
ポドフィロトキシン
【課題】患者への投与による副作用が十分に少ないポドフィロトキシンのプロドラッグを提供する。
【解決手段】ポドフィロトキシンのプロドラッグとして

等を結合した化合物。化学式(IV)において、R1およびR2は各々C1−C10アルキルであり、あるいはR1およびR2およびそれらが結合される炭素とがC5−C6シクロアルキルを示し、あるいはR1がHであり、R2はC1−10アルキル, C2−10アルケニル, C3−5シクロアルキル, フリル, チエニル, C6−10アリルおよびC7−14アラルキルを含む群から選択されること。
【解決手段】ポドフィロトキシンのプロドラッグとして
等を結合した化合物。化学式(IV)において、R1およびR2は各々C1−C10アルキルであり、あるいはR1およびR2およびそれらが結合される炭素とがC5−C6シクロアルキルを示し、あるいはR1がHであり、R2はC1−10アルキル, C2−10アルケニル, C3−5シクロアルキル, フリル, チエニル, C6−10アリルおよびC7−14アラルキルを含む群から選択されること。
【発明の詳細な説明】
【背景技術】
【0001】
ポドフィロトキシンおよびその代謝産物/前駆物質のようなリグナンは、フェニールプロパノイド代謝経路の一部であり、植物界に広く分布している。それらのいくつか、特にポドフィロトキシンそれ自体は、抗がん性、抗真菌性および/あるいは抗菌性を有することが知られている。ポドフィロトキシンは、メイアップル(Podophyllum peltatum)や、樹脂としてLinum album, Linum flavumおよびLinum nodiflorumのようなアマ属(Linum)から抽出され、19世紀後半のアメリカ南部で生殖器疣の治療のため医者によって使用された。生殖器疣はヒト乳頭腫ウイルス(HPV)を病因とする癌のいくつかと関連している。
【0002】
ポドフィロトキシンは、トポイソメラーゼIIの阻害剤であるエトポシド(etoposide)、テニポシド(teniposide)およびetopophosのような化学療法薬の親分子として特に関心が持たれている。現在、ポドフィロトキシンの需要は、野生植物の成長が遅いことや過剰採集によって不十分となっている世界的な供給量を遥かに超えている。ポドフィロトキシンの供給量の減少を補うため、Podophyllum peltatumやLinum albumの細胞を培養する試みが着手され、ある程度の成功を収めており、その半合成誘導体、エトポシドおよびテニポシドは、重要な抗がん剤として広く使用されている。しかし、それらには、水溶性が乏しいこと、代謝不活性、薬剤耐性の発生といったいくつかの問題がある。
【0003】
これらの問題を克服するため、ポドフィロトキシンの誘導体が多くの研究室で合成されている(Yin et al., acta pharm. Sinica 1993, 28,758-761, Wang et al., actachem. Sinica 1992, 50, 698-701, Chang et al., J. med. chem. 1994, 37446-442,Pelter et al., J. nat. prod. 1994, 57, 1598-1602)。しかしながら、これらのいずれにおいても副作用の抑制と効用の両方において大幅な改善には到っていない。
【0004】
エトポシドは、一般的な成人がんと同様に、急性リンパ腫、横紋筋肉腫、神経芽細胞腫のような小児がんを含む広域のがんに対して高い効果を示す抗がん剤として広く使用され、骨髄移植調整療法においても使用されている。しかしながら、治療へのエトポシドの使用は、骨髄抑制を主としたその毒性によって制限される。
【0005】
一般的ながんにおいてだけでなく、特に危険性の高い白血病において化学療法を成功させるための挑戦の一つとして、多剤耐性の克服が挙げられる。患者の多くは、初期において種々の化学療法用薬剤を組み合わせた治療に反応する。しかしながら、多剤化学療法は、細胞傷害性薬剤の存在下において増殖し続ける多剤耐性(MDR)細胞群を誘発するおそれがある(DaltonWS. Mechanism of drug resistance in hematologic malignancies Semin Hematol.1997;34:3-8)。
【0006】
そのような細部群における薬剤感受性の低下は、生体内における最大耐量を超える量の細胞傷害性薬剤の投与を必要とするだろう。白血病や悪性腫瘍における最も特徴的な耐性メカニズムの一つは、P糖蛋白、多剤耐性-1遺伝子(MDR-1)の産生物質によって仲介される薬剤排出(drug extrusion)であり、治療効果の低い結果と関連することが報告されている(Hunault M, Zhou D, Delmer A, et al. Multidrug resistance gene expression inacute myeloid leukemia: major prognosis significance for in vivo drugresistance to induction treatment. Ann Hematol. 1997;74:65-71; Marie JP, ZhouDC, Gurbuxani S, Legrand O, Zittoun R. MDR1/P-glycoprotein in haematologicalneoplasms. Eur J Cancer. 1996;32A:1034-1038.)。
【0007】
多剤化学療法の導入以来、薬剤耐性を回避するために種々の方法が開発されている。
【0008】
患者に生じた薬剤耐性を克服するための努力は、(i)スケジューリング、すなわち、再発白血病における葉酸代謝拮抗薬(antifolates)のような長期間少量投与療法、あるいは短期間多量投与、たとえば、骨肉腫におけるその後の葉酸回復剤(folate -rescue)の使用を伴う葉酸代謝拮抗薬、(ii)MDR仲介耐性の場合におけるMDR-1阻害剤のような化学増感剤の使用を伴う併用療法、(iii) 放射線療法、温熱療法、高圧酸素のような非化学増感剤の使用を伴う併用化学療法(Dalton WS. Mechanisms of drug resistance inhematologic malignancies. Semin Hematol. 1997;34:3-8; Joel SP, Slevin ML.Schedule-dependent topoisomerase II-inhibiting drugs. Cancer ChemotherPharmacol. 1994;34 Suppl:S84-S88; Ishikawa T, Kuo MT, Furuta K, Suzuki M. Thehuman multidrug resistance-associated protein (MRP) gene family: from biologicalfunction to drug molecular design. Clin Chem Lab Med. 2000;38:893-897.)に集中している。
【0009】
また、脱アミノドキソルビシン類似物質(Solary E, Ling YH, Perez-Soler R, Priebe W,Pommier Y. Hydroxyrubicin, a deaminated derivative of doxorubicin, inhibitsmammalianDNA topoisomerase II and partially circumvents multidrug resistance.Int J Cancer. 1994;58:85-94.)や、エトポシドのβアミノ誘導体(ZhangYL, Guo X, Cheng YC, Lee KH. Antitumor agents. 148. Synthesis and biologicalevaluation of novel 4 beta-amino derivatives of etoposide with betterpharmacological profiles. J Med Chem. 1994;37:446-452; Zhang YL, Shen YC, WangZQ, et al. Antitumor agents, 130, Novel 4 beta-arylamino derivatives of 3',4'- didemethoxy-3',4'-dioxo-4-deoxypodophyllotoxinas potent inhibitors of human DNA topoisomerase II. J Nat Prod.1992;55:1100-1111.)のような薬剤耐性機構を積極的に回避する類似物質を見出すために抗悪性腫瘍薬を直接修正する試みもわずかながらなされている。このように、MDRに関連する問題への有効な解決策は、MDRメカニズムによって影響されず、全身毒性の低減と抗がん作用の向上を発揮する薬剤の理論的な設計を含む。
【0010】
エトポシドに対する耐性は、標的酵素トポイソメラーゼIIのダウンレギュレーション、bcl-2のような抗アポトーシス機構のプロレグレーション(pro-regulation)またはアップレグレーションのいずれかのダウンレギュレーション、代謝の増大および/あるいは輸送系によって仲介された細胞からの薬剤の排出を伴いながら細胞レベルで起こる。また、そのような輸送系の誘発は、MDR-1, MRPあるいはLRP仲介多剤耐性において見られるように、しばしば他の抗悪性腫瘍薬に対する交差耐性を招く(Gottesman MM. How cancer cellsevade chemotherapy: sixteenth Richard and Hinda Rosenthal Foundation AwardLecture. Cancer Res. 1993;53:747-754; Borst P, Evers R, Kool M, Wijnholds J. Afamily of drug transporters: the multidrug resistance-associated proteins. JNatl Cancer Inst. 2000;92:1295-1302; Borst P, Evers R, Kool M, Wijnholds J. Themultidrug resistance protein family. Biochim Biophys Acta. 1999;1461:347-357.)。薬剤耐性の主たるメカニズムは、血液悪性腫瘍において発生することが報告されているように、MDR-1遺伝子産物、P糖蛋白の大量発現である。したがって、輸送系仲介多剤耐性を克服するための試みは、MDR-1発現の制御(Liu C, Qureshi IA, Ding X, et al. Modulation ofmultidrug resistance gene (MDR-1) with antisense oligodeoxynucleotides. ClinSci (Colch ). 1996;91:93-98.)や、サイクロスポリン(Sonneveld P, Durie BG, Lokhorst HM, et al. Modulation ofmultidrug-resistant multiple myeloma by cyclosporin. The Leukaemia Group of theEORTC and the HOVON. Lancet. 1992;340:255-259.)、ベラパミル(Joly P, Lallemand A,Oum'Hamed Z, Trentesaux C, Idoine O, Desplaces A. Effects of verapamil andS9788 on MDR-1 mRNA expression studied by in situ hybridization. AnticancerRes. 1996;16:3609-3614.)、あるいはvalspodar (TaiHL. Technology evaluation: Valspodar, Novartis AG. Curr Opin Mol Ther.2000;2:459-467.)のようなMDR-1阻害剤の同時投与に集中しており、これらはみな生体外および生体内においてある程度の有効性を示している。これらの膜輸送蛋白は、おそらく異なる薬剤よりも硫酸塩あるいはグルクロニド、グルタチオンのような一般の代謝産物が特異的に認識されるため、全く異なる構造を有する驚くべき広い範囲の基質を排出する(Zhu BT. A novel hypothesis for the mechanism ofaction of P-glycoprotein as a multidrug transporter. Mol Carcinog.1999;25:1-13.)。したがって、これらの耐性機構は、最初に認識され、次いでプロテアソームによって分解されるように多くの全く異なる蛋白質がユビキチンで”標識”されるユビキチンシステムと似ている。このように、分子”標識”を妨害する薬剤改良から生じた新しい分子は、多剤耐性がん細胞から取り除かれない可能性がある。
【0011】
エトポシドの標的組織へのより特異的なターゲティングを可能にするエトポシドの誘導体を合成するため、いくつかの試みが着手されている。EP0423747は、グリコシル−エトポシド−プロドラッグの合成を報告しており、このプロドラッグは腫瘍−特異酵素複合体の作用によって有効なエトポシドドラッグとグリコシル残留物とに開裂され、それにより、腫瘍−特異酵素複合のためドラッグはその好ましい作用部位でのみ活性化される(さらに米国特許第4975278号を参照)。また、Shabatら(PNAS, vol. 98, 13, 7528-7533)は、4’−フェノール性OH基がアルドールカーバメート(aldol carbamate)化合物によってマスクされた、エトポシドに基づく抗体−プロドラッグ系の合成について記載している。しかしながら、そのようなプロドラッグは、プロドラッグを活性化してエトポシドを産出させる触媒抗体38C2と組み合わされない限り、単独ではいかなる抗がん活性も示さなかった。Shabatに記載されているプロドラッグは、自然界では起こらないretro-Michael/retro-aldol反応によってのみ活性化される。38C2触媒抗体のような人工触媒のみがその転化に触媒作用を及ぼす。触媒作用により活性薬剤に転化させる触媒抗体の併用の付加的要請は、そのようなプロドラッグの使用および取り扱い一層困難にする。したがって、上記の誘導体プロドラッグのいずれも上記がん治療において特に有効であることは証明されていない。
【0012】
生物学的利用性、薬物動力学、水溶解性を改善することを目的として、種々の抗がん剤のプロドラッグが合成されている。例えば、WO99/30561は、ドラッグ成分の遊離と活性化がヌクレオチド成分をドラッグ成分に接合する接合部のエステル結合の加水分解から生じる、ヌクレオチド系プロドラッグを開示している。
【0013】
また、米国特許第49756278号は、腫瘍細胞に結合する腫瘍−特異抗体−触媒複合体の投与、および抗体結合触媒の存在下において腫瘍側で活性な細胞傷害性ドラッグに変換されるプロドラッグの追加投与による細胞傷害性ドラッグの腫瘍細胞への輸送方法を記載している。この概念は、エトポシド−4’−フォスフェイトあるいは、7−(2’−アミノエチル フォスフェイト)−マイトマイシンと同時に使用されるが、これまでのところその利用は限定されている。
【0014】
また、WO94/13324は、対応する官能基を1-O-アルキル-、1-O-アシル-、1-S-アシル-、および1-S-アルキル-sn-グリセロ-3-フォスフェイト誘導体に変換することによるドラッグのプロドラッグへの変換を記載している。しかし、WO94/13324に報告されているプロドラッグが癌治療において特に有効であることは証明されていない。
【0015】
また、WO98/13059は、転移性細胞の表面に位置するペプチド加水分解酵素の基質であるアミノ端末にキャップが形成されたペプチドを含むプロドラッグを報告している。そのために主として使用される抗がん剤は、ドキソルビシン、タクソール、カプサイシン、マイトマイシンC、あるいはesperamycinである。プロドラッグの基質を加水分解するペプチド加水分解酵素は主としてカテプシンBである。
【0016】
また、米国特許第5,977,065号は、アクチノマイシンD、ドキソルビシン、マイトマイシンC、あるいは4−ニトロベンジルクロロフォーメイトとの反応から生じるナイトロジェンマスタードのプロドラッグを開示している。
【0017】
ブリストル・マイヤーズ会社のヨーロッパ特許出願EP0320988は、動物における抗がん活性が報告されている4’−ジメチルエピポドフィロトキシングルコシドの4’− カーバメート、4’−カーボネートおよび4’−エステルを開示している。EP0320988に開示されている化合物は、多剤耐性を克服する能力はない。
【0018】
Nicolaou (Nature1993, 364, 464)およびNiethammer et al. (Bioconj. Chem.2001, 3, 414)は、pH依存性、除放機構によって加水分解的に開裂されるジヒドロキシプロピル側鎖を有するC7水酸基でブロックされたパクリタキセルプロドラッグについて報告している。得られたプロドラッグは、より水溶性であるとともに、3倍多い最大耐量(MTD)で使用できるという点で親ドラッグに対して優れている。パクリタキセルは、乳癌、肺癌および卵巣癌において特に使用されている抗がん剤である。チューブリン不可逆重合を促進し、分裂前期(G2期)における細胞周期停止により細胞分裂を中断させることが知られている。
【0019】
パクリタキセルの第2の細胞傷害メカニズムは、微小管の重合に関係しない現象である、TNF alphaの誘導を援助することである。上記出版物に記載されているパクリタキセルプロドラッグは、水溶液中において不安定であり、自発的に加水分解する。したがって、Niethammer et al.に報告されているプロドラッグの利用は非常に制限される。これまでのところ、親分子に比較して十分に改善された効用と副作用の大幅な低減とを示すエトポシドに関してプロドラッグは報告されていない。
【発明の概要】
【発明が解決しようとする課題】
【0020】
したがって、本発明の目的は、患者への投与による副作用が十分に少ないポドフィロトキシンのプロドラッグを提供することにある。また、本発明の目的は、水溶液中で安定であり、活性薬剤への転化に触媒抗体の使用を必要としないポドフィロトキシンのプロドラッグを提供することにある。また、本発明の目的は、作用の意図された部位、すなわち腫瘍部における薬剤の徐放(slow release)を可能にするドフィロトキシンのプロドラッグを提供することにある。本発明のさらなる目的は、薬剤の親分子が直面する薬剤耐性を克服することが可能なポドフィロトキシンのプロドラッグを提供することにある。さらに、本発明の目的は、そのようなプロドラッグおよび同プロドラッグを含む医薬組成物の調製方法、および同プロドラッグや同医薬組成物の潜在的な使用を提供することにある。
【課題を解決するための手段】
【0021】
上記した課題は、以下の化学式(I)によって示されるポドフィロトキシンによって達成される、
【0022】
【化1】
【0023】
pは2〜100の整数であり、
Bは以下の化学式(II)によって示される:
【0024】
【化2】
【0025】
しかるに、Xは、O、S、およびNR”を含む群から選択され、Yは、O、S、およびNR”を含む群から選択され、R”=アルキル、アリルあるいはHであり、
nは0〜6の整数であり、
Zは、エチレングリコール、プロピレングリコール、グリセロール、ペンタエリスリトール、ポリエチレングリコール、および以下の化学式(III)によって示される化合物を含む群から選択されるポリヒドロキシアルキル基であり、
【0026】
【化3】
【0027】
あるいは、Zは、付加的にジオキソラン基が結合した上記のポリヒドロキシアルキル基であり、
あるいは、Zは、哺乳類のレセプタの標的部位(targetting moieties)、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター探索機能(tumor cell associated receptor finding function)を有するペプチドおよびタンパク質を含む群から選択される。
【0028】
一実施形態において、上記ジオキソラン基は、2, 2-ジアルキル-1, 3-ジオキソランを含む群から選択され、2位置での各アルキルは、無置換および置換メチル、エチル、プロピル、ブチル、ペンチル、ヘキシルを含む群から独立に選択される。
【0029】
一実施形態において、Zは、哺乳類の膜表面レセプタあるいは核内レセプタの標的部位であり、あるいはZは、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター結合機能(tumor cell associated receptor binding function)を有するペプチドあるいはタンパク質であり、好ましくは、Zは、ステロイド、成長因子レセプター阻害タンパク質、模倣性非ペプチドおよびペプチドを含む群から選択される。
【0030】
一実施形態において、Aは、化学式(IV)によって示される化合物を含む群から選択され、
【0031】
【化4】
【0032】
しかるに、R1およびR2は各々C1-C10アルキルであり、あるいはR1およびR2およびそれらが結合される炭素とがC5-C6シクロアルキルを示し、あるいはR1がHであり、R2はC1-10アルキル, C2-10アルケニル, C3-5シクロアルキル, フリル, チエニル, C6-10アリルおよびC7-14アラルキルを含む群から選択される。
【0033】
好ましくは、R1がHであり、R2はメチルもしくはチエニルである。
【0034】
一実施形態において、XがOであり、YがOである。
【0035】
別の実施形態において、XがOであり、YがSである。
【0036】
さらに別の実施形態において、XがOであり、YがNHである。
【0037】
別の実施形態において、ポドフィロトキシンは、以下の群から選択される。
【0038】
【化5】
【0039】
さらに別の実施形態において、ポドフィロトキシンは、以下の群から選択され、
【0040】
【化6】
【0041】
Zは請求項1と同様に定義され、好ましくは、ポドフィロトキシンは、以下の群から選択される。
【0042】
【化7】
【0043】
加水分解によって活性化される不活性前駆体分子としてエトポシドVP-16の加水分解活性プロドラッグを開発するため、4’フェノール性ヒドロキシ基の誘導体が合成された。これまでの研究により、そのOH基に融合させた安定なリンカーは>3 logsで細胞傷害を減少させることから、この4’フェノール性ヒドロキシ基はエトポシドの細胞傷害活性にとって非常に重要であることが示されている(Shabat D, Lode HN, Pertl U, etal. In vivo activity in a catalytic antibody-prodrug system: Antibody catalyzedetoposide prodrug activation for selective chemotherapy. Proc Natl Acad Sci U SA. 2001;98:7528-7533.)。しかしながら、Shabatによって報告されたプロドラッグは、活性薬剤を産出するためにそれらのプロドラッグを活性化する触媒抗体38C2と併用されない限り、細胞毒性の授与において効果がなく、いかなる抗腫瘍活性も示さなかった。また、Shabatによって報告されたプロドラッグは、自然には起こらないretro-Michael-retro-aldol反応によってのみ活性化され、そのため転化に触媒作用を及ぼすことのできる触媒抗体あるいは特定の目的に合わせた酵素が添加されなけばならない。これに対して、本発明のプロドラッグはシンプルな加水分解反応によって活性化されるとともに、普通の生理的条件下、pH値の広い範囲で安定である。
【0044】
パクリタクセルに対して、ポドフィロトキシン、特にエトポシドは、パクリタクセルの利用分野とは識別される固形癌および白血病を含むヒト悪性腫瘍の広い利用範囲においてトポイソメラーゼII阻害剤である。例えば、エトポシドは、急性リンパ性白血病、急性骨髄(性)白血病、神経芽細胞腫、横紋筋肉腫の治療に使用される。上記出版物(特に、Niethammer et al.)に報告されているパクリタクセルプロドラッグに対して、本発明のプロドラッグは、生理的条件下、水溶液中において非常に安定であるとともに、適切なpH変化によって容易に加水分解される。理論によって縛られたくないが、その格別の安定性のひとつの理由は、3’および5’位置それぞれにおける2つの隣接するメトキシ基によって4’位置に形成される疎水性ポケットと共同した、分子(すなわち、フェノール環)の”southern”部の疎水性にあると考えられている。
【0045】
したがって、VP-16の加水分解活性プロドラッグの理論的設計は、所望の効果に合わせた化学反応性4’フェノール性ヒドロキシ基の修飾(modification)を含む。化学修飾の種類に応じて、所望のpH依存率でプロドラッグの活性化が起こるように定義できる。本発明のプロドラッグは、pH値の広い範囲において安定であるとともに、例えばカルボキシルエステラーゼのような自然界に存在する酵素の存在下、生理的条件でのpHの適切な変化によって活性化できる。これについては以下に詳細に示される。
【0046】
したがって、理論的設計により、生体内および生体外において多剤耐性腫瘍細胞に対して十分な抗腫瘍活性を有する、エトポシドの加水分解活性プロドラッグを製造できるという驚くべき発見がなされた。
【0047】
ここに使用される”腫瘍細胞関連レセプター探索機能を有するペプチドあるいはタンパク質”とは、腫瘍細胞と関連するレセプターに結合する能力を有するあらゆるペプチドあるいはタンパク質であることを意味する。
【0048】
また、”哺乳類レセプターの標的部位”とは、哺乳類レセプターに結合する能力を有する群である。”成長因子レセプター阻害タンパク質、模倣性非ペプチドおよびペプチド”とは、メカニズムを問わず、特にそれに結合することによって成長因子レセプターを阻害する能力を有するあらゆるタンパク質、ペプチドあるいは非ペプチド性低分子化合物であることを意味する。”エトポシドおよびテニポシド”とは、以下の化学式の化合物、およびそのプロトン化形態であることを意味する。
【0049】
【化8】
【0050】
”官能基(functionality)”とは、分子に付着し、その分子に化学反応を受けさせるあらゆる化学部位であることを意味する。
(参考例)
好ましい実施形態において、ポドフィロトキシンは、以下の群から選択される。
【0051】
【化9】
【0052】
また、本発明の目的は、ポドフィロトキシンの調製方法によって達成される。すなわち、Aが上記と同様に定義される以下の化学式(V)によって示される化合物を
【0053】
【化10】
【0054】
ハロフォーメート(haloformate)W-(C=X)-(Y)-(CH2)n-Zと反応させる、しかるにX,Y,Zおよびnは上記と同様に定義され、WはCl、F、BrあるいはIである;
あるいは、化学式(V)によって示される化合物をホスゲンあるいはトリクロロメチルクロロフォーメート(trichloromethylchloroformate)と反応させ、4’−フェノールクロロフォーメート(4’-phenol chloroformate)中間生成物を生成し、次いで前記4’−フェノールクロロフォーメート中間生成物をアルコールあるいは化学式ZYHのチオールと反応させ、対応するカーボネートあるいはチオカーボネートを生成する、しかるに、Y=OもしくはSであり、Zは上記と同様に定義される、あるいは、前記4’−フェノールクロロフォーメート中間生成物を化学式HNR”Zのアミンと反応させ、対応するカーバメートを生成する、しかるに、R”およびZは上記と同様に定義される。
【0055】
好ましくは、化学式(V)によって示される化合物を、p−ニトロフェニルソケタールカーボネート(p-nitrophenyl soketalcarbonate)、ソケタールクロロフォーメート(soketalchloroformate)、およびPEG−クロロフォーメート(PEG-chloroformate)を含む群から選択される化合物と反応させる。
【0056】
本発明の方法の一実施形態において、上記した方法により得られる生産物は加水分解される。
【0057】
本発明のさらなる目的は、本発明にかかるポドフィロトキシンを含む医薬組成物および医薬的に許容できるキャリアー(pharmaceutically acceptable carrier)によって達成される。
【0058】
本発明のさらに別の目的は、本発明にかかるポドフィロトキシンの使用、あるいは細胞増多症(cell proliferative disorders)の治療薬の製造のための本発明の医薬組成物の使用によって達成され、幼児期、青年期、あるいは成人期において細胞増多症である場合における使用が有効である。
【図面の簡単な説明】
【0059】
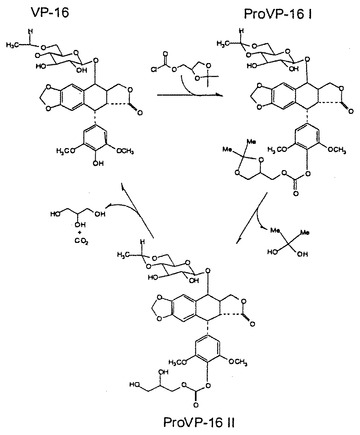
【図1】Pro VP-16IおよびIIの合成の概要を示す図である。
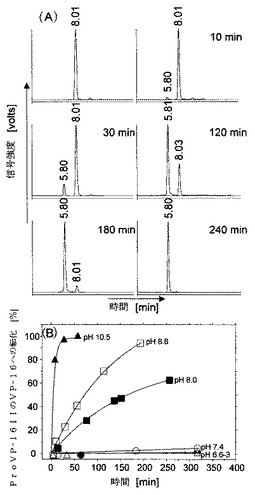
【図2】Pro VP-16IのPro VP-16IIおよびVP-16への転化を示す図である。
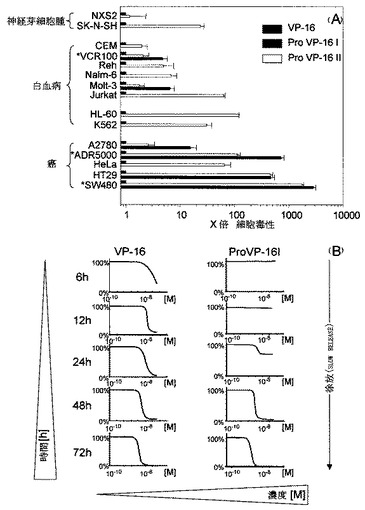
【図3】VP-16と比較した、Pro VP-16IおよびIIの細胞毒性プロファイルを示す図である。
【図4】多剤耐性MOVP-3細胞におけるPro VP-16IおよびIIの効果を示す図である。
【図5】耐性細胞におけるProVP-16IおよびIIによるアポトーシスの導入を示す図である。
【図6】マウスのProVP-16II治療における毒性および抗腫瘍効果を示す図である。
【発明を実施するための形態】
【0060】
各図面についてさらに詳細に説明する。図1は、Pro VP-16IおよびIIの合成と活性化の概要を示している。Pro VP-16Iの合成は、材料および方法において述べるように、ソルケタールクロロフォーメート(solketal chloroformate)とVP-16の反応を含む。化合物Pro VP-16IIは、2,2ジヒドロキシプロパンの除去を伴う酸加水分解によってPro VP-16Iから合成される。ProVP-16 IIのVP-16への活性化は、グリセロールと二酸化炭素の除去を伴って起こる。
【0061】
図2は、Pro VP-16IのPro VP-16IIおよびVP-16への転化(Conversion)を示している。Pro VP-16Iは、THF / 2% 塩酸中でインキュベートされ、(A)に表示される時間においてサンプルをHPLCによって解析した。Pro VP-16IIの加水分解活性は、(B)に示されるpHレベルでリン酸緩衝生理食塩水(PBS)において求めた。Pro VP-16I(3mM)は、PBS溶液中においてインキュベートされ、HPLCによって定期的にサンプルを解析した。また、ピーク積分によって求めた曲線下の面積から転化率(%)を計算した。
【0062】
図3は、VP-16との比較におけるPro VP-16IおよびIIの細胞毒性プロファイルを示している。すなわち、(A)の各細胞株に対するこれらのプロドラッグのIC50濃度測定を3度繰り返すことによってProVP-16IおよびIIの細胞毒性効果を細胞株のパネルに対して評価し、IC50 VP-16 , IC50ProVP-16 IもしくはIIに基づいて、VP-16に関連するプロドラッグによって引き起こされた細胞毒性を計算した。バーは平均値±SDを示す。Pro VP-16IあるいはIIとVP-16との間の差は、NXS2を除くすべての細胞株において統計的に有意(p<0.01)であった。図中、星印はMDR-1発現の増幅をともなった細胞株を示す。
【0063】
また、Molt-3細胞を使用して加水分解活性ProVP-16 Iによる細胞毒性の除放性(slow release kinetics)を調べた(B)。96ウェルプレートにおいて、ProVP-16IおよびVP-16の濃度(10-10 M〜10-6 M)を増加させ、ウェルあたり104個の細胞をインキュベートした。材料および方法に述べるように、表示された時点においてXTTassayによる細胞の生死判別測定を3度繰り返した。450nmでの光学密度測定からOD450サンプル , OD 450未処理 x 100%に基づき細胞生死判別率(%)を計算した。結果をドラッグ濃度の半対数関数としてプロットした。
【0064】
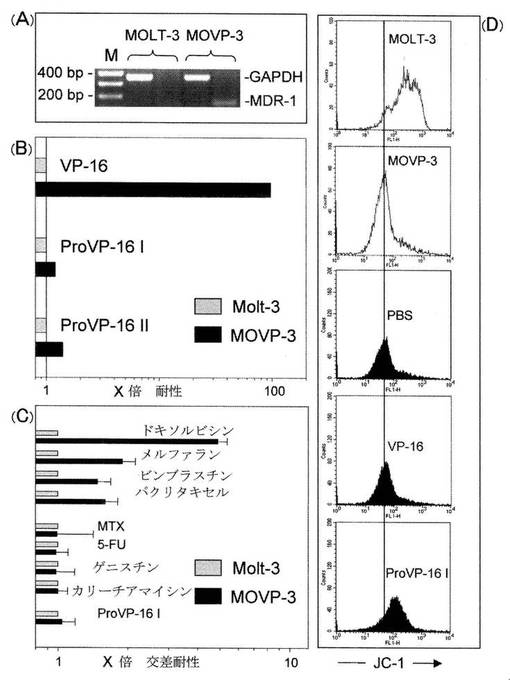
図4は、多剤耐性MOVP-3細胞におけるPro VP-16IおよびIIの効果を示している。新たに生成されたVP-16耐性MOVP-3細胞株を、MDR-1遺伝子発現(A)、VP-16導入細胞毒性に対する耐性(B)、P糖タンパク質の既知の基質であるMDR-1ドラッグに対する交差耐性(C)に関して解析した。MDR-1遺伝子発現は、MOVP-3およびMolt-3細胞それぞれから分離された全RNAにおけるRT-PCR解析によって調べた(6A)。GAPDHの発現をcDNAの完全性のコントロールとして使用した。
【0065】
また、IC50濃度から、IC50 MOVP-3 , IC50 Molt-3(n=3)に基づいてMOVP-3細胞のVP-16に対する耐性を計算し、結果をProVP-16 I およびIIで観察された効果と比較した(6B)。VP-16で認められたMolt-3およびMOVP-3細胞間の差異は、ProVP-16 IおよびII(p>0.05)とは対照的に統計的に有意(p<0.001)であった。また、(6C) MDR-1ドラッグ(ドキソルビシン, メルファラン、ビンブラスチンおよびパクリタキセル)に対するMOVP-3細胞の交差耐性を、(6B)に述べるように、IC50値(n=3)から計算した。コントロールとして、非MDR-1ドラッグ(MTX、5-FU、ゲニスチン、カリーチアマイシンΘ、ProVP-16 I)の結果も示されている。Molt-3およびMOVP-3細胞間におけるMDR-1ドラッグの差異は、非MDR-1ドラッグ(P>0.05)とは対照的にすべて統計的に有意であった(p<0.01)。
【0066】
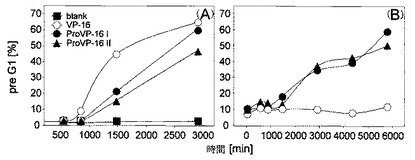
図5は、耐性細胞におけるProVP-16IおよびIIによるアポトーシスの導入を示している。細胞周期におけるProVP-16 IおよびII (5x10-7 M)の効果をMolt-3細胞(A)およびMOVP-3細胞(B)において解析し、結果をVP-16 (5x10-7 M)と比較した。細胞を表示されている時間(n=3)に収穫し、固定し、死細胞染色用蛍光色素 PI(PropidiumIodide)で着色し、材料及び方法に述べるようにFACSによって解析した。pre G1における細胞率(アポトーシス細胞)をDNAヒストグラムから計算した。
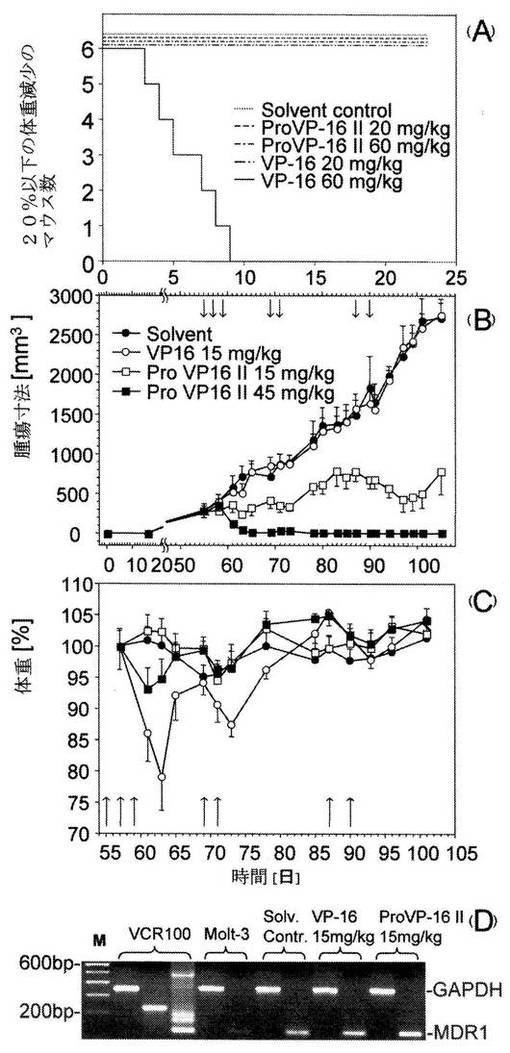
【0067】
図6は、マウスのProVP-16II治療における毒性および抗腫瘍効果を示している。A/Jマウス(n=6)に、1, 3, 5, 7, 9および11日目にProVP-16 II (20および60 mg/kg)、およびVP-16 (20 mg/kg)を、1, 3および5日目にVP-16 (60 mg/kg)を腹腔内投与した(A)。各動物の体重を長期にわたって測定し、0日目に測定した体重のパーセントとして算出した。Kaplan Maierプロットによって示されるように、20%以上の体重減少を重大事の発生と定義した。
【0068】
また、ProVP-16II治療の抗腫瘍効果を多剤耐性異種移植モデルにおいて調べた(B,C)。SCIDマウス(n=7)に5x106のMOVP-3細胞が皮下注射し、平均サイズ250 mm3の一次腫瘍を接種後55日目に確立した。治療は、腫瘍細胞接種後、55、57、59、69、71、87および90日目にPro VP-16 II (45および15 mg/kg), VP-16(15mg/kg)、および溶媒の腹腔内投与により行った。腫瘍の成長をマイクロノギスにて計測し、腫瘍サイズを材料及び方法に述べるようにして計算した(B)。ProVP-16 II (45および15 mg/kg)で治療した動物の実験群と対照群(溶媒およびVP-16)との間に見られた差異は、統計的に有意(63日後、p<0.001)であった(B)。治療を施した動物の体重を長期にわたって測定し、57日目の体重のパーセントとして算出した(C)。
【0069】
治療実験の最後に、残留するs.c.腫瘍を除去し、残留腫瘍のない45mg/kgで治療したマウスを除いて、MDR-1の遺伝子発現をRT-PCRによって解析した。各グループの腫瘍の典型的なシグナルが示され、陽性コントロールとして使用されたMDR-1発現VCR-100細胞と比較された。229もしくは127bpシグナルの存在は、MDR-1の発現を示唆している。
【0070】
本発明にかかるポドフィロトキシンは、細胞毒性の仲介(mediating)において特に効果的であることを証明した。また、エトポシドの種々の誘導体が合成され、加水分解を介してのそれらの活性化メカニズムが明らかになった(以下の実施例を参照)。重要なのは、生理緩衝液条件においてプロドラッグが安定に維持されることである。VP-16の4’ヒドロキシ基をブロックするため安定なカーバメートリンカー(carbamate linker)を有するエトポシドのプロドラッグが生理緩衝液条件において完全に安定であることはすでに示されている(Shabat D, Lode HN, Pertl U, etal. In vivo activity in a catalytic antibody-prodrug system: Antibody catalyzedetoposide prodrug activation for selective chemotherapy. Proc Natl Acad Sci U SA. 2001;98:7528-7533.)。しかし、ProVP-16 IおよびIIによって実証される本発明の加水分解活性プロドラッグとは対照的に、カーバメートプロドラッグは、触媒抗体38C2によって仲介される自然には起こらないretro-aldol retro-Michael反応の触媒作用によってのみ活性化されるように設計されている。強調すべきは、本発明のプロドラッグに対してこのカーバメートプロドラッグは調査したすべての腫瘍細胞株に対して細胞毒性を仲介する効果がなく、それ故に細胞毒性を導入する生物学的中心としてのVP-16の4’ヒドロキシ基の重要な役割を引き出すには到っていなかった。すなわち、ここに述べられたプロドラッグにおいては活性中心がブロックされるが、加水分解活性メカニズムを有する本発明にかかるプロドラッグは細胞毒性を非常に効果的に仲介する。転化されていないProVP-16 IおよびIIの無毒性は、Molt-3細胞(図3)に示される徐放メカニズムによって示されており、インキュベーションの12時間までのプロドラッグの細胞毒性効果の欠如によってVP-16と対照的である。これら両方のVP-16誘導体の長時間にわたる細胞毒性活性の安定した増加は、本発明の新規化合物が初期において安定で無毒性であるが、その後標的細胞の内部で活性になることを明確に示している。また、生体外で観察される徐放メカニズムは、ProVP-16IIが親化合物の最大許容投与量の3倍以上を許容することに示されるように、マウスにおける全身毒性の劇的な減少の説明となる(図6)。
【0071】
本発明にかかるプロドラッグのさらなる特徴は、VP-16に比較して多くのがん細胞群に高い効用を発揮することである。すなわち、MDR-1遺伝子発現の増幅をともなった細胞において(VCR 100, ADR 5000およびSW480)、本発明にかかるプロドラッグの3log以上の優れた効用が観察された(図4)。このような改善は、MDR-1modulatorsによって稀に達成される(Dalton WS. Mechanisms of drug resistance inhematologic malignancies. Semin Hematol. 1997;34:3-8; Sonneveld P, Durie BG,Lokhorst HM, et al. Modulation of multidrug-resistant multiple myeloma bycyclosporin. The Leukaemia Group of the EORTC and the HOVON. Lancet.1992;340:255-259; Joly P, Lallemand A, Oum'Hamed Z, Trentesaux C, Idoine O,Desplaces A. Effects of verapamil and S9788 on MDR-1 mRNA expression studied byin situ hybridization. Anticancer Res. 1996;16:3609-3614; Tai HL. Technologyevaluation: Valspodar, Novartis AG. Curr Opin Mol Ther. 2000;2:459-467; Kang Y,Perry RR. Effect of alpha-interferon on P-glycoprotein expression and functionand on verapamil modulation of doxorubicin resistance. CancerRes. 1994;54:2952-2958; Hofmann J, Gekeler V, Ise W, et al. Mechanismof action of dexniguldipine-HCl (B8509-035), a new potent modulator ofmultidrug resistance. Biochem Pharmacol. 1995;49:603-609.)。
【0072】
さらに、薬効評価は、本発明にかかるプロドラッグがMDR-1仲介による基質排出を抑制することを示している。すなわち、この新しいプロドラッグは、MDR-1p糖蛋白機能を抑制してMDR-1発現がん細胞株に対して優れた活性を発揮する。重要なのは、この生体外における劇的な効果は、生体内における薬剤耐性T細胞白血病異種移植モデルにおいて一次腫瘍を長期にわたって小さく保つ効果として認められることである。このモデルでは、MDR-1遺伝子発現が生体外においてVP-16に対して100x耐性で増幅され(図4)、結果的に最大許容投与量でのVP-16による治療効果が完全に失われた(図6)。興味深いことに、このように人工的に高い薬剤耐性を示すモデルにおいて、本発明にかかるプロドラッグでの治療によれば劇的な抗腫瘍効果を引き出すことができる。これは、上記のような高いMDR-1の増幅が再発悪性腫瘍における多剤化学療法を受けた患者に稀に観察されることから特に重要である(Beck J, Handgretinger R, DopferR, Klingebiel T, Niethammer D, Gekeler V. Expression of mdr1, mrp,topoisomerase II alpha/beta, and cyclin A in primary or relapsed states ofacute lymphoblastic leukaemias. Br J Haematol. 1995;89:356-363; Beck JF, BohnetB, Brugger D, et al. Expression analysis of protein kinase C isozymes andmultidrug resistance associated genes in ovarian cancer cells. Anticancer Res.1998;18:701-705.)。
【0073】
この研究の重要な発見は、生体内および生体外において、本発明のプロドラッグにより多剤耐性モデルで高い抗腫瘍効果が観察され、それらを用いた治療が従来のポドフィロトキシンを用いた化学療法に特筆すべき改善をもたらす点にある。
【0074】
以下の実施例は例示を意図するものであり、本発明の範囲を制限するものではない。
【実施例1】
【0075】
材料:2,3-ビス(2-メトキシ-4-ニトロ-5-スルフォフェニル)-5-[(フェニルアミノ)カルボニル]-2H-テトラゾリウム水酸化物(XTT)( 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazoliumhydroxide (XTT))、VP-16, ソルケタール(solketal)、有機溶媒、および電子捕獲剤(phenazine methosulfate (PMS))がSigma-Aldrich社(Deisenhofen, Germany)から得た。細胞培養試薬。制限酵素、その他の分子生物学用試薬は、Life Technologies社(Karlsruhe, Germany)から得た。
【0076】
細胞:腫瘍細胞群は、100IU/mlのペニシリン/ストレプトマイシン(P/S)の存在下、RPMI,10% FCS (Nalm-6, Reh, Molt-3, Jurkat, HL-60, K562, HeLa, CEM, A2780, SW480)あるいはDMEM, 10% FCS (NXS2, SK-N-SH, HT-29)において成長させ、標準的な組織培養条件(5% CO2, 37oC)において増殖させた。先に述べたNXS2(Lode HN, Xiang R, Varki NM,Dolman CS, Gillies SD, Reisfeld RA. Targeted interleukin-2 therapy forspontaneous neuroblastoma metastases to bone marrow. Journal of the NationalCancer Institute. 1997;89:1586-1594.)、およびJames Beck氏(Greifswald,Germany)により提供されたVCR100, ADR5000およびA2780(Beck J, Handgretinger R,Dopfer R, Klingebiel T, Niethammer D, Gekeler V. Expression of mdr1, mrp,topoisomerase II alpha/beta, and cyclin A in primary or relapsed states ofacute lymphoblastic leukaemias. Br J Haematol. 1995;89:356-363; Beck JF, BohnetB, Brugger D, et al. Expression analysis of protein kinase C isozymes andmultidrug resistance associated genes in ovarian cancer cells. Anticancer Res.1998;18:701-705.)を除くすべての細胞株はATCC, Rockville, MDから得た。多量のVP-16に連続曝露することによりエトポシド耐性subline MOVP-3を生成するためMolt-3細胞を使用した。6ヶ月経過後、MOVP-3細胞は1μM VP-16の存在下において安定に増殖し、これを生体外および生体内実験に使用した。
【0077】
マウス:ドイツのSulzfeldのCharles River研究所から年齢8週間のメスのA/JマウスおよびFOXCHASETM C.B-17/lcrCrl-scid BRマウスを得た。これらのマウスを我々の施設で無菌マウスコロニー内に8匹づつのグループとなるように収容した。マウスには適宜標準的なマウス実験用食を与えた。尚、動物実験は、研究用動物の使用と配慮に関するドイツ指針、すなわち、"Tierschutzgesetz"に基づいて行った。
【0078】
プロエトポシドの分析化学:プロエトポシドの合成およびHPLCによる解析については、Wrasidlo W, Schroeder U, Bernt K, et al. Synthesis, hydrolyticactivation and cytotoxicity of etoposide prodrugs. Bioorg Med Chem Lett. 2002;12. 557-560にすでに報告されており、その全文を参考のためこの明細書に添付する。
【0079】
RNA分離、逆転写、PCR増幅:total cellular RNA、cDNA合成およびRT-PCR条件は、前に記載されている(Lode HN, Xiang R, Varki NM, Dolman CS, Gillies SD, Reisfeld RA. Targetedinterleukin-2 therapy for spontaneous neuroblastoma metastases to bone marrow.Journal of the National Cancer Institute. 1997;89:1586-1594.)。ヒトMDR-1の増幅を、センス5’ GGA GAG ATC CTC ACC AAG CG 3’およびアンチセンス5’GTT GCC AAC CAT AGA TGA AGG 3’を用いて35サイクル(15s 96oC, 30 s60oC, 90 s 72oC)で行い、229bp断片指定MDR-1を得た。また、21サイクル後、センス5’ GCT CAG ACA GGATGT GAG TT 3’およびアンチセンス5’ CTG GGT AAT TAC AGC AAG CC 3’を用いて30サイクルで1.0ml MDR-1のさらに狭い領域を増幅(nestedamplification)することにより高感度検出を行い、127bp断片を得た。また、センス5’ CGGGAA GCT TGT GAT CAA TGG 3’およびアンチセンス5’ GGC AGT GAT GGCATG GAC TG 3’を用いて25サイクルでグリセロール−アルデヒド−リン酸デヒドロゲナーゼ(glycerol-aldehyde-phosphate-dehydrogenase:GAPDH)を増幅することによりcDNA完全性(cDNA integrity)を検証し、358bp断片を得た。すべての断片の特異性は、シークエンシングによって検証した。
【0080】
薬効JC-1評価:染色のため、細胞は2度洗浄され、前に述べたようにJC-1単量体含有PBSに再懸濁した(Legrand O, Perrot JY,Simonin G, Baudard M, Marie JP. JC-1: a very sensitive fluorescent probe totest Pgp activity in adult acute myeloid leukemia. Blood.)。簡単に言えば、0.1 μM JC-1単量体が5x105/mlの細胞数で15分間37℃でインキュベートされ、エトポシドおよびPro-VP-16 IおよびIIの存在する条件および存在しない条件においてインキュベートされた。2μMシクロスポリンAでインキュベートしたサンプルを陽性コントロールとして使用した(データは表示されない)。FACSortフローサイトメーター(BectonDickinson)を用いて細胞蛍光が記録され、JC-1シグナルは、色素単量体の解析用FL1-channel (530 nm フィルター)に検出された。
【0081】
MDR-1の安定トランスフェクション:MDR-1cDNAは、ハンブルグ大学のC. Baum氏によりpUCプラスミド内に提供された。哺乳類発現のため、NotIおよびBamHI制限酵素認識部位を用いて2シストロン性真核細胞用発現プラスミド(bicistronic eukaryotic expression plasmid)pIRESpuroにMDR-1cDNAをクローニングした。pMDR-IRESpuroは、エレクトロポレーション法(960 μF, 250V)によってMolt-3細胞内にトランスフェクトされた。安定トランスフェクタントは、300 ng/mlのピューロマイシで選択された。MDR-1発現は、1μg/ml MDR-1特異mAb(C. Baum, ハンブルグ大学)を用いて、FACS-解析(FACS-calibur,Becton Dickinson, Bedford, MA)により調べた。
【0082】
細胞毒性検査:細胞毒性は、これまでにも報告されているように(Scudiero DA, Shoemaker RH,Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cellgrowth and drug sensitivity in culture using human and other tumor cell lines.Cancer Res. 1988;48:4827-4833.)、XTTテトラゾリウム/ホルマザン評価法(XTT tetrazolium/formazan assay)によって調べた。すなわち、100 μl培地中、104個/ウェルの密度で96の平底ウェルプレートに細胞を蒔き、10-4から10-12Mの範囲の薬剤濃度に曝した。所定の時刻(6〜72時間)において、細胞の生死判別評価を37°Cで4時間インキュベートされた0.2%v/v PMS (PBS中、1.53 mg/ml)で活性化した50μlXTT試薬(無血清RPMI中、1mg/ml in)を添加することにより行った。プレートは、450 nmでThermomax (Molecular Devices社)において解析された。OD値は、薬剤濃度の関数としてプロットされ、softmax softwareを用いて曲線を積分してIC50濃度値を得た。
【0083】
細胞周期分析:細胞周期の特定の時期は、蛍光色素PIを用いた標準的な評価法により調べた。すなわち、24のウェルプレートに105/ウェルで細胞を蒔き、VP-16もしくはProVP-16 IおよびIIでインキュベートした。所定の時刻に細胞を収穫し、4.5 ml エタノール(75%, -20°C)に少なくとも12時間(4°C)固定した。PBS(pH 7.4)内において細胞を洗浄し、RNAse (0.3 mg/ml)および蛍光色素(50 μg/ml)を含有する250 μlのPBS中に再懸濁し、暗室(30min, RT)でインキュベートした。その後、細胞周期の特定の時期を定義するDNAヒストグラムのFACS解析を2度繰り返し、平均した結果をパーセントで表した。
【0084】
マウスにおける毒性研究:10mMエトポシドおよびプロエトポシドの原液を50:50 v/v Cremophor:エタノールに調合し、PBS(pH 7.4)に最終濃度に希釈した。エトポシドあるいはプロエトポシドのいずれかを20 mg/kgあるいは60 mg/kg、あるいは溶媒コントロール(25% DMSO, 12,5% エタノール, 12,5% Cremophor, 50%PBS)を年齢8〜10週間のA/Jマウスに、1, 3, 5, 7, 9および11日目に腹腔内投与した。VP-16 (60 mg/kg)は1,3および5日目にのみ注射した。体重およびその生存を長期にわたり観察した。
【0085】
T−細胞白血病異種移植モデルにおける抗腫瘍効果:100 μl PBS (pH 7.4)中5x106のMOVP 3細胞を各SCIDマウスの左脇腹の皮膚に注射することにより一次腫瘍を導入した。注射後55日目において、一次腫瘍の確立は明白であった。溶媒(25% DMSO, 12,5% エタノール, 12,5% cremophor, 50%PBS), VP-16 (15mg/kg)およびPro VP-16 II (15および45 mg/kg)を全体で200μl腹腔内注射することによりマウス(n=8)に治療を施した。各マウスは、腫瘍細胞接種後55、57、59、69,71、87、90日目の計7回の注射を受けた。一次腫瘍のサイズはマイクロノギスにより長期にわたって測定し、その体積は1/2 x 幅2 x 長さに基づいて算出した。また、体重は標準的なデジタル重量計で測定した。
【0086】
統計値:動物実験群の間における異なる所見の統計的な有意性を両側検定(two-tailed Student’s t test)によって調べ、両側検定p値(two-tailedp values)が<0.01であれば、有意性有りとみなした。
【0087】
エトポシドプロドラッグ活性化の化学:ProVP16-IおよびIIが実際にプロドラッグであることを実証するために、まずHPLCによって生体外におけるそれらの活性化特性を調べた(図1、2)。すなわち、ProVP-16IのProVP-16IIへの転化およびその後のVP-16の放出は2段階活性化機構に従う(図1)。まず、ProVP-16 Iは、酸性条件(THF, 2N HCL)下でのグリコシド(glycoside)部位の一部(約10%)の分解で2時間以内に2,2-ジヒドロキシプロパン(2,2-dihydroxypropane)の除去を伴ってProVP-16 IIに転化する(図2A)。次に、ProVP-16 IIは、塩基性pH条件下でグリセリンの除去を伴ってVP-16に加水分解する(図1、図2B)。すべての実験において、転化−時間曲線は一次反応速度論(first order kinetics)を示した。ここで重要なのは、ProVP-16 IIは生理緩衝液条件(PBS, pH 7.4, 37°C)下において5〜18時間まで<5%の転化率で安定であり、ProVP-16 IはPBS (pH 7.4, 37°C)において測定可能な転化は認められず完全に安定であった。また、ProVP-16 IIとは対照的に、ProVP-16 Iは塩基性緩衝液条件(pH £ 10.0)下でも不活性であった。ProVP-16 Iのこの独特の加水分解安定性は、この分子の”entire southern region”の疎水性、程度はさほどではないもののカーボネート部位を囲む2つのオルトメトキシ(ortho methoxy)基の立体障害に起因している。
【0088】
本発明にかかるプロドラッグの実用性は、自然に存在する触媒の存在下で活性化、すなわち加水分解され、しかもpH値の広い範囲にわたって水溶液中で安定であるという事実からも明らかである。
【0089】
ProPV-16 IおよびIIの活性化は、転化半減期がそれぞれ750.8 minおよび56.1 minである血清の存在下で起こる。また、豚肝臓カルボキシルエステラーゼ(Porcine liver caboxylesterase)は、ProPV-16 IおよびIIのVP-16への転化をそれぞれ14.2 minおよび514.1 minの転化半減期で仲介した。これらの所見は、カルボキシルエステラーゼによるpH7.4での酵素性プロドラッグ活性化を明確に示している(表1)。
【0090】
【表1】
【実施例2】
【0091】
白血病およびがん細胞株に対するProVP-16I および IIの活性度:これらの2つのプロエトポシドの細胞毒性に関する試験を、XTT生体染色抗増殖性評価法(XTT vital stain antiproliferation assay)を用いて腫瘍細胞群のパネルに対して行った(図3A)。試験に用いたヒト腫瘍細胞群の大部分に関して、両方のプロエトポシドとも、SW480大腸癌で1000倍低いIC50値, HT-29大腸癌、ADR5000 卵巣癌およびHL-60 pre B−細胞白血病で100-1000低いIC50値, HeLa子宮頸癌, A2780卵巣癌, K562慢性骨髄性白血病, Jurkat T−細胞白血病、およびSK-N-SH神経芽細胞腫で10-100 倍低いIC50値, およびMolt-3 T−細胞白血病,RehおよびNalm-6 pre B−細胞白血病、VCR100T−細胞性リンパ芽球性白血病およびCEM T−細胞性リンパ芽球性白血病細胞で2-10倍低いIC50値で親化合物よりも十分に活性であった。NXS2マウス神経芽細胞腫(図3A), CHO中国ハムスター卵巣,およびHammsヒト大腸癌細胞の3つの細胞群のみエトポシドとプロエトポシドに等しく良好に反応した。MDR-1発現の増幅を伴ったドキソルビシン耐性ADR5000およびビンクリスチン耐性VCR100の2つの細胞群においては、無耐性親細胞群A2780およびCEMに比べ両プロエトポシドのより高い細胞毒性効果が見られた。
【0092】
Pro-VP16 Iの細胞毒性作用の時間的経過を、Molt-3T−細胞白血病細胞の使用して追跡した(図3B)。結果は、Pro-VP16 Iによる遅発性の細胞毒性作用がインキュベーションの24時間後に始まることを示している。この時点において、親VP-16の細胞毒性効果はすでにほとんど完全に確立されている。ProVP16 Iの細胞毒性効果は、インキュベーション(IC501.0x10-8)後、48〜72時間後に完了し、VP-16(IC506.5 x10-8 M)のそれを越えた。同様の結果は、ProVP-16 IIでも観察された(データは表示されない)。これらの知見は、ProVP-16 IおよびIIによる細胞毒性活動の徐放(slow release)機構を示しており、プロドラッグのコンセプトに完全に合致している。
【実施例3】
【0093】
多剤耐性細胞におけるProVP-16I および IIの効果:ProVP-16 IおよびIIが、自然にあるMDR発現細胞群においてVP-16よりもさらに効果的であるという所見に基づいて(図3)、論点はこれらのプロドラッグが生体外における人工的なMDRを克服可能かどうかに向けられた。この目的のため、MDR-1ネガティブ細胞群Molt-3を使用して耐性サブクローンMOVP-3を生成した(図4A)。MOVP-3細胞におけるMDR-1 mRNA発現をRT-PCRによって調べ(図4A)、一方、細胞表面におけるMDR-1蛋白発現の増加をUIC2モノクローナル抗体を用いてFACS-解析により明らかにした。Bax およびBcl-2に加えてMRP, LRP, トポイソメラーゼI, IIαおよびIIβをさらに調べる拡張された遺伝子発現解析は(データを表示しない)、Molt-3に対比してMOVP-3細胞における薬剤耐性を説明する唯一の差はMDR-1の発現であることを明らかにした。これは、MOVP-3のIC50値が2x10-6 Mで、Molt-3細胞のIC50値が2x10-8 Mでエトポシドに対する100倍耐性を示したsublineの機能解析によってさらに確認された(図4B)。しかしながら、MOVP-3は、ProVP16IおよびIIへの感受性は十分に維持しており、両プロドラッグのIC50値が2x10-8 Mであり、親Molt-3細胞と薬剤耐性MOVP-3細胞の間でプロエトポシド仲介細胞毒性に有意な差異は認められなかった。同様の結果は、エトポシド、ドキソルビシン、パクリタキセルおよびビンブラスチンに対する耐性を生じたpMDR-IRESpuro (Molt-3/MDR-1)を使用してMDR-1 cDNAの安定トランスフェクションを施したMolt-3細胞においても得られた(データは表示されない)。また、Molt-3/MDR-1細胞は、ProVP-16 IおよびIIに対する十分な感受性を有した。エンプティーベクター(pIRESpuro)あるいはGFP含有ベクターで安定トランスフェクトされたコントロールは、試験したいかなる薬剤にも耐性を示さなかった。
【0094】
さらに、MOVP-3sublineにおける耐性のタイプを交差耐性を評価することにより調べた。MOVP-3細胞は、非MDR-1薬剤(MTX,5-FU, ゲニスチン, カリーチアマイシンq)とは対照的に、すべてのMDR-1タイプの薬剤(エトポシド、ドキソルビシン、パクリタキセル, ビンブラスチン)に対して交差耐性を示した(図4C)。これらの所見は、ProVP-16 IおよびIIが生体外においてMDR-1仲介多剤耐性を克服できることを明確に示している。
【0095】
JC-1色素を用いた機能性MDR-1評価においては、Molt-3親細胞と対比して耐性sublineにおけるFL-1信号の減少によって示されるように(図4D)、MOVP-3細胞におけるJC-1排出の増加はMolt-3コントロールとは対照的であった。この減少は、3x10-4 MのPro-VP-16 Iでの共インキュベーション(coincubation)によって排除され(図4D)、10x10-6 M(データは表示されない)までの広い濃度範囲にわたってMDR-1仲介排出を抑制した。興味深いことに、等モル濃度で使用されたVP-16は、PBSコントロールとほぼ同じJC-1信号によって示されるように、MDR-1機能の制御において効果がなかった。これらの知見は、プロドラッグ設計がMDR-1仲介基質排出を直接的に減少させることを明確に示している。
【0096】
多剤耐性MOVP-3細胞においてProVP-16 IおよびIIによって仲介される細胞毒性メカニズムを評価するため、細胞周期へのこれらの薬剤の影響を10nMから1 μMの範囲の濃度において解析するとともに、Molt-3親細胞と比較した。0.5μMのプロドラッグで得られた典型的な結果を図5に示す。具体的には、非同調MOVP-3 (図5B)およびMolt-3(図5A)細胞を72時間VP-16, ProVP-16 IおよびIIとインキュベートした。表示されている時刻において、定期的に細胞周期解析(cellcycle analysis)を行った。その結果、全濃度範囲(10nM-1 μM)と同様に(データは表示されない)、0.5μM(図5B)のPro VP-16 IおよびIIは、24時間後MOVP-3細胞内のpre-G1ピークの増加、すなわち、アポトーシス特性の導入に非常に効果的であることがわかった。プロドラッグとは対照的に、VP-16はアポトーシスの導入に効果がなかった。これは、MDR-1によってVP-16は排除されるが、ProVP-16 IおよびIIは排除されないことと合致する。また、Molt-3親細胞においても全濃度範囲(0.5 μM, 図5A)にわたってアポトーシスの導入が実証された。この場合は、VP-16がProVP-16 IおよびIIとほぼ同じ程度にMolt-3細胞にアポトーシスを導入することがわかった。全体的にみれば、これらの結果はProVP-16IおよびIIはMDR-1の基質ではないことを示唆している。
【実施例4】
【0097】
多剤耐性T−細胞白血病異種移植モデルにおけるProVP-16 I および IIの生体内毒性および有効性:ProVP-16 IおよびII間には差がないことを示した体外所見に基づいて、ProVP-16 IIをその高い水溶性を理由に生体内実験用として選択した。まず、ProVP-16IIの全身毒性をVP-16およびProVP-16 II (図6A)を腹腔内投与したA/Jマウス(n=6)において調べた。20 mg/kgのVP-16を投与されたすべてのマウスは、10%の平均体重減少をともなって生存した。一方、60 mg/kgのVP-16で治療したマウスの5/6は>20%の体重減少を示した。これらの知見は、ProVP-16 IIで得た結果とは極めて対照的であった。すなわち、20および60 mg/kgのProVP-16IIを投与は、いずれの実験群においても死を伴うことなくうまく受け入れられた。60 mg/kgのProVP-16 IIを投与したマウスのみに一時的に<10%の体重減少がみられたが、20mg/kgのPro VP-16 IIの投与では平均体重が安定に維持された。このように、<20%の体重減少によって定義される最大許容投与量は、VP-16において20 mg/kg、ProVP-16IIにおいて60 mg/kgであり、プロドラッグ設計によって全身毒性を少なくとも1/3に減らせることを示している。
【0098】
次に、ProVP-16 IIの抗腫瘍効果をT−細胞白血病の多剤耐性異種移植モデルにおいて調べ、VP-16と比較した。5x106の多剤耐性MOVP-3細胞の皮下投与によって一次腫瘍を導入し、腫瘍の成長を105日間にわたって追跡した。腫瘍細胞接種後55日目に腹腔内投与により治療を開始した。この時、腫瘍の平均サイズは250 mm3であった。全身毒性をさらに低減するため図6Aに示される結果に基づいて、VP-16 (15 mg/kg)およびProVP-16 II (15および45 mg/kg)の投与量を選択した。45 mg/kgのProVP-16 IIでの治療は、治療開始後10日目において7/7の動物に一次腫瘍の縮小をもたらし、それは2ヶ月間にわたって安定であった(図6B)。この治療は、一時的に6%の体重減少を引き起こしただけでうまく受け入れられた(図6C)。この所見は、抗腫瘍効果が見られず、溶媒のみで治療したコントロールマウスと同様に連続的に一次腫瘍の成長が認められた15 mg/kgのVP-16で治療したマウスの観察結果とは対照的であった。また、15 mg/kgのVP-16で治療したマウスには、一時的にではあるが20%の平均体重減少が伴い、重大な細胞毒性が観察された。一方、15 mg/kgのProVP-16 IIを投与したマウスは、測定可能な体重減少を示すことなく(図6C)、15 mg/kgのVP-16で治療したマウスとは対照的に、一次腫瘍成長に劇的な減少をもたらした。MDR-1発現が実験期間中、安定に残存していたかどうかを調べるために、105日目の腫瘍外植片からRNAが分離された。RT-PCRにより、調べたすべての腫瘍がMDR-1シグナルを示した(図6D)。
【0099】
明細書、請求項および/あるいは添付図面に開示された本発明の特徴は、個々におよびその組み合わせにおいて本発明の種々の形態を実現するための材料となる。
【背景技術】
【0001】
ポドフィロトキシンおよびその代謝産物/前駆物質のようなリグナンは、フェニールプロパノイド代謝経路の一部であり、植物界に広く分布している。それらのいくつか、特にポドフィロトキシンそれ自体は、抗がん性、抗真菌性および/あるいは抗菌性を有することが知られている。ポドフィロトキシンは、メイアップル(Podophyllum peltatum)や、樹脂としてLinum album, Linum flavumおよびLinum nodiflorumのようなアマ属(Linum)から抽出され、19世紀後半のアメリカ南部で生殖器疣の治療のため医者によって使用された。生殖器疣はヒト乳頭腫ウイルス(HPV)を病因とする癌のいくつかと関連している。
【0002】
ポドフィロトキシンは、トポイソメラーゼIIの阻害剤であるエトポシド(etoposide)、テニポシド(teniposide)およびetopophosのような化学療法薬の親分子として特に関心が持たれている。現在、ポドフィロトキシンの需要は、野生植物の成長が遅いことや過剰採集によって不十分となっている世界的な供給量を遥かに超えている。ポドフィロトキシンの供給量の減少を補うため、Podophyllum peltatumやLinum albumの細胞を培養する試みが着手され、ある程度の成功を収めており、その半合成誘導体、エトポシドおよびテニポシドは、重要な抗がん剤として広く使用されている。しかし、それらには、水溶性が乏しいこと、代謝不活性、薬剤耐性の発生といったいくつかの問題がある。
【0003】
これらの問題を克服するため、ポドフィロトキシンの誘導体が多くの研究室で合成されている(Yin et al., acta pharm. Sinica 1993, 28,758-761, Wang et al., actachem. Sinica 1992, 50, 698-701, Chang et al., J. med. chem. 1994, 37446-442,Pelter et al., J. nat. prod. 1994, 57, 1598-1602)。しかしながら、これらのいずれにおいても副作用の抑制と効用の両方において大幅な改善には到っていない。
【0004】
エトポシドは、一般的な成人がんと同様に、急性リンパ腫、横紋筋肉腫、神経芽細胞腫のような小児がんを含む広域のがんに対して高い効果を示す抗がん剤として広く使用され、骨髄移植調整療法においても使用されている。しかしながら、治療へのエトポシドの使用は、骨髄抑制を主としたその毒性によって制限される。
【0005】
一般的ながんにおいてだけでなく、特に危険性の高い白血病において化学療法を成功させるための挑戦の一つとして、多剤耐性の克服が挙げられる。患者の多くは、初期において種々の化学療法用薬剤を組み合わせた治療に反応する。しかしながら、多剤化学療法は、細胞傷害性薬剤の存在下において増殖し続ける多剤耐性(MDR)細胞群を誘発するおそれがある(DaltonWS. Mechanism of drug resistance in hematologic malignancies Semin Hematol.1997;34:3-8)。
【0006】
そのような細部群における薬剤感受性の低下は、生体内における最大耐量を超える量の細胞傷害性薬剤の投与を必要とするだろう。白血病や悪性腫瘍における最も特徴的な耐性メカニズムの一つは、P糖蛋白、多剤耐性-1遺伝子(MDR-1)の産生物質によって仲介される薬剤排出(drug extrusion)であり、治療効果の低い結果と関連することが報告されている(Hunault M, Zhou D, Delmer A, et al. Multidrug resistance gene expression inacute myeloid leukemia: major prognosis significance for in vivo drugresistance to induction treatment. Ann Hematol. 1997;74:65-71; Marie JP, ZhouDC, Gurbuxani S, Legrand O, Zittoun R. MDR1/P-glycoprotein in haematologicalneoplasms. Eur J Cancer. 1996;32A:1034-1038.)。
【0007】
多剤化学療法の導入以来、薬剤耐性を回避するために種々の方法が開発されている。
【0008】
患者に生じた薬剤耐性を克服するための努力は、(i)スケジューリング、すなわち、再発白血病における葉酸代謝拮抗薬(antifolates)のような長期間少量投与療法、あるいは短期間多量投与、たとえば、骨肉腫におけるその後の葉酸回復剤(folate -rescue)の使用を伴う葉酸代謝拮抗薬、(ii)MDR仲介耐性の場合におけるMDR-1阻害剤のような化学増感剤の使用を伴う併用療法、(iii) 放射線療法、温熱療法、高圧酸素のような非化学増感剤の使用を伴う併用化学療法(Dalton WS. Mechanisms of drug resistance inhematologic malignancies. Semin Hematol. 1997;34:3-8; Joel SP, Slevin ML.Schedule-dependent topoisomerase II-inhibiting drugs. Cancer ChemotherPharmacol. 1994;34 Suppl:S84-S88; Ishikawa T, Kuo MT, Furuta K, Suzuki M. Thehuman multidrug resistance-associated protein (MRP) gene family: from biologicalfunction to drug molecular design. Clin Chem Lab Med. 2000;38:893-897.)に集中している。
【0009】
また、脱アミノドキソルビシン類似物質(Solary E, Ling YH, Perez-Soler R, Priebe W,Pommier Y. Hydroxyrubicin, a deaminated derivative of doxorubicin, inhibitsmammalianDNA topoisomerase II and partially circumvents multidrug resistance.Int J Cancer. 1994;58:85-94.)や、エトポシドのβアミノ誘導体(ZhangYL, Guo X, Cheng YC, Lee KH. Antitumor agents. 148. Synthesis and biologicalevaluation of novel 4 beta-amino derivatives of etoposide with betterpharmacological profiles. J Med Chem. 1994;37:446-452; Zhang YL, Shen YC, WangZQ, et al. Antitumor agents, 130, Novel 4 beta-arylamino derivatives of 3',4'- didemethoxy-3',4'-dioxo-4-deoxypodophyllotoxinas potent inhibitors of human DNA topoisomerase II. J Nat Prod.1992;55:1100-1111.)のような薬剤耐性機構を積極的に回避する類似物質を見出すために抗悪性腫瘍薬を直接修正する試みもわずかながらなされている。このように、MDRに関連する問題への有効な解決策は、MDRメカニズムによって影響されず、全身毒性の低減と抗がん作用の向上を発揮する薬剤の理論的な設計を含む。
【0010】
エトポシドに対する耐性は、標的酵素トポイソメラーゼIIのダウンレギュレーション、bcl-2のような抗アポトーシス機構のプロレグレーション(pro-regulation)またはアップレグレーションのいずれかのダウンレギュレーション、代謝の増大および/あるいは輸送系によって仲介された細胞からの薬剤の排出を伴いながら細胞レベルで起こる。また、そのような輸送系の誘発は、MDR-1, MRPあるいはLRP仲介多剤耐性において見られるように、しばしば他の抗悪性腫瘍薬に対する交差耐性を招く(Gottesman MM. How cancer cellsevade chemotherapy: sixteenth Richard and Hinda Rosenthal Foundation AwardLecture. Cancer Res. 1993;53:747-754; Borst P, Evers R, Kool M, Wijnholds J. Afamily of drug transporters: the multidrug resistance-associated proteins. JNatl Cancer Inst. 2000;92:1295-1302; Borst P, Evers R, Kool M, Wijnholds J. Themultidrug resistance protein family. Biochim Biophys Acta. 1999;1461:347-357.)。薬剤耐性の主たるメカニズムは、血液悪性腫瘍において発生することが報告されているように、MDR-1遺伝子産物、P糖蛋白の大量発現である。したがって、輸送系仲介多剤耐性を克服するための試みは、MDR-1発現の制御(Liu C, Qureshi IA, Ding X, et al. Modulation ofmultidrug resistance gene (MDR-1) with antisense oligodeoxynucleotides. ClinSci (Colch ). 1996;91:93-98.)や、サイクロスポリン(Sonneveld P, Durie BG, Lokhorst HM, et al. Modulation ofmultidrug-resistant multiple myeloma by cyclosporin. The Leukaemia Group of theEORTC and the HOVON. Lancet. 1992;340:255-259.)、ベラパミル(Joly P, Lallemand A,Oum'Hamed Z, Trentesaux C, Idoine O, Desplaces A. Effects of verapamil andS9788 on MDR-1 mRNA expression studied by in situ hybridization. AnticancerRes. 1996;16:3609-3614.)、あるいはvalspodar (TaiHL. Technology evaluation: Valspodar, Novartis AG. Curr Opin Mol Ther.2000;2:459-467.)のようなMDR-1阻害剤の同時投与に集中しており、これらはみな生体外および生体内においてある程度の有効性を示している。これらの膜輸送蛋白は、おそらく異なる薬剤よりも硫酸塩あるいはグルクロニド、グルタチオンのような一般の代謝産物が特異的に認識されるため、全く異なる構造を有する驚くべき広い範囲の基質を排出する(Zhu BT. A novel hypothesis for the mechanism ofaction of P-glycoprotein as a multidrug transporter. Mol Carcinog.1999;25:1-13.)。したがって、これらの耐性機構は、最初に認識され、次いでプロテアソームによって分解されるように多くの全く異なる蛋白質がユビキチンで”標識”されるユビキチンシステムと似ている。このように、分子”標識”を妨害する薬剤改良から生じた新しい分子は、多剤耐性がん細胞から取り除かれない可能性がある。
【0011】
エトポシドの標的組織へのより特異的なターゲティングを可能にするエトポシドの誘導体を合成するため、いくつかの試みが着手されている。EP0423747は、グリコシル−エトポシド−プロドラッグの合成を報告しており、このプロドラッグは腫瘍−特異酵素複合体の作用によって有効なエトポシドドラッグとグリコシル残留物とに開裂され、それにより、腫瘍−特異酵素複合のためドラッグはその好ましい作用部位でのみ活性化される(さらに米国特許第4975278号を参照)。また、Shabatら(PNAS, vol. 98, 13, 7528-7533)は、4’−フェノール性OH基がアルドールカーバメート(aldol carbamate)化合物によってマスクされた、エトポシドに基づく抗体−プロドラッグ系の合成について記載している。しかしながら、そのようなプロドラッグは、プロドラッグを活性化してエトポシドを産出させる触媒抗体38C2と組み合わされない限り、単独ではいかなる抗がん活性も示さなかった。Shabatに記載されているプロドラッグは、自然界では起こらないretro-Michael/retro-aldol反応によってのみ活性化される。38C2触媒抗体のような人工触媒のみがその転化に触媒作用を及ぼす。触媒作用により活性薬剤に転化させる触媒抗体の併用の付加的要請は、そのようなプロドラッグの使用および取り扱い一層困難にする。したがって、上記の誘導体プロドラッグのいずれも上記がん治療において特に有効であることは証明されていない。
【0012】
生物学的利用性、薬物動力学、水溶解性を改善することを目的として、種々の抗がん剤のプロドラッグが合成されている。例えば、WO99/30561は、ドラッグ成分の遊離と活性化がヌクレオチド成分をドラッグ成分に接合する接合部のエステル結合の加水分解から生じる、ヌクレオチド系プロドラッグを開示している。
【0013】
また、米国特許第49756278号は、腫瘍細胞に結合する腫瘍−特異抗体−触媒複合体の投与、および抗体結合触媒の存在下において腫瘍側で活性な細胞傷害性ドラッグに変換されるプロドラッグの追加投与による細胞傷害性ドラッグの腫瘍細胞への輸送方法を記載している。この概念は、エトポシド−4’−フォスフェイトあるいは、7−(2’−アミノエチル フォスフェイト)−マイトマイシンと同時に使用されるが、これまでのところその利用は限定されている。
【0014】
また、WO94/13324は、対応する官能基を1-O-アルキル-、1-O-アシル-、1-S-アシル-、および1-S-アルキル-sn-グリセロ-3-フォスフェイト誘導体に変換することによるドラッグのプロドラッグへの変換を記載している。しかし、WO94/13324に報告されているプロドラッグが癌治療において特に有効であることは証明されていない。
【0015】
また、WO98/13059は、転移性細胞の表面に位置するペプチド加水分解酵素の基質であるアミノ端末にキャップが形成されたペプチドを含むプロドラッグを報告している。そのために主として使用される抗がん剤は、ドキソルビシン、タクソール、カプサイシン、マイトマイシンC、あるいはesperamycinである。プロドラッグの基質を加水分解するペプチド加水分解酵素は主としてカテプシンBである。
【0016】
また、米国特許第5,977,065号は、アクチノマイシンD、ドキソルビシン、マイトマイシンC、あるいは4−ニトロベンジルクロロフォーメイトとの反応から生じるナイトロジェンマスタードのプロドラッグを開示している。
【0017】
ブリストル・マイヤーズ会社のヨーロッパ特許出願EP0320988は、動物における抗がん活性が報告されている4’−ジメチルエピポドフィロトキシングルコシドの4’− カーバメート、4’−カーボネートおよび4’−エステルを開示している。EP0320988に開示されている化合物は、多剤耐性を克服する能力はない。
【0018】
Nicolaou (Nature1993, 364, 464)およびNiethammer et al. (Bioconj. Chem.2001, 3, 414)は、pH依存性、除放機構によって加水分解的に開裂されるジヒドロキシプロピル側鎖を有するC7水酸基でブロックされたパクリタキセルプロドラッグについて報告している。得られたプロドラッグは、より水溶性であるとともに、3倍多い最大耐量(MTD)で使用できるという点で親ドラッグに対して優れている。パクリタキセルは、乳癌、肺癌および卵巣癌において特に使用されている抗がん剤である。チューブリン不可逆重合を促進し、分裂前期(G2期)における細胞周期停止により細胞分裂を中断させることが知られている。
【0019】
パクリタキセルの第2の細胞傷害メカニズムは、微小管の重合に関係しない現象である、TNF alphaの誘導を援助することである。上記出版物に記載されているパクリタキセルプロドラッグは、水溶液中において不安定であり、自発的に加水分解する。したがって、Niethammer et al.に報告されているプロドラッグの利用は非常に制限される。これまでのところ、親分子に比較して十分に改善された効用と副作用の大幅な低減とを示すエトポシドに関してプロドラッグは報告されていない。
【発明の概要】
【発明が解決しようとする課題】
【0020】
したがって、本発明の目的は、患者への投与による副作用が十分に少ないポドフィロトキシンのプロドラッグを提供することにある。また、本発明の目的は、水溶液中で安定であり、活性薬剤への転化に触媒抗体の使用を必要としないポドフィロトキシンのプロドラッグを提供することにある。また、本発明の目的は、作用の意図された部位、すなわち腫瘍部における薬剤の徐放(slow release)を可能にするドフィロトキシンのプロドラッグを提供することにある。本発明のさらなる目的は、薬剤の親分子が直面する薬剤耐性を克服することが可能なポドフィロトキシンのプロドラッグを提供することにある。さらに、本発明の目的は、そのようなプロドラッグおよび同プロドラッグを含む医薬組成物の調製方法、および同プロドラッグや同医薬組成物の潜在的な使用を提供することにある。
【課題を解決するための手段】
【0021】
上記した課題は、以下の化学式(I)によって示されるポドフィロトキシンによって達成される、
【0022】
【化1】
【0023】
pは2〜100の整数であり、
Bは以下の化学式(II)によって示される:
【0024】
【化2】
【0025】
しかるに、Xは、O、S、およびNR”を含む群から選択され、Yは、O、S、およびNR”を含む群から選択され、R”=アルキル、アリルあるいはHであり、
nは0〜6の整数であり、
Zは、エチレングリコール、プロピレングリコール、グリセロール、ペンタエリスリトール、ポリエチレングリコール、および以下の化学式(III)によって示される化合物を含む群から選択されるポリヒドロキシアルキル基であり、
【0026】
【化3】
【0027】
あるいは、Zは、付加的にジオキソラン基が結合した上記のポリヒドロキシアルキル基であり、
あるいは、Zは、哺乳類のレセプタの標的部位(targetting moieties)、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター探索機能(tumor cell associated receptor finding function)を有するペプチドおよびタンパク質を含む群から選択される。
【0028】
一実施形態において、上記ジオキソラン基は、2, 2-ジアルキル-1, 3-ジオキソランを含む群から選択され、2位置での各アルキルは、無置換および置換メチル、エチル、プロピル、ブチル、ペンチル、ヘキシルを含む群から独立に選択される。
【0029】
一実施形態において、Zは、哺乳類の膜表面レセプタあるいは核内レセプタの標的部位であり、あるいはZは、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター結合機能(tumor cell associated receptor binding function)を有するペプチドあるいはタンパク質であり、好ましくは、Zは、ステロイド、成長因子レセプター阻害タンパク質、模倣性非ペプチドおよびペプチドを含む群から選択される。
【0030】
一実施形態において、Aは、化学式(IV)によって示される化合物を含む群から選択され、
【0031】
【化4】
【0032】
しかるに、R1およびR2は各々C1-C10アルキルであり、あるいはR1およびR2およびそれらが結合される炭素とがC5-C6シクロアルキルを示し、あるいはR1がHであり、R2はC1-10アルキル, C2-10アルケニル, C3-5シクロアルキル, フリル, チエニル, C6-10アリルおよびC7-14アラルキルを含む群から選択される。
【0033】
好ましくは、R1がHであり、R2はメチルもしくはチエニルである。
【0034】
一実施形態において、XがOであり、YがOである。
【0035】
別の実施形態において、XがOであり、YがSである。
【0036】
さらに別の実施形態において、XがOであり、YがNHである。
【0037】
別の実施形態において、ポドフィロトキシンは、以下の群から選択される。
【0038】
【化5】
【0039】
さらに別の実施形態において、ポドフィロトキシンは、以下の群から選択され、
【0040】
【化6】
【0041】
Zは請求項1と同様に定義され、好ましくは、ポドフィロトキシンは、以下の群から選択される。
【0042】
【化7】
【0043】
加水分解によって活性化される不活性前駆体分子としてエトポシドVP-16の加水分解活性プロドラッグを開発するため、4’フェノール性ヒドロキシ基の誘導体が合成された。これまでの研究により、そのOH基に融合させた安定なリンカーは>3 logsで細胞傷害を減少させることから、この4’フェノール性ヒドロキシ基はエトポシドの細胞傷害活性にとって非常に重要であることが示されている(Shabat D, Lode HN, Pertl U, etal. In vivo activity in a catalytic antibody-prodrug system: Antibody catalyzedetoposide prodrug activation for selective chemotherapy. Proc Natl Acad Sci U SA. 2001;98:7528-7533.)。しかしながら、Shabatによって報告されたプロドラッグは、活性薬剤を産出するためにそれらのプロドラッグを活性化する触媒抗体38C2と併用されない限り、細胞毒性の授与において効果がなく、いかなる抗腫瘍活性も示さなかった。また、Shabatによって報告されたプロドラッグは、自然には起こらないretro-Michael-retro-aldol反応によってのみ活性化され、そのため転化に触媒作用を及ぼすことのできる触媒抗体あるいは特定の目的に合わせた酵素が添加されなけばならない。これに対して、本発明のプロドラッグはシンプルな加水分解反応によって活性化されるとともに、普通の生理的条件下、pH値の広い範囲で安定である。
【0044】
パクリタクセルに対して、ポドフィロトキシン、特にエトポシドは、パクリタクセルの利用分野とは識別される固形癌および白血病を含むヒト悪性腫瘍の広い利用範囲においてトポイソメラーゼII阻害剤である。例えば、エトポシドは、急性リンパ性白血病、急性骨髄(性)白血病、神経芽細胞腫、横紋筋肉腫の治療に使用される。上記出版物(特に、Niethammer et al.)に報告されているパクリタクセルプロドラッグに対して、本発明のプロドラッグは、生理的条件下、水溶液中において非常に安定であるとともに、適切なpH変化によって容易に加水分解される。理論によって縛られたくないが、その格別の安定性のひとつの理由は、3’および5’位置それぞれにおける2つの隣接するメトキシ基によって4’位置に形成される疎水性ポケットと共同した、分子(すなわち、フェノール環)の”southern”部の疎水性にあると考えられている。
【0045】
したがって、VP-16の加水分解活性プロドラッグの理論的設計は、所望の効果に合わせた化学反応性4’フェノール性ヒドロキシ基の修飾(modification)を含む。化学修飾の種類に応じて、所望のpH依存率でプロドラッグの活性化が起こるように定義できる。本発明のプロドラッグは、pH値の広い範囲において安定であるとともに、例えばカルボキシルエステラーゼのような自然界に存在する酵素の存在下、生理的条件でのpHの適切な変化によって活性化できる。これについては以下に詳細に示される。
【0046】
したがって、理論的設計により、生体内および生体外において多剤耐性腫瘍細胞に対して十分な抗腫瘍活性を有する、エトポシドの加水分解活性プロドラッグを製造できるという驚くべき発見がなされた。
【0047】
ここに使用される”腫瘍細胞関連レセプター探索機能を有するペプチドあるいはタンパク質”とは、腫瘍細胞と関連するレセプターに結合する能力を有するあらゆるペプチドあるいはタンパク質であることを意味する。
【0048】
また、”哺乳類レセプターの標的部位”とは、哺乳類レセプターに結合する能力を有する群である。”成長因子レセプター阻害タンパク質、模倣性非ペプチドおよびペプチド”とは、メカニズムを問わず、特にそれに結合することによって成長因子レセプターを阻害する能力を有するあらゆるタンパク質、ペプチドあるいは非ペプチド性低分子化合物であることを意味する。”エトポシドおよびテニポシド”とは、以下の化学式の化合物、およびそのプロトン化形態であることを意味する。
【0049】
【化8】
【0050】
”官能基(functionality)”とは、分子に付着し、その分子に化学反応を受けさせるあらゆる化学部位であることを意味する。
(参考例)
好ましい実施形態において、ポドフィロトキシンは、以下の群から選択される。
【0051】
【化9】
【0052】
また、本発明の目的は、ポドフィロトキシンの調製方法によって達成される。すなわち、Aが上記と同様に定義される以下の化学式(V)によって示される化合物を
【0053】
【化10】
【0054】
ハロフォーメート(haloformate)W-(C=X)-(Y)-(CH2)n-Zと反応させる、しかるにX,Y,Zおよびnは上記と同様に定義され、WはCl、F、BrあるいはIである;
あるいは、化学式(V)によって示される化合物をホスゲンあるいはトリクロロメチルクロロフォーメート(trichloromethylchloroformate)と反応させ、4’−フェノールクロロフォーメート(4’-phenol chloroformate)中間生成物を生成し、次いで前記4’−フェノールクロロフォーメート中間生成物をアルコールあるいは化学式ZYHのチオールと反応させ、対応するカーボネートあるいはチオカーボネートを生成する、しかるに、Y=OもしくはSであり、Zは上記と同様に定義される、あるいは、前記4’−フェノールクロロフォーメート中間生成物を化学式HNR”Zのアミンと反応させ、対応するカーバメートを生成する、しかるに、R”およびZは上記と同様に定義される。
【0055】
好ましくは、化学式(V)によって示される化合物を、p−ニトロフェニルソケタールカーボネート(p-nitrophenyl soketalcarbonate)、ソケタールクロロフォーメート(soketalchloroformate)、およびPEG−クロロフォーメート(PEG-chloroformate)を含む群から選択される化合物と反応させる。
【0056】
本発明の方法の一実施形態において、上記した方法により得られる生産物は加水分解される。
【0057】
本発明のさらなる目的は、本発明にかかるポドフィロトキシンを含む医薬組成物および医薬的に許容できるキャリアー(pharmaceutically acceptable carrier)によって達成される。
【0058】
本発明のさらに別の目的は、本発明にかかるポドフィロトキシンの使用、あるいは細胞増多症(cell proliferative disorders)の治療薬の製造のための本発明の医薬組成物の使用によって達成され、幼児期、青年期、あるいは成人期において細胞増多症である場合における使用が有効である。
【図面の簡単な説明】
【0059】
【図1】Pro VP-16IおよびIIの合成の概要を示す図である。
【図2】Pro VP-16IのPro VP-16IIおよびVP-16への転化を示す図である。
【図3】VP-16と比較した、Pro VP-16IおよびIIの細胞毒性プロファイルを示す図である。
【図4】多剤耐性MOVP-3細胞におけるPro VP-16IおよびIIの効果を示す図である。
【図5】耐性細胞におけるProVP-16IおよびIIによるアポトーシスの導入を示す図である。
【図6】マウスのProVP-16II治療における毒性および抗腫瘍効果を示す図である。
【発明を実施するための形態】
【0060】
各図面についてさらに詳細に説明する。図1は、Pro VP-16IおよびIIの合成と活性化の概要を示している。Pro VP-16Iの合成は、材料および方法において述べるように、ソルケタールクロロフォーメート(solketal chloroformate)とVP-16の反応を含む。化合物Pro VP-16IIは、2,2ジヒドロキシプロパンの除去を伴う酸加水分解によってPro VP-16Iから合成される。ProVP-16 IIのVP-16への活性化は、グリセロールと二酸化炭素の除去を伴って起こる。
【0061】
図2は、Pro VP-16IのPro VP-16IIおよびVP-16への転化(Conversion)を示している。Pro VP-16Iは、THF / 2% 塩酸中でインキュベートされ、(A)に表示される時間においてサンプルをHPLCによって解析した。Pro VP-16IIの加水分解活性は、(B)に示されるpHレベルでリン酸緩衝生理食塩水(PBS)において求めた。Pro VP-16I(3mM)は、PBS溶液中においてインキュベートされ、HPLCによって定期的にサンプルを解析した。また、ピーク積分によって求めた曲線下の面積から転化率(%)を計算した。
【0062】
図3は、VP-16との比較におけるPro VP-16IおよびIIの細胞毒性プロファイルを示している。すなわち、(A)の各細胞株に対するこれらのプロドラッグのIC50濃度測定を3度繰り返すことによってProVP-16IおよびIIの細胞毒性効果を細胞株のパネルに対して評価し、IC50 VP-16 , IC50ProVP-16 IもしくはIIに基づいて、VP-16に関連するプロドラッグによって引き起こされた細胞毒性を計算した。バーは平均値±SDを示す。Pro VP-16IあるいはIIとVP-16との間の差は、NXS2を除くすべての細胞株において統計的に有意(p<0.01)であった。図中、星印はMDR-1発現の増幅をともなった細胞株を示す。
【0063】
また、Molt-3細胞を使用して加水分解活性ProVP-16 Iによる細胞毒性の除放性(slow release kinetics)を調べた(B)。96ウェルプレートにおいて、ProVP-16IおよびVP-16の濃度(10-10 M〜10-6 M)を増加させ、ウェルあたり104個の細胞をインキュベートした。材料および方法に述べるように、表示された時点においてXTTassayによる細胞の生死判別測定を3度繰り返した。450nmでの光学密度測定からOD450サンプル , OD 450未処理 x 100%に基づき細胞生死判別率(%)を計算した。結果をドラッグ濃度の半対数関数としてプロットした。
【0064】
図4は、多剤耐性MOVP-3細胞におけるPro VP-16IおよびIIの効果を示している。新たに生成されたVP-16耐性MOVP-3細胞株を、MDR-1遺伝子発現(A)、VP-16導入細胞毒性に対する耐性(B)、P糖タンパク質の既知の基質であるMDR-1ドラッグに対する交差耐性(C)に関して解析した。MDR-1遺伝子発現は、MOVP-3およびMolt-3細胞それぞれから分離された全RNAにおけるRT-PCR解析によって調べた(6A)。GAPDHの発現をcDNAの完全性のコントロールとして使用した。
【0065】
また、IC50濃度から、IC50 MOVP-3 , IC50 Molt-3(n=3)に基づいてMOVP-3細胞のVP-16に対する耐性を計算し、結果をProVP-16 I およびIIで観察された効果と比較した(6B)。VP-16で認められたMolt-3およびMOVP-3細胞間の差異は、ProVP-16 IおよびII(p>0.05)とは対照的に統計的に有意(p<0.001)であった。また、(6C) MDR-1ドラッグ(ドキソルビシン, メルファラン、ビンブラスチンおよびパクリタキセル)に対するMOVP-3細胞の交差耐性を、(6B)に述べるように、IC50値(n=3)から計算した。コントロールとして、非MDR-1ドラッグ(MTX、5-FU、ゲニスチン、カリーチアマイシンΘ、ProVP-16 I)の結果も示されている。Molt-3およびMOVP-3細胞間におけるMDR-1ドラッグの差異は、非MDR-1ドラッグ(P>0.05)とは対照的にすべて統計的に有意であった(p<0.01)。
【0066】
図5は、耐性細胞におけるProVP-16IおよびIIによるアポトーシスの導入を示している。細胞周期におけるProVP-16 IおよびII (5x10-7 M)の効果をMolt-3細胞(A)およびMOVP-3細胞(B)において解析し、結果をVP-16 (5x10-7 M)と比較した。細胞を表示されている時間(n=3)に収穫し、固定し、死細胞染色用蛍光色素 PI(PropidiumIodide)で着色し、材料及び方法に述べるようにFACSによって解析した。pre G1における細胞率(アポトーシス細胞)をDNAヒストグラムから計算した。
【0067】
図6は、マウスのProVP-16II治療における毒性および抗腫瘍効果を示している。A/Jマウス(n=6)に、1, 3, 5, 7, 9および11日目にProVP-16 II (20および60 mg/kg)、およびVP-16 (20 mg/kg)を、1, 3および5日目にVP-16 (60 mg/kg)を腹腔内投与した(A)。各動物の体重を長期にわたって測定し、0日目に測定した体重のパーセントとして算出した。Kaplan Maierプロットによって示されるように、20%以上の体重減少を重大事の発生と定義した。
【0068】
また、ProVP-16II治療の抗腫瘍効果を多剤耐性異種移植モデルにおいて調べた(B,C)。SCIDマウス(n=7)に5x106のMOVP-3細胞が皮下注射し、平均サイズ250 mm3の一次腫瘍を接種後55日目に確立した。治療は、腫瘍細胞接種後、55、57、59、69、71、87および90日目にPro VP-16 II (45および15 mg/kg), VP-16(15mg/kg)、および溶媒の腹腔内投与により行った。腫瘍の成長をマイクロノギスにて計測し、腫瘍サイズを材料及び方法に述べるようにして計算した(B)。ProVP-16 II (45および15 mg/kg)で治療した動物の実験群と対照群(溶媒およびVP-16)との間に見られた差異は、統計的に有意(63日後、p<0.001)であった(B)。治療を施した動物の体重を長期にわたって測定し、57日目の体重のパーセントとして算出した(C)。
【0069】
治療実験の最後に、残留するs.c.腫瘍を除去し、残留腫瘍のない45mg/kgで治療したマウスを除いて、MDR-1の遺伝子発現をRT-PCRによって解析した。各グループの腫瘍の典型的なシグナルが示され、陽性コントロールとして使用されたMDR-1発現VCR-100細胞と比較された。229もしくは127bpシグナルの存在は、MDR-1の発現を示唆している。
【0070】
本発明にかかるポドフィロトキシンは、細胞毒性の仲介(mediating)において特に効果的であることを証明した。また、エトポシドの種々の誘導体が合成され、加水分解を介してのそれらの活性化メカニズムが明らかになった(以下の実施例を参照)。重要なのは、生理緩衝液条件においてプロドラッグが安定に維持されることである。VP-16の4’ヒドロキシ基をブロックするため安定なカーバメートリンカー(carbamate linker)を有するエトポシドのプロドラッグが生理緩衝液条件において完全に安定であることはすでに示されている(Shabat D, Lode HN, Pertl U, etal. In vivo activity in a catalytic antibody-prodrug system: Antibody catalyzedetoposide prodrug activation for selective chemotherapy. Proc Natl Acad Sci U SA. 2001;98:7528-7533.)。しかし、ProVP-16 IおよびIIによって実証される本発明の加水分解活性プロドラッグとは対照的に、カーバメートプロドラッグは、触媒抗体38C2によって仲介される自然には起こらないretro-aldol retro-Michael反応の触媒作用によってのみ活性化されるように設計されている。強調すべきは、本発明のプロドラッグに対してこのカーバメートプロドラッグは調査したすべての腫瘍細胞株に対して細胞毒性を仲介する効果がなく、それ故に細胞毒性を導入する生物学的中心としてのVP-16の4’ヒドロキシ基の重要な役割を引き出すには到っていなかった。すなわち、ここに述べられたプロドラッグにおいては活性中心がブロックされるが、加水分解活性メカニズムを有する本発明にかかるプロドラッグは細胞毒性を非常に効果的に仲介する。転化されていないProVP-16 IおよびIIの無毒性は、Molt-3細胞(図3)に示される徐放メカニズムによって示されており、インキュベーションの12時間までのプロドラッグの細胞毒性効果の欠如によってVP-16と対照的である。これら両方のVP-16誘導体の長時間にわたる細胞毒性活性の安定した増加は、本発明の新規化合物が初期において安定で無毒性であるが、その後標的細胞の内部で活性になることを明確に示している。また、生体外で観察される徐放メカニズムは、ProVP-16IIが親化合物の最大許容投与量の3倍以上を許容することに示されるように、マウスにおける全身毒性の劇的な減少の説明となる(図6)。
【0071】
本発明にかかるプロドラッグのさらなる特徴は、VP-16に比較して多くのがん細胞群に高い効用を発揮することである。すなわち、MDR-1遺伝子発現の増幅をともなった細胞において(VCR 100, ADR 5000およびSW480)、本発明にかかるプロドラッグの3log以上の優れた効用が観察された(図4)。このような改善は、MDR-1modulatorsによって稀に達成される(Dalton WS. Mechanisms of drug resistance inhematologic malignancies. Semin Hematol. 1997;34:3-8; Sonneveld P, Durie BG,Lokhorst HM, et al. Modulation of multidrug-resistant multiple myeloma bycyclosporin. The Leukaemia Group of the EORTC and the HOVON. Lancet.1992;340:255-259; Joly P, Lallemand A, Oum'Hamed Z, Trentesaux C, Idoine O,Desplaces A. Effects of verapamil and S9788 on MDR-1 mRNA expression studied byin situ hybridization. Anticancer Res. 1996;16:3609-3614; Tai HL. Technologyevaluation: Valspodar, Novartis AG. Curr Opin Mol Ther. 2000;2:459-467; Kang Y,Perry RR. Effect of alpha-interferon on P-glycoprotein expression and functionand on verapamil modulation of doxorubicin resistance. CancerRes. 1994;54:2952-2958; Hofmann J, Gekeler V, Ise W, et al. Mechanismof action of dexniguldipine-HCl (B8509-035), a new potent modulator ofmultidrug resistance. Biochem Pharmacol. 1995;49:603-609.)。
【0072】
さらに、薬効評価は、本発明にかかるプロドラッグがMDR-1仲介による基質排出を抑制することを示している。すなわち、この新しいプロドラッグは、MDR-1p糖蛋白機能を抑制してMDR-1発現がん細胞株に対して優れた活性を発揮する。重要なのは、この生体外における劇的な効果は、生体内における薬剤耐性T細胞白血病異種移植モデルにおいて一次腫瘍を長期にわたって小さく保つ効果として認められることである。このモデルでは、MDR-1遺伝子発現が生体外においてVP-16に対して100x耐性で増幅され(図4)、結果的に最大許容投与量でのVP-16による治療効果が完全に失われた(図6)。興味深いことに、このように人工的に高い薬剤耐性を示すモデルにおいて、本発明にかかるプロドラッグでの治療によれば劇的な抗腫瘍効果を引き出すことができる。これは、上記のような高いMDR-1の増幅が再発悪性腫瘍における多剤化学療法を受けた患者に稀に観察されることから特に重要である(Beck J, Handgretinger R, DopferR, Klingebiel T, Niethammer D, Gekeler V. Expression of mdr1, mrp,topoisomerase II alpha/beta, and cyclin A in primary or relapsed states ofacute lymphoblastic leukaemias. Br J Haematol. 1995;89:356-363; Beck JF, BohnetB, Brugger D, et al. Expression analysis of protein kinase C isozymes andmultidrug resistance associated genes in ovarian cancer cells. Anticancer Res.1998;18:701-705.)。
【0073】
この研究の重要な発見は、生体内および生体外において、本発明のプロドラッグにより多剤耐性モデルで高い抗腫瘍効果が観察され、それらを用いた治療が従来のポドフィロトキシンを用いた化学療法に特筆すべき改善をもたらす点にある。
【0074】
以下の実施例は例示を意図するものであり、本発明の範囲を制限するものではない。
【実施例1】
【0075】
材料:2,3-ビス(2-メトキシ-4-ニトロ-5-スルフォフェニル)-5-[(フェニルアミノ)カルボニル]-2H-テトラゾリウム水酸化物(XTT)( 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazoliumhydroxide (XTT))、VP-16, ソルケタール(solketal)、有機溶媒、および電子捕獲剤(phenazine methosulfate (PMS))がSigma-Aldrich社(Deisenhofen, Germany)から得た。細胞培養試薬。制限酵素、その他の分子生物学用試薬は、Life Technologies社(Karlsruhe, Germany)から得た。
【0076】
細胞:腫瘍細胞群は、100IU/mlのペニシリン/ストレプトマイシン(P/S)の存在下、RPMI,10% FCS (Nalm-6, Reh, Molt-3, Jurkat, HL-60, K562, HeLa, CEM, A2780, SW480)あるいはDMEM, 10% FCS (NXS2, SK-N-SH, HT-29)において成長させ、標準的な組織培養条件(5% CO2, 37oC)において増殖させた。先に述べたNXS2(Lode HN, Xiang R, Varki NM,Dolman CS, Gillies SD, Reisfeld RA. Targeted interleukin-2 therapy forspontaneous neuroblastoma metastases to bone marrow. Journal of the NationalCancer Institute. 1997;89:1586-1594.)、およびJames Beck氏(Greifswald,Germany)により提供されたVCR100, ADR5000およびA2780(Beck J, Handgretinger R,Dopfer R, Klingebiel T, Niethammer D, Gekeler V. Expression of mdr1, mrp,topoisomerase II alpha/beta, and cyclin A in primary or relapsed states ofacute lymphoblastic leukaemias. Br J Haematol. 1995;89:356-363; Beck JF, BohnetB, Brugger D, et al. Expression analysis of protein kinase C isozymes andmultidrug resistance associated genes in ovarian cancer cells. Anticancer Res.1998;18:701-705.)を除くすべての細胞株はATCC, Rockville, MDから得た。多量のVP-16に連続曝露することによりエトポシド耐性subline MOVP-3を生成するためMolt-3細胞を使用した。6ヶ月経過後、MOVP-3細胞は1μM VP-16の存在下において安定に増殖し、これを生体外および生体内実験に使用した。
【0077】
マウス:ドイツのSulzfeldのCharles River研究所から年齢8週間のメスのA/JマウスおよびFOXCHASETM C.B-17/lcrCrl-scid BRマウスを得た。これらのマウスを我々の施設で無菌マウスコロニー内に8匹づつのグループとなるように収容した。マウスには適宜標準的なマウス実験用食を与えた。尚、動物実験は、研究用動物の使用と配慮に関するドイツ指針、すなわち、"Tierschutzgesetz"に基づいて行った。
【0078】
プロエトポシドの分析化学:プロエトポシドの合成およびHPLCによる解析については、Wrasidlo W, Schroeder U, Bernt K, et al. Synthesis, hydrolyticactivation and cytotoxicity of etoposide prodrugs. Bioorg Med Chem Lett. 2002;12. 557-560にすでに報告されており、その全文を参考のためこの明細書に添付する。
【0079】
RNA分離、逆転写、PCR増幅:total cellular RNA、cDNA合成およびRT-PCR条件は、前に記載されている(Lode HN, Xiang R, Varki NM, Dolman CS, Gillies SD, Reisfeld RA. Targetedinterleukin-2 therapy for spontaneous neuroblastoma metastases to bone marrow.Journal of the National Cancer Institute. 1997;89:1586-1594.)。ヒトMDR-1の増幅を、センス5’ GGA GAG ATC CTC ACC AAG CG 3’およびアンチセンス5’GTT GCC AAC CAT AGA TGA AGG 3’を用いて35サイクル(15s 96oC, 30 s60oC, 90 s 72oC)で行い、229bp断片指定MDR-1を得た。また、21サイクル後、センス5’ GCT CAG ACA GGATGT GAG TT 3’およびアンチセンス5’ CTG GGT AAT TAC AGC AAG CC 3’を用いて30サイクルで1.0ml MDR-1のさらに狭い領域を増幅(nestedamplification)することにより高感度検出を行い、127bp断片を得た。また、センス5’ CGGGAA GCT TGT GAT CAA TGG 3’およびアンチセンス5’ GGC AGT GAT GGCATG GAC TG 3’を用いて25サイクルでグリセロール−アルデヒド−リン酸デヒドロゲナーゼ(glycerol-aldehyde-phosphate-dehydrogenase:GAPDH)を増幅することによりcDNA完全性(cDNA integrity)を検証し、358bp断片を得た。すべての断片の特異性は、シークエンシングによって検証した。
【0080】
薬効JC-1評価:染色のため、細胞は2度洗浄され、前に述べたようにJC-1単量体含有PBSに再懸濁した(Legrand O, Perrot JY,Simonin G, Baudard M, Marie JP. JC-1: a very sensitive fluorescent probe totest Pgp activity in adult acute myeloid leukemia. Blood.)。簡単に言えば、0.1 μM JC-1単量体が5x105/mlの細胞数で15分間37℃でインキュベートされ、エトポシドおよびPro-VP-16 IおよびIIの存在する条件および存在しない条件においてインキュベートされた。2μMシクロスポリンAでインキュベートしたサンプルを陽性コントロールとして使用した(データは表示されない)。FACSortフローサイトメーター(BectonDickinson)を用いて細胞蛍光が記録され、JC-1シグナルは、色素単量体の解析用FL1-channel (530 nm フィルター)に検出された。
【0081】
MDR-1の安定トランスフェクション:MDR-1cDNAは、ハンブルグ大学のC. Baum氏によりpUCプラスミド内に提供された。哺乳類発現のため、NotIおよびBamHI制限酵素認識部位を用いて2シストロン性真核細胞用発現プラスミド(bicistronic eukaryotic expression plasmid)pIRESpuroにMDR-1cDNAをクローニングした。pMDR-IRESpuroは、エレクトロポレーション法(960 μF, 250V)によってMolt-3細胞内にトランスフェクトされた。安定トランスフェクタントは、300 ng/mlのピューロマイシで選択された。MDR-1発現は、1μg/ml MDR-1特異mAb(C. Baum, ハンブルグ大学)を用いて、FACS-解析(FACS-calibur,Becton Dickinson, Bedford, MA)により調べた。
【0082】
細胞毒性検査:細胞毒性は、これまでにも報告されているように(Scudiero DA, Shoemaker RH,Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cellgrowth and drug sensitivity in culture using human and other tumor cell lines.Cancer Res. 1988;48:4827-4833.)、XTTテトラゾリウム/ホルマザン評価法(XTT tetrazolium/formazan assay)によって調べた。すなわち、100 μl培地中、104個/ウェルの密度で96の平底ウェルプレートに細胞を蒔き、10-4から10-12Mの範囲の薬剤濃度に曝した。所定の時刻(6〜72時間)において、細胞の生死判別評価を37°Cで4時間インキュベートされた0.2%v/v PMS (PBS中、1.53 mg/ml)で活性化した50μlXTT試薬(無血清RPMI中、1mg/ml in)を添加することにより行った。プレートは、450 nmでThermomax (Molecular Devices社)において解析された。OD値は、薬剤濃度の関数としてプロットされ、softmax softwareを用いて曲線を積分してIC50濃度値を得た。
【0083】
細胞周期分析:細胞周期の特定の時期は、蛍光色素PIを用いた標準的な評価法により調べた。すなわち、24のウェルプレートに105/ウェルで細胞を蒔き、VP-16もしくはProVP-16 IおよびIIでインキュベートした。所定の時刻に細胞を収穫し、4.5 ml エタノール(75%, -20°C)に少なくとも12時間(4°C)固定した。PBS(pH 7.4)内において細胞を洗浄し、RNAse (0.3 mg/ml)および蛍光色素(50 μg/ml)を含有する250 μlのPBS中に再懸濁し、暗室(30min, RT)でインキュベートした。その後、細胞周期の特定の時期を定義するDNAヒストグラムのFACS解析を2度繰り返し、平均した結果をパーセントで表した。
【0084】
マウスにおける毒性研究:10mMエトポシドおよびプロエトポシドの原液を50:50 v/v Cremophor:エタノールに調合し、PBS(pH 7.4)に最終濃度に希釈した。エトポシドあるいはプロエトポシドのいずれかを20 mg/kgあるいは60 mg/kg、あるいは溶媒コントロール(25% DMSO, 12,5% エタノール, 12,5% Cremophor, 50%PBS)を年齢8〜10週間のA/Jマウスに、1, 3, 5, 7, 9および11日目に腹腔内投与した。VP-16 (60 mg/kg)は1,3および5日目にのみ注射した。体重およびその生存を長期にわたり観察した。
【0085】
T−細胞白血病異種移植モデルにおける抗腫瘍効果:100 μl PBS (pH 7.4)中5x106のMOVP 3細胞を各SCIDマウスの左脇腹の皮膚に注射することにより一次腫瘍を導入した。注射後55日目において、一次腫瘍の確立は明白であった。溶媒(25% DMSO, 12,5% エタノール, 12,5% cremophor, 50%PBS), VP-16 (15mg/kg)およびPro VP-16 II (15および45 mg/kg)を全体で200μl腹腔内注射することによりマウス(n=8)に治療を施した。各マウスは、腫瘍細胞接種後55、57、59、69,71、87、90日目の計7回の注射を受けた。一次腫瘍のサイズはマイクロノギスにより長期にわたって測定し、その体積は1/2 x 幅2 x 長さに基づいて算出した。また、体重は標準的なデジタル重量計で測定した。
【0086】
統計値:動物実験群の間における異なる所見の統計的な有意性を両側検定(two-tailed Student’s t test)によって調べ、両側検定p値(two-tailedp values)が<0.01であれば、有意性有りとみなした。
【0087】
エトポシドプロドラッグ活性化の化学:ProVP16-IおよびIIが実際にプロドラッグであることを実証するために、まずHPLCによって生体外におけるそれらの活性化特性を調べた(図1、2)。すなわち、ProVP-16IのProVP-16IIへの転化およびその後のVP-16の放出は2段階活性化機構に従う(図1)。まず、ProVP-16 Iは、酸性条件(THF, 2N HCL)下でのグリコシド(glycoside)部位の一部(約10%)の分解で2時間以内に2,2-ジヒドロキシプロパン(2,2-dihydroxypropane)の除去を伴ってProVP-16 IIに転化する(図2A)。次に、ProVP-16 IIは、塩基性pH条件下でグリセリンの除去を伴ってVP-16に加水分解する(図1、図2B)。すべての実験において、転化−時間曲線は一次反応速度論(first order kinetics)を示した。ここで重要なのは、ProVP-16 IIは生理緩衝液条件(PBS, pH 7.4, 37°C)下において5〜18時間まで<5%の転化率で安定であり、ProVP-16 IはPBS (pH 7.4, 37°C)において測定可能な転化は認められず完全に安定であった。また、ProVP-16 IIとは対照的に、ProVP-16 Iは塩基性緩衝液条件(pH £ 10.0)下でも不活性であった。ProVP-16 Iのこの独特の加水分解安定性は、この分子の”entire southern region”の疎水性、程度はさほどではないもののカーボネート部位を囲む2つのオルトメトキシ(ortho methoxy)基の立体障害に起因している。
【0088】
本発明にかかるプロドラッグの実用性は、自然に存在する触媒の存在下で活性化、すなわち加水分解され、しかもpH値の広い範囲にわたって水溶液中で安定であるという事実からも明らかである。
【0089】
ProPV-16 IおよびIIの活性化は、転化半減期がそれぞれ750.8 minおよび56.1 minである血清の存在下で起こる。また、豚肝臓カルボキシルエステラーゼ(Porcine liver caboxylesterase)は、ProPV-16 IおよびIIのVP-16への転化をそれぞれ14.2 minおよび514.1 minの転化半減期で仲介した。これらの所見は、カルボキシルエステラーゼによるpH7.4での酵素性プロドラッグ活性化を明確に示している(表1)。
【0090】
【表1】
【実施例2】
【0091】
白血病およびがん細胞株に対するProVP-16I および IIの活性度:これらの2つのプロエトポシドの細胞毒性に関する試験を、XTT生体染色抗増殖性評価法(XTT vital stain antiproliferation assay)を用いて腫瘍細胞群のパネルに対して行った(図3A)。試験に用いたヒト腫瘍細胞群の大部分に関して、両方のプロエトポシドとも、SW480大腸癌で1000倍低いIC50値, HT-29大腸癌、ADR5000 卵巣癌およびHL-60 pre B−細胞白血病で100-1000低いIC50値, HeLa子宮頸癌, A2780卵巣癌, K562慢性骨髄性白血病, Jurkat T−細胞白血病、およびSK-N-SH神経芽細胞腫で10-100 倍低いIC50値, およびMolt-3 T−細胞白血病,RehおよびNalm-6 pre B−細胞白血病、VCR100T−細胞性リンパ芽球性白血病およびCEM T−細胞性リンパ芽球性白血病細胞で2-10倍低いIC50値で親化合物よりも十分に活性であった。NXS2マウス神経芽細胞腫(図3A), CHO中国ハムスター卵巣,およびHammsヒト大腸癌細胞の3つの細胞群のみエトポシドとプロエトポシドに等しく良好に反応した。MDR-1発現の増幅を伴ったドキソルビシン耐性ADR5000およびビンクリスチン耐性VCR100の2つの細胞群においては、無耐性親細胞群A2780およびCEMに比べ両プロエトポシドのより高い細胞毒性効果が見られた。
【0092】
Pro-VP16 Iの細胞毒性作用の時間的経過を、Molt-3T−細胞白血病細胞の使用して追跡した(図3B)。結果は、Pro-VP16 Iによる遅発性の細胞毒性作用がインキュベーションの24時間後に始まることを示している。この時点において、親VP-16の細胞毒性効果はすでにほとんど完全に確立されている。ProVP16 Iの細胞毒性効果は、インキュベーション(IC501.0x10-8)後、48〜72時間後に完了し、VP-16(IC506.5 x10-8 M)のそれを越えた。同様の結果は、ProVP-16 IIでも観察された(データは表示されない)。これらの知見は、ProVP-16 IおよびIIによる細胞毒性活動の徐放(slow release)機構を示しており、プロドラッグのコンセプトに完全に合致している。
【実施例3】
【0093】
多剤耐性細胞におけるProVP-16I および IIの効果:ProVP-16 IおよびIIが、自然にあるMDR発現細胞群においてVP-16よりもさらに効果的であるという所見に基づいて(図3)、論点はこれらのプロドラッグが生体外における人工的なMDRを克服可能かどうかに向けられた。この目的のため、MDR-1ネガティブ細胞群Molt-3を使用して耐性サブクローンMOVP-3を生成した(図4A)。MOVP-3細胞におけるMDR-1 mRNA発現をRT-PCRによって調べ(図4A)、一方、細胞表面におけるMDR-1蛋白発現の増加をUIC2モノクローナル抗体を用いてFACS-解析により明らかにした。Bax およびBcl-2に加えてMRP, LRP, トポイソメラーゼI, IIαおよびIIβをさらに調べる拡張された遺伝子発現解析は(データを表示しない)、Molt-3に対比してMOVP-3細胞における薬剤耐性を説明する唯一の差はMDR-1の発現であることを明らかにした。これは、MOVP-3のIC50値が2x10-6 Mで、Molt-3細胞のIC50値が2x10-8 Mでエトポシドに対する100倍耐性を示したsublineの機能解析によってさらに確認された(図4B)。しかしながら、MOVP-3は、ProVP16IおよびIIへの感受性は十分に維持しており、両プロドラッグのIC50値が2x10-8 Mであり、親Molt-3細胞と薬剤耐性MOVP-3細胞の間でプロエトポシド仲介細胞毒性に有意な差異は認められなかった。同様の結果は、エトポシド、ドキソルビシン、パクリタキセルおよびビンブラスチンに対する耐性を生じたpMDR-IRESpuro (Molt-3/MDR-1)を使用してMDR-1 cDNAの安定トランスフェクションを施したMolt-3細胞においても得られた(データは表示されない)。また、Molt-3/MDR-1細胞は、ProVP-16 IおよびIIに対する十分な感受性を有した。エンプティーベクター(pIRESpuro)あるいはGFP含有ベクターで安定トランスフェクトされたコントロールは、試験したいかなる薬剤にも耐性を示さなかった。
【0094】
さらに、MOVP-3sublineにおける耐性のタイプを交差耐性を評価することにより調べた。MOVP-3細胞は、非MDR-1薬剤(MTX,5-FU, ゲニスチン, カリーチアマイシンq)とは対照的に、すべてのMDR-1タイプの薬剤(エトポシド、ドキソルビシン、パクリタキセル, ビンブラスチン)に対して交差耐性を示した(図4C)。これらの所見は、ProVP-16 IおよびIIが生体外においてMDR-1仲介多剤耐性を克服できることを明確に示している。
【0095】
JC-1色素を用いた機能性MDR-1評価においては、Molt-3親細胞と対比して耐性sublineにおけるFL-1信号の減少によって示されるように(図4D)、MOVP-3細胞におけるJC-1排出の増加はMolt-3コントロールとは対照的であった。この減少は、3x10-4 MのPro-VP-16 Iでの共インキュベーション(coincubation)によって排除され(図4D)、10x10-6 M(データは表示されない)までの広い濃度範囲にわたってMDR-1仲介排出を抑制した。興味深いことに、等モル濃度で使用されたVP-16は、PBSコントロールとほぼ同じJC-1信号によって示されるように、MDR-1機能の制御において効果がなかった。これらの知見は、プロドラッグ設計がMDR-1仲介基質排出を直接的に減少させることを明確に示している。
【0096】
多剤耐性MOVP-3細胞においてProVP-16 IおよびIIによって仲介される細胞毒性メカニズムを評価するため、細胞周期へのこれらの薬剤の影響を10nMから1 μMの範囲の濃度において解析するとともに、Molt-3親細胞と比較した。0.5μMのプロドラッグで得られた典型的な結果を図5に示す。具体的には、非同調MOVP-3 (図5B)およびMolt-3(図5A)細胞を72時間VP-16, ProVP-16 IおよびIIとインキュベートした。表示されている時刻において、定期的に細胞周期解析(cellcycle analysis)を行った。その結果、全濃度範囲(10nM-1 μM)と同様に(データは表示されない)、0.5μM(図5B)のPro VP-16 IおよびIIは、24時間後MOVP-3細胞内のpre-G1ピークの増加、すなわち、アポトーシス特性の導入に非常に効果的であることがわかった。プロドラッグとは対照的に、VP-16はアポトーシスの導入に効果がなかった。これは、MDR-1によってVP-16は排除されるが、ProVP-16 IおよびIIは排除されないことと合致する。また、Molt-3親細胞においても全濃度範囲(0.5 μM, 図5A)にわたってアポトーシスの導入が実証された。この場合は、VP-16がProVP-16 IおよびIIとほぼ同じ程度にMolt-3細胞にアポトーシスを導入することがわかった。全体的にみれば、これらの結果はProVP-16IおよびIIはMDR-1の基質ではないことを示唆している。
【実施例4】
【0097】
多剤耐性T−細胞白血病異種移植モデルにおけるProVP-16 I および IIの生体内毒性および有効性:ProVP-16 IおよびII間には差がないことを示した体外所見に基づいて、ProVP-16 IIをその高い水溶性を理由に生体内実験用として選択した。まず、ProVP-16IIの全身毒性をVP-16およびProVP-16 II (図6A)を腹腔内投与したA/Jマウス(n=6)において調べた。20 mg/kgのVP-16を投与されたすべてのマウスは、10%の平均体重減少をともなって生存した。一方、60 mg/kgのVP-16で治療したマウスの5/6は>20%の体重減少を示した。これらの知見は、ProVP-16 IIで得た結果とは極めて対照的であった。すなわち、20および60 mg/kgのProVP-16IIを投与は、いずれの実験群においても死を伴うことなくうまく受け入れられた。60 mg/kgのProVP-16 IIを投与したマウスのみに一時的に<10%の体重減少がみられたが、20mg/kgのPro VP-16 IIの投与では平均体重が安定に維持された。このように、<20%の体重減少によって定義される最大許容投与量は、VP-16において20 mg/kg、ProVP-16IIにおいて60 mg/kgであり、プロドラッグ設計によって全身毒性を少なくとも1/3に減らせることを示している。
【0098】
次に、ProVP-16 IIの抗腫瘍効果をT−細胞白血病の多剤耐性異種移植モデルにおいて調べ、VP-16と比較した。5x106の多剤耐性MOVP-3細胞の皮下投与によって一次腫瘍を導入し、腫瘍の成長を105日間にわたって追跡した。腫瘍細胞接種後55日目に腹腔内投与により治療を開始した。この時、腫瘍の平均サイズは250 mm3であった。全身毒性をさらに低減するため図6Aに示される結果に基づいて、VP-16 (15 mg/kg)およびProVP-16 II (15および45 mg/kg)の投与量を選択した。45 mg/kgのProVP-16 IIでの治療は、治療開始後10日目において7/7の動物に一次腫瘍の縮小をもたらし、それは2ヶ月間にわたって安定であった(図6B)。この治療は、一時的に6%の体重減少を引き起こしただけでうまく受け入れられた(図6C)。この所見は、抗腫瘍効果が見られず、溶媒のみで治療したコントロールマウスと同様に連続的に一次腫瘍の成長が認められた15 mg/kgのVP-16で治療したマウスの観察結果とは対照的であった。また、15 mg/kgのVP-16で治療したマウスには、一時的にではあるが20%の平均体重減少が伴い、重大な細胞毒性が観察された。一方、15 mg/kgのProVP-16 IIを投与したマウスは、測定可能な体重減少を示すことなく(図6C)、15 mg/kgのVP-16で治療したマウスとは対照的に、一次腫瘍成長に劇的な減少をもたらした。MDR-1発現が実験期間中、安定に残存していたかどうかを調べるために、105日目の腫瘍外植片からRNAが分離された。RT-PCRにより、調べたすべての腫瘍がMDR-1シグナルを示した(図6D)。
【0099】
明細書、請求項および/あるいは添付図面に開示された本発明の特徴は、個々におよびその組み合わせにおいて本発明の種々の形態を実現するための材料となる。
【特許請求の範囲】
【請求項1】
以下の化学式(I)によって示されるポドフィロトキシン
【化1】
pは2〜100の整数であり、
Bは以下の化学式(II)によって示される:
【化2】
しかるに、Xは、O、S、およびNR”を含む群から選択され、Yは、O、S、およびNR”を含む群から選択され、R”=アルキル、アリルあるいはHであり、
nは0〜6の整数であり、
Zは、エチレングリコール、プロピレングリコール、グリセロール、ペンタエリスリトール、ポリエチレングリコール、および以下の化学式(III)によって示される化合物を含む群から選択されるポリヒドロキシアルキル基であり、
【化3】
あるいは、Zは、付加的にジオキソラン基が結合した上記のポリヒドロキシアルキル基であり、
あるいは、Zは、哺乳類のレセプタの標的部位(targetting moieties)、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター探索機能(tumor cell associated receptor finding function)を有するペプチドおよびタンパク質を含む群から選択される。
【請求項2】
上記ジオキソラン基は、2, 2-ジアルキル-1, 3-ジオキソランを含む群から選択され、2位置での各アルキルは、無置換および置換メチル、エチル、プロピル、ブチル、ペンチル、ヘキシルを含む群から独立に選択されることを特徴とする請求項1に記載のポドフィロトキシン。
【請求項3】
Zは、哺乳類の膜表面レセプタあるいは核内レセプタの標的部位であり、あるいはZは、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター結合機能(tumor cell associated receptor binding function)を有するペプチドあるいはタンパク質であることを特徴とする請求項1乃至2に記載のポドフィロトキシン。
【請求項4】
Zは、ステロイド、成長因子レセプター阻害タンパク質、模倣性非ペプチドおよびペプチドを含む群から選択されることを特徴とする請求項3に記載のポドフィロトキシン。
【請求項5】
Aは、化学式(IV)によって示される化合物を含む群から選択され、
【化4】
しかるに、R1およびR2は各々C1-C10アルキルであり、あるいはR1およびR2およびそれらが結合される炭素とがC5-C6シクロアルキルを示し、あるいはR1がHであり、R2はC1-10アルキル, C2-10アルケニル, C3-5シクロアルキル, フリル, チエニル, C6-10アリルおよびC7-14アラルキルを含む群から選択されることを特徴とする先行する請求項のいずれかに記載のポドフィロトキシン。
【請求項6】
R1がHであり、R2はメチルもしくはチエニルであることを特徴とする請求項5に記載のポドフィロトキシン。
【請求項7】
XがOであり、YがOであることを特徴とする先行する請求項のいずれかに記載のポドフィロトキシン。
【請求項8】
XがOであり、YがSであることを特徴とする請求項1乃至6のいずれかに記載のポドフィロトキシン。
【請求項9】
XがOであり、YがNHであることを特徴とする請求項1乃至6のいずれかに記載のポドフィロトキシン。
【請求項10】
ポドフィロトキシンは、以下の群から選択されることを特徴とする請求項1乃至6および8のいずれかに記載のポドフィロトキシン。
【化5】
【請求項11】
ポドフィロトキシンは、以下の群から選択され、
【化6】
Zは請求項1と同様に定義されることを特徴とする請求項1乃至6および9のいずれかに記載のポドフィロトキシン。
【請求項12】
ポドフィロトキシンは、以下の群から選択されることを特徴とする請求項11に記載のポドフィロトキシン。
【化7】
【請求項1】
以下の化学式(I)によって示されるポドフィロトキシン
【化1】
pは2〜100の整数であり、
Bは以下の化学式(II)によって示される:
【化2】
しかるに、Xは、O、S、およびNR”を含む群から選択され、Yは、O、S、およびNR”を含む群から選択され、R”=アルキル、アリルあるいはHであり、
nは0〜6の整数であり、
Zは、エチレングリコール、プロピレングリコール、グリセロール、ペンタエリスリトール、ポリエチレングリコール、および以下の化学式(III)によって示される化合物を含む群から選択されるポリヒドロキシアルキル基であり、
【化3】
あるいは、Zは、付加的にジオキソラン基が結合した上記のポリヒドロキシアルキル基であり、
あるいは、Zは、哺乳類のレセプタの標的部位(targetting moieties)、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター探索機能(tumor cell associated receptor finding function)を有するペプチドおよびタンパク質を含む群から選択される。
【請求項2】
上記ジオキソラン基は、2, 2-ジアルキル-1, 3-ジオキソランを含む群から選択され、2位置での各アルキルは、無置換および置換メチル、エチル、プロピル、ブチル、ペンチル、ヘキシルを含む群から独立に選択されることを特徴とする請求項1に記載のポドフィロトキシン。
【請求項3】
Zは、哺乳類の膜表面レセプタあるいは核内レセプタの標的部位であり、あるいはZは、抗体、ステロイド、トランスフェリン、腫瘍細胞関連レセプター結合機能(tumor cell associated receptor binding function)を有するペプチドあるいはタンパク質であることを特徴とする請求項1乃至2に記載のポドフィロトキシン。
【請求項4】
Zは、ステロイド、成長因子レセプター阻害タンパク質、模倣性非ペプチドおよびペプチドを含む群から選択されることを特徴とする請求項3に記載のポドフィロトキシン。
【請求項5】
Aは、化学式(IV)によって示される化合物を含む群から選択され、
【化4】
しかるに、R1およびR2は各々C1-C10アルキルであり、あるいはR1およびR2およびそれらが結合される炭素とがC5-C6シクロアルキルを示し、あるいはR1がHであり、R2はC1-10アルキル, C2-10アルケニル, C3-5シクロアルキル, フリル, チエニル, C6-10アリルおよびC7-14アラルキルを含む群から選択されることを特徴とする先行する請求項のいずれかに記載のポドフィロトキシン。
【請求項6】
R1がHであり、R2はメチルもしくはチエニルであることを特徴とする請求項5に記載のポドフィロトキシン。
【請求項7】
XがOであり、YがOであることを特徴とする先行する請求項のいずれかに記載のポドフィロトキシン。
【請求項8】
XがOであり、YがSであることを特徴とする請求項1乃至6のいずれかに記載のポドフィロトキシン。
【請求項9】
XがOであり、YがNHであることを特徴とする請求項1乃至6のいずれかに記載のポドフィロトキシン。
【請求項10】
ポドフィロトキシンは、以下の群から選択されることを特徴とする請求項1乃至6および8のいずれかに記載のポドフィロトキシン。
【化5】
【請求項11】
ポドフィロトキシンは、以下の群から選択され、
【化6】
Zは請求項1と同様に定義されることを特徴とする請求項1乃至6および9のいずれかに記載のポドフィロトキシン。
【請求項12】
ポドフィロトキシンは、以下の群から選択されることを特徴とする請求項11に記載のポドフィロトキシン。
【化7】
【図1】

【図2】
【図3】
【図5】
【図4】
【図6】
【図2】
【図3】
【図5】
【図4】
【図6】
【公開番号】特開2010−120943(P2010−120943A)
【公開日】平成22年6月3日(2010.6.3)
【国際特許分類】
【出願番号】特願2009−269930(P2009−269930)
【出願日】平成21年11月27日(2009.11.27)
【分割の表示】特願2003−549356(P2003−549356)の分割
【原出願日】平成14年12月3日(2002.12.3)
【出願人】(504214431)ウニベルジテートスクリーニクム シャリテー デア フンボルト−ウニベルジテート ツー ベルリン テクノロジー トランスファーステラ (1)
【Fターム(参考)】
【公開日】平成22年6月3日(2010.6.3)
【国際特許分類】
【出願日】平成21年11月27日(2009.11.27)
【分割の表示】特願2003−549356(P2003−549356)の分割
【原出願日】平成14年12月3日(2002.12.3)
【出願人】(504214431)ウニベルジテートスクリーニクム シャリテー デア フンボルト−ウニベルジテート ツー ベルリン テクノロジー トランスファーステラ (1)
【Fターム(参考)】
[ Back to top ]