
ペルオキシソーム増殖物質活性化受容体サブタイプδに対するアゴニスト作用を有する化合物、その調製方法及びその使用
ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)に対するアゴニスト作用を有する式I

の新規化合物、このような化合物を含む医薬組成物、及びその調製方法、並びにPPARδを活性化することにより治療又は予防し得る疾患の治療又は予防のための薬物の製造における使用(前記疾患はメタボリック症候群、肥満、異常脂血症、病的糖血症、インスリン耐性、老人性痴呆及び腫瘍から選ばれる)。前記新規化合物の調製に使用される新規中間体及びその調製方法。
の新規化合物、このような化合物を含む医薬組成物、及びその調製方法、並びにPPARδを活性化することにより治療又は予防し得る疾患の治療又は予防のための薬物の製造における使用(前記疾患はメタボリック症候群、肥満、異常脂血症、病的糖血症、インスリン耐性、老人性痴呆及び腫瘍から選ばれる)。前記新規化合物の調製に使用される新規中間体及びその調製方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)に対するアゴニスト作用を有する新規化合物、その調製方法、これらの化合物を含む薬物、及び心血管疾患等の治療及び予防におけるこれらの化合物の適用に関する。また、本発明は新規化合物の新規中間体及びその中間体の調製方法に関する。
【背景技術】
【0002】
現在の世界では、高速の発展及び生活基準の上昇とともに、人々が肥満、インスリン耐性(型II糖尿病)、脂質代謝障害及び高血圧を特徴とするメタボリック症候群のグローバルな発生をもたらす過度の脂肪及びタンパク質を摂取している。これらはヒトの健康に対する大きな脅威に相当する。個体の一般特性、年齢、性別、生理学的性質、栄養状態、食習慣等に加えて、メタボリック症候群は生体内の脂質代謝、エネルギー代謝及び炭水化物代謝のインバランスと関連している。こうして、メタボリック症候群を治療する有効な方法はエネルギー、脂肪及び炭水化物の生体内のバランスを維持又は回復することを目的とする治療養生法である。核受容体(NR)が細胞内だけでなく、全体の個体の体内のエネルギー、脂質及び炭水化物の生体内のバランスの維持に重要な役割を果たすので、それらは研究の焦点となっている。種々の生理学的リガンド(例えば、飽和脂肪酸、不飽和脂肪酸、これらの代謝産物、及び種々の合成化合物)により活性化される場合にのみ、核受容体が応答遺伝子の転写系を調節することができ、こうしてその生理活性を発揮することができる (Kasuga, J.ら, Bioorg. Med. Chem. 2007, 15, 5177-5190)。
核受容体ファミリーの中で、ペルオキシソーム増殖物質活性化受容体(PPAR)が10年以上にわたって人々の関心を引き付けてきており、これらはそれらのリガンドにより活性化された核転写因子であり、しかもこれらはメタボリック症候群に重要な調節因子として作用する (Guan, Y. J. Am. Soc. Nephrol, 2004, 15, 2801-2815)。それ故、PPARはインスリン耐性、損なわれたグルコーストレランス、型II糖尿病、肥満、高脂血症、高血圧、血管心臓障害、アテローム硬化症等の如き疾患の発生、発達及びコントロールに重要な役割を果たす。
【0003】
PPARは3種のサブタイプPPARα、PPARδ及びPPARγに分類され、これらは遺伝子の特定のDNA配列に結合することにより遺伝子の発現を調節する (Berger, J.ら, The Journal of Biological Chemistry, 1999, 274 (10), 6718-6725)。PPARαは主として肝臓、心臓、腸道、腎臓及びマクロファージ中で発現され、活性化された後に、脂肪酸の代謝を増大し、マクロファージ中の炎症応答を軽減し、低密度リポタンパク質コレステロールを低下し得る。PPARγは脂肪細胞、胎盤腫及びその他の組織中で発現され、活性化された後に、血液グルコースレベルを低下し、インスリン感受性を増大するだけでなく、脂質代謝、サイトカイン拮抗作用、坑炎症、免疫調節及び血圧調節等に重要な役割を果たし得る (Kasuga, J.ら, Bioorg. Med. Chem. 2007, 15, 5177-5190)。その他の2種のサブタイプとは対照的に、PPARδの生理機能は今まで明らかではない。しかしながら、薬理学実験のための動物モデルに関する最近の研究において、PPARδは脂肪組織及び筋肉中で脂肪酸分解代謝及びエネルギー脱共役を増大でき、マクロファージ由来の炎症を抑制し得ることが示されていた。人体の体重獲得を調節し、体の耐久性を高め、インスリン感受性を増大し、アテローム硬化症を改善することにおける種々の機能のために、PPARδのリガンドが高脂血症、肥満、インスリン耐性、及びアテローム硬化症の治療に有効な薬物であるかもしれない。
現在、PPARδ受容体アゴニスト薬のいずれもが薬物として商業上入手し得ない。PPARδアゴニストについての現在の研究の中で、グラクソスミスクラインにより開発されたGW501516 についての臨床研究はGW501516 が高密度リポタンパク質(HDL) コレステロールのレベルを80%まで増大し、低密度リポタンパク質(LDL) コレステロールのレベルを29%まで低下し、トリグリセリド(TG)のレベルを56%まで低下し、インスリンのレベルを48%まで低下し得ることを示していた (Oliver, W.; Jr.; Shenk, J. L. ら, Natl. Acad. Sci. U.S.A. 2001, 98, 5306-5311)。こうして、GW501516 は肥満及び心血管疾患の治療に有効な薬物になり得ると考えられる (WO01/00603A1, Bioorg. Med. Chem. Lett. 2003, 13, 1517)。しかしながら、GW501516 プロジェクトはそのフェーズII臨床試験からの不利な結果のために今一時的に中止されている。
【発明の概要】
【発明が解決しようとする課題】
【0004】
こうして、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)についてのアゴニスト作用及びGW501516のそれよりも極めて良好な血液脂質の調節能力を有する新規化合物を提供することについての緊急の要望がある。
【課題を解決するための手段】
【0005】
本発明の一つの目的は式(I)の化合物及び/又はその医薬上許される塩及び/又はこれらの溶媒和物を提供することである。
【0006】
【化1】
【0007】
式中、
1) XはO、S、N、又は (CH2)n (式中、nは1から4までの整数である)であり、XはO、S、又はCH2であることが好ましく、
2) YはO、S又はN、好ましくはO又はSであり、
3) RはH又はC1-C9アルキル、好ましくはH、メチル、又はエチルであり、
4) R1 及びR2 は互いに独立にH又はC1-C4 アルキルであり、かつR1及びR2 の少なくとも一つがHであり、R1及びR2は互いに独立にメチル、エチル、又はHであることが好ましく、
5) R3 はH、C1-C9 アルキル、好ましくはH又はC1-C4 アルキル、例えば、メチル、エチル、イソプロピル、更に好ましくはメチルであり、
6) R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル、又は置換フェニルであり、その置換フェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6 から選ばれ、R5 及びR6 は互いに独立にH又はC1-C9 アルキルであり、またR4 が置換フェニルである場合、その置換基が4-メトキシ又は4-メチルであることが好ましく、
【0008】
7) G1 及びG4は個々にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G1 及びG4 は個々にメチル又はエチルであることが好ましく、
8) G2 及びG3は個々にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G2 及びG3は個々にメチル、エチル、又はHであることが好ましく、
9) G5、G6、G8、及びG9は個々にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G5、G6、G8、及びG9 は個々にH、F、Cl、Br、メチル、エチル、又はメトキシであることが好ましく、かつ
10) G7 はH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G7 はトリフルオロメチル、イソプロピル、エチル、メチル、又はClであることが好ましく、G7 はトリフルオロメチルであることが更に好ましい。
【0009】
本発明の化合物がエステル形態である場合、それは以下に化合物I(エステル)と称される。本発明の好ましい化合物I(エステル)は下記の化合物を含む:
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (以下、 “E-1”と称される);
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-2”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-3”と称される);
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルチオ)-フェノキシ)-アセテート (以下、“E-4”と称される);
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (以下、 “E-5”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (以下、 “E-6”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルチオ)-フェノキシ)-アセテート (以下、 “E-7”と称される);
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-アセテート (以下、“E-8”と称される);
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-アセテート (以下、“E-9” と称される);
【0010】
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-10”と称される);
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-11”と称される);
エチル 2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-アセテート (以下、“E-12” と称される);
メチル 2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-13” と称される);
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルチオ)-フェノキシ)-アセテート (以下、 “E-14” と称される);
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-15” と称される);
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-16”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、 “E-17” と称される);
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-18” と称される);
エチル 2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-19” と称される)。
【0011】
化合物I(エステル)はその酸性形態(以下、化合物I(酸)と称される)を得るためにアルカリ性条件下で加水分解し得る。アルカリ性条件はアルカリ(これはアルカリ金属水酸化物であり、例えば、水酸化ナトリウム、水酸化リチウム、水酸化カリウム等があげられるが、これらに限定されない)を使用して生じられる。その加水分解に使用される溶媒系はC1-C4 アルコール (例えば、メタノール、エタノール、プロパノール、ブタノール等)-水 (アルコール: 水 の比= 9 -1:1 (容積/容積)), テトラヒドロフラン (THF) -水 (THF: 水 の比= 9 - 1:1 (容積/容積))、又はアルコール-ジクロロメタン-水 (アルコール: ジクロロメタン: 水 の比= 9 - 1: 9 - 1: 1 (容積/容積))であり、その反応温度は0-80℃、好ましくは20-40℃であり、その反応時間は1-12時間、好ましくは2-4 時間である。
本発明の好ましい化合物I(酸)は下記の化合物を含む:
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸 (以下、“A-1” と称される);
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-2” と称される);
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-3” と称される);
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-4” と称される);
【0012】
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸 (以下、“A-5”と称される);
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸 (以下、“A-6” と称される);
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-7” と称される);
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-酢酸 (以下、“A-8” と称される);
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-酢酸 (以下、“A-9” と称される);
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-10” と称される);
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-11” と称される);
【0013】
2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-酢酸 (以下、“A-12” と称される);
2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-13” と称される);
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-14” と称される);
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-15” と称される);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(以下、 “A-16” と称される);
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-17” と称される);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(以下、“A-18” と称される);
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-19” と称される)。
【0014】
本発明の特に好ましい化合物I(酸)は下記の化合物を含む:
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-3);
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-4);
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルオキシ)-フェニルチオ)-酢酸 (A-11);
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-14);
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-15);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-16);
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸(A-17);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-18);
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-19)。
本発明の式Iの化合物の医薬上許される塩はそのアルカリ金属塩又はアルカリ土類金属塩を表す。カリウム塩、ナトリウム塩及びカルシウム塩が好ましい。
本発明の式Iの化合物の溶媒和物はその水和物又は有機溶媒和物、好ましくは水和物及びアルコラートを表す。水和物は1-4個の水分子を含んでもよい。アルコラートはメタノール、エタノール、及びプロパノールで生成されたアルコラートを含んでもよい。
本発明の別の目的は新規化合物、即ち、化合物IIIを提供することである。
【0015】
【化2】
【0016】
式中、
ZはCl又はBrであり、
R3 はH又はC1-C9 アルキルであり、
R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル又は置換フェニルであり、そのフェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6から選ばれ、かつ
G5、G6、G7、G8、及びG9は互いに独立にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6であり、R5 及びR6 は互いに独立にH又はC1-C9 アルキルである。
本発明の好ましい化合物IIIは下記の化合物を含む:
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (以下、“III-1”と称される);
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (以下、“III-2” と称される);
3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オン (以下、“III-3” と称される)。
本発明の化合物IIIは本発明の式Iの化合物を調製するための中間体として使用し得る。
本発明の別の目的は式IIIの化合物の調製方法を提供することである。
本発明の式IIIの化合物は式VIの化合物を塩素化試薬又は臭素化試薬と反応させることにより合成し得る。
【0017】
【化3】
【0018】
式中、
ZはCl又はBrであり、R3、R4、G5-G9 は式IIIの化合物の定義に示されたのと同じである。
塩素化試薬又は臭素化試薬は
(1) N-ブロモスクシンイミド(NBS)/トリフェニルホスフィン(Ph3P);
(2) 塩化チオニル(SOCl2) 又は臭化チオニル (SOBr2);
(3) N-クロロスクシンイミド(NCS)/トリフェニルホスフィン(Ph3P);
(4) 四塩化炭素(CCl4) 又は四臭化炭素(CBr4)/トリフェニルホスフィン(Ph3P);
(5) 五塩化リン(PCl5) 又は五臭化リン(PBr5);
(6) オキシ塩化リン(POCl3) 又はオキシ臭化リン(POBr3); 及び
(7) 三塩化リン(PCl3) 又は三臭化リン (PBr3)
から選ばれる。
溶媒はジクロロメタン、クロロホルム、四塩化炭素、及びこれらのあらゆる混合物から選ばれ、反応温度は10-80℃であり、反応時間は2-8 時間である。
式IIIの化合物は下記のスキームに従って合成されることが好ましい。
【0019】
【化4】
【0020】
上記スキームに、下記の工程が含まれる。
1)テトラヒドロフラン (THF) を溶媒として、また水素化ナトリウムをアルカリとして使用して、ベンジルアルコール 1 がブロミド2 と反応してエーテル3を生成し、
2)エタノール (EtOH) を溶媒として使用して、エーテル3 をアミン 4でアミン化してアミド5を生成し、
3)トルエン (PhCH3) を溶媒として使用して、アミド5 がオキシ塩化リン (POCl3) の作用のもとにフェニルヒドラジン 6 と反応してヒドラゾン 7を生成し、
4)テトラヒドロフラン (THF) を溶媒として使用して、ヒドラゾン 7 がカルボニルジイミダゾール(CDI) と反応して化合物 8を生成し、
5)エタノール(EtOH)を溶媒として、またパラジウム/カーボン(Pd/C) を触媒として使用して、ベンジルを常圧下で水素化により化合物8 から開裂してアルコール 9を生成し、
6)ジクロロメタン (DCM) を溶媒として使用して、アルコール 9 を塩素化試薬又は臭素化試薬の作用のもとに化合物IIIに変換する。
例えば、R4、G5、G6、G8、G9 がHであり、R3 がメチルであり、かつG7がトリフルオロメチルである場合、その合成スキームは以下のとおりである。
【0021】
【化5】
【0022】
上記スキームに、下記の工程が含まれる。
1)テトラヒドロフラン(THF) を溶媒として、また水素化ナトリウム(NaH) をアルカリとして使用して、還流下で加熱すると、ベンジルアルコール 1がエチルブロモアセテートと反応してエチルベンジルオキシアセテート10を生成し、
2)エタノール(EtOH) を溶媒として使用して、エチルベンジルオキシアセテート10を室温でメチルアミン11でアミン化してベンジルオキシアセトメチルアミン12を生成し、
3)トルエン (PhCH3) を溶媒として使用して、ベンジルオキシアセトメチルアミン12が80℃でオキシ塩化リン(POCl3) の作用のもとにp-トリフルオロメチルフェニルヒドラジン13と反応してヒドラゾン14を生成し、
4)テトラヒドロフラン(THF) を溶媒として使用して、ヒドラゾン14がカルボニルジイミダゾール(CDI) と反応して化合物15を生成し、
5)エタノール(EtOH)を溶媒として、またパラジウム/カーボン(Pd/C)を触媒として使用して、ベンジルを室温で常圧で水素化により化合物15から開裂してアルコール16を生成し、
6)ジクロロメタン(DCM) を溶媒として、またN-ブロモスクシンイミド(NBS) を臭素化剤として使用して、アルコール16をトリフェニルホスフィン(Ph3P)の作用のもとに化合物III-3に変換する。
式IIIの化合物はまた式VIIの化合物を塩素化試薬又は臭素化試薬と反応させることにより合成し得る。溶媒はクロロホルム又は四塩化炭素であり、臭素化試薬はN-ブロモスクシンイミド(NBS) であり、塩素化試薬はN-クロロスクシンイミド (NCS) であり、触媒はジベンゾイルペルオキシドであり、反応温度は40-80℃であり、反応時間は2-8 時間である。
【0023】
【化6】
【0024】
式中、
ZはCl又はBrであり、
R3 、G5-G9 は式IIIの化合物の定義に示されたのと同じであり、
G10-G14 は互いに独立に、又は同時にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、
R5 及びR6 は互いに独立にH又はC1-C9 アルキルである。
式IIIの化合物は下記のスキームに従って調製されることが好ましい。
【0025】
【化7】
【0026】
上記スキームに、下記の工程が含まれる。
1)メタノール(MeOH)を溶媒として、また硫酸を触媒として使用して、酸17がエステル化を受けてエステル18を生成し、
2)メタノール(MeOH)を溶媒として使用して、エステル18をアミン19でアミン化してアミド20を生成し、
3)トルエン(PhCH3) を溶媒として使用して、アミド20がオキシ塩化リン(POCl3) の作用のもとにフェニルヒドラジン 6と反応してヒドラゾン22を生成し、
4)テトラヒドロフラン(THF) を溶媒として使用して、ヒドラゾン22がカルボニルジイミダゾール(CDI) と反応して化合物23を生成し、
5)クロロホルムを溶媒として、N-ブロモスクシンイミド(NBS) を臭素化試薬として(又はN-クロロスクシンイミド(NCS) を塩素化試薬として) 、またジベンゾイルペルオキシドを触媒として使用して、化合物23を臭素化(又は塩素化) して化合物IIIを生成する。
例えば、R4 がフェニルであり、G5、G6、G8、G9 がHであり、R3 がメチルであり、かつG7がトリフルオロメチルである場合、その合成スキームは以下のとおりである。
【0027】
【化8】
【0028】
上記スキームに、下記の工程が含まれる。
1)メタノール(MeOH) を溶媒として、また硫酸を触媒として使用して、還流下で加熱すると、酸24がエステル化を受けてエステル25を生成し、
2)メタノール(MeOH)を溶媒として使用して、エステル25を室温でアミン11でアミン化してアミド26を生成し、
3)トルエン(PhCH3) を溶媒として使用して、アミド26が80℃でオキシ塩化リン (POCl3) の作用のもとにp-トリフルオロメチルフェニルヒドラジン13と反応してヒドラゾン27を生成し、
4)テトラヒドロフラン(THF) を溶媒として使用して、ヒドラゾン27が室温でカルボニルジイミダゾール(CDI) と反応して化合物28を生成し、
5)クロロホルムを溶媒として、N-ブロモスクシンイミド(NBS) を臭素化試薬として、またジベンゾイルペルオキシドを触媒として使用して、還流下で加熱すると、化合物28が臭素化されて化合物III-1を生成する。
本発明の別の目的は式(I)の化合物の調製方法を提供することである。
本発明の式(I) の化合物は下記のスキーム1又はスキーム2に示される方法により調製される。
スキーム1:
【0029】
【化9】
【0030】
式中、
X、Y、R1-R4 、G1-G9 は式Iの化合物の定義に示されたのと同じであり、RはH又はC1-C9 アルキル、好ましくはメチル又はエチルである。
スキーム1において、中間体化合物II及びIIIがアルカリの作用のもとにカップリングにかけられて化合物I(エステル)を生成する。アルカリは有機又は無機アルカリであり、無機アルカリはアルカリ金属炭酸塩、可溶性アルカリ土類金属炭酸塩、炭酸アンモニウム等、又はこれらのあらゆる混合物を含んでもよく、無機アルカリの例として、炭酸ナトリウム、炭酸カリウム、炭酸ストロンチウム、炭酸アンモニウム等が挙げられ、有機アルカリはトリエチルアミン等であってもよい。溶媒はアセトニトリル、ジメチルホルムアミド(DMF) 、ジメチルスルホキシド(DMSO) 、テトラヒドロフラン(THF) 、ジオキサン等、又はこれらのあらゆる混合物から選ばれることが好ましい。反応温度は0-100℃、好ましくは40-80℃であり、反応時間は1-12時間、好ましくは4-8 時間であることが好ましい。
上記プロセススキームにおいて、式IIの化合物は商業上入手されてもよく、又は従来技術の文献から知られている典型的な方法(例えば、M. L. Sznaidman, Curt D. Haffner, Patric R. ら, Bioorg. Med. Chem. Lett. 2003, 13, 1517-1521; Zhi-liang wei ら, J. Org. Chem. 2003, 68, 9116-9118; Org. Syn. Coll. Vol 1, 102, 1941; Org. Syn. Coll. Vol 2, 290, 1943; Handbook of Fine Organic Chemical Raw Materials and Intermediate. XU Ke-xun (編集), Scientific &Technological Industry Press, 3-426-3-584に示された方法)により合成されてもよい。
式IIの化合物は下記の化合物を含むが、これらに限定されない:
エチル 2-(2-メチル-4-ヒドロキシ)-フェノキシ-アセテート (以下、“II-1”と称される);
エチル 2-(3-メチル-4-ヒドロキシ)-フェノキシ-アセテート (以下、“II-2” と称される);
エチル 2-(2-エチル-4-ヒドロキシ)-フェノキシ-アセテート (以下、“II-3” と称される);
エチル 2-(3-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (以下、“II-4” と称される);
エチル 2-(2-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (以下、“II-5” と称される);
エチル 2-(2,5-ジメチル-4-ヒドロキシ)-フェニルチオ-アセテート (以下、“II-6” と称される)。
スキーム2
【0031】
【化10】
【0032】
式中、
X、Y、Z、R1-R4 、G1-G9 は式Iの化合物の定義に示されたのと同じであり、
RはH又はC1-C9 アルキル、好ましくはメチル又はエチルである。
スキーム2において、アルカリとしての炭酸塩との連続反応を使用して、化合物IIIを最初に化合物IV次いで化合物Vと反応させるが、反応の経過中に中間体を分離する必要はない。アセトニトリルに加えて、テトラヒドロフラン(THF) 、ジオキサン、等、又はこれらのあらゆる混合物が溶媒として使用されてもよい。
炭酸カリウムに加えて、炭酸ナトリウム、炭酸ストロンチウム、炭酸アンモニウム等、又はこれらのあらゆる混合物が炭酸塩として使用されてもよい。中間体化合物IV及びVの両方が市販されている。
スキーム2において、化合物I(エステル)がアルカリ性条件下で加水分解されて化合物I(酸)を生じることができ(RがC1-C9 アルキルである場合)、前記アルカリ性条件は下記のアルカリ、例えば、アルカリ金属水酸化物(水酸化ナトリウム、水酸化リチウム、水酸化カリウム等、又はこれらのあらゆる混合物が挙げられるが、これらに限定されない)で生成し得る。加水分解に使用される溶媒系はC1-C4 アルコール (例えば、メタノール、エタノール、プロパノール、ブタノール等)-水 (アルコール: 水 = 9 -1:1 (容積/容積))、THF-水 (THF: 水 = 9 - 1:1 (容積/容積))、又はアルコール-ジクロロメタン-水 (アルコール: ジクロロメタン: 水 = 9 - 1: 9 - 1: 1 (容積/容積))であり、反応温度は0-80℃、好ましくは20-40℃であり、反応時間は1-12時間、好ましくは2-4 時間である。
【0033】
【化11】
【0034】
式中、
X、Y、Z、R1-R4 、G1-G9 は式Iの化合物の定義に示されたのと同じであり、
RはH又はC1-C9 アルキル、好ましくはメチル又はエチルである。
本発明の別の目的は式(I)の上記化合物を活性成分として含む医薬組成物を提供することである。
式(I)の上記化合物を含む本発明の医薬組成物は式(I)の化合物及び医薬製剤化に使用される通常のアジュバントを含む。
このような医薬製剤化に使用される通常のアジュバントは薬物管理の担当部門により認可され、医薬アジュバントの基準に合致するものを表す。それらはそれらの異なる機能に基づいて二つのグループに分類されていた:アジュバントの一つのグループは医薬製剤の加工及び製造に必要であり、これらとして、希釈剤、結合剤、滑剤、懸濁剤及び潤滑剤等が挙げられ、アジュバントの別のグループは生体内の薬物の消化及び吸収を促進するように機能し、これらとして、崩壊剤、補助溶媒等が挙げられる。それらは人体の生体内で活性ではなく、治療効果を与えず、また毒性を与えない。
上記アジュバントの中で、希釈剤は下記の物質のいずれか一種又はあらゆる2種以上のあらゆる混合物から選ばれてもよい:澱粉、変性澱粉、蔗糖、ラクトース一水和物、無水ラクトース、グルコース、マンニトール、及び種々の微結晶性セルロース。
上記アジュバントの中で、結合剤は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:ヒドロキシプロピルメチルセルロース、前ゼラチン化澱粉、ポリビドン(ポリビニルピロリドン)、カルボキシメチルセルロース及びその誘導体、メチルセルロース、エチルセルロース、澱粉、炭水化物等、好ましくはヒドロキシプロピルメチルセルロース、前ゼラチン化澱粉、及びポリビドン。
【0035】
上記アジュバントの中で、潤滑剤は下記の物質のいずれか一種又はあらゆる2種以上のあらゆる混合物から選ばれてもよい:ステアリン酸マグネシウム、タルク粉末、及び型I水添植物油。
上記アジュバントの中で、懸濁剤は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:ゼラチン、ペクチン、アラビアゴム、アルギン酸ナトリウム、メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロース、及びメチルセルロース。
上記アジュバントの中で、崩壊剤は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:澱粉、低置換ヒドロキシプロピルセルロース、ナトリウムカルボキシメチル澱粉、カルシウムカルボキシメチルセルロース、架橋ポリビドン、架橋セルロース及び架橋ナトリウムカルボキシメチルセルロース。
上記アジュバントの中で、補助溶媒は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:スパンシリーズ、トゥイーンシリーズ、ポリエチレングリコールシリーズ、大豆レシチン等。
【0036】
上記医薬組成物は下記の経口製剤のあらゆる形態であってもよい: 1, 単純錠剤; 2, フィルム被覆錠剤; 3, 糖剤; 4, 腸被覆錠剤; 5, 分散性錠剤; 6, カプセル; 7, 顆粒; 8, 懸濁液; 及び9, 溶液。
上記製剤形態は通常の製剤化方法により調製し得る。
本発明の別の目的はペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患を治療又は予防するための薬物の製造における式(I)の化合物の使用を提供することである。前記疾患として、メタボリック症候群、肥満、脂質異常血症、病的糖血症、インスリン耐性、老人性痴呆又は腫瘍等が挙げられる。
本発明によれば、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患を治療又は予防するための薬物の製造における式(I)の化合物の使用が提供され、前記疾患がメタボリック症候群、肥満、脂質異常血症、病的糖血症、インスリン耐性、老人性痴呆又は腫瘍等の一種以上から選ばれる。
本発明によれば、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患の治療又は予防方法が提供され、その方法は患者への治療又は予防有効量の式(I)の化合物の投与の工程を含み、前記疾患がメタボリック症候群、肥満、脂質異常血症、病的糖血症、インスリン耐性、老人性痴呆又は腫瘍等の一種以上から選ばれる。
本件出願の明細書を読んだ後に、当業者は本発明のその他の利点及び使用を認めるかもしれない。
【図面の簡単な説明】
【0037】
【図1】動物の体重の経時変化曲線を示し、これは薬物が動物の体重及び食物摂取に関する作用を有することを示す。横軸は計量時間(2日毎に計量)を表し、縦軸は動物の体重を表す。陽性対照はオルリスタット(二つの用量グループ:30mg/kg及び10mg/kg)である。本発明において、薬物製剤(A-3及びA-11)は二つの用量グループ(10mg/kg及び3.3mg/kg)を分離する。
【図2】動物中の血液グルコースの変化曲線を示し、これは経口グルコーストレランス試験からの結果を示す。横軸はグルコースを経口投与された後の血液サンプリング時点、0分、30分、60分、120分を表し、縦軸は相当する血液グルコースレベル(ミリモル/L)を表す。
【図3】本発明のA-3 (HS060098) 及びA-1 (HS060001) の薬物-時間曲線を表す。横軸は血液サンプリングの時点(時間)を表し、縦軸は異なる時間における動物からの血漿中の薬物の濃度(μg/ml) を表す。
【発明を実施するための形態】
【0038】
本発明が以下の特別な実施例を参照して詳しく記載される。本明細書の開示に鑑みて、種々の変更及び改良が本発明の精神及び範囲から逸脱しないで当業者により本発明になし得ることが理解されるべきであり、これらの全てが特許請求の範囲により特定された請求範囲内に入るべきである。更に、本明細書に提示された実施例は説明目的のためにすぎず、本発明の限定と何ら見なされるべきではないことが理解されるべきである。
本件出願において、C1-C9 基(アルキル、アルコキシ、アルキルチオ等) はC1-C2基、C1-C3基、C1-C4基、C1-C5基、C1-C6基、C1-C7基、C1-C8基、及びC1-C9基 (アルキル、アルコキシ、アルキルチオ等)等を含む。
薬物スクリーニングのためのモデル
核受容体活性化剤のin vitroスクリーニング
そのスクリーニングモデルにおける実験操作は以下のとおりである。
1. 核受容体関連スクリーニングモデルの簡単な記載
生細胞中の核受容体アゴニストをスクリーニングするためのスクリーニングモデルは活性化された核受容体がその下流遺伝子の転写を活性化し得るという原理に基づいて設計される。リポーター遺伝子プラスミドが構築され、この場合、核受容体(NRE) のDNA 結合配列がルシフェラーゼ遺伝子の上流に挿入され、その結果、ルシフェラーゼ遺伝子の発現が核受容体の制御下に置かれる。次いで、リポーター遺伝子プラスミド及び核受容体が細胞に同時に移入され、核受容体アゴニストが細胞培地中に存在する場合に核受容体が活性化される。次いで活性化された受容体がルシフェラーゼ遺伝子の発現を誘発することができ、その間にルシフェラーゼの量がその発光基質により測定し得る。この方法で、核受容体に関する化合物による活性化の強さがルミネセンス強さを観察することにより知り得る。トランスフェクション効率、接種された細胞の量、化合物の毒性等の如き因子により生じられる試験誤差の較正のために、GFP プラスミドが内部基準として同時に共トランスフェクトされ、試験ウェルの全てについてのルミネセント値が実験結果を分析する場合にGFP 値で較正される。実験結果が溶媒対照について1の値でもって活性化の相対的倍率として表され、倍率が大きい程、活性化能が高くなる。
【0039】
2. 実験操作
そのスクリーニングモデルに関する実験の詳しいプロトコルが下記の論文に言及されているかもしれない: “PPARa選択的アクチベーターとしての非環式1,3-ジカルボニル化合物の新規クラスの設計、合成及び評価”, Bioorg Med Chem Lett. 2004; 14(13): 3507-11。特別な操作が以下に記載される。
実験試薬:試験すべき化合物 (20 mM, DMSO中, -80℃で貯蔵).
(1) 1日目: 細胞培養及び接種
肝臓癌細胞HepG2 (ATCC (American Type Culture Collection)から) を37℃、100%の相対湿度、及び5%のCO2のインキュベーター中のT-75培養びん (グレイナー、ドイツ) 中の10% 熱不活化ウシ胎児血清(FBS, インビトロゲン, グランド・アイランド, NY, USA) を補給されたDMEM培地中で培養した。培養びん中の細胞培養物が80-90% の集密度に達した場合に、それを0.25%トリプシン (EDTAを含む) で3分間消化し、2000 細胞/100μl/ウェルの接種密度で96ウェル細胞培養プレートに接種した。
(2) 2日目:細胞トランスフェクション
翌日に、96ウェル培養プレート中の細胞が50-80% の集密度まで増殖した場合、細胞トランスフェクションを行なった。細胞共トランスフェクション系はFuGene6 トランスフェクション剤 (Roche Molecular Biochemicals, インジアナポリス, IN, U.S.A.) 及び60 ng DNA (10ngのhRXR, 10ngのpCMV βGal, 10ngの核受容体発現プラスミドRXR/PPARδ, 30ngのGFP 蛍光リポーター遺伝子プラスミドの夫々)を含んでいた。
【0040】
(3) 薬物措置
細胞培地をトランスフェクション24時間後に直ちに捨て、試験すべき薬物を含む新しいDMEM培地(活性炭により処理された10%のFBSを含む)200 μl により交換した。試験すべき薬物の最終濃度は10, 5, 1, 0.1, 0.01, 0.001, 及び0 μMであり、陽性対照は夫々のウェル中でDMSO(最終濃度は0.1%であった)とともに0.05 μM の2-ブロモステアリン酸 (Sigma, USAから購入)である。
(4) キナーゼ活性アッセイ
薬物による措置の24時間後に、細胞を溶解溶液 (Cell Culture Lysis buffer, Promega) で溶解し、遠心分離し、上澄みを集めた。上澄みを蛍光検出キット (Promega) で処理し、蛍光メーター (Ascent Fluoroskan FL reader, Thermo Labsystems, フィンランド) によりカウントし、ルシフェラーゼの相対強さを測定した。内部基準(トランスフェクション効率について較正する内部基準)として実験に使用されたβ-ガラクトシダーゼ活性をアッセイするために、上澄み50 μl を新しいミクロプレートに移し、プロメガキットで処理し、ミクロプレートリーダー (Bio-tech Instruments Inc., Winooski, VT, USA) (Sauerberg, P.; Olsen, G. S.; Jeppesen, L.; Mogensen, J. P. ら, J. Med. Chem., 2007, 50, 1495-1503)から405nmの波長で読み取った。
【0041】
3. 分析:
サンプルのメジアン有効濃度(EC50) はサンプルが50%の薬理学的作用を有する濃度である。この値は化合物の薬理学的作用を評価するのに重要なパラメーターの一つである。本スクリーニング方法において、サンプルのEC50 を6種の異なる濃度のサンプルにより受容体の活性化に従って計算した。
4. スクリーニング結果
PPARδ 受容体を活性化し得る式Iの化合物を得た。
in vitro の式Iの化合物(酸)の活性データ
in vitro の式Iの化合物(酸)の活性を下記の方法により測定した:サンプル [式 (I) の化合物(酸)] を溶解し、異なる濃度に希釈し、PPARδ 受容体を活性化するためのサンプルの活性を異なる濃度で試験した。濃度vs作用の対応を得、次いでメジアン有効濃度 (EC50) についての相当する値を計算した。試験結果を表1に示した。
【0042】
表1 in vitroの式Iの化合物(酸)の活性に関するデータ
【0043】
注: EC50 (nM) 値が低い程、活性が高い。
表1中のデータから、本発明の化合物の全てがPPARδについてアゴニスト活性を有することがわかるかもしれない。
化合物の生体内の薬力学的活性についてのスクリーニング
1.血液脂質を調節する活性についてのスクリーニング
本発明の式Iの化合物(酸)の一部の生体内活性に関するアッセイ
4種の化合物をA-1 〜A-19 の中から選び、それらの生体内の活性に関するアッセイにかけた。薬物の介入作用を高脂肪食誘発高脂血症を有するモデルであるSDラット、ApoE マウス、メソクリセタス・オーラタス(Mesocricetus auratus )等で観察した。結果を表2に示した。
【0044】
表2 式Iの化合物(酸)の生体内の活性データ
【0045】
注: ↓ は減少を表し、また↑ は増加を表す。
選ばれ、生体内の活性アッセイにかけられた4種の化合物がコレステロール(TC)、トリグリセリド(TG)及び低密度リポタンパク質(LDL) を減少することができ、また高密度リポタンパク質(HDL) を増加し得ることが表2からわかり、これは本発明の化合物が血液脂質調節の優れた作用を有することを示した。同時に、4種の化合物はまたSDラットモデル及びApoEマウスモデルで同様の薬理学的作用を有し、GW501516のそれと較べて、血液脂質調節に関して一層良好な作用を示した。
特に、化合物A-3 はコレステロール(TC)レベルを40%まで減少でき、トリグリセリド(TG)を65%まで減少でき、また低密度リポタンパク質(LDL) を51%まで減少できるだけでなく、高密度リポタンパク質(HDL) のレベルを43%まで増加することができる。
その後、A-3 の薬物の効力を高脂血症のマカカ・レサス(Macaca rhesus )モデルで確かめた。投与の3ヶ月後に、血液サンプルをELISAによる血液学的インジケーターの測定、並びにインスリン、アポリポタンパク質A-1 (apoA-1) 及びアポリポタンパク質B-100 (apoB-100) の測定のために大腿静脈から採取した。結果は、高脂血症モデルの対照グループと比較して、A-3 の投与にかけられた動物グループ中の血清全コレステロール(TC)の含量が45%減少され、低密度(LDL) の含量が38%減少され、高密度リポタンパク質(HDLc)の含量が67%増加されることを示した。しばらくして、ELISAアッセイにより、インスリン濃度が有意に減少され、apoA-1の濃度が有意に増加され、apoB-100 の濃度が有意に減少され、またapoA-1 対apoB-100 の比が正常な動物中のそれに近づくことがわかり、これは血液学的測定におけるLDL 及びHDL からの結果と充分に合致した。上記結果は高脂血症を患っていた動物に薬物A-3 を投与した後に、血液学的アッセイ及びELISAアッセイからのインジケーターの全てが有意に反転することを示し、これは順にその薬物の治療作用がGW501516のそれよりも極めて良好であることを実証した。
【0046】
2. 体重損失の薬理学的活性についてのスクリーニング
実験操作
生後3週の雄のC57BL/6J マウスを実験動物として選び、そのうちの15匹をランダムに選んで標準飼料で給餌し、残りを高脂肪飼料で給餌させ、マウスの夫々を耳カッティングによりマークした。マウスの体重及び飼料摂取を毎週計量した。給餌を15週続けた後、高脂肪飼料グループ中のマウスは42gの平均体重を有し、標準飼料グループ中のマウスは28gの平均体重を有し、モデルグループ及び投薬グループの両方中のマウスを高脂肪飼料で更に給餌した。4週にわたる本発明の化合物の投与後に、動物における血液脂質及び血液グルコースのレベル、体重、並びに飼料摂取に関する薬物の作用を観察するとともに、グルコーストレランス試験を行なって動物におけるグルコーストレランスに関する薬物の作用を調べた。
(1) 動物における血液脂質及び血液グルコースのレベルに関する薬物の作用
表3に示されるように、A-3 及びA-11 はモデル動物からの血清中のTG及びGLU のレベルを有意に減少することができ、オルリスタットの作用よりも極めて良好な作用を示す
【0047】
【0048】
正常なグループと比較してΔΔΔP<0.001;
モデルグループと比較して*P<0.05,**P<0.01,***P<0.001
(2) 動物における体重及び食物摂取に関する薬物の作用
表4及び図1に示されるように、A-3 及びA-11は動物の体重を有意に減少でき、動物の摂取に関して重大な作用を有さず、薬物の優れた体重損失機能を示す。
【0049】
表4 動物における食物摂取に関する薬物の作用
【0050】
(3) 経口グルコーストレランス試験
結果を図2に示し、即ち、血液グルコースの初期のレベルは肥満の動物モデルで2時間以内に回復し得ず、グルコーストレランス曲線が異常であり、グルコーストレランスが減少したが、A-3 及びA-11は肥満の動物でグルコーストレランスを有意に改善し得る。
3. 薬物速度論研究
1.血漿濃度-時間曲線
実験操作:
SDラット36匹を1:1の雌対雄の比及び150 -170gの範囲の体重で選び、それらを二つの用量グループに指定し、それらにA-1 及びA-3 を夫々投与し、3種の用量サブグループを50、10、及び2mg/kgである投薬濃度、6匹の動物で夫々の用量グループ、1:1の雌対雄の比の夫々のサブグループで設計した。ラットの全てを12時間にわたって断食させ、随時水に接近させた。投薬容積は10ml/kgであり、0.4-0.5 ml の血液サンプルを胃の洗浄後の異なる時点で目覚めた動物の後眼窩静脈から採取し、5000 rpm で10分間遠心分離した。血漿サンプル200 μL を正確に測定し、次いでアセトニトリル200 μL を補給し、均等にかき混ぜ、5分間放置し、遠心分離した(14000 rpm, 5分間) 。上澄みを集め、次いで再度遠心分離した(14000 rpm, 5分間) 。得られる上澄みを集め、HPLC注入にかけた。血液サンプリングのための時点は夫々0, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 36, 48及び72時間であり、夫々の時点に関するピーク面積は同じ時点における6匹のラットから得られたピーク面積の平均であった。血漿濃度を計算し、薬物濃度-時間曲線をプロットした。
結果を図3に示した。
結果はA-3 のt1/2 が10-12時間であり、A-1 のt1/2 が26-28時間であり、A-3 の生物利用能が86%であり、またA-1 の生物利用能が15%であることを示す。
2.組織分布
SDラット18匹を水に自由に接近させて16時間にわたって断食させ、それらを1:1の雌対雄の比で6匹の動物/グループで3つのグループに指定し、それらにHS060098 (10 mg/kg)を投与し、次いで動物を投薬の1時間後、10時間後、そして24時間後に犠牲にし、夫々の動物の心臓、肝臓、脾臓、肺、腎臓を迅速に取り出した。器官の表面の汚れた血液を生理食塩水で直ちに洗い去った。器官からの組織を別々に切開し、充分に洗浄し、ふき取り、計量し、400 mg/mlの生理食塩水を補給し、氷浴中で1分間にわたって均一にし、3750 rpmで20分間にわたって遠心分離した。上澄み400 μl をピペットで正確に計り取り、続いて内部標準 (5ミリモル/Lのモル濃度の内部標準として使用されたHS060001) 40 μl を添加し、精製水3ml で希釈し、均一にかき混ぜ、カラム (SPE C18 カラムを最初に精製水3ml で洗浄し、続いて活性化のために10% メタノール3ml で洗浄した) に装填した。カラムを精製水3 mlで1 ml/分ですすぎ、次いで溶離剤 (メタノール :精製水 = 9: 1) 3 ml で溶離した。溶離液を集め、45℃で窒素流のもとに送風乾燥した。残渣をアセトニトリル400 μl に溶解し、次いでHPLCインサートを入れ、続いてサンプルを注入した。
結果を表5に示す。結果はA-3 及びA-1 が主として肝臓中に分布し、A-3 が比較的に均一に分布することを示す。
【0051】
表5 器官中のA−3及びA−1の主たる分布
【実施例】
【0052】
中間体化合物IIの調製:
実施例1:化合物II-1の調製
エチル 2-(2-メチル-4-ヒドロキシ)-フェノキシ-アセテート(II-1)の調製
【0053】
【化12】
【0054】
500 mlの1口フラスコに撹拌しながら3-メチル-4-ヒドロキシアセトフェノン (25 g, 166.5 ミリモル)、アセトニトリル250 ml 、及び炭酸カリウム (K2CO3, 26 g, 188.2 ミリモル) を連続して添加した。次いでアセトニトリル (50 ml)中のエチルブロモアセテート (20 ml, 172.2 ミリモル) をそれに滴下して添加した。その反応液を室温で8時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル(200 ml) で希釈し、濾過し、蒸発させて黄色の油を得た。
別の500 mlの1口フラスコ中で、ジクロロメタン (CH2Cl2, 300 ml) 中の上記の黄色の油の撹拌溶液に、3-クロロ過安息香酸 (46 g, 199.9 ミリモル) 及び触媒量の4-メチルベンゼンスルホン酸を添加した。得られる混合物を室温で一夜撹拌した。その反応が完結した後、その反応混合物を濾過し、得られるフィルターケーキをジクロロメタン100 mlで洗浄した。合わせた濾液を次亜硫酸ナトリウム (Na2S2SO4) 及び重炭酸ナトリウム (NaHCO3) の飽和水溶液で洗浄し、次いで蒸発させて淡黄色の油を得た。
上記反応から得られた粗生成物を別の500 mlの1口フラスコに移し、エタノール80ml及び触媒量の4-メチルベンゼンスルホン酸をそれに添加した。その反応溶液を6時間にわたって加熱、還流し、次いで真空で蒸発させて淡黄色の固体を得た。その淡黄色の固体を石油エーテル/酢酸エチル(2:1 v/v) により再結晶してエチル 2-(2-メチル-4-ヒドロキシ)-フェノキシ-アセテート (16 g, 3工程につき45.8%) を白色の固体として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.34 (t, J = 7.14 Hz, 3H), 2.27 (s, 3H), 4.30 (q, J = 7.14 Hz, 2H), 4.61 (s, 2H), 6.61-6.63 (m, 1H), 6.65 (s, 1H), 6.67-6.68 (m, 1H), 7.31 (s, 1H).
実施例2:化合物II-2の調製
3-メチル-4-ヒドロキシアセトフェノンを出発物質として使用して、エチル 2-(3-メチル-4-ヒドロキシ)-フェノキシ-アセテート (II-2) を実施例1に記載された方法と同様の方法に従って40.2%の収率で白色の固体として調製した。
【0055】
【化13】
【0056】
II-2
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 1.23 (t, J = 7.12 Hz, 3H), 2.29 (s, 3H), 4.18 (q, J = 7.14 Hz, 2H), 4.64 (s, 2H), 6.57-6.60 (m, 1H), 6.68 (s, 1H), 6.69-6.70 (m, 1H), 8.87 (s, 1H); 13C NMR (100 MHz, DMSO) δ 14.5, 16.6, 60.9, 65.9, 112.2, 112.8, 115.4, 117.6, 125.2, 150.2, 150.8, 169.6.
実施例3:化合物II-3の調製
3-エチル-4-ヒドロキシアセトフェノンを出発物質として使用して、エチル 2-(2-エチル-4-ヒドロキシ)-フェノキシ-アセテート (II-3) を実施例1に記載された方法と同様の方法に従って74.5%の収率で淡黄色の固体として調製した。
【0057】
【化14】
【0058】
II-3
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.23 (t J = 7.53 Hz, 3H), 1.34 (t J = 7.12 Hz, 3H), 2.69 (q, J = 7.52 Hz, 2H), 4.30 (q, J = 7.14 Hz, 2H), 4.61 (s, 2H), 5.35 (s, 1H), 6.6 (dd, J = 8.65 Hz, 2.84 HZ, 1H), 6.65 (d, J = 8.63 Hz, 1H), 6.72 (d, J =2.84 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 14.1, 23.1, 61.3, 66.7, 112.6, 113.1, 116.5, 134.9, 149.9, 150.4, 169.8.
実施例4:化合物II-4の調製
エチル 2-(3-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (II-4)の調製
【0059】
【化15】
【0060】
500 mlの3口フラスコに、o-クレゾール (15 g, 138.7 ミリモル) 、チオシアン酸ナトリウム(NaSCN, 34 g, 419.3 ミリモル) 、臭化ナトリウム (NaBr, 16 g, 155.5 ミリモル) 及びメタノール (200 ml) を撹拌しながら連続して添加した。その混合物を0℃に冷却し、次いでメタノール(30 ml) 中の臭素 (8.6 ml, 167 ミリモル) の溶液をそれに滴下して添加した。0℃で1時間撹拌した後、その混合物を室温に温め、次いで室温で更に4時間撹拌した。その反応が完結した後、重炭酸ナトリウム (NaHCO3) の飽和水溶液200 mlをその反応混合物に添加し、10分間撹拌した。その混合物を酢酸エチル (2 X 500 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して黄色の油を得た。シリカゲルカラムクロマトグラフィー精製 (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) を行なって3-メチル-4-ヒドロキシ-フェニル チオシアン酸を白色の固体 (16 g, 収率: 69.8%)として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 2.17 (s, 3H), 6.92(d, J = 8.4 Hz, 1H), 7.37 (dd, J = 8.4 Hz, 2.48 HZ, 1H), 7.44 (d, J = 2.48 HZ, 1H), 10.1 (s, 1H); 13C NMR (100 MHz, DMSO) δ 16.2, 111.1, 113.1, 116.8, 127.3, 132.2, 135.4, 158.3。
【0061】
500 mlの3口フラスコ中で、四水素化アルミニウムリチウム(LiAlH4, 3.5 g, 92.2 ミリモル) 及びテトラヒドロフラン (THF, 50 ml) の撹拌混合物に0℃でテトラヒドロフラン (THF, 30 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (6.13 g, 37.1 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6 M 塩酸の添加により3-4 に調節した。次いで水層を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物4-ヒドロキシ-3-メチルチオフェノールを黄色の油として得た。
250 mlの1口フラスコ中で、アセトニトリル (80 ml) 中の4-ヒドロキシ-3-メチルチオフェノールの上記粗生成物の溶液にエチル 2-ブロモアセテート (4.3 ml, 37 ミリモル)を撹拌しながら添加し、続いて炭酸カリウム (5.1 g, 36.9 ミリモル) を添加した。得られる混合物を室温で8時間撹拌した。その反応が完結した後、その反応混合物を酢酸エチル (80 ml)で希釈し、濾過し、蒸発させて残渣を得た。残渣をシリカゲルカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) により精製してエチル 2-(3-メチル-4-ヒドロキシ)-フェニルチオ-アセテート6.38 g を黄色の油 (収率: 76%)として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.24 (t J = 7.2 Hz, 3H), 2.18 (s, 3H), 3.49 (s, 2H), 4.15 (q, J = 7.14 Hz, 2H), 5.74 (s, 1H), 6.63 (d, J = 8.27 Hz, 1H), 7.17 (dd, J = 8.16 Hz, 2.28 HZ, 1H), 7.25 (d, J = 2.28 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 15.7, 38.8, 61.6, 115.6, 124.1, 125.1, 131.9, 135.7, 154.5, 170.6.
実施例5:化合物II-5の調製
m-クレゾールを出発物質として使用して、エチル 2-(2-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (II-5) を実施例4に記載された方法と同様の方法に従って63%の収率で黄色の油として調製した。
【0062】
【化16】
【0063】
II-5
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.23 (t J = 7.1 Hz, 3H), 2.39 (s, 3H), 3.44 (s, 2H), 4.14 (q, J = 7.11 Hz, 2H), 6.38 (s, 1H), 6.54 (dd, J = 8.36 Hz, 2.72 HZ, 1H), 6.64 (d, J = 2.71 Hz, 1H), 7.31 (d, J =8.37 Hz, 1H).
実施例6:化合物II-6の調製
2,5-ジメチルフェノールを出発物質として使用して、エチル 2-(2,5-ジメチル-4-ヒドロキシ)-フェニルチオ-アセテート (II-6) を実施例4に記載された方法と同様の方法に従って72%の収率で白色の固体として調製した。
【0064】
【化17】
【0065】
II-6
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.23 (t J = 7.16 Hz, 3H), 2.16 (s, 3H), 2.38 (s, 3H), 3.43 (s, 2H), 4.13 (q, J = 7.13 Hz, 2H), 5.19 (s, 1H), 6.58 (s, 1H), 7.23 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 15.1, 20.2, 37.9, 61.4, 116.9, 122.1, 123.3, 136.9, 140.0, 154.3, 170.3.
中間体化合物IIIの調製:
実施例7:化合物 III-1の調製
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (化合物 III-1) の調製は下記の反応スキームに従ってなし得る。
【0066】
【化18】
【0067】
N-メチルフェニルアセトアミドの調製
【化19】
【0068】
フェニル酢酸 (10 g, 73.5 ミリモル) 及びメタノール (60 ml) を250 mlの3口フラスコに添加し、続いて10滴の濃硫酸を添加した。得られる溶液を2時間にわたって加熱、還流した。次いで、その溶液を室温に冷却し、続いてメチルアミンの25-30%の水溶液 (50 ml, 約403 ミリモル) を添加し、更に3時間にわたって加熱、還流した。反応が完結した後、それを室温に冷却した。溶媒の殆どを回転真空蒸発により除去し、水30mlをそれに添加し、次いでその混合物を酢酸エチル (3 X 80 ml)で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮してN-メチル フェニルアセトアミド (白色の固体, 収率: 91.8%) 10.1gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.69 (d, J = 4.84 Hz, 3H), 3.51 (s, 3H), 5.95 (wide, 1H), 7.21-7.24 (m, 3H), 7.28-7.32 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 26.4, 43.6, 127.2, 128.9, 129.4, 135.1, 171.8.
3-ベンジル-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オンの調製
【0069】
【化20】
【0070】
1000 ml の3口フラスコ中で、80-90℃のトルエン (250 ml) 中のN-メチルフェニルアセトアミド (25 g, 167.7 ミリモル) 及び(4-トリフルオロメチル) フェニルヒドラジン ( 30g, 170 ミリモル) の撹拌溶液に、トルエン(50 ml) 中のオキシ塩化リン (POCl3, 15 ml,180 ミリモル) の溶液を滴下して添加した。その添加が完結した後、その混合物を80-90℃で5時間撹拌し、次いでその反応が完結した時に室温に冷却し、濾過した。残留フィルターケーキを、それが黄褐色になるまで酢酸エチルで洗浄した。濾液を重炭酸ナトリウムの飽和水溶液で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、再度濾過した。フィルターケーキを酢酸エチルで洗浄した。フィルターケーキを合わせ、真空乾燥した。最後にN-メチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン43.5gを得た (黄褐色の固体, 収率: 84.4%)。
1000 mlの3口フラスコ中で、テトラヒドロフラン (THF, 500 ml) 中のN-メチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン (58.6 g, 190.9 ミリモル) 及びN,N-ジメチルアミノピリジン (DMAP, 0.5 g, 4.0ミリモル) の撹拌混合物に、カルボニルジイミダゾール (CDI, 46.43 g, 286.4 ミリモル) を徐々に添加した。得られる混合物を室温で12時間撹拌した。その反応が完結した後、その混合物を水300 mlを含む2000 ml のビーカーに注ぎ、撹拌下で6N塩酸でpH 2-4に調節し、濾過した。フィルターケーキを蒸留水で洗浄し、次いで真空乾燥して3-ベンジル-4-メチル-1-(4-トリフルオロメチル) -フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (淡黄色の固体, 収率: 89.2%)56.7gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.11 (s, 3H), 4.0 (s, 2H), 7.25-7.38 (m, 5H), 7.67 (d, J = 8.68 Hz, 2H), 8.18 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 27.6, 32.7, 118.0, 122.8, 125.5, 126.6, 127.7, 128.4, 129.1, 133.6, 140.7, 146.8, 152.6.
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1) の調製
【0071】
【化21】
【0072】
500 mlの3口フラスコに、3-ベンジル-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (15 g, 45 ミリモル) 及びクロロホルム (CHCl3, 250 ml) を撹拌しながら添加した。その撹拌溶液に、N-ブロモスクシンイミド (NBS, 12 g, 68 ミリモル) 及びベンゾイルペルオキシド (0.5 g, 2.25 ミリモル) を添加した。その混合物を12時間にわたって慎重に加熱、還流した。その反応が完結した後、その混合物を濾過した。濾液を減圧で濃縮して残渣を得、これをフラッシュシリカゲルカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル = 8:1 v/v) により精製して3-(1’-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン14.6gを赤褐色の油 (収率: 79.7%)として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.28 (s, 3H), 6.07 (s, 1H), 7.25-7.58 (m, 5H), 7.66 (d, J = 8.68 Hz, 2H), 8.13 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 28.4, 42.1, 69.6, 118.3, 126.2, 126.8, 127.3, 129.1, 129.9 133.9, 140.3, 145.7, 152.4.
実施例8:化合物 III-2の調製
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (化合物 III-2) の調製を下記の反応スキームに従って行い得る。
【0073】
【化22】
【0074】
N-n-ブチルフェニルアセトアミドの調製
【化23】
【0075】
フェニル酢酸 (16 g, 117.6 ミリモル) 及びメタノール ( 60 ml) を250 mlの3口フラスコに添加し、続いて5滴の濃硫酸を添加した。得られる溶液を6時間にわたって加熱、還流した。次いで、その混合物を室温に冷却し、続いてn-ブチルアミン (12 ml, 121.4 ミリモル) を添加し、更に3時間にわたって加熱、還流した。その反応が完結した後、それを室温に冷却した。溶媒の殆どを回転真空蒸発により除去し、水30mlをそれに添加し、その混合物を酢酸エチル (3 X 80 ml)で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮してN-n-ブチルフェニルアセトアミド (白色の固体, 収率: 70.4%) 15.9gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 7.28 Hz, 3H), 1.21-1.29 (m, 2H), 1.36-1.44 (m, 2H), 3.2 (q, J = 7.04 Hz, 2H), 5.62 (ブロード, 1H), 7.24-7.29 (m, 3H), 7.32-7.36 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 13.7, 19.9, 31.5, 39.4, 43.8, 127.2, 128.9, 129.4, 135.1, 170.9.
3-ベンジル-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オンの調製
【0076】
【化24】
【0077】
500 mlの3口フラスコ中で、80-90℃のトルエン (150 ml) 中のN-n-ブチルフェニルアセトアミド (11 g, 57.5 ミリモル) の撹拌溶液に、トルエン (20 ml) 中のオキシ塩化リン (POCl3,17 ml, 75.1 ミリモル) の溶液を滴下して添加した。その添加が完結した後、その混合物を80-90℃で5時間撹拌し、次いでその反応が完結した後に室温に冷却し、濾過した。フィルターケーキを、それが黄褐色になるまで酢酸エチルで洗浄した。濾液を重炭酸ナトリウムの飽和水溶液で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、再度濾過した。フィルターケーキを酢酸エチルで再度洗浄した。フィルターケーキを合わせ、真空乾燥した。最後に、N-n-ブチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン15.7gを得た (黄褐色の固体)。
500 mlの3口フラスコ中で、テトラヒドロフラン (THF, 250 ml) 中のN-n-ブチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン (15.7 g) 及びN,N-ジメチルアミノピリジン(DMAP, 0.5 g, 4.0 ミリモル) の撹拌混合物に、カルボニルジイミダゾール (CDI, 7.43 g, 45.8 ミリモル) を室温で徐々に添加した。その反応混合物を室温で12時間撹拌した。その反応が完結した後、その混合物を水100 mlを含む500 mlのビーカーに注ぎ、撹拌下で6N塩酸でpH 2-4に調節し、濾過した。フィルターケーキを蒸留水で洗浄し、次いで真空乾燥して3-ベンジル-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン 6.28g(淡黄色の固体, 2工程の収率: 29.2%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 7.3 Hz, 3H), 1.22-1.29 (m, 2H), 1.37-1.44 (m, 2H), 3.49 (q, J = 7.6 Hz, 2H), 4.0 (s, 2H), 7.26-7.38 (m, 5H), 7.67 (d, J = 8.70 Hz, 2H), 8.19 (d, J = 8.65 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 19.8, 30.6, 32.8, 41.7, 117.9, 126.1, 126.9, 127.2, 127.8, 128.6, 129.1, 134.0, 140.7, 146.6, 152.5; MS (ESI) m/z 376.29 (M+1) +.
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-2)の調製
【0078】
【化25】
【0079】
250 mlの3口フラスコに、3-ベンジル-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (3.75 g, 10 ミリモル) 及びクロロホルム (100 ml) を添加した。その撹拌溶液にN-ブロモスクシンイミド (NBS, 3.56 g, 20 ミリモル) 及びベンゾイルペルオキシド (0.4 g, 1.65 ミリモル) を添加した。その混合物を6時間にわたって慎重に加熱、還流した。その反応が完結した後、溶媒を回転真空蒸発により除去し、残渣をカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) にかけて3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-2) (赤褐色の粘稠な液体, 収率: 85.9%) 3.9gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.36 Hz, 3H), 1.27-1.35 (m, 2H), 1.47-1.53 (m, 1H), 1.63-1.66 (m, 1H), 3.66-3.72 (m, 2H), 6.00 (s, 1H), 7.41-7.46 (m, 3H), 7.61 (dd, J = 7.99 Hz, 1.83 Hz, 2H) 7.66 (d, J = 8.69 Hz, 2H), 8.15 (d, J = 8.57 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 19.9, 30.3, 41.5, 42.4, 118.2, 126.2, 126.8, 127.3, 128.6, 128.9, 129.5, 134.8, 140.4, 145.6, 152.2.
実施例9:化合物 III-3の調製
化合物 III-3の調製を下記の反応スキームに従って達成し得る。
【0080】
【化26】
【0081】
N-メチルベンズオキソアセトアミドの調製
【化27】
【0082】
60% 水素化ナトリウム (3.7 g, 92.5 ミリモル) 及びテトラヒドロフラン (20 ml) を250 mlの3口フラスコに添加し、続いて加熱し、テトラヒドロフラン中のベンジルアルコール (10 g, 96.1 ミリモル) の溶液20mlを滴下して添加した。その添加が完結した後、その反応混合物を1時間にわたって加熱、還流した。次いでエチルブロモアセテート (11ml, 94.8 ミリモル) を添加し、その反応混合物を更に4時間にわたって還流下で撹拌した。室温に冷却した後、メチルアミンの水溶液25mlをそれに添加し、その反応を更に6時間にわたって還流下で行なった。その反応が完結した後、それを減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =4:1 v/v) にかけてN-メチルベンズオキソアセトアミド (白色の固体, 2工程の収率: 32%)5.3gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.83 (d, J = 5.16 Hz, 3H), 3.98 (s, 2H), 4.55 (S, 2h), 6.61 (s, 1H), 7.30-7.39 (m, 5H).
3-ベンズオキソメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オンの調製
【0083】
【化28】
【0084】
250 mlの3口フラスコ中で、80-90℃のトルエン (40 ml) 中のN-メチル ベンジルオキシアセトアミド (6.4 g, 3.57 ミリモル) の撹拌溶液に、トルエン (20 ml) 中のオキシ塩化リン (3.6 ml,38.5 ミリモル) を滴下して添加した。得られる反応混合物を80-90℃で5時間撹拌した。その反応が完結した後、それを室温に冷却し、濾過した。フィルターケーキを、それが肉色になるまで酢酸エチルで洗浄した。濾液を重炭酸ナトリウムの飽和水溶液で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、再度濾過した。フィルターケーキを酢酸エチルで再度洗浄した。フィルターケーキを合わせ、真空乾燥した。最後に、肉色の固体4.5gを得た。
上記肉色の固体4.5gを別の500 mlの3口フラスコに移し、テトラヒドロフラン (THF) 200 mlをそれに添加し、撹拌し、カルボニルジイミダゾール (CDI) (5 g, 30.8 ミリモル) を徐々にそれに添加した。その反応混合物をその添加後に室温で12時間撹拌した。その反応が完結した後、それを減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =4:1 v/v) にかけて3-ベンズオキソ メチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (明るい紅褐色のゼラチン状物, 2工程の収率: 23%) 3gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.35 (s, 3H), 4.50 (s, 2H), 4.59 (s, 2H), 7.32-7.37 (m, 5H), 7.66 (d, J = 8.46 Hz, 2H), 8.14 (d, J = 8.46 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 26.9, 27.8, 62.8, 72.9, 118.1, 126.1, 126.2, 126.3, 128.3, 128.5, 136.6, 140.5, 144.7, 152.5.
3-ヒドロキシメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-
トリアゾール-5-オンの調製
【0085】
【化29】
【0086】
250 mlの3口フラスコ中で、エタノール (60 ml) 中の上記生成物3-ベンズオキソ メチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (3.0 g, 8.3 ミリモル) の撹拌溶液に10% パラジウム/カーボン1gを添加した。そのフラスコ中の10% パラジウム/カーボン1gをN2 で置換し、次いで水素ガスを導入した。その反応混合物を室温で24時間撹拌した。その反応が完結した後、それを濾過し、真空濃縮して3-ヒドロキシメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オン (白色の固体, 収率: 93%)2.1gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.34 (s, 3H), 4.52 (d, J = 5.84 Hz, 2H), 5.77 (t, J = 5.84 Hz, 1H), 7.85 (d, J = 8.59 Hz, 2H), 8.17 (d, J = 8.59 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 27.9, 55.2, 118.0, 125.2, 125.5, 126.0, 141.3, 149.0, 152.5.
3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (III-3)の調製
【0087】
【化30】
【0088】
250 mlの3口フラスコ中で、ジクロロメタン (100 ml) 中の上記生成物 (2.1 g, 7.77 ミリモル) の撹拌溶液にN-ブロモスクシンイミド (NBS, 1.8 g, 10.1 ミリモル) を添加し、続いてトリフェニルホスフィン (2.4 g, 9.15 ミリモル) を徐々に添加した。その反応液を室温で4時間撹拌した。その反応が完結した後、それをカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (淡黄色の固体, 収率: 89%) 2.3gを得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.42 (s, 3H), 4.34 (s, 2H), 7.67 (d, J = 8.63 Hz, 2H), 8.12 (d, J = 8.63 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 19.1, 26.9, 27.9, 118.2, 126.2, 140.2, 143.5, 152.1; MS (ESI) m/z 332.27 (M-4) +.
化合物 (I) (エステル):の調製
実施例10:化合物 E-1の調製
【0089】
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (E-1)の調製
150 mlの1口フラスコ中で、アセトニトリル (150 ml) 中の3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1) (36.3 g, 88.1 ミリモル) の撹拌溶液にエチル (2-メチル-4-ヒドロキシ)-フェノキシ-アセテート (II-1) (17.5 g, 83.3 ミリモル) 、N,N-ジメチルアミノピリジン (DMAP, 0.5 g, 4.09 ミリモル) 、及び炭酸カリウム (K2CO3, 13.8 g, 99.9 ミリモル) を添加した。その反応混合物を室温で8時間撹拌した。その反応が完結した後、それを濾過した。フィルターケーキを酢酸エチル (3X50 ml) で洗浄し、その後に捨てた。濾液を合わせ、回転真空蒸発にかけて溶媒を除去し、次いでカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) にかけて無色のゼラチン状物 (これは放置されている間に徐々に白色の固体になった) (33.5 g,収率: 74.3%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.28 (t, J = 7.11 Hz, 3H), 2.26 (s, 3H), 3.20 (s, 3H), 4.26 (q, J = 7.11 Hz, 2H), 4.61 (s, 2H), 6.28 (s, 1H), 6.65 (d, J = 8.8 Hz, 1H), 6.81 (d, J = 8.75 Hz, 1H), 6.93 (d, J = 3.0 Hz, 1H), 7.25-7.45 (m, 3H), 7.50 (d, J = 7.32, 2H), 7.67 (d, J = 8.48 Hz, 2H), 8.14 (d, J = 8.48 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.7, 28.2, 61.3, 65.9, 75.1, 112.8, 113.2, 118.3, 119.3, 122.7, 125.8, 126.2, 127.1, 128.9, 129.0, 129.4, 135.1, 140.4, 146.3, 151.5, 152.8, 169.1; MS (ESI) m/z 558.9 (M+NH4+).
実施例11:化合物 E-2の調製
【0090】
化合物III-1及びII-5を出発物質として使用して、エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-2) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは60%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.12 Hz, 3H), 2.45 (s, 3H), 3.21 (s, 3H), 3.50 (s, 2H), 4.12 (q, J = 7.12 Hz, 2H), 6.39 (s, 1H), 6.92 (dd, J = 8.59 Hz, 2.82 Hz, 1H), 7.0, (d, J = 2.82 Hz, 1H), 7.40-7.53(m, 3H), 7.52 (d, J = 7.48 Hz, 2H), 7.69 (d, J = 8.76 Hz, 2H), 8.18 (d, J = 8.76 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 20.9, 28.3, 37.1, 61.4, 74.6, 113.6, 117.9, 118.2, 125.4, 125.8, 126.2, 126.9, 127.3, 128.96, 129.1, 134.2, 134.7, 140.5, 142.2, 145.9, 152.6, 169.7; MS (ESI) m/z 558.01(M+H) +.
実施例12:化合物 E-3の調製
【0091】
化合物III-1 及びII-4を出発物質として使用して、エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-3) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは71%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.20 (t, J = 7.10 Hz, 3H), 2.37 (s, 3H), 3.19 (s, 3H), 3.53 (s, 2H), 4.14 (q, J = 7.10 Hz, 2H), 6.39 (s, 1H), 6.94 (d, J = 8.58 Hz, 1H), 7.24 (dd, J = 8.58 Hz, 2.4 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 7.40-7.46(m, 3H), 7.50 (d, J = 7.52 Hz, 2H), 7.69 (d, J = 8.84 Hz, 2H), 8.17 (d, J = 8.84 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.5, 28.2, 38.0, 61.4, 74.5, 113.1, 118.2, 122.7, 125.4, 125.8, 126.2, 126.3, 127.1, 127.4, 128.0, 128.96, 129.1, 130.8, 134.8, 140.4, 145.9, 152.6, 169.8; MS (ESI) m/z 557.97 (M+H) +.
実施例13:化合物 E-4の調製
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-4)の調製
【0092】
【化31】
【0093】
一定圧力の滴下ロートを備えた250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン (30 ml) の撹拌混合物に0℃でテトラヒドロフラン (THF, 30 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.7 g, 10.3 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌そして室温で更に2時間の撹拌後に、その反応を氷水浴中で砕いた氷の添加により停止した。その混合物のpHの値を6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を塩化ナトリウムの飽和溶液 (2 X100 ml) で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物3-メチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
【0094】
150 mlの1口フラスコ中で、アセトニトリル (30 ml) 中の3-(1'-ブロモ-ベンジル)-4-メチル-1- (4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1, 4.01 g, 9.73 ミリモル) の撹拌溶液に、アセトニトリル (30 ml)中の新たに調製された粗生成物3-メチル-4-ヒドロキシ-メルカプトベンゼン、N,N-ジメチルアミノピリジン (DMAP, 0.07 g, 0.57 ミリモル) 、及び炭酸カリウム (K2CO3, 1.28 g, 9.27 ミリモル) を添加した。その反応液を室温で4時間撹拌した後、炭酸カリウム (K2CO3, 1.4 g, 10.1 ミリモル) 及びエチルブロモアセテート (1.1 ml, 9.5 ミリモル) を添加した。その反応混合物を更に8時間撹拌した。その反応が完結した後、それを濾過した。フィルターケーキを酢酸エチル (3X30 ml) で洗浄し、その後に捨てた。濾液を合わせ、回転真空蒸発にかけて溶媒を除去し、次いでカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて白色の固体4.4 g (収率: 81%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.26 (t, J = 7.14 Hz, 3H), 2.19 (s, 3H), 3.15 (s, 3H), 4.23 (q, J = 7.14 Hz, 2H), 4.58 (s, 2H), 5.17 (s, 1H), 6.54 (d, J = 8.44 Hz, 1H), 7.11 (dd, J = 8.32 Hz, 2.28 Hz, 1H), 7.17 (d, J = 2.28 Hz, 1H), 7.33-7.38(m, 5H), 7.66 (d, J = 8.56 Hz, 2H), 8.10 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.1, 28.0, 50.6, 61.4, 65.4, 113.6, 118.2, 122.8, 125.5, 126.2, 126.8, 127.1, 128.2, 128.5, 128.9, 133.8, 135.2, 137.8, 140.5, 146.5, 152.6, 157.1, 168.5; MS (ESI) m/z 558.03(M+H) +.
実施例14:化合物 E-5の調製
化合物III-1 及びII-3を出発物質として使用して、エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (E-5) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは87.3%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.24 Hz, 3H), 1.27 (t, J = 7.24 Hz, 3H), 2.67 (m, 2H), 3.20 (s, 3H), 4.24 (q, J = 7.24 Hz, 2H), 4.57 (s, 2H), 6.29 (s, 1H), 6.81 (dd, J = 8.88 Hz, 3.08 Hz, 1H), 6.95 (d, J = 3.08 Hz, 1H), 7.36-7.45 (m, 3H), 7.52 (d, J = 7.60, 2H), 7.67 (d, J = 8.76 Hz, 2H), 8.16 (d, J = 8.76 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9, 14.1, 23.2, 23.3, 28.3, 61.3, 66.3, 75.3, 112.5, 112.6, 112.96, 113.2, 116.5, 117.6, 118.2, 125.4, 125.9, 126.2, 127.3, 128.8, 128.96, 135.2, 135.3, 140.5, 146.4, 150.6, 151.3, 151.6, 152.8, 169.2;MS (ESI) m/z 556.29(M+H) +.
実施例15:化合物 E-6の調製
【0095】
化合物III-1 及びII-2 を出発物質として使用して、エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (E-6) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは98%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.02 Hz, 3H), 2.24 (s, 3H), 3.25 (s, 3H), 4.29 (q, J = 7.02 Hz, 2H), 4.59 (s, 2H), 6.35 (s, 1H), 6.56-6.75 (m, 1H), 6.88 (d, J = 2.95 Hz, 1H), 6.95 (d, J = 8.94 Hz, 1H), 7.42-7.55(m, 3H), 7.58 (d, J = 7.49 Hz, 2H), 7.72 (d, J = 8.69 Hz, 2H), 8.23 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.7, 28.2, 61.3, 65.96, 75.1, 112.1, 112.8, 113.9, 115.4, 117.8, 118.2, 118.5, 125.5, 125.8, 126.2, 126.9, 127.2, 128.8, 129.0, 135.3, 140.5, 146.4, 149.9, 152.7, 169.1; MS (ESI) m/z 541.9(M+H+); 558.9 (M+NH4+).
実施例16:化合物 E-7の調製
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-7)の調製
【0096】
【化32】
【0097】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 0.8 g, 21 ミリモル) 及びテトラヒドロフラン (30 ml) の撹拌混合物に0℃(冷却水で冷却)のテトラヒドロフラン (THF 30 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.1 g, 6.6 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌そして室温で更に2時間の撹拌後に、その反応を氷水浴中で砕いた氷の添加により停止した。その混合物のpHの値を6N塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を塩化ナトリウムの飽和溶液 (2 X100 ml) で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物3-メチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
別の250 mlの1口フラスコ中で、アセトニトリル (30 ml)中の得られた3-メチル-4-ヒドロキシルメルカプトベンゼンの撹拌溶液に3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1, 2.5 g, 6.06 ミリモル) 、アセトニトリル (30 ml) 、N,N-ジメチルアミノピリジン (DMAP, 0.1 g, 0.82 ミリモル) 及び炭酸カリウム (K2CO3, 0.85 g, 6.15 ミリモル) を添加した。その混合物を室温で5時間撹拌した後、炭酸カリウム (K2CO3,1.6 g, 11.6 ミリモル) 及びエチルブロモアセテート (1.2 ml,10.3 ミリモル) をそれに添加し、得られる反応混合物を室温で更に8時間撹拌した。その反応が完結した後、それを濾過した。フィルターケーキを酢酸エチル (3X50 ml) で洗浄し、その後に捨てた。濾液を合わせ、回転真空蒸発にかけて溶媒を除去し、次いでカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル = 8:1→5:1 v/v) にかけて白色の固体2.0 g (収率: 59%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.27 (t, J = 7.18 Hz, 3H), 2.31 (s, 3H), 3.13 (s, 3H), 4.24 (q, J = 7.18 Hz, 2H), 4.55 (s, 2H), 5.09 (s, 1H), 6.59 (dd, J = 8.56 Hz, 2.88 Hz, 1H), 6.78, (d, J = 2.88 Hz, 1H), 7.24 (d, J = 8.68 Hz, 1H), (7.30-7.34(m, 4H), 7.66 (d, J = 8.66 Hz, 2H), 8.10 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 20.9, 28.1, 49.8, 61.5, 65.2, 112.6, 117.0, 118.2, 123.1, 125.5, 126.2, 126.8, 127.1, 128.0, 128.5, 128.9, 135.1, 137.9, 140.5, 144.5, 146.4, 152.5, 168.5; MS (ESI) m/z 558.05 (M+H) +.
実施例17:化合物 E-8の調製
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-アセテート(E-8)の調製
【0098】
【化33】
【0099】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン (30 ml) の撹拌混合物に0℃のテトラヒドロフラン (20 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.38 g, 8.35 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌の後に、その反応液を室温に温め、室温で更に2時間撹拌した。次いで、その反応をエタノール (10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 80 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、丸1日にわたって空気中に放置し、カラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してカップリングされた化合物3-メチル-4-ヒドロキシ-メルカプトベンゼン1.6g (黄色のゼリー状の液体,収率: 68.8%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.2 (s, 3H), 5.35 (s, 1H), 6.69 (d, J = 8.28 Hz, 1H), 7.18 (dd, J = 8.23 Hz, 2.23 Hz, 1H), 7.24 (d, J = 2.24 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.7, 115.5, 125.0, 128.3, 130.2, 134.0, 154.4; MS (ESI) m/z 278.33 (M+H+).
150 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の3-メチル-4-ヒドロキシ-メルカプトベンゼン (1.6 g, 5.76 ミリモル) の撹拌溶液にエチルブロモアセテート (1.4 ml, 60.7 ミリモル) 及び炭酸カリウム (K2CO3, 1.6 g, 11.6 ミリモル) を添加した。その反応液を室温で12時間撹拌した後、その反応溶液を酢酸エチル60mlの添加により希釈し、濾過し、蒸発させて残渣を得た。残渣をカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製して淡黄色のゼリー状液体2.0 g (収率: 77.5%)を得た。
【0100】
150 mlの3口フラスコ中で、エタノール (15 ml) 中の上記生成物 (2.0 g, 4.44 ミリモル) の撹拌溶液に水 (15 ml) 及び濃塩酸 (7 ml) を添加した。亜鉛粉末 (10 g, 153 ミリモル) を撹拌しながら徐々に添加した。その添加後、その反応混合物を室温で30分間撹拌し、次いでジクロロメタン (3 X60 ml) で抽出した。有機相を合わせ、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して淡黄色の液体を得た。
150 mlの1口フラスコ中で、アセトニトリル30ml中の上記粗生成物の撹拌溶液に3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4- トリアゾール-5(4H)-オンIII-3 (1.3 g, 3.9 ミリモル) の溶液30ml及び炭酸カリウム (K2CO3, 3.4 g, 24.6 ミリモル) を添加した。得られる反応混合物を室温で12時間撹拌した。その反応が完結した後、その反応溶液を酢酸エチル50mlの添加により希釈し、濾過した。濾液を減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =3:1 v/v) にかけて白色の固体1.7 g (収率: 91%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.25 (t, J = 7.12 Hz, 3H), 2.22 (s, 3H), 3.3 7 (s, 3H), 3.87 (s, 2H), 4.21 (q, J = 7.12 Hz, 2H), 4.58 (s, 2H), 6.58 (d, J = 8.37 Hz, 1H), 7.16 (dd, J = 8.37 Hz, 2.34 Hz, 1H), 7.23 (d, J = 2.34 Hz, 1H), 7.63 (d, J = 8.57 Hz, 2H), 7.98 (d, J = 8.57 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.1, 27.8, 31.3, 61.3, 65.5, 111.6, 118.1, 122.7, 123.5, 125.4, 126.1, 128.1, 128.6, 132.4, 136.3, 140.5, 144.6, 152.4, 156.8, 168.5; MS (ESI) m/z 482.49 (M+H)+.
実施例18:化合物 E-9の調製
【0101】
化合物III-3 及びII-2を出発物質として使用して、エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-アセテート (E-9) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは73.5%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.14 Hz, 3H), 2.33 (s, 3H), 3.45 (s, 3H), 4.28 (q, J = 7.14 Hz, 2H), 4.63 (s, 2H), 5.01 (s, 2H), 6.71 (d, J = 8.85 Hz, 1H), 6.8 (dd, J = 8.85 Hz, 3.08 Hz, 1H), 6.88 (d, J = 3.08 Hz, 1H), 7.70 (d, J = 8.56 Hz, 2H), 8.17 (d, J = 8.55 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.2, 61.2, 61.9, 66.4, 112.0, 112.7, 117.7, 118.2, 118.3, 126.2, 126.3, 129.3, 140.5, 143.9, 151.6, 151.8, 152.4, 169.1; MS (ESI) m/z 464.39 (M-1), 466.48 (M+H)+.
実施例19:化合物E-10の調製
【0102】
化合物III-1 及びII-6を出発物質として使用して、エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-10) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは72.2%の収率で黄色のゼリー状液体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.29 (t J = 7.16 Hz, 3H), 2.32 (s, 3H), 2.37 (s, 3H), 3.18 (s, 3H), 3.51 (s, 2H), 4.25 (q, J = 7.16 Hz, 2H), 6.39 (s. 1H), 6.88 (s, 1H), 7.31 (s, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.67 Hz, 2H), 8.16 (d, J = 8.67 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.0, 20.7, 28.2, 37.2, 74.4, 114.7, 118.2, 125.4, 125.6, 125.7, 126.3, 128.9, 129.1, 134.9, 135.7, 139.5, 140.4, 146.1, 152.7, 155.0, 168.7; MS (ESI) m/z 573.07 (M+H)+.
実施例20:化合物E-11の調製
【0103】
化合物III-3 及びII-4を出発物質として使用して、エチル 2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-アセテート (E-11) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは69.7%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.14 Hz, 3H), 2.19 (s, 3H), 3.43 (s, 3H), 3.55 (s, 2H), 4.28 (q, J = 7.14 Hz, 2H), 5.03 (s, 2H), 6.91 (d, J = 8.77 Hz, 3H), 7.29-7.32 (m, 2H), 7.67 (d, J = 8.66 Hz, 2H), 8.12 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.1, 28.1, 38.1, 61.4, 111.8, 118.3, 126.2, 126.3, 127.2, 127.5, 127.9, 130.8, 134.8, 140.3, 143.6, 152.5, 155.4, 169.2; MS (ESI) m/z 481.23 (M-1)-, 482.34 (M)-, 483.35 (M+H)-.
実施例21:化合物E-12の調製
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-12)の調製
【0104】
【化34】
【0105】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン (40 ml) の撹拌混合物に0℃のテトラヒドロフラン20ml中の2,5-ジメチル-4-ヒドロキシ-フェニルチオシアン酸 (1.2 g, 6.7 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌後に、その混合物を室温に温め、次いで室温で更に1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 60 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
別の250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の得られた粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼンの撹拌溶液に3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン(III-1) (1.6 g, 3.88 ミリモル)及び炭酸カリウム (K2CO3, 0.54 g, 3.9 ミリモル)を添加した。その混合物を室温で4時間撹拌した後、エチルブロモアセテート (1.8 ml, 15.5 ミリモル) 及び炭酸カリウム (K2CO3, 0.54 g, 3.9 ミリモル) をそれに添加した。その混合物を室温で一夜撹拌した。反応が完結した後、その反応溶液を酢酸エチル200 mlで希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製して黄色のゼリー状液体1.1g (3工程の収率: 50%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.28 (t J = 7.16 Hz, 3H), 2.12 (s, 3H), 2.25 (s, 3H), 3.13 (s, 3H), 4.25 (q, J = 7.16 Hz, 2H), 4.59 (s, 2H), 5.08 (s, 1H), 6.51 (s, 1H), 7.13 (s, 1H), 7.27-35 (m, 5H), 7.66 (d, J = 8.75 Hz, 2H), 8.10 (d, J =8.75 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 15.5, 20.7, 28.0, 50.0, 61.3, 65.5, 113.1, 117.9, 118.2, 122.4, 125.9, 126.1, 126.2, 126.8, 128.1, 128.4, 128.8, 129.1, 135.2, 138.9, 140.5, 141.5, 146.5, 152.6, 157.0, 168.6; MS (ESI) m/z 588.31 (M+NH4+).
実施例22:化合物E-13の調製
エチル 2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル) -4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-13)の調製
【0106】
【化35】
【0107】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.4 ミリモル) 及びテトラヒドロフラン (THF 30 ml) の撹拌溶液に0℃のテトラヒドロフラン20ml中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.38 g, 8.35 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に2時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 80 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してジスルフィド1.44 g (黄色のゼリー状液体, 収率: 62%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.2 (s, 3H), 5.35 (s, 1H), 6.69 (d, J = 8.28 Hz, 1H), 7.18 (dd, J = 8.23 Hz, 2.23 Hz, 1H), 7.24 (d, J = 2.24 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.7, 115.5, 125.0, 128.3, 130.2, 134.0, 154.4; MS (ESI) m/z 278.33 (M+H) +.
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の上記粗生成物 (0.7 g, 2.59 ミリモル) の撹拌溶液にメチル 2-ブロモ-プロピオネート (0.7 ml, 6.0 ミリモル) 及び炭酸カリウム (K2CO3, 2 g, 14.5 ミリモル) を添加した。得られる混合物を室温で一夜撹拌した。その反応が完結した後、その反応混合物を酢酸エチル (100 ml) で希釈し、濾過した。合わせた溶液を減圧で蒸発させて残渣を得、これをクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製して黄色のゼリー状液体1.0 g (収率: 86%)を得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.63 (d, J = 6.84 Hz, 1H); 2.34 (s, 3H), 3.75 (s, 3H), 4.73 (q, J = 6.84 Hz, 1H), 6.57-6.61 (m, 1H), 7.19-7.27 (m, 2H).
【0108】
250 ml の1口フラスコ中で、エタノール (15 ml) 中の上記生成物 (1.0 g, 2.22 ミリモル) の撹拌溶液に水15 ml 及び濃塩酸5 mlを添加した。亜鉛粉末 (10 g, 153 ミリモル) を徐々に添加した。その添加後、その反応混合物を室温で30分間撹拌し、次いでジクロロメタン (3X50 ml)で抽出した。有機相を合わせ、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して淡黄色の液体を得た。
150 mlの1口フラスコ中で、アセトニトリル30ml中の上記粗生成物の撹拌溶液にアセトニトリル (30 ml) 中の3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H -1,2,4-トリアゾール-5(4H)-オン (III-1) (1.8 g, 4.36 ミリモル) の溶液及び炭酸カリウム (K2CO3, 2.5 g, 18.1 ミリモル) を添加した。その反応混合物を室温で6時間撹拌した。その反応が完結した後、その反応混合物を酢酸エチル100 mlの添加により希釈し、濾過し、減圧で濃縮し、カラムクロマトグラフィー(シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけてE-13(一対のジアステレオマー) (淡黄色のゼリー状液体, 収率: 50%)1.2gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.61 (d, J = 6.84 Hz, 1H); 2.18 (s, 3H), 3.15 (s, 3H), 3.70 (d, J = 12.61 Hz, 3H); 4.70 (q, J = 6.82 Hz, 1H); 5.18 (s, 1H); 6.51 (d, J = 8.46 Hz, 1H), 7.0-7.11 (m, 1H), 7.10-7.15 (m, 1H), 7.34-7.37 (m, 5H), 7.67 (d, J = 8.74 Hz, 2H), 8.11 (d, J = 8.74 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.1, 18.5, 28.1, 50.6, 52.2, 72.7, 112.1, 118.2, 123.0, 125.5, 126.2, 126.8, 127.1, 128.2, 128.5, 128.9, 133.8, 135.2, 137.7, 140.6, 146.5, 152.6, 156.9, 172.2.
実施例23:化合物E-14の調製
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-14)の調製
【0109】
【化36】
【0110】
500 mlの3口フラスコにo-エチルフェノール (5 g, 40.9 ミリモル) 、チオシアン酸ナトリウム(10 g, 123.3 ミリモル) 、臭化ナトリウム (4.3 g, 41.79 ミリモル) 及びメタノール (100 ml) を連続して添加した。その混合物を氷浴中で0℃に冷却し、次いでメタノール (50 ml) 中の臭素 (2.6 ml, 50.6 ミリモル) の溶液をそれに滴下して添加した。0℃で1時間撹拌した後、その混合物を室温に温め、次いで室温で更に4時間撹拌した。重炭酸ナトリウムの飽和水溶液200 mlをその反応混合物に徐々に添加し、10分間撹拌した。その混合物を酢酸エチル (2X300 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過した。濾液を減圧で濃縮して赤褐色の粘稠な液体を得た。カラムクロマトグラフィーを行なって (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) 、3-エチル-4-ヒドロキシ-フェニルチオシアン酸 6.8g(赤褐色の粘稠な液体, 収率: 93%)を得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 1.25 (t J = 7.28 Hz, 3H), 2.64 (q, J = 7.53 Hz, 2H), 5.46 (s, 1H), 6.80 (d, J = 8.36 Hz, 1H), 7.28 (dd, J = 8.36 Hz, 2.52 HZ, 1H), 7.34 (d, J = 2.48.36 Hz, 1H).
150 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.2 ミリモル) 及びテトラヒドロフラン(40 ml) の撹拌混合物に0℃のテトラヒドロフラン20ml中の3-エチル-4-ヒドロキシ-フェニルチオシアン酸 (1.3 g, 7.25 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6N塩酸の添加によりpH 3-4に調節し、次いで水相を酢酸エチル (3X60 ml)で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物3-エチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
別の150 mlの1口フラスコ中で、アセトニトリル (20 ml) 中の得られる粗生成物3-エチル-4-ヒドロキシルメルカプトベンゼンの撹拌溶液に炭酸カリウム (K2CO3, 1.0 g, 7.23 ミリモル) を添加し、続いてアセトニトリル20ml中の 3-(1'-ブロモ-ベンジル)-4-メチル-1- (4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1) (1.6 g, 3.88 ミリモル)の溶液を滴下して添加した。その混合物を室温で6時間撹拌した後、炭酸カリウム (K2CO3, 1.0 g, 7.23ミリモル) 及びエチルブロモアセテート (1.6 ml,13.8 ミリモル) をそれに添加した。その混合物を同温度で更に8時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル (50 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してE-14 1.3g(黄色のゼラチン状物, 3工程の収率: 58.6%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 1.09 (t J = 7.48 Hz, 3H), 1.26 (t J = 7.28 Hz, 3H), 2.59 (q, J = 7.52 Hz, 2H), 3.15 (s, 3H), 4.22 (q, J = 7.20 Hz, 2H), 4.58 (s, 2H), 5.18 (s, 1H), 6.56 (d, J = 8.20 Hz, 1H), 7.15 (dd, J = 8.32 Hz, 2.28 HZ, 1H), 7.31-7.36 (m, 5H), 7.66 (d, J = 8.72 Hz, 1H), 8.11 (d, J = 8.52 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.7, 14.1, 23.0, 28.1, 50.6, 61.3, 65.4, 111.5, 118.2, 123.3, 126.1, 126.8, 127.1, 128.2, 128.5, 128.9, 133.8, 134.3, 135.2, 136.2, 140.5, 146.5, 152.6, 156.7, 168.5; MS (ESI) m/z 572.14 (M+H+); 589.1 (M+ NH4+).
実施例24:化合物E-15の調製
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-15)の調製
【0111】
【化37】
【0112】
150 mlの1口フラスコ中で、アセトニトリル (30 ml) 中の3-メチル-4-ヒドロキシ-メルカプトベンゼン (0.52 g, 1.87 ミリモル) の撹拌溶液にブロミドIII-1 (1.53 g, 3.71 ミリモル) 及び炭酸カリウム(K2CO3, 2.5 g, 18.1 ミリモル) を添加した。その混合物を室温で12時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル(100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて淡黄色のゼリー状液体1.4 g を得た。
150 mlの3口フラスコ中で、エタノール (15 ml) 中の上記生成物 (1.4 g, 1.48 ミリモル) の撹拌溶液に水15ml及び濃塩酸5 mlを添加した。亜鉛粉末 (10 g, 153 ミリモル) を徐々に添加した。その添加後、その反応液を室温で30分間撹拌し、次いでジクロロメタン (3 X50 ml) で抽出した。有機相を合わせ、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して淡黄色の液体を得た。
【0113】
150 ml の1口フラスコ中で、アセトニトリル (30 ml) 中の上記粗生成物の撹拌溶液にアセトニトリル (30 ml) 中のエチル 2-ブロモ-イソプロピオネート (3 ml, 20.2 ミリモル) 及び炭酸カリウム(K2CO3, 2.0 g, 14.4 ミリモル) を添加した。その反応混合物を室温で12時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル(100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル=5:1 v/v) により精製してE-15 0.58g(無色のゼラチン状物, 3工程の収率: 26.6%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.20 Hz, 3H), 1.45 (s, 6H), 2.37 (s, 3H), 3.20 (s, 3H), 4.10 (q, J = 7.20 Hz, 2H), 6.41 (s, 1H), 6.94 (d, J = 8.52 Hz, 1H), 7.25 (dd, J = 8.52 Hz, 1.8 Hz, 1H), 7.32 (d, J = 1.8 Hz, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.52 Hz, 2H), 8.17 (d, J = 8.52 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.4, 25.8, 28.2, 50.9, 61.1, 74.5, 112.4, 118.2, 124.2, 125.7, 126.2, 126.3, 127.4, 129.0, 129.1, 134.7, 136.1, 139.9, 140.4, 145.9, 152.6, 156.2, 173.9; MS (ESI) m/z 584.71(M-H)-.
実施例25:化合物E-16の調製
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(2-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-16)の調製
【0114】
【化38】
【0115】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 2.0 g, 52.7ミリモル) 及びテトラヒドロフラン(40 ml) の撹拌混合物に0℃のテトラヒドロフラン (30 ml )中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (2.5 g, 15.13ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に2時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加によりpH 3-4に調節し、次いで水相を酢酸エチル (3X100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて黄色の液体を得た。
250 mlの1口フラスコ中で、アセトニトリル (30 ml)中の得られる黄色の液体の撹拌溶液にブロミドIII-1 (3.2 g, 7.76 ミリモル)及び炭酸カリウム (K2CO3, 1.15 g, 8.32 ミリモル) を添加した。その混合物を室温で6時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル (100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて黄色のゼリー状液体3.6 g を得た。
【0116】
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の上記生成物 (2.2 g, 4.67 ミリモル) の撹拌溶液にエチル 2-ブロモ-イソプロピオネート (2 ml, 13.5ミリモル) 及び炭酸カリウム (K2CO3) (1.5 g, 10.8 ミリモル)を添加した。その反応混合物を36時間にわたって加熱、還流した。反応が完結した後、その反応混合物を酢酸エチル (100 ml)で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してE-16 1.0g(黄色のゼラチン状物, 3工程の収率: 44%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.20 (t, J = 7.14 Hz, 3H), 1.55 (d, J = 5.76 Hz, 6H), 2.13 (s, 3H), 3.15 (s, 3H), 4.18 (q, J = 7.14 Hz, 2H), 5.19 (s, 1H), 6.49 (d, J = 8.46 Hz, 1H), 7.03 (dd, J = 8.46 Hz, 2.36 Hz, 1H), 7.16 (d, J = 2.36 Hz, 1H), 7.32-7.37 (m, 5H), 7.66 (d, J = 8.74 Hz, 2H), 8.10 (d, J = 8.74 Hz, 2H); MS (ESI) m/z 586.39(M+H+).
実施例26:化合物E-17の調製
【0117】
化合物III-2 及びII-4を出発物質として使用して、エチル 2-(3-メチル-4-(1-(3-(4-
n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-17) を実施例9に記載されたのと同様の化学反応方法に従って調製した。それは81.2%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.76 (t, J = 7.30 Hz, 3H ), 0.92-1.02 (m, 1H), 1.03-1.29 (m, 5H), 1.35-1.48(m, 1H), 2.37 (s, 3H), 3.47-3.56 (m, 3H), 3.63-3.67 (m, 1H), 4.11-4.16 (m, 2H), 6.38 (s, 1H), 6.97 (d, J = 8.59 Hz, 1H), 7.24 (dd, J = 8.57 Hz, 2.25 Hz, 1H), 7.34 (d, J = 2.04 Hz, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.73 Hz, 2H), 8.19 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 14.1, 16.5, 19.9, 30.1, 38.0, 42.4, 61.4, 74.7, 113.1, 118.2, 125.6, 126.2, 126.3, 128.0, 128.9, 129.1, 130.8, 134.9, 135.3, 140.5, 145.9, 152.6, 155.0, 169.8; MS (ESI) m/z 600.0 (M)+, 601.2 (M+1)+, 602.2 (M+2)+, 603.2 (M+3)+ .
実施例27:化合物E-18の調製
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(2-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-18)の調製
【0118】
【化39】
【0119】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.2 g, 31.6 ミリモル) 及びテトラヒドロフラン(50 ml) の撹拌混合物に0℃のテトラヒドロフラン (20 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.4 g, 8.47 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に2時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6N塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて黄色の液体を得た。
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の得られる黄色の液体の撹拌溶液にブロミドIII-2 (1.4 g, 3.08 ミリモル) 及び炭酸カリウム (K2CO3, 0.42 g, 3.04 ミリモル) を添加した。その混合物を室温で6時間撹拌した後、アセトニトリル (10 ml) 中のエチル 2-ブロモ-イソプロピオネート (6 ml, 40.5 ミリモル) 及び炭酸カリウム (K2CO3, 2.4 g, 17.4 ミリモル) をそれに添加した。その混合物を36時間にわたって加熱、還流した。その反応が完結した後、その反応混合物を酢酸エチル (100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =10:1 v/v) にかけてE-18 (黄色のゼラチン状物, 3工程の収率: 28.6 %) 1.52 gを得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.80 (t, J = 7.30 Hz, 3H ), 1.16-1.36(m, 7H), 1.56 (d, J = 2.84 Hz, 6H), 2.12 (s, 3H), 3.48-3.57 (m, 2H), 4.17-4.23 (m, 2H), 5.10 (s, 1H), 6.48 (d, J = 8.44 Hz, 1H), 7.01 (dd, J = 8.46 Hz, 2.28 Hz, 1H), 7.17 (d, J = 2.08 Hz, 1H), 7.30-7.35 (m, 5H), 7.67 (d, J = 8.81 Hz, 2H), 8.15 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 14.0, 16.5, 19.8, 25.3, 29.7, 30.4, 41.9, 50.3, 61.5, 79.2, 116.4, 118.1, 124.5, 126.1, 128.4, 128.5, 128.8, 130.3, 133.3, 135.9, 137.8, 140.6, 146.4, 152.3, 154.9, 174.1; MS (ESI) m/z 626.1 (M-2)-, 627.1 (M-1)-, 628.1 (M)-.
実施例28:化合物E-19の調製
エチル 2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-19)の調製
【0120】
【化40】
【0121】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン(40 ml) の撹拌混合物に0℃のテトラヒドロフラン (20 ml) 中の2,5-ジメチル-4-ヒドロキシ-フェニルチオシアン酸 (1.02 g, 5.69 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 60 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体) を得た。
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の得られる粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼンの撹拌溶液に3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オンIII-2 (1.1 g, 2.42 ミリモル) 及び炭酸カリウム (K2CO3, 0.35 g, 2.35 ミリモル) を添加した。その混合物を室温で4時間撹拌した後、エチルブロモアセテート (1.8 ml, 15.5 ミリモル) 及び炭酸カリウム (K2CO3, 1.5 g, 10.8 ミリモル) をそれに添加し、次いでその反応混合物を室温で一夜撹拌した。その反応が完結した後、その反応混合物を酢酸エチル (200 ml) で希釈し、濾過した。濾液を真空で蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) にかけてE-19 (黄色のゼラチン状物, 3工程の収率: 22.9%) 0.8 g を得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.80 (t, J = 7.26 Hz, 3H ), 1.18-1.33 (m, 7H), 2.13 (s, 3H), 2.22 (s, 3H), 3.50-3.52 (m, 2H), 4.23-4.29 (m, 2H), 4.59 (s, 2H), 5.01 (s, 1H), 6.50 (s, 1H), 7.13 (s, 1H), 7.30-7.32 (m, 5H), 7.68 (d, J = 8.64 Hz, 2H), 8.16 (d, J = 8.52 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 14.2, 15.5, 19.8, 20.8, 29.7, 30.4, 41.8, 49.7, 61.4, 65.5, 112.7, 118.1, 122.7, 125.8, 126.2, 128.3, 128.4, 128.8, 135.9, 139.0, 140.6, 141.6, 146.5, 152.3, 156.9, 168.7.
化合物 (I) (酸)の調製:
実施例29:化合物 A-1の調製
【0122】
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸(A-1)の調製
ジクロロメタン (50 ml) 及びエタノール (50 ml) 中のエチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ-アセテート (28 g, 51.8 ミリモル) の撹拌溶液に水酸化ナトリウム (10 g, 250 ミリモル) の水溶液50mlを添加した。その反応混合物を室温で12時間撹拌した。その反応が完結した後、その混合物のpHの値を6N塩酸の添加により2-3 に調節した。得られる混合物をジクロロメタン (3 X 100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、蒸発させて残渣を得た。残渣をカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =4:1→1:1 v/v) により精製してA-1 18.2 g を白色の固体 (収率: 68.5%)として得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.29 (s, 3H), 3.25 (s, 3H), 4.62 (s, 2H), 6.32 (s, 1H), 6.69 (d, J = 8.8 Hz, 1H), 6.86 (d, J = 8.75 Hz, 1H), 6.98 (s, 1H), 7.43-7.50 (m, 3H), 7.55-7.58 (m, 2H), 7.72 (d, J = 8.48 Hz, 2H), 8.19 (d, J = 8.48 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.4, 18.1, 28.3, 58.4, 65.9, 75.3, 112.8, 113.2, 118.3, 119.3, 122.7, 125.8, 126.2, 127.1, 128.9, 129.0, 129.4, 135.1, 140.4, 146.3, 151.5, 152.8, 173.2; MS (ESI) m/z 514.2 (M+H+); 531 (M+NH4+).
実施例30:化合物 A-2の調製
【0123】
化合物E-2 を出発物質として使用して、2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 A-2) を実施例29に記載されたのと同様の合成方法に従って調製した。それは46%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.42 (s, 3H), 3.18 (s, 3H), 3.51 (s, 2H), 6.76 (s, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 2.76 Hz, 1H), 7.25-7.52 (m, 5H), 7.67 (d, J = 8.67 Hz, 2H), 8.14 (d, J = 8.58 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 20.9, 28.3, 36.9, 74.6, 113.7, 118.0, 118.3, 125.8, 126.2, 126.3, 126.6, 127.1, 128.98, 129.01, 134.1, 134.6, 140.4, 142.2, 145.9, 152.6, 156.8, 174.6; MS (ESI) m/z 528 (M-H)-; 529 (M-); 530 1 (M+H)-.
実施例31:化合物 A-3の調製
【0124】
化合物E-3 を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸(A-3) を実施例29に記載されたのと同様の合成方法に従って調製した。それは24.2%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H), 3.24 (s, 3H), 3.61 (s, 2H), 6.43 (s, 1H), 6.99 (d, J = 8.58 Hz, 1H), 7.22-7.29 (m, 1H), 7.38 (d, J = 2.28 Hz, 1H), 7.38-7.56 (m, 5H), 7.73 (d, J = 8.68 Hz, 2H), 8.21 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.5, 28.2, 37.8, 74.5, 113.2, 118.2, 125.7, 126.2, 126.6, 127.1, 128.2, 128.99, 129.1, 130.8, 134.7, 140.4, 145.9, 152.7, 155.0, 174.2; MS (ESI) m/z 556.9 (M+CO)+.
実施例32:化合物 A-4の調製
【0125】
化合物E-4 を出発物質として使用して、2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-4) を実施例29に記載されたのと同様の合成方法に従って調製した。それは52.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.18 (s, 3H), 3.13, (s, 3H), 4.63 (s, 2H), 5.19 (s, 1H), 6.57 (d, J = 8.43 Hz, 1H), 7.13 (dd, J = 6.07 Hz, 2.26 Hz, 1H), 7.18 (d, J = 1.8 Hz, 1H), 7.32-7.36 (m, 5H), 7.66 (d, J = 8.73 Hz, 2H), 8.10 (d, J = 8.69 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.0, 28.1, 50.5, 64.9, 111.5, 118.3, 123.6, 126.1, 126.2, 128.2, 128.4, 128.6, 128.9, 133.9, 135.0, 137.8, 140.4, 146.5, 152.6, 156.7, 172.6; MS (ESI) m/z 530 (M+H)+, 531 (M+2), 532 (M+3).
実施例33:化合物 A-5の調製
【0126】
化合物E-5 を出発物質として使用して、2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸(A-5) を実施例29に記載されたのと同様の合成方法に従って調製した。それは62.8%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.55 Hz, 3H), 2.66 (q, J = 7.51 Hz, 2H), 3.19 (s, 3H), 4.62 (s, 2H), 6.29 (s, 1H), 6.66 (d, J = 8.88 Hz, 1H), 6.83 (dd, J = 8.87 Hz, 3.09 Hz, 1H), 6.96 (d, J = 3.09 Hz, 1H), 7.38-7.53 (m, 5H), 7.67 (d, J = 8.85 Hz, 2H), 8.15 (d, J = 8.78 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9, 23.2, 28.3, 65.7, 75.2, 112.6, 113.0, 117.7, 118.2, 125.8, 126.2, 127.0, 127.4, 128.8, 129.01, 135.1, 135.2, 140.4, 146.3, 150.9, 151.8, 152.8, 173.3; MS (ESI) m/z 525.9 (M-1)-, 527 (M)-, 528 (M+1)-.
実施例34:化合物 A-6の調製
【0127】
化合物E-6 を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸(A-6) を実施例29に記載されたのと同様の合成方法に従って調製した。それは42.4%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.40 (s, 3H), 3.25 (s, 3H), 4.65 (s, 3H), 6.34 (s, 1H), 6.69 (d d, J = 8.86 Hz, 3.0 Hz, 1H), 6.88 (d, J = 3.0 Hz, 1H), 6.95 (d, J = 8.88 Hz, 2H), 7.43-7.58 (m, 5H), 7.73 (d, J = 8.63 Hz, 2H), 8.21 (d, J = 8.58 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.8, 28.3, 65.4, 75.1, 112.2, 113.8, 118.3, 118.5, 125.7, 126.2, 126.3, 127.1, 128.9, 129.1, 135.1, 140.4, 146.3, 150.2, 152.4, 152.8, 173.1; MS (ESI) m/z 513 (M)-, 512 (M-1)-, 514 (M+1)-.
【0128】
実施例35:化合物 A-7の調製
【0129】
化合物E-7 を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-7) を実施例29に記載されたのと同様の合成方法に従って調製した。それは42%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.33 (s, 3H), 3.11 (s, 3H), 4.65 (s, 2H), 5.11 (s, 1H), 6.62 (dd, J = 8.53 Hz, 2.88 Hz, 1H), 6.78 (d, J = 2.88 Hz, 1H), 7.27-7.34 (m, 5H), 7.66 (d, J = 8.79 Hz, 2H), 8.09 (d, J = 8.71 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.0, 28.1, 49.7, 64.6, 112.6, 116.9, 118.3, 123.1, 126.2, 128.1, 128.6, 129.0, 135.0, 137.9, 140.4, 144.6, 146.5, 152.6, 158.3, 172.1; MS (ESI) m/z 530 (M)+, 531 (M+1)+.
実施例36:化合物 A-8の調製
【0130】
化合物E-8 を出発物質として使用して、2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-酢酸(A-8) を実施例29に記載されたのと同様の合成方法に従って調製した。それは76%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 2.11 (s, 3H), 3.3 (s, 3H), 4.13 (s, 2H), 4.68 (s, 2H), 6.80 (d, J = 8.13 Hz, 1H), 7.25-7.27 (m, 2H), 7.80 (d, J = 8.85 Hz, 2H), 7.99 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, DMSO) δ 16.2, 28.0, 30.4, 62.5, 65.3, 112.5, 118.1, 120.6, 123.3, 123.6, 123.9, 124.9, 125.2, 125.6, 125.9, 126.8, 127.5, 128.7, 132.1, 135.6, 141.1, 146.3, 152.3, 156.6, 170.5; MS (ESI) m/z 452.8 (M).
実施例37:化合物 A-9の調製
【0131】
化合物E-9 を出発物質として使用して、2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-酢酸(A-9) を実施例29に記載されたのと同様の合成方法に従って調製した。それは69.2%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.28 (s, 3H), 3.43 (s, 3H), 4.64 (s, 2H), 4.99 (s, 2H), 6.69 (d, J = 8.40 Hz, 1H), 6.78 (dd, J = 8.82 Hz, 3.06 Hz, 1H), 6.85 (dd, J = 2.96 Hz, 1H), 7.67 (d, J = 8.60 Hz, 2H), 8.13 (d, J = 8.55 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.4, 28.1, 61.9, 65.9, 112.1, 112.8, 118.2, 118.3, 118.4, 126.2, 126.3, 129.3, 143.9, 151.2, 152.0, 152.5, 170.4; MS (ESI) m/z 436 (M-1), 437 (M), 438 (M+1).
実施例38:化合物A-10の調製
【0132】
化合物E-10を出発物質として使用して、2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-10) を実施例29に記載されたのと同様の合成方法に従って調製した。それは88.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.09 (s, 3H), 2.25 (s, 3H), 3.09 (s, 3H), 4.59 (s, 2H),5.09 (s, 1H), 6.52 (s. 1H), 7.13 (s, 1H), 7.31-7.37 (m, 5H), 7.65 (d, J = 8.70 Hz, 2H), 8.09 (d, J = 8.70 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 15.4, 20.7, 28.0, 49.7, 65.2, 113.2, 118.4, 122.7, 126.2, 126.7, 128.2, 128.5, 128.9, 135.1, 139.0, 140.4, 141.6, 146.7, 152.6, 156.8, 173.0; MS (ESI) m/z 542.42 (M-1)-, 543.54 (M)-, 544.43 (M+H)-.
実施例39:化合物A-11の調製
【0133】
化合物E-11を出発物質として使用して、2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェンチオ)-酢酸(A-11) を実施例29に記載されたのと同様の合成方法に従って調製した。それは70.8%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 2.37 (s, 3H), 3.18 (s, 3H), 3.51 (s, 2H), 6.39 (s. 1H), 6.88 (s, 1H), 7.31 (s, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.67 Hz, 2H), 8.16 (d, J = 8.67 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.0, 20.7, 28.2, 37.2, 74.4, 114.7, 118.2, 125.4, 125.6, 125.7, 126.3, 128.9, 129.1, 134.9, 135.7, 139.5, 140.4, 146.1, 152.7, 155.0, 174.7; MS (ESI) m/z 541.96 (M-1)-, 543.04 (M)-, 544.07 (M+H)-.
実施例40:化合物A-12の調製
【0134】
化合物E-12を出発物質として使用して、2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-酢酸(A-12) を実施例29に記載されたのと同様の合成方法に従って調製した。それは47.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.19 (s, 3H), 3.43 (s, 3H), 3.55 (s, 2H), 5.03 (s, 2H), 6.91 (d, J = 8.77 Hz, 3H), 7.29-7.32 (m, 2H), 7.67 (d, J = 8.66 Hz, 2H), 8.12 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.1, 28.1, 38.1, 61.4, 111.8, 118.3, 126.2, 126.3, 127.2, 127.5, 127.9, 130.8, 134.8, 140.3, 143.6, 152.5, 155.4, 174.8; MS (ESI) m/z 452.23 (M-1)-, 453.34 (M)-, 454.35 (M+H)-.
実施例41:化合物A-13の調製
【0135】
化合物E-13を出発物質として使用して、2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-13) を実施例29に記載されたのと同様の合成方法に従って調製した。それは76.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.55 (d, J = 6.56 Hz, 3H), 2.12 (s, 3H), 3.06 (s, 3H), 4.62 (q, J = 6.56 Hz, 1H), 5.17 (s, 1H), 6.56 (d, J = 9.04 Hz, 1H), 7.14-7.17 (m, 2H), 7.31-7.38 (m, 5H), 7.65 (d, J = 8.68 Hz, 2H), 8.09 (d, J = 8.68 Hz, 2H); MS (ESI) m/z 542.24 (M-1)-, 543.26 (M)-, 544.24 (M+H)-.
実施例42:化合物A-14の調製
【0136】
化合物E-14を出発物質として使用して、2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-14) を実施例29に記載されたのと同様の合成方法に従って調製した。それは63.1%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.07 (t, J = 7.56 Hz, 3H), 2.56 (q, J = 7.56 Hz, 2H), 3.09 (s, 3H), 4.59 (s, 2H), 5.18 (s, 1H), 6.56 (d, J = 9.04 Hz, 1H), 7.14-7.17 (m, 2H), 7.31-7.38 (m, 5H), 7.65 (d, J = 8.68 Hz, 2H), 8.09 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.7, 22.9, 28.0, 50.4, 65.0, 111.6, 118.3, 123.6, 125.4, 126.1, 126.2, 127.3, 127.5, 128.3, 128.5, 128.9, 133.9, 134.2, 135.1, 136.3, 140.4, 146.6, 152.5, 156.4, 172.7; MS (ESI) m/z 544.14 (M+H)+, 561.93 (M+NH4)+.
実施例43:化合物A-15の調製
【0137】
化合物E-15を出発物質として使用して、2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-15) を実施例29に記載されたのと同様の合成方法に従って調製した。それは70.6%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 6H), 2.35 (s, 3H), 3.18 (s, 3H), 6.40 (s, 1H), 6.96 (d, J = 8.56 Hz, 1H), 7.26-7.27 (m, 1H),7.35-7.50 (m, 6H), 7.67 (d, J = 8.72 Hz, 2H), 8.15 (d, J = 8.64 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.4, 25.4, 28.2, 50.7, 74.5, 112.6, 118.2, 123.7, 125.7, 126.2, 127.4, 128.1, 128.9, 129.1, 134.7, 136.1, 139.8, 140.4, 145.9, 152.6, 156.4, 179.1; MS (ESI) m/z 556.69 (M)+, 558.04 (M+1)+.
実施例44:化合物A-16の調製
【0138】
化合物E-16を出発物質として使用して、2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-16) を実施例29に記載されたのと同様の合成方法に従って調製した。それは52.6%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.57 (d, J = 5.25 Hz, 6H ), 2.12 (s, 3H), 3.10 (s, 3H), 5.21 (s, 2H), 6.63 (d, J = 8.47 Hz, 1H),7.05 (dd, J = 8.47 Hz, 2.06 Hz, 1H), 7.19 (d, J = 1.99 Hz, 1H), 7.25-7.37 (m, 5H), 7.65 (d, J = 8.76 Hz, 2H), 8.09 (d, J = 8.62 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.6, 25.1, 25.3, 28.1, 50.3, 79.4, 117.3, 118.3, 124.1, 126.2, 126.3, 128.2, 128.5, 128.9, 130.8, 133.2, 134.9, 137.8, 140.4, 146.6, 152.6, 154.5, 177.4; MS (ESI) m/z 558.06 (M)+
実施例45:化合物A-17の調製
【0139】
化合物E-17を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-17) を実施例29に記載されたのと同様の合成方法に従って調製した。それは62%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.75 (t, J = 7.29 Hz, 3H ), 1.03-1.05 (m, 1H), 1.11-1.18(m, 2H),1.19-1.26 (m, 1H), 1.40-1.50 (m, 1H), 2.36 (s, 3H), 3.47-3.53 (m, 1H), 3.56 (s, 2H), 3.62-3.69 (m, 1H), 6.38 (s, 1H), 6.97 (d, J = 8.58 Hz, 1H), 7.24 (dd, J = 8.47 Hz, 1.92 Hz, 1H), 7.33 (d, J = 1.99 Hz, 1H), 7.39-7.51 (m, 5H), 7.68 (d, J = 8.74 Hz, 2H), 8.17 (d, J = 8.61 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 16.6, 19.9, 37.8, 42.4, 74.7, 113.2, 118.2, 125.6, 126.2, 126.3, 128.2, 128.9, 129.1, 130.9, 134.8, 135.1, 140.4, 145.9, 152.6, 155.3, 174.4; MS (ESI) m/z 569.93 (M-2)-, 570.9 (M-1)- .
実施例46:化合物A-18の調製
【0140】
化合物E-18を出発物質として使用して、2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル- 5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-18) を実施例29に記載されたのと同様の合成方法に従って調製した。それは61.3%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.79 (t, J = 7.26 Hz, 3H ), 1.17-1.32(m, 6H), 1.59 (s, 6H), 2.13 (s, 3H), 3.48-3.53 (m, 2H), 5.13 (s, 1H), 6.63 (d, J = 8.44 Hz, 1H), 7.05 (dd, J = 8.47 Hz, 2.04 Hz, 1H), 7.17 (d, J = 2.08 Hz, 1H), 7.31-7.35 (m, 5H), 7.67 (d, J = 8.84 Hz, 2H), 8.14 (d, J = 8.60 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 16.6, 19.8, 25.2, 25.3, 29.7, 30.4, 41.8, 50.1, 79.4, 117.3, 118.3, 124.4, 126.1, 128.4, 128.5, 128.8, 130.7, 133.4, 135.7, 137.9, 140.5, 146.5, 152.4, 154.5, 177.9; MS (ESI) m/z 597.96 (M-2)-, 599.0 (M-1)-, 600.0 (M)-.
実施例47:化合物A-19の調製
【0141】
化合物E-19を出発物質として使用して、2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-19) を実施例29に記載されたのと同様の合成方法に従って調製した。それは31.4%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.79 (t, J = 7.27 Hz, 3H ), 1.13-1.23 (m, 2H), 1.28-1.36 (m, 2H), 2.12 (s, 3H), 2.25 (s, 3H), 3.47-3.55 (m, 2H), 4.66 (s, 2H), 5.03 (s, 1H), 6.53 (s, 1H), 7.15 (s, 1H), 7.29-7.36 (m, 5H), 7.68 (d, J = 8.66 Hz, 2H), 8.16 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 15.5, 19.8, 20.8, 30.4, 41.9, 49.6, 64.9, 112.9, 118.3, 123.1, 125.7, 126.2, 128.3, 128.5, 128.8, 135.8, 139.1, 140.5, 141.7, 146.5, 152.4, 156.6, 173.2; MS (ESI) m/z 584.03 (M-1)-, 584.98 (M)-。
【技術分野】
【0001】
本発明はペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)に対するアゴニスト作用を有する新規化合物、その調製方法、これらの化合物を含む薬物、及び心血管疾患等の治療及び予防におけるこれらの化合物の適用に関する。また、本発明は新規化合物の新規中間体及びその中間体の調製方法に関する。
【背景技術】
【0002】
現在の世界では、高速の発展及び生活基準の上昇とともに、人々が肥満、インスリン耐性(型II糖尿病)、脂質代謝障害及び高血圧を特徴とするメタボリック症候群のグローバルな発生をもたらす過度の脂肪及びタンパク質を摂取している。これらはヒトの健康に対する大きな脅威に相当する。個体の一般特性、年齢、性別、生理学的性質、栄養状態、食習慣等に加えて、メタボリック症候群は生体内の脂質代謝、エネルギー代謝及び炭水化物代謝のインバランスと関連している。こうして、メタボリック症候群を治療する有効な方法はエネルギー、脂肪及び炭水化物の生体内のバランスを維持又は回復することを目的とする治療養生法である。核受容体(NR)が細胞内だけでなく、全体の個体の体内のエネルギー、脂質及び炭水化物の生体内のバランスの維持に重要な役割を果たすので、それらは研究の焦点となっている。種々の生理学的リガンド(例えば、飽和脂肪酸、不飽和脂肪酸、これらの代謝産物、及び種々の合成化合物)により活性化される場合にのみ、核受容体が応答遺伝子の転写系を調節することができ、こうしてその生理活性を発揮することができる (Kasuga, J.ら, Bioorg. Med. Chem. 2007, 15, 5177-5190)。
核受容体ファミリーの中で、ペルオキシソーム増殖物質活性化受容体(PPAR)が10年以上にわたって人々の関心を引き付けてきており、これらはそれらのリガンドにより活性化された核転写因子であり、しかもこれらはメタボリック症候群に重要な調節因子として作用する (Guan, Y. J. Am. Soc. Nephrol, 2004, 15, 2801-2815)。それ故、PPARはインスリン耐性、損なわれたグルコーストレランス、型II糖尿病、肥満、高脂血症、高血圧、血管心臓障害、アテローム硬化症等の如き疾患の発生、発達及びコントロールに重要な役割を果たす。
【0003】
PPARは3種のサブタイプPPARα、PPARδ及びPPARγに分類され、これらは遺伝子の特定のDNA配列に結合することにより遺伝子の発現を調節する (Berger, J.ら, The Journal of Biological Chemistry, 1999, 274 (10), 6718-6725)。PPARαは主として肝臓、心臓、腸道、腎臓及びマクロファージ中で発現され、活性化された後に、脂肪酸の代謝を増大し、マクロファージ中の炎症応答を軽減し、低密度リポタンパク質コレステロールを低下し得る。PPARγは脂肪細胞、胎盤腫及びその他の組織中で発現され、活性化された後に、血液グルコースレベルを低下し、インスリン感受性を増大するだけでなく、脂質代謝、サイトカイン拮抗作用、坑炎症、免疫調節及び血圧調節等に重要な役割を果たし得る (Kasuga, J.ら, Bioorg. Med. Chem. 2007, 15, 5177-5190)。その他の2種のサブタイプとは対照的に、PPARδの生理機能は今まで明らかではない。しかしながら、薬理学実験のための動物モデルに関する最近の研究において、PPARδは脂肪組織及び筋肉中で脂肪酸分解代謝及びエネルギー脱共役を増大でき、マクロファージ由来の炎症を抑制し得ることが示されていた。人体の体重獲得を調節し、体の耐久性を高め、インスリン感受性を増大し、アテローム硬化症を改善することにおける種々の機能のために、PPARδのリガンドが高脂血症、肥満、インスリン耐性、及びアテローム硬化症の治療に有効な薬物であるかもしれない。
現在、PPARδ受容体アゴニスト薬のいずれもが薬物として商業上入手し得ない。PPARδアゴニストについての現在の研究の中で、グラクソスミスクラインにより開発されたGW501516 についての臨床研究はGW501516 が高密度リポタンパク質(HDL) コレステロールのレベルを80%まで増大し、低密度リポタンパク質(LDL) コレステロールのレベルを29%まで低下し、トリグリセリド(TG)のレベルを56%まで低下し、インスリンのレベルを48%まで低下し得ることを示していた (Oliver, W.; Jr.; Shenk, J. L. ら, Natl. Acad. Sci. U.S.A. 2001, 98, 5306-5311)。こうして、GW501516 は肥満及び心血管疾患の治療に有効な薬物になり得ると考えられる (WO01/00603A1, Bioorg. Med. Chem. Lett. 2003, 13, 1517)。しかしながら、GW501516 プロジェクトはそのフェーズII臨床試験からの不利な結果のために今一時的に中止されている。
【発明の概要】
【発明が解決しようとする課題】
【0004】
こうして、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)についてのアゴニスト作用及びGW501516のそれよりも極めて良好な血液脂質の調節能力を有する新規化合物を提供することについての緊急の要望がある。
【課題を解決するための手段】
【0005】
本発明の一つの目的は式(I)の化合物及び/又はその医薬上許される塩及び/又はこれらの溶媒和物を提供することである。
【0006】
【化1】
【0007】
式中、
1) XはO、S、N、又は (CH2)n (式中、nは1から4までの整数である)であり、XはO、S、又はCH2であることが好ましく、
2) YはO、S又はN、好ましくはO又はSであり、
3) RはH又はC1-C9アルキル、好ましくはH、メチル、又はエチルであり、
4) R1 及びR2 は互いに独立にH又はC1-C4 アルキルであり、かつR1及びR2 の少なくとも一つがHであり、R1及びR2は互いに独立にメチル、エチル、又はHであることが好ましく、
5) R3 はH、C1-C9 アルキル、好ましくはH又はC1-C4 アルキル、例えば、メチル、エチル、イソプロピル、更に好ましくはメチルであり、
6) R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル、又は置換フェニルであり、その置換フェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6 から選ばれ、R5 及びR6 は互いに独立にH又はC1-C9 アルキルであり、またR4 が置換フェニルである場合、その置換基が4-メトキシ又は4-メチルであることが好ましく、
【0008】
7) G1 及びG4は個々にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G1 及びG4 は個々にメチル又はエチルであることが好ましく、
8) G2 及びG3は個々にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G2 及びG3は個々にメチル、エチル、又はHであることが好ましく、
9) G5、G6、G8、及びG9は個々にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G5、G6、G8、及びG9 は個々にH、F、Cl、Br、メチル、エチル、又はメトキシであることが好ましく、かつ
10) G7 はH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6は互いに独立にH又はC1-C9 アルキルであり、G7 はトリフルオロメチル、イソプロピル、エチル、メチル、又はClであることが好ましく、G7 はトリフルオロメチルであることが更に好ましい。
【0009】
本発明の化合物がエステル形態である場合、それは以下に化合物I(エステル)と称される。本発明の好ましい化合物I(エステル)は下記の化合物を含む:
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (以下、 “E-1”と称される);
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-2”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-3”と称される);
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルチオ)-フェノキシ)-アセテート (以下、“E-4”と称される);
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (以下、 “E-5”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (以下、 “E-6”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルチオ)-フェノキシ)-アセテート (以下、 “E-7”と称される);
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-アセテート (以下、“E-8”と称される);
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-アセテート (以下、“E-9” と称される);
【0010】
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-10”と称される);
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-11”と称される);
エチル 2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-アセテート (以下、“E-12” と称される);
メチル 2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-13” と称される);
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルチオ)-フェノキシ)-アセテート (以下、 “E-14” と称される);
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、“E-15” と称される);
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-16”と称される);
エチル 2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (以下、 “E-17” と称される);
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-18” と称される);
エチル 2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (以下、“E-19” と称される)。
【0011】
化合物I(エステル)はその酸性形態(以下、化合物I(酸)と称される)を得るためにアルカリ性条件下で加水分解し得る。アルカリ性条件はアルカリ(これはアルカリ金属水酸化物であり、例えば、水酸化ナトリウム、水酸化リチウム、水酸化カリウム等があげられるが、これらに限定されない)を使用して生じられる。その加水分解に使用される溶媒系はC1-C4 アルコール (例えば、メタノール、エタノール、プロパノール、ブタノール等)-水 (アルコール: 水 の比= 9 -1:1 (容積/容積)), テトラヒドロフラン (THF) -水 (THF: 水 の比= 9 - 1:1 (容積/容積))、又はアルコール-ジクロロメタン-水 (アルコール: ジクロロメタン: 水 の比= 9 - 1: 9 - 1: 1 (容積/容積))であり、その反応温度は0-80℃、好ましくは20-40℃であり、その反応時間は1-12時間、好ましくは2-4 時間である。
本発明の好ましい化合物I(酸)は下記の化合物を含む:
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸 (以下、“A-1” と称される);
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-2” と称される);
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-3” と称される);
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-4” と称される);
【0012】
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸 (以下、“A-5”と称される);
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸 (以下、“A-6” と称される);
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-7” と称される);
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-酢酸 (以下、“A-8” と称される);
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-酢酸 (以下、“A-9” と称される);
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-10” と称される);
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-11” と称される);
【0013】
2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-酢酸 (以下、“A-12” と称される);
2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-13” と称される);
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-14” と称される);
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-15” と称される);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(以下、 “A-16” と称される);
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (以下、“A-17” と称される);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(以下、“A-18” と称される);
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (以下、“A-19” と称される)。
【0014】
本発明の特に好ましい化合物I(酸)は下記の化合物を含む:
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-3);
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-4);
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))- ベンジルオキシ)-フェニルチオ)-酢酸 (A-11);
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-14);
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-15);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-16);
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸(A-17);
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-18);
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-19)。
本発明の式Iの化合物の医薬上許される塩はそのアルカリ金属塩又はアルカリ土類金属塩を表す。カリウム塩、ナトリウム塩及びカルシウム塩が好ましい。
本発明の式Iの化合物の溶媒和物はその水和物又は有機溶媒和物、好ましくは水和物及びアルコラートを表す。水和物は1-4個の水分子を含んでもよい。アルコラートはメタノール、エタノール、及びプロパノールで生成されたアルコラートを含んでもよい。
本発明の別の目的は新規化合物、即ち、化合物IIIを提供することである。
【0015】
【化2】
【0016】
式中、
ZはCl又はBrであり、
R3 はH又はC1-C9 アルキルであり、
R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル又は置換フェニルであり、そのフェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6から選ばれ、かつ
G5、G6、G7、G8、及びG9は互いに独立にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6であり、R5 及びR6 は互いに独立にH又はC1-C9 アルキルである。
本発明の好ましい化合物IIIは下記の化合物を含む:
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (以下、“III-1”と称される);
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (以下、“III-2” と称される);
3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オン (以下、“III-3” と称される)。
本発明の化合物IIIは本発明の式Iの化合物を調製するための中間体として使用し得る。
本発明の別の目的は式IIIの化合物の調製方法を提供することである。
本発明の式IIIの化合物は式VIの化合物を塩素化試薬又は臭素化試薬と反応させることにより合成し得る。
【0017】
【化3】
【0018】
式中、
ZはCl又はBrであり、R3、R4、G5-G9 は式IIIの化合物の定義に示されたのと同じである。
塩素化試薬又は臭素化試薬は
(1) N-ブロモスクシンイミド(NBS)/トリフェニルホスフィン(Ph3P);
(2) 塩化チオニル(SOCl2) 又は臭化チオニル (SOBr2);
(3) N-クロロスクシンイミド(NCS)/トリフェニルホスフィン(Ph3P);
(4) 四塩化炭素(CCl4) 又は四臭化炭素(CBr4)/トリフェニルホスフィン(Ph3P);
(5) 五塩化リン(PCl5) 又は五臭化リン(PBr5);
(6) オキシ塩化リン(POCl3) 又はオキシ臭化リン(POBr3); 及び
(7) 三塩化リン(PCl3) 又は三臭化リン (PBr3)
から選ばれる。
溶媒はジクロロメタン、クロロホルム、四塩化炭素、及びこれらのあらゆる混合物から選ばれ、反応温度は10-80℃であり、反応時間は2-8 時間である。
式IIIの化合物は下記のスキームに従って合成されることが好ましい。
【0019】
【化4】
【0020】
上記スキームに、下記の工程が含まれる。
1)テトラヒドロフラン (THF) を溶媒として、また水素化ナトリウムをアルカリとして使用して、ベンジルアルコール 1 がブロミド2 と反応してエーテル3を生成し、
2)エタノール (EtOH) を溶媒として使用して、エーテル3 をアミン 4でアミン化してアミド5を生成し、
3)トルエン (PhCH3) を溶媒として使用して、アミド5 がオキシ塩化リン (POCl3) の作用のもとにフェニルヒドラジン 6 と反応してヒドラゾン 7を生成し、
4)テトラヒドロフラン (THF) を溶媒として使用して、ヒドラゾン 7 がカルボニルジイミダゾール(CDI) と反応して化合物 8を生成し、
5)エタノール(EtOH)を溶媒として、またパラジウム/カーボン(Pd/C) を触媒として使用して、ベンジルを常圧下で水素化により化合物8 から開裂してアルコール 9を生成し、
6)ジクロロメタン (DCM) を溶媒として使用して、アルコール 9 を塩素化試薬又は臭素化試薬の作用のもとに化合物IIIに変換する。
例えば、R4、G5、G6、G8、G9 がHであり、R3 がメチルであり、かつG7がトリフルオロメチルである場合、その合成スキームは以下のとおりである。
【0021】
【化5】
【0022】
上記スキームに、下記の工程が含まれる。
1)テトラヒドロフラン(THF) を溶媒として、また水素化ナトリウム(NaH) をアルカリとして使用して、還流下で加熱すると、ベンジルアルコール 1がエチルブロモアセテートと反応してエチルベンジルオキシアセテート10を生成し、
2)エタノール(EtOH) を溶媒として使用して、エチルベンジルオキシアセテート10を室温でメチルアミン11でアミン化してベンジルオキシアセトメチルアミン12を生成し、
3)トルエン (PhCH3) を溶媒として使用して、ベンジルオキシアセトメチルアミン12が80℃でオキシ塩化リン(POCl3) の作用のもとにp-トリフルオロメチルフェニルヒドラジン13と反応してヒドラゾン14を生成し、
4)テトラヒドロフラン(THF) を溶媒として使用して、ヒドラゾン14がカルボニルジイミダゾール(CDI) と反応して化合物15を生成し、
5)エタノール(EtOH)を溶媒として、またパラジウム/カーボン(Pd/C)を触媒として使用して、ベンジルを室温で常圧で水素化により化合物15から開裂してアルコール16を生成し、
6)ジクロロメタン(DCM) を溶媒として、またN-ブロモスクシンイミド(NBS) を臭素化剤として使用して、アルコール16をトリフェニルホスフィン(Ph3P)の作用のもとに化合物III-3に変換する。
式IIIの化合物はまた式VIIの化合物を塩素化試薬又は臭素化試薬と反応させることにより合成し得る。溶媒はクロロホルム又は四塩化炭素であり、臭素化試薬はN-ブロモスクシンイミド(NBS) であり、塩素化試薬はN-クロロスクシンイミド (NCS) であり、触媒はジベンゾイルペルオキシドであり、反応温度は40-80℃であり、反応時間は2-8 時間である。
【0023】
【化6】
【0024】
式中、
ZはCl又はBrであり、
R3 、G5-G9 は式IIIの化合物の定義に示されたのと同じであり、
G10-G14 は互いに独立に、又は同時にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、
R5 及びR6 は互いに独立にH又はC1-C9 アルキルである。
式IIIの化合物は下記のスキームに従って調製されることが好ましい。
【0025】
【化7】
【0026】
上記スキームに、下記の工程が含まれる。
1)メタノール(MeOH)を溶媒として、また硫酸を触媒として使用して、酸17がエステル化を受けてエステル18を生成し、
2)メタノール(MeOH)を溶媒として使用して、エステル18をアミン19でアミン化してアミド20を生成し、
3)トルエン(PhCH3) を溶媒として使用して、アミド20がオキシ塩化リン(POCl3) の作用のもとにフェニルヒドラジン 6と反応してヒドラゾン22を生成し、
4)テトラヒドロフラン(THF) を溶媒として使用して、ヒドラゾン22がカルボニルジイミダゾール(CDI) と反応して化合物23を生成し、
5)クロロホルムを溶媒として、N-ブロモスクシンイミド(NBS) を臭素化試薬として(又はN-クロロスクシンイミド(NCS) を塩素化試薬として) 、またジベンゾイルペルオキシドを触媒として使用して、化合物23を臭素化(又は塩素化) して化合物IIIを生成する。
例えば、R4 がフェニルであり、G5、G6、G8、G9 がHであり、R3 がメチルであり、かつG7がトリフルオロメチルである場合、その合成スキームは以下のとおりである。
【0027】
【化8】
【0028】
上記スキームに、下記の工程が含まれる。
1)メタノール(MeOH) を溶媒として、また硫酸を触媒として使用して、還流下で加熱すると、酸24がエステル化を受けてエステル25を生成し、
2)メタノール(MeOH)を溶媒として使用して、エステル25を室温でアミン11でアミン化してアミド26を生成し、
3)トルエン(PhCH3) を溶媒として使用して、アミド26が80℃でオキシ塩化リン (POCl3) の作用のもとにp-トリフルオロメチルフェニルヒドラジン13と反応してヒドラゾン27を生成し、
4)テトラヒドロフラン(THF) を溶媒として使用して、ヒドラゾン27が室温でカルボニルジイミダゾール(CDI) と反応して化合物28を生成し、
5)クロロホルムを溶媒として、N-ブロモスクシンイミド(NBS) を臭素化試薬として、またジベンゾイルペルオキシドを触媒として使用して、還流下で加熱すると、化合物28が臭素化されて化合物III-1を生成する。
本発明の別の目的は式(I)の化合物の調製方法を提供することである。
本発明の式(I) の化合物は下記のスキーム1又はスキーム2に示される方法により調製される。
スキーム1:
【0029】
【化9】
【0030】
式中、
X、Y、R1-R4 、G1-G9 は式Iの化合物の定義に示されたのと同じであり、RはH又はC1-C9 アルキル、好ましくはメチル又はエチルである。
スキーム1において、中間体化合物II及びIIIがアルカリの作用のもとにカップリングにかけられて化合物I(エステル)を生成する。アルカリは有機又は無機アルカリであり、無機アルカリはアルカリ金属炭酸塩、可溶性アルカリ土類金属炭酸塩、炭酸アンモニウム等、又はこれらのあらゆる混合物を含んでもよく、無機アルカリの例として、炭酸ナトリウム、炭酸カリウム、炭酸ストロンチウム、炭酸アンモニウム等が挙げられ、有機アルカリはトリエチルアミン等であってもよい。溶媒はアセトニトリル、ジメチルホルムアミド(DMF) 、ジメチルスルホキシド(DMSO) 、テトラヒドロフラン(THF) 、ジオキサン等、又はこれらのあらゆる混合物から選ばれることが好ましい。反応温度は0-100℃、好ましくは40-80℃であり、反応時間は1-12時間、好ましくは4-8 時間であることが好ましい。
上記プロセススキームにおいて、式IIの化合物は商業上入手されてもよく、又は従来技術の文献から知られている典型的な方法(例えば、M. L. Sznaidman, Curt D. Haffner, Patric R. ら, Bioorg. Med. Chem. Lett. 2003, 13, 1517-1521; Zhi-liang wei ら, J. Org. Chem. 2003, 68, 9116-9118; Org. Syn. Coll. Vol 1, 102, 1941; Org. Syn. Coll. Vol 2, 290, 1943; Handbook of Fine Organic Chemical Raw Materials and Intermediate. XU Ke-xun (編集), Scientific &Technological Industry Press, 3-426-3-584に示された方法)により合成されてもよい。
式IIの化合物は下記の化合物を含むが、これらに限定されない:
エチル 2-(2-メチル-4-ヒドロキシ)-フェノキシ-アセテート (以下、“II-1”と称される);
エチル 2-(3-メチル-4-ヒドロキシ)-フェノキシ-アセテート (以下、“II-2” と称される);
エチル 2-(2-エチル-4-ヒドロキシ)-フェノキシ-アセテート (以下、“II-3” と称される);
エチル 2-(3-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (以下、“II-4” と称される);
エチル 2-(2-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (以下、“II-5” と称される);
エチル 2-(2,5-ジメチル-4-ヒドロキシ)-フェニルチオ-アセテート (以下、“II-6” と称される)。
スキーム2
【0031】
【化10】
【0032】
式中、
X、Y、Z、R1-R4 、G1-G9 は式Iの化合物の定義に示されたのと同じであり、
RはH又はC1-C9 アルキル、好ましくはメチル又はエチルである。
スキーム2において、アルカリとしての炭酸塩との連続反応を使用して、化合物IIIを最初に化合物IV次いで化合物Vと反応させるが、反応の経過中に中間体を分離する必要はない。アセトニトリルに加えて、テトラヒドロフラン(THF) 、ジオキサン、等、又はこれらのあらゆる混合物が溶媒として使用されてもよい。
炭酸カリウムに加えて、炭酸ナトリウム、炭酸ストロンチウム、炭酸アンモニウム等、又はこれらのあらゆる混合物が炭酸塩として使用されてもよい。中間体化合物IV及びVの両方が市販されている。
スキーム2において、化合物I(エステル)がアルカリ性条件下で加水分解されて化合物I(酸)を生じることができ(RがC1-C9 アルキルである場合)、前記アルカリ性条件は下記のアルカリ、例えば、アルカリ金属水酸化物(水酸化ナトリウム、水酸化リチウム、水酸化カリウム等、又はこれらのあらゆる混合物が挙げられるが、これらに限定されない)で生成し得る。加水分解に使用される溶媒系はC1-C4 アルコール (例えば、メタノール、エタノール、プロパノール、ブタノール等)-水 (アルコール: 水 = 9 -1:1 (容積/容積))、THF-水 (THF: 水 = 9 - 1:1 (容積/容積))、又はアルコール-ジクロロメタン-水 (アルコール: ジクロロメタン: 水 = 9 - 1: 9 - 1: 1 (容積/容積))であり、反応温度は0-80℃、好ましくは20-40℃であり、反応時間は1-12時間、好ましくは2-4 時間である。
【0033】
【化11】
【0034】
式中、
X、Y、Z、R1-R4 、G1-G9 は式Iの化合物の定義に示されたのと同じであり、
RはH又はC1-C9 アルキル、好ましくはメチル又はエチルである。
本発明の別の目的は式(I)の上記化合物を活性成分として含む医薬組成物を提供することである。
式(I)の上記化合物を含む本発明の医薬組成物は式(I)の化合物及び医薬製剤化に使用される通常のアジュバントを含む。
このような医薬製剤化に使用される通常のアジュバントは薬物管理の担当部門により認可され、医薬アジュバントの基準に合致するものを表す。それらはそれらの異なる機能に基づいて二つのグループに分類されていた:アジュバントの一つのグループは医薬製剤の加工及び製造に必要であり、これらとして、希釈剤、結合剤、滑剤、懸濁剤及び潤滑剤等が挙げられ、アジュバントの別のグループは生体内の薬物の消化及び吸収を促進するように機能し、これらとして、崩壊剤、補助溶媒等が挙げられる。それらは人体の生体内で活性ではなく、治療効果を与えず、また毒性を与えない。
上記アジュバントの中で、希釈剤は下記の物質のいずれか一種又はあらゆる2種以上のあらゆる混合物から選ばれてもよい:澱粉、変性澱粉、蔗糖、ラクトース一水和物、無水ラクトース、グルコース、マンニトール、及び種々の微結晶性セルロース。
上記アジュバントの中で、結合剤は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:ヒドロキシプロピルメチルセルロース、前ゼラチン化澱粉、ポリビドン(ポリビニルピロリドン)、カルボキシメチルセルロース及びその誘導体、メチルセルロース、エチルセルロース、澱粉、炭水化物等、好ましくはヒドロキシプロピルメチルセルロース、前ゼラチン化澱粉、及びポリビドン。
【0035】
上記アジュバントの中で、潤滑剤は下記の物質のいずれか一種又はあらゆる2種以上のあらゆる混合物から選ばれてもよい:ステアリン酸マグネシウム、タルク粉末、及び型I水添植物油。
上記アジュバントの中で、懸濁剤は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:ゼラチン、ペクチン、アラビアゴム、アルギン酸ナトリウム、メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロース、及びメチルセルロース。
上記アジュバントの中で、崩壊剤は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:澱粉、低置換ヒドロキシプロピルセルロース、ナトリウムカルボキシメチル澱粉、カルシウムカルボキシメチルセルロース、架橋ポリビドン、架橋セルロース及び架橋ナトリウムカルボキシメチルセルロース。
上記アジュバントの中で、補助溶媒は下記の物質のいずれか一種又はあらゆる2種以上の混合物から選ばれてもよい:スパンシリーズ、トゥイーンシリーズ、ポリエチレングリコールシリーズ、大豆レシチン等。
【0036】
上記医薬組成物は下記の経口製剤のあらゆる形態であってもよい: 1, 単純錠剤; 2, フィルム被覆錠剤; 3, 糖剤; 4, 腸被覆錠剤; 5, 分散性錠剤; 6, カプセル; 7, 顆粒; 8, 懸濁液; 及び9, 溶液。
上記製剤形態は通常の製剤化方法により調製し得る。
本発明の別の目的はペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患を治療又は予防するための薬物の製造における式(I)の化合物の使用を提供することである。前記疾患として、メタボリック症候群、肥満、脂質異常血症、病的糖血症、インスリン耐性、老人性痴呆又は腫瘍等が挙げられる。
本発明によれば、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患を治療又は予防するための薬物の製造における式(I)の化合物の使用が提供され、前記疾患がメタボリック症候群、肥満、脂質異常血症、病的糖血症、インスリン耐性、老人性痴呆又は腫瘍等の一種以上から選ばれる。
本発明によれば、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患の治療又は予防方法が提供され、その方法は患者への治療又は予防有効量の式(I)の化合物の投与の工程を含み、前記疾患がメタボリック症候群、肥満、脂質異常血症、病的糖血症、インスリン耐性、老人性痴呆又は腫瘍等の一種以上から選ばれる。
本件出願の明細書を読んだ後に、当業者は本発明のその他の利点及び使用を認めるかもしれない。
【図面の簡単な説明】
【0037】
【図1】動物の体重の経時変化曲線を示し、これは薬物が動物の体重及び食物摂取に関する作用を有することを示す。横軸は計量時間(2日毎に計量)を表し、縦軸は動物の体重を表す。陽性対照はオルリスタット(二つの用量グループ:30mg/kg及び10mg/kg)である。本発明において、薬物製剤(A-3及びA-11)は二つの用量グループ(10mg/kg及び3.3mg/kg)を分離する。
【図2】動物中の血液グルコースの変化曲線を示し、これは経口グルコーストレランス試験からの結果を示す。横軸はグルコースを経口投与された後の血液サンプリング時点、0分、30分、60分、120分を表し、縦軸は相当する血液グルコースレベル(ミリモル/L)を表す。
【図3】本発明のA-3 (HS060098) 及びA-1 (HS060001) の薬物-時間曲線を表す。横軸は血液サンプリングの時点(時間)を表し、縦軸は異なる時間における動物からの血漿中の薬物の濃度(μg/ml) を表す。
【発明を実施するための形態】
【0038】
本発明が以下の特別な実施例を参照して詳しく記載される。本明細書の開示に鑑みて、種々の変更及び改良が本発明の精神及び範囲から逸脱しないで当業者により本発明になし得ることが理解されるべきであり、これらの全てが特許請求の範囲により特定された請求範囲内に入るべきである。更に、本明細書に提示された実施例は説明目的のためにすぎず、本発明の限定と何ら見なされるべきではないことが理解されるべきである。
本件出願において、C1-C9 基(アルキル、アルコキシ、アルキルチオ等) はC1-C2基、C1-C3基、C1-C4基、C1-C5基、C1-C6基、C1-C7基、C1-C8基、及びC1-C9基 (アルキル、アルコキシ、アルキルチオ等)等を含む。
薬物スクリーニングのためのモデル
核受容体活性化剤のin vitroスクリーニング
そのスクリーニングモデルにおける実験操作は以下のとおりである。
1. 核受容体関連スクリーニングモデルの簡単な記載
生細胞中の核受容体アゴニストをスクリーニングするためのスクリーニングモデルは活性化された核受容体がその下流遺伝子の転写を活性化し得るという原理に基づいて設計される。リポーター遺伝子プラスミドが構築され、この場合、核受容体(NRE) のDNA 結合配列がルシフェラーゼ遺伝子の上流に挿入され、その結果、ルシフェラーゼ遺伝子の発現が核受容体の制御下に置かれる。次いで、リポーター遺伝子プラスミド及び核受容体が細胞に同時に移入され、核受容体アゴニストが細胞培地中に存在する場合に核受容体が活性化される。次いで活性化された受容体がルシフェラーゼ遺伝子の発現を誘発することができ、その間にルシフェラーゼの量がその発光基質により測定し得る。この方法で、核受容体に関する化合物による活性化の強さがルミネセンス強さを観察することにより知り得る。トランスフェクション効率、接種された細胞の量、化合物の毒性等の如き因子により生じられる試験誤差の較正のために、GFP プラスミドが内部基準として同時に共トランスフェクトされ、試験ウェルの全てについてのルミネセント値が実験結果を分析する場合にGFP 値で較正される。実験結果が溶媒対照について1の値でもって活性化の相対的倍率として表され、倍率が大きい程、活性化能が高くなる。
【0039】
2. 実験操作
そのスクリーニングモデルに関する実験の詳しいプロトコルが下記の論文に言及されているかもしれない: “PPARa選択的アクチベーターとしての非環式1,3-ジカルボニル化合物の新規クラスの設計、合成及び評価”, Bioorg Med Chem Lett. 2004; 14(13): 3507-11。特別な操作が以下に記載される。
実験試薬:試験すべき化合物 (20 mM, DMSO中, -80℃で貯蔵).
(1) 1日目: 細胞培養及び接種
肝臓癌細胞HepG2 (ATCC (American Type Culture Collection)から) を37℃、100%の相対湿度、及び5%のCO2のインキュベーター中のT-75培養びん (グレイナー、ドイツ) 中の10% 熱不活化ウシ胎児血清(FBS, インビトロゲン, グランド・アイランド, NY, USA) を補給されたDMEM培地中で培養した。培養びん中の細胞培養物が80-90% の集密度に達した場合に、それを0.25%トリプシン (EDTAを含む) で3分間消化し、2000 細胞/100μl/ウェルの接種密度で96ウェル細胞培養プレートに接種した。
(2) 2日目:細胞トランスフェクション
翌日に、96ウェル培養プレート中の細胞が50-80% の集密度まで増殖した場合、細胞トランスフェクションを行なった。細胞共トランスフェクション系はFuGene6 トランスフェクション剤 (Roche Molecular Biochemicals, インジアナポリス, IN, U.S.A.) 及び60 ng DNA (10ngのhRXR, 10ngのpCMV βGal, 10ngの核受容体発現プラスミドRXR/PPARδ, 30ngのGFP 蛍光リポーター遺伝子プラスミドの夫々)を含んでいた。
【0040】
(3) 薬物措置
細胞培地をトランスフェクション24時間後に直ちに捨て、試験すべき薬物を含む新しいDMEM培地(活性炭により処理された10%のFBSを含む)200 μl により交換した。試験すべき薬物の最終濃度は10, 5, 1, 0.1, 0.01, 0.001, 及び0 μMであり、陽性対照は夫々のウェル中でDMSO(最終濃度は0.1%であった)とともに0.05 μM の2-ブロモステアリン酸 (Sigma, USAから購入)である。
(4) キナーゼ活性アッセイ
薬物による措置の24時間後に、細胞を溶解溶液 (Cell Culture Lysis buffer, Promega) で溶解し、遠心分離し、上澄みを集めた。上澄みを蛍光検出キット (Promega) で処理し、蛍光メーター (Ascent Fluoroskan FL reader, Thermo Labsystems, フィンランド) によりカウントし、ルシフェラーゼの相対強さを測定した。内部基準(トランスフェクション効率について較正する内部基準)として実験に使用されたβ-ガラクトシダーゼ活性をアッセイするために、上澄み50 μl を新しいミクロプレートに移し、プロメガキットで処理し、ミクロプレートリーダー (Bio-tech Instruments Inc., Winooski, VT, USA) (Sauerberg, P.; Olsen, G. S.; Jeppesen, L.; Mogensen, J. P. ら, J. Med. Chem., 2007, 50, 1495-1503)から405nmの波長で読み取った。
【0041】
3. 分析:
サンプルのメジアン有効濃度(EC50) はサンプルが50%の薬理学的作用を有する濃度である。この値は化合物の薬理学的作用を評価するのに重要なパラメーターの一つである。本スクリーニング方法において、サンプルのEC50 を6種の異なる濃度のサンプルにより受容体の活性化に従って計算した。
4. スクリーニング結果
PPARδ 受容体を活性化し得る式Iの化合物を得た。
in vitro の式Iの化合物(酸)の活性データ
in vitro の式Iの化合物(酸)の活性を下記の方法により測定した:サンプル [式 (I) の化合物(酸)] を溶解し、異なる濃度に希釈し、PPARδ 受容体を活性化するためのサンプルの活性を異なる濃度で試験した。濃度vs作用の対応を得、次いでメジアン有効濃度 (EC50) についての相当する値を計算した。試験結果を表1に示した。
【0042】
表1 in vitroの式Iの化合物(酸)の活性に関するデータ
【0043】
注: EC50 (nM) 値が低い程、活性が高い。
表1中のデータから、本発明の化合物の全てがPPARδについてアゴニスト活性を有することがわかるかもしれない。
化合物の生体内の薬力学的活性についてのスクリーニング
1.血液脂質を調節する活性についてのスクリーニング
本発明の式Iの化合物(酸)の一部の生体内活性に関するアッセイ
4種の化合物をA-1 〜A-19 の中から選び、それらの生体内の活性に関するアッセイにかけた。薬物の介入作用を高脂肪食誘発高脂血症を有するモデルであるSDラット、ApoE マウス、メソクリセタス・オーラタス(Mesocricetus auratus )等で観察した。結果を表2に示した。
【0044】
表2 式Iの化合物(酸)の生体内の活性データ
【0045】
注: ↓ は減少を表し、また↑ は増加を表す。
選ばれ、生体内の活性アッセイにかけられた4種の化合物がコレステロール(TC)、トリグリセリド(TG)及び低密度リポタンパク質(LDL) を減少することができ、また高密度リポタンパク質(HDL) を増加し得ることが表2からわかり、これは本発明の化合物が血液脂質調節の優れた作用を有することを示した。同時に、4種の化合物はまたSDラットモデル及びApoEマウスモデルで同様の薬理学的作用を有し、GW501516のそれと較べて、血液脂質調節に関して一層良好な作用を示した。
特に、化合物A-3 はコレステロール(TC)レベルを40%まで減少でき、トリグリセリド(TG)を65%まで減少でき、また低密度リポタンパク質(LDL) を51%まで減少できるだけでなく、高密度リポタンパク質(HDL) のレベルを43%まで増加することができる。
その後、A-3 の薬物の効力を高脂血症のマカカ・レサス(Macaca rhesus )モデルで確かめた。投与の3ヶ月後に、血液サンプルをELISAによる血液学的インジケーターの測定、並びにインスリン、アポリポタンパク質A-1 (apoA-1) 及びアポリポタンパク質B-100 (apoB-100) の測定のために大腿静脈から採取した。結果は、高脂血症モデルの対照グループと比較して、A-3 の投与にかけられた動物グループ中の血清全コレステロール(TC)の含量が45%減少され、低密度(LDL) の含量が38%減少され、高密度リポタンパク質(HDLc)の含量が67%増加されることを示した。しばらくして、ELISAアッセイにより、インスリン濃度が有意に減少され、apoA-1の濃度が有意に増加され、apoB-100 の濃度が有意に減少され、またapoA-1 対apoB-100 の比が正常な動物中のそれに近づくことがわかり、これは血液学的測定におけるLDL 及びHDL からの結果と充分に合致した。上記結果は高脂血症を患っていた動物に薬物A-3 を投与した後に、血液学的アッセイ及びELISAアッセイからのインジケーターの全てが有意に反転することを示し、これは順にその薬物の治療作用がGW501516のそれよりも極めて良好であることを実証した。
【0046】
2. 体重損失の薬理学的活性についてのスクリーニング
実験操作
生後3週の雄のC57BL/6J マウスを実験動物として選び、そのうちの15匹をランダムに選んで標準飼料で給餌し、残りを高脂肪飼料で給餌させ、マウスの夫々を耳カッティングによりマークした。マウスの体重及び飼料摂取を毎週計量した。給餌を15週続けた後、高脂肪飼料グループ中のマウスは42gの平均体重を有し、標準飼料グループ中のマウスは28gの平均体重を有し、モデルグループ及び投薬グループの両方中のマウスを高脂肪飼料で更に給餌した。4週にわたる本発明の化合物の投与後に、動物における血液脂質及び血液グルコースのレベル、体重、並びに飼料摂取に関する薬物の作用を観察するとともに、グルコーストレランス試験を行なって動物におけるグルコーストレランスに関する薬物の作用を調べた。
(1) 動物における血液脂質及び血液グルコースのレベルに関する薬物の作用
表3に示されるように、A-3 及びA-11 はモデル動物からの血清中のTG及びGLU のレベルを有意に減少することができ、オルリスタットの作用よりも極めて良好な作用を示す
【0047】
【0048】
正常なグループと比較してΔΔΔP<0.001;
モデルグループと比較して*P<0.05,**P<0.01,***P<0.001
(2) 動物における体重及び食物摂取に関する薬物の作用
表4及び図1に示されるように、A-3 及びA-11は動物の体重を有意に減少でき、動物の摂取に関して重大な作用を有さず、薬物の優れた体重損失機能を示す。
【0049】
表4 動物における食物摂取に関する薬物の作用
【0050】
(3) 経口グルコーストレランス試験
結果を図2に示し、即ち、血液グルコースの初期のレベルは肥満の動物モデルで2時間以内に回復し得ず、グルコーストレランス曲線が異常であり、グルコーストレランスが減少したが、A-3 及びA-11は肥満の動物でグルコーストレランスを有意に改善し得る。
3. 薬物速度論研究
1.血漿濃度-時間曲線
実験操作:
SDラット36匹を1:1の雌対雄の比及び150 -170gの範囲の体重で選び、それらを二つの用量グループに指定し、それらにA-1 及びA-3 を夫々投与し、3種の用量サブグループを50、10、及び2mg/kgである投薬濃度、6匹の動物で夫々の用量グループ、1:1の雌対雄の比の夫々のサブグループで設計した。ラットの全てを12時間にわたって断食させ、随時水に接近させた。投薬容積は10ml/kgであり、0.4-0.5 ml の血液サンプルを胃の洗浄後の異なる時点で目覚めた動物の後眼窩静脈から採取し、5000 rpm で10分間遠心分離した。血漿サンプル200 μL を正確に測定し、次いでアセトニトリル200 μL を補給し、均等にかき混ぜ、5分間放置し、遠心分離した(14000 rpm, 5分間) 。上澄みを集め、次いで再度遠心分離した(14000 rpm, 5分間) 。得られる上澄みを集め、HPLC注入にかけた。血液サンプリングのための時点は夫々0, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 36, 48及び72時間であり、夫々の時点に関するピーク面積は同じ時点における6匹のラットから得られたピーク面積の平均であった。血漿濃度を計算し、薬物濃度-時間曲線をプロットした。
結果を図3に示した。
結果はA-3 のt1/2 が10-12時間であり、A-1 のt1/2 が26-28時間であり、A-3 の生物利用能が86%であり、またA-1 の生物利用能が15%であることを示す。
2.組織分布
SDラット18匹を水に自由に接近させて16時間にわたって断食させ、それらを1:1の雌対雄の比で6匹の動物/グループで3つのグループに指定し、それらにHS060098 (10 mg/kg)を投与し、次いで動物を投薬の1時間後、10時間後、そして24時間後に犠牲にし、夫々の動物の心臓、肝臓、脾臓、肺、腎臓を迅速に取り出した。器官の表面の汚れた血液を生理食塩水で直ちに洗い去った。器官からの組織を別々に切開し、充分に洗浄し、ふき取り、計量し、400 mg/mlの生理食塩水を補給し、氷浴中で1分間にわたって均一にし、3750 rpmで20分間にわたって遠心分離した。上澄み400 μl をピペットで正確に計り取り、続いて内部標準 (5ミリモル/Lのモル濃度の内部標準として使用されたHS060001) 40 μl を添加し、精製水3ml で希釈し、均一にかき混ぜ、カラム (SPE C18 カラムを最初に精製水3ml で洗浄し、続いて活性化のために10% メタノール3ml で洗浄した) に装填した。カラムを精製水3 mlで1 ml/分ですすぎ、次いで溶離剤 (メタノール :精製水 = 9: 1) 3 ml で溶離した。溶離液を集め、45℃で窒素流のもとに送風乾燥した。残渣をアセトニトリル400 μl に溶解し、次いでHPLCインサートを入れ、続いてサンプルを注入した。
結果を表5に示す。結果はA-3 及びA-1 が主として肝臓中に分布し、A-3 が比較的に均一に分布することを示す。
【0051】
表5 器官中のA−3及びA−1の主たる分布
【実施例】
【0052】
中間体化合物IIの調製:
実施例1:化合物II-1の調製
エチル 2-(2-メチル-4-ヒドロキシ)-フェノキシ-アセテート(II-1)の調製
【0053】
【化12】
【0054】
500 mlの1口フラスコに撹拌しながら3-メチル-4-ヒドロキシアセトフェノン (25 g, 166.5 ミリモル)、アセトニトリル250 ml 、及び炭酸カリウム (K2CO3, 26 g, 188.2 ミリモル) を連続して添加した。次いでアセトニトリル (50 ml)中のエチルブロモアセテート (20 ml, 172.2 ミリモル) をそれに滴下して添加した。その反応液を室温で8時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル(200 ml) で希釈し、濾過し、蒸発させて黄色の油を得た。
別の500 mlの1口フラスコ中で、ジクロロメタン (CH2Cl2, 300 ml) 中の上記の黄色の油の撹拌溶液に、3-クロロ過安息香酸 (46 g, 199.9 ミリモル) 及び触媒量の4-メチルベンゼンスルホン酸を添加した。得られる混合物を室温で一夜撹拌した。その反応が完結した後、その反応混合物を濾過し、得られるフィルターケーキをジクロロメタン100 mlで洗浄した。合わせた濾液を次亜硫酸ナトリウム (Na2S2SO4) 及び重炭酸ナトリウム (NaHCO3) の飽和水溶液で洗浄し、次いで蒸発させて淡黄色の油を得た。
上記反応から得られた粗生成物を別の500 mlの1口フラスコに移し、エタノール80ml及び触媒量の4-メチルベンゼンスルホン酸をそれに添加した。その反応溶液を6時間にわたって加熱、還流し、次いで真空で蒸発させて淡黄色の固体を得た。その淡黄色の固体を石油エーテル/酢酸エチル(2:1 v/v) により再結晶してエチル 2-(2-メチル-4-ヒドロキシ)-フェノキシ-アセテート (16 g, 3工程につき45.8%) を白色の固体として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.34 (t, J = 7.14 Hz, 3H), 2.27 (s, 3H), 4.30 (q, J = 7.14 Hz, 2H), 4.61 (s, 2H), 6.61-6.63 (m, 1H), 6.65 (s, 1H), 6.67-6.68 (m, 1H), 7.31 (s, 1H).
実施例2:化合物II-2の調製
3-メチル-4-ヒドロキシアセトフェノンを出発物質として使用して、エチル 2-(3-メチル-4-ヒドロキシ)-フェノキシ-アセテート (II-2) を実施例1に記載された方法と同様の方法に従って40.2%の収率で白色の固体として調製した。
【0055】
【化13】
【0056】
II-2
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 1.23 (t, J = 7.12 Hz, 3H), 2.29 (s, 3H), 4.18 (q, J = 7.14 Hz, 2H), 4.64 (s, 2H), 6.57-6.60 (m, 1H), 6.68 (s, 1H), 6.69-6.70 (m, 1H), 8.87 (s, 1H); 13C NMR (100 MHz, DMSO) δ 14.5, 16.6, 60.9, 65.9, 112.2, 112.8, 115.4, 117.6, 125.2, 150.2, 150.8, 169.6.
実施例3:化合物II-3の調製
3-エチル-4-ヒドロキシアセトフェノンを出発物質として使用して、エチル 2-(2-エチル-4-ヒドロキシ)-フェノキシ-アセテート (II-3) を実施例1に記載された方法と同様の方法に従って74.5%の収率で淡黄色の固体として調製した。
【0057】
【化14】
【0058】
II-3
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.23 (t J = 7.53 Hz, 3H), 1.34 (t J = 7.12 Hz, 3H), 2.69 (q, J = 7.52 Hz, 2H), 4.30 (q, J = 7.14 Hz, 2H), 4.61 (s, 2H), 5.35 (s, 1H), 6.6 (dd, J = 8.65 Hz, 2.84 HZ, 1H), 6.65 (d, J = 8.63 Hz, 1H), 6.72 (d, J =2.84 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 14.1, 23.1, 61.3, 66.7, 112.6, 113.1, 116.5, 134.9, 149.9, 150.4, 169.8.
実施例4:化合物II-4の調製
エチル 2-(3-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (II-4)の調製
【0059】
【化15】
【0060】
500 mlの3口フラスコに、o-クレゾール (15 g, 138.7 ミリモル) 、チオシアン酸ナトリウム(NaSCN, 34 g, 419.3 ミリモル) 、臭化ナトリウム (NaBr, 16 g, 155.5 ミリモル) 及びメタノール (200 ml) を撹拌しながら連続して添加した。その混合物を0℃に冷却し、次いでメタノール(30 ml) 中の臭素 (8.6 ml, 167 ミリモル) の溶液をそれに滴下して添加した。0℃で1時間撹拌した後、その混合物を室温に温め、次いで室温で更に4時間撹拌した。その反応が完結した後、重炭酸ナトリウム (NaHCO3) の飽和水溶液200 mlをその反応混合物に添加し、10分間撹拌した。その混合物を酢酸エチル (2 X 500 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して黄色の油を得た。シリカゲルカラムクロマトグラフィー精製 (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) を行なって3-メチル-4-ヒドロキシ-フェニル チオシアン酸を白色の固体 (16 g, 収率: 69.8%)として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 2.17 (s, 3H), 6.92(d, J = 8.4 Hz, 1H), 7.37 (dd, J = 8.4 Hz, 2.48 HZ, 1H), 7.44 (d, J = 2.48 HZ, 1H), 10.1 (s, 1H); 13C NMR (100 MHz, DMSO) δ 16.2, 111.1, 113.1, 116.8, 127.3, 132.2, 135.4, 158.3。
【0061】
500 mlの3口フラスコ中で、四水素化アルミニウムリチウム(LiAlH4, 3.5 g, 92.2 ミリモル) 及びテトラヒドロフラン (THF, 50 ml) の撹拌混合物に0℃でテトラヒドロフラン (THF, 30 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (6.13 g, 37.1 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6 M 塩酸の添加により3-4 に調節した。次いで水層を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物4-ヒドロキシ-3-メチルチオフェノールを黄色の油として得た。
250 mlの1口フラスコ中で、アセトニトリル (80 ml) 中の4-ヒドロキシ-3-メチルチオフェノールの上記粗生成物の溶液にエチル 2-ブロモアセテート (4.3 ml, 37 ミリモル)を撹拌しながら添加し、続いて炭酸カリウム (5.1 g, 36.9 ミリモル) を添加した。得られる混合物を室温で8時間撹拌した。その反応が完結した後、その反応混合物を酢酸エチル (80 ml)で希釈し、濾過し、蒸発させて残渣を得た。残渣をシリカゲルカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) により精製してエチル 2-(3-メチル-4-ヒドロキシ)-フェニルチオ-アセテート6.38 g を黄色の油 (収率: 76%)として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.24 (t J = 7.2 Hz, 3H), 2.18 (s, 3H), 3.49 (s, 2H), 4.15 (q, J = 7.14 Hz, 2H), 5.74 (s, 1H), 6.63 (d, J = 8.27 Hz, 1H), 7.17 (dd, J = 8.16 Hz, 2.28 HZ, 1H), 7.25 (d, J = 2.28 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 15.7, 38.8, 61.6, 115.6, 124.1, 125.1, 131.9, 135.7, 154.5, 170.6.
実施例5:化合物II-5の調製
m-クレゾールを出発物質として使用して、エチル 2-(2-メチル-4-ヒドロキシ)-フェニルチオ-アセテート (II-5) を実施例4に記載された方法と同様の方法に従って63%の収率で黄色の油として調製した。
【0062】
【化16】
【0063】
II-5
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.23 (t J = 7.1 Hz, 3H), 2.39 (s, 3H), 3.44 (s, 2H), 4.14 (q, J = 7.11 Hz, 2H), 6.38 (s, 1H), 6.54 (dd, J = 8.36 Hz, 2.72 HZ, 1H), 6.64 (d, J = 2.71 Hz, 1H), 7.31 (d, J =8.37 Hz, 1H).
実施例6:化合物II-6の調製
2,5-ジメチルフェノールを出発物質として使用して、エチル 2-(2,5-ジメチル-4-ヒドロキシ)-フェニルチオ-アセテート (II-6) を実施例4に記載された方法と同様の方法に従って72%の収率で白色の固体として調製した。
【0064】
【化17】
【0065】
II-6
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.23 (t J = 7.16 Hz, 3H), 2.16 (s, 3H), 2.38 (s, 3H), 3.43 (s, 2H), 4.13 (q, J = 7.13 Hz, 2H), 5.19 (s, 1H), 6.58 (s, 1H), 7.23 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 15.1, 20.2, 37.9, 61.4, 116.9, 122.1, 123.3, 136.9, 140.0, 154.3, 170.3.
中間体化合物IIIの調製:
実施例7:化合物 III-1の調製
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (化合物 III-1) の調製は下記の反応スキームに従ってなし得る。
【0066】
【化18】
【0067】
N-メチルフェニルアセトアミドの調製
【化19】
【0068】
フェニル酢酸 (10 g, 73.5 ミリモル) 及びメタノール (60 ml) を250 mlの3口フラスコに添加し、続いて10滴の濃硫酸を添加した。得られる溶液を2時間にわたって加熱、還流した。次いで、その溶液を室温に冷却し、続いてメチルアミンの25-30%の水溶液 (50 ml, 約403 ミリモル) を添加し、更に3時間にわたって加熱、還流した。反応が完結した後、それを室温に冷却した。溶媒の殆どを回転真空蒸発により除去し、水30mlをそれに添加し、次いでその混合物を酢酸エチル (3 X 80 ml)で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮してN-メチル フェニルアセトアミド (白色の固体, 収率: 91.8%) 10.1gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.69 (d, J = 4.84 Hz, 3H), 3.51 (s, 3H), 5.95 (wide, 1H), 7.21-7.24 (m, 3H), 7.28-7.32 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 26.4, 43.6, 127.2, 128.9, 129.4, 135.1, 171.8.
3-ベンジル-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オンの調製
【0069】
【化20】
【0070】
1000 ml の3口フラスコ中で、80-90℃のトルエン (250 ml) 中のN-メチルフェニルアセトアミド (25 g, 167.7 ミリモル) 及び(4-トリフルオロメチル) フェニルヒドラジン ( 30g, 170 ミリモル) の撹拌溶液に、トルエン(50 ml) 中のオキシ塩化リン (POCl3, 15 ml,180 ミリモル) の溶液を滴下して添加した。その添加が完結した後、その混合物を80-90℃で5時間撹拌し、次いでその反応が完結した時に室温に冷却し、濾過した。残留フィルターケーキを、それが黄褐色になるまで酢酸エチルで洗浄した。濾液を重炭酸ナトリウムの飽和水溶液で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、再度濾過した。フィルターケーキを酢酸エチルで洗浄した。フィルターケーキを合わせ、真空乾燥した。最後にN-メチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン43.5gを得た (黄褐色の固体, 収率: 84.4%)。
1000 mlの3口フラスコ中で、テトラヒドロフラン (THF, 500 ml) 中のN-メチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン (58.6 g, 190.9 ミリモル) 及びN,N-ジメチルアミノピリジン (DMAP, 0.5 g, 4.0ミリモル) の撹拌混合物に、カルボニルジイミダゾール (CDI, 46.43 g, 286.4 ミリモル) を徐々に添加した。得られる混合物を室温で12時間撹拌した。その反応が完結した後、その混合物を水300 mlを含む2000 ml のビーカーに注ぎ、撹拌下で6N塩酸でpH 2-4に調節し、濾過した。フィルターケーキを蒸留水で洗浄し、次いで真空乾燥して3-ベンジル-4-メチル-1-(4-トリフルオロメチル) -フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (淡黄色の固体, 収率: 89.2%)56.7gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.11 (s, 3H), 4.0 (s, 2H), 7.25-7.38 (m, 5H), 7.67 (d, J = 8.68 Hz, 2H), 8.18 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 27.6, 32.7, 118.0, 122.8, 125.5, 126.6, 127.7, 128.4, 129.1, 133.6, 140.7, 146.8, 152.6.
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1) の調製
【0071】
【化21】
【0072】
500 mlの3口フラスコに、3-ベンジル-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (15 g, 45 ミリモル) 及びクロロホルム (CHCl3, 250 ml) を撹拌しながら添加した。その撹拌溶液に、N-ブロモスクシンイミド (NBS, 12 g, 68 ミリモル) 及びベンゾイルペルオキシド (0.5 g, 2.25 ミリモル) を添加した。その混合物を12時間にわたって慎重に加熱、還流した。その反応が完結した後、その混合物を濾過した。濾液を減圧で濃縮して残渣を得、これをフラッシュシリカゲルカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル = 8:1 v/v) により精製して3-(1’-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン14.6gを赤褐色の油 (収率: 79.7%)として得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.28 (s, 3H), 6.07 (s, 1H), 7.25-7.58 (m, 5H), 7.66 (d, J = 8.68 Hz, 2H), 8.13 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 28.4, 42.1, 69.6, 118.3, 126.2, 126.8, 127.3, 129.1, 129.9 133.9, 140.3, 145.7, 152.4.
実施例8:化合物 III-2の調製
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (化合物 III-2) の調製を下記の反応スキームに従って行い得る。
【0073】
【化22】
【0074】
N-n-ブチルフェニルアセトアミドの調製
【化23】
【0075】
フェニル酢酸 (16 g, 117.6 ミリモル) 及びメタノール ( 60 ml) を250 mlの3口フラスコに添加し、続いて5滴の濃硫酸を添加した。得られる溶液を6時間にわたって加熱、還流した。次いで、その混合物を室温に冷却し、続いてn-ブチルアミン (12 ml, 121.4 ミリモル) を添加し、更に3時間にわたって加熱、還流した。その反応が完結した後、それを室温に冷却した。溶媒の殆どを回転真空蒸発により除去し、水30mlをそれに添加し、その混合物を酢酸エチル (3 X 80 ml)で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮してN-n-ブチルフェニルアセトアミド (白色の固体, 収率: 70.4%) 15.9gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 7.28 Hz, 3H), 1.21-1.29 (m, 2H), 1.36-1.44 (m, 2H), 3.2 (q, J = 7.04 Hz, 2H), 5.62 (ブロード, 1H), 7.24-7.29 (m, 3H), 7.32-7.36 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 13.7, 19.9, 31.5, 39.4, 43.8, 127.2, 128.9, 129.4, 135.1, 170.9.
3-ベンジル-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オンの調製
【0076】
【化24】
【0077】
500 mlの3口フラスコ中で、80-90℃のトルエン (150 ml) 中のN-n-ブチルフェニルアセトアミド (11 g, 57.5 ミリモル) の撹拌溶液に、トルエン (20 ml) 中のオキシ塩化リン (POCl3,17 ml, 75.1 ミリモル) の溶液を滴下して添加した。その添加が完結した後、その混合物を80-90℃で5時間撹拌し、次いでその反応が完結した後に室温に冷却し、濾過した。フィルターケーキを、それが黄褐色になるまで酢酸エチルで洗浄した。濾液を重炭酸ナトリウムの飽和水溶液で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、再度濾過した。フィルターケーキを酢酸エチルで再度洗浄した。フィルターケーキを合わせ、真空乾燥した。最後に、N-n-ブチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン15.7gを得た (黄褐色の固体)。
500 mlの3口フラスコ中で、テトラヒドロフラン (THF, 250 ml) 中のN-n-ブチル-2-フェニル-N-(4-トリフルオロメチル) フェニルアセトアミドヒドラゾン (15.7 g) 及びN,N-ジメチルアミノピリジン(DMAP, 0.5 g, 4.0 ミリモル) の撹拌混合物に、カルボニルジイミダゾール (CDI, 7.43 g, 45.8 ミリモル) を室温で徐々に添加した。その反応混合物を室温で12時間撹拌した。その反応が完結した後、その混合物を水100 mlを含む500 mlのビーカーに注ぎ、撹拌下で6N塩酸でpH 2-4に調節し、濾過した。フィルターケーキを蒸留水で洗浄し、次いで真空乾燥して3-ベンジル-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン 6.28g(淡黄色の固体, 2工程の収率: 29.2%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 7.3 Hz, 3H), 1.22-1.29 (m, 2H), 1.37-1.44 (m, 2H), 3.49 (q, J = 7.6 Hz, 2H), 4.0 (s, 2H), 7.26-7.38 (m, 5H), 7.67 (d, J = 8.70 Hz, 2H), 8.19 (d, J = 8.65 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 19.8, 30.6, 32.8, 41.7, 117.9, 126.1, 126.9, 127.2, 127.8, 128.6, 129.1, 134.0, 140.7, 146.6, 152.5; MS (ESI) m/z 376.29 (M+1) +.
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-2)の調製
【0078】
【化25】
【0079】
250 mlの3口フラスコに、3-ベンジル-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (3.75 g, 10 ミリモル) 及びクロロホルム (100 ml) を添加した。その撹拌溶液にN-ブロモスクシンイミド (NBS, 3.56 g, 20 ミリモル) 及びベンゾイルペルオキシド (0.4 g, 1.65 ミリモル) を添加した。その混合物を6時間にわたって慎重に加熱、還流した。その反応が完結した後、溶媒を回転真空蒸発により除去し、残渣をカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) にかけて3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-2) (赤褐色の粘稠な液体, 収率: 85.9%) 3.9gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.36 Hz, 3H), 1.27-1.35 (m, 2H), 1.47-1.53 (m, 1H), 1.63-1.66 (m, 1H), 3.66-3.72 (m, 2H), 6.00 (s, 1H), 7.41-7.46 (m, 3H), 7.61 (dd, J = 7.99 Hz, 1.83 Hz, 2H) 7.66 (d, J = 8.69 Hz, 2H), 8.15 (d, J = 8.57 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 19.9, 30.3, 41.5, 42.4, 118.2, 126.2, 126.8, 127.3, 128.6, 128.9, 129.5, 134.8, 140.4, 145.6, 152.2.
実施例9:化合物 III-3の調製
化合物 III-3の調製を下記の反応スキームに従って達成し得る。
【0080】
【化26】
【0081】
N-メチルベンズオキソアセトアミドの調製
【化27】
【0082】
60% 水素化ナトリウム (3.7 g, 92.5 ミリモル) 及びテトラヒドロフラン (20 ml) を250 mlの3口フラスコに添加し、続いて加熱し、テトラヒドロフラン中のベンジルアルコール (10 g, 96.1 ミリモル) の溶液20mlを滴下して添加した。その添加が完結した後、その反応混合物を1時間にわたって加熱、還流した。次いでエチルブロモアセテート (11ml, 94.8 ミリモル) を添加し、その反応混合物を更に4時間にわたって還流下で撹拌した。室温に冷却した後、メチルアミンの水溶液25mlをそれに添加し、その反応を更に6時間にわたって還流下で行なった。その反応が完結した後、それを減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =4:1 v/v) にかけてN-メチルベンズオキソアセトアミド (白色の固体, 2工程の収率: 32%)5.3gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.83 (d, J = 5.16 Hz, 3H), 3.98 (s, 2H), 4.55 (S, 2h), 6.61 (s, 1H), 7.30-7.39 (m, 5H).
3-ベンズオキソメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オンの調製
【0083】
【化28】
【0084】
250 mlの3口フラスコ中で、80-90℃のトルエン (40 ml) 中のN-メチル ベンジルオキシアセトアミド (6.4 g, 3.57 ミリモル) の撹拌溶液に、トルエン (20 ml) 中のオキシ塩化リン (3.6 ml,38.5 ミリモル) を滴下して添加した。得られる反応混合物を80-90℃で5時間撹拌した。その反応が完結した後、それを室温に冷却し、濾過した。フィルターケーキを、それが肉色になるまで酢酸エチルで洗浄した。濾液を重炭酸ナトリウムの飽和水溶液で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、再度濾過した。フィルターケーキを酢酸エチルで再度洗浄した。フィルターケーキを合わせ、真空乾燥した。最後に、肉色の固体4.5gを得た。
上記肉色の固体4.5gを別の500 mlの3口フラスコに移し、テトラヒドロフラン (THF) 200 mlをそれに添加し、撹拌し、カルボニルジイミダゾール (CDI) (5 g, 30.8 ミリモル) を徐々にそれに添加した。その反応混合物をその添加後に室温で12時間撹拌した。その反応が完結した後、それを減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =4:1 v/v) にかけて3-ベンズオキソ メチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (明るい紅褐色のゼラチン状物, 2工程の収率: 23%) 3gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.35 (s, 3H), 4.50 (s, 2H), 4.59 (s, 2H), 7.32-7.37 (m, 5H), 7.66 (d, J = 8.46 Hz, 2H), 8.14 (d, J = 8.46 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 26.9, 27.8, 62.8, 72.9, 118.1, 126.1, 126.2, 126.3, 128.3, 128.5, 136.6, 140.5, 144.7, 152.5.
3-ヒドロキシメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-
トリアゾール-5-オンの調製
【0085】
【化29】
【0086】
250 mlの3口フラスコ中で、エタノール (60 ml) 中の上記生成物3-ベンズオキソ メチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (3.0 g, 8.3 ミリモル) の撹拌溶液に10% パラジウム/カーボン1gを添加した。そのフラスコ中の10% パラジウム/カーボン1gをN2 で置換し、次いで水素ガスを導入した。その反応混合物を室温で24時間撹拌した。その反応が完結した後、それを濾過し、真空濃縮して3-ヒドロキシメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オン (白色の固体, 収率: 93%)2.1gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.34 (s, 3H), 4.52 (d, J = 5.84 Hz, 2H), 5.77 (t, J = 5.84 Hz, 1H), 7.85 (d, J = 8.59 Hz, 2H), 8.17 (d, J = 8.59 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 27.9, 55.2, 118.0, 125.2, 125.5, 126.0, 141.3, 149.0, 152.5.
3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (III-3)の調製
【0087】
【化30】
【0088】
250 mlの3口フラスコ中で、ジクロロメタン (100 ml) 中の上記生成物 (2.1 g, 7.77 ミリモル) の撹拌溶液にN-ブロモスクシンイミド (NBS, 1.8 g, 10.1 ミリモル) を添加し、続いてトリフェニルホスフィン (2.4 g, 9.15 ミリモル) を徐々に添加した。その反応液を室温で4時間撹拌した。その反応が完結した後、それをカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1,4-二水素-1,2,4-トリアゾール-5-オン (淡黄色の固体, 収率: 89%) 2.3gを得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 3.42 (s, 3H), 4.34 (s, 2H), 7.67 (d, J = 8.63 Hz, 2H), 8.12 (d, J = 8.63 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 19.1, 26.9, 27.9, 118.2, 126.2, 140.2, 143.5, 152.1; MS (ESI) m/z 332.27 (M-4) +.
化合物 (I) (エステル):の調製
実施例10:化合物 E-1の調製
【0089】
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (E-1)の調製
150 mlの1口フラスコ中で、アセトニトリル (150 ml) 中の3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1) (36.3 g, 88.1 ミリモル) の撹拌溶液にエチル (2-メチル-4-ヒドロキシ)-フェノキシ-アセテート (II-1) (17.5 g, 83.3 ミリモル) 、N,N-ジメチルアミノピリジン (DMAP, 0.5 g, 4.09 ミリモル) 、及び炭酸カリウム (K2CO3, 13.8 g, 99.9 ミリモル) を添加した。その反応混合物を室温で8時間撹拌した。その反応が完結した後、それを濾過した。フィルターケーキを酢酸エチル (3X50 ml) で洗浄し、その後に捨てた。濾液を合わせ、回転真空蒸発にかけて溶媒を除去し、次いでカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) にかけて無色のゼラチン状物 (これは放置されている間に徐々に白色の固体になった) (33.5 g,収率: 74.3%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.28 (t, J = 7.11 Hz, 3H), 2.26 (s, 3H), 3.20 (s, 3H), 4.26 (q, J = 7.11 Hz, 2H), 4.61 (s, 2H), 6.28 (s, 1H), 6.65 (d, J = 8.8 Hz, 1H), 6.81 (d, J = 8.75 Hz, 1H), 6.93 (d, J = 3.0 Hz, 1H), 7.25-7.45 (m, 3H), 7.50 (d, J = 7.32, 2H), 7.67 (d, J = 8.48 Hz, 2H), 8.14 (d, J = 8.48 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.7, 28.2, 61.3, 65.9, 75.1, 112.8, 113.2, 118.3, 119.3, 122.7, 125.8, 126.2, 127.1, 128.9, 129.0, 129.4, 135.1, 140.4, 146.3, 151.5, 152.8, 169.1; MS (ESI) m/z 558.9 (M+NH4+).
実施例11:化合物 E-2の調製
【0090】
化合物III-1及びII-5を出発物質として使用して、エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-2) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは60%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.12 Hz, 3H), 2.45 (s, 3H), 3.21 (s, 3H), 3.50 (s, 2H), 4.12 (q, J = 7.12 Hz, 2H), 6.39 (s, 1H), 6.92 (dd, J = 8.59 Hz, 2.82 Hz, 1H), 7.0, (d, J = 2.82 Hz, 1H), 7.40-7.53(m, 3H), 7.52 (d, J = 7.48 Hz, 2H), 7.69 (d, J = 8.76 Hz, 2H), 8.18 (d, J = 8.76 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 20.9, 28.3, 37.1, 61.4, 74.6, 113.6, 117.9, 118.2, 125.4, 125.8, 126.2, 126.9, 127.3, 128.96, 129.1, 134.2, 134.7, 140.5, 142.2, 145.9, 152.6, 169.7; MS (ESI) m/z 558.01(M+H) +.
実施例12:化合物 E-3の調製
【0091】
化合物III-1 及びII-4を出発物質として使用して、エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-3) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは71%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.20 (t, J = 7.10 Hz, 3H), 2.37 (s, 3H), 3.19 (s, 3H), 3.53 (s, 2H), 4.14 (q, J = 7.10 Hz, 2H), 6.39 (s, 1H), 6.94 (d, J = 8.58 Hz, 1H), 7.24 (dd, J = 8.58 Hz, 2.4 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 7.40-7.46(m, 3H), 7.50 (d, J = 7.52 Hz, 2H), 7.69 (d, J = 8.84 Hz, 2H), 8.17 (d, J = 8.84 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.5, 28.2, 38.0, 61.4, 74.5, 113.1, 118.2, 122.7, 125.4, 125.8, 126.2, 126.3, 127.1, 127.4, 128.0, 128.96, 129.1, 130.8, 134.8, 140.4, 145.9, 152.6, 169.8; MS (ESI) m/z 557.97 (M+H) +.
実施例13:化合物 E-4の調製
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-4)の調製
【0092】
【化31】
【0093】
一定圧力の滴下ロートを備えた250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン (30 ml) の撹拌混合物に0℃でテトラヒドロフラン (THF, 30 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.7 g, 10.3 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌そして室温で更に2時間の撹拌後に、その反応を氷水浴中で砕いた氷の添加により停止した。その混合物のpHの値を6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を塩化ナトリウムの飽和溶液 (2 X100 ml) で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物3-メチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
【0094】
150 mlの1口フラスコ中で、アセトニトリル (30 ml) 中の3-(1'-ブロモ-ベンジル)-4-メチル-1- (4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1, 4.01 g, 9.73 ミリモル) の撹拌溶液に、アセトニトリル (30 ml)中の新たに調製された粗生成物3-メチル-4-ヒドロキシ-メルカプトベンゼン、N,N-ジメチルアミノピリジン (DMAP, 0.07 g, 0.57 ミリモル) 、及び炭酸カリウム (K2CO3, 1.28 g, 9.27 ミリモル) を添加した。その反応液を室温で4時間撹拌した後、炭酸カリウム (K2CO3, 1.4 g, 10.1 ミリモル) 及びエチルブロモアセテート (1.1 ml, 9.5 ミリモル) を添加した。その反応混合物を更に8時間撹拌した。その反応が完結した後、それを濾過した。フィルターケーキを酢酸エチル (3X30 ml) で洗浄し、その後に捨てた。濾液を合わせ、回転真空蒸発にかけて溶媒を除去し、次いでカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて白色の固体4.4 g (収率: 81%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.26 (t, J = 7.14 Hz, 3H), 2.19 (s, 3H), 3.15 (s, 3H), 4.23 (q, J = 7.14 Hz, 2H), 4.58 (s, 2H), 5.17 (s, 1H), 6.54 (d, J = 8.44 Hz, 1H), 7.11 (dd, J = 8.32 Hz, 2.28 Hz, 1H), 7.17 (d, J = 2.28 Hz, 1H), 7.33-7.38(m, 5H), 7.66 (d, J = 8.56 Hz, 2H), 8.10 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.1, 28.0, 50.6, 61.4, 65.4, 113.6, 118.2, 122.8, 125.5, 126.2, 126.8, 127.1, 128.2, 128.5, 128.9, 133.8, 135.2, 137.8, 140.5, 146.5, 152.6, 157.1, 168.5; MS (ESI) m/z 558.03(M+H) +.
実施例14:化合物 E-5の調製
化合物III-1 及びII-3を出発物質として使用して、エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (E-5) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは87.3%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.24 Hz, 3H), 1.27 (t, J = 7.24 Hz, 3H), 2.67 (m, 2H), 3.20 (s, 3H), 4.24 (q, J = 7.24 Hz, 2H), 4.57 (s, 2H), 6.29 (s, 1H), 6.81 (dd, J = 8.88 Hz, 3.08 Hz, 1H), 6.95 (d, J = 3.08 Hz, 1H), 7.36-7.45 (m, 3H), 7.52 (d, J = 7.60, 2H), 7.67 (d, J = 8.76 Hz, 2H), 8.16 (d, J = 8.76 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9, 14.1, 23.2, 23.3, 28.3, 61.3, 66.3, 75.3, 112.5, 112.6, 112.96, 113.2, 116.5, 117.6, 118.2, 125.4, 125.9, 126.2, 127.3, 128.8, 128.96, 135.2, 135.3, 140.5, 146.4, 150.6, 151.3, 151.6, 152.8, 169.2;MS (ESI) m/z 556.29(M+H) +.
実施例15:化合物 E-6の調製
【0095】
化合物III-1 及びII-2 を出発物質として使用して、エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート (E-6) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは98%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.02 Hz, 3H), 2.24 (s, 3H), 3.25 (s, 3H), 4.29 (q, J = 7.02 Hz, 2H), 4.59 (s, 2H), 6.35 (s, 1H), 6.56-6.75 (m, 1H), 6.88 (d, J = 2.95 Hz, 1H), 6.95 (d, J = 8.94 Hz, 1H), 7.42-7.55(m, 3H), 7.58 (d, J = 7.49 Hz, 2H), 7.72 (d, J = 8.69 Hz, 2H), 8.23 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.7, 28.2, 61.3, 65.96, 75.1, 112.1, 112.8, 113.9, 115.4, 117.8, 118.2, 118.5, 125.5, 125.8, 126.2, 126.9, 127.2, 128.8, 129.0, 135.3, 140.5, 146.4, 149.9, 152.7, 169.1; MS (ESI) m/z 541.9(M+H+); 558.9 (M+NH4+).
実施例16:化合物 E-7の調製
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-7)の調製
【0096】
【化32】
【0097】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 0.8 g, 21 ミリモル) 及びテトラヒドロフラン (30 ml) の撹拌混合物に0℃(冷却水で冷却)のテトラヒドロフラン (THF 30 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.1 g, 6.6 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌そして室温で更に2時間の撹拌後に、その反応を氷水浴中で砕いた氷の添加により停止した。その混合物のpHの値を6N塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を塩化ナトリウムの飽和溶液 (2 X100 ml) で洗浄し、無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物3-メチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
別の250 mlの1口フラスコ中で、アセトニトリル (30 ml)中の得られた3-メチル-4-ヒドロキシルメルカプトベンゼンの撹拌溶液に3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1, 2.5 g, 6.06 ミリモル) 、アセトニトリル (30 ml) 、N,N-ジメチルアミノピリジン (DMAP, 0.1 g, 0.82 ミリモル) 及び炭酸カリウム (K2CO3, 0.85 g, 6.15 ミリモル) を添加した。その混合物を室温で5時間撹拌した後、炭酸カリウム (K2CO3,1.6 g, 11.6 ミリモル) 及びエチルブロモアセテート (1.2 ml,10.3 ミリモル) をそれに添加し、得られる反応混合物を室温で更に8時間撹拌した。その反応が完結した後、それを濾過した。フィルターケーキを酢酸エチル (3X50 ml) で洗浄し、その後に捨てた。濾液を合わせ、回転真空蒸発にかけて溶媒を除去し、次いでカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル = 8:1→5:1 v/v) にかけて白色の固体2.0 g (収率: 59%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.27 (t, J = 7.18 Hz, 3H), 2.31 (s, 3H), 3.13 (s, 3H), 4.24 (q, J = 7.18 Hz, 2H), 4.55 (s, 2H), 5.09 (s, 1H), 6.59 (dd, J = 8.56 Hz, 2.88 Hz, 1H), 6.78, (d, J = 2.88 Hz, 1H), 7.24 (d, J = 8.68 Hz, 1H), (7.30-7.34(m, 4H), 7.66 (d, J = 8.66 Hz, 2H), 8.10 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 20.9, 28.1, 49.8, 61.5, 65.2, 112.6, 117.0, 118.2, 123.1, 125.5, 126.2, 126.8, 127.1, 128.0, 128.5, 128.9, 135.1, 137.9, 140.5, 144.5, 146.4, 152.5, 168.5; MS (ESI) m/z 558.05 (M+H) +.
実施例17:化合物 E-8の調製
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-アセテート(E-8)の調製
【0098】
【化33】
【0099】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン (30 ml) の撹拌混合物に0℃のテトラヒドロフラン (20 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.38 g, 8.35 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌の後に、その反応液を室温に温め、室温で更に2時間撹拌した。次いで、その反応をエタノール (10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 80 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、丸1日にわたって空気中に放置し、カラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してカップリングされた化合物3-メチル-4-ヒドロキシ-メルカプトベンゼン1.6g (黄色のゼリー状の液体,収率: 68.8%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.2 (s, 3H), 5.35 (s, 1H), 6.69 (d, J = 8.28 Hz, 1H), 7.18 (dd, J = 8.23 Hz, 2.23 Hz, 1H), 7.24 (d, J = 2.24 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.7, 115.5, 125.0, 128.3, 130.2, 134.0, 154.4; MS (ESI) m/z 278.33 (M+H+).
150 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の3-メチル-4-ヒドロキシ-メルカプトベンゼン (1.6 g, 5.76 ミリモル) の撹拌溶液にエチルブロモアセテート (1.4 ml, 60.7 ミリモル) 及び炭酸カリウム (K2CO3, 1.6 g, 11.6 ミリモル) を添加した。その反応液を室温で12時間撹拌した後、その反応溶液を酢酸エチル60mlの添加により希釈し、濾過し、蒸発させて残渣を得た。残渣をカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製して淡黄色のゼリー状液体2.0 g (収率: 77.5%)を得た。
【0100】
150 mlの3口フラスコ中で、エタノール (15 ml) 中の上記生成物 (2.0 g, 4.44 ミリモル) の撹拌溶液に水 (15 ml) 及び濃塩酸 (7 ml) を添加した。亜鉛粉末 (10 g, 153 ミリモル) を撹拌しながら徐々に添加した。その添加後、その反応混合物を室温で30分間撹拌し、次いでジクロロメタン (3 X60 ml) で抽出した。有機相を合わせ、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して淡黄色の液体を得た。
150 mlの1口フラスコ中で、アセトニトリル30ml中の上記粗生成物の撹拌溶液に3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4- トリアゾール-5(4H)-オンIII-3 (1.3 g, 3.9 ミリモル) の溶液30ml及び炭酸カリウム (K2CO3, 3.4 g, 24.6 ミリモル) を添加した。得られる反応混合物を室温で12時間撹拌した。その反応が完結した後、その反応溶液を酢酸エチル50mlの添加により希釈し、濾過した。濾液を減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =3:1 v/v) にかけて白色の固体1.7 g (収率: 91%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.25 (t, J = 7.12 Hz, 3H), 2.22 (s, 3H), 3.3 7 (s, 3H), 3.87 (s, 2H), 4.21 (q, J = 7.12 Hz, 2H), 4.58 (s, 2H), 6.58 (d, J = 8.37 Hz, 1H), 7.16 (dd, J = 8.37 Hz, 2.34 Hz, 1H), 7.23 (d, J = 2.34 Hz, 1H), 7.63 (d, J = 8.57 Hz, 2H), 7.98 (d, J = 8.57 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.1, 27.8, 31.3, 61.3, 65.5, 111.6, 118.1, 122.7, 123.5, 125.4, 126.1, 128.1, 128.6, 132.4, 136.3, 140.5, 144.6, 152.4, 156.8, 168.5; MS (ESI) m/z 482.49 (M+H)+.
実施例18:化合物 E-9の調製
【0101】
化合物III-3 及びII-2を出発物質として使用して、エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-アセテート (E-9) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは73.5%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.14 Hz, 3H), 2.33 (s, 3H), 3.45 (s, 3H), 4.28 (q, J = 7.14 Hz, 2H), 4.63 (s, 2H), 5.01 (s, 2H), 6.71 (d, J = 8.85 Hz, 1H), 6.8 (dd, J = 8.85 Hz, 3.08 Hz, 1H), 6.88 (d, J = 3.08 Hz, 1H), 7.70 (d, J = 8.56 Hz, 2H), 8.17 (d, J = 8.55 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.2, 61.2, 61.9, 66.4, 112.0, 112.7, 117.7, 118.2, 118.3, 126.2, 126.3, 129.3, 140.5, 143.9, 151.6, 151.8, 152.4, 169.1; MS (ESI) m/z 464.39 (M-1), 466.48 (M+H)+.
実施例19:化合物E-10の調製
【0102】
化合物III-1 及びII-6を出発物質として使用して、エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-10) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは72.2%の収率で黄色のゼリー状液体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.29 (t J = 7.16 Hz, 3H), 2.32 (s, 3H), 2.37 (s, 3H), 3.18 (s, 3H), 3.51 (s, 2H), 4.25 (q, J = 7.16 Hz, 2H), 6.39 (s. 1H), 6.88 (s, 1H), 7.31 (s, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.67 Hz, 2H), 8.16 (d, J = 8.67 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.0, 20.7, 28.2, 37.2, 74.4, 114.7, 118.2, 125.4, 125.6, 125.7, 126.3, 128.9, 129.1, 134.9, 135.7, 139.5, 140.4, 146.1, 152.7, 155.0, 168.7; MS (ESI) m/z 573.07 (M+H)+.
実施例20:化合物E-11の調製
【0103】
化合物III-3 及びII-4を出発物質として使用して、エチル 2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-アセテート (E-11) を実施例9に記載されたのと同様の化学反応操作に従って調製した。それは69.7%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.14 Hz, 3H), 2.19 (s, 3H), 3.43 (s, 3H), 3.55 (s, 2H), 4.28 (q, J = 7.14 Hz, 2H), 5.03 (s, 2H), 6.91 (d, J = 8.77 Hz, 3H), 7.29-7.32 (m, 2H), 7.67 (d, J = 8.66 Hz, 2H), 8.12 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.1, 28.1, 38.1, 61.4, 111.8, 118.3, 126.2, 126.3, 127.2, 127.5, 127.9, 130.8, 134.8, 140.3, 143.6, 152.5, 155.4, 169.2; MS (ESI) m/z 481.23 (M-1)-, 482.34 (M)-, 483.35 (M+H)-.
実施例21:化合物E-12の調製
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-12)の調製
【0104】
【化34】
【0105】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン (40 ml) の撹拌混合物に0℃のテトラヒドロフラン20ml中の2,5-ジメチル-4-ヒドロキシ-フェニルチオシアン酸 (1.2 g, 6.7 ミリモル) の溶液を滴下して添加した。0℃で30分間の撹拌後に、その混合物を室温に温め、次いで室温で更に1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 60 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
別の250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の得られた粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼンの撹拌溶液に3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オン(III-1) (1.6 g, 3.88 ミリモル)及び炭酸カリウム (K2CO3, 0.54 g, 3.9 ミリモル)を添加した。その混合物を室温で4時間撹拌した後、エチルブロモアセテート (1.8 ml, 15.5 ミリモル) 及び炭酸カリウム (K2CO3, 0.54 g, 3.9 ミリモル) をそれに添加した。その混合物を室温で一夜撹拌した。反応が完結した後、その反応溶液を酢酸エチル200 mlで希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製して黄色のゼリー状液体1.1g (3工程の収率: 50%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.28 (t J = 7.16 Hz, 3H), 2.12 (s, 3H), 2.25 (s, 3H), 3.13 (s, 3H), 4.25 (q, J = 7.16 Hz, 2H), 4.59 (s, 2H), 5.08 (s, 1H), 6.51 (s, 1H), 7.13 (s, 1H), 7.27-35 (m, 5H), 7.66 (d, J = 8.75 Hz, 2H), 8.10 (d, J =8.75 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 15.5, 20.7, 28.0, 50.0, 61.3, 65.5, 113.1, 117.9, 118.2, 122.4, 125.9, 126.1, 126.2, 126.8, 128.1, 128.4, 128.8, 129.1, 135.2, 138.9, 140.5, 141.5, 146.5, 152.6, 157.0, 168.6; MS (ESI) m/z 588.31 (M+NH4+).
実施例22:化合物E-13の調製
エチル 2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル) -4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-13)の調製
【0106】
【化35】
【0107】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.4 ミリモル) 及びテトラヒドロフラン (THF 30 ml) の撹拌溶液に0℃のテトラヒドロフラン20ml中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.38 g, 8.35 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に2時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 80 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮し、カラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してジスルフィド1.44 g (黄色のゼリー状液体, 収率: 62%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.2 (s, 3H), 5.35 (s, 1H), 6.69 (d, J = 8.28 Hz, 1H), 7.18 (dd, J = 8.23 Hz, 2.23 Hz, 1H), 7.24 (d, J = 2.24 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.7, 115.5, 125.0, 128.3, 130.2, 134.0, 154.4; MS (ESI) m/z 278.33 (M+H) +.
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の上記粗生成物 (0.7 g, 2.59 ミリモル) の撹拌溶液にメチル 2-ブロモ-プロピオネート (0.7 ml, 6.0 ミリモル) 及び炭酸カリウム (K2CO3, 2 g, 14.5 ミリモル) を添加した。得られる混合物を室温で一夜撹拌した。その反応が完結した後、その反応混合物を酢酸エチル (100 ml) で希釈し、濾過した。合わせた溶液を減圧で蒸発させて残渣を得、これをクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製して黄色のゼリー状液体1.0 g (収率: 86%)を得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.63 (d, J = 6.84 Hz, 1H); 2.34 (s, 3H), 3.75 (s, 3H), 4.73 (q, J = 6.84 Hz, 1H), 6.57-6.61 (m, 1H), 7.19-7.27 (m, 2H).
【0108】
250 ml の1口フラスコ中で、エタノール (15 ml) 中の上記生成物 (1.0 g, 2.22 ミリモル) の撹拌溶液に水15 ml 及び濃塩酸5 mlを添加した。亜鉛粉末 (10 g, 153 ミリモル) を徐々に添加した。その添加後、その反応混合物を室温で30分間撹拌し、次いでジクロロメタン (3X50 ml)で抽出した。有機相を合わせ、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して淡黄色の液体を得た。
150 mlの1口フラスコ中で、アセトニトリル30ml中の上記粗生成物の撹拌溶液にアセトニトリル (30 ml) 中の3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H -1,2,4-トリアゾール-5(4H)-オン (III-1) (1.8 g, 4.36 ミリモル) の溶液及び炭酸カリウム (K2CO3, 2.5 g, 18.1 ミリモル) を添加した。その反応混合物を室温で6時間撹拌した。その反応が完結した後、その反応混合物を酢酸エチル100 mlの添加により希釈し、濾過し、減圧で濃縮し、カラムクロマトグラフィー(シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけてE-13(一対のジアステレオマー) (淡黄色のゼリー状液体, 収率: 50%)1.2gを得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.61 (d, J = 6.84 Hz, 1H); 2.18 (s, 3H), 3.15 (s, 3H), 3.70 (d, J = 12.61 Hz, 3H); 4.70 (q, J = 6.82 Hz, 1H); 5.18 (s, 1H); 6.51 (d, J = 8.46 Hz, 1H), 7.0-7.11 (m, 1H), 7.10-7.15 (m, 1H), 7.34-7.37 (m, 5H), 7.67 (d, J = 8.74 Hz, 2H), 8.11 (d, J = 8.74 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.1, 18.5, 28.1, 50.6, 52.2, 72.7, 112.1, 118.2, 123.0, 125.5, 126.2, 126.8, 127.1, 128.2, 128.5, 128.9, 133.8, 135.2, 137.7, 140.6, 146.5, 152.6, 156.9, 172.2.
実施例23:化合物E-14の調製
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-14)の調製
【0109】
【化36】
【0110】
500 mlの3口フラスコにo-エチルフェノール (5 g, 40.9 ミリモル) 、チオシアン酸ナトリウム(10 g, 123.3 ミリモル) 、臭化ナトリウム (4.3 g, 41.79 ミリモル) 及びメタノール (100 ml) を連続して添加した。その混合物を氷浴中で0℃に冷却し、次いでメタノール (50 ml) 中の臭素 (2.6 ml, 50.6 ミリモル) の溶液をそれに滴下して添加した。0℃で1時間撹拌した後、その混合物を室温に温め、次いで室温で更に4時間撹拌した。重炭酸ナトリウムの飽和水溶液200 mlをその反応混合物に徐々に添加し、10分間撹拌した。その混合物を酢酸エチル (2X300 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過した。濾液を減圧で濃縮して赤褐色の粘稠な液体を得た。カラムクロマトグラフィーを行なって (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) 、3-エチル-4-ヒドロキシ-フェニルチオシアン酸 6.8g(赤褐色の粘稠な液体, 収率: 93%)を得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 1.25 (t J = 7.28 Hz, 3H), 2.64 (q, J = 7.53 Hz, 2H), 5.46 (s, 1H), 6.80 (d, J = 8.36 Hz, 1H), 7.28 (dd, J = 8.36 Hz, 2.52 HZ, 1H), 7.34 (d, J = 2.48.36 Hz, 1H).
150 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.2 ミリモル) 及びテトラヒドロフラン(40 ml) の撹拌混合物に0℃のテトラヒドロフラン20ml中の3-エチル-4-ヒドロキシ-フェニルチオシアン酸 (1.3 g, 7.25 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6N塩酸の添加によりpH 3-4に調節し、次いで水相を酢酸エチル (3X60 ml)で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物3-エチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体)を得た。
別の150 mlの1口フラスコ中で、アセトニトリル (20 ml) 中の得られる粗生成物3-エチル-4-ヒドロキシルメルカプトベンゼンの撹拌溶液に炭酸カリウム (K2CO3, 1.0 g, 7.23 ミリモル) を添加し、続いてアセトニトリル20ml中の 3-(1'-ブロモ-ベンジル)-4-メチル-1- (4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン (III-1) (1.6 g, 3.88 ミリモル)の溶液を滴下して添加した。その混合物を室温で6時間撹拌した後、炭酸カリウム (K2CO3, 1.0 g, 7.23ミリモル) 及びエチルブロモアセテート (1.6 ml,13.8 ミリモル) をそれに添加した。その混合物を同温度で更に8時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル (50 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してE-14 1.3g(黄色のゼラチン状物, 3工程の収率: 58.6%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 1.09 (t J = 7.48 Hz, 3H), 1.26 (t J = 7.28 Hz, 3H), 2.59 (q, J = 7.52 Hz, 2H), 3.15 (s, 3H), 4.22 (q, J = 7.20 Hz, 2H), 4.58 (s, 2H), 5.18 (s, 1H), 6.56 (d, J = 8.20 Hz, 1H), 7.15 (dd, J = 8.32 Hz, 2.28 HZ, 1H), 7.31-7.36 (m, 5H), 7.66 (d, J = 8.72 Hz, 1H), 8.11 (d, J = 8.52 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.7, 14.1, 23.0, 28.1, 50.6, 61.3, 65.4, 111.5, 118.2, 123.3, 126.1, 126.8, 127.1, 128.2, 128.5, 128.9, 133.8, 134.3, 135.2, 136.2, 140.5, 146.5, 152.6, 156.7, 168.5; MS (ESI) m/z 572.14 (M+H+); 589.1 (M+ NH4+).
実施例24:化合物E-15の調製
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-15)の調製
【0111】
【化37】
【0112】
150 mlの1口フラスコ中で、アセトニトリル (30 ml) 中の3-メチル-4-ヒドロキシ-メルカプトベンゼン (0.52 g, 1.87 ミリモル) の撹拌溶液にブロミドIII-1 (1.53 g, 3.71 ミリモル) 及び炭酸カリウム(K2CO3, 2.5 g, 18.1 ミリモル) を添加した。その混合物を室温で12時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル(100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて淡黄色のゼリー状液体1.4 g を得た。
150 mlの3口フラスコ中で、エタノール (15 ml) 中の上記生成物 (1.4 g, 1.48 ミリモル) の撹拌溶液に水15ml及び濃塩酸5 mlを添加した。亜鉛粉末 (10 g, 153 ミリモル) を徐々に添加した。その添加後、その反応液を室温で30分間撹拌し、次いでジクロロメタン (3 X50 ml) で抽出した。有機相を合わせ、無水硫酸マグネシウムで乾燥させ、濾過し、減圧で濃縮して淡黄色の液体を得た。
【0113】
150 ml の1口フラスコ中で、アセトニトリル (30 ml) 中の上記粗生成物の撹拌溶液にアセトニトリル (30 ml) 中のエチル 2-ブロモ-イソプロピオネート (3 ml, 20.2 ミリモル) 及び炭酸カリウム(K2CO3, 2.0 g, 14.4 ミリモル) を添加した。その反応混合物を室温で12時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル(100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル=5:1 v/v) により精製してE-15 0.58g(無色のゼラチン状物, 3工程の収率: 26.6%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.20 Hz, 3H), 1.45 (s, 6H), 2.37 (s, 3H), 3.20 (s, 3H), 4.10 (q, J = 7.20 Hz, 2H), 6.41 (s, 1H), 6.94 (d, J = 8.52 Hz, 1H), 7.25 (dd, J = 8.52 Hz, 1.8 Hz, 1H), 7.32 (d, J = 1.8 Hz, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.52 Hz, 2H), 8.17 (d, J = 8.52 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 16.4, 25.8, 28.2, 50.9, 61.1, 74.5, 112.4, 118.2, 124.2, 125.7, 126.2, 126.3, 127.4, 129.0, 129.1, 134.7, 136.1, 139.9, 140.4, 145.9, 152.6, 156.2, 173.9; MS (ESI) m/z 584.71(M-H)-.
実施例25:化合物E-16の調製
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(2-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-16)の調製
【0114】
【化38】
【0115】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 2.0 g, 52.7ミリモル) 及びテトラヒドロフラン(40 ml) の撹拌混合物に0℃のテトラヒドロフラン (30 ml )中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (2.5 g, 15.13ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に2時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加によりpH 3-4に調節し、次いで水相を酢酸エチル (3X100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて黄色の液体を得た。
250 mlの1口フラスコ中で、アセトニトリル (30 ml)中の得られる黄色の液体の撹拌溶液にブロミドIII-1 (3.2 g, 7.76 ミリモル)及び炭酸カリウム (K2CO3, 1.15 g, 8.32 ミリモル) を添加した。その混合物を室温で6時間撹拌した。反応が完結した後、その反応混合物を酢酸エチル (100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) にかけて黄色のゼリー状液体3.6 g を得た。
【0116】
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の上記生成物 (2.2 g, 4.67 ミリモル) の撹拌溶液にエチル 2-ブロモ-イソプロピオネート (2 ml, 13.5ミリモル) 及び炭酸カリウム (K2CO3) (1.5 g, 10.8 ミリモル)を添加した。その反応混合物を36時間にわたって加熱、還流した。反応が完結した後、その反応混合物を酢酸エチル (100 ml)で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =5:1 v/v) により精製してE-16 1.0g(黄色のゼラチン状物, 3工程の収率: 44%)を得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.20 (t, J = 7.14 Hz, 3H), 1.55 (d, J = 5.76 Hz, 6H), 2.13 (s, 3H), 3.15 (s, 3H), 4.18 (q, J = 7.14 Hz, 2H), 5.19 (s, 1H), 6.49 (d, J = 8.46 Hz, 1H), 7.03 (dd, J = 8.46 Hz, 2.36 Hz, 1H), 7.16 (d, J = 2.36 Hz, 1H), 7.32-7.37 (m, 5H), 7.66 (d, J = 8.74 Hz, 2H), 8.10 (d, J = 8.74 Hz, 2H); MS (ESI) m/z 586.39(M+H+).
実施例26:化合物E-17の調製
【0117】
化合物III-2 及びII-4を出発物質として使用して、エチル 2-(3-メチル-4-(1-(3-(4-
n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート (E-17) を実施例9に記載されたのと同様の化学反応方法に従って調製した。それは81.2%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.76 (t, J = 7.30 Hz, 3H ), 0.92-1.02 (m, 1H), 1.03-1.29 (m, 5H), 1.35-1.48(m, 1H), 2.37 (s, 3H), 3.47-3.56 (m, 3H), 3.63-3.67 (m, 1H), 4.11-4.16 (m, 2H), 6.38 (s, 1H), 6.97 (d, J = 8.59 Hz, 1H), 7.24 (dd, J = 8.57 Hz, 2.25 Hz, 1H), 7.34 (d, J = 2.04 Hz, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.73 Hz, 2H), 8.19 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 14.1, 16.5, 19.9, 30.1, 38.0, 42.4, 61.4, 74.7, 113.1, 118.2, 125.6, 126.2, 126.3, 128.0, 128.9, 129.1, 130.8, 134.9, 135.3, 140.5, 145.9, 152.6, 155.0, 169.8; MS (ESI) m/z 600.0 (M)+, 601.2 (M+1)+, 602.2 (M+2)+, 603.2 (M+3)+ .
実施例27:化合物E-18の調製
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(2-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-18)の調製
【0118】
【化39】
【0119】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.2 g, 31.6 ミリモル) 及びテトラヒドロフラン(50 ml) の撹拌混合物に0℃のテトラヒドロフラン (20 ml) 中の3-メチル-4-ヒドロキシ-フェニルチオシアン酸 (1.4 g, 8.47 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に2時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6N塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて黄色の液体を得た。
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の得られる黄色の液体の撹拌溶液にブロミドIII-2 (1.4 g, 3.08 ミリモル) 及び炭酸カリウム (K2CO3, 0.42 g, 3.04 ミリモル) を添加した。その混合物を室温で6時間撹拌した後、アセトニトリル (10 ml) 中のエチル 2-ブロモ-イソプロピオネート (6 ml, 40.5 ミリモル) 及び炭酸カリウム (K2CO3, 2.4 g, 17.4 ミリモル) をそれに添加した。その混合物を36時間にわたって加熱、還流した。その反応が完結した後、その反応混合物を酢酸エチル (100 ml) で希釈し、濾過した。濾液を蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =10:1 v/v) にかけてE-18 (黄色のゼラチン状物, 3工程の収率: 28.6 %) 1.52 gを得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.80 (t, J = 7.30 Hz, 3H ), 1.16-1.36(m, 7H), 1.56 (d, J = 2.84 Hz, 6H), 2.12 (s, 3H), 3.48-3.57 (m, 2H), 4.17-4.23 (m, 2H), 5.10 (s, 1H), 6.48 (d, J = 8.44 Hz, 1H), 7.01 (dd, J = 8.46 Hz, 2.28 Hz, 1H), 7.17 (d, J = 2.08 Hz, 1H), 7.30-7.35 (m, 5H), 7.67 (d, J = 8.81 Hz, 2H), 8.15 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 14.0, 16.5, 19.8, 25.3, 29.7, 30.4, 41.9, 50.3, 61.5, 79.2, 116.4, 118.1, 124.5, 126.1, 128.4, 128.5, 128.8, 130.3, 133.3, 135.9, 137.8, 140.6, 146.4, 152.3, 154.9, 174.1; MS (ESI) m/z 626.1 (M-2)-, 627.1 (M-1)-, 628.1 (M)-.
実施例28:化合物E-19の調製
エチル 2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート (E-19)の調製
【0120】
【化40】
【0121】
250 mlの3口フラスコ中で、四水素化リチウムアルミニウム (LiAlH4, 1.0 g, 26.3 ミリモル) 及びテトラヒドロフラン(40 ml) の撹拌混合物に0℃のテトラヒドロフラン (20 ml) 中の2,5-ジメチル-4-ヒドロキシ-フェニルチオシアン酸 (1.02 g, 5.69 ミリモル) の溶液を滴下して添加した。0℃で30分間撹拌した後、その混合物を室温に温め、次いで室温で更に1時間撹拌した。その反応をエタノール(10 ml) の添加により停止した。その混合物のpHの値を氷水浴中で6M塩酸の添加により3-4 に調節し、次いで水相を酢酸エチル (3 X 60 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、真空で蒸発させて粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼン (黄色の液体) を得た。
250 mlの1口フラスコ中で、アセトニトリル (60 ml) 中の得られる粗生成物2,5-ジメチル-4-ヒドロキシ-メルカプトベンゼンの撹拌溶液に3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル)フェニル-1H-1,2,4-トリアゾール-5(4H)-オンIII-2 (1.1 g, 2.42 ミリモル) 及び炭酸カリウム (K2CO3, 0.35 g, 2.35 ミリモル) を添加した。その混合物を室温で4時間撹拌した後、エチルブロモアセテート (1.8 ml, 15.5 ミリモル) 及び炭酸カリウム (K2CO3, 1.5 g, 10.8 ミリモル) をそれに添加し、次いでその反応混合物を室温で一夜撹拌した。その反応が完結した後、その反応混合物を酢酸エチル (200 ml) で希釈し、濾過した。濾液を真空で蒸発させて残渣を得、これをカラムクロマトグラフィー (シリカゲルH:300-400 メッシュ; 石油エーテル/酢酸エチル =8:1 v/v) にかけてE-19 (黄色のゼラチン状物, 3工程の収率: 22.9%) 0.8 g を得た。
その構造を核磁気共鳴分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.80 (t, J = 7.26 Hz, 3H ), 1.18-1.33 (m, 7H), 2.13 (s, 3H), 2.22 (s, 3H), 3.50-3.52 (m, 2H), 4.23-4.29 (m, 2H), 4.59 (s, 2H), 5.01 (s, 1H), 6.50 (s, 1H), 7.13 (s, 1H), 7.30-7.32 (m, 5H), 7.68 (d, J = 8.64 Hz, 2H), 8.16 (d, J = 8.52 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 14.2, 15.5, 19.8, 20.8, 29.7, 30.4, 41.8, 49.7, 61.4, 65.5, 112.7, 118.1, 122.7, 125.8, 126.2, 128.3, 128.4, 128.8, 135.9, 139.0, 140.6, 141.6, 146.5, 152.3, 156.9, 168.7.
化合物 (I) (酸)の調製:
実施例29:化合物 A-1の調製
【0122】
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸(A-1)の調製
ジクロロメタン (50 ml) 及びエタノール (50 ml) 中のエチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ-アセテート (28 g, 51.8 ミリモル) の撹拌溶液に水酸化ナトリウム (10 g, 250 ミリモル) の水溶液50mlを添加した。その反応混合物を室温で12時間撹拌した。その反応が完結した後、その混合物のpHの値を6N塩酸の添加により2-3 に調節した。得られる混合物をジクロロメタン (3 X 100 ml) で抽出した。合わせた有機層を無水硫酸マグネシウムで乾燥させ、濾過し、蒸発させて残渣を得た。残渣をカラムクロマトグラフィー (シリカゲルH: 300-400 メッシュ; 石油エーテル/酢酸エチル =4:1→1:1 v/v) により精製してA-1 18.2 g を白色の固体 (収率: 68.5%)として得た。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.29 (s, 3H), 3.25 (s, 3H), 4.62 (s, 2H), 6.32 (s, 1H), 6.69 (d, J = 8.8 Hz, 1H), 6.86 (d, J = 8.75 Hz, 1H), 6.98 (s, 1H), 7.43-7.50 (m, 3H), 7.55-7.58 (m, 2H), 7.72 (d, J = 8.48 Hz, 2H), 8.19 (d, J = 8.48 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.4, 18.1, 28.3, 58.4, 65.9, 75.3, 112.8, 113.2, 118.3, 119.3, 122.7, 125.8, 126.2, 127.1, 128.9, 129.0, 129.4, 135.1, 140.4, 146.3, 151.5, 152.8, 173.2; MS (ESI) m/z 514.2 (M+H+); 531 (M+NH4+).
実施例30:化合物 A-2の調製
【0123】
化合物E-2 を出発物質として使用して、2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 A-2) を実施例29に記載されたのと同様の合成方法に従って調製した。それは46%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.42 (s, 3H), 3.18 (s, 3H), 3.51 (s, 2H), 6.76 (s, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 2.76 Hz, 1H), 7.25-7.52 (m, 5H), 7.67 (d, J = 8.67 Hz, 2H), 8.14 (d, J = 8.58 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 20.9, 28.3, 36.9, 74.6, 113.7, 118.0, 118.3, 125.8, 126.2, 126.3, 126.6, 127.1, 128.98, 129.01, 134.1, 134.6, 140.4, 142.2, 145.9, 152.6, 156.8, 174.6; MS (ESI) m/z 528 (M-H)-; 529 (M-); 530 1 (M+H)-.
実施例31:化合物 A-3の調製
【0124】
化合物E-3 を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸(A-3) を実施例29に記載されたのと同様の合成方法に従って調製した。それは24.2%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H), 3.24 (s, 3H), 3.61 (s, 2H), 6.43 (s, 1H), 6.99 (d, J = 8.58 Hz, 1H), 7.22-7.29 (m, 1H), 7.38 (d, J = 2.28 Hz, 1H), 7.38-7.56 (m, 5H), 7.73 (d, J = 8.68 Hz, 2H), 8.21 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.5, 28.2, 37.8, 74.5, 113.2, 118.2, 125.7, 126.2, 126.6, 127.1, 128.2, 128.99, 129.1, 130.8, 134.7, 140.4, 145.9, 152.7, 155.0, 174.2; MS (ESI) m/z 556.9 (M+CO)+.
実施例32:化合物 A-4の調製
【0125】
化合物E-4 を出発物質として使用して、2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-4) を実施例29に記載されたのと同様の合成方法に従って調製した。それは52.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.18 (s, 3H), 3.13, (s, 3H), 4.63 (s, 2H), 5.19 (s, 1H), 6.57 (d, J = 8.43 Hz, 1H), 7.13 (dd, J = 6.07 Hz, 2.26 Hz, 1H), 7.18 (d, J = 1.8 Hz, 1H), 7.32-7.36 (m, 5H), 7.66 (d, J = 8.73 Hz, 2H), 8.10 (d, J = 8.69 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.0, 28.1, 50.5, 64.9, 111.5, 118.3, 123.6, 126.1, 126.2, 128.2, 128.4, 128.6, 128.9, 133.9, 135.0, 137.8, 140.4, 146.5, 152.6, 156.7, 172.6; MS (ESI) m/z 530 (M+H)+, 531 (M+2), 532 (M+3).
実施例33:化合物 A-5の調製
【0126】
化合物E-5 を出発物質として使用して、2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸(A-5) を実施例29に記載されたのと同様の合成方法に従って調製した。それは62.8%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.55 Hz, 3H), 2.66 (q, J = 7.51 Hz, 2H), 3.19 (s, 3H), 4.62 (s, 2H), 6.29 (s, 1H), 6.66 (d, J = 8.88 Hz, 1H), 6.83 (dd, J = 8.87 Hz, 3.09 Hz, 1H), 6.96 (d, J = 3.09 Hz, 1H), 7.38-7.53 (m, 5H), 7.67 (d, J = 8.85 Hz, 2H), 8.15 (d, J = 8.78 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9, 23.2, 28.3, 65.7, 75.2, 112.6, 113.0, 117.7, 118.2, 125.8, 126.2, 127.0, 127.4, 128.8, 129.01, 135.1, 135.2, 140.4, 146.3, 150.9, 151.8, 152.8, 173.3; MS (ESI) m/z 525.9 (M-1)-, 527 (M)-, 528 (M+1)-.
実施例34:化合物 A-6の調製
【0127】
化合物E-6 を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸(A-6) を実施例29に記載されたのと同様の合成方法に従って調製した。それは42.4%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.40 (s, 3H), 3.25 (s, 3H), 4.65 (s, 3H), 6.34 (s, 1H), 6.69 (d d, J = 8.86 Hz, 3.0 Hz, 1H), 6.88 (d, J = 3.0 Hz, 1H), 6.95 (d, J = 8.88 Hz, 2H), 7.43-7.58 (m, 5H), 7.73 (d, J = 8.63 Hz, 2H), 8.21 (d, J = 8.58 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.8, 28.3, 65.4, 75.1, 112.2, 113.8, 118.3, 118.5, 125.7, 126.2, 126.3, 127.1, 128.9, 129.1, 135.1, 140.4, 146.3, 150.2, 152.4, 152.8, 173.1; MS (ESI) m/z 513 (M)-, 512 (M-1)-, 514 (M+1)-.
【0128】
実施例35:化合物 A-7の調製
【0129】
化合物E-7 を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-7) を実施例29に記載されたのと同様の合成方法に従って調製した。それは42%の収率で淡黄色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.33 (s, 3H), 3.11 (s, 3H), 4.65 (s, 2H), 5.11 (s, 1H), 6.62 (dd, J = 8.53 Hz, 2.88 Hz, 1H), 6.78 (d, J = 2.88 Hz, 1H), 7.27-7.34 (m, 5H), 7.66 (d, J = 8.79 Hz, 2H), 8.09 (d, J = 8.71 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.0, 28.1, 49.7, 64.6, 112.6, 116.9, 118.3, 123.1, 126.2, 128.1, 128.6, 129.0, 135.0, 137.9, 140.4, 144.6, 146.5, 152.6, 158.3, 172.1; MS (ESI) m/z 530 (M)+, 531 (M+1)+.
実施例36:化合物 A-8の調製
【0130】
化合物E-8 を出発物質として使用して、2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-酢酸(A-8) を実施例29に記載されたのと同様の合成方法に従って調製した。それは76%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, DMSO) δ 2.11 (s, 3H), 3.3 (s, 3H), 4.13 (s, 2H), 4.68 (s, 2H), 6.80 (d, J = 8.13 Hz, 1H), 7.25-7.27 (m, 2H), 7.80 (d, J = 8.85 Hz, 2H), 7.99 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, DMSO) δ 16.2, 28.0, 30.4, 62.5, 65.3, 112.5, 118.1, 120.6, 123.3, 123.6, 123.9, 124.9, 125.2, 125.6, 125.9, 126.8, 127.5, 128.7, 132.1, 135.6, 141.1, 146.3, 152.3, 156.6, 170.5; MS (ESI) m/z 452.8 (M).
実施例37:化合物 A-9の調製
【0131】
化合物E-9 を出発物質として使用して、2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-酢酸(A-9) を実施例29に記載されたのと同様の合成方法に従って調製した。それは69.2%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.28 (s, 3H), 3.43 (s, 3H), 4.64 (s, 2H), 4.99 (s, 2H), 6.69 (d, J = 8.40 Hz, 1H), 6.78 (dd, J = 8.82 Hz, 3.06 Hz, 1H), 6.85 (dd, J = 2.96 Hz, 1H), 7.67 (d, J = 8.60 Hz, 2H), 8.13 (d, J = 8.55 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.4, 28.1, 61.9, 65.9, 112.1, 112.8, 118.2, 118.3, 118.4, 126.2, 126.3, 129.3, 143.9, 151.2, 152.0, 152.5, 170.4; MS (ESI) m/z 436 (M-1), 437 (M), 438 (M+1).
実施例38:化合物A-10の調製
【0132】
化合物E-10を出発物質として使用して、2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-10) を実施例29に記載されたのと同様の合成方法に従って調製した。それは88.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.09 (s, 3H), 2.25 (s, 3H), 3.09 (s, 3H), 4.59 (s, 2H),5.09 (s, 1H), 6.52 (s. 1H), 7.13 (s, 1H), 7.31-7.37 (m, 5H), 7.65 (d, J = 8.70 Hz, 2H), 8.09 (d, J = 8.70 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 15.4, 20.7, 28.0, 49.7, 65.2, 113.2, 118.4, 122.7, 126.2, 126.7, 128.2, 128.5, 128.9, 135.1, 139.0, 140.4, 141.6, 146.7, 152.6, 156.8, 173.0; MS (ESI) m/z 542.42 (M-1)-, 543.54 (M)-, 544.43 (M+H)-.
実施例39:化合物A-11の調製
【0133】
化合物E-11を出発物質として使用して、2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェンチオ)-酢酸(A-11) を実施例29に記載されたのと同様の合成方法に従って調製した。それは70.8%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 2.37 (s, 3H), 3.18 (s, 3H), 3.51 (s, 2H), 6.39 (s. 1H), 6.88 (s, 1H), 7.31 (s, 1H), 7.39-7.52 (m, 5H), 7.69 (d, J = 8.67 Hz, 2H), 8.16 (d, J = 8.67 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.0, 20.7, 28.2, 37.2, 74.4, 114.7, 118.2, 125.4, 125.6, 125.7, 126.3, 128.9, 129.1, 134.9, 135.7, 139.5, 140.4, 146.1, 152.7, 155.0, 174.7; MS (ESI) m/z 541.96 (M-1)-, 543.04 (M)-, 544.07 (M+H)-.
実施例40:化合物A-12の調製
【0134】
化合物E-12を出発物質として使用して、2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-酢酸(A-12) を実施例29に記載されたのと同様の合成方法に従って調製した。それは47.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 2.19 (s, 3H), 3.43 (s, 3H), 3.55 (s, 2H), 5.03 (s, 2H), 6.91 (d, J = 8.77 Hz, 3H), 7.29-7.32 (m, 2H), 7.67 (d, J = 8.66 Hz, 2H), 8.12 (d, J = 8.66 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.1, 28.1, 38.1, 61.4, 111.8, 118.3, 126.2, 126.3, 127.2, 127.5, 127.9, 130.8, 134.8, 140.3, 143.6, 152.5, 155.4, 174.8; MS (ESI) m/z 452.23 (M-1)-, 453.34 (M)-, 454.35 (M+H)-.
実施例41:化合物A-13の調製
【0135】
化合物E-13を出発物質として使用して、2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-13) を実施例29に記載されたのと同様の合成方法に従って調製した。それは76.9%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.55 (d, J = 6.56 Hz, 3H), 2.12 (s, 3H), 3.06 (s, 3H), 4.62 (q, J = 6.56 Hz, 1H), 5.17 (s, 1H), 6.56 (d, J = 9.04 Hz, 1H), 7.14-7.17 (m, 2H), 7.31-7.38 (m, 5H), 7.65 (d, J = 8.68 Hz, 2H), 8.09 (d, J = 8.68 Hz, 2H); MS (ESI) m/z 542.24 (M-1)-, 543.26 (M)-, 544.24 (M+H)-.
実施例42:化合物A-14の調製
【0136】
化合物E-14を出発物質として使用して、2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-14) を実施例29に記載されたのと同様の合成方法に従って調製した。それは63.1%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.07 (t, J = 7.56 Hz, 3H), 2.56 (q, J = 7.56 Hz, 2H), 3.09 (s, 3H), 4.59 (s, 2H), 5.18 (s, 1H), 6.56 (d, J = 9.04 Hz, 1H), 7.14-7.17 (m, 2H), 7.31-7.38 (m, 5H), 7.65 (d, J = 8.68 Hz, 2H), 8.09 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.7, 22.9, 28.0, 50.4, 65.0, 111.6, 118.3, 123.6, 125.4, 126.1, 126.2, 127.3, 127.5, 128.3, 128.5, 128.9, 133.9, 134.2, 135.1, 136.3, 140.4, 146.6, 152.5, 156.4, 172.7; MS (ESI) m/z 544.14 (M+H)+, 561.93 (M+NH4)+.
実施例43:化合物A-15の調製
【0137】
化合物E-15を出発物質として使用して、2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-15) を実施例29に記載されたのと同様の合成方法に従って調製した。それは70.6%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 6H), 2.35 (s, 3H), 3.18 (s, 3H), 6.40 (s, 1H), 6.96 (d, J = 8.56 Hz, 1H), 7.26-7.27 (m, 1H),7.35-7.50 (m, 6H), 7.67 (d, J = 8.72 Hz, 2H), 8.15 (d, J = 8.64 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.4, 25.4, 28.2, 50.7, 74.5, 112.6, 118.2, 123.7, 125.7, 126.2, 127.4, 128.1, 128.9, 129.1, 134.7, 136.1, 139.8, 140.4, 145.9, 152.6, 156.4, 179.1; MS (ESI) m/z 556.69 (M)+, 558.04 (M+1)+.
実施例44:化合物A-16の調製
【0138】
化合物E-16を出発物質として使用して、2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-16) を実施例29に記載されたのと同様の合成方法に従って調製した。それは52.6%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 1.57 (d, J = 5.25 Hz, 6H ), 2.12 (s, 3H), 3.10 (s, 3H), 5.21 (s, 2H), 6.63 (d, J = 8.47 Hz, 1H),7.05 (dd, J = 8.47 Hz, 2.06 Hz, 1H), 7.19 (d, J = 1.99 Hz, 1H), 7.25-7.37 (m, 5H), 7.65 (d, J = 8.76 Hz, 2H), 8.09 (d, J = 8.62 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.6, 25.1, 25.3, 28.1, 50.3, 79.4, 117.3, 118.3, 124.1, 126.2, 126.3, 128.2, 128.5, 128.9, 130.8, 133.2, 134.9, 137.8, 140.4, 146.6, 152.6, 154.5, 177.4; MS (ESI) m/z 558.06 (M)+
実施例45:化合物A-17の調製
【0139】
化合物E-17を出発物質として使用して、2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸 (A-17) を実施例29に記載されたのと同様の合成方法に従って調製した。それは62%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.75 (t, J = 7.29 Hz, 3H ), 1.03-1.05 (m, 1H), 1.11-1.18(m, 2H),1.19-1.26 (m, 1H), 1.40-1.50 (m, 1H), 2.36 (s, 3H), 3.47-3.53 (m, 1H), 3.56 (s, 2H), 3.62-3.69 (m, 1H), 6.38 (s, 1H), 6.97 (d, J = 8.58 Hz, 1H), 7.24 (dd, J = 8.47 Hz, 1.92 Hz, 1H), 7.33 (d, J = 1.99 Hz, 1H), 7.39-7.51 (m, 5H), 7.68 (d, J = 8.74 Hz, 2H), 8.17 (d, J = 8.61 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 16.6, 19.9, 37.8, 42.4, 74.7, 113.2, 118.2, 125.6, 126.2, 126.3, 128.2, 128.9, 129.1, 130.9, 134.8, 135.1, 140.4, 145.9, 152.6, 155.3, 174.4; MS (ESI) m/z 569.93 (M-2)-, 570.9 (M-1)- .
実施例46:化合物A-18の調製
【0140】
化合物E-18を出発物質として使用して、2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル- 5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸 (A-18) を実施例29に記載されたのと同様の合成方法に従って調製した。それは61.3%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.79 (t, J = 7.26 Hz, 3H ), 1.17-1.32(m, 6H), 1.59 (s, 6H), 2.13 (s, 3H), 3.48-3.53 (m, 2H), 5.13 (s, 1H), 6.63 (d, J = 8.44 Hz, 1H), 7.05 (dd, J = 8.47 Hz, 2.04 Hz, 1H), 7.17 (d, J = 2.08 Hz, 1H), 7.31-7.35 (m, 5H), 7.67 (d, J = 8.84 Hz, 2H), 8.14 (d, J = 8.60 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 16.6, 19.8, 25.2, 25.3, 29.7, 30.4, 41.8, 50.1, 79.4, 117.3, 118.3, 124.4, 126.1, 128.4, 128.5, 128.8, 130.7, 133.4, 135.7, 137.9, 140.5, 146.5, 152.4, 154.5, 177.9; MS (ESI) m/z 597.96 (M-2)-, 599.0 (M-1)-, 600.0 (M)-.
実施例47:化合物A-19の調製
【0141】
化合物E-19を出発物質として使用して、2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸(A-19) を実施例29に記載されたのと同様の合成方法に従って調製した。それは31.4%の収率で白色の固体である。
その構造を核磁気共鳴分析法及び質量分析法からの下記のデータにより特性決定した。
1H NMR (400 MHz, CDCl3) δ 0.79 (t, J = 7.27 Hz, 3H ), 1.13-1.23 (m, 2H), 1.28-1.36 (m, 2H), 2.12 (s, 3H), 2.25 (s, 3H), 3.47-3.55 (m, 2H), 4.66 (s, 2H), 5.03 (s, 1H), 6.53 (s, 1H), 7.15 (s, 1H), 7.29-7.36 (m, 5H), 7.68 (d, J = 8.66 Hz, 2H), 8.16 (d, J = 8.56 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5, 15.5, 19.8, 20.8, 30.4, 41.9, 49.6, 64.9, 112.9, 118.3, 123.1, 125.7, 126.2, 128.3, 128.5, 128.8, 135.8, 139.1, 140.5, 141.7, 146.5, 152.4, 156.6, 173.2; MS (ESI) m/z 584.03 (M-1)-, 584.98 (M)-。
【特許請求の範囲】
【請求項1】
式(I)の化合物又はその医薬上許される塩もしくはこれらの溶媒和物。
【化1】
[式中、
XはO、S、N、又は (CH2)n (式中、nは1から4までの整数である)であり、
YはO、S又はNであり、
RはH又はC1-C9アルキルであり、
R1 及びR2 は互いに独立にH又はC1-C4 アルキルであり、かつR1及びR2 の少なくとも一つがHであり、
R3 はH又はC1-C9 アルキルであり、
R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル又は置換フェニルであり、そのフェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6又はCONR5R6 から選ばれ、
G1、G2、G3、G4、G5、G6、G7、G8及びG9 は互いに独立にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6 、COOR5 、NR5COR6 又はCONR5R6 であり、かつ
R5 及びR6 は独立に互いにH又はC1-C9 アルキルである]
【請求項2】
XがO、S又はCH2である、請求項1記載の化合物。
【請求項3】
YがO又はSである、請求項1記載の化合物。
【請求項4】
RがH、メチル又はエチルである、請求項1記載の化合物。
【請求項5】
R1 がH、メチル又はエチルである、請求項1記載の化合物。
【請求項6】
R2 がH、メチル又はエチルである、請求項1記載の化合物。
【請求項7】
R3 がH又はC1-C4 アルキルである、請求項1記載の化合物。
【請求項8】
R3 がメチルである、請求項1又は7記載の化合物。
【請求項9】
R4 が置換フェニルである場合に、その置換基がC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6 、COOR5 、NR5COR6又はCONR5R6から選ばれ、R5 及びR6 が互いに独立にH又はC1-C9 アルキルである、請求項1記載の化合物。
【請求項10】
G1、G2、G3、及びG4 が互いに独立に、又は同時にH、C1-C9 アルキル、C1-C9 アルコキシ、又はC1-C9 アルキルチオである、請求項1記載の化合物。
【請求項11】
G5、G6、G7、G8、及びG9が互いに独立にH、C1-C9 アルキル、C1-C9 アルコキシ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6 が互いに独立にH又はC1-C9 アルキルである、請求項1記載の化合物。
【請求項12】
下記の化合物
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート;
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート;
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-アセテート;
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-アセテート;
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-アセテート;
メチル 2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸;
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸;
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-酢酸;
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-酢酸;
2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸
から選ばれた、請求項1記載の化合物。
【請求項13】
下記の化合物
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ-フェニルチオ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸
から選ばれた、請求項1記載の化合物。
【請求項14】
請求項1から13のいずれか1項記載の化合物を含むことを特徴とする医薬組成物。
【請求項15】
製剤形態が単純錠剤、フィルム被覆錠剤、糖剤、腸被覆錠剤、分散性粉末、カプセル、顆粒、経口溶液又は経口懸濁液を含む群から選ばれる、請求項14記載の医薬組成物。
【請求項16】
ペルオキソーム増殖物質活性化サブタイプδ (PPARδ) を活性化することにより治療又は予防し得る疾患の治療又は予防のための薬物の製造における請求項1から13のいずれか1項記載の化合物の使用。
【請求項17】
前記疾患がメタボリック症候群、肥満、異常脂血症、病的糖血症、インスリン耐性及び腫瘍の一種以上から選ばれる、請求項16記載の使用。
【請求項18】
スキーム1:
中間体化合物II及びIIIをアルカリの作用のもとに一緒にカップリングして、化合物I(エステル)又は化合物I(酸)を生じ、前記アルカリが有機又は無機アルカリであり、その無機アルカリがアルカリ金属炭酸塩、可溶性アルカリ土類金属炭酸塩、炭酸アンモニウム、又はこれらのあらゆる混合物であり、その有機アルカリがトリエチルアミンであり、前記溶媒がアセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、又はこれらのあらゆる混合物であり、反応温度が0-100℃であり、反応時間が1-12時間であり、
【化2】
(式中、
ZはCl又はBrであり、
X、Y、R、R1、R2、R3、R4、G1、G2、G3、G4、G5、G6、G7、G8、及びG9は請求項1に定義されたのと同じである)
又は
スキーム2:
アルカリとしての炭酸塩を用いる連続反応プロセスにおいて、アセトニトリル、テトラヒドロフラン、ジオキサン及びこれらのあらゆる混合物から選ばれた溶媒を使用し、かつ炭酸カリウム、炭酸ナトリウム、炭酸ストロンチウム、炭酸アンモニウム等を含む群から選ばれた炭酸塩を使用して、化合物IIIを最初に化合物IV次いで化合物Vと反応させて、その反応の過程中に中間体を分離しないで化合物I (エステル) を生じる
【化3】
(式中、
ZはCl又はBrであり、
X、Y、R、R1、R2、R3、R4、G1、G2、G3、G4、G5、G6、G7、G8、及びG9は請求項1に定義されたのと同じである)
に記載の工程を含むことを特徴とする、RがC1-C9 アルキルである請求項1記載の化合物の調製方法。
【請求項19】
下記の加水分解条件(使用されるアルカリが水酸化ナトリウム、水酸化リチウム、水酸化カリウム等を含むアルカリ金属水酸化物であり、使用される溶媒系が9 -1:1 (容積/容積)の範囲のアルコール/水の比を有するC1-C4 アルコール-水系、9 -1:1 (容積/容積)の範囲のTHF/水の比を有するテトラヒドロフラン-水系、及び9 -1:9 -1:1 (容積/容積)の範囲のアルコール/ジクロロメタン/水の比を有するアルコール-ジクロロメタン-水系から選ばれ、反応温度が0-80℃、好ましくは20-40℃であり、反応時間が1-12時間、好ましくは2-4 時間である)を使用する化合物I(エステル)のアルカリ性加水分解の工程を含むことを特徴とする、RがHである、請求項1記載の化合物の調製方法。
【請求項20】
式III
【化4】
の化合物。
[式中、
ZはCl又はBrであり、
R3 はH又はC1-C9 アルキルであり、
R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル又は置換フェニルであり、そのフェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6から選ばれ、
G5、G6、G7、G8、及びG9は互いに独立にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6であり、かつ
R5 及びR6 は互いに独立にH又はC1-C9 アルキルである]
【請求項21】
下記の化合物:
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン;
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン; 及び
3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オン
から選ばれた、請求項20記載の化合物。
【請求項22】
下記の工程:
A)式(VI)の化合物を塩素化試薬もしくは臭素化試薬と反応させる工程、
【化5】
[式中、
ZはCl又はBrであり、R3、R4、G5-G9 は請求項20に定義されたのと同じである]
(その反応条件は以下のとおりである:塩素化試薬又は臭素化試薬は
(1) N-ブロモスクシンイミド/トリフェニルホスフィン;
(2) 塩化チオニル又は臭化チオニル;
(3) N-クロロスクシンイミド/トリフェニルホスフィン;
(4) 四塩化炭素又は四臭化炭素/トリフェニルホスフィン;
(5) 五塩化リン又は五臭化リン;
(6) オキシ塩化リン又はオキシ臭化リン; 及び
(7) 三塩化リン又は三臭化リン
を含む群から選ばれ、
溶媒はジクロロメタン、クロロホルム、四塩化炭素、及びこれらのあらゆる混合物から選ばれ、反応温度は40-80℃であり、反応時間は2-8 時間である)
又は
B)式(VII) の化合物を塩素化試薬又は臭素化試薬と反応させて、式III の化合物を得る工程(その反応溶媒はクロロホルム又は四塩化炭素であり、臭素化試薬はN-ブロモスクシンイミドであり、塩素化試薬はN-クロロスクシンイミドであり、触媒はジベンゾイルペルオキシドであり、反応温度は40-80℃であり、反応時間は2-8 時間である)
【化6】
[式中、
ZはCl又はBrであり、
R3, 、G5-G9 は請求項20に定義されたのと同じであり、
G10-G14 は互いに独立に、又は同時にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、
R5 及びR6 は互いに独立にH又はC1-C9 アルキルである]
を含むことを特徴とする、請求項20記載の式III の化合物の調製方法。
【請求項23】
患者への治療又は予防有効量の式(I) の化合物の投与の工程を含む、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患の治療又は予防方法であって、前記疾患がメタボリック症候群、肥満、異常脂血症、病的糖血症、インスリン耐性、老人性痴呆、又は腫瘍等であることを特徴とする前記治療又は予防方法。
【請求項1】
式(I)の化合物又はその医薬上許される塩もしくはこれらの溶媒和物。
【化1】
[式中、
XはO、S、N、又は (CH2)n (式中、nは1から4までの整数である)であり、
YはO、S又はNであり、
RはH又はC1-C9アルキルであり、
R1 及びR2 は互いに独立にH又はC1-C4 アルキルであり、かつR1及びR2 の少なくとも一つがHであり、
R3 はH又はC1-C9 アルキルであり、
R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル又は置換フェニルであり、そのフェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6又はCONR5R6 から選ばれ、
G1、G2、G3、G4、G5、G6、G7、G8及びG9 は互いに独立にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6 、COOR5 、NR5COR6 又はCONR5R6 であり、かつ
R5 及びR6 は独立に互いにH又はC1-C9 アルキルである]
【請求項2】
XがO、S又はCH2である、請求項1記載の化合物。
【請求項3】
YがO又はSである、請求項1記載の化合物。
【請求項4】
RがH、メチル又はエチルである、請求項1記載の化合物。
【請求項5】
R1 がH、メチル又はエチルである、請求項1記載の化合物。
【請求項6】
R2 がH、メチル又はエチルである、請求項1記載の化合物。
【請求項7】
R3 がH又はC1-C4 アルキルである、請求項1記載の化合物。
【請求項8】
R3 がメチルである、請求項1又は7記載の化合物。
【請求項9】
R4 が置換フェニルである場合に、その置換基がC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6 、COOR5 、NR5COR6又はCONR5R6から選ばれ、R5 及びR6 が互いに独立にH又はC1-C9 アルキルである、請求項1記載の化合物。
【請求項10】
G1、G2、G3、及びG4 が互いに独立に、又は同時にH、C1-C9 アルキル、C1-C9 アルコキシ、又はC1-C9 アルキルチオである、請求項1記載の化合物。
【請求項11】
G5、G6、G7、G8、及びG9が互いに独立にH、C1-C9 アルキル、C1-C9 アルコキシ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、R5 及びR6 が互いに独立にH又はC1-C9 アルキルである、請求項1記載の化合物。
【請求項12】
下記の化合物
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート;
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート;
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-アセテート);
エチル 2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-アセテート;
エチル 2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-アセテート;
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-アセテート;
メチル 2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-アセテート;
エチル 2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
エチル 2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-アセテート;
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸;
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸;
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メチルチオ)-フェノキシ)-酢酸;
2-(2-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(3-メチル-4-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル)-メトキシ)-フェニルチオ)-酢酸;
2-メチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸
から選ばれた、請求項1記載の化合物。
【請求項13】
下記の化合物
2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ-フェニルチオ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2-(2-エチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2,2-ジメチル-2-(3-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-メチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(3-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルオキシ)-フェニルチオ)-酢酸;
2,2-ジメチル-2-(2-メチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸;
2-(2,5-ジメチル-4-(1-(3-(4-n-ブチル-5-オキソ-1-(4-トリフルオロメチル-フェニル)-4,5-二水素-1H-1,2,4-トリアゾリル))-ベンジルチオ)-フェノキシ)-酢酸
から選ばれた、請求項1記載の化合物。
【請求項14】
請求項1から13のいずれか1項記載の化合物を含むことを特徴とする医薬組成物。
【請求項15】
製剤形態が単純錠剤、フィルム被覆錠剤、糖剤、腸被覆錠剤、分散性粉末、カプセル、顆粒、経口溶液又は経口懸濁液を含む群から選ばれる、請求項14記載の医薬組成物。
【請求項16】
ペルオキソーム増殖物質活性化サブタイプδ (PPARδ) を活性化することにより治療又は予防し得る疾患の治療又は予防のための薬物の製造における請求項1から13のいずれか1項記載の化合物の使用。
【請求項17】
前記疾患がメタボリック症候群、肥満、異常脂血症、病的糖血症、インスリン耐性及び腫瘍の一種以上から選ばれる、請求項16記載の使用。
【請求項18】
スキーム1:
中間体化合物II及びIIIをアルカリの作用のもとに一緒にカップリングして、化合物I(エステル)又は化合物I(酸)を生じ、前記アルカリが有機又は無機アルカリであり、その無機アルカリがアルカリ金属炭酸塩、可溶性アルカリ土類金属炭酸塩、炭酸アンモニウム、又はこれらのあらゆる混合物であり、その有機アルカリがトリエチルアミンであり、前記溶媒がアセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、又はこれらのあらゆる混合物であり、反応温度が0-100℃であり、反応時間が1-12時間であり、
【化2】
(式中、
ZはCl又はBrであり、
X、Y、R、R1、R2、R3、R4、G1、G2、G3、G4、G5、G6、G7、G8、及びG9は請求項1に定義されたのと同じである)
又は
スキーム2:
アルカリとしての炭酸塩を用いる連続反応プロセスにおいて、アセトニトリル、テトラヒドロフラン、ジオキサン及びこれらのあらゆる混合物から選ばれた溶媒を使用し、かつ炭酸カリウム、炭酸ナトリウム、炭酸ストロンチウム、炭酸アンモニウム等を含む群から選ばれた炭酸塩を使用して、化合物IIIを最初に化合物IV次いで化合物Vと反応させて、その反応の過程中に中間体を分離しないで化合物I (エステル) を生じる
【化3】
(式中、
ZはCl又はBrであり、
X、Y、R、R1、R2、R3、R4、G1、G2、G3、G4、G5、G6、G7、G8、及びG9は請求項1に定義されたのと同じである)
に記載の工程を含むことを特徴とする、RがC1-C9 アルキルである請求項1記載の化合物の調製方法。
【請求項19】
下記の加水分解条件(使用されるアルカリが水酸化ナトリウム、水酸化リチウム、水酸化カリウム等を含むアルカリ金属水酸化物であり、使用される溶媒系が9 -1:1 (容積/容積)の範囲のアルコール/水の比を有するC1-C4 アルコール-水系、9 -1:1 (容積/容積)の範囲のTHF/水の比を有するテトラヒドロフラン-水系、及び9 -1:9 -1:1 (容積/容積)の範囲のアルコール/ジクロロメタン/水の比を有するアルコール-ジクロロメタン-水系から選ばれ、反応温度が0-80℃、好ましくは20-40℃であり、反応時間が1-12時間、好ましくは2-4 時間である)を使用する化合物I(エステル)のアルカリ性加水分解の工程を含むことを特徴とする、RがHである、請求項1記載の化合物の調製方法。
【請求項20】
式III
【化4】
の化合物。
[式中、
ZはCl又はBrであり、
R3 はH又はC1-C9 アルキルであり、
R4 はH、C1-C9 アルキル、C3-C7 シクロアルキル、フェニル又は置換フェニルであり、そのフェニルの置換基はC1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6から選ばれ、
G5、G6、G7、G8、及びG9は互いに独立にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6 又はCONR5R6であり、かつ
R5 及びR6 は互いに独立にH又はC1-C9 アルキルである]
【請求項21】
下記の化合物:
3-(1'-ブロモ-ベンジル)-4-メチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン;
3-(1'-ブロモ-ベンジル)-4-n-ブチル-1-(4-トリフルオロメチル) フェニル-1H-1,2,4-トリアゾール-5(4H)-オン; 及び
3-ブロモメチル-4-メチル-1-(4-トリフルオロメチル-フェニル)-1, 4-二水素-1,2,4-トリアゾール-5-オン
から選ばれた、請求項20記載の化合物。
【請求項22】
下記の工程:
A)式(VI)の化合物を塩素化試薬もしくは臭素化試薬と反応させる工程、
【化5】
[式中、
ZはCl又はBrであり、R3、R4、G5-G9 は請求項20に定義されたのと同じである]
(その反応条件は以下のとおりである:塩素化試薬又は臭素化試薬は
(1) N-ブロモスクシンイミド/トリフェニルホスフィン;
(2) 塩化チオニル又は臭化チオニル;
(3) N-クロロスクシンイミド/トリフェニルホスフィン;
(4) 四塩化炭素又は四臭化炭素/トリフェニルホスフィン;
(5) 五塩化リン又は五臭化リン;
(6) オキシ塩化リン又はオキシ臭化リン; 及び
(7) 三塩化リン又は三臭化リン
を含む群から選ばれ、
溶媒はジクロロメタン、クロロホルム、四塩化炭素、及びこれらのあらゆる混合物から選ばれ、反応温度は40-80℃であり、反応時間は2-8 時間である)
又は
B)式(VII) の化合物を塩素化試薬又は臭素化試薬と反応させて、式III の化合物を得る工程(その反応溶媒はクロロホルム又は四塩化炭素であり、臭素化試薬はN-ブロモスクシンイミドであり、塩素化試薬はN-クロロスクシンイミドであり、触媒はジベンゾイルペルオキシドであり、反応温度は40-80℃であり、反応時間は2-8 時間である)
【化6】
[式中、
ZはCl又はBrであり、
R3, 、G5-G9 は請求項20に定義されたのと同じであり、
G10-G14 は互いに独立に、又は同時にH、C1-C9 アルキル、ヒドロキシル、C1-C9 アルコキシ、メルカプト、C1-C9 アルキルチオ、トリフルオロメチル、F、Cl、Br、ニトロ、NR5R6、COOR5、NR5COR6、又はCONR5R6であり、
R5 及びR6 は互いに独立にH又はC1-C9 アルキルである]
を含むことを特徴とする、請求項20記載の式III の化合物の調製方法。
【請求項23】
患者への治療又は予防有効量の式(I) の化合物の投与の工程を含む、ペルオキシソーム増殖物質活性化受容体サブタイプδ(PPARδ)を活性化することにより治療又は予防し得る疾患の治療又は予防方法であって、前記疾患がメタボリック症候群、肥満、異常脂血症、病的糖血症、インスリン耐性、老人性痴呆、又は腫瘍等であることを特徴とする前記治療又は予防方法。
【図1】

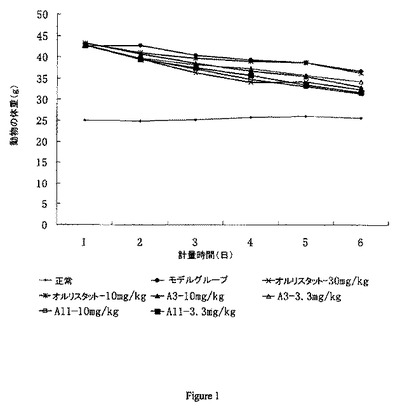
【図2】
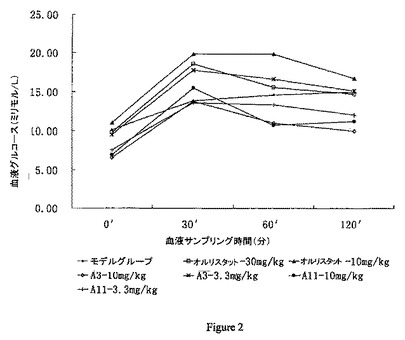
【図3】
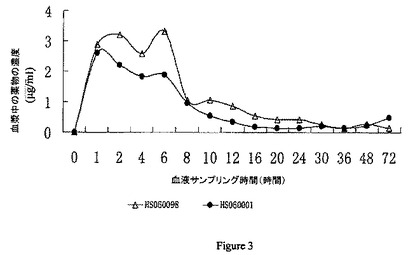
【図2】
【図3】
【公表番号】特表2011−529925(P2011−529925A)
【公表日】平成23年12月15日(2011.12.15)
【国際特許分類】
【出願番号】特願2011−521428(P2011−521428)
【出願日】平成21年8月7日(2009.8.7)
【国際出願番号】PCT/CN2009/073140
【国際公開番号】WO2010/015212
【国際公開日】平成22年2月11日(2010.2.11)
【出願人】(510116819)浙江海正薬業股▲ふん▼有限公司 (4)
【氏名又は名称原語表記】Zhejiang Hisun Pharmaceutical CO.,LTD.
【住所又は居所原語表記】46 Waisha Road,Jiaojiang District,Taizhou City,Zhejiang,318000,P.R.China
【Fターム(参考)】
【公表日】平成23年12月15日(2011.12.15)
【国際特許分類】
【出願日】平成21年8月7日(2009.8.7)
【国際出願番号】PCT/CN2009/073140
【国際公開番号】WO2010/015212
【国際公開日】平成22年2月11日(2010.2.11)
【出願人】(510116819)浙江海正薬業股▲ふん▼有限公司 (4)
【氏名又は名称原語表記】Zhejiang Hisun Pharmaceutical CO.,LTD.
【住所又は居所原語表記】46 Waisha Road,Jiaojiang District,Taizhou City,Zhejiang,318000,P.R.China
【Fターム(参考)】
[ Back to top ]