
ホタル由来ルシフェラーゼ
【課題】高い強度でルシフェリンを発光させるルシフェラーゼを提供する。
【解決手段】オキナワマドボタル(Pyrocoelia matsumurai)に由来し、野生型ルシフェラーゼのアミノ酸配列との間で80%以上の相同性を示し、かつ野生型ルシフェラーゼと比較してより高い強度でルシフェリンを発光させる、変異型ルシフェラーゼ。
【解決手段】オキナワマドボタル(Pyrocoelia matsumurai)に由来し、野生型ルシフェラーゼのアミノ酸配列との間で80%以上の相同性を示し、かつ野生型ルシフェラーゼと比較してより高い強度でルシフェリンを発光させる、変異型ルシフェラーゼ。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はホタル由来ルシフェラーゼに関する。
【背景技術】
【0002】
細胞内のシグナル伝達および遺伝子発現といった細胞の機能を調べるためには、蛍光色素および蛍光タンパク質といった蛍光プローブ並びにルシフェリン・ルシフェラーゼ反応を利用する化学発光プローブが用いられている。特に遺伝子の発現調節の解析には、励起光による細胞のダメージが生じず且つ自家発光の問題が生じない、定量性に優れた発光計測が用いられる。例えば、ルシフェラーゼ遺伝子が導入された細胞を観察する場合、ルシフェラーゼ活性に因る細胞からの発光強度を測定することで、ルシフェラーゼ遺伝子の発現の強さ(具体的には発現量)を調べることができる。具体的には、最初に細胞を溶解して細胞溶解液を作製し、その後この細胞溶解液にルシフェリンおよびアデノシン三リン酸(ATP)等を添加し、光電子増倍管を用いたルミノメーターで発光強度を定量し、その結果に基づいて発現量を特定する。この方法では、発光強度の測定は細胞を溶解した後に行われる。このため、ある時点でのルシフェラーゼ遺伝子の発現量は、測定に使用した全細胞の平均値として得られる。ルシフェラーゼ遺伝子等の発光遺伝子をレポーター遺伝子として導入する方法として、例えばリン酸カルシウム法、リポフェクチン法またはエレクトロポレーション法等を使用でき、各方法は目的および細胞の種類の違いに応じて使い分けられている。細胞に導入するルシフェラーゼ遺伝子の上流または下流に目的のDNA断片を繋ぎ、ルシフェラーゼ遺伝子の発現量を測定することで、当該DNA断片がルシフェラーゼ遺伝子の転写に及ぼす影響を調べることが可能となる。また、細胞に導入するルシフェラーゼ遺伝子と目的の遺伝子とを共発現させることで、当該遺伝子産物がルシフェラーゼ遺伝子の発現に及ぼす影響を調べることが可能となる。
【0003】
発光遺伝子発現量の時間変化を調べるには、生きた細胞からの発光強度を経時的に測定する必要がある。このような測定は、ルミノメーターを備えたインキュベーターにて細胞を培養し、細胞集団全体からの発光強度を一定時間ごとに測定することで行われる。細胞集団全体における発光遺伝子の発現量の経時的な変化を捉えることにより、一定の周期性をもった発現リズム等を分析することができる。
【0004】
近年、生物学および医学の研究において、生命現象を分子レベルで動的に捉える必要性が高まってきている。蛍光観察を利用する研究分野では、試料内のタンパク質分子機能を動的に捉えるために、タイムラプスまたは動画撮像が行われている。従来技術では、蛍光試料を用いた経時的観察(例えば、蛍光分子を付したタンパク質1分子の動画観察)が行われている。
【0005】
一方、経時的観察のために化学発光試料を用いる場合、化学発光試料の発光強度は極めて小さいため、イメージ・インテンシファイアを装着したCCDカメラを用いて観察する必要がある。また最近では、化学発光試料を観察するための光学系を有した顕微鏡も開発されている(特許文献1および2)。
【0006】
発光強度が小さい化学発光試料を顕微鏡により撮像する場合、鮮明な画像を撮影するために必要な露出時間は長くなる。このような化学発光試料は、使用できる研究用途が制限される。例えば、発光強度の小ささのために30分間の露出時間が必要となる場合、30分間隔での経時的撮影は可能であっても、より短い間隔での撮影、さらにはリアルタイムでの撮影は不可能である。また、画像の取得に当って、発光する細胞に焦点を合わせるために複数枚の画像を取得し比較する必要があるが、発光強度の小ささのために長い露出時間を必要とする場合、1枚の画像を取得するだけでも手間と時間を要することになる。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2006−301599号公報
【特許文献2】国際公開第06/088109号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、高い強度でルシフェリンを発光させるルシフェラーゼを提供することにある。
【課題を解決するための手段】
【0009】
本発明の一態様に係る変異型ルシフェラーゼは、オキナワマドボタル(Pyrocoelia matsumurai)に由来し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる。
【発明の効果】
【0010】
本発明によれば、高い発光強度を示すルシフェラーゼが提供されるため、発光像を撮像するために必要な露出時間を短縮することができ、且つ従来よりも時間分解能が高い経時的観察が可能となる。
【図面の簡単な説明】
【0011】
【図1】野生型オキナワマドボタルルシフェラーゼを用いた場合に得られた、各pHにおける発光スペクトルを示す図である。
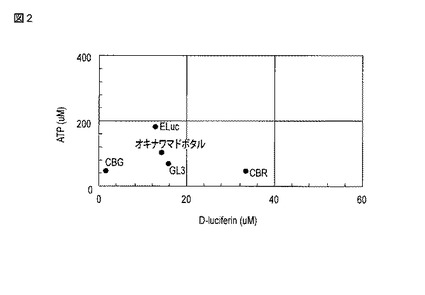
【図2】各ルシフェラーゼのKm値をプロットした図である。
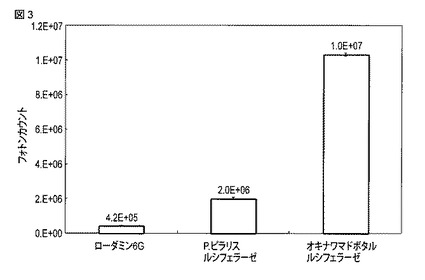
【図3】野生型オキナワマドボタルルシフェラーゼ、P.ピラリスルシフェラーゼおよびローダミン6Gを用いた場合に得られた発光強度を比較する図である。
【図4】哺乳細胞にて発現させた野生型オキナワマドボタルルシフェラーゼおよびP.ピラリスルシフェラーゼを用いた場合に得られた発光強度を比較する図である。
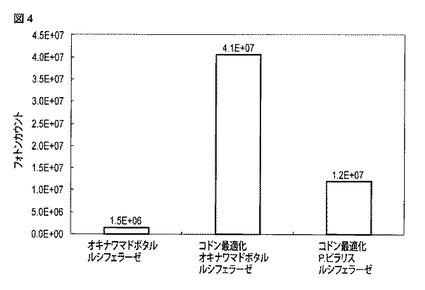
【図5】野生型オキナワマドボタルルシフェラーゼおよび変異型オキナワマドボタルルシフェラーゼを発現する細胞について得られた、D−ルシフェリン添加後の発光強度の時間変化を示す図である。
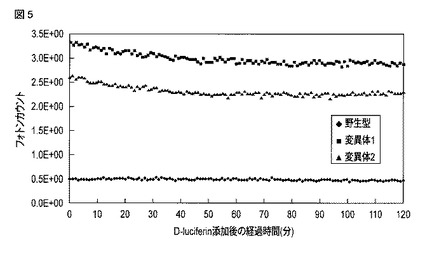
【図6】野生型オキナワマドボタルルシフェラーゼおよび変異型オキナワマドボタルルシフェラーゼを発現する細胞について得られた、セレンテラジン添加後の発光強度の時間変化を示す図である。
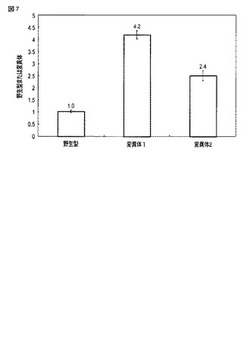
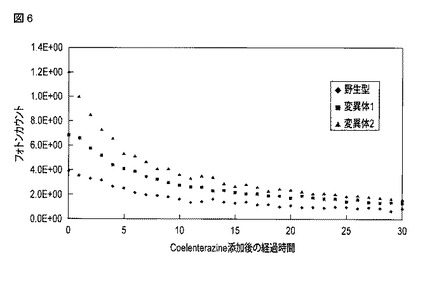
【図7】野生型オキナワマドボタルルシフェラーゼを用いた場合に得られた発光強度と変異型オキナワマドボタルルシフェラーゼを用いた場合に得られた発光強度とを比較した図である。
【発明を実施するための形態】
【0012】
本発明の実施形態は、オキナワマドボタル(Pyrocoelia matsumurai)に由来するルシフェラーゼに関する。
【0013】
「ルシフェラーゼ」とは、一般に、発光が生じる化学反応を触媒する酵素を指す。当該酵素の基質となる物質はルシフェリンと呼ばれる。ATPの存在下、ルシフェラーゼの触媒作用により、ルシフェリンが化学変化を起こす際に発光する。現在、ルシフェラーゼは、ホタルに由来するものおよびバクテリアに由来するものが取得されている。実施形態に係るルシフェラーゼも、上述の通り定義されるルシフェラーゼと同義であるが、後述するホタルから初めて取得された新規のルシフェラーゼである。実施形態に関してルシフェリンとは、好ましくはホタルルシフェラーゼの基質となるホタルルシフェリンを意味する。より好ましくは、ホタルルシフェリンとはD−ルシフェリンである。
【0014】
実施形態に係るルシフェラーゼは、オキナワマドボタル(Pyrocoelia matsumurai)に由来する。オキナワマドボタルとは節足動物門昆虫綱コウチュウ目ホタル科マドボタル属に属すホタルである。本発明の実施形態に関してオキナワマドボタルという場合、特に、主に沖縄本島に生息する基亜種ピロコエリア・マツムライ・マツムライ(Pyrocoelia matsumurai matsumurai)を意味する。なお、ここにいう「由来する」とは、オキナワマドボタルが有する野生型のルシフェラーゼに加えて、その変異体をも含むことを意味する。
【0015】
実施形態に係るルシフェラーゼによれば、既知のルシフェラーゼと比較して格段に高い発光強度が得られる。従って、実施形態に係るルシフェラーゼは、タンパク質のイメージングのためのレポーターとして利用する場合に特に有利な効果を奏する。すなわち、実施形態に係るルシフェラーゼは、少量でも高い発光強度を提供できるため、発現量が低いタンパク質の良好な検出を可能とする。また、実施形態に係るルシフェラーゼは、得られる発光強度が高いことにより検出に必要な露出時間を短縮できる。このため、実施形態に係るルシフェラーゼを経時的観察のためのレポーターとして利用すれば、撮影間隔を短くすることが可能となり、よりリアルタイムに近い観察が可能となる。すなわち、時間分解能が高い経時的観察が可能となる。
【0016】
実施形態に係るルシフェラーゼの一例は、配列番号1に示されるアミノ酸配列を含むルシフェラーゼである。当該ルシフェラーゼは、オキナワマドボタルから得られた野生型のルシフェラーゼである。この明細書および特許請求の範囲では、当該ルシフェラーゼを、野生型ルシフェラーゼ、野生型オキナワマドボタルルシフェラーゼまたは野生型オキナワマドボタル由来ルシフェラーゼ等と称する。
【0017】
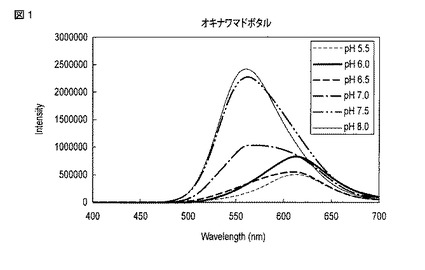
図1に、実施形態に係る野生型オキナワマドボタルルシフェラーゼを用いた場合に得られたルシフェリンの発光スペクトルを示す。この図に示されるとおり、実施形態に係る野生型オキナワマドボタルルシフェラーゼは、pH8において、最大発光波長が560nm付近にある発光を生じさせる。ここで「最大発光波長」とは、ルシフェラーゼが誘起するルシフェリンの発光を測定することによって得られるスペクトルにおいて、発光強度が最大となる波長を意味する。
【0018】
また、図3に、野生型オキナワマドボタルルシフェラーゼを用いた場合に得られたルシフェリンの発光強度、ローダミン(Rhodamine)6Gによる発光強度およびP.ピラリスルシフェラーゼを用いた場合に得られたルシフェリンの発光強度を比較したグラフが示される。この図から、野生型オキナワマドボタルルシフェラーゼを使用した場合、ローダミン6Gによる発光強度の24.4倍以上の発光強度を達成できることがわかる。この発光強度は、300nmから650nmの波長の光を10秒間取得して積算したものである。このように、実施形態に係るルシフェラーゼを用いた場合は、既知の発光物質および従来のホタルルシフェラーゼを用いた場合と比較して、より高い発光強度を達成できる。実施形態に係る野生型オキナワマドボタルルシフェラーゼにより、好ましくはローダミン6Gによる発光強度の5倍以上、10倍以上、15倍以上、20倍以上、21倍以上、22倍以上、23倍以上または24倍以上の発光強度が得られる。
【0019】
実施形態に係るルシフェラーゼは、オキナワマドボタルから取得された野生型のルシフェラーゼだけでなく、野生型ルシフェラーゼのアミノ酸配列の一部に変異が生じた変異型のルシフェラーゼを含む。ここでは、このようなルシフェラーゼを、変異型ルシフェラーゼ、変異型オキナワマドボタルルシフェラーゼまたは変異型オキナワマドボタル由来ルシフェラーゼ等と称する。
【0020】
そのような変異は、ルシフェラーゼの酵素活性を向上させる変異であってよい。すなわち、そのような変異は、野生型オキナワマドボタルルシフェラーゼよりも高い発光強度をもたらす変異であってよい。例えば、実施形態に係る変異型オキナワマドボタルルシフェラーゼを用いた場合に達成される発光強度は、野生型オキナワマドボタルルシフェラーゼを用いた場合に達成される発光強度の2倍以上、2.5倍以上、3倍以上、4倍以上または5倍以上であり得る。また、実施形態に係る変異型オキナワマドボタルルシフェラーゼを用いた場合に達成される発光強度は、ローダミン(Rhodamine)6Gによる発光強度の50倍以上、60倍以上、70倍以上、80倍以上、90倍以上、100倍以上または110倍以上であり得る。
【0021】
発光強度の向上をもたらす変異の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における424番目のイソロイシン(I)をロイシン(L)に置換する変異(I424L)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における531番目のイソロイシン(I)をアルギニン(R)に置換する変異(I531R)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における355番目のグルタミン酸(E)をアルギニン(R)に置換する変異(E355R)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における367番目のバリン(V)をアラニン(A)に置換する変異(V367A)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における446番目のリシン(K)をグルタミン(Q)に置換する変異(K446Q)である。これら計5種の変異は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)に対して、全て同時に、または任意の組合せで導入することができる。
【0022】
あるいは、上記5種の変異は、配列番号1とは異なるアミノ酸配列を有するタンパク質に対しても導入することができる。例えば、そのようなタンパク質のアミノ酸配列と配列番号1との対応関係を考慮して変異を導入することができる。そのようなタンパク質にI424L変異を導入したい場合、配列番号1と導入の対象とするタンパク質の配列とを比較して、配列番号1に示されるアミノ酸配列の424番目のIに対応するアミノ酸残基を決定し、当該アミノ酸をLに置換することでI424L変異を導入することができる。ここにおいて「対応するアミノ酸残基」は、例えば相同性検索といった公知の技術を利用して決定することができる。
【0023】
実施形態に係る変異型オキナワマドボタルルシフェラーゼの一例は、配列番号36に記載のアミノ酸配列を有するルシフェラーゼである。ここでは、この変異型ルシフェラーゼを変異体1と称する。変異体1は、野生型オキナワマドボタルルシフェラーゼ(配列番号1)に対して、I424L、E355R、V367AおよびK446Qの変異を導入して得られた変異型ルシフェラーゼである。変異体1を用いた場合に達成され得る発光強度は、例えば、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度の4.2倍以上である。また、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の24.4倍以上である。以上から、変異体1を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の102.5倍(=24.4倍×4.2倍)以上であると算出される。
【0024】
また、実施形態は、変異体1に対して更なる変異を導入したものも含む。例えば、変異体1に対して、さらなる発光強度の向上を目的として、さらなる変異を導入したものを含む。あるいは、変異体1に対して、発光強度の向上とは別の目的、例えば、可溶性の向上、分解に対する耐性の向上等を目的として、さらなる変異を導入したものを含む。具体的には、実施形態は、変異体1のアミノ酸配列(配列番号36)との間で一定の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼを含む。好ましくは、この変異型ルシフェラーゼは、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、4.2倍以上の強度でルシフェリンを発光させる。ここにおいて、アミノ酸配列における一定の相同性とは、例えば、75%以上、80%以上、85%以上、90%以上、95%以上、96%以上、97%以上、98%以上または99%以上の相同性である。また、好ましくは、変異体1に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列は、配列番号36に示されるアミノ酸配列の1から20のアミノ酸が変異したものであり、1から15のアミノ酸が変異したものであり、1から10のアミノ酸が変異したものであり、1から5のアミノ酸が変異したものであり、1から4のアミノ酸が変異したものであり、1から3のアミノ酸が変異したものであり、1または2のアミノ酸が変異したものである。好ましくは、変異体1に対して更なる変異が導入された変異型ルシフェラーゼでは、I424L、E355R、V367AおよびK446Qの変異が維持されている。すなわち、変異体1に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列において、424番目のアミノ酸(またはそれに対応するアミノ酸)がロイシンであり、355番目のアミノ酸(またはそれに対応するアミノ酸)がアルギニンであり、367番目のアミノ酸(またはそれに対応するアミノ酸)がアラニンであり、および446番目のアミノ酸(またはそれに対応するアミノ酸)がグルタミンである。ここにおいて「対応するアミノ酸」は、例えば相同性検索といった公知の技術を利用して決定することができる。
【0025】
実施形態に係る変異型オキナワマドボタルルシフェラーゼの別の例は、配列番号37に記載のアミノ酸配列を有するルシフェラーゼである。ここでは、この変異型ルシフェラーゼを変異体2と称する。変異体2は、野生型オキナワマドボタルルシフェラーゼ(配列番号1)に対して、I424L、I531R、E355R、V367AおよびK446Qの変異を導入して得られた変異型ルシフェラーゼである。変異体2による発光強度は、例えば、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度の2.4倍以上である。また、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の24.4倍以上である。以上から、変異体2を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の58.7倍(=24.4倍×2.4倍)以上であると算出される。
【0026】
また、実施形態は、変異体2に対して更なる変異を導入したものも含む。例えば、変異体2に対して、さらなる発光強度の向上を目的として、さらなる変異を導入したものを含む。あるいは、変異体2に対して、発光強度の向上とは別の目的、例えば、可溶性の向上、分解に対する耐性の向上等を目的として、さらなる変異を導入したものを含む。具体的には、実施形態は、変異体2のアミノ酸配列(配列番号37)との間で一定の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼを含む。好ましくは、この変異型ルシフェラーゼは、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼの2.4倍以上の強度でルシフェリンを発光させる。ここにおいて、アミノ酸配列における一定の相同性とは、例えば、75%以上、80%以上、85%以上、90%以上、95%以上、96%以上、97%以上、98%以上または99%以上の相同性である。また、好ましくは、変異体2に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列は、配列番号37に示されるアミノ酸配列の1から20のアミノ酸が変異したものであり、1から15のアミノ酸が変異したものであり、1から10のアミノ酸が変異したものであり、1から5のアミノ酸が変異したものであり、1から4のアミノ酸が変異したものであり、1から3のアミノ酸が変異したものであり、1または2のアミノ酸が変異したものである。好ましくは、変異体2に対して更なる変異が導入された変異型ルシフェラーゼでは、I424L、I531R、E355R、V367AおよびK446Qの変異が維持されている。すなわち、変異体1に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列において、424番目のアミノ酸(またはそれに対応するアミノ酸)がロイシンであり、531番目のアミノ酸(またはそれに対応するアミノ酸)がアルギニンであり、355番目のアミノ酸(またはそれに対応するアミノ酸)がアルギニンであり、367番目のアミノ酸(またはそれに対応するアミノ酸)がアラニンであり、および446番目のアミノ酸(またはそれに対応するアミノ酸)がグルタミンである。ここにおいて「対応するアミノ酸」は、例えば相同性検索といった公知の技術を利用して決定することができる。
【0027】
実施形態に係る変異型ルシフェラーゼのその他の例は、実験的な操作性を向上させる変異が導入された変異型ルシフェラーゼである。たとえば、野生型ルシフェラーゼが哺乳細胞内において低い可溶性を示す場合、実施形態に係る変異型ルシフェラーゼは、その可溶性を高める変異が導入されたルシフェラーゼである。
【0028】
変異型ルシフェラーゼとは、実施形態に係るルシフェラーゼの特徴、すなわち従来のルシフェラーゼと比較して高い発光強度が得られる限りにおいて、ルシフェラーゼのアミノ酸配列に変異、例えば、アミノ酸の置換、欠失および/または付加等が生じたルシフェラーゼであってよい。この変異とは、野生型ルシフェラーゼのアミノ酸配列の1以上のアミノ酸の変異または1から数個のアミノ酸の変異であり、好ましくは、野生型ルシフェラーゼのアミノ酸配列の1から20のアミノ酸の変異、1から15のアミノ酸の変異、1から10のアミノ酸の変異、1から5のアミノ酸の変異、1から4のアミノ酸の変異、1から3のアミノ酸の変異、1または2のアミノ酸の変異である。好ましくは、当該変異型ルシフェラーゼのアミノ酸配列は、野生型ルシフェラーゼのアミノ酸配列との間で75%以上、80%以上、85%以上、90%以上、95%以上、96%以上、97%以上、98%以上または99%以上の相同性を有する。
【0029】
本発明の他の実施形態は、上記実施形態に係るルシフェラーゼをコードする塩基配列を含む核酸に関する。すなわち、当該核酸は、オキナワマドボタルに由来するルシフェラーゼ遺伝子を含む核酸である。実施形態において、核酸とは、例えばDNAまたはRNAを指す。また、実施形態において、ルシフェラーゼの「遺伝子」とは、主として、mRNAに転写される領域、すなわち構造遺伝子を意味する。
【0030】
実施形態に係るルシフェラーゼをコードする塩基配列を含む核酸の一例は、配列番号2に示される塩基配列を含む核酸である。この配列を有する遺伝子は、オキナワマドボタルからクローニングされており、野生型オキナワマドボタルルシフェラーゼをコードする。
【0031】
実施形態に係るルシフェラーゼをコードする塩基配列を含む核酸の別の例は、配列番号34または35に示される塩基配列を含む核酸である。配列番号34の配列を有する遺伝子は変異体1をコードする。配列番号35の配列を有する遺伝子は変異体2をコードする。それぞれ、オキナワマドボタルからクローニングされた野生型の塩基配列に対し、哺乳細胞における発現のためのコドン最適化を行った後、特定のアミノ酸の置換に対応した変異を導入することで得られる配列である。
【0032】
実施形態に係る核酸は、野生型ルシフェラーゼ遺伝子の塩基配列を含む核酸であっても、そこにおいて変異が生じた変異型ルシフェラーゼ遺伝子の塩基配列を含む核酸であってもよい。ここにおいて、変異型ルシフェラーゼ遺伝子とは、コードされたルシフェラーゼが、実施形態に係るルシフェラーゼの特徴、すなわち従来のルシフェラーゼと比較して高い発光強度が得られる限りにおいて、塩基配列中の特定の塩基、たとえば数個の塩基の置換、欠失および/または付加等を受けた遺伝子であってよい。また、塩基配列の変異には、コードされるアミノ酸配列に変化が生じない変異が含まれる。すなわち、実施形態に係る核酸には、野生型ルシフェラーゼをコードする変異型ルシフェラーゼ遺伝子を含む核酸が含まれる。
【0033】
そのようなコードされるアミノ酸配列に変化が生じない変異の一例は、遺伝子中に存在する特定の制限酵素の認識配列を無効にする変異である。この変異によって、遺伝子を含む核酸は当該制限酵素によって切断されなくなるものの、当該遺伝子は変異前と同じアミノ酸配列を有するタンパク質をコードできる。このような変異は、制限酵素の認識配列を構成するコドンを、異なる塩基配列の同義コドンに変換することで達成できる。このような変異は、遺伝子組み換えに使用する制限酵素の認識配列が当該遺伝子中に存在する場合に有用である。この場合、予め遺伝子中の認識配列を無効にしておくことで、制限酵素で処理した場合に遺伝子を含む核酸が断片化することを防ぐことができ、結果として組み換えが容易になる。特定の制限酵素の認識配列を無効にした例は、配列番号3に示される塩基配列である。当該塩基配列は、オキナワマドボタルルシフェラーゼ遺伝子の塩基配列からBamHIおよびEcoRIの認識配列を無効にしたものである。
【0034】
そのようなコードされるアミノ酸配列に変化が生じない変異の別の例は、遺伝子のコドンを特定の生物種における発現に最適化させる変異である。ここにいう「最適化」とは、核酸に含まれる遺伝子のコドンを、特定の生物種においてコドン出現頻度が高いコドンに代えることを意味する。最適化を行った場合、特定の生物種における遺伝子の発現は、最適化をしない場合に比べて高まる。実施形態に係るルシフェラーゼ遺伝子はホタルから取得されたものであるため、当該遺伝子を導入しようとする生物種が分類学的にホタルから遠い程、最適化の効果はより高いと考えられる。実施形態において特定の生物種とは、例えば細菌細胞、酵母細胞および哺乳細胞である。更に哺乳細胞は、例えばマウスの細胞、サルの細胞およびヒトの細胞である。
【0035】
実施形態に係るコドンが最適化されたルシフェラーゼをコードする塩基配列を含む核酸の一例は、配列番号4に示される塩基配列を含む核酸である。当該核酸は、BamHIおよびEcoRIの認識配列が無効にされており、且つ哺乳細胞における発現にコドンが最適化されている。
【0036】
実施形態に係る核酸は、Kozak配列が付与されたルシフェラーゼ遺伝子の塩基配列を含む核酸を含む。Kozak配列とは、開始コドンとその前後の複数の塩基配列から成る配列であり、Kozak配列が存在することで、その遺伝子の発現量が増大することがわかっている。Kozak配列は、生物種または生物群ごとに共通配列が見出されている。実施形態に係るKozak配列を含む核酸は、それを導入する生物種に対応したKozak配列を有する。例えば哺乳細胞に導入される場合、核酸は、Kozak配列としてgccrccatgg(配列番号5)(rはグアニンまたはアデニンを意味する)の配列を含む。Kozak配列を付与するルシフェラーゼ遺伝子は、野生型遺伝子であってもよいが、上述のようなコドンの最適化を行った変異型遺伝子であってもよい。
【0037】
本発明のさらに他の実施形態はこれらの核酸を含むベクターに関する。当該ベクターには、ルシフェラーゼをコードする核酸以外に、発現を調節するための配列またはマーカー遺伝子の配列を含む核酸等を含んでよい。
【0038】
また、本発明のさらに他の実施形態は発光プローブに関する。当該発光プローブは、野生型のルシフェラーゼを含むものでもよいが、変異型ルシフェラーゼを含むものでもよい。特に、公知の技術によって、発光プローブとしての利用に適した改変を加えたものが好ましい。さらに実施形態は、上記発光プローブを用いた種々の試料(インビボ、インビトロ)に関する種々の使用(イメージング、測光等)に有効に適用できる。
【0039】
本発明のさらに他の実施形態は、実施形態に係るルシフェラーゼを利用して細胞内の機能を解析する方法に関する。当該方法は、実施形態に係るルシフェラーゼを細胞内に導入する工程、および前記ルシフェラーゼが誘起するルシフェリンの発光をイメージング装置により検出する工程を含む。例えば、DNA中の特定の発現調節領域の下流に実施形態に係るルシフェラーゼ遺伝子を導入し、ルシフェラーゼの発現を、それが誘起するルシフェリンの発光の有無によって検出することで、発現調節領域の機能を調べることが可能である。
【0040】
本発明のさらに他の実施形態は、上記実施形態に係るルシフェラーゼを利用して細胞内タンパク質を解析する方法に関する。当該方法は、実施形態に係るルシフェラーゼと解析の対象とするタンパク質とから成る融合タンパク質を細胞内に導入する工程;および、前記ルシフェラーゼが誘起するルシフェリンの発光をイメージング装置により検出する工程を含む。
【0041】
当該方法は、解析の対象とするタンパク質の細胞内の局在の観察、および、その局在の時間変化の観察(タイムラプス)を含む。また、当該方法は、タンパク質の局在だけでなく、そのタンパク質が単に発現したか否かの確認をも含む。使用される細胞に特別な限定はなく、細胞のイメージングの分野において通常使用できる細胞であってよい。また、解析の対象とするタンパク質も特別に限定はなく、研究の目的に応じたタンパク質を選択することができる。当該タンパク質は、使用する細胞内に本来存在するタンパクであってよく、または細胞内に本来存在しない異種性のまたは改変したタンパク質であってよい。
【0042】
融合タンパク質を細胞内に導入する場合、既知の導入方法を使用することができる。1つの方法は、細胞外で精製した融合タンパク質を細胞内に直接導入する方法である。例えば、マイクロインジェクション法によって融合タンパク質を細胞内に直接注入することができる。または、融合タンパク質を含む培養液にて細胞をインキュベートさせて、エンドサイトーシスによって融合タンパク質を細胞に取り込ませることができる。また別の方法は、まず融合タンパク質をコードする塩基配列を含む核酸を導入し、その後細胞内で融合タンパク質を発現させる方法である。例えば、当該核酸を含む発現ベクターを、リン酸カルシウム法、リポフェクション法またはエレクトロポレーション法等によって細胞内に導入し、発現ベクターから融合タンパク質を発現させることができる。ここにおいて、融合タンパク質の遺伝子は、実施形態に係るルシフェラーゼ遺伝子と解析の対象とするタンパク質の遺伝子とを含む遺伝子であり、ここにおいて、ルシフェラーゼ遺伝子とタンパク質遺伝子とは、それぞれ正常に翻訳されるよう連結されている。
【0043】
ルシフェリンの発光をイメージング装置により検出する工程は、既知の検出方法を使用することができる。例えば、ルシフェラーゼを含む融合タンパク質を発現する細胞に、ルシフェリン、ATPおよびMg2+イオン等を適宜与えてルシフェラーゼにルシフェリンの発光反応を誘起させ、発生した発光をイメージング装置により検出することができる。イメージング装置とは、例えば発光を捉えるためのフィルターを備えた顕微鏡である。顕微鏡を使用することで、細胞内における発光の位置を特定して、この情報をもとにタンパク質の局在を特定することが可能となる。また、イメージング装置として、経時的に撮像できる機能を備えた顕微鏡を使用することができ、この顕微鏡によって経時的観察も可能となる。
【実施例】
【0044】
[実施例1:オキナワマドボタル由来のルシフェラーゼ遺伝子のクローニング]
1.材料
材料として沖縄県沖縄本島で採集したオキナワマドボタル(Pyrocoelia matsumurai)の幼虫を用いた。
【0045】
2.トータルRNAの抽出とcDNAの合成
ホタルの幼虫からハサミを用いて発光器を切り取った。組織および細胞のホモジナイズ用のビーズを含むチューブであるLysing Matrix Dチューブ(MP−Biomedicals社)に、採取した発光器および1mLのトータルRNA抽出試薬TRIzol Reagent(インビトロジェン社)を入れた。このチューブを組織細胞破砕装置FastPrep 24(MP−Biomedicals社)またはFastPrep FP100A(MP−Biomedicals社)に装着し、振動速度6.5m/sおよび振動時間45秒の条件で、ホタルの発光器を試薬中にて破砕した。完了後、チューブを破砕装置から取り出し、30分間氷上に置いた。その後、もう一度同じ条件で破砕を実施した。
【0046】
次に、トータルRNA抽出試薬TRIzol Reagentの説明書に従って、破砕した溶液からトータルRNAの分離精製を行った。得られたmRNA溶液100μlを、エタノール沈殿法によって沈殿濃縮した。次に、完全長cDNA合成試薬GeneRacer(インビトロジェン社)をマニュアルに従って使用して、沈殿濃縮したトータルRNAから完全長cDNAを合成した。得られたcDNA溶液20μlをホタル完全長cDNAライブラリーとして以下の遺伝子実験に用いた。
【0047】
3.ホタルルシフェラーゼ遺伝子の5’末端側の同定
3−1.Rapid Amplification of cDNA End(RACE)法に用いるプライマーの作製
オキナワマドボタルのルシフェラーゼ遺伝子のクローニングをPolymerase chain reaction(PCR)法によって行った。このPCRに使用するプライマーは、既知の近縁生物由来のルシフェラーゼ遺伝子のアミノ酸配列に基づいて、以下の通り作製した。
【0048】
ホタルルシフェラーゼにおいてよく保存されているアミノ酸領域を確認するために、既に公開されている10種類のホタルルシフェラーゼのアミノ酸配列を、配列情報解析ソフトウェアDNASIS Pro(日立ソフトウェアエンジニアリング株式会社)を用いて比較した。比較に用いた近縁生物は、ラムフィリス・ノクティルカ(Lampyris noctiluca)(登録番号CAA61668)、ルキオラ・クルシアタ(Luciola cruciata)(登録番号P13129)、ルキオラ・ラテラリス(Luciola lateralis)(登録番号Q01158)、ルキオラ・ミングレリカ(Luciola mingrelica)(登録番号Q26304)、ホタリア・パルヴラ(Hotaria parvula)(登録番号AAC37253)、フォティヌス・ピラリス(Photinus pyralis)(登録番号BAF48390)、フォトゥリス・ペンシルヴァニカ(Photuris pennsylvanica)(登録番号Q27757)、ピロコエリア・ミヤコ(Pyrocoelia miyako)(登録番号AAC37254)、ピロコエリア・ルファ(Pyrocoelia rufa)(登録番号AAG45439)およびラハゴフタルムス・オーバイ(Rhagophthalmus ohbai)(登録番号BAF34360)である。
【0049】
その結果、ホタルルシフェラーゼのC末端側440残基付近に位置するL−I−K−Y−K−G−Y−Q−V(配列番号6)のアミノ酸配列がよく保存されていることがわかった。この9つのアミノ酸配列をコードするコドンから塩基配列を予測し、5’末端RACE PCRに用いる12種類のホタルルシフェラーゼ特異的混合プライマーを設計した。このプライマーの名称および配列は以下の通りである(プライマー配列中のY、RおよびNは混合塩基を示す):flexLuc5−ATA(5’−ACY TGR TAN CCY TTA TAT TTA AT−3’:配列番号7)、flexLuc5−ATG(5’−ACY TGR TAN CCY TTA TAT TTG AT−3’:配列番号8)、flexLuc5−ATT(5’−ACY TGR TAN CCY TTA TAT TTT AT−3’:配列番号9)、flexLuc5−ACA(5’−ACY TGR TAN CCY TTA TAC TTA AT−3’:配列番号10)、flexLuc5−ACG(5’−ACY TGR TAN CCY TTA TAC TTG AT−3’:配列番号11)、flexLuc5−ACT(5’−ACY TGR TAN CCY TTA TAC TTT AT−3’:配列番号12)、flexLuc5−GTA(5’−ACY TGR TAN CCY TTG TAT TTA AT−3’:配列番号13)、flexLuc5−GTG(5’−ACY TGR TAN CCY TTG TAT TTG AT−3’:配列番号14)、flexLuc5−GTT(5’−ACY TGR TAN CCY TTG TAT TTT AT−3’:配列番号15)、flexLuc5−GCA(5’−ACY TGR TAN CCY TTG TAC TTA AT−3’:配列番号16)、flexLuc5−GCG(5’−ACY TGR TAN CCY TTG TAC TTG AT−3’:配列番号17)、flexLuc5−GCT(5’−ACY TGR TAN CCY TTG TAC TTT AT−3’:配列番号18)。これらのプライマーの合成はライフテクノロジーズジャパン株式会社に委託して行った。
【0050】
3−2.5’−RACE PCRによる、ホタルルシフェラーゼ遺伝子の5’末端側のクローニング
上述の通り作製したホタル完全長cDNAライブラリーを鋳型として用い、上述の通り作製した12種類の特異的混合プライマーおよび5’末端特異的プライマーであるGeneRacer5’Primer(5’−CGA CTG GAG CAC GAG GAC ACT GA−3’:配列番号19)およびGeneRacer5’Nested Primer(5’−GGA CAC TGA CAT GGA CTG AAG GAG TA−3’:配列番号20)を用いて5’−RACE PCRを行った。GeneRacer5’ PrimerおよびGeneRacer5’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれているものを使用した。5’−RACE PCRによって効率的にルシフェラーゼ遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taq(タカラバイオ株式会社)を用いて、マニュアルに従って実施した。
【0051】
一度目のPCRとして、上述の通り作製した12種類の特異的混合プライマーのいずれかとGeneRacer5’ Primerとから成る12通りのプライマー対を用いてルシフェラーゼ遺伝子を増幅した。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度が1.0μMの12種類プライマーの内1つおよび最終濃度が0.3μMのGeneRacer3’ Primerを含む10μlのPCR反応溶液を作製し、そこへホタル完全長cDNAライブラリー溶液を0.2μl加えた。なお、ホタル完全長cDNAライブラリー溶液の濃度は未定量であった。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、45℃30秒および72℃90秒を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%トリス酢酸緩衝液(TAE)アガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。12の反応溶液の全てにおいてわずかに遺伝子増幅が認められたため、これらのPCR反応溶液を鋳型としてそれぞれnested PCR反応を次の通り実施した。
【0052】
nested PCRとして、一度目のPCRで使用した12種類プライマーのうちの4つとGeneRacer3’ Nested Primerとから成る4通りのプライマー対を用いてルシフェラーゼ遺伝子の増幅を行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.005U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度が1.0μMの12種類プライマーの内1つおよび最終濃度が0.3μMのGeneRacer3’ Primerを含む10μlのPCR反応溶液を作製し、そこへ鋳型として1度目のPCR反応溶液を滅菌水で10倍希釈した溶液を1.0μl加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、45℃30秒および72℃90秒を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。約1.4kbp付近に効率よく遺伝子が増幅したプライマーの組み合わせ条件を確認した。
【0053】
3−3.5’−RACEで増幅した遺伝子の塩基配列の決定
5’−RACEで増幅した遺伝子の塩基配列を読み取るため、ゲル抽出によるPCR産物の精製、サブクローニングおよびダイレクトシークエンスを実施した。詳細を以下に示す。
【0054】
約1.4kbp付近に効率よく遺伝子が増幅したプライマーの組み合わせでPCR(最終容量20μl)を実施し、ゲル抽出の手法を用いて目的とする遺伝子片を回収した。ゲル抽出はWizard SV Gel and PCR Clean−Up System(プロメガ株式会社)を用いて、マニュアルに従って実施した。TAクローニングの手法を用いて、ゲルから抽出したPCR産物のサブクローニングを実施した。TAクローニングはpGEM−T Easy Vector System(プロメガ株式会社)を用いて、マニュアルに従って実施した。その後、このベクターDNAを大腸菌(TOP10株またはDH5α株)に形質転換し、青・白スクリーニングの手法を用いてインサートポジティブコロニーを選択した。選択されたコロニーをダイレクトコロニーPCRに供し、遺伝子が導入されていることを確認した。ダイレクトコロニーPCRには、M13−F(−29) Primer(5’−CAC GAC GTT GTA AAA CGA C−3’:配列番号21)とM13 Reverse(5’−GGA TAA CAA TTT CAC AGG−3’:配列番号22)とから成るプライマー対を用いた。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度がそれぞれ0.2μMのプライマー対を含む10μlのPCR反応溶液を作製し、そこへ鋳型として少量の大腸菌コロニーを加えた。PCR反応は、最初に94℃1分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を25サイクル繰り返し、最後に72℃2分間の伸長反応を行った。PCR反応後、2μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0055】
増幅が確認できたPCR反応溶液について、ダイレクトシークエンシング法を用いてその遺伝子の塩基配列の決定を行った。PCR産物精製キットExoSAP−IT(GEヘルスケアバイオサイエンス)を用いて、PCR反応溶液に含まれる余剰なdNTPおよびプライマーを除去し、PCRダイレクトシークエンシングのための鋳型を調製した。BigDye Terminator v3.1 Cycle Sequencing Kit(アプライドバイオシステムズ)を用いて、この鋳型を含むシークエンシング反応溶液を調製し、サーマルサイクラーを用いてシークエンシング反応を行った。PCR産物の精製およびシークエンシングはそれぞれマニュアルに従って実施した。シークエンシング反応後、反応産物の精製を次の通りに行った。反応溶液に2.5倍量の100%エタノールを加え、遠心機を用いて核酸を沈殿させた。次に、上精を取り除いた後、70%エタノールを加えて沈殿を洗浄し、遠心機を用いて核酸を沈殿させた。最後に、上精を取り除いた後、沈殿を乾燥させた。精製した沈殿にHi−Di Formamide(アプライドバイオシステムズ)を15μl加え、溶解させた。この溶液を94℃2分間の熱変性させ、更に氷上で急冷して、塩基配列を決定するためのサンプルとした。このサンプルをApplied Biosystems 3130xl ジェネティックアナライザ(アプライドバイオシステムズ)を用いて塩基配列を読み取った。塩基配列の解析方法はマニュアルに従って実施した。
【0056】
シークエンシングによって得られた遺伝子配列を、配列情報解析ソフトウェア DNASIS Proの「シークエンス連結」機能を用いて解析した。この配列をNational Center for Biotechnology Information(NCBI)が提供するblastxサーチを利用して相同性検索を実施し、既知ホタルルシフェラーゼの塩基配列と高い相同性を示すことを確認した。以上の実験および解析で得られた塩基配列を新規ホタルルシフェラーゼ遺伝子の5’末端側であると決定した。
【0057】
4.ホタルルシフェラーゼ遺伝子の3’Race PCRおよび完全長cDNAの取得
4−1.3’Race PCRに用いるプライマーの設計
5’Race PCRの実験で得られたホタルルシフェラーゼ遺伝子の5’末端側非翻訳領域の配列を基にして、3’RACEに用いるプライマーおよびNested PCRに用いるプライマーを作成した。プライマーの合成はライフテクノロジーズジャパン株式会社に委託した。
【0058】
4−2.ホタルルシフェラーゼ遺伝子の完全長cDNA取得のための3’Race PCR
上述の通り作成したホタル完全長cDNAライブラリーを鋳型として用い、目的とするホタルルシフェラーゼの5’末端側非翻訳領域の塩基配列から作製したプライマーNo6(2/3)−Full−F1(GACACTAACGCGCTAACATCATTGCAAGA:配列番号23)およびGeneRacer3’ Primer(5’−GCT GTC AAC GAT ACG CTA CGT AAC G−3’:配列番号24)およびGeneRacer3’ Nested Primer(5’−CGC TAC GTA ACG GCA TGA CAG TG−3’:配列番号25)を用いて3’−RACE PCRを行った。GeneRacer3’ PrimerおよびGeneRacer3’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれているものを使用した。3’−RACE PCRによって効率的にルシフェラーゼ遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taq(タカラバイオ株式会社)を用いて、マニュアルに従って実施した。
【0059】
一度目のPCRとして、5’末端側非翻訳領域の塩基配列から作製したプライマーとGeneRacer3’ Primerとから成るプライマー対を用いてルシフェラーゼ遺伝子の増幅を行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)および最終濃度がそれぞれ0.3μMのプライマーを含む20μlのPCR反応溶液を作製し、そこへホタル完全長cDNAライブラリー溶液を0.4μl加えた。なお、ホタル完全長cDNAライブラリー溶液の濃度は未定量であった。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。わずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてそれぞれnested PCR反応を実施した。
【0060】
Nested PCRとして、Nested PCR用のプライマーNo6(2/3)−Full−F2(CGCGCTAACATCATTGCAAGAATGGAAGA:配列番号26)とGeneRacer3’ Nested Primerとから成るプライマー対を用いてルシフェラーゼ遺伝子の増幅を行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)および最終濃度がそれぞれ0.3μMのプライマーを含む20μlのNested PCR反応溶液を作製し、そこへ鋳型として1度目のPCR反応溶液を滅菌水で10倍希釈した溶液を1.0μl加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。約2kbp付近に効率よく遺伝子が増幅していることを確認した。
【0061】
4−3.3’−Raceで増幅した遺伝子の塩基配列の決定
3’−RACEで増幅した遺伝子の塩基配列を読み取るため、ゲル抽出によるPCR産物の精製、サブクローニングおよびダイレクトシークエンスを実施した。詳細を以下に示す。
【0062】
約2kbp付近に効率よく遺伝子が増幅したプライマーの組み合わせでPCR(最終容量20μl)を実施し、ゲル抽出の手法を用いて目的とする遺伝子片を回収した。ゲル抽出はWizard SV Gel and PCR Clean−Up System(プロメガ株式会社)を用いて、マニュアルに従って実施した。TAクローニングの手法を用いて、ゲルから抽出したPCR産物のサブクローニングを実施した。TAクローニングはpGEM−T Easy Vector System(プロメガ株式会社)を用いて、マニュアルに従って実施した。その後、このベクターDNAを大腸菌(TOP10株またはDH5α株)に形質転換し、青・白スクリーニングの手法を用いてインサートポジティブコロニーを選択した。選択されたコロニーをダイレクトコロニーPCRに供し、遺伝子が導入されていることを確認した。ダイレクトコロニーPCRには、M13−F(−29) Primer(5’−CAC GAC GTT GTA AAA CGA C−3’:配列番号21)とM13 Reverse(5’−GGA TAA CAA TTT CAC AGG−3’:配列番号22)とから成るプライマー対を用いた。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度がそれぞれ0.2μMのプライマー対を含む10μlのPCR反応溶液を作製し、そこへ鋳型として少量の大腸菌コロニーを加えた。PCR反応は、最初に94℃1分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を25サイクル繰り返し、最後に72℃2分間の伸長反応を行った。PCR反応後、2μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0063】
増幅が確認できたPCR反応溶液について、ダイレクトシークエンシング法を用いてその遺伝子の塩基配列の決定を行った。PCR産物精製キットExoSAP−IT(GEヘルスケアバイオサイエンス)を用いて、PCR反応溶液に含まれる余剰なdNTPおよびプライマーを除去し、PCRダイレクトシークエンシングのための鋳型を調製した。BigDye Terminator v3.1 Cycle Sequencing Kit(アプライドバイオシステムズ)を用いて、この鋳型を含むシークエンシング反応溶液を調製し、サーマルサイクラーを用いてシークエンシング反応を行った。シークエンスに用いたプライマーはベクタープライマーまたは遺伝子特異的なプライマーを用いた。PCR産物の精製およびシークエンシングはそれぞれマニュアルに従って実施した。シークエンシング反応後、反応産物の精製を次の通りに行った。反応溶液に2.5倍量の100%エタノールを加え、遠心機を用いて核酸を沈殿させた。次に、上精を取り除いた後、70%エタノールを加えて沈殿を洗浄し、遠心機を用いて核酸を沈殿させた。最後に、上精を取り除いた後、沈殿を乾燥させた。精製した沈殿にHi−Di Formamide(アプライドバイオシステムズ)を15μl加え、溶解させた。この溶液を94℃2分間の熱変性させ、更に氷上で急冷して、塩基配列を決定するためのサンプルとした。このサンプルをApplied Biosystems 3130xlジェネティックアナライザ(アプライドバイオシステムズ)を用いて塩基配列を読み取った。塩基配列の解析方法はマニュアルに従って実施した。
【0064】
シークエンシングによって完全長ホタルルシフェラーゼ遺伝子を獲得した。この塩基配列(配列番号2)もしくはアミノ酸(配列番号1)へ翻訳した配列について、NCBIが提供するblastxまたはblastpサーチを利用して相同性検索を実施した。それぞれの検索で既知ホタルルシフェラーゼの塩基配列と高い相同性を示すことを確認した。以上の実験および解析で得られた塩基配列を新規ホタルルシフェラーゼ遺伝子の完全長cDNA配列であると決定した。
【0065】
以下、この新規のルシフェラーゼを野生型オキナワマドボタルルシフェラーゼと称する。
【0066】
[実施例2:野生型オキナワマドボタルルシフェラーゼの酵素学的なパラメータの測定]
1.野生型オキナワマドボタルルシフェラーゼ遺伝子のタンパク質発現
野生型オキナワマドボタルルシフェラーゼ遺伝子を大腸菌でタンパク質発現させるため、pRSET−Bベクター(インビトロジェン)に導入した。遺伝子発現ベクターの構築は標準的な方法に従い以下のように実験を実施した。
【0067】
1−1.野生型オキナワマドボタルルシフェラーゼ遺伝子の制限酵素認識部位の改変
上述の通り決定した塩基配列によれば、野生型オキナワマドボタルルシフェラーゼ遺伝子はBamHIおよびEcoRIの制限酵素認識配列を含む。ルシフェラーゼのアミノ酸配列を維持しつつ、これらの塩基配列におけるこれらの認識配列を除去する遺伝子改変を実施した。この処理は、後述するルシフェラーゼ遺伝子の発現ベクターへの導入を容易にするために行った。遺伝子変異の導入は、多比良和誠編「遺伝子の機能阻害実験法―簡単で確実な遺伝子機能解析から遺伝子治療への応用まで」(羊土社、2001年発行、p.17−25)に示される方法に従った。変異導入後の配列は、配列番号3に示される塩基配列である。
【0068】
1−2.野生型オキナワマドボタルルシフェラーゼ遺伝子の遺伝子発現ベクターへの導入
ルシフェラーゼ遺伝子を、pRSET−Bベクターの制限酵素サイトBamHI−EcoRI間に導入するため、開始コドンおよびその前に制限酵素BamHI認識配列GGATCCを含むプライマー、並びに終始コドンおよびその後ろに制限酵素EcoRI認識配列GAATTCを含むプライマーを作成した。このプライマー対を用いて、ルシフェラーゼ遺伝子の両端に上述の制限酵素認識部位を含んだ断片の増幅を行った。PCRにはポリメラーゼKOD−Plis−(東洋紡績株式会社)を用いて、マニュアルに従って実施した。
【0069】
最終濃度が等倍の10×PCR Buffer、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が1.0mMのMgSO4、最終濃度が0.02U/μlのTOYOBO KOD−Plis−(1U/μl)、最終濃度がそれぞれ0.3μMのプライマー対を含む20μlのPCR反応溶液を作製し、そこへ鋳型としてBamHIとEcoRI認識配列を持たないルシフェラーゼ遺伝子の溶液を0.4μl加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、55℃30秒および68℃2分を30サイクル繰り返し、最後に68℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。遺伝子増幅が認められたため、このPCR反応溶液を通常のエタノール沈殿法により沈殿濃縮し、制限酵素処理用10×H Bufferを4μl、制限酵素BamHI(東洋紡績株式会社)および制限酵素EcoRI(東洋紡績株式会社)を各2μlならびに滅菌脱イオン水32μlを加えて溶解し、37℃で2時間保温して制限酵素処理した。その後、この反応溶液をエタノール沈殿法により沈殿濃縮した後、滅菌脱イオン水に溶解した。この溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色を行った。紫外線照射下において確認されるDNAバンドを含むゲルを、ナイフを用いて切り出した。この切り出したゲルからWizard(R) SV Gel and PCR Clean−Up System(プロメガ)を用いてDNAを抽出した。これらの操作はマニュアルに従って実施した。その後、Ligation Pack(ニッポンジーン)をマニュアルに従って用いて、あらかじめ同様の方法で制限酵素BamHIと制限酵素EcoRIで処理したpRSET−Bベクターに抽出したDNAを導入した。このベクターDNAを大腸菌JM109(DE3)株に形質転換し、コロニーを形成させた。
【0070】
得られたコロニーを鋳型としてダイレクトコロニーPCRを実施し、pRSET−Bに導入したルシフェラーゼ遺伝子を増幅した。ダイレクトコロニーPCRは、T7 promoter Primer(5’−TAA TAC GAC TCA CTA TAG GG−3’:配列番号27)およびT7 Reverse Primer(5’−CTA GTT ATT GCT CAG CGG TGG−3’:配列番号28)のプライマー対を用いて行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)および最終濃度がそれぞれ0.2μMのプライマーを含む10μlのPCR反応溶液を作製し、そこへ鋳型として少量の大腸菌コロニーを加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を25サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0071】
増幅が確認できたPCR反応溶液について、ダイレクトシークエンシング法を用いてその遺伝子の塩基配列の決定を行った。PCR産物精製キットExoSAP−ITを用いて、PCR反応溶液に含まれる余剰なdNTPおよびプライマーを除去し、PCRダイレクトシークエンシングのための鋳型を調製した。BigDye Terminator v3.1 Cycle Sequencing Kitを用いて、この鋳型を含むシークエンシング反応溶液を調製し、サーマルサイクラーを用いてシークエンシング反応を行った。シークエンスには、ベクタープライマーまたは遺伝子特異的なプライマーを用いた。PCR産物の精製およびシークエンシングはそれぞれマニュアルに従って実施した。シークエンシング反応後、反応産物の精製を次の通りに行った。反応溶液に2.5倍量の100%エタノールを加え、遠心機を用いて核酸を沈殿させた。次に、上精を取り除いた後、70%エタノールを加えて沈殿を洗浄し、遠心機を用いて核酸を沈殿させた。最後に、上精を取り除いた後、沈殿を乾燥させた。精製した沈殿にHi−Di Formamide(アプライドバイオシステムズ)を15μl加え、溶解させた。この溶液を94℃2分間の熱変性させ、更に氷上で急冷して、塩基配列を決定するためのサンプルとした。このサンプルをApplied Biosystems 3130xl ジェネティックアナライザを用いて塩基配列を読み取り、正常に遺伝子発現ベクターpRSET−Bに導入されていることを確認した。
【0072】
2.発光タンパク質の精製
JM109(DE3)を含む大腸菌溶液50μlにルシフェラーゼ発現ベクター0.5μlを添加し、氷上で10分、その後42℃で1分、最後に氷上で2分インキュベートした。その後、大腸菌溶液50μlをSOC培地200μlに加えた。その大腸菌/SOC培地混合溶液を37℃で20分間振とうしながらインキュベートした。インキュベート後のサンプル100μlをLB培地プレート(100μg/mlアンピシリンを含む)にストリークし、37℃で一晩インキュベートした。翌日得られたコロニーをピックアップし、500mlスケールのLB培地で培養した。培養は37℃で24時間、18℃で24時間行った。合計48時間の培養の後、遠心分離で菌体を回収し、0.1M Tris−HCl溶液(pH8.0)に再度懸濁して超音波破砕した。菌体破砕液を遠心分離(15000rpm、10分)し、沈査を除去して上清を回収した。ベッドボリューム2mlのカラムにNi−Agar懸濁液500μlと0.1M Tris−HCl2mlを加え、カラムを平衡化した。回収した上清をカラムに添加し、自然落下させた。上清の全量がカラムを通過するまでの間の操作は全て4℃の条件で行った。25mMイミダゾール/0.1M Tris−HCl溶液2mlでカラムを洗浄した。洗浄後のカラムに500mMイミダゾール/0.1M Tris−HCl溶液を2ml加え、ルシフェラーゼを溶出した。溶出されたサンプルをゲルろ過カラムPD−10(GEヘルスケア)でろ過し、脱塩した。脱塩後のサンプルをVivaspin6(ザルトリウス)で限界ろ過し、濃縮されたサンプルにグリセリンを添加して、50%グリセリン溶液とした。保存は−20℃で行った。
【0073】
3.発光スペクトルの測定
測定のための装置としてLumiFlSpectroCapture(ATTO)を用い、0.1Mクエン酸/0.1M Na2HPO4 buffer(pH6.0−8.0)に1mM D−ルシフェリン、2mM ATPおよび4mM MgCl2を含む溶液に、精製酵素を1μg/mlから10μg/mlの間の最終濃度で添加し、酵素添加後15秒経過時点で発光スペクトルを測定した。測定結果を図1に示す。
【0074】
図1より、取得された野生型オキナワマドボタルルシフェラーゼは、pH8.0の環境において、560nm付近に最大発光波長を示している。また、pH7.5において563nm付近、pH7.0において571nm付近(および610nm付近により小さなピーク)、pH6.5において608nm付近およびpH6.0において612nm付近、pH5.5において613nm付近となった。
【0075】
4.速度論的解析
4−1.D−ルシフェリンおよびATPの濃度の決定
D−ルシフェリン溶液中のD−ルシフェリン濃度およびATP溶液中のATP濃度を以下の通りに決定した。
【0076】
UV−Visible Spectrometer(Hitachi)を用いて、D−ルシフェリン溶液およびATP溶液の紫外可視吸収スペクトルを測定し、この測定結果と以下のε値とから濃度を算出した。
D−ルシフェリン:λmax 328nm、ε18200、pH5.0
ATP:λmax 259nm、ε15400、pH7.0。
【0077】
測定はそれぞれ10回ずつ行い、吸光度の平均値を濃度算出に用いた。このように濃度を決定したD−ルシフェリン溶液およびATP溶液を用いて、以下のKm値算出を行った。
【0078】
4−2.D−ルシフェリンに対するKm値の測定
様々なD−ルシフェリン濃度の環境下において、得られたルシフェラーゼによる発光の強度を測定した。測定結果に基づいて、D−ルシフェリンに対するKm値を決定した。
【0079】
D−ルシフェリンを0.1M Tris−HCl(pH8.0)に添加して、異なる濃度の12種類のDールシフェリン溶液を作製した。これらの溶液は、D−ルシフェリンの終濃度がそれぞれ0.625、1.25、2.5、5、10、20、40、80、160、320、480および640μMとなるようにD−ルシフェリンを含む。これらのD−ルシフェリン溶液を96穴マイクロプレートに50μlずつ分注した。各種精製ルシフェラーゼ、4mM ATPおよび8mM MgSO4を含む0.1M Tris−HCl(pH8.0)溶液をルミノメーターの標準ポンプに接続し、当該溶液をウェルに50μl添加すると同時に測定を行った。測定にはLuminescensor(ATTO)を使用した。測定は各ルシフェリン濃度について3回ずつ行った。
【0080】
得られたフォトンカウント値のピーク強度を初速度Vとして、ルシフェリン濃度Sに対してプロットした。このプロットにミカエリス・メンテン型のカーブフィッティングを行い、Km値を算出した。カーブフィッティングは非線形の最小二乗法で行い、パラメータの探索にはニュートン法を用いた。
【0081】
4−3.ATPに対するKm値の測定
様々なATP濃度の環境下において、得られたルシフェラーゼによる発光の強度を測定した。測定結果に基づいて、ATPに対するKm値を決定した。
【0082】
ATPを0.1M Tris−HCl(pH8.0)に添加して、異なる濃度の12種類のATP溶液を作製した。これらの溶液は、ATPの終濃度がそれぞれ5、10、20、40、80、160、320、480、640、800、1280、1600および1920μMとなるようにATPを含む。これらのATP溶液をそれぞれ96穴マイクロプレートに50μlずつ分注した。各種精製ルシフェラーゼ、1mM D−ルシフェリン、8mM MgSO4を含む0.1M Tris−HCl(pH8.0)溶液をルミノメーターの標準ポンプに接続し、当該溶液をウェルに50μl添加すると同時に測定を行った。測定は各ATP濃度について3回ずつ行った。
【0083】
得られたフォトンカウント値のピーク強度を初速度Vとして、ATP濃度Sに対してプロットした。このプロットにミカエリス・メンテン型のカーブフィッティングを行い、Km値を算出した。カーブフィッティングは非線形の最小二乗法で行い、パラメータの探索にはニュートン法を用いた。
【0084】
上記のようにして決定されたD−ルシフェリンに対するKm値およびATPに対するKm値を表1に示す。表1には、同様に測定した既知のルシフェラーゼの各Km値も示される。GL3とは既知のホタル由来のルシフェラーゼである。また、ELuc、CBGおよびCBRとは既知のコメツキムシ由来のルシフェラーゼである。これらの既知のルシフェラーゼは市販されるものを使用した。
【0085】
【表1】

【0086】
さらに、これらの結果について、縦軸をATPに対するKm値とし、横軸をD−ルシフェリンに対するKm値としてプロットした図を、図2として示す。
【0087】
[実施例3:発光強度の測定]
野生型オキナワマドボタルルシフェラーゼによる発光強度を、既知のルシフェラーゼおよびローダミン6Gによる発光強度と比較した。
【0088】
1.ローダミン6Gによる化学発光の測定
ローダミン6Gを0.1Mクエン酸/0.2M Na2HPO4(pH4.0)溶液に溶解した。このローダミン6G溶液を15000rpmで1分間遠心分離し、上清を回収した。回収した上清を0.1Mクエン酸/0.2M Na2HPO4溶液で1000倍希釈し、希釈液の吸光度を測定した。測定はNanoVue(GEヘルスケア)で行った。測定は530nmにおいて5回行い、その平均値として0.048の吸光度が得られた。ローダミン6Gのε値(1.16x105mol−1cm−1)を用いて元のローダミン6G溶液の濃度を算出した結果、414μMであった。この溶液を0.1Mクエン酸/0.2M Na2HPO4溶液(pH4.0)で希釈し、30μMの希釈液を作製した。
【0089】
この希釈液を使用して、以下の表2の通り溶液1を調製した。
【0090】
【表2】
【0091】
また、ビス(2,4,6−トリクロロフェニル)オキサレート(TCPO)をアセトニトリルに最終濃度3mMとなるように溶解して溶液2を作製した。
【0092】
溶液1を96穴のマイクロプレートに100μlずつ分注した。そこへ、ポンプを使用して溶液2を50μl添加すると同時に測定を開始して、溶液2の添加から10秒間のフォトンカウントの積算値を取得した。測定にはLuminescensor(ATTO)を使用し、300nmから650nmの波長の光を取得した。同様の測定を10回行った。
【0093】
2.P.ピラリスルシフェラーゼおよび野生型オキナワマドボタルルシフェラーゼによる化学発光の測定
JM109(DE3)株を含む溶液50μlにルシフェラーゼ発現ベクターを含む溶液0.5μlを添加し、氷上で10分、42℃で1分、氷上で2分インキュベートした。その後、この大腸菌溶液50μlをSOC培地200μlに加えた。大腸菌/SOC培地混合溶液を37℃で20分間振とうしながらインキュベートした。インキュベート後のサンプル100μlをLB培地プレート(100μg/mlアンピシリンを含む)にストリークし、37℃で一晩インキュベートした。翌日、できたコロニーをピックアップし、500mlスケールのLB培地で培養した。培養は37℃で24時間、18℃で24時間行った。合計48時間の培養の後、遠心分離によって菌体を回収し、0.1M Tris−HCl溶液(pH8.0)に再度懸濁して超音波破砕した。菌体破砕液を遠心分離し(15000rpm、10分間)、沈査を除去して上清を回収した。ベッドボリューム2mlのカラムにNi−Agar懸濁液500μlおよび0.1M Tris−HCl溶液2mlを加え、カラムを平衡化した。回収した上清をカラムに添加し、自然落下させた。上清の全量がカラムを通過するまでの間の操作は全て4℃の条件で行った。25mMイミダゾール/0.1M Tris−HCl溶液2mlでカラムを洗浄した。洗浄後のカラムに500mMイミダゾール/0.1M Tris−HCl溶液を2ml加え、ルシフェラーゼを溶出した。溶出されたサンプルをゲルろ過カラムPD−10(GEヘルスケア)でろ過し、脱塩した。脱塩後のサンプルをVivaspin6(ザルトリウス)で限界ろ過した。
【0094】
得られたサンプルの濃度を比色法で測定し、P.ピラリスルシフェラーゼおよび野生型オキナワマドボタルルシフェラーゼの濃度を確認した。最終濃度が1μg/mlとなるように上記ルシフェラーゼを0.1Mクエン酸/0.2M Na2HPO4(pH8.0)に希釈した。
【0095】
このルシフェラーゼ溶液を50μlずつ96穴マイクロプレートに分注した。2mM D−ルシフェリン、4mM ATPおよび8mM MgSO4/0.1M Tris−HCl(pH8.0)を含む溶液をルミノメーターの標準ポンプによって50μl添加し、添加と同時に測定を行った。溶液の添加から10秒間のフォトンカウントの積算値を取得した。測定にはLuminescensor(ATTO)を使用し、300nmから650nmの波長の光を取得した。各測定は6回行った。
【0096】
3.測定結果
以上のように測定したローダミン6G、P.ピラリスルシフェラーゼおよび野生型オキナワマドボタルルシフェラーゼによる化学発光の強度を図3に示す。
【0097】
図3から、野生型オキナワマドボタルルシフェラーゼは、ローダミン6Gによる発光の24.4倍の強度の発光を示すことがわかった。また、野生型オキナワマドボタルルシフェラーゼは、P.ピラリスルシフェラーゼによる発光の5倍の強度の発光を示すことがわかった。
【0098】
[実施例4:哺乳細胞における野生型オキナワマドボタルシフェラーゼの発現]
オキナワマドボタルをHeLa細胞に発現させて、細胞内での発光強度を測定した。
【0099】
オキナワマドボタルルシフェラーゼ遺伝子の導入のために2種類の発現ベクターを作製した。オキナワマドボタルから取得したオリジナルのオキナワマドボタルルシフェラーゼ遺伝子においてBamHIおよびEcoRIの認識配列を削除した遺伝子(配列番号3)、さらに哺乳細胞における発現に最適化した遺伝子(配列番号4)をそれぞれ含む核酸を、pcDNA3.1(+)ベクター(Invitorogen)のマルチクローニングサイトのBamHIおよびEcoRIのサイト間に挿入した。また、比較のために、既知であるP.ピラリスルシフェラーゼの遺伝子について哺乳細胞における発現に最適化させた遺伝子を含む核酸を、同様にpcDNA3.1(+)ベクターに挿入した。
【0100】
このようにして得られた3種のプラスミドを、それぞれリポフェクション法によってHeLa細胞に遺伝子導入し、24時間後D−MEM培地を交換した。測定直前にD−ルシフェリンを最終濃度1mMで添加し、Kronos(ATTO)を用いて発光強度を測定した。その結果を図4に示す。なお、図4に示される発光強度は10秒間の積算値を示している。
【0101】
図4から、オキナワマドボタルルシフェラーゼのコドンの最適化によって、HeLa細胞で発現させた場合の発光強度が27倍増大することがわかる。また、コドン最適化後のオキナワマドボタルルシフェラーゼはコドン最適化後のP.ピラリスルシフェラーゼより3.4倍の発光強度を示した。
【0102】
[実施例5:変異型オキナワマドボタルルシフェラーゼの作製]
【0103】
変異型オキナワマドボタルルシフェラーゼを発現する変異型遺伝子を作製した。
【0104】
哺乳細胞における発現に最適化した遺伝子(配列番号4)に対し、次の5種の変異を任意の組合せで導入した。
【0105】
アミノ酸配列上424番目のイソロイシンをロイシンに置換する変異(I424L)
アミノ酸配列上531番目のイソロイシンをアルギニンに置換する変異(I531R)
アミノ酸配列上355番目のグルタミン酸をアルギニンに置換する変異(E355R)
アミノ酸配列上367番目のバリンをアラニンに置換する変異(V367A)
アミノ酸配列上446番目のリシンをグルタミンに置換する変異(K446Q)
【0106】
それぞれの変異の導入のために、以下の5種のプライマーを用いた。
I424L変異:OkinawaMado_I424L(GGCTGGCTGCATTCAGGCGATcTgGCCTACTACGATAAGGACGGC:配列番号29)
I531R変異:OkinawaMado_I531R(CCCAAAGGTCTCACAGGCAAGAgaGACTCTCGAAAAATTCGAGAA:配列番号30)
E355R変異:OkinawaMado_E355R(AGTGCAGTAATCATTACTCCAagGGGCGACGACAAACCAGGAGCC:配列番号31)
V367A変異:OkinawaMado_V367A(CCAGGAGCCTGCGGCAAGGTCGcCCCCTTCTTTTCCGCCAAAATTGTG:配列番号32)
K446Q変異:OkinawaMado_K446Q(CTGAAAAGTCTGATCAAGTACcAGGGCTACCAGGTGCCACCTGCA:配列番号33)
【0107】
変異の導入は文献の記載に基づいて行った(Asako Sawano and Atsushi Miyawaki, Nucleic Acids Research,2000, Vol.28, No.16)。変異導入後にシーケンサーを用いて配列を読み取り、目的とする変異がオキナワマドボタルルシフェラーゼ遺伝子に導入されていることを確認した。
【0108】
作製した種々の変異型オキナワマドボタルルシフェラーゼのうち2種類(変異体1および変異体2と称する)に着目した。これらは、それぞれ以下の変異を有する。
【0109】
変異体1:I424L、E355R、V367AおよびK446Q
変異体2:I424L、I531R、E355R、V367AおよびK446Q
変異体1のアミノ酸配列は配列番号36に記載され、変異体1をコードする遺伝子の塩基配列は配列番号34に記載される。また、変異体2のアミノ酸配列は配列番号37に記載され、変異体2をコードする遺伝子の塩基配列は配列番号35に記載される。
【0110】
次に、変異体1および2による発光強度について、野生型オキナワマドボタルルシフェラーゼと比較した。
【0111】
野生型オキナワマドボタルルシフェラーゼの遺伝子を含む核酸(配列番号4)、変異体1の遺伝子を含む核酸(配列番号34)および変異体2の遺伝子を含む核酸(配列番号35)に対してKozak配列を付与した。なお、3種の配列とも哺乳細胞における発現に対してコドン最適化されている。その後、pF9A CMV hRLuc neo Flexi vector(Promega)のマルチクローニングサイトのSgfIおよびPmeIのサイト間に挿入した。pF9Aベクターは内部コントロールとしてウミシイタケルシフェラーゼ遺伝子(hRLuc)をベクター配列に含んでおり、マルチクローニングサイトに挿入された発光遺伝子による発光強度をウミシイタケルシフェラーゼによる発光強度との比として算出することが可能である。
【0112】
得られた3種のプラスミドを、24穴のプレートに播種したHeLa細胞にそれぞれリポフェクション法で遺伝子導入し、24時間後PBSで細胞を洗浄した。24穴プレートの各ウェルに2mM D−ルシフェリン/CO2 Independent Medium(Invitrogen)を500μlずつ添加し、25℃、各ウェルあたり1秒間測定の条件でLuminescensor(ATTO)を用いて120分間発光強度を測定した。90分経過時点の発光強度を野生型オキナワマドボタルルシフェラーゼ、変異体1および変異体2の発光強度とした。各ウェル内の培養液を除去した後、PBSで各ウェルを3回洗浄した。次に各ウェルに10μMセレンテラジン/CO2 Independent Mediumを500μlずつ添加し、25℃、各ウェルあたり1秒間測定の条件でLuminescensorを用いて30分間発光強度を測定した。セレンテラジン添加後、10分経過時点の発光強度を内部コントロールであるウミシイタケルシフェラーゼによる発光強度とした。野生型オキナワマドボタルルシフェラーゼ、変異体1および変異体2による発光強度をウミシイタケルシフェラーゼによる発光強度でそれぞれ除算した値を算出し、その値を各ルシフェラーゼの発光強度としてグラフ化した。
【0113】
図5には、D−ルシフェリン添加直後から120分経過時点までにおける、各細胞中のオキナワマドボタルルシフェラーゼによる発光強度の時間変化が示される。図6には、セレンテラジン添加直後から30分経過時点までにおける、各細胞中のウミシイタケルシフェラーゼによる発光強度の時間変化が示される。図7には、野生型オキナワマドボタルルシフェラーゼ、変異体1および変異体2による90分経過時点における発光強度を、内部コントロールであるウミシイタケルシフェラーゼによる10分経過時点における発光強度でそれぞれ除算して得られた発光強度比を示す。
【0114】
図7に示されるように、変異体1によって得られた発光強度は、野生型オキナワマドボタルルシフェラーゼによる発光強度と比較して4.2倍高かった。変異体2によって得られた発光強度は、野生型オキナワマドボタルルシフェラーゼによる発光強度と比較して2.4倍高かった。変異の導入によってオキナワマドボタルルシフェラーゼの発光強度が増大することが示された。
【技術分野】
【0001】
本発明はホタル由来ルシフェラーゼに関する。
【背景技術】
【0002】
細胞内のシグナル伝達および遺伝子発現といった細胞の機能を調べるためには、蛍光色素および蛍光タンパク質といった蛍光プローブ並びにルシフェリン・ルシフェラーゼ反応を利用する化学発光プローブが用いられている。特に遺伝子の発現調節の解析には、励起光による細胞のダメージが生じず且つ自家発光の問題が生じない、定量性に優れた発光計測が用いられる。例えば、ルシフェラーゼ遺伝子が導入された細胞を観察する場合、ルシフェラーゼ活性に因る細胞からの発光強度を測定することで、ルシフェラーゼ遺伝子の発現の強さ(具体的には発現量)を調べることができる。具体的には、最初に細胞を溶解して細胞溶解液を作製し、その後この細胞溶解液にルシフェリンおよびアデノシン三リン酸(ATP)等を添加し、光電子増倍管を用いたルミノメーターで発光強度を定量し、その結果に基づいて発現量を特定する。この方法では、発光強度の測定は細胞を溶解した後に行われる。このため、ある時点でのルシフェラーゼ遺伝子の発現量は、測定に使用した全細胞の平均値として得られる。ルシフェラーゼ遺伝子等の発光遺伝子をレポーター遺伝子として導入する方法として、例えばリン酸カルシウム法、リポフェクチン法またはエレクトロポレーション法等を使用でき、各方法は目的および細胞の種類の違いに応じて使い分けられている。細胞に導入するルシフェラーゼ遺伝子の上流または下流に目的のDNA断片を繋ぎ、ルシフェラーゼ遺伝子の発現量を測定することで、当該DNA断片がルシフェラーゼ遺伝子の転写に及ぼす影響を調べることが可能となる。また、細胞に導入するルシフェラーゼ遺伝子と目的の遺伝子とを共発現させることで、当該遺伝子産物がルシフェラーゼ遺伝子の発現に及ぼす影響を調べることが可能となる。
【0003】
発光遺伝子発現量の時間変化を調べるには、生きた細胞からの発光強度を経時的に測定する必要がある。このような測定は、ルミノメーターを備えたインキュベーターにて細胞を培養し、細胞集団全体からの発光強度を一定時間ごとに測定することで行われる。細胞集団全体における発光遺伝子の発現量の経時的な変化を捉えることにより、一定の周期性をもった発現リズム等を分析することができる。
【0004】
近年、生物学および医学の研究において、生命現象を分子レベルで動的に捉える必要性が高まってきている。蛍光観察を利用する研究分野では、試料内のタンパク質分子機能を動的に捉えるために、タイムラプスまたは動画撮像が行われている。従来技術では、蛍光試料を用いた経時的観察(例えば、蛍光分子を付したタンパク質1分子の動画観察)が行われている。
【0005】
一方、経時的観察のために化学発光試料を用いる場合、化学発光試料の発光強度は極めて小さいため、イメージ・インテンシファイアを装着したCCDカメラを用いて観察する必要がある。また最近では、化学発光試料を観察するための光学系を有した顕微鏡も開発されている(特許文献1および2)。
【0006】
発光強度が小さい化学発光試料を顕微鏡により撮像する場合、鮮明な画像を撮影するために必要な露出時間は長くなる。このような化学発光試料は、使用できる研究用途が制限される。例えば、発光強度の小ささのために30分間の露出時間が必要となる場合、30分間隔での経時的撮影は可能であっても、より短い間隔での撮影、さらにはリアルタイムでの撮影は不可能である。また、画像の取得に当って、発光する細胞に焦点を合わせるために複数枚の画像を取得し比較する必要があるが、発光強度の小ささのために長い露出時間を必要とする場合、1枚の画像を取得するだけでも手間と時間を要することになる。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2006−301599号公報
【特許文献2】国際公開第06/088109号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、高い強度でルシフェリンを発光させるルシフェラーゼを提供することにある。
【課題を解決するための手段】
【0009】
本発明の一態様に係る変異型ルシフェラーゼは、オキナワマドボタル(Pyrocoelia matsumurai)に由来し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる。
【発明の効果】
【0010】
本発明によれば、高い発光強度を示すルシフェラーゼが提供されるため、発光像を撮像するために必要な露出時間を短縮することができ、且つ従来よりも時間分解能が高い経時的観察が可能となる。
【図面の簡単な説明】
【0011】
【図1】野生型オキナワマドボタルルシフェラーゼを用いた場合に得られた、各pHにおける発光スペクトルを示す図である。
【図2】各ルシフェラーゼのKm値をプロットした図である。
【図3】野生型オキナワマドボタルルシフェラーゼ、P.ピラリスルシフェラーゼおよびローダミン6Gを用いた場合に得られた発光強度を比較する図である。
【図4】哺乳細胞にて発現させた野生型オキナワマドボタルルシフェラーゼおよびP.ピラリスルシフェラーゼを用いた場合に得られた発光強度を比較する図である。
【図5】野生型オキナワマドボタルルシフェラーゼおよび変異型オキナワマドボタルルシフェラーゼを発現する細胞について得られた、D−ルシフェリン添加後の発光強度の時間変化を示す図である。
【図6】野生型オキナワマドボタルルシフェラーゼおよび変異型オキナワマドボタルルシフェラーゼを発現する細胞について得られた、セレンテラジン添加後の発光強度の時間変化を示す図である。
【図7】野生型オキナワマドボタルルシフェラーゼを用いた場合に得られた発光強度と変異型オキナワマドボタルルシフェラーゼを用いた場合に得られた発光強度とを比較した図である。
【発明を実施するための形態】
【0012】
本発明の実施形態は、オキナワマドボタル(Pyrocoelia matsumurai)に由来するルシフェラーゼに関する。
【0013】
「ルシフェラーゼ」とは、一般に、発光が生じる化学反応を触媒する酵素を指す。当該酵素の基質となる物質はルシフェリンと呼ばれる。ATPの存在下、ルシフェラーゼの触媒作用により、ルシフェリンが化学変化を起こす際に発光する。現在、ルシフェラーゼは、ホタルに由来するものおよびバクテリアに由来するものが取得されている。実施形態に係るルシフェラーゼも、上述の通り定義されるルシフェラーゼと同義であるが、後述するホタルから初めて取得された新規のルシフェラーゼである。実施形態に関してルシフェリンとは、好ましくはホタルルシフェラーゼの基質となるホタルルシフェリンを意味する。より好ましくは、ホタルルシフェリンとはD−ルシフェリンである。
【0014】
実施形態に係るルシフェラーゼは、オキナワマドボタル(Pyrocoelia matsumurai)に由来する。オキナワマドボタルとは節足動物門昆虫綱コウチュウ目ホタル科マドボタル属に属すホタルである。本発明の実施形態に関してオキナワマドボタルという場合、特に、主に沖縄本島に生息する基亜種ピロコエリア・マツムライ・マツムライ(Pyrocoelia matsumurai matsumurai)を意味する。なお、ここにいう「由来する」とは、オキナワマドボタルが有する野生型のルシフェラーゼに加えて、その変異体をも含むことを意味する。
【0015】
実施形態に係るルシフェラーゼによれば、既知のルシフェラーゼと比較して格段に高い発光強度が得られる。従って、実施形態に係るルシフェラーゼは、タンパク質のイメージングのためのレポーターとして利用する場合に特に有利な効果を奏する。すなわち、実施形態に係るルシフェラーゼは、少量でも高い発光強度を提供できるため、発現量が低いタンパク質の良好な検出を可能とする。また、実施形態に係るルシフェラーゼは、得られる発光強度が高いことにより検出に必要な露出時間を短縮できる。このため、実施形態に係るルシフェラーゼを経時的観察のためのレポーターとして利用すれば、撮影間隔を短くすることが可能となり、よりリアルタイムに近い観察が可能となる。すなわち、時間分解能が高い経時的観察が可能となる。
【0016】
実施形態に係るルシフェラーゼの一例は、配列番号1に示されるアミノ酸配列を含むルシフェラーゼである。当該ルシフェラーゼは、オキナワマドボタルから得られた野生型のルシフェラーゼである。この明細書および特許請求の範囲では、当該ルシフェラーゼを、野生型ルシフェラーゼ、野生型オキナワマドボタルルシフェラーゼまたは野生型オキナワマドボタル由来ルシフェラーゼ等と称する。
【0017】
図1に、実施形態に係る野生型オキナワマドボタルルシフェラーゼを用いた場合に得られたルシフェリンの発光スペクトルを示す。この図に示されるとおり、実施形態に係る野生型オキナワマドボタルルシフェラーゼは、pH8において、最大発光波長が560nm付近にある発光を生じさせる。ここで「最大発光波長」とは、ルシフェラーゼが誘起するルシフェリンの発光を測定することによって得られるスペクトルにおいて、発光強度が最大となる波長を意味する。
【0018】
また、図3に、野生型オキナワマドボタルルシフェラーゼを用いた場合に得られたルシフェリンの発光強度、ローダミン(Rhodamine)6Gによる発光強度およびP.ピラリスルシフェラーゼを用いた場合に得られたルシフェリンの発光強度を比較したグラフが示される。この図から、野生型オキナワマドボタルルシフェラーゼを使用した場合、ローダミン6Gによる発光強度の24.4倍以上の発光強度を達成できることがわかる。この発光強度は、300nmから650nmの波長の光を10秒間取得して積算したものである。このように、実施形態に係るルシフェラーゼを用いた場合は、既知の発光物質および従来のホタルルシフェラーゼを用いた場合と比較して、より高い発光強度を達成できる。実施形態に係る野生型オキナワマドボタルルシフェラーゼにより、好ましくはローダミン6Gによる発光強度の5倍以上、10倍以上、15倍以上、20倍以上、21倍以上、22倍以上、23倍以上または24倍以上の発光強度が得られる。
【0019】
実施形態に係るルシフェラーゼは、オキナワマドボタルから取得された野生型のルシフェラーゼだけでなく、野生型ルシフェラーゼのアミノ酸配列の一部に変異が生じた変異型のルシフェラーゼを含む。ここでは、このようなルシフェラーゼを、変異型ルシフェラーゼ、変異型オキナワマドボタルルシフェラーゼまたは変異型オキナワマドボタル由来ルシフェラーゼ等と称する。
【0020】
そのような変異は、ルシフェラーゼの酵素活性を向上させる変異であってよい。すなわち、そのような変異は、野生型オキナワマドボタルルシフェラーゼよりも高い発光強度をもたらす変異であってよい。例えば、実施形態に係る変異型オキナワマドボタルルシフェラーゼを用いた場合に達成される発光強度は、野生型オキナワマドボタルルシフェラーゼを用いた場合に達成される発光強度の2倍以上、2.5倍以上、3倍以上、4倍以上または5倍以上であり得る。また、実施形態に係る変異型オキナワマドボタルルシフェラーゼを用いた場合に達成される発光強度は、ローダミン(Rhodamine)6Gによる発光強度の50倍以上、60倍以上、70倍以上、80倍以上、90倍以上、100倍以上または110倍以上であり得る。
【0021】
発光強度の向上をもたらす変異の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における424番目のイソロイシン(I)をロイシン(L)に置換する変異(I424L)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における531番目のイソロイシン(I)をアルギニン(R)に置換する変異(I531R)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における355番目のグルタミン酸(E)をアルギニン(R)に置換する変異(E355R)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における367番目のバリン(V)をアラニン(A)に置換する変異(V367A)である。別の例は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)における446番目のリシン(K)をグルタミン(Q)に置換する変異(K446Q)である。これら計5種の変異は、野生型オキナワマドボタルルシフェラーゼのアミノ酸配列(配列番号1)に対して、全て同時に、または任意の組合せで導入することができる。
【0022】
あるいは、上記5種の変異は、配列番号1とは異なるアミノ酸配列を有するタンパク質に対しても導入することができる。例えば、そのようなタンパク質のアミノ酸配列と配列番号1との対応関係を考慮して変異を導入することができる。そのようなタンパク質にI424L変異を導入したい場合、配列番号1と導入の対象とするタンパク質の配列とを比較して、配列番号1に示されるアミノ酸配列の424番目のIに対応するアミノ酸残基を決定し、当該アミノ酸をLに置換することでI424L変異を導入することができる。ここにおいて「対応するアミノ酸残基」は、例えば相同性検索といった公知の技術を利用して決定することができる。
【0023】
実施形態に係る変異型オキナワマドボタルルシフェラーゼの一例は、配列番号36に記載のアミノ酸配列を有するルシフェラーゼである。ここでは、この変異型ルシフェラーゼを変異体1と称する。変異体1は、野生型オキナワマドボタルルシフェラーゼ(配列番号1)に対して、I424L、E355R、V367AおよびK446Qの変異を導入して得られた変異型ルシフェラーゼである。変異体1を用いた場合に達成され得る発光強度は、例えば、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度の4.2倍以上である。また、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の24.4倍以上である。以上から、変異体1を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の102.5倍(=24.4倍×4.2倍)以上であると算出される。
【0024】
また、実施形態は、変異体1に対して更なる変異を導入したものも含む。例えば、変異体1に対して、さらなる発光強度の向上を目的として、さらなる変異を導入したものを含む。あるいは、変異体1に対して、発光強度の向上とは別の目的、例えば、可溶性の向上、分解に対する耐性の向上等を目的として、さらなる変異を導入したものを含む。具体的には、実施形態は、変異体1のアミノ酸配列(配列番号36)との間で一定の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼを含む。好ましくは、この変異型ルシフェラーゼは、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、4.2倍以上の強度でルシフェリンを発光させる。ここにおいて、アミノ酸配列における一定の相同性とは、例えば、75%以上、80%以上、85%以上、90%以上、95%以上、96%以上、97%以上、98%以上または99%以上の相同性である。また、好ましくは、変異体1に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列は、配列番号36に示されるアミノ酸配列の1から20のアミノ酸が変異したものであり、1から15のアミノ酸が変異したものであり、1から10のアミノ酸が変異したものであり、1から5のアミノ酸が変異したものであり、1から4のアミノ酸が変異したものであり、1から3のアミノ酸が変異したものであり、1または2のアミノ酸が変異したものである。好ましくは、変異体1に対して更なる変異が導入された変異型ルシフェラーゼでは、I424L、E355R、V367AおよびK446Qの変異が維持されている。すなわち、変異体1に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列において、424番目のアミノ酸(またはそれに対応するアミノ酸)がロイシンであり、355番目のアミノ酸(またはそれに対応するアミノ酸)がアルギニンであり、367番目のアミノ酸(またはそれに対応するアミノ酸)がアラニンであり、および446番目のアミノ酸(またはそれに対応するアミノ酸)がグルタミンである。ここにおいて「対応するアミノ酸」は、例えば相同性検索といった公知の技術を利用して決定することができる。
【0025】
実施形態に係る変異型オキナワマドボタルルシフェラーゼの別の例は、配列番号37に記載のアミノ酸配列を有するルシフェラーゼである。ここでは、この変異型ルシフェラーゼを変異体2と称する。変異体2は、野生型オキナワマドボタルルシフェラーゼ(配列番号1)に対して、I424L、I531R、E355R、V367AおよびK446Qの変異を導入して得られた変異型ルシフェラーゼである。変異体2による発光強度は、例えば、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度の2.4倍以上である。また、野生型オキナワマドボタルルシフェラーゼ(配列番号1)を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の24.4倍以上である。以上から、変異体2を用いた場合に達成され得る発光強度は、例えば、ローダミン6Gによる発光強度の58.7倍(=24.4倍×2.4倍)以上であると算出される。
【0026】
また、実施形態は、変異体2に対して更なる変異を導入したものも含む。例えば、変異体2に対して、さらなる発光強度の向上を目的として、さらなる変異を導入したものを含む。あるいは、変異体2に対して、発光強度の向上とは別の目的、例えば、可溶性の向上、分解に対する耐性の向上等を目的として、さらなる変異を導入したものを含む。具体的には、実施形態は、変異体2のアミノ酸配列(配列番号37)との間で一定の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼを含む。好ましくは、この変異型ルシフェラーゼは、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼの2.4倍以上の強度でルシフェリンを発光させる。ここにおいて、アミノ酸配列における一定の相同性とは、例えば、75%以上、80%以上、85%以上、90%以上、95%以上、96%以上、97%以上、98%以上または99%以上の相同性である。また、好ましくは、変異体2に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列は、配列番号37に示されるアミノ酸配列の1から20のアミノ酸が変異したものであり、1から15のアミノ酸が変異したものであり、1から10のアミノ酸が変異したものであり、1から5のアミノ酸が変異したものであり、1から4のアミノ酸が変異したものであり、1から3のアミノ酸が変異したものであり、1または2のアミノ酸が変異したものである。好ましくは、変異体2に対して更なる変異が導入された変異型ルシフェラーゼでは、I424L、I531R、E355R、V367AおよびK446Qの変異が維持されている。すなわち、変異体1に対して更なる変異が導入された変異型ルシフェラーゼのアミノ酸配列において、424番目のアミノ酸(またはそれに対応するアミノ酸)がロイシンであり、531番目のアミノ酸(またはそれに対応するアミノ酸)がアルギニンであり、355番目のアミノ酸(またはそれに対応するアミノ酸)がアルギニンであり、367番目のアミノ酸(またはそれに対応するアミノ酸)がアラニンであり、および446番目のアミノ酸(またはそれに対応するアミノ酸)がグルタミンである。ここにおいて「対応するアミノ酸」は、例えば相同性検索といった公知の技術を利用して決定することができる。
【0027】
実施形態に係る変異型ルシフェラーゼのその他の例は、実験的な操作性を向上させる変異が導入された変異型ルシフェラーゼである。たとえば、野生型ルシフェラーゼが哺乳細胞内において低い可溶性を示す場合、実施形態に係る変異型ルシフェラーゼは、その可溶性を高める変異が導入されたルシフェラーゼである。
【0028】
変異型ルシフェラーゼとは、実施形態に係るルシフェラーゼの特徴、すなわち従来のルシフェラーゼと比較して高い発光強度が得られる限りにおいて、ルシフェラーゼのアミノ酸配列に変異、例えば、アミノ酸の置換、欠失および/または付加等が生じたルシフェラーゼであってよい。この変異とは、野生型ルシフェラーゼのアミノ酸配列の1以上のアミノ酸の変異または1から数個のアミノ酸の変異であり、好ましくは、野生型ルシフェラーゼのアミノ酸配列の1から20のアミノ酸の変異、1から15のアミノ酸の変異、1から10のアミノ酸の変異、1から5のアミノ酸の変異、1から4のアミノ酸の変異、1から3のアミノ酸の変異、1または2のアミノ酸の変異である。好ましくは、当該変異型ルシフェラーゼのアミノ酸配列は、野生型ルシフェラーゼのアミノ酸配列との間で75%以上、80%以上、85%以上、90%以上、95%以上、96%以上、97%以上、98%以上または99%以上の相同性を有する。
【0029】
本発明の他の実施形態は、上記実施形態に係るルシフェラーゼをコードする塩基配列を含む核酸に関する。すなわち、当該核酸は、オキナワマドボタルに由来するルシフェラーゼ遺伝子を含む核酸である。実施形態において、核酸とは、例えばDNAまたはRNAを指す。また、実施形態において、ルシフェラーゼの「遺伝子」とは、主として、mRNAに転写される領域、すなわち構造遺伝子を意味する。
【0030】
実施形態に係るルシフェラーゼをコードする塩基配列を含む核酸の一例は、配列番号2に示される塩基配列を含む核酸である。この配列を有する遺伝子は、オキナワマドボタルからクローニングされており、野生型オキナワマドボタルルシフェラーゼをコードする。
【0031】
実施形態に係るルシフェラーゼをコードする塩基配列を含む核酸の別の例は、配列番号34または35に示される塩基配列を含む核酸である。配列番号34の配列を有する遺伝子は変異体1をコードする。配列番号35の配列を有する遺伝子は変異体2をコードする。それぞれ、オキナワマドボタルからクローニングされた野生型の塩基配列に対し、哺乳細胞における発現のためのコドン最適化を行った後、特定のアミノ酸の置換に対応した変異を導入することで得られる配列である。
【0032】
実施形態に係る核酸は、野生型ルシフェラーゼ遺伝子の塩基配列を含む核酸であっても、そこにおいて変異が生じた変異型ルシフェラーゼ遺伝子の塩基配列を含む核酸であってもよい。ここにおいて、変異型ルシフェラーゼ遺伝子とは、コードされたルシフェラーゼが、実施形態に係るルシフェラーゼの特徴、すなわち従来のルシフェラーゼと比較して高い発光強度が得られる限りにおいて、塩基配列中の特定の塩基、たとえば数個の塩基の置換、欠失および/または付加等を受けた遺伝子であってよい。また、塩基配列の変異には、コードされるアミノ酸配列に変化が生じない変異が含まれる。すなわち、実施形態に係る核酸には、野生型ルシフェラーゼをコードする変異型ルシフェラーゼ遺伝子を含む核酸が含まれる。
【0033】
そのようなコードされるアミノ酸配列に変化が生じない変異の一例は、遺伝子中に存在する特定の制限酵素の認識配列を無効にする変異である。この変異によって、遺伝子を含む核酸は当該制限酵素によって切断されなくなるものの、当該遺伝子は変異前と同じアミノ酸配列を有するタンパク質をコードできる。このような変異は、制限酵素の認識配列を構成するコドンを、異なる塩基配列の同義コドンに変換することで達成できる。このような変異は、遺伝子組み換えに使用する制限酵素の認識配列が当該遺伝子中に存在する場合に有用である。この場合、予め遺伝子中の認識配列を無効にしておくことで、制限酵素で処理した場合に遺伝子を含む核酸が断片化することを防ぐことができ、結果として組み換えが容易になる。特定の制限酵素の認識配列を無効にした例は、配列番号3に示される塩基配列である。当該塩基配列は、オキナワマドボタルルシフェラーゼ遺伝子の塩基配列からBamHIおよびEcoRIの認識配列を無効にしたものである。
【0034】
そのようなコードされるアミノ酸配列に変化が生じない変異の別の例は、遺伝子のコドンを特定の生物種における発現に最適化させる変異である。ここにいう「最適化」とは、核酸に含まれる遺伝子のコドンを、特定の生物種においてコドン出現頻度が高いコドンに代えることを意味する。最適化を行った場合、特定の生物種における遺伝子の発現は、最適化をしない場合に比べて高まる。実施形態に係るルシフェラーゼ遺伝子はホタルから取得されたものであるため、当該遺伝子を導入しようとする生物種が分類学的にホタルから遠い程、最適化の効果はより高いと考えられる。実施形態において特定の生物種とは、例えば細菌細胞、酵母細胞および哺乳細胞である。更に哺乳細胞は、例えばマウスの細胞、サルの細胞およびヒトの細胞である。
【0035】
実施形態に係るコドンが最適化されたルシフェラーゼをコードする塩基配列を含む核酸の一例は、配列番号4に示される塩基配列を含む核酸である。当該核酸は、BamHIおよびEcoRIの認識配列が無効にされており、且つ哺乳細胞における発現にコドンが最適化されている。
【0036】
実施形態に係る核酸は、Kozak配列が付与されたルシフェラーゼ遺伝子の塩基配列を含む核酸を含む。Kozak配列とは、開始コドンとその前後の複数の塩基配列から成る配列であり、Kozak配列が存在することで、その遺伝子の発現量が増大することがわかっている。Kozak配列は、生物種または生物群ごとに共通配列が見出されている。実施形態に係るKozak配列を含む核酸は、それを導入する生物種に対応したKozak配列を有する。例えば哺乳細胞に導入される場合、核酸は、Kozak配列としてgccrccatgg(配列番号5)(rはグアニンまたはアデニンを意味する)の配列を含む。Kozak配列を付与するルシフェラーゼ遺伝子は、野生型遺伝子であってもよいが、上述のようなコドンの最適化を行った変異型遺伝子であってもよい。
【0037】
本発明のさらに他の実施形態はこれらの核酸を含むベクターに関する。当該ベクターには、ルシフェラーゼをコードする核酸以外に、発現を調節するための配列またはマーカー遺伝子の配列を含む核酸等を含んでよい。
【0038】
また、本発明のさらに他の実施形態は発光プローブに関する。当該発光プローブは、野生型のルシフェラーゼを含むものでもよいが、変異型ルシフェラーゼを含むものでもよい。特に、公知の技術によって、発光プローブとしての利用に適した改変を加えたものが好ましい。さらに実施形態は、上記発光プローブを用いた種々の試料(インビボ、インビトロ)に関する種々の使用(イメージング、測光等)に有効に適用できる。
【0039】
本発明のさらに他の実施形態は、実施形態に係るルシフェラーゼを利用して細胞内の機能を解析する方法に関する。当該方法は、実施形態に係るルシフェラーゼを細胞内に導入する工程、および前記ルシフェラーゼが誘起するルシフェリンの発光をイメージング装置により検出する工程を含む。例えば、DNA中の特定の発現調節領域の下流に実施形態に係るルシフェラーゼ遺伝子を導入し、ルシフェラーゼの発現を、それが誘起するルシフェリンの発光の有無によって検出することで、発現調節領域の機能を調べることが可能である。
【0040】
本発明のさらに他の実施形態は、上記実施形態に係るルシフェラーゼを利用して細胞内タンパク質を解析する方法に関する。当該方法は、実施形態に係るルシフェラーゼと解析の対象とするタンパク質とから成る融合タンパク質を細胞内に導入する工程;および、前記ルシフェラーゼが誘起するルシフェリンの発光をイメージング装置により検出する工程を含む。
【0041】
当該方法は、解析の対象とするタンパク質の細胞内の局在の観察、および、その局在の時間変化の観察(タイムラプス)を含む。また、当該方法は、タンパク質の局在だけでなく、そのタンパク質が単に発現したか否かの確認をも含む。使用される細胞に特別な限定はなく、細胞のイメージングの分野において通常使用できる細胞であってよい。また、解析の対象とするタンパク質も特別に限定はなく、研究の目的に応じたタンパク質を選択することができる。当該タンパク質は、使用する細胞内に本来存在するタンパクであってよく、または細胞内に本来存在しない異種性のまたは改変したタンパク質であってよい。
【0042】
融合タンパク質を細胞内に導入する場合、既知の導入方法を使用することができる。1つの方法は、細胞外で精製した融合タンパク質を細胞内に直接導入する方法である。例えば、マイクロインジェクション法によって融合タンパク質を細胞内に直接注入することができる。または、融合タンパク質を含む培養液にて細胞をインキュベートさせて、エンドサイトーシスによって融合タンパク質を細胞に取り込ませることができる。また別の方法は、まず融合タンパク質をコードする塩基配列を含む核酸を導入し、その後細胞内で融合タンパク質を発現させる方法である。例えば、当該核酸を含む発現ベクターを、リン酸カルシウム法、リポフェクション法またはエレクトロポレーション法等によって細胞内に導入し、発現ベクターから融合タンパク質を発現させることができる。ここにおいて、融合タンパク質の遺伝子は、実施形態に係るルシフェラーゼ遺伝子と解析の対象とするタンパク質の遺伝子とを含む遺伝子であり、ここにおいて、ルシフェラーゼ遺伝子とタンパク質遺伝子とは、それぞれ正常に翻訳されるよう連結されている。
【0043】
ルシフェリンの発光をイメージング装置により検出する工程は、既知の検出方法を使用することができる。例えば、ルシフェラーゼを含む融合タンパク質を発現する細胞に、ルシフェリン、ATPおよびMg2+イオン等を適宜与えてルシフェラーゼにルシフェリンの発光反応を誘起させ、発生した発光をイメージング装置により検出することができる。イメージング装置とは、例えば発光を捉えるためのフィルターを備えた顕微鏡である。顕微鏡を使用することで、細胞内における発光の位置を特定して、この情報をもとにタンパク質の局在を特定することが可能となる。また、イメージング装置として、経時的に撮像できる機能を備えた顕微鏡を使用することができ、この顕微鏡によって経時的観察も可能となる。
【実施例】
【0044】
[実施例1:オキナワマドボタル由来のルシフェラーゼ遺伝子のクローニング]
1.材料
材料として沖縄県沖縄本島で採集したオキナワマドボタル(Pyrocoelia matsumurai)の幼虫を用いた。
【0045】
2.トータルRNAの抽出とcDNAの合成
ホタルの幼虫からハサミを用いて発光器を切り取った。組織および細胞のホモジナイズ用のビーズを含むチューブであるLysing Matrix Dチューブ(MP−Biomedicals社)に、採取した発光器および1mLのトータルRNA抽出試薬TRIzol Reagent(インビトロジェン社)を入れた。このチューブを組織細胞破砕装置FastPrep 24(MP−Biomedicals社)またはFastPrep FP100A(MP−Biomedicals社)に装着し、振動速度6.5m/sおよび振動時間45秒の条件で、ホタルの発光器を試薬中にて破砕した。完了後、チューブを破砕装置から取り出し、30分間氷上に置いた。その後、もう一度同じ条件で破砕を実施した。
【0046】
次に、トータルRNA抽出試薬TRIzol Reagentの説明書に従って、破砕した溶液からトータルRNAの分離精製を行った。得られたmRNA溶液100μlを、エタノール沈殿法によって沈殿濃縮した。次に、完全長cDNA合成試薬GeneRacer(インビトロジェン社)をマニュアルに従って使用して、沈殿濃縮したトータルRNAから完全長cDNAを合成した。得られたcDNA溶液20μlをホタル完全長cDNAライブラリーとして以下の遺伝子実験に用いた。
【0047】
3.ホタルルシフェラーゼ遺伝子の5’末端側の同定
3−1.Rapid Amplification of cDNA End(RACE)法に用いるプライマーの作製
オキナワマドボタルのルシフェラーゼ遺伝子のクローニングをPolymerase chain reaction(PCR)法によって行った。このPCRに使用するプライマーは、既知の近縁生物由来のルシフェラーゼ遺伝子のアミノ酸配列に基づいて、以下の通り作製した。
【0048】
ホタルルシフェラーゼにおいてよく保存されているアミノ酸領域を確認するために、既に公開されている10種類のホタルルシフェラーゼのアミノ酸配列を、配列情報解析ソフトウェアDNASIS Pro(日立ソフトウェアエンジニアリング株式会社)を用いて比較した。比較に用いた近縁生物は、ラムフィリス・ノクティルカ(Lampyris noctiluca)(登録番号CAA61668)、ルキオラ・クルシアタ(Luciola cruciata)(登録番号P13129)、ルキオラ・ラテラリス(Luciola lateralis)(登録番号Q01158)、ルキオラ・ミングレリカ(Luciola mingrelica)(登録番号Q26304)、ホタリア・パルヴラ(Hotaria parvula)(登録番号AAC37253)、フォティヌス・ピラリス(Photinus pyralis)(登録番号BAF48390)、フォトゥリス・ペンシルヴァニカ(Photuris pennsylvanica)(登録番号Q27757)、ピロコエリア・ミヤコ(Pyrocoelia miyako)(登録番号AAC37254)、ピロコエリア・ルファ(Pyrocoelia rufa)(登録番号AAG45439)およびラハゴフタルムス・オーバイ(Rhagophthalmus ohbai)(登録番号BAF34360)である。
【0049】
その結果、ホタルルシフェラーゼのC末端側440残基付近に位置するL−I−K−Y−K−G−Y−Q−V(配列番号6)のアミノ酸配列がよく保存されていることがわかった。この9つのアミノ酸配列をコードするコドンから塩基配列を予測し、5’末端RACE PCRに用いる12種類のホタルルシフェラーゼ特異的混合プライマーを設計した。このプライマーの名称および配列は以下の通りである(プライマー配列中のY、RおよびNは混合塩基を示す):flexLuc5−ATA(5’−ACY TGR TAN CCY TTA TAT TTA AT−3’:配列番号7)、flexLuc5−ATG(5’−ACY TGR TAN CCY TTA TAT TTG AT−3’:配列番号8)、flexLuc5−ATT(5’−ACY TGR TAN CCY TTA TAT TTT AT−3’:配列番号9)、flexLuc5−ACA(5’−ACY TGR TAN CCY TTA TAC TTA AT−3’:配列番号10)、flexLuc5−ACG(5’−ACY TGR TAN CCY TTA TAC TTG AT−3’:配列番号11)、flexLuc5−ACT(5’−ACY TGR TAN CCY TTA TAC TTT AT−3’:配列番号12)、flexLuc5−GTA(5’−ACY TGR TAN CCY TTG TAT TTA AT−3’:配列番号13)、flexLuc5−GTG(5’−ACY TGR TAN CCY TTG TAT TTG AT−3’:配列番号14)、flexLuc5−GTT(5’−ACY TGR TAN CCY TTG TAT TTT AT−3’:配列番号15)、flexLuc5−GCA(5’−ACY TGR TAN CCY TTG TAC TTA AT−3’:配列番号16)、flexLuc5−GCG(5’−ACY TGR TAN CCY TTG TAC TTG AT−3’:配列番号17)、flexLuc5−GCT(5’−ACY TGR TAN CCY TTG TAC TTT AT−3’:配列番号18)。これらのプライマーの合成はライフテクノロジーズジャパン株式会社に委託して行った。
【0050】
3−2.5’−RACE PCRによる、ホタルルシフェラーゼ遺伝子の5’末端側のクローニング
上述の通り作製したホタル完全長cDNAライブラリーを鋳型として用い、上述の通り作製した12種類の特異的混合プライマーおよび5’末端特異的プライマーであるGeneRacer5’Primer(5’−CGA CTG GAG CAC GAG GAC ACT GA−3’:配列番号19)およびGeneRacer5’Nested Primer(5’−GGA CAC TGA CAT GGA CTG AAG GAG TA−3’:配列番号20)を用いて5’−RACE PCRを行った。GeneRacer5’ PrimerおよびGeneRacer5’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれているものを使用した。5’−RACE PCRによって効率的にルシフェラーゼ遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taq(タカラバイオ株式会社)を用いて、マニュアルに従って実施した。
【0051】
一度目のPCRとして、上述の通り作製した12種類の特異的混合プライマーのいずれかとGeneRacer5’ Primerとから成る12通りのプライマー対を用いてルシフェラーゼ遺伝子を増幅した。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度が1.0μMの12種類プライマーの内1つおよび最終濃度が0.3μMのGeneRacer3’ Primerを含む10μlのPCR反応溶液を作製し、そこへホタル完全長cDNAライブラリー溶液を0.2μl加えた。なお、ホタル完全長cDNAライブラリー溶液の濃度は未定量であった。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、45℃30秒および72℃90秒を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%トリス酢酸緩衝液(TAE)アガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。12の反応溶液の全てにおいてわずかに遺伝子増幅が認められたため、これらのPCR反応溶液を鋳型としてそれぞれnested PCR反応を次の通り実施した。
【0052】
nested PCRとして、一度目のPCRで使用した12種類プライマーのうちの4つとGeneRacer3’ Nested Primerとから成る4通りのプライマー対を用いてルシフェラーゼ遺伝子の増幅を行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.005U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度が1.0μMの12種類プライマーの内1つおよび最終濃度が0.3μMのGeneRacer3’ Primerを含む10μlのPCR反応溶液を作製し、そこへ鋳型として1度目のPCR反応溶液を滅菌水で10倍希釈した溶液を1.0μl加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、45℃30秒および72℃90秒を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。約1.4kbp付近に効率よく遺伝子が増幅したプライマーの組み合わせ条件を確認した。
【0053】
3−3.5’−RACEで増幅した遺伝子の塩基配列の決定
5’−RACEで増幅した遺伝子の塩基配列を読み取るため、ゲル抽出によるPCR産物の精製、サブクローニングおよびダイレクトシークエンスを実施した。詳細を以下に示す。
【0054】
約1.4kbp付近に効率よく遺伝子が増幅したプライマーの組み合わせでPCR(最終容量20μl)を実施し、ゲル抽出の手法を用いて目的とする遺伝子片を回収した。ゲル抽出はWizard SV Gel and PCR Clean−Up System(プロメガ株式会社)を用いて、マニュアルに従って実施した。TAクローニングの手法を用いて、ゲルから抽出したPCR産物のサブクローニングを実施した。TAクローニングはpGEM−T Easy Vector System(プロメガ株式会社)を用いて、マニュアルに従って実施した。その後、このベクターDNAを大腸菌(TOP10株またはDH5α株)に形質転換し、青・白スクリーニングの手法を用いてインサートポジティブコロニーを選択した。選択されたコロニーをダイレクトコロニーPCRに供し、遺伝子が導入されていることを確認した。ダイレクトコロニーPCRには、M13−F(−29) Primer(5’−CAC GAC GTT GTA AAA CGA C−3’:配列番号21)とM13 Reverse(5’−GGA TAA CAA TTT CAC AGG−3’:配列番号22)とから成るプライマー対を用いた。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度がそれぞれ0.2μMのプライマー対を含む10μlのPCR反応溶液を作製し、そこへ鋳型として少量の大腸菌コロニーを加えた。PCR反応は、最初に94℃1分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を25サイクル繰り返し、最後に72℃2分間の伸長反応を行った。PCR反応後、2μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0055】
増幅が確認できたPCR反応溶液について、ダイレクトシークエンシング法を用いてその遺伝子の塩基配列の決定を行った。PCR産物精製キットExoSAP−IT(GEヘルスケアバイオサイエンス)を用いて、PCR反応溶液に含まれる余剰なdNTPおよびプライマーを除去し、PCRダイレクトシークエンシングのための鋳型を調製した。BigDye Terminator v3.1 Cycle Sequencing Kit(アプライドバイオシステムズ)を用いて、この鋳型を含むシークエンシング反応溶液を調製し、サーマルサイクラーを用いてシークエンシング反応を行った。PCR産物の精製およびシークエンシングはそれぞれマニュアルに従って実施した。シークエンシング反応後、反応産物の精製を次の通りに行った。反応溶液に2.5倍量の100%エタノールを加え、遠心機を用いて核酸を沈殿させた。次に、上精を取り除いた後、70%エタノールを加えて沈殿を洗浄し、遠心機を用いて核酸を沈殿させた。最後に、上精を取り除いた後、沈殿を乾燥させた。精製した沈殿にHi−Di Formamide(アプライドバイオシステムズ)を15μl加え、溶解させた。この溶液を94℃2分間の熱変性させ、更に氷上で急冷して、塩基配列を決定するためのサンプルとした。このサンプルをApplied Biosystems 3130xl ジェネティックアナライザ(アプライドバイオシステムズ)を用いて塩基配列を読み取った。塩基配列の解析方法はマニュアルに従って実施した。
【0056】
シークエンシングによって得られた遺伝子配列を、配列情報解析ソフトウェア DNASIS Proの「シークエンス連結」機能を用いて解析した。この配列をNational Center for Biotechnology Information(NCBI)が提供するblastxサーチを利用して相同性検索を実施し、既知ホタルルシフェラーゼの塩基配列と高い相同性を示すことを確認した。以上の実験および解析で得られた塩基配列を新規ホタルルシフェラーゼ遺伝子の5’末端側であると決定した。
【0057】
4.ホタルルシフェラーゼ遺伝子の3’Race PCRおよび完全長cDNAの取得
4−1.3’Race PCRに用いるプライマーの設計
5’Race PCRの実験で得られたホタルルシフェラーゼ遺伝子の5’末端側非翻訳領域の配列を基にして、3’RACEに用いるプライマーおよびNested PCRに用いるプライマーを作成した。プライマーの合成はライフテクノロジーズジャパン株式会社に委託した。
【0058】
4−2.ホタルルシフェラーゼ遺伝子の完全長cDNA取得のための3’Race PCR
上述の通り作成したホタル完全長cDNAライブラリーを鋳型として用い、目的とするホタルルシフェラーゼの5’末端側非翻訳領域の塩基配列から作製したプライマーNo6(2/3)−Full−F1(GACACTAACGCGCTAACATCATTGCAAGA:配列番号23)およびGeneRacer3’ Primer(5’−GCT GTC AAC GAT ACG CTA CGT AAC G−3’:配列番号24)およびGeneRacer3’ Nested Primer(5’−CGC TAC GTA ACG GCA TGA CAG TG−3’:配列番号25)を用いて3’−RACE PCRを行った。GeneRacer3’ PrimerおよびGeneRacer3’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれているものを使用した。3’−RACE PCRによって効率的にルシフェラーゼ遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taq(タカラバイオ株式会社)を用いて、マニュアルに従って実施した。
【0059】
一度目のPCRとして、5’末端側非翻訳領域の塩基配列から作製したプライマーとGeneRacer3’ Primerとから成るプライマー対を用いてルシフェラーゼ遺伝子の増幅を行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)および最終濃度がそれぞれ0.3μMのプライマーを含む20μlのPCR反応溶液を作製し、そこへホタル完全長cDNAライブラリー溶液を0.4μl加えた。なお、ホタル完全長cDNAライブラリー溶液の濃度は未定量であった。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。わずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてそれぞれnested PCR反応を実施した。
【0060】
Nested PCRとして、Nested PCR用のプライマーNo6(2/3)−Full−F2(CGCGCTAACATCATTGCAAGAATGGAAGA:配列番号26)とGeneRacer3’ Nested Primerとから成るプライマー対を用いてルシフェラーゼ遺伝子の増幅を行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)および最終濃度がそれぞれ0.3μMのプライマーを含む20μlのNested PCR反応溶液を作製し、そこへ鋳型として1度目のPCR反応溶液を滅菌水で10倍希釈した溶液を1.0μl加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を30サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。約2kbp付近に効率よく遺伝子が増幅していることを確認した。
【0061】
4−3.3’−Raceで増幅した遺伝子の塩基配列の決定
3’−RACEで増幅した遺伝子の塩基配列を読み取るため、ゲル抽出によるPCR産物の精製、サブクローニングおよびダイレクトシークエンスを実施した。詳細を以下に示す。
【0062】
約2kbp付近に効率よく遺伝子が増幅したプライマーの組み合わせでPCR(最終容量20μl)を実施し、ゲル抽出の手法を用いて目的とする遺伝子片を回収した。ゲル抽出はWizard SV Gel and PCR Clean−Up System(プロメガ株式会社)を用いて、マニュアルに従って実施した。TAクローニングの手法を用いて、ゲルから抽出したPCR産物のサブクローニングを実施した。TAクローニングはpGEM−T Easy Vector System(プロメガ株式会社)を用いて、マニュアルに従って実施した。その後、このベクターDNAを大腸菌(TOP10株またはDH5α株)に形質転換し、青・白スクリーニングの手法を用いてインサートポジティブコロニーを選択した。選択されたコロニーをダイレクトコロニーPCRに供し、遺伝子が導入されていることを確認した。ダイレクトコロニーPCRには、M13−F(−29) Primer(5’−CAC GAC GTT GTA AAA CGA C−3’:配列番号21)とM13 Reverse(5’−GGA TAA CAA TTT CAC AGG−3’:配列番号22)とから成るプライマー対を用いた。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)、最終濃度がそれぞれ0.2μMのプライマー対を含む10μlのPCR反応溶液を作製し、そこへ鋳型として少量の大腸菌コロニーを加えた。PCR反応は、最初に94℃1分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を25サイクル繰り返し、最後に72℃2分間の伸長反応を行った。PCR反応後、2μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0063】
増幅が確認できたPCR反応溶液について、ダイレクトシークエンシング法を用いてその遺伝子の塩基配列の決定を行った。PCR産物精製キットExoSAP−IT(GEヘルスケアバイオサイエンス)を用いて、PCR反応溶液に含まれる余剰なdNTPおよびプライマーを除去し、PCRダイレクトシークエンシングのための鋳型を調製した。BigDye Terminator v3.1 Cycle Sequencing Kit(アプライドバイオシステムズ)を用いて、この鋳型を含むシークエンシング反応溶液を調製し、サーマルサイクラーを用いてシークエンシング反応を行った。シークエンスに用いたプライマーはベクタープライマーまたは遺伝子特異的なプライマーを用いた。PCR産物の精製およびシークエンシングはそれぞれマニュアルに従って実施した。シークエンシング反応後、反応産物の精製を次の通りに行った。反応溶液に2.5倍量の100%エタノールを加え、遠心機を用いて核酸を沈殿させた。次に、上精を取り除いた後、70%エタノールを加えて沈殿を洗浄し、遠心機を用いて核酸を沈殿させた。最後に、上精を取り除いた後、沈殿を乾燥させた。精製した沈殿にHi−Di Formamide(アプライドバイオシステムズ)を15μl加え、溶解させた。この溶液を94℃2分間の熱変性させ、更に氷上で急冷して、塩基配列を決定するためのサンプルとした。このサンプルをApplied Biosystems 3130xlジェネティックアナライザ(アプライドバイオシステムズ)を用いて塩基配列を読み取った。塩基配列の解析方法はマニュアルに従って実施した。
【0064】
シークエンシングによって完全長ホタルルシフェラーゼ遺伝子を獲得した。この塩基配列(配列番号2)もしくはアミノ酸(配列番号1)へ翻訳した配列について、NCBIが提供するblastxまたはblastpサーチを利用して相同性検索を実施した。それぞれの検索で既知ホタルルシフェラーゼの塩基配列と高い相同性を示すことを確認した。以上の実験および解析で得られた塩基配列を新規ホタルルシフェラーゼ遺伝子の完全長cDNA配列であると決定した。
【0065】
以下、この新規のルシフェラーゼを野生型オキナワマドボタルルシフェラーゼと称する。
【0066】
[実施例2:野生型オキナワマドボタルルシフェラーゼの酵素学的なパラメータの測定]
1.野生型オキナワマドボタルルシフェラーゼ遺伝子のタンパク質発現
野生型オキナワマドボタルルシフェラーゼ遺伝子を大腸菌でタンパク質発現させるため、pRSET−Bベクター(インビトロジェン)に導入した。遺伝子発現ベクターの構築は標準的な方法に従い以下のように実験を実施した。
【0067】
1−1.野生型オキナワマドボタルルシフェラーゼ遺伝子の制限酵素認識部位の改変
上述の通り決定した塩基配列によれば、野生型オキナワマドボタルルシフェラーゼ遺伝子はBamHIおよびEcoRIの制限酵素認識配列を含む。ルシフェラーゼのアミノ酸配列を維持しつつ、これらの塩基配列におけるこれらの認識配列を除去する遺伝子改変を実施した。この処理は、後述するルシフェラーゼ遺伝子の発現ベクターへの導入を容易にするために行った。遺伝子変異の導入は、多比良和誠編「遺伝子の機能阻害実験法―簡単で確実な遺伝子機能解析から遺伝子治療への応用まで」(羊土社、2001年発行、p.17−25)に示される方法に従った。変異導入後の配列は、配列番号3に示される塩基配列である。
【0068】
1−2.野生型オキナワマドボタルルシフェラーゼ遺伝子の遺伝子発現ベクターへの導入
ルシフェラーゼ遺伝子を、pRSET−Bベクターの制限酵素サイトBamHI−EcoRI間に導入するため、開始コドンおよびその前に制限酵素BamHI認識配列GGATCCを含むプライマー、並びに終始コドンおよびその後ろに制限酵素EcoRI認識配列GAATTCを含むプライマーを作成した。このプライマー対を用いて、ルシフェラーゼ遺伝子の両端に上述の制限酵素認識部位を含んだ断片の増幅を行った。PCRにはポリメラーゼKOD−Plis−(東洋紡績株式会社)を用いて、マニュアルに従って実施した。
【0069】
最終濃度が等倍の10×PCR Buffer、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が1.0mMのMgSO4、最終濃度が0.02U/μlのTOYOBO KOD−Plis−(1U/μl)、最終濃度がそれぞれ0.3μMのプライマー対を含む20μlのPCR反応溶液を作製し、そこへ鋳型としてBamHIとEcoRI認識配列を持たないルシフェラーゼ遺伝子の溶液を0.4μl加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、55℃30秒および68℃2分を30サイクル繰り返し、最後に68℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。遺伝子増幅が認められたため、このPCR反応溶液を通常のエタノール沈殿法により沈殿濃縮し、制限酵素処理用10×H Bufferを4μl、制限酵素BamHI(東洋紡績株式会社)および制限酵素EcoRI(東洋紡績株式会社)を各2μlならびに滅菌脱イオン水32μlを加えて溶解し、37℃で2時間保温して制限酵素処理した。その後、この反応溶液をエタノール沈殿法により沈殿濃縮した後、滅菌脱イオン水に溶解した。この溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色を行った。紫外線照射下において確認されるDNAバンドを含むゲルを、ナイフを用いて切り出した。この切り出したゲルからWizard(R) SV Gel and PCR Clean−Up System(プロメガ)を用いてDNAを抽出した。これらの操作はマニュアルに従って実施した。その後、Ligation Pack(ニッポンジーン)をマニュアルに従って用いて、あらかじめ同様の方法で制限酵素BamHIと制限酵素EcoRIで処理したpRSET−Bベクターに抽出したDNAを導入した。このベクターDNAを大腸菌JM109(DE3)株に形質転換し、コロニーを形成させた。
【0070】
得られたコロニーを鋳型としてダイレクトコロニーPCRを実施し、pRSET−Bに導入したルシフェラーゼ遺伝子を増幅した。ダイレクトコロニーPCRは、T7 promoter Primer(5’−TAA TAC GAC TCA CTA TAG GG−3’:配列番号27)およびT7 Reverse Primer(5’−CTA GTT ATT GCT CAG CGG TGG−3’:配列番号28)のプライマー対を用いて行った。最終濃度が等倍の10×Ex Taq Buffer(20mM Mg2+plus)、最終濃度が各0.2mMのdNTP Mixture(各2.5mM)、最終濃度が0.05U/μlのTaKaRa Ex Taq(5U/μl)および最終濃度がそれぞれ0.2μMのプライマーを含む10μlのPCR反応溶液を作製し、そこへ鋳型として少量の大腸菌コロニーを加えた。PCR反応は、最初に94℃2分間の熱変性を行った後、94℃30秒、50℃30秒および72℃2分を25サイクル繰り返し、最後に72℃5分間の伸長反応を行った。PCR反応後、1μlのPCR反応溶液を、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイド染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0071】
増幅が確認できたPCR反応溶液について、ダイレクトシークエンシング法を用いてその遺伝子の塩基配列の決定を行った。PCR産物精製キットExoSAP−ITを用いて、PCR反応溶液に含まれる余剰なdNTPおよびプライマーを除去し、PCRダイレクトシークエンシングのための鋳型を調製した。BigDye Terminator v3.1 Cycle Sequencing Kitを用いて、この鋳型を含むシークエンシング反応溶液を調製し、サーマルサイクラーを用いてシークエンシング反応を行った。シークエンスには、ベクタープライマーまたは遺伝子特異的なプライマーを用いた。PCR産物の精製およびシークエンシングはそれぞれマニュアルに従って実施した。シークエンシング反応後、反応産物の精製を次の通りに行った。反応溶液に2.5倍量の100%エタノールを加え、遠心機を用いて核酸を沈殿させた。次に、上精を取り除いた後、70%エタノールを加えて沈殿を洗浄し、遠心機を用いて核酸を沈殿させた。最後に、上精を取り除いた後、沈殿を乾燥させた。精製した沈殿にHi−Di Formamide(アプライドバイオシステムズ)を15μl加え、溶解させた。この溶液を94℃2分間の熱変性させ、更に氷上で急冷して、塩基配列を決定するためのサンプルとした。このサンプルをApplied Biosystems 3130xl ジェネティックアナライザを用いて塩基配列を読み取り、正常に遺伝子発現ベクターpRSET−Bに導入されていることを確認した。
【0072】
2.発光タンパク質の精製
JM109(DE3)を含む大腸菌溶液50μlにルシフェラーゼ発現ベクター0.5μlを添加し、氷上で10分、その後42℃で1分、最後に氷上で2分インキュベートした。その後、大腸菌溶液50μlをSOC培地200μlに加えた。その大腸菌/SOC培地混合溶液を37℃で20分間振とうしながらインキュベートした。インキュベート後のサンプル100μlをLB培地プレート(100μg/mlアンピシリンを含む)にストリークし、37℃で一晩インキュベートした。翌日得られたコロニーをピックアップし、500mlスケールのLB培地で培養した。培養は37℃で24時間、18℃で24時間行った。合計48時間の培養の後、遠心分離で菌体を回収し、0.1M Tris−HCl溶液(pH8.0)に再度懸濁して超音波破砕した。菌体破砕液を遠心分離(15000rpm、10分)し、沈査を除去して上清を回収した。ベッドボリューム2mlのカラムにNi−Agar懸濁液500μlと0.1M Tris−HCl2mlを加え、カラムを平衡化した。回収した上清をカラムに添加し、自然落下させた。上清の全量がカラムを通過するまでの間の操作は全て4℃の条件で行った。25mMイミダゾール/0.1M Tris−HCl溶液2mlでカラムを洗浄した。洗浄後のカラムに500mMイミダゾール/0.1M Tris−HCl溶液を2ml加え、ルシフェラーゼを溶出した。溶出されたサンプルをゲルろ過カラムPD−10(GEヘルスケア)でろ過し、脱塩した。脱塩後のサンプルをVivaspin6(ザルトリウス)で限界ろ過し、濃縮されたサンプルにグリセリンを添加して、50%グリセリン溶液とした。保存は−20℃で行った。
【0073】
3.発光スペクトルの測定
測定のための装置としてLumiFlSpectroCapture(ATTO)を用い、0.1Mクエン酸/0.1M Na2HPO4 buffer(pH6.0−8.0)に1mM D−ルシフェリン、2mM ATPおよび4mM MgCl2を含む溶液に、精製酵素を1μg/mlから10μg/mlの間の最終濃度で添加し、酵素添加後15秒経過時点で発光スペクトルを測定した。測定結果を図1に示す。
【0074】
図1より、取得された野生型オキナワマドボタルルシフェラーゼは、pH8.0の環境において、560nm付近に最大発光波長を示している。また、pH7.5において563nm付近、pH7.0において571nm付近(および610nm付近により小さなピーク)、pH6.5において608nm付近およびpH6.0において612nm付近、pH5.5において613nm付近となった。
【0075】
4.速度論的解析
4−1.D−ルシフェリンおよびATPの濃度の決定
D−ルシフェリン溶液中のD−ルシフェリン濃度およびATP溶液中のATP濃度を以下の通りに決定した。
【0076】
UV−Visible Spectrometer(Hitachi)を用いて、D−ルシフェリン溶液およびATP溶液の紫外可視吸収スペクトルを測定し、この測定結果と以下のε値とから濃度を算出した。
D−ルシフェリン:λmax 328nm、ε18200、pH5.0
ATP:λmax 259nm、ε15400、pH7.0。
【0077】
測定はそれぞれ10回ずつ行い、吸光度の平均値を濃度算出に用いた。このように濃度を決定したD−ルシフェリン溶液およびATP溶液を用いて、以下のKm値算出を行った。
【0078】
4−2.D−ルシフェリンに対するKm値の測定
様々なD−ルシフェリン濃度の環境下において、得られたルシフェラーゼによる発光の強度を測定した。測定結果に基づいて、D−ルシフェリンに対するKm値を決定した。
【0079】
D−ルシフェリンを0.1M Tris−HCl(pH8.0)に添加して、異なる濃度の12種類のDールシフェリン溶液を作製した。これらの溶液は、D−ルシフェリンの終濃度がそれぞれ0.625、1.25、2.5、5、10、20、40、80、160、320、480および640μMとなるようにD−ルシフェリンを含む。これらのD−ルシフェリン溶液を96穴マイクロプレートに50μlずつ分注した。各種精製ルシフェラーゼ、4mM ATPおよび8mM MgSO4を含む0.1M Tris−HCl(pH8.0)溶液をルミノメーターの標準ポンプに接続し、当該溶液をウェルに50μl添加すると同時に測定を行った。測定にはLuminescensor(ATTO)を使用した。測定は各ルシフェリン濃度について3回ずつ行った。
【0080】
得られたフォトンカウント値のピーク強度を初速度Vとして、ルシフェリン濃度Sに対してプロットした。このプロットにミカエリス・メンテン型のカーブフィッティングを行い、Km値を算出した。カーブフィッティングは非線形の最小二乗法で行い、パラメータの探索にはニュートン法を用いた。
【0081】
4−3.ATPに対するKm値の測定
様々なATP濃度の環境下において、得られたルシフェラーゼによる発光の強度を測定した。測定結果に基づいて、ATPに対するKm値を決定した。
【0082】
ATPを0.1M Tris−HCl(pH8.0)に添加して、異なる濃度の12種類のATP溶液を作製した。これらの溶液は、ATPの終濃度がそれぞれ5、10、20、40、80、160、320、480、640、800、1280、1600および1920μMとなるようにATPを含む。これらのATP溶液をそれぞれ96穴マイクロプレートに50μlずつ分注した。各種精製ルシフェラーゼ、1mM D−ルシフェリン、8mM MgSO4を含む0.1M Tris−HCl(pH8.0)溶液をルミノメーターの標準ポンプに接続し、当該溶液をウェルに50μl添加すると同時に測定を行った。測定は各ATP濃度について3回ずつ行った。
【0083】
得られたフォトンカウント値のピーク強度を初速度Vとして、ATP濃度Sに対してプロットした。このプロットにミカエリス・メンテン型のカーブフィッティングを行い、Km値を算出した。カーブフィッティングは非線形の最小二乗法で行い、パラメータの探索にはニュートン法を用いた。
【0084】
上記のようにして決定されたD−ルシフェリンに対するKm値およびATPに対するKm値を表1に示す。表1には、同様に測定した既知のルシフェラーゼの各Km値も示される。GL3とは既知のホタル由来のルシフェラーゼである。また、ELuc、CBGおよびCBRとは既知のコメツキムシ由来のルシフェラーゼである。これらの既知のルシフェラーゼは市販されるものを使用した。
【0085】
【表1】
【0086】
さらに、これらの結果について、縦軸をATPに対するKm値とし、横軸をD−ルシフェリンに対するKm値としてプロットした図を、図2として示す。
【0087】
[実施例3:発光強度の測定]
野生型オキナワマドボタルルシフェラーゼによる発光強度を、既知のルシフェラーゼおよびローダミン6Gによる発光強度と比較した。
【0088】
1.ローダミン6Gによる化学発光の測定
ローダミン6Gを0.1Mクエン酸/0.2M Na2HPO4(pH4.0)溶液に溶解した。このローダミン6G溶液を15000rpmで1分間遠心分離し、上清を回収した。回収した上清を0.1Mクエン酸/0.2M Na2HPO4溶液で1000倍希釈し、希釈液の吸光度を測定した。測定はNanoVue(GEヘルスケア)で行った。測定は530nmにおいて5回行い、その平均値として0.048の吸光度が得られた。ローダミン6Gのε値(1.16x105mol−1cm−1)を用いて元のローダミン6G溶液の濃度を算出した結果、414μMであった。この溶液を0.1Mクエン酸/0.2M Na2HPO4溶液(pH4.0)で希釈し、30μMの希釈液を作製した。
【0089】
この希釈液を使用して、以下の表2の通り溶液1を調製した。
【0090】
【表2】
【0091】
また、ビス(2,4,6−トリクロロフェニル)オキサレート(TCPO)をアセトニトリルに最終濃度3mMとなるように溶解して溶液2を作製した。
【0092】
溶液1を96穴のマイクロプレートに100μlずつ分注した。そこへ、ポンプを使用して溶液2を50μl添加すると同時に測定を開始して、溶液2の添加から10秒間のフォトンカウントの積算値を取得した。測定にはLuminescensor(ATTO)を使用し、300nmから650nmの波長の光を取得した。同様の測定を10回行った。
【0093】
2.P.ピラリスルシフェラーゼおよび野生型オキナワマドボタルルシフェラーゼによる化学発光の測定
JM109(DE3)株を含む溶液50μlにルシフェラーゼ発現ベクターを含む溶液0.5μlを添加し、氷上で10分、42℃で1分、氷上で2分インキュベートした。その後、この大腸菌溶液50μlをSOC培地200μlに加えた。大腸菌/SOC培地混合溶液を37℃で20分間振とうしながらインキュベートした。インキュベート後のサンプル100μlをLB培地プレート(100μg/mlアンピシリンを含む)にストリークし、37℃で一晩インキュベートした。翌日、できたコロニーをピックアップし、500mlスケールのLB培地で培養した。培養は37℃で24時間、18℃で24時間行った。合計48時間の培養の後、遠心分離によって菌体を回収し、0.1M Tris−HCl溶液(pH8.0)に再度懸濁して超音波破砕した。菌体破砕液を遠心分離し(15000rpm、10分間)、沈査を除去して上清を回収した。ベッドボリューム2mlのカラムにNi−Agar懸濁液500μlおよび0.1M Tris−HCl溶液2mlを加え、カラムを平衡化した。回収した上清をカラムに添加し、自然落下させた。上清の全量がカラムを通過するまでの間の操作は全て4℃の条件で行った。25mMイミダゾール/0.1M Tris−HCl溶液2mlでカラムを洗浄した。洗浄後のカラムに500mMイミダゾール/0.1M Tris−HCl溶液を2ml加え、ルシフェラーゼを溶出した。溶出されたサンプルをゲルろ過カラムPD−10(GEヘルスケア)でろ過し、脱塩した。脱塩後のサンプルをVivaspin6(ザルトリウス)で限界ろ過した。
【0094】
得られたサンプルの濃度を比色法で測定し、P.ピラリスルシフェラーゼおよび野生型オキナワマドボタルルシフェラーゼの濃度を確認した。最終濃度が1μg/mlとなるように上記ルシフェラーゼを0.1Mクエン酸/0.2M Na2HPO4(pH8.0)に希釈した。
【0095】
このルシフェラーゼ溶液を50μlずつ96穴マイクロプレートに分注した。2mM D−ルシフェリン、4mM ATPおよび8mM MgSO4/0.1M Tris−HCl(pH8.0)を含む溶液をルミノメーターの標準ポンプによって50μl添加し、添加と同時に測定を行った。溶液の添加から10秒間のフォトンカウントの積算値を取得した。測定にはLuminescensor(ATTO)を使用し、300nmから650nmの波長の光を取得した。各測定は6回行った。
【0096】
3.測定結果
以上のように測定したローダミン6G、P.ピラリスルシフェラーゼおよび野生型オキナワマドボタルルシフェラーゼによる化学発光の強度を図3に示す。
【0097】
図3から、野生型オキナワマドボタルルシフェラーゼは、ローダミン6Gによる発光の24.4倍の強度の発光を示すことがわかった。また、野生型オキナワマドボタルルシフェラーゼは、P.ピラリスルシフェラーゼによる発光の5倍の強度の発光を示すことがわかった。
【0098】
[実施例4:哺乳細胞における野生型オキナワマドボタルシフェラーゼの発現]
オキナワマドボタルをHeLa細胞に発現させて、細胞内での発光強度を測定した。
【0099】
オキナワマドボタルルシフェラーゼ遺伝子の導入のために2種類の発現ベクターを作製した。オキナワマドボタルから取得したオリジナルのオキナワマドボタルルシフェラーゼ遺伝子においてBamHIおよびEcoRIの認識配列を削除した遺伝子(配列番号3)、さらに哺乳細胞における発現に最適化した遺伝子(配列番号4)をそれぞれ含む核酸を、pcDNA3.1(+)ベクター(Invitorogen)のマルチクローニングサイトのBamHIおよびEcoRIのサイト間に挿入した。また、比較のために、既知であるP.ピラリスルシフェラーゼの遺伝子について哺乳細胞における発現に最適化させた遺伝子を含む核酸を、同様にpcDNA3.1(+)ベクターに挿入した。
【0100】
このようにして得られた3種のプラスミドを、それぞれリポフェクション法によってHeLa細胞に遺伝子導入し、24時間後D−MEM培地を交換した。測定直前にD−ルシフェリンを最終濃度1mMで添加し、Kronos(ATTO)を用いて発光強度を測定した。その結果を図4に示す。なお、図4に示される発光強度は10秒間の積算値を示している。
【0101】
図4から、オキナワマドボタルルシフェラーゼのコドンの最適化によって、HeLa細胞で発現させた場合の発光強度が27倍増大することがわかる。また、コドン最適化後のオキナワマドボタルルシフェラーゼはコドン最適化後のP.ピラリスルシフェラーゼより3.4倍の発光強度を示した。
【0102】
[実施例5:変異型オキナワマドボタルルシフェラーゼの作製]
【0103】
変異型オキナワマドボタルルシフェラーゼを発現する変異型遺伝子を作製した。
【0104】
哺乳細胞における発現に最適化した遺伝子(配列番号4)に対し、次の5種の変異を任意の組合せで導入した。
【0105】
アミノ酸配列上424番目のイソロイシンをロイシンに置換する変異(I424L)
アミノ酸配列上531番目のイソロイシンをアルギニンに置換する変異(I531R)
アミノ酸配列上355番目のグルタミン酸をアルギニンに置換する変異(E355R)
アミノ酸配列上367番目のバリンをアラニンに置換する変異(V367A)
アミノ酸配列上446番目のリシンをグルタミンに置換する変異(K446Q)
【0106】
それぞれの変異の導入のために、以下の5種のプライマーを用いた。
I424L変異:OkinawaMado_I424L(GGCTGGCTGCATTCAGGCGATcTgGCCTACTACGATAAGGACGGC:配列番号29)
I531R変異:OkinawaMado_I531R(CCCAAAGGTCTCACAGGCAAGAgaGACTCTCGAAAAATTCGAGAA:配列番号30)
E355R変異:OkinawaMado_E355R(AGTGCAGTAATCATTACTCCAagGGGCGACGACAAACCAGGAGCC:配列番号31)
V367A変異:OkinawaMado_V367A(CCAGGAGCCTGCGGCAAGGTCGcCCCCTTCTTTTCCGCCAAAATTGTG:配列番号32)
K446Q変異:OkinawaMado_K446Q(CTGAAAAGTCTGATCAAGTACcAGGGCTACCAGGTGCCACCTGCA:配列番号33)
【0107】
変異の導入は文献の記載に基づいて行った(Asako Sawano and Atsushi Miyawaki, Nucleic Acids Research,2000, Vol.28, No.16)。変異導入後にシーケンサーを用いて配列を読み取り、目的とする変異がオキナワマドボタルルシフェラーゼ遺伝子に導入されていることを確認した。
【0108】
作製した種々の変異型オキナワマドボタルルシフェラーゼのうち2種類(変異体1および変異体2と称する)に着目した。これらは、それぞれ以下の変異を有する。
【0109】
変異体1:I424L、E355R、V367AおよびK446Q
変異体2:I424L、I531R、E355R、V367AおよびK446Q
変異体1のアミノ酸配列は配列番号36に記載され、変異体1をコードする遺伝子の塩基配列は配列番号34に記載される。また、変異体2のアミノ酸配列は配列番号37に記載され、変異体2をコードする遺伝子の塩基配列は配列番号35に記載される。
【0110】
次に、変異体1および2による発光強度について、野生型オキナワマドボタルルシフェラーゼと比較した。
【0111】
野生型オキナワマドボタルルシフェラーゼの遺伝子を含む核酸(配列番号4)、変異体1の遺伝子を含む核酸(配列番号34)および変異体2の遺伝子を含む核酸(配列番号35)に対してKozak配列を付与した。なお、3種の配列とも哺乳細胞における発現に対してコドン最適化されている。その後、pF9A CMV hRLuc neo Flexi vector(Promega)のマルチクローニングサイトのSgfIおよびPmeIのサイト間に挿入した。pF9Aベクターは内部コントロールとしてウミシイタケルシフェラーゼ遺伝子(hRLuc)をベクター配列に含んでおり、マルチクローニングサイトに挿入された発光遺伝子による発光強度をウミシイタケルシフェラーゼによる発光強度との比として算出することが可能である。
【0112】
得られた3種のプラスミドを、24穴のプレートに播種したHeLa細胞にそれぞれリポフェクション法で遺伝子導入し、24時間後PBSで細胞を洗浄した。24穴プレートの各ウェルに2mM D−ルシフェリン/CO2 Independent Medium(Invitrogen)を500μlずつ添加し、25℃、各ウェルあたり1秒間測定の条件でLuminescensor(ATTO)を用いて120分間発光強度を測定した。90分経過時点の発光強度を野生型オキナワマドボタルルシフェラーゼ、変異体1および変異体2の発光強度とした。各ウェル内の培養液を除去した後、PBSで各ウェルを3回洗浄した。次に各ウェルに10μMセレンテラジン/CO2 Independent Mediumを500μlずつ添加し、25℃、各ウェルあたり1秒間測定の条件でLuminescensorを用いて30分間発光強度を測定した。セレンテラジン添加後、10分経過時点の発光強度を内部コントロールであるウミシイタケルシフェラーゼによる発光強度とした。野生型オキナワマドボタルルシフェラーゼ、変異体1および変異体2による発光強度をウミシイタケルシフェラーゼによる発光強度でそれぞれ除算した値を算出し、その値を各ルシフェラーゼの発光強度としてグラフ化した。
【0113】
図5には、D−ルシフェリン添加直後から120分経過時点までにおける、各細胞中のオキナワマドボタルルシフェラーゼによる発光強度の時間変化が示される。図6には、セレンテラジン添加直後から30分経過時点までにおける、各細胞中のウミシイタケルシフェラーゼによる発光強度の時間変化が示される。図7には、野生型オキナワマドボタルルシフェラーゼ、変異体1および変異体2による90分経過時点における発光強度を、内部コントロールであるウミシイタケルシフェラーゼによる10分経過時点における発光強度でそれぞれ除算して得られた発光強度比を示す。
【0114】
図7に示されるように、変異体1によって得られた発光強度は、野生型オキナワマドボタルルシフェラーゼによる発光強度と比較して4.2倍高かった。変異体2によって得られた発光強度は、野生型オキナワマドボタルルシフェラーゼによる発光強度と比較して2.4倍高かった。変異の導入によってオキナワマドボタルルシフェラーゼの発光強度が増大することが示された。
【特許請求の範囲】
【請求項1】
オキナワマドボタル(Pyrocoelia matsumurai)に由来し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼ。
【請求項2】
配列番号1に示されるアミノ酸配列との間で80%以上の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較してより高い強度でルシフェリンを発光させる変異型ルシフェラーゼ。
【請求項3】
前記野生型ルシフェラーゼに対して、2.4倍以上の強度で前記ルシフェリンを発光させる請求項1または2に記載の変異型ルシフェラーゼ。
【請求項4】
前記野生型ルシフェラーゼに対して、4.2倍以上の強度で前記ルシフェリンを発光させる請求項1または2に記載の変異型ルシフェラーゼ。
【請求項5】
請求項1から4の何れか1項に記載の変異型ルシフェラーゼであって、
配列番号1に記載されるアミノ酸配列の424番目のイソロイシンに対応するアミノ酸残基がロイシンであること、
配列番号1に記載されるアミノ酸配列の531番目のイソロイシンに対応するアミノ酸残基がアルギニンであること、
配列番号1に記載されるアミノ酸配列の355番目のグルタミン酸に対応するアミノ酸残基がアルギニンであること、
配列番号1に記載されるアミノ酸配列の367番目のバリンに対応するアミノ酸残基がアラニンであること、および
配列番号1に記載されるアミノ酸配列の446番目のリシンに対応するアミノ酸残基がグルタミンであること
の少なくとも1つを満足するアミノ酸配列を有する変異型ルシフェラーゼ。
【請求項6】
配列番号36または37に記載のアミノ酸配列を有する変異型ルシフェラーゼ。
【請求項7】
配列番号36または37に記載のアミノ酸配列との間で80%以上の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼ。
【請求項8】
請求項1から7の何れか1項に記載の変異型ルシフェラーゼをコードする塩基配列を含む核酸。
【請求項9】
配列番号34または配列番号35に示される塩基配列を含む請求項8に記載の核酸。
【請求項1】
オキナワマドボタル(Pyrocoelia matsumurai)に由来し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼ。
【請求項2】
配列番号1に示されるアミノ酸配列との間で80%以上の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較してより高い強度でルシフェリンを発光させる変異型ルシフェラーゼ。
【請求項3】
前記野生型ルシフェラーゼに対して、2.4倍以上の強度で前記ルシフェリンを発光させる請求項1または2に記載の変異型ルシフェラーゼ。
【請求項4】
前記野生型ルシフェラーゼに対して、4.2倍以上の強度で前記ルシフェリンを発光させる請求項1または2に記載の変異型ルシフェラーゼ。
【請求項5】
請求項1から4の何れか1項に記載の変異型ルシフェラーゼであって、
配列番号1に記載されるアミノ酸配列の424番目のイソロイシンに対応するアミノ酸残基がロイシンであること、
配列番号1に記載されるアミノ酸配列の531番目のイソロイシンに対応するアミノ酸残基がアルギニンであること、
配列番号1に記載されるアミノ酸配列の355番目のグルタミン酸に対応するアミノ酸残基がアルギニンであること、
配列番号1に記載されるアミノ酸配列の367番目のバリンに対応するアミノ酸残基がアラニンであること、および
配列番号1に記載されるアミノ酸配列の446番目のリシンに対応するアミノ酸残基がグルタミンであること
の少なくとも1つを満足するアミノ酸配列を有する変異型ルシフェラーゼ。
【請求項6】
配列番号36または37に記載のアミノ酸配列を有する変異型ルシフェラーゼ。
【請求項7】
配列番号36または37に記載のアミノ酸配列との間で80%以上の相同性を示すアミノ酸配列を有し、配列番号1に示されるアミノ酸配列を有する野生型ルシフェラーゼと比較して、より高い強度でルシフェリンを発光させる変異型ルシフェラーゼ。
【請求項8】
請求項1から7の何れか1項に記載の変異型ルシフェラーゼをコードする塩基配列を含む核酸。
【請求項9】
配列番号34または配列番号35に示される塩基配列を含む請求項8に記載の核酸。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−235756(P2012−235756A)
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願番号】特願2011−108206(P2011−108206)
【出願日】平成23年5月13日(2011.5.13)
【出願人】(000000376)オリンパス株式会社 (11,466)
【Fターム(参考)】
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願日】平成23年5月13日(2011.5.13)
【出願人】(000000376)オリンパス株式会社 (11,466)
【Fターム(参考)】
[ Back to top ]