
ホルモン療法耐性乳癌細胞株及びそれを用いた薬剤スクリーニング方法
【課題】ホルモン療法耐性乳癌細胞株、及びこれを用いた薬剤スクリーニング方法を提供する。
【解決手段】GFP遺伝子を発現しうるホルモン療法耐性乳癌細胞に被験物質を接触させる工程と、この細胞を培養し、細胞の増殖及び/又はGFPの発現を測定する工程と、前記被検物質が細胞の増殖及び/又はGFPの発現の低減をもたらしたかどうかを判定する工程とを含み、前記細胞が、エストロゲン非依存性、かつ、エストロゲンレセプター(ER)活性依存性である細胞株、エストロゲン非依存性、かつ、ER活性非依存性である細胞株、エストロゲン非依存性、ER活性非依存性、かつ、成長因子(GF)依存性であり、HER2及びEGFRを発現する細胞株、アンドロゲン代謝産物依存性である細胞株、及び/又はアロマターゼ阻害剤(AI)非感受性、かつ、ER活性依存性である細胞株、のいずれか1以上であることを特徴とする、乳癌治療剤スクリーニング方法。
【解決手段】GFP遺伝子を発現しうるホルモン療法耐性乳癌細胞に被験物質を接触させる工程と、この細胞を培養し、細胞の増殖及び/又はGFPの発現を測定する工程と、前記被検物質が細胞の増殖及び/又はGFPの発現の低減をもたらしたかどうかを判定する工程とを含み、前記細胞が、エストロゲン非依存性、かつ、エストロゲンレセプター(ER)活性依存性である細胞株、エストロゲン非依存性、かつ、ER活性非依存性である細胞株、エストロゲン非依存性、ER活性非依存性、かつ、成長因子(GF)依存性であり、HER2及びEGFRを発現する細胞株、アンドロゲン代謝産物依存性である細胞株、及び/又はアロマターゼ阻害剤(AI)非感受性、かつ、ER活性依存性である細胞株、のいずれか1以上であることを特徴とする、乳癌治療剤スクリーニング方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ホルモン療法耐性乳癌細胞株、及びそれを用いた乳癌治療剤のスクリーニング方法に関する。
【背景技術】
【0002】
エストロゲンは、乳癌の発生及び増殖に深く関係する女性ホルモンであり、乳癌細胞の多くはエストロゲン依存性に増殖するホルモン依存性腫瘍である。乳癌細胞がエストロゲンを受容すると、エストロゲンレセプター(ER)を介して細胞増殖に関わるシグナル伝達系が活性化され、細胞増殖が促進されて癌が進展する。
【0003】
乳癌患者の70%以上の乳癌はエストロゲンレセプター(ER)陽性であり、ホルモン療法の治療対象である。
【0004】
乳癌治療薬としてのホルモン剤は、大きく2種類に分類される。一つは、エストロゲンレセプターの阻害作用を示す薬剤(抗エストロゲン剤;タモキシフェン、トレミフェン、フルベストラント、ラロキシフェンなど)であり、もう一つは、エストロゲン合成酵素であるアロマターゼを阻害する薬剤(アロマターゼ阻害剤(AI);アナストロゾール、レトロゾール、エグゼメスタンなど)である。
【0005】
しかし、これらの薬剤はいずれも、やがて耐性を獲得されてしまうという問題がある。ホルモン療法を受けた乳癌患者のうち1/3は、癌がホルモン治療に耐性を獲得し、再発する。再発後は、化学療法が行われるが、予後が不良であり、治療が困難である。したがって、ホルモン療法耐性の獲得が、臨床上、重大な問題となっており、耐性機序の解明と新たな治療方法が望まれている。
【0006】
現在、ホルモン療法耐性機構として、(1)増殖因子シグナルカスケードの亢進、(2)HER2の発現増加によるシグナルカスケードの亢進、(3)エストロゲン非依存的なER活性化、(4)アンドロゲン代謝産物によるER活性化、(5)薬物代謝能や薬物排出能の亢進などが考えられている(非特許文献1:医学のあゆみ Vol.230(1)p.31-36(2009))。
【0007】
ホルモン療法耐性機序の解明及び新たな治療剤の開発のため、種々の試みが行われている。例えば、国際公開WO2006−129735号公報(特許文献1;「遺伝子導入細胞及び細胞分析法」)には、エストロゲン応答配列(ERE)を緑色蛍光タンパク質(GFP)遺伝子の転写開始点上流に配置した発現レポーター遺伝子を安定導入したER陽性乳癌細胞株を用いて、患者から採取した組織切片から、当該患者にとってアロマターゼ阻害剤(AI)が有効か否かを評価する方法が開示されている。非特許文献1(医学のあゆみ Vol. 230(1) p.31-36 (2009))には、エストロゲン枯渇条件下で細胞を培養し、エストロゲン枯渇耐性株を取得したことが開示されている。
【0008】
しかし、これらの細胞株の詳細な特徴及び挙動や、ホルモン療法耐性機序に基づく新たな治療剤の開発は記載されていない。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開WO2006−129735号公報
【非特許文献】
【0010】
【非特許文献1】医学のあゆみ Vol.230 (1) p.31-36 (2009)
【非特許文献2】Cancer Sci., vol. 100 (1), p.1773-1778 (2009)
【非特許文献3】Cancer Res, vol.65, p.4653-4662 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、乳癌のホルモン療法耐性について考えられている種々の耐性機序をカバーする細胞株を提供すること、及びこれらの細胞を用いたホルモン療法耐性細胞に効果的な薬剤のスクリーニング方法を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者は、GFP遺伝子を発現レポーターとして安定導入したER陽性乳癌細胞株を親株として、長期間のホルモン療法を模倣した培養方法でGFPを指標としてAI耐性株を得ることにより、異なる耐性機序を有する5種類のホルモン療法耐性乳癌細胞株を樹立した。
【0013】
したがって、本発明によれば、
〔1〕 ホルモン療法に耐性を示す乳癌の治療剤をスクリーニングする方法であって、エストロゲン応答配列の制御下で緑色蛍光タンパク質(GFP)遺伝子を発現しうるホルモン療法耐性乳癌細胞に被験物質を接触させる工程と、この細胞を培養し、細胞の増殖及び/又はGFPの発現を測定する工程と、前記被検物質と接触させていない前記ホルモン療法耐性乳癌細胞を同じ条件下で培養及び測定した結果と比較して前記被検物質が細胞の増殖及び/又はGFPの発現の低減をもたらしたかどうかを判定する工程とを含み、
前記ホルモン療法耐性乳癌細胞が、
エストロゲン非依存性、かつ、エストロゲンレセプター(ER)活性依存性である細胞株(タイプ1)、
エストロゲン非依存性、かつ、ER活性非依存性である細胞株(タイプ2)、
エストロゲン非依存性、ER活性非依存性、かつ、成長因子(GF)依存性であり、HER2及び上皮増殖因子レセプター(EGFR)を発現する細胞株(タイプ3)、
アンドロゲン代謝産物依存性である細胞株(タイプ4)、及び/又は
アロマターゼ阻害剤(AI)非感受性、かつ、ER活性依存性である細胞株(タイプ5)、
のいずれか1以上であることを特徴とする、乳癌治療剤スクリーニング方法;
〔2〕 前記ホルモン療法耐性乳癌細胞として、タイプ1〜タイプ5のうち2以上の細胞株を用いてそれぞれの細胞を別々に被験物質に接触させる、前記〔1〕記載のスクリーニング方法;
〔3〕 前記ホルモン療法耐性乳癌細胞が、
受領番号FERM AP−22142であるタイプ1細胞、
受領番号FERM AP−22143であるタイプ2細胞、
受領番号FERM AP−22144であるタイプ3細胞、
受領番号FERM AP−22145であるタイプ4細胞、及び/又は
受領番号FERM AP−22146であるタイプ5細胞、
である、前記〔1〕又は〔2〕記載のスクリーニング方法;
〔4〕 受領番号FERM AP−22142である細胞;
〔5〕 受領番号FERM AP−22143である細胞;
〔6〕 受領番号FERM AP−22144である細胞;
〔7〕 受領番号FERM AP−22145である細胞;
〔8〕 受領番号FERM AP−22146である細胞、
が提供される。
【発明の効果】
【0014】
本発明の細胞株によれば、ホルモン療法耐性乳癌の各種メカニズムに対して有効な治療方法の選択が可能となる。すなわち、本発明の5種類の細胞株は、乳癌のホルモン療法耐性の獲得に関して考えられる耐性機序のほとんどをカバーするものであり、想定されるAI耐性の機序を解明するツールとして有用である。また、本発明の細胞の増殖阻害やGFP活性阻害を指標にして、従来のホルモン療法耐性細胞又はホルモン療法耐性患者に効果的な薬剤のスクリーニング及び開発が可能となる。さらに、本発明の細胞は、乳癌患者の治療選択(薬物療法)の指針となりうる。具体的には、ホルモン療法耐性乳癌のなかには、他のホルモン療法剤が有効と考えられるケース、他のホルモン療法剤と分子標的薬との併用が有効と考えられるケース、ホルモン療法剤の効果は期待できないが分子標的薬又は化学療法が適切と考えられるケース等が混在している。したがって、ホルモン療法耐性乳癌の患者について、どのようなケースに該当するかを本発明の細胞を指標としてタイピングすることにより、各タイプに対して有効な治療法を選択することができる。
【図面の簡単な説明】
【0015】
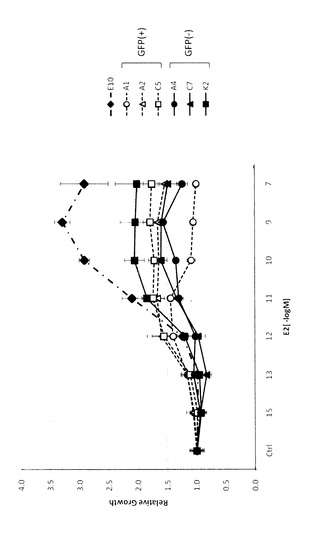
【図1】図1は、タイプ1及び2細胞のエストロゲン非依存性の増殖を表す図である。10%DCC−FCSを含有するRPMI1640培地で3日間培養した後、細胞を5×103個/ウェルの密度で24ウェルプレートに播き、種々の濃度のエストロゲンを含有するエストロゲン枯渇培地で4日間培養し、細胞数をカウントした。コントロールの細胞数に対する相対値(平均±SD)で示す。
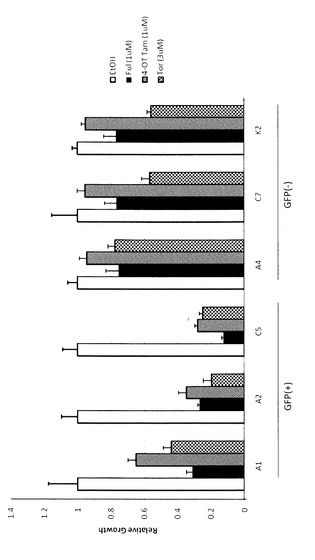
【図2】図2は、タイプ1及び2細胞に対する抗エストロゲン薬の効果を表す図である。10%DCC−FCSを含有するRPMI1640培地で培養した細胞に、各抗エストロゲン薬(1μM フルベストラント(Ful)、1μM 4−ヒドロキシタモキシフェン(4−OHT)又は3μM トレミフェン(Tor))又はコントロールとしてエタノール(EtOH)を添加して4日間培養した後、細胞数をカウントした。コントロールの細胞数に対する相対値で示す。
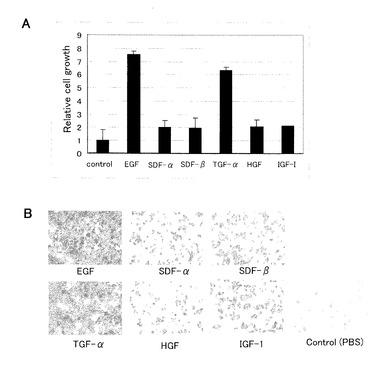
【図3】図3は、タイプ3細胞の増殖因子依存性を表す図である。パネルAは各増殖因子の存在下(100ng/mL)で10日間培養後の細胞数(PBSを添加したコントロールを1とした相対値;平均±SD)、パネルBはそれらの細胞のクリスタルバイオレット染色像を表す。
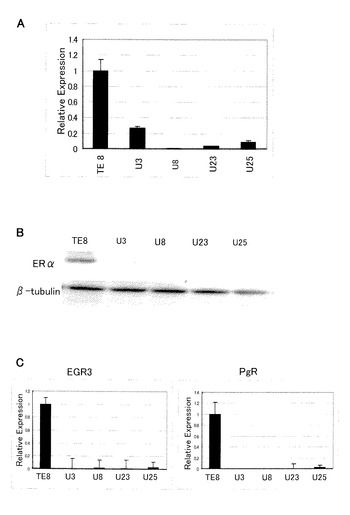
【図4】図4は、タイプ3細胞のER及びエストロゲン応答性遺伝子の発現を表す図である。TE8細胞はエストロゲン枯渇培地で3日間培養後エストロゲンを添加して1日培養したもの、タイプ3細胞は増殖因子培地で培養したものを用いた。パネルAはERαのリアルタイムRT−PCR(mRNA)、パネルBはERαのウェスタンブロッティング(タンパク)、パネルCはエストロゲン応答性遺伝子EGR3及びPgRのリアルタイムRT−PCR(mRNA)の結果をそれぞれ表す。
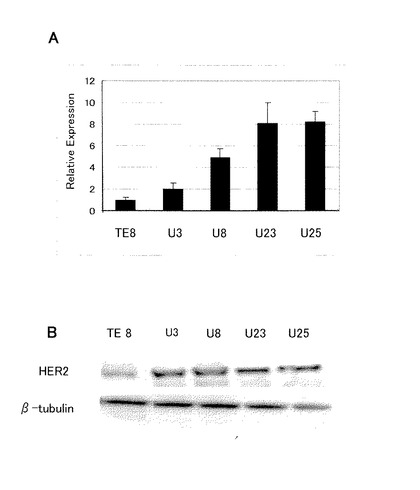
【図5】図5は、タイプ3細胞のHER2の発現を表す図である。パネルAはリアルタイムRT−PCR(mRNA)、パネルBはウェスタンブロッティング(タンパク)の結果をそれぞれ表す。
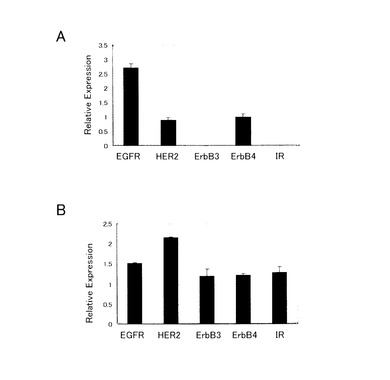
【図6】図6は、タイプ3細胞のヒトホスホRTKアレイキットによるリン酸化膜レセプター解析の結果を表す図である。パネルAはTE8細胞(普通培地)、パネルBはタイプ3(U25)細胞(増殖因子培地)のライセートを用いた結果を、それぞれキットの陽性コントロールに対する相対比で表す。
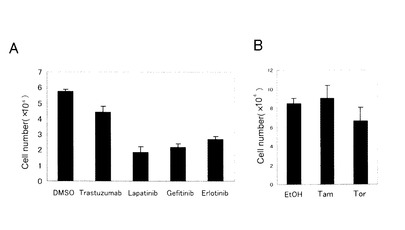
【図7】図7は、タイプ3細胞における分子標的薬及び抗エストロゲン薬の効果を表す図である。パネルA:細胞を1×104個/ウェルの密度で24ウェルに播き、増殖因子培地で培養した。抗HER2薬であるトラスツズマブ(150mg/mL)、抗EGFR及びHER2薬であるラパチニブ(5μM)、抗EGFR薬であるゲフィチニブ(5μM)、抗EGFR薬であるエルロチニブ(5μM)を、それぞれプレートに添加し、3日後に細胞数をカウントした。パネルB:細胞を1.5×104個/ウェルの密度で24ウェルに播き、増殖因子培地で培養した。タモキシフェン(1μM)、トレミフェン(3μM)を抗エストロゲン薬としてそれぞれプレートに添加した。データは平均±SD(n=3)で表す。
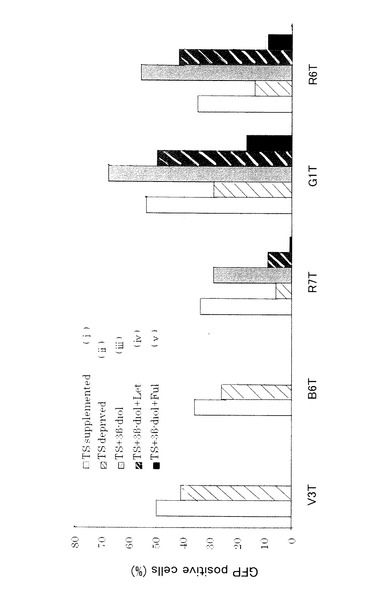
【図8】図8は、アンドロゲン代謝産物依存性及びエストロゲン枯渇耐性細胞の選択を表す図である。TS添加ステロイド枯渇培地で3ヶ月培養した後、GFPを発現する5個のクローンを選択した。各工程でGFP陽性細胞の比率によって評価したER活性を示す。V3T及びB6T細胞株は、ステロイド枯渇培地においてもER活性を示し、ERが構成的に活性化されていることが示された。R7TのER活性はレトロゾールによって抑制され、アロマターゼ依存性であることが示された。G1T及びR6TのER活性はステロイド枯渇培地で阻害されたが、レトロゾールによって阻害されなかった。これらの細胞はタイプ4(AD−EDR)細胞株として樹立された。
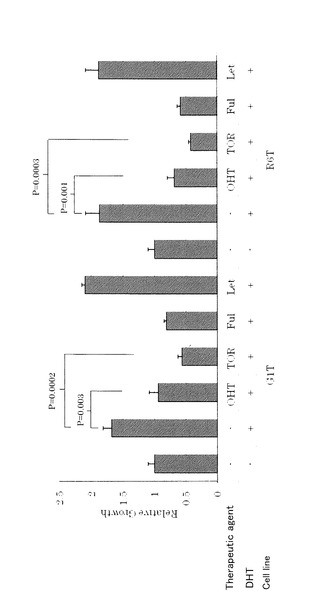
【図9】図9は、タイプ4細胞におけるDHT増殖誘導がSERMによってブロックされることを表す図である。タイプ4細胞においては、100nM DHTによる増殖誘導が100nM タモキシフェン(OHT)又は3μM トレミフェン(TOR)、又は1μM フルベストラント(ICI)によって効率的にブロックされたが、100nM レトロゾール(Let)によってはブロックされなかった。
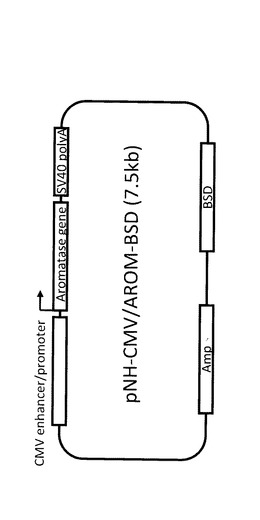
【図10】図10は、アロマターゼ発現ベクターpNH−CMV/AROM−BSDの構造を表す図である。
【図11】図11は、タイプ5細胞のレトロゾール添加時のER活性を表す図である。テストステロン(100nM)、レトロゾール(100nM)添加時のER活性をルシフェラーゼアッセイで測定した。
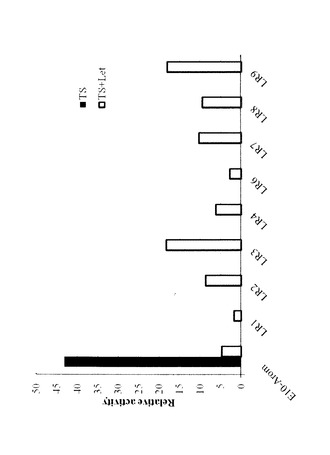
【図12】図12は、タイプ5細胞におけるレトロゾール感受性を表す図である。E10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、テストステロン(0.1〜100nM)、レトロゾール(100nM)を添加し、4日後に細胞数を測定した。
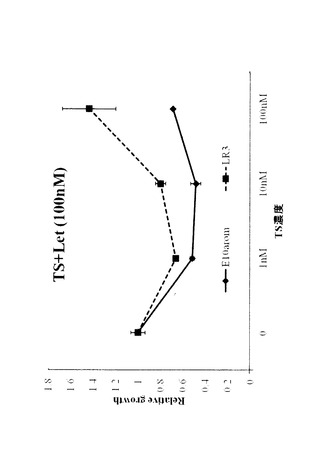
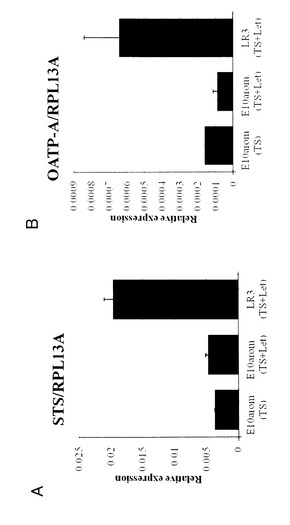
【図13】図13は、タイプ5細胞におけるエストロンスルフェート(E1S)代謝関連遺伝子の発現を表す図である。LR3において、E1SをESに変換する酵素であるSTS(パネルA)と、E1Sを細胞内に取り込むトランスポーターであるOATP−A(パネルB)のmRNA発現を、リアルタイムRT−PCRで測定した。
【図14】図14は、タイプ5細胞におけるE1S及びSTX64(STS阻害剤)感受性を表す図である。E10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、E1S(0、100nM〜10μM)単独あるいはSTX64(100nM)と組み合わせて添加し、4日後に細胞数を測定した。
【発明を実施するための形態】
【0016】
本発明の細胞株は、提唱されている各種のホルモン療法耐性メカニズムを表すものであり、タイプ1〜5に分けられる。
5種類の細胞株のうち、タイプ1、4、5はER発現株(GFP陽性)、タイプ2、3はER非発現株(GFP陰性)に分類される。
【0017】
ホルモン療法耐性細胞の作製
本発明の細胞株は、エストロゲン応答配列(ERE)の下流にレポーター遺伝子としてGFP遺伝子を含むベクターで形質転換した乳癌細胞を、ホルモン療法を模した特定の培養条件下で長期培養し、所望の性質を示すものを選択することにより作製することができる。
これらの細胞は、ERが活性化されるとGFPを発現して蛍光を発するため、細胞が生きたままの状態でERE活性を視覚的に捉えることができる。
本発明のタイプ1〜タイプ5のそれぞれの細胞の取得のための具体的手順は、実施例において詳述するとおりである。
【0018】
タイプ1細胞株
タイプ1細胞株(GFP陽性)は、ERの過剰発現及びいくつかのER標的遺伝子の構成的な高発現を示す。HER2発現の有意な誘導は示さない。総合的なリン酸化プロテオミクス解析により、タイプ1細胞では、細胞内リン酸化シグナル経路が動的変化をしていることがわかった。
抗エストロゲン薬により、タイプ1細胞株の生育は阻害される。
本発明のタイプ1細胞は、ERαの過剰発現を示し、エストロゲンに対し過敏性を示すが、エストロゲンに対する細胞増殖依存性は親細胞よりもはるかに低い。ERαの過剰発現は、タイプ1細胞の生存のためにエストロゲン非依存性を補完すると思われる。
さらに、MAPキナーゼ経路の有意な誘導が観察された。タイプ1(GFP陽性)細胞は、細胞間リン酸化経路の変化が示唆されており、特に、PKA経路は、エストロゲン非依存性ER活性化に重要と考えられる。
【0019】
タイプ2細胞株
タイプ2細胞株(GFP陰性)は、ER及びその標的遺伝子の顕著なダウンレギュレーションを示す。さらに、細胞内シグナル伝達因子c−Jun N末端キナーゼ(JNK)の構成的活性化が観察される。タイプ2細胞株については、MAPキナーゼ経路の有意な誘導が観察された。
【0020】
抗エストロゲン薬により、タイプ2細胞株の生育は阻害されない。一方、タイプ2細胞株では、JNK阻害剤又はEGFレセプター(EGFR)標的薬により生育が阻害される。JNK経路は、ホルモン療法耐性乳癌の新規な治療標的でありうる。
【0021】
タイプ2細胞は、エストロゲン非依存性及びER非依存性増殖メカニズムを獲得した細胞である。これらの細胞株は、リン酸化JNKの強い誘導及びAP-1転写活性の有意な活性化を示した。JNK経路は、種々の癌の増殖及び発展に重要な役割を果たすことが報告されているので、これらの細胞のエストロゲン及びER非依存性は、このシグナル経路の構成的活性化によって獲得された可能性がある。
【0022】
タイプ1及びタイプ2についてのこれらの結果は、ER陽性乳癌細胞が、ER依存性及び非依存性の複数のメカニズムによってエストロゲン枯渇耐性を獲得しうることを示す。
【0023】
タイプ3細胞株
タイプ3細胞株は、エストロゲン枯渇培地では生存できず、増殖因子(特にEGF及びTGF−α)を添加したエストロゲン枯渇培地で生存することができる。当初はGFP蛍光を強く発していたが、短期間のうちにGFP発現を消失した。また、ER発現も劇的に低下し、機能を失った。さらに、増殖因子レセプターによる細胞内シグナル伝達は、PI3キナーゼ(PI3K)/Akt及びJNK経路の分子を増大させた。
タイプ3細胞は、EGFR標的薬により有意に増殖が阻害される。これらの結果から、タイプ3細胞は、エストロゲン非依存性、かつ増殖因子依存性の細胞増殖メカニズムを獲得したことが示唆される。さらに、細胞内シグナル経路は、タイプ3細胞においては、親細胞であるTE8と比較して変化している。
【0024】
タイプ4細胞株
タイプ4細胞株は、アンドロゲン代謝産物依存性、かつエストロゲン枯渇耐性である。タイプ4細胞株は、アンドロゲンからの局所的エストロゲン産生を再現するストローマ細胞(間質細胞)との共培養においてレトロゾール(Let)耐性ER活性を示し、AI耐性モデルとして使用することができる。タイプ4細胞株は、アンドロゲン又は3β−ジオールの存在下で増大したER活性及び増殖を示し、これはレトロゾールによって阻害されない。リアルタイムPCR解析によって、タイプ4細胞におけるジヒドロテストステロン(DHT)代謝酵素3β−ヒドロキシステロイドデヒドロゲナーゼタイプ1(3β−HSDタイプ1:HSD3B1)のmRNA発現の増大及びアンドロゲンレセプター(AR)発現の低減が示された。さらに、タイプ4細胞においては、アンドロゲン又は3β−ジオールによってエストロゲン応答性遺伝子PgR及びEGR3の発現が誘導されるが、アンドロゲン応答性遺伝子KLK3は誘導されない。これらのデータから、増大したDHT代謝及びARシグナルの中断がアロマターゼ阻害剤によって誘導されるエストロゲン枯渇及びアンドロゲン過剰条件に対する適応性を与えることが示唆される。
【0025】
アンドロゲン又は3β−ジオールによって誘導されるER活性の解析により、タイプ4細胞株の3β−ジオールに対する過敏性及びアンドロゲン耐性が示された。mRNA発現の定量的解析により、タイプ4細胞における増大したDHT代謝及びアンドロゲンシグナルの中断のため、アンドロゲン又は3β−ジオールがアロマターゼ非依存的に、よりエストロゲニックな挙動を示すことが示唆された。言い換えれば、エストロゲン性アンドロゲン代謝産物による低減したARシグナル及び増大したERシグナルは、アロマターゼ活性に依存せずに、エストロゲン枯渇条件への適合性をこれらの細胞株に与えうる。タイプ4細胞株に対する、3β−HSDタイプ1の特異的阻害剤であるトリロスタンの有効性は認められなかった。3β−HSDタイプ1は、DHTを3β−ジオールに代謝する唯一の酵素ではないため、DHTは他の酵素で代謝され、トリロスタンによってその活性が抑制される3β−HSDタイプ1が補償されると考えられる。タイプ4細胞についてのインビトロでのデータは、アンドロゲン又はその代謝産物によって誘導されるER活性化及びアンドロゲンシグナル中断がAI耐性において重要な役割を果たすこと、及びAR又はアンドロゲン代謝酵素の発現レベルが臨床的乳癌におけるAI耐性の生物学的マーカーとして有用であることを示唆する。
【0026】
ER依存性を維持するいくつかのクローンがAI処理を模した条件下で樹立され、これらのタイプ4細胞のDHTによる増殖誘導は選択的エストロゲンレセプター調節剤(SERM)によって効率的に阻害されるが、レトロゾールによっては阻害されない。これらの知見から、AIの後にSERMを連続して使用することの有効性が支持される。
【0027】
タイプ4細胞株は、インビトロでのAI耐性のメカニズムを研究するために有用であり、AI耐性進行乳癌についての診断マーカー及び新規治療戦略の開発に有用である
【0028】
タイプ5細胞株
タイプ5細胞株は、エストロゲン依存性、かつER依存性のレトロゾール(Let)耐性細胞である。親細胞であるMCF―7―E10aromは、テストステロン存在下でER活性及び増殖を示し、これがLetによって阻害されるが、タイプ5細胞株は阻害されない。タイプ5細胞株は、エストロン(Estrone;E1)スルフェート(E1S)に対する感受性が親細胞よりも高く、またリアルタイムPCR解析によって、E1Sを細胞内に取り込むトランスポーターであるOATP―A、及びE1SをE1に変換する酵素であるステロイドスルファターゼ(Steroid sulfatase;STS)のmRNAの発現の増大が示された。さらに、E1Sによって誘導される細胞増殖は、STS阻害剤であるSTX64により阻害されることが確認された。これらのデータから、タイプ5細胞株はSTSやOATP―Aの発現を増大させ、アロマターゼ非依存的にエストロゲンを産生する経路を亢進することで、Let耐性を獲得していることが示唆される。また、タイプ5細胞株についてのインビトロでのデータは、STSやOATP―Aによるエストロゲン産生の増大がAI耐性において重要な役割を果たすこと、及びSTS阻害剤のAI耐性患者への有用性や、STSやOATP―Aの発現レベルが臨床的乳癌におけるAI耐性の生物学的マーカーとして有用であることを示唆する。
【0029】
本発明の細胞株のうち、以下のものは、2011年7月8日付けで独立行政法人 産業技術総合研究所 特許微生物寄託センター(茨城県つくば市東1丁目1番地1 つくばセンター中央第6)に寄託された。
タイプ1:クローンA2(受領番号FERM AP−22142)。
タイプ2:クローンA4(受領番号FERM AP−22143)。
タイプ3:クローンU25(受領番号FERM AP−22144)。
タイプ4:クローンG1T(受領番号FERM AP−22145)。
タイプ5:クローンLR3(受領番号FERM AP−22146)。
【0030】
スクリーニング方法
本発明の細胞株を用いて、ホルモン療法耐性乳癌に対して有効な治療剤をスクリーニングすることができる。本発明のスクリーニング方法は、本発明の細胞株に被験物質を接触させる工程と、細胞を培養する工程と、細胞の増殖及び/又はGFPの発現を測定する工程とを含む。その結果、前記被検物質と接触させていない以外は同様に処理した本発明の細胞株と比較して細胞の増殖及び/又はGFPの発現が低減していれば、その被検物質は細胞増殖を抑制したと判定することができる。
【0031】
細胞と被検物質との接触は、培地に被検物質を添加することにより容易に行うことができる。被検物質との接触は、短時間のパルスで行ってもよく、培養期間中を通じて培地に存在させてもよい。したがって、接触と培養とは同時に行うことができる。培地としては、必要に応じて以下に説明する普通培地、エストロゲン枯渇培地、又はこれらに所定の添加物を添加した培地等を使用することができる。細胞の増殖は、常法により細胞数をカウントすること等により、行うことができる。GFPの発現は、例えば、蛍光顕微鏡下で蛍光を発する細胞の比率を測定すること等により測定することができる。
【0032】
スクリーニングに用いる本発明の細胞株としては、タイプ1〜タイプ5のいずれか1以上のタイプであればよいが、2以上を用いて各タイプに対する同じ被検物質の効果の差を調べることができる。また、2以上を用いていずれか1つのタイプにのみ有効な被検物質を選択することができる。
【0033】
本発明の細胞は、上述のようにしてインビトロでの薬剤スクリーニングに用いることができるのに加えて、これらの細胞を動物に移植してそれぞれの機序のモデル動物を作製し、より個体に近い実験系で薬剤の効果を評価するために使用することができる。
【0034】
また、新規な薬剤を見出すことだけでなく、既存薬剤の投与順序や併用の組合せ等に関しても、最適な方法を見出すために、上記と同様のインビトロ又はインビボ実験を行うことができる。
【0035】
治療選択法
また、本発明の細胞は、個々の患者に最適な治療選択を可能にするための情報を提供することができる。具体的には、ホルモン療法耐性乳癌のなかには、他のホルモン療法剤が有効と考えられるケース、他のホルモン療法剤と分子標的薬との併用が有効と考えられるケース、ホルモン療法剤の効果は期待できないが分子標的薬又は化学療法が適切と考えられるケース等が混在していると考えられる。したがって、ホルモン療法耐性乳癌の患者について、どのようなケースに該当するかを本発明の細胞を指標としてタイピングすることにより、各タイプに対して有効な治療法を選択することができる。例えば、タイプ1についてはリン酸化ER、タイプ2についてはpJNK、タイプ3についてはHER2、タイプ4についてはAR、及びタイプ5についてはSTSといったタンパク又は遺伝子の発現を指標として患者から採取した乳癌組織片がどの対応に該当するかを例えば免疫染色等によってタイピングし、該当したタイプの細胞に対して有効性の高い治療法をその患者の最適な治療法として選択することができる。
【実施例】
【0036】
I.ホルモン療法耐性乳癌細胞株の樹立
1.樹立に使用した親細胞
親細胞として、pd2EGFP−1ベクター(Clontech)において半減期の短いGFPのcDNAの上流にエストロゲン応答配列(ERE)を配したレポータープラスミド(特許文献1)を安定的に導入したER陽性ヒト乳癌細胞株MCF−7−E10細胞(「E10細胞」と呼ぶことがある;特許文献1)を、タイプ1、2、4細胞株の樹立のために用いた。同じプラスミドを導入したER陽性ヒト乳癌細胞株T47D−TE8細胞(「TE8細胞」と呼ぶことがある)を、タイプ3細胞株の樹立のために用いた。TE8細胞は以下のIII.に記載する方法で作製した。
タイプ5細胞株の樹立のための親細胞としては、以下のV.に記載するとおりに作製したE10−Aromを用いた。
【0037】
2.細胞培養法
細胞の通常の培養には、10%FCS(Tissue Culture Biologicals)及び1%ペニシリン/ストレプトマイシン(GIBCO)を添加したRPMI1640培地(SIGMA)(「普通培地」と呼ぶことがある)を用いた。
耐性株のエストロゲン枯渇培養には、10%デキストラン被覆チャコール処理FCS(DCC−FCS;Tissue Culture Biologicals)および1%ペニシリン/ストレプトマイシン(GIBCO)を添加したフェノールレッド無含有RPMI1640培地(GIBCO)(「枯渇培地」又は「エストロゲン枯渇培地」と呼ぶことがある)を用いた。
【0038】
細胞は、すべて5%CO2、37℃に調整したCO2インキュベーターで培養した。
【0039】
特に記載がない場合、実験結果は、平均±SD(n=3)で表す。
【0040】
3.試薬等
エストラジオール、タモキシフェン(4−ヒドロキシタモキシフェン(4-hydroxytamoxifen))、テストステロン、DHT、3β−ジオール、デキサメタゾンは、Sigma-Aldrich Corpから購入した。抗ER抗体(H−184)は、Santa Cruz Biotechnology Inc.、抗ヒトerbB2抗体はDako Cytomationからそれぞれ入手した。抗EGFR (V279)、抗Akt、抗リン酸化Akt (Thr308)、抗SAPK/JNK、抗44/42 MAPK (Erk1/2)、抗リン酸化44/42 MAPK(Erk1/2) (Thr202/Tyr204)、抗リン酸化C−Raf (Ser259)、抗β−チューブリン抗体は、Cell Signaling Technologyから入手した。
トラスツズマブ(Trastuzumab ;商品名「ハーセプチン」)及びエルロチニブ(erlotinib;商品名「タルセバ」)は中外製薬 (Tokyo, Japan)から、ラパチニブ(lapatinib)はGlaxoSmithKline plc. (Brentford, UK)から、ゲフィチニブ(gefitinib;商品名「イレッサ」)はアストラゼネカ(Osaka, Japan)から、トレミフェン(toremifen)は日本化薬(Tokyo, Japan)から、それぞれ入手した。レトロゾールはノバルティスファーマ(Tokyo, Japan)から、フルベストラントはアストラゼネカから、それぞれ入手した。トリロスタン(3β−HSDタイプ1阻害剤)はTronto Research Chemicals(North York, Canada)から、及びバイカルタミド(アンドロゲンレセプター阻害剤)はLKT Laboratories(St. Paul, MN, USA)から、それぞれ入手した。
【0041】
4.リアルタイムRT−PCR
それぞれの条件下で培養した細胞から商品名「Isogen」(Nippon Gene)を用いて総RNAを抽出し、商品名「RNA PCR kit」(Takara Shuzo)を用いて第1鎖cDNAを合成した。リアルタイムRT−PCRは、商品名「StepOne real-time PCR system」(Applied Biosystems)を用いて標準プロトコールにしたがって行った。目的の遺伝子の発現を、内部標準としてのGAPDH又はRPL13Aに対する相対値として算出した。データは、平均±SD(n=3)で示した。
【0042】
5.ウェスタンブロッティング
100mmディッシュで増殖させた細胞を氷冷生理食塩水で洗浄し、ホスファターゼ阻害剤カクテル(「Phos STOP」、Roche)及びプロテアーゼ阻害剤カクテル(Roche)を含む1mLのリシスバッファー(「Complete Lysis-M」、Roche Diagnotics GmbH)で溶解させた。5分後、ライセートをパルスソニケーションに供し、14,000rpmで10分間遠心分離した後、10%ポリアクリルアミドゲル(Wako)でSDS−PAGEを行い、タンパク質をPVDF膜(GE Healthcare Bioscience)に転写した。この膜を、一次抗体(5%BSA、0.05% Tween−20含有中)と反応させた。次に、西洋ワサビ結合二次抗体を適用した。化学発光基質(「Immunsutar AP substrate」、 BIO-RAD)と反応させた後、目的のタンパクのバンドを、X線フィルムを感光させることにより可視化した。
【0043】
6.ルシフェラーゼアッセイ
ERE活性を、商品名「Dual-Luciferase Reporter System」(Promega)を用いて、基本的にBiochem Biophys Res Commun 2001, 285:340-347に記載された方法に従って測定した。枯渇培地で3日間培養後、5×104個の細胞を6cmディッシュに播き、同じ培地中で48時間培養した。0.5μgのエストロゲンレポータープラスミド(Tk−ERE−Luci)及び0.05μgのコントロールベクター(pRL―TK, Promega)を300μLの無血清培地中で5μLの商品名「TransIt」LT-1試薬(Takara Bio)と混合し、製造業者の指示書に従ってトランスフェクションを行った。薬剤の存在下又は非存在下で24時間細胞を培養した後、商品名「Dual-Luciferase Reporter System」(Promega)を製造業者の指示書に従って用いてルシフェラーゼ活性を測定し、コントロールに対する相対値を算出した。
【0044】
II. タイプ1細胞株及びタイプ2細胞株の作製及び特徴づけ
1.タイプ1細胞株及びタイプ2細胞株の樹立
普通培地で培養していたE10細胞を枯渇培地に播種し、長期的にエストロゲン枯渇培養した。大部分の細胞は増殖が止まり死に至ったが、一部の細胞は、エストロゲン枯渇耐性を獲得し、増殖してコロニーを形成した。これを蛍光顕微鏡で観察し、より強い蛍光を発しているもの(タイプ1細胞株;クローンA1、A2、C5)と、蛍光を消失しているもの(タイプ2細胞株;クローンA4、C7、K2)とを選択し、顕微鏡下でマイクロピペットを用いて剥離して、96ウェル培養プレートに単離した。その後、24ウェル培養プレート、6cmディッシュ、10cmディッシュ、と段階的に大きな培養容器に移しつつ細胞数を増やし、継続的に枯渇培養を続け、安定的なエストロゲン枯渇耐性細胞株(タイプ1、タイプ2を総称して、「E10−EDR細胞」(estrogen depletion resistant cells)と呼ぶことがある)を樹立した。
【0045】
2.細胞増殖におけるエストロゲン依存性
親株であるE10細胞(枯渇培地で3日間培養し、ステロイドを枯渇させたもの)及びE10−EDR細胞を、5×103細胞/ウェルの密度で24ウェル培養プレートに播き、種々の濃度のエストロゲンの存在下及び非存在下で培養した。4日後、細胞をPBSで1回洗浄し、トリプシン/EDTA処理によって回収した。細胞をパーティクルカウンター(「CDA-500 Sysmex automated cell counter」(Sysmex Corporation)でカウントした。
【0046】
結果を、第1日における細胞数に対する相対値で示す(図1)。タイプ1(A1、A2、C5)、タイプ2(A4、C7、K2)ともに、親株(E10)と比較して細胞増殖におけるエストロゲン(E2)に対する依存性が明らかに低下していた。
【0047】
3.タイプ1及びタイプ2細胞株に対する抗エストロゲン剤の効果
タイプ1及びタイプ2細胞の細胞増殖に対する抗エストロゲン剤の影響を検討した。枯渇培地で培養した細胞に、各抗エストロゲン薬(1μM フルベストラント(Ful)、1μM 4−ヒドロキシタモキシフェン(4−OT Tam)又は3μM トレミフェン(Tor))又はコントロールとしてエタノール(EtOH)を添加して4日間培養した後、細胞数をカウントした。
【0048】
結果を図2に示す。タイプ1細胞(A1、A2及びC5)は、エストロゲン非依存性であるが、ERをブロックする薬剤である抗エストロゲン薬(タモキシフェン、トレミフェン、フルベストラント)によって増殖が阻害された。一方、これらの抗エストロゲン薬は、タイプ2細胞(A4、C7及びK2)に対しては、トレミフェンがC7及びK2の増殖を少し阻害したものの、顕著な増殖抑制効果を示さなかった。
【0049】
これらの結果は、ホルモン療法のための抗エストロゲン薬はタイプ1細胞には有効であるが、タイプ2細胞にはあまり有効ではないことを示す。また、タイプ1細胞においては、抗エストロゲン剤フルベストラントの添加によりGFP活性が強力に阻害されることから、これらのGFP発現においてERαが大きく寄与することが示唆された。
【0050】
4.タイプ1細胞及びタイプ2細胞のその他の特徴
ER活性は、タイプ1細胞において高く、タイプ2細胞において低かった。リアルタイムPCRにより、内在性ERα標的遺伝子の発現を調べたところ、タイプ1細胞(A1、A2及びC5)は実質的にpS2、PgR、IGFBP4及びEGR3を発現したが、タイプ2細胞(A4、K2及びC7)は、これらのER標的遺伝子のmRNAの有意な発現を示さなかった。したがって、タイプ1細胞においては、エストロゲンに依存してER転写活性が構成的に活性化されているが、タイプ2細胞においては、ER転写活性がほとんど失われている。
ERαのmRNA及びタンパク質は、タイプ1細胞においては強く誘導されていたが、タイプ2細胞においてはほとんど認められなかった。タイプ1細胞において、ERαのSer118、Ser167、及びSer305でのリン酸化の顕著な変化は認められなかった。
いずれの細胞株も、HER2発現についてはアップレギュレーションが認められなかった。
【0051】
細胞内リン酸化経路に関与する代表的なタンパク質のウェスタンブロットを行い、タイプ2細胞株(A4及びK2)においてJNKが構成的に活性化されていることがわかった。A4及びK2のAP−1ルシフェラ−ゼ活性は、E10細胞よりも有意に高かった。
【0052】
さらに、JNK阻害剤(SP600125)は、タイプ1細胞よりもタイプ2細胞においてより効果的に増殖を阻害した。これらの結果から、タイプ2細胞においてはJNK関連シグナル経路が生存に重要であることが示唆された。
【0053】
III. タイプ3細胞株の作製及び特徴づけ
1.親株(T47D−TE8細胞)の樹立
T47D−TE8細胞は、ヒト乳癌細胞株T47D(ATCC番号:HTB−133)から、E10細胞の作製に用いたものと同じERE−GFP遺伝子を担持するプラスミドを公知の方法にしたがって導入することにより樹立した(非特許文献3:Yamaguchi Y. et al., Cancer Res, 2005, 65: 4653-4662)。
T47D−TE8細胞は、普通培地で培養した。
【0054】
2.タイプ3細胞株の樹立
エストロゲン枯渇条件下での長期培養によりT47D−TE8細胞から取得した細胞は、ホルモン療法で治療された乳癌のモデルを表す。
親株としてT47D−TE8細胞を用いて、上記IIの1.と同様の方法により、タイプ3細胞株を樹立した。ただし、T47D−TE8細胞は、通常の枯渇培地ではすべて死滅してしまうため、培養時は常に増殖因子カクテル(20ng/ml CXCL12/SDF−1α、20ng/ml CXCL12/SDF−1β、20ng/ml EGF、20ng/ml HGF、20ng/ml IGF−1、20ng/ml TGF−α(R&D systems))を添加した培地(増殖因子培地)を用いて3ヶ月以上培養した。
【0055】
T47D−TE8細胞は、エストロゲン枯渇培地では死滅したが、増殖因子培地では生存し、いくつかのコロニーを形成した。強力なER活性を有するものを得るために、より強い蛍光を発するコロニーを選択し、さらに7〜18週間、増殖因子培地で個別に培養した。この過程で、一部のコロニーは死滅し、最終的に31個のコロニーが得られた(タイプ3細胞;「TE8−EDR細胞」と呼ぶことがある)。これらの細胞は、当初は強力な蛍光を発したが、その後6〜8週間のうちに蛍光を失った。したがって、ER活性を失ったことが示唆された。増殖因子のないエストロゲン枯渇培地で得られ、ER活性を維持したEDR細胞を考慮すると、TE8−EDR細胞は、増殖因子の添加によってER活性を失ったと考えられる。
【0056】
タイプ3細胞として最終的に得られた31個のクローンのうち、U3、U8、U23及びU25細胞の特徴を解析した。
【0057】
3.タイプ3細胞の増殖因子依存性
各増殖因子(100ng/mL)を個別に含むエストロゲン枯渇培地で10日間培養することにより、タイプ3細胞(クローンU25)が増殖因子カクテルに含まれるどの増殖因子に依存しているかを調べた。
【0058】
結果を図3に示す。タイプ3細胞の増殖は、EGFとTGF−αに依存していることがわかった。細胞数は、EGF及びTGF−αを添加した場合に、それぞれコントロールの7.5倍及び6.4倍に増大し、他の増殖因子(SDF−α、SDF−β、HGF、IGF−1)を添加した場合には2倍となった(パネルA)。クリスタルバイオレット染色によっても、EGF及びTGF−αによる細胞数の増大が顕著であることが示された(パネルB)。EGF及びTGF−αは、同じレセプター(EGFR/HER1)を利用して増殖因子のシグナルを細胞内システムに伝達すると考えられている。したがって、タイプ3細胞の生存及び増殖にEGFRが関与することが示された。
【0059】
4.タイプ3細胞におけるER及びHER2の発現
タイプ3細胞においてはERの発現が低下し、乳癌で重要な治療の標的となっているHER2の発現が上昇していることを以下のようにして確認した。
タイプ3細胞におけるERα及びエストロゲン応答性遺伝子の発現を、リアルタイムRT−PCRにより調べた。
【0060】
結果を図4に示す。通常培地のTE8親細胞と比較して、タイプ3細胞(U3、U8、U23、U25)のERαのmRNA発現は、顕著に低減していた(パネルA)。ウェスタンブロッティングにより検出したところ、タイプ3細胞のERαタンパク質発現も低減していた(パネルB)。さらに、エストロゲン応答性遺伝子EGR3及びPgRのmRNAも低減していた(パネルC)。
【0061】
したがって、タイプ3細胞は、ER発現及び機能的活性を失っており、その生存及び増殖はエストロゲン非依存性であると考えられる。
【0062】
次に、EGFRは他のHERファミリーレセプターと2量体を形成するため、同様にHER2の発現を調べた。HER2過剰発現は、乳癌の20〜30%で観察されるため、乳癌の診断、サブタイピング、治療に重要な因子である。
【0063】
結果を図5に示す。タイプ3細胞(U3、U8、U23、U25)におけるHER2 mRNAは、TE8親細胞と比較して有意に増大していた(パネルA)。HERタンパク質の発現も増大していた(パネルB)。これらの結果は、HER2が、EGFRと協働してシグナル伝達する増殖因子により増大したことを示唆する。
【0064】
5.ホスホ−RTKアッセイによる解析
EGFRは種々の膜レセプターと2量体を形成するため、さらにヒトホスホ−RTKアレイアッセイキット(Human Phospho-RTK Array Kits (R&D Systems))を用いて、細胞膜レセプターの網羅的発現解析を行った。サンプルとして、増殖因子培地で培養したタイプ3細胞及び枯渇培地で3日間の後エストロゲンを添加して1日培養した親細胞(TE8細胞)を用いた。このキットは、42項目の膜型レセプターに対する一次抗体が固定されているメンブレンに細胞ライセートを結合させ、細胞に存在するこれらのリン酸化されたレセプターチロシンキナーゼ二次抗体で検出する。
【0065】
結果を図6に示す。タイプ3細胞株におけるHERファミリー全体の発現増加がみられた。
TE8親細胞において、EGFR、HER2及びErbB4のリン酸化レセプターが検出された(パネルA)。これらのレセプターに加えて、タイプ3細胞(クローンU25)では、リン酸化されたErbB3及びインスリンレセプターが検出された(パネルB)。この結果は、タイプ3細胞が、さらなるレセプターを利用して種々の増殖因子依存性経路を確保する可能性を示す。
【0066】
6.タイプ3細胞の増殖に対する分子標的薬及び抗エストロゲン薬の効果
タイプ3細胞は増殖因子依存性が高く、EGFR及びHER2が増殖において重要な役割を果たすので、HER2及び/又はEGFRに対する分子標的薬の細胞増殖に対する効果を調べた。また、抗エストロゲン薬の効果も同様に調べた。
【0067】
増殖因子培地において培養したタイプ3細胞(クローンU25)に、薬剤を加え、3日間培養した。次に、細胞数をカウントした。薬剤として、150mg/mlトラスツズマブ(trastuzumab;抗HER2薬)、5μMラパチニブ(lapatinib;抗EGFR及びHER2薬)、5μMゲフィチニブ(gefitinib;抗EGFR薬)、5μMエルロチニブ(erlotinib ;抗EGFR薬)をそれぞれ用いた。陰性コントロールとしてDMSOを添加した。抗エストロゲン薬であるタモキシフェン及びトレミフェンは、それぞれ1μM及び3μMで用いた。
【0068】
結果を図7に示す。トラスツズマブは、HER2陽性乳癌に臨床的に使用される抗HER2薬であり、臨床試験では3年生存率を改善した。ラパチニブは、EGFR及びHER2を共に標的とする阻害剤であり、乳癌の新薬として期待されている。ゲフィチニブ及びエルロチニブは、EGFRを標的とする薬剤である。パネルAに示すように、EGFRを標的とする薬剤、ラパチニブ、ゲフィチニブ、エルロチニブは、細胞増殖を有意に阻害したが、トラスツズマブの効果はそれより劣っていた。タモキシフェン及びトレミフェンの効果も同様に調べたが、これらの薬剤は効果を示さなかった(パネルB)。これらの結果から、EDR細胞の増殖はEGFRを阻害することにより有意に低減するが、HER2又はER機能の阻害によっては低減せず、EGFRが増殖に重要であることがわかった。
【0069】
結果として、タイプ3細胞は、容易にエストロゲン依存性経路を失い、エストロゲン非依存性かつER非依存性経路を獲得したことが示唆された。タイプ3細胞における細胞内シグナル経路は、親細胞であるTE8におけるそれと異なっており、親細胞がエストロゲン依存性であって主にエストロゲン依存性経路を使うのに対し、タイプ3細胞は、エストロゲン依存性経路を失い、増殖因子依存性経路を使うようになっている。この経路は、主にEGFR及びHER2のような増殖因子レセプターに関与し、PI3K経路が特に重要である。
【0070】
IV. タイプ4細胞株の作製及び特徴づけ
1.タイプ4細胞株の樹立
上記II.のタイプ1細胞の樹立法と同様の手順を、100nMテストステロン(TS)を添加した枯渇培地で行い、E10細胞から複数の安定株を樹立した。GFP発現による蛍光を示すコロニーをピックアップして個別に培養した。このうち、純粋な枯渇培地で蛍光が低下し、テストステロン100nM及び3β−ジオール(Androgen metabolites 5α-androstane-3β, 17β-diol)100nMの添加で再び蛍光を発し、その後のレトロゾール100nMの添加によって蛍光が減弱しないものを、アンドロゲン代謝産物依存性エストロゲン枯渇耐性細胞株(「AD−EDR」(Androgen metabolite dependent EDR cells)と呼ぶことがある)(タイプ4細胞)とした。
【0071】
以下の条件下でER活性の評価を行った。各クローンのER活性は、培養中のGFP陽性細胞の比率によって評価した。まず、ステロイド枯渇TS添加培養におけるER活性を評価した(i)。次に、ステロイド枯渇培養で3日間培養後、同様にER活性を評価し、構成的に活性化されたERを有するGFP陽性クローンを排除した(ii)。継続的に、100nM TS及び100 nM 3β-ジオールを各培地に添加し、24時間の時点でER活性を確認した(iii)。最終的に、100nMレトロゾールの添加後もER活性を有しているクローンを、タイプ4(AD−EDR)細胞株と名づけた(iv)。同時に、これらの細胞株において、ER活性が1μMフルベストラントによって阻害されることを確認した(v)。タイプ4細胞株は、100nM TSを添加したステロイド枯渇培地で維持した。
【0072】
タイプ4細胞株は、アンドロゲン又はその代謝産物に依存してER活性を示すと考えられ、ストローマ細胞との共培養においてレトロゾール耐性ER活性を示した。共培養は、乳癌細胞とストローマ細胞とを可溶性因子が通過しうる膜を隔てて接するようにし、別個に増殖させたもので、ストローマ細胞のアロマターゼによってアンドロゲンから局所的にエストロゲンが合成される微細環境を模したものである。
【0073】
TS添加ステロイド枯渇培地で3ヶ月培養後、GFPを発現する5つのクローンが得られた。各処理工程でのER活性を図8にまとめて示す。
【0074】
5つのうち、G1T及びR6Tは、ER活性がステロイド枯渇培地では阻害されたがレトロゾールによって阻害されなかったため、アンドロゲン又はその代謝産物に依存するER活性を示すものとして、タイプ4細胞として選択された。
樹立されたタイプ4細胞(G1T、R6T)は、アンドロゲンで活性化され、アロマターゼ阻害剤は無効であり、フルベストラントによってER活性が阻害される。このことは、アンドロゲンがエストロゲンに変換されて作用するのではなく、直接エストロゲンレセプターを活性化していることを示唆している。
【0075】
クローンV3T及びB6Tは、増殖因子レセプター経路又はその他のメカニズムにより構成的に活性化されたERを有すると考えられた。これは、3日間のステロイド枯渇条件においてもER活性を維持したためである。クローンR7Tは、ER活性がレトロゾールによって抑制されるため、TSからアロマターゼによって供給されるエストロゲンに依存するER活性を有すると考えられる。
【0076】
2.テストステロン(TS)、ジヒドロテストステロン(DHT)又は3β−ジオール存在下でのER活性及び増殖
定義されたホルモン条件下でのアッセイのために、細胞を数回洗浄した後、枯渇培地で3日間培養した。その後、細胞を枯渇培地中で24ウェル培養プレートに104個/ウェルの密度で播き、所定の濃度のTS、DHT、3β−ジオール、エストラジオール(E2)又はコントロールとしてエタノールを、100nM レトロゾール、1μM フルベストラント、所定の濃度のトリロスタン又はバイカルタミド(bicalutamide)、100nM OHT又は3μM TORの存在下又は非存在下で、各ウェルに添加し、4日間培養した。各ウェルの細胞を回収してカウントし、コントロールに対する相対値を算出した。残りの細胞をER活性の評価に使用した。
【0077】
タイプ4細胞は、TS、DHT又は3β−ジオールの存在下で増大したER活性及び増殖を示した。E2による増殖誘導では親細胞であるE10細胞とタイプ4細胞とで有意差がなかった。低濃度のTS及びDHT(1nM又は10nM)はすべての細胞株の増殖を阻害したが、タイプ4細胞は、100nM TS又は100nM DHTによって親細胞よりも増大した増殖を示した。100nM 3β−ジオールによる増殖誘導は、両細胞株において1nM E2と匹敵するものであった。タイプ4細胞株は、親細胞よりも、低濃度(1nM又は10nM)の3β−ジオールによって増大した増殖を示した。
【0078】
3.タイプ4細胞株におけるDHT代謝酵素の発現及びアンドロゲンシグナル伝達の中断
アンドロゲン産生酵素及びホルモンレセプターのmRNA発現を調べた。また、TS、DHT又は3β−ジオールによるエストロゲン応答性遺伝子及びアンドロゲン応答性遺伝子の誘導も調べた。普通培地又は100nM テストステロンを添加した枯渇培地で3日間培養した細胞から総RNAを抽出し、各種遺伝子について、内部標準としてRPL13Aに対する相対的なmRNA発現量を測定した。
タイプ4細胞株(G1T、R6T)は、DHT代謝酵素である3β−ヒドロキシステロイドジヒドロゲナーゼ1型(HSD3B1)の増大した発現及びアンドロゲンレセプター(AR)の低減した発現を示した。アルド−ケトレダクターゼ(AKR1C3)はG1Tにおいてアップレギュレーションされていた。アロマターゼ(CYP19)及び17β−ヒドロキシステロイドデヒドロゲナーゼ2型(HSD17B2)はすべての細胞株で検出されなかった。
タイプ4細胞は、親細胞であるE10細胞と比較して、TS、DHT又は3β−ジオールにより増大したエストロゲン応答性遺伝子PgR及びEGR3の発現を示した。さらに、タイプ4細胞においては、TS、DHT又は3β−ジオールによってエストロゲン応答性抗アポトーシスタンパクBcl−2が誘導された。
これらの結果から、タイプ4細胞においては、DHT代謝がアップレギュレーションされており、アンドロゲンシグナル伝達がほとんど停止していることが示唆された。さらに、タイプ4細胞においては、TS又はDHTがより効率的に3β−ジオールに代謝されること、及び3β−ジオールがより効率的にエストロゲン様作用を示すことが示唆された。
【0079】
4.タイプ4細胞株に対する抗エストロゲン剤の効果
タイプ4細胞におけるDHTによる増殖誘導は、アロマターゼに依存せずに産生されるDHT代謝産物3β−ジオールによるER活性化に起因する。そこで、臨床適用可能な選択的エストロゲンレセプター調節剤(SERM)であるOHT及びTOR(これらはERαリガンドの結合を阻害する)がフルベストラント(ICI又はFul)と同様にDHT誘導細胞増殖を阻害するかどうかを調べた。具体的には、100nM DHT添加と同時にOHT(1μM)又はTOR(3μM)を、G1T細胞及びR6T細胞にそれぞれ添加して、4日後、細胞数を計測して増殖能を評価した。
結果を図9に示す。予想どおり、エストロゲンレセプターをブロックする抗エストロゲン剤であるOHT又はTORによってICIと同様にタイプ4細胞の増殖を効率的に阻害したが、アロマターゼ阻害剤であるレトロゾール(Let)は無効であることが示された。
これらの結果は、タイプ4細胞株がアンドロゲン耐性及び3β−ジオールに対する過敏性を示すことを示唆する。さらに、これらの特性は、アロマターゼ活性には依存しなかった。
【0080】
5.ER活性
ERE−ルシフェラーゼレポーターアッセイにより、100nM DHTはタイプ4細胞においてER活性を誘導したが、親細胞においては誘導しなかったことがわかった。タイプ4細胞においては、100nM 3β−ジオールにより誘導されたER活性は、親細胞における活性の2倍高かった。TSにより誘導されるER活性はこのアッセイではすべての細胞株で検出されなかったが、ERE−GFPレポーターアッセイではタイプ4細胞において増大したER活性が検出された。
TS、DHT又は3β−ジオールによって誘導されたER活性は、1μM フルベストラントによって阻害されたが、100nM レトロゾールによっては阻害されなかった。同様の結果は、すべての細胞株の増殖において得られた。
【0081】
V. タイプ5細胞株の作製及び特徴づけ
1.親株(E10−Arom細胞)の樹立
E10細胞にブラストシジン(BSD)耐性遺伝子を持つアロマターゼ発現ベクター(pNH−CMV/AROM−BSD;図10)を導入し、ブラストシジン(10μg/mL)添加普通培地で形質転換細胞を選択した。生き残ったコロニーを顕微鏡下でマイクロピペットを用いて剥離し、96ウェル培養プレートに単離した。その後、24ウェル培養プレートに移し、その際に培地をブラストシジン添加普通培地からテストステロン100nM添加枯渇培地に交換した。
この細胞は自身でアロマターゼを産生しテストステロンをエストラジオールに変換できるので、テストステロン添加枯渇培地でも増殖できる。さらに6cmディッシュ、10cmディッシュと段階的に大きな培養容器に移しつつ細胞数を増やし、継続的にテストステロン添加枯渇培地で培養を続け、安定的なアロマターゼ発現E10細胞株(E10−Arom)を樹立した。
【0082】
2.タイプ5耐性株の作製
このE10−Arom細胞を親株として用い、アロマターゼ阻害剤であるレトロゾール100nM及びテストステロン100nM添加枯渇培地で3ヶ月間培養を続け、タイプ1細胞の樹立法と同様にコロニーを形成させた。このうち、レトロゾール100nM及びテストステロン100nM添加枯渇培地で蛍光が低下せず、純粋な枯渇培地で蛍光が低下するものを選択的にクローニングし、レトロゾール耐性MCF−7−E10Arom−LR細胞(Letrozole resistant cells)(タイプ5細胞)とした(クローンLR2、LR3)。
【0083】
3.LR細胞におけるレトロゾール添加時のER活性
樹立したレトロゾール耐性細胞8株について、テストステロン 100nM、レトロゾール 100nM添加時のER活性をルシフェラーゼアッセイで測定した。
【0084】
結果を図11に表す。この中で、特にLR3は親株であるE10−Aromに比べてレトロゾール添加時にも高いER活性を維持していた。
【0085】
4.LR細胞におけるレトロゾール感受性の検討
レトロゾール添加時に特に高いER活性を示していたLR3について、レトロゾール感受性の検討を行った。
親株であるE10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、テストステロン(0.1〜100nM)、レトロゾール(100nM)を添加し、4日後にSysmex CDA-500にて細胞数を測定した。
【0086】
結果を図12に表す。LR3は、E10Aromに比べて、レトロゾール添加時にも高い細胞増殖能を維持していた。
【0087】
5.LR細胞におけるエストロンスルフェート(EIS)代謝関連遺伝子の発現
LR3において、E1SをESに変換する酵素であるSTSと、E1Sを細胞内に取り込むトランスポーターであるOATP−AのmRNA発現を、リアルタイムRT−PCRで測定した。
【0088】
結果を図13に表す。LR3は、E10−Aromと比較してSTS、OATP−A共に発現が上昇していることが確認された。
【0089】
6.LR細胞におけるEIS、STX64(STS阻害剤)感受性の検討
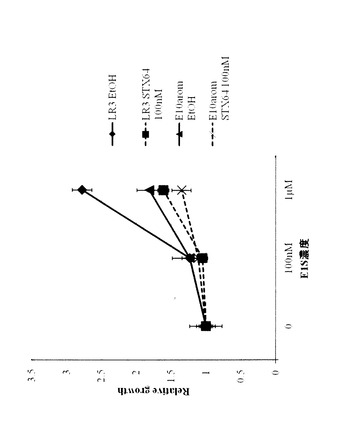
耐性獲得におけるSTSの関与が示唆されたので、LR3のE1SとSTS阻害剤であるSTX64に対する感受性を検討した。
親株であるE10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、E1S(0、100nM〜10μM)単独あるいはSTX64(100nM)と組み合わせて添加し、4日後にSysmex CDA-500にて細胞数を測定した。
【0090】
結果を図14に表す。LR3は、E10AromよりもE1Sに対する感受性が高まっていることが確認された。さらに、LR3においてE1Sによって誘導された細胞増殖は、STX64添加で親株と同程度まで抑制されることが確認された。
【技術分野】
【0001】
本発明は、ホルモン療法耐性乳癌細胞株、及びそれを用いた乳癌治療剤のスクリーニング方法に関する。
【背景技術】
【0002】
エストロゲンは、乳癌の発生及び増殖に深く関係する女性ホルモンであり、乳癌細胞の多くはエストロゲン依存性に増殖するホルモン依存性腫瘍である。乳癌細胞がエストロゲンを受容すると、エストロゲンレセプター(ER)を介して細胞増殖に関わるシグナル伝達系が活性化され、細胞増殖が促進されて癌が進展する。
【0003】
乳癌患者の70%以上の乳癌はエストロゲンレセプター(ER)陽性であり、ホルモン療法の治療対象である。
【0004】
乳癌治療薬としてのホルモン剤は、大きく2種類に分類される。一つは、エストロゲンレセプターの阻害作用を示す薬剤(抗エストロゲン剤;タモキシフェン、トレミフェン、フルベストラント、ラロキシフェンなど)であり、もう一つは、エストロゲン合成酵素であるアロマターゼを阻害する薬剤(アロマターゼ阻害剤(AI);アナストロゾール、レトロゾール、エグゼメスタンなど)である。
【0005】
しかし、これらの薬剤はいずれも、やがて耐性を獲得されてしまうという問題がある。ホルモン療法を受けた乳癌患者のうち1/3は、癌がホルモン治療に耐性を獲得し、再発する。再発後は、化学療法が行われるが、予後が不良であり、治療が困難である。したがって、ホルモン療法耐性の獲得が、臨床上、重大な問題となっており、耐性機序の解明と新たな治療方法が望まれている。
【0006】
現在、ホルモン療法耐性機構として、(1)増殖因子シグナルカスケードの亢進、(2)HER2の発現増加によるシグナルカスケードの亢進、(3)エストロゲン非依存的なER活性化、(4)アンドロゲン代謝産物によるER活性化、(5)薬物代謝能や薬物排出能の亢進などが考えられている(非特許文献1:医学のあゆみ Vol.230(1)p.31-36(2009))。
【0007】
ホルモン療法耐性機序の解明及び新たな治療剤の開発のため、種々の試みが行われている。例えば、国際公開WO2006−129735号公報(特許文献1;「遺伝子導入細胞及び細胞分析法」)には、エストロゲン応答配列(ERE)を緑色蛍光タンパク質(GFP)遺伝子の転写開始点上流に配置した発現レポーター遺伝子を安定導入したER陽性乳癌細胞株を用いて、患者から採取した組織切片から、当該患者にとってアロマターゼ阻害剤(AI)が有効か否かを評価する方法が開示されている。非特許文献1(医学のあゆみ Vol. 230(1) p.31-36 (2009))には、エストロゲン枯渇条件下で細胞を培養し、エストロゲン枯渇耐性株を取得したことが開示されている。
【0008】
しかし、これらの細胞株の詳細な特徴及び挙動や、ホルモン療法耐性機序に基づく新たな治療剤の開発は記載されていない。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開WO2006−129735号公報
【非特許文献】
【0010】
【非特許文献1】医学のあゆみ Vol.230 (1) p.31-36 (2009)
【非特許文献2】Cancer Sci., vol. 100 (1), p.1773-1778 (2009)
【非特許文献3】Cancer Res, vol.65, p.4653-4662 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、乳癌のホルモン療法耐性について考えられている種々の耐性機序をカバーする細胞株を提供すること、及びこれらの細胞を用いたホルモン療法耐性細胞に効果的な薬剤のスクリーニング方法を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者は、GFP遺伝子を発現レポーターとして安定導入したER陽性乳癌細胞株を親株として、長期間のホルモン療法を模倣した培養方法でGFPを指標としてAI耐性株を得ることにより、異なる耐性機序を有する5種類のホルモン療法耐性乳癌細胞株を樹立した。
【0013】
したがって、本発明によれば、
〔1〕 ホルモン療法に耐性を示す乳癌の治療剤をスクリーニングする方法であって、エストロゲン応答配列の制御下で緑色蛍光タンパク質(GFP)遺伝子を発現しうるホルモン療法耐性乳癌細胞に被験物質を接触させる工程と、この細胞を培養し、細胞の増殖及び/又はGFPの発現を測定する工程と、前記被検物質と接触させていない前記ホルモン療法耐性乳癌細胞を同じ条件下で培養及び測定した結果と比較して前記被検物質が細胞の増殖及び/又はGFPの発現の低減をもたらしたかどうかを判定する工程とを含み、
前記ホルモン療法耐性乳癌細胞が、
エストロゲン非依存性、かつ、エストロゲンレセプター(ER)活性依存性である細胞株(タイプ1)、
エストロゲン非依存性、かつ、ER活性非依存性である細胞株(タイプ2)、
エストロゲン非依存性、ER活性非依存性、かつ、成長因子(GF)依存性であり、HER2及び上皮増殖因子レセプター(EGFR)を発現する細胞株(タイプ3)、
アンドロゲン代謝産物依存性である細胞株(タイプ4)、及び/又は
アロマターゼ阻害剤(AI)非感受性、かつ、ER活性依存性である細胞株(タイプ5)、
のいずれか1以上であることを特徴とする、乳癌治療剤スクリーニング方法;
〔2〕 前記ホルモン療法耐性乳癌細胞として、タイプ1〜タイプ5のうち2以上の細胞株を用いてそれぞれの細胞を別々に被験物質に接触させる、前記〔1〕記載のスクリーニング方法;
〔3〕 前記ホルモン療法耐性乳癌細胞が、
受領番号FERM AP−22142であるタイプ1細胞、
受領番号FERM AP−22143であるタイプ2細胞、
受領番号FERM AP−22144であるタイプ3細胞、
受領番号FERM AP−22145であるタイプ4細胞、及び/又は
受領番号FERM AP−22146であるタイプ5細胞、
である、前記〔1〕又は〔2〕記載のスクリーニング方法;
〔4〕 受領番号FERM AP−22142である細胞;
〔5〕 受領番号FERM AP−22143である細胞;
〔6〕 受領番号FERM AP−22144である細胞;
〔7〕 受領番号FERM AP−22145である細胞;
〔8〕 受領番号FERM AP−22146である細胞、
が提供される。
【発明の効果】
【0014】
本発明の細胞株によれば、ホルモン療法耐性乳癌の各種メカニズムに対して有効な治療方法の選択が可能となる。すなわち、本発明の5種類の細胞株は、乳癌のホルモン療法耐性の獲得に関して考えられる耐性機序のほとんどをカバーするものであり、想定されるAI耐性の機序を解明するツールとして有用である。また、本発明の細胞の増殖阻害やGFP活性阻害を指標にして、従来のホルモン療法耐性細胞又はホルモン療法耐性患者に効果的な薬剤のスクリーニング及び開発が可能となる。さらに、本発明の細胞は、乳癌患者の治療選択(薬物療法)の指針となりうる。具体的には、ホルモン療法耐性乳癌のなかには、他のホルモン療法剤が有効と考えられるケース、他のホルモン療法剤と分子標的薬との併用が有効と考えられるケース、ホルモン療法剤の効果は期待できないが分子標的薬又は化学療法が適切と考えられるケース等が混在している。したがって、ホルモン療法耐性乳癌の患者について、どのようなケースに該当するかを本発明の細胞を指標としてタイピングすることにより、各タイプに対して有効な治療法を選択することができる。
【図面の簡単な説明】
【0015】
【図1】図1は、タイプ1及び2細胞のエストロゲン非依存性の増殖を表す図である。10%DCC−FCSを含有するRPMI1640培地で3日間培養した後、細胞を5×103個/ウェルの密度で24ウェルプレートに播き、種々の濃度のエストロゲンを含有するエストロゲン枯渇培地で4日間培養し、細胞数をカウントした。コントロールの細胞数に対する相対値(平均±SD)で示す。
【図2】図2は、タイプ1及び2細胞に対する抗エストロゲン薬の効果を表す図である。10%DCC−FCSを含有するRPMI1640培地で培養した細胞に、各抗エストロゲン薬(1μM フルベストラント(Ful)、1μM 4−ヒドロキシタモキシフェン(4−OHT)又は3μM トレミフェン(Tor))又はコントロールとしてエタノール(EtOH)を添加して4日間培養した後、細胞数をカウントした。コントロールの細胞数に対する相対値で示す。
【図3】図3は、タイプ3細胞の増殖因子依存性を表す図である。パネルAは各増殖因子の存在下(100ng/mL)で10日間培養後の細胞数(PBSを添加したコントロールを1とした相対値;平均±SD)、パネルBはそれらの細胞のクリスタルバイオレット染色像を表す。
【図4】図4は、タイプ3細胞のER及びエストロゲン応答性遺伝子の発現を表す図である。TE8細胞はエストロゲン枯渇培地で3日間培養後エストロゲンを添加して1日培養したもの、タイプ3細胞は増殖因子培地で培養したものを用いた。パネルAはERαのリアルタイムRT−PCR(mRNA)、パネルBはERαのウェスタンブロッティング(タンパク)、パネルCはエストロゲン応答性遺伝子EGR3及びPgRのリアルタイムRT−PCR(mRNA)の結果をそれぞれ表す。
【図5】図5は、タイプ3細胞のHER2の発現を表す図である。パネルAはリアルタイムRT−PCR(mRNA)、パネルBはウェスタンブロッティング(タンパク)の結果をそれぞれ表す。
【図6】図6は、タイプ3細胞のヒトホスホRTKアレイキットによるリン酸化膜レセプター解析の結果を表す図である。パネルAはTE8細胞(普通培地)、パネルBはタイプ3(U25)細胞(増殖因子培地)のライセートを用いた結果を、それぞれキットの陽性コントロールに対する相対比で表す。
【図7】図7は、タイプ3細胞における分子標的薬及び抗エストロゲン薬の効果を表す図である。パネルA:細胞を1×104個/ウェルの密度で24ウェルに播き、増殖因子培地で培養した。抗HER2薬であるトラスツズマブ(150mg/mL)、抗EGFR及びHER2薬であるラパチニブ(5μM)、抗EGFR薬であるゲフィチニブ(5μM)、抗EGFR薬であるエルロチニブ(5μM)を、それぞれプレートに添加し、3日後に細胞数をカウントした。パネルB:細胞を1.5×104個/ウェルの密度で24ウェルに播き、増殖因子培地で培養した。タモキシフェン(1μM)、トレミフェン(3μM)を抗エストロゲン薬としてそれぞれプレートに添加した。データは平均±SD(n=3)で表す。
【図8】図8は、アンドロゲン代謝産物依存性及びエストロゲン枯渇耐性細胞の選択を表す図である。TS添加ステロイド枯渇培地で3ヶ月培養した後、GFPを発現する5個のクローンを選択した。各工程でGFP陽性細胞の比率によって評価したER活性を示す。V3T及びB6T細胞株は、ステロイド枯渇培地においてもER活性を示し、ERが構成的に活性化されていることが示された。R7TのER活性はレトロゾールによって抑制され、アロマターゼ依存性であることが示された。G1T及びR6TのER活性はステロイド枯渇培地で阻害されたが、レトロゾールによって阻害されなかった。これらの細胞はタイプ4(AD−EDR)細胞株として樹立された。
【図9】図9は、タイプ4細胞におけるDHT増殖誘導がSERMによってブロックされることを表す図である。タイプ4細胞においては、100nM DHTによる増殖誘導が100nM タモキシフェン(OHT)又は3μM トレミフェン(TOR)、又は1μM フルベストラント(ICI)によって効率的にブロックされたが、100nM レトロゾール(Let)によってはブロックされなかった。
【図10】図10は、アロマターゼ発現ベクターpNH−CMV/AROM−BSDの構造を表す図である。
【図11】図11は、タイプ5細胞のレトロゾール添加時のER活性を表す図である。テストステロン(100nM)、レトロゾール(100nM)添加時のER活性をルシフェラーゼアッセイで測定した。
【図12】図12は、タイプ5細胞におけるレトロゾール感受性を表す図である。E10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、テストステロン(0.1〜100nM)、レトロゾール(100nM)を添加し、4日後に細胞数を測定した。
【図13】図13は、タイプ5細胞におけるエストロンスルフェート(E1S)代謝関連遺伝子の発現を表す図である。LR3において、E1SをESに変換する酵素であるSTS(パネルA)と、E1Sを細胞内に取り込むトランスポーターであるOATP−A(パネルB)のmRNA発現を、リアルタイムRT−PCRで測定した。
【図14】図14は、タイプ5細胞におけるE1S及びSTX64(STS阻害剤)感受性を表す図である。E10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、E1S(0、100nM〜10μM)単独あるいはSTX64(100nM)と組み合わせて添加し、4日後に細胞数を測定した。
【発明を実施するための形態】
【0016】
本発明の細胞株は、提唱されている各種のホルモン療法耐性メカニズムを表すものであり、タイプ1〜5に分けられる。
5種類の細胞株のうち、タイプ1、4、5はER発現株(GFP陽性)、タイプ2、3はER非発現株(GFP陰性)に分類される。
【0017】
ホルモン療法耐性細胞の作製
本発明の細胞株は、エストロゲン応答配列(ERE)の下流にレポーター遺伝子としてGFP遺伝子を含むベクターで形質転換した乳癌細胞を、ホルモン療法を模した特定の培養条件下で長期培養し、所望の性質を示すものを選択することにより作製することができる。
これらの細胞は、ERが活性化されるとGFPを発現して蛍光を発するため、細胞が生きたままの状態でERE活性を視覚的に捉えることができる。
本発明のタイプ1〜タイプ5のそれぞれの細胞の取得のための具体的手順は、実施例において詳述するとおりである。
【0018】
タイプ1細胞株
タイプ1細胞株(GFP陽性)は、ERの過剰発現及びいくつかのER標的遺伝子の構成的な高発現を示す。HER2発現の有意な誘導は示さない。総合的なリン酸化プロテオミクス解析により、タイプ1細胞では、細胞内リン酸化シグナル経路が動的変化をしていることがわかった。
抗エストロゲン薬により、タイプ1細胞株の生育は阻害される。
本発明のタイプ1細胞は、ERαの過剰発現を示し、エストロゲンに対し過敏性を示すが、エストロゲンに対する細胞増殖依存性は親細胞よりもはるかに低い。ERαの過剰発現は、タイプ1細胞の生存のためにエストロゲン非依存性を補完すると思われる。
さらに、MAPキナーゼ経路の有意な誘導が観察された。タイプ1(GFP陽性)細胞は、細胞間リン酸化経路の変化が示唆されており、特に、PKA経路は、エストロゲン非依存性ER活性化に重要と考えられる。
【0019】
タイプ2細胞株
タイプ2細胞株(GFP陰性)は、ER及びその標的遺伝子の顕著なダウンレギュレーションを示す。さらに、細胞内シグナル伝達因子c−Jun N末端キナーゼ(JNK)の構成的活性化が観察される。タイプ2細胞株については、MAPキナーゼ経路の有意な誘導が観察された。
【0020】
抗エストロゲン薬により、タイプ2細胞株の生育は阻害されない。一方、タイプ2細胞株では、JNK阻害剤又はEGFレセプター(EGFR)標的薬により生育が阻害される。JNK経路は、ホルモン療法耐性乳癌の新規な治療標的でありうる。
【0021】
タイプ2細胞は、エストロゲン非依存性及びER非依存性増殖メカニズムを獲得した細胞である。これらの細胞株は、リン酸化JNKの強い誘導及びAP-1転写活性の有意な活性化を示した。JNK経路は、種々の癌の増殖及び発展に重要な役割を果たすことが報告されているので、これらの細胞のエストロゲン及びER非依存性は、このシグナル経路の構成的活性化によって獲得された可能性がある。
【0022】
タイプ1及びタイプ2についてのこれらの結果は、ER陽性乳癌細胞が、ER依存性及び非依存性の複数のメカニズムによってエストロゲン枯渇耐性を獲得しうることを示す。
【0023】
タイプ3細胞株
タイプ3細胞株は、エストロゲン枯渇培地では生存できず、増殖因子(特にEGF及びTGF−α)を添加したエストロゲン枯渇培地で生存することができる。当初はGFP蛍光を強く発していたが、短期間のうちにGFP発現を消失した。また、ER発現も劇的に低下し、機能を失った。さらに、増殖因子レセプターによる細胞内シグナル伝達は、PI3キナーゼ(PI3K)/Akt及びJNK経路の分子を増大させた。
タイプ3細胞は、EGFR標的薬により有意に増殖が阻害される。これらの結果から、タイプ3細胞は、エストロゲン非依存性、かつ増殖因子依存性の細胞増殖メカニズムを獲得したことが示唆される。さらに、細胞内シグナル経路は、タイプ3細胞においては、親細胞であるTE8と比較して変化している。
【0024】
タイプ4細胞株
タイプ4細胞株は、アンドロゲン代謝産物依存性、かつエストロゲン枯渇耐性である。タイプ4細胞株は、アンドロゲンからの局所的エストロゲン産生を再現するストローマ細胞(間質細胞)との共培養においてレトロゾール(Let)耐性ER活性を示し、AI耐性モデルとして使用することができる。タイプ4細胞株は、アンドロゲン又は3β−ジオールの存在下で増大したER活性及び増殖を示し、これはレトロゾールによって阻害されない。リアルタイムPCR解析によって、タイプ4細胞におけるジヒドロテストステロン(DHT)代謝酵素3β−ヒドロキシステロイドデヒドロゲナーゼタイプ1(3β−HSDタイプ1:HSD3B1)のmRNA発現の増大及びアンドロゲンレセプター(AR)発現の低減が示された。さらに、タイプ4細胞においては、アンドロゲン又は3β−ジオールによってエストロゲン応答性遺伝子PgR及びEGR3の発現が誘導されるが、アンドロゲン応答性遺伝子KLK3は誘導されない。これらのデータから、増大したDHT代謝及びARシグナルの中断がアロマターゼ阻害剤によって誘導されるエストロゲン枯渇及びアンドロゲン過剰条件に対する適応性を与えることが示唆される。
【0025】
アンドロゲン又は3β−ジオールによって誘導されるER活性の解析により、タイプ4細胞株の3β−ジオールに対する過敏性及びアンドロゲン耐性が示された。mRNA発現の定量的解析により、タイプ4細胞における増大したDHT代謝及びアンドロゲンシグナルの中断のため、アンドロゲン又は3β−ジオールがアロマターゼ非依存的に、よりエストロゲニックな挙動を示すことが示唆された。言い換えれば、エストロゲン性アンドロゲン代謝産物による低減したARシグナル及び増大したERシグナルは、アロマターゼ活性に依存せずに、エストロゲン枯渇条件への適合性をこれらの細胞株に与えうる。タイプ4細胞株に対する、3β−HSDタイプ1の特異的阻害剤であるトリロスタンの有効性は認められなかった。3β−HSDタイプ1は、DHTを3β−ジオールに代謝する唯一の酵素ではないため、DHTは他の酵素で代謝され、トリロスタンによってその活性が抑制される3β−HSDタイプ1が補償されると考えられる。タイプ4細胞についてのインビトロでのデータは、アンドロゲン又はその代謝産物によって誘導されるER活性化及びアンドロゲンシグナル中断がAI耐性において重要な役割を果たすこと、及びAR又はアンドロゲン代謝酵素の発現レベルが臨床的乳癌におけるAI耐性の生物学的マーカーとして有用であることを示唆する。
【0026】
ER依存性を維持するいくつかのクローンがAI処理を模した条件下で樹立され、これらのタイプ4細胞のDHTによる増殖誘導は選択的エストロゲンレセプター調節剤(SERM)によって効率的に阻害されるが、レトロゾールによっては阻害されない。これらの知見から、AIの後にSERMを連続して使用することの有効性が支持される。
【0027】
タイプ4細胞株は、インビトロでのAI耐性のメカニズムを研究するために有用であり、AI耐性進行乳癌についての診断マーカー及び新規治療戦略の開発に有用である
【0028】
タイプ5細胞株
タイプ5細胞株は、エストロゲン依存性、かつER依存性のレトロゾール(Let)耐性細胞である。親細胞であるMCF―7―E10aromは、テストステロン存在下でER活性及び増殖を示し、これがLetによって阻害されるが、タイプ5細胞株は阻害されない。タイプ5細胞株は、エストロン(Estrone;E1)スルフェート(E1S)に対する感受性が親細胞よりも高く、またリアルタイムPCR解析によって、E1Sを細胞内に取り込むトランスポーターであるOATP―A、及びE1SをE1に変換する酵素であるステロイドスルファターゼ(Steroid sulfatase;STS)のmRNAの発現の増大が示された。さらに、E1Sによって誘導される細胞増殖は、STS阻害剤であるSTX64により阻害されることが確認された。これらのデータから、タイプ5細胞株はSTSやOATP―Aの発現を増大させ、アロマターゼ非依存的にエストロゲンを産生する経路を亢進することで、Let耐性を獲得していることが示唆される。また、タイプ5細胞株についてのインビトロでのデータは、STSやOATP―Aによるエストロゲン産生の増大がAI耐性において重要な役割を果たすこと、及びSTS阻害剤のAI耐性患者への有用性や、STSやOATP―Aの発現レベルが臨床的乳癌におけるAI耐性の生物学的マーカーとして有用であることを示唆する。
【0029】
本発明の細胞株のうち、以下のものは、2011年7月8日付けで独立行政法人 産業技術総合研究所 特許微生物寄託センター(茨城県つくば市東1丁目1番地1 つくばセンター中央第6)に寄託された。
タイプ1:クローンA2(受領番号FERM AP−22142)。
タイプ2:クローンA4(受領番号FERM AP−22143)。
タイプ3:クローンU25(受領番号FERM AP−22144)。
タイプ4:クローンG1T(受領番号FERM AP−22145)。
タイプ5:クローンLR3(受領番号FERM AP−22146)。
【0030】
スクリーニング方法
本発明の細胞株を用いて、ホルモン療法耐性乳癌に対して有効な治療剤をスクリーニングすることができる。本発明のスクリーニング方法は、本発明の細胞株に被験物質を接触させる工程と、細胞を培養する工程と、細胞の増殖及び/又はGFPの発現を測定する工程とを含む。その結果、前記被検物質と接触させていない以外は同様に処理した本発明の細胞株と比較して細胞の増殖及び/又はGFPの発現が低減していれば、その被検物質は細胞増殖を抑制したと判定することができる。
【0031】
細胞と被検物質との接触は、培地に被検物質を添加することにより容易に行うことができる。被検物質との接触は、短時間のパルスで行ってもよく、培養期間中を通じて培地に存在させてもよい。したがって、接触と培養とは同時に行うことができる。培地としては、必要に応じて以下に説明する普通培地、エストロゲン枯渇培地、又はこれらに所定の添加物を添加した培地等を使用することができる。細胞の増殖は、常法により細胞数をカウントすること等により、行うことができる。GFPの発現は、例えば、蛍光顕微鏡下で蛍光を発する細胞の比率を測定すること等により測定することができる。
【0032】
スクリーニングに用いる本発明の細胞株としては、タイプ1〜タイプ5のいずれか1以上のタイプであればよいが、2以上を用いて各タイプに対する同じ被検物質の効果の差を調べることができる。また、2以上を用いていずれか1つのタイプにのみ有効な被検物質を選択することができる。
【0033】
本発明の細胞は、上述のようにしてインビトロでの薬剤スクリーニングに用いることができるのに加えて、これらの細胞を動物に移植してそれぞれの機序のモデル動物を作製し、より個体に近い実験系で薬剤の効果を評価するために使用することができる。
【0034】
また、新規な薬剤を見出すことだけでなく、既存薬剤の投与順序や併用の組合せ等に関しても、最適な方法を見出すために、上記と同様のインビトロ又はインビボ実験を行うことができる。
【0035】
治療選択法
また、本発明の細胞は、個々の患者に最適な治療選択を可能にするための情報を提供することができる。具体的には、ホルモン療法耐性乳癌のなかには、他のホルモン療法剤が有効と考えられるケース、他のホルモン療法剤と分子標的薬との併用が有効と考えられるケース、ホルモン療法剤の効果は期待できないが分子標的薬又は化学療法が適切と考えられるケース等が混在していると考えられる。したがって、ホルモン療法耐性乳癌の患者について、どのようなケースに該当するかを本発明の細胞を指標としてタイピングすることにより、各タイプに対して有効な治療法を選択することができる。例えば、タイプ1についてはリン酸化ER、タイプ2についてはpJNK、タイプ3についてはHER2、タイプ4についてはAR、及びタイプ5についてはSTSといったタンパク又は遺伝子の発現を指標として患者から採取した乳癌組織片がどの対応に該当するかを例えば免疫染色等によってタイピングし、該当したタイプの細胞に対して有効性の高い治療法をその患者の最適な治療法として選択することができる。
【実施例】
【0036】
I.ホルモン療法耐性乳癌細胞株の樹立
1.樹立に使用した親細胞
親細胞として、pd2EGFP−1ベクター(Clontech)において半減期の短いGFPのcDNAの上流にエストロゲン応答配列(ERE)を配したレポータープラスミド(特許文献1)を安定的に導入したER陽性ヒト乳癌細胞株MCF−7−E10細胞(「E10細胞」と呼ぶことがある;特許文献1)を、タイプ1、2、4細胞株の樹立のために用いた。同じプラスミドを導入したER陽性ヒト乳癌細胞株T47D−TE8細胞(「TE8細胞」と呼ぶことがある)を、タイプ3細胞株の樹立のために用いた。TE8細胞は以下のIII.に記載する方法で作製した。
タイプ5細胞株の樹立のための親細胞としては、以下のV.に記載するとおりに作製したE10−Aromを用いた。
【0037】
2.細胞培養法
細胞の通常の培養には、10%FCS(Tissue Culture Biologicals)及び1%ペニシリン/ストレプトマイシン(GIBCO)を添加したRPMI1640培地(SIGMA)(「普通培地」と呼ぶことがある)を用いた。
耐性株のエストロゲン枯渇培養には、10%デキストラン被覆チャコール処理FCS(DCC−FCS;Tissue Culture Biologicals)および1%ペニシリン/ストレプトマイシン(GIBCO)を添加したフェノールレッド無含有RPMI1640培地(GIBCO)(「枯渇培地」又は「エストロゲン枯渇培地」と呼ぶことがある)を用いた。
【0038】
細胞は、すべて5%CO2、37℃に調整したCO2インキュベーターで培養した。
【0039】
特に記載がない場合、実験結果は、平均±SD(n=3)で表す。
【0040】
3.試薬等
エストラジオール、タモキシフェン(4−ヒドロキシタモキシフェン(4-hydroxytamoxifen))、テストステロン、DHT、3β−ジオール、デキサメタゾンは、Sigma-Aldrich Corpから購入した。抗ER抗体(H−184)は、Santa Cruz Biotechnology Inc.、抗ヒトerbB2抗体はDako Cytomationからそれぞれ入手した。抗EGFR (V279)、抗Akt、抗リン酸化Akt (Thr308)、抗SAPK/JNK、抗44/42 MAPK (Erk1/2)、抗リン酸化44/42 MAPK(Erk1/2) (Thr202/Tyr204)、抗リン酸化C−Raf (Ser259)、抗β−チューブリン抗体は、Cell Signaling Technologyから入手した。
トラスツズマブ(Trastuzumab ;商品名「ハーセプチン」)及びエルロチニブ(erlotinib;商品名「タルセバ」)は中外製薬 (Tokyo, Japan)から、ラパチニブ(lapatinib)はGlaxoSmithKline plc. (Brentford, UK)から、ゲフィチニブ(gefitinib;商品名「イレッサ」)はアストラゼネカ(Osaka, Japan)から、トレミフェン(toremifen)は日本化薬(Tokyo, Japan)から、それぞれ入手した。レトロゾールはノバルティスファーマ(Tokyo, Japan)から、フルベストラントはアストラゼネカから、それぞれ入手した。トリロスタン(3β−HSDタイプ1阻害剤)はTronto Research Chemicals(North York, Canada)から、及びバイカルタミド(アンドロゲンレセプター阻害剤)はLKT Laboratories(St. Paul, MN, USA)から、それぞれ入手した。
【0041】
4.リアルタイムRT−PCR
それぞれの条件下で培養した細胞から商品名「Isogen」(Nippon Gene)を用いて総RNAを抽出し、商品名「RNA PCR kit」(Takara Shuzo)を用いて第1鎖cDNAを合成した。リアルタイムRT−PCRは、商品名「StepOne real-time PCR system」(Applied Biosystems)を用いて標準プロトコールにしたがって行った。目的の遺伝子の発現を、内部標準としてのGAPDH又はRPL13Aに対する相対値として算出した。データは、平均±SD(n=3)で示した。
【0042】
5.ウェスタンブロッティング
100mmディッシュで増殖させた細胞を氷冷生理食塩水で洗浄し、ホスファターゼ阻害剤カクテル(「Phos STOP」、Roche)及びプロテアーゼ阻害剤カクテル(Roche)を含む1mLのリシスバッファー(「Complete Lysis-M」、Roche Diagnotics GmbH)で溶解させた。5分後、ライセートをパルスソニケーションに供し、14,000rpmで10分間遠心分離した後、10%ポリアクリルアミドゲル(Wako)でSDS−PAGEを行い、タンパク質をPVDF膜(GE Healthcare Bioscience)に転写した。この膜を、一次抗体(5%BSA、0.05% Tween−20含有中)と反応させた。次に、西洋ワサビ結合二次抗体を適用した。化学発光基質(「Immunsutar AP substrate」、 BIO-RAD)と反応させた後、目的のタンパクのバンドを、X線フィルムを感光させることにより可視化した。
【0043】
6.ルシフェラーゼアッセイ
ERE活性を、商品名「Dual-Luciferase Reporter System」(Promega)を用いて、基本的にBiochem Biophys Res Commun 2001, 285:340-347に記載された方法に従って測定した。枯渇培地で3日間培養後、5×104個の細胞を6cmディッシュに播き、同じ培地中で48時間培養した。0.5μgのエストロゲンレポータープラスミド(Tk−ERE−Luci)及び0.05μgのコントロールベクター(pRL―TK, Promega)を300μLの無血清培地中で5μLの商品名「TransIt」LT-1試薬(Takara Bio)と混合し、製造業者の指示書に従ってトランスフェクションを行った。薬剤の存在下又は非存在下で24時間細胞を培養した後、商品名「Dual-Luciferase Reporter System」(Promega)を製造業者の指示書に従って用いてルシフェラーゼ活性を測定し、コントロールに対する相対値を算出した。
【0044】
II. タイプ1細胞株及びタイプ2細胞株の作製及び特徴づけ
1.タイプ1細胞株及びタイプ2細胞株の樹立
普通培地で培養していたE10細胞を枯渇培地に播種し、長期的にエストロゲン枯渇培養した。大部分の細胞は増殖が止まり死に至ったが、一部の細胞は、エストロゲン枯渇耐性を獲得し、増殖してコロニーを形成した。これを蛍光顕微鏡で観察し、より強い蛍光を発しているもの(タイプ1細胞株;クローンA1、A2、C5)と、蛍光を消失しているもの(タイプ2細胞株;クローンA4、C7、K2)とを選択し、顕微鏡下でマイクロピペットを用いて剥離して、96ウェル培養プレートに単離した。その後、24ウェル培養プレート、6cmディッシュ、10cmディッシュ、と段階的に大きな培養容器に移しつつ細胞数を増やし、継続的に枯渇培養を続け、安定的なエストロゲン枯渇耐性細胞株(タイプ1、タイプ2を総称して、「E10−EDR細胞」(estrogen depletion resistant cells)と呼ぶことがある)を樹立した。
【0045】
2.細胞増殖におけるエストロゲン依存性
親株であるE10細胞(枯渇培地で3日間培養し、ステロイドを枯渇させたもの)及びE10−EDR細胞を、5×103細胞/ウェルの密度で24ウェル培養プレートに播き、種々の濃度のエストロゲンの存在下及び非存在下で培養した。4日後、細胞をPBSで1回洗浄し、トリプシン/EDTA処理によって回収した。細胞をパーティクルカウンター(「CDA-500 Sysmex automated cell counter」(Sysmex Corporation)でカウントした。
【0046】
結果を、第1日における細胞数に対する相対値で示す(図1)。タイプ1(A1、A2、C5)、タイプ2(A4、C7、K2)ともに、親株(E10)と比較して細胞増殖におけるエストロゲン(E2)に対する依存性が明らかに低下していた。
【0047】
3.タイプ1及びタイプ2細胞株に対する抗エストロゲン剤の効果
タイプ1及びタイプ2細胞の細胞増殖に対する抗エストロゲン剤の影響を検討した。枯渇培地で培養した細胞に、各抗エストロゲン薬(1μM フルベストラント(Ful)、1μM 4−ヒドロキシタモキシフェン(4−OT Tam)又は3μM トレミフェン(Tor))又はコントロールとしてエタノール(EtOH)を添加して4日間培養した後、細胞数をカウントした。
【0048】
結果を図2に示す。タイプ1細胞(A1、A2及びC5)は、エストロゲン非依存性であるが、ERをブロックする薬剤である抗エストロゲン薬(タモキシフェン、トレミフェン、フルベストラント)によって増殖が阻害された。一方、これらの抗エストロゲン薬は、タイプ2細胞(A4、C7及びK2)に対しては、トレミフェンがC7及びK2の増殖を少し阻害したものの、顕著な増殖抑制効果を示さなかった。
【0049】
これらの結果は、ホルモン療法のための抗エストロゲン薬はタイプ1細胞には有効であるが、タイプ2細胞にはあまり有効ではないことを示す。また、タイプ1細胞においては、抗エストロゲン剤フルベストラントの添加によりGFP活性が強力に阻害されることから、これらのGFP発現においてERαが大きく寄与することが示唆された。
【0050】
4.タイプ1細胞及びタイプ2細胞のその他の特徴
ER活性は、タイプ1細胞において高く、タイプ2細胞において低かった。リアルタイムPCRにより、内在性ERα標的遺伝子の発現を調べたところ、タイプ1細胞(A1、A2及びC5)は実質的にpS2、PgR、IGFBP4及びEGR3を発現したが、タイプ2細胞(A4、K2及びC7)は、これらのER標的遺伝子のmRNAの有意な発現を示さなかった。したがって、タイプ1細胞においては、エストロゲンに依存してER転写活性が構成的に活性化されているが、タイプ2細胞においては、ER転写活性がほとんど失われている。
ERαのmRNA及びタンパク質は、タイプ1細胞においては強く誘導されていたが、タイプ2細胞においてはほとんど認められなかった。タイプ1細胞において、ERαのSer118、Ser167、及びSer305でのリン酸化の顕著な変化は認められなかった。
いずれの細胞株も、HER2発現についてはアップレギュレーションが認められなかった。
【0051】
細胞内リン酸化経路に関与する代表的なタンパク質のウェスタンブロットを行い、タイプ2細胞株(A4及びK2)においてJNKが構成的に活性化されていることがわかった。A4及びK2のAP−1ルシフェラ−ゼ活性は、E10細胞よりも有意に高かった。
【0052】
さらに、JNK阻害剤(SP600125)は、タイプ1細胞よりもタイプ2細胞においてより効果的に増殖を阻害した。これらの結果から、タイプ2細胞においてはJNK関連シグナル経路が生存に重要であることが示唆された。
【0053】
III. タイプ3細胞株の作製及び特徴づけ
1.親株(T47D−TE8細胞)の樹立
T47D−TE8細胞は、ヒト乳癌細胞株T47D(ATCC番号:HTB−133)から、E10細胞の作製に用いたものと同じERE−GFP遺伝子を担持するプラスミドを公知の方法にしたがって導入することにより樹立した(非特許文献3:Yamaguchi Y. et al., Cancer Res, 2005, 65: 4653-4662)。
T47D−TE8細胞は、普通培地で培養した。
【0054】
2.タイプ3細胞株の樹立
エストロゲン枯渇条件下での長期培養によりT47D−TE8細胞から取得した細胞は、ホルモン療法で治療された乳癌のモデルを表す。
親株としてT47D−TE8細胞を用いて、上記IIの1.と同様の方法により、タイプ3細胞株を樹立した。ただし、T47D−TE8細胞は、通常の枯渇培地ではすべて死滅してしまうため、培養時は常に増殖因子カクテル(20ng/ml CXCL12/SDF−1α、20ng/ml CXCL12/SDF−1β、20ng/ml EGF、20ng/ml HGF、20ng/ml IGF−1、20ng/ml TGF−α(R&D systems))を添加した培地(増殖因子培地)を用いて3ヶ月以上培養した。
【0055】
T47D−TE8細胞は、エストロゲン枯渇培地では死滅したが、増殖因子培地では生存し、いくつかのコロニーを形成した。強力なER活性を有するものを得るために、より強い蛍光を発するコロニーを選択し、さらに7〜18週間、増殖因子培地で個別に培養した。この過程で、一部のコロニーは死滅し、最終的に31個のコロニーが得られた(タイプ3細胞;「TE8−EDR細胞」と呼ぶことがある)。これらの細胞は、当初は強力な蛍光を発したが、その後6〜8週間のうちに蛍光を失った。したがって、ER活性を失ったことが示唆された。増殖因子のないエストロゲン枯渇培地で得られ、ER活性を維持したEDR細胞を考慮すると、TE8−EDR細胞は、増殖因子の添加によってER活性を失ったと考えられる。
【0056】
タイプ3細胞として最終的に得られた31個のクローンのうち、U3、U8、U23及びU25細胞の特徴を解析した。
【0057】
3.タイプ3細胞の増殖因子依存性
各増殖因子(100ng/mL)を個別に含むエストロゲン枯渇培地で10日間培養することにより、タイプ3細胞(クローンU25)が増殖因子カクテルに含まれるどの増殖因子に依存しているかを調べた。
【0058】
結果を図3に示す。タイプ3細胞の増殖は、EGFとTGF−αに依存していることがわかった。細胞数は、EGF及びTGF−αを添加した場合に、それぞれコントロールの7.5倍及び6.4倍に増大し、他の増殖因子(SDF−α、SDF−β、HGF、IGF−1)を添加した場合には2倍となった(パネルA)。クリスタルバイオレット染色によっても、EGF及びTGF−αによる細胞数の増大が顕著であることが示された(パネルB)。EGF及びTGF−αは、同じレセプター(EGFR/HER1)を利用して増殖因子のシグナルを細胞内システムに伝達すると考えられている。したがって、タイプ3細胞の生存及び増殖にEGFRが関与することが示された。
【0059】
4.タイプ3細胞におけるER及びHER2の発現
タイプ3細胞においてはERの発現が低下し、乳癌で重要な治療の標的となっているHER2の発現が上昇していることを以下のようにして確認した。
タイプ3細胞におけるERα及びエストロゲン応答性遺伝子の発現を、リアルタイムRT−PCRにより調べた。
【0060】
結果を図4に示す。通常培地のTE8親細胞と比較して、タイプ3細胞(U3、U8、U23、U25)のERαのmRNA発現は、顕著に低減していた(パネルA)。ウェスタンブロッティングにより検出したところ、タイプ3細胞のERαタンパク質発現も低減していた(パネルB)。さらに、エストロゲン応答性遺伝子EGR3及びPgRのmRNAも低減していた(パネルC)。
【0061】
したがって、タイプ3細胞は、ER発現及び機能的活性を失っており、その生存及び増殖はエストロゲン非依存性であると考えられる。
【0062】
次に、EGFRは他のHERファミリーレセプターと2量体を形成するため、同様にHER2の発現を調べた。HER2過剰発現は、乳癌の20〜30%で観察されるため、乳癌の診断、サブタイピング、治療に重要な因子である。
【0063】
結果を図5に示す。タイプ3細胞(U3、U8、U23、U25)におけるHER2 mRNAは、TE8親細胞と比較して有意に増大していた(パネルA)。HERタンパク質の発現も増大していた(パネルB)。これらの結果は、HER2が、EGFRと協働してシグナル伝達する増殖因子により増大したことを示唆する。
【0064】
5.ホスホ−RTKアッセイによる解析
EGFRは種々の膜レセプターと2量体を形成するため、さらにヒトホスホ−RTKアレイアッセイキット(Human Phospho-RTK Array Kits (R&D Systems))を用いて、細胞膜レセプターの網羅的発現解析を行った。サンプルとして、増殖因子培地で培養したタイプ3細胞及び枯渇培地で3日間の後エストロゲンを添加して1日培養した親細胞(TE8細胞)を用いた。このキットは、42項目の膜型レセプターに対する一次抗体が固定されているメンブレンに細胞ライセートを結合させ、細胞に存在するこれらのリン酸化されたレセプターチロシンキナーゼ二次抗体で検出する。
【0065】
結果を図6に示す。タイプ3細胞株におけるHERファミリー全体の発現増加がみられた。
TE8親細胞において、EGFR、HER2及びErbB4のリン酸化レセプターが検出された(パネルA)。これらのレセプターに加えて、タイプ3細胞(クローンU25)では、リン酸化されたErbB3及びインスリンレセプターが検出された(パネルB)。この結果は、タイプ3細胞が、さらなるレセプターを利用して種々の増殖因子依存性経路を確保する可能性を示す。
【0066】
6.タイプ3細胞の増殖に対する分子標的薬及び抗エストロゲン薬の効果
タイプ3細胞は増殖因子依存性が高く、EGFR及びHER2が増殖において重要な役割を果たすので、HER2及び/又はEGFRに対する分子標的薬の細胞増殖に対する効果を調べた。また、抗エストロゲン薬の効果も同様に調べた。
【0067】
増殖因子培地において培養したタイプ3細胞(クローンU25)に、薬剤を加え、3日間培養した。次に、細胞数をカウントした。薬剤として、150mg/mlトラスツズマブ(trastuzumab;抗HER2薬)、5μMラパチニブ(lapatinib;抗EGFR及びHER2薬)、5μMゲフィチニブ(gefitinib;抗EGFR薬)、5μMエルロチニブ(erlotinib ;抗EGFR薬)をそれぞれ用いた。陰性コントロールとしてDMSOを添加した。抗エストロゲン薬であるタモキシフェン及びトレミフェンは、それぞれ1μM及び3μMで用いた。
【0068】
結果を図7に示す。トラスツズマブは、HER2陽性乳癌に臨床的に使用される抗HER2薬であり、臨床試験では3年生存率を改善した。ラパチニブは、EGFR及びHER2を共に標的とする阻害剤であり、乳癌の新薬として期待されている。ゲフィチニブ及びエルロチニブは、EGFRを標的とする薬剤である。パネルAに示すように、EGFRを標的とする薬剤、ラパチニブ、ゲフィチニブ、エルロチニブは、細胞増殖を有意に阻害したが、トラスツズマブの効果はそれより劣っていた。タモキシフェン及びトレミフェンの効果も同様に調べたが、これらの薬剤は効果を示さなかった(パネルB)。これらの結果から、EDR細胞の増殖はEGFRを阻害することにより有意に低減するが、HER2又はER機能の阻害によっては低減せず、EGFRが増殖に重要であることがわかった。
【0069】
結果として、タイプ3細胞は、容易にエストロゲン依存性経路を失い、エストロゲン非依存性かつER非依存性経路を獲得したことが示唆された。タイプ3細胞における細胞内シグナル経路は、親細胞であるTE8におけるそれと異なっており、親細胞がエストロゲン依存性であって主にエストロゲン依存性経路を使うのに対し、タイプ3細胞は、エストロゲン依存性経路を失い、増殖因子依存性経路を使うようになっている。この経路は、主にEGFR及びHER2のような増殖因子レセプターに関与し、PI3K経路が特に重要である。
【0070】
IV. タイプ4細胞株の作製及び特徴づけ
1.タイプ4細胞株の樹立
上記II.のタイプ1細胞の樹立法と同様の手順を、100nMテストステロン(TS)を添加した枯渇培地で行い、E10細胞から複数の安定株を樹立した。GFP発現による蛍光を示すコロニーをピックアップして個別に培養した。このうち、純粋な枯渇培地で蛍光が低下し、テストステロン100nM及び3β−ジオール(Androgen metabolites 5α-androstane-3β, 17β-diol)100nMの添加で再び蛍光を発し、その後のレトロゾール100nMの添加によって蛍光が減弱しないものを、アンドロゲン代謝産物依存性エストロゲン枯渇耐性細胞株(「AD−EDR」(Androgen metabolite dependent EDR cells)と呼ぶことがある)(タイプ4細胞)とした。
【0071】
以下の条件下でER活性の評価を行った。各クローンのER活性は、培養中のGFP陽性細胞の比率によって評価した。まず、ステロイド枯渇TS添加培養におけるER活性を評価した(i)。次に、ステロイド枯渇培養で3日間培養後、同様にER活性を評価し、構成的に活性化されたERを有するGFP陽性クローンを排除した(ii)。継続的に、100nM TS及び100 nM 3β-ジオールを各培地に添加し、24時間の時点でER活性を確認した(iii)。最終的に、100nMレトロゾールの添加後もER活性を有しているクローンを、タイプ4(AD−EDR)細胞株と名づけた(iv)。同時に、これらの細胞株において、ER活性が1μMフルベストラントによって阻害されることを確認した(v)。タイプ4細胞株は、100nM TSを添加したステロイド枯渇培地で維持した。
【0072】
タイプ4細胞株は、アンドロゲン又はその代謝産物に依存してER活性を示すと考えられ、ストローマ細胞との共培養においてレトロゾール耐性ER活性を示した。共培養は、乳癌細胞とストローマ細胞とを可溶性因子が通過しうる膜を隔てて接するようにし、別個に増殖させたもので、ストローマ細胞のアロマターゼによってアンドロゲンから局所的にエストロゲンが合成される微細環境を模したものである。
【0073】
TS添加ステロイド枯渇培地で3ヶ月培養後、GFPを発現する5つのクローンが得られた。各処理工程でのER活性を図8にまとめて示す。
【0074】
5つのうち、G1T及びR6Tは、ER活性がステロイド枯渇培地では阻害されたがレトロゾールによって阻害されなかったため、アンドロゲン又はその代謝産物に依存するER活性を示すものとして、タイプ4細胞として選択された。
樹立されたタイプ4細胞(G1T、R6T)は、アンドロゲンで活性化され、アロマターゼ阻害剤は無効であり、フルベストラントによってER活性が阻害される。このことは、アンドロゲンがエストロゲンに変換されて作用するのではなく、直接エストロゲンレセプターを活性化していることを示唆している。
【0075】
クローンV3T及びB6Tは、増殖因子レセプター経路又はその他のメカニズムにより構成的に活性化されたERを有すると考えられた。これは、3日間のステロイド枯渇条件においてもER活性を維持したためである。クローンR7Tは、ER活性がレトロゾールによって抑制されるため、TSからアロマターゼによって供給されるエストロゲンに依存するER活性を有すると考えられる。
【0076】
2.テストステロン(TS)、ジヒドロテストステロン(DHT)又は3β−ジオール存在下でのER活性及び増殖
定義されたホルモン条件下でのアッセイのために、細胞を数回洗浄した後、枯渇培地で3日間培養した。その後、細胞を枯渇培地中で24ウェル培養プレートに104個/ウェルの密度で播き、所定の濃度のTS、DHT、3β−ジオール、エストラジオール(E2)又はコントロールとしてエタノールを、100nM レトロゾール、1μM フルベストラント、所定の濃度のトリロスタン又はバイカルタミド(bicalutamide)、100nM OHT又は3μM TORの存在下又は非存在下で、各ウェルに添加し、4日間培養した。各ウェルの細胞を回収してカウントし、コントロールに対する相対値を算出した。残りの細胞をER活性の評価に使用した。
【0077】
タイプ4細胞は、TS、DHT又は3β−ジオールの存在下で増大したER活性及び増殖を示した。E2による増殖誘導では親細胞であるE10細胞とタイプ4細胞とで有意差がなかった。低濃度のTS及びDHT(1nM又は10nM)はすべての細胞株の増殖を阻害したが、タイプ4細胞は、100nM TS又は100nM DHTによって親細胞よりも増大した増殖を示した。100nM 3β−ジオールによる増殖誘導は、両細胞株において1nM E2と匹敵するものであった。タイプ4細胞株は、親細胞よりも、低濃度(1nM又は10nM)の3β−ジオールによって増大した増殖を示した。
【0078】
3.タイプ4細胞株におけるDHT代謝酵素の発現及びアンドロゲンシグナル伝達の中断
アンドロゲン産生酵素及びホルモンレセプターのmRNA発現を調べた。また、TS、DHT又は3β−ジオールによるエストロゲン応答性遺伝子及びアンドロゲン応答性遺伝子の誘導も調べた。普通培地又は100nM テストステロンを添加した枯渇培地で3日間培養した細胞から総RNAを抽出し、各種遺伝子について、内部標準としてRPL13Aに対する相対的なmRNA発現量を測定した。
タイプ4細胞株(G1T、R6T)は、DHT代謝酵素である3β−ヒドロキシステロイドジヒドロゲナーゼ1型(HSD3B1)の増大した発現及びアンドロゲンレセプター(AR)の低減した発現を示した。アルド−ケトレダクターゼ(AKR1C3)はG1Tにおいてアップレギュレーションされていた。アロマターゼ(CYP19)及び17β−ヒドロキシステロイドデヒドロゲナーゼ2型(HSD17B2)はすべての細胞株で検出されなかった。
タイプ4細胞は、親細胞であるE10細胞と比較して、TS、DHT又は3β−ジオールにより増大したエストロゲン応答性遺伝子PgR及びEGR3の発現を示した。さらに、タイプ4細胞においては、TS、DHT又は3β−ジオールによってエストロゲン応答性抗アポトーシスタンパクBcl−2が誘導された。
これらの結果から、タイプ4細胞においては、DHT代謝がアップレギュレーションされており、アンドロゲンシグナル伝達がほとんど停止していることが示唆された。さらに、タイプ4細胞においては、TS又はDHTがより効率的に3β−ジオールに代謝されること、及び3β−ジオールがより効率的にエストロゲン様作用を示すことが示唆された。
【0079】
4.タイプ4細胞株に対する抗エストロゲン剤の効果
タイプ4細胞におけるDHTによる増殖誘導は、アロマターゼに依存せずに産生されるDHT代謝産物3β−ジオールによるER活性化に起因する。そこで、臨床適用可能な選択的エストロゲンレセプター調節剤(SERM)であるOHT及びTOR(これらはERαリガンドの結合を阻害する)がフルベストラント(ICI又はFul)と同様にDHT誘導細胞増殖を阻害するかどうかを調べた。具体的には、100nM DHT添加と同時にOHT(1μM)又はTOR(3μM)を、G1T細胞及びR6T細胞にそれぞれ添加して、4日後、細胞数を計測して増殖能を評価した。
結果を図9に示す。予想どおり、エストロゲンレセプターをブロックする抗エストロゲン剤であるOHT又はTORによってICIと同様にタイプ4細胞の増殖を効率的に阻害したが、アロマターゼ阻害剤であるレトロゾール(Let)は無効であることが示された。
これらの結果は、タイプ4細胞株がアンドロゲン耐性及び3β−ジオールに対する過敏性を示すことを示唆する。さらに、これらの特性は、アロマターゼ活性には依存しなかった。
【0080】
5.ER活性
ERE−ルシフェラーゼレポーターアッセイにより、100nM DHTはタイプ4細胞においてER活性を誘導したが、親細胞においては誘導しなかったことがわかった。タイプ4細胞においては、100nM 3β−ジオールにより誘導されたER活性は、親細胞における活性の2倍高かった。TSにより誘導されるER活性はこのアッセイではすべての細胞株で検出されなかったが、ERE−GFPレポーターアッセイではタイプ4細胞において増大したER活性が検出された。
TS、DHT又は3β−ジオールによって誘導されたER活性は、1μM フルベストラントによって阻害されたが、100nM レトロゾールによっては阻害されなかった。同様の結果は、すべての細胞株の増殖において得られた。
【0081】
V. タイプ5細胞株の作製及び特徴づけ
1.親株(E10−Arom細胞)の樹立
E10細胞にブラストシジン(BSD)耐性遺伝子を持つアロマターゼ発現ベクター(pNH−CMV/AROM−BSD;図10)を導入し、ブラストシジン(10μg/mL)添加普通培地で形質転換細胞を選択した。生き残ったコロニーを顕微鏡下でマイクロピペットを用いて剥離し、96ウェル培養プレートに単離した。その後、24ウェル培養プレートに移し、その際に培地をブラストシジン添加普通培地からテストステロン100nM添加枯渇培地に交換した。
この細胞は自身でアロマターゼを産生しテストステロンをエストラジオールに変換できるので、テストステロン添加枯渇培地でも増殖できる。さらに6cmディッシュ、10cmディッシュと段階的に大きな培養容器に移しつつ細胞数を増やし、継続的にテストステロン添加枯渇培地で培養を続け、安定的なアロマターゼ発現E10細胞株(E10−Arom)を樹立した。
【0082】
2.タイプ5耐性株の作製
このE10−Arom細胞を親株として用い、アロマターゼ阻害剤であるレトロゾール100nM及びテストステロン100nM添加枯渇培地で3ヶ月間培養を続け、タイプ1細胞の樹立法と同様にコロニーを形成させた。このうち、レトロゾール100nM及びテストステロン100nM添加枯渇培地で蛍光が低下せず、純粋な枯渇培地で蛍光が低下するものを選択的にクローニングし、レトロゾール耐性MCF−7−E10Arom−LR細胞(Letrozole resistant cells)(タイプ5細胞)とした(クローンLR2、LR3)。
【0083】
3.LR細胞におけるレトロゾール添加時のER活性
樹立したレトロゾール耐性細胞8株について、テストステロン 100nM、レトロゾール 100nM添加時のER活性をルシフェラーゼアッセイで測定した。
【0084】
結果を図11に表す。この中で、特にLR3は親株であるE10−Aromに比べてレトロゾール添加時にも高いER活性を維持していた。
【0085】
4.LR細胞におけるレトロゾール感受性の検討
レトロゾール添加時に特に高いER活性を示していたLR3について、レトロゾール感受性の検討を行った。
親株であるE10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、テストステロン(0.1〜100nM)、レトロゾール(100nM)を添加し、4日後にSysmex CDA-500にて細胞数を測定した。
【0086】
結果を図12に表す。LR3は、E10Aromに比べて、レトロゾール添加時にも高い細胞増殖能を維持していた。
【0087】
5.LR細胞におけるエストロンスルフェート(EIS)代謝関連遺伝子の発現
LR3において、E1SをESに変換する酵素であるSTSと、E1Sを細胞内に取り込むトランスポーターであるOATP−AのmRNA発現を、リアルタイムRT−PCRで測定した。
【0088】
結果を図13に表す。LR3は、E10−Aromと比較してSTS、OATP−A共に発現が上昇していることが確認された。
【0089】
6.LR細胞におけるEIS、STX64(STS阻害剤)感受性の検討
耐性獲得におけるSTSの関与が示唆されたので、LR3のE1SとSTS阻害剤であるSTX64に対する感受性を検討した。
親株であるE10−Arom、LR3を3日間エストロゲン枯渇培養後、24ウェル培養プレートに1.0×104個/ウェル播種、E1S(0、100nM〜10μM)単独あるいはSTX64(100nM)と組み合わせて添加し、4日後にSysmex CDA-500にて細胞数を測定した。
【0090】
結果を図14に表す。LR3は、E10AromよりもE1Sに対する感受性が高まっていることが確認された。さらに、LR3においてE1Sによって誘導された細胞増殖は、STX64添加で親株と同程度まで抑制されることが確認された。
【特許請求の範囲】
【請求項1】
ホルモン療法に耐性を示す乳癌の治療剤をスクリーニングする方法であって、エストロゲン応答配列の制御下で緑色蛍光タンパク質(GFP)遺伝子を発現しうるホルモン療法耐性乳癌細胞に被験物質を接触させる工程と、この細胞を培養し、細胞の増殖及び/又はGFPの発現を測定する工程と、前記被検物質と接触させていない前記ホルモン療法耐性乳癌細胞を同じ条件下で培養及び測定した結果と比較して前記被検物質が細胞の増殖及び/又はGFPの発現の低減をもたらしたかどうかを判定する工程とを含み、
前記ホルモン療法耐性乳癌細胞が、
エストロゲン非依存性、かつ、エストロゲンレセプター(ER)活性依存性である細胞株(タイプ1)、
エストロゲン非依存性、かつ、ER活性非依存性である細胞株(タイプ2)、
エストロゲン非依存性、ER活性非依存性、かつ、成長因子(GF)依存性であり、HER2及び上皮増殖因子レセプター(EGFR)を発現する細胞株(タイプ3)、
アンドロゲン代謝産物依存性である細胞株(タイプ4)、及び/又は
アロマターゼ阻害剤(AI)非感受性、かつ、ER活性依存性である細胞株(タイプ5)、
のいずれか1以上であることを特徴とする、乳癌治療剤スクリーニング方法。
【請求項2】
前記ホルモン療法耐性乳癌細胞として、タイプ1〜タイプ5のうち2以上の細胞株を用いてそれぞれの細胞を別々に被験物質に接触させる、請求項1記載のスクリーニング方法。
【請求項3】
前記ホルモン療法耐性乳癌細胞が、
受領番号FERM AP−22142であるタイプ1細胞、
受領番号FERM AP−22143であるタイプ2細胞、
受領番号FERM AP−22144であるタイプ3細胞、
受領番号FERM AP−22145であるタイプ4細胞、及び/又は
受領番号FERM AP−22146であるタイプ5細胞、
である、請求項1又は2記載のスクリーニング方法。
【請求項4】
受領番号FERM AP−22142である細胞。
【請求項5】
受領番号FERM AP−22143である細胞。
【請求項6】
受領番号FERM AP−22144である細胞。
【請求項7】
受領番号FERM AP−22145である細胞。
【請求項8】
受領番号FERM AP−22146である細胞。
【請求項1】
ホルモン療法に耐性を示す乳癌の治療剤をスクリーニングする方法であって、エストロゲン応答配列の制御下で緑色蛍光タンパク質(GFP)遺伝子を発現しうるホルモン療法耐性乳癌細胞に被験物質を接触させる工程と、この細胞を培養し、細胞の増殖及び/又はGFPの発現を測定する工程と、前記被検物質と接触させていない前記ホルモン療法耐性乳癌細胞を同じ条件下で培養及び測定した結果と比較して前記被検物質が細胞の増殖及び/又はGFPの発現の低減をもたらしたかどうかを判定する工程とを含み、
前記ホルモン療法耐性乳癌細胞が、
エストロゲン非依存性、かつ、エストロゲンレセプター(ER)活性依存性である細胞株(タイプ1)、
エストロゲン非依存性、かつ、ER活性非依存性である細胞株(タイプ2)、
エストロゲン非依存性、ER活性非依存性、かつ、成長因子(GF)依存性であり、HER2及び上皮増殖因子レセプター(EGFR)を発現する細胞株(タイプ3)、
アンドロゲン代謝産物依存性である細胞株(タイプ4)、及び/又は
アロマターゼ阻害剤(AI)非感受性、かつ、ER活性依存性である細胞株(タイプ5)、
のいずれか1以上であることを特徴とする、乳癌治療剤スクリーニング方法。
【請求項2】
前記ホルモン療法耐性乳癌細胞として、タイプ1〜タイプ5のうち2以上の細胞株を用いてそれぞれの細胞を別々に被験物質に接触させる、請求項1記載のスクリーニング方法。
【請求項3】
前記ホルモン療法耐性乳癌細胞が、
受領番号FERM AP−22142であるタイプ1細胞、
受領番号FERM AP−22143であるタイプ2細胞、
受領番号FERM AP−22144であるタイプ3細胞、
受領番号FERM AP−22145であるタイプ4細胞、及び/又は
受領番号FERM AP−22146であるタイプ5細胞、
である、請求項1又は2記載のスクリーニング方法。
【請求項4】
受領番号FERM AP−22142である細胞。
【請求項5】
受領番号FERM AP−22143である細胞。
【請求項6】
受領番号FERM AP−22144である細胞。
【請求項7】
受領番号FERM AP−22145である細胞。
【請求項8】
受領番号FERM AP−22146である細胞。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2013−17414(P2013−17414A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2011−152458(P2011−152458)
【出願日】平成23年7月11日(2011.7.11)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度 医薬基盤研究所基礎研究推進事業、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成23年7月11日(2011.7.11)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度 医薬基盤研究所基礎研究推進事業、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
[ Back to top ]