
ホスホジエステラーゼのcAMP基質特異的阻害剤
【課題】 本発明は、ホスホジエステラーゼのcAMP基質特異的阻害作用を有する、ベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分とした薬剤を提供することを目的とする。
【解決手段】 本発明は、
(1) 次の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、ホスホジエステラーゼのcAMP基質特異的阻害剤、
【化3】

(式中、R1及びR2は、同一又は異なって、水素又はハロゲンを表す。)
(2) 上記(1)の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、アトピー性喘息を含む気管支喘息等に対する予防薬又はこれらの治療薬、
に関する。
【解決手段】 本発明は、
(1) 次の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、ホスホジエステラーゼのcAMP基質特異的阻害剤、
【化3】
(式中、R1及びR2は、同一又は異なって、水素又はハロゲンを表す。)
(2) 上記(1)の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、アトピー性喘息を含む気管支喘息等に対する予防薬又はこれらの治療薬、
に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、次の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩(以下、本発明化合物という。)を有効成分として含有する、ホスホジエステラーゼのcAMP基質特異的阻害剤に関する。
【化1】
(式中、R1及びR2は、同一又は異なって、水素又はハロゲンを表す。)
【背景技術】
【0002】
本発明化合物は、優れた抗潰瘍作用、細胞防御作用、胃粘膜血流増加作用を有しており(例えば、特許文献1〜3、非特許文献1〜3参照。)、その中でも2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジン マレイン酸塩(一般名:マレイン酸イルソグラジン)(以下、マレイン酸イルソグラジンという。)は、商品名「ガスロンN」で、胃潰瘍、急性胃炎、慢性胃炎の治療剤として上市されている。また、マレイン酸イルソグラジンは、ラット胃粘膜細胞内cAMPの増加作用を有していることが知られている(例えば、非特許文献4参照。)。
【0003】
多くのホルモン及び神経伝達物質は、cAMP(3’,5’−サイクリックアデノシンモノホスフェート)の細胞内レベルを上昇させることで組織機能を調節している。セカンドメッセンジャーとして機能するcAMPの細胞内レベルは、合成及び分解を制御する機序によって調整される。
cAMPの合成はアデニル酸シクラーゼによって制御され、そのアデニル酸シクラーゼはホルスコリンなどの薬物によって直接活性化される。また、プロスタグランジン(PGE2あるいはPGI2など)やβ−アドレナリン受容体作動薬などは、細胞表面受容体への特異的作働薬であり、間接的にアデニル酸シクラーゼを活性化させて細胞内のcAMPを増加させる引き金としての役割を果たす。
一方、cAMPの分解は、いくつかのホスホジエステラーゼ(以下、PDEという。)アイソザイムによって制御される。これら一連のPDEは、cGMP(グアノシン3’,5’−サイクリックモノホスフェート)の分解も制御する。PDEは、cAMPの加水分解を制御することにより、細胞内情報伝達機構の活性化の程度やその持続に極めて重要な役割を果たしている。また、cAMPの生物学的機能の多様性を決めるコンパートメンテーションにも関与する重要な不活化酵素である。
【0004】
PDEは、その基質特異性、酵素活性調節機構、及び種々の阻害剤の感受性に基づいて機能的に分類され、現在まで、PDEには遺伝子的に異なる11種(PDE1〜PDE11)の既知のファミリーが知られている(例えば、非特許文献5参照)。この中で、PDE4、PDE7及びPDE8は、基質としてcAMPのみを加水分解するcAMP基質特異的PDEである。PDE5、PDE6及びPDE9は、基質としてcGMPのみを加水分解するcGMP特異的PDEである。PDE1〜PDE3、PDE10及びPDE11は、cAMPとcGMPの両環状ヌクレオチドを加水分解するPDEである。なかでもPDE1は、カルモジュリンにより制御されており、PDE2とPDE3はcGMPによって活性の制御が行われている。
PDEは、組織によりその発現が大きく異なることも良く知られている。例えば、PDE10とPDE11は、いずれもcAMP、cGMPの両環状ヌクレオチドを加水分解するPDEであるが、PDE10は脳に非常に多く分布する一方で、PDE11は骨格筋や前立腺など末梢組織に分布する。また、PDE5、PDE6ならびにPDE9はいずれもcGMP特異的PDEであるが、PDE6は網膜にのみ発現しているPDEとして知られている。
【0005】
PDE4阻害による細胞内cAMPの増加は、たとえば、好中球、好酸球、T−リンパ球、マクロファージなどの炎症性細胞の不活性化や、気管平滑筋の弛緩を引き起こすことが報告されている(例えば、非特許文献6、7参照。)。したがって、PDE4阻害剤は、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎などの治療に利用することができる。
PDE7もT−リンパ球に多く発現していることから(例えば、非特許文献8参照。)、その阻害剤は慢性関節リウマチや乾癬などT細胞依存性疾患の治療に利用することができる。
PDE1はカルモジュリン依存性のPDEである。PDE1の阻害剤は尿失禁の治療剤として有用である(例えば、特許文献9参照。)。
また、PDE3はcGMP阻害性のPDEである。PDE3阻害による細胞内cAMPの増加は、たとえば、血管平滑筋の弛緩や血小板凝集の阻害を引き起こすことから(例えば、非特許文献10参照。)、この阻害剤は血栓症の治療剤として有用である。
【0006】
【特許文献1】特開昭50−111085号公報
【特許文献2】特開昭50−111086号公報
【特許文献3】特開昭51−75083号公報
【非特許文献1】Ueda F,et al,Arzneim.−Forsch./Drug Res.,34(I),474(1984)
【非特許文献2】上田房雄ほか,応用薬理,33(1),143(1987)
【非特許文献3】平松新ほか,薬理と治療,11(7),2481(1983)
【非特許文献4】上田房雄ほか,応用薬理,33(1),157(1987)
【非特許文献5】Francis SH,et al,Prog Nucleic Acid Res Mol Biol,65,1−52(2001)
【非特許文献6】Barnes PJ,Nature,402,B31−B38(1999)
【非特許文献7】Mauro M,et al,Trends Pharmacol Sci,18,164−170(1997)
【非特許文献8】Li L,et al,Science,283,848−851(1999)
【非特許文献9】Truss MC,et al,World J Urol,18,439−443(2000)
【非特許文献10】Sudo T,et al,Biochem Pharmacol,59,347−356(2000)
【非特許文献11】Banner KH,et al,Eur Respir J,8,996−1000(1995)
【非特許文献12】Mauro M,et al,Trends Pharmacol Sci,18,164−170(1997)
【非特許文献13】厚生省医薬安全局,「医薬品等安全性情報別冊 平成8年度 新医薬品等の副作用のまとめ」,1997年10月,p12
【非特許文献14】Nichols MR,et al,Mol.Pharmacol.,57,738−45(2000)
【非特許文献15】Oka M,et al,Naunyn Schmiedebergs Arch Pharmacol,356,189−96(1997)
【非特許文献16】Wang P,et al,Mol.Pharmacol.,56,170−174(1999)
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、ベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、PDEのcAMP基質特異的阻害剤を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者は、本発明化合物が有する胃粘膜細胞内cAMPの増加作用について鋭意研究を重ねた結果、本発明化合物が、PDEによるcAMPの加水分解のみを選択的に阻害することを見出し、本発明を完成した。
【0009】
本発明は、
(1) 本発明化合物を有効成分として含有する、PDEのcAMP基質特異的阻害剤、
(2) ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、上記(1)の阻害剤、
(3) ベンゾグアナミン誘導体の医薬上許容される塩がマレイン酸イルソグラジンである、上記(1)の阻害剤、
(4) 本発明化合物を有効成分として含有する、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎、尿失禁若しくは血栓症に対する予防薬又はこれらの治療薬、
(5) ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、上記(4)の予防薬又は治療薬、
(6) ベンゾグアナミン誘導体の医薬上許容される塩がマレイン酸イルソグラジンである、上記(4)の予防薬又は治療薬、
に関する。
【0010】
後述の試験例1に示す通り、本発明化合物は、PDEによるcAMPの加水分解に対して強力な阻害作用を示す一方で、PDEによるcGMPの加水分解に対しては明らかな阻害作用を示さない。さらに詳細に検討した結果、後述の試験例2〜4に示す通り、PDE1、PDE2、PDE3及びPDE4の各アイソザイムに対してほとんど同程度の阻害活性を有する。
【0011】
PDE4阻害剤の有する優れた抗炎症作用などを臨床的に応用する場合、しばしば悪心、嘔吐などの消化器系の副作用の発現が問題になることは良く知られている(例えば、非特許文献11、12参照。)。本発明化合物は、臨床上、悪心、嘔吐などの消化器系の副作用をほとんど発現しないことから、PDE4阻害剤としても非常に有用である(例えば、非特許文献13参照。)。
【0012】
以下に本発明を詳述する。
「ホスホジエステラーゼのcAMP基質特異的阻害剤」とは、PDEによるcAMPの加水分解のみを選択的に阻害し、PDEによるcGMPの加水分解に対しては明らかな阻害作用を有さない薬剤をいう。
「ハロゲン」としては、例えば、フッ素、塩素、臭素、ヨウ素を挙げることができる。
「医薬上許容される塩」としては、例えば、塩酸、臭化水素酸、硫酸、燐酸などの鉱酸の塩、酢酸、クエン酸、酒石酸、マレイン酸、コハク酸、フマル酸、p−トルエンスルホン酸、ベンゼンスルホン酸、メタンスルホン酸などの有機酸の塩を挙げることができる。
【図面の簡単な説明】
【0013】
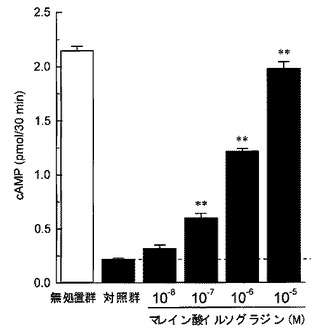
【図1】ウシ脳由来精製PDEによるcAMPの加水分解に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)、横軸はマレイン酸イルソグラジンの濃度(M)を表す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
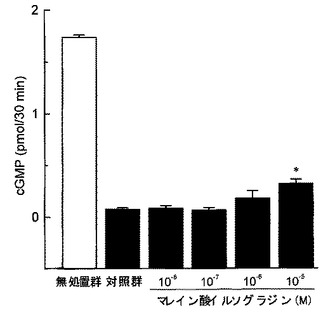
【図2】ウシ脳由来精製PDEによるcGMPの加水分解に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcGMP量(pmol/30min.)、横軸はマレイン酸イルソグラジンの濃度(M)を表す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「*」はダネット検定の結果(*P<0.05)を表す。
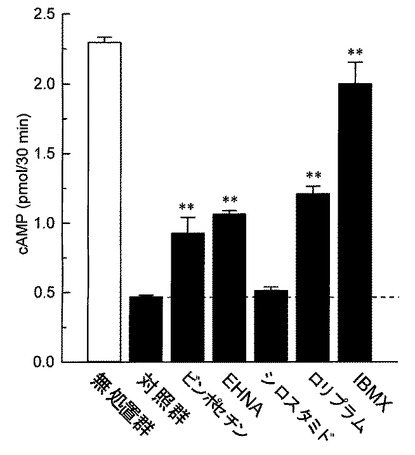
【図3】ウシ脳由来精製PDEによるcAMPの加水分解に対する各種PDE阻害剤の作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
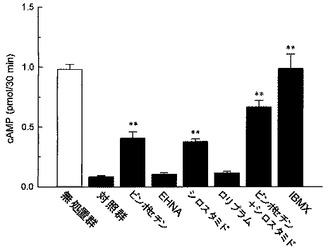
【図4】ウシ心臓由来精製PDEによるcAMPの加水分解に対する各種PDE阻害剤の作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
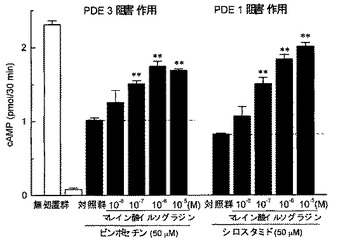
【図5】ビンポセチン(5×10−5M)、又はシロスタミド(5×10−5M)存在下、ウシ心臓由来精製PDEによるcAMPの加水分解に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【図6】ヒト好中球のcAMP産生に対する各種PDE阻害剤の作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【図7】ヒト好中球のcAMP産生に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「*」「**」はダネット検定の結果(*P<0.05、**P<0.01)を表す。
【図8】ヒト好中球のcAMP産生に対するIBMX存在下でのマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を表す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【発明を実施するための最良の形態】
【0014】
本発明化合物は公知化合物であり、公知の方法(例えば、特許文献1〜3参照。)により製造することができる。例えば、モノ若しくはジハロゲノベンゾニトリルとジシアンジアミドとを反応させることにより、又は、モノ若しくはジハロゲノ安息香酸誘導体(カルボン酸、酸ハロゲン化物、エステル、アミド、等)とビグアナイドとを反応させることにより製造することができる。
本発明化合物は、遊離の塩基のまま医薬として用いることができるが、公知の方法により医薬上許容される塩の形にして用いることもできる。例えば、本発明化合物のマレイン酸塩は、ベンゾグアナミン誘導体を、当量のマレイン酸が溶解したアルコール溶液又は酢酸エチル溶液に溶解し、濃縮することにより得ることができる。
【0015】
本発明化合物は、そのまま又は医薬上許容される無毒性かつ不活性の担体中に、例えば0.01〜99.5%、好ましくは0.5〜90%を含有する医薬組成物として、人を含む動物に投与される。
担体としては、固形、半固形又は液状の希釈剤、充填剤及びその他の処方用の助剤一種以上が用いられる。薬物は投与単位形態で投与することが望ましい。また、薬物は、静脈内投与、経口投与、組織内投与、局所投与(経皮投与等)又は経直腸的に投与することができるが、これらに限定されるものではない。これらの投与方法に適した剤型で投与されるのはもちろんである。
PDEのcAMP基質特異的阻害剤としての用量は、疾患・傷害の性質と程度、年齢、体重などの患者の状態、投与経路などを考慮した上で設定することが望ましいが、通常は、成人に対して本発明に係る薬物の有効成分量として、1日あたり、0.1〜1000mg/ヒトの範囲、好ましくは1〜500mg/ヒトの範囲が一般的である。
場合によっては、これ以下で足りるし、また逆にこれ以上の用量を必要とすることもある。また1日2〜4回に分割して投与することもできる。
【0016】
経口投与は固形又は液状の用量単位、例えば、末剤、散剤、錠剤、糖衣剤、カプセル剤、顆粒剤、懸濁剤、液剤、シロップ剤、ドロップ剤、舌下錠その他の剤型によって行うことができる。
末剤は薬物を適当な細かさにすることにより製造される。散剤は薬物を適当な細かさと成し、ついで同様に細かくした医薬用担体、例えば澱粉、マンニトールのような可食性炭水化物その他と混合することにより製造される。必要に応じ風味剤、保存剤、分散剤、着色剤、香料その他のものを混じてもよい。
【0017】
カプセル剤は、まず上述のようにして粉末状となった末剤や散剤又は錠剤の項で述べるように顆粒化したものを、例えばゼラチンカプセルのようなカプセル外皮の中へ充填することにより製造される。滑沢剤や流動化剤、例えばコロイド状のシリカ、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、固形のポリエチレングリコールのようなものを粉末状態のものに混合し、然るのちに充填操作を行うこともできる。崩壊剤や可溶化剤、例えばカルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、低置換度ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、炭酸カルシウム、炭酸ナトリウムを添加すれば、カプセル剤が摂取されたときの医薬の有効性を改善することができる。
【0018】
また、薬物の微粉末を植物油、ポリエチレングリコール、グリセリン、界面活性剤中に懸濁分散し、これをゼラチンシートで包んで軟カプセル剤とすることができる。錠剤は賦形剤を加えて粉末混合物を作り、顆粒化もしくはスラグ化し、ついで崩壊剤又は滑沢剤を加えたのち打錠することにより製造される。粉末混合物は、適当に粉末化された薬物を上述の希釈剤やベースと混合し、必要に応じ結合剤(例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、ゼラチン、ポリビニルピロリドン、ポリビニルアルコールなど)、溶解遅延化剤(例えば、パラフィンなど)、再吸収剤(例えば、四級塩)や吸着剤(例えばベントナイト、カオリン、リン酸ジカルシウムなど)をも併用してもよい。粉末混合物は、まず結合剤、例えばシロップ、澱粉糊、アラビアゴム、セルロース溶液又は高分子物質溶液で湿らせ、攪拌混合し、これを乾燥、粉砕して顆粒とすることができる。このように粉末を顆粒化するかわりに、まず打錠機にかけたのち、得られる不完全な形態のスラグを破砕して顆粒にすることも可能である。
【0019】
このようにして作られる顆粒は、滑沢剤としてステアリン酸、ステアリン酸塩、タルク、ミネラルオイルその他を添加することにより、互いに付着することを防ぐことができる。このように滑沢化された混合物をついで打錠する。こうして製造した素錠にフィルムコーティングや糖衣を施すことができる。
また薬物は、上述のように顆粒化やスラグ化の工程を経ることなく、流動性の不活性担体と混合したのちに直接打錠してもよい。シェラックの密閉被膜からなる透明又は半透明の保護被覆、糖や高分子材料の被覆、及び、ワックスよりなる磨上被覆の如きも用いうる。
【0020】
他の経口投与剤型、例えば溶液、シロップ、エリキシルなどもまたその一定量が薬物の一定量を含有するように用量単位形態にすることができる。シロップは、薬物を適当な香味水溶液に溶解して製造され、またエリキシルは非毒性のアルコール性担体を用いることにより製造される。懸濁剤は、薬物を非毒性担体中に分散させることにより処方される。可溶化剤や乳化剤(例えば、エトキシ化されたイソステアリルアルコール類、ポリオキシエチレンソルビトールエステル類)、保存剤、風味賦与剤(例えば、ペパミント油、サッカリン)その他もまた必要に応じ添加することができる。
必要とあらば、経口投与のための用量単位処方はマイクロカプセル化してもよい。該処方はまた被覆をしたり、高分子・ワックス等中にうめこんだりすることにより作用時間の延長や持続放出をもたらすこともできる。
【0021】
組織内投与は、皮下・筋肉又は静脈内注射用としたところの液状用量単位形態、例えば溶液や懸濁剤の形態を用いることによって行うことができる。これらのものは、薬物の一定量を、注射の目的に適合する非毒性の液状担体、例えば水性や油性の媒体に懸濁し又は溶解し、ついで該懸濁液又は溶液を滅菌することにより製造される。注射液を等張にするために非毒性の塩や塩溶液を添加してもよい。更に、安定剤、保存剤、乳化剤のようなものを併用することもできる。
【0022】
直腸投与は、薬物を低融点の水に可溶又は不溶の固体、例えばポリエチレングリコール、カカオ脂、半合成の油脂(例えば、ウイテプゾール、登録商標)、高級エステル類(例えばパルミチン酸ミリスチルエステル)及びそれらの混合物に溶解又は懸濁させて製造した坐剤等を用いることによって行うことができる。
【実施例】
【0023】
以下に試験例及び製剤例を掲げて本発明を更に詳しく説明するが、本発明はこれらのみに限定されるものではない。
【0024】
試験例1 マレイン酸イルソグラジンがウシ脳由来精製PDEに及ぼす影響についての検討
マレイン酸イルソグラジンが、ウシ脳由来精製PDEによるcAMP及びcGMPの加水分解に及ぼす影響について検討した。
ウシ脳から精製されたPDE(Sigma−RBI,Natick,MA,USA)を、cAMP又はcGMP、及び、マレイン酸イルソグラジン共存下、Krebs−Ringer bicarbonate液(120mM NaCl、3.3mM KCl、1.3mM CaCl2、1.2mM MgSO4、1.2mM KH2PO4、0.03mM EDTA、25mM NaHCO3、11mM glucose、pH7.4)(以下、KRB液という)中で30分間(37℃)のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液は10,000×gにて15分間(4℃)遠心した後、上清をcAMP及びcGMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMP及びcGMPの定量はEIAキット(Amersham,Buckinghamshire,U.K.)(以下同様)を用いて行った。その結果を図1(マレイン酸イルソグラジンがcAMPの加水分解に及ぼす影響)及び図2(マレイン酸イルソグラジンがcGMPの加水分解に及ぼす影響)に示す。
【0025】
cAMPの加水分解に及ぼす影響について、図1に示す通り、マレイン酸イルソグラジンはPDEによるcAMPの加水分解を濃度依存的に、かつ、ほぼ完全に抑制し、その抑制作用は、10−7M以上の濃度で統計学的に有意なものであった。一方、cGMPの加水分解に及ぼす影響については、図2に示す通り、マレイン酸イルソグラジンは、極めて高用量(10−5M)でのみ、cGMPの加水分解をわずかに抑制するにとどまった。
この結果から、マレイン酸イルソグラジンはPDEによるcAMPの加水分解のみを選択的に阻害し、PDEによるcGMPの加水分解に対しては明らかな阻害作用を有さないことが明らかである。
【0026】
試験例2 マレイン酸イルソグラジンのPDE阻害作用とPDEアイソザイムとの関係についての検討
ウシ脳由来精製PDEに含まれるアイソザイムを明らかにし、マレイン酸イルソグラジンのcAMP加水分解阻害作用と各PDEアイソザイムとの関係を検討した。
試験例1で用いたPDEを、cAMP及び各被験薬物共存下、KRB液中で30分間(37℃)のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液は10,000×gにて15分間(4℃)遠心した後、上清をcAMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMPの定量はEIAキットを用いて行った。その結果を図3に示す。
【0027】
図3に示すように、PDE1選択的阻害剤であるビンポセチン(5×10−5M)、PDE2選択的阻害剤であるエリスロ−9−(2−ヒドロキシ−3−ノニル)アデニン塩酸塩(EHNA)(5×10−5M)、及び、PDE4選択的阻害剤であるロリプラム(5×10−5M)は、いずれもウシ脳由来精製PDEによるcAMPの加水分解を部分的に抑制した。一方、PDE3選択的阻害剤であるシロスタミド(5×10−5M)は、ウシ脳由来精製PDEによるcAMPの加水分解に対して影響を及ぼさなかった。非選択的PDE阻害剤である3−イソブチル−1−メチルキサンチン(IBMX、10−3M)は、PDEによる加水分解を完全に抑制した。
この結果と試験例1の結果を総合的に判断すると、ウシ脳由来精製PDEには少なくともPDE1、PDE2及びPDE4のアイソザイムが含まれており、マレイン酸イルソグラジンは、PDE1、PDE2及びPDE4に対し、阻害作用を示すことが明らかである。
【0028】
試験例3 マレイン酸イルソグラジンがウシ心臓由来精製PDEに及ぼす影響についての検討
ウシ脳由来精製PDEの代わりにウシ心臓由来精製PDEを用い、試験例1及び試験例2と同様の検討を行った。
ウシ心臓から精製されたPDE(Sigma−RBI,Natick,MA,USA)を、cAMP及び各被験薬物(図4)又はマレイン酸イルソグラジン(図5)の共存下、KRB液中で30分間(37℃)のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液は10,000×gにて15分間(4℃)遠心した後、上清をcAMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMPの定量はEIAキットを用いて行った。その結果を図4(各種PDE選択的阻害剤がcAMPの加水分解に及ぼす影響)及び図5(マレイン酸イルソグラジンがcAMPの加水分解に及ぼす影響)に示す。
【0029】
図4に示すように、PDE1選択的阻害剤であるビンポセチン(5×10−5M)、及び、PDE3選択的阻害剤であるシロスタミド(5×10−5M)は、いずれもウシ心臓由来精製PDEによるcAMPの加水分解を部分的に抑制し、かつ、ビンポセチンとシロスタミドの併用による効果は相加的なものであった。一方、PDE2選択的阻害剤であるEHNA(5×10−5M)、及び、PDE4選択的阻害剤であるロリプラム(5×10−5M)は、ウシ心臓由来精製PDEによるcAMPの加水分解に対して影響を及ぼさなかった。非選択的PDE阻害剤であるIBMX(10−3M)は、PDEによる加水分解を完全に抑制した。
図5に示すように、マレイン酸イルソグラジンは、PDE1選択的阻害剤であるビンポセチン存在下(5×10−5M)でのPDEによるcAMPの加水分解を濃度依存的に抑制した。また、マレイン酸イルソグラジンは、PDE3選択的阻害剤であるシロスタミド存在下(5×10−5M)でのPDEによるcAMPの加水分解に対しても、濃度依存的な抑制作用を示した。
【0030】
これらの結果より、ウシ心臓由来精製PDEには少なくともPDE1及びPDE3のサブタイプが含まれていることが明らかである。この結果は、ウシ心臓由来PDEは主にPDE1とPDE3で構成されているとの報告(例えば、非特許文献14参照。)と一致する。また、マレイン酸イルソグラジンは、少なくとも、PDE1、及び、PDE3に対し、阻害作用を示すことが明らかである。
【0031】
試験例4 マレイン酸イルソグラジンがヒト好中球のcAMP産生に及ぼす影響についての検討
マレイン酸イルソグラジン、及び各種PDE阻害剤が、ヒト好中球のcAMP産生に及ぼす影響を検討した。
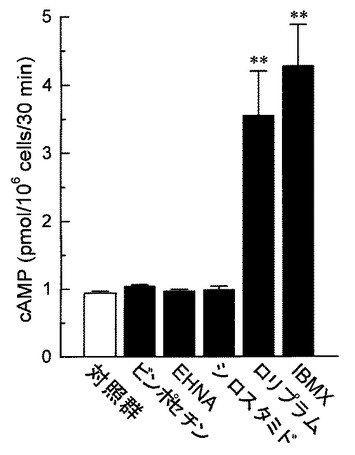
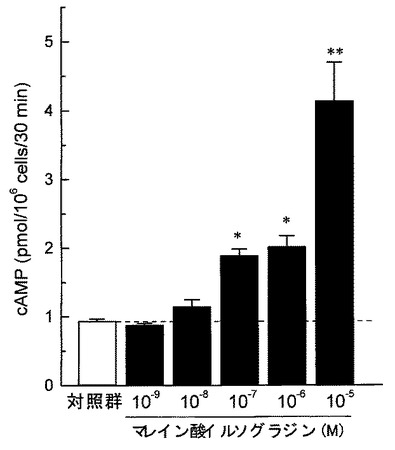
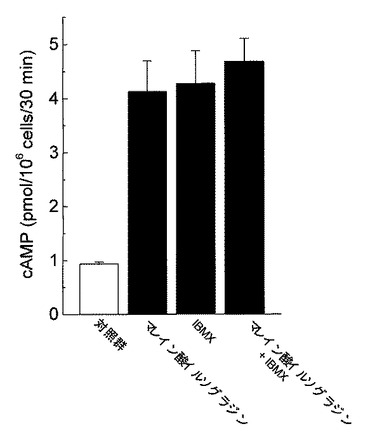
好中球は健常人静脈血より採取した。ヘパリン加血液を生理食塩水で希釈した後、血球分離剤(Polymorphprep,AXIS−Shield PoC AS,Oslo,Norway)に重層し、血液分離剤添付マニュアルに従い好中球を分離採取した。採取した好中球はHank’s balanced salt solution(HBSS)にて所定の濃度に調整した。cAMP含量の測定は、公知の方法に準拠した(例えば、非特許文献15参照。)。分離した好中球(1 assayあたり1×106個)をHBSSにて洗浄後、37℃にてCO2インキュベ−タ−中で10分間プレインキュベ−ションを行った。その後、各被験薬物を添加し、37°Cで30分間のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液を10,000×gにて15分間(4℃)遠心した後、上清をcAMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMP定量はEIAキットを用いて行った。その結果を図6(各種PDE選択的阻害剤が及ぼす影響)、図7(マレイン酸イルソグラジンが及ぼす影響)、図8(マレイン酸イルソグラジン、非選択的PDE阻害剤が及ぼす影響)に示す。
【0032】
図6に示すように、cAMP量は、PDE4選択的阻害剤であるロリプラム(5×10−5M)、又は非選択的PDE阻害剤であるIBMX(10−3M)を作用させた場合に増加し、PDE1選択的阻害剤であるビンポセチン(5×10−5M)、PDE2選択的阻害剤であるEHNA(5×10−5M)、又はPDE3選択的阻害剤であるシロスタミド(5×10−5M)をそれぞれ作用させた場合には、cAMP量の増加は見られなかった。
図7に示すように、マレイン酸イルソグラジンは、濃度依存的に好中球のcAMP産生量を増加させ、その効果は10−7M以上の濃度において統計学的に有意であった。
図8に示すように、マレイン酸イルソグラジンのcAMP産生亢進作用は、非選択的PDE阻害剤であるIBMX(10−3M)を共存させても増強しなかった。
【0033】
これらの結果から、ヒト好中球においては主にPDE4がcAMPの加水分解を担っていることが明らかである。この結果は、ヒト好中球にはPDE4Bが高発現しているとの報告(例えば、非特許文献16参照)と一致する。また、マレイン酸イルソグラジンは、少なくとも、PDE4に対し、阻害作用を示すことが明らかである。
【0034】
製剤例1
錠剤(内服錠)
処方1錠80mg中
マレイン酸イルソグラジン 2.0mg
トウモロコシ澱粉 49.6mg
結晶セルロース 24.0mg
メチルセルロース 4.0mg
ステアリン酸マグネシウム 0.4mg
この割合の混合末を通常の方法により打錠成形し内服錠とする。
製剤例2
錠剤(内服錠)
処方1錠80mg中
マレイン酸イルソグラジン 4.0mg
トウモロコシ澱粉 47.6mg
結晶セルロース 24.0mg
メチルセルロース 4.0mg
ステアリン酸マグネシウム 0.4mg
この割合の混合末を通常の方法により打錠成形し内服錠とする。
【産業上の利用可能性】
【0035】
本発明化合物は、PDEによるcAMPの加水分解のみを選択的に阻害し、PDEによるcGMPの加水分解に対しては明らかな阻害作用を有さないことから、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎、尿失禁若しくは血栓症に対する予防薬又は治療薬として有用である。
【技術分野】
【0001】
本発明は、次の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩(以下、本発明化合物という。)を有効成分として含有する、ホスホジエステラーゼのcAMP基質特異的阻害剤に関する。
【化1】
(式中、R1及びR2は、同一又は異なって、水素又はハロゲンを表す。)
【背景技術】
【0002】
本発明化合物は、優れた抗潰瘍作用、細胞防御作用、胃粘膜血流増加作用を有しており(例えば、特許文献1〜3、非特許文献1〜3参照。)、その中でも2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジン マレイン酸塩(一般名:マレイン酸イルソグラジン)(以下、マレイン酸イルソグラジンという。)は、商品名「ガスロンN」で、胃潰瘍、急性胃炎、慢性胃炎の治療剤として上市されている。また、マレイン酸イルソグラジンは、ラット胃粘膜細胞内cAMPの増加作用を有していることが知られている(例えば、非特許文献4参照。)。
【0003】
多くのホルモン及び神経伝達物質は、cAMP(3’,5’−サイクリックアデノシンモノホスフェート)の細胞内レベルを上昇させることで組織機能を調節している。セカンドメッセンジャーとして機能するcAMPの細胞内レベルは、合成及び分解を制御する機序によって調整される。
cAMPの合成はアデニル酸シクラーゼによって制御され、そのアデニル酸シクラーゼはホルスコリンなどの薬物によって直接活性化される。また、プロスタグランジン(PGE2あるいはPGI2など)やβ−アドレナリン受容体作動薬などは、細胞表面受容体への特異的作働薬であり、間接的にアデニル酸シクラーゼを活性化させて細胞内のcAMPを増加させる引き金としての役割を果たす。
一方、cAMPの分解は、いくつかのホスホジエステラーゼ(以下、PDEという。)アイソザイムによって制御される。これら一連のPDEは、cGMP(グアノシン3’,5’−サイクリックモノホスフェート)の分解も制御する。PDEは、cAMPの加水分解を制御することにより、細胞内情報伝達機構の活性化の程度やその持続に極めて重要な役割を果たしている。また、cAMPの生物学的機能の多様性を決めるコンパートメンテーションにも関与する重要な不活化酵素である。
【0004】
PDEは、その基質特異性、酵素活性調節機構、及び種々の阻害剤の感受性に基づいて機能的に分類され、現在まで、PDEには遺伝子的に異なる11種(PDE1〜PDE11)の既知のファミリーが知られている(例えば、非特許文献5参照)。この中で、PDE4、PDE7及びPDE8は、基質としてcAMPのみを加水分解するcAMP基質特異的PDEである。PDE5、PDE6及びPDE9は、基質としてcGMPのみを加水分解するcGMP特異的PDEである。PDE1〜PDE3、PDE10及びPDE11は、cAMPとcGMPの両環状ヌクレオチドを加水分解するPDEである。なかでもPDE1は、カルモジュリンにより制御されており、PDE2とPDE3はcGMPによって活性の制御が行われている。
PDEは、組織によりその発現が大きく異なることも良く知られている。例えば、PDE10とPDE11は、いずれもcAMP、cGMPの両環状ヌクレオチドを加水分解するPDEであるが、PDE10は脳に非常に多く分布する一方で、PDE11は骨格筋や前立腺など末梢組織に分布する。また、PDE5、PDE6ならびにPDE9はいずれもcGMP特異的PDEであるが、PDE6は網膜にのみ発現しているPDEとして知られている。
【0005】
PDE4阻害による細胞内cAMPの増加は、たとえば、好中球、好酸球、T−リンパ球、マクロファージなどの炎症性細胞の不活性化や、気管平滑筋の弛緩を引き起こすことが報告されている(例えば、非特許文献6、7参照。)。したがって、PDE4阻害剤は、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎などの治療に利用することができる。
PDE7もT−リンパ球に多く発現していることから(例えば、非特許文献8参照。)、その阻害剤は慢性関節リウマチや乾癬などT細胞依存性疾患の治療に利用することができる。
PDE1はカルモジュリン依存性のPDEである。PDE1の阻害剤は尿失禁の治療剤として有用である(例えば、特許文献9参照。)。
また、PDE3はcGMP阻害性のPDEである。PDE3阻害による細胞内cAMPの増加は、たとえば、血管平滑筋の弛緩や血小板凝集の阻害を引き起こすことから(例えば、非特許文献10参照。)、この阻害剤は血栓症の治療剤として有用である。
【0006】
【特許文献1】特開昭50−111085号公報
【特許文献2】特開昭50−111086号公報
【特許文献3】特開昭51−75083号公報
【非特許文献1】Ueda F,et al,Arzneim.−Forsch./Drug Res.,34(I),474(1984)
【非特許文献2】上田房雄ほか,応用薬理,33(1),143(1987)
【非特許文献3】平松新ほか,薬理と治療,11(7),2481(1983)
【非特許文献4】上田房雄ほか,応用薬理,33(1),157(1987)
【非特許文献5】Francis SH,et al,Prog Nucleic Acid Res Mol Biol,65,1−52(2001)
【非特許文献6】Barnes PJ,Nature,402,B31−B38(1999)
【非特許文献7】Mauro M,et al,Trends Pharmacol Sci,18,164−170(1997)
【非特許文献8】Li L,et al,Science,283,848−851(1999)
【非特許文献9】Truss MC,et al,World J Urol,18,439−443(2000)
【非特許文献10】Sudo T,et al,Biochem Pharmacol,59,347−356(2000)
【非特許文献11】Banner KH,et al,Eur Respir J,8,996−1000(1995)
【非特許文献12】Mauro M,et al,Trends Pharmacol Sci,18,164−170(1997)
【非特許文献13】厚生省医薬安全局,「医薬品等安全性情報別冊 平成8年度 新医薬品等の副作用のまとめ」,1997年10月,p12
【非特許文献14】Nichols MR,et al,Mol.Pharmacol.,57,738−45(2000)
【非特許文献15】Oka M,et al,Naunyn Schmiedebergs Arch Pharmacol,356,189−96(1997)
【非特許文献16】Wang P,et al,Mol.Pharmacol.,56,170−174(1999)
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、ベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、PDEのcAMP基質特異的阻害剤を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者は、本発明化合物が有する胃粘膜細胞内cAMPの増加作用について鋭意研究を重ねた結果、本発明化合物が、PDEによるcAMPの加水分解のみを選択的に阻害することを見出し、本発明を完成した。
【0009】
本発明は、
(1) 本発明化合物を有効成分として含有する、PDEのcAMP基質特異的阻害剤、
(2) ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、上記(1)の阻害剤、
(3) ベンゾグアナミン誘導体の医薬上許容される塩がマレイン酸イルソグラジンである、上記(1)の阻害剤、
(4) 本発明化合物を有効成分として含有する、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎、尿失禁若しくは血栓症に対する予防薬又はこれらの治療薬、
(5) ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、上記(4)の予防薬又は治療薬、
(6) ベンゾグアナミン誘導体の医薬上許容される塩がマレイン酸イルソグラジンである、上記(4)の予防薬又は治療薬、
に関する。
【0010】
後述の試験例1に示す通り、本発明化合物は、PDEによるcAMPの加水分解に対して強力な阻害作用を示す一方で、PDEによるcGMPの加水分解に対しては明らかな阻害作用を示さない。さらに詳細に検討した結果、後述の試験例2〜4に示す通り、PDE1、PDE2、PDE3及びPDE4の各アイソザイムに対してほとんど同程度の阻害活性を有する。
【0011】
PDE4阻害剤の有する優れた抗炎症作用などを臨床的に応用する場合、しばしば悪心、嘔吐などの消化器系の副作用の発現が問題になることは良く知られている(例えば、非特許文献11、12参照。)。本発明化合物は、臨床上、悪心、嘔吐などの消化器系の副作用をほとんど発現しないことから、PDE4阻害剤としても非常に有用である(例えば、非特許文献13参照。)。
【0012】
以下に本発明を詳述する。
「ホスホジエステラーゼのcAMP基質特異的阻害剤」とは、PDEによるcAMPの加水分解のみを選択的に阻害し、PDEによるcGMPの加水分解に対しては明らかな阻害作用を有さない薬剤をいう。
「ハロゲン」としては、例えば、フッ素、塩素、臭素、ヨウ素を挙げることができる。
「医薬上許容される塩」としては、例えば、塩酸、臭化水素酸、硫酸、燐酸などの鉱酸の塩、酢酸、クエン酸、酒石酸、マレイン酸、コハク酸、フマル酸、p−トルエンスルホン酸、ベンゼンスルホン酸、メタンスルホン酸などの有機酸の塩を挙げることができる。
【図面の簡単な説明】
【0013】
【図1】ウシ脳由来精製PDEによるcAMPの加水分解に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)、横軸はマレイン酸イルソグラジンの濃度(M)を表す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【図2】ウシ脳由来精製PDEによるcGMPの加水分解に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcGMP量(pmol/30min.)、横軸はマレイン酸イルソグラジンの濃度(M)を表す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「*」はダネット検定の結果(*P<0.05)を表す。
【図3】ウシ脳由来精製PDEによるcAMPの加水分解に対する各種PDE阻害剤の作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【図4】ウシ心臓由来精製PDEによるcAMPの加水分解に対する各種PDE阻害剤の作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【図5】ビンポセチン(5×10−5M)、又はシロスタミド(5×10−5M)存在下、ウシ心臓由来精製PDEによるcAMPの加水分解に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【図6】ヒト好中球のcAMP産生に対する各種PDE阻害剤の作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【図7】ヒト好中球のcAMP産生に対するマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)示す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「*」「**」はダネット検定の結果(*P<0.05、**P<0.01)を表す。
【図8】ヒト好中球のcAMP産生に対するIBMX存在下でのマレイン酸イルソグラジンの作用を検討した結果を示したグラフである。縦軸はcAMP量(pmol/30min.)を表す。各カラムは平均値±標準誤差を示す(n=5)。グラフ中の「**」はダネット検定の結果(**P<0.01)を表す。
【発明を実施するための最良の形態】
【0014】
本発明化合物は公知化合物であり、公知の方法(例えば、特許文献1〜3参照。)により製造することができる。例えば、モノ若しくはジハロゲノベンゾニトリルとジシアンジアミドとを反応させることにより、又は、モノ若しくはジハロゲノ安息香酸誘導体(カルボン酸、酸ハロゲン化物、エステル、アミド、等)とビグアナイドとを反応させることにより製造することができる。
本発明化合物は、遊離の塩基のまま医薬として用いることができるが、公知の方法により医薬上許容される塩の形にして用いることもできる。例えば、本発明化合物のマレイン酸塩は、ベンゾグアナミン誘導体を、当量のマレイン酸が溶解したアルコール溶液又は酢酸エチル溶液に溶解し、濃縮することにより得ることができる。
【0015】
本発明化合物は、そのまま又は医薬上許容される無毒性かつ不活性の担体中に、例えば0.01〜99.5%、好ましくは0.5〜90%を含有する医薬組成物として、人を含む動物に投与される。
担体としては、固形、半固形又は液状の希釈剤、充填剤及びその他の処方用の助剤一種以上が用いられる。薬物は投与単位形態で投与することが望ましい。また、薬物は、静脈内投与、経口投与、組織内投与、局所投与(経皮投与等)又は経直腸的に投与することができるが、これらに限定されるものではない。これらの投与方法に適した剤型で投与されるのはもちろんである。
PDEのcAMP基質特異的阻害剤としての用量は、疾患・傷害の性質と程度、年齢、体重などの患者の状態、投与経路などを考慮した上で設定することが望ましいが、通常は、成人に対して本発明に係る薬物の有効成分量として、1日あたり、0.1〜1000mg/ヒトの範囲、好ましくは1〜500mg/ヒトの範囲が一般的である。
場合によっては、これ以下で足りるし、また逆にこれ以上の用量を必要とすることもある。また1日2〜4回に分割して投与することもできる。
【0016】
経口投与は固形又は液状の用量単位、例えば、末剤、散剤、錠剤、糖衣剤、カプセル剤、顆粒剤、懸濁剤、液剤、シロップ剤、ドロップ剤、舌下錠その他の剤型によって行うことができる。
末剤は薬物を適当な細かさにすることにより製造される。散剤は薬物を適当な細かさと成し、ついで同様に細かくした医薬用担体、例えば澱粉、マンニトールのような可食性炭水化物その他と混合することにより製造される。必要に応じ風味剤、保存剤、分散剤、着色剤、香料その他のものを混じてもよい。
【0017】
カプセル剤は、まず上述のようにして粉末状となった末剤や散剤又は錠剤の項で述べるように顆粒化したものを、例えばゼラチンカプセルのようなカプセル外皮の中へ充填することにより製造される。滑沢剤や流動化剤、例えばコロイド状のシリカ、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、固形のポリエチレングリコールのようなものを粉末状態のものに混合し、然るのちに充填操作を行うこともできる。崩壊剤や可溶化剤、例えばカルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、低置換度ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、炭酸カルシウム、炭酸ナトリウムを添加すれば、カプセル剤が摂取されたときの医薬の有効性を改善することができる。
【0018】
また、薬物の微粉末を植物油、ポリエチレングリコール、グリセリン、界面活性剤中に懸濁分散し、これをゼラチンシートで包んで軟カプセル剤とすることができる。錠剤は賦形剤を加えて粉末混合物を作り、顆粒化もしくはスラグ化し、ついで崩壊剤又は滑沢剤を加えたのち打錠することにより製造される。粉末混合物は、適当に粉末化された薬物を上述の希釈剤やベースと混合し、必要に応じ結合剤(例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、ゼラチン、ポリビニルピロリドン、ポリビニルアルコールなど)、溶解遅延化剤(例えば、パラフィンなど)、再吸収剤(例えば、四級塩)や吸着剤(例えばベントナイト、カオリン、リン酸ジカルシウムなど)をも併用してもよい。粉末混合物は、まず結合剤、例えばシロップ、澱粉糊、アラビアゴム、セルロース溶液又は高分子物質溶液で湿らせ、攪拌混合し、これを乾燥、粉砕して顆粒とすることができる。このように粉末を顆粒化するかわりに、まず打錠機にかけたのち、得られる不完全な形態のスラグを破砕して顆粒にすることも可能である。
【0019】
このようにして作られる顆粒は、滑沢剤としてステアリン酸、ステアリン酸塩、タルク、ミネラルオイルその他を添加することにより、互いに付着することを防ぐことができる。このように滑沢化された混合物をついで打錠する。こうして製造した素錠にフィルムコーティングや糖衣を施すことができる。
また薬物は、上述のように顆粒化やスラグ化の工程を経ることなく、流動性の不活性担体と混合したのちに直接打錠してもよい。シェラックの密閉被膜からなる透明又は半透明の保護被覆、糖や高分子材料の被覆、及び、ワックスよりなる磨上被覆の如きも用いうる。
【0020】
他の経口投与剤型、例えば溶液、シロップ、エリキシルなどもまたその一定量が薬物の一定量を含有するように用量単位形態にすることができる。シロップは、薬物を適当な香味水溶液に溶解して製造され、またエリキシルは非毒性のアルコール性担体を用いることにより製造される。懸濁剤は、薬物を非毒性担体中に分散させることにより処方される。可溶化剤や乳化剤(例えば、エトキシ化されたイソステアリルアルコール類、ポリオキシエチレンソルビトールエステル類)、保存剤、風味賦与剤(例えば、ペパミント油、サッカリン)その他もまた必要に応じ添加することができる。
必要とあらば、経口投与のための用量単位処方はマイクロカプセル化してもよい。該処方はまた被覆をしたり、高分子・ワックス等中にうめこんだりすることにより作用時間の延長や持続放出をもたらすこともできる。
【0021】
組織内投与は、皮下・筋肉又は静脈内注射用としたところの液状用量単位形態、例えば溶液や懸濁剤の形態を用いることによって行うことができる。これらのものは、薬物の一定量を、注射の目的に適合する非毒性の液状担体、例えば水性や油性の媒体に懸濁し又は溶解し、ついで該懸濁液又は溶液を滅菌することにより製造される。注射液を等張にするために非毒性の塩や塩溶液を添加してもよい。更に、安定剤、保存剤、乳化剤のようなものを併用することもできる。
【0022】
直腸投与は、薬物を低融点の水に可溶又は不溶の固体、例えばポリエチレングリコール、カカオ脂、半合成の油脂(例えば、ウイテプゾール、登録商標)、高級エステル類(例えばパルミチン酸ミリスチルエステル)及びそれらの混合物に溶解又は懸濁させて製造した坐剤等を用いることによって行うことができる。
【実施例】
【0023】
以下に試験例及び製剤例を掲げて本発明を更に詳しく説明するが、本発明はこれらのみに限定されるものではない。
【0024】
試験例1 マレイン酸イルソグラジンがウシ脳由来精製PDEに及ぼす影響についての検討
マレイン酸イルソグラジンが、ウシ脳由来精製PDEによるcAMP及びcGMPの加水分解に及ぼす影響について検討した。
ウシ脳から精製されたPDE(Sigma−RBI,Natick,MA,USA)を、cAMP又はcGMP、及び、マレイン酸イルソグラジン共存下、Krebs−Ringer bicarbonate液(120mM NaCl、3.3mM KCl、1.3mM CaCl2、1.2mM MgSO4、1.2mM KH2PO4、0.03mM EDTA、25mM NaHCO3、11mM glucose、pH7.4)(以下、KRB液という)中で30分間(37℃)のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液は10,000×gにて15分間(4℃)遠心した後、上清をcAMP及びcGMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMP及びcGMPの定量はEIAキット(Amersham,Buckinghamshire,U.K.)(以下同様)を用いて行った。その結果を図1(マレイン酸イルソグラジンがcAMPの加水分解に及ぼす影響)及び図2(マレイン酸イルソグラジンがcGMPの加水分解に及ぼす影響)に示す。
【0025】
cAMPの加水分解に及ぼす影響について、図1に示す通り、マレイン酸イルソグラジンはPDEによるcAMPの加水分解を濃度依存的に、かつ、ほぼ完全に抑制し、その抑制作用は、10−7M以上の濃度で統計学的に有意なものであった。一方、cGMPの加水分解に及ぼす影響については、図2に示す通り、マレイン酸イルソグラジンは、極めて高用量(10−5M)でのみ、cGMPの加水分解をわずかに抑制するにとどまった。
この結果から、マレイン酸イルソグラジンはPDEによるcAMPの加水分解のみを選択的に阻害し、PDEによるcGMPの加水分解に対しては明らかな阻害作用を有さないことが明らかである。
【0026】
試験例2 マレイン酸イルソグラジンのPDE阻害作用とPDEアイソザイムとの関係についての検討
ウシ脳由来精製PDEに含まれるアイソザイムを明らかにし、マレイン酸イルソグラジンのcAMP加水分解阻害作用と各PDEアイソザイムとの関係を検討した。
試験例1で用いたPDEを、cAMP及び各被験薬物共存下、KRB液中で30分間(37℃)のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液は10,000×gにて15分間(4℃)遠心した後、上清をcAMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMPの定量はEIAキットを用いて行った。その結果を図3に示す。
【0027】
図3に示すように、PDE1選択的阻害剤であるビンポセチン(5×10−5M)、PDE2選択的阻害剤であるエリスロ−9−(2−ヒドロキシ−3−ノニル)アデニン塩酸塩(EHNA)(5×10−5M)、及び、PDE4選択的阻害剤であるロリプラム(5×10−5M)は、いずれもウシ脳由来精製PDEによるcAMPの加水分解を部分的に抑制した。一方、PDE3選択的阻害剤であるシロスタミド(5×10−5M)は、ウシ脳由来精製PDEによるcAMPの加水分解に対して影響を及ぼさなかった。非選択的PDE阻害剤である3−イソブチル−1−メチルキサンチン(IBMX、10−3M)は、PDEによる加水分解を完全に抑制した。
この結果と試験例1の結果を総合的に判断すると、ウシ脳由来精製PDEには少なくともPDE1、PDE2及びPDE4のアイソザイムが含まれており、マレイン酸イルソグラジンは、PDE1、PDE2及びPDE4に対し、阻害作用を示すことが明らかである。
【0028】
試験例3 マレイン酸イルソグラジンがウシ心臓由来精製PDEに及ぼす影響についての検討
ウシ脳由来精製PDEの代わりにウシ心臓由来精製PDEを用い、試験例1及び試験例2と同様の検討を行った。
ウシ心臓から精製されたPDE(Sigma−RBI,Natick,MA,USA)を、cAMP及び各被験薬物(図4)又はマレイン酸イルソグラジン(図5)の共存下、KRB液中で30分間(37℃)のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液は10,000×gにて15分間(4℃)遠心した後、上清をcAMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMPの定量はEIAキットを用いて行った。その結果を図4(各種PDE選択的阻害剤がcAMPの加水分解に及ぼす影響)及び図5(マレイン酸イルソグラジンがcAMPの加水分解に及ぼす影響)に示す。
【0029】
図4に示すように、PDE1選択的阻害剤であるビンポセチン(5×10−5M)、及び、PDE3選択的阻害剤であるシロスタミド(5×10−5M)は、いずれもウシ心臓由来精製PDEによるcAMPの加水分解を部分的に抑制し、かつ、ビンポセチンとシロスタミドの併用による効果は相加的なものであった。一方、PDE2選択的阻害剤であるEHNA(5×10−5M)、及び、PDE4選択的阻害剤であるロリプラム(5×10−5M)は、ウシ心臓由来精製PDEによるcAMPの加水分解に対して影響を及ぼさなかった。非選択的PDE阻害剤であるIBMX(10−3M)は、PDEによる加水分解を完全に抑制した。
図5に示すように、マレイン酸イルソグラジンは、PDE1選択的阻害剤であるビンポセチン存在下(5×10−5M)でのPDEによるcAMPの加水分解を濃度依存的に抑制した。また、マレイン酸イルソグラジンは、PDE3選択的阻害剤であるシロスタミド存在下(5×10−5M)でのPDEによるcAMPの加水分解に対しても、濃度依存的な抑制作用を示した。
【0030】
これらの結果より、ウシ心臓由来精製PDEには少なくともPDE1及びPDE3のサブタイプが含まれていることが明らかである。この結果は、ウシ心臓由来PDEは主にPDE1とPDE3で構成されているとの報告(例えば、非特許文献14参照。)と一致する。また、マレイン酸イルソグラジンは、少なくとも、PDE1、及び、PDE3に対し、阻害作用を示すことが明らかである。
【0031】
試験例4 マレイン酸イルソグラジンがヒト好中球のcAMP産生に及ぼす影響についての検討
マレイン酸イルソグラジン、及び各種PDE阻害剤が、ヒト好中球のcAMP産生に及ぼす影響を検討した。
好中球は健常人静脈血より採取した。ヘパリン加血液を生理食塩水で希釈した後、血球分離剤(Polymorphprep,AXIS−Shield PoC AS,Oslo,Norway)に重層し、血液分離剤添付マニュアルに従い好中球を分離採取した。採取した好中球はHank’s balanced salt solution(HBSS)にて所定の濃度に調整した。cAMP含量の測定は、公知の方法に準拠した(例えば、非特許文献15参照。)。分離した好中球(1 assayあたり1×106個)をHBSSにて洗浄後、37℃にてCO2インキュベ−タ−中で10分間プレインキュベ−ションを行った。その後、各被験薬物を添加し、37°Cで30分間のインキュベ−ションを行った。反応は、過塩素酸(最終濃度0.2M)を加えることにより停止した。反応液を10,000×gにて15分間(4℃)遠心した後、上清をcAMPの定量に用いた。上清はK2CO3を加えて中和し、10,000×gにて15分間(4℃)遠心した。cAMP定量はEIAキットを用いて行った。その結果を図6(各種PDE選択的阻害剤が及ぼす影響)、図7(マレイン酸イルソグラジンが及ぼす影響)、図8(マレイン酸イルソグラジン、非選択的PDE阻害剤が及ぼす影響)に示す。
【0032】
図6に示すように、cAMP量は、PDE4選択的阻害剤であるロリプラム(5×10−5M)、又は非選択的PDE阻害剤であるIBMX(10−3M)を作用させた場合に増加し、PDE1選択的阻害剤であるビンポセチン(5×10−5M)、PDE2選択的阻害剤であるEHNA(5×10−5M)、又はPDE3選択的阻害剤であるシロスタミド(5×10−5M)をそれぞれ作用させた場合には、cAMP量の増加は見られなかった。
図7に示すように、マレイン酸イルソグラジンは、濃度依存的に好中球のcAMP産生量を増加させ、その効果は10−7M以上の濃度において統計学的に有意であった。
図8に示すように、マレイン酸イルソグラジンのcAMP産生亢進作用は、非選択的PDE阻害剤であるIBMX(10−3M)を共存させても増強しなかった。
【0033】
これらの結果から、ヒト好中球においては主にPDE4がcAMPの加水分解を担っていることが明らかである。この結果は、ヒト好中球にはPDE4Bが高発現しているとの報告(例えば、非特許文献16参照)と一致する。また、マレイン酸イルソグラジンは、少なくとも、PDE4に対し、阻害作用を示すことが明らかである。
【0034】
製剤例1
錠剤(内服錠)
処方1錠80mg中
マレイン酸イルソグラジン 2.0mg
トウモロコシ澱粉 49.6mg
結晶セルロース 24.0mg
メチルセルロース 4.0mg
ステアリン酸マグネシウム 0.4mg
この割合の混合末を通常の方法により打錠成形し内服錠とする。
製剤例2
錠剤(内服錠)
処方1錠80mg中
マレイン酸イルソグラジン 4.0mg
トウモロコシ澱粉 47.6mg
結晶セルロース 24.0mg
メチルセルロース 4.0mg
ステアリン酸マグネシウム 0.4mg
この割合の混合末を通常の方法により打錠成形し内服錠とする。
【産業上の利用可能性】
【0035】
本発明化合物は、PDEによるcAMPの加水分解のみを選択的に阻害し、PDEによるcGMPの加水分解に対しては明らかな阻害作用を有さないことから、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎、尿失禁若しくは血栓症に対する予防薬又は治療薬として有用である。
【特許請求の範囲】
【請求項1】
次の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、ホスホジエステラーゼのcAMP基質特異的阻害剤。
【化2】
(式中、R1及びR2は、同一又は異なって、水素又はハロゲンを表す。)
【請求項2】
ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、請求項1記載の阻害剤。
【請求項3】
ベンゾグアナミン誘導体の医薬上許容される塩が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジン マレイン酸塩である、請求項1記載の阻害剤。
【請求項4】
請求項1記載の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎、尿失禁若しくは血栓症に対する予防薬又はこれらの治療薬。
【請求項5】
ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、請求項4記載の予防薬又は治療薬。
【請求項6】
ベンゾグアナミン誘導体の医薬上許容される塩が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジン マレイン酸塩である、請求項4記載の予防薬又は治療薬。
【請求項1】
次の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、ホスホジエステラーゼのcAMP基質特異的阻害剤。
【化2】
(式中、R1及びR2は、同一又は異なって、水素又はハロゲンを表す。)
【請求項2】
ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、請求項1記載の阻害剤。
【請求項3】
ベンゾグアナミン誘導体の医薬上許容される塩が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジン マレイン酸塩である、請求項1記載の阻害剤。
【請求項4】
請求項1記載の一般式(I)で表されるベンゾグアナミン誘導体又はその医薬上許容される塩を有効成分として含有する、アトピー性喘息を含む気管支喘息、慢性気管支炎、慢性閉塞性肺疾患、成人性呼吸困難症候群、全身性エリテマトーデス、アレルギー性鼻炎、アトピー性皮膚炎、結膜炎、じんま疹、乾癬、ケロイド、歯肉炎、歯周炎、歯槽膿漏、強皮症、慢性関節リウマチ、慢性糸球体腎炎、クローン病、潰瘍性大腸炎、食道炎、尿失禁若しくは血栓症に対する予防薬又はこれらの治療薬。
【請求項5】
ベンゾグアナミン誘導体が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジンである、請求項4記載の予防薬又は治療薬。
【請求項6】
ベンゾグアナミン誘導体の医薬上許容される塩が2,4−ジアミノ−6−(2,5−ジクロロフェニル)−1,3,5−トリアジン マレイン酸塩である、請求項4記載の予防薬又は治療薬。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【国際公開番号】WO2005/026132
【国際公開日】平成17年3月24日(2005.3.24)
【発行日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願番号】特願2005−513980(P2005−513980)
【国際出願番号】PCT/JP2004/013648
【国際出願日】平成16年9月17日(2004.9.17)
【出願人】(000004156)日本新薬株式会社 (46)
【Fターム(参考)】
【国際公開日】平成17年3月24日(2005.3.24)
【発行日】平成19年11月8日(2007.11.8)
【国際特許分類】
【国際出願番号】PCT/JP2004/013648
【国際出願日】平成16年9月17日(2004.9.17)
【出願人】(000004156)日本新薬株式会社 (46)
【Fターム(参考)】
[ Back to top ]