
ペレット製剤
高剪断造粒によってペレットを製造する方法であって、該ペレットはpH依存的水溶性を有する医薬としての有効成分を含み、該ペレットは該方法を用いて得られ、該ペレットを含む医薬としての経口剤形が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、水におけるpH依存的溶解性を有する化合物を含むペレットを製造する方法、及び該方法で得られるペレットに関する。さらに、本発明は、該ペレットを含む経口剤形に関する。
【0002】
さらに、本発明はまた(強力な)pH依存的水溶性を有する有効成分を含む改善された放出プロフィールを有するペレットの形体であるか、又はそれから製造された固体医薬製剤に関し、特に、該有効成分は低水溶性の弱塩基である。
【0003】
医薬としての有効成分は、種々の投薬量で投与されることが必要とされ、カプセル又はサシェなどの剤形又は複数回単位剤形を用いるために適切であり得る。これらの剤形は、適切な担体に調合された必要量の有効成分を含む。
【0004】
ペレットは、複数回単位剤形として一般には定義され、1回単位剤形と比較して、有効性及び安全性などの多数の治療的利点を有すると考えられている。また、ペレットは、それに含まれる有効成分の放出パターンに影響を及ぼすように、適した被覆材料で被覆されてもよい。
【0005】
定常なかつ調節可能な放出を有するために、ペレットは、定常形状として、より具体的には規則的な形状の球として与えられることを必要とする。
【0006】
ペレットからの有効成分の放出を支配する重要因子は、有効成分が放出される媒体と接触する表面積である。不規則な形状のペレットは不規則な表面を有し、結果として有効成分の放出の不規則性を与える。
【0007】
これに関して、粒子の平均径はまた、有効成分の放出とペレットの溶解において重要な役割を果たす。
【0008】
有効成分の放出は、画定された平均径を有する規則的に形状化されたペレットを用いて良好に制御可能である。また、ペレットの多孔性及び粗さは、有効成分の放出、並びにペレッの操作及び実施可能性に関連性のある物理的特徴に影響を及ぼすパラメータでもある。
【0009】
球形のペレットは容易に被覆することができ、より均一な被覆厚は、ペレットが規則的な円形を有する場合に達成され得る。これは、ペレットのサイズ分布が狭い場合にさらにより顕著である。
【0010】
さらに、球形のペレットは、容易に操作され、カプセル、サシェ、又は複数回単位錠剤などの他の応用形態に充填される。
【0011】
したがって、製薬産業において使用されるペレットは、一般に自由流動的であり、狭い粒子サイズ分布を有する0.3〜2.00mmの粒径、及び約10%の多孔性を有する球状粒子である。
【0012】
医薬技術分野における技術状態によれば、このようなペレットの製造に適したプロセス及び装置がいくつかある(Isaac Ghebre−Sellassie: Pharmaceutical Peptization Technology,Marcel Dekker,Inc.,New York,Basel,1989)。
【0013】
ペレットの製造用に開発された最初のプロセスは、いわゆる層化法である。このプロセスでは、非常に良質な球状ペレットが、いくつかの被覆層によって漸次的に完成されたコアを提供することによって調製可能である。コアは、通常、不活性な材料、例えば、糖、スターチ、塩化ナトリウム粒子又はそれらの混合物、及び有効成分を含む。
【0014】
ペレット表面に与えられる小さな剪断力により、上記の最初のプロセスによれば、得られたペレットの表面の滑らかさは満足できない場合がある。結果として、ペレットは互いに結合し、高い比率で廃棄されることになる。層化法のプロセス時間は通常長い。
【0015】
第2に、ペレットの製造に適したより重要なプロセスは、いわゆる押出−球形化プロセスである。このプロセスでは、有効成分及び必要とされる賦形剤は、例えば、ともに粉末形態であり、一緒に混合されて粉末混合物を形成する;適切に均質になった粉末混合物を液体と混合し、均質な湿性塊が得られるまで練られる。この塊を押出し、押出プロセスから得られた押出物は球形にされる(球状形態にされる)。最後に、球状段階から得られた生のペレットを乾燥する。ペレットの質は、主に、押出し及び球形化のプロセス変数によって決定される。押出−球形化プロセスは、例えば、EP1252886及びWO2007/135470に開示されている。
【0016】
押出/球形化によって調製された、微結晶性セルロース(MCC)、テオフィリン及び様々な範囲のレベルであるアルギン酸ナトリウム(すなわち、10〜50%w/w)を含むペレットが知られている(Sriamomsak et al.European Journal of Pharmaceutics and Biopharmaceutics 69(2008)274−284)。したがって、2種のアルギン酸ナトリウムが、酢酸カルシウム又は炭酸カルシウム(0、0.3、3及び10%w/w)を添加して、及び添加しないで評価された。ペレット特性、例えば、サイズ、形状、形態及び薬物放出挙動に及ぼすアルギン酸ナトリウム及びカルシウム塩の量及び種類の効果が調査された。結果は、アルギン酸ナトリウム及びカルシウム塩の量が得られたペレットのサイズ及び形状に影響を及ぼすことを示した。しかしながら、異なる種類のアルギン酸ナトリウム及びカルシウム塩は、異なる程度で修飾に応答した。走査型電子顕微鏡写真に見られるように、ペレット構造において空洞が観察され、これは、球形化プロセスに関与する力に起因していた。大部分のペレット製剤は、60分以内に約75〜85%テオフィリンを放出した。ペレット製剤へのカルシウム塩の組み込みは、使用されたカルシウム塩の溶解性に依存して薬物放出を変更した。
【0017】
ペレットの製造に適した更なるプロセス(ペレット化)は、当該技術分野において知られ、例えば、高剪断ミキサーにおけるペレットの製造が挙げられる。
【0018】
高剪断ミキサーペレット化プロセスは異なる工程:粉末の均質化、造粒、及び乾燥を伴う。その後のペレットのプライマー核は、バインダースプレイ及び撹拌中の分散によって形成される。ペレットは、最終的にふるいにかけることができる。
【0019】
高剪断ミキサーにおけるペレットの形成は多変量プロセスであり、したがって、このようなプロセスにとってプロセス変数を特定し、調節することは重要である。特に、製造物特性は、プロセス変数、例えば、羽根速度及び混練時間の変化に感受性である。したがって、これらのパラメータの最適化はプロセスにとって重要である。
【0020】
大きすぎる粒子の生成を避けるために、適切な撹拌を必要とする。さらに、バインダー液体流速はまた、ペレットの質に影響を及ぼす重要なパラメータであり得る。それは、塊の緻密化が混合及びスプレイ後に行われるためである。ペレット凝集の工程内調節は重要であり、したがって、いくつかのプロセス指標が追随する必要があり、顆粒形成の終点を決定する方法は集中的な調査を必要とする。
【0021】
さらに、多数の有効成分は有効成分の一時的な過剰又は過少投薬を避けるために、特定の放出動力学、例えば、消化管の特定の部分における有効成分の迅速かつ選択的な放出を必要とする。
【0022】
これに関連して、生物薬剤学的分類システム(Amidon et al.,1995;Dressman et al.,1998,2001)のクラスIIに属する有効成分の製剤は、一般に、非常に挑戦的であり、それは、経口生物学的利用能が、消化(GI)管における溶解速度によって決定されるためである。
【0023】
低溶解性又は溶解速度は、多くの場合、pH依存的又は水難溶性薬物のGI管からの吸収において律速段階となることは一般に認められている。これは経口生物学的利用能を危うくする。それは、溶液中の薬物濃度が生物学的膜を通過する大部分の医薬としての有効成分の吸収のために駆動力であるからである。したがって、経口投与後、pH依存的な溶解速度又は水難溶性薬物の増大は、現代薬剤学の最も挑戦的側面の1つである。
【0024】
FDAによって確立された生物学的同等性要件によれば、低溶解性を有する有効成分は、水に対して5mg/mlよりも低い溶解性を有するものと考えられている(Fed.Reg.In 21 CFR,Ch 1;(4/1/87 Ed.)Part 32;320)。
【0025】
さらに、生物学的同等性要件に係る低溶解性を有する弱塩基性の有効成分は、通常、pH依存的な水溶解性を有する。これらの成分は、通常、低pH値で相対的に良好な溶解性を示す。より高いpH(例えば、pH6.8)では、これらの薬物の溶解性は5mg/mlよりも低い。化合物のpH依存的溶解性により、これらの薬物の医薬製剤は、通常、pH依存的溶解挙動を示す。
【0026】
しかしながら、低溶解性有効成分について迅速又は長期のpH非依存的放出を可能にする医薬製剤は、製剤分野において非常に望ましい。
【発明の概要】
【課題を解決するための手段】
【0027】
本発明は、医薬としての有効成分、及びアルギン酸塩を含む固体ペレットを提供すること目的とし、平均フェレ(Feret)径が約300〜800μmであり、破砕強度が約4〜10Nであり、縦横比が約1.0〜1.2であることによって特徴付けられる。
【0028】
本発明の更なる目的は、高剪断造粒によって、医薬としての有効成分及びアルギン酸塩を含む固体ペレットを製造するための方法を提供することであり、該方法は、
−医薬としての有効成分及び必要とされる賦形剤の各粉末が高剪断ミキサーボウルに入れられ、混合して粉末混合物を形成する混合段階、
−造粒液として塩化カルシウム溶液の粉末混合物に添加して開始され、造粒塊が得られる造粒段階、
−該造粒塊がペレットの製造に伴って羽根車(impeller)によって球形化される球形化段階、
−乾燥段階、及び
−最終ふるい段階
を含む。
【0029】
本発明に係るペレットに含まれる有効成分は、特に、一般的に強いpH依存的溶解性を有する有効成分であり、例えば、pHが約3で良好な溶解性を有するが、pHが約6.8又はそれよりも高い場合に5mg/mlより低い溶解性を有する有効成分である。より具体的には、該有効成分は塩基であり、例えば、pKaが約8.5又はそれよりも高い弱塩基である。
【0030】
本発明に係るペレットは、好ましくは、例えば、アルギン酸、アルギン酸ナトリウム、アルギン酸カリウム、アルギン酸アンモニウム、アルギン酸カルシウム、アルギン酸マグネシウム又はそれらの混合物の形態であるアルギン酸塩を含む。アルギン酸ナトリウム及びアルギン酸カルシウムが最も好ましい。
【0031】
本発明に係る固体ペレットは、好ましくは、平均フェレ径が約300〜800μmであり、破砕強度が約4〜8Nである。
【0032】
本発明に係る方法では、混合段階は、有効成分、アルギン酸塩及び任意の他の賦形剤を高剪断ミキサーボウル、例えば、ガラス製又はステンレス製ボウル中で混合することによって行われる。粉末は、約900〜約1100rpmの羽根回転、約900〜約1100rpmのチョッパー回転で約2〜6分間混合される。
【0033】
約900ml体積のボウル、及び約100gの粉末の充填を用いて、羽根回転は、好ましくは約1000rpm、チョッパー回転は約1000rpmの約3分間である。
【0034】
造粒液はまたバインダーとして画定され、造粒段階中、粉末に添加される。本発明によれば、バインダーは、水又は水溶液であり、好ましくは塩化カルシウム溶液又は同等のカルシウム塩の溶液である。該溶液は、通常、約3〜約15%v/v、好ましくは約5〜約10%v/vの塩化カルシウム濃度を有する。塩化カルシウムの代わりに、対応する必要量の同等の医薬として許容されるカルシウム塩を用いることができる。
【0035】
各製剤に必要とされるバインダーの総量は、ボウル内の温度及び羽根、並びにカルシウム塩及び最終のペレット組成物の濃度のようなプロセス変数に依存する。
【0036】
本発明に係るプロセスのために上記で与えられた条件について、約100gの充填では、バインダーの必要な絶対量は、室温で約90〜約130mlであり、さらにボウルを加熱する。加熱されたボウルの温度は約40℃〜約50℃である。バインダーのスプレイ速度は、すぐに、粉末混合物に対して、スプレイされた結合溶液の体積を決定し、通常、上記条件によれば、約8〜30ml/分である。
【0037】
本発明の高剪断造粒プロセスによれば、造粒中、チョッパー速度は一定に保持され、該速度は、約2800〜3200rpmであり、好ましくは3000rpmである。羽根速度は1200〜1400rpmで変化し、好ましくは1300rpmである。
【0038】
球形化段階は、約4〜6分間、ペレットを操作することによって造粒後に行われ、この場合、羽根速度は約450〜約600rpmであり、好ましくは500rpmである。本発明の態様の具体的な形態によれば、球形化段階ではチョッパーは使用されない。
【0039】
続く乾燥段階は、例えば、約5〜20分間、好ましくは15分間、約40〜約55℃の温度(製造物温度)で流動床コーターを用いて行われる。
【0040】
最終的に、ペレットをふるいにかける。平均フェレ径が約300〜800μm、より好ましくは約400〜600μmのペレットサイズ画分は、許容されるペレットサイズ範囲として考えられる。
【0041】
本発明の意味における固体ペレットは、空洞がないペレットである。
【0042】
本発明に係るペレットは、好ましくは水系で、高剪断造粒によって製造される。例えば、押出/球形化によって調製された従来のペレットと比較して、本発明に係るペレットは固体であり、調節された粗さ、並びに低多孔性及び高破砕強度を有する表面を含む。これらの特徴は、ペレットの改善された機械的安定性を与え、それらの均質で安定な被覆を可能にする。さらに、それらは固体であるため、多量の有効成分が該ペレットに充填され得る。
【0043】
上述したように、本発明の態様の具体的形態によれば、ペレットは被覆される。典型的な被覆は、例えば、ポリ酢酸ビニル/ポリビニルピロリドン共重合体(Kollicoat SR 30D)である。
【0044】
本発明に係るペレットの小さい粒子サイズは、カプセルあたりより多量のペレットを有するカプセルを有意に満たすことを可能にし、カプセルあたりの投薬量変動性を減少させる効果を有する。小さいサイズのペレットの更なる利点は、小児科の医薬製剤を製造することができる点である。
【0045】
本発明のペレットは、ペレットに含まれる医薬としての有効成分の放出を改善する。特に、本発明に係るペレットに含まれる有効成分のpH非依存的放出は、本発明に従って達成される。
【0046】
該有効成分は、例えば、強いpH依存的可溶性を有し、例えば、有効成分は、低pH、例えば、pHが約3で良好な溶解性を有するが、高いpH、例えば、約6.8又はそれより高いpHでは5mg/mlより低い溶解性を有する。
【0047】
態様の特定の形態によれば、該有効成分は弱塩基であり、pKaは約8.5又はそれよりも高い。この効果は、溶解媒体のpHが約6.8又はそれより高い場合に特に現れ、該弱塩基は、例えば、塩酸バルデナフィル又は塩酸ベラパミルの場合のような水における低溶解性を有する。
【0048】
強力なpH依存的溶解性が弱塩基性であり、pKaが約8.5又はそれよりも高い有効成分はまた考慮され、好ましくは本発明に係るペレットに含まれ得る。
【0049】
本発明に係るペレットに好ましくは含まれ得る医薬としての有効成分は、例えば、ベータ遮断薬、例えばプロプラノロール、メトプロロール及びアテノロール、カルシウムアンタゴニスト、例えばジルチアゼ及びベラパミル、抗微生物薬、例えばセファレキシン、セファクロル、抗ヒスタミン薬、例えばクロルフェニラミン、シンナリジン、ジフェンヒドラミン、鎮痛薬、例えばジアゼパム、又は抗精神病薬、例えばクロルプロマジン、フルフェナジン、並びに任意の医薬として許容されるその塩が含まれる。
【0050】
医薬としての有効成分についての溶解速度の改善は、アルギン酸塩の使用によって得られる効果である。ペレットにおけるアルギン酸塩成分は、低pH値(例えば、pH=3)より高いpH値(例えば、pH=6.8)で良好に安定である。これは、ペレットの緩徐な崩壊に起因して、医薬としての有効成分としての弱塩基の溶解が低pH(例えば、p=3)で遅れるという効果を有し、一方、これは、有効成分の溶解及びペレットの迅速崩壊によって、高pH値(例えば、pH=6.8)で改善される。
【0051】
アルギン酸塩の使用は、溶解が通常は制限される高pH値(例えば、pH=6.8)で、有効成分の溶解速度を改善することができる。
【0052】
改善された溶解速度の結果として、有効成分の良好な生物学的利用能はまた本発明に係るペレットによって達成される。
【0053】
本発明に係るペレットは、他の賦形剤、例えばマトリックスビルダー又は充填剤、例えば、スクロース、マンニトール、ラクトース、デキストロース及びソルビトールを含む群から選択される水溶性非イオン性基質をさらに含むことができる。
【0054】
ペレットの機械的強度に影響を及ぼす追加の配合剤としてのセルロース又はセルロース誘導体はまた、本発明に係るペレットにおいて使用され得る。微結晶性セルロース(MCC)が特に好都合である。
【0055】
本発明に係るペレットは、例えば、約1%〜40%w/wの医薬としての有効成分からなり、残りの60〜99%が10%〜80%のアルギン酸塩、及び20%〜90%の充填剤及び/又はマトリックスビルダーで作られている。
【0056】
以下の実施例は、例証を目的として提供され、限定されない。要約すると、本発明のペレットは、上記で開示された一般的方法によって調製され得て、一方、以下の実施例は、本発明の態様の典型的な形態の調製を開示している。
【図面の簡単な説明】
【0057】
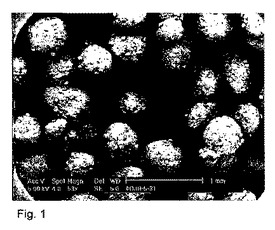
【図1】造粒液中の5%塩化カルシウム及び8ml/分のスプレイ速度で調製されたペレットのSEM(走査型電子顕微鏡)顕微鏡写真(製剤番号7)。
【図2】造粒液中の5%塩化カルシウム及び10ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号8)。
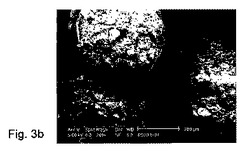
【図3】造粒液中の5%塩化カルシウム及び20ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号10)。
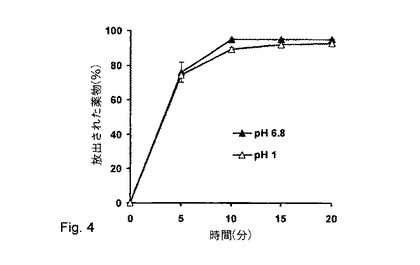
【図4】アルギン酸塩(Protanal LF 120M)マトリックスペレットからの塩酸ベラパミルのpH非依存的放出(製剤番号10)。
【図5】造粒液中の10%塩化カルシウム及び20ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号16)。
【図6】造粒/球形化プロセス中に加熱された(40℃)容器中に調製されたペレットのSEM顕微鏡写真(製剤番号14)。
【図7】造粒液中の20%塩化カルシウム及び20ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号17)。
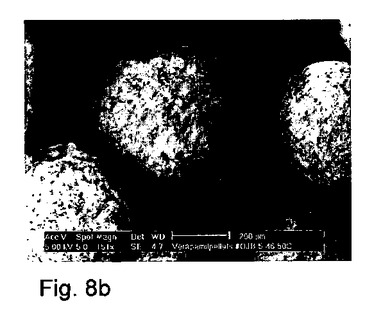
【図8】造粒/球形化プロセス中に加熱された(50℃)容器中に調製されたペレットのSEM顕微鏡写真(製剤番号14)。
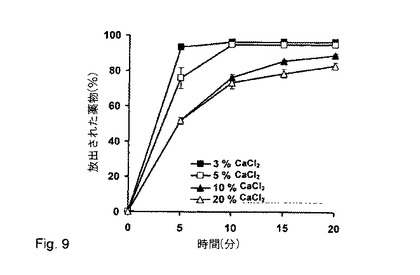
【図9】リン酸緩衝液pH6.8中の塩酸ベラパミルの放出に対する塩化カルシウム濃度の効果(製剤番号4、10、16、17)。
【図10】造粒液として水及び20ml/分のスプレイ速度で調製された純粋MCCペレットのSEM顕微鏡写真(製剤番号23)。
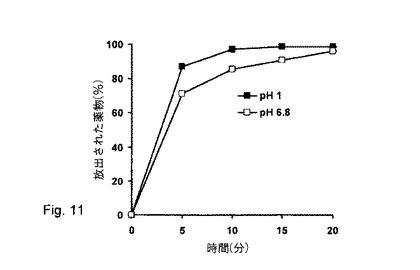
【図11】純粋MCCペレットからの塩酸ベラパミルのpH依存的薬物放出(製剤番号23)。
【図12】造粒液中の5%塩化カルシウム及び20ml/分のスプレイ速度で調製された塩酸バルデナフィル含有ペレットのSEM顕微鏡写真(製剤番号25)。
【図13】従来のMCC(微結晶性セルロース)ペレットと対比したアルギン酸塩/MCCペレットからのpH6.8での非常にpH依存的な可溶性化合物(塩酸バルデナフィル)の放出(製剤番号24及び25)。
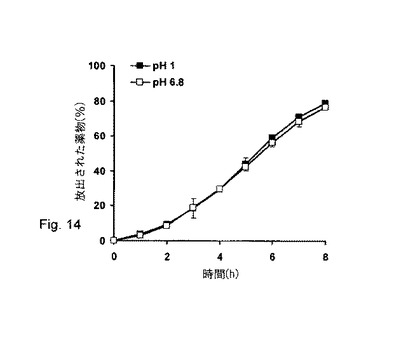
【図14】アルギン酸ナトリウム/MCCからなる被覆されたペレット(1日、60℃にて硬化)からのベラパミルHClのpH非依存的薬物放出。
【発明を実施するための形態】
【0058】
特徴付け方法
収量:
収量は、100、250、355、630、710、800、900、1000、1400μmのふるい器具を用いたふるい分析(Retsch GmbH,Haan,Germany)によって決定された。ペレットサイズ画分の355〜630μmは、使用に適した収量として決定された。
【0059】
pKa:
pKa又はイオン化定数は、化合物の中性及び帯電した形態の平衡係数の負の対数として定義される。典型的には、滴定法において、化合物は、その中性形態に向けて滴定される。
【0060】
難溶性化合物のpKaは、水溶性メタノール溶液において測定され得る。いくつかの滴定が、様々な比率のメタノール:水を用いて行われる場合、Yeasuda−Shedlovsky式は、純粋水溶液における理論的pKaを表すことができる(Avdeef A,Comer J.E.A,Thomson,S.J.“pH−Metric Logp 3.Glass Electrode Calibration in Methanol−Water Applied to pKa Determination of Water Insoluble Subatances by Potentiometric Titration”Anal.Chem.1993,65,pp42−49)。
【0061】
縦横比及び平均フェレ径:
縦横比yA(0<yA≦1)は、最大フェレ径に対する最小フェレ径の比率yA=xフェレ最小/xフェレ最大によって定義される。それは、粒子の伸長の指標を与える。また、いくつかの文献は、球形の定義として1/yAを用いた。フェレ径は、ノギスを用いた粒子の投影面積から推定される。一般に、それは、任意角で粒子の2つの平衡な接線間の距離として定義される。実際には、最小xF,最小及び最大フェレ径xF,最大>、平均フェレ径:
【0062】
【化1】

【0063】
並びに最大及び最小フェレ径の方法に対して90°で得られたフェレ径xF,最大90が用いられる。最小フェレ径は、多くの場合、ふるい分析に同等な径として用いられる。
【0064】
縦横比及び平均フェレ径の両方は、画像分析によって決定された。355〜630μm(高剪断造粒プロセスを通じて調製されたペレット)と1000〜1400μm(押出/球形化プロセスを通じて調製されたペレット)のふるい画分を画像分析に用いた。画像分析は、ビデオカメラ(HV−T20,Hitachi Kokusai Electric Europe GmbH,Erkrath,Germany)を組み合わせた光学顕微鏡(Olympus BX 50,Olympus Deutschland GmbH,Hamburg,Germany)を用いて行われた。ペレットサイズ及び形状測定について、各試料の500を超えるペレットをスライドガラス上で調製し、スキャンし、ビデオカメラによった撮像した。デジタル画像加工は、「Software Analysis five」(Olympus Soft Imaging Solutions GmbH,Munster,Germany)を用いて行われた。各ペレットについて、35子のフェレ径を測定し、平均フェレ径の計算に使用した。最大フェレ径と、その最大フェレ径に直交したフェレ径の比率を縦横比として用いた。
【0065】
多孔性:
ペレットの多孔性(ε)は、式(1):
【0066】
【化2】
【0067】
に従って、ガス比重びん密度(gass pycnometric density)及び水銀ポロシメーターから計算され得る。
【0068】
ペレットのガス比重びん密度(ρg)は、ヘリウムピクノメーター(pycnometer)(Ultrapyk 1000T,Quantachrome,Odelzhausen,Germany)によって決定された。各試験されたペレットバッチについて、3つの試料を分析した。
【0069】
ペレットの見掛け密度(ρg)は、水銀ポロシメーター(Pascal 140 & 440,Fisons−Carlo Erba,Valencia,USA)によって評価された。
【0070】
機械的特性(破砕強度):
15個のペレットの破砕強度は、テクスチャ分析機(TAXT plus,Stable Micro Systems,Godalming,Surrey UK)を用いることによって調査された。各ペレットは、分析機のフラットプレートと上部パンチとの間に置かれた。次に、径が2mmであるパンチを0.1mm/秒の速度で下げた。ペレットが破砕された時点は、最初のピークとして力−時間グラフ上に示される。力−時間プロットは、テクスチャ分析機ソフトウェアExponentを用いて記録された。破砕力の算術平均は、破砕強度として用いられた。
【0071】
走査型電子顕微鏡(SEM)写真:
ペレットは、アルゴン雰囲気下、金−パラジウム(MED 020,Bal−tec AG,Liechtenstein,Germany)を用いて60秒間被覆され、次に走査型電子顕微鏡(SEM)(DSM 982,Zeiss,Oberkochen,Germany)を用いて観察された。
【0072】
薬物放出:
塩酸ベラパミルのインビトロ放出は、31USP回転パドル法(900mlの0.1N HCl又はUSPリン酸緩衝液pH6.8;37℃;75rpm;n=3;Distek Premiere 5100 Dissolution System,Distek Inc.,North Brunswisk,USA)を用いて決定された。所定時間間隔で、10ml試料を採取し(置換ではない)、ろ過し、アッセイした。放出された有効成分の量は、USP31法に従って、HPLCによって測定された。塩酸バルデナフィルの溶解は、75rpmの31USP回転パドル法を用いることによって、01%ラウリル硫酸ナトリウムを含む900mLのリン酸緩衝液pH6.8(10%)中で行われた。所定時間間隔で、10ml試料を採取し(置換ではない)、ろ過し、アッセイした。放出された有効成分の量は、HPLCによって決定された。
【0073】
共焦点レーザー走査顕微鏡(CLSM):
CLSM法は、ペレット表面の粗さ値(Ra値)を決定するために使用された。この方法は、例えば、F.Depypere et al.,Quantification of microparticle coating quality by confocal laser scanning microscopy(CLSM),Eur.J.Pharm.Biopharm.(2009)に報告されている。
【0074】
高剪断造粒:
900mlのガラス製ボウル、3つの羽根、及びチョッパーを備えた小スケールの実験室用高剪断ミキサー(Pro−C− ept 4M8,Zelzate,Belgium)においてペレットを製造した。有効成分及び必要とされる賦形剤は、例えば、ともに粉末形態であって、高剪断ミキサーボウルに入れられ、1000rpmの羽根回転、及び1000rpmのチョッパー回転で一緒に混合され、粉末混合物を形成した。造粒は、造粒液として塩化カルシウム溶液又は水の添加により開始され、湿性粉末塊を形成した。1mmの開口を有するチューブを用いて、ドシマット(dosimat)(765 Dosimat,Metrohm Ltd.,Hensau,Switzerland)を用いて液体放出の速度を調節した。各製剤について必要とされる造粒液の全量は、プロセス変数とペレットの組成に依存していた。造粒中、選ばれた羽根速度は1300rpmであり、チョッパー速度は3000rpmで一定に保持した。造粒後、ペレットは、500rpmの羽根速度(チョッパーなし)を用いて5分間球形化された。流動床を用いて、10分間、40℃の製造物温度でペレットを乾燥させた。ペレットをふるいにかけ、355〜630μmのペレットサイズの画分を回収した。
【0075】
実験番号14の例では、ガラス製バイアルを室温で用い、造粒中に加熱した。この実験で用いられた2つの温度は40℃と50℃であった。
【0076】
高剪断造粒
粉末混合物の組成(表1〜4):
20.0gの塩酸ベラパミル
49.5gの微結晶性セルロース(Avicel PH101)
30.5gのアルギン酸ナトリウム(Protanal LF 120M)
【0077】
【表1】
【0078】
表1は、どのようにして造粒液中の全量の増加が、3%で安定な造粒液の塩化カルシウム含量を用いて、高い破砕強度を有するペレットをもたらしたかを示す。これは、材料のより大きな柔軟性(plasticity)に起因し、より低い多孔率を有するより密度の高いペレットを導く。
【0079】
低い全量の造粒液と高い全量の造粒液を用いて製造された製剤の多孔性値はそれぞれ約35%と10%であった。これは、造粒液の増加がより低い多孔率を有するペレットの密度を高めるようにするという仮説と良好な一致を示す。結果として、より低い全量の造粒液を有するペレットは、微結晶性セルロース、Celletsで構成される市販のペレットと比較すると、より低い破砕強度によって特徴付けられた(3.8N)。しかしながら、より高い全量の造粒液で調製された製剤は、7.4Nより高い破砕強度を有する良好な硬度を示した。
【0080】
【表2】
【0081】
【表3】
【0082】
表2及び3は、造粒液中の塩化カルシウム濃度の増加がペレット形状を改善し、塊の望ましくない画分を減少させ、これは、主要な架橋プロセスによって引き起こされたアルギン酸ナトリウムのより低い接着特性に起因していることを示す。ペレットは許容できると考えられ、それは、縦横比の中央値の値全てが、1.2を下回り、約72.5〜77.5%というより高い収率であったためであった(製剤番号7〜16)。破砕強度は4.7N〜8.7Nで変化し、それは、どんなスプレイ速度が適用されても、全ての製剤の良好な硬度を示す。良好な硬度は、アルギン酸塩とカルシウムイオンとの間の改善された架橋の結果であった。
【0083】
MCCとアルギン酸ナトリウムを含むペレットからの塩酸ベラパミルの薬物放出は、1〜6.8の範囲でほぼpHに非依存的であった(図4、製剤番号10)。アルギン酸塩(Protanal(登録商標)LF 120M)は、逆溶解性により、薬物の溶解度差を弱めることができた。アルギン酸ナトリウムは低pHレベルでよりゆっくりと溶解し、これは、アルギン酸塩が、拡散障壁を作る難溶性アルギン酸の形態で沈殿するためである。リン酸緩衝液中の最速薬物放出は、カルシウムイオンとのナトリウムイオン交換によって影響を受け、このようにして、分解するペレット構造を作製した。
【0084】
【表4】
【0085】
20%の塩化カルシウムを用いて調製されたペレットは、粉末混合物及びスプレイ造粒液から得られた湿性粉末塊の非最適化剛性/柔軟性により、表面構造に亀裂を示した。高い剛性画分は粉末塊の結合特性を減少させ、結果として、ペレットは不安定になった。したがって、5%と10%の塩化カルシウム濃度で調製されたペレットは、それらの特性に関して最適であると考えられた。
【0086】
図9に示され得るように(製剤番号4、10、16、17)、造粒液中のカルシウム濃度は、リン酸緩衝液pH6.8において薬物放出速度に影響を及ぼした。放出プロフィールを比較すると、塩化カルシウム濃度を増加させた場合、薬物放出速度が減少したことは明らかである。より低い放出速度は、ペレットの膨潤比を制限する高分子鎖の可動性における制限の増加と結び付けられる。
【0087】
粉末混合物の組成(表5):
20.0gの塩酸ベラパミル
40.0gの微結晶性セルロース(Avicel PH101)
40.0gのアルギン酸ナトリウム(Protanal LF 120M)
【0088】
【表5】
【0089】
許容され得る収率が54.5〜74.8%である、高剪断造粒機におけるペレット製造は、アルギン酸塩の量が増加したとしても可能である。ペレットは5.6〜7.7Nの間である良好な硬度によって特徴付けられるが、1.2と1.29の範囲を超える高い縦横比が得られた。これは、水との接触で膨潤する高い量のアルギン酸塩に由来する。このようにして、造粒塊の柔軟性/剛性は球体を形成するには最適ではなかった。
【0090】
粉末混合物の組成(表6):
20.0gの塩酸ベラパミル
60.0gの微結晶性セルロース(Avicel PH101)
20.0gのラクトース一水和物(Granulac 200)
【0091】
【表6】
【0092】
MCCに基づくペレットの特徴は、アルギン酸ナトリウムを含むペレットと同程度である(製剤番号7〜16)。
【0093】
しかし、塩酸ベラパミル放出は、pH1で測定した薬物放出速度と比較すると、pH6.8で僅かに遅く、これは、薬物の弱塩基性によって説明され得る(図11)。
【0094】
粉末混合物の組成(表7):
20.0gの塩酸バルデナフィル
60.0gの微結晶性セルロース(Avicel PH101)
20.0gのラクトース一水和物
【0095】
【表7】
【0096】
粉末混合物の組成(表8):
20.0gの塩酸バルデナフィル
49.5gの微結晶性セルロース(Avicel PH101)
30.5gのアルギン酸ナトリウム(Protanal LF 120M)
【0097】
【表8】
【0098】
塩酸ベラパミルペレットの特徴は、塩酸ベラパミルペレットと同等である。製剤のアルギン酸ナトリウムの使用は収率を増大させ、塩酸バルデナフィルを含むペレットを改善した。
【0099】
純粋なMCC系ペレットからの塩酸バルデナフィルの薬物放出は、pH6.8でアルギン酸ナトリウム/MCC系ペレットからの薬物放出と比較して非常に低かった(図13)。アルギン酸ナトリウムはより高いpH環境でより溶解性であり、したがって、それは、この環境における難溶性塩酸バルデナフィルの薬物放出を増加させた。
【0100】
押出/球形化
粉末混合物の組成(表9):
20.0%の塩酸バルデナフィル
40.0%の微結晶性セルロース(Avicel PH101)
40.0%のアルギン酸ナトリウム(Protanal LF 120M)
バッチサイズ:5000g
【0101】
【表9】
【0102】
押出/球形化法を通じたペレットの製造は、高剪断造粒における製造法と比較して、より長いプロセス時間とより高い複雑性によって特徴付けられる。押出された塊の接着特性により、スクリーニングの穿孔径は1500μmに増大した。この径は、高剪断造粒プロセスによって製造されたペレットと比較した場合、より大きなサイズを有するペレットを製造した。そのため、押出/球形化によって調製されたペレットの平均フェレ径は約0.10623mmであった。破砕強度は約23.6Nであったが、押出/球形化及び高剪断造粒によって調製された製剤の破砕強度値は、異なるペレット径であるため、互いに比較され得なかった。
【0103】
さらに、押出/球形化によって得られたより小さなペレットは、空洞が観察され、ペレットの製造へと導くことができた従来技術からの教示と照らして、更なる調査を行わなかった。
【0104】
押出/球形化によって得られたペレットと比較して、高剪断造粒によって得られたペレットの特徴における相対的な差は、ペレット表面の粗さ値(Ra値)であった。ペレット表面の調節された粗さは、このような多粒子医薬形態の最適な流動性及び実施可能性を可能にする。
【0105】
約16.6μmであった表9の製剤26におる高剪断造粒によって調製されたペレットの粗さ値と比較すると、押出/球形化法で調製されたペレットのRa値は相対的に低く、この場合、表2の製剤10に従って調製されたペレットについては6.7μmであった。
【0106】
アルギン酸塩系ペレットの被覆
方法:
30.5%アルギン酸ナトリウム、49.5%MCC及び20%塩酸ベラパミルを含むマトリックスペレットは、新規な高剪断造粒法によって調製された。続く被覆プロセスのために最適なペレット特性(すなわち、良好な硬度及び球形)を得るために、10%塩化カルシウム溶液を造粒液として用いた。次に、ペレットは、水性酢酸ビニル/ポリビニルピロリドン分散液(Kollicoat SR 30 D、15%w/w固体内容物)を用いて被覆された。
【0107】
被覆方法について、ペレットの40gの画分は、理論的な被覆レベルが20%(コアペレットに基づくw/w)に達成されるまで、下部(bottom)スプレイ及びWursterインサートを用いて流動床コーター(Midi−Glatt,Glatt,Binzen,Germany)において被覆された。
【0108】
被覆条件:バッチサイズ:40.0g、入口温度:30〜35℃、ノズル径:0.5mm、スプレイ圧:0.5バール、スプレイ速度:1g/分、及び35℃にて5分間の最終乾燥。
【0109】
被覆プロセス後のペレット接着を避けるために、二酸化珪素(Syloid 244FP)を添加し、硬化段階前にペレットと混合した。ペレットを1日間、60℃にて硬化した。
【0110】
結果:
被覆されたペレットからの塩酸ベラパミルの得られた放出プロフィールはpH非依存的のままであった(図14)。
【技術分野】
【0001】
本発明は、水におけるpH依存的溶解性を有する化合物を含むペレットを製造する方法、及び該方法で得られるペレットに関する。さらに、本発明は、該ペレットを含む経口剤形に関する。
【0002】
さらに、本発明はまた(強力な)pH依存的水溶性を有する有効成分を含む改善された放出プロフィールを有するペレットの形体であるか、又はそれから製造された固体医薬製剤に関し、特に、該有効成分は低水溶性の弱塩基である。
【0003】
医薬としての有効成分は、種々の投薬量で投与されることが必要とされ、カプセル又はサシェなどの剤形又は複数回単位剤形を用いるために適切であり得る。これらの剤形は、適切な担体に調合された必要量の有効成分を含む。
【0004】
ペレットは、複数回単位剤形として一般には定義され、1回単位剤形と比較して、有効性及び安全性などの多数の治療的利点を有すると考えられている。また、ペレットは、それに含まれる有効成分の放出パターンに影響を及ぼすように、適した被覆材料で被覆されてもよい。
【0005】
定常なかつ調節可能な放出を有するために、ペレットは、定常形状として、より具体的には規則的な形状の球として与えられることを必要とする。
【0006】
ペレットからの有効成分の放出を支配する重要因子は、有効成分が放出される媒体と接触する表面積である。不規則な形状のペレットは不規則な表面を有し、結果として有効成分の放出の不規則性を与える。
【0007】
これに関して、粒子の平均径はまた、有効成分の放出とペレットの溶解において重要な役割を果たす。
【0008】
有効成分の放出は、画定された平均径を有する規則的に形状化されたペレットを用いて良好に制御可能である。また、ペレットの多孔性及び粗さは、有効成分の放出、並びにペレッの操作及び実施可能性に関連性のある物理的特徴に影響を及ぼすパラメータでもある。
【0009】
球形のペレットは容易に被覆することができ、より均一な被覆厚は、ペレットが規則的な円形を有する場合に達成され得る。これは、ペレットのサイズ分布が狭い場合にさらにより顕著である。
【0010】
さらに、球形のペレットは、容易に操作され、カプセル、サシェ、又は複数回単位錠剤などの他の応用形態に充填される。
【0011】
したがって、製薬産業において使用されるペレットは、一般に自由流動的であり、狭い粒子サイズ分布を有する0.3〜2.00mmの粒径、及び約10%の多孔性を有する球状粒子である。
【0012】
医薬技術分野における技術状態によれば、このようなペレットの製造に適したプロセス及び装置がいくつかある(Isaac Ghebre−Sellassie: Pharmaceutical Peptization Technology,Marcel Dekker,Inc.,New York,Basel,1989)。
【0013】
ペレットの製造用に開発された最初のプロセスは、いわゆる層化法である。このプロセスでは、非常に良質な球状ペレットが、いくつかの被覆層によって漸次的に完成されたコアを提供することによって調製可能である。コアは、通常、不活性な材料、例えば、糖、スターチ、塩化ナトリウム粒子又はそれらの混合物、及び有効成分を含む。
【0014】
ペレット表面に与えられる小さな剪断力により、上記の最初のプロセスによれば、得られたペレットの表面の滑らかさは満足できない場合がある。結果として、ペレットは互いに結合し、高い比率で廃棄されることになる。層化法のプロセス時間は通常長い。
【0015】
第2に、ペレットの製造に適したより重要なプロセスは、いわゆる押出−球形化プロセスである。このプロセスでは、有効成分及び必要とされる賦形剤は、例えば、ともに粉末形態であり、一緒に混合されて粉末混合物を形成する;適切に均質になった粉末混合物を液体と混合し、均質な湿性塊が得られるまで練られる。この塊を押出し、押出プロセスから得られた押出物は球形にされる(球状形態にされる)。最後に、球状段階から得られた生のペレットを乾燥する。ペレットの質は、主に、押出し及び球形化のプロセス変数によって決定される。押出−球形化プロセスは、例えば、EP1252886及びWO2007/135470に開示されている。
【0016】
押出/球形化によって調製された、微結晶性セルロース(MCC)、テオフィリン及び様々な範囲のレベルであるアルギン酸ナトリウム(すなわち、10〜50%w/w)を含むペレットが知られている(Sriamomsak et al.European Journal of Pharmaceutics and Biopharmaceutics 69(2008)274−284)。したがって、2種のアルギン酸ナトリウムが、酢酸カルシウム又は炭酸カルシウム(0、0.3、3及び10%w/w)を添加して、及び添加しないで評価された。ペレット特性、例えば、サイズ、形状、形態及び薬物放出挙動に及ぼすアルギン酸ナトリウム及びカルシウム塩の量及び種類の効果が調査された。結果は、アルギン酸ナトリウム及びカルシウム塩の量が得られたペレットのサイズ及び形状に影響を及ぼすことを示した。しかしながら、異なる種類のアルギン酸ナトリウム及びカルシウム塩は、異なる程度で修飾に応答した。走査型電子顕微鏡写真に見られるように、ペレット構造において空洞が観察され、これは、球形化プロセスに関与する力に起因していた。大部分のペレット製剤は、60分以内に約75〜85%テオフィリンを放出した。ペレット製剤へのカルシウム塩の組み込みは、使用されたカルシウム塩の溶解性に依存して薬物放出を変更した。
【0017】
ペレットの製造に適した更なるプロセス(ペレット化)は、当該技術分野において知られ、例えば、高剪断ミキサーにおけるペレットの製造が挙げられる。
【0018】
高剪断ミキサーペレット化プロセスは異なる工程:粉末の均質化、造粒、及び乾燥を伴う。その後のペレットのプライマー核は、バインダースプレイ及び撹拌中の分散によって形成される。ペレットは、最終的にふるいにかけることができる。
【0019】
高剪断ミキサーにおけるペレットの形成は多変量プロセスであり、したがって、このようなプロセスにとってプロセス変数を特定し、調節することは重要である。特に、製造物特性は、プロセス変数、例えば、羽根速度及び混練時間の変化に感受性である。したがって、これらのパラメータの最適化はプロセスにとって重要である。
【0020】
大きすぎる粒子の生成を避けるために、適切な撹拌を必要とする。さらに、バインダー液体流速はまた、ペレットの質に影響を及ぼす重要なパラメータであり得る。それは、塊の緻密化が混合及びスプレイ後に行われるためである。ペレット凝集の工程内調節は重要であり、したがって、いくつかのプロセス指標が追随する必要があり、顆粒形成の終点を決定する方法は集中的な調査を必要とする。
【0021】
さらに、多数の有効成分は有効成分の一時的な過剰又は過少投薬を避けるために、特定の放出動力学、例えば、消化管の特定の部分における有効成分の迅速かつ選択的な放出を必要とする。
【0022】
これに関連して、生物薬剤学的分類システム(Amidon et al.,1995;Dressman et al.,1998,2001)のクラスIIに属する有効成分の製剤は、一般に、非常に挑戦的であり、それは、経口生物学的利用能が、消化(GI)管における溶解速度によって決定されるためである。
【0023】
低溶解性又は溶解速度は、多くの場合、pH依存的又は水難溶性薬物のGI管からの吸収において律速段階となることは一般に認められている。これは経口生物学的利用能を危うくする。それは、溶液中の薬物濃度が生物学的膜を通過する大部分の医薬としての有効成分の吸収のために駆動力であるからである。したがって、経口投与後、pH依存的な溶解速度又は水難溶性薬物の増大は、現代薬剤学の最も挑戦的側面の1つである。
【0024】
FDAによって確立された生物学的同等性要件によれば、低溶解性を有する有効成分は、水に対して5mg/mlよりも低い溶解性を有するものと考えられている(Fed.Reg.In 21 CFR,Ch 1;(4/1/87 Ed.)Part 32;320)。
【0025】
さらに、生物学的同等性要件に係る低溶解性を有する弱塩基性の有効成分は、通常、pH依存的な水溶解性を有する。これらの成分は、通常、低pH値で相対的に良好な溶解性を示す。より高いpH(例えば、pH6.8)では、これらの薬物の溶解性は5mg/mlよりも低い。化合物のpH依存的溶解性により、これらの薬物の医薬製剤は、通常、pH依存的溶解挙動を示す。
【0026】
しかしながら、低溶解性有効成分について迅速又は長期のpH非依存的放出を可能にする医薬製剤は、製剤分野において非常に望ましい。
【発明の概要】
【課題を解決するための手段】
【0027】
本発明は、医薬としての有効成分、及びアルギン酸塩を含む固体ペレットを提供すること目的とし、平均フェレ(Feret)径が約300〜800μmであり、破砕強度が約4〜10Nであり、縦横比が約1.0〜1.2であることによって特徴付けられる。
【0028】
本発明の更なる目的は、高剪断造粒によって、医薬としての有効成分及びアルギン酸塩を含む固体ペレットを製造するための方法を提供することであり、該方法は、
−医薬としての有効成分及び必要とされる賦形剤の各粉末が高剪断ミキサーボウルに入れられ、混合して粉末混合物を形成する混合段階、
−造粒液として塩化カルシウム溶液の粉末混合物に添加して開始され、造粒塊が得られる造粒段階、
−該造粒塊がペレットの製造に伴って羽根車(impeller)によって球形化される球形化段階、
−乾燥段階、及び
−最終ふるい段階
を含む。
【0029】
本発明に係るペレットに含まれる有効成分は、特に、一般的に強いpH依存的溶解性を有する有効成分であり、例えば、pHが約3で良好な溶解性を有するが、pHが約6.8又はそれよりも高い場合に5mg/mlより低い溶解性を有する有効成分である。より具体的には、該有効成分は塩基であり、例えば、pKaが約8.5又はそれよりも高い弱塩基である。
【0030】
本発明に係るペレットは、好ましくは、例えば、アルギン酸、アルギン酸ナトリウム、アルギン酸カリウム、アルギン酸アンモニウム、アルギン酸カルシウム、アルギン酸マグネシウム又はそれらの混合物の形態であるアルギン酸塩を含む。アルギン酸ナトリウム及びアルギン酸カルシウムが最も好ましい。
【0031】
本発明に係る固体ペレットは、好ましくは、平均フェレ径が約300〜800μmであり、破砕強度が約4〜8Nである。
【0032】
本発明に係る方法では、混合段階は、有効成分、アルギン酸塩及び任意の他の賦形剤を高剪断ミキサーボウル、例えば、ガラス製又はステンレス製ボウル中で混合することによって行われる。粉末は、約900〜約1100rpmの羽根回転、約900〜約1100rpmのチョッパー回転で約2〜6分間混合される。
【0033】
約900ml体積のボウル、及び約100gの粉末の充填を用いて、羽根回転は、好ましくは約1000rpm、チョッパー回転は約1000rpmの約3分間である。
【0034】
造粒液はまたバインダーとして画定され、造粒段階中、粉末に添加される。本発明によれば、バインダーは、水又は水溶液であり、好ましくは塩化カルシウム溶液又は同等のカルシウム塩の溶液である。該溶液は、通常、約3〜約15%v/v、好ましくは約5〜約10%v/vの塩化カルシウム濃度を有する。塩化カルシウムの代わりに、対応する必要量の同等の医薬として許容されるカルシウム塩を用いることができる。
【0035】
各製剤に必要とされるバインダーの総量は、ボウル内の温度及び羽根、並びにカルシウム塩及び最終のペレット組成物の濃度のようなプロセス変数に依存する。
【0036】
本発明に係るプロセスのために上記で与えられた条件について、約100gの充填では、バインダーの必要な絶対量は、室温で約90〜約130mlであり、さらにボウルを加熱する。加熱されたボウルの温度は約40℃〜約50℃である。バインダーのスプレイ速度は、すぐに、粉末混合物に対して、スプレイされた結合溶液の体積を決定し、通常、上記条件によれば、約8〜30ml/分である。
【0037】
本発明の高剪断造粒プロセスによれば、造粒中、チョッパー速度は一定に保持され、該速度は、約2800〜3200rpmであり、好ましくは3000rpmである。羽根速度は1200〜1400rpmで変化し、好ましくは1300rpmである。
【0038】
球形化段階は、約4〜6分間、ペレットを操作することによって造粒後に行われ、この場合、羽根速度は約450〜約600rpmであり、好ましくは500rpmである。本発明の態様の具体的な形態によれば、球形化段階ではチョッパーは使用されない。
【0039】
続く乾燥段階は、例えば、約5〜20分間、好ましくは15分間、約40〜約55℃の温度(製造物温度)で流動床コーターを用いて行われる。
【0040】
最終的に、ペレットをふるいにかける。平均フェレ径が約300〜800μm、より好ましくは約400〜600μmのペレットサイズ画分は、許容されるペレットサイズ範囲として考えられる。
【0041】
本発明の意味における固体ペレットは、空洞がないペレットである。
【0042】
本発明に係るペレットは、好ましくは水系で、高剪断造粒によって製造される。例えば、押出/球形化によって調製された従来のペレットと比較して、本発明に係るペレットは固体であり、調節された粗さ、並びに低多孔性及び高破砕強度を有する表面を含む。これらの特徴は、ペレットの改善された機械的安定性を与え、それらの均質で安定な被覆を可能にする。さらに、それらは固体であるため、多量の有効成分が該ペレットに充填され得る。
【0043】
上述したように、本発明の態様の具体的形態によれば、ペレットは被覆される。典型的な被覆は、例えば、ポリ酢酸ビニル/ポリビニルピロリドン共重合体(Kollicoat SR 30D)である。
【0044】
本発明に係るペレットの小さい粒子サイズは、カプセルあたりより多量のペレットを有するカプセルを有意に満たすことを可能にし、カプセルあたりの投薬量変動性を減少させる効果を有する。小さいサイズのペレットの更なる利点は、小児科の医薬製剤を製造することができる点である。
【0045】
本発明のペレットは、ペレットに含まれる医薬としての有効成分の放出を改善する。特に、本発明に係るペレットに含まれる有効成分のpH非依存的放出は、本発明に従って達成される。
【0046】
該有効成分は、例えば、強いpH依存的可溶性を有し、例えば、有効成分は、低pH、例えば、pHが約3で良好な溶解性を有するが、高いpH、例えば、約6.8又はそれより高いpHでは5mg/mlより低い溶解性を有する。
【0047】
態様の特定の形態によれば、該有効成分は弱塩基であり、pKaは約8.5又はそれよりも高い。この効果は、溶解媒体のpHが約6.8又はそれより高い場合に特に現れ、該弱塩基は、例えば、塩酸バルデナフィル又は塩酸ベラパミルの場合のような水における低溶解性を有する。
【0048】
強力なpH依存的溶解性が弱塩基性であり、pKaが約8.5又はそれよりも高い有効成分はまた考慮され、好ましくは本発明に係るペレットに含まれ得る。
【0049】
本発明に係るペレットに好ましくは含まれ得る医薬としての有効成分は、例えば、ベータ遮断薬、例えばプロプラノロール、メトプロロール及びアテノロール、カルシウムアンタゴニスト、例えばジルチアゼ及びベラパミル、抗微生物薬、例えばセファレキシン、セファクロル、抗ヒスタミン薬、例えばクロルフェニラミン、シンナリジン、ジフェンヒドラミン、鎮痛薬、例えばジアゼパム、又は抗精神病薬、例えばクロルプロマジン、フルフェナジン、並びに任意の医薬として許容されるその塩が含まれる。
【0050】
医薬としての有効成分についての溶解速度の改善は、アルギン酸塩の使用によって得られる効果である。ペレットにおけるアルギン酸塩成分は、低pH値(例えば、pH=3)より高いpH値(例えば、pH=6.8)で良好に安定である。これは、ペレットの緩徐な崩壊に起因して、医薬としての有効成分としての弱塩基の溶解が低pH(例えば、p=3)で遅れるという効果を有し、一方、これは、有効成分の溶解及びペレットの迅速崩壊によって、高pH値(例えば、pH=6.8)で改善される。
【0051】
アルギン酸塩の使用は、溶解が通常は制限される高pH値(例えば、pH=6.8)で、有効成分の溶解速度を改善することができる。
【0052】
改善された溶解速度の結果として、有効成分の良好な生物学的利用能はまた本発明に係るペレットによって達成される。
【0053】
本発明に係るペレットは、他の賦形剤、例えばマトリックスビルダー又は充填剤、例えば、スクロース、マンニトール、ラクトース、デキストロース及びソルビトールを含む群から選択される水溶性非イオン性基質をさらに含むことができる。
【0054】
ペレットの機械的強度に影響を及ぼす追加の配合剤としてのセルロース又はセルロース誘導体はまた、本発明に係るペレットにおいて使用され得る。微結晶性セルロース(MCC)が特に好都合である。
【0055】
本発明に係るペレットは、例えば、約1%〜40%w/wの医薬としての有効成分からなり、残りの60〜99%が10%〜80%のアルギン酸塩、及び20%〜90%の充填剤及び/又はマトリックスビルダーで作られている。
【0056】
以下の実施例は、例証を目的として提供され、限定されない。要約すると、本発明のペレットは、上記で開示された一般的方法によって調製され得て、一方、以下の実施例は、本発明の態様の典型的な形態の調製を開示している。
【図面の簡単な説明】
【0057】
【図1】造粒液中の5%塩化カルシウム及び8ml/分のスプレイ速度で調製されたペレットのSEM(走査型電子顕微鏡)顕微鏡写真(製剤番号7)。
【図2】造粒液中の5%塩化カルシウム及び10ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号8)。
【図3】造粒液中の5%塩化カルシウム及び20ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号10)。
【図4】アルギン酸塩(Protanal LF 120M)マトリックスペレットからの塩酸ベラパミルのpH非依存的放出(製剤番号10)。
【図5】造粒液中の10%塩化カルシウム及び20ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号16)。
【図6】造粒/球形化プロセス中に加熱された(40℃)容器中に調製されたペレットのSEM顕微鏡写真(製剤番号14)。
【図7】造粒液中の20%塩化カルシウム及び20ml/分のスプレイ速度で調製されたペレットのSEM顕微鏡写真(製剤番号17)。
【図8】造粒/球形化プロセス中に加熱された(50℃)容器中に調製されたペレットのSEM顕微鏡写真(製剤番号14)。
【図9】リン酸緩衝液pH6.8中の塩酸ベラパミルの放出に対する塩化カルシウム濃度の効果(製剤番号4、10、16、17)。
【図10】造粒液として水及び20ml/分のスプレイ速度で調製された純粋MCCペレットのSEM顕微鏡写真(製剤番号23)。
【図11】純粋MCCペレットからの塩酸ベラパミルのpH依存的薬物放出(製剤番号23)。
【図12】造粒液中の5%塩化カルシウム及び20ml/分のスプレイ速度で調製された塩酸バルデナフィル含有ペレットのSEM顕微鏡写真(製剤番号25)。
【図13】従来のMCC(微結晶性セルロース)ペレットと対比したアルギン酸塩/MCCペレットからのpH6.8での非常にpH依存的な可溶性化合物(塩酸バルデナフィル)の放出(製剤番号24及び25)。
【図14】アルギン酸ナトリウム/MCCからなる被覆されたペレット(1日、60℃にて硬化)からのベラパミルHClのpH非依存的薬物放出。
【発明を実施するための形態】
【0058】
特徴付け方法
収量:
収量は、100、250、355、630、710、800、900、1000、1400μmのふるい器具を用いたふるい分析(Retsch GmbH,Haan,Germany)によって決定された。ペレットサイズ画分の355〜630μmは、使用に適した収量として決定された。
【0059】
pKa:
pKa又はイオン化定数は、化合物の中性及び帯電した形態の平衡係数の負の対数として定義される。典型的には、滴定法において、化合物は、その中性形態に向けて滴定される。
【0060】
難溶性化合物のpKaは、水溶性メタノール溶液において測定され得る。いくつかの滴定が、様々な比率のメタノール:水を用いて行われる場合、Yeasuda−Shedlovsky式は、純粋水溶液における理論的pKaを表すことができる(Avdeef A,Comer J.E.A,Thomson,S.J.“pH−Metric Logp 3.Glass Electrode Calibration in Methanol−Water Applied to pKa Determination of Water Insoluble Subatances by Potentiometric Titration”Anal.Chem.1993,65,pp42−49)。
【0061】
縦横比及び平均フェレ径:
縦横比yA(0<yA≦1)は、最大フェレ径に対する最小フェレ径の比率yA=xフェレ最小/xフェレ最大によって定義される。それは、粒子の伸長の指標を与える。また、いくつかの文献は、球形の定義として1/yAを用いた。フェレ径は、ノギスを用いた粒子の投影面積から推定される。一般に、それは、任意角で粒子の2つの平衡な接線間の距離として定義される。実際には、最小xF,最小及び最大フェレ径xF,最大>、平均フェレ径:
【0062】
【化1】
【0063】
並びに最大及び最小フェレ径の方法に対して90°で得られたフェレ径xF,最大90が用いられる。最小フェレ径は、多くの場合、ふるい分析に同等な径として用いられる。
【0064】
縦横比及び平均フェレ径の両方は、画像分析によって決定された。355〜630μm(高剪断造粒プロセスを通じて調製されたペレット)と1000〜1400μm(押出/球形化プロセスを通じて調製されたペレット)のふるい画分を画像分析に用いた。画像分析は、ビデオカメラ(HV−T20,Hitachi Kokusai Electric Europe GmbH,Erkrath,Germany)を組み合わせた光学顕微鏡(Olympus BX 50,Olympus Deutschland GmbH,Hamburg,Germany)を用いて行われた。ペレットサイズ及び形状測定について、各試料の500を超えるペレットをスライドガラス上で調製し、スキャンし、ビデオカメラによった撮像した。デジタル画像加工は、「Software Analysis five」(Olympus Soft Imaging Solutions GmbH,Munster,Germany)を用いて行われた。各ペレットについて、35子のフェレ径を測定し、平均フェレ径の計算に使用した。最大フェレ径と、その最大フェレ径に直交したフェレ径の比率を縦横比として用いた。
【0065】
多孔性:
ペレットの多孔性(ε)は、式(1):
【0066】
【化2】
【0067】
に従って、ガス比重びん密度(gass pycnometric density)及び水銀ポロシメーターから計算され得る。
【0068】
ペレットのガス比重びん密度(ρg)は、ヘリウムピクノメーター(pycnometer)(Ultrapyk 1000T,Quantachrome,Odelzhausen,Germany)によって決定された。各試験されたペレットバッチについて、3つの試料を分析した。
【0069】
ペレットの見掛け密度(ρg)は、水銀ポロシメーター(Pascal 140 & 440,Fisons−Carlo Erba,Valencia,USA)によって評価された。
【0070】
機械的特性(破砕強度):
15個のペレットの破砕強度は、テクスチャ分析機(TAXT plus,Stable Micro Systems,Godalming,Surrey UK)を用いることによって調査された。各ペレットは、分析機のフラットプレートと上部パンチとの間に置かれた。次に、径が2mmであるパンチを0.1mm/秒の速度で下げた。ペレットが破砕された時点は、最初のピークとして力−時間グラフ上に示される。力−時間プロットは、テクスチャ分析機ソフトウェアExponentを用いて記録された。破砕力の算術平均は、破砕強度として用いられた。
【0071】
走査型電子顕微鏡(SEM)写真:
ペレットは、アルゴン雰囲気下、金−パラジウム(MED 020,Bal−tec AG,Liechtenstein,Germany)を用いて60秒間被覆され、次に走査型電子顕微鏡(SEM)(DSM 982,Zeiss,Oberkochen,Germany)を用いて観察された。
【0072】
薬物放出:
塩酸ベラパミルのインビトロ放出は、31USP回転パドル法(900mlの0.1N HCl又はUSPリン酸緩衝液pH6.8;37℃;75rpm;n=3;Distek Premiere 5100 Dissolution System,Distek Inc.,North Brunswisk,USA)を用いて決定された。所定時間間隔で、10ml試料を採取し(置換ではない)、ろ過し、アッセイした。放出された有効成分の量は、USP31法に従って、HPLCによって測定された。塩酸バルデナフィルの溶解は、75rpmの31USP回転パドル法を用いることによって、01%ラウリル硫酸ナトリウムを含む900mLのリン酸緩衝液pH6.8(10%)中で行われた。所定時間間隔で、10ml試料を採取し(置換ではない)、ろ過し、アッセイした。放出された有効成分の量は、HPLCによって決定された。
【0073】
共焦点レーザー走査顕微鏡(CLSM):
CLSM法は、ペレット表面の粗さ値(Ra値)を決定するために使用された。この方法は、例えば、F.Depypere et al.,Quantification of microparticle coating quality by confocal laser scanning microscopy(CLSM),Eur.J.Pharm.Biopharm.(2009)に報告されている。
【0074】
高剪断造粒:
900mlのガラス製ボウル、3つの羽根、及びチョッパーを備えた小スケールの実験室用高剪断ミキサー(Pro−C− ept 4M8,Zelzate,Belgium)においてペレットを製造した。有効成分及び必要とされる賦形剤は、例えば、ともに粉末形態であって、高剪断ミキサーボウルに入れられ、1000rpmの羽根回転、及び1000rpmのチョッパー回転で一緒に混合され、粉末混合物を形成した。造粒は、造粒液として塩化カルシウム溶液又は水の添加により開始され、湿性粉末塊を形成した。1mmの開口を有するチューブを用いて、ドシマット(dosimat)(765 Dosimat,Metrohm Ltd.,Hensau,Switzerland)を用いて液体放出の速度を調節した。各製剤について必要とされる造粒液の全量は、プロセス変数とペレットの組成に依存していた。造粒中、選ばれた羽根速度は1300rpmであり、チョッパー速度は3000rpmで一定に保持した。造粒後、ペレットは、500rpmの羽根速度(チョッパーなし)を用いて5分間球形化された。流動床を用いて、10分間、40℃の製造物温度でペレットを乾燥させた。ペレットをふるいにかけ、355〜630μmのペレットサイズの画分を回収した。
【0075】
実験番号14の例では、ガラス製バイアルを室温で用い、造粒中に加熱した。この実験で用いられた2つの温度は40℃と50℃であった。
【0076】
高剪断造粒
粉末混合物の組成(表1〜4):
20.0gの塩酸ベラパミル
49.5gの微結晶性セルロース(Avicel PH101)
30.5gのアルギン酸ナトリウム(Protanal LF 120M)
【0077】
【表1】
【0078】
表1は、どのようにして造粒液中の全量の増加が、3%で安定な造粒液の塩化カルシウム含量を用いて、高い破砕強度を有するペレットをもたらしたかを示す。これは、材料のより大きな柔軟性(plasticity)に起因し、より低い多孔率を有するより密度の高いペレットを導く。
【0079】
低い全量の造粒液と高い全量の造粒液を用いて製造された製剤の多孔性値はそれぞれ約35%と10%であった。これは、造粒液の増加がより低い多孔率を有するペレットの密度を高めるようにするという仮説と良好な一致を示す。結果として、より低い全量の造粒液を有するペレットは、微結晶性セルロース、Celletsで構成される市販のペレットと比較すると、より低い破砕強度によって特徴付けられた(3.8N)。しかしながら、より高い全量の造粒液で調製された製剤は、7.4Nより高い破砕強度を有する良好な硬度を示した。
【0080】
【表2】
【0081】
【表3】
【0082】
表2及び3は、造粒液中の塩化カルシウム濃度の増加がペレット形状を改善し、塊の望ましくない画分を減少させ、これは、主要な架橋プロセスによって引き起こされたアルギン酸ナトリウムのより低い接着特性に起因していることを示す。ペレットは許容できると考えられ、それは、縦横比の中央値の値全てが、1.2を下回り、約72.5〜77.5%というより高い収率であったためであった(製剤番号7〜16)。破砕強度は4.7N〜8.7Nで変化し、それは、どんなスプレイ速度が適用されても、全ての製剤の良好な硬度を示す。良好な硬度は、アルギン酸塩とカルシウムイオンとの間の改善された架橋の結果であった。
【0083】
MCCとアルギン酸ナトリウムを含むペレットからの塩酸ベラパミルの薬物放出は、1〜6.8の範囲でほぼpHに非依存的であった(図4、製剤番号10)。アルギン酸塩(Protanal(登録商標)LF 120M)は、逆溶解性により、薬物の溶解度差を弱めることができた。アルギン酸ナトリウムは低pHレベルでよりゆっくりと溶解し、これは、アルギン酸塩が、拡散障壁を作る難溶性アルギン酸の形態で沈殿するためである。リン酸緩衝液中の最速薬物放出は、カルシウムイオンとのナトリウムイオン交換によって影響を受け、このようにして、分解するペレット構造を作製した。
【0084】
【表4】
【0085】
20%の塩化カルシウムを用いて調製されたペレットは、粉末混合物及びスプレイ造粒液から得られた湿性粉末塊の非最適化剛性/柔軟性により、表面構造に亀裂を示した。高い剛性画分は粉末塊の結合特性を減少させ、結果として、ペレットは不安定になった。したがって、5%と10%の塩化カルシウム濃度で調製されたペレットは、それらの特性に関して最適であると考えられた。
【0086】
図9に示され得るように(製剤番号4、10、16、17)、造粒液中のカルシウム濃度は、リン酸緩衝液pH6.8において薬物放出速度に影響を及ぼした。放出プロフィールを比較すると、塩化カルシウム濃度を増加させた場合、薬物放出速度が減少したことは明らかである。より低い放出速度は、ペレットの膨潤比を制限する高分子鎖の可動性における制限の増加と結び付けられる。
【0087】
粉末混合物の組成(表5):
20.0gの塩酸ベラパミル
40.0gの微結晶性セルロース(Avicel PH101)
40.0gのアルギン酸ナトリウム(Protanal LF 120M)
【0088】
【表5】
【0089】
許容され得る収率が54.5〜74.8%である、高剪断造粒機におけるペレット製造は、アルギン酸塩の量が増加したとしても可能である。ペレットは5.6〜7.7Nの間である良好な硬度によって特徴付けられるが、1.2と1.29の範囲を超える高い縦横比が得られた。これは、水との接触で膨潤する高い量のアルギン酸塩に由来する。このようにして、造粒塊の柔軟性/剛性は球体を形成するには最適ではなかった。
【0090】
粉末混合物の組成(表6):
20.0gの塩酸ベラパミル
60.0gの微結晶性セルロース(Avicel PH101)
20.0gのラクトース一水和物(Granulac 200)
【0091】
【表6】
【0092】
MCCに基づくペレットの特徴は、アルギン酸ナトリウムを含むペレットと同程度である(製剤番号7〜16)。
【0093】
しかし、塩酸ベラパミル放出は、pH1で測定した薬物放出速度と比較すると、pH6.8で僅かに遅く、これは、薬物の弱塩基性によって説明され得る(図11)。
【0094】
粉末混合物の組成(表7):
20.0gの塩酸バルデナフィル
60.0gの微結晶性セルロース(Avicel PH101)
20.0gのラクトース一水和物
【0095】
【表7】
【0096】
粉末混合物の組成(表8):
20.0gの塩酸バルデナフィル
49.5gの微結晶性セルロース(Avicel PH101)
30.5gのアルギン酸ナトリウム(Protanal LF 120M)
【0097】
【表8】
【0098】
塩酸ベラパミルペレットの特徴は、塩酸ベラパミルペレットと同等である。製剤のアルギン酸ナトリウムの使用は収率を増大させ、塩酸バルデナフィルを含むペレットを改善した。
【0099】
純粋なMCC系ペレットからの塩酸バルデナフィルの薬物放出は、pH6.8でアルギン酸ナトリウム/MCC系ペレットからの薬物放出と比較して非常に低かった(図13)。アルギン酸ナトリウムはより高いpH環境でより溶解性であり、したがって、それは、この環境における難溶性塩酸バルデナフィルの薬物放出を増加させた。
【0100】
押出/球形化
粉末混合物の組成(表9):
20.0%の塩酸バルデナフィル
40.0%の微結晶性セルロース(Avicel PH101)
40.0%のアルギン酸ナトリウム(Protanal LF 120M)
バッチサイズ:5000g
【0101】
【表9】
【0102】
押出/球形化法を通じたペレットの製造は、高剪断造粒における製造法と比較して、より長いプロセス時間とより高い複雑性によって特徴付けられる。押出された塊の接着特性により、スクリーニングの穿孔径は1500μmに増大した。この径は、高剪断造粒プロセスによって製造されたペレットと比較した場合、より大きなサイズを有するペレットを製造した。そのため、押出/球形化によって調製されたペレットの平均フェレ径は約0.10623mmであった。破砕強度は約23.6Nであったが、押出/球形化及び高剪断造粒によって調製された製剤の破砕強度値は、異なるペレット径であるため、互いに比較され得なかった。
【0103】
さらに、押出/球形化によって得られたより小さなペレットは、空洞が観察され、ペレットの製造へと導くことができた従来技術からの教示と照らして、更なる調査を行わなかった。
【0104】
押出/球形化によって得られたペレットと比較して、高剪断造粒によって得られたペレットの特徴における相対的な差は、ペレット表面の粗さ値(Ra値)であった。ペレット表面の調節された粗さは、このような多粒子医薬形態の最適な流動性及び実施可能性を可能にする。
【0105】
約16.6μmであった表9の製剤26におる高剪断造粒によって調製されたペレットの粗さ値と比較すると、押出/球形化法で調製されたペレットのRa値は相対的に低く、この場合、表2の製剤10に従って調製されたペレットについては6.7μmであった。
【0106】
アルギン酸塩系ペレットの被覆
方法:
30.5%アルギン酸ナトリウム、49.5%MCC及び20%塩酸ベラパミルを含むマトリックスペレットは、新規な高剪断造粒法によって調製された。続く被覆プロセスのために最適なペレット特性(すなわち、良好な硬度及び球形)を得るために、10%塩化カルシウム溶液を造粒液として用いた。次に、ペレットは、水性酢酸ビニル/ポリビニルピロリドン分散液(Kollicoat SR 30 D、15%w/w固体内容物)を用いて被覆された。
【0107】
被覆方法について、ペレットの40gの画分は、理論的な被覆レベルが20%(コアペレットに基づくw/w)に達成されるまで、下部(bottom)スプレイ及びWursterインサートを用いて流動床コーター(Midi−Glatt,Glatt,Binzen,Germany)において被覆された。
【0108】
被覆条件:バッチサイズ:40.0g、入口温度:30〜35℃、ノズル径:0.5mm、スプレイ圧:0.5バール、スプレイ速度:1g/分、及び35℃にて5分間の最終乾燥。
【0109】
被覆プロセス後のペレット接着を避けるために、二酸化珪素(Syloid 244FP)を添加し、硬化段階前にペレットと混合した。ペレットを1日間、60℃にて硬化した。
【0110】
結果:
被覆されたペレットからの塩酸ベラパミルの得られた放出プロフィールはpH非依存的のままであった(図14)。
【特許請求の範囲】
【請求項1】
平均フェレ(Feret)径が約300〜800μmであり、破砕強度が約4〜10Nであり、縦横比が約1.0〜1.2であることによって特徴付けられる、医薬としての有効成分及びアルギン酸塩を含む固体ペレット。
【請求項2】
前記医薬としての有効成分がpH依存的安定性、すなわち、約3のpHで5mg/mlより高い良好な溶解性を有するが、約6.8又はそれを超えるpHで5mg/mlより低い安定性を有し、及び/又は該有効成分が約8.5又はそれより高いpKaを有することによって特徴付けられる、請求項1に記載の固体ペレット。
【請求項3】
医薬としての有効成分が、プロプラノロール、メトプロロール、アテノロール、ジルチアゼム、ベラパミル、セファレキシン、セファクロル、クロルフェニラミン、シンナリジン、ジフェンヒドラミン、ジアゼパム・クロルプロマジン、フルフェナジン、ベラパミル、バルデナフィル又は任意の医薬として許容されるその塩であることによって特徴付けられる、請求項1又は2に記載の固体ペレット。
【請求項4】
前記ペレットが、マンニトール、ラクトース、デキストロース及びソルビトールを含む群から選択される可溶性非イオン性基質をさらに含むことによって特徴付けられる、請求項1〜3のいずれか1項に記載の固体ペレット。
【請求項5】
前記ペレットが、セルロース又はセルロース誘導体をさらに含むことによって特徴付けられる、請求項1〜4のいずれか1項に記載の固体ペレット。
【請求項6】
前記ペレットが微結晶性セルロースを含むことによって特徴付けられる、請求項5に記載の固体ペレット。
【請求項7】
前記ペレットが、約1%〜40%w/wの医薬としての有効成分からなり、残りの60〜99%は10%〜80%のアルギン酸塩、及び20%〜90%の充填剤及び/又はマトリックス内容物(matrix builder)、例えば、微結晶性セルロースで作製されていることによって特徴付けられる、請求項1〜6のいずれか1項に記載の固体ペレット。
【請求項8】
前記ペレットが被覆されていることによって特徴付けられる、請求項1〜7のいずれか1項に記載の固体ペレット。
【請求項9】
−医薬としての有効成分と必須の賦形剤の各粉末が、高剪断ミキサーボウルに入れられ、混合して粉末混合物を形成する混合段階、
−造粒液として塩化カルシウム溶液の粉末混合物に添加して開始され、造粒塊が得られる造粒段階、
−該造粒塊がペレットの製造に伴って羽根車(impeller)によって球形化される球形化段階、
−乾燥段階、及び
−最終ふるい段階
を含む高剪断造粒による、医薬としての有効成分とアルギン酸塩とを含む固体ペレットの製造方法。
【請求項10】
前記混合段階中、粉末は、約900〜約1100rpmの羽根回転、及び約900〜約1100rpmのチョッパー回転で約2〜6分間混合された粉末である、請求項9に記載の方法。
【請求項11】
前記造粒液が、約3〜約5%v/v、好ましくは約5〜約10%v/v濃度の塩化カルシウム又は同等の医薬として許容されるカルシウム塩の水溶液である、請求項9又は10に記載の方法。
【請求項12】
前記造粒段階中、チョッパー速度を一定に保持し、該速度が約2800〜3200rpm、好ましくは3000rpmであり、羽根速度が1200〜1400rpm、好ましくは1300rpmである、請求項9〜11のいずれか1項に記載の方法。
【請求項13】
球形化段階が、造粒段階後に、約450〜約600rpm、好ましくは500rpmの羽根速度で、かつチョッパーなしにペレットを約4〜6分間作用させることによって行われる、請求項9〜12にいずれか1項に記載の方法。
【請求項14】
乾燥段階が約5〜20分間、好ましくは15分間かけて、約40℃〜約55℃(製造物温度)で流動床によって行われる、請求項9〜13のいずれか1項に記載の方法。
【請求項15】
該ふるい段階が機械的ふるいによって行われる、請求項9〜14のいずれか1項に記載の方法。
【請求項16】
請求項1〜8のいずれか1項に定義される前記ペレットを含む医薬経口剤形。
【請求項1】
平均フェレ(Feret)径が約300〜800μmであり、破砕強度が約4〜10Nであり、縦横比が約1.0〜1.2であることによって特徴付けられる、医薬としての有効成分及びアルギン酸塩を含む固体ペレット。
【請求項2】
前記医薬としての有効成分がpH依存的安定性、すなわち、約3のpHで5mg/mlより高い良好な溶解性を有するが、約6.8又はそれを超えるpHで5mg/mlより低い安定性を有し、及び/又は該有効成分が約8.5又はそれより高いpKaを有することによって特徴付けられる、請求項1に記載の固体ペレット。
【請求項3】
医薬としての有効成分が、プロプラノロール、メトプロロール、アテノロール、ジルチアゼム、ベラパミル、セファレキシン、セファクロル、クロルフェニラミン、シンナリジン、ジフェンヒドラミン、ジアゼパム・クロルプロマジン、フルフェナジン、ベラパミル、バルデナフィル又は任意の医薬として許容されるその塩であることによって特徴付けられる、請求項1又は2に記載の固体ペレット。
【請求項4】
前記ペレットが、マンニトール、ラクトース、デキストロース及びソルビトールを含む群から選択される可溶性非イオン性基質をさらに含むことによって特徴付けられる、請求項1〜3のいずれか1項に記載の固体ペレット。
【請求項5】
前記ペレットが、セルロース又はセルロース誘導体をさらに含むことによって特徴付けられる、請求項1〜4のいずれか1項に記載の固体ペレット。
【請求項6】
前記ペレットが微結晶性セルロースを含むことによって特徴付けられる、請求項5に記載の固体ペレット。
【請求項7】
前記ペレットが、約1%〜40%w/wの医薬としての有効成分からなり、残りの60〜99%は10%〜80%のアルギン酸塩、及び20%〜90%の充填剤及び/又はマトリックス内容物(matrix builder)、例えば、微結晶性セルロースで作製されていることによって特徴付けられる、請求項1〜6のいずれか1項に記載の固体ペレット。
【請求項8】
前記ペレットが被覆されていることによって特徴付けられる、請求項1〜7のいずれか1項に記載の固体ペレット。
【請求項9】
−医薬としての有効成分と必須の賦形剤の各粉末が、高剪断ミキサーボウルに入れられ、混合して粉末混合物を形成する混合段階、
−造粒液として塩化カルシウム溶液の粉末混合物に添加して開始され、造粒塊が得られる造粒段階、
−該造粒塊がペレットの製造に伴って羽根車(impeller)によって球形化される球形化段階、
−乾燥段階、及び
−最終ふるい段階
を含む高剪断造粒による、医薬としての有効成分とアルギン酸塩とを含む固体ペレットの製造方法。
【請求項10】
前記混合段階中、粉末は、約900〜約1100rpmの羽根回転、及び約900〜約1100rpmのチョッパー回転で約2〜6分間混合された粉末である、請求項9に記載の方法。
【請求項11】
前記造粒液が、約3〜約5%v/v、好ましくは約5〜約10%v/v濃度の塩化カルシウム又は同等の医薬として許容されるカルシウム塩の水溶液である、請求項9又は10に記載の方法。
【請求項12】
前記造粒段階中、チョッパー速度を一定に保持し、該速度が約2800〜3200rpm、好ましくは3000rpmであり、羽根速度が1200〜1400rpm、好ましくは1300rpmである、請求項9〜11のいずれか1項に記載の方法。
【請求項13】
球形化段階が、造粒段階後に、約450〜約600rpm、好ましくは500rpmの羽根速度で、かつチョッパーなしにペレットを約4〜6分間作用させることによって行われる、請求項9〜12にいずれか1項に記載の方法。
【請求項14】
乾燥段階が約5〜20分間、好ましくは15分間かけて、約40℃〜約55℃(製造物温度)で流動床によって行われる、請求項9〜13のいずれか1項に記載の方法。
【請求項15】
該ふるい段階が機械的ふるいによって行われる、請求項9〜14のいずれか1項に記載の方法。
【請求項16】
請求項1〜8のいずれか1項に定義される前記ペレットを含む医薬経口剤形。
【図1】

【図2】

【図3a】

【図3b】
【図4】
【図5a】

【図5b】

【図6a】

【図6b】

【図7a】

【図7b】

【図8a】

【図8b】
【図8c】

【図8d】

【図9】
【図10a】

【図10b】
【図11】
【図12a】

【図12b】

【図13】

【図14】
【図2】
【図3a】
【図3b】
【図4】
【図5a】
【図5b】
【図6a】
【図6b】
【図7a】
【図7b】
【図8a】
【図8b】
【図8c】
【図8d】
【図9】
【図10a】
【図10b】
【図11】
【図12a】
【図12b】
【図13】
【図14】
【公表番号】特表2012−533526(P2012−533526A)
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−519915(P2012−519915)
【出願日】平成22年7月6日(2010.7.6)
【国際出願番号】PCT/EP2010/004113
【国際公開番号】WO2011/006611
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(300049958)バイエル ファーマ アクチエンゲゼルシャフト (357)
【Fターム(参考)】
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成22年7月6日(2010.7.6)
【国際出願番号】PCT/EP2010/004113
【国際公開番号】WO2011/006611
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(300049958)バイエル ファーマ アクチエンゲゼルシャフト (357)
【Fターム(参考)】
[ Back to top ]