
ボルテゾミブおよびその生成のためのプロセス
本願は、ボルテゾミブおよびその中間体を調製するためのプロセス、ならびにボルテゾミブの結晶形態のためのプロセスを提供する。本願によれば、ボルテゾミブの中間体の調製のためのプロセス、およびボルテゾミブの調製のためのプロセス、ならびにこれらのプロセスにより生成された中間体およびボルテゾミブが提供される。さらに、本願はまた、実質的に純粋なボルテゾミブの調製のためのプロセス、および実質的に純粋なボルテゾミブを提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(出願の分野)
本願は、ボルテゾミブおよびその調製のために有用な中間体化合物の調製のためのプロセスに関する。
【0002】
本願はまた、実質的に純粋なボルテゾミブの調製のためのプロセスに関する。
【0003】
本願はさらに、ボルテゾミブの結晶形態AおよびBの調製のためのプロセスに関する。本願はまた、ボルテゾミブの中間体化合物および独特な形態に関する。
【背景技術】
【0004】
(出願の背景)
ボルテゾミブとは、化学名[(1R)−3−メチル−1−[[(2S)−1−オキソ−3−フェニル−2−[(ピラジニルカルボニル)アミノ]プロピル]アミノ]ブチル]ボロン酸を有する薬物化合物について認可された名称であり、そして構造式I
【0005】
【化1】

により表される。
【0006】
ボルテゾミブは、抗腫瘍剤であり、そして市場で商標名「VELCADE(登録商標)」のもとで注射剤の形態で入手可能な治療用プロテオソームインヒビターである。各バイアルは、3.5mgのボルテゾミブを、滅菌凍結乾燥粉末として含む。米国において、ボルテゾミブは、多発性骨髄腫およびマントル細胞リンパ腫の処置のために認可されている。
【0007】
Summary Basis Of Approval for Bortezomib(NDA 21−602)の化学的概説の節に、薬物物質、薬物製品および再構築された薬物製品が、3つの異なる分子形態を有することが開示された。PS−341(ボルテゾミブ)薬物物質は、固相において、三量体のボロキシンとして存在する。水に曝露されると、このボロキシンは、単量体のボロン酸PS−341に加水分解する。凍結乾燥されたSP−341薬物製品の構造は、対称なマンニトールエステルであることが決定された。0.9% NaCl溶液により再構築される間、再構築されたPS−341薬物製品は、マンニトールエステルとPS−341ボロン酸との間の平衡からなる。
【0008】
特許文献1は、ボルテゾミブ、その薬学的に受容可能な塩、薬学的組成物、および哺乳動物におけるプロテオソーム機能を阻害するための使用を開示する。さらに、ボルテゾミブおよびそのアナログの調製のためのプロセスを開示する。
【0009】
特許文献2は、ボルテゾミブエステルの凍結乾燥処方物を開示する。特許文献2によれば、特許文献1に記載されるようなプロセスにより調製されるボルテゾミブは、白色非晶質粉末である。
【0010】
特許文献3は、ルイス酸触媒(転移反応を促進し、そしてα−炭素原子のエピマー化を最小にするため)の存在下での中間体ボロ「ネート」錯体の転移による、ボロン酸エステルの同族体化のためのプロセスを開示する。
【0011】
特許文献4は、ボルテゾミブ中間体(これは、ボロン酸エステル化合物である)およびボルテゾミブそのものを調製するためのプロセスを開示する。
【0012】
特許文献4は、式III
【0013】
【化2】
の中間体化合物の調製のための、式X
【0014】
【化3】
のボロ「ネート」錯体のルイス酸により促進される転移による、以前に報告されたプロセスが、テトラヒドロフラン(水と混和性であるエーテル溶媒)を使用し、そして厳密に乾燥された器具、溶媒、およびルイス酸試薬を必要とするので、このような反応は、高価であり、スケールアップが困難であることを開示する。さらに、特許文献4によれば、先行技術のプロセスのスケールアップの試みは頻繁に、ボロン酸エステル化合物のジアステレオマー比のさらなる悪化をもたらし、このことは、テトラヒドロフラン溶媒を除去して水混和性溶媒で交換するための反応混合物の濃縮中のハロゲン化物イオンへの生成物の曝露、または引き続く水性物質洗浄中にテトラヒドロフランの完全な除去をし損なうことの、いずれかに起因する。
【0015】
特許文献4は、ボロ「ネート」錯体の転移を、水との混和性が低いエーテル溶媒および配位共溶媒中で実施することにより、先行技術の問題に取り組むようである。このプロセスにおいて使用するために特許文献4において確認されている、低水混和性エーテル溶媒の非限定的な例としては、tert−ブチルメチルエーテル、tert−ブチルエチルエーテル、tert−アミルメチルエーテル、およびイソプロピルエーテルが挙げられる。
【0016】
さらに、特許文献4は、
(i)式IX
【0017】
【化4】
の中間体ボロン酸エステル化合物、有機ボロン酸アクセプター、低級アルカノール、C5〜8炭化水素溶媒、および水性鉱酸を含む二相混合物を提供する工程;
(ii)この二相混合物を攪拌してボルテゾミブを得る工程;
(iii)溶媒層を分離する工程;ならびに
(iv)ボルテゾミブまたはそのボロン酸無水物を有機溶媒中に抽出する工程、
を包含する、ボルテゾミブの調製のためのプロセスを開示する。
【0018】
生成物の純度を高めるために、工程(iii)の後に得られた水層が抽出工程(iv)の前に洗浄されて、中性有機不純物が除去される。このようなプロセスは、以下の工程:
1)溶媒層を分離する工程;
2)水層を塩基性pHに調整する工程;
3)この水層を有機溶媒で洗浄する工程;および
4)この水相を約6未満のpHに調整する工程、
を包含する。
【0019】
従って、特許文献4に記載されるプロセスは、酸性条件下および塩基性条件下での複数回の有機溶媒洗浄、引き続いて、化合物を有機溶媒中に抽出する工程、その生成物を単離する工程、ならびにさらに再結晶して、純度が高まったボルテゾミブを得る工程を包含する。
【0020】
ボルテゾミブを水性塩基性溶液に曝露すると、ボルテゾミブの純度が低下することが見出されている。具体的には、このようなプロセスが大規模で行われる場合、ボルテゾミブの水性塩基性条件への長時間にわたる曝露は、回避することが困難であり、従って、このプロセスは、産業規模で使用することに耐えられないかもしれない。
【0021】
特許文献5は、ボルテゾミブの結晶形態Iおよび結晶形態II、ならびにこれらの調製のためのプロセスを開示する。形態Iのボルテゾミブは、溶媒(例えば、アセトン、CHCl3、CH2Cl2、またはニトリルなど)および希釈剤(例えば、ジイソプロピルエーテル、第三級ブチルメチルエーテル、n−ヘキサンおよびn−ヘプタン)を使用することにより調製される。形態IIのボルテゾミブは、酢酸エチルの熱溶液から調製される。特許文献5はまた、形態Iおよび形態IIが上記溶媒を使用することにより相互変換可能であることを開示する。
【先行技術文献】
【特許文献】
【0022】
【特許文献1】米国特許第5,780,454号明細書
【特許文献2】米国特許第6,713,446号明細書
【特許文献3】米国特許第4,525,309号明細書
【特許文献4】米国特許出願公開第2005/0240047号明細書
【特許文献5】国際公開第2008/075376号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0023】
ボルテゾミブおよびその中間体の調製のための、単純かつ好都合なプロセスを提供することが、依然として必要とされている。
【課題を解決するための手段】
【0024】
(発明の要旨)
本願によれば、ボルテゾミブの中間体の調製のためのプロセス、およびボルテゾミブの調製のためのプロセス、ならびにこれらのプロセスにより生成された中間体およびボルテゾミブが提供される。
【0025】
さらに、本願はまた、実質的に純粋なボルテゾミブの調製のためのプロセス、および実質的に純粋なボルテゾミブを提供する。
【0026】
1つの局面において、本願は、式III
【0027】
【化5】
の中間体化合物の調製のためのプロセスを提供し、このプロセスは、ルイス酸触媒、水混和性エーテル溶媒および過剰なジクロロメタンの存在下での、式X
【0028】
【化6】
のボロ「ネート」錯体の転移を包含する。式IIIの中間体化合物の調製のためのプロセスにおける過剰なジクロロメタンの使用は、ボロ「ネート」錯体形成中に反応物質として沈殿するのみでなく、水性酸溶液でのこの反応混合物のクエンチ後の有機層分離もまた補助することが見出された。本発明のプロセスにより得られる式IIIの中間体化合物は、引き続いて、先行技術において公知であるプロセスによって、遊離塩基の形態または酸付加塩形態の、式V
【0029】
【化7】
の中間体化合物の調製のために利用される。式Vの化合物は、引き続いて、式VIIIの中間体化合物との縮合反応による、ボルテゾミブの調製のためのプロセスにおいて利用され得る。式VIIIの中間体化合物を調製するためのプロセス、および中間体VIIIは、本願の特定の実施形態である。
【0030】
1つの実施形態において、本願は、式VIII
【0031】
【化8】
の中間体化合物または塩の調製のためのプロセスを提供する。このプロセスは、縮合剤の存在下での、ピラジンカルボン酸とL−フェニルアラニンとの反応を包含する。本願のプロセスにおいて使用される縮合剤の例は、クロロギ酸アルキル/アリール、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド/HOBtおよびDCC/N−ヒドロキシスクシンイミドから選択される。
【0032】
1つの局面において、本願は、ボルテゾミブの調製のためのプロセスを提供し、このプロセスは、
(i)遊離塩基の形態または酸付加塩の形態の式Vの化合物を、式VIIIの化合物または塩と縮合させて、(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【0033】
【化9】
を生成すること、および
(ii)式IXの化合物のボルテゾミブへの変換、
を包含する。
【0034】
別の局面において、本願は、ボルテゾミブの調製のためのプロセスを提供し、このプロセスは、
a)(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【0035】
【化10】
を、有機ボロン酸アクセプターおよび水性鉱酸と、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させる工程;
b)水層を分離する工程;
c)この水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出する工程;ならびに
d)ボルテゾミブを単離する工程、
を包含する。
【0036】
別の局面において、本願は、ボルテゾミブの精製のためのプロセスを提供し、このプロセスは、
a)ボルテゾミブの有機溶媒中の溶液を提供する工程;
b)反溶媒を添加することにより、生成物を沈殿させる工程;および
c)得られた生成物を分離する工程、
を包含する。
【0037】
本願のプロセスは、いくつかの実施形態において、その立体異性体および/または不純物を含まず、そしてHPLCにより約95%より高い純度を有する、実質的に純粋なボルテゾミブを提供する。
【0038】
本明細書中で議論される形態Aおよび形態B、ならびに事実上任意の形態のボルテゾミブが、本明細書中に記載される溶媒(メタノール、水、酢酸エチル、トルエンおよびジクロロメタンが挙げられるが、これらに限定されない)を使用して精製され得る。従って、形態Aを生成するために使用され得る溶媒は、形態Bを精製するために使用され得、そして形態Bを生成するために使用される溶媒は、形態Aを精製するために使用され得る。
【0039】
さらに、本願はまた、ボルテゾミブの結晶形態AおよびBの調製のためのプロセスを提供する。
【0040】
本願のプロセスにより得られる形態Aの固体は、三量体無水物ではなく、単量体であることが、驚くべきことに見出された。形態Aは、本発明の別の局面である。形態Aは、メタノールおよび水の溶媒系を使用して生成され得る。
【0041】
別の実施形態において、本願は、ボルテゾミブの結晶形態Aの調製のためのプロセスを提供し、このプロセスは、
a)アルコール(特に、メタノール)中のボルテゾミブの溶液を提供する工程;
b)水を添加して固体を沈殿させる工程;および
c)得られた固体を単離する工程、
を包含する。
【0042】
なお別の実施形態において、本願は、ボルテゾミブの結晶形態Bの調製のためのプロセスを提供し、このプロセスは、
a)ハロゲン化アルカン溶媒またはエステル溶媒中のボルテゾミブの溶液を提供する工程;
b)芳香族炭化水素溶媒を添加して固体を沈殿させる工程;および
c)得られた固体を単離する工程、
を包含する。形態Bは、本発明のなお別の実施形態である。1つの実施形態において、形態Bは、トルエンと混合されたジクロロメタンまたは酢酸エチルのうちのいずれか一方の溶媒系を使用して、生成される。
【0043】
別の実施形態において、本願は、本願のプロセスにより得られた、薬学的に有効な量のボルテゾミブ、および少なくとも1つの薬学的に受容可能な賦形剤を含有する、薬学的組成物を提供する。
【0044】
本発明はまた、ボルテゾミブを安定化するための保存システムを提供する。本願の保存システムは、好ましくは、少なくとも1つの密封ポリマーバッグ(例えば、約0.10mm〜約0.50mmの厚さを有する、透明または不透明なポリエチレンバッグ)またはこのようなバッグの組み合わせを備え、これらのバッグは、所望であれば、積層アルミニウムバッグの内側にシールされ得る。必要に応じて、酸素吸収剤および水吸収剤(または乾燥剤)が、このようなバッグのうちの1つ以上の間に含まれ得る。最後に、包装されたサンプルが、HDPE容器内に保存される。
【0045】
本願の規定された薬物包装システムは、長い保存期間にわたって、ボルテゾミブの分解を防止し得る。好ましくは、この保存システムは、大気中の空気および/または水との可能な接触に起因する薬物の不安定性を低下または排除し得る。
【図面の簡単な説明】
【0046】
【図1】実施例8により調製されたボルテゾミブ形態AのX線粉末回折(XRPD)パターンの代表的な例。
【図2】実施例9により調製されたボルテゾミブ形態AのX線粉末回折(XRPD)パターンの代表的な例。
【図3】実施例9により調製されたボルテゾミブ形態Aの示差走査熱量分析(「DSC」)曲線の代表的な例。
【図4】実施例9により調製されたボルテゾミブ形態Aの熱重量分析(TGA)曲線の代表的な例。
【図5】実施例2により調製されたボルテゾミブ形態BのX線粉末回折(XRPD)パターンの代表的な例。
【図6】実施例2により調製されたボルテゾミブ形態Bの熱重量分析(TGA)曲線の代表的な例。
【図7】実施例2により調製されたボルテゾミブ形態Bの赤外吸収スペクトルの代表的な例。
【図8】実施例6により調製された形態BのボルテゾミブのX線粉末回折(XRPD)パターンの代表的な例。
【図9】実施例6により調製されたボルテゾミブの熱重量分析(TGA)曲線の代表的な例。
【図10】実施例6により調製されたボルテゾミブの赤外吸収スペクトルの代表的な例。
【発明を実施するための形態】
【0047】
(発明の詳細な説明)
本明細書は、本発明を具体的に指摘し、明確に特許請求する特許請求の範囲で終わるが、本発明は、以下の説明からよりよく理解されると考えられる。本明細書中で使用されるすべての百分率および比は、全組成物の重量に基づき、そして行われた全ての測定は、他に指定されない限り、25℃で標準圧力におけるものである。全ての温度は、他に特定されない限り、セ氏である。本発明は、本発明の構成要素および本明細書中に記載される他の成分または要素を含み得る(制限なし)か、またはこれらから本質的になり得る。本明細書中で使用される場合、「含む」とは、記載される要素、または構造もしくは機能におけるそれらの均等物に加えて、記載されない他の任意の要素を意味する。用語「有する」および「含む」もまた、文脈が逆のことを示唆しない限り、制限がないと解釈されるべきである。
【0048】
本明細書中で使用される場合、「〜から本質的になる」とは、本発明が、請求項に記載される成分に加えてさらなる成分を含み得ることを意味する。ただし、これらのさらなる成分は、本願発明の基本的な新規特徴を本質的に変更しない場合のみである。好ましくは、このような追加物は、全く存在しないか、または微量でのみ存在する。しかし、化合物の有用性(有用性の程度とは違う)が維持される限り、本発明の基本的な新規特徴を本質的に変更し得る材料を、約10重量%まで含むことが可能であり得る。本明細書中に記載される全ての範囲は、2つの値の「間の」範囲を記載するものを含めて、端点を含む。
【0049】
「約」、「一般に」、「実質的に」などの用語は、用語または値が絶対的ではないが、先行技術には見出されないように、その用語または値を修飾すると解釈されるべきである。このような用語は、状況により規定され、そしてこれらの用語が修飾する用語は、これらの用語と同様に、当業者により理解される。これは少なくとも、値を測定するために使用される所定の技術についての、予測される実験誤差、技術的誤差、および器具誤差の程度を含む。本明細書および特許請求の範囲は、最終製品(例えば、本発明の錠剤または他の投薬形態)が、例えば、特定の粒子サイズまたは粒度分布を有する粒子、あるいは特定の型(例えば、特別な型)の充填剤を含むように記載し得るが、その記載が満足される最終投薬形態を見分けることは困難であり得ることに留意のこと。しかし、このような記載は、例えば、最終製造の前(例えば錠剤の場合、ブレンドおよび錠剤処方)に使用される材料がその記載に合う場合、満足され得る。実際に、投薬形態から直接的には確認され得ない最終製品の任意の特性または特徴に関して、その特性が、最終製造工程の直前に記載される構成要素に存在する場合、十分である。
【0050】
本明細書が物質(例えば、この例においては、ボルテゾミブ)ならびにその独特の結晶形態、塩、溶媒和物および/または光学異性体に、パターン、スペクトルまたは他のグラフデータを参照することにより言及する場合、それらが図または1つ以上のデータ点により「実質的に」示されるかまたは図示されることを限定することにより、なされ得る。この文脈において使用される「実質的に」により、当業者に公知である多数の要因に起因して、パターン、スペクトルおよび他のグラフデータが、それらの位置を、強度または他の値に対して、シフトされ得ることが理解される。例えば、結晶学および粉末X線回折の分野において、パターンの1つ以上のピークのピーク位置または相対強度のシフトは、限定されないが、使用される設備、サンプル調製プロトコル、好ましい充填および配向、放射線源、操作者の誤差、データ収集の方法および長さなどに起因して起こり得る。しかし、当業者は、本明細書中の図を、この場合にはボルテゾミブの未知の形態について生成されたパターンと比較し、そしてその正体を、本明細書中に開示および特許請求される形態のうちの1つであると確認することが可能であるはずである。同じことが、本明細書中に報告され得る他の技術についても事実である。
【0051】
さらに、図が参照される場合、その結晶形態、塩、溶媒和物、および/または光学異性体を、同定の目的で、任意の付随する記載される誤差の範囲内で独特に規定する、図中に図示される任意の数のデータ点の選択が、許容され、そして本明細書は、これを包含し、想定する。ここでまた、例えば、ボルテゾミブの結晶形態について、この形態を独特に同定する図1に表される任意の数のPXRDピーク(しばしば、±0.2°の2θのものを4個〜10個の間)を、この物質の説明および特許請求の手段として選択することが、許容される。
【0052】
分子(例えば、この場合にはボルテゾミブ)に対する言及は、他に特定されない限り、そして一般的な開示と不一致でない限り、その任意の塩、結晶形態もしくは非晶質形態、光学異性体および/または溶媒和物形態をいう。
【0053】
分子または他の物質が、本明細書中で「純粋」であると同定される場合、一般に、他に特定されない限り、この物質が、約99%以上純粋であることを意味する。一般に、これは、所望でない残留溶媒、反応副生成物、不純物および未反応出発物質に関する純度をいう。立体異性体または多型の場合、「純粋」はまた、適切であるように、99%の1つのエナンチオマーもしくはジアステレオマーまたは多型を意味する。
【0054】
用語「実質的に純粋なボルテゾミブ」とは、本明細書中で使用される場合、HPLCにより約95%より高い純度を有し、所望でない立体異性体および/または他の不純物の含有量がほとんどまたは全くない、ボルテゾミブを意味すると理解されるべきである。ボルテゾミブ中の任意の所望でない立体異性体または他の不純物の量は、存在する場合、比較的少量であり、例えば、HPLCにより、ボルテゾミブの重量に基づいて、約5重量%未満、好ましくは、約1重量%未満、より好ましくは、約0.5重量%未満、最も好ましくは、約0.2重量%未満である。
【0055】
本願は、ボルテゾミブの中間体の調製のためのプロセス、およびボルテゾミブの調製のためのプロセスを提供する。
【0056】
1つの局面において、本願は、式III
【0057】
【化11】
の中間体化合物の調製のためのプロセスを提供する。このプロセスは、ルイス酸触媒、水混和性エーテル溶媒およびジクロロメタンの存在下での、式X
【0058】
【化12】
のボロ「ネート」錯体の転移を包含する。
【0059】
1つの実施形態において、このプロセスは、以下の工程を包含する:
I.リチウムジイソプロピルアミド(LDA)を、ジクロロメタンおよび水混和性エーテル溶媒を含む溶媒混合物中の式II
【0060】
【化13】
の化合物の溶液に添加し、引き続いて、得られた溶液を約−40℃〜−70℃の温度に、約10分間〜約60分間、1つの実施形態においては、約30分間にわたって維持することによる、式Xの化合物の形成;
II.塩化亜鉛のテトラヒドロフラン中の混合物を、工程Iの生成物に添加し、引き続いて、この反応塊を約−40℃〜−70℃の温度に、しばしば、約30分間〜約120分間、1つの実施形態においては、約60分間にわたって維持する工程;
III.反応温度を約10℃〜およそ周囲温度(25℃)まで上昇させる工程;
IV.水性酸溶液を添加する工程;ならびに
V.必要に応じて、式IIIの化合物を含む有機層を分離する工程。
【0061】
この後に必要であれば、有機層のブラインでの洗浄および/または有機溶媒の濃縮を行って、式IIIの化合物を単離し得る。
【0062】
式IIIの中間体化合物を調製するためのプロセスにおける、過剰なジクロロメタンの使用は、上記プロセスの好ましい実施形態である。理論により束縛されることを望まないが、式IIIの中間体化合物を調製するためのプロセスにおける過剰なジクロロメタンの使用(これは、本願の特定の局面である)は、ボロ「ネート」錯体の形成中に反応物質として沈殿するのみでなく、反応混合物を水性酸溶液でクエンチした後の有機層分離をまた補助すると考えられる。このプロセスの好ましい実施形態の詳細は、実施例1の工程bに詳述されている。
【0063】
1つの実施形態において、ジクロロメタンは、式IIの化合物1モルあたり約4モル〜約8モルの範囲で利用され得る。この範囲はまた、式IIの化合物1モルあたり約5モル〜約6モルの間であり得る。
【0064】
本願のプロセスにおいて使用される水混和性エーテル溶媒としては、テトラヒドロフランが挙げられ、これは、式IIの化合物1グラムあたり約10倍〜約20倍の範囲で利用され得る。この範囲はまた、式IIの化合物の約15倍〜約17倍、別の実施形態においては、約16倍であり得る。
【0065】
この反応において利用され得る塩化亜鉛の量は、式IIの化合物と比較して、モル過剰であり得る。塩化亜鉛は、式IIの化合物1モルあたり約1.2モル〜約2.0モルの量で存在し得る。塩化亜鉛は、式IIの化合物1モルあたり約1.7モル〜約1.8モルの量で使用され得る。約6%w/wまでの含水量を有する市販の塩化亜鉛が、意図される結果に影響を与えることなく、本願のプロセスにおいて使用され得る。
【0066】
LDA混合物を調製するために利用されるn−ヘキシルリチウムおよびジイソプロピルアミンは、式IIの化合物に対して、それぞれ約1モル〜約1.5モルの量で使用され得る。これらはまた、それぞれ、式IIの化合物1モルあたり約1.2モル〜約1.3モルの量で使用され得る。n−ヘキシルリチウムおよびジイソプロピルアミンは、互いに対して同じモル比で使用される。
【0067】
反応塊をクエンチするために利用され得る酸は、有機酸であっても無機酸であってもよい。無機酸は、塩酸、硫酸またはリン酸から選択され得る。好ましくは、有機酸は、酒石酸、クエン酸から選択され得る。
【0068】
反応塊をクエンチするために使用される水性酸溶液の強度は、約5%w/w〜約20%w/wの範囲であり得る。別の実施形態において、この強度は、約10%w/w〜約12%w/wの酸溶液である。有機層と水層との分離前の反応混合物のpHは、約0.5〜約3であり得る。
【0069】
本願のプロセスにより得られた式IIIの中間体化合物は、引き続いて、遊離塩基の形態または酸付加塩形態の式V
【0070】
【化14】
の中間体化合物の調製のために利用される。この調製は、本明細書中以下のスキーム1に描写されるような、先行技術(Journal Of Biologiacl Chemistry;第259巻、第24号、15106−15114頁、1984年12月25日;米国特許第7,223,745号)において公知であるプロセスと類似のプロセスによる:
【0071】
【化15】
本願のプロセスにおいて使用される式Vの化合物は、トリフルオロ酢酸塩の形態であり得る。
【0072】
式IIIの中間体化合物を式IVの化合物に変換する工程、および式Vの化合物への引き続く変換についての具体的な詳細は、実施例1の工程cおよび工程dに提供されている。
【0073】
遊離塩基の形態または酸付加塩形態の式Vの化合物は、ボルテゾミブの調製のための主要な出発物質であるので、ガスクロマトグラフィー(GC)により測定される場合、ボルテゾミブの収率および純度に影響を与えない、改善された純度を有する式Vの化合物を有することが望ましい。本願のプロセスにより得られる、遊離塩基の形態または酸付加塩の形態の式Vの化合物は、いくつかの実施形態において、GCにより測定される場合、約95%より高い純度、好ましくは、約98%より高い純度、そしてより好ましくは、約99.5%より高い純度を有し得る。
【0074】
遊離塩基の形態または酸付加塩形態の式Vの中間体化合物は、式VIIIの中間体化合物との縮合によるボルテゾミブの調製のための本願のプロセスにおいて、引き続き利用され得る。式VIIIの中間体化合物を調製するためのプロセスは、本願の特定の実施形態のうちの1つである。
【0075】
1つの実施形態において、本願は、式VIII
【0076】
【化16】
の中間体化合物の調製のためのプロセスを提供する。このプロセスは、以下に与えられるスキーム2に描写されるように、ピラジン2−カルボン酸とL−フェニルアラニンまたは塩との、クロロギ酸アルキル/アリールの存在下での反応を包含する。
【0077】
【化17】
ここでRは、必要に応じて置換されたアルキル/アリール基を表す。
【0078】
式VIIIの化合物の調製のために利用され得るクロロギ酸アルキル/アリールとしては、クロロギ酸エチル、クロロギ酸ベンジル、クロロギ酸パラニトロフェニルが挙げられるが、これらに限定されない。
【0079】
カップリング反応は、好ましくは、ケトン溶媒中で、塩基の存在下で、約−20℃〜約40℃の範囲の温度で実施され得る。このケトン溶媒は、アセトン、メチルイソブチルケトン、エチルメチルケトンなどから選択され得る。水が、この反応のための共溶媒として存在し得る。
【0080】
この縮合反応において使用される塩基としては、無機塩基(例えば、水酸化ナトリウム、水酸化カリウムなど);有機塩基(例えば、アルキルアミン(トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、ジメチルアミノピリジン、ジアザビシクロウンデカン、N−メチルモルホリンなどが挙げられる))が挙げられるが、これらに限定されない。上に特定された有機塩基および/または無機塩基のいずれかの混合物もまた、この反応のために使用され得る。
【0081】
本願の実施形態はまた、スキーム3に表されるような、式VIIIのN−(2−ピラジンカルボニル)−L−フェニルアラニンを調製するための代替のプロセスを提供する:
【0082】
【化18】
ここでR’は、必要に応じて置換されたアルキル基を表す。この必要に応じて置換されたアルキル基としては、メチル、エチル、プロピル、tert−ブチル、必要に応じて置換されたベンジルが挙げられるが、これらに限定されない。
【0083】
このプロセスは、
(i)縮合剤の存在下、1つの実施形態においては、塩基の存在下での、ピラジン−2−カルボン酸と、L−フェニルアラニンまたはその塩の必要に応じて置換されたアルキルエステルとの縮合;および
(ii)工程(i)の生成物において得られたエステル官能基の、1つの実施形態においては水性アルカリ溶液を使用する加水分解、
を包含する2つの工程を含む。
【0084】
工程(i)において使用される置換ピラジン2−カルボン酸の量は、L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり、約1.0モル当量〜約1.8モル当量の範囲であり得る。L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり1.2モルもまた、使用され得る。
【0085】
工程(i)において使用される縮合剤は、ジシクロヘキシルカルボジイミド/N−ヒドロキシスクシンイミドおよびエチル−3−(3−ジメチルアミノプロピル)カルボジイミドまたはその塩/N−ヒドロキシベンゾトリアゾールの組み合わせから選択され得る。
【0086】
個々に使用されるジシクロヘキシルカルボジイミド/N−ヒドロキシスクシンイミドおよびエチル−3−(3−ジメチルアミノプロピル)カルボジイミドまたはその塩/N−ヒドロキシベンゾトリアゾールの量は、L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり約1.0モル〜約1.8モルの範囲であり得る。1.2モルが使用され得る。N−ヒドロキシスクシンイミドおよびN−ヒドロキシベンゾトリアゾールは、しばしば、それぞれジシクロヘキシルカルボジイミドおよびエチル−3−(3−ジメチルアミノプロピル)カルボジイミドまたはその塩に対して、同じモル比で使用される。
【0087】
この縮合反応において使用される塩基としては、ジイソプロピルエチルアミン、ピリジン、ジメチルアミノピリジン、ジアザビシクロウンデカン、N−メチルモルホリンが挙げられ得るが、これらに限定されない。使用される塩基の量は、L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり約1.0モル〜約2.0モルの範囲であり得る。好ましくは、1.5モルの塩基もまた使用され得る。
【0088】
工程(i)の縮合反応は、DMF、DMA、またはケトン溶媒(アセトン、メチルイソブチルケトン、エチルメチルケトンなどから選択され得る)などの溶媒中で実施され得る。
【0089】
この反応が実施され得る温度は、約−20℃〜約60℃の範囲であり得る。この反応は、約0℃〜約30℃の温度で実施され得る。
【0090】
この反応は、適切な時間にわたって実施され得る。所望であれば、工程(i)から得られた生成物(中間体エステル)は、加水分解前に、一般的な後処理手順によってか、または本願に開示されるようなプロセスによって、単離され得る。
【0091】
工程(i)において得られる生成物のエステル官能基の加水分解は、好ましくは、水性アルカリ溶液を使用して実施され得る。必要に応じて、有機溶媒もまた、この加水分解工程のための共溶媒として存在し得る。
【0092】
この加水分解のために使用される適切な塩基としては、水酸化ナトリウム、水酸化カリウムなどが挙げられ得るが、これらに限定されない。
【0093】
エステル加水分解のために使用される塩基の量は、当業者により決定され得る。例えば、工程(i)の生成物の中間体エステルが加水分解前に単離される場合、加水分解のために使用される塩基の量は、工程(i)の生成物の単離されたエステル1モルあたり約1.0モル〜約2.0モルの範囲であり得る。1.1モルの塩基もまた使用され得る。
【0094】
工程(ii)の加水分解反応のための共溶媒として使用される有機溶媒は、アセトン、メチルイソブチルケトン、エチルメチルケトン、メタノール、エタノール、イソプロパノールの溶媒から選択され得る。
【0095】
この反応が実施され得る温度は、約0℃〜約60℃の範囲であり得る。この反応はまた、約25℃〜約35℃の温度で実施され得る。この反応は、適切な時間にわたって実施され得、そして得られる生成物(式VIIIの化合物)は、一般的な後処理手順によってか、または本願に開示されるようなプロセスによって、単離され得る。
【0096】
別の局面において、本発明は、HPLCにより95%以上の純度を有する、式VIII
【0097】
【化19】
の化合物N−(2−ピラジンカルボニル)−L−フェニルアラニンを提供する。この化合物は、ボルテゾミブの調製における中間体である。
【0098】
式VIIIの化合物は、ボルテゾミブの調製のための主要な出発物質であるので、ボルテゾミブの収率および純度に影響を与えない、化学的HPLC純度とキラルHPLC純度の両方を有する式VIIIの化合物を有することが望ましい。本願のプロセスにより得られる式VIIIの化合物は、約95%より高い、好ましくは、約99%より高い、より好ましくは、約99.5%より高い、そして最も好ましくは、約99.8%より高い、化学的HPLC純度とキラルHPLC純度との両方を有する。
【0099】
式VIIIの化合物(すなわち、N−(2−ピラジンカルボニル)−L−フェニルアラニンまたはその塩)と、遊離塩基の形態または酸付加塩形態の式Vの化合物との、任意の公知の方法による、式Xの化合物を生成するための縮合、および式IXの化合物のボルテゾミブへの引き続く変換は、スキーム4において本明細書中以下に表されるように、本願の別の局面である。
【0100】
【化20】
式VIIIの化合物または塩と、遊離塩基の形態または酸付加塩形態の式Vの化合物との縮合反応についての具体的な詳細は、実施例1の工程(h)に提供されている。
【0101】
さらなる局面において、本願は、ボルテゾミブの調製のためのプロセスを提供し、このプロセスは、
a)(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【0102】
【化21】
を、有機ボロン酸アクセプターおよび水性鉱酸と、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させる工程;ならびに
b)水層を分離する工程;
c)この水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出する工程;ならびに必要に応じて、
d)ボルテゾミブを単離する工程、
を包含する。
【0103】
本願による、式IXの化合物からのボルテゾミブの調製のプロセスについての全ての工程が、以下に独立して記載される。
【0104】
工程a)
工程a)は、式IX
【0105】
【化22】
の化合物を、有機ボロン酸アクセプターおよび水性鉱酸と、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させて、式Iの化合物を得ることを包含する。
【0106】
工程a)において使用され得る有機ボロン酸アクセプターとしては、ブチルボロン酸、イソブチルボロン酸、フェニルボロン酸、ベンジルボロン酸などが挙げられるが、これらに限定されない。1つの実施形態において、イソブチルボロン酸が、ボロン酸アクセプターとして使用される。
【0107】
工程a)において使用される有機ボロン酸アクセプターの量は、式IXの化合物1モルあたり約1モル当量〜約1.5モル当量の範囲であり得る。式IXの化合物1モルあたり1.2モルが使用され得る。
【0108】
この反応において使用される鉱酸は、塩酸、硫酸、リン酸から選択され得る。1つの実施形態において、塩酸が使用される。使用される水性鉱酸の濃度は、約0.5N〜約3Nの範囲であり得る。1Nの濃度の水性鉱酸もまた使用され得る。この反応のために使用される水性鉱酸の量は、式IXの化合物1グラムあたり約5mlから25mlまで変動し得る。この反応において使用される水性鉱酸の濃度および量は、当業者により容易に決定され得る。
【0109】
工程a)のプロセスにおいて使用され得るアルコール溶媒としては、C1〜C4アルコール(例えば、メタノール、エタノール、イソプロパノール、ブタノール、またはこれらの混合物)が挙げられ得るが、これらに限定されない。工程a)のプロセスにおいて使用され得る脂肪族炭化水素溶媒としては、C5〜C10の、直鎖もしくは分枝鎖のアルカンまたはシクロアルカン(例えば、n−ペンタン、n−ヘキサン、n−ヘプタン、シクロヘキサンまたはこれらの混合物)が挙げられるが、これらに限定されない。いくつかの実施形態において、メタノールおよびn−ヘプタンを含む溶媒混合物が、反応溶媒として使用され得る。
【0110】
工程a)のプロセスは、約25℃から、およそ使用される溶媒の還流温度までの温度で実施され得る。実際に、この反応は、約25℃〜約35℃の温度で実施され得る。
【0111】
工程b)
工程b)は、水層の分離を包含する。
【0112】
反応の完了後、水層が反応混合物から分離され得、そしてその有機層が処分される。この水層は、必要に応じて、好ましくは、C5〜C8脂肪族炭化水素溶媒(例えば、n−ヘプタン)で洗浄され得る。この洗浄は、この水層を脂肪族炭化水素溶媒と一緒に、約10分間〜約15分間激しく攪拌し、そして有機層を分離することにより実施され得る。この有機層は、処分され得る。必要に応じて、このプロセスは、さらに1回〜3回繰り返され得る。
【0113】
必要に応じた洗浄工程後に得られた水層は、減圧ありまたはなしで、濃縮され得る。
【0114】
工程c)
工程c)は、水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出することを包含する。
【0115】
工程b)において得られた水層は、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出され得る。この抽出プロセスは、この溶媒をこの水層に添加し、そして約10分間〜約15分間激しく攪拌し、引き続いて有機層を分離することによって、実施され得る。
【0116】
抽出のために使用され得る水非混和性有機溶媒としては、アルコール溶媒(例えば、イソブタノール、およびt−ブタノール);ハロゲン化溶媒(例えば、ジクロロメタン、1,2−ジクロロエタンおよびクロロホルム);エステル溶媒(例えば、酢酸エチル、酢酸n−プロピル、酢酸イソプロピルおよび酢酸n−ブチル);またはこれらの混合物が挙げられるが、これらに限定されない。抽出のために選択される有機溶媒中への水の溶解度は、約10%w/w未満、好ましくは、約2%w/w未満であるべきである。1つの実施形態において、ハロゲン化アルカンが、抽出溶媒として使用される。なお別の実施形態において、ジクロロメタンが、抽出溶媒として使用される。
【0117】
この抽出プロセスは、ボルテゾミブが有機溶媒中に完全に抽出されるまで繰り返され得る。異なる抽出において得られた有機層が合わせられ、そして必要に応じて、飽和重炭酸ナトリウム溶液、引き続いてブライン溶液で洗浄され、そして減圧下で完全にか、または最小体積まで濃縮されて、ボルテゾミブの残渣または濃縮溶液を得る。この濃縮溶液または残渣は、必要に応じて、25℃〜35℃の温度まで冷却され得る。
【0118】
工程d)
工程d)は、任意であり、生成物の単離を包含する。
【0119】
生成物の単離は、冷却、種結晶の添加、または濃縮溶液もしくは残渣への有機溶媒の添加、あるいはこれらの組み合わせなどの方法により、実施され得る。
【0120】
1つの実施形態において、固体は、工程c)の濃縮溶液または残渣に有機溶媒を添加するなどの方法により、単離され得る。
【0121】
単離のために使用され得る有機溶媒としては、炭化水素溶媒(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);ハロ炭化水素溶媒(例えば、ジクロロメタン、ジクロロエタン);エステル溶媒(例えば、酢酸エチル、酢酸プロピル)、またはこれらの混合物が挙げられ得るが、これらに限定されない。1つの実施形態において、トルエンが、生成物を単離するために使用され得る。トルエンと、ハロ炭化水素溶媒またはエステル溶媒のいずれかとの混合物もまた、使用され得る。しかし、溶媒の混合物が単離のために使用される場合、この混合物中の個々の溶媒の比は、約2%v/v〜約98%v/vの範囲であり得る。
【0122】
溶媒は、工程c)の後に得られたボルテゾミブの濃縮溶液または残渣に、沈殿を起こすために充分な時間(例えば、約15分間〜約2時間、またはより長時間)にわたって添加され得る。適切な温度は、約0℃〜約50℃の範囲であり得る。次いで、得られた反応混合物は、完全な沈殿を起こすために、約30分間〜約5時間、またはより長時間にわたって攪拌され得る。この反応混合物は、25℃〜35℃で、約2時間〜約3時間攪拌され得る。
【0123】
得られた沈殿物は、当該分野において公知である技術により分離され得る。当業者は、固体を不均質混合物から分離するための多くの方法が存在することを理解し得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術を使用することにより、分離され得る。分離後、その固体は、必要に応じて、適切な溶媒で洗浄され得る。
【0124】
このように得られた固体は、乾燥させられ得る。乾燥は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、好ましくは、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、所望の純度を有する生成物を得るために必要な任意の時間(例えば、約1時間〜約25時間、またはより長時間)にわたって実施され得る。
【0125】
別の局面において、本願は、ボルテゾミブの精製のためのプロセスを提供する。このプロセスは、
a)有機溶媒中のボルテゾミブの溶液を提供する工程;
b)反溶媒を添加することにより、生成物を沈殿させる工程;
c)得られた生成物を分離する工程、
を包含する。ボルテゾミブの精製のためのプロセス工程が、本明細書中以下で別々に記載される:
工程a)
工程a)は、有機溶媒中のボルテゾミブの溶液を提供する工程を包含する。
【0126】
有機溶媒中のボルテゾミブの溶液を提供する工程は、必要に応じて窒素雰囲気下での、ボルテゾミブが調製された化学反応の溶液、または有機溶媒へのボルテゾミブの溶解を包含する。約90%以上の純度を有する任意の形態のボルテゾミブが、この溶液を提供するために認容可能である。任意の形態のボルテゾミブ(例えば、非晶質または結晶形態、または任意の方法により得られる任意の割合での非晶質と結晶形態との混合物のボルテゾミブ)が、溶液を提供するために使用され得る。
【0127】
溶解のために使用され得る有機溶媒としては、アルコール溶媒(例えば、メタノール、エタノール、イソプロピルアルコール、n−ブタノール、イソブタノール、およびt−ブタノール);ハロゲン化溶媒(例えば、ジクロロメタン、1,2−ジクロロエタン、クロロホルム);エステル溶媒(例えば、酢酸エチル、酢酸n−プロピル、酢酸イソプロピル、および酢酸n−ブチル);ニトリル溶媒(例えば、アセトニトリル、プロピオニトリル);またはこれらの混合物が挙げられるが、これらに限定されない。好ましくは、メタノール、イソプロピルアルコール、ジクロロメタンまたは酢酸エチルが、ボルテゾミブの精製のために使用され得る。
【0128】
ボルテゾミブの溶液は、約20℃から、使用される溶媒の沸点までの温度で提供され得る。好ましくは、この溶液は、約25℃〜約35℃の温度で提供される。
【0129】
溶解しない沈殿物は、濾過、遠心分離、デカンテーション、および他の技術によって、適切に除去され得る。使用される器具、溶液の濃度および温度に依存して、濾過装置は、尚早な結晶化を回避するために、適切に予熱される必要があり得る。
【0130】
工程b)
工程b)は、反溶媒を添加することによる生成物の沈殿を包含する。
【0131】
工程a)のボルテゾミブ溶媒は、沈殿のために、反溶媒と合わせられ得る。反溶媒の添加は、約5分間〜約1時間、またはより長時間にわたって行われ得る。反溶媒が添加され得る温度は、約0℃〜約45℃の範囲であり得る。使用される温度は、周囲温度(25℃まで)であり得る。
【0132】
得られる懸濁物は、約0℃〜約35℃の温度で維持される。得られた混合物は、完全な沈殿を起こすために、約30分間〜約5時間、またはより長時間攪拌される。1つの実施形態において、ボルテゾミブの懸濁物は、約25℃〜約35℃の温度で、2時間〜3時間維持される。
【0133】
本発明のプロセスにおいて使用され得る反溶媒としては、水、炭化水素(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);エーテル(例えば、ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン、ジメトキシエタン、メチル第三級ブチルエーテル);またはこれらの混合物が挙げられるが、これらに限定されない。特定の実施形態において、トルエンまたはジイソプロピルエーテルのうちのいずれかが、反溶媒として使用される。
【0134】
工程c)
工程c)は、生成物の分離を包含する。
【0135】
得られた沈殿物は、当該分野において公知である技術によって分離され得る。当業者は、固体を混合物から分離するための多くの方法が存在することを理解し得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術により分離され得る。分離後、この固体は、必要に応じて、適切な溶媒で洗浄され得る。
【0136】
湿った固体は、さらに乾燥させられ得る。乾燥は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、1つの実施形態においては、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、所望の純度を有する生成物を得るために必要な任意の時間(例えば、約1時間〜約40時間、またはより長時間)にわたって実施され得る。
【0137】
この精製プロセスは、所望の純度のボルテゾミブが達成されるまで必要に応じて繰り返され得る。例えば、精製は、本質的に純粋なボルテゾミブ、実質的に純粋なボルテゾミブ、または純粋なボルテゾミブが得られるまで、続けられ得る。
【0138】
別の1つの実施形態において、本願は、溶媒がイソプロピルアルコールであり、そして反溶媒がイソプロピルエーテルである、ボルテゾミブの精製のためのプロセスを提供する。
【0139】
なお別の実施形態において、本発明は、実質的に純粋なボルテゾミブの調製のための精製プロセスを提供し、このプロセスは、以下の工程a)〜c)を包含する:
a)アルコール、ハロゲン化溶媒、エステル、ニトリル、炭化水素、エーテルまたはこれらの混合物から選択される有機溶媒中の、ボルテゾミブの溶液を提供する工程;
b)必要であれば、工程a)において得られた溶液に反溶媒を添加する工程;
c)工程a)または工程b)から得られた固体生成物を単離する工程。
【0140】
実質的に純粋なボルテゾミブの調製のためのプロセス工程が、本明細書中以下に別々に記載される:
工程a)は、有機溶媒中のボルテゾミブの溶液を提供する工程を包含する。
【0141】
この溶解のために使用され得る有機溶媒としては、アルコール溶媒(例えば、メタノール、エタノール、イソプロピルアルコール、n−ブタノール、イソブタノール、およびt−ブタノール);ハロゲン化溶媒(例えば、ジクロロメタン、1,2−ジクロロエタン、クロロホルム);エステル溶媒(例えば、酢酸エチル、酢酸n−プロピル、酢酸イソプロピルおよび酢酸n−ブチル);ニトリル溶媒(例えば、アセトニトリル、プロピオニトリル);炭化水素(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);エーテル(例えば、ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン、ジメトキシエタン、メチル第三級ブチルエーテル);またはこれらの混合物が挙げられるが、これらに限定されない。
【0142】
ボルテゾミブの溶液は、ボルテゾミブの精製のためのプロセスの上記実施形態に記載されたようなプロセスにより提供され得る。
【0143】
工程b)は、必要であれば、工程a)において得られた溶液に反溶媒を添加する工程を包含する。
【0144】
工程a)のボルテゾミブ溶液は、必要であれば、沈殿のために反溶媒と合わせられ得る。
【0145】
本発明のプロセスにおいて使用され得る反溶媒としては、水、炭化水素(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);エーテル(例えば、ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン、ジメトキシエタン、メチル第三級ブチルエーテル);またはこれらの混合物が挙げられるが、これらに限定されない。
【0146】
反溶媒を添加して化合物を沈殿させるプロセスは、ボルテゾミブの精製のためのプロセスの上記実施形態に記載されるようなプロセスにより実施され得る。
【0147】
工程c) 工程a)または工程b)から得られた固体生成物を単離する工程。
【0148】
工程a)または工程b)から得られた沈殿物が、ボルテゾミブの精製のためのプロセスの上記実施形態に記載されるようなプロセスによって、分離され得、そして乾燥させられ得る。
【0149】
1つの実施形態において、本願は、ボルテゾミブが、トルエンと組み合わせたジクロロメタンまたは酢酸エチルから選択される有機溶媒の混合物から精製される、ボルテゾミブの精製のためのプロセスを提供する。
【0150】
この混合物中の個々の溶媒は、1%v/v〜99%v/vの範囲であり得る。1つの実施形態において、個々の溶媒は、約5%v/v〜約10%v/vの範囲であり得る。
【0151】
本願の1つの局面は、約95%より高い純度、別の実施形態においては、約99%より高い純度、なお別の実施形態においては、約99.5%より高い純度、そしてなお別の実施形態においては、約99.8%より高い純度(HPLCによる)を有し、そして立体異性体および/または不純物を実質的に含まない、実質的に純粋なボルテゾミブを提供する。
【0152】
1つの実施形態において、本願は、ボルテゾミブの結晶形態Aの調製のためのプロセスを提供し、このプロセスは、
i)アルコール中のボルテゾミブの溶液を提供する工程;
ii)水を添加して固体を沈殿させる工程;および必要に応じて
iii)得られた固体を分離する工程、
を包含する。
【0153】
アルコール中のボルテゾミブの溶液を提供する工程は、ボルテゾミブを、アルコール溶媒中に、必要に応じて窒素雰囲気下で溶解することを包含する。ボルテゾミブの任意の結晶形態もしくは非晶質形態、または結晶形態と非晶質形態との混合物が、溶液を提供するために認容可能である。
【0154】
この溶解のために使用され得るアルコール溶媒としては、メタノール、エタノール、イソプロピルアルコール、n−ブタノール、イソブタノール、およびt−ブタノールが挙げられるが、これらに限定されない。特定の実施形態において、メタノールが、ボルテゾミブの溶解のために使用される。溶解のために使用されるアルコールの量は、ボルテゾミブ1gあたり約2ml〜約10mlで変動し得る。
【0155】
ボルテゾミブの溶液は、約20℃から、使用されるアルコール溶媒の沸点までの温度の範囲の温度で提供され得る。特定の実施形態において、ボルテゾミブの溶液は、約20℃〜約35℃の温度で提供され得る。溶解しない粒子は、濾過、遠心分離、デカンテーション、および他の技術により、適切に除去され得る。
【0156】
工程i)のボルテゾミブ溶液は、沈殿のために水と合わせられる。この水は、約20℃〜約45℃、好ましくは、約25℃〜35℃の温度で、この溶液に添加され得る。この沈殿のために使用される水の量は、ボルテゾミブ1gあたり約2ml〜約10mlで変動し得る。ボルテゾミブ溶液への水の添加は、約5分間〜約1時間、またはより長時間にわたって実施され得る。
【0157】
沈殿のために使用される水の量は、アルコール中のボルテゾミブの濃度、および添加の温度に依存し、そして当業者により容易に決定され得る。
【0158】
この懸濁物は、約0℃〜約35℃の温度で維持される。1つの実施形態において、この懸濁物は、約30℃〜約35℃で維持される。得られた混合物は、完全な沈殿を起こすために、約30分間〜約5時間、またはより長時間にわたって、攪拌され得る。沈殿または完全な沈殿のための他の方法もまた、使用され得る。1つの実施形態において、ボルテゾミブの懸濁物は、約25℃〜約35℃の温度で、2時間〜3時間維持される。
【0159】
得られた沈殿物は、当該分野において公知である技術により分離され得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術を使用することにより、分離され得る。分離後、この固体は、必要に応じて、洗浄され得る。得られた湿った固体は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、1つの実施形態においては、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、ボルテゾミブ形態Aを得るための任意の時間(例えば、約1時間〜約25時間、またはより長時間)にわたって実施され得る。
【0160】
本願のプロセスにより得られた形態Aは、三量体無水物ではなく単量体であることが、驚くべきことに見出された。
【0161】
本発明のプロセスにより得られたボルテゾミブ形態Aのアセトニトリル溶液の質量分析(陽イオン、エレクトロスプレー)は、陰イオンモードで、m/z=383.19においてピークを示した。このことは、この生成物が、三量体ボロキシン(無水物形態)ではなく単量体ボロン酸であることを示す。
【0162】
同じ結晶性生成物についての陽イオンモードは、m/z=367.4を示した。なぜなら、ボルテゾミブのプロトン化分子イオン(M+)+は、不安定であり、そしてインソース脱水(18Da)を起こすからである。さらに、それぞれm/z=1121、1099、および1137における、三量体ボロキシンのナトリウム付加体、プロトン付加体、およびカリウム付加体は観察されず、この化合物の単量体の性質を確認した。
【0163】
1つの実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、実質的に図1に図示されるようなX線回折パターンにより特徴付けられる。
【0164】
1つの実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、約5.82、9.47、9.93、12.80、18.31、20.50、20.90、21.60、22.20、および23.70±0.2°2θの回折角2θに特性ピークを有するX線回折パターンにより特徴付けられる。図2に図示されるこのパターンは、Xcelerator***検出器を有するBragg−Brentanoθ:θゴニオメーターを備えるPANalytical器具を使用して生成された。このパターンを、40kVのチューブ電圧および40mAのチューブ電流で、0.02°のステップサイズおよび1ステップあたりの時間10秒で、3°〜45°の2θの角度範囲にわたって記録した。サンプルを、CuK放射線(=1.5418Å)に曝露した。同じ設備および設定を使用して、図5および図8のパターンを生成した。あるいは、形態Aは、5.82、9.93、11.53、12.80、13.11、15.27、15.47、16.90、17.32、18.31、18.96、19.27、19.85、20.50、21.60、22.24、23.74、24.29、24.65、25.76、26.32、28.03、29.96±0.2°の回折角2θのピークにより特徴付けられ得る。これらのピークを反映するパターンは、図1に見られる。この図は、シンチレーションカウンター検出器を有するRINT2000広角ゴニオメーターを備えるRigaku Dmax 2200器具を使用して生成された。このパターンを、50kVのチューブ電圧および34mAのチューブ電流で、0.02°のステップサイズおよび1ステップあたりの時間3°/分で、3°〜45°の2θの角度範囲にわたって記録した。サンプルを、CuK放射線(=1.5418Å)に曝露した。
【0165】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、実質的に図2に図示されるような、約75.20℃および179.73℃に吸熱ピークを有する、示差走査熱量分析(DSC)サーモグラムにより特徴付けられる。
【0166】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、実質的に図3に図示されるような、約2.88%の重量損失に対応するTGA曲線により特徴付けられる。
【0167】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、KFにより約5%までの含水量により特徴付けられる。
【0168】
1つの実施形態において、本願は、ボルテゾミブの結晶形態Bの調製のためのプロセスを提供し、このプロセスは、
a)ハロゲン化アルカン溶媒またはエステル溶媒中の、ボルテゾミブの溶液を提供する工程;
b)芳香族炭化水素溶媒を添加して固体を沈殿させる工程;および必要に応じて
c)得られた固体を単離する工程、
を包含する。
【0169】
ボルテゾミブの溶液を提供する工程は、好ましくは、ハロゲン化アルカン溶媒またはエステル溶媒のいずれかへの、必要に応じて窒素雰囲気下での化合物の溶解を包含する。ボルテゾミブの任意の結晶形態もしくは非晶質形態、または結晶形態と非晶質形態との混合物が、溶液を提供するために認容可能である。
【0170】
この溶解のために使用される溶媒の量は、ボルテゾミブ1gあたり約2ml〜約10mlで変動し得る。
【0171】
この溶解のために使用され得るハロゲン化アルカン溶媒としては、ジクロロメタン、1,2−ジクロロエタンおよびクロロホルムが挙げられるが、これらに限定されない。この溶解のために使用され得るエステル溶媒としては、酢酸エチル、酢酸イソプロピル、酢酸第三級ブチルが挙げられるが、これらに限定されない。沈殿のために使用され得る芳香族炭化水素溶媒としては、トルエン、キシレンが挙げられるが、これらに限定されない。
【0172】
ボルテゾミブの溶液は、約20℃から、使用される溶媒の沸点までの温度で提供され得る。1つの実施形態において、ボルテゾミブの溶液は、約25℃〜約35℃の温度で提供される。溶解しない沈殿物は、濾過、遠心分離、デカンテーション、および他の技術によって、適切に除去され得る。
【0173】
沈殿は、芳香族炭化水素溶媒をボルテゾミブの溶液に添加することにより実施され得る。添加がなされ得る温度は、約20℃〜約35℃の範囲である。
【0174】
沈殿のために使用される芳香族炭化水素の量は、ハロゲン化アルカン溶媒またはエステル溶媒中のボルテゾミブの濃度、および添加の温度に依存し、そして当業者により容易に決定され得る。
【0175】
懸濁物は、約0℃〜約35℃の温度で、約30分間〜約5時間、またはより長時間にわたり維持され得る。1つの実施形態において、ボルテゾミブの懸濁物は、完全な沈殿を起こすために、約25℃〜約35℃の温度で、2時間〜3時間維持される。
【0176】
得られた沈殿物は、当該分野において公知である技術により分離され得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術を使用することにより、分離され得る。分離後、得られた湿った固体は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、好ましくは、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、ボルテゾミブ形態Bを得るための任意の時間(例えば、約1時間〜約25時間、またはより長時間)にわたって実施され得る。他の技術もまた同様に使用され得る。
【0177】
1つの実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、約4.76、6.30、8.69、9.56、10.72、11.91、12.45、14.64、16.17、17.81、19.21、20.39、21.41、22.70、23.40、24.82、および31.78±0.2°の2θの回折角2θにおける特性ピークを有するX線回折パターンにより特徴付けられる。あるいは、形態Bは、4.76、6.30、8.69、9.56、10.72、11.91、12.45、14.64、16.17、17.81、18.27、19.21、20.39、21.41、22.70、23.40、24.21、26.15、31.78±0.2°の回折角2θにおけるピークにより特徴付けられ得る。
【0178】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、実質的に図5に図示されるようなX線回折パターンにより特徴付けられる。
【0179】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、実質的に図5に図示される、約0.39%の重量損失に対応するTGA曲線により特徴付けられる。
【0180】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、実質的に図6のスペクトルにより図示されるような、臭化カリウム(KBr)ペレット中での赤外吸収スペクトルにより特徴付けられる。
【0181】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、KFにより約3%までの含水量により特徴付けられる。
【0182】
本願のプロセスにより得られたボルテゾミブ(形態Aおよび形態Bを含む)の残留溶媒は、International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use(日米EU医薬品規制調和国際会議:「ICH」)指針により与えられる限度内である。
【0183】
本願のプロセスにより得られたボルテゾミブは、所望の粒子サイズを得るために、必要に応じて粉砕され得る。粉砕または微細化は、生成物の乾燥前、乾燥中、または乾燥の完了後に実施され得る。粉砕操作は、粒子を互いに高速で衝突させることにより、粒子のサイズを減少させ、そして粒子の表面積を増加させる。
【0184】
別の実施形態において、本願は、本発明のプロセスにより得られた、薬学的に有効な量のボルテゾミブの結晶形態Aおよび/またはB、ならびに少なくとも1つの薬学的に受容可能な賦形剤を含有する、薬学的組成物を提供する。
【0185】
本願のプロセスに従って得られたボルテゾミブ結晶形態は、安定であるのみでなく、薬学的処方物を調製する際に使用するためにもまたよく適している。本願による薬学的処方物としては、固体経口投薬形態(例えば、錠剤、カプセル剤、散剤など);液体経口投薬形態(例えば、液剤、分散剤、懸濁剤、乳剤など);非経口投薬形態(筋肉内、皮下、静脈内が挙げられる)(例えば、溶液もしくは懸濁液もしくは分散液、または再形成用の滅菌粉末による、注射可能な投薬物);経皮送達系;標的化送達系などが挙げられるが、これらに限定されない。
【0186】
さらに、本願の発明者らは、密封ポリマーバッグ(例えば、乾燥剤を含まない透明または不透明なポリエチレンバッグ)またはこのようなバッグの組み合わせを備える保存システム内で、通常の温度条件下で保存されたボルテゾミブが、不安定であることを見出した。一般的な保存条件下で保存された場合のボルテゾミブの安定性研究を、表1に要約する。
【0187】
表1:25℃および60%RH(一般的な保存条件であり、本発明の局面であると本明細書中に記載される条件を使用しない)で保存された場合のボルテゾミブ
【0188】
【表1】
*酢酸エチルから
この表において、不純物aは、
【0189】
【化23】
であり、そして不純物bは、ボルテゾミブのRRジアステレオマーとSSジアステレオマーとの組み合わせである。
(S,S)−異性体:
【0190】
【化24】
(R,R)−異性体:
【0191】
【化25】
上記結果は、25℃および60% RHで、上記のような保存システム中で保存されたボルテゾミブが、製品のかなりの分解を示し、これがボルテゾミブの純度の低下を生じることを示す。
【0192】
本願の保存方法に従う、制御された環境中でのボルテゾミブの保存は、ボルテゾミブを安定化し、そして長い保存期間にわたってボルテゾミブの純度を維持することが見出された。いかなる特定の理論にも束縛されることを望まないが、ボルテゾミブに関連する明らかな不安定性の問題は、ボルテゾミブを、低下した湿度レベル、ならびに/あるいは低い大気中酸素レベルおよび/または低い光レベルを有する環境で保存することにより克服され得ると考えられる。
【0193】
さらなる局面において、本願は、ボルテゾミブを安定化するための保存システムを提供する。本願の保存システムは、好ましくは、少なくとも1つの密封ポリマーバッグ(例えば、約0.10mm〜約0.50mmの厚さを有する、透明または不透明なポリエチレンバッグ)、またはこのようなバッグの組み合わせを備え、これらのバッグは、所望であれば、積層アルミニウムバッグの内側にシールされ得る。酸素吸収剤および水吸収剤(または乾燥剤)が、このようなバッグのうちの1つ以上の間に含まれ得る。最後に、包装されたサンプルは、HDPE容器内に保存される。
【0194】
好ましい局面のうちの1つにおいて、本願は、ボルテゾミブを不活性雰囲気下で安定化するための保存システムを提供し、その包装システムは、
a.少なくとも1つの外側密封ポリマーバッグ;
b.ボルテゾミブを含む別のポリマーバッグ、または必要に応じて、このようなバッグの組み合わせであって、これらのバッグは、所望であれば、積層アルミニウムバッグの内側にシールされ得る、バッグ;
c.ポリマーバッグaとポリマーバッグbとの間に配置された、酸素吸収剤および必要に応じて、水吸収剤(または乾燥剤)、
を備える。
【0195】
本願の保存システムは、好ましくは、内部にボルテゾミブが収容された容器を備え、この容器は、好ましくは、外部環境に対して、より低い湿度、酸素レベルおよび光レベル、またはこれらの組み合わせを有する内部環境を提供し得る。本願の保存システムは、好ましくは、保存条件下(2℃〜8℃の温度、およびまた25℃〜35℃の温度、60% RH)で少なくとも約3ヶ月間、ボルテゾミブの純度を維持し得る。本願の保存システムは、より好ましくは、少なくとも約3ヶ月間、そして最も好ましくは、約6ヶ月間、初期純度または分解が全くない状態から約0.2%未満の最大分解で、ボルテゾミブの純度を維持し得る。本願のプロセスにより得られた、制御された環境においてボルテゾミブについて実施した安定性の実験を、表2に要約する。
【0196】
特に好ましい実施形態において、本願の保存システムは、ボルテゾミブ(例えば、実施例5により生成されたボルテゾミブ)を、少なくとも約1ヶ月間、少なくとも約2ヶ月間、または少なくとも約6ヶ月間でさえも、2℃〜8℃またはそれより高温において維持し得る(表2を参照のこと)。
【0197】
本願の保存システムにおいて使用され得る容器は、好ましくは、少なくとも1つの外部密封ポリマーバッグを備える。適切なポリマーバッグとしては、保存の目的に適した1つ以上の市販のバッグ(例えば、ポリエチレンバッグ(例えば、低密度ポリエチレンバッグおよび高密度ポリエチレンバッグ)、ポリプロピレンバッグ、ポリエステルバッグ、ナイロンバッグ、ポリ塩化ビニル(PVC)バッグなど)が挙げられ得る。この保存システムにおいて利用されるポリマーバッグは、約0.10mm〜約0.50mmの厚さを有し得る。
【0198】
特に好ましい実施形態において、この保存システムは、密封積層アルミニウムバッグ、この密封アルミニウムバッグに収容された高密度ポリエチレンバッグ、密封不透明ポリエチレンバッグに収容された密封透明低密度ポリエチレンバッグ、この透明ポリエチレンバッグに含まれたボルテゾミブ、ならびにこの透明ポリエチレンバッグと不透明ポリエチレンバッグとの間に配置された、酸素吸収剤および必要に応じて乾燥剤またはこれらの両方を備える。
【0199】
適切な酸素吸収剤としては、アスコルビン酸ベースの有機物型のもの、および鉄粉含有材料ベースの無機物型のものが挙げられるが、これらに限定されない。好ましくは、AgelessZ200またはAgelessZ100などが、ボルテゾミブの安定性を維持するための酸素吸収剤として利用され得る。
【0200】
適切な乾燥剤としては、酸化アルミニウム、塩化カルシウム、ドライエライト(CaSO4)、モレキュラーシーブ(例えば、活性化モレキュラーシーブ)、シリカゲルなど、およびこれらの組み合わせが挙げられるが、これらに限定されない。好ましくは、シリカゲルが、ボルテゾミブの安定性を維持するための乾燥剤として使用され得る。
【0201】
本願の薬物包装は、長い保存期間にわたって、ボルテゾミブの分解を防止し得る。好ましくは、この保存システムは、酸素および/または水との接触に起因する薬物の不安定性を低下または排除し得る。このシステムは、最低約3ヶ月間保存される場合に、ボルテゾミブの有意な分解の排除を生じ、より好ましくは、初期純度または分解が全くない状態から約0.2%までの最大分解を生じる。
【0202】
本願の保存システムのための例示的な包装はまた、ボルテゾミブが透明ポリエチレンバッグに包装されて密封され、この透明ポリエチレンバッグが不透明(例えば、黒色)ポリエチレンバッグに包装され、次いでこの不透明ポリエチレンバッグが密封され、そして次に、積層アルミニウムバッグに包装および密封され、次いでこの積層アルミニウムバッグが密封される、包装の型を含み得る。酸素吸収剤および乾燥剤が、2つのポリマーバッグ/層の間に配置(例えば、分散)され得る。
【0203】
本願によれば、本明細書中に記載されるような包装は、不活性雰囲気下(例えば、窒素雰囲気下)または周囲空気中で包装され得る。
【0204】
本願の特定の具体的な局面および実施形態が、以下の実施例を参照しながらより詳細に説明される。これらの実施例は、説明のみにより提供されるのであり、本願の範囲をいかなる方法でも限定すると解釈されるべきではない。
【実施例】
【0205】
(実施例1:N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IX)を調製するためのプロセス)
【0206】
【化26】
式IXの化合物を調製するためのプロセスは、工程a)〜工程h)の工程を含み、これらの工程が、以下に個々に示される。
【0207】
工程a)2−(2−メチルプロピル)−(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロール(式II)の調製:
【0208】
【化27】
イソブチルボロン酸(50.0g)の、n−ヘプタン(250ml)中の攪拌溶液に、25℃〜30℃で(+)−ピナンジオール(83.3g)を添加し、そして25℃〜30℃で1時間攪拌した。この反応塊にブライン溶液を添加し、そしてこの混合物を攪拌した。層を分離させ、そしてその有機層を、さらなる溶媒が留去されなくなるまで減圧下で濃縮して、表題化合物(式II)を得た。
【0209】
工程b)2−((1S)−1−クロロ−3−メチルブチル)−(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロール(式III)の調製:
【0210】
【化28】
I.塩化亜鉛とテトラヒドロフランとの混合物を調製する工程
II.LDA混合物を調製する工程
III.ジクロロメタンおよび水混和性エーテル溶媒を含有する溶媒混合物中の、式II
【0211】
【化29】
の化合物の溶液を調製する工程
IV.工程IIの溶液を工程IIIの溶液に添加し、続いてこの溶液を約−40℃〜−70℃の温度で維持する工程
V.工程Iの混合物を工程IVの生成物に添加し、続いてこの反応塊を約−40℃〜−70℃の温度で維持する工程
VI.その反応温度を約10℃〜周囲温度まで上昇させる工程
VII.水性酸溶液を添加する工程
VIII.式IIIの化合物を含有する有機層を分離し、そしてその生成物を単離する工程。
【0212】
I.塩化亜鉛とテトラヒドロフランとの混合物を調製する工程
ZnCl2(115g)を、第一の丸底フラスコ(R.B.フラスコ)内のテトラヒドロフラン(805ml)に、窒素雰囲気下25℃〜35℃で入れ、そして得られた混合物の温度を35℃〜40℃まで上昇させ、3時間〜4時間維持して、ZnCl2溶液を得た。
【0213】
II.LDA混合物を調製する工程
ジイソプロピルアミン(86ml)を、第二のR.B.フラスコ内のテトラヒドロフラン(345ml)に窒素雰囲気下で入れ、そして得られた混合物を−7℃〜−15℃まで冷却し、n−ヘキシルリチウムをこの混合物に入れ、そして30分間〜40分間維持して、LDA混合物を得た。
【0214】
III.式IIの化合物の溶液を調製する工程
式IIの化合物(115.0g)を、第三のR.B.フラスコ内のジクロロメタン(161ml)およびテトラヒドロフラン(690ml)に、窒素雰囲気下25℃〜35℃で入れ、そしてこの混合物を−55℃〜−60℃まで冷却した。
【0215】
IV.工程IIの溶液を工程IIIの溶液に添加する工程
LDA混合物を第二のR.B.フラスコから反応混合物に−55℃〜−60℃で入れ、そして30分間維持した。その温度を−50℃まで上昇させた。
【0216】
V.工程Iの混合物を工程IVの生成物に添加する工程
ZnCl2溶液を第一のR.B.フラスコから−45℃〜−50℃で入れ、そして1時間維持した。
【0217】
VI.その反応温度を約10℃まで上昇させる工程
その反応混合物を10℃まで温めた。
【0218】
VII.水性酸溶液を添加する工程
10% H2SO4を入れ、10分間〜15分間攪拌し、そして有機層を分離した。
【0219】
VIII.式IIIの化合物を含有する有機層を分離し、そしてその生成物を単離する工程
工程VIIにおいて分離した有機層を次の工程に供したが、水相は処分した。
【0220】
この有機層を、水層が約6〜7のpHに達するまで、攪拌しながらブライン溶液で洗浄した。
【0221】
この有機層を減圧下で濃縮して、式IIIの化合物を単離した。
【0222】
工程c)N,N−ビス(トリメチルシリル)−(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロール−2−イル]−3−メチルブチルアミン(式IV)の調製:
【0223】
【化30】
ヘキサメチルジシラザン(101.3ml)をテトラヒドロフラン(414ml)に窒素雰囲気下で入れ、そしてこの混合物を−20℃〜−30℃まで冷却した。n−ヘキシルリチウムをこの混合物に、攪拌しながら、−20℃〜−30℃の温度を維持する程度にゆっくりと入れた。この反応混合物を−20℃〜−25℃で1時間〜2時間攪拌し、式IIIの化合物(138g)を、この新たに調製したTHF中のリチウムHMDSに、−15℃〜−20℃の温度を維持するように入れた。この反応混合物を25℃〜30℃の温度まで温め、そして2時間〜3時間維持した。この反応混合物をシリカ床で濾過し、そしてこの床をジイソプロピルエーテルで洗浄した。その濾液を減圧下で濃縮して残渣にし、表題化合物(式IV)を得た。
【0224】
工程d)4,6−メタノ−1,3,2−ベンゾジオキサボロール−2−メタンアミン,ヘキサヒドロ−3a,5,5−トリメチル−α−(2−メチルプロピル)−,(αR,3aS,4S,6S,7aR)−,トリフルオロアセテート(式V)の調製:
【0225】
【化31】
トリフルオロ酢酸(129ml)をジイソプロピルエーテル(1980ml)に窒素雰囲気下25℃〜30℃で入れ、そしてこの反応塊を−10℃まで冷却した。式IVの化合物(198g)をこの反応塊にゆっくりと−10℃で入れ、そしてこの温度で8時間維持した。この反応塊を濾過し、ジイソプロピルエーテル(198ml)で洗浄し、そして得られた固体を25℃〜30℃で水(1500ml)でスラリー洗浄した。このスラリーを濾過し、水で洗浄し、そして得られた固体を減圧下40℃〜50℃で8時間乾燥させて、74.0gの表題化合物(式V)を得た。
純度(GCによる)=99.54%。
【0226】
工程e)L−フェニルアラニンメチルエステル塩酸塩(式VI)の調製:
【0227】
【化32】
L−フェニルアラニン(25g)のメタノール(125ml)中の攪拌溶液に、25℃〜30℃で、塩化チオニル(13.2ml)を攪拌しながら入れ、そしてこの混合物を55℃〜60℃で2時間〜3時間維持した。この反応塊を25℃〜30℃まで冷却し、そして減圧下で、出発物質に対する2倍体積まで濃縮した。イソプロピルアルコール(125ml)をこの反応塊に入れ、そして出発物質に対する2倍体積まで濃縮した。この反応塊を0℃〜5℃まで冷却し、そして攪拌しながら1時間〜2時間維持した。この反応塊を濾過し、イソプロピルアルコールで洗浄し、30分間吸引乾燥させ、そして得られた固体を45℃〜50℃で3時間〜4時間乾燥させて、28.8gの表題化合物(式VI)を得た。HPLCによるキラル純度:100%。
【0228】
工程f)L−フェニルアラニン,N−(ピラジニルカルボニル)−メチルエステル(式VII)の調製:
【0229】
【化33】
ピラジン−2−カルボン酸(3.45g)のDMF(50ml)中の攪拌混合物に、25℃〜30℃で、N−ヒドロキシスクシンイミド(3.2g)を攪拌しながら入れ、そして0℃〜5℃まで冷却した。N,N’−ジシクロヘキシルカルボジイミド(DCC)(5.75g)をこの反応塊に0℃〜5℃で入れ、そして15分間〜20分間攪拌した。式VIの化合物(5.0g)をこの反応塊に0℃〜5℃で入れ、そして15分間〜20分間攪拌した。さらに、NMM(3.8ml)をこの反応塊に0℃〜5℃で入れ、そして15分間〜20分間攪拌した。この反応混合物を25℃〜30℃まで温め、そして攪拌しながら2時間〜3時間維持した。この反応塊を濾過し、そしてその固体を分離した。得られた濾液を酢酸エチル(100ml)で希釈し、そして脱塩水で洗浄した。有機層を1N HClで洗浄し、続いて重炭酸ナトリウム溶液で洗浄した。その有機層を減圧下で、式VIに対する2倍体積まで濃縮し、そして25℃〜30℃まで冷却した。n−ヘプタン(20ml)を入れて化合物を沈殿させ、この反応塊を0℃〜5℃まで冷却し、1時間〜2時間維持し、そして減圧下で濾過した。得られた固体を40℃〜45℃で3時間〜4時間乾燥させて、5.7gの表題化合物(式VII)を得た。HPLCによる純度:99.73%、HPLCによるキラル純度:99.97%。
【0230】
工程g)N−(ピラジニルカルボニル)−L−フェニルアラニン(式VIII)の調製:
【0231】
【化34】
式VII(100g)のアセトン(500ml)中の攪拌混合物に、25℃〜30℃で、NaOH溶液(15.4gのNaOHを500mlの水に溶解することにより得た)を入れ、そしてこの温度で30分間〜50分間維持した。1N HClを使用することにより、この反応塊のpHを2に調整し、そしてこの反応塊を0℃〜5℃まで冷却した。この反応塊を、攪拌しながら0℃〜5℃で1時間〜2時間維持し、減圧下で濾過し、そして得られた材料を45℃〜50℃で4時間〜5時間乾燥させて、84.4gの表題化合物(式VIII)を得た。
HPLCによる純度:99.94重量%
HPLCによるキラル純度:100%。
【0232】
あるいは、N−(ピラジニルカルボニル)−L−フェニルアラニン(式VIII)はまた、以下により調製され得る:
(a)以下に記載されるようなプロセスに従ってエチルクロロホルメートを使用する:
アセトン(40ml)、ピラジンカルボン酸(5g)およびトリエチルアミン(6.77ml)の混合物を、約−5℃〜約0℃まで冷却し、そしてエチルクロロホルメート(4.76ml)を入れた。この反応塊を、約30分間攪拌した。この反応懸濁物を約25℃〜約30℃の温度に達せさせ、そして約3時間維持した。この反応懸濁物を約0℃〜約5℃まで冷却した。第二のフラスコ中で、水性水酸化ナトリウム(70mlの水中1.68g)溶液を約0℃〜約5℃まで冷却し、そしてこれに、アセトン(30ml)およびL−フェニルアラニン(6.6g)を添加し、そしてこの混合物をその温度で約1時間攪拌した。この第二のフラスコの反応塊を、第一のフラスコの反応塊に、約0℃〜約5℃の温度で添加し、次いで、約2時間攪拌し、続いて約25℃〜約30℃まで温度を上昇させた。この反応塊を、約25℃〜約30℃の温度で約16時間さらに攪拌した。酢酸エチル(150ml)をこの反応溶液に入れ、そして約30分間攪拌した。層を分離し、そして1N塩酸(35ml)を、その分離した水層に添加した。この反応溶液を約0℃〜約5℃まで冷却し、そして約2時間攪拌した。得られた懸濁物を濾過し、そしてその固体を水(10ml)で洗浄した。次いで、その固体を約50℃の温度で約4時間乾燥させて、2.6gの表題化合物を得た。
HPLCによる純度:99.2重量%
HPLCによるキラル純度:100%。
【0233】
(b)または、以下に記載されるようなプロセスに従って、EDC・HClとHOBtとの組み合わせを使用することによる:
ピラジンカルボン酸(168.7g)、ジメチルホルムアミド(1.4リットル)、ヒドロキシベンゾトリアゾール(HOBt:220g)、およびN−メチルモルホリン(221ml)の混合物を、約0℃〜約5℃の温度まで冷却した。EDC塩酸塩(1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド・HCl;278g)をこの反応溶液に約0℃の温度で添加し、そして約30分間攪拌した。上記から得られたL−フェニルアラニンメチルエステル塩酸塩(240g)をDMF(1リットル)に溶解し、次いで、この反応混合物に添加した。N−メチルモルホリン(110ml)をこの反応混合物に添加し、そしてこの反応混合物を、約0℃〜約5℃の温度で約1時間維持した。この反応混合物を、約25℃〜約35℃の温度まで温め、そして水(3.6リットル)で希釈した。この反応塊を酢酸エチル(3×2.4リットル)で抽出した。分離した酢酸エチル層を1N塩酸(1.2リットル)で洗浄し、次いで、2つの層を分離した。その有機層を、飽和重炭酸ナトリウム溶液(4.8リットル)およびブライン溶液(2.4リットル)で洗浄した。その有機層を、約45℃の温度で完全に濃縮して、260gのピラジン−2−カルボニルフェニルアラニンメチルエステルを得た。
【0234】
ピラジン−2−カルボニルフェニルアラニンメチルエステル(5g)をアセトン(25ml)に溶解し、そして約5分間攪拌した。水酸化ナトリウム溶液(25mlの水中701mgの水酸化ナトリウム)をこの反応溶液に添加し、そして約25℃の温度で約3時間攪拌し、次いで、そのpHを、1N塩酸(11ml)を用いて約2のpHに調整した。この反応混合物を約0℃〜約5℃の温度まで冷却し、そして約1時間攪拌した。この懸濁物を濾過し、そして吸引乾燥させて、4.0gのピラジン−2−カルボニルフェニルアラニンを得た。
HPLCによるキラル純度:100%
HPLCによる化学純度:99.88%。
【0235】
工程h)式IXの調製:
【0236】
【化35】
式VIIIの化合物(28.6g)のジクロロメタン(400ml)中の攪拌混合物に、窒素雰囲気下25℃〜30℃で、N−ヒドロキシスクシンイミド(13.3g)およびDCC(23.9g)を入れ、そして10分間〜20分間攪拌した。式Vの化合物(40g)をこの反応塊に入れ、そして15分間〜20分間攪拌した。ジイソプロピルエチルアミン(DIPEA)(27ml)を入れ、そしてこの反応塊を25℃〜30℃で2時間〜3時間維持した。この反応塊を濾過し、そしてこの固体をジクロロメタン(80ml)で洗浄した。得られた濾液を1N HClで洗浄し、続いて重炭酸ナトリウム溶液で洗浄した。有機層を、式Vに対する2倍体積まで濃縮した。メタノール(200ml)を入れ、そして式Vに対する2倍体積まで濃縮した。得られた濃縮塊は、表題化合物(式IX)である。
【0237】
あるいは、式IXの化合物はまた、以下に記載されるようなプロセスによって、EDC・HClおよびヒドロキシベンゾトリアゾールを使用することにより調製され得る:
N−(2−ピラジンカルボニル)−L−フェニルアラニン(500mg)をジクロロメタン(10ml)中に懸濁させ、そして約−5℃〜約0℃まで冷却した。ヒドロキシベンゾトリアゾール(HOBt:310mg)をこの反応塊に入れ、続いて、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC・HCl、385ml)を添加し、そして15分間攪拌した。(1R)−(S)−ピナンジオール1−アンモニウムトリフルオロアセテート−3−メチルブタン−1−ボロネート(695mg)をこの反応混合物に添加し、そして約5℃の温度で約10分間攪拌した。ジイソプロピルエチルアミン(0.6ml)をこの反応混合物に入れ、そして約5℃の温度で約30分間攪拌した。この反応混合物を約25℃〜約30℃の温度まで温め、そして約1時間攪拌し、続いて、1N塩酸(30ml)を添加した。層を分離し、そしてその有機層を、1N塩酸(15ml)および飽和重炭酸ナトリウム溶液(2×30ml)で洗浄した。その有機層を完全に濃縮して、表題化合物を得た。
HPLCによる純度:84.98%。HPLCにより測定された9.01%は、最後のボルテゾミブ工程の前に形成されたボルテゾミブであると考えられることに留意のこと。従って、HPLCにより測定される場合の全体の純度は、84.98%+9.0%、すなわち93.99%であるはずである。
【0238】
(実施例2:ボルテゾミブ(式I)を調製するためのプロセス)
式IXの化合物(13.6g)のメタノール(272ml)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(272ml)およびイソブチルボロン酸(3.2g)を入れた。2N HCl(272ml)をこの反応塊に激しく攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離して処分した。n−ヘプタン(272ml×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下35℃〜48℃で濃縮した。この水層をジクロロメタン(272ml)で激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(272ml×2)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、6mlの反応塊を得、そして25℃〜30℃まで冷却した。
純度:HPLCにより95.13%。
【0239】
トルエン(102ml)を上記反応塊に入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%ジクロロメタンで洗浄し、そして減圧下45℃〜50℃で5時間乾燥させて、粗製ボルテゾミブを得た。
収量:7.0g(70%)
HPLCによる純度:99.22%
HPLCによる不純物B:0.43%
多型の形態:形態B
XRDパターン:図5に図示されるとおり。
【0240】
(実施例3:メタノールおよび水を使用するボルテゾミブの精製のプロセス)
ボルテゾミブ(5.0g、純度99.22%)およびメタノール(15ml)を丸底フラスコに入れ、そして25℃〜30℃で攪拌した。得られた溶液に脱塩水(15ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(30ml;水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、3.4gの表題化合物を得た。
HPLCによる純度:99.57%
HPLCによる不純物B:0.30%。
【0241】
同じプロセスを再現することにより得られた生成物のさらなる精製は、99.6%の純度(HPLCによる)を有するボルテゾミブを生じた。
HPLCによる不純物B:0.23%
HPLCによるキラル純度:99.83%。
【0242】
(実施例4:ボルテゾミブを調製した後に精製するためのプロセス)
式IXの化合物(68.3g)のメタノール(1.22L)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(1.36L)およびイソブチルボロン酸(16.13g)を入れた。1N HCl(13.6L)をこの反応塊に攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離し、そして処分した。n−ヘプタン(1.36L×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下で濃縮した。その水層をジクロロメタン(13.6L)で、激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(13.6L×2)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、粗製ボルテゾミブ(47.0g)を得た。
HPLCによる純度:95.62%
HPLCによる不純物B:0.59%。
【0243】
精製1:ボルテゾミブ(25g、純度:95.62%)およびトルエン中5%酢酸エチル(250ml)を丸底フラスコに入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%酢酸エチルで洗浄し、そして減圧下50℃で5時間乾燥させて、ボルテゾミブを得た。
収量:18.0g(72%)
HPLCによる純度:99.68%
HPLCによる不純物B:0.27%。
【0244】
精製2:ボルテゾミブ(18.0g、純度99.68%)およびメタノール(54ml)を丸底フラスコに入れ、そして攪拌した。この反応塊を焼結式漏斗(sinted funnel)で濾過し、そしてその床を18mlのメタノールで洗浄した。得られた濾液に脱塩水(72ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(108ml:水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、14gの表題化合物を得た。
収量:14.0g(77%)
HPLCによる純度:99.83%
不純物B:0.15%(HPLCによる)
HPLCによるキラル純度:99.85%。
【0245】
(実施例5:ボルテゾミブを調製した後に精製するためのプロセス)
式IXの化合物(10.25g)のメタノール(174.5ml)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(205ml)およびイソブチルボロン酸(2.42g)を入れた。0.5N HCl(205ml)をこの反応塊に攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離し、そして処分した。n−ヘプタン(205ml×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下で濃縮した。その水層をジクロロメタン(205ml)で、激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(205ml)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、粗製ボルテゾミブ(5.8g)を得た。
HPLCによる純度:95.81%
HPLCによる不純物B:0.34%。
【0246】
精製1:ボルテゾミブ(5g、純度:95.81%)およびトルエン中5%ジクロロメタン(40ml)を丸底フラスコに入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%ジクロロメタンで洗浄し、そして減圧下50℃で5時間乾燥させて、ボルテゾミブを得た。
収量:4.2g(84%)
HPLCによる純度:99.12%
HPLCによる不純物B:0.31%。
【0247】
精製2:ボルテゾミブ(4.2g、純度99.12%)およびメタノール(12.6ml)を丸底フラスコに入れ、そして攪拌した。この反応塊に脱塩水(12.6ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(25.2ml:水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、2.95gの表題化合物を得た。
収量:2.95g(70%)
HPLCによる純度:99.70%
HPLCによる不純物B:0.2%
キラル純度:99.83%(HPLCによる)。
【0248】
(実施例6:酢酸エチルおよびトルエンを使用するボルテゾミブの精製のプロセス)
ボルテゾミブ(5.0g、純度96.0%)およびトルエン中5%酢酸エチル(40ml)を丸底フラスコに入れた。この反応混合物を約28℃の温度で3時間攪拌した。この反応混合物を濾過し、そしてその固体をトルエン中5%酢酸エチル(50ml)で洗浄した。得られた固体を50℃で5時間乾燥させて、3.5gの表題化合物を得た。
HPLCによる純度:99.28%
HPLCによる異性体不純物:0.55%
XRDパターン:図8に図示されるとおり。
【0249】
(実施例7:イソプロピルアルコールおよびジイソプロピルエーテルを使用するボルテゾミブの精製のプロセス)
ボルテゾミブ(1.0g、純度:93.46%)およびイソプロピルアルコール(6.0ml)を丸底フラスコに入れ、そして約27℃で溶解するまで攪拌した。得られた溶液にジイソプロピルエーテル(20ml)を添加し、そして約26℃の温度で3時間攪拌した。この反応混合物を濾過し、そしてその固体をジイソプロピルエーテル(5ml)で洗浄した。得られた固体を約15分間吸引乾燥させて、400mgの表題化合物を得た。
HPLCによる純度:99.49%
HPLCによる異性体不純物:0.18%。
【0250】
(実施例8:ボルテゾミブの精製のプロセス)
ボルテゾミブ(0.5g、異性体不純物1.89%;純度:97.47%)およびメタノール(1.5ml)を丸底フラスコに入れた。得られた溶液に水(1.5ml)を添加し、そして約25℃の温度で約2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(12ml:水+メタノール1:1)で洗浄した。最後に、得られた固体を約25℃の温度で約20分間吸引乾燥させて、400mgの表題化合物を得た。
HPLCによる純度:99.35重量%
HPLCによる異性体純度:0.65重量%。
旋光:
SOR(比旋光):5N HClの媒体中(濃度:1%)に基づいて、25℃において−45.00°
SOR:メタノールの媒体中(濃度:1%)に基づいて、25℃の温度において−43.82°
XRDパターン:図1に図示されるとおり。
【0251】
(実施例9:ボルテゾミブの形態Aの調製のためのプロセス)
ボルテゾミブ(3.0g、純度99.57%)およびメタノール(9ml)を丸底フラスコに入れ、そして25℃〜30℃で攪拌した。得られた溶液に脱塩水(9ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(18ml;水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、2.0gの表題化合物を得た。
XRDパターン:図2に図示されるとおり。
【0252】
(実施例10:本発明の例示的な保存システム中でのボルテゾミブの安定性研究)
ボルテゾミブの2つの異なるサンプルを、2℃〜8℃および25℃、60%相対湿度(RH)の保存条件で、保存システム中(約0.10mmの厚さを有する1つのポリマーバッグに包装し、その後、約0.10mmの厚さを有し外部乾燥剤を含む別のポリマーバッグに再度包装する)で保存した。15日後、1ヵ月後(30日目)、2ヵ月後(60日目)および3ヵ月後(90日目)に、各サンプルから少量を取り出し、そしてその純度をHPLCにより確認して、各サンプルのもとのHPLCクロマトグラム(保存期間の開始前)と比較した。各サンプルの化学純度を、HPLCクロマトグラムにより%面積として得、そして初期純度値(保存期間の開始前)と比較した。その結果を表2に要約する。
【0253】
表2:2℃〜8℃および25℃、60% RHで、本発明に記載されるような包装中で保存される場合のボルテゾミブの安定性
【0254】
【表2】
*メタノール/水から(形態A)
**酢酸エチルから。
【0255】
上記結果は、本発明により記載されるような包装条件を使用して、長い保存期間にわたって、ボルテゾミブが安定化され得、同時にその満足な純度を維持し得ることを示す。
【0256】
(実施例11:塩基性媒体中のボルテゾミブの水溶液の安定性研究)
ボルテゾミブの水溶液を、先行技術に記載されるような反応プロセスから得、そして塩基性pH約10.5に調整し(2N NaOHを使用)、そして25℃〜35℃で維持する。1時間後、2時間後および3時間後に、溶液から少量を取り出し、そしてその純度をHPLCにより確認し、そして初期のサンプルのもとのHPLCクロマトグラム(pHを調整した直後)と比較した。各サンプルの純度を、HPLCクロマトグラムにより%面積として得、そして初期純度値と比較した。その結果を表3に要約する。
【0257】
表3:塩基性媒体中のボルテゾミブの時間に伴う分解
【0258】
【表3】
表3において、不純物aは、
【0259】
【化36】
であり、そして不純物bは、ボルテゾミブのRRジアステレオマーとSSジアステレオマーとの組み合わせである。
【0260】
上記データから、ボルテゾミブの水溶液が塩基性媒体中で攪拌状態に維持された場合、時間と共に、化合物の純度は継続的に低下し、そして不純物の含有量は次第に増加したことが明らかである。
【0261】
(実施例12:ボルテゾミブを調製した後に精製するためのプロセス)
式IXの化合物(27.3g)のメタノール(491.4ml)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(546ml)およびイソブチルボロン酸(6.4g)を入れた。1.0N HCl(546ml)をこの反応塊に攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離し、そして処分した。n−ヘプタン(546ml×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下で濃縮して、メタノールを除去した。その水層をジクロロメタン(546ml)で、激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(546ml×2)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、粗製ボルテゾミブ(19.0g)を得た。
HPLCによる純度:96.66%
HPLCによる異性体不純物:0.46%。
【0262】
精製1:ボルテゾミブ(17g、純度:96.66%)およびトルエン中5%酢酸エチル(136ml)を丸底フラスコに入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%酢酸エチルで洗浄し、そして減圧下50℃で5時間乾燥させて、ボルテゾミブを得た。
収量:14.0g(82.3%)
HPLCによる純度:98.61%
HPLCによる異性体不純物:0.34%。
【0263】
精製2:ボルテゾミブ(14g、純度98.61%)およびメタノール(42ml)を丸底フラスコに入れ、そして攪拌した。この反応塊を半融ガラス濾過器(scinted funnel)で濾過し、そしてメタノール(14ml)で洗浄した。その濾液を丸底フラスコに入れ、そしてこの反応塊に脱塩水(56ml)を添加し、そして約27℃の温度で約2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(84ml:水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、8.5gの表題化合物を得た。
収量:8.5g(60%)
HPLCによる純度:99.84%
HPLCによる異性体不純物:0.12%。
【0264】
(HPLC分析)
(方法A(化学純度について))
化学純度についてのボルテゾミブサンプルのHPLC測定を、Waters対称シールドRP−18(250×内径4.6mm、粒子サイズ5μm)および270nmで作動するUV検出器を備えるWatersシステムを使用して実施した。分析を、以下の移動相を使用して、1.0ml/分の流量で、55分間の実行時間で実施した。
移動相A:700mlの水、300mlのアセトニトリルおよび1mlのギ酸を混合し、濾過し、脱気する。
移動相B:800mlのアセトニトリル、200mlの水および1mlのギ酸を混合し、濾過し、脱気する。
溶出:勾配プログラム
【0265】
【表3A】
(方法B(キラル純度について))
キラル純度についてのボルテゾミブサンプルのHPLC測定を、5μmシリカゲルにコーティングしたアミローストリス(3,5ジメチルフェニルカルバメート)250×4.6(ChiralPak AD−H)および270nmで作動するUV検出器を備えるWatersシステムを使用して実施した。分析を、以下の移動相を使用して、1.0ml/分の流量で、25分間の実行時間で実施した。
移動相:n−ヘキサン、イソプロピルアルコールおよび無水アルコールを、8:1:1の比で混合する。
【0266】
上記方法で調製したボルテゾミブの立体異性体および/または不純物を、相対保持時間(「RRT」)により特徴付ける。RRTは、HPLCクロマトグラムにおいて、不純物が検出される保持時間を、ボルテゾミブの保持時間に関して考慮することにより計算され得る。この分析の結果を、表4および表5に示す。
【0267】
表4:方法A(HPLCによる化学純度)
【0268】
【表4】
N.D.:検出不可能
*は、式
【0269】
【化37】
の不純物aを表す。
**は、以下の式:
(S,S)−異性体:
【0270】
【化38】
(R,R)−異性体:
【0271】
【化39】
の構成化合物を含む、ボルテゾミブのジアステレオマー混合物である不純物bを表す。
【0272】
このボルテゾミブのジアステレオマー不純物は、他の異性体不純物、すなわち、
(R,S)−異性体:
【0273】
【化40】
と一緒にキラルHPLC法(方法B)により分離され得る。これらのRRTを、表5に要約する。
【0274】
表5:方法B(HPLCによるキラル純度)
【0275】
【表5】
【技術分野】
【0001】
(出願の分野)
本願は、ボルテゾミブおよびその調製のために有用な中間体化合物の調製のためのプロセスに関する。
【0002】
本願はまた、実質的に純粋なボルテゾミブの調製のためのプロセスに関する。
【0003】
本願はさらに、ボルテゾミブの結晶形態AおよびBの調製のためのプロセスに関する。本願はまた、ボルテゾミブの中間体化合物および独特な形態に関する。
【背景技術】
【0004】
(出願の背景)
ボルテゾミブとは、化学名[(1R)−3−メチル−1−[[(2S)−1−オキソ−3−フェニル−2−[(ピラジニルカルボニル)アミノ]プロピル]アミノ]ブチル]ボロン酸を有する薬物化合物について認可された名称であり、そして構造式I
【0005】
【化1】
により表される。
【0006】
ボルテゾミブは、抗腫瘍剤であり、そして市場で商標名「VELCADE(登録商標)」のもとで注射剤の形態で入手可能な治療用プロテオソームインヒビターである。各バイアルは、3.5mgのボルテゾミブを、滅菌凍結乾燥粉末として含む。米国において、ボルテゾミブは、多発性骨髄腫およびマントル細胞リンパ腫の処置のために認可されている。
【0007】
Summary Basis Of Approval for Bortezomib(NDA 21−602)の化学的概説の節に、薬物物質、薬物製品および再構築された薬物製品が、3つの異なる分子形態を有することが開示された。PS−341(ボルテゾミブ)薬物物質は、固相において、三量体のボロキシンとして存在する。水に曝露されると、このボロキシンは、単量体のボロン酸PS−341に加水分解する。凍結乾燥されたSP−341薬物製品の構造は、対称なマンニトールエステルであることが決定された。0.9% NaCl溶液により再構築される間、再構築されたPS−341薬物製品は、マンニトールエステルとPS−341ボロン酸との間の平衡からなる。
【0008】
特許文献1は、ボルテゾミブ、その薬学的に受容可能な塩、薬学的組成物、および哺乳動物におけるプロテオソーム機能を阻害するための使用を開示する。さらに、ボルテゾミブおよびそのアナログの調製のためのプロセスを開示する。
【0009】
特許文献2は、ボルテゾミブエステルの凍結乾燥処方物を開示する。特許文献2によれば、特許文献1に記載されるようなプロセスにより調製されるボルテゾミブは、白色非晶質粉末である。
【0010】
特許文献3は、ルイス酸触媒(転移反応を促進し、そしてα−炭素原子のエピマー化を最小にするため)の存在下での中間体ボロ「ネート」錯体の転移による、ボロン酸エステルの同族体化のためのプロセスを開示する。
【0011】
特許文献4は、ボルテゾミブ中間体(これは、ボロン酸エステル化合物である)およびボルテゾミブそのものを調製するためのプロセスを開示する。
【0012】
特許文献4は、式III
【0013】
【化2】
の中間体化合物の調製のための、式X
【0014】
【化3】
のボロ「ネート」錯体のルイス酸により促進される転移による、以前に報告されたプロセスが、テトラヒドロフラン(水と混和性であるエーテル溶媒)を使用し、そして厳密に乾燥された器具、溶媒、およびルイス酸試薬を必要とするので、このような反応は、高価であり、スケールアップが困難であることを開示する。さらに、特許文献4によれば、先行技術のプロセスのスケールアップの試みは頻繁に、ボロン酸エステル化合物のジアステレオマー比のさらなる悪化をもたらし、このことは、テトラヒドロフラン溶媒を除去して水混和性溶媒で交換するための反応混合物の濃縮中のハロゲン化物イオンへの生成物の曝露、または引き続く水性物質洗浄中にテトラヒドロフランの完全な除去をし損なうことの、いずれかに起因する。
【0015】
特許文献4は、ボロ「ネート」錯体の転移を、水との混和性が低いエーテル溶媒および配位共溶媒中で実施することにより、先行技術の問題に取り組むようである。このプロセスにおいて使用するために特許文献4において確認されている、低水混和性エーテル溶媒の非限定的な例としては、tert−ブチルメチルエーテル、tert−ブチルエチルエーテル、tert−アミルメチルエーテル、およびイソプロピルエーテルが挙げられる。
【0016】
さらに、特許文献4は、
(i)式IX
【0017】
【化4】
の中間体ボロン酸エステル化合物、有機ボロン酸アクセプター、低級アルカノール、C5〜8炭化水素溶媒、および水性鉱酸を含む二相混合物を提供する工程;
(ii)この二相混合物を攪拌してボルテゾミブを得る工程;
(iii)溶媒層を分離する工程;ならびに
(iv)ボルテゾミブまたはそのボロン酸無水物を有機溶媒中に抽出する工程、
を包含する、ボルテゾミブの調製のためのプロセスを開示する。
【0018】
生成物の純度を高めるために、工程(iii)の後に得られた水層が抽出工程(iv)の前に洗浄されて、中性有機不純物が除去される。このようなプロセスは、以下の工程:
1)溶媒層を分離する工程;
2)水層を塩基性pHに調整する工程;
3)この水層を有機溶媒で洗浄する工程;および
4)この水相を約6未満のpHに調整する工程、
を包含する。
【0019】
従って、特許文献4に記載されるプロセスは、酸性条件下および塩基性条件下での複数回の有機溶媒洗浄、引き続いて、化合物を有機溶媒中に抽出する工程、その生成物を単離する工程、ならびにさらに再結晶して、純度が高まったボルテゾミブを得る工程を包含する。
【0020】
ボルテゾミブを水性塩基性溶液に曝露すると、ボルテゾミブの純度が低下することが見出されている。具体的には、このようなプロセスが大規模で行われる場合、ボルテゾミブの水性塩基性条件への長時間にわたる曝露は、回避することが困難であり、従って、このプロセスは、産業規模で使用することに耐えられないかもしれない。
【0021】
特許文献5は、ボルテゾミブの結晶形態Iおよび結晶形態II、ならびにこれらの調製のためのプロセスを開示する。形態Iのボルテゾミブは、溶媒(例えば、アセトン、CHCl3、CH2Cl2、またはニトリルなど)および希釈剤(例えば、ジイソプロピルエーテル、第三級ブチルメチルエーテル、n−ヘキサンおよびn−ヘプタン)を使用することにより調製される。形態IIのボルテゾミブは、酢酸エチルの熱溶液から調製される。特許文献5はまた、形態Iおよび形態IIが上記溶媒を使用することにより相互変換可能であることを開示する。
【先行技術文献】
【特許文献】
【0022】
【特許文献1】米国特許第5,780,454号明細書
【特許文献2】米国特許第6,713,446号明細書
【特許文献3】米国特許第4,525,309号明細書
【特許文献4】米国特許出願公開第2005/0240047号明細書
【特許文献5】国際公開第2008/075376号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0023】
ボルテゾミブおよびその中間体の調製のための、単純かつ好都合なプロセスを提供することが、依然として必要とされている。
【課題を解決するための手段】
【0024】
(発明の要旨)
本願によれば、ボルテゾミブの中間体の調製のためのプロセス、およびボルテゾミブの調製のためのプロセス、ならびにこれらのプロセスにより生成された中間体およびボルテゾミブが提供される。
【0025】
さらに、本願はまた、実質的に純粋なボルテゾミブの調製のためのプロセス、および実質的に純粋なボルテゾミブを提供する。
【0026】
1つの局面において、本願は、式III
【0027】
【化5】
の中間体化合物の調製のためのプロセスを提供し、このプロセスは、ルイス酸触媒、水混和性エーテル溶媒および過剰なジクロロメタンの存在下での、式X
【0028】
【化6】
のボロ「ネート」錯体の転移を包含する。式IIIの中間体化合物の調製のためのプロセスにおける過剰なジクロロメタンの使用は、ボロ「ネート」錯体形成中に反応物質として沈殿するのみでなく、水性酸溶液でのこの反応混合物のクエンチ後の有機層分離もまた補助することが見出された。本発明のプロセスにより得られる式IIIの中間体化合物は、引き続いて、先行技術において公知であるプロセスによって、遊離塩基の形態または酸付加塩形態の、式V
【0029】
【化7】
の中間体化合物の調製のために利用される。式Vの化合物は、引き続いて、式VIIIの中間体化合物との縮合反応による、ボルテゾミブの調製のためのプロセスにおいて利用され得る。式VIIIの中間体化合物を調製するためのプロセス、および中間体VIIIは、本願の特定の実施形態である。
【0030】
1つの実施形態において、本願は、式VIII
【0031】
【化8】
の中間体化合物または塩の調製のためのプロセスを提供する。このプロセスは、縮合剤の存在下での、ピラジンカルボン酸とL−フェニルアラニンとの反応を包含する。本願のプロセスにおいて使用される縮合剤の例は、クロロギ酸アルキル/アリール、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド/HOBtおよびDCC/N−ヒドロキシスクシンイミドから選択される。
【0032】
1つの局面において、本願は、ボルテゾミブの調製のためのプロセスを提供し、このプロセスは、
(i)遊離塩基の形態または酸付加塩の形態の式Vの化合物を、式VIIIの化合物または塩と縮合させて、(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【0033】
【化9】
を生成すること、および
(ii)式IXの化合物のボルテゾミブへの変換、
を包含する。
【0034】
別の局面において、本願は、ボルテゾミブの調製のためのプロセスを提供し、このプロセスは、
a)(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【0035】
【化10】
を、有機ボロン酸アクセプターおよび水性鉱酸と、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させる工程;
b)水層を分離する工程;
c)この水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出する工程;ならびに
d)ボルテゾミブを単離する工程、
を包含する。
【0036】
別の局面において、本願は、ボルテゾミブの精製のためのプロセスを提供し、このプロセスは、
a)ボルテゾミブの有機溶媒中の溶液を提供する工程;
b)反溶媒を添加することにより、生成物を沈殿させる工程;および
c)得られた生成物を分離する工程、
を包含する。
【0037】
本願のプロセスは、いくつかの実施形態において、その立体異性体および/または不純物を含まず、そしてHPLCにより約95%より高い純度を有する、実質的に純粋なボルテゾミブを提供する。
【0038】
本明細書中で議論される形態Aおよび形態B、ならびに事実上任意の形態のボルテゾミブが、本明細書中に記載される溶媒(メタノール、水、酢酸エチル、トルエンおよびジクロロメタンが挙げられるが、これらに限定されない)を使用して精製され得る。従って、形態Aを生成するために使用され得る溶媒は、形態Bを精製するために使用され得、そして形態Bを生成するために使用される溶媒は、形態Aを精製するために使用され得る。
【0039】
さらに、本願はまた、ボルテゾミブの結晶形態AおよびBの調製のためのプロセスを提供する。
【0040】
本願のプロセスにより得られる形態Aの固体は、三量体無水物ではなく、単量体であることが、驚くべきことに見出された。形態Aは、本発明の別の局面である。形態Aは、メタノールおよび水の溶媒系を使用して生成され得る。
【0041】
別の実施形態において、本願は、ボルテゾミブの結晶形態Aの調製のためのプロセスを提供し、このプロセスは、
a)アルコール(特に、メタノール)中のボルテゾミブの溶液を提供する工程;
b)水を添加して固体を沈殿させる工程;および
c)得られた固体を単離する工程、
を包含する。
【0042】
なお別の実施形態において、本願は、ボルテゾミブの結晶形態Bの調製のためのプロセスを提供し、このプロセスは、
a)ハロゲン化アルカン溶媒またはエステル溶媒中のボルテゾミブの溶液を提供する工程;
b)芳香族炭化水素溶媒を添加して固体を沈殿させる工程;および
c)得られた固体を単離する工程、
を包含する。形態Bは、本発明のなお別の実施形態である。1つの実施形態において、形態Bは、トルエンと混合されたジクロロメタンまたは酢酸エチルのうちのいずれか一方の溶媒系を使用して、生成される。
【0043】
別の実施形態において、本願は、本願のプロセスにより得られた、薬学的に有効な量のボルテゾミブ、および少なくとも1つの薬学的に受容可能な賦形剤を含有する、薬学的組成物を提供する。
【0044】
本発明はまた、ボルテゾミブを安定化するための保存システムを提供する。本願の保存システムは、好ましくは、少なくとも1つの密封ポリマーバッグ(例えば、約0.10mm〜約0.50mmの厚さを有する、透明または不透明なポリエチレンバッグ)またはこのようなバッグの組み合わせを備え、これらのバッグは、所望であれば、積層アルミニウムバッグの内側にシールされ得る。必要に応じて、酸素吸収剤および水吸収剤(または乾燥剤)が、このようなバッグのうちの1つ以上の間に含まれ得る。最後に、包装されたサンプルが、HDPE容器内に保存される。
【0045】
本願の規定された薬物包装システムは、長い保存期間にわたって、ボルテゾミブの分解を防止し得る。好ましくは、この保存システムは、大気中の空気および/または水との可能な接触に起因する薬物の不安定性を低下または排除し得る。
【図面の簡単な説明】
【0046】
【図1】実施例8により調製されたボルテゾミブ形態AのX線粉末回折(XRPD)パターンの代表的な例。
【図2】実施例9により調製されたボルテゾミブ形態AのX線粉末回折(XRPD)パターンの代表的な例。
【図3】実施例9により調製されたボルテゾミブ形態Aの示差走査熱量分析(「DSC」)曲線の代表的な例。
【図4】実施例9により調製されたボルテゾミブ形態Aの熱重量分析(TGA)曲線の代表的な例。
【図5】実施例2により調製されたボルテゾミブ形態BのX線粉末回折(XRPD)パターンの代表的な例。
【図6】実施例2により調製されたボルテゾミブ形態Bの熱重量分析(TGA)曲線の代表的な例。
【図7】実施例2により調製されたボルテゾミブ形態Bの赤外吸収スペクトルの代表的な例。
【図8】実施例6により調製された形態BのボルテゾミブのX線粉末回折(XRPD)パターンの代表的な例。
【図9】実施例6により調製されたボルテゾミブの熱重量分析(TGA)曲線の代表的な例。
【図10】実施例6により調製されたボルテゾミブの赤外吸収スペクトルの代表的な例。
【発明を実施するための形態】
【0047】
(発明の詳細な説明)
本明細書は、本発明を具体的に指摘し、明確に特許請求する特許請求の範囲で終わるが、本発明は、以下の説明からよりよく理解されると考えられる。本明細書中で使用されるすべての百分率および比は、全組成物の重量に基づき、そして行われた全ての測定は、他に指定されない限り、25℃で標準圧力におけるものである。全ての温度は、他に特定されない限り、セ氏である。本発明は、本発明の構成要素および本明細書中に記載される他の成分または要素を含み得る(制限なし)か、またはこれらから本質的になり得る。本明細書中で使用される場合、「含む」とは、記載される要素、または構造もしくは機能におけるそれらの均等物に加えて、記載されない他の任意の要素を意味する。用語「有する」および「含む」もまた、文脈が逆のことを示唆しない限り、制限がないと解釈されるべきである。
【0048】
本明細書中で使用される場合、「〜から本質的になる」とは、本発明が、請求項に記載される成分に加えてさらなる成分を含み得ることを意味する。ただし、これらのさらなる成分は、本願発明の基本的な新規特徴を本質的に変更しない場合のみである。好ましくは、このような追加物は、全く存在しないか、または微量でのみ存在する。しかし、化合物の有用性(有用性の程度とは違う)が維持される限り、本発明の基本的な新規特徴を本質的に変更し得る材料を、約10重量%まで含むことが可能であり得る。本明細書中に記載される全ての範囲は、2つの値の「間の」範囲を記載するものを含めて、端点を含む。
【0049】
「約」、「一般に」、「実質的に」などの用語は、用語または値が絶対的ではないが、先行技術には見出されないように、その用語または値を修飾すると解釈されるべきである。このような用語は、状況により規定され、そしてこれらの用語が修飾する用語は、これらの用語と同様に、当業者により理解される。これは少なくとも、値を測定するために使用される所定の技術についての、予測される実験誤差、技術的誤差、および器具誤差の程度を含む。本明細書および特許請求の範囲は、最終製品(例えば、本発明の錠剤または他の投薬形態)が、例えば、特定の粒子サイズまたは粒度分布を有する粒子、あるいは特定の型(例えば、特別な型)の充填剤を含むように記載し得るが、その記載が満足される最終投薬形態を見分けることは困難であり得ることに留意のこと。しかし、このような記載は、例えば、最終製造の前(例えば錠剤の場合、ブレンドおよび錠剤処方)に使用される材料がその記載に合う場合、満足され得る。実際に、投薬形態から直接的には確認され得ない最終製品の任意の特性または特徴に関して、その特性が、最終製造工程の直前に記載される構成要素に存在する場合、十分である。
【0050】
本明細書が物質(例えば、この例においては、ボルテゾミブ)ならびにその独特の結晶形態、塩、溶媒和物および/または光学異性体に、パターン、スペクトルまたは他のグラフデータを参照することにより言及する場合、それらが図または1つ以上のデータ点により「実質的に」示されるかまたは図示されることを限定することにより、なされ得る。この文脈において使用される「実質的に」により、当業者に公知である多数の要因に起因して、パターン、スペクトルおよび他のグラフデータが、それらの位置を、強度または他の値に対して、シフトされ得ることが理解される。例えば、結晶学および粉末X線回折の分野において、パターンの1つ以上のピークのピーク位置または相対強度のシフトは、限定されないが、使用される設備、サンプル調製プロトコル、好ましい充填および配向、放射線源、操作者の誤差、データ収集の方法および長さなどに起因して起こり得る。しかし、当業者は、本明細書中の図を、この場合にはボルテゾミブの未知の形態について生成されたパターンと比較し、そしてその正体を、本明細書中に開示および特許請求される形態のうちの1つであると確認することが可能であるはずである。同じことが、本明細書中に報告され得る他の技術についても事実である。
【0051】
さらに、図が参照される場合、その結晶形態、塩、溶媒和物、および/または光学異性体を、同定の目的で、任意の付随する記載される誤差の範囲内で独特に規定する、図中に図示される任意の数のデータ点の選択が、許容され、そして本明細書は、これを包含し、想定する。ここでまた、例えば、ボルテゾミブの結晶形態について、この形態を独特に同定する図1に表される任意の数のPXRDピーク(しばしば、±0.2°の2θのものを4個〜10個の間)を、この物質の説明および特許請求の手段として選択することが、許容される。
【0052】
分子(例えば、この場合にはボルテゾミブ)に対する言及は、他に特定されない限り、そして一般的な開示と不一致でない限り、その任意の塩、結晶形態もしくは非晶質形態、光学異性体および/または溶媒和物形態をいう。
【0053】
分子または他の物質が、本明細書中で「純粋」であると同定される場合、一般に、他に特定されない限り、この物質が、約99%以上純粋であることを意味する。一般に、これは、所望でない残留溶媒、反応副生成物、不純物および未反応出発物質に関する純度をいう。立体異性体または多型の場合、「純粋」はまた、適切であるように、99%の1つのエナンチオマーもしくはジアステレオマーまたは多型を意味する。
【0054】
用語「実質的に純粋なボルテゾミブ」とは、本明細書中で使用される場合、HPLCにより約95%より高い純度を有し、所望でない立体異性体および/または他の不純物の含有量がほとんどまたは全くない、ボルテゾミブを意味すると理解されるべきである。ボルテゾミブ中の任意の所望でない立体異性体または他の不純物の量は、存在する場合、比較的少量であり、例えば、HPLCにより、ボルテゾミブの重量に基づいて、約5重量%未満、好ましくは、約1重量%未満、より好ましくは、約0.5重量%未満、最も好ましくは、約0.2重量%未満である。
【0055】
本願は、ボルテゾミブの中間体の調製のためのプロセス、およびボルテゾミブの調製のためのプロセスを提供する。
【0056】
1つの局面において、本願は、式III
【0057】
【化11】
の中間体化合物の調製のためのプロセスを提供する。このプロセスは、ルイス酸触媒、水混和性エーテル溶媒およびジクロロメタンの存在下での、式X
【0058】
【化12】
のボロ「ネート」錯体の転移を包含する。
【0059】
1つの実施形態において、このプロセスは、以下の工程を包含する:
I.リチウムジイソプロピルアミド(LDA)を、ジクロロメタンおよび水混和性エーテル溶媒を含む溶媒混合物中の式II
【0060】
【化13】
の化合物の溶液に添加し、引き続いて、得られた溶液を約−40℃〜−70℃の温度に、約10分間〜約60分間、1つの実施形態においては、約30分間にわたって維持することによる、式Xの化合物の形成;
II.塩化亜鉛のテトラヒドロフラン中の混合物を、工程Iの生成物に添加し、引き続いて、この反応塊を約−40℃〜−70℃の温度に、しばしば、約30分間〜約120分間、1つの実施形態においては、約60分間にわたって維持する工程;
III.反応温度を約10℃〜およそ周囲温度(25℃)まで上昇させる工程;
IV.水性酸溶液を添加する工程;ならびに
V.必要に応じて、式IIIの化合物を含む有機層を分離する工程。
【0061】
この後に必要であれば、有機層のブラインでの洗浄および/または有機溶媒の濃縮を行って、式IIIの化合物を単離し得る。
【0062】
式IIIの中間体化合物を調製するためのプロセスにおける、過剰なジクロロメタンの使用は、上記プロセスの好ましい実施形態である。理論により束縛されることを望まないが、式IIIの中間体化合物を調製するためのプロセスにおける過剰なジクロロメタンの使用(これは、本願の特定の局面である)は、ボロ「ネート」錯体の形成中に反応物質として沈殿するのみでなく、反応混合物を水性酸溶液でクエンチした後の有機層分離をまた補助すると考えられる。このプロセスの好ましい実施形態の詳細は、実施例1の工程bに詳述されている。
【0063】
1つの実施形態において、ジクロロメタンは、式IIの化合物1モルあたり約4モル〜約8モルの範囲で利用され得る。この範囲はまた、式IIの化合物1モルあたり約5モル〜約6モルの間であり得る。
【0064】
本願のプロセスにおいて使用される水混和性エーテル溶媒としては、テトラヒドロフランが挙げられ、これは、式IIの化合物1グラムあたり約10倍〜約20倍の範囲で利用され得る。この範囲はまた、式IIの化合物の約15倍〜約17倍、別の実施形態においては、約16倍であり得る。
【0065】
この反応において利用され得る塩化亜鉛の量は、式IIの化合物と比較して、モル過剰であり得る。塩化亜鉛は、式IIの化合物1モルあたり約1.2モル〜約2.0モルの量で存在し得る。塩化亜鉛は、式IIの化合物1モルあたり約1.7モル〜約1.8モルの量で使用され得る。約6%w/wまでの含水量を有する市販の塩化亜鉛が、意図される結果に影響を与えることなく、本願のプロセスにおいて使用され得る。
【0066】
LDA混合物を調製するために利用されるn−ヘキシルリチウムおよびジイソプロピルアミンは、式IIの化合物に対して、それぞれ約1モル〜約1.5モルの量で使用され得る。これらはまた、それぞれ、式IIの化合物1モルあたり約1.2モル〜約1.3モルの量で使用され得る。n−ヘキシルリチウムおよびジイソプロピルアミンは、互いに対して同じモル比で使用される。
【0067】
反応塊をクエンチするために利用され得る酸は、有機酸であっても無機酸であってもよい。無機酸は、塩酸、硫酸またはリン酸から選択され得る。好ましくは、有機酸は、酒石酸、クエン酸から選択され得る。
【0068】
反応塊をクエンチするために使用される水性酸溶液の強度は、約5%w/w〜約20%w/wの範囲であり得る。別の実施形態において、この強度は、約10%w/w〜約12%w/wの酸溶液である。有機層と水層との分離前の反応混合物のpHは、約0.5〜約3であり得る。
【0069】
本願のプロセスにより得られた式IIIの中間体化合物は、引き続いて、遊離塩基の形態または酸付加塩形態の式V
【0070】
【化14】
の中間体化合物の調製のために利用される。この調製は、本明細書中以下のスキーム1に描写されるような、先行技術(Journal Of Biologiacl Chemistry;第259巻、第24号、15106−15114頁、1984年12月25日;米国特許第7,223,745号)において公知であるプロセスと類似のプロセスによる:
【0071】
【化15】
本願のプロセスにおいて使用される式Vの化合物は、トリフルオロ酢酸塩の形態であり得る。
【0072】
式IIIの中間体化合物を式IVの化合物に変換する工程、および式Vの化合物への引き続く変換についての具体的な詳細は、実施例1の工程cおよび工程dに提供されている。
【0073】
遊離塩基の形態または酸付加塩形態の式Vの化合物は、ボルテゾミブの調製のための主要な出発物質であるので、ガスクロマトグラフィー(GC)により測定される場合、ボルテゾミブの収率および純度に影響を与えない、改善された純度を有する式Vの化合物を有することが望ましい。本願のプロセスにより得られる、遊離塩基の形態または酸付加塩の形態の式Vの化合物は、いくつかの実施形態において、GCにより測定される場合、約95%より高い純度、好ましくは、約98%より高い純度、そしてより好ましくは、約99.5%より高い純度を有し得る。
【0074】
遊離塩基の形態または酸付加塩形態の式Vの中間体化合物は、式VIIIの中間体化合物との縮合によるボルテゾミブの調製のための本願のプロセスにおいて、引き続き利用され得る。式VIIIの中間体化合物を調製するためのプロセスは、本願の特定の実施形態のうちの1つである。
【0075】
1つの実施形態において、本願は、式VIII
【0076】
【化16】
の中間体化合物の調製のためのプロセスを提供する。このプロセスは、以下に与えられるスキーム2に描写されるように、ピラジン2−カルボン酸とL−フェニルアラニンまたは塩との、クロロギ酸アルキル/アリールの存在下での反応を包含する。
【0077】
【化17】
ここでRは、必要に応じて置換されたアルキル/アリール基を表す。
【0078】
式VIIIの化合物の調製のために利用され得るクロロギ酸アルキル/アリールとしては、クロロギ酸エチル、クロロギ酸ベンジル、クロロギ酸パラニトロフェニルが挙げられるが、これらに限定されない。
【0079】
カップリング反応は、好ましくは、ケトン溶媒中で、塩基の存在下で、約−20℃〜約40℃の範囲の温度で実施され得る。このケトン溶媒は、アセトン、メチルイソブチルケトン、エチルメチルケトンなどから選択され得る。水が、この反応のための共溶媒として存在し得る。
【0080】
この縮合反応において使用される塩基としては、無機塩基(例えば、水酸化ナトリウム、水酸化カリウムなど);有機塩基(例えば、アルキルアミン(トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、ジメチルアミノピリジン、ジアザビシクロウンデカン、N−メチルモルホリンなどが挙げられる))が挙げられるが、これらに限定されない。上に特定された有機塩基および/または無機塩基のいずれかの混合物もまた、この反応のために使用され得る。
【0081】
本願の実施形態はまた、スキーム3に表されるような、式VIIIのN−(2−ピラジンカルボニル)−L−フェニルアラニンを調製するための代替のプロセスを提供する:
【0082】
【化18】
ここでR’は、必要に応じて置換されたアルキル基を表す。この必要に応じて置換されたアルキル基としては、メチル、エチル、プロピル、tert−ブチル、必要に応じて置換されたベンジルが挙げられるが、これらに限定されない。
【0083】
このプロセスは、
(i)縮合剤の存在下、1つの実施形態においては、塩基の存在下での、ピラジン−2−カルボン酸と、L−フェニルアラニンまたはその塩の必要に応じて置換されたアルキルエステルとの縮合;および
(ii)工程(i)の生成物において得られたエステル官能基の、1つの実施形態においては水性アルカリ溶液を使用する加水分解、
を包含する2つの工程を含む。
【0084】
工程(i)において使用される置換ピラジン2−カルボン酸の量は、L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり、約1.0モル当量〜約1.8モル当量の範囲であり得る。L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり1.2モルもまた、使用され得る。
【0085】
工程(i)において使用される縮合剤は、ジシクロヘキシルカルボジイミド/N−ヒドロキシスクシンイミドおよびエチル−3−(3−ジメチルアミノプロピル)カルボジイミドまたはその塩/N−ヒドロキシベンゾトリアゾールの組み合わせから選択され得る。
【0086】
個々に使用されるジシクロヘキシルカルボジイミド/N−ヒドロキシスクシンイミドおよびエチル−3−(3−ジメチルアミノプロピル)カルボジイミドまたはその塩/N−ヒドロキシベンゾトリアゾールの量は、L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり約1.0モル〜約1.8モルの範囲であり得る。1.2モルが使用され得る。N−ヒドロキシスクシンイミドおよびN−ヒドロキシベンゾトリアゾールは、しばしば、それぞれジシクロヘキシルカルボジイミドおよびエチル−3−(3−ジメチルアミノプロピル)カルボジイミドまたはその塩に対して、同じモル比で使用される。
【0087】
この縮合反応において使用される塩基としては、ジイソプロピルエチルアミン、ピリジン、ジメチルアミノピリジン、ジアザビシクロウンデカン、N−メチルモルホリンが挙げられ得るが、これらに限定されない。使用される塩基の量は、L−フェニルアラニンまたはその塩のアルキルエステル1モルあたり約1.0モル〜約2.0モルの範囲であり得る。好ましくは、1.5モルの塩基もまた使用され得る。
【0088】
工程(i)の縮合反応は、DMF、DMA、またはケトン溶媒(アセトン、メチルイソブチルケトン、エチルメチルケトンなどから選択され得る)などの溶媒中で実施され得る。
【0089】
この反応が実施され得る温度は、約−20℃〜約60℃の範囲であり得る。この反応は、約0℃〜約30℃の温度で実施され得る。
【0090】
この反応は、適切な時間にわたって実施され得る。所望であれば、工程(i)から得られた生成物(中間体エステル)は、加水分解前に、一般的な後処理手順によってか、または本願に開示されるようなプロセスによって、単離され得る。
【0091】
工程(i)において得られる生成物のエステル官能基の加水分解は、好ましくは、水性アルカリ溶液を使用して実施され得る。必要に応じて、有機溶媒もまた、この加水分解工程のための共溶媒として存在し得る。
【0092】
この加水分解のために使用される適切な塩基としては、水酸化ナトリウム、水酸化カリウムなどが挙げられ得るが、これらに限定されない。
【0093】
エステル加水分解のために使用される塩基の量は、当業者により決定され得る。例えば、工程(i)の生成物の中間体エステルが加水分解前に単離される場合、加水分解のために使用される塩基の量は、工程(i)の生成物の単離されたエステル1モルあたり約1.0モル〜約2.0モルの範囲であり得る。1.1モルの塩基もまた使用され得る。
【0094】
工程(ii)の加水分解反応のための共溶媒として使用される有機溶媒は、アセトン、メチルイソブチルケトン、エチルメチルケトン、メタノール、エタノール、イソプロパノールの溶媒から選択され得る。
【0095】
この反応が実施され得る温度は、約0℃〜約60℃の範囲であり得る。この反応はまた、約25℃〜約35℃の温度で実施され得る。この反応は、適切な時間にわたって実施され得、そして得られる生成物(式VIIIの化合物)は、一般的な後処理手順によってか、または本願に開示されるようなプロセスによって、単離され得る。
【0096】
別の局面において、本発明は、HPLCにより95%以上の純度を有する、式VIII
【0097】
【化19】
の化合物N−(2−ピラジンカルボニル)−L−フェニルアラニンを提供する。この化合物は、ボルテゾミブの調製における中間体である。
【0098】
式VIIIの化合物は、ボルテゾミブの調製のための主要な出発物質であるので、ボルテゾミブの収率および純度に影響を与えない、化学的HPLC純度とキラルHPLC純度の両方を有する式VIIIの化合物を有することが望ましい。本願のプロセスにより得られる式VIIIの化合物は、約95%より高い、好ましくは、約99%より高い、より好ましくは、約99.5%より高い、そして最も好ましくは、約99.8%より高い、化学的HPLC純度とキラルHPLC純度との両方を有する。
【0099】
式VIIIの化合物(すなわち、N−(2−ピラジンカルボニル)−L−フェニルアラニンまたはその塩)と、遊離塩基の形態または酸付加塩形態の式Vの化合物との、任意の公知の方法による、式Xの化合物を生成するための縮合、および式IXの化合物のボルテゾミブへの引き続く変換は、スキーム4において本明細書中以下に表されるように、本願の別の局面である。
【0100】
【化20】
式VIIIの化合物または塩と、遊離塩基の形態または酸付加塩形態の式Vの化合物との縮合反応についての具体的な詳細は、実施例1の工程(h)に提供されている。
【0101】
さらなる局面において、本願は、ボルテゾミブの調製のためのプロセスを提供し、このプロセスは、
a)(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【0102】
【化21】
を、有機ボロン酸アクセプターおよび水性鉱酸と、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させる工程;ならびに
b)水層を分離する工程;
c)この水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出する工程;ならびに必要に応じて、
d)ボルテゾミブを単離する工程、
を包含する。
【0103】
本願による、式IXの化合物からのボルテゾミブの調製のプロセスについての全ての工程が、以下に独立して記載される。
【0104】
工程a)
工程a)は、式IX
【0105】
【化22】
の化合物を、有機ボロン酸アクセプターおよび水性鉱酸と、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させて、式Iの化合物を得ることを包含する。
【0106】
工程a)において使用され得る有機ボロン酸アクセプターとしては、ブチルボロン酸、イソブチルボロン酸、フェニルボロン酸、ベンジルボロン酸などが挙げられるが、これらに限定されない。1つの実施形態において、イソブチルボロン酸が、ボロン酸アクセプターとして使用される。
【0107】
工程a)において使用される有機ボロン酸アクセプターの量は、式IXの化合物1モルあたり約1モル当量〜約1.5モル当量の範囲であり得る。式IXの化合物1モルあたり1.2モルが使用され得る。
【0108】
この反応において使用される鉱酸は、塩酸、硫酸、リン酸から選択され得る。1つの実施形態において、塩酸が使用される。使用される水性鉱酸の濃度は、約0.5N〜約3Nの範囲であり得る。1Nの濃度の水性鉱酸もまた使用され得る。この反応のために使用される水性鉱酸の量は、式IXの化合物1グラムあたり約5mlから25mlまで変動し得る。この反応において使用される水性鉱酸の濃度および量は、当業者により容易に決定され得る。
【0109】
工程a)のプロセスにおいて使用され得るアルコール溶媒としては、C1〜C4アルコール(例えば、メタノール、エタノール、イソプロパノール、ブタノール、またはこれらの混合物)が挙げられ得るが、これらに限定されない。工程a)のプロセスにおいて使用され得る脂肪族炭化水素溶媒としては、C5〜C10の、直鎖もしくは分枝鎖のアルカンまたはシクロアルカン(例えば、n−ペンタン、n−ヘキサン、n−ヘプタン、シクロヘキサンまたはこれらの混合物)が挙げられるが、これらに限定されない。いくつかの実施形態において、メタノールおよびn−ヘプタンを含む溶媒混合物が、反応溶媒として使用され得る。
【0110】
工程a)のプロセスは、約25℃から、およそ使用される溶媒の還流温度までの温度で実施され得る。実際に、この反応は、約25℃〜約35℃の温度で実施され得る。
【0111】
工程b)
工程b)は、水層の分離を包含する。
【0112】
反応の完了後、水層が反応混合物から分離され得、そしてその有機層が処分される。この水層は、必要に応じて、好ましくは、C5〜C8脂肪族炭化水素溶媒(例えば、n−ヘプタン)で洗浄され得る。この洗浄は、この水層を脂肪族炭化水素溶媒と一緒に、約10分間〜約15分間激しく攪拌し、そして有機層を分離することにより実施され得る。この有機層は、処分され得る。必要に応じて、このプロセスは、さらに1回〜3回繰り返され得る。
【0113】
必要に応じた洗浄工程後に得られた水層は、減圧ありまたはなしで、濃縮され得る。
【0114】
工程c)
工程c)は、水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出することを包含する。
【0115】
工程b)において得られた水層は、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出され得る。この抽出プロセスは、この溶媒をこの水層に添加し、そして約10分間〜約15分間激しく攪拌し、引き続いて有機層を分離することによって、実施され得る。
【0116】
抽出のために使用され得る水非混和性有機溶媒としては、アルコール溶媒(例えば、イソブタノール、およびt−ブタノール);ハロゲン化溶媒(例えば、ジクロロメタン、1,2−ジクロロエタンおよびクロロホルム);エステル溶媒(例えば、酢酸エチル、酢酸n−プロピル、酢酸イソプロピルおよび酢酸n−ブチル);またはこれらの混合物が挙げられるが、これらに限定されない。抽出のために選択される有機溶媒中への水の溶解度は、約10%w/w未満、好ましくは、約2%w/w未満であるべきである。1つの実施形態において、ハロゲン化アルカンが、抽出溶媒として使用される。なお別の実施形態において、ジクロロメタンが、抽出溶媒として使用される。
【0117】
この抽出プロセスは、ボルテゾミブが有機溶媒中に完全に抽出されるまで繰り返され得る。異なる抽出において得られた有機層が合わせられ、そして必要に応じて、飽和重炭酸ナトリウム溶液、引き続いてブライン溶液で洗浄され、そして減圧下で完全にか、または最小体積まで濃縮されて、ボルテゾミブの残渣または濃縮溶液を得る。この濃縮溶液または残渣は、必要に応じて、25℃〜35℃の温度まで冷却され得る。
【0118】
工程d)
工程d)は、任意であり、生成物の単離を包含する。
【0119】
生成物の単離は、冷却、種結晶の添加、または濃縮溶液もしくは残渣への有機溶媒の添加、あるいはこれらの組み合わせなどの方法により、実施され得る。
【0120】
1つの実施形態において、固体は、工程c)の濃縮溶液または残渣に有機溶媒を添加するなどの方法により、単離され得る。
【0121】
単離のために使用され得る有機溶媒としては、炭化水素溶媒(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);ハロ炭化水素溶媒(例えば、ジクロロメタン、ジクロロエタン);エステル溶媒(例えば、酢酸エチル、酢酸プロピル)、またはこれらの混合物が挙げられ得るが、これらに限定されない。1つの実施形態において、トルエンが、生成物を単離するために使用され得る。トルエンと、ハロ炭化水素溶媒またはエステル溶媒のいずれかとの混合物もまた、使用され得る。しかし、溶媒の混合物が単離のために使用される場合、この混合物中の個々の溶媒の比は、約2%v/v〜約98%v/vの範囲であり得る。
【0122】
溶媒は、工程c)の後に得られたボルテゾミブの濃縮溶液または残渣に、沈殿を起こすために充分な時間(例えば、約15分間〜約2時間、またはより長時間)にわたって添加され得る。適切な温度は、約0℃〜約50℃の範囲であり得る。次いで、得られた反応混合物は、完全な沈殿を起こすために、約30分間〜約5時間、またはより長時間にわたって攪拌され得る。この反応混合物は、25℃〜35℃で、約2時間〜約3時間攪拌され得る。
【0123】
得られた沈殿物は、当該分野において公知である技術により分離され得る。当業者は、固体を不均質混合物から分離するための多くの方法が存在することを理解し得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術を使用することにより、分離され得る。分離後、その固体は、必要に応じて、適切な溶媒で洗浄され得る。
【0124】
このように得られた固体は、乾燥させられ得る。乾燥は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、好ましくは、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、所望の純度を有する生成物を得るために必要な任意の時間(例えば、約1時間〜約25時間、またはより長時間)にわたって実施され得る。
【0125】
別の局面において、本願は、ボルテゾミブの精製のためのプロセスを提供する。このプロセスは、
a)有機溶媒中のボルテゾミブの溶液を提供する工程;
b)反溶媒を添加することにより、生成物を沈殿させる工程;
c)得られた生成物を分離する工程、
を包含する。ボルテゾミブの精製のためのプロセス工程が、本明細書中以下で別々に記載される:
工程a)
工程a)は、有機溶媒中のボルテゾミブの溶液を提供する工程を包含する。
【0126】
有機溶媒中のボルテゾミブの溶液を提供する工程は、必要に応じて窒素雰囲気下での、ボルテゾミブが調製された化学反応の溶液、または有機溶媒へのボルテゾミブの溶解を包含する。約90%以上の純度を有する任意の形態のボルテゾミブが、この溶液を提供するために認容可能である。任意の形態のボルテゾミブ(例えば、非晶質または結晶形態、または任意の方法により得られる任意の割合での非晶質と結晶形態との混合物のボルテゾミブ)が、溶液を提供するために使用され得る。
【0127】
溶解のために使用され得る有機溶媒としては、アルコール溶媒(例えば、メタノール、エタノール、イソプロピルアルコール、n−ブタノール、イソブタノール、およびt−ブタノール);ハロゲン化溶媒(例えば、ジクロロメタン、1,2−ジクロロエタン、クロロホルム);エステル溶媒(例えば、酢酸エチル、酢酸n−プロピル、酢酸イソプロピル、および酢酸n−ブチル);ニトリル溶媒(例えば、アセトニトリル、プロピオニトリル);またはこれらの混合物が挙げられるが、これらに限定されない。好ましくは、メタノール、イソプロピルアルコール、ジクロロメタンまたは酢酸エチルが、ボルテゾミブの精製のために使用され得る。
【0128】
ボルテゾミブの溶液は、約20℃から、使用される溶媒の沸点までの温度で提供され得る。好ましくは、この溶液は、約25℃〜約35℃の温度で提供される。
【0129】
溶解しない沈殿物は、濾過、遠心分離、デカンテーション、および他の技術によって、適切に除去され得る。使用される器具、溶液の濃度および温度に依存して、濾過装置は、尚早な結晶化を回避するために、適切に予熱される必要があり得る。
【0130】
工程b)
工程b)は、反溶媒を添加することによる生成物の沈殿を包含する。
【0131】
工程a)のボルテゾミブ溶媒は、沈殿のために、反溶媒と合わせられ得る。反溶媒の添加は、約5分間〜約1時間、またはより長時間にわたって行われ得る。反溶媒が添加され得る温度は、約0℃〜約45℃の範囲であり得る。使用される温度は、周囲温度(25℃まで)であり得る。
【0132】
得られる懸濁物は、約0℃〜約35℃の温度で維持される。得られた混合物は、完全な沈殿を起こすために、約30分間〜約5時間、またはより長時間攪拌される。1つの実施形態において、ボルテゾミブの懸濁物は、約25℃〜約35℃の温度で、2時間〜3時間維持される。
【0133】
本発明のプロセスにおいて使用され得る反溶媒としては、水、炭化水素(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);エーテル(例えば、ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン、ジメトキシエタン、メチル第三級ブチルエーテル);またはこれらの混合物が挙げられるが、これらに限定されない。特定の実施形態において、トルエンまたはジイソプロピルエーテルのうちのいずれかが、反溶媒として使用される。
【0134】
工程c)
工程c)は、生成物の分離を包含する。
【0135】
得られた沈殿物は、当該分野において公知である技術によって分離され得る。当業者は、固体を混合物から分離するための多くの方法が存在することを理解し得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術により分離され得る。分離後、この固体は、必要に応じて、適切な溶媒で洗浄され得る。
【0136】
湿った固体は、さらに乾燥させられ得る。乾燥は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、1つの実施形態においては、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、所望の純度を有する生成物を得るために必要な任意の時間(例えば、約1時間〜約40時間、またはより長時間)にわたって実施され得る。
【0137】
この精製プロセスは、所望の純度のボルテゾミブが達成されるまで必要に応じて繰り返され得る。例えば、精製は、本質的に純粋なボルテゾミブ、実質的に純粋なボルテゾミブ、または純粋なボルテゾミブが得られるまで、続けられ得る。
【0138】
別の1つの実施形態において、本願は、溶媒がイソプロピルアルコールであり、そして反溶媒がイソプロピルエーテルである、ボルテゾミブの精製のためのプロセスを提供する。
【0139】
なお別の実施形態において、本発明は、実質的に純粋なボルテゾミブの調製のための精製プロセスを提供し、このプロセスは、以下の工程a)〜c)を包含する:
a)アルコール、ハロゲン化溶媒、エステル、ニトリル、炭化水素、エーテルまたはこれらの混合物から選択される有機溶媒中の、ボルテゾミブの溶液を提供する工程;
b)必要であれば、工程a)において得られた溶液に反溶媒を添加する工程;
c)工程a)または工程b)から得られた固体生成物を単離する工程。
【0140】
実質的に純粋なボルテゾミブの調製のためのプロセス工程が、本明細書中以下に別々に記載される:
工程a)は、有機溶媒中のボルテゾミブの溶液を提供する工程を包含する。
【0141】
この溶解のために使用され得る有機溶媒としては、アルコール溶媒(例えば、メタノール、エタノール、イソプロピルアルコール、n−ブタノール、イソブタノール、およびt−ブタノール);ハロゲン化溶媒(例えば、ジクロロメタン、1,2−ジクロロエタン、クロロホルム);エステル溶媒(例えば、酢酸エチル、酢酸n−プロピル、酢酸イソプロピルおよび酢酸n−ブチル);ニトリル溶媒(例えば、アセトニトリル、プロピオニトリル);炭化水素(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);エーテル(例えば、ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン、ジメトキシエタン、メチル第三級ブチルエーテル);またはこれらの混合物が挙げられるが、これらに限定されない。
【0142】
ボルテゾミブの溶液は、ボルテゾミブの精製のためのプロセスの上記実施形態に記載されたようなプロセスにより提供され得る。
【0143】
工程b)は、必要であれば、工程a)において得られた溶液に反溶媒を添加する工程を包含する。
【0144】
工程a)のボルテゾミブ溶液は、必要であれば、沈殿のために反溶媒と合わせられ得る。
【0145】
本発明のプロセスにおいて使用され得る反溶媒としては、水、炭化水素(例えば、トルエン、キシレン、シクロヘキサン、n−ヘキサン、n−ヘプタン);エーテル(例えば、ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン(THF)、1,4−ジオキサン、ジメトキシエタン、メチル第三級ブチルエーテル);またはこれらの混合物が挙げられるが、これらに限定されない。
【0146】
反溶媒を添加して化合物を沈殿させるプロセスは、ボルテゾミブの精製のためのプロセスの上記実施形態に記載されるようなプロセスにより実施され得る。
【0147】
工程c) 工程a)または工程b)から得られた固体生成物を単離する工程。
【0148】
工程a)または工程b)から得られた沈殿物が、ボルテゾミブの精製のためのプロセスの上記実施形態に記載されるようなプロセスによって、分離され得、そして乾燥させられ得る。
【0149】
1つの実施形態において、本願は、ボルテゾミブが、トルエンと組み合わせたジクロロメタンまたは酢酸エチルから選択される有機溶媒の混合物から精製される、ボルテゾミブの精製のためのプロセスを提供する。
【0150】
この混合物中の個々の溶媒は、1%v/v〜99%v/vの範囲であり得る。1つの実施形態において、個々の溶媒は、約5%v/v〜約10%v/vの範囲であり得る。
【0151】
本願の1つの局面は、約95%より高い純度、別の実施形態においては、約99%より高い純度、なお別の実施形態においては、約99.5%より高い純度、そしてなお別の実施形態においては、約99.8%より高い純度(HPLCによる)を有し、そして立体異性体および/または不純物を実質的に含まない、実質的に純粋なボルテゾミブを提供する。
【0152】
1つの実施形態において、本願は、ボルテゾミブの結晶形態Aの調製のためのプロセスを提供し、このプロセスは、
i)アルコール中のボルテゾミブの溶液を提供する工程;
ii)水を添加して固体を沈殿させる工程;および必要に応じて
iii)得られた固体を分離する工程、
を包含する。
【0153】
アルコール中のボルテゾミブの溶液を提供する工程は、ボルテゾミブを、アルコール溶媒中に、必要に応じて窒素雰囲気下で溶解することを包含する。ボルテゾミブの任意の結晶形態もしくは非晶質形態、または結晶形態と非晶質形態との混合物が、溶液を提供するために認容可能である。
【0154】
この溶解のために使用され得るアルコール溶媒としては、メタノール、エタノール、イソプロピルアルコール、n−ブタノール、イソブタノール、およびt−ブタノールが挙げられるが、これらに限定されない。特定の実施形態において、メタノールが、ボルテゾミブの溶解のために使用される。溶解のために使用されるアルコールの量は、ボルテゾミブ1gあたり約2ml〜約10mlで変動し得る。
【0155】
ボルテゾミブの溶液は、約20℃から、使用されるアルコール溶媒の沸点までの温度の範囲の温度で提供され得る。特定の実施形態において、ボルテゾミブの溶液は、約20℃〜約35℃の温度で提供され得る。溶解しない粒子は、濾過、遠心分離、デカンテーション、および他の技術により、適切に除去され得る。
【0156】
工程i)のボルテゾミブ溶液は、沈殿のために水と合わせられる。この水は、約20℃〜約45℃、好ましくは、約25℃〜35℃の温度で、この溶液に添加され得る。この沈殿のために使用される水の量は、ボルテゾミブ1gあたり約2ml〜約10mlで変動し得る。ボルテゾミブ溶液への水の添加は、約5分間〜約1時間、またはより長時間にわたって実施され得る。
【0157】
沈殿のために使用される水の量は、アルコール中のボルテゾミブの濃度、および添加の温度に依存し、そして当業者により容易に決定され得る。
【0158】
この懸濁物は、約0℃〜約35℃の温度で維持される。1つの実施形態において、この懸濁物は、約30℃〜約35℃で維持される。得られた混合物は、完全な沈殿を起こすために、約30分間〜約5時間、またはより長時間にわたって、攪拌され得る。沈殿または完全な沈殿のための他の方法もまた、使用され得る。1つの実施形態において、ボルテゾミブの懸濁物は、約25℃〜約35℃の温度で、2時間〜3時間維持される。
【0159】
得られた沈殿物は、当該分野において公知である技術により分離され得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術を使用することにより、分離され得る。分離後、この固体は、必要に応じて、洗浄され得る。得られた湿った固体は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、1つの実施形態においては、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、ボルテゾミブ形態Aを得るための任意の時間(例えば、約1時間〜約25時間、またはより長時間)にわたって実施され得る。
【0160】
本願のプロセスにより得られた形態Aは、三量体無水物ではなく単量体であることが、驚くべきことに見出された。
【0161】
本発明のプロセスにより得られたボルテゾミブ形態Aのアセトニトリル溶液の質量分析(陽イオン、エレクトロスプレー)は、陰イオンモードで、m/z=383.19においてピークを示した。このことは、この生成物が、三量体ボロキシン(無水物形態)ではなく単量体ボロン酸であることを示す。
【0162】
同じ結晶性生成物についての陽イオンモードは、m/z=367.4を示した。なぜなら、ボルテゾミブのプロトン化分子イオン(M+)+は、不安定であり、そしてインソース脱水(18Da)を起こすからである。さらに、それぞれm/z=1121、1099、および1137における、三量体ボロキシンのナトリウム付加体、プロトン付加体、およびカリウム付加体は観察されず、この化合物の単量体の性質を確認した。
【0163】
1つの実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、実質的に図1に図示されるようなX線回折パターンにより特徴付けられる。
【0164】
1つの実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、約5.82、9.47、9.93、12.80、18.31、20.50、20.90、21.60、22.20、および23.70±0.2°2θの回折角2θに特性ピークを有するX線回折パターンにより特徴付けられる。図2に図示されるこのパターンは、Xcelerator***検出器を有するBragg−Brentanoθ:θゴニオメーターを備えるPANalytical器具を使用して生成された。このパターンを、40kVのチューブ電圧および40mAのチューブ電流で、0.02°のステップサイズおよび1ステップあたりの時間10秒で、3°〜45°の2θの角度範囲にわたって記録した。サンプルを、CuK放射線(=1.5418Å)に曝露した。同じ設備および設定を使用して、図5および図8のパターンを生成した。あるいは、形態Aは、5.82、9.93、11.53、12.80、13.11、15.27、15.47、16.90、17.32、18.31、18.96、19.27、19.85、20.50、21.60、22.24、23.74、24.29、24.65、25.76、26.32、28.03、29.96±0.2°の回折角2θのピークにより特徴付けられ得る。これらのピークを反映するパターンは、図1に見られる。この図は、シンチレーションカウンター検出器を有するRINT2000広角ゴニオメーターを備えるRigaku Dmax 2200器具を使用して生成された。このパターンを、50kVのチューブ電圧および34mAのチューブ電流で、0.02°のステップサイズおよび1ステップあたりの時間3°/分で、3°〜45°の2θの角度範囲にわたって記録した。サンプルを、CuK放射線(=1.5418Å)に曝露した。
【0165】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、実質的に図2に図示されるような、約75.20℃および179.73℃に吸熱ピークを有する、示差走査熱量分析(DSC)サーモグラムにより特徴付けられる。
【0166】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、実質的に図3に図示されるような、約2.88%の重量損失に対応するTGA曲線により特徴付けられる。
【0167】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Aは、KFにより約5%までの含水量により特徴付けられる。
【0168】
1つの実施形態において、本願は、ボルテゾミブの結晶形態Bの調製のためのプロセスを提供し、このプロセスは、
a)ハロゲン化アルカン溶媒またはエステル溶媒中の、ボルテゾミブの溶液を提供する工程;
b)芳香族炭化水素溶媒を添加して固体を沈殿させる工程;および必要に応じて
c)得られた固体を単離する工程、
を包含する。
【0169】
ボルテゾミブの溶液を提供する工程は、好ましくは、ハロゲン化アルカン溶媒またはエステル溶媒のいずれかへの、必要に応じて窒素雰囲気下での化合物の溶解を包含する。ボルテゾミブの任意の結晶形態もしくは非晶質形態、または結晶形態と非晶質形態との混合物が、溶液を提供するために認容可能である。
【0170】
この溶解のために使用される溶媒の量は、ボルテゾミブ1gあたり約2ml〜約10mlで変動し得る。
【0171】
この溶解のために使用され得るハロゲン化アルカン溶媒としては、ジクロロメタン、1,2−ジクロロエタンおよびクロロホルムが挙げられるが、これらに限定されない。この溶解のために使用され得るエステル溶媒としては、酢酸エチル、酢酸イソプロピル、酢酸第三級ブチルが挙げられるが、これらに限定されない。沈殿のために使用され得る芳香族炭化水素溶媒としては、トルエン、キシレンが挙げられるが、これらに限定されない。
【0172】
ボルテゾミブの溶液は、約20℃から、使用される溶媒の沸点までの温度で提供され得る。1つの実施形態において、ボルテゾミブの溶液は、約25℃〜約35℃の温度で提供される。溶解しない沈殿物は、濾過、遠心分離、デカンテーション、および他の技術によって、適切に除去され得る。
【0173】
沈殿は、芳香族炭化水素溶媒をボルテゾミブの溶液に添加することにより実施され得る。添加がなされ得る温度は、約20℃〜約35℃の範囲である。
【0174】
沈殿のために使用される芳香族炭化水素の量は、ハロゲン化アルカン溶媒またはエステル溶媒中のボルテゾミブの濃度、および添加の温度に依存し、そして当業者により容易に決定され得る。
【0175】
懸濁物は、約0℃〜約35℃の温度で、約30分間〜約5時間、またはより長時間にわたり維持され得る。1つの実施形態において、ボルテゾミブの懸濁物は、完全な沈殿を起こすために、約25℃〜約35℃の温度で、2時間〜3時間維持される。
【0176】
得られた沈殿物は、当該分野において公知である技術により分離され得る。例えば、重力または吸引による濾過、遠心分離、デカンテーションなどの任意の技術を使用することにより、分離され得る。分離後、得られた湿った固体は、棚型乾燥機、真空オーブン、エアオーブン、流動床乾燥機、スピンフラッシュ乾燥機、フラッシュ乾燥機などで、適切に実施され得る。この乾燥は、約35℃〜約70℃、好ましくは、約50℃の温度で、必要に応じて減圧下で実施され得る。この乾燥は、ボルテゾミブ形態Bを得るための任意の時間(例えば、約1時間〜約25時間、またはより長時間)にわたって実施され得る。他の技術もまた同様に使用され得る。
【0177】
1つの実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、約4.76、6.30、8.69、9.56、10.72、11.91、12.45、14.64、16.17、17.81、19.21、20.39、21.41、22.70、23.40、24.82、および31.78±0.2°の2θの回折角2θにおける特性ピークを有するX線回折パターンにより特徴付けられる。あるいは、形態Bは、4.76、6.30、8.69、9.56、10.72、11.91、12.45、14.64、16.17、17.81、18.27、19.21、20.39、21.41、22.70、23.40、24.21、26.15、31.78±0.2°の回折角2θにおけるピークにより特徴付けられ得る。
【0178】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、実質的に図5に図示されるようなX線回折パターンにより特徴付けられる。
【0179】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、実質的に図5に図示される、約0.39%の重量損失に対応するTGA曲線により特徴付けられる。
【0180】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、実質的に図6のスペクトルにより図示されるような、臭化カリウム(KBr)ペレット中での赤外吸収スペクトルにより特徴付けられる。
【0181】
別の実施形態において、本願のプロセスにより得られたボルテゾミブの結晶形態Bは、KFにより約3%までの含水量により特徴付けられる。
【0182】
本願のプロセスにより得られたボルテゾミブ(形態Aおよび形態Bを含む)の残留溶媒は、International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use(日米EU医薬品規制調和国際会議:「ICH」)指針により与えられる限度内である。
【0183】
本願のプロセスにより得られたボルテゾミブは、所望の粒子サイズを得るために、必要に応じて粉砕され得る。粉砕または微細化は、生成物の乾燥前、乾燥中、または乾燥の完了後に実施され得る。粉砕操作は、粒子を互いに高速で衝突させることにより、粒子のサイズを減少させ、そして粒子の表面積を増加させる。
【0184】
別の実施形態において、本願は、本発明のプロセスにより得られた、薬学的に有効な量のボルテゾミブの結晶形態Aおよび/またはB、ならびに少なくとも1つの薬学的に受容可能な賦形剤を含有する、薬学的組成物を提供する。
【0185】
本願のプロセスに従って得られたボルテゾミブ結晶形態は、安定であるのみでなく、薬学的処方物を調製する際に使用するためにもまたよく適している。本願による薬学的処方物としては、固体経口投薬形態(例えば、錠剤、カプセル剤、散剤など);液体経口投薬形態(例えば、液剤、分散剤、懸濁剤、乳剤など);非経口投薬形態(筋肉内、皮下、静脈内が挙げられる)(例えば、溶液もしくは懸濁液もしくは分散液、または再形成用の滅菌粉末による、注射可能な投薬物);経皮送達系;標的化送達系などが挙げられるが、これらに限定されない。
【0186】
さらに、本願の発明者らは、密封ポリマーバッグ(例えば、乾燥剤を含まない透明または不透明なポリエチレンバッグ)またはこのようなバッグの組み合わせを備える保存システム内で、通常の温度条件下で保存されたボルテゾミブが、不安定であることを見出した。一般的な保存条件下で保存された場合のボルテゾミブの安定性研究を、表1に要約する。
【0187】
表1:25℃および60%RH(一般的な保存条件であり、本発明の局面であると本明細書中に記載される条件を使用しない)で保存された場合のボルテゾミブ
【0188】
【表1】
*酢酸エチルから
この表において、不純物aは、
【0189】
【化23】
であり、そして不純物bは、ボルテゾミブのRRジアステレオマーとSSジアステレオマーとの組み合わせである。
(S,S)−異性体:
【0190】
【化24】
(R,R)−異性体:
【0191】
【化25】
上記結果は、25℃および60% RHで、上記のような保存システム中で保存されたボルテゾミブが、製品のかなりの分解を示し、これがボルテゾミブの純度の低下を生じることを示す。
【0192】
本願の保存方法に従う、制御された環境中でのボルテゾミブの保存は、ボルテゾミブを安定化し、そして長い保存期間にわたってボルテゾミブの純度を維持することが見出された。いかなる特定の理論にも束縛されることを望まないが、ボルテゾミブに関連する明らかな不安定性の問題は、ボルテゾミブを、低下した湿度レベル、ならびに/あるいは低い大気中酸素レベルおよび/または低い光レベルを有する環境で保存することにより克服され得ると考えられる。
【0193】
さらなる局面において、本願は、ボルテゾミブを安定化するための保存システムを提供する。本願の保存システムは、好ましくは、少なくとも1つの密封ポリマーバッグ(例えば、約0.10mm〜約0.50mmの厚さを有する、透明または不透明なポリエチレンバッグ)、またはこのようなバッグの組み合わせを備え、これらのバッグは、所望であれば、積層アルミニウムバッグの内側にシールされ得る。酸素吸収剤および水吸収剤(または乾燥剤)が、このようなバッグのうちの1つ以上の間に含まれ得る。最後に、包装されたサンプルは、HDPE容器内に保存される。
【0194】
好ましい局面のうちの1つにおいて、本願は、ボルテゾミブを不活性雰囲気下で安定化するための保存システムを提供し、その包装システムは、
a.少なくとも1つの外側密封ポリマーバッグ;
b.ボルテゾミブを含む別のポリマーバッグ、または必要に応じて、このようなバッグの組み合わせであって、これらのバッグは、所望であれば、積層アルミニウムバッグの内側にシールされ得る、バッグ;
c.ポリマーバッグaとポリマーバッグbとの間に配置された、酸素吸収剤および必要に応じて、水吸収剤(または乾燥剤)、
を備える。
【0195】
本願の保存システムは、好ましくは、内部にボルテゾミブが収容された容器を備え、この容器は、好ましくは、外部環境に対して、より低い湿度、酸素レベルおよび光レベル、またはこれらの組み合わせを有する内部環境を提供し得る。本願の保存システムは、好ましくは、保存条件下(2℃〜8℃の温度、およびまた25℃〜35℃の温度、60% RH)で少なくとも約3ヶ月間、ボルテゾミブの純度を維持し得る。本願の保存システムは、より好ましくは、少なくとも約3ヶ月間、そして最も好ましくは、約6ヶ月間、初期純度または分解が全くない状態から約0.2%未満の最大分解で、ボルテゾミブの純度を維持し得る。本願のプロセスにより得られた、制御された環境においてボルテゾミブについて実施した安定性の実験を、表2に要約する。
【0196】
特に好ましい実施形態において、本願の保存システムは、ボルテゾミブ(例えば、実施例5により生成されたボルテゾミブ)を、少なくとも約1ヶ月間、少なくとも約2ヶ月間、または少なくとも約6ヶ月間でさえも、2℃〜8℃またはそれより高温において維持し得る(表2を参照のこと)。
【0197】
本願の保存システムにおいて使用され得る容器は、好ましくは、少なくとも1つの外部密封ポリマーバッグを備える。適切なポリマーバッグとしては、保存の目的に適した1つ以上の市販のバッグ(例えば、ポリエチレンバッグ(例えば、低密度ポリエチレンバッグおよび高密度ポリエチレンバッグ)、ポリプロピレンバッグ、ポリエステルバッグ、ナイロンバッグ、ポリ塩化ビニル(PVC)バッグなど)が挙げられ得る。この保存システムにおいて利用されるポリマーバッグは、約0.10mm〜約0.50mmの厚さを有し得る。
【0198】
特に好ましい実施形態において、この保存システムは、密封積層アルミニウムバッグ、この密封アルミニウムバッグに収容された高密度ポリエチレンバッグ、密封不透明ポリエチレンバッグに収容された密封透明低密度ポリエチレンバッグ、この透明ポリエチレンバッグに含まれたボルテゾミブ、ならびにこの透明ポリエチレンバッグと不透明ポリエチレンバッグとの間に配置された、酸素吸収剤および必要に応じて乾燥剤またはこれらの両方を備える。
【0199】
適切な酸素吸収剤としては、アスコルビン酸ベースの有機物型のもの、および鉄粉含有材料ベースの無機物型のものが挙げられるが、これらに限定されない。好ましくは、AgelessZ200またはAgelessZ100などが、ボルテゾミブの安定性を維持するための酸素吸収剤として利用され得る。
【0200】
適切な乾燥剤としては、酸化アルミニウム、塩化カルシウム、ドライエライト(CaSO4)、モレキュラーシーブ(例えば、活性化モレキュラーシーブ)、シリカゲルなど、およびこれらの組み合わせが挙げられるが、これらに限定されない。好ましくは、シリカゲルが、ボルテゾミブの安定性を維持するための乾燥剤として使用され得る。
【0201】
本願の薬物包装は、長い保存期間にわたって、ボルテゾミブの分解を防止し得る。好ましくは、この保存システムは、酸素および/または水との接触に起因する薬物の不安定性を低下または排除し得る。このシステムは、最低約3ヶ月間保存される場合に、ボルテゾミブの有意な分解の排除を生じ、より好ましくは、初期純度または分解が全くない状態から約0.2%までの最大分解を生じる。
【0202】
本願の保存システムのための例示的な包装はまた、ボルテゾミブが透明ポリエチレンバッグに包装されて密封され、この透明ポリエチレンバッグが不透明(例えば、黒色)ポリエチレンバッグに包装され、次いでこの不透明ポリエチレンバッグが密封され、そして次に、積層アルミニウムバッグに包装および密封され、次いでこの積層アルミニウムバッグが密封される、包装の型を含み得る。酸素吸収剤および乾燥剤が、2つのポリマーバッグ/層の間に配置(例えば、分散)され得る。
【0203】
本願によれば、本明細書中に記載されるような包装は、不活性雰囲気下(例えば、窒素雰囲気下)または周囲空気中で包装され得る。
【0204】
本願の特定の具体的な局面および実施形態が、以下の実施例を参照しながらより詳細に説明される。これらの実施例は、説明のみにより提供されるのであり、本願の範囲をいかなる方法でも限定すると解釈されるべきではない。
【実施例】
【0205】
(実施例1:N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IX)を調製するためのプロセス)
【0206】
【化26】
式IXの化合物を調製するためのプロセスは、工程a)〜工程h)の工程を含み、これらの工程が、以下に個々に示される。
【0207】
工程a)2−(2−メチルプロピル)−(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロール(式II)の調製:
【0208】
【化27】
イソブチルボロン酸(50.0g)の、n−ヘプタン(250ml)中の攪拌溶液に、25℃〜30℃で(+)−ピナンジオール(83.3g)を添加し、そして25℃〜30℃で1時間攪拌した。この反応塊にブライン溶液を添加し、そしてこの混合物を攪拌した。層を分離させ、そしてその有機層を、さらなる溶媒が留去されなくなるまで減圧下で濃縮して、表題化合物(式II)を得た。
【0209】
工程b)2−((1S)−1−クロロ−3−メチルブチル)−(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロール(式III)の調製:
【0210】
【化28】
I.塩化亜鉛とテトラヒドロフランとの混合物を調製する工程
II.LDA混合物を調製する工程
III.ジクロロメタンおよび水混和性エーテル溶媒を含有する溶媒混合物中の、式II
【0211】
【化29】
の化合物の溶液を調製する工程
IV.工程IIの溶液を工程IIIの溶液に添加し、続いてこの溶液を約−40℃〜−70℃の温度で維持する工程
V.工程Iの混合物を工程IVの生成物に添加し、続いてこの反応塊を約−40℃〜−70℃の温度で維持する工程
VI.その反応温度を約10℃〜周囲温度まで上昇させる工程
VII.水性酸溶液を添加する工程
VIII.式IIIの化合物を含有する有機層を分離し、そしてその生成物を単離する工程。
【0212】
I.塩化亜鉛とテトラヒドロフランとの混合物を調製する工程
ZnCl2(115g)を、第一の丸底フラスコ(R.B.フラスコ)内のテトラヒドロフラン(805ml)に、窒素雰囲気下25℃〜35℃で入れ、そして得られた混合物の温度を35℃〜40℃まで上昇させ、3時間〜4時間維持して、ZnCl2溶液を得た。
【0213】
II.LDA混合物を調製する工程
ジイソプロピルアミン(86ml)を、第二のR.B.フラスコ内のテトラヒドロフラン(345ml)に窒素雰囲気下で入れ、そして得られた混合物を−7℃〜−15℃まで冷却し、n−ヘキシルリチウムをこの混合物に入れ、そして30分間〜40分間維持して、LDA混合物を得た。
【0214】
III.式IIの化合物の溶液を調製する工程
式IIの化合物(115.0g)を、第三のR.B.フラスコ内のジクロロメタン(161ml)およびテトラヒドロフラン(690ml)に、窒素雰囲気下25℃〜35℃で入れ、そしてこの混合物を−55℃〜−60℃まで冷却した。
【0215】
IV.工程IIの溶液を工程IIIの溶液に添加する工程
LDA混合物を第二のR.B.フラスコから反応混合物に−55℃〜−60℃で入れ、そして30分間維持した。その温度を−50℃まで上昇させた。
【0216】
V.工程Iの混合物を工程IVの生成物に添加する工程
ZnCl2溶液を第一のR.B.フラスコから−45℃〜−50℃で入れ、そして1時間維持した。
【0217】
VI.その反応温度を約10℃まで上昇させる工程
その反応混合物を10℃まで温めた。
【0218】
VII.水性酸溶液を添加する工程
10% H2SO4を入れ、10分間〜15分間攪拌し、そして有機層を分離した。
【0219】
VIII.式IIIの化合物を含有する有機層を分離し、そしてその生成物を単離する工程
工程VIIにおいて分離した有機層を次の工程に供したが、水相は処分した。
【0220】
この有機層を、水層が約6〜7のpHに達するまで、攪拌しながらブライン溶液で洗浄した。
【0221】
この有機層を減圧下で濃縮して、式IIIの化合物を単離した。
【0222】
工程c)N,N−ビス(トリメチルシリル)−(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロール−2−イル]−3−メチルブチルアミン(式IV)の調製:
【0223】
【化30】
ヘキサメチルジシラザン(101.3ml)をテトラヒドロフラン(414ml)に窒素雰囲気下で入れ、そしてこの混合物を−20℃〜−30℃まで冷却した。n−ヘキシルリチウムをこの混合物に、攪拌しながら、−20℃〜−30℃の温度を維持する程度にゆっくりと入れた。この反応混合物を−20℃〜−25℃で1時間〜2時間攪拌し、式IIIの化合物(138g)を、この新たに調製したTHF中のリチウムHMDSに、−15℃〜−20℃の温度を維持するように入れた。この反応混合物を25℃〜30℃の温度まで温め、そして2時間〜3時間維持した。この反応混合物をシリカ床で濾過し、そしてこの床をジイソプロピルエーテルで洗浄した。その濾液を減圧下で濃縮して残渣にし、表題化合物(式IV)を得た。
【0224】
工程d)4,6−メタノ−1,3,2−ベンゾジオキサボロール−2−メタンアミン,ヘキサヒドロ−3a,5,5−トリメチル−α−(2−メチルプロピル)−,(αR,3aS,4S,6S,7aR)−,トリフルオロアセテート(式V)の調製:
【0225】
【化31】
トリフルオロ酢酸(129ml)をジイソプロピルエーテル(1980ml)に窒素雰囲気下25℃〜30℃で入れ、そしてこの反応塊を−10℃まで冷却した。式IVの化合物(198g)をこの反応塊にゆっくりと−10℃で入れ、そしてこの温度で8時間維持した。この反応塊を濾過し、ジイソプロピルエーテル(198ml)で洗浄し、そして得られた固体を25℃〜30℃で水(1500ml)でスラリー洗浄した。このスラリーを濾過し、水で洗浄し、そして得られた固体を減圧下40℃〜50℃で8時間乾燥させて、74.0gの表題化合物(式V)を得た。
純度(GCによる)=99.54%。
【0226】
工程e)L−フェニルアラニンメチルエステル塩酸塩(式VI)の調製:
【0227】
【化32】
L−フェニルアラニン(25g)のメタノール(125ml)中の攪拌溶液に、25℃〜30℃で、塩化チオニル(13.2ml)を攪拌しながら入れ、そしてこの混合物を55℃〜60℃で2時間〜3時間維持した。この反応塊を25℃〜30℃まで冷却し、そして減圧下で、出発物質に対する2倍体積まで濃縮した。イソプロピルアルコール(125ml)をこの反応塊に入れ、そして出発物質に対する2倍体積まで濃縮した。この反応塊を0℃〜5℃まで冷却し、そして攪拌しながら1時間〜2時間維持した。この反応塊を濾過し、イソプロピルアルコールで洗浄し、30分間吸引乾燥させ、そして得られた固体を45℃〜50℃で3時間〜4時間乾燥させて、28.8gの表題化合物(式VI)を得た。HPLCによるキラル純度:100%。
【0228】
工程f)L−フェニルアラニン,N−(ピラジニルカルボニル)−メチルエステル(式VII)の調製:
【0229】
【化33】
ピラジン−2−カルボン酸(3.45g)のDMF(50ml)中の攪拌混合物に、25℃〜30℃で、N−ヒドロキシスクシンイミド(3.2g)を攪拌しながら入れ、そして0℃〜5℃まで冷却した。N,N’−ジシクロヘキシルカルボジイミド(DCC)(5.75g)をこの反応塊に0℃〜5℃で入れ、そして15分間〜20分間攪拌した。式VIの化合物(5.0g)をこの反応塊に0℃〜5℃で入れ、そして15分間〜20分間攪拌した。さらに、NMM(3.8ml)をこの反応塊に0℃〜5℃で入れ、そして15分間〜20分間攪拌した。この反応混合物を25℃〜30℃まで温め、そして攪拌しながら2時間〜3時間維持した。この反応塊を濾過し、そしてその固体を分離した。得られた濾液を酢酸エチル(100ml)で希釈し、そして脱塩水で洗浄した。有機層を1N HClで洗浄し、続いて重炭酸ナトリウム溶液で洗浄した。その有機層を減圧下で、式VIに対する2倍体積まで濃縮し、そして25℃〜30℃まで冷却した。n−ヘプタン(20ml)を入れて化合物を沈殿させ、この反応塊を0℃〜5℃まで冷却し、1時間〜2時間維持し、そして減圧下で濾過した。得られた固体を40℃〜45℃で3時間〜4時間乾燥させて、5.7gの表題化合物(式VII)を得た。HPLCによる純度:99.73%、HPLCによるキラル純度:99.97%。
【0230】
工程g)N−(ピラジニルカルボニル)−L−フェニルアラニン(式VIII)の調製:
【0231】
【化34】
式VII(100g)のアセトン(500ml)中の攪拌混合物に、25℃〜30℃で、NaOH溶液(15.4gのNaOHを500mlの水に溶解することにより得た)を入れ、そしてこの温度で30分間〜50分間維持した。1N HClを使用することにより、この反応塊のpHを2に調整し、そしてこの反応塊を0℃〜5℃まで冷却した。この反応塊を、攪拌しながら0℃〜5℃で1時間〜2時間維持し、減圧下で濾過し、そして得られた材料を45℃〜50℃で4時間〜5時間乾燥させて、84.4gの表題化合物(式VIII)を得た。
HPLCによる純度:99.94重量%
HPLCによるキラル純度:100%。
【0232】
あるいは、N−(ピラジニルカルボニル)−L−フェニルアラニン(式VIII)はまた、以下により調製され得る:
(a)以下に記載されるようなプロセスに従ってエチルクロロホルメートを使用する:
アセトン(40ml)、ピラジンカルボン酸(5g)およびトリエチルアミン(6.77ml)の混合物を、約−5℃〜約0℃まで冷却し、そしてエチルクロロホルメート(4.76ml)を入れた。この反応塊を、約30分間攪拌した。この反応懸濁物を約25℃〜約30℃の温度に達せさせ、そして約3時間維持した。この反応懸濁物を約0℃〜約5℃まで冷却した。第二のフラスコ中で、水性水酸化ナトリウム(70mlの水中1.68g)溶液を約0℃〜約5℃まで冷却し、そしてこれに、アセトン(30ml)およびL−フェニルアラニン(6.6g)を添加し、そしてこの混合物をその温度で約1時間攪拌した。この第二のフラスコの反応塊を、第一のフラスコの反応塊に、約0℃〜約5℃の温度で添加し、次いで、約2時間攪拌し、続いて約25℃〜約30℃まで温度を上昇させた。この反応塊を、約25℃〜約30℃の温度で約16時間さらに攪拌した。酢酸エチル(150ml)をこの反応溶液に入れ、そして約30分間攪拌した。層を分離し、そして1N塩酸(35ml)を、その分離した水層に添加した。この反応溶液を約0℃〜約5℃まで冷却し、そして約2時間攪拌した。得られた懸濁物を濾過し、そしてその固体を水(10ml)で洗浄した。次いで、その固体を約50℃の温度で約4時間乾燥させて、2.6gの表題化合物を得た。
HPLCによる純度:99.2重量%
HPLCによるキラル純度:100%。
【0233】
(b)または、以下に記載されるようなプロセスに従って、EDC・HClとHOBtとの組み合わせを使用することによる:
ピラジンカルボン酸(168.7g)、ジメチルホルムアミド(1.4リットル)、ヒドロキシベンゾトリアゾール(HOBt:220g)、およびN−メチルモルホリン(221ml)の混合物を、約0℃〜約5℃の温度まで冷却した。EDC塩酸塩(1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド・HCl;278g)をこの反応溶液に約0℃の温度で添加し、そして約30分間攪拌した。上記から得られたL−フェニルアラニンメチルエステル塩酸塩(240g)をDMF(1リットル)に溶解し、次いで、この反応混合物に添加した。N−メチルモルホリン(110ml)をこの反応混合物に添加し、そしてこの反応混合物を、約0℃〜約5℃の温度で約1時間維持した。この反応混合物を、約25℃〜約35℃の温度まで温め、そして水(3.6リットル)で希釈した。この反応塊を酢酸エチル(3×2.4リットル)で抽出した。分離した酢酸エチル層を1N塩酸(1.2リットル)で洗浄し、次いで、2つの層を分離した。その有機層を、飽和重炭酸ナトリウム溶液(4.8リットル)およびブライン溶液(2.4リットル)で洗浄した。その有機層を、約45℃の温度で完全に濃縮して、260gのピラジン−2−カルボニルフェニルアラニンメチルエステルを得た。
【0234】
ピラジン−2−カルボニルフェニルアラニンメチルエステル(5g)をアセトン(25ml)に溶解し、そして約5分間攪拌した。水酸化ナトリウム溶液(25mlの水中701mgの水酸化ナトリウム)をこの反応溶液に添加し、そして約25℃の温度で約3時間攪拌し、次いで、そのpHを、1N塩酸(11ml)を用いて約2のpHに調整した。この反応混合物を約0℃〜約5℃の温度まで冷却し、そして約1時間攪拌した。この懸濁物を濾過し、そして吸引乾燥させて、4.0gのピラジン−2−カルボニルフェニルアラニンを得た。
HPLCによるキラル純度:100%
HPLCによる化学純度:99.88%。
【0235】
工程h)式IXの調製:
【0236】
【化35】
式VIIIの化合物(28.6g)のジクロロメタン(400ml)中の攪拌混合物に、窒素雰囲気下25℃〜30℃で、N−ヒドロキシスクシンイミド(13.3g)およびDCC(23.9g)を入れ、そして10分間〜20分間攪拌した。式Vの化合物(40g)をこの反応塊に入れ、そして15分間〜20分間攪拌した。ジイソプロピルエチルアミン(DIPEA)(27ml)を入れ、そしてこの反応塊を25℃〜30℃で2時間〜3時間維持した。この反応塊を濾過し、そしてこの固体をジクロロメタン(80ml)で洗浄した。得られた濾液を1N HClで洗浄し、続いて重炭酸ナトリウム溶液で洗浄した。有機層を、式Vに対する2倍体積まで濃縮した。メタノール(200ml)を入れ、そして式Vに対する2倍体積まで濃縮した。得られた濃縮塊は、表題化合物(式IX)である。
【0237】
あるいは、式IXの化合物はまた、以下に記載されるようなプロセスによって、EDC・HClおよびヒドロキシベンゾトリアゾールを使用することにより調製され得る:
N−(2−ピラジンカルボニル)−L−フェニルアラニン(500mg)をジクロロメタン(10ml)中に懸濁させ、そして約−5℃〜約0℃まで冷却した。ヒドロキシベンゾトリアゾール(HOBt:310mg)をこの反応塊に入れ、続いて、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC・HCl、385ml)を添加し、そして15分間攪拌した。(1R)−(S)−ピナンジオール1−アンモニウムトリフルオロアセテート−3−メチルブタン−1−ボロネート(695mg)をこの反応混合物に添加し、そして約5℃の温度で約10分間攪拌した。ジイソプロピルエチルアミン(0.6ml)をこの反応混合物に入れ、そして約5℃の温度で約30分間攪拌した。この反応混合物を約25℃〜約30℃の温度まで温め、そして約1時間攪拌し、続いて、1N塩酸(30ml)を添加した。層を分離し、そしてその有機層を、1N塩酸(15ml)および飽和重炭酸ナトリウム溶液(2×30ml)で洗浄した。その有機層を完全に濃縮して、表題化合物を得た。
HPLCによる純度:84.98%。HPLCにより測定された9.01%は、最後のボルテゾミブ工程の前に形成されたボルテゾミブであると考えられることに留意のこと。従って、HPLCにより測定される場合の全体の純度は、84.98%+9.0%、すなわち93.99%であるはずである。
【0238】
(実施例2:ボルテゾミブ(式I)を調製するためのプロセス)
式IXの化合物(13.6g)のメタノール(272ml)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(272ml)およびイソブチルボロン酸(3.2g)を入れた。2N HCl(272ml)をこの反応塊に激しく攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離して処分した。n−ヘプタン(272ml×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下35℃〜48℃で濃縮した。この水層をジクロロメタン(272ml)で激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(272ml×2)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、6mlの反応塊を得、そして25℃〜30℃まで冷却した。
純度:HPLCにより95.13%。
【0239】
トルエン(102ml)を上記反応塊に入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%ジクロロメタンで洗浄し、そして減圧下45℃〜50℃で5時間乾燥させて、粗製ボルテゾミブを得た。
収量:7.0g(70%)
HPLCによる純度:99.22%
HPLCによる不純物B:0.43%
多型の形態:形態B
XRDパターン:図5に図示されるとおり。
【0240】
(実施例3:メタノールおよび水を使用するボルテゾミブの精製のプロセス)
ボルテゾミブ(5.0g、純度99.22%)およびメタノール(15ml)を丸底フラスコに入れ、そして25℃〜30℃で攪拌した。得られた溶液に脱塩水(15ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(30ml;水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、3.4gの表題化合物を得た。
HPLCによる純度:99.57%
HPLCによる不純物B:0.30%。
【0241】
同じプロセスを再現することにより得られた生成物のさらなる精製は、99.6%の純度(HPLCによる)を有するボルテゾミブを生じた。
HPLCによる不純物B:0.23%
HPLCによるキラル純度:99.83%。
【0242】
(実施例4:ボルテゾミブを調製した後に精製するためのプロセス)
式IXの化合物(68.3g)のメタノール(1.22L)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(1.36L)およびイソブチルボロン酸(16.13g)を入れた。1N HCl(13.6L)をこの反応塊に攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離し、そして処分した。n−ヘプタン(1.36L×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下で濃縮した。その水層をジクロロメタン(13.6L)で、激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(13.6L×2)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、粗製ボルテゾミブ(47.0g)を得た。
HPLCによる純度:95.62%
HPLCによる不純物B:0.59%。
【0243】
精製1:ボルテゾミブ(25g、純度:95.62%)およびトルエン中5%酢酸エチル(250ml)を丸底フラスコに入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%酢酸エチルで洗浄し、そして減圧下50℃で5時間乾燥させて、ボルテゾミブを得た。
収量:18.0g(72%)
HPLCによる純度:99.68%
HPLCによる不純物B:0.27%。
【0244】
精製2:ボルテゾミブ(18.0g、純度99.68%)およびメタノール(54ml)を丸底フラスコに入れ、そして攪拌した。この反応塊を焼結式漏斗(sinted funnel)で濾過し、そしてその床を18mlのメタノールで洗浄した。得られた濾液に脱塩水(72ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(108ml:水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、14gの表題化合物を得た。
収量:14.0g(77%)
HPLCによる純度:99.83%
不純物B:0.15%(HPLCによる)
HPLCによるキラル純度:99.85%。
【0245】
(実施例5:ボルテゾミブを調製した後に精製するためのプロセス)
式IXの化合物(10.25g)のメタノール(174.5ml)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(205ml)およびイソブチルボロン酸(2.42g)を入れた。0.5N HCl(205ml)をこの反応塊に攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離し、そして処分した。n−ヘプタン(205ml×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下で濃縮した。その水層をジクロロメタン(205ml)で、激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(205ml)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、粗製ボルテゾミブ(5.8g)を得た。
HPLCによる純度:95.81%
HPLCによる不純物B:0.34%。
【0246】
精製1:ボルテゾミブ(5g、純度:95.81%)およびトルエン中5%ジクロロメタン(40ml)を丸底フラスコに入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%ジクロロメタンで洗浄し、そして減圧下50℃で5時間乾燥させて、ボルテゾミブを得た。
収量:4.2g(84%)
HPLCによる純度:99.12%
HPLCによる不純物B:0.31%。
【0247】
精製2:ボルテゾミブ(4.2g、純度99.12%)およびメタノール(12.6ml)を丸底フラスコに入れ、そして攪拌した。この反応塊に脱塩水(12.6ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(25.2ml:水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、2.95gの表題化合物を得た。
収量:2.95g(70%)
HPLCによる純度:99.70%
HPLCによる不純物B:0.2%
キラル純度:99.83%(HPLCによる)。
【0248】
(実施例6:酢酸エチルおよびトルエンを使用するボルテゾミブの精製のプロセス)
ボルテゾミブ(5.0g、純度96.0%)およびトルエン中5%酢酸エチル(40ml)を丸底フラスコに入れた。この反応混合物を約28℃の温度で3時間攪拌した。この反応混合物を濾過し、そしてその固体をトルエン中5%酢酸エチル(50ml)で洗浄した。得られた固体を50℃で5時間乾燥させて、3.5gの表題化合物を得た。
HPLCによる純度:99.28%
HPLCによる異性体不純物:0.55%
XRDパターン:図8に図示されるとおり。
【0249】
(実施例7:イソプロピルアルコールおよびジイソプロピルエーテルを使用するボルテゾミブの精製のプロセス)
ボルテゾミブ(1.0g、純度:93.46%)およびイソプロピルアルコール(6.0ml)を丸底フラスコに入れ、そして約27℃で溶解するまで攪拌した。得られた溶液にジイソプロピルエーテル(20ml)を添加し、そして約26℃の温度で3時間攪拌した。この反応混合物を濾過し、そしてその固体をジイソプロピルエーテル(5ml)で洗浄した。得られた固体を約15分間吸引乾燥させて、400mgの表題化合物を得た。
HPLCによる純度:99.49%
HPLCによる異性体不純物:0.18%。
【0250】
(実施例8:ボルテゾミブの精製のプロセス)
ボルテゾミブ(0.5g、異性体不純物1.89%;純度:97.47%)およびメタノール(1.5ml)を丸底フラスコに入れた。得られた溶液に水(1.5ml)を添加し、そして約25℃の温度で約2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(12ml:水+メタノール1:1)で洗浄した。最後に、得られた固体を約25℃の温度で約20分間吸引乾燥させて、400mgの表題化合物を得た。
HPLCによる純度:99.35重量%
HPLCによる異性体純度:0.65重量%。
旋光:
SOR(比旋光):5N HClの媒体中(濃度:1%)に基づいて、25℃において−45.00°
SOR:メタノールの媒体中(濃度:1%)に基づいて、25℃の温度において−43.82°
XRDパターン:図1に図示されるとおり。
【0251】
(実施例9:ボルテゾミブの形態Aの調製のためのプロセス)
ボルテゾミブ(3.0g、純度99.57%)およびメタノール(9ml)を丸底フラスコに入れ、そして25℃〜30℃で攪拌した。得られた溶液に脱塩水(9ml)を添加し、そして約27℃の温度で2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(18ml;水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、2.0gの表題化合物を得た。
XRDパターン:図2に図示されるとおり。
【0252】
(実施例10:本発明の例示的な保存システム中でのボルテゾミブの安定性研究)
ボルテゾミブの2つの異なるサンプルを、2℃〜8℃および25℃、60%相対湿度(RH)の保存条件で、保存システム中(約0.10mmの厚さを有する1つのポリマーバッグに包装し、その後、約0.10mmの厚さを有し外部乾燥剤を含む別のポリマーバッグに再度包装する)で保存した。15日後、1ヵ月後(30日目)、2ヵ月後(60日目)および3ヵ月後(90日目)に、各サンプルから少量を取り出し、そしてその純度をHPLCにより確認して、各サンプルのもとのHPLCクロマトグラム(保存期間の開始前)と比較した。各サンプルの化学純度を、HPLCクロマトグラムにより%面積として得、そして初期純度値(保存期間の開始前)と比較した。その結果を表2に要約する。
【0253】
表2:2℃〜8℃および25℃、60% RHで、本発明に記載されるような包装中で保存される場合のボルテゾミブの安定性
【0254】
【表2】
*メタノール/水から(形態A)
**酢酸エチルから。
【0255】
上記結果は、本発明により記載されるような包装条件を使用して、長い保存期間にわたって、ボルテゾミブが安定化され得、同時にその満足な純度を維持し得ることを示す。
【0256】
(実施例11:塩基性媒体中のボルテゾミブの水溶液の安定性研究)
ボルテゾミブの水溶液を、先行技術に記載されるような反応プロセスから得、そして塩基性pH約10.5に調整し(2N NaOHを使用)、そして25℃〜35℃で維持する。1時間後、2時間後および3時間後に、溶液から少量を取り出し、そしてその純度をHPLCにより確認し、そして初期のサンプルのもとのHPLCクロマトグラム(pHを調整した直後)と比較した。各サンプルの純度を、HPLCクロマトグラムにより%面積として得、そして初期純度値と比較した。その結果を表3に要約する。
【0257】
表3:塩基性媒体中のボルテゾミブの時間に伴う分解
【0258】
【表3】
表3において、不純物aは、
【0259】
【化36】
であり、そして不純物bは、ボルテゾミブのRRジアステレオマーとSSジアステレオマーとの組み合わせである。
【0260】
上記データから、ボルテゾミブの水溶液が塩基性媒体中で攪拌状態に維持された場合、時間と共に、化合物の純度は継続的に低下し、そして不純物の含有量は次第に増加したことが明らかである。
【0261】
(実施例12:ボルテゾミブを調製した後に精製するためのプロセス)
式IXの化合物(27.3g)のメタノール(491.4ml)中の攪拌混合物に、25℃〜30℃で、n−ヘプタン(546ml)およびイソブチルボロン酸(6.4g)を入れた。1.0N HCl(546ml)をこの反応塊に攪拌しながら入れ、そしてこの反応塊を25℃〜30℃で1時間〜2時間維持した。この反応の完了後、n−ヘプタン層を分離し、そして処分した。n−ヘプタン(546ml×2)をその水層に入れ、そして10分間〜15分間激しく攪拌した。n−ヘプタン層を分離し、そして得られた水層を減圧下で濃縮して、メタノールを除去した。その水層をジクロロメタン(546ml)で、激しく攪拌しながら抽出した。この抽出プロセスを繰り返し(546ml×2)、そして得られたジクロロメタン層をプールし、そして飽和重炭酸ナトリウム溶液、続いてブライン溶液で洗浄した。その有機層を分離し、減圧下で濃縮して、粗製ボルテゾミブ(19.0g)を得た。
HPLCによる純度:96.66%
HPLCによる異性体不純物:0.46%。
【0262】
精製1:ボルテゾミブ(17g、純度:96.66%)およびトルエン中5%酢酸エチル(136ml)を丸底フラスコに入れ、そして25℃〜30℃で2時間〜3時間攪拌した。得られた固体を減圧下で濾過し、トルエン中5%酢酸エチルで洗浄し、そして減圧下50℃で5時間乾燥させて、ボルテゾミブを得た。
収量:14.0g(82.3%)
HPLCによる純度:98.61%
HPLCによる異性体不純物:0.34%。
【0263】
精製2:ボルテゾミブ(14g、純度98.61%)およびメタノール(42ml)を丸底フラスコに入れ、そして攪拌した。この反応塊を半融ガラス濾過器(scinted funnel)で濾過し、そしてメタノール(14ml)で洗浄した。その濾液を丸底フラスコに入れ、そしてこの反応塊に脱塩水(56ml)を添加し、そして約27℃の温度で約2時間攪拌した。この反応懸濁物を濾過し、そしてその固体を水性メタノール(84ml:水:メタノール1:1)で洗浄した。得られた固体を約50℃の温度で約5時間乾燥させて、8.5gの表題化合物を得た。
収量:8.5g(60%)
HPLCによる純度:99.84%
HPLCによる異性体不純物:0.12%。
【0264】
(HPLC分析)
(方法A(化学純度について))
化学純度についてのボルテゾミブサンプルのHPLC測定を、Waters対称シールドRP−18(250×内径4.6mm、粒子サイズ5μm)および270nmで作動するUV検出器を備えるWatersシステムを使用して実施した。分析を、以下の移動相を使用して、1.0ml/分の流量で、55分間の実行時間で実施した。
移動相A:700mlの水、300mlのアセトニトリルおよび1mlのギ酸を混合し、濾過し、脱気する。
移動相B:800mlのアセトニトリル、200mlの水および1mlのギ酸を混合し、濾過し、脱気する。
溶出:勾配プログラム
【0265】
【表3A】
(方法B(キラル純度について))
キラル純度についてのボルテゾミブサンプルのHPLC測定を、5μmシリカゲルにコーティングしたアミローストリス(3,5ジメチルフェニルカルバメート)250×4.6(ChiralPak AD−H)および270nmで作動するUV検出器を備えるWatersシステムを使用して実施した。分析を、以下の移動相を使用して、1.0ml/分の流量で、25分間の実行時間で実施した。
移動相:n−ヘキサン、イソプロピルアルコールおよび無水アルコールを、8:1:1の比で混合する。
【0266】
上記方法で調製したボルテゾミブの立体異性体および/または不純物を、相対保持時間(「RRT」)により特徴付ける。RRTは、HPLCクロマトグラムにおいて、不純物が検出される保持時間を、ボルテゾミブの保持時間に関して考慮することにより計算され得る。この分析の結果を、表4および表5に示す。
【0267】
表4:方法A(HPLCによる化学純度)
【0268】
【表4】
N.D.:検出不可能
*は、式
【0269】
【化37】
の不純物aを表す。
**は、以下の式:
(S,S)−異性体:
【0270】
【化38】
(R,R)−異性体:
【0271】
【化39】
の構成化合物を含む、ボルテゾミブのジアステレオマー混合物である不純物bを表す。
【0272】
このボルテゾミブのジアステレオマー不純物は、他の異性体不純物、すなわち、
(R,S)−異性体:
【0273】
【化40】
と一緒にキラルHPLC法(方法B)により分離され得る。これらのRRTを、表5に要約する。
【0274】
表5:方法B(HPLCによるキラル純度)
【0275】
【表5】
【特許請求の範囲】
【請求項1】
以下の工程a)〜c):
a)アルコール、ハロゲン化溶媒、エステルおよび炭化水素、ニトリル、炭化水素、エーテルまたはこれらの混合物から選択される有機溶媒中の、ボルテゾミブの溶液を提供する工程;
b)必要であれば、工程a)において得られた溶液に反溶媒を添加する工程;
c)工程a)または工程b)から得られた固体生成物を単離する工程、
を包含する、実質的に純粋なボルテゾミブの調製のためのプロセス。
【請求項2】
工程b)における前記反溶媒が、水、炭化水素、エーテル、またはこれらの混合物から選択され、ただし、工程a)において使用される溶媒と同じではない、請求項1に記載のプロセス。
【請求項3】
a)(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【化41】
を、有機ボロン酸アクセプターおよび水性HClと、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させる工程;
b)水層を分離する工程;
c)該水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出する工程;ならびに
d)ボルテゾミブを単離する工程、
を包含する、ボルテゾミブの調製のためのプロセス。
【請求項4】
前記有機ボロン酸アクセプターが、ブチルボロン酸、イソブチルボロン酸、フェニルボロン酸、ベンジルボロン酸などから選択される、請求項3に記載の方法。
【請求項5】
工程a)において使用される前記有機ボロン酸の量が、式IXの化合物1モル当量あたり約1モル〜約1.5モル当量の範囲である、請求項3に記載の方法。
【請求項6】
水性HClの濃度が、約0.5N〜約3Nの範囲である、請求項3に記載の方法。
【請求項7】
前記アルコール溶媒が、メタノール、エタノール、イソプロパノールまたはこれらの混合物などのC1〜C4アルコールから選択される、請求項3に記載の方法。
【請求項8】
前記炭化水素溶媒が、n−ペンタン、n−ヘキサン、n−ヘプタン、シクロヘキサンまたはこれらの混合物などの、C4〜C10の直鎖もしくは分枝鎖アルカンまたはシクロアルカンから選択される、請求項3に記載の方法。
【請求項9】
工程c)において使用される有機溶媒が、約10%w/w未満の水溶解度を有し得る、請求項3に記載の方法。
【請求項10】
前記水非混和性有機溶媒が、アルコール(C4〜C7)、ハロゲン化溶媒、エステルまたはこれらの混合物から選択される、請求項3に記載の方法。
【請求項11】
工程(d)における前記単離が、冷却、種結晶の添加、または反応混合物への有機溶媒の添加、またはこれらの組み合わせなどの方法により実施される、請求項3に記載の方法。
【請求項12】
前記反応混合物が、反応溶液または反応残渣であり得る、請求項11に記載の方法。
【請求項13】
前記有機溶媒が、炭化水素、ハロ炭化水素、エステル、またはこれらの混合物である、請求項11に記載の方法。
【請求項14】
アルコール溶媒中にボルテゾミブを含む溶液から結晶性ボルテゾミブを沈殿させる工程を包含し、該結晶性ボルテゾミブが、実質的に図2に一致するX線粉末回折パターンを有する、ボルテゾミブの結晶形態Aの調製のためのプロセス。
【請求項15】
a)アルコール中のボルテゾミブの溶液を提供する工程;
b)水を添加して固体を沈殿させる工程;および
c)実質的に図2に一致するX線粉末回折パターンを有する結晶性ボルテゾミブを単離する工程、
を包含する、請求項14に記載のプロセス。
【請求項16】
前記アルコールがメタノールまたはエタノールである、請求項15に記載の方法。
【請求項17】
a)ハロゲン化アルカン溶媒またはエステル溶媒中のボルテゾミブの溶液を提供する工程;
b)芳香族炭化水素溶媒を添加して固体を沈殿させる工程;および
c)得られた固体を単離する工程、
を包含する、ボルテゾミブの結晶形態Bの調製のためのプロセス。
【請求項18】
前記ボルテゾミブの結晶形態Bが、実質的に図5に一致するX線粉末回折パターンを有する、請求項17に記載のプロセス。
【請求項19】
前記ハロゲン化アルカンが、ジクロロメタン、1,2−ジクロロエタンおよびクロロホルムであり、前記エステル溶媒が、酢酸エチル、酢酸イソプロピル、酢酸第三級ブチルまたはその混合物である、請求項17に記載のプロセス。
【請求項20】
工程b)における前記芳香族炭化水素溶媒が、トルエン、キシレン、またはこれらの混合物である、請求項17に記載のプロセス。
【請求項21】
式III
【化42】
の中間体の調製のためのプロセスであって、該プロセスは、
式X
【化43】
のボロネート錯体化合物を調製する工程であって、式II
【化44】
の化合物を、リチウムジイソプロピルアミド(LDA)と、ルイス酸触媒、水混和性エーテル溶媒および過剰なジクロロメタンの存在下で反応させることによる、工程、ならびに
引き続く式Xのボロ「ネート」錯体の転移、
を包含する、プロセス。
【請求項22】
前記過剰なジクロロメタンが、式IIの化合物1モルあたり約4モル〜約8モルで利用される、請求項21に記載のプロセス。
【請求項23】
前記水混和性エーテル溶媒が、環状エーテル溶媒である、請求項21に記載のプロセス。
【請求項24】
前記環状エーテル溶媒がTHFである、請求項22に記載のプロセス。
【請求項25】
I.LDA混合物を、ジクロロメタンと水混和性エーテル溶媒とを含む溶媒混合物中の式II
【化45】
の化合物の溶液に添加し、続いて得られた溶液を約−40℃〜約−70℃の温度で維持する工程;
II.塩化亜鉛のテトラヒドロフラン中の混合物を、工程Iの生成物に添加し、続いて該反応塊を約−40℃〜約−70℃の温度で維持する工程;
III.反応温度を約10℃〜周囲温度まで上昇させる工程;
IV.水性酸溶液を添加する工程;および
V.必要に応じて、前記式IIIの化合物を含む有機層を分離する工程;
VI.該式IIIの化合物を単離する工程;
を包含する、請求項21に記載のプロセス。
【請求項26】
前記LDA混合物が、ジイソプロピルアミンおよびn−ヘキシルリチウムを使用することにより調製される、請求項25に記載のプロセス。
【請求項27】
工程Vにおける前記有機層が、前記式IIIの化合物を単離するために必要に応じて濃縮される、請求項25に記載のプロセス。
【請求項28】
前記ジクロロメタンが、ボロ「ネート」錯体形成中の反応物質として使用され得、そしてまた、有機層分離を補助する、請求項21または25に記載のプロセス。
【請求項29】
前記塩化亜鉛の量が、好ましくは、前記式IIの化合物1モルあたり約1.2モル〜約2モルの範囲であり得る、請求項21または25に記載のプロセス。
【請求項30】
前記ルイス酸触媒が塩化亜鉛である、請求項21に記載のプロセス。
【請求項31】
前記酸が、有機酸または無機酸である、請求項25に記載のプロセス。
【請求項32】
ピラジンカルボン酸とL−フェニルアラニンとの、クロロギ酸アルキル/アリールの存在下での反応を包含する、式VIII
【化46】
の中間体の調製のためのプロセス。
【請求項33】
前記クロロギ酸アルキル/アリールが、クロロギ酸エチル、クロロギ酸ベンジル、およびクロロギ酸パラニトロフェニルである、請求項32に記載のプロセス。
【請求項34】
式
【化47】
の固相の化合物。
【請求項35】
質量分析の陰イオンモードにおいてm/z=383.19のピークにより特徴付けられる、請求項34に記載の化合物。
【請求項36】
質量分析の陽イオンモードにおいてm/z=367.4のピークにより特徴付けられる、請求項34に記載の化合物。
【請求項37】
質量分析において、三量体ボロキシンのナトリウム付加体、プロトン付加体、およびカリウム付加体に対応する、m/z=1121、1099、および1137のピークの非存在により特徴付けられる、請求項34に記載の化合物。
【請求項38】
ボルテゾミブ形態Aである、請求項34に記載の化合物。
【請求項39】
約5%w/wまでの含水量を有する、請求項34、35および36のいずれか1項に記載の化合物。
【請求項40】
最低約3ヶ月間保存される場合に、初期純度から約0.2%以下の最大分解を示す、ボルテゾミブ。
【請求項41】
a.少なくとも1つの外側密封ポリマーバッグ;
b.該少なくとも1つの外側密封ポリマーバッグ内にシールされた、ボルテゾミブを含む少なくとも1つの内側密封ポリマーバッグ;
c.該外側ポリマーバッグと該内側ポリマーバッグとの間に配置された酸素吸収剤、
を備える、ボルテゾミブのための保存システム。
【請求項42】
前記外側ポリマーバッグと前記内側ポリマーバッグとの間に配置された水吸収剤をさらに備える、請求項41に記載の保存システム。
【請求項43】
少なくとも1つの積層アルミニウムバッグをさらに備え、該積層アルミニウムバッグ内に、前記少なくとも1つの外側密封ポリマーバッグ自体がシールされている、請求項41に記載の保存システム。
【請求項44】
前記ポリマーバッグが、透明または不透明であり、そして約0.10mm〜約0.50mmの厚さを有する、請求項41に記載の保存システム。
【請求項45】
前記酸素吸収剤が、アスコルビン酸、鉄粉含有材料、AGELESS Z200およびAGELESS Z100からなる群より選択される、請求項41に記載の保存システム。
【請求項46】
前記水吸収剤が、酸化アルミニウム、塩化カルシウム、ドライエライト(CaSO4)、モレキュラーシーブ(例えば、活性化モレキュラーシーブ)、およびシリカゲルからなる群より選択される、請求項42に記載の保存システム。
【請求項47】
前記外側ポリマーバッグと前記内側ポリマーバッグとの間に配置された不活性雰囲気をさらに備える、請求項41に記載の包装システム。
【請求項1】
以下の工程a)〜c):
a)アルコール、ハロゲン化溶媒、エステルおよび炭化水素、ニトリル、炭化水素、エーテルまたはこれらの混合物から選択される有機溶媒中の、ボルテゾミブの溶液を提供する工程;
b)必要であれば、工程a)において得られた溶液に反溶媒を添加する工程;
c)工程a)または工程b)から得られた固体生成物を単離する工程、
を包含する、実質的に純粋なボルテゾミブの調製のためのプロセス。
【請求項2】
工程b)における前記反溶媒が、水、炭化水素、エーテル、またはこれらの混合物から選択され、ただし、工程a)において使用される溶媒と同じではない、請求項1に記載のプロセス。
【請求項3】
a)(N−[(1S)−2−[[(1R)−1−[(3aS,4S,6S,7aR)−ヘキサヒドロ−3a,5,5−トリメチル−4,6−メタノ−1,3,2−ベンゾジオキサボロル−2−イル]−3−メチルブチルアミノ]−2−オキソ−1−(フェニルメチル)エチル]ピラジンカルボキサミド(式IXの化合物)
【化41】
を、有機ボロン酸アクセプターおよび水性HClと、アルコール溶媒および脂肪族炭化水素溶媒の存在下で反応させる工程;
b)水層を分離する工程;
c)該水層を、脂肪族炭化水素溶媒以外の水非混和性有機溶媒で抽出する工程;ならびに
d)ボルテゾミブを単離する工程、
を包含する、ボルテゾミブの調製のためのプロセス。
【請求項4】
前記有機ボロン酸アクセプターが、ブチルボロン酸、イソブチルボロン酸、フェニルボロン酸、ベンジルボロン酸などから選択される、請求項3に記載の方法。
【請求項5】
工程a)において使用される前記有機ボロン酸の量が、式IXの化合物1モル当量あたり約1モル〜約1.5モル当量の範囲である、請求項3に記載の方法。
【請求項6】
水性HClの濃度が、約0.5N〜約3Nの範囲である、請求項3に記載の方法。
【請求項7】
前記アルコール溶媒が、メタノール、エタノール、イソプロパノールまたはこれらの混合物などのC1〜C4アルコールから選択される、請求項3に記載の方法。
【請求項8】
前記炭化水素溶媒が、n−ペンタン、n−ヘキサン、n−ヘプタン、シクロヘキサンまたはこれらの混合物などの、C4〜C10の直鎖もしくは分枝鎖アルカンまたはシクロアルカンから選択される、請求項3に記載の方法。
【請求項9】
工程c)において使用される有機溶媒が、約10%w/w未満の水溶解度を有し得る、請求項3に記載の方法。
【請求項10】
前記水非混和性有機溶媒が、アルコール(C4〜C7)、ハロゲン化溶媒、エステルまたはこれらの混合物から選択される、請求項3に記載の方法。
【請求項11】
工程(d)における前記単離が、冷却、種結晶の添加、または反応混合物への有機溶媒の添加、またはこれらの組み合わせなどの方法により実施される、請求項3に記載の方法。
【請求項12】
前記反応混合物が、反応溶液または反応残渣であり得る、請求項11に記載の方法。
【請求項13】
前記有機溶媒が、炭化水素、ハロ炭化水素、エステル、またはこれらの混合物である、請求項11に記載の方法。
【請求項14】
アルコール溶媒中にボルテゾミブを含む溶液から結晶性ボルテゾミブを沈殿させる工程を包含し、該結晶性ボルテゾミブが、実質的に図2に一致するX線粉末回折パターンを有する、ボルテゾミブの結晶形態Aの調製のためのプロセス。
【請求項15】
a)アルコール中のボルテゾミブの溶液を提供する工程;
b)水を添加して固体を沈殿させる工程;および
c)実質的に図2に一致するX線粉末回折パターンを有する結晶性ボルテゾミブを単離する工程、
を包含する、請求項14に記載のプロセス。
【請求項16】
前記アルコールがメタノールまたはエタノールである、請求項15に記載の方法。
【請求項17】
a)ハロゲン化アルカン溶媒またはエステル溶媒中のボルテゾミブの溶液を提供する工程;
b)芳香族炭化水素溶媒を添加して固体を沈殿させる工程;および
c)得られた固体を単離する工程、
を包含する、ボルテゾミブの結晶形態Bの調製のためのプロセス。
【請求項18】
前記ボルテゾミブの結晶形態Bが、実質的に図5に一致するX線粉末回折パターンを有する、請求項17に記載のプロセス。
【請求項19】
前記ハロゲン化アルカンが、ジクロロメタン、1,2−ジクロロエタンおよびクロロホルムであり、前記エステル溶媒が、酢酸エチル、酢酸イソプロピル、酢酸第三級ブチルまたはその混合物である、請求項17に記載のプロセス。
【請求項20】
工程b)における前記芳香族炭化水素溶媒が、トルエン、キシレン、またはこれらの混合物である、請求項17に記載のプロセス。
【請求項21】
式III
【化42】
の中間体の調製のためのプロセスであって、該プロセスは、
式X
【化43】
のボロネート錯体化合物を調製する工程であって、式II
【化44】
の化合物を、リチウムジイソプロピルアミド(LDA)と、ルイス酸触媒、水混和性エーテル溶媒および過剰なジクロロメタンの存在下で反応させることによる、工程、ならびに
引き続く式Xのボロ「ネート」錯体の転移、
を包含する、プロセス。
【請求項22】
前記過剰なジクロロメタンが、式IIの化合物1モルあたり約4モル〜約8モルで利用される、請求項21に記載のプロセス。
【請求項23】
前記水混和性エーテル溶媒が、環状エーテル溶媒である、請求項21に記載のプロセス。
【請求項24】
前記環状エーテル溶媒がTHFである、請求項22に記載のプロセス。
【請求項25】
I.LDA混合物を、ジクロロメタンと水混和性エーテル溶媒とを含む溶媒混合物中の式II
【化45】
の化合物の溶液に添加し、続いて得られた溶液を約−40℃〜約−70℃の温度で維持する工程;
II.塩化亜鉛のテトラヒドロフラン中の混合物を、工程Iの生成物に添加し、続いて該反応塊を約−40℃〜約−70℃の温度で維持する工程;
III.反応温度を約10℃〜周囲温度まで上昇させる工程;
IV.水性酸溶液を添加する工程;および
V.必要に応じて、前記式IIIの化合物を含む有機層を分離する工程;
VI.該式IIIの化合物を単離する工程;
を包含する、請求項21に記載のプロセス。
【請求項26】
前記LDA混合物が、ジイソプロピルアミンおよびn−ヘキシルリチウムを使用することにより調製される、請求項25に記載のプロセス。
【請求項27】
工程Vにおける前記有機層が、前記式IIIの化合物を単離するために必要に応じて濃縮される、請求項25に記載のプロセス。
【請求項28】
前記ジクロロメタンが、ボロ「ネート」錯体形成中の反応物質として使用され得、そしてまた、有機層分離を補助する、請求項21または25に記載のプロセス。
【請求項29】
前記塩化亜鉛の量が、好ましくは、前記式IIの化合物1モルあたり約1.2モル〜約2モルの範囲であり得る、請求項21または25に記載のプロセス。
【請求項30】
前記ルイス酸触媒が塩化亜鉛である、請求項21に記載のプロセス。
【請求項31】
前記酸が、有機酸または無機酸である、請求項25に記載のプロセス。
【請求項32】
ピラジンカルボン酸とL−フェニルアラニンとの、クロロギ酸アルキル/アリールの存在下での反応を包含する、式VIII
【化46】
の中間体の調製のためのプロセス。
【請求項33】
前記クロロギ酸アルキル/アリールが、クロロギ酸エチル、クロロギ酸ベンジル、およびクロロギ酸パラニトロフェニルである、請求項32に記載のプロセス。
【請求項34】
式
【化47】
の固相の化合物。
【請求項35】
質量分析の陰イオンモードにおいてm/z=383.19のピークにより特徴付けられる、請求項34に記載の化合物。
【請求項36】
質量分析の陽イオンモードにおいてm/z=367.4のピークにより特徴付けられる、請求項34に記載の化合物。
【請求項37】
質量分析において、三量体ボロキシンのナトリウム付加体、プロトン付加体、およびカリウム付加体に対応する、m/z=1121、1099、および1137のピークの非存在により特徴付けられる、請求項34に記載の化合物。
【請求項38】
ボルテゾミブ形態Aである、請求項34に記載の化合物。
【請求項39】
約5%w/wまでの含水量を有する、請求項34、35および36のいずれか1項に記載の化合物。
【請求項40】
最低約3ヶ月間保存される場合に、初期純度から約0.2%以下の最大分解を示す、ボルテゾミブ。
【請求項41】
a.少なくとも1つの外側密封ポリマーバッグ;
b.該少なくとも1つの外側密封ポリマーバッグ内にシールされた、ボルテゾミブを含む少なくとも1つの内側密封ポリマーバッグ;
c.該外側ポリマーバッグと該内側ポリマーバッグとの間に配置された酸素吸収剤、
を備える、ボルテゾミブのための保存システム。
【請求項42】
前記外側ポリマーバッグと前記内側ポリマーバッグとの間に配置された水吸収剤をさらに備える、請求項41に記載の保存システム。
【請求項43】
少なくとも1つの積層アルミニウムバッグをさらに備え、該積層アルミニウムバッグ内に、前記少なくとも1つの外側密封ポリマーバッグ自体がシールされている、請求項41に記載の保存システム。
【請求項44】
前記ポリマーバッグが、透明または不透明であり、そして約0.10mm〜約0.50mmの厚さを有する、請求項41に記載の保存システム。
【請求項45】
前記酸素吸収剤が、アスコルビン酸、鉄粉含有材料、AGELESS Z200およびAGELESS Z100からなる群より選択される、請求項41に記載の保存システム。
【請求項46】
前記水吸収剤が、酸化アルミニウム、塩化カルシウム、ドライエライト(CaSO4)、モレキュラーシーブ(例えば、活性化モレキュラーシーブ)、およびシリカゲルからなる群より選択される、請求項42に記載の保存システム。
【請求項47】
前記外側ポリマーバッグと前記内側ポリマーバッグとの間に配置された不活性雰囲気をさらに備える、請求項41に記載の包装システム。
【図1】

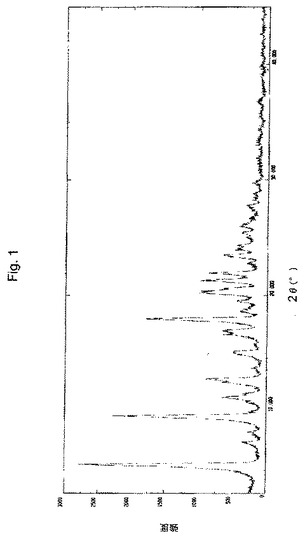
【図2】
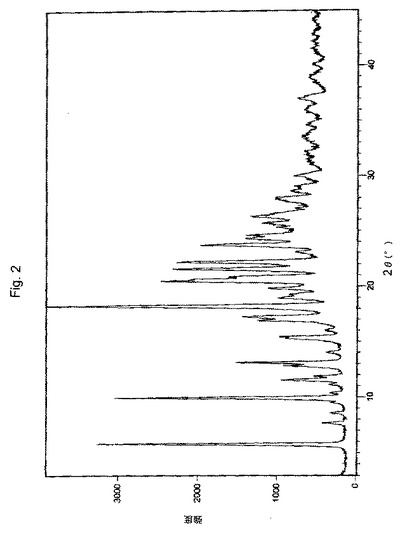
【図3】
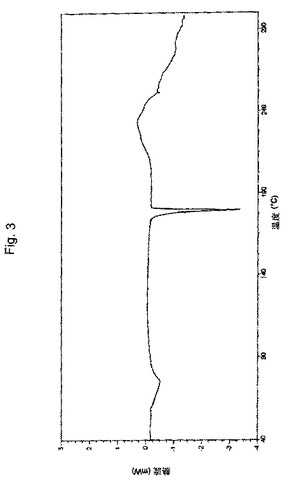
【図4】
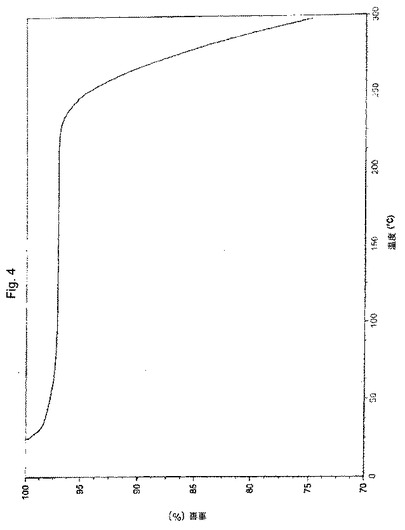
【図5】
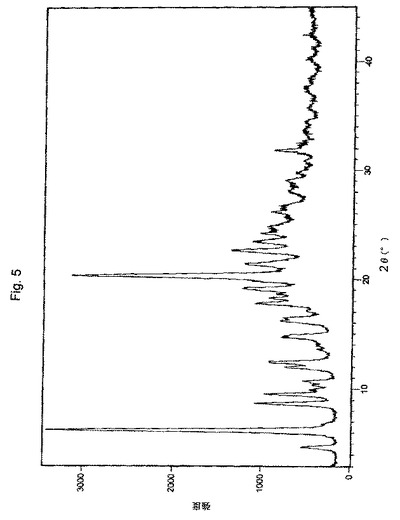
【図6】
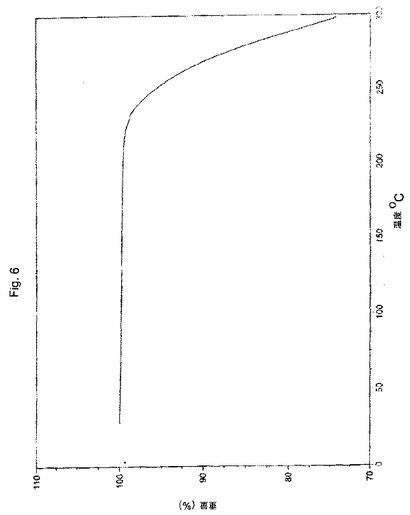
【図7】
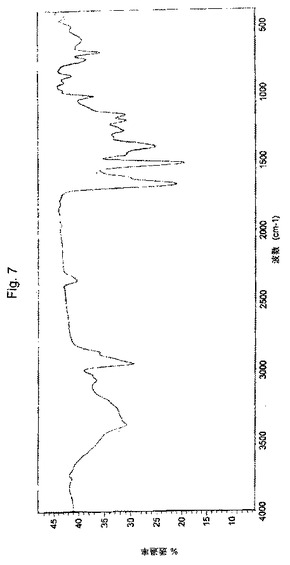
【図8】
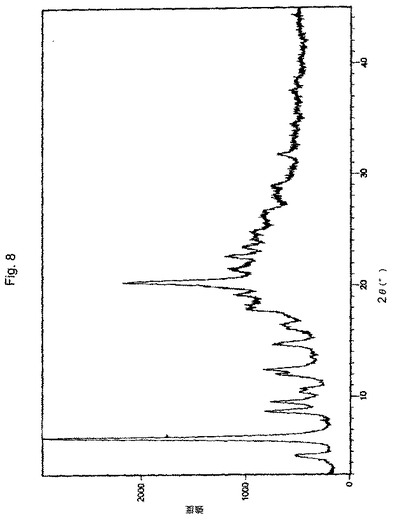
【図9】
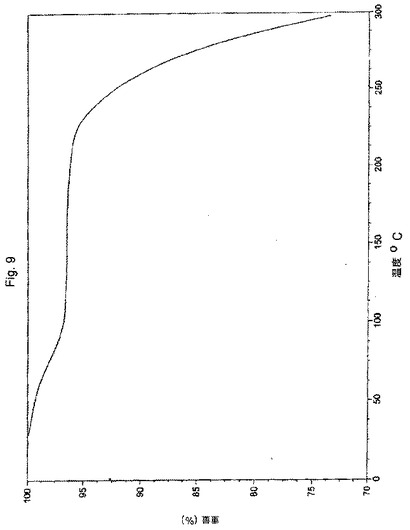
【図10】
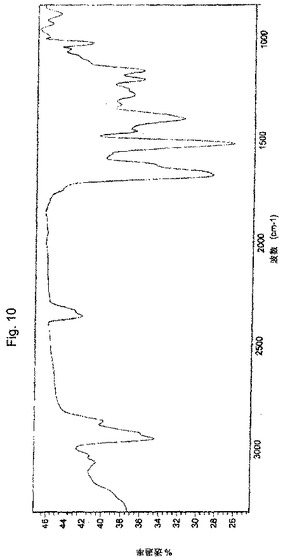
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2010−539183(P2010−539183A)
【公表日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願番号】特願2010−525028(P2010−525028)
【出願日】平成20年9月12日(2008.9.12)
【国際出願番号】PCT/US2008/076178
【国際公開番号】WO2009/036281
【国際公開日】平成21年3月19日(2009.3.19)
【出願人】(399131150)ドクター・レディーズ・ラボラトリーズ・リミテッド (19)
【出願人】(506017137)ドクター レディズ ラボラトリーズ, インコーポレイテッド (24)
【Fターム(参考)】
【公表日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願日】平成20年9月12日(2008.9.12)
【国際出願番号】PCT/US2008/076178
【国際公開番号】WO2009/036281
【国際公開日】平成21年3月19日(2009.3.19)
【出願人】(399131150)ドクター・レディーズ・ラボラトリーズ・リミテッド (19)
【出願人】(506017137)ドクター レディズ ラボラトリーズ, インコーポレイテッド (24)
【Fターム(参考)】
[ Back to top ]