細胞と関心対象でない細胞に分別される。次いで、試料の処理において、収まる領域に従って各細胞が2つのカテゴリーのうちの一方に分類される。高度な検出性能は細胞の適切な特性が得られる場合にのみ期待され得るため、細胞の種類の解析は非常に重要である。
【0077】
フローサイトメトリーによって細胞分析が行われるだけでなく、細胞の分別も行われる。米国特許第3,826,364号において、機能的に異なる細胞種等の粒子を物理的に分離する装置が開示されている。この機械では、レーザーにより、適切なレンズまたはレンズシステムにより粒子の流れに焦点を合わせた照明が提供され、その結果その中の粒子から高度に限局化した散乱が得られる。さらに、流動中の蛍光粒子を励起するために、高輝度源照明が粒子の流れに向けられる。流動中の特定の粒子は選択的に荷電され、次いで、指定の容器に向けて偏向させることによって分離され得る。この分離の古典的な形態は蛍光タグ化抗体によるものであり、この抗体は、分離のために1つまたは複数の細胞種を標識するために使用される。
【0078】
フローサイトメトリー法の他の例には、米国特許第4,284,412号;第4,989,977号;第4,498,766号;第5,478,722号;4,857,451号;第4,774,189号;4,767,206号;第4,714,682号;第5,160,974号;および第4,661,913号に記載されている方法が含まれるが、これらに限定されず、これらの開示はそれぞれ参照として本明細書に明確に組み入れられる。
【0079】
本発明に関して、フローサイトメトリーの重要な局面は、複数ラウンドのスクリーニングを連続して行うことができる点である。選別の最初のラウンドから細胞を単離し、直ちにフローサイトメーターに再導入し、スクリーニングのストリンジェンシーを改善するために再度スクリーニングすることが可能である。当業者に周知である別の恩典は、フローサイトメトリーを用いて非生細胞を回収できる点である。フローサイトメトリーは基本的に粒子選別技術であるため、細胞が成長または増殖する能力は必要ではない。そのような非生細胞から核酸を回収する技法は当技術分野において周知であり、これには、例えばPCRを含む鋳型依存的増幅技法の使用が含まれる。
【0080】
VII. 核酸に基づいた発現系
本発明の特定の態様においては、組換えタンパク質を発現させるために核酸に基づいた発現系を使用できる。例えば、本発明の1つの態様は、選択したリガンドに対する親和性を有する候補抗体または他の結合タンパク質を含む融合ポリペプチドのコード配列によるグラム陰性菌の形質転換、およびグラム陰性菌の細胞質膜上でのそのような分子の発現を含む。本発明の他の態様では、標的リガンドに対する特異性を有する単離された結合タンパク質を調製するために、そのようなコード配列の発現を例えば真核生物宿主細胞において行うことができる。次に、単離したタンパク質を1つまたは複数の治療用途または診断用途に用いることができる。
【0081】
A. 核酸送達の方法
本発明の特定の局面は、標的細胞への核酸の送達を含み得る。例えば、細菌宿主細胞を、標的リガンドに潜在的に結合し得る候補分子をコードする核酸で形質転換することができる。本発明の特定の態様においては、発現を細菌の細胞質膜に標的することが望ましい場合がある。真核生物宿主細胞の形質転換も同様に、標的リガンドを結合し得ると同定された様々な候補分子の発現に使用できる。
【0082】
細胞を形質転換するための核酸送達に適した方法には、核酸(例えばDNA)をそのような細胞またはその細胞小器官にさえ導入し得る実質的に任意の方法が含まれると考えられる。そのような方法には、マイクロインジェクション(HarlanおよびWeintraub、1985;米国特許第5,789,215号、参照として本明細書に組み入れられる)を含む注入(米国特許第5,994,624号、第5,981,274号、第5,945,100号、第5,780,448号、第5,736,524号、第5,702,932号、第5,656,610号。第5,589,466号、および第5,580,859号、それぞれ参照として本明細書に組み入れられる);エレクトロポレーション(米国特許第5,384,253号、参照として本明細書に組み入れられる);リン酸カルシウム沈殿(GrahamおよびVan Der Eb、1973;ChenおよびOkayama、1987;Rippeら、1990);DEAE-デキストランとそれに続くポリエチレングリコールの使用(Gopal、1985);直接的超音波負荷(Fechheimerら、1987);リポソームを介するトランスフェクション(NicolauおよびSene、1982;Fraleyら、1979;Nicolauら、1987;Wongら、1980;Kanedaら、1989;Katoら、1991);微粒子銃(国際公開公報第94/09699号および第95/06128号;米国特許第5,610,042号;第5,322,783号、第5,563,055号、第5,550,318号、第5,538,877号、および第5,538,880号、それぞれ参照として本明細書に組み入れられる);炭化ケイ素繊維との撹拌(Kaepplerら、1990;米国特許第5,302,523号および第5,464,765号、それぞれ参照として本明細書に組み入れられる);アグロバクテリウムを介する形質転換(米国特許第5,591,616号および第5,563,055号、それぞれ参照として本明細書に組み入れられる); またはプロトプラストのPEGを介する形質転換(Omirullehら、1993;米国特許第4,684,611号および第4,952,500号、それぞれ参照として本明細書に組み入れられる);乾燥/抑制を介するDNA取り込み(Potrykusら、1985)等のDNAの直接的送達が含まれるが、これらに限定されない。これらのような技法を適用し、細胞小器官、細胞、組織、または生物体を安定にまたは一過性に形質転換することができる。
【0083】
1. エレクトロポレーション法
本発明の特定の態様では、エレクトロポレーションにより核酸を細胞に導入する。エレクトロポレーションは、細胞懸濁液およびDNAの高圧放電への曝露を含む。この方法のいくつかの変法では、ペクチン分解酵素等のある種の細胞壁分解酵素を用いて、標的レシピエント細胞を未処理の細胞よりもエレクトロポレーションによってより形質転換されやすくする(米国特許第5,384,253号、参照として本明細書に組み入れられる)。または、機械的損傷によって、レシピエント細胞をより形質転換されやすくできる。
【0084】
2. リン酸カルシウム法
本発明の他の態様では、リン酸カルシウム沈殿法により核酸を細胞に導入する。この技法を用いて、ヒトKB細胞にアデノウイルス5 DNAがトランスフェクションされた(GrahamおよびVen Der Eb、1973)。同様にこの方法により、マウスL(A9)、マウスC127、CHO、CV-1、BHK、NIH3T3、およびHeLa細胞に、ネオマイシンマーカー遺伝子がトランスフェクションされ(ChenおよびOkayama、1987)、ラット肝細胞に様々なマーカー遺伝子がトランスフェクションされた(Rippeら、1990)。
【0085】
B. ベクター
例えば、標的リガンドに結合する能力についてスクリーニングすることが望まれる候補ポリペプチドをコードする核酸配列によるグラム陰性菌の形質転換において、本発明によりベクターを使用できる。本発明の1つの態様において、標的ポリペプチドをコードする核酸配列の不均一性「ライブラリー」全体を細菌集団に導入することができ、それにより全ライブラリーのスクリーニングが可能になる。「ベクター」という用語を使用して、細胞内に導入されそこで複製され得るための、核酸配列を挿入し得る担体核酸分子を指す。核酸配列は「外因性」または「異種性」であってよく、これはその配列がベクターを導入する細胞にとって外来性であること、またはその配列が細胞内の配列と相同的であるが、通常はその配列が見出されない宿主細胞核酸内の位置にあることを意味する。ベクターには、プラスミド、コスミド、ウイルス(バクテリオファージ、動物ウイルス、および植物ウイルス)、および人工染色体(例えばYAC)が含まれる。当業者は標準的な組換え技法によりベクターを構築することができ、この技法は、どちらも参照として本明細書に組み入れられるManiatisら、1988、およびAusubelら、1994に記載されている。
【0086】
「発現ベクター」という用語は、転写され得る遺伝子産物の少なくとも一部をコードする核酸配列を含むベクターを指す。場合によっては、次にRNA分子がタンパク質、ポリペプチド、またはペプチドに翻訳される。別の場合には、例えばアンチセンス分子またはリボザイムの産生において、これらの配列は翻訳されない。発現ベクターは様々な「制御配列」を含み得り、「制御配列」とは、特定の宿主生物体において機能的に結合したコード配列の転写および場合によっては翻訳に必要な核酸配列を指す。ベクターおよび発現ベクターは、転写および翻訳を支配する制御配列に加えて、同様に他の機能に役立ち、以下に記載する核酸配列を含んでよい。
【0087】
1. プロモーターおよびエンハンサー
「プロモーター」とは、そこで転写の開始および速度が制御される核酸配列の領域である制御配列である。プロモーターは、RNAポリメラーゼおよび他の転写因子等の制御タンパク質および分子が結合し得る遺伝子エレメントを含み得る。「機能的に位置する」、「機能的に結合した」、「制御下で」、および「転写制御下で」という語句は、配列の転写開始およびまたは発現を制御すべき核酸配列に関して、プロモーターが正しく機能的な位置および/または方向にあることを意味する。プロモーターは「エンハンサー」と共に使用してもしなくてもよく、「エンハンサー」とは、核酸配列の転写活性化に関与するシス作動性の制御配列を指す。
【0088】
プロモーターは、コード部分および/またはエキソンの上流に位置する5'非コード配列を単離することによって得られ得るように、遺伝子または配列と天然で付随しているものであってよい。そのようなプロモーターは「内因性」と称すことができる。同様にエンハンサーも、核酸配列の下流または上流に位置して、核酸配列と天然に付随しているものであってよい。または、組換えまたは異種性プロモーターの制御下にコード核酸部分を置くことにより、ある種の恩典が得られることになるが、そのようなプロモーターとは、その天然環境において核酸配列と通常は付随していないプロモーターを指す。組換えまたは異種性エンハンサーもまた、その天然環境において核酸配列と通常は付随していないエンハンサーを指す。そのようなプロモーターまたはエンハンサーには、他の遺伝子のプロモーターまたはエンハンサー、任意の他の原核生物、ウイルス、または真核生物細胞から単離されたプロモーターまたはエンハンサー、ならびに「天然に存在」しない、すなわち異なる転写制御領域の異なるエレメントおよび/または発現を変化させる変異を含むプロモーターまたはエンハンサーが含まれ得る。合成によりプロモーターおよびエンハンサーの核酸配列を作製することに加えて、本明細書で開示する組成物と関連して、組換えクローニング技法および/またはPCR(商標)を含む核酸増幅技法を用いて配列を産生してもよい(それぞれ参照として本明細書に組み入れられる米国特許第4,683,202号、米国特許第5,928,906号を参照のこと)。さらに、ミトコンドリア、葉緑体等の非核細胞小器官内で配列の転写および/または発現を指示する制御配列も同様に使用し得ることを意図する。
【0089】
当然のことながら、発現させるために選択した細胞種、細胞小器官、および生物体において、DNA部分の発現を効率的に指示するプロモーターおよび/またはエンハンサーを使用することが重要であろう。本発明で使用し得るそのようなプロモーターの一例は、大腸菌アラビノースプロモーターである。分子生物学の分野における当業者は、一般に、タンパク質発現のためのプロモーター、エンハンサー、および細胞種の組み合わせの使用に詳しく、例えば、参照として本明細書に組み入れられるSambrookら(1989)を参照されたい。使用するプロモーターは、構成的、組織特異的、誘導性、および/または組換えタンパク質および/またはペプチドの大量発現において有利であるように、導入したDNA部分の高レベル発現を指示する適切な条件下で有用であってよい。プロモーターは、異種性であっても内因性であってもよい。
【0090】
2. 開始シグナルおよび内部リボソーム結合部位
コード配列の効率的な翻訳には、特異的な開始シグナルも必要とされ得る。これらのシグナルには、ATG開始コドンまたは隣接配列が含まれる。ATG開始コドンを含む外因性翻訳制御シグナルが提供される必要がある場合もある。当業者は、容易にこれを決定し必要なシグナルを提供し得ると考えられる。挿入物全体の翻訳を確実にするため、開始コドンは所望のコード配列のフレームと「インフレーム」でなければならないことは周知である。外因性の翻訳制御シグナルおよび開始コドンは、天然であっても合成であってもよい。発現の効率は、適切な転写エンハンサーエレメントを含めることによって増強され得る。
。
【0091】
3. マルチクローニング部位
ベクターはマルチクローニング部位(MCS)を含み得るが、MCSとは、そのいずれかがベクターを消化するための標準的な組換え技術と併せて使用され得る、複数の制限酵素部位を含む核酸領域である(参照として本明細書に組み入れられるCarbonelliら、1999、Lavensonら、1998、およびCocea、1997を参照のこと)。「制限酵素消化」とは、核酸分子内の特定の位置でのみ機能する酵素による核酸分子の触媒作用的切断を指す。これらの制限酵素の多くは市販されている。そのような酵素の使用は、当業者に理解されている。外因性配列をベクター内に連結できるように、MCS内で切断する制限酵素を用いてベクターを線状化または断片化する場合が多い。「連結」とは、2つの核酸断片間でリン酸ジエステル結合を形成する過程を指し、これらの断片は相互に隣接してもしなくてもよい。制限酵素および連結反応を含む技法は、組換え技術の分野の当業者に周知である。
【0092】
4. 終結シグナル
本発明に従って調製するベクターまたは構築物は、一般に、少なくとも1つの終結シグナルを含むことになる。「終結シグナル」または「ターミネーター」とは、RNAポリメラーゼによるRNA転写の特異的終結に関与するDNA配列からなる。したがって、特定の態様においては、RNA転写産物の産生を終了させる終結シグナルを意図する。ターミネーターは、所望のメッセージレベルを達成するためにインビボで必要とされる場合がある。
【0093】
本発明における使用が意図されるターミネーターには、本明細書に記載するかまたは当業者に周知である任意の周知である転写ターミネーターが含まれ、これには、例えばrhp依存性ターミネーターまたはrho依存性ターミネーターが含まれるが、これらに限定されない。特定の態様において、終結シグナルは、配列切断に起因するなどして、転写可能なまたは翻訳可能な配列を欠いている可能性がある。
【0094】
5. 複製起点
宿主細胞においてベクターを増幅するため、ベクターは、そこで複製が開始される特異的核酸配列である1つまたは複数の複製起点(多くの場合「ori」と称される)を含み得る。または、宿主細胞が酵母である場合は、自己複製配列(ARS)が使用され得る。
【0095】
6. 選択可能なおよびスクリーニング可能なマーカー
本発明の特定の態様では、発現ベクター内にマーカーを含めることによって、本発明の核酸構築物を含む細胞をインビトロまたはインビボで同定することができる。そのようなマーカーによって同定可能な変化が細胞に付与され、発現ベクターを含む細胞の容易な同定が可能となる。一般に、選択可能なマーカーは、選択を可能にする特性を付与するものである。陽性選択可能なマーカーは、マーカーの存在によって選択が可能になるマーカーであるのに対して、陰性選択可能なマーカーは、その存在によって選択が妨げられるマーカーである。陽性選択可能なマーカーの例は、薬剤耐性マーカーである。
【0096】
通常、薬物選択マーカーを含めることは形質転換体のクローニングおよび同定に役立ち、例えば、ネオマイシン、ピューロマイシン、ハイグロマイシン、DHFR、GPT、ゼオシン、およびヒスチジノールに対する耐性を付与する遺伝子は有用な選択可能マーカーである。条件の実行に基づいて形質転換体の識別を可能にする表現型を付与するマーカーに加えて、その基本が比色分析であるGFP等のスクリーニング可能なマーカーを含む、他の種類のマーカーもまた意図する。または、単純ヘルペスウイルスチミジンキナーゼ(tk)またはクロラムフェニコールアセチルトランスフェラーゼ(CAT)等のスクリーニング可能な酵素を使用してもよい。当業者はまた、おそらくFACS解析に関連して、免疫学的マーカーをいかにして使用するかを理解すると考えられる。遺伝子産物をコードする核酸が同時に発現され得る限り、使用するマーカーは重要でないと考えられる。選択可能なまたはスクリーニング可能なマーカーのさらなる例は、当業者に周知である。
【0097】
C. 宿主細胞
本明細書では、「細胞」、「細胞株」、および「細胞培養物」という用語は互換的に用いられ得る。これらの用語すべてには、任意のおよびすべての次の世代であるそれらの子孫も含まれる。すべての子孫は、意図的変異または偶発的変異により同一でない可能性があることが理解される。異種性核酸配列の発現との関連において、「宿主細胞」とは原核細胞を指し、これには、ベクターを複製し得るおよび/またはベクターによりコードされる異種性遺伝子を発現し得る形質転換可能な任意の生物が含まれる。宿主細胞は、ベクターのレシピエントとして使用することができ、現に使用されている。宿主細胞は「トランスフェクション」または「形質転換」することができ、これらの用語は、外因性核酸を宿主細胞内に伝達または導入する過程を指す。形質転換細胞には、最初の被験細胞およびその子孫が含まれる。
【0098】
本発明の特定の態様において、宿主細胞はグラム陰性菌である。この細菌は、内膜と外膜との間にペリプラズム膜を有し、特にペリプラズムと細胞質との間に細胞質膜としても知られている上記の内膜を有するという点で、本発明での使用に適している。そのようなものとして、そのようなペリプラズム空間を有する任意の他の細胞も本発明に従って使用することができる。本発明で使用し得るグラム陰性菌の例には、大腸菌、緑膿菌、コレラ菌、ネズミチフス菌、フレクスナー赤痢菌、インフルエンザ菌、百日咳菌、火傷病菌、根粒菌種が含まれるが、これらに限定されない。グラム陰性菌細胞は、選択したリガンドを結合し得る候補結合ポリペプチドを含む融合ポリペプチドをコードする配列で形質転換した細菌としてさらに定義され得る。ポリペプチドはペリプラズム空間に面した細胞質膜の外面に固定されるが、これは抗体コード配列または別の配列を含み得る。ポリペプチドを発現させるための1つの手段は、そのような方向づけを起こし得るリーダー配列をポリペプチドに結合させることによる。
【0099】
多数の原核生物細胞株および培養物が宿主細胞としての使用に利用でき、これらは、生きた培養物および遺伝物質の保管所としての機能を果たす機関であるアメリカン・タイプ・カルチャー・コレクション(ATCC)(www.atcc.org)を通して入手できる。適切な宿主は、ベクター骨格および所望の結果に基づき、当業者により決定され得る。プラスミドまたはコスミドは、例えば、多くのベクターを複製するために原核宿主細胞内に導入され得る。ベクターを複製および/または発現させるために宿主細胞として使用される細菌細胞には、DH5α、JM109、およびKC8、ならびSURE(登録商標)コンピテント細胞およびSOLOPACK(商標)ゴールド細胞(STRATAGENE(登録商標)、ラ・ホーヤ)等の多くの市販の細菌宿主が含まれる。または、大腸菌LE392等の細菌細胞をバクテリオファージの宿主細胞として使用することも可能である。
【0100】
様々な細胞種および生物に由来する多くの宿主細胞が入手可能であり、当業者に周知であると考えられる。同様に、ウイルスベクターを、原核生物宿主細胞、特にベクターの複製または発現に関して許容状態である原核生物宿主細胞とともに用いてもよい。いくつかのベクターは、原核細胞および真核細胞の両方においてベクターの複製および/または発現を可能にする制御配列を利用できる。当業者はさらに、上記の宿主細胞を維持するためおよびベクターの複製を可能にするためにそれらすべてをインキュベートする条件を理解するものと考えられる。ベクターの大量生産、ならびにベクターによってコードされる核酸、およびその同族ポリペプチド、タンパク質、またはペプチドの産生を可能にする技法および条件もまた理解され、周知である。
【0101】
D. 発現系
上記の組成物の少なくとも一部またはすべてを含む多数の発現系が存在する。そのような系は、例えば、本発明により特定のリガンドに結合し得ると同定されるポリペプチド産物を産生するために使用できる。原核生物に基づく系は、本発明での、核酸配列、またはその同族ポリヌクレオチド、タンパク質、およびペプチドを産生させるための使用に利用することができる。多くのそのような系は広く市販されている。発現系の他の例には、T7、Tac、Trc、BAD、λpL、テトラサイクリン、またはLacプロモーター等の強力な原核生物プロモーターを含むベクター、pET発現系、および大腸菌発現系が含まれる。
【0102】
E. 候補の結合タンパク質および抗体
本発明の特定の局面において、標的リガンドに潜在的に結合し得るタンパク質および短いペプチドを含む候補の抗体または他の組換えポリペプチドは、宿主細菌細胞の細胞質膜上に発現する。そのような抗体または他の結合ポリペプチドの異種性集団を発現させることにより、標的リガンドに対して高い親和性を有する抗体が同定され得る。同定された抗体は、次に、本明細書に記載するように様々な診断用途または治療用途に使用することが可能である。
【0103】
本明細書で使用する「抗体」という用語は、IgG、IgM、IgA、IgD、およびIgE等の任意の免疫学的結合因子を広く指すよう意図される。「抗体」という用語はまた、抗原結合領域を有する任意の抗体様分子を指すためにも使用され、これにはFab'、Fab, F(ab')2、一本鎖ドメイン抗体(DAB)、Fv、scFv(一本鎖Fv)等の抗体断片、ならびに二価抗体、三価抗体、および多価抗体等の操作された多価抗体断片が含まれる。抗体に基づいた様々な構築物および断片を調製および使用するための技法は、当技術分野において周知である。抗体を調製および特徴づけするための手段もまた、当技術分野において周知である(例えば、Antibodies: A Laboratory Manual、Cold Spring Harbor Laboratory、1988;参照として本明細書に組み入れられる、を参照のこと)。
【0104】
標的リガンドに対する親和性を有する抗体が同定されたならば、必要に応じて、ろ過、遠心分離、およびHPLCまたはアフィニティークロマトグラフィー等の様々なクロマトグラフィー法を用いて、抗体またはリガンド結合ポリペプチドを精製することができる。抗体を含むそのようなポリペプチドの断片は、ペプシンまたはパパイン等の酵素による消化を含む方法によるか、または化学的還元によるジスルフィド結合の切断によって、そのように産生された抗体から得ることができる。または、本発明の包含する、タンパク質断片を含む抗体または他のポリペプチドは、自動ペプチド合成機を使用して合成することも可能である。
【0105】
分子クローニングアプローチは、本発明に従ってその後標的リガンドに対する親和性に関してスクリーニングされ得る候補抗体の異種性集団を作製するための1つの適切な方法を含む。本発明の1つの態様では、動物の脾臓から単離されたRNAから、コンビナトリアル免疫グロブリンファージミドを調製することができる。スクリーニングされるべきリガンドで動物を免疫することにより、アッセイを特定の抗原に標的することが可能である。従来技術に勝る本アプローチの恩典は、1ラウンドで約104倍の抗体を産生させスクリーニングすることができる点、およびH鎖およびL鎖の組み合わせによって、適切な抗体の発見の機会をさらに増す新たな特異性が産生される点である。
【0106】
VIII. 核酸の操作および検出
本発明の特定の態様では、核酸を操作、単離、および/または検出するための1つまたは複数の技法を使用することが所望され得る。そのような技法には、例えば、宿主細胞を形質転換するためのベクターの調製、およびトランスジェニック細胞から選択された核酸部分をクローニングする方法が含まれ得る。そのような操作を実施するための方法は、本開示を考慮して当業者に周知であろう。
【0107】
増幅用の鋳型として使用する核酸は、標準的な方法に従って細胞、組織、または他の試料から単離することができる(Sambrookら、1989)。特定の態様においては、鋳型核酸を実質的に精製せずに、細胞全体、または組織ホモジネートまたは生体液試料で解析を行うことが可能である。核酸は、ゲノムDNAまたは分画したRNAもしくは全細胞RNAであってよい。RNAを使用する場合、まずRNAを相補DNAに変換することが望ましい場合がある。
【0108】
本明細書で用いる「プライマー」という用語は、鋳型依存的過程において、新生核酸の合成をプライミングし得る任意の核酸を包含することが意図される。典型的には、プライマーは10〜20および/または30塩基対長のオリゴヌクレオチドであるが、より長い配列も使用できる。プライマーは、二本鎖および/または一本鎖の形態で提供され得るが、一本鎖の形態が好ましい。
【0109】
選択した核酸配列に相当する核酸に選択的にハイブリダイズするように設計したプライマー対を、選択的ハイブリダイゼーションを可能にする条件下で、鋳型核酸と接触させる。所望の用途に応じて、プライマーに完全に相補的な配列へのハイブリダイゼーションのみを可能にする高ストリンジェンシー条件を選択してもよい。他の態様では、プライマー配列との1つまたは複数のミスマッチを含む核酸の増幅を可能にするため、ストリンジェンシーを下げてハイブリダイゼーションを行ってもよい。ハイブリダイズしたならば、鋳型-プライマー複合体を、鋳型依存的核酸合成を促進する1つまたは複数の酵素と接触させる。十分量の増幅産物が産生されるまで、「サイクル」とも称される複数ラウンドの増幅を行う。
【0110】
増幅産物を検出および定量することができる。特定の適用では、検出は視覚的手段によって行われ得る。または、検出は、取り込まれた放射標識または蛍光標識の化学発光、放射性シンチグラフィーを介した、またはさらに電気インパルス信号および/または熱インパルス信号を用いる系を介した、産物の間接的同定を含み得る(Affymax技術;Bellus、1994)。
【0111】
所与の鋳型試料中に存在するオリゴヌクレオチド配列を増幅するために、多くの鋳型依存的過程が利用可能である。最もよく知られている増幅法はポリメラーゼ連鎖反応法(PCR(商標)と称される)であり、これは米国特許第4,683,195号、第4,683,202号、および第4,800,159号、ならびにInnisら、1988に詳細に記載されており、これらはそれぞれその全体が参照として本明細書に組み入れられる。
【0112】
逆転写PCR(商標)増幅手順は、増幅したmRNAの量を定量化するために実施され得る。RNAをcDNAに逆転写する方法は周知である(Sambrookら、1989を参照のこと)。逆転写の別法では、熱安定性DNAポリメラーゼを利用する。これらの方法は、国際公開公報第90/07641号に記載されている。ポリメラーゼ連鎖反応法は、当技術分野で周知である。RT-PCRの代表的な方法は、米国特許第5,882,864号に記載されている。
【0113】
増幅の別の方法はリガーゼ連鎖反応法(「LCR」)であり、これは、その全体が参照として本明細書に組み入れられる欧州出願第320 308号に開示されている。米国特許第4,883,750号では、標的配列にプローブ対を結合させるための、LCRに類似した方法について記載している。米国特許第5,912,148号に開示されている、PCR(商標)およびオリゴヌクレオチドリガーゼアッセイ(OLA)に基づく方法も用いることが可能である。
【0114】
本発明を実施する上で使用できる、標的核酸配列を増幅するための別法は、米国特許第5,843,650号、第5,846,709号、第5,846,783号、第5,849,546号、第5,849,497号、第5,849,547号、第5,858,652号、第5,866,366号、第5,916,776号、第5,922,574号、第5,928,905号、第5,928,906号、第5,932,451号、第5,935,825号、第5,939,291号、および第5,942,391号、英国出願第2202 328号、ならびにPCT出願PCT/US89/01025に開示されており、これらはそれぞれその全体が参照として本明細書に組み入れられる。
【0115】
PCT出願PCT/US87/00880に記載されているQbetaレプリカーゼもまた、本発明における増幅法として使用することが可能である。この方法では、標的の配列に相補的な領域を有するRNAの複製配列を、RNAポリメラーゼの存在下で試料に添加する。ポリメラーゼが複製配列をコピーすることになり、次にこれを検出することができる。
【0116】
制限エンドヌクレアーゼおよびリガーゼを用いて、制限酵素部位の一方の鎖内にヌクレオチド5'-[α-チオ]-三リン酸を含む標的分子の増幅を達成する等温増幅法もまた、本発明における核酸の増幅に有用である場合がある(Walkerら、1992)。米国特許第5,916,779号に開示されている鎖置換増幅(SDA)は、複数ラウンドの鎖置換および合成、すなわちニックトランスレーションを含む、核酸の等温増幅を実施する別の方法である。
【0117】
他の核酸増幅手順には、核酸配列に基づく増幅(NASBA)および3SR(Kwohら、1989;Gingerasら、国際公開公報第88/10315号、その全体が参照として本明細書に組み入れられる)を含む、転写に基づく増幅系(TAS)が含まれる。欧州出願第329 822号では、一本鎖RNA(「ssRNA」)、ssDNA、および二本鎖DNA(dsDNA)を繰り返して合成する段階を含む核酸増幅過程を開示しており、本発明に基づいてこの過程を使用することが可能である。
【0118】
国際公開公報第89/06700号(その全体が参照として本明細書に組み入れられる)では、標的一本鎖DNA(「ssDNA」)に対するプロモーター領域/プライマー配列のハイブリダイゼーション、それに続くその配列の多くのRNAコピーの転写に基づいた核酸配列増幅の図式を開示している。この図式は周期的ではない、すなわち、生じたRNA転写産物から新たな鋳型が産生されない。他の増幅法には、「race」および「片側PCR」が含まれる(Frohman、1990;Oharaら、1989)。
【0119】
IX. 実施例
以下の実施例は、本発明の好ましい態様を実証するために含めるものである。以下の実施例において開示する技法により、本発明者らが発見した技法が本発明の実施に際して十分に機能することが示され、したがって以下の実施例において開示する技法がその実施の好ましい様式を構成すると見なされ得ることが、当業者によって理解されるべきである。しかし当業者は、本開示を踏まえて、開示する特定の態様において多くの変更がなされ得り、本発明の精神および範囲から逸脱することなくなお同様または類似の結果が得られることを理解すべきである。
【0120】
実施例1
固定化ペリプラズム発現が低分子およびペプチドを標的することの実証
APExにより提示されたscFvが低分子およびペプチドを標的する能力を図1A〜図1Bおよび図1Cにそれぞれ示す。NlpAの最初の6アミノ酸(APEx解析のため内膜標的配列として機能する)の、抗メタンフェタミン、抗ジゴキシン、または抗ペプチドscfvのいずれかへの融合体を含む3つの大腸菌培養物を培養し、以下に記載するようにタンパク質発現を誘導した。次に、各構築物の細胞を、5×PBS緩衝液中で200 nM濃度のメタンフェタミン-FL(図1A)、ジゴキシゲニン-bodipy(図1B)、または200 nMペプチド(18 mer)-BodipyFL(図1C)で標識した。提示したデータは、各標識細胞培養物による10,000イベントのヒストグラム表示を示す。結果から、APExによって提示されたscfvが、非特異的リガンドに対する交差反応性を最小限に抑えつつ、その特異的抗原複合化フルオロフォアに結合する能力が実証される。
【0121】
実施例2
固定化ペリプラズム発現によるAb断片の認識の実証
scFvがより大きなタンパク質に接近できることを実証するため、まず、ヒトAb断片またはマウスAb断片に対するポリクローナル抗体血清が、固定化ペリプラズム発現によって大腸菌内膜上に提示されたそれぞれに由来するscFvを認識することを実証した。固定化ペリプラズム発現によりマウス由来scFvを発現する大腸菌(図2A)または固定化ペリプラズム発現によりヒト由来scFvを発現する大腸菌(図2B)を、以下に記載するように、抗マウスポリクローナルIgG(H+L)-Alexa-FLまたは抗ヒトポリクローナルIgG(Fab)-FITCのいずれかで標識した。それぞれ10000イベントのヒストグラム表示形態における結果から(図2A、図2B)、抗ヒトポリクローナル(約150 kDaの大きさ)がヒト由来scFvを特異的に認識し、抗マウスポリクローナル(150 kDa)がマウス由来scFvを認識することが実証された。
【0122】
実施例3
固定化ペリプラズム発現によって提示されるscFvが、大きな抗原複合化フルオロフォアに特異的に結合する能力の実証
固定化ペリプラズム発現によって提示されるscFvが、大きな抗原複合化フルオロフォアに特異的に結合する能力を実証するため、固定化ペリプラズム発現によって抗防御抗原(PA) scFv(PAは炭疽菌毒素の一成分である:83 kDaタンパク質)または抗ジゴキシゲニンscFvを発現する大腸菌を、以下に示すように誘導し標識した。10,000イベントのヒストグラムデータから、抗ジゴキシゲニンscFvを発現する細胞と比較して、PA-Cy5抗原複合化フルオロフォアへの特異的な結合が実証された(図3A)。この点をさらに明らかにするため、ジゴキシゲニンを240 kDa蛍光タンパク質であるフィコエリトリン(PE)に結合した。以下に示すように、細胞をこの複合体で標識した。固定化ペリプラズム発現により抗ジゴキシゲニンscFvを発現する大腸菌(10,000イベント)が大きなPE-ジゴキシゲニン複合体で標識されるのに対し、固定化ペリプラズム発現によって非特異的scFvを発現する大腸菌は蛍光をほとんど示さないことが認められた(図3B)。
【0123】
実施例4
フローサイトメトリー選択によるscFvライブラリーからの改良scFv変種の選択の実証
固定化ペリプラズム発現によってメタンフェタミンに対する親和性を有するscFvの変異ライブラリーを発現するポリクローナル大腸菌のスキャンを行った。メタンフェタミン複合化フルオロフォアを用いた選別および再選別を2ラウンド行うことにより、ライブラリーの亜集団が単離された(図4)。
【0124】
このライブラリーに由来する個々のクローンを同じメタンフェタミンフルオロフォアで標識し、以下のように解析した。親の抗メタンフェタミンscFvよりも高い平均FLシグナルを有する、変異体9と命名したクローンの例を図5に示す。
【0125】
実施例5
材料および方法:
A. ベクターの構築
リーダーペプチドおよび成熟NlpAタンパク質の最初の6アミノ酸は、プライマー
、VENTポリメラーゼ(New England Biolabs)、およびdNTP(Roche)を用いて、XL1-ブルー大腸菌(Stratagene)において全細胞PCR(Perken Elmer)を行うことにより作製した。次いで、これをNde1およびSfi1制限酵素エンドヌクレアーゼで切断し、マルチクローニングサイト(MCS)の下流に、エレメント(mycおよびhisタグ、Cm耐性マーカー、colE1起点、ならびにlac I)を有する大腸菌発現ベクターのlacプロモーターとMCSとの間にクローニングした。次に、関心対象のscFvをMCS内にクローニングし、このベクターをAbleC大腸菌(Stratagene)に形質転換した。
【0126】
B. 発現
TB培地+2%グルコースおよび30 mg/lクロラムフェニコール中に、大腸菌をOD600が0.1になるまで播種する。細胞を37℃で2時間培養した後、30分間で温度を25℃に下げる。次に、1 mM IPTGにより細胞を25℃で4時間誘導する。
【0127】
元のscFv配列を鋳型として利用し、Fromant Mら(1995)によって記載されているように、変異原性PCR法によりscFv配列の変異ライブラリーを構築した。次に、これらの変異産物を上記のAPEx発現ベクターにクローニングし、ABLEC大腸菌に形質転換し、2%グルコースおよび30 ug/mlクロラムフェニコールを含むSOC培地で寒天プレートにプレーティングした。30℃で一晩インキュベートした後、プレートから大腸菌を掻き取り、15%グリセロールアリコート中で凍結し、後にフローサイトメトリー選別するために-80℃で保存した。
【0128】
C. 標識手順
誘導した後、細胞を200 nMプローブを添加した5×PBS中で45分間インキュベートするか、または350μlの0.75 Mショ糖、100 mM Tris中に再懸濁する。次に10 mg/mlリゾチーム35μlを添加し、続いて穏やかに振盪しながら1 mM EDTA 700μlを1滴ずつ添加する。氷上で10分間静置させた後、0.5 M MgCl2を50μl添加する。さらに10分間氷上で静置させた後、懸濁液を13,200gで1分間遠心分離し、上清を除去し、1×PBS 500μl中に再懸濁する。次に細胞を200 nMプローブで45分間標識し、その後フローサイトメトリーにより解析し、改善された蛍光を選択する。
【0129】
D. 株およびプラスミド
APExによるスクリーニングには、株ABLE(商標)C(Stratagene)を使用した。大腸菌株TG1およびHB2151は、Griffinライブラリーと共に提供された。ABLE(商標)CおよびABLE(商標)KはStratageneから購入し、ファージM13K07はPharmaciaから購入した。ファージディスプレイビヒクルのFACS解析の陽性対照を、pHEN2内の既存のscFvを26.10 scFvに置換することにより構築し、pHEN2.digを作製した。陰性対照は、Griffin.1ライブラリーと共に提供された、抗サイログロブリンscFvを有するpHEN.thyである。PtacベクターはpIMS120の誘導体である(Hayhurst、2000)。
【0130】
E. ファージのパニング
Griffin.1ライブラリーは、CDR3ループの一部またはすべてがランダムに変異され、インビボで組換えられたヒト重鎖および軽鎖の大きなレパートリーに由来する半合成scFvライブラリーである(Griffithsら、1994)。このライブラリーは、本発明に基づく固定化ペリプラズム発現によるスクリーニングのための、候補結合ポリペプチドの1つの可能な供給源を示す。以下の実施例9に概説するように、ウェブサイトの取り扱い説明(www.mrc-cpe.cam.ac.uk/~phage/glp.html)に従って、ライブラリーをレスキューし、5ラウンドのパニングに供した。10μgml-1ジゴキシン-BSA複合体でイムノチューブをコーティングし、中和した溶出液を等分し、次のラウンドのファージパニング用にTG-1を、またはFAC解析用にABLE(商標)Cを感染させるために使用した。
【0131】
抗原結合ファージの濃縮を示すため、溶出液の力価をモニタリングした。反応性を確認するため、精製して力価を標準化した、各ラウンドで生じるファージ保存液のポリクローナルファージELISAを、ジゴキシン-オボアルブミン複合体で実施した。第3、第4、および第5ラウンドで生じた陽性クローンの割合は、各ラウンド後の96個の単離体のモノクローナルファージELISAにより決定した。陽性を、バックグラウンドシグナルが0.01を超えることはほとんどない、0.5よりも大きい吸光度と任意に定義した。第3、第4、および第5ラウンドによる24個の陽性クローンに対して、MvaIフィンガープリントを実施した。
【0132】
F. FACSスクリーニング
APEx発現をスキャンするため、B項およびC項に記載したように、APEx構築物を有する大腸菌のグリセロール保存液を培養して標識した。標識した後、細胞をPBSで一度洗浄し、スキャンした。bodipyまたはFL標識抗原を用いた上記の研究では、励起するために488 nmレーザーを使用し、Cy5には633 nmレーザーを使用した。機器を以下のように設定し、FACSCalibur(BD)でスキャンを達成した:側方散乱トリガーV 400、閾値250、前方散乱E01、FL1 V 400 FL2 V400(488 nm ex)、FL4 V 700(633 nm ex)。
【0133】
APEx発現の選別は以下の通りであった。選別はすべて、MoFlo FC(Cytomation)を用いて行った。B項およびC項に記載したように、前記ライブラリーを培養して標識し、PBSで一度洗浄し、FL強度が増加したものを選別した。集団のポリクローナルスキャンが濃縮を示すまで、次のラウンドの選別を行った(図4を参照のこと)。次に、個々のクローンを拾い上げ、FL活性を解析した。
【0134】
他の研究のため、ファージミド含有ABLE(商標)Cグリセロール保存液のアリコートを、600 nmにおけるODが約0.1 cm-1になるように1 mlの2×TY(2%グルコース、100μgml-1アンピシリン)中に掻き取った。37℃で2時間激しく振盪した後、1 mMになるようにIPTGを添加し、培養物を25℃で4時間振盪した。培養物50μlを、室温で1時間、5×PBS 1 ml中で適度に撹拌しながら、100 nM BODIPY(商標)-ジゴキシゲニンで標識した(Daughertyら、1999)。標識の最後の10分間、ヨウ化プロピジウムを2μg/ml-1になるように添加した。細胞をペレット化し、標識混合液100μl中に再懸濁した。Becton-Dickinson FACSortでスキャンを行い、1500s-1に104イベントを回収した。
【0135】
FACSライブラリーを選別するため、テリフィックブロス中で細胞を培養し、0.1 mM IPTGで誘導した。1000 s-1の排除モードで、106イベントに対して(第2ラウンドでは107)選別を実施した。回収された選別溶液を0.7μm膜フィルターに通して、適切な抗生物質を添加したSOC寒天の表面上にフィルターを乗せた後、コロニーを30℃で24時間培養した。
【0136】
E. ファージクローンの解析
ELISAによるファージ粒子のスクリーニングは、以下のように要約される。ELISAにおけるファージの結合を、一次ヒツジ抗M13抗血清(CP laboratoriesまたは5 prime ? 3 prime)、その後の西洋ワサビペルオキシダーゼ(HRP)複合化抗ヒツジ抗体(Sigma)により検出する。または、HRP-抗M13複合体を用いることも可能である(Pharmacia)。2% MPBSまたは3% BSA-PBSでプレートをブロッキングできる。ポリクローナルファージELISAに関しては、その技法は一般に以下の通りである:MicroTest III可塑性アッセイプレート(Falcon)を、ウェルあたり100μlのタンパク質抗原でコーティングする。抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし4℃で一晩コーティングする。ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、37℃で2時間、ウェルを2% MPBSまたは3% BSA-PBSで満たす。PBSでウェルを3回濯ぐ。選択の各ラウンドの終了時に保存したファージのアリコートから、PEG沈殿したファージ10μlを添加する(約1010 tfu)。2% MPBSまたは3% BSA-PBSで100μlに調整する。室温で90分間インキュベートする。被験溶液を除去し、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。2% MPBSまたは3% BSA-PBS中に適切に希釈したHRP-抗M13またはヒツジ抗M13抗血清を添加する。室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。ヒツジ抗M13抗血清を用いる場合には、2% MPBSまたは3% BSA中に適切に希釈したHRP-抗ヒツジ抗血清と共に室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液、pH 6.0、使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0137】
モノクローナルファージELISAは、以下のように要約され得る。モノクローナルファージ抗体を同定するため、pHENファージ粒子をレスキューする必要がある:C10のプレートから(各ラウンドの選択後)、96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを播種し、30℃で振盪しながら(300 rpm)一晩培養する。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび1%グルコースを含む2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。37℃で振盪しながら1時間培養する。最終濃度が15%になるようにグリセロールを添加し、次いでこのプレートを-70℃で保存することによって、元の96ウェルプレートのグリセロール保存液を作製する。(2枚目のプレートの)各ウェルに、感染効率(moi)10となるようにVCS-M13またはM13KO7ヘルパーファージを添加する。37℃で30分間静置する。1,800gで10分間遠心分離した後、上清を吸引除去する。100μg/mlアンピシリンおよび50μg/mlカナマイシンを含む2×TY 200μl中に、ペレットを再懸濁する。30℃で振盪しながら一晩培養する。1,800gで10分間遠心分離し、上清100μlを先に詳述したファージELISAにおいて使用する。
【0138】
抗体断片の産生は以下のように要約される。選択されたpHENは、HB2151に感染させ、次いでELISA用に抗体断片の可溶性発現が起こるように誘導する必要がある。各選択から溶出されたファージ10μl(約105 t.u.)を採取し、対数増殖中のHB2151細菌200μlに37℃で30分間感染させる(湯浴)。100μg/mlアンピシリンおよび1%グルコースを含むTYE上に、1μl、10μl、100μl、および1:10希釈物をプレーティングする。これらのプレートを37℃で一晩インキュベートする。96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを釣菌し、37℃で振盪しながら(300 rpm)一晩培養する。別のプレートに接種するために使用したならば、最終濃度が15%となるようにグリセロールを添加し、-70℃で保存することによって、このプレートのグリセロール保存液を作製できる。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび0.1%グルコースを含む新鮮2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。600 nmにおけるODが約0.9になるまで(約3時間)、37℃で振盪しながら培養する。所要のODに達したならば、100μg/mlアンピシリンおよび9 mM IPTG(最終濃度1 mM IPTG)を含む2×TY 25μlを添加する。30℃でさらに16時間〜24時間振盪し続ける。ウェル当たり100μlのタンパク質抗原で、MicroTest III可塑性アッセイプレート(Falcon)をコーティングする。
【0139】
抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし、室温で一晩コーティングする。翌日、ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、ウェル当たり200μlの3% BSA-PBSで37℃で2時間ブロッキングする。細菌プレートを1,800gで10分間遠心分離し、上清(可溶性scFvを含む)100μlをELISAプレートに添加し、室温で1時間インキュベートする。被験溶液を除去し、PBSで3回洗浄する。1% BSA-PBS中で濃度4μg/mlの精製9E10抗体(mycタグ化抗体断片を検出する)50μl、および1% BSA-PBS中で1:500希釈したHRP-抗マウス抗体50μlを添加する。室温で60分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液(pH 6.0)。使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する。)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0140】
ライブラリー中の挿入物は、
と命名したプライマーを用いてPCRスクリーニングすることにより、スクリーニングできる。VHおよびVLの配列決定に関しては、プライマー
の使用を推奨する。
【0141】
実施例6
抗体親和性成熟
Shortら(1995)は、表面プラズモン共鳴により測定される、ジゴキシンに対する平衡解離定数(KD) が300 pMである、A4-19と命名された26-10変異体を単離した。A4-19は、重鎖CDR1内に3つのアミノ酸置換を含む(VH:T30→P、VH:D31→S、およびVH:T34→Y)。既に非常に堅固な結合を示す抗体から開始する場合でさえも、可溶性ペリプラズム発現/FACSスクリーニングによって、結合親和性の増大した変異体が得られ得るかどうかを試験した。ジゴキシンハプテンに接触するか(VL:T91、VL:P96)または近接する(VL:V94)3つの軽鎖CDR3残基を(Jeffreyら、1993)、NNS(S=GまたはC)戦略を用いてランダム化した(Daughertyら、1998)。pelBリーダーによりペリプラズム中に発現する2.5×106個の形質転換体のライブラリーを作製し、2ラウンドのFACSによりスクリーニングした。1ラウンド目のスクリーニングでは、100 nMの蛍光プローブで標識した細胞をPBSで1度洗浄し、非蛍光粒子が蛍光粒子と同じ液体エレメント内に検出される場合にも、機器がすべての蛍光イベントを回収するという回収様式を用いて選別した。回収様式での操作により、純度は犠牲になるものの、非常に稀な細胞が回収されることが十分に保証された。
【0142】
回収した細胞を再度培養して標識し、洗浄し、次いで50倍過剰(50μM)の遊離ジゴキシンと共に様々な時間(15分〜90分)インキュベートした。同時発生的蛍光イベントおよび非蛍光イベントを拒絶し、それにより高度の純度が得られる排除様式を用いた選別により、所望のレベルの蛍光を保持する細胞を単離した。非蛍光競合物質と様々な時間インキュベートした後に得られた細胞プールの蛍光減衰速度を測定した。初期の時点で(競合物質とのインキュベーション<60分)、開始のA4-19抗体と比較してわずかに速い速度が認められたが、60分および90分の集団では速度が減少した。競合60分後に得られた細胞集団由来の5個のランダムなクローンおよび90分プールに由来する13クローンを無作為に拾い、配列決定した(表1)。強い配列共通性が明らかに明白であった。精製抗体のハプテン結合反応速度をSPRにより決定し、その結果を表1に示す。対応するアミノ酸配列を、配列番号:8〜19に示す。ゲル濾過FPLCによる精製および解析の際に、どの変異体も二量体化することが認められなかったことに留意されたい。試験した変異体はすべて、開始のA4-19抗体の結合速度定数(0.9±0.2x106 M-1)とは識別できない結合速度定数(kon)を示した。競合60分後に単離されたクローンのkdissは、A4-19のkdissと同じか、またはそれよりも速かった。競合90分後に単離されたクローンは、溶液中でより遅いkdissを示した。1つのクローン90.3は、150 pMのKDを生じる2倍遅い解離速度定数を示した。したがって、本発明のライブラリースクリーニング方法によって、既にナノモル以下のKDを示す抗体から開始する場合でも、特異的標識によりさらに優れた変異体の単離が可能となった。驚くほどのことではないが、興味深いことに、4-19に存在する3つの重鎖CDR1変異および軽鎖の残基94および96における2つの変異の効果は、相加的であった。
【0143】
(表1)60分(クローン60.1〜60.4)および90分(クローン90.1〜90.6)オフレート選択により単離された、重鎖および軽鎖CDR3アミノ酸配列(配列番号:8〜20)。括弧内に同一クローン数を示す。ND:未検。
【0144】
実施例7
蛍光シグナルの最大化
ペリプラズム中に可溶型のscFv抗体を発現する細胞の蛍光強度は、用いた大腸菌株および培養条件に強く依存した。26-10抗体を用いると、細胞を25℃で培養した場合に最大蛍光強度が得られた。準生理的温度での培養は、いくつかの有益な効果を有する。低い温度(すなわち25℃)でscFvを発現させることにより、折りたたみ経路を遅くすることによって直接的に、および発現の負荷を低減するためにプラスミドのコピー数を減少することによって間接的に、scFvの適切な折りたたみが促進される。実際に、37℃でのscFvの直接発現では、一般に可溶性タンパク質はほとんどまたは全く産生されない(例えば、Goughら、1999を参照のこと)。外膜の組成もまた非生理的温度で変わり、透過性の増大が生じる(Martinezら、1999)。それどころかむしろ、様々な大腸菌株間の著しい相違に注目した。試験した様々な株の中で、ABLE(商標)Cにおいて最も高い蛍光強度が得られた。この株におけるタンパク質発現および外膜タンパク質特性の予備的な解析から、高い蛍光シグナルはColE1起点プラスミドのコピー数を減少させるpcnB変異に起因せず、むしろ細胞外被のタンパク質組成における相違に起因することが示された。実際に、ABLE(商標)Cのより強力な染色は、ELISAおよびウェスタンブロッティングによって推定されるように、他の株に対する高レベルのタンパク質発現とは関連していなかった。
【0145】
高浸透圧条件下で蛍光標識することにより、有意に高い蛍光が生じた。標識中に細胞を5×PBS中でインキュベートした場合、5〜7倍の蛍光の増加が得られた(通常のPBS中でインキュベートした細胞の平均FL1が20〜30であるのに対し、平均FL1>150)。しかし、細胞の生存度がかなり減少したため、増大したシグナルは損なわれた。タンパク質の高度に多様化したライブラリーをスクリーニングする場合、そのような生存度の減少は所望されない場合があり、その発現は宿主細胞に対する有害な効果を既に有している可能性がある。同様に、M13KO7等の繊維状ファージとの同時感染によってファージショック応答が誘導され、特に外膜透過性の増大が生じる。M13KO7の感染により、集団の平均蛍光において3倍の増大が生じた。しかし、高浸透圧ショックを用いた場合と同様に、ヨウ化プロピジウム染色によって決定される培養物の生存度は若干減少した。
【0146】
蛍光リガンドによる細胞の標識、それに続く大過剰の遊離リガンドとのインキュベーションにより、平均蛍光強度が時間依存的に減少する。蛍光減衰の速度は、抗体-抗原複合体の解離速度を反映する(Daughertyら、2000)。ジゴキシンに関して、蛍光減衰の速度が、BIACOREを使用して精製抗体で測定した解離速度と比較して約3〜4倍遅いことが認められた。インビトロにおける抗体/抗原複合体の解離速度と比較して蛍光減衰の速度が低いことは、リガンドと細胞との間の衝突頻度、ペリプラズムにおける抗体の濃度、および当然、外膜を介する拡散の速度を含む様々な効果に由来する(ペリプラズム空間における反応速度論の解析に関しては、Martinezら、(1996)を参照のこと)。予測され得るように、インビトロで決定したkoff速度に対するペリプラズムにおける蛍光減衰の速度の比は、抗原に依存する。
【0147】
実施例8
ペリプラズム中に可溶型のscFv抗体を発現する細胞の蛍光検出および濃縮
26-10 scFv抗体は、ジゴキシンおよびジゴキシゲニン等の強心配糖体に高い親和性で結合する(ジゴキシンおよびジゴキシゲニンに対する精製抗体のKDは、それぞれ0.9±0.2×10-9 M-1および2.4±0.4×10-9 M-1である。Chenら、1999)。26-10 scFvおよびその変種は、ハプテン結合に及ぼすCDRおよびフレームワーク領域における変異の影響を理解するためのモデル系として広く用いられている(Schilbachら、1992;Shortら、1995;Daughertyら、1998、2000;Chenら、1999)。26-10 scFvの誘導体を、大腸菌アラビノースプロモーター下で、大腸菌ペリプラズムへの分泌を可能にするpelBリーダーペプチドと共に可溶型で発現させた。生じたプラスミドベクター(pBAD30pelB-Dig)をara-大腸菌株LMG194に形質転換し、0.2% w/vアラビノースでタンパク質合成を誘導した。200 nMのジゴキシゲニン-BODIPY(商標)と共にインキュベートした場合、25℃で培養した細胞が強く蛍光を発し、非特異的に結合したリガンドを除去するために十分に洗浄した後でさえも蛍光シグナルが保持されることが認められた。ペリプラズムにおける親水性溶質の透過に関して一般に受容される大きさの限界である約600 Da(DecadおよびNikaido、1976)よりも有意に高い分子量を有するプローブで細胞が標識されることから、蛍光シグナルが主に生存不能な透過可能となった細胞に起因する可能性が生じた。しかし、通常隔絶された核酸にインターカレートすることにより、膜損傷細胞に特異的に結合する生存度染色ヨウ化プロピジウムでの染色から、>90%の細胞がこの色素に透過性でないことが明らかになった。これは、対数期後期に回収した対照大腸菌培養物における無傷の細胞の割合と類似している。
【0148】
ペリプラズム中に可溶型の26-10抗体を発現する細胞を、1ラウンドの選別のみで、ベクターで形質転換した大過剰の大腸菌から濃縮し得た。具体的には、LMG194(pBAD30pelB-Dig)を、空ベクター(pBAD30)を含む10,000倍過剰の大腸菌と混合した。前者の細胞はアンピシリンおよびクロラムフェニコールの両方に耐性であるのに対して(ampr、Cmr)、後者はアンピシリンのみに耐性である(ampr)。0.2% w/vアラビノースで4時間誘導した後、細胞を100 nMのジゴキシゲニン-BODIPY(商標)で1時間標識し、FACSにより蛍光性の細胞を単離した。選別された細胞を再培養し上記のように再標識した後に、この集団は5〜8倍の平均蛍光強度の増加を示した(選別前の細胞混合物についてFL1=4であるのに対してFL1=20)。濃縮された集団におけるscFv発現クローンの画分を、Cmrでもあるamprクローンの数によって測定した。amprコロニーの80%はCmrでもあり、このことから、蛍光標識および細胞選別により1ラウンドで1,000倍をはるかに上回る濃縮が起こることが示された。
【0149】
実施例9
準最適温度における細胞透過性の増加
ペリプラズム中に可溶型のscFv抗体を発現する細胞の蛍光強度は、用いた大腸菌株および培養条件に強く依存した。26-10抗体を用いた場合、細胞を25℃で培養した場合に最大蛍光強度が得られた。準生理的温度での培養は、いくつかの有益な効果を有する。低い温度(すなわち25℃)でscFvを発現させることにより、折りたたみ経路を遅くすることによって直接的に、および発現の負荷を低減するためにプラスミドのコピー数を減少することによって間接的に、scFvの適切な折りたたみが促進される。実際に、37℃でのscFvの直接発現では、一般に可溶性タンパク質はほとんどまたは全く産生されない(例えば、Goughら、1999を参照のこと)。外膜の組成もまた非生理的温度で変わり、透過性の増大が生じる(Martinezら、1999)。それどころかむしろ、様々な大腸菌株間の著しい相違に注目した。試験した様々な株の中で、ABLE(商標)Cにおいて最も高い蛍光強度が得られた。この株におけるタンパク質発現および外膜タンパク質特性の予備的な解析から、高い蛍光シグナルはColE1起点プラスミドのコピー数を減少させるpcnB変異に起因せず、むしろ細胞外被のタンパク質組成における相違に起因することが示された。実際に、ABLE(商標)Cのより強力な染色は、ELISAおよびウェスタンブロッティングによって推定されるように、他の株に対する高レベルのタンパク質発現とは関連していなかった。
【0150】
高浸透圧条件下で蛍光標識することにより、有意に高い蛍光が生じた。標識中に細胞を5×PBS中でインキュベートした場合、5〜7倍の蛍光の増加が得られた(通常のPBS中でインキュベートした細胞の平均FL1が20〜30であるのに対し、平均FL1>150)。しかし、細胞の生存度がかなり減少したため、増大したシグナルは損なわれた。タンパク質の高度に多様化したライブラリーをスクリーニングする場合、そのような生存度の減少は所望されない場合があり、その発現は宿主細胞に対する有害な効果を既に有している可能性がある。同様に、M13KO7等の繊維状ファージとの同時感染によってファージショック応答が誘導され、特に外膜透過性の増大が生じる。M13KO7の感染により、集団の平均蛍光において3倍の増大が生じた。しかし、高浸透圧ショックを用いた場合と同様に、ヨウ化プロピジウム染色によって決定される培養物の生存度は若干減少した。
【0151】
蛍光リガンドによる細胞の標識、それに続く大過剰の遊離リガンドとのインキュベーションにより、平均蛍光強度が時間依存的に減少する。蛍光減衰の速度は、抗体-抗原複合体の解離速度を反映する(Daughertyら、2000)。ジゴキシンに関して、蛍光減衰の速度が、BIACOREを使用して精製抗体で測定した解離速度と比較して約3〜4倍遅いことが認められた。インビトロにおける抗体/抗原複合体の解離速度と比較して蛍光減衰の速度が低いことは、リガンドと細胞との間の衝突頻度、ペリプラズムにおける抗体の濃度、および当然、外膜を介する拡散の速度を含む様々な効果に由来する(ペリプラズム空間における反応速度論の解析に関しては、Martinezら、(1996)を参照のこと)。予測され得るように、インビトロで決定したkoff速度に対するペリプラズムにおける蛍光減衰の速度の比は、抗原に依存する。
【0152】
実施例10
FACSによるレパートリー抗体ライブラリーの解析およびスクリーニング
多種多様な抗体配列を含む大きなレパートリーライブラリーをスクリーニングすることにより、新規に、すなわち動物を免疫することなく、抗体を単離することができる。そのような大きなライブラリーのスクリーニングは、十分に確立されている(Nissimら、1994、Winterら、1994、Griffithら、1994、Knappikら、2000)。これまで、利用可能な大きな抗体レパートリーライブラリーはすべて、ファージディスプレイによる使用のために構築されてきた。しかし、ファージディスプレイ用に構築されたライブラリーは、内膜への固定化または可溶型での、細菌のペリプラズム空間内でのタンパク質発現のためにも使用することが可能である。特に、繊維状バクテリオファージにおける低タンパク質コピー数提示では、組換えポリペプチドをpIIIへのN末端融合物として発現させる。ファージ生合成過程において、pIII融合物はまずペリプラズムに標的され、pIIIの小さなC末端部分によって内膜に固定される。ファージが放出される際に、scFv-pIII融合物はファージの末端で野生型pIIIと同時に組み込まれ、これにより会合過程が完了する(RakonjacおよびModel、1998;Rakonjacら、1999)。ファージディスプレイに関して最も広く用いられるベクターでは、N末端scFvとpIII遺伝子との間にアンバーコドンが配置されている。したがって、適切な大腸菌サプレッサー株では、scFvを提示するために完全長のscFv-pIII融合タンパク質が産生されるが、非サプレッサー株では、可溶性scFvのみが発現する。または、融合物に内膜固定化ペプチドを含めることにより、固定化発現が達成され得る。
【0153】
ファージディスプレイでの抑制の程度はベクターおよび株によって変化するが、10%の読み過ごししか可能にしない傾向がある。したがって、ファージディスプレイの生物学の結果として、アンバーコドンを含むすべてのライブラリーは、宿主に関係なくある程度のペリプラズムの発現を生じる。よって、FACSが、既存の高度に多様化した天然ライブラリーからのリガンド結合タンパク質の単離に役立ち得るかどうかを検討することは、非常に興味深いことであった(Griffithsら、1994;Vaughanら、1996;Sheetsら、1998;Piniら、1998;de Haardら、1999;Knappikら、2000;SblatteroおよびBradbury、2000)。
【0154】
ファージパニングによるファージライブラリーの従来のスクリーニングにより、天然抗体レパートリーライブラリーから、強心配糖体ジゴキシンに特異的なscFvを発現するファージが濃縮された。BSA複合体に対してパニング過程を行い、オボアルブミン複合体に対してスクリーニングを行い、タンパク質およびハプテン-タンパク質界面の結合体の出現を減少させた。パン4由来の24個の陽性単離体は同じフィンガープリントを共有し、6クローンのDNA配列決定から、同じ重鎖配列および軽鎖配列が確認され(「dig1」)、6つのうち1つ(「dig2」)は特有のHCDR3とLCDR3との組み合わせを有した。同一条件下および異なる条件下でファージライブラリーを再度スクリーニングしたところ、同じDNAフィンガープリントを有するクローンの単離のみが生じた。
【0155】
パニングの各ラウンド後に大腸菌ABLE(商標)CでレスキューしたファージのFACS解析から、ファージELISAシグナルに酷似した、3ラウンドにおける平均蛍光の増加が示された。1ラウンドのFACSによる結合クローンの有意な濃縮は、3ラウンド目のファージパニングから得られた集団を用いて開始することにより得られた。この結果は、パニング実験の過程で得られた濃縮特性と一致する。第3、第4、および第5ラウンド由来の106個の細胞をFACSスクリーニングおよび選別することによって、それぞれ30%、80%、および100%の頻度で陽性クローンが単離された。
【0156】
第3ラウンドの集団からFACSによって単離された14クローンのうちの5つは、ジゴキシンに対する結合に関して陽性であることが認められた。重要なことには、このクローンのうちの3つは、ファージパニングで見逃された別の抗体に相当していた(本明細書において「dig3」と称する)。残りの2つは、dig1クローンであった。この結果から、ペリプラズム空間に発現し、蛍光リガンドによって標識されたライブラリーのFACSスクリーニングにより、他のライブラリースクリーニング法によって単離され得ないクローンが単離されることが実証される。
【0157】
実施例11
Griffin.1ライブラリーを使用するための方法の要約
Griffin.1ライブラリーを使用するための方法は、以下のように要約され得る。Griffin.1ライブラリーは、合成V遺伝子断片から作製されたscFvファージミドライブラリーである。このライブラリーは、重鎖および軽鎖可変領域をloxライブラリーベクター(Griffithsら、1994)からファージミドベクターpHEN2に再クローニングすることにより作製された。このライブラリーを使用するためのキットは、合成scFvライブラリーのチューブ(1 ml)、陽性対照(抗サイログロブリンクローンを含むTG1)のグリセロール保存液、陰性対照(pHEN2を含むTG1)のグリセロール保存液、ファージ粒子を増殖させるための大腸菌TG1(Gibson、1984)サプレッサー株(K12、del(lac-pro)、supE、thi、hsdD5/F'traD36、proA+B+、lacIq、lacZdelM15)のグリセロール保存液(供給される株は、このT-ファージ耐性変種である)、抗体断片を発現させるための大腸菌HB2151(Carterら、1985)および非サプレッサー株(K12、ara、del(lac-pro)、thi/F'proA+B+、lacIq、lacZdelM15)のグリセロール保存液を含む。このライブラリーは、必要時まで-70℃で凍結保存する。
【0158】
株をプレーティングし、次に100μg/mlアンピシリンおよび1%グルコースを含む2×TY中のそれぞれの一晩培養物として(37℃で振盪しながら)培養する。100μg/mlアンピシリンおよび1%グルコースを含む2×TY(2×TYは、1リットルあたり、トリプトン16 g、酵母エキス10 g、およびNaCl 5 g)で培養物を1:100に希釈し、以下に記載する手順に従ってファージミドをレスキューする。サイログロブリンでコーティングしたイムノチューブにおける1ラウンドの選択には、陽性対照および陰性対照の1:100混合物を共に用いる。
【0159】
ライブラリーを使用するための手順は、以下のように要約される。ファージ/ファージミドは、性繊毛を介してF+大腸菌に感染する。性繊毛の生成および効率的な感染のためには、大腸菌を37℃で培養し、これが対数期(600 nmにおけるODが0.4〜0.6)になければならない。以下の手順全体にわたり、そのような培養物が必要である。それは以下のように調製し得る:最小培地プレートから2×TY培地5 ml中に細菌コロニーを移し、37℃で振盪しながら一晩培養する。翌日、新鮮な2×TY培地中に1:100希釈することにより継代培養し、OD 0.4〜0.6まで37℃で振盪しながら培養し、次いでファージに感染させる。様々なヘルパーファージが、ファージミドライブラリーのレスキューに利用可能である。VCS-M13(Stratagene)およびM13KO7(Pharmacia)は小アリコートで購入することができ、ファージミドライブラリーをレスキューするために以下のように大量に調製することができる:OD 0.2の大腸菌TG1(または他の適切な株)200μlに、(十分に分離したプラークを得るために)ヘルパーファージの段階希釈物10μlを、37℃(温浴)で振盪せずに30分間感染させる。融解したH-トップアガー(42℃)3 mlを添加し、温めたTYE(注7)プレート上に注ぐ。静置した後、37℃で一晩インキュベートする。TG1の対数増殖培養物(上記参照)3〜4 ml中に、小プラークを採取する。37℃で振盪しながら約2時間培養する。2リットルフラスコ中の2×TY 500 ml中に接種し、上記のように1時間培養し、次いで最終濃度が50〜70μg/mlになるようにカナマイシン(25μg/ml水溶液)を添加する。さらに8〜16時間培養する。細菌を10,800 gで15分間遠心分離する。ファージの上清に1/5量のPEG/NaCl(20%ポリエチレングリコール6000-2.5 M NaCl)を添加し、氷上で最低30分間インキュベートする。10,800 gで15分間遠心分離する。ペレットをTE 2 ml中に再懸濁し、0.45μnフィルター(Minisart NML;Sartorius)を通して保存液をろ過滅菌する。保存液の力価を測定した後、約1×1012 p.f.u./mlになるように希釈する。-20℃でアリコートを保存する。特記されない限り、遠心はすべて4℃で行う。
【0160】
ライブラリーの増殖に関して、その手順は以下のように要約される:細菌ライブラリー保存液のすべて(約1×1010クローン)を、100μg/mlアンピシリンおよび1%グルコースを含む2×TY 500 mlに接種する。600 nmにおけるODが0.5になるまで、37℃で振盪しながら培養するが、これには約1.5〜2時間を要するはずである。1:20の比率でヘルパーファージを添加することにより(600 nmにおけるODが1の細菌=約8x108細菌/mlであることを考慮した、細菌細胞数:ヘルパーファージ粒子数)、この培養物25 ml(1x1010細菌)にVCS-M13またはM13KO7ヘルパーファージを感染させる。
【0161】
感染させた細胞を3,300 gで10分間遠心分離する。100μg/mlアンピシリンおよび25μg/mlカナマイシンを含む2×TY 30 ml中に、ペレットを穏やかに再懸濁する。100μg/mlアンピシリンおよび25μg/mlカナマイシンを含む予め温めた2×TY 470 mlを添加し、30℃で振盪しながら一晩インキュベートする。ポリエチレングリコール(PEG)6000で沈殿させることにより、ファージが濃縮され、あらゆる可溶性抗体が除去され得る(TG1において、抗体遺伝子とgIIIとの連結部においてコードされるアンバー終止コドンの抑制が決して完全ではないので)。A6からの培養物を10,800 gで10分間(または3,300 gで30分間)遠心分離する。上清に1/5量のPEG/NaCl(20%ポリエチレングリコール6000、2.5 M NaCl)を添加する。十分に混合し、4℃で1時間以上静置する。10,800 gで30分間遠心分離する。ペレットを水40 ml中に再懸濁し、PEG/NaCl 8 mlを添加する。混合し、4℃で20分以上静置する。10,800 gで10分間、または3,300 gで30分間遠心分離した後、上清を吸引除去する。短時間再遠心分離した後、残存するすべてのPEG/NaClを吸引除去する。ペレットをPBS 5 ml中に再懸濁し、微量遠心機で11,600 gで10分間遠心分離し、残りの細菌残屑のほとんどを除去する。短期保存の場合は4℃で、または長期保存の場合はPBS、15% グリセロール中で-70℃で、ファージ上清を保存する。ファージ保存液の力価を測定するため、PBS 1 ml中にファージ1μlを希釈し、このうちの1μlを用いて、OD600で0.4〜0.6のTG1 1 mlを感染させる。このうちの50μl、1:102希釈物の50μl、および1:104希釈物の50μlを、100μg/mlアンピシリンおよび1%グルコースを含むTYEプレート上にプレーティングし、37℃で一晩培養する。ファージ保存液は、1012〜1013/mlであるはずである。
【0162】
イムノチューブにおける選択は、以下のように要約される。Nuncイムノチューブ(Maxisorp カタログ番号4-44202)を、所要の抗原4 mlで一晩コーティングする。コーティングの効率は、抗原濃度、緩衝液、および温度に依存できる。通常、PBS中または50 mM炭酸水素ナトリウム、pH 9.6中の抗原10μg/ml〜100μg/mlを室温(rt)で用いる。翌日、PBSでチューブを3回洗浄する(単にチューブにPBSを注ぎ込み、次いでさらに直ちにそのPBSを捨てる)。2% MPBSでいっぱいになるまでチューブを満たす。カバーをし、37℃で(または抗原の安定性に応じて室温で)2時間インキュベートしてブロッキングする。PBSでチューブを3回洗浄する。2% MPBS 4 ml中に、A13からのファージ1012〜1013 cfuを添加する。上下回転台上で連続して回転させながら室温で30分間インキュベートし、次いで室温で少なくともさらに90分間静置する。上清中の非結合ファージを捨てる。1ラウンド目の選択では、0.1% Tween-20を含むPBSでチューブを10回洗浄し、次にPBSで10回洗浄して界面活性剤を除去する。各洗浄段階は、緩衝液を注ぎ込み、直ちに捨てることによって行う。2ラウンド目およびそれに続くラウンドでの選択では、0.1% Tween-20を含むPBSでチューブを20回洗浄し、次にPBSで20回洗浄する。チューブから過剰のPBSを振り出し、100 mMトリエチルアミン1 ml(水50 ml中のトリエチルアミン(7.18 M) 700μl、使用する日に希釈する)を添加することによってファージを溶出し、上下回転台上で10分間連続して回転させる。インキュベーションを行っている間に、1M Tris(pH 7.4)0.5 mlを含むチューブを予め調製しておき、即座に中和するために、7から溶出したファージ1 mlに添加する。ファージは4℃で保存することもできるし、または上記のようにTG1を感染させるために用いることもできる。溶出後、1M Tris(pH 7.4)200μlをイムノチューブにさらに添加し、チューブ内の残ったファージを中和する。TG1の対数増殖培養物を9.25 ml採取し、溶出したファージを0.75 ml添加する。また、TG1培養物4 mlをイムノチューブに添加する。両培養物を37℃(湯浴)で30分間、感染させるために振盪せずにインキュベートする。感染させたTG1細菌10 mlおよび4 mlをプールし、100μl採取して4つ〜5つの100倍段階希釈物を作製する。100μg/mlアンピシリンおよび1%グルコースを含むTYE上に、これらの希釈物をプレーティングする。37℃で一晩培養する。残りの感染TG1培養物を採取し、3,300 gで10分間遠心分離する。ペレット化した細菌を2×TY 1 ml中に再懸濁し、100μg/mlアンピシリンおよび1%グルコースを含むTYEの大きなNuncバイオアッセイディッシュ(Gibco-BRL(注8))上にプレーティングする。一晩またはコロニーが見えるまで、30℃で培養する。
【0163】
さらなる選択ラウンドのため、細胞のバイオアッセイディッシュに5 ml〜6 mlの2×TY、15%グリセロールを添加し、ガラススプレッダーで細胞をほぐす。100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100 mlに、掻き取った細菌50〜100μlを接種した後、残りの細菌を-70℃で保存する。ここでも、600 nmでの開始OD=<0.1であることを確認することが望ましい。600 nmでのODが0.5になるまで(約2時間)、37℃で振盪しながら細菌を培養する。1:20の比率でヘルパーファージを添加することにより(600 nmにおける1 OD細菌=約8×108細菌/mlであることを考慮した、細菌細胞数:ヘルパーファージ粒子数)、この培養物10 mlにVCS-M13またはM13KO7ヘルパーファージを感染させる。37℃の湯浴中、振盪せずに30分間インキュベートする。感染細胞を3,300 gで10分間遠心分離する。100μg/mlアンピシリンおよび25μg/mlカナマイシンを含む2×TY 50 ml中に、ペレットを穏やかに再懸濁し、30℃で振盪しながら一晩インキュベートする。一晩培養物40 mlを採取し、10,800 gで10分間、または3,300 gで30分間遠心分離する。上清に1/5量(8 ml)のPEG/NaCl(20%ポリエチレングリコール6000、2.5 M NaCl)を添加する。十分に混合し、4℃で1時間以上静置する。10,800 gで10分間、または3,300 gで30分間遠心分離した後、上清を吸引除去する。短時間再遠心分離した後、残存するすべてのPEG/NaCl屑を吸引除去する。ペレットをPBS 2 ml中に再懸濁し、微量遠心機で11,600 gで10分間遠心分離し、残りの細菌残屑のほとんどを除去する。このファージ1 mlを4℃で保存し、残りの1 mlアリコートを次の選択ラウンドに使用できる。この選択をさらに2〜3ラウンド繰り返す。
【0164】
ELISAによるファージ粒子のスクリーニングは、以下のように要約される。ELISAにおけるファージの結合を、一次ヒツジ抗M13抗血清(CP laboratoriesまたは5 prime ? 3 prime)、その後の西洋ワサビペルオキシダーゼ(HRP)複合化抗ヒツジ抗体(Sigma)により検出する。または、HRP-抗M13複合体を用いることも可能である(Pharmacia)。2% MPBSまたは3% BSA-PBSでプレートをブロッキングできる。ポリクローナルファージELISAに関しては、その技法は一般に以下の通りである:MicroTest III可塑性アッセイプレート(Falcon)を、ウェルあたり100μlのタンパク質抗原でコーティングする。抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし室温で一晩コーティングする。ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、ウェル当たり200μlの2% MPBSまたは3% BSA-PBSで37℃で2時間ブロッキングする。PBSでウェルを3回濯ぐ。選択の各ラウンドの終了時に保存したファージのアリコートから、PEG沈殿したファージ10μlを添加する(約1010 cfu)。2% MPBSまたは3% BSA-PBSで100μlに調整する。室温で90分間インキュベートする。被験溶液を除去し、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。2% MPBSまたは3% BSA-PBS中に適切に希釈したHRP-抗M13またはヒツジ抗M13抗血清を添加する。室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。ヒツジ抗M13抗血清を用いる場合には、2% MPBSまたは3% BSA中に適切に希釈したHRP-抗ヒツジ抗血清と共に室温で90分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液(pH 6.0)。使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0165】
モノクローナルファージELISAは、以下のように要約され得る。モノクローナルファージ抗体を同定するため、pHENファージ粒子をレスキューする必要がある。C10のプレートから(各ラウンドの選択後)、96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを播種し、37℃で振盪しながら(300 rpm)一晩培養する。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび1%グルコースを含む2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。37℃で振盪しながら1時間培養する。最終濃度が15%になるようにグリセロールを添加し、次いでこのプレートを-70℃で保存することによって、元の96ウェルプレートのグリセロール保存液を作製する。(2枚目のプレートの)各ウェルに、100μg/mlアンピシリン、1%グルコース、および109 pfu VCS-M13またはM13KO7ヘルパーファージを含む2×TY 25μlを添加する。37℃で30分間静置した後、37℃で1時間振盪する。1,800gで10分間遠心分離した後、上清を吸引除去する。100μg/mlアンピシリンおよび50μg/mlカナマイシンを含む2×TY 200μl中に、ペレットを再懸濁する。30℃で振盪しながら一晩培養する。1,800gで10分間遠心分離し、上清100μlを先に詳述したファージELISAにおいて使用する。
【0166】
可溶性抗体断片の産生は以下のように要約される。選択されたpHENは、HB2151に感染させ、次いでELISA用に抗体断片の可溶性発現が起こるように誘導する必要がある。各選択から溶出されたファージ10μl(約105 t.u.)を採取し、対数増殖中のHB2151細菌200μlに37℃で30分間感染させる(湯浴)。100μg/mlアンピシリンおよび1%グルコースを含むTYE上に、1μl、10μl、100μl、および1:10希釈物をプレーティングする。これらのプレートを37℃で一晩インキュベートする。96ウェルプレート(Corning「Cell Well」)中の100μg/mlアンピシリンおよび1%グルコースを含む2×TY 100μl中に個々のコロニーを釣菌し、37℃で振盪しながら(300 rpm)一晩培養する。別のプレートに接種するために使用したならば、最終濃度が15%となるようにグリセロールを添加し、-70℃で保存することによって、このプレートのグリセロール保存液を作製できる。96ウェル移行装置を使用して、このプレートから少量の接種菌液(約2μl)を、100μg/mlアンピシリンおよび0.1%グルコースを含む新鮮2×TYをウェル当たり200μl含む2枚目の96ウェルプレートに移す。600 nmにおけるODが約0.9になるまで(約3時間)、37℃で振盪しながら培養する。所要のODに達したならば、100μg/mlアンピシリンおよび9 mM IPTG(最終濃度1 mM IPTG)を含む2×TY 25μlを添加する。30℃でさらに16時間〜24時間振盪し続ける。ウェル当たり100μlのタンパク質抗原で、MicroTest III可塑性アッセイプレート(Falcon)をコーティングする。抗原は通常、PBSまたは50 mM炭酸水素ナトリウム、pH 9.6のいずれか中で濃度10μg/ml〜100μg/mlとし、室温で一晩コーティングする。翌日、ELISAプレートをひっくり返して過剰な液体を除去することによりPBSでウェルを3回濯ぎ、ウェル当たり200μlの3% BSA-PBSで37℃で2時間ブロッキングする。細菌プレートを1,800gで10分間遠心分離し、上清(可溶性scFvを含む)100μlをELISAプレートに添加し、室温で1時間インキュベートする。被験溶液を除去し、PBSで3回洗浄する。1% BSA-PBS中で濃度4μg/mlの精製9E10抗体(mycタグ化抗体断片を検出する)50μl、および1% BSA-PBS中に1:500希釈したHRP-抗マウス抗体50μlを添加する。室温で60分間インキュベートし、PBS-0.05% Tween20で3回洗浄し、次にPBSで3回洗浄する。基質溶液(100μg/ml TMBの100 mM酢酸ナトリウム溶液(pH 6.0)。使用前にこの溶液50 mlに対して30%過酸化水素10μlを直接添加する。)で発色させる。各ウェルに100μlを添加し、室温で10分間静置する。青色が発色するはずである。1 M硫酸50μlを添加することにより反応を停止する。色が黄色に変化するはずである。450 nmおよび405 nmのODを読み取る。OD 450からOD 405を差し引く。
【0167】
ライブラリー中の挿入物は、
と命名したプライマーを用いてPCRスクリーニングすることにより、スクリーニングできる。VHおよびVLの配列決定に関しては、プライマー
の使用を推奨する。
【0168】
実施例12
レパートリーライブラリーからのTNBに特異的なscFv抗体の単離
本実施例では、リガンドTNB(トリニトロベンゼン)に対するレパートリー抗体ライブラリーのスクリーニングについて要約する。標準的な手順により(実施例9を参照、またwww.mrc-cpe.cam.ac.uk/~phage/glp.htmlにも記載されている)、レパートリーライブラリー(Griffin-1ライブラリー)のファージパニングをまず3ラウンド行うことにより、ライブラリースクリーニングを開始した。様々なラウンドのパニングからレスキューしたファージを用いて、大腸菌ABLE Cを感染させた。この細胞を対数期中期まで培養し、上記のように可溶型のscFv抗体の発現を誘導し、蛍光色素Cy5に複合化した100 nM TNBSで標識した。標識した細胞を、633 nmで放射する5 mMダイオードレーザーを装備したCytomation MoFlo装置を用いて、フローサイトメトリーにより解析した。膜フィルター上で高い蛍光を発するクローンを単離し、さらに解析した。FACSにより単離された10クローンのうち3クローンをさらに解析し、それらがTNBS-BSA複合体への強い結合を示すことが認められた。配列解析により、TNBS特異的クローンのうちの1つは、ファージディスプレイによっても見出されていたことが確認された。しかし、ライブラリーの可溶性ペリプラズム発現およびFACSスクリーニングによって単離された他の2つのクローンは、ファージパニングによって単離されたクローンのいずれにも相当しなかった。
【0169】
実施例13
大腸菌ペリプラズム中に可溶型で発現された抗体によるオリゴヌクレオチドプローブの検出
本実施例は、修飾したオリゴヌクレオチドが細菌の外膜を通して拡散し得ることを示す。5つののA残基間に4つのヌクレアーゼ耐性ホスホロチオエート結合を含む配列5'-ジゴキシゲニン-AAAAA-フルオレセイン-3'(dig-5A-FLと命名、分子量2,384 Da、配列番号:5)を有するオリゴヌクレオチドは、Integrated DNA Technologies、アイオワ州により合成され、精製された(RP HPLC)。このオリゴヌクレオチドのジゴキシゲニン部分は、ジゴキシンに特異的なscFv抗体(抗ジゴキシンscFv)によって認識され得る。プローブ分子が外膜を通して拡散し得るならば、ペリプラズム内に抗ジゴキシンscFvを発現する細胞は5A-Flを結合し得り、次にはこれによって細胞が蛍光性になるはずである。
【0170】
陰性対照としてのアトラジン(Hayhurst 2000)またはジゴキシゲニンに特異的なペリプラズムscFvを発現するABLE(商標)C細胞を、5×強度のPBS中で、100 nMジゴキシゲニン-BODIPY(商標)または100 nM dig-5A-FLのいずれかと共にインキュベートした。生存度染色のため、ヨウ化プロピジウムも添加した。ヨウ化プロピジウム排除(無傷の膜を有する細胞を同定するため)および側方散乱に基づいて、生細胞にゲートをかけた。1秒当たり1,000イベントの速度で、約10,000個の細胞を解析した。得られたデータを図3に示す。プローブに結合しない非関連の抗アトラジン抗体を発現する細胞は、バックグラウンドの蛍光のみを示した。対照的に、抗ジゴキシンscFv抗体を提示する細胞は、ジゴキシゲニン-BODIPY(商標)および5-A-FLの両方で明らかに標識された。後者のプローブは、対照細胞で観察されたシグナルよりも明らかに高いシグナルを生じた。5-A-FLは、より小さく非電荷であるジゴキシゲニン-BODIPYと比較して低い蛍光強度を生じるものの、前者プローブで得られたシグナルはFACSによるscFvライブラリーのスクリーニングに十分であった。
【0171】
実施例14
市販の蛍光基質を用いた、フザリウム・ソラニ(Fusarium solani)のリパーゼクチナーゼを発現する大腸菌の、フローサイトメトリーによる識別
本実施例では、市販の蛍光基質を用いて、ペリプラズム内に関連酵素を提示する大腸菌を特異的に標識し得ることを実証する。驚くべきことに、これらの反応の可溶性蛍光生成物は細胞内に十分に保持され、これにより非酵素発現細菌からの酵素発現大腸菌の識別および選択が可能になる。
【0172】
全遺伝子合成によりフザリウム・ソラニのリパーゼクチナーゼをコードする遺伝子を構築し、プラスミドpBAD18Cm内の強力な誘導性プロモーターpBADの下流に配置した。pBADプロモーターからのタンパク質発現は、FACSによるタンパク質ライブラリーのスクリーニングに有益である(Daughertyら、1999)。クチナーゼ遺伝子をコードする得られたプラスミドを、pKG3-53-1と命名した。pKG3-53-1および対照としてのpBAD18Cmを、DH5αに形質転換した。本実施例では、対照細胞からクチナーゼ発現細胞(DH5α(pKG3-53-1))を識別する能力を、2つの異なる市販の基質:フルオレセインジブチレートまたはLysoSensorグリーンDND-189(LSG)(どちらも、Molecular Probes、オレゴン州による)を用いて判定した。後者は、酵素によるエステル加水分解に起因して起こる細胞内のpH変化を検出する、正に荷電した蛍光プローブである。
【0173】
テリフィックブロス/クロラムフェニコール50:g/ml(TB/Cm)中、37℃で激しく振盪しながら細胞を一晩培養した。TB/Cm(50:g/ml)10 ml中に、一晩培養物100:lから継代培養物を作製した。OD600=0.6になるまで、37℃で激しく振盪しながらこれらの継代培養物を培養した。継代培養物4 mlアリコートを、Beckman Allegra 6R遠心機で3650 rpmで20分間ペレット化した。上清を除去し、0.2%グルコースおよび50:g/mlのクロラムフェニコール(Cm)を含むM9最小培地4 ml中に、ペレットを再懸濁した。最終濃度が0.2%になるように、20%保存液のアラビノースを添加した。この培養物を、25℃で激しく振盪しながら4時間誘導した。続いて、誘導した培養物の2 mlアリコートを、Eppendorf 5415C遠心機で8000 rpmで10分間ペレット化し、新鮮培地で洗浄し、再度8000 rpmで10分間ペレット化した。洗浄したペレットを、吸光度OD600=1.0になるように、グルコースを含まないM9塩培地中に再懸濁した。この保存溶液を1:10希釈し、希釈した細胞懸濁液1 mlを、ジメチルスルホキシド(DMSO)中の0.1 mMフルオレセインジブチレート(FDB)保存溶液0.1 mlと混合した。最終FDP濃度は10:Mであった。37℃で30分間、反応を進行させた。標識した細胞を、Ar 488 nmレーザーを装備したBecton Dickinson FACSortで直ちに解析した。クチナーゼ発現細胞および対照細胞の蛍光分布を図9Aに示す。
【0174】
陽性(酵素発現)細胞と対照細胞との識別に対する第2のプローブの有用性もまた試験した。上記のように、pKG3-53-4プラスミドからクチナーゼを発現する大腸菌および陰性細胞(未改変のpBAD18Cmプラスミドを発現する)を培養して誘導し、洗浄した。ペレットを1%ショ糖で洗浄し、再度ペレット化し、OD600=1.0になるように新鮮な1%ショ糖中に再懸濁した。この細胞の保存溶液を、氷上で保存した。
【0175】
標識用に、LysoSensorグリーンDND-189(LSG、Molecular Probes)保存溶液を、DMSO中で1 mMになるように調製した。1 M 4-ニトロフェニルブチレート保存溶液もまた、DMSO中で調製した。まず細胞保存溶液を希釈し、最終濃度が1μMになるようにLSGを添加し、最終濃度が1μMになるように4-ニトロフェニルブチレートを1:1000希釈することにより、細胞の標識を開始した。細胞による4-ニトロフェニルブチレートの酵素的加水分解を25℃で5分間進行させ、次いで上記のようにBecton Dickinson FACSortで直ちに細胞を解析した。LysoSensorグリーンDND-189プローブで染色したクチナーゼ発現細胞および対照細胞の蛍光分布を、図9Bに示す。
【0176】
実施例15
炭疽菌防御抗原に対する親和性が120倍を上回って改善された抗体を単離するための、固定化ペリプラズム発現の使用
大きなライブラリーのスクリーニングには、遺伝子、それがコードするタンパク質、および所望の機能の間の物理的関連性が必要である。そのような関連性は、機構的研究、生物工学的目的、およびプロテオミクス研究に有益であると判明した様々なインビボ提示技術を用いて確立され得る(Hoess、2001;HayhurstおよびGeorgiou、2001;Wittrup、2000)。
【0177】
APExは、フローサイトメトリー(FC)によるスクリーニングを可能にする別のアプローチである。FCは、ハイスループットと、各ライブラリーメンバーのリアルタイム、定量的、多パラメータ解析を兼ね備える。時間当たり4億個細胞を超える次数の選別速度で市販のFC機械を使用して、微生物形質転換効率の制約内で利用可能な大きさのライブラリーをスクリーニングすることができる。さらに、多パラメータFCにより、リアルタイムでそれぞれのおよびすべてのクローンの機能に関して有益な情報が提供され得り、よってこれはライブラリー構築過程を導き選別条件を最適化するのに役立つ(BoderおよびWittrup、2000;Daughertyら、2000)。
【0178】
細菌および酵母のタンパク質提示はFCと組み合わせて、様々なリガンドに対する高親和性抗体を操作するために用いられている(Daughertyら、1999;Boderら、2000)。しかし、タンパク質を細胞表面に提示するための必要条件から、ライブラリースクリーニングの適用に影響し得る生物学的制約が課される。真核生物における折りたたまれないタンパク質反応またはグラム陰性菌の外膜に対するタンパク質選別のストリンジェンシー等の過程により、実際に表面提示と適合するポリペプチドの多様性が制限される(Sagtら、2002;Sathopoulosら、1996)。さらに、微生物表面は、その巨大分子組成がタンパク質:リガンド認識を妨げ得る化学的に複雑な構造をしている。外膜上のリポ多糖の存在がタンパク質:リガンド認識に対する立体的障壁を示すため、この問題はグラム陰性菌において特に著しく、実際にこれが線毛等の特殊化した付属器の進化に寄与したと思われる(Hultgrenら、1996)。
【0179】
APExは、以前の提示戦略の生物学的制約および抗原の接近限界を克服し、これにより実質的に任意の大きさの抗原に対する抗体の効率的な単離が可能になる。APExでは、タンパク質はN末端またはC末端融合物として大腸菌細胞質膜の外(ペリプラズム)面に係留され、よって細胞表面におけるタンパク質の提示に関連した生物学的制約が除かれる。細菌外膜を化学的/酵素的に透過処理した後、固定化scFv抗体を発現する大腸菌を、少なくとも240 kDaの蛍光抗原で特異的に標識し、FCによって解析することができる。本発明者らは、APExを用いることによって、120倍を超えて親和性が増加した、炭疽菌の防御抗原に対する抗体断片を含む、著しく改善されたリガンド親和性を有する抗体の効率的単離を実証した。
【0180】
A. 固定化ペリプラズム発現およびリガンド結合の検出
スクリーニング用途のためには、理想的な発現系は、異種性ポリペプチドの合成に起因し得る細胞毒性または増殖異常を最小限に抑えるべきである(Daughertyら、2000)。APExを使用することにより、膜貫通タンパク質融合物に関連する複雑な問題が回避される(MirouxおよびWalker、1996;Mingarroら、1997)。細菌リポタンパク質は、膜タンパク質とは異なり、膜固定化のためにSRPまたはYidC経路を必要としないことが知られている(Samuelsonら、2000)。リポタンパク質はSec経路を介して膜を超えて分泌され、ペリプラズムに入ると、シグナル配列のC末端側に存在するシステイン残基にジアシルグリセリド基がチオエーテル結合を介して付着する。その後、シグナルペプチダーゼIIによってシグナルペプチドが切断され、修飾されたシステイン残基においてタンパク質が脂肪アシル化され、最終的に親油性脂肪酸が膜内に挿入し、これによりタンパク質が固定される(Pugsley、1993;Seydelら、1999;Yajushiら、2000)。
【0181】
リーダーペプチドおよび成熟NlpAタンパク質の最初の6アミノ酸をコードする配列(推定上の脂肪アシル化部位および内膜標的部位を含む)を、内膜のペリプラズム面へのscFv抗体の固定化に使用した。NlpAは、内膜に独占的に局在する非必須大腸菌リポタンパク質である(Yuら、1986;Yamaguchiら、1988)。特に注目すべきは、内膜標的のための共通残基と考えられている、脂肪アシル化システイン残基に隣接したアスパラギン酸残基である(Yamaguchiら、1988)。26-10抗ジゴキシン/ジゴキシゲニン(Dig) scFvおよび抗炭疽菌防御抗原(PA)14B7 scFvへのNlpA融合物を構築し、大腸菌においてlacプロモーターから発現させた。IPTGによりNlpA-[scFv]合成を誘導した後、細胞をEDTAおよびリゾチームと共にインキュベートして外膜および細胞壁を破壊した。透過処理した細胞を、蛍光色素BODIPY(商標)(200 nM)に複合化したそれぞれの抗原と混合し、フローサイトメトリーにより細胞の蛍光を測定した。NlpA-[14B7 scFv]およびNlpA-[Dig scFv]を発現する処理細胞は、対照と比較して、それぞれ9倍および16倍高い平均蛍光強度を示した(図7A)。細胞を非関連の蛍光抗原と混合した場合にはバックグラウンドの蛍光のみが検出され、このことから、本研究の条件下ではバックグラウンドの結合が無視できることが示された。
【0182】
細胞質膜上に固定された抗体断片が大きな抗原に結合する能力をさらに評価するため、NlpA-[Dig scFv]が、240 kDA蛍光タンパク質フィコエリトリン(PE)に複合化したジゴキシゲニンを認識する能力について試験した。複合体をNlpA-[Dig scFv]を発現する細胞と混合し、EDTA-リゾチームで処理した。高い細胞蛍光が観察されたことから、膜に固定化された抗体によるジゴキシゲニン-PEの結合が示された(図7B)。全体として、蓄積されたデータから、Tris-EDTA-リゾチームで処理した細胞において、細胞質膜上に固定されたscFvは、低分子から少なくとも最大で分子量240 kDaまでのタンパク質に及ぶリガンドに容易に結合し得ることが実証された。重要なことには、ジゴキシゲニン-PEで標識し、その後フローサイトメトリーを1ラウンド行うことにより、非関連の抗原特異性を有するscFvとの同様の融合物を発現する細胞から、NlpA-[Dig scFv]を発現する細菌が500倍を超えて濃縮された。
【0183】
B. APExによるライブラリースクリーニング
抗PA 14B7 scFvの遺伝子のエラープローンPCRにより1×107個のメンバーのライブラリーを構築し、NlpA膜固定化配列に融合した。無作為に選択した12個のライブラリークローンをDNA配列決定したところ、遺伝子当たり平均して2%のヌクレオチド置換が明らかになった。IPTGによりNlpA-[14B7変異体scFv]合成を融合した後、細胞をTris-EDTA-リゾチームで処理し、洗浄し、200 nM PA-BODIPY(商標)で標識した。内膜の完全性を、ヨウ化プロピジウム(PI)で染色することによりモニタリングした。超ハイスループットCytomation Inc. MoFlo液滴偏向フローサイトメーターを用いて、低いPI蛍光(630 nm放射)および高いBODIPY(商標)蛍光に選択的にゲートをかけて、全部で2×108個の細菌を選別した。最も高い530 nm蛍光(FL1)を有して選別された約5%の細胞を回収し、直ちにPIのみで再度染色し、上記のように再度選別した。この2回目の選別サイクルには抗原を添加しなかったため、解離反応速度の低い抗体を発現する細胞のみが、依然として蛍光性のままである。おそらくは潜在的scFv毒性(Somervilleら、1994;HayhurstおよびHarris、1999)、Tris-EDTA-リゾチーム処理、および高剪断フローサイトメトリー環境への曝露の組み合わせに起因して、この集団のプレーティング効率は低かった。したがって、潜在的高親和性クローンの喪失を回避するため、選別によって回収された約1×104個の蛍光イベントをPCR(商標)増幅することにより、scFvをコードするDNAをレスキューした。PCR(商標)増幅に用いた条件により、標識過程におけるTris-EDTA-リゾチーム処理に起因する、部分的に加水分解された細胞壁を有する細胞から、細胞DNAが量的に放出されたことに留意されたい。30ラウンドのPCR(商標)増幅を行った後、DNAをpAPEx1に連結し、新鮮な大腸菌に形質転換した。選別の2ラウンド目は、この場合には集団のうちの最も蛍光の高い2%のみを回収することを除いては先の通りに行い、次いで直ちに再選別して約5,000蛍光イベントを得た。
【0184】
2ラウンド目によるscFv DNAをPCR(商標)により増幅し、scAb型で可溶型の抗体断片を発現させるためにpMoPac16(Hayhurstら、2003)に連結した。scAb抗体断片は、軽鎖がヒトκ定常領域に融合したscFvである。この抗体断片型は、scFvと比較して優れたペリプラズム溶解性を示す(Maynardら、2002;Hayhurst、2000)。scAb型の20クローンを無作為に拾い、液体培地中で培養した。IPTGで誘導した後、ペリプラズムタンパク質を単離し、表面プラズモン共鳴(SPR)を用いて、相対抗原解離反応速度に関してscAbタンパク質を順位づけした。20クローンのうち11クローンが、14B7親抗体と比較してより低い抗原解離反応速度を示した。抗原解離反応速度が最も低い3つのscAbを大量に産生し、Niクロマトグラフィー、それに続くゲル濾過FPLCによりこれを精製した。興味深いことに、ライブラリーから選択されたクローンはすべて、優れた発現特性を示し、振盪フラスコ培養においてL当たり4 mg〜8 mgの精製タンパク質が得られた。詳細なBIACore解析により、3クローンはすべて親の14B7抗体と比較して、PAに対して実質的に低いKDを示すことが実証された(図8Aおよび図8B)。KDの改善は主に、抗原解離の低下(すなわち、koffの低下)に起因した。最も親和性の高いクローンM18は、(koffが4.2×10-5 M-1 秒-1で)KD 35 pMを示し、これはM18-PA半減期6.6時間に相当する。これは、親抗体14B7(BIACore 3000により決定されたKD=4.3 nM)と比較して、親和性が120倍を超えて改善されたことを表す。同定された変異体を図8Bに示し、1H、M5、M6、およびM18抗体の高次構造を示す略図を図10に示す。M5の変異は以下の通りであった:軽鎖においてQ38R、Q55L、S56P、T74A、Q78L、および重鎖においてK62R。M6に関して変異は以下の通りであった:軽鎖においてS22G、L33S、Q55L、S56P、Q78L、およびL94P、ならびに重鎖においてS7P、K19R、S30N、T68I、およびM80L。M18に関して変異は以下の通りであった:軽鎖においてI21V、L46F、S56P、S76N、Q78L、およびL94P、ならびに重鎖においてS30N、T57S、K64E、およびT68。図11は、可変重鎖および可変軽鎖ならびに形成された変異を示す、14B7 scFv配列(配列番号:21)とM18 scFv配列(配列番号:23)とのアラインメントを示す。これらの配列をコードする核酸を、それぞれ配列番号:20および22に示す。
【0185】
変異体抗体へのNlpA融合物を発現する、Tris-EDTA-リゾチームで透過処理した細胞の蛍光強度は、抗原結合親和性に応じて変化した(図8C)。例えば、バックグラウンド蛍光が約5であるのと比較し、NlpA-[M18 scFv]タンパク質を発現する細胞は平均蛍光強度250を示すのに対して、親の14B7 scFvを発現する細胞は平均蛍光強度30を示した(図8B)。中程度の親和性を有する抗体は、その相対親和性順位と一致して中程度の蛍光強度を示した。35 pM程度の低い解離定数を示す抗体を発現する細胞を分離する能力により、何故3つの独特な非常に高い親和性変種が単離され得たかに関する道理にかなった説明が提供され、フローサイトメトリー解析を用いて得られた優れた分解能が示される。
【0186】
詳細に解析した3つのクローン、M5、M6、およびM18は、それぞれ7個、12個、および11個のアミノ酸置換を含んでいた。ファージディスプレイを用いた以前の研究において(Maynardら、2002)、本発明者らは、1) 変異誘発エラープローンPCR(商標)、2) 5ラウンドのファージパニング、および3) パニング後のクローンのDNAシャッフリングからそれぞれなる3サイクルにより、14B7 scFvの変種を単離した。その研究で単離された最も優れたクローンは、Q55LおよびS56P置換を含み、(BIACore 3000により決定された)KD 150 pMを示す1Hであった。これらの2つの変異は結合ポケットの疎水性を増すようであり、疎水性相互作用の増大が抗体の親和性成熟において優性効果であるという山のような証拠を拡充するものである(Liら、2003)。同じアミノ酸置換は、APExによって単離されたM5およびM6クローンにおいても認められる。しかし、これらの2つのクローンにおけるさらなる変異により、親和性のさらなる増大が付与された。M5、M6、およびM18が単一ラウンドのアセクシャル(asexual)PCR(商標)後に単離され、なおどれも、ファージディスプレイによって実に複数ラウンドのセクシャル(sexual)変異誘発および選択の後に単離され得た最も優れた抗体と比較して、より高い親和性を有していたことは注目に値する。
【0187】
APExによって単離された最も親和性の高いクローンM18は、S56P変異を含んでいたが、1H、M5、およびM6に見出されたQ55L置換を欠いていた。部位特異的変異誘発によりQ55L置換をM18に導入すると、生じたScAbは、kon 1.1×106 M-1 秒-1およびkoff 2.4×10-5 秒-1の抗原結合(KD=21 pM)のさらなる改善を示し、これは複合体半減期11.6時間に相当する。しかし、この変異の導入により、精製タンパク質の収量が5倍を超えて減少し、振盪フラスコ培養で1.2 mg/Lになった。修飾したM18配列を配列番号:25に示し、この配列をコードする核酸を配列番号:24に示す。
【0188】
C. ファージディスプレイされたscFv抗体のAPEx
重要な治療標的および診断標的に対する多くの抗体断片が、ファージディスプレイによりスクリーニングしてレパートリーライブラリーから単離されている。時間を要するサブクローニング段階を必要としない、迅速な抗原結合解析およびそのような抗体の親和性成熟のための手段を開発することが望ましい。抗体は最も一般的には、ファージ遺伝子3マイナーコートタンパク質(g3p)のN末端への融合を介して、繊維状ファージ上に提示される(Barbasら、1991)。ファージの形態形成過程において、g3pはその最C末端を介して内膜に一時的に付着し、その後成長していくビリオンに取り込まれ得る(BoekeおよびModel、1982)。したがって、抗体断片はペリプラズム区画内に固定されかつ提示もされる。よって本発明者らは、APEx形式を用いて、抗体ライブラリーのスクリーニング目的で、g3p融合タンパク質が利用できるかどうかを評価した。上記の高親和性抗PA M18 scFv、抗ジゴキシン/ジゴキシゲニン26-10 scFv、および抗メタンフェタミン scFv(Meth)を、ファージディスプレイの目的で広く用いられ、g3p提示のために遺伝子IIIの短い変種を利用するファージミドpAK200内のlacプロモーターの下流のg3pのN末端にインフレームでクローニングした(Krebberら、1997)。IPTGで誘導した後、scFv-g3p融合物を発現する細胞をTris-EDTA-リゾチームにより透過処理し、それぞれの蛍光抗原で標識した(図9)。3つのscFvいずれに関しても、それぞれの抗原とインキュベートした場合にのみ高い蛍光が得られた。重要なことには、g3pのN末端に融合したscFvの平均蛍光強度が、NlpAアンカーのC末端への融合により得られた平均蛍光強度に匹敵した。図9の結果から、 (i) 大きな可溶性ドメインがN末端で膜アンカーに係留され得ること;(ii) 繊維状ファージ上に提示するためにファージミド内にクローニングした抗体断片が、APEx形式を用いてフローサイトメトリーにより容易に解析され得ること;および(iii) scFv抗体が、N末端またはC末端の融合物として、抗原結合を喪失することなく細胞質膜上に固定され得ることが実証される。
【0189】
D. 考察
本発明者らは、コンビナトリアルライブラリーから高親和性リガンド結合タンパク質、特にscFv抗体の効率的な選択を可能にする方法を開発した。1つの局面において、APExは、内膜の外面へのタンパク質の固定化、次の外膜の破壊、その後の蛍光標識抗原とのインキュベーションおよびFC選別に基づく。この戦略により、以前の細菌ペリプラズムおよび表面提示アプローチを超えるいくつかの恩典が提供される:1) 内膜中にタンパク質を保持するために脂肪アシル化アンカーを利用することにより、提示の成功のために必要とされるのは6アミノ酸程度の短いアミノ酸の融合のみであり、これによって場合によっては大きな融合物が課し得る悪影響が低減される;2) 内膜は、提示された抗体断片への大きな抗原の結合を立体的に妨げ得るLPSまたは他の複雑な糖質等の分子を欠いている;3) 融合物質は、提示される前に膜1枚を横切らなくてはならないだけである;4) N末端およびC末端融合戦略の両方を使用することができる;および5) 一般のファージディスプレイベクターから発現されるタンパク質に対して、APExを直接使用することができる。この最後の点が特に重要であるが、それはこの点によって、サブクローニングする段階を全く必要とせずに、ファージパニング実験によるクローンを量的に解析しまたはフローサイトメトリーによりさらに選別し得るハイブリッドライブラリースクリーニング戦略が可能となるからである。
【0190】
低分子(例えば、ジゴキシゲニンおよびメタンフェタミン <1 kDa)からフィコエリトリン複合体(240 kDa)に及ぶ抗原を検出するために、APExを使用することができる。実際には、より大きなタンパク質を同様に使用し得ることを意図するように、図3Bで使用したフィコエリトリン複合体は、抗原検出の上限を規定することを意図していない。
【0191】
実施例において、蛍光標識抗原に結合するscFvをコードする遺伝子は、PCR(商標)により選別した細胞からレスキューした。このアプローチの恩典は、潜在的scFv毒性、Tris-EDTA-リゾチーム破壊、およびFC剪断力の組み合わせに起因する、もはや生存できないクローンの単離が可能になる点である。このような方法で、単離されたクローンの多様性が最大化される。PCR(商標)レスキューのさらに別の恩典は、サブクローニングする前に、プールした細胞からのDNA増幅を変異誘発条件下で実施し得る点である。したがって、選択の各ラウンド後に、単離した遺伝子にランダム変異を導入することができ、これにより定向進化のさらなるラウンドが簡略化される(HanesおよびPluckthun、1997)。さらに、増幅段階において、プール内のタンパク質コード遺伝子間の鋳型交換に有利に働くPCR(商標)条件を使用することにより、選択されたクローン間の組換えが可能になる。PCR(商標)レスキューは、他のライブラリースクリーニング形式においても同様に有利であると考えられる。
【0192】
いずれのライブラリースクリーニングにも関わる重要な問題は、単離されたクローンを高レベルで発現する能力である。既存の提示形式は、提示されたタンパク質の発現特性に影響し得る大きな固定化配列への融合を含む。したがって、十分に提示されるscFvは、非融合タンパク質としての可溶型での高発現に必ずしも従い得ない(Hayhurstら、2003)。一方、本発明において細胞質膜へのタンパク質のN末端係留に使用し得る短い(6アミノ酸)尾部は、融合物の発現特性に影響しないようである。この仮説と一致して、APExにより単離された炭疽菌PAに対して親和性が増大した3つのクローンはすべて、多くのアミノ酸置換を有するにもかかわらず、優れた可溶性発現特性を示した。同様に、メタンフェタミン抗体の親和性成熟においても十分に発現するクローンが得られたことから、細菌において可溶型で大量に容易に産生され得るクローンの単離は、本発明を用いた選択の固有の特性であることが示唆される。
【0193】
本実施例において、本発明者らは親和性成熟のためにAPExを使用し、21 pM程度に低いKD値を示す、炭疽菌防御抗原に対するscFvを操作した。PAに対して最も高い親和性を示すscFv結合部位をヒト化し、全長IgGに変換し、前臨床試験において、炭疽菌中毒に対するその中和の可能性を評価している。親和性成熟に加えて、ペリプラズムループ内に固定されたscFv抗体が蛍光抗原に結合し得り、蛍光レポーターとして機能する膜タンパク質トポロジーの解析、および亢進した機能を有する酵素変種の選択を含むいくつかの他のタンパク質工学用途に、APExを利用することが可能である。特に、細胞外被によって酵素触媒による産物を保持する部位が提供されるため、酵素ライブラリー選別にAPExを容易に適合化することができ、これにより触媒による代謝回転に直接基づいた選択が可能になる(Olsenら、2000)。本発明者らはまた、膜タンパク質に対するリガンドのスクリーニングへのAPExの利用に関して評価している。要するに、固定化ペリプラズム発現が、コンビナトリアルライブラリースクリーニングおよび他のタンパク質工学用途を促進する可能性を有することが実証された。
【0194】
E. 材料および方法
1. 組換えDNA技法
NdeI部位およびSfiI部位に隣接したリーダーペプチドおよび成熟NlpAタンパク質の最初の6アミノ酸を、プライマー
を用いてXL1-ブルー(Stratagene、カリフォルニア州)の全細胞PCRを行うことにより増幅した。生じたNlpA断片を用いて、NdeIおよびSfiIを介してpMoPac1(Hayhurstら、2003)のpelBリーダー配列を置換し、pAPEx1を作製した。ジゴキシン(Chenら、1999)、炭疽菌防御抗原PA(Maynardら、2002)、およびメタンフェタミンに特異的なscFvを、非適合性SfiI部位を介してpAPEx1内のNlpA断片の下流に挿入した。同じ遺伝子をファージディスプレイベクターpAK200(Krebberら、1997)にクローニングすることにより、scFvの対応するg3p融合物を作製した。
【0195】
2. 培養条件
全体にわたって使用した宿主株は、大腸菌ABLE C(商標)(Stratagene)であった。pAPEx1またはpAK200誘導体で形質転換した大腸菌を、2%グルコースおよび30ug/mlクロラムフェニコールを添加したテリフィックブロス(TB)中に、OD600が0.1になるまで播種した。細胞の培養および誘導は、以前に記載されている通りに行った(Chenら、2001)。誘導した後、細胞の外膜を記載されている通りに透過処理した(NeuおよびHeppel、1965)。手短に説明すると、細胞(OD600=20で約1 mlに相当する)をペレット化し、0.75 Mショ糖、0.1 M Tris-HCl、pH 8.0、100μg/mlニワトリ卵白リゾチームの氷冷溶液350μl中に再懸濁した。氷冷1 mM EDTA 700μlを穏やかに添加し、懸濁液を氷上に10分間置いた。0.5 M MgCl2 50μlを添加し、混合物を氷上にさらに10分間置いた。得られた細胞を穏やかにペレット化し、200 nMプローブを添加したリン酸緩衝食塩水(1×PBS)中に再懸濁し室温で45分間インキュベートしてから、FCにより評価した。
【0196】
3. 蛍光プローブ
ジゴキシゲニン-BODIPYの合成は、以前に記載されている(Daughertyら、1999)。メタンフェタミン-フルオレセイン複合体は、Roche Diagnosticsより分与されたものであった。S. Leppla NIHにより分与された精製PAタンパク質は、製造業者の説明に従って、bodipy FL SE D-2184とのモル比1対7でBODIPY(商標)と複合体化した。複合化されなかったBODIPY(商標)は、透析により除去した。
【0197】
ジゴキシゲニン-フィコエリトリンを合成するため、製造業者の説明に従って、R-フィコエリトリンおよび3-アミノ-3-ジオキシジゴキシゲニンヘミスクシンアミド、スクシニミジルエステル(Molecular Probe)をモル比1対5で複合化した。過剰のPBS中で透析することにより、遊離のジゴキシゲニンを除去した。
【0198】
4. FCによるscFvライブラリーの親和性成熟
標準的な手順によりエラープローンPCRを用いて14B7親scFvからライブラリーを作製し(Fromantら、1995)、pAPEx1発現ベクターにクローニングした。形質転換、誘導、および標識し、次いで内膜の完全性をモニタリングするために細胞をヨウ化プロピジウムで染色した(PI放射 617 nm)。励起に488 nmアルゴンレーザーを用いて、MoFlo(Cytomation)液滴偏向フローサイトメーターで細胞を解析した。530/40バンドパスフィルターを通して検出されるフルオレセイン/Bodipy FL放射スペクトラムの蛍光の改善、および630/40バンドパスフィルターを通して検出されるPI放射における標識の欠除に基づいて、細胞を選択した。
【0199】
1回目の選別後に捕獲された大腸菌を、フローサイトメーターで直ちに再選別した。続いて、選別された細胞懸濁液中のscFv遺伝子をPCR(商標)により増幅した。増幅されたならば、次に変異体scFv遺伝子をpAPEx1ベクターに再クローニングし、細胞に再形質転換し、次いで寒天プレート上で30℃で一晩培養した。得られたクローンを上記のように2ラウンド目の選別および再選別に供し、その後scAbタンパク質を発現させるためにscFv遺伝子をpMoPac16(Hayhurstら、2003)にサブクローニングした。
【0200】
5. 表面プラズモン共鳴解析
以前に記載されている通りに、IMAC/サイズ排除FPLCにより、単量体scAbタンパク質を精製した(Hayhurstら、2003)。BIACore3000機器を用いて、SPRにより親和性測定値を取得した。EDC/NHS化学により、約500RUのPAをCM5チップにカップリングした。同様にBSAもカップリングし、線の減算に使用した。BIA HBS-EP緩衝液中25℃で流速100μl/分にて、反応速度解析を実施した。20 nMから開始した各抗体の2倍希釈物を5種類、3つ組で解析した。
【0201】
本明細書で開示され請求される方法はすべて、本開示の観点から、過度に実験することなく作製および実行することができる。本発明の組成物および方法を好ましい態様に関して記載したが、本発明の概念、精神、および範囲から逸脱することなく、本明細書に記載した方法および方法の段階または方法の段階の順序に変更がなされ得ることは、当業者に明白であろう。さらに具体的には、本明細書に記載した物質の代わりに、化学的および生理学的に関連する特定の物質を代用し得て、同じもしくは類似した結果が得られることは明白であろう。当業者にとって明白であるこのような類似の代用品および変更はすべて、添付の特許請求の範囲によって定義される本発明の精神、範囲、および概念の範囲内であると見なされる。
【0202】
参考文献
以下の参考文献は、例示的な手順の補足または他の詳細な補足を、本明細書に記載したものに提供する範囲内において、参照として本明細書に明確に組み入れられる。
【図面の簡単な説明】
【0203】
添付の図面は本明細書の一部を形成し、本発明の特定の局面をさらに実証するために含めるものである。本明細書に示した特定の態様の詳細な説明と組み合わせてこれらの図面の1つまたは複数を参照することにより、本発明をより良く理解できる。
【図1】図1A〜図1Cは、APExによる抗原標的の選択的同定を示す。APExは、大腸菌において表示されるscFvを発現した。低分子の(A) ジゴキシゲニン-Bodipy FLおよび(B) メタンフェタミン-FLと結合する発現したscFv;または(C) 例えばペプチド18aaといったペプチドに結合する発現したScFvを示す。
【図2】図2Aおよび図2Bは、スフェロプラストの表面上でのScFvの検出を示す。APExは、大腸菌において表示されるscFvを発現した。発現したScFvは、大きな抗原、例えばPA-Cy5(83 kD)、フィコエリトリン-ジゴキシゲニン(240 kD)を結合し得た。APExによって発現したscFvが大きなタンパク質に接近し得るという証拠が提供される。
【図3】図3Aおよび図3Bは、大きな標的抗原を複合化したフルオロフォアについてのScFvの検出を示す。
【図4】Meth-FLプローブに対するメタンフェタミン結合scFvの成熟を示す。
【図5】親の抗メタンフェタミンscFvと比較して高い平均FLシグナルを有する、変異体9と命名したクローンの解析。固定化ペリプラズム発現により発現したscFvを示す。
【図6】高親和性抗体断片をフローサイトメトリーに基づいて単離するための、固定化ペリプラズム発現(APEx)の原理を示す略図である。
【図7】APExにより描出される標的の例。(A) 200 nM Bodipy(商標)複合化蛍光抗原で標識した、PA特異的(14B7)scFvおよびジゴキシゲニン特異的(Dig)scFvを発現するABLEC(商標)の蛍光分布。ヒストグラムは、大腸菌10,000イベントの平均蛍光強度を示す。(B) 200 nMの240 kDaジゴキシゲニン-フィコエリトリン複合体で標識した、14B7 scFvまたはDig scFvを発現する細胞のヒストグラム。
【図8】APExにより選択された抗PA抗体断片の解析を示す。(A) 抗PA scAbとPAとの結合のシグナルプラズモン共鳴(SPR)解析。(B) SPRによって得られた親和性データの表。(C) 大腸菌で発現させ、200 nM PA-Bodipy(商標)複合体で標識した、pAPEx1中の抗PA scFvの、抗メタンフェタミン(Meth)scFv陰性対照と比較してのFCヒストグラム。
【図9】N末端とC末端固定化戦略の比較を示す。(A) 大腸菌においてpAPEx1ベクター中のN末端融合物として発現する抗ジゴキシゲニンDig scfv、抗PA M18 scFv、および抗メタンフェタミンMeth scFvは、200 nMのそれぞれの抗原で特異的に標識される。(B) pAK200ベクター中の同じscFvのC末端融合物も、200 nMのそれぞれの抗原で特異的に標識される。
【図10】1H、M5、M6、およびM18配列の高次構造およびアミノ酸置換を示す、抗体結合ポケットを上から見た図を示す。
【図11】可変重鎖および可変軽鎖ならびに結合親和性を改善するために形成された変異を示す、14B7 scFv配列(配列番号:21)とM18 scFv配列(配列番号:23)とのアラインメントを示す。
【特許請求の範囲】
【請求項1】
以下の段階を含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を含む細菌を取得する方法:
(a) 内膜および外膜およびペリプラズムを含むグラム陰性菌であって、細菌のペリプラズム内に曝露される候補結合ポリペプチドをコードする核酸配列を発現するグラム陰性菌を提供する段階;
(b) 標識リガンドが結合ポリペプチドと接触できる条件下で、細菌を標識リガンドと接触させる段階;ならびに
(c) 該候補結合ポリペプチドに結合した該標識リガンドの存在に基づき、該細菌を選択する段階。
【請求項2】
以下の段階をさらに含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を取得する方法としてさらに定義される、請求項1記載の方法:
(d) 細菌から候補結合ポリペプチドをコードする核酸配列をクローニングする段階。
【請求項3】
融合ポリペプチドの発現を内膜の外側に方向づけ得るリーダー配列に、核酸配列が機能的に結合しているとしてさらに定義される、請求項1記載の方法。
【請求項4】
グラム陰性菌が大腸菌である、請求項1記載の方法。
【請求項5】
段階(a)が、グラム陰性菌の集団を提供する段階を含むとしてさらに定義される、請求項1記載の方法。
【請求項6】
候補結合ポリペプチドが細菌の内膜の外側に固定される、請求項1記載の方法。
【請求項7】
細菌の集団が、複数の候補結合ポリペプチドをコードする複数の核酸配列をまとめて発現するとしてさらに定義される、請求項5記載の方法。
【請求項8】
細菌が生存不能である、請求項2記載の方法。
【請求項9】
細菌が生存可能である、請求項2記載の方法。
【請求項10】
クローニングが核酸配列の増幅を含む、請求項2記載の方法。
【請求項11】
複数の核酸配列が、候補結合ポリペプチドと細菌の内膜の外側に固定されるポリペプチドとを含む融合ポリペプチドをコードするとしてさらに定義される、請求項7記載の方法。
【請求項12】
細菌の集団が以下の段階を含む方法によって得られる、請求項5記載の方法:
(a) 候補結合ポリペプチドおよび内膜アンカーポリペプチドを含む複数の融合ポリペプチドをコードする複数の核酸配列を調製する段階;ならびに
(b) DNA挿入物を用いてグラム陰性菌の集団を形質転換する段階。
【請求項13】
グラム陰性菌の集団を標識リガンドと接触させる、請求項5記載の方法。
【請求項14】
段階(c)の選択段階が、少なくとも2ラウンドの選択を含むとしてさらに定義され、細菌の亜集団が、候補結合ポリペプチドに結合した標識リガンドの存在に基づいて選択され、さらに、亜集団が、候補結合ポリペプチドに結合した標識リガンドの存在に基づく少なくとも1回のさらなる選択に供される、請求項5記載の方法。
【請求項15】
約2ラウンド〜6ラウンドの選択が行われる、請求項14記載の方法。
【請求項16】
フローサイトメトリーまたは磁気分離によって選択が行われる、請求項14記載の方法。
【請求項17】
候補結合ポリペプチドが抗体またはその断片としてさらに定義される、請求項1記載の方法。
【請求項18】
候補結合ポリペプチドがscAb、Fab、またはscFvとしてさらに定義される、請求項17記載の方法。
【請求項19】
候補結合ポリペプチドが、抗体以外の少なくとも40アミノ酸の結合タンパク質としてさらに定義される、請求項1記載の方法。
【請求項20】
候補結合ポリペプチドが39個未満のアミノ酸を含むとしてさらに定義される、請求項1記載の方法。
【請求項21】
候補結合ポリペプチドが酵素としてさらに定義される、請求項1記載の方法。
【請求項22】
標識リガンドが、ペプチド、ポリペプチド、酵素、核酸、低分子、および合成分子からなる群より選択される、請求項1記載の方法。
【請求項23】
標識リガンドが蛍光標識されているとしてさらに定義される、請求項1記載の方法。
【請求項24】
候補結合ポリペプチドをコードする核酸が既知の核酸配列に隣接しているとしてさらに定義される場合、それによって、選択後に該核酸が増幅され得る、請求項1記載の方法。
【請求項25】
標識リガンドに対する細菌の外膜の透過性を増大させるために細菌を処理する段階をさらに含む、請求項1記載の方法。
【請求項26】
処理段階が、高浸透圧条件による処理、物理的ストレスによる処理、ファージによる細菌感染、リゾチームによる処理、EDTAによる処理、消化酵素による処理、および外膜を破壊する化学物質による処理からなる群より選択される方法を含む、請求項25記載の方法。
【請求項27】
処理段階がそのような方法の組み合わせを含む、請求項26記載の方法。
【請求項28】
処理段階が、リゾチームおよびEDTAによる処理を含む、請求項27記載の方法。
【請求項29】
処理段階が、外膜の物理的破壊、化学的破壊、および酵素的破壊の組み合わせによる細菌の処理を含む、請求項25記載の方法。
【請求項30】
細菌が、標識リガンドに対する外膜の透過性の増大を付与する変異を含む、請求項1記載の方法。
【請求項31】
細菌の外膜を除去する段階をさらに含む、請求項1記載の方法。
【請求項32】
細菌を準生理的(sub-physiological)温度で培養する、請求項1記載の方法。
【請求項33】
準生理的温度が約25℃である、請求項32記載の方法。
【請求項34】
候補結合ポリペプチドに結合していない標識リガンドを除去する段階をさらに含む、請求項1記載の方法。
【請求項35】
少なくとも2つの標識リガンドと細菌を接触させる段階を含むとしてさらに定義される、請求項1記載の方法。
【請求項36】
選択段階がフローサイトメトリーを含む、請求項1記載の方法。
【請求項37】
選択段階が磁気分離を含む、請求項1記載の方法。
【請求項38】
リガンドと候補結合ポリペプチドが可逆的に結合する、請求項1記載の方法。
【請求項39】
内膜の外側に固定されるポリペプチドが膜貫通タンパク質またはその断片を含む、請求項6記載の方法。
【請求項40】
内膜の外側に固定されるポリペプチドが、大腸菌NlpA遺伝子によってコードされる最初の2アミノ酸、大腸菌NlpA遺伝子によってコードされる最初の6アミノ酸、繊維状ファージの遺伝子IIIタンパク質またはその断片、内膜リポタンパク質またはその断片からなる群より選択される配列を含む、請求項6記載の方法。
【請求項41】
内膜の外側に固定されるポリペプチドが、ポリペプチドのN末端またはC末端を介して固定される、請求項6記載の方法。
【請求項42】
配列が、
からなる群より選択される内膜リポタンパク質またはその断片である、請求項40記載の方法。
【請求項43】
標的リガンドに対する特異的親和性を有する第1の結合ポリペプチドを少なくとも1つコードする核酸配列を含む細菌を取得する方法であって、以下の段階を含む方法:
(a) 内膜および外膜およびペリプラズムを含むグラム陰性菌であって、細菌のペリプラズム内に曝露される少なくとも1つの候補結合ポリペプチドをコードする核酸配列を発現するグラム陰性菌を提供する段階;
(b) 標識リガンドが結合ポリペプチドと接触できる条件下で、細菌を蛍光標識リガンドと接触させる段階;ならびに
(c) FACSを用いて、蛍光標識リガンドの存在に関して細菌を選択する段階。
【請求項44】
標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を取得する方法としてさらに定義される方法であって、以下の段階をさらに含む、請求項43記載の方法:
(d) 細菌から候補結合ポリペプチドをコードする核酸配列をクローニングする段階。
【請求項45】
グラム陰性菌の集団を提供する段階を含むとしてさらに定義される、請求項43記載の方法。
【請求項46】
細菌の集団が、複数の候補結合ポリペプチドをコードする複数の核酸配列をまとめて発現するとしてさらに定義される、請求項45記載の方法。
【請求項47】
候補結合ポリペプチドが細菌の内膜の外側に固定される、請求項43記載の方法。
【請求項48】
表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合する、単離された抗体またはその断片。
【請求項49】
約96 pMから約21 pMの間の結合親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項50】
約35 pMから約21 pMの間の結合親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項51】
scAb、Fab、またはSFvとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項52】
IgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項53】
ヒト化抗体としてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項54】
抗体がヒト型である、請求項48記載の単離された抗体またはその断片。
【請求項55】
少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む、請求項48記載の単離された抗体またはその断片。
【請求項56】
炭疽菌防御抗原に免疫学的に結合し、かつ、I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、L94P、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lからなる群より選択される改変(ここで、I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、およびL94Pは可変軽鎖内に存在し、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lは可変重鎖内に存在する)を少なくとも3つまとめて含む可変軽鎖および可変重鎖を除く、配列番号:21の可変軽鎖および可変重鎖を含む、単離された抗体またはその断片。
【請求項57】
上記改変を少なくとも5つ含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項58】
上記改変をすべて含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項59】
表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項60】
Q55LおよびS56Pを含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項61】
配列番号:22または24の可変軽鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項62】
配列番号:22または24の可変重鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項63】
配列番号:22の可変軽鎖および可変重鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項64】
配列番号:24の可変軽鎖および可変重鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項65】
scAb、Fab、またはSFvとしてさらに定義される、請求項54記載の単離された抗体またはその断片。
【請求項66】
IgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義される、請求項54記載の単離された抗体またはその断片。
【請求項67】
ヒト化抗体としてさらに定義される、請求項54記載の単離された抗体またはその断片。
【請求項68】
少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む、請求項48記載の単離された抗体またはその断片。
【請求項69】
請求項56記載の抗体またはその断片をコードする、単離された核酸。
【請求項70】
配列番号:23または25の可変軽鎖をコードするとしてさらに定義される、請求項69記載の単離された核酸。
【請求項71】
配列番号:23または25の可変重鎖をコードするとしてさらに定義される、請求項69記載の単離された核酸。
【請求項72】
配列番号:23のポリペプチドをコードするとしてさらに定義される、請求項69記載の単離された核酸。
【請求項73】
配列番号:25のポリペプチドをコードするとしてさらに定義される、請求項69記載の単離された核酸。
【請求項1】
以下の段階を含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を含む細菌を取得する方法:
(a) 内膜および外膜およびペリプラズムを含むグラム陰性菌であって、細菌のペリプラズム内に曝露される候補結合ポリペプチドをコードする核酸配列を発現するグラム陰性菌を提供する段階;
(b) 標識リガンドが結合ポリペプチドと接触できる条件下で、細菌を標識リガンドと接触させる段階;ならびに
(c) 該候補結合ポリペプチドに結合した該標識リガンドの存在に基づき、該細菌を選択する段階。
【請求項2】
以下の段階をさらに含む、標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を取得する方法としてさらに定義される、請求項1記載の方法:
(d) 細菌から候補結合ポリペプチドをコードする核酸配列をクローニングする段階。
【請求項3】
融合ポリペプチドの発現を内膜の外側に方向づけ得るリーダー配列に、核酸配列が機能的に結合しているとしてさらに定義される、請求項1記載の方法。
【請求項4】
グラム陰性菌が大腸菌である、請求項1記載の方法。
【請求項5】
段階(a)が、グラム陰性菌の集団を提供する段階を含むとしてさらに定義される、請求項1記載の方法。
【請求項6】
候補結合ポリペプチドが細菌の内膜の外側に固定される、請求項1記載の方法。
【請求項7】
細菌の集団が、複数の候補結合ポリペプチドをコードする複数の核酸配列をまとめて発現するとしてさらに定義される、請求項5記載の方法。
【請求項8】
細菌が生存不能である、請求項2記載の方法。
【請求項9】
細菌が生存可能である、請求項2記載の方法。
【請求項10】
クローニングが核酸配列の増幅を含む、請求項2記載の方法。
【請求項11】
複数の核酸配列が、候補結合ポリペプチドと細菌の内膜の外側に固定されるポリペプチドとを含む融合ポリペプチドをコードするとしてさらに定義される、請求項7記載の方法。
【請求項12】
細菌の集団が以下の段階を含む方法によって得られる、請求項5記載の方法:
(a) 候補結合ポリペプチドおよび内膜アンカーポリペプチドを含む複数の融合ポリペプチドをコードする複数の核酸配列を調製する段階;ならびに
(b) DNA挿入物を用いてグラム陰性菌の集団を形質転換する段階。
【請求項13】
グラム陰性菌の集団を標識リガンドと接触させる、請求項5記載の方法。
【請求項14】
段階(c)の選択段階が、少なくとも2ラウンドの選択を含むとしてさらに定義され、細菌の亜集団が、候補結合ポリペプチドに結合した標識リガンドの存在に基づいて選択され、さらに、亜集団が、候補結合ポリペプチドに結合した標識リガンドの存在に基づく少なくとも1回のさらなる選択に供される、請求項5記載の方法。
【請求項15】
約2ラウンド〜6ラウンドの選択が行われる、請求項14記載の方法。
【請求項16】
フローサイトメトリーまたは磁気分離によって選択が行われる、請求項14記載の方法。
【請求項17】
候補結合ポリペプチドが抗体またはその断片としてさらに定義される、請求項1記載の方法。
【請求項18】
候補結合ポリペプチドがscAb、Fab、またはscFvとしてさらに定義される、請求項17記載の方法。
【請求項19】
候補結合ポリペプチドが、抗体以外の少なくとも40アミノ酸の結合タンパク質としてさらに定義される、請求項1記載の方法。
【請求項20】
候補結合ポリペプチドが39個未満のアミノ酸を含むとしてさらに定義される、請求項1記載の方法。
【請求項21】
候補結合ポリペプチドが酵素としてさらに定義される、請求項1記載の方法。
【請求項22】
標識リガンドが、ペプチド、ポリペプチド、酵素、核酸、低分子、および合成分子からなる群より選択される、請求項1記載の方法。
【請求項23】
標識リガンドが蛍光標識されているとしてさらに定義される、請求項1記載の方法。
【請求項24】
候補結合ポリペプチドをコードする核酸が既知の核酸配列に隣接しているとしてさらに定義される場合、それによって、選択後に該核酸が増幅され得る、請求項1記載の方法。
【請求項25】
標識リガンドに対する細菌の外膜の透過性を増大させるために細菌を処理する段階をさらに含む、請求項1記載の方法。
【請求項26】
処理段階が、高浸透圧条件による処理、物理的ストレスによる処理、ファージによる細菌感染、リゾチームによる処理、EDTAによる処理、消化酵素による処理、および外膜を破壊する化学物質による処理からなる群より選択される方法を含む、請求項25記載の方法。
【請求項27】
処理段階がそのような方法の組み合わせを含む、請求項26記載の方法。
【請求項28】
処理段階が、リゾチームおよびEDTAによる処理を含む、請求項27記載の方法。
【請求項29】
処理段階が、外膜の物理的破壊、化学的破壊、および酵素的破壊の組み合わせによる細菌の処理を含む、請求項25記載の方法。
【請求項30】
細菌が、標識リガンドに対する外膜の透過性の増大を付与する変異を含む、請求項1記載の方法。
【請求項31】
細菌の外膜を除去する段階をさらに含む、請求項1記載の方法。
【請求項32】
細菌を準生理的(sub-physiological)温度で培養する、請求項1記載の方法。
【請求項33】
準生理的温度が約25℃である、請求項32記載の方法。
【請求項34】
候補結合ポリペプチドに結合していない標識リガンドを除去する段階をさらに含む、請求項1記載の方法。
【請求項35】
少なくとも2つの標識リガンドと細菌を接触させる段階を含むとしてさらに定義される、請求項1記載の方法。
【請求項36】
選択段階がフローサイトメトリーを含む、請求項1記載の方法。
【請求項37】
選択段階が磁気分離を含む、請求項1記載の方法。
【請求項38】
リガンドと候補結合ポリペプチドが可逆的に結合する、請求項1記載の方法。
【請求項39】
内膜の外側に固定されるポリペプチドが膜貫通タンパク質またはその断片を含む、請求項6記載の方法。
【請求項40】
内膜の外側に固定されるポリペプチドが、大腸菌NlpA遺伝子によってコードされる最初の2アミノ酸、大腸菌NlpA遺伝子によってコードされる最初の6アミノ酸、繊維状ファージの遺伝子IIIタンパク質またはその断片、内膜リポタンパク質またはその断片からなる群より選択される配列を含む、請求項6記載の方法。
【請求項41】
内膜の外側に固定されるポリペプチドが、ポリペプチドのN末端またはC末端を介して固定される、請求項6記載の方法。
【請求項42】
配列が、
からなる群より選択される内膜リポタンパク質またはその断片である、請求項40記載の方法。
【請求項43】
標的リガンドに対する特異的親和性を有する第1の結合ポリペプチドを少なくとも1つコードする核酸配列を含む細菌を取得する方法であって、以下の段階を含む方法:
(a) 内膜および外膜およびペリプラズムを含むグラム陰性菌であって、細菌のペリプラズム内に曝露される少なくとも1つの候補結合ポリペプチドをコードする核酸配列を発現するグラム陰性菌を提供する段階;
(b) 標識リガンドが結合ポリペプチドと接触できる条件下で、細菌を蛍光標識リガンドと接触させる段階;ならびに
(c) FACSを用いて、蛍光標識リガンドの存在に関して細菌を選択する段階。
【請求項44】
標的リガンドに対する特異的親和性を有する結合ポリペプチドをコードする核酸配列を取得する方法としてさらに定義される方法であって、以下の段階をさらに含む、請求項43記載の方法:
(d) 細菌から候補結合ポリペプチドをコードする核酸配列をクローニングする段階。
【請求項45】
グラム陰性菌の集団を提供する段階を含むとしてさらに定義される、請求項43記載の方法。
【請求項46】
細菌の集団が、複数の候補結合ポリペプチドをコードする複数の核酸配列をまとめて発現するとしてさらに定義される、請求項45記載の方法。
【請求項47】
候補結合ポリペプチドが細菌の内膜の外側に固定される、請求項43記載の方法。
【請求項48】
表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合する、単離された抗体またはその断片。
【請求項49】
約96 pMから約21 pMの間の結合親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項50】
約35 pMから約21 pMの間の結合親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項51】
scAb、Fab、またはSFvとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項52】
IgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項53】
ヒト化抗体としてさらに定義される、請求項48記載の単離された抗体またはその断片。
【請求項54】
抗体がヒト型である、請求項48記載の単離された抗体またはその断片。
【請求項55】
少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む、請求項48記載の単離された抗体またはその断片。
【請求項56】
炭疽菌防御抗原に免疫学的に結合し、かつ、I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、L94P、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lからなる群より選択される改変(ここで、I21V、S22G、L33S、Q38R、L46F、Q55L、S56P、T74A、S76N、Q78L、およびL94Pは可変軽鎖内に存在し、S7P、K19R、S30N、T57S、K62R、K64E、T68I、およびM80Lは可変重鎖内に存在する)を少なくとも3つまとめて含む可変軽鎖および可変重鎖を除く、配列番号:21の可変軽鎖および可変重鎖を含む、単離された抗体またはその断片。
【請求項57】
上記改変を少なくとも5つ含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項58】
上記改変をすべて含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項59】
表面プラズモン共鳴により決定された約140 pMから約21 pMの間の親和性Kdで炭疽菌防御抗原に免疫学的に結合するとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項60】
Q55LおよびS56Pを含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項61】
配列番号:22または24の可変軽鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項62】
配列番号:22または24の可変重鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項63】
配列番号:22の可変軽鎖および可変重鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項64】
配列番号:24の可変軽鎖および可変重鎖を含むとしてさらに定義される、請求項56記載の単離された抗体またはその断片。
【請求項65】
scAb、Fab、またはSFvとしてさらに定義される、請求項54記載の単離された抗体またはその断片。
【請求項66】
IgA、IgD、IgE、IgG、またはIgMのFcドメインを含むとしてさらに定義される、請求項54記載の単離された抗体またはその断片。
【請求項67】
ヒト化抗体としてさらに定義される、請求項54記載の単離された抗体またはその断片。
【請求項68】
少なくとも40 kDaの一価の抗体部分を形成するscFv断片および抗体定常領域を含む、請求項48記載の単離された抗体またはその断片。
【請求項69】
請求項56記載の抗体またはその断片をコードする、単離された核酸。
【請求項70】
配列番号:23または25の可変軽鎖をコードするとしてさらに定義される、請求項69記載の単離された核酸。
【請求項71】
配列番号:23または25の可変重鎖をコードするとしてさらに定義される、請求項69記載の単離された核酸。
【請求項72】
配列番号:23のポリペプチドをコードするとしてさらに定義される、請求項69記載の単離された核酸。
【請求項73】
配列番号:25のポリペプチドをコードするとしてさらに定義される、請求項69記載の単離された核酸。
【図1】


【図2】
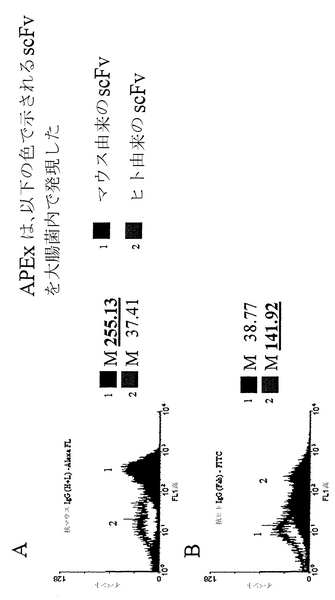
【図3】
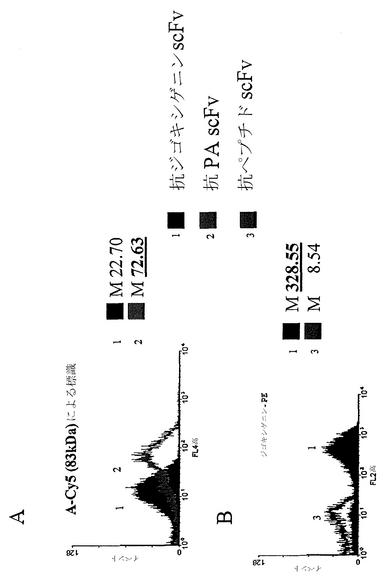
【図4】

【図5】

【図6】
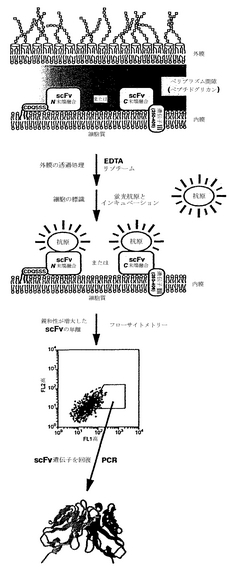
【図7】
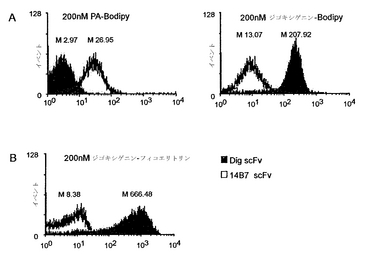
【図8】
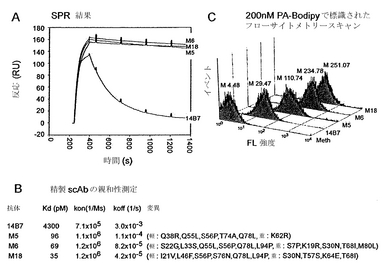
【図9】
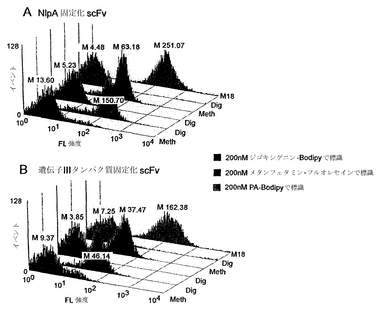
【図10】

【図11】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2006−515514(P2006−515514A)
【公表日】平成18年6月1日(2006.6.1)
【国際特許分類】
【出願番号】特願2005−508230(P2005−508230)
【出願日】平成15年7月15日(2003.7.15)
【国際出願番号】PCT/US2003/021928
【国際公開番号】WO2005/019409
【国際公開日】平成17年3月3日(2005.3.3)
【出願人】(500039463)ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム (115)
【Fターム(参考)】
【公表日】平成18年6月1日(2006.6.1)
【国際特許分類】
【出願日】平成15年7月15日(2003.7.15)
【国際出願番号】PCT/US2003/021928
【国際公開番号】WO2005/019409
【国際公開日】平成17年3月3日(2005.3.3)
【出願人】(500039463)ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム (115)
【Fターム(参考)】
[ Back to top ]