
ポリアミド樹脂組成物及びそれを成形して得た成形体
【課題】従来の脂肪族ポリアミド樹脂に比較して、十分な相対粘度ηrが達成され、成形可能温度幅が広く、耐熱性、溶融成形性、及び成形サイクルが低減でき、低吸水性を損なうことなく、耐薬品性、耐加水分解性、燃料バリア性に優れた成形体を成形できるポリアミド樹脂と離型剤とを含む、成形時における型と成形物との良好なすべり性及び/又は短い成形時間も達成できるポリアミド樹脂組成物と、それを成形して得られる成形体を提供する。
【解決手段】ポリアミド樹脂(成分A)及び特定の離型剤(成分B)を含み、成分Aが、ジカルボン酸由来の単位とジアミン由来の単位とが結合してなり、ジカルボン酸が蓚酸(化合物a)を含み、ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、化合物bと化合物cのモル比が99:1〜50:50であるポリアミド樹脂組成物。
【解決手段】ポリアミド樹脂(成分A)及び特定の離型剤(成分B)を含み、成分Aが、ジカルボン酸由来の単位とジアミン由来の単位とが結合してなり、ジカルボン酸が蓚酸(化合物a)を含み、ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、化合物bと化合物cのモル比が99:1〜50:50であるポリアミド樹脂組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリアミド樹脂組成物及びそれを成形して得た成形体に関する。
【背景技術】
【0002】
以下、ポリアミド樹脂の呼称は、JIS K 6920−1に基づく場合もある。
ナイロン6(PA6)、ナイロン66(PA66)などに代表される結晶性ポリアミドは、その優れた特性と溶融成形の容易さから、衣料用、産業資材用繊維、あるいは汎用のエンジニアリングプラスチックとして広く用いられているが、一方では吸水による物性変化、酸、高温のアルコール、熱水中での劣化などの問題点も指摘されており、より寸法安定性、耐薬品性に優れたポリアミドへの要求が高まっている。
【0003】
ジカルボン酸成分として蓚酸を用いるポリアミド樹脂はポリオキサミド樹脂と呼ばれ、同じアミノ基濃度の他のポリアミド樹脂と比較して融点が高いこと、吸水率が低いことが知られ(特許文献1)、吸水による物性変化が問題となっていた従来のポリアミドが使用困難な分野での活用が期待される。
【0004】
これまでに、ジアミン成分として種々の脂肪族直鎖ジアミンを用いたポリオキサミド樹脂が提案されている。例えば、
非特許文献1には、ジアミン成分として1,6−ヘキサンジアミンを用いたポリオキサミド樹脂が開示され、
非特許文献2には、ジアミン成分が1,9−ノナンジアミンであるポリオキサミド樹脂(以下、PA92ともいう)が開示され、
特許文献2には、種々ジアミン成分と、ジカルボン酸エステルとして蓚酸ジブチルを用いたポリオキサミド樹脂が開示され、
特許文献3には、ジアミン成分として1,9−ノナンジアミン及び2−メチル−1,8−オクタンジアミンの2種のジアミンを特定の比率で用いたポリオキサミド樹脂が開示されている。
【0005】
一方、ポリアミド樹脂の成形性を向上させるために、ポリアミド樹脂と離型剤とを含む樹脂組成物を用いることが従来公知である。
例えば特許文献4には、ポリアミド樹脂又はこれを含む樹脂組成物と、ポリアミド樹脂中に均一に分散された層状珪酸塩及び成形性改良剤とからなるポリアミド樹脂組成物を用いることによって、成形時間が短く、かつ優れた機械的性質を有する成形体を与えるポリアミド組成物を提供することが開示されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2006−57033号公報
【特許文献2】特表平5−506466号公報
【特許文献3】WO2008/072754号公報
【特許文献4】特開平1−301750号公報
【非特許文献】
【0007】
【非特許文献1】S.W.Shalaby,J.Polym.Sci.,11,1(1973)
【非特許文献2】L.Franco,et al.,Macromolecules,31,3912(1998)
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、
非特許文献1に開示されるポリオキサミド樹脂については、融点(約320℃)が熱分解温度(窒素中の1%重量減少温度;約310℃)と近いため、溶融重合、溶融成形が困難であり実用に耐えうるものではなく、
非特許文献2に開示されるポリオキサミド樹脂については、蓚酸源として蓚酸ジエチルを用いた場合の製造法とその結晶構造を開示しているが、ここで得られるPA92は固有粘度が0.97dL/g、融点が246℃のポリマーであり、強靭な成形体が成形出来ない程度の低分子量体しか得られておらず、
特許文献2に開示されるポリオキサミド樹脂については、固有粘度が0.99dL/g、融点が248℃のPA92を製造したことが示されているが、強靭な成形体が成形出来ない程度の低分子量体しか得られていないという問題点があり、
特許文献3に開示されるポリオキサミド樹脂については、ジアミン成分として1,9−ノナンジアミン及び2−メチル−1,8−オクタンジアミンの2種のジアミンを特定の比率で用いたポリオキサミド樹脂が示されているが、これらポリオキサミド樹脂は成形可能温度幅が広く、成形加工性に優れ、かつ低吸水性、耐薬品性、耐加水分解性、燃料バリア性などにも優れるが、融点が240℃前後であり、成形サイクル性と高融点であることが要求される電気・電子機器用途に対しては、耐熱性がやや劣る。
さらに、特許文献4に開示されている技術では、成形時間の短縮及び優れた機械的性質の付与と同時に低吸水性、耐薬品性及び耐加水分解性を満足させるものではない。
【0009】
本発明が解決しようとする課題は、
従来のポリオキサミド樹脂と比較して、
窒素雰囲気下、10℃/分の昇温速度で測定した示差走査熱量法により測定した融点Tm(℃)(以下、融点Tmともいう)と
窒素雰囲気下、10℃/分の昇温速度で測定した熱重量分析における1%重量減少温度Td(℃)(熱分解温度)との温度差(Td−Tm)(℃)(以下、温度差(Td−Tm)ともいう)から見積もられる成形可能温度幅が広く、
融点Tmから見積もられる耐熱性が優れ、
適度な溶融粘度を有し溶融成形性に優れ、及び成形サイクルが低減でき、
脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来の脂肪族ポリアミド樹脂に比較して、耐薬品性、耐加水分解性、燃料バリア性に優れた成形体を成形できるポリアミド樹脂と離型剤とを含む、
成形時における型と成形物との良好なすべり性及び/又は短い成形時間も達成できるポリアミド樹脂組成物とそれを成形して得られる成形体を提供することである。
【課題を解決するための手段】
【0010】
本発明は、
(1)ポリアミド樹脂(成分A)及び離型剤(成分B)を含むポリアミド樹脂組成物であって、
前記成分Aが、ジカルボン酸由来の単位とジアミン由来の単位とが結合してなり、
前記ジカルボン酸が蓚酸(化合物a)を含み、
前記ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、
前記化合物bと前記化合物cのモル比が99:1〜50:50であり、
前記成分Bが、ポリアルキレングリコールの末端変性物、リン酸エステル類、亜リン酸エステル類、高級脂肪酸モノエステル類、高級脂肪酸、高級脂肪酸金属塩、エチレンビスアミド化合物、低分子量ポリエチレン、珪酸マグネシウム及び置換ベンジリデンソルビトール類からなる群から選ばれる少なくとも1以上の化合物であるポリアミド樹脂組成物、及び
(2)上記(1)記載のポリアミド樹脂組成物を成形して得られる成形体。
【発明の効果】
【0011】
本発明によれば、
従来のポリオキサミド樹脂と比較して、
温度差(Td−Tm)から見積もられる成形可能温度幅が広く、
融点Tmから見積もられる耐熱性が優れ、
適度な溶融粘度を有し溶融成形性に優れ、及び成形サイクルが低減でき、
脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来の脂肪族ポリアミド樹脂に比較して、耐薬品性、耐加水分解性、燃料バリア性に優れた成形体を成形できるポリアミド樹脂と離型剤とを含む、
成形時における型と成形物との良好なすべり性及び/又は短い成形時間も達成できるポリアミド樹脂組成物とそれを成形して得られる成形体を提供することができる。
【図面の簡単な説明】
【0012】
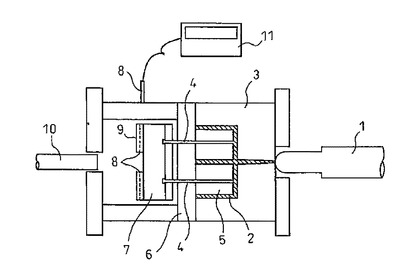
【図1】離型力を測定するために、実施例1〜12及び比較例2〜3で使用した射出成形機の概略を示す断面図である。
【発明を実施するための形態】
【0013】
〔成分A〕
(1)成分Aの構成
本発明におけるポリアミド樹脂である成分Aは、
ジカルボン酸成分が蓚酸であり、ジアミン成分が1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミンからなる、即ち、
ジカルボン酸由来の単位とジアミン由来の単位とが結合してなるポリアミド樹脂であって、
前記ジカルボン酸が蓚酸(化合物a)を含み、
前記ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、
前記化合物bと前記化合物cのモル比が99:1〜50:50であり、
好ましくは、96%硫酸を溶媒とし、濃度が1.0g/dlの前記成分Aの溶液を用いて25℃で測定した相対粘度ηrが1.8〜6.0である。
【0014】
成分Aは、化合物a、b及びcを用いて、好ましくはこれらの混合物を用いて重縮合することで、高分子量で、高融点で、融点と熱分解温度の差が大きく溶融成形性に優れ、さらに直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来のポリアミドに比較して耐薬品性、耐加水分解性ならびに燃料バリア性に優れる。
【0015】
成分Aは、耐薬品性、耐加水分解性及び燃料バリア性を確保する観点から、化合物bと化合物cのモル比は、
好ましくは、99:1〜55:45モル%であり、
より好ましくは、99:1〜60:40モル%である。
なお、以下、化合物b及び化合物cのモル比は、成分A中の化合物b由来の単位と化合物c由来の単位のモル比も意味する。
【0016】
成分Aの製造で、化合物a(蓚酸)を直接原料として使用すると、化合物a(蓚酸)そのものは熱分解してしまい、成分Aの融点がその熱分解温度を超えることから、製造時には蓚酸源化合物(以下、蓚酸源ともいう)を用い、蓚酸源由来の蓚酸とジアミンとを重縮合して得る。この蓚酸は、蓚酸ジエステル等の蓚酸源由来のものであり、これらはアミノ基との反応性を有するものであればよい。
蓚酸源として、重縮合反応における副反応を抑制する観点から蓚酸ジエステルが好ましく、蓚酸ジメチル、蓚酸ジエチル、蓚酸ジn−(またはi−)プロピル、蓚酸ジn−(またはi−、またはt−)ブチル等の脂肪族1価アルコールの蓚酸ジエステル、蓚酸ジシクロヘキシル等の脂環式アルコールの蓚酸ジエステル、蓚酸ジフェニル等の芳香族アルコールの蓚酸ジエステル等が挙げられる。
蓚酸ジエステルの中でも炭素原子数が3を超える脂肪族1価アルコールの蓚酸ジエステル、脂環式アルコールの蓚酸ジエステル、芳香族アルコールの蓚酸ジエステルがさらに好ましく、
その中でも蓚酸ジブチル及び蓚酸ジフェニルがさらに好ましく、
蓚酸ジブチルがさらに好ましい。
【0017】
(2)成分Aの相対粘度
成分Aは、
カルボン酸成分として化合物aである蓚酸を用い、
ジアミン成分として、化合物bである1,6−ヘキサンジアミンと、化合物cである2−メチル−1,5−ペンタンジアミンを重縮合することで、融点が好ましくは200〜330℃の範囲にすることができ、融点が330℃を超える化合物aと化合物bとを重縮合して得られるポリアミド樹脂(以下、比較成分A2ともいう)に比べ、後述する成分Aの後重合工程での溶融重合において、副反応が起こり高分子量化を阻害するような過度の高温条件にする必要がないため、高分子量化(相対粘度を増加させること)が可能である。
従って、成分Aは、従来のポリアミド樹脂に比べて相対粘度を増大できるので、優れた溶融成形性を有する。
溶融成形後の成形物が脆くなり物性が低下する傾向を避けることと、溶融成形時の溶融粘度が高くなり成形加工性が悪くなる傾向を避ける観点と、相対粘度ηrと溶融粘度が一定以上に高く、過度に高くないことが好ましいとされる自動車部材や電気・電子部品のような用途に好適であるという観点から、成分Aの濃度が1.0g/dlの96%濃硫酸溶液を用い、25℃で測定した相対粘度ηrは、
好ましくは1.8〜6.0であり、より好ましくは1.8〜4.5であり、
より好ましくは1.8〜3.0であり、更に好ましくは1.85〜2.5、
更に好ましくは1.85〜2.2であるようにすることができる。
なお、後述する成分Aの後重合工程での溶融重合において、減圧度を上げることで、相対粘度ηrを増大することができる。
【0018】
また、同様の観点から、成分Aの溶融粘度は、好ましくは100〜700Pa・s、より好ましくは110〜600Pa・s、更に好ましくは120〜500Pa・s、更に好ましくは130〜400Pa・s、更に好ましくは150〜300Pa・s、更に好ましくは160〜220Pa・s、更に好ましくは170〜200Pa・sである。
【0019】
さらに、同様の観点から、成分Aの数平均分子量(Mn)は、好ましくは10000〜50000であり、より好ましくは11000〜40000であり、更に好ましくは11000〜35000である。
【0020】
数平均分子量(Mn)は、1H−NMRスペクトルから求めたシグナル強度をもとに、例えば、蓚酸源として蓚酸ジブチル、ジアミン成分として1,6−ヘキサンジアミン(化合物b)と2−メチル−1,5−ペンタンジアミン(化合物c)を90:10のモル%比で用いて製造したポリアミド〔以下、PX6−2(化合物b/化合物c=90/10)と略称する〕の場合は下式により算出した。
Mn=np×170.21+n(NH2)×115.20+n(OBu)×129.13+n(NHCHO)×29.14
なお、1H−NMRの測定条件は以下の通りである。
・使用機種:ブルカー・バイオスピン社製 AVANCE500
・溶媒:重硫酸
・積算回数:1024回
また、前記式中の各項は以下のように規定される。
・np=Np/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・n(NH2)
=N(NH2)/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・n(NHCHO)
=N(NHCHO)/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・n(OBu)
=N(OBu)/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・Np=Sp/sp−N(NHCHO)
・N(NH2)=S(NH2)/s(NH2)
・N(NHCHO)=S(NHCHO)/s(NHCHO)
・N(OBu)=S(OBu)/s(OBu)
但し、各項は以下の意味を有する。
・Np:PA62(化合物B/化合物C=90/10)の末端ユニットを除いた、分子鎖中の繰り返しユニット総数。
・np:分子1本当たりの分子鎖中の繰り返しユニット数。
・Sp:PX6−2(化合物b/化合物c=90/10)の末端を除いた、分子鎖中の繰り返しユニット中のオキサミド基に隣接するメチレン基のプロトンに基づくシグナル(3.1ppm付近)の積分値。
・sp:積分値Spにカウントされる水素数(4個)。
・N(NH2):PX6−2(化合物b/化合物c=90/10)の末端アミノ基の総数。
・n(NH2):分子1本当たりの末端アミノ基の数。
・S(NH2):PX6−2(化合物b/化合物c=90/10)の末端アミノ基に隣接するメチレン基のプロトンに基づくシグナル(2.6ppm付近)の積分値。
・s(NH2):積分値S(NH2)にカウントされる水素数(2個)。
・N(NHCHO):PX6−2(化合物b/化合物c=90/10)の末端ホルムアミド基の総数。
・n(NHCHO):分子1本当たりの末端ホルムアミド基の数。
・S(NHCHO):PX6−2(化合物b/化合物c=90/10)のホルムアミド基のプロトンに基づくシグナル(7.8ppm)の積分値。
・s(NHCHO):積分値S(NHCHO)にカウントされる水素数(1個)。
・N(OBu):PX6−2(化合物b/化合物c=90/10)の末端ブトキシ基の総数。
・n(OBu):分子1本当たりの末端ブトキシ基の数。
・S(OBu):PX6−2(化合物b/化合物c=90/10)の末端ブトキシ基の酸素原子に隣接するメチレン基のプロトンに基づくシグナル(4.1ppm付近)の積分値。
・s(OBu):積分値S(OBu)にカウントされる水素数(2個)。
【0021】
(3)成分Aの物性
成分Aは、さらに、化合物b及びcの重縮合比率を変更することで、
温度差(Td−Tm)を、比較成分A2に比べて大きく、化合物aと化合物cとを重縮合して得られるポリアミド樹脂(以下、比較成分A1ともいう)に比べて小さく、
融点Tmを、比較成分A2に比べて低く、比較成分A1に比べて高く、
1%重量減少温度Tdを、比較成分A1に比べて高く、
飽和吸水率を、比較成分A2に比べて小さく、比較成分A1に比べて大きくすることができる。
【0022】
即ち、成分Aは、従来のポリオキサミド樹脂と比較して、
相対粘度ηr(高分子量化)、
温度差(Td−Tm)から見積もられる成形可能温度幅、
融点Tmから見積もられる耐熱性、
溶融粘度から見積もられる溶融成形性、及び
低吸水性のいずれをも十分に確保することができる。
【0023】
成分Aは、成形可能温度幅、耐熱性、溶融成形性及び低吸水性のいずれをも十分に確保した上で、化合物bの重合比率(モル比)の高さに由来する耐薬品性、耐加水分解性及び燃料バリア性に特に寄与する。
【0024】
成分Aは、成形可能温度幅、耐熱性、溶融成形性及び低吸水性のいずれをも十分に確保する観点から、
Tmは、好ましくは260〜330℃であり、より好ましくは265〜330℃であり、
Tdは、好ましくは341〜370℃、より好ましくは345〜370℃、更に好ましくは350〜365℃であり、
温度差(Td−Tm)は、好ましくは10〜95℃、より好ましくは20〜95℃、更に好ましくは25〜95℃であり、
飽和吸水率は、好ましくは0〜2.4、より好ましくは1〜2.4、更に好ましくは2〜2.4、更に好ましくは2.3〜2.4である。
【0025】
(4)成分Aの製造
成分Aは、ポリアミドを製造する方法として知られている任意の方法を用いて製造することができるが、高分子量化および生産性の観点から、
好ましくは、ジアミン及び蓚酸ジエステルをバッチ式又は連続式で重縮合反応させることにより得ることであり、
より好ましくは、ジアミン及び蓚酸ジエステルを前重縮合工程と後重縮合工程からなる二段重合法もしくは、WO2008−072754公報記載の加圧重合法によって得ることである。
更に好ましい二段重合法及び加圧重合法としては、具体的には、以下の操作で示される。
【0026】
(4−1)二段重合法
(i)前重縮合工程:まず反応器内を窒素置換した後、ジアミン(化合物b及びc)及び化合物aの蓚酸源である蓚酸ジエステルを混合する。混合する場合にジアミン及び蓚酸ジエステルが共に可溶な溶媒を用いても良い。ジアミン成分及び蓚酸源成分が共に可溶な溶媒としては、トルエン、キシレン、トリクロロベンゼン、フェノール、トリフルオロエタノールなどを用いることができ、特にトルエンを好ましく用いることができる。例えば、ジアミンを溶解したトルエン溶液を50℃に加熱した後、これに対して蓚酸ジエステルを加える。
このとき、蓚酸ジエステルと上記ジアミンの仕込み比は、高分子量化の観点から、蓚酸ジエステル/上記ジアミンで、0.8〜1.5(モル比)、好ましくは0.91〜1.1(モル比)、更に好ましくは0.99〜1.01(モル比)である。
【0027】
このように仕込んだ反応器内を攪拌及び/又は窒素バブリングしながら、常圧下で昇温する。反応温度は、最終到達温度が80〜150℃、好ましくは100〜140℃の範囲になるように制御するのが好ましい。最終到達温度での反応時間は3時間〜6時間である。
【0028】
(ii)後重縮合工程:更に高分子量化を図るために、前重縮合工程で生成した重合物を常圧下において反応器内で徐々に昇温する。昇温過程において前重縮合工程の最終到達温度、すなわち好ましくは80〜150℃から、最終的に、
好ましくは295℃以上350℃以下、より好ましくは298℃以上345℃以下、更に好ましくは298℃以上340℃以下の温度範囲にまで到達させる。
昇温時間を含めて好ましくは1〜8時間、より好ましくは2〜6時間保持して反応を行うことが好ましい。さらに後重合工程において、必要に応じて減圧下での重合を行うこともできる。減圧重合を行う場合の好ましい最終到達圧力は13.3Pa〜0.1MPaである。
【0029】
(4−2)加圧重合法
まずジアミンを耐圧容器内に入れ窒素置換した後、封圧下において反応温度まで昇温する。その後、反応温度において封圧状態を保ったまま蓚酸化合物を耐圧容器内に注入し、重縮合反応を開始させる。反応温度は、ジアミンと蓚酸化合物の反応によって生じるポリアミドが、スラリー状、もしくは溶液状態を維持でき、かつ熱分解しない温度であれば特に制限されない。例えば、成分aの場合、上記反応温度は、150℃から250℃が好ましい。ここで、蓚酸ジブチルとジアミンの仕込み比は、蓚酸ジブチルのモル量/ジアミンの総モル量で、0.8〜1.5(モル比)、好ましくは0.91〜1.1(モル比)、更に好ましくは0.99〜1.01(モル比)である。
次に耐圧容器内を封圧状態に保ちながらポリアミド樹脂の融点以上かつ熱分解しない温度以下に昇温する。例えば、成分aの場合、融点は245〜300℃であることから、250℃以上350℃以下、好ましくは255℃以上340℃以下、更に好ましくは260℃以上335℃以下に昇温する。所定温度に到達するまでの耐圧容器内の圧力は、およそ生成するアルコールの飽和蒸気圧から0.1MPa、好ましくは1MPaから0.2MPaに調整する。所定温度に到達後は、生成したアルコールを留去しながら放圧し、必要に応じて常圧窒素気流下もしくは減圧下において継続して重縮合反応を行う。減圧重合を行う場合の好ましい最終到達圧力は13.3Pa〜0.1MPaである。
【0030】
(5)成分Aにおけるジカルボン酸として使用できる成分
成分Aにおいて、本発明の効果を損なわない範囲で化合物a以外の他のジカルボン酸成分を使用する事が出来る。
化合物a(蓚酸)以外の他のジカルボン酸成分としては、
マロン酸、ジメチルマロン酸、コハク酸、グルタル酸、アジピン酸、2−メチルアジピン酸、トリメチルアジピン酸、ピメリン酸、2,2−ジメチルグルタル酸、3,3−ジエチルコハク酸、アゼライン酸、セバシン酸、スベリン酸などの脂肪族ジカルボン酸、
また、1,3−シクロペンタンジカルボン酸、1,4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、
さらに、テレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、2,7−ナフタレンジカルボン酸、1,4−ナフタレンジカルボン酸、1,4−フェニレンジオキシジ酢酸、1,3−フェニレンジオキシジ酢酸、ジ安息香酸、4,4’−オキシジ安息香酸、ジフェニルメタン−4,4’−ジカルボン酸、ジフェニルスルホン−4,4’−ジカルボン酸、4,4’−ビフェニルジカルボン酸などの芳香族ジカルボン酸
などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。
さらに、トリメリット酸、トリメシン酸、ピロメリット酸などの多価カルボン酸を溶融成形が可能な範囲内で用いることもできる。
他のジカルボン酸成分を使用する場合、その割合は、化合物a(蓚酸)に対して、25モル%以下であり、15モル%以下が好ましく、10モル%以下がより好ましく、5モル%以下がさらに好ましく、0モル%(即ち、ジカルボン酸成分が化合物aだけからなること)がさらに好ましい。なお、化合物a(蓚酸)に対する他のジカルボン酸成分のモル比は、成分A中の、化合物a由来の単位と他のジカルボン酸成分由来の単位のモル比も意味する。
【0031】
成分Aにおいて、本発明の効果を損なわない範囲で、化合物b及びc以外の他のジアミン成分を使用する事が出来る。
1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミン以外の他のジアミン成分としては、エチレンジアミン、プロピレンジアミン、1,4−ブタンジアミン、1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン、1,8−オクタンジアミン、1,10−デカンジアミン、1,11−ウンデカンジアミン、
1,12−ドデカンジアミン、3−メチル−1,5−ペンタンジアミン、2,2,4−トリメチル−1,6−ヘキサンジアミン、2,4,4−トリメチル−1,6−ヘキサンジアミン、5−メチル−1,9−ノナンジアミンなどの脂肪族ジアミン、
さらに、シクロヘキサンジアミン、メチルシクロヘキサンジアミン、イソホロンジアミンなどの脂環式ジアミン、
さらにp−フェニレンジアミン、m−フェニレンジアミン、p−キシレンジアミン、m−キシレンジアミン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルエーテルなどの芳香族ジアミン、
などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。
他のジアミン成分を使用する場合、その割合は、化合物b及びcに対して25モル%以下であり、15モル%以下が好ましく、10モル%以下がより好ましく、5モル%以下がさらに好ましく、0モル%(即ち、ジアミン成分が化合物b及びcだけからなること)がさらに好ましい。なお、化合物b及びcに対する他のジアミン成分のモル比は、成分A中の、化合物b及びc由来の単位と他のジアミン成分由来の単位のモル比も意味する。
【0032】
(6)成分Aの成形加工
成分Aの成形方法としては、射出、押出、中空、プレス、ロール、発泡、真空・圧空、延伸などポリアミドに適用できる公知の成形加工法はすべて可能であるが、成形サイクル性を短縮する観点から、中でも、射出成形による成形加工において好適であり、これらの成形法によってフィルム、シート、成形品、繊維などに加工することができる。
【0033】
〔成分B〕
離型剤である成分Bは、ポリアルキレングリコールの末端変性物、リン酸エステル類、亜リン酸エステル類、高級脂肪酸モノエステル類、高級脂肪酸、高級脂肪酸金属塩、エチレンビスアミド化合物、低分子量ポリエチレン、珪酸マグネシウム及び置換ベンジリデンソルビトール類からなる群から選ばれる少なくとも1以上の化合物であり、
成形時における型と成形物との良好なすべり性及び/又は成形時間の抑制を安定に確保する観点から選択される。
【0034】
好ましいポリアルキレングリコールの末端変性物の例としては、ポリエチレングリコールの末端変性物、ポリプロピレングリコールの末端変性物などが挙げられる。末端変性は、アミノ基、カルボキシル基又はメチル基でなされていることが好ましい。より具体的な例としては、下記式(1):
X−R1−(O−CH2−CH2)n−O−R2−X (1)
(式中、XはNH2、又はCOOH、又はHを表し、R1及びR2は、各々独立に炭素数1〜10の直鎖状又は分岐状のアルキレン基を表し、nは4〜1200の数である)
又は下記式(2):
X−R3−(O−CH2−C(CH3)H)n−O−R4−X (2)
(式中、XはNH2、又はCOOH、又はHを表し、R3及びR4は、各々独立に炭素数1〜10の直鎖状又は分岐状のアルキレン基を表し、nは1〜200の数である)で表されるものが挙げられる。
【0035】
好ましいリン酸エステル類の例としては、下記式:
(R5O)nPO(OH)3−n
(式中、nは1又は2であり、R5は炭素数1〜10のアルキル基である)で表されるものが挙げられる。上記式中、n=2である場合の2個のRO基は同一でも異なっていてもよい。Rとしては、エチル基、ブチル基、オクチル基、エチルヘキシル基などが挙げられる。
【0036】
好ましい亜リン酸エステル類の例としては、下記式:
(R6O)3P
(式中、R6は、水素、又は炭素数10〜25、より好ましくは12〜20のアルキル基、もしくはフェニル基、もしくはそれらの基の一部が炭化水素基で置換されている基を表す)で表されるものが挙げられる。上記式中の3個のRO基は同一でも異なっていてもよい。Rとしては、デシル基、ラウリル基、トリデシル基、ステアリル基、オレイル基などの脂肪族基;フェニル基、ビフェニル基などの芳香族基;エチル基、プロピル基、t−ブチル基、ノニル基などの置換基を有する芳香族基などが挙げられる。
【0037】
上記のリン酸エステル及び亜リン酸エステルのより具体的な例としては、ジ(2−エチルヘキシル)ホスフェート、トリデシルホスファイト、トリス(トリデシル)ホスファイト、トリステアリルホスファイトなどの脂肪族リン酸エステル及び脂肪族亜リン酸エステル、トリフェニルホスファイト、ジフェニルモノデシルホスファイトなどの芳香族亜リン酸エステルなどが挙げられる。
【0038】
好ましい高級脂肪酸モノエステル類としては、下記式:
R7−CO−O−R8
(式中、R7及びR8は、各々独立に炭素数8〜32、好ましくは10〜30のアルキル基を表す)
で表されるもの、すなわち高級脂肪酸と高級脂肪族1価アルコールとのエステル化合物が挙げられる。上記式中のR1及びR2としては、デシル基、ラウリル基、トリデシル基、ステアリル基、オレイル基などの脂肪族基などが挙げられる。
【0039】
また、上記高級脂肪酸としては、ミリスチン酸、パルミチン酸、ベヘニン酸、オレイン酸、アラギジン酸などが挙げられる。また、高級脂肪族アルコールとしては、ミリスチルアルコール、ベヘニルアルコール、オレイルアルコール、ステアリルアルコール、ヘキシルデシルアルコールなどを挙げることができる。
【0040】
高級脂肪酸モノエステル類のより具体的な例としては、高級脂肪酸モノアルキルエステル、例えばミリスチン酸ミリスチル、ステアリン酸ステアリル、ベヘニン酸ベヘニル、オレイン酸オレイル、ミリスチン酸ヘキシルデシルなどが挙げられる。
【0041】
好ましい高級脂肪酸と高級脂肪酸金属塩の例としては、下記式:
CH3−(CH2)n−COOX
(式中、nは9〜25、好ましくは11〜20の数を表し、XはH又は周期律表第I〜III族の金属を表す)で表されるものが挙げられる。
【0042】
高級脂肪酸としては、ステアリン酸、パルミチン酸、オレイン酸、アラギジン酸、ベヘニン酸などが挙げられる。また高級脂肪酸の金属塩としては、ステアリン酸亜鉛、ステアリン酸リチウム、ステアリン酸カルシウム、パルミチン酸アルミニウムなどが挙げられる。
【0043】
好ましいエチレンビスアミド化合物の例としては、下記式:
CH3(CH2)mCONH(CH2)2NHCO(CH2)nCH3
(式中、m及びnは各々独立に9〜25、好ましくは10〜20の数である)で表されるものが挙げられる。
【0044】
エチレンビスアミド化合物のより具体的な例としては、エチレンビスステアリルアミド、エチレンビスパルミチルアミドなどが挙げられる。
【0045】
好ましい低分子量ポリエチレンとしては、粘度平均分子量が500〜5000の範囲内であるものが挙げられ、粘度平均分子量が1000〜3000の範囲のものがより好ましい。
粘度平均分子量の測定は、粘度平均分子量の測定は、ウベローデ型粘度計を用いた溶液粘度測定による。
【0046】
好ましい珪酸マグネシウムの例としては、平均粒径1〜10μmのものが挙げられる。平均粒径が1μm以上である場合、成形物表面における白いむらが発生しにくく、10μm以下である場合、成形物の機械的物性、特に引張り破断点伸び及び衝撃強さが低下しにくい。ポリアミド樹脂との密着性を改良する目的で、珪酸マグネシウムにアミノシランなどによる表面処理を行ってもよい。
平均粒径の測定は、動的散乱法による。
【0047】
好ましい置換ベンジリデンソルビトール類の例としては、ソルビトールと置換ベンズアルデヒドとの酸触媒下での脱水縮合により合成される置換ベンジリデンソルビトールが挙げられる。置換ベンズアルデヒドのソルビトールへの縮合割合は、ソルビトール1モルに対して1モル又は2モルが好ましい。従って、これらの置換ベンジリデンソルビトール類は、下記式:
【0048】
【化1】

(式中、R9は、H又はヒドロキシル基又はハロゲン又は炭素数1〜200のアルキル基を表す)
又は、下記式:
【0049】
【化2】
(式中、R10及びR11は、各々独立に、H又はヒドロキシル基又はハロゲン又は炭素数1〜200のアルキル基を表す)で表される。
【0050】
置換ベンジリデンソルビトール類としては、例えば、1,3−ベンジリデンソルビトール、1,3,2,4−ジベンジリデンソルビトール、1,3−モノ(p−ヒドロキシベンジリデン)ソルビトール、1,3,2,4−ジ(p−ヒドロキシベンジリデン)ソルビトール、1,3−モノ(p−クロロベンジリデン)ソルビトール、1,3,2,4−ジ(p−クロロベンジリデン)ソルビトール、1,3−モノ(m−ニトロベンジリデン)ソルビトール、1,3,2,4−ジ(m−ニトロベンジリデン)ソルビトール、1,3−(p−クロロベンジリデン)2,4−(p−エチルベンジリデン)−d−ソルビトールなどが挙げられる。
【0051】
〔ポリアミド樹脂組成物〕
本発明のポリアミド樹脂組成物は、成形可能温度幅が広く、耐熱性、及び溶融成形性に優れ、脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来の脂肪族ポリアミド樹脂に比較して、耐薬品性、耐加水分解性、燃料バリア性、さらに、成形時における型と成形物との良好なすべり性及び/又は成形時間の抑制を安定に確保する観点から、
成分Bを除くポリアミド樹脂組成物中の成分Aの含有量は、好ましくは50〜100質量%、より好ましくは55〜100質量%、更に好ましくは60〜100質量%、更に好ましくは70〜100質量%、更に好ましくは80〜100質量%、更に好ましくは90〜100質量%、更に好ましくは92〜100質量%、更に好ましくは95〜100質量%であり、
成分Bの含有量は、成分A100質量部に対して、好ましくは0.01〜5質量部、より好ましくは0.05〜3質量部、更に好ましくは0.1〜1.5質量部、更に好ましくは0.1〜1質量部、更に好ましくは0.1〜0.5質量部である。
【0052】
ポリアルキレングリコールの末端変性物の場合、成形時の冷却時間の短縮による成形時間の短縮効果と機械的物性の安定性の観点から、
リン酸エステル及び亜リン酸エステルの場合、成形時における型と成形物とのすべり性と成形サイクル時間の短縮効果、リン酸エステル及び亜リン酸エステルとポリアミド樹脂との相溶性、成形物表面における銀状(シルバーマーク)の発生の抑制及び成形物の機械的物性の安定性の観点から、
高級脂肪酸モノエステル類の場合、成形時における型と成形物とのすべり性、高級脂肪酸モノエステル類とポリアミド樹脂との相溶性、成形物表面における銀状の発生の抑制及び成形物の機械的物性の安定性の観点から、
高級脂肪酸及びその金属塩の場合、成形時における型と成形物とのすべり性、高級脂肪酸モノエステル類とポリアミド樹脂との相溶性、成形物表面における銀状の発生の抑制や成形物の機械的物性、特に引張り破断点伸び、衝撃強さの安定性の観点から、
エチレンビスアミド化合物の場合、成形時における型と成形物とのすべり性、成形時間の短縮効果、成形物の表面外観及び機械的物性の安定性の観点から、
低分子量ポリエチレンの場合、成形時における型と成形物とのすべり性、低分子量ポリエチレンとポリアミド樹脂との相溶性、成形物表面における銀状の発生の抑制及び成形物の機械的物性の安定性の観点から、
珪酸マグネシウムの場合、成形性の向上効果、成形物の機械的物性、特に引張り破断点伸び、衝撃強さの安定性の観点から、
置換ベンジリデンソルビトール類の場合、成形時の冷却時間の短縮による成形時間の短縮効果、成形物表面における銀状の発生の抑制、成形物の機械的物性の安定性の観点から、
成分Bは、上記の好適含有量の範囲であることが好ましい。
【0053】
本発明のポリアミド樹脂組成物では、その他の成分として以下を含むことが好ましい。
(1)成分A以外のポリマー
本発明の樹脂組成物には、必要に応じて、各ポリマーの特性を利用するために、
成分A以外の他のポリアミド類、例えば、ポリオキサミド、芳香族ポリアミド、脂肪族ポリアミド及び脂環式ポリアミドからなる群から選ばれる少なくとも1種のポリアミド、及び/又はポリアミド外のポリマー、例えば、熱可塑性ポリマー、エラストマーを含めることができる。
【0054】
成分A以外のポリマーは、本発明の効果を損なわない範囲であれば特に限定されないが、成分A100質量部に対して、好ましくは0.1〜100質量部、より好ましくは0.1〜50質量部、更に好ましくは0.5〜30質量部である。
【0055】
(2)添加剤
本発明の樹脂組成物は、本発明の効果を損なわない範囲において、添加剤を含むことができる。添加剤として、例えば、顔料、染料、着色剤、酸化防止剤、耐候剤、紫外線吸収剤、光安定化剤、滑剤、結晶核剤、結晶化促進剤、耐熱剤、帯電防止剤、可塑剤、銅化合物等の安定剤、帯電防止剤、難燃剤、ガラス繊維、潤滑剤、フィラー、補強繊維、補強粒子、発泡剤等を挙げることができる。
【0056】
上記成分A以外のポリマー及び/又は添加剤の添加方法は、それぞれを成分Aに分散させることができる方法であれば、特に制限されるものではなく、その効果を損なわない任意の時点において、成分Aに添加することができる。
例えば、成分A以外のポリマー及び/又は添加剤を、
成分Aの重縮合反応時、またはその後に添加することもできる。
【0057】
(3)層状珪酸塩
ポリアミド樹脂に優れた機械的特性及び耐熱性を付与する観点から、本発明のポリアミド樹脂組成物に層状珪酸塩をさらに含ませることができる。
【0058】
上記層状珪酸塩は、一辺の長さが0.002〜1μmで、厚さが6〜20Åである平板状のものであることが好ましい。また、上記層状珪酸塩は、成分Aの中で、各層が約20Å以上の層間距離を保ち、均一に分散されるものであることが好ましい。
【0059】
ここで、「層間距離」とは、平板状をなす層状珪酸塩の各重心の間の距離をいい、「均一に分散する」とは、各層が主にランダムな状態で存在し、層状珪酸塩の50質量%以上、好ましくは70質量%以上が、複層物を形成することなく単層に分散していることをいう。
【0060】
上記層状珪酸塩の量は、当該層状珪酸塩の効果が発揮される量であれば、特に制限されるものではないが、射出成形体の剛性、耐候性及び/又は耐熱性、並びに液体又は蒸気に対するバリア性を向上させる観点と、樹脂組成物の成形加工性と耐衝撃性を確保する観点とから、成分A100質量部に対して、好ましくは0.05〜10質量部、より好ましくは0.05〜8質量部、更に好ましくは0.05〜5質量部である。
【0061】
上記層状珪酸塩の原料としては、珪酸マグネシウム又は珪酸アルミニウムの層から構成される層状フィロ珪酸鉱物、すなわち、珪酸アルミニウム質フィロ珪酸塩又は珪酸マグネシウム質フィロ珪酸塩を例示することができる。具体的には、モンモリロナイト、サポナイト、バイデライト、ノントロナイト、ヘクトライト、スティブンサイト等のスメクタイト系粘土鉱物やバーミキュライト、ハロイサイト等を例示することができ、これらは天然のものであっても、合成されたものであってもよい。
【0062】
また、上記層状珪酸塩を成分Aに分散させるために、通常、膨潤化剤が用いられる。当該膨潤化剤は、粘土鉱物の層間を拡げる役割と、粘土鉱物に層間ポリマーを取り込む力を与える役割とを有するものである。上記膨潤化剤としては、本発明の場合には、1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミンを用いることが好ましい。
【0063】
なお、上記層状珪酸塩は、ミキサー、ボールミル、振動ミル、ピンミル、ジェットミル、叩解機等を用いて粉砕し、予め所望の形状及びサイズのものとしておくことが好ましい。
【0064】
上記層状珪酸塩を添加する方法は、上記層状珪酸塩が成分Aに均一に分散し得る方法である限り、特に制限はない。例えば、層状珪酸塩の原料が多層状粘土鉱物である場合には、特開昭62−74957号に開示されるように、層状珪酸塩を塩酸等によりイオン化し、ここに膨潤化剤、例えば、1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミンを添加して、あらかじめ層状珪酸塩の各層の間隔を広げる。次いで、当該層の間に成分Aの原料を導入し、さらに当該層の間で上記原料を重合させることができる。
【0065】
また、膨潤化剤として有機化合物を用いて層間を約100Å以上に予め広げ、これを成分Aと溶融混合して、各層をポリアミド樹脂に分散させてもよい。
【0066】
〔本発明の成形体〕
(1)ポリアミド樹脂組成物から成形体への成形加工
本発明は、上述した本発明のポリアミド樹脂組成物から成形された成形体も提供する。
本発明のポリアミド樹脂組成物から成形体への成形方法としては、射出、押出、中空、プレス、ロール、発泡、真空・圧空、延伸などポリアミドに適用できる公知の成形加工法は全て使用可能であり、これらの成形法によってフィルム、シート、成形品、繊維などの成形物に加工することができる。
【0067】
具体的には、例えば、ポリアミド樹脂、離型剤及び必要に応じて用いる各種添加剤の所定量を、V型ブレンダー、タンブラーなどの低速回転混合機やヘンシェルミキサーなどの高速回転混合機を用いてあらかじめ混合した後、射出成形機や押出成形機を用いて、成形物を直接成形する方法を適用できる。
【0068】
(2)成形物の用途
本発明によって得られる成形体は、従来ポリアミド成形体が用いられてきた各種押出成形品、各種射出成形品、シート、フィルム、パイプ、チューブ、モノフィラメント、繊維、容器などの成形物として、自動車部材、コンピューター及び関連機器、光学機器部材、電気・電子機器、情報・通信機器、精密機器、土木・建築用品、医療用品、家庭用品など広範な用途に好適に使用できる。
【実施例】
【0069】
〔実施例及び比較例〕
製造例1〜5において、本発明のポリアミド樹脂組成物に使用する成分A(PX6−1〜5)を製造した。
製造例1〜5において製造した成分Aと成分B(化合物p〜w)を使用して実施例1〜12の本発明のポリアミド樹脂組成物を製造した。
【0070】
(1)製造例1(成分A:PX6−1)
攪拌機、温度計、トルクメーター、圧力計、ダイアフラムポンプを直結した原料投入口、窒素ガス導入口、放圧口、圧力調節装置及びポリマー抜出し口を備えた内容積が約150リットルの圧力容器に、
化合物a(1,6−ヘキサンジアミン)15.407kg(132.58モル)と
化合物b(2−メチル−1,5−ペンタンジアミン)811.1g(6.98モル)の混合物(化合物aと化合物bのモル比が95:5)を仕込み、
圧力容器の内部を純度が99.9999%の窒素ガスで0.5MPaに加圧した後、
次に常圧まで窒素ガスを放出する操作を5回繰り返し、窒素置換を行った後、
封圧下、攪拌しながら系内を昇温した。
約30分間かけてシュウ酸ジブチルの温度を80℃にした後、
シュウ酸ジブチル28.230kg(139.56モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、2時間かけて温度を330℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が330℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、4.5時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=1.95であった。
【0071】
(2)製造例2(成分A:PX6−2)
化合物a14.717kg(126.64モル)と化合物b1.635kg(14.07モル)の混合物(化合物aと化合物bのモル比が90:10)を仕込み、
シュウ酸ジブチル28.462kg(140.71モル)を仕込んだほかは、
実施例1と同様に反応を行ってポリアミドを得た。
得られたポリアミドは白色の強靭なポリマーで、ηr=2.05であった。
【0072】
(3)製造例3(成分A:PX6−3)
化合物a12.16kg(104.64モル)と化合物b5.212kg(44.85モル)の混合物(化合物aと化合物bのモル比が70:30)を仕込み、
シュウ酸ジブチル30.238kg(149.49モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、1.5時間かけて温度を290℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。重縮合物の温度が290℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を310℃にし、310℃において1.5時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=2.40であった。
【0073】
(4)製造例4(成分A:PX6−4)
化合物a10.294kg(88.583モル)と化合物b6.863kg(59.057モル)の混合物(化合物aと化合物bのモル比が60:40)を仕込み、
シュウ酸ジブチル29.864kg(147.64モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、1.5時間かけて温度を275℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。
重縮合物の温度が270℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を290℃にし、290℃において2時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=2.68であった。
【0074】
(5)製造例5(成分A:PX6−5)
化合物a8.361kg(71.947モル)と化合物b8.361kg(71.947モル)の混合物(化合物aと化合物bのモル比が50:50)を仕込み、
シュウ酸ジブチル29.107kg(143.89モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、1.5時間かけて温度を260℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。
重縮合物の温度が260℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を275℃にし、275℃において3時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=2.61であった。
【0075】
(6)比較製造例1:(ポリアミド樹脂:PX6−6)
(i)前重縮合工程:撹拌機、還流冷却器、窒素導入管、原料投入口を備えた内容積が1Lのセパラブルフラスコの内部を純度が99.9999%の窒素ガスで置換し、
脱水済みトルエン500ml、
1,6−ヘキサンジアミン58.7209g(0.5053モル)を仕込んだ。
このセパラブルフラスコをオイルバス中に設置して50℃に昇温した後、
シュウ酸ジブチル102.1956g(0.5053モル)を仕込んだ。
次にオイルバスの温度を130℃まで昇温し、還流下、5時間反応を行った。
なお、原料仕込みから反応終了までの全ての操作は50ml/分の窒素気流下で行った。
(ii)後重縮合工程:上記操作によって得られた前重合物を撹拌機、空冷管、窒素導入管を備えた直径約35mmφのガラス製反応管に仕込み、反応管内を13.3Pa以下の減圧下に保ち、次に常圧まで窒素ガスを導入する操作を5回繰り返した後、50ml/分の窒素気流下290℃に保った塩浴に移し、直ちに昇温を開始した。1時間かけて塩浴の温度を340℃とした後、容器内を約66.5Paまで減圧し、さらに2時間反応させた。
続いて常圧まで窒素ガスを導入したのち、塩浴から取り出し50ml/分の窒素気流下で室温まで冷却してポリアミド樹脂を得た。
得られたポリマーは黄色のポリマーであり、ηr=1.65であった。
【0076】
(7)比較例2及び3のポリアミド樹脂組成物に使用したポリアミド樹脂
ナイロン6(宇部興産製、UBEナイロン1015B:PA6)(比較樹脂例2)及び
ナイロン66(宇部興産製、UBEナイロン2020B:PA66)(比較樹脂例3)のペレットを使用した。
【0077】
(8)離型剤
化合物p:ポリプロピレングリコールの末端変性物(PPGA)
化合物q:トリス(トリデシルフォスファイト)
化合物r:ミリスチル酸ミリスチル
化合物s:ステアリン酸亜鉛
化合物t:エチレンビスステアリルアミド
化合物u:低分子量PE
化合物v:タルク(珪酸マグネシウム)
化合物w:1,3,2,4−ジベンジリデンソルビトール
【0078】
(9)実施例1〜12及び比較例2〜3
製造例1〜5、PA6及びPA66のポリアミド樹脂と離型剤(化合物p〜w)について、表1に示す組成で、下記射出条件で射出成形した本発明の箱型成形体を製造した。
(9−1)射出成形機(図1)
図1は、離型力を測定するための射出成形機の概略を示す断面図であり、射出成形機のシリンダー1、箱型成形体又はキャビティー2、固定金型3、エジェクターピン4、コア5、移動金型6、エジェクタープレート7、圧力センサー8、圧力センサー固定板9、ノックアウト棒10、記録計11を有する。
図1に示す、離型力測定装置を取付けた金型及び射出成形機を用い、後述の射出条件で成形して得られる箱型成形体の離型力及び変形を測定した。
【0079】
図1において、固定金型3、移動金型6は横80mm、縦100mm、深さ30mm、肉厚2.3mmで内側に十字のリブが入っている箱を成形するように加工されている。
【0080】
(9−2)実施例3〜5、実施例9〜12、比較例2〜3の成形条件
射出成形機:(株)日本製鋼所製 N140BII
シリンダー設定温度:
C1 290℃、C2 295℃、C3 300℃、C4 300℃、
NH(ノズルヘッド) 300℃
射出圧力:1次圧 650kg/cm2
金型温度:移動金型 90℃、固定金型 85℃
射出時間:10秒
冷却時間:15秒、30秒
【0081】
(9−3)実施例1〜2、実施例6〜8の成形条件
射出成形機:(株)日本製鋼所製 N140BII
シリンダー設定温度:
C1 330℃、C2 335℃、C3 340℃、C4 340℃、
NH(ノズルヘッド) 340℃
射出圧力:1次圧 650kg/cm2
金型温度:移動金型 90℃、固定金型 85℃
射出時間:10秒
冷却時間:15秒、30秒
【0082】
製造例1〜5、比較製造例1、PA6及びPA66のポリアミド樹脂、並びに、実施例1〜12及び比較例2〜3のポリアミド樹脂組成物について、相対粘度、融点、1%重量減少温度、溶融粘度、飽和吸水率、耐薬品性、耐加水分解性、ドライ及びウェットにおける物性を後述する条件で測定し、
実施例1〜12及び比較例2〜3で製造した本発明の成形体について、離型力及び成形体の変形を後述する条件で測定し、
表1に結果を示した。
【0083】
〔物性測定、成形、評価条件〕
以下の内容で行った。
【0084】
(1)ポリアミド樹脂の相対粘度ηr
ηrは実施例1〜5、比較製造例1及び比較樹脂例2の各ポリアミド樹脂の96%硫酸溶液(濃度:1.0g/dl)を使用してオストワルド型粘度計を用いて25℃で測定した。
【0085】
(2)ポリアミド樹脂の溶融粘度
製造例1〜5、比較製造例1及び比較樹脂例2の各ポリアミド樹脂の溶融粘度はティー・エイ・インスツルメント・ジャパン社製溶融粘弾性測定装置ARESに25mmのコーン・プレートを装着して、窒素中、
ポリアミド樹脂としてPA6−1〜2及び6を含む場合は340℃、
ポリアミド樹脂としてPA6−3〜5を含む場合は300℃、
ポリアミド樹脂としてPA6を含む場合は260℃、
せん断速度0.1s−1の条件で測定した。
【0086】
(3)ポリアミド樹脂の融点(Tm)
製造例1〜5、比製造較例1及び比較樹脂例2〜3の各ポリアミド樹脂のTmは、PerkinELmer社製PYRIS Diamond DSC用いて窒素雰囲気下で測定した。
ポリアミド樹脂としてPA6−1〜2及び6を含む場合のTmは、
30℃から350℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、
350℃で3分保持したのち、
−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、
次に350℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。
ポリアミド樹脂としてPA6−3〜5、PA6及びPA66を含む場合のTmは、
30℃から310℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、
310℃で3分保持したのち、
−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、
次に310℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。
昇温セカンドランの吸熱ピーク温度をTmとした。
【0087】
(4)ポリアミド樹脂の1%重量減少温度Td
製造例1〜5、比製造較例1及び比較樹脂例2〜3の各ポリアミド樹脂のTdは島津製作所社製THERMOGRAVIMETRIC ANALYZER TGA−50を用い、熱重量分析(TGA)により測定した。
20ml/分の窒素気流下室温から500℃まで10℃/分の昇温速度で昇温し、Tdを測定した。
【0088】
(5)試験用フィルム及びプレートの成形条件
(5−1)飽和吸水率、耐薬品性、耐加水分解性及び吸水率の試験用フィルム
東邦マシナリー社製真空プレス機TMB−10を用いてフィルム成形して実施例1〜5及び比較例1〜3の各ポリアミド樹脂の飽和吸水率、耐薬品性、耐加水分解性及び吸水率の試験用フィルムを得た。
500〜700Paの減圧雰囲気下、
ポリアミド樹脂としてPA6−1〜2及び6を含む場合は340℃、
ポリアミド樹脂としてPA6−3〜5を含む場合は300℃、
ポリアミド樹脂としてPA6を含む場合は260℃、
ポリアミド樹脂としてPA66を含む場合は290℃、
で5分間加熱溶融させた後、
5MPaで1分間プレスを行いフィルム成形した。
次に減圧雰囲気を常圧まで戻したのち室温5MPaで1分間冷却結晶化させて試験用フィルムを得た。
【0089】
(5−2)機械的物性の試験用プレート
樹脂温度
ポリアミド樹脂としてPA6−1〜2及び6を含む場合は340℃、
ポリアミド樹脂としてPA6−3〜5を含む場合は300℃、
ポリアミド樹脂としてPA6を含む場合は260℃、
ポリアミド樹脂としてPA66を含む場合は290℃、
金型温度80℃の射出成形により成形して試験用プレートを得た。
射出成形条件は、射出圧力:一次圧650kg/cm2、射出時間:11秒、冷却時間:20秒とした。
【0090】
(6)ポリアミド樹脂及びポリアミド樹脂組成物の飽和吸水率
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を23℃のイオン交換水に浸漬し、
所定時間ごとに試験用フィルムを取り出し、フィルムの質量を測定した。
試験用フィルムの質量の増加率が0.2%の範囲内で3回続いた場合に試験用フィルムへの水分の吸収が飽和に達したと判断して、
水に浸漬する前の試験用フィルムの質量(Xg)と飽和に達した時の試験用フィルムの質量(Yg)から下記式(1)により飽和吸水率(%)を算出した。
飽和吸水率(%)=100×(Y−X)/X (1)
【0091】
(7)ポリアミド樹脂及びポリアミド樹脂組成物の耐薬品性
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を以下に列挙する薬品中に7日間浸漬した後に、フィルムの重量残存率(%)及び外観の変化を観測した。濃塩酸、64%硫酸、30%NaOH水溶液、5%K2MnO4のそれぞれの溶液においては23℃、ベンジルアルコールでは50℃において浸漬した試料について試験を行った。
【0092】
(8)ポリアミド樹脂の耐加水分解性
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を、オートクレーブに入れ、水(pH=7)、0.5mol/l硫酸(pH=1)又は1mol/l水酸化ナトリウム水溶液(pH=14)内で、121℃、60分間処理した後の重量残存率(%)及び外観変化を調べた。
【0093】
(9)ポリアミド樹脂の機械的物性
試験用プレートを、
成形後直ちに調湿せずに23℃で評価したものをドライ、
成形後に湿度65%、温度23℃で調湿した後に23℃で評価したものをウェットとして表中に記載した。
【0094】
(9−1)引張強度:ASTM D638に記載のTypeIの試験片のダンベル形状に成形した試験用プレートを用いてASTM D638に準拠して測定した。
(9−2)曲げ弾性率:試験用プレートを用いてASTM D790に準拠し測定した。
(9−3)荷重たわみ温度(熱変形温度):試験用プレートを用いてASTM D648に準拠し、荷重1.82MPaで測定した。
【0095】
(10)ポリアミド樹脂の吸水率
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を使用して、23℃及び湿度65%RHの条件下に置いた以外は、(6)飽和吸水率の測定方法に従って、吸水率(平衡吸水率)(%)を算出した。
(11)離型力と成形品の変形
箱型成形体を射出成形後、移動金型6を後退させ、ノックアウト棒10を圧力センサー8に押し付け、エジェクタープレート7及びエジェクターピン4を介して箱を移動金型6から離す際に圧力センサー8にかかる力(離型力)を記録計11によって測定した。また取出された成形品の変形をチェックした。
【0096】
【表1】
【0097】
表1から、本発明のポリアミド樹脂組成物は、ナイロン6及びナイロン66等の材料と比較して低吸水であり、耐薬品性、耐加水分解性に優れ、wet条件下での機械的物性に優れ、そしてジアミン成分として1,6−ヘキサンジアミン単体を用いたポリアミド樹脂(PX6−6)よりも成形可能温度幅が広く溶融成形性に優れ、さらに高分子量化が可能で、耐熱性に優れた耐熱性成形体を製造することができることが分かる。
なお、PX6−6を使用したポリアミド樹脂組成物は、Td−Tmが小さいため、溶融混練できず、成形もできなかった。
【符号の説明】
【0098】
固定金型3、
エジェクターピン4
移動金型6
エジェクタープレート7
圧力センサー8
ノックアウト棒10
記録計11
【技術分野】
【0001】
本発明は、ポリアミド樹脂組成物及びそれを成形して得た成形体に関する。
【背景技術】
【0002】
以下、ポリアミド樹脂の呼称は、JIS K 6920−1に基づく場合もある。
ナイロン6(PA6)、ナイロン66(PA66)などに代表される結晶性ポリアミドは、その優れた特性と溶融成形の容易さから、衣料用、産業資材用繊維、あるいは汎用のエンジニアリングプラスチックとして広く用いられているが、一方では吸水による物性変化、酸、高温のアルコール、熱水中での劣化などの問題点も指摘されており、より寸法安定性、耐薬品性に優れたポリアミドへの要求が高まっている。
【0003】
ジカルボン酸成分として蓚酸を用いるポリアミド樹脂はポリオキサミド樹脂と呼ばれ、同じアミノ基濃度の他のポリアミド樹脂と比較して融点が高いこと、吸水率が低いことが知られ(特許文献1)、吸水による物性変化が問題となっていた従来のポリアミドが使用困難な分野での活用が期待される。
【0004】
これまでに、ジアミン成分として種々の脂肪族直鎖ジアミンを用いたポリオキサミド樹脂が提案されている。例えば、
非特許文献1には、ジアミン成分として1,6−ヘキサンジアミンを用いたポリオキサミド樹脂が開示され、
非特許文献2には、ジアミン成分が1,9−ノナンジアミンであるポリオキサミド樹脂(以下、PA92ともいう)が開示され、
特許文献2には、種々ジアミン成分と、ジカルボン酸エステルとして蓚酸ジブチルを用いたポリオキサミド樹脂が開示され、
特許文献3には、ジアミン成分として1,9−ノナンジアミン及び2−メチル−1,8−オクタンジアミンの2種のジアミンを特定の比率で用いたポリオキサミド樹脂が開示されている。
【0005】
一方、ポリアミド樹脂の成形性を向上させるために、ポリアミド樹脂と離型剤とを含む樹脂組成物を用いることが従来公知である。
例えば特許文献4には、ポリアミド樹脂又はこれを含む樹脂組成物と、ポリアミド樹脂中に均一に分散された層状珪酸塩及び成形性改良剤とからなるポリアミド樹脂組成物を用いることによって、成形時間が短く、かつ優れた機械的性質を有する成形体を与えるポリアミド組成物を提供することが開示されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2006−57033号公報
【特許文献2】特表平5−506466号公報
【特許文献3】WO2008/072754号公報
【特許文献4】特開平1−301750号公報
【非特許文献】
【0007】
【非特許文献1】S.W.Shalaby,J.Polym.Sci.,11,1(1973)
【非特許文献2】L.Franco,et al.,Macromolecules,31,3912(1998)
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、
非特許文献1に開示されるポリオキサミド樹脂については、融点(約320℃)が熱分解温度(窒素中の1%重量減少温度;約310℃)と近いため、溶融重合、溶融成形が困難であり実用に耐えうるものではなく、
非特許文献2に開示されるポリオキサミド樹脂については、蓚酸源として蓚酸ジエチルを用いた場合の製造法とその結晶構造を開示しているが、ここで得られるPA92は固有粘度が0.97dL/g、融点が246℃のポリマーであり、強靭な成形体が成形出来ない程度の低分子量体しか得られておらず、
特許文献2に開示されるポリオキサミド樹脂については、固有粘度が0.99dL/g、融点が248℃のPA92を製造したことが示されているが、強靭な成形体が成形出来ない程度の低分子量体しか得られていないという問題点があり、
特許文献3に開示されるポリオキサミド樹脂については、ジアミン成分として1,9−ノナンジアミン及び2−メチル−1,8−オクタンジアミンの2種のジアミンを特定の比率で用いたポリオキサミド樹脂が示されているが、これらポリオキサミド樹脂は成形可能温度幅が広く、成形加工性に優れ、かつ低吸水性、耐薬品性、耐加水分解性、燃料バリア性などにも優れるが、融点が240℃前後であり、成形サイクル性と高融点であることが要求される電気・電子機器用途に対しては、耐熱性がやや劣る。
さらに、特許文献4に開示されている技術では、成形時間の短縮及び優れた機械的性質の付与と同時に低吸水性、耐薬品性及び耐加水分解性を満足させるものではない。
【0009】
本発明が解決しようとする課題は、
従来のポリオキサミド樹脂と比較して、
窒素雰囲気下、10℃/分の昇温速度で測定した示差走査熱量法により測定した融点Tm(℃)(以下、融点Tmともいう)と
窒素雰囲気下、10℃/分の昇温速度で測定した熱重量分析における1%重量減少温度Td(℃)(熱分解温度)との温度差(Td−Tm)(℃)(以下、温度差(Td−Tm)ともいう)から見積もられる成形可能温度幅が広く、
融点Tmから見積もられる耐熱性が優れ、
適度な溶融粘度を有し溶融成形性に優れ、及び成形サイクルが低減でき、
脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来の脂肪族ポリアミド樹脂に比較して、耐薬品性、耐加水分解性、燃料バリア性に優れた成形体を成形できるポリアミド樹脂と離型剤とを含む、
成形時における型と成形物との良好なすべり性及び/又は短い成形時間も達成できるポリアミド樹脂組成物とそれを成形して得られる成形体を提供することである。
【課題を解決するための手段】
【0010】
本発明は、
(1)ポリアミド樹脂(成分A)及び離型剤(成分B)を含むポリアミド樹脂組成物であって、
前記成分Aが、ジカルボン酸由来の単位とジアミン由来の単位とが結合してなり、
前記ジカルボン酸が蓚酸(化合物a)を含み、
前記ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、
前記化合物bと前記化合物cのモル比が99:1〜50:50であり、
前記成分Bが、ポリアルキレングリコールの末端変性物、リン酸エステル類、亜リン酸エステル類、高級脂肪酸モノエステル類、高級脂肪酸、高級脂肪酸金属塩、エチレンビスアミド化合物、低分子量ポリエチレン、珪酸マグネシウム及び置換ベンジリデンソルビトール類からなる群から選ばれる少なくとも1以上の化合物であるポリアミド樹脂組成物、及び
(2)上記(1)記載のポリアミド樹脂組成物を成形して得られる成形体。
【発明の効果】
【0011】
本発明によれば、
従来のポリオキサミド樹脂と比較して、
温度差(Td−Tm)から見積もられる成形可能温度幅が広く、
融点Tmから見積もられる耐熱性が優れ、
適度な溶融粘度を有し溶融成形性に優れ、及び成形サイクルが低減でき、
脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来の脂肪族ポリアミド樹脂に比較して、耐薬品性、耐加水分解性、燃料バリア性に優れた成形体を成形できるポリアミド樹脂と離型剤とを含む、
成形時における型と成形物との良好なすべり性及び/又は短い成形時間も達成できるポリアミド樹脂組成物とそれを成形して得られる成形体を提供することができる。
【図面の簡単な説明】
【0012】
【図1】離型力を測定するために、実施例1〜12及び比較例2〜3で使用した射出成形機の概略を示す断面図である。
【発明を実施するための形態】
【0013】
〔成分A〕
(1)成分Aの構成
本発明におけるポリアミド樹脂である成分Aは、
ジカルボン酸成分が蓚酸であり、ジアミン成分が1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミンからなる、即ち、
ジカルボン酸由来の単位とジアミン由来の単位とが結合してなるポリアミド樹脂であって、
前記ジカルボン酸が蓚酸(化合物a)を含み、
前記ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、
前記化合物bと前記化合物cのモル比が99:1〜50:50であり、
好ましくは、96%硫酸を溶媒とし、濃度が1.0g/dlの前記成分Aの溶液を用いて25℃で測定した相対粘度ηrが1.8〜6.0である。
【0014】
成分Aは、化合物a、b及びcを用いて、好ましくはこれらの混合物を用いて重縮合することで、高分子量で、高融点で、融点と熱分解温度の差が大きく溶融成形性に優れ、さらに直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来のポリアミドに比較して耐薬品性、耐加水分解性ならびに燃料バリア性に優れる。
【0015】
成分Aは、耐薬品性、耐加水分解性及び燃料バリア性を確保する観点から、化合物bと化合物cのモル比は、
好ましくは、99:1〜55:45モル%であり、
より好ましくは、99:1〜60:40モル%である。
なお、以下、化合物b及び化合物cのモル比は、成分A中の化合物b由来の単位と化合物c由来の単位のモル比も意味する。
【0016】
成分Aの製造で、化合物a(蓚酸)を直接原料として使用すると、化合物a(蓚酸)そのものは熱分解してしまい、成分Aの融点がその熱分解温度を超えることから、製造時には蓚酸源化合物(以下、蓚酸源ともいう)を用い、蓚酸源由来の蓚酸とジアミンとを重縮合して得る。この蓚酸は、蓚酸ジエステル等の蓚酸源由来のものであり、これらはアミノ基との反応性を有するものであればよい。
蓚酸源として、重縮合反応における副反応を抑制する観点から蓚酸ジエステルが好ましく、蓚酸ジメチル、蓚酸ジエチル、蓚酸ジn−(またはi−)プロピル、蓚酸ジn−(またはi−、またはt−)ブチル等の脂肪族1価アルコールの蓚酸ジエステル、蓚酸ジシクロヘキシル等の脂環式アルコールの蓚酸ジエステル、蓚酸ジフェニル等の芳香族アルコールの蓚酸ジエステル等が挙げられる。
蓚酸ジエステルの中でも炭素原子数が3を超える脂肪族1価アルコールの蓚酸ジエステル、脂環式アルコールの蓚酸ジエステル、芳香族アルコールの蓚酸ジエステルがさらに好ましく、
その中でも蓚酸ジブチル及び蓚酸ジフェニルがさらに好ましく、
蓚酸ジブチルがさらに好ましい。
【0017】
(2)成分Aの相対粘度
成分Aは、
カルボン酸成分として化合物aである蓚酸を用い、
ジアミン成分として、化合物bである1,6−ヘキサンジアミンと、化合物cである2−メチル−1,5−ペンタンジアミンを重縮合することで、融点が好ましくは200〜330℃の範囲にすることができ、融点が330℃を超える化合物aと化合物bとを重縮合して得られるポリアミド樹脂(以下、比較成分A2ともいう)に比べ、後述する成分Aの後重合工程での溶融重合において、副反応が起こり高分子量化を阻害するような過度の高温条件にする必要がないため、高分子量化(相対粘度を増加させること)が可能である。
従って、成分Aは、従来のポリアミド樹脂に比べて相対粘度を増大できるので、優れた溶融成形性を有する。
溶融成形後の成形物が脆くなり物性が低下する傾向を避けることと、溶融成形時の溶融粘度が高くなり成形加工性が悪くなる傾向を避ける観点と、相対粘度ηrと溶融粘度が一定以上に高く、過度に高くないことが好ましいとされる自動車部材や電気・電子部品のような用途に好適であるという観点から、成分Aの濃度が1.0g/dlの96%濃硫酸溶液を用い、25℃で測定した相対粘度ηrは、
好ましくは1.8〜6.0であり、より好ましくは1.8〜4.5であり、
より好ましくは1.8〜3.0であり、更に好ましくは1.85〜2.5、
更に好ましくは1.85〜2.2であるようにすることができる。
なお、後述する成分Aの後重合工程での溶融重合において、減圧度を上げることで、相対粘度ηrを増大することができる。
【0018】
また、同様の観点から、成分Aの溶融粘度は、好ましくは100〜700Pa・s、より好ましくは110〜600Pa・s、更に好ましくは120〜500Pa・s、更に好ましくは130〜400Pa・s、更に好ましくは150〜300Pa・s、更に好ましくは160〜220Pa・s、更に好ましくは170〜200Pa・sである。
【0019】
さらに、同様の観点から、成分Aの数平均分子量(Mn)は、好ましくは10000〜50000であり、より好ましくは11000〜40000であり、更に好ましくは11000〜35000である。
【0020】
数平均分子量(Mn)は、1H−NMRスペクトルから求めたシグナル強度をもとに、例えば、蓚酸源として蓚酸ジブチル、ジアミン成分として1,6−ヘキサンジアミン(化合物b)と2−メチル−1,5−ペンタンジアミン(化合物c)を90:10のモル%比で用いて製造したポリアミド〔以下、PX6−2(化合物b/化合物c=90/10)と略称する〕の場合は下式により算出した。
Mn=np×170.21+n(NH2)×115.20+n(OBu)×129.13+n(NHCHO)×29.14
なお、1H−NMRの測定条件は以下の通りである。
・使用機種:ブルカー・バイオスピン社製 AVANCE500
・溶媒:重硫酸
・積算回数:1024回
また、前記式中の各項は以下のように規定される。
・np=Np/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・n(NH2)
=N(NH2)/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・n(NHCHO)
=N(NHCHO)/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・n(OBu)
=N(OBu)/[(N(NH2)+N(NHCHO)+N(OBu))/2]
・Np=Sp/sp−N(NHCHO)
・N(NH2)=S(NH2)/s(NH2)
・N(NHCHO)=S(NHCHO)/s(NHCHO)
・N(OBu)=S(OBu)/s(OBu)
但し、各項は以下の意味を有する。
・Np:PA62(化合物B/化合物C=90/10)の末端ユニットを除いた、分子鎖中の繰り返しユニット総数。
・np:分子1本当たりの分子鎖中の繰り返しユニット数。
・Sp:PX6−2(化合物b/化合物c=90/10)の末端を除いた、分子鎖中の繰り返しユニット中のオキサミド基に隣接するメチレン基のプロトンに基づくシグナル(3.1ppm付近)の積分値。
・sp:積分値Spにカウントされる水素数(4個)。
・N(NH2):PX6−2(化合物b/化合物c=90/10)の末端アミノ基の総数。
・n(NH2):分子1本当たりの末端アミノ基の数。
・S(NH2):PX6−2(化合物b/化合物c=90/10)の末端アミノ基に隣接するメチレン基のプロトンに基づくシグナル(2.6ppm付近)の積分値。
・s(NH2):積分値S(NH2)にカウントされる水素数(2個)。
・N(NHCHO):PX6−2(化合物b/化合物c=90/10)の末端ホルムアミド基の総数。
・n(NHCHO):分子1本当たりの末端ホルムアミド基の数。
・S(NHCHO):PX6−2(化合物b/化合物c=90/10)のホルムアミド基のプロトンに基づくシグナル(7.8ppm)の積分値。
・s(NHCHO):積分値S(NHCHO)にカウントされる水素数(1個)。
・N(OBu):PX6−2(化合物b/化合物c=90/10)の末端ブトキシ基の総数。
・n(OBu):分子1本当たりの末端ブトキシ基の数。
・S(OBu):PX6−2(化合物b/化合物c=90/10)の末端ブトキシ基の酸素原子に隣接するメチレン基のプロトンに基づくシグナル(4.1ppm付近)の積分値。
・s(OBu):積分値S(OBu)にカウントされる水素数(2個)。
【0021】
(3)成分Aの物性
成分Aは、さらに、化合物b及びcの重縮合比率を変更することで、
温度差(Td−Tm)を、比較成分A2に比べて大きく、化合物aと化合物cとを重縮合して得られるポリアミド樹脂(以下、比較成分A1ともいう)に比べて小さく、
融点Tmを、比較成分A2に比べて低く、比較成分A1に比べて高く、
1%重量減少温度Tdを、比較成分A1に比べて高く、
飽和吸水率を、比較成分A2に比べて小さく、比較成分A1に比べて大きくすることができる。
【0022】
即ち、成分Aは、従来のポリオキサミド樹脂と比較して、
相対粘度ηr(高分子量化)、
温度差(Td−Tm)から見積もられる成形可能温度幅、
融点Tmから見積もられる耐熱性、
溶融粘度から見積もられる溶融成形性、及び
低吸水性のいずれをも十分に確保することができる。
【0023】
成分Aは、成形可能温度幅、耐熱性、溶融成形性及び低吸水性のいずれをも十分に確保した上で、化合物bの重合比率(モル比)の高さに由来する耐薬品性、耐加水分解性及び燃料バリア性に特に寄与する。
【0024】
成分Aは、成形可能温度幅、耐熱性、溶融成形性及び低吸水性のいずれをも十分に確保する観点から、
Tmは、好ましくは260〜330℃であり、より好ましくは265〜330℃であり、
Tdは、好ましくは341〜370℃、より好ましくは345〜370℃、更に好ましくは350〜365℃であり、
温度差(Td−Tm)は、好ましくは10〜95℃、より好ましくは20〜95℃、更に好ましくは25〜95℃であり、
飽和吸水率は、好ましくは0〜2.4、より好ましくは1〜2.4、更に好ましくは2〜2.4、更に好ましくは2.3〜2.4である。
【0025】
(4)成分Aの製造
成分Aは、ポリアミドを製造する方法として知られている任意の方法を用いて製造することができるが、高分子量化および生産性の観点から、
好ましくは、ジアミン及び蓚酸ジエステルをバッチ式又は連続式で重縮合反応させることにより得ることであり、
より好ましくは、ジアミン及び蓚酸ジエステルを前重縮合工程と後重縮合工程からなる二段重合法もしくは、WO2008−072754公報記載の加圧重合法によって得ることである。
更に好ましい二段重合法及び加圧重合法としては、具体的には、以下の操作で示される。
【0026】
(4−1)二段重合法
(i)前重縮合工程:まず反応器内を窒素置換した後、ジアミン(化合物b及びc)及び化合物aの蓚酸源である蓚酸ジエステルを混合する。混合する場合にジアミン及び蓚酸ジエステルが共に可溶な溶媒を用いても良い。ジアミン成分及び蓚酸源成分が共に可溶な溶媒としては、トルエン、キシレン、トリクロロベンゼン、フェノール、トリフルオロエタノールなどを用いることができ、特にトルエンを好ましく用いることができる。例えば、ジアミンを溶解したトルエン溶液を50℃に加熱した後、これに対して蓚酸ジエステルを加える。
このとき、蓚酸ジエステルと上記ジアミンの仕込み比は、高分子量化の観点から、蓚酸ジエステル/上記ジアミンで、0.8〜1.5(モル比)、好ましくは0.91〜1.1(モル比)、更に好ましくは0.99〜1.01(モル比)である。
【0027】
このように仕込んだ反応器内を攪拌及び/又は窒素バブリングしながら、常圧下で昇温する。反応温度は、最終到達温度が80〜150℃、好ましくは100〜140℃の範囲になるように制御するのが好ましい。最終到達温度での反応時間は3時間〜6時間である。
【0028】
(ii)後重縮合工程:更に高分子量化を図るために、前重縮合工程で生成した重合物を常圧下において反応器内で徐々に昇温する。昇温過程において前重縮合工程の最終到達温度、すなわち好ましくは80〜150℃から、最終的に、
好ましくは295℃以上350℃以下、より好ましくは298℃以上345℃以下、更に好ましくは298℃以上340℃以下の温度範囲にまで到達させる。
昇温時間を含めて好ましくは1〜8時間、より好ましくは2〜6時間保持して反応を行うことが好ましい。さらに後重合工程において、必要に応じて減圧下での重合を行うこともできる。減圧重合を行う場合の好ましい最終到達圧力は13.3Pa〜0.1MPaである。
【0029】
(4−2)加圧重合法
まずジアミンを耐圧容器内に入れ窒素置換した後、封圧下において反応温度まで昇温する。その後、反応温度において封圧状態を保ったまま蓚酸化合物を耐圧容器内に注入し、重縮合反応を開始させる。反応温度は、ジアミンと蓚酸化合物の反応によって生じるポリアミドが、スラリー状、もしくは溶液状態を維持でき、かつ熱分解しない温度であれば特に制限されない。例えば、成分aの場合、上記反応温度は、150℃から250℃が好ましい。ここで、蓚酸ジブチルとジアミンの仕込み比は、蓚酸ジブチルのモル量/ジアミンの総モル量で、0.8〜1.5(モル比)、好ましくは0.91〜1.1(モル比)、更に好ましくは0.99〜1.01(モル比)である。
次に耐圧容器内を封圧状態に保ちながらポリアミド樹脂の融点以上かつ熱分解しない温度以下に昇温する。例えば、成分aの場合、融点は245〜300℃であることから、250℃以上350℃以下、好ましくは255℃以上340℃以下、更に好ましくは260℃以上335℃以下に昇温する。所定温度に到達するまでの耐圧容器内の圧力は、およそ生成するアルコールの飽和蒸気圧から0.1MPa、好ましくは1MPaから0.2MPaに調整する。所定温度に到達後は、生成したアルコールを留去しながら放圧し、必要に応じて常圧窒素気流下もしくは減圧下において継続して重縮合反応を行う。減圧重合を行う場合の好ましい最終到達圧力は13.3Pa〜0.1MPaである。
【0030】
(5)成分Aにおけるジカルボン酸として使用できる成分
成分Aにおいて、本発明の効果を損なわない範囲で化合物a以外の他のジカルボン酸成分を使用する事が出来る。
化合物a(蓚酸)以外の他のジカルボン酸成分としては、
マロン酸、ジメチルマロン酸、コハク酸、グルタル酸、アジピン酸、2−メチルアジピン酸、トリメチルアジピン酸、ピメリン酸、2,2−ジメチルグルタル酸、3,3−ジエチルコハク酸、アゼライン酸、セバシン酸、スベリン酸などの脂肪族ジカルボン酸、
また、1,3−シクロペンタンジカルボン酸、1,4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、
さらに、テレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、2,7−ナフタレンジカルボン酸、1,4−ナフタレンジカルボン酸、1,4−フェニレンジオキシジ酢酸、1,3−フェニレンジオキシジ酢酸、ジ安息香酸、4,4’−オキシジ安息香酸、ジフェニルメタン−4,4’−ジカルボン酸、ジフェニルスルホン−4,4’−ジカルボン酸、4,4’−ビフェニルジカルボン酸などの芳香族ジカルボン酸
などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。
さらに、トリメリット酸、トリメシン酸、ピロメリット酸などの多価カルボン酸を溶融成形が可能な範囲内で用いることもできる。
他のジカルボン酸成分を使用する場合、その割合は、化合物a(蓚酸)に対して、25モル%以下であり、15モル%以下が好ましく、10モル%以下がより好ましく、5モル%以下がさらに好ましく、0モル%(即ち、ジカルボン酸成分が化合物aだけからなること)がさらに好ましい。なお、化合物a(蓚酸)に対する他のジカルボン酸成分のモル比は、成分A中の、化合物a由来の単位と他のジカルボン酸成分由来の単位のモル比も意味する。
【0031】
成分Aにおいて、本発明の効果を損なわない範囲で、化合物b及びc以外の他のジアミン成分を使用する事が出来る。
1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミン以外の他のジアミン成分としては、エチレンジアミン、プロピレンジアミン、1,4−ブタンジアミン、1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン、1,8−オクタンジアミン、1,10−デカンジアミン、1,11−ウンデカンジアミン、
1,12−ドデカンジアミン、3−メチル−1,5−ペンタンジアミン、2,2,4−トリメチル−1,6−ヘキサンジアミン、2,4,4−トリメチル−1,6−ヘキサンジアミン、5−メチル−1,9−ノナンジアミンなどの脂肪族ジアミン、
さらに、シクロヘキサンジアミン、メチルシクロヘキサンジアミン、イソホロンジアミンなどの脂環式ジアミン、
さらにp−フェニレンジアミン、m−フェニレンジアミン、p−キシレンジアミン、m−キシレンジアミン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルエーテルなどの芳香族ジアミン、
などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。
他のジアミン成分を使用する場合、その割合は、化合物b及びcに対して25モル%以下であり、15モル%以下が好ましく、10モル%以下がより好ましく、5モル%以下がさらに好ましく、0モル%(即ち、ジアミン成分が化合物b及びcだけからなること)がさらに好ましい。なお、化合物b及びcに対する他のジアミン成分のモル比は、成分A中の、化合物b及びc由来の単位と他のジアミン成分由来の単位のモル比も意味する。
【0032】
(6)成分Aの成形加工
成分Aの成形方法としては、射出、押出、中空、プレス、ロール、発泡、真空・圧空、延伸などポリアミドに適用できる公知の成形加工法はすべて可能であるが、成形サイクル性を短縮する観点から、中でも、射出成形による成形加工において好適であり、これらの成形法によってフィルム、シート、成形品、繊維などに加工することができる。
【0033】
〔成分B〕
離型剤である成分Bは、ポリアルキレングリコールの末端変性物、リン酸エステル類、亜リン酸エステル類、高級脂肪酸モノエステル類、高級脂肪酸、高級脂肪酸金属塩、エチレンビスアミド化合物、低分子量ポリエチレン、珪酸マグネシウム及び置換ベンジリデンソルビトール類からなる群から選ばれる少なくとも1以上の化合物であり、
成形時における型と成形物との良好なすべり性及び/又は成形時間の抑制を安定に確保する観点から選択される。
【0034】
好ましいポリアルキレングリコールの末端変性物の例としては、ポリエチレングリコールの末端変性物、ポリプロピレングリコールの末端変性物などが挙げられる。末端変性は、アミノ基、カルボキシル基又はメチル基でなされていることが好ましい。より具体的な例としては、下記式(1):
X−R1−(O−CH2−CH2)n−O−R2−X (1)
(式中、XはNH2、又はCOOH、又はHを表し、R1及びR2は、各々独立に炭素数1〜10の直鎖状又は分岐状のアルキレン基を表し、nは4〜1200の数である)
又は下記式(2):
X−R3−(O−CH2−C(CH3)H)n−O−R4−X (2)
(式中、XはNH2、又はCOOH、又はHを表し、R3及びR4は、各々独立に炭素数1〜10の直鎖状又は分岐状のアルキレン基を表し、nは1〜200の数である)で表されるものが挙げられる。
【0035】
好ましいリン酸エステル類の例としては、下記式:
(R5O)nPO(OH)3−n
(式中、nは1又は2であり、R5は炭素数1〜10のアルキル基である)で表されるものが挙げられる。上記式中、n=2である場合の2個のRO基は同一でも異なっていてもよい。Rとしては、エチル基、ブチル基、オクチル基、エチルヘキシル基などが挙げられる。
【0036】
好ましい亜リン酸エステル類の例としては、下記式:
(R6O)3P
(式中、R6は、水素、又は炭素数10〜25、より好ましくは12〜20のアルキル基、もしくはフェニル基、もしくはそれらの基の一部が炭化水素基で置換されている基を表す)で表されるものが挙げられる。上記式中の3個のRO基は同一でも異なっていてもよい。Rとしては、デシル基、ラウリル基、トリデシル基、ステアリル基、オレイル基などの脂肪族基;フェニル基、ビフェニル基などの芳香族基;エチル基、プロピル基、t−ブチル基、ノニル基などの置換基を有する芳香族基などが挙げられる。
【0037】
上記のリン酸エステル及び亜リン酸エステルのより具体的な例としては、ジ(2−エチルヘキシル)ホスフェート、トリデシルホスファイト、トリス(トリデシル)ホスファイト、トリステアリルホスファイトなどの脂肪族リン酸エステル及び脂肪族亜リン酸エステル、トリフェニルホスファイト、ジフェニルモノデシルホスファイトなどの芳香族亜リン酸エステルなどが挙げられる。
【0038】
好ましい高級脂肪酸モノエステル類としては、下記式:
R7−CO−O−R8
(式中、R7及びR8は、各々独立に炭素数8〜32、好ましくは10〜30のアルキル基を表す)
で表されるもの、すなわち高級脂肪酸と高級脂肪族1価アルコールとのエステル化合物が挙げられる。上記式中のR1及びR2としては、デシル基、ラウリル基、トリデシル基、ステアリル基、オレイル基などの脂肪族基などが挙げられる。
【0039】
また、上記高級脂肪酸としては、ミリスチン酸、パルミチン酸、ベヘニン酸、オレイン酸、アラギジン酸などが挙げられる。また、高級脂肪族アルコールとしては、ミリスチルアルコール、ベヘニルアルコール、オレイルアルコール、ステアリルアルコール、ヘキシルデシルアルコールなどを挙げることができる。
【0040】
高級脂肪酸モノエステル類のより具体的な例としては、高級脂肪酸モノアルキルエステル、例えばミリスチン酸ミリスチル、ステアリン酸ステアリル、ベヘニン酸ベヘニル、オレイン酸オレイル、ミリスチン酸ヘキシルデシルなどが挙げられる。
【0041】
好ましい高級脂肪酸と高級脂肪酸金属塩の例としては、下記式:
CH3−(CH2)n−COOX
(式中、nは9〜25、好ましくは11〜20の数を表し、XはH又は周期律表第I〜III族の金属を表す)で表されるものが挙げられる。
【0042】
高級脂肪酸としては、ステアリン酸、パルミチン酸、オレイン酸、アラギジン酸、ベヘニン酸などが挙げられる。また高級脂肪酸の金属塩としては、ステアリン酸亜鉛、ステアリン酸リチウム、ステアリン酸カルシウム、パルミチン酸アルミニウムなどが挙げられる。
【0043】
好ましいエチレンビスアミド化合物の例としては、下記式:
CH3(CH2)mCONH(CH2)2NHCO(CH2)nCH3
(式中、m及びnは各々独立に9〜25、好ましくは10〜20の数である)で表されるものが挙げられる。
【0044】
エチレンビスアミド化合物のより具体的な例としては、エチレンビスステアリルアミド、エチレンビスパルミチルアミドなどが挙げられる。
【0045】
好ましい低分子量ポリエチレンとしては、粘度平均分子量が500〜5000の範囲内であるものが挙げられ、粘度平均分子量が1000〜3000の範囲のものがより好ましい。
粘度平均分子量の測定は、粘度平均分子量の測定は、ウベローデ型粘度計を用いた溶液粘度測定による。
【0046】
好ましい珪酸マグネシウムの例としては、平均粒径1〜10μmのものが挙げられる。平均粒径が1μm以上である場合、成形物表面における白いむらが発生しにくく、10μm以下である場合、成形物の機械的物性、特に引張り破断点伸び及び衝撃強さが低下しにくい。ポリアミド樹脂との密着性を改良する目的で、珪酸マグネシウムにアミノシランなどによる表面処理を行ってもよい。
平均粒径の測定は、動的散乱法による。
【0047】
好ましい置換ベンジリデンソルビトール類の例としては、ソルビトールと置換ベンズアルデヒドとの酸触媒下での脱水縮合により合成される置換ベンジリデンソルビトールが挙げられる。置換ベンズアルデヒドのソルビトールへの縮合割合は、ソルビトール1モルに対して1モル又は2モルが好ましい。従って、これらの置換ベンジリデンソルビトール類は、下記式:
【0048】
【化1】
(式中、R9は、H又はヒドロキシル基又はハロゲン又は炭素数1〜200のアルキル基を表す)
又は、下記式:
【0049】
【化2】
(式中、R10及びR11は、各々独立に、H又はヒドロキシル基又はハロゲン又は炭素数1〜200のアルキル基を表す)で表される。
【0050】
置換ベンジリデンソルビトール類としては、例えば、1,3−ベンジリデンソルビトール、1,3,2,4−ジベンジリデンソルビトール、1,3−モノ(p−ヒドロキシベンジリデン)ソルビトール、1,3,2,4−ジ(p−ヒドロキシベンジリデン)ソルビトール、1,3−モノ(p−クロロベンジリデン)ソルビトール、1,3,2,4−ジ(p−クロロベンジリデン)ソルビトール、1,3−モノ(m−ニトロベンジリデン)ソルビトール、1,3,2,4−ジ(m−ニトロベンジリデン)ソルビトール、1,3−(p−クロロベンジリデン)2,4−(p−エチルベンジリデン)−d−ソルビトールなどが挙げられる。
【0051】
〔ポリアミド樹脂組成物〕
本発明のポリアミド樹脂組成物は、成形可能温度幅が広く、耐熱性、及び溶融成形性に優れ、脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、従来の脂肪族ポリアミド樹脂に比較して、耐薬品性、耐加水分解性、燃料バリア性、さらに、成形時における型と成形物との良好なすべり性及び/又は成形時間の抑制を安定に確保する観点から、
成分Bを除くポリアミド樹脂組成物中の成分Aの含有量は、好ましくは50〜100質量%、より好ましくは55〜100質量%、更に好ましくは60〜100質量%、更に好ましくは70〜100質量%、更に好ましくは80〜100質量%、更に好ましくは90〜100質量%、更に好ましくは92〜100質量%、更に好ましくは95〜100質量%であり、
成分Bの含有量は、成分A100質量部に対して、好ましくは0.01〜5質量部、より好ましくは0.05〜3質量部、更に好ましくは0.1〜1.5質量部、更に好ましくは0.1〜1質量部、更に好ましくは0.1〜0.5質量部である。
【0052】
ポリアルキレングリコールの末端変性物の場合、成形時の冷却時間の短縮による成形時間の短縮効果と機械的物性の安定性の観点から、
リン酸エステル及び亜リン酸エステルの場合、成形時における型と成形物とのすべり性と成形サイクル時間の短縮効果、リン酸エステル及び亜リン酸エステルとポリアミド樹脂との相溶性、成形物表面における銀状(シルバーマーク)の発生の抑制及び成形物の機械的物性の安定性の観点から、
高級脂肪酸モノエステル類の場合、成形時における型と成形物とのすべり性、高級脂肪酸モノエステル類とポリアミド樹脂との相溶性、成形物表面における銀状の発生の抑制及び成形物の機械的物性の安定性の観点から、
高級脂肪酸及びその金属塩の場合、成形時における型と成形物とのすべり性、高級脂肪酸モノエステル類とポリアミド樹脂との相溶性、成形物表面における銀状の発生の抑制や成形物の機械的物性、特に引張り破断点伸び、衝撃強さの安定性の観点から、
エチレンビスアミド化合物の場合、成形時における型と成形物とのすべり性、成形時間の短縮効果、成形物の表面外観及び機械的物性の安定性の観点から、
低分子量ポリエチレンの場合、成形時における型と成形物とのすべり性、低分子量ポリエチレンとポリアミド樹脂との相溶性、成形物表面における銀状の発生の抑制及び成形物の機械的物性の安定性の観点から、
珪酸マグネシウムの場合、成形性の向上効果、成形物の機械的物性、特に引張り破断点伸び、衝撃強さの安定性の観点から、
置換ベンジリデンソルビトール類の場合、成形時の冷却時間の短縮による成形時間の短縮効果、成形物表面における銀状の発生の抑制、成形物の機械的物性の安定性の観点から、
成分Bは、上記の好適含有量の範囲であることが好ましい。
【0053】
本発明のポリアミド樹脂組成物では、その他の成分として以下を含むことが好ましい。
(1)成分A以外のポリマー
本発明の樹脂組成物には、必要に応じて、各ポリマーの特性を利用するために、
成分A以外の他のポリアミド類、例えば、ポリオキサミド、芳香族ポリアミド、脂肪族ポリアミド及び脂環式ポリアミドからなる群から選ばれる少なくとも1種のポリアミド、及び/又はポリアミド外のポリマー、例えば、熱可塑性ポリマー、エラストマーを含めることができる。
【0054】
成分A以外のポリマーは、本発明の効果を損なわない範囲であれば特に限定されないが、成分A100質量部に対して、好ましくは0.1〜100質量部、より好ましくは0.1〜50質量部、更に好ましくは0.5〜30質量部である。
【0055】
(2)添加剤
本発明の樹脂組成物は、本発明の効果を損なわない範囲において、添加剤を含むことができる。添加剤として、例えば、顔料、染料、着色剤、酸化防止剤、耐候剤、紫外線吸収剤、光安定化剤、滑剤、結晶核剤、結晶化促進剤、耐熱剤、帯電防止剤、可塑剤、銅化合物等の安定剤、帯電防止剤、難燃剤、ガラス繊維、潤滑剤、フィラー、補強繊維、補強粒子、発泡剤等を挙げることができる。
【0056】
上記成分A以外のポリマー及び/又は添加剤の添加方法は、それぞれを成分Aに分散させることができる方法であれば、特に制限されるものではなく、その効果を損なわない任意の時点において、成分Aに添加することができる。
例えば、成分A以外のポリマー及び/又は添加剤を、
成分Aの重縮合反応時、またはその後に添加することもできる。
【0057】
(3)層状珪酸塩
ポリアミド樹脂に優れた機械的特性及び耐熱性を付与する観点から、本発明のポリアミド樹脂組成物に層状珪酸塩をさらに含ませることができる。
【0058】
上記層状珪酸塩は、一辺の長さが0.002〜1μmで、厚さが6〜20Åである平板状のものであることが好ましい。また、上記層状珪酸塩は、成分Aの中で、各層が約20Å以上の層間距離を保ち、均一に分散されるものであることが好ましい。
【0059】
ここで、「層間距離」とは、平板状をなす層状珪酸塩の各重心の間の距離をいい、「均一に分散する」とは、各層が主にランダムな状態で存在し、層状珪酸塩の50質量%以上、好ましくは70質量%以上が、複層物を形成することなく単層に分散していることをいう。
【0060】
上記層状珪酸塩の量は、当該層状珪酸塩の効果が発揮される量であれば、特に制限されるものではないが、射出成形体の剛性、耐候性及び/又は耐熱性、並びに液体又は蒸気に対するバリア性を向上させる観点と、樹脂組成物の成形加工性と耐衝撃性を確保する観点とから、成分A100質量部に対して、好ましくは0.05〜10質量部、より好ましくは0.05〜8質量部、更に好ましくは0.05〜5質量部である。
【0061】
上記層状珪酸塩の原料としては、珪酸マグネシウム又は珪酸アルミニウムの層から構成される層状フィロ珪酸鉱物、すなわち、珪酸アルミニウム質フィロ珪酸塩又は珪酸マグネシウム質フィロ珪酸塩を例示することができる。具体的には、モンモリロナイト、サポナイト、バイデライト、ノントロナイト、ヘクトライト、スティブンサイト等のスメクタイト系粘土鉱物やバーミキュライト、ハロイサイト等を例示することができ、これらは天然のものであっても、合成されたものであってもよい。
【0062】
また、上記層状珪酸塩を成分Aに分散させるために、通常、膨潤化剤が用いられる。当該膨潤化剤は、粘土鉱物の層間を拡げる役割と、粘土鉱物に層間ポリマーを取り込む力を与える役割とを有するものである。上記膨潤化剤としては、本発明の場合には、1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミンを用いることが好ましい。
【0063】
なお、上記層状珪酸塩は、ミキサー、ボールミル、振動ミル、ピンミル、ジェットミル、叩解機等を用いて粉砕し、予め所望の形状及びサイズのものとしておくことが好ましい。
【0064】
上記層状珪酸塩を添加する方法は、上記層状珪酸塩が成分Aに均一に分散し得る方法である限り、特に制限はない。例えば、層状珪酸塩の原料が多層状粘土鉱物である場合には、特開昭62−74957号に開示されるように、層状珪酸塩を塩酸等によりイオン化し、ここに膨潤化剤、例えば、1,6−ヘキサンジアミン及び2−メチル−1,5−ペンタンジアミンを添加して、あらかじめ層状珪酸塩の各層の間隔を広げる。次いで、当該層の間に成分Aの原料を導入し、さらに当該層の間で上記原料を重合させることができる。
【0065】
また、膨潤化剤として有機化合物を用いて層間を約100Å以上に予め広げ、これを成分Aと溶融混合して、各層をポリアミド樹脂に分散させてもよい。
【0066】
〔本発明の成形体〕
(1)ポリアミド樹脂組成物から成形体への成形加工
本発明は、上述した本発明のポリアミド樹脂組成物から成形された成形体も提供する。
本発明のポリアミド樹脂組成物から成形体への成形方法としては、射出、押出、中空、プレス、ロール、発泡、真空・圧空、延伸などポリアミドに適用できる公知の成形加工法は全て使用可能であり、これらの成形法によってフィルム、シート、成形品、繊維などの成形物に加工することができる。
【0067】
具体的には、例えば、ポリアミド樹脂、離型剤及び必要に応じて用いる各種添加剤の所定量を、V型ブレンダー、タンブラーなどの低速回転混合機やヘンシェルミキサーなどの高速回転混合機を用いてあらかじめ混合した後、射出成形機や押出成形機を用いて、成形物を直接成形する方法を適用できる。
【0068】
(2)成形物の用途
本発明によって得られる成形体は、従来ポリアミド成形体が用いられてきた各種押出成形品、各種射出成形品、シート、フィルム、パイプ、チューブ、モノフィラメント、繊維、容器などの成形物として、自動車部材、コンピューター及び関連機器、光学機器部材、電気・電子機器、情報・通信機器、精密機器、土木・建築用品、医療用品、家庭用品など広範な用途に好適に使用できる。
【実施例】
【0069】
〔実施例及び比較例〕
製造例1〜5において、本発明のポリアミド樹脂組成物に使用する成分A(PX6−1〜5)を製造した。
製造例1〜5において製造した成分Aと成分B(化合物p〜w)を使用して実施例1〜12の本発明のポリアミド樹脂組成物を製造した。
【0070】
(1)製造例1(成分A:PX6−1)
攪拌機、温度計、トルクメーター、圧力計、ダイアフラムポンプを直結した原料投入口、窒素ガス導入口、放圧口、圧力調節装置及びポリマー抜出し口を備えた内容積が約150リットルの圧力容器に、
化合物a(1,6−ヘキサンジアミン)15.407kg(132.58モル)と
化合物b(2−メチル−1,5−ペンタンジアミン)811.1g(6.98モル)の混合物(化合物aと化合物bのモル比が95:5)を仕込み、
圧力容器の内部を純度が99.9999%の窒素ガスで0.5MPaに加圧した後、
次に常圧まで窒素ガスを放出する操作を5回繰り返し、窒素置換を行った後、
封圧下、攪拌しながら系内を昇温した。
約30分間かけてシュウ酸ジブチルの温度を80℃にした後、
シュウ酸ジブチル28.230kg(139.56モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、2時間かけて温度を330℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が330℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、4.5時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=1.95であった。
【0071】
(2)製造例2(成分A:PX6−2)
化合物a14.717kg(126.64モル)と化合物b1.635kg(14.07モル)の混合物(化合物aと化合物bのモル比が90:10)を仕込み、
シュウ酸ジブチル28.462kg(140.71モル)を仕込んだほかは、
実施例1と同様に反応を行ってポリアミドを得た。
得られたポリアミドは白色の強靭なポリマーで、ηr=2.05であった。
【0072】
(3)製造例3(成分A:PX6−3)
化合物a12.16kg(104.64モル)と化合物b5.212kg(44.85モル)の混合物(化合物aと化合物bのモル比が70:30)を仕込み、
シュウ酸ジブチル30.238kg(149.49モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、1.5時間かけて温度を290℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。重縮合物の温度が290℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を310℃にし、310℃において1.5時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=2.40であった。
【0073】
(4)製造例4(成分A:PX6−4)
化合物a10.294kg(88.583モル)と化合物b6.863kg(59.057モル)の混合物(化合物aと化合物bのモル比が60:40)を仕込み、
シュウ酸ジブチル29.864kg(147.64モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、1.5時間かけて温度を275℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。
重縮合物の温度が270℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を290℃にし、290℃において2時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=2.68であった。
【0074】
(5)製造例5(成分A:PX6−5)
化合物a8.361kg(71.947モル)と化合物b8.361kg(71.947モル)の混合物(化合物aと化合物bのモル比が50:50)を仕込み、
シュウ酸ジブチル29.107kg(143.89モル)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。
供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。
その後、1.5時間かけて温度を260℃まで昇温した。
その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。
重縮合物の温度が260℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。
常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を275℃にし、275℃において3時間反応させた。
その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.1MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。
紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。
得られたポリアミドは白色の強靭なポリマーであり、ηr=2.61であった。
【0075】
(6)比較製造例1:(ポリアミド樹脂:PX6−6)
(i)前重縮合工程:撹拌機、還流冷却器、窒素導入管、原料投入口を備えた内容積が1Lのセパラブルフラスコの内部を純度が99.9999%の窒素ガスで置換し、
脱水済みトルエン500ml、
1,6−ヘキサンジアミン58.7209g(0.5053モル)を仕込んだ。
このセパラブルフラスコをオイルバス中に設置して50℃に昇温した後、
シュウ酸ジブチル102.1956g(0.5053モル)を仕込んだ。
次にオイルバスの温度を130℃まで昇温し、還流下、5時間反応を行った。
なお、原料仕込みから反応終了までの全ての操作は50ml/分の窒素気流下で行った。
(ii)後重縮合工程:上記操作によって得られた前重合物を撹拌機、空冷管、窒素導入管を備えた直径約35mmφのガラス製反応管に仕込み、反応管内を13.3Pa以下の減圧下に保ち、次に常圧まで窒素ガスを導入する操作を5回繰り返した後、50ml/分の窒素気流下290℃に保った塩浴に移し、直ちに昇温を開始した。1時間かけて塩浴の温度を340℃とした後、容器内を約66.5Paまで減圧し、さらに2時間反応させた。
続いて常圧まで窒素ガスを導入したのち、塩浴から取り出し50ml/分の窒素気流下で室温まで冷却してポリアミド樹脂を得た。
得られたポリマーは黄色のポリマーであり、ηr=1.65であった。
【0076】
(7)比較例2及び3のポリアミド樹脂組成物に使用したポリアミド樹脂
ナイロン6(宇部興産製、UBEナイロン1015B:PA6)(比較樹脂例2)及び
ナイロン66(宇部興産製、UBEナイロン2020B:PA66)(比較樹脂例3)のペレットを使用した。
【0077】
(8)離型剤
化合物p:ポリプロピレングリコールの末端変性物(PPGA)
化合物q:トリス(トリデシルフォスファイト)
化合物r:ミリスチル酸ミリスチル
化合物s:ステアリン酸亜鉛
化合物t:エチレンビスステアリルアミド
化合物u:低分子量PE
化合物v:タルク(珪酸マグネシウム)
化合物w:1,3,2,4−ジベンジリデンソルビトール
【0078】
(9)実施例1〜12及び比較例2〜3
製造例1〜5、PA6及びPA66のポリアミド樹脂と離型剤(化合物p〜w)について、表1に示す組成で、下記射出条件で射出成形した本発明の箱型成形体を製造した。
(9−1)射出成形機(図1)
図1は、離型力を測定するための射出成形機の概略を示す断面図であり、射出成形機のシリンダー1、箱型成形体又はキャビティー2、固定金型3、エジェクターピン4、コア5、移動金型6、エジェクタープレート7、圧力センサー8、圧力センサー固定板9、ノックアウト棒10、記録計11を有する。
図1に示す、離型力測定装置を取付けた金型及び射出成形機を用い、後述の射出条件で成形して得られる箱型成形体の離型力及び変形を測定した。
【0079】
図1において、固定金型3、移動金型6は横80mm、縦100mm、深さ30mm、肉厚2.3mmで内側に十字のリブが入っている箱を成形するように加工されている。
【0080】
(9−2)実施例3〜5、実施例9〜12、比較例2〜3の成形条件
射出成形機:(株)日本製鋼所製 N140BII
シリンダー設定温度:
C1 290℃、C2 295℃、C3 300℃、C4 300℃、
NH(ノズルヘッド) 300℃
射出圧力:1次圧 650kg/cm2
金型温度:移動金型 90℃、固定金型 85℃
射出時間:10秒
冷却時間:15秒、30秒
【0081】
(9−3)実施例1〜2、実施例6〜8の成形条件
射出成形機:(株)日本製鋼所製 N140BII
シリンダー設定温度:
C1 330℃、C2 335℃、C3 340℃、C4 340℃、
NH(ノズルヘッド) 340℃
射出圧力:1次圧 650kg/cm2
金型温度:移動金型 90℃、固定金型 85℃
射出時間:10秒
冷却時間:15秒、30秒
【0082】
製造例1〜5、比較製造例1、PA6及びPA66のポリアミド樹脂、並びに、実施例1〜12及び比較例2〜3のポリアミド樹脂組成物について、相対粘度、融点、1%重量減少温度、溶融粘度、飽和吸水率、耐薬品性、耐加水分解性、ドライ及びウェットにおける物性を後述する条件で測定し、
実施例1〜12及び比較例2〜3で製造した本発明の成形体について、離型力及び成形体の変形を後述する条件で測定し、
表1に結果を示した。
【0083】
〔物性測定、成形、評価条件〕
以下の内容で行った。
【0084】
(1)ポリアミド樹脂の相対粘度ηr
ηrは実施例1〜5、比較製造例1及び比較樹脂例2の各ポリアミド樹脂の96%硫酸溶液(濃度:1.0g/dl)を使用してオストワルド型粘度計を用いて25℃で測定した。
【0085】
(2)ポリアミド樹脂の溶融粘度
製造例1〜5、比較製造例1及び比較樹脂例2の各ポリアミド樹脂の溶融粘度はティー・エイ・インスツルメント・ジャパン社製溶融粘弾性測定装置ARESに25mmのコーン・プレートを装着して、窒素中、
ポリアミド樹脂としてPA6−1〜2及び6を含む場合は340℃、
ポリアミド樹脂としてPA6−3〜5を含む場合は300℃、
ポリアミド樹脂としてPA6を含む場合は260℃、
せん断速度0.1s−1の条件で測定した。
【0086】
(3)ポリアミド樹脂の融点(Tm)
製造例1〜5、比製造較例1及び比較樹脂例2〜3の各ポリアミド樹脂のTmは、PerkinELmer社製PYRIS Diamond DSC用いて窒素雰囲気下で測定した。
ポリアミド樹脂としてPA6−1〜2及び6を含む場合のTmは、
30℃から350℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、
350℃で3分保持したのち、
−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、
次に350℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。
ポリアミド樹脂としてPA6−3〜5、PA6及びPA66を含む場合のTmは、
30℃から310℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、
310℃で3分保持したのち、
−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、
次に310℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。
昇温セカンドランの吸熱ピーク温度をTmとした。
【0087】
(4)ポリアミド樹脂の1%重量減少温度Td
製造例1〜5、比製造較例1及び比較樹脂例2〜3の各ポリアミド樹脂のTdは島津製作所社製THERMOGRAVIMETRIC ANALYZER TGA−50を用い、熱重量分析(TGA)により測定した。
20ml/分の窒素気流下室温から500℃まで10℃/分の昇温速度で昇温し、Tdを測定した。
【0088】
(5)試験用フィルム及びプレートの成形条件
(5−1)飽和吸水率、耐薬品性、耐加水分解性及び吸水率の試験用フィルム
東邦マシナリー社製真空プレス機TMB−10を用いてフィルム成形して実施例1〜5及び比較例1〜3の各ポリアミド樹脂の飽和吸水率、耐薬品性、耐加水分解性及び吸水率の試験用フィルムを得た。
500〜700Paの減圧雰囲気下、
ポリアミド樹脂としてPA6−1〜2及び6を含む場合は340℃、
ポリアミド樹脂としてPA6−3〜5を含む場合は300℃、
ポリアミド樹脂としてPA6を含む場合は260℃、
ポリアミド樹脂としてPA66を含む場合は290℃、
で5分間加熱溶融させた後、
5MPaで1分間プレスを行いフィルム成形した。
次に減圧雰囲気を常圧まで戻したのち室温5MPaで1分間冷却結晶化させて試験用フィルムを得た。
【0089】
(5−2)機械的物性の試験用プレート
樹脂温度
ポリアミド樹脂としてPA6−1〜2及び6を含む場合は340℃、
ポリアミド樹脂としてPA6−3〜5を含む場合は300℃、
ポリアミド樹脂としてPA6を含む場合は260℃、
ポリアミド樹脂としてPA66を含む場合は290℃、
金型温度80℃の射出成形により成形して試験用プレートを得た。
射出成形条件は、射出圧力:一次圧650kg/cm2、射出時間:11秒、冷却時間:20秒とした。
【0090】
(6)ポリアミド樹脂及びポリアミド樹脂組成物の飽和吸水率
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を23℃のイオン交換水に浸漬し、
所定時間ごとに試験用フィルムを取り出し、フィルムの質量を測定した。
試験用フィルムの質量の増加率が0.2%の範囲内で3回続いた場合に試験用フィルムへの水分の吸収が飽和に達したと判断して、
水に浸漬する前の試験用フィルムの質量(Xg)と飽和に達した時の試験用フィルムの質量(Yg)から下記式(1)により飽和吸水率(%)を算出した。
飽和吸水率(%)=100×(Y−X)/X (1)
【0091】
(7)ポリアミド樹脂及びポリアミド樹脂組成物の耐薬品性
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を以下に列挙する薬品中に7日間浸漬した後に、フィルムの重量残存率(%)及び外観の変化を観測した。濃塩酸、64%硫酸、30%NaOH水溶液、5%K2MnO4のそれぞれの溶液においては23℃、ベンジルアルコールでは50℃において浸漬した試料について試験を行った。
【0092】
(8)ポリアミド樹脂の耐加水分解性
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を、オートクレーブに入れ、水(pH=7)、0.5mol/l硫酸(pH=1)又は1mol/l水酸化ナトリウム水溶液(pH=14)内で、121℃、60分間処理した後の重量残存率(%)及び外観変化を調べた。
【0093】
(9)ポリアミド樹脂の機械的物性
試験用プレートを、
成形後直ちに調湿せずに23℃で評価したものをドライ、
成形後に湿度65%、温度23℃で調湿した後に23℃で評価したものをウェットとして表中に記載した。
【0094】
(9−1)引張強度:ASTM D638に記載のTypeIの試験片のダンベル形状に成形した試験用プレートを用いてASTM D638に準拠して測定した。
(9−2)曲げ弾性率:試験用プレートを用いてASTM D790に準拠し測定した。
(9−3)荷重たわみ温度(熱変形温度):試験用プレートを用いてASTM D648に準拠し、荷重1.82MPaで測定した。
【0095】
(10)ポリアミド樹脂の吸水率
試験用フィルム(寸法:20mm×10mm、厚さ0.25mm;質量約0.05g)を使用して、23℃及び湿度65%RHの条件下に置いた以外は、(6)飽和吸水率の測定方法に従って、吸水率(平衡吸水率)(%)を算出した。
(11)離型力と成形品の変形
箱型成形体を射出成形後、移動金型6を後退させ、ノックアウト棒10を圧力センサー8に押し付け、エジェクタープレート7及びエジェクターピン4を介して箱を移動金型6から離す際に圧力センサー8にかかる力(離型力)を記録計11によって測定した。また取出された成形品の変形をチェックした。
【0096】
【表1】
【0097】
表1から、本発明のポリアミド樹脂組成物は、ナイロン6及びナイロン66等の材料と比較して低吸水であり、耐薬品性、耐加水分解性に優れ、wet条件下での機械的物性に優れ、そしてジアミン成分として1,6−ヘキサンジアミン単体を用いたポリアミド樹脂(PX6−6)よりも成形可能温度幅が広く溶融成形性に優れ、さらに高分子量化が可能で、耐熱性に優れた耐熱性成形体を製造することができることが分かる。
なお、PX6−6を使用したポリアミド樹脂組成物は、Td−Tmが小さいため、溶融混練できず、成形もできなかった。
【符号の説明】
【0098】
固定金型3、
エジェクターピン4
移動金型6
エジェクタープレート7
圧力センサー8
ノックアウト棒10
記録計11
【特許請求の範囲】
【請求項1】
ポリアミド樹脂(成分A)及び離型剤(成分B)を含むポリアミド樹脂組成物であって、
前記成分Aが、ジカルボン酸由来の単位とジアミン由来の単位とが結合してなり、
前記ジカルボン酸が蓚酸(化合物a)を含み、
前記ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、
前記化合物bと前記化合物cのモル比が99:1〜50:50であり、
前記成分Bが、ポリアルキレングリコールの末端変性物、リン酸エステル類、亜リン酸エステル類、高級脂肪酸モノエステル類、高級脂肪酸、高級脂肪酸金属塩、エチレンビスアミド化合物、低分子量ポリエチレン、珪酸マグネシウム及び置換ベンジリデンソルビトール類からなる群から選ばれる少なくとも1以上の化合物であるポリアミド樹脂組成物。
【請求項2】
請求項1記載のポリアミド樹脂組成物を成形して得られる成形体。
【請求項1】
ポリアミド樹脂(成分A)及び離型剤(成分B)を含むポリアミド樹脂組成物であって、
前記成分Aが、ジカルボン酸由来の単位とジアミン由来の単位とが結合してなり、
前記ジカルボン酸が蓚酸(化合物a)を含み、
前記ジアミンが1,6−ヘキサンジアミン(化合物b)及び2−メチル−1,5−ペンタンジアミン(化合物c)を含み、
前記化合物bと前記化合物cのモル比が99:1〜50:50であり、
前記成分Bが、ポリアルキレングリコールの末端変性物、リン酸エステル類、亜リン酸エステル類、高級脂肪酸モノエステル類、高級脂肪酸、高級脂肪酸金属塩、エチレンビスアミド化合物、低分子量ポリエチレン、珪酸マグネシウム及び置換ベンジリデンソルビトール類からなる群から選ばれる少なくとも1以上の化合物であるポリアミド樹脂組成物。
【請求項2】
請求項1記載のポリアミド樹脂組成物を成形して得られる成形体。
【図1】

【公開番号】特開2013−95778(P2013−95778A)
【公開日】平成25年5月20日(2013.5.20)
【国際特許分類】
【出願番号】特願2011−237374(P2011−237374)
【出願日】平成23年10月28日(2011.10.28)
【出願人】(000000206)宇部興産株式会社 (2,022)
【Fターム(参考)】
【公開日】平成25年5月20日(2013.5.20)
【国際特許分類】
【出願日】平成23年10月28日(2011.10.28)
【出願人】(000000206)宇部興産株式会社 (2,022)
【Fターム(参考)】
[ Back to top ]