
ペリンドプリルエルブミンの調製方法
本発明は、純粋なペリンドプリルエルブミンの新規の調製方法に関する。本発明は又、ペリンドプリルエルブミンの結晶形Dの新規の調製方法にも関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、純粋なペリンドプリルエルブミンの新規の調製方法に関する。本発明は又、ペリンドプリルエルブミンの結晶形Dの新規調製方法にも関する。
【背景技術】
【0002】
ペリンドプリル及びその薬学的に許容できる塩は、アンジオテンシン変換酵素阻害薬として公知であり、循環器疾患の治療、特に高血圧及び心不全の治療に用いられている。ペリンドプリルは、(2S,3aS,7aS)−((2−(1−(エトキシカルボニル)−(S)−ブチルアミノ)−(S)−プロピオニル)オクタヒドロ−インドール−2−カルボン酸として化学的に公知であり、式(I):
【0003】
【化1】

により表し得る。
【0004】
ペリンドプリルは、EP0049658B1及びUS4,508,729に光学的純粋なS,S,S,S,S異性体として及びナトリウム塩の形態で初めて開示された。その後の数多くの後続特許及び特許出願(EP0308341B1、EP1279665A1、EP1333026A1、WO2004/099236など)が、ペリンドプリルの種々の調製方法を記載している。
【0005】
医薬製品に広く用いられているペリンドプリルエルブミンとして公知のペリンドプリルのtert−ブチルアミン塩は、EP0308341B1に初めて開示された。ペリンドプリルエルブミンは、結晶化条件(例えば、溶媒系、ペリンドプリルエルブミン濃度及び冷却力学など)に依り種々の結晶形で得られる。EP1296947B1は、酢酸エチルからのペリンドプリルエルブミン結晶形αの結晶化を開示し、EP1294689Aは、ジクロロメタン又は酢酸エチルからのペリンドプリルエルブミン結晶形βの結晶化を開示し、EP1296948B1は、クロロホルムからのペリンドプリルエルブミン結晶形γの結晶化を開示し、及びWO2004/113293は、ペリンドプリルエルブミン結晶形δ及び結晶形εの結晶化を開示している。結晶形εは、水1.5から2.5%(容量/容量)を含むtert−ブチルメチルエーテルからの結晶化により得られる一方、結晶形δは、共沸蒸留により結晶形εから得られる。
【0006】
EP0308341B1は、ジシクロヘキシルカルボジイミド及び1−ヒドロキシベンゾトリアゾールの存在下で、保護された(2S,3aS,7aS)−2−オクタヒドロインドール−2−カルボン酸をN−[(S)−1−カルベトキシブチル]−(S)−アラニンと結合することによるペリンドプリルの工業的調製方法を記載している。ペリンドプリルエルブミンは、粗製ペリンドプリルからtert−ブチルアミン添加後の結晶化により得られる。この方法の欠点は、除去することが困難なジシクロヘキシルカルボジイミドから派生し得る副生成物の生成である。この理由で、追加の精製段階が純粋なペリンドプリル及び/又は純粋なペリンドプリルエルブミンを得るために必要である。
【0007】
調製方法中に生成する副生成物に加えて、ペリンドプリルの分解生成物も又、不純物として粗製ペリンドプリル中に存在する。ペリンドプリル及びその塩は、化学的に高度に反応しやすい化合物であり、a)いくつかのキラル中心における異性体化、b)側鎖エステル基の加水分解、及び/又はc)分子内環化によるジケトピペラジンの形成、を介して分解しやすい。2つの最も重要なジケトピペラジンは、ジケトピペラジンIと称される(R)−エチル2−((3S,5aS,9aS,10aS)−3−メチル−1,4−ジオキソ−デカヒドロピラジノ[1,2−a]インドール−2(1H)−イル)ペンタノエート及びジケトピペラジンIIと称される2−((3S,5aS,9aS,10aR)−3−メチル−1,4−ジオキソ−デカヒドロピラジノ[1,2−a]インドール−2(1H)−イル)ペンタン酸(これらの式は下記のとおり)であり、これらは又、欧州局方5.1にもそれぞれ不純物F及びCとして示されている。
【0008】
【化2】
【0009】
WO01/58868は、(2S,3aS,7aS)−オクタヒドロインドール−2−カルボン酸ベンジルパラ−トルエンスルホン酸エステル及びN−[(S)−エトキシカルボニル−1−ブチル]−(S)−アラニンからのジシクロヘキシルカルボジイミド、1−ヒドロキシベンゾトリアゾール及び場合によりトリエチルアミンの存在下でのペリンドプリルの改良された調製方法を記載している。ジシクロヘキシルカルボジイミドから派生し得る副生成物の形成は、この方法に用いられる試薬の量の厳密な制御により低減される。しかしながら、記載された方法自身は、他の不純物の問題を解決しない。
【0010】
ジシクロヘキシルカルボジイミドから派生し得る不純物の問題は、ジシクロヘキシルカルボジイミドの使用を避けるペリンドプリルの調製方法により完全に解決される。例えば、そのような方法が特許出願EP1279665A1及びEP1333026A1中に記載されている。しかしながら、tert−ブチルアミンと、これらの特許出願中に開示されている方法により調製される粗製ペリンドプリルとの反応により得られるペリンドプリルエルブミンは、熱的再結晶又は活性炭処理などの別の精製方法により、更に精製しなければならない。
【0011】
WO2004/046172も又、高純度ペリンドプリルエルブミンの調製方法を記載しているということができ、この方法では、ペリンドプリルの保護された前駆体を塩基(例えば、tert−ブチルアミン)の存在下で脱保護して、粗製ペリンドプリルを単離することなく直接ペリンドプリル塩(例えば、ペリンドプリルエルブミン)を得る。製造方法中のジケトピペラジン不純物の生成は、反応時間が短いため最小限に抑えられる。しかしながら、他の不純物は、得られたペリンドプリルエルブミンから追加の結晶化段階により除去しなければならない。
【0012】
WO2005/019173は、粗製ペリンドプリル又はその塩の水溶液を適した有機溶媒を用いてpH4.0から6.5で抽出した後、有機層を分離し、tert−ブチルアミンを加えてペリンドプリルエルブミンを調製することによる、粗製ペリンドプリルからの純粋なペリンドプリルエルブミンの調製方法を開示している。この方法の欠点は、段階の数が多すぎて低収率になり得ることである。
【発明の開示】
【発明が解決しようとする課題】
【0013】
得られたペリンドプリルエルブミンの追加の精製が必要なく、工業的規模で応用可能な、粗製ペリンドプリルから純粋なペリンドプリルエルブミンを調製するための簡便で効果的な方法を開発する必要性が引き続き存在する。
【0014】
本発明は、粗製ペリンドプリルからの純粋なペリンドプリルエルブミンの改良された調製方法を提供し、前記方法は特にジケトピペラジン不純物の除去に効果的である。更に、前記方法は、簡便であり工業的規模で応用可能である。
【課題を解決するための手段】
【0015】
本発明の第1の対象は、
(a)湿潤脂肪族エステル中の又は複数の湿潤脂肪族エステルの混合物中の粗製ペリンドプリル溶液を提供する段階、
(b)tert−ブチルアミンを該溶液に添加する段階、
(c)ペリンドプリルエルブミンを結晶化する段階、及び
(d)結晶性ペリンドプリルエルブミンを単離する段階、
を含む、結晶性ペリンドプリルエルブミンの調製方法に関する。
【0016】
本発明の方法により、ジケトピペラジン不純物の0.20%(重量/重量)未満を、好ましくはジケトピペラジン不純物の0.10%(重量/重量)未満を含む純粋なペリンドプリルエルブミンを得ることが可能になる。
【0017】
本発明における「湿潤脂肪族エステル」は、含水率を高めた又は水を飽和させた脂肪族エステルを意味する。好ましくは、含水率を高めた湿潤脂肪族エステルは、水1%(容量/容量)から6%(容量/容量)を、より好ましくは水2%(容量/容量)から4%(容量/容量)を、最も好ましくは水2%(容量/容量)から3%(容量/容量)を含む。
【0018】
段階(a)において、湿潤脂肪族エステルは好ましくは、湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルからなる群より選択される。好ましくは湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルは、湿潤酢酸エチル、湿潤酢酸イソプロピル、湿潤酢酸ブチル及び湿潤プロピオン酸エチルを含むが、これらに限定されない。より好ましくは段階(a)で用いる湿潤脂肪族エステルは、湿潤酢酸エチルである。好ましくは、段階(a)で用いる湿潤酢酸エチルは水1%(容量/容量)から6%(容量/容量)を、より好ましくは水2%(容量/容量)から4%(容量/容量)を、最も好ましくは水2%(容量/容量)から3%(容量/容量)を含む。
【0019】
酢酸エチルの飽和は、分液ロート中で水と振り混ぜ、更に水相を分離することにより実施し得る。残留水滴を避けるために、酢酸エチル相を冷却し(好ましくは−20℃から−10℃の温度に)、水滴から注意深くデカントして分離する。そのような酢酸エチルは、室温における飽和濃度より若干低い濃度で水を含み、段階(a)に用い得る好ましい湿潤脂肪族エステルの1つである。
【0020】
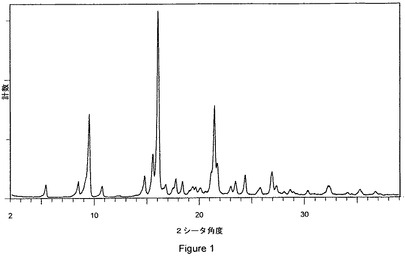
本発明の方法によりペリンドプリルエルブミンの結晶化が湿潤脂肪族エステル中で又は複数の湿潤脂肪族エステルの混合物中で行われる場合、得られるペリンドプリルエルブミン結晶形は、従来技術で公知の結晶形ではなく、異なる粉末X線回折パターンを有するペリンドプリルエルブミンの新規の結晶形に対応することを、驚くべきことに見出した。ペリンドプリルエルブミンの新規結晶形(結晶形Dという名称の)は、次の特徴的な反射角2θ:5.3±0.2°、10.7±0.2°、16.0±0.2°、24.4±0.2°及び26.9±0.2°を含む粉末X線回折パターンを有する。新規結晶形Dは、次の特徴的な2θ角を有する図1に図示する粉末X線回折パターンを有する。
【0021】
【表2】
【0022】
本発明の対象となるもう1つの実施態様は、
(a)湿潤脂肪族エステル中の又は複数の湿潤脂肪族エステルの混合物中の粗製ペリンドプリル溶液を提供する段階、
(b)tert−ブチルアミンを前記溶液に添加する段階、
(c)ペリンドプリルエルブミン結晶形Dを結晶化する段階、及び
(d)ペリンドプリルエルブミン結晶形Dを単離する段階、
を含む、ペリンドプリルエルブミン結晶形Dの調製方法に関する。
【0023】
本発明の方法により、ジケトピペラジン不純物0.20%(重量/重量)未満を、好ましくはジケトピペラジン不純物0.10%(重量/重量)未満を含む純粋なペリンドプリルエルブミン結晶形Dを得ることが可能になる。
【0024】
段階(a)において、該粗製ペリンドプリルの湿潤脂肪族エステル溶液又は湿潤脂肪族エステル混合物溶液は、粗製ペリンドプリルを湿潤脂肪族エステル中に、又は湿潤脂肪族エステル混合物中に溶解し、場合によりその後不溶不純物をろ過により除去することにより提供し得る。もう1つの選択において、段階(a)で、粗製ペリンドプリルの溶液又は懸濁液は、粗製ペリンドプリルの湿潤脂肪族エステル溶液又は湿潤脂肪族エステル混合物溶液を提供するために用い得る。替わりに、粗製ペリンドプリルの溶液は、適切な化学反応により提供され得る。
【0025】
本発明による1つの特定の方法において、段階(a)は、
(a1)粗製ペリンドプリルを水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル又は湿潤脂肪族エステル混合物に溶解する下位段階、及び
(a2)不溶不純物をろ過により除去する下位段階、
を含む。
【0026】
本発明による1つの特定の方法において、段階(a)は、
(a1’)粗製ペリンドプリルを水2%(容量/容量)から4%(容量/容量)を含む湿潤酢酸エチルに溶解する下位段階、及び
(a2’)不溶不純物をろ過により除去する下位段階、
を含む。
【0027】
本発明による1つの特定の方法において、段階(a)は、
(a1’)粗製ペリンドプリルを水を飽和させた湿潤酢酸エチルに溶解する下位段階、及び
(a2’)不溶不純物をろ過により除去する下位段階、
を含む。
【0028】
段階(b)において、tert−ブチルアミンは、好ましくは−20℃及びtert−ブチルアミンの沸点との間の温度で、より好ましくは20℃から40℃の温度で加える。
【0029】
本発明による1つの特定の方法において、段階(c)は、
(c1)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2)得られた沸騰溶液をろ過する下位段階、及び
(c3)得られたろ液を40℃未満に冷却して、結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを得る下位段階、
を含む。
【0030】
下位段階(c3)において、ろ液は好ましくは20℃未満に、より好ましくは−10℃から0℃の温度に冷却する。
【0031】
本発明による1つの特定の方法において、段階(c)は、
(c1’)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2’)得られた沸騰溶液をろ過する下位段階、及び
(c3’)得られたろ液を20℃未満に冷却して、結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを得る下位段階、
を含む。
【0032】
本発明による1つの特定の方法において、段階(c)は、
(c1’’)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2’’)得られた沸騰溶液をろ過する下位段階、及び
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを得る下位段階、
を含む。
【0033】
好ましくは、下位段階(c3)、(c3’)又は(c3’’)により冷却した後得られたペリンドプリルエルブミン結晶形Dを含む混合物は、攪拌せずに又は約15から約60分間、好ましくは約15から約45分間、より好ましくは約30分間放置し、その後ペリンドプリルエルブミン結晶形Dの単離(段階(d))を行う。
【0034】
本発明による1つの特定の方法において、段階(d)は、
(d1)段階(c)で得られた結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dをろ過又は遠心分離により、好ましくはろ過により単離する下位段階、及び
(d2)結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを乾燥する下位段階、
を含む。
【0035】
下位段階(d1)における結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dのろ過は、得られる結晶性ペリンドプリルエルブミンの良好な収率及び品質を保証するために、好ましくは0℃未満の温度で、より好ましくは−20℃から−10℃の温度で行う。
【0036】
下位段階(d2)で行う乾燥の好ましい温度は、25℃から50℃、より好ましくは30℃から40℃である。好ましくは、結晶性ペリンドプリルエルブミンは、恒量となるまで乾燥する。
【0037】
本発明による1つの特定の方法において、段階(d)は、
(d1’)段階(c)で得られた結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを、−20℃から−10℃の温度におけるろ過により単離する下位段階、及び
(d2’)結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する下位段階、
を含む。
【0038】
本発明による1つの好ましい方法は、
(a1)粗製ペリンドプリルを、水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル又は湿潤脂肪族エステル混合物に溶解する下位段階、
(a2)不溶不純物をろ過により除去する下位段階、
(b)tert−ブチルアミンを段階(a2)で得られた溶液に、好ましくは−20℃及びtert−ブチルアミンの沸点との間の温度で加える下位段階、
(c1)段階(b)で得られた混合物を、用いた脂肪族エステル又は湿潤脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2)得られた沸騰溶液をろ過する下位段階、
(c3)得られたろ液を40℃未満に冷却して、結晶性ペリンドプリルエルブミンを得る下位段階、
(d1)段階(c3)で得られた結晶性ペリンドプリルエルブミンをろ過又は遠心分離により、好ましくはろ過により単離する下位段階、及び
(d2)ペリンドプリルエルブミン結晶形Dを乾燥する下位段階、
を含む。
【0039】
本発明による1つの好ましいペリンドプリルエルブミン結晶形D調製方法は、
(a1’)粗製ペリンドプリルを、水を含む湿潤酢酸エチルに溶解する下位段階、
(a2’)不溶不純物をろ過により除去する下位段階、
(b’) tert−ブチルアミンを段階(a2)で得られた溶液に20℃から40℃の温度で加える下位段階、
(c1’’)段階(b’)で得られた混合物を酢酸エチルの沸点まで加熱する下位段階、
(c2’’)得られた沸騰溶液をろ過する下位段階、
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、ペリンドプリルエルブミン結晶形Dを得る下位段階、
(d1’)段階(c3’’)で得られたペリンドプリルエルブミン結晶形Dを−20℃から−10℃の温度におけるろ過により単離する下位段階、及び
(d2’)ペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する下位段階、
を含む。
【0040】
本発明による1つのより好ましいペリンドプリルエルブミン結晶形D調製方法は、
(a1’)粗製ペリンドプリルを、水を含む湿潤酢酸エチルに溶解する下位段階、
(a2’)不溶不純物をろ過により除去する下位段階、
(b’)tert−ブチルアミンを段階(a2)で得られた溶液に20℃から40℃の温度で加える下位段階、
(c1’’)段階(b’)で得られた混合物を酢酸エチルの沸点まで加熱する下位段階、
(c2’’)得られた沸騰溶液をろ過する下位段階、
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、ペリンドプリルエルブミン結晶形Dを得る下位段階、
(c4’’)段階(c3’’)で得られた混合物を攪拌することなく15から60分間放置する下位段階、
(d1’)段階(c4’’)で得られたペリンドプリルエルブミン結晶形Dを−20℃から−10℃の温度におけるろ過により単離する下位段階、及び
(d2’)ペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する下位段階、
を含む。
【0041】
場合により、本発明の方法により得られるペリンドプリルエルブミン結晶形Dは、湿潤脂肪族エステルから、好ましくは湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステル又はその混合物から更に再結晶化し得る。
【0042】
好ましくは新規結晶形Dが得られる、本発明による湿潤脂肪族エステルからのペリンドプリルエルブミンの結晶化は、表1に示すようにジケトピペラジン不純物の大部分を除去するための優れた方法である。
【0043】
【表3】
【0044】
本発明のもう1つの対象は、純粋なペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製、好ましくは高純度ペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製であり前記純粋な又は高純度ペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形がジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含む前記調製のための、ジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミン結晶形Dの使用に関する。
【0045】
本発明のもう1つの対象は、好ましくはジケトピペラジン不純物を0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製のための本発明の方法により調製される結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dの使用に関する。
【0046】
本発明のもう1つの対象は、1つの更なる段階において、段階(d)又は下位段階(d1)、(d1’)、(d2)又は(d2’)後に得られる、ジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミン結晶形Dが、ジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの純粋な結晶形α又はいずれかの他の公知の結晶形に変換される上述した方法のいずれかに関する。
【0047】
本発明のもう1つの対象は、好ましくは水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル若しくは湿潤脂肪族エステル混合物からの、又はより好ましくは2%(容量/容量)から4%(容量/容量)を含む水を飽和させた湿潤脂肪族エステルからのペリンドプリルエルブミンの熱再結晶化を含むペリンドプリルエルブミンの精製方法に関する。
【0048】
ペリンドプリルエルブミンの熱的再結晶に用いる湿潤脂肪族エステルは、好ましくは湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルからなる群より選択される。好ましくは、湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルは湿潤酢酸エチル、湿潤酢酸イソプロピル、湿潤酢酸ブチル及び湿潤プロピオン酸エチルを含むが、これらに限定されない。より好ましくは、ペリンドプリルエルブミンの熱的再結晶に用いる湿潤脂肪族エステルは、湿潤酢酸エチルである。
【0049】
ペリンドプリルエルブミンを精製する好ましい方法は、
(ai)ペリンドプリルエルブミンを、湿潤脂肪族エステル又は湿潤脂肪族エステル混合物に、好ましくは水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル又は水を飽和させた湿潤脂肪族エステル若しくは湿潤脂肪族エステル混合物に溶解する段階、
(bi)場合により、不溶不純物をろ過により除去する段階、
(ci)得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する段階、
(di)場合により、得られた沸騰溶液をろ過する段階、
(ei)得られたろ液を40℃未満に、好ましくは−10℃から0℃の温度に冷却する段階、
(fi)段階(ei)で得られたペリンドプリルエルブミン結晶形Dをろ過又は遠心分離により、好ましくは−20℃から−10℃の温度におけるろ過により単離する段階、及び
(gi)ペリンドプリルエルブミン結晶形Dを、好ましくは30℃から40℃の温度で乾燥する段階、
を含む。
【0050】
本発明のもう1つの対象は、1つの更なる段階において、段階(d)、(d2)、(d2’)又は(gi)後に得られる、結晶性ペリンドプリルエルブミンが、好ましくはペリンドプリルエルブミン結晶形Dが、薬学的に許容できる剤形(特に、前記剤形が錠剤、ピル剤、カプセル剤又は注射用剤である)に製剤される上述した方法のいずれかに関する。
【0051】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミンのいずれかの調製方法により得られる結晶性ペリンドプリルエルブミンである。
【0052】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミンのいずれかの調製方法により得られ得る結晶性ペリンドプリルエルブミンである。
【0053】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミン結晶形Dのいずれかの調製方法により得られるペリンドプリルエルブミン結晶形Dである。
【0054】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミン結晶形Dのいずれかの調製方法により得られ得るペリンドプリルエルブミン結晶形Dである。
【0055】
本発明のもう1つの対象は、ジケトピペラジン不純物を0.20%(重量/重量)未満、好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの治療有効量を、1つ以上の薬学的に許容できる担体又はその他の賦形剤と共に含む医薬組成物に関する。
【0056】
ペリンドプリル塩の治療有効量は、高血圧又は心疾患を治療するために有用な剤形中に適切なペリンドプリルの量を含むペリンドプリル塩の量である。一般に、ペリンドプリルの薬学的有効量は、ペリンドプリルの1から15mg、好ましくは2から8mgである。
【0057】
薬学的に許容できる賦形剤は、結着剤、希釈剤、崩壊剤、安定化剤、防腐剤、滑剤、香料、風味剤、甘味剤、及び製剤技術の分野において公知の他の賦形剤からなる群より選択し得る。好ましくは、担体及び賦形剤は、ヒドロキシプロピルセルロース、ラクトース、微晶質セルロース、炭酸カルシウム、デンプン、コロイド状二酸化ケイ素、デンプングリコール酸ナトリウム、タルク、ステアリン酸マグネシウム、ポリビニルピロリドン、及び製剤技術の分野において公知の他の賦形剤からなる群より選択し得る。
【0058】
場合により、本発明の医薬組成物は、ペリンドプリルに加えて1つ以上の追加の医薬有効成分を含む組み合わせ組成物であり得る。好ましくは、追加の医薬有効成分は、例えばインダパミドなどの利尿薬である。
【0059】
適した医薬組成物は、有効成分の即放性又は徐放性を有する錠剤、発泡性錠剤又は分散錠剤及びカプセル剤などの固形剤形である。
【0060】
該医薬組成物は、製剤技術の分野において公知の方法により調製し得る。
【0061】
もう1つの実施態様において、本発明は、心臓血管疾患(例えば、高血圧又は心不全)の治療における使用のための医薬組成物を調製するためのジケトピペラジン不純物を0.20%(重量/重量)未満、好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの使用に関する。
【0062】
もう1つの実施態様において、本発明は、ジケトピペラジン不純物を0.20%(重量/重量)未満、好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの治療有効量を投与することを含む、心臓血管疾患(例えば、高血圧又は心不全)の治療のための方法に関する。
【発明を実施するための最良の形態】
【0063】
以下の実施例は本発明を例示するが、それらは本発明を限定するものでは決してない。
【実施例1】
【0064】
粗製ペリンドプリルの調製
アセトニトリル225mL中の(2S,3aS,7aS)−2−カルボキシペルヒドロインドールベンジルエステル9.54g、N−((S)−1−カルベトキシブチル)−(S)−アラニン7.26g及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート12.7gの混合物を室温で30分攪拌した後、塩水の560mLを加える。生成物を酢酸エチル400mLで2回抽出し、抽出物を合わせて、最初、水800mLで洗浄し、次に濃塩酸及び水1.5Lで酸性とする。有機相を無水硫酸ナトリウムで乾燥し、真空中40℃で、蒸発させて、ベンジル(2S,3aS,7aS)−((2−(1−(エトキシカルボニル)−(S)−ブチルアミノ)−(S)−プロピオニル)オクタヒドロインドール−2−カルボキシレート(ペリンドプリルのベンジルエステル)13.5g(88%)を得る。
【0065】
粗製ペリンドプリルベンジルエステル(13.5g)をメタノール300mLに溶解する。溶液に触媒(10%活性炭担持パラジウム)1.35gを加える。混合物を、室温で適度な水素気流中で更に5時間攪拌する。次に触媒をろ取してメタノールの50mLで洗浄し、溶液は真空中50℃で蒸発させる。得られる残渣は、無色透明油状化合物としての粗製ペリンドプリル(ジケトピペラジンIの2.33%、ジケトピペラジンIIの0.54%を含む)である。
【実施例2】
【0066】
ペリンドプリルからのペリンドプリルエルブミンの調製
実施例1で得られたペリンドプリル(4.0g、ジケトピペラジンI2.33%、ジケトピペラジンII0.54%を含む)を湿潤酢酸エチル(40mL、含水率4%(容量/容量)に溶解する。不溶不純物をろ別し、ろ液にtert−ブチルアミン(1.5mL)を室温で攪拌しながら加え、混合物を還流まで加熱する。沸騰溶液をろ過し、0℃に冷却する。生成物を沈殿させ、30分後、それをろ過し真空中40℃で24時間乾燥し、結晶形Dのペリンドプリルエルブミンを得る(2.5g、ジケトピペラジンI0.14%、ジケトピペラジンII0.03%を含む)。
【実施例3】
【0067】
飽和湿潤酢酸エチルの調製
酢酸エチル(100mL)を水10mLと共に振とうし、水相を除去する。酢酸エチルを−10℃に冷却し、他の容器に、器壁に付着した水分が室温まで温められるのを避けながらポンプ移送する。
ペリンドプリルからのペリンドプリルエルブミンの調製
実施例1で得られたペリンドプリル(4.0g)を上記で調製した酢酸エチル(40mL)に溶解する。不溶不純物をろ別し、ろ液にtert−ブチルアミン(1.5mL)を室温で攪拌しながら加え、混合物を還流まで加熱する。沸騰溶液をろ過し、0℃に冷却する。生成物を沈殿させ、30分後それをろ過し真空中40℃で24時間乾燥し、結晶形Dのペリンドプリルエルブミンを得る(2.9g)。
【実施例4】
【0068】
ペリンドプリルエルブミン結晶形Dのペリンドプリルエルブミン結晶形αからの調製
実施例3で調製した、ペリンドプリルエルブミン(5g)及び湿潤酢酸エチル(30mL)の混合物を攪拌しながら還流まで加熱する。溶液を場合によりろ過し、0℃に冷却する。生成物を沈殿させる。30分後、得られた懸濁液をろ過し、沈殿物を真空中40℃で24時間乾燥して、ペリンドプリルエルブミン結晶形Dを得る(4.15g)。
【実施例5】
【0069】
ペリンドプリルエルブミン結晶形Dのペリンドプリルエルブミン結晶形αからの調製
ペリンドプリルエルブミン(5g)及び湿潤酢酸イソプロピル(酢酸イソプロピル30mL及び水1mLから調製)の混合物を攪拌しながら還流まで加熱する。溶液を場合によりろ過し、−10℃に冷却した後、攪拌せずに−10℃で1時間放置する。得られた懸濁液をろ過し、沈殿物を真空中40℃で24時間乾燥して、ペリンドプリルエルブミン結晶形Dを得る。
【0070】
実施例中の分析データは、下記ハードウェアにより得る。
【0071】
試料の粉末X線回折スペクトルは、反射技術を備えたSiemens D−5000(CuKα放射線源、測定範囲:2°から37°2θ、ステップ:0.04°2θ、積分時間:1秒)で記録する。
【0072】
ジケトピペラジン定量のクロマトグラフ条件:
1.移動相:
A:ヘプタンスルホン酸ナトリウム0.92gを水1000mLに溶解し、トリエチルアミン1mLを加えて、過塩素酸及び水との混合物でpH2.0に調整する。
B:アセトニトリル
2.カラム:C8,4μm、6nmの孔径、250×4.0mm(Merck Supersphere 60 RP−8)
3.条件:温度:70℃、流速:1.5mL/分、波長:215nm、注入容量:20μL、勾配表:
【0073】
【表4】
4.ペリンドプリルについての相対的保持(約11分)
ジケトピペラジンII−0.56
ジケトピペラジンI−1.7
5.装置:Water Alliance2695分離モジュール、検出器PDA2996、ソフトウェアEmpower5.0
【図面の簡単な説明】
【0074】
【図1】本発明の方法により得られるペリンドプリルエルブミン結晶形DのX線回折グラフの図である。
【技術分野】
【0001】
本発明は、純粋なペリンドプリルエルブミンの新規の調製方法に関する。本発明は又、ペリンドプリルエルブミンの結晶形Dの新規調製方法にも関する。
【背景技術】
【0002】
ペリンドプリル及びその薬学的に許容できる塩は、アンジオテンシン変換酵素阻害薬として公知であり、循環器疾患の治療、特に高血圧及び心不全の治療に用いられている。ペリンドプリルは、(2S,3aS,7aS)−((2−(1−(エトキシカルボニル)−(S)−ブチルアミノ)−(S)−プロピオニル)オクタヒドロ−インドール−2−カルボン酸として化学的に公知であり、式(I):
【0003】
【化1】
により表し得る。
【0004】
ペリンドプリルは、EP0049658B1及びUS4,508,729に光学的純粋なS,S,S,S,S異性体として及びナトリウム塩の形態で初めて開示された。その後の数多くの後続特許及び特許出願(EP0308341B1、EP1279665A1、EP1333026A1、WO2004/099236など)が、ペリンドプリルの種々の調製方法を記載している。
【0005】
医薬製品に広く用いられているペリンドプリルエルブミンとして公知のペリンドプリルのtert−ブチルアミン塩は、EP0308341B1に初めて開示された。ペリンドプリルエルブミンは、結晶化条件(例えば、溶媒系、ペリンドプリルエルブミン濃度及び冷却力学など)に依り種々の結晶形で得られる。EP1296947B1は、酢酸エチルからのペリンドプリルエルブミン結晶形αの結晶化を開示し、EP1294689Aは、ジクロロメタン又は酢酸エチルからのペリンドプリルエルブミン結晶形βの結晶化を開示し、EP1296948B1は、クロロホルムからのペリンドプリルエルブミン結晶形γの結晶化を開示し、及びWO2004/113293は、ペリンドプリルエルブミン結晶形δ及び結晶形εの結晶化を開示している。結晶形εは、水1.5から2.5%(容量/容量)を含むtert−ブチルメチルエーテルからの結晶化により得られる一方、結晶形δは、共沸蒸留により結晶形εから得られる。
【0006】
EP0308341B1は、ジシクロヘキシルカルボジイミド及び1−ヒドロキシベンゾトリアゾールの存在下で、保護された(2S,3aS,7aS)−2−オクタヒドロインドール−2−カルボン酸をN−[(S)−1−カルベトキシブチル]−(S)−アラニンと結合することによるペリンドプリルの工業的調製方法を記載している。ペリンドプリルエルブミンは、粗製ペリンドプリルからtert−ブチルアミン添加後の結晶化により得られる。この方法の欠点は、除去することが困難なジシクロヘキシルカルボジイミドから派生し得る副生成物の生成である。この理由で、追加の精製段階が純粋なペリンドプリル及び/又は純粋なペリンドプリルエルブミンを得るために必要である。
【0007】
調製方法中に生成する副生成物に加えて、ペリンドプリルの分解生成物も又、不純物として粗製ペリンドプリル中に存在する。ペリンドプリル及びその塩は、化学的に高度に反応しやすい化合物であり、a)いくつかのキラル中心における異性体化、b)側鎖エステル基の加水分解、及び/又はc)分子内環化によるジケトピペラジンの形成、を介して分解しやすい。2つの最も重要なジケトピペラジンは、ジケトピペラジンIと称される(R)−エチル2−((3S,5aS,9aS,10aS)−3−メチル−1,4−ジオキソ−デカヒドロピラジノ[1,2−a]インドール−2(1H)−イル)ペンタノエート及びジケトピペラジンIIと称される2−((3S,5aS,9aS,10aR)−3−メチル−1,4−ジオキソ−デカヒドロピラジノ[1,2−a]インドール−2(1H)−イル)ペンタン酸(これらの式は下記のとおり)であり、これらは又、欧州局方5.1にもそれぞれ不純物F及びCとして示されている。
【0008】
【化2】
【0009】
WO01/58868は、(2S,3aS,7aS)−オクタヒドロインドール−2−カルボン酸ベンジルパラ−トルエンスルホン酸エステル及びN−[(S)−エトキシカルボニル−1−ブチル]−(S)−アラニンからのジシクロヘキシルカルボジイミド、1−ヒドロキシベンゾトリアゾール及び場合によりトリエチルアミンの存在下でのペリンドプリルの改良された調製方法を記載している。ジシクロヘキシルカルボジイミドから派生し得る副生成物の形成は、この方法に用いられる試薬の量の厳密な制御により低減される。しかしながら、記載された方法自身は、他の不純物の問題を解決しない。
【0010】
ジシクロヘキシルカルボジイミドから派生し得る不純物の問題は、ジシクロヘキシルカルボジイミドの使用を避けるペリンドプリルの調製方法により完全に解決される。例えば、そのような方法が特許出願EP1279665A1及びEP1333026A1中に記載されている。しかしながら、tert−ブチルアミンと、これらの特許出願中に開示されている方法により調製される粗製ペリンドプリルとの反応により得られるペリンドプリルエルブミンは、熱的再結晶又は活性炭処理などの別の精製方法により、更に精製しなければならない。
【0011】
WO2004/046172も又、高純度ペリンドプリルエルブミンの調製方法を記載しているということができ、この方法では、ペリンドプリルの保護された前駆体を塩基(例えば、tert−ブチルアミン)の存在下で脱保護して、粗製ペリンドプリルを単離することなく直接ペリンドプリル塩(例えば、ペリンドプリルエルブミン)を得る。製造方法中のジケトピペラジン不純物の生成は、反応時間が短いため最小限に抑えられる。しかしながら、他の不純物は、得られたペリンドプリルエルブミンから追加の結晶化段階により除去しなければならない。
【0012】
WO2005/019173は、粗製ペリンドプリル又はその塩の水溶液を適した有機溶媒を用いてpH4.0から6.5で抽出した後、有機層を分離し、tert−ブチルアミンを加えてペリンドプリルエルブミンを調製することによる、粗製ペリンドプリルからの純粋なペリンドプリルエルブミンの調製方法を開示している。この方法の欠点は、段階の数が多すぎて低収率になり得ることである。
【発明の開示】
【発明が解決しようとする課題】
【0013】
得られたペリンドプリルエルブミンの追加の精製が必要なく、工業的規模で応用可能な、粗製ペリンドプリルから純粋なペリンドプリルエルブミンを調製するための簡便で効果的な方法を開発する必要性が引き続き存在する。
【0014】
本発明は、粗製ペリンドプリルからの純粋なペリンドプリルエルブミンの改良された調製方法を提供し、前記方法は特にジケトピペラジン不純物の除去に効果的である。更に、前記方法は、簡便であり工業的規模で応用可能である。
【課題を解決するための手段】
【0015】
本発明の第1の対象は、
(a)湿潤脂肪族エステル中の又は複数の湿潤脂肪族エステルの混合物中の粗製ペリンドプリル溶液を提供する段階、
(b)tert−ブチルアミンを該溶液に添加する段階、
(c)ペリンドプリルエルブミンを結晶化する段階、及び
(d)結晶性ペリンドプリルエルブミンを単離する段階、
を含む、結晶性ペリンドプリルエルブミンの調製方法に関する。
【0016】
本発明の方法により、ジケトピペラジン不純物の0.20%(重量/重量)未満を、好ましくはジケトピペラジン不純物の0.10%(重量/重量)未満を含む純粋なペリンドプリルエルブミンを得ることが可能になる。
【0017】
本発明における「湿潤脂肪族エステル」は、含水率を高めた又は水を飽和させた脂肪族エステルを意味する。好ましくは、含水率を高めた湿潤脂肪族エステルは、水1%(容量/容量)から6%(容量/容量)を、より好ましくは水2%(容量/容量)から4%(容量/容量)を、最も好ましくは水2%(容量/容量)から3%(容量/容量)を含む。
【0018】
段階(a)において、湿潤脂肪族エステルは好ましくは、湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルからなる群より選択される。好ましくは湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルは、湿潤酢酸エチル、湿潤酢酸イソプロピル、湿潤酢酸ブチル及び湿潤プロピオン酸エチルを含むが、これらに限定されない。より好ましくは段階(a)で用いる湿潤脂肪族エステルは、湿潤酢酸エチルである。好ましくは、段階(a)で用いる湿潤酢酸エチルは水1%(容量/容量)から6%(容量/容量)を、より好ましくは水2%(容量/容量)から4%(容量/容量)を、最も好ましくは水2%(容量/容量)から3%(容量/容量)を含む。
【0019】
酢酸エチルの飽和は、分液ロート中で水と振り混ぜ、更に水相を分離することにより実施し得る。残留水滴を避けるために、酢酸エチル相を冷却し(好ましくは−20℃から−10℃の温度に)、水滴から注意深くデカントして分離する。そのような酢酸エチルは、室温における飽和濃度より若干低い濃度で水を含み、段階(a)に用い得る好ましい湿潤脂肪族エステルの1つである。
【0020】
本発明の方法によりペリンドプリルエルブミンの結晶化が湿潤脂肪族エステル中で又は複数の湿潤脂肪族エステルの混合物中で行われる場合、得られるペリンドプリルエルブミン結晶形は、従来技術で公知の結晶形ではなく、異なる粉末X線回折パターンを有するペリンドプリルエルブミンの新規の結晶形に対応することを、驚くべきことに見出した。ペリンドプリルエルブミンの新規結晶形(結晶形Dという名称の)は、次の特徴的な反射角2θ:5.3±0.2°、10.7±0.2°、16.0±0.2°、24.4±0.2°及び26.9±0.2°を含む粉末X線回折パターンを有する。新規結晶形Dは、次の特徴的な2θ角を有する図1に図示する粉末X線回折パターンを有する。
【0021】
【表2】
【0022】
本発明の対象となるもう1つの実施態様は、
(a)湿潤脂肪族エステル中の又は複数の湿潤脂肪族エステルの混合物中の粗製ペリンドプリル溶液を提供する段階、
(b)tert−ブチルアミンを前記溶液に添加する段階、
(c)ペリンドプリルエルブミン結晶形Dを結晶化する段階、及び
(d)ペリンドプリルエルブミン結晶形Dを単離する段階、
を含む、ペリンドプリルエルブミン結晶形Dの調製方法に関する。
【0023】
本発明の方法により、ジケトピペラジン不純物0.20%(重量/重量)未満を、好ましくはジケトピペラジン不純物0.10%(重量/重量)未満を含む純粋なペリンドプリルエルブミン結晶形Dを得ることが可能になる。
【0024】
段階(a)において、該粗製ペリンドプリルの湿潤脂肪族エステル溶液又は湿潤脂肪族エステル混合物溶液は、粗製ペリンドプリルを湿潤脂肪族エステル中に、又は湿潤脂肪族エステル混合物中に溶解し、場合によりその後不溶不純物をろ過により除去することにより提供し得る。もう1つの選択において、段階(a)で、粗製ペリンドプリルの溶液又は懸濁液は、粗製ペリンドプリルの湿潤脂肪族エステル溶液又は湿潤脂肪族エステル混合物溶液を提供するために用い得る。替わりに、粗製ペリンドプリルの溶液は、適切な化学反応により提供され得る。
【0025】
本発明による1つの特定の方法において、段階(a)は、
(a1)粗製ペリンドプリルを水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル又は湿潤脂肪族エステル混合物に溶解する下位段階、及び
(a2)不溶不純物をろ過により除去する下位段階、
を含む。
【0026】
本発明による1つの特定の方法において、段階(a)は、
(a1’)粗製ペリンドプリルを水2%(容量/容量)から4%(容量/容量)を含む湿潤酢酸エチルに溶解する下位段階、及び
(a2’)不溶不純物をろ過により除去する下位段階、
を含む。
【0027】
本発明による1つの特定の方法において、段階(a)は、
(a1’)粗製ペリンドプリルを水を飽和させた湿潤酢酸エチルに溶解する下位段階、及び
(a2’)不溶不純物をろ過により除去する下位段階、
を含む。
【0028】
段階(b)において、tert−ブチルアミンは、好ましくは−20℃及びtert−ブチルアミンの沸点との間の温度で、より好ましくは20℃から40℃の温度で加える。
【0029】
本発明による1つの特定の方法において、段階(c)は、
(c1)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2)得られた沸騰溶液をろ過する下位段階、及び
(c3)得られたろ液を40℃未満に冷却して、結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを得る下位段階、
を含む。
【0030】
下位段階(c3)において、ろ液は好ましくは20℃未満に、より好ましくは−10℃から0℃の温度に冷却する。
【0031】
本発明による1つの特定の方法において、段階(c)は、
(c1’)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2’)得られた沸騰溶液をろ過する下位段階、及び
(c3’)得られたろ液を20℃未満に冷却して、結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを得る下位段階、
を含む。
【0032】
本発明による1つの特定の方法において、段階(c)は、
(c1’’)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2’’)得られた沸騰溶液をろ過する下位段階、及び
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを得る下位段階、
を含む。
【0033】
好ましくは、下位段階(c3)、(c3’)又は(c3’’)により冷却した後得られたペリンドプリルエルブミン結晶形Dを含む混合物は、攪拌せずに又は約15から約60分間、好ましくは約15から約45分間、より好ましくは約30分間放置し、その後ペリンドプリルエルブミン結晶形Dの単離(段階(d))を行う。
【0034】
本発明による1つの特定の方法において、段階(d)は、
(d1)段階(c)で得られた結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dをろ過又は遠心分離により、好ましくはろ過により単離する下位段階、及び
(d2)結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを乾燥する下位段階、
を含む。
【0035】
下位段階(d1)における結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dのろ過は、得られる結晶性ペリンドプリルエルブミンの良好な収率及び品質を保証するために、好ましくは0℃未満の温度で、より好ましくは−20℃から−10℃の温度で行う。
【0036】
下位段階(d2)で行う乾燥の好ましい温度は、25℃から50℃、より好ましくは30℃から40℃である。好ましくは、結晶性ペリンドプリルエルブミンは、恒量となるまで乾燥する。
【0037】
本発明による1つの特定の方法において、段階(d)は、
(d1’)段階(c)で得られた結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを、−20℃から−10℃の温度におけるろ過により単離する下位段階、及び
(d2’)結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する下位段階、
を含む。
【0038】
本発明による1つの好ましい方法は、
(a1)粗製ペリンドプリルを、水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル又は湿潤脂肪族エステル混合物に溶解する下位段階、
(a2)不溶不純物をろ過により除去する下位段階、
(b)tert−ブチルアミンを段階(a2)で得られた溶液に、好ましくは−20℃及びtert−ブチルアミンの沸点との間の温度で加える下位段階、
(c1)段階(b)で得られた混合物を、用いた脂肪族エステル又は湿潤脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2)得られた沸騰溶液をろ過する下位段階、
(c3)得られたろ液を40℃未満に冷却して、結晶性ペリンドプリルエルブミンを得る下位段階、
(d1)段階(c3)で得られた結晶性ペリンドプリルエルブミンをろ過又は遠心分離により、好ましくはろ過により単離する下位段階、及び
(d2)ペリンドプリルエルブミン結晶形Dを乾燥する下位段階、
を含む。
【0039】
本発明による1つの好ましいペリンドプリルエルブミン結晶形D調製方法は、
(a1’)粗製ペリンドプリルを、水を含む湿潤酢酸エチルに溶解する下位段階、
(a2’)不溶不純物をろ過により除去する下位段階、
(b’) tert−ブチルアミンを段階(a2)で得られた溶液に20℃から40℃の温度で加える下位段階、
(c1’’)段階(b’)で得られた混合物を酢酸エチルの沸点まで加熱する下位段階、
(c2’’)得られた沸騰溶液をろ過する下位段階、
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、ペリンドプリルエルブミン結晶形Dを得る下位段階、
(d1’)段階(c3’’)で得られたペリンドプリルエルブミン結晶形Dを−20℃から−10℃の温度におけるろ過により単離する下位段階、及び
(d2’)ペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する下位段階、
を含む。
【0040】
本発明による1つのより好ましいペリンドプリルエルブミン結晶形D調製方法は、
(a1’)粗製ペリンドプリルを、水を含む湿潤酢酸エチルに溶解する下位段階、
(a2’)不溶不純物をろ過により除去する下位段階、
(b’)tert−ブチルアミンを段階(a2)で得られた溶液に20℃から40℃の温度で加える下位段階、
(c1’’)段階(b’)で得られた混合物を酢酸エチルの沸点まで加熱する下位段階、
(c2’’)得られた沸騰溶液をろ過する下位段階、
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、ペリンドプリルエルブミン結晶形Dを得る下位段階、
(c4’’)段階(c3’’)で得られた混合物を攪拌することなく15から60分間放置する下位段階、
(d1’)段階(c4’’)で得られたペリンドプリルエルブミン結晶形Dを−20℃から−10℃の温度におけるろ過により単離する下位段階、及び
(d2’)ペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する下位段階、
を含む。
【0041】
場合により、本発明の方法により得られるペリンドプリルエルブミン結晶形Dは、湿潤脂肪族エステルから、好ましくは湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステル又はその混合物から更に再結晶化し得る。
【0042】
好ましくは新規結晶形Dが得られる、本発明による湿潤脂肪族エステルからのペリンドプリルエルブミンの結晶化は、表1に示すようにジケトピペラジン不純物の大部分を除去するための優れた方法である。
【0043】
【表3】
【0044】
本発明のもう1つの対象は、純粋なペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製、好ましくは高純度ペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製であり前記純粋な又は高純度ペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形がジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含む前記調製のための、ジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミン結晶形Dの使用に関する。
【0045】
本発明のもう1つの対象は、好ましくはジケトピペラジン不純物を0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製のための本発明の方法により調製される結晶性ペリンドプリルエルブミン、好ましくはペリンドプリルエルブミン結晶形Dの使用に関する。
【0046】
本発明のもう1つの対象は、1つの更なる段階において、段階(d)又は下位段階(d1)、(d1’)、(d2)又は(d2’)後に得られる、ジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミン結晶形Dが、ジケトピペラジン不純物を好ましくは0.20%(重量/重量)未満、より好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの純粋な結晶形α又はいずれかの他の公知の結晶形に変換される上述した方法のいずれかに関する。
【0047】
本発明のもう1つの対象は、好ましくは水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル若しくは湿潤脂肪族エステル混合物からの、又はより好ましくは2%(容量/容量)から4%(容量/容量)を含む水を飽和させた湿潤脂肪族エステルからのペリンドプリルエルブミンの熱再結晶化を含むペリンドプリルエルブミンの精製方法に関する。
【0048】
ペリンドプリルエルブミンの熱的再結晶に用いる湿潤脂肪族エステルは、好ましくは湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルからなる群より選択される。好ましくは、湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルは湿潤酢酸エチル、湿潤酢酸イソプロピル、湿潤酢酸ブチル及び湿潤プロピオン酸エチルを含むが、これらに限定されない。より好ましくは、ペリンドプリルエルブミンの熱的再結晶に用いる湿潤脂肪族エステルは、湿潤酢酸エチルである。
【0049】
ペリンドプリルエルブミンを精製する好ましい方法は、
(ai)ペリンドプリルエルブミンを、湿潤脂肪族エステル又は湿潤脂肪族エステル混合物に、好ましくは水1%(容量/容量)から6%(容量/容量)を含む湿潤脂肪族エステル又は水を飽和させた湿潤脂肪族エステル若しくは湿潤脂肪族エステル混合物に溶解する段階、
(bi)場合により、不溶不純物をろ過により除去する段階、
(ci)得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する段階、
(di)場合により、得られた沸騰溶液をろ過する段階、
(ei)得られたろ液を40℃未満に、好ましくは−10℃から0℃の温度に冷却する段階、
(fi)段階(ei)で得られたペリンドプリルエルブミン結晶形Dをろ過又は遠心分離により、好ましくは−20℃から−10℃の温度におけるろ過により単離する段階、及び
(gi)ペリンドプリルエルブミン結晶形Dを、好ましくは30℃から40℃の温度で乾燥する段階、
を含む。
【0050】
本発明のもう1つの対象は、1つの更なる段階において、段階(d)、(d2)、(d2’)又は(gi)後に得られる、結晶性ペリンドプリルエルブミンが、好ましくはペリンドプリルエルブミン結晶形Dが、薬学的に許容できる剤形(特に、前記剤形が錠剤、ピル剤、カプセル剤又は注射用剤である)に製剤される上述した方法のいずれかに関する。
【0051】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミンのいずれかの調製方法により得られる結晶性ペリンドプリルエルブミンである。
【0052】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミンのいずれかの調製方法により得られ得る結晶性ペリンドプリルエルブミンである。
【0053】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミン結晶形Dのいずれかの調製方法により得られるペリンドプリルエルブミン結晶形Dである。
【0054】
本発明のもう1つの対象は、本発明によるペリンドプリルエルブミン結晶形Dのいずれかの調製方法により得られ得るペリンドプリルエルブミン結晶形Dである。
【0055】
本発明のもう1つの対象は、ジケトピペラジン不純物を0.20%(重量/重量)未満、好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの治療有効量を、1つ以上の薬学的に許容できる担体又はその他の賦形剤と共に含む医薬組成物に関する。
【0056】
ペリンドプリル塩の治療有効量は、高血圧又は心疾患を治療するために有用な剤形中に適切なペリンドプリルの量を含むペリンドプリル塩の量である。一般に、ペリンドプリルの薬学的有効量は、ペリンドプリルの1から15mg、好ましくは2から8mgである。
【0057】
薬学的に許容できる賦形剤は、結着剤、希釈剤、崩壊剤、安定化剤、防腐剤、滑剤、香料、風味剤、甘味剤、及び製剤技術の分野において公知の他の賦形剤からなる群より選択し得る。好ましくは、担体及び賦形剤は、ヒドロキシプロピルセルロース、ラクトース、微晶質セルロース、炭酸カルシウム、デンプン、コロイド状二酸化ケイ素、デンプングリコール酸ナトリウム、タルク、ステアリン酸マグネシウム、ポリビニルピロリドン、及び製剤技術の分野において公知の他の賦形剤からなる群より選択し得る。
【0058】
場合により、本発明の医薬組成物は、ペリンドプリルに加えて1つ以上の追加の医薬有効成分を含む組み合わせ組成物であり得る。好ましくは、追加の医薬有効成分は、例えばインダパミドなどの利尿薬である。
【0059】
適した医薬組成物は、有効成分の即放性又は徐放性を有する錠剤、発泡性錠剤又は分散錠剤及びカプセル剤などの固形剤形である。
【0060】
該医薬組成物は、製剤技術の分野において公知の方法により調製し得る。
【0061】
もう1つの実施態様において、本発明は、心臓血管疾患(例えば、高血圧又は心不全)の治療における使用のための医薬組成物を調製するためのジケトピペラジン不純物を0.20%(重量/重量)未満、好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの使用に関する。
【0062】
もう1つの実施態様において、本発明は、ジケトピペラジン不純物を0.20%(重量/重量)未満、好ましくは0.10%(重量/重量)未満含むペリンドプリルエルブミンの治療有効量を投与することを含む、心臓血管疾患(例えば、高血圧又は心不全)の治療のための方法に関する。
【発明を実施するための最良の形態】
【0063】
以下の実施例は本発明を例示するが、それらは本発明を限定するものでは決してない。
【実施例1】
【0064】
粗製ペリンドプリルの調製
アセトニトリル225mL中の(2S,3aS,7aS)−2−カルボキシペルヒドロインドールベンジルエステル9.54g、N−((S)−1−カルベトキシブチル)−(S)−アラニン7.26g及びO−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート12.7gの混合物を室温で30分攪拌した後、塩水の560mLを加える。生成物を酢酸エチル400mLで2回抽出し、抽出物を合わせて、最初、水800mLで洗浄し、次に濃塩酸及び水1.5Lで酸性とする。有機相を無水硫酸ナトリウムで乾燥し、真空中40℃で、蒸発させて、ベンジル(2S,3aS,7aS)−((2−(1−(エトキシカルボニル)−(S)−ブチルアミノ)−(S)−プロピオニル)オクタヒドロインドール−2−カルボキシレート(ペリンドプリルのベンジルエステル)13.5g(88%)を得る。
【0065】
粗製ペリンドプリルベンジルエステル(13.5g)をメタノール300mLに溶解する。溶液に触媒(10%活性炭担持パラジウム)1.35gを加える。混合物を、室温で適度な水素気流中で更に5時間攪拌する。次に触媒をろ取してメタノールの50mLで洗浄し、溶液は真空中50℃で蒸発させる。得られる残渣は、無色透明油状化合物としての粗製ペリンドプリル(ジケトピペラジンIの2.33%、ジケトピペラジンIIの0.54%を含む)である。
【実施例2】
【0066】
ペリンドプリルからのペリンドプリルエルブミンの調製
実施例1で得られたペリンドプリル(4.0g、ジケトピペラジンI2.33%、ジケトピペラジンII0.54%を含む)を湿潤酢酸エチル(40mL、含水率4%(容量/容量)に溶解する。不溶不純物をろ別し、ろ液にtert−ブチルアミン(1.5mL)を室温で攪拌しながら加え、混合物を還流まで加熱する。沸騰溶液をろ過し、0℃に冷却する。生成物を沈殿させ、30分後、それをろ過し真空中40℃で24時間乾燥し、結晶形Dのペリンドプリルエルブミンを得る(2.5g、ジケトピペラジンI0.14%、ジケトピペラジンII0.03%を含む)。
【実施例3】
【0067】
飽和湿潤酢酸エチルの調製
酢酸エチル(100mL)を水10mLと共に振とうし、水相を除去する。酢酸エチルを−10℃に冷却し、他の容器に、器壁に付着した水分が室温まで温められるのを避けながらポンプ移送する。
ペリンドプリルからのペリンドプリルエルブミンの調製
実施例1で得られたペリンドプリル(4.0g)を上記で調製した酢酸エチル(40mL)に溶解する。不溶不純物をろ別し、ろ液にtert−ブチルアミン(1.5mL)を室温で攪拌しながら加え、混合物を還流まで加熱する。沸騰溶液をろ過し、0℃に冷却する。生成物を沈殿させ、30分後それをろ過し真空中40℃で24時間乾燥し、結晶形Dのペリンドプリルエルブミンを得る(2.9g)。
【実施例4】
【0068】
ペリンドプリルエルブミン結晶形Dのペリンドプリルエルブミン結晶形αからの調製
実施例3で調製した、ペリンドプリルエルブミン(5g)及び湿潤酢酸エチル(30mL)の混合物を攪拌しながら還流まで加熱する。溶液を場合によりろ過し、0℃に冷却する。生成物を沈殿させる。30分後、得られた懸濁液をろ過し、沈殿物を真空中40℃で24時間乾燥して、ペリンドプリルエルブミン結晶形Dを得る(4.15g)。
【実施例5】
【0069】
ペリンドプリルエルブミン結晶形Dのペリンドプリルエルブミン結晶形αからの調製
ペリンドプリルエルブミン(5g)及び湿潤酢酸イソプロピル(酢酸イソプロピル30mL及び水1mLから調製)の混合物を攪拌しながら還流まで加熱する。溶液を場合によりろ過し、−10℃に冷却した後、攪拌せずに−10℃で1時間放置する。得られた懸濁液をろ過し、沈殿物を真空中40℃で24時間乾燥して、ペリンドプリルエルブミン結晶形Dを得る。
【0070】
実施例中の分析データは、下記ハードウェアにより得る。
【0071】
試料の粉末X線回折スペクトルは、反射技術を備えたSiemens D−5000(CuKα放射線源、測定範囲:2°から37°2θ、ステップ:0.04°2θ、積分時間:1秒)で記録する。
【0072】
ジケトピペラジン定量のクロマトグラフ条件:
1.移動相:
A:ヘプタンスルホン酸ナトリウム0.92gを水1000mLに溶解し、トリエチルアミン1mLを加えて、過塩素酸及び水との混合物でpH2.0に調整する。
B:アセトニトリル
2.カラム:C8,4μm、6nmの孔径、250×4.0mm(Merck Supersphere 60 RP−8)
3.条件:温度:70℃、流速:1.5mL/分、波長:215nm、注入容量:20μL、勾配表:
【0073】
【表4】
4.ペリンドプリルについての相対的保持(約11分)
ジケトピペラジンII−0.56
ジケトピペラジンI−1.7
5.装置:Water Alliance2695分離モジュール、検出器PDA2996、ソフトウェアEmpower5.0
【図面の簡単な説明】
【0074】
【図1】本発明の方法により得られるペリンドプリルエルブミン結晶形DのX線回折グラフの図である。
【特許請求の範囲】
【請求項1】
(a)湿潤脂肪族エステル中の又は湿潤脂肪族エステルの混合物中の粗製ペリンドプリル溶液を提供する段階、
(b)tert−ブチルアミンを前記溶液に添加する段階、
(c)ペリンドプリルエルブミンを結晶化する段階、及び
(d)結晶性ペリンドプリルエルブミンを単離する段階、
を含む、結晶性ペリンドプリルエルブミンの調製方法。
【請求項2】
前記湿潤脂肪族エステルが、湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルからなる群より選択される、請求項1に記載の方法。
【請求項3】
前記湿潤脂肪族エステルが、湿潤酢酸エチルである、請求項1又は2に記載の方法。
【請求項4】
前記湿潤酢酸エチルが、水1%(容量/容量)から6%(容量/容量)を含む、請求項3に記載の方法。
【請求項5】
前記湿潤酢酸エチルが、水2%(容量/容量)から4%(容量/容量)を含む、請求項3又は4に記載の方法。
【請求項6】
前記湿潤酢酸エチルが、−20℃から−10℃の温度において水を飽和させることにより調製される、請求項3に記載の方法。
【請求項7】
段階(b)において、tert−ブチルアミンが、20℃から40℃の温度で加えられる、請求項1から6のいずれか1項に記載の方法。
【請求項8】
段階(c)が、
(c1)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2)得られた沸騰溶液をろ過する下位段階、及び
(c3)得られたろ液を40℃未満に冷却して、結晶性ペリンドプリルエルブミンを得る下位段階、
を含む、請求項1から7のいずれか1項に記載の方法。
【請求項9】
下位段階(c3)において、前記ろ液を−10℃から0℃の温度に冷却する請求項8に記載の方法。
【請求項10】
段階(d)が、
(d1)段階(c)で得られた結晶性ペリンドプリルエルブミンをろ過又は遠心分離により単離する下位段階、及び
(d2)結晶性ペリンドプリルエルブミンを乾燥する下位段階、
を含む、請求項1から9のいずれか1項に記載の方法。
【請求項11】
下位段階(d1)における前記ろ過が、0℃未満の温度で行われる、請求項10に記載の方法。
【請求項12】
下位段階(d1)における前記ろ過が、−20℃から−10℃の温度で行われる、請求項10又は11に記載の方法。
【請求項13】
段階(d)で得られる前記結晶性ペリンドプリルエルブミンが、ジケトピペラジン不純物約0.20%(重量/重量)未満を含む、請求項1から12のいずれか1項に記載の方法。
【請求項14】
前記結晶性ペリンドプリルエルブミンが、ペリンドプリルエルブミン結晶形Dである、請求項1から13のいずれか1項に記載の方法。
【請求項15】
前記ペリンドプリルエルブミン結晶形Dが、次の特徴的な反射角2θ:5.3±0.2°、10.7±0.2°、16.0±0.2°、24.4±0.2°及び26.9±0.2°を含む粉末X線回折パターンを有する、請求項14に記載の方法。
【請求項16】
前記ペリンドプリルエルブミン結晶形Dが、次の特徴的な2θ角:
【表1】
を含む粉末X線回折パターンを有する、請求項14又は15に記載の方法。
【請求項17】
(a1’)粗製ペリンドプリルを、水を飽和させた湿潤酢酸エチルに溶解する段階、
(a2’)不溶不純物をろ過により除去する段階、
(b’)tert−ブチルアミンを段階(a2)で得られた溶液に20℃から40℃の温度で加える段階、
(c1’’)段階(b’)で得られた混合物を酢酸エチルの沸点まで加熱する段階、
(c2’’)得られた沸騰溶液をろ過する段階、
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、ペリンドプリルエルブミン結晶形Dを得る段階、
(d1’)段階(c3’’)で得られたペリンドプリルエルブミン結晶形Dを−20℃から−10℃の温度におけるろ過により単離する段階、及び
(d2’)ペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する段階、
を含む、ペリンドプリルエルブミン結晶形Dの調製方法。
【請求項18】
ペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製のための、請求項1から18のいずれか1項により調製される結晶性ペリンドプリルエルブミンの使用。
【請求項19】
湿潤脂肪族エステル又は湿潤脂肪族エステル混合物からのペリンドプリルエルブミンの熱再結晶化を含むペリンドプリルエルブミンの精製方法。
【請求項20】
請求項1から13のいずれか1項に記載の方法により得られる結晶性ペリンドプリルエルブミン。
【請求項21】
請求項14から17のいずれか1項に記載の方法により得られるペリンドプリルエルブミン結晶形D。
【請求項22】
請求項14から17のいずれか1項に記載の方法により得られ得るペリンドプリルエルブミン結晶形D。
【請求項23】
1つの更なる段階において、段階(d)後に得られる結晶性ペリンドプリルエルブミンが、薬学的に許容できる剤形に製剤される、請求項1から16のいずれか1項に記載の方法。
【請求項24】
1つの更なる段階において、段階(d2’)後に得られるペリンドプリルエルブミン形晶形Dが、薬学的に許容できる剤形に製剤される、請求項17に記載の方法。
【請求項1】
(a)湿潤脂肪族エステル中の又は湿潤脂肪族エステルの混合物中の粗製ペリンドプリル溶液を提供する段階、
(b)tert−ブチルアミンを前記溶液に添加する段階、
(c)ペリンドプリルエルブミンを結晶化する段階、及び
(d)結晶性ペリンドプリルエルブミンを単離する段階、
を含む、結晶性ペリンドプリルエルブミンの調製方法。
【請求項2】
前記湿潤脂肪族エステルが、湿潤C1−C4脂肪族カルボン酸C1−C4アルキルエステルからなる群より選択される、請求項1に記載の方法。
【請求項3】
前記湿潤脂肪族エステルが、湿潤酢酸エチルである、請求項1又は2に記載の方法。
【請求項4】
前記湿潤酢酸エチルが、水1%(容量/容量)から6%(容量/容量)を含む、請求項3に記載の方法。
【請求項5】
前記湿潤酢酸エチルが、水2%(容量/容量)から4%(容量/容量)を含む、請求項3又は4に記載の方法。
【請求項6】
前記湿潤酢酸エチルが、−20℃から−10℃の温度において水を飽和させることにより調製される、請求項3に記載の方法。
【請求項7】
段階(b)において、tert−ブチルアミンが、20℃から40℃の温度で加えられる、請求項1から6のいずれか1項に記載の方法。
【請求項8】
段階(c)が、
(c1)段階(b)で得られた混合物を、用いた脂肪族エステル又は脂肪族エステル混合物の沸点まで加熱する下位段階、
(c2)得られた沸騰溶液をろ過する下位段階、及び
(c3)得られたろ液を40℃未満に冷却して、結晶性ペリンドプリルエルブミンを得る下位段階、
を含む、請求項1から7のいずれか1項に記載の方法。
【請求項9】
下位段階(c3)において、前記ろ液を−10℃から0℃の温度に冷却する請求項8に記載の方法。
【請求項10】
段階(d)が、
(d1)段階(c)で得られた結晶性ペリンドプリルエルブミンをろ過又は遠心分離により単離する下位段階、及び
(d2)結晶性ペリンドプリルエルブミンを乾燥する下位段階、
を含む、請求項1から9のいずれか1項に記載の方法。
【請求項11】
下位段階(d1)における前記ろ過が、0℃未満の温度で行われる、請求項10に記載の方法。
【請求項12】
下位段階(d1)における前記ろ過が、−20℃から−10℃の温度で行われる、請求項10又は11に記載の方法。
【請求項13】
段階(d)で得られる前記結晶性ペリンドプリルエルブミンが、ジケトピペラジン不純物約0.20%(重量/重量)未満を含む、請求項1から12のいずれか1項に記載の方法。
【請求項14】
前記結晶性ペリンドプリルエルブミンが、ペリンドプリルエルブミン結晶形Dである、請求項1から13のいずれか1項に記載の方法。
【請求項15】
前記ペリンドプリルエルブミン結晶形Dが、次の特徴的な反射角2θ:5.3±0.2°、10.7±0.2°、16.0±0.2°、24.4±0.2°及び26.9±0.2°を含む粉末X線回折パターンを有する、請求項14に記載の方法。
【請求項16】
前記ペリンドプリルエルブミン結晶形Dが、次の特徴的な2θ角:
【表1】
を含む粉末X線回折パターンを有する、請求項14又は15に記載の方法。
【請求項17】
(a1’)粗製ペリンドプリルを、水を飽和させた湿潤酢酸エチルに溶解する段階、
(a2’)不溶不純物をろ過により除去する段階、
(b’)tert−ブチルアミンを段階(a2)で得られた溶液に20℃から40℃の温度で加える段階、
(c1’’)段階(b’)で得られた混合物を酢酸エチルの沸点まで加熱する段階、
(c2’’)得られた沸騰溶液をろ過する段階、
(c3’’)得られたろ液を−10℃から0℃の温度に冷却して、ペリンドプリルエルブミン結晶形Dを得る段階、
(d1’)段階(c3’’)で得られたペリンドプリルエルブミン結晶形Dを−20℃から−10℃の温度におけるろ過により単離する段階、及び
(d2’)ペリンドプリルエルブミン結晶形Dを30℃から40℃の温度で乾燥する段階、
を含む、ペリンドプリルエルブミン結晶形Dの調製方法。
【請求項18】
ペリンドプリルエルブミン結晶形α又はいずれかの他の公知の結晶形の調製のための、請求項1から18のいずれか1項により調製される結晶性ペリンドプリルエルブミンの使用。
【請求項19】
湿潤脂肪族エステル又は湿潤脂肪族エステル混合物からのペリンドプリルエルブミンの熱再結晶化を含むペリンドプリルエルブミンの精製方法。
【請求項20】
請求項1から13のいずれか1項に記載の方法により得られる結晶性ペリンドプリルエルブミン。
【請求項21】
請求項14から17のいずれか1項に記載の方法により得られるペリンドプリルエルブミン結晶形D。
【請求項22】
請求項14から17のいずれか1項に記載の方法により得られ得るペリンドプリルエルブミン結晶形D。
【請求項23】
1つの更なる段階において、段階(d)後に得られる結晶性ペリンドプリルエルブミンが、薬学的に許容できる剤形に製剤される、請求項1から16のいずれか1項に記載の方法。
【請求項24】
1つの更なる段階において、段階(d2’)後に得られるペリンドプリルエルブミン形晶形Dが、薬学的に許容できる剤形に製剤される、請求項17に記載の方法。
【図1】

【公表番号】特表2009−504595(P2009−504595A)
【公表日】平成21年2月5日(2009.2.5)
【国際特許分類】
【出願番号】特願2008−525486(P2008−525486)
【出願日】平成18年8月10日(2006.8.10)
【国際出願番号】PCT/EP2006/007926
【国際公開番号】WO2007/020012
【国際公開日】平成19年2月22日(2007.2.22)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
【公表日】平成21年2月5日(2009.2.5)
【国際特許分類】
【出願日】平成18年8月10日(2006.8.10)
【国際出願番号】PCT/EP2006/007926
【国際公開番号】WO2007/020012
【国際公開日】平成19年2月22日(2007.2.22)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
[ Back to top ]