
ペリンドプリル
【課題】なし
【解決手段】
式(II)の保護された前駆化合物から、式(I)のペリンドプリルの薬剤として許容し得る塩を生産し:
【化9】

(式中、Rは、カルボキシル基の保護基を表す。)、前記方法は、対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合した、該カルボキシル基COORを脱保護することを含み、前記脱保護は、塩基の存在下で行い、前記脱保護により形成される前記遊離酸を有する、薬剤として許容し得る塩を形成する。
【解決手段】
式(II)の保護された前駆化合物から、式(I)のペリンドプリルの薬剤として許容し得る塩を生産し:
【化9】
(式中、Rは、カルボキシル基の保護基を表す。)、前記方法は、対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合した、該カルボキシル基COORを脱保護することを含み、前記脱保護は、塩基の存在下で行い、前記脱保護により形成される前記遊離酸を有する、薬剤として許容し得る塩を形成する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ペリンドプリルの薬剤として許容し得る塩の生産方法、及びその新規多形相に関するものである。
【背景技術】
【0002】
ペリンドプリルとは、国際一般的名称(2S,3aS,7aS)-1-{2-[1-(エトキシカルボニル)-(S)-ブチルアミノ]-(S)-プロピオニル}-オクタヒドロインドール-2-カルボン酸のことである。ペリンドプリルは、アンギオテンシン-転換酵素(ACE)阻害剤のような、治療的応用があることが知られている。ACEは、ペプチジルジペプチターゼであり、アンギオテンシンIからアンギオテンシンIIへの変換を触媒し、かつブラジキニンの分解を引き起こす。アンギオテンシンIIは、血管収縮薬であり、また、該副腎皮質により、アルドステロン分泌を刺激する。従って、ACE阻害は、高血圧症、及び鬱血性心不全のような、疾患状態に苦しむ患者に対して、治療的有用性があることを示している。さらに、ACE阻害剤は、認識力障害の治療に有用であることが見つかっている。
【0003】
ペリンドプリルは、下記構造式(I)を有する。
【化5】
ペリンドプリルは、米国特許第4508729号に記載されている。この米国特許に記載されている生産方法は、アルコール性媒体中、及び中性の脱水剤と有機、又は無機水素化シアノホウ素との存在下で行われる。必要に応じて、脱保護工程は、例えば、加水分解法、及び/又は水素化分解法に準拠して行われる。
【0004】
米国特許第4914214号には、ペリンドプリル、及びそのt-ブチルアミン塩の生産方法が記載されている。該方法は、エステル保護された(2S,3aS,7aS)-2-カルボキシペルヒドロインドールと(S,S)ジアステレオ異性体のN-[(S)-1-カルボエトキシブチル]-(S)-アラニンとを縮合し、続いて、パラジウム5%、及び水を含む木炭を用いて脱保護することを含む。次いで、ターシャリー-ブチルアミンを加え、ペリンドプリルのt-ブチルアミン塩を得る。
【0005】
国際特許出願のWO01/87835には、新規結晶形態(すなわち、α結晶形態)のペリンドプリルt-ブチルアミン塩が記載されており、該塩、及び該塩を含む医薬製剤の生産方法が記載されている。
国際特許出願WO01/87836には、新規結晶形態(すなわち、∃結晶形態)のペリンドプリルt-ブチルアミン塩が記載されており、該塩、及び該塩を含む医薬製剤の生産方法が記載されている。
国際特許出願WO01/87835には、新規結晶形態(すなわち、γ結晶形態)のペリンドプリルt-ブチルアミン塩が記載されており、該塩、及び該塩を含む医薬製剤の生産方法が記載されている。
国際特許出願WO01/58868には、ペリンドプリル、又はその医薬として許容し得る塩の生産方法が記載されており、該方法は、改善された純度のペリンドプリル、又はその塩を提供している。さらに、国際特許出願WO01/58868に従って生産された、ペリンドプリル、又はその塩に関して、公知の不純物水準は、0.2〜0.1重量%よりも少ないことが記載されている。中間の工程段階を、1-ヒドロキシベンゾトリアゾール、ジシクロヘキシルカルボジイミド、及び任意のトリエチルアミンの存在下、20〜77ECの範囲内の温度で行い、続いて、脱保護し、かつ要求される塩に変換する。
【発明の開示】
【発明が解決しようとする課題】
【0006】
一般に、ペリンドプリル、又はその医薬として許容し得る塩を生産する先行技術の方法は、多大な時間を必要とし、かつ多くの場合、ジケトピペラジン類似体のような、望ましくない、関連不純物を生じる。従って、上述の問題を解決するように、ペリンドプリル、又はその医薬として許容し得る塩の生産方法を改善する必要がある。
【課題を解決するための手段】
【0007】
今回、我々は、ペリンドプリルの医薬として許容し得る塩の公知である生産方法と比べて、反応時間の早さに関して、及び高純度生成物となるように、望ましくない不純物の生成を防ぐことに関して有利である、ペリンドプリルの医薬として許容し得る塩の生産方法を開発した。
【発明を実施するための最良の形態】
【0008】
本発明の一態様において、式(II)の保護された前駆化合物から、式(I)のペリンドプリルの医薬として許容し得る塩を生産する方法を提供する:
【化6】
(式中、Rは、カルボキシル基の保護基を表す。)。前記方法は、対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合しているカルボキシル基COORの脱保護を行うことを含み、前記脱保護を塩基の存在下で行い、前記脱保護により形成される前記遊離酸を有する、医薬として許容し得る塩を形成する。通常、Rは、適切なカルボキル基の保護基を表すことができ、本発明の方法により、選択的に除去することができる。好ましくは、Rは、任意に置換されたアラルキル基、特に、任意に置換されたベンジル基を表し得る。従って、通常、Rは、無置換ベンジル基を表し得るが;代わりに、4-ハロ置換、又は4-C1-4アルコキシ置換ベンジル基、特に、4-Clベンジル基、又は4-メトキシベンジル基のような、置換ベンジル基を使用してもよい。
【0009】
適切に、本発明の方法において行う脱保護は、貴金属触媒(好ましくは、パラジウム-オン-チャコール(palladium-on-chacoal))の存在下での水素化分解を含み得る。
本発明の方法は、高純度生成物が得られる点において有利である。好ましくは、本発明の方法により生産される、ペリンドプリルの医薬として許容し得る塩は、純度約99%w/wを超え、かつさらに好ましくは、純度約99.5%w/wを超える。本発明の方法により生産される、ペリンドプリルの医薬として許容し得る塩の純度は、酢酸エチル、又はイソプロパノールなどのような、適切な溶媒中、任意の結晶化工程でさらに向上させることができ、好ましくは、純度約99.8%w/wである、ペリンドプリルの医薬として許容し得る塩を得ることができる。
好ましくは、本明細書中で使用する該塩基を、上述のように、該脱保護により形成される遊離酸を有する、医薬として許容し得る塩を形成するように選択し、それによって、該反応のワークアップから直接的に、ペリンドプリルの医薬として許容し得る塩を得ることができる。本発明の特に好ましい実施態様において、該塩基は、t-ブチルアミンであり、かつ本発明のそのような好ましい方法は、該反応工程から直接的に、ペリンドプリルの高純度t-ブチルアミン塩を与え得る。
【0010】
本発明の上述の好ましい実施態様において、前述したように、実質的に、式(II)の保護された前駆化合物(好ましくは、Rがベンジル基である、式(II)のベンジル保護基を有する前駆化合物)から、ペリンドプリルt-ブチルアミン(当業者にとって、ペリンドプリルエルブミンとして周知のものである。)を生産する方法を提供し、前記方法は、対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合したカルボキシル基COORの脱保護(好ましくは、パラジウム-オン-チャコールのような、貴金属触媒の存在下の水素化分解)を行うことを含み、前記脱保護は、ペリンドプリルのt-ブチルアミン塩を形成するように、t-ブチルアミンの存在下で行われる。
【0011】
適切には、式(II)の前駆化合物を、最初、イソプロパノールなどのような、アルカノール溶媒中に溶解し、続いて、そこに該塩基を加える。さらに続いて、適切に、パラジウム-オン-チャコールを加え、かつ数時間、水素化分解することにより、該カルボキシル基COORを脱保護する。適切には、該アルカノール溶媒を、減圧下で濃縮し、酢酸エチルなどのような、水の不混和性溶媒に置き換える。次いで、該得られた個体を冷やし、かつ濾過して、ペリンドプリルの医薬として許容し得る塩を得ることができる。
実質的に、前述のような本発明の方法は、該方法により得られるペリンドプリルの医薬として許容し得る塩を水和することを含み、式(Ia)の水和ペリンドプリルの医薬として許容し得る塩を得ることができる:
【0012】
【化7】
【0013】
(式中、nは1〜5の整数であり、又は整数2〜5の逆数である。)。該水和は、水を加える方法により、又は空気中で乾燥することにより行うことができる。
好ましくは、nは1であり、それによって、ペリンドプリル一水和物の医薬として許容し得る塩を、本発明の方法により形成する。
また、本発明は、ペリンドプリルの医薬として許容し得る塩の一水和物の生産方法を提供し、前記方法は、ペリンドプリルの医薬として許容し得る塩を水和し、前記一水和物を得ることを含む。水和を、水を加える方法により、又は空気中で乾燥することにより、行うことができ、かつ好ましくは、ペリンドプリルt-ブチルアミンを水和し、ペリンドプリルt-ブチルアミン一水和物を得る。
【0014】
さらに、本発明は、実質的に、前述の方法により生産した、任意に水和した形態のペリンドプリルの医薬として許容し得る塩を提供する。特に、式(Ia)の水和ペリンドプリルの医薬として許容し得る塩を提供する:
【化8】
(式中、nは1〜5の整数であり、又は整数2〜5の逆数である。)。好ましくは、nは1である。式(Ia)の水和ペリンドプリルの好ましい医薬として許容し得る塩は、該t-ブチルアミン塩である。特に好ましい実施態様において、本発明は、ペリンドプリルt-ブチルアミン(又はエルブミン)一水和物を提供する。
【0015】
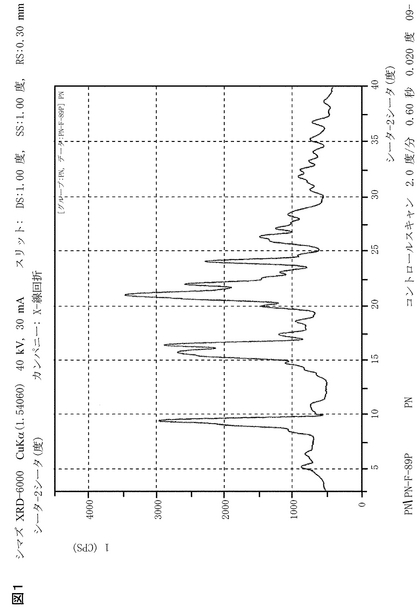
また、本発明は、図1に提示するような、X-線回折特性、又は実質的に同一のX-線回折特性を有する、ペリンドプリルt-ブチルアミン一水和物を提供する。さらに、本発明のペリンドプリルt-ブチルアミン一水和物は、下記特徴的ピーク(2θ)のX-線粉末回折パターンを有することを特徴とし得る:9.5504、14.8600、15.7486、16.5400、20.0400、21.0499、22.0600、24.1744、26.3300、及び27.1600である。
さらに、X-線回折から得られた、本発明のペリンドプリル t-ブチルアミン一水和物の特性データを下記表1に示す。
【0016】
【表1】
【0017】
本発明により提供されるペリンドプリルは、ACE阻害剤のように、治療的有用性を有する。
加えて、さらに、本発明は、ACE阻害が必要な患者のACEの阻害方法を提供し、該方法は、前記患者に、ACE阻害に有効な量の、本発明に従って提供されるペリンドプリル(好ましくは、ペリンドプリルt-ブチルアミン一水和物)を投与することを含む。
また、本発明は、ACE阻害薬の製造において、本発明に従って提供されるペリンドプリル(好ましくは、ペリンドプリルt-ブチルアミン一水和物)の使用を提供する。
【0018】
例えば、患者が、高血圧症、又は慢性鬱血性心不全などに苦しんでいる場合、該患者は、ACE阻害の処置を必要とし得る。ACE阻害は、アンギオテンシンIIの水準を低下させ、かつ従って、それによって誘発される、該血管収縮、血圧上昇、及び高アルドステロン作用を阻害する。また、ACE阻害は、ブラジキニンの内在的水準を増強するであろう。本発明に従って提供されるペリンドプリルのACE阻害に有効な量は、例えば、血圧低下作用を必要とする患者のACE阻害に有効な量である。
患者の有効な処置において、本発明に従って提供されるペリンドプリルを、経口、及び非経口の投与法を含む、有効量において生物的に利用できる該化合物で構成された、すべての形態、又は様式で投与することができる。例えば、本発明に従って提供されるペリンドプリルを、経口、皮下、筋肉内、静脈内、経皮、神経内、及び直腸などに投与することができる。通常、経口投与が好ましい。製剤を生産する当業者は、処置されるべき該疾患状態に依存し、かつ該疾患の段階に依存する投与において、適切な形態及び様式を、容易に選択することができる。
【0019】
本発明に従って提供されるペリンドプリルを、薬剤組成物、又は薬の形態で投与することができ、そのために、これらを、本発明のペリンドプリルと、医薬として許容し得るキャリア、希釈剤、又は賦形剤とを組合わせて生産し、該割合、及びその種類を、選択された投与方法、及び標準的薬務により決定する。
他の態様において、本発明は、一以上の医薬として許容し得るキャリア、希釈剤、又は賦形剤とともに、ACE阻害に有効な量の、本発明に従って提供された該ペリンドプリルを含む、薬剤組成物を提供する。
"医薬として許容し得る"とは、該キャリア、希釈剤、又は賦形剤が、本発明に従って提供された該ペリンドプリルに適合し、かつその受容に関して有害性はないことを意味する。
【0020】
該薬剤組成物、又は薬を、製薬技術において周知である方法により生産する。該キャリア、希釈剤、又は賦形剤は、固体、半固体、又は液体物質であってもよく、ビヒクル、又は該有効成分の媒体として役立ち得る。適切なキャリア、希釈剤、又は賦形剤は、当業者にとって、周知のものである。本発明の薬剤組成物を、経口、又は非経口用に適合させることができ、かつ該患者に、錠剤、カプセル、坐剤、溶液、又は懸濁液などの形態で投与することができる。
【0021】
該薬剤組成物を、例えば、不活性希釈剤とともに、又は食用キャリアとともに、経口投与することができる。これらを、ゼラチンカプセルに囲む、又は錠剤中に包含することができる。経口治療投与のため、本発明の一水和物を、賦形剤と組合わせ、かつ錠剤、カプセル、エリキシル剤、懸濁液、及びシロップ剤などの形態で使用することができる。
また、該錠剤、ピル、及びカプセルなどは、一以上の下記アジュバントを含むことができる:微結晶性セルロース、トラガカントゴム、又はゼラチンのような結合剤;デンプン、又はラクトースのような賦形剤;アルギン酸、及びコーンスターチなどのような崩壊剤;ステアリン酸マグネシウムのような潤滑剤;コロイド状二酸化ケイ素のような流動促進剤;及びサッカロース、又はサッカリンのような甘味剤である。該投与単位形態が、カプセルである場合、上述の種類の物質に加え、ポリエチレングリコール、又は脂肪油のような液体キャリアを含むことができる。他の投与単位形態では、該投与単位の物理的形態を改質する、他の様々な物質(例えば、コーティング剤)を含むことができる。従って、錠剤、又はピルを、糖、ゼラック、又は他の腸溶コーティング剤で覆うことができる。シロップ剤は、該有効成分に加え、甘味剤としてサッカロースを含み、かつ防腐剤を含むことができる。これらの様々な組成物の生産に使用する物質は、薬剤的に純粋であり、かつ使用量において、無毒性であるべきである。
【0022】
本発明に従って提供されるペリンドプリルを、非経口投与するため、溶液、又は懸濁液と組合わせることができる。また、該溶液、又は懸濁液は、一以上の下記アジュバントを含むことができる:注射用蒸留水、食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコール、又は他の合成溶媒のような無菌希釈剤;ベンジルアルコール、又はメチルパラベンのような抗菌剤;アスコルビン酸、又は亜硫酸水素ナトリウムのような酸化防止剤;エチレンジアミン四酢酸のようなキレート剤;及びアセタート、クエン酸塩、又はリン酸塩のような緩衝液である。該非経口的調製物を、ガラス、又はプラスチック製の、アンプル、使い捨てシリンジ、又は多人数用バイアルで囲うことができる。
さらに、今回の本発明を、下記図面、及び実施例により説明するが、形はどうであれ、本発明の範囲を制限するものではない。
【実施例】
【0023】
(実施例1)
(2S,3aS,7aS)-1-{2-[1-(エトキシカルボニル)-(S)-ブチルアミノ]-(S)-プロピオニル}-オクタヒドロインドール-2-カルボン酸のベンジルエステル、すなわち、ベンジルペリンドプリル(10 gms)を、イソプロパノール(100 ml)中に溶解した。該透明な液に、t-ブチルアミン(2.5 gms)、及び10%w/wのパラジウム-オン-チャコール(2 gms)を加えた。該反応混合液を、1 kg/cm2の圧力下で2時間、水素化した。
該反応塊を濾過し、該触媒を除去した。該溶媒を、減圧下で濃縮し、かつイソプロパノールを、酢酸エチルの同時添加により、置き換えた。該得られた固体を、0 ECに冷却し、かつ濾過し、ペリンドプリルエルブミン(7.8 gms)を得た。
【0024】
(実施例2)
ペリンドプリルエルブミン(10 gms)を、アセトン(80 ml)中に懸濁させた。これに、水(0.4 ml)を加え、かつ該内容物を加熱し、該固体を溶解させ、かつ周囲の温度に冷却した。該得られた懸濁液を濾過し、ペリンドプリルエルブミン一水和物(9.4 gms)を得た。
(実施例3)
ペリンドプリルエルブミン(20 gms)を、酢酸エチル(300 ml)中に懸濁させた。これに、水(1.5 ml)を加え、かつ該内容物を加熱し、該固体を溶解させ、かつ10 ECに冷却した。該得られた懸濁液を濾過し、ペリンドプリルエルブミン一水和物(17 gms)を得た。
【0025】
(実施例4)
ペリンドプリルエルブミン(5 gms)を、アセトニトリル(75 ml)中に懸濁させた。これに、水(0.4 ml)を加え、かつ該内容物を加熱し、該固体を溶解させ、かつ0 ECに冷却した。該得られた懸濁液を濾過し、ペリンドプリルエルブミン一水和物(2.9 gms)を得た。
(実施例5)
ペリンドプリルエルブミン(20 gms)を、酢酸エチル(300 ml)中に懸濁させた。該内容物を加熱し、該固体を溶解させ、かつ10 ECに冷却した。該得られた懸濁液を濾過し、かつ少なくとも相対湿度75%の空気中で乾燥させ、ペリンドプリルエルブミン一水和物(17 gms)を得た。
【0026】
(実施例6)
(ペリンドプリルエルブミン一水和物の生産)
原料:−
1.ペリンドプリルエルブミン無水物 = 10 gm
2.イソプロピルアルコール = 70 ml
3.水 = 2 ml
4.酢酸エチル = 85 ml
手順:−
1.ペリンドプリルエルブミン(無水物)10 gmを、丸底フラスコに入れる。イソプロピルアルコール70 mlを加える。1/2時間攪拌する(約95%の生成物が溶解される。)。
2.水2 mlを加える。15分間攪拌する(透明溶液が得られる。)。
3.反応塊を、38〜40℃で2時間攪拌する。
4.イソプロピルアルコールを、減圧下(600 mm未満)、40℃未満で、完全に蒸留する(ゲル型物質が観察される。)。
5.酢酸エチル30 mlを加える。40℃未満で、15分間攪拌する(透明溶液が観察される)減圧下、40℃未満で、蒸留する(半固体が観察される。)。
6.酢酸エチル40 mlを、36〜38℃で加える。15分間攪拌する(自由固体(free solid)が観察される。)。
7.1時間室温(25〜30℃)で攪拌する(遊離結晶性固体が観察される。)。
8.10℃に冷却する。2時間攪拌する。
9.固体を濾過し、かつ酢酸エチル15 mlで洗浄する。2時間搾取して乾燥させる。
10.減圧下、40℃未満で12時間乾燥させる。
含水率 = 3.2〜3.8%
融点 = 145〜150℃
(実施例7)
下記錠剤を製造した。
【0027】
【表2】
【0028】
手順:上の成分を、それぞれのふるいを通して、ふるい分けをする。該成分を、適切なブレンダーで混合する。該錠剤を、適切な成形型に圧縮する。
【表3】
【0029】
手順:
1)ペリンドプリルエルブミン一水和物を、エタノール中に溶解する。
2)硬化ひまし油以外の上の成分を、上の溶液とともに顆粒状にする。該顆粒、及びサイズ剤を乾燥させる。
3)適切なブレンダー内に、硬化ひまし油を用いて滑らかにする。該顆粒を、適切な成形型に圧縮する。
【図面の簡単な説明】
【0030】
【図1】本発明のペリンドプリルエルブミン一水和物のX-線回折パターン。シマズ-6000 X-線回折計(Shimadzu-6000 x-ray diffractometer)を用いて、該試料を分析した。該光源は、波長1.5406Åを有するCuのKα単色光を使用した。該発散スリット(Divergent Slit)は、1°を使用した。該受光スリット(Receiving Slit)は、0.30 mmとした。シンチレーションカウンター(Scintillation counter)は、3°〜40°の範囲で、1分間に2°の走査速度を有する検出器として使用した。
【技術分野】
【0001】
本発明は、ペリンドプリルの薬剤として許容し得る塩の生産方法、及びその新規多形相に関するものである。
【背景技術】
【0002】
ペリンドプリルとは、国際一般的名称(2S,3aS,7aS)-1-{2-[1-(エトキシカルボニル)-(S)-ブチルアミノ]-(S)-プロピオニル}-オクタヒドロインドール-2-カルボン酸のことである。ペリンドプリルは、アンギオテンシン-転換酵素(ACE)阻害剤のような、治療的応用があることが知られている。ACEは、ペプチジルジペプチターゼであり、アンギオテンシンIからアンギオテンシンIIへの変換を触媒し、かつブラジキニンの分解を引き起こす。アンギオテンシンIIは、血管収縮薬であり、また、該副腎皮質により、アルドステロン分泌を刺激する。従って、ACE阻害は、高血圧症、及び鬱血性心不全のような、疾患状態に苦しむ患者に対して、治療的有用性があることを示している。さらに、ACE阻害剤は、認識力障害の治療に有用であることが見つかっている。
【0003】
ペリンドプリルは、下記構造式(I)を有する。
【化5】
ペリンドプリルは、米国特許第4508729号に記載されている。この米国特許に記載されている生産方法は、アルコール性媒体中、及び中性の脱水剤と有機、又は無機水素化シアノホウ素との存在下で行われる。必要に応じて、脱保護工程は、例えば、加水分解法、及び/又は水素化分解法に準拠して行われる。
【0004】
米国特許第4914214号には、ペリンドプリル、及びそのt-ブチルアミン塩の生産方法が記載されている。該方法は、エステル保護された(2S,3aS,7aS)-2-カルボキシペルヒドロインドールと(S,S)ジアステレオ異性体のN-[(S)-1-カルボエトキシブチル]-(S)-アラニンとを縮合し、続いて、パラジウム5%、及び水を含む木炭を用いて脱保護することを含む。次いで、ターシャリー-ブチルアミンを加え、ペリンドプリルのt-ブチルアミン塩を得る。
【0005】
国際特許出願のWO01/87835には、新規結晶形態(すなわち、α結晶形態)のペリンドプリルt-ブチルアミン塩が記載されており、該塩、及び該塩を含む医薬製剤の生産方法が記載されている。
国際特許出願WO01/87836には、新規結晶形態(すなわち、∃結晶形態)のペリンドプリルt-ブチルアミン塩が記載されており、該塩、及び該塩を含む医薬製剤の生産方法が記載されている。
国際特許出願WO01/87835には、新規結晶形態(すなわち、γ結晶形態)のペリンドプリルt-ブチルアミン塩が記載されており、該塩、及び該塩を含む医薬製剤の生産方法が記載されている。
国際特許出願WO01/58868には、ペリンドプリル、又はその医薬として許容し得る塩の生産方法が記載されており、該方法は、改善された純度のペリンドプリル、又はその塩を提供している。さらに、国際特許出願WO01/58868に従って生産された、ペリンドプリル、又はその塩に関して、公知の不純物水準は、0.2〜0.1重量%よりも少ないことが記載されている。中間の工程段階を、1-ヒドロキシベンゾトリアゾール、ジシクロヘキシルカルボジイミド、及び任意のトリエチルアミンの存在下、20〜77ECの範囲内の温度で行い、続いて、脱保護し、かつ要求される塩に変換する。
【発明の開示】
【発明が解決しようとする課題】
【0006】
一般に、ペリンドプリル、又はその医薬として許容し得る塩を生産する先行技術の方法は、多大な時間を必要とし、かつ多くの場合、ジケトピペラジン類似体のような、望ましくない、関連不純物を生じる。従って、上述の問題を解決するように、ペリンドプリル、又はその医薬として許容し得る塩の生産方法を改善する必要がある。
【課題を解決するための手段】
【0007】
今回、我々は、ペリンドプリルの医薬として許容し得る塩の公知である生産方法と比べて、反応時間の早さに関して、及び高純度生成物となるように、望ましくない不純物の生成を防ぐことに関して有利である、ペリンドプリルの医薬として許容し得る塩の生産方法を開発した。
【発明を実施するための最良の形態】
【0008】
本発明の一態様において、式(II)の保護された前駆化合物から、式(I)のペリンドプリルの医薬として許容し得る塩を生産する方法を提供する:
【化6】
(式中、Rは、カルボキシル基の保護基を表す。)。前記方法は、対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合しているカルボキシル基COORの脱保護を行うことを含み、前記脱保護を塩基の存在下で行い、前記脱保護により形成される前記遊離酸を有する、医薬として許容し得る塩を形成する。通常、Rは、適切なカルボキル基の保護基を表すことができ、本発明の方法により、選択的に除去することができる。好ましくは、Rは、任意に置換されたアラルキル基、特に、任意に置換されたベンジル基を表し得る。従って、通常、Rは、無置換ベンジル基を表し得るが;代わりに、4-ハロ置換、又は4-C1-4アルコキシ置換ベンジル基、特に、4-Clベンジル基、又は4-メトキシベンジル基のような、置換ベンジル基を使用してもよい。
【0009】
適切に、本発明の方法において行う脱保護は、貴金属触媒(好ましくは、パラジウム-オン-チャコール(palladium-on-chacoal))の存在下での水素化分解を含み得る。
本発明の方法は、高純度生成物が得られる点において有利である。好ましくは、本発明の方法により生産される、ペリンドプリルの医薬として許容し得る塩は、純度約99%w/wを超え、かつさらに好ましくは、純度約99.5%w/wを超える。本発明の方法により生産される、ペリンドプリルの医薬として許容し得る塩の純度は、酢酸エチル、又はイソプロパノールなどのような、適切な溶媒中、任意の結晶化工程でさらに向上させることができ、好ましくは、純度約99.8%w/wである、ペリンドプリルの医薬として許容し得る塩を得ることができる。
好ましくは、本明細書中で使用する該塩基を、上述のように、該脱保護により形成される遊離酸を有する、医薬として許容し得る塩を形成するように選択し、それによって、該反応のワークアップから直接的に、ペリンドプリルの医薬として許容し得る塩を得ることができる。本発明の特に好ましい実施態様において、該塩基は、t-ブチルアミンであり、かつ本発明のそのような好ましい方法は、該反応工程から直接的に、ペリンドプリルの高純度t-ブチルアミン塩を与え得る。
【0010】
本発明の上述の好ましい実施態様において、前述したように、実質的に、式(II)の保護された前駆化合物(好ましくは、Rがベンジル基である、式(II)のベンジル保護基を有する前駆化合物)から、ペリンドプリルt-ブチルアミン(当業者にとって、ペリンドプリルエルブミンとして周知のものである。)を生産する方法を提供し、前記方法は、対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合したカルボキシル基COORの脱保護(好ましくは、パラジウム-オン-チャコールのような、貴金属触媒の存在下の水素化分解)を行うことを含み、前記脱保護は、ペリンドプリルのt-ブチルアミン塩を形成するように、t-ブチルアミンの存在下で行われる。
【0011】
適切には、式(II)の前駆化合物を、最初、イソプロパノールなどのような、アルカノール溶媒中に溶解し、続いて、そこに該塩基を加える。さらに続いて、適切に、パラジウム-オン-チャコールを加え、かつ数時間、水素化分解することにより、該カルボキシル基COORを脱保護する。適切には、該アルカノール溶媒を、減圧下で濃縮し、酢酸エチルなどのような、水の不混和性溶媒に置き換える。次いで、該得られた個体を冷やし、かつ濾過して、ペリンドプリルの医薬として許容し得る塩を得ることができる。
実質的に、前述のような本発明の方法は、該方法により得られるペリンドプリルの医薬として許容し得る塩を水和することを含み、式(Ia)の水和ペリンドプリルの医薬として許容し得る塩を得ることができる:
【0012】
【化7】
【0013】
(式中、nは1〜5の整数であり、又は整数2〜5の逆数である。)。該水和は、水を加える方法により、又は空気中で乾燥することにより行うことができる。
好ましくは、nは1であり、それによって、ペリンドプリル一水和物の医薬として許容し得る塩を、本発明の方法により形成する。
また、本発明は、ペリンドプリルの医薬として許容し得る塩の一水和物の生産方法を提供し、前記方法は、ペリンドプリルの医薬として許容し得る塩を水和し、前記一水和物を得ることを含む。水和を、水を加える方法により、又は空気中で乾燥することにより、行うことができ、かつ好ましくは、ペリンドプリルt-ブチルアミンを水和し、ペリンドプリルt-ブチルアミン一水和物を得る。
【0014】
さらに、本発明は、実質的に、前述の方法により生産した、任意に水和した形態のペリンドプリルの医薬として許容し得る塩を提供する。特に、式(Ia)の水和ペリンドプリルの医薬として許容し得る塩を提供する:
【化8】
(式中、nは1〜5の整数であり、又は整数2〜5の逆数である。)。好ましくは、nは1である。式(Ia)の水和ペリンドプリルの好ましい医薬として許容し得る塩は、該t-ブチルアミン塩である。特に好ましい実施態様において、本発明は、ペリンドプリルt-ブチルアミン(又はエルブミン)一水和物を提供する。
【0015】
また、本発明は、図1に提示するような、X-線回折特性、又は実質的に同一のX-線回折特性を有する、ペリンドプリルt-ブチルアミン一水和物を提供する。さらに、本発明のペリンドプリルt-ブチルアミン一水和物は、下記特徴的ピーク(2θ)のX-線粉末回折パターンを有することを特徴とし得る:9.5504、14.8600、15.7486、16.5400、20.0400、21.0499、22.0600、24.1744、26.3300、及び27.1600である。
さらに、X-線回折から得られた、本発明のペリンドプリル t-ブチルアミン一水和物の特性データを下記表1に示す。
【0016】
【表1】
【0017】
本発明により提供されるペリンドプリルは、ACE阻害剤のように、治療的有用性を有する。
加えて、さらに、本発明は、ACE阻害が必要な患者のACEの阻害方法を提供し、該方法は、前記患者に、ACE阻害に有効な量の、本発明に従って提供されるペリンドプリル(好ましくは、ペリンドプリルt-ブチルアミン一水和物)を投与することを含む。
また、本発明は、ACE阻害薬の製造において、本発明に従って提供されるペリンドプリル(好ましくは、ペリンドプリルt-ブチルアミン一水和物)の使用を提供する。
【0018】
例えば、患者が、高血圧症、又は慢性鬱血性心不全などに苦しんでいる場合、該患者は、ACE阻害の処置を必要とし得る。ACE阻害は、アンギオテンシンIIの水準を低下させ、かつ従って、それによって誘発される、該血管収縮、血圧上昇、及び高アルドステロン作用を阻害する。また、ACE阻害は、ブラジキニンの内在的水準を増強するであろう。本発明に従って提供されるペリンドプリルのACE阻害に有効な量は、例えば、血圧低下作用を必要とする患者のACE阻害に有効な量である。
患者の有効な処置において、本発明に従って提供されるペリンドプリルを、経口、及び非経口の投与法を含む、有効量において生物的に利用できる該化合物で構成された、すべての形態、又は様式で投与することができる。例えば、本発明に従って提供されるペリンドプリルを、経口、皮下、筋肉内、静脈内、経皮、神経内、及び直腸などに投与することができる。通常、経口投与が好ましい。製剤を生産する当業者は、処置されるべき該疾患状態に依存し、かつ該疾患の段階に依存する投与において、適切な形態及び様式を、容易に選択することができる。
【0019】
本発明に従って提供されるペリンドプリルを、薬剤組成物、又は薬の形態で投与することができ、そのために、これらを、本発明のペリンドプリルと、医薬として許容し得るキャリア、希釈剤、又は賦形剤とを組合わせて生産し、該割合、及びその種類を、選択された投与方法、及び標準的薬務により決定する。
他の態様において、本発明は、一以上の医薬として許容し得るキャリア、希釈剤、又は賦形剤とともに、ACE阻害に有効な量の、本発明に従って提供された該ペリンドプリルを含む、薬剤組成物を提供する。
"医薬として許容し得る"とは、該キャリア、希釈剤、又は賦形剤が、本発明に従って提供された該ペリンドプリルに適合し、かつその受容に関して有害性はないことを意味する。
【0020】
該薬剤組成物、又は薬を、製薬技術において周知である方法により生産する。該キャリア、希釈剤、又は賦形剤は、固体、半固体、又は液体物質であってもよく、ビヒクル、又は該有効成分の媒体として役立ち得る。適切なキャリア、希釈剤、又は賦形剤は、当業者にとって、周知のものである。本発明の薬剤組成物を、経口、又は非経口用に適合させることができ、かつ該患者に、錠剤、カプセル、坐剤、溶液、又は懸濁液などの形態で投与することができる。
【0021】
該薬剤組成物を、例えば、不活性希釈剤とともに、又は食用キャリアとともに、経口投与することができる。これらを、ゼラチンカプセルに囲む、又は錠剤中に包含することができる。経口治療投与のため、本発明の一水和物を、賦形剤と組合わせ、かつ錠剤、カプセル、エリキシル剤、懸濁液、及びシロップ剤などの形態で使用することができる。
また、該錠剤、ピル、及びカプセルなどは、一以上の下記アジュバントを含むことができる:微結晶性セルロース、トラガカントゴム、又はゼラチンのような結合剤;デンプン、又はラクトースのような賦形剤;アルギン酸、及びコーンスターチなどのような崩壊剤;ステアリン酸マグネシウムのような潤滑剤;コロイド状二酸化ケイ素のような流動促進剤;及びサッカロース、又はサッカリンのような甘味剤である。該投与単位形態が、カプセルである場合、上述の種類の物質に加え、ポリエチレングリコール、又は脂肪油のような液体キャリアを含むことができる。他の投与単位形態では、該投与単位の物理的形態を改質する、他の様々な物質(例えば、コーティング剤)を含むことができる。従って、錠剤、又はピルを、糖、ゼラック、又は他の腸溶コーティング剤で覆うことができる。シロップ剤は、該有効成分に加え、甘味剤としてサッカロースを含み、かつ防腐剤を含むことができる。これらの様々な組成物の生産に使用する物質は、薬剤的に純粋であり、かつ使用量において、無毒性であるべきである。
【0022】
本発明に従って提供されるペリンドプリルを、非経口投与するため、溶液、又は懸濁液と組合わせることができる。また、該溶液、又は懸濁液は、一以上の下記アジュバントを含むことができる:注射用蒸留水、食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコール、又は他の合成溶媒のような無菌希釈剤;ベンジルアルコール、又はメチルパラベンのような抗菌剤;アスコルビン酸、又は亜硫酸水素ナトリウムのような酸化防止剤;エチレンジアミン四酢酸のようなキレート剤;及びアセタート、クエン酸塩、又はリン酸塩のような緩衝液である。該非経口的調製物を、ガラス、又はプラスチック製の、アンプル、使い捨てシリンジ、又は多人数用バイアルで囲うことができる。
さらに、今回の本発明を、下記図面、及び実施例により説明するが、形はどうであれ、本発明の範囲を制限するものではない。
【実施例】
【0023】
(実施例1)
(2S,3aS,7aS)-1-{2-[1-(エトキシカルボニル)-(S)-ブチルアミノ]-(S)-プロピオニル}-オクタヒドロインドール-2-カルボン酸のベンジルエステル、すなわち、ベンジルペリンドプリル(10 gms)を、イソプロパノール(100 ml)中に溶解した。該透明な液に、t-ブチルアミン(2.5 gms)、及び10%w/wのパラジウム-オン-チャコール(2 gms)を加えた。該反応混合液を、1 kg/cm2の圧力下で2時間、水素化した。
該反応塊を濾過し、該触媒を除去した。該溶媒を、減圧下で濃縮し、かつイソプロパノールを、酢酸エチルの同時添加により、置き換えた。該得られた固体を、0 ECに冷却し、かつ濾過し、ペリンドプリルエルブミン(7.8 gms)を得た。
【0024】
(実施例2)
ペリンドプリルエルブミン(10 gms)を、アセトン(80 ml)中に懸濁させた。これに、水(0.4 ml)を加え、かつ該内容物を加熱し、該固体を溶解させ、かつ周囲の温度に冷却した。該得られた懸濁液を濾過し、ペリンドプリルエルブミン一水和物(9.4 gms)を得た。
(実施例3)
ペリンドプリルエルブミン(20 gms)を、酢酸エチル(300 ml)中に懸濁させた。これに、水(1.5 ml)を加え、かつ該内容物を加熱し、該固体を溶解させ、かつ10 ECに冷却した。該得られた懸濁液を濾過し、ペリンドプリルエルブミン一水和物(17 gms)を得た。
【0025】
(実施例4)
ペリンドプリルエルブミン(5 gms)を、アセトニトリル(75 ml)中に懸濁させた。これに、水(0.4 ml)を加え、かつ該内容物を加熱し、該固体を溶解させ、かつ0 ECに冷却した。該得られた懸濁液を濾過し、ペリンドプリルエルブミン一水和物(2.9 gms)を得た。
(実施例5)
ペリンドプリルエルブミン(20 gms)を、酢酸エチル(300 ml)中に懸濁させた。該内容物を加熱し、該固体を溶解させ、かつ10 ECに冷却した。該得られた懸濁液を濾過し、かつ少なくとも相対湿度75%の空気中で乾燥させ、ペリンドプリルエルブミン一水和物(17 gms)を得た。
【0026】
(実施例6)
(ペリンドプリルエルブミン一水和物の生産)
原料:−
1.ペリンドプリルエルブミン無水物 = 10 gm
2.イソプロピルアルコール = 70 ml
3.水 = 2 ml
4.酢酸エチル = 85 ml
手順:−
1.ペリンドプリルエルブミン(無水物)10 gmを、丸底フラスコに入れる。イソプロピルアルコール70 mlを加える。1/2時間攪拌する(約95%の生成物が溶解される。)。
2.水2 mlを加える。15分間攪拌する(透明溶液が得られる。)。
3.反応塊を、38〜40℃で2時間攪拌する。
4.イソプロピルアルコールを、減圧下(600 mm未満)、40℃未満で、完全に蒸留する(ゲル型物質が観察される。)。
5.酢酸エチル30 mlを加える。40℃未満で、15分間攪拌する(透明溶液が観察される)減圧下、40℃未満で、蒸留する(半固体が観察される。)。
6.酢酸エチル40 mlを、36〜38℃で加える。15分間攪拌する(自由固体(free solid)が観察される。)。
7.1時間室温(25〜30℃)で攪拌する(遊離結晶性固体が観察される。)。
8.10℃に冷却する。2時間攪拌する。
9.固体を濾過し、かつ酢酸エチル15 mlで洗浄する。2時間搾取して乾燥させる。
10.減圧下、40℃未満で12時間乾燥させる。
含水率 = 3.2〜3.8%
融点 = 145〜150℃
(実施例7)
下記錠剤を製造した。
【0027】
【表2】
【0028】
手順:上の成分を、それぞれのふるいを通して、ふるい分けをする。該成分を、適切なブレンダーで混合する。該錠剤を、適切な成形型に圧縮する。
【表3】
【0029】
手順:
1)ペリンドプリルエルブミン一水和物を、エタノール中に溶解する。
2)硬化ひまし油以外の上の成分を、上の溶液とともに顆粒状にする。該顆粒、及びサイズ剤を乾燥させる。
3)適切なブレンダー内に、硬化ひまし油を用いて滑らかにする。該顆粒を、適切な成形型に圧縮する。
【図面の簡単な説明】
【0030】
【図1】本発明のペリンドプリルエルブミン一水和物のX-線回折パターン。シマズ-6000 X-線回折計(Shimadzu-6000 x-ray diffractometer)を用いて、該試料を分析した。該光源は、波長1.5406Åを有するCuのKα単色光を使用した。該発散スリット(Divergent Slit)は、1°を使用した。該受光スリット(Receiving Slit)は、0.30 mmとした。シンチレーションカウンター(Scintillation counter)は、3°〜40°の範囲で、1分間に2°の走査速度を有する検出器として使用した。
【特許請求の範囲】
【請求項1】
式(II)の保護された前駆化合物から、式(I)のペリンドプリルの医薬として許容し得る塩を生産する方法であって、
【化1】
(式中、Rは、カルボキシル基の保護基を表す。):対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合しているカルボキシル基COORの脱保護を行うことを含み、前記脱保護を塩基の存在下で行い、前記脱保護により形成される前記遊離酸を有する、医薬として許容し得る塩を形成する、前記方法。
【請求項2】
Rが、任意に置換されたアラルキルを表す、請求項1記載の方法。
【請求項3】
Rが、無置換ベンジル基を表す、請求項2記載の方法。
【請求項4】
Rが、4-ハロ置換、又は4-C1-4アルコキシ置換ベンジル基を表す、請求項2記載の方法。
【請求項5】
Rが、4-Clベンジル、又は4-メトキシベンジル基を表す、請求項4記載の方法。
【請求項6】
脱保護が、貴金属触媒の存在下の水素化分解である、請求項1〜5のいずれか1項記載の方法。
【請求項7】
該貴金属触媒が、パラジウム-オン-チャコールである、請求項6記載の方法。
【請求項8】
前記塩基が、t-ブチルアミンである、請求項1〜7のいずれか1項記載の方法。
【請求項9】
式(II)の保護された前駆化合物から、ペリンドプリルt-ブチルアミンを生産する方法であって、
【化2】
(式中、Rは、カルボキシル基の保護基を表す。):対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合しているカルボキシル基COORの脱保護を行うことを含み、前記脱保護を、ペリンドプリルのt-ブチルアミン塩を形成するように、t-ブチルアミンの存在下で行う、前記方法。
【請求項10】
Rが、無置換ベンジル基を表す、請求項9記載の方法。
【請求項11】
脱保護が、パラジウム-オン-チャコールの存在下の水素化分解である、請求項1〜5のいずれか1項記載の方法。
【請求項12】
さらに、前記方法により得られる、ペリンドプリルの医薬として許容し得る塩を、式(Ia)の水和したペリンドプリルの医薬として許容し得る塩が得られるように、水和することを含む、請求項1〜11のいずれか1項記載の方法:
【化3】
(式中、nは、1〜5の整数であり、又は整数2〜5の逆数である。)。
【請求項13】
nが1である、請求項12記載の方法。
【請求項14】
前記一水和物が得られるように、ペリンドプリルの医薬として許容し得る塩を水和することを含む、ペリンドプリルの医薬として許容し得る塩の一水和物を生産する方法。
【請求項15】
ペリンドプリルt-ブチルアミンを水和し、ペリンドプリルt-ブチルアミン一水和物を得る、請求項12〜14のいずれか1項記載の方法。
【請求項16】
請求項1〜15のいずれか1項記載の方法により生産される、任意に水和した形態のペリンドプリルの医薬として許容し得る塩。
【請求項17】
式(Ia)の水和ペリンドプリルの医薬として許容し得る塩:
【化4】
(式中、nは1〜5の整数であり、又は整数2〜5の逆数である。)。
【請求項18】
nが1である、請求項17記載の医薬として許容し得る塩。
【請求項19】
t-ブチルアミン塩である、請求項17、又は18記載の医薬として許容し得る塩。
【請求項20】
ペリンドプリルt-ブチルアミン一水和物。
【請求項21】
図1に示されるような、X-線回折特性、又は実質的に同一のX-線回折特性を有する、ペリンドプリルt-ブチルアミン一水和物。
【請求項22】
下記特徴的ピーク(2θ)のX-線粉末回折パターンを有することを特徴とする、ペリンドプリルt-ブチルアミン一水和物:9.5504、14.8600、15.7486、16.5400、20.0400、21.0499、22.0600、24.1744、26.3300、及び27.1600。
【請求項23】
一以上の医薬として許容し得るキャリア、希釈剤、又は賦形剤とともに、ACE阻害に有効な量の、請求項16〜22のいずれか1項記載のペリンドプリルの医薬として許容し得る塩を含む、医薬組成物。
【請求項24】
ACE阻害薬の製造において、請求項16〜22のいずれか1項記載のペリンドプリルの医薬として許容し得る塩の使用。
【請求項25】
ACE阻害が必要な患者に、ACE阻害に有効な量の、請求項16〜22のいずれか1項記載のペリンドプリルの医薬として許容し得る塩を投与することを含む、ACE阻害方法。
【請求項1】
式(II)の保護された前駆化合物から、式(I)のペリンドプリルの医薬として許容し得る塩を生産する方法であって、
【化1】
(式中、Rは、カルボキシル基の保護基を表す。):対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合しているカルボキシル基COORの脱保護を行うことを含み、前記脱保護を塩基の存在下で行い、前記脱保護により形成される前記遊離酸を有する、医薬として許容し得る塩を形成する、前記方法。
【請求項2】
Rが、任意に置換されたアラルキルを表す、請求項1記載の方法。
【請求項3】
Rが、無置換ベンジル基を表す、請求項2記載の方法。
【請求項4】
Rが、4-ハロ置換、又は4-C1-4アルコキシ置換ベンジル基を表す、請求項2記載の方法。
【請求項5】
Rが、4-Clベンジル、又は4-メトキシベンジル基を表す、請求項4記載の方法。
【請求項6】
脱保護が、貴金属触媒の存在下の水素化分解である、請求項1〜5のいずれか1項記載の方法。
【請求項7】
該貴金属触媒が、パラジウム-オン-チャコールである、請求項6記載の方法。
【請求項8】
前記塩基が、t-ブチルアミンである、請求項1〜7のいずれか1項記載の方法。
【請求項9】
式(II)の保護された前駆化合物から、ペリンドプリルt-ブチルアミンを生産する方法であって、
【化2】
(式中、Rは、カルボキシル基の保護基を表す。):対応する遊離酸が得られるように、式(II)の化合物の該複素環に結合しているカルボキシル基COORの脱保護を行うことを含み、前記脱保護を、ペリンドプリルのt-ブチルアミン塩を形成するように、t-ブチルアミンの存在下で行う、前記方法。
【請求項10】
Rが、無置換ベンジル基を表す、請求項9記載の方法。
【請求項11】
脱保護が、パラジウム-オン-チャコールの存在下の水素化分解である、請求項1〜5のいずれか1項記載の方法。
【請求項12】
さらに、前記方法により得られる、ペリンドプリルの医薬として許容し得る塩を、式(Ia)の水和したペリンドプリルの医薬として許容し得る塩が得られるように、水和することを含む、請求項1〜11のいずれか1項記載の方法:
【化3】
(式中、nは、1〜5の整数であり、又は整数2〜5の逆数である。)。
【請求項13】
nが1である、請求項12記載の方法。
【請求項14】
前記一水和物が得られるように、ペリンドプリルの医薬として許容し得る塩を水和することを含む、ペリンドプリルの医薬として許容し得る塩の一水和物を生産する方法。
【請求項15】
ペリンドプリルt-ブチルアミンを水和し、ペリンドプリルt-ブチルアミン一水和物を得る、請求項12〜14のいずれか1項記載の方法。
【請求項16】
請求項1〜15のいずれか1項記載の方法により生産される、任意に水和した形態のペリンドプリルの医薬として許容し得る塩。
【請求項17】
式(Ia)の水和ペリンドプリルの医薬として許容し得る塩:
【化4】
(式中、nは1〜5の整数であり、又は整数2〜5の逆数である。)。
【請求項18】
nが1である、請求項17記載の医薬として許容し得る塩。
【請求項19】
t-ブチルアミン塩である、請求項17、又は18記載の医薬として許容し得る塩。
【請求項20】
ペリンドプリルt-ブチルアミン一水和物。
【請求項21】
図1に示されるような、X-線回折特性、又は実質的に同一のX-線回折特性を有する、ペリンドプリルt-ブチルアミン一水和物。
【請求項22】
下記特徴的ピーク(2θ)のX-線粉末回折パターンを有することを特徴とする、ペリンドプリルt-ブチルアミン一水和物:9.5504、14.8600、15.7486、16.5400、20.0400、21.0499、22.0600、24.1744、26.3300、及び27.1600。
【請求項23】
一以上の医薬として許容し得るキャリア、希釈剤、又は賦形剤とともに、ACE阻害に有効な量の、請求項16〜22のいずれか1項記載のペリンドプリルの医薬として許容し得る塩を含む、医薬組成物。
【請求項24】
ACE阻害薬の製造において、請求項16〜22のいずれか1項記載のペリンドプリルの医薬として許容し得る塩の使用。
【請求項25】
ACE阻害が必要な患者に、ACE阻害に有効な量の、請求項16〜22のいずれか1項記載のペリンドプリルの医薬として許容し得る塩を投与することを含む、ACE阻害方法。
【図1】

【公表番号】特表2006−520320(P2006−520320A)
【公表日】平成18年9月7日(2006.9.7)
【国際特許分類】
【出願番号】特願2004−552875(P2004−552875)
【出願日】平成15年11月18日(2003.11.18)
【国際出願番号】PCT/GB2003/004981
【国際公開番号】WO2004/046172
【国際公開日】平成16年6月3日(2004.6.3)
【出願人】(505166340)シプラ エルティーディ (2)
【Fターム(参考)】
【公表日】平成18年9月7日(2006.9.7)
【国際特許分類】
【出願日】平成15年11月18日(2003.11.18)
【国際出願番号】PCT/GB2003/004981
【国際公開番号】WO2004/046172
【国際公開日】平成16年6月3日(2004.6.3)
【出願人】(505166340)シプラ エルティーディ (2)
【Fターム(参考)】
[ Back to top ]