
ポリエステルの製造方法
【課題】 ポリエステルの重縮合反応速度を大にして、高分子量かつ高品質のポリエステルを短い重縮合時間で製造する方法を提供する。
【解決手段】 芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程に少なくとも1種のエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
【解決手段】 芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程に少なくとも1種のエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
【発明の詳細な説明】
【技術分野】
【0001】
本発明はポリエステルの製造方法に関する。特に、重縮合速度が大きく効率的な製造方法に関する。
【背景技術】
【0002】
ポリエチレンテレフタレートに代表されるポリエステルは、機械的性質、熱的性質、電気的性質などに優れているため、繊維や、各種用途のフィルム、シート、ボトルなどの成形品に広く使われ、需要も拡大している。
【0003】
ボトル等の包装材料として用いられるような高分子量のポリエステルは、通常、ジカルボン酸及び/又はそのエステル形成性誘導体とジオールとをエステル化及び/又はエステル交換反応を経て、溶融重縮合、固相重縮合することにより製造される。
【0004】
現在主流であるポリエステルの製造方法においては、比較的高分子量のポリエステルプレポリマーを得るべく、溶融重縮合工程に、複雑な攪拌翼を備えた横型のプラグフロー性を有する反応器を用いている。また、固相重縮合の反応時間は、通常、10数時間以上にも及んでいる。そのため、これらを改善した効率的な製造方法が求められている。例えば、比較的低分子量のポリエステルプレポリマーを固相重縮合工程に供することを特徴とするポリエステルの製造方法は、多数開示されている。
【0005】
特許文献1には、固相重縮合速度の触媒量、プレポリマーのカルボキシル末端基含量への依存性に関する詳細な検討結果が開示されている。ここでは、プレポリマーのカルボキシル末端基量を、エステル化反応におけるテレフタル酸とエチレングリコールとの仕込み比率を変化させること、反応の後の段階で初期仕込み量に対して過剰量のエチレングリコールを添加すること、または、重縮合工程に部分真空がかけられた後から触媒を添加することによって調整しているが、具体的な実施方法の記載がない。また、この方法は、必ずしも連続溶融重縮合方法に適した方法ではない。
【0006】
一方、重縮合反応を進めるための触媒として、古くからアンチモン触媒、ゲルマニウム触媒、チタン触媒などが知られており、また、タングステン触媒も重縮合反応活性があるとして知られている(特許文献2、3、4、5)。しかし、このようなタングステン触媒は溶融重縮合反応に関するものであり、また得られるポリエステルの色調は黄味があり必ずしも満足できるものではなかった。
【0007】
ポリエステルの重縮合反応による分子量の増大は、通常、2種類の反応によって進められる。即ち、カルボキシル基とアルコール基とのエステル化反応(脱水縮合反応)、エステル結合とアルコール基とのエステル交換反応(脱ジオール縮合反応)である。特許文献6には、エステル化反応触媒活性とエステル交換反応触媒活性の合計に対するエステル化反応触媒活性の比率に着目し、固相重縮合速度を向上させる技術が開示されている。しかし、該文献に記載の発明は、溶融重縮合の段階でエステル化反応を極力進めることにより末端カルボキシル基濃度が低い、すなわち、末端アルコール基濃度が高いポリエステルプレポリマーを得て、エステル交換反応主体の固相重縮合を行うことを主旨としており、重縮合反応の効率化という点では必ずしも満足できる方法ではなかった。
【0008】
【特許文献1】特開昭55−133421号公報
【特許文献2】特公昭44−19554号公報
【特許文献3】特公昭46−2517号公報
【特許文献4】特公昭48−39235号公報
【特許文献5】特開昭56−74123号公報
【特許文献6】特開2006−265536号公報
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の課題は、ポリエステルの重縮合反応速度を大にして、高分子量かつ高品質のポリエステルを短い重縮合時間で製造する方法を提供することにより、ポリエステルの製造効率を高めることにある。
【課題を解決するための手段】
【0010】
本発明者らは、前述のポリエステルを高分子量化する2種類の反応(エステル化反応、エステル交換反応)のうち特にエステル化反応による分子量増大に着目し、エステル化反応触媒を用い、エステル化率、プレポリマーの末端カルボキシル基数の総末端数に対する割合などを特定の範囲にコントロールすることにより、エステル化反応とエステル交換反応をバランス良く進行させることで分子量増大がなされ、重縮合の時間短縮と効率化が図れることを見出し本発明に至った。即ち本発明の要旨は以下である。
(1)芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程に少なくとも1種のエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25dL/g〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
(2)エステル化触媒として、タングステン触媒を用いることを特徴とする(1)に記載のポリエステルの製造方法。
(3)エステル化触媒として、チタン触媒を用いることを特徴とする(1)または(2)に記載のポリエステルの製造方法。
(4)R0及びR1が下記(式2)を満たすことを特徴とする(1)〜(3)のいずれか1項に記載のポリエステルの製造方法。
(式2) −0.2≦ R0 − R1
(5)エステル化触媒として、タングステン触媒及びチタン触媒を該順に添加することを特徴とする(1)〜(4)のいずれか1項に記載のポリエステルの製造方法。
(6)エステル化工程(a)に先立つ原料調製工程、及び、エステル化工程(a)と溶融重縮合工程(b)の間にオリゴマー移送工程を有し、タングステン触媒をエステル化工程(a)及び/又は原料調製工程において添加し、更にチタン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする(5)に記載のポリエステルの製造方法。
(7)エステル化触媒として、チタン触媒及びタングステン触媒を該順に添加することを特徴とする(1)〜(4)のいずれか1項に記載のポリエステルの製造方法。
(8)エステル化工程(a)に先立つ原料調製工程を有し、チタン触媒をエステル化工程(a)及び/又は原料調製工程に添加し、更にタングステン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする(7)に記載のポリエステルの製造方法。
(9)プレポリマー粒子の平均質量が1.0mg/粒〜50mg/粒であることを特徴とする(1)〜(8)のいずれか1項に記載のポリエステルの製造方法。
(10)ポリエステル中のアンチモン原子の含有量が10質量ppm以下であることを特徴とする(1)〜(9)のいずれか1項に記載のポリエステルの製造方法。
【発明の効果】
【0011】
本発明により、高分子量で、形状が均一で、かつ、アンチモン原子の含有量が少ない高品質のポリエステルを、短い重縮合時間で製造することが可能になった。
【発明を実施するための最良の形態】
【0012】
以下において、本発明の内容について詳細に説明する。尚、本願明細書において「〜」とはその前後に記載される数値を下限値及び上限値として含む意味で使用される。
【0013】
本発明のポリエステルの製造方法は、芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法である。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程にエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25dL/g〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
ここで、連続的な製造方法とは、製品を途切れさせることなく、概略一定の条件で製造し続けることを意味する。
【0014】
本発明のポリエステル製造方法は、好ましくは、ジカルボン酸成分とジオール成分とを混合してスラリーを得る原料調製工程、得られたスラリーをエステル化工程に移送するスラリー移送工程、移送したスラリーをエステル化反応させてエステル化反応生成物(オリゴマー)を得るエステル化工程(a)、得られたオリゴマーを溶融重縮合工程に移送するオリゴマー移送工程、移送したオリゴマーを溶融重縮合反応させてポリエステルのプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーを造粒工程に移送するプレポリマー移送工程、移送したプレポリマーを造粒してプレポリマー粒子を得る造粒工程(c)、得られたプレポリマー粒子を固相重縮合反応させることによりポリエステルを得る固相重縮合工程(d)を有する。
【0015】
本発明において「芳香族ジカルボン酸を主成分とする」とは、通常、ポリエステルを製造するに使用する全ジカルボン酸成分に対して95モル%以上が芳香族ジカルボン酸成分であることをいい、好ましくは97モル%以上である。芳香族ジカルボン酸成分の含有量が前記範囲未満では、耐熱ボトル等の成形体としての耐熱性が劣る傾向にある。また「脂肪族ジオールを主成分とする」とは、通常、ポリエステルを製造するに使用する全ジオール成分に対して脂肪族ジオール成分が95モル%以上であることをいい、好ましくは97モル%以上である。
【0016】
芳香族ジカルボン酸としては、テレフタル酸、フタル酸、イソフタル酸、ジブロモイソフタル酸、スルホイソフタル酸ナトリウム、フェニレンジオキシジカルボン酸、4,4'−ジフェニルジカルボン酸、4,4'−ジフェニルエーテルジカルボン酸、4,4'−ジフェニルケトンジカルボン酸、4,4'−ジフェノキシエタンジカルボン酸、4,4'−ジフェニルスルホンジカルボン酸、2,6−ナフタレンジカルボン酸等の芳香族ジカルボン酸が挙げられる。なかでも、テレフタル酸、2,6−ナフタレンジカルボン酸は耐熱性の良好なポリエステルを得やすく好ましい。さらに工業的入手の容易さからテレフタル酸が特に好ましい。
ここで、芳香族ジカルボン酸以外のジカルボン酸成分としては、例えば、ヘキサヒドロテレフタル酸、ヘキサヒドロイソフタル酸等の脂環式ジカルボン酸、及び、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ウンデカジカルボン酸、ドデカジカルボン酸等の脂肪族ジカルボン酸等が挙げられる。
【0017】
また、脂肪族ジオール成分としては、例えば、エチレングリコール、トリメチレングリコール、テトラメチレングリコール、ペンタメチレングリコール、ヘキサメチレングリコール、オクタメチレングリコール、デカメチレングリコール、ネオペンチルグリコール、2−エチル−2−ブチル−1,3−プロパンジオール、ジエチレングリコール、ポリエチレングリコール、ポリテトラメチレンエーテルグリコール等の脂肪族ジオール、1,2−シクロヘキサンジオール、1,4−シクロヘキサンジオール、1,1−シクロヘキサンジメチロール、1,4−シクロヘキサンジメチロール、2,5−ノルボルナンジメチロール等の脂環式ジオールが挙げられる。
中でも、エチレングリコール、テトラメチレングリコールが好ましく、エチレングリコールが特に好ましい。
【0018】
脂肪族ジオール以外のジオールとしては、キシリレングリコール、4,4’−ジヒドロキシビフェニル、2,2−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−β−ヒドロキシエトキシフェニル)プロパン、ビス(4−ヒドロキシフェニル)スルホン、ビス(4−β−ヒドロキシエトキシフェニル)スルホン酸等の芳香族ジオール、ならびに、2,2−ビス(4’−ヒドロキシフェニル)プロパンのエチレンオキサイド付加物又はプロピレンオキサイド付加物等が挙げられ、これらのうち2種以上を成分としてもよい。
【0019】
さらに、例えば、グリコール酸、p−ヒドロキシ安息香酸、p−β−ヒドロキシエトキシ安息香酸等のヒドロキシカルボン酸やアルコキシカルボン酸、および、ステアリルアルコール、ベンジルアルコール、ステアリン酸、安息香酸、t−ブチル安息香酸、ベンゾイル安息香酸等の単官能成分、トリカルバリル酸、トリメリット酸、トリメシン酸、ピロメリット酸、没食子酸、トリメチロールエタン、トリメチロールプロパン、グリセロール、ペンタエリスリトール等の三官能以上の多官能成分等の一種又は二種以上が、共重合成分として用いられてもよい。
【0020】
本発明では、溶融重縮合工程(b)及び/又はそれ以前の工程において、少なくとも1種のエステル化触媒を添加する。ここで、エステル化触媒とは、反応系に添加することにより、無添加の場合よりエステル化反応が促進される化合物であり、具体的には、ベンゼンスルホン酸、トルエンスルホン酸、硫酸、硝酸、リン酸などのブレーンステッド酸やその部分中和物、ならびに、アルカリ金属、アルカリ土類金属、アンチモン、ゲルマニウム、アルミニウム、亜鉛、スズ、マンガン、コバルト、チタンおよびタングステンから選択される金属を含む化合物が挙げられる。中でも触媒活性が高く、得られるポリエステルの色調などが良好であるチタンを含む化合物若しくは混合物であるチタン触媒及び/又はタングステンを含む化合物若しくは混合物であるタングステン触媒が好ましい。アンチモンを含む化合物若しくは混合物であるアンチモン触媒はポリエステル中に異物が発生しやすいため、ポリエステルの含有量として、アンチモン原子が10質量ppm以下であることが好ましい。
チタン触媒としては、例えば、テトラ−n−プロピルチタネート、テトラ−i−プロピルチタネート、テトラ−n−ブチルチタネート、テトラ−n−ブチルチタネートテトラマー、テトラ−t−ブチルチタネート、アセチル−トリ−i−プロピルチタネートなどのテトラアルコキシチタネート、酢酸チタン、蓚酸チタン、塩化チタン等のチタン含有化合物の他、アルコール、アルカリ土類金属化合物、リン酸エステル化合物、および前記テトラアルコキシチタネート、酢酸チタン、蓚酸チタン、塩化チタン等のチタン含有化合物の少なく2種以上を混合することにより得られる液状物などが挙げられる。これらのチタン触媒の中でも、テトラ−i−プロピルチタネート及び/又はテトラ−n−ブチルチタネートを含むことが好ましく、テトラ−n−ブチルチタネート、アルコール、アルカリ土類金属化合物およびリン酸エステル化合物を混合することにより得られる液状物がさらに好ましい。溶融重縮合工程(b)及び/又はそれ以前の工程に添加する合計量としては得られるポリエステルに対するチタン金属原子の量として通常1質量ppm〜20質量ppmであり、4質量ppm〜10質量ppmが好ましい。
タングステン触媒としては、例えば、三酸化タングステン、パラタングステン酸、メタタングステン酸、タングステン酸、ケイタングステン酸、リンタングステン酸およびそれらの塩が挙げられ、中でも、メタタングステン酸アンモニウム、パラタングステン酸アンモニウム、タングステン酸ナトリウムおよびタングステン酸が好ましく、メタタングステン酸アンモニウムおよびパラタングステン酸アンモニウムがさらに好ましい。これらのタングステン触媒は1種類のみを用いても良いし、2種類以上を用いても良い。溶融重縮合工程(b)及び/又はそれ以前の工程に添加する合計量としては得られるポリエステルに対するタングステン金属原子の量として、通常10質量ppm〜150質量ppmであり、20質量ppm〜100質量ppmがより好ましい。
ここで、エステル化触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加するとは、ジカルボン酸成分とジオール成分とを混合してスラリーを得る原料調製工程、得られたスラリーをエステル化工程に移送するスラリー移送工程、移送したスラリーをエステル化反応させてエステル化反応生成物(オリゴマー)を得るエステル化工程、得られたオリゴマーを溶融重縮合工程に移送するオリゴマー移送工程などに添加することを含み、特にオリゴマー移送工程に添加するのが好ましい。
【0021】
本発明の製造方法において、エステル化工程で得られるエステル化反応生成物(オリゴマー)のエステル化率(原料ジカルボン酸成分の全カルボキシル基のうちジオール成分と反応してエステル化したものの割合)は、下限は86%であり、好ましくは92%であり、また上限は96%であり、好ましくは95%である。エステル化率が下限未満の場合、相対的に末端カルボキシル基が過剰であり、重縮合反応の頭打ちが起こるため、好ましくない。エステル化率が上限超過の場合、その後の重縮合工程においてエステル交換反応が主体的となり、本発明の効果が十分に発揮されないため、好ましくない。
【0022】
本発明において、造粒工程(c)において得られるプレポリマーの総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60である。R0の下限は好ましくは0.25であり、上限は0.55が好ましい。R0が下限未満の場合、固相重縮合反応をエステル交換反応中心で進行させる必要があり、本発明の効果が十分に発揮されないため、好ましくない。R0が上限超過の場合、相対的に末端カルボキシル基が過剰であり、重縮合反応の頭打ちが起こるため、好ましくない。造粒工程(c)において得られるプレポリマーの固有粘度は、0.25dL/g〜0.50dL/gであり、下限は好ましくは0.28dL/g、更に好ましくは0.30dL/g、上限は好ましくは0.48dL/g、更に好ましくは0.46dL/gである。
【0023】
本発明において、固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが(式1)を満たし、かつ固有粘度が0.7〜1.5dL/gである。
(式1) R0 − R1 ≦ 0.1
R0−R1の上限は好ましくは0.05、更に好ましくは0.03である。R0−R1の下限は好ましくは−0.5、より好ましくは−0.3、さらに好ましくは−0.2である。R0−R1が下限未満の場合、固相重縮合工程(d)でエステル交換反応の比率が高いため、特に移動床反応器のような層高の高い反応様式の場合、副生するジオールによる固相重縮合速度低下の影響が大きくなる場合がある。R0−R1が上限超過の場合、固相重縮合反応途中で相対的に末端アルコール基が過剰となり、本発明の効果が十分に発揮されないため、好ましくない。
【0024】
エステル化率は、エステル化工程における反応時間、温度などにより制御できる。
R0は、溶融重縮合工程における反応時間、温度、圧力、混合状態などにより制御できる。
R1は固相重縮合工程における反応時間、温度、雰囲気中の水やジオール成分の濃度などにより制御することができる。
更には、エステル化率、R0、R1は、添加するエステル化触媒の活性や選択率による影響も受けるため、エステル化触媒の種類の選定や添加時期などによっても制御できる。
本発明では特に、エステル化触媒として、タングステン触媒及びチタン触媒を順次添加することが好ましく、タングステン触媒をエステル化工程(a)及び/又は原料調製工程において添加し、更にチタン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加する、または、チタン触媒をエステル化工程(a)及び/又は原料調製工程に添加し、更にタングステン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することによって好ましく制御できる。
【0025】
エステル化率、R0、R1、プレポリマーの固有粘度が上記範囲にあるとエステル化反応触媒によるエステル化反応が促進されかつ高反応率まで反応させる必要が無く、また、溶融重縮合反応、固相重縮合反応が速く全体として非常に効率的である。
【0026】
本発明の固相重縮合工程(b)を経て得られるポリエステルの固有粘度は、0.70dL/g〜1.5dL/gである。好ましい下限値は、0.75dL/g以上である。好ましい上限値は、1.00dL/g以下である。下限未満であるとこれを原料としたボトルなどの成形体の機械強度が劣り、好ましくない。また、上限超過では成形体成形時に溶融粘度が高すぎて成形不良の原因となる場合がある。
【0027】
本発明のポリエステルの製造方法は、通常、芳香族ジカルボン酸を主成分とするジカルボン酸成分と脂肪族ジオールを主成分とするジオール成分からポリエステルを製造するに際して、ジカルボン酸成分とジオール成分とを混合してスラリーを得る原料調製工程、得られたスラリーをエステル化工程に移送するスラリー移送工程、移送したスラリーをエステル化反応させてエステル化反応生成物(オリゴマー)を得るエステル化工程(a)、得られたオリゴマーを溶融重縮合工程に移送するオリゴマー移送工程、移送したオリゴマーを溶融重縮合反応させてポリエステルのプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーを造粒工程に移送するプレポリマー移送工程、移送したプレポリマーを造粒してプレポリマー粒子を得る造粒工程(c)、得られたプレポリマー粒子を固相重縮合反応させることによりポリエステルを得る固相重縮合工程(d)を有する。以下、具体的に、芳香族ジカルボン酸としてテレフタル酸、ジオールとしてエチレングリコールを例に製造方法を説明するが、本発明はこれに限定されるものではない。
【0028】
原料調製工程
本発明において、原料スラリーの調製は、テレフタル酸を主成分とするシカルボン酸成分とエチレングリコールを主成分とするジオール成分、及び必要に応じて用いられる共重合成分等とを、ジカルボン酸成分に対するジオール成分のモル比を、通常、1.0〜2.0として調製する。このモル比は1.05〜1.8とするのが好ましく、1.1〜1.6とするのが更に好ましい。
【0029】
スラリー移送工程、エステル化工程(a)
次いで、調製した原料スラリーを、1つの又は連続する複数のエステル化反応槽を備えたエステル化工程に移送し、常圧〜加圧下、加熱下で、エステル化反応させてポリエステル低分子量体であるオリゴマーとする。
【0030】
エステル化反応における反応条件としては、単一のエステル化反応槽の場合、通常、温度を240℃〜295℃、圧力を通常0kPaG〜400kPaGとし、攪拌下に、通常、1時間〜10時間の反応時間(滞留時間)とする。
また、複数のエステル化反応槽の場合は、第1段目のエステル化反応槽における反応温度を、通常240℃〜295℃、好ましくは245℃〜293℃、圧力を、通常5kPaG〜300kPaG、好ましくは10kPaG〜200kPaGとし、最終段における反応温度を、通常250℃〜295℃、好ましくは255℃〜293℃、圧力を、通常0kPaG〜150kPaG、好ましくは0kPaG〜130kPaGとする。ここでkPaGは大気圧に対する相対圧力をkPa単位で表したものである。滞留時間の合計は、通常、1〜10時間、好ましくは、1.2〜5時間とする。
【0031】
本発明において、エステル化反応生成物としてのオリゴマーのエステル化率は、上記に記載のとおりである。
【0032】
溶融重縮合工程(b)
引き続いて、得られたオリゴマーを、重縮合反応槽を備えた溶融重縮合工程に移送し、減圧下、加熱下で溶融重縮合反応させる。
【0033】
本発明においては、溶融重縮合で得るポリエステルプレポリマーの固有粘度が0.50dL/g以下と低いため、従来よく使用されている攪拌翼を備えた横型プラグフロー型第2段重縮合反応槽および第3段重縮合反応槽は必ずしも必要でなく溶融重縮合工程は単純化され設備コストも低減される。特に、本発明の製造方法は、溶融重縮合反応槽が1つの場合において、従来の製造方法と比較して、より単純化された設備で高分子量のポリエステルを製造できる。
【0034】
溶融重縮合における反応条件は、反応温度が、通常260〜290℃、好ましくは270℃〜280℃、圧力が、通常50kPaA〜0.1kPaA、好ましくは40kPaA〜0.1kPaAである。ここでkPaAは絶対圧力をkPa単位で表したものである。
【0035】
プレポリマー移送工程、造粒工程(c)
前記溶融重縮合により得られた樹脂は、通常、重縮合反応槽の底部に設けられた抜き出し口からストランド状に抜き出して、水冷しながら若しくは水冷後、カッターで切断してポリエステルプレポリマー粒子とする。あるいは、重縮合反応槽の底部に設けられた抜き出し口から水中に吐出して冷却しながら、吐出方向とほぼ平行方向の回転軸を有し、抜き出し口先端部に隣接設置されたカッターで切断してポリエステルプレポリマー粒子とする。または粉砕機で粉砕して所望の平均質量の粒子とすることもできる。プレポリマー粒子の平均質量は、通常0.2mg/粒〜50mg/粒であり、下限は好ましくは0.5mg/粒、より好ましくは1.0mg/粒である。また、上限は好ましくは30mg/粒、より好ましくは20mg/粒である。プレポリマー粒子の平均質量がこの範囲の場合、固相重縮合速度が大きく、また、気力輸送等の取り扱いが容易であるため、一層好ましい。
【0036】
結晶化工程
粒子化後のポリエステルプレポリマーを、通常、結晶化する。結晶化は、日本国特許第3073498号公報等に記載の公知の方法によって行うことができ、例えば、120℃〜180℃の不活性ガス気流中で0.5時間〜12時間流動化させることで行うことができる。結晶化を行うことにより、その後の熱処理工程におけるペレット同士の融着を防止できるという効果が得られる。
【0037】
熱処理
本発明では、粒子化後のポリエステルプレポリマーを、または、該プレポリマーを結晶化した後に、熱処理を行ってもよい。熱処理を行うことにより、プレポリマーが固相重縮合され、高分子量化されやすくなる。熱処理は、WO2007/026841号パンフレット等に記載の公知の方法によって行うことができ、例えば、移動床反応器においてポリエステルを上方から下方へ、加熱された不活性ガスを下方から上方へ、それぞれ連続的に流すことで行うことができる。
【0038】
固相重縮合工程(d)
本発明では、粒子化後のポリエステルプレポリマーを、必要に応じて、結晶化後及び/または熱処理した後に固相重縮合をする。固相重縮合反応は、温度の下限は、好ましくは200℃であり、より好ましくは205℃であり、さらに好ましくは208℃である。温度の上限は、好ましくは、当該ポリエステルの融点よりも5℃低い温度、より好ましくは融点よりも8℃低い温度、さらに好ましくは融点よりも10℃低い温度の不活性ガス雰囲気において実施する。ここで、ポリエステルの融点とは、当該ポリエステルを、示差走査熱量計を用いて、窒素気流下、0℃から、20℃/分の速度で300℃まで昇温した際のDSC曲線における、最も高温側の吸熱ピークの頂点に対応する温度のことである。また、不活性ガスとは、酸素濃度が、通常、0.1体積%以下、好ましくは0.05体積%以下であり、かつ、実質的にポリエステルと反応しない気体のことである。実質的にポリエステルと反応しない気体としては、窒素、ヘリウム、ネオン、アルゴン、キセノン、二酸化炭素等が例示でき、主に経済性の点から窒素が好ましく用いられる。
【0039】
固相重縮合温度が低いと固相重縮合速度が遅く、好ましくない。固相重縮合温度が高いと、固相重縮合時にポリエステル粒子が融着するため、好ましくない。特に、ポリエステルプレポリマー粒子の平均質量が0.1mg/粒以下の場合は、融着を回避するために流動床にて固相重縮合するのが好ましい。固相重縮合時間は、目標固有粘度に応じて設定すればよく、通常、1〜50時間程度である。固相重縮合後の平均質量は、通常、固相重縮合前のプレポリマー粒子の平均質量とほぼ一致する。
【0040】
本発明の製造方法により得られるポリエステルは、射出成形や押出成形によりプリフォームを成形後、延伸ブロー成形により、飲料包装等に用いられるボトルにすることができる。また、ダイレクトブロー成形により、ボトルにすることができる。
【0041】
また、押出成形や延伸成形によりフィルム、シートにして包装材料など各種用途に供することができる。また、押出・延伸成形により、繊維とすることができる。
【実施例】
【0042】
以下、実施例により本発明を更に具体的に説明するが、本発明はその要旨を超えない限り、以下の実施例に限定されるものではない。
なお、実施例及び比較例における物性の測定は、以下の方法により行った。
<固有粘度(以下、「IV」ともいう)>
試料約0.25gを、フェノール/1,1,2,2−テトラクロロエタン(質量比:1/1)の混合溶媒約25mLに、濃度が1.00×10-2kg/Lとなるように、非晶状態のポリエステルは110℃、30分で、固相重縮合後のポリエステルは140℃、30分でそれぞれ溶解させた後、30℃まで冷却し、全自動溶液粘度計(センテック社製、2CH型DJ504)にて、濃度が1.00×10-2kg/Lの試料溶液及び溶媒のみの落下秒数を測定し、下式により算出した。
IV(dL/g)=[(1+4KH・ηsp)0.5−1]/(200KH・C)
ここで、ηsp=η/η0−1であり、ηは試料溶液の落下秒数を表し、η0は溶媒の落下秒数を表し、Cはポリマー溶液濃度(kg/L)を表し、KHはハギンズの定数である。
本実施例では、KHは0.33を採用した。
【0043】
<ポリエステルプレポリマー粒子の平均質量>
精密天秤を用いて、ポリエステルプレポリマー粒子30粒の合計質量を0.1mgの桁まで測定し、測定値を30で除することにより、粒子1粒当たりの平均質量を算出した。
【0044】
<末端カルボキシル基濃度(以下、「AV」ともいう)>
試料を粉砕した後、熱風乾燥機にて140℃で15分間乾燥させ、デシケーター内で室温まで冷却した試料から、0.1gを精秤して試験管に採取し、ベンジルアルコール3mLを加えて、乾燥窒素ガスを吹き込みながら195℃、3分間で溶解させ、次いで、クロロホルム5mLを徐々に加えて室温まで冷却した。この溶液にフェノールレッド指示薬を1〜2滴加え、乾燥窒素ガスを吹き込みながら攪拌下に、0.1規定の水酸化ナトリウムのベンジルアルコール溶液で滴定し、黄色から赤色に変じた時点で終了とした。また、ブランクとして、ポリエステル試料を使用せずに同様の操作を実施し、これらの結果を用いて以下の式により末端カルボキシル基濃度を算出した。
AV(当量/トン)=(A−B)×0.1×f/W
ここで、Aは、試料を用いた場合の滴定に要した0.1規定の水酸化ナトリウムのベンジルアルコール溶液の量(μL)を表し、Bは、試料を加えないブランクでの滴定に要した0.1規定の水酸化ナトリウムのベンジルアルコール溶液の量(μL)を表し、Wは、ポリエステル試料の量(g)を表し、fは、0.1規定の水酸化ナトリウムのベンジルアルコール溶液の力価を表す。
なお、0.1規定の水酸化ナトリウムのベンジルアルコール溶液の力価(f)は、試験管にメタノール5mLを採取し、フェノールレッドのエタノール溶液を指示薬として1〜2滴加え、0.1規定の水酸化ナトリウムのベンジルアルコール溶液0.4mLで変色点まで滴定し、次いで、力価既知の0.1規定の塩酸を標準液として0.2mL採取して加え、再度、0.1規定の水酸化ナトリウムのベンジルアルコール溶液で変色点まで滴定し(以上の操作は、乾燥窒素ガスを吹き込みながら行った。)、以下の式により算出した。
力価(f)=0.1規定の塩酸の力価×0.1規定の塩酸の採取量(μL)/0.1規定の水酸化ナトリウムのベンジルアルコール溶液の滴定量(μL)
【0045】
<ポリエステルの総末端基濃度(以下、「TEV」ともいう)>
ポリエステルの総末端基濃度(TEV)は固有粘度(IV)から以下の式で計算して求めた。
IV(dL/g)から数平均分子量Mnを次式により計算し、
Mn=(IV×10000/7.55)^(1/0.685)
MnからTEV(当量/トン)を次式により計算する。
TEV=2×(1000000/Mn)
【0046】
<総末端基数に対する末端カルボキシル基数の割合(R)>
R=AV/TEVで求めた。
【0047】
<実施例1>
エステル化及び/又は重縮合反応に用いたエステル化触媒の調製方法を以下に示す。
<タングステン触媒(触媒)>
メタタングステン酸アンモニウム水溶液(商品名:MW−2、日本無機化学工業社製、三酸化タングステン(WO3)として濃度50質量%)をエチレングリコールで希釈して、タングステン原子濃度が0.2質量%の触媒液(タングステン触媒)を調製した。
【0048】
<チタン触媒>
(Ti/Mg/P=1/1/1.2のモル比の触媒液の合成)
300mL摺り栓付きの三角フラスコ中に、エタノール(特級、純度99.6%以上)を50g入れ、次に酢酸マグネシウム・4水和物7.51gを添加し、スターラーで20分間攪拌して、ほぼ均一に溶解させた。次に、エチルアシッドフォスフェート(商品名:JP−502、城北化学社製、モノエステル体とジエステル体の質量比0.82:1)を2.95g入れて5分間攪拌し、更にジブチルホスフェート(商品名:DBP、城北化学工業社製)を4.41g入れて5分間攪拌した。続いてテトラ−n−ブチルチタネート11.91gを添加し、10分間攪拌することで均一溶液を得た。得られた混合液にエチレングリコール36.1gを加え、次に300mLのナスフラスコに移してエバポレーターにセットし、60℃、11kPaAの条件でエタノール及び低沸物を留去し、47.9gの均一透明な触媒液(チタン触媒)を調製した。チタン触媒液中の各金属成分の割合はTi/Mg/P=1/1/1.2(モル比)であり、チタン濃度3.5質量%であった。
【0049】
<ポリエステルの製造>
以下にポリエステルの製造方法を示す。
(ポリエステルプレポリマーの製造およびプレポリマーの粒子化)
撹拌機、エチレングリコール仕込み配管及びテレフタル酸仕込み配管を具備するスラリー調製槽;スラリーやエステル化反応物を各エステル化反応槽へ移送する各配管;撹拌機、分離塔、原料受入れ口、触媒仕込み配管、エチレングリコール仕込み配管および反応物移送配管を具備する完全混合型第一エステル化反応槽及び第二エステル化反応槽;エステル化反応物(オリゴマー)を溶融重縮合反応槽へ移送する配管;撹拌機、分離塔、オリゴマー受入れ口及びプレポリマー抜き出し口を具備する完全混合型第一溶融重縮合反応槽;撹拌機、分離塔、プレポリマー受入れ口及びポリマー抜き出し口を具備するプラグフロー型第二溶融重縮合反応槽及び第三溶融重縮合反応槽;プレポリマーを抜き出し口よりギヤポンプを介してダイプレートからストランド状に取り出し水冷下ストランドカットする粒子化装置(ストランドカッターはリーター・オートマチック社製、ペレタイザー、P−USG100)を備えたポリエステルプレポリマー連続製造装置を用いた。
【0050】
前記のポリエステルプレポリマー連続製造装置を用いて、テレフタル酸とエチレングリコールとをエステル化反応し、更に溶融重縮合反応することにより得られた溶融状態のプレポリマーをダイプレートからストランド状に取り出し切断することで、ポリエステルプレポリマー粒子を製造した。具体的には以下の通りに行った。
スラリー調製槽にて、テレフタル酸/イソフタル酸/エチレングリコール(モル比で、0.985:0.015:1.5)スラリーを調製した。また、ビス−(β−ヒドロキシエチル)テレフタレート400質量部をエステル化第一槽に仕込み窒素雰囲気下で溶融し、温度262℃、圧力92kPaGに保たれた中へ、前記のスラリー調製槽で調製されたスラリーを135質量部/時間で、ポリエステルとしての平均滞留時間が4.6時間になるように連続的に仕込み、分離塔から生成する水を留去しながらエステル化反応を行いつつ、反応液を連続的にエステル化第二反応槽へ移送した。
第二エステル化反応槽では、温度260℃、圧力3.5kPaGに設定し、上記で調製したタングステン触媒を、得られるポリエステルプレポリマーに対して、タングステンとして40質量ppmとなる量を連続的に添加し、また、エチレングリコールを4質量部/時間の速度で連続的に添加しながら、平均滞留時間1.9時間でエステル化反応を行った。定常状態における第二エステル化反応槽出口における反応物サンプルのエステル化率は95.6%であった。
続いて移送配管を通じ完全混合型第一溶融重縮合反応槽へ連続的に移送すると同時に、この移送配管に、上記にて調製したチタン触媒を、得られるポリエステルプレポリマーに対して、チタン、マグネシウム、リンが、それぞれ、4質量ppm、2質量ppm、3.1質量ppmとなるように、連続的に添加した。
第一溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間1.5時間にて反応を行い、移送配管を通じ第二溶融重縮合反応槽へ連続的に移送した。
第二溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間1.0時間にて溶融重縮合反応を行い、移送配管を通じ第三溶融重縮合反応槽へ移送した。
第三溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間0.5時間にて溶融重縮合反応を行った。
【0051】
上記で得られた溶融ポリエステルプレポリマーを、そのまま、ギヤポンプ及び抜き出し配管を通じてダイヘッドへ導き、ダイホールからストランド状に取り出し、水冷後、リーター・オートマチック社製、ペレタイザー、P−USG100により造粒した。造粒方法はストランドカット法であり、具体的には、ストランド状ポリエステルプレポリマーを水と接触させて冷却させながら、水と共にカッター方向に搬送し、カッター前に設置された一対の引取ロールにて挟むことで引き取り、カッターに供給し、固定刃と回転刃とを有するカッターにて切断することにより、ポリエステルプレポリマー粒子を得た。
得られたポリエステルプレポリマー粒子は、長さ1.25mm、幅1.2mm、厚さ0.9mmのほぼ直方体の両端に半円柱を付けた形状に近い楕円柱状で、ほぼ均一な形状であった。粒子の平均質量は1.8mg/粒であり、固有粘度(IV)は0.383dL/g、R0は0.38であった。
結果を表1に示す。表1中、添加EG量は、添加したエチレングリコール量(質量部/h)を示している。また、「h」は、時間を表す(表2以下についても同じ)。さらに、Wはポリエステル中のタングステン原子の量(質量ppm)を表し、Tiはポリエステル中のチタン原子の量(質量ppm)を表す。
【0052】
(固相重縮合)
上記のポリエステルプレポリマー粒子2.0gをアルミ製トレイ(縦160mm×横100mm、深さ:30mm)にチップ同士が重ならないように並べ、内温60℃に設定されたイナートオーブン(ヤマト科学社製、I/O DN4101)中の中央部に設置した。30NL/分の窒素流通下(ここで、NLとは0℃1気圧における体積(L)のことである)で、60℃から160℃まで30分で昇温させ、160℃で2時間乾燥、結晶化を行った。その後、30分かけて230℃まで昇温し、230℃で、それぞれ、8時間の固相重縮合及び16時間の固相重縮合を行い、ポリエステルを得た。固相重縮合終了後、30分かけて60℃まで降温した後、チップを回収した。固相重縮合時間が8時間の試料及び16時間の試料のIV、R1、ΔIV上昇速度(ΔIV/h)を、それぞれ、求めた。
溶融重縮合後、固相重縮合を8時間行った試料および固相重縮合を16時間行った試料についての結果を表2に示す。
【0053】
<比較例1>
実施例1において、第二エステル化反応槽の温度を263℃、平均滞留時間を2.4時間に変更した以外は実施例1と同様に行った。結果を表1及び表2に示す。なお、粒子の平均質量は1.8mg/粒であった。
【0054】
<実施例2>
実施例1において、第二エステル化反応槽に連続添加するエチレングリコール量を3.5質量部/時間とした以外は実施例1と同様に行った。結果を表1及び表2に示す。なお、粒子の平均質量は1.8mg/粒で、ほぼ均一な形状であった。
【0055】
<比較例2>
実施例1において、第二エステル化反応槽に連続添加するエチレングリコール量を2質量部/時間とした以外は実施例1と同様に行った。結果を表1及び表2に示す。なお、粒子の平均質量は1.8mg/粒であった。
【0056】
<実施例3>
実施例1とは触媒の添加量と添加位置を、また、第二及び第三重縮合反応槽を不使用とするなどを変更し、具体的には以下のように行った。
【0057】
(ポリエステルプレポリマーの製造およびプレポリマーの粒子化)
スラリー調製槽にて、テレフタル酸/イソフタル酸/エチレングリコール(モル比で、0.985:0.015:1.5)スラリーを調製した。また、ビス−(β−ヒドロキシエチル)テレフタレート400質量部をエステル化第一槽に仕込み窒素雰囲気下で溶融し、温度262℃、圧力92kPaGに保たれた中へ、前記のスラリー調製槽で調製されたスラリーを135質量部/時間で、ポリエステルとしての平均滞留時間が4.8時間になるように連続的に仕込み、分離塔から生成する水を留去しながらエステル化反応を行いつつ、反応液を連続的に第二エステル化反応槽へ移送した。第二エステル化反応槽にエチレングリコールを2.8質量部/時間で連続的に添加しながら、温度260℃、圧力3.7kPaG下、平均滞留時間2.4時間でエステル化反応を行った。定常状態における第二エステル化反応槽出口における反応物サンプルのエステル化率は92.6%であった。
続いて移送配管を通じ完全混合型第一溶融重縮合反応槽へ連続的に移送すると同時に、その移送配管のエステル化反応物に、実施例1にて調製したチタン触媒を、得られるポリエステルプレポリマーに対して、チタン、マグネシウム、リンとしてそれぞれ2質量ppm、1質量ppm、1.6質量ppmとなる量を連続的に添加した。更にその下流に、実施例1にて調製したタングステン触媒を、得られるポリエステルプレポリマーに対して、タングステンとして60質量ppmとなるように連続的に添加した。
第一溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間1.6時間にて溶融重縮合反応を行った。このようにして得られた溶融ポリエステルプレポリマーを、ギヤポンプ及び抜き出し配管を通じてダイヘッドへ導き、実施例1のプレポリマー造粒方法と同様の方法により造粒した。その結果、長さ1.25mm、幅1.2mm、厚さ0.9mmのほぼ直方体の両端に半円柱を付けた形状に近い楕円柱状で、ほぼ均一な形状のPETプレポリマー粒子を得た。粒子の平均質量は1.8mg/粒であった。
【0058】
得られたポリエステルプレポリマー粒子を実施例1と同様に固相重縮合を行った。但し、固相重縮合は、16時間とした。結果を表1および表3に示す。
【0059】
<実施例4>、<比較例3>
実施例3において第二エステル化反応槽の圧力、第一重縮合槽の平均滞留時間をそれぞれ表1に示すように変更した以外は実施例3と同様に行った。結果を表1および表3に示す。なお、実施例4において粒子の平均質量は1.8mg/粒で、ほぼ均一な形状であった。
【0060】
【表1】

【0061】
【表2】
【0062】
【表3】
表2の比較例1、2と実施例1、2との比較より、また、表3の実施例3、4と、比較例3との比較により、R0値等が本発明の範囲を満たす場合は満たさない場合より、ΔIV/hが大きくなることが認められた。
【0063】
<実施例5>
(結晶化工程)
実施例4で得たプレポリマー粒子30gを底面が130mm×170mmの角形で、深さが30mmのステンレス製バットに広げて置き、内部のガス温度が180℃のイナートオーブン(タバイエスペック社製、IPHH−201M型)に入れ、イナートオーブンの内部に流通させる窒素の流量を50NL/分、温度を180℃の窒素流通下として、180℃で1時間の結晶化処理を行った。
【0064】
(熱処理装置)
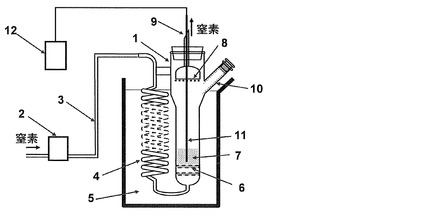
実施例4で得たプレポリマー粒子を結晶化処理した試料を、図1に示すガラス製熱処理装置を用いて熱処理を行った。
以下、該熱処理装置について説明する。
図1に示す熱処理装置において、試料は、試料充填部の内径が45mmのガラス製の熱処理管(1)に充填されている。熱処理管(1)には、ガス流量計(2)、窒素導入管(3)、窒素予熱管(4)を経由して、オイルバス(5)に充填されたオイルにより加熱された窒素が導入される。導入された窒素は、熱処理管(1)下部にある分散板(6)により分散され、熱処理管(1)内部で略均一な線速度を有する上昇流となって、試料層(7)を通過する。試料層(7)を通過した窒素は、熱処理管(1)上部にあるフィルター(8)を経由して、ガスパージ口(9)から熱処理管(1)の外部に排出される。熱処理管(1)は枝管(10)を有しており、その上部にある開口部(通常はガラス栓にて閉止してある)から試料の投入や試料の採取が可能である。また、熱処理管(1)内部の試料の温度は、熱電対(11)を備えた温度計(12)で測定できる。本実施例の範囲の温度、空塔線速度においては、熱処理管(1)の内部温度は、オイルバス中のオイル温度よりも2℃低い温度となるため、目標とする固相重縮合温度に対して、オイルの温度は2℃高い温度に調節した。
【0065】
(第一段固相重縮合工程)
熱処理管(1)の枝管(10)の開口部より、上記結晶化処理後のプレポリマー粒子30gを仕込み、窒素を流通して内部を窒素置換した。その後熱処理管(1)内の窒素の空塔線速度(ここで「空塔線速度」とは、試料層の空塔線速度を意味する(以下、同じ))が210℃で0.30m/秒となるように窒素の流量をガス流量計(2)で設定し、オイルの温度が212℃に調節された第一のオイルバス(5)に熱処理装置を浸漬した。この時点を210℃での第1段固相重縮合の開始とする。2時間後に枝管(10)の開口部より用試料約0.3gを採取した。
【0066】
(昇温工程)
試料採取後、窒素の空塔線速度が235℃で1.0m/秒となるように窒素の流量を変更し、オイルの温度が237℃に調節された第二のオイルバス(5)に熱処理装置を移した。この時点を235℃での昇温工程の開始とした。試料の温度が235℃に到達するまでに、10分を要した。昇温工程の開始から10分後に枝管(10)の開口部より試料を採取した。
<第2段固相重縮合工程>
試料採取後、窒素の空塔線速度が230℃で0.30m/秒となるように窒素の流量を変更し、オイルの温度が232℃に調節された第三のオイルバス(5)に熱処理装置を移した。この時点を230℃での第2段固相重縮合開始とした。第2段固相重縮合開始点から8時間の時点で試料を採取した。第2段固相重縮合の8時間の時点に採取した試料の固有粘度(IV)、R1、ΔIV/hをそれぞれ求めた。結果を表4に示す。
【0067】
<比較例4>
実施例4で得たプレポリマー粒子の代わりに比較例3で得たプレポリマー粒子を用いた以外は実施例5と同様に行った。結果を表4に示す。この例の場合、R0が0.60よりも大きいため、実施例5に比較してΔIV/hが小さい結果だった。
【0068】
【表4】
【0069】
<実施例6>
(ポリエステルの製造)
以下にポリエステルの製造方法を示す。
【0070】
(原料オリゴマーの製造)
テレフタル酸ジメチル2012kg(10.4×103モル)とエチレングリコール1286(20.7×103モル)とをエステル化反応槽に供給して溶解後、エチレングリコールに溶解させた酢酸カルシウムを、カルシウム原子として0.20kg(エステル交換反応により得られる生成物に対して100ppm)となるように添加し、220℃に保持しつつ、生成するメタノールを留出させながらエステル交換反応を行った。エステル交換反応が終了した後、このエステル化反応槽に、テレフタル酸1721kg(10.4×103モル)とエチレングリコール772kg(12.4×103モル)とをスラリー調製槽で攪拌・混合して得られたスラリーを3時間かけて連続的に移送し、常圧下、250℃でエステル化反応を行い、移送開始から4時間反応を行った後に、反応液の50%を系外へ抜き出した。
このエステル化反応槽において、前記と同様にして得られたテレフタル酸とエチレングリコールからなるスラリーを追加してエステル化反応を行い、反応液の50%を抜き出す工程を、計10回繰り返して行い、エステル化反応中の酢酸カルシウムの濃度を0.5質量ppm以下とした。
このようにして、実質的にエステル交換触媒成分を含有しないテレフタル酸とエチレングリコールからなるエステル化反応生成物を製造した。このエステル化反応生成物を、エステル化反応槽から抜き出し、大気下で冷却・固化することにより、以下の実施例で使用する原料オリゴマーを得た。得られた原料オリゴマーのエステル化反応率は89%であった。
【0071】
(ポリエステルプレポリマーの製造)
前記原料オリゴマー104gをトルクメータ付属攪拌装置付き重縮合反応器に移して、系内を窒素で置換した後、常圧下オイルバス(260℃一定)中でオリゴマーの溶解を行った。以下、オリゴマー溶解開始時間を0時間として時間を表記する。
30分後に50rpmで攪拌を開始し、60分後にオリゴマーが完全に溶解していることを確認後、タングステン触媒(メタタングステン酸アンモニウム水溶液(商品名:MW−2、日本無機化学工業社製、三酸化タングステン(WO3)として濃度50質量%))を、得られるポリエステルプレポリマーに対してタングステンとして40質量ppmとなるようにエチレングリコールに希釈して2ml添加した。65分後に減圧を開始し、125分後に0.27kPaAまで減圧した。減圧操作は圧力の対数値が時間に逆比例するように行った。重縮合温度は、65分から145分の間に260℃から280℃まで一定速度で昇温した。到達固有粘度が0.40〜0.50dL/gの範囲に入るように、表5に示す重縮合時間、溶融重縮合反応を行った。なお、重縮合時間は減圧開始から常圧に戻すまでの時間とした。
重縮合終了後、攪拌を停止し、窒素にて常圧に戻し、重縮合反応器をオイルバスから取り出した。重縮合反応器をオイルバスから取り出した後、速やかに該反応器の抜き出し口を開け、窒素で系内を微加圧にすることでポリエステルを抜き出し、水冷・固化させてストランド状のポリエステルを得た。得られたポリエステルは粒子の平均質量が1mg/粒となるように裁断された。得られたポリエステルの固有粘度は0.401dL/g、R0は0.33であった。
【0072】
(固相重縮合)
上記のポリエステルプレポリマー粒子2.0gをアルミ製トレイ(縦160mm×横100mm、深さ:30mm)にチップ同士が重ならないように並べ、内温60℃に設定されたイナートオーブン(ヤマト科学社製、I/O DN4101)中の中央部に設置した。30NL/分の窒素流通下(ここで、NLとは0℃1気圧における体積(L)のことである)で、60℃から160℃まで30分で昇温させ、160℃で2時間乾燥、結晶化を行った。その後、30分かけて230℃まで昇温し、230℃で8時間固相重縮合を行い、ポリエステルを得た。固相重縮合終了後、30分かけて60℃まで降温した後、チップを回収した。固相重縮合時間8時間の試料のIV、R1、ΔIV/hを求めた。結果を表5に示す。
【0073】
<実施例7>
実施例6とは溶融重縮合における触媒の種類、添加操作を変更し、具体的には以下のように行った。
【0074】
(溶融重縮合)
実施例6と同様に製造した原料オリゴマー104gをトルクメータ付属攪拌装置付き重縮合反応器に移して、系内を窒素で置換した後、常圧下オイルバス(260℃一定)中でオリゴマーの溶解を行った。以下、オリゴマー溶解開始時間を0時間として時間を表記する。
30分後に50rpmで攪拌を開始し、60分後にオリゴマーが完全に溶解していることを確認後、チタン触媒(テトラ−n−ブチルチタネート(TBT))を、得られるポリエステルプレポリマーに対してチタンとして4質量ppmとなるようにエチレングリコールに希釈して1mL添加した。次いで65分後にタングステン触媒(メタタングステン酸アンモニウム水溶液(商品名:MW−2、日本無機化学工業社製、三酸化タングステン(WO3)として濃度50質量%))を、得られるポリエステルプレポリマーに対してタングステンとして40質量ppmとなるようにエチレングリコールに希釈して1mL添加した。70分後に減圧を開始し、130分後に0.27kPaAまで減圧した。減圧操作は圧力の対数値が時間に逆比例するように行った。重縮合温度は、70分から150分の間に260℃から280℃まで一定速度で昇温した。到達固有粘度が0.40〜0.50dL/gの範囲に入るように、表5に示す重縮合時間、溶融重縮合反応を行った。なお、重縮合時間は減圧開始から常圧に戻すまでの時間とした。
重縮合終了後、攪拌を停止し、窒素にて常圧に戻し、重縮合反応器をオイルバスから取り出した。重縮合反応器をオイルバスから取り出した後、速やかに該反応器の抜き出し口を開け、窒素で系内を微加圧にすることでポリエステルを抜き出し、水冷・固化させてストランド状のポリエステルを得た。得られたポリエステルは粒子の平均質量が1mg/粒となるように裁断された。得られたポリエステルの固有粘度は0.446dL/g、R0は0.42であった。
得られたポリエステルプレポリマー粒子を実施例6と同様に固相重縮合を行った。結果を表5に示す。
【0075】
<実施例8>
実施例6において、タングステン触媒をチタン触媒(テトラ−n−ブチルチタネート(TBT))に代え、得られるポリエステルプレポリマーに対してチタンとして4質量ppmとなるようにエチレングリコールに希釈して2mL添加した他は、同様の方法で溶融重縮合を行い、ポリエステルを得た。得られたポリエステルの固有粘度は0.439dL/g、R0は0.30であった。得られたポリエステルプレポリマー粒子を実施例6と同様に固相重縮合を行った。結果を表5に示す。
【0076】
<実施例9>
(触媒の調製)
実施例6とは溶融重縮合における触媒の種類、添加操作を変更し、具体的には以下のように行った。
(溶融重縮合)
実施例6と同様に製造した原料オリゴマー104gをトルクメータ付属攪拌装置付き重縮合反応器に移して、系内を窒素で置換した後、常圧下オイルバス(260℃一定)中でオリゴマーの溶解を行った。以下、オリゴマー溶解開始時間を0時間として時間を表記する。
30分後に50rpmで攪拌を開始し、60分後にオリゴマーが完全に溶解していることを確認後、エチルアシッドフォスフェート(商品名:JP−502、城北化学社製、モノエステル体とジエステル体の重量比0.82:1、略称:EAP)を、得られるポリエステルプレポリマーに対してリンとして13質量ppmとなるようにエチレングリコールに希釈して0.7mL添加した。次いで65分後にアンチモン触媒(三酸化アンチモン(略称:SbO)1.8質量%エチレングリコール溶液)を、得られるポリエステルプレポリマーに対してアンチモンとして220質量ppmとなるように1.3mL添加した。70分後に減圧を開始し、130分後に0.27kPaAまで減圧した。減圧操作は圧力の対数値が時間に逆比例するように行った。重縮合温度は、70分から150分の間に260℃から280℃まで一定速度で昇温した。到達固有粘度が0.40〜0.50dL/gの範囲に入るように、表5に示す重縮合時間、溶融重縮合反応を行った。なお、重縮合時間は減圧開始から常圧に戻すまでの時間とした。
重縮合終了後、攪拌を停止し、窒素にて常圧に戻し、重縮合反応器をオイルバスから取り出した。重縮合反応器をオイルバスから取り出した後、速やかに該反応器の抜き出し口を開け、窒素で系内を微加圧にすることでポリエステルを抜き出し、水冷・固化させてストランド状のポリエステルを得た。得られたポリエステルは粒子の平均質量が1mg/粒となるように裁断された。得られたポリエステルの固有粘度は0.447dL/g、R0は0.34であった。ポリエステル中のアンチモン原子含有量は、215質量ppmであった。
得られたポリエステルプレポリマー粒子を実施例6と同様に固相重縮合を行った。結果を表5に示す。
【0077】
【表5】
【0078】
表5の実施例6〜9の結果から、タングステン触媒とチタン触媒を併用する場合に最も良好な触媒活性が得られ、より短い時間で高分子量のポリエステルを製造できることが分かる。
【産業上の利用可能性】
【0079】
本発明により、高分子量で、形状が均一かつアンチモン原子の含有量が少ないポリエステルを、短い重縮合時間で製造することができる。更に、ポリエステルプレポリマーの固有粘度が低いので、複雑で高価な溶融重縮合反応装置を用いる必要が無く効率的な製造方法を提供できる。
【図面の簡単な説明】
【0080】
【図1】本発明で用いるガラス製熱処理装置の一構成例を示す模式図である。
【符号の説明】
【0081】
1: 熱処理管
2: ガス流量計
3: 窒素導入管
4: 窒素予熱管
5: オイルバス
6: 分散板
7: 試料層
8: フィルター
9: ガスパージ口
10: 枝管
11: 熱電対
12: 温度計
【技術分野】
【0001】
本発明はポリエステルの製造方法に関する。特に、重縮合速度が大きく効率的な製造方法に関する。
【背景技術】
【0002】
ポリエチレンテレフタレートに代表されるポリエステルは、機械的性質、熱的性質、電気的性質などに優れているため、繊維や、各種用途のフィルム、シート、ボトルなどの成形品に広く使われ、需要も拡大している。
【0003】
ボトル等の包装材料として用いられるような高分子量のポリエステルは、通常、ジカルボン酸及び/又はそのエステル形成性誘導体とジオールとをエステル化及び/又はエステル交換反応を経て、溶融重縮合、固相重縮合することにより製造される。
【0004】
現在主流であるポリエステルの製造方法においては、比較的高分子量のポリエステルプレポリマーを得るべく、溶融重縮合工程に、複雑な攪拌翼を備えた横型のプラグフロー性を有する反応器を用いている。また、固相重縮合の反応時間は、通常、10数時間以上にも及んでいる。そのため、これらを改善した効率的な製造方法が求められている。例えば、比較的低分子量のポリエステルプレポリマーを固相重縮合工程に供することを特徴とするポリエステルの製造方法は、多数開示されている。
【0005】
特許文献1には、固相重縮合速度の触媒量、プレポリマーのカルボキシル末端基含量への依存性に関する詳細な検討結果が開示されている。ここでは、プレポリマーのカルボキシル末端基量を、エステル化反応におけるテレフタル酸とエチレングリコールとの仕込み比率を変化させること、反応の後の段階で初期仕込み量に対して過剰量のエチレングリコールを添加すること、または、重縮合工程に部分真空がかけられた後から触媒を添加することによって調整しているが、具体的な実施方法の記載がない。また、この方法は、必ずしも連続溶融重縮合方法に適した方法ではない。
【0006】
一方、重縮合反応を進めるための触媒として、古くからアンチモン触媒、ゲルマニウム触媒、チタン触媒などが知られており、また、タングステン触媒も重縮合反応活性があるとして知られている(特許文献2、3、4、5)。しかし、このようなタングステン触媒は溶融重縮合反応に関するものであり、また得られるポリエステルの色調は黄味があり必ずしも満足できるものではなかった。
【0007】
ポリエステルの重縮合反応による分子量の増大は、通常、2種類の反応によって進められる。即ち、カルボキシル基とアルコール基とのエステル化反応(脱水縮合反応)、エステル結合とアルコール基とのエステル交換反応(脱ジオール縮合反応)である。特許文献6には、エステル化反応触媒活性とエステル交換反応触媒活性の合計に対するエステル化反応触媒活性の比率に着目し、固相重縮合速度を向上させる技術が開示されている。しかし、該文献に記載の発明は、溶融重縮合の段階でエステル化反応を極力進めることにより末端カルボキシル基濃度が低い、すなわち、末端アルコール基濃度が高いポリエステルプレポリマーを得て、エステル交換反応主体の固相重縮合を行うことを主旨としており、重縮合反応の効率化という点では必ずしも満足できる方法ではなかった。
【0008】
【特許文献1】特開昭55−133421号公報
【特許文献2】特公昭44−19554号公報
【特許文献3】特公昭46−2517号公報
【特許文献4】特公昭48−39235号公報
【特許文献5】特開昭56−74123号公報
【特許文献6】特開2006−265536号公報
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の課題は、ポリエステルの重縮合反応速度を大にして、高分子量かつ高品質のポリエステルを短い重縮合時間で製造する方法を提供することにより、ポリエステルの製造効率を高めることにある。
【課題を解決するための手段】
【0010】
本発明者らは、前述のポリエステルを高分子量化する2種類の反応(エステル化反応、エステル交換反応)のうち特にエステル化反応による分子量増大に着目し、エステル化反応触媒を用い、エステル化率、プレポリマーの末端カルボキシル基数の総末端数に対する割合などを特定の範囲にコントロールすることにより、エステル化反応とエステル交換反応をバランス良く進行させることで分子量増大がなされ、重縮合の時間短縮と効率化が図れることを見出し本発明に至った。即ち本発明の要旨は以下である。
(1)芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程に少なくとも1種のエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25dL/g〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
(2)エステル化触媒として、タングステン触媒を用いることを特徴とする(1)に記載のポリエステルの製造方法。
(3)エステル化触媒として、チタン触媒を用いることを特徴とする(1)または(2)に記載のポリエステルの製造方法。
(4)R0及びR1が下記(式2)を満たすことを特徴とする(1)〜(3)のいずれか1項に記載のポリエステルの製造方法。
(式2) −0.2≦ R0 − R1
(5)エステル化触媒として、タングステン触媒及びチタン触媒を該順に添加することを特徴とする(1)〜(4)のいずれか1項に記載のポリエステルの製造方法。
(6)エステル化工程(a)に先立つ原料調製工程、及び、エステル化工程(a)と溶融重縮合工程(b)の間にオリゴマー移送工程を有し、タングステン触媒をエステル化工程(a)及び/又は原料調製工程において添加し、更にチタン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする(5)に記載のポリエステルの製造方法。
(7)エステル化触媒として、チタン触媒及びタングステン触媒を該順に添加することを特徴とする(1)〜(4)のいずれか1項に記載のポリエステルの製造方法。
(8)エステル化工程(a)に先立つ原料調製工程を有し、チタン触媒をエステル化工程(a)及び/又は原料調製工程に添加し、更にタングステン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする(7)に記載のポリエステルの製造方法。
(9)プレポリマー粒子の平均質量が1.0mg/粒〜50mg/粒であることを特徴とする(1)〜(8)のいずれか1項に記載のポリエステルの製造方法。
(10)ポリエステル中のアンチモン原子の含有量が10質量ppm以下であることを特徴とする(1)〜(9)のいずれか1項に記載のポリエステルの製造方法。
【発明の効果】
【0011】
本発明により、高分子量で、形状が均一で、かつ、アンチモン原子の含有量が少ない高品質のポリエステルを、短い重縮合時間で製造することが可能になった。
【発明を実施するための最良の形態】
【0012】
以下において、本発明の内容について詳細に説明する。尚、本願明細書において「〜」とはその前後に記載される数値を下限値及び上限値として含む意味で使用される。
【0013】
本発明のポリエステルの製造方法は、芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法である。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程にエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25dL/g〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
ここで、連続的な製造方法とは、製品を途切れさせることなく、概略一定の条件で製造し続けることを意味する。
【0014】
本発明のポリエステル製造方法は、好ましくは、ジカルボン酸成分とジオール成分とを混合してスラリーを得る原料調製工程、得られたスラリーをエステル化工程に移送するスラリー移送工程、移送したスラリーをエステル化反応させてエステル化反応生成物(オリゴマー)を得るエステル化工程(a)、得られたオリゴマーを溶融重縮合工程に移送するオリゴマー移送工程、移送したオリゴマーを溶融重縮合反応させてポリエステルのプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーを造粒工程に移送するプレポリマー移送工程、移送したプレポリマーを造粒してプレポリマー粒子を得る造粒工程(c)、得られたプレポリマー粒子を固相重縮合反応させることによりポリエステルを得る固相重縮合工程(d)を有する。
【0015】
本発明において「芳香族ジカルボン酸を主成分とする」とは、通常、ポリエステルを製造するに使用する全ジカルボン酸成分に対して95モル%以上が芳香族ジカルボン酸成分であることをいい、好ましくは97モル%以上である。芳香族ジカルボン酸成分の含有量が前記範囲未満では、耐熱ボトル等の成形体としての耐熱性が劣る傾向にある。また「脂肪族ジオールを主成分とする」とは、通常、ポリエステルを製造するに使用する全ジオール成分に対して脂肪族ジオール成分が95モル%以上であることをいい、好ましくは97モル%以上である。
【0016】
芳香族ジカルボン酸としては、テレフタル酸、フタル酸、イソフタル酸、ジブロモイソフタル酸、スルホイソフタル酸ナトリウム、フェニレンジオキシジカルボン酸、4,4'−ジフェニルジカルボン酸、4,4'−ジフェニルエーテルジカルボン酸、4,4'−ジフェニルケトンジカルボン酸、4,4'−ジフェノキシエタンジカルボン酸、4,4'−ジフェニルスルホンジカルボン酸、2,6−ナフタレンジカルボン酸等の芳香族ジカルボン酸が挙げられる。なかでも、テレフタル酸、2,6−ナフタレンジカルボン酸は耐熱性の良好なポリエステルを得やすく好ましい。さらに工業的入手の容易さからテレフタル酸が特に好ましい。
ここで、芳香族ジカルボン酸以外のジカルボン酸成分としては、例えば、ヘキサヒドロテレフタル酸、ヘキサヒドロイソフタル酸等の脂環式ジカルボン酸、及び、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ウンデカジカルボン酸、ドデカジカルボン酸等の脂肪族ジカルボン酸等が挙げられる。
【0017】
また、脂肪族ジオール成分としては、例えば、エチレングリコール、トリメチレングリコール、テトラメチレングリコール、ペンタメチレングリコール、ヘキサメチレングリコール、オクタメチレングリコール、デカメチレングリコール、ネオペンチルグリコール、2−エチル−2−ブチル−1,3−プロパンジオール、ジエチレングリコール、ポリエチレングリコール、ポリテトラメチレンエーテルグリコール等の脂肪族ジオール、1,2−シクロヘキサンジオール、1,4−シクロヘキサンジオール、1,1−シクロヘキサンジメチロール、1,4−シクロヘキサンジメチロール、2,5−ノルボルナンジメチロール等の脂環式ジオールが挙げられる。
中でも、エチレングリコール、テトラメチレングリコールが好ましく、エチレングリコールが特に好ましい。
【0018】
脂肪族ジオール以外のジオールとしては、キシリレングリコール、4,4’−ジヒドロキシビフェニル、2,2−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−β−ヒドロキシエトキシフェニル)プロパン、ビス(4−ヒドロキシフェニル)スルホン、ビス(4−β−ヒドロキシエトキシフェニル)スルホン酸等の芳香族ジオール、ならびに、2,2−ビス(4’−ヒドロキシフェニル)プロパンのエチレンオキサイド付加物又はプロピレンオキサイド付加物等が挙げられ、これらのうち2種以上を成分としてもよい。
【0019】
さらに、例えば、グリコール酸、p−ヒドロキシ安息香酸、p−β−ヒドロキシエトキシ安息香酸等のヒドロキシカルボン酸やアルコキシカルボン酸、および、ステアリルアルコール、ベンジルアルコール、ステアリン酸、安息香酸、t−ブチル安息香酸、ベンゾイル安息香酸等の単官能成分、トリカルバリル酸、トリメリット酸、トリメシン酸、ピロメリット酸、没食子酸、トリメチロールエタン、トリメチロールプロパン、グリセロール、ペンタエリスリトール等の三官能以上の多官能成分等の一種又は二種以上が、共重合成分として用いられてもよい。
【0020】
本発明では、溶融重縮合工程(b)及び/又はそれ以前の工程において、少なくとも1種のエステル化触媒を添加する。ここで、エステル化触媒とは、反応系に添加することにより、無添加の場合よりエステル化反応が促進される化合物であり、具体的には、ベンゼンスルホン酸、トルエンスルホン酸、硫酸、硝酸、リン酸などのブレーンステッド酸やその部分中和物、ならびに、アルカリ金属、アルカリ土類金属、アンチモン、ゲルマニウム、アルミニウム、亜鉛、スズ、マンガン、コバルト、チタンおよびタングステンから選択される金属を含む化合物が挙げられる。中でも触媒活性が高く、得られるポリエステルの色調などが良好であるチタンを含む化合物若しくは混合物であるチタン触媒及び/又はタングステンを含む化合物若しくは混合物であるタングステン触媒が好ましい。アンチモンを含む化合物若しくは混合物であるアンチモン触媒はポリエステル中に異物が発生しやすいため、ポリエステルの含有量として、アンチモン原子が10質量ppm以下であることが好ましい。
チタン触媒としては、例えば、テトラ−n−プロピルチタネート、テトラ−i−プロピルチタネート、テトラ−n−ブチルチタネート、テトラ−n−ブチルチタネートテトラマー、テトラ−t−ブチルチタネート、アセチル−トリ−i−プロピルチタネートなどのテトラアルコキシチタネート、酢酸チタン、蓚酸チタン、塩化チタン等のチタン含有化合物の他、アルコール、アルカリ土類金属化合物、リン酸エステル化合物、および前記テトラアルコキシチタネート、酢酸チタン、蓚酸チタン、塩化チタン等のチタン含有化合物の少なく2種以上を混合することにより得られる液状物などが挙げられる。これらのチタン触媒の中でも、テトラ−i−プロピルチタネート及び/又はテトラ−n−ブチルチタネートを含むことが好ましく、テトラ−n−ブチルチタネート、アルコール、アルカリ土類金属化合物およびリン酸エステル化合物を混合することにより得られる液状物がさらに好ましい。溶融重縮合工程(b)及び/又はそれ以前の工程に添加する合計量としては得られるポリエステルに対するチタン金属原子の量として通常1質量ppm〜20質量ppmであり、4質量ppm〜10質量ppmが好ましい。
タングステン触媒としては、例えば、三酸化タングステン、パラタングステン酸、メタタングステン酸、タングステン酸、ケイタングステン酸、リンタングステン酸およびそれらの塩が挙げられ、中でも、メタタングステン酸アンモニウム、パラタングステン酸アンモニウム、タングステン酸ナトリウムおよびタングステン酸が好ましく、メタタングステン酸アンモニウムおよびパラタングステン酸アンモニウムがさらに好ましい。これらのタングステン触媒は1種類のみを用いても良いし、2種類以上を用いても良い。溶融重縮合工程(b)及び/又はそれ以前の工程に添加する合計量としては得られるポリエステルに対するタングステン金属原子の量として、通常10質量ppm〜150質量ppmであり、20質量ppm〜100質量ppmがより好ましい。
ここで、エステル化触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加するとは、ジカルボン酸成分とジオール成分とを混合してスラリーを得る原料調製工程、得られたスラリーをエステル化工程に移送するスラリー移送工程、移送したスラリーをエステル化反応させてエステル化反応生成物(オリゴマー)を得るエステル化工程、得られたオリゴマーを溶融重縮合工程に移送するオリゴマー移送工程などに添加することを含み、特にオリゴマー移送工程に添加するのが好ましい。
【0021】
本発明の製造方法において、エステル化工程で得られるエステル化反応生成物(オリゴマー)のエステル化率(原料ジカルボン酸成分の全カルボキシル基のうちジオール成分と反応してエステル化したものの割合)は、下限は86%であり、好ましくは92%であり、また上限は96%であり、好ましくは95%である。エステル化率が下限未満の場合、相対的に末端カルボキシル基が過剰であり、重縮合反応の頭打ちが起こるため、好ましくない。エステル化率が上限超過の場合、その後の重縮合工程においてエステル交換反応が主体的となり、本発明の効果が十分に発揮されないため、好ましくない。
【0022】
本発明において、造粒工程(c)において得られるプレポリマーの総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60である。R0の下限は好ましくは0.25であり、上限は0.55が好ましい。R0が下限未満の場合、固相重縮合反応をエステル交換反応中心で進行させる必要があり、本発明の効果が十分に発揮されないため、好ましくない。R0が上限超過の場合、相対的に末端カルボキシル基が過剰であり、重縮合反応の頭打ちが起こるため、好ましくない。造粒工程(c)において得られるプレポリマーの固有粘度は、0.25dL/g〜0.50dL/gであり、下限は好ましくは0.28dL/g、更に好ましくは0.30dL/g、上限は好ましくは0.48dL/g、更に好ましくは0.46dL/gである。
【0023】
本発明において、固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが(式1)を満たし、かつ固有粘度が0.7〜1.5dL/gである。
(式1) R0 − R1 ≦ 0.1
R0−R1の上限は好ましくは0.05、更に好ましくは0.03である。R0−R1の下限は好ましくは−0.5、より好ましくは−0.3、さらに好ましくは−0.2である。R0−R1が下限未満の場合、固相重縮合工程(d)でエステル交換反応の比率が高いため、特に移動床反応器のような層高の高い反応様式の場合、副生するジオールによる固相重縮合速度低下の影響が大きくなる場合がある。R0−R1が上限超過の場合、固相重縮合反応途中で相対的に末端アルコール基が過剰となり、本発明の効果が十分に発揮されないため、好ましくない。
【0024】
エステル化率は、エステル化工程における反応時間、温度などにより制御できる。
R0は、溶融重縮合工程における反応時間、温度、圧力、混合状態などにより制御できる。
R1は固相重縮合工程における反応時間、温度、雰囲気中の水やジオール成分の濃度などにより制御することができる。
更には、エステル化率、R0、R1は、添加するエステル化触媒の活性や選択率による影響も受けるため、エステル化触媒の種類の選定や添加時期などによっても制御できる。
本発明では特に、エステル化触媒として、タングステン触媒及びチタン触媒を順次添加することが好ましく、タングステン触媒をエステル化工程(a)及び/又は原料調製工程において添加し、更にチタン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加する、または、チタン触媒をエステル化工程(a)及び/又は原料調製工程に添加し、更にタングステン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することによって好ましく制御できる。
【0025】
エステル化率、R0、R1、プレポリマーの固有粘度が上記範囲にあるとエステル化反応触媒によるエステル化反応が促進されかつ高反応率まで反応させる必要が無く、また、溶融重縮合反応、固相重縮合反応が速く全体として非常に効率的である。
【0026】
本発明の固相重縮合工程(b)を経て得られるポリエステルの固有粘度は、0.70dL/g〜1.5dL/gである。好ましい下限値は、0.75dL/g以上である。好ましい上限値は、1.00dL/g以下である。下限未満であるとこれを原料としたボトルなどの成形体の機械強度が劣り、好ましくない。また、上限超過では成形体成形時に溶融粘度が高すぎて成形不良の原因となる場合がある。
【0027】
本発明のポリエステルの製造方法は、通常、芳香族ジカルボン酸を主成分とするジカルボン酸成分と脂肪族ジオールを主成分とするジオール成分からポリエステルを製造するに際して、ジカルボン酸成分とジオール成分とを混合してスラリーを得る原料調製工程、得られたスラリーをエステル化工程に移送するスラリー移送工程、移送したスラリーをエステル化反応させてエステル化反応生成物(オリゴマー)を得るエステル化工程(a)、得られたオリゴマーを溶融重縮合工程に移送するオリゴマー移送工程、移送したオリゴマーを溶融重縮合反応させてポリエステルのプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーを造粒工程に移送するプレポリマー移送工程、移送したプレポリマーを造粒してプレポリマー粒子を得る造粒工程(c)、得られたプレポリマー粒子を固相重縮合反応させることによりポリエステルを得る固相重縮合工程(d)を有する。以下、具体的に、芳香族ジカルボン酸としてテレフタル酸、ジオールとしてエチレングリコールを例に製造方法を説明するが、本発明はこれに限定されるものではない。
【0028】
原料調製工程
本発明において、原料スラリーの調製は、テレフタル酸を主成分とするシカルボン酸成分とエチレングリコールを主成分とするジオール成分、及び必要に応じて用いられる共重合成分等とを、ジカルボン酸成分に対するジオール成分のモル比を、通常、1.0〜2.0として調製する。このモル比は1.05〜1.8とするのが好ましく、1.1〜1.6とするのが更に好ましい。
【0029】
スラリー移送工程、エステル化工程(a)
次いで、調製した原料スラリーを、1つの又は連続する複数のエステル化反応槽を備えたエステル化工程に移送し、常圧〜加圧下、加熱下で、エステル化反応させてポリエステル低分子量体であるオリゴマーとする。
【0030】
エステル化反応における反応条件としては、単一のエステル化反応槽の場合、通常、温度を240℃〜295℃、圧力を通常0kPaG〜400kPaGとし、攪拌下に、通常、1時間〜10時間の反応時間(滞留時間)とする。
また、複数のエステル化反応槽の場合は、第1段目のエステル化反応槽における反応温度を、通常240℃〜295℃、好ましくは245℃〜293℃、圧力を、通常5kPaG〜300kPaG、好ましくは10kPaG〜200kPaGとし、最終段における反応温度を、通常250℃〜295℃、好ましくは255℃〜293℃、圧力を、通常0kPaG〜150kPaG、好ましくは0kPaG〜130kPaGとする。ここでkPaGは大気圧に対する相対圧力をkPa単位で表したものである。滞留時間の合計は、通常、1〜10時間、好ましくは、1.2〜5時間とする。
【0031】
本発明において、エステル化反応生成物としてのオリゴマーのエステル化率は、上記に記載のとおりである。
【0032】
溶融重縮合工程(b)
引き続いて、得られたオリゴマーを、重縮合反応槽を備えた溶融重縮合工程に移送し、減圧下、加熱下で溶融重縮合反応させる。
【0033】
本発明においては、溶融重縮合で得るポリエステルプレポリマーの固有粘度が0.50dL/g以下と低いため、従来よく使用されている攪拌翼を備えた横型プラグフロー型第2段重縮合反応槽および第3段重縮合反応槽は必ずしも必要でなく溶融重縮合工程は単純化され設備コストも低減される。特に、本発明の製造方法は、溶融重縮合反応槽が1つの場合において、従来の製造方法と比較して、より単純化された設備で高分子量のポリエステルを製造できる。
【0034】
溶融重縮合における反応条件は、反応温度が、通常260〜290℃、好ましくは270℃〜280℃、圧力が、通常50kPaA〜0.1kPaA、好ましくは40kPaA〜0.1kPaAである。ここでkPaAは絶対圧力をkPa単位で表したものである。
【0035】
プレポリマー移送工程、造粒工程(c)
前記溶融重縮合により得られた樹脂は、通常、重縮合反応槽の底部に設けられた抜き出し口からストランド状に抜き出して、水冷しながら若しくは水冷後、カッターで切断してポリエステルプレポリマー粒子とする。あるいは、重縮合反応槽の底部に設けられた抜き出し口から水中に吐出して冷却しながら、吐出方向とほぼ平行方向の回転軸を有し、抜き出し口先端部に隣接設置されたカッターで切断してポリエステルプレポリマー粒子とする。または粉砕機で粉砕して所望の平均質量の粒子とすることもできる。プレポリマー粒子の平均質量は、通常0.2mg/粒〜50mg/粒であり、下限は好ましくは0.5mg/粒、より好ましくは1.0mg/粒である。また、上限は好ましくは30mg/粒、より好ましくは20mg/粒である。プレポリマー粒子の平均質量がこの範囲の場合、固相重縮合速度が大きく、また、気力輸送等の取り扱いが容易であるため、一層好ましい。
【0036】
結晶化工程
粒子化後のポリエステルプレポリマーを、通常、結晶化する。結晶化は、日本国特許第3073498号公報等に記載の公知の方法によって行うことができ、例えば、120℃〜180℃の不活性ガス気流中で0.5時間〜12時間流動化させることで行うことができる。結晶化を行うことにより、その後の熱処理工程におけるペレット同士の融着を防止できるという効果が得られる。
【0037】
熱処理
本発明では、粒子化後のポリエステルプレポリマーを、または、該プレポリマーを結晶化した後に、熱処理を行ってもよい。熱処理を行うことにより、プレポリマーが固相重縮合され、高分子量化されやすくなる。熱処理は、WO2007/026841号パンフレット等に記載の公知の方法によって行うことができ、例えば、移動床反応器においてポリエステルを上方から下方へ、加熱された不活性ガスを下方から上方へ、それぞれ連続的に流すことで行うことができる。
【0038】
固相重縮合工程(d)
本発明では、粒子化後のポリエステルプレポリマーを、必要に応じて、結晶化後及び/または熱処理した後に固相重縮合をする。固相重縮合反応は、温度の下限は、好ましくは200℃であり、より好ましくは205℃であり、さらに好ましくは208℃である。温度の上限は、好ましくは、当該ポリエステルの融点よりも5℃低い温度、より好ましくは融点よりも8℃低い温度、さらに好ましくは融点よりも10℃低い温度の不活性ガス雰囲気において実施する。ここで、ポリエステルの融点とは、当該ポリエステルを、示差走査熱量計を用いて、窒素気流下、0℃から、20℃/分の速度で300℃まで昇温した際のDSC曲線における、最も高温側の吸熱ピークの頂点に対応する温度のことである。また、不活性ガスとは、酸素濃度が、通常、0.1体積%以下、好ましくは0.05体積%以下であり、かつ、実質的にポリエステルと反応しない気体のことである。実質的にポリエステルと反応しない気体としては、窒素、ヘリウム、ネオン、アルゴン、キセノン、二酸化炭素等が例示でき、主に経済性の点から窒素が好ましく用いられる。
【0039】
固相重縮合温度が低いと固相重縮合速度が遅く、好ましくない。固相重縮合温度が高いと、固相重縮合時にポリエステル粒子が融着するため、好ましくない。特に、ポリエステルプレポリマー粒子の平均質量が0.1mg/粒以下の場合は、融着を回避するために流動床にて固相重縮合するのが好ましい。固相重縮合時間は、目標固有粘度に応じて設定すればよく、通常、1〜50時間程度である。固相重縮合後の平均質量は、通常、固相重縮合前のプレポリマー粒子の平均質量とほぼ一致する。
【0040】
本発明の製造方法により得られるポリエステルは、射出成形や押出成形によりプリフォームを成形後、延伸ブロー成形により、飲料包装等に用いられるボトルにすることができる。また、ダイレクトブロー成形により、ボトルにすることができる。
【0041】
また、押出成形や延伸成形によりフィルム、シートにして包装材料など各種用途に供することができる。また、押出・延伸成形により、繊維とすることができる。
【実施例】
【0042】
以下、実施例により本発明を更に具体的に説明するが、本発明はその要旨を超えない限り、以下の実施例に限定されるものではない。
なお、実施例及び比較例における物性の測定は、以下の方法により行った。
<固有粘度(以下、「IV」ともいう)>
試料約0.25gを、フェノール/1,1,2,2−テトラクロロエタン(質量比:1/1)の混合溶媒約25mLに、濃度が1.00×10-2kg/Lとなるように、非晶状態のポリエステルは110℃、30分で、固相重縮合後のポリエステルは140℃、30分でそれぞれ溶解させた後、30℃まで冷却し、全自動溶液粘度計(センテック社製、2CH型DJ504)にて、濃度が1.00×10-2kg/Lの試料溶液及び溶媒のみの落下秒数を測定し、下式により算出した。
IV(dL/g)=[(1+4KH・ηsp)0.5−1]/(200KH・C)
ここで、ηsp=η/η0−1であり、ηは試料溶液の落下秒数を表し、η0は溶媒の落下秒数を表し、Cはポリマー溶液濃度(kg/L)を表し、KHはハギンズの定数である。
本実施例では、KHは0.33を採用した。
【0043】
<ポリエステルプレポリマー粒子の平均質量>
精密天秤を用いて、ポリエステルプレポリマー粒子30粒の合計質量を0.1mgの桁まで測定し、測定値を30で除することにより、粒子1粒当たりの平均質量を算出した。
【0044】
<末端カルボキシル基濃度(以下、「AV」ともいう)>
試料を粉砕した後、熱風乾燥機にて140℃で15分間乾燥させ、デシケーター内で室温まで冷却した試料から、0.1gを精秤して試験管に採取し、ベンジルアルコール3mLを加えて、乾燥窒素ガスを吹き込みながら195℃、3分間で溶解させ、次いで、クロロホルム5mLを徐々に加えて室温まで冷却した。この溶液にフェノールレッド指示薬を1〜2滴加え、乾燥窒素ガスを吹き込みながら攪拌下に、0.1規定の水酸化ナトリウムのベンジルアルコール溶液で滴定し、黄色から赤色に変じた時点で終了とした。また、ブランクとして、ポリエステル試料を使用せずに同様の操作を実施し、これらの結果を用いて以下の式により末端カルボキシル基濃度を算出した。
AV(当量/トン)=(A−B)×0.1×f/W
ここで、Aは、試料を用いた場合の滴定に要した0.1規定の水酸化ナトリウムのベンジルアルコール溶液の量(μL)を表し、Bは、試料を加えないブランクでの滴定に要した0.1規定の水酸化ナトリウムのベンジルアルコール溶液の量(μL)を表し、Wは、ポリエステル試料の量(g)を表し、fは、0.1規定の水酸化ナトリウムのベンジルアルコール溶液の力価を表す。
なお、0.1規定の水酸化ナトリウムのベンジルアルコール溶液の力価(f)は、試験管にメタノール5mLを採取し、フェノールレッドのエタノール溶液を指示薬として1〜2滴加え、0.1規定の水酸化ナトリウムのベンジルアルコール溶液0.4mLで変色点まで滴定し、次いで、力価既知の0.1規定の塩酸を標準液として0.2mL採取して加え、再度、0.1規定の水酸化ナトリウムのベンジルアルコール溶液で変色点まで滴定し(以上の操作は、乾燥窒素ガスを吹き込みながら行った。)、以下の式により算出した。
力価(f)=0.1規定の塩酸の力価×0.1規定の塩酸の採取量(μL)/0.1規定の水酸化ナトリウムのベンジルアルコール溶液の滴定量(μL)
【0045】
<ポリエステルの総末端基濃度(以下、「TEV」ともいう)>
ポリエステルの総末端基濃度(TEV)は固有粘度(IV)から以下の式で計算して求めた。
IV(dL/g)から数平均分子量Mnを次式により計算し、
Mn=(IV×10000/7.55)^(1/0.685)
MnからTEV(当量/トン)を次式により計算する。
TEV=2×(1000000/Mn)
【0046】
<総末端基数に対する末端カルボキシル基数の割合(R)>
R=AV/TEVで求めた。
【0047】
<実施例1>
エステル化及び/又は重縮合反応に用いたエステル化触媒の調製方法を以下に示す。
<タングステン触媒(触媒)>
メタタングステン酸アンモニウム水溶液(商品名:MW−2、日本無機化学工業社製、三酸化タングステン(WO3)として濃度50質量%)をエチレングリコールで希釈して、タングステン原子濃度が0.2質量%の触媒液(タングステン触媒)を調製した。
【0048】
<チタン触媒>
(Ti/Mg/P=1/1/1.2のモル比の触媒液の合成)
300mL摺り栓付きの三角フラスコ中に、エタノール(特級、純度99.6%以上)を50g入れ、次に酢酸マグネシウム・4水和物7.51gを添加し、スターラーで20分間攪拌して、ほぼ均一に溶解させた。次に、エチルアシッドフォスフェート(商品名:JP−502、城北化学社製、モノエステル体とジエステル体の質量比0.82:1)を2.95g入れて5分間攪拌し、更にジブチルホスフェート(商品名:DBP、城北化学工業社製)を4.41g入れて5分間攪拌した。続いてテトラ−n−ブチルチタネート11.91gを添加し、10分間攪拌することで均一溶液を得た。得られた混合液にエチレングリコール36.1gを加え、次に300mLのナスフラスコに移してエバポレーターにセットし、60℃、11kPaAの条件でエタノール及び低沸物を留去し、47.9gの均一透明な触媒液(チタン触媒)を調製した。チタン触媒液中の各金属成分の割合はTi/Mg/P=1/1/1.2(モル比)であり、チタン濃度3.5質量%であった。
【0049】
<ポリエステルの製造>
以下にポリエステルの製造方法を示す。
(ポリエステルプレポリマーの製造およびプレポリマーの粒子化)
撹拌機、エチレングリコール仕込み配管及びテレフタル酸仕込み配管を具備するスラリー調製槽;スラリーやエステル化反応物を各エステル化反応槽へ移送する各配管;撹拌機、分離塔、原料受入れ口、触媒仕込み配管、エチレングリコール仕込み配管および反応物移送配管を具備する完全混合型第一エステル化反応槽及び第二エステル化反応槽;エステル化反応物(オリゴマー)を溶融重縮合反応槽へ移送する配管;撹拌機、分離塔、オリゴマー受入れ口及びプレポリマー抜き出し口を具備する完全混合型第一溶融重縮合反応槽;撹拌機、分離塔、プレポリマー受入れ口及びポリマー抜き出し口を具備するプラグフロー型第二溶融重縮合反応槽及び第三溶融重縮合反応槽;プレポリマーを抜き出し口よりギヤポンプを介してダイプレートからストランド状に取り出し水冷下ストランドカットする粒子化装置(ストランドカッターはリーター・オートマチック社製、ペレタイザー、P−USG100)を備えたポリエステルプレポリマー連続製造装置を用いた。
【0050】
前記のポリエステルプレポリマー連続製造装置を用いて、テレフタル酸とエチレングリコールとをエステル化反応し、更に溶融重縮合反応することにより得られた溶融状態のプレポリマーをダイプレートからストランド状に取り出し切断することで、ポリエステルプレポリマー粒子を製造した。具体的には以下の通りに行った。
スラリー調製槽にて、テレフタル酸/イソフタル酸/エチレングリコール(モル比で、0.985:0.015:1.5)スラリーを調製した。また、ビス−(β−ヒドロキシエチル)テレフタレート400質量部をエステル化第一槽に仕込み窒素雰囲気下で溶融し、温度262℃、圧力92kPaGに保たれた中へ、前記のスラリー調製槽で調製されたスラリーを135質量部/時間で、ポリエステルとしての平均滞留時間が4.6時間になるように連続的に仕込み、分離塔から生成する水を留去しながらエステル化反応を行いつつ、反応液を連続的にエステル化第二反応槽へ移送した。
第二エステル化反応槽では、温度260℃、圧力3.5kPaGに設定し、上記で調製したタングステン触媒を、得られるポリエステルプレポリマーに対して、タングステンとして40質量ppmとなる量を連続的に添加し、また、エチレングリコールを4質量部/時間の速度で連続的に添加しながら、平均滞留時間1.9時間でエステル化反応を行った。定常状態における第二エステル化反応槽出口における反応物サンプルのエステル化率は95.6%であった。
続いて移送配管を通じ完全混合型第一溶融重縮合反応槽へ連続的に移送すると同時に、この移送配管に、上記にて調製したチタン触媒を、得られるポリエステルプレポリマーに対して、チタン、マグネシウム、リンが、それぞれ、4質量ppm、2質量ppm、3.1質量ppmとなるように、連続的に添加した。
第一溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間1.5時間にて反応を行い、移送配管を通じ第二溶融重縮合反応槽へ連続的に移送した。
第二溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間1.0時間にて溶融重縮合反応を行い、移送配管を通じ第三溶融重縮合反応槽へ移送した。
第三溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間0.5時間にて溶融重縮合反応を行った。
【0051】
上記で得られた溶融ポリエステルプレポリマーを、そのまま、ギヤポンプ及び抜き出し配管を通じてダイヘッドへ導き、ダイホールからストランド状に取り出し、水冷後、リーター・オートマチック社製、ペレタイザー、P−USG100により造粒した。造粒方法はストランドカット法であり、具体的には、ストランド状ポリエステルプレポリマーを水と接触させて冷却させながら、水と共にカッター方向に搬送し、カッター前に設置された一対の引取ロールにて挟むことで引き取り、カッターに供給し、固定刃と回転刃とを有するカッターにて切断することにより、ポリエステルプレポリマー粒子を得た。
得られたポリエステルプレポリマー粒子は、長さ1.25mm、幅1.2mm、厚さ0.9mmのほぼ直方体の両端に半円柱を付けた形状に近い楕円柱状で、ほぼ均一な形状であった。粒子の平均質量は1.8mg/粒であり、固有粘度(IV)は0.383dL/g、R0は0.38であった。
結果を表1に示す。表1中、添加EG量は、添加したエチレングリコール量(質量部/h)を示している。また、「h」は、時間を表す(表2以下についても同じ)。さらに、Wはポリエステル中のタングステン原子の量(質量ppm)を表し、Tiはポリエステル中のチタン原子の量(質量ppm)を表す。
【0052】
(固相重縮合)
上記のポリエステルプレポリマー粒子2.0gをアルミ製トレイ(縦160mm×横100mm、深さ:30mm)にチップ同士が重ならないように並べ、内温60℃に設定されたイナートオーブン(ヤマト科学社製、I/O DN4101)中の中央部に設置した。30NL/分の窒素流通下(ここで、NLとは0℃1気圧における体積(L)のことである)で、60℃から160℃まで30分で昇温させ、160℃で2時間乾燥、結晶化を行った。その後、30分かけて230℃まで昇温し、230℃で、それぞれ、8時間の固相重縮合及び16時間の固相重縮合を行い、ポリエステルを得た。固相重縮合終了後、30分かけて60℃まで降温した後、チップを回収した。固相重縮合時間が8時間の試料及び16時間の試料のIV、R1、ΔIV上昇速度(ΔIV/h)を、それぞれ、求めた。
溶融重縮合後、固相重縮合を8時間行った試料および固相重縮合を16時間行った試料についての結果を表2に示す。
【0053】
<比較例1>
実施例1において、第二エステル化反応槽の温度を263℃、平均滞留時間を2.4時間に変更した以外は実施例1と同様に行った。結果を表1及び表2に示す。なお、粒子の平均質量は1.8mg/粒であった。
【0054】
<実施例2>
実施例1において、第二エステル化反応槽に連続添加するエチレングリコール量を3.5質量部/時間とした以外は実施例1と同様に行った。結果を表1及び表2に示す。なお、粒子の平均質量は1.8mg/粒で、ほぼ均一な形状であった。
【0055】
<比較例2>
実施例1において、第二エステル化反応槽に連続添加するエチレングリコール量を2質量部/時間とした以外は実施例1と同様に行った。結果を表1及び表2に示す。なお、粒子の平均質量は1.8mg/粒であった。
【0056】
<実施例3>
実施例1とは触媒の添加量と添加位置を、また、第二及び第三重縮合反応槽を不使用とするなどを変更し、具体的には以下のように行った。
【0057】
(ポリエステルプレポリマーの製造およびプレポリマーの粒子化)
スラリー調製槽にて、テレフタル酸/イソフタル酸/エチレングリコール(モル比で、0.985:0.015:1.5)スラリーを調製した。また、ビス−(β−ヒドロキシエチル)テレフタレート400質量部をエステル化第一槽に仕込み窒素雰囲気下で溶融し、温度262℃、圧力92kPaGに保たれた中へ、前記のスラリー調製槽で調製されたスラリーを135質量部/時間で、ポリエステルとしての平均滞留時間が4.8時間になるように連続的に仕込み、分離塔から生成する水を留去しながらエステル化反応を行いつつ、反応液を連続的に第二エステル化反応槽へ移送した。第二エステル化反応槽にエチレングリコールを2.8質量部/時間で連続的に添加しながら、温度260℃、圧力3.7kPaG下、平均滞留時間2.4時間でエステル化反応を行った。定常状態における第二エステル化反応槽出口における反応物サンプルのエステル化率は92.6%であった。
続いて移送配管を通じ完全混合型第一溶融重縮合反応槽へ連続的に移送すると同時に、その移送配管のエステル化反応物に、実施例1にて調製したチタン触媒を、得られるポリエステルプレポリマーに対して、チタン、マグネシウム、リンとしてそれぞれ2質量ppm、1質量ppm、1.6質量ppmとなる量を連続的に添加した。更にその下流に、実施例1にて調製したタングステン触媒を、得られるポリエステルプレポリマーに対して、タングステンとして60質量ppmとなるように連続的に添加した。
第一溶融重縮合反応槽では、温度270℃、圧力3kPaA下、平均滞留時間1.6時間にて溶融重縮合反応を行った。このようにして得られた溶融ポリエステルプレポリマーを、ギヤポンプ及び抜き出し配管を通じてダイヘッドへ導き、実施例1のプレポリマー造粒方法と同様の方法により造粒した。その結果、長さ1.25mm、幅1.2mm、厚さ0.9mmのほぼ直方体の両端に半円柱を付けた形状に近い楕円柱状で、ほぼ均一な形状のPETプレポリマー粒子を得た。粒子の平均質量は1.8mg/粒であった。
【0058】
得られたポリエステルプレポリマー粒子を実施例1と同様に固相重縮合を行った。但し、固相重縮合は、16時間とした。結果を表1および表3に示す。
【0059】
<実施例4>、<比較例3>
実施例3において第二エステル化反応槽の圧力、第一重縮合槽の平均滞留時間をそれぞれ表1に示すように変更した以外は実施例3と同様に行った。結果を表1および表3に示す。なお、実施例4において粒子の平均質量は1.8mg/粒で、ほぼ均一な形状であった。
【0060】
【表1】
【0061】
【表2】
【0062】
【表3】
表2の比較例1、2と実施例1、2との比較より、また、表3の実施例3、4と、比較例3との比較により、R0値等が本発明の範囲を満たす場合は満たさない場合より、ΔIV/hが大きくなることが認められた。
【0063】
<実施例5>
(結晶化工程)
実施例4で得たプレポリマー粒子30gを底面が130mm×170mmの角形で、深さが30mmのステンレス製バットに広げて置き、内部のガス温度が180℃のイナートオーブン(タバイエスペック社製、IPHH−201M型)に入れ、イナートオーブンの内部に流通させる窒素の流量を50NL/分、温度を180℃の窒素流通下として、180℃で1時間の結晶化処理を行った。
【0064】
(熱処理装置)
実施例4で得たプレポリマー粒子を結晶化処理した試料を、図1に示すガラス製熱処理装置を用いて熱処理を行った。
以下、該熱処理装置について説明する。
図1に示す熱処理装置において、試料は、試料充填部の内径が45mmのガラス製の熱処理管(1)に充填されている。熱処理管(1)には、ガス流量計(2)、窒素導入管(3)、窒素予熱管(4)を経由して、オイルバス(5)に充填されたオイルにより加熱された窒素が導入される。導入された窒素は、熱処理管(1)下部にある分散板(6)により分散され、熱処理管(1)内部で略均一な線速度を有する上昇流となって、試料層(7)を通過する。試料層(7)を通過した窒素は、熱処理管(1)上部にあるフィルター(8)を経由して、ガスパージ口(9)から熱処理管(1)の外部に排出される。熱処理管(1)は枝管(10)を有しており、その上部にある開口部(通常はガラス栓にて閉止してある)から試料の投入や試料の採取が可能である。また、熱処理管(1)内部の試料の温度は、熱電対(11)を備えた温度計(12)で測定できる。本実施例の範囲の温度、空塔線速度においては、熱処理管(1)の内部温度は、オイルバス中のオイル温度よりも2℃低い温度となるため、目標とする固相重縮合温度に対して、オイルの温度は2℃高い温度に調節した。
【0065】
(第一段固相重縮合工程)
熱処理管(1)の枝管(10)の開口部より、上記結晶化処理後のプレポリマー粒子30gを仕込み、窒素を流通して内部を窒素置換した。その後熱処理管(1)内の窒素の空塔線速度(ここで「空塔線速度」とは、試料層の空塔線速度を意味する(以下、同じ))が210℃で0.30m/秒となるように窒素の流量をガス流量計(2)で設定し、オイルの温度が212℃に調節された第一のオイルバス(5)に熱処理装置を浸漬した。この時点を210℃での第1段固相重縮合の開始とする。2時間後に枝管(10)の開口部より用試料約0.3gを採取した。
【0066】
(昇温工程)
試料採取後、窒素の空塔線速度が235℃で1.0m/秒となるように窒素の流量を変更し、オイルの温度が237℃に調節された第二のオイルバス(5)に熱処理装置を移した。この時点を235℃での昇温工程の開始とした。試料の温度が235℃に到達するまでに、10分を要した。昇温工程の開始から10分後に枝管(10)の開口部より試料を採取した。
<第2段固相重縮合工程>
試料採取後、窒素の空塔線速度が230℃で0.30m/秒となるように窒素の流量を変更し、オイルの温度が232℃に調節された第三のオイルバス(5)に熱処理装置を移した。この時点を230℃での第2段固相重縮合開始とした。第2段固相重縮合開始点から8時間の時点で試料を採取した。第2段固相重縮合の8時間の時点に採取した試料の固有粘度(IV)、R1、ΔIV/hをそれぞれ求めた。結果を表4に示す。
【0067】
<比較例4>
実施例4で得たプレポリマー粒子の代わりに比較例3で得たプレポリマー粒子を用いた以外は実施例5と同様に行った。結果を表4に示す。この例の場合、R0が0.60よりも大きいため、実施例5に比較してΔIV/hが小さい結果だった。
【0068】
【表4】
【0069】
<実施例6>
(ポリエステルの製造)
以下にポリエステルの製造方法を示す。
【0070】
(原料オリゴマーの製造)
テレフタル酸ジメチル2012kg(10.4×103モル)とエチレングリコール1286(20.7×103モル)とをエステル化反応槽に供給して溶解後、エチレングリコールに溶解させた酢酸カルシウムを、カルシウム原子として0.20kg(エステル交換反応により得られる生成物に対して100ppm)となるように添加し、220℃に保持しつつ、生成するメタノールを留出させながらエステル交換反応を行った。エステル交換反応が終了した後、このエステル化反応槽に、テレフタル酸1721kg(10.4×103モル)とエチレングリコール772kg(12.4×103モル)とをスラリー調製槽で攪拌・混合して得られたスラリーを3時間かけて連続的に移送し、常圧下、250℃でエステル化反応を行い、移送開始から4時間反応を行った後に、反応液の50%を系外へ抜き出した。
このエステル化反応槽において、前記と同様にして得られたテレフタル酸とエチレングリコールからなるスラリーを追加してエステル化反応を行い、反応液の50%を抜き出す工程を、計10回繰り返して行い、エステル化反応中の酢酸カルシウムの濃度を0.5質量ppm以下とした。
このようにして、実質的にエステル交換触媒成分を含有しないテレフタル酸とエチレングリコールからなるエステル化反応生成物を製造した。このエステル化反応生成物を、エステル化反応槽から抜き出し、大気下で冷却・固化することにより、以下の実施例で使用する原料オリゴマーを得た。得られた原料オリゴマーのエステル化反応率は89%であった。
【0071】
(ポリエステルプレポリマーの製造)
前記原料オリゴマー104gをトルクメータ付属攪拌装置付き重縮合反応器に移して、系内を窒素で置換した後、常圧下オイルバス(260℃一定)中でオリゴマーの溶解を行った。以下、オリゴマー溶解開始時間を0時間として時間を表記する。
30分後に50rpmで攪拌を開始し、60分後にオリゴマーが完全に溶解していることを確認後、タングステン触媒(メタタングステン酸アンモニウム水溶液(商品名:MW−2、日本無機化学工業社製、三酸化タングステン(WO3)として濃度50質量%))を、得られるポリエステルプレポリマーに対してタングステンとして40質量ppmとなるようにエチレングリコールに希釈して2ml添加した。65分後に減圧を開始し、125分後に0.27kPaAまで減圧した。減圧操作は圧力の対数値が時間に逆比例するように行った。重縮合温度は、65分から145分の間に260℃から280℃まで一定速度で昇温した。到達固有粘度が0.40〜0.50dL/gの範囲に入るように、表5に示す重縮合時間、溶融重縮合反応を行った。なお、重縮合時間は減圧開始から常圧に戻すまでの時間とした。
重縮合終了後、攪拌を停止し、窒素にて常圧に戻し、重縮合反応器をオイルバスから取り出した。重縮合反応器をオイルバスから取り出した後、速やかに該反応器の抜き出し口を開け、窒素で系内を微加圧にすることでポリエステルを抜き出し、水冷・固化させてストランド状のポリエステルを得た。得られたポリエステルは粒子の平均質量が1mg/粒となるように裁断された。得られたポリエステルの固有粘度は0.401dL/g、R0は0.33であった。
【0072】
(固相重縮合)
上記のポリエステルプレポリマー粒子2.0gをアルミ製トレイ(縦160mm×横100mm、深さ:30mm)にチップ同士が重ならないように並べ、内温60℃に設定されたイナートオーブン(ヤマト科学社製、I/O DN4101)中の中央部に設置した。30NL/分の窒素流通下(ここで、NLとは0℃1気圧における体積(L)のことである)で、60℃から160℃まで30分で昇温させ、160℃で2時間乾燥、結晶化を行った。その後、30分かけて230℃まで昇温し、230℃で8時間固相重縮合を行い、ポリエステルを得た。固相重縮合終了後、30分かけて60℃まで降温した後、チップを回収した。固相重縮合時間8時間の試料のIV、R1、ΔIV/hを求めた。結果を表5に示す。
【0073】
<実施例7>
実施例6とは溶融重縮合における触媒の種類、添加操作を変更し、具体的には以下のように行った。
【0074】
(溶融重縮合)
実施例6と同様に製造した原料オリゴマー104gをトルクメータ付属攪拌装置付き重縮合反応器に移して、系内を窒素で置換した後、常圧下オイルバス(260℃一定)中でオリゴマーの溶解を行った。以下、オリゴマー溶解開始時間を0時間として時間を表記する。
30分後に50rpmで攪拌を開始し、60分後にオリゴマーが完全に溶解していることを確認後、チタン触媒(テトラ−n−ブチルチタネート(TBT))を、得られるポリエステルプレポリマーに対してチタンとして4質量ppmとなるようにエチレングリコールに希釈して1mL添加した。次いで65分後にタングステン触媒(メタタングステン酸アンモニウム水溶液(商品名:MW−2、日本無機化学工業社製、三酸化タングステン(WO3)として濃度50質量%))を、得られるポリエステルプレポリマーに対してタングステンとして40質量ppmとなるようにエチレングリコールに希釈して1mL添加した。70分後に減圧を開始し、130分後に0.27kPaAまで減圧した。減圧操作は圧力の対数値が時間に逆比例するように行った。重縮合温度は、70分から150分の間に260℃から280℃まで一定速度で昇温した。到達固有粘度が0.40〜0.50dL/gの範囲に入るように、表5に示す重縮合時間、溶融重縮合反応を行った。なお、重縮合時間は減圧開始から常圧に戻すまでの時間とした。
重縮合終了後、攪拌を停止し、窒素にて常圧に戻し、重縮合反応器をオイルバスから取り出した。重縮合反応器をオイルバスから取り出した後、速やかに該反応器の抜き出し口を開け、窒素で系内を微加圧にすることでポリエステルを抜き出し、水冷・固化させてストランド状のポリエステルを得た。得られたポリエステルは粒子の平均質量が1mg/粒となるように裁断された。得られたポリエステルの固有粘度は0.446dL/g、R0は0.42であった。
得られたポリエステルプレポリマー粒子を実施例6と同様に固相重縮合を行った。結果を表5に示す。
【0075】
<実施例8>
実施例6において、タングステン触媒をチタン触媒(テトラ−n−ブチルチタネート(TBT))に代え、得られるポリエステルプレポリマーに対してチタンとして4質量ppmとなるようにエチレングリコールに希釈して2mL添加した他は、同様の方法で溶融重縮合を行い、ポリエステルを得た。得られたポリエステルの固有粘度は0.439dL/g、R0は0.30であった。得られたポリエステルプレポリマー粒子を実施例6と同様に固相重縮合を行った。結果を表5に示す。
【0076】
<実施例9>
(触媒の調製)
実施例6とは溶融重縮合における触媒の種類、添加操作を変更し、具体的には以下のように行った。
(溶融重縮合)
実施例6と同様に製造した原料オリゴマー104gをトルクメータ付属攪拌装置付き重縮合反応器に移して、系内を窒素で置換した後、常圧下オイルバス(260℃一定)中でオリゴマーの溶解を行った。以下、オリゴマー溶解開始時間を0時間として時間を表記する。
30分後に50rpmで攪拌を開始し、60分後にオリゴマーが完全に溶解していることを確認後、エチルアシッドフォスフェート(商品名:JP−502、城北化学社製、モノエステル体とジエステル体の重量比0.82:1、略称:EAP)を、得られるポリエステルプレポリマーに対してリンとして13質量ppmとなるようにエチレングリコールに希釈して0.7mL添加した。次いで65分後にアンチモン触媒(三酸化アンチモン(略称:SbO)1.8質量%エチレングリコール溶液)を、得られるポリエステルプレポリマーに対してアンチモンとして220質量ppmとなるように1.3mL添加した。70分後に減圧を開始し、130分後に0.27kPaAまで減圧した。減圧操作は圧力の対数値が時間に逆比例するように行った。重縮合温度は、70分から150分の間に260℃から280℃まで一定速度で昇温した。到達固有粘度が0.40〜0.50dL/gの範囲に入るように、表5に示す重縮合時間、溶融重縮合反応を行った。なお、重縮合時間は減圧開始から常圧に戻すまでの時間とした。
重縮合終了後、攪拌を停止し、窒素にて常圧に戻し、重縮合反応器をオイルバスから取り出した。重縮合反応器をオイルバスから取り出した後、速やかに該反応器の抜き出し口を開け、窒素で系内を微加圧にすることでポリエステルを抜き出し、水冷・固化させてストランド状のポリエステルを得た。得られたポリエステルは粒子の平均質量が1mg/粒となるように裁断された。得られたポリエステルの固有粘度は0.447dL/g、R0は0.34であった。ポリエステル中のアンチモン原子含有量は、215質量ppmであった。
得られたポリエステルプレポリマー粒子を実施例6と同様に固相重縮合を行った。結果を表5に示す。
【0077】
【表5】
【0078】
表5の実施例6〜9の結果から、タングステン触媒とチタン触媒を併用する場合に最も良好な触媒活性が得られ、より短い時間で高分子量のポリエステルを製造できることが分かる。
【産業上の利用可能性】
【0079】
本発明により、高分子量で、形状が均一かつアンチモン原子の含有量が少ないポリエステルを、短い重縮合時間で製造することができる。更に、ポリエステルプレポリマーの固有粘度が低いので、複雑で高価な溶融重縮合反応装置を用いる必要が無く効率的な製造方法を提供できる。
【図面の簡単な説明】
【0080】
【図1】本発明で用いるガラス製熱処理装置の一構成例を示す模式図である。
【符号の説明】
【0081】
1: 熱処理管
2: ガス流量計
3: 窒素導入管
4: 窒素予熱管
5: オイルバス
6: 分散板
7: 試料層
8: フィルター
9: ガスパージ口
10: 枝管
11: 熱電対
12: 温度計
【特許請求の範囲】
【請求項1】
芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程に少なくとも1種のエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25dL/g〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
【請求項2】
エステル化触媒として、タングステン触媒を用いることを特徴とする請求項1に記載のポリエステルの製造方法。
【請求項3】
エステル化触媒として、チタン触媒を用いることを特徴とする請求項1または2に記載のポリエステルの製造方法。
【請求項4】
R0及びR1が下記(式2)を満たすことを特徴とする請求項1〜3のいずれか1項に記載のポリエステルの製造方法。
(式2) −0.2≦ R0 − R1
【請求項5】
エステル化触媒として、タングステン触媒及びチタン触媒を該順に添加することを特徴とする請求項1〜4のいずれか1項に記載のポリエステルの製造方法。
【請求項6】
エステル化工程(a)に先立つ原料調製工程、及び、エステル化工程(a)と溶融重縮合工程(b)の間にオリゴマー移送工程を有し、タングステン触媒をエステル化工程(a)及び/又は原料調製工程において添加し、更にチタン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする請求項5に記載のポリエステルの製造方法。
【請求項7】
エステル化触媒として、チタン触媒及びタングステン触媒を該順に添加することを特徴とする請求項1〜4のいずれか1項に記載のポリエステルの製造方法。
【請求項8】
エステル化工程(a)に先立つ原料調製工程を有し、チタン触媒をエステル化工程(a)及び/又は原料調製工程に添加し、更にタングステン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする請求項7に記載のポリエステルの製造方法。
【請求項9】
プレポリマー粒子の平均質量が1.0mg/粒〜50mg/粒であることを特徴とする請求項1〜8のいずれか1項に記載のポリエステルの製造方法。
【請求項10】
ポリエステル中のアンチモン原子の含有量が10質量ppm以下であることを特徴とする請求項1〜9のいずれか1項に記載のポリエステルの製造方法。
【請求項1】
芳香族ジカルボン酸を主成分とするジカルボン酸成分と、脂肪族ジオールを主成分とするジオール成分とを原料とし、エステル化触媒を用いてエステル化反応によりオリゴマーを得るエステル化工程(a)、得られたオリゴマーを溶融重縮合反応によりプレポリマーを得る溶融重縮合工程(b)、得られたプレポリマーをプレポリマー粒子とする造粒工程(c)、および得られたプレポリマー粒子を固相重縮合反応によりポリエステルを得る固相重縮合工程(d)、とを含む連続的なポリエステルの製造方法であって、以下の(1)〜(4)を満足することを特徴とするポリエステルの製造方法。
(1)エステル化工程(a)において得られるエステル化反応生成物のエステル化率が86%〜96%。
(2)溶融重縮合工程(b)及び/又はそれ以前の工程に少なくとも1種のエステル化触媒を添加する。
(3)造粒工程(c)において得られるプレポリマー粒子の総末端基数に対する末端カルボキシル基数の割合R0が0.20〜0.60であり、かつ、固有粘度が0.25dL/g〜0.50dL/g。
(4)固相重縮合工程(d)において得られるポリエステルの総末端基数に対する末端カルボキシル基数の割合R1と前記R0とが下記(式1)を満たし、かつ、固有粘度が0.7dL/g〜1.5dL/g。
(式1) R0 − R1 ≦ 0.1
【請求項2】
エステル化触媒として、タングステン触媒を用いることを特徴とする請求項1に記載のポリエステルの製造方法。
【請求項3】
エステル化触媒として、チタン触媒を用いることを特徴とする請求項1または2に記載のポリエステルの製造方法。
【請求項4】
R0及びR1が下記(式2)を満たすことを特徴とする請求項1〜3のいずれか1項に記載のポリエステルの製造方法。
(式2) −0.2≦ R0 − R1
【請求項5】
エステル化触媒として、タングステン触媒及びチタン触媒を該順に添加することを特徴とする請求項1〜4のいずれか1項に記載のポリエステルの製造方法。
【請求項6】
エステル化工程(a)に先立つ原料調製工程、及び、エステル化工程(a)と溶融重縮合工程(b)の間にオリゴマー移送工程を有し、タングステン触媒をエステル化工程(a)及び/又は原料調製工程において添加し、更にチタン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする請求項5に記載のポリエステルの製造方法。
【請求項7】
エステル化触媒として、チタン触媒及びタングステン触媒を該順に添加することを特徴とする請求項1〜4のいずれか1項に記載のポリエステルの製造方法。
【請求項8】
エステル化工程(a)に先立つ原料調製工程を有し、チタン触媒をエステル化工程(a)及び/又は原料調製工程に添加し、更にタングステン触媒を溶融重縮合工程(b)及び/又はそれ以前の工程に添加することを特徴とする請求項7に記載のポリエステルの製造方法。
【請求項9】
プレポリマー粒子の平均質量が1.0mg/粒〜50mg/粒であることを特徴とする請求項1〜8のいずれか1項に記載のポリエステルの製造方法。
【請求項10】
ポリエステル中のアンチモン原子の含有量が10質量ppm以下であることを特徴とする請求項1〜9のいずれか1項に記載のポリエステルの製造方法。
【図1】

【公開番号】特開2009−114234(P2009−114234A)
【公開日】平成21年5月28日(2009.5.28)
【国際特許分類】
【出願番号】特願2007−285564(P2007−285564)
【出願日】平成19年11月1日(2007.11.1)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成21年5月28日(2009.5.28)
【国際特許分類】
【出願日】平成19年11月1日(2007.11.1)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]