
ポリ−β−1→4−N−アセチルグルコサミン
【課題】 その性質を高度に再現できて、しかも簡単に製造できる、純粋な、精製されたキチンおよびキトサンの調製物を提供する。
【解決手段】 タンパク質を含まず、他の有機汚染物質を実質的に含まず、無機汚染物質を実質的に含まない、β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含んでなり、約800,000ダルトン〜約3,000万ダルトンの分子量を有する微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【解決手段】 タンパク質を含まず、他の有機汚染物質を実質的に含まず、無機汚染物質を実質的に含まない、β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含んでなり、約800,000ダルトン〜約3,000万ダルトンの分子量を有する微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【発明の詳細な説明】
【技術分野】
【0001】
1. 序論
本発明は、第一に、簡単に調製および精製されたポリ-β-1→4-N-アセチルグルコサミン (p-GlcNAc) 多糖種に関するものである。本発明のp-GlcNAcは、その構成成分の単糖がβ-1→4 配置で結合されている高分子量のポリマーであって、タンパク質を含まず、また単一アミノ酸や他の有機および無機の汚染物質を実質的に含まないものである。加えて、p-GlcNAcの誘導体および再構成物も記述される。本発明はさらに、微小藻類(好ましくは珪藻植物)出発物質源から本発明のp-GlcNAcを精製する方法に関する。さらにまた、本発明はp-GlcNAcを誘導体化しかつ再構成する方法に関する。その上に、本発明は純化p-GlcNAc、その誘導体および/またはその再構成物の使用に関するものである。
【背景技術】
【0002】
2. 発明の背景
今日では、p-GlcNAcから部分的に構成された多糖類の特性、活性および用途に関する文献は膨大な数にのぼる。こうした物質の仲間は総称して「キチン」と呼ばれており、一方、脱アセチル化されたキチン誘導体は「キトサン」と呼ばれている。これらの用語が最初に用いられたころ、つまり1823年頃、キチンは完全にアセチル化されたもので、キトサンは完全に脱アセチル化された組成のものであるといったように、キチンとキトサンは別個の、明確に規定された、特異で、不変の化学種として常に自然界に存在すると考えられていた。しかし、「キチン」と「キトサン」が実際には非常にあいまいな用語であるということが分かったのは約1世紀後であった。これらの用語は、明確に規定された化合物と言うよりもむしろ、実際には広範に異なる物理的および化学的性質を示す化合物のファミリーを指す用語である。これらの差異は生産物のさまざまな分子量、異なるアセチル化度、そして共有結合された種特異的タンパク質、単一アミノ酸、無機汚染物質といった汚染物質の存在によるものである。今日でさえ、「キチン」と「キトサン」はあいまいに用いられており、実際には、あまり明確には規定されていない、多くの異なる化合物の混合物を指している。
【0003】
例えば、甲殻綱の外殻、菌類の菌蓋などの従来の供給源から単離された「キチン」の特性は予測し得ないほどにバラツキがあった。このようなバラツキは、種の違いばかりでなく、「キチン」産生種の生化学的特性の一部を決定するさまざまな環境的および季節的影響が原因となっている。事実、原料の予測し得ない可変性がキチンを基礎とした産業の進展を大いに阻んでいる。
【0004】
現在、純粋な、完全にアセチル化されたp-GlcNAc、すなわち有機または無機不純物によって汚染されていない生産物、を出発物質源から単離および調製したとする報告は科学文献中にまったく見当たらない。McLachlan ら (非特許文献1:McLachlan, A.G. ら, 1965, Can. J. Botany 43:707-713)はキチンの単離を報告したものの、その後の研究で、得られた「純化」物質は実際にはタンパク質と他の汚染物質を含んでいたことが判明した。
【0005】
脱アセチル化および部分脱アセチル化されたキチン調製物は、高い反応性、濃密な陽電荷、強力な金属キレート化能、タンパク質を共有結合する能力、さらに多くの水性溶媒への溶解性といった有益と思われる化学的性質を示す。ところが、上述したように、これらの調製物の予測できない可変性のため、こうした不均一化合物の有用性は厳しい制限を受ける。例えば、現在入手できる「キチン」および「キトサン」は再現性のないデータおよび許容できないほどに広い実験結果の変動をもたらす。さらに、入手可能な調製物はとても均質または純粋とは言えず、また、調製物の構成成分もこうした調製物が諸用途(特に医学用途)に受け入れられるほどに十分に再現性があるとは言えない。かくして、その性質を高度に再現できて、しかも簡単に製造できる、純粋な、精製されたキチンおよびキトサンの調製物は、非常に望まれているにもかかわらず、目下のところ存在していない。
【非特許文献1】マクラクラン・エー・ジー(McLachlan, A.G.)ら、カナディアン・ジャーナル・オブ・ボタニー(Canadian Journal of Botany, Can. J. Botany)、1965年、第43巻、p.707-713
【発明の開示】
【0006】
3. 発明の概要
本発明は、第一に、単離されかつ簡単に調製された純粋なp-GlcNAc種に関する。本発明のp-GlcNAcは、その構成成分の単糖がβ-1→4 配置で結合されている高分子量のポリマーであって、タンパク質を含まず、また他の有機および無機の汚染物質を実質的に含まないものである。
【0007】
本発明の重要性は、予測できない原料の変動性の問題を解決できた点にある。初めて、生物医学的に純粋で、一致した性質を示す高分子量のp-GlcNAcを簡便な手段でかつ商業規模で調製することが可能となった。本発明において調製された物質は高度に結晶性であり、規定培地で増殖させた多数の海洋微小藻類(好ましくは珪藻植物)のうちの1種の注意深く制御された無菌培養物から得られる。
【0008】
本発明はさらに、p-GlcNAcの誘導体および再構成物ならびに前記誘導体および再構成物の製造方法を提供する。かかる誘導体はポリグルコサミンおよびその誘導体を含むがこれらに限らず、また再構成物は膜、フィラメント、不織繊維、スポンジおよび三次元マトリックスを含むがこれらに限らない。さらにまた、本発明は、微小藻類(好ましくは珪藻植物)源からの本発明のp-GlcNAcの精製方法に関する。その上に、本発明は精製p-GlcNAc、その誘導体および/またはその再構成物の使用に関する。中でも、これらは生物医学、医薬品および化粧品産業(すべて最高純度の出発物質を必要とする)のような産業に関係する新規な商業用途において使用される。例えば、本発明のp-GlcNAc物質は制御可能な生分解性を示すように構成することができ、さらに、徐放性ドラッグデリバリーシステムの一部として、細胞カプセル化システムとして、術後癒着の防止処置として使用することができる。
【0009】
4. 図面の説明
図1. 100% p-GlcNAc の化学構造。「n」は約4,000 〜150,000 の範囲の整数を表し、約4,000 〜15,000が好適である。
【0010】
図2. p-GlcNAcの炭水化物分析、ガスクロマトグラフィー−質量分光分析データ。黒の四角は、以下の第5.3.2 節に記載した、化学/生物学的方法の酸処理/中和変法を用いて精製されたp-GlcNAcを表す。
【0011】
図3A. 純化p-GlcNAcの固体膜 (solid membrane) の円偏光二色性スペクトル。
【0012】
図3B. 脱アセチル化p-GlcNAcの固体膜の円偏光二色性スペクトル。純化p-GlcNAcで観測された211 nmの最小値および195 nmの最大値(図3A)の消失は、以下の第5.4 節に記載したような使用条件下での完全な脱アセチル化を示す。
【0013】
図4A. 機械力精製法(上部)および化学/生物学的精製法(下部)により調製された純化珪藻p-GlcNAcの薄膜の赤外スペクトル分析。
【0014】
図4B. 以下の第5.5 節に詳述した方法に従って膜に注型された市販の2つの「キチン」調製物の赤外スペクトル分析。
【0015】
図4C. 以下の第5.4 節に詳述した方法に従って熱変性(上部)および化学的脱アセチル化(下部)により改質された純化p-GlcNAcの赤外スペクトル分析。
【0016】
図4D. 以下の第5.3.2 節に詳述した化学/生物学的精製法を用いて、珪藻タラシオシラ・フルビアチリス(Thallasiosira fluviatilis) から誘導されたp-GlcNAc膜の赤外スペクトル分析。
【0017】
図4E. オートクレーブ滅菌後に、以下の第5.3.1 節に記載したような機械力精製法により調製されたp-GlcNAc膜の赤外スペクトル分析。
【0018】
図5A. 以下の第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析。チャートはピーク強度、面積、および基準対照に対する比率を示す。ピークの全面積の比率。
【0019】
図5B. 以下の第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析。グラフはピークの全面積の比率を示す。
【0020】
図6A−6B. 以下の第5.3.1 節に記載した機械力精製法によって調製されたp-GlcNAc膜の透過型電子顕微鏡写真 (TEM)。倍率:6A: 4190x ;6B: 16,250x。
【0021】
図7A−7B. 以下の第5.3.2 節で化学/生物学的精製法の説明において記載したHF処理によるp-GlcNAc膜の透過型電子顕微鏡写真 (TEM)。倍率:7A: 5270x;7B: 8150x。
【0022】
図8A−8B. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜の透過型電子顕微鏡写真 (TEM)。倍率:8A:5270x ;8B: 16,700x。
【0023】
図9A. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:200x。
【0024】
図9B. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:1000x。
【0025】
図9C. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:5000x。
【0026】
図9D. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:10000x。
【0027】
図9E. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:20000x。
【0028】
図10A-10B. 以下の第5.3 節に記載した細胞溶解/中和精製法を用いて最初に調製し、ジメチルアセトアミド/塩化リチウム中に溶解し、そして以下の第5.5節に記載したように水中でマットに再沈殿させた物質から作られた純化p-GlcNAc膜の走査電子顕微鏡写真。倍率:10A: 1000x;10B: 10,000x。
【0029】
図11A-11B. 脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真。倍率:11A:1000x ;11B: 10,000x。
【0030】
図12A-12B. 珪藻の写真。珪藻の細胞体から伸びているp-GlcNAc繊維に注目のこと。
【0031】
図13. 本発明の可能なp-GlcNAc誘導体および脱アセチル化p-GlcNAc誘導体の一部を示す略図(S. Hirano, “Production and Application of Chitin and Chitosan in Japan", in “Chitin and Chitosan", 1989, Skjak-Braek, Anthonsen and Sanford 編, Elsevier Science Publishing Co., pp. 37-43からの改作)。
【0032】
図14. p-GlcNAc膜の存在下または不在下で増殖させた細胞の生存能実験。黒丸(黒○):p-GlcNAcマトリックス上で増殖させた細胞;白丸(○):マトリックスの不在下で増殖させた細胞。
【0033】
図15A-15B. p-GlcNAc膜上で増殖させた形質転換マウス繊維芽細胞のSEM顕微鏡写真。倍率:15A:1000x ;15B: 3000x。
【0034】
図16A. 以下の第13.1節に記載した方法に従って調製したコラーゲン単独対照物質の走査電子顕微鏡写真 (SEM)。倍率 100x。
【0035】
図16B. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は3:1に等しく、最終濃度はコラーゲンが7.5 mg/ml で、p-GlcNAcが0.07 mg/mlであった。倍率 100x。
【0036】
図16C. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は1:1に等しく、最終濃度はコラーゲンが5.0 mg/ml で、p-GlcNAcが0.12 mg/mlであった。倍率 100x。
【0037】
図16D. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は2:2に等しく、最終濃度はコラーゲンが10.0 mg/mlで、p-GlcNAcが0.25 mg/mlであった。倍率 100x。
【0038】
図16E. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は1:3に等しく、最終濃度はコラーゲンが2.5 mg/ml で、p-GlcNAcが0.25 mg/mlであった。倍率 100x。
【0039】
図17A. 上記図16A のコラーゲン単独対照物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0040】
図17B. 上記図16B のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0041】
図17C. 上記図16C のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0042】
図17D. 上記図16D のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0043】
図18. 各炭素原子の面積を得るために、次いで CH3(面積)対C原子(面積)の比を計算するために使用した変換NMRデータ曲線。
【0044】
図19. 典型的なp-GlcNAc C13-NMRスペクトル。各ピークはこの分子中の特異な炭素原子の該スペクトルへの寄与を表す。
【0045】
図20. CH3(面積)対C原子(面積)比の計算値を表す変換NMRスペクトルデータ。上図:データのグラフ表示;下図:データの数値表示。
【0046】
図21A-G. 種々の溶媒中で調製された三次元p-GlcNAcマトリックス。詳細には、p-GlcNAcマトリックスは蒸留水(図21A, 図21D)、蒸留水中の10% メタノール(図21B)、蒸留水中の25% メタノール(図21C)、蒸留水中の10% エタノール(図21E)、蒸留水中の25% エタノール(図21F)、および蒸留水中の40% エタノール(図21G)中で調製した。倍率:200x。各図面には200ミクロンの縮尺線を示してある。
【0047】
図22A-G. 蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞。倍率:100x (図22A, 22E) 、500x (図22B)、1000x(図22C, 22F) 、5000x(図22D, 22G)。各図面には5、20、50または200ミクロンの縮尺線を示してある。
【0048】
図23. 以下の第18.1節に記載した手順を用いて得られた典型的な標準曲線。このような標準曲線は、以下の第18.1節に記載したリゾチーム−キチナーゼアッセイにおいて使用した。
【0049】
図24. p-GlcNAcリゾチーム消化データ。ここに示したグラフは、p-GlcNAc膜をリゾチームで消化するときの時間の経過にともなうN-アセチルグルコサミンの蓄積を表す。このグラフは完全アセチル化p-GlcNAcと部分(50%) 脱アセチル化p-GlcNAcとの分解速度を比較したものであり、部分脱アセチル化p-GlcNAcの分解速度が完全アセチル化p-GlcNAc物質の分解速度を実質的に上回ることを実証した。
【0050】
図25. p-GlcNAcリゾチーム消化データ。ここに示したグラフは、p-GlcNAc膜をリゾチームで消化するときの時間の経過にともなうN-アセチルグルコサミンの蓄積を表す。このグラフは2つの部分脱アセチル化p-GlcNAc膜(すなわち、25%および50% 脱アセチル化p-GlcNAc膜)の分解速度を比較したものである。このデータによれば、50% 脱アセチル化p-GlcNAc膜の分解速度が25% 脱アセチル化p-GlcNAc膜よりも実質的に高いことから、脱アセチル化のパーセントが増加するにつれて分解速度は増加することが分かる。
【0051】
図26A-26E. p-GlcNAcのin vivo 生分解性データ。図26A-26C は、以下の第18.1節に記載したように基本型1(完全アセチル化p-GlcNAc)膜を腹部に移植したラットを示す。図26A は移植0日目のラットを、図26B は移植後14日目のラットを、そして図26C は移植後21日目のラットを示す。図26D-26E は、以下の第18.1節に記載したように基本型3A(凍結乾燥および部分脱アセチル化を行ったp-GlcNAc)膜を腹部に移植したラットを示す。図26D は移植0日目のラットを、図26Eは移植後14日目のラットを示す。
【0052】
図27. ここに示したグラフは、以下の第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0053】
図28. ここに示したグラフは、以下の第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0054】
図29. ここに示したグラフは、以下の第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0055】
図30. ここに示したグラフは、以下の第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0056】
図31. ここに示した棒グラフは、p-GlcNAc/5'FU 高用量(棒1)、p-GlcNAc/5'FU 低用量(棒2)、またはp-GlcNAc膜のみ(棒3)で処置した、あるいは処置を施さなかった(棒4)動物の動物あたりの腫瘍の大きさの平均変化パーセントを表す。棒1と棒2についてはn=4 、棒3と棒4についてはn=2 である。
【発明を実施するための最良の形態】
【0057】
5. 詳細な説明
まず初めに、本発明の精製p-GlcNAc種、p-GlcNAc誘導体およびそれらの再構成物の物理的特性について記述する。次に、微小藻類(好ましくは珪藻植物)出発物質源から本発明のp-GlcNAc種を精製する方法を記述する。第三に、p-GlcNAcの誘導体および再構成物、ならびに前記誘導体および再構成物の製造方法を提示する。最後に、本発明のp-GlcNAc、p-GlcNAc誘導体および/またはp-GlcNAc再構成物の使用を提示する。
【0058】
5.1 p-GlcNAc
本発明のp-GlcNAc多糖種は、ゲル浸透クロマトグラフィー測定に基づく重量平均が約800,000ダルトンから約3,000 万ダルトンの範囲にある高分子量のポリマーである。このような分子量は、β-1→4 配置において約4,000から約150,000個の結合されたN-アセチルグルコサミン単糖を有するp-GlcNAc種を表す。N-アセチルグルコサミン単糖の数は約4,000から約15,000個が好ましい(図1参照)。
【0059】
本発明のp-GlcNAcの可変性は非常に低く、またその純度は非常に高い。これら点は両方とも化学および物理的基準によって立証される。これらの基準のなかには、化学組成および非多糖不純物が含まれる。第一に、二つの異なる精製法(これらは両方とも後述の第5.3節に記述する)を用いて生産したp-GlcNAcの化学組成データを下記の表Iに示す。表から分かるように、二つの方法によって生産されたp-GlcNAcの化学組成は、実験誤差の限界内で、p-GlcNAcの処方組成と同一である。第二に、これも表Iから分かるように、生産されたp-GlcNAcは検出可能なタンパク質不純物を含まず、遊離のアミノ酸等の他の有機不純物を実質的に含まず、そして灰分および金属イオン等の無機不純物を実質的に含まない(本発明のp-GlcNAcは約0.05%までの微量金属を含んでもよい)。さらに、本発明のp-GlcNAcは結合水の百分率が非常に低い。
【表1A】

本発明の純粋なp-GlcNAcは、図2に示すものと実質的に類似した炭水化物分析プロフィールを示す。本発明の純粋なp-GlcNAcの主な単糖は、N-アセチルグルコサミンである。さらに、本発明の純粋なp-GlcNAcは単糖グルコサミンを含まない。
【0060】
本発明のp-GlcNAcの円二色性(CD)およびシャープな赤外スペクトル(IR)を図3A、および図4Aならびに4Dにそれぞれ示す。これらの図は、後述の第5.3節に記述する方法を用いて作成した物質の分析を表すものである。このような物理的データは、本発明のp-GlcNAcが高純度で高結晶性であることを確証する。CDおよびIRデータを得るために用いた方法は、後述第6節の実施例に記載する。
【0061】
本発明の純粋なp-GlcNAcのNMR分析は、図5A、5B、18Aおよび18Bに見られるものと実質的に類似のパターンを示す。このようなNMRパターンは、本発明のp-GlcNAcが十分にアセチル化されたポリマーであるということと一致するデータを示すばかりでなく、本発明のp-GlcNAc種においては不純な有機物が存在しないことをも示している。
【0062】
後述の第5.3節に記述する方法を用いて生産し、そして後述の第8および9節に記載の実施例に示す、本発明のp-GlcNAcの電子顕微鏡構造を図6Aから図9Eに掲げる。
【0063】
本発明のp-GlcNAcは高度な生体適合性を示す。生体適合性は種々の技法によって確認することができる。これらの技法は以下のものを含むがそれらだけに限定されない。すなわち、動物被験者への溶出試験、筋肉内移植、皮内または全身注射等の方法である。簡単に述べると、溶出試験(米国薬局方XXII, 1990, pp.1415-1497; 米国薬局方XXII, 1991, 補足5, pp.2702-2703)は試験物質抽出物の生体適合性を評価するために設計されており、抽出可能な細胞毒性物質に感受性の哺乳動物細胞培養系(例えばL929細胞系)の、試験物質に応答しての生物的反応性をアッセイするものである。後述第10節に記載の実施例は、本発明のp-GlcNAcの高い生体適合性を示す。
【0064】
5.2 p-GlcNAcの微小藻類源の生産方法
5.2.1 p-GlcNAcの微小藻類源
本発明のp-GlcNAcは微小藻類、好ましくは珪藻類により生産され、そしてそれより精製されうる。いくつかの属の珪藻類、およびそれらの属に属する多数の種をp-GlcNAcの原料源として使用できる。これらの珪藻類はそれぞれ、細胞体より伸長するp-GlcNAcから成る繊維を生産する。このような珪藻類の写真については、図12A〜12Bを参照されたい。本発明のp-GlcNAcの生産のための原料源として使用可能な珪藻類は、コアミケイソウ属(Coscinodiscus)、タイコケイソウ属(Cyclotella)、およびタラシオシラ属(Thalassiosira)属のメンバーを含み、なかでもタラシオシラ属が好ましいが、これらだけに限定されない。
【0065】
コアミケイソウ属の中で、本発明のp-GlcNAcの生産のための原料源として使用可能な珪藻種は、コンシナス(concinnus) およびラジアタス(radiatus)種を含むが、これらだけに限定されない。タイコケイソウ属のうち使用可能な珪藻は、カスピア(caspia)、クリプティカ(cryptica)およびメネギニアナ(meneghiniana)種を含むが、これらだけに限定されない。本発明のp-GlcNAcの原料源を生産するために使用可能なタラシオシラ属の珪藻は、ニッツショイデス(nitzschoides)、イースティバリス(aestivalis)、アンタークティカ(antarctica)、デシフェンズ(deciphens)、エクセントリカ(eccentrica)、フロリダナ(floridana)、フルビアチリス(fluviatilis) 、グラビダ(gravida)、グイラーディー(guillardii)、ヒアリナ(hyalina)、ミニマ(minima)、ノルデンスキオルディー(nordenskioldii)、オセアニカ(oceanica)、ポリコルダ(polychorda)、シュードナナ(pseudonana); ロチュラ(rotula)、ツビフェラ(tubifera)、ツミダ(tumida) およびワイスフロギ(weissflogii) 種を含み、なかでもフルビアチリスおよびワイスフロギ種が好ましいが、これらだけに限定されない。
【0066】
上記のような珪藻類は、例えば海洋植物プランクトンコレクションセンター(Center for Collection of Marine Phytoplankton)のビグロー海洋科学研究所(Bigelow Laboratory for Ocean Sciences) (McKown Point, West Boothday Harbor, Maine, 04575)の培養コレクションより入手可能である。
【0067】
5.2.2 珪藻類の増殖方法
上記第5.2.1に記載の珪藻類はいずれも、例えば本節に記述する方法を用いて増殖させることができる。新規な珪藻培養は、無菌条件下で栄養培地に成熟珪藻培養物のアリコートを接種することにより始める。この栄養培地は、他のあらゆる微生物を含まないものでなければならない。したがって、この栄養培地の調製に用いられる水、有機成分、および無機成分を含むすべての物質は無菌でなければならない。さらに、この操作に関与するすべての手順は厳正な無菌条件下で実施されることが必須である。すなわち、すべての容器、1つの容器から他の容器への物質の移し替えのすべて、等は無菌の環境で実施されなければならない。一度に調製される栄養培地の量は、新しい培養を始めるのに必要な量を越えてはならない。例えば、珪藻培養用の容器として約1平方フィートの表面を占めるフェーンバック(Fernback)フラスコを用いることができ、そして、そのような容器は珪藻生物の最適な増殖のために1リットルの栄養培地を必要とする。
【0068】
栄養培地の調製は以下の操作を含む:
a)海水の獲得および処理
b)蒸留水および脱イオン水の調製
c)一次栄養ストックの調製
d)栄養作業ストックの調製
e)最終栄養培地の調製。
【0069】
濾過した海水を、例えば海洋生物学研究所(the Marine Biology Laboratory)(Woods Hole, Massachusetts)より得ることができる。海水容器は5℃で保管しなければならない。必要な場合は、孔径が0.45ミクロンのニトロセルロース濾過膜(Millipore, Inc.)を用いて、ブフナー濾過ユニットにより必要量の水を濾過することができる。次に、海水を例えば121℃で、1 リットルにつき15分間オートクレーブにかけることにより滅菌する。この滅菌過程の完了後、好ましくは溶液が約5℃に達するのを可能とする寒い部屋に移して、蓋をした容器を直ちに冷却する。使用するときは、溶液を室温にする。
【0070】
標準的な装置および手順を用いて水道水を蒸留し、脱イオン化して、回収し、無菌の、しっかり蓋をした、好ましくはガラス製の容器に保管する。
【0071】
栄養培地の調製に必要な原液の調製の際に従うことができる処方を以下に掲げる。このような処方は指針として使用されるべきであるが、ここに記述するp-GlcNAcの調製方法に充分な微小藻類珪藻の増殖を維持することが可能な栄養培地の調製に寄与する、これらの処方の日常的な変法もまた、本発明の範囲内にあることが理解されねばならない。
【0072】
I.微量金属一次ストック(TMPS)
a.39 mM CuSO4・5H2O(硫酸銅(II)五水和物)(9.8 g 硫酸銅(II)/L)
b.7.5 mM ZnSO4・7H2O(硫酸亜鉛七水和物)(22 g 硫酸亜鉛/L)
c.42 mM CoCl2・6H2O(塩化コバルト(II)六水和物)(10 g 塩化コバルト(II)/L)
d.91 mM MnCl2・4H2O(塩化マンガン(II)四水和物)(18 g 塩化コバルト(II)/L)
e .26 mM NaMoO4・2H2O(モリブデン酸ナトリウム二水和物)(6.3 g モリブデン酸ナトリウム/L)
f.153.5 mM H2SeO3 (亜セレン酸)(12.9 g 亜セレン酸/L)。
【0073】
各栄養分を孔径が0.2 mm未満のフィルターを用いて無菌濾過する。
【0074】
II. ビタミン一次ストック(VPS)
a. 1 mg/ml ビタミンB12
b. 0.1 mg/ml ビオチン
両方のストックを孔径が0.2 mm未満のフィルターを用いて無菌濾過する。
【0075】
III. ナトリウム塩作業ストック(SSWS)
a.硝酸ナトリウム作業ストック: 0.88 M (75 g NaNO3/L)
b.リン酸ナトリウム一塩基一水和物作業ストック:36.2 mM NaH2PO4・H2O (5 g NaH2PO4・H2O/L)
c.メタケイ酸ナトリウム九水和物作業ストック: 0.11 M Na2SiO3・9H2O (30 g Na2SiO3・9H2O/L)
SSWSのそれぞれを孔径が0.2 mm未満のフィルターを用いて無菌濾過する。
【0076】
IV. 微量金属作業ストック(TMWS)
11.7 mM Na2EDTA ( エチレンジアミン四酢酸、二ナトリウム塩二水和物)(4.36 g/L)
11.7 mM FeCl3・6H2O(塩化鉄(III)六水和物)(3.15 g/L)
1 ml/Lの、上に列挙した6つの一次微量金属ストックのそれぞれ。
【0077】
孔径が0.2 mm未満のフィルターを用いて無菌濾過する。微量金属作業ストックは、新しい栄養培地を調製するたびに新たに調製しなければならない。
【0078】
V.ビタミン作業ストック(VWS)
1.0 μg/ml ビオチン (1.0 ml 一次ビオチンストック/100 ml)
1.0 μg/ml ビタミンB12 (0.1 ml ビタミンB12 一次ストック/100 ml)
20 mg のチアミンHCl (塩酸チアミン/100 ml)
孔径が0.2 mm未満のフィルターを用いて無菌濾過する。新しい栄養培地を調製するたびに、新たなビタミン作業ストックを調製しなければならない。
【0079】
以下に、栄養培地の調製および珪藻培養のために使用することのできる技法を記述する。これらの技法に加えて、ここに記述する調製方法に充分な珪藻の増殖を維持することが可能な栄養培地および手順をもたらし得る、ここに記述する処方および/または手順における日常的変法はすべて、本発明の範囲内に入ることが理解されねばならない。
【0080】
栄養培地は、例えば以下のようにして調製することができる:濾過し、滅菌した海水各1リットルに、1 mlのNaNO3 作業ストック、1 mlのNaH2PO4・H2O作業ストック、1 mlの微量金属作業ストック、および1 mlのNa2SiO3・9H2O 作業ストックを加える。Na2SiO3・9H2O の添加と同時に、2 mlの1 N HCl を加え、溶液を振とうして混合する。次に 1.5 ml の1 N NaOHを加え、溶液を再度振とうして混合する。最後に、0.5 mlのビタミン作業ストックを加える。
【0081】
新しい珪藻培養物を増殖させるため、7 mlの成熟培養物(細胞密度約 1x105 個/ml)を、100 mlの栄養培地(上記の方法にしたがって調製することができる)を含有する滅菌容器に移す。接種を受けた培養物を次に8日間下記の条件下でインキュベートする:
温度:20℃
常時照明
攪拌:毎朝および毎夕1回2〜3秒間フラスコを穏やかに回す。
【0082】
8日間のインキュベーション後、インキュベートした培養物 80 mlを無菌条件下で1000 ml の栄養培地に移す。この培地は、例えばチーズクロスで覆われた綿ウール栓によって保護された2.8 Lのフェーンバックフラスコに入れられている。このような培養物は、所望の細胞密度までインキュベートし、増殖させる。または、新しい珪藻培養物に接種するのに使用される。ひとたび培養物が所望の細胞密度に達したならば、培養物のp-GlcNAc繊維を収穫し、そして後述の第5.3 節に記述するような方法を用いて、本発明のp-GlcNAcを精製することができる。約7〜8、好ましくは7.4の培養pHを維持するため、培養液にCO2を溶解させることができる。このような中性pH環境の維持は、各珪藻培養物から得られるp-GlcNAcの収量を大幅に増大させる。
【0083】
5.3 p-GlcNAc繊維の単離、精製および濃縮方法
本節では、上記第5.2節に記述するような珪藻培養物からのp-GlcNAc繊維の調製に用いることができる方法を提示する。
【0084】
微小藻類、好ましくは珪藻、の原料源からp-GlcNAcを精製する下記の方法のそれぞれは、非常に純度の高い、混入物のない、結晶性p-GlcNAcを生ずる一方、各方法は特異的な特性および有利な特徴を有するp-GlcNAcを産する。例えば、下記の第5.3.1 節に記述する機械力による方法で精製された本発明のp-GlcNAcは、細胞のp-GlcNAcへの結合のためにより優れた基質を提供するp-GlcNAc膜をもたらす。下記の第5.3.2 節に記述する第二の方法、すなわち化学的/生物的方法は、機械力による方法によってもたらされる平均p-GlcNAc収量よりも遙に高い平均収量をもたらす。さらに、第5.3.2 節の化学的/生物的方法の一部として記述されている酸処理/中和変法は、極めて長いp-GlcNAc繊維で(ある繊維は100 μm を越える)、かつ2〜3千万ダルトンという高い分子量を有するp-GlcNAcをもたらす。
【0085】
5.3.1 純粋なp-GlcNAcを調製するための機械力による方法
p-GlcNAc繊維は、培養物の内容物を適切な機械力にかけることによって珪藻細胞体から分離することができる。このような機械力は、例えばコロイドミル、超音装置、または発泡装置により生成される剪断力、または、例えばワーリング(Waring)ブレンダーによって生成される切断力を含むが、これらだけに限定されない。
【0086】
こうして得られた珪藻細胞体およびp-GlcNAc繊維の懸濁液を、次に分離する。例えば、この懸濁液を一連の遠心分離工程にかける。この工程は、細胞体からp-GlcNAcを分離し、もしあったとしても目に見える綿状物質の殆ど無い、透明な上清を生ずる。遠心分離工程には、角度固定ローターおよび約10℃という温度が好ましい。速度、継続時間、および必要とされる遠心分離工程の総数は、例えば使用する特定の遠心分離ローター等によって変わる。しかし、これらのパラメーターの数値決定は、当業者には明白であろう。
【0087】
次に、当業者に周知の技法を用いて上清中のp-GlcNAc繊維を濃縮する。これらの技法は吸引および濾過装置を含むが、これらだけに限定されない。
【0088】
最後に、濃縮したp-GlcNAc繊維を、例えば蒸留脱イオン水、HClおよびエタノール、または他の適切な溶剤、好ましくは有機および無機物質の両方が溶解するアルコール等の溶剤を用いて洗浄する。
【0089】
後述の第7節に記載の実施例は、p-GlcNAc精製のためのこの方法の使用を示す。
【0090】
5.3.2 p-GlcNAc精製のための化学的/生物的方法
この方法においては、p-GlcNAc繊維を以下に詳述する化学的および/または生物学的作用物質にさらすことによって珪藻細胞体より分離する。
【0091】
珪藻培養物を、珪藻細胞壁を弱めることが可能な化学物質で処理することができる。これは、p-GlcNAc繊維の構造を変えずに、その放出をもたらす。このような化学物質はフッ化水素酸(HF)を含むが、これだけに限定されない。または、成熟した珪藻培養物を、生物的過程を変更することが可能な生物的作用物質で処理することができる。このような作用物質を用いてp-GlcNAc繊維の合成を阻害し、その結果すでに存在する繊維を放出させることができる。たとえば、このような作用物質は酵素N-アセチルグルコサミニル-P-トランスフェラーゼの阻害剤であるポリオキシン-Dを含むが、これだけに限定されない。
【0092】
次に、上記の化学的または生物的作用物質の一つで処理した珪藻培養物の細胞体およびp-GlcNAc含有繊維を分離する。例えば、処理した珪藻培養物の内容物を、二つの別個の層の形成を可能とするように、静置することができる。上の層は主にp-GlcNAc繊維を含有し、他方下の層は細胞体を含有する。p-GlcNAc繊維を含有する上の層をサイフォンで吸い上げ、下の層の沈殿した細胞性物質を残すことができる。
【0093】
次に、サイフォンで吸い上げたp-GlcNAc繊維を含有する層をさらに精製して、p-GlcNAc繊維を損なわない洗剤を用いて処理することによりタンパク質および他の望ましくない物質を除去する。このような洗剤は、硫酸ドデシルナトリウム(SDS)を含むが、これだけに限定されない。
【0094】
p-GlcNAc繊維を珪藻細胞体から分離するためにHF処理のような酸処理を用いる場合は、繊維を分散させるための工程を設けることができる。このような工程は、繊維分散のための機械力の使用(例えば、繊維をワーリングブレンダーによる分散に付す工程等)を含むが、これだけに限定されない。
【0095】
または、酸で処理した懸濁液を、洗剤処理によるさらなる精製にかける前に、任意選択の工程において中和してもよい。このような中和は、一般に、懸濁液のpHを約1.8から約7.0に変える。この中和は、例えば適当量の 1 M Tris (pH 8.0)の添加、または適当量の水酸化ナトリウム(NaOH)の添加により行なわれる。一般に、中和は本明細書で論じられている他の精製方法よりも、実質的に長さの長い純粋なp-GlcNAc繊維をもたらす。
【0096】
次に、吸引濾過装置の使用等の当業者に周知の技法を用いて、精製p-GlcNAc繊維を濃縮することができる。最後に、蒸留脱イオン水、HClおよびエタノール、または他の適切な溶剤、好ましくは有機および無機物質の両方が溶解するアルコール等の溶剤を用いた一連の工程により、p-GlcNAc繊維を洗浄する。
【0097】
後述の第8節に記載の実施例は、このような精製方法の上首尾な使用を示す。
【0098】
5.4 p-GlcNAcの誘導体化
種々の制御された条件および手順を用いることにより、本発明の純粋な、十分アセチル化されたp-GlcNAcを、広範な異なる化合物に誘導体化することができる。これらの化合物のいくつかを示す図については、図13を参照されたい。このような誘導体化された化合物は、以下に詳述する化学的および/または酵素的手段により修飾された、部分的に、または完全に脱アセチル化されたp-GlcNAcを含むが、これらだけに限定されない。さらに、p-GlcNAcまたはその脱アセチル化誘導体は、硫酸化、リン酸化および/または硝酸化されることにより誘導体化されうる。さらに、以下に詳しく述べるように、本発明のp-GlcNAcまたは脱アセチル化p-GlcNAcより、O-スルホニル、N-アシル、O-アルキル、N-アルキル、デオキシハロゲン、およびN-アルキリデンおよびN-アリーリデン、および他の誘導体が調製できる。本発明の脱アセチル化p-GlcNAcは、種々の有機塩および/または金属キレートを調製するためにも使用できる。さらに、本発明のp-GlcNAcまたはその誘導体は、種々の分子のうち任意のものを共有または非共有結合により自身に結合させることが可能である。そしてさらに、本発明のp-GlcNAcまたはその誘導体を、均一(uniform)且つそれぞれ別個な(離散的な(discrete))分子量特性を有する分子群を生じる、制御された加水分解条件下に置くことができる。
【0099】
本発明のp-GlcNAcの一つ以上の単糖ユニットを脱アセチル化し、ポリ-β-1→4-N-グルコサミン種を形成することが可能である。ポリ-β-1→4-N-アセチルグルコサミン種の単糖ユニットのそれぞれが脱アセチル化されている本発明のポリ-β-1→4-N-グルコサミン種は、約640,000ダルトンから約2千4百万ダルトンの分子量を有する。約640,000ダルトンから約2.4 百万ダルトンが好ましい。このような分子量範囲を有する種は、β-1→4 配置において約4,000から約150,000個の結合されたグルコサミン単糖を有する種を表す。グルコサミン単糖の数は約4,000から約15,000が好ましい。ポリ-β-1→4-N-グルコサミン種の単糖ユニットの少なくとも一つはアセチル化されたままであることができる。約25%から約75%のアセチル化が好ましく、約30%のアセチル化が最も好ましい。
【0100】
本発明のp-GlcNAcを塩基で処理することにより脱アセチル化し、遊離のアミノ基を有するグルコサミンをもたらすことができる。この加水分解過程は、濃い水酸化ナトリウムまたは水酸化カリウムの溶液を用いて高温で実施することができる。しかし、脱アセチル化の程度を正確にコントロールし、また多糖分子の炭水化物主鎖の分解を避けるため、p-GlcNAc脱アセチル化にはキチンデアセチラーゼ酵素を用いる酵素的方法を使用することが好ましい。このようなデアセチラーゼによる酵素的方法は、当業者に周知であり、また参照としてここにその全体を組み入れている米国特許第5,219,749号に記述されているように実施することができる。
【0101】
本発明のp-GlcNAcの単糖ユニットの一つ、または二つ以上を誘導体化し、以下に示すように、少なくとも一つの硫酸基を含有させるか、またはリン酸化もしくは硝酸化することができる:
【化1】
【0102】
ここで、水素に代わるRおよび/またはR1、および/または-COCH3に代わるR2は、硫酸(-SHO3)、リン酸(-P(OH)2)または硝酸(-NO2)基でありうる。
【0103】
以下にこのようなp-GlcNAc誘導体を調製する方法を記述する。本節に記述の方法を実施する前に、まずp-GlcNAc原料を液体窒素中で凍結乾燥し、凍結し、そして微粉砕することが有利であろう。
【0104】
硫酸化p-GlcNAc誘導体は、例えば二段階工程により生成しうる。第一段階において、例えばTokuraら(Tokura, S.ら,1983, Polym. J. 15:485)が記述する技法を用いて、本発明のp-GlcNAcおよび/またはp-GlcNAc誘導体よりO-カルボキシメチルp-GlcNAcを調製しうる。第二に、例えば N,N-ジメチル-ホルムアミド-三酸化硫黄を用いて、当業者に周知の技法、例えばSchweiger (Schweiger, R.G., 1972, Carbohydrate Res. 21:219) が記述する技法にしたがって、硫酸化工程を実施することができる。こうして得た産物をナトリウム塩として単離することができる。
【0105】
本発明のリン酸化p-GlcNAc誘導体は、当業者に周知の技法、例えば Nishiら(Nishi, N.ら, 1986, "Chitin in Nature and Technology", Muzzarelliら編、Plenum Press, New York, pp. 297-299)が記述する技法を用いて調製することができる。簡単に述べると、p-GlcNAc/メタンスルホン酸混合物を攪拌しながら約0℃から約5℃の温度で五酸化リン(約0.5から4.0モル当量) で処理する。処理は約2時間行なう。次に、当業者に周知の標準的技法を用いて、得られた産物を沈殿させ、洗浄する。例えば、エーテル等の溶剤を用いてサンプルを沈殿させ、遠心分離し、エーテル、アセトン、またはメタノール等の溶剤で洗浄し、乾燥させる。
【0106】
硝酸化p-GlcNAc誘導体は、当業者に周知の技法、例えば SchoriginおよびHalt(Schorigin, R.およびHalt, E., 1934, Chem. Ber. 67:1712) が記述する技法を用いて調製することができる。簡単に述べると、p-GlcNAcおよび/またはp-GlcNAc誘導体を濃硝酸で処理し、安定した硝酸化産物を形成することができる。
【0107】
本発明のp-GlcNAcの単糖ユニットの一つ、または二つ以上は、以下に示すように、スルホニル基を含有することができる:
【化2】
【0108】
ここでR3はアルキル、アリール、アルケニルまたはアルキニルの一部分でありうる。このような誘導体は当業者に周知の方法、例えば Kuritaら (Kuritaら, 1990, Polym. Prep. [Am. Chem. Soc., Div. Polym. Chem.] 31:624-625)が記述する方法を用いて生成させることができる。簡単に述べると、アルカリp-GlcNAc水溶液をトシルクロリドのクロロホルム溶液と反応させ、次に低温でこの反応を円滑に進行させる。
【0109】
本発明のp-GlcNAc、またはその脱アセチル化誘導体の単糖の一つ、または二つ以上は、以下に示すように、一つ、または二つ以上のO-アシル基を含有することができる:
【化3】
【0110】
ここで、水素に代わるR4および/またはR5は、アルキル、アルケニル、またはアルキニルの一部分でありうる。そして、R6はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体の一例は、Komai (Komai, T.ら,1986, "Chitin in Nature and Technology", Muzzarelliら編、Plenum Press, New York, pp. 497-506)が記述する方法等の周知の方法を用いて生成させることができる。簡単に述べると、p-GlcNAcを多数の適切な塩化アシルのうち任意のものとメタンスルホン酸中で反応させ、カプロイル、カプリール、ランロイル、またはベンゾイル誘導体を含むが、それらだけに限定されないp-GlcNAc誘導体を生じさせる。
【0111】
本発明の脱アセチル化p-GlcNAcの一つ、または二つ以上の単糖は、以下に示すようにN-アシル基を含有することができる:
【化4】
【0112】
ここで、R7はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、当業者に周知の技法、例えば Hiranoら (Hirano, S.ら, 1976, Carbohydrate Research 47:315-320)が記述する技法を用いて達成することができる。
【0113】
脱アセチル化p-GlcNAcは、多数の有機酸水溶液において可溶性である。このようなp-GlcNAcを含有する溶液に、選択されたカルボン酸無水物を水性メタノール性酢酸中で添加すると、N-アシルp-GlcNAc誘導体の形成をもたらす。
【0114】
本発明の脱アセチル化p-GlcNAc、またはその脱アセチル化誘導体の一つ、または二つ以上の単糖は、以下に示すようにO-アルキル基を含有することができる:
【化5】
【0115】
ここで、R8はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、当業者に周知の技法を用いて達成することができる。例えば、Mareshら(Maresh, G. ら,"Chitin and Chitosan", Skjak-Braek, G.ら,編、1989, Elsevier Publishing Co., pp. 389-395) が記述する手順を用いる。簡単に述べると、脱アセチル化p-GlcNAcをジメトキシエタン(DME) 中に分散し、過剰な酸化プロピレンと反応させる。反応時間は約24時間で、反応はオ−トクレーブ中で40から90℃で起こる。次に、混合物を水で希釈し、濾過する。DMEは蒸留によって除去しうる。最後に、凍結乾燥によって最終産物を単離することができる。
【0116】
本発明のp-GlcNAcの一つ、または二つ以上の単糖ユニットは、以下に示すようにアルカリ誘導体でありうる:
【化6】
【0117】
このような誘導体は、当業者に周知の技法を用いて得ることができる。例えば、Noguchiら (Noguchi, J.ら,1969, Kogyo Kagaku Zasshi 72:796-799) が記述する方法が使用できる。簡単に述べると、p-GlcNAcを減圧(vacuo)下でNaOH(好ましくは43%)に約2時間約0℃で浸す。次に、例えばバスケット遠心機を用いた遠心および機械的圧縮により過剰のNaOHを除去する。
【0118】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すようにN-アルキル基を含有することができる:
【化7】
【0119】
ここで、R9はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、例えば、前述のMareshら(Maresh, G.ら,"Chitin and Chitosan", Skjak-Brack, G.ら編、1989, Elsevier Publishing Co., pp. 389-395) がO-アルキルp-GlcNAc誘導体の生産のために記述するような手順を用いて達成することができる。
【0120】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すように少なくとも一つのデオキシハロゲン誘導体を含有することができる:
【化8】
【0121】
ここで、R10はF、Cl、Br、またはIでありうるが、Iが好ましい。このような誘導体は当業者に周知の技法を用いて得ることができる。例えば Kuritaら (Kuritaら, 1990, Polym. Prep. [Am. Chem. Soc., Div. Polym. Chem.] 31:624-625)が記述する手順を用いることができる。簡単に述べると、トシル化p-GlcNAcをジメチルスルホキシド中でハロゲン化ナトリウムと反応させる。p-GlcNAcのトシル化は、アルカリp-GlcNAc水溶液を塩化トシルのクロロホルム溶液と反応させることにより実施することができる。このような反応は低温で円滑に進行する。
【0122】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すように塩を形成することができる:
【化9】
【0123】
ここで、R11はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、当業者に周知の技法を用いて達成することができる。例えば、AustinおよびSennett (Austin, P.R. およびSennett, S., "Chitin in Nature and Technology", 1986, Muzzarelli, R.A.A.ら編、Plenum Press, pp. 279-286)が記述する方法等が使用できる。簡単に述べると、脱アセチル化p-GlcNAcを、例えば酢酸エチルまたはイソロパノール等の有機媒体中に懸濁し、そこに適切な有機酸、例えばギ酸、酢酸、グリコール酸、または乳酸等を加える。混合物をある期間( 例えば1〜3時間) 静置する。反応温度および乾燥温度は、約12℃から約35℃の間で変わるが、好ましくは20℃から25℃である。次に、濾過によって塩を分離し、新鮮な媒体で洗浄し、残留媒体を蒸発させる。
【0124】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すように金属キレートを形成することができる:
【化10】
【0125】
ここで、R12は金属イオン、特に遷移金属の一つでありうる。そしてXは、脱アセチル化p-GlcNAcに存在するアミノ基および置換アミノ基に存在する窒素電子によって確立される供与結合である。
【0126】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すようにN-アルキリデン基またはN-アリーリデン基を含有することができる:
【化11】
【0127】
ここで、R13はアルキル、アルケニル、アルキニル、またはアリールの一部分でありうる。このような誘導体化は、当業者に周知の技法を用いて達成することができる。例えば、Hiranoら (Hirano, S.ら, 1981, J. Biomed. Mat. Res. 15:903-911)が記述する手順を用いることができる。簡単に述べると、脱アセチル化p-GlcNAcのN-置換反応をカルボン酸無水物および/またはアリールアルデヒドを用いて実施し、アシル- および/またはアリ−リデン誘導体をもたらすことができる。
【0128】
さらに、本発明のp-GlcNAc、またはその脱アセチル化誘導体を制御された加水分解条件下に置くことができる。これは、均一でそれぞれ別個(離散的)な分子量および他の物理的特性を有する分子群をもたらす。そのような加水分解条件は、例えば、酵素リゾチームを用いた処理を含みうる。加水分解の度合いをコントロールするため、p-GlcNAcを様々な期間リゾチームに暴露する。さらに、加水分解の速度は、リゾチーム処理されつつあるp-GlcNAcがどの程度まで脱アセチル化されたかを関数としてコントロールすることができる。脱アセチル化条件は、本節で先に記述した通りでよい。p-GlcNAc分子がより十分に脱アセチル化されるほど、この分子はより十分に加水分解される。分子量の低下の他に起こる物理的特性の変化は、加水分解および/または脱アセチル化処理から引き出されるのかもしれない。広範な加水分解はp-GlcNAcの液化を引き起こす。加水分解/脱アセチル化手順の結果を後述第9節の実施例に示す。
【0129】
さらに、熱変性はp-GlcNAcの結晶構造を修飾する機能を果たしうる。p-GlcNAc産物結晶構造のこのような修飾は、例えばp-GlcNAcの反応性に有利な影響を及ぼしうる。
【0130】
さらに、種々の分子を共有結合または非共有結合によって、本発明のp-GlcNAcの脱アセチル化誘導体に機能的に結合することができる。このような分子は、以下のものを含むがそれらだけに限定されない。すなわち、神経成長因子等の増殖因子、ペプシン等のプロテアーゼ、ホルモン、などのポリペプチド、または、RGD配列、フィブロネクチン認識配列、などのペプチド認識配列、ラミニン、インテグリン、細胞接着分子、等である。脱アセチル化p-GlcNAcの露出した一次アミンに分子を共有結合させることは、例えば特定の長さの化学的スペーサーとして作用する二官能架橋剤を用いる化学結合によって達成しうる。このような技法は当業者に周知であり、また、例えば DavisおよびPreston (Davis, M.およびPreston, J.F. 1981, Anal. Biochem. 116:404-407) およびStarosら(Staros, J.V.ら,1986, Anal. Biochem. 156:220-222) の方法に類似している。簡単に述べると、脱アセチル化された、または部分的に脱アセチル化された本発明のp-GlcNAcに結合させるべきペプチド上のカルボン酸基を活性化し、次にp-GlcNAcに架橋させる。活性化は、例えばリン酸緩衝液中でカルボジイミドEDC (1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド)等の溶液をペプチド溶液に添加することにより達成しうる。好ましくは、この溶液は結合を促進するためにスルホ-NHS (N-ヒドロキシスルホスクシンイミド) 等の試薬をさらに含有する。活性化ペプチドは、高pHの緩衝液、例えば炭酸塩緩衝液(pH 9.0〜9.2)等の中で混合することにより脱アセチル化p-GlcNAcに架橋させることができる。
【0131】
または、上に記述したような分子を、当業者に周知の技法を用いて、非共有結合により脱アセチル化p-GlcNAcに結合させることができる。例えば、選択した一つ、または複数の分子を、凍結乾燥に先だって脱アセチル化p-GlcNAc溶液と混合することができる。
【0132】
または、p-GlcNAcおよび/またはp-GlcNAc誘導体からなるハイブリッド(hybrid)を形成することができる。このようなハイブリッドは、p-GlcNAcおよび/またはp-GlcNAc誘導体に加えて、多数の天然および/または合成物質のうち任意のものを含有することができる。例えば、ハイブリッドはp-GlcNAcおよび/またはp-GlcNAc誘導体プラス一つ、または二つ以上の細胞外マトリックス(ECM)成分より形成されうる。このようなECM成分は、コラーゲン、フィブロネクチン、グリコサミノグリカン、および/またはペプチドグリカンを含みうるが、これらだけに限定されない。ハイブリッドはまた、p-GlcNAcおよび/またはp-GlcNAc誘導体プラス一つ、または二つ以上の合成物質、例えばポリエチレン等より形成されうる。このようなp-GlcNAc/ポリエチレンまたはp-GlcNAc誘導体/ポリエチレンハイブリッドは、ハイブリッド成分を例えばオートクレーブにかけることにより、熱的に結合させて作成することができる。
【0133】
さらに、新規な塑性物質の製造のために、ヨード-p-GlcNAc誘導体を、例えばスチレンと共重合させることが可能である。ヨード-p-GlcNAcは、KuritaおよびInoue (Kurita, K. およびInoue, S., 1989, "Chitin and Chitosan", Skjak-Braek, G.ら編、1989, Elsevier Science Publishing Co., Inc., p.365)の記述する方法と類似の方法により、p-GlcNAcのトシル化およびヨウ素化によって調製することができる。次に、p-GlcNAcのヨード誘導体をニトロベンゼン中に分散し、触媒として塩化スズ(IV)を用いて、スチレンと反応させることができる。
【0134】
コラーゲン/p-GlcNAcハイブリッドの場合には、簡単に述べると、p-GlcNAc懸濁液およびコラーゲン懸濁液を混合して凍結乾燥し、そして好ましくは脱熱水的に(dehydrothermally)架橋する。このようなハイブリッドのコラーゲン種は、天然のものでも合成のものでもよい。また、ヒトのものでも、または非ヒト、例えばウシ等に由来するものでもよい。p-GlcNAc/コラーゲンおよび/またはp-GlcNAc誘導体/コラーゲンハイブリッド物質は均一な特性を示し、また、例えば細胞の結合および増殖のための効率的な三次元マトリックスとして役立つ多孔性マトリックスを形成する。後述の第13節に記載の実施例は、このようなp-GlcNAc/コラーゲンハイブリッドの形成、特性、および有用性を示す。
【0135】
脱アセチル化p-GlcNAcと、例えばヘパリン、アルギン酸ナトリウム、およびカルボキシメチルp-GlcNAc等の化合物との組合せから成るハイブリッドを、本明細書に記述するような技法を用いて作り出すことができる。このような組合せは、例えば膜および繊維等に形成または再形成することができる。
【0136】
脱アセチル化p-GlcNAcと、正および負の両方の電荷を有するポリアニオン、例えばポリアクリル酸またはペクチン等との複合体を作り出すことができる。このような複合体の形成は、Mirelesら(Mireles, C.ら,1992, "Advances in Chitin and Chitosan", Brine, C.J.ら編、Elsevier Publishers, Ltd.)が記述する方法と類似の方法にしたがって達成することができる。脱アセチル化p-GlcNAcおよび、例えばポリアクリル酸、カラギーナンまたはペクチンをそれぞれHClおよびNaClに溶解し、そして等しいpHを有する反応物溶液を混合する。この操作は、例えば廃水処理等に有用な、陽性および陰性の両方の特徴を備えた、効果的な凝集作用を有する分子をもたらす。
【0137】
5.5 再構成物(Reformulations)
本発明のp-GlcNAc、ならびに第5.4節に記述したその脱アセチル化誘導体および/またはそれらの誘導体化物は、溶解して、種々の形および配置に再構成することができる。
【0138】
本発明のp-GlcNAcの溶解は、ジメチルアセトアミド(DMA)/塩化リチウムで処理することにより達成することができる。p-GlcNAcは、5% LiCl (DMAの重量に対して) を含有するDMA溶液中で攪拌することにより容易に溶解する。p-GlcNAc塩等の水溶性p-GlcNAc誘導体は水に溶解する。少なくとも約75%脱アセチル化されたp-GlcNAcは、例えば1%酢酸等の穏やかな酸性溶液中で溶解しうる。水に不溶性のp-GlcNAc誘導体は、有機溶剤中で溶解しうる。
【0139】
フェニルイソシアナートを用いたDMA:LiCl中でのp-GlcNAcの誘導体化は、カルバニル酸塩を生産するのに用いうる。さらに、トルエン-p-スルホニルクロリドを用いたDMA:LiCl中でのp-GlcNAcの誘導体化は、トルエン-p-スルホネートを生産するのに用いうる。
【0140】
次に、溶解した本発明のp-GlcNAc、その脱アセチル化誘導体、および/またはそれらの誘導体化物を沈殿させ、そしてマット、糸(string)、ロープ、微小球、微小ビーズ、膜、繊維、粉末およびスポンジを含むが、これらだけに限定されない形に再構成することができる。さらに、超薄型(すなわち、厚さ約1ミクロン未満)均一膜を形成することができる。
【0141】
このような再構成物は、例えば、純粋なp-GlcNAcは水およびアルコール、好ましくはエタノール等の溶液に不溶性である事実を利用して達成しうる。例えば注入のような従来の手段により、p-GlcNAcを含有するDMA/LiCl混合物を上記の水またはアルコール、好ましくはエタノール溶液に導入すると、再沈殿をもたらし、それゆえ溶解したp-GlcNAcの再構成をもたらす。このような純粋なp-GlcNAcの再構成物を、後述第11節に記載の実施例に示す。水溶性のp-GlcNAc誘導体の場合は、例えば酢酸エチルまたはイソプロパノール等の有機溶剤中で再沈殿させることにより再構成を達成できる。少なくとも約75%脱アセチル化されたp-GlcNAcの再構成は、アルカリ性溶液中で再沈殿させることにより達成できる。水に不溶性のp-GlcNAc誘導体は、例えば水等の水性溶液中で再沈殿させることにより再構成しうる。
【0142】
脱アセチル化p-GlcNAcを酸化セルロースと共に調製して、紙製品の濡れ強度を改善するp-GlcNAc/セルロースハイブリッド物質を作り出すことができる。綿の鎖に似た平坦なリボン状の形を有する脱アセチル化p-GlcNAc鎖によって、酸化綿基質に非常に近似することができる。このような近似は、吸着を促進する力へのファンデルワールス力の寄与を最大にし、その結果、ハイブリッドp-GlcNAc-セルロース物質の濡れ強度特性を増強する。
【0143】
p-GlcNAc膜および三次元p-GlcNAcマトリックスは、膜またはマトリックス内に制御された平均孔径の形成をもたらす方法により作成することができる。膜およびマトリックスにおいて、孔径は使用するp-GlcNAcの量を変えることにより、またメタノールもしくはエタノール(エタノールが好ましい)等の特定の溶剤を約5%から約40%までの特定の量で、膜および/またはマトリックスの形成の前に添加することによりコントロールすることができる。一般に、溶剤の百分率が高いほど、形成される平均孔径は小さくなる。後述の第15節に記載の実施例は、このような多孔性p-GlcNAc構造物の合成および特徴付けを示す。
【0144】
5.6 用途
本発明のp-GlcNAc、ならびに第5.4節に記述したその脱アセチル化誘導体および/またはそれらの誘導体化物、および第5.5節に記述した再構成物は、種々の用途を有する。本明細書に記載のp-GlcNAcおよびその誘導体の有利な特性に加えて、例えば本発明の分子の非毒性、非発熱性、生物分解性、および生体適合性は、農業、化粧品、バイオ医薬産業、動物の栄養および健康、ならびに食品、化学、写真、および医薬産業等の様々な分野において利用される。
【0145】
5.6.1 p-GlcNAc物質のバイオ医薬用途
5.6.1.1 薬物固定化/デリバリー用途
p-GlcNAc物質のバイオ医薬用途は、例えば、酵素および/または薬物固定化/デリバリー方法を含みうる。例えば、本発明のp-GlcNAcおよびその誘導体は、第5.4節に記述したように、興味のあるペプチド(例えば増殖因子)をそこに共有結合させることができる。ペプチド含有p-GlcNAcを、当業者に周知の標準的方法を用いて患者に投与することができる。これらの方法は、注射、移植、関節鏡、腹腔鏡、または類似の手段を含むが、これらだけに限定されない。ペプチド含有p-GlcNAcを患者に導入すると、本発明のp-GlcNAcは生物分解するので結合ペプチドが患者の血流中に徐々に放出され、その結果、制御された薬物デリバリーの方法を提供する。
【0146】
予測できる生分解性を有する脱アセチル化または部分的に脱アセチル化されたp-GlcNAc種を製造することができる。例えば、脱アセチル化のパーセンテージはp-GlcNAc種が分解する速度に影響を与える。一般的には、脱アセチル化のパーセンテージが大きくなると生分解性及び吸収の速度が早くなる。従って、p-GlcNAc生分解性の程度及びインビボでの吸収速度はp-GlcNAcの製造の間に制御できる。そのようなp-GlcNAc物質の製造及び特性化の例は下記の第18節に示す。そのような制御可能な生分解性速度を有するp-GlcNAc物質は膜、ゲル、スポンジ、微小球、繊維等の中に形成することができる。これらのp-GlcNAc生成物は人体中の柔らかな組織及び固い組織の両方の組織に接着し型にはめられた状態になり、縫合することを要しない。p-GlcNAc物質は、例えば、一般的な手術あるいは侵入を最小限にした手術、例えば腹腔鏡手術等の間に適用することができる。
【0147】
生分解性の制御可能な速度を有するp-GlcNAc物質は、例えば、出血組織、器官、及び血管中の止血を促進するため、歯周手術後の修復過程の間の軟組織及び硬組織の分離のための歯周バリアを与えること、手術空間充填物を提供すること、特に皮膚の皺を減らす目的での皮膚における軟組織の増殖を促進すること、失禁を制御する目的での泌尿器括約筋を増強するために有用である。下記第19節に示した実施例は、そのようなp-GlcNAc物質の上記のような用途の1つ、即ち止血を促進することにおける使用を示すものである。
【0148】
さらに、本発明の分子は徐放薬送達ビヒクルとして有用であり、この場合は対象となる薬剤をp-GlcNAcまたはその誘導体によりカプセル化する。薬剤/p-GlcNAcカプセル化物は、例えば上記第5.3.2節に示した化学的/生物学的精製方法の酸処理/中和変法により製造することができる。p-GlcNAc溶液のpHをほぼ中性のpH範囲 (即ち約7.4)に上昇させるのではなく、p-GlcNAcの精製が完了した後にpHを約9.0 に上昇させて塩基性pH環境を形成することができる。より高い塩基性のpHにおいて、本発明のp-GlcNAcまたはその誘導体の構造はより3次元的、即ち「開放的な」形態となると考えられる。pHが低くなると、分子の配置はよりコンパクトなものに戻り、「閉鎖的な」形態となる。従って、対象となる薬剤を高いpHでp-GlcNAcに加え、その後p-GlcNAc/薬剤懸濁物のpHを低下させ、対象の薬剤をp-GlcNAcマトリックス中に「トラップ」あるいはカプセル化することができる。
【0149】
このようなp-GlcNAcカプセル化物は当分野の技術者によく知られた標準的な技術により患者に投与することができ、投与されるとカプセル化物のp-GlcNAcが分解されるにつれてカプセル化された薬剤が徐々に患者の身体に放出されるようにすることができる。
【0150】
p-GlcNAcをベースとするゲル及び膜は、治療用薬剤の送達システムとして種々の用途を有する。そのような用途としては、例えば抗腫瘍剤送達システムがある。本明細書中に記載する薬剤送達システムは任意の抗腫瘍薬とともに使用することができる。そのような薬剤は当分野の技術者によく知られており、p-GlcNAcゲルあるいは膜中に配合して、例えば、腫瘍あるいは手術後の腫瘍の空間の領域に直接、部位特異的徐放送達を与えることができる。このような固定化徐放p-GlcNAc薬剤生成物は、手術後の重要な初期的防護手段として役立ち得る。このようなp-GlcNAc抗腫瘍薬送達システムは、手術では全く、あるいは場合により接近不可能な腫瘍の治療において特に有用であり、これは例えばある種の脳腫瘍にあてはまる。
【0151】
p-GlcNAc抗腫瘍システムの別のターゲットは、限定するものではないが、皮膚、胃腸管系、膵臓、肺、乳房、泌尿器系、子宮癌、及びHIV-関連カポジ肉腫等である。
【0152】
放射能エンハンサーである抗腫瘍薬は、手術の代わりに、あるいは手術後に放射治療が処方される場合に好ましい。そのような薬剤の例は、例えば、5'- フルオロウラシル、マイトマイシン、シスプラチン及びその誘導体、タキソール、アドリアマイシン、アクチノマイシン、ブレオマイシン、ダウノマイシン、メタマイシン等である。
【0153】
抗腫瘍薬の投与量範囲は、患者の全身治療に処方される典型的な 1日当たり投与量と同等でも、低くても高くてもよい。薬剤が腫瘍の部位に局所的に送達されるのでより高い投与量が許容され得る。従って、血液細胞等のその他の組織は直ちには薬剤に露出されない。そのような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0154】
本発明のp-GlcNAc/薬剤送達システムはさらに、感染の治療に使用できる。そのような用途のためには、水溶性あるいは非水溶性の抗生物質をp-GlcNAcをベースとする物質、例えばゲルあるいは膜中に固定/配合することができる。抗生物質は当分野の技術者によく知られており、例えば、ペニシリン、セファロスポリン、テトラサイクリン、アンピシリン、オーレオマイシン、バシトラシン、クロラムフェニコール、シクロセリン、エリスロマイシン、ゲンタマイシン、グラマシジン、カナマイシン、ネオマイシン、ストレプトマイシン、トブラマイシン、バンコマイシン等がある。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0155】
このようなp-GlcNAc抗生物質生成物は、外部的に起こる細菌感染、例えば皮膚、頭皮、皮膚潰瘍もしくは眼表面に起こる細菌感染、あるいは内部的におこる細菌感染、例えば脳、筋肉、腹部の局所的な感染等の治療に使用できる。特筆すべき用途は、HIV-関連日和見感染症の治療である。
【0156】
本発明のp-GlcNAc/薬剤送達システムは抗炎症薬とともに配合して、炎症及び免疫プロセスの機能不全活性を制御することができる。例えば、p-GlcNAcを非ステロイド系抗炎症薬(NSAID)と配合して、リウマチ関節炎、変形性関節炎、全身性狼瘡等の疾患により誘発された局所痛及び炎症を軽減するのに使用することができる。本発明のp-GlcNAcゲルあるいは膜/薬剤送達システムを使用したそのようなNSAIDの局所送達は、胃過敏症、窒素過剰血症、血小板機能不全、肝機能異常のようなNSAIDの副作用を軽減するのに有用である。NSAIDは当分野の技術者によく知られており、アスピリン、エトドラック、フェノプロフェン、ナプロキセン等のようなシクロオキシゲナーゼの阻害剤を含む。その他の抗炎症薬を本発明のp-GlcNAc/薬剤送達システムの一部として使用することができ、例えば、ロイコトリエンのような脂肪炎症メディエーターの阻害剤である。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0157】
本発明のp-GlcNAc/薬剤送達システムはさらに、特異的な真菌症の治療のために、上記のような技術を使用して抗真菌薬とともに配合することができる。抗真菌薬は当分野の技術者によく知られており、例えば、アンホテリシン、アニソマイシン、アンチフンゴン、ブラストマイシン、グリセオフルビン、ニスタチン等がある。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0158】
本発明のp-GlcNAc/薬剤送達システムはまた、特異的な原虫感染症の治療のために、上記のような技術を使用して抗原虫薬とともに配合することができる。抗原虫薬は当分野の技術者によく知られており、例えば、アンチアメビン、アンチプロトジン、モノマイシン、パロモマイシン、トリコマイシン等がある。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0159】
本発明のp-GlcNAc/薬剤送達システムは、上記のような技術を使用して殺精子剤とともに配合して効果的な避妊薬を製造することができる。適当な殺精子剤は当分野の技術者によく知られている。そのような殺精子剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0160】
本発明のp-GlcNAc/薬剤送達システムはさらに、治療用タンパク質薬剤を使用して配合することができる。そのような配合物は、例えば上記のような技術を使用して製造することができる。そのようなp-GlcNAc治療用タンパク質系を利用することにより、所望のターゲット部位に特異的なタンパク質を直接送達することができ、そのような部位でのそのタンパク質の徐放を行うことができる。可能性のあるタンパク質の例は、限定するものではないが、インシュリン、モノクローナル抗体、乳癌イムノトキシン、腫瘍壊死因子、インターフェロン、ヒト成長ホルモン、リンホカイン、コロニー刺激因子、インターロイキン、ヒト血清アルブミン等がある。このような治療用タンパク質の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0161】
本発明のp-GlcNAc物質はそれ自体免疫的に中性であり、ヒトにおいて免疫反応を生成しないので、上記したような固定化薬剤を保有するp-GlcNAc膜、3D多孔質マトリックス及び/またはゲルを含むp-GlcNAcデバイスは、そのような薬剤を免疫反応なしに送達し得る。ある種の付加的な物質、例えば天然のアルギン酸塩や合成ポリマー等を使用し、p-GlcNAc物質と組み合わせてそのよなデバイスを構築することができる場合もある。
【0162】
上記の薬剤あるいは物質のいずれについても、本明細書に記載するp-GlcNAcをベースとする系と組み合わせたその治療上有効な投与量は、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。「治療上有効な」投与量とは、本明細書中に記載したプロセス及び/または疾患の兆候の軽減をもたらすのに十分なその化合物の量をいうものである。
【0163】
薬剤の毒性及び治療上の効能は細胞培養中あるいは実験動物において標準的な薬学的手法により測定することができ、例えばLD50 (集団の50% に対して致死的な投与量) 及びED50 (集団の50% において治療上有効な投与量) を測定することができる。毒性効果と治療上有効な効果との間の投与量の比率が治療指数であり、LD50/ED50 比で表される。大きい治療指数を示す化合物が好ましい。有害な副作用を示す化合物も使用できるが、非感染細胞に対する重大な障害を最小限にし、それにより副作用を低くするためには、そのような化合物を罹患組織の部位に向かわせる送達システムの設計に注意を払わなくてはならない。
【0164】
細胞培養アッセイ及び動物実験により得られたデータを、ヒトにおける投与量の範囲を計算するのに使用できる。そのような化合物の投与量は、毒性が殆どあるいは全くなく、ED50を含む循環濃度の範囲内にあることが好ましい。投与量は、使用する投与形態及び使用する投与経路に応じてこの範囲内で変化させることができる。本発明の方法に使用される任意の化合物について、治療上有効な投与量を最初に細胞培養アッセイから見積もることができる。細胞培養で決定されたIC50 (即ち、症状の最大阻害の半分の阻害を生じる試験化合物の濃度) を含む循環血漿濃度範囲が得られるように、動物モデル中で投与量を計算することができる。このような情報を使用して、ヒトにおける有用な投与量をより正確に決定することができる。血漿中のレベルは、例えば高速液体クロマトグラフィーにより測定することができる。
【0165】
5.6.1.2. p-GlcNAc細胞カプセル化物の用途
p-GlcNAcカプセル化細胞を製造することができ、そのようなp-GlcNAcカプセル化細胞を、当分野の技術者によく知られた標準的技術により患者に投与することができる。例えば、上記第5.6.1.1 節に記載した投与方法を参照されたい。あるいは例えば、Aebisher et al., (Aebisher, P. et al., “Fundamentals of Animal Cell Encapsulation and Immobilization", 1993, CRC Press, pp. 197-224)を参照されたい。これは引用によりその全体を本明細書の一部とする。細胞は、p-GlcNAcもしくは部分的に脱アセチル化されたp-GlcNAc膜、3次元的p-GlcNAc多孔質マトリックスあるいはp-GlcNAcゲルにより、あるいはその上に、あるいはその中にカプセル化され得る。
【0166】
3次元マトリックスは細胞とともに播種し、さらにカプセル化することなく一定の用途に使用することができる。あるいは、細胞をp-GlcNAcをベースとするポリマーゲル、例えばp-GlcNAc−ラクテートポリマー電解質ポリマー(ポリカチオン性ポリマー)の微小球あるいは小滴中にカプセル化することができる。細胞がその中にカプセル化されたゲル、小滴あるいは微小球はその後逆に荷電した第2のポリマー電解質により被覆し(例えばアルギネートのようなポリアニオンにより)、カプセル化された細胞の免疫的隔離を生じる外側カプセルを形成し、それにより宿主生物の免疫拒絶反応の危険を減らすことができる。
【0167】
さらに別の配合方法においては、p-GlcNAcゲル、3次元p-GlcNAcマトリックス、あるいはその両方にトラップされた細胞を熱可塑性のカプセル中に入れることができる。熱可塑性物質をベースとするカプセルを使用して、宿主生物中に移植された細胞に免疫保護を与えることができる。そのような熱可塑性カプセルは、例えばヒドロキシエチルメチルアクリレート/メチルメタクリレート共重合体(HEMA-MMA)のような物質により形成される。熱可塑性物質から誘導されるマイクロカプセルは、例えば、ポリエチレングリコール中のHEMA-MMAの溶液と細胞含有p-GlcNAcマトリックス及び/またはゲル媒体を、ヘキサデカンのような適当な有機溶媒中に共押出しすることにより形成される。例えば、Aebisher et al., (Aebisher, P. et al., “Fundamentals of Animal Cell Encapsulation and Immobilization", 1993, CRC Press, pp. 197-224)に記載された方法を参照されたい。
【0168】
p-GlcNAc細胞カプセル化物は種々の用途を有する。先ず、膜、マトリックスあるいはゲルに結合しあるいはその中にカプセル化された細胞により合成され、分泌される治療用化合物の送達のために使用できる。例示であって限定するものではないが、糖尿病の治療のためにインシュリンを、アルツハイマー病の治療のために神経成長因子を、血友病の治療のために第VIII因子あるいはその他の凝集因子を、パーキンソン病の治療のためにドーパミンンを、慢性的疼痛の治療のために副腎クロム親和性細胞を介してエンケファリンを、筋ジストロフィーの治療のためにジストロフィンを、あるいは成長異常の治療のためにヒト成長ホルモンを送達するために、p-GlcNAc/細胞カプセル化物を使用することができる。
【0169】
さらに本発明のp-GlcNAc物質はそれ自体免疫的に中性であり、ヒトにおいて免疫反応を生成しないので、結合した細胞を保有するp-GlcNAc膜、3次元多孔質p-GlcNAcマトリックス及び/またはp-GlcNAcゲルからなるデバイスを製造し、構築することができ、それらは細胞が免疫的に隔離された形態、即ち抗細胞宿主免疫反応が生じないような形態で細胞をベースとする治療用物質を送達し得る。ある種の付加的な物質、例えば天然のアルギネートや合成ポリマー等をp-GlcNAc物質自体に加えて使用し、そのようなデバイスを構築することができる。
【0170】
p-GlcNAc/細胞カプセル化組成物はさらに、細胞を送達して組織再生を開始させるために使用することもできる。外傷部位において組織再生を生じる細胞増殖を開始するためにカプセル化される特定の細胞種の用途は、限定するものではないが、皮膚、軟骨、神経、骨、肝臓、血管等の再生である。p-GlcNAc物質中にカプセル化された細胞の組織再生のための応用が有利であることの一つの理由は、p-GlcNAc物質が外傷組織に接着でき、哺乳動物細胞の増殖のための基体を提供し、外傷部位における組織再生プロセスの間に新しい健常な組織の増殖と同時に生体吸収を受ける能力を有することである。限定するものではないが、例として、皮膚、骨、軟骨、肝臓、腱、靱帯組織等の再生が挙げられる。
【0171】
5.6.1.3 手術後の癒着の防止のためのp-GlcNAc物質の使用
さらに、p-GlcNAc膜は、手術後の癒着を防ぐための生分解可能で生体適合性の機械的バリアを与えるために使用できる。下記第17節に示した実施例は、そのようなp-GlcNAcの用途を示すものである。膜あるいはスポンジ中に配合した固体p-GlcNAcまたはp-GlcNAc誘導体をそのような用途に使用できる。好ましくは膜は薄いもので、一般に約1 mm未満の厚さを有するものである。好ましいp-GlcNAc誘導体は、約50〜80% 脱アセチル化されたp-GlcNAc誘導体である。そのようなp-GlcNAc誘導体は、一般に移植後約7〜21日で吸収される。
【0172】
液体のp-GlcNAc誘導体も手術後の癒着の防止の使用に適している。そのような用途に好ましい液体p-GlcNAc誘導体は脱アセチル化p-GlcNAc塩誘導体及びカルボキシメチルp-GlcNAc誘導体である。手術後の癒着の防止に特に好ましいp-GlcNAc誘導体は、p-GlcNAc-ラクテート誘導体、特にp-GlcNAc-ラクテートゲル誘導体である。そのようなp-GlcNAc-ラクテート誘導体は、例えば第17.1節に記載したように、ポリエチレングリコール及び水を使用して配合することができる。p-GlcNAc-ラクテート誘導体は高い粘度を有するもの、あるいは低い粘度を有するものとして製造することができ、対象となる特定の適用症に合致した特性を与えることができる。例えば、シリンジあるいはスプレーにより送達するためには低い粘度を有するp-GlcNAc生成物を使用するのが有用であり、適応症が成形外科的なものである場合には高い粘度を有し、従ってより高い潤滑性を有するp-GlcNAc生成物を使用することが望ましい場合がある。
【0173】
手術後の癒着の防止のためには、明らかに限局性のものである外傷部位には固体p-GlcNAc配合物が適している。そのようなp-GlcNAc配合物は手術の処置後に適用し、材料が外傷を受けた組織を完全に覆うようにする。これは一般的な手術、あるいは侵入を最小限にした手術(例えば腹腔鏡を使用した)において使用することができる。固体p-GlcNAc配合物は、当分野の技術者によく知られた標準的な外科的処置及び設備を使用して切断し、適用することができる。
【0174】
液体のp-GlcNAc配合物は、手術後の癒着の防止のために、そのような手術後の癒着を起こしやすいより大きい領域に適用することができる。例えばp-GlcNAc-ラクテートゲルは、手術手順の前に適用して潤滑性を増し、外傷組織の量を減らすことができる。あるいは、p-GlcNAc-ラクテートのような液体p-GlcNAc配合物を手術手順の後に適用して物理的バリアを形成し、手術後の癒着の形成を防ぐことができる。
【0175】
p-GlcNAc物質は、シリンジデバイスから外傷部位に塗布し、スプレーし、あるいは滴下することができる。腹腔鏡使用処置においては、例えば、低い粘度を有する物質を標準的な吸引洗浄装置により送達することができる。高い粘度を有する物質は目的箇所に到達させるために圧力を要する。圧力は圧縮ガスピストンあるいはシリンジ型の装置により得られる。
【0176】
p-GlcNAc-ラクテートゲル配合物のような液体p-GlcNAc配合物の手術後の癒着を防止するのに要する量は、外傷を受けた組織の範囲に比例する。投与するp-GlcNAc物質は、表面積の平方センチメートルあたり0.1 ml〜1.5 mlの範囲で適用する。
【0177】
5.6.1.4 p-GlcNAcの物質その他の生物医学的使用
p-GlcNAc物質のその他の生物医学的利用としては、例えば細胞培養基体としてのそのような物質の使用が挙げられる。例えば、下記第12節に挙げた実施例に示すように、本発明のp-GlcNAcは培養物中で哺乳動物細胞増殖に非常に有効な基体となる。さらに、3次元的形態のp-GlcNAcは、3次元的細胞培養物増殖を可能とする培地成分として使用することができる。
【0178】
本発明のp-GlcNAcの細胞基体としての能力は生体内においても利用できる。即ち、本明細書に記載する本発明のp-GlcNAc、あるいはその誘導体は、組織の再生を促進するように作用できる(例えば、歯肉線に近い歯を覆う結合組織、移植血管、靱帯、腱、軟骨、骨、皮膚、神経組織の再生)。従って、本発明のp-GlcNAc分子は、例えば、広範な形成外科的用途を有する。
【0179】
脱アセチル化p-GlcNAcは、移植血管のシール材としての使用に好ましい。N-カルボキシメチル及びN-カルボキシブチル脱アセチル化p-GlcNAcのような脱アセチル化p-GlcNAc誘導体は組織再生剤として好ましい。N-カルボキシメチル脱アセチル化p-GlcNAcを、例えば、角膜に接種して血管新生を起こすことができる。
【0180】
本明細書に記載する本発明のp-GlcNAcあるいはその誘導体の別の生物医学的用途は、外傷用包帯、外傷治療軟膏、手術用縫合糸、スポンジ等に該分子を使用することである。
【0181】
さらにそのような分子は、例えば、変形性関節炎の治療、血清コレステロールレベルを低下させること、あるいは抗ウィルス剤として、抗細菌剤として、免疫調節剤として、抗凝集剤として、透析及び限外濾過膜として、抗腫瘍剤として、コンタクトレンズ材料として、腎不全患者に投与してイレミック毒素の吸収剤として使用できる。微結晶p-GlcNAc懸濁物あるいは水溶性p-GlcNAc誘導体は、例えば関節炎の関節に直接注射することにより、関節炎の治療に好ましいものである。
【0182】
p-GlcNAcはさらに、人工皮膚あるいはドナー皮膚の成分としての用途も有する。例えば、p-GlcNAcを好ましくは不織p-GlcNAcフィルムとして、例えばドナー真皮上、薄く剥がした厚さの皮膚ドナー部位にあてることができる。
【0183】
例えばペプシンのようなプロテアーゼを結合した脱アセチル化p-GlcNAcは、そのようなp-GlcNAc/プロテアーゼ化合物にタンパク質を接触させてその制御された消化を行うのに使用できる。
【0184】
本発明のp-GlcNAcの特定の誘導化、即ちその誘導体は、特定の用途に好ましいものである (誘導化については上記第5.4 節に記載した)。例えば、硫酸化、リン酸化及び/またはニトロ化p-GlcNAc誘導体は、抗凝集剤あるいはリポタンパク質リパーゼ活性化剤として好ましい。N-アシルp-GlcNAc誘導体も抗凝集剤に好ましく、さらに例えば、人工血管、抗ウィルス化合物、抗腫瘍 (具体的には癌細胞凝集化合物) 、透析及び限外濾過膜、及び放出が制御された薬物送達システムの製造にも好ましい。O-アルキルp-GlcNAc及びその脱アセチル化誘導体も放出が制御された薬物送達システムの製造に好ましい。N-アルキルp-GlcNAc誘導体は抗細菌剤として好ましい。オキシド脱アミノ誘導体は抗癌剤として好ましく、特に癌細胞の免疫療法と組み合わせて使用することが好ましい。脱アセチル化p-GlcNAc誘導体は外傷治療薬として好ましい。N-アルキリデン及びN-アリーリデンp-GlcNAc誘導体は酵素固定化の用途に好ましい。
【0185】
5.6.2 p-GlcNAc物質の農業用用途
本発明のp-GlcNAcあるいはその誘導体は、種々の農業用用途にも使用できる。そのような用途としては、限定するものではないが、殺昆虫剤、殺真菌剤、殺細菌剤、殺線虫剤の用途がある。N-カルボキシメチル脱アセチル化p-GlcNAc誘導体は、有効な細菌静止薬としての使用に好ましい。N-アルキルp-GlcNAc誘導体は殺真菌剤の用途に好ましい。さらに本発明の分子は、種々の土壌処理用途に使用することができ、限定するものではないが、肥料組成物等がある。さらに農薬を固定化、カプセル化、あるいはこの節で先に記載したような方法によりトラップすることによりそのような農薬の制御された放出を行うことができる。さらに、例えばRhizobium根粒着生因子及び/または窒素固定誘発物質の類縁体を本発明のp-GlcNAc及び/またはp-GlcNAc誘導体上に固定し、またそれを介して施用することができる。
【0186】
5.6.3 p-GlcNAc物質の栄養/食品産業用用途
本明細書に記載した本発明のp-GlcNAc及びその誘導体はさらに、動物及びヒトの栄養物摂取の分野に用途を有する。例えば、本発明の分子を飼料成分として使用し得る。この節で先に記載したような方法を使用して、放出が制御された製品を動物系において製造することができる。さらに、当業者によく知られた日常的な改変を行うことにより、上記のような生物医学的用途を動物系において利用できる。
【0187】
本明細書に記載したような本発明のp-GlcNAc及びその誘導体の食品産業での用途は、限定するものではないが、抗コレステロール物質(即ち、低コレステロール化合物)、脂肪結合化合物、乳化剤、担体、保存剤、調味料、食品生地改良剤(food texturizer) 等があり、さらに果実コーティング、食品パッケージ製品がある。
【0188】
5.6.4 p-GlcNAc物質の化粧品用用途
本発明のp-GlcNAcの化粧品用用途としては、限定するものではないが、ヘアケア及びスキンケア用の製品の製造がある。スキンケア製品としては例えば、脱アセチル化p-GlcNAc塩を利用した化粧品、カルボキシメチルp-GlcNAc含有製品、脱アセチル化p-GlcNAc及びヒドロキシプロピル- 、N-スクシニル- 誘導体及び4級化p-GlcNAc誘導体のような誘導体を含有する化粧用パック等がある。ヘア製品としては例えば、カルボキシメチルp-GlcNAc含有製品、及びフィルム形成性p-GlcNAc誘導体等がある。
【0189】
5.6.5 p-GlcNAc物質の化学工学用用途
本発明のp-GlcNAc及びその誘導体は化学工学工業において有用な種々の用途を有する。例えば、p-GlcNAcは金属のポリマーへの接着のためのカプリング剤として使用でき、グリコールp-GlcNAcにより形成された膜は脱塩用途に使用でき、その他のp-GlcNAc誘導体により形成された膜はハロゲンイオンの輸送に使用できる。その他の用途としては、難燃剤の製造、金属キレート化合物及び痕跡量の重金属並びにPCB のような水溶性の産業汚染物を液体から除去できる化合物の製造等がある。p-GlcNAc及び/またはp-GlcNAc誘導体は写真用途に使用できる。例えば、p-GlcNAc及び/またはp-GlcNAc誘導体の金属、例えばハロゲン化銀をキレート化する能力は、p-GlcNAc及び/またはp-GlcNAc誘導体の例えば薄い膜であるリキャストマットに写真溶液を接触させることにより利用できる。
【実施例】
【0190】
6. 実施例: p-GlcNAcの純粋調製物の物理的特性化
この実施例においては、p-GlcNAc及び脱アセチル化p-GlcNAc膜の円偏光二色性(CD)及び赤外分光スペクトル(IR)分析を示す。
【0191】
6.1 材料及び方法
p-GlcNAc及び市販の「キチン」調製物
CD分析において使用したp-GlcNAcは先に第5.3.1 節に記載した機械力精製法を使用して製造した。
【0192】
市販の「キチン」はNovaChem, Ltd., PO Box 1030 Armdale, Halifax, Nova Scotia, Canada, B3L 4K9から購入した。
【0193】
IR分析に使用したp-GlcNAc膜は、示したように、先に第5.3.1 節に記載した機械力精製法か、第5.3.2 節に記載した化学的/生物学的精製法により製造した。
【0194】
市販の「p-GlcNAc」調製物を、5%の塩化リチウムを含むジメチルアセトアミド溶液中に溶解し、蒸留及び脱イオン化した水上に積層して膜を沈殿させることにより、膜にキャストした。
【0195】
p-GlcNAc誘導体及び処理: CD及びIR分析の両方で使用した脱アセチル化p-GlcNAcは、p-GlcNAcを50% NaOHで60℃において2時間処理することにより製造した。IR分析において使用した熱変性p-GlcNAc膜は、0.2 mM EDTA 中で3分間煮沸することにより変性させた。オートクレーブしたp-GlcNAcは122 ℃で30分間オートクレーブしたものである。
【0196】
CD分析法: 実質的にDomardの方法(Domard, A., 1986, Int. J. Macromol. 8:243-246) に従って固体CD法を行った。
【0197】
6.2 結果
6.2.1 CD分析
未処理のp-GlcNAcから得られたCDスペクトル(図3A)においては、p-GlcNAcのアセチル部分のカルボニル基の存在のために予測されたn-π* 及びπ-π* 光学活性電子項遷移(220-185nM) の存在が観察された。このようなピークは、図3Bに示したように、脱アセチル化p-GlcNAc生成物から得られたCDスペクトルには全く存在しなかった。
【0198】
6.2.2 IRスペクトル分析
この分析において得られたIRスペクトルはp-GlcNAcの化学構造に合致するものであった。さらに、各IRピークが明確に得られたことは、p-GlcNAc繊維中の秩序立った規則正しい (即ち、偽結晶) 構造の存在を示すものである。機械力精製法により精製されたp-GlcNAcのIRスペクトルについては図4Aを、化学的/生物学的方法により精製されたp-GlcNAcのIRスペクトルについては図4Dを参照。比較として、市販の「キチン」調製物のIRスペクトルを示す図4Bを参照。
【0199】
オートクレーブされたp-GlcNAc物質から得られたIRスペクトル(図4E)は図4Aで見られるIRスペクトルと視覚的に異ならない。このデータは、p-GlcNAc物質がオートクレーブによりポリマー構造を損なうことなく滅菌され得ることを示している。
【0200】
7. 実施例: 機械力精製法を使用したp-GlcNAcの精製
この節においては、第5.3.1 節に記載した機械力法を使用してp-GlcNAcを精製した。
【0201】
7.1 材料及び方法/結果
珪藻培養条件: 珪藻の一種タラシオシラ・フルビアチリス(Thallasiosira fluviatilis)を上記第5.1 及び第5.2 節に記載した方法に従った培養で増殖させた。
【0202】
SEM方法: ここで使用したSEM法は下記第12.1節に記載するものである。
【0203】
p-GlcNAc精製法: 上記第5.3.1 節に記載した機械力法を使用して珪藻培養物からp-GlcNAcを精製した。具体的には、培養物の内容をWaringブレンダーのトップスピード混合動作に瞬間的に3回かけ、p-GlcNAc繊維を珪藻菌体から分離した。3回の操作の合計時間は約1秒であった。得られた懸濁物をSorvall GS-4固定角ローターで約10℃において3500 rpmで20分間遠心分離にかけた。上清をデカントし、Sorvall GS-4固定角ローターで約10℃において4000 rpmで20分間再度遠心分離した。再度上清をデカントし、10℃において4000 rpmで遠心分離した。最後の3回目の遠心分離の上清は透明で、ないとはいえないまでも液体中に浮遊する目に見える沈殿は殆どなかった。透明な上清を0.8 μm のポア径のニトロセルロースフィルターを備えたブフナー濾過ユニット中にデカントし、吸引して繊維懸濁物から液体を濾過し、膜上に繊維を回収した。回収された繊維を70℃において蒸留し脱イオン化した1リットルの水により洗浄した。ほとんど全ての水が排出された後、吸引しながら70℃において1リットルの1N HClにより繊維を洗浄した。ほとんど全ての酸溶液が排出された後、70℃において蒸留し脱イオン化した1リットルの水により吸引を使用して繊維を洗浄した。ほとんど全ての洗浄水が排出された後、室温で1リットルの95% エタノールにより繊維を洗浄し、減圧した。その上に白色の繊維膜が回収された濾過膜を濾過ユニットから取り出し、その膜及び膜支持体を58℃の乾燥オーブンで20分間乾燥し、その後膜及びその支持体をデシケーター中に16時間置いた。
【0204】
この精製工程の後において、1000 ml の培養物からのp-GlcNAcの収率は珪藻培養物1リットルあたり6.85 mg であった。この方法によりp-GlcNAc繊維を回収して形成された膜のSEM 写真を図6A〜6Bに示す。
【0205】
8.実施例:生物学的/化学的精製方法を使用したp-GlcNAcの精製
この節において、上記第5.3.2節の2つの化学的/生物学的手法を使用して、p-GlcNAcを精製した。簡単に述べると、1つはHF処理を介して、また2番目には酸処理/中和を介してp-GlcNAcを精製した。
【0206】
8.1.物質および方法/結果
ケイ藻の培養条件:ケイ藻種、タラシオシラ・フルビアチリス(Thallasiosira fluviatilis )を上記第5.1および第5.2節の操作法に従った培養によって増殖させた。
【0207】
SEM操作法:この実験において利用した手法は下記第12.1節と同様である。
【0208】
精製操作法:最初に、p-GlcNAcをHF処理によって精製した。この結果を図7A−7Bに示す。詳細に述べると、室温において換気フード下で、ケイ藻含有培養液に、細胞培養物原液1000ml毎に49%(29N)HF溶液 2.4mlを添加し、0.07M HF溶液とした。その後この混合物を約30秒間激しく振とうし、その結果液体の表面に持続性の気泡を生成させた。この容器を5−6時間静置し、重い粒子を沈降させた。この時間の経過後、気泡層は形成されたままで、一方液体自体は2層に分離した。第1層は容器の底面にある非常に暗い緑色の薄層で、その上がおそらく液体の総容量の85-90 %を占めるずっと明るい灰緑色で曇った第2層であった。ガラス毛細管と真空吸引を使用して、気泡層を注意深く吸引除去した。次に、主として沈降細胞本体からなる暗色の底部層をかき乱さないように注意しながら、灰色の曇った上層を吸引して、別のプラスチック容器に移した。この灰色の曇った上層をさらに16時間静置した。この液体は最初ほとんど無色か明灰色だが透明ではなかった。16時間の沈降後、液体の主要部分の上面に少量の気泡が残り、容器の底部に少量の緑色物質が沈降した。液体の色は薄くなったが依然として透明ではなかった。液体上面の気泡を前と同様に吸引除去した。次に、容器底部の緑色物質を残したままで、液体主要部を注意深く吸引して取り出した。こうして単離した液体は大部分がp-GlcNAc繊維で、他に不純物を少し含んでいた。
【0209】
この繊維含有液体からこれまでの操作段階中にケイ藻から遊離したタンパク質およびその他の不要物質を除去するため、繊維および細胞残留物の懸濁液をドデシル硫酸ナトリウム(SDS)で洗浄した。詳細に述べると、液体の最終濃度がSDS 0.5容量%となるのに必要な量の20%SDS溶液を添加した。この液体を入れた容器を密封し、振とう機上で水平を保つようにして、約100 振とう数/分で24時間撹拌した。振とう開始後まもなく、懸濁液中に白色のp-GlcNAc繊維の大きな固まりが生成し、容器の上部にかなりの量の気泡が集まった。SDS洗浄の終了後、容器の内容物を0.8μmの濾過膜(Supor Filter,Gelman )を着けたブフナー濾過器に移した。液体を吸引濾過し、液体中のp-GlcNAc繊維を濾過膜上に集めた。
【0210】
続いてこの濾過膜上に集めたp-GlcNAcをさらに洗浄した。まず、蒸留脱イオンした熱(70℃)H2Oを当初の懸濁液の3倍容量使用して、繊維を洗浄した。蒸留脱イオンしたH2Oを使用した水流によって、ブフナー濾過器の濾過膜上に集めた白色繊維塊をワーリングブレンダーに移し、約10回の短時間混合バーストによって、繊維塊を崩壊させた。崩壊繊維の懸濁液を上記と同様のニトロセルロース濾過膜を着けたブフナー漏斗に移し、吸引によって液体を除去した。採取した繊維を熱(70℃)1N HCl溶液1000mlで洗浄し、続いてさらに蒸留脱イオンした熱(70℃)H2O 1000mlで洗浄した。最後に、繊維を室温で、95%エタノール1000mlで洗浄し、濾過して乾燥させる。その後、繊維膜とこの繊維膜を担持している濾過膜を58℃の乾燥機で20分間乾燥した。その後、この膜と膜担持材をデシケーター中に16時間入れておいた。その後、膜を濾過膜から注意深くはがした。
【0211】
第2番目に、上記第5.3.2節の酸処理/中和方法を使用して、p-GlcNAcを精製した。詳細に述べると、SDS洗浄段階の前まではこの節の前半の記載と同様にp-GlcNAcを処理し、この段階において、2.9M Tris 溶液の添加によって、溶液のpHを約7.0 に中和した。この精製操作によるp-GlcNAcの収率はケイ藻培養物1l 当たり20.20mg だった。平均して、ケイ藻培養物1l 当たり約60mgが得られる。この精製操作において形成された膜のSEM写真を図8A−8Bおよび9A−9Bに示す。
【0212】
9.実施例:p-GlcNAcの脱アセチル化
p-GlcNAc膜を50%NaOH含有溶液中に懸濁させた。この懸濁液を80℃で2時間加熱した。生成した脱アセチル化膜を乾燥し、走査型電子顕微鏡で調べた。この結果を図11A−11Bに示す。
【0213】
10.実施例:p-GlcNAcの生体適合性
この実施例において、溶離試験、ウサギにおける筋肉内埋め込み、ウサギにおける皮内注入、およびマウスにおける全身注入試験によって、本発明のp-GlcNAcが検出し得る程度の生物学的活性を持たないことが実証される。
【0214】
10.1.物質および方法
10.1.1.溶離試験
溶離試験の条件は、U.S.薬局方 XXII,1990,pp.1415-1497 、およびU.S.薬局方XXII, 補遺5,1991,pp.2702-2703 に示された説明にしたがった。
【0215】
細胞培養物:マウス繊維芽L929細胞系(American Type Culture Collection Rockville,MD;ATCC No.CCL1;NCTC clone 929 )を使用した。24時間目のL929細胞の密集単層を完全な最少必須培地(MEM)中で増殖させた。
【0216】
p-GlcNAc:上記第5.3.1節の機械的精製方法によって調製したp-GlcNAc固体膜から、U.S.薬局方XXII(1990)の要件にしたがって、血清補充MEM20ml中に抽出した。
【0217】
対照:陽性対照として天然ゴムを使用し、陰性対照としてシリコーンを使用した。試験品であるp-GlcNAcと同一の手法で対照を試験した。
【0218】
抽出物:37℃、5%二酸化炭素含有加湿雰囲気中で24時間かけて、抽出物を調製した。抽出物のpH変化を測定し、当初の培地のpH単位の+/−0.2 以内になるようにpHを調整した。抽出物のpHを低下させるのにはHClで、抽出物のpHを上げるのにはNaHCO3 で調整を行なった。抽出物を細胞単層に適用する前に、0.22ミクロンフィルターを通して滅菌濾過した。
【0219】
適用:p-GlcNAcまたは対照3mlずつを使用して細胞培養物の維持培地と入れ替えた。すべての抽出物を2つずつ試験した。
【0220】
評価基準:細胞単層の応答を肉眼または顕微鏡下で評価した。生物学的反応性、すなわち細胞の変性および/または奇形化を下記に示すように0〜4の基準で評点をつけた。陰性対照品に関して細胞反応性の徴候が何ら見られず(等級0)、陽性対照品が軽度の反応性(等級2)以上の反応性を示す場合に、その試験系は好適なものである。試験品で処理した培養物の中に軽度より大きな反応性を示すものがない場合、この試験品(すなわちp-GlcNAc)は生体適合性試験を満足することとなる。
【表1B】
10.1.2.筋肉内埋め込み
動物:健康なニュージーランド白ウサギ、雄および雌(Eastern Rabbit Breeding Laboratory,Taunton,MA )を使用した。ウサギは懸垂型ステンレススチール製カゴに個別に入れた。入手後すぐに、実際の試験と同一の条件下で8日間隔離しておいた。カゴの下の非接触床として、硬材チップ(Sani-chips(登録商標),J.P.Murphy Forest Products,Montvale,NJ )を使用した。動物の環境を、温度68+/-3°F、相対湿度30〜70%、1時間当たり少なくとも10〜13回の完全換気、および全スペクトルの蛍光灯を使用した12時間の明/暗サイクルに維持した。動物に市販の餌(Agway ProLab,Waverly,NY )を制御しながら、そして公共水道水を自由に与えた。餌、床材または水には、試験結果に干渉する可能性がある、既知の汚染物質は何も存在しなかった。この実験のために選定した動物はより多くの動物群から選抜した。ウサギは10g 近傍まで体重を測定し、耳の刺青によつて個体を特定した。
【0221】
p-GlcNAc:使用したp-GlcNAcは上記第10.1.1節と同様である。
【0222】
埋め込み試験:各埋め込み試験毎に2匹のウサギを使用した。試験当日、動物の脊柱の両側の皮膚の毛を刈り取った。筋肉の動きを止めるため、各動物に麻酔した。滅菌皮下注射針とスタイレットを使用して、p-GlcNAc試験片(1mm×1mm×10mm)4枚を2匹のウサギのそれぞれの脊椎の一方の側の脊椎傍筋肉中に(中心線から2.5 〜5cm、脊柱に平行、および互いに約2.5 cm間隔で)埋め込んだ。同様の方式で、各動物の反対側の筋肉にUSP陰性対照プラスチックRS片(1mm×1mm×10mm)2枚を埋め込んだ。動物を7日間そのまま維持した。観察期間の終了時、動物の体重を測定し、注射用バルビツレート、Euthanasia-5(Veterinary Laboratories,Inc.,Lenexa,KS)によって安楽死させた。組織を出血せずに切開することができるまで十分な時間を経過させた。各埋め込み片の中心部分を取り巻く組織一帯を、拡大レンズを使用して肉眼で検査した。以下の基準を使用して、出血、壊死、退色および感染を採点した:0=正常、1=軽度、2=中程度、および3=強度。カプセル化があれば、最初にカプセルの幅(すなわち、埋め込み物の末端からカプセルの末端までの距離)をおよそ0.1mm 近傍まで測定して採点した。このカプセル化は以下のように採点した:
【表1C】
p-GlcNAcと陽性対照品の平均評価点の差を計算した。各ウサギにおいて、4枚のp-GlcNAc試験片の1枚についてだけでも、p-GlcNAcと陽性対照プラスチック埋め込み部位における生体反応の各部門の平均評価点間の差異が1以下ならば、または、そのp-GlcNAc品における生体反応の全部門の平均評価点と全陽性対照プラスチック埋め込み部位における生体反応の全部門の平均評価点間の差異が1以下ならば、その試験は陰性であるとみなされる。
【0223】
10.1.3.皮内注入
動物:上記第10.1.2節のようにして、ニュージーランド白ウサギを使用し、飼育した。
【0224】
p-GlcNAc:上記第5.3.1節の機械的精製方法によって調製したp-GlcNAc固体膜を抽出フラスコに入れ、これに好適な溶媒20mlを添加した。24時間、70℃に加熱することによって抽出を実施した。この操作後、抽出物を室温まで冷却した。投与前に各抽出ビンを激しく振とうした。
【0225】
皮内試験:試験当日、動物の背中側の毛を刈り取った。2匹のウサギのそれぞれの一方の側の5ヵ所に、各p-GlcNAc抽出物0.2ml を注入した。ウサギ1匹当たり1以上のp-GlcNAc抽出物を使用した。各ウサギの他方の側の5ヵ所に、対応する対照品0.2ml を注入した。注入の24、48および72時間後、紅斑、水腫および壊死の徴候に関して、注入部位を観察した。下記表IIに示すような皮膚反応評価に関するDraize Scale(USP 薬局方XXII,1990,1497-1500 ;USP 薬局方XXII, 補遺5,1991,2703-2705)にしたがって、観察を評価した。
【表2A】
対応する対照に対するp-GlcNAcの総合的な平均評価点を決定するため、24、48および72時間における紅斑および水腫の全評価点をそれぞれ合計し、12(すなわち、動物2×評価期間3×評価部門2)で割った。観察期間の終了時に、動物の体重を測定し、バルビツレート、Euthansia-5(Veterinary Laboratories,Inc.,Lenexa,KS) の注射によって安楽死させた。p-GlcNAcと対照の平均反応評価点(紅斑/水腫)の差異が1.0 またはそれ以下であれば、試験結果は満足し得る。
【0226】
10.1.4.全身注入
動物:アルビノスイスマウス(Mus musculus)雌(Charles River Breeding Laboratories,Wilmington,MA )を使用した。マウス5匹のグループをステンレススチール製のふたをしたポリプロピレン製カゴに入れた。カゴの中の接触床として、硬材チップ(Sani-chips(登録商標),J.P.Murphy Forest Products,Montvale,NJ )を使用した。この動物施設を接触制限区域として維持した。動物室を、室温68+/-3°F、相対湿度30〜70%、1時間当たり最低10〜13回完全換気し、全スペクトル蛍光灯を使用して12時間の明/暗サイクルを保持した。マウスに市販の餌と公共水道水を自由に与えた。試験結果に干渉することが予想されるような既知の汚染物質は餌、床材または水に含まれていなかった。この研究のために選定した動物は、より多くの動物群の中から選抜した。マウスの体重を0.1g近傍まで測定し、耳のパンチによって個体を特定した。
【0227】
p-GlcNAc:使用した試料は上記第10.1.1節の通りである。上記第10.1.3節の操作にしたがって抽出物を調製した。
【0228】
全身注入試験:マウス5匹のグループに、p-GlcNAc抽出物または対応する対照品を、下記に示すように同量ずつ同一の経路で注入した。
【表2B】
PEG400 を使用して調製したp-GlcNAc抽出物および対応する対照を0.9%NaClで希釈し、1ml当たりPEG400 200mg となるようにした。皮内試験については、PEG400 を0.9%NaClで希釈し、1ml当たりPEG400 120mg となるようにした。
【0229】
動物を、注入直後、注入後24時間、48時間および72時間後に観察した。観察期間の終了時、動物の体重を測定し、二酸化炭素ガス中にさらすことによって安楽死させた。p-GlcNAc処理した動物の中で対照品で処理した動物よりも生物学的反応性が有意に大きいものがなければ、この試験の要求が満足される。
【0230】
10.2.結果
10.2.1.溶離試験
p-GlcNAc試験品に対する細胞単層の応答を肉眼および顕微鏡下で評価した。評価に際して、細胞化学的染色は利用しなかった。陰性対照品またはp-GlcNAcの被曝48時間後、細胞の生物学的反応の徴候は何ら観察されなかった(等級0)。下記表III に示すように、陽性対照品については、強度の反応性(等級4)が記録された。
【表3】
したがって、本発明のp-GlcNAcは生体適合性に関する溶離試験の要求を満たしており、すなわち、非毒性である。
【0231】
10.2.2.筋肉内埋め込み
試験をしたウサギの両方(AおよびB)の体重が増加し、毒性の徴候は何ら見られなかった。データに関しては表IVを参照されたい。付け加えると、どの動物においても毒性の顕著な徴候は見られなかった。試験および対照品の肉眼での評価では、炎症、被包、出血、壊死または退色は示さなかった。結果については表IVを参照されたい。したがって、この試験では、各ウサギにおいて、すべてのp-GlcNAc埋め込み部位の生物学的反応の全部門の平均評価点とすべての対照埋め込み部位の全部門の平均評価点との差異が1.0 以下であることから、検定したp-GlcNAcは生物学的反応性を有していないことが示される。
【表4】
10.2.3.皮内試験
すべての動物の体重が増加した。データについては表Vを参照されたい。p-GlcNAcまたは対照品部位のいずれにおいても紅斑または水腫の徴候は観察されなかった。どの動物においても毒性の顕著な徴候は観察されなかった。p-GlcNAcおよび対照品の平均反応評価点(紅斑/水腫)の差が1.0 よりも小さかったので、p-GlcNAcは皮内試験の要求を満たしている。結果については表VIを参照されたい。したがって、この試験によって分析したところ、p-GlcNAcは生物学的反応性を示さない。
【表5】
【表6A】
【表6B】
【表6C】
【表6D】
10.2.4.全身試験
p-GlcNAc抽出物または対照品で処理したすべてのマウスの体重が増加した。データについては表VII を参照されたい。付け加えると、p-GlcNAcまたは対照のどの動物においても明白な毒性の徴候はなんら観察されなかった。結果については表VIを参照されたい。したがって、どのp-GlcMAc試験動物も対照品で処理した動物以上に有意に大きな生物学的反応性を示さないことが結論づけられる。
【表7】
11.実施例:p-GlcNAcの再構成
この節において述べる具体例においては、5%LiClを含有するジメチルアセトアミド溶液1mlに、p-GlcNAc膜(16.2mg)を溶解させた。このp-GlcNAc含有溶液を注射器に入れて、純水50ml中に押し出し、繊維を沈澱させた。生成した繊維を走査型電子顕微鏡で調べた。これを図10A−10Bに示す。
【0232】
12.実施例:p-GlcNAcへの細胞の付着
この具体例においては、p-GlcNAcが培養液中での細胞の付着と増殖のための有効な基質であることが実証される。
【0233】
12.1.材料および方法
細胞:形質転換マウス3T3繊維芽細胞株を使用し、10%ウシ胎児血清(FBS)を添加したDMEM中で増殖させた。
【0234】
p-GlcNAc膜:上記第5.3.1節および第8節の方法にしたがって、p-GlcNAcを調製した。
【0235】
最初に、水を1滴使用して、#1(18mm)Corning のカバーグラスにp-GlcNAc膜を貼りつけ、121 ℃で30分間オートクレーブにかけることによってこれを接着させた。次に、この手法によって調製した膜を6ウェル培養プレートの培養ウェル中に入れた。
【0236】
細胞の計数:細胞の数を、培養液中のものは血球計算盤で直接に計数し、マトリックス上のものは、計数前にまず膜を新しい培地(DMEM+10%FBS)で洗浄し、次にトリプシンで処理(10%、37℃で5分間)した後、計数した。
【0237】
SEM走査条件:ツァイス(Zeiss)962 装置を加速電圧10kv、および作用距離15mmで使用した。ポラロイド型55p/n(u4) 型を指示にしたがって各種倍率で使用した。
【0238】
試料のコーティング:炭素コーティング(100Å)および100ÅのAuPd。
【0239】
検体の調製:一次固定のため、培養増殖用培地を血清を含まないイーグルのDMEM中2%グルタルアルデヒドと交換した。増殖用培地から固定液への完全な移行を確実にするため、何度か交換を行った。室温で0.5 時間固定を行なった。カバーグラスを0.1 M ショ糖と0.1 M カコジル酸Na、pH7.2 中2%グルタルアルデヒドを含有する新しいバイアルに移し、さらに室温で1.5 時間固定した。
【0240】
脱水、CPD、マウントおよびスパッタコーティング:
試料を0.1 M カコジル酸Na、pH7.2 で洗浄し、カバーグラスをCPD支持器に移した。エタノールシリーズ(30%、50%、75%、85%、95%および3×100 %、5分ずつ)によって脱水を行ない、試料を臨界点乾燥した。次に、カバーグラスをAlスタブ上にマウントし、真空蒸発器を使用して炭素コーティングし(@100Å)、100ÅのAuPdによってスパッタコーティングを行なった。
【0241】
12.2.結果
p-GlcNAc膜について、この上で培養液中の細胞が増殖することができる基質となる能力を試験した。p-GlcNAc膜の存在または不在ウェル中でマウス繊維芽細胞を増殖させ、培養物の生育力を分析するため、毎日細胞の計数を実施した。このような連日の細胞計数の結果の1つを図14に示す。これによると、接種5日後、p-GlcNAc膜含有ウェルのみが生育する細胞を保持しており、p-GlcNAc膜が培養液中での細胞の持続増殖のための有効な基質として作用することを実証している。さらに、図15A−15Bに示すSEM写真は、p-GlcNAc膜に強固に付着した健康な細胞を表している。
【0242】
13.実施例:p-GlcNAc/コラーゲンハイブリッド
この具体例において示されるのは、p-GlcNAc/コラーゲンハイブリッド材料の形成および特性決定である。
【0243】
13.1.材料および方法
材料:この試験において述べるハイブリッドの調製のために、ウシI型コラーゲンを使用した。p-GlcNAcを上記第5.3.2節の機械的方法によって調製した。
【0244】
ハイブリッドの調製:コラーゲン(10mg/ml)およびp-GlcNAc(0.25mg/ml)の懸濁液を各種割合で混合し、液体N2 中(−80℃)で凍結させ、−9℃に4時間保持し、凍結乾燥した。60℃において真空(約0.030 Torr)下で3日間、材料を脱水熱架橋した。
【0245】
細胞培養:製造したコラーゲン/p-GlcNAcハイブリッド上で、マウス3T3繊維芽細胞を増殖させた。標準的な培養操作を行い、培養8日後、SEM写真を撮った。
【0246】
13.2.結果
コラーゲンおよびp-GlcNAc懸濁液を各種割合(すなわち、コラーゲン:p-GlcNAc懸濁液の比が3:1,1:1,2:2,および1:3)で混合し、凍結し、凍結乾燥し、そして架橋した。この操作によってコラーゲン/p-GlcNAcスラブが得られた。生成した物質のSEM写真を図16B−Eに示す。図16Aはコラーゲン−材料対照のみを示す。ハイブリッド材料の多孔質構造に注目されたい。
【0247】
本発明のコラーゲン/p-GlcNAcハイブリッドは、図17A−DのSEM写真が示すように、細胞が付着して増殖するのに有効な3次元構造を提供する。
【0248】
14.実施例:純粋なp-GlcNAc調製物のNMR特性
この実施例において示されるのは、純粋なp-GlcNAc調製物のNMR(核磁気共鳴)分析である。
【0249】
14.1.材料および方法
p-GlcNAc調製品:上記第5.3.2節の化学的精製方法を使用し、化学試薬としてフッ化水素酸を利用して、ここで述べるNMR試験に使用するp-GlcNAcを調製した。
【0250】
NMR手法:Bruker 500MH NMR分光器を使用して、固体状態のNMRデータを得た。未処理のNMRスペクトルデータからバックグラウンドを除去し、ベースラインを標準化するように変換するため、コンピュータイメージ分析を使用した。こうした変換データの例を図18に示す。図18のような変換NMR曲線を使用して、すべての炭素原子型の領域の値を得て、それからC原子(領域)に対するCH3(領域)の比を計算した。ここに述べたようにして得た数値を図20に示す。
【0251】
14.2.結果
p-GlcNAc試料500mg のC13- NMRスペクトルを測定することによって、固体状態のNMRデータを得た。典型的なNMRスペクトルを図19に示す。それぞれのピークが分子中の各ユニークな特徴ある炭素原子のスペクトルへの寄与に相当する。各炭素種別毎に生じたピークの領域をスペクトル中で得られたすべてのNMRピークの領域の総計で割ることによって、分子中の各炭素原子型の相対百分率を測定した。このようにして、標準原子によって測定した、分子中の各原子の割合を計算することができた。すべてのp-GlcNAc分子は、明確にC1,C2,C3,C4,C5およびC6原子を有するN−アセチル化グルコサミン残基からなっている。ここで、ポリマー中のすべてのグルコサミン残基がN−アセチル化されているならば、上記のいずれのグルコサミン残基炭素原子ピーク領域に対するN−アセチルCH3炭素原子ピーク領域の割合も1.0 となるはずである。CH3(領域)割合の値を得るため、図20においてこれらのデータを使用した。
【0252】
図20において計算した割合は、多くの場合、1.0 に等しいか、または実験誤差の範囲内でほとんど等しい。たとえば、CH3/C2=1.097 、CH3/C6=0.984 、CH3/C5=1.007 、CH3/C1=0.886 である。これらの結果は、本発明のp-GlcNAc材料が不純物を含まず、完全にアセチル化されている(すなわち、本質的に100%のグルコサミン残基がN−アセチル化されている)という結論に一致する。
【0253】
15.実施例:孔径が制御されたp-GlcNAcの3次元マトリックスの合成および生
物学的特性決定
以下に述べるのは、制御された平均孔径を有するp-GlcNAc基剤の3次元多孔質マトリックスの製造方法である。こうしたマトリックスは各種の重要な用途を有する。特に、例えば細胞のカプセル化の用途などである。これらの細胞をカプセル化した組成物は移植用細胞基剤の治療剤として、また軟骨再生などの、その他の細胞および組織の工学的応用において有用である。ここに実証するように、p-GlcNAc材料の形態および3次元構造を操作することができるので、これによって、本発明のp-GlcNAc材料の応用の可能性を広げる上で強力な道具が提供される。
【0254】
15.1.材料および方法
p-GlcNAc出発材料:上記第5.3.2節の化学的精製方法を使用し、化学試薬としてフッ化水素酸を使用して、p-GlcNAcを調製した。
【0255】
マトリックスの形成:凍結乾燥する前に、下記第15.2節に列記する溶媒中でp-GlcNAc試料 20 mgの懸濁液(5 mlずつ)を製造した。次に試料を組織培養皿のウェルに注ぎ入れ、−20℃で凍結した。この凍結試料を凍結乾燥し、生成した3次元マトリックスを取り出した。
【0256】
走査型電子顕微鏡操作:ここで利用した操作法は、上記第12.1節にしたがって実施した。図21A−Gに示す画像は200X倍率のマトリックス材料であり、これらの図のそれぞれに200 ミクロンを示すスケールを表示している。
【0257】
15.2.結果
上記第15.1節にしたがって、以下の各溶媒からp-GlcNAc試料が得られた。
【0258】
A.蒸留水
B.蒸留水中10%メタノール
C.蒸留水中25%メタノール
D.蒸留水のみ
E.蒸留水中10%エタノール
F.蒸留水中25%エタノール
G.蒸留水中40%エタノール
各溶媒を使用して形成したマトリックス試料を、走査型電子顕微鏡分析に供した。これを図21A−Gに示す。これらの図によって、各懸濁液中のメタノールまたはエタノールの比率が増加するにしたがって、マトリックスの平均孔径が小さくなることがわかる。
【0259】
特定すると、2つの水懸濁液(図21Aおよび21D)中の孔径は平均して200 ミクロンに近い。図21Cおよび21F(それぞれ25%メタノールおよび25%エタノール)に示す試料の孔径は平均して30〜50ミクロンである。
【0260】
ここに示した結果によると、p-GlcNAcの孔径を制御するのに、エタノールおよびメタノールの両方が有効に利用され得るが、p-GlcNAcマトリックスの孔径の制御能力において、エタノールの方がメタノールよりも有効であるようである。
【0261】
16. 実施例: 三次元多孔性p-GlcNAcマトリックス上での細胞増殖
本節では、細胞培養の基質として三次元多孔性p-GlcNAcマトリックスを有効に使用したことを実証する結果を記載する。
【0262】
16.1 材料および方法
p-GlcNAc出発物質: p-GlcNAcを、フッ化水素酸を化学試薬として使用して上記の第5.3.2 節に記載の化学的精製法を使用して調製した。
【0263】
マトリックス形成: 三次元p-GlcNAcマトリックスを水、水−エタノールまたは水- メタノール混合物中のp-GlcNAcの懸濁液を凍結乾燥させて製造した。
【0264】
凍結乾燥前に、以下の溶媒中に、p-GlcNAc 20 mgを含有する懸濁液(5 ml)を調製した。
【0265】
1. 蒸留水のみ
2. 10% メタノール含有蒸留水
3. 25% メタノール含有蒸留水
4. 蒸留水のみ
5. 10% エタノール含有蒸留水
6. 25% エタノール含有蒸留水
7. 40% エタノール含有蒸留水
サンプルをプラスチック製の組織培養皿の丸いウェルの中に注ぎ、−20℃で凍結させた。その後、凍結サンプルを凍結乾燥させ、そして得られた三次元マトリックスを取り出した。各マトリックスのサンプルを走査型電子顕微鏡(SEM) 分析にかけた。
【0266】
細胞: ATCCより入手した、マウス胚BALBC/3T3 の繊維芽細胞系( クローンA31)を三次元多孔性p-GlcNAcマトリックス上での培養に使用した。
【0267】
培養条件: 多孔性マトリックスの 1 cm2サンプルを組織培養ウェルに入れ、そして標準の組織培養増殖培地を上から注いだ。各ウェルに細胞を植え、細胞をCO2 インキュベーター(5% CO2)内で37℃で6 日間培養した。
【0268】
SEM 手順: マトリックスサンプルを固定し、そして上記の第12.1節に記載したようにSEM 分析にかけた。マトリックスは蒸留水中の p-GlcNAc を凍結乾燥して製造した。図の解説( 図 22A〜G)に示したように、像は100 倍〜5000倍の倍率で変化する。
【0269】
16.2. 結果
付着しているマウス繊維芽細胞を含んでいるp-GlcNAcマトリックスのSEM 写真を図22A 〜G に示す。これらの写真により三次元p-GlcNAcマトリックスは付着したマウス繊維芽細胞を含むことが分かる。さらに、写真から細胞とp-GlcNAcマトリックス物質との間には密接な相互作用と連結があることも明らかである。また、細胞は球状の三次元形態をしており、プラスチックの培養皿で直接培養した細胞の、平らに広がった形とは異なることも注目に値する。細胞の生存率は95% 以上であった。
【0270】
17. 実施例: p-GlcNAcは術後癒着を有効に防止する
本文の実施例では、p-GlcNAc物質の有効な使用例、詳細には、p-GlcNAc膜およびゲル構成物が一連の癒着動物モデルにおいて、術後癒着の形成を防止することを示す。
【0271】
17.1 材料および方法
p-GlcNAc- 乳酸の合成: p-GlcNAc膜出発物質は、フッ化水素酸を化学試薬として使用して第5.3.2 節に記載のように化学的方法により製造した。
【0272】
p-GlcNAcを以下の方法により脱アセチル化p-GlcNAcに変換した。( 脱アセチル化により生じる質量損失を約15% と仮定すると、p-GlcNAc- 乳酸 1g を生成するために、p-GlcNAc約 1.4 gを必要とすることに注意すべきである。) p-GlcNAc膜物質約200 mgを60% NaOH約200 mlと激しく混合した。p-GlcNAc膜物質をできるかぎり分離させるために激しい振盪を利用した。使用するNaOH溶液は少なくとも使用の12時間前に調製した。p-GlcNAc膜物質を分離させ湿潤させるために断続的に振盪しながら、サンプルを80℃の湯浴に6 時間入れた。6 時間後、サンプルを湯浴から取り出し、そしてNaOH溶液を直ちに取り除いた。室温で膜物質をpH 7になるまで蒸留H2O で洗浄した。膜を水から取り出し、そしてテフロンシート上で乾燥させた。
【0273】
この時点で、脱アセチル化の程度を測定するためにC 、H 、N 分析のためのサンプル 2 mg を採取した。さらに、1%酢酸への溶解度を調べた。溶解度が1 mg/ml である場合はp-GlcNAc物質が適切に脱アセチル化されていることを示す。
【0274】
その後、部分的脱アセチル化p-GlcNAcを以下の方法を使用してp-GlcNAc- 乳酸に変換した。すなわち、部分的脱アセチル化p-GlcNAc物質を全て湿潤させ、そして攪拌を可能にするのに十分量の2-プロパノール( 水を10% 含有) を250 mlのエルレンマイヤーフラスコ中の部分的脱アセチル化p-GlcNAc 1 gに添加した。( 約30 ml の2-プロパノールを必要とした。) 2- プロパノールは試薬級で、そして各合成前に調製したばかりのものでなければならない。攪拌しながら、50% 乳酸水溶液 1.1 mL を加えた。乳酸は試薬級でなければならず、有効な( すなわち非エステル化) 乳酸の正確な存在濃度を測定するために分析する必要がある。そのために、通常0.1 N NaOHを用いてフェノールフタレインの終点(pH 7.0)まで滴定した。各p-GlcNAc合成のためには、使用する乳酸の濃度は一定でなければならず、すなわち、+/−1 パーセントでなければならない。混合物を少なくとも2 時間攪拌した。反応速度を上げるために僅かに加熱することができる。反応を完全に進行させるために、反応時間を延長でき、または50% 乳酸水溶液の量を増加できる。攪拌後、フラスコの内容物を定量用の無灰濾紙を使用してブフナーロートで濾過した。物質を無水2-プロパノール 15 mlで洗浄した。ヒュームフード中で2 時間風乾させ、その後40℃のオーブンに一夜放置した。部分的脱アセチル化p-GlcNAc出発物質 1グラムごとに、約1.4 g の最終p-GlcNAc- 乳酸( すなわち、40% の質量増加) が得られるはずである。
【0275】
p-GlcNAc- 乳酸のゲル構成物: p-GlcNAc- 乳酸物質を以下のようにゲルに構成した。p-GlcNAc- 乳酸出発物質を再蒸留脱イオン水に溶解し0.1 〜4.0 重量% の濃度にした。その後、試薬級のプロピレングリコール(2- プロパンジオール) をプロピレングリコールの最終濃度が1 〜10% の間になるよう添加した。いくつかの場合は、生成物が細菌および/ またはカビで汚染されるのを防ぐために防腐剤を添加した。代表的には、0.1%〜4.0%のp-GlcNAc- 乳酸濃度で調製した。これらの調成物の粘度はp-GlcNAc- 乳酸の割合が増すにつれて急激に増加し、0.5%またはそれ以上のp-GlcNAc- 乳酸を有するものがゲルとなる。
【0276】
動物モデル:
Sprague-Dawley系ラット:このモデルでは、盲腸の奬液膜表面を擦りまたは刺激し、それを腹膜の側壁に付着させておくことにより癒着を生じさせる。この方法を用いて対照動物に癒着を誘導できる成功率は平均 80%と報告されている。
【0277】
詳細には、これらのラットで術後癒着を誘導する手術は以下のとおりであた。動物を仰向けに寝かせ、無菌的外科手術に適するように準備し滅菌した布で覆った。正中線切開して腹腔を露出した。左の腹壁の約0.5 cm×1.0 cmの腹膜側壁の部分を注意深く切開し、腹膜とともに筋肉の薄層を切り取った。
【0278】
その後、盲腸を持ち上げ取り出した。盲腸の近位端の側方の表面上の約0.5 cm×1.0 cmの部分を乾燥ガーゼで10回擦って傷つけた。その後、盲腸をメスの刃でこすって小さな点状出血を生じさせた。盲腸の擦り傷および腹膜の切開を15分間露出させておいた。
【0279】
15分後、試験物質( すなわちp-GlcNAc物質) または対照物質を盲腸の傷に施した。その後、盲腸の擦り傷および腹膜の傷を対置し、そしてさらに15分間アリス組織鉗子で接触させておいた。
【0280】
その後、盲腸の擦り傷の部分を腹膜部に隣接するように盲腸を腹部に戻した。腹部の切開を閉じて、そして動物を麻酔から覚醒させた。
【0281】
術後14日目に、動物を安楽死させ、擦り傷部分の術後癒着の形成を調べた。癒着が存在した場合は、癒着に関係している全ての領域( すなわち、体壁、試験または対照物質そして内部器官) を動物から切り離した。
【0282】
癒着の程度と癒着の付着力を以下の尺度に従って評価した:
癒着程度のスコア:
0. 癒着無し
1. 領域の25% 以下の癒着
2. 領域の50% 以下の癒着
3. 領域の75% 以下の癒着
4. 領域の25% を越える癒着
付着力スコア:
0. 癒着無し
1. 鈍的切開により引き離される
2. 攻撃的な切開により引き離される
3. 鋭的切開を必要とする
別の動物モデル: ブタおよびウマの大型動物の腸モデルを使用して腹膜癒着の防止を評価した。
【0283】
手術: 動物を仰向けに寝かせ、無菌的外科手術に適するように準備し滅菌した布で覆った。正中線で切開して腹腔を露出させた。小腸を持ち上げ、2 cm×2 cm部分を確認し、十分に擦り( メスを使用して約200 回擦る) 、そして10分間乾燥させた。その後、試験物質(すなわちp-GlcNAc物質) または対照物を擦り傷に施し、そして小腸の傷の部分を腹部に戻した。上記の大型動物の腸モデルでは、それぞれが隣接する傷から4 インチ離れているように腸管上に6 箇所の傷を付けると最も癒着を形成する傾向がある。最後に傷を付けた後、腹部の切開を閉じ、そして動物を麻酔から覚醒させた。
【0284】
腹膜癒着の分析: 手術後21日目に、動物を安楽死させ、擦過傷領域を調べ、癒着の形成をSprague-Dawley系ラットの盲腸モデルの場合と同様の手順に従って評価した。
【0285】
17.2 結果
損傷が生じた場合、身体は損傷部を回復させるために、複雑な一連の反応を開始する。治癒の最終段階では、傷の部位に結合組織が形成され、身体構造を再生し、そして患部がさらに損傷を受けるのを防ぐ。ある場合には、この治癒過程の流れが適正にいかず、生命を脅かす事態を招くことがあり得る。
【0286】
例えば、術後の内蔵器官が回復する過程で、手術中に生じたフィブリン塊が隣接する器官の表面に浸入し、繊維芽細胞の移動を生じさせる連結部を形成し得る。この移動がコラーゲンの沈着および組織の増殖を招き、ひいては関係する器官がお互いに癒着することとなる。
【0287】
上記の癒着を術後癒着と呼ぶが、その器官または関係する器官に歪みを生じて疼通、閉塞および機能不全を引き起こし得る。関節の固定化、腸閉塞および不妊はしばしば術後癒着に関連している。さらに、術後癒着が症状を悪化させ、その付近の将来の手術時間を長引かせる原因となるであろう。この最後の問題点は、別の手術が必要であり得る開心術および婦人科の帝王切開の処置で特に問題となる。癒着の形成は腹部、心臓血管および整形外科の処置後に非常に一般的である。
【0288】
癒着が病的になり臓器の機能を著しく損なう場合は、外科的癒着剥離術( 細心の外科技術と関連した癒着の鋭的または鈍的切開) が、癒着を取り除くために現在行われている方法である。1991年にはこの外科的癒着剥離術が約500,000 例実施された。しかしながら、この処置は癒着形成の再発率が90% に上ると報告されており、効果のないことで有名である。さらに、上記の術後癒着の防止に有効とされている他の技術や組成物もない。
【0289】
従って、ここに示した結果は、本発明のp-GlcNAc物質が術後癒着の防止に有効性を示したことにおいて意義深いものである。詳細には、ここに示した結果は、p-GlcNAc物質をベースとする固形および液体処方物が、認可されたラットおよびブタの動物モデル系における腹部の術後癒着の形成のバリヤーとしての効力を示すものである。
【0290】
癒着形成の研究に使用される、認可された動物モデルの1つは、Sprague-Dawlery 系ラットにおける内臓と腹腔壁の癒着を用いている。部分的脱アセチル化p-GlcNAc膜および部分的脱アセチル化p-GlcNAc- 乳酸ゲル構成物の両者とも、未処置対照群またはこの効能を表示している唯一の市販薬であるInterCEED(商標名)(Johnson & Johnson)処置群に比べて癒着の発生を防止および/ またはかなり軽減していた。
【0291】
詳細には、全部で18匹のラットを使用してp-GlcNAc- 乳酸ゲル構成物を試験した。12匹を対照とし、6 匹は0.25%p-GlcNAc-乳酸ゲル、10% プロピレングリコール、水を投与した。p-GlcNAc- 乳酸ゲル処置群はどちらの対照群と比較しても、下記の表VIIIに示したように、有意に術後癒着の形成を減少させた。
【表8】
部分的脱アセチル化p-GlcNAc膜もラット動物モデルにより術後癒着の発症の予防能力を検討した。この実験には全部で22匹のラットを使用した。12匹を対照群に、6 匹を未処置群に、6 匹をInterCEEDTM処置群にした。10匹はそれぞれ約60% 脱アセチル化p-GlcNAcの 1cm×1cm 膜で処置した。下記の表IXに示した通り、部分的脱アセチル化p-GlcNAc膜処置動物は未処置およびInterCEEDTM処置に比べて術後癒着の形成の発生が有意に低下した。
【表9】
腹膜癒着を防止する大型動物の腸モデルもp-GlcNAc組成物の試験に使用した。詳細には、6 匹のブタと1 匹のウマを使用して、部分的脱アセチル化p-GlcNAc膜およびp-GlcNAc- 乳酸ゲルの両者を試験した。部分的脱アセチル化p-GlcNAc膜は 60%脱アセチル化p-GlcNAc膜の2 cm×2 cm片から成り、一方p-GlcNAc- 乳酸ゲルは10% プロピレングリコールおよび水で調製した0.25%p-GlcNAc-乳酸から成っていた。対照動物は傷の部位に何も処置を施さなかった。
【0292】
これらの大型動物の研究結果により、対照の部位は多重癒着を生じ、周りの部位の組織も脅かしていたが、p-GlcNAc膜およびゲル構成物は有効に癒着の形成を防止した。
【0293】
対照および処置した部位からのサンプルを別にSEM を使用して調べたが、処置組織に比較して対照の部位では繊維症の増加を示した。
【0294】
18.実施例: p-GlcNAc物質の生分解能
本節に示す実施例は、in vitroおよびin vivo での生分解能および吸収速度を制御しうる本発明のp-GlcNAc物質を製造できることを明示する。
【0295】
18.1 材料および方法
p-GlcNAc物質: 基本型 I は、フッ化水素酸を化学試薬として利用して化学的方法で、上記の第5.3.2 節に記載の方法により製造した。基本型 Iは100%アセチル化p-GlcNAcであった。
【0296】
基本型 3A のp-GlcNAc出発物質は、フッ化水素酸を化学試薬として利用して化学的方法で上記第5.3.2 節に記載の方法により製造した。その後、p-GlcNAc物質を上記の第5.4 節に記載の方法で脱アセチル化した。詳細には、p-GlcNAc物質を40%NaOH 溶液で60℃で30分処理した。生じた基本型 3A は測定の結果30% 脱アセチル化されていた。
【0297】
基本型 4のp-GlcNAc出発物質は、フッ化水素酸を化学試薬として利用して化学的方法で、上記の第5.3.2 節に記載の方法により製造した。その後p-GlcNAc物質を40%NaOH 溶液で60℃で30分処理して脱アセチル化した。次いで、繊維を蒸留水に懸濁し、−20℃で凍結しそして凍結乾燥した。基本型 4も測定の結果30% 脱アセチル化されていた。
【0298】
腹部移植モデル: Sprague-Dawley系白色ラットを腹部移植モデル実験に使用した。動物を麻酔し外科手術の準備をし、そして皮膚と腹部筋肉を切開した。盲腸の位置を確かめ、そして持ち上げた。p-GlcNAc物質の1 cm×1 cm膜を盲腸の上に置き、そして切開部をナイロン糸で閉じた。盲腸上に物質を置かなかったものを対照動物とした。
【0299】
移植の14および21日後に動物を切開した。内移植と外移植処置の間の写真を撮った( 図23A 〜E)。盲腸のサンプルを移植後に組織病理学のために準備した。
【0300】
in vitro p-GlcNAc 分解リゾチーム- キチナーゼ分析: 分析はN-アセチルグルコサミンの比色分析であり、そして以下のとおりに実施した。反応サンプル 150μl を13×100 mmのガラス使い捨て試験管に2通りずつ分注した。0.25 M リン酸カリウム緩衝液(pH 7.1) 25 μl を各試験管に添加し、その後0.8 M ホウ酸カリウム溶液(pH 9.8) 35 μl を添加した。試験管を2分以内に直ちに氷浴に浸す。その後、サンプルを氷浴から取り出し、新たに調製したDMAB試薬1mlを添加し、そしてサンプルが渦巻くように混合した。DMAB( ジメチルアミノベンズアルデヒド) 試薬は氷酢酸 70 mlおよび11.6 N( 濃)HCl 10 mlをp-ジメチルアミノベンズアルデヒド8 gに添加して製造した。
【0301】
標準曲線を作成するために、以下の手順を実施した。GlcNAc 原液を0.010 M酢酸ナトリウム緩衝液(pH4.5) で0.1 mg/ml に希釈し、そして希釈したGlcNAc溶液 0 μl 、20μl 、30μl 、90μl または120 μl を一組の試験管に添加した。その後、0.010 M 酢酸ナトリウム緩衝液(pH 4.5) 150μl 、130 μl 、60μlまたは30μl をそれぞれ試験管に添加した。次いで、0.25 M リン酸カリウム緩衝液(pH 7.1) 25 μl および0.8 M ホウ酸カリウム緩衝液(pH 9.8) 35 μl を各試験管に添加した。同様の手順により2組目の試験管を調製する。
【0302】
試験管に栓をし、そして100 ℃で正確に3 分間、沸騰させる。その後、試験管を氷浴に浸す。試験管を氷浴から取り出し、新たに上記の節で記載した方法に従って調製したDMAB試薬 1 ml を各試験管に添加する。試験管を37℃で20分間インキュベートする。各試験管の内容物の吸光度を585 nMで読み取る。吸光度はできるかぎり迅速に読み取らねばならない。標準曲線をグラフ用紙に描き、そして反応サンプルのN-アセチルグルコサミンの濃度を測定するのに使用する。典型的な標準曲線を図23に示す。
【0303】
18.2 結果
リゾチームによる分解に対するp-GlcNAc膜物質の相対的感受性を検定する実験により、p-GlcNAc物質のin-vitroでの生分解能を研究した。p-GlcNAc膜を10 mM酢酸緩衝液中の過剰のリゾチームにさらし、そしてN-アセチルグルコサミンのその後の放出を、上記の第18.1節に記載されている検定を使用して測定した。
【0304】
これらの実験の結果により、部分的脱アセチル化膜は、よりリゾチームに消化されやすく( 図24を参照) 、そしてさらに、リゾチームの分解速度は脱アセチル化の程度に直接に関係している(50%と25% の脱アセチル化p-GlcNAc膜の分解速度を比較している図25を参照) ことが示された。
【0305】
さらに、in-vivo でのp-GlcNAc物質の生分解能に関する実験を実施した。これらの実験では腹部移植モデルを利用した。以下に記載する3 種のp-GlcNAc物質を試験した。
【0306】
試験したp-GlcNAc物質:
1) 完全アセチル化p-GlcNAc( 基本型 1 と名付けた);
2) 部分的脱アセチル化p-GlcNAc膜( 基本型 3A と名付けた);および
3) 凍結乾燥した部分的脱アセチル化p-GlcNAc膜( 基本型 4と名付けた)。
【0307】
完全アセチル化p-GlcNAc( 基本型 1) は、図26A 〜26C に示すように21日以内に分解吸収された。部分的脱アセチル化p-GlcNAc膜( 基本型 3A)は図26D 〜26Eに示すように、14日以内に完全に分解吸収された。凍結乾燥した部分的脱アセチル化p-GlcNAc膜( 基本型 4) は移植の21日後でも完全には分解吸収されていなかった。
【0308】
組織病理学的分析により、ひとたびp-GlcNAc物質が分解吸収されれば、処置動物および対照動物から得られた組織サンプルの間で、検知できる組織学的相違は存在しないことが示された。
【0309】
19. 実施例: p-GlcNAc の止血作用の研究
ここに記載する実験は、出血防止に対する、本発明のp-GlcNAc物質の有効性を研究するものである。p-GlcNAc物質の出血防止での有効性をさらに市販の止血製剤と比較する。
【0310】
19.1 材料および方法
p-GlcNAc および対照物質: 部分的脱アセチル化( 約70%)p-GlcNAc膜を、フッ化水素酸を化学試薬として利用して、上記の第5.3.2.節に記載の化学分離技術、および上記の第5.4 節に記載の技術を使用して調製した。2cm ×1cm 片を使用した。p-GlcNAc- 乳酸ゲル( プロピレングリコールおよび水の中で形成した4%p-GlcNAc- 乳酸) を上記の第17.1節に記載の方法に従って製造した。脾臓と肝臓の出血研究に利用した対照物質はGelfoam(商標名)(Upjohn社) であった。小血管(small blood vessel)出血の研究に使用した対照物質はGelfoamTMおよびAvitene(商標名)(Medchem Products社) であった。
【0311】
試験動物: ニュージーランド白色ウサギを使用した。3 匹は脾臓に2 箇所の、そして肝臓に1 箇所の創傷を受けた。4 匹は尾部の腸間膜動脈系の同様の大きさの血管に、5 箇所の外科的創傷を受けた。
【0312】
外科手術の準備: 動物をケタミン(ketamine)HCl およびキシラジン(xylazine)を用いて麻酔した。動物を仰向けに寝かせ、そして腹部の毛を全て除去した。その後、腹部をポビドン- ヨウ素および70% イソプロピルアルコールで洗浄し、そして無菌の外科手術のために滅菌した布で覆った。
【0313】
肝臓/ 脾臓の外科手術: 正中線切開をし、そして脾臓または肝臓のいずれかを露出して体外に出し、そしてモイストラップスポンジ(moist lap sponge)で包んだ。直径3 〜4 mm のコルクボーラーを使用して、器官の一方の端に約2 mmの深さの円形の創傷を作った。ひとたび脾臓組織を除去したら、事前に秤量した4 インチ×4 インチのガーゼスポンジを使用して、脾臓の創傷から失われた全ての血液を1分間吸収した。スポンジを再秤量し、その特定の創傷から失われた血液の量を量った。その後、処置物質の1種を創傷に施して試験動物を処置した。止血までの時間と止血までに失われた血液の量を記録した。
【0314】
最初の創傷の止血が達成された後、脾臓の二番目の創傷と肝臓の1 箇所の創傷を同様の手順に従って作った。
【0315】
小血管の外科手術: 正中線切開をし、そして小腸を体外に出し尾部腸間膜動脈系を露出した。腸をモイストラップスポンジで包み、そしてほぼ同じ大きさの5本の血管を確認した。メスを用いて1 本の血管に約1 mmの深さの創傷を作った。事前に秤量した4 インチ×4 インチのガーゼスポンジを使用して1 分間、血管の創傷から失われた全ての血液を吸収した。スポンジを再秤量して、その特定の創傷から失われた血液の量を測定した。その後、処置物質の1 つを創傷に施して実験動物を処置した。止血までの時間と止血までに失われた血液の量を記録した。
【0316】
最初の創傷の止血が達成された後、さらに4 箇所の創傷を同様の手順に従って作った。
【0317】
19.2 結果
p-GlcNAc物質の、ラット動物モデルの脾臓および肝臓での出血防止能について試験した。試験したp-GlcNAc物質は: 1)部分的脱アセチル化( 約70%)p-GlcNAc;および 2)p-GlcNAc-乳酸ゲル( プロピレングリコールおよび水の中で形成した4% p-GlcNAc-乳酸)。これらのp-GlcNAc物質の効果をGelfoamTM(Upjohn 社) と比較した。
【0318】
各物質を3 回試験した( 脾臓で2回、肝臓で1回)。p-GlcNAcをベースとする物質は両者とも、適用後、最初の1分以内の出血の防止においてGelfoamTMに匹敵する効果を示した。
【0319】
p-GlcNAcをベースとする物質は別の利点を有する。詳細には、処置の間、適所にとどめておく必要がなく、体内に放置でき、体内で2 〜3 週間以内に分解吸収され(GelfoamTMはこの効果を示さない) 、全身および最小限の観血的外科手術の両方に適合する。
【0320】
次に、p-GlcNAcをベースとする物質の小血管での出血を防止する効力を研究し、そして市販の止血製剤と比較した。
【0321】
各物質を5 回(1匹の動物は2 回、そして他の3 匹は1 回) 試験した。p-GlcNAc膜およびゲル構成物は容易に部位に適用され2 分以内に出血を防止した。GelfoamTMは、その機能を発揮するためにその場に保持しなければならないが、p-GlcNAc物質と同じく2分以内に止血が達成された。コラーゲンから成る繊維状物質であるAviteneTMは、扱いにくくそして止血するのに5分以上を要した。
【0322】
それ故、ここに記載する結果により、ここで試験したp-GlcNAc物質は有効で便利な止血剤であることが実証された。
【0323】
20. 実施例: p-GlcNAcドラッグデリバリーシステム
抗腫瘍性薬剤を悪性皮膚癌および結腸癌性腫瘍部位に送りだし、送りだされた抗腫瘍剤が腫瘍に対して治療上の強い影響力を発揮するp-GlcNAc物質の有効な使用例を示す研究をここに記載する。
【0324】
20.1 材料および方法
p-GlcNAc- 乳酸ドラッグデリバリー組成物: 5'- フルオロウラシル(5'-FU)およびp-GlcNAc- 乳酸の混合物を以下のように作成した; 5'-FU(50 mg/mL) 0.5 mLをプロピレングリコール 0.5 mL と混合し、そして4% p-GlcNAc-乳酸 2.0 mL を添加し混合した。p-GlcNAc- 乳酸は上記の節に記載の技術を使用して製造した。広範に混合した後も、5'FUは完全にはp-GlcNAc- 乳酸ゲルに溶解しなかった。完全に混合したと仮定すると、5'-FU の最終濃度は6.25 mg/mLとなろう。
【0325】
マイトマイシン(Mito)とp-GlcNAc- 乳酸の混合物を以下のように作成した; Mito( 凍結乾燥粉末) 0.5 mgをプロピレングリコール 5 ml に溶解し、そしてMito溶液 0.5 ml をMPT の4% p-GlcNAc-乳酸調製物 0.5 mL と混合し最終Mito濃度が0.2 mg/ml であり最終p-GlcNAc- 乳酸の濃度が2%となるようにした。Mitoは容易にp-GlcNAc- 乳酸ゲルに溶解するので、この物質は相溶性であった。
【0326】
p-GlcNAc膜5'FUデリバリー組成物: 5'- フルオロウラシル(5'-FU)のサンプルを、フッ化水素酸を化学試薬として利用し、上記の第5.3.2 節に記載の化学分離法を用いて製造した純粋なp-GlcNAc膜物質のディスク内に固定した。ここに記載する各ディスクは直径1.5 cmであった。
【0327】
高用量(HD)ディスクを製造するために、5'-FU の50 mg/mL溶液 0.64 mLを純粋なp-GlcNAc 約8 mg を含有する懸濁液と混合した。混合物を数時間、静置し5'-FU がp-GlcNAcに吸収されるのを促進し、そしてその後、55℃で3.5 時間乾燥した。生じたHDディスクは、5'-FU を総量で32 mg 含有していたが、これは通常、癌患者に投与する5'-FU の14日間の総用量の約二倍に等しい。
【0328】
低用量(LD)の5'-FU を含有するp-GlcNAcディスクをそのLDディスクが5'-FU 17 mg を含有する以外は同様の方法で製造した。この量はヒトの体重 Kg 当たりの5'-FU の通常量に基づいて、実験マウスの体重に標準化した、通常の14日間のヒトの用量に等しい量である。
【0329】
試験動物: 5'-FU の研究のために、HT-29 結腸癌性腫瘍を発症させるために、SCID( 重症複合型免疫不全症) マウスに、標準組織培養法により得られたHT-29 結腸癌細胞(ATCC; 接種物当たり1 ×105 細胞) を側腹部の皮下注射により接種した。これらの注射により14〜21日で触診できる腫瘍を発生した。腫瘍を切開し、そして壊死組織を切り捨てた。HT-29 結腸癌性腫瘍を3 ×3 ×3 mm片に薄切りした。
【0330】
実験的SCIDマウスを標準用量のアベチン(avetin)で腹腔内注射により麻酔し、そしてHT-29 結腸癌腫瘍の切片を各マウスの盲腸に移植した。詳細には、各マウスの腹部を外科的に切開して盲腸の位置を確かめ、メスで切って小さく切開した。3 ×3 ×3 mm腫瘍薄片を、5.0 絹縫合糸を使用して盲腸の上の切開を覆って縫合した。その後、腹部をクレーアダム釘(Clay Adam staple)を使用して閉じた。
【0331】
全てのマウスを1 匹ずつケージにいれ、そして2 週間摂食した。全てのマウスが健康で、そして2 週間の期間の終わりに閉塞性結腸を有するものはなかった。
【0332】
14日目に、各マウスを麻酔し、そして再び開腹した。腫瘍の成長を測定した(長さおよび水平の寸法)。その後、腫瘍をp-GlcAc/抗腫瘍薬で処置するか、または対照として使用した。
【0333】
6 匹のマウスをp-GlcNAc- 乳酸5'FUの研究に使用し、そして15匹をp-GlcNAc膜5'FUの研究に使用した。
【0334】
マイトマイシンの研究のために、9 匹のSCIDマウスに、A431扁平上皮細胞皮膚癌細胞(ATCC; 接種物当たり1 ×105 細胞) を皮下注射して移植した。14日以内に全てのマウスで腫瘍が生じた。
【0335】
処置: p-GlcNAc- 乳酸5'FUの研究では、5'フルオロウラシル(5'-FU)含有p-GlcNAcゲル混合物を腫瘍塊上の皮膚領域に1 日に1 回「塗布」して動物を処置した。腫瘍の大きさの測定を毎日実施した。対照動物には、p-GlcNAcのみで処置したもの、5'-FU 無し、処置を全く受けていないものを含んでいた。
【0336】
p-GlcNAc膜5'FUの研究では、結腸の上に腫瘍を14日間、成長させた後、薬剤含有p-GlcNAc膜のディスクを外科的に直接その表面に移植して、SCIDマウスのHT29結腸腫瘍を処置した。移植手順後14日目に、マウスを犠牲にした。薬物含有p-GlcNAc膜の移植の直前の0 日および実験の最後の14日目に腫瘍体積を測定した。対照動物には、5'-FU を含まないp-GlcNAc膜で処置したもの、および全く処置を受けていないものを含んでいた。これとは別に、2 匹の動物はHDおよびLD規定食餌法に相当する用量の5'-FU の全身的な注入を毎日受けた。
【0337】
p-GlcNAc- 乳酸Mito研究では、p-GlcNAc- 乳酸5'FUの研究のように、3 匹の動物をMito含有混合物で処置して、動物を毎日処置した。3 匹はMitoを含まないp-GlcNAcで処置し、2 匹は処置をせず、そして1 匹はプロピレングリコールを受けた。
【0338】
20.2 結果
20.1.1 p-GlcNAc - 乳酸5'FU
p-GlcNAc- 乳酸5'FUドラッグデリバリーシステムの腫瘍の大きさに対する効果の研究を意図した実験を、上記の第20.1節に記載のように実施した。
【0339】
各腫瘍の最大の長さと幅の寸法を測定し、そしてこれらの寸法を使用して断面積を計算した。断面積値を以下の表Xに示す。
【表10】
p-GlcNAc- 乳酸5'FU処置動物を対照と比較したデータを図27〜28に示す。表Xおよび図27〜28に要約したデータにより、5'-FU 含有p-GlcNAc- 乳酸ゲルで処置したラットのHT-29 皮下腫瘍は対照と比較して増殖速度が有意に遅滞することが明らかに示唆される。その増殖はp-GlcNAc- 乳酸ゲルの対照と比較して2.5倍、そして未処置の対照と比較して4倍遅くなっている。
【0340】
20.2.2 p-GlcNAc- 乳酸MITO
p-GlcNAc- 乳酸5'FUドラッグデリバリーシステムの腫瘍の大きさに対する効果を研究するように意図された実験も、上記の第20.1節に記載のように実施した。
【0341】
各腫瘍の最大の長さと幅の寸法を測定し、これらの寸法を使用して断面積を計算した。断面積値を下記の表XIに示した。
【表11】
p-GlcNAc- 乳酸Mito処置動物を対照と比較したデータを図29〜30に示す。表XIおよび図29〜30に要約したデータにより、マイトマイシン含有p-GlcNAc- 乳酸ゲルで処置したラットで増殖した腫瘍は増殖速度が有意に遅延することが明らかに示唆される。その増殖はp-GlcNAc- 乳酸ゲル対照と比較して4 倍、そして未処置の対照と比較して4 倍遅くなった。
【0342】
20.2.3 p-GlcNAc膜5'FU
次に、p-GlcNAc膜5'FUドラッグデリバリーシステムの皮膚癌腫瘍の大きさに対する効果を研究することを意図する実験を、上記の第20.1節に記載のように実施した。
【0343】
種々の処置により生じた体積の変化百分率を含む、研究中に得られた腫瘍体積のデータを下記の表XIIに要約する。腫瘍は円柱状の形と仮定した。体積を、幅および長さを測定し、そして次式: V=πr2l[式中、半径r は幅の0.5倍であり、そしてlは長さである]を使用して計算した。
【表12】
図31は、上記の表XIIに表されているデータの一部を要約する。図31に示すように、データにより、高用量(HD)5'-FU 含有p-GlcNAc膜で処置した腫瘍は増殖を停止させ、そして全ての場合に、実際に有意に小さくなったことが強く示唆される。低用量(LD)ポリマー物質により、疾患が安定化し、そして腫瘍の大きさがわずかに減少した。対照的に、対照動物の腫瘍の大きさは急速に増加しつづけた。IVで処置した3 匹の対照動物のうち2 匹が研究中に死亡し、このことは当量の5'-FUの全身的なデリバリーは致死量にあたるが、一方p-GlcNAcポリマーを介する部位特異性デリバリーは動物から疾病を取り除くのに有効であることが示されたのは興味深い。
【0344】
20.3 結果
この節に示したデータにより、抗腫瘍性薬剤の部位特異性デリバリーは腫瘍の増殖を遅らせ後退させる正の効果を有することが強く示唆される。2 種の異なる構成物、すなわち、p-GlcNAc- 乳酸およびp-GlcNAc膜構成物を有するように製造されたp-GlcNAcドラッグデリバリー組成物を使用して有用な結果が得られた。さらに、2 種の異なる抗腫瘍剤、5'-FU およびMitoを使用して有効な結果が得られた。それ故、本発明のp-GlcNAcドラッグデリバリーシステムは、例えば、必要な腫瘍細胞部位に特異的に薬剤を送りだすのに有用な抗腫瘍活性を示す。
【0345】
ここに記載した本発明の多くの修飾と変法がその精神と範囲を逸脱することなく可能であることは明白である。上述の特定の実施態様は例示としてのみ掲載し、本発明は添付された請求の範囲の用語によってのみ制限されるものである。
【図面の簡単な説明】
【0346】
【図1】100% p-GlcNAc の化学構造を示す図である。
【図2】p-GlcNAcの炭水化物分析、ガスクロマトグラフィー−質量分光分析データを示す図である。
【図3A】純化p-GlcNAcの固体膜 (solid membrane) の円偏光二色性スペクトルを示す図である。
【図3B】脱アセチル化p-GlcNAcの固体膜の円偏光二色性スペクトルを示す図である。
【図4A】機械力精製法(上部)および化学/生物学的精製法(下部)により調製された純化珪藻p-GlcNAcの薄膜の赤外スペクトルを示す図である。
【図4B】第5.5 節に詳述した方法に従って膜に注型された市販の2つの「キチン」調製物の赤外スペクトルを示す図である。
【図4C】第5.4 節に詳述した方法に従って熱変性(上部)および化学的脱アセチル化(下部)により改質された純化p-GlcNAcの赤外スペクトルを示す図である。
【図4D】第5.3.2 節に詳述した化学/生物学的精製法を用いて、珪藻タラシオシラ・フルビアチリス(Thallasiosira fluviatilis) から誘導されたp-GlcNAc膜の赤外スペクトルを示す図である。
【図4E】オートクレーブ滅菌後に、第5.3.1 節に記載したような機械力精製法により調製されたp-GlcNAc膜の赤外スペクトルを示す図である。
【図5A】第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析結果を示す図である。
【図5B】第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析結果を示す図である。
【図6A】第5.3.1 節に記載した機械力精製法によって調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:4190x)。
【図6B】第5.3.1 節に記載した機械力精製法によって調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:16,250x)。
【図7A】第5.3.2 節で化学/生物学的精製法の説明において記載したHF処理によるp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:5270x)。
【図7B】第5.3.2 節で化学/生物学的精製法の説明において記載したHF処理によるp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:8150x)。
【図8A】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:5270x)。
【図8B】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:16,700x)。
【図9A】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真を示す図である(倍率:200x)。
【図9B】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:1000x)である。
【図9C】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:5000x)である。
【図9D】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:10000x)である。
【図9E】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:20000x)である。
【図10A】第5.3 節に記載した細胞溶解/中和精製法を用いて最初に調製し、ジメチルアセトアミド/塩化リチウム中に溶解し、そして以下の第5.5節に記載したように水中でマットに再沈殿させた物質から作られた純化p-GlcNAc膜の走査電子顕微鏡写真(倍率:1000x)である。
【図10B】第5.3 節に記載した細胞溶解/中和精製法を用いて最初に調製し、ジメチルアセトアミド/塩化リチウム中に溶解し、そして以下の第5.5節に記載したように水中でマットに再沈殿させた物質から作られた純化p-GlcNAc膜の走査電子顕微鏡写真(倍率10,000x)である。
【図11A】脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真(倍率:1000x)である。
【図11B】脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真(倍率:10,000x)である。
【図12A】珪藻の写真を示す図である。
【図12B】珪藻の写真を示す図である。
【図13】本発明の可能なp-GlcNAc誘導体および脱アセチル化p-GlcNAc誘導体の一部を示す略図である。
【図14】p-GlcNAc膜の存在下または不在下で増殖させた細胞の生存能実験の結果を示す図である。
【図15A】p-GlcNAc膜上で増殖させた形質転換マウス繊維芽細胞のSEM顕微鏡写真(倍率:1000x)である。
【図15B】p-GlcNAc膜上で増殖させた形質転換マウス繊維芽細胞のSEM顕微鏡写真(倍率:3000x)である。
【図16A】第13.1節に記載した方法に従って調製したコラーゲン単独対照物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16B】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16C】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16D】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16E】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図17A】図16A のコラーゲン単独対照物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図17B】図16B のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図17C】図16C のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図17D】図16D のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図18】各炭素原子の面積を得るために、次いで CH3(面積)対C原子(面積)の比を計算するために使用した変換NMRデータ曲線を示す図である。
【図19】典型的なp-GlcNAc C13-NMRスペクトルを示す図である。
【図20】CH3(面積)対C原子(面積)比の計算値を表す変換NMRスペクトルデータを示す図である。上図はデータのグラフ表示であり、下図:データの数値表示である。
【図21A】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21B】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21C】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21D】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21E】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21F】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21G】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図22A】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22B】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22C】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22D】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22E】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22F】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22G】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図23】第18.1節に記載した手順を用いて得られた典型的な標準曲線を示す図である。
【図24】p-GlcNAcリゾチーム消化データを示す図である。
【図25】p-GlcNAcリゾチーム消化データを示す図である。
【図26A】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26B】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26C】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26D】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26E】p-GlcNAcのin vivo 生分解性データを示す図である。
【図27】第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図28】第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図29】第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図30】第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図31】p-GlcNAc/5'FU 高用量(棒1)、p-GlcNAc/5'FU 低用量(棒2)、またはp-GlcNAc膜のみ(棒3)で処置した、あるいは処置を施さなかった(棒4)動物の動物あたりの腫瘍の大きさの平均変化パーセントを表す図である。
【技術分野】
【0001】
1. 序論
本発明は、第一に、簡単に調製および精製されたポリ-β-1→4-N-アセチルグルコサミン (p-GlcNAc) 多糖種に関するものである。本発明のp-GlcNAcは、その構成成分の単糖がβ-1→4 配置で結合されている高分子量のポリマーであって、タンパク質を含まず、また単一アミノ酸や他の有機および無機の汚染物質を実質的に含まないものである。加えて、p-GlcNAcの誘導体および再構成物も記述される。本発明はさらに、微小藻類(好ましくは珪藻植物)出発物質源から本発明のp-GlcNAcを精製する方法に関する。さらにまた、本発明はp-GlcNAcを誘導体化しかつ再構成する方法に関する。その上に、本発明は純化p-GlcNAc、その誘導体および/またはその再構成物の使用に関するものである。
【背景技術】
【0002】
2. 発明の背景
今日では、p-GlcNAcから部分的に構成された多糖類の特性、活性および用途に関する文献は膨大な数にのぼる。こうした物質の仲間は総称して「キチン」と呼ばれており、一方、脱アセチル化されたキチン誘導体は「キトサン」と呼ばれている。これらの用語が最初に用いられたころ、つまり1823年頃、キチンは完全にアセチル化されたもので、キトサンは完全に脱アセチル化された組成のものであるといったように、キチンとキトサンは別個の、明確に規定された、特異で、不変の化学種として常に自然界に存在すると考えられていた。しかし、「キチン」と「キトサン」が実際には非常にあいまいな用語であるということが分かったのは約1世紀後であった。これらの用語は、明確に規定された化合物と言うよりもむしろ、実際には広範に異なる物理的および化学的性質を示す化合物のファミリーを指す用語である。これらの差異は生産物のさまざまな分子量、異なるアセチル化度、そして共有結合された種特異的タンパク質、単一アミノ酸、無機汚染物質といった汚染物質の存在によるものである。今日でさえ、「キチン」と「キトサン」はあいまいに用いられており、実際には、あまり明確には規定されていない、多くの異なる化合物の混合物を指している。
【0003】
例えば、甲殻綱の外殻、菌類の菌蓋などの従来の供給源から単離された「キチン」の特性は予測し得ないほどにバラツキがあった。このようなバラツキは、種の違いばかりでなく、「キチン」産生種の生化学的特性の一部を決定するさまざまな環境的および季節的影響が原因となっている。事実、原料の予測し得ない可変性がキチンを基礎とした産業の進展を大いに阻んでいる。
【0004】
現在、純粋な、完全にアセチル化されたp-GlcNAc、すなわち有機または無機不純物によって汚染されていない生産物、を出発物質源から単離および調製したとする報告は科学文献中にまったく見当たらない。McLachlan ら (非特許文献1:McLachlan, A.G. ら, 1965, Can. J. Botany 43:707-713)はキチンの単離を報告したものの、その後の研究で、得られた「純化」物質は実際にはタンパク質と他の汚染物質を含んでいたことが判明した。
【0005】
脱アセチル化および部分脱アセチル化されたキチン調製物は、高い反応性、濃密な陽電荷、強力な金属キレート化能、タンパク質を共有結合する能力、さらに多くの水性溶媒への溶解性といった有益と思われる化学的性質を示す。ところが、上述したように、これらの調製物の予測できない可変性のため、こうした不均一化合物の有用性は厳しい制限を受ける。例えば、現在入手できる「キチン」および「キトサン」は再現性のないデータおよび許容できないほどに広い実験結果の変動をもたらす。さらに、入手可能な調製物はとても均質または純粋とは言えず、また、調製物の構成成分もこうした調製物が諸用途(特に医学用途)に受け入れられるほどに十分に再現性があるとは言えない。かくして、その性質を高度に再現できて、しかも簡単に製造できる、純粋な、精製されたキチンおよびキトサンの調製物は、非常に望まれているにもかかわらず、目下のところ存在していない。
【非特許文献1】マクラクラン・エー・ジー(McLachlan, A.G.)ら、カナディアン・ジャーナル・オブ・ボタニー(Canadian Journal of Botany, Can. J. Botany)、1965年、第43巻、p.707-713
【発明の開示】
【0006】
3. 発明の概要
本発明は、第一に、単離されかつ簡単に調製された純粋なp-GlcNAc種に関する。本発明のp-GlcNAcは、その構成成分の単糖がβ-1→4 配置で結合されている高分子量のポリマーであって、タンパク質を含まず、また他の有機および無機の汚染物質を実質的に含まないものである。
【0007】
本発明の重要性は、予測できない原料の変動性の問題を解決できた点にある。初めて、生物医学的に純粋で、一致した性質を示す高分子量のp-GlcNAcを簡便な手段でかつ商業規模で調製することが可能となった。本発明において調製された物質は高度に結晶性であり、規定培地で増殖させた多数の海洋微小藻類(好ましくは珪藻植物)のうちの1種の注意深く制御された無菌培養物から得られる。
【0008】
本発明はさらに、p-GlcNAcの誘導体および再構成物ならびに前記誘導体および再構成物の製造方法を提供する。かかる誘導体はポリグルコサミンおよびその誘導体を含むがこれらに限らず、また再構成物は膜、フィラメント、不織繊維、スポンジおよび三次元マトリックスを含むがこれらに限らない。さらにまた、本発明は、微小藻類(好ましくは珪藻植物)源からの本発明のp-GlcNAcの精製方法に関する。その上に、本発明は精製p-GlcNAc、その誘導体および/またはその再構成物の使用に関する。中でも、これらは生物医学、医薬品および化粧品産業(すべて最高純度の出発物質を必要とする)のような産業に関係する新規な商業用途において使用される。例えば、本発明のp-GlcNAc物質は制御可能な生分解性を示すように構成することができ、さらに、徐放性ドラッグデリバリーシステムの一部として、細胞カプセル化システムとして、術後癒着の防止処置として使用することができる。
【0009】
4. 図面の説明
図1. 100% p-GlcNAc の化学構造。「n」は約4,000 〜150,000 の範囲の整数を表し、約4,000 〜15,000が好適である。
【0010】
図2. p-GlcNAcの炭水化物分析、ガスクロマトグラフィー−質量分光分析データ。黒の四角は、以下の第5.3.2 節に記載した、化学/生物学的方法の酸処理/中和変法を用いて精製されたp-GlcNAcを表す。
【0011】
図3A. 純化p-GlcNAcの固体膜 (solid membrane) の円偏光二色性スペクトル。
【0012】
図3B. 脱アセチル化p-GlcNAcの固体膜の円偏光二色性スペクトル。純化p-GlcNAcで観測された211 nmの最小値および195 nmの最大値(図3A)の消失は、以下の第5.4 節に記載したような使用条件下での完全な脱アセチル化を示す。
【0013】
図4A. 機械力精製法(上部)および化学/生物学的精製法(下部)により調製された純化珪藻p-GlcNAcの薄膜の赤外スペクトル分析。
【0014】
図4B. 以下の第5.5 節に詳述した方法に従って膜に注型された市販の2つの「キチン」調製物の赤外スペクトル分析。
【0015】
図4C. 以下の第5.4 節に詳述した方法に従って熱変性(上部)および化学的脱アセチル化(下部)により改質された純化p-GlcNAcの赤外スペクトル分析。
【0016】
図4D. 以下の第5.3.2 節に詳述した化学/生物学的精製法を用いて、珪藻タラシオシラ・フルビアチリス(Thallasiosira fluviatilis) から誘導されたp-GlcNAc膜の赤外スペクトル分析。
【0017】
図4E. オートクレーブ滅菌後に、以下の第5.3.1 節に記載したような機械力精製法により調製されたp-GlcNAc膜の赤外スペクトル分析。
【0018】
図5A. 以下の第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析。チャートはピーク強度、面積、および基準対照に対する比率を示す。ピークの全面積の比率。
【0019】
図5B. 以下の第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析。グラフはピークの全面積の比率を示す。
【0020】
図6A−6B. 以下の第5.3.1 節に記載した機械力精製法によって調製されたp-GlcNAc膜の透過型電子顕微鏡写真 (TEM)。倍率:6A: 4190x ;6B: 16,250x。
【0021】
図7A−7B. 以下の第5.3.2 節で化学/生物学的精製法の説明において記載したHF処理によるp-GlcNAc膜の透過型電子顕微鏡写真 (TEM)。倍率:7A: 5270x;7B: 8150x。
【0022】
図8A−8B. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜の透過型電子顕微鏡写真 (TEM)。倍率:8A:5270x ;8B: 16,700x。
【0023】
図9A. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:200x。
【0024】
図9B. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:1000x。
【0025】
図9C. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:5000x。
【0026】
図9D. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:10000x。
【0027】
図9E. 以下の第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真。倍率:20000x。
【0028】
図10A-10B. 以下の第5.3 節に記載した細胞溶解/中和精製法を用いて最初に調製し、ジメチルアセトアミド/塩化リチウム中に溶解し、そして以下の第5.5節に記載したように水中でマットに再沈殿させた物質から作られた純化p-GlcNAc膜の走査電子顕微鏡写真。倍率:10A: 1000x;10B: 10,000x。
【0029】
図11A-11B. 脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真。倍率:11A:1000x ;11B: 10,000x。
【0030】
図12A-12B. 珪藻の写真。珪藻の細胞体から伸びているp-GlcNAc繊維に注目のこと。
【0031】
図13. 本発明の可能なp-GlcNAc誘導体および脱アセチル化p-GlcNAc誘導体の一部を示す略図(S. Hirano, “Production and Application of Chitin and Chitosan in Japan", in “Chitin and Chitosan", 1989, Skjak-Braek, Anthonsen and Sanford 編, Elsevier Science Publishing Co., pp. 37-43からの改作)。
【0032】
図14. p-GlcNAc膜の存在下または不在下で増殖させた細胞の生存能実験。黒丸(黒○):p-GlcNAcマトリックス上で増殖させた細胞;白丸(○):マトリックスの不在下で増殖させた細胞。
【0033】
図15A-15B. p-GlcNAc膜上で増殖させた形質転換マウス繊維芽細胞のSEM顕微鏡写真。倍率:15A:1000x ;15B: 3000x。
【0034】
図16A. 以下の第13.1節に記載した方法に従って調製したコラーゲン単独対照物質の走査電子顕微鏡写真 (SEM)。倍率 100x。
【0035】
図16B. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は3:1に等しく、最終濃度はコラーゲンが7.5 mg/ml で、p-GlcNAcが0.07 mg/mlであった。倍率 100x。
【0036】
図16C. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は1:1に等しく、最終濃度はコラーゲンが5.0 mg/ml で、p-GlcNAcが0.12 mg/mlであった。倍率 100x。
【0037】
図16D. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は2:2に等しく、最終濃度はコラーゲンが10.0 mg/mlで、p-GlcNAcが0.25 mg/mlであった。倍率 100x。
【0038】
図16E. 以下の第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)。コラーゲン懸濁液:p-GlcNAc懸濁液の比は1:3に等しく、最終濃度はコラーゲンが2.5 mg/ml で、p-GlcNAcが0.25 mg/mlであった。倍率 100x。
【0039】
図17A. 上記図16A のコラーゲン単独対照物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0040】
図17B. 上記図16B のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0041】
図17C. 上記図16C のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0042】
図17D. 上記図16D のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM。倍率 100x。
【0043】
図18. 各炭素原子の面積を得るために、次いで CH3(面積)対C原子(面積)の比を計算するために使用した変換NMRデータ曲線。
【0044】
図19. 典型的なp-GlcNAc C13-NMRスペクトル。各ピークはこの分子中の特異な炭素原子の該スペクトルへの寄与を表す。
【0045】
図20. CH3(面積)対C原子(面積)比の計算値を表す変換NMRスペクトルデータ。上図:データのグラフ表示;下図:データの数値表示。
【0046】
図21A-G. 種々の溶媒中で調製された三次元p-GlcNAcマトリックス。詳細には、p-GlcNAcマトリックスは蒸留水(図21A, 図21D)、蒸留水中の10% メタノール(図21B)、蒸留水中の25% メタノール(図21C)、蒸留水中の10% エタノール(図21E)、蒸留水中の25% エタノール(図21F)、および蒸留水中の40% エタノール(図21G)中で調製した。倍率:200x。各図面には200ミクロンの縮尺線を示してある。
【0047】
図22A-G. 蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞。倍率:100x (図22A, 22E) 、500x (図22B)、1000x(図22C, 22F) 、5000x(図22D, 22G)。各図面には5、20、50または200ミクロンの縮尺線を示してある。
【0048】
図23. 以下の第18.1節に記載した手順を用いて得られた典型的な標準曲線。このような標準曲線は、以下の第18.1節に記載したリゾチーム−キチナーゼアッセイにおいて使用した。
【0049】
図24. p-GlcNAcリゾチーム消化データ。ここに示したグラフは、p-GlcNAc膜をリゾチームで消化するときの時間の経過にともなうN-アセチルグルコサミンの蓄積を表す。このグラフは完全アセチル化p-GlcNAcと部分(50%) 脱アセチル化p-GlcNAcとの分解速度を比較したものであり、部分脱アセチル化p-GlcNAcの分解速度が完全アセチル化p-GlcNAc物質の分解速度を実質的に上回ることを実証した。
【0050】
図25. p-GlcNAcリゾチーム消化データ。ここに示したグラフは、p-GlcNAc膜をリゾチームで消化するときの時間の経過にともなうN-アセチルグルコサミンの蓄積を表す。このグラフは2つの部分脱アセチル化p-GlcNAc膜(すなわち、25%および50% 脱アセチル化p-GlcNAc膜)の分解速度を比較したものである。このデータによれば、50% 脱アセチル化p-GlcNAc膜の分解速度が25% 脱アセチル化p-GlcNAc膜よりも実質的に高いことから、脱アセチル化のパーセントが増加するにつれて分解速度は増加することが分かる。
【0051】
図26A-26E. p-GlcNAcのin vivo 生分解性データ。図26A-26C は、以下の第18.1節に記載したように基本型1(完全アセチル化p-GlcNAc)膜を腹部に移植したラットを示す。図26A は移植0日目のラットを、図26B は移植後14日目のラットを、そして図26C は移植後21日目のラットを示す。図26D-26E は、以下の第18.1節に記載したように基本型3A(凍結乾燥および部分脱アセチル化を行ったp-GlcNAc)膜を腹部に移植したラットを示す。図26D は移植0日目のラットを、図26Eは移植後14日目のラットを示す。
【0052】
図27. ここに示したグラフは、以下の第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0053】
図28. ここに示したグラフは、以下の第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0054】
図29. ここに示したグラフは、以下の第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0055】
図30. ここに示したグラフは、以下の第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す。
【0056】
図31. ここに示した棒グラフは、p-GlcNAc/5'FU 高用量(棒1)、p-GlcNAc/5'FU 低用量(棒2)、またはp-GlcNAc膜のみ(棒3)で処置した、あるいは処置を施さなかった(棒4)動物の動物あたりの腫瘍の大きさの平均変化パーセントを表す。棒1と棒2についてはn=4 、棒3と棒4についてはn=2 である。
【発明を実施するための最良の形態】
【0057】
5. 詳細な説明
まず初めに、本発明の精製p-GlcNAc種、p-GlcNAc誘導体およびそれらの再構成物の物理的特性について記述する。次に、微小藻類(好ましくは珪藻植物)出発物質源から本発明のp-GlcNAc種を精製する方法を記述する。第三に、p-GlcNAcの誘導体および再構成物、ならびに前記誘導体および再構成物の製造方法を提示する。最後に、本発明のp-GlcNAc、p-GlcNAc誘導体および/またはp-GlcNAc再構成物の使用を提示する。
【0058】
5.1 p-GlcNAc
本発明のp-GlcNAc多糖種は、ゲル浸透クロマトグラフィー測定に基づく重量平均が約800,000ダルトンから約3,000 万ダルトンの範囲にある高分子量のポリマーである。このような分子量は、β-1→4 配置において約4,000から約150,000個の結合されたN-アセチルグルコサミン単糖を有するp-GlcNAc種を表す。N-アセチルグルコサミン単糖の数は約4,000から約15,000個が好ましい(図1参照)。
【0059】
本発明のp-GlcNAcの可変性は非常に低く、またその純度は非常に高い。これら点は両方とも化学および物理的基準によって立証される。これらの基準のなかには、化学組成および非多糖不純物が含まれる。第一に、二つの異なる精製法(これらは両方とも後述の第5.3節に記述する)を用いて生産したp-GlcNAcの化学組成データを下記の表Iに示す。表から分かるように、二つの方法によって生産されたp-GlcNAcの化学組成は、実験誤差の限界内で、p-GlcNAcの処方組成と同一である。第二に、これも表Iから分かるように、生産されたp-GlcNAcは検出可能なタンパク質不純物を含まず、遊離のアミノ酸等の他の有機不純物を実質的に含まず、そして灰分および金属イオン等の無機不純物を実質的に含まない(本発明のp-GlcNAcは約0.05%までの微量金属を含んでもよい)。さらに、本発明のp-GlcNAcは結合水の百分率が非常に低い。
【表1A】
本発明の純粋なp-GlcNAcは、図2に示すものと実質的に類似した炭水化物分析プロフィールを示す。本発明の純粋なp-GlcNAcの主な単糖は、N-アセチルグルコサミンである。さらに、本発明の純粋なp-GlcNAcは単糖グルコサミンを含まない。
【0060】
本発明のp-GlcNAcの円二色性(CD)およびシャープな赤外スペクトル(IR)を図3A、および図4Aならびに4Dにそれぞれ示す。これらの図は、後述の第5.3節に記述する方法を用いて作成した物質の分析を表すものである。このような物理的データは、本発明のp-GlcNAcが高純度で高結晶性であることを確証する。CDおよびIRデータを得るために用いた方法は、後述第6節の実施例に記載する。
【0061】
本発明の純粋なp-GlcNAcのNMR分析は、図5A、5B、18Aおよび18Bに見られるものと実質的に類似のパターンを示す。このようなNMRパターンは、本発明のp-GlcNAcが十分にアセチル化されたポリマーであるということと一致するデータを示すばかりでなく、本発明のp-GlcNAc種においては不純な有機物が存在しないことをも示している。
【0062】
後述の第5.3節に記述する方法を用いて生産し、そして後述の第8および9節に記載の実施例に示す、本発明のp-GlcNAcの電子顕微鏡構造を図6Aから図9Eに掲げる。
【0063】
本発明のp-GlcNAcは高度な生体適合性を示す。生体適合性は種々の技法によって確認することができる。これらの技法は以下のものを含むがそれらだけに限定されない。すなわち、動物被験者への溶出試験、筋肉内移植、皮内または全身注射等の方法である。簡単に述べると、溶出試験(米国薬局方XXII, 1990, pp.1415-1497; 米国薬局方XXII, 1991, 補足5, pp.2702-2703)は試験物質抽出物の生体適合性を評価するために設計されており、抽出可能な細胞毒性物質に感受性の哺乳動物細胞培養系(例えばL929細胞系)の、試験物質に応答しての生物的反応性をアッセイするものである。後述第10節に記載の実施例は、本発明のp-GlcNAcの高い生体適合性を示す。
【0064】
5.2 p-GlcNAcの微小藻類源の生産方法
5.2.1 p-GlcNAcの微小藻類源
本発明のp-GlcNAcは微小藻類、好ましくは珪藻類により生産され、そしてそれより精製されうる。いくつかの属の珪藻類、およびそれらの属に属する多数の種をp-GlcNAcの原料源として使用できる。これらの珪藻類はそれぞれ、細胞体より伸長するp-GlcNAcから成る繊維を生産する。このような珪藻類の写真については、図12A〜12Bを参照されたい。本発明のp-GlcNAcの生産のための原料源として使用可能な珪藻類は、コアミケイソウ属(Coscinodiscus)、タイコケイソウ属(Cyclotella)、およびタラシオシラ属(Thalassiosira)属のメンバーを含み、なかでもタラシオシラ属が好ましいが、これらだけに限定されない。
【0065】
コアミケイソウ属の中で、本発明のp-GlcNAcの生産のための原料源として使用可能な珪藻種は、コンシナス(concinnus) およびラジアタス(radiatus)種を含むが、これらだけに限定されない。タイコケイソウ属のうち使用可能な珪藻は、カスピア(caspia)、クリプティカ(cryptica)およびメネギニアナ(meneghiniana)種を含むが、これらだけに限定されない。本発明のp-GlcNAcの原料源を生産するために使用可能なタラシオシラ属の珪藻は、ニッツショイデス(nitzschoides)、イースティバリス(aestivalis)、アンタークティカ(antarctica)、デシフェンズ(deciphens)、エクセントリカ(eccentrica)、フロリダナ(floridana)、フルビアチリス(fluviatilis) 、グラビダ(gravida)、グイラーディー(guillardii)、ヒアリナ(hyalina)、ミニマ(minima)、ノルデンスキオルディー(nordenskioldii)、オセアニカ(oceanica)、ポリコルダ(polychorda)、シュードナナ(pseudonana); ロチュラ(rotula)、ツビフェラ(tubifera)、ツミダ(tumida) およびワイスフロギ(weissflogii) 種を含み、なかでもフルビアチリスおよびワイスフロギ種が好ましいが、これらだけに限定されない。
【0066】
上記のような珪藻類は、例えば海洋植物プランクトンコレクションセンター(Center for Collection of Marine Phytoplankton)のビグロー海洋科学研究所(Bigelow Laboratory for Ocean Sciences) (McKown Point, West Boothday Harbor, Maine, 04575)の培養コレクションより入手可能である。
【0067】
5.2.2 珪藻類の増殖方法
上記第5.2.1に記載の珪藻類はいずれも、例えば本節に記述する方法を用いて増殖させることができる。新規な珪藻培養は、無菌条件下で栄養培地に成熟珪藻培養物のアリコートを接種することにより始める。この栄養培地は、他のあらゆる微生物を含まないものでなければならない。したがって、この栄養培地の調製に用いられる水、有機成分、および無機成分を含むすべての物質は無菌でなければならない。さらに、この操作に関与するすべての手順は厳正な無菌条件下で実施されることが必須である。すなわち、すべての容器、1つの容器から他の容器への物質の移し替えのすべて、等は無菌の環境で実施されなければならない。一度に調製される栄養培地の量は、新しい培養を始めるのに必要な量を越えてはならない。例えば、珪藻培養用の容器として約1平方フィートの表面を占めるフェーンバック(Fernback)フラスコを用いることができ、そして、そのような容器は珪藻生物の最適な増殖のために1リットルの栄養培地を必要とする。
【0068】
栄養培地の調製は以下の操作を含む:
a)海水の獲得および処理
b)蒸留水および脱イオン水の調製
c)一次栄養ストックの調製
d)栄養作業ストックの調製
e)最終栄養培地の調製。
【0069】
濾過した海水を、例えば海洋生物学研究所(the Marine Biology Laboratory)(Woods Hole, Massachusetts)より得ることができる。海水容器は5℃で保管しなければならない。必要な場合は、孔径が0.45ミクロンのニトロセルロース濾過膜(Millipore, Inc.)を用いて、ブフナー濾過ユニットにより必要量の水を濾過することができる。次に、海水を例えば121℃で、1 リットルにつき15分間オートクレーブにかけることにより滅菌する。この滅菌過程の完了後、好ましくは溶液が約5℃に達するのを可能とする寒い部屋に移して、蓋をした容器を直ちに冷却する。使用するときは、溶液を室温にする。
【0070】
標準的な装置および手順を用いて水道水を蒸留し、脱イオン化して、回収し、無菌の、しっかり蓋をした、好ましくはガラス製の容器に保管する。
【0071】
栄養培地の調製に必要な原液の調製の際に従うことができる処方を以下に掲げる。このような処方は指針として使用されるべきであるが、ここに記述するp-GlcNAcの調製方法に充分な微小藻類珪藻の増殖を維持することが可能な栄養培地の調製に寄与する、これらの処方の日常的な変法もまた、本発明の範囲内にあることが理解されねばならない。
【0072】
I.微量金属一次ストック(TMPS)
a.39 mM CuSO4・5H2O(硫酸銅(II)五水和物)(9.8 g 硫酸銅(II)/L)
b.7.5 mM ZnSO4・7H2O(硫酸亜鉛七水和物)(22 g 硫酸亜鉛/L)
c.42 mM CoCl2・6H2O(塩化コバルト(II)六水和物)(10 g 塩化コバルト(II)/L)
d.91 mM MnCl2・4H2O(塩化マンガン(II)四水和物)(18 g 塩化コバルト(II)/L)
e .26 mM NaMoO4・2H2O(モリブデン酸ナトリウム二水和物)(6.3 g モリブデン酸ナトリウム/L)
f.153.5 mM H2SeO3 (亜セレン酸)(12.9 g 亜セレン酸/L)。
【0073】
各栄養分を孔径が0.2 mm未満のフィルターを用いて無菌濾過する。
【0074】
II. ビタミン一次ストック(VPS)
a. 1 mg/ml ビタミンB12
b. 0.1 mg/ml ビオチン
両方のストックを孔径が0.2 mm未満のフィルターを用いて無菌濾過する。
【0075】
III. ナトリウム塩作業ストック(SSWS)
a.硝酸ナトリウム作業ストック: 0.88 M (75 g NaNO3/L)
b.リン酸ナトリウム一塩基一水和物作業ストック:36.2 mM NaH2PO4・H2O (5 g NaH2PO4・H2O/L)
c.メタケイ酸ナトリウム九水和物作業ストック: 0.11 M Na2SiO3・9H2O (30 g Na2SiO3・9H2O/L)
SSWSのそれぞれを孔径が0.2 mm未満のフィルターを用いて無菌濾過する。
【0076】
IV. 微量金属作業ストック(TMWS)
11.7 mM Na2EDTA ( エチレンジアミン四酢酸、二ナトリウム塩二水和物)(4.36 g/L)
11.7 mM FeCl3・6H2O(塩化鉄(III)六水和物)(3.15 g/L)
1 ml/Lの、上に列挙した6つの一次微量金属ストックのそれぞれ。
【0077】
孔径が0.2 mm未満のフィルターを用いて無菌濾過する。微量金属作業ストックは、新しい栄養培地を調製するたびに新たに調製しなければならない。
【0078】
V.ビタミン作業ストック(VWS)
1.0 μg/ml ビオチン (1.0 ml 一次ビオチンストック/100 ml)
1.0 μg/ml ビタミンB12 (0.1 ml ビタミンB12 一次ストック/100 ml)
20 mg のチアミンHCl (塩酸チアミン/100 ml)
孔径が0.2 mm未満のフィルターを用いて無菌濾過する。新しい栄養培地を調製するたびに、新たなビタミン作業ストックを調製しなければならない。
【0079】
以下に、栄養培地の調製および珪藻培養のために使用することのできる技法を記述する。これらの技法に加えて、ここに記述する調製方法に充分な珪藻の増殖を維持することが可能な栄養培地および手順をもたらし得る、ここに記述する処方および/または手順における日常的変法はすべて、本発明の範囲内に入ることが理解されねばならない。
【0080】
栄養培地は、例えば以下のようにして調製することができる:濾過し、滅菌した海水各1リットルに、1 mlのNaNO3 作業ストック、1 mlのNaH2PO4・H2O作業ストック、1 mlの微量金属作業ストック、および1 mlのNa2SiO3・9H2O 作業ストックを加える。Na2SiO3・9H2O の添加と同時に、2 mlの1 N HCl を加え、溶液を振とうして混合する。次に 1.5 ml の1 N NaOHを加え、溶液を再度振とうして混合する。最後に、0.5 mlのビタミン作業ストックを加える。
【0081】
新しい珪藻培養物を増殖させるため、7 mlの成熟培養物(細胞密度約 1x105 個/ml)を、100 mlの栄養培地(上記の方法にしたがって調製することができる)を含有する滅菌容器に移す。接種を受けた培養物を次に8日間下記の条件下でインキュベートする:
温度:20℃
常時照明
攪拌:毎朝および毎夕1回2〜3秒間フラスコを穏やかに回す。
【0082】
8日間のインキュベーション後、インキュベートした培養物 80 mlを無菌条件下で1000 ml の栄養培地に移す。この培地は、例えばチーズクロスで覆われた綿ウール栓によって保護された2.8 Lのフェーンバックフラスコに入れられている。このような培養物は、所望の細胞密度までインキュベートし、増殖させる。または、新しい珪藻培養物に接種するのに使用される。ひとたび培養物が所望の細胞密度に達したならば、培養物のp-GlcNAc繊維を収穫し、そして後述の第5.3 節に記述するような方法を用いて、本発明のp-GlcNAcを精製することができる。約7〜8、好ましくは7.4の培養pHを維持するため、培養液にCO2を溶解させることができる。このような中性pH環境の維持は、各珪藻培養物から得られるp-GlcNAcの収量を大幅に増大させる。
【0083】
5.3 p-GlcNAc繊維の単離、精製および濃縮方法
本節では、上記第5.2節に記述するような珪藻培養物からのp-GlcNAc繊維の調製に用いることができる方法を提示する。
【0084】
微小藻類、好ましくは珪藻、の原料源からp-GlcNAcを精製する下記の方法のそれぞれは、非常に純度の高い、混入物のない、結晶性p-GlcNAcを生ずる一方、各方法は特異的な特性および有利な特徴を有するp-GlcNAcを産する。例えば、下記の第5.3.1 節に記述する機械力による方法で精製された本発明のp-GlcNAcは、細胞のp-GlcNAcへの結合のためにより優れた基質を提供するp-GlcNAc膜をもたらす。下記の第5.3.2 節に記述する第二の方法、すなわち化学的/生物的方法は、機械力による方法によってもたらされる平均p-GlcNAc収量よりも遙に高い平均収量をもたらす。さらに、第5.3.2 節の化学的/生物的方法の一部として記述されている酸処理/中和変法は、極めて長いp-GlcNAc繊維で(ある繊維は100 μm を越える)、かつ2〜3千万ダルトンという高い分子量を有するp-GlcNAcをもたらす。
【0085】
5.3.1 純粋なp-GlcNAcを調製するための機械力による方法
p-GlcNAc繊維は、培養物の内容物を適切な機械力にかけることによって珪藻細胞体から分離することができる。このような機械力は、例えばコロイドミル、超音装置、または発泡装置により生成される剪断力、または、例えばワーリング(Waring)ブレンダーによって生成される切断力を含むが、これらだけに限定されない。
【0086】
こうして得られた珪藻細胞体およびp-GlcNAc繊維の懸濁液を、次に分離する。例えば、この懸濁液を一連の遠心分離工程にかける。この工程は、細胞体からp-GlcNAcを分離し、もしあったとしても目に見える綿状物質の殆ど無い、透明な上清を生ずる。遠心分離工程には、角度固定ローターおよび約10℃という温度が好ましい。速度、継続時間、および必要とされる遠心分離工程の総数は、例えば使用する特定の遠心分離ローター等によって変わる。しかし、これらのパラメーターの数値決定は、当業者には明白であろう。
【0087】
次に、当業者に周知の技法を用いて上清中のp-GlcNAc繊維を濃縮する。これらの技法は吸引および濾過装置を含むが、これらだけに限定されない。
【0088】
最後に、濃縮したp-GlcNAc繊維を、例えば蒸留脱イオン水、HClおよびエタノール、または他の適切な溶剤、好ましくは有機および無機物質の両方が溶解するアルコール等の溶剤を用いて洗浄する。
【0089】
後述の第7節に記載の実施例は、p-GlcNAc精製のためのこの方法の使用を示す。
【0090】
5.3.2 p-GlcNAc精製のための化学的/生物的方法
この方法においては、p-GlcNAc繊維を以下に詳述する化学的および/または生物学的作用物質にさらすことによって珪藻細胞体より分離する。
【0091】
珪藻培養物を、珪藻細胞壁を弱めることが可能な化学物質で処理することができる。これは、p-GlcNAc繊維の構造を変えずに、その放出をもたらす。このような化学物質はフッ化水素酸(HF)を含むが、これだけに限定されない。または、成熟した珪藻培養物を、生物的過程を変更することが可能な生物的作用物質で処理することができる。このような作用物質を用いてp-GlcNAc繊維の合成を阻害し、その結果すでに存在する繊維を放出させることができる。たとえば、このような作用物質は酵素N-アセチルグルコサミニル-P-トランスフェラーゼの阻害剤であるポリオキシン-Dを含むが、これだけに限定されない。
【0092】
次に、上記の化学的または生物的作用物質の一つで処理した珪藻培養物の細胞体およびp-GlcNAc含有繊維を分離する。例えば、処理した珪藻培養物の内容物を、二つの別個の層の形成を可能とするように、静置することができる。上の層は主にp-GlcNAc繊維を含有し、他方下の層は細胞体を含有する。p-GlcNAc繊維を含有する上の層をサイフォンで吸い上げ、下の層の沈殿した細胞性物質を残すことができる。
【0093】
次に、サイフォンで吸い上げたp-GlcNAc繊維を含有する層をさらに精製して、p-GlcNAc繊維を損なわない洗剤を用いて処理することによりタンパク質および他の望ましくない物質を除去する。このような洗剤は、硫酸ドデシルナトリウム(SDS)を含むが、これだけに限定されない。
【0094】
p-GlcNAc繊維を珪藻細胞体から分離するためにHF処理のような酸処理を用いる場合は、繊維を分散させるための工程を設けることができる。このような工程は、繊維分散のための機械力の使用(例えば、繊維をワーリングブレンダーによる分散に付す工程等)を含むが、これだけに限定されない。
【0095】
または、酸で処理した懸濁液を、洗剤処理によるさらなる精製にかける前に、任意選択の工程において中和してもよい。このような中和は、一般に、懸濁液のpHを約1.8から約7.0に変える。この中和は、例えば適当量の 1 M Tris (pH 8.0)の添加、または適当量の水酸化ナトリウム(NaOH)の添加により行なわれる。一般に、中和は本明細書で論じられている他の精製方法よりも、実質的に長さの長い純粋なp-GlcNAc繊維をもたらす。
【0096】
次に、吸引濾過装置の使用等の当業者に周知の技法を用いて、精製p-GlcNAc繊維を濃縮することができる。最後に、蒸留脱イオン水、HClおよびエタノール、または他の適切な溶剤、好ましくは有機および無機物質の両方が溶解するアルコール等の溶剤を用いた一連の工程により、p-GlcNAc繊維を洗浄する。
【0097】
後述の第8節に記載の実施例は、このような精製方法の上首尾な使用を示す。
【0098】
5.4 p-GlcNAcの誘導体化
種々の制御された条件および手順を用いることにより、本発明の純粋な、十分アセチル化されたp-GlcNAcを、広範な異なる化合物に誘導体化することができる。これらの化合物のいくつかを示す図については、図13を参照されたい。このような誘導体化された化合物は、以下に詳述する化学的および/または酵素的手段により修飾された、部分的に、または完全に脱アセチル化されたp-GlcNAcを含むが、これらだけに限定されない。さらに、p-GlcNAcまたはその脱アセチル化誘導体は、硫酸化、リン酸化および/または硝酸化されることにより誘導体化されうる。さらに、以下に詳しく述べるように、本発明のp-GlcNAcまたは脱アセチル化p-GlcNAcより、O-スルホニル、N-アシル、O-アルキル、N-アルキル、デオキシハロゲン、およびN-アルキリデンおよびN-アリーリデン、および他の誘導体が調製できる。本発明の脱アセチル化p-GlcNAcは、種々の有機塩および/または金属キレートを調製するためにも使用できる。さらに、本発明のp-GlcNAcまたはその誘導体は、種々の分子のうち任意のものを共有または非共有結合により自身に結合させることが可能である。そしてさらに、本発明のp-GlcNAcまたはその誘導体を、均一(uniform)且つそれぞれ別個な(離散的な(discrete))分子量特性を有する分子群を生じる、制御された加水分解条件下に置くことができる。
【0099】
本発明のp-GlcNAcの一つ以上の単糖ユニットを脱アセチル化し、ポリ-β-1→4-N-グルコサミン種を形成することが可能である。ポリ-β-1→4-N-アセチルグルコサミン種の単糖ユニットのそれぞれが脱アセチル化されている本発明のポリ-β-1→4-N-グルコサミン種は、約640,000ダルトンから約2千4百万ダルトンの分子量を有する。約640,000ダルトンから約2.4 百万ダルトンが好ましい。このような分子量範囲を有する種は、β-1→4 配置において約4,000から約150,000個の結合されたグルコサミン単糖を有する種を表す。グルコサミン単糖の数は約4,000から約15,000が好ましい。ポリ-β-1→4-N-グルコサミン種の単糖ユニットの少なくとも一つはアセチル化されたままであることができる。約25%から約75%のアセチル化が好ましく、約30%のアセチル化が最も好ましい。
【0100】
本発明のp-GlcNAcを塩基で処理することにより脱アセチル化し、遊離のアミノ基を有するグルコサミンをもたらすことができる。この加水分解過程は、濃い水酸化ナトリウムまたは水酸化カリウムの溶液を用いて高温で実施することができる。しかし、脱アセチル化の程度を正確にコントロールし、また多糖分子の炭水化物主鎖の分解を避けるため、p-GlcNAc脱アセチル化にはキチンデアセチラーゼ酵素を用いる酵素的方法を使用することが好ましい。このようなデアセチラーゼによる酵素的方法は、当業者に周知であり、また参照としてここにその全体を組み入れている米国特許第5,219,749号に記述されているように実施することができる。
【0101】
本発明のp-GlcNAcの単糖ユニットの一つ、または二つ以上を誘導体化し、以下に示すように、少なくとも一つの硫酸基を含有させるか、またはリン酸化もしくは硝酸化することができる:
【化1】
【0102】
ここで、水素に代わるRおよび/またはR1、および/または-COCH3に代わるR2は、硫酸(-SHO3)、リン酸(-P(OH)2)または硝酸(-NO2)基でありうる。
【0103】
以下にこのようなp-GlcNAc誘導体を調製する方法を記述する。本節に記述の方法を実施する前に、まずp-GlcNAc原料を液体窒素中で凍結乾燥し、凍結し、そして微粉砕することが有利であろう。
【0104】
硫酸化p-GlcNAc誘導体は、例えば二段階工程により生成しうる。第一段階において、例えばTokuraら(Tokura, S.ら,1983, Polym. J. 15:485)が記述する技法を用いて、本発明のp-GlcNAcおよび/またはp-GlcNAc誘導体よりO-カルボキシメチルp-GlcNAcを調製しうる。第二に、例えば N,N-ジメチル-ホルムアミド-三酸化硫黄を用いて、当業者に周知の技法、例えばSchweiger (Schweiger, R.G., 1972, Carbohydrate Res. 21:219) が記述する技法にしたがって、硫酸化工程を実施することができる。こうして得た産物をナトリウム塩として単離することができる。
【0105】
本発明のリン酸化p-GlcNAc誘導体は、当業者に周知の技法、例えば Nishiら(Nishi, N.ら, 1986, "Chitin in Nature and Technology", Muzzarelliら編、Plenum Press, New York, pp. 297-299)が記述する技法を用いて調製することができる。簡単に述べると、p-GlcNAc/メタンスルホン酸混合物を攪拌しながら約0℃から約5℃の温度で五酸化リン(約0.5から4.0モル当量) で処理する。処理は約2時間行なう。次に、当業者に周知の標準的技法を用いて、得られた産物を沈殿させ、洗浄する。例えば、エーテル等の溶剤を用いてサンプルを沈殿させ、遠心分離し、エーテル、アセトン、またはメタノール等の溶剤で洗浄し、乾燥させる。
【0106】
硝酸化p-GlcNAc誘導体は、当業者に周知の技法、例えば SchoriginおよびHalt(Schorigin, R.およびHalt, E., 1934, Chem. Ber. 67:1712) が記述する技法を用いて調製することができる。簡単に述べると、p-GlcNAcおよび/またはp-GlcNAc誘導体を濃硝酸で処理し、安定した硝酸化産物を形成することができる。
【0107】
本発明のp-GlcNAcの単糖ユニットの一つ、または二つ以上は、以下に示すように、スルホニル基を含有することができる:
【化2】
【0108】
ここでR3はアルキル、アリール、アルケニルまたはアルキニルの一部分でありうる。このような誘導体は当業者に周知の方法、例えば Kuritaら (Kuritaら, 1990, Polym. Prep. [Am. Chem. Soc., Div. Polym. Chem.] 31:624-625)が記述する方法を用いて生成させることができる。簡単に述べると、アルカリp-GlcNAc水溶液をトシルクロリドのクロロホルム溶液と反応させ、次に低温でこの反応を円滑に進行させる。
【0109】
本発明のp-GlcNAc、またはその脱アセチル化誘導体の単糖の一つ、または二つ以上は、以下に示すように、一つ、または二つ以上のO-アシル基を含有することができる:
【化3】
【0110】
ここで、水素に代わるR4および/またはR5は、アルキル、アルケニル、またはアルキニルの一部分でありうる。そして、R6はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体の一例は、Komai (Komai, T.ら,1986, "Chitin in Nature and Technology", Muzzarelliら編、Plenum Press, New York, pp. 497-506)が記述する方法等の周知の方法を用いて生成させることができる。簡単に述べると、p-GlcNAcを多数の適切な塩化アシルのうち任意のものとメタンスルホン酸中で反応させ、カプロイル、カプリール、ランロイル、またはベンゾイル誘導体を含むが、それらだけに限定されないp-GlcNAc誘導体を生じさせる。
【0111】
本発明の脱アセチル化p-GlcNAcの一つ、または二つ以上の単糖は、以下に示すようにN-アシル基を含有することができる:
【化4】
【0112】
ここで、R7はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、当業者に周知の技法、例えば Hiranoら (Hirano, S.ら, 1976, Carbohydrate Research 47:315-320)が記述する技法を用いて達成することができる。
【0113】
脱アセチル化p-GlcNAcは、多数の有機酸水溶液において可溶性である。このようなp-GlcNAcを含有する溶液に、選択されたカルボン酸無水物を水性メタノール性酢酸中で添加すると、N-アシルp-GlcNAc誘導体の形成をもたらす。
【0114】
本発明の脱アセチル化p-GlcNAc、またはその脱アセチル化誘導体の一つ、または二つ以上の単糖は、以下に示すようにO-アルキル基を含有することができる:
【化5】
【0115】
ここで、R8はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、当業者に周知の技法を用いて達成することができる。例えば、Mareshら(Maresh, G. ら,"Chitin and Chitosan", Skjak-Braek, G.ら,編、1989, Elsevier Publishing Co., pp. 389-395) が記述する手順を用いる。簡単に述べると、脱アセチル化p-GlcNAcをジメトキシエタン(DME) 中に分散し、過剰な酸化プロピレンと反応させる。反応時間は約24時間で、反応はオ−トクレーブ中で40から90℃で起こる。次に、混合物を水で希釈し、濾過する。DMEは蒸留によって除去しうる。最後に、凍結乾燥によって最終産物を単離することができる。
【0116】
本発明のp-GlcNAcの一つ、または二つ以上の単糖ユニットは、以下に示すようにアルカリ誘導体でありうる:
【化6】
【0117】
このような誘導体は、当業者に周知の技法を用いて得ることができる。例えば、Noguchiら (Noguchi, J.ら,1969, Kogyo Kagaku Zasshi 72:796-799) が記述する方法が使用できる。簡単に述べると、p-GlcNAcを減圧(vacuo)下でNaOH(好ましくは43%)に約2時間約0℃で浸す。次に、例えばバスケット遠心機を用いた遠心および機械的圧縮により過剰のNaOHを除去する。
【0118】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すようにN-アルキル基を含有することができる:
【化7】
【0119】
ここで、R9はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、例えば、前述のMareshら(Maresh, G.ら,"Chitin and Chitosan", Skjak-Brack, G.ら編、1989, Elsevier Publishing Co., pp. 389-395) がO-アルキルp-GlcNAc誘導体の生産のために記述するような手順を用いて達成することができる。
【0120】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すように少なくとも一つのデオキシハロゲン誘導体を含有することができる:
【化8】
【0121】
ここで、R10はF、Cl、Br、またはIでありうるが、Iが好ましい。このような誘導体は当業者に周知の技法を用いて得ることができる。例えば Kuritaら (Kuritaら, 1990, Polym. Prep. [Am. Chem. Soc., Div. Polym. Chem.] 31:624-625)が記述する手順を用いることができる。簡単に述べると、トシル化p-GlcNAcをジメチルスルホキシド中でハロゲン化ナトリウムと反応させる。p-GlcNAcのトシル化は、アルカリp-GlcNAc水溶液を塩化トシルのクロロホルム溶液と反応させることにより実施することができる。このような反応は低温で円滑に進行する。
【0122】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すように塩を形成することができる:
【化9】
【0123】
ここで、R11はアルキル、アルケニル、またはアルキニルの一部分でありうる。このような誘導体化は、当業者に周知の技法を用いて達成することができる。例えば、AustinおよびSennett (Austin, P.R. およびSennett, S., "Chitin in Nature and Technology", 1986, Muzzarelli, R.A.A.ら編、Plenum Press, pp. 279-286)が記述する方法等が使用できる。簡単に述べると、脱アセチル化p-GlcNAcを、例えば酢酸エチルまたはイソロパノール等の有機媒体中に懸濁し、そこに適切な有機酸、例えばギ酸、酢酸、グリコール酸、または乳酸等を加える。混合物をある期間( 例えば1〜3時間) 静置する。反応温度および乾燥温度は、約12℃から約35℃の間で変わるが、好ましくは20℃から25℃である。次に、濾過によって塩を分離し、新鮮な媒体で洗浄し、残留媒体を蒸発させる。
【0124】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すように金属キレートを形成することができる:
【化10】
【0125】
ここで、R12は金属イオン、特に遷移金属の一つでありうる。そしてXは、脱アセチル化p-GlcNAcに存在するアミノ基および置換アミノ基に存在する窒素電子によって確立される供与結合である。
【0126】
本発明のp-GlcNAcの脱アセチル化誘導体の一つ、または二つ以上の単糖ユニットは、以下に示すようにN-アルキリデン基またはN-アリーリデン基を含有することができる:
【化11】
【0127】
ここで、R13はアルキル、アルケニル、アルキニル、またはアリールの一部分でありうる。このような誘導体化は、当業者に周知の技法を用いて達成することができる。例えば、Hiranoら (Hirano, S.ら, 1981, J. Biomed. Mat. Res. 15:903-911)が記述する手順を用いることができる。簡単に述べると、脱アセチル化p-GlcNAcのN-置換反応をカルボン酸無水物および/またはアリールアルデヒドを用いて実施し、アシル- および/またはアリ−リデン誘導体をもたらすことができる。
【0128】
さらに、本発明のp-GlcNAc、またはその脱アセチル化誘導体を制御された加水分解条件下に置くことができる。これは、均一でそれぞれ別個(離散的)な分子量および他の物理的特性を有する分子群をもたらす。そのような加水分解条件は、例えば、酵素リゾチームを用いた処理を含みうる。加水分解の度合いをコントロールするため、p-GlcNAcを様々な期間リゾチームに暴露する。さらに、加水分解の速度は、リゾチーム処理されつつあるp-GlcNAcがどの程度まで脱アセチル化されたかを関数としてコントロールすることができる。脱アセチル化条件は、本節で先に記述した通りでよい。p-GlcNAc分子がより十分に脱アセチル化されるほど、この分子はより十分に加水分解される。分子量の低下の他に起こる物理的特性の変化は、加水分解および/または脱アセチル化処理から引き出されるのかもしれない。広範な加水分解はp-GlcNAcの液化を引き起こす。加水分解/脱アセチル化手順の結果を後述第9節の実施例に示す。
【0129】
さらに、熱変性はp-GlcNAcの結晶構造を修飾する機能を果たしうる。p-GlcNAc産物結晶構造のこのような修飾は、例えばp-GlcNAcの反応性に有利な影響を及ぼしうる。
【0130】
さらに、種々の分子を共有結合または非共有結合によって、本発明のp-GlcNAcの脱アセチル化誘導体に機能的に結合することができる。このような分子は、以下のものを含むがそれらだけに限定されない。すなわち、神経成長因子等の増殖因子、ペプシン等のプロテアーゼ、ホルモン、などのポリペプチド、または、RGD配列、フィブロネクチン認識配列、などのペプチド認識配列、ラミニン、インテグリン、細胞接着分子、等である。脱アセチル化p-GlcNAcの露出した一次アミンに分子を共有結合させることは、例えば特定の長さの化学的スペーサーとして作用する二官能架橋剤を用いる化学結合によって達成しうる。このような技法は当業者に周知であり、また、例えば DavisおよびPreston (Davis, M.およびPreston, J.F. 1981, Anal. Biochem. 116:404-407) およびStarosら(Staros, J.V.ら,1986, Anal. Biochem. 156:220-222) の方法に類似している。簡単に述べると、脱アセチル化された、または部分的に脱アセチル化された本発明のp-GlcNAcに結合させるべきペプチド上のカルボン酸基を活性化し、次にp-GlcNAcに架橋させる。活性化は、例えばリン酸緩衝液中でカルボジイミドEDC (1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド)等の溶液をペプチド溶液に添加することにより達成しうる。好ましくは、この溶液は結合を促進するためにスルホ-NHS (N-ヒドロキシスルホスクシンイミド) 等の試薬をさらに含有する。活性化ペプチドは、高pHの緩衝液、例えば炭酸塩緩衝液(pH 9.0〜9.2)等の中で混合することにより脱アセチル化p-GlcNAcに架橋させることができる。
【0131】
または、上に記述したような分子を、当業者に周知の技法を用いて、非共有結合により脱アセチル化p-GlcNAcに結合させることができる。例えば、選択した一つ、または複数の分子を、凍結乾燥に先だって脱アセチル化p-GlcNAc溶液と混合することができる。
【0132】
または、p-GlcNAcおよび/またはp-GlcNAc誘導体からなるハイブリッド(hybrid)を形成することができる。このようなハイブリッドは、p-GlcNAcおよび/またはp-GlcNAc誘導体に加えて、多数の天然および/または合成物質のうち任意のものを含有することができる。例えば、ハイブリッドはp-GlcNAcおよび/またはp-GlcNAc誘導体プラス一つ、または二つ以上の細胞外マトリックス(ECM)成分より形成されうる。このようなECM成分は、コラーゲン、フィブロネクチン、グリコサミノグリカン、および/またはペプチドグリカンを含みうるが、これらだけに限定されない。ハイブリッドはまた、p-GlcNAcおよび/またはp-GlcNAc誘導体プラス一つ、または二つ以上の合成物質、例えばポリエチレン等より形成されうる。このようなp-GlcNAc/ポリエチレンまたはp-GlcNAc誘導体/ポリエチレンハイブリッドは、ハイブリッド成分を例えばオートクレーブにかけることにより、熱的に結合させて作成することができる。
【0133】
さらに、新規な塑性物質の製造のために、ヨード-p-GlcNAc誘導体を、例えばスチレンと共重合させることが可能である。ヨード-p-GlcNAcは、KuritaおよびInoue (Kurita, K. およびInoue, S., 1989, "Chitin and Chitosan", Skjak-Braek, G.ら編、1989, Elsevier Science Publishing Co., Inc., p.365)の記述する方法と類似の方法により、p-GlcNAcのトシル化およびヨウ素化によって調製することができる。次に、p-GlcNAcのヨード誘導体をニトロベンゼン中に分散し、触媒として塩化スズ(IV)を用いて、スチレンと反応させることができる。
【0134】
コラーゲン/p-GlcNAcハイブリッドの場合には、簡単に述べると、p-GlcNAc懸濁液およびコラーゲン懸濁液を混合して凍結乾燥し、そして好ましくは脱熱水的に(dehydrothermally)架橋する。このようなハイブリッドのコラーゲン種は、天然のものでも合成のものでもよい。また、ヒトのものでも、または非ヒト、例えばウシ等に由来するものでもよい。p-GlcNAc/コラーゲンおよび/またはp-GlcNAc誘導体/コラーゲンハイブリッド物質は均一な特性を示し、また、例えば細胞の結合および増殖のための効率的な三次元マトリックスとして役立つ多孔性マトリックスを形成する。後述の第13節に記載の実施例は、このようなp-GlcNAc/コラーゲンハイブリッドの形成、特性、および有用性を示す。
【0135】
脱アセチル化p-GlcNAcと、例えばヘパリン、アルギン酸ナトリウム、およびカルボキシメチルp-GlcNAc等の化合物との組合せから成るハイブリッドを、本明細書に記述するような技法を用いて作り出すことができる。このような組合せは、例えば膜および繊維等に形成または再形成することができる。
【0136】
脱アセチル化p-GlcNAcと、正および負の両方の電荷を有するポリアニオン、例えばポリアクリル酸またはペクチン等との複合体を作り出すことができる。このような複合体の形成は、Mirelesら(Mireles, C.ら,1992, "Advances in Chitin and Chitosan", Brine, C.J.ら編、Elsevier Publishers, Ltd.)が記述する方法と類似の方法にしたがって達成することができる。脱アセチル化p-GlcNAcおよび、例えばポリアクリル酸、カラギーナンまたはペクチンをそれぞれHClおよびNaClに溶解し、そして等しいpHを有する反応物溶液を混合する。この操作は、例えば廃水処理等に有用な、陽性および陰性の両方の特徴を備えた、効果的な凝集作用を有する分子をもたらす。
【0137】
5.5 再構成物(Reformulations)
本発明のp-GlcNAc、ならびに第5.4節に記述したその脱アセチル化誘導体および/またはそれらの誘導体化物は、溶解して、種々の形および配置に再構成することができる。
【0138】
本発明のp-GlcNAcの溶解は、ジメチルアセトアミド(DMA)/塩化リチウムで処理することにより達成することができる。p-GlcNAcは、5% LiCl (DMAの重量に対して) を含有するDMA溶液中で攪拌することにより容易に溶解する。p-GlcNAc塩等の水溶性p-GlcNAc誘導体は水に溶解する。少なくとも約75%脱アセチル化されたp-GlcNAcは、例えば1%酢酸等の穏やかな酸性溶液中で溶解しうる。水に不溶性のp-GlcNAc誘導体は、有機溶剤中で溶解しうる。
【0139】
フェニルイソシアナートを用いたDMA:LiCl中でのp-GlcNAcの誘導体化は、カルバニル酸塩を生産するのに用いうる。さらに、トルエン-p-スルホニルクロリドを用いたDMA:LiCl中でのp-GlcNAcの誘導体化は、トルエン-p-スルホネートを生産するのに用いうる。
【0140】
次に、溶解した本発明のp-GlcNAc、その脱アセチル化誘導体、および/またはそれらの誘導体化物を沈殿させ、そしてマット、糸(string)、ロープ、微小球、微小ビーズ、膜、繊維、粉末およびスポンジを含むが、これらだけに限定されない形に再構成することができる。さらに、超薄型(すなわち、厚さ約1ミクロン未満)均一膜を形成することができる。
【0141】
このような再構成物は、例えば、純粋なp-GlcNAcは水およびアルコール、好ましくはエタノール等の溶液に不溶性である事実を利用して達成しうる。例えば注入のような従来の手段により、p-GlcNAcを含有するDMA/LiCl混合物を上記の水またはアルコール、好ましくはエタノール溶液に導入すると、再沈殿をもたらし、それゆえ溶解したp-GlcNAcの再構成をもたらす。このような純粋なp-GlcNAcの再構成物を、後述第11節に記載の実施例に示す。水溶性のp-GlcNAc誘導体の場合は、例えば酢酸エチルまたはイソプロパノール等の有機溶剤中で再沈殿させることにより再構成を達成できる。少なくとも約75%脱アセチル化されたp-GlcNAcの再構成は、アルカリ性溶液中で再沈殿させることにより達成できる。水に不溶性のp-GlcNAc誘導体は、例えば水等の水性溶液中で再沈殿させることにより再構成しうる。
【0142】
脱アセチル化p-GlcNAcを酸化セルロースと共に調製して、紙製品の濡れ強度を改善するp-GlcNAc/セルロースハイブリッド物質を作り出すことができる。綿の鎖に似た平坦なリボン状の形を有する脱アセチル化p-GlcNAc鎖によって、酸化綿基質に非常に近似することができる。このような近似は、吸着を促進する力へのファンデルワールス力の寄与を最大にし、その結果、ハイブリッドp-GlcNAc-セルロース物質の濡れ強度特性を増強する。
【0143】
p-GlcNAc膜および三次元p-GlcNAcマトリックスは、膜またはマトリックス内に制御された平均孔径の形成をもたらす方法により作成することができる。膜およびマトリックスにおいて、孔径は使用するp-GlcNAcの量を変えることにより、またメタノールもしくはエタノール(エタノールが好ましい)等の特定の溶剤を約5%から約40%までの特定の量で、膜および/またはマトリックスの形成の前に添加することによりコントロールすることができる。一般に、溶剤の百分率が高いほど、形成される平均孔径は小さくなる。後述の第15節に記載の実施例は、このような多孔性p-GlcNAc構造物の合成および特徴付けを示す。
【0144】
5.6 用途
本発明のp-GlcNAc、ならびに第5.4節に記述したその脱アセチル化誘導体および/またはそれらの誘導体化物、および第5.5節に記述した再構成物は、種々の用途を有する。本明細書に記載のp-GlcNAcおよびその誘導体の有利な特性に加えて、例えば本発明の分子の非毒性、非発熱性、生物分解性、および生体適合性は、農業、化粧品、バイオ医薬産業、動物の栄養および健康、ならびに食品、化学、写真、および医薬産業等の様々な分野において利用される。
【0145】
5.6.1 p-GlcNAc物質のバイオ医薬用途
5.6.1.1 薬物固定化/デリバリー用途
p-GlcNAc物質のバイオ医薬用途は、例えば、酵素および/または薬物固定化/デリバリー方法を含みうる。例えば、本発明のp-GlcNAcおよびその誘導体は、第5.4節に記述したように、興味のあるペプチド(例えば増殖因子)をそこに共有結合させることができる。ペプチド含有p-GlcNAcを、当業者に周知の標準的方法を用いて患者に投与することができる。これらの方法は、注射、移植、関節鏡、腹腔鏡、または類似の手段を含むが、これらだけに限定されない。ペプチド含有p-GlcNAcを患者に導入すると、本発明のp-GlcNAcは生物分解するので結合ペプチドが患者の血流中に徐々に放出され、その結果、制御された薬物デリバリーの方法を提供する。
【0146】
予測できる生分解性を有する脱アセチル化または部分的に脱アセチル化されたp-GlcNAc種を製造することができる。例えば、脱アセチル化のパーセンテージはp-GlcNAc種が分解する速度に影響を与える。一般的には、脱アセチル化のパーセンテージが大きくなると生分解性及び吸収の速度が早くなる。従って、p-GlcNAc生分解性の程度及びインビボでの吸収速度はp-GlcNAcの製造の間に制御できる。そのようなp-GlcNAc物質の製造及び特性化の例は下記の第18節に示す。そのような制御可能な生分解性速度を有するp-GlcNAc物質は膜、ゲル、スポンジ、微小球、繊維等の中に形成することができる。これらのp-GlcNAc生成物は人体中の柔らかな組織及び固い組織の両方の組織に接着し型にはめられた状態になり、縫合することを要しない。p-GlcNAc物質は、例えば、一般的な手術あるいは侵入を最小限にした手術、例えば腹腔鏡手術等の間に適用することができる。
【0147】
生分解性の制御可能な速度を有するp-GlcNAc物質は、例えば、出血組織、器官、及び血管中の止血を促進するため、歯周手術後の修復過程の間の軟組織及び硬組織の分離のための歯周バリアを与えること、手術空間充填物を提供すること、特に皮膚の皺を減らす目的での皮膚における軟組織の増殖を促進すること、失禁を制御する目的での泌尿器括約筋を増強するために有用である。下記第19節に示した実施例は、そのようなp-GlcNAc物質の上記のような用途の1つ、即ち止血を促進することにおける使用を示すものである。
【0148】
さらに、本発明の分子は徐放薬送達ビヒクルとして有用であり、この場合は対象となる薬剤をp-GlcNAcまたはその誘導体によりカプセル化する。薬剤/p-GlcNAcカプセル化物は、例えば上記第5.3.2節に示した化学的/生物学的精製方法の酸処理/中和変法により製造することができる。p-GlcNAc溶液のpHをほぼ中性のpH範囲 (即ち約7.4)に上昇させるのではなく、p-GlcNAcの精製が完了した後にpHを約9.0 に上昇させて塩基性pH環境を形成することができる。より高い塩基性のpHにおいて、本発明のp-GlcNAcまたはその誘導体の構造はより3次元的、即ち「開放的な」形態となると考えられる。pHが低くなると、分子の配置はよりコンパクトなものに戻り、「閉鎖的な」形態となる。従って、対象となる薬剤を高いpHでp-GlcNAcに加え、その後p-GlcNAc/薬剤懸濁物のpHを低下させ、対象の薬剤をp-GlcNAcマトリックス中に「トラップ」あるいはカプセル化することができる。
【0149】
このようなp-GlcNAcカプセル化物は当分野の技術者によく知られた標準的な技術により患者に投与することができ、投与されるとカプセル化物のp-GlcNAcが分解されるにつれてカプセル化された薬剤が徐々に患者の身体に放出されるようにすることができる。
【0150】
p-GlcNAcをベースとするゲル及び膜は、治療用薬剤の送達システムとして種々の用途を有する。そのような用途としては、例えば抗腫瘍剤送達システムがある。本明細書中に記載する薬剤送達システムは任意の抗腫瘍薬とともに使用することができる。そのような薬剤は当分野の技術者によく知られており、p-GlcNAcゲルあるいは膜中に配合して、例えば、腫瘍あるいは手術後の腫瘍の空間の領域に直接、部位特異的徐放送達を与えることができる。このような固定化徐放p-GlcNAc薬剤生成物は、手術後の重要な初期的防護手段として役立ち得る。このようなp-GlcNAc抗腫瘍薬送達システムは、手術では全く、あるいは場合により接近不可能な腫瘍の治療において特に有用であり、これは例えばある種の脳腫瘍にあてはまる。
【0151】
p-GlcNAc抗腫瘍システムの別のターゲットは、限定するものではないが、皮膚、胃腸管系、膵臓、肺、乳房、泌尿器系、子宮癌、及びHIV-関連カポジ肉腫等である。
【0152】
放射能エンハンサーである抗腫瘍薬は、手術の代わりに、あるいは手術後に放射治療が処方される場合に好ましい。そのような薬剤の例は、例えば、5'- フルオロウラシル、マイトマイシン、シスプラチン及びその誘導体、タキソール、アドリアマイシン、アクチノマイシン、ブレオマイシン、ダウノマイシン、メタマイシン等である。
【0153】
抗腫瘍薬の投与量範囲は、患者の全身治療に処方される典型的な 1日当たり投与量と同等でも、低くても高くてもよい。薬剤が腫瘍の部位に局所的に送達されるのでより高い投与量が許容され得る。従って、血液細胞等のその他の組織は直ちには薬剤に露出されない。そのような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0154】
本発明のp-GlcNAc/薬剤送達システムはさらに、感染の治療に使用できる。そのような用途のためには、水溶性あるいは非水溶性の抗生物質をp-GlcNAcをベースとする物質、例えばゲルあるいは膜中に固定/配合することができる。抗生物質は当分野の技術者によく知られており、例えば、ペニシリン、セファロスポリン、テトラサイクリン、アンピシリン、オーレオマイシン、バシトラシン、クロラムフェニコール、シクロセリン、エリスロマイシン、ゲンタマイシン、グラマシジン、カナマイシン、ネオマイシン、ストレプトマイシン、トブラマイシン、バンコマイシン等がある。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0155】
このようなp-GlcNAc抗生物質生成物は、外部的に起こる細菌感染、例えば皮膚、頭皮、皮膚潰瘍もしくは眼表面に起こる細菌感染、あるいは内部的におこる細菌感染、例えば脳、筋肉、腹部の局所的な感染等の治療に使用できる。特筆すべき用途は、HIV-関連日和見感染症の治療である。
【0156】
本発明のp-GlcNAc/薬剤送達システムは抗炎症薬とともに配合して、炎症及び免疫プロセスの機能不全活性を制御することができる。例えば、p-GlcNAcを非ステロイド系抗炎症薬(NSAID)と配合して、リウマチ関節炎、変形性関節炎、全身性狼瘡等の疾患により誘発された局所痛及び炎症を軽減するのに使用することができる。本発明のp-GlcNAcゲルあるいは膜/薬剤送達システムを使用したそのようなNSAIDの局所送達は、胃過敏症、窒素過剰血症、血小板機能不全、肝機能異常のようなNSAIDの副作用を軽減するのに有用である。NSAIDは当分野の技術者によく知られており、アスピリン、エトドラック、フェノプロフェン、ナプロキセン等のようなシクロオキシゲナーゼの阻害剤を含む。その他の抗炎症薬を本発明のp-GlcNAc/薬剤送達システムの一部として使用することができ、例えば、ロイコトリエンのような脂肪炎症メディエーターの阻害剤である。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0157】
本発明のp-GlcNAc/薬剤送達システムはさらに、特異的な真菌症の治療のために、上記のような技術を使用して抗真菌薬とともに配合することができる。抗真菌薬は当分野の技術者によく知られており、例えば、アンホテリシン、アニソマイシン、アンチフンゴン、ブラストマイシン、グリセオフルビン、ニスタチン等がある。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0158】
本発明のp-GlcNAc/薬剤送達システムはまた、特異的な原虫感染症の治療のために、上記のような技術を使用して抗原虫薬とともに配合することができる。抗原虫薬は当分野の技術者によく知られており、例えば、アンチアメビン、アンチプロトジン、モノマイシン、パロモマイシン、トリコマイシン等がある。このような薬剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0159】
本発明のp-GlcNAc/薬剤送達システムは、上記のような技術を使用して殺精子剤とともに配合して効果的な避妊薬を製造することができる。適当な殺精子剤は当分野の技術者によく知られている。そのような殺精子剤の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0160】
本発明のp-GlcNAc/薬剤送達システムはさらに、治療用タンパク質薬剤を使用して配合することができる。そのような配合物は、例えば上記のような技術を使用して製造することができる。そのようなp-GlcNAc治療用タンパク質系を利用することにより、所望のターゲット部位に特異的なタンパク質を直接送達することができ、そのような部位でのそのタンパク質の徐放を行うことができる。可能性のあるタンパク質の例は、限定するものではないが、インシュリン、モノクローナル抗体、乳癌イムノトキシン、腫瘍壊死因子、インターフェロン、ヒト成長ホルモン、リンホカイン、コロニー刺激因子、インターロイキン、ヒト血清アルブミン等がある。このような治療用タンパク質の投与量は当分野の技術者によく知られており、あるいは例えば以下のこの節の最後に記載するように、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。
【0161】
本発明のp-GlcNAc物質はそれ自体免疫的に中性であり、ヒトにおいて免疫反応を生成しないので、上記したような固定化薬剤を保有するp-GlcNAc膜、3D多孔質マトリックス及び/またはゲルを含むp-GlcNAcデバイスは、そのような薬剤を免疫反応なしに送達し得る。ある種の付加的な物質、例えば天然のアルギン酸塩や合成ポリマー等を使用し、p-GlcNAc物質と組み合わせてそのよなデバイスを構築することができる場合もある。
【0162】
上記の薬剤あるいは物質のいずれについても、本明細書に記載するp-GlcNAcをベースとする系と組み合わせたその治療上有効な投与量は、当分野の技術者によく知られた標準的な技術を使用して日常的な業務として決定することができる。「治療上有効な」投与量とは、本明細書中に記載したプロセス及び/または疾患の兆候の軽減をもたらすのに十分なその化合物の量をいうものである。
【0163】
薬剤の毒性及び治療上の効能は細胞培養中あるいは実験動物において標準的な薬学的手法により測定することができ、例えばLD50 (集団の50% に対して致死的な投与量) 及びED50 (集団の50% において治療上有効な投与量) を測定することができる。毒性効果と治療上有効な効果との間の投与量の比率が治療指数であり、LD50/ED50 比で表される。大きい治療指数を示す化合物が好ましい。有害な副作用を示す化合物も使用できるが、非感染細胞に対する重大な障害を最小限にし、それにより副作用を低くするためには、そのような化合物を罹患組織の部位に向かわせる送達システムの設計に注意を払わなくてはならない。
【0164】
細胞培養アッセイ及び動物実験により得られたデータを、ヒトにおける投与量の範囲を計算するのに使用できる。そのような化合物の投与量は、毒性が殆どあるいは全くなく、ED50を含む循環濃度の範囲内にあることが好ましい。投与量は、使用する投与形態及び使用する投与経路に応じてこの範囲内で変化させることができる。本発明の方法に使用される任意の化合物について、治療上有効な投与量を最初に細胞培養アッセイから見積もることができる。細胞培養で決定されたIC50 (即ち、症状の最大阻害の半分の阻害を生じる試験化合物の濃度) を含む循環血漿濃度範囲が得られるように、動物モデル中で投与量を計算することができる。このような情報を使用して、ヒトにおける有用な投与量をより正確に決定することができる。血漿中のレベルは、例えば高速液体クロマトグラフィーにより測定することができる。
【0165】
5.6.1.2. p-GlcNAc細胞カプセル化物の用途
p-GlcNAcカプセル化細胞を製造することができ、そのようなp-GlcNAcカプセル化細胞を、当分野の技術者によく知られた標準的技術により患者に投与することができる。例えば、上記第5.6.1.1 節に記載した投与方法を参照されたい。あるいは例えば、Aebisher et al., (Aebisher, P. et al., “Fundamentals of Animal Cell Encapsulation and Immobilization", 1993, CRC Press, pp. 197-224)を参照されたい。これは引用によりその全体を本明細書の一部とする。細胞は、p-GlcNAcもしくは部分的に脱アセチル化されたp-GlcNAc膜、3次元的p-GlcNAc多孔質マトリックスあるいはp-GlcNAcゲルにより、あるいはその上に、あるいはその中にカプセル化され得る。
【0166】
3次元マトリックスは細胞とともに播種し、さらにカプセル化することなく一定の用途に使用することができる。あるいは、細胞をp-GlcNAcをベースとするポリマーゲル、例えばp-GlcNAc−ラクテートポリマー電解質ポリマー(ポリカチオン性ポリマー)の微小球あるいは小滴中にカプセル化することができる。細胞がその中にカプセル化されたゲル、小滴あるいは微小球はその後逆に荷電した第2のポリマー電解質により被覆し(例えばアルギネートのようなポリアニオンにより)、カプセル化された細胞の免疫的隔離を生じる外側カプセルを形成し、それにより宿主生物の免疫拒絶反応の危険を減らすことができる。
【0167】
さらに別の配合方法においては、p-GlcNAcゲル、3次元p-GlcNAcマトリックス、あるいはその両方にトラップされた細胞を熱可塑性のカプセル中に入れることができる。熱可塑性物質をベースとするカプセルを使用して、宿主生物中に移植された細胞に免疫保護を与えることができる。そのような熱可塑性カプセルは、例えばヒドロキシエチルメチルアクリレート/メチルメタクリレート共重合体(HEMA-MMA)のような物質により形成される。熱可塑性物質から誘導されるマイクロカプセルは、例えば、ポリエチレングリコール中のHEMA-MMAの溶液と細胞含有p-GlcNAcマトリックス及び/またはゲル媒体を、ヘキサデカンのような適当な有機溶媒中に共押出しすることにより形成される。例えば、Aebisher et al., (Aebisher, P. et al., “Fundamentals of Animal Cell Encapsulation and Immobilization", 1993, CRC Press, pp. 197-224)に記載された方法を参照されたい。
【0168】
p-GlcNAc細胞カプセル化物は種々の用途を有する。先ず、膜、マトリックスあるいはゲルに結合しあるいはその中にカプセル化された細胞により合成され、分泌される治療用化合物の送達のために使用できる。例示であって限定するものではないが、糖尿病の治療のためにインシュリンを、アルツハイマー病の治療のために神経成長因子を、血友病の治療のために第VIII因子あるいはその他の凝集因子を、パーキンソン病の治療のためにドーパミンンを、慢性的疼痛の治療のために副腎クロム親和性細胞を介してエンケファリンを、筋ジストロフィーの治療のためにジストロフィンを、あるいは成長異常の治療のためにヒト成長ホルモンを送達するために、p-GlcNAc/細胞カプセル化物を使用することができる。
【0169】
さらに本発明のp-GlcNAc物質はそれ自体免疫的に中性であり、ヒトにおいて免疫反応を生成しないので、結合した細胞を保有するp-GlcNAc膜、3次元多孔質p-GlcNAcマトリックス及び/またはp-GlcNAcゲルからなるデバイスを製造し、構築することができ、それらは細胞が免疫的に隔離された形態、即ち抗細胞宿主免疫反応が生じないような形態で細胞をベースとする治療用物質を送達し得る。ある種の付加的な物質、例えば天然のアルギネートや合成ポリマー等をp-GlcNAc物質自体に加えて使用し、そのようなデバイスを構築することができる。
【0170】
p-GlcNAc/細胞カプセル化組成物はさらに、細胞を送達して組織再生を開始させるために使用することもできる。外傷部位において組織再生を生じる細胞増殖を開始するためにカプセル化される特定の細胞種の用途は、限定するものではないが、皮膚、軟骨、神経、骨、肝臓、血管等の再生である。p-GlcNAc物質中にカプセル化された細胞の組織再生のための応用が有利であることの一つの理由は、p-GlcNAc物質が外傷組織に接着でき、哺乳動物細胞の増殖のための基体を提供し、外傷部位における組織再生プロセスの間に新しい健常な組織の増殖と同時に生体吸収を受ける能力を有することである。限定するものではないが、例として、皮膚、骨、軟骨、肝臓、腱、靱帯組織等の再生が挙げられる。
【0171】
5.6.1.3 手術後の癒着の防止のためのp-GlcNAc物質の使用
さらに、p-GlcNAc膜は、手術後の癒着を防ぐための生分解可能で生体適合性の機械的バリアを与えるために使用できる。下記第17節に示した実施例は、そのようなp-GlcNAcの用途を示すものである。膜あるいはスポンジ中に配合した固体p-GlcNAcまたはp-GlcNAc誘導体をそのような用途に使用できる。好ましくは膜は薄いもので、一般に約1 mm未満の厚さを有するものである。好ましいp-GlcNAc誘導体は、約50〜80% 脱アセチル化されたp-GlcNAc誘導体である。そのようなp-GlcNAc誘導体は、一般に移植後約7〜21日で吸収される。
【0172】
液体のp-GlcNAc誘導体も手術後の癒着の防止の使用に適している。そのような用途に好ましい液体p-GlcNAc誘導体は脱アセチル化p-GlcNAc塩誘導体及びカルボキシメチルp-GlcNAc誘導体である。手術後の癒着の防止に特に好ましいp-GlcNAc誘導体は、p-GlcNAc-ラクテート誘導体、特にp-GlcNAc-ラクテートゲル誘導体である。そのようなp-GlcNAc-ラクテート誘導体は、例えば第17.1節に記載したように、ポリエチレングリコール及び水を使用して配合することができる。p-GlcNAc-ラクテート誘導体は高い粘度を有するもの、あるいは低い粘度を有するものとして製造することができ、対象となる特定の適用症に合致した特性を与えることができる。例えば、シリンジあるいはスプレーにより送達するためには低い粘度を有するp-GlcNAc生成物を使用するのが有用であり、適応症が成形外科的なものである場合には高い粘度を有し、従ってより高い潤滑性を有するp-GlcNAc生成物を使用することが望ましい場合がある。
【0173】
手術後の癒着の防止のためには、明らかに限局性のものである外傷部位には固体p-GlcNAc配合物が適している。そのようなp-GlcNAc配合物は手術の処置後に適用し、材料が外傷を受けた組織を完全に覆うようにする。これは一般的な手術、あるいは侵入を最小限にした手術(例えば腹腔鏡を使用した)において使用することができる。固体p-GlcNAc配合物は、当分野の技術者によく知られた標準的な外科的処置及び設備を使用して切断し、適用することができる。
【0174】
液体のp-GlcNAc配合物は、手術後の癒着の防止のために、そのような手術後の癒着を起こしやすいより大きい領域に適用することができる。例えばp-GlcNAc-ラクテートゲルは、手術手順の前に適用して潤滑性を増し、外傷組織の量を減らすことができる。あるいは、p-GlcNAc-ラクテートのような液体p-GlcNAc配合物を手術手順の後に適用して物理的バリアを形成し、手術後の癒着の形成を防ぐことができる。
【0175】
p-GlcNAc物質は、シリンジデバイスから外傷部位に塗布し、スプレーし、あるいは滴下することができる。腹腔鏡使用処置においては、例えば、低い粘度を有する物質を標準的な吸引洗浄装置により送達することができる。高い粘度を有する物質は目的箇所に到達させるために圧力を要する。圧力は圧縮ガスピストンあるいはシリンジ型の装置により得られる。
【0176】
p-GlcNAc-ラクテートゲル配合物のような液体p-GlcNAc配合物の手術後の癒着を防止するのに要する量は、外傷を受けた組織の範囲に比例する。投与するp-GlcNAc物質は、表面積の平方センチメートルあたり0.1 ml〜1.5 mlの範囲で適用する。
【0177】
5.6.1.4 p-GlcNAcの物質その他の生物医学的使用
p-GlcNAc物質のその他の生物医学的利用としては、例えば細胞培養基体としてのそのような物質の使用が挙げられる。例えば、下記第12節に挙げた実施例に示すように、本発明のp-GlcNAcは培養物中で哺乳動物細胞増殖に非常に有効な基体となる。さらに、3次元的形態のp-GlcNAcは、3次元的細胞培養物増殖を可能とする培地成分として使用することができる。
【0178】
本発明のp-GlcNAcの細胞基体としての能力は生体内においても利用できる。即ち、本明細書に記載する本発明のp-GlcNAc、あるいはその誘導体は、組織の再生を促進するように作用できる(例えば、歯肉線に近い歯を覆う結合組織、移植血管、靱帯、腱、軟骨、骨、皮膚、神経組織の再生)。従って、本発明のp-GlcNAc分子は、例えば、広範な形成外科的用途を有する。
【0179】
脱アセチル化p-GlcNAcは、移植血管のシール材としての使用に好ましい。N-カルボキシメチル及びN-カルボキシブチル脱アセチル化p-GlcNAcのような脱アセチル化p-GlcNAc誘導体は組織再生剤として好ましい。N-カルボキシメチル脱アセチル化p-GlcNAcを、例えば、角膜に接種して血管新生を起こすことができる。
【0180】
本明細書に記載する本発明のp-GlcNAcあるいはその誘導体の別の生物医学的用途は、外傷用包帯、外傷治療軟膏、手術用縫合糸、スポンジ等に該分子を使用することである。
【0181】
さらにそのような分子は、例えば、変形性関節炎の治療、血清コレステロールレベルを低下させること、あるいは抗ウィルス剤として、抗細菌剤として、免疫調節剤として、抗凝集剤として、透析及び限外濾過膜として、抗腫瘍剤として、コンタクトレンズ材料として、腎不全患者に投与してイレミック毒素の吸収剤として使用できる。微結晶p-GlcNAc懸濁物あるいは水溶性p-GlcNAc誘導体は、例えば関節炎の関節に直接注射することにより、関節炎の治療に好ましいものである。
【0182】
p-GlcNAcはさらに、人工皮膚あるいはドナー皮膚の成分としての用途も有する。例えば、p-GlcNAcを好ましくは不織p-GlcNAcフィルムとして、例えばドナー真皮上、薄く剥がした厚さの皮膚ドナー部位にあてることができる。
【0183】
例えばペプシンのようなプロテアーゼを結合した脱アセチル化p-GlcNAcは、そのようなp-GlcNAc/プロテアーゼ化合物にタンパク質を接触させてその制御された消化を行うのに使用できる。
【0184】
本発明のp-GlcNAcの特定の誘導化、即ちその誘導体は、特定の用途に好ましいものである (誘導化については上記第5.4 節に記載した)。例えば、硫酸化、リン酸化及び/またはニトロ化p-GlcNAc誘導体は、抗凝集剤あるいはリポタンパク質リパーゼ活性化剤として好ましい。N-アシルp-GlcNAc誘導体も抗凝集剤に好ましく、さらに例えば、人工血管、抗ウィルス化合物、抗腫瘍 (具体的には癌細胞凝集化合物) 、透析及び限外濾過膜、及び放出が制御された薬物送達システムの製造にも好ましい。O-アルキルp-GlcNAc及びその脱アセチル化誘導体も放出が制御された薬物送達システムの製造に好ましい。N-アルキルp-GlcNAc誘導体は抗細菌剤として好ましい。オキシド脱アミノ誘導体は抗癌剤として好ましく、特に癌細胞の免疫療法と組み合わせて使用することが好ましい。脱アセチル化p-GlcNAc誘導体は外傷治療薬として好ましい。N-アルキリデン及びN-アリーリデンp-GlcNAc誘導体は酵素固定化の用途に好ましい。
【0185】
5.6.2 p-GlcNAc物質の農業用用途
本発明のp-GlcNAcあるいはその誘導体は、種々の農業用用途にも使用できる。そのような用途としては、限定するものではないが、殺昆虫剤、殺真菌剤、殺細菌剤、殺線虫剤の用途がある。N-カルボキシメチル脱アセチル化p-GlcNAc誘導体は、有効な細菌静止薬としての使用に好ましい。N-アルキルp-GlcNAc誘導体は殺真菌剤の用途に好ましい。さらに本発明の分子は、種々の土壌処理用途に使用することができ、限定するものではないが、肥料組成物等がある。さらに農薬を固定化、カプセル化、あるいはこの節で先に記載したような方法によりトラップすることによりそのような農薬の制御された放出を行うことができる。さらに、例えばRhizobium根粒着生因子及び/または窒素固定誘発物質の類縁体を本発明のp-GlcNAc及び/またはp-GlcNAc誘導体上に固定し、またそれを介して施用することができる。
【0186】
5.6.3 p-GlcNAc物質の栄養/食品産業用用途
本明細書に記載した本発明のp-GlcNAc及びその誘導体はさらに、動物及びヒトの栄養物摂取の分野に用途を有する。例えば、本発明の分子を飼料成分として使用し得る。この節で先に記載したような方法を使用して、放出が制御された製品を動物系において製造することができる。さらに、当業者によく知られた日常的な改変を行うことにより、上記のような生物医学的用途を動物系において利用できる。
【0187】
本明細書に記載したような本発明のp-GlcNAc及びその誘導体の食品産業での用途は、限定するものではないが、抗コレステロール物質(即ち、低コレステロール化合物)、脂肪結合化合物、乳化剤、担体、保存剤、調味料、食品生地改良剤(food texturizer) 等があり、さらに果実コーティング、食品パッケージ製品がある。
【0188】
5.6.4 p-GlcNAc物質の化粧品用用途
本発明のp-GlcNAcの化粧品用用途としては、限定するものではないが、ヘアケア及びスキンケア用の製品の製造がある。スキンケア製品としては例えば、脱アセチル化p-GlcNAc塩を利用した化粧品、カルボキシメチルp-GlcNAc含有製品、脱アセチル化p-GlcNAc及びヒドロキシプロピル- 、N-スクシニル- 誘導体及び4級化p-GlcNAc誘導体のような誘導体を含有する化粧用パック等がある。ヘア製品としては例えば、カルボキシメチルp-GlcNAc含有製品、及びフィルム形成性p-GlcNAc誘導体等がある。
【0189】
5.6.5 p-GlcNAc物質の化学工学用用途
本発明のp-GlcNAc及びその誘導体は化学工学工業において有用な種々の用途を有する。例えば、p-GlcNAcは金属のポリマーへの接着のためのカプリング剤として使用でき、グリコールp-GlcNAcにより形成された膜は脱塩用途に使用でき、その他のp-GlcNAc誘導体により形成された膜はハロゲンイオンの輸送に使用できる。その他の用途としては、難燃剤の製造、金属キレート化合物及び痕跡量の重金属並びにPCB のような水溶性の産業汚染物を液体から除去できる化合物の製造等がある。p-GlcNAc及び/またはp-GlcNAc誘導体は写真用途に使用できる。例えば、p-GlcNAc及び/またはp-GlcNAc誘導体の金属、例えばハロゲン化銀をキレート化する能力は、p-GlcNAc及び/またはp-GlcNAc誘導体の例えば薄い膜であるリキャストマットに写真溶液を接触させることにより利用できる。
【実施例】
【0190】
6. 実施例: p-GlcNAcの純粋調製物の物理的特性化
この実施例においては、p-GlcNAc及び脱アセチル化p-GlcNAc膜の円偏光二色性(CD)及び赤外分光スペクトル(IR)分析を示す。
【0191】
6.1 材料及び方法
p-GlcNAc及び市販の「キチン」調製物
CD分析において使用したp-GlcNAcは先に第5.3.1 節に記載した機械力精製法を使用して製造した。
【0192】
市販の「キチン」はNovaChem, Ltd., PO Box 1030 Armdale, Halifax, Nova Scotia, Canada, B3L 4K9から購入した。
【0193】
IR分析に使用したp-GlcNAc膜は、示したように、先に第5.3.1 節に記載した機械力精製法か、第5.3.2 節に記載した化学的/生物学的精製法により製造した。
【0194】
市販の「p-GlcNAc」調製物を、5%の塩化リチウムを含むジメチルアセトアミド溶液中に溶解し、蒸留及び脱イオン化した水上に積層して膜を沈殿させることにより、膜にキャストした。
【0195】
p-GlcNAc誘導体及び処理: CD及びIR分析の両方で使用した脱アセチル化p-GlcNAcは、p-GlcNAcを50% NaOHで60℃において2時間処理することにより製造した。IR分析において使用した熱変性p-GlcNAc膜は、0.2 mM EDTA 中で3分間煮沸することにより変性させた。オートクレーブしたp-GlcNAcは122 ℃で30分間オートクレーブしたものである。
【0196】
CD分析法: 実質的にDomardの方法(Domard, A., 1986, Int. J. Macromol. 8:243-246) に従って固体CD法を行った。
【0197】
6.2 結果
6.2.1 CD分析
未処理のp-GlcNAcから得られたCDスペクトル(図3A)においては、p-GlcNAcのアセチル部分のカルボニル基の存在のために予測されたn-π* 及びπ-π* 光学活性電子項遷移(220-185nM) の存在が観察された。このようなピークは、図3Bに示したように、脱アセチル化p-GlcNAc生成物から得られたCDスペクトルには全く存在しなかった。
【0198】
6.2.2 IRスペクトル分析
この分析において得られたIRスペクトルはp-GlcNAcの化学構造に合致するものであった。さらに、各IRピークが明確に得られたことは、p-GlcNAc繊維中の秩序立った規則正しい (即ち、偽結晶) 構造の存在を示すものである。機械力精製法により精製されたp-GlcNAcのIRスペクトルについては図4Aを、化学的/生物学的方法により精製されたp-GlcNAcのIRスペクトルについては図4Dを参照。比較として、市販の「キチン」調製物のIRスペクトルを示す図4Bを参照。
【0199】
オートクレーブされたp-GlcNAc物質から得られたIRスペクトル(図4E)は図4Aで見られるIRスペクトルと視覚的に異ならない。このデータは、p-GlcNAc物質がオートクレーブによりポリマー構造を損なうことなく滅菌され得ることを示している。
【0200】
7. 実施例: 機械力精製法を使用したp-GlcNAcの精製
この節においては、第5.3.1 節に記載した機械力法を使用してp-GlcNAcを精製した。
【0201】
7.1 材料及び方法/結果
珪藻培養条件: 珪藻の一種タラシオシラ・フルビアチリス(Thallasiosira fluviatilis)を上記第5.1 及び第5.2 節に記載した方法に従った培養で増殖させた。
【0202】
SEM方法: ここで使用したSEM法は下記第12.1節に記載するものである。
【0203】
p-GlcNAc精製法: 上記第5.3.1 節に記載した機械力法を使用して珪藻培養物からp-GlcNAcを精製した。具体的には、培養物の内容をWaringブレンダーのトップスピード混合動作に瞬間的に3回かけ、p-GlcNAc繊維を珪藻菌体から分離した。3回の操作の合計時間は約1秒であった。得られた懸濁物をSorvall GS-4固定角ローターで約10℃において3500 rpmで20分間遠心分離にかけた。上清をデカントし、Sorvall GS-4固定角ローターで約10℃において4000 rpmで20分間再度遠心分離した。再度上清をデカントし、10℃において4000 rpmで遠心分離した。最後の3回目の遠心分離の上清は透明で、ないとはいえないまでも液体中に浮遊する目に見える沈殿は殆どなかった。透明な上清を0.8 μm のポア径のニトロセルロースフィルターを備えたブフナー濾過ユニット中にデカントし、吸引して繊維懸濁物から液体を濾過し、膜上に繊維を回収した。回収された繊維を70℃において蒸留し脱イオン化した1リットルの水により洗浄した。ほとんど全ての水が排出された後、吸引しながら70℃において1リットルの1N HClにより繊維を洗浄した。ほとんど全ての酸溶液が排出された後、70℃において蒸留し脱イオン化した1リットルの水により吸引を使用して繊維を洗浄した。ほとんど全ての洗浄水が排出された後、室温で1リットルの95% エタノールにより繊維を洗浄し、減圧した。その上に白色の繊維膜が回収された濾過膜を濾過ユニットから取り出し、その膜及び膜支持体を58℃の乾燥オーブンで20分間乾燥し、その後膜及びその支持体をデシケーター中に16時間置いた。
【0204】
この精製工程の後において、1000 ml の培養物からのp-GlcNAcの収率は珪藻培養物1リットルあたり6.85 mg であった。この方法によりp-GlcNAc繊維を回収して形成された膜のSEM 写真を図6A〜6Bに示す。
【0205】
8.実施例:生物学的/化学的精製方法を使用したp-GlcNAcの精製
この節において、上記第5.3.2節の2つの化学的/生物学的手法を使用して、p-GlcNAcを精製した。簡単に述べると、1つはHF処理を介して、また2番目には酸処理/中和を介してp-GlcNAcを精製した。
【0206】
8.1.物質および方法/結果
ケイ藻の培養条件:ケイ藻種、タラシオシラ・フルビアチリス(Thallasiosira fluviatilis )を上記第5.1および第5.2節の操作法に従った培養によって増殖させた。
【0207】
SEM操作法:この実験において利用した手法は下記第12.1節と同様である。
【0208】
精製操作法:最初に、p-GlcNAcをHF処理によって精製した。この結果を図7A−7Bに示す。詳細に述べると、室温において換気フード下で、ケイ藻含有培養液に、細胞培養物原液1000ml毎に49%(29N)HF溶液 2.4mlを添加し、0.07M HF溶液とした。その後この混合物を約30秒間激しく振とうし、その結果液体の表面に持続性の気泡を生成させた。この容器を5−6時間静置し、重い粒子を沈降させた。この時間の経過後、気泡層は形成されたままで、一方液体自体は2層に分離した。第1層は容器の底面にある非常に暗い緑色の薄層で、その上がおそらく液体の総容量の85-90 %を占めるずっと明るい灰緑色で曇った第2層であった。ガラス毛細管と真空吸引を使用して、気泡層を注意深く吸引除去した。次に、主として沈降細胞本体からなる暗色の底部層をかき乱さないように注意しながら、灰色の曇った上層を吸引して、別のプラスチック容器に移した。この灰色の曇った上層をさらに16時間静置した。この液体は最初ほとんど無色か明灰色だが透明ではなかった。16時間の沈降後、液体の主要部分の上面に少量の気泡が残り、容器の底部に少量の緑色物質が沈降した。液体の色は薄くなったが依然として透明ではなかった。液体上面の気泡を前と同様に吸引除去した。次に、容器底部の緑色物質を残したままで、液体主要部を注意深く吸引して取り出した。こうして単離した液体は大部分がp-GlcNAc繊維で、他に不純物を少し含んでいた。
【0209】
この繊維含有液体からこれまでの操作段階中にケイ藻から遊離したタンパク質およびその他の不要物質を除去するため、繊維および細胞残留物の懸濁液をドデシル硫酸ナトリウム(SDS)で洗浄した。詳細に述べると、液体の最終濃度がSDS 0.5容量%となるのに必要な量の20%SDS溶液を添加した。この液体を入れた容器を密封し、振とう機上で水平を保つようにして、約100 振とう数/分で24時間撹拌した。振とう開始後まもなく、懸濁液中に白色のp-GlcNAc繊維の大きな固まりが生成し、容器の上部にかなりの量の気泡が集まった。SDS洗浄の終了後、容器の内容物を0.8μmの濾過膜(Supor Filter,Gelman )を着けたブフナー濾過器に移した。液体を吸引濾過し、液体中のp-GlcNAc繊維を濾過膜上に集めた。
【0210】
続いてこの濾過膜上に集めたp-GlcNAcをさらに洗浄した。まず、蒸留脱イオンした熱(70℃)H2Oを当初の懸濁液の3倍容量使用して、繊維を洗浄した。蒸留脱イオンしたH2Oを使用した水流によって、ブフナー濾過器の濾過膜上に集めた白色繊維塊をワーリングブレンダーに移し、約10回の短時間混合バーストによって、繊維塊を崩壊させた。崩壊繊維の懸濁液を上記と同様のニトロセルロース濾過膜を着けたブフナー漏斗に移し、吸引によって液体を除去した。採取した繊維を熱(70℃)1N HCl溶液1000mlで洗浄し、続いてさらに蒸留脱イオンした熱(70℃)H2O 1000mlで洗浄した。最後に、繊維を室温で、95%エタノール1000mlで洗浄し、濾過して乾燥させる。その後、繊維膜とこの繊維膜を担持している濾過膜を58℃の乾燥機で20分間乾燥した。その後、この膜と膜担持材をデシケーター中に16時間入れておいた。その後、膜を濾過膜から注意深くはがした。
【0211】
第2番目に、上記第5.3.2節の酸処理/中和方法を使用して、p-GlcNAcを精製した。詳細に述べると、SDS洗浄段階の前まではこの節の前半の記載と同様にp-GlcNAcを処理し、この段階において、2.9M Tris 溶液の添加によって、溶液のpHを約7.0 に中和した。この精製操作によるp-GlcNAcの収率はケイ藻培養物1l 当たり20.20mg だった。平均して、ケイ藻培養物1l 当たり約60mgが得られる。この精製操作において形成された膜のSEM写真を図8A−8Bおよび9A−9Bに示す。
【0212】
9.実施例:p-GlcNAcの脱アセチル化
p-GlcNAc膜を50%NaOH含有溶液中に懸濁させた。この懸濁液を80℃で2時間加熱した。生成した脱アセチル化膜を乾燥し、走査型電子顕微鏡で調べた。この結果を図11A−11Bに示す。
【0213】
10.実施例:p-GlcNAcの生体適合性
この実施例において、溶離試験、ウサギにおける筋肉内埋め込み、ウサギにおける皮内注入、およびマウスにおける全身注入試験によって、本発明のp-GlcNAcが検出し得る程度の生物学的活性を持たないことが実証される。
【0214】
10.1.物質および方法
10.1.1.溶離試験
溶離試験の条件は、U.S.薬局方 XXII,1990,pp.1415-1497 、およびU.S.薬局方XXII, 補遺5,1991,pp.2702-2703 に示された説明にしたがった。
【0215】
細胞培養物:マウス繊維芽L929細胞系(American Type Culture Collection Rockville,MD;ATCC No.CCL1;NCTC clone 929 )を使用した。24時間目のL929細胞の密集単層を完全な最少必須培地(MEM)中で増殖させた。
【0216】
p-GlcNAc:上記第5.3.1節の機械的精製方法によって調製したp-GlcNAc固体膜から、U.S.薬局方XXII(1990)の要件にしたがって、血清補充MEM20ml中に抽出した。
【0217】
対照:陽性対照として天然ゴムを使用し、陰性対照としてシリコーンを使用した。試験品であるp-GlcNAcと同一の手法で対照を試験した。
【0218】
抽出物:37℃、5%二酸化炭素含有加湿雰囲気中で24時間かけて、抽出物を調製した。抽出物のpH変化を測定し、当初の培地のpH単位の+/−0.2 以内になるようにpHを調整した。抽出物のpHを低下させるのにはHClで、抽出物のpHを上げるのにはNaHCO3 で調整を行なった。抽出物を細胞単層に適用する前に、0.22ミクロンフィルターを通して滅菌濾過した。
【0219】
適用:p-GlcNAcまたは対照3mlずつを使用して細胞培養物の維持培地と入れ替えた。すべての抽出物を2つずつ試験した。
【0220】
評価基準:細胞単層の応答を肉眼または顕微鏡下で評価した。生物学的反応性、すなわち細胞の変性および/または奇形化を下記に示すように0〜4の基準で評点をつけた。陰性対照品に関して細胞反応性の徴候が何ら見られず(等級0)、陽性対照品が軽度の反応性(等級2)以上の反応性を示す場合に、その試験系は好適なものである。試験品で処理した培養物の中に軽度より大きな反応性を示すものがない場合、この試験品(すなわちp-GlcNAc)は生体適合性試験を満足することとなる。
【表1B】
10.1.2.筋肉内埋め込み
動物:健康なニュージーランド白ウサギ、雄および雌(Eastern Rabbit Breeding Laboratory,Taunton,MA )を使用した。ウサギは懸垂型ステンレススチール製カゴに個別に入れた。入手後すぐに、実際の試験と同一の条件下で8日間隔離しておいた。カゴの下の非接触床として、硬材チップ(Sani-chips(登録商標),J.P.Murphy Forest Products,Montvale,NJ )を使用した。動物の環境を、温度68+/-3°F、相対湿度30〜70%、1時間当たり少なくとも10〜13回の完全換気、および全スペクトルの蛍光灯を使用した12時間の明/暗サイクルに維持した。動物に市販の餌(Agway ProLab,Waverly,NY )を制御しながら、そして公共水道水を自由に与えた。餌、床材または水には、試験結果に干渉する可能性がある、既知の汚染物質は何も存在しなかった。この実験のために選定した動物はより多くの動物群から選抜した。ウサギは10g 近傍まで体重を測定し、耳の刺青によつて個体を特定した。
【0221】
p-GlcNAc:使用したp-GlcNAcは上記第10.1.1節と同様である。
【0222】
埋め込み試験:各埋め込み試験毎に2匹のウサギを使用した。試験当日、動物の脊柱の両側の皮膚の毛を刈り取った。筋肉の動きを止めるため、各動物に麻酔した。滅菌皮下注射針とスタイレットを使用して、p-GlcNAc試験片(1mm×1mm×10mm)4枚を2匹のウサギのそれぞれの脊椎の一方の側の脊椎傍筋肉中に(中心線から2.5 〜5cm、脊柱に平行、および互いに約2.5 cm間隔で)埋め込んだ。同様の方式で、各動物の反対側の筋肉にUSP陰性対照プラスチックRS片(1mm×1mm×10mm)2枚を埋め込んだ。動物を7日間そのまま維持した。観察期間の終了時、動物の体重を測定し、注射用バルビツレート、Euthanasia-5(Veterinary Laboratories,Inc.,Lenexa,KS)によって安楽死させた。組織を出血せずに切開することができるまで十分な時間を経過させた。各埋め込み片の中心部分を取り巻く組織一帯を、拡大レンズを使用して肉眼で検査した。以下の基準を使用して、出血、壊死、退色および感染を採点した:0=正常、1=軽度、2=中程度、および3=強度。カプセル化があれば、最初にカプセルの幅(すなわち、埋め込み物の末端からカプセルの末端までの距離)をおよそ0.1mm 近傍まで測定して採点した。このカプセル化は以下のように採点した:
【表1C】
p-GlcNAcと陽性対照品の平均評価点の差を計算した。各ウサギにおいて、4枚のp-GlcNAc試験片の1枚についてだけでも、p-GlcNAcと陽性対照プラスチック埋め込み部位における生体反応の各部門の平均評価点間の差異が1以下ならば、または、そのp-GlcNAc品における生体反応の全部門の平均評価点と全陽性対照プラスチック埋め込み部位における生体反応の全部門の平均評価点間の差異が1以下ならば、その試験は陰性であるとみなされる。
【0223】
10.1.3.皮内注入
動物:上記第10.1.2節のようにして、ニュージーランド白ウサギを使用し、飼育した。
【0224】
p-GlcNAc:上記第5.3.1節の機械的精製方法によって調製したp-GlcNAc固体膜を抽出フラスコに入れ、これに好適な溶媒20mlを添加した。24時間、70℃に加熱することによって抽出を実施した。この操作後、抽出物を室温まで冷却した。投与前に各抽出ビンを激しく振とうした。
【0225】
皮内試験:試験当日、動物の背中側の毛を刈り取った。2匹のウサギのそれぞれの一方の側の5ヵ所に、各p-GlcNAc抽出物0.2ml を注入した。ウサギ1匹当たり1以上のp-GlcNAc抽出物を使用した。各ウサギの他方の側の5ヵ所に、対応する対照品0.2ml を注入した。注入の24、48および72時間後、紅斑、水腫および壊死の徴候に関して、注入部位を観察した。下記表IIに示すような皮膚反応評価に関するDraize Scale(USP 薬局方XXII,1990,1497-1500 ;USP 薬局方XXII, 補遺5,1991,2703-2705)にしたがって、観察を評価した。
【表2A】
対応する対照に対するp-GlcNAcの総合的な平均評価点を決定するため、24、48および72時間における紅斑および水腫の全評価点をそれぞれ合計し、12(すなわち、動物2×評価期間3×評価部門2)で割った。観察期間の終了時に、動物の体重を測定し、バルビツレート、Euthansia-5(Veterinary Laboratories,Inc.,Lenexa,KS) の注射によって安楽死させた。p-GlcNAcと対照の平均反応評価点(紅斑/水腫)の差異が1.0 またはそれ以下であれば、試験結果は満足し得る。
【0226】
10.1.4.全身注入
動物:アルビノスイスマウス(Mus musculus)雌(Charles River Breeding Laboratories,Wilmington,MA )を使用した。マウス5匹のグループをステンレススチール製のふたをしたポリプロピレン製カゴに入れた。カゴの中の接触床として、硬材チップ(Sani-chips(登録商標),J.P.Murphy Forest Products,Montvale,NJ )を使用した。この動物施設を接触制限区域として維持した。動物室を、室温68+/-3°F、相対湿度30〜70%、1時間当たり最低10〜13回完全換気し、全スペクトル蛍光灯を使用して12時間の明/暗サイクルを保持した。マウスに市販の餌と公共水道水を自由に与えた。試験結果に干渉することが予想されるような既知の汚染物質は餌、床材または水に含まれていなかった。この研究のために選定した動物は、より多くの動物群の中から選抜した。マウスの体重を0.1g近傍まで測定し、耳のパンチによって個体を特定した。
【0227】
p-GlcNAc:使用した試料は上記第10.1.1節の通りである。上記第10.1.3節の操作にしたがって抽出物を調製した。
【0228】
全身注入試験:マウス5匹のグループに、p-GlcNAc抽出物または対応する対照品を、下記に示すように同量ずつ同一の経路で注入した。
【表2B】
PEG400 を使用して調製したp-GlcNAc抽出物および対応する対照を0.9%NaClで希釈し、1ml当たりPEG400 200mg となるようにした。皮内試験については、PEG400 を0.9%NaClで希釈し、1ml当たりPEG400 120mg となるようにした。
【0229】
動物を、注入直後、注入後24時間、48時間および72時間後に観察した。観察期間の終了時、動物の体重を測定し、二酸化炭素ガス中にさらすことによって安楽死させた。p-GlcNAc処理した動物の中で対照品で処理した動物よりも生物学的反応性が有意に大きいものがなければ、この試験の要求が満足される。
【0230】
10.2.結果
10.2.1.溶離試験
p-GlcNAc試験品に対する細胞単層の応答を肉眼および顕微鏡下で評価した。評価に際して、細胞化学的染色は利用しなかった。陰性対照品またはp-GlcNAcの被曝48時間後、細胞の生物学的反応の徴候は何ら観察されなかった(等級0)。下記表III に示すように、陽性対照品については、強度の反応性(等級4)が記録された。
【表3】
したがって、本発明のp-GlcNAcは生体適合性に関する溶離試験の要求を満たしており、すなわち、非毒性である。
【0231】
10.2.2.筋肉内埋め込み
試験をしたウサギの両方(AおよびB)の体重が増加し、毒性の徴候は何ら見られなかった。データに関しては表IVを参照されたい。付け加えると、どの動物においても毒性の顕著な徴候は見られなかった。試験および対照品の肉眼での評価では、炎症、被包、出血、壊死または退色は示さなかった。結果については表IVを参照されたい。したがって、この試験では、各ウサギにおいて、すべてのp-GlcNAc埋め込み部位の生物学的反応の全部門の平均評価点とすべての対照埋め込み部位の全部門の平均評価点との差異が1.0 以下であることから、検定したp-GlcNAcは生物学的反応性を有していないことが示される。
【表4】
10.2.3.皮内試験
すべての動物の体重が増加した。データについては表Vを参照されたい。p-GlcNAcまたは対照品部位のいずれにおいても紅斑または水腫の徴候は観察されなかった。どの動物においても毒性の顕著な徴候は観察されなかった。p-GlcNAcおよび対照品の平均反応評価点(紅斑/水腫)の差が1.0 よりも小さかったので、p-GlcNAcは皮内試験の要求を満たしている。結果については表VIを参照されたい。したがって、この試験によって分析したところ、p-GlcNAcは生物学的反応性を示さない。
【表5】
【表6A】
【表6B】
【表6C】
【表6D】
10.2.4.全身試験
p-GlcNAc抽出物または対照品で処理したすべてのマウスの体重が増加した。データについては表VII を参照されたい。付け加えると、p-GlcNAcまたは対照のどの動物においても明白な毒性の徴候はなんら観察されなかった。結果については表VIを参照されたい。したがって、どのp-GlcMAc試験動物も対照品で処理した動物以上に有意に大きな生物学的反応性を示さないことが結論づけられる。
【表7】
11.実施例:p-GlcNAcの再構成
この節において述べる具体例においては、5%LiClを含有するジメチルアセトアミド溶液1mlに、p-GlcNAc膜(16.2mg)を溶解させた。このp-GlcNAc含有溶液を注射器に入れて、純水50ml中に押し出し、繊維を沈澱させた。生成した繊維を走査型電子顕微鏡で調べた。これを図10A−10Bに示す。
【0232】
12.実施例:p-GlcNAcへの細胞の付着
この具体例においては、p-GlcNAcが培養液中での細胞の付着と増殖のための有効な基質であることが実証される。
【0233】
12.1.材料および方法
細胞:形質転換マウス3T3繊維芽細胞株を使用し、10%ウシ胎児血清(FBS)を添加したDMEM中で増殖させた。
【0234】
p-GlcNAc膜:上記第5.3.1節および第8節の方法にしたがって、p-GlcNAcを調製した。
【0235】
最初に、水を1滴使用して、#1(18mm)Corning のカバーグラスにp-GlcNAc膜を貼りつけ、121 ℃で30分間オートクレーブにかけることによってこれを接着させた。次に、この手法によって調製した膜を6ウェル培養プレートの培養ウェル中に入れた。
【0236】
細胞の計数:細胞の数を、培養液中のものは血球計算盤で直接に計数し、マトリックス上のものは、計数前にまず膜を新しい培地(DMEM+10%FBS)で洗浄し、次にトリプシンで処理(10%、37℃で5分間)した後、計数した。
【0237】
SEM走査条件:ツァイス(Zeiss)962 装置を加速電圧10kv、および作用距離15mmで使用した。ポラロイド型55p/n(u4) 型を指示にしたがって各種倍率で使用した。
【0238】
試料のコーティング:炭素コーティング(100Å)および100ÅのAuPd。
【0239】
検体の調製:一次固定のため、培養増殖用培地を血清を含まないイーグルのDMEM中2%グルタルアルデヒドと交換した。増殖用培地から固定液への完全な移行を確実にするため、何度か交換を行った。室温で0.5 時間固定を行なった。カバーグラスを0.1 M ショ糖と0.1 M カコジル酸Na、pH7.2 中2%グルタルアルデヒドを含有する新しいバイアルに移し、さらに室温で1.5 時間固定した。
【0240】
脱水、CPD、マウントおよびスパッタコーティング:
試料を0.1 M カコジル酸Na、pH7.2 で洗浄し、カバーグラスをCPD支持器に移した。エタノールシリーズ(30%、50%、75%、85%、95%および3×100 %、5分ずつ)によって脱水を行ない、試料を臨界点乾燥した。次に、カバーグラスをAlスタブ上にマウントし、真空蒸発器を使用して炭素コーティングし(@100Å)、100ÅのAuPdによってスパッタコーティングを行なった。
【0241】
12.2.結果
p-GlcNAc膜について、この上で培養液中の細胞が増殖することができる基質となる能力を試験した。p-GlcNAc膜の存在または不在ウェル中でマウス繊維芽細胞を増殖させ、培養物の生育力を分析するため、毎日細胞の計数を実施した。このような連日の細胞計数の結果の1つを図14に示す。これによると、接種5日後、p-GlcNAc膜含有ウェルのみが生育する細胞を保持しており、p-GlcNAc膜が培養液中での細胞の持続増殖のための有効な基質として作用することを実証している。さらに、図15A−15Bに示すSEM写真は、p-GlcNAc膜に強固に付着した健康な細胞を表している。
【0242】
13.実施例:p-GlcNAc/コラーゲンハイブリッド
この具体例において示されるのは、p-GlcNAc/コラーゲンハイブリッド材料の形成および特性決定である。
【0243】
13.1.材料および方法
材料:この試験において述べるハイブリッドの調製のために、ウシI型コラーゲンを使用した。p-GlcNAcを上記第5.3.2節の機械的方法によって調製した。
【0244】
ハイブリッドの調製:コラーゲン(10mg/ml)およびp-GlcNAc(0.25mg/ml)の懸濁液を各種割合で混合し、液体N2 中(−80℃)で凍結させ、−9℃に4時間保持し、凍結乾燥した。60℃において真空(約0.030 Torr)下で3日間、材料を脱水熱架橋した。
【0245】
細胞培養:製造したコラーゲン/p-GlcNAcハイブリッド上で、マウス3T3繊維芽細胞を増殖させた。標準的な培養操作を行い、培養8日後、SEM写真を撮った。
【0246】
13.2.結果
コラーゲンおよびp-GlcNAc懸濁液を各種割合(すなわち、コラーゲン:p-GlcNAc懸濁液の比が3:1,1:1,2:2,および1:3)で混合し、凍結し、凍結乾燥し、そして架橋した。この操作によってコラーゲン/p-GlcNAcスラブが得られた。生成した物質のSEM写真を図16B−Eに示す。図16Aはコラーゲン−材料対照のみを示す。ハイブリッド材料の多孔質構造に注目されたい。
【0247】
本発明のコラーゲン/p-GlcNAcハイブリッドは、図17A−DのSEM写真が示すように、細胞が付着して増殖するのに有効な3次元構造を提供する。
【0248】
14.実施例:純粋なp-GlcNAc調製物のNMR特性
この実施例において示されるのは、純粋なp-GlcNAc調製物のNMR(核磁気共鳴)分析である。
【0249】
14.1.材料および方法
p-GlcNAc調製品:上記第5.3.2節の化学的精製方法を使用し、化学試薬としてフッ化水素酸を利用して、ここで述べるNMR試験に使用するp-GlcNAcを調製した。
【0250】
NMR手法:Bruker 500MH NMR分光器を使用して、固体状態のNMRデータを得た。未処理のNMRスペクトルデータからバックグラウンドを除去し、ベースラインを標準化するように変換するため、コンピュータイメージ分析を使用した。こうした変換データの例を図18に示す。図18のような変換NMR曲線を使用して、すべての炭素原子型の領域の値を得て、それからC原子(領域)に対するCH3(領域)の比を計算した。ここに述べたようにして得た数値を図20に示す。
【0251】
14.2.結果
p-GlcNAc試料500mg のC13- NMRスペクトルを測定することによって、固体状態のNMRデータを得た。典型的なNMRスペクトルを図19に示す。それぞれのピークが分子中の各ユニークな特徴ある炭素原子のスペクトルへの寄与に相当する。各炭素種別毎に生じたピークの領域をスペクトル中で得られたすべてのNMRピークの領域の総計で割ることによって、分子中の各炭素原子型の相対百分率を測定した。このようにして、標準原子によって測定した、分子中の各原子の割合を計算することができた。すべてのp-GlcNAc分子は、明確にC1,C2,C3,C4,C5およびC6原子を有するN−アセチル化グルコサミン残基からなっている。ここで、ポリマー中のすべてのグルコサミン残基がN−アセチル化されているならば、上記のいずれのグルコサミン残基炭素原子ピーク領域に対するN−アセチルCH3炭素原子ピーク領域の割合も1.0 となるはずである。CH3(領域)割合の値を得るため、図20においてこれらのデータを使用した。
【0252】
図20において計算した割合は、多くの場合、1.0 に等しいか、または実験誤差の範囲内でほとんど等しい。たとえば、CH3/C2=1.097 、CH3/C6=0.984 、CH3/C5=1.007 、CH3/C1=0.886 である。これらの結果は、本発明のp-GlcNAc材料が不純物を含まず、完全にアセチル化されている(すなわち、本質的に100%のグルコサミン残基がN−アセチル化されている)という結論に一致する。
【0253】
15.実施例:孔径が制御されたp-GlcNAcの3次元マトリックスの合成および生
物学的特性決定
以下に述べるのは、制御された平均孔径を有するp-GlcNAc基剤の3次元多孔質マトリックスの製造方法である。こうしたマトリックスは各種の重要な用途を有する。特に、例えば細胞のカプセル化の用途などである。これらの細胞をカプセル化した組成物は移植用細胞基剤の治療剤として、また軟骨再生などの、その他の細胞および組織の工学的応用において有用である。ここに実証するように、p-GlcNAc材料の形態および3次元構造を操作することができるので、これによって、本発明のp-GlcNAc材料の応用の可能性を広げる上で強力な道具が提供される。
【0254】
15.1.材料および方法
p-GlcNAc出発材料:上記第5.3.2節の化学的精製方法を使用し、化学試薬としてフッ化水素酸を使用して、p-GlcNAcを調製した。
【0255】
マトリックスの形成:凍結乾燥する前に、下記第15.2節に列記する溶媒中でp-GlcNAc試料 20 mgの懸濁液(5 mlずつ)を製造した。次に試料を組織培養皿のウェルに注ぎ入れ、−20℃で凍結した。この凍結試料を凍結乾燥し、生成した3次元マトリックスを取り出した。
【0256】
走査型電子顕微鏡操作:ここで利用した操作法は、上記第12.1節にしたがって実施した。図21A−Gに示す画像は200X倍率のマトリックス材料であり、これらの図のそれぞれに200 ミクロンを示すスケールを表示している。
【0257】
15.2.結果
上記第15.1節にしたがって、以下の各溶媒からp-GlcNAc試料が得られた。
【0258】
A.蒸留水
B.蒸留水中10%メタノール
C.蒸留水中25%メタノール
D.蒸留水のみ
E.蒸留水中10%エタノール
F.蒸留水中25%エタノール
G.蒸留水中40%エタノール
各溶媒を使用して形成したマトリックス試料を、走査型電子顕微鏡分析に供した。これを図21A−Gに示す。これらの図によって、各懸濁液中のメタノールまたはエタノールの比率が増加するにしたがって、マトリックスの平均孔径が小さくなることがわかる。
【0259】
特定すると、2つの水懸濁液(図21Aおよび21D)中の孔径は平均して200 ミクロンに近い。図21Cおよび21F(それぞれ25%メタノールおよび25%エタノール)に示す試料の孔径は平均して30〜50ミクロンである。
【0260】
ここに示した結果によると、p-GlcNAcの孔径を制御するのに、エタノールおよびメタノールの両方が有効に利用され得るが、p-GlcNAcマトリックスの孔径の制御能力において、エタノールの方がメタノールよりも有効であるようである。
【0261】
16. 実施例: 三次元多孔性p-GlcNAcマトリックス上での細胞増殖
本節では、細胞培養の基質として三次元多孔性p-GlcNAcマトリックスを有効に使用したことを実証する結果を記載する。
【0262】
16.1 材料および方法
p-GlcNAc出発物質: p-GlcNAcを、フッ化水素酸を化学試薬として使用して上記の第5.3.2 節に記載の化学的精製法を使用して調製した。
【0263】
マトリックス形成: 三次元p-GlcNAcマトリックスを水、水−エタノールまたは水- メタノール混合物中のp-GlcNAcの懸濁液を凍結乾燥させて製造した。
【0264】
凍結乾燥前に、以下の溶媒中に、p-GlcNAc 20 mgを含有する懸濁液(5 ml)を調製した。
【0265】
1. 蒸留水のみ
2. 10% メタノール含有蒸留水
3. 25% メタノール含有蒸留水
4. 蒸留水のみ
5. 10% エタノール含有蒸留水
6. 25% エタノール含有蒸留水
7. 40% エタノール含有蒸留水
サンプルをプラスチック製の組織培養皿の丸いウェルの中に注ぎ、−20℃で凍結させた。その後、凍結サンプルを凍結乾燥させ、そして得られた三次元マトリックスを取り出した。各マトリックスのサンプルを走査型電子顕微鏡(SEM) 分析にかけた。
【0266】
細胞: ATCCより入手した、マウス胚BALBC/3T3 の繊維芽細胞系( クローンA31)を三次元多孔性p-GlcNAcマトリックス上での培養に使用した。
【0267】
培養条件: 多孔性マトリックスの 1 cm2サンプルを組織培養ウェルに入れ、そして標準の組織培養増殖培地を上から注いだ。各ウェルに細胞を植え、細胞をCO2 インキュベーター(5% CO2)内で37℃で6 日間培養した。
【0268】
SEM 手順: マトリックスサンプルを固定し、そして上記の第12.1節に記載したようにSEM 分析にかけた。マトリックスは蒸留水中の p-GlcNAc を凍結乾燥して製造した。図の解説( 図 22A〜G)に示したように、像は100 倍〜5000倍の倍率で変化する。
【0269】
16.2. 結果
付着しているマウス繊維芽細胞を含んでいるp-GlcNAcマトリックスのSEM 写真を図22A 〜G に示す。これらの写真により三次元p-GlcNAcマトリックスは付着したマウス繊維芽細胞を含むことが分かる。さらに、写真から細胞とp-GlcNAcマトリックス物質との間には密接な相互作用と連結があることも明らかである。また、細胞は球状の三次元形態をしており、プラスチックの培養皿で直接培養した細胞の、平らに広がった形とは異なることも注目に値する。細胞の生存率は95% 以上であった。
【0270】
17. 実施例: p-GlcNAcは術後癒着を有効に防止する
本文の実施例では、p-GlcNAc物質の有効な使用例、詳細には、p-GlcNAc膜およびゲル構成物が一連の癒着動物モデルにおいて、術後癒着の形成を防止することを示す。
【0271】
17.1 材料および方法
p-GlcNAc- 乳酸の合成: p-GlcNAc膜出発物質は、フッ化水素酸を化学試薬として使用して第5.3.2 節に記載のように化学的方法により製造した。
【0272】
p-GlcNAcを以下の方法により脱アセチル化p-GlcNAcに変換した。( 脱アセチル化により生じる質量損失を約15% と仮定すると、p-GlcNAc- 乳酸 1g を生成するために、p-GlcNAc約 1.4 gを必要とすることに注意すべきである。) p-GlcNAc膜物質約200 mgを60% NaOH約200 mlと激しく混合した。p-GlcNAc膜物質をできるかぎり分離させるために激しい振盪を利用した。使用するNaOH溶液は少なくとも使用の12時間前に調製した。p-GlcNAc膜物質を分離させ湿潤させるために断続的に振盪しながら、サンプルを80℃の湯浴に6 時間入れた。6 時間後、サンプルを湯浴から取り出し、そしてNaOH溶液を直ちに取り除いた。室温で膜物質をpH 7になるまで蒸留H2O で洗浄した。膜を水から取り出し、そしてテフロンシート上で乾燥させた。
【0273】
この時点で、脱アセチル化の程度を測定するためにC 、H 、N 分析のためのサンプル 2 mg を採取した。さらに、1%酢酸への溶解度を調べた。溶解度が1 mg/ml である場合はp-GlcNAc物質が適切に脱アセチル化されていることを示す。
【0274】
その後、部分的脱アセチル化p-GlcNAcを以下の方法を使用してp-GlcNAc- 乳酸に変換した。すなわち、部分的脱アセチル化p-GlcNAc物質を全て湿潤させ、そして攪拌を可能にするのに十分量の2-プロパノール( 水を10% 含有) を250 mlのエルレンマイヤーフラスコ中の部分的脱アセチル化p-GlcNAc 1 gに添加した。( 約30 ml の2-プロパノールを必要とした。) 2- プロパノールは試薬級で、そして各合成前に調製したばかりのものでなければならない。攪拌しながら、50% 乳酸水溶液 1.1 mL を加えた。乳酸は試薬級でなければならず、有効な( すなわち非エステル化) 乳酸の正確な存在濃度を測定するために分析する必要がある。そのために、通常0.1 N NaOHを用いてフェノールフタレインの終点(pH 7.0)まで滴定した。各p-GlcNAc合成のためには、使用する乳酸の濃度は一定でなければならず、すなわち、+/−1 パーセントでなければならない。混合物を少なくとも2 時間攪拌した。反応速度を上げるために僅かに加熱することができる。反応を完全に進行させるために、反応時間を延長でき、または50% 乳酸水溶液の量を増加できる。攪拌後、フラスコの内容物を定量用の無灰濾紙を使用してブフナーロートで濾過した。物質を無水2-プロパノール 15 mlで洗浄した。ヒュームフード中で2 時間風乾させ、その後40℃のオーブンに一夜放置した。部分的脱アセチル化p-GlcNAc出発物質 1グラムごとに、約1.4 g の最終p-GlcNAc- 乳酸( すなわち、40% の質量増加) が得られるはずである。
【0275】
p-GlcNAc- 乳酸のゲル構成物: p-GlcNAc- 乳酸物質を以下のようにゲルに構成した。p-GlcNAc- 乳酸出発物質を再蒸留脱イオン水に溶解し0.1 〜4.0 重量% の濃度にした。その後、試薬級のプロピレングリコール(2- プロパンジオール) をプロピレングリコールの最終濃度が1 〜10% の間になるよう添加した。いくつかの場合は、生成物が細菌および/ またはカビで汚染されるのを防ぐために防腐剤を添加した。代表的には、0.1%〜4.0%のp-GlcNAc- 乳酸濃度で調製した。これらの調成物の粘度はp-GlcNAc- 乳酸の割合が増すにつれて急激に増加し、0.5%またはそれ以上のp-GlcNAc- 乳酸を有するものがゲルとなる。
【0276】
動物モデル:
Sprague-Dawley系ラット:このモデルでは、盲腸の奬液膜表面を擦りまたは刺激し、それを腹膜の側壁に付着させておくことにより癒着を生じさせる。この方法を用いて対照動物に癒着を誘導できる成功率は平均 80%と報告されている。
【0277】
詳細には、これらのラットで術後癒着を誘導する手術は以下のとおりであた。動物を仰向けに寝かせ、無菌的外科手術に適するように準備し滅菌した布で覆った。正中線切開して腹腔を露出した。左の腹壁の約0.5 cm×1.0 cmの腹膜側壁の部分を注意深く切開し、腹膜とともに筋肉の薄層を切り取った。
【0278】
その後、盲腸を持ち上げ取り出した。盲腸の近位端の側方の表面上の約0.5 cm×1.0 cmの部分を乾燥ガーゼで10回擦って傷つけた。その後、盲腸をメスの刃でこすって小さな点状出血を生じさせた。盲腸の擦り傷および腹膜の切開を15分間露出させておいた。
【0279】
15分後、試験物質( すなわちp-GlcNAc物質) または対照物質を盲腸の傷に施した。その後、盲腸の擦り傷および腹膜の傷を対置し、そしてさらに15分間アリス組織鉗子で接触させておいた。
【0280】
その後、盲腸の擦り傷の部分を腹膜部に隣接するように盲腸を腹部に戻した。腹部の切開を閉じて、そして動物を麻酔から覚醒させた。
【0281】
術後14日目に、動物を安楽死させ、擦り傷部分の術後癒着の形成を調べた。癒着が存在した場合は、癒着に関係している全ての領域( すなわち、体壁、試験または対照物質そして内部器官) を動物から切り離した。
【0282】
癒着の程度と癒着の付着力を以下の尺度に従って評価した:
癒着程度のスコア:
0. 癒着無し
1. 領域の25% 以下の癒着
2. 領域の50% 以下の癒着
3. 領域の75% 以下の癒着
4. 領域の25% を越える癒着
付着力スコア:
0. 癒着無し
1. 鈍的切開により引き離される
2. 攻撃的な切開により引き離される
3. 鋭的切開を必要とする
別の動物モデル: ブタおよびウマの大型動物の腸モデルを使用して腹膜癒着の防止を評価した。
【0283】
手術: 動物を仰向けに寝かせ、無菌的外科手術に適するように準備し滅菌した布で覆った。正中線で切開して腹腔を露出させた。小腸を持ち上げ、2 cm×2 cm部分を確認し、十分に擦り( メスを使用して約200 回擦る) 、そして10分間乾燥させた。その後、試験物質(すなわちp-GlcNAc物質) または対照物を擦り傷に施し、そして小腸の傷の部分を腹部に戻した。上記の大型動物の腸モデルでは、それぞれが隣接する傷から4 インチ離れているように腸管上に6 箇所の傷を付けると最も癒着を形成する傾向がある。最後に傷を付けた後、腹部の切開を閉じ、そして動物を麻酔から覚醒させた。
【0284】
腹膜癒着の分析: 手術後21日目に、動物を安楽死させ、擦過傷領域を調べ、癒着の形成をSprague-Dawley系ラットの盲腸モデルの場合と同様の手順に従って評価した。
【0285】
17.2 結果
損傷が生じた場合、身体は損傷部を回復させるために、複雑な一連の反応を開始する。治癒の最終段階では、傷の部位に結合組織が形成され、身体構造を再生し、そして患部がさらに損傷を受けるのを防ぐ。ある場合には、この治癒過程の流れが適正にいかず、生命を脅かす事態を招くことがあり得る。
【0286】
例えば、術後の内蔵器官が回復する過程で、手術中に生じたフィブリン塊が隣接する器官の表面に浸入し、繊維芽細胞の移動を生じさせる連結部を形成し得る。この移動がコラーゲンの沈着および組織の増殖を招き、ひいては関係する器官がお互いに癒着することとなる。
【0287】
上記の癒着を術後癒着と呼ぶが、その器官または関係する器官に歪みを生じて疼通、閉塞および機能不全を引き起こし得る。関節の固定化、腸閉塞および不妊はしばしば術後癒着に関連している。さらに、術後癒着が症状を悪化させ、その付近の将来の手術時間を長引かせる原因となるであろう。この最後の問題点は、別の手術が必要であり得る開心術および婦人科の帝王切開の処置で特に問題となる。癒着の形成は腹部、心臓血管および整形外科の処置後に非常に一般的である。
【0288】
癒着が病的になり臓器の機能を著しく損なう場合は、外科的癒着剥離術( 細心の外科技術と関連した癒着の鋭的または鈍的切開) が、癒着を取り除くために現在行われている方法である。1991年にはこの外科的癒着剥離術が約500,000 例実施された。しかしながら、この処置は癒着形成の再発率が90% に上ると報告されており、効果のないことで有名である。さらに、上記の術後癒着の防止に有効とされている他の技術や組成物もない。
【0289】
従って、ここに示した結果は、本発明のp-GlcNAc物質が術後癒着の防止に有効性を示したことにおいて意義深いものである。詳細には、ここに示した結果は、p-GlcNAc物質をベースとする固形および液体処方物が、認可されたラットおよびブタの動物モデル系における腹部の術後癒着の形成のバリヤーとしての効力を示すものである。
【0290】
癒着形成の研究に使用される、認可された動物モデルの1つは、Sprague-Dawlery 系ラットにおける内臓と腹腔壁の癒着を用いている。部分的脱アセチル化p-GlcNAc膜および部分的脱アセチル化p-GlcNAc- 乳酸ゲル構成物の両者とも、未処置対照群またはこの効能を表示している唯一の市販薬であるInterCEED(商標名)(Johnson & Johnson)処置群に比べて癒着の発生を防止および/ またはかなり軽減していた。
【0291】
詳細には、全部で18匹のラットを使用してp-GlcNAc- 乳酸ゲル構成物を試験した。12匹を対照とし、6 匹は0.25%p-GlcNAc-乳酸ゲル、10% プロピレングリコール、水を投与した。p-GlcNAc- 乳酸ゲル処置群はどちらの対照群と比較しても、下記の表VIIIに示したように、有意に術後癒着の形成を減少させた。
【表8】
部分的脱アセチル化p-GlcNAc膜もラット動物モデルにより術後癒着の発症の予防能力を検討した。この実験には全部で22匹のラットを使用した。12匹を対照群に、6 匹を未処置群に、6 匹をInterCEEDTM処置群にした。10匹はそれぞれ約60% 脱アセチル化p-GlcNAcの 1cm×1cm 膜で処置した。下記の表IXに示した通り、部分的脱アセチル化p-GlcNAc膜処置動物は未処置およびInterCEEDTM処置に比べて術後癒着の形成の発生が有意に低下した。
【表9】
腹膜癒着を防止する大型動物の腸モデルもp-GlcNAc組成物の試験に使用した。詳細には、6 匹のブタと1 匹のウマを使用して、部分的脱アセチル化p-GlcNAc膜およびp-GlcNAc- 乳酸ゲルの両者を試験した。部分的脱アセチル化p-GlcNAc膜は 60%脱アセチル化p-GlcNAc膜の2 cm×2 cm片から成り、一方p-GlcNAc- 乳酸ゲルは10% プロピレングリコールおよび水で調製した0.25%p-GlcNAc-乳酸から成っていた。対照動物は傷の部位に何も処置を施さなかった。
【0292】
これらの大型動物の研究結果により、対照の部位は多重癒着を生じ、周りの部位の組織も脅かしていたが、p-GlcNAc膜およびゲル構成物は有効に癒着の形成を防止した。
【0293】
対照および処置した部位からのサンプルを別にSEM を使用して調べたが、処置組織に比較して対照の部位では繊維症の増加を示した。
【0294】
18.実施例: p-GlcNAc物質の生分解能
本節に示す実施例は、in vitroおよびin vivo での生分解能および吸収速度を制御しうる本発明のp-GlcNAc物質を製造できることを明示する。
【0295】
18.1 材料および方法
p-GlcNAc物質: 基本型 I は、フッ化水素酸を化学試薬として利用して化学的方法で、上記の第5.3.2 節に記載の方法により製造した。基本型 Iは100%アセチル化p-GlcNAcであった。
【0296】
基本型 3A のp-GlcNAc出発物質は、フッ化水素酸を化学試薬として利用して化学的方法で上記第5.3.2 節に記載の方法により製造した。その後、p-GlcNAc物質を上記の第5.4 節に記載の方法で脱アセチル化した。詳細には、p-GlcNAc物質を40%NaOH 溶液で60℃で30分処理した。生じた基本型 3A は測定の結果30% 脱アセチル化されていた。
【0297】
基本型 4のp-GlcNAc出発物質は、フッ化水素酸を化学試薬として利用して化学的方法で、上記の第5.3.2 節に記載の方法により製造した。その後p-GlcNAc物質を40%NaOH 溶液で60℃で30分処理して脱アセチル化した。次いで、繊維を蒸留水に懸濁し、−20℃で凍結しそして凍結乾燥した。基本型 4も測定の結果30% 脱アセチル化されていた。
【0298】
腹部移植モデル: Sprague-Dawley系白色ラットを腹部移植モデル実験に使用した。動物を麻酔し外科手術の準備をし、そして皮膚と腹部筋肉を切開した。盲腸の位置を確かめ、そして持ち上げた。p-GlcNAc物質の1 cm×1 cm膜を盲腸の上に置き、そして切開部をナイロン糸で閉じた。盲腸上に物質を置かなかったものを対照動物とした。
【0299】
移植の14および21日後に動物を切開した。内移植と外移植処置の間の写真を撮った( 図23A 〜E)。盲腸のサンプルを移植後に組織病理学のために準備した。
【0300】
in vitro p-GlcNAc 分解リゾチーム- キチナーゼ分析: 分析はN-アセチルグルコサミンの比色分析であり、そして以下のとおりに実施した。反応サンプル 150μl を13×100 mmのガラス使い捨て試験管に2通りずつ分注した。0.25 M リン酸カリウム緩衝液(pH 7.1) 25 μl を各試験管に添加し、その後0.8 M ホウ酸カリウム溶液(pH 9.8) 35 μl を添加した。試験管を2分以内に直ちに氷浴に浸す。その後、サンプルを氷浴から取り出し、新たに調製したDMAB試薬1mlを添加し、そしてサンプルが渦巻くように混合した。DMAB( ジメチルアミノベンズアルデヒド) 試薬は氷酢酸 70 mlおよび11.6 N( 濃)HCl 10 mlをp-ジメチルアミノベンズアルデヒド8 gに添加して製造した。
【0301】
標準曲線を作成するために、以下の手順を実施した。GlcNAc 原液を0.010 M酢酸ナトリウム緩衝液(pH4.5) で0.1 mg/ml に希釈し、そして希釈したGlcNAc溶液 0 μl 、20μl 、30μl 、90μl または120 μl を一組の試験管に添加した。その後、0.010 M 酢酸ナトリウム緩衝液(pH 4.5) 150μl 、130 μl 、60μlまたは30μl をそれぞれ試験管に添加した。次いで、0.25 M リン酸カリウム緩衝液(pH 7.1) 25 μl および0.8 M ホウ酸カリウム緩衝液(pH 9.8) 35 μl を各試験管に添加した。同様の手順により2組目の試験管を調製する。
【0302】
試験管に栓をし、そして100 ℃で正確に3 分間、沸騰させる。その後、試験管を氷浴に浸す。試験管を氷浴から取り出し、新たに上記の節で記載した方法に従って調製したDMAB試薬 1 ml を各試験管に添加する。試験管を37℃で20分間インキュベートする。各試験管の内容物の吸光度を585 nMで読み取る。吸光度はできるかぎり迅速に読み取らねばならない。標準曲線をグラフ用紙に描き、そして反応サンプルのN-アセチルグルコサミンの濃度を測定するのに使用する。典型的な標準曲線を図23に示す。
【0303】
18.2 結果
リゾチームによる分解に対するp-GlcNAc膜物質の相対的感受性を検定する実験により、p-GlcNAc物質のin-vitroでの生分解能を研究した。p-GlcNAc膜を10 mM酢酸緩衝液中の過剰のリゾチームにさらし、そしてN-アセチルグルコサミンのその後の放出を、上記の第18.1節に記載されている検定を使用して測定した。
【0304】
これらの実験の結果により、部分的脱アセチル化膜は、よりリゾチームに消化されやすく( 図24を参照) 、そしてさらに、リゾチームの分解速度は脱アセチル化の程度に直接に関係している(50%と25% の脱アセチル化p-GlcNAc膜の分解速度を比較している図25を参照) ことが示された。
【0305】
さらに、in-vivo でのp-GlcNAc物質の生分解能に関する実験を実施した。これらの実験では腹部移植モデルを利用した。以下に記載する3 種のp-GlcNAc物質を試験した。
【0306】
試験したp-GlcNAc物質:
1) 完全アセチル化p-GlcNAc( 基本型 1 と名付けた);
2) 部分的脱アセチル化p-GlcNAc膜( 基本型 3A と名付けた);および
3) 凍結乾燥した部分的脱アセチル化p-GlcNAc膜( 基本型 4と名付けた)。
【0307】
完全アセチル化p-GlcNAc( 基本型 1) は、図26A 〜26C に示すように21日以内に分解吸収された。部分的脱アセチル化p-GlcNAc膜( 基本型 3A)は図26D 〜26Eに示すように、14日以内に完全に分解吸収された。凍結乾燥した部分的脱アセチル化p-GlcNAc膜( 基本型 4) は移植の21日後でも完全には分解吸収されていなかった。
【0308】
組織病理学的分析により、ひとたびp-GlcNAc物質が分解吸収されれば、処置動物および対照動物から得られた組織サンプルの間で、検知できる組織学的相違は存在しないことが示された。
【0309】
19. 実施例: p-GlcNAc の止血作用の研究
ここに記載する実験は、出血防止に対する、本発明のp-GlcNAc物質の有効性を研究するものである。p-GlcNAc物質の出血防止での有効性をさらに市販の止血製剤と比較する。
【0310】
19.1 材料および方法
p-GlcNAc および対照物質: 部分的脱アセチル化( 約70%)p-GlcNAc膜を、フッ化水素酸を化学試薬として利用して、上記の第5.3.2.節に記載の化学分離技術、および上記の第5.4 節に記載の技術を使用して調製した。2cm ×1cm 片を使用した。p-GlcNAc- 乳酸ゲル( プロピレングリコールおよび水の中で形成した4%p-GlcNAc- 乳酸) を上記の第17.1節に記載の方法に従って製造した。脾臓と肝臓の出血研究に利用した対照物質はGelfoam(商標名)(Upjohn社) であった。小血管(small blood vessel)出血の研究に使用した対照物質はGelfoamTMおよびAvitene(商標名)(Medchem Products社) であった。
【0311】
試験動物: ニュージーランド白色ウサギを使用した。3 匹は脾臓に2 箇所の、そして肝臓に1 箇所の創傷を受けた。4 匹は尾部の腸間膜動脈系の同様の大きさの血管に、5 箇所の外科的創傷を受けた。
【0312】
外科手術の準備: 動物をケタミン(ketamine)HCl およびキシラジン(xylazine)を用いて麻酔した。動物を仰向けに寝かせ、そして腹部の毛を全て除去した。その後、腹部をポビドン- ヨウ素および70% イソプロピルアルコールで洗浄し、そして無菌の外科手術のために滅菌した布で覆った。
【0313】
肝臓/ 脾臓の外科手術: 正中線切開をし、そして脾臓または肝臓のいずれかを露出して体外に出し、そしてモイストラップスポンジ(moist lap sponge)で包んだ。直径3 〜4 mm のコルクボーラーを使用して、器官の一方の端に約2 mmの深さの円形の創傷を作った。ひとたび脾臓組織を除去したら、事前に秤量した4 インチ×4 インチのガーゼスポンジを使用して、脾臓の創傷から失われた全ての血液を1分間吸収した。スポンジを再秤量し、その特定の創傷から失われた血液の量を量った。その後、処置物質の1種を創傷に施して試験動物を処置した。止血までの時間と止血までに失われた血液の量を記録した。
【0314】
最初の創傷の止血が達成された後、脾臓の二番目の創傷と肝臓の1 箇所の創傷を同様の手順に従って作った。
【0315】
小血管の外科手術: 正中線切開をし、そして小腸を体外に出し尾部腸間膜動脈系を露出した。腸をモイストラップスポンジで包み、そしてほぼ同じ大きさの5本の血管を確認した。メスを用いて1 本の血管に約1 mmの深さの創傷を作った。事前に秤量した4 インチ×4 インチのガーゼスポンジを使用して1 分間、血管の創傷から失われた全ての血液を吸収した。スポンジを再秤量して、その特定の創傷から失われた血液の量を測定した。その後、処置物質の1 つを創傷に施して実験動物を処置した。止血までの時間と止血までに失われた血液の量を記録した。
【0316】
最初の創傷の止血が達成された後、さらに4 箇所の創傷を同様の手順に従って作った。
【0317】
19.2 結果
p-GlcNAc物質の、ラット動物モデルの脾臓および肝臓での出血防止能について試験した。試験したp-GlcNAc物質は: 1)部分的脱アセチル化( 約70%)p-GlcNAc;および 2)p-GlcNAc-乳酸ゲル( プロピレングリコールおよび水の中で形成した4% p-GlcNAc-乳酸)。これらのp-GlcNAc物質の効果をGelfoamTM(Upjohn 社) と比較した。
【0318】
各物質を3 回試験した( 脾臓で2回、肝臓で1回)。p-GlcNAcをベースとする物質は両者とも、適用後、最初の1分以内の出血の防止においてGelfoamTMに匹敵する効果を示した。
【0319】
p-GlcNAcをベースとする物質は別の利点を有する。詳細には、処置の間、適所にとどめておく必要がなく、体内に放置でき、体内で2 〜3 週間以内に分解吸収され(GelfoamTMはこの効果を示さない) 、全身および最小限の観血的外科手術の両方に適合する。
【0320】
次に、p-GlcNAcをベースとする物質の小血管での出血を防止する効力を研究し、そして市販の止血製剤と比較した。
【0321】
各物質を5 回(1匹の動物は2 回、そして他の3 匹は1 回) 試験した。p-GlcNAc膜およびゲル構成物は容易に部位に適用され2 分以内に出血を防止した。GelfoamTMは、その機能を発揮するためにその場に保持しなければならないが、p-GlcNAc物質と同じく2分以内に止血が達成された。コラーゲンから成る繊維状物質であるAviteneTMは、扱いにくくそして止血するのに5分以上を要した。
【0322】
それ故、ここに記載する結果により、ここで試験したp-GlcNAc物質は有効で便利な止血剤であることが実証された。
【0323】
20. 実施例: p-GlcNAcドラッグデリバリーシステム
抗腫瘍性薬剤を悪性皮膚癌および結腸癌性腫瘍部位に送りだし、送りだされた抗腫瘍剤が腫瘍に対して治療上の強い影響力を発揮するp-GlcNAc物質の有効な使用例を示す研究をここに記載する。
【0324】
20.1 材料および方法
p-GlcNAc- 乳酸ドラッグデリバリー組成物: 5'- フルオロウラシル(5'-FU)およびp-GlcNAc- 乳酸の混合物を以下のように作成した; 5'-FU(50 mg/mL) 0.5 mLをプロピレングリコール 0.5 mL と混合し、そして4% p-GlcNAc-乳酸 2.0 mL を添加し混合した。p-GlcNAc- 乳酸は上記の節に記載の技術を使用して製造した。広範に混合した後も、5'FUは完全にはp-GlcNAc- 乳酸ゲルに溶解しなかった。完全に混合したと仮定すると、5'-FU の最終濃度は6.25 mg/mLとなろう。
【0325】
マイトマイシン(Mito)とp-GlcNAc- 乳酸の混合物を以下のように作成した; Mito( 凍結乾燥粉末) 0.5 mgをプロピレングリコール 5 ml に溶解し、そしてMito溶液 0.5 ml をMPT の4% p-GlcNAc-乳酸調製物 0.5 mL と混合し最終Mito濃度が0.2 mg/ml であり最終p-GlcNAc- 乳酸の濃度が2%となるようにした。Mitoは容易にp-GlcNAc- 乳酸ゲルに溶解するので、この物質は相溶性であった。
【0326】
p-GlcNAc膜5'FUデリバリー組成物: 5'- フルオロウラシル(5'-FU)のサンプルを、フッ化水素酸を化学試薬として利用し、上記の第5.3.2 節に記載の化学分離法を用いて製造した純粋なp-GlcNAc膜物質のディスク内に固定した。ここに記載する各ディスクは直径1.5 cmであった。
【0327】
高用量(HD)ディスクを製造するために、5'-FU の50 mg/mL溶液 0.64 mLを純粋なp-GlcNAc 約8 mg を含有する懸濁液と混合した。混合物を数時間、静置し5'-FU がp-GlcNAcに吸収されるのを促進し、そしてその後、55℃で3.5 時間乾燥した。生じたHDディスクは、5'-FU を総量で32 mg 含有していたが、これは通常、癌患者に投与する5'-FU の14日間の総用量の約二倍に等しい。
【0328】
低用量(LD)の5'-FU を含有するp-GlcNAcディスクをそのLDディスクが5'-FU 17 mg を含有する以外は同様の方法で製造した。この量はヒトの体重 Kg 当たりの5'-FU の通常量に基づいて、実験マウスの体重に標準化した、通常の14日間のヒトの用量に等しい量である。
【0329】
試験動物: 5'-FU の研究のために、HT-29 結腸癌性腫瘍を発症させるために、SCID( 重症複合型免疫不全症) マウスに、標準組織培養法により得られたHT-29 結腸癌細胞(ATCC; 接種物当たり1 ×105 細胞) を側腹部の皮下注射により接種した。これらの注射により14〜21日で触診できる腫瘍を発生した。腫瘍を切開し、そして壊死組織を切り捨てた。HT-29 結腸癌性腫瘍を3 ×3 ×3 mm片に薄切りした。
【0330】
実験的SCIDマウスを標準用量のアベチン(avetin)で腹腔内注射により麻酔し、そしてHT-29 結腸癌腫瘍の切片を各マウスの盲腸に移植した。詳細には、各マウスの腹部を外科的に切開して盲腸の位置を確かめ、メスで切って小さく切開した。3 ×3 ×3 mm腫瘍薄片を、5.0 絹縫合糸を使用して盲腸の上の切開を覆って縫合した。その後、腹部をクレーアダム釘(Clay Adam staple)を使用して閉じた。
【0331】
全てのマウスを1 匹ずつケージにいれ、そして2 週間摂食した。全てのマウスが健康で、そして2 週間の期間の終わりに閉塞性結腸を有するものはなかった。
【0332】
14日目に、各マウスを麻酔し、そして再び開腹した。腫瘍の成長を測定した(長さおよび水平の寸法)。その後、腫瘍をp-GlcAc/抗腫瘍薬で処置するか、または対照として使用した。
【0333】
6 匹のマウスをp-GlcNAc- 乳酸5'FUの研究に使用し、そして15匹をp-GlcNAc膜5'FUの研究に使用した。
【0334】
マイトマイシンの研究のために、9 匹のSCIDマウスに、A431扁平上皮細胞皮膚癌細胞(ATCC; 接種物当たり1 ×105 細胞) を皮下注射して移植した。14日以内に全てのマウスで腫瘍が生じた。
【0335】
処置: p-GlcNAc- 乳酸5'FUの研究では、5'フルオロウラシル(5'-FU)含有p-GlcNAcゲル混合物を腫瘍塊上の皮膚領域に1 日に1 回「塗布」して動物を処置した。腫瘍の大きさの測定を毎日実施した。対照動物には、p-GlcNAcのみで処置したもの、5'-FU 無し、処置を全く受けていないものを含んでいた。
【0336】
p-GlcNAc膜5'FUの研究では、結腸の上に腫瘍を14日間、成長させた後、薬剤含有p-GlcNAc膜のディスクを外科的に直接その表面に移植して、SCIDマウスのHT29結腸腫瘍を処置した。移植手順後14日目に、マウスを犠牲にした。薬物含有p-GlcNAc膜の移植の直前の0 日および実験の最後の14日目に腫瘍体積を測定した。対照動物には、5'-FU を含まないp-GlcNAc膜で処置したもの、および全く処置を受けていないものを含んでいた。これとは別に、2 匹の動物はHDおよびLD規定食餌法に相当する用量の5'-FU の全身的な注入を毎日受けた。
【0337】
p-GlcNAc- 乳酸Mito研究では、p-GlcNAc- 乳酸5'FUの研究のように、3 匹の動物をMito含有混合物で処置して、動物を毎日処置した。3 匹はMitoを含まないp-GlcNAcで処置し、2 匹は処置をせず、そして1 匹はプロピレングリコールを受けた。
【0338】
20.2 結果
20.1.1 p-GlcNAc - 乳酸5'FU
p-GlcNAc- 乳酸5'FUドラッグデリバリーシステムの腫瘍の大きさに対する効果の研究を意図した実験を、上記の第20.1節に記載のように実施した。
【0339】
各腫瘍の最大の長さと幅の寸法を測定し、そしてこれらの寸法を使用して断面積を計算した。断面積値を以下の表Xに示す。
【表10】
p-GlcNAc- 乳酸5'FU処置動物を対照と比較したデータを図27〜28に示す。表Xおよび図27〜28に要約したデータにより、5'-FU 含有p-GlcNAc- 乳酸ゲルで処置したラットのHT-29 皮下腫瘍は対照と比較して増殖速度が有意に遅滞することが明らかに示唆される。その増殖はp-GlcNAc- 乳酸ゲルの対照と比較して2.5倍、そして未処置の対照と比較して4倍遅くなっている。
【0340】
20.2.2 p-GlcNAc- 乳酸MITO
p-GlcNAc- 乳酸5'FUドラッグデリバリーシステムの腫瘍の大きさに対する効果を研究するように意図された実験も、上記の第20.1節に記載のように実施した。
【0341】
各腫瘍の最大の長さと幅の寸法を測定し、これらの寸法を使用して断面積を計算した。断面積値を下記の表XIに示した。
【表11】
p-GlcNAc- 乳酸Mito処置動物を対照と比較したデータを図29〜30に示す。表XIおよび図29〜30に要約したデータにより、マイトマイシン含有p-GlcNAc- 乳酸ゲルで処置したラットで増殖した腫瘍は増殖速度が有意に遅延することが明らかに示唆される。その増殖はp-GlcNAc- 乳酸ゲル対照と比較して4 倍、そして未処置の対照と比較して4 倍遅くなった。
【0342】
20.2.3 p-GlcNAc膜5'FU
次に、p-GlcNAc膜5'FUドラッグデリバリーシステムの皮膚癌腫瘍の大きさに対する効果を研究することを意図する実験を、上記の第20.1節に記載のように実施した。
【0343】
種々の処置により生じた体積の変化百分率を含む、研究中に得られた腫瘍体積のデータを下記の表XIIに要約する。腫瘍は円柱状の形と仮定した。体積を、幅および長さを測定し、そして次式: V=πr2l[式中、半径r は幅の0.5倍であり、そしてlは長さである]を使用して計算した。
【表12】
図31は、上記の表XIIに表されているデータの一部を要約する。図31に示すように、データにより、高用量(HD)5'-FU 含有p-GlcNAc膜で処置した腫瘍は増殖を停止させ、そして全ての場合に、実際に有意に小さくなったことが強く示唆される。低用量(LD)ポリマー物質により、疾患が安定化し、そして腫瘍の大きさがわずかに減少した。対照的に、対照動物の腫瘍の大きさは急速に増加しつづけた。IVで処置した3 匹の対照動物のうち2 匹が研究中に死亡し、このことは当量の5'-FUの全身的なデリバリーは致死量にあたるが、一方p-GlcNAcポリマーを介する部位特異性デリバリーは動物から疾病を取り除くのに有効であることが示されたのは興味深い。
【0344】
20.3 結果
この節に示したデータにより、抗腫瘍性薬剤の部位特異性デリバリーは腫瘍の増殖を遅らせ後退させる正の効果を有することが強く示唆される。2 種の異なる構成物、すなわち、p-GlcNAc- 乳酸およびp-GlcNAc膜構成物を有するように製造されたp-GlcNAcドラッグデリバリー組成物を使用して有用な結果が得られた。さらに、2 種の異なる抗腫瘍剤、5'-FU およびMitoを使用して有効な結果が得られた。それ故、本発明のp-GlcNAcドラッグデリバリーシステムは、例えば、必要な腫瘍細胞部位に特異的に薬剤を送りだすのに有用な抗腫瘍活性を示す。
【0345】
ここに記載した本発明の多くの修飾と変法がその精神と範囲を逸脱することなく可能であることは明白である。上述の特定の実施態様は例示としてのみ掲載し、本発明は添付された請求の範囲の用語によってのみ制限されるものである。
【図面の簡単な説明】
【0346】
【図1】100% p-GlcNAc の化学構造を示す図である。
【図2】p-GlcNAcの炭水化物分析、ガスクロマトグラフィー−質量分光分析データを示す図である。
【図3A】純化p-GlcNAcの固体膜 (solid membrane) の円偏光二色性スペクトルを示す図である。
【図3B】脱アセチル化p-GlcNAcの固体膜の円偏光二色性スペクトルを示す図である。
【図4A】機械力精製法(上部)および化学/生物学的精製法(下部)により調製された純化珪藻p-GlcNAcの薄膜の赤外スペクトルを示す図である。
【図4B】第5.5 節に詳述した方法に従って膜に注型された市販の2つの「キチン」調製物の赤外スペクトルを示す図である。
【図4C】第5.4 節に詳述した方法に従って熱変性(上部)および化学的脱アセチル化(下部)により改質された純化p-GlcNAcの赤外スペクトルを示す図である。
【図4D】第5.3.2 節に詳述した化学/生物学的精製法を用いて、珪藻タラシオシラ・フルビアチリス(Thallasiosira fluviatilis) から誘導されたp-GlcNAc膜の赤外スペクトルを示す図である。
【図4E】オートクレーブ滅菌後に、第5.3.1 節に記載したような機械力精製法により調製されたp-GlcNAc膜の赤外スペクトルを示す図である。
【図5A】第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析結果を示す図である。
【図5B】第5.3.2 節に記載した化学/生物学的精製法を用いて精製したp-GlcNAcのNMR分析結果を示す図である。
【図6A】第5.3.1 節に記載した機械力精製法によって調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:4190x)。
【図6B】第5.3.1 節に記載した機械力精製法によって調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:16,250x)。
【図7A】第5.3.2 節で化学/生物学的精製法の説明において記載したHF処理によるp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:5270x)。
【図7B】第5.3.2 節で化学/生物学的精製法の説明において記載したHF処理によるp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:8150x)。
【図8A】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:5270x)。
【図8B】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である(倍率:16,700x)。
【図9A】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真を示す図である(倍率:200x)。
【図9B】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:1000x)である。
【図9C】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:5000x)である。
【図9D】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:10000x)である。
【図9E】第5.3.2 節に記載した化学/生物学的精製法の酸処理/中和変法により調製されたp-GlcNAc膜を表す走査電子顕微鏡写真(倍率:20000x)である。
【図10A】第5.3 節に記載した細胞溶解/中和精製法を用いて最初に調製し、ジメチルアセトアミド/塩化リチウム中に溶解し、そして以下の第5.5節に記載したように水中でマットに再沈殿させた物質から作られた純化p-GlcNAc膜の走査電子顕微鏡写真(倍率:1000x)である。
【図10B】第5.3 節に記載した細胞溶解/中和精製法を用いて最初に調製し、ジメチルアセトアミド/塩化リチウム中に溶解し、そして以下の第5.5節に記載したように水中でマットに再沈殿させた物質から作られた純化p-GlcNAc膜の走査電子顕微鏡写真(倍率10,000x)である。
【図11A】脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真(倍率:1000x)である。
【図11B】脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真(倍率:10,000x)である。
【図12A】珪藻の写真を示す図である。
【図12B】珪藻の写真を示す図である。
【図13】本発明の可能なp-GlcNAc誘導体および脱アセチル化p-GlcNAc誘導体の一部を示す略図である。
【図14】p-GlcNAc膜の存在下または不在下で増殖させた細胞の生存能実験の結果を示す図である。
【図15A】p-GlcNAc膜上で増殖させた形質転換マウス繊維芽細胞のSEM顕微鏡写真(倍率:1000x)である。
【図15B】p-GlcNAc膜上で増殖させた形質転換マウス繊維芽細胞のSEM顕微鏡写真(倍率:3000x)である。
【図16A】第13.1節に記載した方法に従って調製したコラーゲン単独対照物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16B】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16C】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16D】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図16E】第13.1節に記載した方法に従って調製したコラーゲン/p-GlcNAcハイブリッド物質の走査電子顕微鏡写真 (SEM)(倍率 100x)である。
【図17A】図16A のコラーゲン単独対照物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図17B】図16B のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図17C】図16C のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図17D】図16D のコラーゲン/p-GlcNAc物質上で培養したマウス3T3 繊維芽細胞のSEM(倍率 100x)である。
【図18】各炭素原子の面積を得るために、次いで CH3(面積)対C原子(面積)の比を計算するために使用した変換NMRデータ曲線を示す図である。
【図19】典型的なp-GlcNAc C13-NMRスペクトルを示す図である。
【図20】CH3(面積)対C原子(面積)比の計算値を表す変換NMRスペクトルデータを示す図である。上図はデータのグラフ表示であり、下図:データの数値表示である。
【図21A】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21B】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21C】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21D】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21E】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21F】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図21G】種々の溶媒中で調製された三次元p-GlcNAcマトリックスを示す写真を示す図である。
【図22A】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22B】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22C】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22D】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22E】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22F】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図22G】蒸留水中のp-GlcNAcを凍結乾燥することにより調製した三次元p-GlcNAcマトリックス上で増殖させた繊維芽細胞を示す写真を示す図である。
【図23】第18.1節に記載した手順を用いて得られた典型的な標準曲線を示す図である。
【図24】p-GlcNAcリゾチーム消化データを示す図である。
【図25】p-GlcNAcリゾチーム消化データを示す図である。
【図26A】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26B】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26C】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26D】p-GlcNAcのin vivo 生分解性データを示す図である。
【図26E】p-GlcNAcのin vivo 生分解性データを示す図である。
【図27】第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図28】第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/5'フルオロウラシル(FU)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図29】第20.1節に記載したように、処置を施さなかった動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図30】第20.1節に記載したように、p-GlcNAc-乳酸のみで処置した動物(黒○)またはp-GlcNAc-乳酸/マイトマイシン(mito)で処置した動物(○)の腫瘍の大きさの増加パーセントに関するデータを表す図である。
【図31】p-GlcNAc/5'FU 高用量(棒1)、p-GlcNAc/5'FU 低用量(棒2)、またはp-GlcNAc膜のみ(棒3)で処置した、あるいは処置を施さなかった(棒4)動物の動物あたりの腫瘍の大きさの平均変化パーセントを表す図である。
【特許請求の範囲】
【請求項1】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項2】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項3】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がマウスにおける全身注射によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項4】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性が溶出試験によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項5】
溶出試験のスコアが0である、請求項4に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項6】
溶出試験のスコアが1である、請求項4に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項7】
溶出試験のスコアが2である、請求項4に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項8】
約300万ダルトンまでの分子量を有する、請求項1〜7のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項9】
β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含む、請求項1〜8のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項10】
β-1→4配置で共有結合された約4,000〜約15,000のN-アセチルグルコサミン単糖を含む、請求項1〜9のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項11】
少なくとも1つのN-アセチルグルコサミン単糖が脱アセチル化されている、請求項1〜10のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項12】
少なくとも約25〜75%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項1〜11のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項13】
少なくとも約70%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項1〜12のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項14】
免疫的に中性(immunoneutral)である、請求項1〜13のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項15】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-N-アセチルグルコサミン分子が得られる、請求項1〜14のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項16】
酵素処理によって加水分解される、請求項15に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項17】
酵素がリゾチームである、請求項16に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項18】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定されるポリ-β-1→4-グルコサミン。
【請求項19】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項20】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性がマウスにおける全身注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項21】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性が溶出試験によって決定されるポリ-β-1→4-グルコサミン。
【請求項22】
溶出試験のスコアが0である、請求項21に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項23】
溶出試験のスコアが1である、請求項21に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項24】
溶出試験のスコアが2である、請求項21に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項25】
約240万ダルトンまでの分子量を有する、請求項18〜24のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項26】
β-1→4配置で共有結合された約4,000〜約150,000のグルコサミン単糖を含む、請求項18〜25のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項27】
β-1→4配置で共有結合された約4,000〜約15,000のグルコサミン単糖を含む、請求項18〜26のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項28】
少なくとも1つのグルコサミン単糖がアセチル化されている、請求項18〜27のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項29】
少なくとも約25〜75%のグルコサミン単糖がアセチル化されている、請求項18〜28のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項30】
少なくとも約30%のグルコサミン単糖がアセチル化されている、請求項18〜29のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項31】
免疫的に中性(immunoneutral)である、請求項18〜23のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項32】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-グルコサミン分子が得られる、請求項18〜31のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項33】
酵素処理によって加水分解される、請求項32に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項34】
酵素がリゾチームである、請求項33に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項35】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項36】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項37】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がマウスにおける全身注射によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項38】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性が溶出試験によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項39】
溶出試験のスコアが0である、請求項38に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項40】
溶出試験のスコアが1である、請求項38に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項41】
溶出試験のスコアが2である、請求項38に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項42】
約300万ダルトンまでの分子量を有する、請求項35〜41のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項43】
β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含む、請求項35〜42のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項44】
β-1→4配置で共有結合された約4,000〜約15,000のN-アセチルグルコサミン単糖を含む、請求項35〜43のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項45】
少なくとも1つのN-アセチルグルコサミン単糖が脱アセチル化されている、請求項35〜44のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項46】
少なくとも約25〜75%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項35〜45のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項47】
少なくとも約70%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項35〜46のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項48】
免疫的に中性(immunoneutral)である、請求項35〜47のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項49】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-N-アセチルグルコサミン分子が得られる、請求項35〜48のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項50】
酵素処理によって加水分解される、請求項49に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項51】
酵素がリゾチームである、請求項50に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項52】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定されるポリ-β-1→4-グルコサミン。
【請求項53】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項54】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性がマウスにおける全身注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項55】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性が溶出試験によって決定されるポリ-β-1→4-グルコサミン。
【請求項56】
溶出試験のスコアが0である、請求項55に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項57】
溶出試験のスコアが1である、請求項55に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項58】
溶出試験のスコアが2である、請求項55に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項59】
約240万ダルトンまでの分子量を有する、請求項52〜58のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項60】
β-1→4配置で共有結合された約4,000〜約150,000のグルコサミン単糖を含む、請求項52〜59のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項61】
β-1→4配置で共有結合された約4,000〜約15,000のグルコサミン単糖を含む、請求項52〜60のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項62】
少なくとも1つのグルコサミン単糖がアセチル化されている、請求項52〜61のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項63】
少なくとも約25〜75%のグルコサミン単糖がアセチル化されている、請求項52〜62のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項64】
少なくとも約30%のグルコサミン単糖がアセチル化されている、請求項52〜63のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項65】
免疫的に中性(immunoneutral)である、請求項52〜57のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項66】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-グルコサミン分子が得られる、請求項52〜65のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項67】
酵素処理によって加水分解される、請求項66に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項68】
酵素がリゾチームである、請求項67に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項69】
治療上有効な量の微小藻類ポリ-β-1→4-N-アセチルグルコサミンおよび製薬上許容される担体を含む医薬組成物。
【請求項70】
微小藻類が珪藻源である、請求項69に記載の医薬組成物。
【請求項71】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項69に記載の医薬組成物。
【請求項72】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項69に記載の医薬組成物。
【請求項73】
止血に有効な量の微小藻類ポリ-β-1→4-N-アセチルグルコサミンを含む止血用医薬組成物であって、微小藻類ポリ-β-1→4-N-アセチルグルコサミンが止血に適した形状または構造に構成されている医薬組成物。
【請求項74】
微小藻類が珪藻源である、請求項73に記載の医薬組成物。
【請求項75】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項73に記載の医薬組成物。
【請求項76】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項73に記載の医薬組成物。
【請求項77】
形状または構造が、ゲル、スポンジ、フィルムまたは膜である、請求項73に記載の医薬組成物。
【請求項78】
治療上有効な量の薬剤と、本質的に微小藻類ポリ-β-1→4-N-アセチルグルコサミンからなる製薬上許容される担体とを含む医薬組成物。
【請求項79】
微小藻類が珪藻源である、請求項78に記載の医薬組成物。
【請求項80】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項78に記載の医薬組成物。
【請求項81】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項78に記載の医薬組成物。
【請求項82】
微小藻類に由来しないポリ-β-1→4-N-アセチルグルコサミン医薬担体をさらに含む、請求項78に記載の医薬組成物。
【請求項83】
タンパク質を含まず、他の有機汚染物質を実質的に含まず、無機汚染物質を実質的に含まない、β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含んでなり、約800,000ダルトン〜約3,000万ダルトンの分子量を有する微小藻類ポリ-β-1→4-N-アセチルグルコサミンの製造方法であって、
(a) 細胞体およびポリ-β-1→4-N-アセチルグルコサミン繊維を含む微小藻類を、ポリ-β-1→4-N-アセチルグルコサミン繊維を合成することができる生物的作用物質で、ポリ-β-1→4-N-アセチルグルコサミン繊維を細胞体から遊離させるのに十分な時間処理し、
(b) 細胞体からポリ-β-1→4-N-アセチルグルコサミン繊維を分離し、そして
(c) 分離したポリ-β-1→4-N-アセチルグルコサミン繊維から全てのタンパク質、実質的に全ての他の有機汚染物質、および実質的に全ての無機汚染物質を取り除き、ポリ-β-1→4-N-アセチルグルコサミン種を単離する、
ことを含んでなる方法。
【請求項84】
単離されたポリ-β-1→4-N-アセチルグルコサミン(p-GlcNAc)種が約4,000〜約15,000のN-アセチルグルコサミン単糖を含んでなり、約800,000ダルトン〜約300万ダルトンの分子量を有する、請求項83に記載の方法。
【請求項85】
生物的作用物質がポリオキシン-Dである、請求項83に記載の方法。
【請求項86】
微小藻類が珪藻源である、請求項83に記載の方法。
【請求項87】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項86に記載の方法。
【請求項88】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項87に記載の方法。
【請求項1】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項2】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項3】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がマウスにおける全身注射によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項4】
約3,000万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性が溶出試験によって決定されるポリ-β-1→4-N-アセチルグルコサミン。
【請求項5】
溶出試験のスコアが0である、請求項4に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項6】
溶出試験のスコアが1である、請求項4に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項7】
溶出試験のスコアが2である、請求項4に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項8】
約300万ダルトンまでの分子量を有する、請求項1〜7のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項9】
β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含む、請求項1〜8のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項10】
β-1→4配置で共有結合された約4,000〜約15,000のN-アセチルグルコサミン単糖を含む、請求項1〜9のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項11】
少なくとも1つのN-アセチルグルコサミン単糖が脱アセチル化されている、請求項1〜10のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項12】
少なくとも約25〜75%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項1〜11のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項13】
少なくとも約70%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項1〜12のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項14】
免疫的に中性(immunoneutral)である、請求項1〜13のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項15】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-N-アセチルグルコサミン分子が得られる、請求項1〜14のいずれか1項に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項16】
酵素処理によって加水分解される、請求項15に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項17】
酵素がリゾチームである、請求項16に記載の生体適合性ポリ-β-1→4-N-アセチルグルコサミン。
【請求項18】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定されるポリ-β-1→4-グルコサミン。
【請求項19】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項20】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性がマウスにおける全身注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項21】
約2,400万ダルトンまでの分子量を有する生体適合性ポリ-β-1→4-グルコサミンであって、その生体適合性が溶出試験によって決定されるポリ-β-1→4-グルコサミン。
【請求項22】
溶出試験のスコアが0である、請求項21に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項23】
溶出試験のスコアが1である、請求項21に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項24】
溶出試験のスコアが2である、請求項21に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項25】
約240万ダルトンまでの分子量を有する、請求項18〜24のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項26】
β-1→4配置で共有結合された約4,000〜約150,000のグルコサミン単糖を含む、請求項18〜25のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項27】
β-1→4配置で共有結合された約4,000〜約15,000のグルコサミン単糖を含む、請求項18〜26のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項28】
少なくとも1つのグルコサミン単糖がアセチル化されている、請求項18〜27のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項29】
少なくとも約25〜75%のグルコサミン単糖がアセチル化されている、請求項18〜28のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項30】
少なくとも約30%のグルコサミン単糖がアセチル化されている、請求項18〜29のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項31】
免疫的に中性(immunoneutral)である、請求項18〜23のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項32】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-グルコサミン分子が得られる、請求項18〜31のいずれか1項に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項33】
酵素処理によって加水分解される、請求項32に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項34】
酵素がリゾチームである、請求項33に記載の生体適合性ポリ-β-1→4-グルコサミン。
【請求項35】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項36】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項37】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性がマウスにおける全身注射によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項38】
約3,000万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミンであって、その生体適合性が溶出試験によって決定される微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項39】
溶出試験のスコアが0である、請求項38に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項40】
溶出試験のスコアが1である、請求項38に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項41】
溶出試験のスコアが2である、請求項38に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項42】
約300万ダルトンまでの分子量を有する、請求項35〜41のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項43】
β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含む、請求項35〜42のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項44】
β-1→4配置で共有結合された約4,000〜約15,000のN-アセチルグルコサミン単糖を含む、請求項35〜43のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項45】
少なくとも1つのN-アセチルグルコサミン単糖が脱アセチル化されている、請求項35〜44のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項46】
少なくとも約25〜75%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項35〜45のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項47】
少なくとも約70%のN-アセチルグルコサミン単糖が脱アセチル化されている、請求項35〜46のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項48】
免疫的に中性(immunoneutral)である、請求項35〜47のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項49】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-N-アセチルグルコサミン分子が得られる、請求項35〜48のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項50】
酵素処理によって加水分解される、請求項49に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項51】
酵素がリゾチームである、請求項50に記載の生体適合性の微小藻類ポリ-β-1→4-N-アセチルグルコサミン。
【請求項52】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける筋肉内移植によって決定されるポリ-β-1→4-グルコサミン。
【請求項53】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性がウサギにおける皮内注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項54】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性がマウスにおける全身注射によって決定されるポリ-β-1→4-グルコサミン。
【請求項55】
約2,400万ダルトンまでの分子量を有する生体適合性の微小藻類ポリ-β-1→4-グルコサミンであって、その生体適合性が溶出試験によって決定されるポリ-β-1→4-グルコサミン。
【請求項56】
溶出試験のスコアが0である、請求項55に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項57】
溶出試験のスコアが1である、請求項55に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項58】
溶出試験のスコアが2である、請求項55に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項59】
約240万ダルトンまでの分子量を有する、請求項52〜58のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項60】
β-1→4配置で共有結合された約4,000〜約150,000のグルコサミン単糖を含む、請求項52〜59のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項61】
β-1→4配置で共有結合された約4,000〜約15,000のグルコサミン単糖を含む、請求項52〜60のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項62】
少なくとも1つのグルコサミン単糖がアセチル化されている、請求項52〜61のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項63】
少なくとも約25〜75%のグルコサミン単糖がアセチル化されている、請求項52〜62のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項64】
少なくとも約30%のグルコサミン単糖がアセチル化されている、請求項52〜63のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項65】
免疫的に中性(immunoneutral)である、請求項52〜57のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項66】
加水分解することによって均一且つ離散的な(discrete)分子量を有するポリ-β-1→4-グルコサミン分子が得られる、請求項52〜65のいずれか1項に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項67】
酵素処理によって加水分解される、請求項66に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項68】
酵素がリゾチームである、請求項67に記載の生体適合性の微小藻類ポリ-β-1→4-グルコサミン。
【請求項69】
治療上有効な量の微小藻類ポリ-β-1→4-N-アセチルグルコサミンおよび製薬上許容される担体を含む医薬組成物。
【請求項70】
微小藻類が珪藻源である、請求項69に記載の医薬組成物。
【請求項71】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項69に記載の医薬組成物。
【請求項72】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項69に記載の医薬組成物。
【請求項73】
止血に有効な量の微小藻類ポリ-β-1→4-N-アセチルグルコサミンを含む止血用医薬組成物であって、微小藻類ポリ-β-1→4-N-アセチルグルコサミンが止血に適した形状または構造に構成されている医薬組成物。
【請求項74】
微小藻類が珪藻源である、請求項73に記載の医薬組成物。
【請求項75】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項73に記載の医薬組成物。
【請求項76】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項73に記載の医薬組成物。
【請求項77】
形状または構造が、ゲル、スポンジ、フィルムまたは膜である、請求項73に記載の医薬組成物。
【請求項78】
治療上有効な量の薬剤と、本質的に微小藻類ポリ-β-1→4-N-アセチルグルコサミンからなる製薬上許容される担体とを含む医薬組成物。
【請求項79】
微小藻類が珪藻源である、請求項78に記載の医薬組成物。
【請求項80】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項78に記載の医薬組成物。
【請求項81】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項78に記載の医薬組成物。
【請求項82】
微小藻類に由来しないポリ-β-1→4-N-アセチルグルコサミン医薬担体をさらに含む、請求項78に記載の医薬組成物。
【請求項83】
タンパク質を含まず、他の有機汚染物質を実質的に含まず、無機汚染物質を実質的に含まない、β-1→4配置で共有結合された約4,000〜約150,000のN-アセチルグルコサミン単糖を含んでなり、約800,000ダルトン〜約3,000万ダルトンの分子量を有する微小藻類ポリ-β-1→4-N-アセチルグルコサミンの製造方法であって、
(a) 細胞体およびポリ-β-1→4-N-アセチルグルコサミン繊維を含む微小藻類を、ポリ-β-1→4-N-アセチルグルコサミン繊維を合成することができる生物的作用物質で、ポリ-β-1→4-N-アセチルグルコサミン繊維を細胞体から遊離させるのに十分な時間処理し、
(b) 細胞体からポリ-β-1→4-N-アセチルグルコサミン繊維を分離し、そして
(c) 分離したポリ-β-1→4-N-アセチルグルコサミン繊維から全てのタンパク質、実質的に全ての他の有機汚染物質、および実質的に全ての無機汚染物質を取り除き、ポリ-β-1→4-N-アセチルグルコサミン種を単離する、
ことを含んでなる方法。
【請求項84】
単離されたポリ-β-1→4-N-アセチルグルコサミン(p-GlcNAc)種が約4,000〜約15,000のN-アセチルグルコサミン単糖を含んでなり、約800,000ダルトン〜約300万ダルトンの分子量を有する、請求項83に記載の方法。
【請求項85】
生物的作用物質がポリオキシン-Dである、請求項83に記載の方法。
【請求項86】
微小藻類が珪藻源である、請求項83に記載の方法。
【請求項87】
珪藻がタラシオシラ(Thallasiosira)属のものである、請求項86に記載の方法。
【請求項88】
タラシオシラ属の珪藻がタラシオシラ・フルビアチリス(Thallasiosira fluviatilis)またはタラシオシラ・ワイスフロギ(Thallasiosira weissflogii)である、請求項87に記載の方法。
【図1】

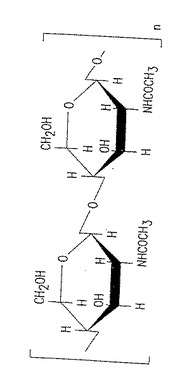
【図2】
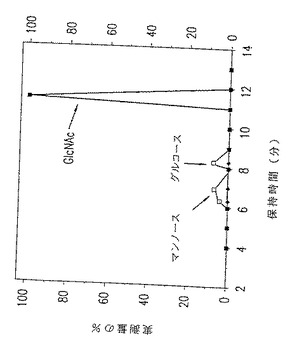
【図3A】
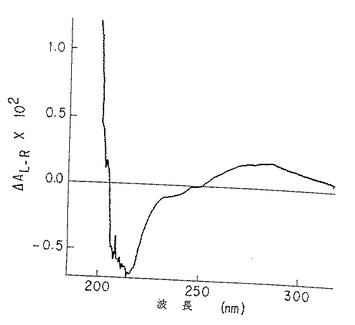
【図3B】
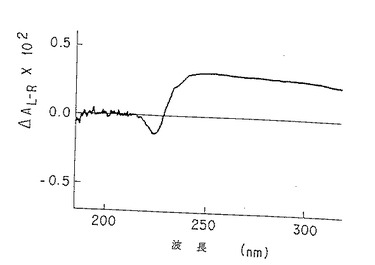
【図4A】
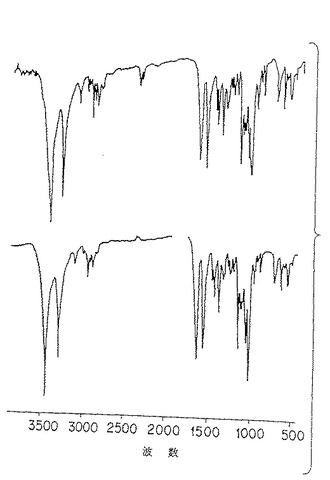
【図4B】
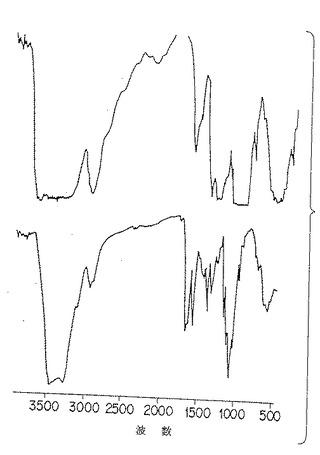
【図4C】
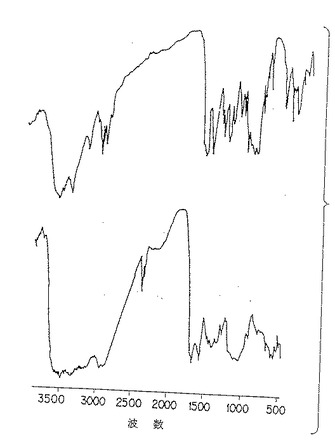
【図4D】
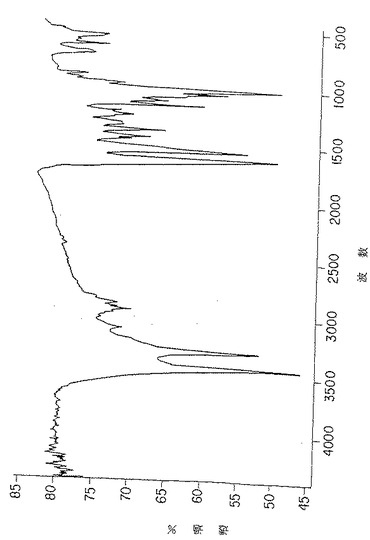
【図4E】
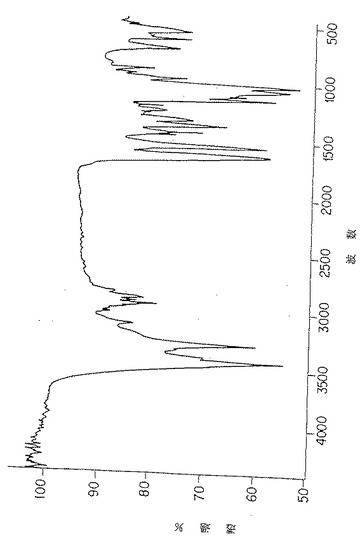
【図5A】
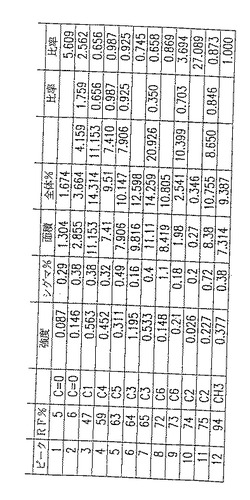
【図5B】
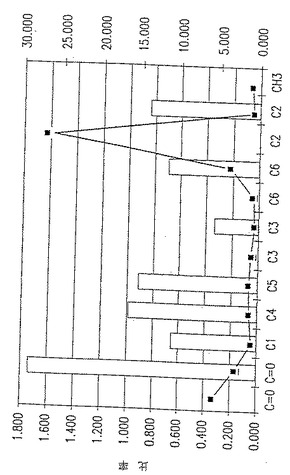
【図6A】

【図6B】

【図7A】

【図7B】

【図8A】

【図8B】

【図9A】

【図9B】

【図9C】

【図9D】

【図9E】

【図10A】

【図10B】

【図11A】

【図11B】

【図12A】

【図12B】

【図13】
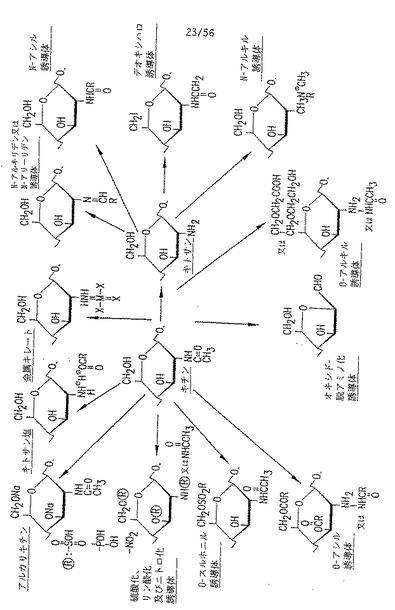
【図14】
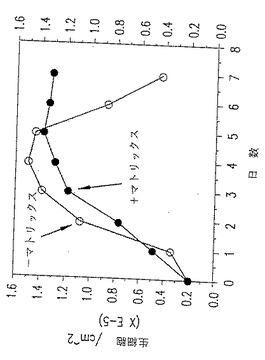
【図15A】
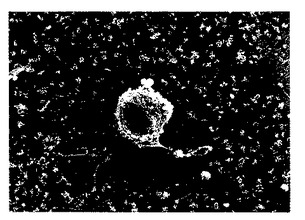
【図15B】

【図16A】

【図16B】

【図16C】
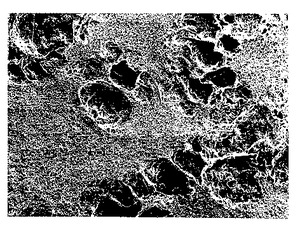
【図16D】

【図16E】

【図17A】
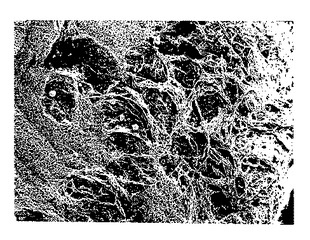
【図17B】

【図17C】

【図17D】
【図18】
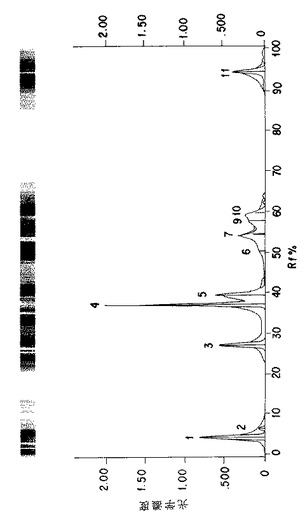
【図19】
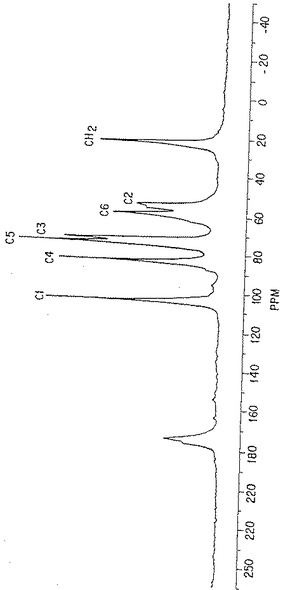
【図20】
【図21A】

【図21B】

【図21C】

【図21D】

【図21E】

【図21F】
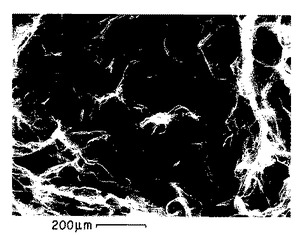
【図21G】
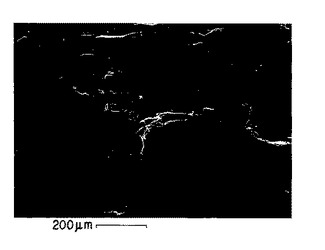
【図22A】

【図22B】

【図22C】
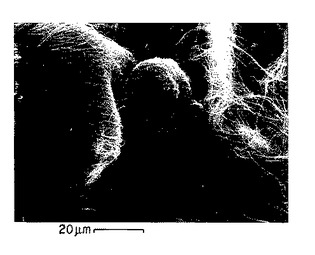
【図22D】

【図22E】

【図22F】

【図22G】

【図23】
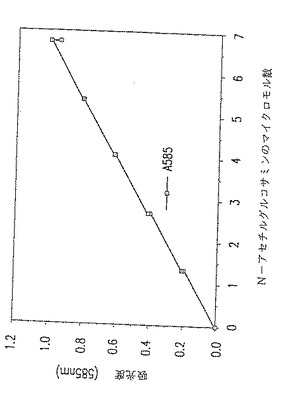
【図24】
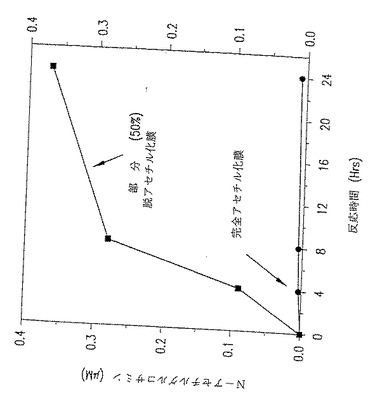
【図25】
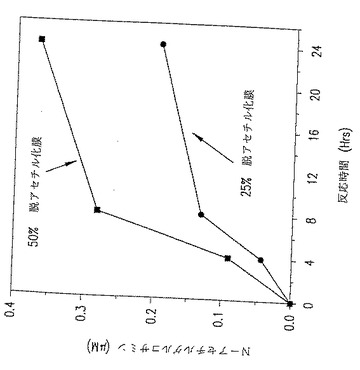
【図26A】
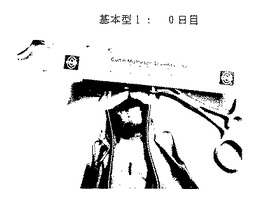
【図26B】
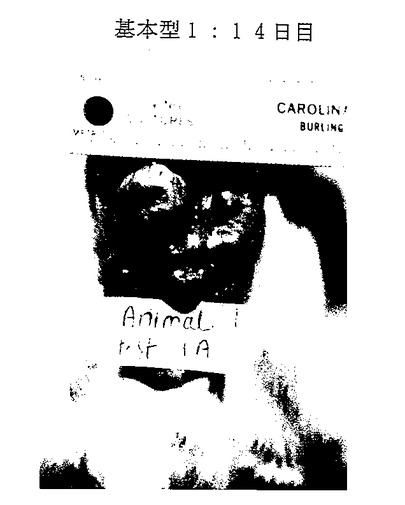
【図26C】

【図26D】
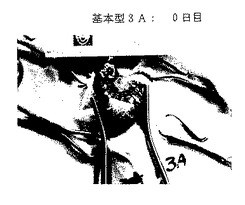
【図26E】
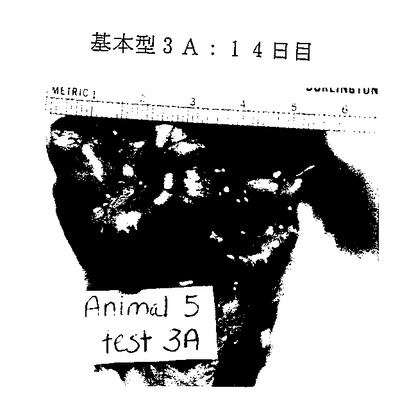
【図27】
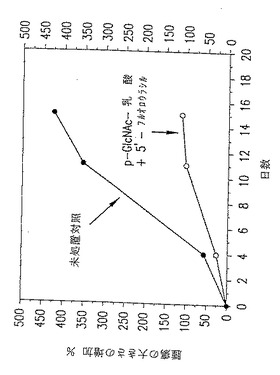
【図28】
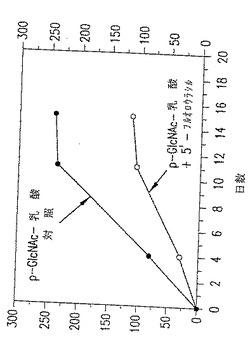
【図29】
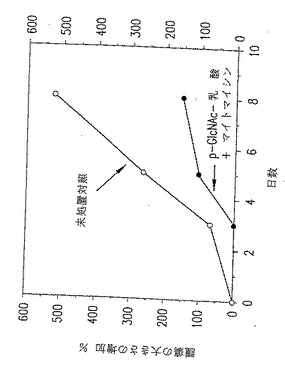
【図30】
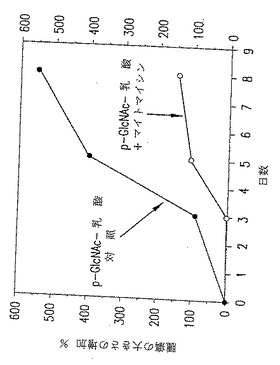
【図31】
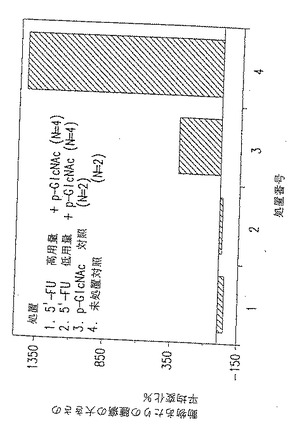
【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図5A】
【図5B】
【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図9A】
【図9B】
【図9C】
【図9D】
【図9E】
【図10A】
【図10B】
【図11A】
【図11B】
【図12A】
【図12B】
【図13】
【図14】
【図15A】
【図15B】
【図16A】
【図16B】
【図16C】
【図16D】
【図16E】
【図17A】
【図17B】
【図17C】
【図17D】
【図18】
【図19】
【図20】
【図21A】
【図21B】
【図21C】
【図21D】
【図21E】
【図21F】
【図21G】
【図22A】
【図22B】
【図22C】
【図22D】
【図22E】
【図22F】
【図22G】
【図23】
【図24】
【図25】
【図26A】
【図26B】
【図26C】
【図26D】
【図26E】
【図27】
【図28】
【図29】
【図30】
【図31】
【公開番号】特開2009−79223(P2009−79223A)
【公開日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願番号】特願2008−267438(P2008−267438)
【出願日】平成20年10月16日(2008.10.16)
【分割の表示】特願2004−59902(P2004−59902)の分割
【原出願日】平成6年12月1日(1994.12.1)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.ポラロイド
2.テフロン
【出願人】(501250337)マリン ポリマー テクノロジーズ,インコーポレーテッド (8)
【Fターム(参考)】
【公開日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願日】平成20年10月16日(2008.10.16)
【分割の表示】特願2004−59902(P2004−59902)の分割
【原出願日】平成6年12月1日(1994.12.1)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.ポラロイド
2.テフロン
【出願人】(501250337)マリン ポリマー テクノロジーズ,インコーポレーテッド (8)
【Fターム(参考)】
[ Back to top ]