
ポリオレフィン中のナノチューブ及び/又はナノ小板の分散
本発明は、A)ナノチューブ、ナノ小板又はその両方を含む溶液を調製する工程;B)工程(A)により得られた溶液を攪拌する工程;C)工程(B)で攪拌された溶液中に1種以上のポリマー材料を溶解し、さらにその溶液から沈殿物を単離する工程;及びD)1種以上のポリオレフィンと前記沈殿物溶融混合する工程を含む、ポリオレフィン中でナノチューブ及び/又はナノ小板を分散する方法、それにより製造されたナノコンポジット、並びに、前記ナノコンポジットから形成される品物を提供するものである。
【発明の詳細な説明】
【技術分野】
【0001】
[関連出願の参照]
本出願は、2009年12月28日に出願された米国の仮出願61/290,465の優先権を主張し、その内容全体は参照として本明細書に取り込まれる。
本発明はポリマー系ナノコンポジットに関する。より詳細には、本発明は、ポリオレフィン中に細かく分散されたナノチューブ及び/又はナノ小板等のナノ粒子を含むポリマー系ナノコンポジットに関する。
【背景技術】
【0002】
ポリオレフィンは、商業的に生産された最も広く使用されているポリマーの1つである。工学的な応用において、ポリプロピレン(PP)は、その高い融点、比較的高い弾性、低コスト及びリサイクル性のために魅力的であると考えられている。PPの応用範囲をさらに広げるために、それら特性を改良する多くの試みがある。改良を遂行するためのそのような戦略の1つは、ナノサイズのフィラーをPPに含有させることによる。改良できる材料の特性は、利用されるナノフィラーのタイプに依存する。一般的に使用されるフィラーには、モンモリロナイト等の珪酸塩ナノクレイがある。珪酸塩ナノクレイは、ポリマーの剛性、強度、気体遮断性、熱変性温度及び難燃性を改良するために使用される。珪酸塩ナノクレイは特に、ポリアミドの改良において有用であることがわかっており、中でも注目すべきは、ナイロン6[文献27及び28]、ポリイミド又はアミド若しくはイミド基を含むポリマーの改良である。ポリマーマトリックス中でナノクレイが良好に薄片化されたときに優れた改良が見られる。しかしながら、珪酸塩ナノクレイをPP中で薄片化することは、あまり成功していない。注目に値する例は、ジオクタデシルジメチルアンモニウムイオンでイオン交換されたモンモリロナイトクレイの集合体に挿入するための、ヒドロキシテレケリック基を有するポリオレフィンオリゴマーの使用である[文献29]。オリゴマー量の増加が、ナノクレイの良好な薄片化をもたらすことが分かった。この方法のさらなる改良は、ステアリルアンモニウムで交換したモンモリロナイトの使用や、無水マレイン酸で変性したPP(PP−MA)の使用である。PP−MAはニートPP(neat PP)の相溶化剤として作用する[文献30−32]。この方法を使用することによって、PP中でナノクレイをほとんど薄片化することができる。ナノクレイに対するPP−MAの比率が極めて重要であり、薄片化を最も多く得られる割合は3:1であった。PPの中で薄片化は達成されたが、ニートPPの物理的特性の改良の点では、ナイロン−クレイのハイブリッドで見られたものと比べものにならなかった。これは実際のところ、クレイの不完全な薄片化とPP−MAの存在による可能性が極めて高い。PPは疎水性が高くクレイは親水性が高いので、このような非常に非相溶性である材料間の相互作用を調節するためには、中間体が必要であると認識されている。そのような相溶化剤の使用を無くすか、使用量を著しく減少させるための、何らかの方法が非常に望まれている。
【0003】
カーボンナノチューブ(CNT)等の異なる種類のナノフィラーもPPに使用できる。カーボンナノチューブ(CNT)は、優れた機械的、電気的及び熱的特性を有するが[文献1及び2]、ポリマーマトリックスにこれらの特性を移す試験の実験結果では、低分散であり、さらにはCNTとポリマーマトリックス間の界面接着が不十分であるために、成功例は限られている[文献3]。また、CNTはナノクレイと同様にPPの難燃性を改良することがわかった[文献33]。良好な分散を達成するための最も一般的なアプローチには、界面活性物質での被覆[文献4及び5]、共有結合官能基化[文献6−9]及び非共有結合官能基化[文献10−16]が挙げられる。これらのうち、CNT表面への長鎖アルキルの付加を用いた酸塩基に基づく非共有結合官能基化では、他の方法に比べて、高い収率が得られて高効率であることが示されており、異なった数種類のポリマーに応用できる。一般的に、CNTへのアルキル鎖の付加は、酸化されたCNT表面と脂肪族アミンの官能基との間のイオン結合によって達成される[文献12及び13]。アミンの官能基が、CNT表面において、カルボン酸の官能基とイオン結合を介して相互作用した強い親和力を有することは、よく立証されている。酸処理されたCNTとオクタデシルアミン間の非共有結合は、双性イオンの構造を介して、有機溶媒中でのCNTの安定した分散をもたらすことが示された[文献12−16]。
【0004】
また、多層カーボンナノチューブ(MWCNT)を解きほぐすためにいくつかの方法が試みられたが、それらの方法では個々のレベルで良好な分散状態を示すことができなかった。Koval’chukらは、CNT表面上でのアルキル化の実現に脂肪族アミンを使用し、PP中でのMWCNTの良好な分散を実現した[文献17及び18]。この手法は簡単で空気と反応しにくく、その結果、高い官能基化を示すが、依然として、複合物中にもつれた構造のMWCNTが含まれている。Jungらは、官能基化のためにオクタデシルアミンを使用し、分散のためには長鎖アルキルがより有用であることを示したが、PPと混合した後では単独の分散が達成できなかった[文献19]。上記のアプローチは、PPマトリクスとCNTの相溶性を改良するが、ナノコンポジット材料中では解きほぐされたCNTの分散状態を十分に示すことができなかった。
【0005】
BaoとTjongは、二軸押出機で、MWCNTとPPの溶融混合の効果を調べ、0.3重量%のMWCNTでの引張係数(33%増加)及び引張強度(16%増加)において有意な改善を認めた[文献34]。しかしながら、MWCNT添加をさらに増量しても、ほとんど改善がされなかった。Fereidoonらは、単層CNT(SWCNT)とPPとの溶融混合状態を調べ、1重量%のSWCNTで引張係数の82%増加及び引張強度の22%増加を達成することができた。Blakeらが用いたより高度な手法は、n−ブチルリチウムで官能基化したMWCNTを作製し、続いて、塩素化したPP(CL−PP)とカップリング反応を行うものである[文献35]。この方法は、CL−PP層でコーティングされたMWCNTを生産し、その結果、CL−PP中での混和性を高めた。引張係数と引張強度はそれぞれ209%と277%と目覚しい改良が報告された。この研究は、達成可能な増強効果がCNTの分散状態に強く依存していることを示唆する。しかしながら、ホストポリマーとしてのCL−PPの使用は、そのような目覚しい結果を達成するためにホストポリマーを変性する必要があることを示している。他では、空気中の加熱によって官能基化されたMWCNTが、PP−MA等の相溶化剤の使用条件下でPPと良好な相溶性を示すことがわかった[文献36]。この場合、MWCNTのミクロサイズの凝集体が形成された。研究では、CNTはポリオレフィンの導電性を改善するために使用できることが示された[文献36及び37]。PP中の約1体積%のMWCNTの含有により、体積導電率の7桁増加を引き起こすことができる[文献38]。また、10重量%のMWCNTでは、体積抵抗率を16桁減少させる[文献39]。このように、CNTはポリオレフィンの電気特性を改良するための優れた材料である。
【0006】
上記背景から、PP等の未変性ポリオレフィン中で、ナノ粒子又はCNT等のナノチューブの分散を高める実用的な方法は依然として不十分であり、意味のある化学変性や大量の相溶化剤を用いることなく、ナノ粒子等のナノ分散を実現できる技術を開発する必要があると結論づけられる。
【発明の概要】
【発明が解決しようとする課題】
【0007】
従って、本発明の目的の1つは、ポリオレフィン中でのナノ小板、ナノチューブ又はその両方について、効率の高い分散方法を提供することである。
【0008】
さらなる本発明の目的は、ナノ小板、ナノチューブ又はポリオレフィンの表面改質によって、ポリオレフィン中のナノ小板、ナノチューブ又はその両方を分散する方法を提供することである。
【0009】
さらなる本発明の目的は、本発明に従って作製されたナノコンポジットを提供することである。
【0010】
さらなる本発明の目的は、ナノコンポジットから作製される製品を提供することである。
【課題を解決するための手段】
【0011】
本発明のこれら及び他の目的は、各々又はこれらの組み合わせについて、ポリオレフィン中でナノチューブ及び/又はナノ小板を分散する以下の工程を含む方法並びに、そこから形成されたナノコンポジットの発見によって達成された。
A)ナノチューブ、ナノ小板又はその両方を含む溶液を調製する工程;
B)工程(A)により得られた溶液を攪拌する工程;
C)工程(B)で攪拌された溶液中に1種以上のポリマー材料を溶解し、さらにその溶液から沈殿物を単離する工程;及び、
D)1種以上のポリオレフィンと前記沈殿物を溶融混合する工程。
【図面の簡単な説明】
【0012】
本発明及び本発明に付随するその多くの利点について、以下の詳細な記載を参照し、添付の図面と共に考察することでより良く理解されることによって、完全な理解が容易に得られる。
【0013】
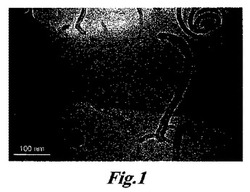
【図1】図1は、溶液からCNT/ZrPとPPとの沈殿により得られたCNT/ZrP/PPのマスターバッチの透過型電子顕微鏡画像である。CNTとZrPナノ小板は共に、PPホスト中でよく分散されている。沈殿物の組成は、重量比でCNT/テトラ(n−ブチルアンモニウム)水酸化物(TBA)/ZrP/PP=1/2/3/4である。
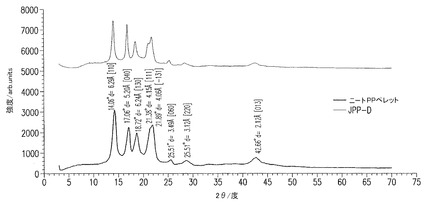
【図2】図2は、ニートPPペレットとナノコンポジットのサンプルJPP−DのX線回折スペクトルである。チャートはPP結晶のα相の存在を示す。積層ZrPナノ小板からの回折ピークが無いことは、ナノ小板がおそらく薄片化されていることを示す。
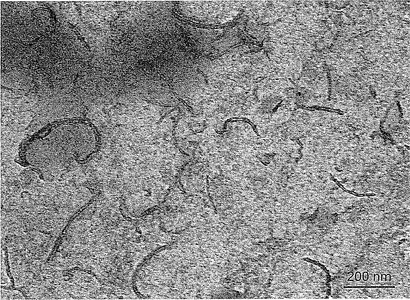
【図3】図3は、PP中に分散した0.05重量%のMWCNTの透過型電子顕微鏡画像である。MWCNTは凝集することなく、非常に高分散状態である。
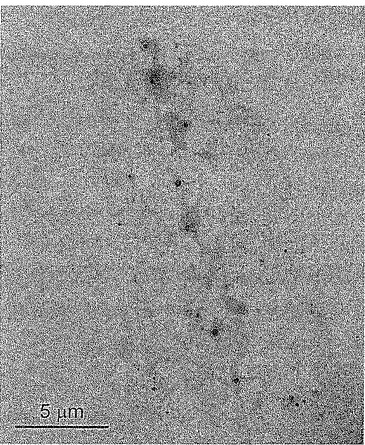
【図4】図4は、プラズマ処理されたPP粒子の電界放出走査型電子顕微鏡写真である。粒子サイズは約100ミクロンである。
【図5】図5は、プラズマ処理されたPP粒子(P−PP−1)の表面にZrPナノ小板が付着している状態の透過型電子顕微鏡画像である。
【図6】図6は、高温で圧縮して薄板にしたZrPナノコンポジットサンプルP−PP−1の透過型電子顕微鏡画像である。単独に分散しているナノ小板が観測された。
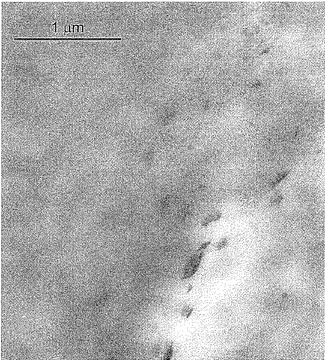
【図7】図7は、0.015重量%ZrPナノコンポジットサンプルP−PP−8の透過型電子顕微鏡画像であり、約100nmサイズのナノ小板の均一な分散を示している。
【図8】図8は、0.015重量%ZrPナノコンポジットサンプルP−PP−8の透過型電子顕微鏡画像であり、100nmサイズのナノ小板の均一な分散を示している。
【図9】図9a−9dは、わずかに酸化した後の著しい絡み合いを示すMWCNT(a,b)と、ナノ小板が分散工程を補助した後の良好に分散したMWCNT(c,d)のTEM顕微鏡画像である。
【図10】図10a−10dは、オクタデシルアミンによって表面改質された単独のMWCNTの製造方法(a,b)と、PP中に良好に分散したMWCNTの製造方法(c,d)についての本発明の方法の概念的な説明図である。
【図11】図11は、わずかに酸化されたMWCTN(上部)と、F−MWCNT(下部)のFTIRスペクトルを示す。
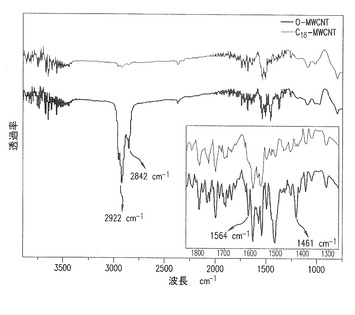
【図12】図12aと12bは、キシレン溶液からの回収後の良好に分散したFD−MWCNTのTEM顕微鏡画像である。
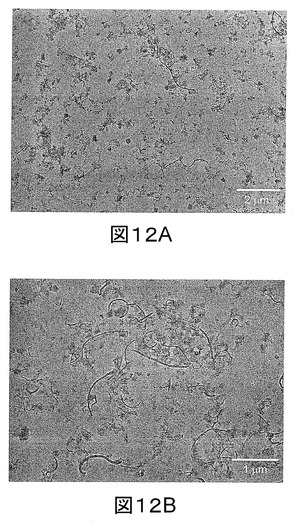
【図13】図13aと13bは、キシレン溶液から作製したPP/FD−MWCNTナノコンポジットのTEM顕微鏡画像である。
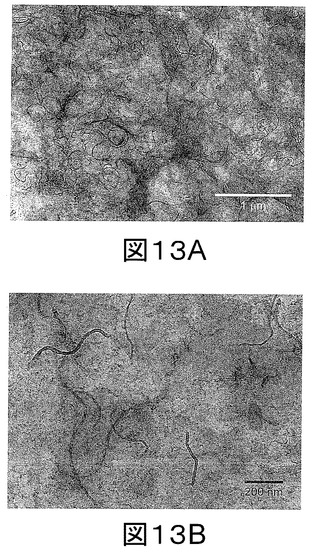
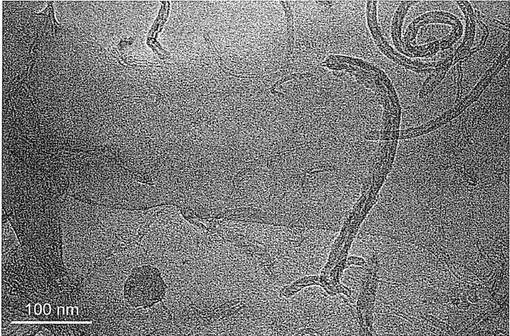
【図14】図14a−14hは、a,b)0.1重量%;c,d)0.6重量%;e,f)1重量%及びg,h)2重量%のMWCNTを含むPPナノコンポジットのTEM画像である。
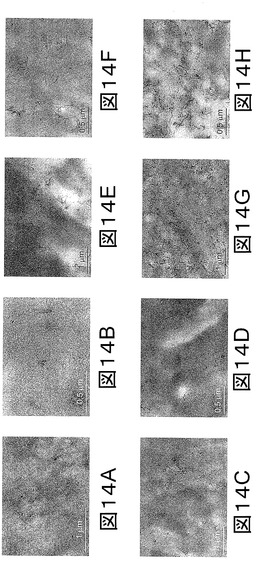
【図15】図15は、ニートPPと良好に分散したMWCNTを含有するナノコンポジットの工学応力−真歪み曲線を示す。
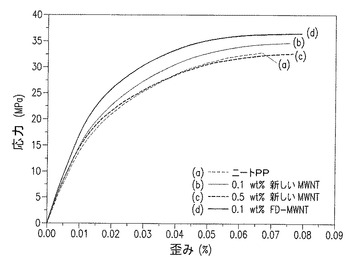
【図16】図16a−16dは、a,b)ニートPP及びc,d)0.1重量%F−MWNTナノコンポジットの、SEMによる破断面図である。
【図17】図17は、F−MWCNTを様々な濃度で含むPPナノコンポジットの表面電気伝導率を示す。
【図18】図18a−18cは、(a)ニートPP,(b)0.1重量%MWCNTを含むPP,及び(c)0.4重量%MWCNTを含むPPの寸法安定性(すなわち、厚さ方向への収縮)の測定値を示す。
【発明を実施するための形態】
【0014】
本発明は、ポリオレフィン中、特にPP中でのナノ粒子又はナノチューブ、特にMWCNTの非常に改良された分散性を達成するための簡単な方法に関する。本発明は、より詳細には、(i)PP等のポリオレフィン中でのMWCNT等のナノチューブ、(ii)PP等のポリオレフィン中でのZrP等のクレイ若しくはナノ小板、又は(iii)PP等のポリオレフィン中でのナノチューブとクレイ若しくはナノ小板の組み合わせにおいて、非常に改良された分散性を達成することに関する。
【0015】
本発明者らの以前の研究において、CNTの電子状態に対して最小限のダメージでMWCNTとSWCNTの両方の解きほぐし及び脱結束を達成するために、わずかに酸化したCNTの表面にナノ小板を電気的に拘束した[文献20]。有機溶媒及びポリマーマトリクス中で薄片化したCNTの分散と解きほぐし状態を維持するために、CNT表面への親有機性の変性が特に必要である。
【0016】
本発明の組成物であるポリオレフィンは、好ましくはポリエチレン(PE)、ポリプロピレン(PP)、ポリブチレン(PB)又はそれらの混合物若しくはコポリマーである。より好ましくは、ポリオレフィンはPPである。以下の考察では、ポリオレフィンがポリプロピレン(PP)であるとして本発明を説明する。しかしながら、これは、本発明の範囲を限定することを意図するものではなく、ポリエチレンやポリブチレンなどの他のポリオレフィンをPPの代わりに使用することができる。
【0017】
本発明において、長鎖脂肪族アミンと無水マレイン酸で変性されたポリプロピレン(PP−MA:文献30−32を参照)から成る群から選択される少なくとも1種によって親有機性の変性が提供できる。好ましくは、中鎖〜長鎖脂肪族アミン(以下、単に「長鎖脂肪族アミン」という)への媒体の使用によって親有機性の変性が提供される。ここでアミンは、より好ましくは第一アミンである。中鎖〜長鎖脂肪族アミンに対する媒体は、変性されるナノチューブ又はナノ小板に対して所望の親有機性を提供するための炭素原子の数が十分である限り、各脂肪族鎖の炭素原子数は任意の規定数とすることができる。
【0018】
好ましくは、長鎖脂肪族アミンは、C4−C30脂肪族基を有し、より好ましくはC6−C30基、さらに好ましくはC10−C30基、さらに好ましくはC14−C24基、さらに好ましくはC16−C20基、そして最も好ましくはC18基を有する。親有機性の変性は、ナノチューブの表面、クレイ若しくはナノ小板の表面、又はポリオレフィンの表面に行うことが出来る。最も好適な実施態様では、官能基化された多層炭素ナノチューブ(F−MWCNT)を生産するために、オクタデシルアミンを選択し、キシレン、デカリン、ブタノール、ジクロロベンゼン、トリクロロベンゼン、N,N−ジメチルホルムアミド及びイソプロパノール等の有機溶媒中で、軽い超音波処理を用いて容易に分散することができる。次いで、その溶液をPPペレット等のポリオレフィンと直接混合し、乾燥することで、低い添加量で有意に改善された導電性及び引張係数でポリオレフィン/F−MWCNTナノコンポジットを生産することができる。また、上述と同様の溶媒は官能基化されたナノ小板又はクレイの分散にも使用できる。
【0019】
本発明で有用なナノチューブは、どのようなナノチューブであっても良い。好ましくは、ナノチューブは、カーボンナノチューブ、二酸化タングステンナノチューブ、シリコンナノチューブ、無機ナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である。より好ましくは、ナノチューブはカーボンナノチューブであり、最も好ましくはSWCNT又はMWCNTである。必要に応じて、ナノチューブは、これには限定されないが、乾燥酸化、放射酸化、プラズマ酸化、熱酸化、拡散酸化又はこれらの組み合わせを含む既存の酸化方法を用いて表面を酸化することができる。
【0020】
本発明に使用されるナノ小板は、これに限定されないが、クレイ(モンモリロナイト等)、ナノクレイ、グラフェン、無機結晶、有機結晶及びこれらの組み合わせを含む、クレイ又は他の形態のナノ小板である。特に、ナノ小板は、好ましくはα−リン酸ジルコニウム(ZrP)である。ZrPは、モンモリロナイトのような周知の天然クレイと同様の層状構造を有するので、合成クレイと見なすことができる。クレイの起源によってカチオン成分が異なりうる天然クレイとは違い、ZrPは化学構造Zr(HPO4)2・H2Oで明確に定義される。また、ZrPのサイズとアスペクト比は、様々な合成条件によって容易に制御され、天然クレイよりも均一のサイズ分布で得ることができる[文献40]。モンモリロナイトと同様の方法でZrPにオニウムイオンを挿入することができ、そして、水溶液中での薄片化は、TBA+OH−を導入してテトラ(n−ブチルアンモニウム)イオン(TBA+)を形成し、ZrPに挿入して次に薄片化することで容易に達成できる[文献41]。本発明では天然クレイの代用としてZrPを使用するが、同様の化学的、物理的性質であることから、ここで開発された方法は天然クレイでも応用することができる。
【0021】
本発明は、良好に分散されたナノチューブ又はナノ小板を含むポリオレフィンナノコンポジットを製造するための簡単ではあるが効果的な方法を提供する。特に、好ましくは、本発明は良好に分散されたMWCNTを含むポリオレフィンナノコンポジット製造するための簡単で効果的な方法を提供する。わずかに酸化されたMWCNTはナノ小板を用いて解きほぐされ、ナノ小板を除去した後でも、高い安定性を示す。好ましくは、良好に分散されたMWCNTはオクタデシルアミンで官能基化され、TEMとSEMの観察で証明されるように、単独の分散状態においても有機溶媒中での安定性を増す。良好に分散されたポリオレフィン/MWCNTのナノコンポジットは、高濃度のMWCNTを含むキシレン溶液等の有機溶媒とポリオレフィンペレットを直接混合することによって作製できる。乾燥し、ニートPP中で希釈するためのマスターバッチとして粉末を使用して、任意のMWCNT濃度でナノコンポジットを形成した。ナノコンポジットは、素晴らしい分散を示し、少ないチューブ添加量で、係数、強度、及び導電性が有意に増加した。機械的特性の強化メカニズムは明確には分かっていないが、部分的には、MWCNTが(1)結晶成長のための核生成剤、(2)ポリオレフィンマトリクスを効率的に強化するためのマトリクスの球状間領域(inter−spherulitic region)の強化剤として働いているためであると考えられる。
【0022】
本発明の実施態様の一つは、ポリオレフィン中でナノ小板及び/又はナノチューブを分散する方法であり、以下の工程を含む:
A)ナノチューブ及びナノ小板を含む溶液を調製する工程;
B)工程(A)により得られた溶液を攪拌する工程;
C)工程(B)で攪拌された溶液中に1種以上のポリマー材料を溶解し、さらにその溶液から沈殿物を単離する工程;
D)1種以上のポリオレフィンと前記沈殿物を溶融混合する工程。
【0023】
本発明者らは、以前に、水溶液中でカーボンナノチューブ及びZrP等のナノ小板を共分散するための新しい方法を開発した[文献20]。この溶液は、ポリオレフィンナノコンポジットを作製する本発明の方法において、上記の実施態様における最初の工程で使用できる。好ましくは、その水溶液はゲル様の稠度を有する粘性のスラリーになるまで加熱される。そして、N,N−ジメチルホルムアミド(DMF)等の溶媒中で再分散される。PP/デカリン溶液が調製され、CNT/ZrPのDMF溶液とイソプロパノールと共に混合される。この溶液は80℃の湯浴で超音波処理し、その後、90℃で30分間攪拌し、最後に室温で冷却される。冷却の間に形成される黒い沈殿物を回収し、イソプロパノールで洗浄し、真空オーブンで乾燥する。好ましくは、10%のCNT、20%のTBA、30%のZrP及び40%のPPを含む黒い沈殿物は、PPを用いて混合溶解するマスターバッチとして使用され、ポリマーナノコンポジットが作製される。イソプロパノールで再分散されたマスターバッチのTEM画像は、MWCNTとZrPのナノ小板がポリマーマトリックス中に単独で分散されることを示す(図1)。1重量%MWCNT/3重量%ZrPを有するナノコンポジットを射出成形試験片にした。サンプルをX線回折(XRD)で分析し、ニートPPペレットの結果と比較した。XRD分光では、非薄片化ZrPの特徴のある回折ピークは無く、PP結晶のα相のみが検出されることを示している(図2)。CNT/ZrP溶液と混合されるPP/デカリン溶液の代わりに、これには限定されないが、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエステルカーボネートコポリマー、ポリ(エステル−カーボネート)樹脂、ポリアミド、高温ポリアミド、ポリエチレン、ポリプロピレン、オレフィンのコポリマー、官能基化ポリオレフィン、ハロゲン化ビニルポリマー、ビニリデンポリマー、ポリ塩化ビニリデン、ポリフッ化ビニル、ポリフッ化ビニリデン、ポリアミドコポリマー、ポリアクリロニトリル、ポリエーテル、ポリケトン、熱可塑性ポリイミド、変性セルロース及び前述のポリマー材料の1種以上を含む混合物を含む他のポリマー材料を使用することができる。次に、CNT/ZrP溶液を用いて得られた混合物は、処理されてCNT/ZrP/ポリマー材料の沈殿を形成し、最終的なナノコンポジットを得るために、ポリオレフィンを用いて溶融混合するマスターバッチとして使用できる。当然、ポリマー材料がPPのみであるナノコンポジットを提供するためには、1種以上のポリマー材料としてPPを使用することが好ましい。
【0024】
あるいはまた、別の実施態様では、ZrPナノ小板は、Xiらによって述べられている方法を用いて水中で分散した後、CNTから分離される[43]CNTは、オクタデシルアミン等の長鎖脂肪族アミンを用いた親有機性変性の後、デカリン等の無極性溶媒中で再分散できる。CNT/キシレン溶液は、高温で攪拌されているPP(又は他のポリマー材料)/キシレン溶液にゆっくりと加えられ、PPとCNTの均一混合を確実にする。溶液の攪拌を停止し、冷却すると、溶液中でPPとCNTが共沈殿する。その沈殿を溶液から分離し、乾燥して、良好に分散されたPP/CNTナノコンポジットを形成する(図3)。
【0025】
TBAの除去と、オクタデシルアミン等の長鎖脂肪族アミンによる変性後、キシレン又はデカリン等の非極性溶媒系にZrPを分散することができる。ZrP/キシレン溶液は、高温で攪拌されているPP/デカリン溶液にゆっくりと加えられ、PP中でのZrPの良好な分散を確実にする。その後、デカリン中でPPとZrPを共沈殿させるために溶液を冷却する。溶液から沈殿物を分離し、乾燥して、良好に分散されたPP/ZrPナノコンポジットを形成する。
【0026】
また、ZrP/キシレンとCNT/キシレンの2種の良好に分散された溶液を一緒に混ぜて、均一な懸濁液を形成する。次に、その混合液を高温で攪拌されているポリマー材料、好ましくはPP/デカリン溶液にゆっくりと加え、ポリマー材料、好ましくはPP中のZrP/CNTの良好な分散を確実にする。その後、その溶液をデカリン中でPPとZrP/CNTを共沈殿させるために冷却する。その沈殿物を溶液から分離し乾燥して、良好に分散されたPP/CNT/ZrPナノコンポジットを形成する。
【0027】
本発明のナノコンポジットは、ナノチューブ及び/又はナノ小板のいずれの所望の添加物も含むことができる。好ましくは、ナノチューブ又はナノ小板の量は、0.1〜20重量%の範囲であり、より好ましくは0.1〜10重量%、最も好ましくは0.3〜5重量%である。より好ましい実施態様では、本発明のナノコンポジットは、ポリオレフィンを95〜99.7重量%含み、ナノチューブ、好ましくはMWCNTを0.3〜5重量%含む。パーコレーション濃度は、CNTのアスペクト比を用いて変えることができる。上記の最も好ましい実施態様では、MWCNTが0.3〜5重量%の濃度であり、その組成物は、10−6S/mを超える表面電気伝導率を有する。
【0028】
本発明のさらなる実施態様では、100ミクロンのサイズのプラズマ処理されたPP(PT−PP)粒子(図4)を水溶液中でZrPと混合する。PT−PP粒子を空気及び窒素の存在下でプラズマによって処理し、その結果、粒子の表面に、COOH、C=O、C−O、NO2及びNO3等の化学官能基が付加される。これらの化学官能基は、一般的に電子が豊富であり、特にカルボン酸基は水中で容易に脱プロトン化してカルボン酸イオンを形成し、その粒子に負の電荷を与える。TBAで変性されたZrPナノ小板は、正の電荷を帯びている。
【0029】
粒子とナノ小板間の静電引力は、強制的に各粒子を囲むZrP層を形成させる。ZrPで一旦処理されたPT−PP粒子は、水中で安定な懸濁液を形成する。また、この溶液にTBA+OH−を添加して、pHを上昇させることができる。pHの上昇は、PT−PP粒子の表面上で脱プロトン化されたカルボキシレート基の濃度を高め、粒子に対するZrPの引力を増加させると考えられる。過剰なアセトン添加は、安定な懸濁液を破壊し、ZrPでコーティングされたポリマー粒子を沈殿させる。その粒子を回収し、90℃のオーブンで乾燥させる。いくつかの粒子がエポキシ中に埋め込まれており、ZrPでコーティングされたポリマー粒子の形態を観察するためのTEM用の薄片の作製に使用される。残りの粒子を高温で圧縮して薄板を形成すると、圧縮されたシートの断面で薄片を形成するためにこれらはエポキシ中に埋め込まれる。これらの薄片は、TEMの画像処理に使用される。TEM画像の解析から、発明者らはPT−PP粒子の表面にZrPナノ小板が付加している証拠を見つけた(図5)。圧縮された薄板の断面のTEM画像では、個々のナノ小板がポリマーマトリックス中で分散されることが示された(図6)。
【0030】
本発明のナノコンポジットは、任意に、1種以上の通常の添加物を通常量含むことができる。好ましくは、1種以上の添加物は、これには限定されないが、フィラー、強化剤、可塑剤、酸化防止剤、熱安定剤、紫外線安定剤、強靭化剤、帯電防止剤、難燃剤、着色剤及び上述の添加物を1種以上含む組み合わせから選択される1種以上の添加物を含む。
【0031】
本発明のナノコンポジットは、フィルム、スポンジ(foams)、繊維及び他の構造形態等の様々な物の形成に使用できる。これらの物は、これには限定されないが、熱成形、押出成形、ブロー成形、延伸ブロー成形、押出ブロー成形等の従来のいかなる方法によっても形成できる。
【0032】
本発明については一般的に記載されたが、説明のために本明細書に記載された特定の実施例を参照することによって、さらなる理解を得ることができる。これら実施例は、特に規定のない限り、それらに限定されないことを意味する。
【実施例】
【0033】
・材料
ZrPナノ小板を使用して、MWCNTを水溶液中で解きほぐして分散した。MWCNTを解きほぐすためのZrPの合成、薄片化及び使用については以前に報告されている[20,21]。簡潔には、15.0gのZrOCl2・8H2O(Fluka)を150.0mlの3.0M H3PO4(EM Science)中で機械的に攪拌しながら100℃で24時間還流した。次に、生成物を遠心分離と再分散によって3回洗浄し、85℃のオーブンで24時間乾燥した後、優しく乳鉢ですりつぶして微粉末にした。ZrP粉末をTBA+OH−(Aldrich,メタノール中1mol/L)を用いて、分子比ZrP:TBA=1:0.8として水中で薄片化した。新しいMWCNT(P−MWCNT)(純度90%、平均直径<10nm、長さ範囲0.1−10μm)をAldrichから購入した。市販のオクタデシルアミン(CH3(CH2)17NH2,Sigma−Aldrichケミカルズ、97%)を得て使用した。メルトフローインデックス(MFI)が1.9g/10minの市販グレードのPP(記号4204)は日本の日本ポリプロ株式会社(JPP)から供給された。
【0034】
<CNT/ZrPナノコンポジットの作製>
・ZrPナノ小板の合成と薄片化
本研究におけるZrPナノ小板の合成と薄片化は、以前に報告された方法と類似している[文献40及び42]。ZrPナノ小板は、還流により合成された:パイレックス製の丸底フラスコ内で、ZrOCl2・8H2O(Fluka)20.0gを200.0rnL 3.0Mで、100℃、24時間攪拌しながら、還流した。反応後、生成物を3回の遠心分離で洗浄し、回収した。その後、ZrPを85℃のオーブン内で24時間乾燥した。乾燥したZrPは乳鉢と乳棒のセットですりつぶして微粉末にした。
【0035】
作製したZrPを、TBA+OH−(Aldrich)により、分子比α−ZrP:TBA=1:0.8で水中で薄片化した。TBAを分散したZrPに加え、少なくとも2時間攪拌し、ナノ小板中にTBAの挿入を行った。その後、分散物を1時間以上超音波処理し、溶液中で完全に薄片化した(分散体の量によって、より長時間の処理が必要な場合がある)。
【0036】
・CNTの酸処理
CNTを酸で処理し、ナノチューブ表面上にカルボキシル基を導入した。硫酸と硝酸の混合溶液(体積比36ml/12ml)を調製した。調製した酸性の混合溶液を0.2gのカーボンナノチューブ(純度90%、平均直径<10nm、長さ範囲0.1−10μm、Aldrich社製)に加え、2時間超音波処理を行った。超音波処理装置の水は一定の水温を維持するために循環させた。次に、152mlの脱イオン水を酸/CNT混合溶液に加え、この溶液を循環水中で1時間超音波処理した。次に、CNTを二フッ化ポリビニリデンのフィルター膜(Millipore,細孔径0.45μm)を用いてろ過し、残留したすべての酸を除くために脱イオン水で十分に洗浄した。洗浄したCNTを脱イオン水に再分散し、3時間超音波処理をした。通常、水中のCNTの最終濃度は0.002g/ml〜0.005g/mlである。
【0037】
・CNT/ZrP分散物の作製
CNT/ZrP分散物を重量比1〜3で作製する。例として、0.2gのCNTが安定した分散状態を形成するためには0.6gのZrPが必要である。通常、1gのZrP分散物に対しては100mlの水が用意され、前述の方法に従って薄片化する。0.6gのZrPサンプルに対しては、CNT/ZrP分散物の作製のためには60mlの分散物を使用する。CNT/水分散物を完全に薄片化したZrP/水分散物に加え、安定した分散状態を形成するために1時間以上超音波処理する。水中で安定したCNT/ZrP分散物を加熱し、CNT/ZrPがゲル状に凝縮するまで水分のほとんどを除去した。次に、25mlのN,N−ジメチルホルムアミド(DMF)(Alfa Aesar)をゲルに混合した。その混合物を1時間以上超音波処理し、CNT/ZrPをDMF中に再分散した。
【0038】
直接的な混合手法を用いたCNT/ZrPナノコンポジットの作製では、すべての水が完全に除去されるまでCNT/ZrPの水分散物を加熱した。乾燥した残留物をオーブンに入れ、90℃で一晩乾燥した。乾燥したCNT/ZrPの残留物を乳鉢ですりつぶして微粉末にした。
【0039】
・CNT/ZrP PP分散物の作製
0.8gのPP(Novatec、JPP)を200mlのデカリン(Sigma Aldrich)に加え、PPペレットが全て溶解するまで130℃のオイルバス中で加熱した。25mlのイソプロパノールをその溶液に加え、次に前のセクションで作製したDMF中のCNT/ZrP分散物を加えて122℃で10分間攪拌した。その溶液を含むフラスコを超音波槽(Bransonic(登録商標)1510)に移し、80℃の浴温で20分間超音波処理した。フラスコをオイルバスに移し、90℃で30分間一定の速度で攪拌した。フラスコの底にすぐに固まった黒い沈殿物が生じたので、透明な溶液を除去し、イソプロパノール(EMD Chem)中でそれらを再分散することによって、残存する沈殿物を回収した。沈殿物を遠心分離し、上清を除去した。イソプロパノール中での再分散と遠心分離後の上清の除去の工程を3回繰り返した。その後、沈殿物を80℃で24時間真空乾燥した。沈殿物の組成は重量比でCNT/TBA/ZrP/PP=1/2/3/4である。
【0040】
・CNT/ZrP PPナノコンポジットの作製
前のセクションで得られた沈殿物と粉末は、ニートPP(Novatec,JPP)で希釈するマスターバッチとして使用し、所望のCNT/ZrP添加によりナノコンポジットを形成した。マスターバッチを一定量のPPと事前に混合し、その混合物を2軸バッチミキサー(Haake Rheocord System 40)の混合槽に添加した。表1にナノコンポジットの作製に用いられた組成を示す。ミキサースクリューを用いて、60rpm、180℃で10分間溶融混合を行った。その後、小型射出成形機(CS−183 MMX,CSI)を用いてナノコンポジットを75mm×12.5mm×3.15mmの角柱状に射出成形した。溶融漕を180℃で維持し、成形型を80℃で維持した。ニートPPの試験片を作製するためには、溶融漕を210℃で維持し、成形型を80℃で維持した。
【0041】
【表1】

【0042】
・CNT PPナノコンポジットの作製−溶解法
Xiらの方法に従って、水溶液中で、薄片化したMWCNTの作製を行ったが、ここでは詳細には説明しない。MWCNT0.002gを含む水溶液15gを調製し、0.02gのオクタデシルアミン(CH3(CH2)17NH2)の粉末を加えた。その混合物を85〜90℃で1時間連続的に攪拌し、カーボンナノチュ−ブをオクタデシルアミンで変性した。一旦攪拌を停止すると、アミン変性されたMWCNT(F−MWCNT)が水溶液から沈殿する。この沈殿物を回収し、80℃のオーブンで2時間乾燥した。完全な分散を達成するために1時間超音波処理した沈殿に15gのキシレンを加えた。1gのPPを15gのデカリンに170℃で溶解した。均一な混合物を生成するために、F−MWCNT/キシレン溶液を攪拌しながら、PP/デカリン溶液に滴下して加えた。さらに、混合物を170℃で30分間攪拌し、溶媒を一部蒸発させた。最終生成物は、PP中にF−MWCNTが分散された粘性のあるゲルである。F−MWCNT/PPナノコンポジットは、デカリンのゲルから完全に乾燥することによって得られる。
【0043】
・PP/ZrPナノコンポジットの作製−溶解法
水溶液中で薄片化したZrP/TBAの作製は、早期に報告されている[文献41]。ZrPは、0.01gのZrP/TBAを含む水溶液の対数において0.6mlのHCl(pH=1)を加えることによってTBAから分離できる。精製したZrPナノ小板の沈殿を遠心分離で回収し、超音波処理を用いて水中で再分散した。その後、10重量%オクタデシルアミノ塩(CH3(CH2)17NH3+)を1g、水溶液中に添加することで、水溶液10g中、0.01gの精製されたZrPが変性された。その混合物を室温で1時間連続的に攪拌し、ZrPの表面をオクタデシルアミノ塩で完全に変性した。一旦、攪拌を停止すると、アミノ変性されたZrP(F−ZrP)が水溶液から沈殿した。次に、キシレン15gを水溶液中の沈殿に加え、F−ZrPをキシレン中に完全に分散させるために1時間超音波処理し、水をデカンテーションした。その後、1gのPPを15gのデカリン中に170℃で溶解した。均一な混合物を生成するために、F−ZrP/キシレン溶液をPP/デカリン溶液に攪拌しながら滴下した。溶媒を一部蒸発させて、その混合物をさらに30分間、170℃で攪拌した。最終生成物は、PP中にF−ZrPが分散した粘性のゲルである。ゲルを完全に乾燥することによって、F−ZrP/PPナノコンポジットを得ることができる。
【0044】
・PP/ZrP/CNTナノコンポジットの作製−溶解法
F−ZrP/キシレンとF−MWCNT/キシレンを分散する工程は上述した。約15gのキシレンをF−ZrPとF−MWCNT(固形分:ZrP0.01g、MWCNT0.0022g)に別々に加え、完全に分散させるために1時間超音波処理した。その後、2つの分散物をお互いに混合し、完全に分散させるために1時間超音波処理した。次に、1gのPPを15gのデカリン中に170℃で溶解した。均一混合物を形成するために、F−ZrP/F−MWCNT/キシレン溶液を攪拌しながらPP/デカリン溶液に滴下した。混合物を170℃でさらに30分間攪拌して溶媒を一部蒸発させた。最終生成物は、F−ZrP/F−MWCNTがPP中に分散した粘性のゲルである。ゲルを完全に乾燥することによって、F−ZrP/F−MWCNT/PPナノコンポジットが得られる。
【0045】
・透過型電子顕微鏡
CNT/ZrP PPナノコンポジットの顕微鏡観察用に、イソプロパノール中にマスターバッチを再分散し、微細分散物を得るために24時間超音波処理した。分散物を1滴、TEMのために炭素フィルムで被覆した銅格子上に置いた。ナノコンポジットの薄片を、射出成形試験片からReinzcutウルトラミクロトームを用いて切り出し、銅格子上に置いた。
CNT PPナノコンポジット用に、MWCNT/PPデカリン溶液の液滴を炭素フィルムで被覆した銅格子上に置いた。溶媒が全て除去されるまで、銅格子をホットプレート上で熱をかけて乾燥した。
JEOL 1200 EXを用いて透過電子顕微鏡観察(TEM)を実施した。
【0046】
・X線回折
ナノコンポジットのサンプルは、X線粉末回折計Bruker−AXS D8を用いて分析した。
【0047】
<ZrPナノコンポジットの作製>
・プラズマ処理されたポリプロピレン(PP)
100ミクロンのマイクロ粒子を含むポリプロピレン粉末を、大気圧下の空気及び窒素中、プラズマで処理した。処理工程の間に、COOH、C=O、C−O、NO2及びNO等の極性基を粒子の表面に導入した。次に、プラズマ処理したポリプロピレン(PT−PP)粒子をZrPナノ小板で変性した。
【0048】
・ZrP/プラズマ処理したPP分散物の作製
水中で薄片化したZrPナノ小板の原液を、上述のように、100mlの水に1gのZrPの濃度で作製した。サンプルP−PP−1用に、0.05gのZrPと5mlの原液をバイアルに作製した。0.1gのPT−PP粒子を、薄片化したα−ZrPナノ小板の溶液に加えた。サンプルP−PP−2用には、0.1ミリモルのTBAもさらに溶液に加えることを除き、同様の手順に従った。PT−PP粒子を含む溶液を0.5時間超音波処理し、その後、常温で2時間以上連続して攪拌した。底面に粒子を沈下させるために、水の3倍体積量のアセトンを溶液に加えた。通常、1時間後に溶液から粒子が完全に分離される。次に、上清を排出し、残った粒子を90℃での穏やかな加熱により乾燥した。乾燥した粒子は後に、特性評価及び熱加工に使用する。
【0049】
・ZrPナノコンポジットの作製
前のセクションに記載した方法で作製した乾燥PT−PP粒子を2枚の鋼板の間に挟み、加熱圧縮機(Dake)を用いて170℃で5分間圧縮し、厚さ200〜400ミクロンのポリマー薄板を形成した。
【0050】
・溶融混合したZrP PPナノコンポジットの作製
ZrPの分散をさらに改善するために、以下のようにZrP(ZrP−m−PTPP)によって変性されたPT−PP粒子をPPと混合した。バッチ式ミキサー内でZrP−m−PTPPをニートPPに加え、180℃で以下の通りに混合し、ZrPの凝集体を分散した:前記の手順に従って、ZrPによって変性されたPT−PP粒子(P−PP−1)とPPをHaakeミキサーを用いて60rpmで20分間混合した。0.06 2のP−PP−1粉末を40gのPPに加え、0.015重量%ZrP PPナノコンポジットを得た。このナノコンポジットはP−PP−8と表した。
【0051】
・電界放射型走査型電子顕微鏡
粒子を炭素テープで覆われたアルミニウムスタブの表面に置き、その粒子を、スパッタコーター(Cressington)を用いてアルゴン下で薄さ4nmのプラチナで被覆した。そのサンプルを電界放射型走査型電子顕微鏡によって画像化した(Quanta 600,FEI)。
【0052】
・透過型電子顕微鏡
ZrPで処理されたPT−PP粉末と1体積%3−グリシドキシプロピルトリメトキシシラン(Z−6040 Dow Chem.)のメタノール溶液10mlを遠心チューブに入れ、5分間遠心分離した。次に、溶液を吸い出し、遠心チューブの底に粉末を残した。5mlの酸化プロピレンを粉末に加えて振盪した後、遠心分離して上清を除去した。エポキシ樹脂は以下の処方に従って作製した:5.67gのドデシル無水コハク酸、2.48gのアラルダイト(商品名)502及び1.85gのQuetol(商品名)651(すべてElectron Microscopy Science EMS社製)。この処方を良く攪拌して均一に混合した。次に、0.2mlのベンジルジメチルアミン(EMS)を攪拌しながらその処方に加えた。エポキシ樹脂をシラン処理した粉末を含有する遠心チューブに注ぎ、55℃で一晩硬化させた。
【0053】
熱加圧されたZrPナノコンポジット薄板については、適当なサイズの試料を切り取り、下記記載のように3−グリシドキシプロピルトリメトキシシランで処理した。1体積%3−グリシドキシプロピルトリメトキシシラン(Z−6040 Dow Chem.)のメタノール溶液を調整した。この溶液を約10mlペトリ皿に注ぎ、ガラス容器に置いた。この試料をガラス容器に入れ、その容器をシールし、40℃で30分間加熱した。これによって、シラン溶液は蒸発し、この容器内は飽和状態になる。試料の表面は、エポキシ樹脂との結合を助けるシラン薄層でコーティングされる。次に、シラン処理した試料を遠心チューブに入れ、エポキシ樹脂をそのチューブに注ぐ。そして、エポキシ樹脂を55℃で一晩硬化させる。硬化したエポキシのブロックから薄片を作製し、銅格子上に置いた。
【0054】
P−PP−8については、圧縮成型したブロックを作製し、ウルトラミクロトームで薄片を作製した。その薄片を炭素フィルムで被覆した銅格子上に置いた。Cressingtonカーボンコーターを用いて、その薄片を10nmの炭素層でコーティングした。
【0055】
ライヘルト−ユングウルトラカットEウルトラミクロトームを用いてその薄片を切り出し、銅格子上に置いた。JEOL 1200 EXを用いて透過型電子顕微鏡観察(TEM)を行った。
P−PP−8のTEM画像ではマトリクス中でのZrPの均一な分布を示し(図7)、ZrP凝集体の崩壊の証拠が示された(図8)。
【0056】
<CNTナノコンポジットの作製>
・MWCNTの解きほぐし
本発明者らの以前の研究[文献20及び21]に記載された手順に従い、新しいMWCNTを酸化した。完全に薄片化したZrPのナノ小板を、わずかに酸化したMWCNT水溶液にCNT:ZrP=1:5の重量割合で加え、MWCNTを解きほぐし、分散させた。この混合物を室温で30分間超音波処理した(Branson 2510)。次に、酸を添加することによって、その溶液からZrPを除去し、MWCNTが残っている混合物の分離物を界面活性剤溶液で懸濁する。この方法で、最大500パーツ・パー・ミリオン(ppm)の濃度の調製に成功した。
【0057】
・F−MWCNTの作製
オクタデシルアミン粉末と良好に分散されたMWCNT溶液を直接混合することによってMWCNTを官能基化した。完全に反応させるために、その混合物を85−90℃で1時間継続的に攪拌し、オクタデシルアミンによって変性されたMWCNT(F−MWCNT)を水溶液から沈殿させた。その沈殿物を回収し、80℃のオーブンで一晩乾燥させた。
【0058】
・解きほぐされたMWCNT/PPナノコンポジットの作製
15グラムのキシレンを沈殿したF−MWCNTに加え、キシレン中でF−MWCNTを単独で分散するために1時間超音波処理した。次に、1グラムのPPを機械的に攪拌しながらそのF−MWNT/キシレン溶液に加えた。均一な混合物を生成するために、混合物を125℃で1時間攪拌した。エタノールを加えて、溶液からPP/F−MWCNTを強制的に沈殿させた。また、エタノールを使用して何度か表面を洗浄し、残留したキシレンを除去した。次に、最終的に得られたPP/F−MWNT粉末を80℃の真空オーブンで12時間乾燥させた。その粉末を180℃で1分間熱加圧し、電気伝導度測定用のPP/F−MWNTナノコンポジットのプラークを作製した。
【0059】
・形態的特性
透過型電子顕微鏡(TEM)の観察は、高解像度透過型電子顕微鏡JEOL 2010を用いて200kVで実施した。薄い炭素被膜を有する銅格子を溶液サンプルで覆い、室温で乾燥した。TEM画像解析用に、ライヘルトユングウルトラカット−Eミクロトームを用いて、ナノコンポジットバルクのサンプルを約80nmの薄さの薄切切片にした。Leo Zeiss 1530 VP Field Emission−SEM(FE−SEM)を用いてSEM画像を得た。
【0060】
・機械的試験
張力試験の試料は、ニートPPペレットと混合した溶液から得られたPP/F−MWCNT混合物を作製し、2分間、180℃で60rpmとしたHaakeミキサー(System40)を用いて、PP中のMWCNTを規定量にした。混合後に、その混合物を室温でゆっくり冷却した。張力試料は、溶融温度及び成形温度をそれぞれ195℃と90℃に固定し、射出速度0.25cm3/sで小型射出成形機(CS−183MMX)を用いて成形した。張力試験用に、射出成形試験片を機械加工し、ASTM D638−08規格に従い特性評価を行なった。クロスヘッド速度を5mm/分として、MTSスクリュー駆動試験機で室温での引張試験を実施した。校正したMTS伸縮計(model632.12B−50)を用いて真の張力を測定した。1サンプルにつき最低5試料の測定に基づいて計測した平均弾性係数と引張強度を標準誤差と共に報告する。
【0061】
・MWCNTの分散
通常、MWCNTは、MWCNTのチューブの長さとチューブの欠陥による特有の湾曲のために、合成後に高密度な絡み合いを形成する。以前、溶液中及びポリマーマトリクス中の両方で、完全に薄片化したZrPナノ小板をCNTの分散と薄片化に使用することに成功した[文献20及び21]。ナノ小板の静電荷を遮断するために酸を添加することによって溶液からナノ小板を容易に除去できる。アセトンと水でチューブを洗浄した後、MWCNTを水中に再分散し、高い解きほぐし状態を維持する。図9に本発明のMWCNT製造前後のTEM画像を示す。図9a−bでは、わずかに酸化したMWCNTが絡み合ったままで残っている。ここで説明した解きほぐしの処理後、TEMを用いた直接観察では、MWCNTの凝集又は絡み合いの証拠は一切なかった。MWCNTは良好に分散し、およそ0.5〜10μmの長さの曲線形を有した(図9c−d)。
【0062】
・オクタデシルアミンの官能基化
良好な分散を達成し、ポリマーマトリックを用いてMWNTの湿潤を促進するために、オクタデシルアミン粉末をMWCNT水溶液に直接混合によって加えた。図10a−bに示すように、良好に分散されたMWCNT水溶液中へのオクタデシルアミン粉末の添加は、MWCNT表面とアルキル基との間の特徴的な−COO−+NH3−結合によるオクタデシルアミン側鎖のイオン付加をもたらす。また、わずかに酸化したMWCNTとF−MWCNTの比較により双性イオン形成を示すIRスペクトルを得た。図11に示すように、1564cm−1のピークは、カルボン酸陰イオンの伸縮振動を示す。また、2842cm−1と2922cm−1で示されたピークは、オクタデシルアミンアルキル鎖中のC−H伸縮振動による。したがって、PP中のF−MWCNTの良好な分散が期待できる。以前の報告[文献17−19]とは対照的に、F−MWCNTは、ほんの少しの超音波処理によって有機溶媒中で容易に再分散でき、ポリマーマトリックスを混合する前に、均一な分散状態を示す。キシレン中のF−MWNTのTEM画像は、100ppmの濃度で、非常に良好な分散と完全な解きほぐし状態を示す(図12)。MWNTの良好な分散は、オクタデシルアミンの官能基の親有機性の増加のためであると考えられる。また、MWCNT表面のアルキル末端(alkyl tail)もPP中の分散に役立つ。
【0063】
・PP/F−MWCNTナノコンポジットの作製
PP/F−MWCNTナノコンポジットは、PPペレットを125℃のF−MWNT/キシレン溶液に直接添加することによって作製した(図10b−c)。MWCNTの濃度は、0.1〜2.0重量%の間でコントロールした。PPペレットを機械的に連続攪拌して溶解した。F−MWCNTが0.5重量%のMWCNTにおいてPP薄膜中で良好に分散されることが、TEM画像により確認された(図13)。反対に、他の研究では、ポリマー中のCNTの安定性と分散性を改良するためにアルキル短鎖をMWCNT表面に結合できることが示されたが[文献17及び18]、ポリマーマトリクス中でのMWCNTの良好な分散又は解きほぐしに関する証拠は文献中では示されていない。図10c−dに示すように、蒸発によってキシレン溶液を除去することによりPP/F−MWCNTナノコンポジットを得た。サンプルは80℃で一晩乾燥した。導電性とTEM顕微鏡観察用のナノコンポジット薄膜は、乾燥後のサンプルを熱加圧することによって作製した。
【0064】
PP/F−MWCNT薄片のTEM画像は、溶媒の除去後でも分散性の質が維持されることを示している。図14に示されるように、PP中に0.1、0.6、1及び2重量%のMWCNTを含むPPは非常に良好な分散性を示し、本発明の手法が、各々のチューブレベルで、PP中でのMWCNT単独の分散を促進するのに効果的であることを強く示唆する。
【0065】
・PP/CNTナノコンポジットの機械的特性
単軸引張の下で検出されるPP/F−MWCNTナノコンポジットの機械的特性を図15に示す。これらの結果は、ヤング率の50%増加と測定引張り強度の17%増加により、0.1重量%の低さのMWCNT濃度で著しい機械的強化が実現されることを示している(表2)。また、未処理のMWCNTのPP中での分散性を用いて対照実験を実施した。これらの系は大きな凝集を示し、ヤング率と引張り強度においてわずかな改善を示した(ニートPPに対してそれぞれ14%と5%の改善)。文献[22、23]で報告されたものと比べて、上記の結果は重要である。
【0066】
MWCNTの少量の添加が、係数と強度における顕著な改善に寄与している強化のメカニズムを調べるために、ニートPPとPP/MWCNTナノコンポジットの両方の引張破断面のSEM画像を得た(図16)。ニートPPは高延性を示し、完全なネッキングが起こるまで破砕しない(図16a,b)。PP/F−MWCNTは、かなり異なった性質を示す。それは、ネック形成の間に裂け、0.1重量%F−MWCNTでは、直径約5μmの破断表面上に広がったミクロサイズの継ぎ目(patch)を示す。慎重な調査によって、MWCNTの引き抜きがないことから、MWCNTとPPマトリクス間の強い界面結合が示唆されることが示された(図16c,d)。また、射出成形の工程の間にもたらされる配置構造や結晶化の間の核生成に起因する強化もあり得る。
【0067】
さらに、本発明のナノコンポジットは、低いCNT添加量で、係数の高い改良を示す(後述の表2)。また、本発明の組成物は、射出成形の後に形状を保持する優れた寸法安定性を示す。CNTを含有するPPは、成形型のほぼ角柱形状を示すが、一方、ニートPPは中央部に大きな収縮がある。Keyence VK−9700共焦点レーザー顕微鏡を使用した共焦点レーザー顕微鏡法で、(a)ニートPP、(b)0.1重量%MWCNTを含むPP、及び(c)0.4重量%MWCNTを含むPPについて、長さ74mm×幅13mm×厚さ3mmの棒状ブランクの成形収縮の測定を行い、厚さ方向の収縮を得て(棒状ブランクの端部と、棒状ブランクの中央部を比較して、その間の厚さの違いを百分率で評価した;棒状ブランクは、上記の機械的試験のものと同様の方法で作製した)、以下の表に値を示し、それぞれ、図18(a)−(c)に表した。
【0068】
【表2】
【0069】
したがって、本発明のナノコンポジットの好適な実施態様は、95〜99.7重量%のポリオレフィン(最も好ましくはポリプロピレン)、及び0.3〜5重量%のナノチューブを含有し、ヤング率が2.0GPaを超え、そして厚さ方向の成形収縮が、純粋なポリオレフィンの成形収縮の4分の1よりも小さいことである。
【0070】
・PP/CNTナノコンポジットの導電性
未処理のMWCNTとF−MWCNTの存在に起因した濃度変化による電気伝導性の変化は、1Vでの表面伝導率に基づいて測定された(図17)。PP/MWCNTナノコンポジットは、0.1〜2重量%の間で、溶液を蒸発させた後のサンプルの直接的な熱加圧により作製された。結晶化の間、MWCNTの凝集と弱いネットワークの破壊のために、PP/P−MWCNT混合物の電気的パーコレーションは、ほぼ2重量%となった。一方、PP/F−MWCNTナノコンポジットは、2.3×10−6S/mの導電率を示す0.6重量%において、絶縁体−導体パーコレーション転移を示した。図17中の挿入画は、MWCNT分散状態の概念的な解釈を提供する。
【0071】
低濃度では、PPマトリクスを介して導電性パスを形成するための十分なMWCNTがない。浸透限界では、単一の電気経路が形成され、PPマトリクス中のチューブが結合したネットワークに沿って電子が動き回る。さらに濃度が高くなると電気経路がさらに形成されて、系内の他の経路に接続し始め、システムが結合されることにより、べき乗則の挙動を示す。電気的パーコレーションにおける充填量は、[24]によれば、非溶融状態のPP/CNT複合体の値が最も低い。このことは、通常、電気転移のための凝集したネットワークに依存する非結晶質系における所見と対照的である[25,26]。これは、F−MWCNTが、結晶化の間にPPのラメラ構造に組み込まれて、核生成剤として働くことを示しており、表2の結晶化度の測定値によって裏付けられている。また、この挙動は、観測された弾性係数と引張り強度の大幅な増加を、部分的に説明することができる。
【0072】
【表3】
【先行技術文献】
【非特許文献】
【0073】
【非特許文献1】R.Saito,G.Dresselhaus,M.Dresselhaus,Physical properties of carbon nanotubes,Imperial College Pr,1998.
【非特許文献2】M.Dresselhaus,G.Dresselhaus,P Avouris,Carbon nanotubes:synthesis,structure,properties,and applications,Springer Verlag,2001.
【非特許文献3】X.Xie,Y.Mai,X.Zhou,Materials Science and Engineering:R:Reports,49(2005)89−112.
【非特許文献4】X.Gong,J.Liu,S.Baskaran,R.Voise,J.Young,Chem.Mater,12(2000)1049−1052.
【非特許文献5】M.O’Connell,P.Boul,L.Ericson,C.Huffman,Y.Wang,E.Haroz,C.Kuper,J.Tour,K.Ausman,R.Smalley,Chemical Physics Letters,342(2001)265−271.
【非特許文献6】V.Georgakilas,K.Kordatos,M.Prato,D.Guldi,M.Holzinger,A.Hirsch,J.Am.Chem.Soc,124(2002)760−761.
【非特許文献7】C.Dyke,J.Tour,J.Phys.Chem.A,108(2004)11151−11159.
【非特許文献8】R.Blake,Y.Gun’ko,J.Coleman,M.Cadek,A.Fonseca,J.Nagy,W.Blau,J.Am.Chem.Soc,126(2004)10226−10227.
【非特許文献9】D.Tasis,N.Tagmatarchis,A.Bianco,M.Prato,Chem.Rev,106(2006)1105−1136.
【非特許文献10】R.Chen,Y.Zhang,D.Wang,H.Dai,J.Am.Chem.Soc,123(2001)3838−3839.
【非特許文献11】J.Chen,H.Liu,W.Weimer,M.Halls,D.Waldeck,G.Walker,J.Am.Chem.Soc,124(2002)9034−9035.
【非特許文献12】J.Chen,M.A.Hamon,H.Hu,Y.Chen,A.M.Rao,P.C.Eklund,R.C.Haddon,Science,282(1998)95−98.
【非特許文献13】M.A.Hamon,J.Chen,H.Hu,Y.Chen,M.E.Itkis,A.M.Rao,P.C.Eklund,R.C.Haddon,Adv.Mater.11(1999)834−840.
【非特許文献14】D.Chattopadhyay,S.Lastella,S.Kim,F.Papadimitrakopoulos,124(2002)728−829.
【非特許文献15】D.Chattopadhyay,I.Galeska,F.Papadimitrakopoulos,J.Am.Chem.Soc.125(2003)3370−3375.
【非特許文献16】J.Chen,A.M.Rao,S.Lyuksyutov,M.E.Itkis,M.A.Hamon,H.Hu,R.W.Cohn,P.C.Eklund,D.T.Colbert,R.E.Smalley,R.C.Haddon,J.Phys.Chem.B105(2001)2525−2528.
【非特許文献17】A.Koval’chuk,V.Shevchenko,A.Shchegolikhin,P.Nedorezova,A.Klyamkina,A.Aladyshev,Macromolecules,41(2008)7536−7542.
【非特許文献18】A.Koval’chuk,A.Shchegolikhin,V.Shevchenko,P.Nedorezova,A.Klyamkina,A.Aladyshev,Macromolecules,41(2008)3149−3156.
【非特許文献19】J.Lee,S.Yang,H.Jung,Macromolecules,42(2009)8328−8334.
【非特許文献20】D.Sun,W.Everett,C.Chu,H.−J.Sue,Small,5(2009)2692−2697.
【非特許文献21】D.Sun,C.Chu,and H.−J.Sue,Chem.Mater.,22(2010)3773−3778.
【非特許文献22】G.Lee,S.Jagannathan,H.Chae,M.Minus,S.Kumar,Polymer,49(2008)1831−1840.
【非特許文献23】B.Yang,J.Shi,K.Pramoda,S.Goh,Composites Science and Technology,68(2008)2490−2497.
【非特許文献24】W.Bauhofer,J.Z.Kovacs,Composites Science and Technology,69(2009)1486−1498.
【非特許文献25】J.Sandler,J.Kirk,I.Kinloch,M.Shaffer,A.Windle,Polymer,44(2003)5893−5899.
【非特許文献26】C.Martin,J.Sandler,M.Shaffer,M.Schwarz,W.Bauhofer,K.Schulte,A.Windle,Composites Science and Technology,64(2004)2309−2316.
【非特許文献27】A.Usuki et al.,J Mater Res 8,1 179(1993).
【非特許文献28】Y.Kojima et al.,J Mater Res 8,1 185(1993).
【非特許文献29】A.Usuki et al.,Journal of Applied Polymer Science 63,137(1997).
【非特許文献30】M.Kawasumi et al.,Macromolecules 30,6333(1997).
【非特許文献31】N.Hasegawa et al.,Journal of Applied Polymer Science 67,87(1998).
【非特許文献32】M.Kato,A.Usuki,and A.Okada,Journal of Applied Polymer Science 66,1781(1997).
【非特許文献33】T.Kashiwagi et al.,Macromolecular Rapid Communications 23,761(2002).
【非特許文献34】S.P.Bao,and S.C.Tjong,Mat Sci Eng a−Struct 485,508(2008).
【非特許文献35】R.Blake et al.,Journal of the American Chemical Society 126,10226(2004).
【非特許文献36】S.H.Lee et al.,Carbon 45,281 0(2007).
【非特許文献37】S.C.Tjong,G.D.Liang,and S.P.Bao,Scripta Materialia 57,461(2007).
【非特許文献38】S.B.Kharchenko et al.,Nature Materials 3,564(2004).
【非特許文献39】T.McNally et al.,Polymer 46,8222(2005).
【非特許文献40】L.Y.Sun et al.,New Journal of Chemistry 31,39(2007).
【非特許文献41】D.M.Kaschak et al.,Journal of the American Chemical Society 120,10887(1998).
【非特許文献42】L.Y.Sun et al.,Chemistry of Materials 19,1749(2007).
【非特許文献43】H.−J.Sue et al.,US Published Patent Application 2009/0035469,filed April 30,2008.
【0074】
本発明のさらなる修正及び変形は、上記の教示の観点から明らかに可能である。したがって、添付されたクレームの範囲内において、本明細書に具体的に記載されたもの以外の方法で本発明を実施できることが理解される。
【技術分野】
【0001】
[関連出願の参照]
本出願は、2009年12月28日に出願された米国の仮出願61/290,465の優先権を主張し、その内容全体は参照として本明細書に取り込まれる。
本発明はポリマー系ナノコンポジットに関する。より詳細には、本発明は、ポリオレフィン中に細かく分散されたナノチューブ及び/又はナノ小板等のナノ粒子を含むポリマー系ナノコンポジットに関する。
【背景技術】
【0002】
ポリオレフィンは、商業的に生産された最も広く使用されているポリマーの1つである。工学的な応用において、ポリプロピレン(PP)は、その高い融点、比較的高い弾性、低コスト及びリサイクル性のために魅力的であると考えられている。PPの応用範囲をさらに広げるために、それら特性を改良する多くの試みがある。改良を遂行するためのそのような戦略の1つは、ナノサイズのフィラーをPPに含有させることによる。改良できる材料の特性は、利用されるナノフィラーのタイプに依存する。一般的に使用されるフィラーには、モンモリロナイト等の珪酸塩ナノクレイがある。珪酸塩ナノクレイは、ポリマーの剛性、強度、気体遮断性、熱変性温度及び難燃性を改良するために使用される。珪酸塩ナノクレイは特に、ポリアミドの改良において有用であることがわかっており、中でも注目すべきは、ナイロン6[文献27及び28]、ポリイミド又はアミド若しくはイミド基を含むポリマーの改良である。ポリマーマトリックス中でナノクレイが良好に薄片化されたときに優れた改良が見られる。しかしながら、珪酸塩ナノクレイをPP中で薄片化することは、あまり成功していない。注目に値する例は、ジオクタデシルジメチルアンモニウムイオンでイオン交換されたモンモリロナイトクレイの集合体に挿入するための、ヒドロキシテレケリック基を有するポリオレフィンオリゴマーの使用である[文献29]。オリゴマー量の増加が、ナノクレイの良好な薄片化をもたらすことが分かった。この方法のさらなる改良は、ステアリルアンモニウムで交換したモンモリロナイトの使用や、無水マレイン酸で変性したPP(PP−MA)の使用である。PP−MAはニートPP(neat PP)の相溶化剤として作用する[文献30−32]。この方法を使用することによって、PP中でナノクレイをほとんど薄片化することができる。ナノクレイに対するPP−MAの比率が極めて重要であり、薄片化を最も多く得られる割合は3:1であった。PPの中で薄片化は達成されたが、ニートPPの物理的特性の改良の点では、ナイロン−クレイのハイブリッドで見られたものと比べものにならなかった。これは実際のところ、クレイの不完全な薄片化とPP−MAの存在による可能性が極めて高い。PPは疎水性が高くクレイは親水性が高いので、このような非常に非相溶性である材料間の相互作用を調節するためには、中間体が必要であると認識されている。そのような相溶化剤の使用を無くすか、使用量を著しく減少させるための、何らかの方法が非常に望まれている。
【0003】
カーボンナノチューブ(CNT)等の異なる種類のナノフィラーもPPに使用できる。カーボンナノチューブ(CNT)は、優れた機械的、電気的及び熱的特性を有するが[文献1及び2]、ポリマーマトリックスにこれらの特性を移す試験の実験結果では、低分散であり、さらにはCNTとポリマーマトリックス間の界面接着が不十分であるために、成功例は限られている[文献3]。また、CNTはナノクレイと同様にPPの難燃性を改良することがわかった[文献33]。良好な分散を達成するための最も一般的なアプローチには、界面活性物質での被覆[文献4及び5]、共有結合官能基化[文献6−9]及び非共有結合官能基化[文献10−16]が挙げられる。これらのうち、CNT表面への長鎖アルキルの付加を用いた酸塩基に基づく非共有結合官能基化では、他の方法に比べて、高い収率が得られて高効率であることが示されており、異なった数種類のポリマーに応用できる。一般的に、CNTへのアルキル鎖の付加は、酸化されたCNT表面と脂肪族アミンの官能基との間のイオン結合によって達成される[文献12及び13]。アミンの官能基が、CNT表面において、カルボン酸の官能基とイオン結合を介して相互作用した強い親和力を有することは、よく立証されている。酸処理されたCNTとオクタデシルアミン間の非共有結合は、双性イオンの構造を介して、有機溶媒中でのCNTの安定した分散をもたらすことが示された[文献12−16]。
【0004】
また、多層カーボンナノチューブ(MWCNT)を解きほぐすためにいくつかの方法が試みられたが、それらの方法では個々のレベルで良好な分散状態を示すことができなかった。Koval’chukらは、CNT表面上でのアルキル化の実現に脂肪族アミンを使用し、PP中でのMWCNTの良好な分散を実現した[文献17及び18]。この手法は簡単で空気と反応しにくく、その結果、高い官能基化を示すが、依然として、複合物中にもつれた構造のMWCNTが含まれている。Jungらは、官能基化のためにオクタデシルアミンを使用し、分散のためには長鎖アルキルがより有用であることを示したが、PPと混合した後では単独の分散が達成できなかった[文献19]。上記のアプローチは、PPマトリクスとCNTの相溶性を改良するが、ナノコンポジット材料中では解きほぐされたCNTの分散状態を十分に示すことができなかった。
【0005】
BaoとTjongは、二軸押出機で、MWCNTとPPの溶融混合の効果を調べ、0.3重量%のMWCNTでの引張係数(33%増加)及び引張強度(16%増加)において有意な改善を認めた[文献34]。しかしながら、MWCNT添加をさらに増量しても、ほとんど改善がされなかった。Fereidoonらは、単層CNT(SWCNT)とPPとの溶融混合状態を調べ、1重量%のSWCNTで引張係数の82%増加及び引張強度の22%増加を達成することができた。Blakeらが用いたより高度な手法は、n−ブチルリチウムで官能基化したMWCNTを作製し、続いて、塩素化したPP(CL−PP)とカップリング反応を行うものである[文献35]。この方法は、CL−PP層でコーティングされたMWCNTを生産し、その結果、CL−PP中での混和性を高めた。引張係数と引張強度はそれぞれ209%と277%と目覚しい改良が報告された。この研究は、達成可能な増強効果がCNTの分散状態に強く依存していることを示唆する。しかしながら、ホストポリマーとしてのCL−PPの使用は、そのような目覚しい結果を達成するためにホストポリマーを変性する必要があることを示している。他では、空気中の加熱によって官能基化されたMWCNTが、PP−MA等の相溶化剤の使用条件下でPPと良好な相溶性を示すことがわかった[文献36]。この場合、MWCNTのミクロサイズの凝集体が形成された。研究では、CNTはポリオレフィンの導電性を改善するために使用できることが示された[文献36及び37]。PP中の約1体積%のMWCNTの含有により、体積導電率の7桁増加を引き起こすことができる[文献38]。また、10重量%のMWCNTでは、体積抵抗率を16桁減少させる[文献39]。このように、CNTはポリオレフィンの電気特性を改良するための優れた材料である。
【0006】
上記背景から、PP等の未変性ポリオレフィン中で、ナノ粒子又はCNT等のナノチューブの分散を高める実用的な方法は依然として不十分であり、意味のある化学変性や大量の相溶化剤を用いることなく、ナノ粒子等のナノ分散を実現できる技術を開発する必要があると結論づけられる。
【発明の概要】
【発明が解決しようとする課題】
【0007】
従って、本発明の目的の1つは、ポリオレフィン中でのナノ小板、ナノチューブ又はその両方について、効率の高い分散方法を提供することである。
【0008】
さらなる本発明の目的は、ナノ小板、ナノチューブ又はポリオレフィンの表面改質によって、ポリオレフィン中のナノ小板、ナノチューブ又はその両方を分散する方法を提供することである。
【0009】
さらなる本発明の目的は、本発明に従って作製されたナノコンポジットを提供することである。
【0010】
さらなる本発明の目的は、ナノコンポジットから作製される製品を提供することである。
【課題を解決するための手段】
【0011】
本発明のこれら及び他の目的は、各々又はこれらの組み合わせについて、ポリオレフィン中でナノチューブ及び/又はナノ小板を分散する以下の工程を含む方法並びに、そこから形成されたナノコンポジットの発見によって達成された。
A)ナノチューブ、ナノ小板又はその両方を含む溶液を調製する工程;
B)工程(A)により得られた溶液を攪拌する工程;
C)工程(B)で攪拌された溶液中に1種以上のポリマー材料を溶解し、さらにその溶液から沈殿物を単離する工程;及び、
D)1種以上のポリオレフィンと前記沈殿物を溶融混合する工程。
【図面の簡単な説明】
【0012】
本発明及び本発明に付随するその多くの利点について、以下の詳細な記載を参照し、添付の図面と共に考察することでより良く理解されることによって、完全な理解が容易に得られる。
【0013】
【図1】図1は、溶液からCNT/ZrPとPPとの沈殿により得られたCNT/ZrP/PPのマスターバッチの透過型電子顕微鏡画像である。CNTとZrPナノ小板は共に、PPホスト中でよく分散されている。沈殿物の組成は、重量比でCNT/テトラ(n−ブチルアンモニウム)水酸化物(TBA)/ZrP/PP=1/2/3/4である。
【図2】図2は、ニートPPペレットとナノコンポジットのサンプルJPP−DのX線回折スペクトルである。チャートはPP結晶のα相の存在を示す。積層ZrPナノ小板からの回折ピークが無いことは、ナノ小板がおそらく薄片化されていることを示す。
【図3】図3は、PP中に分散した0.05重量%のMWCNTの透過型電子顕微鏡画像である。MWCNTは凝集することなく、非常に高分散状態である。
【図4】図4は、プラズマ処理されたPP粒子の電界放出走査型電子顕微鏡写真である。粒子サイズは約100ミクロンである。
【図5】図5は、プラズマ処理されたPP粒子(P−PP−1)の表面にZrPナノ小板が付着している状態の透過型電子顕微鏡画像である。
【図6】図6は、高温で圧縮して薄板にしたZrPナノコンポジットサンプルP−PP−1の透過型電子顕微鏡画像である。単独に分散しているナノ小板が観測された。
【図7】図7は、0.015重量%ZrPナノコンポジットサンプルP−PP−8の透過型電子顕微鏡画像であり、約100nmサイズのナノ小板の均一な分散を示している。
【図8】図8は、0.015重量%ZrPナノコンポジットサンプルP−PP−8の透過型電子顕微鏡画像であり、100nmサイズのナノ小板の均一な分散を示している。
【図9】図9a−9dは、わずかに酸化した後の著しい絡み合いを示すMWCNT(a,b)と、ナノ小板が分散工程を補助した後の良好に分散したMWCNT(c,d)のTEM顕微鏡画像である。
【図10】図10a−10dは、オクタデシルアミンによって表面改質された単独のMWCNTの製造方法(a,b)と、PP中に良好に分散したMWCNTの製造方法(c,d)についての本発明の方法の概念的な説明図である。
【図11】図11は、わずかに酸化されたMWCTN(上部)と、F−MWCNT(下部)のFTIRスペクトルを示す。
【図12】図12aと12bは、キシレン溶液からの回収後の良好に分散したFD−MWCNTのTEM顕微鏡画像である。
【図13】図13aと13bは、キシレン溶液から作製したPP/FD−MWCNTナノコンポジットのTEM顕微鏡画像である。
【図14】図14a−14hは、a,b)0.1重量%;c,d)0.6重量%;e,f)1重量%及びg,h)2重量%のMWCNTを含むPPナノコンポジットのTEM画像である。
【図15】図15は、ニートPPと良好に分散したMWCNTを含有するナノコンポジットの工学応力−真歪み曲線を示す。
【図16】図16a−16dは、a,b)ニートPP及びc,d)0.1重量%F−MWNTナノコンポジットの、SEMによる破断面図である。
【図17】図17は、F−MWCNTを様々な濃度で含むPPナノコンポジットの表面電気伝導率を示す。
【図18】図18a−18cは、(a)ニートPP,(b)0.1重量%MWCNTを含むPP,及び(c)0.4重量%MWCNTを含むPPの寸法安定性(すなわち、厚さ方向への収縮)の測定値を示す。
【発明を実施するための形態】
【0014】
本発明は、ポリオレフィン中、特にPP中でのナノ粒子又はナノチューブ、特にMWCNTの非常に改良された分散性を達成するための簡単な方法に関する。本発明は、より詳細には、(i)PP等のポリオレフィン中でのMWCNT等のナノチューブ、(ii)PP等のポリオレフィン中でのZrP等のクレイ若しくはナノ小板、又は(iii)PP等のポリオレフィン中でのナノチューブとクレイ若しくはナノ小板の組み合わせにおいて、非常に改良された分散性を達成することに関する。
【0015】
本発明者らの以前の研究において、CNTの電子状態に対して最小限のダメージでMWCNTとSWCNTの両方の解きほぐし及び脱結束を達成するために、わずかに酸化したCNTの表面にナノ小板を電気的に拘束した[文献20]。有機溶媒及びポリマーマトリクス中で薄片化したCNTの分散と解きほぐし状態を維持するために、CNT表面への親有機性の変性が特に必要である。
【0016】
本発明の組成物であるポリオレフィンは、好ましくはポリエチレン(PE)、ポリプロピレン(PP)、ポリブチレン(PB)又はそれらの混合物若しくはコポリマーである。より好ましくは、ポリオレフィンはPPである。以下の考察では、ポリオレフィンがポリプロピレン(PP)であるとして本発明を説明する。しかしながら、これは、本発明の範囲を限定することを意図するものではなく、ポリエチレンやポリブチレンなどの他のポリオレフィンをPPの代わりに使用することができる。
【0017】
本発明において、長鎖脂肪族アミンと無水マレイン酸で変性されたポリプロピレン(PP−MA:文献30−32を参照)から成る群から選択される少なくとも1種によって親有機性の変性が提供できる。好ましくは、中鎖〜長鎖脂肪族アミン(以下、単に「長鎖脂肪族アミン」という)への媒体の使用によって親有機性の変性が提供される。ここでアミンは、より好ましくは第一アミンである。中鎖〜長鎖脂肪族アミンに対する媒体は、変性されるナノチューブ又はナノ小板に対して所望の親有機性を提供するための炭素原子の数が十分である限り、各脂肪族鎖の炭素原子数は任意の規定数とすることができる。
【0018】
好ましくは、長鎖脂肪族アミンは、C4−C30脂肪族基を有し、より好ましくはC6−C30基、さらに好ましくはC10−C30基、さらに好ましくはC14−C24基、さらに好ましくはC16−C20基、そして最も好ましくはC18基を有する。親有機性の変性は、ナノチューブの表面、クレイ若しくはナノ小板の表面、又はポリオレフィンの表面に行うことが出来る。最も好適な実施態様では、官能基化された多層炭素ナノチューブ(F−MWCNT)を生産するために、オクタデシルアミンを選択し、キシレン、デカリン、ブタノール、ジクロロベンゼン、トリクロロベンゼン、N,N−ジメチルホルムアミド及びイソプロパノール等の有機溶媒中で、軽い超音波処理を用いて容易に分散することができる。次いで、その溶液をPPペレット等のポリオレフィンと直接混合し、乾燥することで、低い添加量で有意に改善された導電性及び引張係数でポリオレフィン/F−MWCNTナノコンポジットを生産することができる。また、上述と同様の溶媒は官能基化されたナノ小板又はクレイの分散にも使用できる。
【0019】
本発明で有用なナノチューブは、どのようなナノチューブであっても良い。好ましくは、ナノチューブは、カーボンナノチューブ、二酸化タングステンナノチューブ、シリコンナノチューブ、無機ナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である。より好ましくは、ナノチューブはカーボンナノチューブであり、最も好ましくはSWCNT又はMWCNTである。必要に応じて、ナノチューブは、これには限定されないが、乾燥酸化、放射酸化、プラズマ酸化、熱酸化、拡散酸化又はこれらの組み合わせを含む既存の酸化方法を用いて表面を酸化することができる。
【0020】
本発明に使用されるナノ小板は、これに限定されないが、クレイ(モンモリロナイト等)、ナノクレイ、グラフェン、無機結晶、有機結晶及びこれらの組み合わせを含む、クレイ又は他の形態のナノ小板である。特に、ナノ小板は、好ましくはα−リン酸ジルコニウム(ZrP)である。ZrPは、モンモリロナイトのような周知の天然クレイと同様の層状構造を有するので、合成クレイと見なすことができる。クレイの起源によってカチオン成分が異なりうる天然クレイとは違い、ZrPは化学構造Zr(HPO4)2・H2Oで明確に定義される。また、ZrPのサイズとアスペクト比は、様々な合成条件によって容易に制御され、天然クレイよりも均一のサイズ分布で得ることができる[文献40]。モンモリロナイトと同様の方法でZrPにオニウムイオンを挿入することができ、そして、水溶液中での薄片化は、TBA+OH−を導入してテトラ(n−ブチルアンモニウム)イオン(TBA+)を形成し、ZrPに挿入して次に薄片化することで容易に達成できる[文献41]。本発明では天然クレイの代用としてZrPを使用するが、同様の化学的、物理的性質であることから、ここで開発された方法は天然クレイでも応用することができる。
【0021】
本発明は、良好に分散されたナノチューブ又はナノ小板を含むポリオレフィンナノコンポジットを製造するための簡単ではあるが効果的な方法を提供する。特に、好ましくは、本発明は良好に分散されたMWCNTを含むポリオレフィンナノコンポジット製造するための簡単で効果的な方法を提供する。わずかに酸化されたMWCNTはナノ小板を用いて解きほぐされ、ナノ小板を除去した後でも、高い安定性を示す。好ましくは、良好に分散されたMWCNTはオクタデシルアミンで官能基化され、TEMとSEMの観察で証明されるように、単独の分散状態においても有機溶媒中での安定性を増す。良好に分散されたポリオレフィン/MWCNTのナノコンポジットは、高濃度のMWCNTを含むキシレン溶液等の有機溶媒とポリオレフィンペレットを直接混合することによって作製できる。乾燥し、ニートPP中で希釈するためのマスターバッチとして粉末を使用して、任意のMWCNT濃度でナノコンポジットを形成した。ナノコンポジットは、素晴らしい分散を示し、少ないチューブ添加量で、係数、強度、及び導電性が有意に増加した。機械的特性の強化メカニズムは明確には分かっていないが、部分的には、MWCNTが(1)結晶成長のための核生成剤、(2)ポリオレフィンマトリクスを効率的に強化するためのマトリクスの球状間領域(inter−spherulitic region)の強化剤として働いているためであると考えられる。
【0022】
本発明の実施態様の一つは、ポリオレフィン中でナノ小板及び/又はナノチューブを分散する方法であり、以下の工程を含む:
A)ナノチューブ及びナノ小板を含む溶液を調製する工程;
B)工程(A)により得られた溶液を攪拌する工程;
C)工程(B)で攪拌された溶液中に1種以上のポリマー材料を溶解し、さらにその溶液から沈殿物を単離する工程;
D)1種以上のポリオレフィンと前記沈殿物を溶融混合する工程。
【0023】
本発明者らは、以前に、水溶液中でカーボンナノチューブ及びZrP等のナノ小板を共分散するための新しい方法を開発した[文献20]。この溶液は、ポリオレフィンナノコンポジットを作製する本発明の方法において、上記の実施態様における最初の工程で使用できる。好ましくは、その水溶液はゲル様の稠度を有する粘性のスラリーになるまで加熱される。そして、N,N−ジメチルホルムアミド(DMF)等の溶媒中で再分散される。PP/デカリン溶液が調製され、CNT/ZrPのDMF溶液とイソプロパノールと共に混合される。この溶液は80℃の湯浴で超音波処理し、その後、90℃で30分間攪拌し、最後に室温で冷却される。冷却の間に形成される黒い沈殿物を回収し、イソプロパノールで洗浄し、真空オーブンで乾燥する。好ましくは、10%のCNT、20%のTBA、30%のZrP及び40%のPPを含む黒い沈殿物は、PPを用いて混合溶解するマスターバッチとして使用され、ポリマーナノコンポジットが作製される。イソプロパノールで再分散されたマスターバッチのTEM画像は、MWCNTとZrPのナノ小板がポリマーマトリックス中に単独で分散されることを示す(図1)。1重量%MWCNT/3重量%ZrPを有するナノコンポジットを射出成形試験片にした。サンプルをX線回折(XRD)で分析し、ニートPPペレットの結果と比較した。XRD分光では、非薄片化ZrPの特徴のある回折ピークは無く、PP結晶のα相のみが検出されることを示している(図2)。CNT/ZrP溶液と混合されるPP/デカリン溶液の代わりに、これには限定されないが、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエステルカーボネートコポリマー、ポリ(エステル−カーボネート)樹脂、ポリアミド、高温ポリアミド、ポリエチレン、ポリプロピレン、オレフィンのコポリマー、官能基化ポリオレフィン、ハロゲン化ビニルポリマー、ビニリデンポリマー、ポリ塩化ビニリデン、ポリフッ化ビニル、ポリフッ化ビニリデン、ポリアミドコポリマー、ポリアクリロニトリル、ポリエーテル、ポリケトン、熱可塑性ポリイミド、変性セルロース及び前述のポリマー材料の1種以上を含む混合物を含む他のポリマー材料を使用することができる。次に、CNT/ZrP溶液を用いて得られた混合物は、処理されてCNT/ZrP/ポリマー材料の沈殿を形成し、最終的なナノコンポジットを得るために、ポリオレフィンを用いて溶融混合するマスターバッチとして使用できる。当然、ポリマー材料がPPのみであるナノコンポジットを提供するためには、1種以上のポリマー材料としてPPを使用することが好ましい。
【0024】
あるいはまた、別の実施態様では、ZrPナノ小板は、Xiらによって述べられている方法を用いて水中で分散した後、CNTから分離される[43]CNTは、オクタデシルアミン等の長鎖脂肪族アミンを用いた親有機性変性の後、デカリン等の無極性溶媒中で再分散できる。CNT/キシレン溶液は、高温で攪拌されているPP(又は他のポリマー材料)/キシレン溶液にゆっくりと加えられ、PPとCNTの均一混合を確実にする。溶液の攪拌を停止し、冷却すると、溶液中でPPとCNTが共沈殿する。その沈殿を溶液から分離し、乾燥して、良好に分散されたPP/CNTナノコンポジットを形成する(図3)。
【0025】
TBAの除去と、オクタデシルアミン等の長鎖脂肪族アミンによる変性後、キシレン又はデカリン等の非極性溶媒系にZrPを分散することができる。ZrP/キシレン溶液は、高温で攪拌されているPP/デカリン溶液にゆっくりと加えられ、PP中でのZrPの良好な分散を確実にする。その後、デカリン中でPPとZrPを共沈殿させるために溶液を冷却する。溶液から沈殿物を分離し、乾燥して、良好に分散されたPP/ZrPナノコンポジットを形成する。
【0026】
また、ZrP/キシレンとCNT/キシレンの2種の良好に分散された溶液を一緒に混ぜて、均一な懸濁液を形成する。次に、その混合液を高温で攪拌されているポリマー材料、好ましくはPP/デカリン溶液にゆっくりと加え、ポリマー材料、好ましくはPP中のZrP/CNTの良好な分散を確実にする。その後、その溶液をデカリン中でPPとZrP/CNTを共沈殿させるために冷却する。その沈殿物を溶液から分離し乾燥して、良好に分散されたPP/CNT/ZrPナノコンポジットを形成する。
【0027】
本発明のナノコンポジットは、ナノチューブ及び/又はナノ小板のいずれの所望の添加物も含むことができる。好ましくは、ナノチューブ又はナノ小板の量は、0.1〜20重量%の範囲であり、より好ましくは0.1〜10重量%、最も好ましくは0.3〜5重量%である。より好ましい実施態様では、本発明のナノコンポジットは、ポリオレフィンを95〜99.7重量%含み、ナノチューブ、好ましくはMWCNTを0.3〜5重量%含む。パーコレーション濃度は、CNTのアスペクト比を用いて変えることができる。上記の最も好ましい実施態様では、MWCNTが0.3〜5重量%の濃度であり、その組成物は、10−6S/mを超える表面電気伝導率を有する。
【0028】
本発明のさらなる実施態様では、100ミクロンのサイズのプラズマ処理されたPP(PT−PP)粒子(図4)を水溶液中でZrPと混合する。PT−PP粒子を空気及び窒素の存在下でプラズマによって処理し、その結果、粒子の表面に、COOH、C=O、C−O、NO2及びNO3等の化学官能基が付加される。これらの化学官能基は、一般的に電子が豊富であり、特にカルボン酸基は水中で容易に脱プロトン化してカルボン酸イオンを形成し、その粒子に負の電荷を与える。TBAで変性されたZrPナノ小板は、正の電荷を帯びている。
【0029】
粒子とナノ小板間の静電引力は、強制的に各粒子を囲むZrP層を形成させる。ZrPで一旦処理されたPT−PP粒子は、水中で安定な懸濁液を形成する。また、この溶液にTBA+OH−を添加して、pHを上昇させることができる。pHの上昇は、PT−PP粒子の表面上で脱プロトン化されたカルボキシレート基の濃度を高め、粒子に対するZrPの引力を増加させると考えられる。過剰なアセトン添加は、安定な懸濁液を破壊し、ZrPでコーティングされたポリマー粒子を沈殿させる。その粒子を回収し、90℃のオーブンで乾燥させる。いくつかの粒子がエポキシ中に埋め込まれており、ZrPでコーティングされたポリマー粒子の形態を観察するためのTEM用の薄片の作製に使用される。残りの粒子を高温で圧縮して薄板を形成すると、圧縮されたシートの断面で薄片を形成するためにこれらはエポキシ中に埋め込まれる。これらの薄片は、TEMの画像処理に使用される。TEM画像の解析から、発明者らはPT−PP粒子の表面にZrPナノ小板が付加している証拠を見つけた(図5)。圧縮された薄板の断面のTEM画像では、個々のナノ小板がポリマーマトリックス中で分散されることが示された(図6)。
【0030】
本発明のナノコンポジットは、任意に、1種以上の通常の添加物を通常量含むことができる。好ましくは、1種以上の添加物は、これには限定されないが、フィラー、強化剤、可塑剤、酸化防止剤、熱安定剤、紫外線安定剤、強靭化剤、帯電防止剤、難燃剤、着色剤及び上述の添加物を1種以上含む組み合わせから選択される1種以上の添加物を含む。
【0031】
本発明のナノコンポジットは、フィルム、スポンジ(foams)、繊維及び他の構造形態等の様々な物の形成に使用できる。これらの物は、これには限定されないが、熱成形、押出成形、ブロー成形、延伸ブロー成形、押出ブロー成形等の従来のいかなる方法によっても形成できる。
【0032】
本発明については一般的に記載されたが、説明のために本明細書に記載された特定の実施例を参照することによって、さらなる理解を得ることができる。これら実施例は、特に規定のない限り、それらに限定されないことを意味する。
【実施例】
【0033】
・材料
ZrPナノ小板を使用して、MWCNTを水溶液中で解きほぐして分散した。MWCNTを解きほぐすためのZrPの合成、薄片化及び使用については以前に報告されている[20,21]。簡潔には、15.0gのZrOCl2・8H2O(Fluka)を150.0mlの3.0M H3PO4(EM Science)中で機械的に攪拌しながら100℃で24時間還流した。次に、生成物を遠心分離と再分散によって3回洗浄し、85℃のオーブンで24時間乾燥した後、優しく乳鉢ですりつぶして微粉末にした。ZrP粉末をTBA+OH−(Aldrich,メタノール中1mol/L)を用いて、分子比ZrP:TBA=1:0.8として水中で薄片化した。新しいMWCNT(P−MWCNT)(純度90%、平均直径<10nm、長さ範囲0.1−10μm)をAldrichから購入した。市販のオクタデシルアミン(CH3(CH2)17NH2,Sigma−Aldrichケミカルズ、97%)を得て使用した。メルトフローインデックス(MFI)が1.9g/10minの市販グレードのPP(記号4204)は日本の日本ポリプロ株式会社(JPP)から供給された。
【0034】
<CNT/ZrPナノコンポジットの作製>
・ZrPナノ小板の合成と薄片化
本研究におけるZrPナノ小板の合成と薄片化は、以前に報告された方法と類似している[文献40及び42]。ZrPナノ小板は、還流により合成された:パイレックス製の丸底フラスコ内で、ZrOCl2・8H2O(Fluka)20.0gを200.0rnL 3.0Mで、100℃、24時間攪拌しながら、還流した。反応後、生成物を3回の遠心分離で洗浄し、回収した。その後、ZrPを85℃のオーブン内で24時間乾燥した。乾燥したZrPは乳鉢と乳棒のセットですりつぶして微粉末にした。
【0035】
作製したZrPを、TBA+OH−(Aldrich)により、分子比α−ZrP:TBA=1:0.8で水中で薄片化した。TBAを分散したZrPに加え、少なくとも2時間攪拌し、ナノ小板中にTBAの挿入を行った。その後、分散物を1時間以上超音波処理し、溶液中で完全に薄片化した(分散体の量によって、より長時間の処理が必要な場合がある)。
【0036】
・CNTの酸処理
CNTを酸で処理し、ナノチューブ表面上にカルボキシル基を導入した。硫酸と硝酸の混合溶液(体積比36ml/12ml)を調製した。調製した酸性の混合溶液を0.2gのカーボンナノチューブ(純度90%、平均直径<10nm、長さ範囲0.1−10μm、Aldrich社製)に加え、2時間超音波処理を行った。超音波処理装置の水は一定の水温を維持するために循環させた。次に、152mlの脱イオン水を酸/CNT混合溶液に加え、この溶液を循環水中で1時間超音波処理した。次に、CNTを二フッ化ポリビニリデンのフィルター膜(Millipore,細孔径0.45μm)を用いてろ過し、残留したすべての酸を除くために脱イオン水で十分に洗浄した。洗浄したCNTを脱イオン水に再分散し、3時間超音波処理をした。通常、水中のCNTの最終濃度は0.002g/ml〜0.005g/mlである。
【0037】
・CNT/ZrP分散物の作製
CNT/ZrP分散物を重量比1〜3で作製する。例として、0.2gのCNTが安定した分散状態を形成するためには0.6gのZrPが必要である。通常、1gのZrP分散物に対しては100mlの水が用意され、前述の方法に従って薄片化する。0.6gのZrPサンプルに対しては、CNT/ZrP分散物の作製のためには60mlの分散物を使用する。CNT/水分散物を完全に薄片化したZrP/水分散物に加え、安定した分散状態を形成するために1時間以上超音波処理する。水中で安定したCNT/ZrP分散物を加熱し、CNT/ZrPがゲル状に凝縮するまで水分のほとんどを除去した。次に、25mlのN,N−ジメチルホルムアミド(DMF)(Alfa Aesar)をゲルに混合した。その混合物を1時間以上超音波処理し、CNT/ZrPをDMF中に再分散した。
【0038】
直接的な混合手法を用いたCNT/ZrPナノコンポジットの作製では、すべての水が完全に除去されるまでCNT/ZrPの水分散物を加熱した。乾燥した残留物をオーブンに入れ、90℃で一晩乾燥した。乾燥したCNT/ZrPの残留物を乳鉢ですりつぶして微粉末にした。
【0039】
・CNT/ZrP PP分散物の作製
0.8gのPP(Novatec、JPP)を200mlのデカリン(Sigma Aldrich)に加え、PPペレットが全て溶解するまで130℃のオイルバス中で加熱した。25mlのイソプロパノールをその溶液に加え、次に前のセクションで作製したDMF中のCNT/ZrP分散物を加えて122℃で10分間攪拌した。その溶液を含むフラスコを超音波槽(Bransonic(登録商標)1510)に移し、80℃の浴温で20分間超音波処理した。フラスコをオイルバスに移し、90℃で30分間一定の速度で攪拌した。フラスコの底にすぐに固まった黒い沈殿物が生じたので、透明な溶液を除去し、イソプロパノール(EMD Chem)中でそれらを再分散することによって、残存する沈殿物を回収した。沈殿物を遠心分離し、上清を除去した。イソプロパノール中での再分散と遠心分離後の上清の除去の工程を3回繰り返した。その後、沈殿物を80℃で24時間真空乾燥した。沈殿物の組成は重量比でCNT/TBA/ZrP/PP=1/2/3/4である。
【0040】
・CNT/ZrP PPナノコンポジットの作製
前のセクションで得られた沈殿物と粉末は、ニートPP(Novatec,JPP)で希釈するマスターバッチとして使用し、所望のCNT/ZrP添加によりナノコンポジットを形成した。マスターバッチを一定量のPPと事前に混合し、その混合物を2軸バッチミキサー(Haake Rheocord System 40)の混合槽に添加した。表1にナノコンポジットの作製に用いられた組成を示す。ミキサースクリューを用いて、60rpm、180℃で10分間溶融混合を行った。その後、小型射出成形機(CS−183 MMX,CSI)を用いてナノコンポジットを75mm×12.5mm×3.15mmの角柱状に射出成形した。溶融漕を180℃で維持し、成形型を80℃で維持した。ニートPPの試験片を作製するためには、溶融漕を210℃で維持し、成形型を80℃で維持した。
【0041】
【表1】
【0042】
・CNT PPナノコンポジットの作製−溶解法
Xiらの方法に従って、水溶液中で、薄片化したMWCNTの作製を行ったが、ここでは詳細には説明しない。MWCNT0.002gを含む水溶液15gを調製し、0.02gのオクタデシルアミン(CH3(CH2)17NH2)の粉末を加えた。その混合物を85〜90℃で1時間連続的に攪拌し、カーボンナノチュ−ブをオクタデシルアミンで変性した。一旦攪拌を停止すると、アミン変性されたMWCNT(F−MWCNT)が水溶液から沈殿する。この沈殿物を回収し、80℃のオーブンで2時間乾燥した。完全な分散を達成するために1時間超音波処理した沈殿に15gのキシレンを加えた。1gのPPを15gのデカリンに170℃で溶解した。均一な混合物を生成するために、F−MWCNT/キシレン溶液を攪拌しながら、PP/デカリン溶液に滴下して加えた。さらに、混合物を170℃で30分間攪拌し、溶媒を一部蒸発させた。最終生成物は、PP中にF−MWCNTが分散された粘性のあるゲルである。F−MWCNT/PPナノコンポジットは、デカリンのゲルから完全に乾燥することによって得られる。
【0043】
・PP/ZrPナノコンポジットの作製−溶解法
水溶液中で薄片化したZrP/TBAの作製は、早期に報告されている[文献41]。ZrPは、0.01gのZrP/TBAを含む水溶液の対数において0.6mlのHCl(pH=1)を加えることによってTBAから分離できる。精製したZrPナノ小板の沈殿を遠心分離で回収し、超音波処理を用いて水中で再分散した。その後、10重量%オクタデシルアミノ塩(CH3(CH2)17NH3+)を1g、水溶液中に添加することで、水溶液10g中、0.01gの精製されたZrPが変性された。その混合物を室温で1時間連続的に攪拌し、ZrPの表面をオクタデシルアミノ塩で完全に変性した。一旦、攪拌を停止すると、アミノ変性されたZrP(F−ZrP)が水溶液から沈殿した。次に、キシレン15gを水溶液中の沈殿に加え、F−ZrPをキシレン中に完全に分散させるために1時間超音波処理し、水をデカンテーションした。その後、1gのPPを15gのデカリン中に170℃で溶解した。均一な混合物を生成するために、F−ZrP/キシレン溶液をPP/デカリン溶液に攪拌しながら滴下した。溶媒を一部蒸発させて、その混合物をさらに30分間、170℃で攪拌した。最終生成物は、PP中にF−ZrPが分散した粘性のゲルである。ゲルを完全に乾燥することによって、F−ZrP/PPナノコンポジットを得ることができる。
【0044】
・PP/ZrP/CNTナノコンポジットの作製−溶解法
F−ZrP/キシレンとF−MWCNT/キシレンを分散する工程は上述した。約15gのキシレンをF−ZrPとF−MWCNT(固形分:ZrP0.01g、MWCNT0.0022g)に別々に加え、完全に分散させるために1時間超音波処理した。その後、2つの分散物をお互いに混合し、完全に分散させるために1時間超音波処理した。次に、1gのPPを15gのデカリン中に170℃で溶解した。均一混合物を形成するために、F−ZrP/F−MWCNT/キシレン溶液を攪拌しながらPP/デカリン溶液に滴下した。混合物を170℃でさらに30分間攪拌して溶媒を一部蒸発させた。最終生成物は、F−ZrP/F−MWCNTがPP中に分散した粘性のゲルである。ゲルを完全に乾燥することによって、F−ZrP/F−MWCNT/PPナノコンポジットが得られる。
【0045】
・透過型電子顕微鏡
CNT/ZrP PPナノコンポジットの顕微鏡観察用に、イソプロパノール中にマスターバッチを再分散し、微細分散物を得るために24時間超音波処理した。分散物を1滴、TEMのために炭素フィルムで被覆した銅格子上に置いた。ナノコンポジットの薄片を、射出成形試験片からReinzcutウルトラミクロトームを用いて切り出し、銅格子上に置いた。
CNT PPナノコンポジット用に、MWCNT/PPデカリン溶液の液滴を炭素フィルムで被覆した銅格子上に置いた。溶媒が全て除去されるまで、銅格子をホットプレート上で熱をかけて乾燥した。
JEOL 1200 EXを用いて透過電子顕微鏡観察(TEM)を実施した。
【0046】
・X線回折
ナノコンポジットのサンプルは、X線粉末回折計Bruker−AXS D8を用いて分析した。
【0047】
<ZrPナノコンポジットの作製>
・プラズマ処理されたポリプロピレン(PP)
100ミクロンのマイクロ粒子を含むポリプロピレン粉末を、大気圧下の空気及び窒素中、プラズマで処理した。処理工程の間に、COOH、C=O、C−O、NO2及びNO等の極性基を粒子の表面に導入した。次に、プラズマ処理したポリプロピレン(PT−PP)粒子をZrPナノ小板で変性した。
【0048】
・ZrP/プラズマ処理したPP分散物の作製
水中で薄片化したZrPナノ小板の原液を、上述のように、100mlの水に1gのZrPの濃度で作製した。サンプルP−PP−1用に、0.05gのZrPと5mlの原液をバイアルに作製した。0.1gのPT−PP粒子を、薄片化したα−ZrPナノ小板の溶液に加えた。サンプルP−PP−2用には、0.1ミリモルのTBAもさらに溶液に加えることを除き、同様の手順に従った。PT−PP粒子を含む溶液を0.5時間超音波処理し、その後、常温で2時間以上連続して攪拌した。底面に粒子を沈下させるために、水の3倍体積量のアセトンを溶液に加えた。通常、1時間後に溶液から粒子が完全に分離される。次に、上清を排出し、残った粒子を90℃での穏やかな加熱により乾燥した。乾燥した粒子は後に、特性評価及び熱加工に使用する。
【0049】
・ZrPナノコンポジットの作製
前のセクションに記載した方法で作製した乾燥PT−PP粒子を2枚の鋼板の間に挟み、加熱圧縮機(Dake)を用いて170℃で5分間圧縮し、厚さ200〜400ミクロンのポリマー薄板を形成した。
【0050】
・溶融混合したZrP PPナノコンポジットの作製
ZrPの分散をさらに改善するために、以下のようにZrP(ZrP−m−PTPP)によって変性されたPT−PP粒子をPPと混合した。バッチ式ミキサー内でZrP−m−PTPPをニートPPに加え、180℃で以下の通りに混合し、ZrPの凝集体を分散した:前記の手順に従って、ZrPによって変性されたPT−PP粒子(P−PP−1)とPPをHaakeミキサーを用いて60rpmで20分間混合した。0.06 2のP−PP−1粉末を40gのPPに加え、0.015重量%ZrP PPナノコンポジットを得た。このナノコンポジットはP−PP−8と表した。
【0051】
・電界放射型走査型電子顕微鏡
粒子を炭素テープで覆われたアルミニウムスタブの表面に置き、その粒子を、スパッタコーター(Cressington)を用いてアルゴン下で薄さ4nmのプラチナで被覆した。そのサンプルを電界放射型走査型電子顕微鏡によって画像化した(Quanta 600,FEI)。
【0052】
・透過型電子顕微鏡
ZrPで処理されたPT−PP粉末と1体積%3−グリシドキシプロピルトリメトキシシラン(Z−6040 Dow Chem.)のメタノール溶液10mlを遠心チューブに入れ、5分間遠心分離した。次に、溶液を吸い出し、遠心チューブの底に粉末を残した。5mlの酸化プロピレンを粉末に加えて振盪した後、遠心分離して上清を除去した。エポキシ樹脂は以下の処方に従って作製した:5.67gのドデシル無水コハク酸、2.48gのアラルダイト(商品名)502及び1.85gのQuetol(商品名)651(すべてElectron Microscopy Science EMS社製)。この処方を良く攪拌して均一に混合した。次に、0.2mlのベンジルジメチルアミン(EMS)を攪拌しながらその処方に加えた。エポキシ樹脂をシラン処理した粉末を含有する遠心チューブに注ぎ、55℃で一晩硬化させた。
【0053】
熱加圧されたZrPナノコンポジット薄板については、適当なサイズの試料を切り取り、下記記載のように3−グリシドキシプロピルトリメトキシシランで処理した。1体積%3−グリシドキシプロピルトリメトキシシラン(Z−6040 Dow Chem.)のメタノール溶液を調整した。この溶液を約10mlペトリ皿に注ぎ、ガラス容器に置いた。この試料をガラス容器に入れ、その容器をシールし、40℃で30分間加熱した。これによって、シラン溶液は蒸発し、この容器内は飽和状態になる。試料の表面は、エポキシ樹脂との結合を助けるシラン薄層でコーティングされる。次に、シラン処理した試料を遠心チューブに入れ、エポキシ樹脂をそのチューブに注ぐ。そして、エポキシ樹脂を55℃で一晩硬化させる。硬化したエポキシのブロックから薄片を作製し、銅格子上に置いた。
【0054】
P−PP−8については、圧縮成型したブロックを作製し、ウルトラミクロトームで薄片を作製した。その薄片を炭素フィルムで被覆した銅格子上に置いた。Cressingtonカーボンコーターを用いて、その薄片を10nmの炭素層でコーティングした。
【0055】
ライヘルト−ユングウルトラカットEウルトラミクロトームを用いてその薄片を切り出し、銅格子上に置いた。JEOL 1200 EXを用いて透過型電子顕微鏡観察(TEM)を行った。
P−PP−8のTEM画像ではマトリクス中でのZrPの均一な分布を示し(図7)、ZrP凝集体の崩壊の証拠が示された(図8)。
【0056】
<CNTナノコンポジットの作製>
・MWCNTの解きほぐし
本発明者らの以前の研究[文献20及び21]に記載された手順に従い、新しいMWCNTを酸化した。完全に薄片化したZrPのナノ小板を、わずかに酸化したMWCNT水溶液にCNT:ZrP=1:5の重量割合で加え、MWCNTを解きほぐし、分散させた。この混合物を室温で30分間超音波処理した(Branson 2510)。次に、酸を添加することによって、その溶液からZrPを除去し、MWCNTが残っている混合物の分離物を界面活性剤溶液で懸濁する。この方法で、最大500パーツ・パー・ミリオン(ppm)の濃度の調製に成功した。
【0057】
・F−MWCNTの作製
オクタデシルアミン粉末と良好に分散されたMWCNT溶液を直接混合することによってMWCNTを官能基化した。完全に反応させるために、その混合物を85−90℃で1時間継続的に攪拌し、オクタデシルアミンによって変性されたMWCNT(F−MWCNT)を水溶液から沈殿させた。その沈殿物を回収し、80℃のオーブンで一晩乾燥させた。
【0058】
・解きほぐされたMWCNT/PPナノコンポジットの作製
15グラムのキシレンを沈殿したF−MWCNTに加え、キシレン中でF−MWCNTを単独で分散するために1時間超音波処理した。次に、1グラムのPPを機械的に攪拌しながらそのF−MWNT/キシレン溶液に加えた。均一な混合物を生成するために、混合物を125℃で1時間攪拌した。エタノールを加えて、溶液からPP/F−MWCNTを強制的に沈殿させた。また、エタノールを使用して何度か表面を洗浄し、残留したキシレンを除去した。次に、最終的に得られたPP/F−MWNT粉末を80℃の真空オーブンで12時間乾燥させた。その粉末を180℃で1分間熱加圧し、電気伝導度測定用のPP/F−MWNTナノコンポジットのプラークを作製した。
【0059】
・形態的特性
透過型電子顕微鏡(TEM)の観察は、高解像度透過型電子顕微鏡JEOL 2010を用いて200kVで実施した。薄い炭素被膜を有する銅格子を溶液サンプルで覆い、室温で乾燥した。TEM画像解析用に、ライヘルトユングウルトラカット−Eミクロトームを用いて、ナノコンポジットバルクのサンプルを約80nmの薄さの薄切切片にした。Leo Zeiss 1530 VP Field Emission−SEM(FE−SEM)を用いてSEM画像を得た。
【0060】
・機械的試験
張力試験の試料は、ニートPPペレットと混合した溶液から得られたPP/F−MWCNT混合物を作製し、2分間、180℃で60rpmとしたHaakeミキサー(System40)を用いて、PP中のMWCNTを規定量にした。混合後に、その混合物を室温でゆっくり冷却した。張力試料は、溶融温度及び成形温度をそれぞれ195℃と90℃に固定し、射出速度0.25cm3/sで小型射出成形機(CS−183MMX)を用いて成形した。張力試験用に、射出成形試験片を機械加工し、ASTM D638−08規格に従い特性評価を行なった。クロスヘッド速度を5mm/分として、MTSスクリュー駆動試験機で室温での引張試験を実施した。校正したMTS伸縮計(model632.12B−50)を用いて真の張力を測定した。1サンプルにつき最低5試料の測定に基づいて計測した平均弾性係数と引張強度を標準誤差と共に報告する。
【0061】
・MWCNTの分散
通常、MWCNTは、MWCNTのチューブの長さとチューブの欠陥による特有の湾曲のために、合成後に高密度な絡み合いを形成する。以前、溶液中及びポリマーマトリクス中の両方で、完全に薄片化したZrPナノ小板をCNTの分散と薄片化に使用することに成功した[文献20及び21]。ナノ小板の静電荷を遮断するために酸を添加することによって溶液からナノ小板を容易に除去できる。アセトンと水でチューブを洗浄した後、MWCNTを水中に再分散し、高い解きほぐし状態を維持する。図9に本発明のMWCNT製造前後のTEM画像を示す。図9a−bでは、わずかに酸化したMWCNTが絡み合ったままで残っている。ここで説明した解きほぐしの処理後、TEMを用いた直接観察では、MWCNTの凝集又は絡み合いの証拠は一切なかった。MWCNTは良好に分散し、およそ0.5〜10μmの長さの曲線形を有した(図9c−d)。
【0062】
・オクタデシルアミンの官能基化
良好な分散を達成し、ポリマーマトリックを用いてMWNTの湿潤を促進するために、オクタデシルアミン粉末をMWCNT水溶液に直接混合によって加えた。図10a−bに示すように、良好に分散されたMWCNT水溶液中へのオクタデシルアミン粉末の添加は、MWCNT表面とアルキル基との間の特徴的な−COO−+NH3−結合によるオクタデシルアミン側鎖のイオン付加をもたらす。また、わずかに酸化したMWCNTとF−MWCNTの比較により双性イオン形成を示すIRスペクトルを得た。図11に示すように、1564cm−1のピークは、カルボン酸陰イオンの伸縮振動を示す。また、2842cm−1と2922cm−1で示されたピークは、オクタデシルアミンアルキル鎖中のC−H伸縮振動による。したがって、PP中のF−MWCNTの良好な分散が期待できる。以前の報告[文献17−19]とは対照的に、F−MWCNTは、ほんの少しの超音波処理によって有機溶媒中で容易に再分散でき、ポリマーマトリックスを混合する前に、均一な分散状態を示す。キシレン中のF−MWNTのTEM画像は、100ppmの濃度で、非常に良好な分散と完全な解きほぐし状態を示す(図12)。MWNTの良好な分散は、オクタデシルアミンの官能基の親有機性の増加のためであると考えられる。また、MWCNT表面のアルキル末端(alkyl tail)もPP中の分散に役立つ。
【0063】
・PP/F−MWCNTナノコンポジットの作製
PP/F−MWCNTナノコンポジットは、PPペレットを125℃のF−MWNT/キシレン溶液に直接添加することによって作製した(図10b−c)。MWCNTの濃度は、0.1〜2.0重量%の間でコントロールした。PPペレットを機械的に連続攪拌して溶解した。F−MWCNTが0.5重量%のMWCNTにおいてPP薄膜中で良好に分散されることが、TEM画像により確認された(図13)。反対に、他の研究では、ポリマー中のCNTの安定性と分散性を改良するためにアルキル短鎖をMWCNT表面に結合できることが示されたが[文献17及び18]、ポリマーマトリクス中でのMWCNTの良好な分散又は解きほぐしに関する証拠は文献中では示されていない。図10c−dに示すように、蒸発によってキシレン溶液を除去することによりPP/F−MWCNTナノコンポジットを得た。サンプルは80℃で一晩乾燥した。導電性とTEM顕微鏡観察用のナノコンポジット薄膜は、乾燥後のサンプルを熱加圧することによって作製した。
【0064】
PP/F−MWCNT薄片のTEM画像は、溶媒の除去後でも分散性の質が維持されることを示している。図14に示されるように、PP中に0.1、0.6、1及び2重量%のMWCNTを含むPPは非常に良好な分散性を示し、本発明の手法が、各々のチューブレベルで、PP中でのMWCNT単独の分散を促進するのに効果的であることを強く示唆する。
【0065】
・PP/CNTナノコンポジットの機械的特性
単軸引張の下で検出されるPP/F−MWCNTナノコンポジットの機械的特性を図15に示す。これらの結果は、ヤング率の50%増加と測定引張り強度の17%増加により、0.1重量%の低さのMWCNT濃度で著しい機械的強化が実現されることを示している(表2)。また、未処理のMWCNTのPP中での分散性を用いて対照実験を実施した。これらの系は大きな凝集を示し、ヤング率と引張り強度においてわずかな改善を示した(ニートPPに対してそれぞれ14%と5%の改善)。文献[22、23]で報告されたものと比べて、上記の結果は重要である。
【0066】
MWCNTの少量の添加が、係数と強度における顕著な改善に寄与している強化のメカニズムを調べるために、ニートPPとPP/MWCNTナノコンポジットの両方の引張破断面のSEM画像を得た(図16)。ニートPPは高延性を示し、完全なネッキングが起こるまで破砕しない(図16a,b)。PP/F−MWCNTは、かなり異なった性質を示す。それは、ネック形成の間に裂け、0.1重量%F−MWCNTでは、直径約5μmの破断表面上に広がったミクロサイズの継ぎ目(patch)を示す。慎重な調査によって、MWCNTの引き抜きがないことから、MWCNTとPPマトリクス間の強い界面結合が示唆されることが示された(図16c,d)。また、射出成形の工程の間にもたらされる配置構造や結晶化の間の核生成に起因する強化もあり得る。
【0067】
さらに、本発明のナノコンポジットは、低いCNT添加量で、係数の高い改良を示す(後述の表2)。また、本発明の組成物は、射出成形の後に形状を保持する優れた寸法安定性を示す。CNTを含有するPPは、成形型のほぼ角柱形状を示すが、一方、ニートPPは中央部に大きな収縮がある。Keyence VK−9700共焦点レーザー顕微鏡を使用した共焦点レーザー顕微鏡法で、(a)ニートPP、(b)0.1重量%MWCNTを含むPP、及び(c)0.4重量%MWCNTを含むPPについて、長さ74mm×幅13mm×厚さ3mmの棒状ブランクの成形収縮の測定を行い、厚さ方向の収縮を得て(棒状ブランクの端部と、棒状ブランクの中央部を比較して、その間の厚さの違いを百分率で評価した;棒状ブランクは、上記の機械的試験のものと同様の方法で作製した)、以下の表に値を示し、それぞれ、図18(a)−(c)に表した。
【0068】
【表2】
【0069】
したがって、本発明のナノコンポジットの好適な実施態様は、95〜99.7重量%のポリオレフィン(最も好ましくはポリプロピレン)、及び0.3〜5重量%のナノチューブを含有し、ヤング率が2.0GPaを超え、そして厚さ方向の成形収縮が、純粋なポリオレフィンの成形収縮の4分の1よりも小さいことである。
【0070】
・PP/CNTナノコンポジットの導電性
未処理のMWCNTとF−MWCNTの存在に起因した濃度変化による電気伝導性の変化は、1Vでの表面伝導率に基づいて測定された(図17)。PP/MWCNTナノコンポジットは、0.1〜2重量%の間で、溶液を蒸発させた後のサンプルの直接的な熱加圧により作製された。結晶化の間、MWCNTの凝集と弱いネットワークの破壊のために、PP/P−MWCNT混合物の電気的パーコレーションは、ほぼ2重量%となった。一方、PP/F−MWCNTナノコンポジットは、2.3×10−6S/mの導電率を示す0.6重量%において、絶縁体−導体パーコレーション転移を示した。図17中の挿入画は、MWCNT分散状態の概念的な解釈を提供する。
【0071】
低濃度では、PPマトリクスを介して導電性パスを形成するための十分なMWCNTがない。浸透限界では、単一の電気経路が形成され、PPマトリクス中のチューブが結合したネットワークに沿って電子が動き回る。さらに濃度が高くなると電気経路がさらに形成されて、系内の他の経路に接続し始め、システムが結合されることにより、べき乗則の挙動を示す。電気的パーコレーションにおける充填量は、[24]によれば、非溶融状態のPP/CNT複合体の値が最も低い。このことは、通常、電気転移のための凝集したネットワークに依存する非結晶質系における所見と対照的である[25,26]。これは、F−MWCNTが、結晶化の間にPPのラメラ構造に組み込まれて、核生成剤として働くことを示しており、表2の結晶化度の測定値によって裏付けられている。また、この挙動は、観測された弾性係数と引張り強度の大幅な増加を、部分的に説明することができる。
【0072】
【表3】
【先行技術文献】
【非特許文献】
【0073】
【非特許文献1】R.Saito,G.Dresselhaus,M.Dresselhaus,Physical properties of carbon nanotubes,Imperial College Pr,1998.
【非特許文献2】M.Dresselhaus,G.Dresselhaus,P Avouris,Carbon nanotubes:synthesis,structure,properties,and applications,Springer Verlag,2001.
【非特許文献3】X.Xie,Y.Mai,X.Zhou,Materials Science and Engineering:R:Reports,49(2005)89−112.
【非特許文献4】X.Gong,J.Liu,S.Baskaran,R.Voise,J.Young,Chem.Mater,12(2000)1049−1052.
【非特許文献5】M.O’Connell,P.Boul,L.Ericson,C.Huffman,Y.Wang,E.Haroz,C.Kuper,J.Tour,K.Ausman,R.Smalley,Chemical Physics Letters,342(2001)265−271.
【非特許文献6】V.Georgakilas,K.Kordatos,M.Prato,D.Guldi,M.Holzinger,A.Hirsch,J.Am.Chem.Soc,124(2002)760−761.
【非特許文献7】C.Dyke,J.Tour,J.Phys.Chem.A,108(2004)11151−11159.
【非特許文献8】R.Blake,Y.Gun’ko,J.Coleman,M.Cadek,A.Fonseca,J.Nagy,W.Blau,J.Am.Chem.Soc,126(2004)10226−10227.
【非特許文献9】D.Tasis,N.Tagmatarchis,A.Bianco,M.Prato,Chem.Rev,106(2006)1105−1136.
【非特許文献10】R.Chen,Y.Zhang,D.Wang,H.Dai,J.Am.Chem.Soc,123(2001)3838−3839.
【非特許文献11】J.Chen,H.Liu,W.Weimer,M.Halls,D.Waldeck,G.Walker,J.Am.Chem.Soc,124(2002)9034−9035.
【非特許文献12】J.Chen,M.A.Hamon,H.Hu,Y.Chen,A.M.Rao,P.C.Eklund,R.C.Haddon,Science,282(1998)95−98.
【非特許文献13】M.A.Hamon,J.Chen,H.Hu,Y.Chen,M.E.Itkis,A.M.Rao,P.C.Eklund,R.C.Haddon,Adv.Mater.11(1999)834−840.
【非特許文献14】D.Chattopadhyay,S.Lastella,S.Kim,F.Papadimitrakopoulos,124(2002)728−829.
【非特許文献15】D.Chattopadhyay,I.Galeska,F.Papadimitrakopoulos,J.Am.Chem.Soc.125(2003)3370−3375.
【非特許文献16】J.Chen,A.M.Rao,S.Lyuksyutov,M.E.Itkis,M.A.Hamon,H.Hu,R.W.Cohn,P.C.Eklund,D.T.Colbert,R.E.Smalley,R.C.Haddon,J.Phys.Chem.B105(2001)2525−2528.
【非特許文献17】A.Koval’chuk,V.Shevchenko,A.Shchegolikhin,P.Nedorezova,A.Klyamkina,A.Aladyshev,Macromolecules,41(2008)7536−7542.
【非特許文献18】A.Koval’chuk,A.Shchegolikhin,V.Shevchenko,P.Nedorezova,A.Klyamkina,A.Aladyshev,Macromolecules,41(2008)3149−3156.
【非特許文献19】J.Lee,S.Yang,H.Jung,Macromolecules,42(2009)8328−8334.
【非特許文献20】D.Sun,W.Everett,C.Chu,H.−J.Sue,Small,5(2009)2692−2697.
【非特許文献21】D.Sun,C.Chu,and H.−J.Sue,Chem.Mater.,22(2010)3773−3778.
【非特許文献22】G.Lee,S.Jagannathan,H.Chae,M.Minus,S.Kumar,Polymer,49(2008)1831−1840.
【非特許文献23】B.Yang,J.Shi,K.Pramoda,S.Goh,Composites Science and Technology,68(2008)2490−2497.
【非特許文献24】W.Bauhofer,J.Z.Kovacs,Composites Science and Technology,69(2009)1486−1498.
【非特許文献25】J.Sandler,J.Kirk,I.Kinloch,M.Shaffer,A.Windle,Polymer,44(2003)5893−5899.
【非特許文献26】C.Martin,J.Sandler,M.Shaffer,M.Schwarz,W.Bauhofer,K.Schulte,A.Windle,Composites Science and Technology,64(2004)2309−2316.
【非特許文献27】A.Usuki et al.,J Mater Res 8,1 179(1993).
【非特許文献28】Y.Kojima et al.,J Mater Res 8,1 185(1993).
【非特許文献29】A.Usuki et al.,Journal of Applied Polymer Science 63,137(1997).
【非特許文献30】M.Kawasumi et al.,Macromolecules 30,6333(1997).
【非特許文献31】N.Hasegawa et al.,Journal of Applied Polymer Science 67,87(1998).
【非特許文献32】M.Kato,A.Usuki,and A.Okada,Journal of Applied Polymer Science 66,1781(1997).
【非特許文献33】T.Kashiwagi et al.,Macromolecular Rapid Communications 23,761(2002).
【非特許文献34】S.P.Bao,and S.C.Tjong,Mat Sci Eng a−Struct 485,508(2008).
【非特許文献35】R.Blake et al.,Journal of the American Chemical Society 126,10226(2004).
【非特許文献36】S.H.Lee et al.,Carbon 45,281 0(2007).
【非特許文献37】S.C.Tjong,G.D.Liang,and S.P.Bao,Scripta Materialia 57,461(2007).
【非特許文献38】S.B.Kharchenko et al.,Nature Materials 3,564(2004).
【非特許文献39】T.McNally et al.,Polymer 46,8222(2005).
【非特許文献40】L.Y.Sun et al.,New Journal of Chemistry 31,39(2007).
【非特許文献41】D.M.Kaschak et al.,Journal of the American Chemical Society 120,10887(1998).
【非特許文献42】L.Y.Sun et al.,Chemistry of Materials 19,1749(2007).
【非特許文献43】H.−J.Sue et al.,US Published Patent Application 2009/0035469,filed April 30,2008.
【0074】
本発明のさらなる修正及び変形は、上記の教示の観点から明らかに可能である。したがって、添付されたクレームの範囲内において、本明細書に具体的に記載されたもの以外の方法で本発明を実施できることが理解される。
【特許請求の範囲】
【請求項1】
ポリオレフィン中でナノチューブ及び/又はナノ小板を分散する方法であって、以下の工程を含む方法:
A)ナノチューブもしくはナノ小板又はその両方を含む溶液を調製する工程;
B)工程(A)により得られた溶液を攪拌する工程;
C)工程(B)で攪拌された溶液中に1種以上のポリマー材料を溶解し、さらにその溶液から沈殿物を単離する工程;及び、
D)1種以上のポリオレフィンと前記沈殿物を溶融混合する工程。
【請求項2】
前記工程(A)の溶液が、さらに、長鎖脂肪族アミンと無水マレイン酸で変性されたポリプロピレンオリゴマーから成る群から選択される少なくとも1種の分散剤を含む請求項1に記載の方法。
【請求項3】
前記分散剤が、少なくとも1種の長鎖脂肪族アミンである請求項2に記載の方法。
【請求項4】
前記工程(A)の溶液が、ナノチューブを含む請求項1に記載の方法。
【請求項5】
前記工程(A)の溶液が、ナノ小板を含む請求項1に記載の方法。
【請求項6】
前記工程(A)の溶液が、ナノチューブとナノ小板の両方を含む請求項1に記載の方法。
【請求項7】
前記ナノチューブが、カーボンナノチューブ、二酸化タングステンナノチューブ、シリコンナノチューブ、無機ナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項4に記載の方法。
【請求項8】
前記ナノチューブが、カーボンナノチューブ、二酸化タングステンナノチューブ、シリコンナノチューブ、無機ナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項6に記載の方法。
【請求項9】
前記ナノチューブが、乾燥酸化、放射酸化、プラズマ酸化、熱酸化、拡散酸化及びこれらの組み合わせから成る群から選択されるいずれかの方法により酸化される請求項4に記載の方法。
【請求項10】
前記ナノチューブが、カーボンナノチューブである請求項7に記載の方法。
【請求項11】
前記カーボンナノチューブが、多層カーボンナノチューブ、単層カーボンナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項10に記載の方法。
【請求項12】
前記ナノ小板が、クレイ、ナノクレイ、グラフェン、無機結晶、有機結晶及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項5に記載の方法。
【請求項13】
前記ナノ小板が、クレイ、ナノクレイ、グラフェン、無機結晶、有機結晶及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項6に記載の方法。
【請求項14】
前記工程(C)の溶解の前に、前記工程(B)で攪拌された溶液中から前記ナノ小板を回収する工程を含む請求項6に記載の方法。
【請求項15】
前記1種以上のポリマー材料が、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエステルカーボネートコポリマー、ポリ(エステル−カーボネート)樹脂、ポリアミド、高温ポリアミド、ポリエチレン、ポリプロピレン、オレフィンのコポリマー、官能基化ポリオレフィン、ハロゲン化ビニルポリマー、ビニリデンポリマー、ポリ塩化ビニリデン、ポリフッ化ビニル、ポリフッ化ビニリデン、ポリアミドコポリマー、ポリアクリロニトリル、ポリエーテル、ポリケトン、熱可塑性ポリイミド、変性セルロース及びこれらポリマー材料を1種以上含む混合物から成る群から選択される少なくとも1種である請求項1に記載の方法。
【請求項16】
前記工程(D)の溶融混合が、沈殿物を、少なくとも1種のポリオレフィンと、フィラー、強化剤、可塑剤、酸化防止剤、熱安定剤、紫外線安定剤、強靭化剤、帯電防止剤、難燃剤、着色剤及びこれら添加物を1種以上含む組み合わせから成る群から選択される1種以上の添加物と共に溶融混合する請求項1に記載の方法。
【請求項17】
前記少なくとも1種のポリオレフィンが、ポリエチレン、ポリプロピレン、それらの混合物及びそれらのコポリマーから成る群から選択される少なくとも1種である請求項1に記載の方法。
【請求項18】
前記工程(C)の溶解工程が、さらに、溶液に超音波をかけ、次いで冷却して沈殿物を生成する工程を含む請求項1に記載の方法。
【請求項19】
溶融混合の前に、前記沈殿物を乾燥する工程をさらに含む請求項18に記載の方法。
【請求項20】
前記1種以上のポリマー材料が、ポリプロピレンである請求項1に記載の方法。
【請求項21】
前記工程(A)で作製される溶液が、表面改質されたポリプロピレンをさらに含む請求項1に記載の方法。
【請求項22】
前記表面改質されたポリプロピレンがプラズマ処理されたポリプロピレンである請求項21に記載の方法。
【請求項23】
前記少なくとも1種のポリオレフィンが、ポリプロピレンである請求項17に記載の方法。
【請求項24】
前記少なくとも1種のポリオレフィンが、粒子状、繊維状又はチューブ状である請求項1に記載の方法。
【請求項25】
前記工程(C)の溶解工程が、前記1種以上のポリマー材料の溶解の前に、さらに、非極性溶媒を添加する工程を含む請求項1に記載の方法。
【請求項26】
前記ナノチューブ及び前記ナノ小板が、長鎖脂肪族アミン及び無水マレイン酸で変性されたポリプロピレンオリゴマーから成る群から選択される少なくとも1種の分散剤との反応により、それぞれ表面改質されている請求項6に記載の方法。
【請求項27】
前記工程(A)の溶液が、キシレン、デカリン、ブタノール、ジクロロベンゼン、トリクロロベンゼン、N,N−ジメチルホルムアミド及びイソプロパノール等の有機溶剤を含む請求項1に記載の方法。
【請求項28】
請求項1に記載の方法により製造されるナノコンポジット。
【請求項29】
請求項27に記載のナノコンポジットを押出成形、射出成形、延伸ブロー成形又は熱成形する工程を含む、製品の形成方法。
【請求項30】
95〜99.7重量%のポリオレフィンと、0.3〜5重量%のナノチューブ及び/又はナノ小板とを含み、10−6S/mより高い表面電気伝導率を有するナノコンポジット。
【請求項31】
95〜99.7重量%のポリオレフィンと、0.3〜5重量%のナノチューブ及び/又はナノ小板とを含み、2.0GPaより高いヤング率を有するナノコンポジット。
【請求項32】
厚さ方向の成形収縮が、純粋なポリオレフィンの成形収縮の4分の1よりも小さい請求項31に記載のナノコンポジット。
【請求項33】
ポリオレフィン中にカーボンナノチューブを分散する方法であって、
−カーボンナノチューブを含む水分散液を調製する工程;
−少なくとも1種の長鎖脂肪族アミンをカーボンナノチューブを含む前記水分散液と混合することにより前記カーボンナノチューブを官能基化し、官能基化カーボンナノチューブの水分散液を調製する工程;
−前記水分散液から水を除去し、前記官能基化カーボンナノチューブを単離する工程;
−単離された前記官能基化カーボンナノチューブと非極性有機溶媒とを混合し、官能基化カーボンナノチューブの有機溶液を調製する工程;
−前記官能基化カーボンナノチューブの有機溶液と少なくとも1種のポリマー材料を混合し、前記有機溶媒を除去し、少なくとも1種のポリマー材料中の官能基化カーボンナノチューブの沈澱を単離する工程;及び
−少なくとも1種のポリオレフィンと前記沈澱とを溶融混合する工程
を含む方法。
【請求項34】
ポリオレフィン中にナノ小板を分散する方法であって、
−ナノ小板を含む水分散液を調製する工程;
−少なくとも1種の長鎖脂肪族アミンを前記ナノ小板を含む水分散液と混合することによりナノ小板を官能基化し、官能基化ナノ小板の水分散液を調製する工程;
−前記水分散液から水を除去し、前記官能基化ナノ小板を単離する工程;
−単離された前記官能基化ナノ小板と非極性有機溶媒とを混合し、官能基化ナノ小板の有機溶液を調製する工程;
−前記官能基化ナノ小板の有機溶液と少なくとも1種のポリマー材料とを混合し、前記有機溶媒を除去し、少なくとも1種の前記ポリマー材料中の官能基化ナノ小板の沈澱を単離する工程;及び
−少なくとも1種のポリオレフィンと前記沈澱とを溶融混合する工程
を含む方法。
【請求項1】
ポリオレフィン中でナノチューブ及び/又はナノ小板を分散する方法であって、以下の工程を含む方法:
A)ナノチューブもしくはナノ小板又はその両方を含む溶液を調製する工程;
B)工程(A)により得られた溶液を攪拌する工程;
C)工程(B)で攪拌された溶液中に1種以上のポリマー材料を溶解し、さらにその溶液から沈殿物を単離する工程;及び、
D)1種以上のポリオレフィンと前記沈殿物を溶融混合する工程。
【請求項2】
前記工程(A)の溶液が、さらに、長鎖脂肪族アミンと無水マレイン酸で変性されたポリプロピレンオリゴマーから成る群から選択される少なくとも1種の分散剤を含む請求項1に記載の方法。
【請求項3】
前記分散剤が、少なくとも1種の長鎖脂肪族アミンである請求項2に記載の方法。
【請求項4】
前記工程(A)の溶液が、ナノチューブを含む請求項1に記載の方法。
【請求項5】
前記工程(A)の溶液が、ナノ小板を含む請求項1に記載の方法。
【請求項6】
前記工程(A)の溶液が、ナノチューブとナノ小板の両方を含む請求項1に記載の方法。
【請求項7】
前記ナノチューブが、カーボンナノチューブ、二酸化タングステンナノチューブ、シリコンナノチューブ、無機ナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項4に記載の方法。
【請求項8】
前記ナノチューブが、カーボンナノチューブ、二酸化タングステンナノチューブ、シリコンナノチューブ、無機ナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項6に記載の方法。
【請求項9】
前記ナノチューブが、乾燥酸化、放射酸化、プラズマ酸化、熱酸化、拡散酸化及びこれらの組み合わせから成る群から選択されるいずれかの方法により酸化される請求項4に記載の方法。
【請求項10】
前記ナノチューブが、カーボンナノチューブである請求項7に記載の方法。
【請求項11】
前記カーボンナノチューブが、多層カーボンナノチューブ、単層カーボンナノチューブ及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項10に記載の方法。
【請求項12】
前記ナノ小板が、クレイ、ナノクレイ、グラフェン、無機結晶、有機結晶及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項5に記載の方法。
【請求項13】
前記ナノ小板が、クレイ、ナノクレイ、グラフェン、無機結晶、有機結晶及びこれらの組み合わせから成る群から選択される少なくとも1種である請求項6に記載の方法。
【請求項14】
前記工程(C)の溶解の前に、前記工程(B)で攪拌された溶液中から前記ナノ小板を回収する工程を含む請求項6に記載の方法。
【請求項15】
前記1種以上のポリマー材料が、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエステルカーボネートコポリマー、ポリ(エステル−カーボネート)樹脂、ポリアミド、高温ポリアミド、ポリエチレン、ポリプロピレン、オレフィンのコポリマー、官能基化ポリオレフィン、ハロゲン化ビニルポリマー、ビニリデンポリマー、ポリ塩化ビニリデン、ポリフッ化ビニル、ポリフッ化ビニリデン、ポリアミドコポリマー、ポリアクリロニトリル、ポリエーテル、ポリケトン、熱可塑性ポリイミド、変性セルロース及びこれらポリマー材料を1種以上含む混合物から成る群から選択される少なくとも1種である請求項1に記載の方法。
【請求項16】
前記工程(D)の溶融混合が、沈殿物を、少なくとも1種のポリオレフィンと、フィラー、強化剤、可塑剤、酸化防止剤、熱安定剤、紫外線安定剤、強靭化剤、帯電防止剤、難燃剤、着色剤及びこれら添加物を1種以上含む組み合わせから成る群から選択される1種以上の添加物と共に溶融混合する請求項1に記載の方法。
【請求項17】
前記少なくとも1種のポリオレフィンが、ポリエチレン、ポリプロピレン、それらの混合物及びそれらのコポリマーから成る群から選択される少なくとも1種である請求項1に記載の方法。
【請求項18】
前記工程(C)の溶解工程が、さらに、溶液に超音波をかけ、次いで冷却して沈殿物を生成する工程を含む請求項1に記載の方法。
【請求項19】
溶融混合の前に、前記沈殿物を乾燥する工程をさらに含む請求項18に記載の方法。
【請求項20】
前記1種以上のポリマー材料が、ポリプロピレンである請求項1に記載の方法。
【請求項21】
前記工程(A)で作製される溶液が、表面改質されたポリプロピレンをさらに含む請求項1に記載の方法。
【請求項22】
前記表面改質されたポリプロピレンがプラズマ処理されたポリプロピレンである請求項21に記載の方法。
【請求項23】
前記少なくとも1種のポリオレフィンが、ポリプロピレンである請求項17に記載の方法。
【請求項24】
前記少なくとも1種のポリオレフィンが、粒子状、繊維状又はチューブ状である請求項1に記載の方法。
【請求項25】
前記工程(C)の溶解工程が、前記1種以上のポリマー材料の溶解の前に、さらに、非極性溶媒を添加する工程を含む請求項1に記載の方法。
【請求項26】
前記ナノチューブ及び前記ナノ小板が、長鎖脂肪族アミン及び無水マレイン酸で変性されたポリプロピレンオリゴマーから成る群から選択される少なくとも1種の分散剤との反応により、それぞれ表面改質されている請求項6に記載の方法。
【請求項27】
前記工程(A)の溶液が、キシレン、デカリン、ブタノール、ジクロロベンゼン、トリクロロベンゼン、N,N−ジメチルホルムアミド及びイソプロパノール等の有機溶剤を含む請求項1に記載の方法。
【請求項28】
請求項1に記載の方法により製造されるナノコンポジット。
【請求項29】
請求項27に記載のナノコンポジットを押出成形、射出成形、延伸ブロー成形又は熱成形する工程を含む、製品の形成方法。
【請求項30】
95〜99.7重量%のポリオレフィンと、0.3〜5重量%のナノチューブ及び/又はナノ小板とを含み、10−6S/mより高い表面電気伝導率を有するナノコンポジット。
【請求項31】
95〜99.7重量%のポリオレフィンと、0.3〜5重量%のナノチューブ及び/又はナノ小板とを含み、2.0GPaより高いヤング率を有するナノコンポジット。
【請求項32】
厚さ方向の成形収縮が、純粋なポリオレフィンの成形収縮の4分の1よりも小さい請求項31に記載のナノコンポジット。
【請求項33】
ポリオレフィン中にカーボンナノチューブを分散する方法であって、
−カーボンナノチューブを含む水分散液を調製する工程;
−少なくとも1種の長鎖脂肪族アミンをカーボンナノチューブを含む前記水分散液と混合することにより前記カーボンナノチューブを官能基化し、官能基化カーボンナノチューブの水分散液を調製する工程;
−前記水分散液から水を除去し、前記官能基化カーボンナノチューブを単離する工程;
−単離された前記官能基化カーボンナノチューブと非極性有機溶媒とを混合し、官能基化カーボンナノチューブの有機溶液を調製する工程;
−前記官能基化カーボンナノチューブの有機溶液と少なくとも1種のポリマー材料を混合し、前記有機溶媒を除去し、少なくとも1種のポリマー材料中の官能基化カーボンナノチューブの沈澱を単離する工程;及び
−少なくとも1種のポリオレフィンと前記沈澱とを溶融混合する工程
を含む方法。
【請求項34】
ポリオレフィン中にナノ小板を分散する方法であって、
−ナノ小板を含む水分散液を調製する工程;
−少なくとも1種の長鎖脂肪族アミンを前記ナノ小板を含む水分散液と混合することによりナノ小板を官能基化し、官能基化ナノ小板の水分散液を調製する工程;
−前記水分散液から水を除去し、前記官能基化ナノ小板を単離する工程;
−単離された前記官能基化ナノ小板と非極性有機溶媒とを混合し、官能基化ナノ小板の有機溶液を調製する工程;
−前記官能基化ナノ小板の有機溶液と少なくとも1種のポリマー材料とを混合し、前記有機溶媒を除去し、少なくとも1種の前記ポリマー材料中の官能基化ナノ小板の沈澱を単離する工程;及び
−少なくとも1種のポリオレフィンと前記沈澱とを溶融混合する工程
を含む方法。
【図2】

【図3】
【図4】

【図5】
【図6】
【図7】

【図8】

【図9】

【図10】

【図11】
【図12】
【図13】
【図14】
【図15】
【図16】

【図17】

【図18】

【図1】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図1】
【公表番号】特表2013−515847(P2013−515847A)
【公表日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願番号】特願2012−547240(P2012−547240)
【出願日】平成22年12月28日(2010.12.28)
【国際出願番号】PCT/US2010/062236
【国際公開番号】WO2011/082169
【国際公開日】平成23年7月7日(2011.7.7)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.パイレックス
【出願人】(596133485)日本ポリプロ株式会社 (577)
【Fターム(参考)】
【公表日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願日】平成22年12月28日(2010.12.28)
【国際出願番号】PCT/US2010/062236
【国際公開番号】WO2011/082169
【国際公開日】平成23年7月7日(2011.7.7)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.パイレックス
【出願人】(596133485)日本ポリプロ株式会社 (577)
【Fターム(参考)】
[ Back to top ]