
ホモ3量体融合タンパク質の製造
本発明は、同族のリガンドの強力な阻害剤である3量体の腫瘍壊死因子受容体の製造方法を供する。
【発明の詳細な説明】
【技術分野】
【0001】
腫瘍壊死因子(TNF)受容体スーパーファミリーは、宿主の防衛、炎症、および自己免疫に関する分子のラージファミリーであり、そしてヒトの疾患に関与している。TNFの阻害を狙った治療薬剤は、炎症性疾患、例えば、リューマチ性関節炎および炎症性腸炎の制御において有効である。追加的なTNF/TNF受容体スーパーファミリーのメンバーは、現在のところ、自己免疫疾患、アテローム硬化、骨粗鬆症、移植拒絶反応および癌に対する治療を対象としている。
【0002】
TNFおよびTNF受容体ファミリーはともに、自己組織化3量体として活性であるが、機能上、2量体分子、例えば、抗体または受容体IgG融合物だけが治療薬剤として使用されてきた。TNFスーパーファミリー中のリガンドおよび受容体の両方により表される3回回転対称は、拮抗物質に基づく3量体の受容体が増加した結合活性を表すはずであり、このため、2量体分子と比較して増加した有効性を示す。
【0003】
したがって、安定な3量体としてのTNFリガンドおよびTNF受容体の簡単な発現方法のための必要性は依然として存在する。
【発明の開示】
【0004】
本発明は、2量体の受容体分子と比較した場合、それらの同種のリガンドの生物学的活性のより効力のある阻害剤である、3量体のTNF受容体の生産方法を供する。
【0005】
以下に説明するように、本発明は、(1)膜貫通性活性化因子かつCAML(カルシウム信号変調シクロフィリンリガンド)相互作用因子(TACI)の細胞外ドメイン、および(2)3量体化ポリペプチド、を含んで成るポリペプチドを供する。適当なTACIの細胞外ドメインは、SEQ ID NO:4のアミノ酸残基30から110、および(2)SEQ ID NO:4のアミノ酸残基1から110、(3)SEQ ID NO:4のアミノ酸残基30から154、および(4)SEQ ID NO:4のアミノ酸残基1から154、を含む。実例となる3量体化ポリペプチドは、ヒトのコラーゲンXのNC-1断片および熱ショック結合タンパク質-1(Heat Shock Binding Protein-1)の3量体化断片を含む。本発明はさらに、TACIの細胞外ドメインおよび3量体化ポリペプチドを含んで成る融合タンパク質のホモ3量体の複合体を供する。
【0006】
本発明のこれらおよび他の観点は、以下の詳細な説明および図の参酌により明らかとなるであろう。付け加えると、様々な引用を以下に示し、そして、それら全体における引用により組み入れられている。
【0007】
発明の簡単な説明
1.定義
以下の説明において、番号の語は広範囲に使用される。本発明の理解を容易にするために、以下の定義を与える。
【0008】
本明細書で使用される「核酸」または「核酸分子」はポリヌクレオチド、例えば、デオキシリボ核酸(DNA)またはリボ核酸(RNA)、オリゴヌクレオチド、ポリメラーゼ連反応(PCR)により生成された断片、および何らかの連結、切断、エンドヌクレアーゼの作用およびエキソヌクレアーゼの作用により生成された断片を意味する。核酸分子は、自然に発生するヌクレオチドの単量体(例えば、DNAおよびRNA)、自然に発生するヌクレオチドの類似体(例えば、自然に発生するヌクレオチドのα-光学異性体)、または両方の組み合わせである単量体で構成されてよい。修飾されたヌクレオチドは、糖の一部および/またはピリミジンもしくはプリン塩基の一部において変化を有してもよい。糖修飾は、例えば、ハロゲン、アルキル基、アミンおよびアジド基での1以上のヒドロキシル基の置換を含み、あるいは、糖はエーテルまたはエステルとして官能性を持たせることができる。さらに、すべての糖の部分は、立体配置的および電子的に類似した構造体、例えば、アザ糖および炭素環式糖の類似体により置換してもよい。塩基部分の修飾の例は、アルキル化プリンもしくはピリミジン、アシル化プリンもしくはピリミジン、または他の周知の複素環の置換を含む。核酸単量体は、ホスホジエステル結合またはこれらの類似体の結合により連鎖させることができる。ホスホジエステル類似体の結合は、ホスホロチオエート、ホスホロジチオエート、ホスホロセレノエート、ホスホロジセレノエート、ホスホロアニロチオエート、ホスホラニリデート、ホスホラミデート等を含む。「核酸分子」の語もまた、自然に発生し、あるいは修飾された、ポリアミドの主軸を形成する核酸塩基を含んで成るいわゆる「ペプチド核酸」を含む。核酸は1本鎖または2本鎖のいずれかでよい。
【0009】
「核酸分子の補体」の語は、相補ヌクレオチド配列および関連するヌクレオチド配列と比較して逆配向を有する核酸分子を意味する。例えば、配列5’ATGCACGGG3’は5’CCCGTGCAT3’に相補的である。
【0010】
「コンティグ」の語は、他の核酸分子と同一または相補的な配列の連続鎖を有する核酸分子を意味する。コンティグ配列は、核酸分子の所定の鎖と当該核酸分子の全部または部分的な伸展のいずれかにおいて、「オーバーラップ」と言われる。例えば、ポリヌクレオチド5’ATGGAGCTT3’に対する代表的なコンティグは5’AGCTTgagt3’および3’tcgacTACC5’である。
【0011】
「構造遺伝子」の語は、メッセンジャーRNA(mRNA)へと転写される核酸分子を意味し、それから特異的なポリペプチドのアミノ酸配列に特徴的に翻訳される。「注目の遺伝子」は構造遺伝子であってよい。
【0012】
「相補的DNA(cDNA)」は逆転写酵素によりmRNAの鋳型から形成される1本鎖DNA分子である。一般的に、mRNAの一部に相補的なプライマーは逆転写の最初に必要とされる。また、これらの当業者はこれらの1本鎖DNA分子およびその相補的DNA鎖からなる2本鎖DNAを意味して「cDNA」を使用する。「cDNA]の語はまた、RNAの鋳型から合成されるcDNA分子のクローンも意味する。
【0013】
「単離核酸分子」は生物のゲノムDNAに組み込まれない核酸分子である。例えば、細胞のゲノムDNAから分離された成長因子をコードするDNA分子は、単離DNA分子である。単離核酸分子の他の例は、生物のゲノムに組み込まれない化学合成した核酸分子である。特定の種から単離された核酸分子はその種由来の染色体の完全DNA分子よりも小さい。
【0014】
「核酸分子作成物」は天然には存在しない配列に混合あるいは配列した核酸のセグメントを含ませるために、人工的に修飾された1本鎖または2本鎖のいずれかの核酸分子である。
【0015】
「直鎖状DNA」は、非環状の5’および3’末端を有する非環状DNA分子を意味する。直鎖状DNAは、酵素消化または物理的な破壊により、閉環状DNA分子、例えば、プラスミドから調製できる。
【0016】
「プロモーター」は構造遺伝子の転写を指示するヌクレオチド配列である。一般的に、プロモーターは、遺伝子の5’非コード領域、すなわち、構造遺伝子の転写開始部位の近くに存在する。転写の開始に作用するプロモーター中の配列因子は、しばしばコンセンサスヌクレオチド配列により特徴づけられる。これらのプロモーター因子は、RNAポリメラーゼ結合部位、TATA配列、CAAT配列、分化特異的因子(McGehee et al., Mol.Endocrinol.7:551(1993))、サイクリックAMP応答因子、血清応答因子(Treisman,Seminars in Cancer Biol.1:47(1990))、グルココルチコイド応答因子および他の転写因子の結合部位、例えば、CRE/ATF(O'Reilly et al.,J.Boil.Chem.267:19938(1992))、AP2(Ye et al.,J.Boil.Chem.269:25728(1994))、SP1、タンパク質結合cAMP応答因子(Loeken,Gene Exper.3:253(1993))およびオクタマー因子(一般に、Watson et al.,eds.,Molecular Biology of the Gene,4th ed.(The Benjamin/Cummings Publishing Company,Inc.1987)およびLemaigre and Rousseau,Biochem.J.303:1(1994)参照)を含む。仮にプロモーターが誘導性プロモーターであれば、転写の割合は誘発剤に対する応答において増加する。反対に、仮にそのプロモーターが、恒常的プロモーターであれば、転写の割合は誘発剤によって調節されない。また、制止プロモーターも知られる。
【0017】
「コアプロモーター」は、TATAボックスおよび転写の開始点を含む、プロモーターの機能に不可欠なヌクレオチド配列を含む。この定義により、コアプロモーターは、活性を増強し、あるいは組織に特異的な活性を与えるであろう特異的な配列の欠如において、検知可能な活性を有しても、あるいは有さなくてもよい。
【0018】
「調節因子」はコアプロモーターの活性を変化させるヌクレオチド配列である。例えば、調節因子は、排他的に、あるいは選択的に、特に、細胞、組織、またはオルガネラでの転写を可能にする細胞因子と結合するヌクレオチド配列を含んでよい。これらの調節因子のタイプは通常、「細胞に特異的な」、「組織に特異的な」あるいは「オルガネラに特異的な」方法において発現する遺伝子に関係する。
【0019】
「エンハンサー」は、転写開始部位に対するエンハンサーの距離または方向に関係なく、転写の効率を増大させることができる型の調節型因子である。
【0020】
「異種DNA」とは、与えられた宿主細胞中に天然には存在しないDNA分子またはDNA分子群を意味する。宿主のDNAが非宿主のDNAと結合している限り、特定の宿主細胞に対する異種DNA分子は、宿主細胞種(即ち、内生DNA)から派生したDNAを含んでもよい。例えば、転写プロモーターを含んで成る宿主のDNAセグメントと機能するように結合したポリペプチドをコードする非宿主のDNAセグメントを含むDNA分子は、異種DNA分子と考えられる。反対に、異種DNAは、非宿主の遺伝子から派生するプロモーターと機能するように結合する内生遺伝子を含んで成ってよい。他の説明によれば、野生型遺伝子が欠失した突然変異細胞にDNA分子を導入するとすれば、野生型細胞由来の遺伝子を含んで成るDNA分子は異種DNAと考えられる。
【0021】
「ポリペプチド」は、天然的にあるいは合成的に生成されようとも、ペプチド結合により結合したアミノ酸残基のポリマーである。約10以下のアミノ酸残基のポリペプチドは一般に、「ペプチド」を意味する。
【0022】
「タンパク質」は、1以上のポリペプチド鎖を含んで成る巨大分子である。また、タンパク質は、非ペプチド成分、例えば糖群を含んで成ってよい。糖および他の非ペプチド置換物は、タンパク質を生産する細胞によりタンパク質に加えてもよく、細胞のタイプで変化するであろう。タンパク質は、それらのアミノ酸の主鎖構造として本明細書において定義される。置換基、例えば炭水化物基は、一般に、具体的に挙げられていないが、存在してもよい。
【0023】
細胞中で、異種核酸分子から合成されたペプチドまたはポリペプチドは、「異種」ペプチドまたはポリペプチドである。
【0024】
「組み込まれた遺伝子因子」は、宿主細胞の染色体に組み入れたDNAセグメントであり、その後、因子は人工的に細胞に導入される。本発明中、もっとも一般的には、組み込まれた遺伝子因子は、電気穿孔法または他の技術により細胞に導入した線状プラスミドに由来する。組み込まれた遺伝子因子は、親の宿主細胞からその子孫に受け継がれる。
【0025】
「クローニングベクター」は、宿主細胞において、独立して自己複製する能力を有する核酸分子、例えば、プラスミド、コスミドまたはバクテリオファージである。一般的に、クローニングベクターは、ベクターの必須の生物学的機能を失わない測定可能な方法において核酸分子の挿入を許容する1または少数の制限エンドヌクレアーゼ認識部位、並びに、クローニングベクターで形質転換した細胞の識別および選択における使用に適当であるマーカー遺伝子をコードするヌクレオチド配列を含む。マーカー遺伝子は、一般的に、テトラサイクリン耐性またはアンピシリン耐性を供する遺伝子を含む。
【0026】
「発現ベクター」は宿主細胞中で発現する遺伝子をコードする核酸分子である。一般的に、発現ベクターは、転写プロモーター、遺伝子および転写ターミネーターを含んで成る。遺伝子発現は通常、プロモーターの制御下におかれ、そしてこれらの遺伝子は、プロモーターと「機能するように結合」するべきとされる。同様に、調節因子がコアプロモーターの活性を変化させた場合、調節因子およびコアプロモーターは機能するように結合している。
【0027】
「組み換え宿主」は、異種核酸分子、例えば、クローニングベクターまたは発現ベクターを含む細胞である。
【0028】
「組み込み形質変換体」は、異種DNAを細胞のゲノムDNAに組み入れた組み換え宿主細胞である。
【0029】
「発現」の語は、遺伝子生産物の生合成を意味する。例えば、構造遺伝子の場合、発現は、構造遺伝子のmRNAへの転写およびmRNAの1以上のペプチドへの翻訳を含む。
【0030】
「分泌シグナル配列」は、細胞の分泌経路を通して、より大きなポリペプチドペプチドがそこで合成されることを導く、より大きなポリペプチドの成分としてのペプチド(「分泌ペプチド」)をコードするDNA配列を意味する。
【0031】
「単離ポリペプチド」は夾雑している細胞成分、例えば、糖、脂質、または天然にタンパク質に会合する他のタンパク質不純物を本質的に含まないポリペプチドである。一般的に、単離ポリペプチドの調製は、高純度、すなわち、少なくとも約80%の純度、少なくとも約90%の純度、少なくとも約95%の純度、95%以上の純度または99%以上の純度のポリペプチドを含む。特定のタンパク質の調製が単離ポリペプチドを含むことを示す一つの方法は、タンパク質の調製のドデシル硫酸ナトリウム(SDS)-ポリアシルアミドゲル電気泳動およびそのゲルのクマシーブリリアントブルー染色後の一本のバンドの出現によるものである。しかしながら、「単離」の語は、他の物理的形態、例えば、2量体、あるいは、グリコシル化または誘導体化の形態における同一のポリペプチドの存在を除外するわけではない。
【0032】
「アミノ末端」および「カルボキシル末端」はポリペプチド中の位置を示すために本明細書において使用する。文脈が許す場合、これらの語は、近接のまたは相対位置を示すためのポリペプチドの特定の配列または部分に対する引用とともに使用する。例えば、ポリペプチド中の関連する配列に対するカルボキシル末端に位置する配列は、関連する配列のカルボキシルの終端に対して近接するが、完全ポリペプチドのカルボキシルの終端にある必要はない。
【0033】
「融合タンパク質」は、2以上の遺伝子のヌクレオチド配列を含んで成る核酸分子により発現した雑種タンパク質である。このように、融合タンパク質は、天然には互いに会合しない2以上のアミノ酸配列を含んで成る。
【0034】
発現ベクターの成分を説明するために使用する場合、「遺伝子または遺伝子断片」はポリペプチドまたはペプチドをコードするヌクレオチド配列を意味する。その遺伝子または遺伝子断片は、ゲノムDNA、cDNAまたはin vitro 合成技術(例えば、ポリメラーゼ連鎖反応、化学合成等)により得ることができる。
【0035】
「アフィニティタグ」の語は、本明細書において、2番目のポリペプチドの精製または検出を供するため、あるいは、基質に対する2番目のポリペプチドの結合のための部位を供するために2番目のポリペプチドと結合できるポリペプチドセグメントを意味する。原則として、抗体または他の特異的な結合剤として入手可能ないかなるペプチドまたはタンパク質もアフィニティタグとして使用できる。アフィニティタグは、ポリヒスチジン系、プロテインA(Nilsson et al.,EMBO J.4:1075(1985);Nilsson et al.,Method Enzymol.198:3(1991))、グルタチオンSトランスフェラーゼ(Smith and Johnson,Gene 67:31(1988))、Glu-Gluアフィニティタグ(Grussenmeyer et al.,Proc.Natl.Acad.Sci.USA 82:7952(1985))、サブスタンスP、FLAGペプチド(Hopp et al.,Biotechnology 6:1204(1988))、ストレプトアビジン結合ペプチド、または他の抗原性エピトープもしくは結合ドメインを含む。一般には、Ford et al.,Protein Expression and Purification 2:95(1991)を参照のこと。アフィニティタグをコードするDNA分子は、商業者から入手可能である(例えば、Pharmacia Biotech,Piscataway,NJ)。
【0036】
本明細書で使用する、「免疫調節因子」は、サイトカイン、幹細胞成長因子、リンホトキシン、共-刺激分子、造血因子、およびこれらの分子の合成類似体を含む。免疫調節物質の例は、腫瘍壊死因子、インターロイキン、コロニー刺激因子、インターフェロン、幹細胞成長因子、エリトロポイエチンおよびトロンボポイエチンを含む。
【0037】
「免疫グロブリン部分」は、免疫グロブリンの定常部を含んで成るポリペプチドを意味する。例えば、免疫グロブリン部分はH鎖の定常部を含んで成ってよい。
【0038】
「補体/抗補体の対」は、非共有結合的に結合した、非同一部分であり、適当な条件下で安定な対を意味する。例えば、ビオチンおよびアビジン(またはストレプトアビジン)は補体/抗補体の対のプロトタイプメンバーである。他の例としての補対/抗補体の対は、受容体/リガンドの対、抗体/抗原の対(または、ハプテンもしくはエピトープ)の対、センス/アンチセンスポリヌクレオチドの対等を含む。
【0039】
「抗体断片」は、抗体、例えばF(ab’)2、F(ab)2、Fab’、Fab等の一部である。構造に関わらず、抗体断片は、無処置の抗体により認識される同じ抗原と結合する。
【0040】
また、「抗体断片」は、特異的な抗原と結合する、合成の、あるいは遺伝子改変したポリペプチド、例えば、L鎖可変部からなるポリペプチド、H鎖およびL鎖の可変部から成る「Fv」断片、その中のL鎖およびH鎖の可変部がペプチドリンカー(「scFvタンパク質」)により結合される組み換え1本鎖ポリペプチド分子、および高頻度可変部を模倣するアミノ酸残基からなる最小認識ユニットを含む。
【0041】
「検出標識」は、結合しているポリペプチドのパートナーを発現する細胞を識別するための有用な分子を生成するためにポリペプチドと接合できる分子または原子である。検出標識の例は、キレート剤、感光剤、放射性同位体、蛍光試薬、常磁性イオンまたは他のマーカー部分を含む。
【0042】
標準的な分析方法の不正確さにより、ポリマーの分子量および長さは、おおよその値として理解される。これらの値が「およそ」Xまたは「約」Xとして表現している場合、記載されたXの値は、正確には±10%として理解すべきであろう。
【0043】
2.ホモ3量体ペプチドの生産のための発現ベクター
本発明はホモ3量体タンパク質の生産方法を供する。それぞれのホモ3量体のタンパク質は注目のポリペプチドおよび3量体化アミノ酸配列を含んで成る融合タンパク質である。注目のポリペプチドは受容体の細胞外ドメインを含み、それはそれらの同属のリガンドと結合するために使用できる。適当な受容体は腫瘍壊死因子受容体、例えば、TNFRSF1A(また、「p55」、「TNFR-60」および「TNF-R」と称される。例えば、Genbank No.M75866 を参照のこと)、TNFRSF1B(また、「p75」、および「TNFR2」と称される。例えば、Genbank No.M32315 を参照のこと)、TNFRSF13B(「TACI」としても知られる)、TNFRSF13C(「BAFFR」および「Ztnfr12」としても知られる。例えば、Genbank No.AF373846 を参照のこと)、TNFRSF17(「BCMA」としても知られる。例えば、Genbank No.Z29574 を参照のこと)等を含む。他の有用な腫瘍壊死因子受容体は当業者に知られている。
【0044】
本実施例は、膜貫通性活性化因子かつCAML(サイクロフィリンリガンドを変調するカルシウムシグナル)相互作用因子(TACI)の細胞外ドメインを含んで成る融合タンパク質の作成を示す。TACI核酸およびアミノ酸配列は、Bram and Gotz、米国特許番号5,969,102号により説明され、そして本明細書において、SEQ ID NO.3および4として含まれる。説明のTACI細胞外ドメインはSEQ ID NO:4の30から110のアミノ酸残基、SEQ ID NO:4の1から110のアミノ酸残基、SEQ ID NO:4の30から154のアミノ酸残基、およびSEQ ID NO:4の1から110のアミノ酸残基を含んで成るアミノ酸配列を有するポリペプチドを含む。
【0045】
また、本実施例は、2つのタイプの3量体化アミノ酸配列の使用を示す。:カルボキシ末端、ヒトのコラーゲンXの151アミノ酸のNC-1領域、およびタンパク質HSBP-1と結合しているヒトの熱ショック因子の1から65アミノ酸。NC-1ドメインは、Frischholz et al.,J.Biol.Chem.273:4547(1998)により説明されている。ヌクレオチドおよびアミノ酸配列は、本明細書中のSEQ ID NO.19および20として供されている。HSBP-1は、Tai et al.J.Biol.Chem.277:735(2002)により説明されている。HSBP-1の有用な断片のヌクレオチドおよびアミノ酸配列は、SEQ ID NO.21および22として供されている。
【0046】
3量体化アミノ酸配列に加えて、融合タンパク質は、さらに、そのタンパク質を可溶にするために、免疫グロブリン部分を含んで成ることができる。その免疫グロブリン部分は、H鎖定常部、例えば、ヒトのH鎖定常部を含んで成ってよい。IgG1H鎖定常部は、適当なH鎖定常部の一例である。説明のIgG1H鎖定常部はCH2およびCH3ドメインを含んで成るIgG1Fc断片である。そのIgG1Fc断片は、野生型のIgG1Fc断片または突然変異されたIgG1Fc断片であってよい。
【0047】
発現ベクターは注目のポリペプチドおよび3量体化アミノ酸配列を含んで成る融合タンパク質をコードするように作成できる。真核細胞におけるタンパク質の生産のために適当な発現ベクターは、一般的に(1)細菌の複製開始点並びに細菌宿主中の発現ベクターの成長および選択を供するための抗生物質耐性マーカーをコードする原核生物のDNA因子;(2)転写の開始を制御する真核生物のDNA因子、例えば、プロモーター;および(3)転写のプロセッシングを制御するDNA因子、例えば、転写終了/ポリアデニル化シグナル配列、を含む。
【0048】
遺伝子を発現するため、タンパク質をコードする核酸分子を転写の発現を制御する調節配列と機能するように結合させ、それから宿主細胞に導入しなければならない。転写調節配列、例えば、プロモーターおよびエンハンサーに追加して、発現ベクターは転写および翻訳調節配列を含むことができる。説明のように、哺乳類宿主に適当なその転写および翻訳調節シグナルは、ウイルス源、例えば、アデノウイルス、ウシのパピローマウイルス、サルのウイルス等に由来してもよく、その調節シグナルは、高レベルの発現を有する特定の遺伝子に関連する。適当な転写および翻訳調節配列はまた、哺乳類遺伝子、例えば、アクチン、コラーゲン、ミヨシンおよびメタロチオネインの遺伝子から得てもよい。
【0049】
適当な転写調節配列は、RNA合成の開始を指示するのに十分なプロモーター領域を含む。説明の真核生物のプロモーターはマウスのメタロチオネインI(metallothionein I)遺伝子のプロモーター(Hamer et al.,Molec.Appl.Genet.1:273(1982))、ヘルペス(Herpes)ウイルスのTKプロモーター(McKnight,Cell 31:355(1982))、SV40アーリープロモーター(Benoist et al.,Nature 290:304(1981))、ラウス(Rous)肉腫ウイルスプロモーター(Gorman et al.,Proc.Nat'l Acad.Sci.USA 79:6777(1982))、サイトメガロウイルスプロモーター(Foecking et al.,Gene 45:101(1980))、およびマウス乳腺腫瘍ウイルスプロモーター(一般的に、Etcheverry,"Expression of Engeered Proteins in Mammalian Cell Culture,"in Protein Engineering:Principles and Practice,Cleland et al.(eds.),ページ163-181(John Wiley&Sons,Inc.1996)参照)を含む。
【0050】
一方、仮に、原核生物のプロモーターが真核生物のプロモーターにより調節される場合、原核生物のプロモーター、例えば、バクテリオファージ(bacteriophage)T3RNAポリメラーゼプロモーターは哺乳類細胞中の注目の遺伝子の発現を制御するために使用できる(Zhou et al.,Mol.Cell.Biol.10:4529(1990)、およびKaufman et al.,Nucl.Acids Res.19:4485(1991))。
【0051】
アフィニティタグの包含は、融合タンパク質を示す細胞の認識または選択のために有用である。アフィニティタグの例は、ポリヒスチジンタグ(ニッケルキレート化樹脂と親和性を有する)、抗-myc抗体で検出されるc-mycタグ(例えば、EQKLI SEEDL;SEQ ID NO:1)、カルモジュリン結合タンパク質(カルモジュリンアフィニティクロマトグラフィーで単離される)、サブスタンスP、RYIRSタグ(抗-RYIRS抗体と結合する)、抗体で検出される赤血球凝集素Aエピトープタグ(例えば、YPYDVPDYA;SEQ ID NO:2)、Glu-GluタグおよびFLAGタグ(抗-FLAG抗体と結合する)を含む。例えば、Luo et al.,Arch.Biochem.Biophys.329:215(1996)、Morganti et al.,Biotechnol.Appl.Biochem.23:67(1996)、およびZheng et al.,Gene 186:55(1997)を参照のこと。これらのペプチドタグをコードする核酸分子は、例えば、Sigma-Aldrich Corporation(St.Louis,MO)から入手できる。
【0052】
クローニング部位はマルチクローニング部位であってよい。いかなるマルチクローニング部位も使用でき、そして多くが商業的に入手可能である。特に有用なマルチクローニング部位は、3つのすべての読み枠において遺伝子または遺伝子断片のクローニングを許容する。
【0053】
発現ベクターは、選択マーカーをコードするヌクレオチド配列を含んでもよい。広範な種々の選択マーカー遺伝子は入手可能である(例えば、Kaufman,Meth.Enzymol.185:487(1990);Kaufman,Meth.Enzymol.185:537(1990)参照のこと)。例えば、1つの適当な選択マーカーは、抗生物質ネオマイシンへの耐性を供する遺伝子である。この場合、選択はネオマイシン型の薬剤、例えば、G-418等の存在中で行われる。ブレオマイシン耐性遺伝子、例えば、Sh ble遺伝子もまた、今説明の方法に有用な選択マーカー遺伝子である。これらの遺伝子はブレオマイシン/フレオマイシン型薬剤、例えば、ZEOCINの活性を阻害するタンパク質を生産する(Gatignol et al.,Mol.Gen.Genet.207:342(1987);Drocourt et al.,Nucl.Acids Res.18:4009(1990)。ZEOCINは、細菌、真菌、植物、トリ、昆虫および哺乳類の細胞を含む細胞の広範囲における毒素である。追加的な選択マーカーはヒグロマイシンB-ホスホトランスフェラーゼ、AUR1遺伝子生産物、アデノシンデアミナーゼ、アミノグルコシドホスホトランスフェラーゼ、ジヒドロホレートレダクターゼ、チミジンキナーゼ、およびキサンチン-グアニンホスホリボシルトランスフェラーゼを含む(例えば、Srivastava and Schlessinger,Gene 103:53(1991);Romanos et al.,"Expression of Cloned Genes in Yeast,"in DNA Cloning 2:Expression Systems,2nd Edition,ページ123-167(IRL Press 1995);Markie,Methods Mol.Biol.54:359(1996);Pfeifer et al.,Gene 188:183(1997);Tucker and Burke,Gene 199:25(1997);Hashida-Okado et al.,FEBS Letters 425:117(1998)を参照)。選択マーカー遺伝子は、公表されたヌクレオチド配列を使用してクローンされ、あるいは合成されてもよく、あるいは、マーカー遺伝子は商業的に入手可能である。
【0054】
発現ベクターはSV40開始点を含んでもよい。この因子は、SV40ラージT抗原を発現する細胞株中でエピソームの複製および救出のために使用できる。
【0055】
発現ベクターへの挿入に適当な遺伝子または遺伝子断片はcDNAから得ることができる。それは、いかなる公知技術方法によっても調製される。例えば、cDNA分子は、ランダムプライミングにより合成できる。さらに、このようなプライマーは、ベクターにおいて発見される制限エンドヌクレアーゼ部位と結合できる。あるいは、cDNA分子はオリゴd(T)プライミングにより調製できる。また、遺伝子または遺伝子断片は、ゲノムDNAから、あるいは化学合成により得ることができる。適当な遺伝子または遺伝子断片の調製のための標準的な方法は、当業者に知られている(例えば、Ausubel et al.(eds.),Short Protocols in Molecular Biology,3rd Edition(John Wiley&Sons 1995)["Ausubel 1995"])。
【0056】
発現ベクターを作成後、核酸ポリマーの産生のための核酸分子の合成のために、宿主細胞中でそのベクターを増殖させることができる。しばしば「シャトルベクター」と称するベクターは、2以上の無関係な発現系において複製することができる。このような複製を促進するため、そのベクターは、各複製系において各々が有効な、2以上の複製開始点含むべきである。一般的に、シャトルベクターは真核生物系および原核生物系において複製することが可能である。これは真核生物の宿主である「発現細胞型」中のタンパク質の発現の検出および原核生物の宿主である「増幅細胞型」中のベクターの増幅を可能にする。説明のように、一方の複製開始点はSV40に由来してよく、他方の複製開始点はpBR322に由来してよい。当業者は、多数の適当な複製開始点を知っている。
【0057】
ベクターの増殖は、原核生物の宿主細胞、例えば、大腸菌(E.coli)または枯草菌(Bacillus subtilus)中で簡単に行うことができる。適当な大腸菌(E.coli)株は、BL21(DE3)、BL21(DE3)pLysS、BL21(DE3)pLysE、DH1、DH4I、DH5、DH5I、DH5IF'、DH5IMCR、DH10B、DH10B/p3、DH11S、C600、HB101、JM101、JM105、JM109、JM110、K38、RR1、Y1088、Y1089、CSH18、ER1451、およびER1647を含む(例えば、Brown(ed.),Molecular Biology Labfax(Academic Press 1991)参照のこと)。適当な枯草菌(Bacillus subtilus)株は、BR151、YB886、MI119、MI120、およびB170を含む(例えば、Hardy,"Bacillus Cloning Methods,"in DNA Cloning:A Practical Approach,Glover(ed.)(IRL Press 1985)参照のこと)。原核生物の宿主中のベクターの増殖のための標準的な技術は、当業者にとって周知である(例えば、Ausubel 1995;Wu et al.,Methods in Gene Biotechnology(CRC press,Inc.1997)を参照のこと)。
【0058】
3.宿主細胞による組み換えタンパク質の生産
発現ベクターは、いかなる真核生物細胞、例えば、哺乳動物の細胞、昆虫の細胞、トリの細胞、真菌の細胞等にも導入することができる。適当な哺乳動物の宿主細胞の例は、アフリカミドリザルの腎臓細胞(Vero;ATCC CRL 1587)、ヒト胎児の腎臓細胞(293-HEK;ATCC CRL 1573)、ベビーハムスターの腎臓細胞(BHK-21、BHK-570;ATCC CRL 8544、ATCC CRL 10314)、イヌの腎臓細胞(MDCK;ATCC CCL 34)、中国ハムスターの卵巣細胞(CHO-K1;ATCC CCL 61;CHO DG44(Chasin et al.,Som.Cell.Molec.Genet.12:555(1986))、ラットの下垂体細胞(GH1;ATCC CCL82)、HeLa S3細胞(ATCC CCL 2.2)、ラットの肝臓細胞(H-4-II-E;ATCC CRL 1548)、SV40-形質転換化したサルの腎臓細胞(COS-1;ATCC CRL 1650)およびマウス胎児の細胞(NIH-3T3;ATCC CRL 1658)を含む。
【0059】
バキュロウイルス系は、クローン化した注目の遺伝子を昆虫の細胞中に導入するために有効な手段を供する。適当な発現ベクターはオートグラファ・カリフォルニカ・マルチプルヌクレアポリヘドロシスウイルス(Autographa californica multiple nuclear polyhedrosis virus)(AcMNPV)に基づき、そして周知のプロモーター、例えば、ショウジョウバエ(Drosophila)の熱ショックタンパク質(hsp)の70プロモーター、オートグラファ・カリフォルニカ・ヌクレアポリヘドロシスウイルス(Autographa californica nuclear polyhedrosis virus)の前初期遺伝子のプロモーター(ie-1)および遅延性の初期39Kプロモーター、バキュロウイルスのp10プロモーター、およびショウジョウバエのメタロチオネイン(Drosophila metallothionein)プロモーターを含む。組み換えバキュロウイルスを生成する2つ目の方法は、Luckow(Luckow,et al.,J.Virol.67:4566(1993))により説明されるトランスポゾン基準系を利用する。トランスファーベクターを利用するこの系はBAC-to-BACキットとして販売されている(Life Technologies,Rockville,MD)。この系は、遺伝子または遺伝子断片を、「バクミド」と呼ばれる巨大プラスミドとして大腸菌(E.coli)中に維持されるバキュロウイルスのゲノムに移動させるために、トランスファーベクターであるTn7トランスポソンを含むPFASTBAC(Life Technologies)を利用する。Hill-Perkins and Possee,J.Gen.Virol.71:971(1990)、Bonning,et al.,J.Gen.Virol.75:1551(1994)、およびChazenbalk,and Rapoport,J.Biol.Chem.270:1543(1995)を参照のこと。これらのベクターは上記の説明に従い改変できる。
【0060】
組み換えウイルスまたはバクミドは、宿主細胞にトランスフェクションするために使用される。適当な昆虫宿主細胞は、IPLB-sf-21由来の細胞株、スポドプテラ・フルギペルダ(Spodoptera frugiperda)の卵巣細胞株、例えば、Sf9(ATCC CRL 1711)、Sf21AE、およびSf21(Invitrogen Corporation;San Diego,CA)、並びにショウジョウバエのシュナイダー2(Drosophila Schneider-2)細胞、およびイラクサギンウワバ(Trichoplusia ni)由来のHIGHFIVEO細胞株(Invitrogen)(米国特許番号5,300,435号)を含む。商業的に入手可能な無血清培地は細胞を成長させるため、および維持するために使用できる。適当な培地は、Sf9細胞にはSf900IITM(Life Technologies)またはESF921TM(Expression Systems);そしてT.ni細胞にはEx-cellO405TM(JPH Biosciences,Lenexa,KS)またはExpress FiveOTM(Life Technologies)である。組み換えウイルスを使用する場合、その細胞は、約2-5×105の細胞密度から1-2×106の細胞密度に成長させる。その場合、組み替えウイルスのストックは、0.1から10の多重感染度(MOI)、さらに一般的には3付近で加えられる。
【0061】
バキュロウイルス系中の組み換えタンパク質を生産するための確立された技術は、Bailey et al.,"Manipulation of Baculovirus Vector,"in Methods in Molecular Biology,Volume 7: Gene Transfer and Expression Protocols,Murray(ed.),ページ147-168(The Humana Press,Inc.1991),by Patel et al.,"The baculovirus expression system,"in DNA Cloning 2:Expression Systems,2nd Edition,Glover et al.(eds.), ページ205-244(Oxford University Press 1995),by Ausubel(1995) ページ16-37から16-57,by Richardson (ed.),Baculovirus Expression Protocols(The Humana Press,Inc.1995),and by Lucknow,"Insect Cell Expression Technology,"in Protein Engineering:Principles and Practice,Cleland et al., ページ183-218(John Wiley&Sons,Inc.1996)により供される。
【0062】
また、本明細書において説明する発現ベクターは、酵母菌の細胞を含む真菌類細胞にトランスフェクションするために使用できる。この点に関して特に注目の酵母菌種は、出芽酵母(Saccharomyces cervisiae)、ピチア・パストリス(Pichia pastoris)、およびピチア・メタノリカ(Pichia methanolica)を含む。酵母菌中の発現に適当なプロモーターは、GAL1(ガラクトース)、PGK(ホスホグリセレートキナーゼ)、ADH(アルコールデヒドロゲナーゼ)、AOX1(アルコールオキシダーゼ)、HIS4(ヒスチジノールデヒドロゲナーゼ)等由来のプロモーターを含む。多くの酵母菌クローニングベクターは容易に入手可能であり、そして上記の説明に従い改変できる。これらのベクターは、YIp系ベクター、例えば、YIp5、YRpベクター、例えば、YIp17、YEpベクター、例えば、YEp13およびYCpベクター、例えば、YCp19を含む。外来性DNAでの出芽酵母(S.cerevisiae)の形質転換およびそれからの組み替えポリペプチドの生成方法は、例えば、Kawasaki、米国特許番号4,599,311号、Kawasaki et al.、米国特許番号4,931,373号、Brake、米国特許番号4,870,008号、Welch et al.、米国特許番号5,037,743号、およびMurray et al.、米国特許番号4,845,075号により開示されている。形質転換化細胞は、選択マーカーにより決定される表現型、一般的に、薬剤耐性または特定の栄養素(例えば、ロイシン)の不在における成長能により選択される。出芽酵母(Saccharomyces cervisiae)中の使用のための好ましいベクター系は、Kawasaki et al.(米国特許番号4,931,373)により開示されたPOT1ベクター系であり、それは、形質転換化細胞がグルコース含有媒地における成長により選択されることを許容する。酵母菌での使用のための追加的な適当なプロモーターおよびターミネーターは、解糖酵素遺伝子(例えば、Kawasaki、米国特許番号4,599,311号、Kawasaki et al.、 米国特許番号4,615,974号およびBitter、米国特許番号4,977,092号を参照のこと)およびアルコールデヒドロゲナーゼ遺伝子由来のものを含む。また、米国特許番号4,990,446号、5,063,154号、5,139,936号、および4,661,454号を参照のこと。
【0063】
ハンセヌラ・ポリモルファ(Hansenula polymorpha)、スキゾサッカロマイセス・プロムベ(Schizosaccharomyces prombe)、クルイベロマイセス・ラクチス( Kluyveromyces lactis)、クルイベロマイセス・フラギリス(Kluyveromyces fragilis)、ウスチラゴ・マイジス(Ustilago maydis)、ピチア・パストリス(Pichia pastoris)、ピチア・メタノリカ(Pichia methanolica)、ピチア・グイレルモンジイ(Pichia guillermondii)、およびカンジダ・マルトーサ(Candida maltosa)を含む他の酵母菌のための形質転換系は、当業界において知られている。例えば、Gleeson et al.,J.Gen.Microbiol.132:3459(1986)、およびGregg,米国特許番号4,882,279号を参照のこと。アスペルギルス(Aspergillus)細胞は、McKnight et al.、米国特許番号4,935,349号の方法に従い利用できる。アクレモニウム・クリソゲヌム(Acremonium chrysogenum)の形質転換の方法は、Sumino et al.、米国特許番号5,162,228号により開示されている。ネウロスポラ(Neurospora)の形質転換の方法は、Lambowitz、米国特許番号4,486,533号により開示されている。
【0064】
例えば、組み換えタンパク質の生産のための宿主としてのピチア・メタノリカ(Pichia methanolica)の使用は、Raymond、米国特許番号5,716,808号、Raymond、米国特許番号5,736,383号、Raymond et al.,Yeast 14:11-23(1998)、および国際公開番号WO97/17450、WO97/17451、WO98/02536、およびWO98/02565により開示されている。ピチア・メタノリカ(Pichia methanolica)の形質転換に使用のDNA分子は、一般的に2本鎖の環状プラスミドとして調製されるものと思われ、好ましくは形質転換の前に直線化される。P・メタノリカ(P methanolica)におけるポリペプチドの生産のために、プラスミド中のプロモーターおよびターミネーターは、P・メタノリカ(P methanolica)遺伝子のもの、例えば、P・メタノリカ(P methanolica)アルコール利用遺伝子(AUG1またはAUG2)であることが好ましい。他の有用なプロモーターは、ジヒドロキシアセトンシンターゼ(DHAS)、ホルメートデヒドロゲナーゼ(FMD)、およびカタラーゼ(CAT)のものを含む。宿主染色体へのDNAの組み込みを促進するため、宿主のDNA配列により両末端に隣接するプラスミドのすべての発現セグメントを有することが好ましい。メタノールの使用を最小化することが望ましい大規模な工業的工程には、そこに両方のメタノール利用遺伝子(AUG1およびAUG2)が欠失した宿主細胞を使用することが好ましい。選択されたタンパク質の生産には、液胞においてプロテアーゼ遺伝子(PEP4およびPRB1)が欠乏した宿主細胞が好ましい。注目のポリペプチドをコードするDNAを含むプラスミドの、P・メタノリカ(P methanolica)の細胞への導入を促進するために電気穿孔法が使用される。P・メタノリカ(P methanolica)の細胞は、電界強度が、2.5から4.5kV/cm、好ましくは約3.75kV/cm、および時定数(t)が1から40ミリ秒、最も好ましくは約20ミリ秒である、指数関数的に減衰する電場を使用した電気穿孔法により形質転換することができる。
【0065】
発現ベクターは、カラムホスフェイトトランスフェクション、リポソーム性トランスフェクション、微粒子性伝達、電気穿孔法等を含む種々の標準的な技術を使用して宿主細胞へと導入できる。
【0066】
発現ベクターを哺乳動物、酵母菌、および昆虫の細胞へ導入するための標準的な方法は、例えば、Ausubel(1995)により供される。哺乳動物の細胞系により生産された外来タンパク質の発現および回収方法は、例えば、Etcheverry,"Expression of Engineered Proteins in Mammalian Cell Culture,"in Protein Engineering:Principles and Practice、Cleland et al.(eds.)、ページ163(Wiley-Liss,Inc.1996)により供される。バキュロウイルス系から組み換えタンパク質を単離するための確立された方法は、Richardson(ed.)、Baculovirus Expression Protocols(The Humana Press,Inc.1995)により説明されている。
【0067】
発現ベクターは、注目のポリペプチドを生産する細胞から単離することができる。所望すれば、発現ベクターを同定可能なポリペプチドの発現に基づく選択の他の部分に服従させることあるいは増幅細胞にトランスフェクションすることができる。それから、トランスフェクションした増幅細胞を選択マーカーにより選択し、そのベクターを精製し、そして、当業界におけるいかなる既知の方法により遺伝子または遺伝子断片のヌクレオチド配列を配列決定する。仮にヌクレオチド配列が完全ポリペプチド部分のみをコードするならば、そのヌクレオチド配列は、その全遺伝子を検索するために当業界における既知の方法によりプローブとして使用することができる。
【0068】
本発明、このように一般的な説明は、以下の実施例を参照により、さらに容易に理解されるだろう。これらは説明の方法により供され、そして、本発明を制限することを意図するものではない。
【実施例1】
【0069】
TACI-NC1の作成
発現ベクターは、膜貫通性活性化因子かつ(カルシウム信号変調シクロフィリンリガンド)相互作用因子(TACI)のタンパク質の細胞外ドメインおよびヒトのコラーゲンXのNC1ドメインを含んで成る融合物をコードするように作成する。TACIの核酸配列は、Bram and Gotz,米国特許番号5,969,102号により説明され、そして本明細書中にSEQ ID NO.3および4として含まれる。NC-1ドメインは、Frischholz et al.,J.Biol.Chem.273:4547(1998)により説明される。:ヌクレオチドおよびアミノ酸配列は、本明細書中、SEQ ID NO19および20として供される。本作成において、TACIの細胞外ドメインを、c-終端におけるGlu-GluタグおよびTACIとNC1の間に設計された8つのアミノ酸のGly-SerスペーサーでNC1と融合した。
【0070】
NC1は、オリゴヌクレオチド zc40219(5’GGGCCTCCAG GCCCACCAGG T3’;SEQ ID NO:5)およびzc40205(5’TCACATTGGA GCCACTAGGA A3’;SEQ ID NO:6)を使用してヒトのゲノムDNA(Clontech)からPCRにより増幅した。TACIの細胞外の部分は、94℃で1分、55℃で1分および72℃で2分の30サイクルの条件で、オリゴヌクレオチドzc40915(5’ACAGGTGTCC AGGGAATTCA TATAGGCCGG CCACCATGGA TGCAATGAAG AGAGGG3’;SEQ ID NO:7)およびzc40917(5’ACCCTCAGGC ATCGAACCCG AACCCGAACC GGATCC3’;SEQ ID NO:8)でTACI免疫グロブリン融合タンパク質をコードするクローンからPCRにより増幅した。PCR生産物を沈殿させ、そして10μlの水に再懸濁し、それから出芽酵母(S.cerevisiae)中のBglIIで消化したpZMP21へ組み換えた。その組み換え結果の大腸菌(E.coli)のクローンを、適切な組み入れのために、AscI消化によりスクリーニングし、そして配列決定のために3つのポジティブクローンを使用した。1つのクローンをさらなる実験のために選択した。このクローンは、NC1中でグリシンからアルギニンへの突然変異を含み、C末端のGlu-Gluタグから4つのアミノ酸が欠失していた。
【0071】
TACI/NC1-EEをコードするベクターは、電気穿孔のために、20μgのQiagen精製化DNAをPvuIで消化することより直線化した。この直線化DNAをPF-CHO細胞中で電気穿孔した。その細胞を、10日間のHT培地における栄養素の選択の前に24時間回復させた。栄養素の選択から回復後、細胞をさらに10日間、50nmのメトトリキサート(methotrexate)選択に移動した。
【0072】
トランスフェクションした細胞を細胞工場(cell factories)に播種し、そして2リットルの工場調整培地(CM)を単離した。CMを1.5mgの抗-TACIモノクローナル抗体と混合し、4℃で一昼夜インキュベートした。CM-抗体混合物を、流速2mL/minで1.6mLの総容積のPOROS A50カラムに適用した。加えた後、カラムを100カラム容量のpH7.2のPBSで洗浄した。その結合タンパク質をそれから直接、pH8.0の2MのトリスとpH2.5の200mlのグリシンへと溶出した。1ミリリットルのフラクションを収集した。ウェスタンブロット分析に基づき、TACI-NC1を含むフラクションを貯蔵した。その貯蔵したフラクションを300μlに濃縮し、そして、バッファーをpH7.2の14mLのPBSで3回交換し、それから3交換のpH7.2の4LのPBSに対して透析した。
【実施例2】
【0073】
哺乳動物細胞中のTACI-HSB1の発現
HSB1遺伝子の合成
ヒトの熱ショック結合タンパク質(HSBP-1)はTai et al.J.Biol.Chem.277:735(2002)により説明されている。HSBP-1のヌクレオチドおよびアミノ酸配列はSEQ ID NO21および22として供される。4つのオーバーラップしているオリゴヌクレオチド(ヒトの熱ショック結合タンパク質(HSBP-1)のセンスおよびアンチセンス鎖の両方をコードした)を以下のプライマーを使用して固相合成により合成した。:5’GATCGGATCC ATGGCCGAAA CTGATCCTAA AACAGTTCAA GACCTTACCA GCGTAGTCCA GACGCTCCTG CAAGAGATCG AAGATAAGTT TCAGACTATG AGCGACCAAA TCATTGAG3’(SEQ ID NO:9);5’AGAATGCATG ACATGAGCTC CAGGATAGAT GACCTTGAGA AAAATATAGC AGATTTAATG ACGCAAGCTG GTGTGGAAGA GTTGGAAGGA AGTGGTTCTA3’(SEQ ID NO:10);5’GATCTAGAAC CACTTCCTTC CAACTCTTCC ACACCAGCTT GCGTCATTAA ATCTGCTATA TTTTTCTCAA GGTCATCTAT CCTGGAGCTC ATGTCATCGA TTCTCTCAAT3’(SEQ ID NO:11);および5’GATTTGGTCG CTCATAGTCT GAAACTTATC TTGCATCTCT TGCAGGAGCG TCTGGACTAC GCTGGTAAGG TCTTGAACTG TTTTAGGATC AGTTTCGGCC ATGGATCC3’ (SEQ ID NO:12)。各オリゴヌクレオチドの5末端は、120pモルの各オリゴヌクレオチド、1.6μlの100mM ATP、34μlの5×T4-キナーゼバッファー(Life Technologies,Bethesda,MD)、6.4μlの水、および1μlのT4-ポリヌクレオチドキナーゼ(Life Technologies)の混合によりリン酸化し、そして37℃で20分間インキュベートした。そのリン酸化反応は、沸騰したウォーターバス中に置いて、それから、アニーリングを促進させるためにゆっくりと25℃まで冷却した。その断片を20μlの10×T4-リガーゼバッファー(Life Technologies)、0.5μlの100mM ATP、および2μlのT4-DNAリガーゼ(Life Technologies)を加えることにより連結させ、そして、16℃で一昼夜インキュベートした。連結後、そのDNAをアルコール沈殿により採取した。その単離DNAを、3.2μlのB制限バッファー(Promega,Madison,WI)、26.8μlの水、1.5μlのBglII(Life Technologies)、および1.5μlのAsp718(Life Technologies)中で再懸濁し、そして、37℃で2時間インキュベートした。そのリガーゼ反応生成物を15%アガロースゲル上で分画し、そして、ヒトのHSBP-1をコードする220ヌクレオチド断片を、製造業者の手順(Quiagen)に従い Quiagen ゲル単離キットを使用して単離した。ヒトのHSBP-1をT4-DNAリガーゼおよび製造業者のガイドライン(Life Technologies)を使用してAsp718-BamHIの切断したベクターに挿入した。
【0074】
TACIの細胞外ドメインをコードする断片を、オリゴヌクレオチドzc41712(5’CACACGTACG AAGATGGATG CAATGAAGAG AGG3’;SEQ ID NO:13)およびzc41638(5’GGTTAGATCT CGAACCCGAA CCCGAACCGG3’;SEQ ID NO:14)を使用してpTACI-NC1ベクターから増幅させた。そのPCR生成物をBsiW1およびBglIIで切断し、そして1.5%アガロースゲル上で、増幅させたDNAを分画し、それから、製造業者の手順(Quiagen)に従い Quiagen ゲル単離キットを使用して単離した。その単離したDNAを、製造業者のガイドラインに従い(Life Technologies)、T4-DNAリガーゼを使用してHSBP-1をコードする配列を含むAsp718-BglIIの切断したベクターに挿入した。DNAの配列決定により予想したベクターの配列を確認し、それを「pHZTACI-HSBP.9」と命名した。
【0075】
B.TACI-HSBP-1の発現および精製
pHZTACI-HSBP.9ベクターを、TransTransfected を使用してBHK570中にトランスフェクションし、そして、トランスフェクタント耐性のために培養液を10μMのメトトレキサートに選択した。耐性コロニーを組織培養皿に移動し、拡大させ、そして抗-His(C-末端)抗体(Invitrogen,Carlsbad,CA)でのウェスタンブロット分析によりTACI-HSBP-His6の分泌を分析した。生じた細胞株「BHK.TACI-HSBP.2」を拡大した。
【0076】
BHK.TACI-HSBP.2細胞を、細胞工場中に播種し、そして12リットルの工場-条件培地を単離した。その培地を、流速2ml/minで25ミリリットルの総容積のNi-NTA(Invitrogen)に適用した。追加後、カラムをpH7.2の50カラム容量のPBSおよび20カラム容量のリン酸緩衝食塩水、pH6.0の50mMのイミジゾールで洗浄した。それから、その結合タンパク質を、pH6.0のPBS中、50mMから800mMへのイミジゾール濃度の増加の直線勾配で、1ml/minで60分間溶出した。1ミリリットルの分画を採取した。ウェスタンブロット分析に基づき、TACI-HSBP-1を含む分画を貯蔵した。その貯蔵した分画を300μlに濃縮し、そしてバッファーを14mlのリン酸緩衝食塩水(pH7.2)で3回交換し、それから3交換の4リットルのpH7.2のリン酸緩衝食塩水に対して透析した。ウェスタンブロット分析およびクマシー染色ゲルは75%以上の純度および20kDaの単量体の分子量を示した。
【実施例3】
【0077】
TACI融合タンパク質のための増殖アッセイ
アフェレーシス由来の末梢血の単核細胞を、Ficol-Hypaque における密度勾配遠心分離により単離し、そしてリン酸緩衝食塩水中で洗浄した。一般的に、1ドナーから約1010の末梢血の単核細胞を単離することができる。1バイアル当たり約108の細胞を90%のウシ胎児血清および10%のジメチルスルホキド中で凍結させた。
【0078】
複数のバイアルを解凍し、細胞の生存度を測定した。B細胞を、CD19磁気ビーズおよび VarioMacs 磁気分離システム(Miltenyi Biotec;Aubum,CA)を使用して末梢血の単核細胞から単離した。丸底96ウェルプレートをリン酸緩衝食塩水中の5μg/mlのヤギ抗-ヒトIgMで、4℃で24時間にわたり、前コーティングした(Southern Biotechnology Assoc.Inc.;Birmingham AL)。10ng/mLのヒトのIL-4(Pharmingen)および20ng/mLのzTNF4(TACIと結合するリガンド)の存在下において、1ウェル当たり精製したB細胞を105培養した。zTNF4が刺激したB細胞の増殖を阻害する能力を比較するために、1μg/mlで開始したTACI-Fc融合タンパク質、TACI-HSBP-1またはTACI-NC1の3倍希釈系を含ませた。その細胞を、zTNF4、ヒトIL-4の存在下、および阻害因子アリまたはナシで、4日間インキュベートし、それから1ウェル当たり1μCiのH3チミジン(Amersham)で、一昼夜パルスした。プレートはPackardプレートハーベスターを使用して収集し、そして、Packardリーダーを使用してカウントした。本アッセイにおいて、zTNF4が刺激したヒトのB細胞の増殖の阻害で、TACI-HSBP-1は3倍以上の有効性し、そしてTACI-NC1は10倍以上の有効性を示した。図2において示すように、TACI-NC1は、同じ濃度のTACI-Fc5よりもTNF4が誘発したB細胞の増殖を大きく減少させた。この違いは、1.1nmの値を有するTACI-Fc5および57pMの値を有するTACI-NC1により、図中に示されたEC50値において表され、TACI-NC1が約19倍有効であることを示している。
【実施例4】
【0079】
細菌の細胞中のTACI-HSB1の発現
ヒトのTACI-HSBP-1をコードするヌクレオチド配列を含む発現プラスミドを酵母菌のホモログ組み換えを介して、G10エンハンサー配列の後ろに挿入した。ヒトのTACI-HSBP-1のDNA断片はPCRを使用して単離した。PCR反応中のヒトのTACI-HSBP-1の生産において2つのプライマーを使用した。ベクターのフランキング配列の40塩基対を含むプライマーzc42,728(5’CTAGAAATAA TTTTGTTTAA CTTTAAGAAG GAGATATATA TATGGCTATG AGATCCTGCC3’;SEQ ID NO:15)は、G10エンハンサー配列およびヒトのTACI-HSBP-1のアミノ終端に対応する24塩基対から成った。プライマーzc42,731(5’TCTGTATCAG GCTGAAAATC TTATCTCATC CGCCAAAACA CTAGTGATGG TGATGGTGAT GGCC3’;SEQ ID NO:16)はベクターのフランキング配列の40塩基対およびTACI-HSBP-1配列のカルボキシル終端に対応する24塩基対を含んだ。その鋳型はpH2-TACI-HSBP9であった。そのPCR反応条件は以下とした。:94℃で30秒間、50℃で30秒間、そして72℃で1分間の25サイクル;続いて4℃で浸漬した。分析のために、2から4μl容量のPCR試料を1×TBEバッファーを伴う1%アガロースゲル上に流し、そして約550塩基対断片の予想したバンド
【化1】

を観察した。100μlの残留容量の反応物を無水エタノールで沈殿させた。そのペレットを、SmaI-切断化レシピエントベクターpTAP238への組み込みに使用するために10μlの水に再懸濁し、TACI-HSBP-1をコードする作成物を生成した。
【0080】
100マイクロリットルの形質転換受容性酵母菌細胞(S.cerevisiae)を、約1μgのヒトのTACI-HSBP-1フラグメント(PCR生成物)および100ngのSmaI-切断化pTAP238ベクターを含む10μlの混合物と混合し、そして0.2cmの電気穿孔キュベットに移動した。その酵母菌/DNA混合物を、0.75kV(5kV/cm)、無限オーム、25μFにセットした機器を使用して電気パルスし、それから600μlの1.2Mソルビトールをキュベットに加えた。それからその酵母菌を、2つの300μlのURA D(ウラシル欠乏グルコース-含有培地)プレート上で培養し、30℃でインキュベートした。およそ48時間後、1つのプレート由来のUra+酵母菌の形質転換体を1mlの水中で再懸濁し、細胞をペレットにするために素早くスパンした。その細胞ペレットを1mlの溶解バッファー中で再懸濁した。DNAを上記に開示したように回収した。そのDNAペレットを100μlの水中で再懸濁した。
【0081】
40μlの電気形質転換受容性大腸菌(E.coli)のMC1061細胞を1μlの酵母菌DNAで形質転換した。その細胞は2.0kV、25μFおよび400オームで電気パルスした。電気穿孔後、0.6mlのSOC(2% BactoTM Tryptone(Difco,Detroit,MI)、0.5%酵母菌抽出物(Difco)、10mMのNaCl、2.5mMのKCl、10mMのMgCl2、10mMのMgSO4、20mMのグルコース)を細胞に加えた。その細胞を37℃で1時間回復させ、それから、LB+カナマイシンプレート(LBブロス(Lennox)、1.8% BactoTM Agar(Difco)、30mg/Lのカナマイシン)上で培養した。
【0082】
ヒトのTACI-HSBP-1のための正しい発現作成物を宿す個々のクローンは、プラスミドDNAの特徴的な消化により認識される。細胞は30μg/mlのカナマイシンを伴うSuper Broth II(Becton Dickinson)中で一昼夜成長させた。翌日、その細胞を採取し、そしてプラスミドDNAを、スピンカラム(QIAprep(登録商標)Spin Miniprep Kit;Quiagen Inc.,Valencia,CA)を使用して調製した。それから、そのDNAをNotIおよびXbaIで切断した。その正しい制限パターンを伴うクローンをpTAP415と命名し、そして配列決定した。pTAP415中のTACI-HSBP-1のポリヌクレオチド配列をSEQ ID NO:17に示す。
【0083】
10マイクロリットルのpTAP415を、3μlの商業的に入手可能なバッファー(バッファー3;New England Biolabs) および15μlの水中の2マイクロリットルのNotIで37℃、1時間で切断した。7マイクロリットルの反応混合物を2マイクロリットルの5×T4DNAリガーゼバッファー(Life Technologies;Gaitherburg,MD)および1マイクロリットルのT4DNAリガーゼと混合し、室温で1時間インキュベートした。1マイクロリットルのそのライゲージョン混合物を、大腸菌(E.coli)株W3110(ATCC 27325)を形質転換するために使用した。その細胞を2.0kV、25μF、および400オームで電気パルスした。電気穿孔後、0.6mlのSOCを細胞に加えた。その細胞を37℃で1時間成長させ、それからLB+カナマイシンプレート上で培養した。
【0084】
個々のクローンを採取し、成長させた。スピンカラムを使用してプラスミドDNAを調製した。酵母菌のURA3およびCEN/ARS因子の減少を確認するため、そのDNAを、特異的にPvuIIおよびHindIII で切断した。個々のコロニーを採取した。細胞を30μg/mlのカナマイシンを含むSuperbrothII(Becton Dickinson)で一昼夜成長させた。30μg/mlのカナマイシンを含む2ミリリットルの新鮮なSuperbrothIIを接種するために、100マイクロリットルの一昼夜培養液(overnight culture)を使用した。培養液は15mlのコニカルチューブ中で約2時間振りながら37℃で成長させた。1mlの培養液を1mMのIPTGで誘導した。2時間15分後、同量の培養液を5%のβMEを伴う250μlの Thorner バッファー(8Mの尿素、pH7.0で100mMのトリス、10%グリセロール、2mMのEDTA、5%SDS)と混合し、そして、染色した。試料は5分間沸騰させた。20μlの試料を4%-12%PAGEゲル(NOVEX)に充填した。1×MESバッファー中でゲルを流した。発現はクマシーブルー染色により分析した。
【0085】
細菌細胞をフレンチプレスを使用して溶解し、そして細胞の溶菌液中の封入体を、低速の遠心分離によりペレットにした。細胞膜および細胞壁材料を含む夾雑物を除去するために、ペレット分画を2Mの尿素で洗浄した。それから、TACI-HSBP-1大腸菌(E.coli)の封入体を、0.1Mの硫酸ナトリウムおよび0.05Mのナトリウムテトラチオネートを含むpH8の50mMのトリス中の7MのグアニジンHCl中、4℃で撹拌しながら一昼夜抽出した。変性剤/亜硫酸分解剤を同時に伴う抽出物は、タンパク質-タンパク質相互作用を分解し、還元状態およびスルホン化状態において、タンパク質をスルフドリル基を伴う単量体に展開する。再度の折りたたみの前に、試料を35,000×g、4℃で30分間遠心分離し、そして0.2μmのフィルターでろ過した。濃度をRP HPLCアッセイにより測定した。
【実施例5】
【0086】
TACI融合タンパク質の生物学的アッセイ
zTNF4活性を制するための様々な3量体化TACI作成物の能力を試験するために、ヒトのTACIおよびKZ142ルシフェラーゼでトランスフェクションした Jurkat 細胞株を使用した。その Jurkat TACI KZ142ルシフェラーゼ細胞を成長させ、そして育成培地(L-グルタミン、ピルビン酸ナトリウム、0.5mg/mlのG418、および2μg/mlのプロマイシンを伴うRPMI/10%FBS)においてアッセイした。これらの細胞は100μlの育成培地において、40,000細胞/96ウェルで培養した。100ng/mlのzTNF4の存在下において、1ウェル当たり100マイクロリットルの3量体阻害因子を加えた。
【0087】
アッセイ細胞株は、1000ng/mlのzTNF4に対して最大約18倍のルシフェラーゼ応答性を示した。バックグランドのコントロールウェルと比較して、1ミリリットル当たり100ナノグラムのzTNF4は約10倍のルシフェラーゼ応答性を与えた。総量200μlの育成培地中のzTNF4および阻害物質の組み合わせを5%CO2インキュベータ中、37℃で6時間インキュベートした。それから、Beckman GS-6KR 遠心分離機において、96ウェルプレートを2000×gで遠心分離し、そして、プレートを反転させることにより培地を廃棄した。25マイクロリットルの溶解バッファー(Promega E153A)を各ウェルに加え、そして室温で15分間インキュベートした。それから、照度系を読むために、その溶解した細胞を不透明な96ウェルプレートに移動した。1本のルシフェラーゼアッセイバッファー(Promega 152A)を、1本のルシフェラーゼアッセイ基質(Promega E151A)に加え、そして40μlのこの組み合わせを各ウェルに加えた。5秒に統一して、照度計(EG&EG Berthold Microlumat Plus)により各ウェルを読んだ。
【0088】
図1に示すように、コントロール免疫グロブリン融合タンパク質(hwsx11-IgG)はzTNF4誘発ルシフェラーゼ活性においてほとんど有効性を持たなかった。TACI-Fc免疫グロブリン融合タンパク質(TACI-IgG)はルシフェラーゼ活性を阻害したが、より大きな阻害は、哺乳動物細胞または大腸菌(E.coli)中で生産されたTACI-HSBP-1タンパク質で達成された。
【図面の簡単な説明】
【0089】
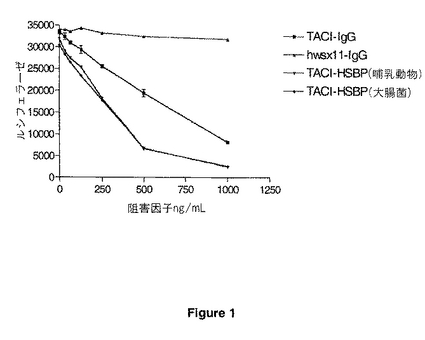
【図1】図1は、TACI-Fc融合タンパク質(「TACI-IgG」)、哺乳動物細胞中で生産されたTACI-HSBP-1タンパク質(「TACI-HSBP(哺乳動物)」)、大腸菌(E.coli)中で生産されたTACI-HSBP-1タンパク質(「TACI-HSBP(大腸菌)」)による、並びに、免疫グロブリン融合タンパク質(「hwsx11-IgG」)の制御によるzTNF4誘発化ルシフェラーゼ活性の阻害を示す。
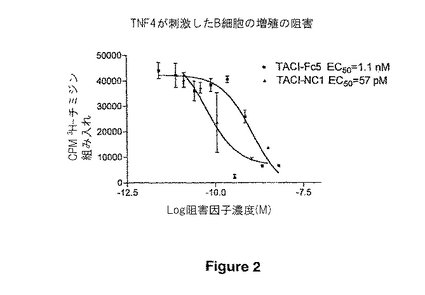
【図2】図2は、3H-チミジン、TACI-NC13量体およびTACI-Fc融合タンパク質(「TACI-Fc5」)の組み入れを通して、B細胞の増殖の阻害を示す。TACI-Fc5は、TACI-FcまたはTACI-IgGのもう1つの名前である。また、この図において示すものは2分子のEC50値である(すなわち、50%のコントロールの指標を阻害する濃度)。
【技術分野】
【0001】
腫瘍壊死因子(TNF)受容体スーパーファミリーは、宿主の防衛、炎症、および自己免疫に関する分子のラージファミリーであり、そしてヒトの疾患に関与している。TNFの阻害を狙った治療薬剤は、炎症性疾患、例えば、リューマチ性関節炎および炎症性腸炎の制御において有効である。追加的なTNF/TNF受容体スーパーファミリーのメンバーは、現在のところ、自己免疫疾患、アテローム硬化、骨粗鬆症、移植拒絶反応および癌に対する治療を対象としている。
【0002】
TNFおよびTNF受容体ファミリーはともに、自己組織化3量体として活性であるが、機能上、2量体分子、例えば、抗体または受容体IgG融合物だけが治療薬剤として使用されてきた。TNFスーパーファミリー中のリガンドおよび受容体の両方により表される3回回転対称は、拮抗物質に基づく3量体の受容体が増加した結合活性を表すはずであり、このため、2量体分子と比較して増加した有効性を示す。
【0003】
したがって、安定な3量体としてのTNFリガンドおよびTNF受容体の簡単な発現方法のための必要性は依然として存在する。
【発明の開示】
【0004】
本発明は、2量体の受容体分子と比較した場合、それらの同種のリガンドの生物学的活性のより効力のある阻害剤である、3量体のTNF受容体の生産方法を供する。
【0005】
以下に説明するように、本発明は、(1)膜貫通性活性化因子かつCAML(カルシウム信号変調シクロフィリンリガンド)相互作用因子(TACI)の細胞外ドメイン、および(2)3量体化ポリペプチド、を含んで成るポリペプチドを供する。適当なTACIの細胞外ドメインは、SEQ ID NO:4のアミノ酸残基30から110、および(2)SEQ ID NO:4のアミノ酸残基1から110、(3)SEQ ID NO:4のアミノ酸残基30から154、および(4)SEQ ID NO:4のアミノ酸残基1から154、を含む。実例となる3量体化ポリペプチドは、ヒトのコラーゲンXのNC-1断片および熱ショック結合タンパク質-1(Heat Shock Binding Protein-1)の3量体化断片を含む。本発明はさらに、TACIの細胞外ドメインおよび3量体化ポリペプチドを含んで成る融合タンパク質のホモ3量体の複合体を供する。
【0006】
本発明のこれらおよび他の観点は、以下の詳細な説明および図の参酌により明らかとなるであろう。付け加えると、様々な引用を以下に示し、そして、それら全体における引用により組み入れられている。
【0007】
発明の簡単な説明
1.定義
以下の説明において、番号の語は広範囲に使用される。本発明の理解を容易にするために、以下の定義を与える。
【0008】
本明細書で使用される「核酸」または「核酸分子」はポリヌクレオチド、例えば、デオキシリボ核酸(DNA)またはリボ核酸(RNA)、オリゴヌクレオチド、ポリメラーゼ連反応(PCR)により生成された断片、および何らかの連結、切断、エンドヌクレアーゼの作用およびエキソヌクレアーゼの作用により生成された断片を意味する。核酸分子は、自然に発生するヌクレオチドの単量体(例えば、DNAおよびRNA)、自然に発生するヌクレオチドの類似体(例えば、自然に発生するヌクレオチドのα-光学異性体)、または両方の組み合わせである単量体で構成されてよい。修飾されたヌクレオチドは、糖の一部および/またはピリミジンもしくはプリン塩基の一部において変化を有してもよい。糖修飾は、例えば、ハロゲン、アルキル基、アミンおよびアジド基での1以上のヒドロキシル基の置換を含み、あるいは、糖はエーテルまたはエステルとして官能性を持たせることができる。さらに、すべての糖の部分は、立体配置的および電子的に類似した構造体、例えば、アザ糖および炭素環式糖の類似体により置換してもよい。塩基部分の修飾の例は、アルキル化プリンもしくはピリミジン、アシル化プリンもしくはピリミジン、または他の周知の複素環の置換を含む。核酸単量体は、ホスホジエステル結合またはこれらの類似体の結合により連鎖させることができる。ホスホジエステル類似体の結合は、ホスホロチオエート、ホスホロジチオエート、ホスホロセレノエート、ホスホロジセレノエート、ホスホロアニロチオエート、ホスホラニリデート、ホスホラミデート等を含む。「核酸分子」の語もまた、自然に発生し、あるいは修飾された、ポリアミドの主軸を形成する核酸塩基を含んで成るいわゆる「ペプチド核酸」を含む。核酸は1本鎖または2本鎖のいずれかでよい。
【0009】
「核酸分子の補体」の語は、相補ヌクレオチド配列および関連するヌクレオチド配列と比較して逆配向を有する核酸分子を意味する。例えば、配列5’ATGCACGGG3’は5’CCCGTGCAT3’に相補的である。
【0010】
「コンティグ」の語は、他の核酸分子と同一または相補的な配列の連続鎖を有する核酸分子を意味する。コンティグ配列は、核酸分子の所定の鎖と当該核酸分子の全部または部分的な伸展のいずれかにおいて、「オーバーラップ」と言われる。例えば、ポリヌクレオチド5’ATGGAGCTT3’に対する代表的なコンティグは5’AGCTTgagt3’および3’tcgacTACC5’である。
【0011】
「構造遺伝子」の語は、メッセンジャーRNA(mRNA)へと転写される核酸分子を意味し、それから特異的なポリペプチドのアミノ酸配列に特徴的に翻訳される。「注目の遺伝子」は構造遺伝子であってよい。
【0012】
「相補的DNA(cDNA)」は逆転写酵素によりmRNAの鋳型から形成される1本鎖DNA分子である。一般的に、mRNAの一部に相補的なプライマーは逆転写の最初に必要とされる。また、これらの当業者はこれらの1本鎖DNA分子およびその相補的DNA鎖からなる2本鎖DNAを意味して「cDNA」を使用する。「cDNA]の語はまた、RNAの鋳型から合成されるcDNA分子のクローンも意味する。
【0013】
「単離核酸分子」は生物のゲノムDNAに組み込まれない核酸分子である。例えば、細胞のゲノムDNAから分離された成長因子をコードするDNA分子は、単離DNA分子である。単離核酸分子の他の例は、生物のゲノムに組み込まれない化学合成した核酸分子である。特定の種から単離された核酸分子はその種由来の染色体の完全DNA分子よりも小さい。
【0014】
「核酸分子作成物」は天然には存在しない配列に混合あるいは配列した核酸のセグメントを含ませるために、人工的に修飾された1本鎖または2本鎖のいずれかの核酸分子である。
【0015】
「直鎖状DNA」は、非環状の5’および3’末端を有する非環状DNA分子を意味する。直鎖状DNAは、酵素消化または物理的な破壊により、閉環状DNA分子、例えば、プラスミドから調製できる。
【0016】
「プロモーター」は構造遺伝子の転写を指示するヌクレオチド配列である。一般的に、プロモーターは、遺伝子の5’非コード領域、すなわち、構造遺伝子の転写開始部位の近くに存在する。転写の開始に作用するプロモーター中の配列因子は、しばしばコンセンサスヌクレオチド配列により特徴づけられる。これらのプロモーター因子は、RNAポリメラーゼ結合部位、TATA配列、CAAT配列、分化特異的因子(McGehee et al., Mol.Endocrinol.7:551(1993))、サイクリックAMP応答因子、血清応答因子(Treisman,Seminars in Cancer Biol.1:47(1990))、グルココルチコイド応答因子および他の転写因子の結合部位、例えば、CRE/ATF(O'Reilly et al.,J.Boil.Chem.267:19938(1992))、AP2(Ye et al.,J.Boil.Chem.269:25728(1994))、SP1、タンパク質結合cAMP応答因子(Loeken,Gene Exper.3:253(1993))およびオクタマー因子(一般に、Watson et al.,eds.,Molecular Biology of the Gene,4th ed.(The Benjamin/Cummings Publishing Company,Inc.1987)およびLemaigre and Rousseau,Biochem.J.303:1(1994)参照)を含む。仮にプロモーターが誘導性プロモーターであれば、転写の割合は誘発剤に対する応答において増加する。反対に、仮にそのプロモーターが、恒常的プロモーターであれば、転写の割合は誘発剤によって調節されない。また、制止プロモーターも知られる。
【0017】
「コアプロモーター」は、TATAボックスおよび転写の開始点を含む、プロモーターの機能に不可欠なヌクレオチド配列を含む。この定義により、コアプロモーターは、活性を増強し、あるいは組織に特異的な活性を与えるであろう特異的な配列の欠如において、検知可能な活性を有しても、あるいは有さなくてもよい。
【0018】
「調節因子」はコアプロモーターの活性を変化させるヌクレオチド配列である。例えば、調節因子は、排他的に、あるいは選択的に、特に、細胞、組織、またはオルガネラでの転写を可能にする細胞因子と結合するヌクレオチド配列を含んでよい。これらの調節因子のタイプは通常、「細胞に特異的な」、「組織に特異的な」あるいは「オルガネラに特異的な」方法において発現する遺伝子に関係する。
【0019】
「エンハンサー」は、転写開始部位に対するエンハンサーの距離または方向に関係なく、転写の効率を増大させることができる型の調節型因子である。
【0020】
「異種DNA」とは、与えられた宿主細胞中に天然には存在しないDNA分子またはDNA分子群を意味する。宿主のDNAが非宿主のDNAと結合している限り、特定の宿主細胞に対する異種DNA分子は、宿主細胞種(即ち、内生DNA)から派生したDNAを含んでもよい。例えば、転写プロモーターを含んで成る宿主のDNAセグメントと機能するように結合したポリペプチドをコードする非宿主のDNAセグメントを含むDNA分子は、異種DNA分子と考えられる。反対に、異種DNAは、非宿主の遺伝子から派生するプロモーターと機能するように結合する内生遺伝子を含んで成ってよい。他の説明によれば、野生型遺伝子が欠失した突然変異細胞にDNA分子を導入するとすれば、野生型細胞由来の遺伝子を含んで成るDNA分子は異種DNAと考えられる。
【0021】
「ポリペプチド」は、天然的にあるいは合成的に生成されようとも、ペプチド結合により結合したアミノ酸残基のポリマーである。約10以下のアミノ酸残基のポリペプチドは一般に、「ペプチド」を意味する。
【0022】
「タンパク質」は、1以上のポリペプチド鎖を含んで成る巨大分子である。また、タンパク質は、非ペプチド成分、例えば糖群を含んで成ってよい。糖および他の非ペプチド置換物は、タンパク質を生産する細胞によりタンパク質に加えてもよく、細胞のタイプで変化するであろう。タンパク質は、それらのアミノ酸の主鎖構造として本明細書において定義される。置換基、例えば炭水化物基は、一般に、具体的に挙げられていないが、存在してもよい。
【0023】
細胞中で、異種核酸分子から合成されたペプチドまたはポリペプチドは、「異種」ペプチドまたはポリペプチドである。
【0024】
「組み込まれた遺伝子因子」は、宿主細胞の染色体に組み入れたDNAセグメントであり、その後、因子は人工的に細胞に導入される。本発明中、もっとも一般的には、組み込まれた遺伝子因子は、電気穿孔法または他の技術により細胞に導入した線状プラスミドに由来する。組み込まれた遺伝子因子は、親の宿主細胞からその子孫に受け継がれる。
【0025】
「クローニングベクター」は、宿主細胞において、独立して自己複製する能力を有する核酸分子、例えば、プラスミド、コスミドまたはバクテリオファージである。一般的に、クローニングベクターは、ベクターの必須の生物学的機能を失わない測定可能な方法において核酸分子の挿入を許容する1または少数の制限エンドヌクレアーゼ認識部位、並びに、クローニングベクターで形質転換した細胞の識別および選択における使用に適当であるマーカー遺伝子をコードするヌクレオチド配列を含む。マーカー遺伝子は、一般的に、テトラサイクリン耐性またはアンピシリン耐性を供する遺伝子を含む。
【0026】
「発現ベクター」は宿主細胞中で発現する遺伝子をコードする核酸分子である。一般的に、発現ベクターは、転写プロモーター、遺伝子および転写ターミネーターを含んで成る。遺伝子発現は通常、プロモーターの制御下におかれ、そしてこれらの遺伝子は、プロモーターと「機能するように結合」するべきとされる。同様に、調節因子がコアプロモーターの活性を変化させた場合、調節因子およびコアプロモーターは機能するように結合している。
【0027】
「組み換え宿主」は、異種核酸分子、例えば、クローニングベクターまたは発現ベクターを含む細胞である。
【0028】
「組み込み形質変換体」は、異種DNAを細胞のゲノムDNAに組み入れた組み換え宿主細胞である。
【0029】
「発現」の語は、遺伝子生産物の生合成を意味する。例えば、構造遺伝子の場合、発現は、構造遺伝子のmRNAへの転写およびmRNAの1以上のペプチドへの翻訳を含む。
【0030】
「分泌シグナル配列」は、細胞の分泌経路を通して、より大きなポリペプチドペプチドがそこで合成されることを導く、より大きなポリペプチドの成分としてのペプチド(「分泌ペプチド」)をコードするDNA配列を意味する。
【0031】
「単離ポリペプチド」は夾雑している細胞成分、例えば、糖、脂質、または天然にタンパク質に会合する他のタンパク質不純物を本質的に含まないポリペプチドである。一般的に、単離ポリペプチドの調製は、高純度、すなわち、少なくとも約80%の純度、少なくとも約90%の純度、少なくとも約95%の純度、95%以上の純度または99%以上の純度のポリペプチドを含む。特定のタンパク質の調製が単離ポリペプチドを含むことを示す一つの方法は、タンパク質の調製のドデシル硫酸ナトリウム(SDS)-ポリアシルアミドゲル電気泳動およびそのゲルのクマシーブリリアントブルー染色後の一本のバンドの出現によるものである。しかしながら、「単離」の語は、他の物理的形態、例えば、2量体、あるいは、グリコシル化または誘導体化の形態における同一のポリペプチドの存在を除外するわけではない。
【0032】
「アミノ末端」および「カルボキシル末端」はポリペプチド中の位置を示すために本明細書において使用する。文脈が許す場合、これらの語は、近接のまたは相対位置を示すためのポリペプチドの特定の配列または部分に対する引用とともに使用する。例えば、ポリペプチド中の関連する配列に対するカルボキシル末端に位置する配列は、関連する配列のカルボキシルの終端に対して近接するが、完全ポリペプチドのカルボキシルの終端にある必要はない。
【0033】
「融合タンパク質」は、2以上の遺伝子のヌクレオチド配列を含んで成る核酸分子により発現した雑種タンパク質である。このように、融合タンパク質は、天然には互いに会合しない2以上のアミノ酸配列を含んで成る。
【0034】
発現ベクターの成分を説明するために使用する場合、「遺伝子または遺伝子断片」はポリペプチドまたはペプチドをコードするヌクレオチド配列を意味する。その遺伝子または遺伝子断片は、ゲノムDNA、cDNAまたはin vitro 合成技術(例えば、ポリメラーゼ連鎖反応、化学合成等)により得ることができる。
【0035】
「アフィニティタグ」の語は、本明細書において、2番目のポリペプチドの精製または検出を供するため、あるいは、基質に対する2番目のポリペプチドの結合のための部位を供するために2番目のポリペプチドと結合できるポリペプチドセグメントを意味する。原則として、抗体または他の特異的な結合剤として入手可能ないかなるペプチドまたはタンパク質もアフィニティタグとして使用できる。アフィニティタグは、ポリヒスチジン系、プロテインA(Nilsson et al.,EMBO J.4:1075(1985);Nilsson et al.,Method Enzymol.198:3(1991))、グルタチオンSトランスフェラーゼ(Smith and Johnson,Gene 67:31(1988))、Glu-Gluアフィニティタグ(Grussenmeyer et al.,Proc.Natl.Acad.Sci.USA 82:7952(1985))、サブスタンスP、FLAGペプチド(Hopp et al.,Biotechnology 6:1204(1988))、ストレプトアビジン結合ペプチド、または他の抗原性エピトープもしくは結合ドメインを含む。一般には、Ford et al.,Protein Expression and Purification 2:95(1991)を参照のこと。アフィニティタグをコードするDNA分子は、商業者から入手可能である(例えば、Pharmacia Biotech,Piscataway,NJ)。
【0036】
本明細書で使用する、「免疫調節因子」は、サイトカイン、幹細胞成長因子、リンホトキシン、共-刺激分子、造血因子、およびこれらの分子の合成類似体を含む。免疫調節物質の例は、腫瘍壊死因子、インターロイキン、コロニー刺激因子、インターフェロン、幹細胞成長因子、エリトロポイエチンおよびトロンボポイエチンを含む。
【0037】
「免疫グロブリン部分」は、免疫グロブリンの定常部を含んで成るポリペプチドを意味する。例えば、免疫グロブリン部分はH鎖の定常部を含んで成ってよい。
【0038】
「補体/抗補体の対」は、非共有結合的に結合した、非同一部分であり、適当な条件下で安定な対を意味する。例えば、ビオチンおよびアビジン(またはストレプトアビジン)は補体/抗補体の対のプロトタイプメンバーである。他の例としての補対/抗補体の対は、受容体/リガンドの対、抗体/抗原の対(または、ハプテンもしくはエピトープ)の対、センス/アンチセンスポリヌクレオチドの対等を含む。
【0039】
「抗体断片」は、抗体、例えばF(ab’)2、F(ab)2、Fab’、Fab等の一部である。構造に関わらず、抗体断片は、無処置の抗体により認識される同じ抗原と結合する。
【0040】
また、「抗体断片」は、特異的な抗原と結合する、合成の、あるいは遺伝子改変したポリペプチド、例えば、L鎖可変部からなるポリペプチド、H鎖およびL鎖の可変部から成る「Fv」断片、その中のL鎖およびH鎖の可変部がペプチドリンカー(「scFvタンパク質」)により結合される組み換え1本鎖ポリペプチド分子、および高頻度可変部を模倣するアミノ酸残基からなる最小認識ユニットを含む。
【0041】
「検出標識」は、結合しているポリペプチドのパートナーを発現する細胞を識別するための有用な分子を生成するためにポリペプチドと接合できる分子または原子である。検出標識の例は、キレート剤、感光剤、放射性同位体、蛍光試薬、常磁性イオンまたは他のマーカー部分を含む。
【0042】
標準的な分析方法の不正確さにより、ポリマーの分子量および長さは、おおよその値として理解される。これらの値が「およそ」Xまたは「約」Xとして表現している場合、記載されたXの値は、正確には±10%として理解すべきであろう。
【0043】
2.ホモ3量体ペプチドの生産のための発現ベクター
本発明はホモ3量体タンパク質の生産方法を供する。それぞれのホモ3量体のタンパク質は注目のポリペプチドおよび3量体化アミノ酸配列を含んで成る融合タンパク質である。注目のポリペプチドは受容体の細胞外ドメインを含み、それはそれらの同属のリガンドと結合するために使用できる。適当な受容体は腫瘍壊死因子受容体、例えば、TNFRSF1A(また、「p55」、「TNFR-60」および「TNF-R」と称される。例えば、Genbank No.M75866 を参照のこと)、TNFRSF1B(また、「p75」、および「TNFR2」と称される。例えば、Genbank No.M32315 を参照のこと)、TNFRSF13B(「TACI」としても知られる)、TNFRSF13C(「BAFFR」および「Ztnfr12」としても知られる。例えば、Genbank No.AF373846 を参照のこと)、TNFRSF17(「BCMA」としても知られる。例えば、Genbank No.Z29574 を参照のこと)等を含む。他の有用な腫瘍壊死因子受容体は当業者に知られている。
【0044】
本実施例は、膜貫通性活性化因子かつCAML(サイクロフィリンリガンドを変調するカルシウムシグナル)相互作用因子(TACI)の細胞外ドメインを含んで成る融合タンパク質の作成を示す。TACI核酸およびアミノ酸配列は、Bram and Gotz、米国特許番号5,969,102号により説明され、そして本明細書において、SEQ ID NO.3および4として含まれる。説明のTACI細胞外ドメインはSEQ ID NO:4の30から110のアミノ酸残基、SEQ ID NO:4の1から110のアミノ酸残基、SEQ ID NO:4の30から154のアミノ酸残基、およびSEQ ID NO:4の1から110のアミノ酸残基を含んで成るアミノ酸配列を有するポリペプチドを含む。
【0045】
また、本実施例は、2つのタイプの3量体化アミノ酸配列の使用を示す。:カルボキシ末端、ヒトのコラーゲンXの151アミノ酸のNC-1領域、およびタンパク質HSBP-1と結合しているヒトの熱ショック因子の1から65アミノ酸。NC-1ドメインは、Frischholz et al.,J.Biol.Chem.273:4547(1998)により説明されている。ヌクレオチドおよびアミノ酸配列は、本明細書中のSEQ ID NO.19および20として供されている。HSBP-1は、Tai et al.J.Biol.Chem.277:735(2002)により説明されている。HSBP-1の有用な断片のヌクレオチドおよびアミノ酸配列は、SEQ ID NO.21および22として供されている。
【0046】
3量体化アミノ酸配列に加えて、融合タンパク質は、さらに、そのタンパク質を可溶にするために、免疫グロブリン部分を含んで成ることができる。その免疫グロブリン部分は、H鎖定常部、例えば、ヒトのH鎖定常部を含んで成ってよい。IgG1H鎖定常部は、適当なH鎖定常部の一例である。説明のIgG1H鎖定常部はCH2およびCH3ドメインを含んで成るIgG1Fc断片である。そのIgG1Fc断片は、野生型のIgG1Fc断片または突然変異されたIgG1Fc断片であってよい。
【0047】
発現ベクターは注目のポリペプチドおよび3量体化アミノ酸配列を含んで成る融合タンパク質をコードするように作成できる。真核細胞におけるタンパク質の生産のために適当な発現ベクターは、一般的に(1)細菌の複製開始点並びに細菌宿主中の発現ベクターの成長および選択を供するための抗生物質耐性マーカーをコードする原核生物のDNA因子;(2)転写の開始を制御する真核生物のDNA因子、例えば、プロモーター;および(3)転写のプロセッシングを制御するDNA因子、例えば、転写終了/ポリアデニル化シグナル配列、を含む。
【0048】
遺伝子を発現するため、タンパク質をコードする核酸分子を転写の発現を制御する調節配列と機能するように結合させ、それから宿主細胞に導入しなければならない。転写調節配列、例えば、プロモーターおよびエンハンサーに追加して、発現ベクターは転写および翻訳調節配列を含むことができる。説明のように、哺乳類宿主に適当なその転写および翻訳調節シグナルは、ウイルス源、例えば、アデノウイルス、ウシのパピローマウイルス、サルのウイルス等に由来してもよく、その調節シグナルは、高レベルの発現を有する特定の遺伝子に関連する。適当な転写および翻訳調節配列はまた、哺乳類遺伝子、例えば、アクチン、コラーゲン、ミヨシンおよびメタロチオネインの遺伝子から得てもよい。
【0049】
適当な転写調節配列は、RNA合成の開始を指示するのに十分なプロモーター領域を含む。説明の真核生物のプロモーターはマウスのメタロチオネインI(metallothionein I)遺伝子のプロモーター(Hamer et al.,Molec.Appl.Genet.1:273(1982))、ヘルペス(Herpes)ウイルスのTKプロモーター(McKnight,Cell 31:355(1982))、SV40アーリープロモーター(Benoist et al.,Nature 290:304(1981))、ラウス(Rous)肉腫ウイルスプロモーター(Gorman et al.,Proc.Nat'l Acad.Sci.USA 79:6777(1982))、サイトメガロウイルスプロモーター(Foecking et al.,Gene 45:101(1980))、およびマウス乳腺腫瘍ウイルスプロモーター(一般的に、Etcheverry,"Expression of Engeered Proteins in Mammalian Cell Culture,"in Protein Engineering:Principles and Practice,Cleland et al.(eds.),ページ163-181(John Wiley&Sons,Inc.1996)参照)を含む。
【0050】
一方、仮に、原核生物のプロモーターが真核生物のプロモーターにより調節される場合、原核生物のプロモーター、例えば、バクテリオファージ(bacteriophage)T3RNAポリメラーゼプロモーターは哺乳類細胞中の注目の遺伝子の発現を制御するために使用できる(Zhou et al.,Mol.Cell.Biol.10:4529(1990)、およびKaufman et al.,Nucl.Acids Res.19:4485(1991))。
【0051】
アフィニティタグの包含は、融合タンパク質を示す細胞の認識または選択のために有用である。アフィニティタグの例は、ポリヒスチジンタグ(ニッケルキレート化樹脂と親和性を有する)、抗-myc抗体で検出されるc-mycタグ(例えば、EQKLI SEEDL;SEQ ID NO:1)、カルモジュリン結合タンパク質(カルモジュリンアフィニティクロマトグラフィーで単離される)、サブスタンスP、RYIRSタグ(抗-RYIRS抗体と結合する)、抗体で検出される赤血球凝集素Aエピトープタグ(例えば、YPYDVPDYA;SEQ ID NO:2)、Glu-GluタグおよびFLAGタグ(抗-FLAG抗体と結合する)を含む。例えば、Luo et al.,Arch.Biochem.Biophys.329:215(1996)、Morganti et al.,Biotechnol.Appl.Biochem.23:67(1996)、およびZheng et al.,Gene 186:55(1997)を参照のこと。これらのペプチドタグをコードする核酸分子は、例えば、Sigma-Aldrich Corporation(St.Louis,MO)から入手できる。
【0052】
クローニング部位はマルチクローニング部位であってよい。いかなるマルチクローニング部位も使用でき、そして多くが商業的に入手可能である。特に有用なマルチクローニング部位は、3つのすべての読み枠において遺伝子または遺伝子断片のクローニングを許容する。
【0053】
発現ベクターは、選択マーカーをコードするヌクレオチド配列を含んでもよい。広範な種々の選択マーカー遺伝子は入手可能である(例えば、Kaufman,Meth.Enzymol.185:487(1990);Kaufman,Meth.Enzymol.185:537(1990)参照のこと)。例えば、1つの適当な選択マーカーは、抗生物質ネオマイシンへの耐性を供する遺伝子である。この場合、選択はネオマイシン型の薬剤、例えば、G-418等の存在中で行われる。ブレオマイシン耐性遺伝子、例えば、Sh ble遺伝子もまた、今説明の方法に有用な選択マーカー遺伝子である。これらの遺伝子はブレオマイシン/フレオマイシン型薬剤、例えば、ZEOCINの活性を阻害するタンパク質を生産する(Gatignol et al.,Mol.Gen.Genet.207:342(1987);Drocourt et al.,Nucl.Acids Res.18:4009(1990)。ZEOCINは、細菌、真菌、植物、トリ、昆虫および哺乳類の細胞を含む細胞の広範囲における毒素である。追加的な選択マーカーはヒグロマイシンB-ホスホトランスフェラーゼ、AUR1遺伝子生産物、アデノシンデアミナーゼ、アミノグルコシドホスホトランスフェラーゼ、ジヒドロホレートレダクターゼ、チミジンキナーゼ、およびキサンチン-グアニンホスホリボシルトランスフェラーゼを含む(例えば、Srivastava and Schlessinger,Gene 103:53(1991);Romanos et al.,"Expression of Cloned Genes in Yeast,"in DNA Cloning 2:Expression Systems,2nd Edition,ページ123-167(IRL Press 1995);Markie,Methods Mol.Biol.54:359(1996);Pfeifer et al.,Gene 188:183(1997);Tucker and Burke,Gene 199:25(1997);Hashida-Okado et al.,FEBS Letters 425:117(1998)を参照)。選択マーカー遺伝子は、公表されたヌクレオチド配列を使用してクローンされ、あるいは合成されてもよく、あるいは、マーカー遺伝子は商業的に入手可能である。
【0054】
発現ベクターはSV40開始点を含んでもよい。この因子は、SV40ラージT抗原を発現する細胞株中でエピソームの複製および救出のために使用できる。
【0055】
発現ベクターへの挿入に適当な遺伝子または遺伝子断片はcDNAから得ることができる。それは、いかなる公知技術方法によっても調製される。例えば、cDNA分子は、ランダムプライミングにより合成できる。さらに、このようなプライマーは、ベクターにおいて発見される制限エンドヌクレアーゼ部位と結合できる。あるいは、cDNA分子はオリゴd(T)プライミングにより調製できる。また、遺伝子または遺伝子断片は、ゲノムDNAから、あるいは化学合成により得ることができる。適当な遺伝子または遺伝子断片の調製のための標準的な方法は、当業者に知られている(例えば、Ausubel et al.(eds.),Short Protocols in Molecular Biology,3rd Edition(John Wiley&Sons 1995)["Ausubel 1995"])。
【0056】
発現ベクターを作成後、核酸ポリマーの産生のための核酸分子の合成のために、宿主細胞中でそのベクターを増殖させることができる。しばしば「シャトルベクター」と称するベクターは、2以上の無関係な発現系において複製することができる。このような複製を促進するため、そのベクターは、各複製系において各々が有効な、2以上の複製開始点含むべきである。一般的に、シャトルベクターは真核生物系および原核生物系において複製することが可能である。これは真核生物の宿主である「発現細胞型」中のタンパク質の発現の検出および原核生物の宿主である「増幅細胞型」中のベクターの増幅を可能にする。説明のように、一方の複製開始点はSV40に由来してよく、他方の複製開始点はpBR322に由来してよい。当業者は、多数の適当な複製開始点を知っている。
【0057】
ベクターの増殖は、原核生物の宿主細胞、例えば、大腸菌(E.coli)または枯草菌(Bacillus subtilus)中で簡単に行うことができる。適当な大腸菌(E.coli)株は、BL21(DE3)、BL21(DE3)pLysS、BL21(DE3)pLysE、DH1、DH4I、DH5、DH5I、DH5IF'、DH5IMCR、DH10B、DH10B/p3、DH11S、C600、HB101、JM101、JM105、JM109、JM110、K38、RR1、Y1088、Y1089、CSH18、ER1451、およびER1647を含む(例えば、Brown(ed.),Molecular Biology Labfax(Academic Press 1991)参照のこと)。適当な枯草菌(Bacillus subtilus)株は、BR151、YB886、MI119、MI120、およびB170を含む(例えば、Hardy,"Bacillus Cloning Methods,"in DNA Cloning:A Practical Approach,Glover(ed.)(IRL Press 1985)参照のこと)。原核生物の宿主中のベクターの増殖のための標準的な技術は、当業者にとって周知である(例えば、Ausubel 1995;Wu et al.,Methods in Gene Biotechnology(CRC press,Inc.1997)を参照のこと)。
【0058】
3.宿主細胞による組み換えタンパク質の生産
発現ベクターは、いかなる真核生物細胞、例えば、哺乳動物の細胞、昆虫の細胞、トリの細胞、真菌の細胞等にも導入することができる。適当な哺乳動物の宿主細胞の例は、アフリカミドリザルの腎臓細胞(Vero;ATCC CRL 1587)、ヒト胎児の腎臓細胞(293-HEK;ATCC CRL 1573)、ベビーハムスターの腎臓細胞(BHK-21、BHK-570;ATCC CRL 8544、ATCC CRL 10314)、イヌの腎臓細胞(MDCK;ATCC CCL 34)、中国ハムスターの卵巣細胞(CHO-K1;ATCC CCL 61;CHO DG44(Chasin et al.,Som.Cell.Molec.Genet.12:555(1986))、ラットの下垂体細胞(GH1;ATCC CCL82)、HeLa S3細胞(ATCC CCL 2.2)、ラットの肝臓細胞(H-4-II-E;ATCC CRL 1548)、SV40-形質転換化したサルの腎臓細胞(COS-1;ATCC CRL 1650)およびマウス胎児の細胞(NIH-3T3;ATCC CRL 1658)を含む。
【0059】
バキュロウイルス系は、クローン化した注目の遺伝子を昆虫の細胞中に導入するために有効な手段を供する。適当な発現ベクターはオートグラファ・カリフォルニカ・マルチプルヌクレアポリヘドロシスウイルス(Autographa californica multiple nuclear polyhedrosis virus)(AcMNPV)に基づき、そして周知のプロモーター、例えば、ショウジョウバエ(Drosophila)の熱ショックタンパク質(hsp)の70プロモーター、オートグラファ・カリフォルニカ・ヌクレアポリヘドロシスウイルス(Autographa californica nuclear polyhedrosis virus)の前初期遺伝子のプロモーター(ie-1)および遅延性の初期39Kプロモーター、バキュロウイルスのp10プロモーター、およびショウジョウバエのメタロチオネイン(Drosophila metallothionein)プロモーターを含む。組み換えバキュロウイルスを生成する2つ目の方法は、Luckow(Luckow,et al.,J.Virol.67:4566(1993))により説明されるトランスポゾン基準系を利用する。トランスファーベクターを利用するこの系はBAC-to-BACキットとして販売されている(Life Technologies,Rockville,MD)。この系は、遺伝子または遺伝子断片を、「バクミド」と呼ばれる巨大プラスミドとして大腸菌(E.coli)中に維持されるバキュロウイルスのゲノムに移動させるために、トランスファーベクターであるTn7トランスポソンを含むPFASTBAC(Life Technologies)を利用する。Hill-Perkins and Possee,J.Gen.Virol.71:971(1990)、Bonning,et al.,J.Gen.Virol.75:1551(1994)、およびChazenbalk,and Rapoport,J.Biol.Chem.270:1543(1995)を参照のこと。これらのベクターは上記の説明に従い改変できる。
【0060】
組み換えウイルスまたはバクミドは、宿主細胞にトランスフェクションするために使用される。適当な昆虫宿主細胞は、IPLB-sf-21由来の細胞株、スポドプテラ・フルギペルダ(Spodoptera frugiperda)の卵巣細胞株、例えば、Sf9(ATCC CRL 1711)、Sf21AE、およびSf21(Invitrogen Corporation;San Diego,CA)、並びにショウジョウバエのシュナイダー2(Drosophila Schneider-2)細胞、およびイラクサギンウワバ(Trichoplusia ni)由来のHIGHFIVEO細胞株(Invitrogen)(米国特許番号5,300,435号)を含む。商業的に入手可能な無血清培地は細胞を成長させるため、および維持するために使用できる。適当な培地は、Sf9細胞にはSf900IITM(Life Technologies)またはESF921TM(Expression Systems);そしてT.ni細胞にはEx-cellO405TM(JPH Biosciences,Lenexa,KS)またはExpress FiveOTM(Life Technologies)である。組み換えウイルスを使用する場合、その細胞は、約2-5×105の細胞密度から1-2×106の細胞密度に成長させる。その場合、組み替えウイルスのストックは、0.1から10の多重感染度(MOI)、さらに一般的には3付近で加えられる。
【0061】
バキュロウイルス系中の組み換えタンパク質を生産するための確立された技術は、Bailey et al.,"Manipulation of Baculovirus Vector,"in Methods in Molecular Biology,Volume 7: Gene Transfer and Expression Protocols,Murray(ed.),ページ147-168(The Humana Press,Inc.1991),by Patel et al.,"The baculovirus expression system,"in DNA Cloning 2:Expression Systems,2nd Edition,Glover et al.(eds.), ページ205-244(Oxford University Press 1995),by Ausubel(1995) ページ16-37から16-57,by Richardson (ed.),Baculovirus Expression Protocols(The Humana Press,Inc.1995),and by Lucknow,"Insect Cell Expression Technology,"in Protein Engineering:Principles and Practice,Cleland et al., ページ183-218(John Wiley&Sons,Inc.1996)により供される。
【0062】
また、本明細書において説明する発現ベクターは、酵母菌の細胞を含む真菌類細胞にトランスフェクションするために使用できる。この点に関して特に注目の酵母菌種は、出芽酵母(Saccharomyces cervisiae)、ピチア・パストリス(Pichia pastoris)、およびピチア・メタノリカ(Pichia methanolica)を含む。酵母菌中の発現に適当なプロモーターは、GAL1(ガラクトース)、PGK(ホスホグリセレートキナーゼ)、ADH(アルコールデヒドロゲナーゼ)、AOX1(アルコールオキシダーゼ)、HIS4(ヒスチジノールデヒドロゲナーゼ)等由来のプロモーターを含む。多くの酵母菌クローニングベクターは容易に入手可能であり、そして上記の説明に従い改変できる。これらのベクターは、YIp系ベクター、例えば、YIp5、YRpベクター、例えば、YIp17、YEpベクター、例えば、YEp13およびYCpベクター、例えば、YCp19を含む。外来性DNAでの出芽酵母(S.cerevisiae)の形質転換およびそれからの組み替えポリペプチドの生成方法は、例えば、Kawasaki、米国特許番号4,599,311号、Kawasaki et al.、米国特許番号4,931,373号、Brake、米国特許番号4,870,008号、Welch et al.、米国特許番号5,037,743号、およびMurray et al.、米国特許番号4,845,075号により開示されている。形質転換化細胞は、選択マーカーにより決定される表現型、一般的に、薬剤耐性または特定の栄養素(例えば、ロイシン)の不在における成長能により選択される。出芽酵母(Saccharomyces cervisiae)中の使用のための好ましいベクター系は、Kawasaki et al.(米国特許番号4,931,373)により開示されたPOT1ベクター系であり、それは、形質転換化細胞がグルコース含有媒地における成長により選択されることを許容する。酵母菌での使用のための追加的な適当なプロモーターおよびターミネーターは、解糖酵素遺伝子(例えば、Kawasaki、米国特許番号4,599,311号、Kawasaki et al.、 米国特許番号4,615,974号およびBitter、米国特許番号4,977,092号を参照のこと)およびアルコールデヒドロゲナーゼ遺伝子由来のものを含む。また、米国特許番号4,990,446号、5,063,154号、5,139,936号、および4,661,454号を参照のこと。
【0063】
ハンセヌラ・ポリモルファ(Hansenula polymorpha)、スキゾサッカロマイセス・プロムベ(Schizosaccharomyces prombe)、クルイベロマイセス・ラクチス( Kluyveromyces lactis)、クルイベロマイセス・フラギリス(Kluyveromyces fragilis)、ウスチラゴ・マイジス(Ustilago maydis)、ピチア・パストリス(Pichia pastoris)、ピチア・メタノリカ(Pichia methanolica)、ピチア・グイレルモンジイ(Pichia guillermondii)、およびカンジダ・マルトーサ(Candida maltosa)を含む他の酵母菌のための形質転換系は、当業界において知られている。例えば、Gleeson et al.,J.Gen.Microbiol.132:3459(1986)、およびGregg,米国特許番号4,882,279号を参照のこと。アスペルギルス(Aspergillus)細胞は、McKnight et al.、米国特許番号4,935,349号の方法に従い利用できる。アクレモニウム・クリソゲヌム(Acremonium chrysogenum)の形質転換の方法は、Sumino et al.、米国特許番号5,162,228号により開示されている。ネウロスポラ(Neurospora)の形質転換の方法は、Lambowitz、米国特許番号4,486,533号により開示されている。
【0064】
例えば、組み換えタンパク質の生産のための宿主としてのピチア・メタノリカ(Pichia methanolica)の使用は、Raymond、米国特許番号5,716,808号、Raymond、米国特許番号5,736,383号、Raymond et al.,Yeast 14:11-23(1998)、および国際公開番号WO97/17450、WO97/17451、WO98/02536、およびWO98/02565により開示されている。ピチア・メタノリカ(Pichia methanolica)の形質転換に使用のDNA分子は、一般的に2本鎖の環状プラスミドとして調製されるものと思われ、好ましくは形質転換の前に直線化される。P・メタノリカ(P methanolica)におけるポリペプチドの生産のために、プラスミド中のプロモーターおよびターミネーターは、P・メタノリカ(P methanolica)遺伝子のもの、例えば、P・メタノリカ(P methanolica)アルコール利用遺伝子(AUG1またはAUG2)であることが好ましい。他の有用なプロモーターは、ジヒドロキシアセトンシンターゼ(DHAS)、ホルメートデヒドロゲナーゼ(FMD)、およびカタラーゼ(CAT)のものを含む。宿主染色体へのDNAの組み込みを促進するため、宿主のDNA配列により両末端に隣接するプラスミドのすべての発現セグメントを有することが好ましい。メタノールの使用を最小化することが望ましい大規模な工業的工程には、そこに両方のメタノール利用遺伝子(AUG1およびAUG2)が欠失した宿主細胞を使用することが好ましい。選択されたタンパク質の生産には、液胞においてプロテアーゼ遺伝子(PEP4およびPRB1)が欠乏した宿主細胞が好ましい。注目のポリペプチドをコードするDNAを含むプラスミドの、P・メタノリカ(P methanolica)の細胞への導入を促進するために電気穿孔法が使用される。P・メタノリカ(P methanolica)の細胞は、電界強度が、2.5から4.5kV/cm、好ましくは約3.75kV/cm、および時定数(t)が1から40ミリ秒、最も好ましくは約20ミリ秒である、指数関数的に減衰する電場を使用した電気穿孔法により形質転換することができる。
【0065】
発現ベクターは、カラムホスフェイトトランスフェクション、リポソーム性トランスフェクション、微粒子性伝達、電気穿孔法等を含む種々の標準的な技術を使用して宿主細胞へと導入できる。
【0066】
発現ベクターを哺乳動物、酵母菌、および昆虫の細胞へ導入するための標準的な方法は、例えば、Ausubel(1995)により供される。哺乳動物の細胞系により生産された外来タンパク質の発現および回収方法は、例えば、Etcheverry,"Expression of Engineered Proteins in Mammalian Cell Culture,"in Protein Engineering:Principles and Practice、Cleland et al.(eds.)、ページ163(Wiley-Liss,Inc.1996)により供される。バキュロウイルス系から組み換えタンパク質を単離するための確立された方法は、Richardson(ed.)、Baculovirus Expression Protocols(The Humana Press,Inc.1995)により説明されている。
【0067】
発現ベクターは、注目のポリペプチドを生産する細胞から単離することができる。所望すれば、発現ベクターを同定可能なポリペプチドの発現に基づく選択の他の部分に服従させることあるいは増幅細胞にトランスフェクションすることができる。それから、トランスフェクションした増幅細胞を選択マーカーにより選択し、そのベクターを精製し、そして、当業界におけるいかなる既知の方法により遺伝子または遺伝子断片のヌクレオチド配列を配列決定する。仮にヌクレオチド配列が完全ポリペプチド部分のみをコードするならば、そのヌクレオチド配列は、その全遺伝子を検索するために当業界における既知の方法によりプローブとして使用することができる。
【0068】
本発明、このように一般的な説明は、以下の実施例を参照により、さらに容易に理解されるだろう。これらは説明の方法により供され、そして、本発明を制限することを意図するものではない。
【実施例1】
【0069】
TACI-NC1の作成
発現ベクターは、膜貫通性活性化因子かつ(カルシウム信号変調シクロフィリンリガンド)相互作用因子(TACI)のタンパク質の細胞外ドメインおよびヒトのコラーゲンXのNC1ドメインを含んで成る融合物をコードするように作成する。TACIの核酸配列は、Bram and Gotz,米国特許番号5,969,102号により説明され、そして本明細書中にSEQ ID NO.3および4として含まれる。NC-1ドメインは、Frischholz et al.,J.Biol.Chem.273:4547(1998)により説明される。:ヌクレオチドおよびアミノ酸配列は、本明細書中、SEQ ID NO19および20として供される。本作成において、TACIの細胞外ドメインを、c-終端におけるGlu-GluタグおよびTACIとNC1の間に設計された8つのアミノ酸のGly-SerスペーサーでNC1と融合した。
【0070】
NC1は、オリゴヌクレオチド zc40219(5’GGGCCTCCAG GCCCACCAGG T3’;SEQ ID NO:5)およびzc40205(5’TCACATTGGA GCCACTAGGA A3’;SEQ ID NO:6)を使用してヒトのゲノムDNA(Clontech)からPCRにより増幅した。TACIの細胞外の部分は、94℃で1分、55℃で1分および72℃で2分の30サイクルの条件で、オリゴヌクレオチドzc40915(5’ACAGGTGTCC AGGGAATTCA TATAGGCCGG CCACCATGGA TGCAATGAAG AGAGGG3’;SEQ ID NO:7)およびzc40917(5’ACCCTCAGGC ATCGAACCCG AACCCGAACC GGATCC3’;SEQ ID NO:8)でTACI免疫グロブリン融合タンパク質をコードするクローンからPCRにより増幅した。PCR生産物を沈殿させ、そして10μlの水に再懸濁し、それから出芽酵母(S.cerevisiae)中のBglIIで消化したpZMP21へ組み換えた。その組み換え結果の大腸菌(E.coli)のクローンを、適切な組み入れのために、AscI消化によりスクリーニングし、そして配列決定のために3つのポジティブクローンを使用した。1つのクローンをさらなる実験のために選択した。このクローンは、NC1中でグリシンからアルギニンへの突然変異を含み、C末端のGlu-Gluタグから4つのアミノ酸が欠失していた。
【0071】
TACI/NC1-EEをコードするベクターは、電気穿孔のために、20μgのQiagen精製化DNAをPvuIで消化することより直線化した。この直線化DNAをPF-CHO細胞中で電気穿孔した。その細胞を、10日間のHT培地における栄養素の選択の前に24時間回復させた。栄養素の選択から回復後、細胞をさらに10日間、50nmのメトトリキサート(methotrexate)選択に移動した。
【0072】
トランスフェクションした細胞を細胞工場(cell factories)に播種し、そして2リットルの工場調整培地(CM)を単離した。CMを1.5mgの抗-TACIモノクローナル抗体と混合し、4℃で一昼夜インキュベートした。CM-抗体混合物を、流速2mL/minで1.6mLの総容積のPOROS A50カラムに適用した。加えた後、カラムを100カラム容量のpH7.2のPBSで洗浄した。その結合タンパク質をそれから直接、pH8.0の2MのトリスとpH2.5の200mlのグリシンへと溶出した。1ミリリットルのフラクションを収集した。ウェスタンブロット分析に基づき、TACI-NC1を含むフラクションを貯蔵した。その貯蔵したフラクションを300μlに濃縮し、そして、バッファーをpH7.2の14mLのPBSで3回交換し、それから3交換のpH7.2の4LのPBSに対して透析した。
【実施例2】
【0073】
哺乳動物細胞中のTACI-HSB1の発現
HSB1遺伝子の合成
ヒトの熱ショック結合タンパク質(HSBP-1)はTai et al.J.Biol.Chem.277:735(2002)により説明されている。HSBP-1のヌクレオチドおよびアミノ酸配列はSEQ ID NO21および22として供される。4つのオーバーラップしているオリゴヌクレオチド(ヒトの熱ショック結合タンパク質(HSBP-1)のセンスおよびアンチセンス鎖の両方をコードした)を以下のプライマーを使用して固相合成により合成した。:5’GATCGGATCC ATGGCCGAAA CTGATCCTAA AACAGTTCAA GACCTTACCA GCGTAGTCCA GACGCTCCTG CAAGAGATCG AAGATAAGTT TCAGACTATG AGCGACCAAA TCATTGAG3’(SEQ ID NO:9);5’AGAATGCATG ACATGAGCTC CAGGATAGAT GACCTTGAGA AAAATATAGC AGATTTAATG ACGCAAGCTG GTGTGGAAGA GTTGGAAGGA AGTGGTTCTA3’(SEQ ID NO:10);5’GATCTAGAAC CACTTCCTTC CAACTCTTCC ACACCAGCTT GCGTCATTAA ATCTGCTATA TTTTTCTCAA GGTCATCTAT CCTGGAGCTC ATGTCATCGA TTCTCTCAAT3’(SEQ ID NO:11);および5’GATTTGGTCG CTCATAGTCT GAAACTTATC TTGCATCTCT TGCAGGAGCG TCTGGACTAC GCTGGTAAGG TCTTGAACTG TTTTAGGATC AGTTTCGGCC ATGGATCC3’ (SEQ ID NO:12)。各オリゴヌクレオチドの5末端は、120pモルの各オリゴヌクレオチド、1.6μlの100mM ATP、34μlの5×T4-キナーゼバッファー(Life Technologies,Bethesda,MD)、6.4μlの水、および1μlのT4-ポリヌクレオチドキナーゼ(Life Technologies)の混合によりリン酸化し、そして37℃で20分間インキュベートした。そのリン酸化反応は、沸騰したウォーターバス中に置いて、それから、アニーリングを促進させるためにゆっくりと25℃まで冷却した。その断片を20μlの10×T4-リガーゼバッファー(Life Technologies)、0.5μlの100mM ATP、および2μlのT4-DNAリガーゼ(Life Technologies)を加えることにより連結させ、そして、16℃で一昼夜インキュベートした。連結後、そのDNAをアルコール沈殿により採取した。その単離DNAを、3.2μlのB制限バッファー(Promega,Madison,WI)、26.8μlの水、1.5μlのBglII(Life Technologies)、および1.5μlのAsp718(Life Technologies)中で再懸濁し、そして、37℃で2時間インキュベートした。そのリガーゼ反応生成物を15%アガロースゲル上で分画し、そして、ヒトのHSBP-1をコードする220ヌクレオチド断片を、製造業者の手順(Quiagen)に従い Quiagen ゲル単離キットを使用して単離した。ヒトのHSBP-1をT4-DNAリガーゼおよび製造業者のガイドライン(Life Technologies)を使用してAsp718-BamHIの切断したベクターに挿入した。
【0074】
TACIの細胞外ドメインをコードする断片を、オリゴヌクレオチドzc41712(5’CACACGTACG AAGATGGATG CAATGAAGAG AGG3’;SEQ ID NO:13)およびzc41638(5’GGTTAGATCT CGAACCCGAA CCCGAACCGG3’;SEQ ID NO:14)を使用してpTACI-NC1ベクターから増幅させた。そのPCR生成物をBsiW1およびBglIIで切断し、そして1.5%アガロースゲル上で、増幅させたDNAを分画し、それから、製造業者の手順(Quiagen)に従い Quiagen ゲル単離キットを使用して単離した。その単離したDNAを、製造業者のガイドラインに従い(Life Technologies)、T4-DNAリガーゼを使用してHSBP-1をコードする配列を含むAsp718-BglIIの切断したベクターに挿入した。DNAの配列決定により予想したベクターの配列を確認し、それを「pHZTACI-HSBP.9」と命名した。
【0075】
B.TACI-HSBP-1の発現および精製
pHZTACI-HSBP.9ベクターを、TransTransfected を使用してBHK570中にトランスフェクションし、そして、トランスフェクタント耐性のために培養液を10μMのメトトレキサートに選択した。耐性コロニーを組織培養皿に移動し、拡大させ、そして抗-His(C-末端)抗体(Invitrogen,Carlsbad,CA)でのウェスタンブロット分析によりTACI-HSBP-His6の分泌を分析した。生じた細胞株「BHK.TACI-HSBP.2」を拡大した。
【0076】
BHK.TACI-HSBP.2細胞を、細胞工場中に播種し、そして12リットルの工場-条件培地を単離した。その培地を、流速2ml/minで25ミリリットルの総容積のNi-NTA(Invitrogen)に適用した。追加後、カラムをpH7.2の50カラム容量のPBSおよび20カラム容量のリン酸緩衝食塩水、pH6.0の50mMのイミジゾールで洗浄した。それから、その結合タンパク質を、pH6.0のPBS中、50mMから800mMへのイミジゾール濃度の増加の直線勾配で、1ml/minで60分間溶出した。1ミリリットルの分画を採取した。ウェスタンブロット分析に基づき、TACI-HSBP-1を含む分画を貯蔵した。その貯蔵した分画を300μlに濃縮し、そしてバッファーを14mlのリン酸緩衝食塩水(pH7.2)で3回交換し、それから3交換の4リットルのpH7.2のリン酸緩衝食塩水に対して透析した。ウェスタンブロット分析およびクマシー染色ゲルは75%以上の純度および20kDaの単量体の分子量を示した。
【実施例3】
【0077】
TACI融合タンパク質のための増殖アッセイ
アフェレーシス由来の末梢血の単核細胞を、Ficol-Hypaque における密度勾配遠心分離により単離し、そしてリン酸緩衝食塩水中で洗浄した。一般的に、1ドナーから約1010の末梢血の単核細胞を単離することができる。1バイアル当たり約108の細胞を90%のウシ胎児血清および10%のジメチルスルホキド中で凍結させた。
【0078】
複数のバイアルを解凍し、細胞の生存度を測定した。B細胞を、CD19磁気ビーズおよび VarioMacs 磁気分離システム(Miltenyi Biotec;Aubum,CA)を使用して末梢血の単核細胞から単離した。丸底96ウェルプレートをリン酸緩衝食塩水中の5μg/mlのヤギ抗-ヒトIgMで、4℃で24時間にわたり、前コーティングした(Southern Biotechnology Assoc.Inc.;Birmingham AL)。10ng/mLのヒトのIL-4(Pharmingen)および20ng/mLのzTNF4(TACIと結合するリガンド)の存在下において、1ウェル当たり精製したB細胞を105培養した。zTNF4が刺激したB細胞の増殖を阻害する能力を比較するために、1μg/mlで開始したTACI-Fc融合タンパク質、TACI-HSBP-1またはTACI-NC1の3倍希釈系を含ませた。その細胞を、zTNF4、ヒトIL-4の存在下、および阻害因子アリまたはナシで、4日間インキュベートし、それから1ウェル当たり1μCiのH3チミジン(Amersham)で、一昼夜パルスした。プレートはPackardプレートハーベスターを使用して収集し、そして、Packardリーダーを使用してカウントした。本アッセイにおいて、zTNF4が刺激したヒトのB細胞の増殖の阻害で、TACI-HSBP-1は3倍以上の有効性し、そしてTACI-NC1は10倍以上の有効性を示した。図2において示すように、TACI-NC1は、同じ濃度のTACI-Fc5よりもTNF4が誘発したB細胞の増殖を大きく減少させた。この違いは、1.1nmの値を有するTACI-Fc5および57pMの値を有するTACI-NC1により、図中に示されたEC50値において表され、TACI-NC1が約19倍有効であることを示している。
【実施例4】
【0079】
細菌の細胞中のTACI-HSB1の発現
ヒトのTACI-HSBP-1をコードするヌクレオチド配列を含む発現プラスミドを酵母菌のホモログ組み換えを介して、G10エンハンサー配列の後ろに挿入した。ヒトのTACI-HSBP-1のDNA断片はPCRを使用して単離した。PCR反応中のヒトのTACI-HSBP-1の生産において2つのプライマーを使用した。ベクターのフランキング配列の40塩基対を含むプライマーzc42,728(5’CTAGAAATAA TTTTGTTTAA CTTTAAGAAG GAGATATATA TATGGCTATG AGATCCTGCC3’;SEQ ID NO:15)は、G10エンハンサー配列およびヒトのTACI-HSBP-1のアミノ終端に対応する24塩基対から成った。プライマーzc42,731(5’TCTGTATCAG GCTGAAAATC TTATCTCATC CGCCAAAACA CTAGTGATGG TGATGGTGAT GGCC3’;SEQ ID NO:16)はベクターのフランキング配列の40塩基対およびTACI-HSBP-1配列のカルボキシル終端に対応する24塩基対を含んだ。その鋳型はpH2-TACI-HSBP9であった。そのPCR反応条件は以下とした。:94℃で30秒間、50℃で30秒間、そして72℃で1分間の25サイクル;続いて4℃で浸漬した。分析のために、2から4μl容量のPCR試料を1×TBEバッファーを伴う1%アガロースゲル上に流し、そして約550塩基対断片の予想したバンド
【化1】
を観察した。100μlの残留容量の反応物を無水エタノールで沈殿させた。そのペレットを、SmaI-切断化レシピエントベクターpTAP238への組み込みに使用するために10μlの水に再懸濁し、TACI-HSBP-1をコードする作成物を生成した。
【0080】
100マイクロリットルの形質転換受容性酵母菌細胞(S.cerevisiae)を、約1μgのヒトのTACI-HSBP-1フラグメント(PCR生成物)および100ngのSmaI-切断化pTAP238ベクターを含む10μlの混合物と混合し、そして0.2cmの電気穿孔キュベットに移動した。その酵母菌/DNA混合物を、0.75kV(5kV/cm)、無限オーム、25μFにセットした機器を使用して電気パルスし、それから600μlの1.2Mソルビトールをキュベットに加えた。それからその酵母菌を、2つの300μlのURA D(ウラシル欠乏グルコース-含有培地)プレート上で培養し、30℃でインキュベートした。およそ48時間後、1つのプレート由来のUra+酵母菌の形質転換体を1mlの水中で再懸濁し、細胞をペレットにするために素早くスパンした。その細胞ペレットを1mlの溶解バッファー中で再懸濁した。DNAを上記に開示したように回収した。そのDNAペレットを100μlの水中で再懸濁した。
【0081】
40μlの電気形質転換受容性大腸菌(E.coli)のMC1061細胞を1μlの酵母菌DNAで形質転換した。その細胞は2.0kV、25μFおよび400オームで電気パルスした。電気穿孔後、0.6mlのSOC(2% BactoTM Tryptone(Difco,Detroit,MI)、0.5%酵母菌抽出物(Difco)、10mMのNaCl、2.5mMのKCl、10mMのMgCl2、10mMのMgSO4、20mMのグルコース)を細胞に加えた。その細胞を37℃で1時間回復させ、それから、LB+カナマイシンプレート(LBブロス(Lennox)、1.8% BactoTM Agar(Difco)、30mg/Lのカナマイシン)上で培養した。
【0082】
ヒトのTACI-HSBP-1のための正しい発現作成物を宿す個々のクローンは、プラスミドDNAの特徴的な消化により認識される。細胞は30μg/mlのカナマイシンを伴うSuper Broth II(Becton Dickinson)中で一昼夜成長させた。翌日、その細胞を採取し、そしてプラスミドDNAを、スピンカラム(QIAprep(登録商標)Spin Miniprep Kit;Quiagen Inc.,Valencia,CA)を使用して調製した。それから、そのDNAをNotIおよびXbaIで切断した。その正しい制限パターンを伴うクローンをpTAP415と命名し、そして配列決定した。pTAP415中のTACI-HSBP-1のポリヌクレオチド配列をSEQ ID NO:17に示す。
【0083】
10マイクロリットルのpTAP415を、3μlの商業的に入手可能なバッファー(バッファー3;New England Biolabs) および15μlの水中の2マイクロリットルのNotIで37℃、1時間で切断した。7マイクロリットルの反応混合物を2マイクロリットルの5×T4DNAリガーゼバッファー(Life Technologies;Gaitherburg,MD)および1マイクロリットルのT4DNAリガーゼと混合し、室温で1時間インキュベートした。1マイクロリットルのそのライゲージョン混合物を、大腸菌(E.coli)株W3110(ATCC 27325)を形質転換するために使用した。その細胞を2.0kV、25μF、および400オームで電気パルスした。電気穿孔後、0.6mlのSOCを細胞に加えた。その細胞を37℃で1時間成長させ、それからLB+カナマイシンプレート上で培養した。
【0084】
個々のクローンを採取し、成長させた。スピンカラムを使用してプラスミドDNAを調製した。酵母菌のURA3およびCEN/ARS因子の減少を確認するため、そのDNAを、特異的にPvuIIおよびHindIII で切断した。個々のコロニーを採取した。細胞を30μg/mlのカナマイシンを含むSuperbrothII(Becton Dickinson)で一昼夜成長させた。30μg/mlのカナマイシンを含む2ミリリットルの新鮮なSuperbrothIIを接種するために、100マイクロリットルの一昼夜培養液(overnight culture)を使用した。培養液は15mlのコニカルチューブ中で約2時間振りながら37℃で成長させた。1mlの培養液を1mMのIPTGで誘導した。2時間15分後、同量の培養液を5%のβMEを伴う250μlの Thorner バッファー(8Mの尿素、pH7.0で100mMのトリス、10%グリセロール、2mMのEDTA、5%SDS)と混合し、そして、染色した。試料は5分間沸騰させた。20μlの試料を4%-12%PAGEゲル(NOVEX)に充填した。1×MESバッファー中でゲルを流した。発現はクマシーブルー染色により分析した。
【0085】
細菌細胞をフレンチプレスを使用して溶解し、そして細胞の溶菌液中の封入体を、低速の遠心分離によりペレットにした。細胞膜および細胞壁材料を含む夾雑物を除去するために、ペレット分画を2Mの尿素で洗浄した。それから、TACI-HSBP-1大腸菌(E.coli)の封入体を、0.1Mの硫酸ナトリウムおよび0.05Mのナトリウムテトラチオネートを含むpH8の50mMのトリス中の7MのグアニジンHCl中、4℃で撹拌しながら一昼夜抽出した。変性剤/亜硫酸分解剤を同時に伴う抽出物は、タンパク質-タンパク質相互作用を分解し、還元状態およびスルホン化状態において、タンパク質をスルフドリル基を伴う単量体に展開する。再度の折りたたみの前に、試料を35,000×g、4℃で30分間遠心分離し、そして0.2μmのフィルターでろ過した。濃度をRP HPLCアッセイにより測定した。
【実施例5】
【0086】
TACI融合タンパク質の生物学的アッセイ
zTNF4活性を制するための様々な3量体化TACI作成物の能力を試験するために、ヒトのTACIおよびKZ142ルシフェラーゼでトランスフェクションした Jurkat 細胞株を使用した。その Jurkat TACI KZ142ルシフェラーゼ細胞を成長させ、そして育成培地(L-グルタミン、ピルビン酸ナトリウム、0.5mg/mlのG418、および2μg/mlのプロマイシンを伴うRPMI/10%FBS)においてアッセイした。これらの細胞は100μlの育成培地において、40,000細胞/96ウェルで培養した。100ng/mlのzTNF4の存在下において、1ウェル当たり100マイクロリットルの3量体阻害因子を加えた。
【0087】
アッセイ細胞株は、1000ng/mlのzTNF4に対して最大約18倍のルシフェラーゼ応答性を示した。バックグランドのコントロールウェルと比較して、1ミリリットル当たり100ナノグラムのzTNF4は約10倍のルシフェラーゼ応答性を与えた。総量200μlの育成培地中のzTNF4および阻害物質の組み合わせを5%CO2インキュベータ中、37℃で6時間インキュベートした。それから、Beckman GS-6KR 遠心分離機において、96ウェルプレートを2000×gで遠心分離し、そして、プレートを反転させることにより培地を廃棄した。25マイクロリットルの溶解バッファー(Promega E153A)を各ウェルに加え、そして室温で15分間インキュベートした。それから、照度系を読むために、その溶解した細胞を不透明な96ウェルプレートに移動した。1本のルシフェラーゼアッセイバッファー(Promega 152A)を、1本のルシフェラーゼアッセイ基質(Promega E151A)に加え、そして40μlのこの組み合わせを各ウェルに加えた。5秒に統一して、照度計(EG&EG Berthold Microlumat Plus)により各ウェルを読んだ。
【0088】
図1に示すように、コントロール免疫グロブリン融合タンパク質(hwsx11-IgG)はzTNF4誘発ルシフェラーゼ活性においてほとんど有効性を持たなかった。TACI-Fc免疫グロブリン融合タンパク質(TACI-IgG)はルシフェラーゼ活性を阻害したが、より大きな阻害は、哺乳動物細胞または大腸菌(E.coli)中で生産されたTACI-HSBP-1タンパク質で達成された。
【図面の簡単な説明】
【0089】
【図1】図1は、TACI-Fc融合タンパク質(「TACI-IgG」)、哺乳動物細胞中で生産されたTACI-HSBP-1タンパク質(「TACI-HSBP(哺乳動物)」)、大腸菌(E.coli)中で生産されたTACI-HSBP-1タンパク質(「TACI-HSBP(大腸菌)」)による、並びに、免疫グロブリン融合タンパク質(「hwsx11-IgG」)の制御によるzTNF4誘発化ルシフェラーゼ活性の阻害を示す。
【図2】図2は、3H-チミジン、TACI-NC13量体およびTACI-Fc融合タンパク質(「TACI-Fc5」)の組み入れを通して、B細胞の増殖の阻害を示す。TACI-Fc5は、TACI-FcまたはTACI-IgGのもう1つの名前である。また、この図において示すものは2分子のEC50値である(すなわち、50%のコントロールの指標を阻害する濃度)。
【特許請求の範囲】
【請求項1】
(1)膜貫通性活性化因子かつCAML(カルシウム信号変調シクロフィリンリガンド)相互作用因子(TACI)の細胞外ドメイン、および(2)3量体化ポリペプチド、を含んで成る単離ポリペプチド。
【請求項2】
請求項1に記載のポリペプチドを含んで成るホモ3量体タンパク質の複合体。
【請求項3】
TACIの細胞外ドメインが、(1)SEQ ID NO:4のアミノ酸残基30から110、(2)SEQ ID NO:4のアミノ酸残基1から110、(3)SEQ ID NO:4のアミノ酸残基30から154、および(4)SEQ ID NO:4のアミノ酸残基1から154、から成る群から選択される、請求項1に記載の単離ポリペプチド。
【請求項4】
3量体化ポリペプチドが、ヒトのコラーゲンXのNC-1断片を含んで成る、請求項1に記載の単離ポリペプチド。
【請求項5】
3量体化ポリペプチドがSEQ ID NO:20のアミノ酸配列を含んで成る、請求項4に記載の単離ポリペプチド。
【請求項6】
TACIの細胞外ドメインがSEQ ID NO:4のアミノ酸残基30から110を含んで成る、請求項5に記載の単離ポリペプチド。
【請求項7】
請求項6に記載のポリペプチドを含んで成る、ホモ3量体タンパク質の複合体。
【請求項8】
3量体化ポリペプチドが、熱ショック結合タンパク質-1の3量体化断片である、請求項1に記載の単離ポリペプチド。
【請求項9】
3量体化ポリペプチドがSEQ ID NO:22のアミノ酸配列を有する、請求項8に記載の単離ポリペプチド。
【請求項10】
TACIの細胞外ドメインがSEQ ID NO:4のアミノ酸残基30から110を含んで成る、請求項9に記載の単離ポリペプチド。
【請求項11】
請求項10に記載のポリペプチドを含んで成る、ホモ3量体タンパク質の複合体。
【請求項12】
以下の機能するように結合した因子:
転写プロモーター;
請求項1に記載のポリペプチドをコードする核酸配列および
転写ターミネーター、
を含んで成る、発現ベクター。
【請求項13】
上記請求項に記載のポリペプチドを発現する、請求項12に記載の発現ベクターを導入した培養細胞。
【請求項14】
請求項13に記載の細胞の培養、および上記請求項に記載のポリペプチドを含んで成るホモ3量体タンパク質の複合体を回収する工程を含んで成る、ホモ3量体タンパク質の複合体の生産方法。
【請求項15】
(1)TACIの細胞外ドメインおよび(2)3量体化ポリペプチドを含んで成るポリペプチドを含んで成るホモ3量体タンパク質の複合体への、B細胞の暴露を含んで成る、TNF4誘発式B細胞増殖の阻害方法。
【請求項16】
上記請求項に記載のホモ3量体の複合体が請求項7に記載の複合体である、請求項15に記載の方法。
【請求項17】
上記請求項に記載のホモ3量体の複合体が請求項11に記載の複合体である、請求項15に記載の方法。
【請求項1】
(1)膜貫通性活性化因子かつCAML(カルシウム信号変調シクロフィリンリガンド)相互作用因子(TACI)の細胞外ドメイン、および(2)3量体化ポリペプチド、を含んで成る単離ポリペプチド。
【請求項2】
請求項1に記載のポリペプチドを含んで成るホモ3量体タンパク質の複合体。
【請求項3】
TACIの細胞外ドメインが、(1)SEQ ID NO:4のアミノ酸残基30から110、(2)SEQ ID NO:4のアミノ酸残基1から110、(3)SEQ ID NO:4のアミノ酸残基30から154、および(4)SEQ ID NO:4のアミノ酸残基1から154、から成る群から選択される、請求項1に記載の単離ポリペプチド。
【請求項4】
3量体化ポリペプチドが、ヒトのコラーゲンXのNC-1断片を含んで成る、請求項1に記載の単離ポリペプチド。
【請求項5】
3量体化ポリペプチドがSEQ ID NO:20のアミノ酸配列を含んで成る、請求項4に記載の単離ポリペプチド。
【請求項6】
TACIの細胞外ドメインがSEQ ID NO:4のアミノ酸残基30から110を含んで成る、請求項5に記載の単離ポリペプチド。
【請求項7】
請求項6に記載のポリペプチドを含んで成る、ホモ3量体タンパク質の複合体。
【請求項8】
3量体化ポリペプチドが、熱ショック結合タンパク質-1の3量体化断片である、請求項1に記載の単離ポリペプチド。
【請求項9】
3量体化ポリペプチドがSEQ ID NO:22のアミノ酸配列を有する、請求項8に記載の単離ポリペプチド。
【請求項10】
TACIの細胞外ドメインがSEQ ID NO:4のアミノ酸残基30から110を含んで成る、請求項9に記載の単離ポリペプチド。
【請求項11】
請求項10に記載のポリペプチドを含んで成る、ホモ3量体タンパク質の複合体。
【請求項12】
以下の機能するように結合した因子:
転写プロモーター;
請求項1に記載のポリペプチドをコードする核酸配列および
転写ターミネーター、
を含んで成る、発現ベクター。
【請求項13】
上記請求項に記載のポリペプチドを発現する、請求項12に記載の発現ベクターを導入した培養細胞。
【請求項14】
請求項13に記載の細胞の培養、および上記請求項に記載のポリペプチドを含んで成るホモ3量体タンパク質の複合体を回収する工程を含んで成る、ホモ3量体タンパク質の複合体の生産方法。
【請求項15】
(1)TACIの細胞外ドメインおよび(2)3量体化ポリペプチドを含んで成るポリペプチドを含んで成るホモ3量体タンパク質の複合体への、B細胞の暴露を含んで成る、TNF4誘発式B細胞増殖の阻害方法。
【請求項16】
上記請求項に記載のホモ3量体の複合体が請求項7に記載の複合体である、請求項15に記載の方法。
【請求項17】
上記請求項に記載のホモ3量体の複合体が請求項11に記載の複合体である、請求項15に記載の方法。
【図1】

【図2】
【図2】
【公表番号】特表2006−502715(P2006−502715A)
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願番号】特願2004−543793(P2004−543793)
【出願日】平成15年10月10日(2003.10.10)
【国際出願番号】PCT/US2003/032878
【国際公開番号】WO2004/033486
【国際公開日】平成16年4月22日(2004.4.22)
【出願人】(500049831)ザイモジェネティクス,インコーポレイティド (37)
【Fターム(参考)】
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願日】平成15年10月10日(2003.10.10)
【国際出願番号】PCT/US2003/032878
【国際公開番号】WO2004/033486
【国際公開日】平成16年4月22日(2004.4.22)
【出願人】(500049831)ザイモジェネティクス,インコーポレイティド (37)
【Fターム(参考)】
[ Back to top ]