
ボリコナゾールおよび抗真菌性CYP2C19阻害薬の組合わせ
本発明は、ボリコナゾールおよび抗真菌性CYP2C19阻害薬を特定の量および重量比で含む組合わせ薬剤を提供する。ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含む医薬組成物、単位剤形およびキット、ならびに真菌感染症の処置におけるそれらの使用も提供する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
発明の分野
本発明は、ボリコナゾール(voriconazole)を含む新規な併用療法に関する。
発明の背景
(2R,3S)-2-(2,4-ジフルオロフェニル)-3-(5-フルオロ-4-ピリミジニル)-1-(1H-1,2,4-トリアゾール-1-イル)-ブタン-2-オール(ボリコナゾールとしても知られる)は、EP-A-440372に開示されている;特に例7を参照。ボリコナゾールは下記の構造:
【0002】
【化1】

【0003】
をもち、真菌感染症の処置に有用である。
ボリコナゾールの薬物動態は、投与レベルの増加に伴う曝露の増大が非直線的になる飽和型の代謝を特徴とする。さらに、薬物曝露は対象間で著しく変動する。ボリコナゾールはシトクロムP450のアイソザイムCYP2C19、CYP2C9およびCYP3A4により代謝される。主な循環性代謝産物(その構造を下記に示す)は、N-酸化により生成する。
【0004】
【化2】
【0005】
本発明者らは、ボリコナゾールの代謝は処置される対象の遺伝子型に大幅に依存することを見いだした。ある遺伝子型はボリコナゾールを高度に代謝してボリコナゾールを体内から急速にクリアランスし、その結果、血漿ボリコナゾール濃度は低下する(約0.6〜約1.4μg/ml)。本明細書中ではこの遺伝子型を”高代謝型(extensive metaboliser)”と呼ぶ。第2の遺伝子型は、ボリコナゾール低代謝型と特徴づけることができる:すなわちこの遺伝子型の場合、ボリコナゾールがクリアランスされる速度がはるかに遅く、したがってより高濃度で体内に留まる(約3.5〜約5.5μg/ml)。本明細書中ではこの遺伝子型を”低代謝型(poor metaboliser)”と呼ぶ。この遺伝子型は、劣性遺伝子によるものと考えられる:すなわちホモ接合性の高代謝型は、ほぼ白人集団の73%、日本人集団の35%を占める;ヘテロ接合性の高代謝型は、ほぼ白人集団の25%、日本人集団の46%を占める。これに対し、低代謝型は、ほぼ白人集団の2%、日本人集団の19%を占めるにすぎない。
【0006】
ボリコナゾールの代謝に関する遺伝子型間の変動は体内の酵素シトクロムP450 2C19(以下、CYP2C19と呼ぶ)の存在度に依存することが現在分かっている:すなわち、高代謝型には酵素CYP2C19が存在し、一方、低代謝型はこの機能性酵素を欠如する。たとえば、M. de Morais, G.Wilkinson, J.Blaisdell et al., J Biol Chem (1994), 269, 15419-15422を参照(その全体を本明細書に援用する)。
【0007】
実際には、ボリコナゾールは高代謝型と低代謝型の両方に用量調整なしに投与されている。しかし、高代謝型に療法効果を及ぼすのに十分な量でボリコナゾールが血漿中に存在するためには、高用量の薬物が必要である:すなわち通常の推奨1日量は400 mg(200 mgを1日2回)である。この用量は、低代謝型では全身曝露がより高くなり、このため望ましくない副作用の起きる可能性がある。さらに、高代謝型による急速な薬物クリアランスのため、血漿中濃度を1日中維持して療法効果を及ぼすには、この化合物を1日2回投与する必要がある。ボリコナゾールを患者が自己投与する場合、1日2回の療法はコンプライアンス問題を提起する。
【0008】
M. Ghannoum, N. Isham, M. Hossain and D. Sheehan, Int. J. Infect. Dis. (2002), Vol.6, Supp.2, 2S50には、ボリコナゾールと、アンホテリシンB(amphotericin B)、Abelcet(商標)、5-フルオロシトシンおよびフルコナゾール(fluconazole)を含めた抗真菌薬とのインビトロ組合わせが示され、広範な生物に対するそれらの相乗作用機序が調べられている。ボリコナゾールとフルコナゾールの組合わせは39%が相加的、61%が無関係と記載されている。
【0009】
M. Ghannoum, N. Isham and D. Sheehan, Abstracts of the Interscience Conference on Antimicrobial Agents and Chemotherapy (2002), 42, 385にも、アンホテリシンB、Abelcet(商標)、フルコナゾール、ミカフンジン(micafungin)、ラブコナゾール(ravuconazole)およびカスポフンジン(caspofungin)を含めた抗真菌薬とのインビトロ組合わせが示されている。ボリコナゾールとフルコナゾールの組合わせは100%が無関係、すなわち相乗作用機序はないと記載されている。
【0010】
H.J. Scherpbier, M.I. Hilhorst and T.W. Kuijpers, Clin. Infect. Dis. (2003), 37, 828には、抗レトロウイルス薬の組合わせによるエイズ患者の治療が示され、食道カンジダ症の治療のために患者の療法にボリコナゾールを加えた場合のプロテアーゼ阻害薬とボリコナゾールの相互作用が報告されている。この相互作用は、ロピナビル(lopinavir)、ネビラピン(nevirapine)およびアンプレナビル(amprenavir)について肝機能不全および血漿中濃度上昇を伴った。それらの患者において血漿ボリコナゾール濃度は測定されなかった。
【0011】
N. Wood, K. Tan, L. Purkins, G. Layton, J. Hamlin, D. Kleinermans and D. Nichols, Br. J. Clin. Pharmacol. (2003) 56, 56には、プロトンポンプ阻害薬オメプラゾール(omeprazole)、すなわちCYP2C19阻害薬がボリコナゾールの定常状態薬物動態に及ぼす影響を調べる研究が記載されている。この研究では、オメプラゾールはボリコナゾール曝露に対して臨床関連作用をもたないと結論された。これは、オメプラゾール療法を開始する患者についてボリコナゾールの用量調整が不必要なことを示唆する。
【0012】
対象間変動を排除または低減するボリコナゾール療法を提供することが望ましいであろう。さらに、ボリコナゾールの投与量が減少したボリコナゾール療法を提供することが望ましいであろう。さらに、血漿からの薬物クリアランスを低下させ、これによりボリコナゾールを1日1回投与すればよいボリコナゾール療法を提供することが望ましいであろう。
【0013】
本発明者らは意外にも、ボリコナゾールとCYP2C19の活性を阻害しうる第2の異なる抗真菌薬との共投与によりCYP2C19酵素を阻害することによって、ボリコナゾール高代謝型において代謝が顕著に低下し、それらの対象における薬物動態プロフィールが低代謝型に近づくことを見いだした。その結果、対象間変動が顕著に低下し、はるかに低用量のボリコナゾールで療法用血漿中濃度に達する。さらに、体内からのボリコナゾールのクリアランスが低下し、ボリコナゾールは1日1回投与した場合に1日中、療法効果を達成しうるのに十分な血漿中濃度で存在する。
【0014】
発明の概要
本発明は、第1観点において、ボリコナゾールおよび抗真菌性CYP2C19阻害薬を後記にさらに詳細に定める特定の量または重量比で含む、組合わせ薬剤を提供する。
【0015】
本発明は、第2観点において、療法有効量のボリコナゾールおよび抗真菌性CYP2C19阻害薬を医薬的に許容できるキャリヤーまたは希釈剤と共に含む、医薬組成物をも提供する。
【0016】
本発明は、第3観点において、療法有効量のボリコナゾールおよび療法有効量の抗真菌性CYP2C19阻害薬を含む、単位剤形をも提供する。
本発明はさらに、第4観点において、複数の個別容器を含み、少なくとも1つの容器がボリコナゾールを収容し、少なくとも1つの異なる容器が抗真菌性CYP2C19阻害薬を収容した、キットを提供する。
【0017】
本発明はさらに、第5観点において、哺乳動物において真菌感染症を処置するための医薬の製造における、前記の組合わせ薬剤、組成物、キットまたは単位剤形の使用を提供する。
【0018】
本発明は、第6観点において、哺乳動物において真菌感染症を処置する方法であって、その処置を必要とする哺乳動物に、有効量の前記の組合わせ薬剤、組成物、キットまたは単位剤形を投与することを含む方法を提供する。
【0019】
以下において、本発明の組合わせ薬剤、医薬組成物、単位剤形、キット、使用および方法を合わせて”本発明の組合わせ”という。
好ましい態様の詳細な記述
本発明の組合わせの一方の成分はボリコナゾールである。ボリコナゾールはEP-A-440372に開示されている(その全体を本明細書に援用する);特に例7を参照。後記に詳述するように、ボリコナゾールは遊離塩基として、またはその塩、溶媒和物もしくはプロドラッグの形で投与できる。
【0020】
本発明の組合わせの他方の成分は、抗真菌性CYP2C19阻害薬である。この抗真菌薬が抗真菌活性を示し、かつ哺乳動物CYP2C19酵素の阻害薬として作用しうる限り、抗真菌薬の厳密な性質は特に限定されない。本明細書において”CYP2C19阻害薬”には、哺乳動物CYP2C19酵素の作用を阻害しうる化合物がいずれも含まれる;Desta et al. (2002) Clinical Pharmacokinetics 41(12), 913-958による概説に記載(その全体を本明細書に援用する)。一般的な療法用量でのボリコナゾール代謝を確実に阻害するために、好ましくは10μM未満のKj値が必要である。Kjを測定するための標準的なインビトロ試験には、プローブ基質として(S)-メフェニトイン((S)-mephenytoin)を用い、4'-ヒドロキシル化を測定する−Meier et al. (1985) Anal. Biochem. 151, 286-291を参照(その全体を本明細書に援用する)。抗真菌性CYP2C19阻害薬の例にはフルコナゾールが含まれ、これは2μMのKjをもつ−L.C. Winkers, C.J. Wurden, E. Storch et al., Drug Metab Dispos (1996) 24, 610-4を参照。CYP2C19阻害薬の他の例は、Desta et al. (2002)(前記)に記載されている。
【0021】
抗真菌性CYP2C19阻害薬は、酵素CYP3A4より酵素CYP2C19によるボリコナゾール代謝を選択的に阻害できることが好ましい。これらのアイソザイムに対する選択性は、(S)-メフェニトイン4-ヒドロキシラーゼ活性およびテストステロン6ベータヒドロキシラーゼ活性に対する相対阻害力価により判定できる:後者の活性の測定についてはFuna and Imaoka (1987) Biochem. Biophys. Acta. 926, 349-358に記載されている(その全体を本明細書に援用する)。より具体的には、選択性はボリコナゾールN-酸化に対する作用により証明でき、それぞれ25および2500μMの基質濃度を用いてCYP2C19(高親和性酵素)および3A4(低親和性酵素)に対する作用を評価する。シトクロムP450アイソザイムによるボリコナゾールN-酸化を測定する方法は、Hyland, Jones and Smith (2003) Drug Metabolism and Disposition, 31(5), 540-547に記載されている(その全体を本明細書に援用する)。要求される選択性の厳密なレベルは、抗真菌性CYP2C19阻害薬ならびにその薬物動態および対象間変動に依存するが、抗真菌性CYP2C19阻害薬がCYP3A4よりCYP2C19に対して2〜10、好ましくは3〜6の選択性を示すことが好ましい(前記のHylandらの報文中におけるそれらの相対IC50により測定)。理論により拘束されるつもりはないが、CYP3A4よりCYP2C19を選択的に阻害すると、対象間変動を低下させ、より低いボリコナゾール用量で療法用血漿中濃度に到達でき、一方、現在の推奨用量方式では患者集団に起きる可能性のある望ましくない副作用を軽減することができると考えられる。低濃度ではCYP2C19が主なクリアランス経路であるが、高濃度ではCYP3A4経路の代謝、すなわち非飽和型のボリコナゾールクリアランスが主体となる。
【0022】
抗真菌性CYP2C19阻害薬は、ボリコナゾール/抗真菌性CYP2C19阻害薬の組合わせを1日1回投与した場合に1日中、血漿中濃度を維持して療法効果を達成しうるのに適切な、薬物動態半減期をもつことが好ましい。適切な半減期は、6〜72時間、好ましくは12〜48時間、より好ましくは18〜36時間である。
【0023】
抗真菌性CYP2C19阻害薬は、主に未変化薬物として身体から排出されることが好ましい。理論により拘束されるつもりはないが、主に未変化薬物として身体から排出される抗真菌性CYP2C19阻害薬が、抗真菌性CYP2C19阻害薬およびボリコナゾール両方の対象間変動を低下させると考えられる。好ましくは50〜99%、より好ましくは70〜80%、最も好ましくは約75%の阻害薬が未変化のまま身体から排出される。
【0024】
抗真菌性CYP2C19阻害薬は抗真菌活性を示す。抗真菌薬でもあるCYP2C19阻害薬の例には、アゾール類、たとえばフルコナゾールが含まれる。これは、この組合わせが抗真菌作用以外の作用を示さないという利点をもつ。この第2の抗真菌薬は、好ましくは抗真菌活性に対する相加作用をもち、好ましくは同一患者集団に使用するのに安全であることが証明できるものである。全体的な抗真菌効果は、もちろん処置される個々の感染症、ボリコナゾールおよび抗真菌性CYP2C19阻害薬の投与量、ならびに処置される患者の年齢、性別、体重および状態に依存する。
【0025】
抗真菌性CYP2C19阻害薬はフルコナゾールであることが特に好ましい。
本発明の組合わせは、ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含む。ボリコナゾールおよび抗真菌性CYP2C19阻害薬は、遊離の酸もしくは塩基として、またはその医薬的に許容できる塩、溶媒和物もしくはプロドラッグの形で投与できる。
【0026】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬の医薬的に許容できる塩には、その酸付加塩および塩基塩が含まれる。
適切な酸付加塩は、無毒性の塩を形成する酸から形成できる。例には、酢酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、炭酸水素塩/炭酸塩、硫酸水素塩/硫酸塩、ホウ酸塩、カムシル酸塩、クエン酸塩、エジシル酸塩、エシル酸塩、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンズ酸塩、塩酸塩/クロリド、臭化水素酸塩/ブロミド、ヨウ化水素酸塩/ヨージド、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシル酸塩、メチル硫酸塩、ナフチル酸塩、2-ナプシル酸塩、ニコチン酸塩、硝酸塩、オロット酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素塩/リン酸二水素塩、サッカリン酸塩、ステアリン酸塩、コハク酸塩、酒石酸塩、トシル酸塩およびトリフルオロ酢酸塩が含まれる。
【0027】
適切な塩基塩は、無毒性の塩を形成する塩基から形成できる。例には、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオラミン、グリシン、リシン、マグネシウム、メグルミン、オーラミン、カリウム、ナトリウム、トロメタミンおよび亜鉛の塩が含まれる。
【0028】
適切な塩類の概説については、”Handbook of Pharmaceutical Salts : Properties, Selection, and Use”, Stahl and Wermuth (Wiley-VCH, ドイツ、ワインハイム, 2002)を参照されたい。
【0029】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬の医薬的に許容できる塩は、ボリコナゾールまたは抗真菌性CYP2C19阻害薬の溶液を目的の酸または塩基と混和することによって、容易に調製できる。塩は、溶液から沈殿させて濾過により回収でき、または溶媒の蒸発により回収できる。塩のイオン化度は完全イオン化からほぼ非イオン化まで変動する可能性がある。
【0030】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬は、溶媒和していない形および溶媒和した形の両方で存在することができる。本明細書において用語’溶媒和物’は、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬と1以上の医薬的に許容できる溶媒分子、たとえばエタノールを含む、分子複合体を記載するために用いられる。用語’水和物’は、溶媒が水である場合に用いられる。
【0031】
以下において、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬という表現はすべて、その塩類、溶媒和物および複合体、ならびにその塩類の溶媒和物および複合体という表現を含む。
【0032】
本発明の組合わせには、前記に定めたボリコナゾールおよび抗真菌性CYP2C19阻害薬、その多形およびプロドラッグが含まれる。
上記のように、本発明は前記に定めたボリコナゾールおよび/または抗真菌性CYP2C19阻害薬のすべての多形を含む。
【0033】
本発明の範囲には、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬のいわゆる’プロドラッグ’も含まれる。たとえば、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬のある種の誘導体(それ自体はほとんどまたは全く薬理活性をもたなくてもよい)が、体内または身体上に投与した際に、たとえば加水分解開裂により変換されて目的活性をもつ化合物になることができる。そのような誘導体を’プロドラッグ’と呼ぶ。プロドラッグの使用に関する情報はさらに、’Pro-drugs as Novel Delivery Systems’, Vol. 14, ACS Symposium Series (T Higuchi and W Stella)、および’Bioreversible Carriers in Drug Design’, Pergamon Press, 1987 (編者E B Roche, American Pharmaceutical Association)中にみられる。
【0034】
本発明によるプロドラッグは、たとえばボリコナゾールおよび/または抗真菌性CYP2C19阻害薬中に存在する適切な官能基を、当業者に’プロ部分’として知られる特定の分子と交換することにより製造できる:たとえば”Design of Prodrugs”, 編者H Bundgaard (Elsevier, 1985)に記載。
【0035】
ボリコナゾールはアルコール官能基(-OH)を含み、したがって本発明によるプロドラッグのある例はそのエステルまたはエーテルを含むことができる:たとえば、その水素をリン酸化により置換してリン酸二水素(2R, 3S)-2-(2,4-ジフルオロフェニル)-3-(5-フルオロ-4-ピリミジニル)-1-(1H-1,2,4-トリアゾール-1-イル)-2-ブチルを得ることによる;WO 97/28169に記載(その全体を本明細書に援用する);特に例3を参照。
【0036】
同様に、抗真菌性CYP2C19阻害薬がフルコナゾールである場合、この化合物もアルコール官能基(-OH)を含み、したがって本発明によるプロドラッグのある例は下記のものを含むことができる:そのエステルもしくはエーテル、たとえばその水素をリン酸化により置換してリン酸二水素2-(2,4-ジフルオロフェニル)-1,3-ビス(1H-1,2,4-トリアゾール-1-イル)-2-プロピルを得ることによる;WO 97/28169;特に例1を参照;またはその医薬的に許容できる塩、特に二ナトリウム塩(Prodif(登録商標))。上記の例による置換基の他の例および他のタイプのプロドラッグの例は、前記の参考文献中にみられる。
【0037】
抗真菌性CYP2C19阻害薬は1個以上の不斉炭素原子を含む場合があり、したがって2種類以上の立体異性体として存在する可能性がある。抗真菌性CYP2C19阻害薬がアルケニルまたはアルケニレン基を含む場合、シス/トランス(またはZ/E)幾何異性体の可能性がある。これらの化合物がたとえばケトもしくはオキシム基または芳香族部分を含む場合、互変異性(tautomeric isomerism、'tautomerism')が起きる可能性がある。その結果、単一化合物が1より多いタイプの異性を示す場合がある。
【0038】
本発明の範囲には、すべての立体異性体、幾何異性体および互変異性体の形の抗真菌性CYP2C19阻害薬が含まれ、これには1より多いタイプの異性を示す化合物、およびその1種類以上の混合物も含まれる。対イオンが光学活性である酸付加塩または塩基塩、たとえばD-乳酸塩もしくはL-リシン塩、またはラセミ体、たとえばDL-酒石酸塩もしくはDL-アルギニン塩も含まれる。
【0039】
シス/トランス異性体は、当業者に周知の常法、たとえばクロマトグラフィーおよび分別結晶化により分離できる。
個々の鏡像異性体を製造/単離するための常法には、適切な光学的に純粋な前駆物質からのキラル合成、またはたとえばキラル高速液体クロマトグラフィー(HPLC)によるラセミ体(または塩もしくは誘導体のラセミ体)の分割が含まれる。
【0040】
あるいは、ラセミ体(またはラセミ前駆物質)を適切な光学活性化合物、たとえばアルコール類と反応させ、または抗真菌性CYP2C19阻害薬が酸性もしくは塩基性部分を含む場合は酸もしくは塩基、たとえば酒石酸もしくは1-フェニルエチルアミンと反応させることができる。得られたジアステレオマー混合物をクロマトグラフィーおよび/または分別結晶化により分離し、一方または両方のジアステレオ異性体を当業者に周知の方法で対応する純粋な鏡像異性体に変換することができる。
【0041】
本発明の組合わせに用いるキラル化合物(およびそのキラル前駆物質)は、クロマトグラフィー、一般にHPLCにより、不斉樹脂上で、一般に0〜50 %(一般に2〜20 %)のイソプロパノールおよび0〜5 %のアルキルアミン(0.1 %のジエチルアミン)を含有する炭化水素、一般にヘプタンまたはヘキサンからなる移動相を用いて、鏡像異性体を富化した形で得ることができる。溶出液の濃縮により、富化した混合物が得られる。
【0042】
立体異性体の集合体は、当業者に周知の常法により分離することができる−たとえば、”Stereochemistry of Organic Compounds”, E L Eliel (Wiley, ニューヨーク, 1994)を参照。
【0043】
本発明の組合わせは、抗真菌薬として有効な、かつCYP2C19酵素の作用を阻害するのに有効な、好ましくはこの酵素をCYP3A4酵素より選択的に阻害する、ある量の抗真菌性CYP2C19阻害薬を含む。厳密な投与量は、処置される患者の年齢、性別、体重および状態など、多様な要因に依存する。しかし、投与量は約5〜約500 mg、より好ましくは約10〜約250 mg、さらに好ましくは約50〜約200 mg、最も好ましくは約75〜約125 mgであることが好ましい。好ましくは、抗真菌性CYP2C19阻害薬はフルコナゾールである。
【0044】
本発明の組合わせは、抗真菌薬として作用するある量のボリコナゾールを含む。厳密な投与量は、処置される患者の年齢、性別、体重および状態など、多様な要因に依存する。しかし、投与量は約5〜約600 mg、より好ましくは約10〜約500 mg、さらに好ましくは約20〜約300 mg、最も好ましくは約25〜約250 mgであることが好ましい。約200 mgの投与量が特に好ましい;これは、ボリコナゾールの投与量を通常の400 mgレベルから半減でき、したがって望ましくない副作用を軽減できるという点で、本発明による著しい利点をもたらす。
【0045】
好ましい態様において、本発明の組合わせは患者に1日1回投与される。これは、患者のコンプライアンスがより良好であるという格別の利点をもたらす。しかし、1日2、3または4回の投与も本発明の範囲に含まれるものとする。ただし、各回当たりの用量はさらに少なくなるであろう。
【0046】
本発明の組合わせの成分の重量比は、処置される患者の年齢、性別、体重および状態など、多様な要因に依存する。しかし、ボリコナゾールと抗真菌性CYP2C19阻害薬を約1:4〜約6:1、好ましくはが約1:2〜約3:1、より好ましくは約3:2〜約5:2の重量比で投与することが好ましい。好ましくは、抗真菌性CYP2C19阻害薬はフルコナゾールである。
【0047】
本発明の組合わせにより処置できる真菌感染症は、EP-A-440372を含めた文献中に十分に記載されており、局所感染症、粘膜感染症、たとえば膣カンジダ症、食道および口腔咽頭カンジダ症、ならびに全身感染症を含む。さらに、本発明の組合わせはアレルギー反応、たとえばアレルギー性鼻副鼻腔炎の処置にも使用できる。本発明の組合わせにより処置できる真菌感染症には、特に下記のものにより起きる感染症が含まれる:カンジダ属菌種(Candida spp)、トリコフィトン属菌種(Trichophyton spp)、ミクロスポルム属菌種(Microsporum spp)、有毛表皮糸状菌(Epidermophyton floccosum)、クリプトコックス-ネオフォルマンス(Cryptococcus neoformans)、アスペルギルス属菌種(Aspergillus spp)、フザリウム属菌種(Fusarium spp)、スセドスポリウム属菌種(Scedosporium spp)、コクシジオイデス-イミチス(Coccidioides immitis)、パラコクシジオイデス-ブラジリエンシス(Paracoccidioides brasiliensis)、ヒストプラズマ属菌種(Histoplasma spp)、ブラストマイセス-デルマチジチス(Blastomyces dermatiditis)、アルテルナリア属菌種(Alternaria spp)、エキソフィアラ属菌種(Exophiala spp)、フォンセカ-ペドロソイ(Fonsecaea pedrosoi)、ペニシリウム-マルネフェイ(Penicillium marneffei)、フィアロフォラ属菌種(Phialophora spp)またはペシロミセス-リラシヌス(Paecilomyces lilacinus)。処置という表現が予防および樹立した症状の軽減を含むものであることは自明であろう。
【0048】
本発明の組合わせにおいて、ボリコナゾールおよび抗真菌性CYP2C19阻害薬は、剤形に関しては別個もしくは互いに一緒に、またそれらの投与時間に関しては同時もしくは逐次、投与することができる。たとえば、ボリコナゾールの投与が抗真菌性CYP2C19阻害薬の投与前、投与と同時、または投与後であってよい。投与間の時間は24時間以内の投与間隔で変更できる。
【0049】
本発明の単位剤形は、ボリコナゾールおよび抗真菌性CYP2C19阻害薬の両方が存在する剤形である。それは、経口投与用の固体配合物、たとえば錠剤、カプセル剤(粒子、液体もしくは粉末を収容したもの)、トローチ剤(液体充填型を含む)、咀しゃく剤、ゲル剤、固溶体、リポソーム剤、フィルム剤(粘膜接着型を含む)、卵形錠、スプレー剤または液体配合物、非経口配合物(一般に、後記に定める賦形剤を含有してもよい水性液剤)、あるいは皮膚または粘膜に局所投与(すなわち皮膚投与または経皮投与)するための配合物、たとえばヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散粉剤、包帯剤、発泡剤、皮膚パッチ、カシェ剤、埋込み剤、スポンジ、繊維、包帯またはマイクロエマルションであってよい。好ましくは本発明の単位剤形は、ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含有する錠剤またはカプセル剤、特に錠剤である。
【0050】
医薬として使用するための本発明の組合わせの化合物は、結晶質または非晶質生成物として投与できる。それらは、たとえば固体プラグ、粉末またはフィルムとして、たとえば沈殿、結晶化、凍結乾燥、噴霧乾燥または蒸発乾燥などの方法により得ることができる。マイクロ波乾燥または高周波乾燥も、この目的に使用できる。
【0051】
一般に本発明化合物は、1種類以上の医薬的に許容できる賦形剤と一緒に配合物として投与される。用語”賦形剤”は、本明細書中で本発明化合物以外の成分を記載するために用いられる。賦形剤の選択は、個々の投与様式、賦形剤が溶解度および安定性に及ぼす影響、ならびに剤形の性質などの要因に大幅に依存するであろう。
【0052】
本発明化合物の送達に適切な医薬組成物およびそれらの調製方法は、当業者に自明であろう。そのような組成物およびそれらの調製方法は、たとえば’Remington's Pharmaceutical Sciences’,第19版(Mack Publishing Company, 1995)にみられる。
【0053】
本発明化合物は経口投与することができる。経口投与は化合物が消化管に入るように嚥下を伴い、あるいは化合物が口から血流に直接入る口腔投与または舌下投与を採用できる。
【0054】
経口投与に適切な配合物には、固体配合物、たとえば錠剤、カプセル剤(粒子、液体もしくは粉末を収容したもの)、トローチ剤(液体充填型を含む)、咀しゃく剤、マルチ粒子およびナノ粒子、ゲル剤、固溶体、リポソーム剤、フィルム剤(粘膜接着型を含む)、卵形錠、スプレー剤ならびに液体配合物が含まれる。
【0055】
液体配合物には、懸濁液剤、液剤、シロップ剤およびエリキシル剤が含まれる。それらの配合物は、軟または硬カプセル剤中の充填物としても使用でき、一般にキャリヤー、たとえば水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロース、または適切な油、ならびに1種類以上の乳化剤および/または懸濁化剤を含む。液体配合物は固体の再構成により、たとえばサッシェ剤から調製することもできる。
【0056】
本発明化合物は、たとえばExpert Opinion in Therapeutic Patents, 11(6), 981-986, Liang and Chen (2001)に記載の即溶性、即時崩壊性の剤形でも使用できる。
錠剤剤形について、薬物は用量に応じて剤形の1〜80重量%、より一般的には剤形の5〜60重量%を構成する。錠剤は、薬物のほかに一般に崩壊剤を含有する。崩壊剤の例には、グリコール酸デンプンナトリウム、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、プレゲル化デンプンおよびアルギン酸ナトリウムが含まれる。一般に、崩壊剤は剤形の1〜25重量%、好ましくは5〜20重量%を構成する。
【0057】
錠剤配合物に凝集性を付与するために、一般に結合剤が用いられる。適切な結合剤には、微結晶性セルロース、ゼラチン、糖類、ポリエチレングリコール、天然ゴムおよび合成ゴム、ポリビニルピロリドン、プレゲル化デンプン、ヒドロキシプロピルセルロースおよびヒドロキシプロピルメチルセルロースが含まれる。錠剤は、希釈剤、たとえば乳糖(1水和物、噴霧乾燥した1水和物、無水物など)、マンニトール、キシリトール、デキストロース、ショ糖、ソルビトール、微結晶性セルロース、デンプンおよび二塩基性リン酸カルシウム2水和物を含有することもできる。
【0058】
錠剤は、所望により界面活性剤、たとえばラウリル硫酸ナトリウムおよびポリソルベート80、ならびに流動促進剤、たとえば二酸化ケイ素およびタルクを含有してもよい。界面活性剤が存在する場合、これらは錠剤の0.2〜5重量%を構成することができ、流動促進剤は錠剤の0.2〜1重量%を構成することができる。
【0059】
錠剤は一般に滑沢剤、たとえばステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウム、およびステアリン酸マグネシウムとラウリル硫酸ナトリウムの混合物をも含有する。滑沢剤は、一般に錠剤の0.25〜10重量%、好ましくは0.5〜3重量%を構成する。
【0060】
使用できる他の成分には、酸化防止剤、着色剤、着香剤、保存剤および味覚隠蔽剤が含まれる。
錠剤の例は、最高で約80 %の薬物、約10〜約90重量%の結合剤、約0〜約85重量%の希釈剤、約2〜約10重量%の崩壊剤、および約0.25〜約10重量%の滑沢剤を含有する。
【0061】
錠剤ブレンドを直接またはローラーにより圧縮して錠剤を成形することができる。錠剤ブレンドまたはブレンドの一部を、錠剤製造前に湿式-、乾式-もしくは溶融-造粒、溶融凝固、または押出しすることができる。最終配合物は1以上の層を含むことができ、コーティングされてもされなくてもよい;それはカプセル封入されてもよい。
【0062】
錠剤配合物は、”Pharmaceutical Dosage Forms : Tablets, Vol. 1”, H.Lieberman and L. Lachman, Marcel Dekker, ニューヨーク州ニューヨーク, 1980 (ISBN0-8247-6918-X)中に考察されている。
【0063】
経口投与用の固体配合物は、即時放出および/または改変放出であるように配合できる。改変放出配合物には、遅延放出、持続放出、パルス放出、制御放出、ターゲティングおよびプログラミングした放出が含まれる。
【0064】
本発明の目的に適切な改変放出配合物は、USP No.6,106,864に記載されている。他の適切な放出技術、たとえば高エネルギー分散液ならびに浸透圧粒子およびコーティング粒子についての詳細は、Verma et al, Pharmaceutical Technology On-line, 25 (2), 1-14 (2001)中にみられる。制御放出を達成するためのチューインガムの使用が、WO 00/35298に記載されている。
【0065】
本発明化合物は、血流、筋肉または内部臓器中へ直接投与することもできる。非経口投与に適切な手段には、静脈内、動脈内、腹腔内、クモ膜下、脳室内、尿道内、胸骨内、頭蓋内、筋肉内および皮下が含まれる。非経口投与に適切な器具には、針(マイクロニードルを含む)付き注射器、無針注射器、および注入法が含まれる。無針注射器の例はPowderject(商標)である。
【0066】
非経口配合物は一般に、塩類、炭水化物および緩衝剤(好ましくはpH 3〜9に)などの賦形剤を含有してもよい水性液剤であるが、ある用途には、それらを無菌の非水性液剤または粉末状の乾燥剤形として配合して、適切なビヒクル、たとえば発熱物質を含まない無菌水と組み合わせて使用するのがより適切である。
【0067】
無菌条件下で、たとえば凍結乾燥による非経口配合物の調製は、当業者に周知の医薬標準法を用いて容易に達成できる。
本発明の非経口液剤の調製に用いるボリコナゾールおよび抗真菌性CYP2C19阻害薬の溶解度は、適切な配合法により、たとえば溶解促進剤を含有させることにより、高めることができる。無針注射投与に用いる配合物は、粉末状の本発明化合物を、適切なビヒクル、たとえば発熱物質を含まない無菌水と組み合わせたものを含む。たとえばボリコナゾールの水溶解度は、英国特許出願No. 0327390.1(本明細書に援用する)に記載される1種類以上のポロキサマーをボリコナゾールに配合することによって高めることができる。あるいは、ボリコナゾールの水溶解度は、WO 91/11172および WO 94/02518(本明細書に援用する)に記載される1種類以上のスルホブチルエーテルシクロデキストリンをボリコナゾールに配合することによって高めることができる。ボリコナゾールとスルホブチルエーテルシクロデキストリンの配合物はWO 98/58677に記載されている(本明細書に援用する)。
【0068】
非経口投与用の配合物は、即時放出および/または改変/制御放出であるように配合できる。制御/改変放出配合物には、遅延放出、持続放出、パルス放出、制御放出、ターゲティングおよびプログラミングした放出が含まれる。たとえば本発明化合物は、有効化合物を改変放出する埋込みデポー剤として投与するための固体、半固体、またはチキソトロピー液として配合できる。そのような配合物の例には、薬物コーティングしたステントおよびPGLAマイクロスフェアが含まれる。
【0069】
本発明化合物は、局所的に皮膚または粘膜に、すなわち皮膚投与または経皮投与することもできる。この目的のための代表的な配合物には、ゲル剤、ヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散粉剤、包帯剤、発泡剤、フィルム剤、皮膚パッチ、カシェ剤、埋込み剤、スポンジ、繊維、包帯およびマイクロエマルションが含まれる。リポソームも使用できる。一般的なキャリヤーには、アルコール、水、鉱油、流動パラフィン、白色ワセリン、グリセリン、ポリエチレングリコールおよびプロピレングリコールが含まれる。透過促進剤を含有させてもよい−たとえばJ Pharm Sci, 88 (10), 955-958, Finnin and Morgan (October 1999)を参照。局所投与は、パッチ、たとえば経皮イオントフォレーゼパッチを用いて行うこともできる。
【0070】
局所投与のための他の手段には、エレクトロポレーション、イオントフォレーゼ、ホノフォレーゼ、ソノフォレーゼ、およびマイクロニードルまたは無針注射(たとえばPowderject(商標)、Bioject(商標)など)が含まれる。
【0071】
局所投与用の配合物は、即時放出および/または改変/制御放出であるように配合できる。制御/改変放出配合物には、遅延放出、持続放出、パルス放出、制御放出、ターゲティングおよびプログラミングした放出が含まれる。ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含有する組成物を、組成物の投与に適切なキットの形で好都合に組み合わせたものも本発明の範囲に含まれる。
【0072】
たとえば本発明のキットは、少なくとも1つがボリコナゾールを含有し、他のものは抗真菌性CYP2C19阻害薬を含有する、2以上の別個の医薬組成物、およびそれらの組成物を個別に収容する手段、たとえば容器、分割されたボトル、または分割されたホイルパケットを含む。そのようなキットの一例は、錠剤、カプセル剤などの包装に慣用されるブリスターパックである。
【0073】
本発明のキットは、異なる剤形、たとえば経口剤形と非経口剤形を投与するために、別個の組成物を異なる投与間隔で投与するために、または別個の組成物相互の力価を判定するために、特に適切である。コンプライアンスを助成するために、キットは一般に投与のための指示を含み、いわゆるメモリーエイドを備えていてもよい。
【0074】
実施例
実施例1
フルコナゾールによるヒト肝ミクロソームにおけるシトクロムP450活性の阻害
下記の表1のデータは、Meier et al. (1985) Anal. Biochem. 151, 286-291、およびFuna and Imaoka (1987) Biochem. Biophys. Acta. 926, 349-358に記載の方法により測定された。
【0075】
【表1】
【0076】
上記のデータにより証明されるように、フルコナゾールは酵素CYP3A4よりCYP2C19の選択的阻害を示す。
実施例2
ヒト肝ミクロソームならびにrCYP2C19およびrCYP23A4におけるボリコナゾールのインビトロ代謝:フルコナゾールによる選択的な酵素阻害
下記のインキュベーションミックス(最終濃度)を、この試験に記載するすべてのアッセイに用いた;50 mMのリン酸緩衝液(pH 7.4)、5 mMのMgCl2、5mMのイソクエン酸、1 U/mLのイソクエン酸デヒドロゲナーゼ。P450の代謝に必要な対応する還元剤はNADPHにより供給され、これはイソクエン酸/イソクエン酸デヒドロゲナーゼにより再生された。
【0077】
時間経過試験
下記のヒト肝ミクロソーム調製物中でインキュベーションを行った;対照バッチHL-MIX-101(60人のドナーから調製);CYP2C19低代謝型としてドナー2人の遺伝子型(HH-92、HH-112)、および高いCYP2C19活性をもつCYP2C19高代謝型(HH-100)。CYP2C19とCYP3A4の相対活性を下記の表2に示す。
【0078】
【表2】
【0079】
(PM = 低代謝型; EM = 高代謝型)
組換えCYP2C19およびCYP3A4でトランスフェクションした昆虫細胞から調製したミクロソーム(それぞれ、BDGENTEST(登録商標)supersomeおよびPanvera baculosome)を用いて、さらにインキュベーションを行った。
【0080】
上記の反応ミックスを用いたインキュベーション期間中のN-オキシド代謝産物の形成が直線的であることを確認するために、一連の予備試験が必要であった。その際、ヒト肝ミクロソーム(1 mg/mL)を基質ボリコナゾール(最終濃度25μM)の存在下でプレインキュベートした後、NADPHを添加した。rCYP2C19およびrCYP3A4を用いる試験については、NADPHと共にプレインキュベーションを行い、基質ボリコナゾールの添加により反応を開始することを留意する。アリコート(1 mL)の反応混合物を経時的に(0〜60分間)採集し、50μLの内標準(2-(2,4-ジフルオロフェニル)-3-(4-ピリミジル)-1-(1H-1,2,4-トリアゾール-1-イル)-2-ブタノール; 15μg/mL)を含有するジクロロメタン4mLを添加した。試料を10分間、回転混合し、次いで3000 rpmで5分間(4℃)遠心した。上部の水層を分離して廃棄した。下部の有機層を37℃でN2流下に蒸発乾固した。続いて試料を100μLの移動相A (下記を参照)中に再生し、25μLをHPLCに注入した。
【0081】
試料をAgilent 1100系列UV-HPLCで分析した。Hichrom 100-5C18カラム(150×4.6 mm)を用い、1 mL/分の低速で、254 nmに設定したUV-λによりクロマトグラフィー分離を実施した。用いた移動相は下記のものであった;(A) 70:30 H20:MeOH中の0.1 Mリン酸アンモニウム(pH 7.0に調整、MeOH添加前)、および(B) MeOH;下記の濃度勾配を使用;0〜2分(0%のB)、2〜20分(0〜100%のB)、20〜23分(100%のB)、23.1分(0%のB)。次の注入の前7分間、カラムを再平衡化した。一般的な反応時間は、フルコナゾールについては7.9分間;N-オキシド代謝産物については11.7分間;内標準については13.1分間;ボリコナゾールについては14.5分間であった。
【0082】
ボリコナゾールN-酸化に対するフルコナゾールの阻害能を、ヒト肝ミクロソームならびにrCYP2C19およびrCYP3A4調製物において調べた。試験したフルコナゾールの最終濃度は、0 0.1、1、10、100および1000μMであった。
【0083】
ヒト肝ミクロソームを1 mg/mLで60分間、rCYP2C19を1O pmol CYP/mLで60分間、rCYP3A4を100 pmol CYP/mLで20分間、インキュベートした。各マトリックスについて、アリコート(4mL)の反応混合物を水浴中で37℃に予熱し、続いて200μLのNADPH (20 mM)および40μLの各フルコナゾール溶液(0〜100 mM)を添加した。50μLのボリコナゾール (2 mM)の添加により反応を開始した。インキュベーション時間の終わりにアリコート(n=3)を各インキュベーションミックスから取り出し、4mLのジクロロメタンおよび50μLの内標準(2-(2,4-ジフルオロフェニル)-3-(4-ピリミジル)-1-(1H-1,2,4-トリアゾール-1-イル)-2-ブタノール; 15μg/mL)を入れた試験管に添加した。続いて前記の抽出および分析操作を行った。
【0084】
報告した濃度で、N-オキシド代謝産物の形成はヒト肝ミクロソーム調製物およびrCYP2C19においては60分間、直線的であった。rCYP3A4においては20分間、直線性がみられた。
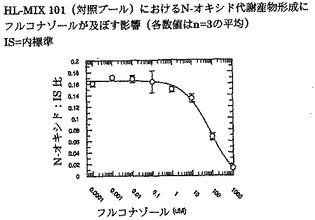
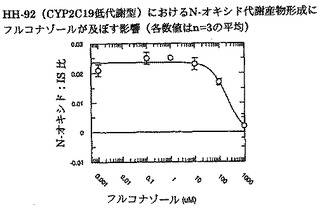
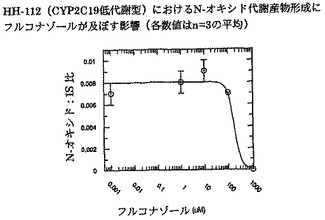
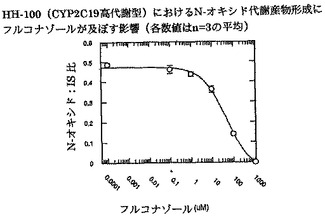
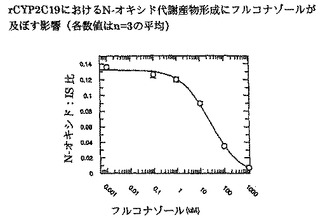
ボリコナゾールからそのN-オキシド代謝産物への代謝に対するフルコナゾールの阻害能(IC50)を、ヒト肝ミクロソームプール(HL-MIX-101)、およびCYP2C19低代謝型(HH-92およびHH-112)または高代謝型(HH-100)として遺伝子型判定された個々のドナーにおいて調べた。さらに、CYP2C19低代謝型を、CYP3A4代謝能の高いものと低いものに小分割した。CYP3A4仲介またはCYP2C19仲介によるボリコナゾール代謝の阻害に対するフルコナゾールの選択性をより良く判定するために、rCYP3A4またはrCYP2C19も調べた。結果を図3〜8にまとめ、表3に照合する。
【0085】
【表3】
【0086】
CYP2C19低代謝型のヒト肝ミクロソームにおいて、フルコナゾールはボリコナゾールN-酸化に対する弱い阻害薬である(IC50は約175μMおよび214μM)。より有効な阻害は、CYP2C19高代謝型ミクロソームにおいてみられた(IC50は約50μM)。rCYP2C19およびrCYP3A4を用いた実験は、フルコナゾールがCYP3A4(IC50は約106μM)と比較してCYP2C19仲介N-酸化に対するより有効な阻害薬である(IC50は約29μM)ことを証明した。
【0087】
これらの実験で得た全般的所見から、CYP3A4仲介によるボリコナゾールN-酸化よりCYP2C19仲介によるボリコナゾールN-酸化に対するフルコナゾールの選択性が3〜4倍であることが明らかである。
【0088】
実施例3
臨床試験
健康な男性被験者に共投与したフルコナゾールがボリコナゾールの定常状態薬物動態に及ぼす影響を調べ、フルコナゾールとボリコナゾールの共投与の安全性および耐容性を評価する試験を行った。年齢21〜55歳の健康な男性被験者10人を集め、2人のCYP2C19低代謝型を含めた少なくとも8人の被験者が確実に試験を完了した。2種類の処置を行った:
1.1日目にボリコナゾール400 mgを1日2回経口投与、続いて2〜3日目に200 mgを1日2回経口投与、4日目の朝に200 mgを1回経口投与;
2.1日目にボリコナゾール400 mgを1日2回経口投与、続いて2〜3日目に200 mgを1日2回経口投与、4日目の朝に200 mgを1回経口投与 + 1日目の朝のボリコナゾール投与と共に400 mgのフルコナゾールを1回経口投与、2〜5日目の朝に200 mgのフルコナゾールを1日1回経口投与。
【0089】
ボリコナゾールに続いてボリコナゾール+フルコナゾールの順序、またはボリコナゾール+フルコナゾールに続いてボリコナゾールの順序のいずれかに、被験者をランダムに帰属させた。
【0090】
血液試料を各処置期間の4、5および6日目に採取して、各処置期間の4日目について薬物動態パラメーターCmax、Tmax、AUCr、AUCtおよびAUCを計算するための血漿ボリコナゾール濃度を求めた。次いで、CYP2C19高代謝型と分類された被験者について、これらのパラメーターを用いてボリコナゾール単独とボリコナゾール+フルコナゾールを比較した。また、CYP2C19低代謝型と分類された被験者についての同じパラメーターを、高代謝型から得たデータとの比較に用いた。
【0091】
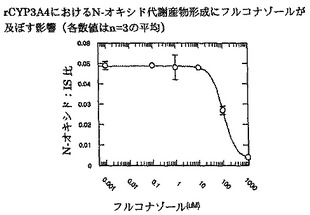
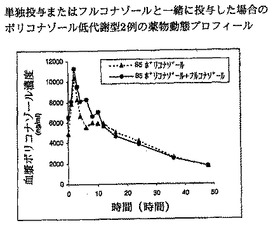
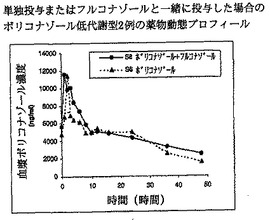
結果を図9および10(低代謝型)ならびに11および12(高代謝型)に示す。図9および10は、低代謝型ではフルコナゾールがボリコナゾールの薬物動態にほとんどまたは全く影響を及ぼさないことを明瞭に示す。
【0092】
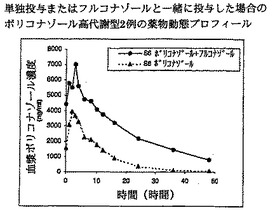
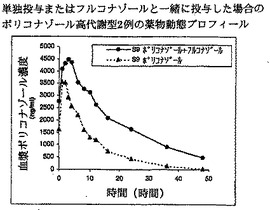
ところが図11および12は、高代謝型において200 mg、1日1回のフルコナゾールが200 mg、1日2回の標準用量ボリコナゾールに影響を及ぼすことを示す。最終投与の24時間後に、ボリコナゾール濃度が有効性に必要な血漿中濃度を超えると考えられる>2000 ng/mlに維持されていることに注目すべきである。さらに、高代謝型におけるボリコナゾールの半減期が>18時間に延長している。
【0093】
これは、ボリコナゾールとフルコナゾールの共投与の結果、高代謝型の体内からのボリコナゾールのクリアランスが低下したことを示す。これによりボリコナゾールは、高代謝型において1日1回投与した場合に療法効果を達成しうるのに十分な血漿中濃度で1日中存在することができる。
【0094】
さらにこのデータは、ボリコナゾールとフルコナゾールの共投与により、はるかに低い用量のボリコナゾールで血漿ボリコナゾール濃度を達成しうること、すなわち1日1回の投与について2倍、1日2回の投与について4倍の低下を予想できることを指摘する。さらにこのデータは、患者集団内の変動を有意に低下させうることを指摘する。
【0095】
このデータは、はるかに低い用量のボリコナゾールで療法用の血漿ボリコナゾール濃度を達成しうることを示す。これにより、低代謝型またはヘテロ接合性の高代謝型について、ボリコナゾールの投与量および血中濃度を2〜4倍低下させうることができるであろう。全身曝露の低下が達成されると、不都合な副作用を最小限に抑えることができる。
【図面の簡単な説明】
【0096】
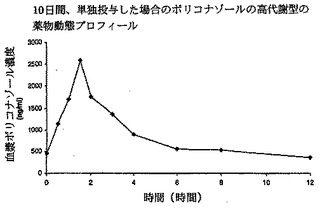
【図1】単独投与した場合のボリコナゾールの高代謝型の薬物動態プロフィールを示す。
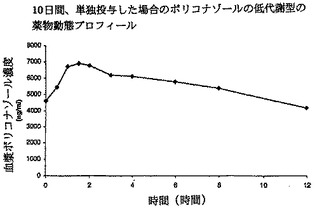
【図2】単独投与した場合のボリコナゾールの低代謝型の薬物動態プロフィールを示す。
【図3】インビトロでヒト肝ミクロソームHL-MIX 101(対照プール)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図4】インビトロでヒト肝ミクロソームHH-92(CYP2C19低代謝型)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図5】インビトロでヒト肝ミクロソームHH-112(CYP2C19低代謝型)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図6】インビトロでヒト肝ミクロソームHH-100(CYP2C19高代謝型)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図7】インビトロでrCYP2C19におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図8】インビトロでrCYP3A4におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図9】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール低代謝型2例の薬物動態プロフィールを示す。
【図10】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール低代謝型2例の薬物動態プロフィールを示す。
【図11】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール高代謝型2例の薬物動態プロフィールを示す。
【図12】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール高代謝型2例の薬物動態プロフィールを示す。
【発明の詳細な説明】
【0001】
発明の分野
本発明は、ボリコナゾール(voriconazole)を含む新規な併用療法に関する。
発明の背景
(2R,3S)-2-(2,4-ジフルオロフェニル)-3-(5-フルオロ-4-ピリミジニル)-1-(1H-1,2,4-トリアゾール-1-イル)-ブタン-2-オール(ボリコナゾールとしても知られる)は、EP-A-440372に開示されている;特に例7を参照。ボリコナゾールは下記の構造:
【0002】
【化1】
【0003】
をもち、真菌感染症の処置に有用である。
ボリコナゾールの薬物動態は、投与レベルの増加に伴う曝露の増大が非直線的になる飽和型の代謝を特徴とする。さらに、薬物曝露は対象間で著しく変動する。ボリコナゾールはシトクロムP450のアイソザイムCYP2C19、CYP2C9およびCYP3A4により代謝される。主な循環性代謝産物(その構造を下記に示す)は、N-酸化により生成する。
【0004】
【化2】
【0005】
本発明者らは、ボリコナゾールの代謝は処置される対象の遺伝子型に大幅に依存することを見いだした。ある遺伝子型はボリコナゾールを高度に代謝してボリコナゾールを体内から急速にクリアランスし、その結果、血漿ボリコナゾール濃度は低下する(約0.6〜約1.4μg/ml)。本明細書中ではこの遺伝子型を”高代謝型(extensive metaboliser)”と呼ぶ。第2の遺伝子型は、ボリコナゾール低代謝型と特徴づけることができる:すなわちこの遺伝子型の場合、ボリコナゾールがクリアランスされる速度がはるかに遅く、したがってより高濃度で体内に留まる(約3.5〜約5.5μg/ml)。本明細書中ではこの遺伝子型を”低代謝型(poor metaboliser)”と呼ぶ。この遺伝子型は、劣性遺伝子によるものと考えられる:すなわちホモ接合性の高代謝型は、ほぼ白人集団の73%、日本人集団の35%を占める;ヘテロ接合性の高代謝型は、ほぼ白人集団の25%、日本人集団の46%を占める。これに対し、低代謝型は、ほぼ白人集団の2%、日本人集団の19%を占めるにすぎない。
【0006】
ボリコナゾールの代謝に関する遺伝子型間の変動は体内の酵素シトクロムP450 2C19(以下、CYP2C19と呼ぶ)の存在度に依存することが現在分かっている:すなわち、高代謝型には酵素CYP2C19が存在し、一方、低代謝型はこの機能性酵素を欠如する。たとえば、M. de Morais, G.Wilkinson, J.Blaisdell et al., J Biol Chem (1994), 269, 15419-15422を参照(その全体を本明細書に援用する)。
【0007】
実際には、ボリコナゾールは高代謝型と低代謝型の両方に用量調整なしに投与されている。しかし、高代謝型に療法効果を及ぼすのに十分な量でボリコナゾールが血漿中に存在するためには、高用量の薬物が必要である:すなわち通常の推奨1日量は400 mg(200 mgを1日2回)である。この用量は、低代謝型では全身曝露がより高くなり、このため望ましくない副作用の起きる可能性がある。さらに、高代謝型による急速な薬物クリアランスのため、血漿中濃度を1日中維持して療法効果を及ぼすには、この化合物を1日2回投与する必要がある。ボリコナゾールを患者が自己投与する場合、1日2回の療法はコンプライアンス問題を提起する。
【0008】
M. Ghannoum, N. Isham, M. Hossain and D. Sheehan, Int. J. Infect. Dis. (2002), Vol.6, Supp.2, 2S50には、ボリコナゾールと、アンホテリシンB(amphotericin B)、Abelcet(商標)、5-フルオロシトシンおよびフルコナゾール(fluconazole)を含めた抗真菌薬とのインビトロ組合わせが示され、広範な生物に対するそれらの相乗作用機序が調べられている。ボリコナゾールとフルコナゾールの組合わせは39%が相加的、61%が無関係と記載されている。
【0009】
M. Ghannoum, N. Isham and D. Sheehan, Abstracts of the Interscience Conference on Antimicrobial Agents and Chemotherapy (2002), 42, 385にも、アンホテリシンB、Abelcet(商標)、フルコナゾール、ミカフンジン(micafungin)、ラブコナゾール(ravuconazole)およびカスポフンジン(caspofungin)を含めた抗真菌薬とのインビトロ組合わせが示されている。ボリコナゾールとフルコナゾールの組合わせは100%が無関係、すなわち相乗作用機序はないと記載されている。
【0010】
H.J. Scherpbier, M.I. Hilhorst and T.W. Kuijpers, Clin. Infect. Dis. (2003), 37, 828には、抗レトロウイルス薬の組合わせによるエイズ患者の治療が示され、食道カンジダ症の治療のために患者の療法にボリコナゾールを加えた場合のプロテアーゼ阻害薬とボリコナゾールの相互作用が報告されている。この相互作用は、ロピナビル(lopinavir)、ネビラピン(nevirapine)およびアンプレナビル(amprenavir)について肝機能不全および血漿中濃度上昇を伴った。それらの患者において血漿ボリコナゾール濃度は測定されなかった。
【0011】
N. Wood, K. Tan, L. Purkins, G. Layton, J. Hamlin, D. Kleinermans and D. Nichols, Br. J. Clin. Pharmacol. (2003) 56, 56には、プロトンポンプ阻害薬オメプラゾール(omeprazole)、すなわちCYP2C19阻害薬がボリコナゾールの定常状態薬物動態に及ぼす影響を調べる研究が記載されている。この研究では、オメプラゾールはボリコナゾール曝露に対して臨床関連作用をもたないと結論された。これは、オメプラゾール療法を開始する患者についてボリコナゾールの用量調整が不必要なことを示唆する。
【0012】
対象間変動を排除または低減するボリコナゾール療法を提供することが望ましいであろう。さらに、ボリコナゾールの投与量が減少したボリコナゾール療法を提供することが望ましいであろう。さらに、血漿からの薬物クリアランスを低下させ、これによりボリコナゾールを1日1回投与すればよいボリコナゾール療法を提供することが望ましいであろう。
【0013】
本発明者らは意外にも、ボリコナゾールとCYP2C19の活性を阻害しうる第2の異なる抗真菌薬との共投与によりCYP2C19酵素を阻害することによって、ボリコナゾール高代謝型において代謝が顕著に低下し、それらの対象における薬物動態プロフィールが低代謝型に近づくことを見いだした。その結果、対象間変動が顕著に低下し、はるかに低用量のボリコナゾールで療法用血漿中濃度に達する。さらに、体内からのボリコナゾールのクリアランスが低下し、ボリコナゾールは1日1回投与した場合に1日中、療法効果を達成しうるのに十分な血漿中濃度で存在する。
【0014】
発明の概要
本発明は、第1観点において、ボリコナゾールおよび抗真菌性CYP2C19阻害薬を後記にさらに詳細に定める特定の量または重量比で含む、組合わせ薬剤を提供する。
【0015】
本発明は、第2観点において、療法有効量のボリコナゾールおよび抗真菌性CYP2C19阻害薬を医薬的に許容できるキャリヤーまたは希釈剤と共に含む、医薬組成物をも提供する。
【0016】
本発明は、第3観点において、療法有効量のボリコナゾールおよび療法有効量の抗真菌性CYP2C19阻害薬を含む、単位剤形をも提供する。
本発明はさらに、第4観点において、複数の個別容器を含み、少なくとも1つの容器がボリコナゾールを収容し、少なくとも1つの異なる容器が抗真菌性CYP2C19阻害薬を収容した、キットを提供する。
【0017】
本発明はさらに、第5観点において、哺乳動物において真菌感染症を処置するための医薬の製造における、前記の組合わせ薬剤、組成物、キットまたは単位剤形の使用を提供する。
【0018】
本発明は、第6観点において、哺乳動物において真菌感染症を処置する方法であって、その処置を必要とする哺乳動物に、有効量の前記の組合わせ薬剤、組成物、キットまたは単位剤形を投与することを含む方法を提供する。
【0019】
以下において、本発明の組合わせ薬剤、医薬組成物、単位剤形、キット、使用および方法を合わせて”本発明の組合わせ”という。
好ましい態様の詳細な記述
本発明の組合わせの一方の成分はボリコナゾールである。ボリコナゾールはEP-A-440372に開示されている(その全体を本明細書に援用する);特に例7を参照。後記に詳述するように、ボリコナゾールは遊離塩基として、またはその塩、溶媒和物もしくはプロドラッグの形で投与できる。
【0020】
本発明の組合わせの他方の成分は、抗真菌性CYP2C19阻害薬である。この抗真菌薬が抗真菌活性を示し、かつ哺乳動物CYP2C19酵素の阻害薬として作用しうる限り、抗真菌薬の厳密な性質は特に限定されない。本明細書において”CYP2C19阻害薬”には、哺乳動物CYP2C19酵素の作用を阻害しうる化合物がいずれも含まれる;Desta et al. (2002) Clinical Pharmacokinetics 41(12), 913-958による概説に記載(その全体を本明細書に援用する)。一般的な療法用量でのボリコナゾール代謝を確実に阻害するために、好ましくは10μM未満のKj値が必要である。Kjを測定するための標準的なインビトロ試験には、プローブ基質として(S)-メフェニトイン((S)-mephenytoin)を用い、4'-ヒドロキシル化を測定する−Meier et al. (1985) Anal. Biochem. 151, 286-291を参照(その全体を本明細書に援用する)。抗真菌性CYP2C19阻害薬の例にはフルコナゾールが含まれ、これは2μMのKjをもつ−L.C. Winkers, C.J. Wurden, E. Storch et al., Drug Metab Dispos (1996) 24, 610-4を参照。CYP2C19阻害薬の他の例は、Desta et al. (2002)(前記)に記載されている。
【0021】
抗真菌性CYP2C19阻害薬は、酵素CYP3A4より酵素CYP2C19によるボリコナゾール代謝を選択的に阻害できることが好ましい。これらのアイソザイムに対する選択性は、(S)-メフェニトイン4-ヒドロキシラーゼ活性およびテストステロン6ベータヒドロキシラーゼ活性に対する相対阻害力価により判定できる:後者の活性の測定についてはFuna and Imaoka (1987) Biochem. Biophys. Acta. 926, 349-358に記載されている(その全体を本明細書に援用する)。より具体的には、選択性はボリコナゾールN-酸化に対する作用により証明でき、それぞれ25および2500μMの基質濃度を用いてCYP2C19(高親和性酵素)および3A4(低親和性酵素)に対する作用を評価する。シトクロムP450アイソザイムによるボリコナゾールN-酸化を測定する方法は、Hyland, Jones and Smith (2003) Drug Metabolism and Disposition, 31(5), 540-547に記載されている(その全体を本明細書に援用する)。要求される選択性の厳密なレベルは、抗真菌性CYP2C19阻害薬ならびにその薬物動態および対象間変動に依存するが、抗真菌性CYP2C19阻害薬がCYP3A4よりCYP2C19に対して2〜10、好ましくは3〜6の選択性を示すことが好ましい(前記のHylandらの報文中におけるそれらの相対IC50により測定)。理論により拘束されるつもりはないが、CYP3A4よりCYP2C19を選択的に阻害すると、対象間変動を低下させ、より低いボリコナゾール用量で療法用血漿中濃度に到達でき、一方、現在の推奨用量方式では患者集団に起きる可能性のある望ましくない副作用を軽減することができると考えられる。低濃度ではCYP2C19が主なクリアランス経路であるが、高濃度ではCYP3A4経路の代謝、すなわち非飽和型のボリコナゾールクリアランスが主体となる。
【0022】
抗真菌性CYP2C19阻害薬は、ボリコナゾール/抗真菌性CYP2C19阻害薬の組合わせを1日1回投与した場合に1日中、血漿中濃度を維持して療法効果を達成しうるのに適切な、薬物動態半減期をもつことが好ましい。適切な半減期は、6〜72時間、好ましくは12〜48時間、より好ましくは18〜36時間である。
【0023】
抗真菌性CYP2C19阻害薬は、主に未変化薬物として身体から排出されることが好ましい。理論により拘束されるつもりはないが、主に未変化薬物として身体から排出される抗真菌性CYP2C19阻害薬が、抗真菌性CYP2C19阻害薬およびボリコナゾール両方の対象間変動を低下させると考えられる。好ましくは50〜99%、より好ましくは70〜80%、最も好ましくは約75%の阻害薬が未変化のまま身体から排出される。
【0024】
抗真菌性CYP2C19阻害薬は抗真菌活性を示す。抗真菌薬でもあるCYP2C19阻害薬の例には、アゾール類、たとえばフルコナゾールが含まれる。これは、この組合わせが抗真菌作用以外の作用を示さないという利点をもつ。この第2の抗真菌薬は、好ましくは抗真菌活性に対する相加作用をもち、好ましくは同一患者集団に使用するのに安全であることが証明できるものである。全体的な抗真菌効果は、もちろん処置される個々の感染症、ボリコナゾールおよび抗真菌性CYP2C19阻害薬の投与量、ならびに処置される患者の年齢、性別、体重および状態に依存する。
【0025】
抗真菌性CYP2C19阻害薬はフルコナゾールであることが特に好ましい。
本発明の組合わせは、ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含む。ボリコナゾールおよび抗真菌性CYP2C19阻害薬は、遊離の酸もしくは塩基として、またはその医薬的に許容できる塩、溶媒和物もしくはプロドラッグの形で投与できる。
【0026】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬の医薬的に許容できる塩には、その酸付加塩および塩基塩が含まれる。
適切な酸付加塩は、無毒性の塩を形成する酸から形成できる。例には、酢酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、炭酸水素塩/炭酸塩、硫酸水素塩/硫酸塩、ホウ酸塩、カムシル酸塩、クエン酸塩、エジシル酸塩、エシル酸塩、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンズ酸塩、塩酸塩/クロリド、臭化水素酸塩/ブロミド、ヨウ化水素酸塩/ヨージド、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシル酸塩、メチル硫酸塩、ナフチル酸塩、2-ナプシル酸塩、ニコチン酸塩、硝酸塩、オロット酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素塩/リン酸二水素塩、サッカリン酸塩、ステアリン酸塩、コハク酸塩、酒石酸塩、トシル酸塩およびトリフルオロ酢酸塩が含まれる。
【0027】
適切な塩基塩は、無毒性の塩を形成する塩基から形成できる。例には、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオラミン、グリシン、リシン、マグネシウム、メグルミン、オーラミン、カリウム、ナトリウム、トロメタミンおよび亜鉛の塩が含まれる。
【0028】
適切な塩類の概説については、”Handbook of Pharmaceutical Salts : Properties, Selection, and Use”, Stahl and Wermuth (Wiley-VCH, ドイツ、ワインハイム, 2002)を参照されたい。
【0029】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬の医薬的に許容できる塩は、ボリコナゾールまたは抗真菌性CYP2C19阻害薬の溶液を目的の酸または塩基と混和することによって、容易に調製できる。塩は、溶液から沈殿させて濾過により回収でき、または溶媒の蒸発により回収できる。塩のイオン化度は完全イオン化からほぼ非イオン化まで変動する可能性がある。
【0030】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬は、溶媒和していない形および溶媒和した形の両方で存在することができる。本明細書において用語’溶媒和物’は、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬と1以上の医薬的に許容できる溶媒分子、たとえばエタノールを含む、分子複合体を記載するために用いられる。用語’水和物’は、溶媒が水である場合に用いられる。
【0031】
以下において、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬という表現はすべて、その塩類、溶媒和物および複合体、ならびにその塩類の溶媒和物および複合体という表現を含む。
【0032】
本発明の組合わせには、前記に定めたボリコナゾールおよび抗真菌性CYP2C19阻害薬、その多形およびプロドラッグが含まれる。
上記のように、本発明は前記に定めたボリコナゾールおよび/または抗真菌性CYP2C19阻害薬のすべての多形を含む。
【0033】
本発明の範囲には、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬のいわゆる’プロドラッグ’も含まれる。たとえば、ボリコナゾールおよび/または抗真菌性CYP2C19阻害薬のある種の誘導体(それ自体はほとんどまたは全く薬理活性をもたなくてもよい)が、体内または身体上に投与した際に、たとえば加水分解開裂により変換されて目的活性をもつ化合物になることができる。そのような誘導体を’プロドラッグ’と呼ぶ。プロドラッグの使用に関する情報はさらに、’Pro-drugs as Novel Delivery Systems’, Vol. 14, ACS Symposium Series (T Higuchi and W Stella)、および’Bioreversible Carriers in Drug Design’, Pergamon Press, 1987 (編者E B Roche, American Pharmaceutical Association)中にみられる。
【0034】
本発明によるプロドラッグは、たとえばボリコナゾールおよび/または抗真菌性CYP2C19阻害薬中に存在する適切な官能基を、当業者に’プロ部分’として知られる特定の分子と交換することにより製造できる:たとえば”Design of Prodrugs”, 編者H Bundgaard (Elsevier, 1985)に記載。
【0035】
ボリコナゾールはアルコール官能基(-OH)を含み、したがって本発明によるプロドラッグのある例はそのエステルまたはエーテルを含むことができる:たとえば、その水素をリン酸化により置換してリン酸二水素(2R, 3S)-2-(2,4-ジフルオロフェニル)-3-(5-フルオロ-4-ピリミジニル)-1-(1H-1,2,4-トリアゾール-1-イル)-2-ブチルを得ることによる;WO 97/28169に記載(その全体を本明細書に援用する);特に例3を参照。
【0036】
同様に、抗真菌性CYP2C19阻害薬がフルコナゾールである場合、この化合物もアルコール官能基(-OH)を含み、したがって本発明によるプロドラッグのある例は下記のものを含むことができる:そのエステルもしくはエーテル、たとえばその水素をリン酸化により置換してリン酸二水素2-(2,4-ジフルオロフェニル)-1,3-ビス(1H-1,2,4-トリアゾール-1-イル)-2-プロピルを得ることによる;WO 97/28169;特に例1を参照;またはその医薬的に許容できる塩、特に二ナトリウム塩(Prodif(登録商標))。上記の例による置換基の他の例および他のタイプのプロドラッグの例は、前記の参考文献中にみられる。
【0037】
抗真菌性CYP2C19阻害薬は1個以上の不斉炭素原子を含む場合があり、したがって2種類以上の立体異性体として存在する可能性がある。抗真菌性CYP2C19阻害薬がアルケニルまたはアルケニレン基を含む場合、シス/トランス(またはZ/E)幾何異性体の可能性がある。これらの化合物がたとえばケトもしくはオキシム基または芳香族部分を含む場合、互変異性(tautomeric isomerism、'tautomerism')が起きる可能性がある。その結果、単一化合物が1より多いタイプの異性を示す場合がある。
【0038】
本発明の範囲には、すべての立体異性体、幾何異性体および互変異性体の形の抗真菌性CYP2C19阻害薬が含まれ、これには1より多いタイプの異性を示す化合物、およびその1種類以上の混合物も含まれる。対イオンが光学活性である酸付加塩または塩基塩、たとえばD-乳酸塩もしくはL-リシン塩、またはラセミ体、たとえばDL-酒石酸塩もしくはDL-アルギニン塩も含まれる。
【0039】
シス/トランス異性体は、当業者に周知の常法、たとえばクロマトグラフィーおよび分別結晶化により分離できる。
個々の鏡像異性体を製造/単離するための常法には、適切な光学的に純粋な前駆物質からのキラル合成、またはたとえばキラル高速液体クロマトグラフィー(HPLC)によるラセミ体(または塩もしくは誘導体のラセミ体)の分割が含まれる。
【0040】
あるいは、ラセミ体(またはラセミ前駆物質)を適切な光学活性化合物、たとえばアルコール類と反応させ、または抗真菌性CYP2C19阻害薬が酸性もしくは塩基性部分を含む場合は酸もしくは塩基、たとえば酒石酸もしくは1-フェニルエチルアミンと反応させることができる。得られたジアステレオマー混合物をクロマトグラフィーおよび/または分別結晶化により分離し、一方または両方のジアステレオ異性体を当業者に周知の方法で対応する純粋な鏡像異性体に変換することができる。
【0041】
本発明の組合わせに用いるキラル化合物(およびそのキラル前駆物質)は、クロマトグラフィー、一般にHPLCにより、不斉樹脂上で、一般に0〜50 %(一般に2〜20 %)のイソプロパノールおよび0〜5 %のアルキルアミン(0.1 %のジエチルアミン)を含有する炭化水素、一般にヘプタンまたはヘキサンからなる移動相を用いて、鏡像異性体を富化した形で得ることができる。溶出液の濃縮により、富化した混合物が得られる。
【0042】
立体異性体の集合体は、当業者に周知の常法により分離することができる−たとえば、”Stereochemistry of Organic Compounds”, E L Eliel (Wiley, ニューヨーク, 1994)を参照。
【0043】
本発明の組合わせは、抗真菌薬として有効な、かつCYP2C19酵素の作用を阻害するのに有効な、好ましくはこの酵素をCYP3A4酵素より選択的に阻害する、ある量の抗真菌性CYP2C19阻害薬を含む。厳密な投与量は、処置される患者の年齢、性別、体重および状態など、多様な要因に依存する。しかし、投与量は約5〜約500 mg、より好ましくは約10〜約250 mg、さらに好ましくは約50〜約200 mg、最も好ましくは約75〜約125 mgであることが好ましい。好ましくは、抗真菌性CYP2C19阻害薬はフルコナゾールである。
【0044】
本発明の組合わせは、抗真菌薬として作用するある量のボリコナゾールを含む。厳密な投与量は、処置される患者の年齢、性別、体重および状態など、多様な要因に依存する。しかし、投与量は約5〜約600 mg、より好ましくは約10〜約500 mg、さらに好ましくは約20〜約300 mg、最も好ましくは約25〜約250 mgであることが好ましい。約200 mgの投与量が特に好ましい;これは、ボリコナゾールの投与量を通常の400 mgレベルから半減でき、したがって望ましくない副作用を軽減できるという点で、本発明による著しい利点をもたらす。
【0045】
好ましい態様において、本発明の組合わせは患者に1日1回投与される。これは、患者のコンプライアンスがより良好であるという格別の利点をもたらす。しかし、1日2、3または4回の投与も本発明の範囲に含まれるものとする。ただし、各回当たりの用量はさらに少なくなるであろう。
【0046】
本発明の組合わせの成分の重量比は、処置される患者の年齢、性別、体重および状態など、多様な要因に依存する。しかし、ボリコナゾールと抗真菌性CYP2C19阻害薬を約1:4〜約6:1、好ましくはが約1:2〜約3:1、より好ましくは約3:2〜約5:2の重量比で投与することが好ましい。好ましくは、抗真菌性CYP2C19阻害薬はフルコナゾールである。
【0047】
本発明の組合わせにより処置できる真菌感染症は、EP-A-440372を含めた文献中に十分に記載されており、局所感染症、粘膜感染症、たとえば膣カンジダ症、食道および口腔咽頭カンジダ症、ならびに全身感染症を含む。さらに、本発明の組合わせはアレルギー反応、たとえばアレルギー性鼻副鼻腔炎の処置にも使用できる。本発明の組合わせにより処置できる真菌感染症には、特に下記のものにより起きる感染症が含まれる:カンジダ属菌種(Candida spp)、トリコフィトン属菌種(Trichophyton spp)、ミクロスポルム属菌種(Microsporum spp)、有毛表皮糸状菌(Epidermophyton floccosum)、クリプトコックス-ネオフォルマンス(Cryptococcus neoformans)、アスペルギルス属菌種(Aspergillus spp)、フザリウム属菌種(Fusarium spp)、スセドスポリウム属菌種(Scedosporium spp)、コクシジオイデス-イミチス(Coccidioides immitis)、パラコクシジオイデス-ブラジリエンシス(Paracoccidioides brasiliensis)、ヒストプラズマ属菌種(Histoplasma spp)、ブラストマイセス-デルマチジチス(Blastomyces dermatiditis)、アルテルナリア属菌種(Alternaria spp)、エキソフィアラ属菌種(Exophiala spp)、フォンセカ-ペドロソイ(Fonsecaea pedrosoi)、ペニシリウム-マルネフェイ(Penicillium marneffei)、フィアロフォラ属菌種(Phialophora spp)またはペシロミセス-リラシヌス(Paecilomyces lilacinus)。処置という表現が予防および樹立した症状の軽減を含むものであることは自明であろう。
【0048】
本発明の組合わせにおいて、ボリコナゾールおよび抗真菌性CYP2C19阻害薬は、剤形に関しては別個もしくは互いに一緒に、またそれらの投与時間に関しては同時もしくは逐次、投与することができる。たとえば、ボリコナゾールの投与が抗真菌性CYP2C19阻害薬の投与前、投与と同時、または投与後であってよい。投与間の時間は24時間以内の投与間隔で変更できる。
【0049】
本発明の単位剤形は、ボリコナゾールおよび抗真菌性CYP2C19阻害薬の両方が存在する剤形である。それは、経口投与用の固体配合物、たとえば錠剤、カプセル剤(粒子、液体もしくは粉末を収容したもの)、トローチ剤(液体充填型を含む)、咀しゃく剤、ゲル剤、固溶体、リポソーム剤、フィルム剤(粘膜接着型を含む)、卵形錠、スプレー剤または液体配合物、非経口配合物(一般に、後記に定める賦形剤を含有してもよい水性液剤)、あるいは皮膚または粘膜に局所投与(すなわち皮膚投与または経皮投与)するための配合物、たとえばヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散粉剤、包帯剤、発泡剤、皮膚パッチ、カシェ剤、埋込み剤、スポンジ、繊維、包帯またはマイクロエマルションであってよい。好ましくは本発明の単位剤形は、ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含有する錠剤またはカプセル剤、特に錠剤である。
【0050】
医薬として使用するための本発明の組合わせの化合物は、結晶質または非晶質生成物として投与できる。それらは、たとえば固体プラグ、粉末またはフィルムとして、たとえば沈殿、結晶化、凍結乾燥、噴霧乾燥または蒸発乾燥などの方法により得ることができる。マイクロ波乾燥または高周波乾燥も、この目的に使用できる。
【0051】
一般に本発明化合物は、1種類以上の医薬的に許容できる賦形剤と一緒に配合物として投与される。用語”賦形剤”は、本明細書中で本発明化合物以外の成分を記載するために用いられる。賦形剤の選択は、個々の投与様式、賦形剤が溶解度および安定性に及ぼす影響、ならびに剤形の性質などの要因に大幅に依存するであろう。
【0052】
本発明化合物の送達に適切な医薬組成物およびそれらの調製方法は、当業者に自明であろう。そのような組成物およびそれらの調製方法は、たとえば’Remington's Pharmaceutical Sciences’,第19版(Mack Publishing Company, 1995)にみられる。
【0053】
本発明化合物は経口投与することができる。経口投与は化合物が消化管に入るように嚥下を伴い、あるいは化合物が口から血流に直接入る口腔投与または舌下投与を採用できる。
【0054】
経口投与に適切な配合物には、固体配合物、たとえば錠剤、カプセル剤(粒子、液体もしくは粉末を収容したもの)、トローチ剤(液体充填型を含む)、咀しゃく剤、マルチ粒子およびナノ粒子、ゲル剤、固溶体、リポソーム剤、フィルム剤(粘膜接着型を含む)、卵形錠、スプレー剤ならびに液体配合物が含まれる。
【0055】
液体配合物には、懸濁液剤、液剤、シロップ剤およびエリキシル剤が含まれる。それらの配合物は、軟または硬カプセル剤中の充填物としても使用でき、一般にキャリヤー、たとえば水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロース、または適切な油、ならびに1種類以上の乳化剤および/または懸濁化剤を含む。液体配合物は固体の再構成により、たとえばサッシェ剤から調製することもできる。
【0056】
本発明化合物は、たとえばExpert Opinion in Therapeutic Patents, 11(6), 981-986, Liang and Chen (2001)に記載の即溶性、即時崩壊性の剤形でも使用できる。
錠剤剤形について、薬物は用量に応じて剤形の1〜80重量%、より一般的には剤形の5〜60重量%を構成する。錠剤は、薬物のほかに一般に崩壊剤を含有する。崩壊剤の例には、グリコール酸デンプンナトリウム、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、プレゲル化デンプンおよびアルギン酸ナトリウムが含まれる。一般に、崩壊剤は剤形の1〜25重量%、好ましくは5〜20重量%を構成する。
【0057】
錠剤配合物に凝集性を付与するために、一般に結合剤が用いられる。適切な結合剤には、微結晶性セルロース、ゼラチン、糖類、ポリエチレングリコール、天然ゴムおよび合成ゴム、ポリビニルピロリドン、プレゲル化デンプン、ヒドロキシプロピルセルロースおよびヒドロキシプロピルメチルセルロースが含まれる。錠剤は、希釈剤、たとえば乳糖(1水和物、噴霧乾燥した1水和物、無水物など)、マンニトール、キシリトール、デキストロース、ショ糖、ソルビトール、微結晶性セルロース、デンプンおよび二塩基性リン酸カルシウム2水和物を含有することもできる。
【0058】
錠剤は、所望により界面活性剤、たとえばラウリル硫酸ナトリウムおよびポリソルベート80、ならびに流動促進剤、たとえば二酸化ケイ素およびタルクを含有してもよい。界面活性剤が存在する場合、これらは錠剤の0.2〜5重量%を構成することができ、流動促進剤は錠剤の0.2〜1重量%を構成することができる。
【0059】
錠剤は一般に滑沢剤、たとえばステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウム、およびステアリン酸マグネシウムとラウリル硫酸ナトリウムの混合物をも含有する。滑沢剤は、一般に錠剤の0.25〜10重量%、好ましくは0.5〜3重量%を構成する。
【0060】
使用できる他の成分には、酸化防止剤、着色剤、着香剤、保存剤および味覚隠蔽剤が含まれる。
錠剤の例は、最高で約80 %の薬物、約10〜約90重量%の結合剤、約0〜約85重量%の希釈剤、約2〜約10重量%の崩壊剤、および約0.25〜約10重量%の滑沢剤を含有する。
【0061】
錠剤ブレンドを直接またはローラーにより圧縮して錠剤を成形することができる。錠剤ブレンドまたはブレンドの一部を、錠剤製造前に湿式-、乾式-もしくは溶融-造粒、溶融凝固、または押出しすることができる。最終配合物は1以上の層を含むことができ、コーティングされてもされなくてもよい;それはカプセル封入されてもよい。
【0062】
錠剤配合物は、”Pharmaceutical Dosage Forms : Tablets, Vol. 1”, H.Lieberman and L. Lachman, Marcel Dekker, ニューヨーク州ニューヨーク, 1980 (ISBN0-8247-6918-X)中に考察されている。
【0063】
経口投与用の固体配合物は、即時放出および/または改変放出であるように配合できる。改変放出配合物には、遅延放出、持続放出、パルス放出、制御放出、ターゲティングおよびプログラミングした放出が含まれる。
【0064】
本発明の目的に適切な改変放出配合物は、USP No.6,106,864に記載されている。他の適切な放出技術、たとえば高エネルギー分散液ならびに浸透圧粒子およびコーティング粒子についての詳細は、Verma et al, Pharmaceutical Technology On-line, 25 (2), 1-14 (2001)中にみられる。制御放出を達成するためのチューインガムの使用が、WO 00/35298に記載されている。
【0065】
本発明化合物は、血流、筋肉または内部臓器中へ直接投与することもできる。非経口投与に適切な手段には、静脈内、動脈内、腹腔内、クモ膜下、脳室内、尿道内、胸骨内、頭蓋内、筋肉内および皮下が含まれる。非経口投与に適切な器具には、針(マイクロニードルを含む)付き注射器、無針注射器、および注入法が含まれる。無針注射器の例はPowderject(商標)である。
【0066】
非経口配合物は一般に、塩類、炭水化物および緩衝剤(好ましくはpH 3〜9に)などの賦形剤を含有してもよい水性液剤であるが、ある用途には、それらを無菌の非水性液剤または粉末状の乾燥剤形として配合して、適切なビヒクル、たとえば発熱物質を含まない無菌水と組み合わせて使用するのがより適切である。
【0067】
無菌条件下で、たとえば凍結乾燥による非経口配合物の調製は、当業者に周知の医薬標準法を用いて容易に達成できる。
本発明の非経口液剤の調製に用いるボリコナゾールおよび抗真菌性CYP2C19阻害薬の溶解度は、適切な配合法により、たとえば溶解促進剤を含有させることにより、高めることができる。無針注射投与に用いる配合物は、粉末状の本発明化合物を、適切なビヒクル、たとえば発熱物質を含まない無菌水と組み合わせたものを含む。たとえばボリコナゾールの水溶解度は、英国特許出願No. 0327390.1(本明細書に援用する)に記載される1種類以上のポロキサマーをボリコナゾールに配合することによって高めることができる。あるいは、ボリコナゾールの水溶解度は、WO 91/11172および WO 94/02518(本明細書に援用する)に記載される1種類以上のスルホブチルエーテルシクロデキストリンをボリコナゾールに配合することによって高めることができる。ボリコナゾールとスルホブチルエーテルシクロデキストリンの配合物はWO 98/58677に記載されている(本明細書に援用する)。
【0068】
非経口投与用の配合物は、即時放出および/または改変/制御放出であるように配合できる。制御/改変放出配合物には、遅延放出、持続放出、パルス放出、制御放出、ターゲティングおよびプログラミングした放出が含まれる。たとえば本発明化合物は、有効化合物を改変放出する埋込みデポー剤として投与するための固体、半固体、またはチキソトロピー液として配合できる。そのような配合物の例には、薬物コーティングしたステントおよびPGLAマイクロスフェアが含まれる。
【0069】
本発明化合物は、局所的に皮膚または粘膜に、すなわち皮膚投与または経皮投与することもできる。この目的のための代表的な配合物には、ゲル剤、ヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散粉剤、包帯剤、発泡剤、フィルム剤、皮膚パッチ、カシェ剤、埋込み剤、スポンジ、繊維、包帯およびマイクロエマルションが含まれる。リポソームも使用できる。一般的なキャリヤーには、アルコール、水、鉱油、流動パラフィン、白色ワセリン、グリセリン、ポリエチレングリコールおよびプロピレングリコールが含まれる。透過促進剤を含有させてもよい−たとえばJ Pharm Sci, 88 (10), 955-958, Finnin and Morgan (October 1999)を参照。局所投与は、パッチ、たとえば経皮イオントフォレーゼパッチを用いて行うこともできる。
【0070】
局所投与のための他の手段には、エレクトロポレーション、イオントフォレーゼ、ホノフォレーゼ、ソノフォレーゼ、およびマイクロニードルまたは無針注射(たとえばPowderject(商標)、Bioject(商標)など)が含まれる。
【0071】
局所投与用の配合物は、即時放出および/または改変/制御放出であるように配合できる。制御/改変放出配合物には、遅延放出、持続放出、パルス放出、制御放出、ターゲティングおよびプログラミングした放出が含まれる。ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含有する組成物を、組成物の投与に適切なキットの形で好都合に組み合わせたものも本発明の範囲に含まれる。
【0072】
たとえば本発明のキットは、少なくとも1つがボリコナゾールを含有し、他のものは抗真菌性CYP2C19阻害薬を含有する、2以上の別個の医薬組成物、およびそれらの組成物を個別に収容する手段、たとえば容器、分割されたボトル、または分割されたホイルパケットを含む。そのようなキットの一例は、錠剤、カプセル剤などの包装に慣用されるブリスターパックである。
【0073】
本発明のキットは、異なる剤形、たとえば経口剤形と非経口剤形を投与するために、別個の組成物を異なる投与間隔で投与するために、または別個の組成物相互の力価を判定するために、特に適切である。コンプライアンスを助成するために、キットは一般に投与のための指示を含み、いわゆるメモリーエイドを備えていてもよい。
【0074】
実施例
実施例1
フルコナゾールによるヒト肝ミクロソームにおけるシトクロムP450活性の阻害
下記の表1のデータは、Meier et al. (1985) Anal. Biochem. 151, 286-291、およびFuna and Imaoka (1987) Biochem. Biophys. Acta. 926, 349-358に記載の方法により測定された。
【0075】
【表1】
【0076】
上記のデータにより証明されるように、フルコナゾールは酵素CYP3A4よりCYP2C19の選択的阻害を示す。
実施例2
ヒト肝ミクロソームならびにrCYP2C19およびrCYP23A4におけるボリコナゾールのインビトロ代謝:フルコナゾールによる選択的な酵素阻害
下記のインキュベーションミックス(最終濃度)を、この試験に記載するすべてのアッセイに用いた;50 mMのリン酸緩衝液(pH 7.4)、5 mMのMgCl2、5mMのイソクエン酸、1 U/mLのイソクエン酸デヒドロゲナーゼ。P450の代謝に必要な対応する還元剤はNADPHにより供給され、これはイソクエン酸/イソクエン酸デヒドロゲナーゼにより再生された。
【0077】
時間経過試験
下記のヒト肝ミクロソーム調製物中でインキュベーションを行った;対照バッチHL-MIX-101(60人のドナーから調製);CYP2C19低代謝型としてドナー2人の遺伝子型(HH-92、HH-112)、および高いCYP2C19活性をもつCYP2C19高代謝型(HH-100)。CYP2C19とCYP3A4の相対活性を下記の表2に示す。
【0078】
【表2】
【0079】
(PM = 低代謝型; EM = 高代謝型)
組換えCYP2C19およびCYP3A4でトランスフェクションした昆虫細胞から調製したミクロソーム(それぞれ、BDGENTEST(登録商標)supersomeおよびPanvera baculosome)を用いて、さらにインキュベーションを行った。
【0080】
上記の反応ミックスを用いたインキュベーション期間中のN-オキシド代謝産物の形成が直線的であることを確認するために、一連の予備試験が必要であった。その際、ヒト肝ミクロソーム(1 mg/mL)を基質ボリコナゾール(最終濃度25μM)の存在下でプレインキュベートした後、NADPHを添加した。rCYP2C19およびrCYP3A4を用いる試験については、NADPHと共にプレインキュベーションを行い、基質ボリコナゾールの添加により反応を開始することを留意する。アリコート(1 mL)の反応混合物を経時的に(0〜60分間)採集し、50μLの内標準(2-(2,4-ジフルオロフェニル)-3-(4-ピリミジル)-1-(1H-1,2,4-トリアゾール-1-イル)-2-ブタノール; 15μg/mL)を含有するジクロロメタン4mLを添加した。試料を10分間、回転混合し、次いで3000 rpmで5分間(4℃)遠心した。上部の水層を分離して廃棄した。下部の有機層を37℃でN2流下に蒸発乾固した。続いて試料を100μLの移動相A (下記を参照)中に再生し、25μLをHPLCに注入した。
【0081】
試料をAgilent 1100系列UV-HPLCで分析した。Hichrom 100-5C18カラム(150×4.6 mm)を用い、1 mL/分の低速で、254 nmに設定したUV-λによりクロマトグラフィー分離を実施した。用いた移動相は下記のものであった;(A) 70:30 H20:MeOH中の0.1 Mリン酸アンモニウム(pH 7.0に調整、MeOH添加前)、および(B) MeOH;下記の濃度勾配を使用;0〜2分(0%のB)、2〜20分(0〜100%のB)、20〜23分(100%のB)、23.1分(0%のB)。次の注入の前7分間、カラムを再平衡化した。一般的な反応時間は、フルコナゾールについては7.9分間;N-オキシド代謝産物については11.7分間;内標準については13.1分間;ボリコナゾールについては14.5分間であった。
【0082】
ボリコナゾールN-酸化に対するフルコナゾールの阻害能を、ヒト肝ミクロソームならびにrCYP2C19およびrCYP3A4調製物において調べた。試験したフルコナゾールの最終濃度は、0 0.1、1、10、100および1000μMであった。
【0083】
ヒト肝ミクロソームを1 mg/mLで60分間、rCYP2C19を1O pmol CYP/mLで60分間、rCYP3A4を100 pmol CYP/mLで20分間、インキュベートした。各マトリックスについて、アリコート(4mL)の反応混合物を水浴中で37℃に予熱し、続いて200μLのNADPH (20 mM)および40μLの各フルコナゾール溶液(0〜100 mM)を添加した。50μLのボリコナゾール (2 mM)の添加により反応を開始した。インキュベーション時間の終わりにアリコート(n=3)を各インキュベーションミックスから取り出し、4mLのジクロロメタンおよび50μLの内標準(2-(2,4-ジフルオロフェニル)-3-(4-ピリミジル)-1-(1H-1,2,4-トリアゾール-1-イル)-2-ブタノール; 15μg/mL)を入れた試験管に添加した。続いて前記の抽出および分析操作を行った。
【0084】
報告した濃度で、N-オキシド代謝産物の形成はヒト肝ミクロソーム調製物およびrCYP2C19においては60分間、直線的であった。rCYP3A4においては20分間、直線性がみられた。
ボリコナゾールからそのN-オキシド代謝産物への代謝に対するフルコナゾールの阻害能(IC50)を、ヒト肝ミクロソームプール(HL-MIX-101)、およびCYP2C19低代謝型(HH-92およびHH-112)または高代謝型(HH-100)として遺伝子型判定された個々のドナーにおいて調べた。さらに、CYP2C19低代謝型を、CYP3A4代謝能の高いものと低いものに小分割した。CYP3A4仲介またはCYP2C19仲介によるボリコナゾール代謝の阻害に対するフルコナゾールの選択性をより良く判定するために、rCYP3A4またはrCYP2C19も調べた。結果を図3〜8にまとめ、表3に照合する。
【0085】
【表3】
【0086】
CYP2C19低代謝型のヒト肝ミクロソームにおいて、フルコナゾールはボリコナゾールN-酸化に対する弱い阻害薬である(IC50は約175μMおよび214μM)。より有効な阻害は、CYP2C19高代謝型ミクロソームにおいてみられた(IC50は約50μM)。rCYP2C19およびrCYP3A4を用いた実験は、フルコナゾールがCYP3A4(IC50は約106μM)と比較してCYP2C19仲介N-酸化に対するより有効な阻害薬である(IC50は約29μM)ことを証明した。
【0087】
これらの実験で得た全般的所見から、CYP3A4仲介によるボリコナゾールN-酸化よりCYP2C19仲介によるボリコナゾールN-酸化に対するフルコナゾールの選択性が3〜4倍であることが明らかである。
【0088】
実施例3
臨床試験
健康な男性被験者に共投与したフルコナゾールがボリコナゾールの定常状態薬物動態に及ぼす影響を調べ、フルコナゾールとボリコナゾールの共投与の安全性および耐容性を評価する試験を行った。年齢21〜55歳の健康な男性被験者10人を集め、2人のCYP2C19低代謝型を含めた少なくとも8人の被験者が確実に試験を完了した。2種類の処置を行った:
1.1日目にボリコナゾール400 mgを1日2回経口投与、続いて2〜3日目に200 mgを1日2回経口投与、4日目の朝に200 mgを1回経口投与;
2.1日目にボリコナゾール400 mgを1日2回経口投与、続いて2〜3日目に200 mgを1日2回経口投与、4日目の朝に200 mgを1回経口投与 + 1日目の朝のボリコナゾール投与と共に400 mgのフルコナゾールを1回経口投与、2〜5日目の朝に200 mgのフルコナゾールを1日1回経口投与。
【0089】
ボリコナゾールに続いてボリコナゾール+フルコナゾールの順序、またはボリコナゾール+フルコナゾールに続いてボリコナゾールの順序のいずれかに、被験者をランダムに帰属させた。
【0090】
血液試料を各処置期間の4、5および6日目に採取して、各処置期間の4日目について薬物動態パラメーターCmax、Tmax、AUCr、AUCtおよびAUCを計算するための血漿ボリコナゾール濃度を求めた。次いで、CYP2C19高代謝型と分類された被験者について、これらのパラメーターを用いてボリコナゾール単独とボリコナゾール+フルコナゾールを比較した。また、CYP2C19低代謝型と分類された被験者についての同じパラメーターを、高代謝型から得たデータとの比較に用いた。
【0091】
結果を図9および10(低代謝型)ならびに11および12(高代謝型)に示す。図9および10は、低代謝型ではフルコナゾールがボリコナゾールの薬物動態にほとんどまたは全く影響を及ぼさないことを明瞭に示す。
【0092】
ところが図11および12は、高代謝型において200 mg、1日1回のフルコナゾールが200 mg、1日2回の標準用量ボリコナゾールに影響を及ぼすことを示す。最終投与の24時間後に、ボリコナゾール濃度が有効性に必要な血漿中濃度を超えると考えられる>2000 ng/mlに維持されていることに注目すべきである。さらに、高代謝型におけるボリコナゾールの半減期が>18時間に延長している。
【0093】
これは、ボリコナゾールとフルコナゾールの共投与の結果、高代謝型の体内からのボリコナゾールのクリアランスが低下したことを示す。これによりボリコナゾールは、高代謝型において1日1回投与した場合に療法効果を達成しうるのに十分な血漿中濃度で1日中存在することができる。
【0094】
さらにこのデータは、ボリコナゾールとフルコナゾールの共投与により、はるかに低い用量のボリコナゾールで血漿ボリコナゾール濃度を達成しうること、すなわち1日1回の投与について2倍、1日2回の投与について4倍の低下を予想できることを指摘する。さらにこのデータは、患者集団内の変動を有意に低下させうることを指摘する。
【0095】
このデータは、はるかに低い用量のボリコナゾールで療法用の血漿ボリコナゾール濃度を達成しうることを示す。これにより、低代謝型またはヘテロ接合性の高代謝型について、ボリコナゾールの投与量および血中濃度を2〜4倍低下させうることができるであろう。全身曝露の低下が達成されると、不都合な副作用を最小限に抑えることができる。
【図面の簡単な説明】
【0096】
【図1】単独投与した場合のボリコナゾールの高代謝型の薬物動態プロフィールを示す。
【図2】単独投与した場合のボリコナゾールの低代謝型の薬物動態プロフィールを示す。
【図3】インビトロでヒト肝ミクロソームHL-MIX 101(対照プール)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図4】インビトロでヒト肝ミクロソームHH-92(CYP2C19低代謝型)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図5】インビトロでヒト肝ミクロソームHH-112(CYP2C19低代謝型)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図6】インビトロでヒト肝ミクロソームHH-100(CYP2C19高代謝型)におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図7】インビトロでrCYP2C19におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図8】インビトロでrCYP3A4におけるN-オキシド代謝産物形成にフルコナゾールが及ぼす影響を示す。
【図9】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール低代謝型2例の薬物動態プロフィールを示す。
【図10】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール低代謝型2例の薬物動態プロフィールを示す。
【図11】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール高代謝型2例の薬物動態プロフィールを示す。
【図12】単独投与またはフルコナゾールと一緒に投与した場合のボリコナゾール高代謝型2例の薬物動態プロフィールを示す。
【特許請求の範囲】
【請求項1】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含み、ボリコナゾールが約5〜約600 mgの量で存在し、抗真菌性CYP2C19阻害薬が約5〜約500 mgの量で存在する、組合わせ薬剤。
【請求項2】
ボリコナゾールが約20〜約300 mgの量で存在し、抗真菌性CYP2C19阻害薬が約50〜約200 mgの量で存在する、請求項1に記載の組合わせ薬剤。
【請求項3】
ボリコナゾールが約25〜約250 mgの量で存在し、抗真菌性CYP2C19阻害薬が約75〜約125 mgの量で存在する、請求項1に記載の組合わせ薬剤。
【請求項4】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含み、ボリコナゾールと抗真菌性CYP2C19阻害薬が約1:4〜約6:1の重量比で存在する、組合わせ薬剤。
【請求項5】
ボリコナゾールと抗真菌性CYP2C19阻害薬が約1:2〜約3:1の重量比で存在する、請求項4に記載の組合わせ薬剤。
【請求項6】
ボリコナゾールと抗真菌性CYP2C19阻害薬が約3:2〜約5:2の重量比で存在する、請求項4に記載の組合わせ薬剤。
【請求項7】
抗真菌性CYP2C19阻害薬がフルコナゾールである、前記のいずれか1項に記載の組合わせ薬剤。
【請求項8】
療法有効量のボリコナゾールおよび抗真菌性CYP2C19阻害薬を、医薬的に許容できるキャリヤーまたは希釈剤と共に含む、医薬組成物。
【請求項9】
抗真菌性CYP2C19阻害薬がフルコナゾールであり、ボリコナゾールとフルコナゾールが請求項1〜3のいずれか1項に記載の量または請求項4〜6のいずれか1項に記載の比率で存在する、請求項8に記載の組成物。
【請求項10】
複数の個別容器を含み、少なくとも1つの容器がボリコナゾールを収容し、少なくとも1つの異なる容器が抗真菌性CYP2C19阻害薬を収容した、キット。
【請求項11】
抗真菌性CYP2C19阻害薬がフルコナゾールであり、ボリコナゾールとフルコナゾールが請求項1〜3のいずれか1項に記載の量または請求項4〜6のいずれか1項に記載の比率で存在する、請求項10に記載のキット。
【請求項12】
療法有効量のボリコナゾールおよび療法有効量の抗真菌性CYP2C19阻害薬を含む、単位剤形。
【請求項13】
抗真菌性CYP2C19阻害薬がフルコナゾールであり、ボリコナゾールとフルコナゾールが請求項1〜3のいずれか1項に記載の量または請求項4〜6のいずれか1項に記載の比率で存在する、請求項12に記載の単位剤形。
【請求項14】
哺乳動物において真菌感染症を処置するための医薬の製造における、請求項1〜7のいずれか1項に記載の組合わせ薬剤、請求項8もしくは9に記載の組成物、請求項10もしくは11に記載のキット、または請求項12もしくは13に記載の単位剤形の使用。
【請求項15】
哺乳動物において真菌感染症を処置する方法であって、その処置を必要とする哺乳動物に、有効量の請求項1〜7のいずれか1項に記載の組合わせ薬剤、請求項8もしくは9に記載の組成物、請求項10もしくは11に記載のキット、または請求項12もしくは13に記載の単位剤形を投与することを含む方法。
【請求項1】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含み、ボリコナゾールが約5〜約600 mgの量で存在し、抗真菌性CYP2C19阻害薬が約5〜約500 mgの量で存在する、組合わせ薬剤。
【請求項2】
ボリコナゾールが約20〜約300 mgの量で存在し、抗真菌性CYP2C19阻害薬が約50〜約200 mgの量で存在する、請求項1に記載の組合わせ薬剤。
【請求項3】
ボリコナゾールが約25〜約250 mgの量で存在し、抗真菌性CYP2C19阻害薬が約75〜約125 mgの量で存在する、請求項1に記載の組合わせ薬剤。
【請求項4】
ボリコナゾールおよび抗真菌性CYP2C19阻害薬を含み、ボリコナゾールと抗真菌性CYP2C19阻害薬が約1:4〜約6:1の重量比で存在する、組合わせ薬剤。
【請求項5】
ボリコナゾールと抗真菌性CYP2C19阻害薬が約1:2〜約3:1の重量比で存在する、請求項4に記載の組合わせ薬剤。
【請求項6】
ボリコナゾールと抗真菌性CYP2C19阻害薬が約3:2〜約5:2の重量比で存在する、請求項4に記載の組合わせ薬剤。
【請求項7】
抗真菌性CYP2C19阻害薬がフルコナゾールである、前記のいずれか1項に記載の組合わせ薬剤。
【請求項8】
療法有効量のボリコナゾールおよび抗真菌性CYP2C19阻害薬を、医薬的に許容できるキャリヤーまたは希釈剤と共に含む、医薬組成物。
【請求項9】
抗真菌性CYP2C19阻害薬がフルコナゾールであり、ボリコナゾールとフルコナゾールが請求項1〜3のいずれか1項に記載の量または請求項4〜6のいずれか1項に記載の比率で存在する、請求項8に記載の組成物。
【請求項10】
複数の個別容器を含み、少なくとも1つの容器がボリコナゾールを収容し、少なくとも1つの異なる容器が抗真菌性CYP2C19阻害薬を収容した、キット。
【請求項11】
抗真菌性CYP2C19阻害薬がフルコナゾールであり、ボリコナゾールとフルコナゾールが請求項1〜3のいずれか1項に記載の量または請求項4〜6のいずれか1項に記載の比率で存在する、請求項10に記載のキット。
【請求項12】
療法有効量のボリコナゾールおよび療法有効量の抗真菌性CYP2C19阻害薬を含む、単位剤形。
【請求項13】
抗真菌性CYP2C19阻害薬がフルコナゾールであり、ボリコナゾールとフルコナゾールが請求項1〜3のいずれか1項に記載の量または請求項4〜6のいずれか1項に記載の比率で存在する、請求項12に記載の単位剤形。
【請求項14】
哺乳動物において真菌感染症を処置するための医薬の製造における、請求項1〜7のいずれか1項に記載の組合わせ薬剤、請求項8もしくは9に記載の組成物、請求項10もしくは11に記載のキット、または請求項12もしくは13に記載の単位剤形の使用。
【請求項15】
哺乳動物において真菌感染症を処置する方法であって、その処置を必要とする哺乳動物に、有効量の請求項1〜7のいずれか1項に記載の組合わせ薬剤、請求項8もしくは9に記載の組成物、請求項10もしくは11に記載のキット、または請求項12もしくは13に記載の単位剤形を投与することを含む方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2007−520542(P2007−520542A)
【公表日】平成19年7月26日(2007.7.26)
【国際特許分類】
【出願番号】特願2006−551940(P2006−551940)
【出願日】平成17年1月13日(2005.1.13)
【国際出願番号】PCT/IB2005/000098
【国際公開番号】WO2005/084671
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
【公表日】平成19年7月26日(2007.7.26)
【国際特許分類】
【出願日】平成17年1月13日(2005.1.13)
【国際出願番号】PCT/IB2005/000098
【国際公開番号】WO2005/084671
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
[ Back to top ]