
ポリエステルフィルムの製造方法、太陽電池用ポリエステルフィルム、及び、太陽電池モジュール
【課題】高い耐加水分解性を有するポリエステルフィルムを製造するポリエステルフィルム製造方法、高い耐加水分解性を有する太陽電池用ポリエステルフィルム、及び、発電効率の安定性に優れる太陽電池モジュールを提供する。
【解決手段】嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、前記ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、前記溶融樹脂をフィルム状に成形する成形工程と、を含むポリエステルフィルム製造方法である。
【解決手段】嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、前記ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、前記溶融樹脂をフィルム状に成形する成形工程と、を含むポリエステルフィルム製造方法である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリエステルフィルムの製造方法、太陽電池用ポリエステルフィルム、及び、太陽電池モジュールに関する。
【背景技術】
【0002】
近年、種々の用途に適用できるように、樹脂の合成方法、加工方法、及び成膜方法等により、種々の特性や機能性をもつポリエステルフィルムの開発が求められている。例えば、太陽電池の用途の樹脂フィルムは、屋根の上などに置かれ雨曝しになる太陽電池の使用環境に対応した耐久性や、太陽電池の発電効率を妨げないための透明性等の性質が求められている。また、太陽電池の用途の樹脂フィルムとしては、太陽電池素子(セル)を封止する太陽電池用封止材(単に「封止材」ともいう)や、前記封止材を外部から保護する太陽電池用バックシートなどが知られている。
【0003】
ポリエステルには、通常はその表面にカルボキシル基や水酸基が多く存在しており、水分が存在する環境では加水分解を起こしやすく、経時で劣化する傾向がある。そのため、屋外等の常に風雨に曝されるような環境におかれる太陽電池モジュール等に用いられるポリエステルは、その加水分解性が抑えられていることが求められる。
ポリエステル樹脂に耐加水分解性を付与するには、加水分解反応の触媒となる末端COOHを低減することが考えられる。末端COOHは溶融時の加熱で発生するため、可塑化溶融押出機で溶融成膜する場合には、溶融時の発熱を緩和することが重要になる。
【0004】
かかる問題に対して、例えば、発熱を抑えて大量生産を達成するため、バレルの内径が140mm以上のベント式二軸押出機を使用し、単位時間当たりの押出量Qとスクリュー回転数Nとの比Q/Nが一定の範囲内となる条件下で溶融押出しを行うポリエステルシートの製造方法が開示されている(例えば、特許文献1参照)。
また、原料供給口からポリエステルの少なくとも一部と脂肪族カルボン酸の金属塩をそれぞれ独立に添加するポリエステル組成物の製造方法において、配管表面に付着する前記金属塩をかき落とすために、ポリエステルの一部の嵩密度を0.6以下とすることが開示されている(例えば、特許文献2参照)。
ポリエステルとして、ポリエチレンテレフタラート(PET)を用いる場合には、PETの溶融成形の際に生じるアセトアルデヒドの発生量を抑制するため、ポリエステル樹脂中の含水率を60〜500ppmに調整し、溶融成形に供するポリエステル樹脂の成形方法が知られている(例えば、特許文献3参照)。
さらに、押出機内での滞留による劣化を抑制するとともに、色調に優れたポリトリメチレンテレフタレート樹脂組成物を製造するために、予めポリトリメチレンテレフタレート及び改良材を供給する設備を不活性ガスで置換すると共に、ポリトリメチレンテレフタレート及び改良材それぞれの供給量(体積量)以上の不活性ガスを連続的に該設備に供給しながら、製造することを特徴とする、ポリトリメチレンテレフタレートの製造方法が開示されている(例えば、特許文献4参照)
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特許第3577178号
【特許文献2】特開平10−329188号公報
【特許文献3】特開平07−205257号公報
【特許文献4】特開2004−307552号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかし、特許文献1〜4に記載の各製造方法では、雨曝し環境に耐え得る耐候性、特に、高い耐加水分解性を有するポリエステルフィルムを得ることが困難であった。
【0007】
本発明は、高い耐加水分解性を有するポリエステルフィルムを製造するポリエステルフィルム製造方法、高い耐加水分解性を有する太陽電池用ポリエステルフィルム、及び、発電効率の安定性に優れる太陽電池モジュールを提供することを目的とする。
【課題を解決するための手段】
【0008】
前記目的を達成するため、以下の発明が提供される。
<1> 嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、二軸押出機から溶融樹脂を、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、溶融樹脂をフィルム状に成形する成形工程と、を含むポリエステルフィルム製造方法である。
【0009】
なお、本発明におけるポリエステル原料樹脂の融点Tmとは、示差走査熱量測定により求められる値をいうが、ポリエステル原料樹脂が複数種のポリエステル樹脂で構成されることにより、2つ以上の融点を有する場合は、最も高い融点を指す。したがって、ポリエステル原料樹脂を複数種類用いる場合には、Tmは、高融点成分の融点を意味する。例えば、ポリエステル原料樹脂として、ポリエチレンテレフタレート〔PET〕と、CHDM系ポリエステル樹脂〔例えば、ポリシクロヘキサンジメチレンテレフタレート(PCT)〕とを用いた場合、PETの融点は255℃であり、PCTの融点は278℃であるが、PETとPCTとを混合して用いる場合、排出温度の基準となる「ポリエステル原料樹脂の融点Tm」=278℃となる。
【0010】
<2> 二軸押出機が、少なくとも1つのベント孔と、原料供給口に最も近いベント孔よりも原料供給口に近い位置に、ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給する第1の窒素ガス供給口と、を備える<1>に記載のポリエステルフィルム製造方法である。
【0011】
<3> 二軸押出機が、ポリエステル原料樹脂を可塑化するニーディング部を有するスクリューと、ニーディング部に位置し、ポリエステル原料樹脂に対して窒素ガスを供給する第2の窒素ガス供給口と、を備える<1>または<2>に記載のポリエステルフィルム製造方法である。
【0012】
<4> 窒素ガス供給口における窒素ガスの供給速度が、2m/分〜50m/分である<2>または<3>に記載のポリエステルフィルム製造方法である。
【0013】
<5> ニーディング部は、ニーディングクリアランスにおける剪断速度が500s−1〜2000s−1である<3>または<4>に記載のポリエステルフィルム製造方法である。
【0014】
<6> ポリエステル原料樹脂に対して、カルボジイミド基を1個有し、カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む環状カルボジイミド化合物を、0.05質量%〜20質量%で混合し、溶融混練して溶融樹脂とする<1>〜<5>のいずれか1つに記載のポリエステルフィルム製造方法である。
【0015】
<7> <1>〜<6>のいずれか1つに記載のポリエステルフィルム製造方法により製造された太陽電池用ポリエステルフィルムである。
【0016】
<8> 1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%含むCHDM系ポリエステル樹脂を含有する層を少なくとも1層有する<7>に記載のポリエステルフィルムである。
【0017】
<9> CHDM系ポリエステルを含有する層は、前記1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜20モル%または80モル%〜100モル%含む<8>に記載のポリエステルフィルムである。
【0018】
<10> 太陽光が入射する透明性のフロント基板と、フロント基板の上に設けられ、太陽電池素子及び太陽電池素子を封止する封止材を有するセル構造部分と、セル構造部分のフロント基板が位置する側と反対側に設けられ、封止材と隣接して配置された<7>〜<9>のいずれか1つに記載の太陽電池用ポリエステルフィルムと、
を備えた太陽電池モジュールである。
【発明の効果】
【0019】
本発明によれば、高い耐加水分解性を有するポリエステルフィルムを製造するポリエステルフィルム製造方法、高い耐加水分解性を有する太陽電池用ポリエステルフィルム、及び、発電効率の安定性に優れる太陽電池モジュールを提供することができる。
【図面の簡単な説明】
【0020】
【図1】本発明に係るポリエステルフィルムの製造方法を実施するための二軸押出機の構成例を示す概略図である。
【図2】本発明に係るポリエステルフィルムの製造方法を実施するフローの一例を示す図である。
【発明を実施するための形態】
【0021】
以下、本発明のポリエステルフィルム製造方法について詳細に説明する。なお、本願明細書において「〜」とはその前後に記載される数値を下限値及び上限値として含む意味で使用される。
<ポリエステルフィルム製造方法>
本発明のポリエステルフィルム製造方法は、嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、 前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、前記溶融樹脂をフィルム状に成形する成形工程と、を含んで構成される。
【0022】
ポリエステル原料樹脂(以下、単に「原料樹脂」ともいう)の熱劣化を抑えてポリエステルを成形することで、耐候性に優れるフィルムを得ることができる。また、原料樹脂の熱劣化の要因は、ポリエステルの加水分解、または酸化分解である。
従って、原料樹脂の発熱量を抑制しつつ、加水分解反応の触媒となる末端COOHの量(Acid Value;AV)を低減したり、原料樹脂に含まれる水分や酸素の量をできる限り減らすことが重要である。特に、原料樹脂の末端COOH量と溶融押出しされたフィルムの末端COOH量との差ΔAVを小さく〔例えば3eq/t(当量/トン;以下同じ)以下に〕することが好ましい。
【0023】
しかし、原料樹脂の含水率を乾燥により100ppm以下にすると、二軸押出機(以下、単に「押出機」とも称する)での原料樹脂の発熱が大きくなり、樹脂温度が上昇することがある。
よって、原料樹脂の乾燥をしすぎると、原料樹脂の水分量は減少したとしても、温度上昇による原料樹脂の熱劣化が起き、ΔAVが増大してしまう傾向にある。
【0024】
一方、押出機の温度を下げた場合、溶融した原料樹脂(溶融樹脂)を排出する押出機の出口(排出口)の温度は低下するが、溶融樹脂が最も発熱する可塑化部(ニーディング部)での発熱を抑えることができず、溶融樹脂の熱劣化が進行してしまうことがあった。
【0025】
ポリエステルフィルムの製造にあっては、押出機内での原料樹脂の加水分解を抑制するために、一般に、固有粘度が0.70dl/g以上の高い粘度の原料樹脂が用いられる。しかし、原料樹脂の固有粘度が大きいほど、原料樹脂の分子量は大きくなる傾向にあり、固有粘度が高いほど、押出機内における混練による摩擦が大きく、原料樹脂が発熱し易い。
【0026】
このように、原料樹脂の耐加水分解を目的に、含水率を下げたり、固有粘度を高くしても、押出機内での原料樹脂の発熱により、原料樹脂のAVが増加し易かった。
したがって、原料樹脂の含水率を下げたり、固有粘度を高くした上で、さらに、原料樹脂のAV低下ないしΔAVの低下を達成する必要があった。
【0027】
かかる問題に対し、本発明のポリエステルフィルム製造方法においては、ポリエステル原料樹脂に、所定の嵩密度を有するフラフを特定量添加し、押出機に供給するときのポリエステル原料樹脂の温度を特定の温度にすることで、押出機内部、特に可塑化部における溶融樹脂の摩擦を軽減し、樹脂温度を低減することができることを見出した。
すなわち、原料樹脂が、嵩高いフラフを含んでいることで、押出機内における原料樹脂の摩擦が低減し、さらに、原料樹脂の温度を所定の温度に加熱しておくことで、樹脂が軟化し易く摩擦を生じ難いため、押出機内での発熱を抑制することができる。
【0028】
一方、原料樹脂は、押出機内にて溶融混練されることにより、溶融樹脂となり、押出機の排出口から外部へ排出されるが、このとき、原料樹脂の発熱を抑制することによって、原料樹脂の溶融不足があると、溶融樹脂中に、未溶融の原料樹脂が異物として残存してしまう。押出機から排出された溶融樹脂が、異物を含んでいると、溶融樹脂をフィルム状に成膜する時、特に、フィルムを延伸するときに、異物の存在に起因して、延伸ムラを生じることがある。
そのため、本発明のポリエステルフィルムの製造方法では、溶融樹脂を所定の温度に加熱して押出機から排出することで、溶融樹脂に、かかる異物が残存することを防止することができる。
【0029】
従って、本発明のポリエステルフィルム製造方法を、上記構成とすることで、高い耐加水分解性を有するポリエステルフィルムを製造することができる。
まず、樹脂供給工程から説明する。
【0030】
〔原料樹脂供給工程〕
原料樹脂供給工程は、嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給して構成される。
つまり、二軸押出機の原料供給口に供給されるときのポリエステル原料樹脂が、嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であることを意味する。
まず、フラフについて説明し、次いで、ポリエステル原料樹脂について説明する。
【0031】
−フラフ−
フラフとは、ポリエステルフィルムの粉砕屑、特に、ポリエステルフィルムの成膜途中に生じたフィルムの粉砕屑、及び、リサイクルのための使用済みポリエステルフィルムの粉砕屑等をいう。
本発明のポリエステルフィルムの製造方法においては、特に、嵩密度が0.2〜0.7であるフラフが用いられ、原料樹脂の全質量に対して10質量%〜50質量%の範囲で原料樹脂に含まれる。
【0032】
嵩密度とは、粉末を一定容積の容器の中に一定状態で入れる等して、所定形状にした粉末の質量を、そのときの体積で除算して求められる密度(単位体積あたりの質量)をいい、嵩密度が小さいほど嵩張る。フラフの嵩密度は、JIS K7365:1999の「プラスチック−規定漏斗から注ぐことができる材料の見掛け密度の求め方」に準拠した方法により測定することができる。
【0033】
本発明のポリエステルフィルムの製造方法において、低含水率であり、高固有粘度の原料樹脂に、嵩密度が0.2〜0.7であるフラフを、原料樹脂の全質量に対して10質量%〜50質量%含むことで、原料樹脂が低含水率であること及び高固有粘度を有することのメリットを維持したまま、押出機内での発熱を抑制することができる。
フラフの嵩密度が、0.2未満であると、押出機の原料供給口で原料樹脂が詰まり(ブリッジともいう)、原料樹脂を溶融することができず、フラフの嵩密度が0.7を超えると、嵩が小さく、原料樹脂の発熱抑制を発現することができない。
【0034】
嵩密度が0.2〜0.7であるフラフは、使用済みポリエステルフィルムを破砕することにより得られる。また、ポリエステルフィルムの製造時に生じる破砕片を用いてもよい。
フラフの嵩密度は、原料樹脂の押出し安定性(ブッリジ抑制)、及び剪断発熱抑制の観点から、0.2〜0.6であることが好ましい。
【0035】
また、フラフは、原料樹脂の全質量に対して、10質量%〜50質量%の範囲で原料樹脂に含まれる。フラフの含有量が、10質量%未満であると、押出機内での原料樹脂の発熱を抑制することができず、50質量%を超えると、原料樹脂のAVが増加する。
フラフの原料樹脂中の含有量は、ΔAVを小さくする観点からは20質量%〜50質量%が好ましく、破断伸度の観点からは15質量%〜40質量%であることが好ましい。
【0036】
フラフのサイズとしては、フラフが上記範囲の嵩密度であれば制限はないが、厚みが20〜5000μmであるものが好ましい。中でも、嵩密度が大きくなり過ぎて充満率が低下しすぎないようにし、溶融不足を回避する観点から、100〜1000μmの範囲、更には100〜500μmの範囲がより好ましい。
【0037】
また、成膜されるポリエステルフィルムの末端COOH量をより低減する点で、フラフのサイズのばらつきは小さい方が好ましく、例えば粉砕片の厚みでは、ばらつきは±100%以内であるのが好ましく、より好ましくは±50%以内であり、更には±10%以内である。粉砕片を用いる場合、厚みなどサイズばらつきを小さく抑えることで、得られるポリエステルフィルムの末端COOH量の変動を低く抑えることができる。
【0038】
‐ポリエステル原料樹脂‐
本発明のポリエステルフィルムの製造方法で用いられるポリエステル原料樹脂(単に、原料樹脂ともいう)は、既述のフラフを含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を用いる。
原料樹脂としては、ポリエステルであれば特に制限されず、例えば、PET(ポリエチレンテレフタレート)、PBT(ポリブチレンテレフタレート)等が挙げられる。
まず、含水率、固有粘度などの、原料樹脂の物性について説明し、次いで、原料樹脂を構成するポリエステル樹脂について説明する。
【0039】
(含水率)
原料樹脂は、含水率が20ppm〜100ppmである。
原料樹脂の含水率が20ppm未満であると、押出機内での原料樹脂の発熱が大きくなり、原料樹脂の末端COOH量と溶融押出しされたフィルムの末端COOH量との差ΔAVが増加し、100ppmを超えると原料樹脂の加水分解によりΔAVが増加する。
原料樹脂の含水率は、30ppm〜70ppmであることが好ましい。
原料樹脂の含水率は、原料樹脂の乾燥温度、乾燥時間等によって調整することができる。
【0040】
(固有粘度、IV)
原料樹脂の固有粘度(Interisic Viscosity;IV)は、0.70dl/g〜1.2dl/gである。原料樹脂のIVが0.70未満であると、原料樹脂の耐加水分解性が低下し、IVが1.2を超えると発熱を十分に抑えることができない。
特に原料樹脂が、ポリエチレンテレフタレート(PET)であるとき、原料PET樹脂のIVは0.70dl/g〜0.85dl/gであることが好ましく、0.70dl/g〜0.80dl/gであることがより好ましい。
【0041】
原料樹脂のIVは、ポリエステル樹脂の重合方式および重合条件によって調整することができ、液相重合の後に固相重合を行うことによって原料となる固有粘度IVが0.70dl/g〜1.2dl/gのポリエステル樹脂を得ることができる。
【0042】
なお、固有粘度(IV)は、溶液粘度(η)と溶媒粘度(η0)の比ηr(=η/η0;相対粘度)から1を引いた比粘度(ηsp=ηr−1)濃度で割った値を濃度がゼロの状態に外挿した値である。IVは、ウベローデ型粘度計を用い、ポリエステルを1,1,2,2−テトラクロルエタン/フェノール(=2/3[質量比])混合溶媒に溶解させ、25℃の溶液粘度から求められる。
【0043】
(結晶化温度)
また、後述する固相重合に先立ち、原料樹脂の融着を防止するために結晶化することが好ましく、結晶化する際の好ましい結晶化温度としては、140℃以上である。より好ましくは140℃以上175℃以下であり、さらに好ましくは140℃以上170℃以下である。また、結晶化時間は、10分以上10時間未満が好ましく、より好ましくは20分以上8時間以下であり、さらに好ましくは40分以上7時間以下である。このとき、窒素等の不活性ガスを流すことが好ましい。
【0044】
(末端COOH量、AV)
また、原料樹脂は、末端COOH量(Acid Value;AV)が25eq/t(当量/トン)以下であることが好ましく、15eq/t以下がより好ましい。本発明の方法により原料樹脂を溶融押出ししてフィルムを製造する際、末端COOH量の増加は3eq/t以下に抑制されるため、末端COOH量が25eq/t以下の原料樹脂を用いれば、末端COOH量が少なく、高い耐加水分解性を有するポリエステルフィルムが得られる。ただし、例えば被着物との間の密着性が得られる観点から、原料樹脂の末端COOH量は2eq/t以上であることが望ましい。なお、「eq/t」は、1トンあたりのモル当量を表す。
【0045】
原料樹脂の末端COOH量は、原料樹脂の含水率、押出機内での溶融温度、混練時間等によって調整することができる。
末端COOH量は、以下の方法により測定される値である。すなわち、原料樹脂0.1gをベンジルアルコール10mlに溶解後、さらにクロロホルムを加えて混合溶液を得、これにフェノールレッド指示薬を滴下する。この溶液を、基準液(0.01N、KOH−ベンジルアルコール混合溶液)で滴定し、滴下量から末端カルボキシル基量を求める。
【0046】
なお、複数の種類の樹脂を混合して用いる場合は、前記原料樹脂の末端COOH量は、混合状態での量を表す。例えば、ポリエチレンテレフタレート(PET)として、そのペレットの1種又は2種以上やPETフィルムの粉砕屑であるチップ材などを混合する場合、ペレットの末端COOH量の総量、又はペレットの末端COOH量とチップの末端COOH量との合計量である。
【0047】
(融点、Tm)
また、原料樹脂の融点Tmは、250℃〜290℃の範囲であることが好ましい。前記融点Tmは示差走査熱量測定により求められる値である。以下、原料樹脂を複数種用いることにより、Tmが複数ある場合は、最も高い融点を原料樹脂の融点Tmという。原料樹脂が複数の樹脂の混合であるときは各樹脂の融点の平均値が上記範囲内にあることが好ましい。
【0048】
(嵩密度)
原料樹脂の嵩密度としては、原料樹脂の押出し安定性、及び剪断発熱抑制の観点から、0.7〜0.9以下の範囲が好ましい。この嵩密度が0.7以上であると、押出しをより安定的に行なうことができ、0.9以下であると、局所的な発熱を効果的に抑制することができる。
【0049】
(樹脂温度)
原料樹脂は、100℃〜160℃に加熱して、押出機の原料供給口(押出機入口)に供給する。
すなわち、押出機の原料供給口における原料樹脂の温度が、100℃〜160℃である。原料樹脂の温度を上記範囲とすることで、原料樹脂の摩擦を抑制し、押出機内での発熱を抑制することができる。
原料樹脂の温度が100℃未満であると、原料樹脂の溶融時の摩擦発熱が増加し、160℃を超えると、原料の分解反応が進行する。
原料樹脂の温度は、120℃〜150℃であることが好ましい。
次に、原料樹脂を構成するポリエステル樹脂について説明する。
【0050】
(ポリエステル樹脂)
原料樹脂を構成するポリエステル樹脂としては、ジカルボン酸又はそのエステル誘導体と、ジオール化合物とを公知の方法でエステル化反応及び/又はエステル交換反応させることによって得ることができる。
ジカルボン酸又はそのエステル誘導体としては、例えば、マロン酸、コハク酸、グルタル酸、アジピン酸、スベリン酸、セバシン酸、ドデカンジオン酸、ダイマー酸、エイコサンジオン酸、ピメリン酸、アゼライン酸、メチルマロン酸、エチルマロン酸等の脂肪族ジカルボン酸類、アダマンタンジカルボン酸、ノルボルネンジカルボン酸、イソソルビド、シクロヘキサンジカルボン酸、デカリンジカルボン酸、などの脂環族ジカルボン酸、テレフタル酸、イソフタル酸、フタル酸、1,4−ナフタレンジカルボン酸、1,5−ナフタレンジカルボン酸、2,6−ナフタレンジカルボン酸、1,8−ナフタレンジカルボン酸、4,4’−ジフェニルジカルボン酸、4,4’−ジフェニルエーテルジカルボン酸、5−ナトリウムスルホイソフタル酸、フェニルエンダンジカルボン酸、アントラセンジカルボン酸、フェナントレンジカルボン、9,9’−ビス(4−カルボキシフェニル)フルオレン酸等の芳香族ジカルボン酸などのジカルボン酸又はそのエステル誘導体が挙げられる。
【0051】
ジカルボン酸は、芳香族ジカルボン酸の少なくとも1種が用いられる場合が好ましい。より好ましくは、ジカルボン酸のうち、芳香族ジカルボン酸を主成分として含有する。好ましい芳香族ジカルボン酸として、テレフタル酸(TPA)、2,6−ナフタレンジカルボン酸(2,6−NDCA)が挙げられ、これらが主成分であるものが好ましい。なお、「主成分」とは、ジカルボン酸成分に占める芳香族ジカルボン酸の割合が各々80質量%以上であることをいう。
ポリエステル樹脂の合成にあたっては、2,6−NDCAおよびTPA以外のジカルボン酸を含んでもよい。より好ましいジカルボン酸としては、イソフタル酸(IPA)等を挙げることができる。IPAの好ましい添加量は、全ジカルボン酸中0モル%〜15モル%が好ましく、より好ましくは0モル%〜12モル%、さらに好ましくは0モル%〜9モル%である。
【0052】
ジオール化合物としては、例えば、エチレングリコール、1,2−プロパンジオール、1,3−プロパンジオール、1,4−ブタンジオール、1,2−ブタンジオール、1,3−ブタンジオール等の脂肪族ジオール類、1,4−シクロヘキサンジメタノール、スピログリコール、イソソルビドなどの脂環式ジオール類、ビスフェノールA、1,3―ベンゼンジメタノール,1,4−ベンセンジメタノール、9,9’−ビス(4−ヒドロキシフェニル)フルオレン、などの芳香族ジオール類等が挙げられる。
【0053】
ジオール化合物は、脂肪族ジオールの少なくとも1種が用いられることが好ましい。脂肪族ジオールは、エチレングリコール(EG)、または1,4−シクロヘキサンジメタノール(CHDM)が好ましく、エチレングリコールおよび1,4−シクロヘキサンジメタノールの少なくとも一方を主成分として含有する。
なお、主成分とは、ジオール化合物に占めるエチレングリコールおよび1,4−シクロヘキサンジメタノールの和の割合が80質量%以上であることをいう。
【0054】
エステル化反応及び/又はエステル交換反応には、従来から公知の反応触媒を用いることができる。反応触媒としては、アルカリ金属化合物、アルカリ土類金属化合物、亜鉛化合物、鉛化合物、マンガン化合物、コバルト化合物、アルミニウム化合物、アンチモン化合物、チタン化合物、リン化合物などが挙げられる。通常は、ポリエステルの製造方法が完結する以前の任意の段階において、重合触媒としてアンチモン化合物、ゲルマニウム化合物、チタン化合物を添加することが好ましい。このような方法としては、例えば、ゲルマニウム化合物を例に挙げると、ゲルマニウム化合物粉体をそのまま添加することが好ましい。
【0055】
好ましいポリエステル樹脂は、ポリエチレンテレフタレート(PET)、ポリエチレン−2,6−ナフタレート(PEN)であり、より好ましくはPETである。PETは、ゲルマニウム(Ge)系触媒、アンチモン(Sb)系触媒、アルミニウム(Al)系触媒、及びチタン(Ti)系触媒から選ばれる1種又は2種以上を用いて重合されるものが好ましく、より好ましくはTi系触媒である。
【0056】
−CHDM系ポリエステル樹脂−
さらに、ポリエステルフィルムの力学強度を上げ、耐熱性を向上する観点からは、ポリエステル樹脂が、1,4−シクロヘキサンジメタノール由来の構造を含むCHDM系ポリエステル樹脂であることが好ましい。
CHDM系ポリエステル樹脂とは、ポリエステル樹脂を得るために用いるジオール化合物の一部または全部として、1,4−シクロヘキサンジメタノール(CHDM)を用いたポリエステル樹脂をいい、分子構造内に、1,4−シクロヘキサンジメタノール由来の構造を含む。具体的には、例えば、ポリシクロヘキサンジメチレンテレフタレート(PCT)等が挙げられる。
【0057】
CHDM系ポリエステル樹脂は、分子構造内に1,4−シクロヘキサンジメタノール由来(CHDM由来ともいう)の構造を有していれば、製造されるポリエステルフィルムの耐候性を向上することができるが、1,4−シクロヘキサンジメタノール由来の構造の割合が次の範囲であると、より耐候性に優れる。
ポリエステル樹脂中の1,4−シクロヘキサンジメタノール由来の構造の割合は、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%であることが好ましく、0.1モル%〜20モル%または80〜100モル%であることがより好ましく、0.5モル%〜16モル%または83モル%〜98モル%であることがさらに好ましく、1モル%〜12モル%または86モル%〜96モル%であることが特に好ましい。
【0058】
より好ましい態様においてはCHDM由来の構造が低い領域(0.1〜20モル%)、高い領域(80〜100モル%)の二つの領域が存在するのは、この領域において、特に、結晶を形成し易く、結晶間に取り込まれた非晶が橋渡しする「タイチェーン」を形成し易いためである。この二つの領域に於いてポリエステル樹脂が結晶構造を取りやすく、高い力学強度および高い耐熱性を発揮し易くなる。
なお、ポリエステル樹脂の分子構造内に、このようなCHDM由来の構造が存在することで、ポリエステル分子の配向性が増加し、タイチェーンの生成を促す。これは以下の理由によるものと考えられる。
CHDMは環状構造であるためEG(エチレングリコール)のように屈曲し難く、剛直である。このため、ポリエステル樹脂の延伸等でポリエステル樹脂に負荷される外力で、ポリエステル分子が配向し易い。配向したポリエステル分子は結晶を形成し易いため、タイチェーンを形成し易い。
【0059】
CHDM系ポリエステル樹脂を合成するときは、ジオール化合物として、少なくとも1,4−シクロヘキサンジメタノール(CHDM)用いるが、さらに、CHDM以外のジオール化合物を用いてもよい。このとき、CHDM以外のジオール化合物については、既述のジオール化合物が挙げられる。中でも、エチレングリコールを用いることが好ましい。
【0060】
CHDM系ポリエステル樹脂を合成するときに用いるジカルボン酸は、既述のジカルボン酸又はそのエステル誘導体が用いられる。
CHDM系ポリエステル樹脂を得る場合は、ジカルボン酸として、少なくともテレフタル酸を用いることが好ましい。
また、ジカルボン酸は、テレフタル酸以外にイソフタル酸(IPA)を加えてもよい。好ましいIPA量は、全ジカルボン酸中0モル%〜15モル%が好ましく、0モル%〜12モル%であることがより好ましく、0モル%〜9モル%であることがさらに好ましい。
【0061】
CHDMは、一般に溶融粘度がPET樹脂よりも高く、固有粘度が0.70dl/g〜1.20dl/gであることが好ましい。
【0062】
1,4−シクロヘキサンジメタノール(CHDM)構造を含有するポリシクロヘキサンジメチレンテレフタレート(PCT)は、例えばWO2009/125701の段落番号[0089]〜[0090]、[0120]〜[0121]に記載の方法も好適に用いることができる。
また、ポリエチレン−2,6−ナフタレート(PEN)は、例えば特開2011−153209の段落番号[0170]、特開2008−39803の段落番号[0046]、および[0060]に記載の方法も好適に用いることができる。
【0063】
前記Ti系触媒は、反応活性が高く、重合温度を低くすることができる。そのため、特に重合反応中にPETが熱分解し、COOHが発生するのを抑制することが可能である。本発明においては、ポリエステルフィルムの末端COOH量を30eq/トン以下の範囲に調整するのに好適である。
【0064】
Ti系触媒を用いた重合により得たTi触媒系PETの製造には、例えば、特開2005−340616号公報、特開2005−239940号公報、特開2004−319444号公報、特許3436268号公報、特許3979866号公報、特許3780137号、特開2007−204538号公報等に記載の重合方法を用いることができる。
【0065】
チタン(Ti)系化合物を、1ppm以上30ppm以下、より好ましくは2ppm以上20ppm以下、さらに好ましくは3ppm以上15ppm以下の範囲で用いて重合を行なうことが好ましい。この場合、本発明の方法によって製造されるポリエステルフィルムには、1ppm以上30ppm以下のチタンが含まれる。
Ti系触媒の量は、1ppm以上であると好ましいIVが得られ、30ppm以下であると、末端COOHを低く抑えることができ、耐加水分解性の向上に有利である。
【0066】
−固相重合−
本発明においては、エステル化反応及び/又はエステル交換反応に加えて更に、ポリエステルを固相重合してもよい。固相重合は、既述のエステル化反応等により重合したポリエステル又は市販のポリエステルをペレット状などの小片形状にし、これを用いて好適に行なえる。具体的には、固相重合として、特許第2621563号、特許第3121876号、特許第3136774号、特許第3603585号、特許第3616522号、特許第3617340号、特許第3680523号、特許第3717392号、特許第4167159号等に記載の方法を用いることができる。
【0067】
固相重合は、150℃以上250℃以下、より好ましくは170℃以上240℃以下、さらに好ましくは190℃以上230℃以下で5時間以上100時間以下、より好ましくは10時間以上80時間以下、さらに好ましくは15時間以上60時間以下の条件で行なうのが好ましい。また、固相重合は、真空中あるいは窒素(N2)気流中で行なうことが好ましい。更に、多価アルコール(エチレングリコール等)を1ppm以上1%以下混合してもよい。
【0068】
固相重合は、バッチ式(容器内に樹脂を入れ、この中で所定の時間熱を与えながら撹拌する方式)で実施してもよく、連続式(加熱した筒の中に樹脂を入れ、これを加熱しながら所定の時間滞流させながら筒中を通過させて、順次送り出す方式)で実施してもよい。
【0069】
本発明においては、原料樹脂として用いるポリエステルの重合度は、ポリエステルの使用用途の要求特性に合わせて適宜選択すればよいが、一般には、溶融重縮合で0.3≦IV≦0.65のポリエステルを得て、溶融重縮合で得られたポリエステルを固相重縮合により0.70≦IV≦0.85に上昇させるのが好ましい。
【0070】
−末端封止剤−
ポリエステルフィルムは、末端封止剤を含有していることが好ましい。
ポリエステルフィルムは、ポリエステル結晶間を橋架けする分子(タイチェーン)を有する構造であることで、強固な構造となり、耐候性に優れる。ポリエステルフィルムが末端封止剤を含有していることで、タイチェーンが発達し過ぎることがなく、脆化を抑えつつも耐熱性を高めることができる。これにより、成膜した際に生じやすい弊害の懸念も小さくなる。
従って、本発明のポリエステルフィルム製造方法では、ポリエステルフィルムに末端封止剤が含まれるようにポリエステルフィルムを製造することが好ましい。末端封止剤は、二軸押出機の原料供給口に供給する前に、ポリエステル原料樹脂と混合してもよいし、二軸押出機でポリエステル原料樹脂を溶融混練しているときに混合してもよいし、二軸押出機から溶融樹脂を排出してから溶融樹脂に混合してもよい。
【0071】
以上の中でも、末端封止剤の添加方法としては、ポリエステル原料樹脂と末端封止剤とを事前に、全量に対して10質量%以上60質量%以下となるように溶融混合し、マスターペレットを作成した上で二軸押出機に投入して用いることが好ましい。
【0072】
なお、末端封止剤とは、ポリエステルの末端のカルボキシル基と反応し、ポリエステルの末端カルボキシ基量を減少させる添加剤である。
末端封鎖剤としては、カルボジイミド化合物、オキサゾリン化合物、エポキシ化合物、カーボネート化合物などが挙げられる。本発明のポリエステルフィルムは、イソシアネート化合物、カルボジイミド化合物およびエポキシ化合物のうちの少なくとも1つの末端封止剤を含むことが好ましく、2種類のカルボジイミド化合物を含むことが好ましい。末端封止剤は、単独で使用してもよく、組合せて使用してもよい。
末端封止剤は、以上の中でも、特に、カルボジイミド基を1個有し、前記カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む環状カルボジイミド化合物が好ましい。
【0073】
−環状カルボジイミド化合物−
環状カルボジイミド化合物は、カルボジイミド基を1個有し、カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む化合物である。
ここで、第一窒素とは、カルボジイミド基(−N=C=N−)が有する2つの窒素原子のうち、一方の窒素原子を指し、第二窒素とは、他方の窒素原子を指す。
環状カルボジイミド化合物は、末端封止剤として、ポリエステルの末端カルボキシル基を封止するため、本発明のポリエステルフィルムが環状カルボジイミド化合物を含有することにより、ポリエステルフィルムの耐候性、特に湿熱耐久性を改善することができる。
【0074】
環状カルボジイミド化合物を用いることによりポリエステルフィルムの耐候性が向上するのは、次の理由によるものと考えられる。
カルボジイミド化合物を環状構造にすることにより、下記のように、ポリエステルに、より一層タイチェーンの形成を促すことができる。
・環状カルボジイミドが解裂し、ポリエステル(PET−1という)の末端カルボン酸と反応する。
・解裂したカルボジイミドの他の一端はイソシアネート基となり、他のポリエステル(PET−2という)の末端水酸基と反応する。
・環状カルボジイミド化合物は環状構造のため、水酸基と反応した部位とカルボン酸と反応した部位は繋がっている。この結果、2本のPET分子鎖(PET−1およびPET−2)が環状カルボジイミドを介し、繋がったタイチェーン構造を形成する。
環状カルボジイミド化合物は、ポリエステル原料樹脂に対して0.05質量%〜20質量%の割合で用いることが好ましい。
以下、環状カルボジイミド化合物の詳細について説明する。
【0075】
環状カルボジイミド化合物は、重量平均分子量(Mw)が400以上であることが好ましく、500〜1500であることがより好ましい。
【0076】
また、環状カルボジイミド化合物は、環状構造を複数有していてもよい。
具体的には、環状カルボジイミド化合物の環状構造は、カルボジイミド基(−N=C=N−)を1個有しその第一窒素と第二窒素とが結合基により結合されている。一つの環状構造中には、1個のカルボジイミド基のみを有するが、例えば、スピロ環など、分子中に複数の環状構造を有する場合にはスピロ原子に結合するそれぞれの環状構造中に1個のカルボジイミド基を有していれば、化合物として複数のカルボジイミド基を有していてよいことはいうまでもない。環状構造中の原子数は、好ましくは8〜50、より好ましくは10〜30、さらに好ましくは10〜20、特に、10〜15が好ましい。
【0077】
ここで、環状構造中の原子数とは、環状構造を直接構成する原子の数を意味し、例えば、8員環であれば8、50員環であれば50である。環状構造中の原子数が8より小さいと、環状カルボジイミド化合物の安定性が低下して、保管、使用が困難となる場合があるためである。また反応性の観点よりは環員数の上限値に関しては特別の制限はないが、50を超える原子数の環状カルボジイミド化合物は合成上困難となり、コストが大きく上昇する場合が発生するためである。かかる観点より環状構造中の原子数は好ましくは、10〜30、より好ましくは10〜20、特に好ましくは10〜15の範囲が選択される。
【0078】
環状構造は、下記式(1)で表される構造であることが好ましい。
【0079】
【化1】

【0080】
式(1)中、Q(以下、結合基Qともいう)は、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの2〜4価の結合基、または、脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである2〜4価の結合基である。なお、2つ以上の基の組み合わせは、芳香族基と芳香族基のように、同種の基を組み合わせた態様であってもよい。
Qを構成する脂肪族基と脂環族基と芳香族基とは、各々独立にヘテロ原子または1価の置換基を含んでいてもよい。ヘテロ原子とはこの場合、O、N、S、Pを指す。結合基の価のうち2つの価は環状構造を形成するために使用される。Qが3価または4価の結合基である場合、環状構造は、単結合、二重結合、原子、または原子団を介して、ポリマーまたは他の環状構造と結合している。
【0081】
結合基Qは、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、もしくは2〜4価の炭素数5〜15の芳香族基、または、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、及び2〜4価の炭素数5〜15の芳香族基から選択される2つ以上の基の組み合わせであることが好ましい。
結合基Qを構成する脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせの例としては、アルキレン基とアリーレン基が結合した、アルキレン−アリーレン基のような構造などが挙げられる。
結合基Qは、下記式(1−1)、式(1−2)または式(1−3)で表される2〜4価の結合基であることが好ましい。
【0082】
【化2】
【0083】
式(1−1)中、Ar1およびAr2は各々独立に、2〜4価の炭素数5〜15の芳香族基である。Ar1およびAr2は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
Ar1またはAr2として表される芳香族基としては、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基(2価)として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これらの芳香族基は置換されていてもよい。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0084】
式(1−2)中、R1およびR2は、各々独立に、2〜4価の炭素数1〜20の脂肪族基、もしくは2〜4価の炭素数3〜20の脂環族基、または、2〜4価の炭素数1〜20の脂肪族基と2〜4価の炭素数3〜20の脂環族基との組み合わせ、あるいは、2〜4価の炭素数1〜20の脂肪族基と2〜4価の炭素数3〜20の脂環族基と2〜4価の炭素数5〜15の芳香族基とから選択される2つ以上の基の組み合わせである。R1およびR2を構成する脂肪族基、脂環族基、および芳香族基は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
【0085】
R1またはR2として表される脂肪族基としては、炭素数1〜20のアルキレン基、炭素数1〜20のアルカントリイル基、炭素数1〜20のアルカンテトライル基などが挙げられる。アルキレン基として、メチレン基、エチレン基、プロピレン基、ブチレン基、ペンチレン基、ヘキシレン基、へプチレン基、オクチレン基、ノニレン基、デシレン基、ドデシレン基、へキサデシレン基などが挙げられる。アルカントリイル基として、メタントリイル基、エタントリイル基、プロパントリイル基、ブタントリイル基、ペンタントリイル基、ヘキサントリイル基、ヘプタントリイル基、オクタントリイル基、ノナントリイル基、デカントリイル基、ドデカントリイル基、ヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、メタンテトライル基、エタンテトライル基、プロパンテトライル基、ブタンテトライル基、ペンタンテトライル基、ヘキサンテトライル基、ヘプタンテトライル基、オクタンテトライル基、ノナンテトライル基、デカンテトライル基、ドデカンテトライル基、ヘキサデカンテトライル基などが挙げられる。これらの脂肪族基は置換されていても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0086】
脂環族基として、炭素数3〜20のシクロアルキレン基、炭素数3〜20のシクロアルカントリイル基、炭素数3〜20のシクロアルカンテトライル基が挙げられる。シクロアルキレン基として、シクロプロピレン基、シクロブチレン基、シクロペンチレン基、シクロヘキシレン基、シクロへプチレン基、シクロオクチレン基、シクロノニレン基、シクロデシレン基、シクロドデシレン基、シクロへキサデシレン基などが挙げられる。アルカントリイル基として、シクロプロパントリイル基、シクロブタントリイル基、シクロペンタントリイル基、シクロヘキサントリイル基、シクロヘプタントリイル基、シクロオクタントリイル基、シクロノナントリイル基、シクロデカントリイル基、シクロドデカントリイル基、シクロヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、シクロプロパンテトライル基、シクロブタンテトライル基、シクロペンタンテトライル基、シクロヘキサンテトライル基、シクロヘプタンテトライル基、シクロオクタンテトライル基、シクロノナンテトライル基、シクロデカンテトライル基、シクロドデカンテトライル基、シクロヘキサデカンテトライル基などが挙げられる。これらの脂環族基は置換されていても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0087】
芳香族基として、それぞれへテロ原子を含んで複素環構造を持っていてもよい、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これら芳香族基は置換されていても良い。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0088】
上記式(1−1)および式(1−2)において、X1およびX2は、各々独立に、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、もしくは、2〜4価の炭素数5〜15の芳香族基、または、2〜4価の炭素数1〜20の脂肪族基と2〜4価の炭素数3〜20の脂環族基と2〜4価の炭素数5〜15の芳香族基とから選択される2つ以上の基の組み合わせである。X1およびX2を構成する脂肪族基、脂環族基、および芳香族基は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
【0089】
脂肪族基として、炭素数1〜20のアルキレン基、炭素数1〜20のアルカントリイル基、炭素数1〜20のアルカンテトライル基などが挙げられる。アルキレン基として、メチレン基、エチレン基、プロピレン基、ブチレン基、ペンチレン基、ヘキシレン基、へプチレン基、オクチレン基、ノニレン基、デシレン基、ドデシレン基、へキサデシレン基などが挙げられる。アルカントリイル基として、メタントリイル基、エタントリイル基、プロパントリイル基、ブタントリイル基、ペンタントリイル基、ヘキサントリイル基、ヘプタントリイル基、オクタントリイル基、ノナントリイル基、デカントリイル基、ドデカントリイル基、ヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、メタンテトライル基、エタンテトライル基、プロパンテトライル基、ブタンテトライル基、ペンタンテトライル基、ヘキサンテトライル基、ヘプタンテトライル基、オクタンテトライル基、ノナンテトライル基、デカンテトライル基、ドデカンテトライル基、ヘキサデカンテトライル基などが挙げられる。これらの脂肪族基は置換されていても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0090】
脂環族基として、炭素数3〜20のシクロアルキレン基、炭素数3〜20のシクロアルカントリイル基、炭素数3〜20のシクロアルカンテトライル基が挙げられる。シクロアルキレン基として、シクロプロピレン基、シクロブチレン基、シクロペンチレン基、シクロヘキシレン基、シクロへプチレン基、シクロオクチレン基、シクロノニレン基、シクロデシレン基、シクロドデシレン基、シクロへキサデシレン基などが挙げられる。アルカントリイル基として、シクロプロパントリイル基、シクロブタントリイル基、シクロペンタントリイル基、シクロヘキサントリイル基、シクロヘプタントリイル基、シクロオクタントリイル基、シクロノナントリイル基、シクロデカントリイル基、シクロドデカントリイル基、シクロヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、シクロプロパンテトライル基、シクロブタンテトライル基、シクロペンタンテトライル基、シクロヘキサンテトライル基、シクロヘプタンテトライル基、シクロオクタンテトライル基、シクロノナンテトライル基、シクロデカンテトライル基、シクロドデカンテトライル基、シクロヘキサデカンテトライル基などが挙げられる。これらの脂環族基は置換されていても良い。
脂環族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0091】
芳香族基として、それぞれへテロ原子を含んで複素環構造を持っていてもよい、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これらの芳香族基は置換されていても良い。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0092】
上記式(1−1)および式(1−2)においてsおよびkは、各々独立に、0〜10の整数であり、好ましくは0〜3の整数であり、より好ましくは0〜1の整数である。
s及びkが10を超えると、環状カルボジイミド化合物は合成上困難となり、コストが大きく上昇する場合が発生するためである。かかる観点より整数は好ましくは0〜3の範囲が選択される。なお、sまたはkが2以上であるとき、繰り返し単位としてのX1、あるいはX2が、他のX1、あるいはX2と異なっていてもよい。
【0093】
上記式(1−3)においてX3は、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、もしくは2〜4価の炭素数5〜15の芳香族基、またはこれらの組み合わせである。
X3を構成する脂肪族基、脂環族基、および芳香族基は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
【0094】
脂肪族基として、炭素数1〜20のアルキレン基、炭素数1〜20のアルカントリイル基、炭素数1〜20のアルカンテトライル基などが挙げられる。アルキレン基として、メチレン基、エチレン基、プロピレン基、ブチレン基、ペンチレン基、ヘキシレン基、へプチレン基、オクチレン基、ノニレン基、デシレン基、ドデシレン基、へキサデシレン基などが挙げられる。アルカントリイル基として、メタントリイル基、エタントリイル基、プロパントリイル基、ブタントリイル基、ペンタントリイル基、ヘキサントリイル基、ヘプタントリイル基、オクタントリイル基、ノナントリイル基、デカントリイル基、ドデカントリイル基、ヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、メタンテトライル基、エタンテトライル基、プロパンテトライル基、ブタンテトライル基、ペンタンテトライル基、ヘキサンテトライル基、ヘプタンテトライル基、オクタンテトライル基、ノナンテトライル基、デカンテトライル基、ドデカンテトライル基、ヘキサデカンテトライル基などが挙げられる。これら脂肪族基は1価の置換基を含んでいても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0095】
脂環族基として、炭素数3〜20のシクロアルキレン基、炭素数3〜20のシクロアルカントリイル基、炭素数3〜20のシクロアルカンテトライル基が挙げられる。シクロアルキレン基として、シクロプロピレン基、シクロブチレン基、シクロペンチレン基、シクロヘキシレン基、シクロへプチレン基、シクロオクチレン基、シクロノニレン基、シクロデシレン基、シクロドデシレン基、シクロへキサデシレン基などが挙げられる。アルカントリイル基として、シクロプロパントリイル基、シクロブタントリイル基、シクロペンタントリイル基、シクロヘキサントリイル基、シクロヘプタントリイル基、シクロオクタントリイル基、シクロノナントリイル基、シクロデカントリイル基、シクロドデカントリイル基、シクロヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、シクロプロパンテトライル基、シクロブタンテトライル基、シクロペンタンテトライル基、シクロヘキサンテトライル基、シクロヘプタンテトライル基、シクロオクタンテトライル基、シクロノナンテトライル基、シクロデカンテトライル基、シクロドデカンテトライル基、シクロヘキサデカンテトライル基などが挙げられる。これら脂環族基は1価の置換基を含んでいても良い。
脂環族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリーレン基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0096】
芳香族基として、それぞれへテロ原子を含んで複素環構造を持っていてもよい、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これらの芳香族基は置換されていても良い。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0097】
また、Ar1、Ar2、R1、R2、X1、X2およびX3はヘテロ原子を含有していてもよい、また、Qが2価の結合基であるときは、Ar1、Ar2、R1、R2、X1、X2およびX3は全て2価の基である。Qが3価の結合基であるときは、Ar1、Ar2、R1、R2、X1、X2およびX3の内の一つが3価の基である。Qが4価の結合基であるときは、Ar1、Ar2、R1、R2、X1、X2およびX3の内の一つが4価の基であるか、二つが3価の基である。
【0098】
環状カルボジイミド化合物としては、次の環状カルボジイミド化合物(a)〜環状カルボジイミド化合物(c)が挙げられる。
【0099】
[環状カルボジイミド化合物(a)]
環状カルボジイミド化合物(a)は、下記式(2)で表される化合物である。
【0100】
【化3】
【0101】
式(2)中、Qaは、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの2価の結合基または脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである2価の結合基であり、さらにヘテロ原子を含有していてもよい。脂肪族基、脂環族基、および芳香族基は、式(1)で説明したものと同じである。但し、式(2)の化合物においては、脂肪族基、脂環族基、および芳香族基は全て2価である。Qaは、下記式(2−1)、式(2−2)または式(2−3)で表される2価の結合基であることが好ましい。
【0102】
【化4】
【0103】
式(2−1)〜式(2−3)中、Ara1、Ara2、Ra1、Ra2、Xa1、Xa2、Xa3、sおよびkは、各々式(1−1)〜式(1−3)中のAr1、Ar2、R1、R2、X1、X2、X3、sおよびkと同じである。但し、これらは全て2価である。
かかる環状カルボジイミド化合物(a)としては、以下の化合物が挙げられる。
【0104】
【化5】
【0105】
[環状カルボジイミド化合物(b)]
環状カルボジイミド化合物(b)は、下記式(3)で表される化合物である。
【0106】
【化6】
【0107】
式(3)中、Qbは、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの3価の結合基または脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである3価の結合基であり、さらにヘテロ原子を含有していてもよい。脂肪族基、脂環族基、および芳香族基は、式(1)で説明したものと同じである。但し、式(3)の化合物においては、Qbを構成する基の内一のつは3価である。
式(3)中、Yは、環状カルボジイミド化合物の環状構造を担持する担体である。
Qbは、下記式(3−1)、式(3−2)または式(3−3)で表される3価の結合基であることが好ましい。
【0108】
【化7】
【0109】
式(3−1)〜式(3−3)中、Arb1、Arb2、Rb1、Rb2、Xb1、Xb2、Xb3、sおよびkは、各々式(1−1)〜式(1−3)のAr1、Ar2、R1、R2、X1、X2、X3、sおよびkと同じである。但しこれらの内の一つは3価の基である。
Yは、単結合、二重結合、原子、原子団またはポリマーであることが好ましい。Yは結合部であり、複数の環状構造がYを介して結合し、式(3)で表される構造を形成している。
かかる環状カルボジイミド化合物(b)としては、下記化合物が挙げられる。
【0110】
【化8】
【0111】
[環状カルボジイミド化合物(c)]
環状カルボジイミド化合物(c)は、下記式(4)で表されるである。
【0112】
【化9】
【0113】
式中、Qcは、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの4価の結合基または脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである4価の結合基であり、さらにヘテロ原子を保有していてもよい。Z1およびZ2は、環状構造を担持する担体である。Z1およびZ2は、互いに結合して環状構造を形成していてもよい。
脂肪族基、脂環族基、および芳香族基は、式(1)で説明したものと同じである。但し、式(4)の化合物において、Qcは4価である。従って、これらの基の内の一つが4価の基であるか、二つが3価の基である。
Qcは、下記式(4−1)、式(4−2)または式(4−3)で表される4価の結合基であることが好ましい。
【0114】
【化10】
【0115】
式(4−1)〜式(4−3)中の、Arc1、Arc2、Rc1、Rc2、Xc1、Xc2、Xc3、sおよびkは、各々式(1−1)〜式(1−3)の、Ar1、Ar2、R1、R2、X1、X2、X3、sおよびkと同じである。但し、Arc1、Arc2、Rc1、Rc2、Xc1、Xc2およびXc3は、これらの内の一つが4価の基であるか、二つが3価の基である。Z1およびZ2は各々独立に、単結合、二重結合、原子、原子団またはポリマーであることが好ましい。Z1およびZ2は結合部であり、複数の環状構造がZ1およびZ2を介して結合し、式(4)で表される構造を形成している。
かかる環状カルボジイミド化合物(c)としては、下記化合物を挙げることができる。
【0116】
【化11】
【0117】
(環状カルボジイミド化合物の製造方法)
環状カルボジイミド化合物は、特開2011−256337号公報の段落番号[0075]に記載の方法などに基づいて合成することができる。
【0118】
〔溶融樹脂排出工程〕
溶融樹脂排出工程は、前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する。
既述のように、ポリエステル原料樹脂の融点Tmは、ポリエステル原料樹脂の融点が1つである場合はその融点を指すが、ポリエステル原料樹脂が複数の融点を有する場合には、最も高い融点をポリエステル原料樹脂の融点Tmとする。より具体的には、ポリエステル原料樹脂について示差走査熱量測定(DSC)を行ったときに、横軸を温度、縦軸を熱量とするDSC曲線において、1つの吸熱ピークP1を有するときは、吸熱ピークP1のピーク温度をポリエステル原料樹脂の融点とする。DSC曲線において、2つ以上の吸熱ピークP2−1、P2−2、・・・P2−nを有するときは、吸熱ピークP2−1〜P2−nのうち、最も高温で生じた吸熱ピークのピーク温度をポリエステル原料樹脂の融点とする。
【0119】
原料樹脂供給工程により、押出機内に供給された原料樹脂は、溶融樹脂排出工程において、溶融混練され、溶融樹脂(メルトともいう)とされると共に、所定の温度で押出機から排出される。
押出機出口における溶融樹脂の温度が「ポリエステル原料樹脂の融点Tm+20℃」未満であると、原料樹脂の溶融不足により、溶融樹脂中に未溶融異物が残存し、「ポリエステル原料樹脂の融点Tm+30℃」を超えるとΔAVが増加する。
【0120】
押出機出口における溶融樹脂の温度は、押出機出口の設定温度を制御することにより調整することができる。
押出機出口における溶融樹脂の温度は、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+27℃であることが好ましい。
次に、原料樹脂を供給し、溶融混練する二軸押出機の詳細について説明する。
【0121】
(二軸押出機)
まず、本発明で用いる二軸押出機について説明する。
二軸押出機は、主として押出機に供給された原料樹脂を溶融し、溶融樹脂を混練するものであり、一般的に、バレル(シリンダーともいう)を備える。バレルは、原料樹脂を供給するための原料供給口と、溶融樹脂を排出する出口(排出口)とを有し、バレル内には、溶融樹脂を混練するために、スクリューが備えられている。押出機は、大別して、スクリューが1本である単軸と、スクリューが複数本ある多軸とがあり、本発明においては、二軸押出機(二軸スクリュー押出機)を用いる。二軸押出機は、ポリエステル原料樹脂を可塑化するニーディング部を有するスクリューを備えていることが好ましい。
【0122】
なお、ニーディング部とは、スクリューの部位であって、ポリエステル原料樹脂を可塑化する機能を有するセグメント(ニーディングディスクの位置)を意味し、可塑部とは、押出機内の領域であって、ポリエステル原料樹脂が可塑化される領域を指す。従って、押出機の可塑部にスクリューのニーディング部が位置する。
【0123】
以下、押出機の構成を、図1および図2を用いて説明する。
図1は、押出機の構成例を概略的に示している。
図2は、本発明に係るポリエステルフィルム製造方法を実施するフローの一例を示している。
【0124】
図1に示す押出機100は、スクリューを二本(二軸)有する二軸押出機である。押出機100は、供給口12及び押出機出口14を有するバレル10(シリンダー)と、バレル10内で回転する2つのスクリュー20A、20Bと、バレル10の周囲に配置され、バレル10内の温度を制御する温度制御手段30と、を備えている。また、図2に示すように、供給口12の手前には原料供給装置46が設けられている。押出機出口14の先にはギアポンプ44と、フィルター42と、ダイ40が設けられている。原料供給装置46は、第1の樹脂を収納する第1の原料供給装置と、第2の樹脂を収納する第2の原料供給装置とを少なくとも含む複数の原料供給装置で構成されていてもよいし、複数の樹脂を個別に収納可能な複数の収納容器を備えた1つの原料供給装置であってもよい。
【0125】
−バレル−
バレル10は原料樹脂を供給するための供給口12と、加熱溶融された原料樹脂が押し出される押出機出口14を有する。
バレル10の内壁面は、耐熱、耐磨耗性、及び腐食性に優れ、樹脂との摩擦が確保可能な素材を用いることが必要である。一般的には内面を窒化処理した窒化鋼が使用されているが、クロムモリブデン鋼、ニッケルクロムモリブデン鋼、ステンレス鋼を窒化処理して用いることもできる。特に耐摩耗性、耐食性を要求される用途では、遠心鋳造法によりニッケル、コバルト、クロム、タングステン等の耐腐食性、耐磨耗性素材合金をバレル10の内壁面にライニングさせたバイメタリックバレルを用いることや、セラミックの溶射皮膜を形成させることが有効である。
【0126】
バレル10には真空を引くためのベント16A、16Bも設けられている。ベント16A、16Bを通じて真空引きをすることでバレル10内の樹脂中の水分等の揮発成分を効率的に除去することができる。ベント16A、16Bを適正に配置することにより、未乾燥状態の原料(ペレット、パウダー、フレークなど)や成膜途中で出たフィルムの粉砕屑(フラフ)等をそのまま原料樹脂として使用することができる。
ベント16A、16Bは脱気効率との関係で、開口面積やベントの数を適正にすることが求められる。本発明で用いる二軸押出機100は、1箇所以上のベント16A、16Bを有することが望ましい。なお、ベント16A、16Bの数が多過ぎると、溶融樹脂がベントから溢れ出るおそれ、滞留劣化異物増加の懸念があるので、ベントは1箇所又は2箇所設けることが好ましい。
また、ベント付近の壁面に滞留した樹脂や析出した揮発成分が押出機100(バレル10)の内部に落下すると、製品に異物として顕在化する可能性があり、注意が必要である。滞留については、ベント蓋の形状の適正化や、上部ベント、側面ベントの適正な選定が有効であり、揮発成分の析出は、配管等の加熱で析出を防止する手法が一般的に用いられる。
【0127】
例えば、PETを押出す場合、加水分解、熱分解、酸化分解の抑制が製品(フィルム)の品質に大きな影響を及ぼす。
例えば、樹脂供給口12を真空化したり、後述する窒素ガス供給口からの窒素供給(窒素パージともいう)を行うことで酸化分解を抑えることができる。
また、剪断発熱による樹脂分解を抑えるため、押出と脱気が両立できる範囲にはニーディング部等のセグメントは極力設けないことが好ましい。
また、スクリュー出口(押出機出口)14の圧力が大きいほど剪断発熱が大きくなるため、ベント16A、16Bによる脱気効率と押出の安定性が確保できる範囲内で、押出機出口14の圧力は極力低くすることが好ましい。
【0128】
−スクリュー−
バレル10内には、モータおよびギアを含む駆動手段21によって回転する2つのスクリュー20A、20Bが設けられている。
本発明のポリエステルフィルム製造方法においては、押出機100は、スクリューの直径をD〔mm〕とするとき、スクリュー径Dが、60mm以上であることが好ましく、140mm以上であることがより好ましい。スクリュー径Dを60mm以上とすることで押出機内における溶融樹脂の滞留を抑制することができ、ポリエステルフィルムの生産性を向上することができる。
大量生産の観点から、スクリュー径Dは、さらに好ましくは160mm以上である。一方、樹脂の溶融ムラを抑制する観点から、スクリュー径Dは200mm以下であることが好ましい。
【0129】
引き続き、図1および図2を用いて、押出機が備えるスクリューの好ましい態様について説明する。
二軸押出機は、2つのスクリュー20A、20Bの噛み合い型と非噛み合い型に大別され、噛み合い型のほうが、非噛み合い型よりも混練効果が大きい。本発明では、噛み合い型と非噛み合い型のいずれのタイプでも良いが、原料樹脂を十分混練して溶融ムラを抑制する観点から、噛み合い型を用いることが好ましい。
2つのスクリュー20A、20Bの回転方向もそれぞれ同方向と異方向に分かれる。異方向回転スクリュー20A、20Bは同方向回転型よりも混練効果が高く、同方向回転型は自己清掃効果を持っているため、押出機内の滞留防止には有効である。
さらに軸方向も平行と斜交があり、強いせん断を付与する場合に用いられるコニカルタイプの形状もある。
【0130】
本発明で用いる二軸押出機では、様々な形状のスクリューセグメントが用いられる。スクリュー20A、20Bの形状としては、例えば、等ピッチの1条のらせん状フライト22が設けられたフルフライトスクリューが用いられる。
ポリエステル原料樹脂を可塑化する可塑部に、ニーディングディスクやローターなどの剪断を付与するセグメントを用いることで、原料樹脂をより確実に溶融することができる。また、逆スクリューやシールリングを用いることにより、樹脂をせき止め、ベント16A、16Bを引く際のメルトシールを形成することができる。例えば、図1に示すように、ベント16A、16B付近に、上記のような原料樹脂の溶融を促進するニーディング部(混練部)24A、24Bを設けることができる。
【0131】
スクリューの部位のうち、ニーディングディスクが設けられているニーディング部は、ニーディングクリアランスにおける剪断速度が500s−1〜2000s−1であることが好ましい。
ニーディングクリアランスとは、隣接するニーディングディスク間の間隔をいう。
剪断速度が500s−1以上であることで、原料樹脂の溶融不足による未溶融異物の残存を抑制することができ、剪断速度が2000s−1以下であることで、原料樹脂の発熱によるΔAVの増加を抑制することができる。
ニーディングクリアランスにおける剪断速度は、500s−1〜1500s−1であることがより好ましく、500s−1〜800s−1であることが特に好ましい。
【0132】
押出機100の後半では溶融樹脂を冷却するための温調ゾーン(冷却部)が有効である。剪断発熱よりもバレル10の伝熱効率が高い場合は、温調ゾーン(冷却部)にピッチの短いスクリュー28を設けることで、バレル10壁面の樹脂移動速度が高まり、温調効率を上げることができる。冷却効果を高める観点から、冷却部に位置するスクリュー28のピッチは、スクリュー径Dに対し、0.5D〜0.8Dであることが好ましい。
【0133】
−窒素ガス供給口−
押出機100は、原料供給口に最も近いベント孔よりも前記原料供給口に近い位置に、前記ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給する第1の窒素ガス供給口を備えることが好ましい。
なお、「ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガス」とは、ポリエステル原料樹脂の供給量をQ〔L/min〕としたとき、窒素ガスの供給量が1.1Q〔L/min〕〜10.0Q〔L/min〕であることを意味する。
図1においては、ベント16Aよりも原料樹脂の供給口12に近い位置に、窒素ガス供給口を備え、かかる窒素ガス供給口から、ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給することが好ましい。
バレル10内において原料樹脂は、上流側の原料供給口12から、下流側の押出機出口14へと溶融混練されながら進行する。第1の窒素ガス供給口は、原料供給口12に最も近いベント16Aよりも上流に位置する。
【0134】
押出機内の溶融樹脂の酸化分解を抑制するために、バレル10内に窒素ガスを供給することは、従来から行なわれていたが、従来は、バレル10から窒素ガスを供給したり、スクリュー20A、20Bの駆動手段21から供給するものであった。しかしながら、押出機内の原料樹脂の酸化分解を抑制するには、溶融している原料樹脂自体を窒素置換する必要があり、バレル10から窒素ガスを供給したり、駆動手段21から供給する手法では、十分に、溶融樹脂に窒素ガスが供給されず、溶融樹脂の酸化分解を抑制することができなかった。
従って、第1の窒素ガス供給口を、原料供給口に最も近いベント孔よりも前記原料供給口に近い位置に備えることで、押出機内の原料樹脂に窒素ガスを供給することができる。
例えば、図1においては、N1またはN2の位置に窒素ガス供給口を設けることが好ましい。
【0135】
特に、窒素ガス供給量を、ポリエステル原料樹脂の供給量の1.1倍以上とすることで、原料樹脂の酸化を十分に抑制することができ、10.0倍以下とすることで、原料供給口に位置する原料樹脂を吹き飛ばしにくく、原料樹脂の供給不良を抑制することができる。窒素ガス供給量は、ポリエステル原料樹脂の供給量の1.1倍〜8.0倍とすることが好ましい。
【0136】
また、押出機100は、押出機100が備えるスクリュー20A、20Bのニーディング部(24Aおよび24Bの少なくとも一方)に位置し、ポリエステル原料樹脂に対して窒素ガスを供給する第2の窒素ガス供給口を備えていることが好ましい。
原料樹脂の酸化分解や熱分解は、原料樹脂が溶融し始める段階〜溶融している最中に起こり易く、原料樹脂がかかる状態であるときに、原料樹脂に対して窒素ガスを供給することが好ましい。原料樹脂は、原料を混練するニーディング部において、溶融し、熱分解を起こしやすくなるため、かかる原料樹脂に対して直接窒素ガスを供給し易いように、ニーディング部に窒素ガス供給口を供えていることが好ましい。
【0137】
原料樹脂の熱分解は、特に、溶融し始める段階において生じやすいため、第2の窒素ガス供給口は、ニーディング部24A、24Bの中央(ニーディング部24Aにおいては、図1におけるN2)よりも、ニーディング部の上流側端部(ニーディング部24Aにおいては、図1におけるN1)に備えられていることが好ましい。
【0138】
図1において、ニーディング部は、原料樹脂の進行方向上流側に位置する24Aと下流側に位置する24Bとが示されているが、本発明において、ニーディング部に位置する第2の窒素ガス供給口は、1つのみでも2つ以上あってもよい。すなわち、図1においては、第2の窒素ガス供給口は、ニーディング部24Aの位置および、ニーディング部24Bの少なくとも一方に備えられていればよい。
【0139】
第1の窒素ガス供給口および第2の窒素ガス供給口における窒素ガスの供給速度は、2m/分〜50m/分であることが好ましい。窒素ガスの供給速度が、2m/分以上であることで原料樹脂の酸化抑制を十分に行なうことができ、50m/分以下とすることで、樹原料樹脂の樹脂温度の低下を抑制し、原料樹脂の可塑化発熱を抑制することができる。
窒素ガスの供給速度は、3m/分〜45m/分であることがより好ましい。
【0140】
−温度制御手段−
バレル10の周囲には、温度制御手段30が設けられている。図1に示す押出機100では、原料供給口12から押出機出口14に向けて長手方向に9つに分割された加熱/冷却装置C1〜C9が温度制御手段30を構成している。このようにバレル10の周囲に分割して配置された加熱/冷却装置C1〜C9によって、例えば可塑部C1〜C7と冷却部C8、C9の各領域(ゾーン)に区画し、バレル10内を領域ごとに所望の温度に制御することができる。
【0141】
加熱は、通常バンドヒーターまたはシーズ線アルミ鋳込みヒーターが用いられるが、これらに限定されず、例えば熱媒循環加熱方法も用いることができる。一方、冷却はブロワーによる空冷が一般的であるが、バレル10の周囲に巻き付けたパイプに水または油を流す方法もある。
【0142】
‐原料樹脂の溶融混練‐
押出機100は、温度制御手段30によりバレル10を加熱するとともにスクリューを回転させ、供給口12から原料樹脂を供給する。なお、供給口12は、原料樹脂のペレット等が加熱されて融着しないようにすることと、モータなどのスクリュー駆動設備を保護するため、伝熱防止として冷却することが好ましい。
【0143】
バレル内に供給された原料樹脂は、温度制御手段30による加熱のほか、スクリュー20A、20Bの回転に伴う樹脂同士の摩擦、樹脂とスクリュー20A、20Bやバレル10との摩擦などによる発熱によって溶融されるとともに、スクリューの回転に伴って押出機出口14に向けて徐々に移動する。
バレル内に供給された原料樹脂は融点Tm℃以上の温度に加熱されるが、樹脂温度が低過ぎると溶融押出時の溶融が不足し、ダイ40からの排出が困難になるおそれがあり、樹脂温度が高過ぎると熱分解によって末端COOHが著しく増加して耐加水分解性の低下を招くおそれがある。なお、既述のように、原料樹脂の融点Tmは、原料樹脂を複数種用いることにより、Tmが複数ある場合は、最も高い融点をいう。
これらの観点から、温度制御手段30による加熱温度及びスクリュー20A、20Bの回転数を調整することにより、二軸押出機内の長手方向における最大樹脂温度Tmaxを(Tm+40)℃〜(Tm+60)℃にすることが好ましく、(Tm+40)℃〜(Tm+55)℃とすることがより好ましく、(Tm+45)℃〜(Tm+50)℃とすることがさらに好ましい。
【0144】
二軸押出機内の長手方向における最大樹脂温度Tmaxは、二軸押出機100のスクリュー20A、20Bが配設されたバレル内で加熱されている原料樹脂の温度であり、剪断発熱があるときはその発熱による局所的高温部を含む温度である。Tmaxはバレル内の樹脂温度の測定により得られる。上記のTm及びTmaxの関係式において、PET樹脂の場合、Tmax[℃]は、末端COOHの増加を抑える観点から、290℃以下が好ましく、280℃以下がより好ましい。また、Tmaxの下限温度は、樹脂の溶融不足を防止する観点から260℃とすることが好ましい。
【0145】
‐ベント圧力‐
ベント16A、16Bを通じて真空引きをすることでバレル内の樹脂中の水分等の揮発成分を効率的に除去することができる。ベント圧力が低過ぎると溶融樹脂がバレル10の外に溢れ出るおそれがあり、ベント圧力が高過ぎると揮発成分の除去が不十分となり、得られたフィルムの加水分解が生じ易くなるおそれがある。溶融樹脂がベント16A、16Bから溢れ出ることを防ぐとともに揮発成分を選択的に除去する観点から、ベント圧力は0.01Torr〜5Torr(1.333Pa〜666.5Pa)とすることが好ましく、0.01Torr〜4Torr(1.333Pa〜533.2Pa)とすることがより好ましい。
【0146】
‐平均滞留時間‐
バレル内で原料樹脂を加熱溶融し、押出機出口14を出た後、ダイ40からフィルム状に押出されるまでの平均滞留時間を10分〜20分とする。原料樹脂を加熱溶融して、押出機100の押出機出口14を出てからダイ40から押出されるまでの平均滞留時間が10分未満では未溶融樹脂が残留し易く、一方、20分を超えると、熱分解によって末端COOH量が増加して耐加水分解性が低下する。このような観点から、原料樹脂を加熱溶融して押出機出口14から押出され後の上記平均滞留時間は、10分〜20分が好ましく、10分〜15分がより好ましい。
ここで、平均滞留時間は、下記式で定義される。
平均滞留時間(秒)=押出機下流配管容積(cm3)×溶融体密度(g/cm3)×3600/1000÷押出量(kg/hr)
【0147】
‐冷却‐
上記のように原料樹脂をバレル内で加熱溶融する一方、温度制御手段30によりバレル10の押出機出口14側の内壁がポリエステル樹脂(原料樹脂)の融点Tm(℃)以下の冷却部となるように制御する。バレル10の押出機出口14側の内壁を冷却部として原料樹脂の融点Tm(℃)以下に制御すれば、樹脂が過剰に加熱されて末端COOH量が増加することを抑制することができる。末端COOH量の増加を確実に抑制する観点から、かかる冷却部における温度は、(Tm−100)℃〜Tm℃の範囲内が好ましく、(Tm−50)℃〜(Tm−10)℃の範囲内がより好ましい。
【0148】
冷却部の長さは、スクリュー径Dに対し、4D〜11Dにすることが好ましい。冷却部の長さが4D以上であれば、溶融加熱された樹脂を効果的に冷却して末端COOHの増加を抑制する。一方、冷却部の長さが11D以下であれば、樹脂を冷却し過ぎて固化することを防ぎ、溶融押出しを円滑に行うことができる。
なお、押出機出口14における樹脂温度は、既述のように、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃にする。
【0149】
〔成形工程〕
本発明のポリエステルフィルム製造方法は、押出機から押出した溶融樹脂をフィルム状に成膜する成形工程を有する。
本発明のポリエステルフィルム製造方法においては、上記構成の樹脂供給工程を経た後、押出機から押出した溶融樹脂をフィルム状に成膜する。
押出機から押出した溶融樹脂は、図2に示すように、ギアポンプ44、フィルター42を介してダイ40から排出される。ダイ40の開口部の形状が長尺状の幅広な形状をしていることで、ダイ40から排出される溶融樹脂は、フィルム状に加工される。
【0150】
−ダイ−
図1に示すバレル10の押出機出口14には、押出機出口14から押出された溶融樹脂をフィルム状(帯状)に排出するためのダイ40(図2)が設けられている。また、バレル10の押出機出口14とダイ40との間には、フィルムに未溶融樹脂や異物が混入することを防ぐためのフィルター42が設けられている。
【0151】
−ギアポンプ−
厚み精度を向上させるためには、押出量の変動を極力減少させることが重要である。押出量の変動を極力減少させるために押出機100とダイ40との間にギアポンプ44を設けてもよい。ギアポンプ44から一定量の樹脂を供給することにより、厚み精度を向上させることができる。特に、二軸スクリュー押出機を用いる場合には、押出機自身の昇圧能力が低いため、ギアポンプ44による押出安定化を図ることが好ましい。
【0152】
ギアポンプ44を用いることにより、ギアポンプ44の2次側の圧力変動を1次側の1/5以下にすることも可能であり、樹脂圧力変動幅を±1%以内にできる。その他のメリットとしては、スクリュー先端部の圧力を上げることなしにフィルターによる濾過が可能なことから、樹脂温度の上昇の防止、輸送効率の向上、及び押出機内での滞留時間の短縮が期待できる。また、フィルターの濾圧上昇が原因で、スクリューから供給される樹脂量が経時変動することも防止できる。ただし、ギアポンプ44を設置すると、設備の選定方法によっては設備の長さが長くなり、樹脂の滞留時間が長くなることと、ギアポンプ部のせん断応力によって分子鎖の切断を引き起こすことがあり注意が必要である。
【0153】
ギアポンプ44は1次圧力(入圧)と2次圧力(出圧)の差を大きくし過ぎると、ギアポンプ44の負荷が大きくなり、せん断発熱が大きくなる。そのため、運転時の差圧は20MPa以内、好ましくは15MPa、更に好ましくは10MPa以内とする。また、フィルム厚みの均一化のために、ギアポンプ44の一次圧力を一定にするために、押出機のスクリュー回転を制御したり、圧力調節弁を用いたりすることも有効である。
【0154】
なお、本発明のポリエステルフィルムの製造方法においては、固有粘度(IV)が0.70dl/g以上の原料樹脂を用いるため、成形工程においては、原料樹脂をバレル内で加熱溶融して押出機出口14から押出された後、10分〜20分の平均滞留時間を経て、スクリュー径Dを考慮してスクリュー回転数N〔rpm〕と押出量Q〔kg/hr〕を制御することで下記式(i)を満たす条件下でフィルム状に溶融押出しを行うことが好ましい。
6.0×10−6×D3≦Q/N≦1.1×10−5×D3 ・・・式(i)
IVが0.7dl/g以上の原料樹脂を溶融する場合、Nを低下させることで溶融と脱気、樹脂冷却を同時に満たし易い。また、押出機出口14での樹脂温度を特に290℃以下に制御することで、特にその下流の配管滞留での末端COOHの増加抑制に大きな効果がある。
Q/Nが6.0×10−6×D3以上とすることで、スクリュー20A、20Bの高回転による原料樹脂の過発熱を抑制し、押出機出口14における樹脂温度を290℃以下にし易く、ΔAVを3eq/t以下にし易い。また、Q/Nが1.1×10−5×D3以下であることで、ベント直下の樹脂充填率が増加しにくく、ベント16A、16Bから溶融樹脂が溢れにくくなるほか、ベント圧が低下しにくいため、押出機内部での樹脂の加水分解が進行しにくく、末端COOHの発生を抑制し易い。さらに、未溶融樹脂がフィルムに混入しにくくなり、ポリエステルフィルムの強度が低下することを抑制することができるので、延伸工程におけるフィルム破断を抑制することができる。
なお、原料樹脂の押出機への供給量〔kg/hr〕と押出機内の押出量とは同様に扱うことができ、原料樹脂の押出機への供給量がQ〔kg/hr〕であるとき、押出機内の押出量はQ〔kg/hr〕であると考えてよい。
【0155】
上記溶融押出しは、下記式(ii)に示す条件で行なうことがより好ましく、下記式(iii)に示す条件下で行うことがさらに好ましい。
7×10−6×D3≦Q/N≦1×10−5×D3 ・・・式(ii)
8×10−6×D3≦Q/N≦9×10−6×D3 ・・・式(iii)
【0156】
スクリュー回転数Nが低過ぎると、温度制御手段30によって温度ムラが生じて未溶融樹脂が生じ易く、スクリュー回転数Nが高過ぎると、過度に発熱して末端COOH量の増加につながるため、スクリュー回転数Nは1.9×102×D−0.5rpm〜8.4×102×D−0.5rpmが好ましく、6.3×102×D−0.5rpm〜7.9×102×D−0.5rpmがより好ましい。
また、押出量Qが少な過ぎると過度に加熱され易くなり、多過ぎると未溶融樹脂が生じ易くなるため、押出量Qは1.1×10−3×D2.5kg/hr〜7.6×10−3×D2.5kg/hrが好ましく、3.8×10−3×D2.5kg/hr〜7.1×10−3×D2.5kg/hrがより好ましい。
【0157】
バレル10の押出機出口14から押し出された樹脂をフィルター42に通してダイ40から(例えば冷却ロールに)押し出してフィルム状に成形する。
ダイ40からメルト(溶融樹脂)を押出した後、冷却ロールに接触させるまでの間(エアギャップ)は、湿度を5%RH〜60%RHに調整することが好ましく、15%RH〜50%RHに調整することがより好ましい。エアギャップでの湿度を上記範囲にすることで、フィルム表面のCOOH量やOH量を調節することが可能であり、低湿度に調節することで、フィルム表面のカルボン酸量を減少させることができる。
【0158】
また、本発明の方法によれば、樹脂温度を一度上げてから冷却部で下げることで、末端COOH量の増加を抑制するとともに、未溶融異物の発生を抑制することができるほか、フィルムのヘイズ上昇を抑制する効果が得られる。特に厚手成膜をする際は冷却速度不足より、ヘイズ上昇しやすいが、その対策方法として用いることが可能である。
なお、フィルム厚は、2mm〜8mmが好ましく、より好ましくは2.5mm〜7mmであり、さらに好ましくは3mm〜6mmである。厚みを厚くすることで、押出されたメルトがガラス転移温度(Tg)以下に冷却するまでの所要時間を長くすることができる。この間に、フィルム表面のCOOH基はポリエステル内部に拡散され、表面COOH量を低減することができる。
【0159】
上記工程により、原料樹脂の末端COOH量と溶融押出しされたフィルムの末端COOH量との差ΔAVが3eq/t以下のポリエステルフィルムを製造することができ、例えば、末端COOH量が25eq/t(トン)以下であるポリエステルフィルムが得られる。末端COOH量が25eq/トン以下であると、耐加水分解性に優れており、長期耐久性が得られる。末端COOH量は、耐加水分解の点では低いことが望ましいが、フィルムを被着物に密着させる場合の密着性向上の点から、2eq/トン以上が好ましい。中でも、10〜20eq/トンの範囲がより好ましい。
末端COOH量の測定は、既述の方法と同様にして行なうことができる。
【0160】
<太陽電池用ポリエステルフィルム>
本発明の太陽電池用ポリエステルフィルムは、既述の本発明のポリエステルフィルム製造方法により製造することができる。
本発明の太陽電池用ポリエステルフィルムは、本発明のポリエステルフィルム製造方法により製造されるため、高い耐加水分解性を有することができる。
【0161】
また、太陽電池用ポリエステルフィルムは、1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%含むCHDM系ポリエステル樹脂を含有する層を少なくとも1層有することで、フィルム耐候性をさらに向上することができる。
CHDM系ポリエステル樹脂を含有する層は、本発明のポリエステルフィルム製造方法により製造されたポリエステルフィルムであってもよいし、本発明のポリエステルフィルム製造方法により製造されたポリエステルフィルム上に押出成形した層であってもよいし、本発明のポリエステルフィルム製造方法により製造されたポリエステルフィルムに貼り付けたフィルムないしシート状の部材であってもよい。
【0162】
このように、太陽電池用ポリエステルフィルムは、CHDM系ポリエステル樹脂を含有する層を少なくとも1層有していればよく、単層であっても、2以上の層を有していてもよい。
特に、CHDM系ポリエステル樹脂が有するCHDM由来の構造が、ジオール化合物由来の構造の全量に対して80モル%〜100モル%であるときは、太陽電池用ポリエステルフィルムは、積層構成にすることが好ましい。これは、CHDM系ポリエステル樹脂中のジオール化合物由来の構造の全量に対するCHDM由来の構造の比率が高くなると、ポリエチレンテレフタレート(PET)に対し、耐候性(耐加水分解性)は高くなり易いが、力学強度が弱くなり易い。 このため、他のポリエステル(例えばPET)と積層することで相補することができ、好ましい。
【0163】
本発明の太陽電池用ポリエステルフィルムは、CHDM系ポリエステル樹脂を含有する層(P1層と称する)と、ポリエチレンテレフタレートを主成分とするポリエステルを含有する層(P2層と称する)とが積層された態様も好ましい。
なお、本発明の太陽電池用ポリエステルフィルムは、P1層とP2層とをそれぞれ2層以上有していてもよい。
【0164】
P2層は、ジカルボン酸由来の構造中にテレフタル酸由来の構造を95モル%以上有し、かつジオール化合物由来の構造中にエチレングリコール由来の構造を95モル%以上含むポリエステル樹脂を含む層を指す。
またP2層のIVは0.7〜0.9が好ましく、より好ましくは0.72〜0.85、さらに好ましくは0.74〜0.82である。このようにP2層のIVを高めにすることでwetサーモ、および、dryサーモでの分解(ポリエステル樹脂の分子量低下)を抑制することができる。
【0165】
本発明の太陽電池用ポリエステルフィルムは、P1層とP2層の層数の和は、2層以上が好ましく、より好ましくは2層以上5層以下、さらに好ましくは2層以上4層以下である。中でも好ましいのが、P2層の両側をP1層で挟んだ3層構造、あるいはP1層の両側をP2層で挟んだ3層構造、P2層とP1層を積層した2層構造である。
【0166】
本発明の太陽電池用ポリエステルフィルムが2層以上で構成される場合、太陽電池用ポリエステルフィルムの厚みは、P1層の総和が全厚みの5%〜40%であることが好ましく、より好ましくは7%〜38%、さらに好ましくは10%〜35%である。この下限値以上にすることで高い耐候性を発現でき、この上限値以下にすることで高い力学強度を発現し易い。
ポリエステルフィルムの各層の厚みは、フィルムの断面を、SIMS(Secondary Ion-microprobe Mass Spectrometer;二次イオン質量分析計)を用い測定し、P1層の特徴フラグメント、および、P2層の特徴フラグメントでイメージングすることで求めることができる。
【0167】
このような太陽電池用ポリエステルフィルムの積層構造は定法により得ることができる。例えば、P1層を構成するポリエステル樹脂をある押出機に供給し、P2層を構成するポリエステル樹脂を他の押出機に供給する等して、複数の押出し機から供給されたメルト(樹脂の融体)をマルチマニフォールドダイ、フィードブロックダイを用い積層し押出してもよい。また、予め用意したP1層およびP2層のどちらか一方となるポリエステルフィルム上に、他方の層を構成するポリエステル樹脂を押し出して積層してもよいし、他の層を貼り付けて積層してもよい。
【0168】
本発明の方法により製造される太陽電池用ポリエステルフィルムは、光安定化剤、酸化防止剤などの添加剤を更に含有することができる。
【0169】
光安定化剤を含有すると、紫外線劣化を防ぐことができる。光安定化剤とは、紫外線などの光線を吸収して熱エネルギーに変換する化合物、樹脂が光吸収して分解して発生したラジカルを捕捉し、分解連鎖反応を抑制する材料などが挙げられる。光安定化剤として好ましくは、紫外線などの光線を吸収して熱エネルギーに変換する化合物である。このような光安定化剤を含有することで、長期間継続的に紫外線の照射を受けても、部分放電電圧の向上効果を長期間高く保つことが可能になったり、樹脂中の紫外線による色調変化、強度劣化等が防止される。
【0170】
例えば紫外線吸収剤は、ポリエステルの他の特性が損なわれない範囲であれば、有機系紫外線吸収剤、無機系紫外線吸収剤、及びこれらの併用のいずれも、特に限定されることなく好適に用いることができる。一方、紫外線吸収剤は、耐湿熱性に優れ、樹脂中に均一分散できることが望まれる。
【0171】
紫外線吸収剤の例としては、有機系の紫外線吸収剤として、サリチル酸系、ベンゾフェノン系、ベンゾトリアゾール系、シアノアクリレート系等の紫外線吸収剤及びヒンダードアミン系等の紫外線安定剤などが挙げられる。具体的には、例えば、サリチル酸系のp−t−ブチルフェニルサリシレート、p−オクチルフェニルサリシレート、ベンゾフェノン系の2,4−ジヒドロキシベンゾフェノン、2−ヒドロキシ−4−メトキシベンゾフェノン、2−ヒドロキシ−4−メトキシ−5−スルホベンゾフェノン、2,2’,4,4’−テトラヒドロキシベンゾフェノン、ビス(2−メトキシ−4−ヒドロキシ−5−ベンゾイルフェニル)メタン、ベンゾトリアゾール系の2−(2’−ヒドロキシ−5’−メチルフェニル)ベンゾトリアゾール、2−(2’−ヒドロキシ−5’−メチルフェニル)ベンゾトリアゾール、2,2’−メチレンビス[4−(1,1,3,3−テトラメチルブチル)−6−(2Hベンゾトリアゾール−2−イル)フェノール]、シアノアクリレート系のエチル=α−シアノ−β,β−ジフェニルアクリレート)、トリアジン系として2−(4,6−ジフェニル−1,3,5−トリアジン−2−イル)−5−[(ヘキシル)オキシ]−フェノール、ヒンダードアミン系のビス(2,2,6,6−テトラメチル−4−ピペリジル)セバケート、コハク酸ジメチル・1−(2−ヒドロキシエチル)−4−ヒドロキシ−2,2,6,6−テトラメチルピペリジン重縮合物、そのほかに、ニッケルビス(オクチルフェニル)サルファイド、及び2,4−ジ・t−ブチルフェニル−3’,5’−ジ・t−ブチル−4’−ヒドロキシベンゾエート、などが挙げられる。
これらの紫外線吸収剤のうち、繰り返し紫外線吸収に対する耐性が高いという点で、トリアジン系紫外線吸収剤がより好ましい。なお、これらの紫外線吸収剤は、上述の紫外線吸収剤単体でフィルムに添加してもよいし、有機系導電性材料や、非水溶性樹脂に紫外線吸収剤能を有するモノマーを共重合させた形態で導入してもよい。
【0172】
光安定化剤のポリエステルフィルム中における含有量は、ポリエステルフィルムの全質量に対して、0.1質量%以上10質量%以下が好ましく、より好ましくは0.3質量%以上7質量%以下であり、さらに好ましくは0.7質量%以上4質量%以下である。これにより、長期経時での光劣化によるポリエステルの分子量低下を抑止でき、その結果発生するフィルム内の凝集破壊に起因する密着力低下を抑止できる。
【0173】
更に、本発明のポリエステルフィルムは、前記光安定化剤の他にも、例えば、易滑剤(微粒子)、紫外線吸収剤、着色剤、核剤(結晶化剤)、難燃化剤などを添加剤として含有することができる。
【0174】
<太陽電池用バックシート>
本発明の太陽電池用ポリエステルフィルムは、太陽電池モジュールの太陽光入射側とは反対側の裏面に配置される裏面保護シート(太陽電池用バックシート)、バリアフィルム基材等の用途に好適である。
【0175】
<太陽電池モジュール>
本発明の太陽電池モジュールは、太陽光が入射する透明性のフロント基板と、前記フロント基板の上に設けられ、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分と、前記セル構造部分の前記フロント基板が位置する側と反対側に設けられ、前記封止材と隣接して配置された、本発明の太陽電池用ポリエステルフィルム(例えば、太陽電池用バックシート)と、を備えて構成される。
【0176】
太陽電池モジュールは、例えば、電気を取り出すリード配線で接続された発電素子(太陽電池素子)をエチレン・酢酸ビニル共重合体系(EVA系)樹脂等の封止剤で封止し、これを、ガラス等の透明基板と、本発明のポリエステルフィルムの製造方法によって得られたポリエステルフィルム(バックシート)との間に挟んで互いに張り合わせることによって構成してもよい。
太陽電池素子の例としては、単結晶シリコン、多結晶シリコン、アモルファスシリコンなどのシリコン系、銅−インジウム−ガリウム−セレン、銅−インジウム−セレン、カドミウム−テルル、ガリウム−砒素などのIII−V族やII−VI族化合物半導体系など、各種公知の太陽電池素子を適用することができる。
【実施例】
【0177】
以下、本発明を実施例により更に具体的に説明するが、本発明はその主旨を越えない限り、以下の実施例に限定されるものではない。
【0178】
〔実施例1〜実施例16、比較例1〜比較例11〕
‐二軸押出機‐
押出機として、図1に示すように2箇所にベントが設けられたバレル内に、下記構成のスクリューを備え、バレルの周囲には長手方向に9つのゾーンに分割して温度制御を行うことができるヒータ(温度制御手段)を備えたダブルベント式同方向回転噛合型の二軸押出機を準備した。
スクリュー径D:110mm
スクリュー長L:スクリュー径Dとの比(L/D)が31.5となる長さ[mm]
(1ゾーンの幅:3.5D)
スクリュー形状:第1ベント直前に可塑化ニーディング部を設置
第2ベント直前に脱気促進ニーディング部を設置
可塑化ニーディング部の剪断速度は、表1に示す速度〔s−1〕とした。
ベント位置:第1ベント・・・図1のC5の位置
第2ベント・・・図1のC7の位置
【0179】
二軸押出機の押出機出口以降には、図2に示すように、下記構成のギアポンプ、金属繊維フィルターおよびダイを接続し、ダイを加熱するヒーターの設定温度は280℃とし、平均滞留時間は10分とした。
ギアポンプ:2ギアタイプ
フィルター:金属繊維焼結フィルター(孔径20μm)
【0180】
‐原料樹脂‐
AVが13eq/tであるポリエチレンテレフタラート樹脂(PET樹脂)のペレットと、及び、AVが15eq/tであり、表1に示す嵩密度を有するフラフとを用いて、原料樹脂とした。なお、比較例1及び比較例2では、フラフを含まず、嵩密度が0.8のPETペレットのみを用いて、表1に示すAVの原料樹脂とした。原料樹脂の融点Tmは、いずれも255℃であった。
また、原料樹脂中のフラフの含有量は表1に示す量である。なお、表1に示すフラフの含有率は、原料樹脂の全質量に対するフラフの割合(質量%)である。
【0181】
押出機は、図1における供給口12側の1番目のゾーン(C1)は70℃に、2〜8番目のゾーン(C2〜C8)は270℃に、9番目のゾーン(C9)は250℃にそれぞれ温度設定を行った。
スクリューの回転数を77rpmに設定し、供給口12から、表1の「押出機入口樹脂温度」に示す温度〔℃〕で、原料樹脂を供給して加熱溶融し、押出量を350kg/hrに設定して溶融押出を行った。
【0182】
押出機内の原料樹脂に対しては、図1のN1に示す位置に窒素ガス供給口を設け、原料樹脂の供給量Q〔L/min〕に対して、表1に示す倍率の供給量〔L/min〕の窒素ガスを供給した。例えば、実施例1においては、原料樹脂の供給量Qの0.5倍(0.5Q)〔L/min〕である。
また、窒素ガスの供給速度は、表1に示す速度〔m/min〕とした。
【0183】
溶融樹脂は、表1の「押出機出口樹脂温度」に示す温度〔℃〕で、押出機出口から押出し、押出された溶融体(メルト)をギアポンプ、金属繊維フィルター(孔径20μm)を通した後、ダイから冷却(チル)ロールに押出した。押出されたメルトは、静電印加法を用いて冷却ロールに密着させた。冷却ロールは、中空のチルロールを用い、この中に熱媒として水を通して温調できるようになっている。
なお、ダイ出口から冷却ロールまでの搬送域(エアギャップ)は、この搬送域を囲い、この中に調湿空気を導入することにより、湿度を30%RHに調節してある。押出機の押出量の調整及びダイの開口部の形状を上記構成とすることにより、メルト厚みを3000μmとした。
以上のようにして、実施例1〜実施例16および比較例1〜比較例12の各PETフィルムの製造を試みた。
【0184】
<PETフィルムの評価>
PETフィルムの破断伸度およびΔAVから、PETフィルムの耐候性を評価した。
【0185】
‐破断伸度の測定‐
冷却ロールにより冷却されたPETフィルムを二軸延伸(3.4×3.8倍)して、二軸延伸後のフィルムの湿熱条件下(120℃×100RH%)での破断伸度が半減する時間で判定する。85時間以上で良好な耐候性(耐加水分解性)を示すものと判定する。結果を表1に示す。
【0186】
−ΔAVの算出−
原料樹脂のAVと溶融押出しされたフィルムのAVとを測定し、原料樹脂のAV(AV1)と溶融押出しされたフィルムのAV(AV2)との差であるΔAV(|AV1−AV2|)を算出した。ΔAVが3eq/t以下であるほど良好な耐候性を示すとして判定した。結果を表1に示す。
【0187】
【表1】
【0188】
〔実施例17〜実施例22〕
末端封止剤として、以下の化合物を各実施例に用いた。
【0189】
1.環状カルボジイミド化合物
1)環状カルボジイミド化合物(1)(表2中の「環状」)
既述の環状カルボジイミド化合物(c)であり、下記構造の化合物(重量平均分子量Mw=516)を用いた。環状カルボジイミド化合物(1)は、特開2011−258641号公報の参考例1に記載の合成方法を参考に合成した。
【0190】
【化12】
【0191】
2.非環状カルボジイミド化合物
1)線状カルボジイミド(1)(表2中の「線状」)
Stabaxol P400(ラインケミー社製、重量平均分子量Mw=約2000)を用いた。
2)モノカルボジイミド(1)(表2中の「モノ」)
Stabaxol I(ラインケミー社製、重量平均分子量Mw=362.5)を用いた。
これらのカルボジイミド末端封止剤の構造を以下に示す。
【0192】
【化13】
【0193】
−末端封止剤入りのPETペレットの作成−
表2に示す末端封止剤と、AVが13eq/tであるポリエチレンテレフタラート樹脂(PET樹脂)と事前に溶融混合し、末端封止剤の濃度が、溶融混合物全量の50質量%となるよう調整して、実施例17〜実施例22で用いる各マスターペレット(末端封止剤入りのPETペレット)を作成した。
【0194】
−PETフィルムの作成−
実施例1のPETフィルムの製造において、実施例1で用いたPET樹脂のペレットに代えて、得られた各マスターペレットを用いたほかは、実施例1と同様にしてPETフィルムを作成した。
【0195】
<PETフィルムの評価>
得られた実施例17〜実施例22のPETフィルムについて、実施例1のPETフィルムと同様の方法で、破断伸度およびΔAVから、PETフィルムの耐候性を評価した。評価結果を表2に示す。なお、表2には、参考のため、実施例1の条件詳細および評価結果も示した。
【0196】
【表2】
【0197】
〔実施例23〜実施例30、および比較例12〕
(1)CHDM系ポリエステル樹脂(PCT樹脂)の作成
・第1工程
ジカルボン酸化合物としてイソフタル酸(IPA)とテレフタル酸(TPA)、ジオール化合物としてシクロヘキサンジメタノール(CHDM)、エチレングリコール(EG)を用い、触媒として酢酸マグネシウム、三酸化アンチモンを150℃、窒素雰囲気下で溶融後、攪拌しながら230℃まで3時間かけて昇温し、メタノールを留出させ、エステル交換反応を終了した。この際、IPA、TPA、CHDM、およびEGの添加量を変えることで、表3に示す組成のCHDM系ポリエステル樹脂(CHDM−1〜CHDM−9)を得た。
【0198】
・第2工程
エステル交換反応終了後、リン酸をエチレングリコールに溶解したエチレングリコール溶液を添加した。
【0199】
・第3工程
重合反応を最終到達温度285℃、真空度0.1Torrで行い、ポリエステルを得、これをペレット化した。
【0200】
・第4工程
上記で得られたポリエステルペレットを、160℃で6時間乾燥、結晶化した。
【0201】
(2)PET系ポリエステル樹脂(PET樹脂)の作成
表3に示すように、CHDMとIPAを添加しない以外は、CHDM系ポリエステル樹脂と同様にして、PET系ポリエステル樹脂(PET−1)を作成した。
【0202】
上記にて得られたCHDM系ポリエステル樹脂について、ジオール化合物中のシクロヘキサンジメタノール(CHDM)含率およびジカルボン酸化合物中のイソフタル酸含率を以下の方法で測定した。
【0203】
(組成測定法)
CHDM系ポリエステルペレットをヘキサフルオロイソプロパノール(HFIP)に溶解した後、1H−NMRにより定量した。標品(CHDM、テレフタル酸、EG、およびイソフタル酸)を予め測定し、これを用いシグナルを同定した。
得られたイソフタル酸残基の量およびCHDM残基の量を表3に記載した。
なお、100モル%−イソフタル酸(IPA)含率(モル%)がテレフタル酸(TPA)含率(モル%)であり、100(モル%)−CHDM率(モル%)がEG含率(モル%)である。
表3に示すように、CHDM−3、CHDM−7、およびPET−1の各ポリエステル樹脂は、乾燥した後、窒素気流中210℃で24時間、固相重合した
なお、これらのCHDM系ポリエステルペレットおよびPET系ポリエステルペレットのIVおよびAVは、実施例1で用いたPETペレットと同様の方法で測定し、表3に記載した。
【0204】
【表3】
【0205】
(3)溶融製膜
(3−1)押出し
PET−1およびCHDM−1〜CHDM−9の各樹脂を乾燥した後、2軸押出機で真空下、溶融混練した。このとき、溶融混練の温度は、PET−1は280℃、CHDM−1〜CHDM−4は285℃、CHDM−5〜CHDM−8は305℃、CHDM−9は、305℃とした。
得られた溶融樹脂を、フィードブロックダイを用い25℃のキャストドラム上に押出し、表4に示す層構成の単層ポリエステルフィルムまたは積層ポリエステルフィルムを作成した。各ポリエステルフィルムを作成する上での押出機での押出条件および排出条件は、表5に示したとおりである。
【0206】
【表4】
【0207】
得られた単層ポリエステルフィルムおよび積層ポリエステルフィルムについて、実施例1と同様にして耐候性を評価した。評価結果を表5に示す。
なお、フラフは、PETのフラフを用い、全PET成分に対する表5に示す量〔質量%〕で添加した。積層ポリエステルフィルムの作成においては、PET−1を溶融混練した押出機にのみ、フラフを添加した。
【0208】
【表5】
【0209】
〔実施例31〜実施例60〕
<太陽電池モジュールの作製>
実施例1〜実施例30の各ポリエステルフィルムを太陽電池用バックシートとして用い、次のようにして、実施例31〜実施例60の太陽電池モジュールを作製した。
厚さ3.2mmの強化ガラスと、EVAシート〔三井化学ファブロ社製のSC50B〕と、結晶系太陽電池セルと、EVAシート〔三井化学ファブロ社製のSC50B〕と、実施例1〜実施例30のポリエステルフィルムのいずれか1枚とを、この順に重ね合わせ、真空ラミネータ〔日清紡社製、真空ラミネート機〕を用いてホットプレスすることにより、各部材とEVAシートとを接着させた。
【0210】
作製した各太陽電池モジュールについて、発電運転をしたところ、いずれも太陽電池として良好な発電性能を示した。
【符号の説明】
【0211】
10 バレル
12 供給口
14 押出機出口
16A、16B ベント
20A、20B スクリュー
30 温度制御手段
40 ダイ
42 フィルター
44 ギアポンプ
46 原料供給装置
100 二軸押出機
C1〜C9 加熱/冷却装置
【技術分野】
【0001】
本発明は、ポリエステルフィルムの製造方法、太陽電池用ポリエステルフィルム、及び、太陽電池モジュールに関する。
【背景技術】
【0002】
近年、種々の用途に適用できるように、樹脂の合成方法、加工方法、及び成膜方法等により、種々の特性や機能性をもつポリエステルフィルムの開発が求められている。例えば、太陽電池の用途の樹脂フィルムは、屋根の上などに置かれ雨曝しになる太陽電池の使用環境に対応した耐久性や、太陽電池の発電効率を妨げないための透明性等の性質が求められている。また、太陽電池の用途の樹脂フィルムとしては、太陽電池素子(セル)を封止する太陽電池用封止材(単に「封止材」ともいう)や、前記封止材を外部から保護する太陽電池用バックシートなどが知られている。
【0003】
ポリエステルには、通常はその表面にカルボキシル基や水酸基が多く存在しており、水分が存在する環境では加水分解を起こしやすく、経時で劣化する傾向がある。そのため、屋外等の常に風雨に曝されるような環境におかれる太陽電池モジュール等に用いられるポリエステルは、その加水分解性が抑えられていることが求められる。
ポリエステル樹脂に耐加水分解性を付与するには、加水分解反応の触媒となる末端COOHを低減することが考えられる。末端COOHは溶融時の加熱で発生するため、可塑化溶融押出機で溶融成膜する場合には、溶融時の発熱を緩和することが重要になる。
【0004】
かかる問題に対して、例えば、発熱を抑えて大量生産を達成するため、バレルの内径が140mm以上のベント式二軸押出機を使用し、単位時間当たりの押出量Qとスクリュー回転数Nとの比Q/Nが一定の範囲内となる条件下で溶融押出しを行うポリエステルシートの製造方法が開示されている(例えば、特許文献1参照)。
また、原料供給口からポリエステルの少なくとも一部と脂肪族カルボン酸の金属塩をそれぞれ独立に添加するポリエステル組成物の製造方法において、配管表面に付着する前記金属塩をかき落とすために、ポリエステルの一部の嵩密度を0.6以下とすることが開示されている(例えば、特許文献2参照)。
ポリエステルとして、ポリエチレンテレフタラート(PET)を用いる場合には、PETの溶融成形の際に生じるアセトアルデヒドの発生量を抑制するため、ポリエステル樹脂中の含水率を60〜500ppmに調整し、溶融成形に供するポリエステル樹脂の成形方法が知られている(例えば、特許文献3参照)。
さらに、押出機内での滞留による劣化を抑制するとともに、色調に優れたポリトリメチレンテレフタレート樹脂組成物を製造するために、予めポリトリメチレンテレフタレート及び改良材を供給する設備を不活性ガスで置換すると共に、ポリトリメチレンテレフタレート及び改良材それぞれの供給量(体積量)以上の不活性ガスを連続的に該設備に供給しながら、製造することを特徴とする、ポリトリメチレンテレフタレートの製造方法が開示されている(例えば、特許文献4参照)
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特許第3577178号
【特許文献2】特開平10−329188号公報
【特許文献3】特開平07−205257号公報
【特許文献4】特開2004−307552号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかし、特許文献1〜4に記載の各製造方法では、雨曝し環境に耐え得る耐候性、特に、高い耐加水分解性を有するポリエステルフィルムを得ることが困難であった。
【0007】
本発明は、高い耐加水分解性を有するポリエステルフィルムを製造するポリエステルフィルム製造方法、高い耐加水分解性を有する太陽電池用ポリエステルフィルム、及び、発電効率の安定性に優れる太陽電池モジュールを提供することを目的とする。
【課題を解決するための手段】
【0008】
前記目的を達成するため、以下の発明が提供される。
<1> 嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、二軸押出機から溶融樹脂を、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、溶融樹脂をフィルム状に成形する成形工程と、を含むポリエステルフィルム製造方法である。
【0009】
なお、本発明におけるポリエステル原料樹脂の融点Tmとは、示差走査熱量測定により求められる値をいうが、ポリエステル原料樹脂が複数種のポリエステル樹脂で構成されることにより、2つ以上の融点を有する場合は、最も高い融点を指す。したがって、ポリエステル原料樹脂を複数種類用いる場合には、Tmは、高融点成分の融点を意味する。例えば、ポリエステル原料樹脂として、ポリエチレンテレフタレート〔PET〕と、CHDM系ポリエステル樹脂〔例えば、ポリシクロヘキサンジメチレンテレフタレート(PCT)〕とを用いた場合、PETの融点は255℃であり、PCTの融点は278℃であるが、PETとPCTとを混合して用いる場合、排出温度の基準となる「ポリエステル原料樹脂の融点Tm」=278℃となる。
【0010】
<2> 二軸押出機が、少なくとも1つのベント孔と、原料供給口に最も近いベント孔よりも原料供給口に近い位置に、ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給する第1の窒素ガス供給口と、を備える<1>に記載のポリエステルフィルム製造方法である。
【0011】
<3> 二軸押出機が、ポリエステル原料樹脂を可塑化するニーディング部を有するスクリューと、ニーディング部に位置し、ポリエステル原料樹脂に対して窒素ガスを供給する第2の窒素ガス供給口と、を備える<1>または<2>に記載のポリエステルフィルム製造方法である。
【0012】
<4> 窒素ガス供給口における窒素ガスの供給速度が、2m/分〜50m/分である<2>または<3>に記載のポリエステルフィルム製造方法である。
【0013】
<5> ニーディング部は、ニーディングクリアランスにおける剪断速度が500s−1〜2000s−1である<3>または<4>に記載のポリエステルフィルム製造方法である。
【0014】
<6> ポリエステル原料樹脂に対して、カルボジイミド基を1個有し、カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む環状カルボジイミド化合物を、0.05質量%〜20質量%で混合し、溶融混練して溶融樹脂とする<1>〜<5>のいずれか1つに記載のポリエステルフィルム製造方法である。
【0015】
<7> <1>〜<6>のいずれか1つに記載のポリエステルフィルム製造方法により製造された太陽電池用ポリエステルフィルムである。
【0016】
<8> 1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%含むCHDM系ポリエステル樹脂を含有する層を少なくとも1層有する<7>に記載のポリエステルフィルムである。
【0017】
<9> CHDM系ポリエステルを含有する層は、前記1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜20モル%または80モル%〜100モル%含む<8>に記載のポリエステルフィルムである。
【0018】
<10> 太陽光が入射する透明性のフロント基板と、フロント基板の上に設けられ、太陽電池素子及び太陽電池素子を封止する封止材を有するセル構造部分と、セル構造部分のフロント基板が位置する側と反対側に設けられ、封止材と隣接して配置された<7>〜<9>のいずれか1つに記載の太陽電池用ポリエステルフィルムと、
を備えた太陽電池モジュールである。
【発明の効果】
【0019】
本発明によれば、高い耐加水分解性を有するポリエステルフィルムを製造するポリエステルフィルム製造方法、高い耐加水分解性を有する太陽電池用ポリエステルフィルム、及び、発電効率の安定性に優れる太陽電池モジュールを提供することができる。
【図面の簡単な説明】
【0020】
【図1】本発明に係るポリエステルフィルムの製造方法を実施するための二軸押出機の構成例を示す概略図である。
【図2】本発明に係るポリエステルフィルムの製造方法を実施するフローの一例を示す図である。
【発明を実施するための形態】
【0021】
以下、本発明のポリエステルフィルム製造方法について詳細に説明する。なお、本願明細書において「〜」とはその前後に記載される数値を下限値及び上限値として含む意味で使用される。
<ポリエステルフィルム製造方法>
本発明のポリエステルフィルム製造方法は、嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、 前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、前記溶融樹脂をフィルム状に成形する成形工程と、を含んで構成される。
【0022】
ポリエステル原料樹脂(以下、単に「原料樹脂」ともいう)の熱劣化を抑えてポリエステルを成形することで、耐候性に優れるフィルムを得ることができる。また、原料樹脂の熱劣化の要因は、ポリエステルの加水分解、または酸化分解である。
従って、原料樹脂の発熱量を抑制しつつ、加水分解反応の触媒となる末端COOHの量(Acid Value;AV)を低減したり、原料樹脂に含まれる水分や酸素の量をできる限り減らすことが重要である。特に、原料樹脂の末端COOH量と溶融押出しされたフィルムの末端COOH量との差ΔAVを小さく〔例えば3eq/t(当量/トン;以下同じ)以下に〕することが好ましい。
【0023】
しかし、原料樹脂の含水率を乾燥により100ppm以下にすると、二軸押出機(以下、単に「押出機」とも称する)での原料樹脂の発熱が大きくなり、樹脂温度が上昇することがある。
よって、原料樹脂の乾燥をしすぎると、原料樹脂の水分量は減少したとしても、温度上昇による原料樹脂の熱劣化が起き、ΔAVが増大してしまう傾向にある。
【0024】
一方、押出機の温度を下げた場合、溶融した原料樹脂(溶融樹脂)を排出する押出機の出口(排出口)の温度は低下するが、溶融樹脂が最も発熱する可塑化部(ニーディング部)での発熱を抑えることができず、溶融樹脂の熱劣化が進行してしまうことがあった。
【0025】
ポリエステルフィルムの製造にあっては、押出機内での原料樹脂の加水分解を抑制するために、一般に、固有粘度が0.70dl/g以上の高い粘度の原料樹脂が用いられる。しかし、原料樹脂の固有粘度が大きいほど、原料樹脂の分子量は大きくなる傾向にあり、固有粘度が高いほど、押出機内における混練による摩擦が大きく、原料樹脂が発熱し易い。
【0026】
このように、原料樹脂の耐加水分解を目的に、含水率を下げたり、固有粘度を高くしても、押出機内での原料樹脂の発熱により、原料樹脂のAVが増加し易かった。
したがって、原料樹脂の含水率を下げたり、固有粘度を高くした上で、さらに、原料樹脂のAV低下ないしΔAVの低下を達成する必要があった。
【0027】
かかる問題に対し、本発明のポリエステルフィルム製造方法においては、ポリエステル原料樹脂に、所定の嵩密度を有するフラフを特定量添加し、押出機に供給するときのポリエステル原料樹脂の温度を特定の温度にすることで、押出機内部、特に可塑化部における溶融樹脂の摩擦を軽減し、樹脂温度を低減することができることを見出した。
すなわち、原料樹脂が、嵩高いフラフを含んでいることで、押出機内における原料樹脂の摩擦が低減し、さらに、原料樹脂の温度を所定の温度に加熱しておくことで、樹脂が軟化し易く摩擦を生じ難いため、押出機内での発熱を抑制することができる。
【0028】
一方、原料樹脂は、押出機内にて溶融混練されることにより、溶融樹脂となり、押出機の排出口から外部へ排出されるが、このとき、原料樹脂の発熱を抑制することによって、原料樹脂の溶融不足があると、溶融樹脂中に、未溶融の原料樹脂が異物として残存してしまう。押出機から排出された溶融樹脂が、異物を含んでいると、溶融樹脂をフィルム状に成膜する時、特に、フィルムを延伸するときに、異物の存在に起因して、延伸ムラを生じることがある。
そのため、本発明のポリエステルフィルムの製造方法では、溶融樹脂を所定の温度に加熱して押出機から排出することで、溶融樹脂に、かかる異物が残存することを防止することができる。
【0029】
従って、本発明のポリエステルフィルム製造方法を、上記構成とすることで、高い耐加水分解性を有するポリエステルフィルムを製造することができる。
まず、樹脂供給工程から説明する。
【0030】
〔原料樹脂供給工程〕
原料樹脂供給工程は、嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給して構成される。
つまり、二軸押出機の原料供給口に供給されるときのポリエステル原料樹脂が、嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であることを意味する。
まず、フラフについて説明し、次いで、ポリエステル原料樹脂について説明する。
【0031】
−フラフ−
フラフとは、ポリエステルフィルムの粉砕屑、特に、ポリエステルフィルムの成膜途中に生じたフィルムの粉砕屑、及び、リサイクルのための使用済みポリエステルフィルムの粉砕屑等をいう。
本発明のポリエステルフィルムの製造方法においては、特に、嵩密度が0.2〜0.7であるフラフが用いられ、原料樹脂の全質量に対して10質量%〜50質量%の範囲で原料樹脂に含まれる。
【0032】
嵩密度とは、粉末を一定容積の容器の中に一定状態で入れる等して、所定形状にした粉末の質量を、そのときの体積で除算して求められる密度(単位体積あたりの質量)をいい、嵩密度が小さいほど嵩張る。フラフの嵩密度は、JIS K7365:1999の「プラスチック−規定漏斗から注ぐことができる材料の見掛け密度の求め方」に準拠した方法により測定することができる。
【0033】
本発明のポリエステルフィルムの製造方法において、低含水率であり、高固有粘度の原料樹脂に、嵩密度が0.2〜0.7であるフラフを、原料樹脂の全質量に対して10質量%〜50質量%含むことで、原料樹脂が低含水率であること及び高固有粘度を有することのメリットを維持したまま、押出機内での発熱を抑制することができる。
フラフの嵩密度が、0.2未満であると、押出機の原料供給口で原料樹脂が詰まり(ブリッジともいう)、原料樹脂を溶融することができず、フラフの嵩密度が0.7を超えると、嵩が小さく、原料樹脂の発熱抑制を発現することができない。
【0034】
嵩密度が0.2〜0.7であるフラフは、使用済みポリエステルフィルムを破砕することにより得られる。また、ポリエステルフィルムの製造時に生じる破砕片を用いてもよい。
フラフの嵩密度は、原料樹脂の押出し安定性(ブッリジ抑制)、及び剪断発熱抑制の観点から、0.2〜0.6であることが好ましい。
【0035】
また、フラフは、原料樹脂の全質量に対して、10質量%〜50質量%の範囲で原料樹脂に含まれる。フラフの含有量が、10質量%未満であると、押出機内での原料樹脂の発熱を抑制することができず、50質量%を超えると、原料樹脂のAVが増加する。
フラフの原料樹脂中の含有量は、ΔAVを小さくする観点からは20質量%〜50質量%が好ましく、破断伸度の観点からは15質量%〜40質量%であることが好ましい。
【0036】
フラフのサイズとしては、フラフが上記範囲の嵩密度であれば制限はないが、厚みが20〜5000μmであるものが好ましい。中でも、嵩密度が大きくなり過ぎて充満率が低下しすぎないようにし、溶融不足を回避する観点から、100〜1000μmの範囲、更には100〜500μmの範囲がより好ましい。
【0037】
また、成膜されるポリエステルフィルムの末端COOH量をより低減する点で、フラフのサイズのばらつきは小さい方が好ましく、例えば粉砕片の厚みでは、ばらつきは±100%以内であるのが好ましく、より好ましくは±50%以内であり、更には±10%以内である。粉砕片を用いる場合、厚みなどサイズばらつきを小さく抑えることで、得られるポリエステルフィルムの末端COOH量の変動を低く抑えることができる。
【0038】
‐ポリエステル原料樹脂‐
本発明のポリエステルフィルムの製造方法で用いられるポリエステル原料樹脂(単に、原料樹脂ともいう)は、既述のフラフを含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を用いる。
原料樹脂としては、ポリエステルであれば特に制限されず、例えば、PET(ポリエチレンテレフタレート)、PBT(ポリブチレンテレフタレート)等が挙げられる。
まず、含水率、固有粘度などの、原料樹脂の物性について説明し、次いで、原料樹脂を構成するポリエステル樹脂について説明する。
【0039】
(含水率)
原料樹脂は、含水率が20ppm〜100ppmである。
原料樹脂の含水率が20ppm未満であると、押出機内での原料樹脂の発熱が大きくなり、原料樹脂の末端COOH量と溶融押出しされたフィルムの末端COOH量との差ΔAVが増加し、100ppmを超えると原料樹脂の加水分解によりΔAVが増加する。
原料樹脂の含水率は、30ppm〜70ppmであることが好ましい。
原料樹脂の含水率は、原料樹脂の乾燥温度、乾燥時間等によって調整することができる。
【0040】
(固有粘度、IV)
原料樹脂の固有粘度(Interisic Viscosity;IV)は、0.70dl/g〜1.2dl/gである。原料樹脂のIVが0.70未満であると、原料樹脂の耐加水分解性が低下し、IVが1.2を超えると発熱を十分に抑えることができない。
特に原料樹脂が、ポリエチレンテレフタレート(PET)であるとき、原料PET樹脂のIVは0.70dl/g〜0.85dl/gであることが好ましく、0.70dl/g〜0.80dl/gであることがより好ましい。
【0041】
原料樹脂のIVは、ポリエステル樹脂の重合方式および重合条件によって調整することができ、液相重合の後に固相重合を行うことによって原料となる固有粘度IVが0.70dl/g〜1.2dl/gのポリエステル樹脂を得ることができる。
【0042】
なお、固有粘度(IV)は、溶液粘度(η)と溶媒粘度(η0)の比ηr(=η/η0;相対粘度)から1を引いた比粘度(ηsp=ηr−1)濃度で割った値を濃度がゼロの状態に外挿した値である。IVは、ウベローデ型粘度計を用い、ポリエステルを1,1,2,2−テトラクロルエタン/フェノール(=2/3[質量比])混合溶媒に溶解させ、25℃の溶液粘度から求められる。
【0043】
(結晶化温度)
また、後述する固相重合に先立ち、原料樹脂の融着を防止するために結晶化することが好ましく、結晶化する際の好ましい結晶化温度としては、140℃以上である。より好ましくは140℃以上175℃以下であり、さらに好ましくは140℃以上170℃以下である。また、結晶化時間は、10分以上10時間未満が好ましく、より好ましくは20分以上8時間以下であり、さらに好ましくは40分以上7時間以下である。このとき、窒素等の不活性ガスを流すことが好ましい。
【0044】
(末端COOH量、AV)
また、原料樹脂は、末端COOH量(Acid Value;AV)が25eq/t(当量/トン)以下であることが好ましく、15eq/t以下がより好ましい。本発明の方法により原料樹脂を溶融押出ししてフィルムを製造する際、末端COOH量の増加は3eq/t以下に抑制されるため、末端COOH量が25eq/t以下の原料樹脂を用いれば、末端COOH量が少なく、高い耐加水分解性を有するポリエステルフィルムが得られる。ただし、例えば被着物との間の密着性が得られる観点から、原料樹脂の末端COOH量は2eq/t以上であることが望ましい。なお、「eq/t」は、1トンあたりのモル当量を表す。
【0045】
原料樹脂の末端COOH量は、原料樹脂の含水率、押出機内での溶融温度、混練時間等によって調整することができる。
末端COOH量は、以下の方法により測定される値である。すなわち、原料樹脂0.1gをベンジルアルコール10mlに溶解後、さらにクロロホルムを加えて混合溶液を得、これにフェノールレッド指示薬を滴下する。この溶液を、基準液(0.01N、KOH−ベンジルアルコール混合溶液)で滴定し、滴下量から末端カルボキシル基量を求める。
【0046】
なお、複数の種類の樹脂を混合して用いる場合は、前記原料樹脂の末端COOH量は、混合状態での量を表す。例えば、ポリエチレンテレフタレート(PET)として、そのペレットの1種又は2種以上やPETフィルムの粉砕屑であるチップ材などを混合する場合、ペレットの末端COOH量の総量、又はペレットの末端COOH量とチップの末端COOH量との合計量である。
【0047】
(融点、Tm)
また、原料樹脂の融点Tmは、250℃〜290℃の範囲であることが好ましい。前記融点Tmは示差走査熱量測定により求められる値である。以下、原料樹脂を複数種用いることにより、Tmが複数ある場合は、最も高い融点を原料樹脂の融点Tmという。原料樹脂が複数の樹脂の混合であるときは各樹脂の融点の平均値が上記範囲内にあることが好ましい。
【0048】
(嵩密度)
原料樹脂の嵩密度としては、原料樹脂の押出し安定性、及び剪断発熱抑制の観点から、0.7〜0.9以下の範囲が好ましい。この嵩密度が0.7以上であると、押出しをより安定的に行なうことができ、0.9以下であると、局所的な発熱を効果的に抑制することができる。
【0049】
(樹脂温度)
原料樹脂は、100℃〜160℃に加熱して、押出機の原料供給口(押出機入口)に供給する。
すなわち、押出機の原料供給口における原料樹脂の温度が、100℃〜160℃である。原料樹脂の温度を上記範囲とすることで、原料樹脂の摩擦を抑制し、押出機内での発熱を抑制することができる。
原料樹脂の温度が100℃未満であると、原料樹脂の溶融時の摩擦発熱が増加し、160℃を超えると、原料の分解反応が進行する。
原料樹脂の温度は、120℃〜150℃であることが好ましい。
次に、原料樹脂を構成するポリエステル樹脂について説明する。
【0050】
(ポリエステル樹脂)
原料樹脂を構成するポリエステル樹脂としては、ジカルボン酸又はそのエステル誘導体と、ジオール化合物とを公知の方法でエステル化反応及び/又はエステル交換反応させることによって得ることができる。
ジカルボン酸又はそのエステル誘導体としては、例えば、マロン酸、コハク酸、グルタル酸、アジピン酸、スベリン酸、セバシン酸、ドデカンジオン酸、ダイマー酸、エイコサンジオン酸、ピメリン酸、アゼライン酸、メチルマロン酸、エチルマロン酸等の脂肪族ジカルボン酸類、アダマンタンジカルボン酸、ノルボルネンジカルボン酸、イソソルビド、シクロヘキサンジカルボン酸、デカリンジカルボン酸、などの脂環族ジカルボン酸、テレフタル酸、イソフタル酸、フタル酸、1,4−ナフタレンジカルボン酸、1,5−ナフタレンジカルボン酸、2,6−ナフタレンジカルボン酸、1,8−ナフタレンジカルボン酸、4,4’−ジフェニルジカルボン酸、4,4’−ジフェニルエーテルジカルボン酸、5−ナトリウムスルホイソフタル酸、フェニルエンダンジカルボン酸、アントラセンジカルボン酸、フェナントレンジカルボン、9,9’−ビス(4−カルボキシフェニル)フルオレン酸等の芳香族ジカルボン酸などのジカルボン酸又はそのエステル誘導体が挙げられる。
【0051】
ジカルボン酸は、芳香族ジカルボン酸の少なくとも1種が用いられる場合が好ましい。より好ましくは、ジカルボン酸のうち、芳香族ジカルボン酸を主成分として含有する。好ましい芳香族ジカルボン酸として、テレフタル酸(TPA)、2,6−ナフタレンジカルボン酸(2,6−NDCA)が挙げられ、これらが主成分であるものが好ましい。なお、「主成分」とは、ジカルボン酸成分に占める芳香族ジカルボン酸の割合が各々80質量%以上であることをいう。
ポリエステル樹脂の合成にあたっては、2,6−NDCAおよびTPA以外のジカルボン酸を含んでもよい。より好ましいジカルボン酸としては、イソフタル酸(IPA)等を挙げることができる。IPAの好ましい添加量は、全ジカルボン酸中0モル%〜15モル%が好ましく、より好ましくは0モル%〜12モル%、さらに好ましくは0モル%〜9モル%である。
【0052】
ジオール化合物としては、例えば、エチレングリコール、1,2−プロパンジオール、1,3−プロパンジオール、1,4−ブタンジオール、1,2−ブタンジオール、1,3−ブタンジオール等の脂肪族ジオール類、1,4−シクロヘキサンジメタノール、スピログリコール、イソソルビドなどの脂環式ジオール類、ビスフェノールA、1,3―ベンゼンジメタノール,1,4−ベンセンジメタノール、9,9’−ビス(4−ヒドロキシフェニル)フルオレン、などの芳香族ジオール類等が挙げられる。
【0053】
ジオール化合物は、脂肪族ジオールの少なくとも1種が用いられることが好ましい。脂肪族ジオールは、エチレングリコール(EG)、または1,4−シクロヘキサンジメタノール(CHDM)が好ましく、エチレングリコールおよび1,4−シクロヘキサンジメタノールの少なくとも一方を主成分として含有する。
なお、主成分とは、ジオール化合物に占めるエチレングリコールおよび1,4−シクロヘキサンジメタノールの和の割合が80質量%以上であることをいう。
【0054】
エステル化反応及び/又はエステル交換反応には、従来から公知の反応触媒を用いることができる。反応触媒としては、アルカリ金属化合物、アルカリ土類金属化合物、亜鉛化合物、鉛化合物、マンガン化合物、コバルト化合物、アルミニウム化合物、アンチモン化合物、チタン化合物、リン化合物などが挙げられる。通常は、ポリエステルの製造方法が完結する以前の任意の段階において、重合触媒としてアンチモン化合物、ゲルマニウム化合物、チタン化合物を添加することが好ましい。このような方法としては、例えば、ゲルマニウム化合物を例に挙げると、ゲルマニウム化合物粉体をそのまま添加することが好ましい。
【0055】
好ましいポリエステル樹脂は、ポリエチレンテレフタレート(PET)、ポリエチレン−2,6−ナフタレート(PEN)であり、より好ましくはPETである。PETは、ゲルマニウム(Ge)系触媒、アンチモン(Sb)系触媒、アルミニウム(Al)系触媒、及びチタン(Ti)系触媒から選ばれる1種又は2種以上を用いて重合されるものが好ましく、より好ましくはTi系触媒である。
【0056】
−CHDM系ポリエステル樹脂−
さらに、ポリエステルフィルムの力学強度を上げ、耐熱性を向上する観点からは、ポリエステル樹脂が、1,4−シクロヘキサンジメタノール由来の構造を含むCHDM系ポリエステル樹脂であることが好ましい。
CHDM系ポリエステル樹脂とは、ポリエステル樹脂を得るために用いるジオール化合物の一部または全部として、1,4−シクロヘキサンジメタノール(CHDM)を用いたポリエステル樹脂をいい、分子構造内に、1,4−シクロヘキサンジメタノール由来の構造を含む。具体的には、例えば、ポリシクロヘキサンジメチレンテレフタレート(PCT)等が挙げられる。
【0057】
CHDM系ポリエステル樹脂は、分子構造内に1,4−シクロヘキサンジメタノール由来(CHDM由来ともいう)の構造を有していれば、製造されるポリエステルフィルムの耐候性を向上することができるが、1,4−シクロヘキサンジメタノール由来の構造の割合が次の範囲であると、より耐候性に優れる。
ポリエステル樹脂中の1,4−シクロヘキサンジメタノール由来の構造の割合は、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%であることが好ましく、0.1モル%〜20モル%または80〜100モル%であることがより好ましく、0.5モル%〜16モル%または83モル%〜98モル%であることがさらに好ましく、1モル%〜12モル%または86モル%〜96モル%であることが特に好ましい。
【0058】
より好ましい態様においてはCHDM由来の構造が低い領域(0.1〜20モル%)、高い領域(80〜100モル%)の二つの領域が存在するのは、この領域において、特に、結晶を形成し易く、結晶間に取り込まれた非晶が橋渡しする「タイチェーン」を形成し易いためである。この二つの領域に於いてポリエステル樹脂が結晶構造を取りやすく、高い力学強度および高い耐熱性を発揮し易くなる。
なお、ポリエステル樹脂の分子構造内に、このようなCHDM由来の構造が存在することで、ポリエステル分子の配向性が増加し、タイチェーンの生成を促す。これは以下の理由によるものと考えられる。
CHDMは環状構造であるためEG(エチレングリコール)のように屈曲し難く、剛直である。このため、ポリエステル樹脂の延伸等でポリエステル樹脂に負荷される外力で、ポリエステル分子が配向し易い。配向したポリエステル分子は結晶を形成し易いため、タイチェーンを形成し易い。
【0059】
CHDM系ポリエステル樹脂を合成するときは、ジオール化合物として、少なくとも1,4−シクロヘキサンジメタノール(CHDM)用いるが、さらに、CHDM以外のジオール化合物を用いてもよい。このとき、CHDM以外のジオール化合物については、既述のジオール化合物が挙げられる。中でも、エチレングリコールを用いることが好ましい。
【0060】
CHDM系ポリエステル樹脂を合成するときに用いるジカルボン酸は、既述のジカルボン酸又はそのエステル誘導体が用いられる。
CHDM系ポリエステル樹脂を得る場合は、ジカルボン酸として、少なくともテレフタル酸を用いることが好ましい。
また、ジカルボン酸は、テレフタル酸以外にイソフタル酸(IPA)を加えてもよい。好ましいIPA量は、全ジカルボン酸中0モル%〜15モル%が好ましく、0モル%〜12モル%であることがより好ましく、0モル%〜9モル%であることがさらに好ましい。
【0061】
CHDMは、一般に溶融粘度がPET樹脂よりも高く、固有粘度が0.70dl/g〜1.20dl/gであることが好ましい。
【0062】
1,4−シクロヘキサンジメタノール(CHDM)構造を含有するポリシクロヘキサンジメチレンテレフタレート(PCT)は、例えばWO2009/125701の段落番号[0089]〜[0090]、[0120]〜[0121]に記載の方法も好適に用いることができる。
また、ポリエチレン−2,6−ナフタレート(PEN)は、例えば特開2011−153209の段落番号[0170]、特開2008−39803の段落番号[0046]、および[0060]に記載の方法も好適に用いることができる。
【0063】
前記Ti系触媒は、反応活性が高く、重合温度を低くすることができる。そのため、特に重合反応中にPETが熱分解し、COOHが発生するのを抑制することが可能である。本発明においては、ポリエステルフィルムの末端COOH量を30eq/トン以下の範囲に調整するのに好適である。
【0064】
Ti系触媒を用いた重合により得たTi触媒系PETの製造には、例えば、特開2005−340616号公報、特開2005−239940号公報、特開2004−319444号公報、特許3436268号公報、特許3979866号公報、特許3780137号、特開2007−204538号公報等に記載の重合方法を用いることができる。
【0065】
チタン(Ti)系化合物を、1ppm以上30ppm以下、より好ましくは2ppm以上20ppm以下、さらに好ましくは3ppm以上15ppm以下の範囲で用いて重合を行なうことが好ましい。この場合、本発明の方法によって製造されるポリエステルフィルムには、1ppm以上30ppm以下のチタンが含まれる。
Ti系触媒の量は、1ppm以上であると好ましいIVが得られ、30ppm以下であると、末端COOHを低く抑えることができ、耐加水分解性の向上に有利である。
【0066】
−固相重合−
本発明においては、エステル化反応及び/又はエステル交換反応に加えて更に、ポリエステルを固相重合してもよい。固相重合は、既述のエステル化反応等により重合したポリエステル又は市販のポリエステルをペレット状などの小片形状にし、これを用いて好適に行なえる。具体的には、固相重合として、特許第2621563号、特許第3121876号、特許第3136774号、特許第3603585号、特許第3616522号、特許第3617340号、特許第3680523号、特許第3717392号、特許第4167159号等に記載の方法を用いることができる。
【0067】
固相重合は、150℃以上250℃以下、より好ましくは170℃以上240℃以下、さらに好ましくは190℃以上230℃以下で5時間以上100時間以下、より好ましくは10時間以上80時間以下、さらに好ましくは15時間以上60時間以下の条件で行なうのが好ましい。また、固相重合は、真空中あるいは窒素(N2)気流中で行なうことが好ましい。更に、多価アルコール(エチレングリコール等)を1ppm以上1%以下混合してもよい。
【0068】
固相重合は、バッチ式(容器内に樹脂を入れ、この中で所定の時間熱を与えながら撹拌する方式)で実施してもよく、連続式(加熱した筒の中に樹脂を入れ、これを加熱しながら所定の時間滞流させながら筒中を通過させて、順次送り出す方式)で実施してもよい。
【0069】
本発明においては、原料樹脂として用いるポリエステルの重合度は、ポリエステルの使用用途の要求特性に合わせて適宜選択すればよいが、一般には、溶融重縮合で0.3≦IV≦0.65のポリエステルを得て、溶融重縮合で得られたポリエステルを固相重縮合により0.70≦IV≦0.85に上昇させるのが好ましい。
【0070】
−末端封止剤−
ポリエステルフィルムは、末端封止剤を含有していることが好ましい。
ポリエステルフィルムは、ポリエステル結晶間を橋架けする分子(タイチェーン)を有する構造であることで、強固な構造となり、耐候性に優れる。ポリエステルフィルムが末端封止剤を含有していることで、タイチェーンが発達し過ぎることがなく、脆化を抑えつつも耐熱性を高めることができる。これにより、成膜した際に生じやすい弊害の懸念も小さくなる。
従って、本発明のポリエステルフィルム製造方法では、ポリエステルフィルムに末端封止剤が含まれるようにポリエステルフィルムを製造することが好ましい。末端封止剤は、二軸押出機の原料供給口に供給する前に、ポリエステル原料樹脂と混合してもよいし、二軸押出機でポリエステル原料樹脂を溶融混練しているときに混合してもよいし、二軸押出機から溶融樹脂を排出してから溶融樹脂に混合してもよい。
【0071】
以上の中でも、末端封止剤の添加方法としては、ポリエステル原料樹脂と末端封止剤とを事前に、全量に対して10質量%以上60質量%以下となるように溶融混合し、マスターペレットを作成した上で二軸押出機に投入して用いることが好ましい。
【0072】
なお、末端封止剤とは、ポリエステルの末端のカルボキシル基と反応し、ポリエステルの末端カルボキシ基量を減少させる添加剤である。
末端封鎖剤としては、カルボジイミド化合物、オキサゾリン化合物、エポキシ化合物、カーボネート化合物などが挙げられる。本発明のポリエステルフィルムは、イソシアネート化合物、カルボジイミド化合物およびエポキシ化合物のうちの少なくとも1つの末端封止剤を含むことが好ましく、2種類のカルボジイミド化合物を含むことが好ましい。末端封止剤は、単独で使用してもよく、組合せて使用してもよい。
末端封止剤は、以上の中でも、特に、カルボジイミド基を1個有し、前記カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む環状カルボジイミド化合物が好ましい。
【0073】
−環状カルボジイミド化合物−
環状カルボジイミド化合物は、カルボジイミド基を1個有し、カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む化合物である。
ここで、第一窒素とは、カルボジイミド基(−N=C=N−)が有する2つの窒素原子のうち、一方の窒素原子を指し、第二窒素とは、他方の窒素原子を指す。
環状カルボジイミド化合物は、末端封止剤として、ポリエステルの末端カルボキシル基を封止するため、本発明のポリエステルフィルムが環状カルボジイミド化合物を含有することにより、ポリエステルフィルムの耐候性、特に湿熱耐久性を改善することができる。
【0074】
環状カルボジイミド化合物を用いることによりポリエステルフィルムの耐候性が向上するのは、次の理由によるものと考えられる。
カルボジイミド化合物を環状構造にすることにより、下記のように、ポリエステルに、より一層タイチェーンの形成を促すことができる。
・環状カルボジイミドが解裂し、ポリエステル(PET−1という)の末端カルボン酸と反応する。
・解裂したカルボジイミドの他の一端はイソシアネート基となり、他のポリエステル(PET−2という)の末端水酸基と反応する。
・環状カルボジイミド化合物は環状構造のため、水酸基と反応した部位とカルボン酸と反応した部位は繋がっている。この結果、2本のPET分子鎖(PET−1およびPET−2)が環状カルボジイミドを介し、繋がったタイチェーン構造を形成する。
環状カルボジイミド化合物は、ポリエステル原料樹脂に対して0.05質量%〜20質量%の割合で用いることが好ましい。
以下、環状カルボジイミド化合物の詳細について説明する。
【0075】
環状カルボジイミド化合物は、重量平均分子量(Mw)が400以上であることが好ましく、500〜1500であることがより好ましい。
【0076】
また、環状カルボジイミド化合物は、環状構造を複数有していてもよい。
具体的には、環状カルボジイミド化合物の環状構造は、カルボジイミド基(−N=C=N−)を1個有しその第一窒素と第二窒素とが結合基により結合されている。一つの環状構造中には、1個のカルボジイミド基のみを有するが、例えば、スピロ環など、分子中に複数の環状構造を有する場合にはスピロ原子に結合するそれぞれの環状構造中に1個のカルボジイミド基を有していれば、化合物として複数のカルボジイミド基を有していてよいことはいうまでもない。環状構造中の原子数は、好ましくは8〜50、より好ましくは10〜30、さらに好ましくは10〜20、特に、10〜15が好ましい。
【0077】
ここで、環状構造中の原子数とは、環状構造を直接構成する原子の数を意味し、例えば、8員環であれば8、50員環であれば50である。環状構造中の原子数が8より小さいと、環状カルボジイミド化合物の安定性が低下して、保管、使用が困難となる場合があるためである。また反応性の観点よりは環員数の上限値に関しては特別の制限はないが、50を超える原子数の環状カルボジイミド化合物は合成上困難となり、コストが大きく上昇する場合が発生するためである。かかる観点より環状構造中の原子数は好ましくは、10〜30、より好ましくは10〜20、特に好ましくは10〜15の範囲が選択される。
【0078】
環状構造は、下記式(1)で表される構造であることが好ましい。
【0079】
【化1】
【0080】
式(1)中、Q(以下、結合基Qともいう)は、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの2〜4価の結合基、または、脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである2〜4価の結合基である。なお、2つ以上の基の組み合わせは、芳香族基と芳香族基のように、同種の基を組み合わせた態様であってもよい。
Qを構成する脂肪族基と脂環族基と芳香族基とは、各々独立にヘテロ原子または1価の置換基を含んでいてもよい。ヘテロ原子とはこの場合、O、N、S、Pを指す。結合基の価のうち2つの価は環状構造を形成するために使用される。Qが3価または4価の結合基である場合、環状構造は、単結合、二重結合、原子、または原子団を介して、ポリマーまたは他の環状構造と結合している。
【0081】
結合基Qは、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、もしくは2〜4価の炭素数5〜15の芳香族基、または、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、及び2〜4価の炭素数5〜15の芳香族基から選択される2つ以上の基の組み合わせであることが好ましい。
結合基Qを構成する脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせの例としては、アルキレン基とアリーレン基が結合した、アルキレン−アリーレン基のような構造などが挙げられる。
結合基Qは、下記式(1−1)、式(1−2)または式(1−3)で表される2〜4価の結合基であることが好ましい。
【0082】
【化2】
【0083】
式(1−1)中、Ar1およびAr2は各々独立に、2〜4価の炭素数5〜15の芳香族基である。Ar1およびAr2は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
Ar1またはAr2として表される芳香族基としては、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基(2価)として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これらの芳香族基は置換されていてもよい。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0084】
式(1−2)中、R1およびR2は、各々独立に、2〜4価の炭素数1〜20の脂肪族基、もしくは2〜4価の炭素数3〜20の脂環族基、または、2〜4価の炭素数1〜20の脂肪族基と2〜4価の炭素数3〜20の脂環族基との組み合わせ、あるいは、2〜4価の炭素数1〜20の脂肪族基と2〜4価の炭素数3〜20の脂環族基と2〜4価の炭素数5〜15の芳香族基とから選択される2つ以上の基の組み合わせである。R1およびR2を構成する脂肪族基、脂環族基、および芳香族基は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
【0085】
R1またはR2として表される脂肪族基としては、炭素数1〜20のアルキレン基、炭素数1〜20のアルカントリイル基、炭素数1〜20のアルカンテトライル基などが挙げられる。アルキレン基として、メチレン基、エチレン基、プロピレン基、ブチレン基、ペンチレン基、ヘキシレン基、へプチレン基、オクチレン基、ノニレン基、デシレン基、ドデシレン基、へキサデシレン基などが挙げられる。アルカントリイル基として、メタントリイル基、エタントリイル基、プロパントリイル基、ブタントリイル基、ペンタントリイル基、ヘキサントリイル基、ヘプタントリイル基、オクタントリイル基、ノナントリイル基、デカントリイル基、ドデカントリイル基、ヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、メタンテトライル基、エタンテトライル基、プロパンテトライル基、ブタンテトライル基、ペンタンテトライル基、ヘキサンテトライル基、ヘプタンテトライル基、オクタンテトライル基、ノナンテトライル基、デカンテトライル基、ドデカンテトライル基、ヘキサデカンテトライル基などが挙げられる。これらの脂肪族基は置換されていても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0086】
脂環族基として、炭素数3〜20のシクロアルキレン基、炭素数3〜20のシクロアルカントリイル基、炭素数3〜20のシクロアルカンテトライル基が挙げられる。シクロアルキレン基として、シクロプロピレン基、シクロブチレン基、シクロペンチレン基、シクロヘキシレン基、シクロへプチレン基、シクロオクチレン基、シクロノニレン基、シクロデシレン基、シクロドデシレン基、シクロへキサデシレン基などが挙げられる。アルカントリイル基として、シクロプロパントリイル基、シクロブタントリイル基、シクロペンタントリイル基、シクロヘキサントリイル基、シクロヘプタントリイル基、シクロオクタントリイル基、シクロノナントリイル基、シクロデカントリイル基、シクロドデカントリイル基、シクロヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、シクロプロパンテトライル基、シクロブタンテトライル基、シクロペンタンテトライル基、シクロヘキサンテトライル基、シクロヘプタンテトライル基、シクロオクタンテトライル基、シクロノナンテトライル基、シクロデカンテトライル基、シクロドデカンテトライル基、シクロヘキサデカンテトライル基などが挙げられる。これらの脂環族基は置換されていても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0087】
芳香族基として、それぞれへテロ原子を含んで複素環構造を持っていてもよい、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これら芳香族基は置換されていても良い。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0088】
上記式(1−1)および式(1−2)において、X1およびX2は、各々独立に、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、もしくは、2〜4価の炭素数5〜15の芳香族基、または、2〜4価の炭素数1〜20の脂肪族基と2〜4価の炭素数3〜20の脂環族基と2〜4価の炭素数5〜15の芳香族基とから選択される2つ以上の基の組み合わせである。X1およびX2を構成する脂肪族基、脂環族基、および芳香族基は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
【0089】
脂肪族基として、炭素数1〜20のアルキレン基、炭素数1〜20のアルカントリイル基、炭素数1〜20のアルカンテトライル基などが挙げられる。アルキレン基として、メチレン基、エチレン基、プロピレン基、ブチレン基、ペンチレン基、ヘキシレン基、へプチレン基、オクチレン基、ノニレン基、デシレン基、ドデシレン基、へキサデシレン基などが挙げられる。アルカントリイル基として、メタントリイル基、エタントリイル基、プロパントリイル基、ブタントリイル基、ペンタントリイル基、ヘキサントリイル基、ヘプタントリイル基、オクタントリイル基、ノナントリイル基、デカントリイル基、ドデカントリイル基、ヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、メタンテトライル基、エタンテトライル基、プロパンテトライル基、ブタンテトライル基、ペンタンテトライル基、ヘキサンテトライル基、ヘプタンテトライル基、オクタンテトライル基、ノナンテトライル基、デカンテトライル基、ドデカンテトライル基、ヘキサデカンテトライル基などが挙げられる。これらの脂肪族基は置換されていても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0090】
脂環族基として、炭素数3〜20のシクロアルキレン基、炭素数3〜20のシクロアルカントリイル基、炭素数3〜20のシクロアルカンテトライル基が挙げられる。シクロアルキレン基として、シクロプロピレン基、シクロブチレン基、シクロペンチレン基、シクロヘキシレン基、シクロへプチレン基、シクロオクチレン基、シクロノニレン基、シクロデシレン基、シクロドデシレン基、シクロへキサデシレン基などが挙げられる。アルカントリイル基として、シクロプロパントリイル基、シクロブタントリイル基、シクロペンタントリイル基、シクロヘキサントリイル基、シクロヘプタントリイル基、シクロオクタントリイル基、シクロノナントリイル基、シクロデカントリイル基、シクロドデカントリイル基、シクロヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、シクロプロパンテトライル基、シクロブタンテトライル基、シクロペンタンテトライル基、シクロヘキサンテトライル基、シクロヘプタンテトライル基、シクロオクタンテトライル基、シクロノナンテトライル基、シクロデカンテトライル基、シクロドデカンテトライル基、シクロヘキサデカンテトライル基などが挙げられる。これらの脂環族基は置換されていても良い。
脂環族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0091】
芳香族基として、それぞれへテロ原子を含んで複素環構造を持っていてもよい、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これらの芳香族基は置換されていても良い。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0092】
上記式(1−1)および式(1−2)においてsおよびkは、各々独立に、0〜10の整数であり、好ましくは0〜3の整数であり、より好ましくは0〜1の整数である。
s及びkが10を超えると、環状カルボジイミド化合物は合成上困難となり、コストが大きく上昇する場合が発生するためである。かかる観点より整数は好ましくは0〜3の範囲が選択される。なお、sまたはkが2以上であるとき、繰り返し単位としてのX1、あるいはX2が、他のX1、あるいはX2と異なっていてもよい。
【0093】
上記式(1−3)においてX3は、2〜4価の炭素数1〜20の脂肪族基、2〜4価の炭素数3〜20の脂環族基、もしくは2〜4価の炭素数5〜15の芳香族基、またはこれらの組み合わせである。
X3を構成する脂肪族基、脂環族基、および芳香族基は、各々独立に、さらに、ヘテロ原子または1価の置換基を含んでいてもよい。
【0094】
脂肪族基として、炭素数1〜20のアルキレン基、炭素数1〜20のアルカントリイル基、炭素数1〜20のアルカンテトライル基などが挙げられる。アルキレン基として、メチレン基、エチレン基、プロピレン基、ブチレン基、ペンチレン基、ヘキシレン基、へプチレン基、オクチレン基、ノニレン基、デシレン基、ドデシレン基、へキサデシレン基などが挙げられる。アルカントリイル基として、メタントリイル基、エタントリイル基、プロパントリイル基、ブタントリイル基、ペンタントリイル基、ヘキサントリイル基、ヘプタントリイル基、オクタントリイル基、ノナントリイル基、デカントリイル基、ドデカントリイル基、ヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、メタンテトライル基、エタンテトライル基、プロパンテトライル基、ブタンテトライル基、ペンタンテトライル基、ヘキサンテトライル基、ヘプタンテトライル基、オクタンテトライル基、ノナンテトライル基、デカンテトライル基、ドデカンテトライル基、ヘキサデカンテトライル基などが挙げられる。これら脂肪族基は1価の置換基を含んでいても良い。
脂肪族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0095】
脂環族基として、炭素数3〜20のシクロアルキレン基、炭素数3〜20のシクロアルカントリイル基、炭素数3〜20のシクロアルカンテトライル基が挙げられる。シクロアルキレン基として、シクロプロピレン基、シクロブチレン基、シクロペンチレン基、シクロヘキシレン基、シクロへプチレン基、シクロオクチレン基、シクロノニレン基、シクロデシレン基、シクロドデシレン基、シクロへキサデシレン基などが挙げられる。アルカントリイル基として、シクロプロパントリイル基、シクロブタントリイル基、シクロペンタントリイル基、シクロヘキサントリイル基、シクロヘプタントリイル基、シクロオクタントリイル基、シクロノナントリイル基、シクロデカントリイル基、シクロドデカントリイル基、シクロヘキサデカントリイル基などが挙げられる。アルカンテトライル基として、シクロプロパンテトライル基、シクロブタンテトライル基、シクロペンタンテトライル基、シクロヘキサンテトライル基、シクロヘプタンテトライル基、シクロオクタンテトライル基、シクロノナンテトライル基、シクロデカンテトライル基、シクロドデカンテトライル基、シクロヘキサデカンテトライル基などが挙げられる。これら脂環族基は1価の置換基を含んでいても良い。
脂環族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリーレン基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0096】
芳香族基として、それぞれへテロ原子を含んで複素環構造を持っていてもよい、炭素数5〜15のアリーレン基、炭素数5〜15のアレーントリイル基、炭素数5〜15のアレーンテトライル基が挙げられる。アリーレン基として、フェニレン基、ナフタレンジイル基などが挙げられる。アレーントリイル基(3価)として、ベンゼントリイル基、ナフタレントリイル基などが挙げられる。アレーンテトライル基(4価)として、ベンゼンテトライル基、ナフタレンテトライル基などが挙げられる。これらの芳香族基は置換されていても良い。
芳香族基が有し得る1価の置換基としては、炭素数1〜20のアルキル基、炭素数6〜15のアリール基、ハロゲン原子、ニトロ基、アミド基、ヒドロキシル基、エステル基、エーテル基、アルデヒド基などが挙げられる。
【0097】
また、Ar1、Ar2、R1、R2、X1、X2およびX3はヘテロ原子を含有していてもよい、また、Qが2価の結合基であるときは、Ar1、Ar2、R1、R2、X1、X2およびX3は全て2価の基である。Qが3価の結合基であるときは、Ar1、Ar2、R1、R2、X1、X2およびX3の内の一つが3価の基である。Qが4価の結合基であるときは、Ar1、Ar2、R1、R2、X1、X2およびX3の内の一つが4価の基であるか、二つが3価の基である。
【0098】
環状カルボジイミド化合物としては、次の環状カルボジイミド化合物(a)〜環状カルボジイミド化合物(c)が挙げられる。
【0099】
[環状カルボジイミド化合物(a)]
環状カルボジイミド化合物(a)は、下記式(2)で表される化合物である。
【0100】
【化3】
【0101】
式(2)中、Qaは、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの2価の結合基または脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである2価の結合基であり、さらにヘテロ原子を含有していてもよい。脂肪族基、脂環族基、および芳香族基は、式(1)で説明したものと同じである。但し、式(2)の化合物においては、脂肪族基、脂環族基、および芳香族基は全て2価である。Qaは、下記式(2−1)、式(2−2)または式(2−3)で表される2価の結合基であることが好ましい。
【0102】
【化4】
【0103】
式(2−1)〜式(2−3)中、Ara1、Ara2、Ra1、Ra2、Xa1、Xa2、Xa3、sおよびkは、各々式(1−1)〜式(1−3)中のAr1、Ar2、R1、R2、X1、X2、X3、sおよびkと同じである。但し、これらは全て2価である。
かかる環状カルボジイミド化合物(a)としては、以下の化合物が挙げられる。
【0104】
【化5】
【0105】
[環状カルボジイミド化合物(b)]
環状カルボジイミド化合物(b)は、下記式(3)で表される化合物である。
【0106】
【化6】
【0107】
式(3)中、Qbは、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの3価の結合基または脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである3価の結合基であり、さらにヘテロ原子を含有していてもよい。脂肪族基、脂環族基、および芳香族基は、式(1)で説明したものと同じである。但し、式(3)の化合物においては、Qbを構成する基の内一のつは3価である。
式(3)中、Yは、環状カルボジイミド化合物の環状構造を担持する担体である。
Qbは、下記式(3−1)、式(3−2)または式(3−3)で表される3価の結合基であることが好ましい。
【0108】
【化7】
【0109】
式(3−1)〜式(3−3)中、Arb1、Arb2、Rb1、Rb2、Xb1、Xb2、Xb3、sおよびkは、各々式(1−1)〜式(1−3)のAr1、Ar2、R1、R2、X1、X2、X3、sおよびkと同じである。但しこれらの内の一つは3価の基である。
Yは、単結合、二重結合、原子、原子団またはポリマーであることが好ましい。Yは結合部であり、複数の環状構造がYを介して結合し、式(3)で表される構造を形成している。
かかる環状カルボジイミド化合物(b)としては、下記化合物が挙げられる。
【0110】
【化8】
【0111】
[環状カルボジイミド化合物(c)]
環状カルボジイミド化合物(c)は、下記式(4)で表されるである。
【0112】
【化9】
【0113】
式中、Qcは、脂肪族基と脂環族基と芳香族基とから選択されるいずれか1つの4価の結合基または脂肪族基と脂環族基と芳香族基とから選択される2つ以上の基の組み合わせである4価の結合基であり、さらにヘテロ原子を保有していてもよい。Z1およびZ2は、環状構造を担持する担体である。Z1およびZ2は、互いに結合して環状構造を形成していてもよい。
脂肪族基、脂環族基、および芳香族基は、式(1)で説明したものと同じである。但し、式(4)の化合物において、Qcは4価である。従って、これらの基の内の一つが4価の基であるか、二つが3価の基である。
Qcは、下記式(4−1)、式(4−2)または式(4−3)で表される4価の結合基であることが好ましい。
【0114】
【化10】
【0115】
式(4−1)〜式(4−3)中の、Arc1、Arc2、Rc1、Rc2、Xc1、Xc2、Xc3、sおよびkは、各々式(1−1)〜式(1−3)の、Ar1、Ar2、R1、R2、X1、X2、X3、sおよびkと同じである。但し、Arc1、Arc2、Rc1、Rc2、Xc1、Xc2およびXc3は、これらの内の一つが4価の基であるか、二つが3価の基である。Z1およびZ2は各々独立に、単結合、二重結合、原子、原子団またはポリマーであることが好ましい。Z1およびZ2は結合部であり、複数の環状構造がZ1およびZ2を介して結合し、式(4)で表される構造を形成している。
かかる環状カルボジイミド化合物(c)としては、下記化合物を挙げることができる。
【0116】
【化11】
【0117】
(環状カルボジイミド化合物の製造方法)
環状カルボジイミド化合物は、特開2011−256337号公報の段落番号[0075]に記載の方法などに基づいて合成することができる。
【0118】
〔溶融樹脂排出工程〕
溶融樹脂排出工程は、前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃で排出する。
既述のように、ポリエステル原料樹脂の融点Tmは、ポリエステル原料樹脂の融点が1つである場合はその融点を指すが、ポリエステル原料樹脂が複数の融点を有する場合には、最も高い融点をポリエステル原料樹脂の融点Tmとする。より具体的には、ポリエステル原料樹脂について示差走査熱量測定(DSC)を行ったときに、横軸を温度、縦軸を熱量とするDSC曲線において、1つの吸熱ピークP1を有するときは、吸熱ピークP1のピーク温度をポリエステル原料樹脂の融点とする。DSC曲線において、2つ以上の吸熱ピークP2−1、P2−2、・・・P2−nを有するときは、吸熱ピークP2−1〜P2−nのうち、最も高温で生じた吸熱ピークのピーク温度をポリエステル原料樹脂の融点とする。
【0119】
原料樹脂供給工程により、押出機内に供給された原料樹脂は、溶融樹脂排出工程において、溶融混練され、溶融樹脂(メルトともいう)とされると共に、所定の温度で押出機から排出される。
押出機出口における溶融樹脂の温度が「ポリエステル原料樹脂の融点Tm+20℃」未満であると、原料樹脂の溶融不足により、溶融樹脂中に未溶融異物が残存し、「ポリエステル原料樹脂の融点Tm+30℃」を超えるとΔAVが増加する。
【0120】
押出機出口における溶融樹脂の温度は、押出機出口の設定温度を制御することにより調整することができる。
押出機出口における溶融樹脂の温度は、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+27℃であることが好ましい。
次に、原料樹脂を供給し、溶融混練する二軸押出機の詳細について説明する。
【0121】
(二軸押出機)
まず、本発明で用いる二軸押出機について説明する。
二軸押出機は、主として押出機に供給された原料樹脂を溶融し、溶融樹脂を混練するものであり、一般的に、バレル(シリンダーともいう)を備える。バレルは、原料樹脂を供給するための原料供給口と、溶融樹脂を排出する出口(排出口)とを有し、バレル内には、溶融樹脂を混練するために、スクリューが備えられている。押出機は、大別して、スクリューが1本である単軸と、スクリューが複数本ある多軸とがあり、本発明においては、二軸押出機(二軸スクリュー押出機)を用いる。二軸押出機は、ポリエステル原料樹脂を可塑化するニーディング部を有するスクリューを備えていることが好ましい。
【0122】
なお、ニーディング部とは、スクリューの部位であって、ポリエステル原料樹脂を可塑化する機能を有するセグメント(ニーディングディスクの位置)を意味し、可塑部とは、押出機内の領域であって、ポリエステル原料樹脂が可塑化される領域を指す。従って、押出機の可塑部にスクリューのニーディング部が位置する。
【0123】
以下、押出機の構成を、図1および図2を用いて説明する。
図1は、押出機の構成例を概略的に示している。
図2は、本発明に係るポリエステルフィルム製造方法を実施するフローの一例を示している。
【0124】
図1に示す押出機100は、スクリューを二本(二軸)有する二軸押出機である。押出機100は、供給口12及び押出機出口14を有するバレル10(シリンダー)と、バレル10内で回転する2つのスクリュー20A、20Bと、バレル10の周囲に配置され、バレル10内の温度を制御する温度制御手段30と、を備えている。また、図2に示すように、供給口12の手前には原料供給装置46が設けられている。押出機出口14の先にはギアポンプ44と、フィルター42と、ダイ40が設けられている。原料供給装置46は、第1の樹脂を収納する第1の原料供給装置と、第2の樹脂を収納する第2の原料供給装置とを少なくとも含む複数の原料供給装置で構成されていてもよいし、複数の樹脂を個別に収納可能な複数の収納容器を備えた1つの原料供給装置であってもよい。
【0125】
−バレル−
バレル10は原料樹脂を供給するための供給口12と、加熱溶融された原料樹脂が押し出される押出機出口14を有する。
バレル10の内壁面は、耐熱、耐磨耗性、及び腐食性に優れ、樹脂との摩擦が確保可能な素材を用いることが必要である。一般的には内面を窒化処理した窒化鋼が使用されているが、クロムモリブデン鋼、ニッケルクロムモリブデン鋼、ステンレス鋼を窒化処理して用いることもできる。特に耐摩耗性、耐食性を要求される用途では、遠心鋳造法によりニッケル、コバルト、クロム、タングステン等の耐腐食性、耐磨耗性素材合金をバレル10の内壁面にライニングさせたバイメタリックバレルを用いることや、セラミックの溶射皮膜を形成させることが有効である。
【0126】
バレル10には真空を引くためのベント16A、16Bも設けられている。ベント16A、16Bを通じて真空引きをすることでバレル10内の樹脂中の水分等の揮発成分を効率的に除去することができる。ベント16A、16Bを適正に配置することにより、未乾燥状態の原料(ペレット、パウダー、フレークなど)や成膜途中で出たフィルムの粉砕屑(フラフ)等をそのまま原料樹脂として使用することができる。
ベント16A、16Bは脱気効率との関係で、開口面積やベントの数を適正にすることが求められる。本発明で用いる二軸押出機100は、1箇所以上のベント16A、16Bを有することが望ましい。なお、ベント16A、16Bの数が多過ぎると、溶融樹脂がベントから溢れ出るおそれ、滞留劣化異物増加の懸念があるので、ベントは1箇所又は2箇所設けることが好ましい。
また、ベント付近の壁面に滞留した樹脂や析出した揮発成分が押出機100(バレル10)の内部に落下すると、製品に異物として顕在化する可能性があり、注意が必要である。滞留については、ベント蓋の形状の適正化や、上部ベント、側面ベントの適正な選定が有効であり、揮発成分の析出は、配管等の加熱で析出を防止する手法が一般的に用いられる。
【0127】
例えば、PETを押出す場合、加水分解、熱分解、酸化分解の抑制が製品(フィルム)の品質に大きな影響を及ぼす。
例えば、樹脂供給口12を真空化したり、後述する窒素ガス供給口からの窒素供給(窒素パージともいう)を行うことで酸化分解を抑えることができる。
また、剪断発熱による樹脂分解を抑えるため、押出と脱気が両立できる範囲にはニーディング部等のセグメントは極力設けないことが好ましい。
また、スクリュー出口(押出機出口)14の圧力が大きいほど剪断発熱が大きくなるため、ベント16A、16Bによる脱気効率と押出の安定性が確保できる範囲内で、押出機出口14の圧力は極力低くすることが好ましい。
【0128】
−スクリュー−
バレル10内には、モータおよびギアを含む駆動手段21によって回転する2つのスクリュー20A、20Bが設けられている。
本発明のポリエステルフィルム製造方法においては、押出機100は、スクリューの直径をD〔mm〕とするとき、スクリュー径Dが、60mm以上であることが好ましく、140mm以上であることがより好ましい。スクリュー径Dを60mm以上とすることで押出機内における溶融樹脂の滞留を抑制することができ、ポリエステルフィルムの生産性を向上することができる。
大量生産の観点から、スクリュー径Dは、さらに好ましくは160mm以上である。一方、樹脂の溶融ムラを抑制する観点から、スクリュー径Dは200mm以下であることが好ましい。
【0129】
引き続き、図1および図2を用いて、押出機が備えるスクリューの好ましい態様について説明する。
二軸押出機は、2つのスクリュー20A、20Bの噛み合い型と非噛み合い型に大別され、噛み合い型のほうが、非噛み合い型よりも混練効果が大きい。本発明では、噛み合い型と非噛み合い型のいずれのタイプでも良いが、原料樹脂を十分混練して溶融ムラを抑制する観点から、噛み合い型を用いることが好ましい。
2つのスクリュー20A、20Bの回転方向もそれぞれ同方向と異方向に分かれる。異方向回転スクリュー20A、20Bは同方向回転型よりも混練効果が高く、同方向回転型は自己清掃効果を持っているため、押出機内の滞留防止には有効である。
さらに軸方向も平行と斜交があり、強いせん断を付与する場合に用いられるコニカルタイプの形状もある。
【0130】
本発明で用いる二軸押出機では、様々な形状のスクリューセグメントが用いられる。スクリュー20A、20Bの形状としては、例えば、等ピッチの1条のらせん状フライト22が設けられたフルフライトスクリューが用いられる。
ポリエステル原料樹脂を可塑化する可塑部に、ニーディングディスクやローターなどの剪断を付与するセグメントを用いることで、原料樹脂をより確実に溶融することができる。また、逆スクリューやシールリングを用いることにより、樹脂をせき止め、ベント16A、16Bを引く際のメルトシールを形成することができる。例えば、図1に示すように、ベント16A、16B付近に、上記のような原料樹脂の溶融を促進するニーディング部(混練部)24A、24Bを設けることができる。
【0131】
スクリューの部位のうち、ニーディングディスクが設けられているニーディング部は、ニーディングクリアランスにおける剪断速度が500s−1〜2000s−1であることが好ましい。
ニーディングクリアランスとは、隣接するニーディングディスク間の間隔をいう。
剪断速度が500s−1以上であることで、原料樹脂の溶融不足による未溶融異物の残存を抑制することができ、剪断速度が2000s−1以下であることで、原料樹脂の発熱によるΔAVの増加を抑制することができる。
ニーディングクリアランスにおける剪断速度は、500s−1〜1500s−1であることがより好ましく、500s−1〜800s−1であることが特に好ましい。
【0132】
押出機100の後半では溶融樹脂を冷却するための温調ゾーン(冷却部)が有効である。剪断発熱よりもバレル10の伝熱効率が高い場合は、温調ゾーン(冷却部)にピッチの短いスクリュー28を設けることで、バレル10壁面の樹脂移動速度が高まり、温調効率を上げることができる。冷却効果を高める観点から、冷却部に位置するスクリュー28のピッチは、スクリュー径Dに対し、0.5D〜0.8Dであることが好ましい。
【0133】
−窒素ガス供給口−
押出機100は、原料供給口に最も近いベント孔よりも前記原料供給口に近い位置に、前記ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給する第1の窒素ガス供給口を備えることが好ましい。
なお、「ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガス」とは、ポリエステル原料樹脂の供給量をQ〔L/min〕としたとき、窒素ガスの供給量が1.1Q〔L/min〕〜10.0Q〔L/min〕であることを意味する。
図1においては、ベント16Aよりも原料樹脂の供給口12に近い位置に、窒素ガス供給口を備え、かかる窒素ガス供給口から、ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給することが好ましい。
バレル10内において原料樹脂は、上流側の原料供給口12から、下流側の押出機出口14へと溶融混練されながら進行する。第1の窒素ガス供給口は、原料供給口12に最も近いベント16Aよりも上流に位置する。
【0134】
押出機内の溶融樹脂の酸化分解を抑制するために、バレル10内に窒素ガスを供給することは、従来から行なわれていたが、従来は、バレル10から窒素ガスを供給したり、スクリュー20A、20Bの駆動手段21から供給するものであった。しかしながら、押出機内の原料樹脂の酸化分解を抑制するには、溶融している原料樹脂自体を窒素置換する必要があり、バレル10から窒素ガスを供給したり、駆動手段21から供給する手法では、十分に、溶融樹脂に窒素ガスが供給されず、溶融樹脂の酸化分解を抑制することができなかった。
従って、第1の窒素ガス供給口を、原料供給口に最も近いベント孔よりも前記原料供給口に近い位置に備えることで、押出機内の原料樹脂に窒素ガスを供給することができる。
例えば、図1においては、N1またはN2の位置に窒素ガス供給口を設けることが好ましい。
【0135】
特に、窒素ガス供給量を、ポリエステル原料樹脂の供給量の1.1倍以上とすることで、原料樹脂の酸化を十分に抑制することができ、10.0倍以下とすることで、原料供給口に位置する原料樹脂を吹き飛ばしにくく、原料樹脂の供給不良を抑制することができる。窒素ガス供給量は、ポリエステル原料樹脂の供給量の1.1倍〜8.0倍とすることが好ましい。
【0136】
また、押出機100は、押出機100が備えるスクリュー20A、20Bのニーディング部(24Aおよび24Bの少なくとも一方)に位置し、ポリエステル原料樹脂に対して窒素ガスを供給する第2の窒素ガス供給口を備えていることが好ましい。
原料樹脂の酸化分解や熱分解は、原料樹脂が溶融し始める段階〜溶融している最中に起こり易く、原料樹脂がかかる状態であるときに、原料樹脂に対して窒素ガスを供給することが好ましい。原料樹脂は、原料を混練するニーディング部において、溶融し、熱分解を起こしやすくなるため、かかる原料樹脂に対して直接窒素ガスを供給し易いように、ニーディング部に窒素ガス供給口を供えていることが好ましい。
【0137】
原料樹脂の熱分解は、特に、溶融し始める段階において生じやすいため、第2の窒素ガス供給口は、ニーディング部24A、24Bの中央(ニーディング部24Aにおいては、図1におけるN2)よりも、ニーディング部の上流側端部(ニーディング部24Aにおいては、図1におけるN1)に備えられていることが好ましい。
【0138】
図1において、ニーディング部は、原料樹脂の進行方向上流側に位置する24Aと下流側に位置する24Bとが示されているが、本発明において、ニーディング部に位置する第2の窒素ガス供給口は、1つのみでも2つ以上あってもよい。すなわち、図1においては、第2の窒素ガス供給口は、ニーディング部24Aの位置および、ニーディング部24Bの少なくとも一方に備えられていればよい。
【0139】
第1の窒素ガス供給口および第2の窒素ガス供給口における窒素ガスの供給速度は、2m/分〜50m/分であることが好ましい。窒素ガスの供給速度が、2m/分以上であることで原料樹脂の酸化抑制を十分に行なうことができ、50m/分以下とすることで、樹原料樹脂の樹脂温度の低下を抑制し、原料樹脂の可塑化発熱を抑制することができる。
窒素ガスの供給速度は、3m/分〜45m/分であることがより好ましい。
【0140】
−温度制御手段−
バレル10の周囲には、温度制御手段30が設けられている。図1に示す押出機100では、原料供給口12から押出機出口14に向けて長手方向に9つに分割された加熱/冷却装置C1〜C9が温度制御手段30を構成している。このようにバレル10の周囲に分割して配置された加熱/冷却装置C1〜C9によって、例えば可塑部C1〜C7と冷却部C8、C9の各領域(ゾーン)に区画し、バレル10内を領域ごとに所望の温度に制御することができる。
【0141】
加熱は、通常バンドヒーターまたはシーズ線アルミ鋳込みヒーターが用いられるが、これらに限定されず、例えば熱媒循環加熱方法も用いることができる。一方、冷却はブロワーによる空冷が一般的であるが、バレル10の周囲に巻き付けたパイプに水または油を流す方法もある。
【0142】
‐原料樹脂の溶融混練‐
押出機100は、温度制御手段30によりバレル10を加熱するとともにスクリューを回転させ、供給口12から原料樹脂を供給する。なお、供給口12は、原料樹脂のペレット等が加熱されて融着しないようにすることと、モータなどのスクリュー駆動設備を保護するため、伝熱防止として冷却することが好ましい。
【0143】
バレル内に供給された原料樹脂は、温度制御手段30による加熱のほか、スクリュー20A、20Bの回転に伴う樹脂同士の摩擦、樹脂とスクリュー20A、20Bやバレル10との摩擦などによる発熱によって溶融されるとともに、スクリューの回転に伴って押出機出口14に向けて徐々に移動する。
バレル内に供給された原料樹脂は融点Tm℃以上の温度に加熱されるが、樹脂温度が低過ぎると溶融押出時の溶融が不足し、ダイ40からの排出が困難になるおそれがあり、樹脂温度が高過ぎると熱分解によって末端COOHが著しく増加して耐加水分解性の低下を招くおそれがある。なお、既述のように、原料樹脂の融点Tmは、原料樹脂を複数種用いることにより、Tmが複数ある場合は、最も高い融点をいう。
これらの観点から、温度制御手段30による加熱温度及びスクリュー20A、20Bの回転数を調整することにより、二軸押出機内の長手方向における最大樹脂温度Tmaxを(Tm+40)℃〜(Tm+60)℃にすることが好ましく、(Tm+40)℃〜(Tm+55)℃とすることがより好ましく、(Tm+45)℃〜(Tm+50)℃とすることがさらに好ましい。
【0144】
二軸押出機内の長手方向における最大樹脂温度Tmaxは、二軸押出機100のスクリュー20A、20Bが配設されたバレル内で加熱されている原料樹脂の温度であり、剪断発熱があるときはその発熱による局所的高温部を含む温度である。Tmaxはバレル内の樹脂温度の測定により得られる。上記のTm及びTmaxの関係式において、PET樹脂の場合、Tmax[℃]は、末端COOHの増加を抑える観点から、290℃以下が好ましく、280℃以下がより好ましい。また、Tmaxの下限温度は、樹脂の溶融不足を防止する観点から260℃とすることが好ましい。
【0145】
‐ベント圧力‐
ベント16A、16Bを通じて真空引きをすることでバレル内の樹脂中の水分等の揮発成分を効率的に除去することができる。ベント圧力が低過ぎると溶融樹脂がバレル10の外に溢れ出るおそれがあり、ベント圧力が高過ぎると揮発成分の除去が不十分となり、得られたフィルムの加水分解が生じ易くなるおそれがある。溶融樹脂がベント16A、16Bから溢れ出ることを防ぐとともに揮発成分を選択的に除去する観点から、ベント圧力は0.01Torr〜5Torr(1.333Pa〜666.5Pa)とすることが好ましく、0.01Torr〜4Torr(1.333Pa〜533.2Pa)とすることがより好ましい。
【0146】
‐平均滞留時間‐
バレル内で原料樹脂を加熱溶融し、押出機出口14を出た後、ダイ40からフィルム状に押出されるまでの平均滞留時間を10分〜20分とする。原料樹脂を加熱溶融して、押出機100の押出機出口14を出てからダイ40から押出されるまでの平均滞留時間が10分未満では未溶融樹脂が残留し易く、一方、20分を超えると、熱分解によって末端COOH量が増加して耐加水分解性が低下する。このような観点から、原料樹脂を加熱溶融して押出機出口14から押出され後の上記平均滞留時間は、10分〜20分が好ましく、10分〜15分がより好ましい。
ここで、平均滞留時間は、下記式で定義される。
平均滞留時間(秒)=押出機下流配管容積(cm3)×溶融体密度(g/cm3)×3600/1000÷押出量(kg/hr)
【0147】
‐冷却‐
上記のように原料樹脂をバレル内で加熱溶融する一方、温度制御手段30によりバレル10の押出機出口14側の内壁がポリエステル樹脂(原料樹脂)の融点Tm(℃)以下の冷却部となるように制御する。バレル10の押出機出口14側の内壁を冷却部として原料樹脂の融点Tm(℃)以下に制御すれば、樹脂が過剰に加熱されて末端COOH量が増加することを抑制することができる。末端COOH量の増加を確実に抑制する観点から、かかる冷却部における温度は、(Tm−100)℃〜Tm℃の範囲内が好ましく、(Tm−50)℃〜(Tm−10)℃の範囲内がより好ましい。
【0148】
冷却部の長さは、スクリュー径Dに対し、4D〜11Dにすることが好ましい。冷却部の長さが4D以上であれば、溶融加熱された樹脂を効果的に冷却して末端COOHの増加を抑制する。一方、冷却部の長さが11D以下であれば、樹脂を冷却し過ぎて固化することを防ぎ、溶融押出しを円滑に行うことができる。
なお、押出機出口14における樹脂温度は、既述のように、ポリエステル原料樹脂の融点Tmに対してTm+20℃〜Tm+30℃にする。
【0149】
〔成形工程〕
本発明のポリエステルフィルム製造方法は、押出機から押出した溶融樹脂をフィルム状に成膜する成形工程を有する。
本発明のポリエステルフィルム製造方法においては、上記構成の樹脂供給工程を経た後、押出機から押出した溶融樹脂をフィルム状に成膜する。
押出機から押出した溶融樹脂は、図2に示すように、ギアポンプ44、フィルター42を介してダイ40から排出される。ダイ40の開口部の形状が長尺状の幅広な形状をしていることで、ダイ40から排出される溶融樹脂は、フィルム状に加工される。
【0150】
−ダイ−
図1に示すバレル10の押出機出口14には、押出機出口14から押出された溶融樹脂をフィルム状(帯状)に排出するためのダイ40(図2)が設けられている。また、バレル10の押出機出口14とダイ40との間には、フィルムに未溶融樹脂や異物が混入することを防ぐためのフィルター42が設けられている。
【0151】
−ギアポンプ−
厚み精度を向上させるためには、押出量の変動を極力減少させることが重要である。押出量の変動を極力減少させるために押出機100とダイ40との間にギアポンプ44を設けてもよい。ギアポンプ44から一定量の樹脂を供給することにより、厚み精度を向上させることができる。特に、二軸スクリュー押出機を用いる場合には、押出機自身の昇圧能力が低いため、ギアポンプ44による押出安定化を図ることが好ましい。
【0152】
ギアポンプ44を用いることにより、ギアポンプ44の2次側の圧力変動を1次側の1/5以下にすることも可能であり、樹脂圧力変動幅を±1%以内にできる。その他のメリットとしては、スクリュー先端部の圧力を上げることなしにフィルターによる濾過が可能なことから、樹脂温度の上昇の防止、輸送効率の向上、及び押出機内での滞留時間の短縮が期待できる。また、フィルターの濾圧上昇が原因で、スクリューから供給される樹脂量が経時変動することも防止できる。ただし、ギアポンプ44を設置すると、設備の選定方法によっては設備の長さが長くなり、樹脂の滞留時間が長くなることと、ギアポンプ部のせん断応力によって分子鎖の切断を引き起こすことがあり注意が必要である。
【0153】
ギアポンプ44は1次圧力(入圧)と2次圧力(出圧)の差を大きくし過ぎると、ギアポンプ44の負荷が大きくなり、せん断発熱が大きくなる。そのため、運転時の差圧は20MPa以内、好ましくは15MPa、更に好ましくは10MPa以内とする。また、フィルム厚みの均一化のために、ギアポンプ44の一次圧力を一定にするために、押出機のスクリュー回転を制御したり、圧力調節弁を用いたりすることも有効である。
【0154】
なお、本発明のポリエステルフィルムの製造方法においては、固有粘度(IV)が0.70dl/g以上の原料樹脂を用いるため、成形工程においては、原料樹脂をバレル内で加熱溶融して押出機出口14から押出された後、10分〜20分の平均滞留時間を経て、スクリュー径Dを考慮してスクリュー回転数N〔rpm〕と押出量Q〔kg/hr〕を制御することで下記式(i)を満たす条件下でフィルム状に溶融押出しを行うことが好ましい。
6.0×10−6×D3≦Q/N≦1.1×10−5×D3 ・・・式(i)
IVが0.7dl/g以上の原料樹脂を溶融する場合、Nを低下させることで溶融と脱気、樹脂冷却を同時に満たし易い。また、押出機出口14での樹脂温度を特に290℃以下に制御することで、特にその下流の配管滞留での末端COOHの増加抑制に大きな効果がある。
Q/Nが6.0×10−6×D3以上とすることで、スクリュー20A、20Bの高回転による原料樹脂の過発熱を抑制し、押出機出口14における樹脂温度を290℃以下にし易く、ΔAVを3eq/t以下にし易い。また、Q/Nが1.1×10−5×D3以下であることで、ベント直下の樹脂充填率が増加しにくく、ベント16A、16Bから溶融樹脂が溢れにくくなるほか、ベント圧が低下しにくいため、押出機内部での樹脂の加水分解が進行しにくく、末端COOHの発生を抑制し易い。さらに、未溶融樹脂がフィルムに混入しにくくなり、ポリエステルフィルムの強度が低下することを抑制することができるので、延伸工程におけるフィルム破断を抑制することができる。
なお、原料樹脂の押出機への供給量〔kg/hr〕と押出機内の押出量とは同様に扱うことができ、原料樹脂の押出機への供給量がQ〔kg/hr〕であるとき、押出機内の押出量はQ〔kg/hr〕であると考えてよい。
【0155】
上記溶融押出しは、下記式(ii)に示す条件で行なうことがより好ましく、下記式(iii)に示す条件下で行うことがさらに好ましい。
7×10−6×D3≦Q/N≦1×10−5×D3 ・・・式(ii)
8×10−6×D3≦Q/N≦9×10−6×D3 ・・・式(iii)
【0156】
スクリュー回転数Nが低過ぎると、温度制御手段30によって温度ムラが生じて未溶融樹脂が生じ易く、スクリュー回転数Nが高過ぎると、過度に発熱して末端COOH量の増加につながるため、スクリュー回転数Nは1.9×102×D−0.5rpm〜8.4×102×D−0.5rpmが好ましく、6.3×102×D−0.5rpm〜7.9×102×D−0.5rpmがより好ましい。
また、押出量Qが少な過ぎると過度に加熱され易くなり、多過ぎると未溶融樹脂が生じ易くなるため、押出量Qは1.1×10−3×D2.5kg/hr〜7.6×10−3×D2.5kg/hrが好ましく、3.8×10−3×D2.5kg/hr〜7.1×10−3×D2.5kg/hrがより好ましい。
【0157】
バレル10の押出機出口14から押し出された樹脂をフィルター42に通してダイ40から(例えば冷却ロールに)押し出してフィルム状に成形する。
ダイ40からメルト(溶融樹脂)を押出した後、冷却ロールに接触させるまでの間(エアギャップ)は、湿度を5%RH〜60%RHに調整することが好ましく、15%RH〜50%RHに調整することがより好ましい。エアギャップでの湿度を上記範囲にすることで、フィルム表面のCOOH量やOH量を調節することが可能であり、低湿度に調節することで、フィルム表面のカルボン酸量を減少させることができる。
【0158】
また、本発明の方法によれば、樹脂温度を一度上げてから冷却部で下げることで、末端COOH量の増加を抑制するとともに、未溶融異物の発生を抑制することができるほか、フィルムのヘイズ上昇を抑制する効果が得られる。特に厚手成膜をする際は冷却速度不足より、ヘイズ上昇しやすいが、その対策方法として用いることが可能である。
なお、フィルム厚は、2mm〜8mmが好ましく、より好ましくは2.5mm〜7mmであり、さらに好ましくは3mm〜6mmである。厚みを厚くすることで、押出されたメルトがガラス転移温度(Tg)以下に冷却するまでの所要時間を長くすることができる。この間に、フィルム表面のCOOH基はポリエステル内部に拡散され、表面COOH量を低減することができる。
【0159】
上記工程により、原料樹脂の末端COOH量と溶融押出しされたフィルムの末端COOH量との差ΔAVが3eq/t以下のポリエステルフィルムを製造することができ、例えば、末端COOH量が25eq/t(トン)以下であるポリエステルフィルムが得られる。末端COOH量が25eq/トン以下であると、耐加水分解性に優れており、長期耐久性が得られる。末端COOH量は、耐加水分解の点では低いことが望ましいが、フィルムを被着物に密着させる場合の密着性向上の点から、2eq/トン以上が好ましい。中でも、10〜20eq/トンの範囲がより好ましい。
末端COOH量の測定は、既述の方法と同様にして行なうことができる。
【0160】
<太陽電池用ポリエステルフィルム>
本発明の太陽電池用ポリエステルフィルムは、既述の本発明のポリエステルフィルム製造方法により製造することができる。
本発明の太陽電池用ポリエステルフィルムは、本発明のポリエステルフィルム製造方法により製造されるため、高い耐加水分解性を有することができる。
【0161】
また、太陽電池用ポリエステルフィルムは、1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%含むCHDM系ポリエステル樹脂を含有する層を少なくとも1層有することで、フィルム耐候性をさらに向上することができる。
CHDM系ポリエステル樹脂を含有する層は、本発明のポリエステルフィルム製造方法により製造されたポリエステルフィルムであってもよいし、本発明のポリエステルフィルム製造方法により製造されたポリエステルフィルム上に押出成形した層であってもよいし、本発明のポリエステルフィルム製造方法により製造されたポリエステルフィルムに貼り付けたフィルムないしシート状の部材であってもよい。
【0162】
このように、太陽電池用ポリエステルフィルムは、CHDM系ポリエステル樹脂を含有する層を少なくとも1層有していればよく、単層であっても、2以上の層を有していてもよい。
特に、CHDM系ポリエステル樹脂が有するCHDM由来の構造が、ジオール化合物由来の構造の全量に対して80モル%〜100モル%であるときは、太陽電池用ポリエステルフィルムは、積層構成にすることが好ましい。これは、CHDM系ポリエステル樹脂中のジオール化合物由来の構造の全量に対するCHDM由来の構造の比率が高くなると、ポリエチレンテレフタレート(PET)に対し、耐候性(耐加水分解性)は高くなり易いが、力学強度が弱くなり易い。 このため、他のポリエステル(例えばPET)と積層することで相補することができ、好ましい。
【0163】
本発明の太陽電池用ポリエステルフィルムは、CHDM系ポリエステル樹脂を含有する層(P1層と称する)と、ポリエチレンテレフタレートを主成分とするポリエステルを含有する層(P2層と称する)とが積層された態様も好ましい。
なお、本発明の太陽電池用ポリエステルフィルムは、P1層とP2層とをそれぞれ2層以上有していてもよい。
【0164】
P2層は、ジカルボン酸由来の構造中にテレフタル酸由来の構造を95モル%以上有し、かつジオール化合物由来の構造中にエチレングリコール由来の構造を95モル%以上含むポリエステル樹脂を含む層を指す。
またP2層のIVは0.7〜0.9が好ましく、より好ましくは0.72〜0.85、さらに好ましくは0.74〜0.82である。このようにP2層のIVを高めにすることでwetサーモ、および、dryサーモでの分解(ポリエステル樹脂の分子量低下)を抑制することができる。
【0165】
本発明の太陽電池用ポリエステルフィルムは、P1層とP2層の層数の和は、2層以上が好ましく、より好ましくは2層以上5層以下、さらに好ましくは2層以上4層以下である。中でも好ましいのが、P2層の両側をP1層で挟んだ3層構造、あるいはP1層の両側をP2層で挟んだ3層構造、P2層とP1層を積層した2層構造である。
【0166】
本発明の太陽電池用ポリエステルフィルムが2層以上で構成される場合、太陽電池用ポリエステルフィルムの厚みは、P1層の総和が全厚みの5%〜40%であることが好ましく、より好ましくは7%〜38%、さらに好ましくは10%〜35%である。この下限値以上にすることで高い耐候性を発現でき、この上限値以下にすることで高い力学強度を発現し易い。
ポリエステルフィルムの各層の厚みは、フィルムの断面を、SIMS(Secondary Ion-microprobe Mass Spectrometer;二次イオン質量分析計)を用い測定し、P1層の特徴フラグメント、および、P2層の特徴フラグメントでイメージングすることで求めることができる。
【0167】
このような太陽電池用ポリエステルフィルムの積層構造は定法により得ることができる。例えば、P1層を構成するポリエステル樹脂をある押出機に供給し、P2層を構成するポリエステル樹脂を他の押出機に供給する等して、複数の押出し機から供給されたメルト(樹脂の融体)をマルチマニフォールドダイ、フィードブロックダイを用い積層し押出してもよい。また、予め用意したP1層およびP2層のどちらか一方となるポリエステルフィルム上に、他方の層を構成するポリエステル樹脂を押し出して積層してもよいし、他の層を貼り付けて積層してもよい。
【0168】
本発明の方法により製造される太陽電池用ポリエステルフィルムは、光安定化剤、酸化防止剤などの添加剤を更に含有することができる。
【0169】
光安定化剤を含有すると、紫外線劣化を防ぐことができる。光安定化剤とは、紫外線などの光線を吸収して熱エネルギーに変換する化合物、樹脂が光吸収して分解して発生したラジカルを捕捉し、分解連鎖反応を抑制する材料などが挙げられる。光安定化剤として好ましくは、紫外線などの光線を吸収して熱エネルギーに変換する化合物である。このような光安定化剤を含有することで、長期間継続的に紫外線の照射を受けても、部分放電電圧の向上効果を長期間高く保つことが可能になったり、樹脂中の紫外線による色調変化、強度劣化等が防止される。
【0170】
例えば紫外線吸収剤は、ポリエステルの他の特性が損なわれない範囲であれば、有機系紫外線吸収剤、無機系紫外線吸収剤、及びこれらの併用のいずれも、特に限定されることなく好適に用いることができる。一方、紫外線吸収剤は、耐湿熱性に優れ、樹脂中に均一分散できることが望まれる。
【0171】
紫外線吸収剤の例としては、有機系の紫外線吸収剤として、サリチル酸系、ベンゾフェノン系、ベンゾトリアゾール系、シアノアクリレート系等の紫外線吸収剤及びヒンダードアミン系等の紫外線安定剤などが挙げられる。具体的には、例えば、サリチル酸系のp−t−ブチルフェニルサリシレート、p−オクチルフェニルサリシレート、ベンゾフェノン系の2,4−ジヒドロキシベンゾフェノン、2−ヒドロキシ−4−メトキシベンゾフェノン、2−ヒドロキシ−4−メトキシ−5−スルホベンゾフェノン、2,2’,4,4’−テトラヒドロキシベンゾフェノン、ビス(2−メトキシ−4−ヒドロキシ−5−ベンゾイルフェニル)メタン、ベンゾトリアゾール系の2−(2’−ヒドロキシ−5’−メチルフェニル)ベンゾトリアゾール、2−(2’−ヒドロキシ−5’−メチルフェニル)ベンゾトリアゾール、2,2’−メチレンビス[4−(1,1,3,3−テトラメチルブチル)−6−(2Hベンゾトリアゾール−2−イル)フェノール]、シアノアクリレート系のエチル=α−シアノ−β,β−ジフェニルアクリレート)、トリアジン系として2−(4,6−ジフェニル−1,3,5−トリアジン−2−イル)−5−[(ヘキシル)オキシ]−フェノール、ヒンダードアミン系のビス(2,2,6,6−テトラメチル−4−ピペリジル)セバケート、コハク酸ジメチル・1−(2−ヒドロキシエチル)−4−ヒドロキシ−2,2,6,6−テトラメチルピペリジン重縮合物、そのほかに、ニッケルビス(オクチルフェニル)サルファイド、及び2,4−ジ・t−ブチルフェニル−3’,5’−ジ・t−ブチル−4’−ヒドロキシベンゾエート、などが挙げられる。
これらの紫外線吸収剤のうち、繰り返し紫外線吸収に対する耐性が高いという点で、トリアジン系紫外線吸収剤がより好ましい。なお、これらの紫外線吸収剤は、上述の紫外線吸収剤単体でフィルムに添加してもよいし、有機系導電性材料や、非水溶性樹脂に紫外線吸収剤能を有するモノマーを共重合させた形態で導入してもよい。
【0172】
光安定化剤のポリエステルフィルム中における含有量は、ポリエステルフィルムの全質量に対して、0.1質量%以上10質量%以下が好ましく、より好ましくは0.3質量%以上7質量%以下であり、さらに好ましくは0.7質量%以上4質量%以下である。これにより、長期経時での光劣化によるポリエステルの分子量低下を抑止でき、その結果発生するフィルム内の凝集破壊に起因する密着力低下を抑止できる。
【0173】
更に、本発明のポリエステルフィルムは、前記光安定化剤の他にも、例えば、易滑剤(微粒子)、紫外線吸収剤、着色剤、核剤(結晶化剤)、難燃化剤などを添加剤として含有することができる。
【0174】
<太陽電池用バックシート>
本発明の太陽電池用ポリエステルフィルムは、太陽電池モジュールの太陽光入射側とは反対側の裏面に配置される裏面保護シート(太陽電池用バックシート)、バリアフィルム基材等の用途に好適である。
【0175】
<太陽電池モジュール>
本発明の太陽電池モジュールは、太陽光が入射する透明性のフロント基板と、前記フロント基板の上に設けられ、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分と、前記セル構造部分の前記フロント基板が位置する側と反対側に設けられ、前記封止材と隣接して配置された、本発明の太陽電池用ポリエステルフィルム(例えば、太陽電池用バックシート)と、を備えて構成される。
【0176】
太陽電池モジュールは、例えば、電気を取り出すリード配線で接続された発電素子(太陽電池素子)をエチレン・酢酸ビニル共重合体系(EVA系)樹脂等の封止剤で封止し、これを、ガラス等の透明基板と、本発明のポリエステルフィルムの製造方法によって得られたポリエステルフィルム(バックシート)との間に挟んで互いに張り合わせることによって構成してもよい。
太陽電池素子の例としては、単結晶シリコン、多結晶シリコン、アモルファスシリコンなどのシリコン系、銅−インジウム−ガリウム−セレン、銅−インジウム−セレン、カドミウム−テルル、ガリウム−砒素などのIII−V族やII−VI族化合物半導体系など、各種公知の太陽電池素子を適用することができる。
【実施例】
【0177】
以下、本発明を実施例により更に具体的に説明するが、本発明はその主旨を越えない限り、以下の実施例に限定されるものではない。
【0178】
〔実施例1〜実施例16、比較例1〜比較例11〕
‐二軸押出機‐
押出機として、図1に示すように2箇所にベントが設けられたバレル内に、下記構成のスクリューを備え、バレルの周囲には長手方向に9つのゾーンに分割して温度制御を行うことができるヒータ(温度制御手段)を備えたダブルベント式同方向回転噛合型の二軸押出機を準備した。
スクリュー径D:110mm
スクリュー長L:スクリュー径Dとの比(L/D)が31.5となる長さ[mm]
(1ゾーンの幅:3.5D)
スクリュー形状:第1ベント直前に可塑化ニーディング部を設置
第2ベント直前に脱気促進ニーディング部を設置
可塑化ニーディング部の剪断速度は、表1に示す速度〔s−1〕とした。
ベント位置:第1ベント・・・図1のC5の位置
第2ベント・・・図1のC7の位置
【0179】
二軸押出機の押出機出口以降には、図2に示すように、下記構成のギアポンプ、金属繊維フィルターおよびダイを接続し、ダイを加熱するヒーターの設定温度は280℃とし、平均滞留時間は10分とした。
ギアポンプ:2ギアタイプ
フィルター:金属繊維焼結フィルター(孔径20μm)
【0180】
‐原料樹脂‐
AVが13eq/tであるポリエチレンテレフタラート樹脂(PET樹脂)のペレットと、及び、AVが15eq/tであり、表1に示す嵩密度を有するフラフとを用いて、原料樹脂とした。なお、比較例1及び比較例2では、フラフを含まず、嵩密度が0.8のPETペレットのみを用いて、表1に示すAVの原料樹脂とした。原料樹脂の融点Tmは、いずれも255℃であった。
また、原料樹脂中のフラフの含有量は表1に示す量である。なお、表1に示すフラフの含有率は、原料樹脂の全質量に対するフラフの割合(質量%)である。
【0181】
押出機は、図1における供給口12側の1番目のゾーン(C1)は70℃に、2〜8番目のゾーン(C2〜C8)は270℃に、9番目のゾーン(C9)は250℃にそれぞれ温度設定を行った。
スクリューの回転数を77rpmに設定し、供給口12から、表1の「押出機入口樹脂温度」に示す温度〔℃〕で、原料樹脂を供給して加熱溶融し、押出量を350kg/hrに設定して溶融押出を行った。
【0182】
押出機内の原料樹脂に対しては、図1のN1に示す位置に窒素ガス供給口を設け、原料樹脂の供給量Q〔L/min〕に対して、表1に示す倍率の供給量〔L/min〕の窒素ガスを供給した。例えば、実施例1においては、原料樹脂の供給量Qの0.5倍(0.5Q)〔L/min〕である。
また、窒素ガスの供給速度は、表1に示す速度〔m/min〕とした。
【0183】
溶融樹脂は、表1の「押出機出口樹脂温度」に示す温度〔℃〕で、押出機出口から押出し、押出された溶融体(メルト)をギアポンプ、金属繊維フィルター(孔径20μm)を通した後、ダイから冷却(チル)ロールに押出した。押出されたメルトは、静電印加法を用いて冷却ロールに密着させた。冷却ロールは、中空のチルロールを用い、この中に熱媒として水を通して温調できるようになっている。
なお、ダイ出口から冷却ロールまでの搬送域(エアギャップ)は、この搬送域を囲い、この中に調湿空気を導入することにより、湿度を30%RHに調節してある。押出機の押出量の調整及びダイの開口部の形状を上記構成とすることにより、メルト厚みを3000μmとした。
以上のようにして、実施例1〜実施例16および比較例1〜比較例12の各PETフィルムの製造を試みた。
【0184】
<PETフィルムの評価>
PETフィルムの破断伸度およびΔAVから、PETフィルムの耐候性を評価した。
【0185】
‐破断伸度の測定‐
冷却ロールにより冷却されたPETフィルムを二軸延伸(3.4×3.8倍)して、二軸延伸後のフィルムの湿熱条件下(120℃×100RH%)での破断伸度が半減する時間で判定する。85時間以上で良好な耐候性(耐加水分解性)を示すものと判定する。結果を表1に示す。
【0186】
−ΔAVの算出−
原料樹脂のAVと溶融押出しされたフィルムのAVとを測定し、原料樹脂のAV(AV1)と溶融押出しされたフィルムのAV(AV2)との差であるΔAV(|AV1−AV2|)を算出した。ΔAVが3eq/t以下であるほど良好な耐候性を示すとして判定した。結果を表1に示す。
【0187】
【表1】
【0188】
〔実施例17〜実施例22〕
末端封止剤として、以下の化合物を各実施例に用いた。
【0189】
1.環状カルボジイミド化合物
1)環状カルボジイミド化合物(1)(表2中の「環状」)
既述の環状カルボジイミド化合物(c)であり、下記構造の化合物(重量平均分子量Mw=516)を用いた。環状カルボジイミド化合物(1)は、特開2011−258641号公報の参考例1に記載の合成方法を参考に合成した。
【0190】
【化12】
【0191】
2.非環状カルボジイミド化合物
1)線状カルボジイミド(1)(表2中の「線状」)
Stabaxol P400(ラインケミー社製、重量平均分子量Mw=約2000)を用いた。
2)モノカルボジイミド(1)(表2中の「モノ」)
Stabaxol I(ラインケミー社製、重量平均分子量Mw=362.5)を用いた。
これらのカルボジイミド末端封止剤の構造を以下に示す。
【0192】
【化13】
【0193】
−末端封止剤入りのPETペレットの作成−
表2に示す末端封止剤と、AVが13eq/tであるポリエチレンテレフタラート樹脂(PET樹脂)と事前に溶融混合し、末端封止剤の濃度が、溶融混合物全量の50質量%となるよう調整して、実施例17〜実施例22で用いる各マスターペレット(末端封止剤入りのPETペレット)を作成した。
【0194】
−PETフィルムの作成−
実施例1のPETフィルムの製造において、実施例1で用いたPET樹脂のペレットに代えて、得られた各マスターペレットを用いたほかは、実施例1と同様にしてPETフィルムを作成した。
【0195】
<PETフィルムの評価>
得られた実施例17〜実施例22のPETフィルムについて、実施例1のPETフィルムと同様の方法で、破断伸度およびΔAVから、PETフィルムの耐候性を評価した。評価結果を表2に示す。なお、表2には、参考のため、実施例1の条件詳細および評価結果も示した。
【0196】
【表2】
【0197】
〔実施例23〜実施例30、および比較例12〕
(1)CHDM系ポリエステル樹脂(PCT樹脂)の作成
・第1工程
ジカルボン酸化合物としてイソフタル酸(IPA)とテレフタル酸(TPA)、ジオール化合物としてシクロヘキサンジメタノール(CHDM)、エチレングリコール(EG)を用い、触媒として酢酸マグネシウム、三酸化アンチモンを150℃、窒素雰囲気下で溶融後、攪拌しながら230℃まで3時間かけて昇温し、メタノールを留出させ、エステル交換反応を終了した。この際、IPA、TPA、CHDM、およびEGの添加量を変えることで、表3に示す組成のCHDM系ポリエステル樹脂(CHDM−1〜CHDM−9)を得た。
【0198】
・第2工程
エステル交換反応終了後、リン酸をエチレングリコールに溶解したエチレングリコール溶液を添加した。
【0199】
・第3工程
重合反応を最終到達温度285℃、真空度0.1Torrで行い、ポリエステルを得、これをペレット化した。
【0200】
・第4工程
上記で得られたポリエステルペレットを、160℃で6時間乾燥、結晶化した。
【0201】
(2)PET系ポリエステル樹脂(PET樹脂)の作成
表3に示すように、CHDMとIPAを添加しない以外は、CHDM系ポリエステル樹脂と同様にして、PET系ポリエステル樹脂(PET−1)を作成した。
【0202】
上記にて得られたCHDM系ポリエステル樹脂について、ジオール化合物中のシクロヘキサンジメタノール(CHDM)含率およびジカルボン酸化合物中のイソフタル酸含率を以下の方法で測定した。
【0203】
(組成測定法)
CHDM系ポリエステルペレットをヘキサフルオロイソプロパノール(HFIP)に溶解した後、1H−NMRにより定量した。標品(CHDM、テレフタル酸、EG、およびイソフタル酸)を予め測定し、これを用いシグナルを同定した。
得られたイソフタル酸残基の量およびCHDM残基の量を表3に記載した。
なお、100モル%−イソフタル酸(IPA)含率(モル%)がテレフタル酸(TPA)含率(モル%)であり、100(モル%)−CHDM率(モル%)がEG含率(モル%)である。
表3に示すように、CHDM−3、CHDM−7、およびPET−1の各ポリエステル樹脂は、乾燥した後、窒素気流中210℃で24時間、固相重合した
なお、これらのCHDM系ポリエステルペレットおよびPET系ポリエステルペレットのIVおよびAVは、実施例1で用いたPETペレットと同様の方法で測定し、表3に記載した。
【0204】
【表3】
【0205】
(3)溶融製膜
(3−1)押出し
PET−1およびCHDM−1〜CHDM−9の各樹脂を乾燥した後、2軸押出機で真空下、溶融混練した。このとき、溶融混練の温度は、PET−1は280℃、CHDM−1〜CHDM−4は285℃、CHDM−5〜CHDM−8は305℃、CHDM−9は、305℃とした。
得られた溶融樹脂を、フィードブロックダイを用い25℃のキャストドラム上に押出し、表4に示す層構成の単層ポリエステルフィルムまたは積層ポリエステルフィルムを作成した。各ポリエステルフィルムを作成する上での押出機での押出条件および排出条件は、表5に示したとおりである。
【0206】
【表4】
【0207】
得られた単層ポリエステルフィルムおよび積層ポリエステルフィルムについて、実施例1と同様にして耐候性を評価した。評価結果を表5に示す。
なお、フラフは、PETのフラフを用い、全PET成分に対する表5に示す量〔質量%〕で添加した。積層ポリエステルフィルムの作成においては、PET−1を溶融混練した押出機にのみ、フラフを添加した。
【0208】
【表5】
【0209】
〔実施例31〜実施例60〕
<太陽電池モジュールの作製>
実施例1〜実施例30の各ポリエステルフィルムを太陽電池用バックシートとして用い、次のようにして、実施例31〜実施例60の太陽電池モジュールを作製した。
厚さ3.2mmの強化ガラスと、EVAシート〔三井化学ファブロ社製のSC50B〕と、結晶系太陽電池セルと、EVAシート〔三井化学ファブロ社製のSC50B〕と、実施例1〜実施例30のポリエステルフィルムのいずれか1枚とを、この順に重ね合わせ、真空ラミネータ〔日清紡社製、真空ラミネート機〕を用いてホットプレスすることにより、各部材とEVAシートとを接着させた。
【0210】
作製した各太陽電池モジュールについて、発電運転をしたところ、いずれも太陽電池として良好な発電性能を示した。
【符号の説明】
【0211】
10 バレル
12 供給口
14 押出機出口
16A、16B ベント
20A、20B スクリュー
30 温度制御手段
40 ダイ
42 フィルター
44 ギアポンプ
46 原料供給装置
100 二軸押出機
C1〜C9 加熱/冷却装置
【特許請求の範囲】
【請求項1】
嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、
前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、前記ポリエステル原料樹脂Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、
前記溶融樹脂をフィルム状に成形する成形工程と、
を含むポリエステルフィルム製造方法。
【請求項2】
前記二軸押出機が、
少なくとも1つのベント孔と、
前記原料供給口に最も近いベント孔よりも前記原料供給口に近い位置に、前記ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給する第1の窒素ガス供給口と、
を備える請求項1に記載のポリエステルフィルム製造方法。
【請求項3】
前記二軸押出機が、
前記ポリエステル原料樹脂を可塑化するニーディング部を有するスクリューと、
前記ニーディング部に位置し、前記ポリエステル原料樹脂に対して窒素ガスを供給する第2の窒素ガス供給口と、
を備える請求項1または請求項2に記載のポリエステルフィルム製造方法。
【請求項4】
前記窒素ガス供給口における前記窒素ガスの供給速度が、2m/分〜50m/分である請求項2または請求項3に記載のポリエステルフィルム製造方法。
【請求項5】
前記ニーディング部は、ニーディングクリアランスにおける剪断速度が500s−1〜2000s−1である請求項3または請求項4に記載のポリエステルフィルム製造方法。
【請求項6】
前記ポリエステル原料樹脂に対して、カルボジイミド基を1個有し、前記カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む環状カルボジイミド化合物を、0.05質量%〜20質量%で混合し、溶融混練して溶融樹脂とする請求項1〜請求項5のいずれか1項に記載のポリエステルフィルム製造方法。
【請求項7】
請求項1〜請求項6のいずれか一項に記載のポリエステルフィルム製造方法により製造された太陽電池用ポリエステルフィルム。
【請求項8】
1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%含むCHDM系ポリエステル樹脂を含有する層を少なくとも1層有する請求項7に記載のポリエステルフィルム。
【請求項9】
前記CHDM系ポリエステルを含有する層は、前記1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜20モル%または80モル%〜100モル%含む請求項8に記載の太陽電池用ポリエステルフィルム。
【請求項10】
太陽光が入射する透明性のフロント基板と、
前記フロント基板の上に設けられ、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分と、
前記セル構造部分の前記フロント基板が位置する側と反対側に設けられ、前記封止材と隣接して配置された、請求項7〜請求項9のいずれか1項に記載の太陽電池用ポリエステルフィルムと、
を備えた太陽電池モジュール。
【請求項1】
嵩密度が0.2〜0.7であるフラフを10質量%〜50質量%含み、含水率が20ppm〜100ppmであり、固有粘度が0.70dl/g〜1.2dl/gであり、かつ、温度が100℃〜160℃であるポリエステル原料樹脂を、二軸押出機の原料供給口に供給する原料樹脂供給工程と、
前記ポリエステル原料樹脂を溶融混練して溶融樹脂とすると共に、前記二軸押出機から前記溶融樹脂を、前記ポリエステル原料樹脂Tmに対してTm+20℃〜Tm+30℃で排出する溶融樹脂排出工程と、
前記溶融樹脂をフィルム状に成形する成形工程と、
を含むポリエステルフィルム製造方法。
【請求項2】
前記二軸押出機が、
少なくとも1つのベント孔と、
前記原料供給口に最も近いベント孔よりも前記原料供給口に近い位置に、前記ポリエステル原料樹脂の供給量の1.1倍〜10.0倍の窒素ガスを供給する第1の窒素ガス供給口と、
を備える請求項1に記載のポリエステルフィルム製造方法。
【請求項3】
前記二軸押出機が、
前記ポリエステル原料樹脂を可塑化するニーディング部を有するスクリューと、
前記ニーディング部に位置し、前記ポリエステル原料樹脂に対して窒素ガスを供給する第2の窒素ガス供給口と、
を備える請求項1または請求項2に記載のポリエステルフィルム製造方法。
【請求項4】
前記窒素ガス供給口における前記窒素ガスの供給速度が、2m/分〜50m/分である請求項2または請求項3に記載のポリエステルフィルム製造方法。
【請求項5】
前記ニーディング部は、ニーディングクリアランスにおける剪断速度が500s−1〜2000s−1である請求項3または請求項4に記載のポリエステルフィルム製造方法。
【請求項6】
前記ポリエステル原料樹脂に対して、カルボジイミド基を1個有し、前記カルボジイミド基の第一窒素と第二窒素とが結合基により結合されている環状構造を含む環状カルボジイミド化合物を、0.05質量%〜20質量%で混合し、溶融混練して溶融樹脂とする請求項1〜請求項5のいずれか1項に記載のポリエステルフィルム製造方法。
【請求項7】
請求項1〜請求項6のいずれか一項に記載のポリエステルフィルム製造方法により製造された太陽電池用ポリエステルフィルム。
【請求項8】
1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜100モル%含むCHDM系ポリエステル樹脂を含有する層を少なくとも1層有する請求項7に記載のポリエステルフィルム。
【請求項9】
前記CHDM系ポリエステルを含有する層は、前記1,4−シクロヘキサンジメタノール由来の構造を、ジオール化合物由来の構造全量に対して0.1モル%〜20モル%または80モル%〜100モル%含む請求項8に記載の太陽電池用ポリエステルフィルム。
【請求項10】
太陽光が入射する透明性のフロント基板と、
前記フロント基板の上に設けられ、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分と、
前記セル構造部分の前記フロント基板が位置する側と反対側に設けられ、前記封止材と隣接して配置された、請求項7〜請求項9のいずれか1項に記載の太陽電池用ポリエステルフィルムと、
を備えた太陽電池モジュール。
【図1】

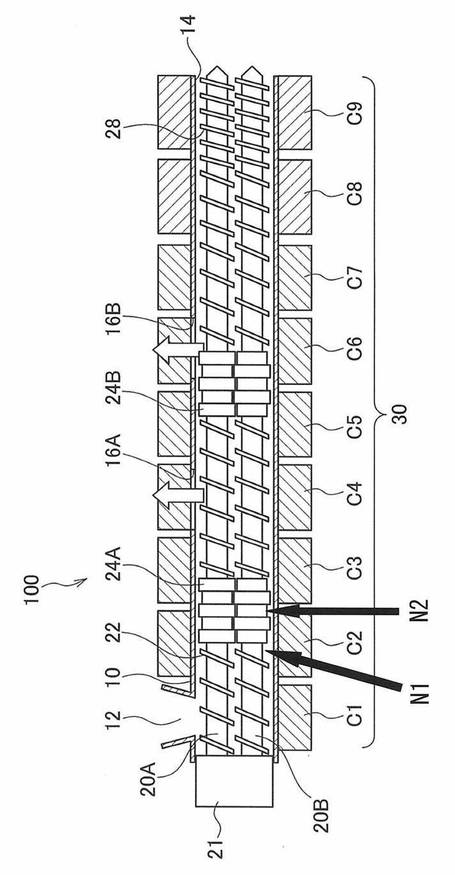
【図2】
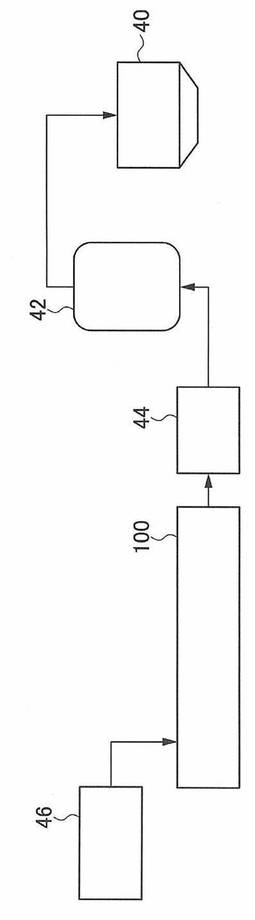
【図2】
【公開番号】特開2013−18976(P2013−18976A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2012−135018(P2012−135018)
【出願日】平成24年6月14日(2012.6.14)
【出願人】(306037311)富士フイルム株式会社 (25,513)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成24年6月14日(2012.6.14)
【出願人】(306037311)富士フイルム株式会社 (25,513)
【Fターム(参考)】
[ Back to top ]