
ポリエステルの製造方法
【課題】環状2量体含有量が低減され、繊維、フィルム、シート、その他の成形体に成形された時に、優れた熱安定性を有し、環状2量体に由来する表面析出物が実用上問題とならず、後加工や用途分野において環状2量体による障害が少なく、かつ経済的なポリエステルの製造方法を提供する。
【解決手段】脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕を0.15以上とするポリエステルの製造方法。
【解決手段】脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕を0.15以上とするポリエステルの製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリエステルの製造方法に関する。詳しくは、ポリエステル中の環状2量体含有量の低減された、ポリエステルの製造方法に関する。
【背景技術】
【0002】
ポリブチレンサクシネートに代表される脂肪族ポリエステルは、その原料を植物資源由来のものに求めることができること、良好な物性および生分解性を有することなどから、農業資材、土木資材、植生資材、包装材等の製品に加工され、広範に利用されつつある。
【0003】
しかしながら、上記脂肪族ポリエステルの成形品は、成形後一定期間放置すると、その表面に曇り(ブリードアウト、白化現象と同義。)が生じて表面光沢が消失するという問題があった。こうした問題は、脂肪族ポリエステルの製造時、ポリエステルの生成と同時に生成される環状2量体が、成形後、一定期間経過した後に成形品の表面に白色物として析出することによるものである。このため、脂肪族ポリエステルから環状2量体を除去する方法が研究されている。
【0004】
例えば、特許文献1には、数平均分子量10,000以上の脂肪族ポリエステルの粉末、ペレット、成形品を、脂肪族ケトン、環状脂肪族エーテル及び脂肪族モノエステル等の有機溶媒を用いて脂肪族ポリエステルの融点よりも低い温度で洗浄して環状2量体(以後、CDと表すことがある)を含むオリゴマーを抽出除去する方法が記載されている。一方、特許文献2には、脂肪族ポリエステル製造において、重縮合反応時に反応触媒の3〜3000倍のリン化合物を添加することによるCD低減方法が記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2002−3606号公報。
【特許文献2】国際公開第WO2006/001084号パンフレット。
【発明の概要】
【発明が解決しようとする課題】
【0006】
特許文献1に記載の技術では、大量の有機溶媒を用いるため、その処理や回収にエネルギーや特別な装置を要するという問題があり、一方、特許文献2において具体的に開示されている技術では、触媒に比べて大量の(28倍、280倍)リン化合物を2mmHg以下の高真空反応の途中で、反応系を常圧に戻して添加するなど工業的生産方法としては必ずしも適当ではなかった。
本発明は、処理や回収にエネルギーや特別な装置を要することなく、工業的に適した効率的な生産方法を用いて環状2量体含有量を低減し、繊維、フィルム、シート、その他の成形体に成形された時に、優れた熱安定性を有し、環状2量体に由来する表面析出物が実用上問題とならず、後加工や用途分野において環状2量体による障害が少なく、かつ経済的なポリエステルの製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは上記課題を解決すべく検討を行い、重縮合反応途中の反応物の末端カルボキシル基量が特定の範囲にあるとき、得られるポリエステル中のCD含有量が低減されることを見出し本発明に到達した。即ち本発明の要旨は、脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕が下記(1)式を満足するポリエステルの製造方法に存する。
0.15≦R (1)
【発明の効果】
【0008】
本発明により、CD抽出などの複雑な工程を経なくてもCD含有量の低減されたポリエステルを得ることができる。
【図面の簡単な説明】
【0009】
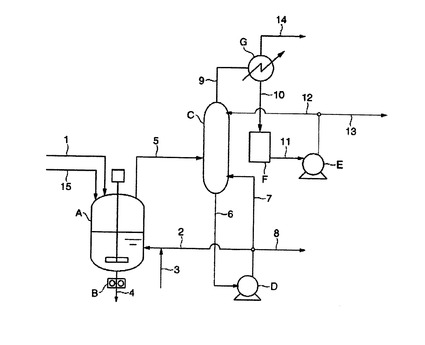
【図1】本発明で採用するエステル化工程の一例の説明図である。
【図2】本発明で採用する重縮合工程の一例の説明図である。
【発明を実施するための形態】
【0010】
本発明は、脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕が0.15以上であるポリエステルの製造方法である。
【0011】
重縮合反応はエステル化反応物分子のOH末端基とCOOH末端基との脱水エステル化反応、OH末端基同士の脱ジオール反応により進み、逐次高分子化が行われる。一方、一部分は高分子化しないで環状2量体(CD)となる。重縮合反応後期に反応物の、総末端基量に対する末端カルボキシル基量の比〔R〕が特定の範囲(0.15≦R)にあるとCDは低減される。CDの生成は末端OH基量に依存するため、末端OH基量の割合を減少させる、つまり総末端基量に対する末端カルボキシル基量の割合〔R〕を増加させることで生成反応を抑制できると考えられる。
【0012】
本発明のポリエステルの製造は、脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として行う。本発明において、脂肪族ジカルボン酸及び/又はそのアルキルエステルを主原料とすることとは、使用されるジカルボン酸及び/又はそのアルキルエステルにおいて、脂肪族ジカルボン酸及び/又はそのアルキルエステルが該成分中最大量であることを意味し、好ましくは、全ジカルボン酸及び/又はそのアルキルエステルの50モル%以上が脂肪族ジカルボン酸及び/又はそのアルキルエステルであることを意味する。同様に、本発明において、脂肪族ジオールを主原料とすることとは、使用されるジオールにおいて、脂肪族ジオールが該成分中最大量であることを意味し、好ましくは、全ジオールの50モル%以上が脂肪族ジオールであることを意味する。
【0013】
<ジカルボン酸>
脂肪族ジカルボン酸としては、1, 2−シクロヘキサンジカルボン酸、1, 3−シクロヘキサンジカルボン酸、1, 4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、マロン酸、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸などの脂肪族ジカルボン酸などを挙げることができ、機械的物性や用途の広さ、原料の入手容易さ等の観点からは、脂肪族ジカルボン酸の中ではコハク酸、アジピン酸が好ましい。
【0014】
その他のジカルボン酸としては、テレフタル酸、イソフタル酸、4, 4' −ジフェニルジカルボン酸、4, 4' −ジフェニルエーテルジカルボン酸、4, 4' −ベンゾフェノンジカルボン酸、4, 4' −ジフェノキシエタンジカルボン酸、4, 4' −ジフェニルスルホンジカルボン酸、2, 6−ナフタレンジカルボン酸などの芳香族ジカルボン酸があげられる
【0015】
これらジカルボン酸成分は、ジカルボン酸として、又はジカルボン酸無水物として、又はジカルボン酸のアルキルエステル、好ましくはジアルキルエステルとして反応に供することができ、ジカルボン酸とジカルボン酸アルキルエステルの混合物としてもよい。ジカルボン酸アルキルエステルのアルキル基に特に制限はないが、アルキル基が長いとエステル交換反応時に生成するアルキルアルコールの沸点の上昇を招き反応液中から揮発せず、結果的に末端停止剤として働き重合を阻害するため、炭素数4以下のアルキル基が好ましく、中でもメチル基が好適である。
【0016】
<ジオール>
脂肪族ジオールとしては、エチレングリコール、ジエチレングリコール、ポリエチレングリコール、1,2−プロパンジオール、1,3−プロパンジオール、1,4- ブタンジオール、ポリプロピレングリコール、ポリテトラメチレングリコール、ジブチレングリコール、1, 5−ペンタンジオール、ネオペンチルグリコール、1, 6−ヘキサンジオール、1, 8−オクタンジオールなどの脂肪族ジオール、1, 2−シクロヘキサンジオール、1, 4−シクロヘキサンジオール、1, 1−シクロヘキサンジメチロール、1, 4−シクロヘキサンジメチロールなどの脂環式ジオールがあげられ、その他のジオールとしてはキシリレングリコール、4, 4' −ジヒドロキシビフェニル、2, 2−ビス( 4−ヒドロキシフェニル) プロパン、ビス( 4−ヒドロキシフェニル) スルホンなどの芳香族ジオール、イソソルビド、イソマンニド、イソイデット、エリトリタンなどの植物原料由来のジオール等を挙げることができ、又、エチレングリコール、1,3−プロパンジオール、1,4−ブタンジオールなども植物原料由来のものを使用することができる。これらの中で得られるポリエステルの物性の面から、エチレングリコール、1,3−プロパンジオール、1,4−ブタンジオール、1,4−シクロヘキサンジメタノール等が好ましい。尚、これらは2種以上が併用されていてもよい。1,4−ブタンジオールは得られる脂肪族ポリエステルの融点(耐熱性)、生分解性、力学特性の観点から好ましく用いられる。
【0017】
<その他の共重合成分>
本発明のポリエステルのその他の構成成分となる共重合成分としては、乳酸、グリコール酸、ヒドロキシ酪酸、ヒドロキシカプロン酸、2−ヒドロキシ−3,3−ジメチル酪酸、2−ヒドロキシ−3−メチル酪酸、2−ヒドロキシイソカプロン酸、リンゴ酸、マレイン酸、クエン酸、フマル酸等のオキシカルボン酸、及びこれらオキシカルボン酸のエステルやラクトン、オキシカルボン酸重合体等、或いはグリセリン、トリメチロールプロパン、ペンタエリスリトール等の3官能以上の多価アルコール、或いは、プロパントリカルボン酸、ピロメリット酸、トリメリット酸、ベンゾフェノンテトラカルボン酸及びこれらの無水物などの3官能以上の多価カルボン酸又はその無水物等が挙げられる。また、3官能以上のオキシカルボン酸、3官能以上のアルコール、3官能以上のカルボン酸などは少量加えることにより高粘度のポリエステルを得やすい。中でも、リンゴ酸、クエン酸、フマル酸などのオキシカルボン酸が好ましく、特にはリンゴ酸が好ましく用いられる。3官能以上の多官能化合物は全ジカルボン酸成分に対して、上限は好ましくは5モル%、より好ましくは0.5モル%であり、下限は好ましくは0.001モル%、より好ましくは0.05モル%である。この範囲の上限超過ではゲル(未溶融物)が生成しやすく、下限未満では粘度上昇の効果が得にくい。
【0018】
<ポリエステルの製造>
以下、本発明のポリエステルの製造方法として、脂肪族ジカルボン酸としてコハク酸を、脂肪族ジオールとして1,4- ブタンジオールを、その他の成分としてリンゴ酸を原料としたポリブチレンサクシネートの製造方法の例を示すが本発明はこれに限定されるものではない。
【0019】
<エステル化工程>
ポリブチレンサクシネートの製造方法は大きく分けてコハク酸又はコハク酸無水物と1,4- ブタンジオールとを主原料として用いるいわゆる直接重合法と、コハク酸ジアルキルエステル、好ましくはコハク酸ジメチルと、1,4- ブタンジオールとを主原料として用いるエステル交換法がある。前者は初期のエステル化反応で主に水が生成し、後者は初期のエステル交換反応で主にアルコールが生成するという違いがあるが、反応留出物の処理の容易さ、原料原単位の高さという観点からは直接重合法が好ましい。ジカルボン酸成分とジオール成分とのエステル化反応又はエステル交換反応は連続する複数の反応槽で行うことができるが、一槽でも行うことができる。また、品質の安定化、エネルギー効率の観点からは、原料を連続的に供給し、連続的にポリブチレンサクシネートを得るいわゆる連続法が好ましく、本発明では連続方法を採用する。
【0020】
直接重合法の一例としては、コハク酸に対する1,4−ブタンジオールの仕込みモル比は通常0.95〜2.0、好ましくは1.0〜1.7、より好ましくは1.05〜1.6である。また、コハク酸に対するリンゴ酸の仕込みモル%は0.05〜0.50モル%が好ましい。エステル化反応は1つのエステル化反応槽でも、連続する複数の反応槽でも行うこともできる。反応温度は、下限が180℃、好ましくは200℃、上限は260℃、好ましくは250℃、より好ましくは240℃である。下限未満であるとエステル化反応速度が遅く反応時間を長時間必要とし、脂肪族ジオールの脱水分解など好ましくない反応が多くなる。上限超過では脂肪族ジオール、脂肪族ジカルボン酸の分解が多くなり、また反応槽内に飛散物が増加し異物発生原因となりやすく反応物に濁り(ヘーズ)を生じやすくなる。又、エステル化温度は一定温度であることが好ましい。一定温度であることによりエステル化率が安定する。一定温度は設定温度±5℃、好ましくは±2℃である。反応雰囲気は、通常、窒素、アルゴン等の不活性ガス雰囲気下である。反応圧力は、50kPa〜200kPaであり下限は好ましくは60kPa、更に好ましくは70kPa、上限は好ましくは130kPa、更に好ましくは110kPaである。下限未満では反応槽内に飛散物が増加し反応物のヘーズが高くなり異物増加の原因となりやすく、又脂肪族ジオールの反応系外への留出が多くなり重縮合反応速度の低下を招きやすい。上限超過では脂肪族ジオールの脱水分解が多くなり、重縮合速度の低下を招きやすい。反応時間は、通常1時間以上であり、上限が通常10時間以下、好ましくは、4時間以下である。
【0021】
一方、エステル交換法の一例としては、コハク酸のジアルキルエステルを主成分とする前記ジカルボン酸エステル成分と1,4−ブタンジオールを主成分とする前記ジオール成分とを、1段又は多段のエステル交換反応槽内で、好ましくはエステル交換触媒の存在下に、通常110〜260℃、好ましくは140〜245℃、特に好ましくは180〜220℃の温度、また、通常10〜133kPa、好ましくは13〜120kPa、特に好ましくは60〜101kPaの圧力下で、0.5〜5時間、好ましくは1〜3時間で行う。
【0022】
<重縮合工程>
次に、得られたエステル化反応生成物は、最終段のエステル化反応槽以降配管を通じて、第一段重縮合反応槽に移される。重縮合反応は、通常、減圧下で行われる。重縮合反応は連続する複数の反応槽を用い減圧下で行う。
【0023】
最終重縮合反応槽の反応圧力は、下限が通常0.01kPa以上、好ましくは0.03kPa以上であり、上限が通常1.4kPa以下、好ましくは0.4kPa以下として行う。重縮合反応時の圧力が高すぎると、重縮合時間が長くなり、それに伴いポリエステルの熱分解による分子量低下や着色が引き起こされ、実用上充分な特性を示すポリエステルの製造が難しくなる傾向がある。一方、超高真空重縮合設備を用いて製造する手法は重縮合反応速度を向上させる観点からは好ましい態様であるが、極めて高額な設備投資が必要となるため、経済的には不利である。反応温度は、下限が通常215℃、好ましくは220℃であり、上限が通常270℃、好ましくは260℃の範囲である。下限未満であると、重縮合反応速度が遅く、高重合度のポリエステル製造に長時間を要するばかりでなく、高動力の撹拌機も必要となる為、経済的に不利である。一方、上限超過であると製造時のポリエステルの熱分解が引き起こされやすく、高重合度のポリエステルの製造が難しくなる傾向がある。反応時間は、下限が通常1時間以上であり、上限が通常15時間以下、好ましくは8時間以下、より好ましくは6時間以下である。反応時間が短すぎると反応が不十分で高重合度のポリエステルが得にくく、その成形品の機械物性が劣る傾向となる。一方、反応時間が長すぎると、ポリエステルの熱分解による分子量低下が顕著となり、その成形品の機械物性が劣る傾向となるばかりでなく、ポリエステルの耐久性に悪影響を与えるカルボキシル基末端量が熱分解により増加する場合がある。
【0024】
<重縮合反応触媒>
本発明において重縮合反応促進のために触媒を用いることが好ましい。触媒としてチタン化合物、アルカリ土類金属化合物、リン化合物が好ましく用いられる。最終段の重縮合槽に供給される重縮合反応物は、チタン原子、アルカリ土類金属原子、及びリン原子を含有し、チタン原子の含有量T(モル/トン)、アルカリ土類金属原子の含有量M(モル/トン)、ならびにリン原子の含有量P(モル/トン)が、下記(2)及び(3)を満足することが好ましい。
0.5≦P/T≦2.0 (2)
0.5≦M/P≦2.0 (3)
【0025】
P/Tが上限超過では重縮合反応が進みにくい傾向となる。下限未満では重縮合反応は進むが得られるポリエステルに黄味の着色が発生しやすい傾向となる。又、P/Tは増加すると前記Rは増大する傾向である。P/Tの上限はより好ましくは1.8 更に好ましくは1.6であり、下限はより好ましくは、0.7である。尚、ここで重縮合反応物中に含まれるチタン原子、アルカリ土類金属原子、及びリン原子の量は最終重縮合反応槽以降で更なるこれら金属及びリンの化合物を添加しない場合、本発明のポリエステル中に概ねこの比で含まれることとなる。又、M/Pが上限超過では得られるポリエステルに黄味の着色が発生しやすい傾向となる。下限未満では重縮合反応が進みにくい傾向となる。又、M/Pが減少すると前記Rは増大傾向となる。上限はより好ましくは1.8、更に好ましくは1.6である。下限はより好ましくは0.7である。
【0026】
触媒としては、チタン化合物、アルカリ土類金属化合物、及びリン化合物を予め混合させた触媒を用いることが好ましく、該触媒が含有するチタン原子、アルカリ土類金属原子、及びリン原子の含有量t、m、pがモル基準で、下記(4)及び(5)式を満足する触媒であることが好ましい。
0.5≦p/t≦2.0 (4)
0.5≦m/p≦2.0 (5)
【0027】
p/tが上限超過では重縮合反応が進みにくい傾向となる。下限未満では重縮合反応は進むが得られるポリエステルに黄味の着色が発生しやすい傾向となる。又、p/tは増加すると前記Rは増大する方向である。p/tの上限はより好ましくは1.8 更に好ましくは1.6であり、下限はより好ましくは、0.7である。又、m/pが上限超過では得られるポリエステルに黄味の着色が発生しやすい傾向となる。下限未満では重縮合反応が進みにくい傾向となる。又、m/pが減少すると前記Rは増大傾向となる。上限はより好ましくは1.3である。下限はより好ましくは0.6である。
【0028】
ここで、チタン化合物としては、チタン元素を含むカルボン酸塩、アルコキシ塩、有機スルホン酸塩又はβ―ジケトナート塩等の有機基を含む化合物、更には酸化物、ハロゲン化物等の無機化合物及びそれらの混合物が好ましく用いられる。これらの中で、テトラアルキルチタネート及びその加水分解物が好ましく、具体的には、例えば、テトラ−n−プロピルチタネート、テトライソプロピルチタネート、テトラ−n−ブチルチタネート、テトラ−t−ブチルチタネート、テトラフェニルチタネート、テトラシクロヘキシルチタネート、テトラベンジルチタネート及びこれらの混合チタネート、及びこれらの加水分解物が挙げられる。また、チタン(オキシ)アセチルアセトネート、チタンテトラアセチルアセトネート、チタン(ジイソプロキシド)アセチルアセトネート、チタンビス(アンモニウムラクテイト)ジヒドロキシド、チタンビス(エチルアセトアセテート)ジイソプロポキシド、チタン(トリエタノールアミネート)イソプロポキシド、ポリヒドロキシチタンステアレート、チタンラクテート、チタントリエタノールアミネート、ブチルチタネートダイマー等も好んで用いられる。
【0029】
又、アルカリ土類金属元素としては、ベリリウム、マグネシウム、カルシウム、ストロンチウム、バリウム等が挙げられ、これらの中では、マグネシウム、カルシウムが好ましく、マグネシウムが特に好ましい。又、そのアルカリ土類金属化合物としては、アルカリ土類金属元素を含むカルボン酸塩、アルコキシ塩、有機スルホン酸塩又はβ―ジケトナート塩等の有機基を含む化合物、更には酸化物、ハロゲン化物等の無機化合物及びそれらの混合物が好ましく用いられる。具体的には、例えば、酸化マグネシウム、水酸化マグネシウム、マグネシウムアルコキシド、酢酸マグネシウム、炭酸マグネシウム、酸化カルシウム、水酸化カルシウム、酢酸カルシウム、炭酸カルシウム等が挙げられ、特に好ましいマグネシウムの化合物の中では、酢酸マグネシウム、及びその水和物が好ましい。
【0030】
又、燐化合物としては、具体的には、例えば、正燐酸、ポリ燐酸、及び、トリメチルホスフェート、トリエチルホスフェート、トリ−n−ブチルホスフェート、トリオクチルホスフェート、トリフェニルホスフェート、トリクレジルホスフェート、トリス(トリエチレングリコール)ホスフェート、メチルアシッドホスフェート、エチルアシッドホスフェート、イソプロピルアシッドホスフェート、ブチルアシッドホスフェート、モノブチルホスフェート、ジブチルホスフェート、ジオクチルホスフェート、トリエチレングリコールアシッドホスフェート等の燐酸エステル等の5価の燐化合物、並びに、亜燐酸、次亜燐酸、及び、トリメチルホスファイト、ジエチルホスファイト、トリエチルホスファイト、トリスドデシルホスファイト、トリスノニルデシルホスファイト、エチルジエチルホスホノアセテート、トリフェニルホスファイト等の亜燐酸エステル、リチウム、ナトリウム、カリウム等の金属塩等の3価の燐化合物等が挙げられ、中で、重縮合速度制御性の面から、5価の燐化合物の燐酸エステルが好ましく、トリメチルホスフェート、エチルアシッドホスフェートがより好ましく、エチルアシッドフォスフェートが特に好ましい。
【0031】
これらの重縮合触媒の添加量は、生成するポリエステルに対する金属量として、下限値が通常、0.1ppm以上、好ましくは0.5ppm以上、より好ましくは1ppm以上であり、上限値が通常、3000ppm以下、好ましくは1000ppm以下、より好ましくは250ppm以下、特に好ましくは130ppm以下である。
【0032】
触媒の反応系への添加位置は、重縮合反応工程以前であれば特に限定されず、原料仕込み時に添加しておいてもよいが、水が多く存在、もしくは発生している状況下で触媒が共存すると触媒が失活し、異物が析出する原因となり製品の品質を損なう場合がある為、エステル化反応工程以後に添加するのが好ましい。
【0033】
本発明においては、触媒は、重合時に溶融或いは溶解した状態であると重合速度が高くなる理由から、重合時に液状であるか、エステル低重合体やポリエステルに溶解する化合物が好ましい。また、重縮合は無溶媒で行うことが好ましいが、これとは別に、触媒を溶解させる為に少量の溶媒を使用しても良い。この触媒溶解用の溶媒としては、メタノール、エタノール、イソプロパノール、ブタノールなどのアルコール類、エチレングリコール、ブタンジオール、ペンタンジオールなどの前述のジオール類、ジエチルエーテル、テトラヒドロフラン等のエーテル類、アセトニトリル等のニトリル類、ヘプタン、トルエン等の炭化水素化合物、水ならびにそれらの混合物等が挙げられ、その使用量は、触媒濃度が、通常0.0001重量%以上、99重量%以下となるように使用する。
【0034】
<重縮合反応槽>
本発明において、重縮合反応槽は反応物を減圧下、加熱下、溶融状態で重縮合反応を行う装置であり重縮合反応槽の型式に特に制限はなく、例えば、縦型攪拌重合槽、横型攪拌重合槽、薄膜蒸発式重合槽、押出機型反応機、などがある。
【0035】
<末端カルボキシル基量>
本発明のポリエステルの製造方法は、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕を0.15以上とする。
【0036】
ここで、総末端基量は概ね末端OH基量と末端COOH基量と末端ビニル基量との和である。Rが下限未満であるとCDの低減効果が少ない。上限は特に制限されないが1.0に近いと重縮合反応による高分子化が進みにくい傾向となる。下限は好ましくは0.2、更に好ましくは0.35である。上限は好ましくは0.6、更に好ましくは0.45である。
【0037】
R値をコントロールする方法としては、最終段の重縮合反応槽の直前の重縮合反応槽における反応温度、反応時間(滞留時間)、反応物中の金属原子含有量、最終段の重縮合反応槽とその直前の反応槽に連結する反応物移送配管の温度、滞留時間などの各条件の適宜選択により行うことができる。即ち、反応温度は高いと、又反応時間が長いとRは増大する方向であり、反応物移送配管の温度が高いと、又滞留時間が長いとRは増大する方向である。反応物中の金属原子含有量については前述した通りである。そして、最終段の重縮合反応槽の入口でのポリエステルの固有粘度を0.8dL/g以上とするのが好ましく、0.9dL/g以上とするのが更に好ましく、1.0dL/g以上とするのが特に好ましい。
【0038】
又、本発明において、重縮合工程の最終段の重縮合反応槽より一つ前の重縮合反応槽の出口における重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R1 〕と、最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔R〕とが下記(6)式を満足することが好ましい。
R−R1 ≧0.1 (6)
下限は好ましくは0.2 である。(R−R1 )が下限未満であるとCD含有量の低減されたポリエステルを得にくい傾向となる。上限は好ましくは0.5、更に好ましくは0.4である。上限超過では重縮合反応が進みにくい傾向となる。
【0039】
(R−R1 )を0.1以上にするには、重縮合工程の最終段の最終段の重縮合反応槽の一つ前の重縮合反応槽から最終段の重縮合反応槽に反応物を移送する配管のジャケット温度を反応温度より高くする方法、配管内の滞留時間を長くする方法、配管途中に熱交換器を組み込み、反応物を昇温する方法など、或いはこれらの方法を組み合わせることにより行うことができる。最終重縮合反応槽に供給される反応物の温度は通常270℃以下、好ましくは260℃以下、より好ましくは250℃以下である。
【0040】
<製造ライン例>
以下に脂肪族ジカルボン酸としてコハク酸、脂肪族ジオールとして1,4−ブタンジオール、多官能化合物としてリンゴ酸を原料とするポリエステルの製造方法の好ましい実施態様を説明するが本発明はこれに限定されるものではない。
【0041】
以下、添付図面に基づき、ポリエステルの製造方法の好ましい実施態様を説明する。図1は、本発明で採用するエステル化工程の一例の説明図、図2は、本発明で採用する重縮合工程の一例の説明図である。図1において、原料のコハク酸及びリンゴ酸は、通常、原料混合槽(図示せず)で1,4−ブタンジオール(BGと表すことがある)と混合され、原料供給ライン(1)からスラリー又は液体の形態でエステル化反応槽(A)に供給される。また、エステル化反応時に触媒を添加する場合は、触媒調整槽(図示せず)でBGの溶液とした後、BG供給ライン(3)に溶液を供給してなされる。図1では再循環1,4−ブタンジオールの再循環ライン(2)にBG供給ライン(3)を連結し、両者を混合した後、エステル化反応槽(A)の液相部に供給する態様を示した。
【0042】
エステル化反応槽(A)から留出するガスは、留出ライン(5)を経て精留塔(C)で高沸成分と低沸成分とに分離される。通常、高沸成分の主成分は1,4−ブタンジオールであり、低沸成分の主成分は、水およびBGの分解物であるテトラヒドロフラン(THFと表すことがある)である。精留塔(C)で分離された高沸成分は抜出ライン(6)から抜き出され、ポンプ(D)を経て、一部はBG再循環ライン(2)からエステル化反応槽(A)に循環され、一部は循環ライン(7)から精留塔(C)に戻される。また、余剰分は抜出ライン(8)から外部に抜き出される。一方、精留塔(C)で分離された軽沸成分はガス抜出ライン(9)から抜き出され、コンデンサ(G)で凝縮され、凝縮液ライン(10)を経てタンク(F)に一時溜められる。タンク(F)に集められた軽沸成分の一部は、抜出ライン(11)、ポンプ(E)及び循環ライン(12)を経て精留塔(C)に戻され、残部は、抜出ライン(13)を経て外部に抜き出される。コンデンサ(G)はベントライン(14)を経て排気装置(図示せず)に接続されている。エステル化反応槽(A)内で生成したエステル化反応物は、抜出ポンプ(B)及びエステル化反応物の抜出ライン(4)を経て図2第1重縮合反応槽(a)に供される。
【0043】
図1に示す工程においては、再循環ライン(2)にBG供給ライン(3)が連結されているが、両者は独立していてもよい。また、原料供給ライン(1)はエステル化反応槽(A)の液相部に接続されていてもよい。重縮合反応槽に供給される前のエステル化反応物に触媒を添加する場合は、触媒調製槽(図示せず)で所定濃度に調製した後、図2における触媒供給ライン(L7)を経て、前述の図1に示すエステル化反応物の抜出ライン(4)に供給される。
【0044】
次に、エステル化反応物の抜出ライン(4)からフィルター(p)を経て第1重縮合反応槽(a)に供給されたエステル化反応物は、減圧下に重縮合されてポリエステル低重合体となりその後、抜出用ギヤポンプ(c)及び出口流路である抜出ライン(L1)、フィルター(q)を経て第2重縮合反応槽(d)に供給される。第2重縮合反応槽(d)では、通常、第1重縮合反応槽(a)よりも低い圧力で更に重縮合反応が進む。得られた重縮合物は、抜出用ギヤポンプ(e)及び出口流路である抜出ライン(L3)、フィルター(r)を経て、第3重縮合槽(k)に供給される。第3重縮合反応槽(k)は、本説明例では本発明の最終重縮合反応槽となる。抜出ライン(L3)を通じて第2重縮合反応槽(d)から第3重縮合反応槽(k)に導入された重縮合反応物は、ここで更に重縮合反応が進められた後、ギヤポンプ(m)、ダイスヘッド(g)、回転式カッター(h)を経てペレット化される。また、フィルターp、q、r、sは必ずしも設置する必要はなく、後述の実施例においてはrは設置されていない。
【0045】
<ポリエステル>
本発明で得られるポリエステルの固有粘度(IV、dL/g)は、下限が1.4dL/gであることが好ましく、特に好ましくは、1.6dL/gである。上限は2.8dL/gが好ましく、更に好ましくは2.5dL/gであり、特に好ましくは2.3dL/gである。固有粘度が下限未満であると、成形品にしたとき 十分な機械強度が得にくい。固有粘度が上限超過であると、成形時に溶融粘度が高く成形しにくい。
【0046】
本発明のポリエステルの末端カルボキシル基濃度(当量/トン)は通常80以下であり、好ましくは60以下、更に好ましくは40以下、特に好ましくは25以下である。下限は低いほど熱安定性、耐加水分解性がよいが、通常5である。上限を超えると、熱安定性が悪く成形時などに熱分解が多くなる。
【0047】
本発明で得られるポリエステルの環状二量体含有量は、上限が通常7,000質量ppm以下であり、好ましくは6,000質量ppm以下である。上限を超えると、成形後一定期間放置すると、その表面に曇り(ブリードアウト、白化現象と同義。)が生じて表面光沢が消失しやすい傾向となる。
【0048】
<ポリエステル組成物>
本発明のポリエステルに、芳香族−脂肪族共重合ポリエステル、及び脂肪族オキシカルボン酸等を配合させてもよい。更に必要に応じて用いられるカルボジイミド化合物、充填材、可塑剤以外に、本発明の効果を阻害しない範囲で他の生分解性樹脂、例えば、ポリカプロラクトン、ポリアミド、ポリビニルアルコール、セルロースエステル等や、澱粉、セルロース、紙、木粉、キチン・キトサン質、椰子殻粉末、クルミ殻粉末等の動物/植物物質微粉末、或いはこれらの混合物を配合することが出来る。更に、成形体の物性や加工性を調整する目的で、熱安定剤、可塑剤、滑剤、ブロッキング防止剤、核剤、無機フィラー、着色剤、顔料、紫外線吸収剤、光安定剤等の添加剤、改質剤、架橋剤等を含有させてもよい。
【0049】
本発明のポリエステル組成物の製造方法は、特に限定されないが、ブレンドしたポリエステルの原料チップを同一の押出機で溶融混合する方法、各々別々の押出機で溶融させた後に混合する方法、一軸押出機、二軸押出機、バンバリーミキサー、ロールミキサー、ブラベンダープラストグラフ、ニーダーブレンダー等の通常の混練機を用いて混練することによって混合する等が挙げられる。また、各々の原料チップを直接成形機に供給して組成物を調製すると同時に、その成形体を得ることも可能である。
【実施例】
【0050】
以下、実施例により本発明を更に詳細に説明するが、本発明は、その要旨を超えない限り、以下の実施例に何ら限定されるものではない。なお、以下の諸例で採用した物性及び評価項目の測定方法は次の通りである。
【0051】
<固有粘度(IV) dL/g>
ウベローデ型粘度計を使用し次の要領で求めた。すなわち、フェノール/テトラクロロエタン(質量比1/1)の混合溶媒を使用し、30℃において、濃度0.5g/dLのポリマー溶液及び溶媒のみの落下秒数を測定し、以下の式(1)より求めた。
IV=((1+4KH ηSP)0.5 −1)/(2KH C) ・・・(1)
(ただし、ηSP=η/η0 −1であり、ηは試料溶液落下秒数、η0 は溶媒の落下秒数、Cは試料溶液濃度(g/dL)、KH はハギンズの定数である。KH は0.33を採用した。)
【0052】
<エステル化反応物の末端カルボキシル基濃度 当量/トン>
エステル化反応物試料0.3gをベンジルアルコール40mLに180℃で20分間加熱溶解させ、10分間冷却した後、0.1mol・L-1のKOH/メタノール溶液で滴定して求めた値を当量/トンで表したものである。
【0053】
<エステル化率(%)>
エステル化率(%)=〔(ケン化価−酸価)/ケン化価〕×100
なお、ここで、酸価はエステル化反応物をベンジルアルコールに溶解し0.1mol・L-1のKOH/メタノール溶液で滴定により得た反応物中の酸当量値であり、ケン化価はオリゴマーを水ーエタノール中水酸化カリウムでアルカリ加水分解し0.5N塩酸で逆滴定して得た反応物中の酸およびエステル化された酸の合計当量値である。
【0054】
<ポリエステルの末端カルボキシル基量(AV) 当量/トン>
ペレット状ポリエステルを粉砕した後、熱風乾燥機にて140℃で15分間乾燥し、デシケーター内で室温まで冷却した試料から、0.1gを精秤して試験管に採取し、ベンジルアルコール3mLを加えて、乾燥窒素ガスを吹き込みながら195℃、3分間で溶解させた。次いで、クロロホルム5cm3 を徐々に加えて室温まで冷却した。この溶液にフェノールレッド指示薬を1〜2滴加え、乾燥窒素ガスを吹き込みながら撹拌下に、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液で滴定し、黄色から赤色に変じた時点で終了とした。また、ブランクとして、ポリエステル試料を加えずに同様の操作を実施し、以下の式(2)によって末端カルボキシル基量(AV)を算出した。
末端カルボキシル量(当量/トン)=(a−b)×0.1×f/W・・・(2)
ここで、aは、滴定に要した0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の量(μL)、bは、ブランクでの滴定に要した0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の量(μL)、wはポリエステルの試料の量(g)、fは、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の力価である。
【0055】
なお、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の力価(f)は、以下の方法で求めた。
試験管にメタノール5cm3 を採取し、フェノールレッドのエタノール溶液の指示薬として1〜2滴加え、0.lmol・L-1の水酸化ナトリウムのベンジルアルコール溶液0.4cm3 で変色点まで滴定し、次いで力価既知の0.1mol・L-1の塩酸水溶液を標準液として0.2cm3 採取して加え、再度、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液で変色点まで滴定した(以上の操作は、乾燥窒素ガス吹き込み下で行った。)。そして、以下の式(3)によって力価(f)を算出した。
力価(f)=0.1mol・L-1の塩酸水溶液の力価×0.1Nの塩酸水溶液の採取量(μL)/0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の滴定量(μL)・・・(3)
【0056】
<ポリエステル総末端基量(TEV)>
試料約40mgを外径5mmのNMR試料管にはかりとり、重クロロホルム0.75mLを加えて溶かした。Bruker社製AVANCE400分光計を用い、室温で 1H−NMRスペクトルを測定した。化学シフトの基準は、TMS(トリメチルシラン)のシグナルを0.00ppmとした。1,4−ブタンジオール由来の末端OH基、末端ビニル基に対応するシグナル強度から末端OH基量(当量/トン)、末端ビニル基量(当量/トン)を算出し、これらと前記の末端カルボキシル基量(当量/トン)とを合算したものを総末端基量(TEV)(当量/トン)とした。
【0057】
<ペレット中の環状二量体(CD)含有量の測定>
試料0.5gを精秤量し、クロロホルム10mLを加え、室温で溶解後、エタノール/水混合液(容量比4/1)30mLを攪拌下ゆっくりと滴下し、ポリマー成分を沈殿させた。15分後、攪拌を止め、90分間静置分離を行った。次いで、上澄み液を2mL採取し、蒸発乾固させた後、アセトニトリルを2mLを加え溶解させた。0.45μmのフィルターでろ過した後、島津製作所製液体クロマトグラフィー「LC―10」を用い、移動相をアセトニトリル/水(容量比=4/6)とし、カラムは資生堂社製「SHISEIDOCAPCELL PAK C−18 TYPE MG」を用いて定量した。環状二量体純粋品を用いた絶対検量線法で決定した。
【0058】
環状二量体純粋品は下記のようにして得られた。すなわち、コハク酸と1,4−ブタンジオールを重合して得られたポリマーペレットをアセトン中50℃で12時間撹拌して、オリゴマー成分を抽出した。抽出終了後、ペレットを濾別し、オリゴマー成分を抽出したアセトン溶液から、アセトンを揮発させて固形物を得た。この固形物をアセトン中50℃で飽和溶液となるように溶解した後、徐冷し、上澄みを捨て、針状の析出物を取り出し、更に数回この再結晶操作を繰り返して精製した。この針状析出物は、 1H−NMR分析及び高速液体クロマトグラフ分析にて環状二量体であることが確認された。
【0059】
<触媒中の金属元素分析>
試料0.1gをケルダールフラスコ中で硫酸存在下、過酸化水素で湿式分解の後、蒸留水にて定容したものについて、プラズマ発光分光分析装置(JOBINYVON社製ICP−AES ULtrace JY−138U型)を用いて定量分析し、触媒中の金属含量(質量%)に換算した。
【0060】
<ポリエステル中の金属濃度分析>
ケルダールフラスコに試料2.0gを秤量し、硫酸を12mLと過酸化水素を添加(過酸化水素は適宜添加する)し、完全に溶解するまで湿式分解を行った後、超純水で所定濃度に希釈した。この溶液中の金属元素をICP(JOBINYVON社製 JY46P)を用いて定量を行い、試料当たりの量(質量ppm)に換算した。
【0061】
〔実施例1〕
[重縮合用触媒の調製]
撹拌装置付きのガラス製ナス型フラスコに酢酸マグネシウム・4水和物を100質量部入れ、更に1500質量部の無水エタノール(純度99質量%以上)を加えた。更にエチルアシッドホスフェート(モノエステル体とジエステル体の混合質量比は45:55)を65.3質量部加え、23℃で撹拌を行った。15分後に酢酸マグネシウムが完全に溶解したことを確認後、テトラ−n−ブチルチタネートを206.2質量部添加した。更に10分間撹拌を継続し、均一混合溶液を得た。この混合溶液を、ナス型フラスコに移し、60℃のオイルバス中でエバポレーターによって減圧下で濃縮を行った。1時間後に殆どのエタノールが留去され、半透明の粘稠な液体を得た。オイルバスの温度を更に80℃まで上昇させ、5Torrの減圧下で更に濃縮を行い粘稠な液体を得た。この液体状の触媒を、1,4−ブタンジオールに溶解させ、チタン原子含有量が3.36質量%となるよう調製した。
【0062】
[ポリエステルの製造]
図1に示すエステル化工程と図2に示す重縮合工程により、以下のようにしてポリエステルを製造した。先ず、リンゴ酸を0.18質量%含有したコハク酸(表中SAと表す)1.00モルに対して、1,4−ブタンジオールを1.30モルおよびリンゴ酸を総量0.002モルの割合となるように混合した50℃のスラリーを、スラリー調製槽(図示せず)から原料供給ライン(1)を通じ、予め、窒素雰囲気下エステル化率99%のポリエステル低分子量体(エステル化反応物)を充填した攪拌機を有するエステル化反応槽(A)に、生成するポリエステルとして、33kg/hとなる様に連続的に供給した。エステル化反応槽(A)を内温230℃、圧力101kPaとし、生成する水、テトラヒドロフラン及び余剰の1,4−ブタンジオールを、留出ライン(5)から留出させ、精留塔(C)で高沸成分と低沸成分とに分離した。系が安定した後の塔底の高沸成分は精留塔(C)の液面が一定になる様に、抜出ライン(8)を通じて、その一部を外部に抜き出した。一方、水とテトラヒドロフランを主体とする低沸成分は、塔頂よりガスの形態で抜き出し、コンデンサ(G)で凝縮させ、タンク(F)の液面が一定になる様に、抜出ライン(13)より外部に抜き出した。同時に、BG再循環ライン(2)より100℃の精留塔(C)の塔底成分(98質量%以上が1,4−ブタンジオール)全量を、また、BG供給ライン(3)より、エステル化反応槽で発生したテトラヒドロフランと等モルの1,4−ブタンジオールを併せて供給し、エステル化反応槽内のコハク酸に対する1,4−ブタンジオールモル比が1.30となるように調整した。供給量は、再循環ライン(2)とBG供給ライン(3)合わせて3.8kg/hであった。また、1,4−ブタンジオールがテトラヒドロフランに転化した量はコハク酸1.00モルに対し、0.042モルであった。(THF化率4.2モル%対コハク酸)
【0063】
エステル化反応槽(A)で生成したエステル化反応物は、ポンプ(B)を使用し、エステル化反応物の抜出ライン(4)から連続的に抜き出し、エステル化反応槽(A)内液のコハク酸ユニット換算での平均滞留時間が3時間になる様に液面を制御した。抜出ライン(4)から抜き出したエステル化反応物は、図2第1重縮合反応槽(a)に連続的に供給した。系が安定した後、エステル化反応槽(A)の出口で採取したエステル化反応物のエステル化率は92.4%であり末端カルボキシル濃度は基884当量/トンであった。予め前述手法で調製した触媒溶液を、触媒調製槽において、チタン原子としての濃度が0.120質量%となる様に、1,4−ブタンジオールで希釈した触媒溶液を調製した後、触媒供給ライン(L7)を通じて、1.416kg/hで連続的にエステル化反応物の抜出ライン(4)に供給した(触媒は反応液の液相に添加された)。供給量は運転期間中安定していた。
【0064】
第1重縮合反応槽(a)の内温は240℃、圧力2.7kPaとし、滞留時間が120分間になる様に、液面制御を行った。減圧機(図示せず)に接続されたベントライン(L2)から、水、テトラヒドロフラン、1,4−ブタンジオールを抜き出しながら、初期重縮合反応を行った。抜き出した反応液は第2重縮合反応槽(d)に連続的に供給した。第2重縮合反応槽(d)の内温は245℃、圧力400Paとし、滞留時間が150分間になる様に、液面制御を行い、減圧機(図示せず)に接続されたベントライン(L4)から、水、テトラヒドロフラン、1,4−ブタンジオールを抜き出しながら、更に重縮合反応を進めた。得られたポリエステルは、抜出用ギヤポンプ(e)により抜出ライン(L3:配管ジャケット温度245℃)を経由し、第3重縮合反応槽(k)に連続的に供給した。L3及び(e)における重縮合反応物の平均滞留時間は10分である。最終重縮合槽である第3重縮合反応槽に入る直前のポリマーの固有粘度(IV0 )は1.05dL/g、CD含有量(CD0 )は8000質量ppm、R(末端カルボキル基量/総末端基量)は0.23であった。
【0065】
第3重縮合反応槽(k)の内温は240℃、圧力は130Pa、滞留時間は60分間とし、更に、重縮合反応を進めた。得られたポリエステルは、ダイスヘッド(g)からストランド状に連続的に抜き出し、回転式カッター(h)でカッティングしペレットとした。連続運転24時間後に評価用サンプルを採取し、得られたポリエステルの物性測定結果を表1に示す。ここで得られたポリエステル中の金属含有量は最終重縮合槽(k)に供給される反応物中の金属含有量と等しいとした。CD除去率は最終重縮合反応槽入り口における反応物のCD含有量(CD0 )から評価用サンプルのCD含有量(CD)を引いて、低減されたCD量を算出しこれを最終重縮合反応槽入り口における反応物のCD含有量(CD0 )で除し、百分率表示したものである。
【0066】
〔実施例2〕
[重縮合用触媒の調製]
撹拌装置付きのガラス製ナス型フラスコに酢酸マグネシウム・4水和物を100質量部入れ、更に1500質量部の無水エタノール(純度99質量%以上)を加えた。更にエチルアシッドホスフェート(モノエステル体とジエステル体の混合質量比は45:55)を80.6質量部加え、23℃で撹拌を行った。15分後に酢酸マグネシウムが完全に溶解したことを確認後、テトラ−n−ブチルチタネートを122.2質量部添加した。更に10分間撹拌を継続し、均一混合溶液を得た。この混合溶液を、ナス型フラスコに移し、60℃のオイルバス中でエバポレーターによって減圧下で濃縮を行った。1時間後に殆どのエタノールが留去され、半透明の粘稠な液体を得た。オイルバスの温度を更に80℃まで上昇させ、5Torrの減圧下で更に濃縮を行い粘稠な液体を得た。この液体状の触媒を、1,4−ブタンジオールに溶解させ、チタン原子含有量が3.36質量%となるよう調製した。
【0067】
[ポリエステルの製造]
上記のように触媒調製方法を変更した以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1表に示す。
【0068】
〔実施例3〕
第2重縮合反応槽(d)から第3重縮合反応槽(k)への移送配管を延長し、そのときの配管内温を260℃、平均滞留時間を15分とするように加熱配管を設置した。その時の、第2重縮合反応槽(d)出口ポリマーのR1 (AV1 /TEV1 )は0.14であり、延長された配管を経由して第3重縮合反応槽(k)に入る直前のポリマーのR(AV/TEV)は0.34であった。それ以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表に示す。
【0069】
〔実施例4〕
第2重縮合反応槽(d)から第3重縮合反応槽(k)への移送配管途中に熱交換機を設置し(図示せず)、移送ポリマー温度が245℃から260℃となるように制御した。このときのL3及び(e)及び熱交換器における重縮合反応物の平均滞留時間は10分である。その時の、第2重縮合反応槽(d)出口ポリマーのR1 (AV1 /TEV1 )は0.14であり、熱交換機を経由して第3重縮合反応槽(k)に入る直前のポリマーのR(AV/TEV)は0.36であった。それ以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1に示す。
【0070】
〔比較例1〕
第2重縮合反応槽(d)の反応温度を240℃、滞留時間を90分とした以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1表に示す。
〔比較例2〕
第2重縮合反応槽(d)の反応温度を250℃、滞留時間を60分とし、第3重縮合反応槽の反応温度を250℃、滞留時間を60分とした以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1表に示す。
【0071】
【表1】

【産業上の利用可能性】
【0072】
本発明により、CD抽出などの複雑な工程を経なくてもCD含有量の低減された脂肪族ポリエステルを得ることができる。
【符号の説明】
【0073】
1:原料供給ライン
2:BG再循環ライン
3:BG供給ライン
4:エステル化反応物の抜出ライン
5:留出ライン
6:抜出ライン
7:循環ライン
8:抜出ライン
9:ガス抜出ライン
10:凝縮液ライン
11:抜出ライン
12:循環ライン
13:抜出ライン
14:ベントライン
15:供給ライン
A:エステル化反応槽
B:抜出ポンプ
C:精留塔
D、E:ポンプ
F:タンク
G:コンデンサ
L1、L3、L5:重縮合反応物抜出ライン
L2、L4、L6:ベントライン
L7:触媒供給ライン
a:第1重縮合反応槽
d:第2重縮合反応槽
k:第3重縮合反応槽(最終重縮合槽)
c、e、m:抜出用ギヤポンプ
g:ダイスヘッド
h:回転式カッター
p、q、r、s:フィルター
【技術分野】
【0001】
本発明は、ポリエステルの製造方法に関する。詳しくは、ポリエステル中の環状2量体含有量の低減された、ポリエステルの製造方法に関する。
【背景技術】
【0002】
ポリブチレンサクシネートに代表される脂肪族ポリエステルは、その原料を植物資源由来のものに求めることができること、良好な物性および生分解性を有することなどから、農業資材、土木資材、植生資材、包装材等の製品に加工され、広範に利用されつつある。
【0003】
しかしながら、上記脂肪族ポリエステルの成形品は、成形後一定期間放置すると、その表面に曇り(ブリードアウト、白化現象と同義。)が生じて表面光沢が消失するという問題があった。こうした問題は、脂肪族ポリエステルの製造時、ポリエステルの生成と同時に生成される環状2量体が、成形後、一定期間経過した後に成形品の表面に白色物として析出することによるものである。このため、脂肪族ポリエステルから環状2量体を除去する方法が研究されている。
【0004】
例えば、特許文献1には、数平均分子量10,000以上の脂肪族ポリエステルの粉末、ペレット、成形品を、脂肪族ケトン、環状脂肪族エーテル及び脂肪族モノエステル等の有機溶媒を用いて脂肪族ポリエステルの融点よりも低い温度で洗浄して環状2量体(以後、CDと表すことがある)を含むオリゴマーを抽出除去する方法が記載されている。一方、特許文献2には、脂肪族ポリエステル製造において、重縮合反応時に反応触媒の3〜3000倍のリン化合物を添加することによるCD低減方法が記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2002−3606号公報。
【特許文献2】国際公開第WO2006/001084号パンフレット。
【発明の概要】
【発明が解決しようとする課題】
【0006】
特許文献1に記載の技術では、大量の有機溶媒を用いるため、その処理や回収にエネルギーや特別な装置を要するという問題があり、一方、特許文献2において具体的に開示されている技術では、触媒に比べて大量の(28倍、280倍)リン化合物を2mmHg以下の高真空反応の途中で、反応系を常圧に戻して添加するなど工業的生産方法としては必ずしも適当ではなかった。
本発明は、処理や回収にエネルギーや特別な装置を要することなく、工業的に適した効率的な生産方法を用いて環状2量体含有量を低減し、繊維、フィルム、シート、その他の成形体に成形された時に、優れた熱安定性を有し、環状2量体に由来する表面析出物が実用上問題とならず、後加工や用途分野において環状2量体による障害が少なく、かつ経済的なポリエステルの製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは上記課題を解決すべく検討を行い、重縮合反応途中の反応物の末端カルボキシル基量が特定の範囲にあるとき、得られるポリエステル中のCD含有量が低減されることを見出し本発明に到達した。即ち本発明の要旨は、脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕が下記(1)式を満足するポリエステルの製造方法に存する。
0.15≦R (1)
【発明の効果】
【0008】
本発明により、CD抽出などの複雑な工程を経なくてもCD含有量の低減されたポリエステルを得ることができる。
【図面の簡単な説明】
【0009】
【図1】本発明で採用するエステル化工程の一例の説明図である。
【図2】本発明で採用する重縮合工程の一例の説明図である。
【発明を実施するための形態】
【0010】
本発明は、脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕が0.15以上であるポリエステルの製造方法である。
【0011】
重縮合反応はエステル化反応物分子のOH末端基とCOOH末端基との脱水エステル化反応、OH末端基同士の脱ジオール反応により進み、逐次高分子化が行われる。一方、一部分は高分子化しないで環状2量体(CD)となる。重縮合反応後期に反応物の、総末端基量に対する末端カルボキシル基量の比〔R〕が特定の範囲(0.15≦R)にあるとCDは低減される。CDの生成は末端OH基量に依存するため、末端OH基量の割合を減少させる、つまり総末端基量に対する末端カルボキシル基量の割合〔R〕を増加させることで生成反応を抑制できると考えられる。
【0012】
本発明のポリエステルの製造は、脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として行う。本発明において、脂肪族ジカルボン酸及び/又はそのアルキルエステルを主原料とすることとは、使用されるジカルボン酸及び/又はそのアルキルエステルにおいて、脂肪族ジカルボン酸及び/又はそのアルキルエステルが該成分中最大量であることを意味し、好ましくは、全ジカルボン酸及び/又はそのアルキルエステルの50モル%以上が脂肪族ジカルボン酸及び/又はそのアルキルエステルであることを意味する。同様に、本発明において、脂肪族ジオールを主原料とすることとは、使用されるジオールにおいて、脂肪族ジオールが該成分中最大量であることを意味し、好ましくは、全ジオールの50モル%以上が脂肪族ジオールであることを意味する。
【0013】
<ジカルボン酸>
脂肪族ジカルボン酸としては、1, 2−シクロヘキサンジカルボン酸、1, 3−シクロヘキサンジカルボン酸、1, 4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、マロン酸、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸などの脂肪族ジカルボン酸などを挙げることができ、機械的物性や用途の広さ、原料の入手容易さ等の観点からは、脂肪族ジカルボン酸の中ではコハク酸、アジピン酸が好ましい。
【0014】
その他のジカルボン酸としては、テレフタル酸、イソフタル酸、4, 4' −ジフェニルジカルボン酸、4, 4' −ジフェニルエーテルジカルボン酸、4, 4' −ベンゾフェノンジカルボン酸、4, 4' −ジフェノキシエタンジカルボン酸、4, 4' −ジフェニルスルホンジカルボン酸、2, 6−ナフタレンジカルボン酸などの芳香族ジカルボン酸があげられる
【0015】
これらジカルボン酸成分は、ジカルボン酸として、又はジカルボン酸無水物として、又はジカルボン酸のアルキルエステル、好ましくはジアルキルエステルとして反応に供することができ、ジカルボン酸とジカルボン酸アルキルエステルの混合物としてもよい。ジカルボン酸アルキルエステルのアルキル基に特に制限はないが、アルキル基が長いとエステル交換反応時に生成するアルキルアルコールの沸点の上昇を招き反応液中から揮発せず、結果的に末端停止剤として働き重合を阻害するため、炭素数4以下のアルキル基が好ましく、中でもメチル基が好適である。
【0016】
<ジオール>
脂肪族ジオールとしては、エチレングリコール、ジエチレングリコール、ポリエチレングリコール、1,2−プロパンジオール、1,3−プロパンジオール、1,4- ブタンジオール、ポリプロピレングリコール、ポリテトラメチレングリコール、ジブチレングリコール、1, 5−ペンタンジオール、ネオペンチルグリコール、1, 6−ヘキサンジオール、1, 8−オクタンジオールなどの脂肪族ジオール、1, 2−シクロヘキサンジオール、1, 4−シクロヘキサンジオール、1, 1−シクロヘキサンジメチロール、1, 4−シクロヘキサンジメチロールなどの脂環式ジオールがあげられ、その他のジオールとしてはキシリレングリコール、4, 4' −ジヒドロキシビフェニル、2, 2−ビス( 4−ヒドロキシフェニル) プロパン、ビス( 4−ヒドロキシフェニル) スルホンなどの芳香族ジオール、イソソルビド、イソマンニド、イソイデット、エリトリタンなどの植物原料由来のジオール等を挙げることができ、又、エチレングリコール、1,3−プロパンジオール、1,4−ブタンジオールなども植物原料由来のものを使用することができる。これらの中で得られるポリエステルの物性の面から、エチレングリコール、1,3−プロパンジオール、1,4−ブタンジオール、1,4−シクロヘキサンジメタノール等が好ましい。尚、これらは2種以上が併用されていてもよい。1,4−ブタンジオールは得られる脂肪族ポリエステルの融点(耐熱性)、生分解性、力学特性の観点から好ましく用いられる。
【0017】
<その他の共重合成分>
本発明のポリエステルのその他の構成成分となる共重合成分としては、乳酸、グリコール酸、ヒドロキシ酪酸、ヒドロキシカプロン酸、2−ヒドロキシ−3,3−ジメチル酪酸、2−ヒドロキシ−3−メチル酪酸、2−ヒドロキシイソカプロン酸、リンゴ酸、マレイン酸、クエン酸、フマル酸等のオキシカルボン酸、及びこれらオキシカルボン酸のエステルやラクトン、オキシカルボン酸重合体等、或いはグリセリン、トリメチロールプロパン、ペンタエリスリトール等の3官能以上の多価アルコール、或いは、プロパントリカルボン酸、ピロメリット酸、トリメリット酸、ベンゾフェノンテトラカルボン酸及びこれらの無水物などの3官能以上の多価カルボン酸又はその無水物等が挙げられる。また、3官能以上のオキシカルボン酸、3官能以上のアルコール、3官能以上のカルボン酸などは少量加えることにより高粘度のポリエステルを得やすい。中でも、リンゴ酸、クエン酸、フマル酸などのオキシカルボン酸が好ましく、特にはリンゴ酸が好ましく用いられる。3官能以上の多官能化合物は全ジカルボン酸成分に対して、上限は好ましくは5モル%、より好ましくは0.5モル%であり、下限は好ましくは0.001モル%、より好ましくは0.05モル%である。この範囲の上限超過ではゲル(未溶融物)が生成しやすく、下限未満では粘度上昇の効果が得にくい。
【0018】
<ポリエステルの製造>
以下、本発明のポリエステルの製造方法として、脂肪族ジカルボン酸としてコハク酸を、脂肪族ジオールとして1,4- ブタンジオールを、その他の成分としてリンゴ酸を原料としたポリブチレンサクシネートの製造方法の例を示すが本発明はこれに限定されるものではない。
【0019】
<エステル化工程>
ポリブチレンサクシネートの製造方法は大きく分けてコハク酸又はコハク酸無水物と1,4- ブタンジオールとを主原料として用いるいわゆる直接重合法と、コハク酸ジアルキルエステル、好ましくはコハク酸ジメチルと、1,4- ブタンジオールとを主原料として用いるエステル交換法がある。前者は初期のエステル化反応で主に水が生成し、後者は初期のエステル交換反応で主にアルコールが生成するという違いがあるが、反応留出物の処理の容易さ、原料原単位の高さという観点からは直接重合法が好ましい。ジカルボン酸成分とジオール成分とのエステル化反応又はエステル交換反応は連続する複数の反応槽で行うことができるが、一槽でも行うことができる。また、品質の安定化、エネルギー効率の観点からは、原料を連続的に供給し、連続的にポリブチレンサクシネートを得るいわゆる連続法が好ましく、本発明では連続方法を採用する。
【0020】
直接重合法の一例としては、コハク酸に対する1,4−ブタンジオールの仕込みモル比は通常0.95〜2.0、好ましくは1.0〜1.7、より好ましくは1.05〜1.6である。また、コハク酸に対するリンゴ酸の仕込みモル%は0.05〜0.50モル%が好ましい。エステル化反応は1つのエステル化反応槽でも、連続する複数の反応槽でも行うこともできる。反応温度は、下限が180℃、好ましくは200℃、上限は260℃、好ましくは250℃、より好ましくは240℃である。下限未満であるとエステル化反応速度が遅く反応時間を長時間必要とし、脂肪族ジオールの脱水分解など好ましくない反応が多くなる。上限超過では脂肪族ジオール、脂肪族ジカルボン酸の分解が多くなり、また反応槽内に飛散物が増加し異物発生原因となりやすく反応物に濁り(ヘーズ)を生じやすくなる。又、エステル化温度は一定温度であることが好ましい。一定温度であることによりエステル化率が安定する。一定温度は設定温度±5℃、好ましくは±2℃である。反応雰囲気は、通常、窒素、アルゴン等の不活性ガス雰囲気下である。反応圧力は、50kPa〜200kPaであり下限は好ましくは60kPa、更に好ましくは70kPa、上限は好ましくは130kPa、更に好ましくは110kPaである。下限未満では反応槽内に飛散物が増加し反応物のヘーズが高くなり異物増加の原因となりやすく、又脂肪族ジオールの反応系外への留出が多くなり重縮合反応速度の低下を招きやすい。上限超過では脂肪族ジオールの脱水分解が多くなり、重縮合速度の低下を招きやすい。反応時間は、通常1時間以上であり、上限が通常10時間以下、好ましくは、4時間以下である。
【0021】
一方、エステル交換法の一例としては、コハク酸のジアルキルエステルを主成分とする前記ジカルボン酸エステル成分と1,4−ブタンジオールを主成分とする前記ジオール成分とを、1段又は多段のエステル交換反応槽内で、好ましくはエステル交換触媒の存在下に、通常110〜260℃、好ましくは140〜245℃、特に好ましくは180〜220℃の温度、また、通常10〜133kPa、好ましくは13〜120kPa、特に好ましくは60〜101kPaの圧力下で、0.5〜5時間、好ましくは1〜3時間で行う。
【0022】
<重縮合工程>
次に、得られたエステル化反応生成物は、最終段のエステル化反応槽以降配管を通じて、第一段重縮合反応槽に移される。重縮合反応は、通常、減圧下で行われる。重縮合反応は連続する複数の反応槽を用い減圧下で行う。
【0023】
最終重縮合反応槽の反応圧力は、下限が通常0.01kPa以上、好ましくは0.03kPa以上であり、上限が通常1.4kPa以下、好ましくは0.4kPa以下として行う。重縮合反応時の圧力が高すぎると、重縮合時間が長くなり、それに伴いポリエステルの熱分解による分子量低下や着色が引き起こされ、実用上充分な特性を示すポリエステルの製造が難しくなる傾向がある。一方、超高真空重縮合設備を用いて製造する手法は重縮合反応速度を向上させる観点からは好ましい態様であるが、極めて高額な設備投資が必要となるため、経済的には不利である。反応温度は、下限が通常215℃、好ましくは220℃であり、上限が通常270℃、好ましくは260℃の範囲である。下限未満であると、重縮合反応速度が遅く、高重合度のポリエステル製造に長時間を要するばかりでなく、高動力の撹拌機も必要となる為、経済的に不利である。一方、上限超過であると製造時のポリエステルの熱分解が引き起こされやすく、高重合度のポリエステルの製造が難しくなる傾向がある。反応時間は、下限が通常1時間以上であり、上限が通常15時間以下、好ましくは8時間以下、より好ましくは6時間以下である。反応時間が短すぎると反応が不十分で高重合度のポリエステルが得にくく、その成形品の機械物性が劣る傾向となる。一方、反応時間が長すぎると、ポリエステルの熱分解による分子量低下が顕著となり、その成形品の機械物性が劣る傾向となるばかりでなく、ポリエステルの耐久性に悪影響を与えるカルボキシル基末端量が熱分解により増加する場合がある。
【0024】
<重縮合反応触媒>
本発明において重縮合反応促進のために触媒を用いることが好ましい。触媒としてチタン化合物、アルカリ土類金属化合物、リン化合物が好ましく用いられる。最終段の重縮合槽に供給される重縮合反応物は、チタン原子、アルカリ土類金属原子、及びリン原子を含有し、チタン原子の含有量T(モル/トン)、アルカリ土類金属原子の含有量M(モル/トン)、ならびにリン原子の含有量P(モル/トン)が、下記(2)及び(3)を満足することが好ましい。
0.5≦P/T≦2.0 (2)
0.5≦M/P≦2.0 (3)
【0025】
P/Tが上限超過では重縮合反応が進みにくい傾向となる。下限未満では重縮合反応は進むが得られるポリエステルに黄味の着色が発生しやすい傾向となる。又、P/Tは増加すると前記Rは増大する傾向である。P/Tの上限はより好ましくは1.8 更に好ましくは1.6であり、下限はより好ましくは、0.7である。尚、ここで重縮合反応物中に含まれるチタン原子、アルカリ土類金属原子、及びリン原子の量は最終重縮合反応槽以降で更なるこれら金属及びリンの化合物を添加しない場合、本発明のポリエステル中に概ねこの比で含まれることとなる。又、M/Pが上限超過では得られるポリエステルに黄味の着色が発生しやすい傾向となる。下限未満では重縮合反応が進みにくい傾向となる。又、M/Pが減少すると前記Rは増大傾向となる。上限はより好ましくは1.8、更に好ましくは1.6である。下限はより好ましくは0.7である。
【0026】
触媒としては、チタン化合物、アルカリ土類金属化合物、及びリン化合物を予め混合させた触媒を用いることが好ましく、該触媒が含有するチタン原子、アルカリ土類金属原子、及びリン原子の含有量t、m、pがモル基準で、下記(4)及び(5)式を満足する触媒であることが好ましい。
0.5≦p/t≦2.0 (4)
0.5≦m/p≦2.0 (5)
【0027】
p/tが上限超過では重縮合反応が進みにくい傾向となる。下限未満では重縮合反応は進むが得られるポリエステルに黄味の着色が発生しやすい傾向となる。又、p/tは増加すると前記Rは増大する方向である。p/tの上限はより好ましくは1.8 更に好ましくは1.6であり、下限はより好ましくは、0.7である。又、m/pが上限超過では得られるポリエステルに黄味の着色が発生しやすい傾向となる。下限未満では重縮合反応が進みにくい傾向となる。又、m/pが減少すると前記Rは増大傾向となる。上限はより好ましくは1.3である。下限はより好ましくは0.6である。
【0028】
ここで、チタン化合物としては、チタン元素を含むカルボン酸塩、アルコキシ塩、有機スルホン酸塩又はβ―ジケトナート塩等の有機基を含む化合物、更には酸化物、ハロゲン化物等の無機化合物及びそれらの混合物が好ましく用いられる。これらの中で、テトラアルキルチタネート及びその加水分解物が好ましく、具体的には、例えば、テトラ−n−プロピルチタネート、テトライソプロピルチタネート、テトラ−n−ブチルチタネート、テトラ−t−ブチルチタネート、テトラフェニルチタネート、テトラシクロヘキシルチタネート、テトラベンジルチタネート及びこれらの混合チタネート、及びこれらの加水分解物が挙げられる。また、チタン(オキシ)アセチルアセトネート、チタンテトラアセチルアセトネート、チタン(ジイソプロキシド)アセチルアセトネート、チタンビス(アンモニウムラクテイト)ジヒドロキシド、チタンビス(エチルアセトアセテート)ジイソプロポキシド、チタン(トリエタノールアミネート)イソプロポキシド、ポリヒドロキシチタンステアレート、チタンラクテート、チタントリエタノールアミネート、ブチルチタネートダイマー等も好んで用いられる。
【0029】
又、アルカリ土類金属元素としては、ベリリウム、マグネシウム、カルシウム、ストロンチウム、バリウム等が挙げられ、これらの中では、マグネシウム、カルシウムが好ましく、マグネシウムが特に好ましい。又、そのアルカリ土類金属化合物としては、アルカリ土類金属元素を含むカルボン酸塩、アルコキシ塩、有機スルホン酸塩又はβ―ジケトナート塩等の有機基を含む化合物、更には酸化物、ハロゲン化物等の無機化合物及びそれらの混合物が好ましく用いられる。具体的には、例えば、酸化マグネシウム、水酸化マグネシウム、マグネシウムアルコキシド、酢酸マグネシウム、炭酸マグネシウム、酸化カルシウム、水酸化カルシウム、酢酸カルシウム、炭酸カルシウム等が挙げられ、特に好ましいマグネシウムの化合物の中では、酢酸マグネシウム、及びその水和物が好ましい。
【0030】
又、燐化合物としては、具体的には、例えば、正燐酸、ポリ燐酸、及び、トリメチルホスフェート、トリエチルホスフェート、トリ−n−ブチルホスフェート、トリオクチルホスフェート、トリフェニルホスフェート、トリクレジルホスフェート、トリス(トリエチレングリコール)ホスフェート、メチルアシッドホスフェート、エチルアシッドホスフェート、イソプロピルアシッドホスフェート、ブチルアシッドホスフェート、モノブチルホスフェート、ジブチルホスフェート、ジオクチルホスフェート、トリエチレングリコールアシッドホスフェート等の燐酸エステル等の5価の燐化合物、並びに、亜燐酸、次亜燐酸、及び、トリメチルホスファイト、ジエチルホスファイト、トリエチルホスファイト、トリスドデシルホスファイト、トリスノニルデシルホスファイト、エチルジエチルホスホノアセテート、トリフェニルホスファイト等の亜燐酸エステル、リチウム、ナトリウム、カリウム等の金属塩等の3価の燐化合物等が挙げられ、中で、重縮合速度制御性の面から、5価の燐化合物の燐酸エステルが好ましく、トリメチルホスフェート、エチルアシッドホスフェートがより好ましく、エチルアシッドフォスフェートが特に好ましい。
【0031】
これらの重縮合触媒の添加量は、生成するポリエステルに対する金属量として、下限値が通常、0.1ppm以上、好ましくは0.5ppm以上、より好ましくは1ppm以上であり、上限値が通常、3000ppm以下、好ましくは1000ppm以下、より好ましくは250ppm以下、特に好ましくは130ppm以下である。
【0032】
触媒の反応系への添加位置は、重縮合反応工程以前であれば特に限定されず、原料仕込み時に添加しておいてもよいが、水が多く存在、もしくは発生している状況下で触媒が共存すると触媒が失活し、異物が析出する原因となり製品の品質を損なう場合がある為、エステル化反応工程以後に添加するのが好ましい。
【0033】
本発明においては、触媒は、重合時に溶融或いは溶解した状態であると重合速度が高くなる理由から、重合時に液状であるか、エステル低重合体やポリエステルに溶解する化合物が好ましい。また、重縮合は無溶媒で行うことが好ましいが、これとは別に、触媒を溶解させる為に少量の溶媒を使用しても良い。この触媒溶解用の溶媒としては、メタノール、エタノール、イソプロパノール、ブタノールなどのアルコール類、エチレングリコール、ブタンジオール、ペンタンジオールなどの前述のジオール類、ジエチルエーテル、テトラヒドロフラン等のエーテル類、アセトニトリル等のニトリル類、ヘプタン、トルエン等の炭化水素化合物、水ならびにそれらの混合物等が挙げられ、その使用量は、触媒濃度が、通常0.0001重量%以上、99重量%以下となるように使用する。
【0034】
<重縮合反応槽>
本発明において、重縮合反応槽は反応物を減圧下、加熱下、溶融状態で重縮合反応を行う装置であり重縮合反応槽の型式に特に制限はなく、例えば、縦型攪拌重合槽、横型攪拌重合槽、薄膜蒸発式重合槽、押出機型反応機、などがある。
【0035】
<末端カルボキシル基量>
本発明のポリエステルの製造方法は、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕を0.15以上とする。
【0036】
ここで、総末端基量は概ね末端OH基量と末端COOH基量と末端ビニル基量との和である。Rが下限未満であるとCDの低減効果が少ない。上限は特に制限されないが1.0に近いと重縮合反応による高分子化が進みにくい傾向となる。下限は好ましくは0.2、更に好ましくは0.35である。上限は好ましくは0.6、更に好ましくは0.45である。
【0037】
R値をコントロールする方法としては、最終段の重縮合反応槽の直前の重縮合反応槽における反応温度、反応時間(滞留時間)、反応物中の金属原子含有量、最終段の重縮合反応槽とその直前の反応槽に連結する反応物移送配管の温度、滞留時間などの各条件の適宜選択により行うことができる。即ち、反応温度は高いと、又反応時間が長いとRは増大する方向であり、反応物移送配管の温度が高いと、又滞留時間が長いとRは増大する方向である。反応物中の金属原子含有量については前述した通りである。そして、最終段の重縮合反応槽の入口でのポリエステルの固有粘度を0.8dL/g以上とするのが好ましく、0.9dL/g以上とするのが更に好ましく、1.0dL/g以上とするのが特に好ましい。
【0038】
又、本発明において、重縮合工程の最終段の重縮合反応槽より一つ前の重縮合反応槽の出口における重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R1 〕と、最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔R〕とが下記(6)式を満足することが好ましい。
R−R1 ≧0.1 (6)
下限は好ましくは0.2 である。(R−R1 )が下限未満であるとCD含有量の低減されたポリエステルを得にくい傾向となる。上限は好ましくは0.5、更に好ましくは0.4である。上限超過では重縮合反応が進みにくい傾向となる。
【0039】
(R−R1 )を0.1以上にするには、重縮合工程の最終段の最終段の重縮合反応槽の一つ前の重縮合反応槽から最終段の重縮合反応槽に反応物を移送する配管のジャケット温度を反応温度より高くする方法、配管内の滞留時間を長くする方法、配管途中に熱交換器を組み込み、反応物を昇温する方法など、或いはこれらの方法を組み合わせることにより行うことができる。最終重縮合反応槽に供給される反応物の温度は通常270℃以下、好ましくは260℃以下、より好ましくは250℃以下である。
【0040】
<製造ライン例>
以下に脂肪族ジカルボン酸としてコハク酸、脂肪族ジオールとして1,4−ブタンジオール、多官能化合物としてリンゴ酸を原料とするポリエステルの製造方法の好ましい実施態様を説明するが本発明はこれに限定されるものではない。
【0041】
以下、添付図面に基づき、ポリエステルの製造方法の好ましい実施態様を説明する。図1は、本発明で採用するエステル化工程の一例の説明図、図2は、本発明で採用する重縮合工程の一例の説明図である。図1において、原料のコハク酸及びリンゴ酸は、通常、原料混合槽(図示せず)で1,4−ブタンジオール(BGと表すことがある)と混合され、原料供給ライン(1)からスラリー又は液体の形態でエステル化反応槽(A)に供給される。また、エステル化反応時に触媒を添加する場合は、触媒調整槽(図示せず)でBGの溶液とした後、BG供給ライン(3)に溶液を供給してなされる。図1では再循環1,4−ブタンジオールの再循環ライン(2)にBG供給ライン(3)を連結し、両者を混合した後、エステル化反応槽(A)の液相部に供給する態様を示した。
【0042】
エステル化反応槽(A)から留出するガスは、留出ライン(5)を経て精留塔(C)で高沸成分と低沸成分とに分離される。通常、高沸成分の主成分は1,4−ブタンジオールであり、低沸成分の主成分は、水およびBGの分解物であるテトラヒドロフラン(THFと表すことがある)である。精留塔(C)で分離された高沸成分は抜出ライン(6)から抜き出され、ポンプ(D)を経て、一部はBG再循環ライン(2)からエステル化反応槽(A)に循環され、一部は循環ライン(7)から精留塔(C)に戻される。また、余剰分は抜出ライン(8)から外部に抜き出される。一方、精留塔(C)で分離された軽沸成分はガス抜出ライン(9)から抜き出され、コンデンサ(G)で凝縮され、凝縮液ライン(10)を経てタンク(F)に一時溜められる。タンク(F)に集められた軽沸成分の一部は、抜出ライン(11)、ポンプ(E)及び循環ライン(12)を経て精留塔(C)に戻され、残部は、抜出ライン(13)を経て外部に抜き出される。コンデンサ(G)はベントライン(14)を経て排気装置(図示せず)に接続されている。エステル化反応槽(A)内で生成したエステル化反応物は、抜出ポンプ(B)及びエステル化反応物の抜出ライン(4)を経て図2第1重縮合反応槽(a)に供される。
【0043】
図1に示す工程においては、再循環ライン(2)にBG供給ライン(3)が連結されているが、両者は独立していてもよい。また、原料供給ライン(1)はエステル化反応槽(A)の液相部に接続されていてもよい。重縮合反応槽に供給される前のエステル化反応物に触媒を添加する場合は、触媒調製槽(図示せず)で所定濃度に調製した後、図2における触媒供給ライン(L7)を経て、前述の図1に示すエステル化反応物の抜出ライン(4)に供給される。
【0044】
次に、エステル化反応物の抜出ライン(4)からフィルター(p)を経て第1重縮合反応槽(a)に供給されたエステル化反応物は、減圧下に重縮合されてポリエステル低重合体となりその後、抜出用ギヤポンプ(c)及び出口流路である抜出ライン(L1)、フィルター(q)を経て第2重縮合反応槽(d)に供給される。第2重縮合反応槽(d)では、通常、第1重縮合反応槽(a)よりも低い圧力で更に重縮合反応が進む。得られた重縮合物は、抜出用ギヤポンプ(e)及び出口流路である抜出ライン(L3)、フィルター(r)を経て、第3重縮合槽(k)に供給される。第3重縮合反応槽(k)は、本説明例では本発明の最終重縮合反応槽となる。抜出ライン(L3)を通じて第2重縮合反応槽(d)から第3重縮合反応槽(k)に導入された重縮合反応物は、ここで更に重縮合反応が進められた後、ギヤポンプ(m)、ダイスヘッド(g)、回転式カッター(h)を経てペレット化される。また、フィルターp、q、r、sは必ずしも設置する必要はなく、後述の実施例においてはrは設置されていない。
【0045】
<ポリエステル>
本発明で得られるポリエステルの固有粘度(IV、dL/g)は、下限が1.4dL/gであることが好ましく、特に好ましくは、1.6dL/gである。上限は2.8dL/gが好ましく、更に好ましくは2.5dL/gであり、特に好ましくは2.3dL/gである。固有粘度が下限未満であると、成形品にしたとき 十分な機械強度が得にくい。固有粘度が上限超過であると、成形時に溶融粘度が高く成形しにくい。
【0046】
本発明のポリエステルの末端カルボキシル基濃度(当量/トン)は通常80以下であり、好ましくは60以下、更に好ましくは40以下、特に好ましくは25以下である。下限は低いほど熱安定性、耐加水分解性がよいが、通常5である。上限を超えると、熱安定性が悪く成形時などに熱分解が多くなる。
【0047】
本発明で得られるポリエステルの環状二量体含有量は、上限が通常7,000質量ppm以下であり、好ましくは6,000質量ppm以下である。上限を超えると、成形後一定期間放置すると、その表面に曇り(ブリードアウト、白化現象と同義。)が生じて表面光沢が消失しやすい傾向となる。
【0048】
<ポリエステル組成物>
本発明のポリエステルに、芳香族−脂肪族共重合ポリエステル、及び脂肪族オキシカルボン酸等を配合させてもよい。更に必要に応じて用いられるカルボジイミド化合物、充填材、可塑剤以外に、本発明の効果を阻害しない範囲で他の生分解性樹脂、例えば、ポリカプロラクトン、ポリアミド、ポリビニルアルコール、セルロースエステル等や、澱粉、セルロース、紙、木粉、キチン・キトサン質、椰子殻粉末、クルミ殻粉末等の動物/植物物質微粉末、或いはこれらの混合物を配合することが出来る。更に、成形体の物性や加工性を調整する目的で、熱安定剤、可塑剤、滑剤、ブロッキング防止剤、核剤、無機フィラー、着色剤、顔料、紫外線吸収剤、光安定剤等の添加剤、改質剤、架橋剤等を含有させてもよい。
【0049】
本発明のポリエステル組成物の製造方法は、特に限定されないが、ブレンドしたポリエステルの原料チップを同一の押出機で溶融混合する方法、各々別々の押出機で溶融させた後に混合する方法、一軸押出機、二軸押出機、バンバリーミキサー、ロールミキサー、ブラベンダープラストグラフ、ニーダーブレンダー等の通常の混練機を用いて混練することによって混合する等が挙げられる。また、各々の原料チップを直接成形機に供給して組成物を調製すると同時に、その成形体を得ることも可能である。
【実施例】
【0050】
以下、実施例により本発明を更に詳細に説明するが、本発明は、その要旨を超えない限り、以下の実施例に何ら限定されるものではない。なお、以下の諸例で採用した物性及び評価項目の測定方法は次の通りである。
【0051】
<固有粘度(IV) dL/g>
ウベローデ型粘度計を使用し次の要領で求めた。すなわち、フェノール/テトラクロロエタン(質量比1/1)の混合溶媒を使用し、30℃において、濃度0.5g/dLのポリマー溶液及び溶媒のみの落下秒数を測定し、以下の式(1)より求めた。
IV=((1+4KH ηSP)0.5 −1)/(2KH C) ・・・(1)
(ただし、ηSP=η/η0 −1であり、ηは試料溶液落下秒数、η0 は溶媒の落下秒数、Cは試料溶液濃度(g/dL)、KH はハギンズの定数である。KH は0.33を採用した。)
【0052】
<エステル化反応物の末端カルボキシル基濃度 当量/トン>
エステル化反応物試料0.3gをベンジルアルコール40mLに180℃で20分間加熱溶解させ、10分間冷却した後、0.1mol・L-1のKOH/メタノール溶液で滴定して求めた値を当量/トンで表したものである。
【0053】
<エステル化率(%)>
エステル化率(%)=〔(ケン化価−酸価)/ケン化価〕×100
なお、ここで、酸価はエステル化反応物をベンジルアルコールに溶解し0.1mol・L-1のKOH/メタノール溶液で滴定により得た反応物中の酸当量値であり、ケン化価はオリゴマーを水ーエタノール中水酸化カリウムでアルカリ加水分解し0.5N塩酸で逆滴定して得た反応物中の酸およびエステル化された酸の合計当量値である。
【0054】
<ポリエステルの末端カルボキシル基量(AV) 当量/トン>
ペレット状ポリエステルを粉砕した後、熱風乾燥機にて140℃で15分間乾燥し、デシケーター内で室温まで冷却した試料から、0.1gを精秤して試験管に採取し、ベンジルアルコール3mLを加えて、乾燥窒素ガスを吹き込みながら195℃、3分間で溶解させた。次いで、クロロホルム5cm3 を徐々に加えて室温まで冷却した。この溶液にフェノールレッド指示薬を1〜2滴加え、乾燥窒素ガスを吹き込みながら撹拌下に、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液で滴定し、黄色から赤色に変じた時点で終了とした。また、ブランクとして、ポリエステル試料を加えずに同様の操作を実施し、以下の式(2)によって末端カルボキシル基量(AV)を算出した。
末端カルボキシル量(当量/トン)=(a−b)×0.1×f/W・・・(2)
ここで、aは、滴定に要した0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の量(μL)、bは、ブランクでの滴定に要した0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の量(μL)、wはポリエステルの試料の量(g)、fは、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の力価である。
【0055】
なお、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の力価(f)は、以下の方法で求めた。
試験管にメタノール5cm3 を採取し、フェノールレッドのエタノール溶液の指示薬として1〜2滴加え、0.lmol・L-1の水酸化ナトリウムのベンジルアルコール溶液0.4cm3 で変色点まで滴定し、次いで力価既知の0.1mol・L-1の塩酸水溶液を標準液として0.2cm3 採取して加え、再度、0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液で変色点まで滴定した(以上の操作は、乾燥窒素ガス吹き込み下で行った。)。そして、以下の式(3)によって力価(f)を算出した。
力価(f)=0.1mol・L-1の塩酸水溶液の力価×0.1Nの塩酸水溶液の採取量(μL)/0.1mol・L-1の水酸化ナトリウムのベンジルアルコール溶液の滴定量(μL)・・・(3)
【0056】
<ポリエステル総末端基量(TEV)>
試料約40mgを外径5mmのNMR試料管にはかりとり、重クロロホルム0.75mLを加えて溶かした。Bruker社製AVANCE400分光計を用い、室温で 1H−NMRスペクトルを測定した。化学シフトの基準は、TMS(トリメチルシラン)のシグナルを0.00ppmとした。1,4−ブタンジオール由来の末端OH基、末端ビニル基に対応するシグナル強度から末端OH基量(当量/トン)、末端ビニル基量(当量/トン)を算出し、これらと前記の末端カルボキシル基量(当量/トン)とを合算したものを総末端基量(TEV)(当量/トン)とした。
【0057】
<ペレット中の環状二量体(CD)含有量の測定>
試料0.5gを精秤量し、クロロホルム10mLを加え、室温で溶解後、エタノール/水混合液(容量比4/1)30mLを攪拌下ゆっくりと滴下し、ポリマー成分を沈殿させた。15分後、攪拌を止め、90分間静置分離を行った。次いで、上澄み液を2mL採取し、蒸発乾固させた後、アセトニトリルを2mLを加え溶解させた。0.45μmのフィルターでろ過した後、島津製作所製液体クロマトグラフィー「LC―10」を用い、移動相をアセトニトリル/水(容量比=4/6)とし、カラムは資生堂社製「SHISEIDOCAPCELL PAK C−18 TYPE MG」を用いて定量した。環状二量体純粋品を用いた絶対検量線法で決定した。
【0058】
環状二量体純粋品は下記のようにして得られた。すなわち、コハク酸と1,4−ブタンジオールを重合して得られたポリマーペレットをアセトン中50℃で12時間撹拌して、オリゴマー成分を抽出した。抽出終了後、ペレットを濾別し、オリゴマー成分を抽出したアセトン溶液から、アセトンを揮発させて固形物を得た。この固形物をアセトン中50℃で飽和溶液となるように溶解した後、徐冷し、上澄みを捨て、針状の析出物を取り出し、更に数回この再結晶操作を繰り返して精製した。この針状析出物は、 1H−NMR分析及び高速液体クロマトグラフ分析にて環状二量体であることが確認された。
【0059】
<触媒中の金属元素分析>
試料0.1gをケルダールフラスコ中で硫酸存在下、過酸化水素で湿式分解の後、蒸留水にて定容したものについて、プラズマ発光分光分析装置(JOBINYVON社製ICP−AES ULtrace JY−138U型)を用いて定量分析し、触媒中の金属含量(質量%)に換算した。
【0060】
<ポリエステル中の金属濃度分析>
ケルダールフラスコに試料2.0gを秤量し、硫酸を12mLと過酸化水素を添加(過酸化水素は適宜添加する)し、完全に溶解するまで湿式分解を行った後、超純水で所定濃度に希釈した。この溶液中の金属元素をICP(JOBINYVON社製 JY46P)を用いて定量を行い、試料当たりの量(質量ppm)に換算した。
【0061】
〔実施例1〕
[重縮合用触媒の調製]
撹拌装置付きのガラス製ナス型フラスコに酢酸マグネシウム・4水和物を100質量部入れ、更に1500質量部の無水エタノール(純度99質量%以上)を加えた。更にエチルアシッドホスフェート(モノエステル体とジエステル体の混合質量比は45:55)を65.3質量部加え、23℃で撹拌を行った。15分後に酢酸マグネシウムが完全に溶解したことを確認後、テトラ−n−ブチルチタネートを206.2質量部添加した。更に10分間撹拌を継続し、均一混合溶液を得た。この混合溶液を、ナス型フラスコに移し、60℃のオイルバス中でエバポレーターによって減圧下で濃縮を行った。1時間後に殆どのエタノールが留去され、半透明の粘稠な液体を得た。オイルバスの温度を更に80℃まで上昇させ、5Torrの減圧下で更に濃縮を行い粘稠な液体を得た。この液体状の触媒を、1,4−ブタンジオールに溶解させ、チタン原子含有量が3.36質量%となるよう調製した。
【0062】
[ポリエステルの製造]
図1に示すエステル化工程と図2に示す重縮合工程により、以下のようにしてポリエステルを製造した。先ず、リンゴ酸を0.18質量%含有したコハク酸(表中SAと表す)1.00モルに対して、1,4−ブタンジオールを1.30モルおよびリンゴ酸を総量0.002モルの割合となるように混合した50℃のスラリーを、スラリー調製槽(図示せず)から原料供給ライン(1)を通じ、予め、窒素雰囲気下エステル化率99%のポリエステル低分子量体(エステル化反応物)を充填した攪拌機を有するエステル化反応槽(A)に、生成するポリエステルとして、33kg/hとなる様に連続的に供給した。エステル化反応槽(A)を内温230℃、圧力101kPaとし、生成する水、テトラヒドロフラン及び余剰の1,4−ブタンジオールを、留出ライン(5)から留出させ、精留塔(C)で高沸成分と低沸成分とに分離した。系が安定した後の塔底の高沸成分は精留塔(C)の液面が一定になる様に、抜出ライン(8)を通じて、その一部を外部に抜き出した。一方、水とテトラヒドロフランを主体とする低沸成分は、塔頂よりガスの形態で抜き出し、コンデンサ(G)で凝縮させ、タンク(F)の液面が一定になる様に、抜出ライン(13)より外部に抜き出した。同時に、BG再循環ライン(2)より100℃の精留塔(C)の塔底成分(98質量%以上が1,4−ブタンジオール)全量を、また、BG供給ライン(3)より、エステル化反応槽で発生したテトラヒドロフランと等モルの1,4−ブタンジオールを併せて供給し、エステル化反応槽内のコハク酸に対する1,4−ブタンジオールモル比が1.30となるように調整した。供給量は、再循環ライン(2)とBG供給ライン(3)合わせて3.8kg/hであった。また、1,4−ブタンジオールがテトラヒドロフランに転化した量はコハク酸1.00モルに対し、0.042モルであった。(THF化率4.2モル%対コハク酸)
【0063】
エステル化反応槽(A)で生成したエステル化反応物は、ポンプ(B)を使用し、エステル化反応物の抜出ライン(4)から連続的に抜き出し、エステル化反応槽(A)内液のコハク酸ユニット換算での平均滞留時間が3時間になる様に液面を制御した。抜出ライン(4)から抜き出したエステル化反応物は、図2第1重縮合反応槽(a)に連続的に供給した。系が安定した後、エステル化反応槽(A)の出口で採取したエステル化反応物のエステル化率は92.4%であり末端カルボキシル濃度は基884当量/トンであった。予め前述手法で調製した触媒溶液を、触媒調製槽において、チタン原子としての濃度が0.120質量%となる様に、1,4−ブタンジオールで希釈した触媒溶液を調製した後、触媒供給ライン(L7)を通じて、1.416kg/hで連続的にエステル化反応物の抜出ライン(4)に供給した(触媒は反応液の液相に添加された)。供給量は運転期間中安定していた。
【0064】
第1重縮合反応槽(a)の内温は240℃、圧力2.7kPaとし、滞留時間が120分間になる様に、液面制御を行った。減圧機(図示せず)に接続されたベントライン(L2)から、水、テトラヒドロフラン、1,4−ブタンジオールを抜き出しながら、初期重縮合反応を行った。抜き出した反応液は第2重縮合反応槽(d)に連続的に供給した。第2重縮合反応槽(d)の内温は245℃、圧力400Paとし、滞留時間が150分間になる様に、液面制御を行い、減圧機(図示せず)に接続されたベントライン(L4)から、水、テトラヒドロフラン、1,4−ブタンジオールを抜き出しながら、更に重縮合反応を進めた。得られたポリエステルは、抜出用ギヤポンプ(e)により抜出ライン(L3:配管ジャケット温度245℃)を経由し、第3重縮合反応槽(k)に連続的に供給した。L3及び(e)における重縮合反応物の平均滞留時間は10分である。最終重縮合槽である第3重縮合反応槽に入る直前のポリマーの固有粘度(IV0 )は1.05dL/g、CD含有量(CD0 )は8000質量ppm、R(末端カルボキル基量/総末端基量)は0.23であった。
【0065】
第3重縮合反応槽(k)の内温は240℃、圧力は130Pa、滞留時間は60分間とし、更に、重縮合反応を進めた。得られたポリエステルは、ダイスヘッド(g)からストランド状に連続的に抜き出し、回転式カッター(h)でカッティングしペレットとした。連続運転24時間後に評価用サンプルを採取し、得られたポリエステルの物性測定結果を表1に示す。ここで得られたポリエステル中の金属含有量は最終重縮合槽(k)に供給される反応物中の金属含有量と等しいとした。CD除去率は最終重縮合反応槽入り口における反応物のCD含有量(CD0 )から評価用サンプルのCD含有量(CD)を引いて、低減されたCD量を算出しこれを最終重縮合反応槽入り口における反応物のCD含有量(CD0 )で除し、百分率表示したものである。
【0066】
〔実施例2〕
[重縮合用触媒の調製]
撹拌装置付きのガラス製ナス型フラスコに酢酸マグネシウム・4水和物を100質量部入れ、更に1500質量部の無水エタノール(純度99質量%以上)を加えた。更にエチルアシッドホスフェート(モノエステル体とジエステル体の混合質量比は45:55)を80.6質量部加え、23℃で撹拌を行った。15分後に酢酸マグネシウムが完全に溶解したことを確認後、テトラ−n−ブチルチタネートを122.2質量部添加した。更に10分間撹拌を継続し、均一混合溶液を得た。この混合溶液を、ナス型フラスコに移し、60℃のオイルバス中でエバポレーターによって減圧下で濃縮を行った。1時間後に殆どのエタノールが留去され、半透明の粘稠な液体を得た。オイルバスの温度を更に80℃まで上昇させ、5Torrの減圧下で更に濃縮を行い粘稠な液体を得た。この液体状の触媒を、1,4−ブタンジオールに溶解させ、チタン原子含有量が3.36質量%となるよう調製した。
【0067】
[ポリエステルの製造]
上記のように触媒調製方法を変更した以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1表に示す。
【0068】
〔実施例3〕
第2重縮合反応槽(d)から第3重縮合反応槽(k)への移送配管を延長し、そのときの配管内温を260℃、平均滞留時間を15分とするように加熱配管を設置した。その時の、第2重縮合反応槽(d)出口ポリマーのR1 (AV1 /TEV1 )は0.14であり、延長された配管を経由して第3重縮合反応槽(k)に入る直前のポリマーのR(AV/TEV)は0.34であった。それ以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表に示す。
【0069】
〔実施例4〕
第2重縮合反応槽(d)から第3重縮合反応槽(k)への移送配管途中に熱交換機を設置し(図示せず)、移送ポリマー温度が245℃から260℃となるように制御した。このときのL3及び(e)及び熱交換器における重縮合反応物の平均滞留時間は10分である。その時の、第2重縮合反応槽(d)出口ポリマーのR1 (AV1 /TEV1 )は0.14であり、熱交換機を経由して第3重縮合反応槽(k)に入る直前のポリマーのR(AV/TEV)は0.36であった。それ以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1に示す。
【0070】
〔比較例1〕
第2重縮合反応槽(d)の反応温度を240℃、滞留時間を90分とした以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1表に示す。
〔比較例2〕
第2重縮合反応槽(d)の反応温度を250℃、滞留時間を60分とし、第3重縮合反応槽の反応温度を250℃、滞留時間を60分とした以外は、実施例1と同様にしてポリエステルを得た。得られたポリエステルの測定結果を表1表に示す。
【0071】
【表1】
【産業上の利用可能性】
【0072】
本発明により、CD抽出などの複雑な工程を経なくてもCD含有量の低減された脂肪族ポリエステルを得ることができる。
【符号の説明】
【0073】
1:原料供給ライン
2:BG再循環ライン
3:BG供給ライン
4:エステル化反応物の抜出ライン
5:留出ライン
6:抜出ライン
7:循環ライン
8:抜出ライン
9:ガス抜出ライン
10:凝縮液ライン
11:抜出ライン
12:循環ライン
13:抜出ライン
14:ベントライン
15:供給ライン
A:エステル化反応槽
B:抜出ポンプ
C:精留塔
D、E:ポンプ
F:タンク
G:コンデンサ
L1、L3、L5:重縮合反応物抜出ライン
L2、L4、L6:ベントライン
L7:触媒供給ライン
a:第1重縮合反応槽
d:第2重縮合反応槽
k:第3重縮合反応槽(最終重縮合槽)
c、e、m:抜出用ギヤポンプ
g:ダイスヘッド
h:回転式カッター
p、q、r、s:フィルター
【特許請求の範囲】
【請求項1】
脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕が下記(1)式を満足することを特徴とする脂肪族ポリエステルの製造方法。
0.15≦R (1)
【請求項2】
重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物が金属元素としてチタン、アルカリ土類金属、及びリンを含有し、チタン原子の含有量T(モル/トン)、アルカリ土類金属原子の含有量M(モル/トン)、ならびにリン原子の含有量P(モル/トン)が、下記(2)及び(3)式を満足することを特徴とする、請求項1に記載のポリエステルの製造方法。
0.5≦P/T≦2.0 (2)
0.5≦M/P≦2.0 (3)
【請求項3】
重縮合工程において、触媒として、チタン化合物、アルカリ土類金属化合物、及びリン化合物を予め混合させた触媒であって、チタン原子の含有量t、アルカリ土類金属原子の含有量m、ならびにリン原子の含有量pがモル基準で、下記(4)及び(5)式を満足する触媒を用いることを特徴とする、請求項1又は2に記載のポリエステルの製造方法。
0.5≦p/t≦2.0 (4)
0.5≦m/p≦2.0 (5)
【請求項4】
重縮合工程の最終段の重縮合反応槽より一つ前の重縮合反応槽の出口における重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R1 〕と、最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔R〕とが下記(6)式を満足することを特徴とする、請求項1〜3のいずれか一項に記載のポリエステルの製造方法。
R−R1 ≧0.1 (6)
【請求項1】
脂肪族ジカルボン酸及び/又はそのアルキルエステルと脂肪族ジオールとを主原料として、エステル化反応及び/又はエステル交換反応を行い、エステル化反応物を得るエステル化工程、及び該エステル化反応物を連続する複数段の重縮合反応槽を用いて、溶融状態で連続的に重縮合反応を行いポリエステルを得る重縮合工程を有するポリエステルの製造方法において、重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R〕が下記(1)式を満足することを特徴とする脂肪族ポリエステルの製造方法。
0.15≦R (1)
【請求項2】
重縮合工程の最終段の重縮合反応槽に供給される重縮合反応物が金属元素としてチタン、アルカリ土類金属、及びリンを含有し、チタン原子の含有量T(モル/トン)、アルカリ土類金属原子の含有量M(モル/トン)、ならびにリン原子の含有量P(モル/トン)が、下記(2)及び(3)式を満足することを特徴とする、請求項1に記載のポリエステルの製造方法。
0.5≦P/T≦2.0 (2)
0.5≦M/P≦2.0 (3)
【請求項3】
重縮合工程において、触媒として、チタン化合物、アルカリ土類金属化合物、及びリン化合物を予め混合させた触媒であって、チタン原子の含有量t、アルカリ土類金属原子の含有量m、ならびにリン原子の含有量pがモル基準で、下記(4)及び(5)式を満足する触媒を用いることを特徴とする、請求項1又は2に記載のポリエステルの製造方法。
0.5≦p/t≦2.0 (4)
0.5≦m/p≦2.0 (5)
【請求項4】
重縮合工程の最終段の重縮合反応槽より一つ前の重縮合反応槽の出口における重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔末端カルボキシル基量(当量/トン)/総末端基量(当量/トン)=R1 〕と、最終段の重縮合反応槽に供給される重縮合反応物の総末端基量に対する末端カルボキシル基量の比〔R〕とが下記(6)式を満足することを特徴とする、請求項1〜3のいずれか一項に記載のポリエステルの製造方法。
R−R1 ≧0.1 (6)
【図1】

【図2】

【図2】
【公開番号】特開2010−209268(P2010−209268A)
【公開日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願番号】特願2009−59029(P2009−59029)
【出願日】平成21年3月12日(2009.3.12)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願日】平成21年3月12日(2009.3.12)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]